Embed Size (px)
DESCRIPTION
高中化学竞赛. 【 第十九讲 分析化学 】. 河南省太康县第一高级中学 ---- 乔纯杰. 【 竞赛基本要求 】. 1 、在化学计算和化学实验中正确使用有效数字。 2 、定量仪器(天平、量筒、移液管、滴定管、容量瓶等等)测量数据的有效数字。运算结果的有效数字。常见容量分析的基本概念(被测物、基准物质、标准溶液、滴定反应等)。 3 、酸碱滴定的滴定曲线(酸碱强度、浓度、溶剂极性对滴定突跃影响的定性关系)。酸碱滴定指示剂的选择。高锰酸钾、重铬酸钾、硫代硫酸钠、 EDTA 为标准溶液的基本滴定反应。 4 、常见容量分析的基本操作、基本反应及分析结果的计算。容量分析的误差分析。. - PowerPoint PPT Presentation
Citation preview

河南省太康县第一高级中学河南省太康县第一高级中学 -------- 乔纯乔纯杰杰
高中化学竞赛高中化学竞赛 【第十九讲 分析化学】【第十九讲 分析化学】

【竞赛基本要求】【竞赛基本要求】
1 、在化学计算和化学实验中正确使用有效数字。2 、定量仪器(天平、量筒、移液管、滴定管、容量瓶等等)测量数据的有效数字。运算结果的有效数字。常见容量分析的基本概念(被测物、基准物质、标准溶液、滴定反应等)。3 、酸碱滴定的滴定曲线(酸碱强度、浓度、溶剂极性对滴定突跃影响的定性关系)。酸碱滴定指示剂的选择。高锰酸钾、重铬酸钾、硫代硫酸钠、 EDTA 为标准溶液的基本滴定反应。4 、常见容量分析的基本操作、基本反应及分析结果的计算。容量分析的误差分析。

【知识点击】一、容量分析的基本概念和原理 待测物质中有关组分含量的测定是定量分析要作的工作。
测定的化学原理是化学反应:待测组分 X + 试剂 R = 产物 P 容量分析是通过消耗试剂体积从而知道物质的量,按以上
反应的化学计量关系求算待测量的。 作滴定分析时,将一定体积的待测溶液置于锥形瓶中,将
已知浓度的试剂通过滴定管滴加到锥形瓶中,当达到滴定终点时,准确读得所加试剂的体积,求得所加试剂的物质的量,从而求出待测组分的物质的量浓度。也可以倒过来把待测溶液从滴定管中滴入锥形瓶中的标准溶液中。那么,滴定终点如何确定呢?这是在滴定反应中最为关心的问题——指示剂的选择。滴定终点即指示剂的变色点。由于指示剂的变色点和化学反应的计量点往往不一致,由此造成的误差称为终点误差或系统误差。
由于滴定分析中要测体积,故把滴定分析又叫容量分析。滴定分析的方法通常有:直接滴定法、返滴定法、间接滴定法。
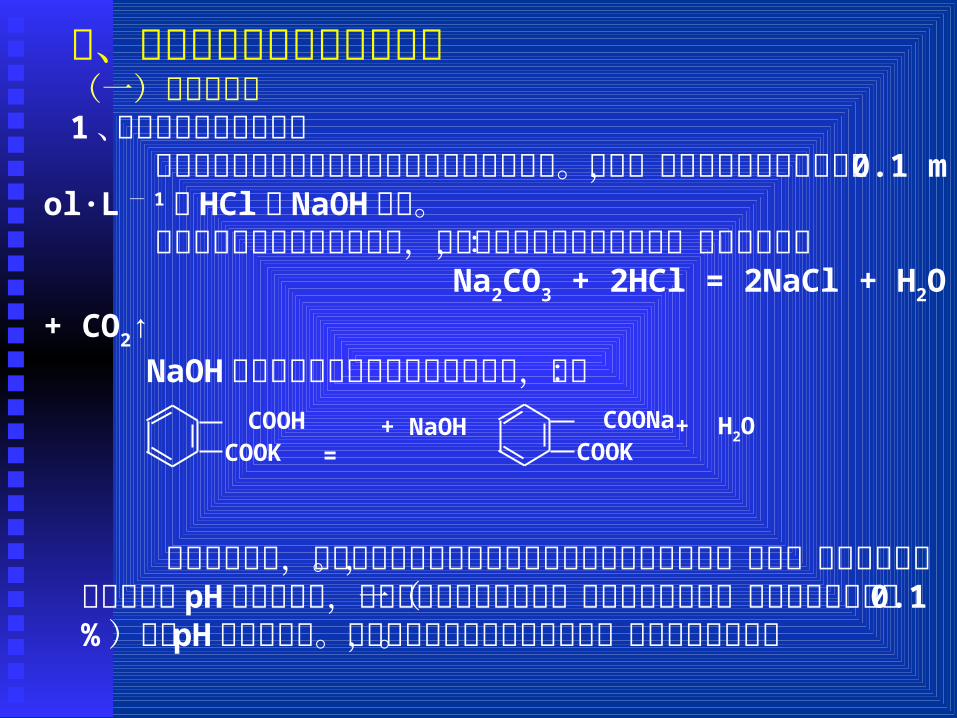
二、常见容量分析方法及其应用(一)酸碱滴定法1、酸碱滴定法的基本原理 酸碱滴定法是以酸碱反应为基础的滴定分析方法。其中,常用的标准溶液是浓度为 0.1 mol·L - 1的 HCl和 NaOH溶液。
盐酸是由基准物碳酸钠标定的,碳酸钠的量是由称量而来的,标定反应为:
Na2CO3 + 2HCl = 2NaCl + H2O + CO2↑ NaOH是用称得的邻苯二甲酸氢钾标定的,即:
COOH
COOK + NaOH = COONa
COOK + H2O
在酸碱滴定中,最重要的是选择最合适的指示剂来指示滴定终点。为此,必须了解滴定过程中溶液 pH 的变化情况,特别是化学计量点前后一定准确度范围内(如相对误差为正负 0.1% )溶液 pH 的变化情况。下面通过实例来说明滴定原理,并说明滴定曲线。

酸起始的物质的量加入碱的物质的量
总
总
Vc
Vc
HCl
b
HCl
b
c
c
][
1HK t
2
1
122
)1(4
)1(2
tHClHCl Ka
ca
c
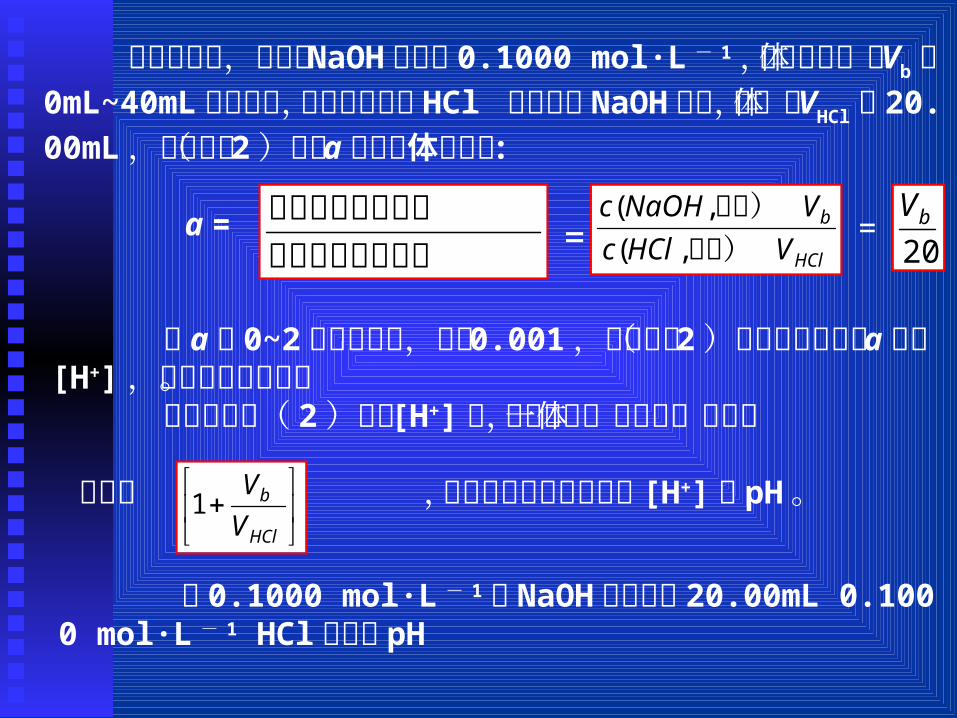
( 1)强碱滴定强酸(或强酸滴定强碱)用标准 NaOH溶液滴定 HCl,滴定反应为: H+ + OH– = H2O滴定常数 Kt = 1/Kw = 1014.00。根据质子条件:
[H+] = cHCl + [OH–] - cb ( 1)
式中 cHCl为滴定过程中盐酸的浓度, cb为 NaOH加入到被滴溶液后的瞬时浓度, cHCl和 cb均随滴定反应的进行而不断变化着,可用滴定分数 a 来衡量滴定反应进行的程度,在酸碱滴定中
a =
可推导强碱滴定强酸的滴定曲线方程为:Kt [H+]2 + Kt cHCl (a- 1)[H+]- 1 = 0 (2)
或 [H+] =
= =
将 a 代入( 1 )式,并使用 [OH–]=
酸起始的物质的量加入碱的物质的量
HCl
b
VHClc
VNaOHc
起始)标准)
,(
,(
20bV
为简便起见,设标准 NaOH的浓度 0.1000 mol·L - 1,其滴入的体积 Vb在 0mL~40mL之间变动,又设被滴定的 HCl 的浓度与 NaOH相同,其体积 VHCl为 20.00mL,于是式( 2)中的 a就转化体积之比 : a = = =
令 a在 0~2范围内取值,步长 0.001,根据式( 2)可解出对应每个 a值的 [H+],并绘出滴定曲线。
注意在用式( 2)解出 [H+]后,还需除以一个反映体积变化
的因子 ,才是实际滴定过程中的 [H+]或 pH。
HCl
b
V
V1
用 0.1000 mol·L - 1 的 NaOH 溶液滴定 20.00mL 0.1000 mol·L -
1 HCl 溶液的 pH

从滴定开始到加入 19.98 mLNaOH溶液,即 99.9%的 HCl被滴定,溶液 pH总共只变化了 3.30个 pH单位, pH变化比较缓慢。从加入19.98mL到 20.02mL NaOH溶液,相对误差从不足 0.1%到过量 0.1%,只用了 NaOH溶液 0.04mL,约一滴,溶液 pH从 4.30急剧增大到 9.70变化了 5.4个 pH 单位,即 [H+] 改变了 25万倍,溶液由酸性变
为碱性。即在化学计量点( pH =7.00)前后 ±0.1%相对误差范围内滴定曲线出现呈近似垂直的一段,溶液的 pH发生突变,这种 pH的突变称为滴定突跃,突跃所在的 pH范围称为突跃范围,简称突跃范围。此后若继续加入 NaOH溶液则进入强碱的缓冲区,溶液的pH变化逐渐减小,曲线又比较平坦。

