Embed Size (px)
Citation preview

THESETHESEEn vue de l'obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSEDOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par l'Université Toulouse III - Paul SabatierDiscipline ou spécialité : Biologie cellulaire et moléculaire
JURYMr J-C. MARTINOU, Professeur, Université Sciences III Genève (Rapporteur)
Mr F. VALLETTE, Directeur de Recherches INSERM, UMRS892 Nantes. (Rapporteur)Mr J. ESTAQUIER, Directeur de Recherches INSERM, U955 Créteil. (Examinateur)
Mme M. PRIAULT, Directeur de Recherches CNRS, UMR5095 Bordeaux. (Examinateur)Mme L. ARNAUNE, Maître de conférences, UMR5241 Toulouse. (co-Directeur de thèse)
Mme P. BELENGUER, Maître de conférences, UMR5241 Toulouse. (co-Directeur de thèse)
Ecole doctorale : ED Biologie Santé Biotechnologies de ToulouseUnité de recherche : UMR5241 /UMR5088
Directeur(s) de Thèse : L.ARNAUNE et P. BELENGUER
Présentée et soutenue par LANDES ThomasLe 16 Octobre 2009
Titre : Dynamique mitochondriale et apoptose:rôle de l'interaction entre Opa1 et Bnip3


Je remercie tout d’abord les membres du jury Mme Priault, Mr Estaquier, Mr Martinou et Mr
Vallette d’avoir accepté d’évaluer ce travail de thèse.
Merci à tous les membres du LBCMCP et du MPM pour leur accueil, et leur gentillesse.
Merci plus particulièrement à ma « maman » au labo, Laetitia. Tu as toujours été disponible
quelquesoit le moment et cela vaut tout l’or du monde lorsque l’on débute. Je te remercie pour
l’encadrement de qualité que tu m’as offert mais également pour le soutien et les
encouragements que tu m’as donnés au cours de ces années.
Pascale et Laurent, merci pour votre disponibilité, votre attention, vos remarques et conseils
toujours très judicieux, ainsi que pour votre aide prépondérante notamment sur la fin de ma
thèse.
Merci Marlène pour ton travail dans l’ombre, indispensable au bon fonctionnement de
l’équipe.
Merci Delphine pour ton œil expert en microscopie et pour les diverses discussions que nous
avons pu avoir.
Merci à tout le reste de l’équipe, ceux qui sont encore présents à l’heure actuelle (Alan,
Ambre, Farnoosh, Marie-Christine, Noélie et Valérie) ou ceux qui sont partis (Ingrid et Manu
notamment).
Enfin, merci à mes proches ; avec, bien entendu, une mention toute particulière pour les deux
personnes qui partagent mon quotidien et qui ont la dure tâche de me supporter. En effet,
merci Marie et Paul, pour m’avoir encouragé et m’avoir changé les idées dans certains
moments parfois un peu difficiles.


1
Introduction ......................................................................................................................... 7PREAMBULE.................................................................................................................... 11I) L’APOPTOSE ................................................................................................................ 17
A) La voie extrinsèque ..................................................................................................... 19B) La voie intrinsèque...................................................................................................... 19
1) Les protéines apoptogènes libérées de la mitochondrie au cours de l’apoptose ......... 192) Le mécanisme de perméabilisation de la membrane externe mitochondriale............. 23
II) LA STRUCTURE DYNAMIQUE DES MITOCHONDRIES .................................... 27A) La distribution subcellulaire et la mobilité des mitochondries...................................... 27B) La dynamique des membranes mitochondriales ........................................................... 31
1) Les dynamines ......................................................................................................... 31a) La structure des dynamines................................................................................... 31b) Le rôle des dynamines.......................................................................................... 33c) Le fonctionnement des dynamines........................................................................ 35
2) Les acteurs de la fission mitochondriale ................................................................... 35a) Les protéines essentielles...................................................................................... 35
Drp1/Dnm1 .......................................................................................................... 35hFis1 .................................................................................................................... 41
b) Les protéines régulatrices ..................................................................................... 41Endophiline B1..................................................................................................... 41Ganglioside-induced Differentiation Associated Protein 1 (GDAP1) .................... 43
c) Existe-t-il une machinerie de fission de la membrane interne ? ............................. 433) La fusion mitochondriale.......................................................................................... 45
a) Les protéines essentielles...................................................................................... 45Les mitofusines 1 et 2 / Fzo1 ................................................................................ 45Opa1/Mgm1/Msp1/Eat-3 ...................................................................................... 49
b) Le couplage de la fusion des deux membranes...................................................... 53c) Les protéines régulatrices ..................................................................................... 55
mitoPLD............................................................................................................... 55MIB...................................................................................................................... 55SLP-2 ................................................................................................................... 55
4) L’architecture des crêtes mitochondriales................................................................. 57C) Les fonctions biologiques de la dynamique mitochondriale ......................................... 59
1) La transmission des mitochondries au cours de la division cellulaire ........................ 592) Le maintien des fonctions mitochondriales............................................................... 613) La distribution des mitochondries............................................................................. 634) La mitophagie .......................................................................................................... 65
D) Dynamique mitochondriale et développement ............................................................. 69E) Dynamique mitochondriale et neurodégénérescence .................................................... 69
III) LES RELATIONS ENTRE LA DYNAMIQUE MITOCHONDRIALE ET L’APOPTOSE.................................................................................................................... 73
A) Le rôle des acteurs de la dynamique mitochondriale dans l’apoptose........................... 73B) Le rôle des protéines de la famille de Bcl-2 dans la dynamique mitochondriale ........... 79
Résultats ............................................................................................................................. 83Première Partie Interaction entre Opa1 et Bnip3................................................................. 85I) RECHERCHE DE PARTENAIRES DE LA PROTÉINE OPA1 ................................ 87
A) Description du système double hybride ....................................................................... 87B) Validation de l’approche expérimentale....................................................................... 89C) Criblage de la banque .................................................................................................. 93

2
II) CARACTÉRISATION DE L’INTERACTION ENTRE LES PROTÉINES OPA1 ET BNIP3 ................................................................................................................................. 99
A) Validation de l’interaction entre Opa1 et Bnip3......................................................... 1011) in vitro ................................................................................................................... 1012) in cellulo, en surexpression ectopique des deux partenaires .................................... 1033) in cellulo, à un taux physiologique des deux partenaires......................................... 105
B) Caractérisation des domaines requis pour l’interaction .............................................. 1091) Bnip3 ..................................................................................................................... 1092) Opa1 ...................................................................................................................... 111
C) Caractérisation fonctionnelle de l’interaction entre Opa1 et Bnip3............................. 1131) Étude de l’effet de Bnip3 sur la dynamique mitochondriale.................................... 1132) Étude de l’effet de Bnip3 sur la viabilité cellulaire ................................................. 1213) Étude de la relevance de l’interaction entre Opa1 et Bnip3..................................... 125
a) Étude de l’effet des mutants de Bnip3................................................................. 125b) Étude de l’effet d’Opa1 ...................................................................................... 127
4) Étude du mode d’action de Bnip3........................................................................... 129III) CONCLUSION.......................................................................................................... 137Deuxième Partie Mutants pathogènes de OPA1 ................................................................ 151I) PUBLICATION ........................................................................................................... 153
Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis....................................................................................................... 153
II) RÉSULTATS SUPPLÉMENTAIRES ....................................................................... 165III) RÉSUMÉ ET DISCUSSION DE L’ARTICLE ........................................................ 167
Matériel et méthodes........................................................................................................ 181I) CULTURE CELLULAIRE, TRANSFECTIONS ET TRAITEMENTS ................... 183II) TECHNIQUES UTILISÉES POUR LA MISE EN ÉVIDENCE ET LA VALIDATION DE L’INTERACTION........................................................................... 183
A) Double hybride ......................................................................................................... 1831) Les vecteurs d’expression....................................................................................... 1842) Transformation....................................................................................................... 1843) Extraction protéique en conditions détergentes....................................................... 1864) Essai �galactosidase sur filtre................................................................................. 1865) Préparation de la banque ........................................................................................ 1876) Transformation de la banque et sélection................................................................ 1887) Elimination des “ faux positifs ”............................................................................. 189
B) GST pull-down.......................................................................................................... 1911) Préparation des protéines recombinantes protéines recombinantes.......................... 191
a) Production.......................................................................................................... 191b) Purification ........................................................................................................ 191c) Élution ............................................................................................................... 192
2) Traduction in vitro en présence de méthionine 35S. ................................................. 1923) Préparation des mitochondries................................................................................ 1924) Interaction.............................................................................................................. 193
C) Immuno-précipitation................................................................................................ 194III) TECHNIQUES UTILISÉES POUR L’ANALYSE DE LA DYNAMIQUE MITOCHONDRIALE ..................................................................................................... 195
A) Marquage des mitochondries..................................................................................... 195B) Tests de fusion au polyéthylèneglycol ....................................................................... 195C) Photoconversion........................................................................................................ 196

3
1) Préparation............................................................................................................. 1962) Acquisition des données......................................................................................... 1963) Traitement des données.......................................................................................... 196
IV) TECHNIQUES UTILISÉES POUR L’ANALYSE DE LA MORT CELLULAIRE.......................................................................................................................................... 197
A) Analyse de l’apoptose ............................................................................................... 1971) Observation des noyaux ......................................................................................... 1972) Observation des oligomères d’Opa1....................................................................... 1983) Observation du potentiel de membrane................................................................... 198
B) Analyse de l’autophagie ............................................................................................ 1991) Par microscopie...................................................................................................... 1992) Par Western-blot .................................................................................................... 199
V) TECHNIQUES COURANTES RELATIVES AUX PROTÉINES........................... 199A) Extraction de protéines totales cellulaires.................................................................. 199B) Electrophorèse et immunodétection........................................................................... 200C) Immunofluorescence indirecte................................................................................... 200
VI) TECHNIQUES COURANTES RELATIVES À L’ADN......................................... 201A) Clonages et sous-clonages de Bnip3.......................................................................... 201B) Sous-clonages d’Opa1............................................................................................... 202C) Souches..................................................................................................................... 202
1) Escherichia coli ..................................................................................................... 2022) Saccharomyces cerevisiae ...................................................................................... 204
D) siARN....................................................................................................................... 204E) Anticorps................................................................................................................... 205
Bibliographie.................................................................................................................... 207
Annexes ............................................................................................................................ 235Publication 1...................................................................................................................... 237
The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fission and apoptosis by distinct mechanisms................................................................................... 237
Publication 2...................................................................................................................... 267Mitochondrial dynamics and disease, OPA1................................................................... 267
Publication 3...................................................................................................................... 277Metalloprotease-mediated Opa1 processing is modulated by the mitochondrial membrane potential ......................................................................................................................... 277

4

5
Liste des figuresFigure 1. Reconstructions tomographiques de mitochondries de foie de souris. .................... 10Figure 2. La phosphorylation oxydative. .............................................................................. 12Figure 3. Structure de l’ADN mitochondrial humain. ........................................................... 14Figure 4. Les différentes voies d’import mitochondrial......................................................... 15Figure 5. Les voies extrinsèque et intrinsèque de l’apoptose................................................. 16Figure 6. Perméabilisation de la membrane externe mitochondriale...................................... 22Figure 7. Représentation schématique des protéines de la famille de Bcl-2.......................... 24Figure 8. Morphologie des mitochondries dans différents types cellulaires........................... 26Figure 9. Le transport des mitochondries.............................................................................. 28Figure 10. La dynamique des membranes mitochondriales. .................................................. 30Figure 11. Structure des protéines de la famille des dynamines. ........................................... 30Figure 12. Les motifs de liaison au GTP............................................................................... 30Figure 13. Les différents modèles du mécanisme d’action des dynamines dans la fission des
membranes. .................................................................................................................. 34Figure 14. Effet d’une mutation à effet dominant négatif de Drp1 sur la morphologie des
mitochondries............................................................................................................... 36Figure 15. L’auto-assemblage sous forme de spirales de Dnm1............................................ 36Figure 16. L’auto-assemblage sous forme d’anneaux de Drp1. ............................................. 36Figure 17. Effet de la perte en Mitofusines sur la morphologie des mitochondries................ 44Figure 18. Représentation schématique du test de fusion après fusion cellulaire au
polyéthylèneglycol (PEG). ........................................................................................... 46Figure 19. Représentation schématique des 8 variants d’épissage de Opa1. .......................... 48Figure 20. Les différentes isoformes protéiques d’Opa1. ...................................................... 48Figure 21. Effet de la surexpression et de l’extinction d’Opa1 sur la morphologie des
mitochondries............................................................................................................... 50Figure 22. Changements de topologie de la membrane interne mitochondriale lors de la
transition orthodoxe-condensée. ................................................................................... 56Figure 23. Effet de la délétion des sous-unités e et g de l’ATP synthase sur la topologie de la
membrane interne mitochondriale chez Saccharomyces cerevisiae. .............................. 56Figure 24. Effet de l’extinction de l’expression du gène Mitofiline sur la topologie de la
membrane interne mitochondriale................................................................................. 58Figure 25. Effet de la perte de Eat-3, Opa1 et Mgm1 sur la topologie de la membrane interne
mitochondriale. ............................................................................................................ 58Figure 26. La macroautophagie et sa régulation par les protéines Atg................................... 66Figure 27. La libération du cytochrome c dans le cytosol. .................................................... 76Figure 28. Principe expérimental du double hybride............................................................. 86Figure 29. Validation de l'approche expérimentale. .............................................................. 88Figure 30. Criblage de la banque à la recherche de partenaires d’Opa1................................. 94Figure 31. Séquence du clone 11 isolé en double hybride. ................................................... 98Figure 32. Interaction in vitro entre Opa1 et Bnip3............................................................. 102Figure 33. Expression et localisation de HA-Bnip3. ........................................................... 102Figure 34. Validation in cellulo de l’interaction entre Opa1 et Bnip3 après surexpression dans
les cellules HeLa. ....................................................................................................... 104Figure 35. Expression de HIF1� et de Bnip3 lors de traitements CoCl2 et DFO. ................ 106Figure 36. Validation in cellulo de l’interaction entre Opa1 et Bnip3 après traitement des
cellules HeLa au chlorure de cobalt (CoCl2)............................................................... 106Figure 37. Représentation schématique et expression des mutants de Bnip3....................... 108Figure 38. Caractérisation des domaines de Bnip3 requis pour l’interaction. ...................... 110

6
Figure 39. Caractérisation des domaines d’Opa1 requis pour l’interaction.......................... 112Figure 40. Morphologie mitochondriale des cellules HeLa surexprimant Bnip3. ................ 112Figure 41. Observation du potentiel de membrane mitochondrial dans des cellules
surexprimant Bnip3. ................................................................................................... 114Figure 42. Morphologie mitochondriale des cellules HeLa surexprimant Bnip3 et/ou Drp1
et/ou Drp1K38A......................................................................................................... 116Figure 43. Effet de Bnip3 sur la fusion mitochondriale après fusion cytoplasmique au
polyéthylèneglycol. .................................................................................................... 116Figure 44. Validation du test de fusion par photoconversion............................................... 118Figure 45. Effet de Bnip3 sur la fusion mitochondriale visualisée par photoconversion. ..... 120Figure 46. Effet de Bnip3 sur la morphologie des mitochondries en hypoxie...................... 122Figure 47. Effet de la surexpression de Bnip3 sur la mort cellulaire.................................... 122Figure 48. Effet de la surexpression de Bnip3 sur l’activation de l’autophagie.................... 124Figure 49. Effet de la surexpression des mutants de Bnip3 sur la morphologie des
mitochondries et la mort cellulaire.............................................................................. 126Figure 50. Caractérisation des clones surexprimant Opa1 de manière stable et inductible... 128Figure 51. Effet de la surexpression d’Opa1 sur les effets induits par Bnip3....................... 128Figure 52. Mise au point des conditions optimales d’extinction de l’expression de Mfn1. .. 130Figure 53. Effet de la surexpression d’Opa1 sur les effets induits par Bnip3 en absence de
Mfn1. ......................................................................................................................... 130Figure 54. Oligomérisation d’Opa1 en présence de tBid..................................................... 132Figure 55. Oligomérisation d’Opa1 en présence de Bnip3. ................................................. 132Figure 56. Oligomérisation d’Opa1 en présence de tBid et Bnip3 dans des mitochondries
isolées de fibroblastes embryonnaires de souris invalidées pour les gènes codant pour Bax et Bak.................................................................................................................. 134
Figure 57. Oligomérisation d’Opa1 en présence des mutants de Bnip3............................... 134Figure 58. Oligomérisation d’Opa1 dans des cellules HeLa cultivées en présence de chlorure
de cobalt..................................................................................................................... 136Figure 59. Morphologie mitochondriale des cellules HeLa surexprimant Opa1 ou Opa1G300E
et/ou Drp1 et/ou Drp1K38A. ...................................................................................... 164

7
Introduction

8

9
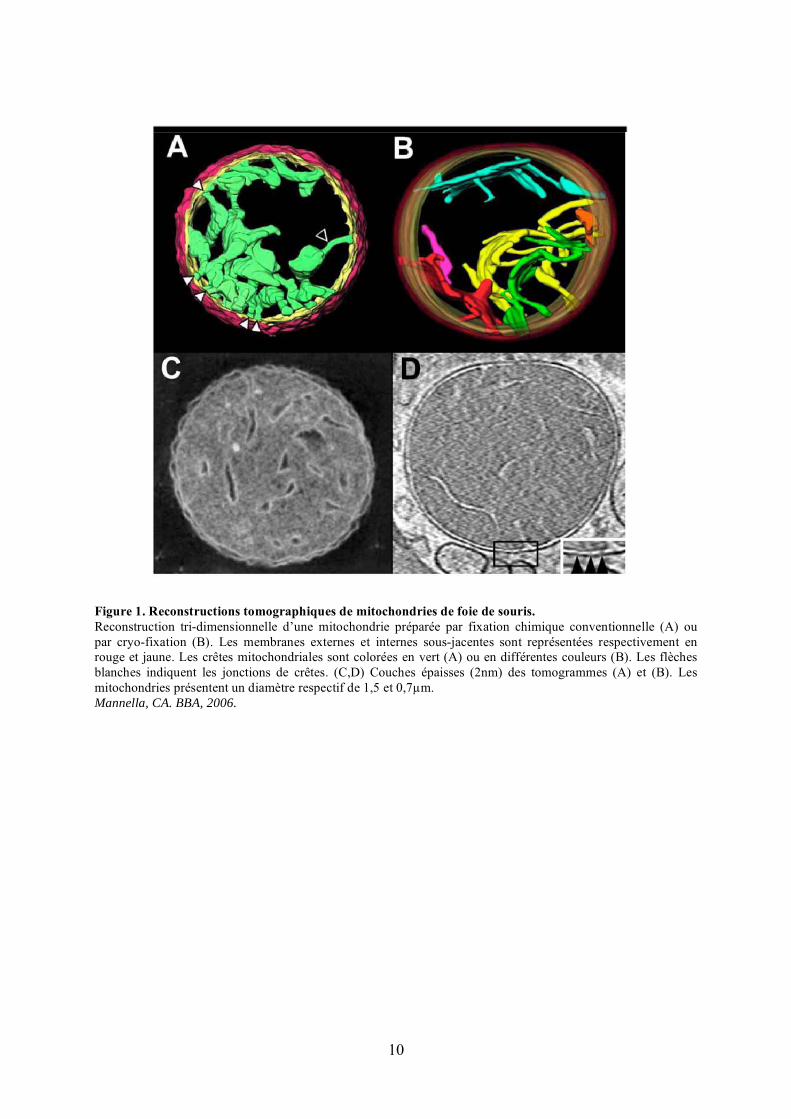
10
Figure 1. Reconstructions tomographiques de mitochondries de foie de souris.Reconstruction tri-dimensionnelle d’une mitochondrie préparée par fixation chimique conventionnelle (A) ou par cryo-fixation (B). Les membranes externes et internes sous-jacentes sont représentées respectivement en rouge et jaune. Les crêtes mitochondriales sont colorées en vert (A) ou en différentes couleurs (B). Les flèches blanches indiquent les jonctions de crêtes. (C,D) Couches épaisses (2nm) des tomogrammes (A) et (B). Les mitochondries présentent un diamètre respectif de 1,5 et 0,7�m.Mannella, CA. BBA, 2006.

11
PREAMBULE
Il y a environ 150 ans, en 1858, un histologiste et physiologiste suisse, nommé
Kölliker, décrit pour la première fois, des structures particulières présentes sous forme de
grains (« chondros » en grec) dans des cellules musculaires. Les progrès réalisés dans les
techniques de coloration et en microscopie au cours des années suivantes ont permis de
constater que, suivant les tissus, ces structures pouvaient également apparaître comme des
filaments (« mitos ») et ont conduit par conséquent à la naissance du terme « mitochondrie »
(proposé initialement par Benda en 1898). Au début des années 1940, un biochimiste belge,
Albert Claude, parvient à isoler et purifier, par centrifugation différentielle, des mitochondries
de cellules hépatiques. Cette étape s’avéra cruciale pour la suite des recherches concernant les
mitochondries qui connurent alors une période faste.
Dans les années 1950, l’ultrastructure mitochondriale fut tout d’abord abordée. Les
premiers clichés obtenus à l’époque, par microscopie électronique à transmission, montrèrent
que les mitochondries étaient entourées par deux membranes, une membrane externe et une
membrane interne, qui délimitent deux sous-compartiments : l’espace inter-membranaire et la
matrice, cette dernière renfermant les crêtes mitochondriales. Les crêtes ont été
successivement décrites comme des repliements de la membrane interne (modèle de Palade,
1952), des chambres fermées individualisées (modèle de Sjostrand, 1953), ou des chambres
reliées à la membrane interne par de fins tubules (modèle de Daems et Wisse, 1966).
L’apparition de la tomographie électronique donnant accès à la structure tridimensionnelle
des mitochondries a permis de proposer dans les années 2000 un modèle, assez proche de
celui proposé par Daems et Wisse (Frey and Mannella, 2000; Perkins and Frey, 2000), dans
lequel la membrane interne est divisée en deux domaines distincts : la membrane interne
« sous-jacente », séparée de la membrane externe par l’espace inter-membranaire, et la
membrane interne des crêtes délimitant l’espace intra-crête. Les crêtes mitochondriales
constituent donc des chambres aux morphologies variables rattachées à la membrane interne
sous-jacente par des structures appelées jonctions de crêtes (Figure 1).
Les années 1950 furent également marquées par la mise en évidence de plusieurs
fonctions métaboliques de la mitochondrie. Outre son rôle dans le cycle de Krebs ou la ß-
oxydation, la mitochondrie est dès lors apparue comme le premier fournisseur d’énergie à la
cellule via la phosphorylation oxydative (Figure 2). Les années qui suivirent furent rythmées
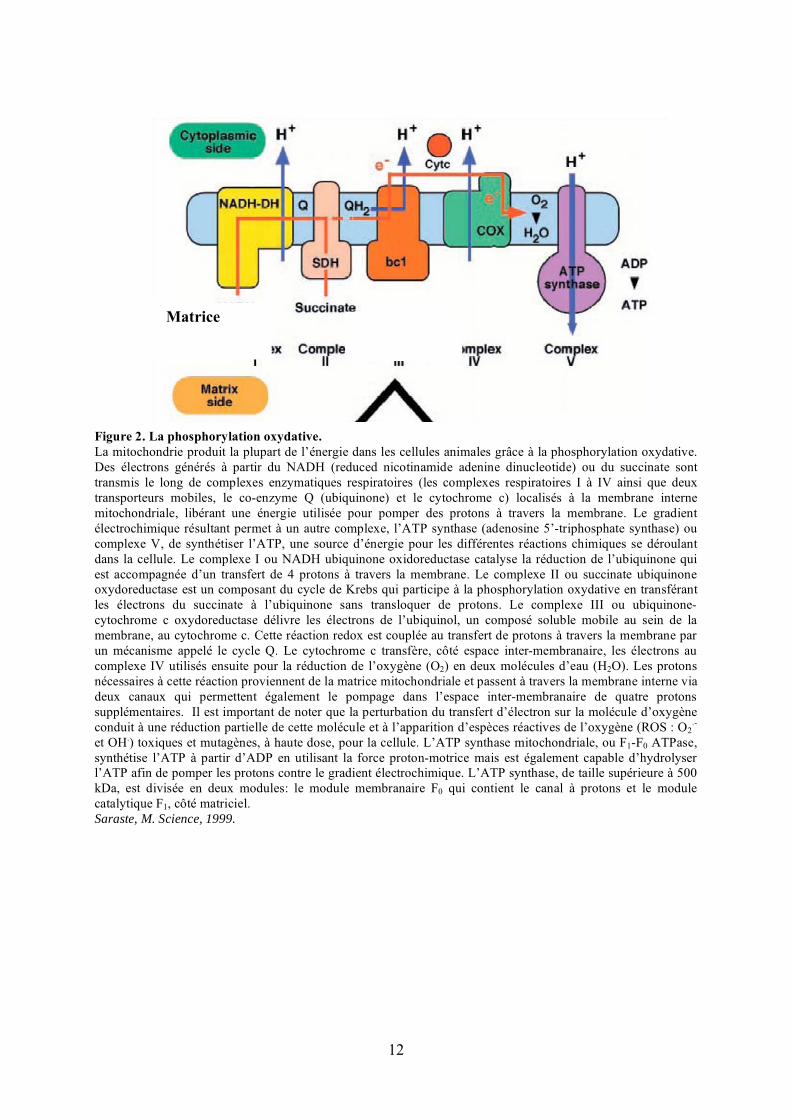
12
Figure 2. La phosphorylation oxydative.La mitochondrie produit la plupart de l’énergie dans les cellules animales grâce à la phosphorylation oxydative. Des électrons générés à partir du NADH (reduced nicotinamide adenine dinucleotide) ou du succinate sont transmis le long de complexes enzymatiques respiratoires (les complexes respiratoires I à IV ainsi que deux transporteurs mobiles, le co-enzyme Q (ubiquinone) et le cytochrome c) localisés à la membrane interne mitochondriale, libérant une énergie utilisée pour pomper des protons à travers la membrane. Le gradient électrochimique résultant permet à un autre complexe, l’ATP synthase (adenosine 5’-triphosphate synthase) ou complexe V, de synthétiser l’ATP, une source d’énergie pour les différentes réactions chimiques se déroulant dans la cellule. Le complexe I ou NADH ubiquinone oxidoreductase catalyse la réduction de l’ubiquinone qui est accompagnée d’un transfert de 4 protons à travers la membrane. Le complexe II ou succinate ubiquinone oxydoreductase est un composant du cycle de Krebs qui participe à la phosphorylation oxydative en transférant les électrons du succinate à l’ubiquinone sans transloquer de protons. Le complexe III ou ubiquinone-cytochrome c oxydoreductase délivre les électrons de l’ubiquinol, un composé soluble mobile au sein de la membrane, au cytochrome c. Cette réaction redox est couplée au transfert de protons à travers la membrane par un mécanisme appelé le cycle Q. Le cytochrome c transfère, côté espace inter-membranaire, les électrons au complexe IV utilisés ensuite pour la réduction de l’oxygène (O2) en deux molécules d’eau (H2O). Les protons nécessaires à cette réaction proviennent de la matrice mitochondriale et passent à travers la membrane interne via deux canaux qui permettent également le pompage dans l’espace inter-membranaire de quatre protons supplémentaires. Il est important de noter que la perturbation du transfert d’électron sur la molécule d’oxygène conduit à une réduction partielle de cette molécule et à l’apparition d’espèces réactives de l’oxygène (ROS : O2
.-
et OH.) toxiques et mutagènes, à haute dose, pour la cellule. L’ATP synthase mitochondriale, ou F1-F0 ATPase, synthétise l’ATP à partir d’ADP en utilisant la force proton-motrice mais est également capable d’hydrolyser l’ATP afin de pomper les protons contre le gradient électrochimique. L’ATP synthase, de taille supérieure à 500 kDa, est divisée en deux modules: le module membranaire F0 qui contient le canal à protons et le module catalytique F1, côté matriciel. Saraste, M. Science, 1999.
Matrice

13
par l’identification et la caractérisation, aussi bien fonctionnelle que structurale, des différents
complexes de la chaîne respiratoire, et se conclurent par le décernement de deux prix Nobel :
l’un à P. Mitchell en 1978 pour sa contribution à l'explication du transfert de l'énergie
biologique par la formulation de la théorie chimiostatique, et l’autre à J. Walker et P. Boyer
en 1997 pour leurs travaux sur l’adenosine triphosphate.
Un autre tournant important de l’Histoire de la mitochondrie survint lors de la mise en
évidence de la présence d’ADN au sein de cette organelle en 1963 et le séquençage complet
de l’ADN mitochondrial bovin au tout début des années 1980 (Anderson et al., 1982). Il fut
alors déterminé que l’ADN mitochondrial (ADNmt), présent en multicopies dans la cellule,
consistait en une molécule circulaire double-brin (Figure 3), complexée avec plusieurs
protéines dans la matrice pour former une structure appelée nucléoïde. La recherche sur la
mitochondrie connut par la suite plusieurs années de « disette », apparaissant aux yeux de
beaucoup comme exotique et ne renfermant plus aucun secret. Cependant, le début des années
1990 fut marqué par un net regain d’intérêt pour cette organelle de la part aussi bien de
biochimistes, que de biologistes cellulaires et moléculaires, de généticiens ou de cliniciens.
L’ADNmt ne codant que pour 13 protéines, il était donc évident que la quasi-totalité des
protéines mitochondriales provenait de l’expression du génome nucléaire. À cette époque, les
premières mutations de l’ADNmt responsables de pathologies furent également décrites et
soulevèrent des questions encore non résolues, relatives par exemple à l’hétéroplasmie,
définie par la cohabitation de différentes séquences d’ADNmt (sauvages et mutées) chez un
même individu, à la difficulté d’établir des corrélations entre génotype et phénotype, ou aux
raisons de la spécificité tissulaire (pour revue Dimauro and Davidzon, 2005). L’étude de la
biogénèse mitochondriale débuta donc et permit notamment de mettre en évidence un système
très complexe d’import des protéines dans la mitochondrie (Figure 4) (pour revue, Neupert
and Herrmann, 2007).
En 1996, la mitochondrie montra un visage tout à fait inattendu lorsqu’une protéine
indispensable à la phosphorylation oxydative, le cytochrome c, apparut également contrôler
un processus létal essentiel pour le développement normal des organismes multicellulaires,
l’apoptose (Liu et al., 1996). Ce domaine fut par la suite profondément étudié et permit
finalement de reconstituer une succession d’évènements abrités par la mitochondrie
constituant la voie intrinsèque apoptotique. Dans les années 1990, la notion de dynamique
mitochondriale commença à émerger. La mise en évidence d’interactions entre les
mitochondries et le cytosquelette (évènements dits extrinsèques) et des processus de fusion et
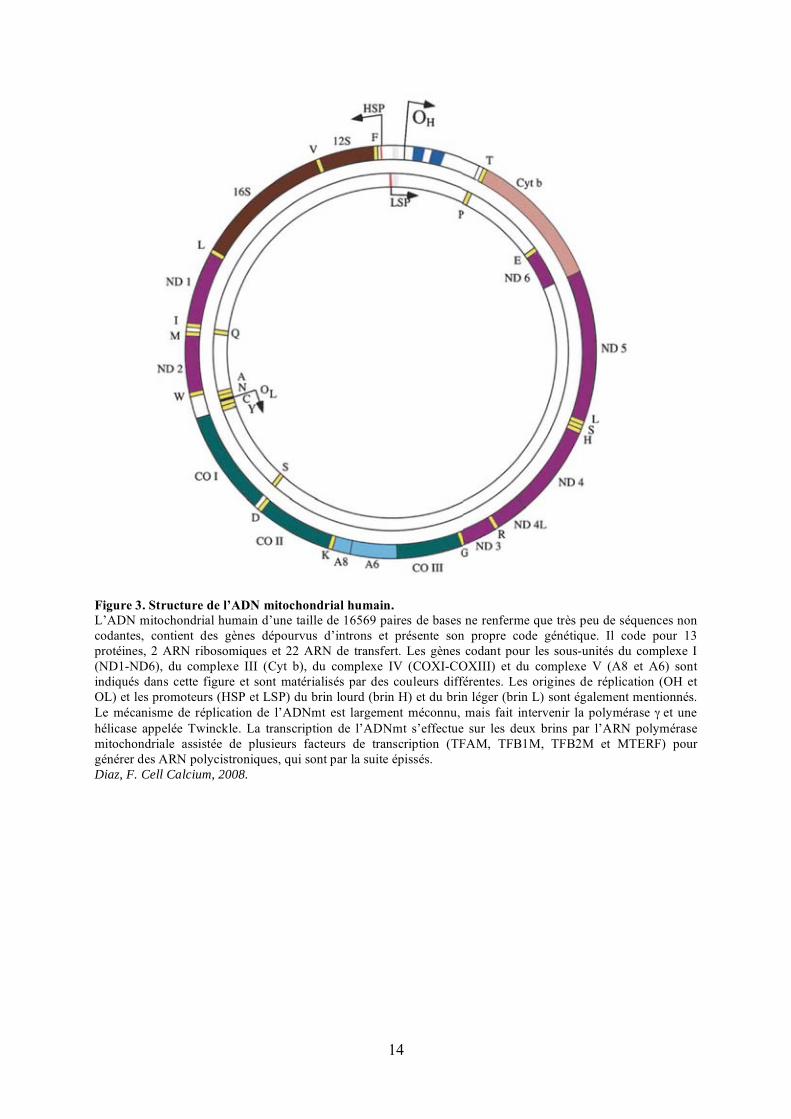
14
Figure 3. Structure de l’ADN mitochondrial humain.L’ADN mitochondrial humain d’une taille de 16569 paires de bases ne renferme que très peu de séquences non codantes, contient des gènes dépourvus d’introns et présente son propre code génétique. Il code pour 13 protéines, 2 ARN ribosomiques et 22 ARN de transfert. Les gènes codant pour les sous-unités du complexe I (ND1-ND6), du complexe III (Cyt b), du complexe IV (COXI-COXIII) et du complexe V (A8 et A6) sont indiqués dans cette figure et sont matérialisés par des couleurs différentes. Les origines de réplication (OH et OL) et les promoteurs (HSP et LSP) du brin lourd (brin H) et du brin léger (brin L) sont également mentionnés. Le mécanisme de réplication de l’ADNmt est largement méconnu, mais fait intervenir la polymérase � et une hélicase appelée Twinckle. La transcription de l’ADNmt s’effectue sur les deux brins par l’ARN polymérase mitochondriale assistée de plusieurs facteurs de transcription (TFAM, TFB1M, TFB2M et MTERF) pour générer des ARN polycistroniques, qui sont par la suite épissés. Diaz, F. Cell Calcium, 2008.
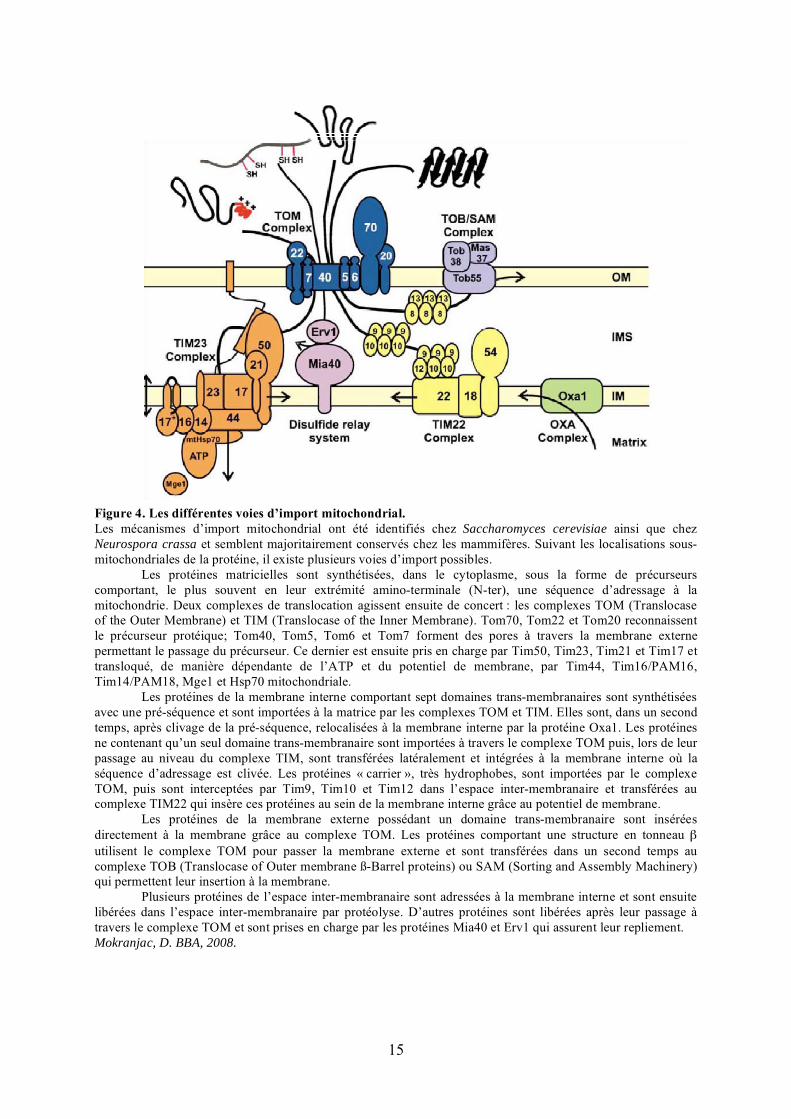
15
Figure 4. Les différentes voies d’import mitochondrial.Les mécanismes d’import mitochondrial ont été identifiés chez Saccharomyces cerevisiae ainsi que chez Neurospora crassa et semblent majoritairement conservés chez les mammifères. Suivant les localisations sous-mitochondriales de la protéine, il existe plusieurs voies d’import possibles.
Les protéines matricielles sont synthétisées, dans le cytoplasme, sous la forme de précurseurs comportant, le plus souvent en leur extrémité amino-terminale (N-ter), une séquence d’adressage à la mitochondrie. Deux complexes de translocation agissent ensuite de concert : les complexes TOM (Translocase of the Outer Membrane) et TIM (Translocase of the Inner Membrane). Tom70, Tom22 et Tom20 reconnaissent le précurseur protéique; Tom40, Tom5, Tom6 et Tom7 forment des pores à travers la membrane externe permettant le passage du précurseur. Ce dernier est ensuite pris en charge par Tim50, Tim23, Tim21 et Tim17 et transloqué, de manière dépendante de l’ATP et du potentiel de membrane, par Tim44, Tim16/PAM16, Tim14/PAM18, Mge1 et Hsp70 mitochondriale.
Les protéines de la membrane interne comportant sept domaines trans-membranaires sont synthétisées avec une pré-séquence et sont importées à la matrice par les complexes TOM et TIM. Elles sont, dans un second temps, après clivage de la pré-séquence, relocalisées à la membrane interne par la protéine Oxa1. Les protéines ne contenant qu’un seul domaine trans-membranaire sont importées à travers le complexe TOM puis, lors de leur passage au niveau du complexe TIM, sont transférées latéralement et intégrées à la membrane interne où la séquence d’adressage est clivée. Les protéines « carrier », très hydrophobes, sont importées par le complexe TOM, puis sont interceptées par Tim9, Tim10 et Tim12 dans l’espace inter-membranaire et transférées au complexe TIM22 qui insère ces protéines au sein de la membrane interne grâce au potentiel de membrane.
Les protéines de la membrane externe possédant un domaine trans-membranaire sont insérées directement à la membrane grâce au complexe TOM. Les protéines comportant une structure en tonneau �utilisent le complexe TOM pour passer la membrane externe et sont transférées dans un second temps au complexe TOB (Translocase of Outer membrane ß-Barrel proteins) ou SAM (Sorting and Assembly Machinery) qui permettent leur insertion à la membrane.
Plusieurs protéines de l’espace inter-membranaire sont adressées à la membrane interne et sont ensuite libérées dans l’espace inter-membranaire par protéolyse. D’autres protéines sont libérées après leur passage à travers le complexe TOM et sont prises en charge par les protéines Mia40 et Erv1 qui assurent leur repliement.Mokranjac, D. BBA, 2008.
16
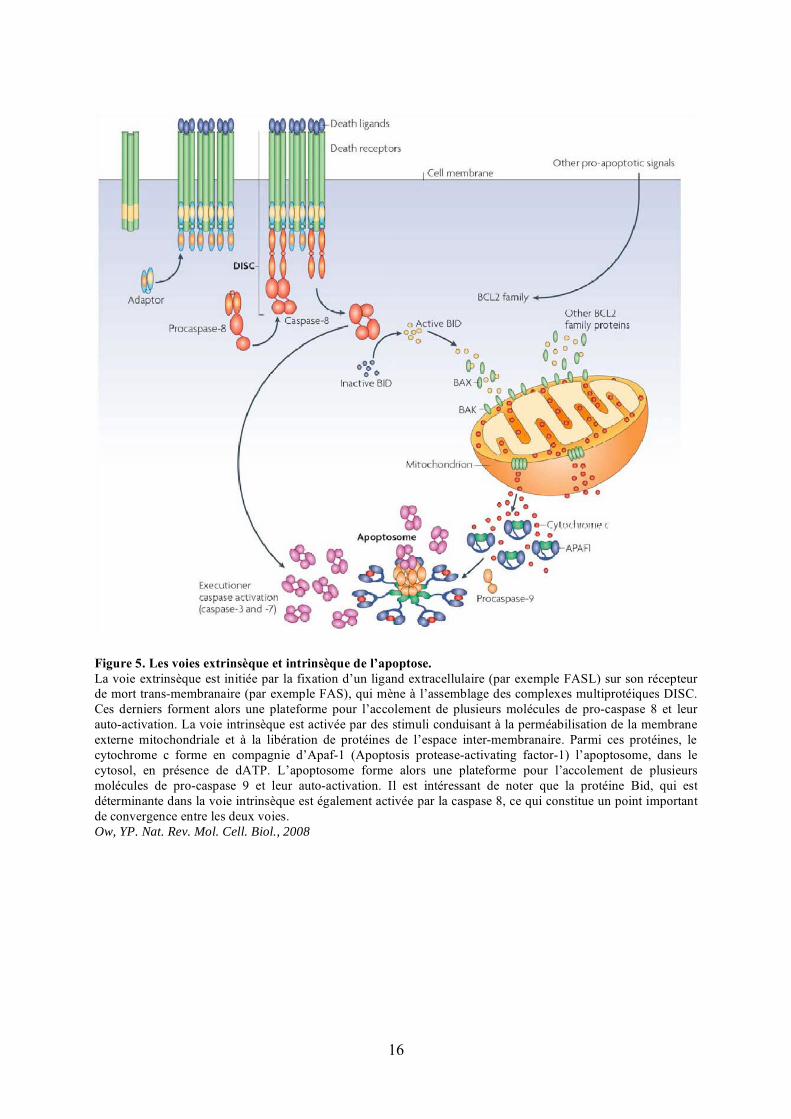
Figure 5. Les voies extrinsèque et intrinsèque de l’apoptose.La voie extrinsèque est initiée par la fixation d’un ligand extracellulaire (par exemple FASL) sur son récepteur de mort trans-membranaire (par exemple FAS), qui mène à l’assemblage des complexes multiprotéiques DISC. Ces derniers forment alors une plateforme pour l’accolement de plusieurs molécules de pro-caspase 8 et leur auto-activation. La voie intrinsèque est activée par des stimuli conduisant à la perméabilisation de la membrane externe mitochondriale et à la libération de protéines de l’espace inter-membranaire. Parmi ces protéines, le cytochrome c forme en compagnie d’Apaf-1 (Apoptosis protease-activating factor-1) l’apoptosome, dans le cytosol, en présence de dATP. L’apoptosome forme alors une plateforme pour l’accolement de plusieurs molécules de pro-caspase 9 et leur auto-activation. Il est intéressant de noter que la protéine Bid, qui est déterminante dans la voie intrinsèque est également activée par la caspase 8, ce qui constitue un point important de convergence entre les deux voies.Ow, YP. Nat. Rev. Mol. Cell. Biol., 2008

17
de fission des membranes mitochondriales (évènements dits intrinsèques) a dès lors permis de
ne plus considérer les mitochondries comme des entités séparées au sein de la cellule mais
plutôt comme un réseau de membranes interconnectées. Dans l’introduction de ce mémoire, je
ne développerai que ces deux derniers points, l’apoptose et la dynamique mitochondriale, qui
ont bouleversé la recherche concernant la mitochondrie et qui ont constitué les thèmes
majeurs abordés au cours de mes travaux de thèse.
I) L’APOPTOSE
L’apoptose est un processus de mort cellulaire actif génétiquement programmé dont
l’existence a été initialement démontrée lors de l’étude de l’embryogénèse du nématode
Caenorhabditis elegans qui au cours de son développement voit mourir un nombre constant
de cellules (131 sur 1090) (Horvitz, 1999). De même, elle joue un rôle prépondérant dans le
développement embryonnaire et la morphogénèse chez les vertébrés (Meier et al., 2000), mais
également dans le système immunitaire (Rathmell and Thompson, 2002). Par conséquent, des
anomalies quantitatives de l'apoptose, par excès ou par défaut, ont été décrites dans des
pathologies aussi diverses que les cancers ou des maladies auto-immunes (de Bruin and
Medema, 2008; Maniati et al., 2008). Plusieurs critères morphologiques permettent de
détecter une cellule apoptotique. Elle est généralement arrondie, et présente une réduction du
volume cellulaire, une condensation de la chromatine, une fragmentation nucléaire et un
bourgeonnement de la membrane plasmique. Les molécules essentielles à l’apparition de ces
caractéristiques et donc au démantèlement de la cellule apoptotique sont des cystéines
protéases appelées caspases. Au nombre de 11 chez l’homme, elles sont capables de cliver
spécifiquement plusieurs substrats cruciaux pour la vie de la cellule. Les caspases sont
présentes dans le cytosol sous forme de pro-enzymes inactives (pro-caspases) qui sont
activées de manière hiérarchique, en cascade, à la suite de clivages réalisés par des caspases
capables de s’autoactiver, les caspases initiatrices (Riedl and Salvesen, 2007). La nature des
caspases activées est déterminée par l’engagement de voies apoptotiques spécifiques qui
communiquent et coopèrent dans certains cas. On distingue d’une part une voie extrinsèque,
ou voie des récepteurs de mort, et d’autre part une voie intrinsèque ou voie mitochondriale
(Figure 5).

18

19
A) La voie extrinsèque
La voie extrinsèque est induite par des ligands de mort, comme FASL (Fas ligand),
TNF� (tumor-necrosis-factor-�) ou Apo-2/TRAIL (Apo-2 ligand ou tumour necrosis factor-
related apoptosis-inducing ligand) (Ashkenazi and Dixit, 1998). En se fixant à leur récepteur
de mort trans-membranaire, FASL, TNF� et Apo-2/TRAIL entraînent la formation de
complexes multiprotéiques, les complexes DISC (death inducing signaling complex), qui
conduisent à l’activation de la procaspase-8. La caspase-8 activée clive ensuite les caspases
effectrices 3 et 7 pour orchestrer la mort cellulaire (Figure 5).
B) La voie intrinsèque
La voie intrinsèque ou voie mitochondriale consiste en la perméabilisation de la
membrane externe mitochondriale qui permet la libération, vers le cytosol, de molécules
situées initialement dans l’espace inter-membranaire, capables d’initier et de réguler
l’activation des caspases (Figure 5).
1) Les protéines apoptogènes libérées de la mitochondrie au cours de l’apoptose
Cinq protéines de l’espace inter-membranaire sont libérées au cours du processus de
perméabilisation de la membrane externe.
Le cytochrome c assure, en conditions normales, le transport des électrons entre les
complexes III et IV de la chaîne respiratoire au niveau de la membrane interne de la
mitochondrie. Après la perméabilisation de la membrane externe de cet organite, le
cytochrome c s’échappe de l’espace inter-membranaire dans le cytosol où il s’associe avec du
dATP et une protéine cytoplasmique, Apaf-1 (Apoptosis protease-activating factor-1), pour
constituer le cœur d’un complexe multimoléculaire appelé apoptosome (Riedl and Salvesen,
2007). L’apoptosome conduit au recrutement et à l’auto-activation de la caspase-9 qui active
les caspases effectrices 3 et 7. Environ 85% du cytochrome c réside dans l’espace intra-crête,
la mobilisation préalable de la protéine de ce compartiment vers l’espace inter-membranaire
est donc requise (Scorrano et al., 2002; Scorrano and Korsmeyer, 2003). Le cytochrome c est

20

21
associé à la membrane interne des crêtes via des interactions électrostatiques et hydrophobes
avec la cardiolipine. La mobilisation de la protéine au cours de l’apoptose pourrait consister,
dans un premier temps, au décrochage de la membrane interne du cytochrome c via la rupture
de ces interactions. En effet, la cardiolipine oxydée, qui pourrait résulter de l’activité de la
phospholipase A2, de la présence d’espèces actives de l’oxygène ou d’une augmentation du
calcium cytosolique, présente une affinité réduite pour le cytochrome c en comparaison avec
la forme non oxydée (Gonzalvez and Gottlieb, 2007; Ott et al., 2007). De plus, des travaux
récents indiquent qu’une modulation subtile des jonctions des crêtes est essentielle pour la
redistribution du cytochrome c dans l’espace inter-membranaire (Frezza et al., 2006;
Yamaguchi et al., 2008). Opa1, la protéine étudiée au laboratoire, serait directement
impliquée dans ce processus (cf Introduction IIB3 et IIIA).
Smac/Diablo (Second Mitochondrial Activator of Caspases / Direct IAP Binding
protein of Low PI) est également relarguée de l’espace inter-membranaire vers le cytosol suite
à la perméabilisation de la membrane externe. Smac/Diablo interagit dans le cytosol avec les
IAPs (Inhibitors of Apoptosis Proteins), des inhibiteurs des caspases. Cette interaction a pour
effet de titrer l’activité inhibitrice des IAPs et de favoriser ainsi l’activation des caspases
(Vaux and Silke, 2003).
OMI/HTRA-2 a été impliquée dans des morts cellulaires aussi bien dépendantes
qu’indépendantes des caspases. Il s’agit d’une sérine-protéase capable de dégrader des
substrats très différents comme des éléments du cytosquelette (actine, �- et �-tubuline,
vimentine) ou des facteurs de traduction (eIF4G et EF-1�) (Hegde et al., 2002; Vande Walle
et al., 2008) mais également d’interagir avec les IAPs (Vaux and Silke, 2003).
AIF (Apoptosis Inducing Factor) est une oxydoréductase mitochondriale jouant un
rôle dans la phosphorylation oxydative ainsi que dans les défenses anti-oxydantes
(Modjtahedi et al., 2006). Dans certaines conditions apoptotiques, une fois relarguée de la
mitochondrie, AIF est transloquée au noyau pour participer à la condensation et à la
fragmentation de la chromatine (Susin et al., 1999; Susin et al., 1996). Néanmoins, cette
contribution dans la mort cellulaire ne semble pas être constante et pourrait dépendre des
types cellulaires et des stimuli apoptotiques utilisés. Par exemple, l’extinction du gène codant
pour AIF n’a aucun effet sur la mort induite par des inhibiteurs des topoisomérases malgré
22
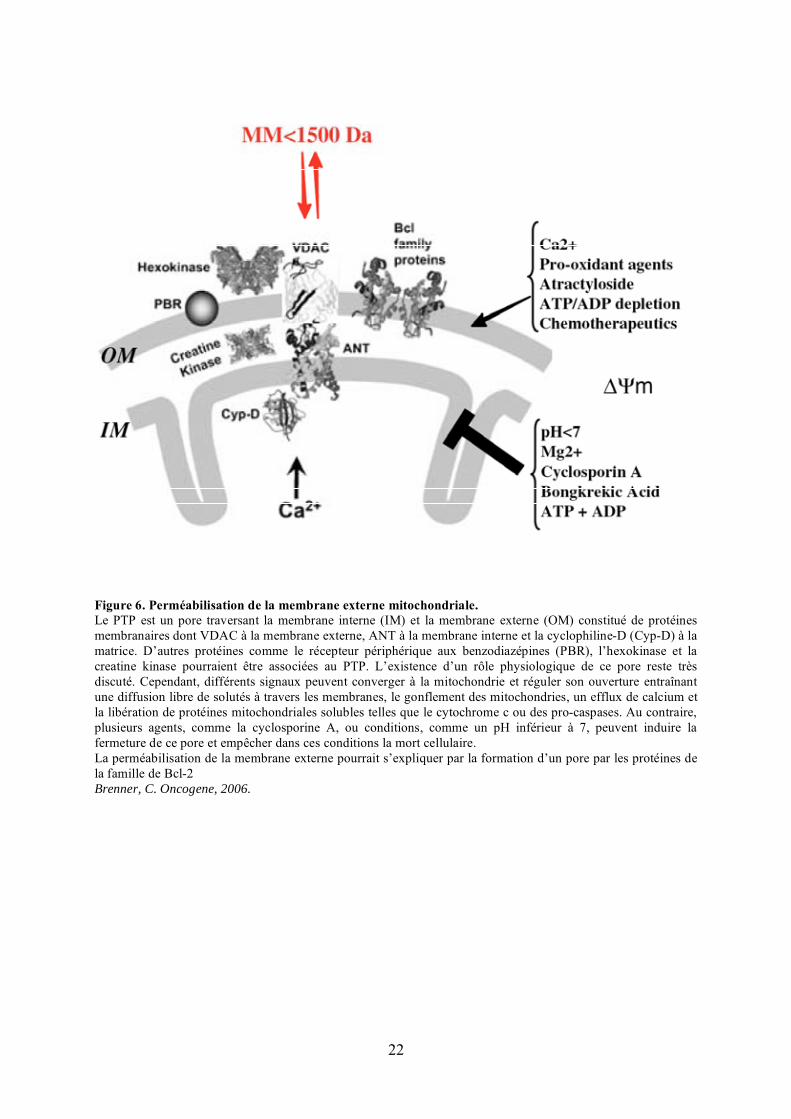
Figure 6. Perméabilisation de la membrane externe mitochondriale.Le PTP est un pore traversant la membrane interne (IM) et la membrane externe (OM) constitué de protéines membranaires dont VDAC à la membrane externe, ANT à la membrane interne et la cyclophiline-D (Cyp-D) à la matrice. D’autres protéines comme le récepteur périphérique aux benzodiazépines (PBR), l’hexokinase et la creatine kinase pourraient être associées au PTP. L’existence d’un rôle physiologique de ce pore reste très discuté. Cependant, différents signaux peuvent converger à la mitochondrie et réguler son ouverture entraînant une diffusion libre de solutés à travers les membranes, le gonflement des mitochondries, un efflux de calcium et la libération de protéines mitochondriales solubles telles que le cytochrome c ou des pro-caspases. Au contraire, plusieurs agents, comme la cyclosporine A, ou conditions, comme un pH inférieur à 7, peuvent induire la fermeture de ce pore et empêcher dans ces conditions la mort cellulaire.La perméabilisation de la membrane externe pourrait s’expliquer par la formation d’un pore par les protéines de la famille de Bcl-2Brenner, C. Oncogene, 2006.

23
l’activation de la voie intrinsèque de l’apoptose (Joza et al., 2001; Urbano et al., 2005). Les
différentes données contradictoires accumulées dans la littérature, associées au fait que AIF
n’est relarguée que très tardivement de la mitochondrie au cours du processus apoptotique
(Arnoult et al., 2003), ont conduit bon nombre de scientifiques à considérer le rôle de AIF
négligeable dans la mort cellulaire.
L’endonucléase G une fois libérée de la mitochondrie est transloquée au noyau où
elle induit la fragmentation de l’ADN indépendamment des caspases (Li et al., 2001; van Loo
et al., 2001). L’impact réel de l’endonucléase G sur la mort cellulaire reste néanmoins
controversé. Deux souris invalidées pour le gène codant pour cette protéine ont été générées
et conduisent à des résultats contradictoires. Alors que Zhang et ses collaborateurs montrent
que l’endonucléase G est impliquée à un stade précoce de l’embryogénèse (Zhang et al.,
2003b), la souris générée par Irvine et ses collaborateurs ne présente ni de défauts
anatomiques ni de défauts histopathologiques (Irvine et al., 2005).
2) Le mécanisme de perméabilisation de la membrane externe mitochondriale
Le mécanisme de perméabilisation de la mitochondrie a été très discuté ces dernières
années. Deux modèles ont été successivement proposés: l’ouverture d’un canal, le pore de
transition de perméabilité (PTP), qui permet la dilatation de la matrice et la rupture de la
membrane externe et la formation d’un pore au niveau de la membrane externe par des
protéines de la famille de Bcl-2.
L’ouverture du PTP localisé au niveau de sites de contact entre membrane externe et
membrane interne mitochondriale conduit à une entrée d’eau et de petits solutés au sein de la
matrice (Figure 6). Cet influx provoque la dilatation de la matrice menant à la rupture de la
membrane externe avec pour effet la libération des protéines de l’espace inter-membranaire
dans le cytosol. La nature du PTP reste à ce jour très débattue même si plusieurs études
semblent impliquer les protéines VDAC à la membrane externe, ANT (Adenine Nucleotide
Translocator) à la membrane interne et la cyclophiline-D à la matrice (Forte and Bernardi,
2006). L’implication du PTP dans l’apoptose est à l’heure actuelle extrêmement controversée.
Alors que plusieurs études tendaient à considérer le PTP comme un acteur majeur du
déclenchement de la voie mitochondriale apoptotique, notamment car il entraîne la libération
du cytochrome c dans le cytosol (Broekemeier et al., 1992; Broekemeier and Pfeiffer, 1989;
24
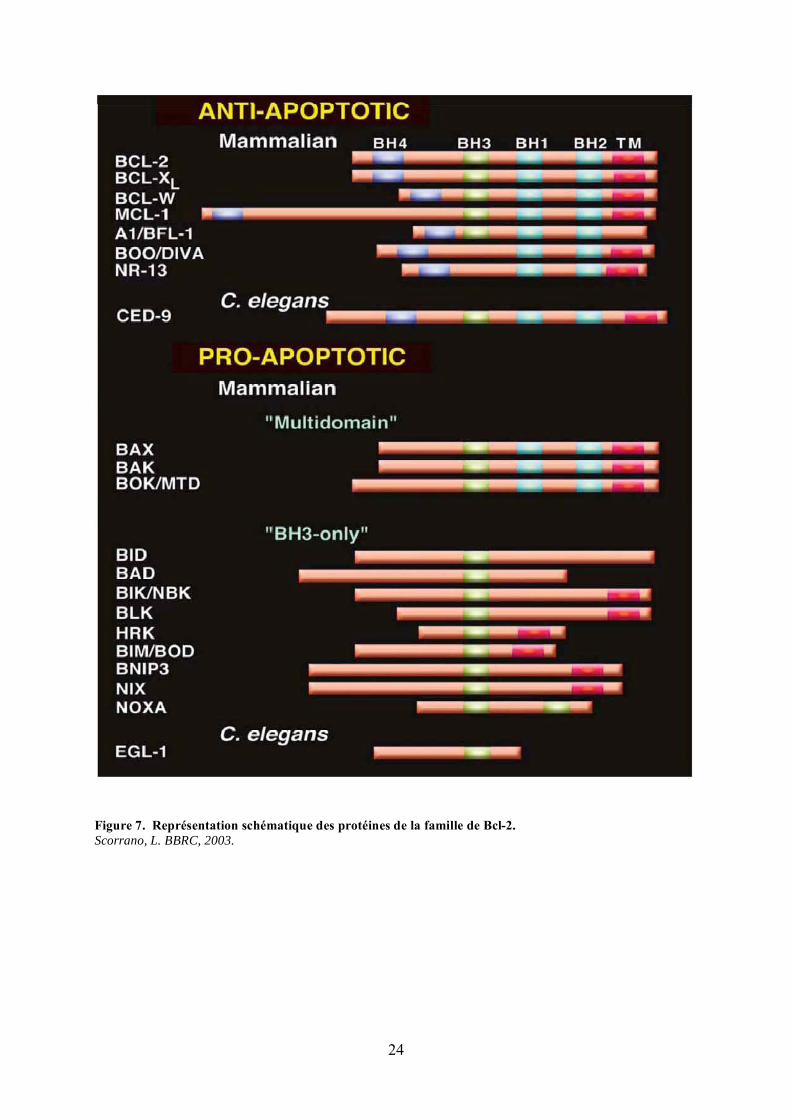
Figure 7. Représentation schématique des protéines de la famille de Bcl-2.Scorrano, L. BBRC, 2003.

