Embed Size (px)
Citation preview

J. M. RODRÍGUEZ
C. AGUIRRE
M. GARCÍA
R. PALOP
2.10. Farmacovigilancia2.10. Farmacovigilancia
INTRODUCCIÓN
Existen actividades propias de la Farmacia Hos-pitalaria que han ido adquiriendo progresiva impor-tancia en los últimos veinte años (por ejemplo, la aten-ción farmacéutica proporcionada a pacientesexternos), pero resulta curioso que precisamente laFarmacovigilancia, una actividad eminentemente clí-nica, haya interesado relativamente poco a los practi-cantes de esta especialidad, que se ha desarrollado endicho periodo de tiempo clara y definitivamenteorientada hacia el paciente.
Cuando a comienzos de la década de los 80 (loque representa una eternidad, observando el progresoy evolución que han tenido las actividades farmacéuticasdesde entonces) inicia su desarrollo el Sistema Españolde Farmacovigilancia, no existían muchos motivos pa-ra el optimismo en relación con la posible participa-ción de los farmacéuticos en tales actividades: las mis-mas parecían reservadas a médicos. Sin embargo, laFEFH promueve la participación de los farmacéuti-cos de hospital mediante la realización de unos talleres
de Farmacovigilancia realizados en 1987 titulados Far-macovigilancia en los hospitales. Estos talleres fueronseguidos del compromiso de la FEFH con la Farma-covigilancia con la firma de dos convenios con el Mi-nisterio de Sanidad y Consumo, el primero de ellos en1989 y posteriormente un segundo convenio en 1993.Entre tanto, la Farmacovigilancia es una actividad in-cluida en todas las actividades de la FEFH, que cul-minan en 1993 con la creación de unas becas para es-tancias en el Centro Coordinador deFarmacovigilancia y que se mantienen en la actuali-dad. Es de destacar que, a partir de 1992, la FEFHpromueve un curso sobre Farmacovigilancia en cadauna de las zonas en la que se organiza aquélla, con laparticipación de los centros autonómicos de Farma-covigilancia, impartiendo un total de ocho cursos. Asícomo la convocatoria ese mismo año del premio deFarmacovigilancia. La actividad en materia de Farma-covigilancia que los farmacéuticos de hospital han re-alizado en estos años hasta el año 1998 fue publicadaen la revista Farmacia Hospitalaria, vol. 23 nº 3, en laque se describen 2.550 tarjetas amarillas.
1

Con posterioridad, las regulaciones legales (esta-tales o autonómicas) que se han venido publicando so-bre Farmacovigilancia, o incluso de la farmacia demodo más general, han recogido no solamente el de-recho que asiste a cualquier profesional sanitario pa-ra notificar reacciones adversas causadas por medi-camentos, sino el deber existente de notificarlas. Laparticipación de los farmacéuticos en dichas tareasquedaba, de este modo, totalmente garantizada.
Sin embargo, si se estudian las Memorias anua-les de los Centros autonómicos de Farmacovigilancia,inmediatamente salta a la vista que, de forma bas-tante universal, el grueso de las notificaciones reci-bidas proviene de la asistencia primaria. El facultati-vo hospitalario (médico o farmacéutico) tiene menostendencia a notificar que el de primaria; a la vista detales datos (y lo que ellos representan), cabe pregun-tarse si, al menos, sus notificaciones son de mejor ca-lidad (o de mayor interés para el sistema) que las deasistencia primaria.
Porque el potencial notificador debe preguntarse:¿qué tipo de reacciones adversas busca, sobre todo, lared internacional de la que el sistema español formaparte? La respuesta es clara: aquellas que han desen-cadenado la muerte del paciente; del mismo modo,aquellas que le han motivado ingreso hospitalario, oque han puesto en peligro su vida; asimismo, aquellasno descritas antes y, finalmente, las producidas pormedicamentos de reciente comercialización. El cen-tro de la diana, a la vista de dichas demandas, se en-cuentra, la mayor parte de las veces en el hospital,pero los clínicos hospitalarios, al parecer, se encuen-tran demasiado ocupados como para cumplimentaruna tarjeta amarilla, actividad que no parece tancomplicada en tiempos de la receta electrónica o de lahistoria clínica informatizada.
Hay que pensar que existe otra explicación, o talvez un conjunto de circunstancias que han llevado auna aparente desidia por parte de los farmacéuticoshospitalarios en dicho campo. Recientemente. C.Planells(1) abundaba en el mismo planteamiento, de-nunciando una situación que consideraba incom-prensible: señalaba que apenas existen unidades activasen el campo de la Farmacovigilancia en los Serviciosde Farmacia españoles; ni tampoco Manuales deProcedimiento al respecto; ni tan siquiera se produ-ce una rotación de residentes por los Centros Auto-nómicos (parece que habiéndose incrementado enun año el periodo formativo de los FIR, dicha estan-
cia de, al menos, tres meses debiera ser consideradaobligatoria). Finalmente, la autora señalaba que enmuy pocas Memorias anuales de los Servicios deFarmacia Hospitalaria se menciona el número denotificaciones enviadas.
Abundando en dicha información, puede cons-tatarse analizando la base de datos del Sistema Es-pañol de Farmacovigilancia (FEDRA) que la mismacontiene, en los doce años que van desde 1989 al2001 (octubre), un total de 4.204 notificaciones pro-cedentes explícitamente de Servicios de FarmaciaHospitalaria. Actualmente todas las ComunidadesAutónomas han creado sus Centros, algunos de loscuales son muy activos en la comunicación con losprofesionales sanitarios, por lo que en modo algunopuede argumentarse desconocimiento para justificarla escasa participación existente. En efecto: los Ser-vicios de Farmacia ¿tan sólo pueden detectar un to-tal de 350 reacciones anuales, casi una por hospital?Conviene en tal sentido tener en cuenta que deacuerdo con un estudio recientemente publicado porLacoste-Roussillon et al(2), cada facultativo de pri-maria observa, al menos, 2,6 reacciones adversas gra-ves cada año, siendo lícito preguntarse si se está lle-vando a cabo alguna actividad que trate de recogerlasdonde se producen o registran: el hospital y su en-torno.
Adicionalmente, los mismos datos del SistemaEspañol de Farmacovigilancia (SEFV) indican quede las 4.204 notificaciones 721 fueron graves, casi 60cada año, bastante menos de una sola por hospital,información que contrasta fuertemente con los datosde Lacoste-Roussillon. O acerca de que 93 fueronmortales (menos de 8 anuales). De dichas reaccio-nes, 428 se referían a asociaciones fármaco-reacciónadversa que no habían sido descritas antes.
No puede afirmarse que el grado de participa-ción que tiene la farmacia hospitalaria en las activi-dades de Farmacovigilancia sea, ni tan siquiera, acep-table; sobre todo si se tiene en cuenta el enormepotencial informativo que poseen los Servicios deFarmacia hospitalarios, a través de procesos pasivosde obtención de datos (por ejemplo, la unidosis in-formatizada on line), o también a través de procedi-mientos activos de búsqueda (algunos de los cuales sedescribirán más adelante) o mediante procesos mix-tos.
En las siguientes páginas se exponen algunosprocedimientos, a través de los cuales es posible de-
576 FARMACIA HOSPITALARIA

tectar sospechas de reacciones adversas, y que ademásson potencialmente útiles para los servicios de farma-cia hospitalaria. Dado que existe un capítulo en estaobra que, específicamente, versa sobre farmacoepide-miología, aquellos procedimientos en los que se utilicenherramientas epidemiológicas se describirán con bas-tante menos extensión que otros que no las empleen,para evitar reiteraciones.
Sin embargo, cualquiera de ellos, adaptado a las ca-racterísticas de cada centro o en función de las activi-dades que se realicen, permite obtener un rendimien-to (en términos de detección de reacciones adversas)que seguramente resultará muy superior (varios cientosde veces superior) al de la pasada década, no simple-mente recogiendo un gran número de reacciones si-no, asimismo, recopilando datos de gran calidad, don-de ya se ha citado antes cuál es el tipo de informaciónque se busca: reacciones graves, desconocidas o pormedicamentos de comercialización reciente. En estesentido debe también tenerse en cuenta que la llegadade nuevos conceptos farmacoterapéuticos, como los an-ticuerpos monoclonales, o la comercialización de mo-léculas innovadoras en la terapia de enfermedades cró-nicas coloca, una vez más, a los Servicios de Farmaciaen el centro del proceso y sería lamentable que laoportunidad de intervenir activamente se perdiese.
El proceso de recogida de datos funciona y resul-ta muy rentable en términos de salud: los ejemplos dela retirada del mercado de algunos fármacos (Tabla 1)es un paradigma de que merece la pena colaborar conel sistema de Farmacovigilancia y ser solidario con lospacientes que aún no han recibido el medicamento, di-
ficultando así que puedan sufrir una reacción adversa enel futuro. El establecimiento de sistemas de preven-ción de reacciones adversas, que quizá resulta el enfo-que más positivamente rentable, conviene plantearloen los Servicios de Farmacia hospitalaria como un co-rolario de la actividad farmacéutica en general, dondela intervención creciente del farmacéutico en las acti-vidades de farmacoterapia tiene como objetivo su op-timización. La misma debe entenderse no únicamentecomo un intento de participar en la curación del en-fermo, sino de una forma más integral, logrando unamayor rentabilidad de la terapia en términos mixtos deeficacia y seguridad.
Pero no cabe hablar de seguridad si no se conoce elperfil de reacciones adversas de los medicamentos, so-bre todo de los nuevos, en el medio autóctono; losejemplos que se aportan en este capítulo (ebrotidina ycerivastatina) muestran la enorme potencialidad quetiene la actividad de Farmacovigilancia, que bastantes ve-ces consiste en tener los ojos abiertos ante señales quese producen continuamente en el hospital.
Del mismo modo, el conocimiento exacto del per-fil de seguridad en los pacientes de nuestro medio per-mite no tener que mirar hacia otros países a la hora deestablecer el mismo, partiendo de datos propios, delmismo modo que se emplean los enfoques bayesianosen farmacocinética clínica. Pero, una vez más, dichoplanteamiento es impensable con una intervencióntan escasa de los farmacéuticos hospitalarios, quienestienen una importante responsabilidad en dicho cam-po.
FARMACOVIGILANCIA
2.1 Organización de la Farmacovigilancia en España
La Farmacovigilancia en España se articula entorno a dos instituciones básicas, como son el Sis-tema Español de Farmacovigilancia (que agrupa losesfuerzos de las Comunidades Autónomas y de laAgencia Española del Medicamento en esta materia)y el Comité de Seguridad de Medicamentos de UsoHumano (CSMUH), un órgano consultivo de laAgencia Española del Medicamento. Para tener unaidea completa acerca del panorama habría queagregar a estos resortes las variadas proyeccionesinternacionales que la Farmacovigilancia realiza enEspaña, en especial el ámbito europeo (Figura 1).
2
577FARMACOVIGILANCIA
Tabla 1. Algunos riesgos identificados por el SEFV que dieron lugar a la retirada del medicamento.
Medicamento Problema
Bendazaco Hepatotoxicidad Cápsulas Dr. Bogas® HipertiroidismoBIOSTAR® crema Quemaduras localesCerivastatina RabdomiolisisCincofeno HepatotoxicidadCinepazida AgranulocitosisDroxicam HepatotoxicidadEbrotidina HepatotoxicidadGlafenina HipersensibilidadGangliósidos Síndrome Guillain-Barré