滴定的突跃范围很重要,它是选择指示剂的依据,指示剂的变色范围必须全部或部分落在滴定曲线、的突跃范围内,这是选用指示剂的必要条件。当然选用指示剂还需要考虑其他因素,例如,石蕊的变色范围虽然也在此突跃范围内,但由于石蕊的颜色变化由紫色到红色或蓝色均不明显,不便于观察,所以不能选用。
如果用用 0.1000 mol·L - 1盐酸滴定 0.1000 mol·L - 1 NaOH溶液,其滴定曲线方向相反,或对称。
强酸强碱恰好完全反应达化学计量点, pH为 7,此时选用酚酞作指示剂,当溶液由无色变为浅红色或由浅红色变为无色即达滴定终点。
滴定突跃范围的大小与滴定剂和被滴定溶液的浓度有关。用不同浓度的 NaOH标准溶液滴定不同浓度的盐酸所得的滴定曲线不同。
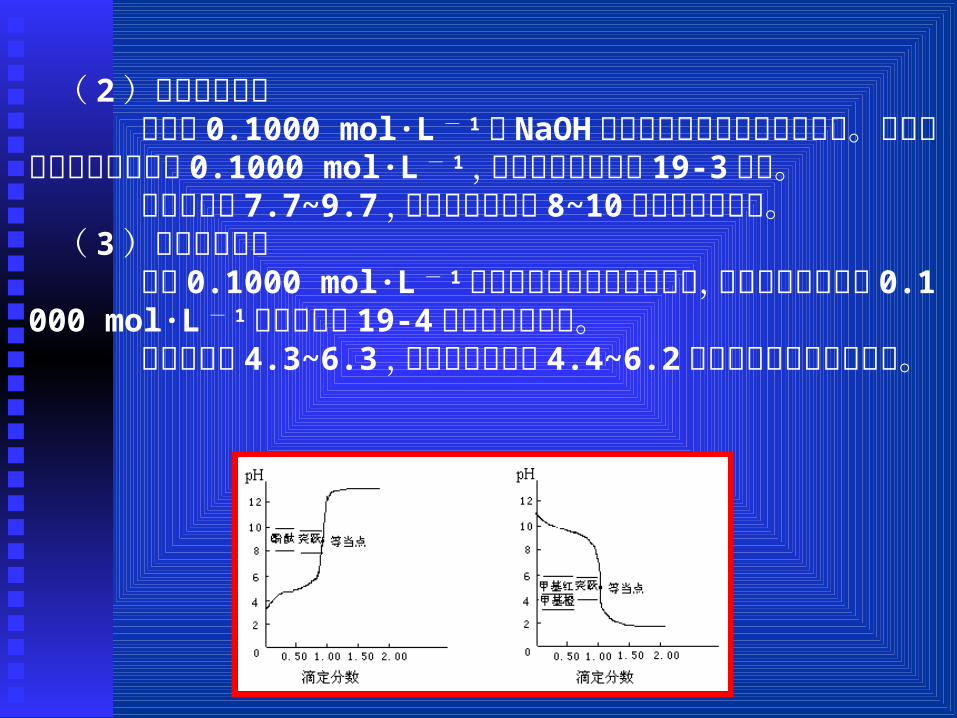
( 2)强碱滴定弱酸 若用用 0.1000 mol·L - 1的 NaOH溶液滴定未知浓度的醋酸溶液。如果醋酸溶液的浓度为用 0.1000 mol·L - 1,所得滴定曲线如图 19-3所示。
突跃范围为 7.7~9.7,选用变色范围为 8~10的酚酞作指示剂。( 3)强酸滴定弱碱 若用 0.1000 mol·L - 1的盐酸滴定未知浓度的氨水,如果氨水的浓度为 0.1000 mol·L - 1也可得如图 19-4所示的滴定曲线。
突跃范围为 4.3~6.3,选用变色范围为 4.4~6.2的甲基红作指示剂最合适。

2 、酸碱滴定法的应用( 1)双指示剂测烧碱 NaOH, Na2CO3的含量准确称取一定量的试样加水溶解,先以酚酞为指示剂,
用已知浓度的盐酸标准溶液滴定至红色刚消去,记下盐酸的体积为 V1(mL)设其浓度为 c。这时 NaOH被全部中和,而 NaCO3
则被中和到 NaHCO3。然后再加入甲基橙,继续用盐酸滴定至溶液由黄色变为橙色,这一滴定过程用盐酸体积为 V2(mL)。由酸碱反应知,把 Na2CO3滴定到 NaHCO3和 NaHCO3滴定到 NaCl消耗盐酸的量是相等的。已知 Na2CO3的式量是 106.0 g·mol - 1,NaOH的式量为 40.00 g·mol - 1,则有:
Na2CO3( %) =
NaOH( %) = %100
1000
00.4021
)试样质量(
)(y
VVc
%1001000
0.1062
)试样质量(g
cV

( 2)双指示剂测纯碱中 Na2CO3和 NaHCO3的含量 用酚酞作指示剂,把 Na2CO3滴定到 NaHCO3,所用盐酸的体积为 V1,再用甲基橙作指示剂,把 NaHCO3滴定到 NaCl,消耗盐酸的体积为 V2。那么原样品混合物中 NaHCO3消耗盐酸的体积是 V2 – V1。据此可求得 Na2CO3和 NaHCO3的含量。( 3)铵盐中氮含量的测定 铵盐加浓 NaOH蒸馏,使 NH4+转化成 NH3,再用硼酸吸收 NH3。 NH3 + H3BO3 = NH4
++ H2BO3—
然后用甲基橙作指示剂,用标准盐酸溶液滴定, H2BO3—到
H3BO3,溶液呈粉红色为滴定终点。硼酸作为 NH3的吸收剂,只要求过量,把 NH3全部吸收就行了,不要求准确的量。
铵盐加浓 NaOH蒸馏出的 NH3,也可用过量的已知浓度的盐酸或硫酸吸收,再用甲基橙作指示剂用标准 NaOH溶液滴定过量的酸来求氮的含量。

(二)络合滴定法(配位滴定法)1、络合滴定法的基本原理络合滴定法是以络合反应为基础的滴定分析方法。络合反
应也是路易士酸碱反应,所以,络合滴定法与酸碱滴定法有许多相似之处。若以乙二胺四乙酸(简称 EDTA)为滴定剂,大多数金属离子M与 Y形成 1︰ 1型络合物,可视M为酸, Y为碱,与一元酸碱滴定类似。但是,M有络合效应和水解效应, Y有酸效应和共存离子效应,所以络合滴定要比酸碱滴定复杂。酸碱滴定中,酸的 Ka或碱的 Kb是不变的,而络合滴定MY的 K 稳′是随滴定体系中反应的条件而变化。欲使滴定过程中 KMY′基本不变,常用酸碱缓冲溶液控制酸度。
设金属离子M的初始浓度为 cM,体积为 VM(mL)等浓度的滴定剂 Y滴定,滴入的体积为 VY(mL)则滴定分数
a =VY/VM

MBE [M]+[MY] = cM ①
[Y]+[MY] = cY = acM ②
由物料平衡方程 :
由络合平衡方程:]][[
][
YM
MYK MY ③
tMM
MMY K
McacM
McK
][]([
][
由①及②式可得:[MY] = cM- [M] = acM- [Y] ④[Y] = acM- cM + [M] ⑤将④⑤式代入③式得:
⑥

展开 cM - [M] = Kt[M]2 - Kt[M] cM + Kt[M]a cM
整理得 Kt[M]2 + [Kt cM( a- 1) +1][M] - cM = 0
此即络合滴定曲线方程,与强酸强碱滴定的滴定曲线方程十分相似。
在化学计量点时, a = 1.00,⑥式可简化为KMY[M]SP
2+ [M]sp - cM = 0
[M]sp =
MY
MMY
K
cK
2
411 2
一般络合滴定要求 KMY≥107,若 cM =10 - 2 mol·L–1,即 KMYcM ≥10 - 5。由于 4KMYcM>>1,
因此, [M]sp ≈
MY
M
MY
MMY
K
c
K
cK
2
4
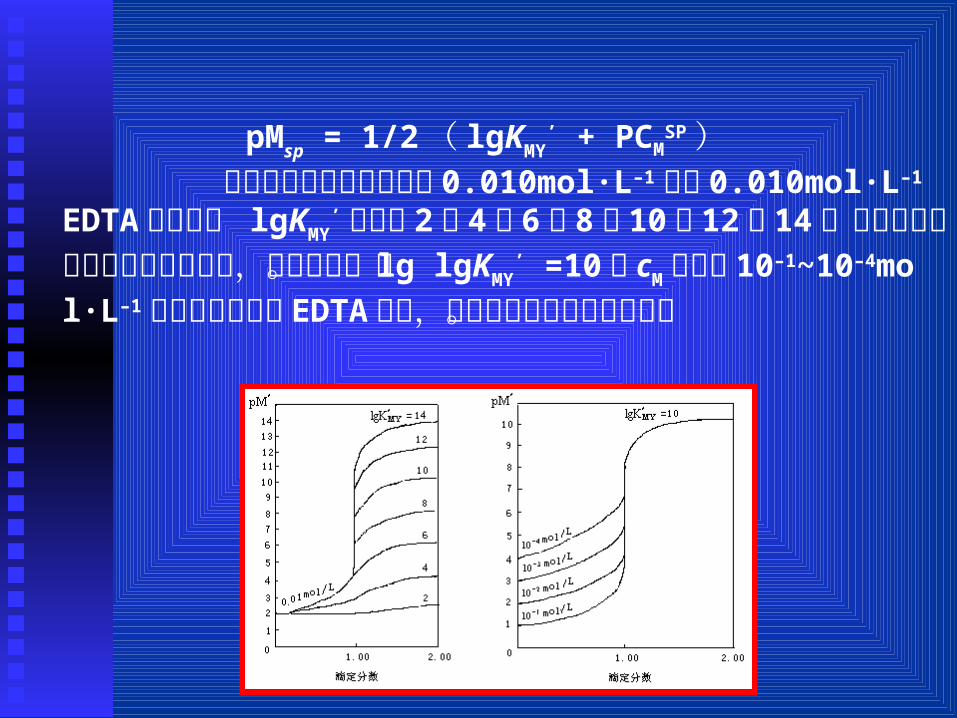
pMsp = 1/2( lgKMY’ + PCM
SP ) 设金属离子的初始浓度为 0.010mol·L–1,用 0.010mol·L–1 EDTA 滴定,若 lgKMY
’分别是 2, 4, 6, 8, 10, 12, 14,应用上式计算出相应的滴定曲线,如图所示。当 lg lgKMY
’ =10,cM分别是 10–1~10–4mol·L–1分别用等浓度的 EDTA滴定,所得的滴定曲线如图所示。
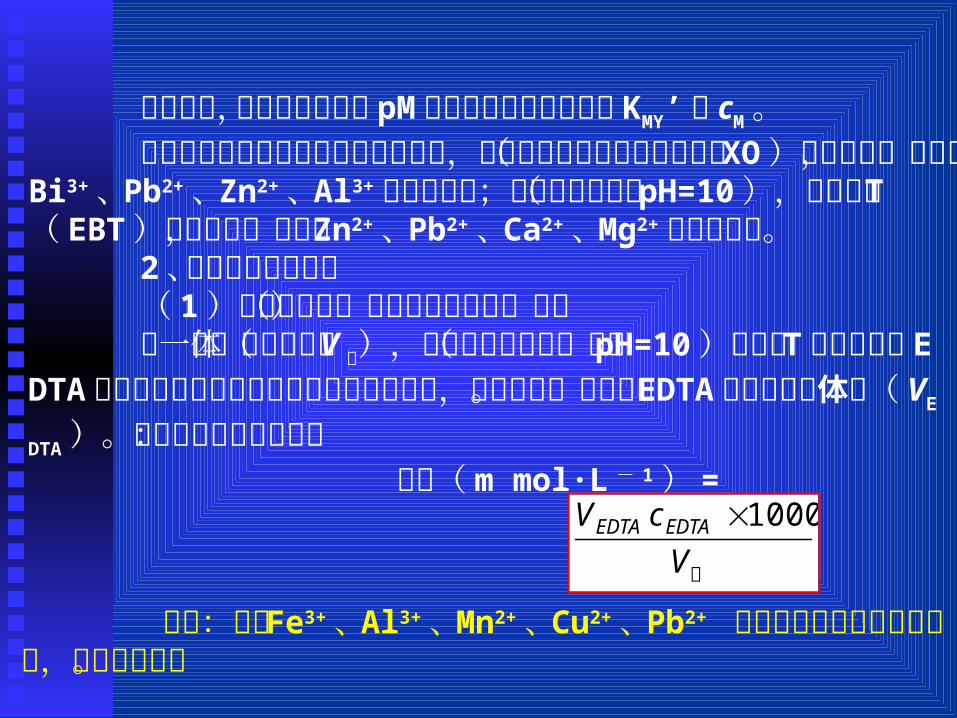
由图可知,影响络合滴定中 pM突跃大小的主要因素是 KM
Y’和 cM。 络合滴定所用的指示剂是金属指示剂,在酸性条件下可用
二甲酚橙( XO)作指示剂,可测定 Bi3+、 Pb2+、 Zn2+、 Al3+等金属离子;在碱性条件下( pH=10),可用铬黑 T( EBT)作指示剂,可测定 Zn2+、 Pb2+、 Ca2+、Mg2+等金属离子。
2、络合滴定法的应用( 1)水的总硬度(钙和镁离子总量)测定取一定体积的水样( V 水),加入氨缓冲溶液(控制 pH=1
0)和铬黑 T指示剂,用 EDTA标准溶液滴定至溶液由酒红色变为纯蓝色,即达终点。记下所用 EDTA标准溶液的体积( VEDTA)。照下式计算水的硬度:
硬度(m mol·L - 1) =水V
cV EDTAEDTA 1000
注意:水中 Fe3+ 、 Al3+ 、 Mn2+ 、 Cu2+ 、 Pb2+ 等离子量略大时会发生干扰,需加掩蔽剂。

