Embed Size (px)
Citation preview

CÉLULAS MADRE HEMATOPOYÉTICAS Y LEUCEMIA MIELOIDE CRÓNICA
ANGÉLICA ROA LARA
COD. 200113686
UNIVERSIDAD DE LOS ANDES FACULTAD DE CIENCIAS
DEPARTAMENTO DE CIENCIAS BIOLÓGICAS PROGRAMA DE MICROBIOLOGÍA
BOGOTÁ, CUNDINAMARCA 2005

CÉLULAS MADRE HEMATOPOYÉTICAS Y LEUCEMIA MIELOIDE CRÓNICA
ANGÉLICA ROA LARA COD. 200113686
Monografía para optar al título de Microbiólogo
Director GLORIA URIBE
Bacterióloga, M.Sc
Codirector JUAN CARLOS BRICEÑO
Ingeniero Mecánico, M.Sc, PhD
UNIVERSIDAD DE LOS ANDES FACULTAD DE CIENCIAS
DEPARATAMENTO DE CIENCIAS BIOLÓGICAS PROGRAMA DE MICROBIOLOGÍA
BOGOTÁ, CUNDINAMARCA 2004

Nota de Aceptación:
Bogotá, 29 de Julio de 2005
Firma del jurado
Firma del jurado

A Dios por premiarme con la bendición de una nueva familia A Santiago, porque desde el momente en que supe que existía todo mi trabajo fue por él y para él A Nelson por su apoyo incondicional A mi familia por enseñarme a ser lo que hoy soy

AGRADECIMIENTOS
Agradezco a:
Mi familia por darme su amor y apoyo incondicional y proporcionarme todos los medios necesarios para que este trabajo pudiera llevarse a cabo en un ambiente de paz y tranquilidad. Nelson, por ser mi soporte espiritual y con su amor hacer que todas las dificultades pudieran ser superadas. Gloria Uribe, por dirigirme y orientarme en el desarrollo de los diferentes temas y a nivel personal por apoyarme y brindarme su mayor colaboración. Juan Carlos Briceño, por darme su apoyo y respaldo en todo momento. Claudia y Paola, porque gracias a sus consejos y a su amistad, este trabajo es aún más valioso. Mis amigos, porque su amistad incondicional me ha hecho una mejor persona. Viviana Rodriguez, por su colaboración y asesoramiento.

CONTENIDO
pag 0. INTRODUCCIÓN.......................................................................................................12 1. CÉLULAS MADRE HEMATOPOYÉTICAS..........................................................14
1.1 HEMATOPOYESIS ...........................................................................................15 1.1.1 Ontogenia. ..................................................................................................15 1.1.2 Microambiente medular............................................................................16
1.1.2.1 Células del estroma).........................................................................16 1.1.2.2 Matriz extracelular.............................................................................17
1.2 BIOLOGÍA E IDENTIFICACIÓN DE LAS CÉLULAS MADRE HEMATOPOYÉTICAS ..................................................................................................19
1.2.1 Biología de las células madre hematopoyéticas..................................19 1.2.2 Propiedades Funcionales.........................................................................20 1.2.3 Identificación...............................................................................................22
1.2.3.1 Marcadores de Superficie ................................................................22 1.2.3.2 Otras características.........................................................................25
1.2.4 Métodos de Separación y Aislamiento de Células Madre Hematopoyéticas.......................................................................................................25
1.2.4.1 Métodos de selección sin anticuerpos...........................................26 1.2.4.2 Métodos de selección basados en el uso de anticuerpos..........26 1.2.4.3 Ensayos funcionales .........................................................................31
2. CRECIMIENTO Y AUTO-RENOVACIÓN DE LAS CÉLULAS MADRE HEMATOPOYÉTICAS......................................................................................................33
2.1 CICLO CELULAR..............................................................................................35 2.1.1 Introducción................................................................................................35 2.1.2 Regulación del ciclo celular. ....................................................................37
2.1.2.1 Ciclinas y Quinasas Dependientes de Ciclinas (Cdks). ..............37 2.1.2.2 Regulación de las Quinasas Dependientes de Ciclinas..............39
2.1.3 Ciclo celular en las células madre hematopoyéticas...........................41 2.2 MECANISMOS DE REGULACIÓN DE AUTO-RENOVACIÓN .................42
2.2.1 Vía Hedgehog............................................................................................43 2.2.1.1 Proteínas Hedgehog.........................................................................43 2.2.1.2 Receptores de Hedgehog................................................................43 2.2.1.3 Transducción de la señal..................................................................44 2.2.1.4 Hedgehog en las Células Madre Hematopoyéticas.....................45

2.2.2 Vía Notch. ...................................................................................................46 2.2.2.1 Estructura general de Notch............................................................46 2.2.2.2 Ligandos, moléculas efectoras y moléculas blancos de Notch..47 2.2.2.3 Activación de Notch...........................................................................49 2.2.2.4 Notch en las Células Madre Hematopoyéticas.............................50
2.2.3 Vía Wnt........................................................................................................51 2.2.3.1 Proteínas Wnt.....................................................................................51 2.2.3.2 Proteoglicanos....................................................................................52 2.2.3.3 Receptores Wnt. ................................................................................52 2.2.3.4 Proteínas efectoras de la vía Wnt ...................................................52 2.2.3.5 Activación de Wnt..............................................................................53 2.2.3.6 Señalización en el núcleo.................................................................54 2.2.3.7 Wnt y las células madre hematopoyéticas....................................56
2.2.4 Otros factores implicados en el mecanismo de auto-renovación......56 2.2.4.1 Proteínas BMP. ..................................................................................56 2.2.4.2 HOXB4 ................................................................................................57 2.2.4.3 Bmi-1....................................................................................................57
2.2.5 Integración de Wnt, Hedgehog, Notch y BMP en las células madre hematopoyéticas........................................................................................................58
3. CÉLULAS MADRE LEUCÉMICAS........................................................................60
3.1 APOPTOSIS .......................................................................................................61 3.2 TELÓMEROS .....................................................................................................63
4. LEUCEMIA MIELOIDE CRÓNICA.........................................................................68
4.1 PRESENTACIÓN CLÍNICA..............................................................................69 4.1.1 Fase crónica...............................................................................................69 4.1.2 Fase acelerada..........................................................................................69 4.1.3 Crisis blástica.............................................................................................70
4.2 BIOLOGÍA MOLECULAR DE LA LEUCEMIA MIELOIDE CRÓNICA.......70 4.2.1 Expresión de BCR y ABL .........................................................................70 4.2.2 Translocación 9;22....................................................................................72 4.2.3 Vías de señalización activadas ...............................................................74
4.2.3.1 Vías RAS y MAP quinasa.................................................................74 4.2.3.2 Vía JAK/STAT ....................................................................................75 4.2.3.3 Vía de la PI-3 quinasa.......................................................................77 4.2.3.4 Vía MYC. .............................................................................................77
4.3 CÉLULA MADRE LEUCÉMICA (Ph+, BCR-ABL+) .......................................78 4.3.1 Origen, fenotipo y morfología..................................................................78 4.3.2 Propiedades alteradas..............................................................................79
4.3.2.1 Proliferación........................................................................................79 4.3.2.2 Adhesión.............................................................................................81 4.3.2.3 Apoptosis............................................................................................82

4.4 TRATAMIENTOS...............................................................................................83 4.4.1 Quimioterapia citotóxica...........................................................................84 4.4.2 Interferón alfa .............................................................................................84 4.4.3 Imatinib mesilato (ST1571)......................................................................85
4.4.3.1 Mecanismo de acción.......................................................................87 4.4.4 Transplante de células madre.................................................................88
4.4.4.1 Efecto injerto vs. leucemia...............................................................89 4.4.4.2 Transplante autólogo........................................................................90
5. IMPLICACIONES ÉTICAS Y LEGALES DE LA INVESTIGACIÓN CON CÉLULAS MADRE ...........................................................................................................92
5.1 EL PROBLEMA..................................................................................................92 5.2 DEFINICIONES BÁSICAS ...............................................................................92 5.3 PROS Y CONTRAS ..........................................................................................93 5.4 TIPOS DE LEGISLACIÓN EN EL MUNDO...................................................96 5.5 INTERROGANTES............................................................................................99
6. CONCLUSIONES Y PERSPECTIVAS A FUTURO ..........................................100 BIBLIOGRAFÍA....................................................................................................104

LISTA DE FIGURAS
pag
Figura 1.1 Selección positiva para la separación de células humanas CD34+ usando EasySepTM. .........................................................................................................29 Figura 1.2 Selección negativa mediante formación de inmuno-rosetas usando RosetteSepTM. ..................................................................................................................30 Figura 2.1 Posibles destinos de la célula madre hematopoyética. ...........................34 Figura 2.2 Regulación de la entrada a la fase M por el factor promotor de maduración o MPF (p34cdc2/ciclina B)..................................................................40 Figura 2.3 Vía de señalización Hedgehog..............................................................46 Figura 2.4 Vía de señalización Notch....................................................................50 Figura 2.5 Vía de señalización WNT.....................................................................56 Figura 3.1 Horquilla-T.......................................................................................................64 Figura 3.2 Problema de la replicación de los extremos..............................................65 Figura 4.1 Proteína p145 ABL.........................................................................................71 Figura 4.2 Proteína p160 BCR........................................................................................72 Figura 4.3 Proteína BCR-ABL .........................................................................................73 Figura 4.4 Vías de señalización activadas por BCR-ABL...........................................75

10
JUSTIFICACIÓN
En las últimas décadas un gran auge de investigaciones sobre células madre ha planteado la posibilidad de que enfermedades degenerativas puedan llegar a tener una cura. Una de las fuentes más disponibles para la obtención de células madre es la sangre o tejido hematopoyético. A partir de éste es posible aislar células madre hematopoyéticas con capacidad de producir cualquier tipo de célula sanguínea. Los transplantes de células madre hematopoyéticas proporcionan una esperanza para la cura de enfermedades como el cáncer del sistema hematopoyético mejor conocido como leucemia. Es por esto que surge la necesidad de realizar una revisión para conocer a fondo la biología de las células madre hematopóyeticas, las técnicas de aislamiento y purificación, cómo es regulado el crecimiento y la fascinante capacidad de auto-renovarse de estas células. Teniendo amplios conocimientos sobre estos temas también es posible analizar cómo una célula madre hematopoyética puede llegar a convertirse en una célula leucémica y cómo dichas células pueden ser utilizadas en el tratamiento de una de las leucemias mejor caracterizadas: la leucemia mieloide crónica.

11
OBJETIVO GENERAL El principal objetivo de esta monografía es realizar una revisión sobre las células madre hematopoyéticas, entender su biología, principales características y mecanismos de regulación de su crecimiento y auto-renovación. Asimismo, se pretende hacer una revisión de la leucemia mieloide crónica, su biología celular y molecular y los tratamientos utilizados para combatirla, todo esto con el fin de comprender el papel que juegan las células madre hematopoyéticas en el desarrollo de la leucemia mieloide crónica y su posible aplicación al tratamiento de dicha enfermedad. OBJETIVOS ESPECÍFICOS
Entender la biología y características de las células madre hematopoyéticas. Conocer los métodos utilizados en la identificación y aislamiento de las
células madre hematopoyéticas. Comprender cómo se regula el crecimiento y auto-renovación de las células
madre hematopoyéticas. Establecer cuáles son los factores que pueden contribuir a que una célula
madre hematopoyética normal pueda transformase en una célula madre leucémica. Definir en qué consiste la leucemia mieloide crónica y cuáles son sus
características citogenéticas y moleculares. Analizar los mecanismos intracelulares que conllevan a la alteración propia
de las células malignas en la leucemia mieloide crónica. Investigar qué tratamientos se usan actualmente para combatir el desarrollo
de esta enfermedad. Determinar el papel que juegan las células madre hematopoyéticas en el
desarrollo y tratamiento de la leucemia mieloide crónica. Analizar las implicaciones éticas y legales del estudio con células madre.

12
0. INTRODUCCIÓN Desde las primeras décadas del siglo veinte, se sugería la existencia de células residentes en la médula ósea, primitivas, indiferenciadas y capaces de dar origen a todos los tipos celulares sanguíneos. Pero fue solo hasta 1961 cuando James Till y Ernest McCulloch pudieron identificar una rara subpoblación de células de la médula ósea capaz de reconstituir el sistema hematopoyético de ratones irradiados y luego, demostraron que estas células a las que llamaron unidad formadora de colonia de bazo tenían el potencial de dar origen a células de múltiples linajes y la habilidad de auto-renovarse. Desde entonces las investigaciones al respecto no han cesado y la identificación de marcadores de superficie específicos de célula ha conllevado a identificar y purificar diferentes células primitivas hematopoyéticas. Asimismo, debido al descubrimiento de muchos reguladores hematopoyéticos y sus respectivos genes, ha sido posible utilizar técnicas de ADN recombinante para producir dichas proteínas reguladoras, lo que ha permitido alcanzar grandes logros en el estudio y expansión in vitro de las células madre hematopoyéticas. Los primeros reportes de leucemia mieloide crónica se dieron a conocer a mediados del siglo diecinueve gracias a la observación de detalles microscópicos a nivel clínico y a nivel post-mortem acuñados por Robert Virchow y Jhon Huges Bennett. Posteriormente, en 1870 Neumann sugirió que la médula ósea jugaba un papel importante dentro de la hematopoyesis y por lo tanto podría ser la generadora de células blancas sanguíneas malignas. Pero fue Paul Erlich en 1891 quien introdujo métodos de tinción para células sanguíneas mediante reacciones específicas, logrando de esta forma un importante avance en la clasificación de las leucemias. A partir de entonces y a lo largo del siglo veinte estudios más profundos han permitido dar una mayor aproximación a las características diagnósticas y a las causas subyacentes a esta enfermedad. Con el descubrimiento del cromosoma Filadelfia en 1960 y trece años más tarde, de la translocación cromosomal recíproca t(9;22) causante de la anormalidad cromosómica, y que genera la proteína de fusión BCR-ABL, fue posible conocer claramente las características citogenéticas de la leucemia mieloide crónica. Las nuevas técnicas de biología molecular han permitido identificar los sitios exactos de rompimiento de los genes involucrados, así como los mecanismos moleculares que conllevan a la transformación de las células progenitoras hematopoyéticas. Además del tratamiento con interferón alfa y la reciente aparición del compuesto Imatinib Mesilato (STI571) con alentadores resultados, uno de los tratamientos utilizados actualmente contra el desarrollo de la leucemia mieloide crónica es el

13
transplante de células madre. En su mayoría, los transplantes que se han realizado han sido de tipo alogénico, es decir con células provenientes de otra persona que no tenga la enfermedad. Infortunadamente este tipo de transplantes se asocia con altas tasas de mortalidad debido a que se desarrolla la enfermedad injerto vs. huésped de forma aguda o crónica. Otra opción es el transplante autogénico, realizado con células progenitoras y células madre benignas provenientes del mismo paciente. Este aún se encuentra en etapas de experimentación, por lo cual hacen falta más estudios concluyentes sobre el tema. A pesar de que la leucemia mieloide crónica se puede ver como un proceso hematopoyético aberrante que se inicia por una célula madre hematopoyética leucémica rara que ha adquirido la capacidad de dividirse indefinidamente por un cambio epigenético, todavía hay mucho por aprender sobre el origen de este tipo de célula y los mecanismos responsables de su aparición en el curso de la enfermedad, así como la aplicación del estudio de células madre hematopoyéticas en el tratamiento de este desorden clonal mieloproliferativo.

14
1. CÉLULAS MADRE HEMATOPOYÉTICAS
Célula madre es el término dado a las células con un potencial muy amplio de diferenciación, que mantienen la capacidad de auto-renovarse indefinidamente. Las células madre o células troncales pueden ser totipotentes, pluripotentes o multipotentes dependiendo de su capacidad de diferenciación. La célula totipotente corresponde al huevo fertilizado, ya que tiene la habilidad de dar origen a un organismo completo. Las células madre pluripotentes pueden producir cualquier tipo de célula de diferentes linajes más no un organismo completo. Y las células madre multipotentes son capaces de dar origen a todos los tipos de células dentro de un linaje específico común. Las células madre embrionarias son células pluripotentes obtenidas a partir de la masa interna de células del embrión temprano en la etapa de blastocisto, antes de que ocurra la gastrulación, aproximadamente a los 14 días post coitum en humanos. Estas células son capaces de diferenciarse en cualquier tipo de célula somática encontrada en un organismo adulto (Draper JS et al., 2003). Sus tres características principales son: primero, se derivan del embrión de pre-implantación, segundo, son capaces de proliferar in vitro de manera indefinida sin diferenciarse, y tercero, tienen potencial de desarrollo estable para formar las tres capas embrionarias (endodermo, mesodermo y ectodermo) después de cultivarlas de forma prolongada (Thomson JA et al., 1998). Entre las células madre multipotentes encontramos las células madre neuronales, hematopoyéticas y mesenquimales. Las células madre neuronales son la fuente de todos los tipos de neuronas del sistema nervioso central y periférico, y aparecen durante la formación de la placa neural, precursora en el desarrollo del tubo neural. Estas células tienen la habilidad de generar los principales tres linajes del sistema nervioso: neuronas, astrocitos y oligodendrocitos (Baizabal JM et al., 2003). Dentro de la médula ósea se encuentran dos poblaciones de células madre adultas: las células madre mesenquimales y las células madre hematopoyéticas. Las células madre mesenquimales, también llamadas células del estroma de la médula ósea, tienen la capacidad de diferenciarse en los linajes encontrados en el estroma de la médula, es decir, los elementos no-hematopoyéticos del tejido conectivo: hueso, adipocitos y cartílago (Short B et al., 2003). Las células madre hematopoyéticas son aquellas que dan origen a todos los linajes de células encontrados en la sangre: linfocitos, eritrocitos, plaquetas, macrófagos, monocitos, eosinófilos, basófilos y neutrófilos (Szilvassy et al., 2003).

15
1.1 HEMATOPOYESIS La hematopoyesis es el proceso fisiológico que da origen a los diferentes elementos celulares de la sangre a partir de una célula madre hematopoyética (CMH) que puede auto-renovarse y diferenciarse a células progenitoras hematopoyéticas comprometidas con un linaje linfoide o mieloide. Estos progenitores posteriormente se multiplicarán varias veces para terminar diferenciándose en elementos maduros de la sangre.
1.1.1 Ontogenia. Hasta finales de la década de los 1990s se creía que la hematopoyesis se originaba extraembrionariamente en las células mesenquimales del saco vitelino y de allí, la CMH migraba a diferentes localizaciones anatómicas como el hígado fetal, el timo y la medula ósea, en donde se realizaría la hematopoyesis definitiva de la vida embriónica, fetal y adulta. Sin embargo, estudios en ratones indican que la CMH se puede encontrar a los 9 y 10 días de desarrollo en la región aórtica-gonadal-mesonefros (AGM) proveniente de células hemangioblásticas (Medvinsky AD et al., 1996; Sanchez MJ et al., 1996), lo cual ha llevado a pensar que luego de allí las CMHs colonizan el hígado fetal, el timo y la medula ósea para llevar a cabo la hematopoyesis definitiva en la vida adulta. Algunos estudios sugieren que el saco vitelino definitivamente no contribuye a la hematopoyesis del adulto, y en otros estudios sin embargo, se ha demostrado que se pueden obtener CMHs del saco vitelino a los 9 y 10 días de desarrollo capaces de reestablecer la hematopoyesis en recién nacidos transplantados (Yoder MC et al., 1997; Yoder MC & Hiatt K 1997.). Por todo esto, aún no se sabe si la CMH de la región AGM coloniza el saco vitelino o si ocurre de forma contraria, nuevos estudios al respecto serán necesarios para aclararlo. Las CMHs colonizan el hígado fetal en humanos a las 5 semanas de gestación y la hematopoyesis hepática se desarrolla a partir de la sexta semana de gestación con predominio de eritropoyesis. Este órgano se convierte en el órgano hematopoyético dominante durante la vida fetal y la actividad hematopoyética del hígado disminuye gradualmente en los dos últimos meses de la vida intrauterina para que al momento del nacimiento sólo queden pequeños islotes hematopoyéticos. A las 8 semanas de gestación las CMHs llegan a la medula ósea y allí se desarrolla la hematopoyesis principalmente de tipo mieloide que contribuye en menor grado a la generación del pool de células sanguíneas en la vida fetal, aunque tanto la medula ósea como el hígado son órganos importantes en la linfopoyesis ya que el desarrollo de células B se inicia en el hígado fetal a las 7 semanas de gestación y cambia a la medula ósea entre las 12-14 semanas. Las CMHs colonizan el bazo a las 12 semanas y el rudimento tímico a las 8 semanas,

16
en éste último ocurre la diferenciación de las células madre del hígado a linfocitos T maduros, los cuales van a poblar los nódulos linfáticos, el hígado y el intestino en el feto hacia la décimo segunda semana de gestación, y posteriormente otros tejidos linfoides periféricos entre la semana 14 y 15 de gestación. También se ha reportado actividad de células NK (natural killer) en hígado fetal humano en las semanas 8-12 de gestación. Las razones y los mecanismos que subyacen a la localización, migración y asentamiento de las CMHs a los diferentes órganos aún no están claramente entendidos. En el adulto la hematopoyesis se desarrolla exclusivamente en la medula ósea (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000).
1.1.2 Microambiente medular.
1.1.2.1 Células del estroma. Durante los dos primeros años de vida, la medula ósea activa (medula roja) se localiza en todos los huesos y gradualmente es reemplazada por tejido medular inactivo (medula amarilla o grasa). El microambinte hematopoyético de la medula ósea contiene células de la estroma cuyo origen puede ser mesenquimal, como es el caso de las células endoteliales, los fibroblastos, los adipocitos y los osteoblastos o puede ser hematopoyético, no-mesenquimal como lo es en los macrófagos y las células dendríticas. Todas estas células de la estroma producen y depositan elementos de la matriz extracelular (MEC) y además de esto, son capaces de producir y concentrar citoquinas locales hematopoyéticas capaces de inducir o inhibir la proliferación y diferenciación de células progenitoras, formando así el "nicho de la célula madre/progenitora"(6). Se sugiere que la diferenciación hacia un linaje específico puede depender de interacciones especializadas entre células del estroma y células progenitoras (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000). Fibroblastos. Los fibroblastos son las células del estroma más estudiadas y
en su mayoría, las líneas celulares clonadas son obtenidas de medula ósea o hígado fetal humano o de ratón. Las células primitivas y en menor grado los progenitores maduros se adhieren a los fibroblastos a través de ligandos que expresan en su superficie y que se unen de forma específica a citoquinas, factores de crecimiento y componentes de la matriz extracelular secretados por los fibroblastos. Aunque las concentraciones de citoquinas producidas por los diferentes fibroblastos clonados son similares, existen diferencias entre los tipos de progenitores que cada línea celular puede mantener, ya que algunas mantienen progenitores comprometidos con linaje linfoide o mieloide y otros ni siquiera son capaces de mantener la hematopoyésis o lo hacen pobremente (Wineman J et al., 1995; Sutherland HJ et al., 1991; Henderson AJ et al., 1990; Breems DA et al., 1997; Isaad CC et al., 1993; Burroughs J et al., 1994).

17
Células endoteliales. Las células endoteliales se ubican alineadas en los
sinusoides de la medula ósea. Los progenitores y los precursores hematopoyéticos, para migrar de la medula ósea, pasan a través de la barrera endotelial de los sinusoides. Al igual que se ha hecho con los fibroblastos, también se han podido purificar células endoteliales de medula ósea. Se ha comprobado que las células CD34+ se adhieren a las células endoteliales (Rafii S et al., 1994) y además son capaces de atravesar esta barrera. Este proceso de migración depende de la interacción de las integrinas-�2 sobre las células CD34+ y las moléculas ICAM-1 (del inglés Intercellular Cell Adhesion Molecule) que expresan las células endoteliales (Mohle R et al., 1997) Osteoblastos. Se ha comprobado que los osteoblastos son capaces de
mantener la hematopoyesis gracias a la producción de factores estimulantes de colonias granulocíticas y monocíticas (FEC-G, FEC-GM) y de interleuquina-6 (IL-6), y que las células humanas CD34+ se adhieren fuertemente a los osteoblastos y al ser co-cultivados, células primitivas pueden mantenerse y expandirse de igual o mejor manera que cuando se cultivan con otras células de la estroma de la medula ósea (Taichman RS et al., 1995; Taichman RS et al., 1994), lo cual sugiere que los osteoblastos tienen un papel muy importante durante el proceso de hematopoyesis.
1.1.2.2 Matriz extracelular. Las células del estroma medular ejercen su acción hematopoyética a través del contacto directo con las CMHs y las células progenitoras, así como por la producción de citoquinas y de proteínas de la matriz extracelular como la fibronectina, la trombospondina, los proteoglicanos y glicosaminoglicanos, varios tipos de colágeno, la laminina y la hemonectina. Aunque no es conocida la forma exacta como actúan estos componentes sobre la hematopoyesis, se sabe que afectan directamente la proliferación y diferenciación de las células progenitoras y pueden alterar la respuesta de éstas a las citoquinas. Fibronectina. Es una glicoproteína de 450kDa presente de forma abundante
en la médula ósea adulta y es producida por las células endoteliales y los fibroblastos (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000). La fibronectina parece tener un papel de gran importancia en el asentamiento de las CMHs y afectar la proliferación y diferenciación de las CMHs y las células progenitoras. Se ha demostrado que la adhesión de la célula progenitora a la fibronectina es importante en la regulación del crecimiento de los progenitores (Levesque JP et al., 1996; Hurley RW et al., 1995; Frisch SM et al., 1996), y su interacción también se requiere para la diferenciación de los progenitores eritroides (Patel VP et al., 1987) y la supervivencia y diferenciación de los progenitores linfoides B (Miyake K et al., 1991).

18
Trombospondina. Es una glicoproteína de 450 kDa producida por las plaquetas, las células endoteliales y los fibroblastos (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000). Está presente de forma abundante en el microambiente medular de los megacariocitos, fibroblastos, y la matriz extracelular asociada con la hematopoyesis activa (Beckstead JH et al., 1986). La trombospondina sirve como ligando para los progenitores comprometidos (Long MW et al., 1990) y su adhesión con las células progenitoras hematopoyéticas podría modular la respuesta de los progenitores a las citoquinas. El co-cultivo de unidades formadoras de colonia (UFC) con factor de célula madre y trombospondina amplifica la respuesta de las UFC a citoquinas solubles como IL-3 y FEC-GM (Long MW et al., 1992). Proteoglicanos y glicosaminoglicanos. Los proteoglicanos presentes en la
médula ósea incluyen heparán sulfato, dermatán sulfato, condroitin sulfato y ácido hialurónico. Están compuestos de una proteína central a la cual se le unen uno o más glicosaminoglicanos (GAGs). Los GAGs son cadenas de polisacáridos largas, cargadas negativamente y sin ramificar, compuestas de repeticiones de unidades disacárido sulfatadas (Hoffman R et al., Anatomy and physiology of hematopoiesis 2000). Los GAGs de heparán-sulfato específicos de la médula ósea permiten la adhesión de células CD34+ (Siczkowski M et al., 1992) y la concentración de factores de crecimiento como IL-3 y FEC-GM (Gupta P et al., 1998; Roberts R et al., 1988; Gallagher JT et al., 1994). Esta habilidad de localizar selectivamente factores de crecimiento y células progenitoras podría ser de gran importancia en la regulación, proliferación y diferenciación de las células progenitoras hematopoyéticas, e implicaría que los GAGs de heparán-sulfato se comportan como "orquestadores" de los nichos de células madre y células progenitoras ya que tienen múltiples funciones: mantienen la adhesión de células, se unen a otros componentes de la matriz extracelular, y permiten la unión de citoquinas (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000). Colágenos y laminina. Los colágenos son fibras de proteínas que se
encuentran en el espacio extracelular. Existen varios tipos de colágeno, de los cuales los más comunes son el tipo I, II, III y IV. Los tipos I-III constituyen el esqueleto estructural del espacio extracelular, mientras que el tipo IV forma la mayor parte de la membranas basales (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000). La laminina es un complejo polipeptídico de 850kDa compuesto de cadenas largas unidas entre sí por puentes disulfuro. Posee dominios funcionales que pueden unir colágeno tipo IV, proteoglicanos y receptores de superficie celular (Engvall E et al., 1995). Aún no se sabe el papel específico que juegan estas proteínas en los procesos de localización, proliferación o diferenciación de progenitores hematopoyéticos, pero la salida de células hematopoyéticas maduras y algunas veces inmaduras, del microambiente medular al torrente sanguíneo requiere el paso de estas células a través de las membranas basales (Tuszynski GP et al., 1997).

19
Hemonectina. Es una proteína de 60kDa encontrada exclusivamente en el ambiente de la médula ósea. El papel exacto de la hemonectina en la hematopoyesis aún es desconocido pero se cree que mantiene la unión de progenitores mieloides de forma preferencial (Campbell AD et al., 1990).
1.2 BIOLOGÍA E IDENTIFICACIÓN DE LAS CÉLULAS MADRE HEMATOPOYÉTICAS
1.2.1 Biología de las células madre hematopoyéticas. El sistema hematopoyético es aquel capaz de dar origen a todos los tipos de células sanguíneas y se origina a partir de una célula madre hematopoyética (CMH). A través del proceso de división asimétrica, una sola división puede resultar en la formación de una célula madre idéntica y una célula mucho más madura. La CMH posee tres características básicas: primero las CMHs son pluripotentes, es decir, que tienen el potencial de generar progenie de todos los linajes sanguíneos: linfocitos B y T, eritrocitos, megacariocitos/plaquetas, basófilos/mastocitos, eosinófilos, neutrófilos/granulocitos, y monocitos/macrófagos. Segundo, las CMHs tienen un alto potencial proliferativo, es decir que son capaces de dar origen a un gran número de células maduras durante el periodo de vida normal del organismo. Y tercero, las CMHs tienen alta capacidad de auto-renovación, manteniendo una división de tipo más simétrico que conduce a la generación de nuevas células madre idénticas. Esta propiedad es muy importante debido al estrés fisiológico al que están sujetas constantemente y que puede conllevar a la reducción en su población (Hoffman R et al., 2000; Szilvassy SJ 2003). Durante la hematopoyesis, células madre y células progenitoras hematopoyéticas dan origen a células hijas que pueden ser reconocidas morfológicamente y son llamadas precursores hematopoyéticos. La transición de las células pluripotentes con capacidad de auto-renovarse, del compartimento de célula madre al compartimento de célula progenitora, involucra un proceso llamado compromiso, el cual aún no está completamente dilucidado pero que se caracteriza por la restricción en la capacidad de diferenciación y de proliferación de la célula madre. Los compartimentos de la célula progenitora están compuestos en su mayoría de células con capacidad de diferenciarse hacia un linaje (progenitores unipotentes), y en muy baja frecuencia de células primitivas bipotentes o multipotentes. Es por ello que el compromiso implica adquirir ciertos receptores de factores de crecimiento específicos y perder otros. Las células progenitoras generalmente se definen por la capacidad que tienen de formar colonias en experimentos in vitro. La mayoría de células de la medula ósea forman el compartimento de precursores y exhiben características morfológicas tanto nucleares como citoplasmáticas que

20
son fácilmente reconocibles y que pueden ser usadas para clasificar el linaje al cual se ha comprometido la célula progenitora (Hoffman R et al., Stem cell model of hematopoiesis, 2000). En el sistema hematopoyético, las células madre son heterogéneas con respecto a su habilidad de auto-renovarse. Los progenitores multipotentes constituyen el 0.05% de las células de medula ósea de ratón, y pueden ser divididas en tres poblaciones diferentes: CMHs con capacidad de auto-renovación a largo plazo, CMHs con capacidad de auto-renovación a corto plazo, y progenitores multipotentes sin potencial detectable de auto-renovación. Estas poblaciones forman un linaje en el cual las CMHs de largo plazo dan origen a las CMHs de corto plazo, que a su vez dan origen a los progenitores multipotentes (Morrison SJ et al., 1997). A medida que las CMHs maduran, progresivamente van perdiendo su potencial de auto-renovarse pero se vuelven mitóticamente más activas, es decir, que en los compartimentos de célula progenitora hay muy poca capacidad de auto-renovación pero es más frecuente encontrar células mitóticamente activas allí que en los compartimentos de célula madre (Hoffman R et al, Stem cell model of hematopoiesis, 2000; Reya T et al., 2001).
1.2.2 Propiedades Funcionales Existe una gran heterogeneidad entre subpoblaciones idénticas de CMHs, razón por la cual múltiples estudios realizados con el fin de identificar algunas de las propiedades funcionales de estas células arrojan resultados diversos aunque complementarios entre sí. El primer estudio cuantitativo de células madre fue realizado por Till y McCulloch en 1961 (Till JE et al., 1961). En éste, se obtuvieron células hematopoyéticas de la medula o el bazo de ratones donadores y se transplantaron por vía intravenosa a ratones previamente irradiados con dosis letales con el fin de destruir la hematopoyesis endógena y prevenir la generación de CMHs del hospedero. Algunas células inyectadas (aproximadamente 10%) se alojaban en el bazo y allí proliferaban y formaban clonas de colonias macroscópicas que podían ser contadas y a las cuales se les llamó unidades formadoras de colonia de bazo (UFC-B). Posteriores estudios de UFC-B se realizaron con el fin de determinar el potencial de proliferación y diferenciación de la célula iniciadora y de cuantificar CMHs in vivo. Actualmente es ampliamente aceptado que la única prueba que define una CMH es aquella en la cual se demuestra su capacidad de completar y mantener (más de 6 meses) la regeneración del sistema linfohematopoyético después de haber realizado transplante (Szilvassy SJ 2003). Los cultivos de medula a largo plazo (LTC [del inglés long-term cultures]) fueron desarrollados por Dexter et al., (Dexter TM et al., 1977) y en ellos, medula ósea

21
humana o de ratón es cultivada en un medio con suero en frascos de cultivo plásticos, las células de la estroma forman una capa alimentadora adherente en la cual las CMHs y las células progenitoras en el inóculo proliferan y se diferencian por semanas y meses en ausencia de factores exógenos (Coloumbel L et al., 1983). Las células de iniciación de cultivo a largo plazo (LTC-IC [del inglés long-term culture initiating cells]) son células progenitoras más primitivas que las células formadoras de colonia (CFC), se pueden encontrar entre o bajo las células de la estroma, son capaces de iniciar y mantener la hematopoyesis por largos periodos de tiempo en cultivo y pueden reiniciar LTC secundarias. Menos del 0.1% de CMHs de la medula ósea son capaces de proliferar a largo plazo y de auto-renovarse (Szilvassy SJ 2003). La proporción de LTC-IC con respecto a CFC en sangre periférica es de 1:50 mientras que en médula ósea es de 1:20 (39). Sin embargo, no se han encontrado diferencias significativas en el potencial de proliferación o diferenciación de las LTC-IC de médula ósea y de sangre en estado normal (Benboubker L et al., 2002). Los ratones diabéticos no-obesos con inmunodeficiencia combinada severa (NOD/SCID) son modelos quiméricos in vivo que llevan por un lado la mutación scid/scid, la cual representa un defecto en las células T y B, y por otro lado, defectos en la vía del complemento y en la función de los macrófagos (ratones NOD). Estas deficiencias inmunológicas permiten evitar el rechazo inmune de células xenogénicas, razón por la cual dichos modelos son ampliamente utilizados para realizar ensayos con células hematopoyéticas primitivas humanas inyectadas y determinar así, el potencial de repoblación que tienen las células (Pflumio F et al., 1996). Variaciones muy sutiles en los modelos in vivo utilizados para identificar poblaciones únicas de CMHs pueden tener efectos significativos en las frecuencias y propiedades funcionales encontradas y es importante considerarlas cuando se evalúan datos provenientes de diferentes estudios (Szilvassy SJ 2003). Se ha encontrado que el número de CMHs humanas transplantables que pueden ser recuperadas de medula ósea de ratones NOD/SCID puede ser expandido hasta 10 veces tratándolos con factor de célula madre, factor estimulante de colonia granulocito/monocito (FEC-GM) y eritropoyetina (EPO), justo antes de sacrificarlos (Cashman JD et al., 1999). Estas citoquinas aparentemente aumentan la probabilidad de auto-renovación de las CMHs humanas estimuladas para proliferar en modelos in vivo. Asimismo, recientemente, se desarrollaron ratones NOD/SCID con un silenciamiento en el gen de la β2 microglobulina, que además de poseer las características de los ratones NOD/SCID también carecen de células asesinas naturales (NK), facilitando así la diferenciación a multilinaje de 10 veces más células de cordón umbilical que las que son requeridas para lograr un nivel de implantación similar en ratones NOD/SCID (Kollet O et al., 2000).

22
1.2.3 Identificación
1.2.3.1 Marcadores de Superficie. A lo largo del proceso de maduración, las CMHs ganan y pierden ciertos marcadores de superficie, que permiten su aislamiento e identificación. A pesar del gran número de estudios realizados, aún no se ha podido encontrar un único marcador específico de las CMHs, sin embargo, el uso de combinaciones de marcadores permite agrupar mezclas de células funcionalmente heterogéneas en subgrupos un poco más homogéneos. Marcadores de linaje. Debido a que las células hematopoyéticas primitivas
no expresan marcadores de linaje propios de células más maduras específicas de la sangre, la ausencia de marcadores de linaje (lin) permite distinguir células inmaduras del resto de células diferenciadas que son más abundantes. La selección de células lin- proporciona un enriquecimiento típico de CMHs de 20 a 500 veces, dependiendo de la combinación de marcadores usados. Algunos antígenos de linaje usados son marcadores eritroides como la glicoforina A, antígeno leucocitario humano-DR (HLA-DR), TER-119, marcadores linfoides como CD45, CD2, CD3, CD4, CD8, CD19, CD20 y mieloides como CD14, CD56 y CD33 (Wognum AW et al., 2003). CD34. Esta sialomucina, fue el primer marcador de diferenciación
reconocido en las células hematopoyéticas primitivas humanas y se continúa usando para aislar progenitores y CMHs para uso clínico (Wognum AW et al., 2003). Se expresa sobre CMHs y células progenitoras hematopoyéticas así como en células endoteliales de pequeños vasos y fibroblastos embriónicos. La densidad del antígeno CD34 es mucho más alta en progenitores tempranos, y la densidad disminuye progresivamente en células maduras. Las células completamente diferenciadas son CD34- (Sutherland DR et al., 1992; Young PE et al., 1995). CD34 se expresa aproximadamente en 1-4% de las células nucleadas en aspirados de medula ósea humana normal y en <1% de las células nucleadas en sangre periférica humana en estado normal (Wognum AW et al., 2003). La movilización y/o terapia citotóxica aumenta el nivel de células CD34+ en la sangre en >1% y se ha vuelto un buen método para aislar suficientes CMHs para transplantes mediante leucoferesis (Körbling M et al., 2001). Algunas más no todas las isoformas de CD34 podrían estar involucradas en la adhesión de CMHs, aunque de forma indirecta a través de la activación de otros receptores de adhesión de la superficie celular (Hoffman R et al., Anatomy and physiology of hematopoiesis, 2000). No se sabe si las interacciones dependientes de CD34 afectan a las CMHs o a las células progenitoras hematopoyéticas pero ratones CD34-/- han disminuido el número de progenitores en la medula (Cheng J et al., 1996), lo cual indicaría que este antígeno está involucrado en el crecimiento de los progenitores.