578 FARMACIA HOSPITALARIA
Figura 1. Organigrama del Sistema Español de Farmacovigilancia.
Centro de la OMS Uppsala
Agencia Española del Medicamento
Comité de Seguridad deluso humano
de Medicamentos
Comité Técnico deFarmacovigilancia
Centros Autonómicos deFarmacovigilancia
MédicosFarmacéuticos
y otros profesionales dela Salud
Decisiones
Alertas Propuestas
Evaluación
Recogida
Industria Farmacéutica
Centro Coordinador deFarmacovigilancia
Agencia Europeadel Medicamento
Comité de Especialidades
Farmacéuticas de la UE (CPMP)

El Comité de Seguridad de medicamentos de UsoHumano es un órgano colegiado, que asesora a laAgencia Española del Medicamento (AEM) en mate-ria de seguridad de medicamentos. Está formado por15 vocales: 3 por razón del cargo (Director y Subdirec-tores Generales de Evaluación y Seguridad de la Agen-cia Española del Medicamento), 6 nombrados por lasadministraciones sanitarias de las Comunidades Autó-nomas y 6 de libre designación. En su seno se evalúan losproblemas de seguridad que surgen con los medica-mentos comercializados, proponiendo medidas enca-minadas a reducir el riego detectado. Para cada uno de lostemas se designa a un ponente, (experto que puede sermiembro del Comité o ajeno al mismo), quien elabora uninforme de evaluación que expone en el seno del Co-mité para su discusión.
Tal como establece el Estatuto de la AEM, cuandoel Comité recomienda que se lleve a cabo una modifi-cación sustancial, revocación o suspensión de la autori-zación de comercialización de una especialidad farma-céutica, es competencia de la Secretaría el informar deoficio al laboratorio farmacéutico interesado sobre suderecho a audiencia ante el Comité. En caso de que el la-boratorio farmacéutico desee ejercer este derecho esconvocado a la reunión del Comité, en la que debe rea-lizar una exposición oral sobre el asunto objeto de debate.
Los acuerdos que se hayan alcanzado en el seno delComité, una vez adoptados por la dirección de la Agen-cia Española del Medicamento, se notifican por escritoa los laboratorios farmacéuticos afectados para su eje-cución.
Además de la evaluación de la relación beneficio-riesgo de medicamentos motivada por problemas deseguridad, el Comité tiene otras funciones entre las quepueden citarse proponer estudios e investigaciones enmateria de Farmacovigilancia, colaborar en la coordi-nación, planificación y desarrollo del Sistema Español deFarmacovigilancia y en la evaluación de estudios post-au-torización y prestar asesoramiento técnico a los repre-sentantes de la AEM que asisten a los grupos de traba-jo y reuniones de la Unión Europea sobre asuntos deFarmacovigilancia.
2.2. El Sistema español de Farmacovigilancia
El Sistema Español de Farmacovigilancia integralas actividades que las Administraciones Sanitarias realizanen España para recoger y elaborar información sobrereacciones adversas a los medicamentos. Se coordina
por el Ministerio de Sanidad y Consumo, a través de laAgencia Española del Medicamento (AEM).
El SEFV se organiza mediante centros, ubicadosen cada una de las 17 Comunidades Autónomas, de ca-da una de las cuales dependen orgánicamente. Desdelos primeros pasos, dados en los años 80, ha sido en1999 cuando finalmente se ha completado el SEFV.Todos los Centros Autonómicos de Farmacovigilancia(CAFV) integran el Comité Técnico de Farmacovigi-lancia, foro de discusión científica sobre nuevas seña-les, aspectos metodológicos, etc. La coordinación se re-aliza a través de la División de Farmacoepidemiología yFarmacovigilancia de la AEM.
Desde 1990 se puso en marcha una base de datoscentral, denominada FEDRA (Farmacovigilancia Es-pañola, Datos de Reacciones Adversas), con acceso te-lemático desde cada centro. Permite acumular la infor-mación que se notifica, una vez evaluada y codificada.Toda la información es accesible en línea, desde cadaCentro de Farmacovigilancia.
Los Laboratorios farmacéuticos, cumpliendo lasnormativas europeas y españolas sobre Farmacovigi-lancia, notifican las sospechas de reacción adversa(RAM) que reciben de los profesionales sanitarios, dela literatura científica y de los estudios que se realicen. Sila RAM cumple los requisitos como reacción adversa“grave”, se notifica en un plazo máximo de 15 días a laAEM. Su incorporación, aunque tardía, es en la actua-lidad relevante; así, durante 1998 y 1999, su participa-ción ha superado el 10% del total de notificaciones re-cibidas en el SEFV, que fueron alrededor de unas 6.000anuales.
Los CAFV centran su actividad en la interlocucióncon los profesionales sanitarios de su correspondienteárea. Este acercamiento al profesional de la salud es el quejustifica el mayor atractivo de este sistema descentralizado.
La actividad principal de los CAFV consiste enproporcionar a los profesionales sanitarios los medios pa-ra notificar las sospechas de RAM, y en crear el am-biente científico apropiado para conseguir su colabo-ración en la identificación de nuevas reaccionesadversas. Los centros editan y distribuyen los formula-rios de notificación, llamados “tarjetas amarillas”. Es-tos formularios, con franqueo en destino, facilitan a losprofesionales sanitarios la notificación de sospechas deRAM a medicamentos. Hoy en día, los CAFV tambiénofrecen (entre otros servicios) números de teléfono, defax, direcciones de correo electrónico, o páginas en In-ternet con el fin de facilitar la notificación.
579FARMACOVIGILANCIA

Los Centros publican boletines periódicos, con in-formación sobre Farmacovigilancia, reacciones adversasy divulgan las decisiones reguladoras tomadas por motivosde seguridad. Se trata de una información de retorno,queintenta mejorar día a día el conocimiento y la formación delos profesionales de la salud que manejan los medica-mentos.Los Centros también organizan, colaboran y par-ticipan en cursos, conferencias, sesiones, etc., sobre Far-macovigilancia que se llevan a cabo en universidades,centros de salud, hospitales, asociaciones científicas y co-legios profesionales.
De forma resumida, en este periodo de más de 15años de actividad del SEFV, hasta septiembre de 2001, seha recogido un total de 65.122 notificaciones de sospe-chas de RAM (Figura 2). Contienen información sobre116.349 sospechas de reacciones adversas, asociadas conun número total de 78.782 fármacos sospechosos. Un63% de las RAM han sido evaluadas como “leves”, un28% como “moderadas”, un 8% se han valorado como“graves” y el 1% restante han sido “mortales”. Del nú-mero total de notificaciones, un 70% se han notificadopor médicos generalistas; un 20% ha sido por médicosde otras especialidades; un 9% por farmacéuticos y el 1%restante se ha notificado por personal de enfermería. Encuanto al nivel asistencial, un 65% han tenido origen ex-trahospitalario y el restante 35% ha sido intrahospitalario.
Como resultado de las actividades de Farmacovi-gilancia llevadas a cabo en España, más adelante en elapartado 3.1 se describen los principales problemasde seguridad de medicamentos (aquellos que llevarona la retirada) en los que la información remitida porlos profesionales sanitarios a través del SEFV, me-diante la tarjeta amarilla, fue determinante de la alerta.
2.3. La integración internacional
España, como miembro de la Unión Europea,debe hacer partícipe a los demás Estados miembrosde todos aquellos problemas de seguridad relacionadoscon medicamentos en los que pueda verse involucra-do al menos otro país de la Unión. Existen foros dediscusión (el Pharmacovigilance Working Party, cons-tituido por expertos en Farmacovigilancia de todas lasagencias reguladoras nacionales), y el Comité de Es-pecialidades Farmacéuticas de la UE (Committe ofProprietary Medicinal Products, CPMP), al que re-porta el primero, y en el que se adoptan las decisionesde alcance comunitario, los procedimientos elabora-dos para tal finalidad y se viene desarrollando un mar-co legal cada vez más preciso (la persona interesadapuede consultar las referencias citadas en la bibliogra-fía)(3).
580 FARMACIA HOSPITALARIA
Figura 2. Evolución anual del número de notificaciones al SEFV 1982-2001 (octubre). En el interior de cada co-lumna se indica el número de Centros de Farmacovigilancia existentes.
7.000Nº de notificaciones
6.000
5.000
4.000
3.000
2.000
1.000
01982 1983 1984 1985 1986 1987 1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001
189 514 679
1.059
1.773
2.444 2.429 2.299
3.597
4.992
5.6995.986
5.047 5.0015.411 5.466
5.925 5.784 5.6885.515
1 1 3 5 5 5 7 12 12 13 15 16 16 16 16 17 18 18 18