( 2 )石灰石中钙和镁的测定 试样经酸溶解,在 pH=10 时,直接滴定溶液中钙和镁离子总量。在 pH> 12.5 时,镁离子生成氢氧化物沉淀,可单独滴定钙离子。方法如下:
取一定体积的试样,加适量掩蔽剂( 5%酒石酸钾钠和 1:2三乙醇胺溶液),加 20%NaOH 溶液,控制 pH> 12.5 ,再加一定量的铜试剂。摇匀后,加入钙指示剂,用 EDTA 标准溶液滴定至溶液由红色变为纯蓝色,即达终点。记下所用 EDTA 标准溶液的体积。计算试样中含 CaO 的质量分数。
( 3 )铝盐中铝的测定 铝离子与 EDTA络合速率甚慢,故不能直接滴定。测定时都是在 pH =3 的条件下,加入过量的 EDTA ,使铝离子完全络合,此时其他金属离子(如铁等)也络合。再调整 pH = 5~6 ,用铜盐(或锌盐)标准溶液返滴过量的 EDTA 。然后利用氟离子能与铝离子生成更稳定的络合物这一性质,加入氟化铵,置换出与铝络合的 EDTA ,再用铜盐标准溶液滴定。

(三)氧化还原滴定法 1、氧化还原滴定法的基本原理 氧化还原滴定法是以氧化还原反应为基础的滴定分析方法。同酸碱滴定法和络合滴定法一样,氧化还原滴定法也有反映滴定过程的滴定曲线,只不过在此滴定过程中发生变化的、反映氧化还原反应(滴定反应)进行程度的是电极电势。在氧化还原滴定中,随着滴定剂的不断滴入,物质的氧化型和还原型的浓度逐渐改变,有关电对的电势也逐渐改变,在化学计量点附近发生突变。现以 0.1000 mol·L–1Ce(SO4)2标准溶液滴定 20.00mL0.100 mol·L–1 Fe2+溶液为例:
Ce2+ + Fe2+ = Ce3+ + Fe3+
( 1)滴定前 由于空气中氧的作用,在 0.1000 mol·L–1的 Fe2+溶液中,必有少量的 Fe3+存在,组成电对 Fe3+/ Fe2+,由于 Fe3+不知道,故此时的电势无法计算。
滴定开始后,体系中就同时存在两个电对。在滴定过程中,任何一点达到平衡时,两电对的电势均相等。

'0/ 23 FeFe
2
3
Fe
Fe
c
c '0/ 34 CeCe
3
4
Ce
Ce
c
cE= E +0.0592 lg = E +0.0592 lg
因此在滴定开始后的不同阶段,可任选一电对计算体系的电势。
3
2.023 FeFe
cc
'0/ 23 FeFe
2
3
Fe
Fe
c
c
( 2)滴定开始至化学计量点前 Ce4+几乎全部被还原成 Ce3+, Ce4+的浓度极小,可根据 Fe3+/
Fe2+电对来计算电位。若滴入 10mLCe4+ 溶液。则
mol·L–1
同理可计算出滴入 Ce4+ 溶液 1.00、 2.00、 4.00、 8.00、18.00、 19.80、 19.98mL的电势。
E= E +0.0592 lg = 0.68V

'0/ 23 FeFe
2
3
Fe
Fe
c
c
'0/ 34 CeCe
3
4
Ce
Ce
c
c
此时, Ce4+和 Fe2+都定量地变成 Ce3+和 Fe3+, Ce4+和 CFe3+的浓度很小不便求得,故不能单独用某一电对来计算电势值,而需要由两电对的奈斯特方程联立求解:
+0.0592 lg
+0.0592 lg
Esp = E
Esp = E
2
'0/
'0/ 3423
CeCeFeFeEE
2
0592.0
32
43
CeFe
CeFe
cc
cc
两式相加得
Esp = + lg
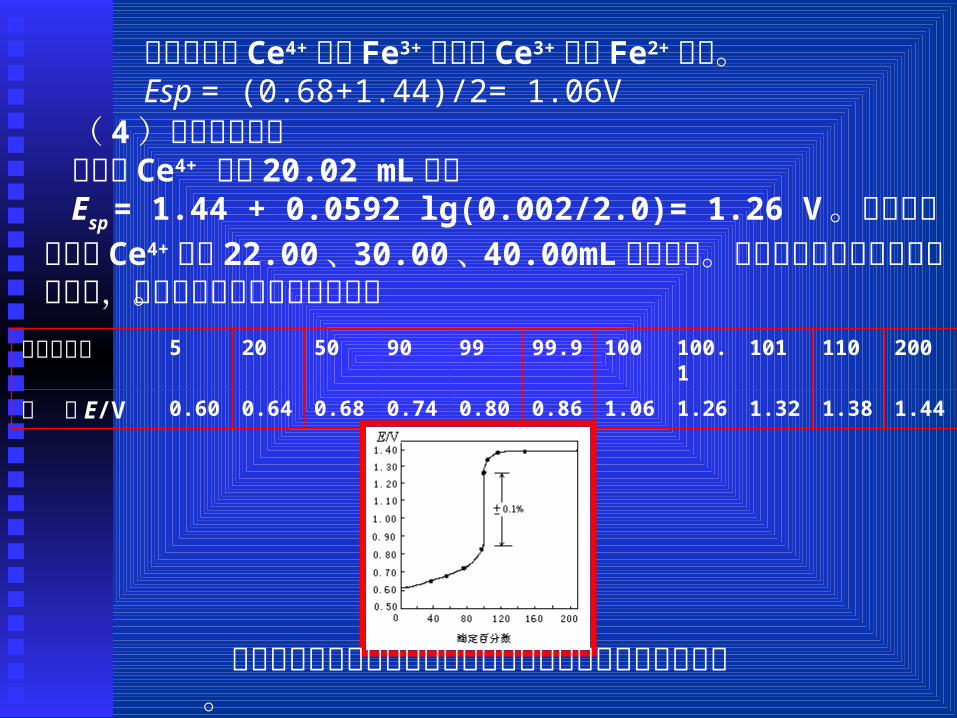
在计量点时 Ce4+ 等于 Fe3+ 浓度, Ce3+ 等于 Fe2+ 浓度。Esp = (0.68+1.44)/2= 1.06V( 4)化学计量点后当滴入 Ce4+ 溶液 20.02 mL时:Esp = 1.44 + 0.0592 lg(0.002/2.0)= 1.26 V 。同样可计算滴入 Ce4+
溶液 22.00、 30.00、 40.00mL时的电势。将不同滴定点的电势列于下表中,并绘制成滴定曲线如图所示。滴定百分数 5 20 50 90 99 99.9 100 100.1 101 110 200
电 势 E/V 0.60 0.64 0.68 0.74 0.80 0.86 1.06 1.26 1.32 1.38 1.44
滴定的终点可用指示剂或通过反应物自身的颜色变化来确定。

2、氧化还原滴定法的应用( 1)高锰酸钾作氧化剂进行的滴定反应 高锰酸钾是一种强的氧化剂,本身又具有颜色,在一
些滴定反应中不需要再加指示剂。高锰酸钾作氧化剂,其还原产物与溶液的酸度有关。在强酸溶液中,还原到Mn2+;在焦磷酸盐或氟化物存在下,还原到Mn( III)的配合物;在弱酸性、中性、弱碱性溶液中,还原到MnO2;在强碱溶液中,还原到MnO4
2-。KMnO4标准溶液浓度以草酸钠来标定。在 H2SO4溶液中反应如下:
2 MnO4- + 5 C2O4
2- + 16H+ = 2Mn2+ + 10CO2 + 8H2O滴定终点时 KMnO4的紫色在 0.5 min~1 min不消失。高锰酸钾标准溶液可滴定 H2O2, C2O4
2-, NO2-, Fe2+等。
用高锰酸钾作标准溶液可测定甲醇、甲醛、苯酚、柠檬酸、甘油、葡萄糖等一些有机化合物。对这些有机物的测定采用了返滴定法,即在弱碱性溶液中加入一定量的过量的标准 KMnO4,它能与这些有机物发生定量的氧化还原反应。如 KMnO4与甲酸的反应: HCOO– + 2MnO4
- + 3OH– = CO32-+ 2MnO4
2-+ 2H2O

反应后再将溶液酸化,加入过量标准 Fe2+溶液,使溶液中的锰还原到Mn2+。再以标准 KMnO4溶液滴定过量的 Fe2+。某些不能与 KMnO4直接反应的物质也可用 KMnO4作标准液通过间接方法来测定。如 Ca2+、 Th4+等在溶液中不与 KMnO4反应,但它们却能与 C2O4
2-发生反应生成草酸盐沉淀,将沉淀从溶液中分离出来后,可用 KMnO4滴定 C2O4
2-,通过测 C2O42-间接的测
出了 Ca2+、 Th4+的含量来。( 2)重铬酸钾作氧化剂进行的滴定反应 K2Cr2O7也是一种强氧化剂,在酸性溶液中, Cr2O7
2-被还原成Cr3+。
重铬酸钾法的优点: K2Cr2O7试剂易于提纯,可以直接称量后配制称标准溶液,不必进行标定; K2Cr2O7相当稳定,可以长期保存;在酸性溶液中, Cr2O7
2-作氧化剂,不会把 Cl–氧化,故不受溶液中的 Cl -干扰。使用 K2Cr2O7滴定时,需使用氧化还原指示剂,如二苯胺磺酸钠等。

( 3)碘量法 碘量法是以 I2作为氧化剂或以 I– 作为还原剂进行滴定分析的方法。 I2是较弱的氧化剂,能于一些较强的还原剂作用; I– 是中等强度的还原剂,能被许多氧化剂氧化。因此,碘量法分为直接和间接两种方法。
直接碘量法:一些还原性物质可用 I2标准溶液直接滴定。例如,亚硫酸盐可用直接电量法测定。反应为:
I2 + SO32-+ H2O = 2I– + SO4
2-+ 2H+
间接碘量法:氧化性物质把 I–氧化为 I2,然后用 Na2S2O3标准溶液滴定生成的 I2。这种方法叫间接碘量法,其反应为:
I2 + 2S2O32- = 2I - + S4O6
2-
用一定量的过量的 I2还可氧化甲醛、葡萄糖、硫脲、丙酮等有机物质,再用标准 Na2S2O3溶液滴定过量 I2,从而可求有机物含量。碘量法中常使用的标准溶液有 Na2S2O3和 I2。硫代硫酸钠用 KIO3, KBrO3, K2Cr2O7等基物质进行标定。如 KIO3作基准物时,它在酸性溶液中于 KI发生反应为: IO3
- + 5I - + 6H+ = 3I2 + 3H2O I2以淀粉作指示剂,用 Na2S2O3滴定。计算 Na2S2O3的浓度。

(四)沉淀滴定法1、沉淀滴定法的基本原理沉淀滴定法是以沉淀反应为基础的滴定分析法。用于沉淀
滴定法的沉淀反应必须符合下列条件:①沉淀的溶解度很小;②沉淀反应必须定量且迅速的发生;③能采用适当的方法确定滴定终点。目前,比较有实际意义的、应用较多的是利用生成微溶性银盐的沉淀反应,如 Ag+ + Cl–=AgCl↓和 Ag+ + SCN– = AgSCN↓进行的沉淀滴定法,此法称为银量法。银量法主要用于测定 Cl–、 Br–、I–、 Ag+及 CN– 等。
2、沉淀滴定法的应用( 1 ——)莫尔法 用铬酸钾作指示剂 此法是在中性或弱碱性溶液中,以 K2CrO4为指示剂,用
AgNO3标准溶液直接滴定 Cl -(或 Br -)。 AgCl的溶解度比 Ag2CrO4小,据分步沉淀原理,当向含 Cl
-的溶液中滴入 NgNO3时,首先沉淀析出的是 AgCl;当 AgCl定量沉淀后,稍过量 AgNO3溶液与 CrO4
2-生成砖红色 Ag2CrO4沉淀,即为滴定终点。反应 : Ag+ + Cl–=AgCl↓(白色)
Ag+ + CrO42- = Ag CrO4↓(砖红色)