23
Células purificadas CD34+ son capaces de reconstituir la hematopoyesis multilinaje en ratones NOD/SCID irradiados (Larochelle A et al., 1996) y ha sido posible demostrar el potencial de implantación de células humanas CD34+ en varios transplantes autólogos y alogénicos (Civin CI et al., 1996; Vogel W et al., 2000). Sin embargo, la frecuencia de células con actividad de progenitoras es <20%, la frecuencia de LTC-ICs es <1% y de CMHs <0.1%, en poblaciones de células CD34+ altamente purificadas (>90%) (Wognum AW et al., 2003). La distinción entre la población de CMHs, células progenitoras y células más maduras CD34+, es posible gracias al patrón de expresión de otros marcadores como CD38, un antígeno que se puede expresar entre un nivel intermedio y alto (>90%) en CMHs CD34+ que incluyen células formadoras de colonia, y se expresa aproximadamente en el 60% de LTC-ICs (Wognum AW et al., 2003). Las células CD34+CD38+ son más abundantes que las CD34+CD38-, pero se ha comprobado que estas últimas pueden reconstituir y mantener la hematopoyesis multilinaje en ratones inmunodeficientes después de realizar transplante (Verstegen MMA et al., 1998). A pesar de utilizar el CD34 como un marcador de CMHs primitivas, también se han detectado células CD34- con potencial linfopoyético en cordón umbilical humano y en fuentes hematopoyéticas adultas, capaces de reconstituir la hematopoyesis multilinaje en ratones inmunodeficientes y de generar células CD34+ in vitro e in vivo. Sin embargo las CD34- son células raras detectables en células formadoras de colonia y sus precursores más no en LTC-ICs, por lo tanto el fenotipo de CMHs CD34- también se caracteriza por la ausencia de CD38, marcadores específicos de linaje y la expresión de CD133 (Wognum AW et al., 2003; Bhatia M et al., 1998; Zanjani E et al., 1998; Nakamura Y et al., 1999; Zanjani ED et al., 2003). Al comparar células CD34-CD38-lin- con células CD34+CD38-lin-, solo una pequeña fracción de las primeras (0.2%) expresaba el marcador CD133 y precisamente esas células fueron capaces de repoblar ratones NOD/SCID aunque la frecuencia de las células y el nivel de repoblamiento fue 100 veces menor que el de las células con fenotipo CD34+CD38-lin- (Gallacher L et al., 2000). Se sugiere que este bajo nivel de implantación puede deberse a que las células CD34- son incapaces de alcanzar la medula ósea después de transplante intravenoso, ya que se ha demostrado que la inyección directa de células CD34-lin- en la medula ósea de ratones NOD/SCID conlleva a niveles elevados de implantación, en comparación con células CD34+ (Wang J et al., 2003). CD133. También conocido como AC133 (Yin AH et al., 1997; Miraglia S et
al., 1997), se expresa en la mayoría más no en todas las células CD34+, incluyendo células repobladoras, progenitores inmaduros y progenitores monocito/granulocito, pero no en la mayoría de progenitores eritroides (Wognum AW et al., 2003). Se cree que representa el homólogo humano de las glicoproteínas prominina 5-transmembranal (PROML 1). La función específica y los ligandos potenciales de la familia de las promininas aún no han sido

24
caracterizados, sin embargo las promininas humanas y de ratón comparten asociación de membrana selectiva y perfiles de expresión en tejido similares (Bhatia M 2001). Las células CD133+ están presentes en sangre de cordón umbilical y en la medula ósea (Pasino M et al., 2000; Matsumoto K et al., 2000). Se ha demostrado la presencia de células CD133+CD34-CD38-Lin- en sangre periférica de adultos movilizada mediante FEC-G + factor de célula madre, con potencial de diferenciación a células CD34+ similar al de células derivadas de cordón umbilical (Hess DA et al., 2002). Aunque CD133 representa un importante marcador de superficie celular para la identificación de células madre y células progenitoras humanas, aún no es claro si el uso de este marcador otorga alguna ventaja sobre el uso de CD34 para el aislamiento o la expansión celular de CMHs y células progenitoras hematopoyéticas (Bhatia M 2001). C-KIT. Conocido también como CD117, corresponde al receptor para el
factor de célula madre, el cual promueve la proliferación y diferenciación de células progenitoras primitivas hematopoyéticas a células progenitoras comprometidas (Hoffman R et al., Growth factors, Cytokines, and the Control of Hematopoiesis, 2000). C-kit se expresa sobre aproximadamente 2/3 de las células CD34+ incluyendo las células progenitoras más comprometidas con linaje, pero está ausente de las células sanguíneas maduras circulantes. Sin embargo, el nivel de expresión de c-kit sobre células CD34+ a menudo es lo suficientemente alto como para permitir su uso en los métodos de aislamiento de células hematopoyéticas (Wognum AW et al., 2003). CDP1. Fue identificado recientemente (proteína que contiene el dominio
cub) y tiene un patrón de expresión similar al de CD133 (Conze T et al., 2003). No se expresa sobre células sanguíneas maduras y su expresión sobre células hematopoyéticas normales está restringido a células CD34+ incluyendo aquellas con actividad repobladora NOD/SCID. Al igual que CD34 y CD133, CDCP1 puede ser útil para el enriquecimiento de CMHs y células progenitoras pero no para su discriminación (Wognum AW et al., 2003). KDR. También conocido como receptor del factor de crecimiento vascular-
endotelial-2 (VEGFR-2) o como Flk-1 en ratones. Se expresa aproximadamente en 0.1-0.5% de las células CD34+ en la medula ósea humana adulta (Wognum AW et al., 2003). Se ha encontrado que células CD34+KDR+ son ricas en LTC-ICs así como en células repobladoras capaces de implantarse en ratones NOD/SCID, a diferencia de células CD34+KDR- que muestran poca habilidad de implantación (Hoffman R et al., Growth factors, Cytokines, and the Control of Hematopoiesis, 2000). La activación de este receptor es importante en el mantenimiento de la viabilidad de CMHs aunque no promueve la proliferación celular (Wognum AW et al., 2003). VEGFR1. Es otro receptor VEGF, se expresa aproximadamente en el 5%
de las células CD34+ de cordón umbilical e hígado fetal humano y en la mayoría

25
de células repobladoras de ratones NOD/SCID. La estimulación de VEGFR1 por su ligando promueve el ciclo de CMH (Hattori K et al., 2002).
1.2.3.2 Otras características. Además de la expresión de antígenos de superficie, las células primitivas hematopoyéticas también pueden ser identificadas por su habilidad de bombear fuera de ellas tinciones fluorescentes mitocondriales o de unión al ADN, como Rhodamina-123 (Rho) y Hoechst 33342 (Ho) (Wognum AW et al., 2003). Se ha encontrado que la mayoría de CMHs de tejidos adultos de humano y de ratón son Rho-/lo (Udomsakdi C et al., 1991; Spangrude GJ 1990), y que dicho fenotipo está determinado por la actividad de la glicoproteína-P, un transportador ABC que se expresa en la superficie celular (Chaudhary PM et al., 1991). Las células de población lateral (SP [del inglés side population]) son una población de células Ho-/lo que ha sido designadas así ya que al analizarlas mediante la técnica de citometría de flujo, forman una agrupación de eventos característica hacia el lado izquierdo inferior de los perfiles de diagrama de puntos de células teñidas con Ho (Wognum AW et al., 2003). Este tipo de células ha sido identificado en cordón umbilical y medula ósea adulta y en medula ósea de monos (Goodell MA et al., 1997). Células SP de médula ósea de ratón pueden diferenciarse a cardiomiocitos y células endoteliales después de ser transplantadas (Jackson KA et al., 2001), lo cual indicaría que el fenotipo SP también es característico de células madre no hematopoyéticas. El fenotipo SP ha sido atribuido a una alta expresión de transportadores de membrana responsables también de la resistencia a múltiples drogas de las células tumorales, una de estas moléculas transportadoras es la ABCG2 (o BCRP1) otro miembro de la familia del gen transportador ABC, necesaria para mediar la salida de la tinción Hoechst en las células SP (Zhou S et al., 2001; Zhou S et al., 2002), por lo tanto ABCG2 podría ser un marcador útil en la identificación y aislamiento de CMHs primitivas. Aunque la purificación también podría lograrse aislando células con fenotipo CD34+CD38-
SP+, esta tinción es bastante tóxica para las CMHs, lo cual podría contrarrestar el grado de pureza (Wognum AW et al., 2003).
1.2.4 Métodos de Separación y Aislamiento de Células Madre Hematopoyéticas.
Debido a la baja frecuencia frecuencia de las CMHs en los tejidos, a que carecen de un marcador de superficie único que las identifique, y a que muchas de sus propiedades fenotípicas y características físicas son compartidas con otras células progenitoras y células maduras es muy difícil separarlas con la pureza suficiente para poder utilizarlas para transplantes clínicos o con fines investigativos. Por ello, se han desarrollado diferentes métodos para identificar las CMHs ya sea por sus

26
características físicas o por su funcionalidad. Estos métodos pueden variar en el grado de dificultad, en el tiempo requerido y en la disponibilidad de herramientas necesarias para desarrollarlos, y aunque con cada técnica es posible obtener CMHs con alto grado de pureza, éstas también pueden ser usadas en combinación para obtener mejores resultados.
1.2.4.1 Métodos de selección sin anticuerpos. Las células hematopoyéticas varían en tamaño y densidad, y son capaces de moverse a través de un medio al aplicarle una fuerza centrífuga de acuerdo a la densidad del medio y al tamaño y densidad de la célula. Debido a esto se utiliza la técnica de centrifugar suspensiones de células hematopoyéticas en un medio de densidad como percoll o mezclas de ficol-hipaque, sin más enriquecimientos, para remover las células maduras más densas como los glóbulos rojos y los granulocitos, aunque es posible que se presente alguna pérdida (10-30%) de CMHs. Como la mayoría de técnicas de purificación de CMHs mediadas por anticuerpos están diseñadas para suspensiones de células de baja densidad, es necesario un primer paso de separación por densidad (Wognum AW et al., 2003). Bajo condiciones constantes la mayoría de CMHs permanecen inactivas mientras que el resto de células maduras se están dividiendo constantemente, es por ello que el uso in vivo o in vitro de drogas citotóxicas, como 5-fluorouracil (5-FU) e hidroxiurea, conllevan a la muerte selectiva de células dividiéndose y es posible recuperar poblaciones de CMHs antes de usar otro método de selección (Wognum AW et al., 2003). Se ha encontrado que las células hematopoyéticas poseen una resistencia relativa a agentes alquilantes como los derivados de la ciclofosfamida (Ej. 4-hidroxiperoxiciclofosfamida y mafosfamida) (Gordon MY et al., 1985) debido a la expresión selectiva de la enzima aldehído deshidrogenasa (ALDH) (Sahovic EA et al., 1998; Moreb JS et al., 1995). Con el fin de identificar y aislar células hematopoyéticas humanas y de ratón se han usado sustratos fluorescentes para ALDH (Storms RW et al., 1999) y la escogencia de células ALDH+ ha llevado a encontrar poblaciones enriquecidas de células muy primitivas así como de células comprometidas con linaje. También se ha encontrado gran expresión de ALDH en células CD34+CD38- y en una pequeña población de células CD34-lin-CD38-, lo cual sugiere que la ALDH puede ser útil para aislar CMHs CD34-, aunque se necesitan más estudios que lo confirmen (Wognum AW et al., 2003).
1.2.4.2 Métodos de selección basados en el uso de anticuerpos. Debido a la alta especificidad y diversidad de los anticuerpos monoclonales, usándolos en combinaciones adecuadas estos son de gran utilidad para separar diferentes subgrupos de células. Los anticuerpos se han usado acoplándolos con toxinas para eliminar células de forma específica, al igual que con antígenos de superficie con el fin de activar la cascada del complemento y lisar así poblaciones celulares.

27
También pueden acoplarse a matrices sólidas para permitir la adherencia celular a frascos, columnas y partículas magnéticas, así como a fluorocromos para usarlos en procedimientos de FACS (del inglés Fluoroescence Activated Cell Sorting). Estos métodos de separación basados en FACS presentan grandes ventajas ya que pueden generar suspensiones de alta pureza, aunque no son capaces de procesar grandes números de células, por lo cual esta tecnología se aplica típicamente a suspensiones en las cuales previamente se han removido células más maduras y el volumen de la muestra ha sido reducido (Wognum AW et al., 2003). La selección de células usando anticuerpos que reconozcan antígenos expresados en la membrana celular puede realizarse de dos formas, la selección positiva se refiere al uso de anticuerpos específicos de marcadores de superficie presentes en las células que se desean aislar. El ejemplo más común es el aislamiento de células CD34+ con el fin de seleccionar células humanas hematopoyéticas primitivas. Por otro lado, también pueden ser usados anticuerpos que reconozcan antígenos que no están presentes en las células deseadas, esto se denomina selección negativa. El acoplamiento de anticuerpos a inmunotoxinas y a moléculas activadoras del complemento es una técnica de selección negativa, mientras que la técnica de FACS puede ser acoplada a selección positiva o negativa o de manera simultánea a las dos (Wognum AW et al., 2003). Para lograr un mayor grado de pureza en la separación de CMHs de una suspensión celular se utilizan varios pasos de selección positiva y negativa, aunque en la selección negativa es necesario utilizar varios anticuerpos para remover diferentes tipos de células no deseadas, mientras que la selección positiva puede realizarse con un solo anticuerpo. Sin embargo, en la selección positiva es necesario remover las células marcadas con el anticuerpo de la matriz de separación. Recientes avances en la separación celular magnética han permitido superar esta desventaja mediante el de partículas magnéticas muy pequeñas en un sistema que no requiere columna de separación (EasySepTM, StemCell Technologies, Inc., Vancouver, BC, Canada) (Thomas TE et al., 1997). Entre las ventajas que presenta este método esta el hecho de que las partículas magnéticas son muy pequeñas de manera que pueden unirse fácilmente a las células marcadas y no requieren ser removidas ya que no interfieren con el posterior análisis del cultivo o con la citometría de flujo, además las células deseadas quedan adheridas al tubo de separación y el resto de células son removidas. Antes del desarrollo de esta nueva técnica, las células marcadas con partículas magnéticas de tamaño submicra debían ser separadas magnéticamente en una columna con matriz magnetizable, luego de esto se requería un paso extra para remover las células purificadas de la columna (Ej. StemSepTM, StemCell Technologies, Inc., y MACS, Miltenyi Bistec, Auburn, CA, USA). Una desventaja que presentan todos los métodos de selección positiva es que pueden remover células más primitivas que no expresen el marcador utilizado, como las CMHs

28
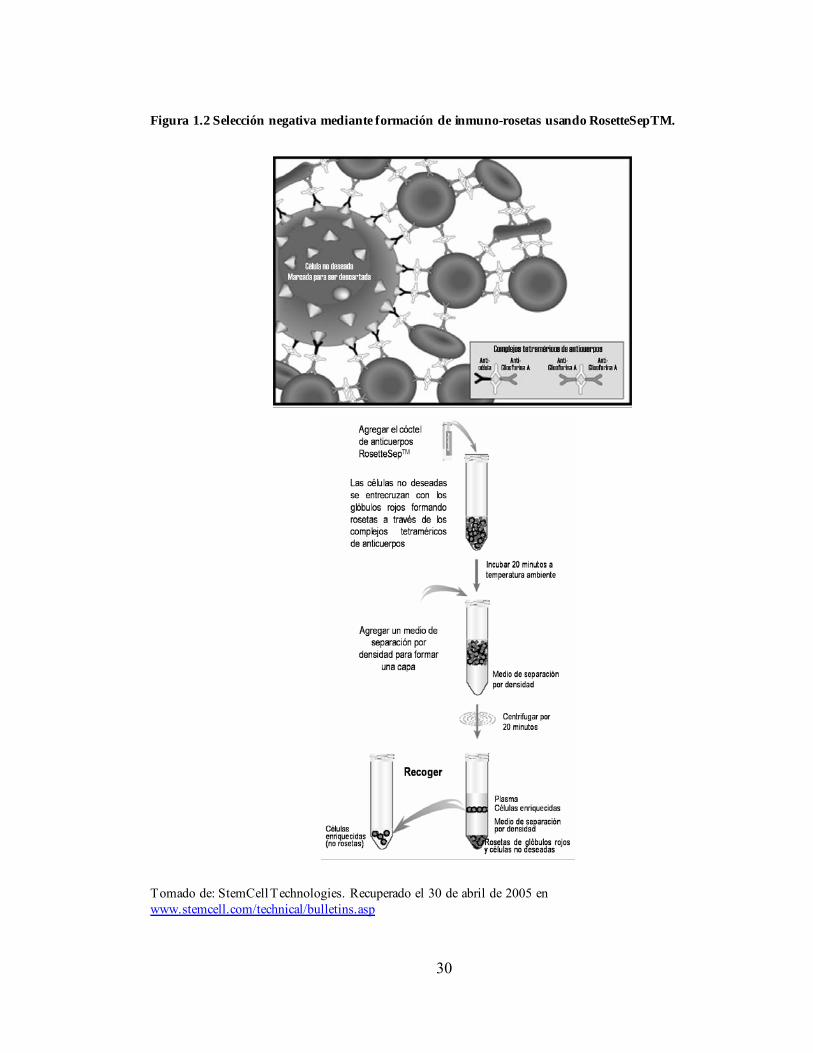
CD34-, además, al remover células maduras se pueden estar eliminando poblaciones celulares que podrían ser tener importantes funciones accesorias necesarias para el asentamiento y la implantación celular (Wognum AW et al., 2003; Bonnet D et al., 1999; Verstegen MMA et al., 1998). En los métodos de selección positiva, el marcador más usado es el CD34 y puede proporcionar un enriquecimiento de 25 a 100 veces de CMHs y células progenitoras (Wognum AW et al., 2003). Actualmente, todos los métodos disponibles se basan en principios inmunomagnéticos mediante el uso de abundantes micro-gotas (Ej. Dynabeads , Dynal, Oslo, Norway), pequeñas nanopartículas con una columna magnética (StemSepTM, MACS), y nanopartículas sin columna (EasySepTM). La figura 1.1 es una representación esquemática del método de selección inmunomagnético sin columna EasySepTM para el enriquecimiento de células CD34+. Los sistemas CliniMACS (Miltenyi Biotec) y Isolex (Baxter Healthcar Corp., Deerfield, IL, USA) han sido adaptados para llevar a cabo la selección a gran escala de células CD34+ de medula ósea o para cultivar células de sangre inmovilizada con el fin de usarlas en transplantes clínicos (Wognum AW et al., 2003). El uso del marcador CD133 para aislar CMHs solamente ofrece un enriquecimiento 2 a 5 veces mayor de CMHs que la selección positiva de células CD34+, sin embargo, la selección de células CD133+ podría ayudar a retener ese extraño grupo de CMHs que son CD34- (Wognum AW et al., 2003). En los métodos de selección negativa la eliminación de células maduras se realiza utilizando un cóctel de anticuerpos específicos para marcadores de linaje, mediante el uso de frascos de poliestireno, partículas densas, nanopartículas paramagnéticas o glóbulos rojos presentes en la muestra. En la técnica RosetteSepTM (figura 1.2), los glóbulos rojos de la muestra se unen selectivamente a las células que se desea eliminar a través de complejos de anticuerpos tetraméricos formando inmuno-rosetas, las cuales son removidas de la muestra por sedimentación en un procedimiento estándar de centrifugación por densidad usando Ficoll-PaqueTM. La selección negativa permite mayor flexibilidad en la selección del tipo de célula deseada, al igual que en el grado de enriquecimiento y recuperación de la misma. Dependiendo de la combinación y las concentraciones de los anticuerpos usados, un cóctel puede ser hecho a la medida con el fin de desechar subgrupos de células específicas (Wognum AW et al., 2003). Los marcadores CD38, CD45RA y CD71 se expresan en precursores tempranos hematopoyéticos de diferentes linajes pero no en CMHs. El uso de estos marcadores permite de 5 a 10 veces mayor enriquecimiento de LTC-ICs desechando progenitores mieloides CD38+CD45RA+ y progenitores eritroides CD71-alto (Thomas TE et al., 1997).

29
Figura 1.1 Selección positiva para la separación de células humanas CD34+ usando EasySepTM.
Tomado de: StemCell Technologies. Recuperado el 30 de abril de 2005 en www.stemcell.com/technical/bulletins.asp
30
Figura 1.2 Selección negativa mediante formación de inmuno-rosetas usando RosetteSepTM.
Tomado de: StemCell Technologies. Recuperado el 30 de abril de 2005 en www.stemcell.com/technical/bulletins.asp

31
1.2.4.3 Ensayos funcionales. El objetivo de los ensayos funcionales consiste en identificar por separado las diferentes clases de células progenitoras hematopoyéticas que son producidas secuencialmente durante el proceso de maduración de células madre a células funcionalmente diferenciadas. Sin importar si el ensayo es in vivo o in vitro, se deben tener en cuenta dos requisitos importantes: uno es la interpretación de los resultados, que solo dependerá de la función de las células, y el segundo es evaluar de forma precisa el número y el potencial de proliferación de dichas células en ensayos cuantitativos. Todos los ensayos miden proliferación celular a través del número de células producidas, y potencial de diferenciación estimando el número de linajes diferentes representados en la progenie (Coulombel L 2004). Existen ensayos de células hematopoyéticas in vitro e in vivo. Entre las pruebas in vitro se incluyen los ensayos a largo plazo en los cuales células madre logran más de 15 divisiones en un periodo que excede las 5 semanas. Una característica común en estos ensayos es que los sistemas requieren la presencia de células alimentadoras capaces de proveer un sustrato y una fuente de factores reguladores con el fin de reconstituir la complejidad del microambiente medular in vivo. Inicialmente las células alimentadoras provenían de células mesenquimales derivadas de medula ósea y constituían la capa adherente, mientras que las células que se deseaba evaluar estaban escondidas en esta capa y proliferaban en la fracción no adherente (Dexter TM et al., 1977). Subsecuentemente, se han ido introduciendo variaciones en las cuales las células alimentadoras y las células progenitoras se cultivan por separado. Las primeras, provienen de líneas celulares de estroma derivadas de medula ósea de ratón e inmortalizadas espontáneamente, las más utilizadas son MS-5, S17, AFT024, M210B4. Las segundas, son células fenotípicamente purificadas de diferentes tejidos humanos y adultos, las LTC-ICs más comunes son CD34+CD38bajo/-. Con el fin de evaluar proliferación celular se realizan experimentos de diluciones limitantes usando estadística de Poisson (Coulombel L 2004). Los ensayos a corto plazo son otro tipo de pruebas in vitro, en donde las células progenitoras comprometidas con linaje logran de 5-10 divisiones en menos de 3 semanas. En este tipo de ensayos los factores de crecimiento que se requieren ya han sido ampliamente caracterizados y la mayoría están disponibles como proteínas recombinantes, a diferencia de los ensayos a largo plazo, en los cuales se sabe muy poco acerca de la compleja red de células y moléculas accesorias necesarias para el crecimiento óptimo de las células madre. Es por esto que, mientras los ensayos a corto plazo han sido fácilmente estandarizados, todavía se usan múltiples condiciones en los ensayos a largo plazo, por lo cual se debe tener precaución al comparar datos obtenidos de diferentes laboratorios, a pesar de que los ensayos usados compartan el mismo nombre (Coulombel L 2004). Los ensayos in vivo se han usado para definir una jerarquía de células

32
hematopoyéticas transplantables en ratones y en menor medida en humanos. Mediante estos ensayos es posible identificar diferentes tipos de células madre y células progenitoras. Dos parámetros importantes son medidos, longevidad y multipotencialidad. Mientras que en los ensayos in vitro la longevidad es expresada en semanas, para los ensayos in vivo, ésta es medida en meses, permitiendo identificar verdaderas células madre en el ratón gracias a su funcionalidad, como producir progenie de los linajes linfoide y mieloide. La habilidad de las células madre para asentarse en la medula ósea de receptores de transplantes era una variable difícil de controlar anteriormente, pero gracias a los ensayos in vivo ahora es posible utilizar este criterio para definir las células reconstituyentes a largo plazo, capaces de producir células diferenciadas de múltiples linajes hematopoyéticos durante meses tanto en medula ósea como en órganos linfoides periféricos de ratón (Coulombel L 2004). La identificación de células progenitoras humanas primitivas se realiza mediante el uso de ratones inmunodeficientes. Las cepas SCID (Kamel-Reid S & Dick JE 198878-80) y NOD/SCID (41,79,81) pueden aceptar transplantes humanos. A pesar de su inmunodeficiencia e hipersensibilidad a la radiación ionizante, los hospederos inmunodeficientes requieren de radiación moderada (3.5 Gy) para permitir así la implantación de células humanas en tejidos de ratones, y la dosis de radiación se correlaciona positivamente con la proporción de células humanas implantadas (Coulombel L 2004). Además, se ha comprobado que no hay necesidad de suplementar a los ratones con citoquinas humanas si se han inyectado más de 5000 células (Bonnet D et al., 1999) Todos estos métodos de selección y purificación pueden ser usados en combinación para lograr mejores resultados en la identificación y aislamiento de poblaciones biológicamente homogéneas de CMHs, con el fin de alcanzar mayores avances en el campo de las investigaciones funcionales y moleculares de los mecanismos que regulan la auto-renovación, restricción de linaje, y diferenciación, así como en la expansión ex vivo, y en otro tipo de manipulación encaminada a promover la recuperación hematopoyética después de terapia citoreductiva o transplante.

33
2. CRECIMIENTO Y AUTO-RENOVACIÓN DE LAS CÉLULAS MADRE HEMATOPOYÉTICAS
Los procesos de auto-renovación y diferenciación de las células madre hematopoyéticas están ampliamente regulados por diferentes factores que permiten mantener un balance entre el número de células primitivas capaces de mantener la hematopoyesis y el número de células maduras con funciones especializadas. Aunque los mecanismos de regulación de auto-renovación no están completamente elucidados, se cree que cuando el crecimiento de las células hematopoyéticas inmaduras no está siendo controlado de manera adecuada, puede llegar a generarse un proceso maligno que conlleve a la producción de células tumorales.
El proceso de la hematopoyesis involucra la auto-renovación de células madre, la expansión de células progenitoras comprometidas con linaje y la maduración de las células a los elementos terminales de la sangre. Una célula madre hematopoyética (CMH) puede tener diferentes destinos (Figura 2.1): a través de la división asimétrica una sola división puede dar origen a dos células madre idénticas, una permanece en estado de quiescencia en fase G0 y la otra prolifera y se diferencia en células más maduras. Mediante la división simétrica puede producir dos células madre idénticas que permanecerán en fase G0 del ciclo celular, o dos células diferenciadas. También puede entrar en apoptosis o puede permanecer en estado de quiescencia. Las células madre hematopoyéticas se caracterizan porque permanecen en estado de quiescencia durante largos períodos de tiempo. Actualmente, es bastante aceptado que el comportamiento de una célula es el resultado de interacciones complejas entre redes de trabajo genéticas, de señalización y metabólicas. Las CMHs expresan genes relacionados con linaje, que pueden ser consolidados o reprimidos justo antes de la diferenciación (Bruno L et al., 2004), es decir que una célula diferenciándose progresa de un programa de expresión génica complejo a programas menos complejos (Hoang T, 2004). Además de estos factores intrínsecos, las células hematopoyéticas requieren de estimulación constante de su entorno para sobrevivir. Estas señales ambientales pueden presentarse a través de interacción directa entre células, o a por la acción de mediadores solubles. En ausencia de estas señales las células pueden entrar en apoptosis. Se han propuesto dos modelos opuestos que conllevan a la diferenciación de las CMHs. El primero es el modelo estocástico, el cual implica que la elección de linaje es un proceso intrínseco de la célula que ocurre por el azar. Un número fijo

34
de células madre es generado durante la embriogénesis, estas células sucesivamente y de forma azarosa empiezan a proliferar y a diferenciarse a células maduras y a medida que las células proliferan, empiezan a perder de forma progresiva su habilidad de auto-renovarse. El segundo modelo es el inductivo, el cual se refiere a la existencia de estímulos externos que dirigen el destino celular. En este modelo, un pequeño número de células madre se desarrollan durante la embriogénesis y pueden reproducirse y auto-renovarse de forma extensa para generar células hijas con el mismo potencial proliferativo y de desarrollo que el de las células parentales. Solo una proporción de estas últimas células será la que se diferencie y madure (Bellantuono I 2004).
Figura 2.1 Posibles destinos de la célula madre hematopoyética.
Tomado de: Attar, E.C., & Scadden, D.T. (2004). Regulation of hematopoietic stem cell growth. Leukemia, 18, 1760–1768. Hasta el momento no hay evidencia definitiva que confirme alguno de los dos modelos. Respaldando el primer modelo están experimentos en los cuales transplantes en serie de la medula ósea de ratones conllevaron a la pérdida progresiva del potencial regenerativo de las células madre (Siminovitch L et al., 1964). Sin embargo, aún no es claro si la disminución del potencial regenerativo es el resultado del procedimiento de transplante, del envejecimiento del ambiente

35
en el cual las células se alojan y empiezan el proceso de hematopoyesis, o de un descenso en la capacidad regenerativa. A favor del modelo inductivo se ha mostrado que un solo ratón contiene suficientes células madre para re-poblar al menos mil ratones y son posibles como mínimo cinco transferencias sucesivas sin que ocurra pérdida aparente de la habilidad de re-poblar ratones anémicos W/Ww (Harrison DE 1979). Esto implica que las células madre son capaces de producir más células madre. Quienes se oponen al modelo estocástico proponen la existencia de un microambiente inductivo en el bazo, es decir que las células hematopoyéticas pueden ser re-programadas por su ambiente. Por ejemplo, durante el desarrollo fetal la expresión del gen de la globina cambia de un programa embrionario que ocurre en los islotes eritrocitarios del saco vitelino a un programa adulto cuando la hematopoyesis se establece en el hígado fetal y luego en la medula ósea. Se ha encontrado que al inyectar CMHs adultas en blastocistos, se expresaban globinas embrionarias derivadas del donante en los estados apropiados del desarrollo, y de forma contraria, células progenitoras embrionarias y fetales transplantadas a receptores adultos expresaban globinas de tipo adulto (Geiger H et al., 1998). Estos dos modelos podrían unirse en un solo punto de vista, desde el cual se considerara al sistema hematopoyético como una población heterogénea de células madre que pueden ser diferentes a nivel genético, siendo las más inmaduras relativamente resistentes a los estímulos de proliferación y diferenciación con respecto a las más maduras, las cuales tienen mayor probabilidad de comprometerse con algún linaje. Sin embargo, ninguna de estas células tiene suficiente potencial proliferativo para repoblar todo un hospedero al que se le han eliminado las células hematopoyéticas y seguir siendo útiles como fuente de células repobladoras para diferentes aplicaciones clínicas (Bellantuono I 2004).
2.1 CICLO CELULAR
2.1.1 Introducción Una célula somática vive y funciona hasta que se divide o muere. Cuando es estimulada para dividirse, pasa a través de una serie de transformaciones cíclicas que dan origen a dos células hijas idénticas al final de cada ciclo de división celular. Este ciclo celular agrupa una serie de criterios bioquímicos y morfológicos, y cuando se repite una y otra vez se convierte en una fuente constante de nuevas células. El ciclo celular se divide en las fases secuenciales S, G2, M y G1.

36
Fase S. En la fase S ocurre la síntesis de ADN y la célula replica su contenido genético. Una célula diploide somática comienza esta fase siendo 2N en cuanto a su contenido de ADN y se convierte en 4N al término de esta. La duración de la fase S puede variar entre un par de minutos en la división rápida de las células embrionarias tempranas, hasta un par de horas en la mayoría de células somáticas. Las células en estados de desarrollo tardíos y en organismos maduros deben transcribir activamente subgrupos de genes para sobrevivir y mantener funciones especializadas. El máximo tiempo requerido por que estas células completen la fase S debe permitirles coordinar la replicación del ADN con la transcripción y preservar así la información estructural de orden superior la cual está influida por la expresión génica que será transmitida a las células de la progenie. Fase M. La fase M o mitosis es el periodo de división nuclear y celular
durante el cual los cromosomas duplicados se dividen equitativamente entre las dos células de la progenie. Microscópicamente es el período de la condensación cromosómica, el rompimiento de la cubierta nuclear, la reorganización del citoesqueleto para formar el huso mitótico, la segregación de cromosomas a polos opuestos, la re-formación de las cubiertas nucleares (con lo cual se completa la división nuclear o cariocinesis), y la separación física de las dos células hijas (con la cual se completa la división celular o citocinesis). Una célula entra a fase M con contenido de ADN 4N y la finaliza como dos células, cada una con contenido de ADN 2N. Fases G1 y G2. Inicialmente estas fases fueron consideradas intervalos (en
inglés “gaps”) entre las fases M y S del ciclo celular. G2 es el período entre S y M cuando las células han terminado de replicar su ADN, se preparan para dividirse, y su contenido de ADN es 4N. Para la mayoría de células que entran a la fase S, el paso a través de G2 es automático, y la duración de esta fase es fija, excepto bajo circunstancias inusuales. La duración de G2 puede ser extremadamente corta y puede no ser detectada en células embrionarias que se dividen rápidamente. G1 corresponde al período entre M y S, es el intervalo entre la finalización de un ciclo de división y el inicio del siguiente. En la mayoría de los casos su duración es variable, puede ser prolongada dependiendo del tipo de célula, y está sujeta a la regulación por parte de factores ambientales como la disponibilidad de factores de crecimiento y nutrientes. Este es el período de crecimiento celular ya que usualmente se requiere cierto incremento en la masa celular antes de iniciar la siguiente fase S. Se podría decir que la cantidad de tiempo que una célula gasta en G1 es inversamente proporcional a su tasa de proliferación. Cuando no hay condiciones adecuadas para la proliferación, las células se detienen en G1, y aquellas que están en S, G2 o M usualmente completan el ciclo y se detienen cuando llegan a G1.

37
La fase G1 ha sido subdividida en segmentos y puntos reguladores basados en estudios acerca de la respuesta proliferativa de células a la aplicación secuencial de diferentes factores de crecimiento, nutrientes e inhibidores metabólicos. El punto de restricción R, es un punto importante en la regulación del ciclo celular y ocurre cerca de la unión G1-S. Este es un punto en el cual las células se comprometen a entrar en la fase S sin importar la estimulación por factores de crecimiento o la disponibilidad de aminoácidos esenciales, y es análogo al punto de compromiso en las levaduras llamado Inicio (Start). Fase G0. Esta fase corresponde a un estado no proliferativo en el cual
pueden permanecer las células viables durante largos períodos de tiempo. Estas células tienen contenido de ADN 2N y se pueden distinguir bioquímicamente de las células en G1, ya que tienen diferencias en sus proteínas y en el metabolismo del ARN. Un ejemplo de estas células son los granulocitos neutrófilos, unas células terminalmente diferenciadas que han salido de forma irreversible del ciclo celular. Sin embargo, hay otras células que entran a G0 de forma reversible y pueden ser inducidas con estímulos apropiados para que vuelvan a G1 y comiencen el ciclo de nuevo.
2.1.2 Regulación del ciclo celular. Existen puntos de control que regulan el orden y el tiempo de las transiciones del ciclo celular asegurando que eventos críticos como la replicación del ADN y la segregación de los cromosomas sean llevados a cabo con alta fidelidad. Además, estos puntos responden al daño deteniendo el ciclo celular con el fin de darle tiempo a la célula para que induzca la trascripción de genes que faciliten la reparación. La pérdida de la regulación da como resultado inestabilidad genómica y se ha asociado con la evolución de células normales a células cancerosas. El paso de las células a través del ciclo celular está dirigido por quinasas serina/treonina de la familia Cdk (quinasa dependiente de ciclina), cuyas actividades catalíticas están bajo el control estricto de proteínas reguladoras asociadas llamadas ciclinas. El control de la actividad ciclina/Cdk se da por la aparición y desaparición de las ciclinas en fases específicas del ciclo celular, por la modificación pos-transcripcional de las Cdks, y por la asociación con inhibidores de Cdk. A su vez, los complejos ciclina/Cdks regulan el ciclo celular modulando la actividad de factores de transcripción críticos.
2.1.2.1 Ciclinas y Quinasas Dependientes de Ciclinas (Cdks). Una quinasa es una enzima que cataliza la transferencia de un grupo fosfato del ATP a otra molécula. Esta transferencia de fosfato es llamada fosforilación. La Cdk es una quinasa que cataliza la fosforilación de ciertos aminoácidos en algunas proteínas, cambiando la estructura tridimensional y la función de la proteína sobre la que

38
actúa. La primera Cdk encontrada fue Cdc2 (ciclo de división celular 2) en Schizosaccharomyces pombe, una proteína serina/treonina de 34-kd. Se observó que cuando esta proteína estaba defectuosa las células se detenían en la fase G1 o G2 del ciclo celular y no entraban a S o M. En humanos también fue hallada una proteína con similar a Cdc2 en cuanto a estructura y función llamada Cdk1, y luego de esta se han encontrado otros miembros de esta familia de quinasas designadas de Cdk2 hasta Cdk8 (Elledge SJ 1996, Hoffman R et al., Control of Cell Growth and Differentiation, 2000). La entrada a la fase M es regulada por el factor promotor de regulación o MPF. Este factor contiene dos proteínas: una es p34cdc2 (Cdk1) y la otra es una ciclina tipo B. La ciclina B no tiene activadad enzimática conocida pero se comporta como un regulador del complejo MPF, ya que p34cdc2 tiene actividad quinasa/MPF solamente cuando se encuentra asociada a la ciclina B. Los niveles de ciclina B aumentan durante las fases S y G2 y justo antes de la entrada a M se alcanzan los niveles suficientes del complejo ciclina B/p34cdc2 para que ocurra la transición G2-M. Este complejo se acumula en su forma inactiva como una proteína fosforilada en múltiples sitios (treonina 161, treonina 14 y tirosina 15) con el fin de evitar que la célula esté en mitosis permanentemente. Cuando el complejo es defosforilado en la treonina 14 y tirosina 15 por la acción de la fosfatasa Cdc25, MPF se vuelve activo y ocurre una retroalimentación ya que puede fosforilar a Cdc25 para amplificar así su propia actividad. A medida que la fase M progresa, el complejo p34cdc2/ciclina B es inactivado por la degradación de la ciclina B a través de la vía de la ubiquitina. Esta inactivación es importante para que las células puedan salir de la fase M (Figura 2.2) (Hoffman R et al., Control of Cell Growth and Differentiation, 2000). En las otras fases del ciclo celular también intervienen complejos Cdk/ciclina. La progresión de G1 a S está regulada principalmente por la asociación entre Cdk2 y Cdk4 con ciclinas D y E. Los Complejos de Cdk4 y Cdk6 con ciclinas tipo D (ciclina D1, D2 y D3) tienen un rol crítico en la progresión a través del punto de restricción que existe en G1. La ciclina E se expresa en un estado tardío de G1, y se requiere de los complejos Cdk2/cilclina E para la transición de G1 a S y la posterior iniciación de la síntesis de ADN. La ciclina A aparece al comienzo de S y decae en G2 y M, y tiene un patrón de expresión paralelo pero que precede al de la ciclina B. Complejos de Cdk2 con ciclina A intervienen en el paso de las células a través de la fase S. Asimismo, la ciclina A en complejo con p32cdc2 puede desencadenar la transición de G2 a M a través de la fosforilación de Cdc25 que inicia la activación del MPF (Hoffman R et al., Control of Cell Growth and Differentiation, 2000, Cooper GM 2000).

39
Figura 2.2 Regulación de la entrada a la fase M por el factor promotor de maduración o MPF (p34cdc2/ciclina B).
Modificado de Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Control of Cell Growth and Di fferentiation. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 57 71). Philadelphia, EE.UU.: Churchill Livingstone
2.1.2.2 Regulación de las Quinasas Dependientes de Ciclinas. La regulación de las Cdks se puede llevar a cabo al menos por cuatro mecanismos moleculares diferentes. El primero es la síntesis y degradación de ciclinas, ya que las Cdks se asocian con éstas. El segundo es la fosforilación de un residuo treonina conservado de la Cdk cerca de la posición 160, esta fosforilación es catalizada por un complejo compuesto por una CAK (quinasa activadora de Cdk), designada Cdk7 asociada con Ciclina H. El tercer mecanismo es la fosforilación inhibitoria de residuos tirosina en las Cdk cerca al aminoácido terminal, catalizada por la proteína quinasa Wee1. El cuarto mecanismo de regulación de las Cdks es la unión de complejos Cdk/ciclina a proteínas inhibitorias llamadas inhibidores de quinasas dependientes de ciclina o CDKIs. En mamíferos hay dos familias de CDKIs encargadas de regular los diferentes complejos ciclina/Cdk, la familia Cip/Kip que incluye a p21Cip1/Waf1, p27Kip1 y p57Kip2, y la familia Ink 4 que incluye a p16Ink4a, p15Ink4b, p18Ink4c y p19Ink4d. Estas subfamilias se distinguen por su especificidad en la Cdk que inhiben (Attar EC & Scadden DT 2004).

