Embed Size (px)
Citation preview

ANTECEDENTES. EL GLOMERULO RENAL
1
1 ANTECEDENTES. ENFERMEDAD RENAL PROGRESIVA.
1.1 El glomérulo renal.
La unidad morfofuncional del riñón es la nefrona. En un hombre adulto existen de 1,5 a 2 millones
de nefronas repartidas por toda la corteza renal, y en ellas se pueden distinguir dos componentes
estructurales principales: el glomérulo y el sistema tubular córtico-medular.
Figura 1. Esquema de la nefrona.
Las nefronas se encuentran en la corteza renal siguiendo un patrón establecido que se repite
periódicamente y que se denomina lobulillo renal. Este lobulillo está constituido por una subunidad
de la corteza comprendida entre dos arterias interlobulillares contiguas, y está centrado por un rayo
medular que, a modo de eje, aparece surcado por un conducto colector principal que desciende
verticalmente hacia las pirámides, recibiendo la orina concentrada en las nefronas situadas a ambos
lados del rayo medular. Se reconocen cuatro subdivisiones en la porción tubular de la nefrona: el
túbulo proximal, el túbulo intermedio (porción delgada del asa de Henle), el túbulo distal y el
sistema colector (figura 1).
El extremo ciego de la porción proximal del sistema tubular aparece dilatado e invaginado,
formando una estructura hueca, de finas paredes epiteliales, denominada cápsula de Bowman. La
concavidad de esta cápsula está ocupada por un ovillo de capilares contorneados que se conoce
como glomérulo. El glomérulo y su cápsula epitelial de doble pared constituyen juntos el
corpúsculo renal estructura que, junto al sistema tubular, completan la nefrona1.

INTRODUCCIÓN
2
El corpúsculo renal posee una forma esférica de un diámetro de 100-150 µm. Éste está formado
por el glomérulo y la cápsula de bowman, la cual se encuentra revestida interiormente por un
epitelio aplanado y posee dos aberturas; el polo vascular, por donde penetra la arteriola aferente y
sale la eferente y el polo urinario que se comunica con el túbulo renal.
El glomérulo procede de la ramificación de la arteria aferente, la cual se subdivide a partir del polo
vascular formando unas redes de capilares independientes denominadas lóbulos glomerulares. Cada
lóbulo está formado por varios capilares dispuestos alrededor de una región de soporte llamada
mesangio glomerular. Este está constituido por células mesangiales y algún monocito/macrófago
infiltrado, incluidas en un material de estructura fibrilar llamado matriz mesangial.
La pared de los capilares glomerulares está formada por la membrana basal glomerular (MBG),
revestida interiormente por el endotelio fenestrado y externamente por las células epiteliales
llamadas podocitos. La MBG no rodea por completo la pared de los capilares glomerulares, sino
que después de un recorrido más o menos circular, se refleja sobre sí misma pasando a formar parte
del capilar adyacente.
Figura 2. A) Esquema microscópico de un glomérulo; b) porción de tres capilares glomerulares; c)
estructura de la membrana basal glomerular, pedicelos y células mesangiales

ANTECEDENTES. EL GLOMERULO RENAL
3
Es esta disposición la que contribuye a delimitar un espacio central ocupado por el mesangio
glomeurlar, donde las células mesangiales están separadas de la luz capilar únicamente por el
endotelio fenestrado y del espacio urinario por la MBG. Esta disposición hace que el mesangio esté
constantemente perfundido de macromoléculas y partículas procedentes de la circulación que
pueden modificar la función glomerular. (Figura 2b).
En el polo vascular del glomérulo se encuentra el aparato yuxtaglomerular, concretamente en el
área de contacto entre la arteriola aferente, la eferente y la mácula densa (una porción del túbulo
distal con células diferenciadas). El aparato yuxtaglomerular es rico en terminaciones adrenérgicas
y juega un papel muy importante en la conservación del sodio, el control de la presión arterial
mediante la secreción de renina y la regulación del filtrado glomerular (retroalimentación túbulo-
glomerular).2
Las células que recubren los capilares se denominan podocitos. Éstos tienen varias prolongaciones
primarias radiales que se disponen paralelas a los capilares subyacentes y dan lugar a numerosas
ramificaciones secundarias denominadas pedicelos. Estos pedicelos se interdigitan con los
pedicelos de los podocitos adyacentes dejando una separación entre ellos de unos 25nm de anchura,
que permiten la salida del filtrado plasmático de los capilares glomerulares y su entrada en el
espacio capsular.
Figura 3. Representación esquemática del patrón interdigitado de las prolongaciones secundarias
(pedicelos) de los podocitos, en la superficie externa de un asa capilar glomerular

INTRODUCCIÓN
4
Estos espacios reciben el nombre de Hendiduras de Filtración. Aunque las prolongaciones
secundarias de los podocitos están íntimamente unidas a la MBG del capilar subyacente, el cuerpo
celular suele estar separado del capilar de 1 a 3 µm. Esto permite que casi toda la superficie del asa
capilar esté tapizada por pedicelos densamente interdigitados. Esta disposición máximiza la
superficie total de las hendiduras intercelulares disponibles facilitando el paso del filtrado
glomerular (Figura 3).
Si observamos la ultraestructura de la MBG se distinguen tres capas: la lámina rara interna
(adyacente al endotelio), la lámina densa intermedia, y la lámina rara externa (adyacente a los
podocitos). La lamina densa está formada por una malla de fibrillas de colágeno tipo IV y V y
glicoproteínas como laminina. Las láminas raras son ricas en el proteoglicano sulfato de heparán y
fibronectina. Estos compuestos actúan como anclaje del endotelio y de los podocitos. La lámina
basal glomerular y el diafragma de la hendidura de filtración forman una barrera física selectiva
que excluye del filtrado a las moléculas con un diámetro superior a los 10 nm. Los radicales
sulfato de heparán, cargados negativamente son los responsables de la barrera electrostática del
filtro glomerular. Se ha constatado en diferentes nefropatías, entre ellas la Nefropatía Diabética, un
engrosamiento de la membrana basal glomerular, además de una disminución del contenido de
sulfato de heparán, hechos que llevan a una alteración en la selectividad del filtro (Figura 4).3
Figura 4. Representación esquemática de la barrera de filtración glomerular.
Fisiológicamente, la ultrafiltración del plasma sanguíneo en los glomerulos renales tiene una gran
importancia en el control del volumen del líquido extracelular, del volumen plasmático, del
volumen minuto cardíaco y de la presión arterial sistémica. Los componentes estructurales de la
barrera de filtración situados entre la sangre que atraviesa los capilares glomerulares y el espacio
corpuscular son: (1) el endotelio fenestrado, (2) la MBG y (3) las fisuras de filtración entre las

ANTECEDENTES. EL GLOMERULO RENAL
5
interdigitaciones de los pedicelos. Se trata de una barrera de filtración selectiva por tamaño y carga
eléctrica, de manera que cualquier cambio que modifique la estructura o función de estos tres
componentes altera la función renal.
A continuación se detallan las características principales de los tres tipos celulares que conforman
el glomérulo y su implicación en la lesión glomerular: las células epiteliales (podocitos), las
endoteliales y las mesangiales (incluidas en una matriz extracelular).
1.1.1 Las células epiteliales
Estructura básica de los podocitos
Los podocitos son células únicas con una organización celular compleja. Presentan una arquitectura
celular dividida en tres segmentos: cuerpo celular, prolongaciones primarias y prolongaciones
secundarias (pedicelos). En general el cuerpo celular y las prolongaciones primarias no están
directamente conectadas con la MBG sino que se hallan flotando libremente en el espacio urinario.
Así, el podocito se halla fijado a los capilares subyacentes únicamente mediante los pedicelos.
Como consecuencia hay un espacio entre el cuerpo celular y los pedicelos. Las prolongaciones
primarias nacen en el cuerpo celular, el cual directamente, o después de bifurcarse, se divide en los
pedicelos. Éstos cubren la cara externa de la MBG y establecen un patrón típico de
interdigitaciones con los pedicelos de los podocitos vecinos, dejando entre ellos unos espacios
denominados hendiduras de filtración. En estas hendiduras de filtración se encuentra el diafragma
de la hendidura, el cual describiremos posteriormente. Reseñar que los pedicelos de un mismo
podocitos no se encuentran en posiciones contiguas, sino que siempre se disponen de manera
alternada con los pedicelos de otros podocitos.
Los podocitos están organizados de una manera polarizada mostrando unos dominios apical y
basolateral. El dominio basolateral incluye a los pedicelos que están en contacto con la MBG. Éstos
presentan numerosas vesículas que reflejan la alta tasa de endocitosis que caracteriza a estas
células. La superficie del dominio de la membrana apical, que se encuentra por encima del
diafragma de la hendidura, contiene una capa muy desarrollada de glicoproteínas cargadas
negativamente, a modo de glicocalix. Esta carga negativa causa una repulsión a las proteinas
cargadas negativamente que atraviesan el capilar glomerular reduciendo la tasa de filtración
glomerular. El glicocalix confiere principalmente la carga negativa a la barrera de filtración
glomerular y es esencial para el mantenimiento de la estructura de los pedicelos. La carga global
negativa es debida principalmente al ácido siálico y a los residuos sulfato de heparán que contienen
las proteínas localizadas en la MBG y los podocitos.

INTRODUCCIÓN
6
Este glicocalix está compuesto por podoendina y varias sialoglicoproteínas entre ellas la
podocalixina (principal glicoproteína), la GLEPP-1 (situada en los pedicelos, interactúa
directamente con podocalixina), la SGP-115/107 y otras. Esta superficie contribuye a conferir una
carga negativa a la barrera de filtración, además de tener una importancia crítica en la formación y
preservación de la arquitectura de los podocitos, previniendo también la unión de éstos al epitelio
parietal de la cápsula de Bowman.
Estructura básica del diafragma de la hendidura
En 1970, Rodewald y Karnosky4 postularon como estructura del diafragma de la hendidura un
modelo semejante al de unas varillas conectadas perpendicularmente a una barra central, formando
un patrón en forma de cremallera. Los poros rectangulares resultantes de este modelo tenían un
tamaño aproximado al de la molécula de albúmina. Además, amparados en la presencia de la
proteína zonula occludens (ZO-1) y en la asociación de esta proteína con el tipo de unión celular
tight junction , asumieron que el diafragma de la hendidura representaba una modificación de este
tipo de unión celular. Sin embargo, un estudio reciente de Reiser y colaboradores5 demuestra que
otras proteínas características de la unión celular tight junction como la simplequina y la occludina
no se encuentran en los podocitos. Además, la ZO-1, que inicialmente se creyó específica de las
tight junctions, se ha visto en otros tipos de uniones como la adherens junction. Ellos han detectado
la expresión de las moléculas P-cadenina y cateninas. También, han observado que el diafragma de
la hendidura presenta algunas características morfológicas típicas del tipo de unión adherens
junction (amplio hueco intercelular y la presencia de una línea densa central). Y proponen el
siguiente modelo de organización molecular:
Figura 5. Organización molecular del diafragma de la hendidura
P-C: P-cadenina, : -catenina, : -catenina, : -catenina, Z: ZO-1, 3: 3-integrina, 1: 1-integrina,
V: vinculina, T: talina, P: paxilina.

ANTECEDENTES. EL GLOMERULO RENAL
7
La P-cadenina representa el núcleo de la proteína con su dominio extracelular formando el
diafragma de la hendidura. Los dominios intracelulares están conectados a la β-catenina y/o la
placoglobina (ϕ catenina). La unión de este complejo al citoesqueleto de α-actina está mediado por
la α-catenina, una proteína homóloga a la vinculina. Alternativamente, la unión a la α-actina se
logra a través de la interacción de la α-catenina con la ZO-1. La molécula nefrina, antígeno 5-1-6,
parece estar asociada también al complejo del diafragma de la hendidura. Estos autores especulan
que la P-cadenina actúa como andamio básico del diafragma de la hendidura, mientras que otras
propiedades específicas como la permeoselectividad son conferidas por otras moléculas (nefrina,
antígeno 5-1-6). Creencia basada en el hecho de que la inyección del anticuerpo contra el antígeno
5-1-6 induce proteinuria masiva sin detectarse alteraciones en la membrana de la hendidura (Figura
5).
Organización del citoesqueleto de los podocitos:
Además de las células mesangiales, los podocitos son cruciales para el soporte del ovillo
glomerular. La segmentación de los podocitos en el cuerpo celular, prolongaciones primarias y
pedicelos puede observarse al nivel de citoesqueleto. Los filamentos intermedios vimentina y
desmina (cuya presencia es variable en función de la especie y cepa animal), típicos de las células
mesenquimatosas, se encuentran principalmente en el cuerpo celular. En las prolongaciones
primarias el citoesqueleto está compuesto principalmente por microtúbulos los cuales se entretejen
con los filamentos intermedios. Han sido descritas en asociación con estos microtúbulos, proteínas
tales como MAP-3 y MAP-4. En contraste con la proteínas del citoesqueleto del cuerpo celular y
de las prolongaciones primarias, los pedicelos presentan una estructura contráctil compuesta por
actina, miosina II, α-actinina, talina y vinculina. Esta maquinaria está anclada a la membrana basal
mediante el complejo α3β1 –integrina. Además de estas proteínas se ha encontrado sinaptopodina
(PP44) asociada a los filamentos actina en los pedicelos. El sistema de microfilamentos de los está
conectado directamente a los microtúbulos de las prolongaciones primarias mediante la proteína
tau, unida a la tubulina y la actina.6
Respuesta de los podocitos a la lesión:
Los podocitos se dañan en muchas enfermedades glomerulares sea el origen de la lesión
inmunitario (nefropatía membranosa), metabólico (diabetes) y/o hemodinámico (hipertensión
glomerular). Con excepción de la nefropatía asociada al virus de inmunodeficiencia adquirida y de
la glomerulosclerosis colapsada idiopática focal y segmentaria, los cambios ocasionados son
secundarios a la lesión podocitaria.7

INTRODUCCIÓN
8
La lesión principal consiste en una alteración de la configuración del diafragma de la hendidura y
de los pedicelos, dando como resultado la fusión de la MBG con las células parietales de la
cápsula de Bowman y el inicio de la proteinuria, que está acompañada de una reorganización del
citoesqueleto de actina. Estos cambios iniciales son totalmente reversibles. Sin embargo, si la
agresión no cesa, el lesión glomerular progresa. Esto implica la vacuolización de los podocitos, la
formación de pseudocistos y el desprendimiento de los podocitos de la MBG. Esto culmina con la
sinequia mediante la fusión de las células epiteliales parietales de la cápsula de Bowman y las áreas
desnudas de la MBG.
¿Por qué los podocitos se desenganchan de la MBG? :
Existe una serie de posibilidades entre las cuales se incluyen la pérdida de las proteínas de unión, el
aumento de muerte celular (apoptosis), la alteración de la membrana podocitaria o el aumento de la
presión hidrostática. Se ha visto que si se induce la agregación de cadena β1 de las integrinas
mediante un anticuerpo específico se produce la fusión de los pedicelos y la proteinuria. A nivel
génico, la fusión de los pedicelos puede ser estimulada por la inactivación del gen de la cadena α3
de las integrinas o por la inactivación del gen de la s-laminina/laminina-β2.
Resiser y colaboradores 8 han publicado recientemente, que la unión de los podocitos a la matriz
extracelular está regulada por tirosin fosfatasas. Estos autores observaron cambios estructurales
como la reorganización del citoesqueleto de actina y algunos contactos focales estaban
acompañados por un aumento de la fosforilación de la tirosina. Añadiendo el policatión protamina
sulfato (PS) o el aminonucleósido puromicina (PA) se provocaron alteraciones morfológicas,
llegando al desprendimiento de los podocitos de la matriz extracelular. Los mismos efectos se
conseguían si se añadía vanadato, un inhibidor de las tirosinas fosfatasas (PTPs), sugiriendo que las
PTPs regulan la estructura de los podocitos. Utilizando técnicas de RT-PCR demostraron la
expresión de cuatro PTPs en el citoplasma de los podocitos: SHP-2, PTP-PEST, PTP-1B y PTP-36.
Sería necesario, por tanto, identificar las dianas de los enzimas PTPs para la comprensión
molecular del desprendimiento de las células epiteliales a causa de cambios en la fosforilación.
Como potenciales candidatos proponen a la paxilina y a la p130CAS.
La consecuencia del desprendimiento de los podocitos es el desarrollo de la glomerulosclerosis,
especialmente en ciertas enfermedades de podocitos. En concreto en las formas secundarias de
FSGS.

ANTECEDENTES. EL GLOMERULO RENAL
9
1.1.2 La célula endotelial
Los capilares glomerulares están formados por un endotelio muy fino, de 40nm, compuesto por
células planas que presentan aberturas o fenestraciones de 40 a 100nm en su pared. A diferencia de
otros endotelios fenestrados, éste no presenta diafragma que lo aisle del exterior. Los núcleos
endoteliales presentan una protusión hacia la luz vascular y el citoplasma posee pocas organelas9.
Estudios in vitro han puesto de manifiesto que las células endoteliales no proliferan en presencia de
otras células glomerulares, ya sean endoteliales o epiteliales. Posiblemente, estas células sintetizan
determinados polipéptidos que inhiben el crecimiento endotelial.
La situación intermedia entre la sangre y los tejidos es un reflejo de muchas de las funciones del
endotelio vascular entre las que hallamos:
1. Construcción de la barrera de filtración glomerular.
2. Regulación del tono vasomotor a través de la secreción de la endotelina y del factor derivado
de las plaquetas, los cuales estimulan la contracción de las células musculares lisas a través del
óxido nítrico y prostaciclina, que favorecen la relajación de las mismas.
3. Mantenimiento de la fluidez sanguínea, mediante secreción de la proteína S, expresión de
anticoagulantes (ej heparán sulfato), activación del plasminógeno a plasmina y secreción de
prostaciclina10.
4. Iniciación de focos inflamatorios a través de la secreción de sustancias vasoactivas que
modifican la hemodinámica vascular y el reclutamiento de leucocitos. Las moléculas que
facilitan la adhesión de los leucocitos al endotelio son selectinas, integrinas e
inmunoglobulinas.
5. Activación de la respuesta inmunitaria. Dirigen la recirculación de los linfocitos y pueden
actuar como células presentadoras de antígenos.
6. Participan en la neoformación de vasos sanguíneos.
Las diversas funciones de las células endoteliales sugieren que éstas pueden afectar, (o a su vez ser
afectadas por ellas) a algunos procesos patológicos contribuyendo al desarrollo de la lesión
glomerular. Éstos incluyen estados de hiperfiltración, la hipercoagulación dentro del capilar
glomerular, y el aumento de interacciones con los leucocitos.

INTRODUCCIÓN
10
1. Efectos de las citocinas y sustancias procoagulantes en el endotelio endotelial.
Se ha observado en modelos de nefrectomía subtotal que la lesión de las células endoteliales es
paralela a la adhesión y agregación plaquetaria. Estos focos de trombosis podrían participar en la
oclusión capilar y en la glomerulosclerosis. Actualmente, sabemos que el endotelio lesionado en
presencia de citocinas como la IL-1, el TNF y la linfotoxina, puede expresar un gran número de
sustancias procoagulantes como el factor V, el inhibidor del plasminógeno y el factor de Willebran
y disminuir la trombomodulina y otros factores no-trombogénicos. Todos estos factores favorecen
la deposición de fibrina y la coagulación intravascular, hechos característicos de las lesiones
glomerulares avanzadas.
2. Daño endotelial mediado por linfocitos.
La presencia de leucocitos dentro del glomérulo en algunas enfermedades inflamatorias o inmunes
aumenta la susceptibilidad de la células endoteliales a ser activadas por citocinas u otros
mediadores inflamatorios expresando en la membrana moléculas de adhesión tales como la ICAM-
1, la VCAM-1 y la ELAM-1, que facilitan la interacción de leucocitos, neutrófilos y macrófagos en
el endotelio glomerular.
3. Daño endotelial mediado por neutrófilos
Los neutrófilos normalmente son activados en condiciones de isquemia, estimulando la secreción
de radicales libres de oxígeno, enzimas proteolíticos y productos derivados de la vía de la
fosfolipasa que contribuyen a la lesión glomerular.
4. Daño endotelial mediado por macrófagos
Los macrófagos participan activamente en la lesión glomerular, concretamente dañan el endotelio
mediante la liberación de radicales libres de oxigeno, secretando citocinas y sustancias vasoactivas
que activan a las células endoteliales y sustancias quimiotácticas que facilitan una infiltración
continua de moléculas en el glomérulo.
5. Daño endotelial mediado por anticuerpos.
Así mismo, se ha observado en numerosas enfermedades de origen inmunológico (lupus sistémico,
esclerodermia y vasculitis) que el daño endotelial glomerular es mediado a través de la activación
del complemento por las cadenas constantes de las inmunoglobulinas (Fc) o por anticuerpos anti-
células endoteliales.

ANTECEDENTES. EL GLOMERULO RENAL
11
1.1.3 El mesangio glomerular
El mesangio glomerular ocupa una posición intracapilar en el glomérulo. Está compuesto por
células mesangiales y por un material fibrilar amorfo, la matriz mesangial, situado en el espacio
existente entre las células que forman los canales mesangiales (Figura 6). Anatómicamente, el
mesangio está separado del lumen capilar únicamente por el endotelio y no por la MBG. Debido al
contacto directo del endotelio fenestrado entre la luz del capilar glomerular y el mesangio, éste se
encuentra constantemente perfundido por macromoléculas y residuos de filtración que se acumulan
en el mesangio11. Este hecho es de gran importancia cuando se trata de complejos inmunológicos,
citocinas y factores de crecimiento, ya que estas sustancias son capaces de modificar las respuestas
fisiológicas y la biología de las propias células glomerulares12.
Figura 6. Esquema del mesangio glomerular.
Composición de la matriz mesangial
La matriz mesangial (ECM) está principalmente compuesta por colágeno de tipos IV y V. También
está formada por glicoproteínas como la laminina, la fibronectina y los proteoglicanos,
especialmente sulfato de heparán (perlecan) y sulfatos de coidritin/dermatan (versican, decorina y
biglicanos). Estos componentes de la matriz son sintetizados y secretados por las propias células
glomerulares, especialmente por las células mesangiales. Además, estas células segregan proteasas
e inhibidores de las mismas regulando de esta manera el recambio de la matriz y en último término,
la cantidad de material depositado en la misma (Tabla 1).

INTRODUCCIÓN
12
La cantidad y composición de la matriz extracelular son determinantes para las propiedades
funcionales y estructurales del glomérulo. La Tabla 1 resume el perfil sintético de las células
mesangiales enfatizando las moléculas de la matriz extracelular en cultivo y en modelos de
enfermedades glomerulares. Necesariamente los componentes de la ECM se sintetizan
continuamente por un proceso sometido a una regulación muy precisa. La pérdida de esta
regulación tan coordinada aumenta la deposición de la ECM, situación que ocurre en el
envejecimiento glomerular y en el desarrollo de la glomerulosclerosis. Eng E y col13 revisaron
numerosas enfermedades renales demostrando que la expansión de la ECM y el consiguiente
desarrollo de la glomerulosclerosis era un hallazgo destacado. Además, se ha visto que la alteración
de la ECM influye en el fenotipo de las células mesangiales especialmente en la replicación celular
y en la síntesis anormal de ECM, perpetuando, de esta manera, la enfermedad glomerular14.
In vivo/normal
In vivo / enfermo In vitro
COLAGENOS I - ⇑ Thy1.1, PAN, nefrectomía,
transferencia de TGB-β glom.⇑ FCS
⇑ - ⇓ GH III - ⇑ PAN, transferencia de TGB-β glom. ⇑ FCS IV + ⇑ Thy1.1, PAN, nefrectomía,
transgénicos GH, CSS, transferenciade TGB-β glom.
⇑ TGF – β, TXA2 , GH⇓ PGE2
V + VI + ⇑ mGvHD ⇑ GH VIII + +GLICOPROTEINAS Fibronectina + ⇑ Thy1.1, PAN, nefrectomía,
transgénicos GH, CSS, transferenciade TGB-β glom., mGvHD, nódulos
diabéticos
⇑ TGF-β, GH, TXA2, Ang II, AVP,LDL
Nidógeno/entactina + ⇑ Thy1.1, nefrectomía Laminina + ⇑ Thy1.1, PAN, nefrectomía,
transgénicos GH, mGvHD⇑ GH, FCS, TXA2
Tenascina + ⇑ en la mayoría de GN humanas,cuando hay expansión de la matriz
extracelular
+
Trombospodina + +PROTEOGLICANOS Sulfato de Heparán + ⇑ Thy1.1, PAN, nefrectomía,
transgénicos GH, nódulos diabéticos⇑ - ⇓ GH
Condroitín/dermatán sulfatos (versican, decorina, biglicano)
+ ⇑ Thy1.1,ratas diabéticas (STZ)
⇑ - ⇓ GH⇑ TGF-β, IL-1, TNF-α
Abreviaturas: Thy1.1. nefritis anti-Thy1.1; PAN, nefrosis aminonucleosisdos; CSS, enfermedad del
suero crónica; mGvHD, enfermedad murina injerto contra huésped; GH, hormona del crecimiento;
FCS, suero bobino fetal; TXA2, tromboxano A2; HG, medio rico en glucosa; TGF- , factor
transformante del crecimiento; Ang II, angiotensina II; AVP, arginina-vasopresina; LDL, lipoproteina
de baja densidad; IL-1, inteleucina 1; TNF- , factor de necrosis tumoral alfa; STZ: estreptozotozina.
Tabla 1. Moléculas de la matriz extracelular, sintetizadas por las células mesangiales in vivo e in vitro.