Además, España forma también parte del Pro-grama Internacional de Farmacovigilancia de la Or-ganización Mundial de la Salud, junto a otros 55países, y como tal envía periódicamente todas lasnotificaciones de reacciones adversas detectadas en nuestro país (eincluidas en la base de datos FEDRA) a la base dedatos informatizada de la Organización Mundial dela Salud en Uppsala.
2.4. Generación de una alerta.
Alerta o señal se define como “información co-municada de una posible relación causal entre unacontecimiento adverso y un medicamento, cuandopreviamente esta relación era desconocida o estabadocumentaba de forma incompleta”. Habitualmen-te se requiere más de una notificación para generaruna señal, dependiendo de la gravedad del aconteci-miento y de la calidad de la información(4). Las se-ñales de alarma surgen de la acumulación, en númeromayor al esperado, de notificaciones espontáneas so-bre algún medicamento o problema concreto, y tam-bién de la aparición de fenómenos nuevos o graves. Pa-ra detectar estas circunstancias, la informacióncontenida en la base de datos del Sistema Español deFarmacovigilancia es evaluada periódicamente porlos técnicos del Sistema Español de Farmacovigilan-cia con el fin de identificar, de forma precoz, posiblesproblemas de seguridad derivados del uso de los me-dicamentos (generación de señales de alerta). Dichasseñales son discutidas en las reuniones del ComitéTécnico de Farmacovigilancia, donde también se de-be valorar la pertinencia de proponerlas, como asun-to para discusión, en el Comité de Seguridad de Me-dicamentos de Uso Humano. Cuando un CentroAutonómico de Farmacovigilancia considera que laseñal que ha generado constituye un problema inmi-nente de salud pública, debe ponerlo en conocimien-to inmediato de todos los Centros Autonómicos y dela Agencia Española del Medicamento (AEM) a travésde la División de Farmacoepidemiología y Farmaco-vigilancia.
A continuación se describen dos casos que resul-tan ejemplares de cómo la generación de una señal oalerta debido a notificaciones espontáneas al SistemaEspañol de Farmacovigilancia llevó a la retirada dedos medicamentos: ebrotidina y cerivastatina.
EbrotidinaLa ebrotidina (Ebrocit)®) es un antagonista de los
receptores H2, con propiedades antisecretoras (como elresto de antiH2) y citoprotectoras. Fue autorizado parael tratamiento de la úlcera gastroduodenal. La suspensiónde su comercialización fue debido a una posible aso-ciación con casos de hepatitis grave.
La sospecha se suscitó a partir de la notificación alCentro de Farmacovigilancia de Andalucía (CFA) deuna agrupación temporo espacial (cluster) de 4 casosde hepatitis, posiblemente asociada a ebrotidina, pro-cedentes de un mismo hospital. Al tratarse de unareacción adversa a medicamentos (RAM) grave (ingre-so hospitalario) y desconocida, y ante la posibilidad deque fuera producida por un medicamento de reciente co-mercialización, dicho Centro analizó la base de datosdel Sistema Español de Farmacovigilancia, observan-do que a fecha de 4-mayo-98 la proporción de RAMde naturaleza hepática notificadas para ebrotidina eradel 83% (15/18 TA) mientras que la proporción deRAM hepáticas para cimetidina, ranitidina y famotidinaera del 5%(5).
Estos datos, junto a un análisis descriptivo de losprimeros 15 casos notificados al Sistema Español deFarmacovigilancia fueron presentados por el CFA alComité Técnico del SEFV, donde se planteó la poten-cial trascendencia sanitaria del tema como posible nue-va señal.
Desde entonces, la aparición progresiva de más no-tificaciones fue reforzando la sospecha inicial, bien porparte de las notificaciones espontáneas procedentes dediferentes Centros de Farmacovigilancia de Comuni-dades Autónomas, así como por los 9 casos recogidosgracias al registro sistemático de RAM hepáticas aso-ciadas a medicamentos que lleva a cabo el Grupo An-daluz para el Estudio de la Hepatotoxicidad.
En 22 casos se dispuso de la suficiente informaciónpara poder clasificar el tipo de lesión hepática: hepato-celular (o citolítico) 13 (59%); colestásico 4 (18%) ymixto en 5 (23%). En cuanto a los fármacos sospecho-sos: ebrotidina 9 (33%); ebrotidina + antiinflamatoriosno esteroideos (AINE) 3 (11%); ebrotidina + AINE +otros fármacos 11 (41%) y ebrotidina + otros fárma-cos no AINE 4 (15%). La mayoría fueron graves, puesen 21 casos (78%) la RAM motivó ingreso hospitala-rio y el resto precisó asistencia extrahospitalaria.
Con carácter extraordinario se reunió el ComitéPermanente de la Comisión Nacional de Farmacovigi-lancia (actualmente llamado Comité de Seguridad de
581FARMACOVIGILANCIA

Medicamentos de Uso Humano) para evaluar el pro-blema y el propio laboratorio fabricante, de acuerdocon la Dirección General de Farmacia y ProductosSanitarios, decidieron la suspensión de comercializa-ción de la especialidad Ebrocit®, “debido a las noti-ficaciones al SEFV de sospechas de posibles reaccio-nes adversas de tipo hepático en tratamientosprolongados y administrado conjuntamente con AI-NE, aunque no se ha demostrado totalmente la im-putabilidad del producto en los mismos”.Cerivastatina
La cerivastatina es un hipolipemiante, inhibidorde la 3-hidroxi-3-metil glutaril coenzima A reductasa(HMG-CoA reductasa), enzima que cataliza el paso li-mitante en la síntesis del colesterol. Fue comerciali-zada para el “tratamiento de la hipercolesterolemiaprimaria en pacientes que no responden adecuada-mente a una dieta apropiada”. La suspensión de lacomercialización fue debida a casos de rabdomioli-sis, especialmente cuando fue utilizada en asociacióncon gemfibrozilo.
El Centro de Farmacovigilancia de Andalucía, araíz de un caso recibido, “revisó las notificacionesexistentes en la base de datos del Sistema Español deFarmacovigilancia y detectó (inicialmente) 34 casosde rabdomiolisis asociados a cerivastatina. En el 65%de los casos, el paciente estaba en tratamiento con-comitante con gemfibrozilo”* y lo puso en conoci-miento de la Agencia Española del Medicamento, lacual en una nota informativa de Comunicación sobreriesgos de medicamentos señalaba: “Aunque la rab-domiolisis es una reacción adversa bien conocida,que aparece asociada al uso de las estatinas, el núme-ro de rabdomiolisis recogidas en España a través de lanotificación espontánea para cerivastatina es superioral recogido para otras estatinas. El Comité de Segu-ridad de Medicamentos de Uso Humano y el Grupode Trabajo de Farmacovigilancia de la Agencia Eu-ropea de Evaluación de Medicamentos están actual-mente evaluando si esto se debe a un mayor riesgode rabdomiolisis asociado a cerivastatina”. Ademásañadía que “la cerivastatina no debe utilizarse en com-
binación con gemfibrozilo, ya que esta asociación au-menta el riesgo de rabdomiolisis”, y que “se deberá te-ner especial cuidado cuando se administre junto conmedicamentos que inhiben la isoenzima 3A4 del cito-cromo P450” e ir: “aumentando gradualmente la dosisdiaria no sobrepasando la dosis máxima diaria (0,4mg)” **.
La Agencia Española del Medicamento (AEM)emitió un segunda Comunicación sobre riesgos de me-dicamentos afirmando: “en el marco de un procedi-miento coordinado con el resto de Autoridades Regu-ladoras de los Estados Miembros de la UniónEuropea, ha procedido a modificar de forma urgentela información incluida en las fichas técnicas y pros-pectos de las especialidades farmacéuticas formuladascon cerivastatina. Se incluye como contraindicación ab-soluta la administración concomitante de cerivastatina ygemfibrozilo”***.
Finalmente la AEM, a solicitud del laboratorio far-macéutico Bayer, suspendió temporalmente la comer-cialización de las especialidades farmacéuticas que con-tienen el principio activo cerivastatina, señalándose en unnueva nota de comunicación de riesgos****: “el motivoradica en que, a pesar de las precauciones adoptadaspor las autoridades sanitarias de los diferentes países enque se comercializa cerivastatina, se sigue detectando eluso asociado de cerivastatina y gemfibrozilo. La asocia-ción de estos dos principios activos aumenta el riesgo derabdomiolisis, trastorno cuya gravedad hace que el ba-lance beneficio/riesgo del tratamiento con cerivastatinasea desfavorable”.
En estos dos ejemplos hay que señalar algunas di-ferencias; cabe destacar que al ser la ebrotidina un me-dicamento de origen español y no comercializado enotros países, las decisiones se tomaron por la Agencia Es-pañola del Medicamento; en cambio, la cerivastatina fuecomercializada en la Unión Europea mediante un sis-tema de registro conocido como “ReconocimientoMutuo”, en el que un país europeo actúa de país de re-ferencia para el resto. En este caso, el Reino Unido eva-luó inicialmente la autorización solicitada, y posterior-mente todos los países europeos admitieron las
582 FARMACIA HOSPITALARIA
* Diario “El Correo”, 24 de agosto de 2001. Página 58.** Agencia Española del Medicamento. Comunicación sobre riesgos de medicamentos. Cerivastatina y casos de rabdomiolisis. 30 de ma-
yo de 2001.*** Agencia Española del Medicamento. Comunicación sobre riesgos de medicamentos. Cerivastatina y casos de rabdomiolisis. 2 de ju-
lio de 2001.**** Agencia Española del Medicamento. Comunicación sobre riesgos de medicamentos. Cerivastatina y casos de rabdomiolisis. 8 de
agosto de 2001.

condiciones de autorización. En la fase posautoriza-ción, el Reino Unido siguió siendo el país de referenciapara evaluar los datos de Farmacovigilancia.
Conclusiones de carácter general:1) La Farmacovigilancia tiene un papel fundamental
en la protección de la salud pública.2) El valor de la notificación de series de casos en la
generación de nuevas hipótesis (RAM descono-cidas).
3) La utilidad del sistema de notificación espontá-nea de RAM (tarjeta amarilla) para identificar po-sibles nuevas señales (particularmente, en RAMde baja incidencia) y canalizar de forma sistemáticatoda información disponible, a partir de la cualpuedan tomarse eventualmente medidas infor-mativas o reguladoras.
4) De especial interés son las notificaciones deRAM graves e inesperadas por fármacos de re-ciente comercialización.
2.5. La evaluación de la relación de causalidad en Farmacovigilancia
El diagnóstico etiológico del origen medicamentosode una reacción adversa no dispone de pruebas especí-ficas; por ello, en general, suele realizarse por exclusión yno pasa del carácter de sospecha. La única certidumbrea este respecto la da la reexposición del paciente y la re-aparición del cuadro (positive rechallange). Esta infor-mación, por diversas razones (fundamentalmente éti-cas) sólo está disponible en un número limitado decasos.
Con la finalidad de analizar la fuerza de la relacióncausal entre el medicamento sospechoso y la reacciónadversa, y a la vez conseguir la mayor concordancia po-sible entre diferentes evaluadores, se han publicado di-ferentes algoritmos. Un algoritmo es, en síntesis, unconjunto de preguntas que pretenden “calificar” cadasospecha de relación medicamento-reacción con un de-terminado grado de probabilidad. Hay publicados has-ta 20 diferentes, si bien esta abundancia, en la práctica, noconstituye un problema ya que cada país utiliza uno só-lo, que se ha convertido en el algoritmo oficial del siste-ma y la integración de los datos en la base internacionalde Uppsala es posible ya que existe una correspondenciabastante estrecha entre los diversos algoritmos.
Para realizar la imputabilidad (relación de causalidadfármaco-reacción adversa) el SEFV utiliza cinco crite-rios:
1. Cronología entre el comienzo del tratamiento y laaparición del efecto indeseable.
2. Criterio bibliográfico (efecto indeseable conocidoo no).
3. Evolución tras la retirada del medicamento.4. Efecto de la readministración.5. Existencia de causa alternativa, explicación no
medicamentosa.En los Anexos de este capítulo se incluye el algo-
ritmo oficial del SEFV.
INTEGRACIÓN DE LOS SERVICIOS DE FARMACIA HOSPITALARIA EN LA ACTIVIDAD DE FARMACOVIGILANCIA
3.1 Sistemas de notificación voluntaria de reacciones adversas asociadas a medicamentos en el hospital
De acuerdo con Vallvé(6), puede decirse que existendos tipos de notificación voluntaria: la comunicaciónsistematizada y la no sistematizada. Hasta el año 1964 enque por primera vez un estado, Gran Bretaña, introdu-jo el programa de la “tarjeta amarilla” (que puede con-siderarse como el primer intento de comunicación sis-temática de reacciones adversas asociadas amedicamentos), la única forma de comunicación vo-luntaria fue la no sistematizada. Históricamente, en ge-neral la mayoría de las revistas biomédicas han inclui-do, e incluyen, en alguno de sus apartados, en forma decartas al director, notas clínicas u otras, la descripciónde efectos indeseables asociados a los medicamentos.Excede el interés de este capítulo el analizar este tipode comunicación, en forma de publicación, pero síconviene precisar, entre otras matizaciones, que no es in-compatible con su comunicación sistematizada, así co-mo el hecho de que muchas revistas incluso exigen pa-ra su publicación que previamente se comunique a losCentros de Farmacovigilancia; también interesa rese-ñar que éstos, al menos en España, revisan periódica-mente los casos comunicados por profesionales espa-ñoles en las revistas y extraen de ellas los que no les hansido comunicados, cuando contienen la informaciónmínima para hacerlo.
En general, se considera a la notificación espontáneasistematizada de sospechas de reacciones adversas porparte de los profesionales sanitarios (“tarjeta amarilla”)como el método más eficiente para la identificación deriesgos de los medicamentos previamente no conoci-
3
583FARMACOVIGILANCIA