)( AgClspK 10108.1
莫尔法中指示剂的用量和溶液的酸度是两个主要问题。①指示剂的用量据溶度积原理,可计算出计量点时 Ag+ 和 Cl -的浓度为:
[Ag+]sp= = =1.2 × 10–5 mol·L–1
2
)(
][4
sp
AgCrOsp
Ag
K 25
12
)103.1(
100.2
在计量点时要刚好析出 Ag2CrO4沉淀以指示终点,此时 CrO4
2-的浓度应为:
[CrO42-] = = = 1.2 × 10–2 mol·L–1
在实际工作中,若 CrO42-的浓度太高,颜色太深,会影响终点
的判断;但 CrO42-的浓度太低,又会使终点出现过缓,影响滴定
的准确度。实验证明加入 K2CrO4的浓度以 5.0 × 10–3 mol·L–1为宜。这样实际使用的 CrO4
2-的浓度较计算值偏低,那么要使 Ag2CrO4沉淀析出,必然要使 AgNO3溶液过量得稍多一点。这样是否满足准确度的要求?我们说计算结果表明,这样不会引起较大的误差,完全可以满足滴定分析的要求。

②溶液的酸度 H2CrO4的 Ka2为 3.2×10 –7,酸性较弱,易溶于酸,即 Ag2CrO4
+ H+ = 2Ag+ + HCrO4- 。所以滴定不能在酸性溶液中进行。但如果溶
液的碱性太强,又将产生 Ag2O沉淀。 莫尔法测定的最适宜的 pH值范围为 6.5 ~10.5之间。若试液碱性太强,可用稀 HNO3中和,酸性太强,可用 NaHCO3、 CaCO3
或 Na2B4O7等中和。当溶液中的铵盐存在时,要求溶液的酸度范围更窄, pH值为 6.5 ~7.2之间。这是因为 pH值再大时,便有相当数量的 NH3释放出来, Ag+ 生成 Ag(NH3)2
+,使 AgCl和 Ag2CrO4的溶解度增大,影响滴定。③莫尔法的适用范围 能直接滴定 Cl -或 Br -,共存时滴定的是总量。原则上讲可应用于滴定 I -及 SCN -,但由于 AgI及 AgSCN沉淀能强烈地吸附 I-或 SCN - 而使终点过早出现,且终点变化不明显,误差较大。
没不适用以 NaCl标准溶液直接滴定 Ag+,因滴入指示剂即生成 Ag2CrO4沉淀,在用 NaCl标准溶液滴定时, Ag2CrO4沉淀十分缓慢地转化为 AgCl沉淀,误差大。如果要用此法测定 Ag+,应在试液中加入过量 NaCl标准液,用 AgNO3标准溶液滴定过量 Cl -。

莫尔法的选择性较差,凡能与 Ag+ 生成微溶物(如 S2–)或配阴离子或能与 CrO4
2-生成微溶性化合物的阳离子(如 Ba2+、 Pb2+、 Hg2+等)以及在中性或弱碱性溶液中易发生水解的离子(如 Fe3+、Bi3+、 Sn4+、 Al3+等)均干扰滴定,应预先分离。( 2 ——)佛尔哈德法 用铁铵矾 [NH4Fe(SO4)2·12H2O]作指示剂 佛尔哈德法分为直接滴定法和返滴定法。 ①直接滴定法测定 Ag+
此法是在含有 Ag+ 的酸性溶液中,以铁铵矾作指示剂,用 NH4
SCN(或 KSCN、 NaSCN)标准溶液滴定 Ag+ 的方法。随着滴定剂的加入,首先析出 AgSCN沉淀,当 Ag+ 定量沉淀后,稍微过量的一滴 NH4SCN与 Fe3+ 反应生成红色配合物,即为终点。其反应如下:
Ag+ + SCN–= AgSCN↓ (白色) Ksp = 1.0 ×10 –12
Fe3+ + SCN–= (FeSCN)2+ (红色) K 稳 = 200 滴定时,溶液的酸度一般控制在 pH值为 0 ~1之间。此时, F
e3+ 主要以 Fe(H2O)63+的形式存在,颜色较浅。如果酸度较低, Fe3+
水解形成棕色 [Fe(H2O)5OH]2+ 等颜色较深,影响终点观察。

][
][3
2
FeK
FeSCN
稳 015.0200
100.6 6
= 2.0×10 –6 mol·L - 1[SCN - ]= =
)( AgSCNspK 12100.1
若被滴定的 Ag+ 溶液的体积为 VmL, SCN -和 Ag+ 的浓度均为 0.1 mol·L - 1,在计量点时
[SCN - ]sp = = = = 1.0×10 –6 mol·L - 1
在计量点后0.1% 时
[SCN - ]’=V
V
2
%1.01.0
[SCN - ]sp< [SCN - ] <[SCN - ]’
= 5.0×10 –5 mol·L - 1
由此可见能够观察到滴定终点颜色时,相对误差小于 0.1% ,准确度符合滴定要求。由于 AgSCN沉淀能吸附部分 Ag+ ,因此此法在滴定过程中应充分摇动溶液,使被吸附的 Ag+ 及时地释放出来,否则易产生终点出现过早而造成误差。

②返滴定法测定卤素离子 先向试液中加入过量 AgNO3标准液,再加铁铵矾指示剂,用
NH4SCN标准溶液返滴定过量 Ag+,此时和直接滴定 Ag+ 一样。此法滴定氯化物,由于 AgCl的溶解度比 AgSCN大,过量的 SCN -将与 AgCl反应,使 AgCl沉淀转化为溶解度更小的 AgSCN。
AgCl + SCN - = AgSCN + Cl -。采用下列措施避免误差: a.当加入过量的 AgNO3标准液后,将溶液加热煮沸,使 AgCl沉淀凝聚,以减小 AgCl沉淀对 Ag+ 的吸附。过滤,将 AgCl沉淀滤去,并用稀 HNO3洗涤沉淀,洗涤液并入滤液中。然后用 NH4SCN标准溶液滴定滤液中过量的 Ag+。
b.加入一定过量的 AgNO3标准溶液后,加入 1~2mL有机溶剂,如 1, 2 – 二氯乙烷,用力摇动,使有机溶剂将 AgCl沉淀包裹,使它与溶液隔开。这样便阻止了 SCN -与 AgCl的反应,此法比较简便。用返滴定法测定溴化物或碘化物时,由于 AgBr、 AgI的溶解度均比 AgSCN的溶解度小,不会出现沉淀转化反应,所以不必采用上述措施。但在测定碘化物时,必须在加入过量的 AgNO3溶液后加入指示剂,否则会发生如下反应: 2Fe3+ + 2I–= 2Fe2+ + I2
佛尔哈德法的最大优点是可以在酸性溶液中进行滴定,许多弱酸根离子如 PO4
3-、 AsO43-、 CrO4
2-等都不干扰测定,因此,此法的选择性较高。但滴定不能在中性或碱性溶液中进行,因为此时, Fe3+ 易生成羟基配合物或氢氧化物沉淀。另外强氧化剂、氮的低价氧化物以及铜盐、汞盐等能与 SCN -起作用,干扰测定,必须预先除去。

三、误差与数据处理1、准确度与精密度 准确度是表示测定值与真实值接近的程度,用误差来衡量。
准确度越高则表明测定值与真实值之间的差值越小。 精密度是表示几次平行测定结果相互接近的程度,它与真实值无关,用极差或偏差来衡量。如果在相同条件下,对同一样品几次平行实验的测定值彼此较接近,则说明测定结果精密度较高;如果实验测定值彼此相差很多,则测定结果的精密度就低。
如何从准确度与精密度两方面来衡量分析结果的好坏呢?例如,甲、乙、丙、丁四人同时分析一瓶 KOH溶液的浓度(真实值为 0.1244 mol·L - 1),每人分别平行测定 3次,结果如下:
甲 乙 丙 丁
测定结果 0.1241 0.1220 0.1230 0.12140.1242 0.1221 0.1284 0.12480.1243 0.1222 0.1257 0.1273
平均值 0.1242 0.1221 0.1257 0.1244真实值 0.1244 0.1244 0.1244 0.1244差值 0.0002 0.0023 0.0013 0.0000

甲的分析结果准确度与精密度均好,结果可靠;乙的精密度虽很高,但准确度太低;丙的精密度与准确度均很差;丁的平均值虽也接近于真实值,但是他的精密度很差, 3次测量数值彼此相差很远,仅仅是由于正负误差相互抵消才使结果凑巧等于(或接近)真实值,而每次测量结果都与真实值相差很大,因而丁的测量结果是不可靠的。由此可见:
( 1)精密度是保证准确度的先决条件,精密度低说明所测结果不可靠,就失去了衡量准确度的前提。
( 2)精密度高,不一定能保证准确度高。2、误差与极差、偏差 误差( E)表示测定值( x)与真实值( T)之间的差
值。误差越小,表示测定值与真实值越接近,准确度越高;反之,误差越大,准确度越低。当测定值大于真实值时,误差为正值,表示测定值偏高;反之误差为负值,表示测定值偏低。
误差可用绝对误差( AE)和相对误差( RE)表示。AE = x - T RE = (AE/T)X100%
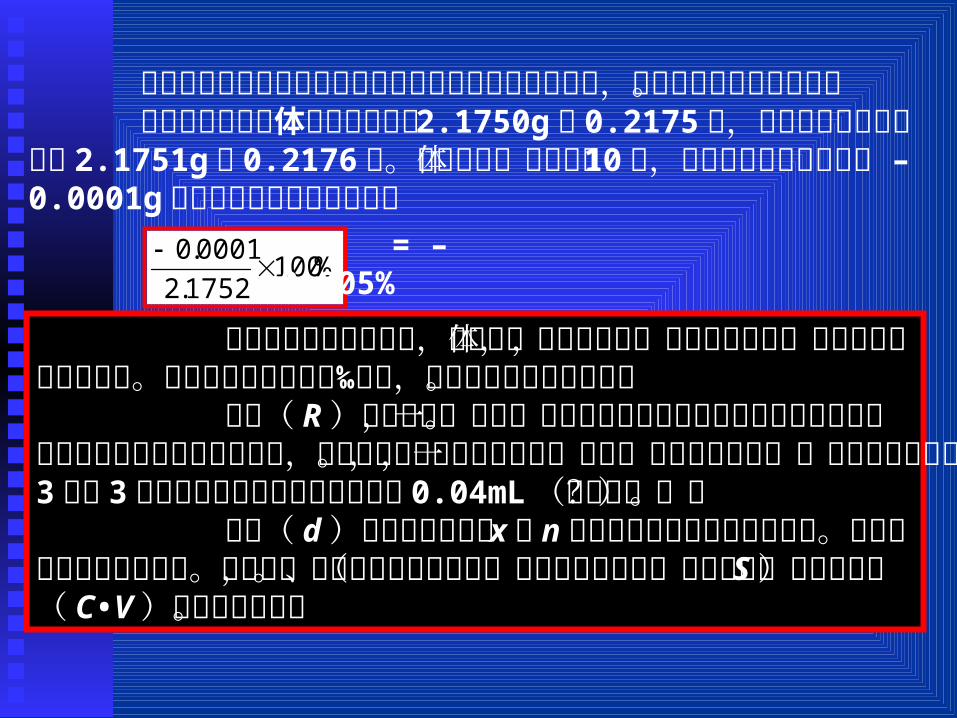
%1001752.2
0001.0
%1002716.0
0001.0
因为绝对误差不能反映它对测定结果准确度的影响程度,所以常用相对误差表示。
例如称得两个物体的质量分别为 2.1750g和 0.2175克,它们的真实质量分别为 2.1751g和 0.2176克。这两个物体质量相差 10 – 倍,但测定的绝对误差都为 0.0001g,它们的相对误差分别为: =
– 0.005%
= – 0. 05%
可见当绝对误差相同时,称量物体的质量越大,相对误差越小,测定的准确度就比较高。相对误差常用千分率‰表示,以免与百分含量相混淆。
极差( R )又叫全距,是指一组平行测定结果中最大值和最小值之差。在平行测定次数较少的情况下,用极差表示精密度比较方便。例如,在滴定分析中,一般要求平行滴定 3次,3次滴定管读数的极差要求不大于 0.04mL (为什么?)。
偏差( d )是指个别测定值 x与 n次测定值的算术平均值的差值。是衡量精密度高低的尺度。偏差小,表示测定的精密度高。偏差有平均偏差、标准偏差( S )和变异系数( C•V )三种表示方法。

dn
xxn
ii
1
1
1
2
n
xxn
ii
x
S ×100 = S = C•V% =
3 、系统误差与偶然误差系统误差是由某种固定的原因所造成的,在多次重复测定中会
重复出现。它具有单向性,即正负、大小都有一定的规律性,是可以测定的,故又称可测误差。一般由以下几种原因引起。
( 1 )方法误差是由采用的分析方法本身造成的。例如滴定分析中指示剂选用不够恰当而造成的误差;重量分析中由于沉淀溶解损失或是共沉淀、后沉淀现象引起的误差等等。
( 2 )仪器误差是仪器本身缺陷造成的。例如天平、砝码不够准确等。
( 3 )试剂误差是试剂不纯带来的。例如,试剂和蒸馏水中含有被测物质或干扰物质等,使分析结果系统偏高或偏低。
( 4 )主观误差是操作者主观原因造成的。例如对终点颜色的辨别不同,有人偏浅或偏深。