ANTECEDENTES. EL GLOMERULO RENAL
13
Células mesangiales
Las células mesangiales forman parte del componente vascular del glomérulo. Son células de
naturaleza mioepitelial, lo cual queda demostrado por el hecho de que a nivel del polo vascular
glomerular se contínuan con las células musculares lisas de la arteriola aferente y además por la
presencia en el citoplasma de filamentos de actina-miosina arreladas a su membrana Generalmente,
estas células se caracterizan por un citoesqueleto intracelular de fibrillas de actina, miosina (propio
de células contráctiles), desmina y vimentina.
Al exámen mediante el microscopio electrónico, las células mesangiales presentan una forma
irregular con numerosos pseudópodos de diferentes longitudes. Es frecuente el hallazgo en el
interior de estas células de vacuolas fagocíticas dilatadas por el material ingerido, sugiriendo
propiedades fagocíticas. En el citoplasma, los orgánulos implicados en la síntesis y secreción de
proteínas (ribosomas, retículo endoplasmático y aparato de Golgi) son relativamente escasos.
También se ha encontrado una pequeña población de células residentes del tipo macrófagos
/monocitos15.
Algunas de las atribuciones del mesangio en la función glomerular son16:
ü Soporte estructural del glomérulo, especialmente de sus capilares
ü Generación y recambio de la matriz mesangial, mediante la secreción de tromboxano (aumento
de producción de matriz) y prostaglandinas (disminución de la ECM).
ü Diana de diferentes agentes vasoactivos: (a) vasoconstrictores, como la angiotensina II,
endotelina, vasopresina, y norepinefrina; (b) vasodilatadores, como la atriopeptina, óxido
nítrico, prostaglandinas PGE2 y PGI2 y dopamina.
ü Diana de mediadores inflamatorios, factores de crecimiento, y citocinas con efectos en la
hemodinámica local, proliferación celular, recambio de la matriz y otros.
ü Producción de mediadores vasoactivos y agentes de crecimiento como las prostaglandinas,
tromboxanos, productos lipoxigenados, factor activante plaquetario (PAF), óxido nítrico, etc.
ü Lugar de producción de factores de crecimiento y citocinas tales como PDGF, IL-1, TGF-β,
etc.
ü Generador de los activadores e inhibidores del plasminógeno.

INTRODUCCIÓN
14
ü Junto con los macrófagos glomerulares, las células mesangiales eliminan macromoléculas e
inmunocomplejos del glomérulo debido a su actividad fagocítica. En concreto, se ha
encontrado que las células mesangiales tanto en humanos como en ratas expresan el receptor
clásico para el LDL, mientras que sólo las ratas poseen el receptor scavenger. La exposición de
estas células al LDL inicia un pequeño incremento en la proliferación.
Muchos estudios que sostienen la hipótesis de que la proliferación glomerular precede, y continúa
incluso, durante la acumulación de la ECM y el desarrollo de la glomerulosclerosis. En esta línea,
Floege y col17 estudiaron la cronología de la acumulación de los componentes de la matriz
extracelular en un modelo de nefrectomia de 5/6 partes de riñón. Los resultados que obtuvieron son
los siguientes: (1) La proliferación celular glomerular precedía al acúmulo de producción de matriz,
(2) el área glomerular no aumentaba en los primeros días de la nefrectomía a pesar de la presencia
de proliferación celular glomerular. Sin embargo, en periodos más tardíos la área glomerular
aumentaba progresivamente (3) La entrada de monocitos y macrófagos en el glomérulo presentaba
una correlación significativa respecto al índice de esclerosis glomerular. También parecía existir
una correlación entre el área glomerular y el índice de esclerosis, aunque no llegaba a ser
estadísticamente significativa. En esta misma línea, se han estudiado otros modelos como la
glomerulonefritis mesagio-proliferativa18 inducida con anti-Thy-1 en rata, donde se ha visto un
aumento de producción y acumulación de ECM, observándose que cuando se inyecta un anticuerpo
anti-PDGF no sólo se disminuía la proliferación mesangial sino que también se reducía la
acumulación de numerosas proteínas de ECM. Lo mismo sucedía, en este mismo modelo, cuando
se dirigían anticuerpos anti-TGF-β (potente inductor de la acumulación de ECM). Todos estos
datos sugieren que la inducción de proliferación celular in vivo por PDGF o por hormonas de
crecimiento conducen a un aumento de la producción de la matriz, la cual comporta la
glomerulosclerosis. Por otro lado, también se ha asociado la merma en réplica de las células
mesangiales a una marcada reducción de la matriz extracelular mesangial y a una menor deposición
del colágeno IV, de la laminina y de la fibronectina.19
Control molecular de la progresión del ciclo celular en las células mesangiales:
En una población celular, la replicación y división de una célula en dos células hijas genéticamente
idénticas depende de dos fases funcionales (S y M) y dos fases preparatorias (G1 y G2).
La fase G1 es previa a la síntesis o replicación del ADN. La célula dobla normalmente su tamaño y
su masa, debido a que activa toda la maquinaria que será necesaria para llevar a cabo la replicación.
Hay células que pueden parar su progresión hacia la división en este estadío tardio y permanecer
durante días, meses o años en estado de reposo en lo que se denomina fase G0. Durante la fase G1,

ANTECEDENTES. EL GLOMERULO RENAL
15
existe también un punto de control de restricción R en que la célula comprueba que toda la
maquinaria esté a punto para continuar la división e incluso que las condiciones ambientales
(nutrientes, sales minerales, temperatura, factores de crecimiento actuando sobre los receptores de
membrana,...) sean las adecuadas20.
La fase S (de síntesis de ADN) corresponde al periodo durante el cual se replica el material
genético o ADN. Cada cromosoma pasa a tener dos cromátidas, es decir, dos moléculas de ADN de
cadena doble. Se dice, por tanto, que una célula diploide con dos juegos de cromosomas, al
terminar la fase S, presenta cuatro copias idénticas del material genético (tetraploide), destinadas a
segregarse durante la mitosis a las dos células hijas para mantener así su estado de diploidía20.
La fase G2 comprende el período entre la finalización de la replicación del ADN y el inicio de la
división. En este momento existe otro punto de control, en el que la célula debe cumplir dos
condiciones: que ya se ha duplicado la masa, de modo que puede dar lugar a dos células hijas, y
que se ha completado con éxito y fidelidad la duplicación del ADN20.
Finalmente, las células entran en la fase M o de mitosis, donde se produce el proceso físico de
división. La fase de mitosis se subdivide en cuatro etapas: profase, metafase, anafase y telofase.
Se sabe que en situaciones de hipertrofia, las células mesangiales quedan retenidas en fase G1,
activando consiguientemente la inducción de la muerte celular programada (apoptosis)21(Figura 7).
G G S G M20 1
Hipertrofia Apoptosis
Quiescente Sintesis DNA Proliferación
Diferenciación
Figura 7: Fases del ciclo celular
Cada una de estas fases del ciclo celular en las células eucariotas está controlada es ejercida por
unas enzimas denominadas quinasas dependientes de ciclinas (CDK) que son capaces de fosforilar
otras proteínas mediante la transferencia de grupos fosfato a aminoácidos específicos. La actividad
de las CDK depende de la formación de complejos con otras proteínas específicas del ciclo, como
son las ciclinas, y habitualmente otras proteínas inhibidoras, denominadas CDKi. La activación de
las CDK por su unión a ciclinas genera, por tanto, una cascada de señales por
fosforilación/desfosforilación que en último término perminten la transcripción y traducción de

INTRODUCCIÓN
16
aquellos genes involucrados en la progresión del ciclo celular. Cuando una señal mitogénica
estimula a la célula, ésta se prepara para entrar en el ciclo y completar su división. Las señales
mitogénicas rescatan a la célula de su quiescencia, a través de la activación de las ciclinas de tipo D
(D1, D2, D3). Estas ciclinas forman complejos de activación con la CDK 4 y CDK 6, que así
ensamblados pueden actuar fosforilando a la proteína retinoblastoma (Rb, proteína codificada por
un gen supresor tumoral que retiene a la célula en G1 delante de la falta de un estímulo
mitogénico). Rb en su estado nativo no está fosforilado y permanece unido al factor de
transcripción E2F, responsable de la activación de muchos genes de proliferación. La unión de Rb-
E2F provoca la represión de la activación. Ante la llegada de un estímulo mitogénico, el complejo
activado de ciclina D / CDK 4 o CDK 6 es capaz de fosforilar Rb, lo que a su vez permite la
liberación del factor E2F, debido al cambio conformacional de la proteína. Una vez E2F se ha
liberado actúa sobre los genes que codifican las proteínas necesarias para entrar en fase S y replicar
el ADN20.
Además de este complejo debe formarse también el complejo CDK 2 / ciclina E, que asegura el
estado de fosforilación de Rb y probablente actúe sobre otros sustratos requeridos para la
transición.
Otro nivel de regulación de las CDK durante el tránsito de G1 lo ejercen las CDKi. Una primera
familia de CDKi la constituyen los inhibidores P21waf-1, p-27 kip1 y p57 kip2 , todos los productos de
estos genes supresores tumorales, actúan anulando la actividad quinasa de los complejos descritos.
Un ejemplo de esta regulación negativa es el que aparece después de la inducción del daño sobre el
ADN de las células (por ejemplo a través de la irradiación UV), que provoca el detenimiento en
G1. El daño sobre el ADN provoca el incremento de p53, que actúa activando la transcripción de
P21waf-1, la cual inhibe el complejo CDK 2 / ciclina E y secuestra el factor PCNA (factor
indispensable para que se inicie y se desarrolle la replicación.
Una segunda familia de proteínas inhibidoras CDKi la constituyen P15ink-4B , P16ink-4, P19ink-4D.
Todas estas proteínas inhibidoras actúan sobre los complejos CDK 4 o 6 / ciclina D, y provocan
igualmente una retención de la célula en fase G1 debido a la inactivación que ejercen. Un ejemplo
de esta inactivaciónl lo proporciona la señal inhibitoria del TGF-β, que directamente actúa
incrementando los niveles de p15, por tanto asegura el efecto inhibidor del ciclo celular20.
En el modelo de glomerulonefritis por anti-Thy1 se produce una proliferación de las células
mesangiales asociado a un aumento de la ciclina A, la cual se une a CDK 2. En la nefropatía
diabética, a diferencia de lo que sucede en numerosas enfermedades glomerulares donde se da una
proliferación marcada, ocurre la hipertrofia de las células mesangiales. Ésta se define como el

ANTECEDENTES. EL GLOMERULO RENAL
17
aumento patofisiológico de la relación proteína-ADN. En esta situación la célula queda retenida en
fase G1, incrementando los niveles proteicos en ausencia de síntesis de ADN. Tal y como
demuestran muchos estudios in vitro como los modelos experimentales de diabetes, se ha
observado que este aumento proteico está asociado a un incremento de los niveles de p21 y p27 y
no a la elevación de los niveles de ciclinas E y A o de la CDK221
Los factores de crecimiento actúan regulando la progresión del ciclo celular. Así, encontramos
reguladores positivos como el PDGF que promueve la progresión de la fase G1 y reguladores
negativos como el TGF-β que bloquea el ciclo celular. Uno de los mecanismos utilizados por el
TGF-β para detener a la célula de esta fase es la liberación de la proteína p27. Esta CDKi actúa
inhibiendo la formación de los complejos ciclina E / cdk2 en presencia de mitógenos22. Existen
numerosas evidencias que demuestran que el TGF-β causa la hipertrofia de las células renales
incluyendo a las células mesangiales. Así Wolf y col23 vieron que la glucosa induce la expresión de
p27 causando la hipertrofia de las células mesangiales. Esta inducción de p27 involucra a parte de
la autoinducción del TGF-β en dichas células.
Figura 8. Regulación del ciclo celular

INTRODUCCIÓN
18
1.2 Enfermedad renal progresiva.
La Enfermedad Renal Progresiva (ERP) es la disminución paulatina del filtrado glomerular (FG)
debido a una reducción progresiva del número de nefronas funcionales. Para su diagnóstico se
requiere que el deterioro de la función renal sea persistente (>3 meses) y que se descarten factores
reversibles del mismo. La ERP es un síndrome que puede estar causado por múltiples
enfermedades. La rapidez en la evolución de la ERP está en función de la enfermedad base y del
tratamiento aplicados, teniendo en cuenta que el insulto inicial puede ser suficiente como detonante
de la ERP a pesar de haber cesado el agente etiológico causante24.
En la ERP, el riñón adopta ante la pérdida de nefronas funcionales una serie de mecanismos
compensadores para conservar sus funciones vitales. Las nefronas sanas incrementan su función, lo
que da como resultado un aumento del flujo que acaba produciendo una hiperfiltración por cada
unidad de nefrona. En fases iniciales, este mecanismo de adaptación de las nefronas permite
soslayar los efectos de la insuficiencia renal. Así se produce un aumento de la excreción fraccional
de sodio y potasio y una intensificación de la reabsorción de bicarbonato para compensar la
tendencia a la acidosis metabólica. La primera función alterada será la capacidad de concentración
de la orina que conlleva a la aparición de nicturia, poliuria y polidipsia. En fases más avanzadas se
detecta una acumulación de toxinas urémicas, materializado en urea, creatinina y un aumento de la
retención del fósforo plasmático, entre otros metabolitos derivados del metabolismo proteico.
Finalmente, los mecanismos de compensación renal no son efectivos detectándose además, acidosis
metabólica e hiperpotasemia24.
Independientemente de la causa etiológica que determine el daño renal inicial y a medida que
disminuye la masa renal se dan adaptaciones estructurales y funcionales que son capaces por sí
mismas de producir lesión glomerular y perpetuar la progresión de la insuficiencia glomerular.
Además, estas adaptaciones aparecen independientemente de la causa que originó la nefropatía y
son más acentuadas cuanto menor es el número de nefronas residuales. Por ejemplo, la extirpación
de un riñón en donadores sanos para un transplante renal, produce hipertrofia e incremento de la
función renal hasta el 70% en el riñón no extirpado. En estudios experimentales de micropuntura en
los que se ha analizado la respuesta de nefronas individuales a la reducción experimental de la
masa renal, se ha observado una elevación marcada de la filtración glomerular por el aumento del
flujo plasmático glomerular y de la presión intracapilar glomerular. Paralelamente a los aumentos
de flujo y filtración glomerular hay hipertrofia del glomérulo y en menor grado de los segmentos
tubulares. La magnitud de estos cambios es proporcional a la masa renal perdida. En términos
generales estas adaptaciones son útiles pues permiten suplir parcialmente la función que se ha
perdido. Sin embargo, cuando la masa renal se ha reducido más allá de un nivel crítico alrededor

ENFERMEDAD RENAL PROGRESIVA
19
del 70-80%, estas adaptaciones son capaces por sí mismas de producir daño renal progresivo. Esta
hipótesis propuesta por Hostteter y col25 se basa en estudios en los que se produjo una disminución
de más del 75% de la masa renal en ratas, observándose que nefronas previamente sanas al ser
expuestas súbitamente a un aumento de presión y flujo glomerular desarrollaban lesiones
glomerulares focales que derivaron en esclerosis paralelamente a la aparición de uremia en las
ratas. Cuando se previno la elevación de la presión y flujo glomerular, administrando a un grupo de
ratas dieta hipoproteica, se previno también la lesión glomerular demostrando la participación
predominante de los cambios hemodinámicos en el desarrollo de los cambios estructurales. Esta
asociación entre hiperfunción glomerular y lesión renal se ha confirmado posteriormente en otros
modelos experimentales como la hipertensión experimental con extirpación parcial de la masa renal
y en la diabetes mellitus experimental.
La ERP se caracteriza por una especial relevancia de los fenómenos de fibrosis y esclerosis en el
parénquima renal. Estas alteraciones se localizan preferentemente en dos niveles: las estructuras
glomerulares y el intersticio renal. La esclerosis glomerular ha sido considerada, durante muchos
años, como uno de los mecanismos fundamentales de la ERP. En sus fases iniciales se caracteriza
por la aparición de lesiones escleróticas, acelulares, con distribución focal. Posteriormente, la
esclerosis afecta a todo el ovillo glomerular, desapareciendo progresivamente las estructuras
celulares y las asas capilares. En este contexto de desaparición continua de la superficie de
filtración aparecería la disminución lenta de la filtración glomerular26.
Los mecanismos responsables de la progresión de las nefropatías aún no han sido identificados
completamente, sin embargo se acepta que la hipertensión glomerular y la hipertensión arterial
sistémica participan significativamente en la evolución de la lesión renal a la glomerulosclerosis.
Las nefropatías primarias y la pérdida de masa renal se acompañan de hipertensión sistémica e
hipertensión glomerular. Otros factores como edad, ingesta proteica y diabetes mellitus pueden
inducir hipertensión por aumento de flujo en el capilar glomerular, sin elevación de la presión
arterial. Una vez establecida, la hipertensión glomerular ejerce efectos nocivos para todas las
células renales. La hipertrofia glomerular, a través de la dilatación de asas capilares, amplifica el
efecto de la hipertensión glomerular en el endotelio, produciéndose una liberación de sustancias
vasoactivas y trombosis intracapilar. La liberación de factores de crecimiento promueve la
proliferación celular y el aumento de matriz mesangial. Se da una sinergia entre la hiperlipidemia y
la hipertensión glomerular que facilita el depósito de lípidos en el endotelio, acentúa la
proliferación mesangial y produce efectos tóxicos en células mesangiales, además de neutralizar la
electronegatividad de los proteoglicanos de la pared capilar. La lesión de las células epiteliales
comporta proteinuria y la pérdida de su efecto inhibitorio sobre el crecimiento de células
mesangiales, además de acentuar la proliferación, el aumento de la matriz mesangial y la pérdida
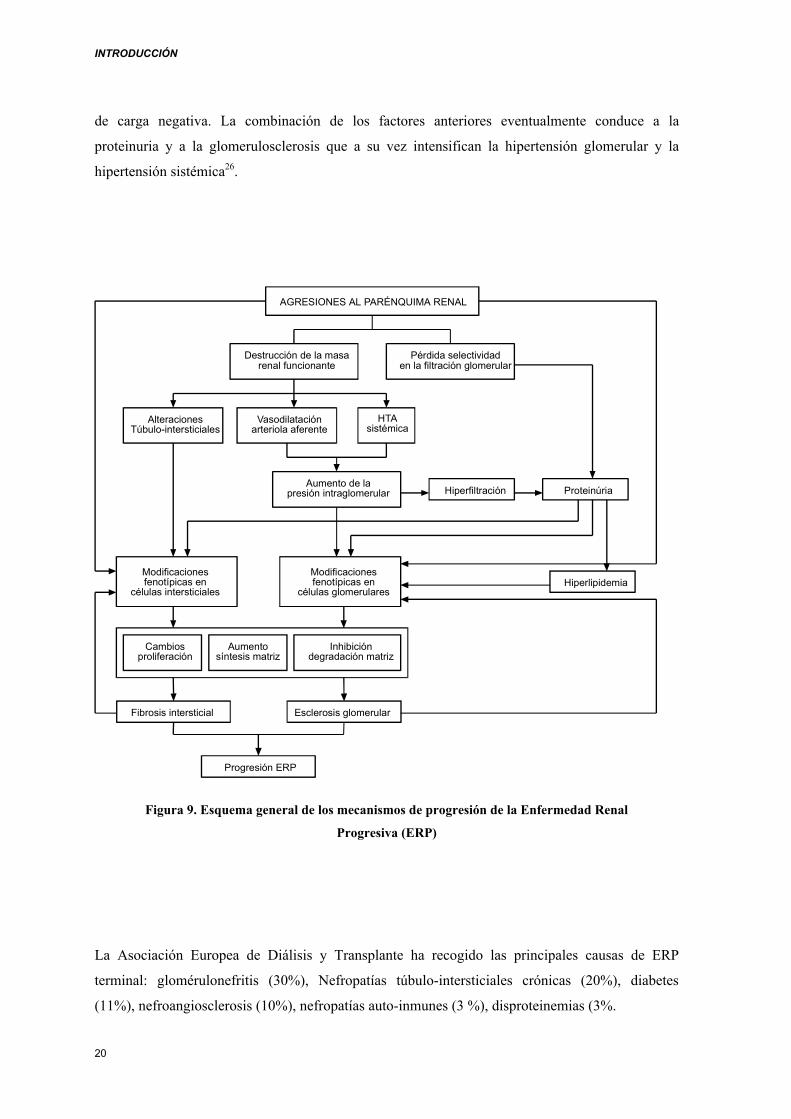
INTRODUCCIÓN
20
de carga negativa. La combinación de los factores anteriores eventualmente conduce a la
proteinuria y a la glomerulosclerosis que a su vez intensifican la hipertensión glomerular y la
hipertensión sistémica26.
AGRESIONES AL PARÉNQUIMA RENAL
Destrucción de la masarenal funcionante
Pérdida selectividaden la filtración glomerular
AlteracionesTúbulo-intersticiales
Vasodilataciónarteriola aferente
HTAsistémica
Aumento de lapresión intraglomerular Hiperfiltración Proteinúria
Modificacionesfenotípicas en
células intersticiales
Modificacionesfenotípicas en
células glomerularesHiperlipidemia
Cambiosproliferación
Aumentosíntesis matriz
Inhibicióndegradación matriz
Fibrosis intersticial Esclerosis glomerular
Progresión ERP
Figura 9. Esquema general de los mecanismos de progresión de la Enfermedad Renal
Progresiva (ERP)
La Asociación Europea de Diálisis y Transplante ha recogido las principales causas de ERP
terminal: glomérulonefritis (30%), Nefropatías túbulo-intersticiales crónicas (20%), diabetes
(11%), nefroangiosclerosis (10%), nefropatías auto-inmunes (3 %), disproteinemias (3%.

ENFERMEDAD RENAL PROGRESIVA
21
1.2.1 Glomerulosclerosis primaria focal y segmentaria
Definición
El término glomerulosclerosis designa a la enfermedad glomerular clínica asociada al síndrome
nefrótico (presencia de proteínas en orina en concentración mayor a 3g/24h) con lesiones
morfológicas focales y segmentarias sin complejos inmunes o alguna causa de origen identificable.
Se la denomina focal porque inicialmente solo afecta a algunos glomérulos y segmentaria porque la
lesión se encuentra en una porción del glomérulo y no en su totalidad (lesión difusa global). Es una
enfermedad no proliferativa ya que no acarrea un incremento de células24.
Se ha detectado que el aumento del tamaño del glomérulo antecede a la glomerulosclerosis, debido
a un incremento de factores de crecimiento que están asociados con el aumento de la matriz
extracelular, piedra angular de la esclerosis. En el desarrollo de la glomerulosclerosis contribuyen
las células glomerulares y las infiltradas, las cuales son estimuladas por los ambientes local y
sistémico constituidos por factores de crecimiento, radicales libres de oxigeno y factores
hemodinámicos y metabólicos. La interacción de todos estos factores junto con el balance de
proliferación en relación con la muerte celular y de la acumulación de matriz respecto a su
degradación condiciona el desarrollo futuro de la lesión27.
En el avance de la glomerulosclerosis se produce una lesión glomerular que está asociado a la
expresión de proteínas del citoesqueleto. Así, las células mesangiales expresan α-actina y las
células epiteliales vimentina y desmina en respuesta al daño celular, confiriendo un cambio
fenotípico a estas células. La explicación completa de estos cambios y de su contribución en el
desarrollo de la glomerulosclerosis queda aún por dilucidar.
Las células tubulares modulan el comportamiento de los fibroblastos intersticiales quiescentes. La
activación de éstos conlleva la adquisición de las características miofibroblásticas, incluyendo la
expresión de α- actina y de las proteínas del citoesqueleto. La capa adventicia vascular es otra
fuente de fibroblastos activados, llegándose a demostrar en algunos modelos experimentales de
fibrosis que estas células producen colágeno intersticial, difundiéndose desde los vasos renales al
intersticio27.
Microscopía óptica y electrónica
Durante los primeros estadios de la FSGS, la patología afecta a los glomérulos yuxtamedulares. La
observación de los glomérulos al microscopio óptico muestra esclerosis mesangial segmentaria con
colapso de los capilares, obliteración del lumen de los vasos e hiperplasia de las células epiteliales

INTRODUCCIÓN
22
viscerales. En los segmentos esclerosados se puede ver un espacio entre las membranas basales de
los capilares glomerulares y las células epiteliales viscerales. A menudo se produce sinequia,
adhesión del segmento esclerosado a la cápsula de Bowman. Con la progresión de la enfermedad,
un mayor número de glomérulos se van esclerosando, incluidos los del córtex interior y exterior.
Los segmentos escleróticos contienen células espumosas cargadas de lípidos y lóbulos eosinofílicos
que son positivos al ácido de Shiff (hialinosis) y que aparecen teñidos de rojo con la tinción de
Masson Tricrome. También se encuentra atrofia tubular, fibrosis intersticial e infiltrado intersticial
crónico de células inflamatorias mononucleares. Las arterias y arteriolas muestran depósitos
hialinos subendoteliales similares a los depósitos glomerulares28.
Al microscopio electrónico, los segmentos esclerosados muestran un aumento de la matriz
mesangial y colapso de los capilares. En estas áreas, las células mesangiales desaparecen dejando
espacios escleróticos hipocelulares. Se encuentran depósitos que corresponden a la hialinosis y
células espumosas con lisosomas repletos de lípidos, ya observados al microscopio óptico. Se
encuentran fusiones podocitarias de las células epiteliales viscerales en todos los glomérulos28.
Papel de los podocitos en la degeneración del glomérulo renal
La FSGS presenta un patrón estereotipado de la degeneración del glomérulo común en una gran
variedad de glomerulopatías. Lo más notable es la pérdida progresiva de nefronas seguida de una
disminución de la función renal que conduce hacia el fallo renal. La secuencia de estos cambios
estructurales desemboca en una esclerosis segmentaria. Así, el desarrollo de la FSGS sigue
aparentemente un patrón muy uniforme basado en la lesión severa podocitaria y en la destrucción
de la arquitectura glomerular, ambas independiente de la naturaleza del insulto inicial. Todo esto es
debido a la naturaleza no replicativa de los podocitos. Si en el curso de la enfermedad renal un
podocito se muere, la única vía para compensar la función que estaba realizando es mediante la
hipertrofia del podocito lateral, aumentando la vulnerabilidad de éstos en futuras agresiones. Este
hecho inicia un círculo vicioso que será el responsable del carácter progresivo de la degeneración
glomerular así como del fallo renal29.
En situación normal, los capilares glomerulares están constantemente expuestos a una alta presión
hidrostática de aproximadamente 80mm Hg. Tal gradiente de presión ejerce una fuerza de
expansión en la pared capilar, que obliga a que el glomérulo tenga un sistema de sostén que le
confiere estabilidad. La MBG y el mesangio representan el sistema básico generador de la tensión
interna en la pared de la MBG. Así, las células mesangiales extienden puentes contráctiles hasta los
lados opuestos de la MBG. Este sistema es capaz de desarrollar tensión en la pared facilitando la
adaptación del glomérulo a los cambios de magnitud de cualquier fuerza mecánica29. Por otro lado,