25
Forte and Bernardi, 2006; Murphy et al., 1996), des travaux récents permettent de proposer
que l’ouverture de ce pore interviendrait plutôt au cours de la nécrose. En effet, l’invalidation
du gène codant pour la cyclophiline-D n’entraîne pas de défauts majeurs de développement
chez la souris, contrairement à celle des gènes codant pour la plupart des membres de la
famille de Bcl-2. De plus, les fibroblastes isolés de ces souris sont sensibles à l’activation de
la voie intrinsèque de l’apoptose, mais sont par contre résistants à la mort cellulaire
nécrotique induite par les ROS ou l’influx calcique (Baines et al., 2005; Baines et al., 2007;
Basso et al., 2005; Nakagawa et al., 2005).
Le second mécanisme proposé consiste en la formation de pore par deux pro-
apoptotiques de la famille de Bcl-2, Bax et Bak (Figure 6). Alors que Bak est
constitutivement ancrée à la membrane externe mitochondriale (Wei et al., 2000), Bax est
principalement présente sous forme monomérique dans le cytosol ou faiblement associée à la
membrane externe. Après une étape d’activation, l’extrémité carboxy-terminale hydrophobe
de la protéine est dévoilée, permettant un changement conformationnel qui facilite sa
translocation et son insertion au niveau de la membrane externe mitochondriale (Billen et al.,
2008; Hsu et al., 1997; Wolter et al., 1997). Bax et Bak oligomérisent ensuite et induisent la
formation de pores à travers cette membrane par lesquels sont libérées diverses protéines de
l’espace inter-membranaire.
La succession des évènements menant à l’activation et à l’oligomérisation de Bax et
Bak demeure encore assez incertaine. Il est toutefois établi qu’elle met en jeu d’autres
protéines de la famille de Bcl-2 (Figure 7). Certaines, comme Bcl-2, Bcl-XL ou Mcl-1, qui
possèdent les 4 régions d’homologie à Bcl-2 (BH1 à BH4), présentent une activité anti-
apoptotique. D’autres, dites à BH3-only car elles ne présentent qu’un seul domaine BH (BH3)
possèdent une activité pro-apoptotique. Ces dernières sont nombreuses (Bid, Bad, Bim, Bik,
Puma, Noxa, Bnip3, Nix, …) et peuvent être activées de différentes manières en réponse à des
signaux apoptotiques de nature variée. Par exemple, l’expression de Puma, Bim et Bnip3 est
induite transcriptionnellement, respectivement par p53 en réponse à des dommages à l’ADN
(Nakano and Vousden, 2001), par FOXO3A (Forkhead box O3A) lors d’une privation en
facteurs de croissance (Dijkers et al., 2000) et HIF1 (Hypoxia inducing factor-1) en réponse à
l’hypoxie (Bruick, 2000). Leur activation peut aussi résulter de modifications post-
traductionnelles. En effet, Bad est activée par déphosphorylation suite à une privation en
facteurs de croissance (Zha et al., 1996), Bid est clivée par la caspase-8 (Li et al., 1998; Luo
26
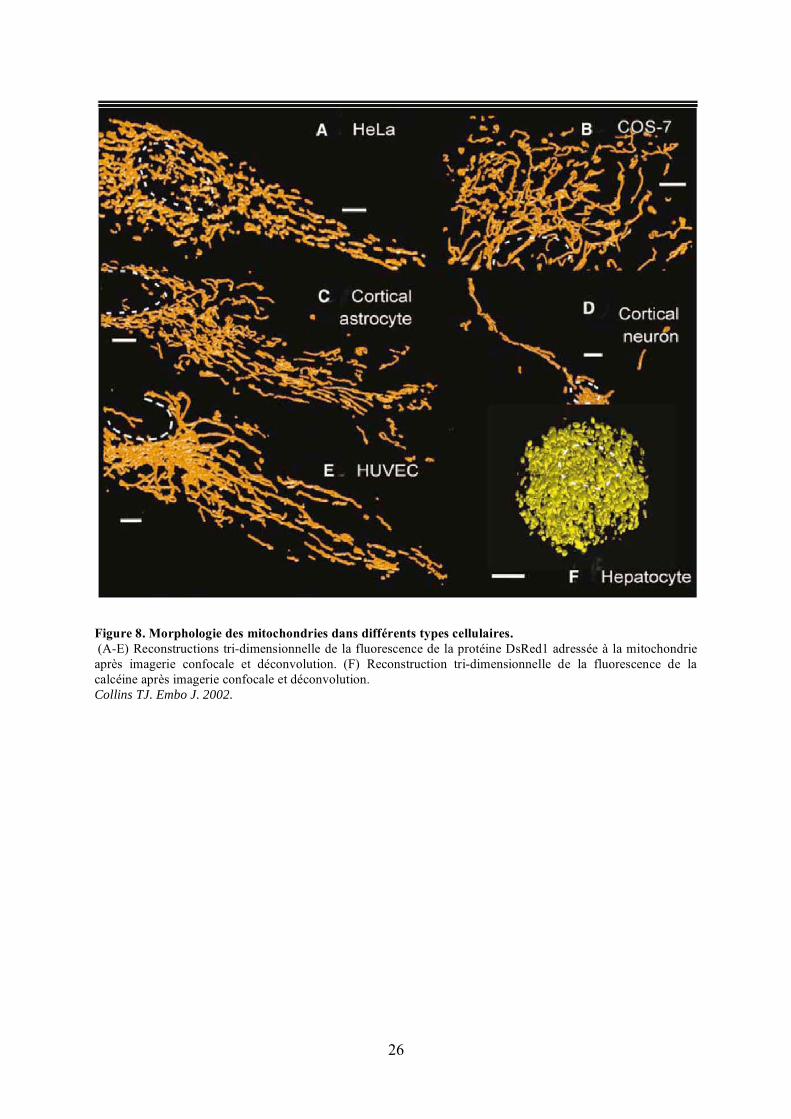
Figure 8. Morphologie des mitochondries dans différents types cellulaires. (A-E) Reconstructions tri-dimensionnelle de la fluorescence de la protéine DsRed1 adressée à la mitochondrie après imagerie confocale et déconvolution. (F) Reconstruction tri-dimensionnelle de la fluorescence de la calcéine après imagerie confocale et déconvolution. Collins TJ. Embo J. 2002.

27
et al., 1998) et Bim, séquestrée au niveau du cytosquelette, est relarguée en réponse à
plusieurs stimuli apoptotiques (Puthalakath et al., 1999). De nombreuses interactions entre les
différentes protéines de la famille de Bcl-2 ont à ce jour été révélées et ont permis de dégager
deux modèles d’activation de Bax et Bak. Dans le modèle indirect, récemment soutenu par
Willis (Willis et al., 2007), Bax et Bak sont constamment complexées avec les protéines anti-
apoptotiques telles que Bcl-2 ou Mcl-1. Pour induire la mort, les protéines BH3-only doivent
interagir avec les protéines anti-apoptotiques et les neutraliser, afin d’entraîner la libération et
l’activation de Bax et Bak. Dans le modèle direct, des protéines activatrices, interagissent
directement avec Bax et Bak et induisent leur changement conformationnel (Gallenne et al.,
2009; Kim et al., 2006; Lovell et al., 2008). Dans ce cas, les protéines anti-apoptotiques
bloquent l’apoptose en se fixant aux protéines activatrices et en les séquestrant. Cela signifie
donc qu’il existerait deux catégories de protéines BH3-only : les protéines activatrices,
comme Bid, Bim et Puma, et les protéines sensibilisatrices (Letai et al., 2002), incapables
d’interagir avec Bax et Bak, mais pouvant, par contre, se fixer aux protéines anti-apoptotiques
pour lever l’inhibition des protéines activatrices.
II) LA STRUCTURE DYNAMIQUE DES MITOCHONDRIES
L’observation, en microscopie optique, des mitochondries à partir de cellules fixées
révèle que ces organites, suivant le type cellulaire, se présentent sous forme de grains, de
bâtonnets ou de filaments de longueur variable (Figure 8). Leur visualisation en vidéo-
microscopie montre qu’elles se déplacent constamment le long du cytosquelette. Lorsqu’elles
se rencontrent, les mitochondries peuvent fusionner et mélanger aussi bien leurs membranes
que leur contenu soluble. À l’inverse, une mitochondrie peut se diviser et générer des
mitochondries « filles » de taille inférieure (Bereiter-Hahn, 1990; Bereiter-Hahn and Voth,
1994). Je débuterai cette partie par une brève description des interactions qui existent entre les
mitochondries et le cytosquelette et je présenterai ensuite plus longuement les bases
moléculaires de la fusion et de la fission mitochondriale.
A) La distribution subcellulaire et la mobilité des mitochondries
Le transport des mitochondries, chez les mammifères, s’effectue le long du réseau de
microtubules. Des points de contact entre les mitochondries et les microtubules ont été mis en
28
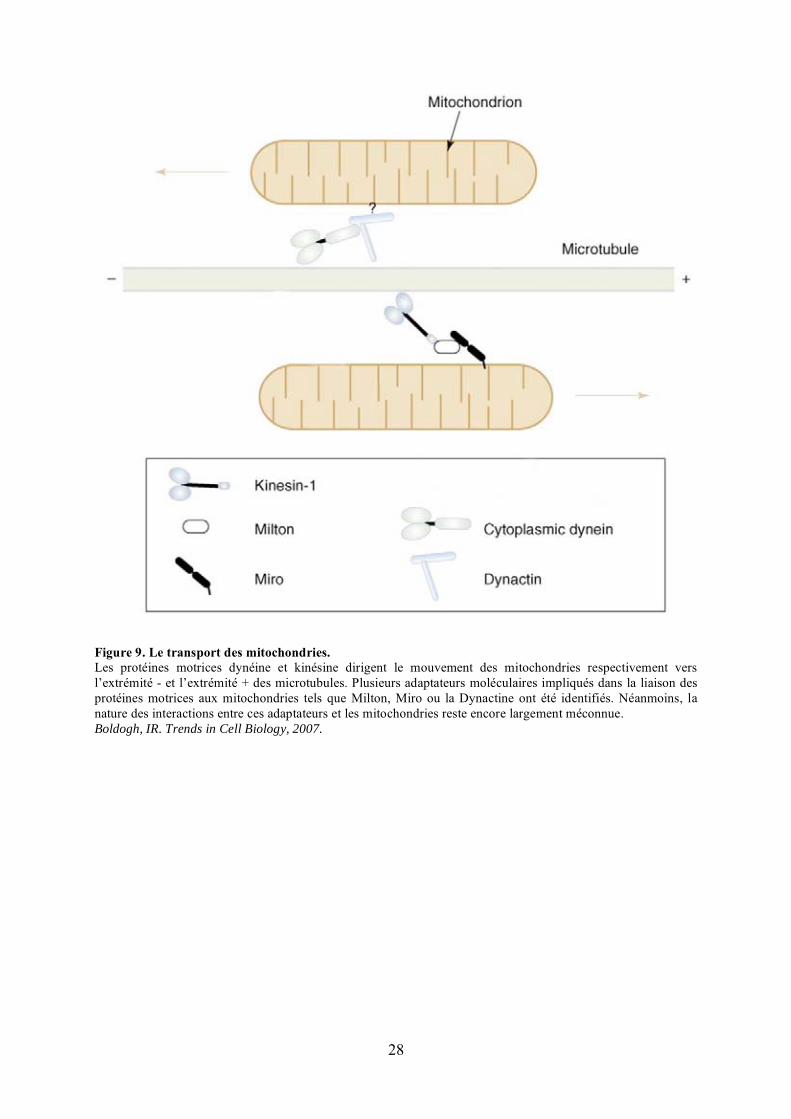
Figure 9. Le transport des mitochondries.Les protéines motrices dynéine et kinésine dirigent le mouvement des mitochondries respectivement vers l’extrémité - et l’extrémité + des microtubules. Plusieurs adaptateurs moléculaires impliqués dans la liaison des protéines motrices aux mitochondries tels que Milton, Miro ou la Dynactine ont été identifiés. Néanmoins, la nature des interactions entre ces adaptateurs et les mitochondries reste encore largement méconnue. Boldogh, IR. Trends in Cell Biology, 2007.

29
évidence par tomographie électronique (Hoog et al., 2007; Yaffe et al., 2003). L’utilisation de
drogues comme le nocodazole ou la vinblastine, perturbant spécifiquement le réseau de
microtubules, affecte la distribution des mitochondries (Ligon and Steward, 2000; Morris and
Hollenbeck, 1995; Yaffe et al., 1996). Le transport des mitochondries dans les axones des
neurones, réalisé très rapidement (de l’ordre du micromètre par seconde) sur de longues
distances, a fait l’objet de nombreuses études et a ainsi permis de mieux appréhender la
distribution des mitochondries (Hollenbeck and Saxton, 2005). L’enrichissement de
mitochondries sur leur lieu d’utilisation, en particulier au niveau des synapses, des cônes de
croissance ou des nœuds de Ranvier, fait appel à un mouvement dit antérograde qui a lieu
depuis le soma (extrémité + des microtubules) vers les régions distales. Le mouvement
rétrograde s’effectue au contraire en sens inverse, des zones distales (extrémité - des
microtubules) vers le soma. Les mitochondries sont associées, par l’intermédiaire de protéines
adaptatrices spécifiques, à des protéines motrices qui, par hydrolyse de l’ATP, fournissent les
forces nécessaires aux mouvements le long des microtubules (Figure 9). Chez les
mammifères, les kinésines 1 (Kif1B) et 3 (Kif3B) assurent le transport antérograde des
mitochondries, alors que le transport rétrograde est médié par la dynéine cytoplasmique
(Nangaku et al., 1994; Schroer et al., 1989; Tanaka et al., 1998; Varadi et al., 2004). Les
protéines adaptatrices Milton et Miro lient Kif1B à la mitochondrie lors du transport
antérograde (Brickley et al., 2005; Glater et al., 2006; Smith et al., 2006). Milton interagit
avec la partie carboxy-terminale de la chaîne lourde de Kif1B et interagit également avec
Miro (Glater et al., 2006), une protéine GTPase Rho-like de la membrane externe
mitochondriale (Fransson et al., 2003). Concernant le transport rétrograde, un complexe
multi-protéique, appelé Dynactine, lie la Dynéine à la surface des mitochondries (Schroer,
2004).
Plusieurs études menées sur des neurones ou sur des cellules musculaires suggèrent
également l’intervention du cytosquelette d’actine dans le transport des mitochondries via
l’utilisation des myosines, en particulier dans le cadre de déplacements plus courts et plus
lents, mais cela demeure encore très controversé (Kuznetsov et al., 1992; Langford, 2002;
Ligon and Steward, 2000; Morris and Hollenbeck, 1995). Néanmoins, il est essentiel chez la
levure à bourgeon pour la transmission des mitochondries au cours de la division cellulaire
(Fehrenbacher et al., 2004; Simon et al., 1997). Les mouvements antérogrades, vers le
bourgeon en formation, et rétrogrades, vers la cellule mère, sont bloqués par un traitement à la
30
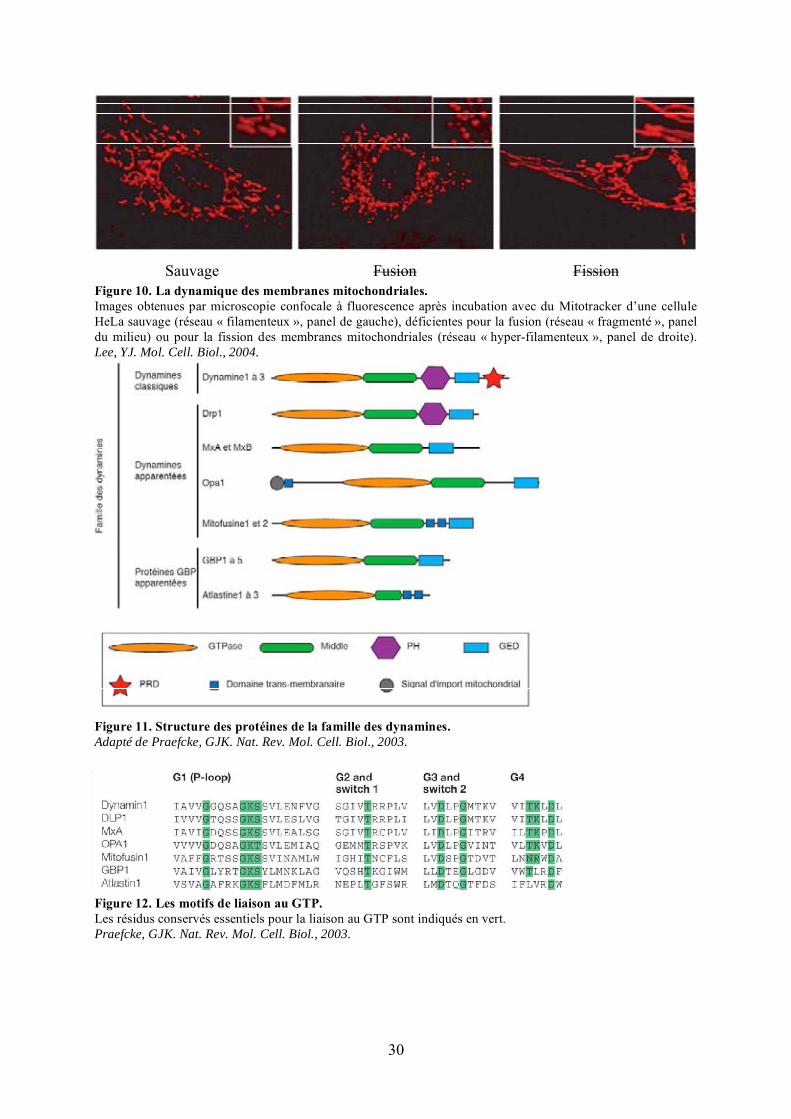
Figure 10. La dynamique des membranes mitochondriales.Images obtenues par microscopie confocale à fluorescence après incubation avec du Mitotracker d’une cellule HeLa sauvage (réseau « filamenteux », panel de gauche), déficientes pour la fusion (réseau « fragmenté », panel du milieu) ou pour la fission des membranes mitochondriales (réseau « hyper-filamenteux », panel de droite). Lee, YJ. Mol. Cell. Biol., 2004.
Figure 11. Structure des protéines de la famille des dynamines.Adapté de Praefcke, GJK. Nat. Rev. Mol. Cell. Biol., 2003.
Figure 12. Les motifs de liaison au GTP.Les résidus conservés essentiels pour la liaison au GTP sont indiqués en vert. Praefcke, GJK. Nat. Rev. Mol. Cell. Biol., 2003.
Sauvage Fusion Fission

31
Latrunculin-A (Boldogh et al., 1998). L’association de la mitochondrie au cytosquelette
d’actine est dans ce cas assurée par trois protéines intégrales de la membrane externe
mitochondriale, Mmm1p (maintenance of mitochondrial morphology), Mdm10p et Mdm12p
(mitochondrial distribution and morphology) (Berger et al., 1997; Burgess et al., 1994; Sogo
and Yaffe, 1994). La délétion des gènes codant pour ces protéines induit l’inhibition de la
mobilité des mitochondries, des défauts de morphologie mitochondriale, mais également la
perte de l’ADN mitochondrial. Le complexe formé par ces trois protéines a été baptisé
« mitochore » (Boldogh et al., 1998; Boldogh and Pon, 2006), en référence au rôle joué par le
kinétochore lors de la mitose.
B) La dynamique des membranes mitochondriales
La morphologie mitochondriale est très variable d’un type cellulaire à l’autre et est
déterminée par l’équilibre entre des évènements de fusion et de fission des membranes
mitochondriales. Les mitochondries dans des cellules présentant un taux de fusion
mitochondriale plus élevé que le taux de fission apparaissent plus ou moins longues et
interconnectées ; on dit alors que le réseau mitochondrial est filamenteux. À l’inverse, les
cellules comportant un taux de fission mitochondriale supérieur à celui de fusion présentent
des mitochondries de petites tailles, ponctiformes ; on qualifie alors le réseau mitochondrial
de fragmenté (Figure 8). Lorsque cet équilibre fusion/fission est perturbé, des changements
spectaculaires peuvent être observés (Figure 10). Vers la fin des années 1990, des cribles
génétiques réalisés chez Saccharomyces cerevisiae ont grâce à cela permis d’identifier les
premiers protagonistes de la dynamique mitochondriale. Il s’agit de grandes GTPases
conservées chez les eucaryotes au cours de l’évolution appartenant à la famille des
dynamines. Avant de les passer en revue, j’exposerai brièvement les caractéristiques
principales de cette famille de protéines.
1) Les dynamines
a) La structure des dynamines
La famille des dynamines est divisée en 3 groupes : les dynamines classiques ou
conventionnelles, les dynamines apparentées et les protéines apparentées à GBP (Figure 11).
Les dynamines classiques (dynamines 1, 2 et 3) ont été les premières caractérisées. Elles

32

33
contiennent cinq domaines caractéristiques:
- Le domaine GTPase, plus long que celui des petites GTPases (300 acides aminés
environ contre 160), qui présente des séquences très conservées essentielles pour la fixation
du GTP (Figure 12). Contrairement aux petites GTPases, les dynamines se caractérisent par
une faible affinité pour le GTP et une forte capacité à hydrolyser ce nucléotide. L’activité
GTPase des dynamines est stimulée par leur oligomérisation et ne nécessite pas l’intervention
de protéines GAP (GTPase Activating Protein) ni de GEF (GTPase Exchange Factor) pour
échanger le GDP par le GTP.
- Le domaine « middle » qui pourrait être impliqué dans l’oligomérisation des
dynamines puisqu’il interagit in vitro et en double-hybride avec le domaine GED (GTPase
Effector Domain) (Okamoto et al., 1999; Smirnova et al., 1999).
- Le domaine d’homologie à la Pleckstrin (domaine PH) qui compte environ 100
acides aminés permettant l’interaction directe avec des phospholipides chargés négativement
et donc l’adressage de la protéine aux membranes (Klein et al., 1998).
- Le domaine GED (GTPase effector domain) qui se comporte comme une GAP
« interne ». Grâce à un motif coiled-coil, le GED interagit de manière intra- ou inter-
moléculaire avec le domaine GTPase et le domaine Middle. Ce domaine dirige ainsi
l’oligomérisation des dynamines qui, in vitro, stimule de près de 50 fois l’activité GTPase
(Sever et al., 1999; Smirnova et al., 1999).
- Le domaine carboxy-terminal PRD (Proline Rich Domain) qui interagit avec des
protéines à domaines SH3 (Src-homology-3) comme l’amphiphysine.
Les dynamines apparentées ne comprennent que les domaines GTPase, Middle et
GED. Des domaines supplémentaires sont toutefois présents au sein de ces protéines comme
par exemple des régions permettant leur adressage subcellulaire (Figure 11) (Praefcke and
McMahon, 2004).
b) Le rôle des dynamines
Les dynamines 1 et 2, ont été initialement impliquées chez les mammifères dans la
libération des vésicules couvertes de clathrine au cours de l’endocytose. Elles ont été par la
suite associées à d’autres évènements cellulaires comme le bourgeonnement des vésicules
couvertes de cavéoline, la phagocytose, la cytokinèse et la formation de lamellipodes ou de
podosomes (Clark et al., 1997; Gold et al., 1999; Henley et al., 1998; Ochoa et al., 2000; Oh
34
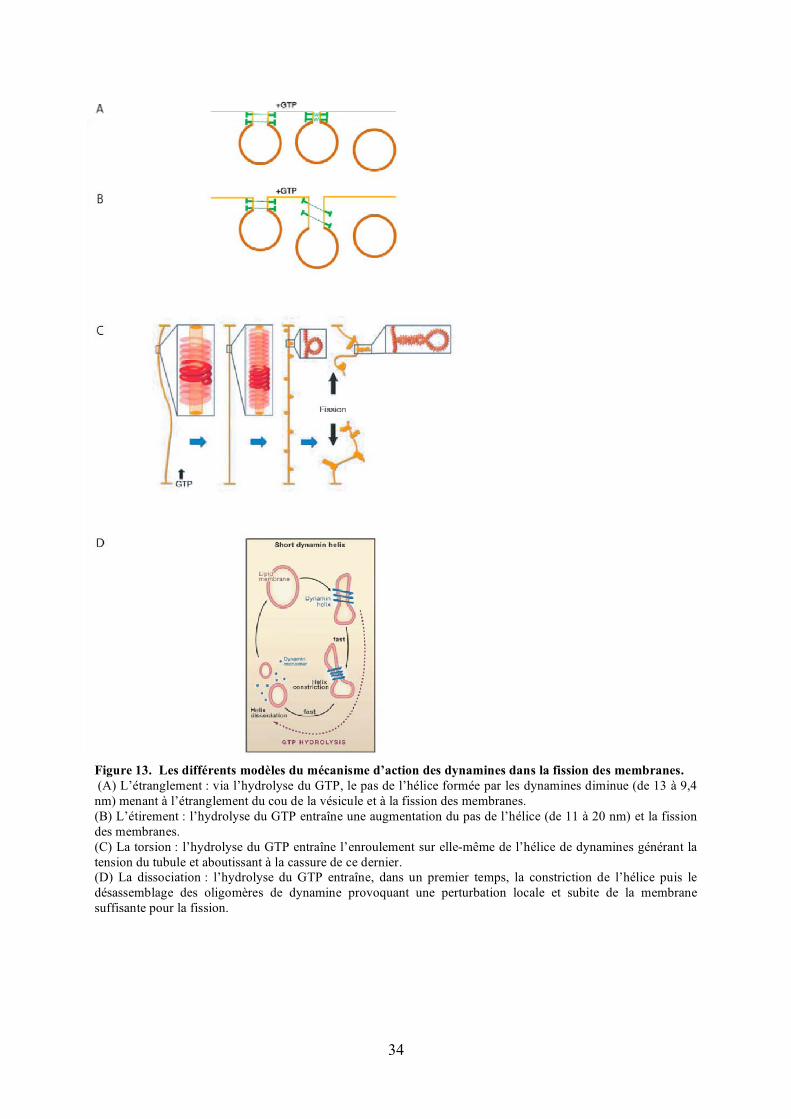
Figure 13. Les différents modèles du mécanisme d’action des dynamines dans la fission des membranes. (A) L’étranglement : via l’hydrolyse du GTP, le pas de l’hélice formée par les dynamines diminue (de 13 à 9,4 nm) menant à l’étranglement du cou de la vésicule et à la fission des membranes. (B) L’étirement : l’hydrolyse du GTP entraîne une augmentation du pas de l’hélice (de 11 à 20 nm) et la fission des membranes. (C) La torsion : l’hydrolyse du GTP entraîne l’enroulement sur elle-même de l’hélice de dynamines générant la tension du tubule et aboutissant à la cassure de ce dernier.(D) La dissociation : l’hydrolyse du GTP entraîne, dans un premier temps, la constriction de l’hélice puis le désassemblage des oligomères de dynamine provoquant une perturbation locale et subite de la membrane suffisante pour la fission.

35
et al., 1998; Orth and McNiven, 2003; Pelissier et al., 2003; van Dam and Stoorvogel, 2002).
Les dynamines apparentées et les GBP apparentées interviennent dans divers
processus cellulaires fondamentaux. Les protéines Mx, présentes uniquement chez les
vertébrés, et les protéines liant le guanylate, les GBPs sont induites en réponse aux interférons
et sont impliquées dans diverses réponses antivirales (Anderson et al., 1999; Kochs and
Haller, 1999; Weber et al., 2000). Les Atlastines participent au maintien de la morphologie de
l’appareil de Golgi et du reticulum endoplasmique (Rismanchi et al., 2008). Les dynamines
apparentées Dnm1 et Vps1 sont essentielles pour la division des péroxysomes chez
Saccharomyces cerevisiae. Enfin, les protéines Drp1, Opa1 et les mitofusines constituent les
principaux acteurs de la dynamique mitochondriale chez les mammifères.
c) Le fonctionnement des dynamines
Les premiers travaux concernant le mécanisme d’action des dynamines ont été menés sur la
dynamine conventionnelle 1. Dans la cellule, la dynamine 1 polymérise sous la forme d’une
hélice ou d’un anneau autour des membranes du cou des vésicules d’endocytose (Takei et al.,
1995). In vitro, cette protéine polymérise spontanément formant des spirales en présence ou
en absence de membranes (Sweitzer and Hinshaw, 1998). Plusieurs modèles menant à la
fission des membranes dans lesquels la dynamine agirait comme une mécano-enzyme ont été
décrits (Figure 13). Il a successivement été proposé qu’un étranglement (Sweitzer and
Hinshaw, 1998) ou un étirement (Stowell et al., 1999) ou une torsion de l’hélice (Roux et al.,
2006) consécutifs à l’hydrolyse du GTP soit à l’origine du décrochage de la vésicule de la
membrane plasmique. Néanmoins, très récemment, deux travaux indépendants menés in vitro
proposent un mécanisme dans lequel l’oligomérisation de la dynamine entraîne l’étirement du
cou de la vésicule, et la dépolymérisation de la dynamine, découlant de l’hydrolyse du GTP,
induit finalement la fission des membranes (Bashkirov et al., 2008; Pucadyil and Schmid,
2008).
2) Les acteurs de la fission mitochondriale
a) Les protéines essentielles
Drp1/Dnm1
La fission mitochondriale nécessite chez les mammifères la dynamine apparentée
Drp1 (dynamin-related protein 1) (Figure 11) (Praefcke and McMahon, 2004). Drp1 est une
36
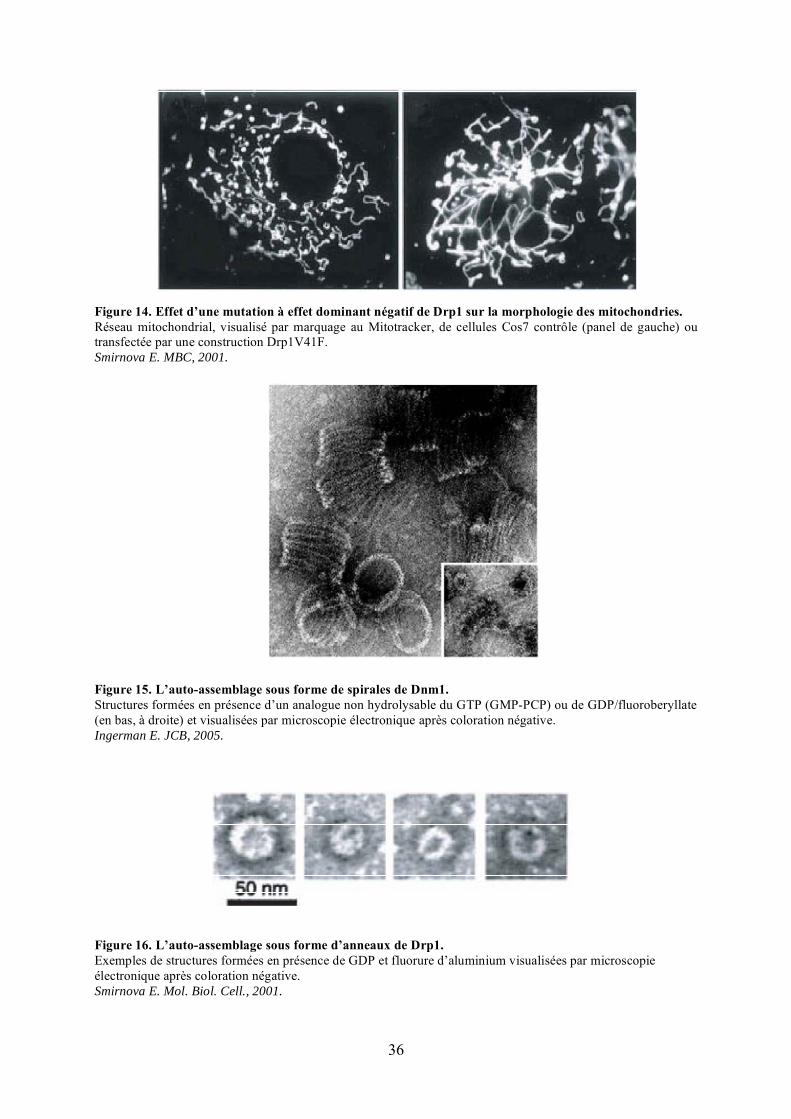
Figure 14. Effet d’une mutation à effet dominant négatif de Drp1 sur la morphologie des mitochondries.Réseau mitochondrial, visualisé par marquage au Mitotracker, de cellules Cos7 contrôle (panel de gauche) ou transfectée par une construction Drp1V41F.Smirnova E. MBC, 2001.
Figure 15. L’auto-assemblage sous forme de spirales de Dnm1.Structures formées en présence d’un analogue non hydrolysable du GTP (GMP-PCP) ou de GDP/fluoroberyllate (en bas, à droite) et visualisées par microscopie électronique après coloration négative.Ingerman E. JCB, 2005.
Figure 16. L’auto-assemblage sous forme d’anneaux de Drp1.Exemples de structures formées en présence de GDP et fluorure d’aluminium visualisées par microscopie électronique après coloration négative.Smirnova E. Mol. Biol. Cell., 2001.

37
protéine cytoplasmique, recrutée de façon transitoire aux futurs sites de fission mitochondriale
sur la membrane externe mitochondriale (Labrousse et al., 1999; Legesse-Miller et al., 2003;
Sesaki and Jensen, 1999; Smirnova et al., 2001). L’extinction de Drp1 par interférence à
l’ARN ou la surexpression de mutants dominants négatifs de la protéine dans différentes
lignées cellulaires entraîne un réseau mitochondrial hyper-filamenteux et très interconnecté
(Figure 14).
La caractérisation biochimique de Dnm1, l’homologue de Drp1 chez Saccharomyces
cerevisiae, semble indiquer un mode d’action de cette protéine dans la fission mitochondriale
relativement proche de celui des dynamines classiques dans l’endocytose. In vitro, en
présence de GTP ou d’un analogue non hydrolysable du GTP, Dnm1 oligomérise sous forme
de grandes spirales (Figure 15) (Fukushima et al., 2001; Ingerman et al., 2005; Naylor et al.,
2006) qui présentent un diamètre largement supérieur à celui des spirales formées par les
dynamines classiques (environ 109 nm versus 50 nm) (Chen et al., 2004; Hinshaw and
Schmid, 1995; Ingerman et al., 2005; Zhang and Hinshaw, 2001) correspondant exactement
au diamètre moyen des sites de constriction des mitochondries. Une réduction du diamètre de
ces spirales consécutive à l’hydrolyse du GTP semble permettre la scission des membranes
mitochondriales. En effet, le mutant Dnm1K41A, qui fixe le GTP mais ne l’hydrolyse pas, est
capable de s’autoassembler in vitro sous forme de spirales, est recruté à la mitochondrie
lorsqu’il est surexprimé chez la levure, mais est incapable de fissionner les mitochondries
entraînant un réseau mitochondrial hyperfilamenteux (Naylor et al., 2006). La fixation du
GTP semble donc suffisante pour l’auto-assemblage de la protéine et son adressage à la
mitochondrie, alors que son hydrolyse semble essentielle pour achever la division
mitochondriale en effectuant la fission des membranes.
Les caractéristiques biochimiques de Drp1 sont beaucoup moins documentées. Drp1
oligomérise in vitro sous forme d’anneaux présentant un diamètre plus réduit que les
structures formées par Dnm1 et similaire à celui des dynamines (environ 50 nm) (Figure 16)
(Smirnova et al., 2001; Yoon et al., 2001). Cette différence entre Drp1 et Dnm1 pourrait
signifier la nécessité de l’intervention de facteurs supplémentaires chez les mammifères dans
l’auto-assemblage de Drp1 in vivo pour la formation de structures de diamètre adéquat
pouvant générer la constriction des mitochondries.
Plusieurs types de modifications post-traductionnelles de Drp1 (phosphorylation,

38

39
ubiquitinylation, sumoylation, nitrosylation) influençant son activité de fission mitochondriale
ont été récemment décrites.
La sérine 637 de la protéine humaine, située dans le domaine GED, est phosphorylée
par une protéine kinase dépendante de l’AMP cyclique (Chang and Blackstone, 2007; Cribbs
and Strack, 2007). Cette modification diminue les interactions intra-moléculaires de Drp1,
inhibe l’activité GTPase de la protéine et par conséquent inhibe la fission mitochondriale.
Cette phosphorylation est réversible puisque, suite à une augmentation de la concentration
cytosolique de calcium et à une dépolarisation mitochondriale, la calcineurine déphosphoryle
ce résidu ce qui active la fission des mitochondries (Cereghetti et al., 2008; Cribbs and Strack,
2007). La sérine 600 de Drp1 est également phosphorylée par la protéine kinase 1�
dépendante de la calmoduline en réponse à l’activation de canaux calciques voltage-
dépendant (VDCCs). Cette phosphorylation augmente l’affinité de la protéine pour un de ses
partenaires hFis (voir ci-dessous) et la translocation de la protéine à la mitochondrie,
stimulant la fission des membranes (Han et al., 2008). Enfin, la sérine 618 chez l’homme,
située dans le GED, est phosphorylée par le complexe cdk1/cyclineB au cours de la mitose, de
la prophase à l’anaphase, et entraîne une stimulation de la fission des mitochondries (Taguchi
et al., 2007). Cette phosphorylation n’ayant aucun effet apparent sur l’activité GTPase de
Drp1, elle pourrait influencer l’interaction de la protéine avec des partenaires.
Drp1 peut également être mono-sumoylée avec SUMO-1 (Small Ubiquitin-Like
Modifier 1) suite à l’intervention de l’enzyme de conjugaison E2 Ubc9 (Ubiquitin-
conjugating enzyme 9) et de la SUMO E3 ligase MAPL (mitochondrial-anchored protein
ligase). Cette modification, observée au cours de l’apoptose, contribue à l’activation de la
fission mitochondriale par stabilisation de Drp1 à la mitochondrie (Braschi et al., 2009;
Harder et al., 2004; Wasiak et al., 2007; Zunino et al., 2007). SENP5, une sentrin/SUMO
protéase est capable de dé-sumoyler Drp1 et de restaurer une morphologie mitochondriale
sauvage (Zunino et al., 2007).
L’E3 ubiquitine ligase mitochondriale MARCH V, aussi appelée MITOL, dont la
perte de fonction conduit à des défauts de fission mitochondriale, interagit avec Drp1 et
régule son recrutement au niveau des sites de fission sans toutefois affecter la stabilité de la
protéine (Karbowski et al., 2007).
Enfin, Drp1 est nitrosylée sur la cystéine 644, dans des neurones cérébro-corticaux en
culture, en réponse à l’oxyde nitrique. Cette modification post-traductionnelle dans le
domaine GED conduit à une augmentation de l’activité GTPase de la protéine et à la fission

40

41
des mitochondries entraînant une diminution du nombre d’épines dendritiques (Cho et al.,
2009).
hFis1
La protéine hFis1, second composant de la machinerie de fission, est uniformément
ancrée à la membrane externe mitochondriale par ses 26 derniers acides aminés qui
constituent un domaine trans-membranaire (James et al., 2003; Mozdy et al., 2000; Yoon et
al., 2003). La surexpression de la protéine dans des cellules HeLa ou Cos7 induit la fission
des mitochondries (James et al., 2003; Yoon et al., 2003). À l’inverse, l’extinction de la
protéine par interférence à l’ARN conduit à un réseau mitochondrial très filamenteux et
interconnecté (Mozdy et al., 2000; Yoon et al., 2003). Les structures RMN et
cristallographiques de hFis1 démontrent que la partie N-ter de la protéine, située dans le
cytosol, est composée de 6 hélices � organisées en motifs de type « TPR » (tetratricopeptide
repeat (Blatch and Lassle, 1999)) qui génèrent une structure hydrophobe concave pouvant
servir de plateforme pour le recrutement de Drp1 à la mitochondrie (Dohm et al., 2004;
Suzuki et al., 2003; Suzuki et al., 2005; Yoon et al., 2003; Yu et al., 2005; Zhang and Chan,
2007). Cependant, bien que les deux protéines interagissent (Yoon et al., 2003), une
modification du niveau d’expression de hFis1 ne semble pas modifier l’adressage
mitochondrial de Drp1 (Jofuku et al., 2005; Lee et al., 2004). Ces données pourraient suggérer
l’intervention de protéines adaptatrices entre hFis1 et Drp1 comme décrit chez la levure avec
Mdv1 et Caf4 (Griffin et al., 2005; Karren et al., 2005; Tieu et al., 2002).
b) Les protéines régulatrices
Endophiline B1
L’Endophiline B1, aussi appelée SH3GLB1 (SH3 domain GRB2-like endophilin B1)
ou Bif1 (Bax interacting factor 1) appartient à la famille des endophilines, protéines capables
de modifier des phospholipides et ainsi de changer la courbure des membranes (Cuddeback et
al., 2001; Gallop et al., 2006; Pierrat et al., 2001). L’extinction de la protéine dans des cellules
HeLa ou Cos7 par interférence à l’ARN entraîne un réseau mitochondrial très interconnecté
en accord avec un rôle de la protéine dans la fission mitochondriale. Comme Drp1, la majeure
partie de la protéine est soluble dans le cytosol alors qu’une petite fraction est retrouvée au
niveau de la membrane externe mitochondriale. Lors de l’activation de la fission
mitochondriale, l’Endophiline B1 est transloquée de manière transitoire à la mitochondrie et
est enrichie aux sites de fission. Le rôle de l’Endophilin B1 dans ce processus est confirmée

42

43
par l’étude de l’homologue de la protéine chez Caenorhabditis elegans, la protéine ERP1
(Karbowski et al., 2004b).
Ganglioside-induced Differentiation Associated Protein 1 (GDAP1)
GDAP1 (Ganglioside-induced Differentiation Associated Protein 1) est une protéine
ancrée à la membrane externe mitochondriale (Wagner et al., 2009). Sa surexpression dans
des cellules Cos7 entraîne la fragmentation du réseau mitochondrial pouvant être inhibée par
la surexpression d’un mutant dominant négatif de Drp1, Drp1K38A ou la surexpression des
acteurs de fusion Mfn1 et Mfn2. L’extinction de la protéine induit à l’inverse l’élongation des
mitochondries. Les cellules surexprimant GDAP1 présentent une activité de fusion intacte
impliquant plutôt GDAP1 dans la fission mitochondriale (Niemann et al., 2005).
c) Existe-t-il une machinerie de fission de la membrane interne ?
Les protéines impliquées dans la fission mitochondriale décrites jusqu’à présent sont
toutes associées à la membrane externe, ce qui permet d’envisager que la fission de la
membrane externe entraîne la fission de la membrane interne. Cependant, plusieurs données
dans la littérature ne sont pas en accord avec cette hypothèse. En effet, la fission des deux
membranes se déroule de manière asynchrone dans des cellules HeLa déficientes pour
l’Endophiline B1 (Karbowski et al., 2004b). Chez Caenorhabditis elegans la surexpression
d’un mutant de Drp1 incapable d’hydrolyser le GTP inhibe la fission de la membrane externe
sans affecter celle de la membrane interne (Labrousse et al., 1999). Des études réalisées chez
Saccharomyces cerevisiae démontrent que la membrane interne subit de fréquents
évènements de constrictions et de scission indépendamment de la fission de la membrane
externe et de la présence de Dnm1 (Jakobs et al., 2003; Legesse-Miller et al., 2003). De plus,
certains travaux suggèrent l’existence d’une machinerie de fission de la membrane interne.
Un candidat possible chez les mammifères est MTP18 qui influence la fission mitochondriale
et présente une localisation au niveau de l’espace inter-membranaire (Tondera et al., 2005).
Chez la levure, Mdm33, qui ne possède pas d’homologue chez les mammifères, pourrait être
impliquée dans ce processus. En effet, la surexpression de Mdm33 mène à l’apparition de
septa matriciels, ce qui est interprété par les auteurs comme le résultat de l’activation de la
fission de la membrane interne (Messerschmitt et al., 2003). Une analyse fonctionnelle plus
approfondie de ces protéines et des relations qu’elles entretiennent avec la machinerie de
fission de la membrane externe semble toutefois nécessaire pour affirmer l’existence de
machineries de fission sur chaque membrane mitochondriale.
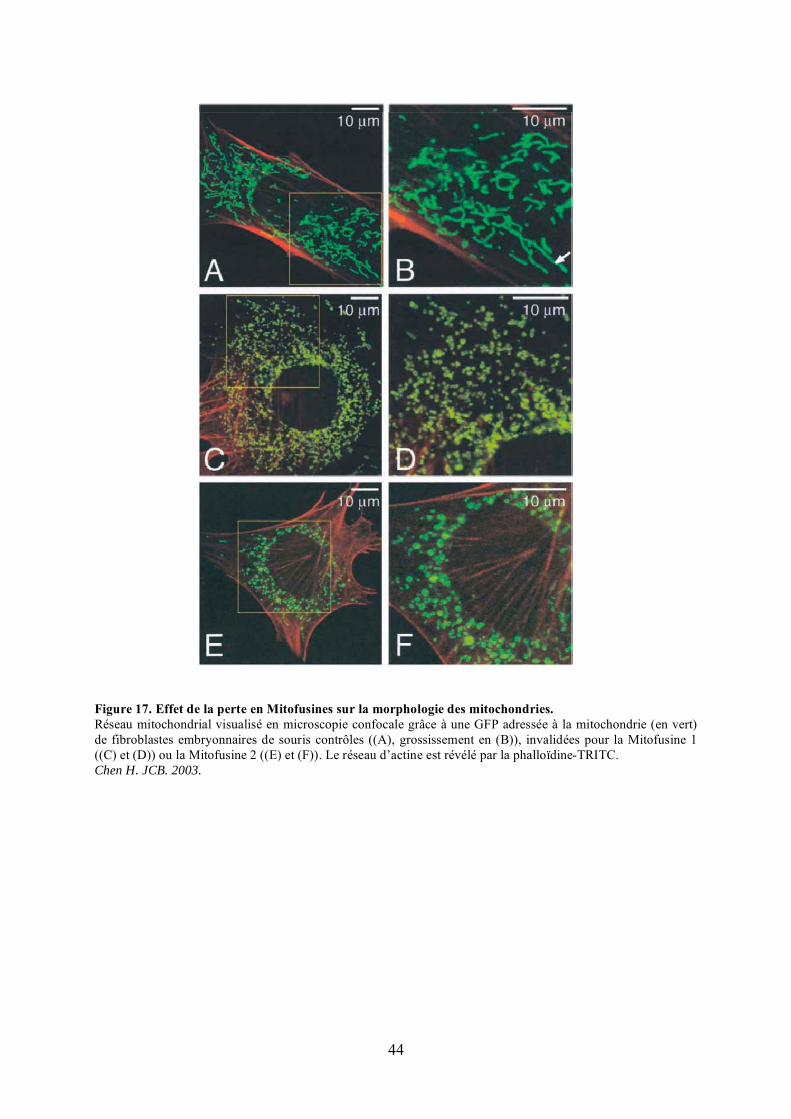
44
Figure 17. Effet de la perte en Mitofusines sur la morphologie des mitochondries.Réseau mitochondrial visualisé en microscopie confocale grâce à une GFP adressée à la mitochondrie (en vert) de fibroblastes embryonnaires de souris contrôles ((A), grossissement en (B)), invalidées pour la Mitofusine 1 ((C) et (D)) ou la Mitofusine 2 ((E) et (F)). Le réseau d’actine est révélé par la phalloïdine-TRITC.Chen H. JCB. 2003.

45
3) La fusion mitochondriale
Les acteurs essentiels de la fusion mitochondriale chez les mammifères se regroupent
en deux machineries, l’une comprenant Mfn1 et Mfn2 agissant sur la membrane externe, et
l’autre composée de Opa1 agissant sur la membrane interne. Ces deux machineries sont en
étroite collaboration pour assurer la coordination des fusions des deux membranes et
maintenir l’intégrité et la fonctionnalité des deux bicouches lipidiques ainsi que des
compartiments qu’elles délimitent.
a) Les protéines essentielles
Les mitofusines 1 et 2 / Fzo1
Fzo1, pour fuzzy onions 1, a été initialement découvert chez Drosophila melanogaster
grâce à l’identification d’une mutation au sein de son gène induisant, au cours de la
spermatogénèse, juste après la méiose, une organisation aberrante des mitochondries
(structures « en pelures d’oignons ») et la stérilité des mâles (Hales and Fuller, 1997). Fzo1
possède deux homologues chez les mammifères, les mitofusines 1 et 2 (Mfn1, Mfn2). Ces
deux protéines, qui présentent un taux de similarité de 81%, sont ancrées à la membrane
externe mitochondriale grâce à deux domaines transmembranaires situés en partie carboxy-
terminale de la protéine (Rojo et al., 2002; Santel and Fuller, 2001). Une grande partie amino-
terminale de la protéine comprenant le domaine GTPase et un motif coiled-coil HR1 ainsi
qu’une courte extrémité carboxy-terminale comprenant un motif coiled-coil HR2 sont
localisées dans le cytosol (Figure 11) (Rojo et al., 2002).
Les fibroblastes embryonnaires de souris invalidées pour les gènes MFN1 et MFN2
(Mfn1-/- et Mfn2-/-), contrairement à des fibroblastes contrôles, présentent un réseau
mitochondrial fragmenté impliquant ces protéines dans la morphologie mitochondriale
(Figure 17) (Chen et al., 2003). Grâce à un test de fusion des mitochondries réalisé sur des
fibroblastes embryonnaires de souris invalidées pour chacune des deux mitofusines, l’impact
de ces protéines sur la fusion mitochondriale a été analysé. Cette approche consiste à
mélanger et à fusionner, par du PEG, deux lignées cellulaires de mammifères aux
mitochondries préalablement marquées par une RFP et une GFP matricielles. Une fois la
fusion cellulaire effectuée, les mitochondries de chacune des cellules constituant
l’hétéropolycaryon sont redistribuées et fusionnent entre elles à leur tour. La fusion
mitochondriale est ainsi évaluée en microscopie à fluorescence par la présence de

46
Figure 18. Représentation schématique du test de fusion après fusion cellulaire au polyéthylèneglycol (PEG).Le test de fusion consiste à mélanger et fusionner, par du polyéthylèneglycol deux lignées cellulaires aux mitochondries préalablement marquées par une RFP ou une GFP matricielles. Une fois la fusion cellulaire effectuée, les mitochondries de chacune des cellules constituant l’hétéropolycaryon sont redistribuées et fusionnent à leur tour de façon naturelle. La fusion mitochondriale peut ainsi être évaluée par la présence de mitochondries abritant les deux protéines fluorescentes, visualisées par microscopie à fluorescence. En revanche, une absence de co-localisation des deux fluorophores indique une inhibition de ce processus.

47
mitochondries abritant les deux fluorescences. En revanche, une absence de co-localisation
des deux fluorophores indique une altération de ce processus (Figure 18) (Legros et al., 2002).
Le taux de fusion des mitochondries de cellules Mfn1-/- (Mfn1-/- x Mfn1-/-), de cellules Mfn2-/-
(Mfn2-/- x Mfn2-/-) ou de cellules Mfn1-/- avec des cellules Mfn2-/- (Mfn1-/- x Mfn2-/-) est
réduite indiquant un rôle de ces deux protéines dans la fusion mitochondriale. Les
hétéropolycaryons issus de la fusion de cellules invalidées pour les deux mitofusines (Mfn1-/-
Mfn2-/- x Mfn1-/- Mfn2-/-) ou de cellules Mfn1-/- Mfn2-/- avec des cellules sauvages ne
présentent aucune activité de fusion, révélant la nécessité pour la fusion de la présence d’au
moins une des deux mitofusines sur chacune des mitochondries adjacentes. En accord avec
cela, les mitofusines sont capables d’interagir en trans de manière homotypique (Mfn1-Mfn1,
Mfn2-Mfn2) et hétérotypique (Mfn1-Mfn2) grâce à leur domaine HR2 (Heptad Repeat 2) et
permettent ainsi la juxtaposition des mitochondries, étape clé précédant la fusion des
bicouches lipidiques (Chen et al., 2005; Detmer and Chan, 2007; Koshiba et al., 2004). Mfn1
est capable de restaurer la fusion des mitochondries dans les cellules Mfn2-/-, et inversement
laissant penser à une possible redondance fonctionnelle entre les deux protéines. Néanmoins,
plusieurs différences sont observées entre ces deux protéines. L’invalidation des gènes MFN1
et MFN2 n’a pas la même conséquence (cf Introduction IID). Les patrons d’expressions de
Mfn1 et Mfn2 sont « différents » : alors que la protéine Mfn1 est détectée à des niveaux
comparables entre les tissus, Mfn2 est présente en quantité supérieure dans le cœur, les
muscles squelettiques et le cerveau (Santel et al., 2003). L’extinction de Mfn1 conduit à la
formation de mitochondries sphériques, libres dans le cytosol, de tailles homogènes
correspondant au diamètre des tubules alors que l’extinction de Mfn2 conduit à des
mitochondries sphériques, associées au cytosquelette, de tailles très hétérogènes et capables
de fusionner faiblement (Figure 17) (Chen et al., 2003). La protéine recombinante Mfn1
hydrolyse très rapidement le GTP et présente une très faible affinité pour le GTP, alors que
Mfn2 présente une activité GTPase très faible mais une très grande affinité pour ce nucléotide
(Ishihara et al., 2004). In vitro, Mfn1 semble jouer un rôle plus important que Mfn2 dans la
juxtaposition des mitochondries, une étape GTP dépendante (Ishihara et al., 2004). Enfin, une
petite fraction de Mfn2 (environ 7% de la quantité totale), et non de Mfn1, est localisée au
réticulum endoplasmique (RE) et joue un rôle essentiel dans l’interaction physique entre le
RE et les mitochondries (de Brito and Scorrano, 2008). L’absence de Mfn2 abolit les contacts
RE-mitochondries, perturbe l’homéostasie calcique et induit la fragmentation du RE.
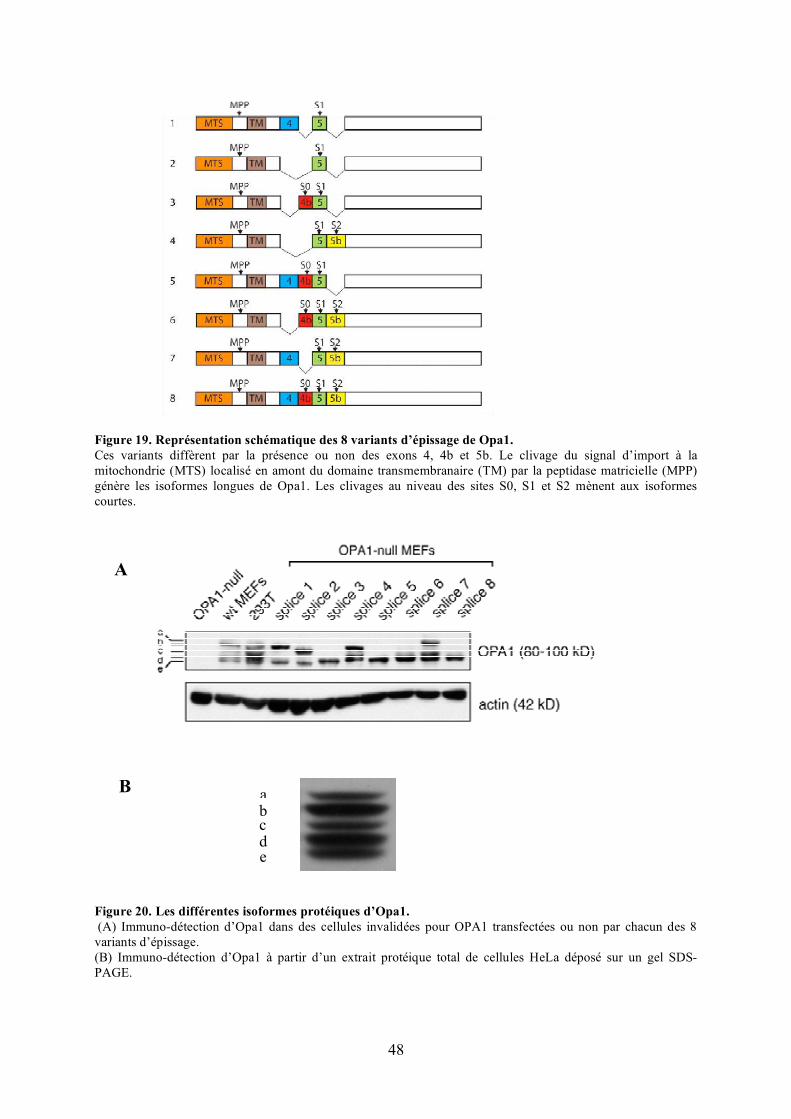
48
Figure 19. Représentation schématique des 8 variants d’épissage de Opa1.Ces variants diffèrent par la présence ou non des exons 4, 4b et 5b. Le clivage du signal d’import à la mitochondrie (MTS) localisé en amont du domaine transmembranaire (TM) par la peptidase matricielle (MPP) génère les isoformes longues de Opa1. Les clivages au niveau des sites S0, S1 et S2 mènent aux isoformes courtes.
Figure 20. Les différentes isoformes protéiques d’Opa1. (A) Immuno-détection d’Opa1 dans des cellules invalidées pour OPA1 transfectées ou non par chacun des 8 variants d’épissage.(B) Immuno-détection d’Opa1 à partir d’un extrait protéique total de cellules HeLa déposé sur un gel SDS-PAGE.
abcde
A
B

49
La régulation de la stabilité de l’homologue des mitofusines, Fzo1, chez
Saccharomyces cerevisiae, a été étudiée. Mdm30, une protéine à séquence F-box, interagit
avec Fzo1 pour induire son ubiquitinylation et sa dégradation via le protéasome (Cohen et al.,
2008; Escobar-Henriques et al., 2006; Fritz et al., 2003). Une dégradation brutale de Fzo1 via
le protéasome mais indépendante de Mdm30 a également été observée après un traitement
avec la phéromone sexuelle � qui entraîne un arrêt des levures en phase G1 (Neutzner and
Youle, 2005). Cependant, à l’heure actuelle, aucune donnée n’est disponible chez les
mammifères.
Opa1/Mgm1/Msp1/Eat-3
Nous avons identifié au laboratoire le gène codant pour Opa1 (Optic atrophy 1) lors de
la recherche de l’homologue humain de Mgm1p chez Saccharomyces cerevisiae et Msp1 chez
Schizosaccharomyces pombe, deux dynamines apparentées impliquées dans la fusion de la
membrane interne mitochondriale mais également dans le maintien du génome mitochondrial
(Guillou et al., 2005; Jones and Fangman, 1992; Pelloquin et al., 1998; Wong et al., 2000).
Nous avons mis en évidence que des mutations au sein de ce gène sont responsables d’une
neuropathie optique : l’atrophie optique dominante de type 1 (cf Introduction IIE) (Alexander
et al., 2000; Delettre et al., 2000). L’expression de ce gène est très complexe. Il comprend 31
exons, dont les exons 4, 4b et 5b qui sont alternativement épissés (Delettre et al., 2001),
générant 8 ARN messagers différents (Figure 19). Ces transcrits sont ubiquitairement
exprimés même si des variations de leur abondance ont été décrites suivant les tissus
(Alexander et al., 2000; Delettre et al., 2000; Misaka et al., 2002). Chacun des 8 variants
d’épissage génère 1 à 3 isoformes protéiques (Figure 20, A). Dans les cellules HeLa, les
variants 1 et 7 sont majoritairement exprimés et cinq isoformes protéiques d’Opa1 (de 84 à
96kDa), deux isoformes longues (isoformes a et b) et trois isoformes courtes (isoformes c, d
et e), sont détectées par Western-blot en condition physiologique dans des cellules HeLa
(Figure 20, B). Ces isoformes proviennent de différents clivages protéolytiques. En effet,
Opa1 possède dans sa partie amino-terminale une pré-séquence d’adressage à la mitochondrie
(Griparic et al., 2004; Misaka et al., 2002; Olichon et al., 2002) qui est clivée au cours de
l’import mitochondrial par la peptidase matricielle, générant les deux isoformes longues de la
protéine (Akepati et al., 2008; Ishihara et al., 2006). Ces isoformes a et b sont ensuite
soumises à des clivages protéolytiques qui produisent pour chacune, une ou deux formes
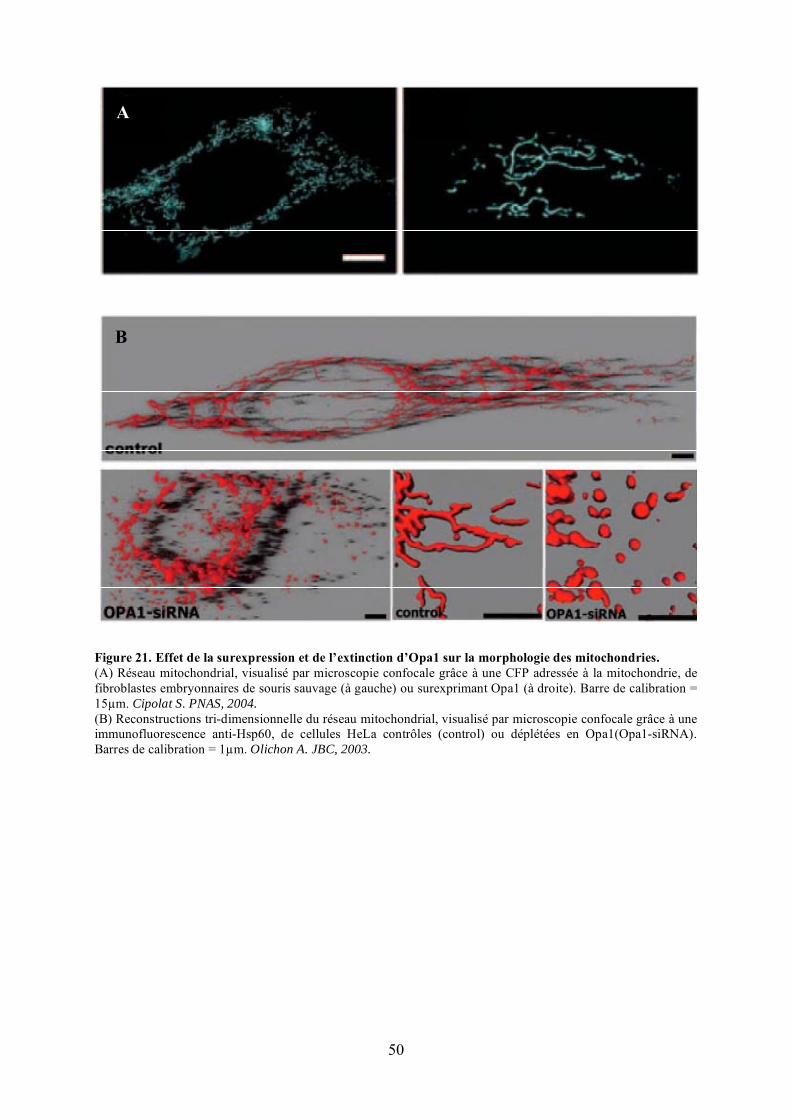
50
Figure 21. Effet de la surexpression et de l’extinction d’Opa1 sur la morphologie des mitochondries.(A) Réseau mitochondrial, visualisé par microscopie confocale grâce à une CFP adressée à la mitochondrie, de fibroblastes embryonnaires de souris sauvage (à gauche) ou surexprimant Opa1 (à droite). Barre de calibration = 15�m. Cipolat S. PNAS, 2004.(B) Reconstructions tri-dimensionnelle du réseau mitochondrial, visualisé par microscopie confocale grâce à une immunofluorescence anti-Hsp60, de cellules HeLa contrôles (control) ou déplétées en Opa1(Opa1-siRNA). Barres de calibration = 1�m. Olichon A. JBC, 2003.
A A
B

51
courtes. À l’heure actuelle, trois sites de clivage ont été proposés : le site S1 situé dans l’exon
5, le site S2 situé dans l’exon 5b (Ishihara et al., 2006) et le site S0 présent dans l’exon 4b
(Figure 19) (Griparic et al., 2007). La nature des protéases impliquées dans le clivage d’Opa1
reste encore incertaine. Les m-AAA protéases, paraplégine et AFG3L (site S1) (Duvezin-
Caubet et al., 2007; Guillery et al., 2008; Ishihara et al., 2006), mais également l’i-AAA
protéase YME1L (sites S0 et S2) (Griparic et al., 2007; Guillery et al., 2008; Song et al.,
2007) semblent, au moins partiellement, jouer un rôle dans ce processus. L’implication de
PARL dans cette maturation a également été proposée (Cipolat et al., 2006), mais reste
cependant extrêmement controversée (Duvezin-Caubet et al., 2007; Griparic et al., 2007;
Guillery et al., 2008; Ishihara et al., 2006). PARL est l’homologue humain de Pcp1 qui, chez
la levure Saccharomyces cerevisiae, est responsable du clivage de la forme longue de Mgm1
en une forme courte (Herlan et al., 2003; McQuibban et al., 2003).
Après clivage par la peptidase matricielle, les formes longues d’Opa1 seraient ancrées
à la membrane interne grâce à une région hydrophobe amino-terminale prédite pour être
trans-membranaire (Figures 11 et 19) avec la majeure partie de la protéine située dans
l’espace inter-membranaire. Les différents clivages protéolytiques décrits ci-dessus
modifieraient la topologie de certaines isoformes courtes de la protéine qui deviendraient dès
lors solubles dans l’espace inter-membranaire ou plus faiblement associées aux membranes
(Cipolat et al., 2006; Ishihara et al., 2006). De plus, l’immuno-détection de la protéine, en
microscopie électronique, indique une localisation préférentielle de la protéine au niveau des
crêtes mitochondriales (Griparic et al., 2004; Olichon et al., 2002; Satoh et al., 2003).
La surexpression ectopique d’Opa1 dans des fibroblastes embryonnaires de souris, qui
présentent des mitochondries ponctiformes, conduit à la formation d’un réseau filamenteux et
interconnecté (Figure 21, A) (Cipolat et al., 2004). Au contraire, l’extinction de l’expression
du gène OPA1, par interférence à l’ARN, entraîne la fragmentation du réseau mitochondrial,
dans des cellules HeLa (ou Cos-7) qui présentent habituellement un réseau mitochondrial
filamenteux (Figure 21, B) (Griparic et al., 2004; Olichon et al., 2003) et se traduit par une
incapacité des mitochondries à fusionner lors de tests de fusion (Arnoult et al., 2005a; Chen et
al., 2005). L’ensemble de ces résultats révèle un rôle de la dynamine dans la fusion
mitochondriale. Bien que cela ne soit pas conservé chez les mammifères (Song et al., 2009),
des expériences de complémentation inter-génique et de co-immuno-précipitation chez

52

53
Saccharomyces cerevisiae montrent que l’oligomérisation en trans de deux protéines Mgm1
localisées sur des mitochondries adjacentes est requise pour cette fonction fusogène (Meeusen
et al., 2006). La maturation protéolytique d’Opa1 apparaît prépondérante dans la régulation de
l’activité fusogène de la protéine. En effet, la disparition des formes longues de la protéine
lors de traitement avec l’agent découplant CCCP par exemple conduit à la fragmentation des
mitochondries et suggère donc que les formes longues sont essentielles dans ce processus
(Guillery et al., 2008). En outre, la mutation des sites de clivages S1 et S2 modifie la
morphologie des mitochondries et souligne également l’importance des formes courtes de la
protéine (Duvezin-Caubet et al., 2006; Griparic et al., 2007; Song et al., 2007). Finalement, la
réintroduction de différents variants sauvages ou qui ne peuvent pas être clivés dans des
fibroblastes embryonnaires de souris invalidées pour le gène OPA1, indique qu’au moins une
forme longue et une forme courte de la protéine sont nécessaires pour la fusion des
mitochondries (Song et al., 2007).
b) Le couplage de la fusion des deux membranes
La fusion de la membrane interne et celle de la membrane externe se déroulent de
manière indépendante (Malka et al., 2005; Meeusen et al., 2004). Cependant, les deux
évènements interviennent de manière hautement coordonnée afin de garantir l’intégrité des
compartiments sous-mitochondriaux. Il a été proposé qu’Ugo1, initialement identifié comme
une protéine essentielle à la fusion mitochondriale chez Saccharomyces cerevisiae (Sesaki
and Jensen, 2001), joue le rôle d’adaptateur pour assurer cette coordination. En effet,
plusieurs mutants thermo-sensibles d’Ugo1 présentent in vitro des défauts de fusion des
membranes externes ou internes mitochondriales (Hoppins et al., 2009). De plus, la topologie
de la protéine semble en accord avec ce rôle éventuel. Elle est en effet ancrée à la membrane
externe par trois domaines trans-membranaires (Hoppins et al., 2009). Son extrémité carboxy-
terminale ainsi que la boucle entre les deuxième et troisième domaines trans-membranaires
sont localisées dans le cytosol alors que la boucle entre les premiers et deuxième domaines
transmembranaires et son extrémité amino-terminale sont situées dans l’espace inter-
membranaire (Hoppins et al., 2009). Ugo1 est co-immuno-précipitée avec les deux autres
acteurs majeurs de la fusion mitochondriale chez la levure, Fzo1 localisée à la membrane
externe et Mgm1 à la membrane interne (Sesaki and Jensen, 2004; Sesaki et al., 2003; Wong
et al., 2003).
Jusqu’à présent, aucun homologue humain d’Ugo1 n’a été identifié. Cependant, la

54

55
coordination de la fusion des deux membranes pourrait être conservée chez les mammifères.
En effet, d’une part, nous avons montré que les mitofusines et Opa1 co-immunoprécipitent
dans des cellules HeLa (Guillery et al., 2008) et, d’autre part, Cipolat et ses collaborateurs ont
mis en évidence une interdépendance fonctionnelle entre Opa1 et Mfn1 (Cipolat et al., 2004).
c) Les protéines régulatrices
Plusieurs protéines non essentielles pour la fusion mitochondriale mais pouvant
cependant la réguler ont récemment été mises en évidence.
mitoPLD
L’extinction par shRNA de MitoPLD, une phospholipase D mitochondriale, dans des
cellules HeLa entraîne la fragmentation du réseau mitochondrial et affecte la fusion des
mitochondries. MitoPLD induit, in vitro et in vivo dans des cellules NIH3T3, une
modification du profil des lipides mitochondriaux avec une diminution de la quantité de la
cardiolipine au profit de l’acide phosphatidique (Choi et al., 2006). Il est donc proposé que
MitoPLD puisse modifier la composition et la courbure des bicouches lipidiques pour les
rendre plus propices à la fusion, après juxtaposition des membranes externes de
mitochondries voisines par les Mitofusines.
MIB
La protéine MIB (Mitofusin Binding Protein) appartient à une superfamille de
réductases/deshydrogénases dont le rôle est encore largement méconnu. Elle a été identifiée
comme partenaire des mitofusines au cours d’un crible double-hybride et pourrait réguler leur
activité. En effet, la surexpression ectopique de MIB conduit à la fragmentation des
mitochondries en inhibant la fusion mitochondriale dans des cellules HeLa alors que, à
l’inverse, l’extinction de la protéine conduit à l’allongement des mitochondries (Eura et al.,
2006).
SLP-2
En réponse à plusieurs stress cellulaires, tels qu’une irradiation aux UV, un traitement
à l’actinomycine D ou à la cycloheximide, les mitochondries fusionnent intensément,
générant un réseau mitochondrial hyper-filamenteux. Cette hyperfusion mitochondriale
induite par le stress (SIMH) permet à la cellule de surmonter un stress transitoire via une
augmentation de la production d’ATP (Tondera et al., 2009). SLP-2 (Stomatin-Like Protein2),
une protéine située dans l’espace intermembranaire appartenant à la famille SPFH
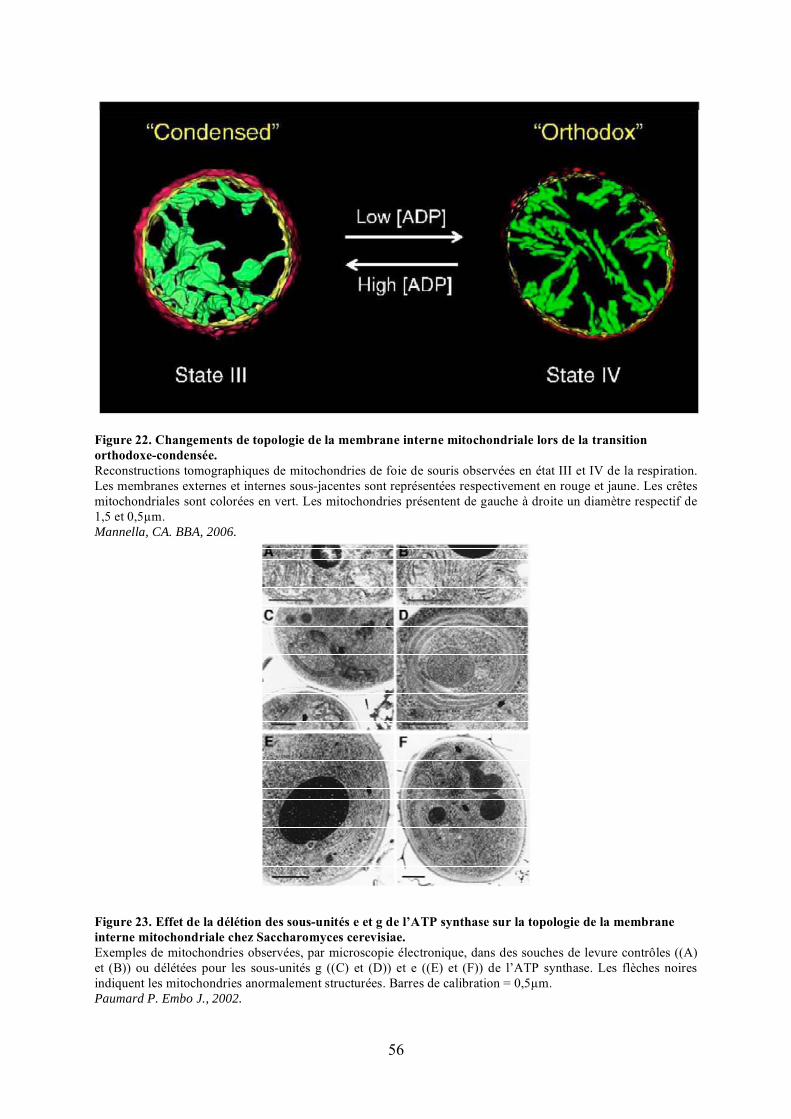
56
Figure 22. Changements de topologie de la membrane interne mitochondriale lors de la transition orthodoxe-condensée.Reconstructions tomographiques de mitochondries de foie de souris observées en état III et IV de la respiration. Les membranes externes et internes sous-jacentes sont représentées respectivement en rouge et jaune. Les crêtes mitochondriales sont colorées en vert. Les mitochondries présentent de gauche à droite un diamètre respectif de 1,5 et 0,5�m.Mannella, CA. BBA, 2006.
Figure 23. Effet de la délétion des sous-unités e et g de l’ATP synthase sur la topologie de la membrane interne mitochondriale chez Saccharomyces cerevisiae.Exemples de mitochondries observées, par microscopie électronique, dans des souches de levure contrôles ((A) et (B)) ou délétées pour les sous-unités g ((C) et (D)) et e ((E) et (F)) de l’ATP synthase. Les flèches noires indiquent les mitochondries anormalement structurées. Barres de calibration = 0,5�m.Paumard P. Embo J., 2002.