偶然误差又称随机误差,它是又某些难以控制、无法避免的偶然原因而引起的,其大小和正负都不固定。偶然误差的大小,决定分析结果的精密度。偶然误差虽然不能通过校正而减小或消除,但它的分布符合统计规律,即正态分布规律,可得出如下的结论:
①大小相等符号相反的偏差出现的几率基本相等; ②偏差小的测定值出现的几率多,偏差大的小; ③测定值的平均值比个别测定值可靠。4 、提高分析结果准确度的方法 ( 1 )认真、仔细地操作、记录、计算,避免过失。 ( 2 )适当增加平行测量的次数,可以减少偶然误差。 ( 3 )采用如下方法来消除系统误差:①空白实验可消除试剂、蒸馏水等引入的杂质所造成的系统误
差;②用标准方法或可靠的分析方法进行对照实验,对照实验是检
验系统误差的有效方法;③使用校准的仪器可消除因仪器不准所引进的系统误差;④用标准加入法来测定回收率。
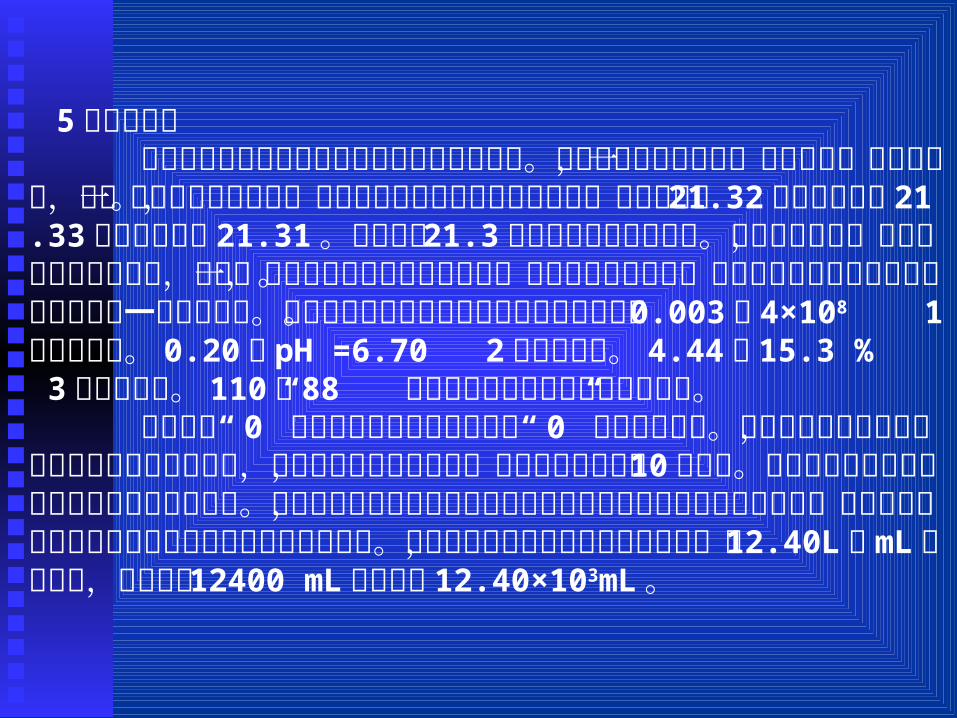
5、有效数字 有效数字是指在实验工作实际能测量到的数字。在实验记录的数据中,只有最后一位是估计的,这一位数字叫不定数字。例如读滴定管中的液面位置数时,甲可能读为 21.32,乙可能读为 21.33,丙可能读为 21.31。由此可见 21.3是滴定管上显示出来的。因实验者不同,可能得到不同的估计值,但这一位估计数字却是客观存在的,因此它是有效数字。也就是说有效数字是实际测到的数字加一位估读数字。通过下面几个有效数字的位数确定来说明。 0.003, 4×108 1位有效数字。 0.20, pH =6.70 2位有效数字。 4.44, 15.3 % 3位有效数字。 110, 88 准确数字或有效数字位数不定数字。
“数字前的 0” “不作为有效数字,数字后的 0”则为有效数字。对数值数的有效数字,只由小数点后的位数决定,与小数点前的位数无关,因小数点前的数是 10的次方。有效数字位数不确定的数字可认为是准确数字。测量所得到的有效数字的位数是由测量对象和测量仪器所决定的,运算所得到的有效数字的位数是由被运算数字决定的。单位转换时有效数字位数不能改变,如 12.40L用mL作单位时,不能写成 12400 mL而应写成 12.40×103mL。

( 1)有效数字的运算①有效数字的修约 由于实验测得的有效数字的位数可能不同,因此在计算时,就要将那些有效数字位数过多的有效数字进行修约,舍弃过多的位数,使得运算简单且计算结果仍然准确。
有效数字修约规则:一次到位,四舍六入五成双。例如: 3.4747 修约到三位有效数字是 3.47 2.760 修约到二位有效数字是 2.8 2.535 修约到三位有效数字是 2.54 2.865 修约到三位有效数字是 2.863.4747 修约到三位有效数字时,不能先修约到四位,再修约到三位,即 3.4747 (五位)→ 3.475 (四位)→ 3.48 (三位)的修约是错误的。
②有效数字的加减运算 有效数字是只含一位可疑数字的数。有效数字相加减所得
到的数字也只能是含一位可疑数字的数。故加减运算时的修约是以小数点后的位数最少的数字来决定,将小数点后多余的有效数字修约舍弃。

例 1: 0.5356 + 4.72- 3.2 = ?3.2是二位有效数字,小数点后只有一位小数,故应以此为
根据,将其他数也修约到小数点后只有一位有效数字。即:0.5356 + 4.72- 3.2 = 0.5 + 4.7 - 3.2 =2.0③有效数字的乘除运算以有效数字位数最少的数为根据,将其他有效数字化为与
此位数相同的数再乘除运算。计算结果的有效数字的位数也和有效数字位数最少的那个数位数相同,在运算过程中可多保留一位有效数字。还应特别注意,在有效数字的乘除运算中,当遇到 9以上的大数时,可多算一位有效数字。如 9.00, 9.24等,将它们看作四位有效数字。因为它们的相对误差和 10.××四位有效数字的相对误差相接近。
例 2: 0.0121×25.64×5 =? 5是一个有效数字位数不确定数字,或是一个准确数字,则有效数字位数最少的数是 0.0121三位有效数字,则把 25.64也修约位三位有效数字,即 25.6。
0.0121×25.64×5=0.0121×25.6×5 =1.55有效数字虽经修约,可是运算结果只能用等号,不得用约
等号。

( 2)有效数字与化学实验测量读数 有效数字是实际测到的数字加上一位估读数字。估读数字会因实验者不同得到不同的估读值,但这一位估读数字是客观存在的,因此它是有效数字。按有效数字的定义,用游码为 5g的台秤称量时,因游码分为 5大格,每大格为 1g,每大格间又分为 10小格,每小格 0.1g,且每小格间的宽度远比 25mL的滴定管的每小格间的宽度还要大。因此用这样的台秤可称取 29.25g的物质。果真能称取 29.25g吗?
让我们以半自动电光分析天平的称量读数过程为例来看称量读数。在进行称量前先要调整天平零点,使标尺的读数线与投影屏上的零线位置重合。零点调整后应检查天平的灵敏度,即加 1mg砝码时,投影屏上微分标尺是否在 1mg位置,如果不在,应调节感量调节圈。因投影屏上每毫克间分为 10小格,又因这种天平的精度所限,它的灵敏度为 10格 /1mg,则天平的感量为 0.1mg/格,即投影屏每偏转 1小格相当于 0.1mg。
若用此天平称量某一重物时所加的砝码和圈码为 5.23g ,这时投影屏的位置如图所
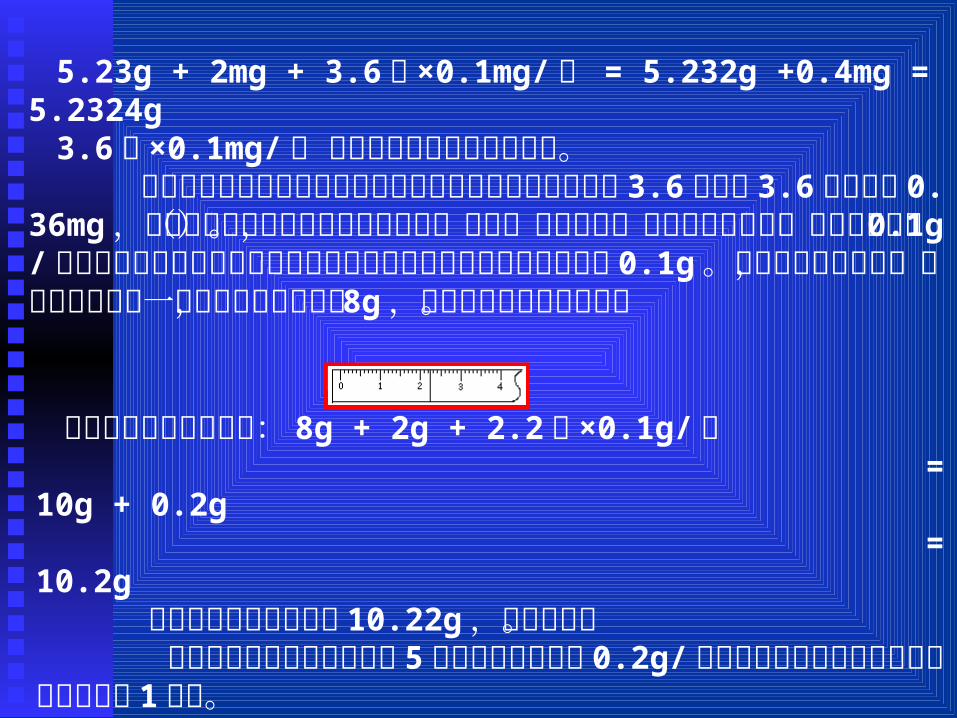
5.23g + 2mg + 3.6格 ×0.1mg/ 格 = 5.232g +0.4mg = 5.2324g3.6格 ×0.1mg/ 格是根据有效数字运算得来的。 通过这个称量过程,我们可以看出在读投影时,虽能读到 3.6格,但 3.6格并不是 0.36mg,格数到质量数要通过天平的灵敏度(感量)运算而来。与分析天平类似,台秤的感量为 0.1g/格,由于中学阶段没有学感量这以概念,说成了托盘天平能称准到 0.1g。照分析天平的读数,如果用台秤称某一物质时,右盘加砝码 8g,游码所在位置如图所示。
则该被称物质的质量为: 8g + 2g + 2.2格 ×0.1g/格 = 10g + 0.2g = 10.2g 若将此物质的质量读作 10.22g ,那就错了。 有的台秤游码标尺每大格有 5小格,则其感量为 0.2g/格,
用此台秤称量时也只能读到小数点后 1位数。

跟台秤同理,用 10mL量筒量液体时,由于 10mL的量筒的精度为 0.2mL/格,如图所示,液体的读数为:
2mL + 2.4格 ×0.2mL/ 格 = 2mL + 0.5mL = 2.5mL 量筒是和台秤相匹配的实验仪器,因之,它们测量体积、称量质量读数时,小数点后都只能保留 1位数。
滴定管是和分析天平相匹配的实验仪器,它的精度较高为 0.10mL/格,用滴定管读数计到小数点后第二位,如 15.36mL。
用容量瓶配制溶液,所得溶液体积为一常数,例如用 50mL的容量瓶配制一定浓度的 NaCl溶液所得的体积为 50mL,它是一个常数,不能看作是两位有效数字,也不能改为 50.0mL,按三位有效数字计。
配制 1 L0.5 mol·L - 1的 NaCl “溶液时, 天平(指托盘天平)上称出 29.3g ”氯化钠 这里的 29.3g是根据托盘天平的称量精度得来的,而计算需要的 NaCl质量 29.25g。
在化学实验中,科学计数至关重要,因为它最能反映实验的真实性和科学性。