dos. La finalidad de estos sistemas es: a) facilitar al pro-fesional la notificación mediante un formulario senci-llo, que contenga todos los aspectos informativos rele-vantes, b) recoger y validar dicha información, y c)registrarla en una base de datos común que posibilite lageneración de “señales”. En todo el proceso se asegurala confidencialidad (del paciente y del notificador).
Las ventajas y limitaciones de la notificación es-pontánea se muestran en la Tabla 2. Los principales va-lores de este método son por una parte su sencillez ypor otra su carácter universal, ya que potencialmenteabarca a toda la población, a todas las reacciones ad-versas y a todos los medicamentos desde el comienzomismo de la comercialización. Su principal desventaja esla infranotificación; así, por ejemplo, en el Reino Unido,el número de médicos que notifican no supera el 10% yen algún estudio se ha visto que sólo el 4% de los ca-sos de ingreso hospitalario asociado a medicamentosfue espontáneamente comunicado al Centro de Far-macovigilancia correspondiente(7). Esta circunstancia ha-ce que, por definición, el número de casos registrados deuna asociación medicamento-reacción represente sólouna pequeña parte de lo que existe en la realidad. El sis-tema da prioridad a la notificación de sospechas de re-acciones adversas graves y a las que involucran a medi-camentos nuevos (primeros cinco años desde suautorización), sin que por ello rechace las que no cum-plen estas condiciones. Por otra parte, es importante se-ñalar que al profesional sanitario únicamente se le pidela sospecha de que el medicamento ha podido participar
en la aparición de cualquier cuadro clínico. El centrocorrespondiente se encargará de evaluar el grado de re-lación causal, de acuerdo con el algoritmo oficial delSistema Español de Farmacovigilancia, descrito en elapartado anterior.
Es importante subrayar que, a pesar de todas las li-mitaciones (señaladas anteriormente) de las que adole-ce el sistema de “tarjeta amarilla”, su contribución enEspaña ha sido fundamental (y prácticamente única) entodos los riesgos identificados por el SEFV que han da-do lugar a la retirada del medicamento. En la Tabla 1 sedescriben todas ellas, pudiendo afirmarse que la identi-ficación del riesgo y la decisión de retirada se tomó en to-dos los casos (quizás con la excepción de la cinepazi-da), sobre la base de la notificación espontánea,mediante tarjeta amarilla, sin que otros métodos de Far-macovigilancia con mayor fuerza de asociación en tér-minos epidemiológicos, como los que se describen en elapartado 4, contribuyeran a la generación de la alerta oa la decisión.
3.2. Estudios de monitorización intensiva en hospitales
A lo largo de las diferentes exposiciones de este ca-pítulo se ha puesto de manifiesto el valor elevado de loshospitales como observatorio capaz de recoger datosde Farmacovigilancia de gran trascendencia para el sis-tema. No es de extrañar, por consiguiente, que desdehace muchos años se hayan venido implantando nu-
584 FARMACIA HOSPITALARIA
Tabla 2. Ventajas y limitaciones de la notificación espontánea.
Ventajas Limitaciones
Método sencillo Infranotificación. La principal consecuencia es la disminución de la sensibilidad
Abarca a toda la población No permite cuantificar incidencias
Rapidez en la detección La tasa de notificación no es constante
Abarca a todos lo medicamentos desde Difícil detección de reacciones adversas el comienzo de su comercialización de aparición retardada
No interfiere con los hábitos de prescripción Sesgo de selección. Medicamentos nuevos
Permite detectar reacciones adversas poco frecuentes

merosas herramientas destinadas a lograr dicho objeti-vo, algunas de las cuales han resultado ser enorme-mente prolíficas en término de cantidad de reacciones ad-versas graves, o de perfiles comparativos de reaccionesadversas entre miembros de un mismo grupo farma-cológico, o incluso de reacciones adversas no descritasantes.
Sin embargo, muchas veces se ha cuestionado larentabilidad de dichas actividades, llevadas a cabo de unmodo sistemático, ya que implican la existencia de unared estable de observadores, vinculada a Servicios clí-nicos que realizan básicamente otras actividades, quepodrían considerarse prioritarias. En este sentido, elHospital con sus pacientes se convertiría en una gigan-tesca cohorte, cuyo manejo resulta a veces complejo.
El denominado Boston Collaborative Drug Sur-veillance Program (BCDSP) surgió en la década de los70, recogiendo información sobre diagnósticos al altay detalles de las historias de 25.000 pacientes consecutivosen hospitales de Massachusetts. Formaron parte del es-tudio 24 hospitales, que cubrían el 45% de las camasdel área de Boston, con casi tres millones de habitan-tes. Dichos datos permitieron llevar a cabo un númeroamplio de subanálisis epidemiológicos (casos y contro-les) para asociar tratamientos con patologías yatrogéni-cas(8).
Se trató de un planteamiento pionero para la época,pero que tuvo que evolucionar a causa de los elevadoscostes económicos relacionados con intervenciones di-rectas; fue evolucionando hacia análisis de bases de da-tos, aunque con las limitaciones tecnológicas de la épo-ca.
Mucha de la información que se obtuvo ha pasadoa formar parte del acervo de la Farmacovigilancia: san-grado por heparina, potenciado por acetilsalicílico; se-dación excesiva por flurazepam en ancianos; fenitoína ehipoalbuminemia; tetraciclinas y aumentos en la ureaplasmática; interacciones con anticoagulantes orales;erupción cutánea y metamizol; y finalmente hemorragiasgastrointestinales producidas por medicamentos, temaque se encuentra lejos de estar cerrado a pesar del tiem-po transcurrido.
Con todo, conviene resaltar que la información ge-nerada a partir de dicho estudio supone, debido a su di-seño observacional, una fuente de señales con las limi-taciones inherentes al mismo.
El análisis de los medicamentos recibidos tres me-ses antes del ingreso también aportó informaciones úti-les (por ejemplo, en relación con el abuso crónico de
analgésicos); un hallazgo colateral consistió en la rela-ción negativa hallada entre el consumo regular de ácidoacetilsalicílico y el infarto, que más tarde ha tenido tan-ta trascendencia. Asimismo se descubrieron datos acer-ca del efecto negativo del tabaco, del alcohol o del café.
Como se ha mencionado antes, el planteamientodel BCDSP evolucionó hacia el análisis de bases de da-tos de prescripciones; se estudiaron los datos delGroup Health Cooperative (localizado en el área de Se-attle, del que formaban parte 280.000 miembros, conacceso libre a medicamentos y hospitales). Se obtuvoinformación acerca de la relación entre reemplaza-miento hormonal y cáncer, o entre fármacos y malfor-maciones congénitas.
Muchos de los datos citados antes condujeron aldiseño de estudios de casos y controles ad hoc (porejemplo, el que relaciona la toma de estrógenos y la en-fermedad vascular, tema que todavía en 2001 sigue vi-gente).
Un planteamiento similar, pero con ciertas particu-laridades, partió de hospitales británicos, donde seconstituyó el grupo denominado MEMO (MedicinesEvaluation Monitoring Group), que se encargó de ela-borar una base de datos conteniendo pacientes y fár-macos, de tal modo que permitiese la identificación de:pacientes que hayan recibido un determinado medica-mento y, de forma subsecuente, aquellos que hayan su-frido una reacción adversa asociada con dicha adminis-tración.
La existencia de métodos que recojan las prescrip-ciones y que validen la administración por enfermería,asociada a la actividad de los farmacéuticos de sala alreponer los stocks, permitían que la base de datos re-cogiese la situación real; sin embargo rápidamente sepuso de manifiesto que la información a tratar era rele-vante tan sólo para aquellos medicamentos prescritoscon suficiente frecuencia, siendo preciso crear una redde hospitales con metodología idéntica para garantizarun suficiente número de datos referentes a productos po-co prescritos. Así surgió el denominado “sistema Aber-deen-Dundee” que puede ser un modelo igualmenteválido que el BCDSP. En este sentido, una vez más se po-ne de manifiesto la necesidad de llevar a cabo programascoordinados, que abarquen un número suficiente de ca-mas hospitalarias.
El valor de una base de datos está relacionado conla calidad de los datos con los que la misma se alimenta;es fundamental que los informes de los pacientes con-tengan toda la información relevante y que exista algún
585FARMACOVIGILANCIA