40
El primer inhibidor identificado y clonado en células de mamíferos fue p21 (Waf1, Cip1, Sdi1), clasificado como inhibidor universal, el cual se une a diferentes complejos de ciclina/Cdk2 o ciclina/Cdk4 e inhibe su actividad, regulando así la progresión del ciclo celular en G1. Esta proteína puede interactuar con el antígeno nuclear de célula proliferante o PCNA, éste es un factor de procesividad durante la síntesis de ADN cuya actividad se ve inhibida al unirse a p21 para facilitar la función de la ADN polimerasa-δ, la polimerasa responsable de la replicación del ADN, por lo tanto p21 también tiene un papel regulador en la replicación y reparación de daño al ADN que ocurren en la fase S. La expresión de p21 está controlada por la proteína supresora de tumor p53, cuya activación es inducida por daño en el ADN, lo cual sugiere que p21 es responsable de la detención del ciclo celular después de que ocurre un daño en el ADN para así dar tiempo a la valoración y reparación del daño (Hoffman R et al., Control of Cell Growth and Differentiation, 2000). Sin embargo, también existen vías de expresión de p21 independientes de p53 y p21 puede tener un rol dual en la detención del ciclo celular inducido por daño al ADN, no solo bloqueando la progresión del ciclo celular mediante la inhibición de Cdks sino también inhibiendo directamente la replicación del ADN en células en fase S. p27 (Kip1) y p57 (Kip2) son otros inhibidores de Cdk similares en estructura y función a p21, que se unen a múltiples complejos ciclina/Cdk. p57 tiene acción inhibitoria sobre los complejos ciclina E/Cdk 2, ciclina D2/Cdk 4 y ciclina A/Cdk 2. Otros inhibidores de Cdk que difieren estructuralmente de los inhibidores universales son p16 (INK4a, MTS1, Cdk41) y p15 (INK4b, MTS2). Estas proteínas se unen exclusivamente a Cdk4 y Cdk6 e inhiben su asociación con la ciclina D y su actividad quinasa. Rb es la proteína producida por el gen de sensibilidad al retinoblastoma y se cree que esta proteína es un blanco de la actividad quinasa de los complejos ciclina D/Cdk4 y ciclina D/Cdk 6, y su fosforilación evita la detención del ciclo celular en G1. Por lo tanto, p15 y p16 pueden inhibir la proliferación celular previniendo la fosforilación de Rb. En la línea germinal de pacientes con melanoma familiar y en el genoma de varias líneas celulares tumorales se ha encontrado que los genes p15 y p16 en la región del cromosoma 9, estaban mutados, lo cual destaca su importancia en la prevención de la proliferación de células neoplásicas in vivo (Hoffman R et al., Control of Cell Growth and Differentiation, 2000). El inhibidor extracelular de la proliferación celular en animales mejor caracterizado es el Factor de crecimiento transformante β o TGF-β. Esta molécula es un homodímero unido por puentes disulfuro que dirige la actividad de complejos ciclina/Cdk a través de la inhibición de Cdk 4, induciendo la expresión de p27, p16 y p15 y bloqueando la fosforilación de la proteína Rb (Xiong Y et al., 1993; Harper JW et al., 1993; Quelle DE et al., 1995; Laiho M et al., 1990)

41
Calculando la expresión relativa de diferentes factores de transcripción y de moléculas reguladoras del ciclo celular en CMHs, se determinó que células primitivas, quiescentes tenían niveles de p21 bastante altos con respecto a células progenitoras más diferenciadas (Cheng T et al., 2001). Para probar que las CMHs primitivas se mantienen en estado de quiescencia gracias a la acción inhibidora de CDKIs, se han realizado análisis in vivo e in vitro de la función de células madre y células progenitoras. Estos estudios demostraron que la medula ósea de ratones deficientes en p21 tiene una cantidad elevada de CMHs primitivas y un incremento en el ciclo celular de CMHs (Chent T et al., 2000), lo cual identifica a p21 como un regulador negativo de la proliferación de CMH. Asimismo, se ha encontrado que la médula ósea de ratones deficientes en p27 tiene aumentada (5.7 veces) la frecuencia de células madre y un pool expandido de CMHs quiescentes (Walkley CR et al., 2005). No se ha encontrado que la pérdida de p21 o p27 de como resultado la leucemia en ratones y ninguna de estas moléculas se ha asociado con leucemia en humanos, aunque las alteraciones en p21 y p27 han estado implicadas en varias malignidades como cáncer de pulmón, de colon y de seno (Alkarain A et al., 2004; Chetty R 2003; Blain SW et al., 2003; Slingerland J et al., 2000).
2.1.3 Ciclo celular en las células madre hematopoyéticas Las CMHs deben suplir todas las líneas celulares de la sangre requeridas a través de la vida de un individuo. Para ello poseen capacidad de auto-renovación y multipotencialidad en la diferenciación con el fin de satisfacer la demanda de células sanguíneas maduras y proteger contra el agotamiento celular. Esta actividad está estrictamente regulada con el fin de mantener la homeóstasis hematopoyética. Se propone que las CMHs utilizan el mecanismo de sucesión clonal, en el cual uno o pocos clones de CMHs se diferencian y generan células sanguíneas maduras cuando son necesitadas, como en los procesos infecciosos en donde hay una inducción en la activación de los mecanismos celulares de defensa que involucran a la mayor parte de las células de la sangre, o cuando por algún motivo las reservas de glóbulos rojos no son suficientes para suplir las necesidades del cuerpo. Mientras que las CMHs restantes permanecen en estado de quiescencia sin contribuir a la hematopoyesis, hasta que la capacidad proliferativa de las células activas se agote (Bell DR et al., 2004). Una característica que comparten las células madre adultas de varios tejidos es su relativa quiescencia. Las células madre generalmente tienen una baja tasa de recambio mientras sus descendientes casi siempre denotados como células progenitoras en el sistema hematopoyético son las que dirigen la proliferación a gran escala. Estas células progenitoras proveen billones de células sanguíneas por día necesarias para mantener la homeostasis (Attar EC & Scadden DT, 2004). Se ha comprobado que, aunque la mayoría de CMHs permanecen quiescentes en

42
fase G0, en algún momento todas las CMHs entran activamente al ciclo celular (Ezoe S et al., 2004). La baja tasa de proliferación característica de las CMHs, es esencial para el mantenimiento de la población de células madre durante la vida de un individuo y es un elemento clave para su identificación en la población de células hematopoyéticas. La dinámica que regula cuales células se auto-renuevan para mantener la población, y cuales proliferan y llegan a comprometerse para generar elementos maduros en la sangre, aun no está clara (Marone M et al., 2002). Se cree que el papel de las citoquinas en el destino de las CMHs, además de la regulación de la entrada al ciclo celular, es inducir un fenómeno complejo llamado activación de las CMHs, el cual implica un número de cambios celulares que causan el paso de fase G0 a fase G1. Esta activación conlleva a la expresión del marcador CD34 y también a una pérdida gradual de la multipotencialidad a medida que la célula va proliferando (Marone M et al., 2002). La relativa quiescencia de las células madre las hace capaces de retornar a un estado prolongado de G0 después de cada ciclo celular. Estas células pueden entrar nuevamente al ciclo cuando son estimuladas después de un largo período de tiempo. Al final del ciclo celular pueden entrar a una vía de no retorno o pueden desviarse a un ciclo proliferativo que es mucho más corto, excluyendo el paso a través de la fase G0. En esta fase la célula retiene multipotencialidad únicamente para un número definido de ciclos. Cuando las células llegan a un estado de mayor compromiso, cambian a un modo de proliferación aun más rápido (Marone M et al., 2002).
2.2 MECANISMOS DE REGULACIÓN DE AUTO-RENOVACIÓN El proceso de auto-renovación es crucial para la función de la célula madre ya que gracias a este proceso la célula puede persistir durante el tiempo de vida del organismo. Además, las células madre deben regular el balance relativo entre la auto-renovación y la diferenciación. Entender la regulación de este proceso es fundamental para entender también la regulación de la proliferación celular del cáncer, ya que el cáncer puede ser considerado como una enfermedad de auto-renovación no regulada. Los mecanismos moleculares que dirigen y regulan la auto-renovación en las CMHs aún no son claros, pero se han propuesto varias vías de señalización que podrían regular este proceso, entre las cuales sobresalen la vía de Hedgehog (Hh), la vía Notch y la vía Wingless (Wnt), además de las proteínas BMP, factores y represores transcripcionales como HOXB4 y Bmi-1.

43
2.2.1 Vía Hedgehog
La vía Hh es una cascada que juega un papel muy importante en la proliferación de la CMH. Los genes hedgehog codifican proteínas que son modificadas por la adición del colesterol. Las proteínas precursoras Hh entran a la vía secretora y experimentan un clivaje intramolecular y una reacción catalizada por la porción carboxi-terminal de la proteína. Como resultado de esto se genera un polipéptido amino-terminal (HhNp) cuya masa molecular aproximada es de 19 kD, que contiene todas las funciones de señalización conocidas de la proteína Hh. Durante este proceso de clivaje, una parte de colesterol se une covalentemente a la glicina del extremo terminal C de su dominio de señalización (Porter JA et al 1996, Peters C et al., 2004). Esta modificación por el colesterol parece anclar los polipéptidos Hh a la superficie celular y restringir su rango de actividad señalizadora.
2.2.1.1 Proteínas Hedgehog. La familia de proteínas Hedgehog (Hh) está involucrada en la organización del mesodermo temprano y en la especialización embrionaria de tejido no hematopoyético (Bhardwaj G et al., 2001). Existen tres genes Hedgehog en mamíferos: Sonic hedgehog (Shh), Indian hedgehog (Ihh) y Desert hedgehog (Dhh). Entre las actividades en las que Shh participa, está el desarrollo de la simetría derecha-izquierda y la regulación de la morfogénesis de gran variedad de órganos incluyendo el ojo, el cabello y los pulmones. Por otro lado, Dhh actúa en la regulación de la espermatogénesis y en la organización del perineurio, el cual envuelve a los nervios periféricos, e Ihh está involucrada en coordinar la proliferación y maduración de los condrocitos durante el desarrollo del esqueleto endocondral (McMahon AP 2000). Las señales Hedgehog parecen tener rangos de actividades tanto cortos como amplios y, adicionalmente actúan como morfogenes para inducir diferentes destinos en la célula de acuerdo a umbrales de concentración específicos. Las moléculas Hh son expresadas sobre la superficie celular como proteínas transmembranales y pueden tener dos modos de señalización diferentes. Pueden mediar la señalización a través del contacto célula-célula entre células adyacentes que expresen el receptor Ptc, y de manera alterna, el clivaje del extremo animo-terminal de Hh puede generar un ligando soluble que es capaz de difundirse a través del microambiente e interactuar con células distales que expresen Ptc (Bhardwaj G et al., 2001).
2.2.1.2 Receptores de Hedgehog. El receptor funcional para Hh está conformado por dos proteínas transmembranales que controlan la señal, el supresor de tumor Patched (Ptc) y el proto-oncogen Smoothed (Smo). Ptc es una proteína compuesta de 2 horquillas extracelulares y 12 dominios transmembranales y Smo es miembro de la familia de los siete receptores

44
transmembranales que exhiben similitud en su secuencia con respecto a los miembros de la familia de proteínas Frizzled, algunas de las cuales actúan como receptores de las proteínas Wnt (Bhardwaj G et al., 2001; Taipale J et al., 2001; Ingham PW, 1998). La función normal de Ptc es inhibir la activación de Smo en ausencia de su ligando Hh, es decir, en células que no han sido estimuladas por Hh. Esta función la realiza a través de la interacción con Smo (Hahn H et al., 1996). Aunque no se ha encontrado que se una directamente a los ligandos Hh, Smo es un componente integral de la vía de señalización Hh que se activa cuando las proteínas Hh se unen al receptor Ptc. Luego de que se han unido ocurre un cambio conformacional en este complejo receptor que libera a Smo de la influencia represora de Ptc. También se plantea que Hh disocia directamente a Ptc de Smo y nuevos datos confirman que la proteína Smo no está en un complejo con Ptc luego de la estimulación por Hh o, según la interpretación de estos datos, se sugiere que Ptc puede reprimir la actividad de Smo sin contactarla directamente. Sin embargo, estos resultados aún siguen siendo cuestionados ya que nuevos estudios dirigidos a esclarecer cuál de estos modelos de regulación es el correcto arrojan diferentes resultados (Kalderon D 2000).
2.2.1.3 Transducción de la señal. A diferencia de la mayoría de vías de señalización, la transducción de señal intracelular Hh actúa en gran parte debido a interacciones represivas secuenciales. En las células que reaccionan a Hh, la liberación y subsiguiente activación de Smo inicia los eventos de señalización, aunque aún no es muy claro cómo la activación de Smo está acoplada a las proteínas citoplasmáticas involucradas con la señalización Hh, las cuales incluyen la proteína quinasa serina/treonina Fused (Fu), el Supresor de Fused (Su(Fu)), la proteína Costal-2 (Cos2) la cual tiene un dominio amino-terminal que comparte bastante similitud en su secuencia con los dominios motores de las proteínas de la superfamilia quinesina (Ingham PW 1998), y el factor de transcripción Cubitus interruptus (Ci) o Gli en mamíferos. Estas proteínas forman un complejo citoplásmico de alto peso molecular que está anclado a microtúbulos, aparentemente a través de Cos2. En ausencia de Hh, Ci (Ci155) es fosforilado por la proteína quinasa A (PKA) y subsecuentemente procesado en el proteasoma para liberar un represor transcripcional amino-terminal (Ci75). Luego de la estimulación por Hh, el gran complejo citoplasmático se disocia de los microtúbulos y como no hay proteólisis de Ci155, el activador transcripcional de longitud completa se transloca al núcleo conduciendo a la activación transcripcional de los genes blanco de Hh. Uno de estos genes es Ptc, que al activarse, da como resultado inhibición por retroalimentación (Taipale J et al., 2001), otros genes cuya expresión se ve modificada son los genes que codifican

45
proteínas como Wnt2a y un inhibidor específico de BMP-4 llamado Noggin (Figura 2.3) (Bhardwaj G et al., 2001).
2.2.1.4 Hedgehog en las Células Madre Hematopoyéticas. Shh, Ptc y Smo se expresan en células humanas primitivas CD34+CD38-Lin-, células mieloides maduras (CD33+), linfocitos B (CD19+) y T (CD3+), células de la estroma de la médula ósea, y células endoteliales de vena umbilical (HUVEC). Las células CD34+CD38-Lin-, las de estroma y las HUVEC, pero no las células comprometidas de linaje mieloide y linfoide, también expresan tres factores transcripcionales Gli (Gli-1, -2, y -3) que actúan corriente abajo de la cascada de señalización Hh (Bhardwaj G et al., 2001). Esto sugiere que la señalización Hh no es esencial para la maduración hematopoyética.
Figura 2.3 Vía de señalización Hedgehog.
Modificado de Ingham, P.W. (1998). Trandsucing Hedgehog: the story so far. The EMBO Journal, 17, 3505-3511. Se ha comprobado que la proliferación de células sanguíneas purificadas, inducida por citoquinas podría ser inhibida por la adición de anti-Hh, lo cual muestra que las proteínas Hh producidas endógenamente juegan un papel importante en la
Componentes que regulan de forma negativa Componentes que regulan de forma positiva
VÍA Hh NO ACTIVADA VÍA Hh ACTIVADA

46
expansión de células hematopoyéticas humanas primitivas. Asímismo, la adición de formas solubles de Shh incrementa el número de células sanguíneas humanas detectables con capacidad pluripotente de repoblar ratones NOD-SCID. En Drosophila, la actividad Shh está asociada con el control de señalización de la proteína morfogénica de hueso (BMP). Noggin es capaz de inhibir la proliferación inducida por Shh de células hematopoyéticas primitivas de una forma similar a un anticuerpo anti-Hh, aunque anti-Hh no tiene efecto sobre la proliferación inducida por BMP-4. Así, Shh parece funcionar como regulador de CMHs, aunque en combinación con otros factores de crecimiento, a través de mecanismos dependientes de señales BMP corriente abajo (Peters C et al., 2004; Szilvassy SJ 2003).
2.2.2 Vía Notch. La vía de señalización Notch y por ende las señales transmitidas a través de ella están involucradas junto con otros factores celulares en la proliferación, diferenciación y en los eventos apoptóticos en casi todos los estados del desarrollo (Artavanis-Tsakonas S et al., 1999). Por lo tanto, los miembros de la familia Notch tienen un papel crítico en la determinación de los destinos celulares y en el mantenimiento de progenitores en muchos sistemas. La familia Notch comprende un grupo de proteínas altamente conservadas que funcionan como receptores en la superficie celular y como reguladores directos de la transcripción génica. Estas moléculas representan un paso único para la transducción de señal desde la superficie celular hasta el núcleo, permitiendo a las células influir directamente en la expresión génica de sus vecinos. En general, la activación de Notch conduce a la supresión transcripcional de los genes específicos de linaje, inhibiendo la diferenciación en respuesta a señales inductivas. La señalización de Notch limita el número de células que adoptan un destino particular y deja a algunos progenitores sin comprometerse pero competentes para adoptar destinos alternativos (Milner LA et al., 1999).
2.2.2.1 Estructura general de Notch. En los mamíferos existen cuatro genes Notch (Notch1, Notch2, Notch3 y Notch4), que se expresan ampliamente durante la embriogénesis y juegan un papel fundamental en el desarrollo. Los receptores Notch son largos polipétptidos transmembranales compuestos de diferentes dominios estructurales múltiples. El dominio extracelular de Notch contiene un número variable de repeticiones en tandem similares a las del factor de crecimiento epidermal (EGF) y tres repeticiones Lin12/Notch (LNR), que funcionan en la unión al ligando y en la activación de Notch. Entre el dominio LNR y el dominio transmembranal (TM) se ubican cisteínas conservadas involucradas en la formación de puentes disulfuro del receptor heterodimérico. También posee sitios

47
putativos de clivaje involucrados en la generación del receptor funcional Notch y en la liberación de Notch a nivel intracelular después de la activación (Milner LA et al., 1999; Aster JC et al., 2001; Bigas A et al., 1998; Artavanis-Tsakonas S et al., 1999). El dominio intracelular de Notch (Notch-IC) incluye una región altamente conservada de seis repeticiones cdc 10/anquirina, características de moléculas involucradas en las interacciones proteína-proteína, la cual es esencial para la transducción de la señal. La proteína Notch no tiene ninguna actividad enzimática conocida, pero en lugar de ello, transmite las señales a través de interacciones moleculares directas. La región que se ubica entre el extremo C-terminal de Notch y las repeticiones cdc 10 ha sido asociada con distintas interacciones de proteína y con la transactivación. El dominio PEST, que también se ubica en la región C-terminal, regula la rotación de la proteína. La mayor divergencia entre los receptores Notch en mamíferos ocurre entre las repeticiones de anquirina y las secuencias C-terminales PEST, en Notch 1, esta región divergente contiene un dominio de activación transcripcional (TAD) rico en prolina y glutamina. La molécula Notch de Drosophila y Notch 1, 2, y 3 en mamíferos también contiene señales de localización nuclear (nls) y secuencias OPA conservadas ricas en glutamina. Otras regiones descritas incluyen el dominio RAM, el cual se une a CSL (CBF1/Supressor de Hairless/Lag-1) efectores de la transducción de señal Notch y una región de respuesta a la citoquina de Notch (NCR), compuesta aproximadamente por 60 aminoácidos y que incluye una secuencia de localización de señal y sitios putativos de fosforilación regulatoria, asociados con diversos efectos de Notch 1, 2 y 3 sobre la diferenciación mieloide (Milner LA et al., 1999; Aster JC et al., 2001; Bigas A et al., 1998).
2.2.2.2 Ligandos, moléculas efectoras y moléculas blancos de Notch. Además de los receptores Notch, en la vía de transducción de señal también figuran otros componentes primarios como son los ligandos, las moléculas efectoras y los blancos de Notch. En Drosophila, dos proteínas transmembranales llamadas Delta y Serrate son ligandos de Notch al igual que Lag-2 y Apx-1 en Caenorhabditis elegans, (proteínas DSL [Delta/Serrate/Lag-2]). En vertebrados se han identificado múltiples ligandos que corresponden a alguna de estas clases. En mamíferos, aquellos ligandos que tienen alta homología a Delta son identificados como Delta o Delta-like (Dll), y aquellos homólogos a Serrate son llamados Serrate o Jagged (Milner LA et al., 1999; Artavanis-Tsakonas S et al., 1999). Por ser proteínas transmembranales, los ligandos DSL poseen un dominio extracelular que contiene un número variable de repeticiones similares a EGF. En este dominio también se ubica una región conservada única de esta familia de moléculas: un dominio DSL, requerido para la unión y activación de Notch. Serrate y Jagged también contienen una región conservada rica en cisteína que no está

48
presente en los homólogos de Delta. Los dominios intracelulares de las proteínas DSL están compuestos de diversas secuencias cortas pero de función desconocida que podrían estar involucradas en la multimerización (Milner LA et al., 1999). El gen de humanos Jagged-1 fue clonado de una librería de ADNc de médula ósea, lo expresan un subset de células del estroma de la médula ósea (Li L et al., 1998) y se ha comprobado que funciona como un factor de crecimiento de células madre hematopoyéticas humanas (Karanu FN et al., 2000). La coexpresión de Jagged-2 y Notch-1 en el timo en desarrollo sugiere que Jagged-2 es un ligando del receptor Notch-1 en este tejido (Li L et al., 1998). Dll-1 y Dll-3 son los genes ortólogos a Delta de Drosophila identificados en ratones y humanos (Bettenhausen B et al., 1995; Dunwoodie SL et al., 1997) y cuyos patrones de expresión aunque son diferentes se sobrelapan, lo cual sugiere que trabajan de forma cooperativa en el proceso de señalización. Dll-1 se expresa en la estroma de la médula ósea y en el bazo, por lo tanto podría jugar un papel en la hematopoyesis y la regulación de células B (Milner LA et al., 1999). La molécula delta-like (dlk) es expresada por la línea celular de estroma de hígado fetal AFT024 y es otro ligando de Notch capaz de mantener progenitores hematopoyéticos transplantables in vitro, también actúa como regulador positivo del crecimiento de células madre en ratón, células pre-B y timocitos (Li L et al., 2005). Entre las moléculas efectoras intracelulares está el factor de transcripción Suppressor of Hairless [Su(H)] en Drosophila y las proteínas homólogas a éste o proteínas CSL (CBF1/RBP-Jk en mamíferos, LAG-1 en C. elegans). Estas proteínas altamente conservadas interactúan físicamente con Notch-IC y median la transducción de señal del citoplasma al núcleo después de la activación de Notch uniéndose específicamente a secuencias de ADN para regular la expresión génica. Las proteínas CSL se unen al dominio RAM de Notch con alta afinidad, aunque también interactúan con el dominio cdc 10 tal vez con diferentes efectos. La unión de CSL a promotores específicos puede conducir a la activación transcripcional cuando Notch y las proteínas CSL se unen como un complejo a secuencias reguladoras del ADN actuando de forma sinergística, o pueden generar supresión transcripcional, ya que en ausencia de Notch, las proteínas CSL funcionan como represores, en este caso Notch y CSL trabajan de forma antagonista (Milner LA et al., 1999). Los principales blancos de la señalización Notch en la mosca son los genes del locus Enhancer of split [E(spl)], un complejo que codifica factores de transcripción hélice-loop-hélice básicos (bHLH). En vertebrados, los genes homólogos son Hairy/Enhancer of split (HES) que al igual que los [E(spl)] se activan por las proteínas CSL CBF1 e inhiben la actividad de otras proteínas bHLH, suprimiendo así la transcripción de genes específicos de linaje (Milner LA et al., 1999).

49
2.2.2.3 Activación de Notch. El dominio extracelular de Notch se activa cuando los ligandos DSL ubicados sobre células adyacentes se unen a las repeticiones EGF de Notch, esta activación da como resultado el clivaje proteolítico, mediado por una metaloproteasa de la familia ADAM y por proteínas de membrana como Presenilina y Nicastrina, y la liberación y translocación al núcleo del dominio intracelular de Notch (Notch-IC) (Figura 2.4). Notch-IC actúa con diferentes proteínas citoplásmicas y nucleares y la transducción de señal se puede dar mediante dos vía diferentes, una dependiente de proteínas CSL (Su[H], CBF1) y otra independiente de éstas. Al interactuar Notch-IC con las proteínas CSL a través de la unión de éstas al dominio RAM de Notch-IC y a sus repeticiones de anquirina, se produce la activación transcripcional dependiente de CSL por el desplazamiento de co-represores (N-CoR/SMRT y CIR) de CSL, y por el reclutamiento de co-activadores (PCAF y GCN5) al extremo C-terminal TAD, dando como resultado la activación de los genes E(spl)/HES, los cuales, como ya se mencionó, funcionan como reguladores negativos de la expresión génica específica de linaje (Milner LA et al., 1999; Aster JC et al., 2001). Figura 2.4 Vía de señalización Notch.

50
Por otro lado, en la vía independiente de CSL el resultado también es la regulación transcripcional, mediada por diferentes moléculas efectoras. En esta vía, las repeticiones de anquirina de Notch-IC se pueden unir a otras proteínas que incluyen la proteína nuclear de Drosophila Mastermind, la cual se asocia con regiones específicas de cromatina y aumenta o suprime la transcripción (Milner LA et al., 1999), con EMB5, SKIP y Deltex (Aster JC et al., 2001), ésta última, una proteína citoplásmica que al interactuar con Notch-IC da como resultado la inhibición de la proteína bHLH E47 (Ordentlich P et al., 1998). Esta vía de señalización, sin embargo, está poco caracterizada y requiere de más estudios. Los mecanismos moleculares involucrados en la regulación transcripcional en el núcleo aún son objeto de investigaciones. Por un lado hay estudios que demuestran actividad nuclear de Notch a pesar de la no visualización de la proteína, lo cual sugiere que pequeñas cantidades de Notch pueden ser suficientes para la activación transcripcional. Sin embargo, otros estudios en los cuales se han encontrado formas activas de Notch-IC unida a la membrana, llevarían a pensar que no es necesaria la traslocación de Notch al núcleo para su función. A pesar de las evidencias sobre el funcionamiento de Notch a nivel nuclear, aún no es claro si la traslocación de Notch-IC al núcleo es necesaria para la función de Notch (Milner LA et al., 1999).
2.2.2.4 Notch en las Células Madre Hematopoyéticas. La vía de señalización Notch es un importante regulador de la auto-renovación de las CMHs. A lo largo de la maduración hematopoyética se expresan los receptores Notch-1 y Notch-2 y cualquiera de los dos es capaz de inhibir la diferenciación mieloide en células 32D, Notch-1 en respuesta a G-CSF y Notch-2 en respuesta a GM-CSF (Bigas A et al., 1998). Jones et al., demostraron que al aislar células CD34+c-kit- de la región aorta-gonadal-mesonefros (AGM) de embriones de ratón y cultivarlas en células de estroma S17 para que expresaran Jagged1, el número de CFCs aumentó cuatro veces comparado con el control de estroma (Jones P et al., 1998).
También se ha demostrado que el ligando en humanos Jagged-1 funciona como factor de crecimiento en CMHs, ya que células humanas de cordón umbilical con fenotipo CD34+CD38-Lin- cultivadas con Jagged-1 y en ausencia de otros factores de crecimiento fueron capaces de reconstituir ratones NOD/SCID a niveles mayores que células recién aisladas (Karanu FN et al., 2000). Varnum-Finney et al., al transducir células de médula ósea Sca-1+c-kit+Lin- con un retrovirus que expresaba constitutivamente una forma activa de Notch-1 fueron capaces de generar líneas de "células madre" inmortalizadas que conservaran la morfología y fenotipo primitivos y la capacidad de diferenciarse in vitro a progenie linfoide o mieloide (Varnum-Finney et al., 2003). Todos estos datos sugieren que la vía Notch puede jugar un papel de gran importancia como regulador de la proliferación

51
y la capacidad de auto-renovación de las CMHs, y es capaz de influir directamente en las decisiones sobre el destino de dichas células.
2.2.3 Vía Wnt La vía Wnt ha sido de gran importancia en el estudio del desarrollo animal. Sin embargo, solo hasta hace pocos años se le ha prestado atención desde el campo de la inmunología, esto debido a que se han desarrollado muchos estudios que indican que la proliferación y diferenciación de la mayoría de progenitores más primitivos comprometidos con linaje está regulada in vitro por moléculas señalizadoras que tienen un papel instructivo durante el desarrollo embrionario. Al parecer, la vía WNT proporciona importantes señales de mantenimiento y/o proliferación para las poblaciones de células madre y células progenitoras. El término Wnt se deriva de la contracción de los nombres de los genes wingless en Drosophila melanogaster y su ortólogo en ratones Int1, un proto-oncogen descubierto como sitio de integración para los virus de tumor mamario de ratón (Nusse R et al., 1982). Hay 19 genes Wnt en el genoma humano y de ratón, los cuales codifican glicoproteínas secretadas modificadas por lípidos de gran importancia en los diferentes fenómenos biológicos de las células en especies que van desde D. melanogaster y el nematodo Caenorhabditis elegans, hasta los ratones y el hombre (Staal FJT et al., 2005). Aunque se han propuesto al menos tres vías de señalización intracelular para Wnt (Staal FJT et al., 2005), la mayoría de estudios del sistema hematopoyético están restringidos a la vía canónica de la β-catenina y TCF/LEF, en la cual el evento final es la translocación nuclear de la β-catenina y su unión física y activación de los factores de transcripción TCF y/o LEF.
2.2.3.1 Proteínas Wnt. Todas las proteínas Wnt son secretadas por células y actúan a través de receptores en la superficie celular ya sea sobre las células productoras o sobre células adyacentes, para determinar el destino celular u otros parámetros de diferenciación. Solamente hasta hace poco fue posible purificar las proteínas Wnt (Willert K et al., 2003), con lo cual se reveló que las moléculas Wnt están palmitoiladas y por lo tanto son mucho más hidrofóbicas de lo que se pensaba. Parece ser que el aminoácido modificado de estas proteínas es la cisteína conservada C77, un residuo que está presente en todas las proteínas Wnt y que es esencial para su función. El palmitato es de gran importancia para la señalización y las proteínas Wnt por lo general están glicosiladas sobre los sitios de glicosilación unidos al extremo N-terminal, aunque es posible que tengan otras

52
modificaciones o que diferentes formas de Wnt estén palmitoadas en sitios diferentes (Nusse R 2003).
2.2.3.2 Proteoglicanos. Poco se sabe acerca del papel de los proteoglicanos pero se propone que pueden ser co-receptores de baja afinidad para Wnts, que servirían para aumentar la concentración local del ligando disponible para la unión a los receptores de alta afinidad. Proteoglicanos en la matriz extracelular pueden entrecruzarse con Wnts para inducir un agrupamiento de los receptores de Wnt en la membrana plasmática (Wordarz A et al., 1998).
2.2.3.3 Receptores Wnt. Los receptores más importantes de la vía de señalización Wnt son los pertenecientes a la familia Frizzled (Fz). Estructuralmente, todos los miembros de esta familia tienen las siguientes características comenzando desde el extremo N-terminal: una secuencia señalizadora principal, seguida de una secuencia de 120 aminoácidos que contiene residuos ricos en cisteína (CRD o dominio rico en cisteína), una región altamente divergente de 40-100 aminoácidos, siete segmentos transmembranales separados por horquillas cortas extracelulares y citoplasmáticas, y una cola citoplasmática. El CRD parece ser el sitio de unión al ligando de las proteínas Frizzled (Wodarz A et al., 1998). Existen otros receptores requeridos para que se lleve a cabo la señalización a través de Wnt. La molécula transmembranal perteneciente a la familia LRP (Proteína relacionada con el receptor de lipoproteína de baja densidad) está codificada por el gen arrow en Drosophila y por Lrp5 o Lrp6 en vertebrados. Se propone que las moléculas Wnt se pueden unir a LRP para formar un complejo tetramérico con Frizzled (46). Además, la cola citoplasmática de LRP puede ineractuar con Axina, uno de los componentes de la vía Wnt (Mao J et al., 2001). Las FRPs (proteínas relacionadas con Frizzled) son proteínas estructuralmente relacionadas con el dominio rico en cisteína de las Frizzleds y actúan como antagonistas secretados de Wnts. Se sugiere que las FRPs pueden unirse directamente a las proteínas Wnt y que el CRD es necesario y suficiente para esta interacción (Nusse R 2003).
2.2.3.4 Proteínas efectoras de la vía Wnt La β-catenina es el ortólogo en vertebrados de la proteína armadillo en
Drosophila melanogaster y es el mediador clave de la vía canónica de Wnt. Fue identificada originalmente como una proteína citoplasmática de importancia en las uniones adherentes, en donde se une al extremo C-terminal de las caderinas, formando un puente entre estas moléculas de adhesión con la α-catenina y por

53
ende, con el citoesqueleto. Sin embargo, existe otro pool de β-catenina citoplasmática con un tiempo de vida extremadamente corto, debido a la actividad de un complejo de destrucción multiproteico, involucrado en la vía de señalización Wnt (44). Dsh codifica para una proteína citoplasmática expresada de forma ubicuita
llamada Dishevelled (conocida como DSH en D. melanogaster y DVL en mamíferos), la cual contiene cuatro dominios altamente conservados. En el extremo N-terminal, DVL contiene una extensión de 50 aminoácidos, las otras tres regiones conservadas son: un dominio básico corto, un dominio PDZ localizado en el centro, y un dominio DEP en el extremo C-terminal, el cual también se encuentra en muchas proteínas que interactúan con la proteína quinasa C (Wodarz A et al., 1998). DVL es una fosfoproteína que se vuelve más fosforilada en los residuos serina y treonina cuando la señal Wnt es activada. La proteína quinasa glicógeno sintasa-3β (GSK3β) es una proteína quinasa
serina-treonina que actúa como represor constitutivo de la actividad señalizadora de la β-catenina, al fomentar la fosforilación de muchos sitios en la porción N-terminal de la β-catenina. Esto implica que la activación de Wnt conlleva a la represión de la actividad constitutiva de GSK3β, permitiendo la activación de la β-catenina (Wodarz A et al., 1998). La axina (inhibidor de eje de los productos del gen supresor de tumor) es
una proteína con similitud en su secuencia al dominio conservado de la región N-terminal de Dsh, que puede unirse directamente a GSK3β y a la β-catenina, promoviendo la fosforilación de la β-catenina en este complejo (Wodarz A et al., 1998). APC (adenomatous polyposis coli) actúa en conjunto con GSK3β para
regular a la β-catenina. Puede unirse a la ésta y promover su degradación. APC puede estar regulada por GSK3β, ya que APC, GSK3β, axina y β-catenina se asocian para formar un complejo. APC es un buen sustrato para GSK3β in vitro y la asociación de la β-catenina con APC parece depender de la fosforilación de APC por GSK3β (Wodarz A et al., 1998).
2.2.3.5 Activación de Wnt. En ausencia de estimulación de la vía Wnt, la proteína β-catenina es desestabilizada por un complejo citoplasmático compuesto por los productos del gen supresor de tumor Axina y APC, por la quinasa caseína 1 (CK1) que es una quinasa de tipo serina/treonina y por (GSK3β) (Behrens J et al., 1998). Después de que se unen Axina y APC, la β-catenina es fosforilada de forma secuencial por CK1 y GSK3β al menos en cuatro residuos conservados del extremo amino-terminal. CK1 fosforila a β−catenina en el aminoácido Ser45,

54
creando así un sitio de preparación para que GSK3β fosforile a los otros residuos, Thr41, Ser37 y Ser33. Esto crea un motivo de reconocimiento para el complejo E3-ubiquitin-ligasa, el cual está compuesto de β-TRCP (proteína que contiene la repetición β-transducina), esta proteína marca a la β-catenina con moléculas de ubiquitina, dejándola lista para la degradación proteasomal (Staal FJT et al., 2005). La acción de este complejo es antagonizada por Dishevelled (DVL), proteína citoplasmática que se activa por un mecanismo desconocido cuando Wnt se une con su receptor. DVL es reclutada a la membrana celular, permitiendo que GSK3β se disocie de la Axina. Cuando GSK3β ya no está unida a la β-catenina, ésta no se fosforila. Gracias a ello la β-catenina no fosforilada puede acumularse y translocarse al núcleo (Figura 2.5).
2.2.3.6 Señalización en el núcleo. En la mayoría de modelos, la principal función de la vía de señalización Wnt es la regulación de las decisiones del destino de la célula. Esta función es llevada a cabo mediante la alteración de forma instructiva del programa transcripcional de cada célula. Es por ello que la mutación en genes Wnt o su expresión inapropiada por lo general conducen a que el destino celular cambie, y esto se refleja en la expresión génica alterada. Muchos genes directa o indirectamente regulados por Wnt son factores de transcripción o moléculas de señalización secretadas que parecen tener un rol clave en la jerarquía de los genes reguladores (Wodarz A et al., 1998). Una vez en el núcleo, la β-catenina actúa como un co-activador transcripcional asociándose con los factores de transcripción de la familia TCF/LEF (Taipale J et al., 2001). El mecanismo mediante el cual realiza esta labor es reemplazando a los co-represores que pertenecen a la familia del gen relacionado con groucho (GRG), los cuales se unen a TCFs (Roose J et al., 1998). El extremo C-terminal de la β-catenina provee un dominio de transactivación que está unido al ADN a través de la caja-HMG (grupo de alta movilidad) del TCF y a través de factores de transcripción LEF (Wodarz A et al., 1998). Luego, la β-catenina llama a CBP (proteína de unión al elemento de respuesta al AMP cíclico) y/o a acetilasas histona p300 y a BRG1 (gen relacionado con brama – 1) (Barker N et al., 2001) para que se unan a promotores blanco TCF, facilitando de esta manera la transcripción de esos genes (Staal FJT et al., 2005). En ausencia de estimulación de la vía, los niveles reducidos de β-catenina permiten la represión de los genes blanco de Wnt mediante la asociación de co-represores transcripcionales con TCF/LEF (Taipale J et al., 2001). En el núcleo, la actividad transcripcional del complejo β-catenina-TCF puede ser modulada por proteínas que compiten con los miembros de la familia TCF/LEF uniéndose a la β-catenina. Chibby, inhibe la activación transcripcional mediada por β-catenina, compitiendo con LEF1 por la unión a la β-catenina (Takemaru K et al.,

55
2003). ICAT es otro inhibidor similar de la β-catenina y de TCF, que también inhibe la vía de señalización Wnt, obstruyendo la interacción entre la β-catenina y los miembros de la familia TCF/LEF (Daniels DL et al., 2002). Figura 2.5 Vía de señalización WNT.
Modificado de: Staal, F.J.T., & Clevers, H.C. (2005). WNT signalling and haematopoiesis: a WNT situation. Nature Reviews in Immunology, 5, 21-30.
VIA
Wnt
NO
ACTIV
ADA
VIA
Wnt
ACT
IVAD
A

56
2.2.3.7 Wnt y las células madre hematopoyéticas. Hay estudios que muestran que las señales Wnt pueden proveer señales proliferativas para células progenitoras inmaduras, incluyendo las CMHs. Células de estroma transfectadas con las proteínas de ratón Wnt1, Wnt2b, Wnt5a o Wnt10b inducían la proliferación de células de hígado fetal con fenotipo AA4+c-kit+Sca1+, una población altamente enriquecida en CMHs (Austin TW et al., 1997). Estas células además, aumentaban la frecuencia de células Lin-CD34+ en co-cultivos que expresaban Wnt, y la morfología celular indicó que estos cultivos dan como resultado la presencia de gran número de células hematopoyéticas poco diferenciadas y solo unas cuantas maduras, en comparación con cultivos control (van den Berg DJ et al., 1998). Asimismo, usando células CD34+ de cordón umbilical y células de estroma transducidas con Wnt5A se observó que, mientras que la proliferación in vitro no se afectaba en los cultivos estromales, en ratones transplantados con células humanas CD34+ la inyección intraperitoneal de medio acondicionado con Wnt5A incrementó la reconstitución multilinaje en más de tres veces comparada con los controles (Murdoch B et al., 2003). Otros estudios demuestran que la vía de señalización Wnt juega un papel importante en la regulación de la auto-renovación de las CMHs. La adición de la proteína Wnt purificada, a células de la médula ósea de ratón con fenotipo c-kit+Thy-1loSca-1+Lin-, aumentó aproximadamente seis veces la frecuencia de células que respondían a dosis limitantes de SCF (factor de célula madre), y el 30% de estas células mantuvieron el fenotipo (Willert K et al., 2003). Por otro lado, la sobreexpresión mediada por transducción retroviral de β-catenina constitutivamente activa en cultivos a largo plazo de CMHs de ratón (con fenotipo c-kit+Thy-1.1loSca-1+Lin-/lo), expandió el pool de células madre transplantables. Esto fue determinado tanto por el fenotipo como por la habilidad de repoblar (Reya T et al., 2003). En este mismo studio, se demostró que la expresión ectópica de axina o de un dominio de unión al ligando de la proteína frizzled, los cuales son inhibidores de la señalización Wnt, conllevaba a la inhibición de la proliferación de CMHs, a muerte aumentada de CMHs in vitro, y a reconstitución in vivo reducida.
2.2.4 Otros factores implicados en el mecanismo de auto-renovación
2.2.4.1 Proteínas BMP. Las proteínas morfogenéticas de hueso o BMPs, son miembros de la superfamilia TGF-β, a la cual también pertenecen los factores transformantes de crecimiento (TGFs), las inhibinas y las activinas. Esta familia participa en gran cantidad de respuestas biológicas como crecimiento celular, apoptosis y diferenciación en varios tipos celulares. Uno de los miembros de la familia BMP es BMP-4, esta proteína se expresa en el mesodermo ventral e induce la diferenciación hematopoyética en embriones de Xenopus (Maeno M et