3 、实验数据的处理( 1 )表格法 表格法具有简明、便于比较等优点。实验完成后,从观察、实验和测定得到的实验数据尽可能整齐地、有规律地用表格的形式表示出来,使得全部数据能一目了然,能看出一些物质间的变化关系,能够了解事物变化的本质,这样便于处理和运算。
( 2 )图解法 图解法能直接显示出变化规律与特点,并能利用图形作进一步的处理,求得斜率、截距、内插值、外推值等等 。注意:
①用直角毫米坐标纸作图时,常以自变量作横坐标,因变量作纵坐标。坐标的起点不一定是零。坐标轴上还应注明所代表的变量的名称及单位。
②坐标比例的选择要能表示全部有效数字,使其与测量的准确度相一致。
③标绘数据时,可用符号代表点。若必须在一张坐标纸画几条线,则对不同组的点应选用不同的符号代表。
④配线描点后即可用直尺或曲线板连成圆滑的曲线。所配的线不应穿过代表测量值及其精密度的圆圈、三角等符号。
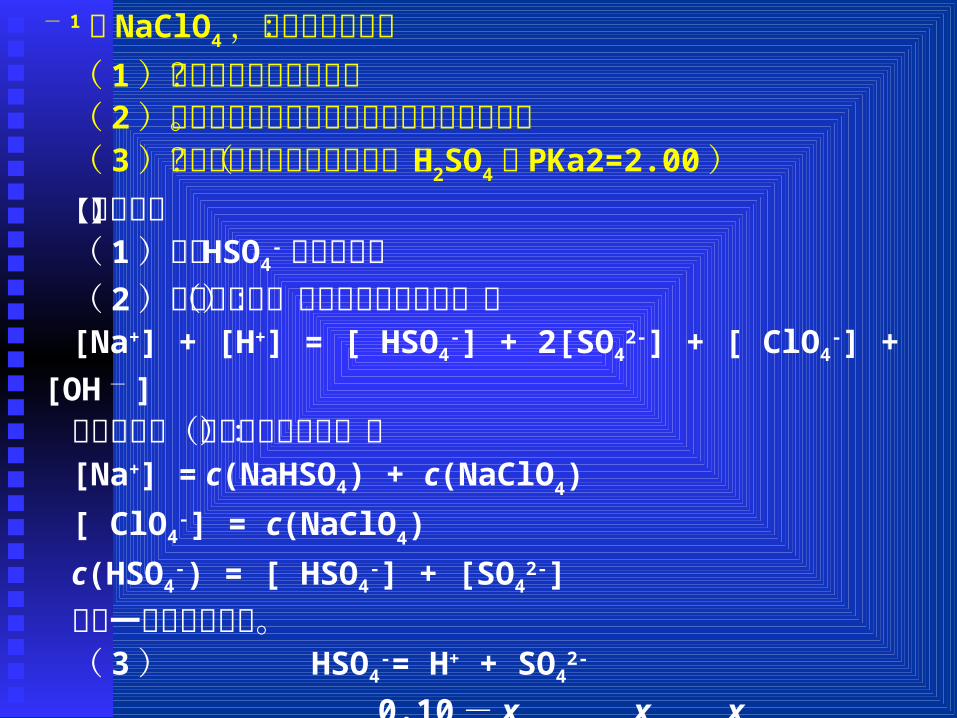
【例题 1】某水溶液中含有 0.10 mol·L - 1的 NaHSO4和 0.50 mo
l·L - 1的 NaClO4,回答以下问题:( 1)本题中什么物质是酸?( 2)给出该水溶液的电荷平衡式和物料平衡式。( 3)溶液中氢离子浓度有多大?( H2SO4的 PKa2=2.00)【解析】:( 1)只有 HSO4
-离子是酸;( 2)电荷平衡式(又叫电中性条件式):[Na+] + [H+] = [ HSO4
-] + 2[SO42-] + [ ClO4
-] + [OH - ]物料平衡式(又叫质量平衡式):[Na+] = c(NaHSO4) + c(NaClO4)
[ ClO4-] = c(NaClO4)
c(HSO4-) = [ HSO4
-] + [SO42-]
最后一式是最重要的。( 3 ) HSO4
-= H+ + SO42-
0.10 - x x x [H+] = 0.027 mol·L - 1
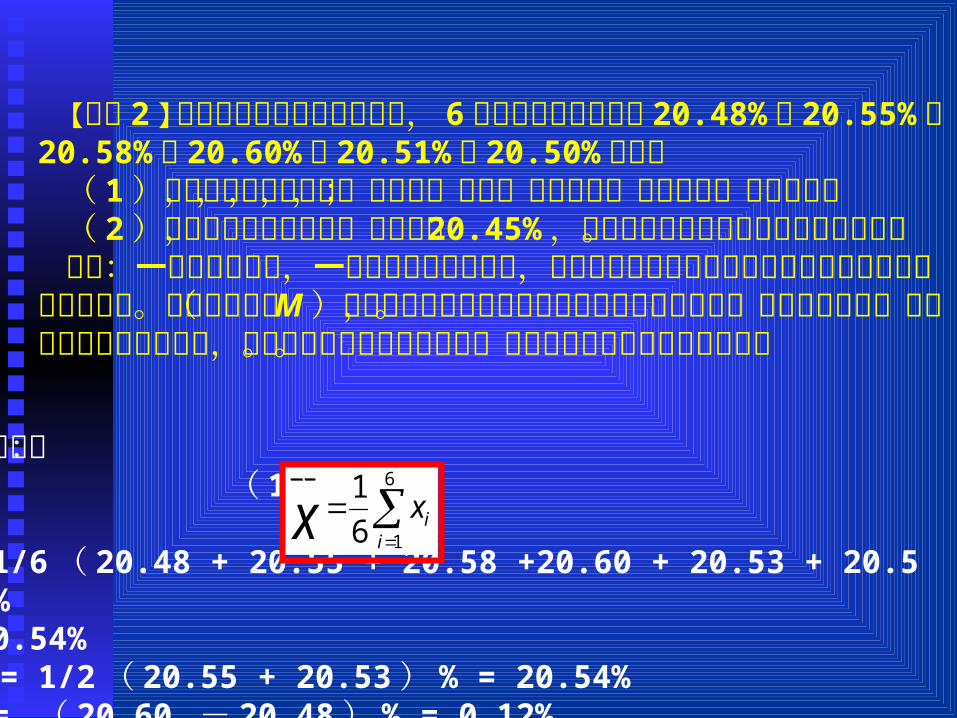
【例题 2 】某同学测定样品中的含氮量, 6次平行测定的结果是 20.48% , 20.55% , 20.58% , 20.60% , 20.51% , 20.50% ,计算
( 1 )这组测量值的平均值,中位数,极差,平均偏差,标准偏差,变异系数;
( 2 )若此样品为标准样品,含氮量为 20.45% ,计算测定结果的绝对误差和相对误差。
分析:一个样品的分析,一般要作几次平行测定,测定结果以几次平行测定所得数据的平均值或中位数表示。其中中位数( M )是指将平行测定的几个结果按大小顺序排列后,处于中间的数。若平行测定的次数为偶次,则为正中间的两数之平均值。其他的均可根据公式进行计算。
x【解析】: ( 1 )
= 1/6( 20.48 + 20.55 + 20.58 +20.60 + 20.53 + 20.50)%
=20.54%M = 1/2( 20.55 + 20.53) % = 20.54%R = ( 20.60 - 20.48) % = 0.12%
6
16
1
iix

d =
n
xxn
ii
1
= 1/6 54.2048.20 ( + 54.2055.20 54.2058.20
++
+
54.2060.20 54.2053.20 54.2050.20 + ) % = 0.037%
= {[(20.48- 20.54)2 + (20.55- 20.54)2+ (20.58- 20.54)2
+ (20.60- 20.54)2+ (20.53- 20.54)2+ (20.50- 20.54)2]×1/5}1/2
= 0.046 (%)
1
1
2
n
xxS
n
ii
C•V %= x
S×100 =
%54.20
%046.0×100 = 0.22
( 2 ) AE = x – T = (20.54 - 20. 45)% = +0.09%
RE = %100T
AE ≈ %100x
AE = %10045.20
09.0
= +0.44%
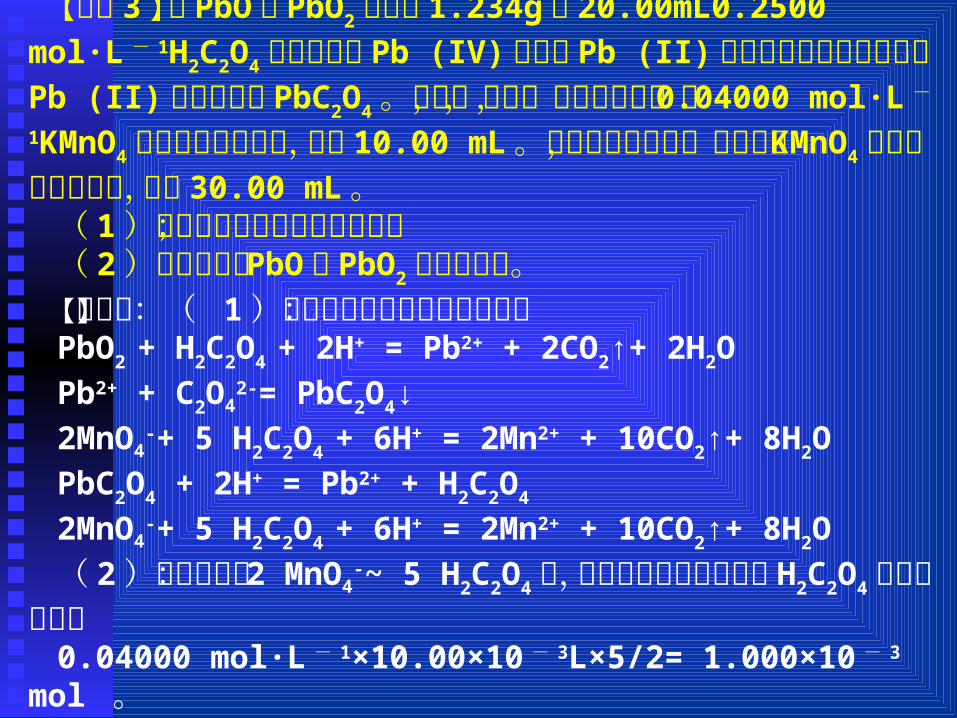
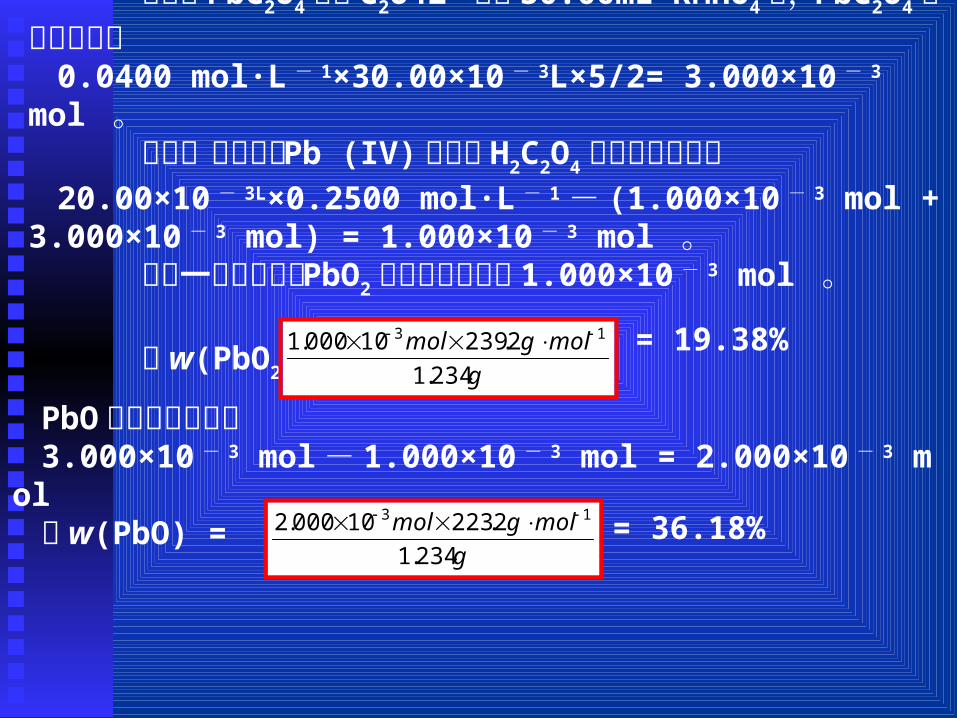
【例题 3】含 PbO 和 PbO2的试样 1.234g 用 20.00mL0.2500 mol·L - 1H2C2O4处理,此时 Pb (IV)还原为 Pb (II),降低溶液酸度,使全部 Pb (II)定量沉淀为 PbC2O4。过滤,洗涤,滤液酸化后,用0.04000 mol·L - 1KMnO4溶液滴定至终点时,用去 10.00 mL。沉淀用酸溶解后,用同样的 KMnO4溶液滴定至终点时,用去 30.00 mL。( 1)写出有关的化学反应方程式;( 2)计算试样中 PbO和 PbO2的百分含量。【解析】:( 1)本题涉及的五步反应依次为:PbO2 + H2C2O4 + 2H+ = Pb2+ + 2CO2↑+ 2H2OPb2+ + C2O4
2-= PbC2O4↓2MnO4
-+ 5 H2C2O4 + 6H+ = 2Mn2+ + 10CO2↑+ 8H2OPbC2O4 + 2H+ = Pb2+ + H2C2O4
2MnO4-+ 5 H2C2O4 + 6H+ = 2Mn2+ + 10CO2↑+ 8H2O
( 2)由关系式: 2 MnO4-~ 5 H2C2O4知,反应后的滤液中剩余
的 H2C2O4物质的量为:0.04000 mol·L - 1×10.00×10 - 3L×5/2= 1.000×10 - 3 mol 。
由滴定 PbC2O4中的 C2O42-用去 30.00mL KMnO4知, PbC2O4
物质的量为:0.0400 mol·L - 1×30.00×10 - 3L×5/2= 3.000×10 - 3 mol 。 则第一个反应被 Pb (IV)氧化的 H2C2O4的物质的量为:20.00×10 - 3L×0.2500 mol·L - 1- (1.000×10 - 3 mol + 3.000×10 - 3
mol) = 1.000×10 - 3 mol 。 由第一个反应可知 PbO2的物质的量亦为 1.000×10 - 3 mol 。
故 w(PbO2) = g
molgmol
234.1
2.23910000.1 13 = 19.38%
PbO的物质的量为:3.000×10 - 3 mol- 1.000×10 - 3 mol = 2.000×10 - 3
mol故 w(PbO) =
g
molgmol
234.1
2.22310000.2 13 = 36.18%