57
(Stomatin/Prohibitin/Flotillin/Hflk) joue un rôle essentiel au cours de ce processus en
empêchant le clivage des formes longues de Opa1 et ce indépendamment des Prohibitines 1 et
2 avec lesquelles elle peut interagir (Da Cruz et al., 2008). Il est à noter que SLP-2 avait
précédemment été identifiée comme partenaire de Mfn2 (Hajek et al., 2007).
4) L’architecture des crêtes mitochondriales
Au cours des dix dernières années, les progrès effectués dans le domaine de la
microscopie électronique ont permis de révéler que l’architecture interne des mitochondries
peut être très variée. Les changements de morphologie des crêtes mitochondriales observés
dans certains cas reposent sur des remodelages dynamiques et réversibles de la membrane
interne et ne résultent pas simplement de repliements aléatoires influencés par la seule
pression osmotique environnante. Des travaux menés par Hackenbrock dans les années 1960
ont permis à partir de mitochondries isolées de foie de souris fixées chimiquement, de définir
deux états morphologiques qui diffèrent surtout par la topologie de leur membrane interne
(Figure 22) (Hackenbrock, 1966). L’état orthodoxe est caractérisé par des crêtes
mitochondriales fines et tubulaires reliées à la membrane interne par une, voire deux
connexions appelées jonctions de crête et par une matrice expansée. À l’inverse, les
mitochondries condensées ont une matrice de volume beaucoup plus réduit et présentent des
crêtes assimilables à des « citernes » interconnectées présentant de multiples jonctions de
crêtes. Le passage d’un état à l’autre serait contrôlé par des évènements de fusion et de fission
(Mannella et al., 2001). La condensation de la matrice serait accomplie par la fusion des
crêtes tubulaires en des compartiments plus larges alors que l’expansion de la matrice
impliquerait la fission des larges « citernes » en crêtes tubulaires individualisées.
Le rôle de la topologie de la membrane interne, à l’instar de celui exercé par la
morphologie globale des mitochondries (cf Introduction IIC) dans les fonctions
mitochondriales commence à émerger. En effet, la diffusion de métabolites et de protéines
entre les différents compartiments mitochondriaux semble être altérée par certains
remaniements de cette membrane. Des simulations informatiques indiquent qu’une déplétion
en ADP pourrait avoir lieu au sein d’espaces intra-crêtes volumineux observés dans les
mitochondries condensées (Mannella et al., 2001). Le passage d’un état condensé à un état
orthodoxe permettrait alors de rétablir la concentration en ADP et par conséquent la
production d’ATP. En accord avec cette hypothèse, ce changement de morphologie est
observé au cours du passage de l’état respiratoire III à l’état IV, état où la concentration en
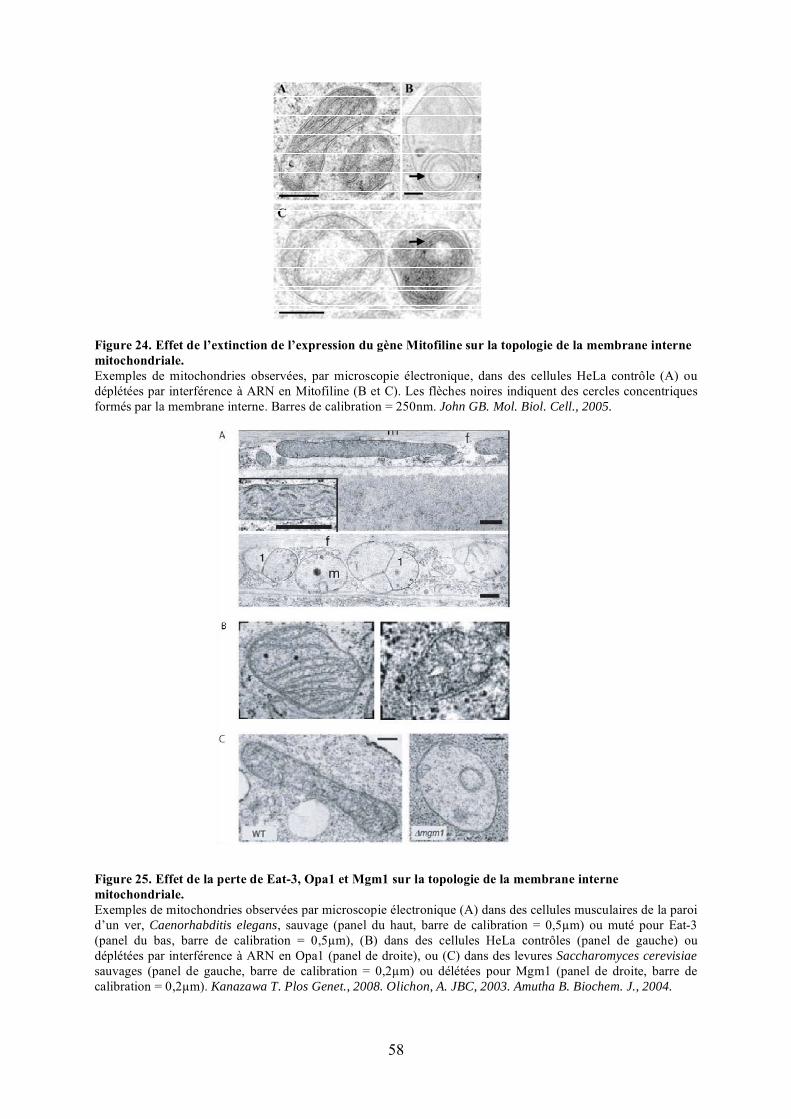
58
Figure 24. Effet de l’extinction de l’expression du gène Mitofiline sur la topologie de la membrane interne mitochondriale.Exemples de mitochondries observées, par microscopie électronique, dans des cellules HeLa contrôle (A) ou déplétées par interférence à ARN en Mitofiline (B et C). Les flèches noires indiquent des cercles concentriques formés par la membrane interne. Barres de calibration = 250nm. John GB. Mol. Biol. Cell., 2005.
Figure 25. Effet de la perte de Eat-3, Opa1 et Mgm1 sur la topologie de la membrane interne mitochondriale.Exemples de mitochondries observées par microscopie électronique (A) dans des cellules musculaires de la paroi d’un ver, Caenorhabditis elegans, sauvage (panel du haut, barre de calibration = 0,5�m) ou muté pour Eat-3(panel du bas, barre de calibration = 0,5�m), (B) dans des cellules HeLa contrôles (panel de gauche) ou déplétées par interférence à ARN en Opa1 (panel de droite), ou (C) dans des levures Saccharomyces cerevisiaesauvages (panel de gauche, barre de calibration = 0,2�m) ou délétées pour Mgm1 (panel de droite, barre de calibration = 0,2�m). Kanazawa T. Plos Genet., 2008. Olichon, A. JBC, 2003. Amutha B. Biochem. J., 2004.

59
ADP limite le taux de respiration (Hackenbrock, 1966). Des données récentes démontrent
clairement que la topologie de la membrane interne joue un rôle majeur au cours de
l’apoptose. Le remodelage des jonctions de crêtes permet la mobilisation dans l’espace inter-
membranaire du cytochrome c, initialement situé dans l’espace intra-crête (Frezza et al.,
2006; Scorrano et al., 2002; Yamaguchi et al., 2008). Nous discuterons plus loin et de manière
plus approfondie de cette étape essentielle de l’apoptose du fait de l’implication d’Opa1 dans
ce processus.
Plusieurs protéines modifiant la topologie de la membrane interne ont été identifiées.
L’ATP synthase influencerait directement la morphologie de la membrane interne. Des
mutations au niveau de deux sous-unités non essentielles de ce complexe, e et g, empêchent la
dimérisation et l’oligomérisation de la protéine et entraînent la disparition des crêtes
mitochondriales. La membrane interne s’organise alors sous la forme de pelures d’oignons
avec deux ou trois bicouches lipidiques disposées de manière concentriques (Figure 23)
(Paumard et al., 2002). Le même phénotype est observé dans des cellules HeLa après
extinction de l’expression par siRNA d’une autre protéine de la membrane interne, la
Mitofiline (Figure 24), qui pourrait être impliquée dans la formation et la stabilisation des
crêtes en régulant de l’oligomérisation de l’ATP synthase (John et al., 2005). La morphologie
de la membrane interne faisant intervenir des évènements de fission et de fusion, il n’est pas
étonnant de voir que des mutations ou une diminution de l’expression d’Opa1 dans les
cellules HeLa ou de ses homologues Mgm1 et EAT3 respectivement chez Saccharomyces
cerevisiae et chez Caenorhabditis elegans entraînent la déstructuration des crêtes (Figure 25)
(Amutha et al., 2004; Kanazawa et al., 2008; Meeusen et al., 2006; Olichon et al., 2003;
Sesaki et al., 2003; Wong et al., 2003). De plus, Mgm1 pourrait servir de chaperonne à la
sous-unité e de l’ATP synthase. En effet, la délétion de son gène inhibe la formation de
dimères de l’ATP synthase (Amutha et al., 2004).
C) Les fonctions biologiques de la dynamique mitochondriale
1) La transmission des mitochondries au cours de la division cellulaire
Un crible génétique visant à isoler des suppresseurs multicopies d’un mutant de
DNM1 chez Saccharomyces cerevisiae a permis d’établir un premier lien entre morphologie
mitochondriale et cycle cellulaire à travers la mise en évidence de l’interaction entre

60

61
Dnm1p et Num1p, une protéine impliquée dans la migration nucléaire au cours de la division
cellulaire (Cerveny et al., 2007; Farkasovsky and Kuntzel, 1995). La protéine Num1p est
localisée à la membrane plasmique mais une fraction de la protéine est également retrouvée à
la mitochondrie, où elle participe à la fission des mitochondries en influençant probablement
l’ancrage à la mitochondrie de Dnm1p. Les auteurs de cette étude montrent également que
Num1p et Dnm1p sont nécessaires pour une répartition équitable des mitochondries entre les
cellules mères et filles au cours de la mitose. En effet, dans des levures délétées pour DNM1
et NUM1, l’intégralité des mitochondries est retrouvée dans le bourgeon avec une forte
proportion de mitochondries hyper-filamenteuses. Chez les mammifères, à l’heure actuelle,
l’existence d’une machinerie spécifique pour assurer la transmission correcte des
mitochondries au cours de la division cellulaire reste incertaine. Compte tenu du nombre
important de mitochondries dans la plupart des types cellulaires, il a été proposé que la
répartition des mitochondries se fasse de manière aléatoire. Néanmoins, des changements de
morphologie mitochondriale sont observés durant le cycle cellulaire chez les mammifères et
soutiennent l’existence d’un lien entre dynamique mitochondriale et cycle cellulaire. En effet,
le réseau mitochondrial devient très filamenteux au cours de la phase G1. Ce changement
apparaît essentiel pour la progression du cycle cellulaire puisque l’inhiber en dépolarisant les
mitochondries provoque un arrêt des cellules au niveau de la transition G1/S et le favoriser en
surexprimant un mutant dominant négatif de Drp1 entraîne une augmentation de la quantité
protéique de la cycline E, précipitant au contraire les cellules en phase G2 (Mitra et al., 2009).
De plus, une fragmentation transitoire des mitochondries, dépendante de Drp1, a lieu au cours
de la mitose de la prophase à l’anaphase. Cette modification est corrélée à la phosphorylation
de Drp1 par le complexe cdk1/cyclineB au niveau du GED, ce qui entraîne une stimulation de
la fission des mitochondries (Taguchi et al., 2007). Si à l’heure actuelle, ces données
demandent à être approfondies, l’impact de la morphologie mitochondriale sur la progression
du cycle cellulaire pourrait s’avérer intéressant et offrir des perspectives en thérapeutique
anti-cancéreuse.
2) Le maintien des fonctions mitochondriales
L’extinction de l’expression des gènes codant pour les acteurs de la fusion des
mitochondries, Opa1, Mfn1 ou Mfn2, dans des fibroblastes embryonnaires de souris, entraîne
la fragmentation des mitochondries mais également d’importants défauts de croissance, une
diminution considérable de l’activité de l’ensemble des complexes de la chaîne respiratoire et

62

63
une grande hétérogénéité dans le potentiel de membrane des mitochondries (Chen et al.,
2005). De plus, l’abolition de la fusion mitochondriale mène à l’accumulation de
mitochondries dépourvues d’ADNmt (Chen et al., 2007) ce qui pourrait expliquer les
dysfonctions mitochondriales observées. La fusion mitochondriale est considérée comme un
moyen pour les mitochondries de coopérer les unes avec les autres afin de complémenter les
déficiences fonctionnelles de certaines d’entre elles en restaurant des éléments manquants ou
défectueux par échange de leur membrane et de leur contenu. En accord avec cela, la
coexistence, dans un hybride cellulaire, de mitochondries provenant de deux lignées
cellulaires, déficientes pour la respiration, portant des mutations différentes au niveau de
gènes situés sur l’ADN mitochondrial, l’une sur l’ARNt Leu et l’autre sur l’ARNt Ile, restaure
une respiration efficace par complémentation inter-génique (Ono et al., 2001). De plus, des
travaux visant à étudier la pathogénèse d’un mutant de délétion de l’ADNmt dans un modèle
murin confirme l’existence in vivo de la complémentation inter-mitochondriale et souligne la
nécessité de l’intégrité de la dynamique mitochondriale dans ce contexte d’hétéroplasmie
pour maintenir une mutation silencieuse (Nakada et al., 2001). Néanmoins, le suivi
individualisé des mitochondries dans des cellules ß-pancréatiques démontre que chaque
mitochondrie subit en moyenne 5 évènements de fusion immédiatement suivie de fission par
heure, menant à la ségrégation de mitochondries peu actives. Ces mitochondries, caractérisées
par un potentiel de membrane réduit, présentent une très faible probabilité de fusionner de
nouveau (6 fois inférieure aux autres mitochondries) et sont ensuite dégradées par autophagie
(Twig et al., 2008). Ces travaux récents iraient donc à l’encontre de la théorie énoncée
précédemment puisque la fusion mitochondriale semble s’effectuer préférentiellement entre
des mitochondries fonctionnelles.
3) La distribution des mitochondries
La dynamique mitochondriale revêt une importance toute particulière dans les
neurones, certainement de par leurs besoins énergétiques élevés mais également de
l’importance de la distribution de cet organelle dans ces cellules. Les mitochondries ne sont
pas distribuées uniformément dans les neurones et sont concentrées dans des régions bien
précises de la cellule notamment dans les espaces pré- et post-synaptiques (Chang et al., 2006;
Li et al., 2004). La localisation mitochondriale au niveau des terminaisons axonales peut être
influencée par la dynamique mitochondriale. En effet, chez la drosophile, des mutations de
Drp1, mais également de Milton et Miro (Guo et al., 2005; Stowers et al., 2002), affectent la

64

65
distribution cellulaire des mitochondries alors largement absentes des synapses, inhibent la
mobilisation du pool vésiculaire de réserve, un procédé hautement dépendant de l’ATP, et par
conséquent inhibent les transmissions synaptiques (Verstreken et al., 2005). De plus,
l’absence de mitochondries aux synapses dans ces cellules altère la régulation de
l’homéostasie calcique, se traduisant par des niveaux résiduels élevés de calcium après une
haute activité synaptique. La distribution mitochondriale dans les dendrites semble elle aussi
affectée par la fission et la fusion mitochondriale. En effet, dans des neurones d’hippocampe
de rat, les mitochondries sont recrutées au niveau des épines dendritiques après stimulation
neuronale (Li et al., 2004). L’inhibition de la fission mitochondriale ou l’activation de la
fusion mitochondriale, respectivement par la surexpression d’un mutant dominant négatif de
Drp1, Drp1K38A, ou la surexpression d’Opa1, affecte la distribution dendritique des
mitochondries et la densité des épines dendritiques. À l’inverse, l’augmentation de la fission
mitochondriale, par la surexpression de Drp1 sauvage, entraîne une présence accrue des
mitochondries au niveau dendritique et résulte en une augmentation du nombre d’épines
dendritiques.
La dynamique mitochondriale affecte la distribution des mitochondries dans d’autres
types cellulaires notamment dans des cellules du système immunitaire. En effet, au cours de la
chimiotaxie lymphocytaire, les mitochondries se concentrent au niveau de l’uropode, étape
qui nécessite la fission des mitochondries. L’inhibition de la fission mitochondriale par la
surexpression de Drp1K38A, ou l’activation de la fusion, par la surexpression d’Opa1 ou
Mfn1, empêche la relocalisation des mitochondries et par conséquent la polarisation et la
migration des lymphocytes en présence de chimioattractants (Campello et al., 2006).
4) La mitophagie
L’autophagie, désignant généralement la macroautophagie, est active à un niveau basal
dans toutes les cellules afin de renouveler des protéines à durée de vie longue et certains
organites. Cependant, elle est stimulée en situation de stress et constitue alors un mécanisme
de survie cellulaire et d’adaptation via la dégradation des composants cellulaires altérés
(Mizushima, 2007). L’autophagie débute par la formation d'une double membrane dont
l’origine reste débattue, appelée « vacuole initiale » ou « autophagosome » (APs), qui
séquestre le matériel cytoplasmique à dégrader, suivi d’une étape de maturation de
l'autophagosome en vacuole dégradative après fusion avec des compartiments endocytiques,
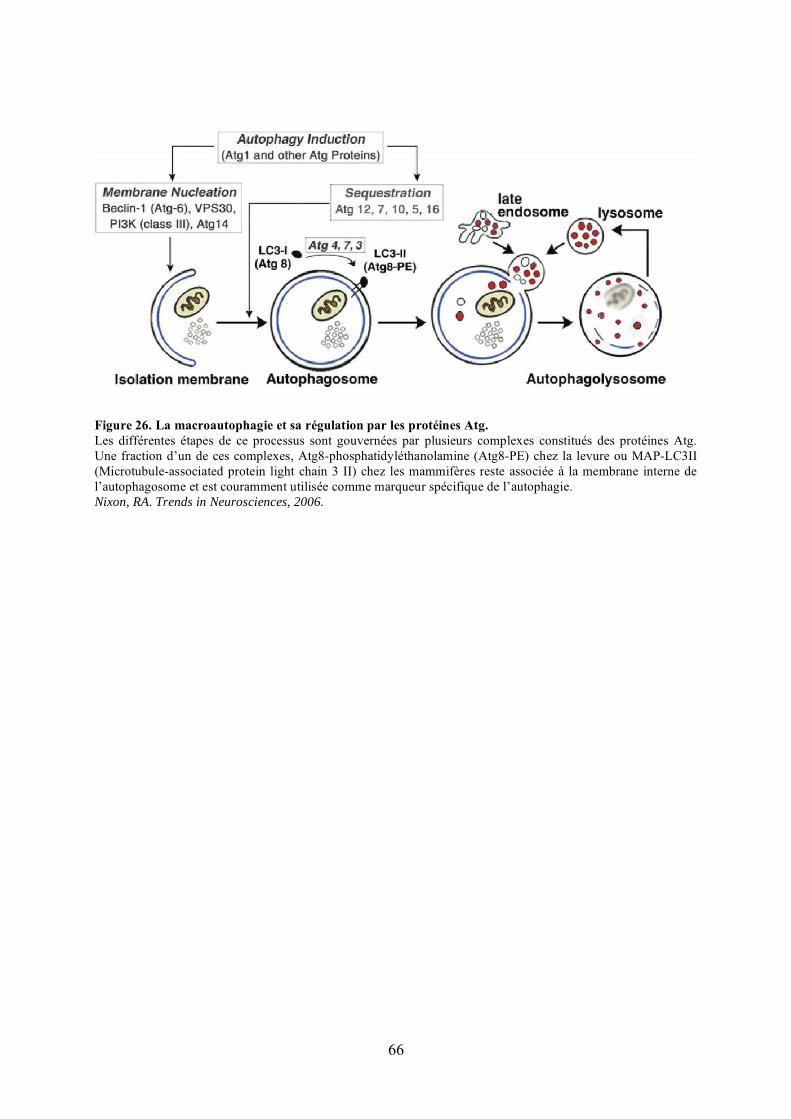
66
Figure 26. La macroautophagie et sa régulation par les protéines Atg.Les différentes étapes de ce processus sont gouvernées par plusieurs complexes constitués des protéines Atg. Une fraction d’un de ces complexes, Atg8-phosphatidyléthanolamine (Atg8-PE) chez la levure ou MAP-LC3II (Microtubule-associated protein light chain 3 II) chez les mammifères reste associée à la membrane interne de l’autophagosome et est couramment utilisée comme marqueur spécifique de l’autophagie.Nixon, RA. Trends in Neurosciences, 2006.

67
et se termine enfin par la dégradation complète du matériel séquestré après fusion avec le
lysosome (Figure 26). Ce processus fait intervenir chez les mammifères plus d’une trentaine
de protéines initialement identifiées chez Saccharomyces cerevisiae, appelées les protéines
Atg (Autophagy related proteins). Les bases moléculaires de la sélectivité de ce processus vis-
à-vis de certaines organelles commencent à être élucidées. L’élimination sélective des
mitochondries par autophagie, appelée mitophagie (Kim et al., 2007; Lemasters, 2005), a été
décrite en réponse à différents stress cellulaires comme des carences en nutriments (Elmore et
al., 2001), des conditions hypoxiques (Zhang et al., 2008) ou apoptotiques (Arnoult et al.,
2005b), mais également au cours de processus de différenciation cellulaire comme lors de la
maturation des érythrocytes (Takano-Ohmuro et al., 2000), ou durant la fécondation des
ovocytes par les spermatozoïdes (Shitara et al., 2000). Concernant les mitochondries, il est
désormais établi que la dissipation du potentiel de membrane interne mitochondrial (��m)
est un pré-requis essentiel pour leur incorporation au sein des APs (Kim et al., 2007;
Rodriguez-Enriquez et al., 2006).
Dernièrement, la mitophagie a été étudiée dans des cellules ß-pancréatiques qui
présentent une forte accumulation de mitochondries dépolarisées menant à des dommages
oxydatifs et au développement de diabètes (Twig et al., 2008). Le suivi individualisé du
devenir des mitochondries dans ces cellules indique que la fission mitochondriale joue un rôle
essentiel au cours de ce processus. Elle permet d’isoler les mitochondries peu actives,
présentant un potentiel de membrane réduit et dans certains cas des capacités respiratoires
altérées ou une absence de nucléoïdes (Arimura et al., 2004), des autres organites et
d’alimenter ainsi la machinerie d’autophagie. L’extinction de l’expression de hFIS1 ou la
surexpression du mutant dominant négatif de Drp1 (Drp1K38A) réduisent spécifiquement
l’autophagie mitochondriale dans ces cellules (Twig et al., 2008), ainsi que dans des neurones
exposés à l’oxyde nitrique (Barsoum et al., 2006). Au contraire, la surexpression de Drp1
facilite l’élimination des mitochondries en réponse à différents stimuli apoptotiques (Arnoult
et al., 2005b). Enfin, il est probable que la fission mitochondriale favorise également
l’élimination des mitochondries en réduisant la taille de ces organites puisque la formation
d’un autophagosome de grande taille consomme beaucoup plus d’énergie que celle d’un
autophagosome de plus petite taille (Terman and Brunk, 2004; Terman et al., 2003).

68

69
D) Dynamique mitochondriale et développement
La pertinence fonctionnelle de la dynamique mitochondriale a clairement été
démontrée par le développement d’animaux modèles invalidés pour les gènes codant pour les
différents acteurs des processus de fusion et de fission. Les souris déficientes pour les
Mitofusines 1 et 2 ne sont pas viables et meurent respectivement aux 12e et au 11e jour du
développement embryonnaire. Alors qu’aucun défaut n’a été constaté dans les souris
invalidées pour MFN1, un nombre insuffisant de cellules trophoblastiques géantes et par
conséquent une insuffisance placentaire est à l’origine de la létalité des souris déficientes pour
Mfn2 (Chen et al., 2003). L’invalidation conditionnelle du gène MFN2 dans l’intégralité de
l’embryon à l’exception du placenta, induit une réduction dramatique du nombre d’épines
dendritiques des cellules Purkinje et de la taille du cervelet dûe à la dégénérescence ultérieure
de ces cellules (Chen et al., 2007). Ces altérations, qui soulignent l’importance de Mfn2 dans
le développement post-natal cérébral des souris, sont responsables d’importants troubles
posturaux et locomoteurs chez ces animaux. Récemment, deux modèles murins portant
chacun une mutation troncative au sein des exons 8 et 10 du gène OPA1 et menant à une
réduction de 50% de la quantité de la protéine ont été décrits (Alavi et al., 2007; Davies et al.,
2007). Alors que les souris homozygotes pour cette mutation meurent in utero au cours de
l’embryogénèse (E12,5), les souris hétérozygotes développent une neuropathie optique à
évolution progressive et lente, une perte partielle des cellules ganglionnaires de la rétine au
bout de 6 mois et une morphologie anormale des axones résiduels du nerf optique avec des
défauts de myélinisation à l’âge de neuf mois. Enfin, Drp1, est également essentiel pour le
développement embryonnaire chez la souris. En effet, les souris invalidées pour le gène
codant pour DRP1 meurent au stade embryonnaire (E12,5) et présentent notamment un
développement anormal du prosencéphale (Ishihara et al., 2009).
E) Dynamique mitochondriale et neurodégénérescence
Des mutations au sein de gènes codant pour les acteurs de la dynamique
mitochondriale ont été associées à des maladies neurodégénératives.
Notre équipe a identifié en 2000 le gène codant pour Opa1, localisé sur le
chromosome 3q28-q29, qui est responsable de la plus fréquente des neuropathies optiques

70

71
héréditaires (incidence 1/50000 à 1/12000), l’atrophie optique dominante de type 1 (ADOA)
(Delettre et al., 2000). L’ADOA qui présente une très grande variabilité dans la sévérité des
symptômes inter- et intra-familiale, conduit à une diminution progressive bilatérale de l’acuité
visuelle, pouvant aller jusqu’à la cécité, résultant d’une dégénérescence des cellules
ganglionnaires de la rétine. La mort de ces cellules pourrait impliquer un processus
apoptotique (cf Résultats, III). L’analyse de la localisation spatiale de la protéine et du patron
d’expression du gène par immunohistochimie ne permet pas d’expliquer la spécificité
tissulaire de la maladie. En effet, au sein de la rétine, Opa1 est exprimée aussi bien dans les
cellules ganglionnaires que dans les photorécepteurs et dans les couches plexiforme externe et
plexiforme interne, de même que dans les régions pré- et post-laminaire du nerf optique
(Aijaz et al., 2004; Ju et al., 2005; Pesch et al., 2004; Wang et al., 2006). Opa1 est de plus
détectée dans toutes les zones cérébrales, sans différence notable dans l’expression de ses
différentes isoformes (Bette et al., 2005; Kamei et al., 2005). Récemment, plusieurs patients
présentant des atteintes ne se limitant pas aux cellules ganglionnaires de la rétine ont été
décrits (Amati-Bonneau et al., 2003; Li et al., 2005; Liguori et al., 2008). Une forme
syndromique de la maladie appelée « ADOA plus », caractérisée par une altération de l’ADN
mitochondrial dans les fibres musculaires des patients menant au développement, outre de
l’atrophie optique, d’ophtalmoplégie, de ptosis, de myopathie ou d’ataxie a également été
mise en évidence (Amati-Bonneau et al., 2008; Ferraris et al., 2008; Hudson et al., 2008;
Verny et al., 2008).
À ce jour, plus de 200 mutations différentes du gène OPA1 ont été recensées, elles se
concentrent principalement dans les régions codant pour le domaine GTPase (exons 8 à 15) et
le domaine carboxy-terminal GED (base de données eOpa1) (Ferre et al., 2005). La moitié
des mutations, incluant des mutations non-sens, des insertions, des délétions ou des mutations
au niveau des sites d’épissage changeant le cadre de lecture, mène à l’apparition de codons
stop prématurés. Les faibles niveaux protéiques de Opa1 observés dans ces cas-là permettent
de proposer que l’haploinsuffisance est à l’origine de la pathologie (Marchbank et al., 2002;
Pesch et al., 2001; Schimpf et al., 2008; Schimpf et al., 2006). Néanmoins, environ 30% des
mutations sont des mutations faux-sens, proches ou au sein du domaine GTPase, ce qui
suggère également un effet dominant négatif des protéines mutées (cf Résultats, III) (Amati-
Bonneau et al., 2008; Ferraris et al., 2008; Hudson et al., 2008).
Une soixantaine de mutations du gène codant pour la Mitofusine 2 sont responsables

72

73
d’une forme axonale (forme 2A) à transmission autosomique dominante de la maladie de
Charcot-Marie-Tooth (CMT), une des maladies neurologiques héréditaires les plus courantes
(1 personne sur 2 500) (Cartoni and Martinou, 2009; Zuchner et al., 2004). Elle est
caractérisée par une atteinte évolutive des nerfs périphériques transmettant les influx nerveux
de la moelle épinière aux muscles, entraînant une amyotrophie distale généralement plus
prononcée aux jambes et parfois associée à des troubles sensitifs. Il est à noter que des
mutations au sein du gène codant pour GDAP1, protéine probablement impliquée dans la
fission mitochondriale entraînent une autre forme de CMT, la forme 4A (Cuesta et al., 2002).
Il s’agit d’une forme démyélinisante autosomique récessive de la maladie.
Waterham et ses collaborateurs ont récemment identifié une mutation létale de Drp1
chez un nouveau-né qui présentait une microcéphalie, une acidose lactique et une atrophie
optique (Waterham et al., 2007).
III) LES RELATIONS ENTRE LA DYNAMIQUE MITOCHONDRIALE
ET L’APOPTOSE
A) Le rôle des acteurs de la dynamique mitochondriale dans l’apoptose
Précocement au cours de l’apoptose, dans plusieurs types cellulaires, une activation de
la fission mitochondriale couplée à une inhibition de la fusion (Karbowski et al., 2004a)
entraîne la fragmentation du réseau mitochondrial. Les mitochondries alors ponctiformes, de
taille réduite, s’agrègent parfois dans un deuxième temps de manière péri-nucléaire,
probablement à cause de l’inactivation de la kinésine associée aux mitochondries (De Vos et
al., 1998; Frank et al., 2001). La fragmentation du réseau mitochondrial au cours de
l’apoptose est dépendante de Drp1 puisqu’elle est bloquée par la surexpression d’un mutant
dominant négatif de la protéine (Drp1K38A) ou l’utilisation de siARN dirigés contre cette
protéine (Arnoult et al., 2005b; Breckenridge et al., 2003; Frank et al., 2001; Lee et al., 2004;
Sugioka et al., 2004). De plus, la perte de fonction de la dynamine retarde l’apoptose induite
par différents stimuli en inhibant partiellement mais spécifiquement la libération du

74

75
cytochrome c (Arnoult et al., 2005b; Barsoum et al., 2006; Estaquier and Arnoult, 2007;
Frank et al., 2001; Parone et al., 2006). Ces données ont, dans un premier temps, été
interprétées comme une preuve du rôle essentiel de la dynamique mitochondriale au cours de
l’apoptose. Néanmoins, s’il n’est pas exclu qu’il puisse faciliter la mort cellulaire, ce
changement de morphologie des mitochondries ne pourrait être qu’une conséquence de
l’intervention de plusieurs protéines de la dynamique mitochondriale dans la voie
mitochondriale de l’apoptose. En effet, des études récentes démontrent que Drp1 possède un
rôle dans la perméabilisation de la membrane externe, en aval de la translocation de Bax mais
avant la libération du cytochrome c, indépendamment de son activité dans la fission
mitochondriale. L’identification d’un inhibiteur spécifique de Drp1, Mdivi-1 a permis de
l’impliquer directement dans l’apoptose (Cassidy-Stone et al., 2008). Mdivi-1 se fixe à Drp1
et bloque son oligomérisation, entraînant une diminution de son activité GTPase. De façon
remarquable, Mdivi-1 bloque la libération du cytochrome c in vitro à partir de mitochondries
isolées de foie de souris incubées avec une forme clivée de Bid recombinante démontrant un
rôle propre de cette dernière dans la perméabilisation de la membrane externe, en amont des
protéines de la famille de Bcl-2. Des travaux réalisés chez Caenorhabditis elegans viennent
renforcer l’existence d’un rôle propre de Drp1 dans l’apoptose. En effet, Drp1, dont la perte
est embryonnaire létale dans cet organisme, est impliquée dans l’élimination des
mitochondries par mitophagie, un processus contribuant probablement à la mort cellulaire
programmée par l’arrêt de la production d’énergie (Arnoult et al., 2005b; Skulachev et al.,
2004). Ce rôle pro-apoptotique de Drp1 n’est activé qu’après clivage de la protéine par la
caspase zymogène CED-3 et est ainsi indépendant de son activité de fission mitochondriale
(Breckenridge et al., 2008). Le rôle de Drp1 dans l’apoptose chez les mammifères semble être
régulé via des modifications post-traductionnelles. En effet, la surexpression d’un mutant
phospho-mimétique de Drp1, Drp1 S656D, augmente la résistance des cellules à l’apoptose
induite par l’étoposide ou la staurosporine. À l’inverse, l’élimination de ce site de
phosphorylation par la PKA via la mutation S656A sensibilise les cellules à l’apoptose
(Cribbs and Strack, 2007). De plus, au cours de l’apoptose, après la fragmentation
mitochondriale mais avant le relargage du cytochrome c, Drp1 est mono-sumoylée et est ainsi
stabilisée à la membrane externe mitochondriale (Wasiak et al., 2007).
Comme Drp1, hFis pourrait également avoir un rôle pro-apoptotique indépendant de
sa fonction dans la dynamique mitochondriale. La perte de fonction de hFis1, en présence
d’un stimulus apoptotique, diminue la fragmentation du réseau mitochondrial et retarde
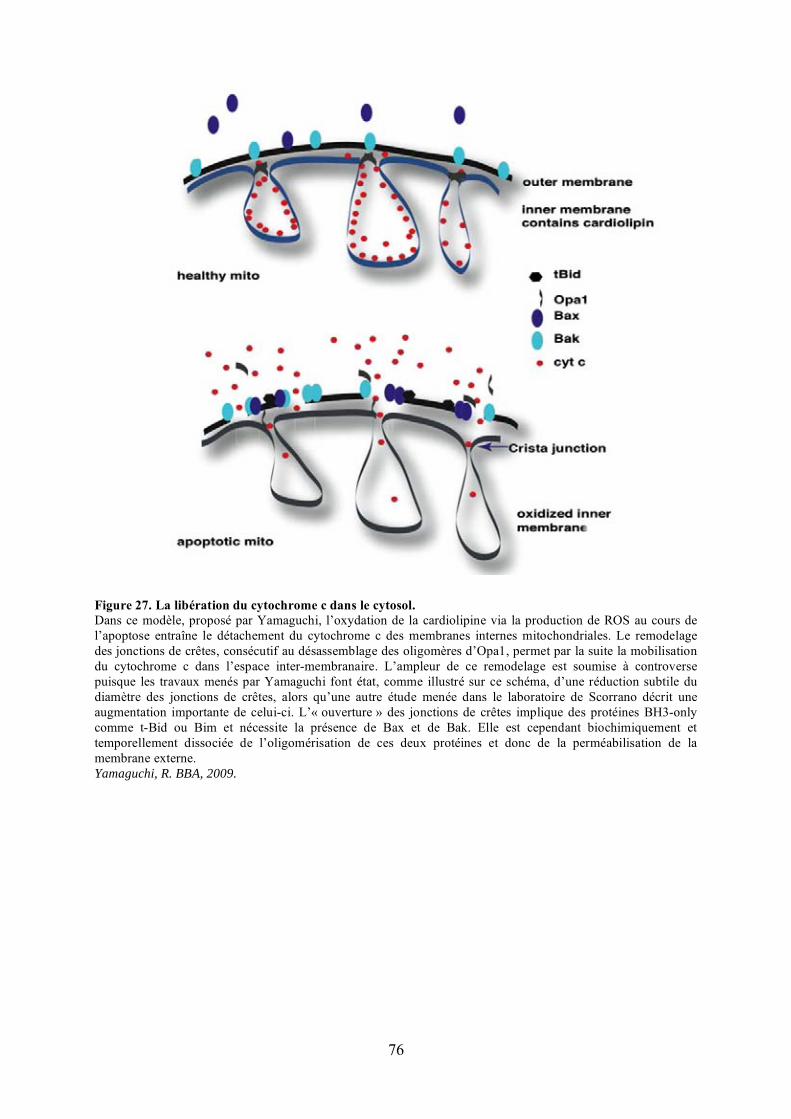
76
Figure 27. La libération du cytochrome c dans le cytosol.Dans ce modèle, proposé par Yamaguchi, l’oxydation de la cardiolipine via la production de ROS au cours de l’apoptose entraîne le détachement du cytochrome c des membranes internes mitochondriales. Le remodelage des jonctions de crêtes, consécutif au désassemblage des oligomères d’Opa1, permet par la suite la mobilisation du cytochrome c dans l’espace inter-membranaire. L’ampleur de ce remodelage est soumise à controverse puisque les travaux menés par Yamaguchi font état, comme illustré sur ce schéma, d’une réduction subtile du diamètre des jonctions de crêtes, alors qu’une autre étude menée dans le laboratoire de Scorrano décrit une augmentation importante de celui-ci. L’« ouverture » des jonctions de crêtes implique des protéines BH3-only comme t-Bid ou Bim et nécessite la présence de Bax et de Bak. Elle est cependant biochimiquement et temporellement dissociée de l’oligomérisation de ces deux protéines et donc de la perméabilisation de la membrane externe. Yamaguchi, R. BBA, 2009.

77
l’apoptose (Lee et al., 2004). Au contraire, la surexpression de Fis1 induit spontanément la
fission des mitochondries et l’apoptose (James et al., 2003). La mutation ponctuelle d’un
résidu de la protéine, localisé dans l’espace inter-membranaire, K148R, empêche la
fragmentation des mitochondries mais n’affecte pas la capacité de la protéine à induire la mort
des cellules, suggérant que les deux fonctions de hFis1, dans la dynamique mitochondriale et
dans l’apoptose, sont indépendantes l’une de l’autre (Alirol et al., 2006). hFis1 agit
différemment de Drp1 au cours de l’apoptose puisque l’extinction du gène hFis1 inhibe la
translocation de Bax et son changement conformationnel (Lee et al., 2004).
Enfin, des travaux récents montrent que l’oligomérisation d’Opa1 contrôle la
redistribution du cytochrome c de l’espace intra-crête vers l’espace inter-membranaire via une
modulation subtile des jonctions des crêtes (Frezza et al., 2006; Yamaguchi et al., 2008). En
réponse à un stimulus apoptotique, les protéines « BH3-only » de la famille de Bcl-2 comme
la forme tronquée de Bid ou Bim déstabilisent, par un mécanisme restant encore à définir, les
oligomères d’Opa1, entraînant l’« ouverture » des jonctions de crêtes et par conséquent la
mobilisation du cytochrome c dans l’espace inter-membranaire (Figure 27) (Frezza et al.,
2006; Yamaguchi et al., 2008). En accord avec l’implication d’Opa1 dans ce processus, une
mutation mimant une conformation liée au GTP de la protéine (Opa1Q297V) (Misaka et al.,
2002) empêche la dissociation des oligomères lors de l’apoptose, la modification du diamètre
des jonctions de crêtes, bloque le relargage du cytochrome c et protège les cellules de
l’apoptose (Yamaguchi et al., 2008). L’analyse de la nature de ces oligomères a permis de
proposer l’implication dans cette fonction pro-apoptotique d’Opa1 d’une forme minoritaire de
la protéine (environ 4% de la quantité totale), soluble dans l’espace inter-membranaire, et
issue du clivage par la sérine protéase de la famille rhomboïde, PARL (Cipolat et al., 2006;
Frezza et al., 2006). Ce rôle explique des résultats antérieurs démontrant que l’extinction de
l’expression d’OPA1 s’accompagne, sinon d’une mort spontanée des cellules, d’une
sensibilité accrue de différentes lignées cellulaires à l’apoptose induite via des stimuli de la
voie intrinsèque de l’apoptose et, qu’à l’inverse, la surexpression ectopique de la protéine
protège les cellules (Arnoult et al., 2005a; Lee et al., 2004; Olichon et al., 2003). Il est à noter
que la surexpression ectopique d’Opa1 a toujours un effet anti-apoptotique dans des
fibroblastes embryonnaires de souris invalidées pour le gène MFN1 (Cipolat et al., 2004) et
que l’extinction spécifique des variants d’épissage d’Opa1 contenant l’exon 4 conduit à une
inhibition de l’activité fusogène de la protéine sans affecter sa fonction anti-apoptotique

78

79
(Olichon et al., 2007a). Ces deux études démontrent que les deux fonctions de la protéine,
fusion des mitochondries et maintien de la conformation des crêtes, sont indépendantes.
B) Le rôle des protéines de la famille de Bcl-2 dans la dynamique
mitochondriale
Plusieurs protéines de la famille de Bcl-2, chez les mammifères, semblent pouvoir
influencer la morphologie des mitochondries indépendamment de leur action dans la
perméabilisation de la membrane externe, et pourraient donc également contribuer à la
fragmentation du réseau mitochondrial au cours de l’apoptose.
En conditions physiologiques, Bax et Bak sont nécessaires pour la fusion
mitochondriale en permettant l’assemblage de Mfn2 en complexes de haut poids moléculaire
(Karbowski et al., 2006). En effet, ces complexes sont de taille plus réduite et la fusion est
altérée dans des fibroblastes embryonnaires de souris déficients pour Bax et Bak présentant de
ce fait des mitochondries moins longues et inter-connectées que des cellules sauvages. La
surexpression de Bax dans des fibroblastes embryonnaires de souris invalidées pour BAX et
BAK rétablit la fusion des mitochondries en restaurant l’assemblage de Mfn2 en complexes
de haut poids moléculaire et sa localisation au niveau des sites de fusion de mitochondries
(Karbowski et al., 2006). Paradoxalement, la surexpression de Bax ou de Bak dans des
cellules HeLa entraîne la fragmentation des mitochondries et l’apoptose (Karbowski et al.,
2004a; Sheridan et al., 2008). Comment ces deux protéines peuvent elle jouer des rôles
opposés dans la dynamique mitochondriale en conditions physiologiques et en apoptose ? Il
est possible que les changements de conformation et de localisation subis par Bax et Bak au
cours de l’apoptose modulent les interactions des deux protéines avec les acteurs de la
dynamique mitochondriale comme les Mitofusines (Brooks et al., 2007) ou l’Endophilin B1
(Cuddeback et al., 2001; Karbowski et al., 2004b; Pierrat et al., 2001). En accord avec cette
hypothèse, en conditions physiologiques, Bak interagit avec les Mitofusines 1 et 2 alors que,
au cours de l’apoptose, Bak et Mfn2 sont dissociés au profit d’une interaction entre Bak et
Mfn1 (Brooks et al., 2007).
Bcl-XL semble également capable de modifier la morphologie des mitochondries.
Cette protéine induit dans des neurones en culture ainsi que dans des lignées cellulaires,
suivant son niveau d’expression, la fission ou la fusion des mitochondries (Berman et al.,
2009; Li et al., 2008). En accord avec ces données, Bcl-XL est capable d’interagir avec Mfn2

80

81
(Delivani et al., 2006), mais aussi avec Drp1 pour permettre, via la stimulation de l’activité
GTPase de la dynamine, la localisation des mitochondries aux synapses et la stimulation de la
synaptogénèse dans des neurones d’hippocampe de rat (Li et al., 2008). Il est à noter que le
seul BH3-only de la famille de Bcl-2 chez Caenorhabditis elegans, EGL-1 ainsi que
l’homologue de Bcl-2, CED-9, ne sont pas capables de modifier la morphologie
mitochondriale dans cet organisme, suggérant que le rôle des protéines de la famille de Bcl-2
dans la dynamique mitochondriale est restreint aux mammifères (Breckenridge et al., 2009).

82

83
Résultats

84

85
Première Partie
Interaction entre Opa1 et Bnip3
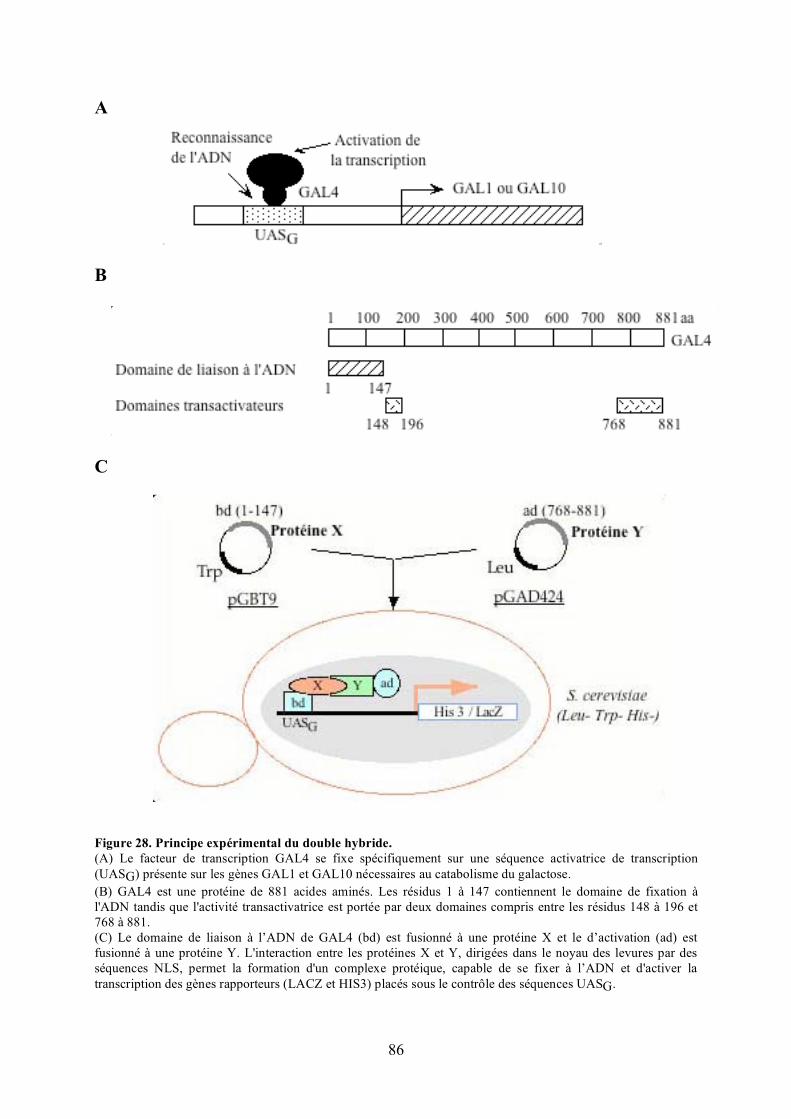
86
A
B
C
Figure 28. Principe expérimental du double hybride.(A) Le facteur de transcription GAL4 se fixe spécifiquement sur une séquence activatrice de transcription (UASG) présente sur les gènes GAL1 et GAL10 nécessaires au catabolisme du galactose.(B) GAL4 est une protéine de 881 acides aminés. Les résidus 1 à 147 contiennent le domaine de fixation à l'ADN tandis que l'activité transactivatrice est portée par deux domaines compris entre les résidus 148 à 196 et 768 à 881.(C) Le domaine de liaison à l’ADN de GAL4 (bd) est fusionné à une protéine X et le d’activation (ad) est fusionné à une protéine Y. L'interaction entre les protéines X et Y, dirigées dans le noyau des levures par des séquences NLS, permet la formation d'un complexe protéique, capable de se fixer à l’ADN et d'activer la transcription des gènes rapporteurs (LACZ et HIS3) placés sous le contrôle des séquences UASG.

87
I) RECHERCHE DE PARTENAIRES DE LA PROTÉINE OPA1
Au début de ce travail, mise à part son implication dans l’atrophie optique dominante
de type 1 (Alexander et al., 2000; Delettre et al., 2000) et sa localisation mitochondriale
(Olichon et al., 2002), très peu de choses étaient connues sur Opa1. Notre équipe a décidé de
rechercher des protéines interagissant avec cette dynamine. Il a été choisi pour cela de mettre
en oeuvre un criblage par double hybride. Cette approche donne directement accès à l’ADNc
codant pour une protéine capable d’interagir avec la protéine d’intérêt in vivo dans la levure
bourgeonnante Saccharomyces cerevisiae. N’ayant pas réalisé l’ensemble des expériences, je
décrirai rapidement la mise en place de cette approche et les résultats obtenus.
A) Description du système double hybride
Le système du double hybride a été initialement développé par Fields et Song en 1989
(Fields and Song, 1989). Il repose sur les propriétés intrinsèques du facteur de transcription
GAL4 de la levure Saccharomyces cerevisiae. GAL4 est une protéine de 881 acides aminés
nécessaire au catabolisme du galactose. Sa fixation spécifique sur une séquence d’ADN de 17
paires de base (UASGAL4 pour "upstream activating séquence" de GAL4) permet l’activation
des gènes GAL1 ou GAL10 (Figure 28, A). Les propriétés de liaison à l’ADN et d’activation
de transcription de GAL4 sont portées par des domaines structuraux différents et
physiquement séparables (Figure 28, B) (Ma and Ptashne, 1987a, b). Les 147 premiers résidus
de GAL4 se lient spécifiquement à l’ADN mais ne peuvent activer la transcription, alors
qu’au contraire les deux régions acides de GAL4 (résidus 148 à 196 et 768 à 881) activent la
transcription mais ne peuvent se lier à l’ADN.
Dans le système double hybride que nous avons utilisé (Figure 28, C), le vecteur
pGBT9 porte le domaine de liaison à l’ADN de GAL4 (bd) placé en fusion traductionnelle
avec un ADNc codant pour la protéine X (permettant l’expression de la protéine de fusion bd-
X) et le gène de prototrophie au tryptophane. Le vecteur pGAD424 porte le domaine
d’activation de transcription de GAL4 (ad) placé en fusion traductionnelle avec un ADNc
codant pour la protéine Y (permettant l’expression de la protéine de fusion ad-Y) et le gène de
prototrophie à la leucine. Les deux plasmides sont introduits dans la souche de levure YGH1
auxotrophe pour le tryptophane et pour la leucine. Cette souche également auxotrophe pour
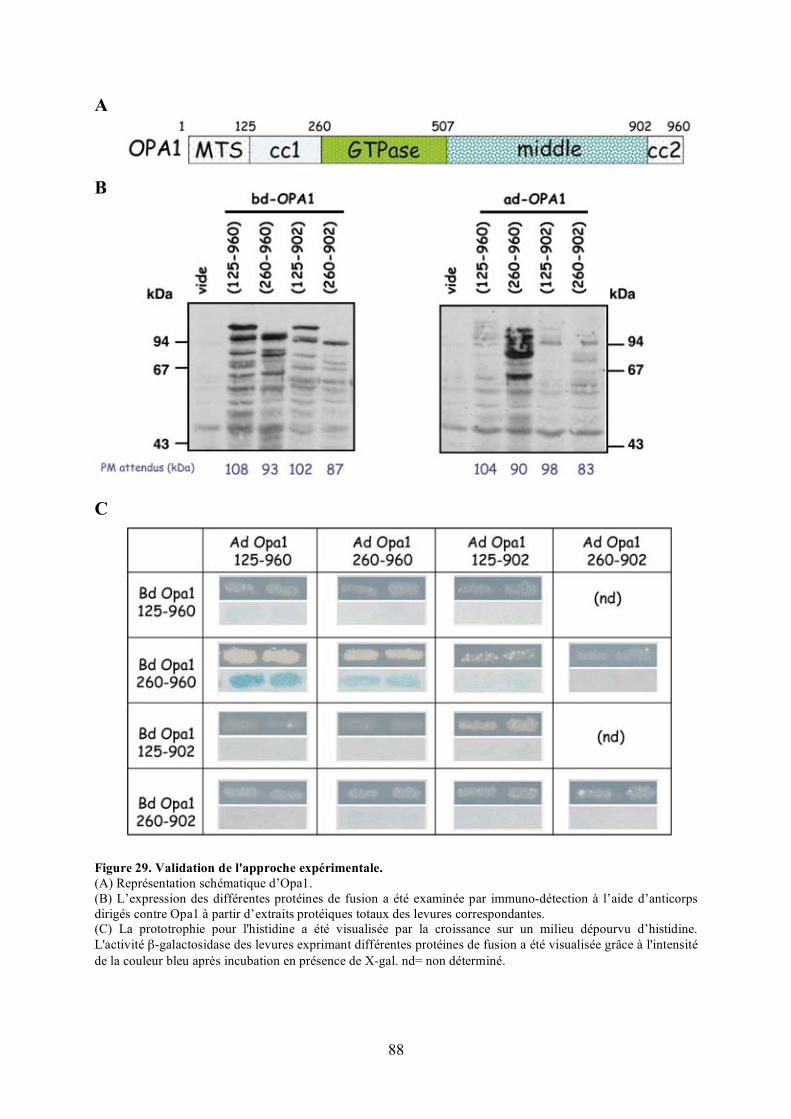
88
A
B
C
Figure 29. Validation de l'approche expérimentale.(A) Représentation schématique d’Opa1.(B) L’expression des différentes protéines de fusion a été examinée par immuno-détection à l’aide d’anticorps dirigés contre Opa1 à partir d’extraits protéiques totaux des levures correspondantes.(C) La prototrophie pour l'histidine a été visualisée par la croissance sur un milieu dépourvu d’histidine. L'activité �-galactosidase des levures exprimant différentes protéines de fusion a été visualisée grâce à l'intensité de la couleur bleu après incubation en présence de X-gal. nd= non déterminé.