ENFERMEDAD RENAL PROGRESIVA
23
los podocitos son la segunda estructura estabilizadora superpuesta al sistema célula mesangial-
MBG. Los dos mecanismos a través de los que actúan son: (1) los podocitos estabilizan a los
capilares glomeruales fijando los puntos de inflexión de la MBG situados entre capilares vecinos.
En concreto, estos espacios están ocupados por prolongaciones primarias y pedicelos que contienen
una gran proporción de elementos del citoesqueleto. (2) Los podocitos contribuyen en la estabilidad
estructural de los capilares glomerulares a través de un mecanismo similar al desarrollado por los
pedicitos (células aplanadas que rodean a las células endoteliales con función contractil). Así, los
podocitos unidos a la cara externa de la MBG por los pedicelos actúan como una estructura elástica
que subyace al capilar glomerular amortiguando los gradientes de presión externos ejercidos a
través de la pared del capilar 29.
Desde este punto de vista, el sistema contráctil de los podocitos consiste en numerosas fuerzas
pequeñas y estabilizadoras, fijadas a varios ángulos de la superficie basal de la MBG. Cada fuerza
es capaz de contrarrestar localmente la distensión de pequeñas áreas de la MBG. Todo esto, junto
con el tono contráctil que caracteriza a los podocitos se compensa la expansión elástica pericapilar
de la MBG29.
.
Figura 10. Dibujo esquemático de la lesión podocitaria. A) Podocito sano. B) Podocito dañado con
fusión podocitaria, atenuación del cuerpo celular y formación de seudocistos, y desunión de la
membrana basal glomerular

INTRODUCCIÓN
24
Cuando se produce un insulto renal de cualquier naturaleza, los podocitos son incapaces de
mantener su fenotipo diferenciado normal, cambiando su apariencia de una manera más o menos
estereotipada. Estos cambios incluyen hipertrofia, sinequia, atenuación del cuerpo celular,
formación de pseudocistos, absorción de gotas lipídicas y finalmente, se desenganchan de la MBG.
La adhesión/sinequia de los podocitos a la MBG ha sido interpretada como un cambio adaptativo
de la célula que incluye la hipertrofia del sistema contráctil del podocito, reforzando su papel de
mantenimiento de la estabilidad del glomérulo. La atenuación del cuerpo celular y la formación de
pseudocistos son consecuencia del incremento de la extensión mecánica. El resto de cambios
fenotípicos podrían ser debidos a una hiperfiltración (hipertrofia), absorción de gotas (lisosomas) y
degradación de las proteínas filtradas (proteinuria). Cuando disminuye el número de podocitos, la
sustitución de éstos, solo es posible mediante la hipertrofia de otros podocitos intactos.
Obviamente, este mecanismo está limitado especialmente en la enfermedad glomerular cuando
todos los podocitos están más o menos afectados (Figura 9).
Las siguientes figuras nos muestran el posible papel de los podocitos en la degeneración del
glomérulo renal (figura 10) y un esquema del desarrollo de la adhesión del capilar glomerular a la
cápsula de Bowman (figura 11).

ENFERMEDAD RENAL PROGRESIVA
25
Figura 11. Papel de los podocitos en la degeneración del glomérulo renal
A, Arquitectura normal del glomérulo. B, expansión mesangial (mesangiolisis). Pérdida del soporte
mecánico del mesangio (separando las uniones célula-MBG mediante la muerte celular o la lisis de
la matriz mesangial). C, capillar balloning. Desestabilización del soporte mecánico podocitario
como consecuencia de la expansión del lumen capilar. La mesangiolisis llega a lograr que los
capilares se distancien entre sí. D, Desplegamiento de los capilares. La falta de soporte por parte de
las células mesangiales y los podocitos hace que el patrón normal de los capilares se pierda
afectando por completo a la porción lobular. Las lesiones de la arquitectura glomerular conducen a
una mayor destrucción de podocitos. Este suele ser un punto frecuente de partida para el desarrollo
de la esclerosis. E, microaneurisma. La extensión del mesangio y la disrupción endotelial
(posiblemente en conjunción con la elevada presión glomerular) lleva a un desplegamiento de la
MBG de todo el lóbulo asociado con la pérdida de todo el sistema capilar, procediendo a la
degeneración glomerular mediante la trombosis del mismo.

EXPERIMENTAL IN VITRO
26
Figura 12. Esquema del desarrollo de la
adhesión del capilar glomerular a la cápsula de
Bowman.
A. Situación normal. Los capilares
periféricos y el epitelio parietal están
separados por el espacio de Bowman. B.
Aposición. Los capilares dilatados y en parte
libres de células podocitarias, se doblan hacia
la cápsula de Bowman y se yuxtaponen al
epitelio parietal. C. Adhesión. Las células
parietales se disuelven unas con otras
uniéndose a los lados de los capilares
desprovistos de cubrimiento podocitario. Así
un vacío en el epitelio parietal puede
representar una ruta hacia el intersticio
cortical. D. Crecimiento de la adhesión. Los
podocitos situados en los flancos de la
adhesión sufren cambios degenerativos, y
finalmente mueren. La adhesión crece
invadiendo a las células parietales a lo largo
de la MBG hacia los capilares vecinos. La
membrana basal parietal se dilata,
estableciendo un espacio voluminoso que
está separado del intersticio cortical por una
capa de fibroblastos corticales, y del espacio
urinario por un epitelio parietal adherido a los
costados de la adhesión. Los podocitos
desaparecen. Los capilares se colapsan o se
obstruyen por hialinosis o por
microtrombosis. Los macrófagos invaden los
capilares y las áreas mesangiales.
¿Cómo conduce a la lesión tubular y consecuentemente a glomerulosclerosis la lesión podocitaria?
La lesión podocitaria severa incluye el desprendimiento de los podocitos de la MBG llevando a la
adhesión del capilar glomerular a la cápsula de bowman. Los capilares adheridos liberan su filtrado
al intersticio no muy lejos de la cápsula de bowman. En repuesta, los fibroblastos intersticiales
crean una cubierta celular alrededor del nuevo foco de filtración intentando prevenir la entrada de
filtrado en el intersticio. Esto resulta en la formación de un espacio paraglomerular repleto de
fluido incrementando la lesión glomerular segmentaria. La extensión de este espacio por el

ENFERMEDAD RENAL PROGRESIVA
27
glomérulo entero lleva a la esclerosis global y la extensión por el espacio urinario hacia el túbulo
correspondiente lleva a la degeneración del túbulo. Resumiendo, el fallo de la filtración glomerular
y el aumento del filtrado es el mecanismo principal en la progresión de la lesión en la
glomerulosclerosis focal y segmentaria clásica. La característica estructural más importante de la
FSGS es la unión del ovillo glomerular al intersticio, materializada más tarde en la sinequia con la
cápsula de Bowman (Figura 12).
Figura 13. Degeneración de una nefrona. Filtración incorrecta del glomérulo y expansión del filtrado
glomerular
¿Cómo causan glomerulosclerosis los miofibroblastos intersticiales? Puede actuar por vía directa e
indirecta; directamente, ocupando el espacio de Bowman, al que los fibroblastos son atraídos
mediante factores de crecimiento o componentes quimiotácticos de la ECM tales como la
fibronectina. Indirectamente, los miofibroblastos intersticiales pueden acelerar la
glomerulosclerosis estrechando las arteriolas renales30
La posibilidad más plausible es que la glomerulosclerosis sea el resultado final de interacciones
muy complejas entre el glomérulo, los túbulos renales, el intersticio y los vasos.

INTRODUCCIÓN
28
1.2.2 Nefropatía diabética
La nefropatía diabética es una enfermedad glomerular secundaria a la diabetes mellitus. El
principal síntoma clínico de la glomerulosclerosis diabética es la proteinuria. Al principio es
intermitente, pero acaba siendo constante y cuantitativamente muy abundante, llegando a alcanzar
niveles nefróticos. La proteinuria esta frecuentemente acompañada de hematuria microscópica y de
hipertensión moderada. Generalmente desemboca en una enfermedad progresiva renal que acaba en
fallo renal33.
Esta enfermedad abarca una multitud de alteraciones estructurales caracterizadas por una
hipertrofia temprana tanto del glomérulo como de los túbulos, un engrosamiento de la Membrana
Basal Glomerular (MBG) y de la Membrana Basal Tubular (MBT), y una progresiva acumulación
de los principales componentes de la matriz extracelular (ECM) en el mesangio glomerular. Estas
lesiones glomerulares son las responsables de los trastornos funcionales que caracterizan a la
nefropatía diabética, como son la proteinuria, la hipertensión, y el fallo renal. Se ha establecido una
alta correlación entre la expansión de la matriz mesangial y el declive de la superficie de filtración.
Por consiguiente, la aparición de glomerulosclerosis difusa (o nodular) en estadios avanzados de la
nefropatía diabética se cree debida al progresivo declive de la tasa de filtración glomerular31.
La fase temprana de la nefropatía se acompaña de una hipertrofia glomerular y tubuloepitelial en la
que se produce un aumento de la área o el volumen glomerular sin engrosar la MBG, como
consecuencia de la estimulación de la tasa de síntesis de los constituyentes de la membrana. En una
fase tardía de la enfermedad se puede observar un engrosamiento progresivo de la MBG y la
MBT31. Este engrosamiento es un marcador muy sensible del estado de la nefropatía, el cual está
altamente correlacionado con la aparición de microalbuminuria y con la expansión de la matriz
mesangial. Otros estudios realizados en glomérulos aislados de ratas diabéticas se observaron un
aumento significativo de los niveles de mARN de la cadena alfa-1 del colágeno tipo IV, laminina
B1 y B2, y colágenos intersticiales del tipo I y III que producían un aumento del grosor de la MBG
y la MBT32.
Junto con el aumento del grosor de la MBG tiene lugar la hialinosis exudativa que provoca un daño
isquémico y, como consecuencia, obliteración de las arteriolas de pequeño y medio tamaño. El
grado de obstrucción de los vasos se correlaciona directamente con la severidad de la
glomerulosclerosis y la fibrosis es inversa al grado de la función renal. Además, se sabe que la
diabetes mellitus cursa funcionalmente con hiperfiltración, aumento del flujo plasmático renal y
nefromegalia33.

ENFERMEDAD RENAL PROGRESIVA
29
Morgensen y col. han estratificado la progresión de la ND de la DMID en cinco estadios, que
probablemente son extrapolables a la DMNID 34,35:
Estadio 1. Hipertrofia renal e hiperfunción en ausencia de lesiones morfológicas
No se observan anomalías morfológicas significativas en el tejido renal de esos pacientes. Sin
embargo, el tamaño renal y el filtrado glomerular (FG) aumentan prácticamente en todos los
pacientes ya en el momento del diagnóstico. Histológicamente se detecta un aumento del volumen
glomerular de la superficie de los capilares glomerulares, cambios reversibles con un control
estricto de la glucemia mediante el inicio del tratamiento insulínico. La hiperfiltración glomerular
se correlaciona con el aumento de la superficie capilar. El incremento de la presión intracapilar
parece ser un factor fundamental en el inicio de la progresión de la nefropatía. Esta fase suele tener
una duración de unos cinco años.
Estadio 2. Lesiones glomerulares incipientes en ausencia de microalbuminuria
Durante esta fase, también denominada “fase silenciosa” y que se puede extender hasta 15 años
aproximadamente, las alteraciones funcionales detectadas en la etapa 1 persisten, aunque un control
metabólico más riguroso puede reducir la filtración glomerular y el filtro plasmático renal a valores
próximos al normal. La excreción urinaria de albúmina aparece en general dentro de los límites
normales. Es posible, sin embargo, observar en esta fase la excreción de cantidades anormalmente
elevadas de albúmina después de la realización de ejercicio físico. En general, esa albuminuria que
sigue al ejercicio no es detectable por métodos clínicos corrientes, exigiendo la utilización de
técnicas más sensibles para su determinación. Por esta razón, este fenómeno se denomina,
“microalbuminuria”, tanto si aparece espontáneo como provocado por el ejercicio. Se han descrito
en esta fase lesiones glomerulares incipientes, que consisten básicamente en un engrosamiento de
la membrana basal y en un aumento del volumen mesangial, con depósito de proteínas, albúmina,
IgG, fibrina y productos de degradación plaquetaria.
No es frecuente la observación de hipertensión arterial franca durante esta fase, aunque la presión
arterial puede estar ligeramente elevada en estos individuos. Esta hipertensión incipiente podría
influir teóricamente, mediante una sobrecarga hemodinámica, en el desarrollo ulterior de la
nefropatía diabética.
Estadio 3. Nefropatía incipiente
Esta etapa se caracteriza principalmente por la aparición, en el 40% aproximadamente de los
pacientes con diabetes mellitus tipo I, de microalbuminuria persistente, cuya magnitud es muy
variable (30-300 mg/24h). No obstante, la mayoría de los investigadores considera como anómalos

INTRODUCCIÓN
30
los niveles de 20mg/día. La presencia de microalbuminuria puede estar asociada a la aparición de
otras anomalías, tales como una discreta hipertensión arterial e incluso una caída del ritmo de
filtración glomerular, especialmente en aquellos pacientes cuya excreción urinaria de albúmina ya
excede de 100mg/24h.
El aumento de la excreción urinaria de albúmina (EUA) parece estar relacionado con la pérdida del
proteoglicano heparán sulfato de la membrana basal glomerular con la consiguiente alteración de
las características eléctricas de la membrana, permitiendo el paso de la albúmina y otras
macromoléculas electronegativas. Los componentes estructurales de la barrera de filtración situada
entre la sangre que pasa por los capilares glomerulares y el espacio corpuscular son: endotelio
fenestrado, la Membrana Basal Glomerular y las fisuras de filtración entre las interdigitaciones de
los pedicelos. Se trata de una barrera selectiva por tamaño y por carga eléctrica de tal manera que
cualquier cambio que modifique la estructura o función de estos tres componentes puede favorecer
la aparición de proteinuria.
Según Wang y col 36 mediante la proteinuria las moléculas filtradas pueden activar las células
tubulares induciéndolas a secretar quimioquinas que atraerían a los macrófagos al intersticio
peritubular y actuarían sobre los miofibroblastos aumentando la síntesis intersticial de colágenos y
fibronectina, produciéndose fibrosis renal intersticial.
Estadio 4. Nefropatía diabética establecida
Después de 10 o 15 años desde el inicio de la diabetes, el 40 % aproximadamente de los pacientes
diabéticos del tipo I desarrollan proteinuria progresiva, en niveles superiores a 500mg /día,
fácilmente detectable por métodos clínicos rutinarios. El ritmo de filtración glomerular, que se
encuentra principalmente en niveles próximos al normal, disminuye continuamente a una tasa
aproximadamente de 1ml /min /mes. Un 75 % de los pacientes desarrollará en los próximos 10
años insuficiencia renal terminal. Esta progresión quizá sea inferior en el paciente con DMNID,
aunque la proteinuria persistente es un factor potente de predicción de insuficiencia renal crónica
terminal (IRCT).
La presencia de retinopatía diabética es un hecho universal en los pacientes con nefropatía
diabética establecida. En los pacientes diabéticos tipo I con proteinuria sin retinopatía se debe
destacar la existencia de otra nefropatía no relacionada con la diabetes. El riesgo de afectación
coronaria y de mortalidad por esta causa es más elevado.

ENFERMEDAD RENAL PROGRESIVA
31
Estudio 5. Insuficiencia renal terminal
Tras un periodo que se extiende típicamente por 10 años desde la aparición de la proteinuria
clínicamente detectable, los pacientes diabéticos con nefropatía llegan a la etapa de la enfermedad
renal terminal. La proteinuria se incrementa, llegando a rango nefrótico. La existencia de
proteinuria nefrótica no siempre se acompaña de síndrome nefrótico completo, clínico y biológico.
Es frecuente constatar una proteinuria superior a 3g /día, pero manteniendo la cifra de albuminuria
superior a 30g /l, incluso hasta el momento del deterioro final de la función renal. La presencia de
proteinuria de rango nefrótico condiciona un rápido descenso del filtrado glomerular. El pronóstico
de estos pacientes una vez alcanzada esta etapa es pesimista, incluso cuando se instituyen
terapéuticas de sustitución tales como la diálisis y el trasplante renal.
1.2.3 Hiperlipidemia y la progresión de la ERP
Datos experimentales sugieren la idea de que los niveles altos de lípidos pueden provocar daño
renal37,38. Este daño se acentúa cuando se da la presencia de otros factores como la hipertensión39,
la reducción en el número de nefronas40 o la existencia de un daño glomerular previo41. Por otro
lado, los de agentes hipolipemiantes retardan y previenen el desarrollo de la glomerulosclerosis42,43.
Los mecanismos por los cuales los lípidos inducen glomerulosclerosis no están del todo
dilucidados, aunque algunos estudios experimentales apuntan a la infiltración de macrófagos44, a la
alteración de la biología de las células mesangiales45 y a la modificación de las lipoproteínas de
baja densidad (LDL) entre otras causas 46.
1.2.3.1 Hiperlipidemia en el síndrome nefrótico
La mayor asociación de las anormalidades lipídicas en la enfermedad renal se encuentra en el
síndrome nefrótico, donde se dan niveles elevados de colesterol en el plasma y en menor
proporción, de triglicéridos y lipoproteína (a). El síndrome nefrótico muestra unos niveles de
colesterol HDL reducidos y una disminución de la fracción de HDL-2. Estudios en la literatura
apuntan a un incremento de las proteínas que transfieren ésteres de colesterol que contribuyen
como lanzaderas de los ésteres de colesterol (HDL2) a lipoproteínas de baja densidad47.
En el síndrome nefrótico, la concentración en plasma de apolipoproteínas generalmente refleja el
metabolismo de las lipoproteínas. Así, se dan niveles elevados de apo B, C-II, y E, las cuales están
asociadas a las VLDL y las LDL. Por otro lado, los niveles principales de lipoproteínas asociadas a
las HDL, las apo A-I y A-II son usualmente normales.

INTRODUCCIÓN
32
Los dos trastornos lipídicos más frecuentes en el síndrome nefrótico son la hipercolesterolemia e
hipertrigliceridemia.
Hipercolesterolemia: En un estudio de 100 pacientes con síndrome nefrótico se encontraron
niveles de colesterol en plasma que excedían en 200mg/ dl en 87% de los pacientes; 300 mg/ dl en
el 53% y 400mg/ dl en el 25%48. Se ha visto que la respuesta hipercolesterolémica es
desencadenada en parte por la reducción de la presión oncótica y la severidad de ésta es
inversamente proporcional a dicha presión49. Además, se ha visto que si se aumenta la presión
oncótica del plasma con albúmina o dextrano, se revierten los cambios produciéndose una rápida
reducción de los lípidos en los pacientes con síndrome nefrótico50.
Por otro lado, estudios in vitro han mostrado que una baja presión oncótica estimula directamente
la transcripción génica de la apoproteína B hepática49. La razón por la que la presión oncótica
estimula la producción hepática de lipoproteínas se desconoce todavía. Nuevas técnicas sugieren
que el principal responsable de la hipercolesterolemia en pacientes con síndrome nefrótico es un
catabolismo disminuido, en detrimento de una intensificación de la síntesis hepática de
lipoproteínas.51.
Hipertrigliceridemia: El defecto en el catabolismo de los triglicéridos está íntimamente
relacionado con el aclaramiento de la albúmina renal, y no con la presión oncótica del plasma52.
Estos hallazgos sugieren un papel principal en la perdida por la orina de algún regulador del
metabolismo de los lípidos. Se ha visto que los niveles de triglicéridos plasmáticos son más altos en
las ratas con síndrome nefrótico que en ratas con albuminemia a pesar de tener tasas de producción
de triglicéridos y presiones oncóticas similares53. Así, un catabolismo reducido de los triglicéridos
debería jugar un papel importante en la hipertrigliceridemia, efecto que no puede ser explicado con
la disminución de la presión oncótica54.
1.2.3.2 Lípidos y diabetes mellitus
La diabetes mellitus se asocia a un incremento de los lípidos plasmáticos (principalmente de
triglicéridos) en pacientes no tratados o con un control metabólico pobre. Al producirse un déficit
agudo de insulina se movilizan los ácidos grasos libres (AGL) desde el tejido adiposo al hígado, lo
que comporta un aumento en la secreción hepática de lipoproteínas de muy baja densidad (VLDL).
Al mismo tiempo disminuye la actividad de la LPL, produciéndose un déficit en el aclaramiento
plasmático de las VLDL y de los quilomicrones.
En pacientes con la DMID, con un control metabólico pobre, se dan modificaciones en la cantidad
de lipoproteínas además de tener alterada su composición: Las VLDL contienen un relativo exceso

ENFERMEDAD RENAL PROGRESIVA
33
de apolipoproteínas C y E mientras que las LDL son partículas más pequeñas y densas enriquecidas
en triglicéridos pero deplecionadas en ésteres de colesterol.
El aumento de la concentración de las VLDL en la DMID está estrechamente relacionado con el
control glicémico y es secundario tanto a una alteración en su catabolismo (disfunción de la
actividad de la LPL) como a un incremento de su síntesis (los hepatocitos tienen una mayor
disponibilidad de sustrato glúcido y de AGL). Los mecanismos postulados como responsables del
incremento de las LDL en plasma son independientes del déficit de actividad de las LPL y están
más relacionados con las alteraciones en la composición que experimentan las lipoproteínas. Así,
las VLDL sintetizadas durante una situación de descompensación cetoacidótica son más ricas en
colesterol libre y esterificado, dando lugar a unas LDL más pequeñas y densas que al no ser
reconocidas por el receptor apoB-E no pueden ser eliminadas del plasma. Además, durante su larga
permanencia en el plasma y debido a la hiperglucemia la apoB de las LDL puede sufrir una
glucosilación no enzimática hecho que impide su interacción con el receptor. Por otra parte, el
déficit de insulina disminuye la actividad del receptor apoB-E, dificultando aún más la unión de las
LDL con su receptor específico.
La insulina interfiere en el metabolismo lipídico en diferentes puntos, siendo fundamental en la
regulación del metabolismo postprandial. Inhibe la movilización de los AGL del tejido adiposo,
sobretodo en períodos postprandiales, y aumenta la reesterificación de los ácidos grasos dentro del
tejido adiposo, acciones mediadas por la inhibición de la LPL intracelular. Al no llegar los AGL al
hígado se produce una disminución de la síntesis hepática de las VLDL. Por otra parte, la insulina
estimula la LPL endotelial, dando lugar a la cascada lipolítica de las lipopartículas ricas en
triglicéridos, con el consiguiente intercambio de lípidos que se produce entre estas lipoproteínas y
las HDL mediante la acción transferidora de ésteres de colesterol (CETP).
Las alteraciones más frecuentes que se producen en el metabolismo lipídico de los pacientes con la
DMNID consisten en un incremento de las concentraciones plasmáticas de triglicéridos y las
VLDL y una disminución de las HDL. El aumento de colesterol total, aunque menos constante que
la hipertrigliceridemia merece una especial atención, ya que una importante proporción depende de
la acumulación de partículas de densidad intermedia (IDL), de alto poder aterogénico. Como
sucedía con la DMID, en la DMNID también se aprecian modificaciones de las lipoproteínas. Las
VLDL e IDL son más ricas en colesterol no esterificado y apo B y E, mientras que las LDL son
más ricas en colesterol, pequeñas y densas.
En la DMNID la fisiopatología del aumento de las VLDL deriva de la presencia de obesidad y de la
resistencia a la insulina concomitantes en ese tipo de diabetes y son conocidos como sídrome X.

INTRODUCCIÓN
34
Reaven55 definió más concretamente las características de este síndrome, llamado también
síndrome plurimetabólico, que consiste en hiperinsulinismo, resistencia a la insulina, obesidad
central, hipertensión arterial, elevaciones de las concentraciones plasmáticas de triglicéridos y
descenso del colesterol-HDL e intolerancia hidrocarbonada o DMNID.
En el síndrome de resistencia insulínica está acelerado el proceso de movilización de los AGL
desde el tejido adiposo al hígado y aumentada la síntesis hepática de la VLDL. También está
disminuida la actividad de la LPL endotelial, por lo que las lipoproteínas ricas en triglicéridos no
pueden ser catabolizadas, permaneciendo en el plasma y contribuyendo así al aumento de
triglicéridos que se produce en este proceso durante los períodos postprandiales.
En la DMNID, el incremento de la síntesis hepática de las VLDL condiciona un aumento de las
LDL plasmáticas, más aterogénicas que las LDL de los individuos normales, ya que pueden sufrir
modificaciones en su composición. Al no poder ser reconocidas por los receptores específicos
tienen que ser eliminadas del torrente circulatorio por la vía del receptor “scavenger” de los
macrófagos, dando lugar a la formación de células espumosas. Igualmente, las HDL pueden sufrir
una glicación, y aunque no se modifica su composición lipídica pierden eficacia en su función de
productora de aflujo del colesterol celular y participación en el transporte reverso del colesterol.
Por otra parte, el descenso de la actividad de la LPL está relacionado con una disminución irregular
de las concentraciones plasmáticas de estas partículas.