57
al., 1996). En humanos, las BMPs se expresan en medula ósea adulta y son esenciales en la remodelación del hueso y el crecimiento. Se ha demostrado que BMP-4 juega un papel importante en el crecimiento, diferenciación y capacidad de repoblar de CMHs humanas CD34+CD38-Lin-, mientras que altas concentraciones de BMP-2 y BMP-7 actúan de forma similar al TGF-β, inhibiendo la proliferación de estas células (Bhatia M et al., 1999).
2.2.4.2 HOXB4. Miembros de la familia de genes homeobox Hox fueron los primeros implicados en la regulación de la especificación de linaje y el desarrollo de las células madre en un número de tejidos, incluyendo el sistema hematopoyético. El factor de transcripción HOXB4 es el que más se ha estudiado. Se han encontrado altos niveles de expresión de este factor en células de medula ósea CD34+CD38-/loCD45RA-CD71- que contienen gran cantidad de células iniciadoras de cultivo a largo plazo (LTC-ICs), pero no se ha encontrado HOXB4 en la mayoría de poblaciones de células progenitoras adultas (Sauvageau G et al., 1994). La diferenciación de células Lin- en los linajes eritroide y granulocítico está acompañada por un aumento en el ARNm de HOXB4. Asimismo, un descenso en los niveles de HOXB4 inhibe la formación de colonias de estos linajes (Giampaolo A et al., 1994). Se ha demostrado que al manipular células de estroma para que secreten HOXB4 y co-cultivarlas con células humanas CD34+CD38lo hay un incremento en el número de células madre (Amsellem S et al., 2003; Krosl J et al., 2003). Además, células de medula ósea de ratón que sobre-expresan HOXB4 aumentan el número de CFU-S (unidades formadoras de colonia de bazo) y CFCs (células formadoras de colonia) capaces de generar gran cantidad de colonias en cultivos primarios y secundarios. Al transplantar estas células transducidas con HOXB4 a ratones irradiados letalmente, se encontró que el compartimento de CRU (unidad repobladora competidora) se regenera hasta alcanzar niveles normales (Sauvageau G et al., 1995). Un hallazgo importante es que CMHs transducidas con HOXB4 no se expanden por encima de los niveles observados en ratones no manipulados, lo cual indica que la sobre-expresión no supera los mecanismos reguladores que mantienen el tamaño del pool de CMHs dentro de los niveles normales (Thorsteinsdottir U et al., 1999).
2.2.4.3 Bmi-1. Bmi-1 es un proto-oncogen perteneciente al grupo Polycomb, el cual representa a los represores transcripcionales. Se expresa en CMHs de ratones y humanos. Se ha encontrado que embriones de ratón que carecen de Bmi-1 son hipocelulares y mueren antes de alcanzar los 2 meses de vida debido a una disminución progresiva de todas las células sanguíneas, incluyendo las más

58
primitivas. Células de medula ósea e hígado fetal obtenidas de ratones con genotipo Bmi-1-/-, al ser transplantadas a ratones irradiados letalmente tienen una capacidad disminuida de repoblar todos los linajes hematopoyéticos (Park IK et al., 2003). Esto indica que Bmi-1 juega un papel importante en el mantenimiento del pool de CMHs. Células de la médula ósea cultivadas de estos ratones homocigotos y transplantadas a ratones secundarios Bmi-1-/-, prácticamente no se detectaban después de 8 semanas del transplante, lo cual sugiere un defecto en la maquinaria de auto-renovación. Se sugiere que los genes p16Ink4a and p19Arf, conocidos por detener el ciclo celular y aumentar la apoptosis, respectivamente, son algunos de los genes encargados de modular la proliferación celular en la medula ósea de estos ratones (Lessard J et al., 2003). Estos estudios sugieren que la manipulación de Bmi-1 con el fin de alterar la proliferación celular, podría ser de gran utilidad en el manejo de células madre leucémicas (Lessard J et al., 2003).
2.2.5 Integración de Wnt, Hedgehog, Notch y BMP en las células madre hematopoyéticas.
Las primeras CMHs que se producen durante el desarrollo son derivadas del mesodermo. Por lo tanto, sería de esperarse que genes que son importantes en el diseño y formación del mesodermo como los componentes de las vías de señalización Hedgehog, Notch y Wnt, también sean esenciales en el compromiso, diferenciación y auto-renovación de las CMHs. Sin embargo, si estas señales influyen en diferentes elementos de la auto-renovación (como proliferación o inhibición de la diferenciación), y cómo están integradas unas con otras aún no es claro, pero se han encontrado evidencias de que juntas, son necesarias para que se lleve a cabo de manera adecuada el mecanismo de auto-renovación en las CMHs. Entre las características que comparten Wnt y Shh están las señales modificadas por lípidos y la participación de los receptores de superficie Frizzled y Smoothened que se relacionan entre sí. Adicionalmente, tanto las señales Wnt como las señales Hh usan las proteínas quinasas GSK3 y Ck1α para facilitar la proteólisis de los efectores transcripcionales: β-catenina para Wnt y Cubitus interruptus para Shh (Kalderon D 2002). Wnt y Shh también comparten características en sus proteínas y receptores. Aunque las proteínas Wnt y Hh no están relacionadas entre sí, ambas llevan palmitatos unidos covalentemente y están destinadas a la secreción, por lo tanto, pueden actuar sobre células lejanas a su fuente. Los receptores Frizzled para Wnt están relacionados a la proteína Smoothened. Ambos receptores tienen siete dominios transmembranales y un dominio rico en cisteína en el extremo N-terminal. Como grupo, estas moléculas están más cercanamente relacionadas

59
entre sí que otras familias de receptores serpenteantes. Sin embargo, aunque los receptores están relacionados, los mecanismos de activación de Fz y Smo son fundamentalmente diferentes (Nusse R 2003). La relación entre Wnt y Notch también ha sido estudiada. Se ha encontrado que CMHs transducidas con β-catenina expresaban de tres a cuatro veces más niveles de HOXB4 y Notch1, genes reguladores de la auto-renovación de CMHs pueden estar relacionados y actuar en una jerarquía molecular (Reya T et al., 2003). En otro estudio, se detectaron señales Wnt y Notch activas en la mayoría de células del nicho de CMHs y una gran fracción de estas células usaban ambas vías simultáneamente. En el mismo estudio, se determinó que la estimulación con proteínas Wnt aumenta la expresión de genes blanco de la vía Notch en CMHs, lo cual demuestra que Wnt contribuye a la expresión diferencial de blancos de la vía Notch. Al inhibir la señal de Notch, en presencia de Wnt3a y SLF (Steel factor), la capacidad de las CMHs de mantener un estado indiferenciado se disminuye pero hay normal proliferación y supervivencia, es decir, que la habilidad de señalización de Notch es dominante con respecto a las proteínas Wnt y SLF (Duncan AW et al., 2005). También se ha visto relación entre Shh y BMP, ya que que Shh regula la proliferación de células sanguíneas primitivas a través de mecanismos que dependen de señales BMP (Bhardwaj G et al., 2001). Entender cómo se relacionan funcionalmente estas vías de señalización con el fin de controlar la regulación de las células madre y explorar las similitudes entre ellas y la manera como operan en diferentes organismos, puede ser un punto crítico para dilucidar el papel de estos factores de señalización en el desarrollo y renovación de células oncogénicas.

60
3. CÉLULAS MADRE LEUCÉMICAS Las células madre juegan un papel fundamental en el desarrollo fisiológico normal de los organismos. Sin embargo, una célula madre normal puede convertirse en una célula madre leucémica (CML) cuando ocurren alteraciones en sus propiedades intrínsecas que conllevan al desarrollo de ciertos tipos de cáncer, particularmente de leucemias mieloides. Estas alteraciones pueden hacer que las CMLs adquieran ciertas características y propiedades similares a las de las células madre hematopoyéticas como la auto-renovación y algunos mecanismos de evasión de la apoptosis que les confieren una ventaja proliferativa sobre el resto de células normales. La principal causa por la cual una célula madre puede llegar a convertirse en una CML es que gracias a su gran capacidad de auto-renovación y proliferación, es posible que, a lo largo del gran número de replicaciones de ADN experimentadas, los sistemas de reparación de daños en éste, empiecen a fallar con el tiempo, acumulando así errores en su código genético. También pueden ocurrir daños intrínsecos o extrínsecos capaces de alterar el ADN. Para contrarrestar estos efectos, las células madre utilizan el mecanismo llamado sucesión clonal, en el cual la mayor parte de la población permanece en estado de quiescencia y solo unas pocas proliferan y se diferencian para suplir las necesidades de células maduras. A medida que el organismo requiera más células maduras y que la población de células madre proliferantes empiece a disminuir, nuevas células madre que permanecían quiescentes, empezarán a proliferar (Bell DR et al., 2004). Gracias a los experimentos en los cuales se transplantan CMHs se ha encontrado que estas células no son capaces de mantener la hematopoyesis de manera indefinida, lo cual es un indicio de que las CMHs envejecen. Un ejemplo de que las reservas de células hematopoyéticas envejecen con la edad del organismo es que, en personas mayores las tasas de morbilidad y mortalidad son muy altas después de realizar terapias mielo-ablasivas contra el cáncer. La leucemia o cáncer del sistema hematopoyético, se origina y se mantiene por CMLs. Se podría entonces suponer que las CMLs surgen por transformaciones en las CMHs y por el envejecimiento propio de éstas, por ello, es importante identificar cómo se comportan ciertos mecanismos como la apoptosis y la senescencia celular mediada por el acortamiento de los telómeros, en las CMHs y las CMLs.

61
3.1 APOPTOSIS La apoptosis o muerte celular programada, es el proceso mediante el cual las células son eliminadas bajo condiciones normales cuando completan su periodo de vida o cuando ocurre algún daño en ellas. Este es un proceso normal en el desarrollo y la homeóstasis del organismo. Morfológicamente, en las células que entran en apoptosis las membranas plasmática y nuclear se arrugan, inflan y condensan, y después ocurre la agregación de la cromatina nuclear. La mitocondria y los ribosomas mantienen su estructura gruesa y parte de su función. Hay una disrupción de la arquitectura del citoesqueleto: la célula se contrae y se fragmenta en cuerpos apoptóticos encerrados en una membrana que rápidamente son ingeridos por macrófagos adyacentes u otras células fagocíticas vecinas. Debido a que estos cuerpos apoptóticos no inducen de forma significativa la liberación de citoquinas, este proceso no implica respuesta inflamatoria (Israels LG & Israels ED, 1999). A diferencia de lo que ocurre en la necrosis, en la apoptosis no hay degradación completa del ADN sino un clivaje mediado por endonucleasas que da como resultado fragmentos de ADN de 180 pares de bases. A nivel molecular, el proceso puede ser resumido en varios pasos: (a) los genes responden al daño en el ADN, (b) las señales de muerte son recibidas a través de la membrana celular mediante el ligando Fas, o (c) mediante enzimas proteolíticas llamadas granzimas, que entran directamente a la célula. Los eventos finales como los cambios en la estructura y ensamblaje celular, están mediados por proteasas específicas llamadas caspasas. Uno de los mecanismos encargados de la reparación del ADN cuando ocurren daños durante la replicación, es la activación del gen p53, el cual detiene el ciclo celular en el punto de restricción de la fase G1 protegido por la proteína Retinoblastoma (Rb). En caso de que el daño en el ADN sea irreparable, p53 lleva a cabo su función alternativa de inducir la apoptosis en la célula a través de la vía BCL-2/ (Israels LG & Israels ED, 1999). Existen diferentes proteínas pertenecientes a la familia BCL-2/BAX encargadas de inducir o inhibir la apoptosis a través de la vía mitocondrial. Entre estas proteínas se encuentran las proteínas inhibidoras de apoptosis BCL-2 y BCL-XL, y las proteínas pro-apoptóticas BAX, BAD y BID. Las primeras están ubicadas en la membrana externa de la mitocondria regulando el transporte de iones y protegiendo del daño a la membrana. Las proteínas BAX se encuentran en el citosol y ante una señal apoptótica como daño en el ADN, migran a la membrana mitocondrial, cambiando la permeabilidad iónica e induciendo la liberación al citosol del contenido interno de la membrana, incluyendo citocromo-C y factor inductor de apoptosis (AIF). AIF se mueve directamente al núcleo e induce la

62
condensación de la cromatina y la fragmentación nuclear, mientras que el citocromo-C se une al factor activador de la proteasa apoptótica-1 (APAF-1), y esta unión a su vez, activa a la pro-caspasa-9 convirtiéndola en caspasa-9, la cual activa a la caspasa-3, responsable de los cambios citológicos propios de la apoptosis (Israels LG & Israels ED, 1999). Además de la vía de activación mitocondrial, existen dos sistemas adicionales a través de los cuales células citotóxicas pueden regular la activación de la apoptosis en células dañadas. Estos son: el sistema de granzimas, y el sistema Fas-receptor/Fas-ligando. El sistema de granzimas trabaja a través de la creación de un poro similar al complejo de ataque a la membrana del sistema complemento. Este poro es creado en células infectadas por patógenos o en células tumorales a través de la liberación de perforinas contenidas en los gránulos secretores de linfocitos citotóxicos y células NK. Estos gránulos también contienen proteínas llamadas granzimas. Luego de la formación del poro, la granzima B entra a la célula blanco mediante endocitosis, encerrada en una vesícula que una vez adentro, libera la proteína e induce la activación de pro-caspasas, la fragmentación del ADN y finalmente la apoptosis (Israels LG & Israels ED, 1999). El sistema Fas-receptor/Fas-ligando está compuesto de una proteína receptora llamada Fas o CD95, perteneciente a la familia del receptor del factor de necrosis tumoral (TNF-R), que se comporta como un transductor de la señal apoptótica; y un ligando de Fas expresado sobre células T citotóxicas y células NK, perteneciente a la familia del factor de necrosis tumoral (TNF). Este ligando se une y se entrecruza con el receptor, poniendo en marcha el proceso apoptótico. Cuando el ligando y el receptor se unen, ya sean Fas con Fas-ligando, o TNF-R con TNF, la parte intracelular de Fas y de TNF-R, que contiene dominios de muerte, interactúa con la proteína FADD (proteína asociada a Fas con un dominio de muerte) y TRADD (proteína asociada a TNF-R con un dominio de muerte) respectivamente, esta unión conduce al reclutamiento y activación de la pro-caspasa-8 y a la subsiguiente activación de caspasas efectoras que conllevan a la apoptosis (Israels LG & Israels ED, 1999). Se ha demostrado que precursores hematopoyéticos primitivos (CD34+CD38-
/bajoHLA-DR-/bajo) expresan altos niveles de las proteínas inhibidoras de apoptosis BCL-X L y BCL-2 (BCL-XL en más altos niveles que BCL-2), y niveles muy bajos de las proteínas pro-apoptóticas BAX, BAD y BAK (Peters R et al., 1998; Park JR et al., 1995). Además, la sobre-expresión de BCL-2 aumenta el número de CMHs en la médula ósea (Domen J et al., 2000), y a medida que las CMHs entran en un proceso de diferenciación los niveles de expresión de BCL-2 descienden mientras que aumenta la expresión del receptor Fas, responsable de inducir la apoptosis

63
(Takenaka K et al., 1996). Esto indica que en las CMHs existe una regulación que asegura el balance entre la auto-renovación, diferenciación y muerte celular. Las CMLs pueden desarrollarse cuando ocurre algún tipo de alteración en cualquiera de los factores implicados en la apoptosis. La sobre-expresión de proteínas inhibidoras de la apoptosis, o, por el contrario, la no-expresión o el mal funcionamiento de proteínas pro-apoptóticas pueden generar células leucémicas incapaces de entrar en apoptosis y con alta actividad proliferativa. Ejemplos de estas alteraciones se pueden ver en el linfoma folicular, en el cual la translocación t(14:18) conlleva a una alteración en el gen BCL-2 (Tsujimoto Y et al., 1985), o en la policitemia vera, en donde se ha encontrado una sobre-expresión de BCL-XL (Silva M et al., 1998). También se han reportado mutaciones en BAX en algunas líneas celulares de leucemia linfoide aguda en humanos (Meijerink JPP et al., 1998). Sin embargo, no solamente las alteraciones en genes involucrados en la apoptosis pueden llegar a generar células neoplásicas, también es posible que oncoproteínas producto de translocaciones en genes que no están relacionados de manera directa en la apoptosis puedan inducir el bloqueo de las vías apoptóticas. Este es el caso de BCR-ABL, una oncoproteína característica de la leucemia mieloide crónica (LMC) resultado de la translocación 9;22 que da origen al cromosoma Filadelfia (Ph). La función de tirosina-quinasa de BCR-ABL le permite activar vías de transducción de señales como la PI-3-quinasa, la vía RAS o la vía STAT. En las células leucémicas BCR-ABL+ se ha encontrado que la expresión de esta oncoproteína es responsable de la inhibición de la apoptosis (Bedi A et al., 1994; McGahon A et al., 1994). No se sabe a cuál es el mecanismo exacto, si es que solamente hay uno, mediante el cual BCR-ABL puede proteger contra la apoptosis pero se cree que está relacionado con la activación de BCL-XL (Amarante-Mendes GP, McGahon AJ, et al., 1998; Brady HJM, 2003), BCL-2 (Sanchez-Garcia I et al., 1995; Delia D et al., 1992) y la inactivación de BAD (Neshat MS et al., 2000), ya sea a través de la vía PI-3K, RAS o STAT (Nieborowska-Skorska M et al., 1999; Franke TF et al., 1997; Neshat MS et al., 2000; Cortez D et al., 1996).
3.2 TELÓMEROS El telómero es un complejo de secuencias de ADN ubicado en el extremo de los cromosomas eucariotas. En vertebrados, estas secuencias están hechas de repeticiones en tándem de (TTAGGG)n cuyos grupos de guanina están ubicados hacia el extremo del cromosoma en la hebra que va en dirección 5’ a 3’. Los telómeros sirven para preservar la integridad nuclear y cromosómica evitando la fusión entre los extremos de los cromosomas y los eventos de recombinación en o cerca de ellos, además protegen contra la pérdida de ADN durante la división celular. Además, sirven como sitio de unión para diferentes proteínas de unión al ADN. La longitud del ADN telomérico humano varía entre cada individuo y de

64
acuerdo al origen, edad e historia proliferativa de las células estudiadas oscila entre 5 y 20 Kpb (Greenwood MJ & Landscorp PM, 2003). La mayor parte del telómero es ADN de doble cadena, sin embargo, el extremo del telómero está compuesto de una proyección de cadena sencilla que se dobla sobre sí misma para prevenir la degradación por exonucleasas, formando una estructura protectora especial mediante la unión inusual a través de puentes de hidrógeno entre guanina y guanina. Esta estructura es conocida como “horquilla-T” (en inglés: “T-loop”) (figura 3.1) (Griffith JD et al., 1999) Figura 3.1 Horquilla-T
Para que se lleve a cabo la replicación del ADN de manera adecuada, las polimerasas de ADN tienen dos requerimientos importantes antes de iniciar el proceso replicativo. Primero, debe tener iniciadores o primers de ARN que marquen el sitio en el ADN donde la enzima debe comenzar la replicación. Como segundo parámetro, la única dirección en la cual puede replicar la polimerasa de ADN es de 5’ a 3’ sobre la hebra recién sintetizada. Debido a que las polimerasas de ADN son incapaces de iniciar la síntesis de ADN de la hebra rezagada desde el extremo del ADN linear y ya que no existen mecanismos conocidos para replicar los primers de ARN de los extremos, el extremo 5’ de la nueva hebra queda replicado de forma incompleta y se genera una pérdida de aproximadamente 50 a 200 pb de ADN telomérico por cada población de células humanas duplicándose. Esto es lo que se conoce como el problema de la replicación de los extremos (figura 3.2) (Watson JD, 1972, Olovnikov AM, 1973). Con el fin de mantener la capacidad proliferativa y evitar la senescencia replicativa de las células, es necesario mantener la longitud del telómero, esto se logra a través de la acción de la telomerasa. La telomerasa humana es una ribonucleoproteína compuesta como su nombre lo dice, por una parte proteica y una parte de ARN. La parte proteica es una transcriptasa reversa que le da la actividad enzimática a la telomerasa. La parte de ARN contiene repeticiones AAUCCC, complementarias a la secuencia del telómero, esto le permite unirse a éste y servir como templado para llevar a cabo la síntesis de ADN y extender la longitud del telómero en dirección 5’-3’ sobre el ADN. A medida que el telómero adquiere nuevas repeticiones la telomerasa se mueve al extremo recién
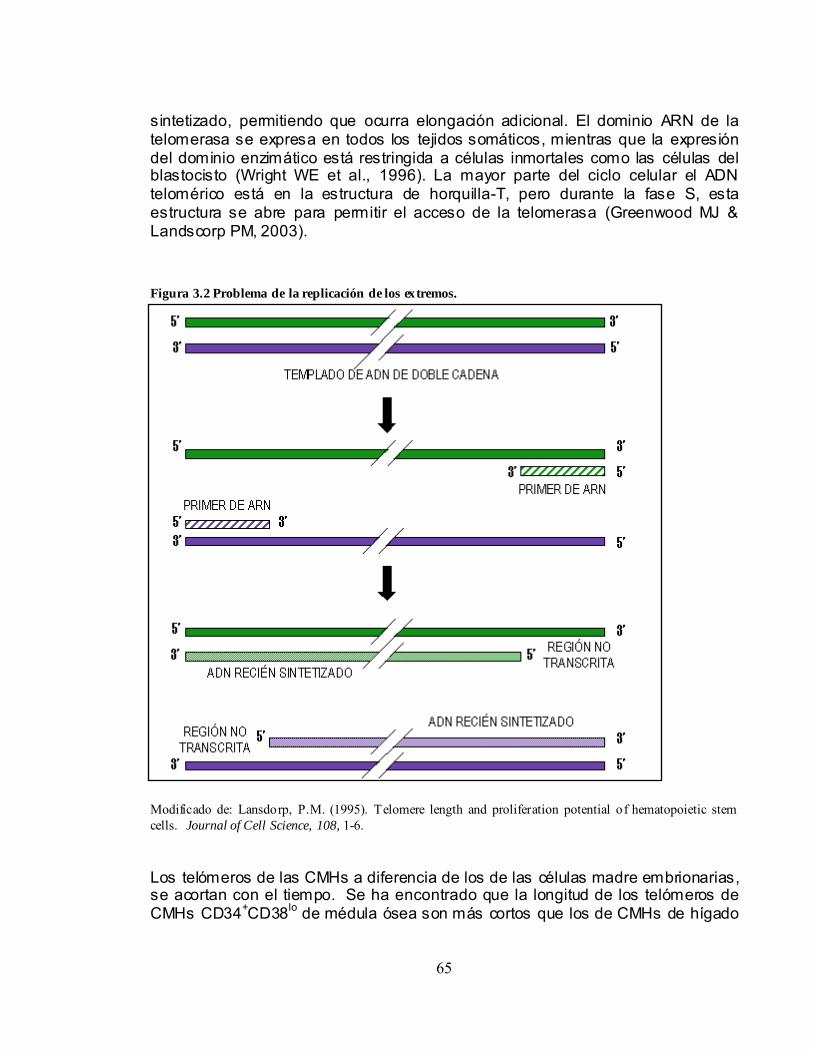
65
sintetizado, permitiendo que ocurra elongación adicional. El dominio ARN de la telomerasa se expresa en todos los tejidos somáticos, mientras que la expresión del dominio enzimático está restringida a células inmortales como las células del blastocisto (Wright WE et al., 1996). La mayor parte del ciclo celular el ADN telomérico está en la estructura de horquilla-T, pero durante la fase S, esta estructura se abre para permitir el acceso de la telomerasa (Greenwood MJ & Landscorp PM, 2003). Figura 3.2 Problema de la replicación de los extremos.
Modificado de: Lansdorp, P.M. (1995). Telomere length and proliferation potential of hematopoietic stem cells. Journal of Cell Science, 108, 1-6. Los telómeros de las CMHs a diferencia de los de las células madre embrionarias, se acortan con el tiempo. Se ha encontrado que la longitud de los telómeros de CMHs CD34+CD38lo de médula ósea son más cortos que los de CMHs de hígado

66
fetal o cordón umbilical, asimismo parece haber una pérdida telomérica asociada con la edad (Vaziri H et al., 1994). Sin embargo, la mayoría de CMHs de médula ósea tienen actividad de telomerasa y esta actividad está asociada con el potencial de auto-renovación de las células, ya que la actividad telomerasa disminuye a medida que las células maduran (Morrison SJ et al., 1996). Aunque no está claramente entendido el papel de la telomerasa en las CMHs, se propone que la telomerasa es reprimida en células madre quiescentes, es activada durante la proliferación, expansión, entrada al ciclo celular y la progresión al compartimento de célula progenitora, y es reprimida de nuevo en la diferenciación a célula terminal (Engelhardt M et al., 1997). A pesar de que la telomerasa se expresa en bajos niveles en CMHs, esto no es suficiente para evitar el acortamiento de los telómeros durante la proliferación (Yui J et al., 1998), aunque recientemente fue demostrado que ratones deficientes en telomerasa mostraban un mayor acortamiento de los telómeros de sus CMHs y menor potencial de reconstitución después de transplantes seriados, en comparación con ratones normales, lo cual sugiere que la telomerasa le da mayor capacidad de replicación a las CMHs gracias a que disminuye la tasa de acortamiento de sus telómeros (Allsopp RC et al., 2003). Los telómeros pueden contribuir en la transformación de las CMHs ya sea a través de un aumento en la actividad de la enzima telomerasa, que conllevaría a que las células tuvieran mayor capacidad de auto-renovarse y pudieran llegar a convertirse en células neoplásicas, o por el contrario, a través del acortamiento de los telómeros que daría lugar a la senescencia celular, causando inestabilidades genómicas que originen mutaciones y la activación de oncogenes. En la leucemia mieloide crónica se ha observado que la longitud de los telómeros es menor en células leucémicas que en células normales (Brümmendorf TH et al., 2000). Asimismo, se ha encontrado que a medida que la enfermedad progresa, la longitud de los telómeros disminuye mientras que la actividad de la telomerasa aumenta (Ohyashiki K et al., 1997; Broccoli D et al., 1995; Boultwood J et al., 1999). Estos estudios indican que las células de leucemia mieloide crónica se replican de manera acelerada, dando lugar al acortamiento de los telómeros e incentivando a un aumento en la actividad de la telomerasa con el fin de suplir esta pérdida. Sin embargo, parece ser que los niveles de expresión de esta enzima no son suficientes para prevenir el acortamiento cromosómico y eventualmente, los telómeros del clon neoplásico disminuyen hasta llegar a un punto crítico en el cual se precipita la progresión de fase crónica a crisis blástica, y tal vez a leucemia mieloide aguda. La inestabilidad cromosómica generada por el aumento en la proliferación, sería la responsable de la adquisición de nuevas anormalidades causantes de alteraciones citogenéticas adicionales (Brümmendorf TH et al., 2000). Dada la complejidad de los mecanismos utilizados por las CMHs para auto-renovarse y proliferar, muchas podrían ser las causas de que una CMH se

67
convierta en una CML. Sin embargo, todas estas causas están enmarcadas dentro de dos procesos principales que afectan el destino de las células: la supervivencia y el envejecimiento celular mediados por la muerte celular programada o apoptosis y por el acortamiento de los telómeros. Por lo tanto, cualquier alteración que intervenga en el balance de estos procesos puede ser responsable de que una célula madre hematopoyética normal llegue a convertirse en una célula neoplásica con capacidad de dar origen a toda una población de células transformadas, y la identificación de estas alteraciones puede ayudarnos a comprender mejor la biología del cáncer, específicamente de las leucemias, y a desarrollar nuevas terapias para combatirlas.

68
4. LEUCEMIA MIELOIDE CRÓNICA
La leucemia mieloide crónica (LMC) es un desorden originado en las células madre hematopoyéticas que da como resultado la expansión clonal de células de linaje principalmente mieloide, y en menor frecuencia linfoide. El marcador patognomónico de la LMC es la presentación del cromosoma Filadelfia (Ph), como resultado de la translocación recíproca entre los cromosomas 9 y 22, que da origen a la expresión del oncogen BCR-ABL el cual codifica una proteína de fusión oncogénica. La edad promedio de los pacientes al momento de la presentación de la enfermedad está entre 45 y 55 años, y 12-30% de los pacientes son mayores de 60 años (Faderl S et al., 1999), un parámetro importante de considerar con respecto a las estrategias terapéuticas como el transplante de células madre y el tratamiento con interferón alfa. Aunque no hay claridad sobre la etiología de la enfermedad, se encontró que la incidencia de la LMC fue mayor en las personas expuestas a la bomba atómica de Hiroshima y Nagasaki (Heyssel T et al., 1960; Lange R et al., 1954), y se ha descrito LMC en pacientes sometidos a radioterapia para el tratamiento de otro tipo de neoplasias (Brown WMC et al., 1965; Boice JD et al., 1985; Corso A et al., 1995). Esto implica que la radiación puede contribuir de forma potencial al desarrollo de la enfermedad. También se han asociado agentes químicos como el benceno y otros solventes orgánicos como causa de la LMC, sin embargo, usualmente no se han identificado factores predisponentes en pacientes que presentan la enfermedad. Los primeros reportes de leucemia mieloide crónica se dieron a conocer a mediados del siglo diecinueve gracias a la observación de detalles microscópicos a nivel clínico y a nivel post-mortem acuñados por Robert Virchow y Jhon Huges Bennett. Posteriormente, en 1870 Neumann sugirió que la médula ósea jugaba un papel importante dentro de la hematopoyesis y por lo tanto podría ser la generadora de células blancas sanguíneas malignas. Pero fue Paul Erlich quien introdujo en 1891 métodos de tinción para células sanguíneas mediante reacciones específicas, logrando de esta forma un importante avance en la clasificación de las leucemias (Geary CG 2000). A partir de entonces y a lo largo del siglo veinte estudios más profundos permitieron dar una mayor aproximación a las características diagnósticas y causas subyacentes a esta enfermedad. Sin embargo, la clave de la LMC fue hallada en 1960 gracias al desarrollo de técnicas para el estudio de células en mitosis que permitieron a Nowell y Hungerford, quienes trabajaban en Filadelfia, descubrir un cromosoma anormal en células cultivadas a partir de sangre de pacientes con LMC. Este cromosoma fue

69
designado por consenso “cromosoma Filadelfia” o “Ph” (Nowell PC et al., 1960). Trece años más tarde, Janet Rowley observó que este cromosoma identificado como el 22q-, era el resultado de la translocación recíproca con el cromosoma 9 (Rowley J, 1973). Esta anormalidad fue designada como t(9;22)(q34;q11).
4.1 PRESENTACIÓN CLÍNICA
4.1.1 Fase crónica La LMC se presenta como una enfermedad de fases, con una fase inicial crónica benigna que progresa a una fase acelerada y posteriormente a una crisis blástica rápidamente fatal. Los síntomas que se presentan en pacientes con fase crónica incluyen: anemia, esplenomegalia, hepatomegalia, sangrado, púrpura y síntomas constitucionales como fatiga, anorexia, pérdida de peso, letargo y fiebre baja o sudoración (Hoffman R et al., 2000). Sin embargo, 20-40% de los pacientes son asintomáticos y en ellos el diagnóstico está basado únicamente en los recuentos anormales de sangre, en los cuales se observa leucocitosis o trombocitosis, aunque el 85-90% de los pacientes también presenta esplenomegalia. Cerca del 50% de los casos de LMC en fase crónica son diagnosticados por exámenes de rutina. En sangre periférica es común encontrar leucocitosis en la fase crónica de la LMC, con recuentos de más de 25 x 109 / L o incluso 100-300 x 109/L. En los extendidos se pueden encontrar granulocitos en todos los estados de maduración y basofilia con o sin eosinofilia. También se presenta un conteo elevado de plaquetas en 30-50% de los casos y la actividad de la fosfatasa alcalina en los leucocitos está disminuida (Savage DG et al., 1997). Los hallazgos en medula ósea son: hipercelularidad, contenido de grasa disminuido, aumento en la proporción de células mieloides con respecto a las eritroides y aumento de los megacariocitos. Los blastos y promielocitos constituyen menos del 10% de todas las células. La morfología en la línea mieloide puede aparecer normal o con una ligera displasia (Sawyers CL 1999).
4.1.2 Fase acelerada La fase acelerada de la LMC aparece durante la progresión entre la fase crónica y la crisis blástica en un periodo de 3-4 años desde el momento de la presentación de la enfermedad. Se caracteriza por una detención progresiva en la maduración mieloide, aumentan los blastos y los basófilos en medula ósea y sangre periférica, hay enfermedad extramedular, trombocitosis o trombocitopenia, se necesitan mayores dosis del medicamento usado en la terapia, hay evolución citogenética

70
clonal es decir, cambios adicionales a nivel cromosomal, aumenta la reticulina o el colágeno en la medula, y se presenta esplenomegalia en aumento a pesar de la terapia. Algunos pacientes presentan fiebre, dolor de huesos inexplicable, pérdida de peso o anemia (Kantarjian H et al., 1988; Sokal JE et al., 1988).
4.1.3 Crisis blástica La crisis blástica es la fase terminal de la LMC y semeja a la leucemia aguda. Se desarrolla en 75-85% de los pacientes con LMC y es de pésimo pronóstico ya que el tiempo promedio de supervivencia es 3-6 meses. Se presentan síntomas constitucionales como dolor de huesos, sudoración, fiebre, pérdida de peso y anorexia, de forma más marcada que en las fases crónica o acelerada de la LMC y se pueden presentar complicaciones que incluyen infección, anemia, trombocitopenia, y enfermedad extramedular. Los sitios más comunes de enfermedad extramedular son: nódulos linfáticos, piel, hueso, y sistema nervioso central. Ocasionalmente, la enfermedad extramedular puede ser la primera manifestación de la crisis blástica, sin evidencia de progresión en la medula ósea (Hoffman R et al., Chronic Myelogenous Leukemia, 2000). . La presencia de 30% o más células leucémicas en sangre periférica o medula, o la presencia de infiltrados extramedulares de células blásticas, son el criterio utilizado por consenso para definir la crisis blástica (Kantarijan HM et al., 1987). En un tercio de los casos, los blastos tienen morfología linfoide y expresan marcadores de este linaje, mientras que los otros dos tercios de los casos tienen un fenotipo similar al de la leucemia mieloblástica aguda y forman un grupo heterogéneo. Es por ello que para pacientes con crisis blástica, el diagnóstico diferencial incluye leucemia mieloide aguda o leucemia linfoblástica aguda, ambas con presencia del cromosoma Filadelfia (Faderl S et al., 1999; Hoffman R et al., Chronic Myelogenous Leukemia, 2000).
4.2 BIOLOGÍA MOLECULAR DE LA LEUCEMIA MIELOIDE CRÓNICA
4.2.1 Expresión de BCR y ABL El gen ABL es el homólogo en humanos del oncogen v-abl presente en el virus de la leucemia de ratones Abelson (A-MuLV) (Abelson HT et al., 1970) y está ubicado en el brazo largo del cromosoma 9, en la banda 9q34. Codifica una proteína tirosina quinasa no-receptora de 145 Kd (p145ABL) cuya función aún no está completamente dilucidada, pero se sugiere que participa en la regulación del ciclo celular (Sawyers CL et al., 1994; Wang JY 1993; Kipreos ET et al., 1990) inhibiendo su progresión en la interfase G1/S, en la inducción de la apoptosis y la

71
respuesta al estrés genotóxico (Wen ST et al., 1996; Agami R et al., 1999; Yuan ZM et al., 1999; Kharbanda S et al., 1997). La actividad y localización intracelular de ABL está regulada por integrinas, y se ha postulado que esta proteína interviene en la transmisión de información acerca del ambiente celular a través de la señalización con integrinas (Lewis JM et al., 1998). El fenotipo de ratones ABL-/- es complejo, con algunos defectos autónomos de la célula en cuanto al desarrollo linfoide y algunos problemas generalizados como baja fecundidad y muerte temprana (Schwartzberg PL et al., 1991; Tybulewicz VL et al., 1991). La proteína p145ABL se expresa de forma ubicua y tiene dos isoformas (Ia y Ib) que salen del splicing alternativo del primer exón. Esta proteína se compone de muchos dominios estructurales. (Figura 4.1) La isoforma tipo Ia es ligeramente más corta que la tipo Ib, la cual contiene miríada de reguladores potenciales (myr) que le permite adherirse a la membrana plasmática. Hacia el extremo N-terminal se localizan tres dominios (SH1-SH3) con homología SRC. El dominio SH1 tiene función tirosina quinasa, mientras que los dominios SH2 y SH3 permiten la interacción con otras proteínas. Dentro del dominio quinasa, Y393 es el mayor sitio de auto-fosforilación, y fenilalanina 401 (F401) está altamente conservado En el centro de la molécula se ubican secuencias ricas en prolina (PxxP) que pueden a su vez, interactuar con los dominios SH3 de otras proteínas como Crk. Hacia el extremo 3’, se encuentran señales de localización nuclear (SLN) y dominios de unión a ADN y a actina (DU-ADN, DU-Actina). Esta proteína puede ser fosforilada por Atm, cdc2 y PKC (Deininger MWN et al., 2000; Stamatoyannopoulos G et al., 2001).
Figura 4.1Proteína p145 ABL
Modificado de: Deininger, M.W.N., Goldman, J.M., & Melo, J.V. (2000). The molecular biology of chronic myeloid leukemia. Blood, 96, 3343-3356. El gen BCR (del inglés Breakpoint Cluster Region) está ubicado en la banda q11 del cromosoma 22, generalmente se expresa en todos los tejidos y codifica una proteína de 160 Kd sin características de receptor de membrana pero con capacidad de fosforilar residuos de serina o treonina. Su importancia biológica aún

72
no está determinada, aunque parece ser que juega algún papel en la transducción de señal. Ratones con genotipo BCR-/- son viables y el único problema que presentan es el aumento en el estallido oxidativo por parte de los neutrófilos, causando lesión al tejido luego de shock endotóxico (Voncken JW et al., 1995). La proteína p160BCR Posee varios dominios estructurales. (Figura 4.2) El primer exón del extremo N-terminal codifica una quinasa serina-treonina. En este mismo extremo se ubica un dominio súper enrollado que permite la formación de un dímero (DD o dominio de dimerización). En el centro de la molécula hay una región con un dominio similar al protooncogen dbl y un dominio con homología a plecstrina (HP), que estimulan el intercambio de guanidina trifosfato (GTP) por guanidina difosfato (GDP) sobre los factores de intercambio de guanidina (FIG) de la proteína G Rho, estos factores a su vez, pueden activar factores de transcripción como NF-kβ. El extremo C-terminal tiene actividad GTPasa para Rac, una GTPasa de la superfamilia Ras que regula la polimerización de la actina y la actividad de una NADPH oxidasa en células fagocíticas. BCR también puede ser fosforilada sobre varios residuos de tirosina, especialmente Y177 (Deininger MWN et al., 2000). Figura 4.2 Proteína p160 BCR
Modificado de: Deininger, M.W.N., Goldman, J.M., & Melo, J.V. (2000). The molecular biology of chronic myeloid leukemia. Blood, 96, 3343-3356.
4.2.2 Translocación 9;22 El gen de fusión BCR-ABL resulta de la translocación recíproca de los genes normales BCR y ABL. (Figura 4.3) A lo largo del gen ABL se pueden encontrar varios puntos de ruptura, ya sea hacia el extremo 5’ del exón alternativo Ib, hacia el extremo 3’ del exón alternativo Ia, y de manera más frecuente entre los exones Ib y Ia. Los puntos de ruptura dentro del gen BCR se ubican dentro de 3 regiones bcr (breakpoint cluster regions). La región mayor o M-bcr comprende 5.8 Kb en donde se ubican los exones 12-16 también denominados b1-b5, es allí donde más

73
frecuentemente ocurren las rupturas que originan los genes quiméricos, b2a2 (e13a2) o b3a2 (e14a2), los cuales codifican para una proteína de 210-Kd (p210BCR-ABL) que posee una elevada actividad tirosina quinasa. La región menor o m-bcr se ubica entre los exones e2’ y e2, y contiene 54.4 Kb. Cuando el punto de ruptura ocurre en esta región, se genera ARNm con un tipo de unión e1a2, que se traduce en una proteína de 190 Kd (p190BCR-ABL). Existe una tercera región llamada µ-bcr, ubicada hacia el extremo 3’ con respecto a la región mayor. La ruptura en esta región genera una proteína de 230 Kd (p230BCR-ABL) codificada por el gen de fusión e19a2. También se han descrito otras variantes ocasionales del gen de fusión BCR-ABL que involucran exones como e2 y e6, en pacientes con LMC (Hoffman R et al., Chronic Myelogenous Leukemia, 2000; Deininger MWN et al., 2000) La proteína de fusión p210BCR-ABL tiene la actividad tirosina quinasa aumentada comparada con la proteína normal producida por el gen ABL (p145ABL). En la proteína quimérica, la actividad tirosina quinasa de ABL se activa constitutivamente por las secuencias traspuestas de BCR. BCR actúa promoviendo la dimerización de la oncoproteína, de tal forma que dos moléculas BCR-ABL adyacentes se fosforilan entre sí sobre los residuos de tirosina en sus horquillas de activación de quinasa. El dominio SH1 del Figura 4.3 Proteína BCR-ABL
Modificado de: Deininger, M.W.N., Goldman, J.M., & Melo, J.V. (2000). The molecular biology of chronic myeloid leukemia. Blood, 96, 3343-3356.