89
l’histidine porte dans son génome deux gènes rapporteurs, LACZ et HIS3, sous la dépendance
trancriptionnelle de GAL4 dans un contexte de deux promoteurs différents : le gène LACZ est
sous la dépendance d’un promoteur synthétique contenant une TATA box classique associée à
l'UAS de GAL4 alors que le gène HIS3 est cloné en aval du promoteur du gène GAL1. Le
gène LACZ code pour la �-galactosidase facilement détectée par une coloration blanc/bleu en
présence de X-gal. Le gène HIS3 code pour l’imidazole glycérol phosphate dehydratase
(IGPD) qui permet la prototrophie à l’histidine même à faible dose. Si et seulement si les
protéines X et Y interagissent, un complexe protéique est formé et reconstitue un facteur de
transcription fonctionnel capable de se lier à l’ADN sur les séquences UAS de GAL4 et
d’activer la transcription. Les colonies à la fois prototrophes pour le tryptophane, la leucine et
l’histidine et produisant la �-galactosidase seront donc des bonnes candidates pour la
production de deux protéines capables d’interagir physiquement. Une telle approche peut être
utilisée pour étudier l'interaction entre deux protéines connues. Elle est surtout très puissante
pour isoler parmi les protéines codées par une banque d'ADNc fusionnée au domaine
transactivateur de GAL4, des partenaires d'une protéine d'intérêt codée par une fusion
transcriptionnelle avec la séquence du domaine de liaison à l'ADN de GAL4.
B) Validation de l’approche expérimentale
Avant de cribler une banque à la recherche de partenaires d’Opa1, nous avons vérifié
que cette protéine pouvait être utilisée dans le système double hybride. Aucun partenaire
d’Opa1 n’étant pour l’heure connu, nous avons émis l’hypothèse que, comme d’autres
dynamines, elle devait être capable d’oligomériser ; cela nous permettant d’envisager
d’étudier en double hybride l’interaction de Opa1 avec elle-même.
Quatres versions de l'ADNc Opa1 codant pour une protéine dépourvue de son
domaine d’adressage mitochondrial (résidus 1 à 125), afin de ne pas interférer avec la
localisation nucléaire imposée par le système du double hybride et dépourvue d’un de ses
domaines coiled coil ou des deux (CC1 et CC2 Figure 29, A) ont été clonées dans le vecteur
pGBT9 et pGAD424 pour donner des protéines de fusion bd-Opa1 ou ad-Opa1,
respectivement. Ces plasmides, ainsi que des plasmides contrôles, ont été introduits dans la
souche YGH1.
L’expression d’Opa1 dans les différentes souches de levure générées a
dans un premier temps été vérifiée. Pour cela, les protéines ont été extraites puis séparées par

90

91
SDS-PAGE et transférées sur membrane de nitrocellulose (Figure 29, B). Une immuno-
détection avec l’anticorps anti-Opa1 a permis de montrer que les protéines de fusion bd-Opa1
tronquées, migrant à un poids moléculaire apparent comparable au poids moléculaire calculé,
sont exprimées à des taux similaires facilement détectables. Par contre, les formes ad-Opa1
sont plus difficilement détectées, à l’exception de la forme d’Opa1 tronquée de son premier
domaine coiled-coil (ad-Opa1 260-960) qui apparait plus fortement exprimée.
Nous avons ensuite déterminé dans les différentes souches de levures la transcription
du gène LACZ par un test colorimétrique sur filtre en présence de X-gal et l’acquisition de la
prototrophie vis à vis de l’histidine par examen de la croissance de cellules sur un milieu sans
histidine. Les domaines de GAL4 produits seuls ou en fusion avec des protéines connues pour
ne pas interagir (lamine et antigène T de SV40) ne conduisent ni à la production de �-
galactosidase ni à celle d’histidine (témoins négatifs). A l’inverse, deux protéines de fusion
capables d’interagir, p53 et AgT, sont capables d’induire la transcription des gènes
rapporteurs LACZ et HIS3 (témoins positifs). Enfin, les protéines Opa1 tronquées exprimées
seules dans la levure sous forme fusionnées à bd ou à ad, n’activent pas la transcription des
gènes rapporteurs (non montré).
La co-expression des protéines de fusion bd-Opa1 et ad-Opa1 conduit à la détection
d’une activité �-galactosidase et d’une prototrophie à l’histidine (Figure 29, C). Des
différences, potentiellement intéressantes, dans la capacité à induire la transcription des gènes
rapporteurs sont observées selon la construction de la protéine Opa1 utilisée. En effet,
l'activité �-galactosidase n'apparaît pas avec la même intensité, et la prototrophie vis-à-vis de
l’histidine dans le milieu de culture n’est pas identique pour toutes les souches. Ainsi, bd-
Opa1 260-960 interagit moins avec ad-Opa1 125-902 qu’avec ad-Opa1 260-960. Ces
différences de niveau de transcription des gènes rapporteurs pourraient refléter des différences
d’expression des formes de la protéine Opa1 dans les levures. En effet, la protéine de fusion
ad-Opa1 125-902 est beaucoup moins produite dans la levure que la protéine ad-Opa1 260-
960. Cependant l’expression différentielle des protéines de fusion ad-OPA1 ne permet pas
d’expliquer l’interaction plus forte de bd-Opa1 260-960 avec ad-Opa1 125-960 qu’avec ad-
Opa1 260-960. Nous proposons donc que les différences d’interaction mais aussi l’absence
d’interaction (par exemple entre bd-opa1 125-960 et ad-Opa1 260-960 alors que bd-opa1 260-
960 et ad-Opa1 125-960 interagissent) puissent être attribuées à des différences d'affinité
entre les différentes formes Opa1 mais également puissent être le reflet de la capacité de

92

93
Opa1, à l’image de ce qui existe pour d’autres dynamines, à engager des domaines coiled-coil
dans des interactions inter-moléculaires et intra-moléculaires. Ainsi par exemple, les résidus
125-260 de Opa1 associés au domaine bd seraient impliqués dans une interaction intra-
moléculaire empêchant les protéines de fusion bd-Opa1 125-960 ou 125-902 d’interagir avec
ad-Opa1.
Nous avons donc établi que la protéine Opa1 tronquée de son domaine d’adressage
mitochondrial et de son premier domaine coiled-coil pouvait être utilisée dans le système du
double hybride. En effet, l’expression de la protéine Opa1 (résidus 260-960), en fusion avec
le domaine de liaison à l'ADN de GAL4, ne conduit pas à l’activation de gènes rapporteurs,
seule ou en présence de protéines témoins avec lesquelles elle ne s’associe pas "in vivo". De
plus, la co-expression de bd-Opa1 (résidus 260-960) et de ad-Opa1 permet de reconstituer un
facteur de transcription fonctionnel, démontrant que Opa1 interagit avec elle-même "in vivo".
C) Criblage de la banque
La protéine bd-Opa1 260-960 a été utilisée dans le système du double hybride pour
rechercher des partenaires de la dynamine parmi les protéines codées par une banque dont les
ADNc ont été fusionnés au domaine transactivateur de GAL4. Afin d’isoler des partenaires de
la dynamine qui pourraient rendre compte, de par leur spécificité d’expression, de la
territorialité de l’atrophie optique dominante de type 1, nous avons utilisé une banque
d'expression (pGADGE-ADNc), préparée à partir de rétine humaine. Cette banque possède
possède 5 105 clones indépendants et l’analyse de 8 clones pris au hasard a permis d’estimer
que 50% des plasmides contiennent un ADNc, d’une taille supérieure à 2kb. Il est à noter que
seulement un tiers des clones codent potentiellement pour une protéine de fusion ad-protéine
de rétine, en raison du respect de la phase ouverte de lecture.
Des cellules YGH1 exprimant la protéine de fusion bd-Opa1 260-960 à partir du
vecteur pGBT9 ont été transformées par la banque pGADGE-ADNc de rétine humaine, puis
étalées sur un milieu sélectionnant à la fois la présence des deux plasmides (milieu dépourvu
de tryptophane et de leucine) mais également la transcription du gène rapporteur HIS3 (milieu
dépourvu d’histidine et complémenté par un inhibiteur chimique de l’IPGD, le 3
AminoTriazole qui permet d’augmenter la sensibilité de la sélection). 1,5 105 clones
indépendants de la banque ont été criblés pour la recherche de protéines interagissant avec

94
Figure 30. Criblage de la banque à la recherche de partenaires d’Opa1.Le plasmide pGADGH-ADNc clone 11 a été re-transformé dans S. cerevisiae en présence de plasmides contrôles pour vérifier la spécificité d'interaction entre ce partenaire isolé et Opa1. La croissance en présence d’histidine indique que les deux plasmides exprimant l’appât (fusionné à bd) ou la proie (fusionnée à ad) sont présents. La croissance en absence d’histine et en présence de 3-amino-1,2,4-triazole (3AT 2,5 à 25 mM) démontrent une interaction entre les protéines correspondantes, la forte concentration sélectionnant les levures qui produisent une grande quantité du produit du gène HIS3. La lamine est utilisée comme contrôle négatif alors que la forte interaction entre p53 et l’AgT de SV40 sert de témoin positif.

95
Opa1. 336 colonies capables de pousser sur un milieu dépourvu de leucine, de tryptophane et
d’histidine et contenant 2,5 mM 3 AT ont été isolées. Parmi ces colonies, 145 ont produit la
�-galactosidase. Ces dernières ont ensuite été cultivées en présence de tryptophane de manière
à perdre le plasmide pGBT9-Opa1 au cours des divisions cellulaires. Les colonies
Tryptophane- et Leucine+ ont été de nouveau testées pour la prototrophie à l’histidine. Les 129
colonies Histidine+ ont été abandonnées puisqu’elles possédaient soit une protéine ne
nécessitant pas la présence d’Opa1 pour conduire à l’activation du gène rapporteur, soit une
protéine de la chaîne de synthèse de l’histidine capable de complémenter le défaut lié à la
délétion du gène HIS3. Les cellules Histidine- provenant de 16 colonies indépendantes ont été
conservées puisqu’elles produisaient une protéine de fusion liée au domaine transactivateur de
GAL4 pouvant interagir spécifiquement avec bd-Opa1 et conduire à la reconstitution d'un
facteur de transcription fonctionnel. Les plasmides pGADGE-ADNc ont alors été extraits de
ces levures puis retransformés dans Saccharomyces cerevisiae avec différents plasmides
contrôles pour vérifier la spécificité de l’interaction avec Opa1. Ces 16 plasmides se sont
comportés de manière comparable lors de ces expériences. Un exemple des résultats obtenus
avec le clone 11 pour la prototrophie vis-à-vis de l’histidine en milieu contenant du 3AT est
présenté en Figure 30. La protéine de fusion exprimée par le vecteur pGADGE ne conduit pas
à une activation des gènes rapporteurs après coexpression avec le domaine de liaison à l’ADN
seul ou en fusion avec la lamine, alors qu'elle permet la transactivation du gène rapporteur en
présence de la protéine de fusion bd-Opa1 260-960. L’interaction bien que spécifique ne
résiste pas à l’ajout de 25 mM de 3AT dans le milieu de culture contrairement à la très forte
interaction entre p53 et AgT.
Dans un second temps, nous avons établi la carte physique des 16 plasmides codant
pour une protéine interagissant spécifiquement avec Opa1. Ils correspondent à 9 ADNc dont 4
d’entre eux sont retrouvés 2 ou 3 fois. L’analyse de la séquence des 16 plasmides a ainsi
confirmé l’isolement de 9 nouveaux partenaires d’Opa1.
En utilisant une approche de double hybride, nous avons pu isoler 9 nouvelles
protéines interagissant avec Opa1. Ces partenaires sont de nature assez variée puisqu’il
s’agit entre autre de facteurs de transcription, de protéines impliquées dans le trafic
membranaire ou de pro-apoptotiques. Il est à noter qu’aucun de ces partenaires ne présente
de spécificité d’expression cellulaire ou tissulaire connue qui aurait pu nous permettre
d’établir un lien direct avec l’atrophie optique dominante de type 1. Toutefois, les gènes

96

97
codant pour ces protéines pourraient constituer des gènes modificateurs de la pathologie ou
des gènes candidats pour des maladies neurodégénératives ou des mitochondriopathies.

98
domaine activateur de transcription de GAL4 EcoRI
1 GATGATGAAGTTACCCCACCAAACCCAAAAAAAGAGATCCTAGAACTAGGCGAATTCGCG 60 D D E V T P P N P K K E I L E L G E F A
NotI SalI SmaI PstI61 GCCGCGTCGACCGCCATGTCGCAGAACGGAGCGCCCGGGATGCAGGAGGAGAGCCTGCAG 120 A A S T A M S Q N G A P G M Q E E S L Q
121 GGCTCCTGGGTAGAACTGCACTTCAGCAATAATGGGAACGGGGGCAGCGTTCCAGCCTCG 180 G S W V E L H F S N N G N G G S V P A S
181 GTTTCTATTTATAATGGAGACATGGAAAAAATACTGCTGGACGCACAGCATGAGTCTGGA 240 V S I Y N G D M E K I L L D A Q H E S G
SacI241 CGGAGTAGCTCCAAGAGCTCTCACTGTGACAGCCCACCTCGCTCGCAGACACCACAAGAT 300 R S S S K S S H C D S P P R S Q T P Q D
301 ACCAACAGAGCTTCTGAAACAGATACCCATAGCATTGGAGAGAAAAACAGCTCACAGTCT 360 T N R A S E T D T H S I G E K N S S Q S
361 GAGGAAGATGATATTGAAAGAAGGAAAGAAGTTGAAAGCATCTTGAAGAAAAACTCAGAT 420 E E D D I E R R K E V E S I L K K N S D
421 TGGATATGGGATTGGTCAAGTCGGCCGGAAAATATTCCCCCCAAGGAGTTCCTCTTTAAA 480 W I W D W S S R P E N I P P K E F L F K
BbvCI481 CACCCGAAGCGCACGGCCACCCTCAGCATGAGGAACACGAGCGTCATGAAGAAAGGGGGC 540 H P K R T A T L S M R N T S V M K K G G
PstI541 ATATTCTCTGCAGAATTTCTGAAAGTTTTCCTTCCATCTCTGCTGCTCTCTCATTTGCTG 600 I F S A E F L K V F L P S L L L S H L L
601 GCCATCGGATTGGGGATCTATATTGGAAGGCGTCTGACAACCTCCACCAGCACCTTTTGA 660 A I G L G I Y I G R R L T T S T S T F *
661 TGAAGAACTGGAGTCTGACTTGGTTCGTTAGTGGATTACTTCTGAGCTTGCAACATAGCT 720
Figure 31. Séquence du clone 11 isolé en double hybride.La séquence du clone a été obtenue à l'aide de l’oligonucléotide AD GAL4 5' (TACCACTACAATGGATG) qui hybride dans le domaine transactivateur de GAL4 36 pb en amont. Les sites de restriction sont mentionnés. La phase de lecture de la protéine codée par cet ADNc est donnée par la fusion avec le domaine activateur de transcription de GAL4. Cette séquence nucléique code pour la totalité de la protéine Bnip3 (en encadré).

99
II) CARACTÉRISATION DE L’INTERACTION ENTRE LES
PROTÉINES OPA1 ET BNIP3
Mon projet de thèse s’est focalisé sur l’un des 9 partenaires d’Opa1, Bnip3 (Figure
31), un membre pro-apoptotique à domaine BH3 de la famille Bcl-2. Le choix de ce
partenaire a été motivé par la mise en évidence, au début de ce travail, par l’équipe, de
l’implication d’Opa1 dans la voie mitochondriale de l’apoptose (Olichon et al., 2003) et de
l’émergence des liens entre la dynamique mitochondriale et l’apoptose (Perfettini et al.,
2005). Avant de passer à la description des résultats, je présenterai brièvement l’état des
connaissances concernant Bnip3 en 2005 lorsque ce travail a débuté.
La protéine Bnip3 a été identifiée comme interagissant avec la protéine virale E1B 19
kDa et l’anti-apoptotique Bcl-2 (Boyd et al., 1994; Yasuda et al., 1998; Zhang et al., 2003a).
Elle est exprimée de manière ubiquitaire mais à un niveau endogène faible (Zhang et al.,
2003a). Cependant, l’expression de Bnip3 est induite transcriptionnellement de façon HIF1
dépendante lors de la mise en hypoxie de lignées épithéliales tumorales (cellules HeLa,
MCF7) (Guo et al., 2001; Sowter et al., 2001) ou de cellules normales en culture
(cardiomyocytes) (Kubasiak et al., 2002). Cette induction permet l’accumulation de la
protéine Bnip3 et conduit à la mort des cellules. Dans des expériences similaires réalisées en
présence d’ARNm antisens de Bnip3, les cellules restent viables, démontrant que Bnip3 est
responsable de la mort des cellules en conditions hypoxiques (Kothari et al., 2003; Kubasiak
et al., 2002).
Bnip3, exprimée à un taux endogène, et en condition normale de culture, est associée à
la membrane externe des mitochondries (Vande Velde et al., 2000). Son accumulation dans la
cellule, suite à sa surexpression ectopique ou après un traitement hypoxique, conduit à son
ancrage dans la membrane externe via son domaine transmembranaire C-terminal localisant
les 10 derniers résidus de Bnip3 dans l’espace inter-membranaire (Figure 32) (Regula et al.,
2002; Vande Velde et al., 2000). L’accumulation de ce « BH3-only » semble donc suffisante
pour conduire à son activation et à l’induction de la mort cellulaire.
Les mécanismes de mort cellulaire induite par Bnip3 sont complexes. En effet,
certains auteurs proposent que Bnip3 puisse titrer les protéines anti-apoptotiques telles que

100

101
Bcl-2 au niveau de la membrane externe des mitochondries pour conduire à la mort des
cellules par apoptose (Chen et al., 1997; Lamy et al., 2003; Ray et al., 2000; Regula et al.,
2002; Wan et al., 2003). Cependant, dans d’autres travaux, la mort cellulaire induite par
Bnip3 ne semble pas s’accompagner d’évènements caractéristiques d’une apoptose (libération
du cytochrome c, implication des caspases) mais plutôt d’un dysfonctionnement des
mitochondries (perte du potentiel de membrane, ouverture du pore de perméabilité
mitochondriale) conduisant à une mort plus proche de la nécrose pouvant être associée à des
processus d’autophagie (Daido et al., 2004; Kanzawa et al., 2005; Vande Velde et al., 2000).
Bnip3 pourrait ainsi être capable d’induire une mort cellulaire qui, suivant le type cellulaire et
le microenvironnement cellulaire, pourrait être soit de l’apoptose, soit de la nécrose associée à
des évènements d’autophagie, soit un type de mort comprenant des caractéristiques des deux
parfois appelé aponécrose (Bacon and Harris, 2004).
Enfin, il est intéressant de noter que la présence du messager et de la protéine Bnip3 a
été rapportée dans des tumeurs solides d’origine épithéliale, parfois associée aux régions
périnécrotiques, permettant d’envisager d’utiliser cette protéine comme un marqueur tumoral
d’hypoxie (Giatromanolaki et al., 2004; Sowter et al., 2003). En raison de son rôle dans
l’induction de la mort cellulaire, il est également envisagé que des dérégulations de son
expression puissent être associées à la progression tumorale ou à l’acquisition de résistance
aux traitements thérapeutiques. En accord avec ce dernier point, il a été montré qu’une
diminution de l’expression de Bnip3 était associée à une résistance aux sels biliaires dans le
cas de carcinomes colorectaux ou à la Gemcitabine dans le cas d’adénomes pancréatiques
(Akada et al., 2005; Crowley-Weber et al., 2002). Cette diminution d’expression pourrait être
en partie due à l’hyperméthylation de son promoteur (Murai et al., 2005; Okami et al., 2004).
A) Validation de l’interaction entre Opa1 et Bnip3
Pour s’assurer de la validité de l’association, mise en évidence en double hybride,
entre Opa1 et Bnip3, il est nécessaire de la vérifier par d’autres approches expérimentales
telles que la production et la purification de protéines suivie de test d’interaction in vitro, ou
des expériences de co-immuno-précipitation.
1) in vitro
Pour valider l’interaction entre Bnip3 et Opa1, nous avons dans un premier temps
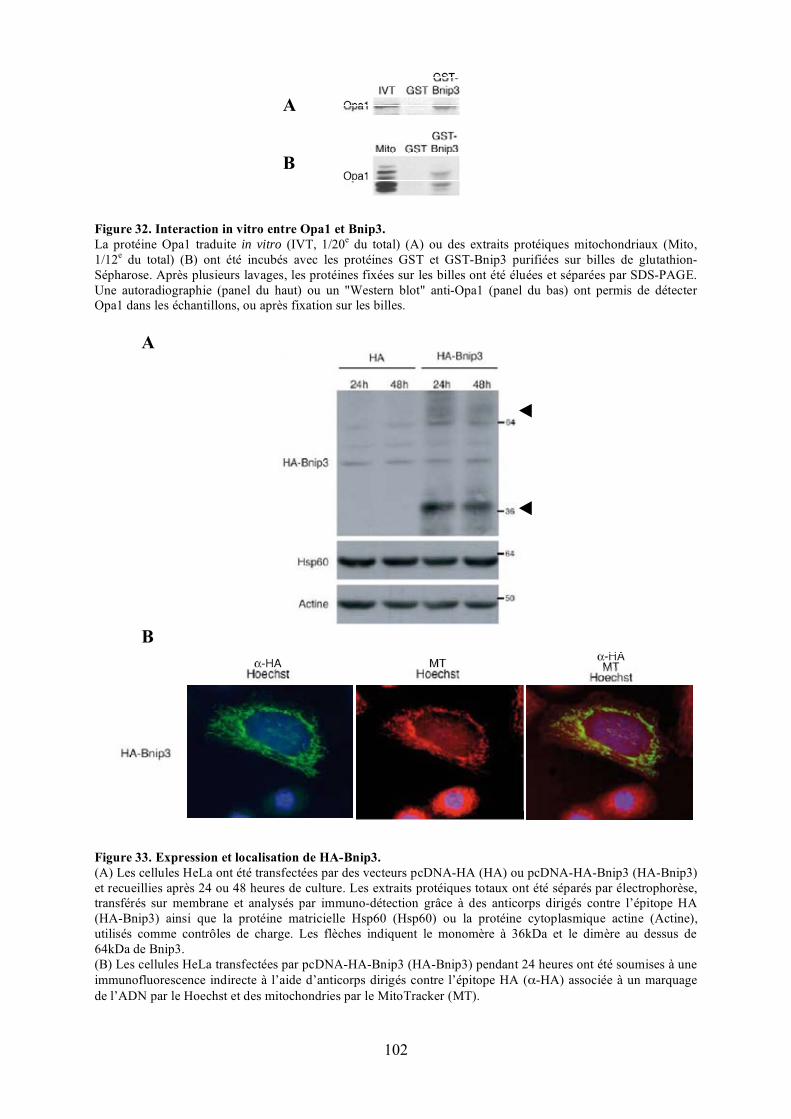
102
A
B
Figure 32. Interaction in vitro entre Opa1 et Bnip3.La protéine Opa1 traduite in vitro (IVT, 1/20e du total) (A) ou des extraits protéiques mitochondriaux (Mito, 1/12e du total) (B) ont été incubés avec les protéines GST et GST-Bnip3 purifiées sur billes de glutathion-Sépharose. Après plusieurs lavages, les protéines fixées sur les billes ont été éluées et séparées par SDS-PAGE. Une autoradiographie (panel du haut) ou un "Western blot" anti-Opa1 (panel du bas) ont permis de détecter Opa1 dans les échantillons, ou après fixation sur les billes.
A
B
Figure 33. Expression et localisation de HA-Bnip3.(A) Les cellules HeLa ont été transfectées par des vecteurs pcDNA-HA (HA) ou pcDNA-HA-Bnip3 (HA-Bnip3) et recueillies après 24 ou 48 heures de culture. Les extraits protéiques totaux ont été séparés par électrophorèse, transférés sur membrane et analysés par immuno-détection grâce à des anticorps dirigés contre l’épitope HA (HA-Bnip3) ainsi que la protéine matricielle Hsp60 (Hsp60) ou la protéine cytoplasmique actine (Actine), utilisés comme contrôles de charge. Les flèches indiquent le monomère à 36kDa et le dimère au dessus de 64kDa de Bnip3.(B) Les cellules HeLa transfectées par pcDNA-HA-Bnip3 (HA-Bnip3) pendant 24 heures ont été soumises à une immunofluorescence indirecte à l’aide d’anticorps dirigés contre l’épitope HA (�-HA) associée à un marquage de l’ADN par le Hoechst et des mitochondries par le MitoTracker (MT).

103
traduit in vitro le variant 1 d’Opa1 (Figure 19) en présence de méthionine 35S dans un lysat de
réticulocytes puis nous l’avons incubée en présence de protéines GST-Bnip3 ou GST seule
produites au préalable chez Escherichia coli et purifiées sur des billes de glutathion-
Sépharose. Le matériel retenu sur les billes a ensuite été analysé par SDS-PAGE et
autoradiographie (Figure 32, A). Opa1 traduite in vitro migre dans ces conditions sous la
forme d’une bande de poids moléculaire 98kDa qui correspond au précurseur de la dynamine,
ainsi que plusieurs petites bandes (non montrées ici) provenant de l’arrêt prématuré ou d’une
initiation interne de la traduction. Opa1 s’associe spécifiquement à la protéine GST-Bnip3 et
non à la GST seule.
Nous avons, de plus, incubé les protéines GST-Bnip3 et GST immobilisées sur des
billes de glutathion-Sépharose avec un extrait protéique mitochondrial de cellules HeLa
(Figure 32, B). Après élution et migration sur SDS-PAGE du matériel retenu sur la colonne,
une immuno-détection réalisée avec des anticorps dirigés contre Opa1 montre que la
dynamine interagit spécifiquement avec GST-Bnip3, mais pas avec la GST seule. Il est à noter
que les cinq isoformes d’Opa1 s’associent à Bnip3 dans des proportions similaires aux
proportions observées dans les extraits cellulaires. Il faut toutefois signaler que la capacité
d’Opa1 à oligomériser pourrait masquer l’existence d’une interaction de Bnip3 avec une
isoforme spécifique d’Opa1.
2) in cellulo, en surexpression ectopique des deux partenaires
Bnip3 est une protéine présente en très faible quantité dans les cellules et n’est, de ce
fait, pas ou peu détectable. Par conséquent, pour caractériser l’interaction entre Bnip3 et
Opa1, nous avons travaillé, dans un premier temps, en conditions de surexpression ectopique
des deux partenaires. Pour cela, un vecteur permettant d’exprimer de manière constitutive
(promoteur fort CMV) une forme étiquetée HA de Bnip3 (pcDNA-HA-Bnip3) a été construit.
L’expression de la protéine a été vérifiée par des expériences d’immuno-détection menées sur
des extraits protéiques totaux de cellules HeLa transfectées par le vecteur pcDNA-HA-Bnip3.
La protéine HA-Bnip3 est fortement détectée dès 24 heures après transfection et est, en
revanche, moins présente après 48 heures (Figure 33, A). La localisation cellulaire de la
protéine de fusion a également été examinée par des expériences d’immuno-fluorescence
indirecte avec les anticorps anti-HA associées à un marquage des mitochondries au
MitoTracker (Figure 33, B). Le MitoTracker diffuse à travers la membrane plasmique,
s’accumule dans les mitochondries suivant le potentiel de membrane et fluoresce lorsqu’il est

104
A
B
Figure 34. Validation in cellulo de l’interaction entre Opa1 et Bnip3 après surexpression dans les cellules HeLa.Les cellules HeLa ont été co-transfectées par des vecteurs pCCEY-Opa1 et pcDNA3-HA-Bnip3 (HA-Bnip3). 24 heures après la transfection, les mitochondries ont été isolées, et un pontage au DSP a été réalisé. Les lysats mitochondriaux ont ensuite été immuno-précipités grâce à des anticorps dirigés contre Opa1 (IP Opa1, A, piste 2) ou contre l’épitope HA (IP HA, B, piste 2). Des contrôles ont été effectués en présence de protéine A-Sépharose mais en absence d’anticorps (-, pistes 3). Des extraits mitochondriaux (1/20e des lysats) ont également été déposés (Mito, pistes 1). Les protéines ont été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre l’épitope HA (HA-Bnip3), la protéine Opa1 (Opa1), et la protéine matricielle Hsp60 (Hsp60).

105
oxydé dans des mitochondries fonctionnelles. HA-Bnip3 co-localise avec le marquage
MitoTracker et présente donc une localisation mitochondriale attendue. Pour sa part, Opa1
(variant 1) est exprimé à partir du plasmide pCCEY. Ce vecteur, utilisé et caractérisé lors de
travaux précédents au laboratoire, conduit à une augmentation de la quantité des isoformes b
et d de la protéine qui se localisent à la mitochondrie (Figure 20, B) (Olichon et al., 2007b).
Des cellules HeLa ont été transfectées par les vecteurs pcDNA-HA-Bnip3 et pCCEY-Opa1.
Une double immunofluorescence réalisée à l’aide d’anticorps anti-Opa1 et anti-HA nous a
permis de définir un pourcentage de co-transfection de l’ordre de 25%. Les mitochondries de
ces cellules ont été isolées 24 heures après transfection par centrifugation différentielle et un
pontage des protéines au DiSuccinimidylPropionate (DSP) a été réalisé. Le pontage fige des
complexes et consolide des interactions faibles et labiles qui pourraient être rompues dans les
conditions détergentes de l’extraction de ces protéines membranaires. Il permet aussi de
s’affranchir d’interactions non spécifiques qui pourraient survenir entre des protéines
artificiellement mises en présence lors du fractionnement cellulaire et de la rupture des
différents compartiments. Après cette étape, les protéines mitochondriales ont été extraites en
présence de détergents (DOC-NP40-Triton) et ont été soumises à des immuno-précipitations
réalisées avec des anticorps dirigés contre Opa1 ou contre l’épitope HA. Les protéines
précipitées ont été séparées par SDS-PAGE après rupture des pontages, transférées sur
membrane de nitrocellulose puis soumises à une immuno-détection avec des anticorps anti-
HA et anti-Opa1 (Figure 34). Dans l’extrait mitochondrial, nous détectons la présence de HA-
Bnip3 et des cinq isoformes d’Opa1. Lorsque les protéines mitochondriales sont incubées en
présence d’anticorps anti-Opa1 (Figure 34, A), nous pouvons détecter la présence de la
dynamine dans le produit d’immuno-précipitation, alors qu’elle n’est pas observée en absence
d’anticorps. La protéine Bnip3 est également détectée dans l’immuno-précipitât, uniquement
en présence d’anticorps anti-Opa1. La protéine mitochondriale Hsp60 est en revanche absente
des immuno-précipitations réalisées en présence ou en absence d’anticorps anti-Opa1.
L’ensemble de ces résultats indique qu’Opa1 est spécifiquement immuno-précipité et que
Bnip3 est spécifiquement co-immuno-précipité. L’expérience inverse a également été réalisée
(Figure 34, B). La forme étiquetée de Bnip3 est spécifiquement immuno-précipitée par des
anticorps anti-HA et les 5 isoformes d’Opa1 spécifiquement co-immuno-précipitées.
3) in cellulo, à un taux physiologique des deux partenaires
L’expression du gène codant pour Bnip3 étant fortement induite en conditions
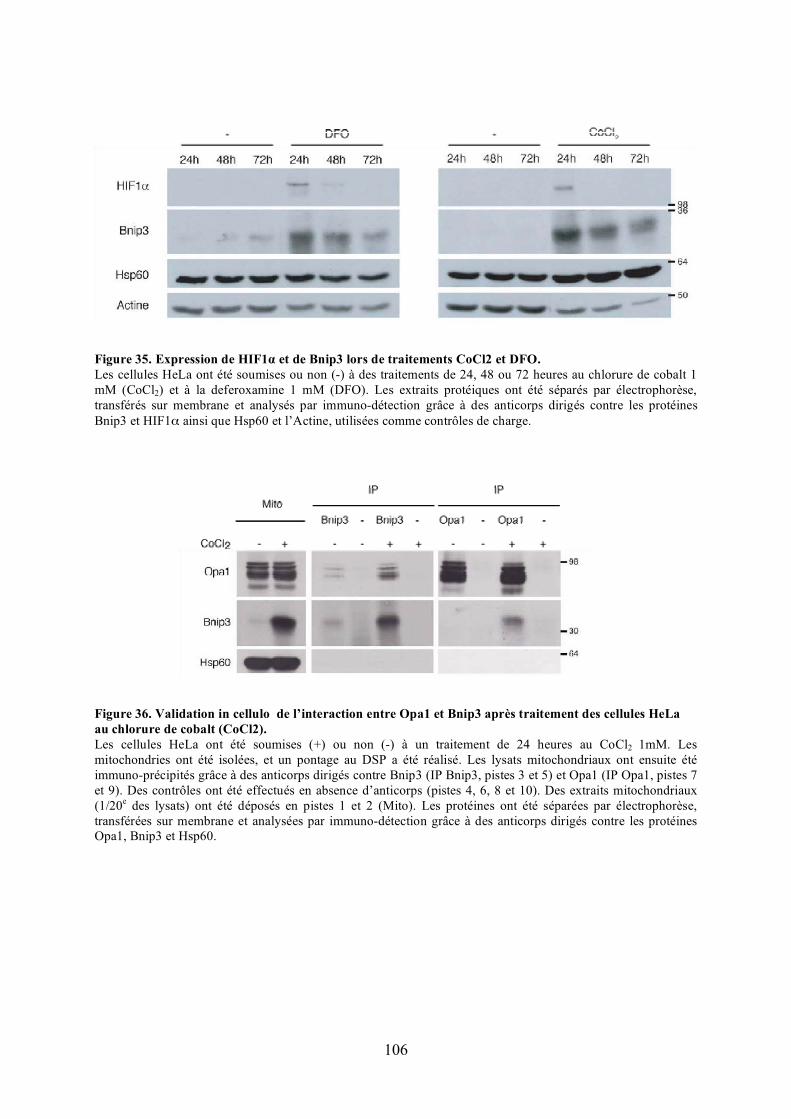
106
Figure 35. Expression de HIF1� et de Bnip3 lors de traitements CoCl2 et DFO.Les cellules HeLa ont été soumises ou non (-) à des traitements de 24, 48 ou 72 heures au chlorure de cobalt 1 mM (CoCl2) et à la deferoxamine 1 mM (DFO). Les extraits protéiques ont été séparés par électrophorèse, transférés sur membrane et analysés par immuno-détection grâce à des anticorps dirigés contre les protéines Bnip3 et HIF1� ainsi que Hsp60 et l’Actine, utilisées comme contrôles de charge.
Figure 36. Validation in cellulo de l’interaction entre Opa1 et Bnip3 après traitement des cellules HeLa au chlorure de cobalt (CoCl2).Les cellules HeLa ont été soumises (+) ou non (-) à un traitement de 24 heures au CoCl2 1mM. Les mitochondries ont été isolées, et un pontage au DSP a été réalisé. Les lysats mitochondriaux ont ensuite été immuno-précipités grâce à des anticorps dirigés contre Bnip3 (IP Bnip3, pistes 3 et 5) et Opa1 (IP Opa1, pistes 7 et 9). Des contrôles ont été effectués en absence d’anticorps (pistes 4, 6, 8 et 10). Des extraits mitochondriaux (1/20e des lysats) ont été déposés en pistes 1 et 2 (Mito). Les protéines ont été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1, Bnip3 et Hsp60.

107
d’hypoxie (Guo et al., 2001; Kubasiak et al., 2002; Sowter et al., 2001), nous avons alors
cherché à démontrer l’interaction entre Opa1 et Bnip3 dans ces conditions. Ne disposant pas
d’enceinte à hypoxie à ce moment-là, nous avons utilisé le chlorure de cobalt (CoCl2) et la
deferoxamine (DFO), déjà utilisés dans la littérature pour induire Bnip3 dans diverses lignées
cellulaires.
Dans un premier temps, nous avons testé la capacité de ces drogues à induire
l’expression de Bnip3 à différentes concentrations et à différents temps. Pour cela, nous avons
analysé par immuno-détection à l’aide d’anticorps dirigés contre Bnip3 et contre la protéine
HIF1� (facteur de transcription responsable de l’induction de l’expression de Bnip3 dans ces
conditions) des extraits protéiques totaux de cellules HeLa traitées durant 24, 48 ou 72 heures
avec 0.1mM, 0.5mM et 1mM de CoCl2 et de DFO. Les conditions les plus reproductibles
correspondant à des traitements effectués à 1mM, je ne présenterai que les résultats relatifs à
cette concentration (Figure 35). Comme attendu, la protéine HIF1�, absente des extraits
contrôles, est induite lors des deux traitements dès 24 heures et de manière transitoire
puisqu’elle n’est plus détectée à 48 ou 72 heures. En absence de tout signal mimant l’hypoxie,
une très faible quantité de Bnip3 est visualisée. Elle pourrait provenir d’un niveau faible
d’expression du pro-apoptotique dans la totalité des cellules ou correspondre à un fort niveau
d’expression dans quelques cellules en train de mourir. Après 24 heures de traitement, Bnip3
est très fortement accumulée, à un poids moléculaire apparent d’environ 36 kDa, qui
correspond au poids moléculaire attendu. Le niveau d’expression de Bnip3 reste similaire
après 48 heures de traitement et diminue au point 72 heures.
L’induction de Bnip3 étant de façon reproductible plus forte et moins transitoire en
CoCl2 qu’en DFO, nous avons dans la suite du travail utilisé le CoCl2 à une concentration de
1mM durant 24 heures. Les mitochondries ont été isolées de cellules HeLa ainsi traitées puis
des immuno-précipitations après pontage des protéines au DSP ont été réalisées (Figure 36).
Dans les extraits mitochondriaux, nous détectons une induction importante de la protéine
Bnip3 en réponse à l’exposition des cellules au CoCl2. Lors de l’immuno-précipitation
d’Opa1 et de Bnip3, nous pouvons observer respectivement Bnip3 et Opa1 co-immuno-
précipiter spécifiquement. En outre, l’analyse de l’immuno-précipitation de Bnip3 à partir de
mitochondries isolées de cellules non hypoxiques indique que la faible quantité de Bnip3
présente de manière endogène en absence de signal mimant l’hypoxie dans les cellules HeLa
est déjà associée à Opa1. En revanche, Bnip3 n’est pas détectée dans ces conditions lors de
l’immuno-précipitation d’Opa1, compte tenu du large excès de la dynamine par rapport à

108
B
Figure 37. Représentation schématique et expression des mutants de Bnip3.(A) Bnip3 contient une séquence PEST (PEST), servant éventuellement pour sa dégradation, le domaine 3 d’homologie à Bcl-2 (BH3) et un domaine trans-membranaire (TM) permettant l’ancrage à la mitochondrie. Bnip3�C est dépourvu des 10 derniers acides aminés carboxy-terminaux. Dans le mutant Bnip3-TMBcl-2 le domaine TM est remplacé par celui de Bcl-2 (TMBcl-2).(B) Les cellules HeLa ont été transfectées par des vecteurs pcDNA-HA (-), pcDNA-HA-Bnip3 (HA-Bnip3), pcDNA-HA-Bnip3�C (HA-Bnip3�C) ou pcDNA-HA-Bnip3TMBcl-2 (HA-Bnip3TMBcl-2) et recueillies après 24 heures de culture. Les extraits protéiques totaux ont été séparés par électrophorèse, transférés sur membrane et analysés par immuno-détection grâce à des anticorps dirigés contre l’épitope HA (HA-Bnip3), et les protéines matricielle Hsp60 (Hsp60) ou cytoplasmique actine (Actine) utilisées comme contrôles de charge.
A

109
Bnip3 dans les cellules.
Nous avons confirmé in vitro et in cellulo l’existence d’une association entre Opa1 et
Bnip3. Les expériences de co-immuno-précipitation laissent supposer que la surexpression
ectopique de Bnip3 pourra être utilisée pour la caractérisation fonctionnelle de l’interaction.
Elle permet en effet de reconstituer l’interaction visualisée à partir des protéines endogènes
dans des cellules traitées au chlorure de cobalt. Nous cherchons actuellement à déterminer si
l’association entre Opa1 et Bnip3 est également induite en hypoxie « vraie ».
B) Caractérisation des domaines requis pour l’interaction
1) Bnip3
Bnip3 et Opa1 sont deux protéines associées aux membranes. Pour cette raison, nous
avons jugé plus opportun de déterminer les domaines impliqués dans l’interaction in cellulo
par co-immuno-précipitation.
L’accumulation de Bnip3 dans la cellule conduit à son ancrage dans la membrane
externe via son domaine trans-membranaire C-terminal localisant les 10 derniers résidus de la
protéine dans l’espace inter-membranaire (Vande Velde et al., 2000). Opa1 étant également
situé dans l’espace inter-membranaire, nous avons émis l’hypothèse que l’interaction pourrait
avoir lieu grâce à ces acides aminés. Néanmoins, comme pour Mgm1 chez Saccharomyces
cerevisiae, l’existence d’isoformes d’Opa1 associées à la membrane externe a été proposée
(Satoh et al., 2003). De ce fait, il est également envisageable que la dynamine puisse interagir
au niveau de la membrane externe avec le domaine trans-membranaire de Bnip3, comme
décrit pour trois partenaires de Bnip3, dont Bnip3 lui-même (Aoyagi et al., 2003; Lamy et al.,
2003; Sulistijo et al., 2003). Pour tester ces hypothèses, nous avons construit des vecteurs
permettant d’exprimer une forme étiquetée HA d’un mutant de délétion des 10 derniers acides
aminés carboxy-terminaux de Bnip3 (Bnip3�C), ainsi que d’un mutant dans lequel le
domaine trans-membranaire de Bnip3 est remplacé par celui de Bcl-2 (Bnip3-TMBcl-2) (Figure
37, A). Ces deux mutants ont déjà été décrits dans la littérature pour présenter une localisation
mitochondriale. Il est cependant nécessaire de préciser que Bnip3-TMBcl-2 est également
présent au niveau du reticulum endoplasmique (Kim et al., 2002; Ray et al., 2000).
L’expression de Bnip3�C et Bnip3-TMBcl-2 a été vérifiée par des expériences d’immuno-
détection menées sur des extraits protéiques totaux de cellules HeLa transfectées par les

110
Figure 38. Caractérisation des domaines de Bnip3 requis pour l’interaction.Les cellules HeLa ont été co-transfectées par des vecteurs pCCEY-Opa1 et pcDNA-HA-Bnip3 (HA-Bnip3), ou pCCEY-Opa1 et pcDNA-HA-Bnip3�C (HA-Bnip3�C), ou pCCEY-Opa1 et pcDNA-HA-Bnip3-TMBcl2 (HA-Bnip3-TMBcl2). 24 heures après la transfection, les mitochondries ont été isolées, et un pontage au DSP a été réalisé. Les lysats mitochondriaux ont ensuite été immuno-précipités grâce à des anticorps dirigés contre Opa1 (IP Opa1, panels du haut, pistes 2, 5 et 8) ou contre l’épitope HA (IP HA, panels du bas, pistes 2, 5 et 8). Des contrôles ont été effectués en présence de protéine A-Sépharose mais en absence d’anticorps et déposés en pistes 3, 6 et 9 (-). Des extraits mitochondriaux (1/20e des lysats) ont été déposés en pistes 1, 4 et 7 (Mito). Les protéines ont été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre l’épitope HA (HA-Bnip3), la protéine Opa1 (Opa1), et la protéine matricielle Hsp60 (Hsp60).

111
vecteurs pcDNA-HA-Bnip3�C et pcDNA-HA-Bnip3TMBcl-2 (Figure 37, B). HA-Bnip3�C et
HA-Bnip3TMBcl-2 sont détectées dans des quantités similaires à HA-Bnip3. Il est toutefois à
noter que, dans cette figure, seule cette dernière dimérise. Néanmoins, dans d’autres
expériences, comme lors des expériences de co-immuno-précipitation (voir ci-dessous), les 3
formes de la protéine dimérisent très faiblement dans les mêmes proportions. Ces données
contradictoires viennent s’ajouter à de nombreuses autres présentes dans la littérature,
conduisant certains observateurs à considérer la dimérisation de la protéine comme un
artéfact. Des cellules HeLa ont été co-transfectées avec les plasmides pCCEY-Opa1 et
pcDNA-HA-Bnip3, pcDNA-HA-Bnip3�C ou pcDNA-HA-Bnip3-TMBcl-2. Les mitochondries
ont été isolées et un pontage des protéines a été réalisé. Après solubilisation, les protéines
mitochondriales ont été soumises à des immuno-précipitations réalisées avec des anticorps
dirigés contre Opa1 ou contre l’épitope HA (Figure 38). La présence des formes mutées de
Bnip3 révélée par l’immuno-détection avec des anticorps anti-HA dans des extraits de
protéines mitochondriales confirme leur localisation mitochondriale. L’immuno-précipitation
d’Opa1 suivie d’un « Western blot » anti-HA ainsi que l’immuno-précipitation des formes de
Bnip3 suivie d’un « Western blot » anti-Opa1 révèlent, après analyse densitométrique, une
diminution de 35% de l’interaction d’Opa1 avec la forme tronquée de Bnip3 et une abolition
totale avec le mutant comprenant le domaine trans-membranaire de Bcl-2 (n=3).
2) Opa1
Nous avons également testé la capacité à interagir avec Bnip3 de deux formes
mutantes d’Opa1. Il s’agit d’allèles pathogènes de la protéine déjà utilisés et caractérisés au
laboratoire (cf Résultats, Deuxième partie), l’un comprenant une délétion des 58 acides
aminés carboxy-terminaux de la dynamine (Opa1�58) et l’autre une mutation ponctuelle au
sein du domaine GTPase (Opa1G300E). Dans le but de s’affranchir de la présence de la
protéine sauvage endogène, des vecteurs permettant d’exprimer de manière constitutive des
formes étiquetées GFP de ces deux mutants, pEGFP-C1-Opa1�58 et pEGFP-C1-Opa1G300E,
ont été construits. L’expression de ces deux constructions s’est avérée toxique pour les
cellules et les expériences de co-immuno-précipitation ont nécessité d’importantes quantités
de matériel. Plusieurs dizaines de millions de cellules HeLa ont ainsi été transfectées par les
vecteurs pcDNA-HA-Bnip3 et pEGFP-C1-Opa1, pEGFP-C1-Opa1�58 ou pEGFP-C1-
Opa1G300E. Après isolement des mitochondries, les protéines mitochondriales,
préalablement pontées au DSP et extraites en présence de détergents, ont été soumises à des
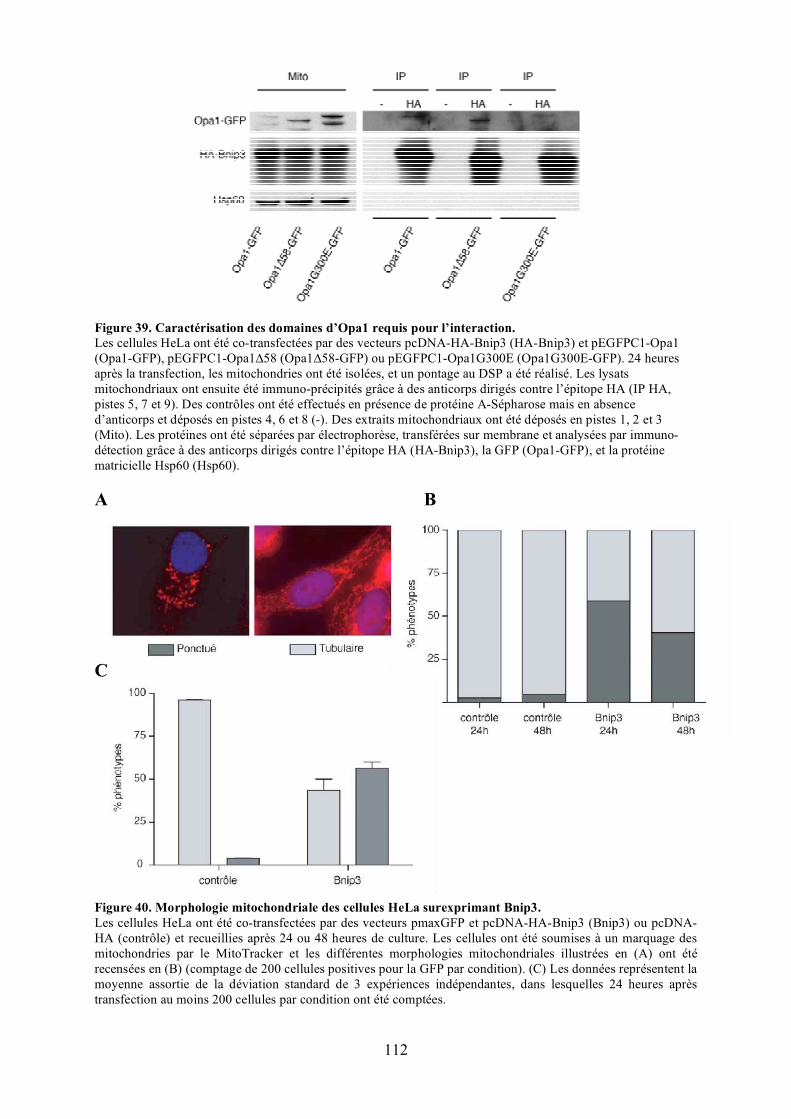
112
Figure 39. Caractérisation des domaines d’Opa1 requis pour l’interaction.Les cellules HeLa ont été co-transfectées par des vecteurs pcDNA-HA-Bnip3 (HA-Bnip3) et pEGFPC1-Opa1 (Opa1-GFP), pEGFPC1-Opa1�58 (Opa1�58-GFP) ou pEGFPC1-Opa1G300E (Opa1G300E-GFP). 24 heures après la transfection, les mitochondries ont été isolées, et un pontage au DSP a été réalisé. Les lysats mitochondriaux ont ensuite été immuno-précipités grâce à des anticorps dirigés contre l’épitope HA (IP HA, pistes 5, 7 et 9). Des contrôles ont été effectués en présence de protéine A-Sépharose mais en absence d’anticorps et déposés en pistes 4, 6 et 8 (-). Des extraits mitochondriaux ont été déposés en pistes 1, 2 et 3 (Mito). Les protéines ont été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre l’épitope HA (HA-Bnip3), la GFP (Opa1-GFP), et la protéine matricielle Hsp60 (Hsp60).
A B
C
Figure 40. Morphologie mitochondriale des cellules HeLa surexprimant Bnip3.Les cellules HeLa ont été co-transfectées par des vecteurs pmaxGFP et pcDNA-HA-Bnip3 (Bnip3) ou pcDNA-HA (contrôle) et recueillies après 24 ou 48 heures de culture. Les cellules ont été soumises à un marquage des mitochondries par le MitoTracker et les différentes morphologies mitochondriales illustrées en (A) ont été recensées en (B) (comptage de 200 cellules positives pour la GFP par condition). (C) Les données représentent la moyenne assortie de la déviation standard de 3 expériences indépendantes, dans lesquelles 24 heures après transfection au moins 200 cellules par condition ont été comptées.

113
immuno-précipitations réalisées avec des anticorps dirigés contre l’épitope HA. Une immuno-
détection anti-GFP révèle qu’Opa1�58 est présent dans des proportions similaires à Opa1
dans les immuno-précipitâts. En revanche, Opa1-G300E, pourtant plus fortement détectée que
la forme sauvage de la protéine dans les extraits mitochondriaux, n’interagit pas, ou très peu,
avec Bnip3 (Figure 39). Devant la difficulté à obtenir ces résultats, nous avons décidé
d’insérer les séquences codantes des différentes formes d’Opa1 étiquetées HA dans le vecteur
pCCEY, constructions qui semblent moins toxiques pour les cellules. Nous utiliserons
prochainement ces plasmides pour répéter nos expériences de co-immuno-précipitation et
ainsi approfondir cette partie de l’étude.
Nous avons établi que le domaine trans-membranaire de Bnip3 est essentiel pour son
association avec Opa1. De plus, son extrémité carboxy-terminale semble stabiliser
l’interaction. Concernant Opa1, même si cela reste à confirmer, il semblerait que le domaine
coiled-coil de la protéine n’intervienne pas dans l’interaction. Il sera intéressant d’examiner
également l’importance du domaine « middle » de la dynamine dans l’association
Opa1/Bnip3. En effet, des travaux récents sur Mgm1, l’homologue de la protéine chez
Saccharomyces cerevisiae, ont mis en évidence dans ce domaine une région conservée de
fixation aux lipides (Rujiviphat et al., 2009). Cette région pourrait favoriser l’association de
la protéine au niveau de la membrane externe et ainsi permettre l’interaction entre les deux
protéines. Notons cependant qu’une mutation ponctuelle au sein du domaine GTPase de la
dynamine semble affecter sa capacité à se fixer au pro-apoptotique. Bien que l’on ne
connaisse pas l’impact réel de cette mutation sur la faculté d’Opa1 à fixer et hydrolyser le
GTP, ces résultats suggèrent que le statut GTP/GDP de la dynamine pourrait tenir un rôle
important dans son association avec Bnip3.
C) Caractérisation fonctionnelle de l’interaction entre Opa1 et Bnip3
1) Étude de l’effet de Bnip3 sur la dynamique mitochondriale
Opa1 étant un acteur de la fusion des membranes mitochondriales, nous avons
caractérisé l’impact de Bnip3 sur la morphologie des mitochondries. Compte tenu du fait que
Bnip3 n’est pas ou très peu exprimée en conditions normales dans les cellules, une approche
d’extinction de son expression par interférence à l’ARN n’était pas judicieuse. D’autre part
comme l’action de Bnip3 nécessite une augmentation de sa quantité, nous avons
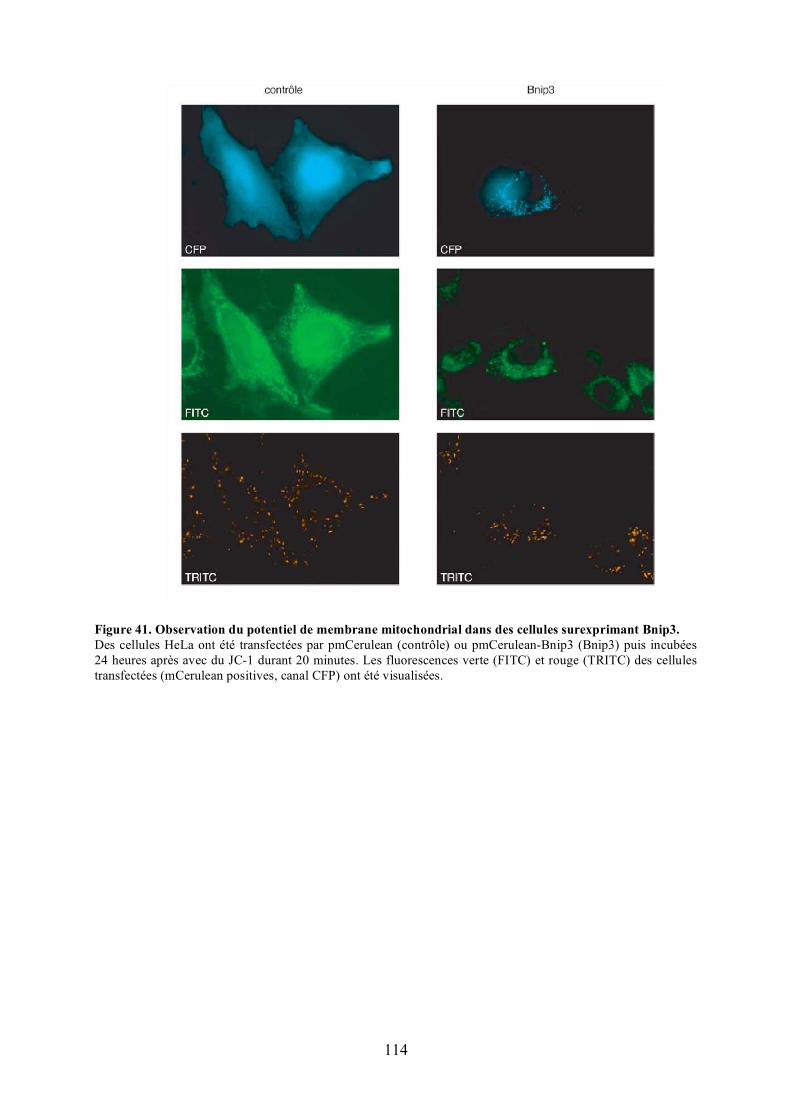
114
Figure 41. Observation du potentiel de membrane mitochondrial dans des cellules surexprimant Bnip3.Des cellules HeLa ont été transfectées par pmCerulean (contrôle) ou pmCerulean-Bnip3 (Bnip3) puis incubées 24 heures après avec du JC-1 durant 20 minutes. Les fluorescences verte (FITC) et rouge (TRITC) des cellules transfectées (mCerulean positives, canal CFP) ont été visualisées.

115
étudié l’effet d’une surexpression du pro-apoptotique. Pour cela, un marquage des
mitochondries au MitoTracker a été réalisé sur des cellules co-transfectées avec pcDNA-HA-
Bnip3 ou pcDNA-HA vide et pmaxGFP (ratio 5 : 1) pour repérer les cellules transfectées.
Nous avons, dans un premier temps, examiné la morphologie des mitochondries au
microscope à fluorescence 24 heures et 48 heures après transfection. Les cellules HeLa
transfectées avec pcDNA-HA vide présentent un réseau tubulaire et interconnecté (Figure 40,
A et B). La surexpression d’HA-Bnip3 génère une fragmentation massive du réseau
mitochondrial puisque 58% des cellules possèdent des mitochondries totalement ponctiformes
24 heures après transfection. Ce chiffre diminue à 48 heures pour atteindre environ 40%,
probablement à cause du rôle pro-apoptotique de Bnip3 et donc de la mort d’une partie des
cellules transfectées (cf, Résultats Première Partie IIC2). Nous avons donc répété ces
expériences 24 heures après transfection, ce qui a permis de valider statistiquement l’effet du
pro-apoptotique sur la morphologie des mitochondries (Figure 40, C).
Compte tenu du fait que Bnip3 affecte dans certaines conditions le potentiel de
membrane (��m) et que l’intégrité de ce dernier est requise pour la fusion des mitochondries,
nous avons voulu vérifier qu’une baisse du ��m n’était pas à l’origine de la fragmentation
induite par la surexpression de Bnip3. Pour cela, nous avons observé le ��m dans les cellules
surexprimant une protéine de fusion mCerulean-Bnip3 et présentant un réseau mitochondrial
fragmenté en utilisant la sonde JC-1 (5,5’,6,6’-tétrachloro-1,1’,3,3’-tétraéthylbenzimidazole
carbocyanide iodide). La sonde JC-1 est un composé fluorescent qui existe sous forme de
monomères lorsqu’il est à faible concentration et sous forme d’agrégats à de fortes
concentrations. La mitochondrie ayant un potentiel membranaire intact concentre JC-1 sous
forme d’agrégats. Au contraire une mitochondrie ayant un potentiel altéré ne peut concentrer
JC-1 (Smiley et al., 1991). La fluorescence des monomères, qui est verte, nous a permis de
visualiser la morphologie des mitochondries alors que la fluorescence des agrégats, qui est
rouge nous a permis de vérifier l’intégrité du ��m. La grande majorité des mitochondries
ponctiformes observées dans les cellules surexprimant Bnip3 apparaissent toujours polarisées
(Figure 41). La fragmentation des mitochondries observée dans ces conditions semble donc
bien relever d’un rôle propre de la protéine sur la morphologie mitochondriale et ne pas
simplement résulter d’un dysfonctionnement des mitochondries relatif à sa fonction pro-
apoptotique.
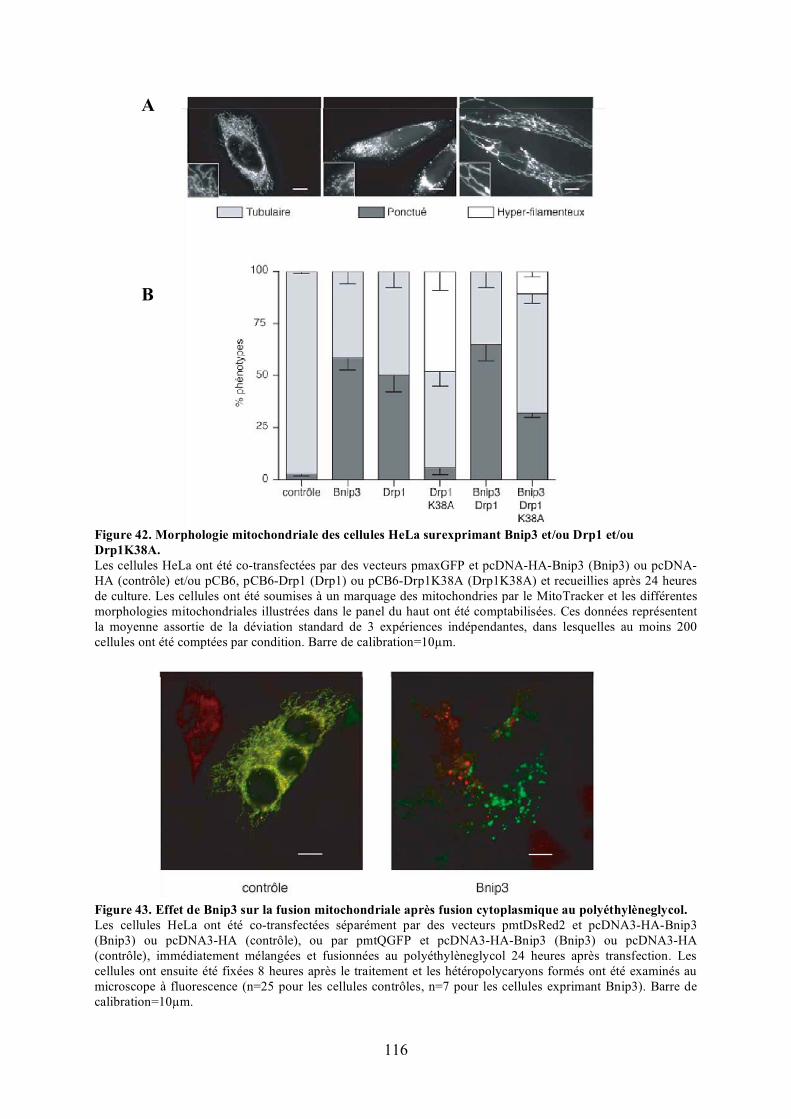
116
A
B
Figure 42. Morphologie mitochondriale des cellules HeLa surexprimant Bnip3 et/ou Drp1 et/ou Drp1K38A.Les cellules HeLa ont été co-transfectées par des vecteurs pmaxGFP et pcDNA-HA-Bnip3 (Bnip3) ou pcDNA-HA (contrôle) et/ou pCB6, pCB6-Drp1 (Drp1) ou pCB6-Drp1K38A (Drp1K38A) et recueillies après 24 heures de culture. Les cellules ont été soumises à un marquage des mitochondries par le MitoTracker et les différentes morphologies mitochondriales illustrées dans le panel du haut ont été comptabilisées. Ces données représentent la moyenne assortie de la déviation standard de 3 expériences indépendantes, dans lesquelles au moins 200 cellules ont été comptées par condition. Barre de calibration=10�m.
Figure 43. Effet de Bnip3 sur la fusion mitochondriale après fusion cytoplasmique au polyéthylèneglycol.Les cellules HeLa ont été co-transfectées séparément par des vecteurs pmtDsRed2 et pcDNA3-HA-Bnip3 (Bnip3) ou pcDNA3-HA (contrôle), ou par pmtQGFP et pcDNA3-HA-Bnip3 (Bnip3) ou pcDNA3-HA (contrôle), immédiatement mélangées et fusionnées au polyéthylèneglycol 24 heures après transfection. Les cellules ont ensuite été fixées 8 heures après le traitement et les hétéropolycaryons formés ont été examinés au microscope à fluorescence (n=25 pour les cellules contrôles, n=7 pour les cellules exprimant Bnip3). Barre de calibration=10�m.