mecanismo para garantizar el control de calidad en elproceso de transcripción de las órdenes, cuando la mis-ma se lleve a cabo, que es lo habitual en la actualidad;son los mismos problemas con los que se encuentracualquier base de datos en los hospitales españoles yponerlo de manifiesto fue trascendente para el MEMO.
El sistema Aberden-Dundee se desarrolló, al re-vés del BCDSP, para validar hipótesis, no para detec-t a rreacciones adversas que previamente no se sospecha-ran, aunque la base de datos existente también ha sidoexplotada en ocasiones con dicho objetivo. Tambiénhan podido verificarse informaciones, tan simples yadmitidas generalmente, como son que cuanto mayores el número de medicamentos que recibe un pacien-te, tanto más probable resulta que desarrolle unareacción adversa; o que las reacciones adversas sonuna de las causas más frecuentemente productoras dealargamiento de las estancias.
Existe, finalmente, la posibilidad de monitorizar, deun modo continuo, todos los acontecimientos que lessuceden a enfermos de una determinada unidad oplanta; dicha actividad puede llevarse a cabo de mu-chos modos, a través de personal de enfermería espe-cíficamente entrenado, mediante farmacéuticos de sa-la, o empleando enfoques mixtos. Del mismo modo, sepueden monitorizar de forma más o menos directa apacientes atendidos en Servicios de Urgencia, en Ra-diología, en Nefrología o en cualquier otro área; y elenfoque puede consistir en analizar a todos los aten-didos, o solamente a determinados grupos, que tienenen común el recibir algún medicamento o grupo deellos (por ejemplo, antirretrovirales), o sufrir de algunapatología concreta (diagnósticos alertantes).
Dicha monitorización puede plantearse de diversasformas: visitas diarias del monitor, alertas a través demedios informáticos más o menos automatizados,etc. Lógicamente, cuanto mayor es la necesidad depersonal para realizar la tarea, los costes aumentan es-pectacularmente y el rendimiento global de la activi-dad se resiente. En consecuencia, los diversos progra-mas que se han implantado en diferentes hospitaleshan tenido casi siempre un proyecto de investigacióndetrás (FIS, etc.), sin que los planteamientos hayan in-tentado el mantenimiento de la actividad en periodosdilatados de tiempo, o integrada dentro de la dinámi-ca puramente asistencial.
3.3. Explotación de datos informatizados
3.3.1. Unidosis
Actualmente, gracias a la implantación, más o menosgeneralizada, del sistema de distribución de medica-mentos en dosis unitaria, los Servicios de Farmacia dis-ponen de información inmediata y directa sobre el uso demedicamentos en el hospital. Disponer de un progra-ma informático que respalde el sistema de distribución demedicamentos en dosis unitarias, donde se transcriban lasórdenes médicas de tratamiento farmacológico, permi-te seguir la calidad de la farmacoterapia, tener un histo-rial farmacológico del paciente, detección de RAM yotros procesos. La idea consiste en aprovechar el cono-cimiento que tiene el farmacéutico de la farmacoterapiaque reciben los pacientes, desde la infraestructura queproporcionan los sistemas de distribución de medica-mentos en dosis unitaria para la detección de RAM.
Entre los distintos métodos para llevar a cabo laFarmacovigilancia en los hospitales como son: métodosepidemiológicos, vigilancia intensiva y notificación vo-luntaria, existen métodos indirectos que permiten iden-tificar RAM y de esta forma aumentar los índices de de-tección y notificación procedente de hospitales.
Cuanto mayor sea la cantidad de información obte-nida de la orden médica que se transcriba al programa in-formático de distribución de medicamentos de dosisunitaria (alergias, diagnóstico, medicación administradapuntualmente al paciente, etc.) mayores serán los méto-dos que podremos utilizar para la detección de RAM.
Monitorización de fármacos alertantes de posibles efectos adversos
Este método consiste en la identificación de pres-cripciones alertantes al revisarlas en el proceso de vali-dación y seguimiento del paciente en la dispensación endosis unitarias. Por “prescripciones alertantes” se en-tienden aquellas cuya prescripción puede estar motivadasecuencialmente por el tratamiento de un efecto adver-so en un paciente concreto y entre las que se puede incluirlas prescripciones de antídotos de fármacos, de antihis-tamínicos, la interrupción brusca de tratamientos y re-ducción de la dosis(1). Gracias al programa informáti-co de distribución de medicamentos en dosis unitariaspodemos obtener listados de pacientes a los que se haprescrito fármacos alertantes (Tabla 3) de efectos ad-versos, posteriormente, se revisa la órden médica del pa-ciente para ver si el fármaco se ha empleado para tratar
586 FARMACIA HOSPITALARIA

un efecto adverso. Las sospechas de RAM se comprue-ban con la historia clínica acudiendo a planta, teniendo encuenta causas alternativas a la prescripción alertante y a lasmanifestaciones clínicas del paciente.
Monitorización de diagnósticos alertantesOtra técnica para la detección de RAM consiste en
seleccionar, de todos los diagnósticos de pacientes que in-gresan a diario por urgencias, aquellos correspondien-tes a ciertos “diagnósticos alertantes”. Estos diagnósticosse definen como aquellos más sospechosos de estar re-lacionados con una RAM y, por tanto, susceptibles deser investigados posteriormente. De acuerdo con estecriterio de selección de pacientes, se acude a la planta dehospitalización donde se revisa exhaustivamente la his-toria clínica (enfermedad actual, hoja de enfermería, ór-denes de tratamiento, resultados de laboratorio, etc). Encaso positivo se comenta con el personal sanitario im-plicado y se entrevista al paciente, con el objeto de co-nocer la historia medicamentosa previa al ingreso; este pa-so es muy importante debido a que, a veces, algunosmedicamentos no aparecen recogidos en la historia clínicay otros en cambio figuran de modo incompleto. Por es-te motivo, un estudio que no sea prospectivo pierde va-lidez debido a los importantes sesgos cometidos a la ho-ra de relacionar medicamentos como responsables delas manifestaciones clínicas que padece el paciente(1).También se recogen pruebas analíticas y pruebas diag-nósticas que permitan descartar otras causas alternati-vas distintas a los medicamentos implicados. En todo
este proceso existen una serie de pasos intermedios, queincluyen documentación del caso y consulta con los clí-nicos para la confirmación o no de la sospecha. La Tabla4 recoge un listado de algunos diagnósticos alertantes(7).
Monitorización de alergiasEn esta fase de implantar sistemas que ayuden a
identificar a pacientes con RAM durante la hospitaliza-ción, se puede señalar como fuente de detección la mo-nitorización de alergias. Las alergias, anotadas en las ór-denes médicas de tratamiento farmacológico, seregistran en el sistema informático de unidosis, de talmanera que al introducir el número de historia del pa-ciente siempre aparezca este dato en la pantalla (inclusoen posteriores ingresos). Así, diariamente se obtiene unlistado en el que se refleja todos los pacientes con alergiasjunto con la medicación que están recibiendo. Un far-macéutico (o un sistema experto) revisa el listado y envíaun informe al médico responsable en caso de detectar al-guna probabilidad de reacción alérgica. El inconvenien-te de este método es que se depende de que el médicoanote la alergia en la orden médica de tratamiento far-macológico.
Monitorización de interacciones medicamentosas de importancia clínica
Consiste en detectar el mayor número de posiblesinteracciones medicamentosas existentes en el trata-miento farmacológico de los pacientes, bien a través del
587FARMACOVIGILANCIA
Tabla 3. Fármacos alertantes.
– Adrenalina– Dantroleno– Antihistamínicos– Diazepam IV– Corticoides IV o tópicos– Fenitoína IV– Fitomenadiona– Naloxona– Protamina– Resinas de intercambio iónico– Loperamida– Acetilcisteína-antídoto– Flumazenilo
Tabla 4. Relación de algunos diagnósticos alertan-tes.
– Eritema multiforme – Melenas– Erupción cutánea – Rectorragia– Urticaria – Hemorragia digestiva– Prurito – Hematemesis– Mialgia – Pancreatitis– Miopatía – Hiponatremia– Ataxia – Hiper e hipopotase-mia– Confusión – Hiper e hipoglucemia– Distonía, disquinesia – Edema– Temblor – Arritmia– Vértigo, mareo – Trombocitopenia– Cefalea – Leucopenia– Náuseas, vómitos – Shock anafiláctico– Intoxicación digitálica – Broncoespasmo

sistema informatizado de distribución de medicamen-tos en dosis unitaria o con la ayuda de un programa au-xiliar, de tal forma que se pueda detectar aquellas inter-acciones predeterminadas, teniendo como base datosfiables de la literatura. Es bien conocido que, a medida queaumenta el número de medicamentos que recibe un pa-ciente de forma concomitante, se incrementa el riesgo desufrir una reacción adversa. Del mismo modo sucede amedida que los pacientes están sujetos a estancias másprolongadas, o cuando la muestra se refiere a personas deedad avanzada. De esta forma parece que la utilización deposibles interacciones entre medicamentos como señalpodría resultar un método válido para detectar pacientessusceptibles de sufrir reacciones adversas en el hospital.Algunos ejemplos serían: ácido acetilsalicílico-heparina;los dos principios activos usados concomitantementeaumentan el riesgo de hemorragia. Amiodarona-digo-xina y digoxina-furosemida; el uso conjunto potencia lacardiotoxicidad.
Uso de los datos del laboratorio para la detec-ción y prevención de reacciones adversas a medica-mentos
El laboratorio de bioquímica es un departamen-to del hospital que se puede aprovechar para la de-tección y prevención de RAM. Llevando un controldiario automatizado de los tratamientos, a través dela unidosis, cruzándolos con datos proporcionadospor el laboratorio (creatinina, potasio, etc.) se puedenidentificar prescripciones de dosis excesivas: porejemplo, de un aminoglucósido en un paciente confunción renal deteriorada; o de benzodiazepinas enpacientes de edad avanzada. Puede asimismo ayu-dar a identificar aquellos pacientes que tengan pres-crita una dosis inapropiada, o un medicamento in-adecuado en función de sus característicasfisiopatológicas. Otra aplicación puede ser, porejemplo, identificar pacientes con niveles elevadosde potasio y en tratamiento con IECA, lo que puededar lugar a hiperpotasemia.
En la sección de farmacocinética del Servicio deFarmacia, el análisis de los pacientes que presentenconcentraciones tóxicas de fármacos (digoxina, fe-nitoína, gentamicina, etc.) puede ayudar a identifi-car posibles RAM y sobre todo a acometer el desa-rrollo de medidas preventivas para evitarlas.
Otro sistema de identificar RAM es a través de lasolicitud de pruebas diagnósticas para ciertos tipos deefectos adversos, como la solicitud de toxina Clos-
tridium difficile en el servicio de Microbiología, an-te la sospecha clínica en pacientes con posible diarreaasociada a antimicrobianos. La información se reci-be a partir de los resultados de las solicitudes de re-alización de la prueba de Clostridium difficile, com-binándola con el tratamiento antimicrobiano querecibe el paciente, proporcionado por la unidosis. Sihay una sospecha de RAM se revisa la historia clíni-ca y se lleva un seguimiento del paciente(1).
Otros métodosDetección de tratamientos duplicados. A través
de los datos informatizados de unidosis se puedeobtener listados de tratamientos de pacientes quetienen dos o más medicamentos de un mismo grupoterapéutico. Ejemplo: paciente asmático que ingresapor urgencias y le prescriben salbutamol sin sus-penderle el formoterol que trae de casa; o varios AI-NE de forma concomitante.
Seguimiento de determinados efectos adversosdiana y la revisión de fármacos habitualmente im-plicados en su producción (por ejemplo, detección deconvulsiones en pacientes en tratamiento con imi-penem o quinolonas).
3.3.2. Utilización del Conjunto Mínimo Básico de Datos (CMBD) en Farmacovigilancia
El Conjunto Mínimo Básico de Datos (CMBD) esun conjunto de variables obtenidas en el momento delalta, que recoge datos administrativos, clínicos, demo-gráficos y proporciona datos sobre el paciente, su en-torno, la institución que lo atiende y su proceso asis-tencial. Las variables de carácter médico (diagnósticoprincipal, otros diagnósticos, procedimiento quirúrgi-co y otros procedimientos diagnósticos-terapéuticos)se codifican según la Clasificación Internacional de lasEnfermedades-9-Modificación Clínica (CIE-9-MC).Su uso es obligado en el conjunto de los hospitales delsistema público de salud (SNS).
Entre las 22 variables (48 campos) de que consta elCMBD, se encuentran algunas de mayor interés relativopor sus potenciales aplicaciones en Farmacovigilancia.Existe una variable que registra la causa externa que haprovocado el diagnóstico principal o los otros diagnós-ticos. Siempre se utiliza como clasificación comple-mentaria al diagnóstico principal y a los otros diagnós-ticos; según el rango de valores E930-E949.9, es
588 FARMACIA HOSPITALARIA