MODELOS EXPERIMENTALES
35
1.3 Modelos experimentales de Enfermedad renal progresiva
1.3.1 Modelos animales asociados a diabetes tipo II
En el presente capítulo, se va a enumerar los modelos animales actuales disponibles de diabetes
tipo II. Muchos de estos modelos, la progresión de hiperinsulinemia al de insulinopenia ofrece una
difícil interpretación. Algunas cepas, tales como los ratones NON o las ratas diabéticas Cohen o
OLEFT, presentan, en términos de mecanismos patogénicos, una combinación de diabetes tipo I y
II. También se observan otros problemas. En particular, en los ratones, la información de la presión
sanguínea generalmente no está disponible y esto hace que el análisis de los mecanismos
patogénicos sea más arduo. En muchos modelos de roedores tipo II, en particular en aquellos que
muestran síntomas de síndrome metabólico, la hiperlipidemia se encuentra combinada con
hiperinsulinemia y/o diabetes. Únicamente, como se discutirá en detalle para la rata Zucker Obese,
la hiperlipidemia representa un mecanismo importante de daño renal independiente de la diabetes.
Además, el perfil lipídico de los roedores no es comparable con el del hombre, lo cual impone
restricciones a la hora de extrapolar la diabetes II asociada a la hiperlipidemia. En conclusión, los
mecanismos patogénicos de los cambios renales en muchos de estos modelos son difíciles de
identificar y separar unos de otros. Por lo tanto, la utilidad de estos modelos animales para el
estudio de la implicación de la DMNID en la lesión renal es comprometida.
1.3.1.1 Ratón Kuo Kondo (KK)
El ratón KK fue una de las diferentes cepas que establecieron Kondo y col56 en 1957. Debido la
herencia poligénica del fenotipo diabético no existe un control apropiado de este ratón. La cepa se
caracteriza metabólicamente por una hiperglicemia moderada con niveles de glucosa máximos a los
cinco meses de edad, así como hiperinsulinemia y una mala tolerancia de la glucosa a partir de dos
meses. El ratón KK es moderadamente obeso. Las bases moleculares de esta obesidad no están
claras, pero parece que no se debe a ninguna mutación en el gen ob. La proteinuria incrementa con
la edad. Histológicamente se observa proliferación mesangial y un incremento continuo de
glomerulosclerosis focal y nodular. Sin embargo, estos nódulos son diferentes a los que se
producen en la nefropatía diabética humana, los cuales se localizan cerca del hilo y son menos
prominentes y extensos. Inmunohistológicamente, hallamos deposiciones de inmunoglobulina y del
complemento en el mesangio y a lo largo de la pared del capilar glomerular. Muestra una lesión
exudativa llamada cápsula fibrinoide.

INTRODUCCIÓN
36
1.3.1.2 Ratón KKAy
Ratón KK que expresa el gen Ay, y desarrolla obesidad, hiperfagia, resistencia a insulina, y una
hiperlipidemia severa. El modelo se caracteriza por un inicio temprano y prolongado de niveles
severos de hiperinsulinemia e hiperglicemia. El ratón KKAy desarrolla rápidamente un
engrosamiento de la membrana basal glomerular visible a los tres meses de edad. También, se
observan un aumento de tamaño glomerular y una dilatación de los capilares glomerulares. El ratón
KKAy desarrolla un aumento de la matriz extracelular mesangial. La hidronefrosis mostrada por la
mayoría de estos ratones entre los 6 y 7 meses de edad limita la viabilidad de este modelo. Este
desorden causa una distensión en la vejiga y una alta tasa de mortalidad a los 7 meses de edad
debido al fallo renal57.
1.3.1.3 Ratón obeso New Zealand (NZO)
Modelo animal de enfermedad autoinmune que desarrolla anemia hemolítica de Coomb y
glomérulonefritis. El ratón NZO desarrolla hiperfagia, obesidad, hiperglicemia moderada,
intolerancia a la glucosa, hiperinsulinemia y resistencia a la insulina58. La obesidad es debida a la
resistencia a la leptina. Los cambios renales incluyen proliferación glomerular severa,
engrosamiento de la membrana basal glomerular, glomerulosclerosis, nódulos eosinofílicos en
algunos glomérulos, hialinización ocasional de algunas arteriolas.
1.3.1.4 Ratón ob/ob
El ratón ob/ob tiene una mutación en el gen ob que comporta una deficiencia en el gen de la
leptina. Este ratón fue descubierto inicialmente como una mutación espontánea somática recesiva
que desarrollaba obesidad e hiperglicemia. La cepa del ratón ob/ob C57BL/Ksj desarrolla diabetes
severa. Exhibe una hiperinsulinemia inicial seguida de un rápido desarrollo de la insulinopenia a
causa de la atrofia de las células-β, así como una hiperglicemia severa, provocando la muerte
temprana del animal. La nefropatía no se desarrolla en todos los ratones ob/ob59, mientras que se ha
descrito una susceptibilidad particular de estos ratones a la nefrotoxidad inducida por metales
pesados60.
1.3.1.5 Ratón diabético db/db
Ratón con una mutación del gen recesivo db, que codifica para el receptor de la leptina, dando
como resultado una obesidad marcada y diabetes tipo II. El síndrome diabético se caracteriza por
una hiperinsulinemia temprana, hiperfagia, obesidad e hiperglicemia progresiva61. La
hiperinsulinemia aparece a los 10 días de edad, alcanzando su pico máximo a los tres meses de
edad, a partir del cual va disminuyendo hasta registrar niveles normales. Después de varios meses

MODELOS EXPERIMENTALES
37
el ratón deja de ganar peso, y desarrolla una atrofia de las células-β paralelamente a una
disminución de los niveles de insulina, muriendo antes de llegar a los 10 meses de edad.
1.3.1.6 Rata diabética Cohen
La rata diabética Cohen fue desarrollada mediante selección genética de ratas albinas a partir de
una cepa de la Universidad de Hebrew ya que desarrollaban síndrome diabético cuando eran
alimentadas con concentrados de sucrosa pobres en cobre62. La cepa presenta hiperglicemia,
glucosuria, hiperinsulinemia con un desarrollo de hipoinsulinemia y resistencia a la insulina. La
dieta rica en sucrosa y pobre en cobre es prerrequisito para el desarrollo de la diabetes y sus
posteriores complicaciones.
1.3.1.7 Rata hipertensa diabética Cohen-Rosenthal
Esta cepa se desarrolló con el cruce de ratas diabéticas Cohen y ratas espontáneamente hipertensas.
El emparejamiento fue llevado a cabo entre animales con mayores niveles de glucosa en sangre y
con mayor presión sanguínea, creando así una cepa con DMNID e hipertensión genética. Como en
la rata diabética Cohen, se requiere una dieta rica en sucrosa y pobre en cobre para inducir el
síndrome diabético en esta cepa. Además de una glomerulosclerosis severa se han encontrado
cambios en las arterias y arteriolas que no aparecían en la rata diabética Cohen, consistentes en
necrosis fibrinoides y en una hiperplasia de las células musculares lisas63.
1.3.1.8 Rata Otsuka Long-Evans Tokushima fatty (OLEFT)
Rata espontáneamente diabética con poliuria, polidipsia y obesidad moderada que fue descubierta
en 1984 en una colonia no consanguínea (outbred) de ratas Long-Evans. Las ratas macho OLEFT
se caracterizan por una obesidad moderada y una mala tolerancia a la glucosa a partir de las 8
semanas de edad. También se observan hipertrigliceridemia y una leve hipercolesterolemia a las 8
semanas de edad. A las 24 semanas, desarrollan hiperinsulinemia. Recientemente, se ha visto que
desarrollan hipertensión a partir de las 10 semanas de edad64.
Los machos muestran una albuminuria elevada a las 6 semanas de edad con anterioridad a la
diabetes. Se observa un incremento del tamaño del riñón. La nefropatía en las ratas OLEFT se
caracteriza por la presencia de glomerulosclerosis y lesiones nodulares.
1.3.1.9 Rata hipertensa espontáneamente hipertensa (SHR)
La rata hipertensa espontáneamente hipertensa (o rata Koleski) se generó en 1970 después de una
mutación espontánea que sufrió la descendencia de un cruce entre una hembra Wistar Kyoto

INTRODUCCIÓN
38
espontáneamente hipertensa y un macho normoteso Sprague-Dawley. El fenotipo obeso es debido a
una mutación autosómica recesiva denominada corpulenta (cp) la cual protagoniza el mismo gen
que actúa en la mutación de la rata Zucker obese. La rata SHR está caracterizada por hiperfagia y
obesidad. Entre los 5y 8 meses de edad muestra una marcada trigliceridemia y una
hipercolesterolemia moderada. Además, tiene una mala tolerancia a la glucosa, hiperinsulinemia
alta y resistencia a la insulina. La proteinuria se manifiesta a edades muy tempranas y es
acompañada de hipoproteinemia.
1.3.1.10 Rata corpulenta-NIH/ espontáneamente hipertensa
Es el cruce de un macho heterocigoto Kotetsky para el gen corpulento (cp/+) y una hembra SHR de
una cepa Okamoto. Después de varios cruces, las ratas homocigotas (cp/cp) mostraron ser obesas,
hiperglicémicas, hipertriglicémicas, hiperinsulinémicas e intolerantes a la glucosa65. Los machos
ostentan una mayor intolerancia a la glucosa que las hembras, desarrollando glucosuria. La
hipertensión en los machos de esta cepa desaparece cuando la obesidad entra en escena. Los islotes
pancreáticos ofrecen una hiperplasia de las células-β. Las ratas obesas SHR/N-cp desarrollan
proteinuria y nefropatía a partir de los 5 meses de edad. Tienen los riñones grandes, muestran una
expansión del mesangio y un aumento del número de células. Algunos glomérulos presentan
lesiones nodulares y glomerulosclerosis focal y segmentaria.
1.3.1.11 Rata fatty Zucker diabética
Esta cepa deriva de algunas ratas Zucker que desarrollaban de manera especial altos niveles de
glucosa mediante una selección a lo largo de varias generaciones de cruzamientos. Afecta a los
machos obesos que desarrollan hiperglicemia e hiperlipidemia con niveles de insulina altos. Estos
animales no desarrollan hipertensión. La albuminuria ocurre muy tempranamente y se desarrolla
cronológicamente. Las ratas diabéticas desarrollan hipertrofia renal y glomerulosclerosis focal y
segmentaria. Sin embargo, la presencia de lesiones renales no diabéticas dificulta la interpretación
de la morfología renal y compromete la utilidad de este modelo en la nefropatía diabética.
1.3.1.12 Rata Wistar fatty (fa/fa)
La rata Wistar fatty se desarrolló mediante la transferencia del gen fa de la rata Zucker obese a la
rata Wistar-Kyoto, la cual es menos sensible a la insulina y menos tolerante a la glucosa que la
ratas Zucker lean. Las ratas fatty Wistar-Kyoto muestran obesidad, hiperfagia, hiperinsulinemia,
hiperlipidemia y resistencia a la insulina periférica. Se observa hipertrofia de los islotes
pancreáticos y la degranulación de las células-β pancreáticas. La liberación de insulina pancreática
disminuye en los machos. Únicamente los machos, desarrollan síntomas diabéticos como

MODELOS EXPERIMENTALES
39
hiperglicemia progresiva, polidipsia y glucosuria a las 8 semanas de edad. Por su parte, las hembras
desarrollan hiperglicemia cuando se las alimenta con una dieta alta en sucrosa.66
1.3.1.13 Rata Goto-Kakizaki (GK)
La rata Goto-Kakizaki es una cepa no-obesa y moderadamente diabética que se desarrolló a partir
de un stock de ratas Wistar mediante una selección de aquellos animales que presentaban niveles
de glucosa mayores durante el test oral de tolerancia a la glucosa a lo largo de más generaciones. El
establecimiento de la cepa como DMNID ocurre entre las 3 y 4 semanas de edad, y es el resultado
de varios patomecanismos que incluyen el empeoramiento ontogenético del desarrollo de los
islotes, la liberación anormal de insulina, resistencia a insulina, hiperinsulinemia basal así como
metabolismo anormal de la glucosa. Se produce un engrosamiento de la membrana basal
glomerular a partir de las 12 semanas de edad. Sin embargo, se conoce poco de los cambios
estructurales y funcionales renales de esta rata67.
EL desarrollo de la DMNID, pe hiperglicemia, en las ratas GK no está asociado a ningún hecho del
síndrome metabólico, en particular a la obesidad, hiperlipidemia e hipertensión. En este aspecto, las
ratas GK se diferencian de otros modelos de roedores de DMNID. Se ha observado que la
hiperglicemia e hiperinsulinemia prolongada que se dan en este modelo, no está asociado a ningún
cambio funcional renal, en particular con la albuminuria. A pesar de la ausencia de cambios
funcionales, se pueden observar modificaciones estructurales como: la hipertrofia glomerular
temprana y el engrosamiento de las membranas tubulares y glomerulares. Estos cambios son
similares a los que ocurren en la fase preclínica de la diabetes humana. Por el contrario, no se han
encontrado signos de activación mesangial o de los fibroblastos intersticiales, puestos de manifiesto
por la expresión de novo del filamento intermedio α-actina, proliferación celular o acumulación de
matriz extracelular68.

INTRODUCCIÓN
40
1.3.2 Rata Zucker Obese: Modelo de obesidad, resistencia a la insulina e hiperlipidemia
La rata Zucker obese (RZO) es el modelo de obesidad más idóneo para estudiar la Diabetes
Mellitus II humana69. Esta cepa es el resultado de una mutación espontánea en un cruce entre las
ratas Merck Stock M y Sherman70.
La obesidad parece ser debida a un gen autosómico recesivo denominado fa, donde Fa representa el
alelo normal, que se hereda por herencia mendeliana. Estas ratas, a diferencia de las Zucker lean,
los controles sanos, son obesas, hiperfágicas e hiperinsulinémicas, aunque los niveles de glucosa en
sangre se mantienen dentro de la normalidad.
La RZO es un modelo clave para el estudio de la obesidad y la resistencia a la insulina, así como la
relación de ambos procesos con la patogénesis de la hipertensión sistémica. Además, debido al
desarrollo de la proteinuria y de la glomerulosclerosis previo al establecimiento de la
hiperlipidemia, nos permite analizar la participación de los factores de riesgo asociados a la
enfermedad vascular en el desarrollo de la lesión glomerular71. Estudios experimentales han
demostrado que la edad y la dieta tienen un efecto directo en la respuesta fisiológica de estas
ratas72.
Después del nacimiento, los homocigotos fa/fa no se diferencian de los heterocigotos hasta el tercer
o cuarto día. En la 5ª semana se evidencian diferencias palpables en cuanto al contenido de grasa
del cuerpo; las RZO son hiperfágicas y presentan una actividad física muy baja. Es a partir de este
momento cuando aparecen las anomalías metabólicas que caracterizan a esta cepa: incremento del
contenido de lipoproteínas en el suero, hipertrigliceridemia y hipercolesterolemia. Entre las
semanas 10 y 12 se produce una acumulación de VLDL en ausencia de lesiones glomerulares
aparentes y de microalbuminuria. La glomerulosclerosis empieza aproximadamente a los 6 meses
de edad y entre las 24 y 48 semanas los depósitos de fibronectina, laminina, colágeno IV y ApoB
en el mesangio glomerular son más frecuentes.
Las características más importantes de la RZO se detallan a continuación:
1.3.2.1 Obesidad
Se ha demostrado que la RZO incrementa mucho más el depósito de grasa por gramo de ingesta
que sus homólogas, las ratas Zucker lean. Este hecho indicaría un mayor aprovechamiento de la
comida por parte de la RZO. Además, la hiperfagia suele ir acompañada de la disminución de
actividad física70.

MODELOS EXPERIMENTALES
41
Algunos investigadores han relacionado la obesidad a efectos hipotalámicos73. Estos efectos
explicarían algunas de las anomalías encontradas en el sistema nervioso simpático, así como el bajo
recambio de la norepinefrina en el tejido adiposo marrón. Una reducción en la regulación de este
tejido por parte del sistema nervioso simpático podría ser el causante de la termogénesis incompleta
que contribuiría a la obesidad de las RZO.
Los adipositos de las RZO tienen una medida superior al de los ratas Zucker lean y se acumulan
mayoritariamente formando importantes depósitos subcutáneos70.
1.3.2.2 Características metabólicas y hormonales
La hiperinsulinemia es la característica más conocida de la RZO, por la implicación que tiene como
modelo experimental de DMNID. Está influida por la concentración de carbohidratos de la dieta y
suele aparecer entre las 3 y 4 semanas de edad, manteniéndose los niveles de insulina elevados
independientemente de las condiciones nutricionales. Se incrementa la síntesis de insulina por parte
del páncreas y secreción por las células-β. Asimismo, las RZO son insulino-resistentes.
Estudios recientes parecen demostrar que la resistencia a la insulina es específica del tejido, siendo
mayor en el músculo, donde se detectan niveles bajos de autofosforilación de la Tirosina quinasa C.
Asimismo, en el hígado y el corazón se detecta una actividad baja de la Proteína quinasa C71. Estos
estudios sugieren que la resistencia a insulina puede ser debida a anomalías en el receptor.
También se detectó una especificidad concerniente a la captación de la glucosa; ésta es alta en el
tejido adiposo blanco y se mantiene baja en el marrón y en el músculo. Así, la energía tiende a ser
almacenada en lugar de ser consumida por el tejido adiposo marrón, pudiendo contribuir a la
patogénesis de la obesidad.
Los niveles de glucagon en el páncreas suelen ser normales, aunque su secreción es inferior. Tiene
una función tiroidea alterada, siendo hipotiroideas. La pituitaria de las RZO tiene una tamaño
inferior al de las ratas Zucker lean. La función reproductora está alterada; las hembras tienen el
tracto genital atrofiado y los machos presentan una disminución del tamaño testicular. La secreción
de gonadotropinas es defectuosa y se incrementa la producción de esteroides suprarrenales70.
Las RZO desarrollan hígados grasos. La albúmina sérica se encuentra en mayor concentración.
Asimismo, no se encuentran diferencias en el hematocrito ni en el número de células blancas
sanguíneas. La agregación plaquetaria y la producción de tromboxano también se mantienen en
niveles normales74.

INTRODUCCIÓN
42
1.3.2.3 Hipertensión
Estudios comparativos de la RZO y la RZL ponen de manifiesto que los dos tipos de rata tienen
una presión muy similar y únicamente tiende a aumentar en las RZO adultas (de más de 6 meses de
edad). De estos estudios se puede concluir que el ligero aumento de presión que se detecta en la
RZO adulta se da con posterioridad al desarrollo de la lesión renal y es independiente de la
obesidad y la resistencia a la insulina73.
1.3.2.4 Hiperlipemia
En la RZO los triglicéridos son los lípidos que más influyen en el desarrollo de la obesidad y el
establecimiento de la lesión glomerular. La hipertrigliceridemia, que ocurre en la RZO a las dos
semanas de nacimiento, con la edad llega a alcanzar niveles 10 veces superiores a las RZL. El
colesterol, en cambio llegará a ser el doble. Además de los cambios que se producen en la
composición de las lipoproteínas, recuerdan a las anomalías que se produce en la DMNID: las
lipoproteínas VLDL son menos densas y con un elevado contenido de triglicéridos75.
El hígado es el órgano implicado en el incremento de lipoproteínas circulantes. El aumento de la
lipogénesis es característico de las ratas jóvenes, y suele estar asociado a una disminución de la
oxidación hepática de los ácidos grasos. El resultado final es el incremento de la síntesis de
lipoproteínas ricas en triglicéridos. Además, el catabolismo de los triglicéridos circulantes es
inferior que en las RZL. A pesar de todo esto (el tejido adiposo marrón, el tejido principal de
oxidación de los ácidos grasos), la actividad de la lipoproteína lipasa disminuye, aumentando en el
tejido adiposo blanco, lugar de almacenamiento76.
La patogénesis de las anomalías lipoproteícas es desconocida. Su papel en la lesión glomerular
parece estar relacionado con las características aterogénicas de las VLDL, las cuales debido a su
tamaño y composición, facilitan su unión a los receptores situados en la membrana plasmática de
los macrófagos. Se forman así las células espumosas. Además, parece que el pequeño tamaño de
las partículas de las VLDL facilite la filtración a través de la pared arterial, aportando así grandes
cantidades de colesterol a la pared y al mesangio glomerular. A pesar de esta facilidad de paso del
colesterol a través de la pared arterial, parece que la arteriosclerosis en la RZO no se desarrolla de
forma espontánea. Esta resistencia a la génesis de la arteriosclerosis es compartida por la mayoría
de cepas de rata.

MODELOS EXPERIMENTALES
43
1.3.2.5 Lesión Renal
En las RZO, la proteinuria se inicia a los 3 meses de edad y progresa hasta que la
glomerulosclerosis se hace evidente a los 6 meses y se produce hipoalbuminemia. Las RZL, en
cambio, presentan niveles de proteinuria y lesión renal muy inferiores y, como en muchas cepas de
ratas depende de la edad.
Encontramos un paralelismo en las lesiones histológicas presentes en las RZO y las RZL
envejecidas, mostrando las primeras un desarrollo de la lesión renal más acelerado.
El mesangio se expande como consecuencia de la proliferación celular y el acumulo de
macromoléculas y proteínas. Estudios realizados valorando la concentración de fibronectina en el
mesangio glomerular han mostrado el acumulo de proteínas junto con la disminución de la
actividad proteinasa en la matriz mesangial77.
Las células epiteliales se desenganchan de la MBG y se fusionan sus podocitos
Dilatación de los túbulos distales con depósitos de eosinófilos, infiltraciones de células
mononucleares y fibrosis
En las arterias y arteriolas se detecta una proliferación de las células musculares lisas de la capa
media y una pequeña proliferación de la íntima.
Actualmente, se desconocen cuales son las causas que originan las lesiones glomerulares, asimismo
diferentes autores han intentado relacionarla con la hemodinámica glomerular, la hiperlipidemia,
los niveles de lipoproteínas circulantes o la reducción de los niveles de PUFAs tisulares71.
Los factores hemodinámicos como la presión glomerular, la tensión de la pared capilar y el tamaño
glomerular parecen influir en la progresión de la lesión renal y no en su origen, ya que sus valores
incrementan cuando ésta ya esta establecida. Se ha visto que el tratamiento de RZO con IECAs
reduce la presión sanguínea, los lípidos séricos, la área glomerular, la albuminuria y la
glomerulosclerosis focal, en antítesis a una elevada presión capilar78.
Para probar si la hiperlipidemia contribuía al inicio y progresión de la glomerulosclerosis, RZO se
trataron con diferentes hipolipemiantes79. Tanto la lovastatina como el ácido clofibrico redujeron
los lípidos séricos y la lesión glomerular.

INTRODUCCIÓN
44
1.4 Mecanismos de progresión de la lesión renal
Los mecanismos que subyacen en la progresión de la ERP son difíciles de establecer debido a
varias razones. En primer lugar, el glomérulo y el intersticio tienen un repertorio limitado de
respuestas ante un insulto, por lo tanto el riñón actúa de una manera más o menos similar frente a
una gran variedad de agresiones diferentes. La principal manifestación de estas causas de lesión
renal es la glomerulosclerosis focal y segmentaria. En ésta, las estructuras celulares especializadas
son reemplazadas por fibroblastos, colágeno y matriz mesenquimal, con interrupción de las
funciones renales como la filtración, la secreción y la reabsorción. Además, cuando se examina un
riñón en fases finales es imposible elucidar la causa de la esclerosis o adivinar el insulto inicial.
En segundo lugar, el riñón dañado tiene tendencia a deteriorarse. En modelos experimentales, se ha
demostrado que la insuficiencia renal establecida puede progresar incluso después de haber cesado
el insulto inicial. Por lo tanto, el riñón por sí mismo contribuye a su deterioro como resultado de
sus respuestas a la lesión, respuestas que sólo son identificables inicialmente80.
Los cambios que caracterizan a las fases iniciales son una disfunción de capilares en el glomérulo,
una proliferación celular en áreas localizadas y un scarring progresivo, alcacanzando el colapso de
la luz capilar. Los túbulos cercanos a los glomérulos lesionados suelen atrofiarse, siendo rodeados
por células inflamatorias. Se produce una fibrosis difusa caracterizada por depósitos de colágeno,
matriz mesenquimal, lípidos y fibroblastos. La membrana basal glomerular de los túbulos y
glomérulos aumenta su grosor durante las primeras fases de la nefropatía, y se condensa en un
material amorfo al final de la misma81.
Una gran variedad de enfermedades progresa hacia disfunciones renales terminales, como la
glomérulonefritis crónica, la nefropatía diabética, y la enfermedad del riñón poliquístico. Aunque el
problema subyacente a menudo no puede ser tratado, existen estudios experimentales en animales y
estudios preliminares en humanos que sugieren que el avance de la enfermedad renal progresiva
puede deberse principalmente a factores hemodinámicos como la hipertensión glomerular sistémica
e intraglomerular, la hipertrofia glomerular y a factores no hemodinámicos como la precipitación
intrarrenal de fosfatos cálcicos, la hiperlipidemia, la angiotensina II y los factores de crecimiento
relacionados con la expansión de la matriz extracelular82.
1.4.1 Cambios hemodinámicos
1.4.1.1 Hipertensión glomerular
La pérdida de masa renal por extirpación o por el curso de una nefropatía trae consigo adaptaciones
funcionales y estructurales en las nefronas remanentes que permiten suplir en parte la función de

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
45
las que se han perdido. Se ha establecido que el factor predominante por el cual la hiperfunción
glomerular produce lesión renal es la elevación de la presión intraglomerular en detrimento del
aumento en filtración y flujo83. Se han realizado estudios84 en ratas con reducción de 2/3 de la masa
renal izquierda e hipertensión sistémica causada por estenosis de la arteria renal derecha,
observándose un aumento significativo del flujo plasmático glomerular de 98-254 nl /min y de la
filtración glomerular por nefrona de 30 a 75 nl /min con cambios en la presión capilar glomerular
(44 vs 43 mm Hg). Cuando se añadía hipertensión arterial, el flujo plasmático y la filtración
aumentaban en la misma proporción (258- y 77 nl /min), sin embargo la presión capilar glomerular
resultaba ser significativamente más alta que en los grupos control (52mm Hg). Además, el estudio
histológico de los glomérulos mostró francas alteraciones estructurales. que consistían en una
expansión y proliferación mesangial, una dilatación de asas capilares, una formación de
microaneurismas y una esclerosis glomerular segmentaria y global. Resultados semejantes se han
obtenido en otros estudios en ratas con ablación extensa de la masa renal y en ratas con diabetes
experimental en las que la disminución de la presión glomerular, inducida con inhibidores de la
enzima conversora de la angiotensina, prevenía la esclerosis glomerular y el flujo glomerular84.
Wardle y col85 interpretaron el papel de la hipertensión glomerular como inductora de una tensión
mecánica en las paredes de los capilares que contribuiría a crear la lesión glomerular y provocaría
una respuesta de las células glomerulares al estrés. También comprobaron que con la hipertensión,
la tensión de las paredes podía aumentar un 500%, siendo normal la pérdida de proteínas a través
del endotelio fenestrado. Además, las células endoteliales son sensibles al estrés, respondiendo a
éste con la secreción de substancias como el factor de Willebrand, la tromboplastina y la trombina
(importantes en la agregación plaquetaria) y diferentes factores de crecimiento como el PDGF,
EGF, FGF, IGF y TFG-β.
En condiciones normales la presión del capilar glomerular está determinada por las resistencias
arteriolares pre y postglomerulares y no varía con los cambios de presión arterial; esta capacidad de
autorregulación es determinada principalmente por los vasos preglomerulares. La elevación
sostenida de la presión capilar glomerular en modelos experimentales que progresan a la esclerosis
glomerular sugiere la existencia de una disfunción de los vasos preglomerulares. La importancia de
la integridad funcional de estos vasos se ha puesto de manifiesto en ratas con hipertensión genética
que acreditan una alta resistencia de la arteria eferente en respuesta a la hipertensión arterial,
manteniendo normal la presión en el capilar glomerular y sin que muestren lesiones estructurales
glomerulares. Por el contrario, si a estas ratas se les extirpa un riñón, la resistencia arteriolar
disminuye, permitiendo la trasmisión de una mayor proporción de presión sistémica al capilar
glomerular dando como resultado la esclerosis del glomérulo.