117
La fragmentation des mitochondries induite par Bnip3 pourrait être la conséquence
d’une diminution de la fusion des membranes mitochondriales comme par exemple lors de
l’extinction de l’expression d’Opa1 par siRNA (Olichon et al., 2003). Au contraire, elle
pourrait résulter d’une augmentation de la fission des membranes mitochondriales observée
par exemple lors de la surexpression de l’acteur majeur de ce processus Drp1. Si Bnip3
modifie la morphologie mitochondriale par une activation de la fission mitochondriale, un
blocage de cette dernière devrait permettre le maintien d’un réseau mitochondrial tubulaire et
interconnecté. Pour tester cette hypothèse, nous avons co-transfecté des cellules HeLa avec
pmaxGFP, pcDNA-HA-Bnip3 et un plasmide codant pour un mutant dominant-négatif de
Drp1, pCB6-Drp1K38A et observé leur réseau mitochondrial. La surexpression de Drp1-
K38A diminue de moitié le nombre de cellules présentant un réseau ponctué et ne restaure
donc que partiellement une morphologie sauvage dans des cellules exprimant Bnip3 (Figure
42). De plus, la surexpression de Drp1K38A, qui conduit généralement à l’apparition de
mitochondries hyper-filamenteuses et très inter-connectées, conséquence de l’accumulation
d’évènements de fusion mitochondriale en absence d’évènements de fission (Smirnova et al.,
2001), ne mène que dans de très faibles proportions à l’apparition d’un telle morphologie en
présence de Bnip3. Ces résultats montrent que Drp1 est impliquée dans la fragmentation
induite par Bnip3 et suggère également que la fusion mitochondriale est altérée dans des
cellules surexprimant Bnip3.
Pour confirmer ce dernier point, nous avons utilisé le test in cellulo de fusion dit « au
PEG » (Legros et al., 2002). Un groupe de cellules HeLa a été transfecté avec un plasmide
codant pour une GFP adressée à la mitochondrie (pmtQGFP) et pcDNA-HA et un second
groupe de cellules HeLa a été transfecté avec un plasmide codant pour une RFP adressée à la
mitochondrie (pmtDsRed2) et pcDNA-HA. Ces cellules ont ensuite été mélangées puis après
24 heures fusionnées par du PEG. Les cellules ont été fixées 8 heures après la fusion
cellulaire et les hétéropolycaryons examinés par microscopie à fluorescence. Dans ces cellules
contrôles (n=25), de nombreuses mitochondries, parfois la totalité comme illustré en Figure
43, apparaissent doublement marquées, indiquant une activité de fusion mitochondriale
totalement intègre. Cette même expérience a été réalisée en utilisant le vecteur pcDNA-HA-
Bnip3. En présence du pro-apoptotique, des mitochondries ponctiformes marquées
différemment sont mêlées les unes aux autres, en particulier dans la région centrale du
polycaryon, avec plusieurs mitochondries « rouges » localisées dans la région du polycaryon
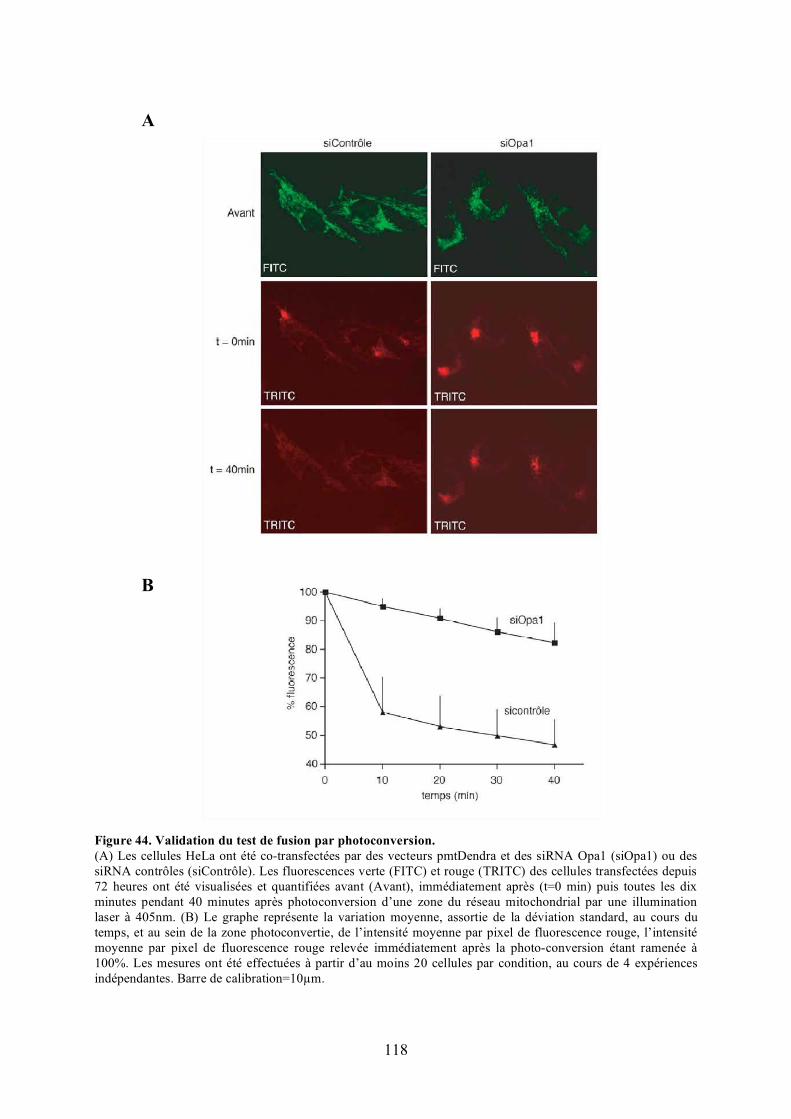
118
A
B
Figure 44. Validation du test de fusion par photoconversion.(A) Les cellules HeLa ont été co-transfectées par des vecteurs pmtDendra et des siRNA Opa1 (siOpa1) ou des siRNA contrôles (siContrôle). Les fluorescences verte (FITC) et rouge (TRITC) des cellules transfectées depuis 72 heures ont été visualisées et quantifiées avant (Avant), immédiatement après (t=0 min) puis toutes les dix minutes pendant 40 minutes après photoconversion d’une zone du réseau mitochondrial par une illumination laser à 405nm. (B) Le graphe représente la variation moyenne, assortie de la déviation standard, au cours du temps, et au sein de la zone photoconvertie, de l’intensité moyenne par pixel de fluorescence rouge, l’intensité moyenne par pixel de fluorescence rouge relevée immédiatement après la photo-conversion étant ramenée à 100%. Les mesures ont été effectuées à partir d’au moins 20 cellules par condition, au cours de 4 expériences indépendantes. Barre de calibration=10�m.

119
contenant des mitochondries « vertes » et vice-versa. Cependant, en dépit de leur proximité et
d’une mobilité tout à fait normale (observée par microscopie en temps réel), ces
mitochondries n’ont pas mélangé leurs protéines fluorescentes mettant en évidence une
inhibition de la fusion mitochondriale par Bnip3. Étant donné la toxicité inhérente à la
surexpression de Bnip3 et au traitement par le PEG, cette approche ne nous a permis d’isoler
et d’analyser qu’un nombre restreint d’hétéropolycaryons surexprimant Bnip3 (n=7).
Pour confirmer ces résultats, nous avons utilisé une approche expérimentale
complémentaire mise au point en 2004 par Karbowski et ses collaborateurs (Karbowski et al.,
2004a). Ce test fait appel à un variant photoactivable de la GFP, isolé chez Aequora victoria,
qui présente une substitution d’un acide aminé permettant, après photoactivation par une
illumination laser, une augmentation de plus de 100 fois de la fluorescence. Suite à
l’adressage de cette protéine à la mitochondrie, puis à la photoactivation d’une zone restreinte
de la cellule, un suivi du taux de dilution de fluorescence qui représente la diffusion de la
fluorescence à d’autres mitochondries voisines situées dans des zones non photoactivées
permise par la fusion mitochondriale est réalisé. Une dissipation de la fluorescence est ainsi
attribuée à des évènements de fusion des mitochondries qui conduit à la dilution de leur
contenu, alors que la fluorescence reste stable au cours du temps si les mitochondries ne
fusionnent plus. Nous avons utilisé pour cette approche non pas une GFP photoactivable mais
une GFP photoconvertible, nommée Dendra, isolée de Dendronephthya sp (Gurskaya et al.,
2006). Dendra est une GFP qui, après illumination laser à 405nm, fluoresce également dans le
rouge. Elle présente l’avantage par rapport aux GFP photoactivables de faciliter le repérage
des cellules transfectées car elle fluoresce dans le vert de manière constitutive, et d’offrir une
augmentation de fluorescence dans le rouge supérieure d’au moins 10 fois à celle décrite pour
les protéines utilisées jusqu’ici. Afin de tester notre système expérimental, nous avons dans
un premier temps observé l’effet de l’extinction d’Opa1 sur la fusion des mitochondries. Pour
cela, nous avons transfecté des cellules HeLa avec un plasmide que nous avons construit
codant pour Dendra, adressée à la mitochondrie par fusion avec la sous-unité VIII de la
cytochrome c oxydase (pmtDendra), en présence d’un ARN interférent dirigé contre Opa1 ou
d’un ARN interférent contrôle. 72 heures après transfection, temps nécessaire à la perte totale
de la protéine Opa1 (Olichon et al., 2003), une région d’intérêt par cellule a été
photoconvertie via une illumination laser à 488nm, entraînant la fluorescence d’une zone du
réseau mitochondrial dans le rouge. La dilution de cette fluorescence a été suivie au cours du
temps pendant 40 minutes puis quantifiée (Figure 44, A et B). Dans les cellules contrôles, la
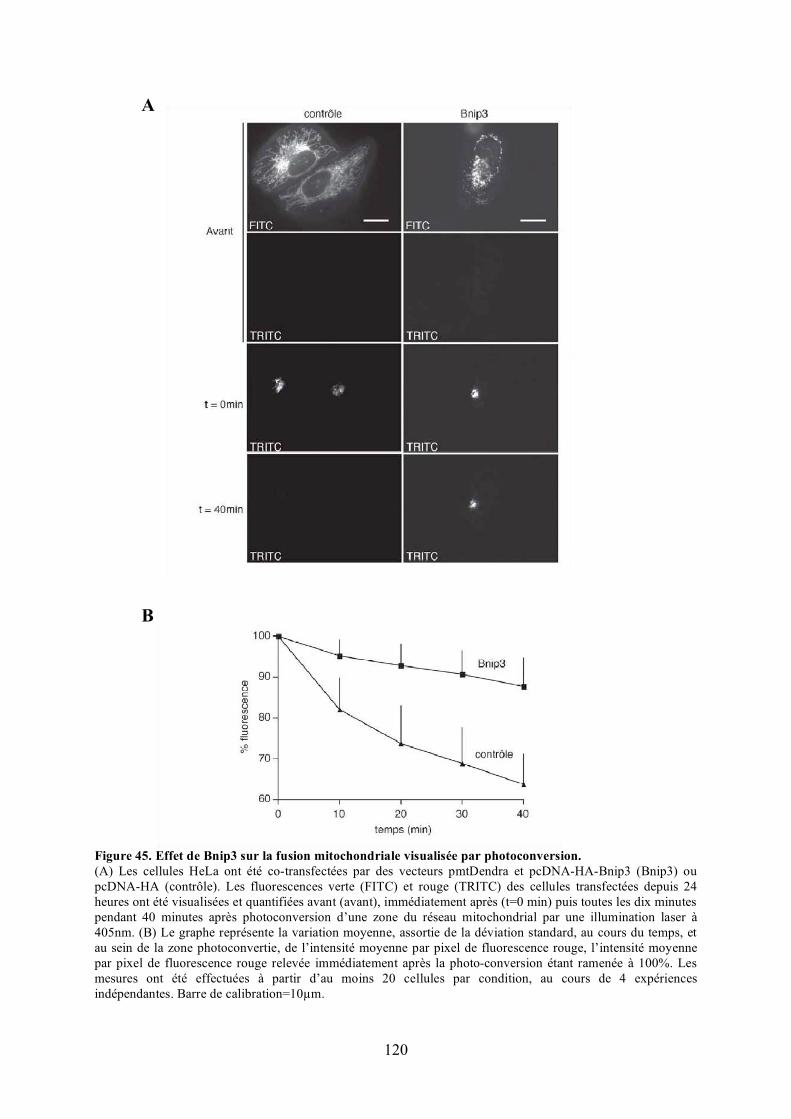
120
A
B
Figure 45. Effet de Bnip3 sur la fusion mitochondriale visualisée par photoconversion.(A) Les cellules HeLa ont été co-transfectées par des vecteurs pmtDendra et pcDNA-HA-Bnip3 (Bnip3) ou pcDNA-HA (contrôle). Les fluorescences verte (FITC) et rouge (TRITC) des cellules transfectées depuis 24 heures ont été visualisées et quantifiées avant (avant), immédiatement après (t=0 min) puis toutes les dix minutes pendant 40 minutes après photoconversion d’une zone du réseau mitochondrial par une illumination laser à 405nm. (B) Le graphe représente la variation moyenne, assortie de la déviation standard, au cours du temps, etau sein de la zone photoconvertie, de l’intensité moyenne par pixel de fluorescence rouge, l’intensité moyenne par pixel de fluorescence rouge relevée immédiatement après la photo-conversion étant ramenée à 100%. Les mesures ont été effectuées à partir d’au moins 20 cellules par condition, au cours de 4 expériences indépendantes. Barre de calibration=10�m.

121
plupart des mitochondries photoconverties subissent des évènements de fusion résultant après
40 minutes en une diminution quasiment de moitié de la quantité de fluorescence au sein de la
région d’intérêt. En revanche, comme attendu, les cellules déplétées pour Opa1, qui possèdent
des mitochondries ponctiformes, ne présentent, en fin de cinétique, qu’une disparition
mineure, parfois inexistante, de fluorescence au niveau des mitochondries photoconverties
reflétant une altération de la fusion des mitochondries. Une fois validée, nous avons utilisé
cette approche pour examiner l’effet de Bnip3 sur la fusion mitochondriale. Des cellules HeLa
ont été transfectées durant 24 heures avec pmtDendra et pcDNA3-HA-Bnip3 ou pcDNA3-HA
puis analysées comme précédemment (Figure 45, A et B). Contrairement aux cellules
contrôles, et de façon similaire à l’extinction d’Opa1, la surexpression du pro-apoptotique
conduit à une diminution importante, de l’ordre de 40%, de la dilution de la fluorescence
présente dans les zones photoconverties. Ces données confirment donc les résultats décrits
précédemment et illustrent la capacité de Bnip3 à inhiber la fusion mitochondriale.
L’ensemble des résultats présentés ci-dessus ont été acquis lors de la surexpression
ectopique Bnip3. Nous avons entrepris de les confirmer en présence d’un taux physiologique
de la protéine. Pour cela, nous avons analysé l’effet d’un traitement au chlorure de cobalt ou
de l’hypoxie sur la morphologie des mitochondries et évalué la contribution de Bnip3 dans ce
changement par interférence à l’ARN. Je ne présenterai ici qu’une expérience préliminaire
(Figure 46) au cours de laquelle des cellules HeLa transfectées avec des ARN interférents
dirigés contre Bnip3 ou des ARN interférents contrôles ont été cultivées en conditions
normales ou en hypoxie (1% d’oxygène) durant 24 heures. L’expression de Bnip3 a été
vérifiée par des expériences d’immuno-détection et la morphologie des mitochondries a été
examinée par immuno-fluorescence à l’aide d’anticorps dirigés contre la sous-unité IV de la
Cytochrome c oxydase. Comme décrit dans la littérature (Zanelli et al., 2006), l’hypoxie
génère une fragmentation massive du réseau mitochondrial puisque environ 40% des cellules
possèdent des mitochondries ponctiformes. L’extinction de l’expression de Bnip3 réduit de
moité ce nombre, ce qui confirme l’implication de Bnip3 dans la dynamique mitochondriale.
2) Étude de l’effet de Bnip3 sur la viabilité cellulaire
De nombreux travaux ont montré que la surexpression de Bnip3 conduit à la mort des
cellules. Cependant, pour déterminer si elle induit également la mort dans nos conditions et si
oui par quel processus, des cellules HeLa ont été transfectées par les plasmides pmCerulean
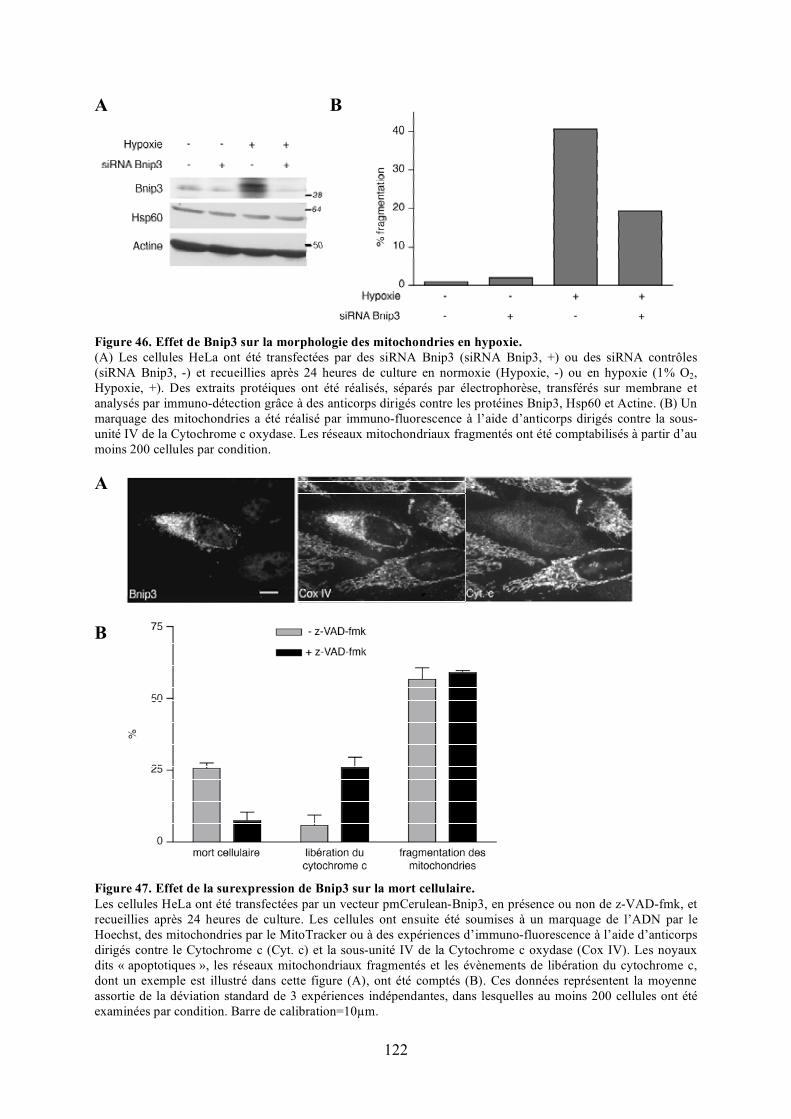
122
A B
B
Figure 46. Effet de Bnip3 sur la morphologie des mitochondries en hypoxie.(A) Les cellules HeLa ont été transfectées par des siRNA Bnip3 (siRNA Bnip3, +) ou des siRNA contrôles (siRNA Bnip3, -) et recueillies après 24 heures de culture en normoxie (Hypoxie, -) ou en hypoxie (1% O2,Hypoxie, +). Des extraits protéiques ont été réalisés, séparés par électrophorèse, transférés sur membrane et analysés par immuno-détection grâce à des anticorps dirigés contre les protéines Bnip3, Hsp60 et Actine. (B) Unmarquage des mitochondries a été réalisé par immuno-fluorescence à l’aide d’anticorps dirigés contre la sous-unité IV de la Cytochrome c oxydase. Les réseaux mitochondriaux fragmentés ont été comptabilisés à partir d’au moins 200 cellules par condition.
A
B
Figure 47. Effet de la surexpression de Bnip3 sur la mort cellulaire.Les cellules HeLa ont été transfectées par un vecteur pmCerulean-Bnip3, en présence ou non de z-VAD-fmk, et recueillies après 24 heures de culture. Les cellules ont ensuite été soumises à un marquage de l’ADN par le Hoechst, des mitochondries par le MitoTracker ou à des expériences d’immuno-fluorescence à l’aide d’anticorps dirigés contre le Cytochrome c (Cyt. c) et la sous-unité IV de la Cytochrome c oxydase (Cox IV). Les noyaux dits « apoptotiques », les réseaux mitochondriaux fragmentés et les évènements de libération du cytochrome c, dont un exemple est illustré dans cette figure (A), ont été comptés (B). Ces données représentent la moyenne assortie de la déviation standard de 3 expériences indépendantes, dans lesquelles au moins 200 cellules ont étéexaminées par condition. Barre de calibration=10�m.

123
(vecteur contrôle) ou pmCerulean-Bnip3 qui code pour une forme sauvage de Bnip3 étiquetée
dans sa partie amino-terminale avec la protéine fluorescente mCerulean. Cette construction
nous a permis de repérer les cellules transfectées tout en conservant les canaux de
fluorescence FITC et TRITC pour réaliser des expériences d’immunofluorescence indirecte
que je décrirai par la suite. La mortalité a été analysée après marquage de l’ADN au Hoechst
et est basée sur l’apparition de noyaux présentant un phénotype dit « apoptotique », c’est-à-
dire des noyaux situés au-dessus du plan focal avec une condensation extrême et une
fragmentation de la chromatine, ce critère étant classiquement utilisé dans la littérature. Les
cellules transfectées par pmCerulean présentent plus de 95 % de noyaux normaux. En
revanche, comme attendu, l’expression de Bnip3 conduit après 24 heures à une forte
augmentation du nombre de noyaux « apoptotiques » représentant plus de 25% des noyaux
comptés (Figure 47, A). Les observations n’ont été effectuées que 24 heures après
transfection, le pourcentage de cellules transfectées chutant nettement au-delà, probablement
à cause du fort niveau d’expression de CFP-Bnip3 obtenu dans ces conditions.
En présence de z-VAD-fmk, un inhibiteur à large spectre des caspases, l’apparition de
noyaux dits « apoptotiques » est considérablement diminuée, ce qui montre donc que Bnip3
induit une mort dépendante des caspases (Figure 47, A). Le blocage de la mort cellulaire par
le z-VAD-fmk nous a également permis d’examiner la localisation sub-cellulaire du
cytochrome c dans les cellules exprimant Bnip3 via des expériences d’immunofluorescence
indirecte. De manière intéressante, plus de 25% des cellules transfectées présentent un
marquage diffus et cytosolique du cytochrome c, contrastant avec le marquage de la sous-
unité IV de la cytochrome c oxydase qui demeure toujours localisé à la mitochondrie (Figure
47, A et B). L’ensemble de nos résultats démontre que la surexpression de Bnip3 conduit dans
les cellules HeLa à une mort apoptotique dépendante des caspases.
Nous avons, de plus, examiné la morphologie des mitochondries dans les cellules
surexprimant Bnip3, en présence ou non de z-VAD-fmk (Figure 47, A). Le pourcentage de
cellules présentant des mitochondries ponctiformes reste inchangé en présence ou non de
l’inhibiteur, ce qui exclue dans ces conditions une contribution des caspases dans la
fragmentation des mitochondries.
La nature de la mort induite par Bnip3 reste très débattue et pourrait mener suivant les
types cellulaires et les stimuli à des types de mort apoptotiques, nécrotiques ou
autophagiques. Afin de déterminer une éventuelle contribution de l’autophagie dans la mort
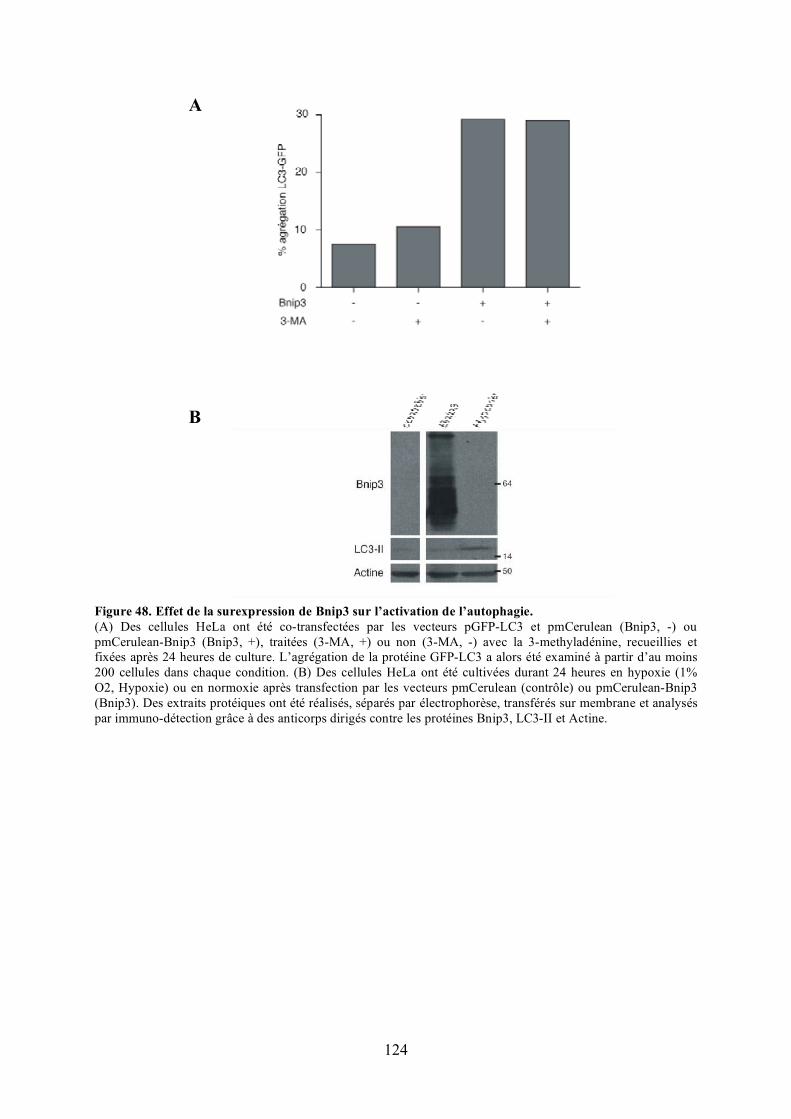
124
A
B
Figure 48. Effet de la surexpression de Bnip3 sur l’activation de l’autophagie.(A) Des cellules HeLa ont été co-transfectées par les vecteurs pGFP-LC3 et pmCerulean (Bnip3, -) ou pmCerulean-Bnip3 (Bnip3, +), traitées (3-MA, +) ou non (3-MA, -) avec la 3-methyladénine, recueillies et fixées après 24 heures de culture. L’agrégation de la protéine GFP-LC3 a alors été examiné à partir d’au moins 200 cellules dans chaque condition. (B) Des cellules HeLa ont été cultivées durant 24 heures en hypoxie (1% O2, Hypoxie) ou en normoxie après transfection par les vecteurs pmCerulean (contrôle) ou pmCerulean-Bnip3 (Bnip3). Des extraits protéiques ont été réalisés, séparés par électrophorèse, transférés sur membrane et analysés par immuno-détection grâce à des anticorps dirigés contre les protéines Bnip3, LC3-II et Actine.

125
cellulaire observée, nous avons, dans un premier temps, co-transfecté des cellules HeLa avec
les constructions codant pour Bnip3 et un marqueur étiqueté GFP de l’autophagie, LC3
(Figure 48, A). En absence d’autophagie, LC3-GFP présente un marquage diffus cytosolique.
En revanche, dans des conditions induisant l’autophagie, comme par exemple une mise en
carence en acides aminés ou en hypoxie des cellules (Elmore et al., 2001; Zhang et al., 2008),
la protéine est clivée, insérée dans les membranes des autophagosomes et est présente de ce
fait sous forme de nombreux aggrégats dans le cytoplasme (Tanida et al., 2004). La
surexpression de Bnip3 induit dans 29% des cellules l’agrégation de la protéine de fusion
contre environ 8% au sein des cellules contrôles suggérant l’activation de l’autophagie dans
ces conditions. Néanmoins, un traitement avec la 3-methyl-adénine, un inhibiteur spécifique
de l’autophagie ne réduit pas cet effet, ce qui semble écarter une intervention de ce processus
et indique vraisemblablement une agrégation non-spécifique de GFP-LC3 dans ces
conditions. Afin de confirmer cela, nous avons, dans un second temps, préparé des extraits
protéiques de cellules HeLa transfectées par les plasmides pmCerulean ou pmCerulean-Bnip3
et examiné par immuno-détection l’apparition de la forme clivée de LC3 (LC3-II) (Figure 48,
B). Alors que LC3-II est assez fortement détectée dans des cellules cultivées dans une étuve à
hypoxie en présence de 1% d’oxygène (témoin positif), cette protéine n’est par contre pas
détectée dans des cellules transfectées par CFP-Bnip3 comme dans des cellules transfectées
par la CFP seule (témoin négatif). L’ensemble de ces résultats indique donc que, dans nos
conditions expérimentales, l’autophagie ne participe pas à la mort cellulaire induite par Bnip3.
3) Étude de la relevance de l’interaction entre Opa1 et Bnip3
a) Étude de l’effet des mutants de Bnip3
Nous avons montré que Bnip3 était capable d’induire la fragmentation des
mitochondries par inhibition de la fusion mitochondriale et la mort cellulaire par apoptose,
deux processus faisant intervenir Opa1. Nous avons donc cherché dans un premier temps à
déterminer si ces effets dépendaient de l’interaction du pro-apoptotique avec la dynamine.
Pour cela, nous avons analysé les conséquences de la surexpression des mutants de Bnip3
présentant une capacité d’interaction altérée avec Opa1 sur la morphologie des mitochondries
et l’apoptose. Nous avons co-transfecté les cellules Hela avec pmaxGFP et pcDNA-HA vide,
pcDNA3-HA-Bnip3�C ou pcDNA3-HA-Bnip3-TMBcl-2. 24 heures après transfection, le
marquage des mitochondries au MitoTracker, de l’ADN au Hoechst puis la fixation des
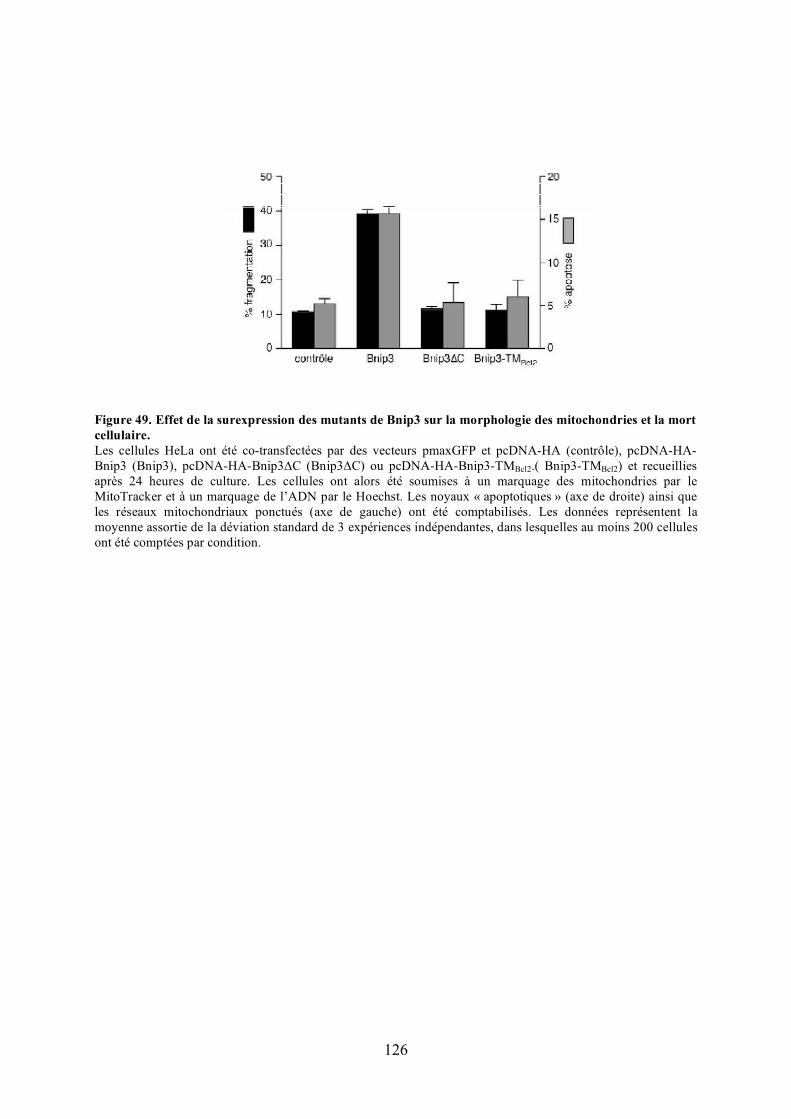
126
Figure 49. Effet de la surexpression des mutants de Bnip3 sur la morphologie des mitochondries et la mort cellulaire.Les cellules HeLa ont été co-transfectées par des vecteurs pmaxGFP et pcDNA-HA (contrôle), pcDNA-HA-Bnip3 (Bnip3), pcDNA-HA-Bnip3�C (Bnip3�C) ou pcDNA-HA-Bnip3-TMBcl2.( Bnip3-TMBcl2) et recueillies après 24 heures de culture. Les cellules ont alors été soumises à un marquage des mitochondries par le MitoTracker et à un marquage de l’ADN par le Hoechst. Les noyaux « apoptotiques » (axe de droite) ainsi que les réseaux mitochondriaux ponctués (axe de gauche) ont été comptabilisés. Les données représentent la moyenne assortie de la déviation standard de 3 expériences indépendantes, dans lesquelles au moins 200 cellules ont été comptées par condition.

127
cellules ont été réalisés (Figure 49). Contrairement à la forme sauvage de Bnip3, les mutants
présentant une délétion de dix acides aminés à l’extrémité carboxy-terminale (Bnip3�C) ou
une substitution du domaine trans-membranaire (Bnip3-TMBcl-2) ne perturbent ni la
morphologie des mitochondries ni la mort cellulaire. L’ensemble de ces données supportent
donc la nécessité pour Bnip3 d’interagir pleinement avec Opa1 pour induire la fragmentation
des mitochondries et pour exercer son activité pro-apoptotique.
b) Étude de l’effet d’Opa1
Plusieurs expériences ont été par la suite envisagées dans le but de renforcer le lien
physiologique entre les deux protéines.
Une première approche consistait à analyser la morphologie des mitochondries et la
mort cellulaire dans des cellules surexprimant Bnip3, en absence de la protéine Opa1.
Cependant le fait que des cellules traitées par un ARN interférent dirigé contre Opa1
présentent une fragmentation de leur réseau mitochondrial, à cause de la perte d’un acteur de
fusion, et une mort cellulaire spontanée, ne nous a pas permis de suivre une telle approche.
Nous avons alors cherché à déterminer si une surexpression de la dynamine pouvait
contrecarrer une surexpression du pro-apoptotique. Des expériences réalisées en
surexpression transitoire des deux partenaires se sont avérées encourageantes mais ont été
difficilement reproductibles du fait qu’Opa1, malgré son implication dans la fusion, lorsqu’il
est fortement surexprimé, induit dans les cellules HeLa non pas l’allongement des
mitochondries mais leur fragmentation (cf Résultats Deuxième partie). Ce paradoxe
s’explique par une activation de la fission des organites consécutive à une induction trop
conséquente de la fusion. Par conséquent, nous avons choisi de générer une lignée de cellules
HeLa « Tet-Off » surexprimant le variant 1 d’Opa1 de façon stable et inductible. Nous avons
isolé plusieurs clones et nous avons analysé pour chacun d’entre eux, l’induction d’Opa1 en
absence de tétracycline par immuno-détection à partir d’extraits cellulaires totaux ainsi que la
morphologie des mitochondries après marquage au MitoTracker et la mort cellulaire après
marquage au Hoechst. Des clones « faux-positifs » ne présentant pas d’augmentation du
niveau d’expression d’Opa1, ainsi que des clones présentant une mortalité trop importante ou
une morphologie mitochondriale aberrante ont été éliminés. La morphologie mitochondriale
des clones restants a ensuite été attentivement examinée après 48 heures d’induction, un
temps nous permettant d’envisager raisonnablement la réalisation de transfections transitoires
par des plasmides ou des ARN interférents. L’analyse de deux de ces clones, les clones 4 et 7,
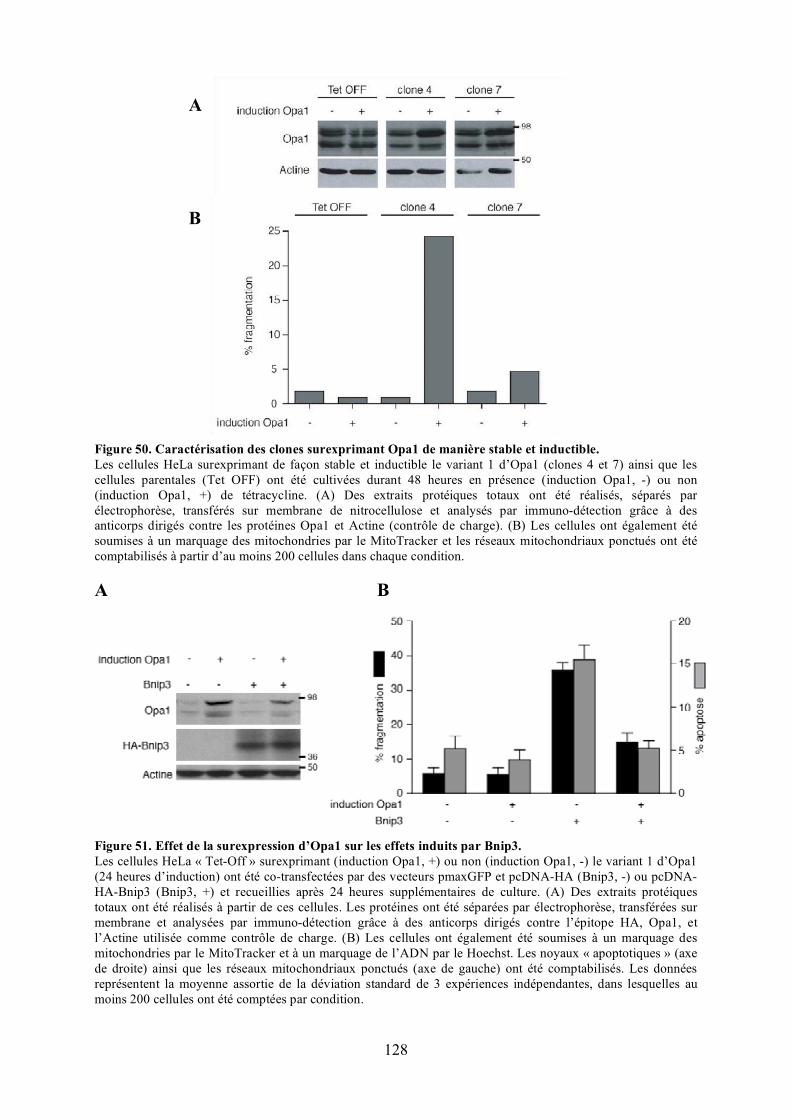
128
A
B
Figure 50. Caractérisation des clones surexprimant Opa1 de manière stable et inductible.Les cellules HeLa surexprimant de façon stable et inductible le variant 1 d’Opa1 (clones 4 et 7) ainsi que les cellules parentales (Tet OFF) ont été cultivées durant 48 heures en présence (induction Opa1, -) ou non (induction Opa1, +) de tétracycline. (A) Des extraits protéiques totaux ont été réalisés, séparés par électrophorèse, transférés sur membrane de nitrocellulose et analysés par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1 et Actine (contrôle de charge). (B) Les cellules ont également été soumises à un marquage des mitochondries par le MitoTracker et les réseaux mitochondriaux ponctués ont été comptabilisés à partir d’au moins 200 cellules dans chaque condition.
A B
Figure 51. Effet de la surexpression d’Opa1 sur les effets induits par Bnip3.Les cellules HeLa « Tet-Off » surexprimant (induction Opa1, +) ou non (induction Opa1, -) le variant 1 d’Opa1 (24 heures d’induction) ont été co-transfectées par des vecteurs pmaxGFP et pcDNA-HA (Bnip3, -) ou pcDNA-HA-Bnip3 (Bnip3, +) et recueillies après 24 heures supplémentaires de culture. (A) Des extraits protéiques totaux ont été réalisés à partir de ces cellules. Les protéines ont été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre l’épitope HA, Opa1, et l’Actine utilisée comme contrôle de charge. (B) Les cellules ont également été soumises à un marquage des mitochondries par le MitoTracker et à un marquage de l’ADN par le Hoechst. Les noyaux « apoptotiques » (axe de droite) ainsi que les réseaux mitochondriaux ponctués (axe de gauche) ont été comptabilisés. Les données représentent la moyenne assortie de la déviation standard de 3 expériences indépendantes, dans lesquelles au moins 200 cellules ont été comptées par condition.

129
est présentée en Figure 50. L’immuno-détection d’Opa1 après 48 heures d’induction montre
dans les deux clones une quantité plus importante des isoformes b et d de la protéine. Les
cellules du clone 4 surexpriment fortement Opa1, ce qui entraine la fragmentation des
mitochondries dans environ 25% d’entre elles. Le clone 7 surexprime des niveaux modérés
d’Opa1 qui n’entraînent pas la fragmentation des mitochondries (Figure 50, A et B). Nous
avons donc réalisé nos expériences à partir du clone 7. Une co-transfection de ce clone a été
réalisée après 24 heures d’induction ou non de l’expression d’Opa1 par pmaxGFP et pcDNA-
HA-Bnip3 ou pcDNA-HA vide. 24 heures après transfection, le marquage des mitochondries
au MitoTracker et de l’ADN au Hoechst ont été réalisés (Figure 51, B). La surexpression
d’Opa1 et de Bnip3 a été vérifiée par immuno-détection à partir d’extraits cellulaires totaux
(Figure 51, A). La surexpression de Bnip3, en absence d’induction d’Opa1, mène à la
fragmentation mitochondriale, dans 36% des cellules, et à la mort, dans 15% d’entre elles. La
surexpression de la dynamine réduit considérablement ces valeurs, qui atteignent
respectivement 14% et 5% et se rapprochent des valeurs observées pour la transfection avec le
vecteur contrôle pcDNA-HA vide (6% et 5% respectivement). Ces résultats indiquent donc
que la surexpression d’Opa1 inhibe la fragmentation des mitochondries et l’apoptose induite
par Bnip3.
4) Étude du mode d’action de Bnip3
Le rôle protecteur d’Opa1 vis-à-vis aussi bien de la fragmentation mitochondriale que
de l’apoptose dans des cellules surexprimant Bnip3 pourrait s’expliquer de différentes façons.
Durant ces dernières années, il a été montré qu’un blocage artificiel de la fragmentation des
mitochondries, par inhibition de la fission ou par augmentation de la fusion de ces organelles,
inhibe le processus apoptotique. La fonction fusogène d’Opa1 pourrait donc à elle seule
empêcher la fragmentation du réseau mitochondrial et protéger les cellules de l’apoptose.
Cependant, le rôle protecteur d’Opa1 pourrait passer par sa fonction dans la structuration des
crêtes mitochondriales et la rétention du cytochrome c dans l’espace intra-crête (Yamaguchi
et al., 2008; Frezza et al., 2006). Dans ce cas-là, Bnip3 pourrait donc inhiber à la fois les
activités fusogène et anti-apoptotique de la dynamine. Pour trancher entre ces deux
hypothèses, nous avons répété les expériences décrites ci-dessus en présence ou non d’ARN
interférents dirigés contre Mfn1. En effet, si la fonction fusogène d’Opa1 requiert la présence
de cette protéine, sa fonction anti-apoptotique perdure dans des cellules déficientes pour
Mfn1. Nous avons donc dans un premier temps mis au point l’extinction de l’expression de
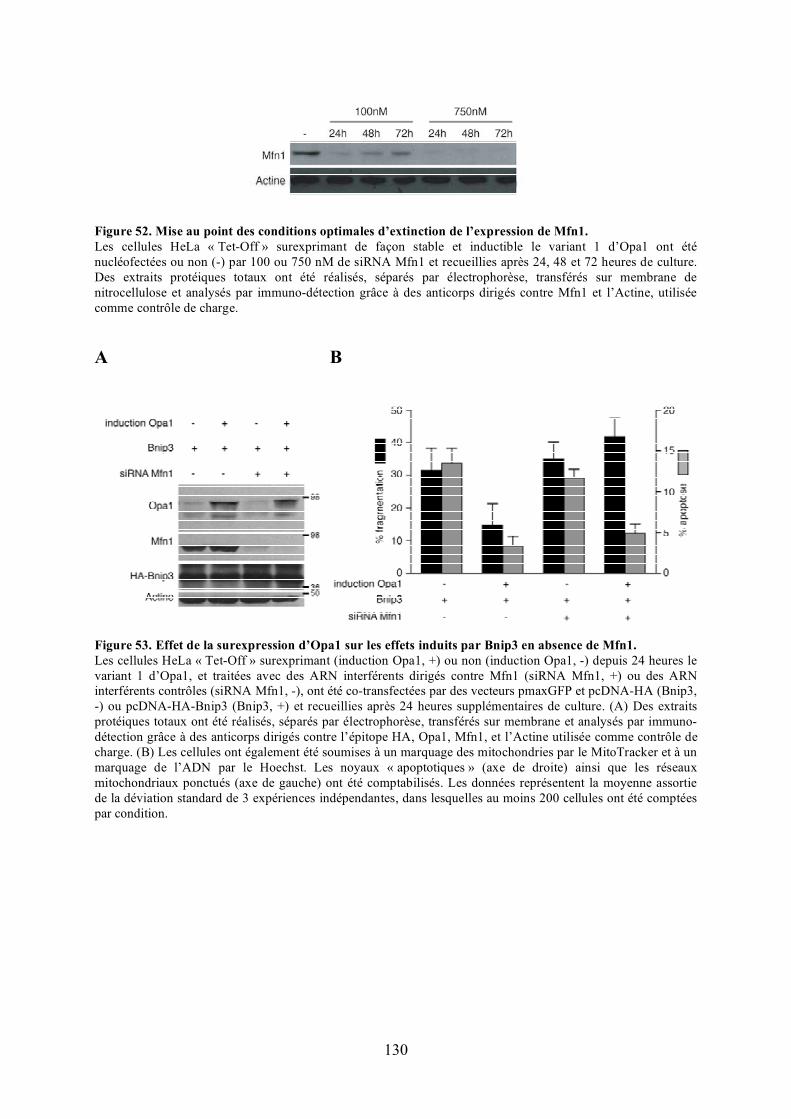
130
Figure 52. Mise au point des conditions optimales d’extinction de l’expression de Mfn1.Les cellules HeLa « Tet-Off » surexprimant de façon stable et inductible le variant 1 d’Opa1 ont été nucléofectées ou non (-) par 100 ou 750 nM de siRNA Mfn1 et recueillies après 24, 48 et 72 heures de culture. Des extraits protéiques totaux ont été réalisés, séparés par électrophorèse, transférés sur membrane de nitrocellulose et analysés par immuno-détection grâce à des anticorps dirigés contre Mfn1 et l’Actine, utilisée comme contrôle de charge.
A B
Figure 53. Effet de la surexpression d’Opa1 sur les effets induits par Bnip3 en absence de Mfn1.Les cellules HeLa « Tet-Off » surexprimant (induction Opa1, +) ou non (induction Opa1, -) depuis 24 heures le variant 1 d’Opa1, et traitées avec des ARN interférents dirigés contre Mfn1 (siRNA Mfn1, +) ou des ARN interférents contrôles (siRNA Mfn1, -), ont été co-transfectées par des vecteurs pmaxGFP et pcDNA-HA (Bnip3, -) ou pcDNA-HA-Bnip3 (Bnip3, +) et recueillies après 24 heures supplémentaires de culture. (A) Des extraits protéiques totaux ont été réalisés, séparés par électrophorèse, transférés sur membrane et analysés par immuno-détection grâce à des anticorps dirigés contre l’épitope HA, Opa1, Mfn1, et l’Actine utilisée comme contrôle de charge. (B) Les cellules ont également été soumises à un marquage des mitochondries par le MitoTracker et à un marquage de l’ADN par le Hoechst. Les noyaux « apoptotiques » (axe de droite) ainsi que les réseaux mitochondriaux ponctués (axe de gauche) ont été comptabilisés. Les données représentent la moyenne assortie de la déviation standard de 3 expériences indépendantes, dans lesquelles au moins 200 cellules ont été comptées par condition.

131
Mfn1 (Figure 52). Pour cela, nous avons transfecté par nucléofection durant 24, 48 et 72
heures les cellules HeLa avec deux concentrations différentes (100 nM et 750 nM) d’un
mélange de 4 ARN interférents dirigés contre Mfn1. L’immuno-détection de Mfn1 à partir
des extraits cellulaires totaux indique une baisse de la quantité de la protéine observable dès
24 heures pour les deux concentrations. Nous avons décidé, après plusieurs tests et après
détermination de la quantité de protéine résiduelle pour chaque condition, de réaliser nos
expériences en présence de la concentration la plus forte d’ARN interférents (750nm) et de
débuter l’extinction de Mfn1 au moment où nous débutons l’induction d’Opa1, soit 24 heures
avant la transfection par pcDNA-HA-Bnip3 et 48 heures avant la fixation des cellules (Figure
53). L’extinction de l’expression du gène codant pour Mfn1 (s’élevant alors à plus de 90%)
n’induit pas de changement spectaculaire au niveau du réseau mitochondrial mais seulement
un léger raccourcissement des organites (Ishihara et al., 2004). Dans des cellules surexprimant
Bnip3, l’extinction de Mfn1 empêche Opa1 d’inhiber la fragmentation mitochondriale mais
pas l’apoptose. Ces résultats indiquent, d’une part, que la protection exercée par Opa1 sur les
cellules vis-à-vis de Bnip3 repose à la fois sur la fonction fusogène et sur la fonction anti-
apoptotique de la dynamine et d’autre part que la fragmentation induite par Bnip3 ne constitue
pas en soi un signal de mort.
Pour déterminer si Bnip3 peut inhiber la capacité d’Opa1 à structurer les crêtes
mitochondriales et à contrôler la mobilisation du cytochrome c (Frezza et al., 2006;
Yamaguchi et al., 2008), nous avons étudié l’effet du pro-apoptotique sur l’oligomérisation de
la dynamine. En effet, il a été montré que deux autres protéines « BH3-only » de la famille de
Bcl-2, la forme tronquée active de Bid (tBid) et Bim, altèrent, par un mécanisme restant
encore à éclaircir, les complexes formés par Opa1. Avant de réaliser les expériences avec
Bnip3, nous avons tenté de reproduire les données publiées précédemment par les groupes de
Luca Scorrano et de Don Newmeyer (Frezza et al., 2006; Yamaguchi et al., 2008). Pour cela,
les mitochondries de cellules HeLa ont été isolées par centrifugation différentielle puis
incubées avec une forme recombinante de Bid tronquée. Les protéines mitochondriales ont
ensuite été pontées à l’EDC (1-éthyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride)
puis ont été séparées par électrophorèse sans rupture des pontages et transférées sur
membrane de nitrocellulose. Une immuno-détection de la protéine Opa1 permet de mettre en
évidence qu’en conditions normales, plus de la moitié de la protéine est complexée sous
forme d’oligomères migrant aux environs de 190 kDa (Figure 54, A). En présence de tBid, un
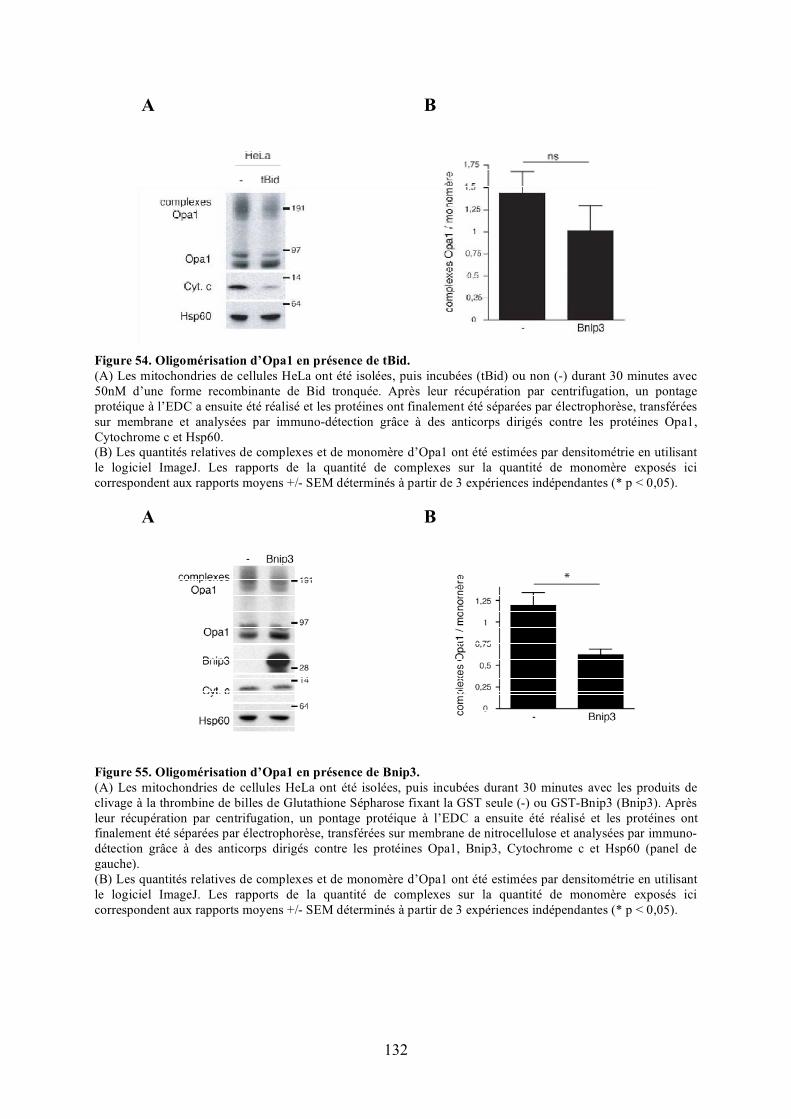
132
A B
Figure 54. Oligomérisation d’Opa1 en présence de tBid.(A) Les mitochondries de cellules HeLa ont été isolées, puis incubées (tBid) ou non (-) durant 30 minutes avec 50nM d’une forme recombinante de Bid tronquée. Après leur récupération par centrifugation, un pontage protéique à l’EDC a ensuite été réalisé et les protéines ont finalement été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1, Cytochrome c et Hsp60. (B) Les quantités relatives de complexes et de monomère d’Opa1 ont été estimées par densitométrie en utilisant le logiciel ImageJ. Les rapports de la quantité de complexes sur la quantité de monomère exposés ici correspondent aux rapports moyens +/- SEM déterminés à partir de 3 expériences indépendantes (* p < 0,05).
A B
Figure 55. Oligomérisation d’Opa1 en présence de Bnip3.(A) Les mitochondries de cellules HeLa ont été isolées, puis incubées durant 30 minutes avec les produits de clivage à la thrombine de billes de Glutathione Sépharose fixant la GST seule (-) ou GST-Bnip3 (Bnip3). Après leur récupération par centrifugation, un pontage protéique à l’EDC a ensuite été réalisé et les protéines ont finalement été séparées par électrophorèse, transférées sur membrane de nitrocellulose et analysées par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1, Bnip3, Cytochrome c et Hsp60 (panel de gauche). (B) Les quantités relatives de complexes et de monomère d’Opa1 ont été estimées par densitométrie en utilisant le logiciel ImageJ. Les rapports de la quantité de complexes sur la quantité de monomère exposés ici correspondent aux rapports moyens +/- SEM déterminés à partir de 3 expériences indépendantes (* p < 0,05).

133
désassemblage des complexes d’Opa1 est observé et coïncide avec la libération du
cytochrome c dans le cytosol suite à la perméabilisation de la membrane externe également
induite par le pro-apoptotique. Comme décrit dans la littérature (Arnoult et al., 2005a), la
libération du cytochrome c est très rapidement suivie de celle du monomère d’Opa1. Ceci
explique que, malgré une disruption massive des oligomères, l’augmentation de la forme
monomérique n’est que très faible conduisant à une variation non significative du rapport de
la quantité de complexes sur la quantité de monomère de la dynamine (Figure 54, B). Pour
étudier l’effet de Bnip3 sur les oligomères d’Opa1, nous avons produit chez Escherichia coli
puis purifié une forme recombinante de Bnip3 et suivi la même démarche que décrite ci-
dessus (Figure 55). L’incubation des mitochondries avec Bnip3 recombinant conduit à la
dissociation des complexes d’Opa1, visualisée par la diminution significative d’un facteur 2
du rapport de la quantité de complexes sur la quantité de monomère (Figure 55). Cependant,
au contraire de tBid, Bnip3 n’induit ni la libération du cytochrome c de la mitochondrie, ni
celle du monomère d’Opa1. Ce résultat nous amène à proposer que, dans ces conditions,
Bnip3 est incapable d’induire la perméabilisation de la membrane externe mitochondriale et
que le cytochrome c et le monomère d’Opa1 restent localisés dans l’espace inter-
membranaire. L’accumulation du monomère de la dynamine dans la mitochondrie supporte ce
modèle et sera vérifiée prochainement en perméabilisant artificiellement la membrane externe
pour libérer le cytochrome c et Opa1.
Ces expériences ont été effectuées dans des fibroblastes embryonnaires de souris
sauvages et invalidées pour les gènes BAX et BAK . Alors que dans les mitochondries isolées
de fibroblastes sauvages, Bnip3 est capable de dissocier les complexes d’Opa1, aucune
différence n’est observée entre des mitochondries de fibroblastes embryonnaires de souris
invalidées pour BAX et BAK contrôles ou traitées avec la forme recombinante du pro-
apoptotique. Donc, bien qu’une interaction directe ait lieu entre Bnip3 et Opa1, Bnip3,
comme tBid (Yamaguchi et al., 2008), requiert la présence de Bax et Bak (Figure 56) pour
dissocier les complexes formés par la dynamine.
Nous avons de plus incubé dans des expériences préliminaires des mitochondries de
cellules HeLa avec les formes recombinantes purifiées des deux mutants de Bnip3, Bnip3�C
et Bnip3-TM-Bcl-2. Comme le montre la Figure 57, les niveaux d’oligomères d’Opa1 restent
inchangés par rapport au contrôle après le traitement avec les deux mutants suggérant que
l’interaction entre Opa1 et Bnip3 est nécessaire pour conduire à la dissociation des oligomères
de la dynamine.
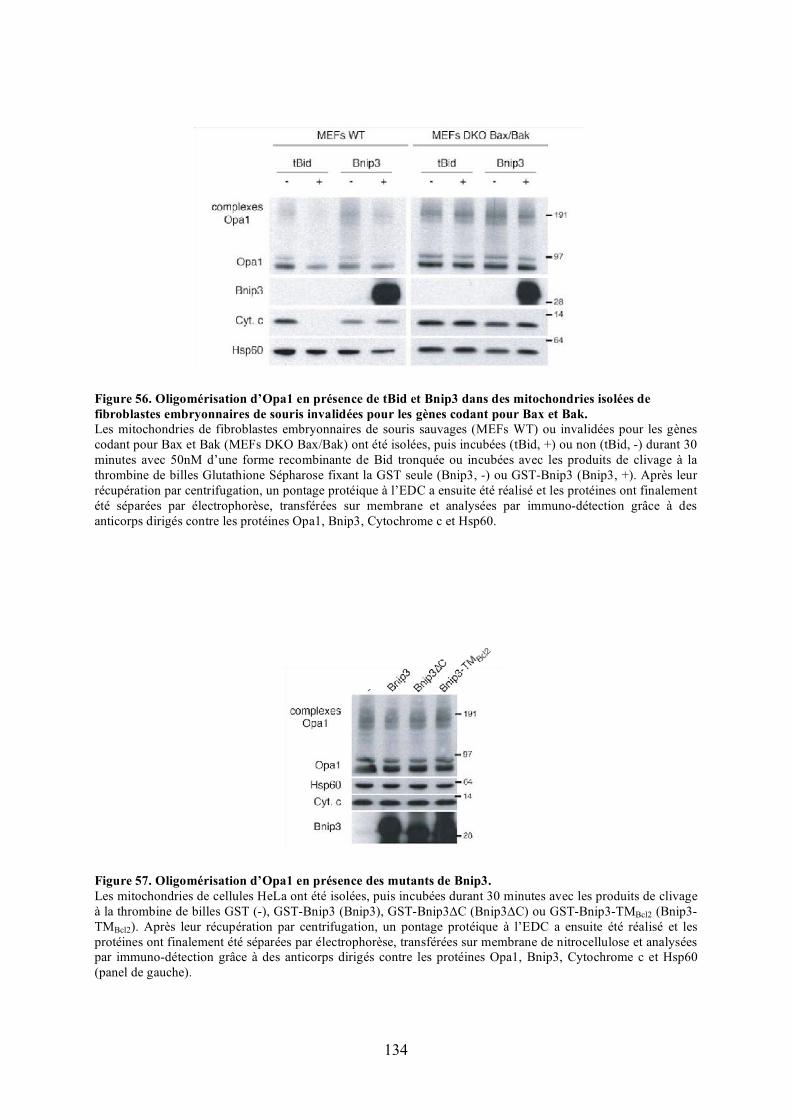
134
Figure 56. Oligomérisation d’Opa1 en présence de tBid et Bnip3 dans des mitochondries isolées de fibroblastes embryonnaires de souris invalidées pour les gènes codant pour Bax et Bak.Les mitochondries de fibroblastes embryonnaires de souris sauvages (MEFs WT) ou invalidées pour les gènes codant pour Bax et Bak (MEFs DKO Bax/Bak) ont été isolées, puis incubées (tBid, +) ou non (tBid, -) durant 30 minutes avec 50nM d’une forme recombinante de Bid tronquée ou incubées avec les produits de clivage à la thrombine de billes Glutathione Sépharose fixant la GST seule (Bnip3, -) ou GST-Bnip3 (Bnip3, +). Après leur récupération par centrifugation, un pontage protéique à l’EDC a ensuite été réalisé et les protéines ont finalement été séparées par électrophorèse, transférées sur membrane et analysées par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1, Bnip3, Cytochrome c et Hsp60.
Figure 57. Oligomérisation d’Opa1 en présence des mutants de Bnip3.Les mitochondries de cellules HeLa ont été isolées, puis incubées durant 30 minutes avec les produits de clivage à la thrombine de billes GST (-), GST-Bnip3 (Bnip3), GST-Bnip3�C (Bnip3�C) ou GST-Bnip3-TMBcl2 (Bnip3-TMBcl2). Après leur récupération par centrifugation, un pontage protéique à l’EDC a ensuite été réalisé et les protéines ont finalement été séparées par électrophorèse, transférées sur membrane de nitrocellulose et analysées par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1, Bnip3, Cytochrome c et Hsp60 (panel de gauche).