74
componente ABL de la oncoproteína BCR-ABL es el más importante para la transformación oncogénica. Otros motivos importantes en esta porción son el dominio SH2 y los dominios de unión a actina. Sobre la parte BCR, el dominio super enrollado es el responsable por la dimerización de la oncoproteína, y la tirosina Y177 es crucial para la unión de proteínas adaptadoras. Los residuos de fosfoserina y fosfotreonina hacia el extremo N-terminal se requieren para la interacción con proteínas que contengan SH2, incluyendo a ABL (Goldman JM et al., 2003). La actividad quinasa incontrolada de BCR-ABL hace que esta proteína adopte las funciones fisiológicas de la enzima normal ABL al interactuar con diferentes proteínas efectoras, actuando como intermediarios en las cascadas señalizadoras de tirosina quinasa para regular la expresión de genes que afectan el crecimiento y diferenciación celular. Esto da como resultado proliferación celular alterada, disminución en la adherencia de las células leucémicas al estroma de la medula ósea, y disminución en la respuesta apoptótica al estímulo mutagénico (Goldman JM et al., 2003). Se han identificado muchas moléculas que actúan como sustratos de BCR-ABL para transmitir señales citoplasmáticas al núcleo y estimular la actividad transformante. Entre estas moléculas se incluyen factores reguladores importantes como GRB-2 (del inglés: growth factor receptor – binding protein 2), SHC (SRC del inglés: homology 2–containing protein), CRKL (del inglés: CRK-oncogene–like protein), PI-3 quinasa y CBL (del inglés: casitas B-lineage lymphoma protein), entre otras. Una de las mayores diferencias entre la proteína de fusión y la proteína normal ABL es la localización celular, ya que mientras ABL se puede encontrar en el núcleo y en el citoplasma, BCR-ABL es únicamente citoplasmática. Cuando ABL está en el núcleo es una proteína pro-apoptótica que tiene un papel importante en la respuesta al estrés genotóxico. En contraste, BCR-ABL es antiapoptótica y parece ser incapaz de entrar al núcleo (Goldman JM et al., 2003).
4.2.3 Vías de señalización activadas
4.2.3.1 Vías RAS y MAP quinasa. RAS, una proteína G pequeña, y las proteínas relacionadas, son componentes altamente conservados en la transducción de señales para conectar varias clases de receptores a vías citoplasmáticas. Estas proteínas regulan casi todos los aspectos de la vida celular, incluyendo la proliferación en respuesta a factores de crecimiento, cambios citoesqueléticos, programas de diferenciación, y muerte celular, dependiendo del contexto específico. La unión directa y la activación de varios receptores de tipo tirosina quinasa y de otras familias de receptores pueden estar mediadas por proteínas conectoras celulares como SHC y GRB-2. Las proteínas RAS son

75
activadas e inactivadas para que cumplan su función en la transducción de señal gracias a la conformación diferencial que asumen para intercambiar GTP por GDP en su bolsillo de unión a nucleótidos y por la subsiguiente hidrólisis que sufren. La forma activada puede interactuar con un amplio rango de moléculas efectoras, incluyendo aquellas que son importantes en la activación de varias cascadas de tipo quinasa como RAF y MEKK1, en la producción de PI y con otros mediadores lipídicos como PI3 quinasa y algunas isoformas de PKC, y otros miembros de la familia de proteínas G pequeñas. Este ciclo está regulado por una multitud de factores celulares que pueden acelerar la activación de RAS facilitando el intercambio de nucleótidos, así como aquellos que aceleran su inactivación aumentando la hidrólisis de GTP (Stamatoyannopoulos G et al., 2001). Se han definido muchas asociaciones entre BCR-ABL y RAS. (Figura 4.4) La autofosforilación en la tirosina 177 provee un sitio para que se una la molécula GRB-2 (Goldman JM et al., 2003), la cual después de unirse a la proteína SOS, estabiliza a RAS en su forma activa unida al GTP. Otras dos moléculas adaptadoras, SHC y CRKL, también pueden activar a RAS. Ambas son sustratos de BCR-ABL y se unen a ésta a través de sus dominios SH, SHC se une al dominio SH2 y CRKL se une al dominio SH3 de BCR-ABL (Deininger MWN et al., 2000). Las cascadas que involucran a las proteínas quinasas altamente conservadas denominadas proteínas quinasas activadas por mitógenos o MAPKs y sus quinasas activadoras intervienen con el fin de responder de manera apropiada a señales externas que regulan el crecimiento, diferenciación y nivel de estrés en las células. Estas proteínas activan vías de transducción de señales que conllevan a activar factores de transcripción en el núcleo y otros efectores en la célula (Elion EA 1998). Se ha encontrado en células Ph(+) que luego de la activación de RAS, ésta recluta a la quinasa serina-treonina RAF hacia la membrana celular, una vez allí, RAF inicia la cascada de las MAPKs a través de las quinasas serina-treonina MEK1/MEK2 y ERK, esta última conduce a la activación de la transcripción génica (Deininger MWN et al., 2000) (Figura 4.4). También hay evidencias de que BCR-ABL es capaz de activar la vía de JNK/SAPK (otra vía MAPK) y que se requiere para la transformación maligna (Raitano AB et al., 1995).
4.2.3.2 Vía JAK/STAT. Una vía alternativa, conocida como la vía JAK/STAT, proporciona una conexión más inmediata entre las proteínas tirosina quinasas y los factores de transcripción. En esta vía, la fosforilación de las proteínas tirosinas afecta directamente la localización y función de los factores de transcripción. Los elementos clave de esta vía son las proteínas STAT (transductores de señal y activadores de transcripción), una familia de factores de transcripción que contienen dominios SH2, a través de los cuales, luego de su activación, se unen a las fosfotirosinas que se encuentran en el dominio citoplasmático de los
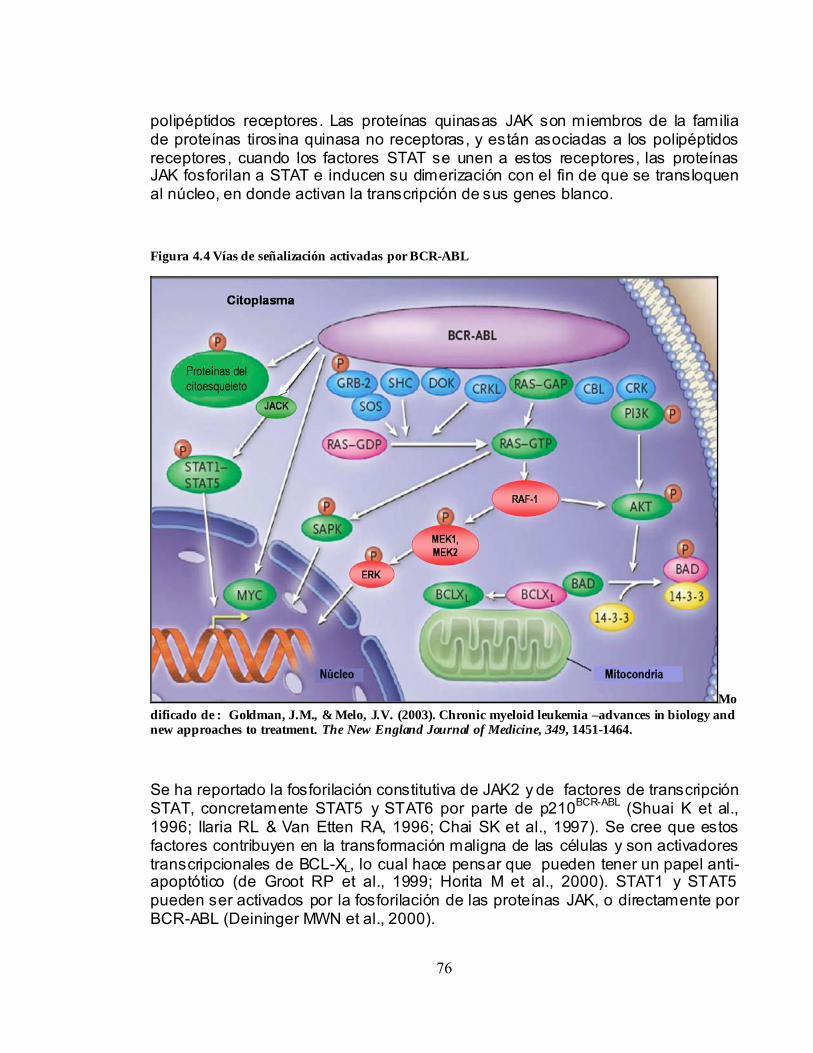
76
polipéptidos receptores. Las proteínas quinasas JAK son miembros de la familia de proteínas tirosina quinasa no receptoras, y están asociadas a los polipéptidos receptores, cuando los factores STAT se unen a estos receptores, las proteínas JAK fosforilan a STAT e inducen su dimerización con el fin de que se transloquen al núcleo, en donde activan la transcripción de sus genes blanco.
Figura 4.4 Vías de señalización activadas por BCR-ABL
Modificado de : Goldman, J.M., & Melo, J.V. (2003). Chronic myeloid leukemia –advances in biology and new approaches to treatment. The New England Journal of Medicine, 349, 1451-1464.
Se ha reportado la fosforilación constitutiva de JAK2 y de factores de transcripción STAT, concretamente STAT5 y STAT6 por parte de p210BCR-ABL (Shuai K et al., 1996; Ilaria RL & Van Etten RA, 1996; Chai SK et al., 1997). Se cree que estos factores contribuyen en la transformación maligna de las células y son activadores transcripcionales de BCL-XL, lo cual hace pensar que pueden tener un papel anti-apoptótico (de Groot RP et al., 1999; Horita M et al., 2000). STAT1 y STAT5 pueden ser activados por la fosforilación de las proteínas JAK, o directamente por BCR-ABL (Deininger MWN et al., 2000).

77
4.2.3.3 Vía de la PI-3 quinasa. La proteína fosfatidilinositol-3 quinasa o PI-3K fosforila a los fosfoinositoles en la posición D-3’ de su anillo de inositol. PI-3K puede ser activada por diferentes mecanismos, por proteínas G, por la asociación de sus dominios SH2 con residuos tirosina presentes en las proteínas tirosina quinasa receptoras, o puede interacturar con dominios SH3 de proteínas tirosina quinasa, a través de su región rica en prolina. Este tipo de asociaciones provee una señal que activa la subunidad catalítica de la PI-3K. Otras proteínas como RAS, SHC, CRK, CRKL, GRB-2, GAP o CBL también pueden influir en la activación de PI-3K. Se ha demostrado que la actividad de la PI3 quinasa es requerida para la proliferación de células BCR-ABL+ (Skorski T et al., 1995). La quinasa serina-treonina AKT, implicada en la señalización anti-apoptótica, parece ser un sustrato importante de PI-3K en la cascada de activación (Skorski T et al., 1997). La proteína pro-apoptótica BAD es el sustrato de AKT en la fosforilación inducida por la IL-3 (del Peso L et al., 1998). Cuando BAD está fosforilada es inactiva debido a que ya no es capaz de unirse a proteínas anti-apoptóticas como BCLXL y porque es atrapada por proteínas citoplasmáticas de tipo 14-3-3. (Figura 4.4) Esto sugiere que BCR-ABL podría imitar la señal de supervivencia dada por la IL-3, de una manera dependiente de PI-3K.
4.2.3.4 Vía MYC. El proto-oncogen c-Myc es el homólogo celular a las secuencias transformantes del retrovirus de mielocitomatosis aviar. Este oncogen activado es un agente transformante clave en la etiología del linfoma de Burkitt en humanos y se ha encontrado sobre-expresado en muchas malignidades humanas (Facchini LM et al., 1998). Se cree que actúa como un factor de transcripción, aunque sus genes blanco se desconocen. La activación de MYC por parte de BCR-ABL es dependiente de los dominios SH2 (Sawyers et al., 1992), pero la vía que une a MYC con SH2 aún no se conoce, aunque se cree que la señal es transducida a través de RAS/RAF, quinasas dependientes de ciclina (cdks), y factores de transcripción E2F que activan al promotor de MYC (Zou X et al., 1997). La expresión regulada de MYC es crítica para la proliferación celular controlada, mientras que la expresión des-regulada caracteriza la activación oncogénica y es frecuente en células derivadas de tumores. Por ello, dependiendo del contexto celular, MYC podría ser una señal proliferativa o apoptótica (Bissonnette RP et al., 1992; Evan GI et al., 1992), y en células de LMC su función apoptótica podría estar contrarrestada por otros mecanismos como la vía de la PI-3K (Deininger MWN et al., 2000).

78
4.3 CÉLULA MADRE LEUCÉMICA (Ph+, BCR-ABL+)
4.3.1 Origen, fenotipo y morfología La LMC es un desorden mieloproliferativo, en el cual las células progenitoras en varios estados de maduración se expanden y son liberadas a sangre periférica de forma prematura, asentándose en locaciones extramedulares. Se propone que la célula que da origen a la LMC es una célula madre pluripotente, capaz de originar las diferentes células del sistema hematopoyético. Esto se ha podido confirmar mediante el uso de citogenética, hibridización in situ por fluorescencia (FISH), y marcadores de expansión clonal ligados al cromosoma X (Fialkow PJ et al., 1977; Raskind WH et al., 1987; Takahashi N et al., 1998). Estos estudios han mostrado que células eritroides, megacariocitos, y precursores de células B y T, son producidos comúnmente por el clon neoplásico. Por otro lado, el gen BCR-ABL fue encontrado en células endoteliales de un paciente con LMC (Gunsilius E et al., 2000). Esto podría ser un indicio de que la célula madre mutada causante de la enfermedad, es una célula más temprana capaz de dar origen no sólo a células hematopoyéticas, sino a otro tipo de células como las endoteliales. La translocación del cromosoma Ph y subsiguiente expresión de la tirosina quinasa BCR-ABL es el evento inductor de la LMC (Daley GQ et al., 1990). Sin embargo, las células malignas Ph+ no pueden ser distinguidas morfológicamente de las células normales y es por ello que el diagnóstico definitivo de la LMC está basado en la demostración citogenética o molecular del cromosoma Ph o del gen BCR-ABL, presente en células sanguíneas o de médula ósea. Asimismo, debido a que las células Ph+, BCR-ABL+ se diferencian de forma normal, el análisis de los cambios en marcadores celulares como CD34, CD38, CD71 o Thy-1, que acompañan este proceso en las células normales, no permite distinguir entre células Ph+ y Ph- en uno u otro estado de diferenciación (Petzer AL et al., 1996; Eaves C et al., 1993; Udomsakdi C et al., 1992). Sin embargo, se ha demostrado que células CD34+ que carecían del marcador HLA-DR estaban altamente enriquecidas en células normales (Ph-, BCR-ABL-), a diferencia de células CD34+HLA-DR+, obtenidas de médula ósea de pacientes con LMC (Verfaillie CM et al., 1992). Aunque se ha demostrado que la presencia del gen BCR-ABL es necesario no solo para inducir sino para mantener la LMC en diferentes modelos animales (Huettner CS et al., 2000), también se han podido detectar células normales (Ph+, BCR-ABL+) en pacientes en fase crónica de LMC. Estudios con métodos como FISH, RT-PCR o citogenética convencional, han mostrado que en estadíos tempranos de la enfermedad, las células iniciadoras de cultivo a largo plazo (LTC-IC), residuales normales (Ph-, BCR-ABL-) pueden sobrepasar el número de LTC-IC leucémicas (Ph+, BCR-ABL+), pero en estadíos más tardíos de la enfermedad,

79
la mayoría de células son derivadas del clon leucémico (Guyotat D et al., 1999; Coulombel L et al., 1983). Esto podría indicar que tal vez la translocación no sea el evento iniciador de la LMC, o que en caso de serlo, los clones Ph+ desplazan a la hematopoyesis normal más no destruyen las células madre hematopoyéticas normales. Por lo tanto, en la médula ósea o en sangre periférica de pacientes con LMC en fases tempranas todavía puede ser posible encontrar remanentes de células progenitoras y células madre normales, esto debido a las anormalidades en la regulación del ciclo celular, proliferación, adhesión y apoptosis. También se han detectado transcritos de BCR-ABL en personas normales que tal vez nunca lleguen a desarrollar LMC (Bose S et al., 1998; Biernaux C et al., 1995). Este hallazgo plantea la posibilidad de que, aún siendo la translocación t(9;22) el evento iniciador de la LMC, tal vez no sea el único evento que desencadena el desarrollo de la enfermedad y otra serie de anormalidades cromosómicas sean necesarias antes de que se manifieste la leucemia.
4.3.2 Propiedades alteradas
4.3.2.1 Proliferación. Se puede decir que hay una perturbación en la regulación del ciclo celular de las células madre Ph+, BCR-ABL+ debido a la detección de cambios que afectan a diferentes poblaciones de estas células neoplásicas. Esta detección ha sido posible gracias a ensayos con LTC-ICs y células formadoras de colonias (CFCs) en los cuales se define su alto potencial proliferativo o su habilidad multipotente de diferenciarse. Las poblaciones de células que tienen esta última característica se conocen como unidades formadoras de colonia granulocito/ eritrocito/ macrófago/ megacariocito o UFC-GEMMs (Holyoake TL et al., 2002). Sin embargo ha sido difícil el estudio de estas poblaciones ya que son muy escasas en sangre periférica y médula ósea de pacientes con LMC, porque carecen de un fenotipo único que diferencie a estas células neoplásicas, y porque de todas formas existen remanentes de células normales. Se han utilizado diferentes metodologías con el fin de establecer cómo está alterada la proliferación en las células madre BCR-ABL+ y para determinar en qué estado del ciclo celular se encuentran estas células. El primer método se refiere a la exposición de las células a la actividad de un agente que es tóxico específicamente para células que se encuentren en fase S del ciclo celular, (ej. 3H-timidina o hidroxiúrea). Esta exposición se realiza durante 15-30 minutos. Al principio se utilizó esta metodología para demostrar que las CFCs con alto potencial proliferativo en la médula de adultos normales, están quiescentes, al igual que las CFCs que normalmente se encuentran en la sangre. En contraste, estos mismos estudios aplicados a CFCs de pacientes con LMC mostraron que la mayoría de CFCs leucémicas, sin importar si están en sangre o médula, pueden

80
hallarse en cualquier estado del ciclo (Eaves CJ et al., 1987). Otros estudios con tiempo de exposición mayor a 3H-timidina, mostraron que en adultos normales la mayoría de LTC-ICs están quiescentes, mientras que en pacientes con LMC, la mayoría de sus LTC-ICs leucémicas están proliferando y se encuentran en fase S del ciclo celular (Ponchio L et al., 1995; Ponchio L, Cashman J et al., 1995; Holyoake T et al., 1999). Una segunda metodología para determinar en qué estado del ciclo celular se encuentran las células primitivas hematopoyéticas, recurre al uso de la tecnología de clasificación de células activadas por fluorescencia o FACS (del inglés “fluorescence activated cell sorting”) para aislar células viables en estado G0, G1 y S/G2/M, basada en las diferencias de sus contenidos de ADN y ARN visualizado mediante tinción con Hoechst 33342 y Pironina Y. Estos experimentos confirmaron que hay un aumento en la proporción de células leucémicas primitivas, tanto CFCs como LTC-ICs, obtenidas de pacientes en fase crónica de LMC, que se encuentran en fase S/G2/M del ciclo celular (65). Estos estudios también probaron que dentro de esta misma población de células leucémicas hay una fración detectable de células en G0. En estudios in vivo también se ha observado que relativamente pocas células leucémicas (105) CD34+ en G0 respondían después de ser transplantadas a ratones NOD/SCID, produciendo números detectables de progenie leucémica (Holyoake T et al., 1999), lo cual indica que la quiescencia de estas células leucémicas transplantables es reversible in vivo, resaltando la importancia de dichas células en el mantenimiento y diseminación de la enfermedad en los pacientes. La tercera metodología usada se basa en la comparación de la longitud de los telómeros en los granulocitos de pacientes con LMC y de individuos normales. Debido a que la longitud del telómero en células somáticas disminuye con cada división celular, éste puede servir como indicador de la historia mitótica y como un indicador de senescencia. La actividad de la telomerasa juega un papel importante en la elongación del telómero, por ello, esta enzima normalmente se expresa en mayores cantidades en células primitivas, y su sobreexpresión puede aumentar el número de divisiones celulares y contribuir a la transformación maligna. Al medir la longitud de los telómeros de granulocitos de individuos normales de diferentes edades, se ha encontrado que las células madre de adultos se dividen en promedio una vez cada 1-2 años (Rufer N et al., 1999). Muchos estudios han reportado que las células de LMC tienen telómeros más cortos (~1Kb) que los de los granulocitos normales en la médula ósea (Engelhardt M et al., 2000, Brummendorf TH et al., 2000; Boultwood J et al., 1999). Mediante métodos cuantitativos se ha observado una pérdida mayor en la longitud del telómero en células de fase blástica de LMC (Boultwood J 2000). Estos estudios sugieren que las células leucémicas BCR-ABL+ se dividen muchas más veces que las células normales, y que la longitud del telómero puede ser un punto crítico en la transición de la fase crónica a la crisis blástica.

81
Todas las investigaciones anteriores aportan evidencias de que las células madre en la LMC se encuentran más activas dentro del ciclo celular que las células Ph-. Esto puede deberse a mecanismos como la activación de vías asociadas con respuestas mitogénicas como son RAS, PI-3K, JAK-STAT, y MAP-K (Cortez D et al., 1997). También puede ser consecuencia de la producción autocrina de factores de crecimiento, en especial IL-3 y factor estimulante de colonia granulocitica (FEC-G) por parte de las células leucémicas (Jiang X et al., 1999; Zhang X et al., 1998; Holyoake TL et al., 2001). Además, se ha encontrado que BCR-ABL físicamente se asocia a c-kit, el receptor del factor de célula madre (SCF) (Hallek M et al., 1996). Se sugiere que este factor puede permitir o tal vez inducir la proliferación de células primitivas Ph+, y que esta respuesta anormal puede deberse a la activación constitutiva de una o más vías de señalización (Moore S et al., 1998). La respuesta alterada de las poblaciones de células madre y células progenitoras a factores que promueven o inhiben el crecimiento, puede contribuir a la expansión celular en la fase crónica de la LMC. Más que proveer un estímulo mitogénico directo, BCR-ABL puede influir en los mecanismos de control de proliferación y supervivencia estimulando a las citoquinas y a los receptores de factores de crecimiento (Kabarowski JHS et al., 2000).
4.3.2.2 Adhesión. Normalmente, las células progenitoras hematopoyéticas se adhieren a la matriz extracelular o a citoquinas inmobilizadas que regulan el crecimiento. Esta adhesión está mediada por receptores de la superficie celular sobre las células progenitoras, en especial por integrinas. Las integrinas son glicoproteínas de la superficie celular compuestas de dos subunidades α y β asociadas no covalentemente. Mientras la cadena � determina la especificidad del ligando, la cadena β inicia vías de transducción de señal después de la unión al ligando. Se ha encontrado que células progenitoras primitivas leucémicas (BCR-ABL+) son defectuosas con respecto a su habilidad de interactuar con células del estroma de la médula ósea, a través de moléculas de adhesión celular y sus ligandos (Gordon MY et al., 1987; Verfaillie CM et al., 1992). La adherencia defectuosa de progenitores hematopoyéticos de LMC a los elementos del estroma de la médula ósea puede facilitar su liberación a la sangre y se sugiere que el contacto con células del estroma puede reducir la probabilidad de que células progenitoras primitivas quiescentes, entren al ciclo celular de forma activa (Gordon MY et al., 1997; Gordon MY et al., 1999). Se cree que estas deficiencias en la adherencia al estroma medular son causadas por un problema en la función más no en la expresión de los receptores de adhesión celular, ya que se ha encontrado que en progenitores de LMC, los niveles de expresión de receptores de adhesión de la superficie celular como los receptores de integrinas α4β1 y α5β1, o receptores de CD44 que en progenitores normales pueden cooperar con α4β1 para establecer la adhesión a la fibronectina, son similares a los niveles encontrados en células progenitoras normales (Petzer

82
AL et al., 2003). Además, algunos receptores de integrina como α2β1 y α6β1, los cuales pueden interactuar con laminina y colágenos, se expresan sobre sub-poblaciones de progenitores de LMC pero no sobre células normales (Lundell BI et al., 1997). Esto sugiere que la liberación prematura de células progenitoras en la LMC se debe por un lado a una pérdida de interacciones adhesivas con el estroma y/o con la fibronectina, y por otro lado a la adquisición de interacciones adhesivas con componentes de la membrana basal. La expresión de la molécula de adhesión LFA-3 (CD58) sobre células progenitoras de LMC, también es deficiente pero puede restablecerse a través de tratamiento con interferón-α (82), el cual también puede reestablecer la adhesión al estroma de CFCs y LTC-ICs, dependiendo de la dosis (Bhatia R et al., 1994). BCR-ABL también es capaz de inducir anormalidades citoesqueléticas severas en células hematopoyéticas primitivas, como aumento en la movilidad espontánea, alteraciones en la membrana, formación de largas extensiones de actina (filopodios) y acelerada tasa de alargamiento y retracción de pseudopodios sobre superficies cubiertas de fibronectina (Salgia R et al., 1997).
4.3.2.3 Apoptosis. La supresión de vías de muerte celular programada o apoptosis, es un mecanismo que se ha asociado a la patogénesis de la LMC (Bedi A et al., 1994). A pesar de que existen estudios que indican que la expresión de BCR-ABL en líneas celulares p210BCR-ABL, proteje de la apoptosis inducida por estrés físico y químico (Akhtar AJ et al., 1995; Cortez D et al., 1995; Nishii K et al., 1996), también hay reportes de respuestas apoptóticas normales de progenitores de LMC en ausencia de factores de crecimiento y con tratamiento genotóxico (Albrecht T et al., 1996; Amos TA et al., 1995). Múltiples mecanismos contribuyen a generar resistencia contra la apoptosis, entre ellos se encuentran mecanismos relacionados con proteínas de la familia BCL-2. BCL-XL y BCL-2 son proteínas homólogas con función anti-apoptótica que protegen contra la muerte celular programada en respuesta a diferentes estímulos. Otros miembros de la familia BCL-2 son BAX y BAD, dos proteínas pro-apoptóticas que inducen la muerte celular uniéndose a BCL-2 y a BCL-XL. La fosforilación y activación de la vía PI-3K es una de las vías por la cual BCR-ABL ejerce su efecto antiapoptótico ya que la activación de la PI-3K da como resultado la fosforilación e inactivación de BAD (Franke TF et al., 1997; Neshat MS et al., 2000). Adicionalmente, la vía RAS puede conllevar a la activación de BAD debido a que, después de activar a AKT, RAF-1 puede fosforilar a BAD sobre dos de sus residuos de serina. (Figura 4.4) (Cortez D et al., 1996; Zha J et al., 1996). También se ha encontrado en células que tienen el oncogen BCR-ABL, un aumento en la expresión de BCL-XL, que contribuye a una disminución en la apoptosis (Horita M et al., 2000; de Groot RP et al., 2000). A través de estas vías, BCR-ABL puede bloquear la liberación de citocromo C y la activación de las

83
caspasas, especialmente de la caspasa 3 (Amarante-Mendes GP, Naekyung Kim C et al., 1998; Dubrez L et al., 1998). El factor de transcripción NF-κB se transloca al núcleo después de la activación celular y su actividad generalmente se asocia con la protección contra la apoptosis. Hay evidencia de que NF-κB se activa en modelos transgénicos de LMC, esta activación es otro mecanismo que puede proteger contra la apoptosis (Reuther JY et al., 1998). También es posible que BCR-ABL inhiba la apoptosis a través de la regulación negativa de la proteína de unión a la secuencia consenso del interferón (ICSBP) (Deininger MWN et al., 2000), ya que se ha encontrado que ratones noqueados en esta proteína desarrollan un síndrome mieloproliferativo, y que células progenitoras hematopoyéticas de ratones ICSBP-/- tienen una alteración en su respuesta a citoquinas (Hao SX et al., 2000; Holtschke T et al., 1996; Scheller M et al., 1999). Se ha observado que células progenitoras de LMC son más sensibles a la radiación comparadas con células normales (Santucci MA et al., 1993), por lo tanto a pesar de que la disminución en la apoptosis es una característica de la LMC, este mecanismo por sí solo no es suficiente para la transformación y progresión de la enfermedad y puede ser que para esto sean requeridas anormalidades adicionales como por ejemplo, pérdida de un gen supresor de tumor (Gordon MY et al., 1999; Shet AS et al., 2002).
4.4 TRATAMIENTOS El primer agente quimioterapéutico utilizado contra la LMC fue el busulfan (Galton DAG 1953). Luego, en 1966 se reportó por primera vez el uso de hidroxiúrea (Kennedy BJ et al., 1966) y aún en la actualidad se siguen usando estos dos agentes para controlar la fase crónica de la LMC. A mediados de los 1980s fue implementado el uso de interferón alfa (Talpaz M et al., 1986; Talpaz M et al., 1987) y éste demostró prolongar la supervivencia, retrasar la transición a fase acelerada y lograr la remisión citogenética en algunos pacientes. El transplante de células madre para el tratamiento de pacientes jóvenes con hermanos compatibles como donantes, se empezó a utilizar en la misma década, y más tarde fue introducido el transplante autólogo a partir de células madre de sangre periférica del mismo paciente luego de realizar quimioterapia. En 1996, fue descrito un nuevo compuesto llamado Imatinib mesilato (STI571) diseñado específicamente para inhibir la proteína tirosina quinasa ABL (Druker BJ et al., 1996), dos años después este compuesto fue aprobado por la FDA para el tratamiento de la LMC logrando mejores respuestas que la terapia con inerferón-α, aunque su eficiencia aún es materia de debate.

84
4.4.1 Quimioterapia citotóxica Durante la fase crónica de la LMC se utiliza terapia citorreductora en la mayoría de los pacientes para evitar complicaciones trombóticas producto de los altos niveles de células blancas en la sangre. Para ello se puede utilizar un solo agente como la hidroxiúrea o el busulfán. La hidroxiúrea inhibe la enzima ribonuleótido reductasa, interfiriendo con la síntesis de ADN. El uso de esta droga puede generar aplasia transitoria que usualmente es reversible a los pocos días de descontinuar el tratamiento. Otras complicaciones son ulceraciones orales y lesiones en las uñas, y si es administrada en altas dosis, puede provocar náuseas, vómito y diarrea (Hoffman R et al., Chronic Myelogenous Leukemia, 2000). El busulfán es un agente alquilante que tiene efecto inmunosupresor selectivo sobre la médula ósea. Sin embargo, de acuerdo con el Séptimo Reporte Anual sobre Carcinogénesis (PB95-109781, 1985), el busulfán está en la lista de agentes carcinógenos (The Merck Index 1996). A diferencia de la hidroxiúrea, la terapia con busulfán puede provocar aplasia prolongada y en ocasiones irreversible. Otros efectos tóxicos incluyen aspermia, amenorrea, y un síndrome pulmonar tardío caracterizado por tos, fiebre, infiltrados pulmonares, y falla respiratoria (Hoffman R et al., Chronic Myelogenous Leukemia, 2000). El 99% de los pacientes tratados con hidroxiúrea o busulfán tiene remisiones hematológicas parciales o completas (Hehlmann R et al., 1994). Se ha demostrado la superioridad de la hidroxiúrea sobre el busulfán debido a que la duración media de la fase crónica y la supervivencia promedio son mejores en el tratamiento con hidroxiúrea que con busulfán (Hehlmann R et al., 1994). Otros agentes quimioterapéuticos usados incluyen la homoharringtonina (113) y la citarabina, ésta última provoca daño en el ADN ya sea inhibiendo su síntesis, interfiriendo en su reparación o incorporándose directamente a él (Sokal JE et al., 1987; Robertson MJ et al., 1993; Guilhot F et al., 1997). Sin embargo, ningún agente quimioterapéutico tiene efectos significativos en la progresión hacia la crisis blástica y es bastante inusual lograr la eliminación completa de células Ph+ de la médula, por lo tanto deben ser considerados solamente como tratamientos paliativos.
4.4.2 Interferón alfa Los interferones son glicoproteínas producidas por células eucariotas en respuesta a estímulos antigénicos como los que ocurren en las infecciones virales y enfermedades malignas. Tienen efectos pleiotrópicos, incluyendo actividades antivirales, inmuno-moduladoras, antiproliferativas y antiangiogénicas. El interferón alfa (IFN-α) es el que más se ha usado en el tratamiento de tumores malignos sólidos y hematológicos (Faderl S et al., 1999).

85
El mecanismo de acción exacto del IFN-α no es muy claro. Estudios in vitro sugieren que este agente tiene efectos antiproliferativos, debido a que es capaz de reestablecer la adhesión defectuosa en la interacción entre las células progenitoras y el estroma medular, posiblemente a través de la restauración de la función de la integrina-β1 (Bhatia R et al., 1996) y de la expresión de la molécula de adhesión celular LFA-3 (CD58) (Upadhyaha G et al., 1991). Además, promueve la actividad de células citotóxicas NK y modula, a través de sistemas locales paracrinos, la liberación de factores de crecimiento en el ambiente del estroma, con un incremento en las citoquinas inhibidoras como el receptor antagonista de la IL-1, el factor transformante de crecimiento (TGF), y MIP-1α (Hoffman R et al., Chronic Myelogenous Leukemia, 2000). El IFN-α también puede inducir la apoptosis a través de la estimulación del sistema Fas-ligando/Fas-receptor (Selleri C et al., 1997). Las remisiones citogenéticas inducidas por el interferón son durables en una gran proporción de pacientes, algunas veces incluso después de que se descontinúa el tratamiento con este agente (Bonifazi F et al., 2001). Se han realizado diferentes estudios que confirman la ventaja del uso del IFN-α sobre la quimioterapia convencional usada en pacientes con fase crónica de LMC y se ha encontrado que, en contraste con otros tratamientos con drogas, el IFN-α produce respuestas citogenéticas mantenidas (22-24 meses) hasta en un tercio de los pacientes, y tiempo promedio de respuesta hematológica de 6-8 meses (Guilhot F et al., 1997; Talpaz M et al., 1991; Italian Cooperative Study Group on Chronic Myeloid Leukemia, 1994; Ohnishi K et al., 1995; Hehlmann R et al., 1994). Un análisis reciente confirma la ventaja de supervivencia para pacientes con fase crónica de LMC tratados con IFN-α (CML Trialists Collaborative Group, 1997). Aunque con RT-PCR, todavía es posible detectar ARNm de BCR-ABL (Wandl UB et al., 1994), estas remisiones a largo plazo son importantes para una cura de la enfermedad a nivel biológico aunque no molecular. El debate acerca del papel preciso que tiene el interferón y su interacción con otras terapias incluyendo el transplante de células madre, continúa. Además, a pesar de su valor para los pacientes en fase crónica de LMC, la terapia con IFN-α puede llegar a ser hasta 200 veces más costosa que la quimioterapia convencional (Hoffman R et al., Chronic Myelogenous Leukemia, 2000).
4.4.3 Imatinib mesilato (ST1571) Los inhibidores de transducción de señal (STI), son compuestos que bloquean o evitan que una proteína ejerza su rol en una vía oncogénica. Debido a que la transformación oncogénica de las células de LMC es llevada a cabo gracias a la actividad constitutiva de la proteína tirosina-quinasa BCR-ABL, lo más lógico sería

86
pensar que inhibiendo esta actividad es posible silenciar la oncoproteína. Para ello se han usado varios tipos de inhibidores de proteínas tipo tirosina-quinasa. Los primeros que se usaron fueron los aislados de fuentes naturales como el isofalvonoide genisteína (Carlo-Stella C et al., 1996) y el antibiótico herbimicina A, derivado de Streptomyces hygroscopicus. Luego, se desarrollaron compuestos sintéticos mediante el diseño de estructuras químicas capaces de competir con el centro catalítico de la quinasa (Deininger MWN et al., 2000). Dentro de estos compuestos, el más prometedor es Imatinib Mesilato (STI571), conocido comercialmente como Glivec ® o Gleevec ®. Fue desarrollado en 1996 por Druker y sus colegas (Druker BJ et al., 1996) con el fin de inhibir específicamente la actividad de la tirosina quinasa ABL y así conducir a las células Ph+ a la apoptosis. El efecto de este compuesto fue confirmado por otros grupos mediante estudios in vitro en líneas celulares (Deininger M et al., 1997; Gambacorti-Passerini C et al., 1997) y en 1998 se llevó a cabo la primera fase de un estudio clínico con pacientes en fase crónica de LMC resistentes a la terapia con IFN-α que fueron tratados con imatinib mesilato (Druker BJ, TalpazM, et al., 2001). Este estudio mostró que la droga tenía baja toxicidad y era efectiva, ya que en todos aquellos pacientes tratados con dosis de 300mg/d se logró la remisión hematológica completa después de cuatro semanas de tratamiento, y en más del 30% se lograron remisiones citogenéticas mayores o completas. Lo más importante de estas respuestas es que eran duraderas en el 96% de los casos. Asímismo, esta droga fue utilizada para tratar pacientes con LMC en crisis blástica y con la misma dosis de 300mg/d se lograron respuestas hematológicas en el 55% de pacientes con fenotipo mieloide y en el 70% de pacientes con fenotipo linfoide. Sin embargo, en contraste con los pacientes tratados en fase crónica, casi el 50% de pacientes con enfermedad mieloide y todos menos un paciente con enfermedad linfoide recayeron entre 42 y 193 días después de haber iniciado el tratamiento con Imatinib mesilato (Druker BJ, Sawyers CL, et al., 2001). Una segunda fase de los estudios clínicos iniciada a mediados de 1999 confirmó los resultados obtenidos en la primera fase, y además, en estos estudios fueron incluidos pacientes en fase acelerada de LMC, en los cuales se observaron resultados que se encontraban en el intermedio de los resultados obtenidos para pacientes en crisis blástica mieloide y en fase crónica (Deininger MWN et al., 2003). En junio de 2000, como parte de la tercera fase de estudios clínicos, se inició un estudio aleatorio de comparación entre Imatinib Mesilato vs. INF-α en combinación con citarabina, para tratar pacientes recién diagnósticados con LMC en fase crónica. Los resultados obtenidos mostraron que Imatinib es ampliamente superior con respecto a las tasas de remisión hematológica y remisión citogenética mayor y completa. Lo más importante es que se observó una diferencia altamente significativa en la tasa de progresión a fase acelerada o crisis blástica (O’Brien SG et al., 2003). Luego de estos resultados, en diciembre de 2002 la FDA (Food and Drug Administration) aprobó al Imatinib Mesilato como el tratamiento de primera línea para LMC recién diagnosticada, y aunque en general esta droga es bien

87
tolerada y el nivel de toxicidad es bajo, es posible que se presenten efectos secundarios en fases avanzadas de la enfermedad, que pueden ser de tipo hematológico como la mielosupresión, o no hematológico como: edema y retención de fluídos, efectos gastrointestinales, reacciones en la piel, artralgia, mialgia, dolor de huesos y toxicidad hepática (Deininger MWN et al., 2003).
4.4.3.1 Mecanismo de acción. Las quinasas son activadas por la fosforilación de los residuos tirosina o serina/treonina. En el caso de ABL, la tirosina 393 (Y393) está fosforilada y permite que los sustratos se unan. En la forma inactiva de ABL, Y393 no está fosforilada e imita al sustrato mediante la formación de un puente de hidrógeno con la asparagina 363. En esta conformación, la “boca” de la quinasa está cerrada, evitando así que los sustratos se unan (Deininger MWN et al., 2003). Imatinib funciona como un inhibidor competitivo de la unión del ATP. El compuesto se une a la forma inactiva de ABL, en donde contacta a 21 aminoácidos y provoca un cambio que le confiere mayor especificidad. Una consecuencia de este cambio inducido es la formación de una especie de jaula hidrofóbica que rodea a la molécula de imatinib, encerrándola por las interacciones de van der Waals con los residuos tirosina 253, leucina 370 y fenilalanina 382. Además, imatinib forma varios puentes de hidrógeno con el dominio quinasa de la proteína. Los residuos metionina 318, treonina 315, metionina 290, glutamina 286, lisina 270 y asparagina 381, junto con las moléculas de agua, forman una red de puentes de hidrógeno alrededor de la molécula de imatinib (Deininger MWN et al., 2003). La proteína ABL fosforilada en el aminoácido tirosina 393 es mucho menos sensible a imatinib ya que esta fosforilación estabiliza la forma activa de la quinasa. BCR-ABL está fosforilada constitutivamente por tirosina y por ello en esta conformación sería incapaz de unirse a imatinib. Sin embargo, gracias a experimentos con líneas celulares BCR-ABL+ tratadas con imatinib, se sabe que la exposición al compuesto resulta en la inhibición de la actividad quinasa en unos minutos. Esto indica que hay un cambio rápido entre la forma fosforilada y la forma no fosforilada, la cual es capaz de unirse a imatinib (Deininger MWN et al., 2003). Aunque es extraño observar resistencia al imatinib en pacientes tratados en fase crónica de la enfermedad, si es posible que se desarrolle en la mayoría de pacientes tratados en fases avanzadas debido a la complejidad genética que presenta la enfermedad en este punto por la cantidad de mutaciones acumuladas que activan otras vías además de aquellas mediadas por BCR-ABL. Esta resistencia puede ser el producto de mutaciones puntuales en el sitio de unión de BCR-ABL a imatinib. Las variaciones en el sitio activo de la enzima además de generar resistencia contra la droga, preservan la unión al ATP, la catálisis y por lo tanto la actividad transformante de la oncoproteína (Nardi V et al., 2003).