INTRODUCCIÓN
46
La alteración de los vasos preglomerulares y la hipertensión glomerular resultante están presentes
durante la progresión de la lesión renal incluso cuando no existe hipertensión arterial sistémica.
Estudios experimentales en nefropatía diabética, en nefropatía membranosa y en nefropatía por
puromicina han demostrado que el desarrollo de la esclerosis glomerular es precedido por la
hipertensión del glomérulo a pesar de tener cifras normales de tensión arterial.
Estudios realizados durante los años treinta en ratas con nefritis experimental, demostraron que
dietas hipoproteicas atenuaban las lesiones histológicas y retardaban su progresión hacia la
insuficiencia renal. También, se puso de manifiesto que el efecto beneficioso de dicha dieta se
debía a que la resistencia de los vasos preglomerulares era elevada y en consecuencia la presión
glomerular permanecía normal. Por el contrario, la dieta normal inducía vasodilatación renal,
aumento de la presión glomerular y esclerosis progresiva. Estudios con otras nefropatias como la
nefritis por suero nefrotóxico y la diabetes mellitus confirmaban este efecto de la dieta
hipoproteica86. Más tarde Mizuri y col87 demostraron que la dieta baja en proteínas aumentaba la
expresión del mARN de la renina y del enzima convertidor de la angiotensina (ACE), siendo estos
factores pronósticos de la progresión de la nefropatía diabética.
El tratamiento antihipertensivo con inhibidores de la enzima de conversión de la angiotensina ha
resultado eficaz para disminuir la hipertensión glomerular y prevenir la esclerosis, tanto en los
modelos experimentales de nefropatía asociada a hipertensión arterial como en la nefrectomia
extensa de la masa renal. Se ha sugerido que la eficacia de este tratamiento se debe a la supresión
de los efectos de la angiotensina II, tanto al nivel de circulación periférica como en la disminución
de la presión de los capilares intraglomerulares88 y a la ausencia del efecto hipertrofiante que la
angiotensina ejerce en las células glomerulares89.
El empleo de la heparina también ha resultado ser eficaz en la prevención de la lesión renal. Su
efecto parece ser independiente de su acción anticoagulante y de cambios en la presión glomerular.
Por otra parte, la heparina inhibe el crecimiento de células glomerulares in vitro, por lo que se ha
sugerido que la protección renal se obtiene al inhibir la proliferación de células mesangiales
estimuladas por el daño endotelial que causa la hipertensión glomerular.
1.4.1.2 Hipertensión sistémica
La hipertensión sistémica tiene efectos adversos en el riñón e inicia el desarrollo de nefropatías
(como la nefrosclerosis hipertensa) o acelera la pérdida de función renal en nefropatias ya
establecidas. Los mecanismos por los cuales la hipertensión daña el riñón en la enfermedad renal
no están del todo claros. Se cree que produce hipertrofia vascular, la cual a su vez ocasiona una
esclerosis vascular isquémica. Además, la hipertensión sistémica provoca esclerosis glomerular

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
47
mediante la inducción de presión intraglomerular, con aumento del flujo y tasa de filtración por
nefrona, la cual intensifica la lesión endotelial90.
1.4.1.3 Hipertrofia glomerular
La progresión de las lesiones glomerulares hacia la esclerosis se acompaña invariablemente de
hipertrofia glomerular, tanto en condiciones experimentales como en clínicas. Así por ejemplo, la
extirpación extensa de la masa renal produce hipertrofia acentuada en las nefronas remanentes
antes de que aparezca esclerosis glomerular. La presencia de un estímulo hipertrofico como la
uninefrectomia acelera el proceso glomerulosclerótico en distintas condiciones experimentales,
como en el caso de la glomérulonefritis por suero nefrotóxico. En la diabetes mellitus, la hipertrofia
de las células mesangiales y tubulares es una característica común y muy temprana y precede a la
esclerosis91.
Figura 14. Papel central del p27kip1 en la inducción de la hipertrofia en las células renales
En un estudio clásico de Pabst y Sterzel92 se investigó la tasa de renovación que tenían las células
glomerulares en una rata mediante estudios de autoradiografia. Encontraron que las células
endoteliales tenían una tasa de renovación de un 1% mientras que las células mesangiales exhibían
una tasa de proliferación muy baja. Sin embargo, la presencia de estímulos patofisiológicos
inducían el crecimiento de las células glomerulares y tubulares. En concreto, las células renales en
reposo pueden tener dos tipos de respuesta al crecimiento: la hiperplasia o la hipertrofia. Las

INTRODUCCIÓN
48
células que están programadas para proliferar progresan a través de su ciclo celular normal,
replican su contenido de ADN y se dividen durante la mitosis. En cambio, las células que se
hipertrofian interrumpen su crecimiento en la fase G1 del ciclo celular; aumentan su tamaño, su
contenido de ARN y de proteínas y no pueden duplicar su ADN porque todavía no han pasado a la
fase S del ciclo celular.
Recientemente Wolf93, trabajando con células tubulares proximales estimuladas con angiotensina II
y con células mesangiales incubadas en glucosa, observó una acumulación del inhibidor de la
quinasa dependiente de ciclinas, p27kip1 (punto de transición del ciclo G1/S) como mecanismo
molecular de la hipertrofia renal94. Así propuso que la detención de las células en fase G1 da lugar
a hipertrofia y probablemente a la acumulación de proteínas de matriz extracelular. En concreto,
observó que las células mesangiales cultivadas en un medio rico en glucosa adquirían la expresión
estimulada de p27kip1a través de la activación de la proteína quinasa C (PKC). Por otro lado, la
hipertrofia inducida por la angiotensina II también dependía de la expresión de p27kip1, la cual
estaba estimulada por radicales libres de oxígeno. Así mismo, vió que tanto la glucosa como la
angiotensina II inducían la expresión de TGF-β, la cual también activaba p27kip1, que interactuaba
con quinasas dependientes de ciclinas inhibiendo su actividad (figura 13).
1.4.2 Cambios no hemodinámicos.
1.4.2.1 Factores de crecimiento.
1.4.2.1.1 Características generales de los factores de crecimiento.
Los factores de crecimiento (FC) son polipéptidos producidos y liberados por diferentes tipos de
células. Éstos forman un grupo de moléculas solubles que regulan una gran variedad de eventos
inmunes y respuestas inflamatorias que incluyen la inflamación, el crecimiento celular,
cicatrizaciones y respuestas al deterioro celular. A pesar de la importancia crucial de estos
mediadores, el estudio de este subtipo de citocinas es muy complejo. Esto es debido principalmente
a que estas moléculas tienen un amplio espectro de efectos con múltiples interacciones y efectos
sinérgicos, antagónicos o solapados, hechos que complican considerablemente la comprensión de
sus papeles biológicos95.
Propiedades generales:
Ø La secreción de FC es un proceso breve y limitado. En presencia de estímulos específicos,
las citocinas son rápidamente sintetizadas y liberadas por exocitosis en pequeñas
cantidades (nanomolar). Generalmente no son almacenadas, y su producción cesa cuando
desaparece el estimulo. La regulación de la producción de FC ocurre generalmente a nivel

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
49
transcripcinal, aunque el control a nivel postranscripcional también interviene con
frecuencia. El Mari de las citocinas contiene una región 3’ rica en AU que le confiere
inestabilidad y una vida media corta. Los polipéptidos de las citocinas también tienen una
vida media corta. Por lo que la regulación de estos factores es rápida y precisa.
Ø Las FC inician su acción uniéndose a receptores específicos en la superficie de las células
diana. La función más importante que realizan es una regulación local de las células
vecinas, la regulación paracrina. También pueden actuar de una manera autocrina,
estimulándose ellas mismas. Algunos FC tales como IL-1, IL-6 y TNF-α también tienen
efectos endocrinos, ejerciendo efectos en células diana a través de la circulación, tal y
como lo hacen las hormonas.
Ø Los FC tienen una acción pleyotrópica. Los FC no sólo son producidos por diferentes tipos
de células sino que también actúan sobre diversos tipos celulares.
Ø Muchas acciones de los FC son redundantes. Existen muchas funciones originalmente
atribuidas a una citocina que comparten con otros FC.
Ø Los FC a menudo influyen en la síntesis de otros FC, formándose cascadas en las cuales la
segunda o tercera citocina actúa sobre la primera.
Ø Los FC a menudo influyen en la acción de otros FC. Dos FC pueden interactuar para
antagonizar la acción de la otro FC, para producir efectos aditivos o sinérgicos.
Familias de los factores de crecimiento:
Se clasifican en 8 familias: (1) del PDGF; (2) del EGF; (3) del FGF; (4) del IGF; (6) del TGF; (7)
del NGF; (7) del HGF y (8) algunas citocinas: familia que incluye a las interleuquinas, los
interferones, los factores de necrosis tumoral (TNF), los factores estimuladores de colonias (CSF),
las quimoquinas y otros.
1. FAMILIA del PDGF (factor de crecimiento derivado de plaquetas): PDGF-AA, PDGF-BB,
PDGF-AB.
2. FAMILIA del EGF (factor de crecimiento epidérmico, urogastroma): TGF-α, VGF, SGF.
3. FAMILIA del FGF (GBGF: factor de crecimiento de unión a la heparina): FGF, HBGF,
FGFa, FGFb, FGF-3, FGF-8, FGF-9, MGSA.
4. FAMILIA del IGF: Insulina, IGF-1 (factor de crecimiento similar a la insulina,
somatomedina C), IGF2 (somatomedina A)

INTRODUCCIÓN
50
5. FAMILIA del TGF-β: superfamilia de los factores transformante de crecimiento (TGFβ1-3
y 21 miembros más como activinas, inhibinas, etc).
6. FAMILIA del NGF (factor de crecimiento nervioso, neurotrofinas): BDNF, NT 4/5
7. FAMILIA del HGF-SF: HGF (factor de crecimiento de los hepatocitos), SF (factor de
diseminación)
8. CITOCINAS: nombre general de los factores de crecimiento que afectan principalmente a
las células sanguíneas (hematopoyéticas) y que se dividen en: interleuquinas, interferones,
factores de necrosis tumoral (TNF), factores estimulante de colonias (CSF) y quimocinas.
Ø Interleuquinas: IL-1α y IL-1β, IL-2, IL-3 a la IL-18.
Ø Interferones: IFNα, IFNβ, IFNγ.
Ø Factores de necrosis tumoral (TNF): TNFα (caquetina).
Ø Factores estimuladores de colonias: G-CSF (de granulocitos), M- CSF(de
macrófagos)
Ø Eritropoyetina (EPO)
Ø Trombopoyetina
Ø LIF: Factor inhibidor de leucemia
Ø Quimocinas
1.4.2.1.2 Rutas intracelulares activadas por los factores de crecimiento
Componentes de las rutas de señalización
Los factores de crecimiento ejercen su función a través de la interacción con receptores específicos
en la superficie de las células. Se trata de proteínas localizadas en la membrana, en las que se
pueden distinguir tres regiones: (1) una región extracelular, orientada hacia el exterior de la célula,
encargada de la unión del ligando, (2) una región que atraviesa la membrana y (3) una región
intracelular, orientada hacia el citoplasma, responsable de la transmisión de la señal (Figura 14).
Cuando un factor estimulador del crecimiento liberado al espacio intercelular por parte de células
vecinas, se une al dominio extracelular de su receptor situado en la célula diana, éste se activa y
transmite una serie de señales al interior de la célula. Estas señales suponen la activación de

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
51
proteínas citoplasmáticas que, a su vez, emiten señales a toda una sucesión de proteínas distintas,
en una cascada dirigida hacia el núcleo celular. Aquí, otras proteínas, los factores de transcripción,
responden activando la expresión de un conjunto de genes, que son los que permiten que la célula
entre en el ciclo celular. Las señales negativas recibidas desde el exterior actúan mediante rutas de
señalización similares a la descrita y, de hecho, existen múltiples puntos de interacción entre ambos
tipos de señales, lo que permite la integración de informaciones diversas.
En esencia, únicamente hay dos mecanismos bioquímicos por los que las proteínas citoplasmáticas
reciben la señal de los receptores. Uno de los mecanismos implica la fosforilación de proteínas en
residuos de serina, treonina o tirosina. Diversas proteínas señalizadoras de esta categoría
denominadas quinasas transfieren grupos fosfato de la molécula donadora ATP a los aminoácidos,
mencionados en la proteína receptora. La fosforilación de proteínas tiene dos funciones
fundamentales en transducción de señales. En primer lugar, puede cambiar la conformación y,
como consecuencia, provocar una alteración de la actividad biológica de la proteína fosforilada. En
segundo lugar, la fosforilación de los residuos de tirosina genera sitios de reconocimiento para
proteínas señalizadoras. La fosforilación de determinados residuos de una proteína puede regular su
actividad enzimática, su capacidad de interacción con otras proteínas (o con el ADN en el caso de
los factores de transcripción), su localización subcelular, etc. En su control están implicadas
quinasas (enzimas que catalizan la transferencia de grupos fosfato a sustratos específicos) y
fosfatasas (enzimas que catalizan la eliminación de estos fosfatos). Se trata por lo tanto, de un
mecanismo de señalización reversible y dinámico. La fosforilación reversible de proteínas
constituye uno de los principales mecanismos de señalización implicados en el control de la
fisiología celular.

INTRODUCCIÓN
52
Receptores deMembrana
Tirosina QuinasasCitoplasmáticas
Adaptadores
Proteínas G
Efectores
Proteínas CitoplasmáticasTransductoras de señañes
Señal extracelular
Expresión de Genes
ADN
Alteración delcomportamiento celular
Citoplasma
Nucleo
Proteínas Nuclearesde unión al ADN
Figura 15. Componentes de la ruta de señalización.
El segundo mecanismo por el que se transmite la señal implica la actividad enzimática GTPasa. Las
proteínas que se unen a GTP actúan como interruptores moleculares y se denominan proteínas G.
La unión de GTP provoca una alteración en la conformación que mantiene la proteína G en estado
activo, capaz de transmitir la señal mediante la interacción con proteínas efectoras. La hidrólisis del
GTP a GDP, catalizada por la GTPasa intrínseca, devuelve la proteína G a un estado inactivo.
Existen reguladores positivos que promueven la transición de la proteína G a su estado activo,
unida a GTP, a través de una actividad permutadora de nucleótidos, y de reguladores negativos o
efectores que devuelven la proteína G a su estado inactivo, unida a GDP, mediante la estimulación
de la actividad GTPasa intrínseca
Rutas metabólicas activadas por receptores de factores de crecimiento
La actividad enzimática de los receptores reside en un dominio catalítico presente en la región
intracelular. Para que la enzima sea activa es necesaria la unión del ligando en el exterior de la
célula; estos receptores funcionan como enzimas alostéricas asociadas a la membrana. La
activación con frecuencia requiere la oligomerización de los receptores (asociación funcional de
varias moléculas del receptor). La unión del factor de crecimiento al dominio extracelular origina
un cambio conformacional en el receptor. En muchos casos, los propios factores de crecimiento son

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
53
capaces de unirse a dos moléculas del receptor al mismo tiempo, facilitando así la dimerización
necesaria para que el receptor se active. Este fenómeno de oligomerización es común a la mayoría
de los receptores para factores de crecimiento, incluso para aquellos sin actividad quinasa
intrínseca.
Una de las consecuencias de la dimerización de los receptores es su fosforilación cruzada. Cada
una de las moléculas del receptor fosforila determinados residuos de tirosina en la molécula
adyacente y viceversa. Este mecanismo cumple dos funciones: por un lado, mantiene el dominio
catalítico de los receptores en una conformación activa y, por otro, crea sitios de anclaje para
“reclutar” y asociar al receptor proteínas citosólicas que reconocen tirosinas fosforiladas. Algunas
de las proteínas que son reclutadas por el receptor activado y fosforilado serán activadas, a su vez,
por un nuevo fenómeno de fosforilación. En otros casos, la asociación al receptor sirve para atraer
diversas proteínas solubles presentes en el citoplasma de la célula a una zona determinada proxima
a la membrana.
En los últimos tiempos se han descubierto una gran parte de los componentes de las vías de
transducción de señales y se ha determinado que las proteínas de la familia Ras son reguladoras
cruciales de los procesos que parten de los receptores de factores de crecimiento y controlan la
proliferación y diferenciación celular. La activación de Ras en respuesta a la estimulación de los
receptores de factores de crecimiento es favorecida por la unión de la proteína adaptadora Grb2,
cuyo dominio SH2 reconoce residuos de tirosina fosforilados en el receptor. A su vez, Grb2
interacciona a través de sus dominios SH3 con el factor de intercambio de nucleótidos de guanina
(Guanine nucleotide Exchange Factor, GEF) Sos, localizándolo en la membrana plasmática, donde
se encuentra situado su sustrato Ras. De esta manera, Sos puede estimular el intercambio de GDP
por GTP en Ras, lo que lleva a Ras desde su forma inactiva a su forma activa con capacidad de
interacción con diversas moléculas efectoras/sustratos.
La conversión de Ras a su forma soluble unida a GTP le permite interaccionar con otras proteínas
que funcionan como efectores. Un efector de Ras es la serina/treonina quinasa Raf. La activación
de Ras promueve su interacción con Raf y localiza esta última en la membrana, donde es activada y
actúa. La proteínas Raf así activadas fosforilan y activan MEK, que es una tirosina/treonina
quinasa. MEK a su vez fosforila las proteínas ERK (Extracellular-signal-Regulated Kinases) que
son serín/treonín quinasas. Las proteínas ERK fosforilan entonces varias proteínas, entre las que
cabe destacar el factor de transcripición Elk-1. Esta cascada de señalización se conoce como la
ruta de las proteína quinasas activadas por mitógenos (Mitogen Activated Protein Kinases,
MAPK). La respuesta celular provocada por esta ruta de señalización incluye el incremento en la
trancripción de genes precursores, como fos, que a su vez regulan la expresión de genes cuyos

INTRODUCCIÓN
54
productos controlan la progresión en el ciclo celular, lo que finalmente da lugar a la síntesis de
ADN y a la división celular (Fig5).
La ruta de la quinasa de lípidos PI3K es otra importante vía de señalización activada por
receptores de membrana en respuesta a la unión con sus ligandos. La actividad PI3K se incrementa
en respuesta a numerosos estímulos extracelulares, especialmente aquellos que implican la
activación de receptores con actividad tirosina quinasa. La proteína PI3K está compuesta por dos
subunidades, una subunidad reguladora con dos dominios SH2 y un dominio SH3, denominada
p85, responsable de la unión con el receptor fosforilado tirosina, y una subunidad catalítica
denominada p110. La actividad de la PI3K genera fosfoinosítidos PI3, 4,5P3, 4P2 y PI3P, y su
activación incrementa los niveles de los dos primeros en la membrana plasmática. Así, la
estimulación de la actividad PI3K induce la activación de la quinasa Akt. Akt es una proteína
serín/treonín quinasa con un dominio PHD de unión a fosfoinosítidos en la región aminoterminal.
La activación de Akt en la célula depende de la inducción de su unión con el fosfoinosítido PI3, 4,
5P3 en la membrana plasmática, donde la proteína translocada es activada por fosforilación por
parte de la senina/treonina quinasa Pdk1, que también posee un dominio PH y cuya activación
también depende, por tanto, de PI3K. Se ha descrito que Akt y Pdk1 están implicadas,
probablemente de manera indirecta, en la activación de la quinasa S6k, que participa en el control
de la síntesis de proteínas y del ciclo celular. Además, recientemente se ha visto que Akt juega un
papel en la protección de la apoptosis mediante la fosforilación de la proteína proapoptótica Bad y
de la caspasa 9, así como del factor de transcripción FKHR1.
1.4.2.1.3 Participación de los factores de crecimiento en la esclerosis glomerular.
La mayoría de enfermedades renales comparten los cambios histológicos que caracterizan las
últimas etapas: en el glomérulo, el número de capilares disminuye, se da una proliferación celular y
scarring progresivo en áreas localizadas que termina con un colapso progresivo de la luz de los
capilares. Los túbulos cercanos a los glomérulos lesionados suelen atrofiarse y se rodean de un gran
número de células inflamatorias. Se produce una fibrosis difusa caracterizada por depósitos de
colágeno, matriz mesenquimal, lípidos y fibroblastos. La MBG de los glomérulos y túbulos suele
engrosar durante las primeras fases de la nefropatía y se condensa en un material amorfo al final de
la misma. Al inicio de la glomerulosclerosis, aparece proteinuria precedida de reducciones en la
filtración glomerular y en el flujo sanguíneo renal. La atrofia tubular se manifiesta con un
empeoramiento en la capacidad de concentrar orina y excretar ácido.
Gran número de nefropatías y su progresión hacia estados finales son mediadas, en parte, por los
efectos de la angiotensina II, que estimula la expresión de factores de crecimiento y citocinas, tales
como el factor transformante del crecimiento (TGF-β1), el factor de necrosis tumoral (TNF-α), el

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
55
factor de crecimiento derivado de las plaquetas (PDGF), el factor de crecimiento fibroblástico
básico (bFGF) y el factor de crecimiento “similar” a la insulina (IGF). La mayoría de estos
componentes promueven el crecimiento celular y la fibrosis. A continuación vamos a enumerar los
principales factores que influyen en la progresión de la enfermedad renal crónica, dedicando la
última parte del capítulo a desarrollar de forma más detallada las dos citocinas estudiadas en el
presente estudio experimental, el TGF-β y el PDGF-B.
HGF (Hepatocyte Growth Factor)
Estudios recientes han demostrado que la expresión del HGF es uno de los principales efectores
que median la regeneración del hígado y el crecimiento del riñón, llevando en algunos casos al
daño tisular. Se ha visto en riñones de ratas diabéticas, que tanto el HGF, su receptor tirosin
quinasa como el producto del oncogen c-met, muestran un incremento en su expresión96. De todas
maneras, quedan muchos aspectos a estudiar para determinar el papel que desempeña exactamente
el HGF en la hipertrofia renal durante el desarrollo de la diabetes.
Por otro lado, se ha visto que el producto de c-met es inducido por una gran variedad de factores de
crecimiento tales como el TGF-β, el PDGF, el IGF-1 y el HGF. Un mecanismo potenciador del
HGF y la inducción de c-met juegan un papel restaurador de la función tubular y de la integridad
renal con posterioridad a la lesión renal. El papel protector del HGF parece estar relacionado con
una supresión de la apoptosis de las células epiteliales97
IGF-1 (Insulin like growth factor)
Se produce en situaciones como la hipertrofia renal compensadora, la diabetes y también en el
tejido renal normal adyacente al infartado. Los lugares de producción más relevantes de este factor
de crecimiento son: el tubo colector, la rama ascendente gruesa del asa de Henle, el túbulo
proximal y el mesangio.
Se cree que la acumulación renal del IGF-1 contribuye al desarrollo de la nefropatía diabética. Esta
acumulación depende directamente del nivel de glucosa en sangre, o indirectamente de sistemas
vasodilatadores como el NO y/o los eicosanoides, provocando una disminución de las resistencias
vasculares renales y aumentando la función y el tamaño del riñón en el diabético. Por otra parte, el
IGF-1, al igual que la insulina, aumenta la síntesis de ADN en células mesangiales y la tasa de
síntesis de los componentes de la matriz extracelular como la laminina, la fibronectina y el
colágeno IV. Se ha comprobado, además, que refuerza los efectos proliferativos de otros factores
de crecimiento, por lo que podría considerarse un factor de progresión del ciclo celular98.