135
Les expériences présentées jusqu’ici qui tendent à montrer que Bnip3 est capable de
modifier la conformation des jonctions de crêtes en interagissant avec la dynamine ont toutes
été réalisées sur des mitochondries isolées. Nous avons donc cherché à confirmer nos résultats
dans des cellules en culture. Pour cela, nous avons cultivé des cellules HeLa en présence ou
non de chlorure de cobalt, en éteignant ou non l’expression du gène codant pour Bnip3 par
interférence à l’ARN. Pour cela, comme pour l’extinction de Mfn1, les cellules ont été
nucléofectées juste avant leur ensemencement avec 750 nM d’un mélange d’ARN interférents
dirigés contre Bnip3 ou d’ARN interférents contrôles. Après 48 heures de culture, dont 24
heures en présence de chlorure de cobalt, nous avons isolé les mitochondries de ces cellules
puis procédé au pontage des protéines à l’aide d’EDC. Les protéines ont ensuite été séparées
par électrophorèse, transférées sur membrane de nitrocellulose puis analysées par immuno-
détection (Figure 58). Dans des cellules HeLa, transfectées par des siRNA contrôles ou
dirigés contre Bnip3, mais n’ayant subi aucun traitement, le monomère et les complexes
d’Opa1 sont présents dans des proportions similaires, le rapport quantité de complexes sur
quantité de monomère étant proche de 1 dans les deux cas. Le traitement des cellules au
chlorure de cobalt conduit à une réduction significative d’environ 50% de ce rapport
coïncidant cette fois avec la libération du cytochrome c et d’Opa1 vu la nette diminution de la
quantité totale des deux protéines. Ces deux effets sont dépendants de la présence de Bnip3
puisque l’extinction de l’expression du pro-apoptotique, qui s’élève à plus de 95%, empêche
le désassemblage des complexes d’Opa1 ainsi que la libération dans le cytosol du cytochrome
c et de Opa1. Ces données obtenues in cellulo suggèrent qu’en présence de chlorure de cobalt,
Bnip3 induit la mobilisation du cytochrome c dans l’espace inter-membranaire via la
dissociation des oligomères d’Opa1 puis la libération du cytochrome c .
Nous avons démontré que l’interaction entre Bnip3 et Opa1 conduit à l’inhibition de
manière indépendante des deux activités de la dynamine, à savoir son activité de fusion des
mitochondries et son activité anti-apoptotique. Concernant cette dernière, nous avons pu
démontrer que Bnip3 est capable d’affecter l’oligomérisation de la protéine et ainsi
d’induire, indépendamment d’un rôle éventuel dans la perméabilisation de la membrane
externe mitochondriale, la mobilisation du cytochrome c dans l’espace inter-membranaire.
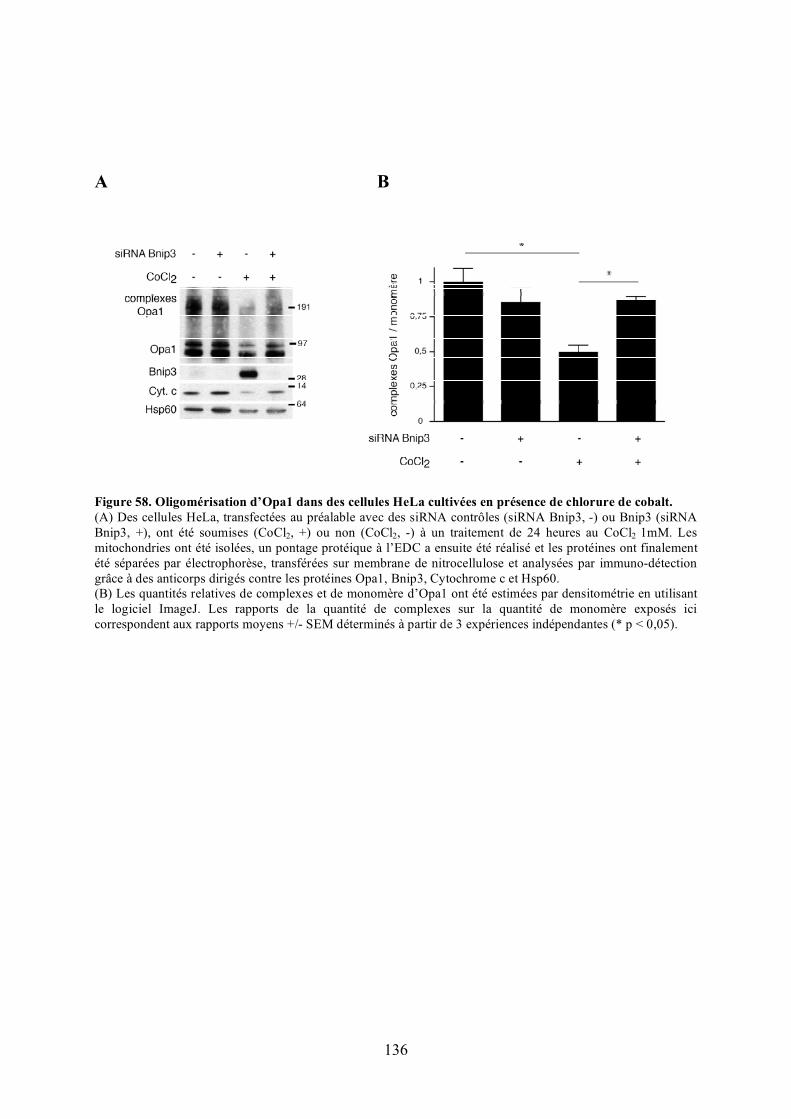
136
A B
Figure 58. Oligomérisation d’Opa1 dans des cellules HeLa cultivées en présence de chlorure de cobalt.(A) Des cellules HeLa, transfectées au préalable avec des siRNA contrôles (siRNA Bnip3, -) ou Bnip3 (siRNA Bnip3, +), ont été soumises (CoCl2, +) ou non (CoCl2, -) à un traitement de 24 heures au CoCl2 1mM. Les mitochondries ont été isolées, un pontage protéique à l’EDC a ensuite été réalisé et les protéines ont finalement été séparées par électrophorèse, transférées sur membrane de nitrocellulose et analysées par immuno-détection grâce à des anticorps dirigés contre les protéines Opa1, Bnip3, Cytochrome c et Hsp60. (B) Les quantités relatives de complexes et de monomère d’Opa1 ont été estimées par densitométrie en utilisant le logiciel ImageJ. Les rapports de la quantité de complexes sur la quantité de monomère exposés ici correspondent aux rapports moyens +/- SEM déterminés à partir de 3 expériences indépendantes (* p < 0,05).

137
III) CONCLUSION
Afin de mieux comprendre le rôle d’Opa1 dans la fusion des mitochondries, dans la
mort cellulaire, nous avons mis en œuvre une recherche de protéines interagissant avec la
dynamine avec, comme stratégie de criblage, le système double-hybride. Cette approche a
finalement permis l’isolement de 9 nouveaux partenaires d’Opa1 de fonctions assez variées. Il
s’agit notamment de facteurs de transcription, de protéines impliquées dans le trafic
membranaire ou de pro-apoptotiques.
Compte tenu, à l’époque où débutait ce travail, de l’émergence d’un lien entre
dynamique mitochondriale et apoptose, nous nous sommes concentrés sur un de ces
partenaires, Bnip3, un membre pro-apoptotique de la famille de Bcl-2. Avant d’étudier les
possibles répercussions fonctionnelles de l’interaction entre Opa1 et Bnip3, nous en avons
vérifié dans un premier temps la validité par d’autres techniques (GST pull-down et co-
immuno-précipitation). La protéine Bnip3, associée à la mitochondrie en condition basale,
s’accumule après un stress cellulaire et s’ancre à la mitochondrie grâce à son domaine trans-
membranaire, ses 10 derniers résidus résidant dans l’espace inter-membranaire (Vande Velde
et al., 2000). Opa1 est ancrée à la membrane interne, la majeure partie de la protéine étant
située dans l’espace inter-membranaire (Griparic et al., 2004; Olichon et al., 2002). Étant
donnée leur topologie, une interaction directe entre les deux protéines pourrait paraître à
première vue difficile. Toutefois, de nombreuses données dans la littérature semblent en
mesure de conforter nos résultats. D’une part, l’association d’Opa1 aux membranes
mitochondriales reste relativement débattue. Différents travaux menés sur la maturation
protéolytique d’Opa1 suggèrent l’existence d’isoformes solubles dans l’espace inter-
membranaire de la protéine (Cipolat et al., 2006; Ishihara et al., 2006). De plus, des
expériences de séparation des deux membranes sur des coussins de sucrose indiquent que
certaines isoformes d’Opa1, et de Mgm1 chez la levure, co-sédimentent avec la membrane
externe (Satoh et al., 2003; Sesaki et al., 2003; Shepard and Yaffe, 1999). Dans ce cas, il n’est
pas exclu que la protéine puisse pénétrer, à l’image de plusieurs protéines virales (Lins et al.,
2008), au sein du feuillet interne de la membrane externe pour se fixer au domaine trans-
membranaire de Bnip3 qui est essentiel pour l’interaction. Des travaux visant à re-étudier la
topologie de la dynamine mais aussi à analyser l’association des différentes isoformes d’Opa1

138

139
avec Bnip3 vont être réalisés. D’autre part, même si leur existence reste discutée, des points
de contact ont été mis en évidence entre la membrane externe et la membrane interne
(Hackenbrock, 1968). Ces domaines ont été proposés comme étant des sites potentiels de
transport de métabolites entre la matrice et le cytoplasme, d’attachement de l’ADN
mitochondrial ou de transfert pour le transport de protéines traduites dans le cytosol reliant les
translocases des membranes externe et interne (Reichert and Neupert, 2002). Leur rôle dans le
couplage de la dynamique fission/fusion des deux membranes et dans la mort cellulaire a
également été suggéré. Bid, un autre BH3-only de la famille de Bcl-2, est retrouvé au niveau
de ces zones de contact. Cette localisation lui permettrait de s’associer à la cardiolipine et de
favoriser la libération du cytochrome c au cours du processus de mort (Kim et al., 2004;
Lutter et al., 2000; Lutter et al., 2001). Il est donc tout à fait concevable que les points de
contact entre la membrane externe et la membrane interne constituent le siège des interactions
entre Opa1 et Bnip3.
Nous avons établi que la surexpression de Bnip3 entraîne la fragmentation du réseau
mitochondrial par inhibition des forces de fusion dans les cellules HeLa. Cet événement
précède l’apparition de dysfonctions mitochondriales (perte du ��m), l’activation des
caspases et la mort des cellules par apoptose. L’ensemble de ces phénotypes ne sont pas
observés lors de la surexpression d’un mutant de Bnip3 ayant perdu la capacité à interagir
avec Opa1 (Bnip3-TMBcl-2) et sont contrebalancés par la surexpression d’Opa1. Ceci nous
permet de proposer que l’effet du pro-apoptotique dans les deux processus, l’apoptose et la
dynamique mitochondriale, repose sur son association avec la dynamine. Les fonctions
fusogène et anti-apoptotique d’Opa1 sont indépendantes l’une de l’autre : la surexpression
ectopique de la dynamine est incapable d’augmenter l’état de fusion des mitochondries dans
des fibroblastes embryonnaires de souris invalidées pour le gène codant pour Mfn1, alors que
l’absence de Mfn1 n’a aucune répercussion sur l’effet anti-apoptotique d’Opa1 (Cipolat et al.,
2004; Frezza et al., 2006). Nous avons utilisé cette propriété pour démontrer, d’une part, que
l’inhibition de la fusion mitochondriale par Bnip3 ne constitue pas à elle seule un signal
apoptotique et, d’autre part, que le « BH3-only » possède deux activités distinctes, dans la
mort cellulaire et dans la dynamique des membranes mitochondriales. Ces observations
viennent s’associer à plusieurs travaux publiés au cours de mes deux dernières années de
thèse visant à clarifier l’impact de la dynamique mitochondriale sur l’apoptose. En effet, au
début de ma thèse, beaucoup d’équipes s’accordaient à exprimer la nécessité de la

140

141
fragmentation des mitochondries dans le déroulement de la voie intrinsèque ou
mitochondriale de ce processus. À l’image de ce que nous avons démontré au cours de ce
travail sur Bnip3, le changement de morphologie pourrait finalement résulter de la capacité
décrite très récemment de certaines protéines de la famille de Bcl-2, dont Bcl-2 elle-même,
Bcl-XL, Bax ou Bak, à réguler l’activité des acteurs de la dynamique mitochondriale (Berman
et al., 2009; Brooks et al., 2007; Delivani et al., 2006; Karbowski et al., 2004; Karbowski et
al., 2006; Li et al., 2008; Sheridan et al., 2008). Si des travaux ont démontré que la
fragmentation des mitochondries n’est pas indispensable au déroulement de la voie
intrinsèque de l’apoptose (Sheridan et al., 2008), le fait que les protéines de la famille de Bcl-
2 aient acquis au cours de l’évolution un rôle supplémentaire dans la dynamique
mitochondriale pourrait suggèrer tout de même l’existence d’un intérêt à modifier la
morphologie des mitochondries pour arriver à la mort cellulaire.
Bnip3 forme avec Nix (ou Bnip3-L) une sous-famille de « BH3-only ». Cette protéine
n’a pas toujours été considérée comme un « BH3-only » à part entière, ce qui lui a valu d’être
laissé pour compte pendant plusieurs années. Néanmoins, elle connaît depuis quelque temps
un net regain d’intérêt car, contrairement à d’autres « BH3-only », le ou les mécanisme(s)
moléculaire(s) par le(s)quel(s) elle induit la mort reste(nt), à l’heure actuelle, totalement
incompris. Bnip3 provoque, selon le type et le micro-environnement cellulaires, des morts
aussi-bien apoptotiques que non-apoptotiques (Burton and Gibson, 2009). Le domaine BH3
de Bnip3, qui diffère de la séquence consensus ((L/V/M)1XXXG5D6(D/E)7F8E9R10) au niveau
notamment d’un résidu hautement conservé (W7 au lieu de D/E7) (Aouacheria et al., 2005), ne
semble que dans quelques cas responsable du pouvoir pro-apoptotique de la protéine et de son
interaction, bien que très faible par rapport aux autres « BH3-only », avec Bcl-2 et Bcl-XL
(Ray et al., 2000; Yasuda et al., 1998). Notre travail constitue une avancée concernant la
compréhension de son rôle pro-apoptotique. L’ensemble de nos données démontrent que
Bnip3 interagit avec Opa1 pour induire l’apoptose. Cette association et l’inhibition de la
dynamine qui en résulte passe par le domaine trans-membranaire et l’extrémité carboxy-
terminale de Bnip3. Nos résultats sont en accord avec plusieurs travaux publiés dans la
littérature durant ma thèse qui désignent ces deux régions de la protéine comme responsables
de son activité pro-apoptotique (Chen et al., 1997; Kim et al., 2002; Ray et al., 2000; Yasuda
et al., 1998). Nous avons démontré qu’une forme recombinante de Bnip3 sauvage,
contrairement aux formes mutantes ayant perdu la capacité à interagir avec Opa1

142

143
(Bnip3TMBcl-2 et Bnip3�C), est capable in organello d’induire la dissociation des oligomères
d’Opa1 connus pour assurer la séquestration du cytochrome c au sein de l’espace intra-crête
(Frezza et al., 2006; Yamaguchi et al., 2008). Il est intéressant de noter que même si une
interaction directe a lieu entre Opa1 et Bnip3, cet effet reste dépendant de la présence des pro-
apoptotiques à multi-domaines Bax et Bak. Des travaux réalisés in vitro, à partir de
membranes artificielles ou liposomes, montrent que Bax est capable d’influencer et de
modifier la conformation des membranes (Antonsson et al., 1997; Basanez et al., 1999;
Basanez et al., 2002). Compte tenu de cela, et du fait que le rôle de Bnip3 semble totalement
indépendant de son domaine BH3, ce que nous chercherons prochainement à confirmer par
l’utilisation de mutants de ce domaine, nous proposons un modèle dans lequel Bax et Bak
favorisent une topologie membranaire propice à l’interaction entre Bnip3 et Opa1.
Nous avons montré que l’interaction entre Opa1 et Bnip3 conduit à la fois à
l’inhibition de la fusion des mitochondries et à l’apoptose. Par quel(s) mécanisme(s) le pro-
apoptotique peut-il contrôler l’activité fusogène d’Opa1 et sa capacité à contrôler la
séquestration du cytochrome c ? Pour assumer ses deux rôles, Opa1 engage des interactions,
homotypiques ou hétérotypiques. En effet, d’une part, la présence d’isoformes longues et
courtes ainsi que leur association avec les Mitofusines sont essentielles pour la fusion des
mitochondries (Cipolat et al., 2004; Guillery et al., 2008; Hoppins et al., 2009; Song et al.,
2007). D’autre part, des isoformes longues et courtes de la protéine oligomérisent pour
permettre le contrôle de la mobilisation du cytochrome c (Frezza et al., 2006; Yamaguchi et
al., 2008). Compte tenu de l’effet démontré au cours de notre étude de Bnip3 sur
l’oligomérisation d’Opa1, la faculté du pro-apoptotique à inhiber la dynamine pourrait reposer
sur la modification de la composition de tels complexes soit directement en exerçant une
compétition moléculaire soit indirectement en altérant l’activité GTPase d’Opa1. En faveur de
ce dernier point, il a récemment été démontré que Bnip3 inhibe en hypoxie la voie de
signalisation mTOR et la croissance cellulaire en interagissant avec la petite GTPase Rheb (Li
et al., 2007). Cette interaction a pour résultat la réduction de la fraction GTP lié, la fraction
active, de Rheb. De plus, nos expériences bien que préliminaires indiquent que l’interaction
entre Bnip3 et Opa1 est fortement diminuée en présence d’une mutation au sein du domaine
GTPase (mutation G300E) de la dynamine, ce qui laisse supposer que l’association des deux
protéines est conditionnée par le statut en GTP d’Opa1. Le manque d’information concernant
ce mutant d’Opa1 ne nous permet cependant d’aller plus loin dans notre raisonnement (la

144

145
mutation pourrait inhiber/stimuler l’activité GTPase ou inhiber/stimuler la fixation du GTP).
De plus, il sera intéressant de déterminer in vitro si le pro-apoptotique affecte la capacité
d’Opa1 à fixer et à hydrolyser le GTP.
Nous avons établi que la surexpression de Bnip3 entraîne la mort des cellules HeLa
par apoptose sans induire l’activation de l’autophagie. Même si le rôle de Bnip3 passe par une
augmentation de sa quantité, le niveau d’expression atteint en condition de surexpression
ectopique pourrait être assez éloigné d’un niveau d’expression physiologique. Pour cette
raison, nous avons décidé d’induire l’expression de Bnip3 par un des inducteurs connus à
savoir l’hypoxie. Dans un premier temps, faute d’enceinte à hypoxie, nous avons utilisé un
mimétique, le chlorure de cobalt utilisé dans la littérature pour induire Bnip3 dans diverses
lignées cellulaires. Nos résultats montre qu’une incubation des cellules HeLa avec cette
drogue entraîne, comme la surexpression ectopique de Bnip3, la libération du cytochrome c et
la mort des cellules. Les travaux en condition d’hypoxie « vraie » viennent juste de débuter.
D’après plusieurs publications, ainsi que d’après certaines expériences préliminaires réalisées
au laboratoire, l’hypoxie « vraie » conduit en premier lieu à une phase d’adaptation cellulaire
(Zhang et al., 2008). Durant cette période, plus ou moins longue suivant le type cellulaire
(environ 72 heures dans des fibroblastes embryonnaires de souris et parfois au delà dans des
cellules HeLa), les cellules tentent de survivre au stress généré en mettant en place un
processus d’autophagie, plus spécifiquement d’autophagie mitochondriale ou mitophagie, la
libération du cytochrome c et la mort ne survenant alors que dans un second temps, une fois
les dommages cellulaires devenus insurmontables. Si Bnip3 est présent et joue un rôle
essentiel lors de cette deuxième phase de mort cellulaire, la protéine est, de façon étonnante
vu son caractère pro-apoptotique, également détectée dès les premières baisses du niveau
d’oxygène (Bruick, 2000). Ainsi, durant plusieurs années, de nombreux groupes ont cherché à
expliquer les raisons de cette présence si précoce de la protéine dans des conditions ne menant
pas en premier lieu à la disparition des cellules. Ce n’est finalement que très récemment que
cette énigme a été élucidée par la mise en évidence d’un rôle de Bnip3 dans l’induction de la
mitophagie. L’établissement de complexes entre Beclin 1, une protéine depuis peu considérée
comme un membre de la famille de Bcl2, et hVps34 est un événement déclencheur de la
formation des autophagosomes (APs) au cours de ce processus (Pattingre et al., 2008). En
absence de stimulus autophagique, cette association est contrôlée grâce à la séquestration de
la Bécline1. En hypoxie, Bnip3 (c’est également le cas pour Bnip3L) par son domaine BH3

146

147
est capable de déplacer les interactions pré-existantes en s’associant avec Bcl2 et BclXL, au
sein de complexes de faible affinité, de libérer la Bécline1 et ainsi de permettre la vésiculation
des APs (Bellot et al., 2009; Zhang et al., 2008). Etant donné le double-jeu de Bnip3, qui
participe à l’adaptation des cellules en hypoxie et au contraire à leur élimination quand les
conditions deviennent trop drastiques, il apparaît intéressant de regarder si l’association entre
Bnip3 et Opa1 a lieu dès les premières heures d’hypoxie et la mise en place de la mitophagie
ou intervient uniquement lors de la mort des cellules quand les conditions deviennent trop
drastiques. Plusieurs données laissent penser que cette interaction est effective dès les
premières baisses en oxygène. En effet, d’une part, nous avons mis en évidence dans des
expériences préliminaires que les mitochondries subissent une fragmentation dépendante de
Bnip3 dès 24 heures d’hypoxie. Ce changement pourrait favoriser l’élimination des
mitochondries en réduisant la taille de ces organites car la formation d’un AP de grande taille
demande beaucoup plus d’énergie que celle d’un AP de plus petite taille (Terman and Brunk,
2004; Terman et al., 2003). D’autre part, des travaux récents réalisés dans des cellules �-
pancréatiques suggèrent une implication essentielle d’Opa1 dans l’élimination des
mitochondries à travers un rôle de senseur donnant aux APs si nécessaire le signal
d’incorporer un organite endommagé (Twig et al., 2008). En effet, l’intégration des
mitochondries au sein des APs est corrélée à une diminution de l’activité d’Opa1. Dans ce
contexte, il est tentant d’imaginer que l’interaction entre Bnip3 et Opa1 rende compétentes les
mitochondries pour leur incorporation au sein des autophagosomes. Pour cela, nous avons
émis une hypothèse de travail que nous nous proposons de vérifier ou d’infirmer. L’existence
de l’interaction entre Bnip3 et Opa1 en hypoxie conduirait à la présence de deux populations
du pro-apoptotique, l’une interagissant avec la dynamine via le domaine trans-membranaire et
l’autre avec Bcl2 et BclXL via le domaine BH3. La destinée de chaque molécule du pro-
apoptotique pourrait être dictée par des modifications post-traductionnelles, des
phosphorylations (Graham et al., 2007) ou des glycosylations (Manka and Millhorn, 2006) de
la protéine ayant été décrites dans la littérature. La dissociation des oligomères d’Opa1 par
Bnip3 serait l’élément déclencheur de l’incorporation des mitochondries dans les APs, la
déplétion du cytochrome c de l’espace intra-crête et sa re-localisation dans l’espace inter-
membranaire, et non dans le cytosol puisque la perméabilisation de la membrane externe
mitochondriale ne semble pas avoir lieu dans ces conditions (Erler et al., 2004), indiquant à la
machinerie d’autophagie que les fonctions métaboliques essentielles au sein d’une
mitochondrie sont irrémédiablement perdues. Ce modèle bien qu’hypothétique est appuyé par

148

149
le fait que, dans différents types cellulaires, lorsque pour diverses raisons les caspases sont
inactivées, les mitochondries ayant libéré le cytochrome c sont sélectivement éliminées par
mitophagie (Xue et al., 2001). Il sera nécessaire d’établir dans un premier temps si, dès la
mise en place de la mitophagie durant les premières heures d’hypoxie, l’oligomérisation
d’Opa1 est affectée. Dans un second temps, l’importance de cette altération dans l’élimination
des mitochondries devra être démontrée. Pour cela, l’étude de l’effet de la mutation Q297V
d’Opa1, qui rend les oligomères de la protéine insensibles au désassemblage au cours de
l’apoptose (Yamaguchi et al., 2008), sur l’intégration des mitochondries au sein des APs
pourrait s’avérer déterminante. Nous pouvons enfin imaginer dans ce modèle que la
disponibilité du cytochrome c dans l’espace inter-membranaire mitochondrial puisse
permettre une réponse plus rapide et efficace de la cellule vis à vis de signaux pro-
apoptotiques lors du passage de la phase d’adaptation à la phase de mort.

150

151
Deuxième Partie
Mutants pathogènes de OPA1

152

153
I) PUBLICATION
Effects of OPA1 mutations on mitochondrial morphology and
apoptosis: relevance to ADOA pathogenesis.

154

155

156
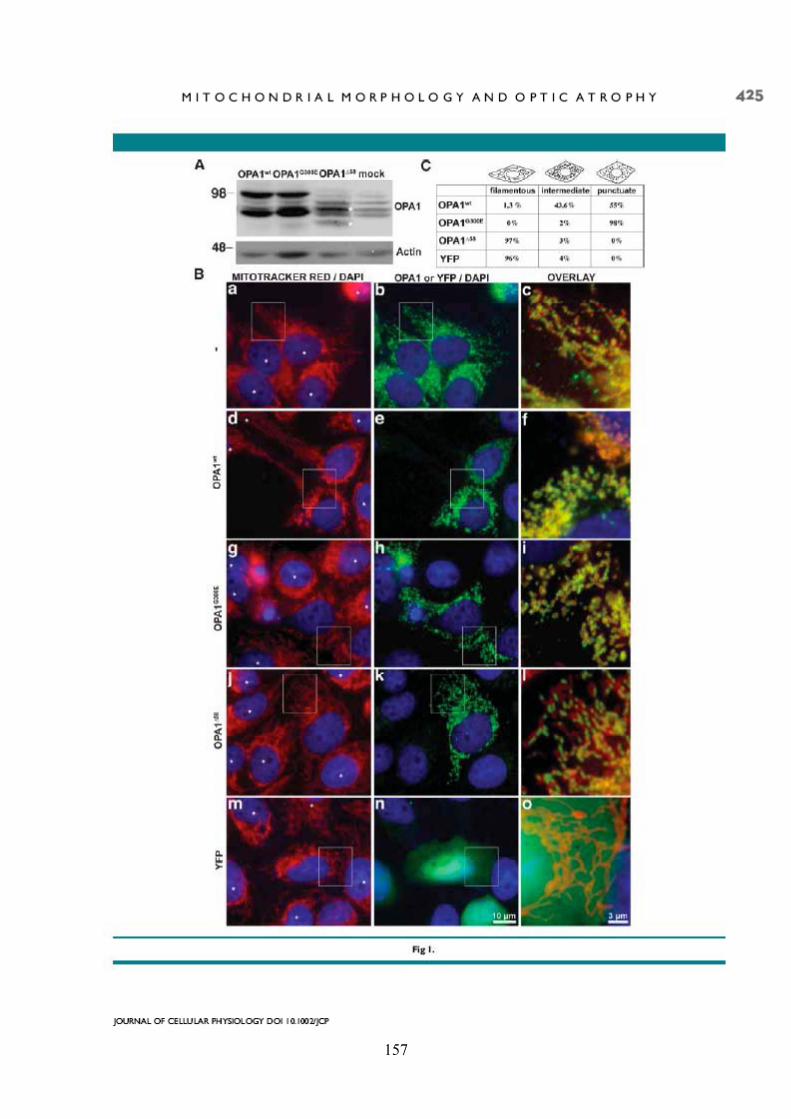
157

158
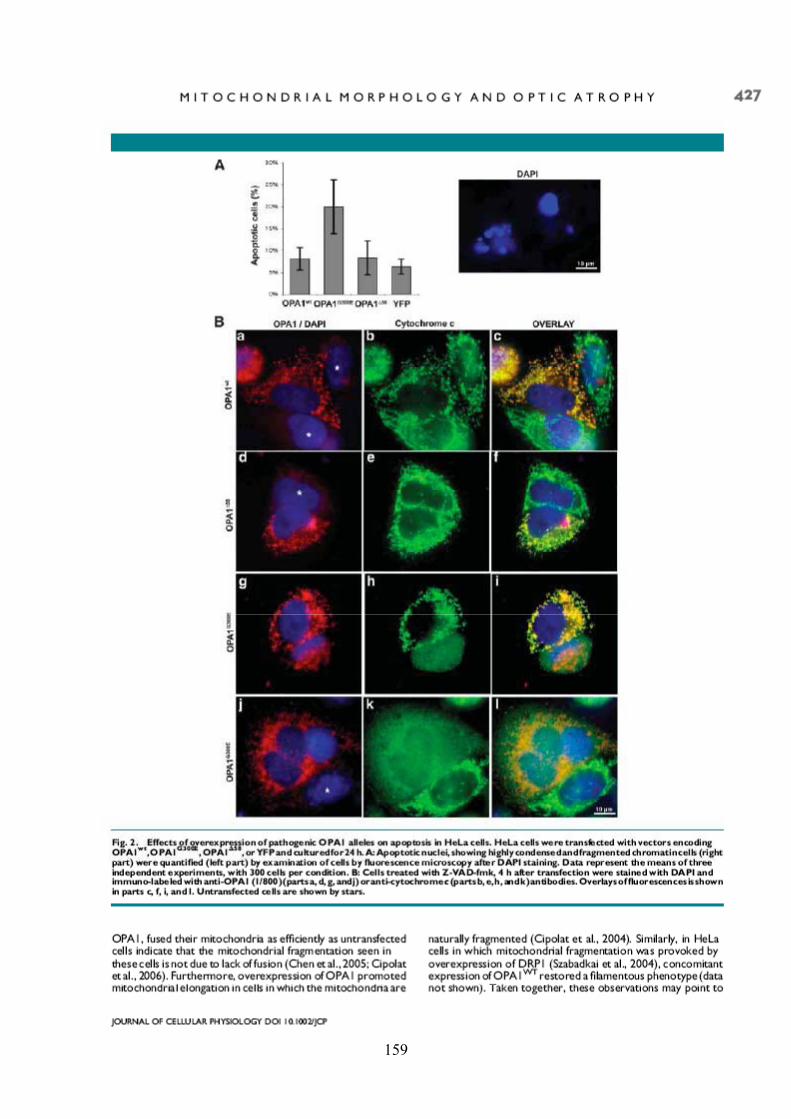
159
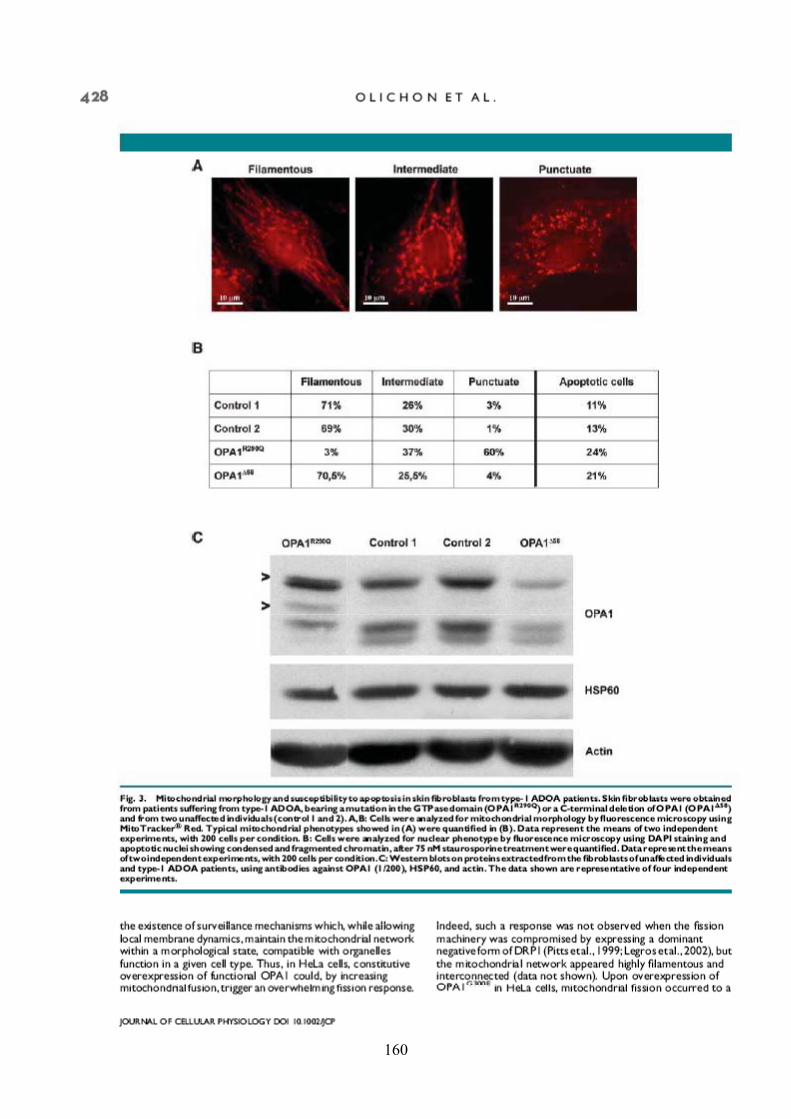
160

161

162

163
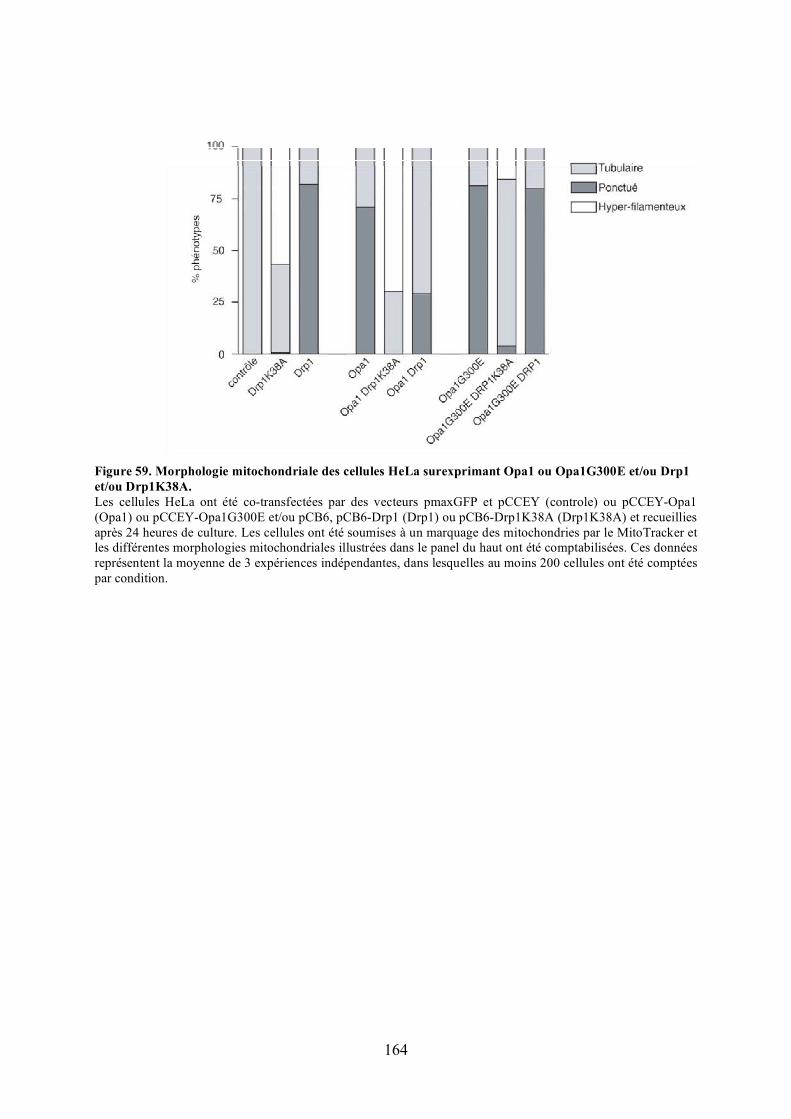
164
Figure 59. Morphologie mitochondriale des cellules HeLa surexprimant Opa1 ou Opa1G300E et/ou Drp1 et/ou Drp1K38A.Les cellules HeLa ont été co-transfectées par des vecteurs pmaxGFP et pCCEY (controle) ou pCCEY-Opa1 (Opa1) ou pCCEY-Opa1G300E et/ou pCB6, pCB6-Drp1 (Drp1) ou pCB6-Drp1K38A (Drp1K38A) et recueillies après 24 heures de culture. Les cellules ont été soumises à un marquage des mitochondries par le MitoTracker et les différentes morphologies mitochondriales illustrées dans le panel du haut ont été comptabilisées. Ces données représentent la moyenne de 3 expériences indépendantes, dans lesquelles au moins 200 cellules ont été comptées par condition.

165
II) RÉSULTATS SUPPLÉMENTAIRES
Les études de gain de fonction d’Opa1 donne des résultats contradictoires. Des travaux
menés dans des fibroblastes embryonnaires de souris montrent que la surexpression ectopique
d’Opa1 entraîne l’allongement des mitochondries. À l’inverse, des cellules HeLa ou Cos7
surexprimant la dynamine présentent un réseau mitochondrial fragmenté. La morphologie des
mitochondries est hétérogène dans les cellules en culture et il semblerait que des cellules
présentant à l’état sauvage un réseau fissionné, comme les fibroblastes embryonnaires de
souris, soient plus propices à l’observation de l’activation de la fusion. La fragmentation
observée dans les autres types cellulaires pourrait résulter d’une stimulation de la fission en
réponse à une activation trop importante de la fusion due à la surexpression. Pour le vérifier,
nous avons co-transfecté des cellules HeLa avec pCCEY-Opa1 ou pCCEY-Opa1G300E et
pCB6-Drp1 ou pCB6-Drp1K38A et observé leur réseau mitochondrial (Figure 59). La
surexpression ectopique de Drp1, d’Opa1 ou d’Opa1G300E entraîne dans la majorité des
cellules la fragmentation du réseau mitochondrial. La surexpression concommitante d’Opa1,
mais pas d’Opa1G300E, dans les cellules surexprimant Drp1 restaure un réseau mitochondrial
filamenteux. La surexpression de Drp1K38A conduit à l’apparition de mitochondries hyper-
filamenteuses et très inter-connectées, s’expliquant par l’accumulation d’évènements de
fusion mitochondriale en absence d’évènements de fission (Smirnova et al., 2001). La
surexpression à la fois de Drp1K38A et du mutant dominant négatif d’Opa1, Opa1G300E, ne
mène pas à l’apparition d’un telle morphologie. L’ensemble de ces résultats montre que la
fission des mitochondries est activée en réponse à une stimulation trop conséquente de la
fusion et permet de lever toute ambiguïté concernant l’implication d’Opa1 dans ce dernier
processus.

166

167
III) RÉSUMÉ ET DISCUSSION DE L’ARTICLE
Des mutations au sein du gène codant pour Opa1 sont responsables chez l’homme
d’une des deux plus fréquentes des neuropathies optiques héréditaires, l’atrophie optique
dominante de type 1 (ADOA). La plupart des mutations sont des délétions ou des
modifications de l’extrémité carboxy-terminale d’Opa1 comportant le domaine
potentiellement GED, reflétant l’importance de cette région de la dynamine pour sa fonction.
La mutation la plus en aval dans la séquence codante induit la délétion des 7 derniers acides
aminés à l’extrémité carboxy-terminale, une altération suffisante pour induire la pathologie.
Les autres mutations sont réparties sur toute la longueur du gène touchant quasiment tous les
exons. Deux mutations, l’une entraînant la délétion totale du gène et l’autre la délétion totale
du cadre ouvert de lecture, conduisent à une réduction de 50% du taux de protéine chez les
patients, ce qui montre qu’une situation d’haplo-insuffisance peut conduire à la pathologie.
Cependant, de nombreuses mutations induisent des substitutions d’acides aminés, la plupart
concentrées dans le domaine GTPase. Ces mutations du domaine catalytique, pourraient
induire une inactivation de la fonction de la protéine et dans ce cas interférer, selon un
mécanisme dominant négatif, avec l’activité de la protéine sauvage codée par l’allèle normal.
En effet, un tel mécanisme a été décrit pour des mutants des dynamines conventionnelles
déficients pour l’activité GTPase (Damke et al., 2001; Marks et al., 2001) ainsi que pour des
mutants des homologues d’Opa1 chez Saccharomyces cerevisiae et Schizosaccharomyces
pombe Mgm1 et Msp1 (Guillou et al., 2005; Shepard and Yaffe, 1999; Wong et al., 2003).
Nos travaux permettent de proposer qu’à la fois des processus de dominance négativité
et d’haplo-insuffisance peuvent être mis en jeu dans l’atrophie optique dominante de type 1.
La surexpression dans les cellules HeLa de l’allèle G300E, muté dans le domaine GTPase,
induit un effet délétère assimilable à une perte totale de la protéine Opa1. En effet, une
fragmentation du réseau mitochondrial et la mort des cellules par apoptose sont observées.
Ces effets s’exercent selon un mode de dominance négativité car la protéine sauvage Opa1 est
exprimée à un taux normal dans les cellules HeLa. Les protéines Opa1 mutées au niveau du
domaine GTPase pourraient donc former des oligomères avec des formes sauvages de la
protéine, et ainsi altérer leur fonction dans la fusion des mitochondries et l’apoptose. Il sera
intéressant d’essayer de confirmer cette hypothèse par des expériences de co-

168

169
immunoprécipitation avec des formes sauvages et mutées de la dynamine. Des résultats
similaires ont été obtenus dans des fibroblastes de peau d’un patient portant une mutation du
domaine GTPase (R290Q). Le réseau mitochondrial apparaît ponctué en comparaison au
réseau filamenteux observé chez des individus contrôles, et les cellules présentent une
sensibilité accrue à l’apoptose induite par la staurosporine.
Le second allèle étudié, tronqué pour la région carboxy-terminale (OPA1�58), n’a pas d’effet
sur la morphologie des mitochondries et sur l’apoptose lors de sa surexpression dans les
cellules HeLa. Il n’exercerait donc pas d’effet dominant négatif sur l’allèle sauvage présent en
quantité normale dans les cellules HeLa. Ceci pourrait s’expliquer par sa plus faible
accumulation dans les cellules, et/ou son incapacité en absence de GED à former des
oligomères avec la protéine sauvage. Les fibroblastes de patients porteurs de la même
mutation présentent comme les cellules HeLa un réseau mitochondrial normal mais sont plus
sensibles à l’apoptose induite par la staurosporine. De façon intéressante, l’analyse par
immuno-détection d’Opa1 dans les fibroblastes mutés a révélé que la seule forme présente de
la protéine correspondait à l’allèle sauvage. L’absence de détection du mutant Opa1�58
associée à sa faible accumulation dans les cellules HeLa nous a donc conduit à proposer que
cette protéine n’était pas stable. Les fibroblastes de patients porteurs de la mutation Opa1�58
ne possèdent donc qu’une dose d’Opa1, ce qui semble affecter la sensibilité à l’apoptose mais
pas la morphologie des mitochondries.
L’ensemble de ces données nous permet de proposer un modèle selon lequel, suivant
les mutations, des mécanismes d’haplo-insuffisance ou de dominance négativité sont mis en
jeux. Les mutations du domaine GTPase exerceraient un effet dominant négatif alors que les
mutations tronquantes de la partie carboxy-terminale ou d’autres mutations déstabilisant la
protéine, agiraient selon un mode d’haplo-insuffisance. Cette hypothèse est renforcée par la
mise en évidence que des mutations d’Opa1 génèrent l’apparition de codons stop prématurés
provoquant une diminution de la quantité d’ARN messagers probablement par un mécanisme
de « non sense decay » au contraire des mutations faux-sens et des mutations respectant le
cadre de lecture pour lesquelles le taux d’ARN messagers muté est identique à celui de l’ARN
sauvage.
Nos travaux permettent de proposer que les cellules ganglionnaires de la rétine
pourraient dégénérer par un processus impliquant l’apoptose comme proposé pour la
neuropathie optique héréditaire de Leber (ou LHON). En effet, des études montrent que des

170

171
cellules cybrides, portant des mutations de l’ADNmt responsables du LHON, présentent une
susceptibilité accrue à l’apoptose induite par le Fas ligand (Danielson et al., 2002), et entrent
en apoptose lorsque l’activité de la chaîne respiratoire est stimulée par le maintien des cellules
dans un milieu dépourvu de glucose (Ghelli et al., 1997). De plus, l’apoptose des cellules
ganglionnaires de la rétine a été démontrée dans d’autres situations pathologiques comme lors
de glaucomes (Kerrigan et al., 1997), d’ischémie de la partie antérieure du nerf optique, située
à proximité directe de la rétine (Levin and Louhab, 1996), ou encore après axotomie du nerf
optique (Bahr, 2000).
D’autres pistes ont été explorées pour expliquer le mécanisme pathogène à l’origine de
l’ADOA, comme l’altération de la forme des mitochondries ou des problèmes énergétiques.
Néanmoins, les fibroblastes de patients porteurs de mutations d’Opa1 présentent un réseau
mitochondrial normal ou fragmenté suivant les études (Chevrollier et al., 2008; Olichon et al.,
2007b; Spinazzi et al., 2008; Zanna et al., 2008). De même, des défauts de phosphorylations
oxydatives ne sont pas toujours révélés ou sont de nature différente. Par conséquent, aucune
image claire n’a pour l’heure été obtenue probablement aussi car peu d’études ont réalisé une
analyse complète (examen de la morphologie mitochondriale, de l’apoptose et bilan
énergétique).
Même si la mise en évidence de formes syndromiques de l’ADOA non restreintes aux
cellules ganglionnaires de la rétine nous incite à en moduler l’importance, une question
restant encore en suspens. Pourquoi les cellules ganglionnaires de la rétine sont-elles
préférentiellement atteintes dans l’ADOA ? Il est possible que les cellules ganglionnaires
soient particulièrement sensibles à des altérations de structure des mitochondries ou à des
dysfonctions mitochondriales. Elles présentent en effet une organisation mitochondriale
structurale et fonctionnelle particulière : le long de leur axone qui émerge vers le nerf optique,
la majorité des mitochondries est regroupée dans des régions dépourvues de myéline, au
niveau desquelles le potentiel d’action est généré. Par conséquent, il est envisageable que ces
regroupements de mitochondries aient une fonction de relais énergétiques. Des altérations
mitochondriales pourraient alors avoir dans les cellules ganglionnaires plus que dans d’autres
types de neurones des conséquences sévères sur le flux axonal ou sur l’activité électro-
physiologique de ces cellules. D’autre part, comme pour d’autres neurones de l’organisme, au
cours de leur maturation, le nombre de cellules ganglionnaires diminue de moitié dès les
premiers mois de la vie consécutivement à une activation de l’apoptose. L’apoptose induite au
cours des neuropathies optiques est-elle une réactivation de ce phénomène ou bien résulte-t-

172

173
elle de l’accumulation de stress particuliers ? En effet, dans le cas de l’ADOA de type I, la
neurodégénérescence est progressive, et des facteurs environnementaux comme l’exposition
au rayonnement UV par exemple pourrait contribuer à la susceptibilité de ces cellules à la
perte de fonction d’Opa1 et conduire à une mort cellulaire par apoptose à partir d’un certain
seuil de dommages. Il est également possible que les proportions des différents variants
d’Opa1 dans ces cellules soient différentes de celles observées dans d’autres tissus. De plus,
des partenaires spécifiques de la protéine, impliquant des mécanismes moléculaires propres à
ces cellules pourraient exister. Leur recherche ou la recherche de régulateurs d’Opa1
spécifiques des cellules ganglionnaires pourrait permettre de comprendre pourquoi cette
pathologie mitochondriale affecte essentiellement les cellules ganglionnaires de la rétine
contrairement à de nombreuses pathologies mitochondriales qui affectent plusieurs types de
tissus. Pour examiner ce dernier point, nous avons recherché des protéines interagissant avec
la dynamine dans une banque d’ADN de rétine totale, faute de pouvoir utiliser une banque de
cellules ganglionnaires. Les 9 partenaires d’Opa1, de fonctions assez variées, ne présentent
pas de spécificité cellulaire ou tissulaire d’expression décrite qui aurait pu nous permettre de
mieux comprendre la territorialité de l’ADOA.

174

175
Discussion Générale

176

177
J’ai participé, au cours de ma thèse, à l’étude de l’effet d’allèles d’OPA1 impliqués
dans l’atrophie optique dominante de type 1 sur la morphologie des mitochondries ainsi que
sur l’apoptose. Ce travail nous a permis de proposer un modèle dans lequel les cellules
ganglionnaires de la rétine chez les patients atteints d’ADOA dégénèrent par apoptose par des
mécanismes mettant en jeu des processus de dominance négativité et d’haplo-insuffisance.
Une fois encore ces travaux ont permis de découpler les fonctions apoptose et dynamique
mitochondriale de Opa1. En effet, des fibroblastes de patients portant la mutation Opa1�58
présentent des mitochondries filamenteuses et sont plus sensibles à l’apoptose. Il serait
intéressant de regarder dans des cellules HeLa surexprimant les deux formes pathogènes
d’Opa1 et dans les fibroblastes de patients correspondants, l’état d’oligomérisation de la
dynamine et ses variations en condition apoptotique. La majeure partie de ma thèse a porté sur
la caractérisation de l’association entre Opa1 et Bnip3, une protéine pro-apoptotique de la
famille de Bcl-2. Nous avons montré que Bnip3 inhibe de façon indépendante les deux
fonctions d’Opa1, dans la fusion des mitochondries et dans l’apoptose. Le fait que des acteurs
majeurs de la voie intrinsèque de l’apoptose régulent indépendamment de leur activité pro-
apoptotique la dynamique des mitochondries suggère que la fragmentation des mitochondries,
bien que n’étant pas un pré-requis essentiel pour mourir, est un événement permettant de
sensibiliser les cellules à un signal de mort.
Bnip3 ayant été impliquée à la fois dans la mort par apoptose lors de conditions
hypoxiques drastiques et dans l’adaptation des cellules par mitophagie lors d’hypoxie moins
sévère et/ou transitoire, il sera intéressant d’étudier les modalités de l’interaction entre Opa1
et Bnip3 dans des cellules ganglionnaires de la rétine. En effet, ces cellules apparaissent
particulièrement exposées et sensibles à des diminutions légères et transitoires de la pression
en oxygène (Kaur et al., 2008; Kergoat et al., 2006). Il est alors envisageable que lors de
variations légères et de courtes durées en oxygène, l’interaction entre Bnip3 et Opa1 favorise
la mitophagie afin de permettre l’adaptation des cellules et qu’au contraire, lors de variations
plus drastiques, elle conduise à l’apoptose. Dans ce contexte, des mutations d’Opa1 affectant
l’interaction avec le pro-apoptotique pourraient perturber la réponse adaptative menant à
l’accumulation progressive de dommages et inévitablement à la mort des cellules. Les
conclusions de l’examen par microscopie électronique de la couche de cellules ganglionnaires
de rétine de souris déficientes pour Opa1 semblent en parfait accord avec nos spéculations. En
effet, une augmentation de la quantité de mitochondries « anormales » assortie d’une
augmentation de la quantité d’autophagosomes caractérise les axones formant le nerf optique

178

179
de ces souris (White et al., 2009). En d’autres mots, bien que la formation des
autophagosomes se déroule normalement (grâce en partie à l’interaction de Bnip3 avec Bcl-2
et Bcl-XL), l’incorporation des mitochondries au sein de ces structures ne semble pas ou peu
s’effectuer (du fait de l’absence d’interaction entre Opa1 et Bnip3 selon notre modèle). La
mise en évidence de l’interaction entre Opa1 et Bnip3 ouvre ainsi des perspectives
intéressantes aussi bien pour améliorer nos connaissances des liens existant entre dynamique
mitochondriale et apoptose, du mode d’action d’un membre BH3-only atypique et peu
caractérisé, que pour mieux comprendre les mécanismes physio-pathologiques conduisant à
l’atrophie optique dominante de type 1 ou le rôle de Bnip3 dans certains cancers.

180

181
Matériel et méthodes

182

183
I) CULTURE CELLULAIRE, TRANSFECTIONS ET TRAITEMENTS
Les cellules HeLa, lignée établie d’un adénocarcinome du col utérin, ainsi que les
fibroblastes embryonnaires de souris ont été cultivées à 37°C en atmosphère humide 95% /
5% CO2 dans du milieu Dulbecco’s Modified Eagle’s Medium (DMEM Gibco) complémenté
en Sérum de veau fœtal (SVF, 10%) et Antibiotiques (Pénicilline 100 U/ml, Streptomycine
0,1mg/ml).
Les cellules ont été transfectées avec des plasmides ou des siARN par la
Lipofectamine 2000 (Invitrogen), la Lipofectine (Invitrogen) ou par nucléofection (Amaxa
Biosystems) suivant les instructions des fournisseurs. Les cellules surexprimant Opa1 de
manière stable et inductible ont été générées à l’aide du Tet-Off Advanced Inducible Gene
Expression System (Clontech). Les clones ont été sélectionnés en présence d’hygromycine B
(100mg/ml) et de généticine (100mg/ml) et ont été cultivés en présence d’hygromycine B, de
généticine et de tétracycline (4mg/ml). Opa1 a été induit en maintenant les cellules en absence
de tétracycline après les avoir lavées 2 fois avec du PBS 1X.
Pour les traitements au chlorure de cobalt (Sigma-Aldrich), 24 heures après
l’ensemencement, les cellules ont été rincées par du PBS 1X puis incubées dans du DMEM–
SVF+Antibiotiques. Le chlorure de cobalt a été préparé extemporanément puis dilué dans le
milieu de culture à une concentration finale de 1mM.
Quand indiqué, les cellules ont été incubées dans une enceinte à hypoxie en présence
de 1% d’oxygène.
Quand cela était nécessaire, les cellules ont été traitées avec du z-VAD-fmk
(Calbiochem), dilué à 100�M dans du DMEM+SVF+Antibiotiques, 4 heures après
transfection.
II) TECHNIQUES UTILISÉES POUR LA MISE EN ÉVIDENCE ET LA
VALIDATION DE L’INTERACTION
A) Double hybride
Nous avons utilisé le système double-hybride MatchmakerTM commercialisé par

184
Clontech (PT1265-1).
1) Les vecteurs d’expression
Dans cette étude, nous avons utilisé les vecteurs d’expression pGBT9 et pGAD424
adaptés à Saccharomyces cerevisiae. Ce sont des vecteurs navettes, et à ce titre, ils portent
une origine de réplication bactérienne et un marqueur de sélection procaryote (gène de
résistance à l’ampicilline). Ces vecteurs possèdent une séquence autonome de réplication (2�)
et un marqueur de sélection levure (LEU2 pour pGAD424 ou TRP1 pour pGBT9) permettant
de complémenter des cellules auxotrophes pour la leucine ou le tryptophane. Ces vecteurs
permettent l’expression constitutive modérée à partir du promoteur ADH1 de protéines
fusionnées au domaine de liaison à l’ADN de GAL4 pour pGBT9 ou au domaine
transactivateur de GAL4 pour pGAD424.
Les sous clonages, dans ces différents vecteurs, des séquences permettant l’expression de
Opa1 ont été réalisé par des techniques classiques de génie génétique. Les vecteurs obtenus
sont répertoriés dans la partie VI C. Des vecteurs contrôles ont également été utilisés:
- pVA3 code pour p53 murin fusionné au domaine de liaison à l’ADN de GAL4
- pTD1 code pour l’AgT de SV40 en fusion au domaine activateur de GAL4.
- pLAM5’ code pour la lamineC humaine en fusion au domaine de liaison à l’ADN de GAL4.
2) Transformation
La transformation des levures par différents vecteurs d’expression a été réalisée par
une technique de chimio-transformation. La souche YGH1 a été utilisée.
- inoculer les cellules dans 20 ml de milieu YPD et incuber pendant 18 à 24 h sous agitation à
30°C jusqu'à DO600nm de 1 à 2.
- diluer la culture pour obtenir 300 ml de milieu YPD à DO de 0,2, et incuber à 30°C sous
agitation pendant 3 heures.
- centrifuger à 1 000 g pendant 5 minutes à température ambiante. Remettre en suspension le
culot dans 25-50 ml d'eau.
- centrifuger à 1 000 g pendant 5 minutes à température ambiante. Remettre le culot cellulaire
en suspension dans 1,5 ml de TE/LiAc 1X.
- à 100 �l de levures compétentes ainsi obtenues, ajouter la solution d’ADN (0.1 �g de
plasmide à transformer + 100 �g d'ADN de sperme de saumon).

185
- ajouter après homogénéisation, 0,6 ml de la solution PEG/LiAc stérile.
- vortexer le mélange de transformation et l’incuber sous agitation à 30°C pendant 30
minutes.
- ajouter 70 �l de DMSO.
- après homogénéisation douce, soumettre les cellules à un choc thermique (42°C pendant 15
minutes).
- après refroidissement dans la glace, centrifuger les cellules à 1 500 g pendant 5 secondes.
Remettre en suspension le culot cellulaire dans 0,5 ml de tampon TE.
- étaler les cellules sur des boîtes contenant le milieu synthétique de sélection approprié, et les
incuber à 30°C jusqu'à l'apparition des colonies.
Milieux:
riche YPD Difco peptone 20 g/l
yeast extract 10 g/l
glucose 20 g/l
adénine 40 mg/l
minimum SD Difco Yeast Nitrogen base without amino acids 6,7 g/l
glucose 20 g/l
adénine 40 mg/l
acides aminées requis ("Drop out " Bio 101)
ajuster le pH à 5,8.
Pour la préparation des des milieux solides, ajouter à la composition, 20g/l d'agar
bactériologique.
Pour la stérilisation des milieux, autoclaver à 115°C pendant 20 minutes.
Solutions:
-TE/LiAc 1X: à faire au moment à partir des solutions TE 10X et LiAc 10X
TE 10X : 0.1 M Tris-HCl, 10 mM EDTA, pH 7.5
LiAc 10X : acétate de lithium 1 M ajusté à pH 7.5 avec de l'acide acétique
- PEG/LiAc: à faire au moment
PEG 4000 40% (polyéthylène glycol PM=3.350 )
TE 1X

186
LiAc 1X
3) Extraction protéique en conditions détergentes
- cultiver les levures dans un volume de 50 ml en phase exponentielle de croissance (DO600nm
de 0,4-0,6),
- centrifuger (2 000 g pendant 5 minutes). Laver le culot cellulaire par 2 ml de tampon de lyse
LB+ complété avec 10% DOC, 10% NP40, 10% Triton 0,2% SDS.
- centrifuger (2 000 g pendant 5 minutes à 4°C). Congeler le culot cellulaire sec dans l’azote
liquide et le conserver à -80°C.
- ajouter pour casser les cellules au volume de culot cellulaire, 200 �l de LB+ et un volume de
billes de verre (425-600 �m, Sigma, G 8772).
- incuber de 4 x 20 secondes à la Fast Prep.
- lorsque le pourcentage de casse cellulaire, estimé après observation au microscope à
contraste de phase, est supérieur à 50%, centrifuger les échantillons pendant 10 minutes à 15
000 g à 4°C. Prélever le surnageant et le re-centrifuger, 2 à 3 fois si nécessaire.
- doser les protéines présentes dans l’extrait selon la méthode Biorad, et les séparer par SDS-
PAGE
Solutions:
- LB+ (tampon de lyse): Tris HCl : 50 mM pH 7,4
NaCl : 250 mM
EDTA : 5 mM
EGTA : 5 mM
Triton : 0,1%
DTT : 1mM
1X inhibiteurs de protéases: (Cocktail Boehringer)
4) Essai ��galactosidase sur filtre
- répliquer sur un papier Whatman rond stérile les cellules à tester, cultivées sur milieu gélosé
approprié. Pour permettre la prolifération cellulaire, poser ce papier sur une nouvelle boîte de
milieu nutritif et incuber 2-3 jours à 30°C.
- perméabiliser les cellules par congélation-décongélation successives (3-4 fois le papier est

187
placé, cellules vers le haut, sur un "bateau" de papier aluminium flottant sur de l'azote liquide
pour la congélation et laissé à température ambiante pour la décongélation).
- déposer sur un papier pré-trempé avec 2 ml de TamponZ/X-Gal, dans une boîte de pétri
vide, le papier Whatman, cellules vers le haut, en évitant de former des bulles d'air entre les
papiers.
- parafilmer la boîte de Pétri et incuber à 30°C.
- contrôler l'apparition/l’intensité de la couleur bleue (30 minutes à 30 heures).
Solutions:
- TamponZ 16,1 g/l Na2HPO4, 7H2O
5,5 g/l NaH2PO4, H2O
0,75 g/l KCl
0,246 g/l MgSO4, 7H2O
Vérifier que le pH est de 7.0.
- Solution stock XGal
Dissoudre le 5-bromo-4-chloro-3-indolyl-�-galactopyranoside (Xgal) (Euromedex B 7150)
dans du N,N-diméthylformamide à la concentration de 20 mg/ml. Conserver à -20° C.
- Solution TamponZ/XGal 100 ml de TamponZ
0,27 ml de �mercaptoéthanol
1,27 ml de solution XGal stock.
5) Préparation de la banque
La banque que nous avons utilisée est une banque donnée par C. Petit (Paris) d’ADNc
de rétine humaine fusionnés avec le domaine activateur de transcription de GAL4 (pGADGE-
ADNc qui diffère du plasmide pGAD424 seulement au niveau du site multiple de clonage) (5
105 clones indépendants).
- inbuber 1 �g de plasmide dans la glace pendant 30 minutes avec des bactéries Top10F’
chimio-compétentes commerciales (109 colonies/�g d’ADN).
- effectuer un choc thermique à 42°C pendant 1 minute 30.
- refroidir les cellules dans la glace pendant 2 minutes.
- ajouter 1 ml de SOC et incuber 1 heure à 37°C sous agitation.
- étaler un aliquot de la transformation sur LB + ampicilline 100 mM (100 �l des dilutions 10-
3 à 10-7) et incuber sur la nuit à 37°C afin de calculer l’efficacité de transformation.