88
4.4.4 Transplante de células madre El transplante alogénico, es decir proveniente de un donante diferente al paciente pero de la misma especie de éste, de células madre hematopoyéticas, se ha convertido en la mejor opción para tratar personas enfermas con LMC. En este, hay una infusión de CMHs ya sea de médula ósea o de sangre periférica del donante movilizadas por citoquinas (como factor estimulante de colonia granulocito [G-CSF]), después de haber realizado quimioterapia o radioterapia intensiva. Sin embargo, este procedimiento presenta un alto riesgo de morbilidad y mortalidad asociado a diferentes factores que hacen que su disponibilidad esté limitada solamente a algunos de los pacientes que presentan la enfermedad. Se han identificado cinco factores de riesgo que pueden tener un impacto significativo en las tasas de supervivencia luego del transplante y en las complicaciones y las tasas de mortalidad asociadas a éste (Gratwohl A et al., 1998). El primer factor es el grado de histocompatibilidad del paciente con el donante. Para realizar el transplante alogénico de células madre y evitar el rechazo inmune, es necesario encontrar un donante cuyas células sean compatibles en su antígeno HLA con las del receptor del transplante. Dicho donante puede ser un familiar o hermano, o puede ser una persona que no sea de la familia del paciente. Sin embargo, aunque la respuesta al transplante es superior cuando el donante es un hermano con HLA idéntico, algunas poblaciones de pacientes obtienen resultados similares ya sea con un donante hermano o con un donante que no es de la familia (Weisdorf DJ et al., 2002). Se ha encontrado que las probabilidad de desarrollar la enfermedad injerto-versus- huésped (GVHD) después del transplante aumentan cuando el donante no es de la familia, mientras que el riesgo de recidivas hematológicas es bajo tanto en donantes hermanos con HLA idéntico como en donantes no familiares (Weisdorf DJ et al., 2002). Después de tantos años de experiencia en transplantes de médula ósea en humanos, muchos estudios han descrito resultados de transplantes compatibles y no compatibles para HLA (McGlave PB et al., 2000; Szydlo R et al., 1997; Balduzzi A et al., 1995), y se ha demostrado que la incompatibilidad serológica HLA-A o –B entre donante y receptor tiene un efecto adverso independiente sobre la supervivencia (Davies SM et al., 1995; Spencer A et al., 1995; Devergie A et al., 1997). Alrededor del mundo se han creado bases de datos centralizadas como el registro del Grupo Europeo para el Transplante de Sangre y Médula (EBMT) (creado en los 1970s), o el Programa Nacional de Donantes de Médula (NMDP) de los E.U, que han permitido llevar un registro de donantes con el fin de buscar aquellos que tengan un alto grado de histocompatibilidad con el paciente y así aumentar las

89
probabilidades de recibir un transplante efectivo contra la enfermedad que no aumente el riesgo de muerte para el paciente. Sin embargo, un número indefinido de pacientes sucumben a la enfermedad mientras esperan por un donante compatible disponible. Además cerca de la mitad de búsquedas iniciadas fallan en encontrar el donante adecuado y la cifra es mayor para pacientes de minorías étnicas o raciales. Inlcuso, si un donante es identificado por ser compatible en su HLA, no todos los donantes están disponibles cuando son necesitados, ya sea porque no es posible contactar al donante por un cambio de residencia o de nombre, por pérdida de motivación con el paso del tiempo, porque ya no califican debido al estado de salud o a la edad, o simplemente porque han muerto (Grewal SS et al., 2003). Otros factores de riesgo que se han identificado para el transplante alogénico de células madre son: la fase de la enfermedad al momento del transplante, el intervalo de tiempo desde el momento del diagnóstico hasta el transplante, la edad del receptor del transplante, y el sexo del donante. Aproximadamente sólo la mitad de los pacientes transplantados en fase acelerada de LMC tienen un tiempo largo de supervivencia, sin embargo este número puede aumentar si el transplante es realizado antes de un año desde el diagnóstico de la enfermedad (Clift RA et al., 1994). Igualmente, se ha reportado que el retraso de más de un año entre el diagnóstico y el transplante ya sea con donante familiar o no-familiar, compromete el resultado clínico (van Rhee F et al., 1997). Además de esto, los efectos benéficos son más evidentes en pacientes jóvenes transplantados de manera temprana en el curso de la enfermedad, ya que más del 70% de este grupo de pacientes sobrevive durante largo tiempo sin la enfermedad, y en algunos estudios a largo plazo, la supervivencia promedio para los pacientes más jóvenes después del transplante alogénico se aproxima a 10 años (Enright H et al., 1996; Gratwohl A et al., 1993). También se ha encontrado que el sexo del donante y del receptor del transplante puede afectar la tasa de supervivencia y de recaída del paciente (Weiden PL et al., 1981).
4.4.4.1 Efecto injerto vs. leucemia. A pesar de que en transplantes provenientes de un donante no-familiar la mortalidad asociada al transplante es alta y el tiempo de supervivencia es bajo, la tasa de recaídas hematológicas o citogenéticas en estos pacientes también es baja. Esto se debe al efecto injerto vs. leucemia asociado a la enfermedad injerto-versus-huésped (GVHD del inglés Graft-versus-Host Desease), que se desarrolla por la alo-reactividad entre el donante y el receptor y que ha demostrado tener efectos anti-leucémicos en los transplantes alogénicos de células madre en pacientes con LMC (Weiden PL et al., 1981). Un transplante alogénico de células madre requiere de un régimen terapéutico cito-reductor previo al transplante con el fin de erradicar las células leucémicas y

90
así crear un espacio en la médula ósea mediante quimioterapia (usualmente con busulfán o ciclofosfamida) o radioterapia. Estos regímenes producen efectos tóxicos en el paciente que podrían llegar a ser reducidos gracias a la introducción de una nueva metodología para el transplante alogénico llamada “transplante sin mielo-ablación”, “transplante alogénico de intensidad reducida” o “transplante de células madre mini-alogénico”. Este procedimiento involucra evitar exponer al paciente a regimenes preparativos altamente tóxicos mediante reducciones substanciales en la dosis de las drogas citotóxicas o de la radiación usada, haciendo énfasis en el uso de agentes inmunosupresores. Al mismo tiempo, el número de células madre hematopoyéticas y linfocitos transfundidos al paciente es maximizado para asegurar la inserción y un óptimo efecto injerto-versus-leucemia. Es decir, se usan los linfocitos T del donante que van en el injerto para combatir las células malignas del paciente, ya que se ha encontrado que la reducción en el pool de células T del injerto está asociada a un menor riesgo de GVHD pero a un incremento en el riesgo de recidividas luego del transplante (Goldman JM et al., 1988). Con los protocolos que se han desarrollado se ha comprobado buena tolerancia al transplante, remisión molecular durable y probabilidad de supervivencia sin la enfermedad de más del 80% en los pacientes que se han sometido a este tipo de transplante (Uzunel M et al., 2003; Or R et al., 2003; Carella AM et al., 2000), aunque todavía es muy prematuro hablar de resultados a largo plazo. La principal ventaja del transplante mini-alogénico de células madre es que, al no tener que someter al paciente a regímenes tan tóxicos, este procedimiento puede ser aplicado a personas mayores, para quienes estaba restringido un transplante convencional.
4.4.4.2 Transplante autólogo. Debido a que en la médula ósea y en sangre periférica de pacientes con LMC es posible encontrar poblaciones de células progenitoras Ph-, BCR-ABL- junto con células Ph+, BCR-ABL+ (Guyotat D et al., 1999; Coulombel L et al., 1983; Verfaillie CM et al., 1996), y dada la poca disponibilidad de donantes HLA-compatibles, y la toxicidad asociada a transplantes alogénicos de células madre, el transplante autólogo ha sido empleado como un tratamiento alternativo en el cual se utilizan las células madre del paciente para ser autotransplantadas luego de que a éste se le ha administrado quimioterapia citotóxica. Se cree que la eficiencia del transplante autólogo se debe a la reducción en el tamaño de la población de células leucémicas y a que los progenitores malignos de LMC están en desventaja con sus contrapartes normales (Bhatia R et al., 1997). Antes del transplante, se realiza una selección negativa o “purga” de la médula con el fin de enriquecer el injerto autólogo de células madre en progenitores hematopoyéticos benignos. Para ello, se han utilizado técnicas ex-vivo en las que se pueden usar agentes anti-leucémicos como el IFN-γ o derivados de la

91
ciclofosfamida (Carlo-Stella C et al., 1994; McGlave P et al., 1994), o cultivos a largo plazo (LTC), en los cuales se ha observado pérdida de progenitores primitivos Ph+ y enriquecimiento de Ph- (Barnett MJ et al., 1994). Estos estudios indican que hay remisión citogenética en los pacientes pero esta remisión es de corta duración, por lo que se podría llegar a pensar que el proceso de selección puede estar asociado a pérdida o daño de células madre (Bhatia R et al., 1997). También es posible utilizar técnicas in vivo para la selección de células benignas de médula. En este tipo de técnicas, se realiza tratamiento con altas dosis de quimioterapia y factor estimulante de colonia granulocítica (FEC-G), seguido de la recolección de células madre de sangre periférica para la re-infusión autóloga (Carella AM et al., 1994; Talpaz M et al., 1995). A pesar de las diferencias en los resultados, parece ser que la selección in vivo puede reducir la carga leucémica en el autoinjerto, y se asocia con mayor obtención de células Ph- después del transplante. Sin embargo, también es posible detectar células BCR-ABL+ (Deisseroth AB et al., 1994) incluso en personas con remisión citogenética completa, lo cual indica que estos procedimientos no eliminan por completo las células leucémicas (Bhatia R et al., 1997). En un análisis retrospectivo, McGlave et al., evaluaron la recuperación de 200 pacientes con LMC que recibieron autotransplante en 8 centros diferentes (McGlave PB et al., 1994). El tiempo promedio de supervivencia en 142 pacientes transplantados durante la fase crónica, acelerada y crisis blástica fue de 42, 35.9, y 4.1 meses, respectivamente. También se confirmó la importancia de factores como la edad, el intervalo de tiempo entre el diagnóstico y el transplante, y la fase de la enfermedad en el resultado del transplante. Sin embargo, el autotransplante solo o en combinación con estrategias de selección efectivas es poco efectivo para lograr remisiones a largo plazo en la mayoría de los pacientes, tal vez porque el efecto injerto-versus-leucemia o la enfermedad injerto-versus-huésped no llegan a desarrollarse. Por lo tanto, es posible que se requiera de tratamiento post-transplante con IFN-α o incluso de otro transplante alogénico para manejar las recaídas (Steegmann JL et al., 1999; Giralt S et al., 1995; Higano S et al., 1997).

92
5. IMPLICACIONES ÉTICAS Y LEGALES DE LA INVESTIGACIÓN CON CÉLULAS MADRE
5.1 EL PROBLEMA El estudio de células madre constituye uno de los mayores retos impuestos para la ciencia en las últimas décadas. El simple hecho de imaginar que a partir de una sola célula podamos llegar a obtener un ser humano completamente formado con características únicas, nos asombra y nos llena de inquietudes pero sobre todo nos entusiasma de manera peligrosa a aventurarnos en la manipulación de una célula de la misma naturaleza con el fin de obtener nuevas células, tejidos y órganos que en un momento dado podrían ser de gran utilidad para darle solución a un sinnúmero de enfermedades que día a día aquejan a más personas en el mundo. Ejemplo de estas enfermedades son las enfermedades degenerativas del sistema nervioso como la enfermedad de Parkinson o la esclerosis múltiple, del corazón como el infarto al miocardio, del hígado como la hepatitis o del páncreas como la diabetes. Sin embargo, los beneficios del desarrollo de tecnologías que permitan lograr mayores avances en este campo también implican ciertos sacrificios. Aunque las células madre pueden provenir de tejido adulto como es el caso de las células madre hematopoyéticas, éstas pueden encontrarse en mayor cantidad y con más facilidad en una fuente más primitiva como el tejido embrionario. Es aquí donde se abre el debate sobre qué tan ético desde el punto de vista biológico y moral es obtener células madre de embriones humanos que no van a llegar a desarrollarse como personas, o dicho de otra forma, ¿qué implicaciones tiene el hecho de usar y destruir embriones humanos como un medio para beneficiar a otros?
5.2 DEFINICIONES BÁSICAS Para entrar a discutir el tema, es necesario dejar claro algunos aspectos científicos básicos como ¿qué es una célula madre?, ¿de dónde puede ser obtenida?, ¿en qué consiste la clonación y con qué fines puede realizarse? Una célula madre es aquella que tiene la capacidad de auto-renovarse de manera indefinida, puede proliferar y generar células maduras especializadas de cualquier tipo y dependiendo de qué tipos de células pueda generar, una célula madre puede ser totipotente, cuando puede llegar a generar un organismo completo, pluripotente

93
cuando es capaz de dar origen a cualquier clase de célula más no a un organismo completo, o multipotente cuando las células que produce pueden ser de cualquier tipo dentro de un tejido especializado. Las células madre pueden ser obtenidas de diferentes fuentes, en tejido adulto es posible encontrarlas en menos del 0.1% de las células de la medula ósea, también en sangre, cerebro y piel pero su capacidad de expansión es muy limitada y se encuentran en cantidades muy pequeñas. En el tejido fetal se han recogido células de fetos abortados o de sangre de cordón umbilical de bebés recién nacidos. Aunque esta última fuente está fácilmente disponible, debido a que las células serán utilizadas para realizar transplantes a personas enfermas, la mayor dificultad radica en que puede presentarse incompatibilidad genética con la persona que requiera el transplante, a menos que la sangre de cordón fuera almacenada en el nacimiento con el fin de utilizarla posteriormente en el mismo paciente. La tercera fuente de células madre es el tejido embrionario. Las células obtenidas a partir de embriones tempranos son pluripotentes y pueden diferenciarse a cualquier tipo de tejido. Existen dos formas de obtener embriones tempranos, una es a partir de los embriones sobrantes de los tratamientos de fertilización in vitro, y otra es creándolos mediante clonación. En cualquiera de los dos casos el embrión no llegará a formar un feto, y el único propósito de utilizarlos será obtener células madre con fines investigativos. El término clonación se refiere a la reproducción de fragmentos de ADN, genes, células o individuos idénticos a los originales. La forma como se lleva a cabo este procedimiento es extrayendo el núcleo de una célula somática, es decir, una célula diploide con un set completo de cromosomas obtenida de un organismo en cualquier estado del desarrollo, e implantándolo en un oocito fertilizado o no fertilizado, cuyo material nuclear ha sido removido o inactivado, el clon formado puede crecer en unos días bajo ciertas condiciones de laboratorio y llegar a generar un embrión temprano, este procedimiento también es llamado transferencia nuclear de célula somática. La clonación puede ser reproductiva cuando el embrión temprano se implanta en el útero de un animal de la misma especie para lograr un embarazo y posterior nacimiento normal, o puede ser terapéutica cuando se obtienen células madre en el estado de blastocisto y posteriormente se cultivan y diferencian en varios tejidos normales (Lisker R, 2003).
5.3 PROS Y CONTRAS En la investigación con células madre embrionarias el único tipo de clonación involucrada es la clonación terapéutica, la cual está sujeta a numerosas discusiones respecto a la obtención y utilización de estas células. El primer problema se refiere a la obtención de células madre a partir de embriones

94
tempranos y está basado en la definición del momento en el cual empieza la vida o ¿a partir de qué momento se es persona? Algunos piensan que la vida comienza en el mismo momento de la fecundación, cuando el óvulo y el espermatozoide se unen. Una contraparte afirma que no se puede hablar de individualidad humana antes de que ocurra la gastrulación, proceso durante el cual se forman las tres capas celulares: endodermo, mesodermo y ectodermo, justo antes de la formación del tubo neural el cual dará origen al sistema nervioso. Esta etapa ocurre hacia el día 14 después de la fecundación. Otros más osados hablan del comienzo de la vida cuando se empieza a sentir al feto aproximadamente hacia el cuarto mes de embarazo, e incluso hay algunos que consideran que se empieza a ser persona cuando se es capaz de vivir por sus propios medios, lo cual podría ser hacia el sexto mes de embarazo. Una última propuesta es que el único criterio de aceptación debería ser el momento del nacimiento. Algo que si está claro en el ámbito científico es que los embriones tempranos a partir de los cuales se obtendrían las células madre para fines investigativos en ningún caso deben exceder los 14 días de desarrollo. Es por ello que existe un gran debate acerca de si la remoción y cultivo de células de un embrión donado indica o no falta de respeto hacia el mismo. En un extremo están quienes creen que un embrión temprano no es más que una colección de células sin mayores derechos que servir para la recolección de células humanas. Pero también podría considerarse que un embrión es un ser humano en potencia y el nivel de respeto dependería del grado de desarrollo. Para juzgar si la investigación mediante el uso de embriones es éticamente aceptable, tal vez deberían ser considerados no solamente el estado de desarrollo sino también el objeto de la investigación y entonces el uso de embriones con el fin de beneficiar a otros, podría llegar a ser moralmente justificable, lo cual no significa que un embrión sea considerado un ser sin valor, y pueda ser tratado como cualquier tejido humano o como objeto sin significado que carece de derechos. Por ejemplo, muchas personas rechazan la visión del embrión como una persona pero creen que éste es diferente del tejido humano ordinario ya que tiene el potencial único de desarrollarse en un ser humano. Por otro lado, hay otros que consideran que el embrión temprano es una persona o sujeto con derechos e intereses, por lo tanto su destrucción intencional equivale al asesinato, agravado por el hecho de que en este nivel de desarrollo un embrión no tiene medios para defenderse. Desde este punto de vista ninguna vida debería sacrificarse para salvar o prolongar la vida de una o más personas. El segundo problema se genera por el uso de embriones “sobrantes” de tratamientos de reproducción asistida. Las parejas que se someten a tratamientos de fertilización in vitro usualmente tienen embriones que no van a necesitar más y en este caso tienen la opción de dejarlos morir, donarlos a otras parejas, o donarlos para la realización de investigaciones. En el caso de optar por la investigación la donación de estos embriones se debe realizar con el consentimiento informado de los donantes, los cuales tienen derecho a saber con certeza el área de investigación y los protocolos (si es que los hay) que se

95
utilizarán en los procesos involucrados, con el fin de autorizar los posibles usos que se le puedan dar al embrión donado. Además debe garantizarse que en caso de que se rehusaran a realizar la donación, esto no afectaría la calidad de los cuidados futuros que pudieran solicitar los donantes. También debe ser claro para ellos que la investigación involucra la destrucción del embrión y en ningún caso está encaminada a proveerle algún tipo de beneficio. Y sobre todo es de gran importancia garantizar que a los embriones no se les dará otro uso diferente al acordado, no podrán ser implantados en el útero de otra mujer para fines reproductivos (Nuffield Council on Bioethics, 2000). Frente a la utilización de embriones con fines investigativos la posición de la Iglesia Católica es clara: la vida comienza desde el momento de la concepción y la utilización de embriones supernumerarios para investigaciones bajo el pretexto del progreso científico o médico, reducen en realidad la vida humana a simple "material biológico" del que se puede disponer libremente (Ioannes Paulus PP. II, 1995). Una tercera preocupación es la creación de embriones mediante clonación terapéutica, ya que si hubiera embriones disponibles frescos o congelados que fueran suficientes y apropiados para uso investigativo, ¿sería necesario permitir la creación de embriones adicionales únicamente para aumentar el número de embriones disponibles para investigación con células madre embrionarias o para terapia? Uno de los mayores beneficios sería que la clonación se realizaría con células somáticas del paciente y de esta forma, al realizar un transplante no se presentarían problemas de rechazo inmunológico, sin embargo, se necesitaría la disponibilidad de muchos oocitos para la producción de líneas celulares, esto podría tener implicaciones sociales si la obtención de los oocitos tuviera remuneración de tipo económico y el procedimiento para obtenerlos presentara algún riesgo para la donante. Además se ha comprobado que en mamíferos no-humanos la clonación ha sido muy ineficiente ya que se necesitan casi 100 oocitos para obtener un clon viable (Robertson JA, 2001). Con este tipo de tratamientos también existe el riesgo de las demandas que se generarían por acceder a ellos dentro del plan obligatorio de salud, aunque los costos deberían ser manejables si las terapias de reemplazo de células tuvieran una relación costo/beneficio baja y fueran más eficientes que las terapias existentes. Por otro lado, algunos piensan que el hecho de crear y desagregar un embrión humano conllevaría a la instrumentalización de la vida humana (Lanza RP et al., 2000). Entonces, ¿hay alguna distinción entre los embriones donados para la derivación de células madre y el uso de embriones creados para este propósito? Está claro que con los embriones provenientes de los tratamientos de fertilización in vitro existía una finalidad previa para la cual fueron creados y era la generación de una nueva vida, pero de todas formas el destino de los embriones no utilizados sería su destrucción o utilización en la investigación. Mientras que con la creación de nuevos embriones lo único que se buscaría sería la derivación de células

96
madre. En ninguno de los dos casos se beneficiaría el embrión, pero en ambos el beneficio sería para otras personas en el futuro. También existe preocupación de que una vez exista la tecnología para crear embriones humanos mediante transferencia nuclear de células somáticas, se fomentara su abuso con el propósito de realizar clonación reproductiva humana. Esto podría evitarse en cierta medida con una adecuada regulación de la clonación terapéutica de acuerdo a las leyes aprobadas en los diferentes países. La cuarta discusión está en torno a la utilización de líneas celulares. Una de las alternativas en la obtención de las células es la creación de líneas de células madre embrionarias que puedan ser utilizadas por diferentes investigadores para evitar así la creación de nuevos embriones con la única finalidad de obtener de éstos células madre. En teoría las líneas celulares pueden ser patentadas y un ejemplo es la patente concedida a Wisconsin Alumni Research Foundation en Estados Unidos, por células madre pluripotentes humanas derivadas de embriones sobrantes. Sin embargo, aún sigue siendo discutido si se deben otorgar patentes a células derivadas de tejido embrionario ya que además de la discusión sobre el estado moral del embrión, las células aisladas son tan cercanas al cuerpo humano, feto o embrión del cual han sido aisladas, que patentarlas podría ser considerado como una forma de comercialización del cuerpo humano ya que se estaría sacando beneficio económico de éste. Además, ya que una patente le confiere control a quien la tiene de decidir quién puede trabajar con la línea celular y con qué propósito, las patentes pueden impedir el acceso al cuidado de la salud debido a la necesidad de una licencia para usar las líneas celulares y por los derechos que se deben pagar a quien tiene la patente (Opinion of the european group on ethics in science and new technologies to the european commission, 2002). Por eso, antes de comenzar a trabajar con líneas celulares embrionarias es necesario establecer políticas que regulen el uso de éstas con fines terapéuticos para asegurar así un balance entre los intereses del inventor y los intereses de la sociedad.
5.4 TIPOS DE LEGISLACIÓN EN EL MUNDO Alrededor del mundo existen diferentes legislaciones que permiten o prohíben la investigación con embriones humanos, y en algunos casos la ley especifica los propósitos de la investigación clínica permitida. Los Estados Unidos, el Reino Unido y muchos otros países permiten el uso de tejido humano fetal para investigación cuando la decisión de una mujer de donar el tejido fetal está claramente separada de la decisión de abortar. La clonación reproductiva está específicamente prohibida por la ley en la mayoría de los países.

97
En el Reino Unido desde 2001 se permite el uso de embriones para la investigación de enfermedades graves y sus respectivos tratamientos. Estos embriones deben ser destruidos en el plazo de 14 días después de la fecundación. En este país es legal crear un embrión para la investigación médica, y para producir células madre destinadas a la curación de enfermedades graves, todo esto con el consentimiento de los donantes (Nuffield Council on Bioethics, 2000; Legislación propia en cada país sobre la investigación con células madre embrionarias, 2003). En Alemania el uso de embriones en la investigación médica es ilegal desde que entró en vigencia en enero de 1991 la Ley de Protección del Embrión. Según esta norma, cualquier persona que fertilice artificialmente un óvulo para cualquier fin distinto a un embarazo en la misma mujer que donó el óvulo puede ser procesada. En este país se permite la importación de líneas celulares bajo ciertas condiciones. Los investigadores que deseen importar células madre embrionarias deben pedir la aprobación del Instituto Robert Koch, las aplicaciones son revisadas por el Comité de Ética Central para la Investigación en Células Madre, el cual representa los campos de la biología, ética, medicina y teología, y este comité emite una recomendación. Esta nueva ley ha llevado según los propios investigadores del país, a que trasladen sus investigaciones a otros países menos restrictivos en el tema (Stafford N, 2003). En el estado de Victoria en Australia también está permitida la investigación en líneas de células madre embrionarias importadas de otros países. En Francia una ley de bioética aprobada en Agosto de 2004 permite la investigación con células madre embrionarias humanas siempre y cuando esté encaminada al desarrollo de tratamientos para enfermedades graves. Bajo esta ley, los investigadores pueden utilizar oocitos fertilizados que hayan sido descartados en las clínicas de fertilización in vitro, siempre y cuando los padres hayan dado su consentimiento. Asimismo, el 5 de Octubre de 2004, el Ministro de Salud francés Philippe Douste-Blazy autorizó la importación de células madre embrionarias humanas (Burgermeister J, 2004). En Japón la clonación con fines investigativos fue autorizada a partir del 2004 (Cyranoski D, 2004). En España a partir del año 2003 es permitida la investigación con los embriones sobrantes de los procesos de reproducción asistida que después de cinco años congelados no hayan sido utilizados para llevar a cabo una gestación. Además se fijó un número mínimo de embriones (entre 2 y 3) que pueden implantarse a las mujeres que decidan someterse a procesos de fecundación. Todo esto con el fin de evitar la producción de embriones destinados a uso científico (El Gobierno autoriza el uso médico de células madre embrionarias, 2003). Estados Unidos permite la investigación con embriones en el sector privado pero la prohíbe o limita de manera estricta cuando los fondos provienen del gobierno, aunque algunos estados tienen leyes que prohíben cualquier tipo de investigación

98
con embriones humanos. En la política propuesta por el Presidente George W. Bush, se pide que la investigación con fondos federales se realice haciendo uso únicamente de las más de 60 líneas de células madre embrionarias que existen en los Estados Unidos y que no se utilice ninguna otra creada después del 9 de Agosto de 2001 (McLaren A, 2001; Opinion of the european group on ethics in science and new technologies to the european commission, 2002). En Canadá la ley prohíbe la clonación con fines reproductivos, de investigación o terapéuticos y las células madre embrionarias solamente pueden ser obtenidas a partir de embriones descartados de tratamientos de fertilización in vitro con el consentimiento de los donantes (Hébert M et al., 2003). En Marzo de 2005 fue aprobada en Brasil la Ley de Bioseguridad, con la cual se autoriza el uso con fines terapéuticos de células madre extraídas de embriones humanos fecundados in vitro y conservados por congelamiento por lo menos durante tres años, si su implantación se revela inviable y la operación cuenta con la aprobación de los donantes (Svartzman J, 2005). En México al igual que en Colombia aún no existe una legislación al respecto, sin embargo, está establecido según la Corte Constitucional de Colombia, que se tienen derechos desde el momento de la concepción (Colombia, Corte constitucional. Sentencia C-133 de 17 de Marzo de 1994; Colombia. Corte constitucional. Sentencia T-179 de mayo 7 de 1993), por lo cual se considera al aborto como un delito y existe una pena señalada para quien incurra en él. En este momento hay radicados en el Congreso dos proyectos de ley por los senadores Jairo Clopatosky y Carlos Moreno de Caro, que de ser aprobados permitirían el uso de células madre provenientes de embriones humanos naturales, tejidos adultos, cordón umbilical y embriones clonados, en procedimientos de carácter investigativo o terapéutico, con el fin de realizar tratamientos de enfermedades que la medicina tradicional considere como incurables (Radican proyecto sobre clonación terapéutica, 2005). También están siendo estudiados tres proyectos más que buscan reglamentar y limitar los alcances de la reproducción asistida en Colombia, así como el uso de los embriones sobrantes de los procesos de fertilización in vitro (Ley en embrión, s.f.). Siendo éste uno de los pocos países con políticas restrictivas tan radicales en temas como el aborto no se pueden comparar las leyes de un país como el nuestro con las de países con gran nivel de desarrollo y sobre todo de apoyo a la investigación científica y que tienen claramente establecido un sistema que regule la creación y el uso de embriones para propósitos de reproducción asistida, tratamiento de enfermedades e investigación médica, como es el caso del Reino Unido, por lo tanto, y a pesar de que este sistema legal esté lo suficientemente fundamentado y sea lo bastante lógico, no deberían extrapolarse de manera exacta estas políticas a un país como el nuestro. Primero es necesario llevar a cabo más investigaciones acerca de la obtención, caracterización y los alcances de las células madre extraídas de tejido adulto, para que poco a poco puedan crearse políticas sobre el uso de células madre embrionarias acordes con los nuevos avances con el fin de establecer una legislación fuerte que no presente vacíos al momento de su aplicación. Aunque existen muchas implicaciones éticas

99
y sociales, las nuevas investigaciones y tecnologías basadas en el uso de células madre deben apuntar al desarrollo científico sin dejar nunca de lado la protección del ser humano.
5.5 INTERROGANTES La terapia con células madre embrionarias parece una prometedora opción para lograr grandes avances en el campo de la medicina y proporciona nuevas esperanzas para aquellos que padecen enfermedades degenerativas y necesitan de un transplante para reemplazar tejidos, órganos o células inservibles, pero cuyas oportunidades de recibirlo se ven opacadas por problemas de rechazo inmunológico o de disponibilidad del material a transplantar. Sin embargo, aún es necesario realizar muchos más estudios para lograr comprender la biología y el funcionamiento de las células madre, ya que no es posible implementar terapias de reemplazo con dichas células si todavía no se sabe cómo lograr que éstas se diferencien en la célula o el tipo de tejido requerido. Además de la parte científica, también hay otras consideraciones a nivel ético y social frente al uso de embriones para la obtención de células madre con fines investigativos y terapéuticos. Surgen interrogantes como ¿en qué momento empieza la vida?, ¿se justifica la destrucción de embriones en el estado de blastocisto para lograr que otras personas se beneficien?, ¿es necesario crear embriones con el único objetivo de obtener células madre de éstos y cómo podría evitarse que nuevas tecnologías como la clonación terapéutica puedan ser usadas para la creación indiscriminada de seres humanos? o si ¿la comercialización de líneas de células madre embrionarias implicaría convertir al ser humano en una mercancía? Aún no existe un consenso en la respuesta a estas preguntas pero si bastantes puntos de vista muy diversos entre sí, lo cual hace más difícil establecer una regulación común. A pesar de que en muchos países ya existen leyes que regulan el estudio con células madre y la utilización de embriones con fines investigativos o reproductivos, en otros países como el nuestro es necesario crear políticas que controlen no solo este tipo de investigaciones relativamente nuevas en el mundo sino otras como la reproducción asistida que a pesar de haber sido implementada desde hace más de diez años la ley aún no ha limitado sus alcances. La creación de políticas públicas que permitan la investigación con células madre embrionarias y que sean éticamente aceptables y legalmente disponibles no cierra la discusión sino que al contrario deja muchas preguntas por responder.

100
6. CONCLUSIONES Y PERSPECTIVAS A FUTURO Las células madre hematopoyéticas son capaces de dar origen a todas las
células del linaje hematopoyético, por ello, su biología es bastante compleja e involucra diferentes procesos que le perimiten desarrollar su capacidad de auto-renovación y proliferación a múltiples linajes. La identificación de las células madre hematopoyéticas es un proceso que
requiere de varios pasos previos de purificación ya que no hay un marcador único y específico capaz de identificar con certeza células lo suficientemente primitivas como para asegurar que son verdaderas células madre hematopoyéticas. Es por ello que es necesario realizar combinaciones tanto en los métodos de selección como en los anticuerpos utilizados con el fin de obtener el mayor grado de pureza. A través de los años se han desarrollado nuevas metodologías que
permiten aislar cada vez con más precisión células primitivas, y a pesar de que existen técnicas más rápidas, fáciles de llevar a cabo y con disponibilidad en el mercado, no se deben dejar de lado los ensayos que evalúan la funcionalidad de las células ya que éstos proporcionan datos importantes acerca del desarrollo y mantenimiento in vivo e in vitro de éstas. Por lo general, la mayoría de células madre hematopoyéticas permanecen
en estado de quiescencia, aunque debe haber una pequeña población capaz de empezar a proliferar en el momento en que aumente la demanda de células sanguíneas en el organismo, como en los procesos infecciosos o cuando las reservas de glóbulos rojos no son suficientes para suplir las necesidades del cuerpo. Es en esos momentos en que las células madre hematopoyéticas son capaces de producir nuevas células maduras con el fin de mantener la homeóstasis. Esta activación está ampliamente regulada por diferentes moléculas que intervienen en el ciclo celular sin embargo, aún no son claros los mecanismos exactos por los cuales estas células deciden si proliferan y dan lugar a células más maduras o permanecen en estado de quiescencia, manteniendo el pool de células primitivas. Hasta el momento se conocen diferentes mecanismos que intervienen en la
regulación de la auto-renovación de las células madre hematopoyéticas como son las vías de transducción de señal Hedgehog, Notch, Wnt, los genes de la familia Hox, las proteínas BMP y el gen Bmi-1. Hay evidencias de que todos estos factores son importantes en en el mantenimiento de la población de células madre hematopoyéticas y actúan de forma colectiva, aunque no se puede decir con

101
certeza en qué momento lo hacen o si unos son más importantes que otros. Dada la complejidad de las vías de señalización, es de suponer que
cualquier desbalance en alguno de estos procesos pueda afectar de forma crítica las propiedades de las células. Por esto y debido a que las leucemias se originan en células hematopoyéticas es importante comprender los procesos que gobiernan la maquinaria interna de las células madre hematopoyéticas, con el fin de entender cómo una alteración en dichos procesos puede afectar el desarrollo de células neoplásicas dando lugar a enfermedades como la leucemia mieloide crónica. A pesar de su gran potencial de auto-renovación, las células madre
hematopoyéticas envejecen, tal vez no con la misma rapidez con que lo hacen las células somáticas, pero si experimentan senescencia celular de forma progresiva a través del tiempo, debido al gran número de divisiones que pueden llevar a cabo, por daños asociados a la replicación del ADN. Sin embargo, estas células son capaces de contrarrestar de forma controlada mecanismos de eliminación de células defectuosas como la muerte célular programada o apoptosis. Cuando las células madre hematopoyéticas evaden la apoptosis de forma descontrolada ya sea por alteraciones como sobreexpresión, mal funcionamiento o no expresión de las proteínas involucradas, o por la expresión de oncogenes, como en el caso de la leucemia mieloide crónica, ocurre la transformación de célula madre hematopoyética a célula madre leucémica. La leucemia mieloide crónica es un desorden mieloproliferativo originado en
una célula madre hematopoyética que sufre una translocación entre los cromosomas 9 y 22. Como producto de esta fusión aparece el oncogen BCR-ABL cuya expresión da lugar a la oncoproteína de tipo tirosina-quinasa BCR-ABL, la cual puede activar diferentes vías de señalización capaces de afectar procesos en la célula como son la adhesión, proliferación y apoptósis, responsables del gran número de cambios característicos que afectan células normales para convertirlas en células leucémicas. La leucemia mieloide crónica se observa con mayor frecuencia en personas
mayores de 50 años, esto podría ser el resultado de la relación entre el cáncer y la senescencia celular. A medida que las células madre hematopoyéticas envejecen se vuelven más susceptibles a la inestabilidad genómica, dando lugar a translocaciones como la t(9;22) o a nuevas mutaciones que contribuyen al desarrollo de la enfermedad. A pesar de que han surgido varios tipos de terapias para combatir la
leucemia mieloide crónica en sus diferentes fases, uno de los tratamientos más eficientes que disminuye el uso de drogas citotóxicas es el transplante de células madre ya sea de tipo alogénico o autólogo. Es por ello que una adecuada comprensión del comportamiento de estas células in vivo e in vitro puede lograr el

102
mejoramiento en las técnicas de separación y análisis de células madre hematopoyéticas con el fin de realizar transplantes exitosos de células sanas que mejoren la calidad de vida de los pacientes que sufren de esta enfermedad. Debido a que es necesario realizar varios pasos de aislamiento y
separación, y ya que el número de células madre hematopoyéticas que puede ser encontrado en tejidos adultos como la médula ósea, sangre periférica o cordón umbilical es bastante bajo, se considera más fácil la obtención de células madre a partir de tejido embrionario. Sumado a esto está el hecho de que trabajar con células embrionarias ha mostrado ser una opción bastante esperanzadora en el tratamiento de enfermedades que involucran no solo al linaje hematopoyético sino cualquier clase de tejido. Sin embargo, los procedimientos involucrado en la obtención de células a partir de embriones tempranos presentan serias implicaciones a nivel ético y legal, y su práctica debe ser llevada a cabo con mucho más cuidado que las investigaciones realizadas con células madre adultas. Además de esto, no todos los países están preparados a nivel legislativo para regular prácticas de este tipo. Teniendo en cuenta el estado actual de la regulación del estudio con células madre en Colombia, las investigaciones en este país deberían ir encaminadas al mejoramiento de las técnicas de aislamiento y separación de células madre adultas, principalmente de células madre hematopoyéticas, dada la disponibilidad de tejidos como sangre periférica y cordón umbilical.