【例题 4】测定土壤中 SO42-的含量的主要步骤如下:称取 50g
风干土样,用水浸取,过滤,滤液移入 250mL容量瓶中,定容。用移液管移取 25.00mL浸取液,加入 1︰ 4盐酸 8滴,加热至沸,用吸量管缓慢地加入过量钡镁混合液(浓度各为 0.0200 mol·L -1) V1(mL)。继续煮沸 5分钟,冷却后,加入氨缓冲溶液( pH=10) 2 mL,以铬黑 T为指示剂,用 0.00200 mol·L - 1的 EDTA标准溶液滴定至溶液由红色变蓝色即为终点,消耗 V2(mL)。另取 25.00 mL蒸馏水,加入 1︰ 4盐酸 8滴,加热至沸,用吸量管缓慢地加入过量钡镁混合液 V2(mL),同前述步骤处理,滴定消耗 EDTA V3(mL)。另取 25.00 mL浸取液,加入 1︰ 4盐酸8滴,加热至沸,冷却后,加入氨缓冲溶液( pH=10) 2 mL,同前述步骤处理,滴定消耗 EDTA V4(mL)。计算每 100g干土样中 SO4
2-的质量。【分析】:这一实验分 3 步进行:① SO42-转化为沉淀后,剩余
的 Ba2+、Mg2+用 EDTA滴定;②未取试样,而是用蒸馏水作空白实验,可认为是测水样中的 SO4
2-;③用了试样,而未用钡、镁试液,可认为是测试样中的 Ba2+、Mg2+ 含量。
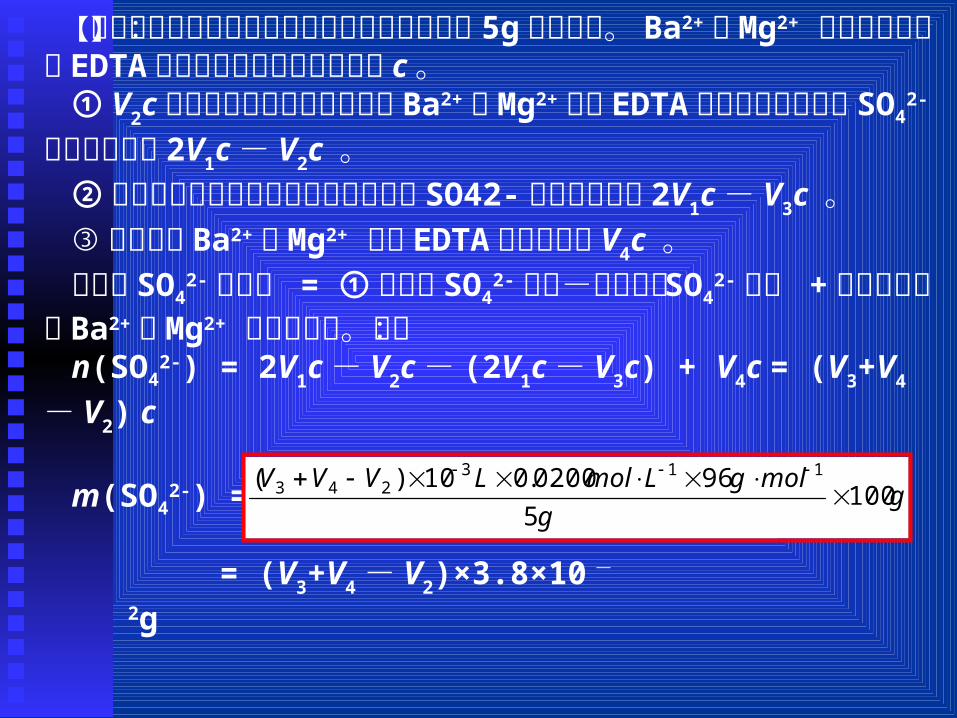
【解析】:此实验过程中所取的干土浸取液实为 5g的干土样。Ba2+、Mg2+ 混合液的浓度和 EDTA的浓度相等,设它的浓度为 c。
① V2c为生成硫酸盐沉淀后剩余的 Ba2+、Mg2+消耗 EDTA的物质的量,那么 SO4
2-的物质的量为 2V1c- V2c 。②用蒸馏水作空白实验,测出蒸馏水中 SO42-的物质的量为 2
V1c- V3c 。③试样中的 Ba2+、Mg2+ 消耗 EDTA的物质的量 V4c 。试样中 SO4
2- 的含量 = ①测得的 SO42-的量-蒸馏水中 SO4
2-的 量 +试样中所含的 Ba2+、Mg2+ 的物质的量。即:n(SO4
2-) = 2V1c- V2c- (2V1c- V3c) + V4c = (V3+V4- V2) c
m(SO42-) = g
g
molgLmolLVVV100
5
960200.010)( 113243
= (V3+V4 - V2)×3.8×10 -
2g

【例题 5】海坜正常生长所需最低的氯离子浓度是 8 mol·L - 1。( 1)一周大雨之后,对海湾的水进行分析。向 50.00mL海湾的水样中加几滴 K2CrO4指示剂,用 16.16mL0.00164 mol·L - 1的 AgNO3溶液滴定,终点时形成明显的砖红色沉淀。已知: Ksp(AgCl=1.78×10-10) , Ksp(Ag2CrO4)=1.00×10-12
①样品中氯离子物质的量浓度是多少?②水样中是否含有足够的氯离子以供海坜正常生长?写出计算过程。③写出滴定剂与样品反应的化学方程式。④写出滴定终点颜色变化的离子方程式,指出反应式中的砖红色化合物。⑤在滴定终点, CrO4
2-的浓度是 0.020mol·L - 1 ,计算当砖红色沉淀出现时溶液中 Cl -的浓度。⑥为使滴定更有效,被滴定液必须呈中性或弱碱性。写出用来描述在酸性介质中所发生的竞争反应的化学方程式(这个反应影响终点的观察)。

( 2)如果开始滴定时样品溶液是酸性的,通常向被滴溶液中加入缓冲溶液以控制 pH。假定海湾水的 pH为 5.10,则由于酸性太强而不能进行准确分析。①从列出体系中选择一个缓冲剂,此缓冲剂能使你建立并维持
pH = 7.20的水溶液介质。(假定缓冲剂不与样品和滴定剂反应) 缓冲体系 弱酸的 Ka(25℃)
a. 0.1mol·L - 1 乳酸 ~ 0.1mol·L - 1 乳酸钠 1.4×10 - 4
b. 0.1mol·L - 1 醋酸 ~ 0.1mol·L - 1 醋酸钠 1.8×10 - 5
c. 0.1mol·L - 1NaH2PO4 ~ 0.1mol·L - 1Na2HPO4 6.2×10 - 8
d. 0.1mol·L - 1 硝酸铵 ~ 0.1mol·L - 1 氨水 5.6×10 - 10
写出使你做出这种选择的计算过程。②用①选出的缓冲体系计算溶解在蒸馏水中以配制 500mLpH = 7
.20的缓冲溶液所需的弱酸及其共轭碱的质量。

( 3)在另一个 50.00mL海湾水样中的氯离子浓度由佛尔哈德法测定。将过量的 AgNO3加到样品中,过量的 Ag+ 用标准 KSCN
溶液滴定,生成 AgSCN沉淀。若加入 50.00mL0.00129 mol·L - 1 AgNO3溶液到水样后引起的过量 Ag+ 需要 27.46mL1.41×10 - 3 mol·L- 1 KSCN溶液来滴定,计算海湾水样中氯离子的浓度。( 4)在具有更高氯离子浓度的天然水中,氯离子可以通过沉淀为 AgCl的重量法来测定。此法的缺点之一是 AgCl易发生分解反应: AgCl(s)= Ag(s) + 1/2Cl2(g)
如果这一光解反应在过量 Ag+ 存在下发生,则伴随另一反应:3Cl2(g) + 2H2O(l) + 5 Ag+ (aq)
= 5AgCl(s) + ClO3-(aq)+ 6H+(aq)
如果 3.000 g AgCl样品(这些样品同含有 Ag+的溶液接触)中有0.0100g发生了光解反应,请计算由这些反应所产生的固体的最后总质量。