posible conocer el subgrupo terapéutico que ha oca-sionado el efecto adverso (a las dosis habituales en te-rapéutica). Con los datos actuales del Sistema Españolde Farmacovigilancia se puede comprobar una infra-notificación de RAM graves, como consecuencia de laescasa notificación de origen hospitalario; a la vista de di-cha situación cabe utilizar la herramienta CBMD para ladetección de dichas reacciones(9).
Las posibles utilidades del CBMD en Farmacovi-gilancia se describen a continuación:
1. Detección de las reacciones adversas. Identifica-ción de pacientes que tienen un código de diag-nóstico CIE-9-MC de efecto adverso a un fár-maco. Consiste en seleccionar historias clínicasen las que figuren algunos de los códigos(E995.2, E930.0-E949.9), que son los que co-rresponden a los efectos adversos producidospor medicamentos cuando se utilizan en dosisterapéuticas; es decir, detección de reacciones ad-versas a medicamentos a través de los diagnósti-cos de alta. El CMBD tiene un carácter comple-mentario respecto a la notificación espontáneade reacciones adversas.
2. Pueden contribuir a aumentar el número de no-tificaciones graves registradas en FEDRA.
3. Generación de señales. El CMBD también per-mite realizar consultas más sensibles, p. e., identi-ficar una patología con independencia de queconste o no como causa externa una RAM. Si apartir de dicha búsqueda se procede a una revisiónsistemática retrospectiva de las historias clínicas,pueden encontrarse asociaciones fármaco-reac-ción que resulten desconocidas en aquel mo-mento.
4. Amplificación de señales y estimación de inci-dencia. Cuando se detecta en el SEFV una posi-ble nueva señal, con objeto de poder amplificar-la o de efectuar de forma ágil una aproximaciónsobre la incidencia de la misma puede ser de granutilidad la identificación de potenciales casos me-diante el CMBD, para proceder de forma siste-mática a la revisión de las correspondientes his-torias clínicas. Ej.: a partir de la supervisión de lashistorias clínicas con diagnóstico de coagulopatíaobstétrica en el CMBD se identificaron en 1998los casos expuestos a dinoprostona; al conocerel número de casos y el consumo de dinopros-tona durante dicho periodo, pudo estimarse la in-cidencia de coagulación intravascular disemina-
da, asociada a dinoprostona, en el Hospital Ma-ternal Virgen del Rocío de Sevilla.
5. Realización de estudios en fase IV. El CMBD secodifica una vez el paciente ha sido dado de alta;por lo tanto, el diseño del estudio tendrá un ca-rácter retrospectivo y limitante, particularmente encuanto a la exposición a los fármacos preingre-so.
Por otra parte, muchos hospitales tienen tambiéninformatizados los diagnósticos que motivan el ingresohospitalario desde que éste se produce; a partir de esta in-formación se han desarrollado distintos programas es-pecíficos de Farmacovigilancia: supervisión sistemáti-ca de una selección de diagnósticos de ingreso o depatologías raras, como agranulocitosis, síndrome deLyell, con un diseño caso-población. En este tipo deestudios se consigue obtener una anamnesis farmaco-lógica completa y, por consiguiente, mayor calidad deinformación, pero son más costosos de llevar a cabo.
6. Evaluación económica de los ingresos hospita-larios por RAM.
Es posible estimar el coste que generan los ingresospor RAM utilizando el CMBD, así como para determi-nadas RAM (como, por ejemplo, la hemorragia gas-trointestinal)(10); dado que se conocen los riesgos re-lativos de los distintos AINE, pueden hacerseestimaciones sobre la proporción de los ingresos (y cos-tes) potencialmente evitables, en caso de haberse pres-crito fármacos alternativos más seguros.
Las RAM que causan ingreso o prolongación deestancia hospitalaria son las que tienen mayor impactosanitario y económico; sin embargo, existe una marca-da infranotificación de las mismas motivada en partepor la baja participación de la mayoría de profesionalesde hospital. En este contexto, además de promover la no-tificación espontánea de RAM graves, tanto desde aten-ción primaria como sobre todo desde el ámbito hospi-talario, debería rentabilizarse el CMBD como fuente deinformación de RAM graves.
OTROS MÉTODOS DE FARMACOVIGILANCIA
4.1. Estudio de casos y controles
En este tipo de estudios, siguiendo a De Abajo etal.(11), los pacientes son seleccionados según presen-ten o no una enfermedad determinada. Los casos se-rán pacientes con la enfermedad y los controles pa-
4
589FARMACOVIGILANCIA

cientes seleccionados aleatoriamente de la misma po-blación fuente de la que surgen los casos y que no pre-sentan la enfermedad en el momento de su selección. Enambos grupos se estudia la exposición a los medica-mentos de interés en un intervalo de tiempo (ventana deexposición), previo al inicio de la enfermedad (día ín-dice), para los casos (o un día aleatorio para los contro-les). La determinación del día índice y de la ventana deexposición es crucial, y debe obedecer a criterios clínicosy epidemiológicos.
La exposición previa a los medicamentos se puedeobtener mediante entrevistas al paciente (a través deun cuestionario estructurado) o bien a través de la re-visión de su historia clínica. El método de obtenciónde dicha información deberá ser, en todo, igual en los ca-sos que en los controles, para evitar sesgos de infor-mación.
Este diseño es especialmente útil cuando se quiereestudiar reacciones adversas poco frecuentes o que re-quieren periodos largos de exposición o inducción pa-ra producirse, ya que se garantiza la inclusión de un nú-mero suficiente de casos sin necesidad de seguir atodos los sujetos de la población fuente de la que deri-van, como ocurriría si se eligiera un diseño de tipo co-horte. Otra ventaja de los estudios de casos y controleses que permiten analizar la asociación de la enfermedadcon diversos factores simultáneamente.
Su principal dificultad estriba en la selección ade-cuada del grupo control. Como se ha dicho, los con-troles deben ser una muestra de la población fuenteque da origen a los casos, pero lo difícil, a veces, es tras-ladar esta idea a un procedimiento operativo de selec-ción. Es interesante comprobar que los estudios de ca-sos y controles se pueden conceptualizar como unestudio de cohorte en el que la experiencia de exposiciónpersona-tiempo de los denominadores de incidenciase ha muestreado, en vez de haberla contabilizado en sutotalidad. Si la distribución de la exposición y de losposibles factores de confusión entre los controles esrepresentativa de su distribución en la población fuen-te (lo cual sólo se puede asegurar si el muestreo es ale-atorio), se puede hacer una estimación no sesgada delriesgo relativo de una enfermedad asociada a un me-dicamento en la población fuente, sin conocer los de-nominadores de incidencia en dicha población, ya quela fracción de muestreo es la misma entre los expuestosy los no expuestos. Es frecuente utilizar en los estudiosde casos y controles una medida de asociación, conocidacomo razón de ventaja, o más comúnmente por su tér-
mino inglés odds ratio (OR); pero si los controles sehan muestreado de forma aleatoria de la poblaciónfuente, se demuestra fácilmente que OR y RR coinciden.
Ie a / P1 a / P1 • ƒ a / P a • PRR= ––– = –––––– = –––––––– = –––––– = ––––– =OR
Io c / Po c / Po • ƒ c / P c • P
Donde a = casos expuestos y c = casos no expues-tos; P1 = Población fuente expuesta; Po = Poblaciónfuente no expuesta; ƒ = Fracción de muestreo; b =controles expuestos y d = controles no expuestos.
El problema surge cuando no se cuenta con unapoblación fuente identificada, desde la que realizar unmuestreo aleatorio. Para estas situaciones se recurre aestrategias subrogadas de selección de controles, asu-miendo que el procedimiento es independiente de laexposición; es decir, que la fracción de muestreo es lamisma entre los expuestos que entre los no expuestos.La validez del estudio queda depositada, pues, en la va-lidez de dicha asunción, lo cual con frecuencia no es fá-cilmente demostrable.
Como se deduce de la discusión precedente, los es-tudios de casos y controles no permiten estimar medi-das de frecuencia (incidencia o riesgo absoluto) de for-ma directa, ya que se desconoce la fracción de muestreoy, por tanto, el denominador.
4.2. Estudio de cohortes
Una cohorte es un conjunto de individuos que sonpartícipes de un determinado acontecimiento en unmomento dado; en Farmacovigilancia hay que hablarde un conjunto de pacientes, identificados, tratados conmedicamentos o no pero seguidos desde el principiocon el objetivo de recoger efectos adversos(12).
La población que la forma puede pertenecer a unárea geográfica (por ejemplo, la cohorte de Framing-ham o los pacientes ingresados en un hospital, que pro-vienen de su área de influencia), tener en común unadeterminada patología (por ejemplo, hemorragia diges-tiva), o el ser tratados con un determinado medica-mento o grupo de ellos (por ejemplo, los tratados con co-xib).
En consecuencia, muchos estudios de cohorte sesuperponen o resultan homologables con aquellas in-vestigaciones epidemiológicas que emplean bases dedatos (de prescripción, hospitalarias, etc.). En este sen-tido, existen cohortes sin intervención (puramente ob-
590 FARMACIA HOSPITALARIA