188
Maintenir en milieu liquide à 4°C les bactéries restantes.
- étaler la banque l’après-midi sur milieu sélectif à une densité telle que les colonies soient
quasiment à confluence (20 000–40 000 colonies par boîte de 150 mm de diamètre).
- préparer 37 boîtes afin d’étaler 3 fois le nombre de clones indépendants de la banque
d’ADN. Incuber 18 à 20 heures à 37°C.
- recouvrir chaque boîte de 5 ml de milieu LB contenant 25% glycérol, et remettre en
suspension les cellules dans ce liquide à l’aide d’un « étaleur ».
- centrifuger la suspension obtenue. Éventuellement l’aliquoter et la garder à -70°C.
- extraire l’ADN plasmidique selon les recommendations du fabriquant (MaxiPrep de
Macherey-Nalgen).
6) Transformation de la banque et sélection
- cultiver des levures YGH1 contenant le plasmide pGBT9 Opa1 260-960 dans 100 ml de
milieu minimum dépourvu de tryptophane, sur la nuit à 30°C (DO600nm > 1).
- ensemencer avec cette culture 1 litre de même milieu à DO600nm comprise entre 0,3-0,5. A
- après incubation pendant 4 heures à 30°C sous agitation, centrifuger la culture à 1 500 g
pendant 5 minutes à température ambiante. Remettre en suspension les cellules dans 500 ml
de TE 1X.
- re-centrifuger à 1 500 g pendant 5 minutes à température ambiante. Remettre en suspension
les cellules dans 20 ml de LiAc/TE 1X et incuber 10 minutes à température ambiante.
- ajouter à la suspension de cellules compétentes le mélange contenant 1 à 2 ml d'ADN de
sperme de saumon dénaturé (10 mg/ml) et 250 à 500 �g d'ADN de la banque.
- après homogénéisation, ajouter 140 ml de la solution PEG/LiAc stérile et incuber le mélange
de transformation sous agitation à 30°C pendant 30 minutes.
- ajouter 17,6 ml de DMSO.
- après homogénéisation douce, soumettre les cellules à un choc thermique (42°C pendant 6
minutes).
- après avoir été refroidies dans un bain à température ambiante, centrifuger les cellules à 1
500 g pendant 5 secondes. Remettre en suspension le culot cellulaire dans 50 ml de tampon
TE 1x.
- centrifuger de nouveau les cellules à 1 500 g pendant 5 secondes. Remettre le culot
cellulaire en suspension dans 1 litre de YPD.
- après une incubation de 1 heure à 30°C sous agitation, centrifuger la culture à 1 500 g

189
pendant 5 minutes à température ambiante.
- laver les cellules par 50 ml de TE 1X.
- re-centrifuger et reprendre dans un volume final de 7,5 ml de TE 1x.
- étaler 7,5 �l de suspension cellulaire sur une boîte de milieu minimum dépourvue de leucine
et de tryptophane afin de calculer l'efficacité de transformation.
- étaler 350 �l de la suspension cellulaire sur 21 boîtes de milieu minimum dépourvu de
leucine, de tryptophane, d’histine et contenant 2,5 mM 3 AT (Sigma A 8056). Ce milieu
permet de sélectionner directement les colonies contenant les deux plasmides (portant les
gènes LEU2 et TRP1) et produisant deux protéines de fusion qui interagissent (gène
rapporteur HIS3). Incuber à 30°C, jusqu'à l'apparition des colonies.
- garder les colonies qui poussent sur milieu contenant le 3 AT pendant 8 jours (au-delà, des
colonies apparaissent pour des souches témoins produisant deux protéines qui n’interagissent
pas), et les tester pour leur activité �Gal.
Les colonies HIS+ et �Gal+ sont des colonies recherchées. Elles peuvent contenir un plasmide
pGADGH-ADNc codant pour une protéine interagissant avec notre protéine d’intérêt.
Cependant comme il est possible que plusieurs plasmides pGADGH-ADNc codant pour
différentes protéines hybrides soient entrés dans la même cellule, strier la colonie sélectionnée
pour permettre la ségrégation des plasmides et re-tester la croissance sur milieu +3AT et la
production de �Galactosidase.
7) Elimination des “ faux positifs ”
L’obtention de cellules dépourvues de plasmide pGBT9-OPA1 260-960 mais ayant
conservé le plasmide pGADGH-ADNc à partir des cellules HIS+ �Gal+ permet d’éliminer un
certain nombre de partenaires candidats en vérifiant la nécessité de la présence du plasmide
pGBT9 OPA1 260-960 pour la production des gènes rapporteurs.
Pour cela :
- cultiver les colonies HIS+ �Gal+ dans 2 ml de milieu minimum dépourvu de leucine mais
contenant du tryptophane (la fréquence de perte de plasmide pGBT9-Opa1 260-960 portant le
gène TRP1 est d'environ 10-20%), pendant plusieurs jours avec une dilution quotidienne.
- au bout de 5 jours, étaler 200 cellules sur milieu minimum dépourvu de leucine. Incuber 3

190
jours à 30°C puis répliquer sur milieu minimum sans leucine avec ou sans tryptophane.
- tester les colonies LEU+ TRP- (YGH1 pGAD GE ADNc) pour la prototrophie vis-à-vis de
l'histidine et pour la production de �Galactosidase. À ce stade, garder seulement les cellules
HIS- �GAL-.
Le vecteur pGAD GE ADNC peut être extrait de levures ayant subi l'étape de perte de
pGBT9 Opa1 260-960. Pour cela :
- cultiver les levures dans 2 ml de milieu pendant 1 jour à 30°C.
- après centrifugation, reprendre les cellules par 0,2 ml de tampon de lyse LB dépourvu
d’inhibiteurs de protéases.
- à cette suspension cellulaire ajouter 0,2 ml de phénol/chloroforme et 0,3 g de billes de verre
de 425-600 microns de diamètre (Sigma, G 8772).
- vortexer le mélange pendant 2 minutes à 4°C.
- centrifuger à 15 000 g pendant 5 minutes.
- précipiter le surnageant toute la nuit à -20°C après addition de 1/10 de volume d’acétate de
sodium 3M et 2,5 volumes d'éthanol.
- après une centrifugation de 30 minutes à 15 000 g et à 4°C, laver le culot par 1 ml l'éthanol
70%.
- centrifuger de nouveau. Reprendre le culot par 20 �l de TE 1X.
- utiliser un aliquot de cette préparation pour transformer des bactéries compétentes E.coli
(DH5�).
Les plasmides pGAD GE ADNc sont alors re-transformés dans la levure YGH1 avec
différentes combinaisons et obtenir les résultats suivants :
Transformations Plasmide 1 Plasmide 2 HIS �Gal
1 aucun pGAD-GE-ADNc - Blanc
2 pGBT9 pGAD-GE-ADNc - Blanc
3 pVA3 pGAD-GE-ADNc - Blanc
4 pGBT9- Opa1 260-960 pGAD-GE-ADNc + BLEU
Transformation 1: pour confirmer que pGADGH-ADNc seul ne permet pas la prototrophie à
l’histidine et l’activité �Galactosidase. Garder les HIS- �Gal-.
Transformation 2: une interaction fortuite entre le candidat Y et le domaine de liaison à
l’ADN de GAL4 peut permettre l’activation des gènes rapporteurs. Garder les HIS-�Gal-.

191
Transformation 3: une liaison de la protéine Y au promoteur des gènes rapporteurs
dépendante de la présence de protéines hybrides (sans égard pour leur nature) avec le domaine
de liaison de GAL4 ou dépendante de la structure de la chromatine peut permettre l’existence
de faux positifs de cette catégorie. Garder les HIS- �Gal-.
Transformation 4: cette transformation correspond à celle qui a permis l'isolement du
plasmide à partir de la banque. L’obtention de colonies HIS+ �Gal+ suggère que pGADGH-
ADNc code pour une protéine Y capable de lier spécifiquement la protéine Opa1.
B) GST pull-down
1) Préparation des protéines recombinantes protéines recombinantes.
Le système de fusion à la Glutathion-S-Transferase (GST) a été utilisé pour produire
et purifier les protéines Bnip3 sauvage ou mutées. Ce système permet de produire chez
Escherichia coli des protéines en fusion traductionnelle avec la GST de Schistosoma
japonicus. Dans les vecteurs d’expression pGEX utilisés, l’expression de la protéine de fusion
(GST en partie amino-terminale) est placée sous le contrôle du promoteur lac, inductible par
l’IPTG (isopropyl-�-D-thiogalactoside). Ces protéines de fusion peuvent ensuite être
facilement purifiées par chromatographie d’affinité sur une matrice de glutathion-Sépharose
et si nécessaire éluées par une digestion à la thrombine qui coupe un site situé entre la GST et
la protéine d’intérêt.
a) Production
- cultiver les bactéries BL21 possédant les plasmides pGEX portant ou non les inserts Bnip3,
Bnip3�C ou Bnip3TMBcl-2 dans du milieu LB complémenté avec de l’ampicilline à 37°C.
- induire l’expression à DO=1 par addition de 1mM d’IPTG pendant 4 à 6 heures à 25°C.
b) Purification
- centrifuger la culture à 4 000g pendant 15 minutes à 4°C.
- reprendre le culot de bactéries par du PBS 1X complémenté avec de la benzonase (Merck,
2,5 unités/ml), du lysozyme 20�g/ml et des inhibiteurs de protéases (Roche).
- incuber 30 minutes à 4°C.
- soniquer jusqu’à disparition de la viscosité.
- ajouter du Triton X-100 1% final.

192
- incuber 30 minutes à 4°C.
- centrifuger à 30 000g pendant 30 minutes à 4°C pour séparer le culot, insoluble, du
surnageant soluble.
- incuber sur la nuit à 4°C la fraction soluble avec des billes de Glutathione Sépharose
(Amersham) préalablement équilibrées dans du PBS 1X 1% Triton X-100 et complété avec
des inhibiteurs de protéases (Roche) (centrifuger à 800g pendant 3 minutes à 4°C entre les
lavages).
- conserver à 4°C les billes dans du PBS 1X complémenté avec des inhibiteurs de protéases
(Roche) et de l’azide de sodium 0,02% après des lavages dans du PBS 1X 1% Triton X-100
complémenté avec des inhibiteurs de protéases (Roche) (centrifuger à 800g pendant 3 minutes
à 4°C entre les lavages).
c) Élution
- laver les billes avec du PBS 1X pour enlever les inhibiteurs protéases (centrifuger à 800g
pendant 3 minutes à 4°C entre les lavages).
- incuber 2 heures à 16°C avec agitation dans du PBS 1X complémenté avec de la thrombine
(Amersham, 1 unité/ 100�g de protéine purifiée dans un volume de 100�l).
- centrifuger à 800g pendant 3 minutes à 4°C.
- conserver à 4°C le surnageant contenant la protéine recombinante.
- incuber, si nécessaire, 30 minutes à 4°C avec agitation avec résine de Benzamidine
Sépharose pour enlever la thrombine.
- centrifuger à 1000g pendant 1 minutes à 4°C.
- conserver à 4°C le surnageant contenant la protéine recombinante.
2) Traduction in vitro en présence de méthionine 35S.
Nous avons produit la protéine Opa1 par traduction in vitro en lysat de réticulocytes,
en utilisant le kit de transcription / traduction couplées TNT (Proméga) en suivant les
indications du fabriquant. Nous avons pour cela utilisé le vecteur pcDNA-Opa1 avec la
polymérase T7.
3) Préparation des mitochondries
- gratter les cellules HeLa dans le milieu de culture (10 boîtes de diamètre 100mm à sous-

193
confluence).
- centrifuger à 600g pendant 5 minutes à 4°C.
- rincer les culots cellulaires par du PBS 1X.
- centrifuger à 600g pendant 5 minutes à 4°C.
- remettre en suspension les cellules dans du tampon MB.
- fractionner les cellules au potter (50 dounces).
- centrifuger à 800g pendant 10 minutes à 4°C pour faire sédimenter les cellules non lysées et
les noyaux.
- prélever le surnageant et le centrifuger à 10 000g, 15 min, à 4°C afin de séparer le culot de
mitochondries du cytosol et des membranes dites légères.
- laver le culot de mitochondries par du tampon MB.
- doser les protéines mitochondriales par la méthode de Bradford (Biorad) après solubilisation
par ajout de Triton X-100 0,2% final (obtention d’environ 3mg de protéines).
- Tampon MB mannitol 210 mM
sucrose 70 mM
EDTA 1mM
HEPES 10 mM
inhibiteurs de protéases (Roche)
4) Interaction
Concernant l’extrait mitochondrial, les mitochondries doivent être remises en suspension et
lysées par une incubation de 30 minutes dans le tampon d’interaction 1% Triton X-100. Après
centrifugation à 100 000g pendant 30 minutes à 4°C, les protéines solubles sont diluées 10
fois dans le tampon d’interaction dépourvu de Triton X-100.
- incuber la protéine Opa1 traduite in vitro ou l’extrait mitochondrial avec les billes de GST-
Bnip3 sauvage ou mutée (ou de GST seule) dans le tampon d’interaction 0,1% Triton X-100
pendant 6 heures à 4°C sous agitation.
- laver les billes 3 fois dans le tampon d’interaction 0,1% Triton X-100 (centrifuger à 800g
pendant 3 minutes à 4°C entre les lavages).
- reprendre les billes par un volume de LSB 2X.
- chauffer pendant 10 minutes à 80°C avant SDS-PAGE.

194
- Tampon d’interaction Tris-HCl pH7.5 50 mM
NaCl 50 mM
EDTA 5mM
EGTA 5mM
inhibiteurs de protéases (Roche)
C) Immuno-précipitation
- isoler des mitochondries de cellules HeLa (15 boîtes de diamètre 100mm ensemencées avec
2 millions de cellules 48 heures avant l’expérience).
- les reprendre et les incuber à une concentration de 6 mg/ml dans du tampon MB contenant
un agent pontant, le DithiobisSuccinimidylPropionate (1 mM final, Molecular Probes) durant
2 heures à 4°C.
- arrêter la réaction par ajout de Glycine à une concentration finale de 125 mM.
- centrifuger à 12000g, 5 minutes, à 4°C.
- incuber 15 minutes à 4°C, avec homogénéisation par vortex, les mitochondries dans le
tampon d’interaction 1% Triton X-100.
- soniquer 3 fois 6 secondes à amplitude 40 (Ultrasonic Processor 130 Watts).
- centrifuger à 25 000g, 15 minutes, à 4°C.
- doser les protéines mitochondriales présentes dans la fraction soluble par la méthode de
Bradford (Biorad).
- les diluer dans du tampon d’interaction 0% Triton jusqu’à l’obtention d’une concentration
finale de Triton X-100 égale à 0,25%.
- incuber les échantillons avec les anticorps anti-Opa1, anti-Bnip3 ou anti-HA, durant 2
heures 30 minutes à 4°C sous agitation.
- incuber durant 1 heure 30 minutes à 4°C en présence de billes protéine A Sépharose
préalablement équilibrées dans du tampon d’interaction contenant 0,1% Triton X-100.
- laver 3 fois les billes par du tampon d’interaction 0,1% Triton X-100.
- les reprendre par un volume de tampon LSB.
- chauffer 10 min à 70°C pour séparer les complexes anticorps-antigènes des billes.
- centrifugation à 12000g, 1 minute, à 4°C.
- dénaturer le surnageant à 95°C durant 30 minutes pour rompre les pontages effectués par le

195
DSP.
- Tampon d’interaction Tris-HCl pH7.5 50 mM
NaCl 250 mM
EDTA 5mM
EGTA 5mM
inhibiteurs de protéases (Roche)
III) TECHNIQUES UTILISÉES POUR L’ANALYSE DE LA
DYNAMIQUE MITOCHONDRIALE
A) Marquage des mitochondries
Les cellules HeLa ensemencées sur lamelles de verre sont incubées avant la fixation
dans le milieu de culture pendant 1 heure à 37°C et dans l’obscurité avec le MitoTracker
(Molecular Probes) à une concentration finale de 100 nM.
B) Tests de fusion au polyéthylèneglycol
- co-transfecter par nucléofection des cellules HeLa avec un plasmide codant pour une GFP
adressée à la mitochondrie (pmtQGFP) et un plasmide codant ou non pour Bnip3.
- co-transfecter par nucléofection des cellules HeLa avec un plasmide codant pour une RFP
adressée à la mitochondrie (pmtDsRed2) et un plasmide codant ou non pour Bnip3.
- mélanger immédiatement après la transfection les cellules transfectées suivant un rapport
1 :1.
- ensemencer 1,5 X 105 cellules dans une boîte de culture de diamètre 35mm contenant une
lamelle de verre de diamètre 25mm.
- 24 heures après l’ensemencement, laver 3 fois les cellules avec du DMEM seul afin
d’enlever le sérum de veau fœtal.
- recouvrir la lamelle d’une solution préchauffée à 37°C de polyéthylèneglycol 50% (1g/1ml
DMEM) et incuber 45 secondes à température ambiante.
- aspirer délicatement la solution de polyéthylèneglycol.

196
- laver délicatement 3 fois avec du DMEM complémenté en sérum de veau fœtal et
antibiotiques.
- laver toutes les 10 minutes durant 30 minutes avec du DMEM complémenté en sérum de
veau fœtal et antibiotiques en replaçant à chaque fois les cellules à l’incubateur.
- 8 heures après le traitement au polyéthylèneglycol, fixer les cellules par 3,7% Formaldéhyde
dans PBS 1X pendant 30 minutes à température ambiante.
- plonger la lamelle dans du PBS 1X puis dans de l’eau.
- monter la lamelle sur une lame avec du Mowiol.
C) Photoconversion
1) Préparation
- co-transfecter par nucléofection des cellules HeLa avec un plasmide codant pour la protéine
Dendra adressée à la mitochondrie (pmtDendra) et un plasmide codant ou non pour Bnip3.
- ensemencer, dans des chambres « LabTek », ces cellules avec de l’OptiMEM (Invitrogen),
10% SVF, 2 mM de L-glutamine, et complémenté avec des antibiotiques.
2) Acquisition des données
Cette étape a été réalisée, 24 heures après transfection, à l’aide d’un microscope DMI6000B
(Leica Microsystems, Wetzlar, Germany) avec un objectif 63x et une ouverture numérique de
1,4 couplé à une caméra COOLsnap HQ2 (Ropper Scientific, Tucson, AZ, USA) et équipé
d’une diode laser.
- choisir une cellule transfectée, présentant un marquage fluorescent du réseau mitochondrial,
observable avec le filtre FITC (vert) du microscope,.
- photoconvertir une zone du réseau mitochondrial par une illumination à 405nm de 10
millisecondes avec la diode laser.
- acquérir, avec le filtre TRITC (rouge), des « stacks » tous les 0,3�m et sur une profondeur
de 3�m, immédiatement après la photoconversion puis toutes les deux minutes pendant 40
minutes.
3) Traitement des données

197
Cette étape a été réalisée avec le logiciel Metamorph (Universal Image, Downingtown, PA,
USA).
- définir à partir de la projection des « stacks » acquis immédiatement après l’illumination
laser une région d’intérêt 1 correspondant à la zone photoconvertie ; mesurer ensuite
l’intensité moyenne de fluorescence (IMF) au sein de cette zone à tous les temps, de 0 à 40
minutes.
- définir également une région d’intérêt 2 délimitant la cellule et mesurer l’IMF au sein de
cette zone à tous les temps, de 0 à 40 minutes.
- définir enfin une région d’intérêt 3 correspondant à une zone ne contenant pas la cellule ;
mesurer l’IMF au sein de cette zone à tous les temps, de 0 à 40 minutes.
- soustraire, à chaque temps, l’IMF de la région d’intérêt 3 aux IMF des régions 1 et 2 afin de
s’affranchir du bruit de fond.
- diviser à chaque temps l’IMF ainsi corrigée de la région 1 par l’IMF corrigée de la région 2
afin de s’affranchir de la perte de fluorescence inhérente à l’acquisition (« photobleaching »).
Les valeurs obtenues aux différents temps sont enfin normalisées par rapport à la valeur
obtenue immédiatement après l’illumination laser considérée elle comme le « 100% de
fluorescence ».
IV) TECHNIQUES UTILISÉES POUR L’ANALYSE DE LA MORT
CELLULAIRE
A) Analyse de l’apoptose
1) Observation des noyaux
L’apparition de noyaux présentant un phénotype dit « apoptotique », c’est-à-dire des
noyaux situés au-dessus du plan focal avec une condensation extrême et une fragmentation de
la chromatine, a été observée après un marquage au Hoechst. Pour cela :
- fixer les cellules préalablement ensemencées sur lamelles de verre durant 5 minutes par
3,7% Formaldéhyde dans du PBS 1X.
- perméabiliser par ajout de Triton X-100 à une concentration finale de 0,125% durant 5
minutes à température ambiante.

198
- incuber les lamelles avec 0,5 mg/ml de Hoechst pendant 15 minutes à 37°C.
- rincer au PBS puis à l’eau.
- monter sur lames avec du Mowiol.
2) Observation des oligomères d’Opa1
- incuber ou non 100�g de mitochondries isolées dans le tampon KCl pendant 30 minutes à
30°C, en présence de la protéine tBid recombinante (50 nM) ou des produits de clivage à la
thrombine des billes GST, GST-Bnip3, GST-Bnip3�C ou GST-Bnip3-TMBcl-2.
- centrifuger les échantillons à 12 000g, pendant 5 minutes à 4°C.
- incuber, pendant 30 minutes à 37°C, à une concentration protéique de 10mg/ml, le culot de
mitochondries dans du PBS 1X complémenté avec de l’EDC 10 mM final (Pierce).
- dénaturer 10 minutes à 80°C dans du tampon NuPAGE LDS.
- analyser sur gels pré-coulés NuPAGE Bis-Tris gradient 4%-12% (Invitrogen).
Pour l’analyse des oligomères d’Opa1 à partir de cellules cultivées ou non en présence de
chlorure de cobalt, les mitochondries ont été isolées, incubées directement avec l’EDC et
analysées sur gel d’électrophorèse dans les conditions décrites ci-dessus.
- Tampon KCl Hepes-KOH pH7.4 15mM
KCl 125mM
MgCl24mM
mannitol 210mM
NaH2PO4 5mM
EGTA 0.5mM
succinate 5mM
ATP 2mM
phosphocreatine 10mM
creatine kinase 10μg/ml
1X inhibiteurs de protéases
3) Observation du potentiel de membrane
- ensemencer les cellules dans des chambres « LabTek ».
- préparer une solution à 5�g/ml de JC1 dans 2ml de milieu de culture.

199
- aspirer le milieu de culture et placer les 2ml de milieu + JC1 sur les cellules.
- replacer la boîte à l’incubateur 37°C pendant 20min.
- aspirer le milieu + JC1 et ajouter 2ml de milieu OptiMEM.
- Aller immédiatement observer au microscope (filtres rouge et vert).
B) Analyse de l’autophagie
1) Par microscopie
-transfecter par nucléofection les cellules avec le vecteur codant pour GFP-LC3.
- avant récolte des cellules, les incuber durant 4 heures dans du DMEM en présence de
PepstatineA (10�g/ml) et de E64d (10�g/ml).
- fixer et monter les cellules.
2) Par Western-blot
- avant récolte des cellules, les incuber durant 4 heures dans du DMEM en présence de
PepstatineA (10�g/ml) et de E64d (10�g/ml).
V) TECHNIQUES COURANTES RELATIVES AUX PROTÉINES
A) Extraction de protéines totales cellulaires
- gratter les cellules HeLa dans le milieu de culture (1 boite de diamètre 100mm).
- centrifuger à 600g, 5 minutes, à 4°C.
- rincer les culots au PBS 1X.
- centrifuger à 600g, 5 minutes, à 4°C.
- remettre en suspension les culots dans le tampon de lyse et incuber 30 minutes avec
homogénéisation au vortex toutes les 10 minutes.
- centrifuger à 20 000g, 10 minutes, à 4°C.
- doser les protéines présentes dans la fraction soluble par la méthode de Bradford (Biorad).
- dénaturer les échantillons protéiques à 80°C durant 10 minutes dans du tampon LSB .
- Tampon de lyse Tris HCl 50 mM pH 7,5

200
NaCl 250 mM
EDTA 5 mM
EGTA 5 mM
Triton X-100 0,1%
DTT 1mM
SDS 0,1%
DOC 0,1%
NP40 1%
1X inhibiteurs de protéases: (Cocktail Roche)
- Tampon LSB (Laemmli Sample Buffer)
Tris-HCl pH 6.8 62,5 mM
SDS 2%
�-mercaptoéthanol 5%
glycérol 10%
bleu de bromophénol 0,01%
B) Electrophorèse et immunodétection
Les protéines ont été séparées par électrophorèse sur gel polyacrylamide, en présence
de SDS, et transférées sur membrane de Nitrocellulose. Après saturation dans du TBS 1X,
0,2% Tween, 5% Lait (TBST-Lait) pendant 1 heure, les membranes ont été incubées avec les
anticorps primaires dilués dans du TBST-Lait, sur la nuit, à 4°C, sous agitation. Après 3
lavages TBST et une incubation 1 heure à température ambiante avec des anticorps
secondaires couplés HRP dilués dans TBST-Lait, la révélation a été réalisée par
chimioluminescence (ECL, PerkinElmer Life Sciences).
Quand nécessaire, une analyse densitométrique des signaux obtenus a été réalisée à l’aide
d’ImageJ (National Institutes of Health; http://rsbweb.nih.gov/ij/).
C) Immunofluorescence indirecte
L’ensemble de ce protocole s’effectue à température ambiante.
- ensemencer des cellules sur des lamelles de verre stériles.
- laver les lamelles avec du PBS 1X.

201
- incuber avec 3,7% Formaldéhyde dans du PBS 1X durant 45 minutes.
- laver 2 fois par du PBS 1X.
- incuber pendant 10 minutes dans du PBS 1X 0,25% Triton X-100.
- laver 2 fois par du PBS 1X.
- incuber pendant 1 minute avec du méthanol 100% maintenu à -20°C.
- incuber pendant 20 minutes avec du PBS 1X 3% BSA.
- incuber 1 heure avec les anticorps primaires dilués dans du PBS 1X 3% BSA.
- laver 2 fois avec du PBS 1X 3% BSA.
- incuber 40 minutes avec les anticorps secondaires dilués dans du PBS 1X 3% BSA.
- laver 1 fois avec du PBS 1X 3% BSA.
- laver 1 fois avec du PBS 1X.
- incuber avec du Hoechst dilué dans du PBS 1X (0,5 mg/ml) pendant 10 minutes.
- laver 1 fois avec du PBS 1X.
- laver 1 fois avec de l’eau.
- après séchage, monter les lamelles sur lames avec du Mowiol.
VI) TECHNIQUES COURANTES RELATIVES À L’ADNLes sous-clonages effectués au cours de cette étude ont fait appel à des techniques
classiques de biologie moléculaire (transformation, préparation d’ADN plasmidique,
digestion par des enzymes de restriction, utilisation d’enzymes de modification, séparation de
fragments d’ADN sur gel d’agarose, …) que je ne décrirai pas ici mais qui peuvent être
retrouvées dans des ouvrages de référence comme « Molecular Cloning : a Laboratory
Manual » (Maniatis T., Fritsh EF., Sambrook J.) ou « Current Protocols in Molecular
Biology » (John Wiley & Sons).
A) Clonages et sous-clonages de Bnip3
La séquence pleine taille de Bnip3 a été obtenue par PCR à partir du vecteur pGAD-
GE clonne11 isolé en double hybride, en utilisant les oligonucléotides sens suivants: 5’ AGA
TCT TGC GGC CGC CAT ATG TCG CAG AAC GGA GCG 3’ et antisens: 5’ GAA TTC
TCT AGA TCA AAA GGT GCT GGT GGA GGT TGT CAG ACG ). Le fragment PCR a été
cloné dans le vecteur Topo-pCRII, et sequencé. La séquence codante de Bnip3 a été sous-
clonée dans différents vecteurs d’expression eucaryotes supérieurs grâce aux sites de

202
restriction présents dans les oligonucléotides (NotI-XbaI pour pcDNA-HA, NdeI-EcoRI pour
pGEX2T’6 et BglII-EcoRI pour pEGFP-C1 ou pECFP-C1).
Pour déléter l’extrémité C-Terminal de Bnip3 (aa185-194), nous avons réalisé une i-
PCR sur pcDNA-HA-BNIP3 en utilisant les oligonucléotides suivants : sens: 5’
CTTTTGATCTAGAGAATTCAAGGG 3’ et antisens 5’ CTTCCAATATAGATCCCCAAT
C 3’). La jonction de ces oligonucléotides créé un site HindIII site. La séquence entière de
Bnip3 a été séquencée. Le vecteur pGEX 2T’6 BNIP3�C a été obtenu par remplacement de
l’insert StyI-EcoRI (188 bp) dans pGEX 2T’6 BNIP3.
Pour remplacer le domaine TM de Bnip3 par celui de Bcl-2, nous avons procédé à une
synthèse d’ADN (CCTCAGCATGAGGAACACGAGCGTCATGAAGAAAGGGGGC
ATATTCTCTGCAGAATTTCTGAAAACTCTGCTAAGCTTGGCCCTGGTGGGAGCTT
GCATCACCCTGGGTGCCTATCTGAGCAGGCGCCTGACAACCTCCACCAGCACCTT
TTGACTCTAGA) insérée dans le site SmaI du vecteur pUC57. Les vecteurs pGEX 2T’6
BNIP3 TMBcl-2 et pcDNA-HA-BNIP3 TMBcl-2 ont été obtenus en remplaçant l’insert BbvCI-
XbaI dans pGEX 2T’6 BNIP3 et pcDNA-HA-BNIP3.
B) Sous-clonages d’Opa1
Afin de générer les vecteurs du double hybride produisant des formes tronquées de
Opa1 (vecteurs #1213 à 1221), les séquences codantes pour 4 formes de OPA1 sont préparées
sous forme d’inserts NdeI klenow/BamHI à partir des vecteurs pET15b Opa1 125-960
(#987) , pET15b Opa1 260-960 (#989), pET15b Opa1 125-902 (#1024), pET15b Opa1 260-
902 (#988), et clonées en phase avec les domaines de GAL4 dans les vecteurs du double
hybride pGBT9 ou pGAD424 (#149, #150) ouverts aux sites SmaI et BamHI.
La construction permettant une expression de Opa1 inductible par la tétracycline
(#MC81) est obtenue en sous-clonant la séquence codante de Opa1 sauvage (insert XbaI traité
klenow/SalI obtenu de pCCEY Opa1, #MC72) dans le vecteur pTRE2hyg (#MC27) ouvert
par PvuII.
C) Souches
1) Escherichia coli
#149 : DH5� pGBT9

203
#150 : DH5� pGAD242#987 : DH5� pET15b- OPA1 125-960#988 : DH5� pET15b-OPA1 260-902#989 : DH5� pEt15b-OPA1 260-960#1213 : DH5� pGBT9 OPA1 125-960#1214 : DH5� pGBT9 OPA1 260-960#1215 : DH5� pGBT9 OPA1 125-902#1216 : DH5� pGBT9 OPA1 260-902#1218 : DH5� pGAD424 OPA1 125-960#1219 : DH5� pGAD424 OPA1 260-960#1220 : DH5� pGAD424 OPA1 125-902#1221 : DH5� pGAD424 OPA1 260-902#1024 : DH5� pET15b OPA1 125-902#1035 : DH5� pEGFPN3 OPA1G300E#1042 : DH5� pEGFPN3 OPA1�58#1043 : DH5� pEGFPN3 OPA1#1279 : DH5� pCCEY#1319 : GM2163 pCRII BNIP3#1321 : DH5� pCDNA3-HA BNIP3#1325 : DH5� pGEX2T'6 BNIP3#1369 : DH5� pC-OPA1-A-iresYFP#1372 : DH5� pC-OPA1-A-G300E_iresYFP#1410 : Top10F’ pCRII BNIP3�C#1414 : Top10F’ pGEX2T'6 BNIP3�C#1420 : BL21 codon+ pGEX2T'6 BNIP3#1422 : BL21 codon+ pGEX2T'6 BNIP3�C#1423 : GM2163 pCRII BNIP3�C#1433 : Top10F' pcDNAHA BNIP3�CMC18 : GM 2163 pcDNA3-HA BNIP3 MC19 : GM 2163 pGEX2T6' BNIP3 MC27 : Top 10F' pTRE2hyg, MC65 : Top10F' pUC57-BNIP3 TMBcl-2MC72 : Top10F' pCEY-OPA1 WT variant A MC73 : Top10F' pCEY-OPA1 Delta58 MC77 : Top10F' pcDNA HA BNIP3 TMBcl-2MC81 : Top10F' TRE OPA1 WT variant A MC97 : Top10F' pGEX2T'6 BNIP3 TMBcl-2MC100 : Top10F' pmCeruleanMC101 : Top10F' pmaxGFPMC102 : Top10F' pCB6-mtQGFPMC103 : Top10F' pDsRed2-MitoMC104 : Top10F' pCB6-Myc Drp1wtMC105 : Top10F' pCB6-Myc Drp1 K38AMC135 : Top10F' pEGFPC1 BNIP3 wtMC136 : Top10F' pEGFPC1 BNIP3�CMC137 : Top10F' pEGFPC1 BNIP3 TMBcl-2MC143 : Top10F' pGFP-LC3 MC144 : Top10F' pDendra2-MISMC180 : Top10F' pmcerulean BNIP3

204
2) Saccharomyces cerevisiae
SC#178 souche parentale YGH1 Mat � ura 3-52, his 3-200, lys 2-801, ade 2-101, trp1-901,leu 2-3, gal 4-542, gal 80-538, can-resistant LYS::gal1UAS-gal1 TATA-HIS 3 URA 3::gal1-lacZSC#305 : #178 pGBT9 pGAD424SC#306 : #178 pVA3 pTD1SC#307 : #178 pLAM5' pTD1SC#308 : #178 pGBT9 OPA1 260-960SC#309 : #178 pGBT9 OPA1 260-960 pTD1SC#310 : #178 pGBT9 OPA1 260-960 pGAD424 OPA1 125-960SC#311 : #178 pGBT9 OPA1 260-960 pGAD424 OPA1 260-960SC#312 : #178 pGBT9 OPA1 260-960 pGAD424 OPA1 125-902SC#313 : #178 pGBT9 OPA1 260-960 pGAD424 OPA1 260-902SC#315 : #178 pGBT9 OPA1 260-902SC#316 : #178 pGBT9 OPA1 260-902 pTD1SC#317 : #178 pGBT9 OPA1 260-902 pGAD424 OPA1 125-960SC#318 : #178 pGBT9 OPA1 260-902 pGAD424 OPA1 260-960SC#319 : #178 pGBT9 OPA1 260-902 pGAD424 OPA1 125-902SC#320 : #178 pGBT9 OPA1 260-902 pGAD424 OPA1 260-902SC#329 : #178 pLAM5' pGAD424 OPA1 125-960SC#330 : #178 pLAM5' pGAD424 OPA1 260-960SC#331 : #178 pLAM5' pGAD424 OPA1 125-902SC#332 : #178 pLAM5' pGAD424 OPA1 260-902SC#334 : #178 pGBT9 OPA1 125-960SC#335 : #178 pGBT9 OPA1 125-960 pGAD424 OPA1 125-960SC#336 : #178 pGBT9 OPA1 125-960 pGAD424 OPA1 260-960SC#337 : #178 pGBT9 OPA1 125-960 pGAD424 OPA1 125-902SC#339 : #178 pGBT9 OPA1 125-902SC#340 : #178 pGBT9 OPA1 125-902 pGAD424 OPA1 125-960SC#341 : #178 pGBT9 OPA1 125-902 pGAD424 OPA1 260-960SC#342: #178 pGBT9 OPA1 125-902 pGAD424 OPA1 125-902SC#344: #178 pGAD424 OPA1 125-960SC#345: #178 pGAD424 OPA1 260-960SC#346: #178 pGAD424 OPA1 125-902SC#347: #178 pGAD424 OPA1 260-902
D) siARN
SiARN BNIP3
duplex (01), D-004636-01séquence sens séquence anti-sensGAACUGCACUUCAGCAAUAUU 5’P UAUUGCUGAAGUGCAGUUCUU
SiARN MFN1 (pool de 4 duplex)
duplex (1), D-010670-01

205
séquence sens séquence anti-sensGAAGAGCUCUGUUAUCAAUUU 5’P AUUGAUAACAGAGCUCUUCUU
duplex (2), D-010670-02séquence sens séquence anti-sensGCACAGAUGUCACUACAGAUU 5’P UCUGUAGUGACAUCUGUGCUU
duplex (3), D-010670-03séquence sens séquence anti-sensGAUACUAGCUACUGUGAAAUU 5’P UUUCACAGUAGCUAGUAUCUU
duplex (4), D-010670-04séquence sens séquence anti-sensCUGGAUAGCUGGAUUGAUAUU 5’P UAUCAAUCCAGCUAUCCAGUU
siARN contrôle, D-001210-02-05
séquences non communiquées
E) Anticorps
#2 mouse anti-Hsp60 humaine Sigma, H-3524
#7 rabbit anti-Opa1Laboratoire, SE 3512
#16 mouse anti-BNIP3 clone ANa40Sigma, B7931
#18 mouse anti Cytochrome CPromega, G7421
# 29 mouse anti-HA (clone 12CA5)Roche, 1 666 606
#31 mouse anti-ActineChemicon, MAB1501
#34 mouse anti-BNIP3 Ana40 Abcam, 10433
#43 mouse anti-Opa1BD Biosciences, 612606
# 45 rabbit anti-BNIP3BD Biosciences, 559690
#46 mouse anti-HIF1 alphaBD Biosciences, 610958
#47 mouse anti-Cytochrome cBD Biosciences, 556433
#48 rabbit anti-BaxSanta Cruz, sc-493
#49t rabbit anti-Mfn1don M. Rojo
#58 rabbit anti COXIVCell Signaling, 4850
#61 mouse anti-GFPRoche, 11814460001
#63 rabbit anti-LC3-IICell Signalling, 2775S

206

207
Bibliographie

208

209
Aijaz, S., Erskine, L., Jeffery, G., Bhattacharya, S.S., and Votruba, M. (2004). Developmental expression profile of the optic atrophy gene product: OPA1 is not localized exclusively in the mammalian retinal ganglion cell layer. Invest Ophthalmol Vis Sci 45, 1667-1673.
Akada, M., Crnogorac-Jurcevic, T., Lattimore, S., Mahon, P., Lopes, R., Sunamura, M., Matsuno, S., and Lemoine, N.R. (2005). Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin Cancer Res 11,3094-3101.
Akepati, V.R., Muller, E.C., Otto, A., Strauss, H.M., Portwich, M., and Alexander, C. (2008). Characterization of OPA1 isoforms isolated from mouse tissues. J Neurochem 106, 372-383.
Alavi, M.V., Bette, S., Schimpf, S., Schuettauf, F., Schraermeyer, U., Wehrl, H.F., Ruttiger, L., Beck, S.C., Tonagel, F., Pichler, B.J., et al. (2007). A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 130, 1029-1042.
Alexander, C., Votruba, M., Pesch, U.E., Thiselton, D.L., Mayer, S., Moore, A., Rodriguez, M., Kellner, U., Leo-Kottler, B., Auburger, G., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26, 211-215.
Alirol, E., James, D., Huber, D., Marchetto, A., Vergani, L., Martinou, J.C., and Scorrano, L. (2006). The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell 17, 4593-4605.
Amati-Bonneau, P., Odent, S., Derrien, C., Pasquier, L., Malthiery, Y., Reynier, P., and Bonneau, D. (2003). The association of autosomal dominant optic atrophy and moderate deafness may be due to the R445H mutation in the OPA1 gene. Am J Ophthalmol 136,1170-1171.
Amati-Bonneau, P., Valentino, M.L., Reynier, P., Gallardo, M.E., Bornstein, B., Boissiere, A., Campos, Y., Rivera, H., de la Aleja, J.G., Carroccia, R., et al. (2008). OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain 131, 338-351.
Amutha, B., Gordon, D.M., Gu, Y., and Pain, D. (2004). A novel role of Mgm1p, a dynamin-related GTPase, in ATP synthase assembly and cristae formation/maintenance. Biochem J381, 19-23.
Anderson, S., de Bruijn, M.H., Coulson, A.R., Eperon, I.C., Sanger, F., and Young, I.G. (1982). Complete sequence of bovine mitochondrial DNA. Conserved features of the mammalian mitochondrial genome. J Mol Biol 156, 683-717.
Anderson, S.L., Carton, J.M., Lou, J., Xing, L., and Rubin, B.Y. (1999). Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology 256, 8-14.
Antonsson, B., Conti, F., Ciavatta, A., Montessuit, S., Lewis, S., Martinou, I., Bernasconi, L., Bernard, A., Mermod, J.J., Mazzei, G., et al. (1997). Inhibition of Bax channel-forming

210
activity by Bcl-2. Science 277, 370-372.
Aouacheria, A., Brunet, F., and Gouy, M. (2005). Phylogenomics of life-or-death switches in multicellular animals: Bcl-2, BH3-Only, and BNip families of apoptotic regulators. Mol Biol Evol 22, 2395-2416.
Aoyagi, M., Higashino, F., Yasuda, M., Takahashi, A., Sawada, Y., Totsuka, Y., Kohgo, T., Sano, H., Kobayashi, M., and Shindoh, M. (2003). Nuclear export of adenovirus E4orf6 protein is necessary for its ability to antagonize apoptotic activity of BH3-only proteins. Oncogene 22, 6919-6927.
Arimura, S., Yamamoto, J., Aida, G.P., Nakazono, M., and Tsutsumi, N. (2004). Frequent fusion and fission of plant mitochondria with unequal nucleoid distribution. Proc Natl Acad Sci U S A 101, 7805-7808.
Arnoult, D., Gaume, B., Karbowski, M., Sharpe, J.C., Cecconi, F., and Youle, R.J. (2003). Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 22, 4385-4399.
Arnoult, D., Grodet, A., Lee, Y.J., Estaquier, J., and Blackstone, C. (2005a). Release of OPA1 during Apoptosis Participates in the Rapid and Complete Release of Cytochrome c and Subsequent Mitochondrial Fragmentation. J Biol Chem 280, 35742-35750.
Arnoult, D., Rismanchi, N., Grodet, A., Roberts, R.G., Seeburg, D.P., Estaquier, J., Sheng, M., and Blackstone, C. (2005b). Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol 15, 2112-2118.
Ashkenazi, A., and Dixit, V.M. (1998). Death receptors: signaling and modulation. Science281, 1305-1308.
Bacon, A.L., and Harris, A.L. (2004). Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann Med 36, 530-539.
Bahr, M. (2000). Live or let die - retinal ganglion cell death and survival during development and in the lesioned adult CNS. Trends Neurosci 23, 483-490.
Baines, C.P., Kaiser, R.A., Purcell, N.H., Blair, N.S., Osinska, H., Hambleton, M.A., Brunskill, E.W., Sayen, M.R., Gottlieb, R.A., Dorn, G.W., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658-662.
Baines, C.P., Kaiser, R.A., Sheiko, T., Craigen, W.J., and Molkentin, J.D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9, 550-555.
Barsoum, M.J., Yuan, H., Gerencser, A.A., Liot, G., Kushnareva, Y., Graber, S., Kovacs, I., Lee, W.D., Waggoner, J., Cui, J., et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. Embo J 25, 3900-3911.
Basanez, G., Nechushtan, A., Drozhinin, O., Chanturiya, A., Choe, E., Tutt, S., Wood, K.A., Hsu, Y., Zimmerberg, J., and Youle, R.J. (1999). Bax, but not Bcl-xL, decreases the

211
lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc Natl Acad Sci U S A 96, 5492-5497.
Basanez, G., Sharpe, J.C., Galanis, J., Brandt, T.B., Hardwick, J.M., and Zimmerberg, J. (2002). Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem 277, 49360-49365.
Bashkirov, P.V., Akimov, S.A., Evseev, A.I., Schmid, S.L., Zimmerberg, J., and Frolov, V.A. (2008). GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell 135, 1276-1286.
Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M.A., and Bernardi, P. (2005). Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 280, 18558-18561.
Bellot, G., Garcia-Medina, R., Gounon, P., Chiche, J., Roux, D., Pouyssegur, J., and Mazure, N.M. (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29, 2570-2581.
Bereiter-Hahn, J. (1990). Behavior of mitochondria in the living cell. Int Rev Cytol 122, 1-63.
Bereiter-Hahn, J., and Voth, M. (1994). Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech 27, 198-219.
Berger, K.H., Sogo, L.F., and Yaffe, M.P. (1997). Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J Cell Biol136, 545-553.
Berman, S.B., Chen, Y.B., Qi, B., McCaffery, J.M., Rucker, E.B., 3rd, Goebbels, S., Nave, K.A., Arnold, B.A., Jonas, E.A., Pineda, F.J., et al. (2009). Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol 184, 707-719.
Bette, S., Schlaszus, H., Wissinger, B., Meyermann, R., and Mittelbronn, M. (2005). OPA1,associated with autosomal dominant optic atrophy, is widely expressed in the human brain. Acta Neuropathol 109, 393-399.
Billen, L.P., Kokoski, C.L., Lovell, J.F., Leber, B., and Andrews, D.W. (2008). Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 6, e147.
Blatch, G.L., and Lassle, M. (1999). The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21, 932-939.
Boldogh, I., Vojtov, N., Karmon, S., and Pon, L.A. (1998). Interaction between mitochondria and the actin cytoskeleton in budding yeast requires two integral mitochondrial outer membrane proteins, Mmm1p and Mdm10p. J Cell Biol 141, 1371-1381.
Boldogh, I.R., and Pon, L.A. (2006). Interactions of mitochondria with the actin cytoskeleton. Biochim Biophys Acta 1763, 450-462.
Boyd, J.M., Malstrom, S., Subramanian, T., Venkatesh, L.K., Schaeper, U., Elangovan, B., D'Sa-Eipper, C., and Chinnadurai, G. (1994). Adenovirus E1B 19 kDa and Bcl-2 proteins

212
interact with a common set of cellular proteins. Cell 79, 341-351.
Braschi, E., Zunino, R., and McBride, H.M. (2009). MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep.
Breckenridge, D.G., Kang, B.H., Kokel, D., Mitani, S., Staehelin, L.A., and Xue, D. (2008). Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell 31, 586-597.
Breckenridge, D.G., Kang, B.H., and Xue, D. (2009). Bcl-2 Proteins EGL-1 and CED-9 Do Not Regulate Mitochondrial Fission or Fusion in Caenorhabditis elegans. Curr Biol.
Breckenridge, D.G., Stojanovic, M., Marcellus, R.C., and Shore, G.C. (2003). Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160, 1115-1127.
Brickley, K., Smith, M.J., Beck, M., and Stephenson, F.A. (2005). GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem 280, 14723-14732.
Broekemeier, K.M., Carpenter-Deyo, L., Reed, D.J., and Pfeiffer, D.R. (1992). Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett 304, 192-194.
Broekemeier, K.M., and Pfeiffer, D.R. (1989). Cyclosporin A-sensitive and insensitive mechanisms produce the permeability transition in mitochondria. Biochem Biophys Res Commun 163, 561-566.
Brooks, C., Wei, Q., Feng, L., Dong, G., Tao, Y., Mei, L., Xie, Z.J., and Dong, Z. (2007). Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A 104, 11649-11654.
Bruick, R.K. (2000). Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A 97, 9082-9087.
Burgess, S.M., Delannoy, M., and Jensen, R.E. (1994). MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J Cell Biol 126, 1375-1391.
Burton, T.R., and Gibson, S.B. (2009). The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ 16, 515-523.
Campello, S., Lacalle, R.A., Bettella, M., Manes, S., Scorrano, L., and Viola, A. (2006). Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J Exp Med 203,2879-2886.
Cartoni, R., and Martinou, J.C. (2009). Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol 218, 268-273.
Cassidy-Stone, A., Chipuk, J.E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., Kurth, M.J., Shaw, J.T., Hinshaw, J.E., Green, D.R., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer

213
membrane permeabilization. Dev Cell 14, 193-204.
Cereghetti, G.M., Stangherlin, A., Martins de Brito, O., Chang, C.R., Blackstone, C., Bernardi, P., and Scorrano, L. (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105, 15803-15808.
Cerveny, K.L., Studer, S.L., Jensen, R.E., and Sesaki, H. (2007). Yeast mitochondrial division and distribution require the cortical num1 protein. Dev Cell 12, 363-375.
Chang, C.R., and Blackstone, C. (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282, 21583-21587.
Chang, D.T., Honick, A.S., and Reynolds, I.J. (2006). Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26, 7035-7045.
Chen, G., Ray, R., Dubik, D., Shi, L., Cizeau, J., Bleackley, R.C., Saxena, S., Gietz, R.D., and Greenberg, A.H. (1997). The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med 186, 1975-1983.
Chen, H., Chomyn, A., and Chan, D.C. (2005). Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280, 26185-26192.
Chen, H., Detmer, S.A., Ewald, A.J., Griffin, E.E., Fraser, S.E., and Chan, D.C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160, 189-200.
Chen, H., McCaffery, J.M., and Chan, D.C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548-562.
Chen, Y.J., Zhang, P., Egelman, E.H., and Hinshaw, J.E. (2004). The stalk region of dynamin drives the constriction of dynamin tubes. Nat Struct Mol Biol 11, 574-575.
Chevrollier, A., Guillet, V., Loiseau, D., Gueguen, N., de Crescenzo, M.A., Verny, C., Ferre, M., Dollfus, H., Odent, S., Milea, D., et al. (2008). Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann Neurol 63, 794-798.
Cho, D.H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., and Lipton, S.A. (2009). S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102-105.
Choi, S.Y., Huang, P., Jenkins, G.M., Chan, D.C., Schiller, J., and Frohman, M.A. (2006). A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 8, 1255-1262.
Cipolat, S., de Brito, O.M., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 101, 15927-15932.
Cipolat, S., Rudka, T., Hartmann, D., Costa, V., Serneels, L., Craessaerts, K., Metzger, K., Frezza, C., Annaert, W., D'Adamio, L., et al. (2006). Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling.

214
Cell 126, 163-175.
Clark, S.G., Shurland, D.L., Meyerowitz, E.M., Bargmann, C.I., and van der Bliek, A.M. (1997). A dynamin GTPase mutation causes a rapid and reversible temperature- inducible locomotion defect in C. elegans. Proc Natl Acad Sci U S A 94, 10438-10443.
Cohen, M.M., Leboucher, G.P., Livnat-Levanon, N., Glickman, M.H., and Weissman, A.M. (2008). Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell 19, 2457-2464.
Cribbs, J.T., and Strack, S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8, 939-944.
Crowley-Weber, C.L., Payne, C.M., Gleason-Guzman, M., Watts, G.S., Futscher, B., Waltmire, C.N., Crowley, C., Dvorakova, K., Bernstein, C., Craven, M., et al. (2002). Development and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholate. Carcinogenesis 23, 2063-2080.
Cuddeback, S.M., Yamaguchi, H., Komatsu, K., Miyashita, T., Yamada, M., Wu, C., Singh, S., and Wang, H.G. (2001). Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J Biol Chem 276, 20559-20565.
Cuesta, A., Pedrola, L., Sevilla, T., Garcia-Planells, J., Chumillas, M.J., Mayordomo, F., LeGuern, E., Marin, I., Vilchez, J.J., and Palau, F. (2002). The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet 30, 22-25.
Da Cruz, S., Parone, P.A., Gonzalo, P., Bienvenut, W.V., Tondera, D., Jourdain, A., Quadroni, M., and Martinou, J.C. (2008). SLP-2 interacts with prohibitins in the mitochondrial inner membrane and contributes to their stability. Biochim Biophys Acta1783, 904-911.
Daido, S., Kanzawa, T., Yamamoto, A., Takeuchi, H., Kondo, Y., and Kondo, S. (2004). Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 64, 4286-4293.
Damke, H., Muhlberg, A.B., Sever, S., Sholly, S., Warnock, D.E., and Schmid, S.L. (2001). Expression, purification, and functional assays for self-association of dynamin-1. MethodsEnzymol 329, 447-457.
Danielson, S.R., Wong, A., Carelli, V., Martinuzzi, A., Schapira, A.H., and Cortopassi, G.A. (2002). Cells bearing mutations causing Leber's hereditary optic neuropathy are sensitized to Fas-Induced apoptosis. J Biol Chem 277, 5810-5815.
Davies, V.J., Hollins, A.J., Piechota, M.J., Yip, W., Davies, J.R., White, K.E., Nicols, P.P., Boulton, M.E., and Votruba, M. (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet 16, 1307-1318.
de Brito, O.M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to

215
mitochondria. Nature 456, 605-610.
de Bruin, E.C., and Medema, J.P. (2008). Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat Rev 34, 737-749.
De Vos, K., Goossens, V., Boone, E., Vercammen, D., Vancompernolle, K., Vandenabeele, P., Haegeman, G., Fiers, W., and Grooten, J. (1998). The 55-kDa tumor necrosis factor receptor induces clustering of mitochondria through its membrane-proximal region. J Biol Chem 273, 9673-9680.
Delettre, C., Griffoin, J.M., Kaplan, J., Dollfus, H., Lorenz, B., Faivre, L., Lenaers, G., Belenguer, P., and Hamel, C.P. (2001). Mutation spectrum and splicing variants in the OPA1 gene. Human Genetics 109, 584-591.
Delettre, C., Lenaers, G., Griffoin, J.M., Gigarel, N., Lorenzo, C., Belenguer, P., Pelloquin, L., Grosgeorge, J., Turc-Carel, C., Perret, E., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet26, 207-210.
Delivani, P., Adrain, C., Taylor, R.C., Duriez, P.J., and Martin, S.J. (2006). Role for CED-9and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell 21, 761-773.
Detmer, S.A., and Chan, D.C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol176, 405-414.
Dijkers, P.F., Medema, R.H., Lammers, J.W., Koenderman, L., and Coffer, P.J. (2000). Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 10, 1201-1204.
Dimauro, S., and Davidzon, G. (2005). Mitochondrial DNA and disease. Ann Med 37, 222-232.
Dohm, J.A., Lee, S.J., Hardwick, J.M., Hill, R.B., and Gittis, A.G. (2004). Cytosolic domain of the human mitochondrial fission protein fis1 adopts a TPR fold. Proteins 54, 153-156.
Duvezin-Caubet, S., Jagasia, R., Wagener, J., Hofmann, S., Trifunovic, A., Hansson, A., Chomyn, A., Bauer, M.F., Attardi, G., Larsson, N.G., et al. (2006). Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem 281, 37972-37979.
Duvezin-Caubet, S., Koppen, M., Wagener, J., Zick, M., Israel, L., Bernacchia, A., Jagasia, R., Rugarli, E.I., Imhof, A., Neupert, W., et al. (2007). OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell 18, 3582-3590.
Elmore, S.P., Qian, T., Grissom, S.F., and Lemasters, J.J. (2001). The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 15, 2286-2287.
Erler, J.T., Cawthorne, C.J., Williams, K.J., Koritzinsky, M., Wouters, B.G., Wilson, C., Miller, C., Demonacos, C., Stratford, I.J., and Dive, C. (2004). Hypoxia-mediated

216
down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol24, 2875-2889.
Escobar-Henriques, M., Westermann, B., and Langer, T. (2006). Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J Cell Biol 173, 645-650.
Estaquier, J., and Arnoult, D. (2007). Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 14,1086-1094.
Eura, Y., Ishihara, N., Oka, T., and Mihara, K. (2006). Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J Cell Sci119, 4913-4925.
Farkasovsky, M., and Kuntzel, H. (1995). Yeast Num1p associates with the mother cell cortex during S/G2 phase and affects microtubular functions. J Cell Biol 131, 1003-1014.
Fehrenbacher, K.L., Yang, H.C., Gay, A.C., Huckaba, T.M., and Pon, L.A. (2004). Live Cell Imaging of Mitochondrial Movement along Actin Cables in Budding Yeast. Curr Biol 14,1996-2004.
Ferraris, S., Clark, S., Garelli, E., Davidzon, G., Moore, S.A., Kardon, R.H., Bienstock, R.J., Longley, M.J., Mancuso, M., Gutierrez Rios, P., et al. (2008). Progressive external ophthalmoplegia and vision and hearing loss in a patient with mutations in POLG2 and OPA1. Arch Neurol 65, 125-131.
Ferre, M., Amati-Bonneau, P., Tourmen, Y., Malthiery, Y., and Reynier, P. (2005). eOPA1: an online database for OPA1 mutations. Hum Mutat 25, 423-428.
Fields, S., and Song, O. (1989). A novel genetic system to detect protein-protein interactions. Nature 340, 245-246.
Forte, M., and Bernardi, P. (2006). The permeability transition and BCL-2 family proteins in apoptosis: co-conspirators or independent agents? Cell Death Differ 13, 1287-1290.
Frank, S., Gaume, B., Bergmann-Leitner, E.S., Leitner, W.W., Robert, E.G., Catez, F., Smith, C.L., and Youle, R.J. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1, 515-525.
Fransson, A., Ruusala, A., and Aspenstrom, P. (2003). Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278, 6495-6502.
Frey, T.G., and Mannella, C.A. (2000). The internal structure of mitochondria. Trends Biochem Sci 25, 319-324.
Frezza, C., Cipolat, S., Martins de Brito, O., Micaroni, M., Beznoussenko, G.V., Rudka, T., Bartoli, D., Polishuck, R.S., Danial, N.N., De Strooper, B., et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177-189.
Fritz, S., Weinbach, N., and Westermann, B. (2003). Mdm30 is an F-box protein required for

217
maintenance of fusion-competent mitochondria in yeast. Mol Biol Cell 14, 2303-2313.
Fukushima, N.H., Brisch, E., Keegan, B.R., Bleazard, W., and Shaw, J.M. (2001). The GTPase effector domain sequence of the Dnm1p GTPase regulates self-assembly and controls a rate-limiting step in mitochondrial fission. Mol Biol Cell 12, 2756-2766.
Gallenne, T., Gautier, F., Oliver, L., Hervouet, E., Noel, B., Hickman, J.A., Geneste, O., Cartron, P.F., Vallette, F.M., Manon, S., et al. (2009). Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J Cell Biol 185, 279-290.
Gallop, J.L., Jao, C.C., Kent, H.M., Butler, P.J., Evans, P.R., Langen, R., and McMahon, H.T. (2006). Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J25, 2898-2910.
Ghelli, A., Degli Esposti, M., Carelli, V., and Lenaz, G. (1997). Changes in mitochondrial complex I activity and coenzyme Q binding site in Leber's hereditary optic neuropathy (LHON). Mol Aspects Med 18 Suppl, S263-267.
Giatromanolaki, A., Koukourakis, M.I., Sowter, H.M., Sivridis, E., Gibson, S., Gatter, K.C., and Harris, A.L. (2004). BNIP3 expression is linked with hypoxia-regulated protein expression and with poor prognosis in non-small cell lung cancer. Clin Cancer Res 10,5566-5571.
Glater, E.E., Megeath, L.J., Stowers, R.S., and Schwarz, T.L. (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173, 545-557.
Gold, E.S., Underhill, D.M., Morrissette, N.S., Guo, J., McNiven, M.A., and Aderem, A. (1999). Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 190, 1849-1856.
Gonzalvez, F., and Gottlieb, E. (2007). Cardiolipin: setting the beat of apoptosis. Apoptosis12, 877-885.
Graham, R.M., Thompson, J.W., Wei, J., Bishopric, N.H., and Webster, K.A. (2007). Regulation of Bnip3 death pathways by calcium, phosphorylation, and hypoxia-reoxygenation. Antioxid Redox Signal 9, 1309-1315.
Griffin, E.E., Graumann, J., and Chan, D.C. (2005). The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol170, 237-248.
Griparic, L., Kanazawa, T., and van der Bliek, A.M. (2007). Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol 178, 757-764.
Griparic, L., van der Wel, N.N., Orozco, I.J., Peters, P.J., and van der Bliek, A.M. (2004). Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279, 18792-18798.
Guillery, O., Malka, F., Landes, T., Guillou, E., Blackstone, C., Lombes, A., Belenguer, P., Arnoult, D., and Rojo, M. (2008). Metalloprotease-mediated OPA1 processing

218
is modulated by the mitochondrial membrane potential. Biol Cell 100, 315-325.
Guillou, E., Bousquet, C., Daloyau, M., Emorine, L.J., and Belenguer, P. (2005). Msp1p is an intermembrane space dynamin-related protein that mediates mitochondrial fusion in a Dnm1p-dependent manner in S. pombe. FEBS Lett 579, 1109-1116.
Guo, K., Searfoss, G., Krolikowski, D., Pagnoni, M., Franks, C., Clark, K., Yu, K.T., Jaye, M., and Ivashchenko, Y. (2001). Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ 8, 367-376.
Guo, X., Macleod, G.T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M., Marin, L., Charlton, M.P., Atwood, H.L., and Zinsmaier, K.E. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379-393.
Gurskaya, N.G., Verkhusha, V.V., Shcheglov, A.S., Staroverov, D.B., Chepurnykh, T.V., Fradkov, A.F., Lukyanov, S., and Lukyanov, K.A. (2006). Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol 24,461-465.
Hackenbrock, C. (1966). Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. Journal of Cell Biology 30, 269-297.
Hackenbrock, C.R. (1968). Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc Natl Acad Sci U S A 61, 598-605.
Hajek, P., Chomyn, A., and Attardi, G. (2007). Identification of a novel mitochondrial complex containing mitofusin 2 and stomatin-like protein 2. J Biol Chem 282, 5670-5681.
Hales, K.G., and Fuller, M.T. (1997). Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 90, 121-129.
Han, X.J., Lu, Y.F., Li, S.A., Kaitsuka, T., Sato, Y., Tomizawa, K., Nairn, A.C., Takei, K., Matsui, H., and Matsushita, M. (2008). CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182, 573-585.
Harder, Z., Zunino, R., and McBride, H. (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14, 340-345.
Hegde, R., Srinivasula, S.M., Zhang, Z., Wassell, R., Mukattash, R., Cilenti, L., DuBois, G., Lazebnik, Y., Zervos, A.S., Fernandes-Alnemri, T., et al. (2002). Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem 277, 432-438.
Henley, J.R., Krueger, E.W.A., Oswald, B.J., and McNiven, M.A. (1998). Dynamin-mediated internalization of caveolae. J Cell Biol 141, 85-99.
Herlan, M., Vogel, F., Bornhovd, C., Neupert, W., and Reichert, A.S. (2003). Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem 278, 27781-27788.
Hinshaw, J.E., and Schmid, S.L. (1995). Dynamin self-assembles into rings suggesting a

219
mechanism for coated vesicle budding. Nature 374, 190-192.
Hollenbeck, P.J., and Saxton, W.M. (2005). The axonal transport of mitochondria. J Cell Sci118, 5411-5419.
Hoog, J.L., Schwartz, C., Noon, A.T., O'Toole, E.T., Mastronarde, D.N., McIntosh, J.R., and Antony, C. (2007). Organization of interphase microtubules in fission yeast analyzed by electron tomography. Dev Cell 12, 349-361.
Hoppins, S., Horner, J., Song, C., McCaffery, J.M., and Nunnari, J. (2009). Mitochondrial outer and inner membrane fusion requires a modified carrier protein. J Cell Biol 184, 569-581.
Horvitz, H.R. (1999). Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res 59, 1701s-1706s.
Hsu, Y.T., Wolter, K.G., and Youle, R.J. (1997). Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A 94, 3668-3672.
Hudson, G., Amati-Bonneau, P., Blakely, E.L., Stewart, J.D., He, L., Schaefer, A.M., Griffiths, P.G., Ahlqvist, K., Suomalainen, A., Reynier, P., et al. (2008). Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain131, 329-337.
Ingerman, E., Perkins, E.M., Marino, M., Mears, J.A., McCaffery, J.M., Hinshaw, J.E., and Nunnari, J. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 170, 1021-1027.
Irvine, R.A., Adachi, N., Shibata, D.K., Cassell, G.D., Yu, K., Karanjawala, Z.E., Hsieh, C.L., and Lieber, M.R. (2005). Generation and characterization of endonuclease G null mice. Mol Cell Biol 25, 294-302.
Ishihara, N., Eura, Y., and Mihara, K. (2004). Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117, 6535-6546.
Ishihara, N., Fujita, Y., Oka, T., and Mihara, K. (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. Embo J 25, 2966-2977.
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S.O., Masuda, K., Otera, H., Nakanishi, Y., Nonaka, I., Goto, Y.I., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol.
Jakobs, S., Martini, N., Schauss, A.C., Egner, A., Westermann, B., and Hell, S.W. (2003). Spatial and temporal dynamics of budding yeast mitochondria lacking the divisioncomponent Fis1p. J Cell Sci 116, 2005-2014.
James, D.I., Parone, P.A., Mattenberger, Y., and Martinou, J.C. (2003). hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278, 36373-36379.
Jofuku, A., Ishihara, N., and Mihara, K. (2005). Analysis of functional domains of rat