【分析】:该题为沉淀滴定法计算题,内容涉及莫尔法和佛尔哈德法滴定的结果计算,沉淀平衡浓度计算及有关的平衡方程式,还涉及缓冲溶液的 pH 计算及配制等问题。解题时需弄清:①沉淀滴定反应的计量关系均为 1︰ 1 。莫尔法测定氯离子时,氯离子物质的量与所消耗硝酸银物质的量相等;佛尔哈德法测定氯离子时,氯离子物质的量为加入的硝酸银物质的量与消耗的硫氰化钾物质的量之差。②当一种沉淀剂同时与两种组分形成沉淀,且均达到沉淀平衡
时,可根据一组分的平衡浓度,通过两个沉淀的溶度积常数求得另一组分的平衡浓度。③对于弱酸及其共轭碱组成用于控制溶液酸度的缓冲溶液,通
常采用最简式计算其 pH 。
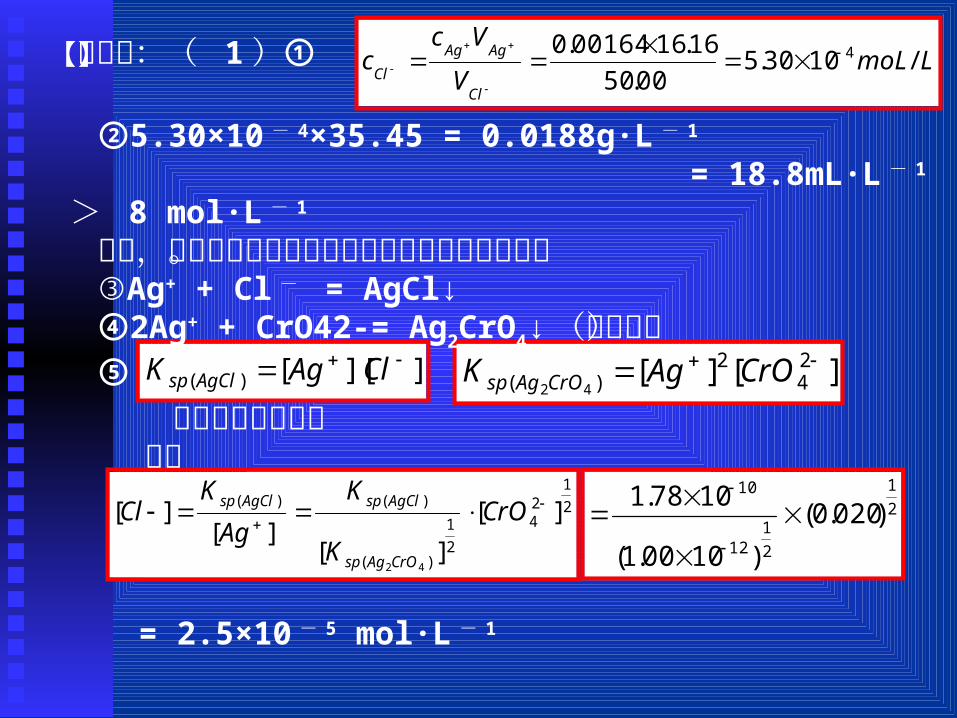
【解析】:( 1)① LmoLV
Vcc
Cl
AgAg
Cl/1030.5
00.50
16.1600164.0 4
]][[)( ClAgK AgClsp ][][ 2
42
)( 42
CrOAgK CrOAgsp
②5.30×10 - 4×35.45 = 0.0188g·L - 1
= 18.8mL·L - 1 > 8 mol·L - 1
可见,水样中含有足够的氯离子以供海坜正常生长。③Ag+ + Cl - = AgCl↓④2Ag+ + CrO42-= Ag2CrO4↓(砖红色)⑤
2
124
2
1
)(
)()( ][
][][
][
42
CrO
K
K
Ag
KCl
CrOAgsp
AgClspAgClsp 2
1
2
112
10
)020.0(
)1000.1(
1078.1
两个沉淀均达到平衡时
= 2.5×10 - 5 mol·L - 1
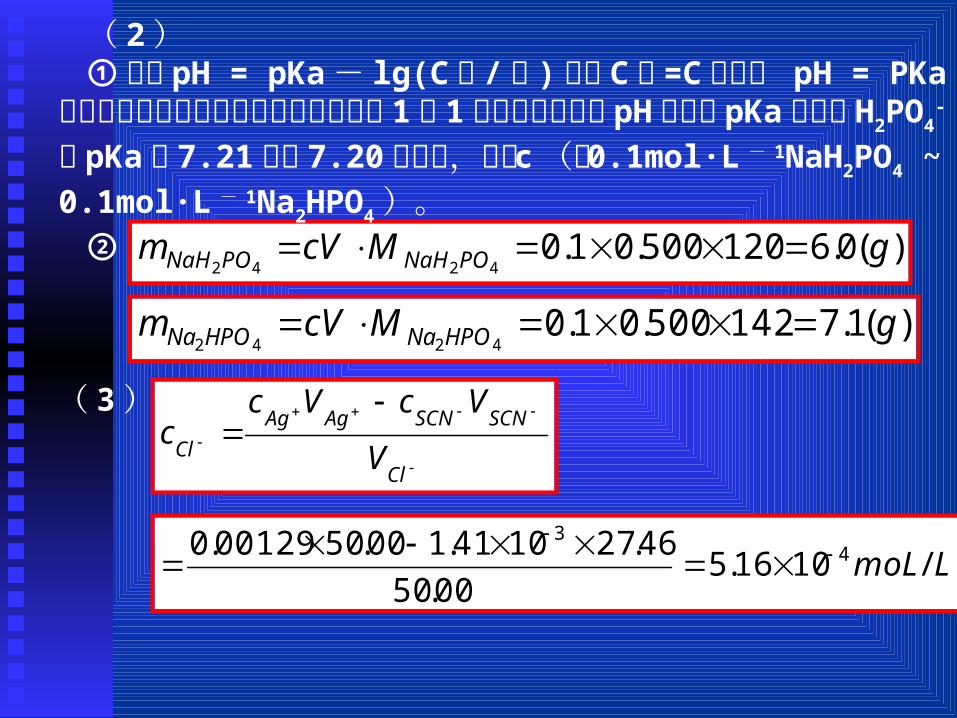
)(0.6120500.01.04242
gMcVm PONaHPONaH
)(1.7142500.01.04242
gMcVm HPONaHPONa
Cl
SCNSCNAgAg
Cl V
VcVcc
LmoL /1016.500.50
46.271041.100.5000129.0 43
( 2)①根据 pH = pKa - lg(C 酸 / 碱 ),当 C 酸 =C 碱时, pH = PKa,题给四种缓冲溶液的缓冲组分浓度均为 1 ︰ 1,故所能控制的pH均与其 pKa相同, H2PO4
- 的 pKa 为 7.21,与 7.20最接近,故选 c(即 0.1mol·L - 1NaH2PO4 ~ 0.1mol·L - 1Na2HPO4)。
②
( 3)
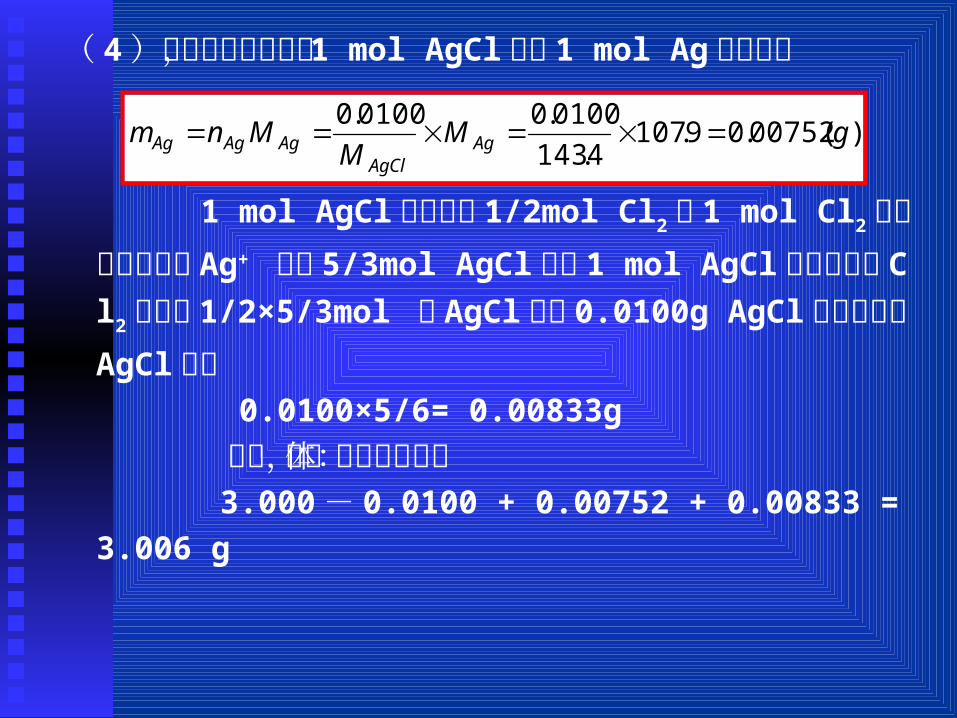
( 4)由光解反应可知, 1 mol AgCl产生 1 mol Ag ,所以:
)(00752.09.1074.143
0100.00100.0gM
MMnm Ag
AgClAgAgAg
1 mol AgCl光解产生 1/2mol Cl2, 1 mol Cl2又可与溶液中的 Ag+ 生成 5/3mol AgCl,故 1 mol AgCl分解产生的 Cl2
可生成 1/2×5/3mol 的 AgCl,由 0.0100g AgCl光解产生的AgCl为:
0.0100×5/6= 0.00833g 所以,固体的总质量为: 3.000- 0.0100 + 0.00752 + 0.00833 = 3.006 g
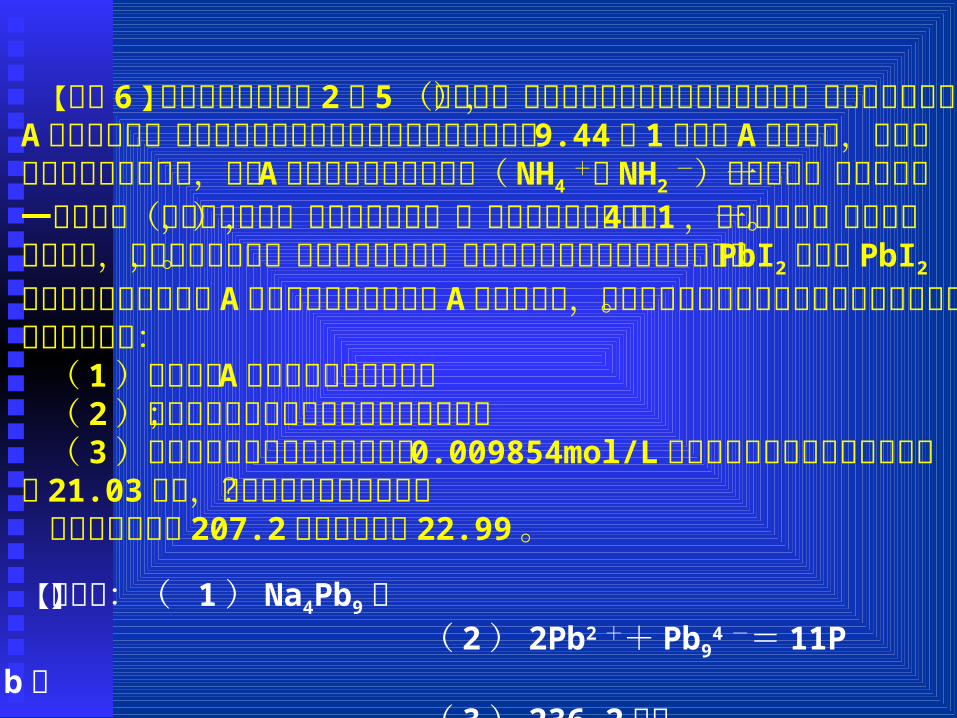
【例题 6 】金属钠和金属铅的 2︰ 5 (摩尔比)的合金可以部分地溶解于液态氨,得到深绿色的溶液 A ,残留的固体是铅,溶解的成分和残留的成分的质量比为 9.44︰ 1 ,溶液 A 可以导电,摩尔电导率的测定实验证实,溶液 A 中除液氨原有少量离子( NH4
+
和 NH2-)外只存在一种阳离子和一种阴离子(不考虑溶剂合,即
氨合的成分),而且它们的个数比是 4︰ 1 ,阳离子只带一个电荷。通电电解,在阳极上析出铅,在阴极上析出钠。用可溶于液氨并在液氨中电离的盐 PbI2配制的 PbI2 的液氨溶液来滴定溶液 A ,达到等当点时,溶液 A 的绿色褪尽,同时溶液里的铅全部以金属铅的形式析出。回答下列问题:
( 1 )写出溶液 A 中的电解质的化学式;( 2 )写出上述滴定反应的配平的离子方程式;( 3 )已知用于滴定的碘化铅的浓度为 0.009854mol/L ,达到等
当点时消耗掉碘化铅溶液 21.03毫升,问共析出金属铅多少克?附:铅的原子量 207.2;钠的原子量 22.99 。
【解析】:( 1 ) Na4Pb9; ( 2 ) 2Pb2 ++ Pb9
4 -= 11Pb; ( 3 ) 236.2毫克。


![Jobs by Walter 史蒂夫•乔布斯传€Š史蒂夫·乔布斯传》官方...Steve Jobs by Walter Isaacson | 史蒂夫•乔布斯传[美]沃尔特•艾萨克森著 CHINA 中信出版社](https://img.pdfslide.tips/doc/110x75/6064f473017f695e2e727f66/jobs-by-walter-eaf-ef-steve.jpg)












![【外国文学名著丛书】坎特伯雷故事[英]杰弗雷·乔叟 方重](https://img.pdfslide.tips/doc/110x75/568cc6881a28ab8c668b4b15/-568cc6881a28ab8c668b4b15.jpg)





![[习惯的力量] the power of habit [美]杰克霍吉 2004 (美)杰克·霍吉](https://img.pdfslide.tips/doc/110x75/568bd4a51a28ab2034958b7a/-the-power-of-habit-2004-.jpg)