servacionales) y aquellas que se crean artificialmente(por ejemplo, cuando se lleva a cabo la incitación a laprescripción por parte de los laboratorios farmacéuticos).Incluso puede recurrirse a la selección de ciertos miem-bros de la cohorte (cuando ésta resulta excesivamente nu-merosa en términos de cantidad de pacientes) por me-dio de un sorteo; cuando dicho sorteo incluye apacientes tratados y no tratados, el método se acerca alensayo clínico, si bien entonces puede hablarse de dos co-hortes: pacientes expuestos y no expuestos.
Las cohortes pueden ser prospectivas, donde losmiembros se identifican y posteriormente se les sigue, ohistóricas (antes llamadas retrospectivas); un segui-miento continuado desde el pasado, que continúa en elfuturo, puede caracterizar una cohorte llamada ambis-pectiva.
La duración del seguimiento debe plantearse enfunción del problema que define la cohorte, es decir,aquella cuestión de seguridad que se desea aclarar. Eltema resulta crucial en el momento del diseño del se-guimiento, ya que existen reacciones adversas casi ins-tantáneas (por ejemplo, un shock anafiláctico, la apariciónde convulsiones, la aparición de trastornos del ritmocardiaco o la irritación local causada por la administra-ción parenteral), mientras que otras se desarrollan con ra-pidez (en días o en horas), como es el caso de la insufi-ciencia renal aguda o los trastornos hidroelectrolíticos;sin embargo, deben transcurrir meses o años para que seproduzca una insuficiencia renal crónica o una osteo-porosis; por no hablar de la producción de tumores,que requiere muchos años de seguimiento.
En Farmacovigilancia, por lo general puede inten-tarse responder mediante estudios de cohortes a cues-tiones como: identificación de reacciones adversas, des-cripción de la forma en que se produjeron y los factoresque aparecían asociados, comparación entre la toleran-cia de varios medicamentos o evaluación de una posiblerelación entre la exposición y el efecto adverso.
Las cohortes presentan escaso valor para recogerreacciones adversas nuevas; son mas útiles para detectarreacciones sospechadas o temidas a partir de datos pre-vios (por ejemplo, de tarjeta amarilla) o del propio me-canismo de acción farmacológico del producto.
Las cohortes observacionales resultan, por tanto, elmejor método para estimar con precisión el riesgo ab-soluto de que se produzca una reacción (o, al menos, latasa por unidad de tiempo). El riesgo relativo podría es-timarse posteriormente, a partir de dicha información;la cohorte permite saber si un producto determinado
puede producir una reacción adversa determinada; lafrecuencia con la que lo hace o su comparación, en su ca-so, con un tratamiento de referencia.
Como sucede a menudo en programas de investi-gación (y un estudio de cohorte debe plantearse comotal), es preciso responder antes de su inicio a cuestio-nes acerca de la calidad en su diseño, de la pertinencia delos datos que se desea obtener y si enrealidad no existe otro modo de contestar a la pregun-ta que se encuentra en el origen de la investigación.
4.3. Sistemas relacionados con el prescription-event monitoring (PEM)
El sistema PEM nació en Gran Bretaña, como tan-tos otros métodos de Farmacovigilancia(8). Es un sis-tema activo de búsqueda de acontecimientos relacio-nados con la prescripción, que se basa en el manejo decopias de las prescripciones de determinados medica-mentos que se desea monitorizar. Según el esquemaempleado, de cada prescripción se obtienen dos copias,una de las cuales se destina a la agencia que monitoriza,mientras que la segunda se une a un documento máscomplejo, en el que se registran los acontecimientosque ha tenido el paciente, previamente identificado porla primera copia.
Para PEM se debe definir lo que es un aconteci-miento (event): cualquier diagnóstico nuevo, o razónpara acudir a la consulta, o cualquier deterioro o mejo-ra de una determinada patología, o una reacción adver-sa, o cualquier otro tipo de queja que el médico considereimportante.
En el fondo, se trata de confeccionar una base de da-tos de prescripciones/pacientes/acontecimientos, di-ferente a aquellas que explotan simplemente las deprescripciones, en el sentido de que únicamente se fija laatención en determinados productos y se hace de formaprospectiva.
Permite comparaciones entre medicamentos conefectos terapéuticos similares, así como conformar hi-pótesis generadas a través de otros sistemas de obtenciónde señales.
4.4. Utilización de bases de datos en Farmacovigilancia
El término bases de datos resulta confuso, ya que ad-mite diversas acepciones y se emplea en cada contextoen muy diferentes sentidos. A los efectos de su utilización
591FARMACOVIGILANCIA

como método de Farmacovigilancia conviene precisarque se refiere a su uso para la investigación de losefectos adversos de los medicamentos. Para que unabase de datos automatizada pueda ser usada en la in-vestigación en Farmacovigilancia es imprescindibleque contenga, siguiendo el criterio de García Rodrí-guez(13), tres tipos de datos: demográficos (fecha denacimiento, sexo, periodo de seguimiento y estado vi-tal), del consumo de medicamentos (medicamento,dosis, presentación, fechas de comienzo y final) y deacontecimientos clínicos (diagnósticos, consultas aespecialistas, ingresos hospitalarios). Con una pers-pectiva de desarrollo histórico es preciso decir queestos tres tipos de datos han estado, y están, general-mente separados en la mayor parte de los registrosinformatizados y a su vez contenidos dentro de ba-ses de datos diferentes y no diseñadas para su uso far-macoepidemiológico. Por ello ha sido difícil utilizarloscon fines de Farmacovigilancia. Inicialmente, se des-arrollaron técnicas de conexión de registros (record-linkage) para realizar dichos estudios, las cuales sedescriben a continuación. Posteriormente, en parale-lo al desarrollo de la informatización de las consultasmédicas y en el registro informatizado de la docu-mentación clínica, se ha producido un cambio sus-tancial en las técnicas de trabajo farmacoepidemioló-gico, ya que en el diseño de algunas de estas bases setuvo en cuenta su utilización para la investigación. Deentre las diversas bases de datos existentes en el mun-do se ha elegido, como ejemplo para una descripciónmás detallada, la General Practice Research Database(GPRD) de Gran Bretaña y el proyecto BIFAP, por sereste último de origen y ámbito español.Record-linkage
La aparición de reacciones adversas inesperadasdurante la década de los 70 hizo emerger numerosossistemas de vinculación prescripción-reacción adver-sa, basados en la integración en una sola historia de to-dos los acontecimientos que le sucedían a una deter-minada persona; inicialmente, el record linkagejuntaba registros de nacimientos, muertes, admisio-nes hospitalarias, etc., en una base de datos localizadaen Oxford desde 1962. Posteriormente, la misma seamplió a casi 2 millones de personas y se planteó suempleo para, también, poderse aplicar a la detecciónd ereacciones adversas por medicamentos.
La existencia misma de las bases de datos de pa-cientes y prescripciones no es una garantía de que
puedan emplearse con el objetivo citado; de aquí quelos primeros estudios se llevaran a cabo en países conun elevado nivel de organización en la sanidad, comolos países escandinavos (donde apenas se han lleva-do a cabo últimamente análisis al respecto) o GranBretaña; también cumplían con dicho perfil determi-nadas organizaciones de seguros médicos pre-pagados,como el Kaiser-Permanente Medical Center de SanFrancisco, que disponen de farmacias propias; elGroup Health Co-operative de Puget Sound (en Se-attle, Estados Unidos) también dispone de una base dedatos utilizable, y también se han llevado a cabo es-tudios en la beneficencia pública americana (Medi-caid), siempre con un planteamiento similar.
Este último programa es el denominado COM-PASSTM (Computerized On-Line Medicaid Phar-maceutical Analysis and Surveillance System), quecruza datos de prescripciones con características delpaciente, analíticas y patologías. Permite llevar a ca-bo estudios de cohorte, análisis de reexposiciones po-sitivas, secuencia temporal de la toma del medica-mento (con las lógicas limitaciones de tratarse de unregistro de recetas), etc. Permite mantener una co-horte pediátrica (más de 12 millones de niños), otra deancianos y la valoración del riesgo de malformacio-nes fetales, por poner algunos ejemplos.
Las limitaciones del planteamiento son las mis-mas de cualquier estudio que emplee bases de datos:no es una buena herramienta para estudiar reaccio-nes adversas muy raras o por medicamentos pocoprescritos; no refleja la influencia de factores comoel hábito de fumar, la ocupación o la dieta; existe un re-traso (a veces muy marcado) entre la utilización delmedicamento y el manejo de los datos; la poblaciónque atiende no es representativa, al tratarse en este ca-so de pacientes pobres aunque otras bases de datosadolecen de sesgos parecidos. Finalmente, es necesa-rio depurar a menudo la base de datos, que contieneerrores.
En Gran Bretaña pudo aprovecharse la conexiónentre la facturación de recetas y las bases de datos depacientes; los datos recogidos de los mismos consis-tían en datos demográficos y las recetas se grababan apartir de calcos. Se introducían asimismo informa-ciones sobre morbilidad y mortalidad, ingresos hos-pitalarios o fallecimientos.
Como sucede siempre que se llevan a cabo estasactividades, los cruces informativos entre bases dedatos pueden conducir:
592 FARMACIA HOSPITALARIA