220
mitochondrial Fis1, the mitochondrial fission-stimulating protein. Biochem Biophys Res Commun 333, 650-659.
John, G.B., Shang, Y., Li, L., Renken, C., Mannella, C.A., Selker, J.M., Rangell, L., Bennett, M.J., and Zha, J. (2005). The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 16, 1543-1554.
Jones, B.A., and Fangman, W.L. (1992). Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev 6,380-389.
Joza, N., Susin, S.A., Daugas, E., Stanford, W.L., Cho, S.K., Li, C.Y., Sasaki, T., Elia, A.J., Cheng, H.Y., Ravagnan, L., et al. (2001). Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410, 549-554.
Ju, W.K., Misaka, T., Kushnareva, Y., Nakagomi, S., Agarwal, N., Kubo, Y., Lipton, S.A., and Bossy-Wetzel, E. (2005). OPA1 expression in the normal rat retina and optic nerve. J Comp Neurol 488, 1-10.
Kamei, S., Chen-Kuo-Chang, M., Cazevieille, C., Lenaers, G., Olichon, A., Belenguer, P., Roussignol, G., Renard, N., Eybalin, M., Michelin, A., et al. (2005). Expression of the opa1 mitochondrial protein in retinal ganglion cells: its downregulation causes aggregation of the mitochondrial network. Invest Ophthalmol Vis Sci 46, 4288-4294.
Kanazawa, T., Zappaterra, M.D., Hasegawa, A., Wright, A.P., Newman-Smith, E.D., Buttle, K.F., McDonald, K., Mannella, C.A., and van der Bliek, A.M. (2008). The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet 4,e1000022.
Kanzawa, T., Zhang, L., Xiao, L., Germano, I.M., Kondo, Y., and Kondo, S. (2005). Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 24, 980-991.
Karbowski, M., Arnoult, D., Chen, H., Chan, D.C., Smith, C.L., and Youle, R.J. (2004a). Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol 164, 493-499.
Karbowski, M., Jeong, S.Y., and Youle, R.J. (2004b). Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol 166, 1027-1039.
Karbowski, M., Neutzner, A., and Youle, R.J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178, 71-84.
Karbowski, M., Norris, K.L., Cleland, M.M., Jeong, S.Y., and Youle, R.J. (2006). Role of Bax and Bak in mitochondrial morphogenesis. Nature 443, 658-662.
Karren, M.A., Coonrod, E.M., Anderson, T.K., and Shaw, J.M. (2005). The role of Fis1p-Mdv1p interactions in mitochondrial fission complex assembly. J Cell Biol 171, 291-301.
Kaur, C., Foulds, W.S., and Ling, E.A. (2008). Hypoxia-ischemia and retinal ganglion cell

221
damage. Clin Ophthalmol 2, 879-889.
Kergoat, H., Herard, M.E., and Lemay, M. (2006). RGC sensitivity to mild systemic hypoxia. Invest Ophthalmol Vis Sci 47, 5423-5427.
Kerrigan, L.A., Zack, D.J., Quigley, H.A., Smith, S.D., and Pease, M.E. (1997). TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol 115,1031-1035.
Kim, H., Rafiuddin-Shah, M., Tu, H.C., Jeffers, J.R., Zambetti, G.P., Hsieh, J.J., and Cheng, E.H. (2006). Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2subfamilies. Nat Cell Biol 8, 1348-1358.
Kim, I., Rodriguez-Enriquez, S., and Lemasters, J.J. (2007). Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462, 245-253.
Kim, J.Y., Cho, J.J., Ha, J., and Park, J.H. (2002). The carboxy terminal C-tail of BNip3 is crucial in induction of mitochondrial permeability transition in isolated mitochondria. Arch Biochem Biophys 398, 147-152.
Kim, T.H., Zhao, Y., Ding, W.X., Shin, J.N., He, X., Seo, Y.W., Chen, J., Rabinowich, H., Amoscato, A.A., and Yin, X.M. (2004). Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell 15, 3061-3072.
Klein, D.E., Lee, A., Frank, D.W., Marks, M.S., and Lemmon, M.A. (1998). The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J Biol Chem 273, 27725-27733.
Kochs, G., and Haller, O. (1999). Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc Natl Acad Sci U S A 96, 2082-2086.
Koshiba, T., Detmer, S.A., Kaiser, J.T., Chen, H., McCaffery, J.M., and Chan, D.C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858-862.
Kothari, S., Cizeau, J., McMillan-Ward, E., Israels, S.J., Bailes, M., Ens, K., Kirshenbaum, L.A., and Gibson, S.B. (2003). BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene 22, 4734-4744.
Kubasiak, L.A., Hernandez, O.M., Bishopric, N.H., and Webster, K.A. (2002). Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci U S A 99, 12825-12830.
Kuznetsov, S.A., Langford, G.M., and Weiss, D.G. (1992). Actin-dependent organelle movement in squid axoplasm. Nature 356, 722-725.
Labrousse, A.M., Zappaterra, M.D., Rube, D.A., and van der Bliek, A.M. (1999). C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4, 815-826.
Lamy, L., Ticchioni, M., Rouquette-Jazdanian, A.K., Samson, M., Deckert, M., Greenberg, A.H., and Bernard, A. (2003). CD47 and the 19 kDa interacting protein-3 (BNIP3) in T

222
cell apoptosis. J Biol Chem 278, 23915-23921.
Langford, G.M. (2002). Myosin-V, a versatile motor for short-range vesicle transport. Traffic3, 859-865.
Lee, Y.J., Jeong, S.Y., Karbowski, M., Smith, C.L., and Youle, R.J. (2004). Roles of the Mammalian mitochondrial fission and fusion mediators fis1, drp1, and opa1 in apoptosis. Mol Biol Cell 15, 5001-5011.
Legesse-Miller, A., Massol, R.H., and Kirchhausen, T. (2003). Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol Biol Cell 14, 1953-1963.
Legros, F., Lombes, A., Frachon, P., and Rojo, M. (2002). Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13, 4343-4354.
Lemasters, J.J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res8, 3-5.
Letai, A., Bassik, M.C., Walensky, L.D., Sorcinelli, M.D., Weiler, S., and Korsmeyer, S.J. (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183-192.
Levin, L.A., and Louhab, A. (1996). Apoptosis of retinal ganglion cells in anterior ischemic optic neuropathy. Arch Ophthalmol 114, 488-491.
Li, C., Kosmorsky, G., Zhang, K., Katz, B.J., Ge, J., and Traboulsi, E.I. (2005). Optic atrophy and sensorineural hearing loss in a family caused by an R445H OPA1 mutation. Am J Med Genet A 138, 208-211.
Li, H., Chen, Y., Jones, A.F., Sanger, R.H., Collis, L.P., Flannery, R., McNay, E.C., Yu, T., Schwarzenbacher, R., Bossy, B., et al. (2008). Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A 105, 2169-2174.
Li, H., Zhu, H., Xu, C.J., and Yuan, J. (1998). Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491-501.
Li, L.Y., Luo, X., and Wang, X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95-99.
Li, Y., Wang, Y., Kim, E., Beemiller, P., Wang, C.Y., Swanson, J., You, M., and Guan, K.L. (2007). Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem 282, 35803-35813.
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873-887.
Ligon, L.A., and Steward, O. (2000). Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J

223
Comp Neurol 427, 351-361.
Liguori, M., La Russa, A., Manna, I., Andreoli, V., Caracciolo, M., Spadafora, P., Cittadella, R., and Quattrone, A. (2008). A phenotypic variation of dominant optic atrophy and deafness (ADOAD) due to a novel OPA1 mutation. J Neurol 255, 127-129.
Lins, L., Decaffmeyer, M., Thomas, A., and Brasseur, R. (2008). Relationships between the orientation and the structural properties of peptides and their membrane interactions. Biochim Biophys Acta 1778, 1537-1544.
Liu, X., Kim, C.N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147-157.
Lovell, J.F., Billen, L.P., Bindner, S., Shamas-Din, A., Fradin, C., Leber, B., and Andrews, D.W. (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074-1084.
Luo, X., Budihardjo, I., Zou, H., Slaughter, C., and Wang, X. (1998). Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94, 481-490.
Lutter, M., Fang, M., Luo, X., Nishijima, M., Xie, X., and Wang, X. (2000). Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol 2, 754-761.
Lutter, M., Perkins, G.A., and Wang, X. (2001). The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol 2, 22.
Ma, J., and Ptashne, M. (1987a). A new class of yeast transcriptional activators. Cell 51, 113-119.
Ma, J., and Ptashne, M. (1987b). Deletion analysis of GAL4 defines two transcriptional activating segments. Cell 48, 847-853.
Malka, F., Guillery, O., Cifuentes-Diaz, C., Guillou, E., Belenguer, P., Lombes, A., and Rojo, M. (2005). Separate fusion of outer and inner mitochondrial membranes. EMBO Rep 6,853-859.
Maniati, E., Potter, P., Rogers, N.J., and Morley, B.J. (2008). Control of apoptosis in autoimmunity. J Pathol 214, 190-198.
Manka, D., and Millhorn, D.E. (2006). A potential molecular link between aerobic glycolysis and cancer. Cell Cycle 5, 343-344.
Mannella, C.A., Pfeiffer, D.R., Bradshaw, P.C., Moraru, II, Slepchenko, B., Loew, L.M., Hsieh, C.E., Buttle, K., and Marko, M. (2001). Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. Dig Liver Dis 33, 795-802.
Marchbank, N.J., Craig, J.E., Leek, J.P., Toohey, M., Churchill, A.J., Markham, A.F., Mackey, D.A., Toomes, C., and Inglehearn, C.F. (2002). Deletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet 39, e47.

224
Marks, B., Stowell, M.H., Vallis, Y., Mills, I.G., Gibson, A., Hopkins, C.R., and McMahon, H.T. (2001). GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410, 231-235.
McQuibban, G.A., Saurya, S., and Freeman, M. (2003). Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423, 537-541.
Meeusen, S., DeVay, R., Block, J., Cassidy-Stone, A., Wayson, S., McCaffery, J.M., and Nunnari, J. (2006). Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127, 383-395.
Meeusen, S., McCaffery, J.M., and Nunnari, J. (2004). Mitochondrial fusion intermediates revealed in vitro. Science 305, 1747-1752.
Meier, P., Finch, A., and Evan, G. (2000). Apoptosis in development. Nature 407, 796-801.
Messerschmitt, M., Jakobs, S., Vogel, F., Fritz, S., Dimmer, K.S., Neupert, W., and Westermann, B. (2003). The inner membrane protein Mdm33 controls mitochondrial morphology in yeast. J Cell Biol 160, 553-564.
Misaka, T., Miyashita, T., and Kubo, Y. (2002). Primary structure of a dynamin-related mouse mitochondrial GTPase and its distribution in brain, subcellular localization, and effect on mitochondrial morphology. J Biol Chem 277, 15834-15842.
Mitra, K., Wunder, C., Roysam, B., Lin, G., and Lippincott-Schwartz, J. (2009). A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A 106, 11960-11965.
Mizushima, N. (2007). Autophagy: process and function. Genes Dev 21, 2861-2873.
Modjtahedi, N., Giordanetto, F., Madeo, F., and Kroemer, G. (2006). Apoptosis-inducing factor: vital and lethal. Trends Cell Biol 16, 264-272.
Morris, R.L., and Hollenbeck, P.J. (1995). Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol 131, 1315-1326.
Mozdy, A.D., McCaffery, J.M., and Shaw, J.M. (2000). Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151, 367-380.
Murai, M., Toyota, M., Satoh, A., Suzuki, H., Akino, K., Mita, H., Sasaki, Y., Ishida, T., Shen, L., Garcia-Manero, G., et al. (2005). Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br J Cancer 92, 1165-1172.
Murphy, A.N., Bredesen, D.E., Cortopassi, G., Wang, E., and Fiskum, G. (1996). Bcl-2potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci U S A 93, 9893-9898.
Nakada, K., Inoue, K., Ono, T., Isobe, K., Ogura, A., Goto, Y.I., Nonaka, I., and Hayashi, J.I. (2001). Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7, 934-940.

225
Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H., Kubo, T., and Tsujimoto, Y. (2005). Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434,652-658.
Nakano, K., and Vousden, K.H. (2001). PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7, 683-694.
Nangaku, M., Sato-Yoshitake, R., Okada, Y., Noda, Y., Takemura, R., Yamazaki, H., and Hirokawa, N. (1994). KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 79, 1209-1220.
Naylor, K., Ingerman, E., Okreglak, V., Marino, M., Hinshaw, J.E., and Nunnari, J. (2006). Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J Biol Chem 281,2177-2183.
Neupert, W., and Herrmann, J.M. (2007). Translocation of proteins into mitochondria. Annu Rev Biochem 76, 723-749.
Neutzner, A., and Youle, R.J. (2005). Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J Biol Chem.
Niemann, A., Ruegg, M., La Padula, V., Schenone, A., and Suter, U. (2005). Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol 170, 1067-1078.
Ochoa, G.C., Slepnev, V.I., Neff, L., Ringstad, N., Takei, K., Daniell, L., Kim, W., Cao, H., McNiven, M., Baron, R., et al. (2000). A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol 150, 377-389.
Oh, P., McIntosh, D.P., and Schnitzer, J.E. (1998). Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol 141, 101-114.
Okami, J., Simeone, D.M., and Logsdon, C.D. (2004). Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res 64, 5338-5346.
Okamoto, P.M., Tripet, B., Litowski, J., Hodges, R.S., and Vallee, R.B. (1999). Multiple distinct coiled-coils are involved in dynamin self-assembly. J Biol Chem 274, 10277-10286.
Olichon, A., Baricault, L., Gas, N., Guillou, E., Valette, A., Belenguer, P., and Lenaers, G. (2003). Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278, 7743-7746.
Olichon, A., Elachouri, G., Baricault, L., Delettre, C., Belenguer, P., and Lenaers, G. (2007a). OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ 14, 682-692.
Olichon, A., Emorine, L.J., Descoins, E., Pelloquin, L., Brichese, L., Gas, N., Guillou, E., Delettre, C., Valette, A., Hamel, C.P., et al. (2002). The human dynamin-related

226
protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 523, 171-176.
Olichon, A., Landes, T., Arnaune-Pelloquin, L., Emorine, L.J., Mils, V., Guichet, A., Delettre, C., Hamel, C., Amati-Bonneau, P., Bonneau, D., et al. (2007b). Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol 211, 423-430.
Ono, T., Isobe, K., Nakada, K., and Hayashi, J.I. (2001). Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28, 272-275.
Orth, J.D., and McNiven, M.A. (2003). Dynamin at the actin-membrane interface. Curr Opin Cell Biol 15, 31-39.
Ott, M., Zhivotovsky, B., and Orrenius, S. (2007). Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ 14, 1243-1247.
Parone, P.A., James, D.I., Da Cruz, S., Mattenberger, Y., Donze, O., Barja, F., and Martinou, J.C. (2006). Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol 26, 7397-7408.
Pattingre, S., Espert, L., Biard-Piechaczyk, M., and Codogno, P. (2008). Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 90, 313-323.
Paumard, P., Vaillier, J., Coulary, B., Schaeffer, J., Soubannier, V., Mueller, D.M., Brethes, D., di Rago, J.P., and Velours, J. (2002). The ATP synthase is involved in generating mitochondrial cristae morphology. Embo J 21, 221-230.
Pelissier, A., Chauvin, J.P., and Lecuit, T. (2003). Trafficking through Rab11 endosomes is required for cellularization during Drosophila embryogenesis. Curr Biol 13, 1848-1857.
Pelloquin, L., Belenguer, P., Menon, Y., and Ducommun, B. (1998). Identification of a fission yeast dynamin-related protein involved in mitochondrial DNA maintenance. Biochem Biophys Res Commun 251, 720-726.
Perfettini, J.L., Roumier, T., and Kroemer, G. (2005). Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol 15, 179-183.
Perkins, G.A., and Frey, T.G. (2000). Recent structural insight into mitochondria gained by microscopy. Trends Biochem Sci 25, 319-324.
Pesch, U.E., Fries, J.E., Bette, S., Kalbacher, H., Wissinger, B., Alexander, C., and Kohler, K. (2004). OPA1, the Disease Gene for Autosomal Dominant Optic Atrophy, Is Specifically Expressed in Ganglion Cells and Intrinsic Neurons of the Retina. Invest Ophthalmol Vis Sci 45, 4217-4225.
Pesch, U.E., Leo-Kottler, B., Mayer, S., Jurklies, B., Kellner, U., Apfelstedt-Sylla, E., Zrenner, E., Alexander, C., and Wissinger, B. (2001). OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum Mol Genet 10, 1359-1368.

227
Pierrat, B., Simonen, M., Cueto, M., Mestan, J., Ferrigno, P., and Heim, J. (2001). SH3GLB, a new endophilin-related protein family featuring an SH3 domain. Genomics 71, 222-234.
Praefcke, G.J., and McMahon, H.T. (2004). The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 5, 133-147.
Pucadyil, T.J., and Schmid, S.L. (2008). Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell 135, 1263-1275.
Puthalakath, H., Huang, D.C., O'Reilly, L.A., King, S.M., and Strasser, A. (1999). The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3, 287-296.
Rathmell, J.C., and Thompson, C.B. (2002). Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 109 Suppl, S97-107.
Ray, R., Chen, G., Vande Velde, C., Cizeau, J., Park, J.H., Reed, J.C., Gietz, R.D., and Greenberg, A.H. (2000). BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem 275, 1439-1448.
Regula, K.M., Ens, K., and Kirshenbaum, L.A. (2002). Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91, 226-231.
Reichert, A.S., and Neupert, W. (2002). Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim Biophys Acta 1592, 41-49.
Riedl, S.J., and Salvesen, G.S. (2007). The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 8, 405-413.
Rismanchi, N., Soderblom, C., Stadler, J., Zhu, P.P., and Blackstone, C. (2008). Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet 17,1591-1604.
Rodriguez-Enriquez, S., Kim, I., Currin, R.T., and Lemasters, J.J. (2006). Tracker dyes toprobe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2, 39-46.
Rojo, M., Legros, F., Chateau, D., and Lombes, A. (2002). Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115, 1663-1674.
Roux, A., Uyhazi, K., Frost, A., and De Camilli, P. (2006). GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature 441, 528-531.
Rujiviphat, J., Meglei, G., Rubinstein, J.L., and McQuibban, G.A. (2009). Phospholipid association is essential for the dynamin-related protein Mgm1 to function in mitochondrial membrane fusion. J Biol Chem.
Santel, A., Frank, S., Gaume, B., Herrler, M., Youle, R.J., and Fuller, M.T. (2003). Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J

228
Cell Sci 116, 2763-2774.
Santel, A., and Fuller, M.T. (2001). Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114, 867-874.
Satoh, M., Hamamoto, T., Seo, N., Kagawa, Y., and Endo, H. (2003). Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem Biophys Res Commun 300, 482-493.
Schimpf, S., Fuhrmann, N., Schaich, S., and Wissinger, B. (2008). Comprehensive cDNA study and quantitative transcript analysis of mutant OPA1 transcripts containing premature termination codons. Hum Mutat 29, 106-112.
Schimpf, S., Schaich, S., and Wissinger, B. (2006). Activation of cryptic splice sites is a frequent splicing defect mechanism caused by mutations in exon and intron sequences of the OPA1 gene. Hum Genet 118, 767-771.
Schroer, T.A. (2004). Dynactin. Annu Rev Cell Dev Biol 20, 759-779.
Schroer, T.A., Steuer, E.R., and Sheetz, M.P. (1989). Cytoplasmic dynein is a minus end-directed motor for membranous organelles. Cell 56, 937-946.
Scorrano, L., Ashiya, M., Buttle, K., Weiler, S., Oakes, S.A., Mannella, C.A., and Korsmeyer, S.J. (2002). A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2, 55-67.
Scorrano, L., and Korsmeyer, S.J. (2003). Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun 304, 437-444.
Sesaki, H., and Jensen, R.E. (1999). Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol 147, 699-706.
Sesaki, H., and Jensen, R.E. (2001). UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol 152, 1123-1134.
Sesaki, H., and Jensen, R.E. (2004). Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem 279, 28298-28303.
Sesaki, H., Southard, S.M., Yaffe, M.P., and Jensen, R.E. (2003). Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell 14,2342-2356.
Sever, S., Muhlberg, A.B., and Schmid, S.L. (1999). Impairment of dynamin's GAP domain stimulates receptor-mediated endocytosis. Nature 398, 481-486.
Shepard, K.A., and Yaffe, M.P. (1999). The yeast dynamin-like protein, Mgm1p, functions on the mitochondrial outer membrane to mediate mitochondrial inheritance. J Cell Biol 144,711-720.
Sheridan, C., Delivani, P., Cullen, S.P., and Martin, S.J. (2008). Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell 31, 570-585.

229
Shitara, H., Kaneda, H., Sato, A., Inoue, K., Ogura, A., Yonekawa, H., and Hayashi, J.I. (2000). Selective and continuous elimination of mitochondria microinjected into mouse eggs from spermatids, but not from liver cells, occurs throughout embryogenesis. Genetics156, 1277-1284.
Simon, V.R., Karmon, S.L., and Pon, L.A. (1997). Mitochondrial inheritance: cell cycle and actin cable dependence of polarized mitochondrial movements in Saccharomyces cerevisiae. Cell Motil Cytoskeleton 37, 199-210.
Skulachev, V.P., Bakeeva, L.E., Chernyak, B.V., Domnina, L.V., Minin, A.A., Pletjushkina, O.Y., Saprunova, V.B., Skulachev, I.V., Tsyplenkova, V.G., Vasiliev, J.M., et al. (2004). Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis. Mol Cell Biochem 256-257, 341-358.
Smiley, S.T., Reers, M., Mottola-Hartshorn, C., Lin, M., Chen, A., Smith, T.W., Steele, G.D., Jr., and Chen, L.B. (1991). Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A 88, 3671-3675.
Smirnova, E., Griparic, L., Shurland, D.L., and van Der Bliek, A.M. (2001). Dynamin-related protein drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12,2245-2256.
Smirnova, E., Shurland, D.L., Newman-Smith, E.D., Pishvaee, B., and van der Bliek, A.M. (1999). A model for dynamin self-assembly based on binding between three different protein domains. J Biol Chem 274, 14942-14947.
Smith, M.J., Pozo, K., Brickley, K., and Stephenson, F.A. (2006). Mapping the GRIF-1binding domain of the kinesin, KIF5C, substantiates a role for GRIF-1 as an adaptor protein in the anterograde trafficking of cargoes. J Biol Chem 281, 27216-27228.
Sogo, L.F., and Yaffe, M.P. (1994). Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J Cell Biol 126, 1361-1373.
Song, Z., Chen, H., Fiket, M., Alexander, C., and Chan, D.C. (2007). OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178, 749-755.
Song, Z., Ghochani, M., McCaffery, J.M., Frey, T.G., and Chan, D.C. (2009). Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol Biol Cell.
Sowter, H.M., Ferguson, M., Pym, C., Watson, P., Fox, S.B., Han, C., and Harris, A.L. (2003). Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol 201, 573-580.
Sowter, H.M., Ratcliffe, P.J., Watson, P., Greenberg, A.H., and Harris, A.L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61, 6669-6673.
Spinazzi, M., Cazzola, S., Bortolozzi, M., Baracca, A., Loro, E., Casarin, A., Solaini, G., Sgarbi, G., Casalena, G., Cenacchi, G., et al. (2008). A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution,

230
but not in function. Hum Mol Genet 17, 3291-3302.
Stowell, M.H., Marks, B., Wigge, P., and McMahon, H.T. (1999). Nucleotide-dependent conformational changes in dynamin: evidence for a mechanochemical molecular spring. Nat Cell Biol 1, 27-32.
Stowers, R.S., Megeath, L.J., Gorska-Andrzejak, J., Meinertzhagen, I.A., and Schwarz, T.L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063-1077.
Sugioka, R., Shimizu, S., and Tsujimoto, Y. (2004). Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279, 52726-52734.
Sulistijo, E.S., Jaszewski, T.M., and MacKenzie, K.R. (2003). Sequence-specific dimerization of the transmembrane domain of the "BH3-only" protein BNIP3 in membranes and detergent. J Biol Chem 278, 51950-51956.
Susin, S.A., Lorenzo, H.K., Zamzami, N., Marzo, I., Snow, B.E., Brothers, G.M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., et al. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441-446.
Susin, S.A., Zamzami, N., Castedo, M., Hirsch, T., Marchetti, P., Macho, A., Daugas, E., Geuskens, M., and Kroemer, G. (1996). Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med 184, 1331-1341.
Suzuki, M., Jeong, S.Y., Karbowski, M., Youle, R.J., and Tjandra, N. (2003). The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J Mol Biol 334, 445-458.
Suzuki, M., Neutzner, A., Tjandra, N., and Youle, R.J. (2005). Novel structure of the N terminus in yeast Fis1 correlates with a specialized function in mitochondrial fission. J Biol Chem 280, 21444-21452.
Sweitzer, S.M., and Hinshaw, J.E. (1998). Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell 93, 1021-1029.
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282, 11521-11529.
Takano-Ohmuro, H., Mukaida, M., Kominami, E., and Morioka, K. (2000). Autophagy in embryonic erythroid cells: its role in maturation. European Journal of Cell Biology 79,759-764.
Takei, K., McPherson, P., Schmid, S.L., and De Camilli, P. (1995). Tubular membrane invaginations coated by dynamin rings are induced by GTP-�S in nerve terminals. Nature374, 186-190.
Tanaka, Y., Kanai, Y., Okada, Y., Nonaka, S., Takeda, S., Harada, A., and Hirokawa, N. (1998). Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93, 1147-1158.

231
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36, 2503-2518.
Terman, A., and Brunk, U.T. (2004). Aging as a catabolic malfunction. Int J Biochem Cell Biol 36, 2365-2375.
Terman, A., Dalen, H., Eaton, J.W., Neuzil, J., and Brunk, U.T. (2003). Mitochondrial recycling and aging of cardiac myocytes: the role of autophagocytosis. Exp Gerontol 38,863-876.
Tieu, Q., Okreglak, V., Naylor, K., and Nunnari, J. (2002). The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J Cell Biol 158, 445-452.
Tondera, D., Czauderna, F., Paulick, K., Schwarzer, R., Kaufmann, J., and Santel, A. (2005). The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci 118, 3049-3059.
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., Da Cruz, S., Clerc, P., Raschke, I., Merkwirth, C., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J.
Twig, G., Elorza, A., Molina, A.J., Mohamed, H., Wikstrom, J.D., Walzer, G., Stiles, L., Haigh, S.E., Katz, S., Las, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J 27, 433-446.
Urbano, A., Lakshmanan, U., Choo, P.H., Kwan, J.C., Ng, P.Y., Guo, K., Dhakshinamoorthy, S., and Porter, A. (2005). AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J 24, 2815-2826.
van Dam, E.M., and Stoorvogel, W. (2002). Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol Biol Cell 13, 169-182.
van Loo, G., Schotte, P., van Gurp, M., Demol, H., Hoorelbeke, B., Gevaert, K., Rodriguez, I., Ruiz-Carrillo, A., Vandekerckhove, J., Declercq, W., et al. (2001). Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ 8, 1136-1142.
Vande Velde, C., Cizeau, J., Dubik, D., Alimonti, J., Brown, T., Israels, S., Hakem, R., and Greenberg, A.H. (2000). BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 20, 5454-5468.
Vande Walle, L., Lamkanfi, M., and Vandenabeele, P. (2008). The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ 15, 453-460.
Varadi, A., Johnson-Cadwell, L.I., Cirulli, V., Yoon, Y., Allan, V.J., and Rutter, G.A. (2004). Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci 117, 4389-4400.
Vaux, D.L., and Silke, J. (2003). Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun 304, 499-504.

232
Verny, C., Loiseau, D., Scherer, C., Lejeune, P., Chevrollier, A., Gueguen, N., Guillet, V., Dubas, F., Reynier, P., Amati-Bonneau, P., et al. (2008). Multiple sclerosis-like disorder in OPA1-related autosomal dominant optic atrophy. Neurology 70, 1152-1153.
Verstreken, P., Ly, C.V., Venken, K.J., Koh, T.W., Zhou, Y., and Bellen, H.J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365-378.
Wagner, K.M., Ruegg, M., Niemann, A., and Suter, U. (2009). Targeting and function of the mitochondrial fission factor GDAP1 are dependent on its tail-anchor. PLoS ONE 4, e5160.
Wan, J., Martinvalet, D., Ji, X., Lois, C., Kaech, S.M., Von Andrian, U.H., Lieberman, J., Ahmed, R., and Manjunath, N. (2003). The Bcl-2 family pro-apoptotic molecule, BNIP3 regulates activation-induced cell death of effector cytotoxic T lymphocytes. Immunology110, 10-17.
Wang, A.G., Fann, M.J., Yu, H.Y., and Yen, M.Y. (2006). OPA1 expression in the human retina and optic nerve. Exp Eye Res 83, 1171-1178.
Wasiak, S., Zunino, R., and McBride, H.M. (2007). Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177,439-450.
Waterham, H.R., Koster, J., van Roermund, C.W., Mooyer, P.A., Wanders, R.J., and Leonard, J.V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356,1736-1741.
Weber, F., Haller, O., and Kochs, G. (2000). MxA GTPase blocks reporter gene expression of reconstituted Thogoto virus ribonucleoprotein complexes. J Virol 74, 560-563.
Wei, M.C., Lindsten, T., Mootha, V.K., Weiler, S., Gross, A., Ashiya, M., Thompson, C.B., and Korsmeyer, S.J. (2000). tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 14, 2060-2071.
White, K.E., Davies, V.J., Hogan, V.E., Piechota, M.J., Nichols, P.P., Turnbull, D.M., and Votruba, M. (2009). OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest Ophthalmol Vis Sci 50,2567-2571.
Willis, S.N., Fletcher, J.I., Kaufmann, T., van Delft, M.F., Chen, L., Czabotar, P.E., Ierino, H., Lee, E.F., Fairlie, W.D., Bouillet, P., et al. (2007). Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856-859.
Wolter, K.G., Hsu, Y.T., Smith, C.L., Nechushtan, A., Xi, X.G., and Youle, R.J. (1997).Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139,1281-1292.
Wong, E.D., Wagner, J.A., Gorsich, S.W., McCaffery, J.M., Shaw, J.M., and Nunnari, J. (2000). The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol 151, 341-352.
Wong, E.D., Wagner, J.A., Scott, S.V., Okreglak, V., Holewinske, T.J., Cassidy-

233
Stone, A., and Nunnari, J. (2003). The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol 160, 303-311.
Xue, L., Fletcher, G.C., and Tolkovsky, A.M. (2001). Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol 11, 361-365.
Yaffe, M.P., Harata, D., Verde, F., Eddison, M., Toda, T., and Nurse, P. (1996). Microtubules mediate mitochondrial distribution in fission yeast. Proc Natl Acad Sci USA 93, 11664-11668.
Yaffe, M.P., Stuurman, N., and Vale, R.D. (2003). Mitochondrial positioning in fission yeast is driven by association with dynamic microtubules and mitotic spindle poles. Proc Natl Acad Sci U S A 100, 11424-11428.
Yamaguchi, R., Lartigue, L., Perkins, G., Scott, R.T., Dixit, A., Kushnareva, Y., Kuwana, T., Ellisman, M.H., and Newmeyer, D.D. (2008). Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell 31, 557-569.
Yasuda, M., Theodorakis, P., Subramanian, T., and Chinnadurai, G. (1998). Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem 273, 12415-12421.
Yoon, Y., Krueger, E.W., Oswald, B.J., and McNiven, M.A. (2003). The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23, 5409-5420.
Yoon, Y., Pitts, K.R., and McNiven, M.A. (2001). Mammalian dynamin-like protein dlp1 tubulates membranes. Mol Biol Cell 12, 2894-2905.
Yu, T., Fox, R.J., Burwell, L.S., and Yoon, Y. (2005). Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J Cell Sci 118, 4141-4151.
Zanelli, S.A., Trimmer, P.A., and Solenski, N.J. (2006). Nitric oxide impairs mitochondrial movement in cortical neurons during hypoxia. J Neurochem 97, 724-736.
Zanna, C., Ghelli, A., Porcelli, A.M., Karbowski, M., Youle, R.J., Schimpf, S., Wissinger, B., Pinti, M., Cossarizza, A., Vidoni, S., et al. (2008). OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain131, 352-367.
Zha, J., Harada, H., Yang, E., Jockel, J., and Korsmeyer, S.J. (1996). Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87, 619-628.
Zhang, H., Bosch-Marce, M., Shimoda, L.A., Tan, Y.S., Baek, J.H., Wesley, J.B., Gonzalez, F.J., and Semenza, G.L. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283, 10892-10903.
Zhang, H.M., Cheung, P., Yanagawa, B., McManus, B.M., and Yang, D.C. (2003a).

234
BNips: a group of pro-apoptotic proteins in the Bcl-2 family. Apoptosis 8, 229-236.
Zhang, J., Dong, M., Li, L., Fan, Y., Pathre, P., Dong, J., Lou, D., Wells, J.M., Olivares-Villagomez, D., Van Kaer, L., et al. (2003b). Endonuclease G is required for early embryogenesis and normal apoptosis in mice. Proc Natl Acad Sci U S A 100, 15782-15787.
Zhang, P., and Hinshaw, J.E. (2001). Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol 3, 922-926.
Zhang, Y., and Chan, D.C. (2007). Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc Natl Acad Sci U S A 104, 18526-18530.
Zuchner, S., Mersiyanova, I.V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E.L., Zappia, M., Nelis, E., Patitucci, A., Senderek, J., et al. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36, 449-451.
Zunino, R., Schauss, A., Rippstein, P., Andrade-Navarro, M., and McBride, H.M. (2007). The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci 120, 1178-1188.

235
Annexes

236

237
Publication 1The BH3-only Bnip3 binds to the dynamin Opa1 to promote
mitochondrial fission and apoptosis by distinct mechanisms

238

239

240

241

242

243

244

245

246

247

248

249

250

251

252

253

254

255
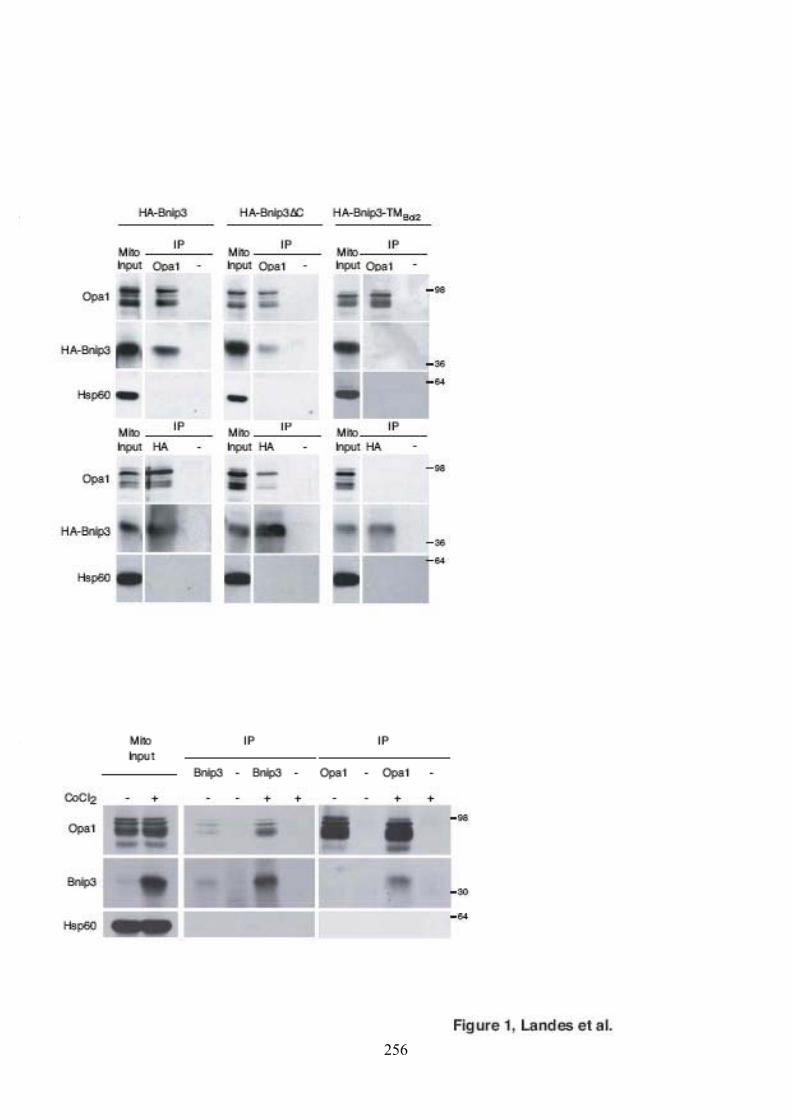
256
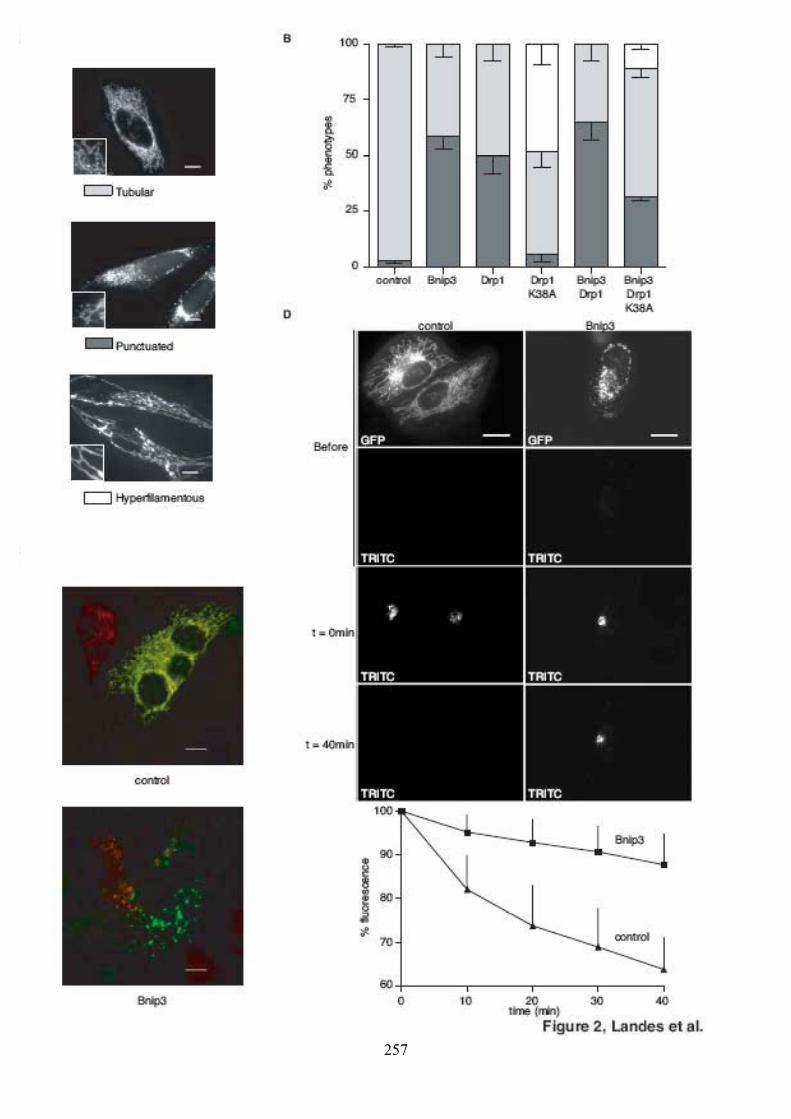
257
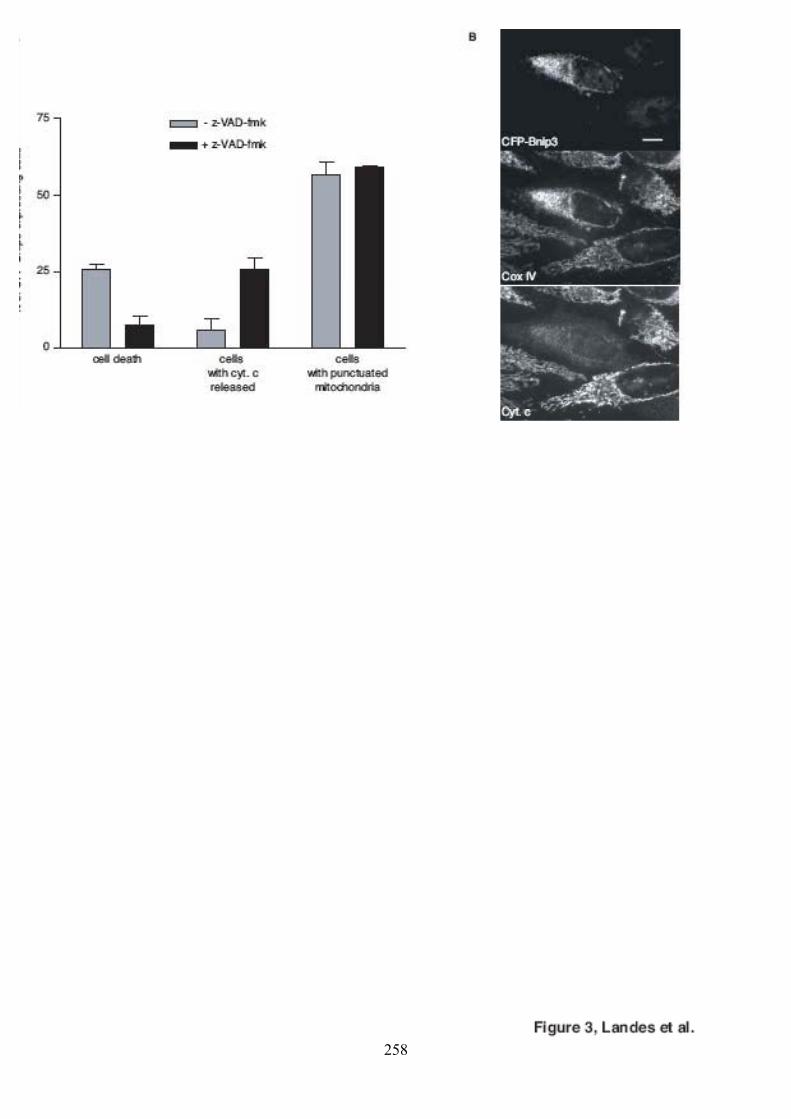
258
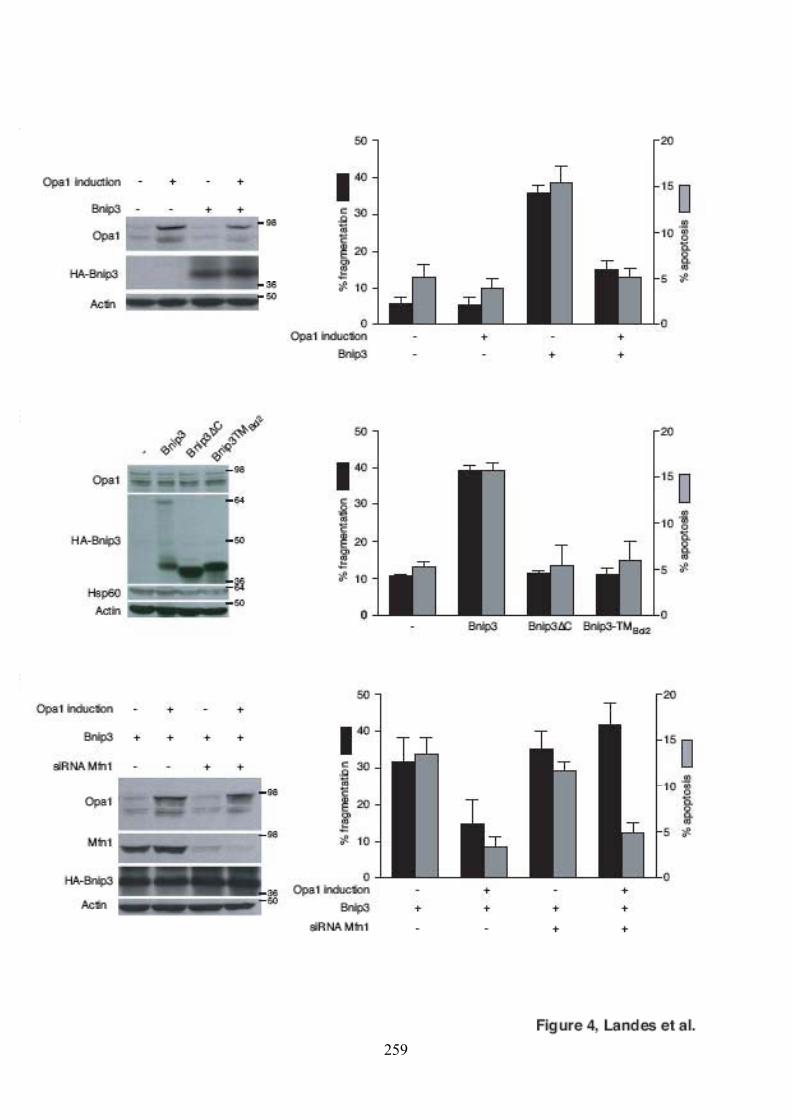
259
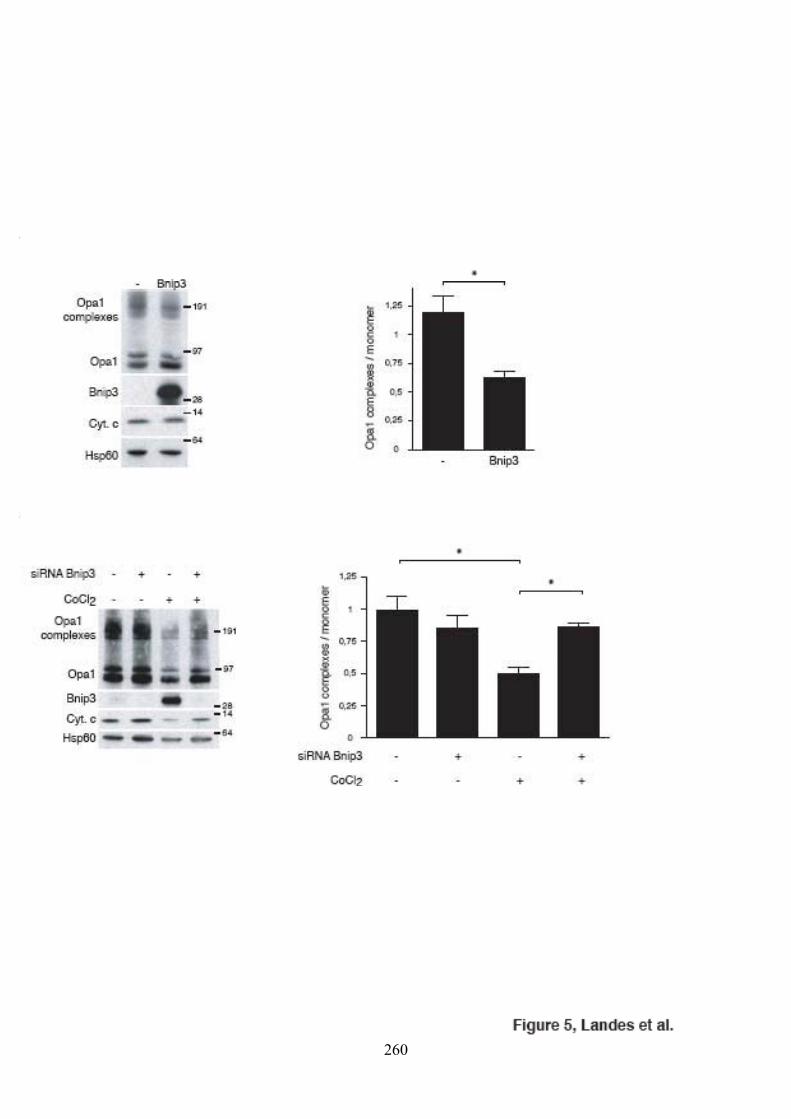
260

261
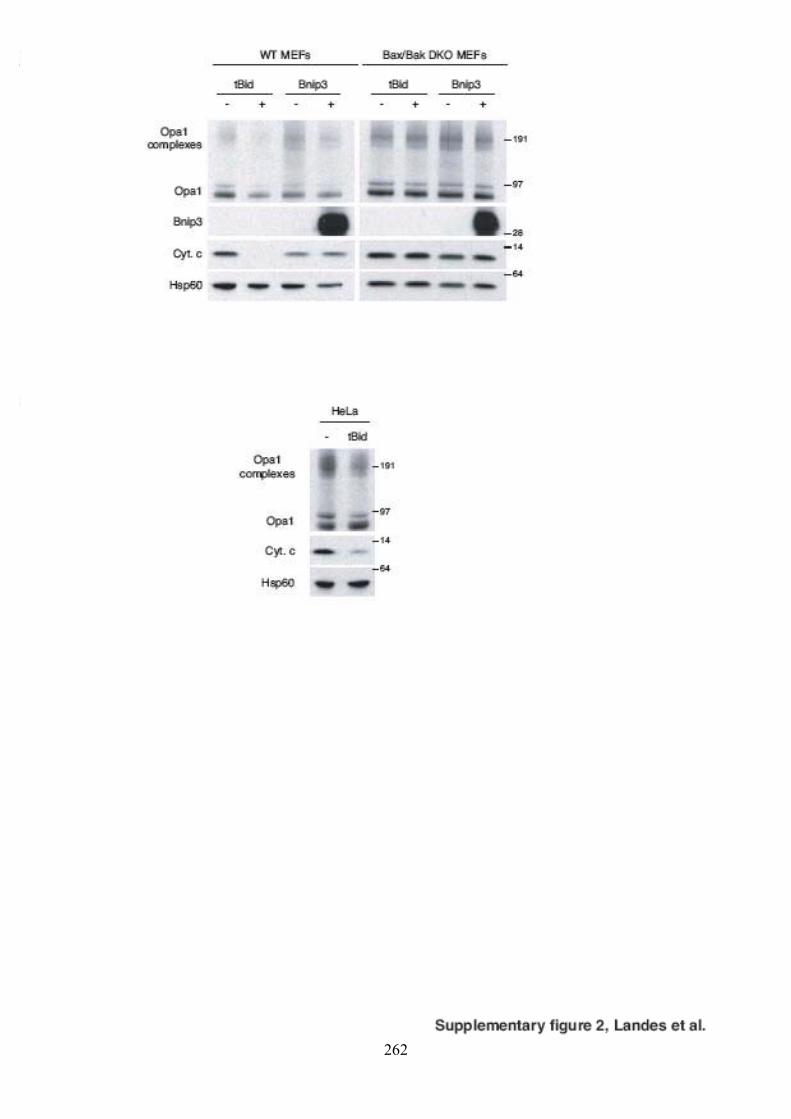
262

263

264

265

266

267
Publication 2Mitochondrial dynamics and disease, OPA1

268

269

270

271
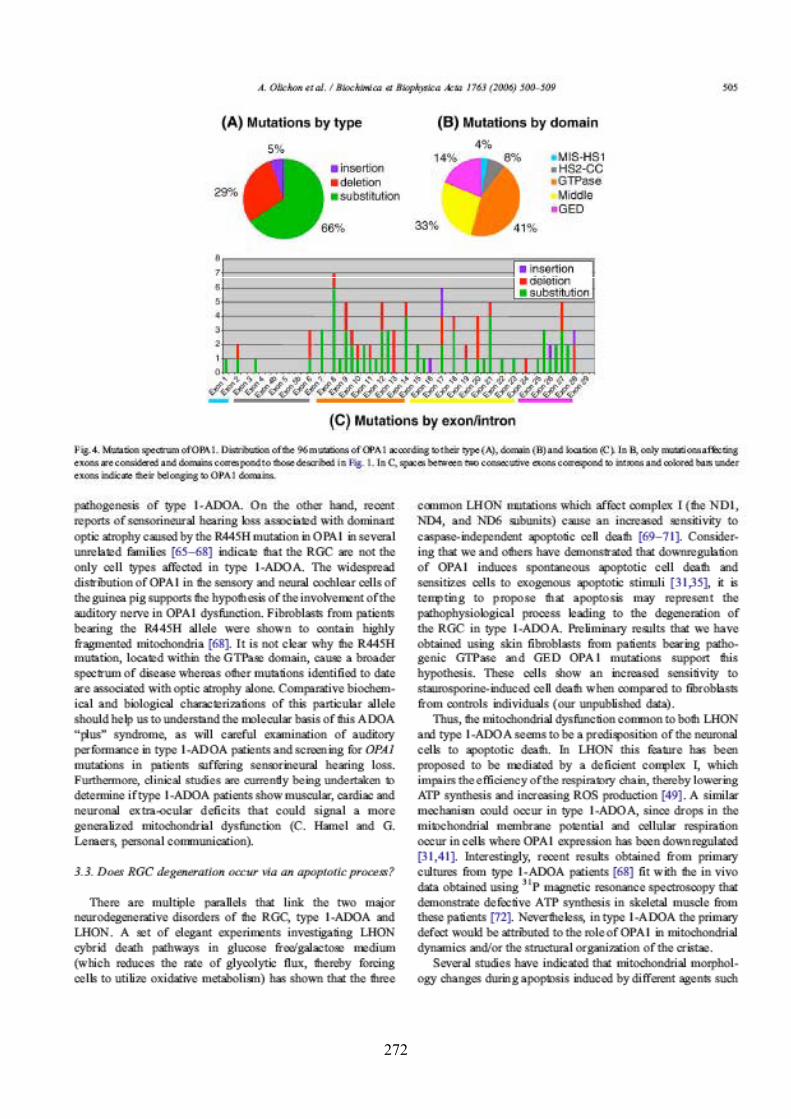
272

273

274

275

276

277
Publication 3Metalloprotease-mediated Opa1 processing is modulated by the
mitochondrial membrane potential

278

279

280

281
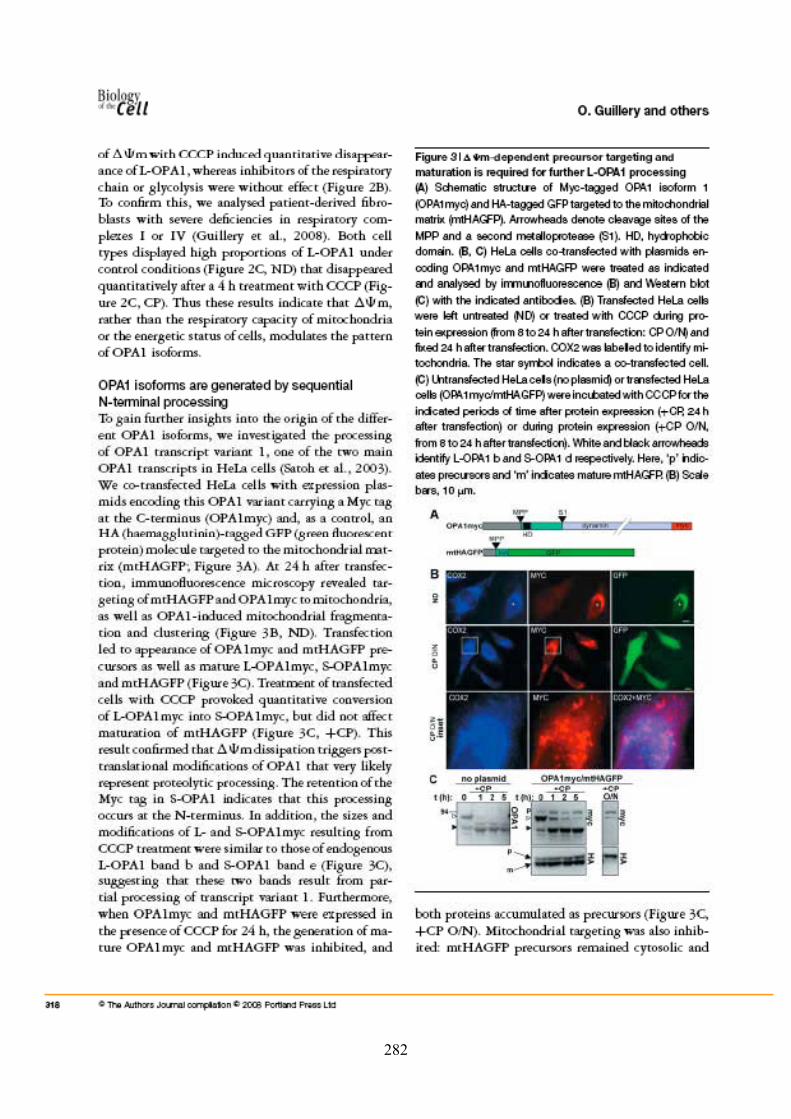
282

283

284

285

286

287

288

289

290

AUTHOR: Landes ThomasTITLE: Mitochondrial dynamics and apoptosis : role of the Opa1/Bnip3 interactions
ABSTRACT :Mitochondria are highly dynamic organelles that continually fuse and divide, yielding
mitochondrial morphologies that range from an interconnected filamentous network to small spherical
grains in adaptation to metabolic demands and state of proliferation or differentiation. Several studies
suggest a link between mitochondrial dynamics and the intrinsic pathway of apoptosis, mediated by
members of the Bcl-2 family, leading to the release of apoptogenic proteins, including cytochrome c,
from the mitochondrial intermembrane space into the cytosol. Although distinguishable, machineries
executing the two processes may use common elements to promote rearrangements of the two
mitochondrial membranes. We have identified Opa1 a dynamin of the inner membrane that regulates
mitochondrial fusion. More recently, Opa1 has also been proposed to control, during apoptosis,
cytochrome c redistribution in the mitochondria (prior to translocation through the outer mitochondrial
membrane) through its capacity to locally form oligomers at mitochondrial cristae junctions. In this study,
we identified Bnip3, a mitochondrial pro-apoptotic BH3-only protein of the Bcl-2 family, as a binding
partner of Opa1. The interaction was first substantiated in vitro and in cellulo. We then demonstrated that,
through this association, Bnip3 induces mitochondrial fragmentation by inhibiting Opa1-mediated
mitochondrial fusion but also triggers apoptosis by disrupting Opa1 oligomers. Both effects are alleviated
by up-regulation of Opa1. However, while counteraction of fragmentation by Opa1 is not observed upon
extinction of its fusion partner Mfn1, the anti-apoptotic effects of Opa1 still occurs. Thus, our results
reveal a direct link between Opa1 and an outer-membrane component of the apoptotic machinery that
modulate inner-membrane structure and fusion by separate mechanisms. These findings may help to
understand the aetiology of autosomal dominant optic atrophy (ADOA1), a leading cause of blindness
caused by Opa1 mutations, since retinal ganglion cells, primarily affected in ADOA, are particularly
prone to hypoxic stress, a potent inductor of Bnip3 expression.
KEY WORDS: Mitochondria, morphology, dynamics, apoptosis
ADDRESS: Université Paul Sabatier - CNRS UMR5241 / UMR5088 - IFR109Bâtiment 4R3 B1 - 118, route de Narbonne - 31062 TOULOUSE

AUTEUR: Landes Thomas
TITRE: Dynamique mitochondriale et apoptose : rôle de l’interaction entre Opa1 et Bnip3
DIRECTEUR DE THÈSE : ARNAUNE Laëtitia / BELENGUER Pascale
LIEU ET DATE DE SOUTENANCE : 16 Octobre 2009, salle de conférences de l’IFR 109 Toulouse
RESUME
La morphologie mitochondriale, qui est variable suivant le type cellulaire, les besoins énergétiques, l’état de prolifération ou de différenciation de la cellule, résulte d’un équilibre dynamique entre des évènements de fission et de fusion des membranes mitochondriales. Plusieurs données dans la littérature indiquent une relation étroite entre la dynamique mitochondriale et l’initiation de la voie mitochondriale de l’apoptose. Cette dernière consiste, grâce à l’intervention des protéines de la famille de Bcl-2, à la perméabilisation de la membrane externe mitochondriale et au relargage de molécules apoptogènes, dont le cytochrome c, de l'espace inter-membranaire vers le cytoplasme. Les travaux menés par notre équipe ces dernières années ont conduit à isoler Opa1 et à démontrer son implication dans la fusion de la membrane interne mitochondriale. Des travaux récents montrent qu’Opa1, via sa capacité à oligomériser, contrôle également la conformation des crêtes mitochondriales qui renferment la majeure partie du cytochrome c. Afin de mieux appréhender les fonctions fusogène et anti-apoptotique de Opa1, une recherche de partenaires a été réalisée, au laboratoire, par une approche de double hybride chez la levure. Mon travail de thèse s’est focalisé sur la caractérisation fonctionnelle de l’interaction entre Opa1 et l’un de ses partenaires, Bnip3, un membre pro-apoptotique de la famille de Bcl-2 localisé à la membrane externe mitochondriale. L’interaction entre Opa1 et Bnip3 a été validée in vitro par des expériences de « GST-pull down » et in cellulo par co-immunoprécipitation après co-expression des deux protéines dans des cellules HeLa ou après culture des cellules en hypoxie, condition menant à l’accumulation de Bnip3. Par les mêmes approches, j’ai démontré que cette interaction requiert l’extrémité carboxy-terminale de Bnip3, incluant un domaine transmembranaire et 10 acides aminés situés dans l’espace inter-membranaire mitochondrial. Dans un second temps, j’ai caractérisé les conséquences de l’interaction sur la morphologie des mitochondries et sur l’apoptose. Cette association provoque la fragmentation du réseau mitochondrial par inhibition de la fusion des membranes mitochondriales et conduit également, de manière indépendante, à l’apoptose par le désassemblage de complexes oligomériques d’Opa1. Ces deux effets sont contrebalancés par la surexpression d’Opa1, ce qui renforce le lien physiologique entre les deux protéines. L’interaction entre Opa1 et Bnip3 constitue le premier lien direct entre Opa1 et la machinerie apoptotique et pourrait permettre de mieux comprendre les mécanismes physiopathologiques de l’atrophie optique dominante de type 1, causée par des mutations du gène codant pour Opa1 et caractérisée par une dégénérescence des cellules ganglionnaires de la rétine.
MOTS-CLES : Mitochondrie, morphologie, dynamique, apoptose
DISCIPLINE : Biologie cellulaire et moléculaire
ADRESSE DU LABORATOIRE : Université Paul Sabatier - CNRS UMR5241 / UMR5088 - IFR109Bâtiment 4R3 B1 - 118, route de Narbonne - 31062 TOULOUSE















![[ APPENDICES ]open_jicareport.jica.go.jp/pdf/12291308_02.pdfMr. Hidetaka SAKABE Mr. Masahiko TSUNODA Mr. Katsufumi MATSUZAWA Mr. Yasuhiro NOZUE Mr. Takeshi YOSHIDA Mr. Tadahiro FUKUDA](https://img.pdfslide.tips/doc/110x75/5e62439f4c60ba60e27d4dd6/-appendices-open-mr-hidetaka-sakabe-mr-masahiko-tsunoda-mr-katsufumi-matsuzawa.jpg)


![THESEthesesups.ups-tlse.fr/923/1/Garner-Bienvenu_Meghyn.pdf · de minimisation de Quine-McCluskey [McC56] commence par calculer l’intégra-lité des implicants/impliqués premiers](https://img.pdfslide.tips/doc/110x75/6060b7c49284332e0e7c9ca2/de-minimisation-de-quine-mccluskey-mcc56-commence-par-calculer-laintgra-lit.jpg)