103
BIBLIOGRAFÍA Abelson, H.T., & Rabstein, L.S. (1970). Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Research, 30, 2213-2222. Agami, R., Blandino, G., Oren, M., & Shaul, Y. (1999). Interaction of c-Abl and p73 alpha and their collaboration to induce apoptosis. Nature, 399, 809–813. Akhtar, A.J., Hilton, J., & Jones, R.J. (1995). BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: a mechanism of resistance to multiple anticancer agents. Blood, 86, 1148–1158. Albrecht, T., Schwab, R., Henkes, M., Peschel, C., Huber, C., & Aulitzky, W.E. (1996). Primary proliferating immature myeloid cells from CML patients are not resistant to induction of apoptosis by DNA damage and growth factor withdrawal. British journal of haematology, 95, 501–507. Alkarain, A., & Slingerland, J. (2004). Deregulation of p27 by oncogenic signaling and its prognostic significance in breast cancer. Breast Cancer Research, 6, 13–21. Allsopp, R.C., Morin, G.B., DePinho, R., Harley, C.B. & Weissman, I.L. (2003). Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood, 102, 517–520. Amarante-Mendes, G.P., A.J. McGahon, W.K. Nishioka, D.E. Afar, O.N. Witte, & D.R. Green. (1998). Bcl-2-independent Bcr-Abl-mediated resistance to apoptosis: protection is correlated with up regulation of Bcl-XL. Oncogene, 16, 1383–1390. Amarante-Mendes, G.P., Naekyung, Kim, C., Liu, L., Huang, Y., Perkins, C.L., Green, D.R., & Bhalla, K. (1998). Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood, 91, 1700–1705. Amos, T.A., Lewis, J.L., Grand, F.H., Gooding, R.P., Goldman, J.M., & Gordon, M.Y. (1995). Apoptosis in chronic myeloid leukaemia: normal responses by progenitor cells to growth factor deprivation, Xirradiation and glucocorticoids. British journal of haematology, 91, 387–393. Amsellem, S., Pflumio, F., Bardinet, D., Izac, B., Charneau, P., Romeo, P.H., Dubart-Kupperschmitt, A. &, Fichelson, S. (2003). Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nature

104
Medicine, 9, 1423–1427. Artavanis-Tsakonas, S., Rand, M.D., & Lake, R.J. (1999). Notch signaling: cell fate control and signal integration in development. Science, 284, 770-776. Aster, J.C., & Pear, W.S. (2001). Notch signaling in leukemia. Current Opinion in Hematology, 8, 237-244. Attar, E.C., & Scadden, D.T. (2004). Regulation of hematopoietic stem cell growth. Leukemia, 18, 1760–1768. Austin, T. W., Solar, G.P., Ziegler, F. C., Liem, L. & Matthews, W. (1997). A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood, 89, 3624–3635. Baizabal, J.M., Furlan-Magaril, Santa-Olalla, J., & Covarrubias, L. (2003). Neural stem cells in development and regenerative medicine. Archives of Medical Research, 34, 572-588. Balduzzi, A., Gooley, T., Anasetti, C., et al. (1995). Unrelated donor marrow transplantation in children. Blood, 86, 3247-3256. Barker, N. Hurlstone, A., Musisi, H., Miles, A., Bienz, M., & Clevers, H. (2001). The chromatin remodeling factor Brg-1 interacts with β-catenin to promote target gene activation. The EMBO Journal, 20, 4935–4943. Barnett, M.J., Eaves, C.J., Phillips, G.L., Gascoyne, R.D., Hogge, D.E., Horsman, D.E., Humphries, R.K., Klingeman, H-G., Lansdorp, P.M., Nantel, S.H., Reece, D.E., Shepherd, J.D., Spinelli, J.G., Sutherland, H.J., & Eaves, A.C. (1994). Autografting with cultured marrow in chronic myelogenous leukemia: Results of a pilot study. Blood, 84, 724-732. Beckstead, J.H., Stenberg, P.E., & McEver, R.P. (1986). Immunohistochemical localization of membrane and alpha-granule proteins in plastic-embedded mouse bone marrow megakaryocytes and murine megakaryocyte colonies. Blood, 68, 696-702. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Bedi, A., Zehnbauer, B.A., Barber, J.P., Sharkis, S.J., & Jones, R.J. (1994). Inhibition of apoptosis by BCR-ABL in Chronic Myeloid Leukemia. Blood, 83, 2038-2044. Behrens, J. Jerchow, B.A., Wurtele, M., Grimm, J., Asbrand, C., Wirtz, R., Kuhl, M.,

105
Wedlich, D., & Birchmeier, W. (1998). Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science, 280, 596–599 (1998). Bell, D.R., & Van Zant, G. (2004). Stem cells, aging, and cancer: invevitabilities and outcomes. Oncogene, 23, 7290-7296. Bellantuono, I. (2004). Haemopoietic stem cells. The International Journal of Biochemistry & Cell Biology, 36, 607–620. Benboubker, L., Binet, C., Cartron, G., Bernard, M-C., Clement, N., Delain, M., Degenne, M., Desbois, I., Colombat, P., & Domenech, J. (2002). Frequency and differentiation capacity of circulating LTC-IC mobilized by G-CSF or GM-CSF following chemotherapy: a comparison with steady-state bone marrow and peripheral blood. Experimental Hematology, 30, 74-81. Bettenhausen, B., Hrabe de Angelis, M., Simon, D., Guenet, J., & Gossler, A. (1995). Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development, 121, 2407-2418. Bhardwaj, G., Murdoch, B., Wu, D., Baker, D.P., Williams, K.P., Chadwick, K., Ling, L.E., Karanu, F.N., & Bhatia, M. (2001). Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nature Immunology, 2, 172-180. Bhatia, M. (2001). AC133 expression in human stem cells. Leukemia, 15, 1685-1688. Bhatia, M., Bonnet, D., Murdoch, B., Gan, O.I., & Dick, J.E. (1998). A newly discovered class of human hematopoietic cells with SCID-repopulating activity. Nature Medicine, 4, 1038-1045. Bhatia, M., Bonnet, D., Wu, D., Murdoch, B., Wrana, J., Gallacher, L. & Dick, J.E. (1999). Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells. The Journal of Experimental Medicine, 189, 1139–1148. Bhatia, R., McCarthy, J.B., & Verfaillie, C.M. (1996). Interferon-α restores normal β1 integin-mediated inhibition of hematopoietic progenitor proliferation by the marrow microenvironment in chronic myelogenous leukemia. Blood, 87, 3883-3891. Bhatia, R., Verfaillie, C.M., Miller, J.S., & McGlave, P.B. (1997). Autologous transplantation therapy for chronic myelogenous leukemia. Blood, 89, 2623–2634.

106
Bhatia, R., Wayner, E.A., McGlave, P.B., & Verfaillie, C.M. (1994). Interferon-alpha restores normal adhesion of chronic myelogenous leukemia hematopoietic progenitors to bone marrow stroma by correcting impaired beta 1 integrin receptor function. The Journal of Clinical Investigation, 94, 384–391. Biernaux, C., Loos, M., Sels, A. Huez, G., & Stryckmans, P. (1995). Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood, 86, 3118-3122. Bigas, A., Martin, D.I., & Milner, L.A. (1998). Notch1 and Notch2 inhibit myeloid differentiation in response to different cytokines. Molecular Cell Biology, 18, 2324-2333. Bissonnette, R.P., Echeverri, F., Mahboubi, A., & Green, D.R. (1992). Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature, 359, 552-554.) Blain, S.W., Scher, H.I., Cordon-Cardo, C., & Koff, A. (2003). p27 as a target for cancer therapeutics. Cancer Cell, 3, 111–115. Boice, J.D., Day, N.E., Andersen, A., Brinton LA, Brown R, Choi NW, Clarke EA, Coleman MP, Curtis RE, Flannery JT, et al. (1985). Second cancer following radiation treatment for cervical cancer. An international collaboration among cancer registries. Journal of the National Cancer Institute, 74, 955-975. Bonifazi, F., de Vivo, A., Rosti, G., Guilhot, F., Guilhot, J., Trabacchi, E., Hehlmann, R., Hochhaus, A., Shepherd, P.C., Steegmann, J.L., et al. (2001). Chronic myeloid leukemia and interferon-alpha: a study of complete cytogenetic responders. Blood, 98, 3074–3081. Bonnet D, Bhatia M, Wang JC, Kapp U, Dick JE. (1999). Cytokine treatment or accessory cells are required to initiate engraftment of purified primitive human hematopoietic cells transplanted at limiting doses into NOD/SCID mice. Bone Marrow Transplantation, 23, 203-209. Bose, S., Deininger, M., Gora-Tybor, J., Goldman, J.M., & Melo, J.V. (1998). The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood, 92, 3362-3367. Boultwood, J., Fidler, C., Shepherd, P., Watkins, F., Snowball, J., Haynes, S., Kusec, R., Gaiger, A., Littlewood, T.J., Peniket, A.J., & Wainscoat, J.S. (1999). Telomere length shortening is associated with disease evolution in chronic myelogenous leukemia. American Journal of Hematology, 61, 5–9. Boultwood, J., Peniket, A., Watkins, F., Shepherd, P., McGale, P., Richards, S., Fidler, C., Littlewood, T.J., & Wainscoat, J.S. (2000). Telomere length shortening in

107
chronic myelogenous leukemia is associated with reduced time to accelerated phase. Blood, 96, 358–361. Brady, H.J.M. (2003). Apoptosis and leukaemia. British Journal of Haematology, 123, 577-585. Breems, D.A., Blokland, E.A., & Ploemacher, R.E. (1997). Stroma-conditioned media improve expansion of human primitive hematopoietic stem cells and progenitor cells. Leukemia, 11, 142-150. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Broccoli, D., Young, J.W., & de Lange, T. (1995). Telomerase activity in normal and malignant hematopoietic cells. Proc Natl Acad Sci USA, 92, 9082-9086. Brown, W.M.C., & Doll, R. (1965). Mortality from cancer and other causes after radiotherapy for ankylosing spondylitis. British medical journal, 5474, 1327-1332. Brümmendorf, T.H., Hoyoake, T.L., Rufer, N., Barnett, M.J., Schulzer, M., Eaves, C.J., Eaves, A.C., & Lansdorp, P.M. (2000). Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood, 95, 1883-1890. Bruno, L., Hoffmann, R., McBlane, F., Brown, J., Gupta, R., Joshi, C., Pearson, S., Seidl, T., Heyworth, C., & Enver, T. (2004). Molecular signatures of self-renewal, differentiation, and lineage choice in multipotential hemopoietic progenitor cells in vitro. Molecular and Cellular Biology, 24, 741–756. Burgermeister, J. (2004, Octubre). France green lights stem cells. The Scientist. Recuperado el 13 de mayo en http://www.the-scientist.com/news/20041011/02 Burroughs, J., Gupta, P., Blazar, B.R., & Verfaillie, C.M. (1994). Diffusible factors from the murine cell line M2-10B4 support human in vitro hematopoiesis. Experimental Hematology, 22, 1095-1101. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Campbell, A.D., Long, M.W., & Wicha, M.S. (1990). Developmental regulation of granulocytic cell binding to hemonectin. Blood, 76, 1758-1764. Carella, A.M., Champlin, R., Slavin, S., McSweeney, P., & Storb, R. (2000). Miniallografts: ongoing trial|s in humans. Bone Marrow Transplantation, 25, 345–350.

108
Carella, A.M., Frassoni, F., Podesta, M., Pungolino, E., Pollicardo, N., Ferrero, R., & Soracco, M. (1994). Idarubicin, intermediate-dose cytarabine, etoposide, and granulocyte-colony-stimulating factor are able to recruit CD34+/HLA-DR- cells during early hematopoietic recovery in accelerated and chronic phases of chronic myeloid leukemia. Journal of hematotherapy, 3, 199-202 Carlo-Stella, C., Dotti, G., Mangoni, L., Regazzi, E., Garau, D., Bonati, A., Almici, C., Sammarelli, G., Savoldo, B., Rizzo, M.T. (1996) Selection of myeloid progenitors lacking BCR/ABL mRNA in chronic myelogenous leukemia patients after in vitro treatment with the tyrosine kinase inhibitor genistein. Blood, 88, 3091–3100. Carlo-Stella, C., Mangoni, L., Almici, C., Caramatti, C., Cottafavi, L., Dotti, G.P., & Rizzoli, V. (1994). Autologous transplant for chronic myelogenous leukemia using marrow treated ex vivo with mafosfamide. Bone Marrow Transplantation, 14, 425-432. Cashman, J., Bockhold, K., Hogge, D.E., Eaves, A.C. & Eaves, C.J. (1997). Sustained proliferation, multi-lineage differentiation and maintenance of primitive human haemopoietic cells in NOD/SCID mice transplanted with human cord blood. British journal of haematology, 98, 1026-1036. Cashman, J.D., & Eaves, C.J (1999). Human growth factor-enhanced regeneration of transplantable human hematopoietic stem cells in nonobese diabetic/severe combined immunodeficient mice. Blood, 93, 481-487. Chai, S.K., Nichols, G.L., & Rothman, P. (1997). Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. Journal of immunology, 159, 4720-4728. Chaudhary, P.M., & Roninson, I.B. (1991). Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell, 66, 85-94. Cheng, J., Baumhueter, S., Cacalano, G et al. (1996). Hematopoietic defects in mice lacking the sialomucin CD34. Blood, 87, 479-490. Cheng, T., Rodrigues, N., Shen, H., Yang, Y., Dombkowski, D., Sykes, M. Scadden, D.T. (2000). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science, 287, 1804–1808. Cheng, T., Shen, H., Rodrigues, N., Stier, S., & Scadden, D.T. (2001). Transforming growth factor beta 1 mediates cell-cycle arrest of primitive hematopoietic cells independent of p21(Cip1/Waf1) or p27(Kip1). Blood 2001, 98,

109
3643–3649. Chetty, R. (2003). p27 Protein and cancers of the gastrointestinal tract and liver: an overview. Journal of Clinical Gastroenterology, 37, 23–27. Civin, C.I., Trischmann, T., Kadan, N.S., Davis, J., Noga, S., Cohen, K., Duffy, B., Groenewegen, I., Wiley, J., Law, P., Hardwick, A., Oldham, F., & Gee, A. (1996). Highly purified CD34-positive cells reconstitute hematopoiesis. Journal of Clinical Oncology, 14, 2224-2233. Clift, R.A., Buckner, C.D., Thomas, E.D., Bryant, E., Anasetti, C., Bensinger, W.I., Bowden, R., Deeg, H.J., Doney, K.C., Fisher, L.D., et al. (1994). Marrow transplantation for patients in accelerated phase of chronic myeloid leukemia. Blood, 84, 4368-4373. CML Trialists Collaborative Group: Interferon alfa versus chemotherapy for chronic myeloid leukemia: a meta-analysis of seven randomized trials. (1997). Journal of the National Cancer Institute, 89, 1616-1620. Colombia. Corte constitucional. Sentencia C-133 de 17 de Marzo de 1994. Magistrado Ponente: Antonio Barrera Carbonell. Colombia. Corte constitucional. Sentencia T-179 de mayo 7 de 1993. Magistrado Ponente: Alejandro Martínez Caballero. Coloumbel, L., Eaves, A.C., & Eaves, C.J. (1983). Enzymatic treatment of long-term marrow cultures reveals the preferential location of primitive hematopoietic progenitors in the adherent layer. Blood, 62, 291-297. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU: Churchill Livingstone. Conze, T., Lammers, R., Kuci, S., Scherl-Mostageer, M., Schwefer, N., Kanz, L., & Bühring, H-J. (2003). CDCP1 is a novel marker for hematopoietic stem cells. Annals of the New York Academy of Sciences, 996, 222-226. Cooper, G.M. (2000). The Cell Cycle. [Versión Electrónica]. En The Cell: a Molecular Aproach (2a. Ed.). Corso, A., Lazzarino, M., Morra, E., Merante S, Astori C, Bernasconi P, Boni M, Bernasconi C. (1995). Chronic myelogenous leukemia and exposure to ionizing radiation— a retrospective study of 443 patients. Annals of hematology, 70, 79-82. Cortez, D., Reuther, G., & Pendergast, A.M. (1997). The Bcr-Abl tyrosine kinase

110
activates mitogenic signaling pathways and stimulates G1-to-S phase transition in hematopoietic cells. Oncogene, 15, 2333-2342. Cortez, D., Stoica, G., Pierce, J.H., & Pendergast, A.M. (1996). The BCR-ABL tyrosine kinase inhibits apoptosis by activating a Ras-dependent signaling pathway. Oncogene, 13, 2589–2594. Cortez, D., Kadlec, L., Pendergast, & A.M.. (1995). Structural and signaling requirements for BCR-ABL-mediated transformation and inhibition of apoptosis. Molecular and Cellular Biology, 15, 5531–5541. Coulombel, L. (2004). Identification of hematopoietic stem/progenitor cells: strength and drawbacks of functional assays. Oncogene, 23, 7210-7222. Coulombel, L., Kalousek, D.K., Eaves, C.J., Gupta, C.M., & Eaves, A.C. (1983). Long-term marrow culture reveals chromosomally normal hematopoietic progenitor cells in patients with Philadelphia chromosome-positive chronic myelogenous leukemia. The New England journal of medicine, 308, 1493-1498. Cyranoski, D. (2004). Research cloning gets green light from Japanese ethicists. Nature, 430, 5-5. Daley, G.Q.,Van Etten, R.A., & Baltimore, D. (1990). Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science, 247, 824–830. Daniels, D.L. & Weis, W. I. (2002). ICAT inhibits β-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Molecular Cell, 10, 573–584. Davies, S.M., Shu, X.O., Blazar, B.R., et al (1995). Unrelated donor bone marrow transplantation: influence of HLA A and B incompatibility on outcome. Blood, 86, 1636-1642. de Groot, R.P., Raaijmakers, J.A., Lammers, J.W., & Koenderman, L. (2000). STAT5-dependent cyclinD1 and Bcl-xL expression in Bcr-Abl transformed cells. Molecular Cell Biology Research Communications 3, 299–305. de Groot, R.P., Raaijmakers, J.A., Lammers, J.W., Jove, R., & Koenderman, L. (1999). STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood, 94, 1108-1112. Deininger, M.W.N., & Druker, B.J. (2003). Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacological Reviews, 55, 401-423. Deininger, M.W.N., Goldman, J.M., & Melo, J.V. (2000). The molecular biology of

111
chronic myeloid leukemia. Blood, 96, 3343-3356. Deininger, M., Goldman, J.M., Lydon, N.B., & Melo, J.V. (1997). The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL positive cells. Blood, 90, 3691–3698. Deisseroth, A.B., Zu, Z., Claxton, D., Hanania, E.G., Fu, S., Ellerson, D., Goldberg, L., Thomas, M., Janicek, K., & Anderson, W.F. (1994). Genetic marking shows that Ph+ cells present in autologous transplants of chronic myelogenous leukemia (CML) contribute to relapse after autologous bone marrow in CML. Blood, 83, 3068–3076. del Peso, L., Gonzalez-Garcia, M., Page, C., Herrera, R., Nunez, G. (1998). Interleukin-3-induced phosphorylation of bad through the protein kinase akt. Science, 278, 687-689. Delia, D., Aiello, A., Soligo, D., Fontanella, E., Melani, C., Pezzella, F., Pierotti, M.A., & Porta, G:D. (1992). Bcl-2 proto- oncogene expression in normal and neoplastic human myeloid cells. Blood, 79, 1291-1298. Devergie, A., Apperley, J.F., Labopin, M. (1997). European results of matched unrelated donor bone marrow transplantation for chronic myeloid leukemia. Impact of HLA class II matching. Bone Marrow Transplantation, 20, 11-19. Dexter, T.M., Allen, T.D., & Lajtha, L.G. (1977). Conditions controlling the proliferation of haemopoietic stem cells in vitro. Journal of Cellular Physiology, 91, 335-344. Domen, J., Cheshier, S.H., & Wiessman, I.L. (2000). The Role of Apoptosis in the Regulation of Hematopoietic Stem Cells: Overexpression of BCL-2 Increases Both Their Number and Repopulation Potential. The Journal of Experimental Medicine, 191, 253-263. Draper, J.S., & Fox, V. (2003). Human embryonic stem cells: multilineage differentiation and mechanisms of self-renewal. Archives of Medical Research, 34, 558-564. Druker, B.J., Sawyers, C.L., Kantarjian, H., Resta, D.J., Reese, S.F., Ford, J.M., Capdeville, R., & Talpaz, M. (2001). Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. The New England Journal of Medicine, 344, 1038–1042. Druker, B.J., Talpaz, M., Resta, D.J., Peng, B., Buchdunger, E., Ford, J.M., Lydon, N.B., Kantarjian, H., Capdeville, R., Ohno-Jones, S., & Sawyers, C.L. (2001).

112
Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England Journal of Medicine, 344, 1031–1037. Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., Lydon, N.B. (1996). Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine, 2, 561–566. Dubrez L, Eymin B, Sordet O, Droin N, Turhan AG, Solary E. (1998). BCR-ABL delays apoptosis upstream of procaspase-3 activation. Blood, 91, 2415–2422. Duncan, A.W., Rattis, F.M., DiMascio, L.N., Congdon, K.L., Pazianos, G., Zhao, C., Yoon, K., Cook, J.M., Willert, K., Gaiano, N., & Reya, T. (2005). Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nature Immunology, 6, 314-322. Dunwoodie, S.L., Henrique, D., Harrison, S.M., & Beddington, R.S.P. (1997). Mouse Dll3: A novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development, 124, 3065-3076. Eaves, C., Udomsakdi, C., Cashman, J., Barnett, M., & Eaves, A. (1993). The biology of normal and neoplastic stem cells in CML. Leukemia & lymphoma, 11 (Suppl 1), 245–253. Eaves, C.J., Eaves, A.C. Cell culture studies in CML. In: Hinton K (ed.). Baillie`re’s Clinical Haematology, 1st edn. Bailliere Tindall/WB, Saunders: London, 1987, pp 931–961. Citado por: Holyoake, T.L., Jiang, X., Drummond, M.W., Eaves, A.C., & Eaves, C.J. (2002). Elucidating critical mechanisms of deregulated stem cell turnover in the chronic myeloid leukemia. Leukemia, 16, 549-558. El Gobierno autoriza el uso médico de células madre embrionarias. (2003, Julio). El Mundo. Recuperado el 13 de mayo de 2005 en http://elmundosalud.elmundo.es/elmundosalud/2003/07/25/biociencia/1059129759.html Elion, E.A. (1998). Routing MAP kinases cascades. Science, 281, 1625-1626. Elledge, S.J. (1996). Cell Cycle Checkpoints: Preventing an Identity Crisis. Science, 274, 1664-1672. Engelhardt, M., Mackenzie, K., Drullinsky, P., Silver, R.T., & Moore, M.A.S. (2000). Telomerase activity and telomere length in acute and chronic leukemia, pre and post-ex vivo culture. Cancer research, 60, 610–617.

113
Engelhardt, M., Kumar, R., Albanell, J., Pettengell, R., Han, W., & Moore, M.A.S. (1997). Telomerase regulation, cell cycle, and telomere stability in primitive hematopoietic cells. Blood, 90, 182-193. Engvall, E. (1995). Structure and function of basement membranes. The International Journal of Deveolopmental Biology, 39, 781-787. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Enright, H., Daniels, K., Arthur, D.C. et al. (1996). Related donor marrow transplant for chronic myeloid leukemia: patient characteristics predictive of outcome. Bone Marrow Transplantation, 17, 537-542. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone. Evan, G.I., Wyllie, A.H., Gilbert, C.S., Littlewood, T.D., Land, H., Brooks, M., Waters, C.M., Penn, L.Z., & Hancock, D.C. (1992). Induction of apoptosis in fibroblasts by c-myc protein. Cell, 69, 119-128. Ezoe, S. Matsumura, I., Satoh, Y., Tanaka, H., & Kanakura, Y. (2004). Cell Cycle Regulation in Hematopoietic Stem/Progenitor Cells. Cell Cycle, 3, 314-318. Facchini, L.M., & Penn, L.Z. (1998). The molecular role of Myc in growth and transformation: recent discoveries lead to new insights. The FASEB Journal, 12, 633-651. Faderl, S., Talpaz, M., Estrov, Z., O’Brien, S., Kurzrock, R., & Kantarjian, H.M. (1999). The biology of chronic myeloid leukemia. The New England Journal of Medicine, 341, 164-172. Fialkow, P.J., Jacobson, R.J., & Papayannopoulou, T. (1977). Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. The American journal of medicine, 6, 125-130. Franke, T.F., Kaplan, D.R., & Cantley, L.C. (1997). PI3K: downstream AKT ion blocks apoptosis. Cell, 88, 435–437. Frisch, S.M., Vuori, K., Ruoslahti, E., & Chan-Hui, P.Y. (1996). Control of adhesion-dependent cell survival by focal adhesion kinase. The Journal of Cell Biology, 134, 793-799. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139

114
154) Philadelphia, EE.UU.: Churchill Livingstone. Gallacher, L., Murdoch, B., Wu, D.M., Karanu, F.N., Keeney, M., & Bhatia, M. (2000). Isolation and characterization of human CD34-Lin- and CD34+Lin- hematopoietic stem cells using cell surface markers AC133 and CD7. Blood, 95, 2813-2820. Gallagher, J.T. (1994). Heparan sulphates as membrane receptors for the fibroblast growth factors. Eur J Clin Chem Clin Biochem, 32, 239-247. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Galton, D.A.G. (1953). Myeleran in chronic myeloid leukaemia: results of treatment. Lancet, 1, 208-213. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone. Gambacorti-Passerini, C., le Coutre, P., Mologni, L., Fanelli, M., Bertazzoli, C., Marchesi, E., di Nicola, M., Biondi, A., Corneo, G.M., Belotti, D, et al. (1997). Inhibition of the ABL kinase activity blocks the proliferation of BCR/ABL+ leukemic cells and induces apoptosis. Blood cells, molecules & diseases, 23, 380–394. Geary, C.G. (2000). The story of chronic myeloid leukaemia. Historical review. British Journal of Haematology, 110, 2-11. Geiger, H., Sick, S., Bonifer, C., & Muller, AM. (1998). Globin gene expression is reprogrammed in chimeras generated by injecting adult hematopoietic stem cells into mouse blastocysts. Cell, 93, 1055–1065. Giampaolo, A., Sterpetti, P., Bulgarini, D., Samoggia, P., Pelosi, E., Valtieri, M. & Peschle, C. (1994). Key functional role and lineage-specific expression of selected HOXB genes in purified hematopoietic progenitor differentiation. Blood, 84, 3637–3647. Giralt, S., O’Brien, S., Talpaz, M., Van Besien, K., Chan, K.W., Rondon, G., Andersson, B., Mehra, R., Khouri, I., & Estey, E. (1995). Interferon-alpha and interleukin-2 as treatment for leukemia relapse after allogeneic bone marrow transplantation. Cytokines and molecular therapy, 1, 115–122. Goldman, J.M., & Melo, J.V. (2003). Chronic myeloid leukemia –advances in biology and new approaches to treatment. The New England Journal of Medicine, 349, 1451-1464.

115
Goldman, J.M., Gale, R.P., Horowitz, M.M., Biggs, J.C., Champlin, R.E., Gluckman, E., Hoffmann, R.G., Jacobsen, S.J., Marmont, A.M., & McGlave, P.B. (1988). Bone marrow transplantation for chronic myelogenous leukemia in chronic phase. Increased risk for relapse associated with T-cell depletion. Annals of Internal Medicine, 108, 806–814. Goodell, M.A., Rosenzweig, M., Kim, H., Marks, D.F., DeMaria, M., Paradis, G., Grupp, S.A., Sieff, C.A., Mulligan, R.C., & Johnson, R.P. (1997). Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nature Medicine, 3, 1337-1345. Gordon, M.Y., Dazzi, F., Marley, S.B., Lewis, J.L., Nguyen, D., Grand, F.H., Davidson, R.J., & Goldman, J.M. (1999). Cell biology of CML cells. Leukemia, 13, S65-S71. Gordon, M.Y., Lewis, J.L., Marley, S.B., Grand, F.H., & Goldman, J.M. (1997). Stromal cells negatively regulate primitive haemopoietic progenitor cell activation via a phosphatidylinositol-anchored cell adhesion/signalling mechanism. British Journal of Haematology, 96, 647–653. Gordon, M.Y., Dowding, C.R., Riley, G.P., Goldman, J.M., & Greaves, M.F. (1987). Altered adhesive interactions with marrow stroma of haematopoietic progenitor cells in chronic myeloid leukaemia. Nature, 328, 342–344. Gordon, M.Y., Goldman, J.M., Gordon-Smith, E.C. (1985). 4-hydroperoxycyclophosphamide inhibits proliferation by human granulocyte-macrophage colony-forming cells (GM-CFC) but spares more primitive progenitor cells. Leukemia Research, 9, 1017-1021. Gratwohl, A., Hermans, J., Goldman, J.M., Arcese, W., Carreras, E., Devergie, A., Frassoni, F., Gahrton, G., Kolb, H.J., Niederwieser, D., et al. (1998). Risk assessment for patients with chronic myeloid leukaemia before allogeneic blood or marrow transplantation. Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Lancet, 352, 1087–1092. Gratwohl, A., Hermans, J., Niederwieser, D., et al (1993). Bone marrow transplantation for chornic myeloid leukemia: long term results. Bone Marrow Transplantation, 12, 509-516. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone. Greenwood, M.J., & Landscorp, P.M. (2003). Telomeres, telomerase, and hematopoietic stem cell biology. Archives of Medical Research, 34, 489-495.

116
Grewal, S.S., Barker, J.N. Davies, S.M., & Wagner, J.E. (2003). Unrelated donor hematopoietic cell transplantation: marrow or umbilical cord blood? Blood, 101, 4233-4244. Griffith, J.D., Comeau, L., Rosenfield, S., Stansel, R.M., Bianchi, A., Moss, H., & de Lange, T. (1999). Mammalian telomeres end in a large duplex loop. Cell, 97, 503-514. Guilhot, F., Chastang, C., Michallet, M., Guerci, A., Harousseau, J., Maloisel, F., Bouabdallah, R., Guyotat, D., Cheron, N., Nicolini, F., Abgrall, J., & Tanzer, J. (1997). Interferon alpha-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. The New England Journal of Medicine, 337, 223-229. Gunsilius, E., Duba, H-C., Petzer, A.L., Kahler, C.M., Grunewald, K., Stockhammer, G., Gabl, C., Dimhofer, S., Clausen, J., & Gastl, G. (2000). Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet, 355, 1688–1691. Gupta, P., Oegema, T.R., Brazil, J.J., Dudek, A.Z., Slungaard, A., & Verfaillie, C.M. (1998). Structurally specific heparan sulfates support primitive human hematopoiesis by formation of a multimolecular stem cell niche. Blood, 92, 4641-4651. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Guyotat, D., Wahbi, K., Viallet, A. et al. (1999). Selection of BCR/ABLnegative stem cells from marrow or blood of patients with chronic myeloid leukemia. Leukemia, 13, 991-998. Hahn, H., Christiansen, J., Wicking, C., Zaphiropoulos, P.G., Chidambaram, A., Gerrard, B., Vorechovsky, I., Bale, A.E., Toftgard, R., Dean, M., & Wainwright, B. (1996). A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. Journal of Biological Chemistry, 271, 12125-12128. Hallek, M., Danhauser-Riedl, S., Herbst, R. et al. (1996). Interaction of the receptor tyrosine kinase p145c-kit with the p210bcr/abl kinase in myeloid cells. British Journal of Haematology, 94, 5-16. Hao, S.X., & Ren, R. (2000). Expression of interferon consensus sequence binding protein (ICSBP) is downregulated in Bcr-Abl-induced murine chronic myelogenous leukemia-like disease, and forced coexpression of ICSBP inhibits Bcr-Abl-induced myeloproliferative disorder. Molecular and cellular b iology, 20, 1149-1161.

117
Harper, J.W., Adami, G.R., Wei, N., Keyomarsi, K., & Elledge, S.J. (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell, 75, 805-816. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Growth Factors, Cytokines, and the Control of Hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 154 202). Philadelphia, EE.UU.: Churchill Livingstone. Harrison, D.E. (1979). Mouse erythropoietic stem cell lines function normally 100 months: Loss related to number of transplantations. Mechanisms of Ageing and Development, 9, 427–433. Hattori, K., Heissig, B., Wu, Y., Dias, S., Tejada, R., Ferris, B., Hicklin, D.J., Zhu, A., Bohlen, P., Witte, L., Hendrikx, J., Hackett, N.R., Crystal, R.G., Moore, M.A.S., Werb, Z., Lyden, D., & Rafii, S. (2002). Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1 (+) stem cells from bone-marrow microenvironment. Nature Medicine, 8, 841-849. Hébert, M., Chenier, N.M., & Norris, S. (2003, Abril). BILL C-13: Assisted human reproduction act. Recuperado el 14 de mayo de 2005 en http://www.parl.gc.ca/common/bills_ls.asp?Parl=37&Ses=2&ls=c13 Hehlmann, R., Heimpel, H., Hasford, J., Kolb, H.J., Pralle, H., Hossfeld, D.K., QueiBer, W., Loffler, H., Hochhaus, A., Heinze, B., et al. (1994). Randomized comparison of interferon-α with busulfan and hydroxyurea in chronic myelogenous leukemia. Blood, 84, 4064-77. Henderson, A.J., Jhonson, A., & Dorshkind, K. (1990). Functional characterization of two stromal cell lines that support B lymphopoiesis. Immunology, 145, 423-428. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Hess, D.A., Levac, K.D., Karanu, F.N., Rosu-Myles, M., White, M.J., Gallacher, L., Murdoch, B., Keeney, M., Ottowski, P., Foley, R., Chin-Yee, I., & Bhatia, M. (2002). Functional analysis of human hematopoietic repopulating cells mobilized with granulocyte colony-stimulating factor alone versus granulocyte colony-stimulating factor in combination with stem cell factor. Blood, 100, 869-878. Heyssel, T., Brill, A.A., Woodbury, L.A. et al (1960). Leukemia in Hiroshima atomic bomb survivors. Blood, 15, 313-131. Higano, S., Chielens, D., Raskind, W., Bryant, E., Flowers, M.E., Radich, J., Clift, R., & Appelbaum, F. (1997). Use of alpha-2a-interferon to treat cytogenetic relapse

118
of chronic myeloid leukemia after marrow transplantation. Blood, 90, 2549–2554.) Hoang, T. (2004). The origin of hematopoietic cell type diversity. Oncogene, 23, 7188–7198. Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Stem cell model of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 126 138) Philadelphia, EE.UU.: Churchill Livingstone Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Growth factors, Cytokines, and the Control of Hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU. Churchill Livingstone. Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Control of Cell Growth and Differentiation. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 57 71). Philadelphia, EE.UU.: Churchill Livingstone. Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone. Holtschke, T., Lohler, J., Kanno, Y., Fehr, T., Giese, N., Rosenbauer, F., Lou, J., Knobeloch, K.P., Gabriele, L., Waring, J.F., et al. (1996). Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell ,87, 307-317. Holyoake, T.L., Jiang, X., Drummond, M.W., Eaves, A.C., & Eaves, C.J. (2002). Elucidating critical mechanisms of deregulated stem cell turnover in the chronic myeloid leukemia. Leukemia, 16, 549-558. Holyoake, T.L., Jiang, X., Jorgensen, H.G., Graham, S., Alcorn, M.J., Laird, C., Eaves, A.C., Eaves, C.J. (2001). Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood, 97, 720-728.

119
Holyoake, T., Jiang, X., Eaves, C., & Eaves, A. (1999). Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood, 94, 2056–2064. Horita, M., Andreu, E.J., Benito, A., Arbona, C., Sanz, C., Benet, I., Prosper, F., & Fernandez-Luna, J.L. (2000). Blockade of the Bcr-Abl kinase -activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5- dependent expression of Bcl-xL. The Journal of experimental medicine, 191, 977–984. Huettner, C.S., Zhang, P., Van Etten, R.A., & Tenen, D.G. (2000). Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nature genetics, 24, 57–60. Hurley, R.W., McCarthy, J.B., & Verfaillie, C.M. (1995). Direct adhesion to bone marrow stroma via fibronectin receptors inhibits hematopoietic progenitor proliferation. The Journal of Clinical Investigation, 96, 511-519. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Ilaria, R.L. Jr, & van Etten, R.A. (1996). P210 and P190 (BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. The Journal of b iological chemistry, 271, 31704-31710. Ingham, P.W. (1998). Trandsucing Hedgehog: the story so far. The EMBO Journal, 17, 3505-3511. Ioannes Paulus PP. II. (1995, Marzo). Evangelium vitae. Recuperado el 13 de mayo de 2005 en http://www.vatican.va/holy_father/john_paul_ii/encyclicals/documents/hf_jp-ii_enc_25031995_evangelium-vitae_sp.html Isaad, C., Croisille, L., Katz, A., Vainchenker W, & Coulombel L. (1993). A murine stromal cell line allows the proliferation of very primitive human CD34++/CD38- progenitor cells in long-term cultures and semisolid assays. Blood, 82, 2916-2924. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Israels, L.G., & Israels, E.D. (1999). Apoptosis. Stem Cells, 17, 306-313. Italian Cooperative Study Group on Chronic Myeloid Leukemia: Interferon alfa-2a as compared with conventional chemotherapy for the treatment of chronic myeloid

120
leukemia. (1994). The New England Journal of Medicine, 330, 820-825. Jackson, K.A., Majka, S.M., Wang, H., Pocius, J., Hartley, C.J., Majesky, M.W., Entman, M.L., Michael, L.H., Hirschi, K.K., & Goodell, M.A. (2001). Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. The Journal of Clinical Investigation, 107, 1395-1402. Jiang, X., Lopez, A., Holyoake, T., Eaves, A., & Eaves, C. (1999). Autocrine production and action of IL-3 and granulocyte colony-stimulating factor in chronic myeloid leukemia. Proc Natl Acad Sci USA, 96,12804–12809. Jones, P., May, G., Healy, L., Brown, J., Hoyne, G., Delassus, S., & Enver, T. (1998). Stromal expression of Jagged 1 promotes colony formation by fetal hematopoietic progenitor cells. Blood, 92, 1505-1511. Kabarowski, J.H.S., & Witte, O.N. (2000). Consequences of BCR-ABL expression within the hematopoietic stem cell in chronic myeloid leukemia. Stem Cells, 18, 399-408. Kalderon, D. (2002). Similarities between the Hedgehog and Wnt signaling pathways. Trends in Cell Biology, 12, 523-531. Kalderon, D. (2000). Transducing the Hedgehog signal. Cell, 103, 371-374. Kamel-Reid, S., & Dick, J.E. (1988). Engraftment of immune-deficient mice with human hematopoietic stem cells. Science, 242, 1706-1709. Kantarjian, H., Dixon, D., Keating, M.J., Talpaz, M., Walters, R.S., McCredie, K.B., & Freireich, E.J. (1988). Characteristics of accelerated disease in chronic myelogenous leukemia. Cancer, 61, 1441-1446. Kantarjian, H.M., Keating, M.J., Talpaz, M., Walters RS, Smith TL, Cork A, McCredie KB, & Freireich EJ. (1987). Chronic myelogenous leukemia in blast crisis: analysis of 242 patients. The American journal of medicine, 83, 445-54. Karanu, F.N., Murdoch, B., Gallacher, L., Wu, D.M., Koremoto, M., Sakano, S., & Bhatia, M. (2000). The Notch ligand Jagged-1 represents a novel growth factor of human hematopoietic stem cells. The Journal of Experimental Medicine, 192, 1365-1372. Kennedy, B.J., & Yarbro, K.W. (1966). Metabolic and therapeutic effects of hydroxyurea in chronic myeloid leukemia. JAMA: The journal of the American Medical Association, 195, 1038-1043. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone.