INTRODUCCIÓN
56
bFGF (basic fibroblast growth factor)
Es un mitógeno potente que estimula la proliferación de múltiples tipos celulares en cultivo como
son mioblastos, células endoteliales vasculares, fibroblastos, y células musculares lisas99. Este
efecto mitogénico es el responsable, en parte, de la proliferación durante el desarrollo y de la
respuesta celular en múltiples procesos patológicos. Karpen y col100 observaron que los niveles de
mARN de bFGF en el glomérulo de las ratas diabéticas aumentaban en el tiempo además de
incrementar paralelamente el marcador de proliferación celular, PCNA101. Por otro lado, estudios
en cultivos celulares han demostrado que el bFGF es un factor de competencia del ciclo celular.
EGF (Epidermal growth factor)
Es otro factor de crecimiento implicado en la fisiopatología renal. Así por ejemplo, es considerado
mitogénico para las células tubulares102. Este factor estimula el transporte de Na+/K+, activa la
glicólisis e incrementa la síntesis proteica, produciendo una vasoconstricción arteriolar aferente y
eferente.
TNF- umor Necrosis Factor) y IL-1 (interleuquina 1):
La activación del gen del TNF-α lleva a la producción de una proteina de 230 aminoácidos. La
molécula de TNF-α madura está formada por 157 aa con un peso molecular de 17 Kd. Los
macrófagos son la fuente principal de TNF-α, aunque las células residentes del glomérulo (células
mesangiales y las células del epitelio tubular) son productoras también de esta molécula. Actúan
activando factores de transcipción, citocinas, receptores, moléculas de adhesión, mediadores de
procesos inflamatorios y proteinas del complejo de inmuhistocompatibilidad. El TNF-α tiene un
papel en el reclutamiento de células inflamatorias, mediante la estimulación de quimocinas como
MCP-1, RANTES, ICAM-1, ECAM-1, VCAM-1103.
VEGF (vascular endotelial growth factor)
Anteriormente se le denominó factor de permeabilidad vascular (VPF). Es una citocina muy
potente que induce angiogénesis y aumenta la permeabilidad endotelial104. Estudios in vitro han
mostrado que el VEGF es estimulado en condiciones de hiperglicemia, hipoxia, hipertensión, y en
presencia de angiotensina II e IGF-1. Así, se ha encontrado que los niveles de VEGF son elevados
en el curso de la diabetes experimental y humana105.
CTGF (Connective tissue growth factor):
Es uno de los mediadores inducidos por el TGF-β y entre otros estímulos, modula el crecimiento de
los fibroblastos y la secreción de ECM de las células mesangiales. Este factor de crecimiento fue

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
57
inicialmente descubierto por Masson106. Recientemente se ha visto en fibrosis renales humanas y
experimentales que su expresión parece estar correlacionada con el grado de fibrosis
túbulointersticial107.
1.4.2.1.3.1 Factor de crecimiento transformante – (TGF )
En humanos existen tres formas de TGF, el β1, β2 y β3 que corresponden a los productos de tres
genes distintos aunque con gran homología que están presentes como hodímeros. Estas tres
proteínas tienen a veces efectos similares mientras que otros casos los efectos difieren. El TGF-β es
producido por múltiples células entre las que se incluyen linfocitos T y B, fagocitos
mononucleares, plaquetas, fibroblastos y células endoteliales. Cada cadena es sintetizada como un
precursor de 390 aminoácidos que tienen las características de un péptido secretorio. El precursor
dímero es secretado y cortado en cadenas de 112 aminoacidos haciéndose una forma activa.
Las funciones del TGF-β son múltiples: a) dependiendo del tipo celular o del estadio de
maduración, el TGF-β puede inhibir o aumentar la proliferación celular; b) según los tipos
celulares el TGF-β induce la diferenciación celular; c) el TGF-β tienen un efecto inhibidor sobre
las células NK; d) sobre los linfocitos B, el TGF-β disminuye la producción de inmunoglobulinas;
e) el TGF-β inhibe los efectos del IFN-γ en los fagocitos mononucleares; f) el TGF-β induce la
reparación de los tejidos, produciendo la aparición de nuevos vasos sanguíneos (angiogénesis); g)
el TGF-β induce la expresión de receptores de integrinas aumentando la adhesión celular a la
matriz extracelular y el contacto célula-célula y h) el TGF-β induce la síntesis de las proteínas de
ECM, propiedad que desarrollaremos de una manera más profunda debido a la implicación que se
le atribuye en el desarrollo de la nefropatía diabética.
El TGF-β se caracteriza por ser una citocina con propiedades fibrogénicas muy potentes ya que es
capaz de realizar tres acciones simultáneas: 1) estimulación de la síntesis de matriz, 2) inhibición
de la degradación de matriz, y 3) modulación de la expresión de los receptores de matriz para
facilitar las interacciones entre célula y matriz108. Finalmente, el TGF-β autoinduce su propia
producción, la cual tiene muchas consecuencias biológicas109.
De la unión del TGF-β con su receptor se derivan: la producción de fibronectina y de colágeno I y
III; la síntesis de fibronectina con un dominio extra A (fibronectina EDA) y de tenascina, el
aumento de expresión de PAI-I (inhibidor del activador del plasminógeno); la inhibición de la
secreción del plasminógeno; la estimulación de la producción de inhibidores de las
metaloproteínas; la inducción del aumento del número de receptores de integrinas expresados en el
glomérulo (integrinas que se unen a fibronectina y colágeno I). El TGF-β inhibe la producción de
colagenasas, y aumenta la producción de inhibidores de metaloproteinasas. Además, en el tejido

INTRODUCCIÓN
58
normal el TGF-β no actúa solo en la reparación tisular. La interacción más importante se da con el
PDGF y el FGF. Ninguna de estas dos citocinas posee propiedades fibrogénicas únicas. La primera
es el más potente inductor de la proliferación celular y la segunda induce angiogénesis. Por otro
lado, el TGF-β inhibe el ciclo celular de muchas células, provocando un aumento de volumen
celular, del ADN y del contenido de proteínas, es decir, hipertrofiándolas.
La importancia del TGF-β en la fibrosis tisular ha sido demostrada en varias líneas de investigación
constatando que: 1) La inyección intravascular del TGF-β en ratas o conejos causa rápidamente
glomerulosclerosis. 2) La transfección del TGF-β en glomérulos causa glomerulosclerosis. 3) La
utilización conjunta del gen del TGF-β y del promotor del gen de la albúmina en la producción de
ratones transgénicos causa una hiperproducción del TGF-β en el hígado de estos animales,
viéndose como evidencia el aumento de los niveles plasmáticos de TGF-β 4) La inyección de
anticuerpos anti-TGF-β in vivo bloquea la fibrosis.
Existen tres isoformas similares al TGF-β en mamíferos: TGF-β1, TGF-β2 y TGF-β3. Una de
ellas, la TGF-β1, es la más importante en la inducción de fibrosis. Inicialmente, el TGF-β es
sintetizado como un precursor inactivo que contiene un prodominio amino terminal, también
llamado proteína asociada de latencia (LAP). Ésta permanece unida de manera no covalente al
TGF-β, previniendo por lo tanto, la unión de TGF-β con su receptor. El complejo latente de TGF-β
y LAP está asociado a su vez a una proteína de unión del TGF-β latente (LTBPs) que permite
asociar el TGF-β a la superficie celular. Después de su secreción, la mayoría del TGF-β es
almacenado en la matriz extracelular como un complejo inactivo asociado al LAP y al LTBP110.
El TGF-β es en parte un inhibidor del crecimiento celular. En algunos casos, como en las células
mesangiales, el efecto mitogénico o antimitogénico del TGF-β depende de la concentración en que
se encuentra111. Se ha visto que la pérdida de la inhibición del crecimiento del TGF-β está
acompañada de la perdida de otras respuestas como el control de la producción de matriz
extracelular112. La inhibición del crecimiento está relacionada con el estado de fosforilación de la
proteína del retinoblastoma. El TGF-β moviliza las células en G1 mediante la reducción de la
fosforilación de la proteína de retinoblastoma. Esta modificación post-traduccional de la
fosforilación de la proteína de retinoblastoma hace que la célula no entre en la fase S del ciclo
celular. Además, el TGF-β puede alterar el estado de fosforilación de la P53, así como reducir el
nivel de la proteína c-myc, conduciéndose de nuevo a la prevención de la proliferación.

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
59
1.4.2.1.3.2 Rutas de señalización del TGF-
El complejo latente de TGF-β es activado mediante la acción de proteasas, tales como la plasmina
o la trombospodina-1. Después de liberarse del complejo latente, dímeros del TGF-β inician su
efecto celular mediante la unión a los receptores de membrana (que son del tipo serín/treonín
quinasa). Existen tres receptores de membrana implicados en la transducción de la señal. El TGF-β
primero se une al receptor tipo III, el cual presenta el ligando al receptor tipo II. El receptor tipo I
es reclutado para formar un heterotetrámero que consiste en dos receptores tipo I y dos receptores
tipo II. Una vez formado este heterotetrámero el receptor tipo II fosforila residuos serina y treonina
del receptor tipo I, iniciando su actividad quinasa. La activación del receptor tipo I lleva al
reclutamiento y fosforilación de algunos miembros de la familia de señales de transducción Smad.
Smad 2 y Smad 3 son fosforiladas por el receptor 1, homodimerizadas y unidas a Smad 4. El
complejo heteromérico de Smad 2, Smad 3 y Smad 4 se transloca al núcleo donde regula la
transcripción de algunos genes diana. La regulación de estos genes por los complejos Smad implica
la interacción de otros factores de crecimiento como FAST y Sp1, e interacciones con otras
cascadas de señalización como MAP quinasas113,114,115 (Figura 15)
Las vías de señalización implicadas en la regulación de matriz están peor definidas que las vías
responsables del control del ciclo celular. Estudios recientes señalan a la protein quinasa A (PKA)
como intermediaria entre el TGF-β y la estimulación de la producción de la cadena α1 del
colágeno1 y de la fibronectina. El mecanismo de activación de la PKA en las células mesangiales
no cursa a través del AMPc sino que implica la fosforilación o la degradación de las moléculas
inhibitorias que interactúan con la subunidad catalítica de PKA. La inhibición de la vía de PKA
mediante el péptido inhibidor de la PKA (PKI) en las células mesangiales atenúa la estimulación
de la expresión del mRNA de fibronectina inducida por el TGF-β116 .

INTRODUCCIÓN
60
Figura 16. TGF- . Mecanismos de activación y transducción de la señal
TGF- en la diabetes mellitus:
La concentración alta de glucosa estimula la síntesis de colágeno tipo I y IV en cultivos de células
mesangiales, atenuándose estos efectos cuando se administran anticuerpos anti-TGF-β. La
concentración alta de glucosa estimula al TGF-β a nivel transcripcional en las células mesangiales,
sin alterarse la vida media del mARN del TGF-β. Estudios del promotor del TGF-β murino han
puesto de manifiesto presenta una región sensible a los niveles de glucosa, región que explica la
regulación de esta citocina por la glucosa.
También, se ha visto en modelos de diabetes en ratones inducida por estreptozotocina que el
mRNA del TGF-β y su receptor II están aumentados ya a los tres días siguientes del inicio de la
hiperglicemia, imcrementando su expresión progresivamente hasta las 24 semanas de diabetes. La
intensificación de la regulación de estas moléculas precede a la expresión del colágeno IV y de la
fibronectina. En estados más avanzados de la diabetes, es probable que el TGF-β se estimule
mediante la hiperglicemia, glicación de proteínas, activación de la protein quinasa C, angiotensina
II y mediante una retroalimentación positiva. Así, el aumento en la regulación de la expresión del
TGF-β juega un papel importante en la acumulación de la matriz mesangial y en la insuficiencia
renal de la nefropatía diabética (Figura 16).

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
61
Figura 17. Papel central del TGF- en la patogénesis de la nefropatía diabética.
Aparte de la deposición de matriz mesangial, se produce una desregulación vascular en estados
iniciales tanto como terminales de la nefropatía diabética. En concreto, en los estados iniciales de la
nefropatía aumenta el flujo glomerular por dilatación de la arteriola aferente. Si nos planteamos
estudiar cual es el mediador de la vasodilatación en estos estados sería importante estudiar
diferentes factores, además del TGF-β, que están estimulados en el glomérulo (sistema
vasoconstrictor de la endotelina y la Angiotensina II, así como factores mitogénicos como PDGF y
bFGF) y cabría esperar que la microcirculacióon glomerular mostrara un aumento en el tono
vascular y una proliferación de las células mesangiales. Sorprendentemente, aparece una dilatación
de los vasos, una hipertrofia glomerular, y una estimulación de las moléculas de la matriz
mesangial. Así, parece que las características del TGF-β, la estimulación de la matriz y la
hipertrofia celular, son fenotipos dominantes. Varias explicaciones podrían contribuir a este hecho:
(1) una disminución en la expresión de los receptores de los agentes vasoconstrictores, (2) el TGF-
β podría ejercer una acción inhibitoria en las células reguladoras del ciclo celular, (3) el TGF-β
interferiría en alguna vía común de los factores mitogénicos y vasoconstrictores disminuyendo sus
efectos.
La angiotensina II, la endotelina, el PDGF y el FGF inducían la movilización de calcio a través de
la vía 1,4,5-trifofato (IP3), liberando calcio intracelular a través de los receptores de IP3. Estos
poseen diferentes receptores de membrana y activan diversas isoformas de la fosfolipasa PLC para
generar IP3. También se ha observado que el tratamiento de células mesangiales o musculares lisas

INTRODUCCIÓN
62
con TGF-β lleva a regulación negativa del Receptor tipo I del Inositol 1,4,5 trifosfato (IP3) tanto al
nivel de proteína como de mARN, hecho que también sucede en la nefropatía diabética. Con todo
esto, Sharma y col117 sugirieron que la producción crónica del TGF-β regula negativamente el
Receptor I de IP3 en las células contráctiles que ordenan la microcirculación glomerular. El déficit
relativo de dicho receptor podría comportar la disminución en la movilización del calcio
intracelular (activada en respuesta a vasoconstrictores como AII, endotelina, PDGF y FGF)
manteniendo al glomérulo y sus arteriolas vasodilatados.
1.4.2.1.3.3 Factor derivado de plaquetas (PDGF)
El PDGF fue identificado como un factor secretado por las plaquetas y que inducía al crecimiento
de los fibroblastos in vitro. En los últimos años, otros tejidos han sido implicados en su síntesis y
secreción: células endoteliales derivadas de vasos sanguíneos, monocito- macrófagos activados, la
placenta y las neuronas de mamíferos durante las fases de desarrollo118. Además, es considerado
uno de los mitógenos más potentes y mejor caracterizados para las células mesangiales humanas,
de rata y ratón119. La siguiente Tabla 2 resume los papales fisiológicos del PDGF:
Proceso Origen EfectosProcesos reparativos Plaquetas, macrófagos, células endotelilales,
células musculares lisas, fibroblastosReparación del daño mediante la migración yproliferación de las células del tejido conectivo
Arteriosclerosis Plaquetas, macrófagos, células endotelilales,células musculares lisas
Excesiva migración y proliferación de lascélulas musculares lisas y macrófagos.
Tumorogénesis Células tumorales Proliferación de las células tumorales(sarcomas) y/o células del estroma(carcinomas)
Diferenciación Mioblastos, células de la glía Estimula la proliferación y retrasa ladiferenciación de los precursores (mioblastos,células O-2)
Embriogénesis Receptores de células positivas Estimula la proliferaciónHemodinámica Células musculares lisas Induce vasoconstricción
Tabla 2. Funciones del PDGF
Estructuralmente el PDGF está constituido por cadenas A (16kd) y B(14kd) unidas por puentes
disulfuro. Por lo que respecta a las tres isoformas aisladas de diferentes tejidos hemos de citar el
heterodímero AB en el hombre, mientras que en la mayoría de sueros de otras especies se ha
aislado el homodímero BB. Finalmente, el homodímero AA se ha aislado en una línea celular de
glioma120.
Las diferentes formas de PDGF se unen y activan dos receptores tirosina quinasas, PDGFR-α, y -
β121. El receptor α tiene una alta afinidad con las tres isoformas de PDGF (PDGF-AA, PDGF-BB,
PDGF-AB) y el receptor β únicamente presenta una alta afinidad con las PDGF-BB. En los
fibroblastos humanos se expresan los dos tipos de receptores en cantidades similares. Mientras que

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
63
los PDGF-AB y BB tienen un efecto mitogénico potente, la mitogenicidad de PDGF-AA es muy
baja. Los receptores de PDGF están localizados sobre todo en las células del tejido conectivo122.
En la Figura 17 se representa la estructura del receptor del PDGF. Éste tiene cuatro dominios: (1)
porción extracelular de la molécula formada por cinco dominios semejantes al de las
inmunoglobulinas; (2) el dominio transmembrana; (3) la región citoplasmática que contiene
secuencias homólogas a otras tirosina quinasas interrumpidas por un dominio KI; (4) en el extremo
carboxi-terminal se encuentran los dominios de autofosforilación.
Figura 18. Receptor del PDGF.
Las interacciones específicas del receptor PDGF-β se encuentran en la región citoplasmática. Estas
asociaciones están ubicadas en los residuos de tirosina autofosforilados y se producen con la
quinasa 3’-fofatidilinositol (PI-3´-K), la proteína de activación GTPasa (GAP) y la fosfolipasa C-γ
(PLC-γ). Si consideramos el hecho de que la TK del PDGF es la única capaz de unirse a las
proteínas anteriores y que el PDGF parece ser el único factor de crecimiento capaz de inducir por sí
mismo proliferación de las células mesenquimales, se podría pensar en la posibilidad de que la
unión de todas estas proteínas sea necesaria para la inducción de los efectos mitogénicos
característicos del PDGF.
El receptor del PDGF contiene al menos nueve sitios de autofosforilación. Uno de ellos, la tirosina
857, se encuentra en el dominio catalítico y su fosforilación mantiene al receptor en la
conformación activa. Los otros ocho residuos de tirosina fosforilados restantes no influyen en la
actividad enzimática del receptor, sino que crean sitios de reconocimiento y unión de proteínas
señalizadoras, como la fosfolipasa Cγ (PLCγ) o Src, o de proteínas adaptadoras sin actividad
enzimática que permiten la asociación al receptor y la activación de nuevas moléculas efectoras,
como la subunidad reguladora de la fosfatidilinositol-3’-quinasa (PI3K) o Grb-2. Estas proteínas se
unen a la TK mediante sus dominios SH y una vez fosforiladas, actúan de transductoras de la señal

INTRODUCCIÓN
64
produciendo cambios en la bioquímica celular. Estas modificaciones son: alcalinización
citoplasmática, incremento de concentración de AMPc e iones calcio y la activación de la proteína
quinasa c (PKC) a través de la degradación de los fosfoinositoles.
Figura 19. Vía de transducción de señal del PDGF
El incremento del pH intracelular se debe a la entrada masiva de iones de sodio mediante la
activación del antiporte Na+/H+ y la subsiguiente salida de Na+ por activación de la ATPasa Na+-
K+. El incremento en la concentración de AMPc implica la activación de la fosfolipasa A2, con la
consiguiente liberación de ácido araquidónico de la membrana plasmática y formación de
prostaglandina E (PGE). Además, la unión del PDGF con su receptor comporta la degradación de
fosfoinositoles asociados a la membrana a través de la fosfolipasa Cγ. A través de la proteína G, se
forma IP3 y diacilglicerol (DAG), causantes de la secreción de iones de calcio del retículo
endoplasmático y la activación de la PKC. El aumento en la concentración intracelular de AMPc y
la activación de la PKC son procesos independientes pero que convergen en la activación

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
65
transcripcional de los early response genes, en primer lugar, y posteriormente del c-myc (Figura
18).
1.4.2.2 Angiotensina II.
1.4.2.2.1 Expresión del Sistema renina-angiotensina sistémica
El sistema renina-angiotensina (RAS) es un sistema de vasoconstricción que podría considerarse
destinado principalmente a garantizar la circulación renal dentro de unos valores de presión
adecuados, aunque su activación tenga efectos hemodinámicos en la circulación interna.
Al examinar el RAS se observa que la secreción de renina se produce en células epiteloides o
yuxtaglomerulares, integrantes de la pared de la arteriola aferente del glomérulo renal. Estas células
forman parte de un complejo estructural denominado aparato yuxtaglomerular, situado en la
proximidad de polo vascular del glomérulo renal y constituido por la arteriola aferente y la
eferente, formando un ángulo dentro del cual se encuentra el tramo inicial del túbulo contorneado
distal, perteneciente a la misma nefrona. Además de las células epiteloides o yuxtaglomerulares,
localizamos también la mácula densa, un grupo de células altamente especializadas que se halla en
el origen del túbulo contorneado distal y cuya denominación se debe a la gran condensación de
núcleos ricos en cromatina. Las estructuras vasculares y tubulares están separadas por un espacio
cuya amplitud varía entre 1.000 y 2.000 å. La variabilidad del área de contacto entre las estructuras
del aparato yuxtaglomerular constituiría la base anatómica para el control de la secreción de la
renina.
Según la teoría de la mácula densa (que defiende la relación inversa entre la secreción de renina y
el contenido de sodio en el túbulo distal), un contenido bajo de sodio en el túbulo distal provocaría
una reducción del volumen en su interior, con lo que disminuiría la superficie de contacto con las
células arteriolares y se estimularía la secreción de renina. También la estimulación adrenérgica, a
través de las vías simpáticas renales y de catecolaminas circulantes, interviene en la secreción de
renina: los receptores β de las células yuxtaglomerulares la liberan y los receptores α, la inhiben.
Por último, en la secreción de renina pueden influir también otros factores humorales como las
prostaglandinas.
La renina es un enzima que una vez secretada en la circulación, actúa sobre otra proteína que es
producida y liberada de forma mantenida en la circulación por el hígado: el angiotensinógeno.
El angiotensinógeno es prácticamente inactivo, pero por acción de la renina se transforma en el
decapéptido angiotensina I. La angiotensina I, que también es inactiva, es modificada
posteriormente y convertida en angiotensina II por la acción de una enzima importantísima, que se

INTRODUCCIÓN
66
denomina enzima de conversión de la angiotensina (ECA) y se encuentra en las células
endoteliales, sobre todo de la circulación pulmonar.
La angiotensina II es el verdadero principio hipertensivo (uno de los más potentes que se conocen)
y actúa a través de dos mecanismos:
ü Una acción vasoconstrictora potente, principalmente en arteriolas.
ü Un efecto estimulante de la secreción de aldosterona por la corteza suprarrenal.
Figura 20. Efecto de la angiotensina II en las células vasculares de la secreción de aldosterona.
La acción de vasoconstricción de la AII se explica por su estímulo directo en las células musculares
vasculares: el mecanismo de acción puede atribuirse a una interacción con los receptores de
membrana, que probablemente activa los canales lentos del calcio (canales controlados por el
receptor) y que ciertamente libera iones de calcio de las reservas intracelulares por otra vía.
Esta segunda modalidad de acción comporta la formación, tras la interacción de la AII con el
receptor; de un mensajero intracelular (el inositoltrifosfato o IP3), capaz de liberar el calcio de las
reservas intracelulares. Esta liberación de calcio activa los mecanismos contráctiles.
A diferencia de la liberación de aldosterona que actúa en el riñón estimulando la secreción de
potasio y la retención de sodio, con lo que se produce una retención hídrica con un incremento
relativo de la volemia (Figura 19).