1) A producir hipótesis sobre reacciones adver-sas, al detectar efectos insospechados, queproceden de la búsqueda sistemática.
2) A validar hipótesis, a través de estudios adhoc.
3) A comprobar la seguridad de ciertos medica-mentos.
4) A analizar la utilización de los medicamentos(hábitos de prescripción).
5) A valorar la eficacia de ciertos medicamen-tos, o beneficios inesperados de los mismos.
Del mismo modo, para extraer de la actividadtodos sus potenciales beneficios, se requiere:1. Cooperación de los médicos.2. Confidencialidad.3. Importante aporte de fondos, para crear y
mantener la base de datos.GPRD
La General Practice Research Database(GPRD) es un archivo de historias clínicas de la po-blación atendida por médicos de asistencia primariaen Gran Bretaña, que comenzó a desarrollarse a fi-nales de los años ochenta; inicialmente surgió co-mo aplicación informática desarrollada por unaempresa privada, y desde octubre de 1999 está ges-tionada por la Medicines Control Agency, equiva-lente británico de la Agencia Española del Medica-mento. Actualmente, introducen datos en la GPRDalrededor de 1.500 médicos generalistas, pertene-cientes a unos 400 equipos de primaria, que cubrenuna población de más de tres millones de personas.El médico informatiza la mayor parte de la infor-mación clínica de sus pacientes, la cual en un siste-ma clásico es habitualmente guardada en papel, in-cluyendo datos demográficos, diagnósticosrealizados por el médico de primaria y especialistas,diagnósticos derivados de hospitalizaciones y datosdetallados de los tratamientos prescritos. La infor-mación introducida es siempre de carácter anóni-mo. Utilizando esta base de datos se han realizado,entre otros, estudios de Farmacovigilancia comoson la estimación del riesgo de afectación hepáticarelacionada con amoxicilina (0,3 por 10.000 pres-cripciones) y amoxicilina-ácido clavulánico (1,7 por10.000 prescripciones)(11). Otro estudio estimó elriesgo de hemorragia digestiva asociada a inhibido-res selectivos de la recaptación de serotonina(ISRS), riesgo que por sí sólo se consideró bajo (1por cada 8.000 prescripciones) pero que aumentaba
de forma importante si a la vez se consumía algúnAINE(14). Por otra parte, la GPRD también haservido para realizar estudios de efectividad. Así,por ejemplo, en un estudio comparativo de raniti-dina, cimetidina y omeprazol en el tratamiento demantenimiento de pacientes con un episodio pre-vio de hemorragia digestiva alta (HDA) por ulcus, sepudo comprobar que el omeprazol era la alternati-va más efectiva en la reducción de recurrencias deHDA.
BIFAPEl proyecto BIFAP (Base de Datos para la Investigación
Farmacoepidemiológica en Atención Primaria) pretendecrear una base de datos similar a la descrita anteriormente,pero en el ámbito de la asistencia primaria española. Es pro-movido conjuntamente por la Agencia Española del Medi-camento (www.agemed.es) y el Centro Español de Investi-gación Farmacoepidemiológica (CEIFE) y cuenta con elapoyo de dos de las sociedades españolas de atención pri-maria, SEMFYC y SEMERGEN, con las que se ha firmadoun convenio de colaboración y que están representadas enel Comité Científico. Un aspecto importante es la total ga-rantía de confidencialidad, para lo cual se aplica un procedi-miento disociativo que rompe el vínculo entre los datos deidentificación personal y los datos clínicos relativos a la salud.El proyecto está abierto a cualquier médico de primaria quepuede participar, de forma personal, individual y voluntaria-mente. Existe un formulario de petición de información enla página web (www.bifap.org).
ANÁLISIS DE LA CONTRIBUCIÓN DE LOS SERVICIOS DE FARMACIA A LA FARMACOVIGILANCIA EN ESPAÑA (1989-2001 hasta octubre)
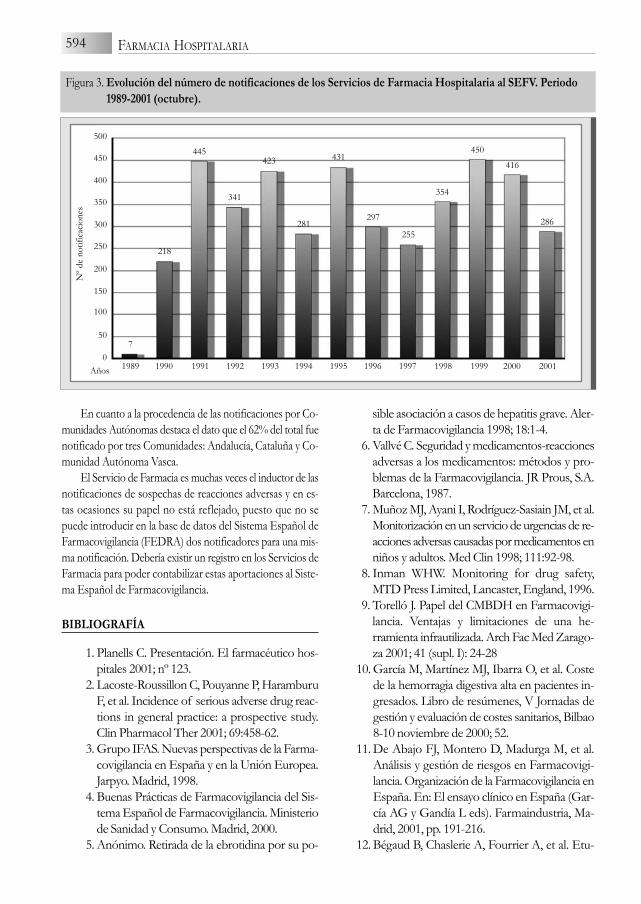
En el periodo desde 1989 al 2001 (octubre), la contri-bución de los Servicios de Farmacia al SEFV fue de un totalde 4.204 notificaciones de sospechas de RAM, Figura 3, so-bre el total de 65.122 notificaciones registradas en FEDRA(6,45%). De ellas, 1.535 (37%) fueron evaluadas como “leves”,1.855 (44%) como “moderadas”, 721 (17%) como “graves”y 93 (2%) como “mortales”. En cuanto al grado de conoci-miento en la literatura de la asociación medicamento-reac-ción, 3.479 (83%) fueron conocidas, de 293 (7%) existían re-ferencias de casos, 428 (10%) eran desconocidas en elmomento de la notificación y en 4 la información farmaco-lógica estaba en contra de dicha asociación.
5
593FARMACOVIGILANCIA
En cuanto a la procedencia de las notificaciones por Co-munidades Autónomas destaca el dato que el 62% del total fuenotificado por tres Comunidades: Andalucía, Cataluña y Co-munidad Autónoma Vasca.
El Servicio de Farmacia es muchas veces el inductor de lasnotificaciones de sospechas de reacciones adversas y en es-tas ocasiones su papel no está reflejado, puesto que no sepuede introducir en la base de datos del Sistema Español deFarmacovigilancia (FEDRA) dos notificadores para una mis-ma notificación. Debería existir un registro en los Servicios deFarmacia para poder contabilizar estas aportaciones al Siste-ma Español de Farmacovigilancia.
BIBLIOGRAFÍA
1. Planells C. Presentación. El farmacéutico hos-pitales 2001; nº 123.
2. Lacoste-Roussillon C, Pouyanne P, HaramburuF, et al. Incidence of serious adverse drug reac-tions in general practice: a prospective study.Clin Pharmacol Ther 2001; 69:458-62.
3. Grupo IFAS. Nuevas perspectivas de la Farma-covigilancia en España y en la Unión Europea.Jarpyo. Madrid, 1998.
4. Buenas Prácticas de Farmacovigilancia del Sis-tema Español de Farmacovigilancia. Ministeriode Sanidad y Consumo. Madrid, 2000.
5. Anónimo. Retirada de la ebrotidina por su po-
sible asociación a casos de hepatitis grave. Aler-ta de Farmacovigilancia 1998; 18:1-4.
6. Vallvé C. Seguridad y medicamentos-reaccionesadversas a los medicamentos: métodos y pro-blemas de la Farmacovigilancia. JR Prous, S.A.Barcelona, 1987.
7. Muñoz MJ, Ayani I, Rodríguez-Sasiain JM, et al.Monitorización en un servicio de urgencias de re-acciones adversas causadas por medicamentos enniños y adultos. Med Clin 1998; 111:92-98.
8. Inman WHW. Monitoring for drug safety,MTD Press Limited, Lancaster, England, 1996.
9. Torelló J. Papel del CMBDH en Farmacovigi-lancia. Ventajas y limitaciones de una he-rramienta infrautilizada. Arch Fac Med Zarago-za 2001; 41 (supl. I): 24-28
10. García M, Martínez MJ, Ibarra O, et al. Costede la hemorragia digestiva alta en pacientes in-gresados. Libro de resúmenes, V Jornadas degestión y evaluación de costes sanitarios, Bilbao8-10 noviembre de 2000; 52.
11. De Abajo FJ, Montero D, Madurga M, et al.Análisis y gestión de riesgos en Farmacovigi-lancia. Organización de la Farmacovigilancia enEspaña. En: El ensayo clínico en España (Gar-cía AG y Gandía L eds). Farmaindustria, Ma-drid, 2001, pp. 191-216.
12. Bégaud B, Chaslerie A, Fourrier A, et al. Etu-
594 FARMACIA HOSPITALARIA
Figura 3. Evolución del número de notificaciones de los Servicios de Farmacia Hospitalaria al SEFV. Periodo1989-2001 (octubre).
500
450
400
350
300
250
200
150
100
50
01989
7
218
445
341
423
281
431
297
255
354
450
416
286
1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001
Nº d
e no
tifica
cione
s
Años

des de cohortes en pharmacovigilance, 2ª ed.ARME- Pharmacovigilance editions. Borde-aux, 1995.
13. Elguero J, Pérez Gutthann S. Bases de datos enfarmacología y terapéutica. Monografías Dr.Antonio Esteve (nº 18). Ediciones Doyma,
Barcelona, 1996.14. De Abajo FJ. Rodríguez LA. Montero D. Asso-
ciation between selective serotonin reuptake in-hibitors and upper gastrointestinal bleeding: po-pulation based case-control study. BMJ 1999;319 (7217):1106-9.
595FARMACOVIGILANCIA

ANEXO 1
596 FARMACIA HOSPITALARIA
Facsímil de la tarjeta amarilla.

597FARMACOVIGILANCIA
ANEXO 2
Algoritmo del Sistema Español de Farmacovigilancia.
Puntuación
1 Cronología
1 +2Compatible
2 +1No totalmente compatible
3 0No información
Intervalo entre la administración del medicamento
y el efecto indeseable
4 -1Cronología incompatible
5 +2Caso particular de un síndrome de abstinencia
1 +2Mejora con la retirada
2 -2Retirado no mejora
4 -2No retirado mejora la reacción
3 +1No retirado no mejora la reacción
5 0No hayinformación
6 0Muerteo efecto irreversible
7 +1No retirado pero hay tolerancia al efecto
8 +1No retirado mejora con eltratamiento sintomático
2 Bibliografía
1 +2Conocida en la literaturade referencia
2 +1Conocida ocasionalmente
3 0Desconocida
Grado de conocimiento en la Bibliografía de la
relación medicamento-efecto
adverso
4 -1Sin relación con el medicamento
3 Retirada del me-dicamento
Evolución del efecto indeseable

598 FARMACIA HOSPITALARIA
Algoritmo del Sistema Español de Farmacovigilancia (continuación).
Puntuación
2 -1Negativa: el efecto no reaparece
3 0No hay reexposición o no hay información
1 +3Positiva: reaparición del efecto
4 0Muerte o efecto irreversible
5 +1Positiva para una
especialidad distinta con el mismo principio
activo
2 -1Verosimilitud parecida para
el medicamento y otras causas
1 -3Sí: patología u otro medicamento
3 0Falta de información
4 +1No existe
4 Readministración
6 +1Positiva para una
especialidad distinta con elmismo mecanismo
de acción o reactividad cru-zada
Existencia de causaalternativa al medi-
camento
La puntuación total respecto de las categorías de probalidad se establece de acuerdo a las cinco categorías siguientes:
– No clasificada falta – Improbable < 0– Condicional 1 - 3 – Posible 4 - 5– Probable 6 - 7 – Definida > 8
5
1 2 3 4 5
lllll



![149.ppt [Modo de compatibilidad] - SEFH | Sociedad ... · 6Causalidad de la RAM segggún algoritmo de Karch-Lasagna del Sistema Español de Farmacovigilancia: ... bradicardias e hipoglucemias](https://img.pdfslide.tips/doc/110x75/5bc1944d09d3f2fb5b8de331/149ppt-modo-de-compatibilidad-sefh-sociedad-6causalidad-de-la-ram.jpg)

