121
Kharbanda, S., Yuan, Z.M., Weichselbaum, R., & Kufe, D. (1997). Functional role for the c-Abl protein tyrosine kinase in the cellular response to genotoxic stress. Biochimica et b iophysica acta, 1333, 01–07. Kipreos, E.T., & Wang, J.Y. (1990). Differential phosphorylation of c-Abl in cell cycle determined by cdc2 kinase and phosphatase activity. Science, 248, 217-220. Kollet, O., Peled, A., Byk, T., Ben-Hur, H., Greiner, D., Shultz, L., & Lapidot, T. (2000). β2 microglobulin-deficient (β2mnull) NOD/SCID mice are excellent recipients for studying human stem cell function. Blood, 95, 3102-3105. Körbling, M., & Anderlini, P. (2001). Peripheral blood stem cell versus bone marrow allotransplantation: does the source of hematopoietic stem cells matter? Blood, 98, 2900-2908. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU. Churchill Livingstone. Krosl, J., Austin, P., Beslu, N., Kroon, E., Humphries, R.K., & Sauvageau, G. (2003). In vitro expansion of hematopoietic stem cells by recombinant TAT-HOXB4 protein. Nature Medicine, 9, 1428–1432. Laiho, M., DeCaprio, J.A., Ludlow, J.W., Livingston, D.M., Massague, J. (1990). Growth inhibition by TGF-beta linked to suppression of retinoblastoma protein phosphorylation. Cell, 62, 175-185. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Growth Factors, Cytokines, and the Control of Hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 154 202). Philadelphia, EE.UU.: Churchill Livingstone. Lange, R., Moloney, M., & Yamawaki, T. (1954). Leukemia in atomic bomb survivors. 1. General observations. Blood, 9, 574-585. Lanza, R.P., Caplan, A.L., Silver, L.M., Cibelli, J.B., West, W.D., & Green, R.M. (2000). The ethical validity of using nuclear transfer in human transplantation. The journal of the American Medical Association, 284, 3175-3179. Lapidot, T., Pflumio, F., Doedens, M., Murdoch, B., Williams, D.E. & Dick, J.E. (1992). Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science, 255, 1137-1141. Larochelle, A., Vormoor, J., Hanenberg, H., Wang, J.C.Y., Bhatia, M., Lapidot, T., Moritz, T., Murdoch, B., Xiao, X.L., Kato, I., Williams, D.A., & Dick, J.E. (1996). Identification of primitive human hematopoietic cells capable of repopulating

122
NOD/SCID mouse bone marrow: implications for gene therapy. Nature Medicine, 2, 1329-1336. Legislación propia en cada país sobre la investigación con células madre embrionarias. (2003, Julio). El Mundo. Recuperado el 13 de mayo de 2005, en http://www.el-mundo.es/elmundosalud/2003/07/25/biociencia/1059128677.html Lessard, J., & Sauvageau, G. (2003). Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature, 423, 255–260. Levesque, J.P., Haylock, D.N., & Simmons, P.J. (1996). Cytokine regulation of proliferation and cell adhesion are correlated events in human CD34+ hemopoietic progenitors. Blood, 88, 1168-1176. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Lewis JM, & Schwartz MA. (1998). Integrins regulate the association and phosphorylation of paxillin by c-Abl. The Journal of Biological Chemistry, 273, 14225-14230. Ley en embrión. (s.f.). Revista Cambio. Recuperado el 13 de mayo en http://www.revistacambio.com/html/saber_vivir/articulos/1620/ Li, L., Forman, S.J., & Bhatia, R. (2005). Expression of DLK1 in hematopoietic cells results in inhibition of differentiation and proliferation. Oncogene, 24, 4472-4476. Li, L., Milner, L.A., Deng, Y., Iwata, M., Banta, A., Graf, L., Marcovina, S., Friedman, C., Trask, B., Hood, L., & Torok-Storb, B. (1998). The human homolog of rat Jagged, hJagged1, is expressed by marrow stroma and inhibits differentiation of 32D cells through interaction with Notch1. Immunity, 8, 43-55. Lisker, R. (2003). Ethical and legal issues in therapeutic cloning and the study of stem cells. Archives of Medical Research, 34, 607-611. Long, M.W., Briddell, R., Walter, A.W., Bruno, E., & Hoffman, R. (1992). Human hematopoietic stem cell adherence to cytokines and matrix molecules. The Journal of Clinical Investigation, 90, 251-255. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Long, M.W., & Dixit, V.M. (1990). Thrombospondin functions as a cytoadhesion molecule for human hematopoietic progenitor cells. Blood, 75, 2311-2318. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E.,

123
& McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Lundell, B.I., McCarthy, J.B., Kovach, N.L., & Verfaillie, C.M. (1997). Activation of beta1 integrins onCMLprogenitors reveals cooperation between beta1 integrins and CD44 in the regulation of adhesion and proliferation. Leukemia, 11, 822–829. Ma, G., Lu, D., Wu, Y., Liu, J., & Arlinghaus, R.B. (1997). Bcr phosphorylated on tyrosine 177 binds Grb2. Oncogene, 14, 2367–2372. Maeno, M., Mead, P.E., Kelley, C., Xu, R.H., Kung, H.F., Suzuki, A. Ueno, N., & Zon, L.I. (1996). The role of BMP-4 and GATA-2 in the induction and differentiation of hematopoietic mesoderm in Xenopus laevis. Blood, 88, 1965–1972. Mao, J., Wang, J., Liu, B., Pan, W., Farr, G. H., 3rd, Flynn, C., Yuan, H., Takada, S., Kimelman, D., Li, L. Wu, D. (2001). Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Molecular Cell, 7, 801-809. Marone, M., De Ritis, D., Bonanno, G., Mozzetti, S., Rutella, S., Scambia, G., & Pierelli, L. (2002). Cell Cycle Regulation in Human Hematopoietic Stem Cells: From Isolation to Activation. Leukemia and Lymphoma, 43, 493-501. Matsumoto, K., Yasui, K., Yamashita, N., Horie, Y., Yamada, T., Tani, Y., Shibata, H., & Nakano, T. (2000). In vitro proliferation potential of AC133 positive cells in peripheral blood. Stem Cells, 18, 196–203. Mayani, H., Alvarado-Moreno, J.A., & Flores-Guzmán, P., (2003). Biology of human hematopoietic stem and progenitor cells present in circulation. Archives of Medical Research, 34, 476-488. McGahon, A., R. Bissonnette, M. Schmitt, K.M. Cotter, D.R. Green, & T.G. Cotter. (1994). BCR/ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood, 83, 1179–1187. McGlave, P.B., Shu, X.O., Wen, W., et al. (2000). Unrelated donor marrow transplantation for chronic myelogenous leukemia: 9 years’ experience of the national marrow donor program. Blood, 95, 2219-2225. McGlave, P.B., De Fabritiis, P., Deisseroth, A., Goldman, J., Barnett, M., Reiffers, J., Simonsson, B., Carella, A., & Aeppli. D. (1994). Autologous transplants for chronic myelogenous leukaemia: results from eight transplant groups. Lancet, 343, 1486–1488.

124
McGlave, P., Miller, J., Miller, W., Perry, E., Fautsch, S., Ramsay, N.K.C., Verfaillie, C.M., & Weisdorf, D.W. (1994). Autologous marrow transplant therapy for CML using marrow treated ex vivo with human recombinant interferon gamma. Blood, 84, 537. (abstr, suppl 1). Citado por: Bhatia, R., Verfaillie, C.M., Miller, J.S., & McGlave, P.B. (1997). Autologous transplantation therapy for chronic myelogenous leukemia. Blood, 89, 2623–2634. McLaren, A. (2001). Ethical and social considerations of stem cell research. Nature insight, 414, 129-131. McMahon, A.P. (2000). More surprises in the Hedgehog signaling pathway. Cell, 100, 185-188. Medvinsky, A., Dzierzak, E. (1996). Definitive hematopoiesis is autonomously initiated by the AGM region. Cell, 86, 897-906. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Meijerink, J.P.P., Mensink, E.J.B.M., Wang, K., Sedlak, T.W., Slöetjes, A.W., Witte, T.D., Waskman, G., & Korsmeyer, S.J. (1998). Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood, 91, 2991-2997. Milner, L.A., & Bigas, A. (1999). Notch as a mediator of cell fate determination in hematopiesis: evidence and speculation. Blood, 93, 2431-2448. Miraglia, S., Godfrey, W., Yin, A.H., Atkins, K., Warnke, R., Holden, J.T., Bray, R.A., Waller, E.K. & Buck, D.W. (1997). A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood, 90, 5013-5021. Miyake, K., Wissman, I.L., Greenberger, J.S.S., & Kincade, P.W. (1991). Evidence of a role of the integrin VLA-4 in lympho-hemopoiesis. The Journal of Experimental Medicine, 173, 599-607. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Mohle, R., Moore, M.A., Nachman, R.L., & Rafii, S. (1997). Transendothelial migration of CD34+ and mature hematopoietic cells: an in vitro study using a human bone marrow endothelial cell line. Blood, 89, 72-80. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone.

125
Moore S, Haylock DN, Levesque J.P., McDiarmid, L.A., Samels, L.M., Bik to, L., Simmons, P.J., & Hughes, T.P. (1998). Stem cell factor as a single agent induces selective proliferation of the Philadelphia chromosome positive fraction of chronic myeloid leukemia CD34(+) cells. Blood, 92, 2461-2470. Moreb, J.S., Turner, C., Sreerama, L., Zucali, J.R., Sladek, N.E., & Schweder, M. (1995). Interleukin-1 and tumor necrosis factor alpha induce class 1 aldehyde dehydrogenase mRNA and protein in bone marrow cells. Leukemia & Lymphoma, 20, 77–84. Morrison, S.J., Wandyez, A.M., Hemmati, H.D., Wright, D.E., & Weissman, I.L. (1997). Identification of a lineage of multipotent hematopoietic progenitors. Development, 124, 1929-1939. Morrison, S.J., Prowse, K. R., Ho, P., & Weissman, I. L. (1996). Telomerase activity in haemopoietic cells is associated with self-renewal potential. Immunity, 5, 207–216. Murdoch, B. Chadwick, K., Martin, M., Shojaei, F., Shah, K.V., Gallacher, L., Moon, R.T., & Bhatia, M. (2003). Wnt-5A augments repopulating capacity and primitive hematopoietic development of human blood stem cells in vivo. Proc. Natl Acad. Sci. USA, 100, 3422–3427. Nakamura, Y., Ando, K., Chargui, J., Kawada, H., Sato, T., Tsuji, T., Hotta, T., & Kato, S. (1999). Ex vivo generation of CD34+ cells from CD34- hematopoietic cells. Blood, 94, 4053-4059. Nardi, V., Azam, M., & Daley, G.Q. (2003). Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Current Opinion in Hematology, 14, 35-43. Neshat, M.S., Raitano, A.B., Wang, H.G. et al. (2000). The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Molecular and Cellular Biology, 20, 1179-1186. Nieborowska-Skorska, M., Wasik, M.A., Slupianek, A., Salomoni, P., Kitamura, T., Calabretta, B. & Skorski, T. (1999). Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. Journal of Experimental Medicine, 189, 1229–1242. Nishii, K., Kabarowski, J.H., Gibbons, D.L., Griffiths, S.D., Titley, I., Wiedemann, L.M., & Greaves, M.F. (1996). ts BCR-ABL kinase activation confers increased

126
resistance to genotoxic damage via cell cycle block. Oncogene, 13, 2225–2234. Nowell, P.C., & Hungerford, D.A. (1960). A minute chromosome in human chronic granulocytic leukemia. Science, 132, 1497. Nuffield Council on Bioethics, Stem cell therapy: the ethical issues. London: Nuffield Council on Bioethics; 2000. Nusse, R. (2003). Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development, 130, 5297-5305. Nusse, R. & Varmus, H. E. (1982). Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell, 31, 99–109. O’Brien, S.G., Guilhot, F., Larson, R.A, Gathmann, I., Baccarani, M., Cervantes, F., Cornelissen, J.J., Fischer, T., Hochhaus, A., Hughes, T, et al. (2003). Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. The New England Journal of Medicine, 348, 994–1004. O’Brien, S., Kantarjian, H., Keating, M., Beran, M., Koller, C., Robertson, L.E., Hester, J., Rios, M.B., Andreeff, M., & Talpaz, M. (1995). Homoharringtonine therapy induces long-term responses in patients with chronic myelogenous leukemia in late chronic phase. Blood, 86, 3322-3326. Ohnishi, K., Ohno, R., Tomonaga, M., Kamada, N., Onozawa, K., Kuramoto, A., Dohy, H., Mizoguchi, H., Miyawaki, S., Tsubaki, K., et al. (1995). A randomized trial comparing interferon-alpha with busulfan for newly diagnosed chronic myelogenous leukemia in chronic phase. Blood, 86, 906-916. Ohyashiki, K., Ohyashiki, J.H., Iwama, H., Hayashi, S., Shay, J.W., & Toyama, K. (1997). Telomerase activity and cytogenetic changes in chronic myeloid leukemia with disease progression. Leukemia, 11, 190-194. Olovnikov, A.M. (1973). A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. Journal of theoretical b iology, 41, 181–190. Opinion of the European group on ethics in science and new technologies to the European commission. Ethical aspects of patenting inventions involving human stem cells. (2002, Mayo). Recuperado el 13 de mayo de 2005 en http://europa.eu.int/comm/european_group_ethics/avis3_en.htm Or, R., Shapira, M.Y., Resnick, I., Amar, A., Ackerstein, A., Samuel, S., Aker, M., Naparstek, E., Nagler, A., & Slavin, S. (2003). Nonmyeloablative allogeneic stem

127
cell transplantation for the treatment of chronic myeloid leukemia in first chronic phase. Blood, 101, 441-445. Ordentlich, P., Lin, A., Shen, C., Blaumueller, C., Matsuno, K., Artavanis-Tsakonas, S., & Kadesch, T. (1998). Notch inhibition of E47 supports the existence of a novel signaling pathway. Molecular and Cellular Biology, 18, 2230-2239. Park, I.K., Qian, D., Kiel, M., Becker, M.W., Pihalja, M., Weissman, I.L., Morrison, S.J., & Clarke, M.F. (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature, 423, 302–305. Park, J.R., Bernstein, I.D., & Hockenbery, D.M. (1995). Primitive human hematopoietic precursors express Bcl-x but not Bcl-2. Blood, 86, 868-876. Pasino. M., Lanza, T., Marotta, F., Scarso, L., De Biasio, P., Amato, S., Corcione, A., Pistoia, V., & Mori, P.G. (2000). Flow cytometric and functional characterization of AC133+ cells from human umbilical cord blood. British Journal of Haematology, 108, 793–800. Patel, V.P., & Lodish, H.F. (1987). A fibronectin matrix is required for differentiation of murine erythroleukemia cells into reticulocytes. The Journal Cell Biology, 105, 3105-3118. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Peters, C., Wolf, Al, Wagner, M., Kuhlmann, J., & Waldmann, H. (2004). The cholesterol membrane anchor of the Hedgehog protein confers stable membrane association to lipid-modified proteins. Proc Natl Acad Sci U S A, 101, 8531-8536. Peters, R., Leyvraz, S., & Perey, L. (1998). Apoptotic regulation in primitive hematopoietic precursors. Blood, 92, 2041-2052. Petzer, A.L., & Gunsilius, E. (2003). Hematopoietic stem sells in chronic myeloid leukemia. Archives of Medical Research, 34, 496-506. Petzer, A.L., Eaves, C.J., Lansdorp, P.M., Ponchio, L., Barnett, M.J., & Eaves, A.C. (1996). Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood, 88, 2162–2171. Pflumio, F., Izac, B., Katz, A., Shultz, L.D., Vainchenker, W., & Coloumbel, L. (1996). Phenotype and function of human hematopoietic cells engrafting immune-deficient CB17-severe combined immunodeficiency mice and non-obese diabetic-

128
severe combined immunodeficiency mice after transplantation of human cord blood mononuclear cells. Blood, 88, 3731-3740. Ponchio, L., Cashman, J., Zoumbos, N., Eaves, C.J., & Eaves, A. (1995). Primitive CML cells show a deregulation of their cycling status both in vivo and in long-term cultures which is not normalized in the presence of interferon-α. Blood, 86, 493a. Ponchio, L., Conneally, E., Eaves, C. (1995). Quantitation of the quiescent fraction of long-term culture-initiating cells in normal human blood and marrow and the kinetics of their growth factor-stimulated entry into S-phase in vitro. Blood, 86, 3314–3321. Porter, J.A., Young, K.E., & Beachy, P.A. (1996). Cholesterol modification of Hedgehog signaling proteins in animal development. Science, 274, 255-259. Quelle, D.E., Ashmun, R.A., Hannon, G.J. et al (1995). Cloning and characterization of murine p16INK4a and p15INK4b genes. Oncogene, 11, 635-645. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Growth Factors, Cytokines, and the Control of Hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 154 202). Philadelphia, EE.UU.: Churchill Livingstone. Quesenberry, P.J., Crittenden, R.B., Lowry, P., Kittler, E.W., Rao, S., Peters, S., Ramshaw, H., & Stewart, F.M. (1994). In vitro and in vivo studies of stromal niches. Blood Cells, 2, 97-104. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Radican proyecto sobre clonación terapéutica. (2005, Marzo). Diario El País. Recuperado el 12 de mayo de 2005 en http://elpais-cali.terra.com.co/HOY/NAL/A324N1.html Rafii, S., Shapiro, F., Rimarachin, J., Nachman, R.L., Ferris, B., Weksler, B., Moore, M.A., & Asch, A.S. (1994). Isolation and characterization of human bone marrow microvascular endothelial cells: hematopoietic progenitor cell adhesion. Blood, 84, 10-19. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Raitano, A.B., Halpern, J.R., Hambuch, T.M., & Sawyers, C.L. (1995). The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci U S A, 92, 11746-1150.

129
Raskind, W.H., & Fialkow, P.J. (1987). The use of cell markers in the study of human hematopoietic neoplasia. Advances in cancer research, 49, 127-167 Remarks by the President on Stem Cell Research. (2001, Agosto). Recuperado el 12 de mayo de 2005 en http://www.whitehouse.gov/news/releases/2001/08/20010809-2.html Reuther, J.Y., Reuther, G.W., Cortez, D., Pendergast, A.M., Baldwin, A.S. Jr. (1998). A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes & development, 12, 968–981. Reya, T., Duncan, A.W., Ailles, L., Domen, J., Scherer, D.C., Willert, K., Hintz, L., Nusse, R., & Weissman, I.L. (2003). A role for Wnt signaling in selfrenewal of haematopoietic stem cells. Nature, 423, 409–414. Reya, T., Morrison, S.J., Clarke, M.F., & Weissman, I.L. (2001). Stem cells, cancer, and cancer stem cells. Nature insight, 414, 105-111. Roberts, R., Gallagher, J., Spooncer, E. Allen, T.D., Bloomfield, F., & Dexter, T.M. (1988). Heparan sulfate bound growth factors: a mechanism for stromal cell mediated haemopoiesis. Nature, 332, 376-378. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Robertson, J.A. (2001). Human embryonic stem cell research: ethical and legal issues. Nature Reviews Genetics, 2, 74-78. Robertson, M.J., Tantravahi, R., Griffen, J.D. Canellos, G.P., & Cannistra, S.A. (1993) Hematologic remission and cytogenetic improvement after treatment of stable-phase chronic myelogenous leukemia with continuous infusion of low-dose cytarabine. American journal of hematology, 43, 95-102. Roose, J. Molenaar, M., Peterson, J., Hurenkamp, J., Brantjes, H., Moerer, P., van de Wetering, M., Destree, O., & Clevers, H. (1998). The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature, 395, 608–612. Rowley, J. (1973). A new consistent chromosomal abnormality in chronic myeloid leukemia identified by quinacrine fluorescence and Giemsa staining. Nature, 243, 290-293. Rufer, N., Brummendorf, T.H., Kolvraa, S., Bischoff, C., Christensen, K., Wadsworth, L., Schultzer, M., & Lansdorp, P.M. (1999). Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. The Journal of

130
experimental medicine, 190, 157–167. Sahovic, E.A., Colvin, M., Hilton, J., & Ogawa, M. (1998). Role of aldehyde dehydrogenase in survival of progenitors for murine blast cell colonies after treatment with 4-hydroperoxycyclophosphamide in vitro. Cancer Research, 48, 1223–1226. Salgia, R., Li, J.L., Ewaniuk, D.S., Pear, W., Pisick, E., Burky, S.A., Ernst, T., Sattler, M., Chen, L.B., & Griffin, J.D. (1997). BCR/ABL induces multiple abnormalities of cytoskeletal function. The Journal of Clinical Investigation, 100, 46–57. Sanchez, M.J., Holmes, A., Miles, C., & Dzierzak, E. (1996). Caracterization of the first definitive hematopoietic stem cells in the AGM and liver of the mouse embryo. Immunity, 5, 513-525. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Sanchez-Garcia, I., and G. Grutz. (1995). Tumorigenic activity of the BCR/ABL oncogenes is mediated by BCL-2. Proc. Natl. Acad. Sci. USA, 92, 5287–5291. Santucci, M.A., Anklesaria, P., Laneuville, P., Das, I.J., Fitzgerald, T.J., & Greenberger, J.S. (1993). Expression of p210 BCR/ABL increases hematopoietic progenitor cell radiosensitivity. International journal of radiation oncology, biology, physics, 26, 881–889. Sauvageau, G., Lansdorp, P.M., Eaves, C.J., Hogge, D.E., Dragowska, W.H., Reid, D.S., Largman, C., Lawrence, H.J., & Humphries, R.K. (1994). Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci USA, 91, 12223–12227. Sauvageau, G., Thorsteinsdottir, U., Eaves, C.J., Lawrence, H.J., Largman, C., Lansdorp, P.M. & Humphries, R.K. (1995). Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes & Development, 9, 1753–1765. Savage, D.G., Szydlo, R.M., & Goldman, J.M. (1997). Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period. British Journal of Haematology, 96, 111-116. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone. Sawyers, C.L. (1999). Chronic myeloid leukemia. The New England Journal of

131
Medicine, 340, 1330-1340. Sawyers, C.L., McLaughlin, J., Goga, A., Havlik, M., & Witte, O. (1994). The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell, 77, 121-131. Sawyers, C.L., Callahan, W., & Witte, O.N. (1992). Dominant negative MYC blocks transformation by ABL oncogenes. Cell, 70, 901-910. Scheller, M., Foerster, J., Heyworth, C.M., et al. (1999). Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood, 94, 3764-3771. Schwartzberg, P.L., Stall, A.M., Hardin, J.D., Bowdish, L.S., Humaran, T., Boast, S., Harbison, M.L., Robertson, E.J., & Goff, S.P. (1991). Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell, 65, 1165-1175. Selleri, C., Sato, T., Del Vecchio, L., Luciano, L., Barrett, A.J., Rotoli, B., Young, N.S., & Maciejewski, J.P. (1997). Involvement of Fas-mediated apoptosis in the inhibitory effects of interferon-α in chronic myelogenous leukemia. Blood, 89, 957-964. Shet, A.S., Jahagirdar, B.N., & Verfaillie, C.M. (2002). Chronic myelogenous leukemia: mechanisms underlying disease progression. Leukemia, 16, 1402-1411. Short, B., Brouard, N., Occhiodoro-Scott, T., Ramakrishnan, A., & Simmons, P.J. (2003). Mesenchymal stem cells. Archives of Medical Research, 34, 565-571. Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. (1996). Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene, 13, 247-254. Siczkowski, M., Clarke, D., & Gordon, M.Y. (1992). Binding of primitive hematopoietic progenitor cells to marrow stromal cells involves heparan sulfate. Blood, 80, 912-919. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Silva, M., Richar, C., Benito, A., Sanz, C., Olalla, I., & Fernandez-Luna, J.L. (1998). Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. The New England Journal of Medicine, 338, 564-571. Siminovitch, L., Till, J. E., & McCulloch, E. A. (1964). Decline in colony forming ability of marrow cells subjected to serial ransplantation into irradiated mice.

132
Journal of Cell and Complex Physiology, 64, 23–31. Skorski, T., Bellacosa, A., Nieborowska-Skorska, M.M., Martinez, R., Choi, J.K., Trotta, R., Wlodarski, P., Perrotti, D., Chan, T.O., Wasik, M.A., Tsichlis, P.N., & Calabretta, B. (1997). Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. The EMBO Journal, 16, 6151-6161. Skorski, T., Kanakaraj, P., Nieborowska-Skorska, M., Ratajczak, M.Z., Wen, S., Zon, G., Gewirtz, A.M., Perussia, B., & Calabretta, B. (1995). Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood, 86, 726-736. Slingerland, J., & Pagano, M. (2000). Regulation of the cdk inhibitor p27 and its deregulation in cancer. Journal of Cellular Physiology, 183, 10–17. Sokal, J.E., Baccarani, M., Russo, D., & Tura, S. (1988). Staging and prognosis in chronic myelogenous leukemia. Seminars in hematology, 25, 49-61. Sokal, J.E., Leong, S.S., & Gomez, G.A. (1987). Preferential inhibition by cytarabine of CFU-GM from patients with chronic granulocytic leukemia. Cancer, 59,197-202. Spangrude, G.J. & Johnson, G.R. (1990). Resting and activated subsets of mouse multipotent hematopoietic stem cells. Proc Natl Acad Sci USA, 87, 7433-7437. Spencer, A., Szydlo, R.M., Brookes, P.A., et al (1995). Bone marrow transplantation for chronic myeloid leukemia with volunteer unrelated donors using ex vivo or in vivo T-cell depletion: major prognostic impact of HLA class I identity between donor and recipient. Blood, 86, 3590-3597. Staal, F.J.T., & Clevers, H.C. (2005). WNT signalling and haematopoiesis: a WNT situation. Nature Reviews in Immunology, 5, 21-30. Stafford, N. (2003, Octubre). Stem cells trickle into Germany. The Scientist. Recuperado el 13 de mayo de 2005 en http://www.the-scientist.com/news/20031031/02 Stamatoyannopoulos, G., Majerus, P.W., Perlmutter, R.M., & Varmus, H. (2001). Mechanisms of Leukemogenesis. En The Molecular Basis of Blood Diseases. (3ra. Ed.)(pp. 832 860). New York, EE.UU.: W.B. Saunders Company. Steegmann, J.L., Casado, L.F., Tomas, J.F., Sanz-Rodríguez, C., Granados, E., de la Camara, R., Alegre, A., Vázquez, L., Ferro, M.T., Figuera, A., et al. (1999). Interferon alpha for chronic myeloid leukemia relapsing after allogeneic bone marrow transplantation. Bone Marrow Transplantation, 23, 483–488.

133
Storms, R.W., Trujillo, A.P., Springer, J.B., Shah, L., Colvin, O.M., Ludeman, S.M., & Smith, C. (1999). Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA, 96, 9118–9123. Sutherland, D.R., & Keating, A. (1992). The CD34 antigen: structure, biology, and potential clinical applications. Journal of Hematotherapy, 1, 115-129. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU. Churchill Livingstone. Sutherland, H.J., Eaves, C.J., Lansdorp, P.M., Thacker, J.D., & Hogge, D.E. (1991). Differential regulation of primitive human hematopoietic cells in long-term cultures maintained on genetically engineered murine stromal cells. Blood, 78, 666-672. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Svartzman, J. (2005, Marzo). Brasil aprueba uso terapéutico de células madre. La Prensa. Recuperado el 13 de Mayo de 2005 en http://www-ni.laprensa.com.ni/archivo/2005/marzo/04/elmundo/elmundo-20050304-02.html Szilvassy, S.J. (2003). The biology of hematopoietic stem cells. Archives of Medical Research, 34, 446-460. Szydlo, R., Goldman, J.M., Klein, J.P., et al. (1997). Results of allogeneic bone marrow transplants for leukemia using donors other than HLA-identical siblings. Journal of clinical oncology, 15, 1767-1777. Taichman, R.S., Reilly, M.J., & Emerson, S.G. (1995). Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood, 87, 518-524. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Taichman, R.S., & Emerson, S.G. (1994). Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. The Journal of Experimental Medicine, 179, 1677-1682. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone.

134
Taipale, J., & Beachy, P. (2001). The Hedgehog and Wnt signalling pathways in cancer. Nature, 411, 349-354. Takahashi, N., Miura, I., Saitoh, K. et al. (1998). Lineage involvement of stem cells bearing the Philadelphia chromosome in chronic myeloid leukemia in the chronic phase as shown by a combination of fluorescence-activated cell sorting and fluorescence in situ hybridization. Blood, 92, 4758-4763. Takemaru, K. Yamaguchi, S., Lee, Y.S., Zhang, Y., Carthew, R.W., & Moon, R.T. (2003). Chibby, a nuclear β-catenin associated antagonist of the Wnt/Wingless pathway. Nature, 422, 905–909. Takenaka, K., Nagafuji, K., Harada, M., Mizuno, S., Miyamoto, T., Makino, S., Gondo, H., Okamura, T., & Niho, Y. (1996). In Vitro Expansion of Hematopoietic Progenitor Cells Induces Functional Expression of Fas Antigen (CD95). Blood, 88, 2871-2877. Talpaz, M., Kantarjian, H., Liang, J., Calver, M., Hamer, J., Tibbits, P., Durett, A., Claxton, D., Giralt, S., Khouri, I., et al. (1995). Percentage of Philadelphia chromosome (Ph)-negative and Ph Positive cells found after autologous transplantation for chronic myelogenous leukemia depends on percentage of diploid cells induced by conventional-dose chemotherapy before collection of autologous cells. Blood, 85, 3257-3263. Talpaz, M., Kantarjian, H., Kurzrock, R., Trujillo, J.M., & Gutterman, J.U. (1991). Interferon-alpha produces sustained cytogenetic responses in chronic myelogenous leukemia. Philadelphia chromosome-positive patients. Annals of internal medicine, 114, 532–538. Talpaz, M., Kantarjian, H.M., McCredie, K.B., et al (1987). Clinical investigation of human alpha-interferon in chronic myelogenous leukemia. Blood, 69, 1280-1288. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone. Talpaz, M., Kantarjian, H.M., McCredie, K. et al (1986). Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha-A in chronic myelogenous leukemia. The New England Journal of Medicine, 314, 1065-1069. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Chronic Myelogenous Leukemia. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 1155 1171). Philadelphia, EE.UU.: Churchill Livingstone.

135
The Merck Index. An encyclopedia of Chemicals, drugs, and biologicasls (12da ed). (1996). Rahway, NJ, EE.UU: Merck Research Laboratories, Division of MERCK & Co. Inc., Whitehouse Station. Thomas, T.E., Fairhurst, M.A., & Lansdorp, P.M. (1997). Rapid single step immuno-magnetic isolation of highly enriched primitive human hematopoietic progenitors. Blood, 90, 347b. Citado por: Wognum, A.W., Eaves, A.C., & Thomas, T.E. (2003). Identification and isolation of hematopoietic stem cells. Archives of Medical Research, 34, 461-475. Thomson, J.A., Itskovitz-Eldor, J., Shapiro, S.S., Waknitz, M.A., Swiergiel, J.J., Marshall, V.S., & Jones, J.M. (1998). Embryonic stem cell lines derived from human blastocysts. Science, 282, 1145–1147. Thorsteinsdottir, U., Sauvageau, G., & Humphries, R.K. (1999). Enhanced in vivo regenerative potential of HOXB4-transduced hematopoietic stem cells with regulation of their pool size. Blood, 94, 2605–2612. Till, J.E., & McCulloch, E.A. (1961). A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiation Research, 14, 213-222. Tsujimoto, Y., Cossman, J., Jaffe, E., & Croce, C.M. (1985). Involvement of the bcl-2 gene in human follicular lymphoma. Science, 228, 1440-1443. Tuszynski, G.P., Wang, T.N., & Berger, D. (1997). Adhesive proteins and the hematogenous spread of cancer. Acta Haematologica, 97, 29-39. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Tybulewicz, V.L., Crawford, C.E., Jackson, P.K., Bronson, R.T., & Mulligan, R.C. (1991). Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell, 65, 1153-1163. Udomsakdi, C., Eaves, C.J., Lansdorp, P.M., & Eaves, A.C. (1992). Phenotypic heterogeneity of primitive leukemic hematopoietic cells in patients with chronic myeloid leukemia. Blood, 80, 2522–2530. Udomsakdi, C., Eaves, C.J., Sutherland, H.J., & Lansdorp, P.M. (1991). Separation of functionally distinct subpopulations of primitive human hematopoietic cells using rhodamine-123. Experimental Hematology, 19, 338-342. Upadhyaha, G., Guba, S., Sih, S., Feinberg, A., Talpaz, M., Kantajian, H., Deisseroth, A., & Emerson, S. (1991). Interferon-alpha restores the deficient

136
expression of the cyto-adhesion molecule lymphocyte function antigen-3 by chronic myelogenous leukemia progenitor cells. The Journal of Clinical Investigation, 88, 2131–2136. Uzunel, M., Mattsson, J., Brune, M., Johansson, J.E., Aschan, J., & Ringden, O. (2003). Kinetics of minimal residual disease and chimerism in patients with chronic myeloid leukemia after nonmyeloablative conditioning and allogeneic stem cell transplantation. Blood, 101, 469-72. van den Berg, D.J., Sharma, A. K., Bruno, E. & Hoffman, R. (1998). Role of members of the Wnt gene family in human hematopoiesis. Blood, 92, 3189–3202. van Rhee, F., Szydlo, R.M., Hermans, J., Devergie, A., Frassoni, F., Arcese, W., de Witte, T., Kolb, H.J., Niederwiser, D., Jacobsen, N., et al. (1997). Long-term results after allogeneic bone marrow transplantation for chronic myelogenous leukemia in chronic phase: a report from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplantation, 20, 553-60. Varnum-Finney, B., Xu, L., Brashem-Stein, C., Nourigat, C., Flowers, D., Bakkour, S., Pear, W.S., & Bernstei, I.D. (2003). Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch 1 signaling. Nature Medicine, 6, 1278-1281. Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB and Lansdorp PM. (1994). Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age. Proc. Natl. Acad. Sci. USA, 91, 9857–9860. Verfaillie CM, Bhatia R, Miller W, et al. (1996). BCR/ABL-negative primitive progenitors suitable for transplantation can be selected from the marrow of most early-chronic phase but not accelerated-phase chronic myelogenous leukemia patients. Blood, 87, 4770-4779. Verfaillie CM, McCarthy JB, McGlave PB. (1992). Mechanisms underlying abnormal trafficking of malignant progenitors in chronic myelogenous leukemia. Blood, 90, 1232–1241. Verfaillie, C.M., Miller, W.J., Boylan, K., & McGlave, P.B. (1992). Selection of benign primitive hematopoietic progenitors in chronic myelogenous leukemia on the basis of HLA-DR antigen expression. Blood, 79, 1003–1010. Verstegen, M.M.A., Hennik, P.B., Terpstra, W., van den Bos, C., Wielenga, J.J., van Rooijen, N., Ploemacher, R.E., Wagemaker, G., & Wognum, A.W. (1998). Transplantation of human umbilical cord blood cells in macrophage-depleted SCID mice: evidence for accessory cell involvement in expansion of immature

137
CD34+CD38- cells. Blood, 91, 1966-1976. Vogel, W., Scheding, S., Kanz, L., & Brugger, W. (2000). Clinical applications of CD34 (+) peripheral blood progenitor cells (PBPC). Stem Cells, 18, 87-92. Voncken JW, van Schaick H, Kaartinen V, Deemer, K., Coates, T., Landing, B., Pattengale, P., Dorseuil, O., Bokoch, G.M., Groffen, J., & Heisterkamp, N. (1995). Increased neutrophil respiratory burst in bcr-null mutants. Cell, 80, 719-728. Vormoor, J., Lapidot, T., Pflumio, F., Risdon, G., Patterson, B., Broxmeyer, H.E., & Dick, J.E. (1994). Immature human cord blood progenitors engraft and proliferate to high levels in severe combined immunodeficient mice. Blood, 83, 2489-2497. Walkley, C.R., Fero, M.L., Chien, W., Purton, L.E., & McArthur, G.A. (2005). Negative cell-cycle regulators cooperatively control self-renewal and differentiation of haematopoietic stem cells. Nature Cell Biology, 7, 172-178. Wandl, U.B., Buzler, R., Niederle, N., Kloke, O., Mengelkoch, B., Becher, R., Seeber, S., Opalka, B. (1994). BCR/ABL-positive and –negative clonogenic cells in CML patients undergoing long-term interferon treatment. Leukemia, 8, 776-779. Wang, J., Kimura, T., Asada, R., Harada, S., Yolota, S., Kawamoto, Y., Fujimura, Y., Tsuji, T., Ikehara, S., & Sonoda, Y. (2003). SCID-repopulating cell activity of human cord blood-derived CD34- cells assured by intrabone marrow injection. Blood, 101, 2924-2931. Wang, J.Y. (1993). Abl tyrosine kinase in signal transduction and cell cycle regulation. Current Opinion in Genetics & Development, 3, 35–43. Watson, J.D. (1972). Origin of concatameric T4 DNA. Nature: New Biology, 239, 197–201. Weiden, P.L., Sullivan, K.M., Fluornoy, N., Storb, R., & Thomas, E.D. (1981). Antileukemic effect of chronic graft versus host disease. Contribution to improved survival after allogeneic marrow transplantation. The New England Journal of Medicine, 304, 1529–1533. Weisdorf, D.J., Anasetti, C., Antin, J.H., Kernan, N.A., Kollman, C., Snyder, D., Petersdorf, E., Nelson, G., McGlave, P. (2002). Allogeneic bone marrow transplantation for chronic myelogenous leukemia: comparative analysis of unrelated versus matched sibling donor transplantation. Blood, 99, 1971–1977. Wen, S.T., Jackson, P.K., & Van Etten, R.A. (1996). The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. The EMBO journal, 15, 1583–1595.

138
Willert, K., Brown, J. D., Danenberg, E., Duncan, A. W., Weissman, I. L., Reya, T., Yates, J. R. and Nusse, R. (2003). Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature, 423, 448-452. Wineman, J., Moore, K., Lemischka, I., & Muller-Sieburg, C. (1995). Functional heterogeneity of the meatopietic hematopoietic microenvironment: rare stromal elements maintain long-term repopulating stem cells. Blood, 87, 4082-4090. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Wodarz, A., & Nusse, R. (1998). Mechanisms of Wnt signaling in development. Annual Review of Cell and Developmental Biology, 14, 59–88. Wognum, A.W., Eaves, A.C., & Thomas, T.E. (2003). Identification and isolation of hematopoietic stem cells. Archives of Medical Research, 34, 461-475. Wright, W.E., Piatyszek, M.A., Rainey, W.E., Byrd, W., & Shay, J.W. (1996). Telomerase activity in human germline and embryonic tissues and cells. Developmental genetics, 18, 173-179. Xiong, Y., Hannon, G.J., Zhang, H., Casso, D., Kobayashi, R., & Beach, D. (1993). p21 is a universal inhibitor of cyclin kinases. Nature, 366, 701- Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Growth Factors, Cytokines, and the Control of Hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 154 202). Philadelphia, EE.UU.: Churchill Livingstone Yin, A.H., Miraglia, S., Zanjani, E., Almeida-Porada, G., Ogawa, M., Leary, A.G., Olweus, J., Kearney, J., & Buck, D.W. (1997). AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood, 90, 5002-5012. Yoder, M.C., & Hiatt, K. (1997). Engraftment of embryonic hematopoietic cells in conditioned newborn recipients. Blood, 89, 2176-2183. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Yoder, M.C., Hiatt, K., & Mukherjee, P. (1997). In vivo repopulating hematopoietic stem cells are present in the murine yolk sac at day 9.0 postcoitus. Proc Natl Acad Sci USA, 94, 6776-6780. Citado por: Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology

139
of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU.: Churchill Livingstone. Young, P.E., Baumhueter, S., & Lasky, L.A. (1995). The sialomucin CD34 is expressed on hematopoietic cells and blood vessels during murine development. Blood, 85, 96-105. Citado por Hoffman, R., Benz, E.J., Shattil, S.J., Furie, B., Cohen, H.J., Silberstein, L.E., & McGlave, P. (2000). Anatomy and physiology of hematopoiesis. En Hematology: Basic principles and practice (3ra. Ed.)(pp. 139 154) Philadelphia, EE.UU. Churchill Livingstone. Yuan ZM, Shioya H, Ishiko T, et al. (1999). p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature, 399, 814-817. Yui, J., Chiu, C., & Lansdorp, P.M. (1998). Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood, 91, 3255-3262. Zanjani, E., Almeida-Porada, G., Livingston, A.G., Flake, A.W., & Ogawa, M. (1998). Human bone marrow CD34- cells engraft in vivo and undergo multilineage expression that includes giving rise to CD34+ cells. Experimental Hematology, 26, 353-360. Zanjani, E.D., Almeida-Porada, G., Livingston, A.G., Zeng, H., & Ogawa, M. (2003). Reversible expression of CD34 by adult human bone marrow long-term engrafting hematopoietic stem cells. Experimental Hematology, 31, 406-412. Zha, J., Harada, H., Yang, E., Jockel, J., & Korsmeyer, S.J. (1996). Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell, 87, 619-628. Zhang, X., & Ren, R. (1998). Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood, 92, 3829–3840. Zhou, S., Morris, J.J., Barnes, Y., Lan, L., Schuetz, J.D., & Sorrentino, B.P. (2002). Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci USA, 99, 12339-12344. Zhou, S., Schuetz, J.D., Bunting, K.D., Colapietro, A-M., Sampath, J., Morris, J.J., Lagutina, I., Grosveld, G.C., Osawa, M., Nakauchi, H., & Sorrentino, B.P. (2001). The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nature Medicine, 7, 1028-1038.

140
Ziegler, B.L., Valtieri, M., Porada, G.A., De Maria, R., Muller, R., Masella, B., Gabbianelli, M., Casella, I., Pelosi, E., Bock, T., Zanjan, E.D., & Peschle, C. (1999). KDR receptor: a key marker defining hematopoietic stem cells. Science, 285, 1553-1558. Zou, X., Rudchenko, S., Wong, K., & Calame, K. (1997). Induction of c-myc transcription by the v-Abl tyrosine kinase requires Ras, Raf1, and cyclin-dependent kinases. Genes & development, 11, 654-662.