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
67
Ambas acciones van en la misma dirección, pero se manifiestan de manera diversa en el tiempo: la
vasoconstricción tiene un efecto inmediato, mientras que el incremento de la volemia requiere un
periodo de tiempo más prolongado.
Existen otras vías a través de las cuales la AII puede explicar la intensificación que ejerce sobre la
presión, que son las siguientes:
ü Una acción directa sobre el sistema nervioso central (SNC), con activación de las vías
adrenérgicas y el consecuente aumento del gasto cardíaco y la vasoconstricción. En el SNC
la AII estimula también el centro de la sed, facilitando el aumento de la volemia.
ü Un estímulo para la liberación de catecolaminas por parte de la médula suprarrenal y de las
terminaciones adrenérgicas.
ü Una acción antrinatriurética directa (retención de sodio) en el riñón que se sumaría a la
acción de la aldosterona.
Estos mecanismos parecen tener, de todos modos, una importancia secundaria en comparación con
los dos descritos anteriormente.
1.4.2.2.2 Papel central de la Angiotensina II en la patogénesis en la Enfermedad Renal Progresiva
Inicialmente, se creía que el sistema renina-angiotensina (RAS) tenía una función únicamente
sistémica. Actualmente, se sabe que existe una acción del RAS sobre muchos tejidos de función
enteramente paracrina. La angiotensina II (AII) es estimulante de la proliferación o la producción
de matriz. Aparece como el agente clave en muchos estados asociados a hipertensión arterial y
glomerulosclerosis tales como la nefrosis de puromicina crónica y la diabetes mellitus no tratada.
Todos ellos se previenen y tratan con inhibidores de la enzima de conversión de la Angiotensina123.
Se ha demostrado que la infusión de AII en riñones aislados perfundidos provoca una pérdida de
permeoselectividad y aumento de la proteinuria. Este hecho se atribuyó a los efectos
hemodinámicos de la AII en la elevación de la presión hidrostática capilar, y al efecto directo en la
permeoselectividad glomerular. Estudios realizados in vitro, han puesto de manifiesto la propiedad
que posee la AII de estimular la proliferación de las células mesangiales y de inducir la expresión
del TGF-β, dando como resultado un aumento de la matriz extracelular (ECM)124. La AII también
estimula a las células endoteliales y células musculares lisas en la producción del gen activador de
plasminógeno (PAI-1).
Mediante la activación de los macrófagos y la fagocitosis, la AII activa los componentes
inflamatorios asociados al daño renal progresivo. Finalmente, la AII induce la producción adrenal

INTRODUCCIÓN
68
de la aldosterona, la cual contribuye independientemente de la AII a la producción de lesión
renal125 (Figura 20).
Figura 21. Papel central de la angiotensina II en la patogénesis de la ERP: efectos hemodinámicos y no
hemodinámicos
EL SISTEMA RENINA-ANGIOTENSINA y TGF-
El sistema renina-angiotensia (RAS) y el TGF-β actúan como mecanismo de emergencia cuando se
produce cualquier tipo de lesión renal con el objetivo de mantener la homeostasis. Cuando la
agresión es persistente la relación entre el RAS y el TGF-β causa una activación continua que
genera una hipertensión crónica y una fibrosis que lleva a la disfunción del órgano. En este sentido,
existen evidencias de que una combinación en el bloqueo de RAS con un agente independiente que
suprima el TGF-β sería superior al bloqueo único de RAS126.
En una respuesta sistémica, RAS genera rápidamente Ang II que actúa por vasoconstricción,
manteniendo así la presión sanguínea. Posteriormente facilita la secreción de aldosterona, dando
como resultado final el aumento del volumen intravascular. El TGF-β es liberado rápidamente por
degranulación plaquetaria, produciendo: (1) autoinducción de la producción de TGF-β de las
células vecinas, amplificándose los efectos biológicos, (2) atracción de monocitos/ macrófagos que
limpian y esterilizan la herida y activación de los fibroblastos que inician la síntesis de matriz, (3)

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
69
deposición de nueva matriz: estimulando la síntesis de nueva matriz, inhibiendo las proteasas que
degradan la matriz y modulando el número de receptores de integrinas, para facilitar la adhesión
celular a la matriz nueva, (4) supresión de los efectos pro-inflamatorios de la IL-1 y del TNF, (5)
regulación de la acción del PDGF y del FGF, coordinando la proliferación celular y la angiogénesis
con la deposición en la matriz y (6) finalización del proceso cuando el tejido está totalmente
reparado127.
Ang II induce la producción del TGF-β en cultivos de células y en vivo128. Se ha observado en el
laboratorio que la infusión de Ang II estimula fuertemente la producción y activación de TGF-β en
el riñón. El bloqueo de la Ang II reduce la sobre expresión del TGF-β en riñón y corazón. Así, los
efectos fibrogénicos atribuidos a la Ang II es probable que sean mediados por el TGF-β. Igual que
la AII, el TGF-β estimula la contracción vascular de las células musculares lisas y las células
mesangiales129. Esto sugiere que la liberación de TGF-β por el aparato yuxtaglomerular podría
modular la microcirculación del glomérulo. De hecho, la microinyección intravenosa de dosis altas
de TGF-β en una rata produce una reducción de la volemia y por consiguiente una reducción de la
tasa de filtración glomerular. La razón de que el TGF-β adquiera este protagonismo en la reducción
de la volemia no está muy claro pero podría ser debido a una sinergia entre TGF-β y la activación
de RAS130.
Otra interacción entre el RAS y el TGF-β es el nivel de aldosterona. AII estimula la producción y
la liberación de aldosterona por la corteza suprarrenal. En contraste, el TGF-β suprime la
producción de aldosterona de la glándula adrenal y reduce la acción de la AII disminuyendo el
número de receptores de ésta expresados en la glándula adrenal131. Además, el TGF-β actúa
bloqueando los efectos de la aldosterona en la reabsorción de sodio en cultivos de células de los
túbulos renales. La infusión de aldosterona en ratas a las que se les ha practicado una nefrectomía
parcial produce un aumento de la presión sanguínea, proteinuria y glomerulosclerosis resultando
ineficaz el bloqueo de AII132.
La expresión en niveles superiores a los normales del TGF-β ha sido confirmada en muchos
modelos de fibrosis renal. También se ha constatado que el bloqueo de la AII no es suficiente para
bloquear la fibrosis renal133. Se ha comprobado que la AII es una molécula fibrogénica porque
cuando se añade en cultivos de células mesangiales activa la producción de colágeno134.
Angiotensina II y Plasmina
La plasmina es un enzima fibrinolítico importante en la disolución de los coágulos subsiguientes a
la herida. Se genera desde el plasminógeno y se bloquea con los inhibidores de la proteína de la
plasmina (PAIs). El PAI, como la AII y el TGF-β, se puede incluir dentro del conjunto de

INTRODUCCIÓN
70
moléculas que acuden rápidamente a los lugares que han sido dañados. Es un enzima de amplio
espectro que ayuda a degradar a la fibrina y a muchas proteínas de la ECM. También rompe
algunas procolagenasas generando moléculas activas que degradan colágeno. El recambio de la
matriz mesangial está relacionada con la generación de plasmina. Los niveles de PAI-1 son los
principales determinantes de la generación de plasmina. Se ha visto que la Ang II inicialmente
aumenta los niveles de mARN de PAI-1 independientemente del TGF-β. Posteriormente, los
niveles de mARN de PAI-1 se incrementan de forma dependiente al TGF-β135.
Aldosterona y fibrosis renal:
La sobreproducción de aldosterona está ligada a la hipertensión y a la glomerulosclerosis. Así, la
administración de aldosterona aumenta la severidad de la enfermedad renal. En concreto, la
aldosterona tiene efectos fibrogénicos independientes de la Ang II aunque está por dilucidar si esta
molécula regula la expresión del TGF-β, ya que la administración de un agonista del receptor de la
aldosterona no bloquea la glomerulosclerosis pero sí disminuye de manera transitoria la
proteinuria, la presión arterial, y la hipertrofia cardíaca (acciones fibróticas de la aldosterona que
podrían ser mediadas por el TGF-β a través de canales diferentes a los clásicos situados en el tubo
distal) 136.
Renina y prorenina como moléculas fibrogénicas:
Como en el caso de la aldosterona, recientes estudios sugieren que la prorenina o la renina tienen
acciones independientes de la AII en la producción de fibrosis. Se ha comprobado que animales
transgénicos normotensos que sobreexpresan prorenina desarrollan glomerulosclerosis severa137.
Además, la renina recombinante humana añadida a un cultivo de células mesangiales humanas
induce una producción aumentada del PAI-1, el cual no sólo es independiente del la AII sino que
actúa a través del receptor en las células mesangiales138.
Varios artículos sugieren que el TGF-β aumenta la liberación de renina. En este sentido, la adición
de TGF-β en cultivos de células yuxtaglomerulares muestra un aumento de renina. Así, podemos
deducir que el TGF-β es un factor importante que libera renina aunque no se desconoce su relación
con la enfermedad fibrótica.
En resumen, se sugiere que la AII ejerce efectos independientes de la presión en la fibrosis renal a
través de la regulación del TGF-β, conduciendo a la fibrosis. La AII tiene efectos directos en la
producción de PAI-1, la cual debe intervenir en la acumulación patológica de ECM limitando la
degradación de la ECM y la activación de colagenasas. Aunque este extremo ofrece más dudas, los
componentes de RAS, aldosterona, prorenina y renina parecen estar íntimamente conectados con la
producción y acumulación de ECM (Figura 19).

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
71
Efectos de la glucosa y la angiotensina
Los efectos de la glucosa en el riñón (especialmente en las células mesangiales) están mediados a
través de la estimulación de la producción de AII, la cual lleva a un incremento de la matriz
extracelular dependiente de TGF-β y a una disminución en la degradación de la matriz139. Es
interesante remarcar, que tanto AII como la glucosa comparten una señalización intracelular similar
y que el mediador del efecto es la activación del TGF-β, factor de crecimiento profibrótico por
excelencia140. La activación de la proteín quinasa (PKC) en ambientes con una alta concentración
de glucosa mediante AII es el punto de regulación principal de la activación de esta citocina.
(Figura 21)
Figura 22. Posibles mecanismos de acumulación de matriz inducida por la glucosa

INTRODUCCIÓN
72
1.4.2.2.3 Importancia de los inhibidores de la enzima de conversión de la angiotensina II
(IECAs).
El descubrimiento y la puesta a punto de estos fármacos tiene su origen en estudios de finales de
los años sesenta e inicios de los setenta realizados sobre una serie de péptidos obtenidos de una
serpiente brasileña (Botropos jararaca), que poseen una actividad antihipertensiva y potenciadora
de la bradiquinina.
Se comprobó que su acción farmacológica estaba ligada a la secuencia de aminoácidos, que
presentaba una analogía con la secuencia de la angiotensina I, de ahí su capacidad de interacción
con la ECA, “ocupándola” e inhibiendo su actividad enzimática. La naturaleza peptídica de estas
sustancias las hacía ineficaces por vía oral. Para superar esta limitación terapéutica se han
sintetizado posteriormente moléculas, de naturaleza no peptídica, capaces de interactuar con la
ECA de manera análoga a los péptidos iniciales. El resultado de estas investigaciones fue la
obtención del captopril, el primer inhibidor de la ECA activo por vía oral y el primer fármaco de
esta nueva clase que se comercializó (1981). En el presente estudio, hemos utilizado el Quinapril,
un IECA caracterizado por la presencia de un grupo fosfórico que le permite unirse al ECA. La
figura siguiente muestra un esquema de su estructura química (Figura 22).
Figura 23. Fórmula química del quinapril
Los IECAs son particularmente efectivos porque producen la dilatación de la arteriola eferente, la
cual permite la salida de la sangre del glomérulo disminuyendo la presión intraglomerular
independientemente de cualquier cambio en la presión arterial sistémica. En el modelo de
nefrectomia parcial cuando se produce ERP, se ha encontrado que el daño endotelial estaba
asociado a un aumento de la expresión de mARN de angiotensinógeno, el cual conduce a un
aumento de los niveles de angiotensina I141. Además, se incrementa el coeficiente de ultrafiltración,

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
73
el cual, junto con el flujo plasmático del glomérulo impide que la filtración individual de cada
nefrona disminuya. Se ha sugerido, también, que los IECAs podrían estimular la secreción de
prostaglandinas vasodilatadoras como PGE2142
. En relación con la hemodinámica glomerular es
importante remarcar que los IECAs, contrariamente a la AII, están directamente relacionados con
la permeabilidad glomerular143, al disminuir el diámetro del poro de la MBG. Como consecuencia
recuperan la barrera de filtración glomerular reduciendo la proteinuria y la hipertrofia glomerular
provocada por el tránsito masivo de macromoléculas a través de los capilares.
Además de actuar a través de mecanismos hemodinámicos, estudios experimentales sugieren un
efecto beneficioso adicional de los IECAs, la disminución de la hipertrofia glomerular, a través de
un efecto directo en el incremento del volumen mesangial inducido por la AII (disminución en la
expresión de los componentes de la matriz mesangial como la cadena α1del colágeno de tipo IV, la
cadena B1 y B2 de la laminina y la cadena α1 del colágeno tipo I y III, características del
glomérulo diabético)144. También se ha sugerido que la atenuación de la expansión mesangial podía
estar relacionada con la expresión del mARN de la ET-1, un activador de las células mesangiales
cuya síntesis está relacionada con la AII145. Así mismo, se ha visto un claro efecto renoprotector de
los IECAs a través de la disminución del TGF-β tanto en la diabetes mellitus experimental como en
la humana146.
1.4.2.3 Papel de los lípidos.
1.4.2.3.1 Clasificación de las lipoproteinas, y del metabolismo y papel en la arteriosclerosis
Los lípidos, tales como el colesterol y triglicéridos, son insolubles en plasma. Por tanto, la
circulación de los lípidos ha de estar ligada a proteínas que los transporten por la sangre hacia los
tejidos para su posterior utilización energética, deposición lipídica, producción de hormonas
esteroideas y formación de ácidos biliares. Al conjunto de la proteína transportadora y porción
lipídica (colesterol esterificado o sin esterificar, triglicéridos y fosfolípidos) se la conoce como
lipoproteína. El componente proteico de las lipoproteínas se denomina apolipoproteína o
apoproteína. Las diferentes apoproteínas sirven como cofactores de los enzimas y como ligandos de
los receptores147.
Lipoproteínas:
Se pueden diferenciar cinco grupos principales de lipoproteínas, cada uno de los cuales tiene una
función diferente:
Quilomicrones: Son partículas muy grandes que transportan los lípidos de la dieta. Están asociados
a diferentes variedades de apolipoproteínas, incluidas la A-I, A-II, A-IV, B-48, C-I, CII, CIII y E.

INTRODUCCIÓN
74
VLDL: Estas partículas transportan triglicéridos de síntesis endógena y en menor proporción
colesterol. Las principales apolipoproteínas con las que se encuentran asociadas las VLDL son la
B-100, C-I, C-II, CIII y E.
IDL: Transporta colesterol y triglicéridos. Está asociada a las apolipoproteínas B-100, C-III y E.
LDL: Llevan ésteres de colesterol y están asociados a B-100.
HDL: Contienen ésteres de colesterol. Están asociados a las lipoproteínas A-I, A-II, C-I, C-II, C-
III, D y E.
Apolipoproteínas:
La principal función de las diferentes proteínas puede resumirse en el siguiente cuadro. Entender
estas funciones es importante clínicamente, porque defectos en el metabolismo de las
apolipoproteínas comportan anormalidades en el metabolismo de los lípidos.
ApolipoproteinaA-I Proteína estructural para las HDL; activa la lecitin-colesterol-acil transferasa (LCAT).A-II Proteína estructural de las HDL; activa la lipasa hepática.A-IV Activador de la lipoproteína lipasa y la LCAT.B-100 Proteína estructural de las VLDL, IDL, LDL, y Lp (a); ligando para el receptor del LDL; es
necesaria para la reunión y secreción de la VLDL.B-48 Contiene el 48% de B-100; se requiere para la reunión y secreción de quilomicrones; no se
une al receptor de la LDL.C-I Activador de la LCATC-II Cofactor esencial de la LPL.C-III Interfiere en el aclaramiento mediado por apo E de las lipoproteínas ricas en triglicéridos
mediante receptores celulares; inhibe la hidrólisis de los triglicéridos por la lipoproteína lipasay la lipasa hepática.
D Cofactor para la proteína que transfiere los ésteres de colesterol.E Ligando del quilomicrón hepático y del receptor del VLDL remanente, dirige el aclaramiento
de estas proteínas de la circulación; ligando del receptor para las LDL.Apo (a) Proteína estructural para la Lp (a); Inhibidor de la activación del plasminógeno en la Lp (a).
Tabla 3. Apolipoproteínas
El metabolismo lipoproteico es susceptible de la siguiente división:
ü Transporte exógeno de triglicéridos
ü Transporte endógeno de triglicéridos
ü Transporte de colesterol

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
75
1.4.2.3.1.1 Transporte exógeno de los triglicéridos
El estado nutricional del individuo determina cuál de las dos rutas se sigue en el metabolismo de
los triglicéridos. En condiciones extremas de ayuno, prácticamente todos los triglicéridos son
sintetizados endógenamente y secretados por el hígado en forma de VLDL. Después de una comida
rica en grasas, los quilomicrones y las VLDL son secretadas por el intestino.
En los enterocitos, los ácidos grasos se combinan con el glicerol para formar triglicéridos, y el
colesterol es esterificado por la enzima Colesterol Acil Transferasa (LCAT) para formar ésteres de
colesterol. Estos lípidos son reunidos intracelularmente en forma de quilomicrones. La principal
apolipoproteína es la B-48, pero la C-II y E son adquiridas cuando los quilomicrones entran en
circulación. La Apo B-48 permite la unión de los lípidos al quilomicrón pero no es capaz de
adherirse al receptor de las LDL, previniendo el aclaramiento prematuro de los quilomicrones en la
circulación antes de que actúe la lipoprotein lipasa.
La Apo C-II es un cofactor de la lipoprotein lipasa que hace que los quilomicrones mengüen
progresivamente gracias a la hidrolis del núcleo de los triglicéridos y a la liberación de ácidos
grasos. Los ácidos grasos son utilizados como fuente de energía, convirtiéndose en triglicéridos o
almacenándose en el tejido adiposo. Los productos finales del metabolismo de los quilomicrones
son los quilomicrones remanentes aclarados en la circulación por sus receptores hepáticos a través
de la ApoE (ligando de alta afinidad). Los quilomicrones remanentes tienen un pequeño núcleo de
lípidos que está envuelto por componentes de superficie. Estos componentes de superficie son
transferidos por el quilomicrón remanente para la formación de HDL.
1.4.2.3.1.2 Transporte endógeno de los triglicéridos
La vía endógena del metabolismo de los lípidos empieza con la síntesis hepática de VLDL. Estas
moléculas contienen un núcleo de triglicéridos (60 % de la masa) y ésteres de colesterol (20% de la
masa). La superficie de las VLDL está formada por apolipoproteínas: Apo C-II (cofactor de la
lipoprotein lipasa), Apo B-100 y Apo E (ligando del receptor de la apolipoproteina B/E).
El nucleo de los triglicéridos de las partículas VLDL nacientes es hidrolizado por la lipoprotein
lipasa. Durante la lipólisis, el nucleo de las VLDL es reducido, generándose remanentes de VLDL
(deminados también, lipoproteínas de densidad intermedia o IDL). Algunos de los componentes de
la superficie de las partículas de los remanentes, incluyendo los fosfolípidos, colesterol no
esterificado y Apolipoproteínas A, C y E, son transferidos a la HDL. Los remanentes de las VLDL
pueden ser clarificados de la circulación por la apo B/E (LDL) o los receptores de los remanentes,
remodelado por las lipasas hepáticas para formar partículas de LDL.

INTRODUCCIÓN
76
1.4.2.3.1.3 Transporte del colesterol
Lipoproteínas de baja densidad- Las partículas de LDL contienen un núcleo rico en ésteres de
colesterol y triglicéridos en menor proporción, enriquecidas además en apolipoproteina B-100
(ligando de apo E/B). La LDL es internalizada en los tejidos hepáticos y no hepáticos. En los
primeros el colesterol LDL es convertido en ácidos biliares y entrar en el lumen intestinal. El LDL
colesterol es internalizado por los tejidos hepáticos y es utilizado para la producción de hormonas,
síntesis de membrana celular o almacenamiento de formas esterificadas.
La internalización de LDL es regulada por los requerimientos de colesterol celular a través de una
vía de retroalimentación negativa que suprime la expresión del receptor de la apo B/E, receptor de
la LDL. Por otro lado, la disminución de la actividad de la HMG-CoA Reductasa, el enzima que
controla la síntesis de novo del colesterol celular, conduce a una caída secuencial del colesterol
celular, aumentando la expresión de los receptores de la apo B/E, incrementando la captación del
colesterol existente en circulación y reduciendo la concentración de colesterol en plasma.
Muchas células hepáticas, incluidos los monocitos, los macrófagos, las células musculares lisas y
las células endoteliales, tienen un receptor “escavenger”. Este receptor se une únicamente a las
LDL modificadas por radicales de oxígeno o por peróxidos lipídicos liberados al espacio
subendotelial por macrófagos o plaquetas activadas. Este mecanismo podría ser el responsable de
la conversión de macrófagos y células musculares lisas en células espumosas de las estrías grasas,
la lesión más temprana en la arteriosclerosis.
La importancia del receptor de las LDL en la regulación del metabolismo del colesterol ha sido
demostrada tanto en modelos experimentales humanas o como animales. Animales knockout para
el gen del receptor de las LDL muestran un aumento en los niveles de colesterol total, defecto que
es corrgid por el gen del receptor de la LDL. En humanos, la hipercolesterolemia familiar está
asociada a un defecto en dicho receptor.
Lipoproteínas de alta densidad- la formación y el metabolismo de las HDL implica las siguientes
etapas:
ü Síntesis hepática e intestinal de pequeñas partículas nacientes de HDL compuestas por
fosfolípidos y apolipoproteínas.
ü Obtención de componentes de superficie (foslípidos, colesterol y apolipoproteínas) de
quilomicrones remanentes y de VLDL remanentes.
ü Adquisición de colesterol libre de tejidos y otras lipoproteínas, como partículas de HDL
iniciales que contienen pocas cantidades de colesterol.

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
77
ü Proteínas que transfieren lípidos que facilitan el movimiento de los ésteres sintetizados a
lipoproteínas que contengan apolipoproteína B. De esta manera, el colesterol puede ser
repartido por los tejidos para la síntesis de esteroides o su almacenamiento.
Las HDL, contrariamente a las LDL transportan el exceso de colesterol de las células hasta el
hígado. Además, son la principal fuente de apolipoproteina CII (cofactor de la lipoproteína lipasa)
y apolipoproteina E (implicada en la toma y catabolismo hepáticos de la VLDL y la IDL). El
efecto neto de los dos últimos pasos es retirar el exceso de colesterol de las células, lo que
constituye el mayor efecto anti-aterogénico atribuido a las HDL.
Lipoproteína (a) o Lp (a)- La Apo (a) se une a la apo B-100 en la superficie de las LDL mediante
puentes disulfuro. La integridad estructural de las LDLs y la formación de la Lp(a) están
moduladas por la enzima lecitin colesterol acil transferasa (LCAT).
Una de las regiones de la Lp(a) es homóloga a los dominios que se unen al plasminógeno. Esta
semejanza estructural al plasminógeno hace que la Lp(a) interfiera en la fibrinolisis mediante la
competición de ambas moléculas por los receptores del plasminógeno, fibrinógeno y fibrina. Como
consecuencia se produce una atenuación de la trombolisis debido a la menor activación del
plasminógeno. Además, la Lp(a) se une a los macrófagos a través del receptor de alta afinidad
promoviendo la formación de células espumosas, siendo fácil su localización en las placas
arterioscleróticas.
1.4.2.3.2 Efectos nefrotóxicos de los lípidos
1.4.2.3.2.1 Mecanismos hemodinámicos
1.4.2.3.2.1.1 Hemodinámica sistémica
Existen estudios que destacan a la hipertensión en el desarrollo de las lesiones renales. En esta
línea, Bnaa KH y Thelle DS148 realizaron un estudio epidemiológico en el que demostraban que
pacientes con hipertensión esencial tenían niveles de colesterol y triglicéridos mayores que sus
respectivos controles. Esta asociación fue observada tanto en hombres como en mujeres.
Paralelamente, Koletsky149 desarrolló un modelo especial de rata hipertensa que desarrollaba
hiperlipidemia y lesión renal. También se observó un efecto renoprotector del ácido linoleico que
era acompañado de una disminución de la presión en ratas a la cuales se les había practicado una
nefrectomia subtotal150.
Otros estudios han mostrado que la presión sistémica elevada parece no jugar ningún papel en la
patogénesis de la esclerosis glomerular inducida por lípidos. Así, se ha observado que los fármacos

INTRODUCCIÓN
78
hipolipemiantes administrados a ratas a las que se les había practicado una nefrectomia de 5/6
partes experimentaban una reducción de la esclerosis glomerular sin verse modificada la presión
sistémica151. También se ha apreciado que la hipertensión desarrollada por las ratas
espontáneamente hipertensas no está asociada a la esclerosis glomerular152.
1.4.2.3.2.1.2 Hemodinámica glomerular
Estudios de micropunción han demostrado un modesto incremento de la presión capilar glomerular
durante la administración de dietas suplementadas con colesterol. Estos cambios ocurren en
ausencia de un aumento de la presión sistémica y están estrechamente correlacionados con la
presión oncótica del plasma. La elevación de la presión oncótica del plasma parece ser un hecho
únicamente relacionado con el modelo de hipertrigliceridemia. Además, se ha evidenciado una
intensificación de la resistencia arteriolar y una alteración significativa de la tasa de la resistencia
arteriolar eferente/aferente, la cual podría contribuir a las fluctuaciones de las presiones
hidrostáticas153.
Por el contrario, hay estudios que defienden que la utilización de hipolipemiantes o fibratos en
animales de experimentación reduce la albuminuria y la glomerulosclerosis pero sin modificar la
presión intraglomerular ni sistémica154.
Se ha sugerido que la hipercolesterolemia reduce la producción vascular de sustancias
vasodilatadoras tales como las prostaciclinas y la sustancia relajante derivada del endotelio,
mientras que aumentan la producción de sustancias vasoconstrictoras tales como el tromboxano A2
y la endotelina155.
1.4.2.3.2.1.3 Hipertrofia glomerular
Kasiske y col156, demostraron que previamente a la aparición de la glomerulosclerosis, la dieta rica
en colesterol inducía el desarrollo de la hipertrofia glomerular. En otros capítulos ya hemos
comentado que la hipertrofia glomerular es una característica común en la progresión de la ERP,
siendo considerada como un mecanismo patogénico importante en el desarrollo de la
glomerulosclerosis.

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
79
1.4.2.3.2.2 Mecanismos biológicos
Deposición de lípidos en el mesangio renal
El endotelio fenestrado que caracteriza a los capilares glomerulares y peritubulares, así como la
porción ascendente de los vasos rectos, hace que el glomérulo y el intersticio renal sean los lugares
idóneos para la acumulación de lípidos. Además, la ausencia de una membrana basal que separe la
luz del mesangio glomerular proporciona un microambiente favorable para la interacción entre las
proteínas y el mesangio.
Por otro lado, en el síndrome nefrótico, se han encontrado niveles plasmáticos altos de VLDL, IDL,
LDL, colesterol y lipoproteína así como de triglicéridos. El enriquecimiento de las VLDL y LDL
en colesterol es debido al aumento de la transferencia de éste y es un reflejo de la larga
permanencia de estas partículas en la circulación. Éstas se acumulan y penetran en el endotelio
vascular sufriendo modificación oxidativa y causando daño renal157.
Un esquema de los factores patogénicos por los cuales los lípidos inducen el daño renal sería el
siguiente:
Deposición de lípidosen el mesangio
Aumento de macrófagosen el mesangio
Oxidación de las LDL
Unión de los macrófagosa los receptores
Internalización de los receptoresunidos a LDL
Transformación de los macrófagosen células espumosas
Biología mesangial alterada
Hiperlipidemia
Efecto deletéreodirecto
Daño renal
Metabolismo alteradode los ácidos grasos
polinsaturados
Figura 24. Esquema de los factores patogénicos en una nefropatía inducida por lípidos

INTRODUCCIÓN
80
Aumento de macrófagos mesangiales
Se ha demostrado que niveles altos de VLDL contribuyen a la infiltración de monocitos en el
mesangio glomerular158. En concreto, los macrófagos tienen un papel importante en la patogénesis
de la arteriosclerosis. Estas células están implicadas en la fagocitosis y degradación de los
depósitos lipídicos. En el riñón, los macrófagos repletos de lípidos (células espumosas) se
encuentran frecuentemente en las etapas iniciales de glomerulonefritis en humanos159.
Así, la hipercelularidad mesangial es debida al influjo de monocitos de la circulación que una vez
llegan al mesangio son transformados en macrófagos residentes del tipo Ia. Además, las células
mesangiales (responsables de modificar las células musculares lisas) reaccionan de una manera
análoga con los depósitos lipídicos y con los productos de los macrófagos, proliferando y
aumentando la formación de matriz extracelular.
Oxidación de las LDL
Las células mesangiales, los macrófagos glomerulares derivados de los monocitos y las células
espumosas producen radicales de oxígeno que oxidan a las LDL y a otros lípidos. Por ejemplo, se
ha comprobado que existe una peroxidación de los ácidos grasos polinsaturados de la membrana
que interfiere en las funciones de la misma.
Ishiyama y col160 comprobaron que el radical libre de oxígeno, anión superoxido O2-, incrementa su
expresión en la hiperlipidemia y en las enfermedades renales. La toxicidad de este radical se debe a
la habilidad que tiene en reaccionar no solo con proteínas y ADN sino también con los lípidos. El
radical O2- se metaboliza en peróxido de hidrógeno (H2O2) y en radicales hidroxilo (OH
-). El
radical hidroxilo es altamente reactivo y puede oxidar fácilmente a los lípidos, formando peróxidos
lipídicos (LOOH).
Efectos directos de las LDL oxidadas
En el riñón, las LDL oxidadas ejercen un gran número de efectos deletéreos: aumento de la
fosfatidilcolina (quimioactante de los monocitos)161, incremento de la actividad de las protein
tirosin quinasas específicas (PTKs) que juegan un papel crítico en la adhesión de los monocitos a
las células endoteliales162, estimulación de la expresión de TGF-β y fibronectina163, regulación de la
expresión del colágeno a través de la modificación de proteínas responsables de la expresión de los
genes que codifican para el colágeno164, intensificación de la producción de matriz mesangial165,
formación de radicales libres de oxígeno O2- que conducen a la muerte celular de las células de la
pared vascular y del glomérulo166, e inducción de la vasoconstricción disminuyendo la actividad del

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
81
óxido nítrico (acción contraria a la de la superóxido dismutasa, catalasa y la L-arginina),
proporcionando protección frente la vasoconstricción inducida por la LDL oxidada167.
Unión de las LDL oxidadas a los receptores de los macrófagos e internalización del receptor unido
a las LDL: transformación de los macrófagos en células espumosas.
Las células mesangiales humanas poseen receptores “scavenger” tipo A inducibles en situaciones
de inflamación. Estos receptores son proteínas integrales capaces de internalizar un amplio espectro
de ligandos, incluidas las LDL-acetiladas y LDL-oxidadas. La acumulación de lípidos a partir de
este proceso lleva a la formación de las células espumosas típicas de las placas arterioscleróticas
contribuyendo al desarrollo de la glomerulosclerosis. Así, se ha sugerido que la expresión de dichos
receptores en las células mesangiales humanas podría ser un marcador de activación de las células
mesangiales durante el desarrollo de la glomerulosclerosis168.
Alteración de la biología del mesangio
La acumulación de macrófagos y células espumosas en el mesangio produce sustancias que alteran
los procesos biológicos locales. Estos cambios son nocivos para el glomérulo renal y tejido
tubulointersticial. La presencia de VLDL, IDL, LDL o una alta concentración de HDL en cultivos
de células mesangiales humanas incrementa la secreción de IL-6, PDGF-AB, y TGF-β, mientras
que la secreción de TNF-α es estimulada por las LDL-oxidadas169.
Las LDL incrementan selectivamente la síntesis de prostaglandinas específicas y hialuronato en las
células mesangiales. Este efecto contribuye a la expansión de matriz mesangial y a la modificación
de las interacciones de las células con la matriz que producen daño renal170.
Efecto directo de los lípidos
Los depósitos de lípidos en el mesangio renal ejercen efectos deletéreos directos tales como la
proliferación celular y la formación de matriz mesangial. Se ha apreciado en cultivos de células
mesangiales humanas que las lipoproteínas ricas en lípidos VLDL, IDL y LDL causan proliferación
mesangial mientras que las LDL-oxidadas tienen un efecto citotóxico171.
Las lipoproteínas tienen una alta afinidad por los glicosaminoglicanos de la membrana basal
glomerular (MBG). La apoB se une in vitro al colágeno y al glicosaminoglicano sulfato de heparán,
el principal constituyente de la MBG in vivo. Esta unión puede ser debida a una región de 130kda
que tiene una secuencia homóloga al receptor de las LDL. Es posible, aunque no está garantizado,
que la unión de la apoB neutralice la carga glomerular y disminuya la permeoselectividad,
aumentando la pérdida de proteínas. La combinación de las LDL y de los glicosaminoglicanos de la
MBG puede producir un complejo que favorezca la internalización de los macrófagos172.

INTRODUCCIÓN
82
Las lipoproteínas aterogénicas (Lp(a) y LDL) estimulan la formación de radicales libres de oxígeno
no solo en las arterias, sino también en las células glomerulares y yuxtaglomerulares, causando una
inhibición de la vasodilatación dependiente de óxido nítrico, fomentando la liberación de renina,
modulando el crecimiento y proliferación de las células mesangiales, además de introducir efectos
potencialmene previsores de los enzimas antioxidativos y las lipoproteínas de alta densidad173.
1.4.2.3.3 Inhibidores del HMG-CoA Reductasa
Los inhibidores de la 3-hidroxi-metil-glutaril coenzima A (HMG-CoA) reductasa, también
llamados estatinas, inducen reducciones importantes en los niveles de colesterol plasmático
estableciéndose como tratamiento de la hipercolesterolemia.
La figura 24 es una representación de la estructura química de la atorvastatina, inhibidor de la
HMG-CoA reductasa que hemos utilizado en el presente estudio experimental.
Figura 25. Estructura química de la atorvastatina
Actualmente, los inhibidores de la HMG-CoA Reductasa, denominados también estatinas, incluyen
a la lovastatina, pravastatina, simvastatina, fluvastatina, atorvastatina y cerivastatina. Estos agentes
son inhibidores competitivos de la hidroxi-metilglutaril-CoA reductasa. Los efectos beneficiosos de
estos enzimas radican en la habilidad que tienen para reducir la síntesis del colesterol por dicha vía.
El producto de la reacción catalizada por este enzima es el mevalonato (MVA). Esta molécula tiene
una gran importancia ya que además de ser el precursor del colesterol lo es también de algunos
componentes isoprenoides no esteroides. Esto confiere a los inhibidores de la HMG-CoA
propiedades pleyotrópicas.

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
83
Acetil CoA
Acetoacetil CoA
HMG CoA
Mevalonato
Mevalonato PP
Isopentenil PP
Geranil PP
Farnesil PP
HMG-CoA reductasa
Mevalonato PP descarboxilasa
Escualeno sintasa
Escualeno epoxidasa
Farnesil transferasa
Escualeno
Colesterol
Dimetilalil PP
Isopentenil Adenosina
2-cis-geranilgeranil PP
trans-geranilgeranil PP
Dolicoles
Ubiquinona
Proteínas preniladas
Geranilgeranil transferasa I, II
Estatinas
Figura 26. Vía metabólica de los productos de la HMG-CoA Reductasa.
Las propiedades de la ruta del mevalonato producen una serie de isoprenoides vitales en el
desarrollo de diversas funciones celulares. Entre estos isoprenoides se encuentran el adenosin
isopentenil (presente en algunos ARN de transferencia), los dolicoles (requeridos en la síntesis de
glicoproteinas), polisoprenoides (cadenas laterales de la ubiquinona) y el anillo hemoA (implicado
en el transporte de electrones) (Figura 25).
Se han identificado varias proteínas que son modificadas post-transduccionalmente mediante
uniones covalentes con grupos isoprenoides derivados del MVA, como el farnesil o geranil
pirofosfato. Estas proteínas necesitan ser preniladas para su unión a la membrana. Los miembros de
esta familia están implicados en una serie de procesos celulares que incluyen la señalización
celular, la diferenciación y proliferación celular, la mielinación, la dinámica del citoesqueleto y el
transporte endocitótico y exocitótico. Como consecuencia, las estatinas mediante la inhibición de la

INTRODUCCIÓN
84
HMG-CoA reductasa pueden afectar a varios procesos que explican las propiedades no
hipolipemiantes de estos fármacos174.
1.4.2.3.3.1 Efectos renoprotectores de los inhibidores de la HMG-CoA reductasa
Efecto en el metabolismo lipídico
Aunque la enzima de la HMG-CoA reductasa es ubicua, la acción predominante de este fármaco se
encuentra en el hígado, donde disminuye la síntesis de las VLDL, precursores de las LDL. Como
resultado de la atenuación de los niveles de colesterol intracelular, los hepatocitos aumentan el
número de receptores de las LDL, produciéndose una aclaración del colesterol plasmático y
activándose la excreción del colesterol vía biliar175.
La atorvastatina reduce la producción de las VLDL mediante la secreción hepática de la apoB. A la
par, está asociada con la disminución de la tasa de recuperación de la actividad de la HMG CoA
reductasa después del tratamiento. Las estatinas tienen una propiedad modesta, aumentando el
colesterol- HDL. Además, disminuyen la concentración de triglicéridos como consecuencia de la
disminución de las VLDL.
Otro mecanismo potencial beneficioso de los inhibidores de la HMG-CoA reductasa está
relacionado con la disminución de la expresión de la molécula MCP-1 en el mesangio cuyo
metabolismo depende de la MMV176.
Efecto en la oxidación de las LDL oxidadas
Las estatinas reducen la susceptibilidad a la oxidación de las LDL al menos a través de cuatro
mecanismos:
Efecto hipolipemiante: reducen el contenido de ácidos grasos y colesterol en las lipoproteínas177.
Disminución de la producción de oxigeno en los macrófagos. Las proteínas preniladas,
probablemente las que pertenecen a la familia Rac, están implicadas en la producción de radicales
superóxido178. También se ha demostrado que las estatinas (a través de la prevención de la
prenilación de p21Rac) atenúan la producción endotelial de aniones superoxido179.
Unión a los fosfolípidos superficiales de las lipoproteínas: se ha establecido que las estatinas, como
la fluvastatina y lovastatina, se unen a la fracción fosfolipídica de las LDL previniendo la difusión
de los radicales libres180.
Metabolitos antioxidantes: se ha demostrado que la atorvastatina y la fluvastatina poseen un
potencial antioxidativo protegiendo a las lipoproteínas de la oxidación181.

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
85
Efectos en la función endotelial
La disfunción endotelial es un evento precoz que ocurre en la hipercolesterolemia. Este efecto está
relacionado con la habilidad de las células endoteliales para producir óxido nítrico (NO) y
probablemente, con la disminución de la L-arginina disponible (substrato de la sintasa de óxido
nítrico, NOS)182. Se ha observado que cuando se suplementa una dieta con hipercolesterolémica
con L-arginina se previene la arteriosclerosis, presuntamente a causa del restablecimiento de la
producción endotelial de NO. Además, estudios realizados in vitro muestran que las estatinas
incrementan la expresión del NOS endotelial183. Wagner y col184 encontraron otros efectos
beneficiosos para las estatinas, como la inhibición de la formación endotelial de O2- que provoca
alteraciones del equilibrio NO y O2-.
Roullet y col185 observaron que la lovastatina incrementaba la sensibilidad a algunos
vasoconstrictores (noradrenalina y serotonina) ejerciendo un efecto negativo en la relajación del
endotelio de la aorta, acción que se prevenía con la adición de mevalonato. Este hallazgo cuestiona
la administración de estatinas como efecto desfavorable en la ruta de la NO vascular.
Efecto en la inflamación
Las células mesangiales son estimuladas por la deposición de lipoproteínas nativas y oxidadas
liberando quimioactantes que provocan la acumulación de monocitos y macrófagos en el
glomérulo. Así, Keane demostró que las estatinas podían reducir la expresión de algunos factores
de crecimiento (M-CSF), quimiocinas (MCP-1) y de algunas moléculas de adhesión (VACM-1,
ICAM-1) implicadas en la adhesión de monocitos en el endotelio y mesangio, hecho asociado a la
disminución del injurio renal186.
Efecto en la proliferación celular
Las células mesangiales proliferan en respuesta a varios factores de crecimiento tales como el
PDGF y el IGF-1 así como ante lipoproteinas tales como la LDL, la IDL y la VLDL. Experimentos
in vitro han demostrado que las estatinas inhiben la proliferación de las células mesangiales, células
del epitelio tubular renal y células musculares lisas186.
Por otro lado, en cultivos de células mesangiales de rata se puso de manifiesto el papel de los
isoprenoides en la proliferación celular. En concreto, se vio que la lovastatina causaba una
inhibición dosis-dependiente en la replicación del ADN. Esta inhibición podía ser rápidamente
revertida por el mevalonato. Similarmente, la adición del isoprenoide farnesol era capaz de invertir
nuevamente los efectos de la lovastatina. Estos datos sugiereron que la disminución de la síntesis
de isoprenoides ejercía un efecto antiproliferativo independientemente a los niveles de colesterol187.

INTRODUCCIÓN
86
1.4.2.3.3.2 Otros efectos de las estatinas
Efecto en la actividad de las plaquetas
Las plaquetas de los pacientes hipercolesterolémicos son más sensibles a los agentes agregantes,
perfil que presentan acompañado de una composición lipídica alterada. En concreto, las estatinas
reducen la agregación plaquetaria modificando el contenido en colesterol de la membrana de las
plaquetas, la cual altera la fluidez de ésta188.
Efecto en los procesos de coagulación:
La terapia con estatinas disminuye significativamente la formación de trombos y mejora el perfil
fibrinolítico (descenso de los niveles del inhibidor del activador del plasminógeno (PAI-1) y
aumento de los niveles del activador de plasminógeno tisular)189.
Efecto en el crecimiento tumoral:
Algunos experimentos in vitro han aportado datos que indican que las estatinas pueden inhibir el
crecimiento tumoral190. Esto es debido en parte al protagonismo del mevalonato en la proliferación
celular y también, al hecho de que muchas células tumorales tienen una alta actividad de la HMG-
CoA reductasa.
1.4.2.4 Quimocinas
Jones en 1950 irrumpía en el campo de las quimocinas de la siguiente manera: “El glomérulo
dañado aumenta su permeabilidad capilar, hecho asociado, como en otros modelos de inflamación,
a lo que se denomina un aumento de la adherencia de las células endoteliales. Los neutrófilos
polimorfonucleares circulantes se unen a estas paredes adherentes de las células endoteliales y así
se acumulan en el glomérulo”191
1.4.2.4.1 Quimocinas: clasificación y mecanismos de acción
Se han descrito más de 40 miembros y 17 receptores de quimocinas hasta el momento192. Las
quimocinas son unas proteínas pequeñas con cuatro cisteínas conservadas que forman dos puentes
disulfuro (Cys1-Cys2 y Cys3-Cys4). Inicialmente las quimocinas se subdividieron en dos grandes
familias basadas en la posición de los dos primeros residuos de cisteína: la familia CXC tiene las
dos primeros residuos de cisteinas de la secuencia primaria separados por un solo aminoácido (la X
representa el aminoácido que interviene)193 y la familia CC con los residuos de cisteína ubicados
consecutivamente. Aunque la mayoría de quimocinas pertenecen a estas dos familias, existen otras
dos adicionales que contienen únicamente un miembro: La quimocina C, linfotactina, que carece de
la primera y tercera cisteína de los cuatro residuos conservados. Éstos comparten el extremo

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
87
carboxi terminal de la familia de las quimocinas CC. Por último, la Fractalquina, quimocina que
presenta 3 aminoácidos entre los dos primeros residuos de cisteína (CX3C). Esta quimocina está
unida a una larga mucina, asociada a su vez a la membrana celular con funciones de quimocina y
de molécula de adhesión.
Algunas quimocinas CXC muestran una denominación estructural adicional, denominada E-L-R-
CXC (Ác. Glutámico-leucina-arginina-cisteína-X-cisteína). Estos aminoácidos están ubicados en
los dos primeros residuos de cisteína. Las quimocinas E-L-R-CXC actúan principalmente como
factores quimiotácticos de neutrófilos, en contraste con las quimocinas exentas del motivo E-L-R,
que se unen de una manera diferente al receptor y son activas en los linfocitos. Así mismo, las
quimocinas de la familia CC actúan principalmente en monocitos, aunque pueden activar linfocitos,
basófilos y eosinófilos, pero no neutrófilos.
Las quimocinas CXC como la IL-8, ENA-78 (el quimioactante 78 de neutrófilos derivado de
células epiteliales), y GRO-α,β,que contienen los aminoácidos E-L-R-CXC, actúan como agentes
angiogénicos , mientras que la PF4, IP-10 y SDF-1 (factor –1 derivado de células del estroma),
privadas de la secuencia de aminoácidos E-L-R, pueden intervenir como factores angiostáticos.
Durante la embriogénesis, la respuesta reparadora y la inflamación crónica, la expresión de
quimocinas puede determinar la microvascularización del tejido194.
Distribución de las quimocinas y sus receptores en el riñón:
En las tablas 4 y 5 mostramos la distribución de las principales quimocinas y sus receptores en el
tejido renal.
Principios de acción de las quimocinas
Ø Las quimocinas son liberadas por células que actúan localmente.
Ø Son producidas por células intrínsecas así como por células inflamatorias
Ø Las quimocinas actúan a través de receptores transmembrana que atraviesan la membrana
siete veces y acoplados a proteínas G.

INTRODUCCIÓN
88
Tabla 4. Receptores de quimocina CC, distribución tisular y sus ligandos
Quimocina HCC-1MCP-2MCP-3MIP-1 aRANTES
MCP-1MCP-2MCP-3MCP-4MCP-5
MCP-3MCP-4EotaxinaEotaxina-2RANTES
MDCTARC
RANTESMIP-1aMIP-1bMCP-2
Receptor CCR1 CCR2 CCR3 CCR4 CCR5Tipo celular queexpresa elreceptor
MonocitosCélulasDendríticas(inmaduras)Células T Th-1NeutrófilosEosinófilosCélulasmesangiales
MonocitosCélulasDendríticas(inmaduras)BasófilosCélulas TCélulas NK
EosinófilosBasófilosCélulas T Th-2CélulasDendríticas
CélulasDendríticas(maduras)BasófilosCélulas T Th-2
Células T Th-1CélulasDendríticas(inmaduras)MonocitosCélulas NK
Quimocina MIP-3α (LARC) ELCSLC
I-309TCA-3
TECK MCP1MCP3
Receptor CCR6 CCR7 CCR8 CCR9 CCR10Tipo celular queexpresa elreceptor
CélulasDendríticas(inmaduras)Células TCélulas B
CélulasDendríticas(maduras)Células TCélulas B
Monocitos Células T
Tabla 5. Receptores de las quimocinas DARC, CXC, CX3C y C, distribución tisular y sus ligandos
Quimiocina IL-8GCP-2
IL-8GCP-2GRO-αGRO-βGRO-γENA-78NAP-2LIX
I-TACIP-10MIG
SDF-1αSDF-1β
Receptor CXCR1 CXCR2 CXCR3 CXCR4Tipo celular queexpresa el receptor
NeutrófilosCélulas Dendríticas(inmaduras)
Neutrófilos Células T Th-1Células BCélulas NK
Células TCélulas Dendríticas(maduras)MonocítosCélulas B
Quimocina BCA-1 Linfotactina Fractalquina Quimocinas CC, CXC(RANTES, MCP1,GRO-a, IL-8, NAP-2,PF-4)
Receptor CXCR5 XCR1 CX3CR1 DARCTipo celular queexpresa el receptor
Células TCélulas B
Células T Células NKCélulas T
EritrocítosCélulas Endoteliales

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
89
Ø La expresión de los receptores de las quimocinas es específico del tipo celular.
Ø Las quimocinas se unen a proteoglicanos y a la matriz extracelular.
Ø Las quimocinas inducen a la haplotaxis (migración celular a través de gradientes en la
superficie).
Ø Pueden actuar como moléculas de adhesión.
Ø Son susceptibles de estimular en los leucocitos la producción de oxígeno, la liberación de
gránulos y de proteasas.
Ø Las quimocinas controlan aspectos de inmunomodulación, angiogénesis, hematopoyesis y
desarrollo.
1.4.2.4.2 Modelo de actuación de las quimocinas en las Enfermedades Renales:
En una fase inicial (Figura 26B), la presencia de un insulto patológico en el glomérulo o
tubulointersticio activa a las células renales intrínsecas y genera mediadores proinflamatorios. Este
insulto puede ser inmunológico, toxico, isquémico, o mecánico, pudiendo ser dirigido hacia una
célula diana renal específica (ej, célula endotelial, epitelial, mesangial o del intersticio). La
expresión de determinadas quimocinas y receptores, junto con la liberación local de mediadores
inflamatorios (tales como la IL-1, el TNF-α, el IFN-γ, especies reactivas de oxígeno, etc),
promueven la regulación positiva y activación de selectinas e integrinas en los leucocitos y células
endoteliales, llevando a la adhesión, migración trasendotelial e infiltración de poblaciones
específicas de leucocitos. Las células T, los monocitos, y los leucocitos polimorfonucleares parecen
adherirse preferencialmente a los compartimentos microvasculares del riñón. Por ejemplo, las
células T son escasas en el glomérulo pero son muy comunes en los infiltrados intersticiales.
Una consecuencia adicional de la activación de células locales es la inducción a la expresión de
receptores de quimocinas tales como CCR1 y CCR3 por parte de las células mesangiales y otros
tipos celulares. La expresión en las células mesangiales de CXCR3 en concierto con la producción
local de la quimocina IP-10 podría llevar a la proliferación mesangial195.
Durante la “fase de amplificación” (Figura 26C), el glomérulo afectado expresa un exceso de
factores proinflamatorios que pueden llegar a la circulación capilar peritubular, así como al lumen
tubular mediante la ultrafiltración, especialmente cuando se produce proteinuria con una pérdida de
la barrera de ultrafiltración. Además, las proteínas y lípidos se escapan a través del endotelio
dañado pudiendo inducir “stres celular” a las células tubulares proximales.

INTRODUCCIÓN
90
Figura 27. Modelo hipotético del papel de algunas quimiocinas durante la E.R.P. (A) Tejido normal,
(B) Fase de iniciación, (C) Fase de amplificación, (D) Fase de progresión.
En complicidad con los mediadores proinflamatorios, esto podría llevar a una activación de las
células tubulares e intersticiales provocando la producción de quimocinas adicionales y la
infiltración de células mononucleares. De forma similar, las células parietales de la cápsula de
Bowman podrían llegar a activarse y así liberar citocinas al intersticio. Este evento podría explicar
el infiltrado periglomerular acentuado que sucede en algunas enfermedades renales. El infiltrado
periglomerular podría jugar un papel en la ruptura progresiva de la cápsula de Bowman, la cual
abriría a su vez una vía de acceso para las células T, macrófagos, y fibroblastos para invadir el
espacio urinario hasta dañar las células parietales y viscerales. Esto podría ser un punto de no-

MECANISMOS DE PROGRESION DE LA LESIÓN RENAL
91
retorno que contemplaría el tránsito del daño glomerular amplificado a una esclerosis irreversible
(Fig 26D).196
Por supuesto, la inflamación renal no siempre acaba con una lesión crónica. Los eventos iniciados
durante la inflamación normalmente acaban en la resolución del proceso. Estudios recientes
sugieren que algunas citocinas y sus receptores pueden colaborar en disminuir la modulación de la
respuesta inflamatoria, hipótesis que está todavía bajo investigación.
1.4.2.4.3 Factor de transcripción NF-κB
El factor de transcripción NF-κB parece controlar en el control la expresión de los genes que
codifican para quimocinas y otros genes que intervienen en la inflamación y respuesta inmune. Se
ha observado la presencia de lugares de unión para NF-κB en muchos promotores de genes, entre
los cuales se encuentran: la molécula de adhesión vascular (VCAM-1), la molécula de adhesión
intracelular (ICAM-1), quimocinas como la proteína quimoactante de monocitos (MCP1), la IP-10
y la IL-8, citocinas proinflamatorias como la IL-1β, enzimas proinflamatorios como la sintasa de
óxido nítrico inducible (iNOS) y la fosfolipasa A2 (PLA2), etc. Por otro lado, estudios in vivo e in
vitro indican el protagonismo del NF-κB en la proliferación de las células musculares lisas.197.
Figura 28. Esquema de activación del NF- B

INTRODUCCIÓN
92
NF-κB se identificó como regulador de la expresión del gen de la cadena ligera “kappa” en los
linfocitos B murinos, pero se ha encontrado en muchos tipos de células. La forma activada del NF-
κB es un heterodímero, que generalmente se compone de dos proteínas, las subunidades p65 y p50.
En células en reposo, el NF-κB se encuentra en el citoplasma unido al IκB, lo que le impide
penetrar en el núcleo. Cuando estas células se estimulan, diversas quinasas específicas fosforilan al
IκB, produciendo su rápida degradación por proteosomas. La disociación entre el IκB y el NF-κB
origina el paso del NF-κB al núcleo, donde se une a secuencias específicas de las regiones
promotoras de los genes diana. El ΙκΒ-α acoge la unión del κB, que es activado por el NF-κB,
incitando al aumento de la síntesis del IκB-α y a intensificar la activación de NF-κB 198.





























