Embed Size (px)
Citation preview

1
INTRODUÇÃO A MÉTODOS CROMATOGRÁFICOS CROMATOGRAFIA EM CAMADA DELGADA E EM COLUNA
OBJETIVOS
� Estudar o comportamento de substâncias orgânicas em termos de estrutura química versus fenômeno adsorção/partição cromatográfica.
� Demonstrar experimentalmente princípios fundamentais da cromatografia. � Empregar a técnica de cromatografia em camada delgada na análise uma amostra e utilizar o
Rf na identificação de compostos orgânicos usualmente presentes em diversas formulações farmacêuticas.
� Empregar a técnica de cromatografia em coluna na purificação de compostos orgânicos. LEITURA RECOMENDADA Métodos de separação, cromatografia, fator de retenção, em livros de Química Analítica ou Química Orgânica. INTRODUÇÃO
Os termos cromatografia, cromatograma e método cromatográfico são atribuídos ao
botânico russo Mikhael S. Tswett, que em 1906 utilizou estes termos para descrever suas experiências com extratos de folhas e gema de ovo. Tswett usou colunas de vidro contendo vários sólidos finamente divididos através das quais filtrou os extratos, lavando-os a seguir com éter de petróleo. Obteve assim, uma separação dos componentes em bandas coloridas ao longo das colunas. Os termos derivam das palavras gregas chrom (cor) e graphe (escrever), embora o processo não dependa da cor, exceto para facilitar a identificação dos componentes separados.
Existem várias técnicas cromatográficas, desde as mais simples, como cromatografia em coluna ou camada fina, até as mais sofisticadas, como a cromatografia líquida de alta eficiência (CLAE)1, que exigem aparelhagem complexa. Entre os métodos de análises as técnicas cromatográficas ocupam um lugar de destaque na química e bioquímica devido à eficiência e facilidade na separação, identificação e quantificação das espécies químicas presentes em uma amostra, mesmo que constituída de misturas complexas. A cromatografia é, fundamentalmente, um método físico-químico de separação dos componentes de uma mistura, realizada por meio da distribuição dos mesmos entre duas fases em íntimo contato. Uma das fases permanece estacionária enquanto a outra se move através dela. Durante a passagem da fase móvel pela estacionária, os componentes de uma mistura se distribuem entre elas, de tal forma que cada um dos componentes é retido seletivamente na fase estacionária, de acordo com sua maior ou menor interação, resultando em migrações diferenciadas. A cromatografia de adsorção baseia-se no grau de partição relativa das substâncias entre uma fase líquida móvel (o eluente) e uma fase sólida estacionária (o adsorvente) geralmente a sílica-gel (SiO
2.xH
2O) ou o óxido de alumínio (Al
2O
3). Outros adsorventes ocasionalmente usados são o
Florisil (um silicato de magnésio), o carvão ativo, o hidróxido de magnésio, o carbonato de cálcio, certas poli-amidas e até mesmo o açúcar. O grau de adsorção depende de vários fatores, como a quantidade de água presente na fase sólida (que será mais reativa quanto mais sêca estiver), da estrutura da substância adsorvida (presença de grupos funcionais polares, capacidade de formar ligações hidrogênio, etc.), da ocupação dos sítios ativos da fase sólida pelo solvente e da força atrativa entre o soluto e o solvente (Figura 1).

2
N
Al2O3
HH
R
O Al
O
O H O
R
C
R
RO
O Al
C
O
ORHOAl
δδ
δ δ
Figura 1. Algumas formas de visualizar as interações entre alumina e componentes orgânicos
de uma amostra. Estruturas similares poderiam ser escritas para a sílicagel. Cromatografia em camada delgada (CCD) A cromatografia em camada delgada (CCD) é uma técnica de fácil execução, rotineiramente usada no laboratório de química orgânica. Amostras muito pequenas (1-100µg) podem ser rapidamente analisadas. Devido à conveniência e rapidez usamos a técnica para: estabelecer se dois compostos são idênticos, verificar a pureza de um composto, determinar o número de componentes em uma mistura, determinar o solvente apropriado para separação em uma coluna cromatográfica, monitorar a separação de uma mistura em uma coluna cromatográfica ou acompanhar o progresso de uma reação.
A técnica consiste em aplicar uma camada fina do adsorvente finamente pulverizado (geralmente sílica ou óxido de alumínio, às vezes adicionados de material fluorescente à luz ultra-violeta) sobre uma placa lisa e plana (vidro ou alumínio). O adsorvente é fixado à placa por adição de gesso ou álcool poli-vinílico. Alguns micro-litros de solução da amostra a ser examinada são aplicados próximos a uma das bordas da placa, e a mesma imersa alguns milímetros em um eluente mantido em recipiente fechado. O eluente, por força da capilaridade, percorre a fase fixa em movimento ascendente, carreando consigo os componentes da amostra. Diferentes compostos ascendem a diferentes alturas dependendo de suas estruturas moleculares. A técnica é especialmente útil no caso de compostos pouco voláteis ou sensíveis ao calor, isto é, substâncias para as quais a cromatografia em fase gasosa é inadequada. Instruções gerais sobre a técnica CCD
A escolha do solvente de eluição (desenvolvimento) adequado é realizada da seguinte maneira: uma solução da amostra nos vários solventes que se deseja testar é aplicada, por meio de tubos capilares, sobre uma camada do adsorvente a ser usado (Figura 2). Quando o círculo do solvente tiver atingido a posição final, o solvente apropriado é aquele que tiver arrastado a amostra a cerca de 0,3 a 0,5 do raio do círculo do solvente (caso a amostra seja incolor é obviamente necessário revelar a posição da substância eluída mediante uso de um reagente adequado!).
Figura 2. Escolha de solvente para a cromatografia em camada delgada
(a) Bom desenvolvimento. (b) Superdesenvolvimento. (c) Subdesenvolvimento. Na Tabela 1 encontramos uma relação, em ordem crescente de polaridade, dos solventes mais usados em cromatografia. Pode-se usar misturas de dois ou mais solventes como eluente, de maneira a modular a polaridade. A mistura mais usada é a de acetato de etila com éter de petróleo ou hexano, por ser constituída de solventes razoavelmente baratos, voláteis e de regular ou baixa toxicidade.

3
Éter de petróleo Ciclohexano Tetracloreto de carbono Benzeno Cloreto de metileno Clorofórmio Éter etílico Acetato de etila Piridina Acetona Álcool n-propílico Etanol Água
Ácido acético Tabela 1: Solventes cromatográficos em ordem crescente de polaridadea. a (polaridade, neste contexto, só tem sentido para a cromatografia e não coincide, necessariamente, com a polaridade medida, por exemplo, pela constante dielétrica). As amostras são aplicadas nas cromatoplacas em forma de soluções (a 1-10% em solvente volátil) a cerca de 1cm da base inferior (Figura 3.1), mediante uso de um tubo capilar. Alguns cuidados devem ser tomados para evitar que a camada do adsorvente seja alterada, ou que o halo de difusão da amostra seja maior que 2 mm. Soluções diluídas da amostra podem ser aplicadas repetidamente, esperando-se a evaporação do solvente antes de nova aplicação, a fim de obter maior concentração no ponto de partida. Mais de uma amostra pode ser aplicada em uma placa, mantendo-se uma distância de 1 cm entre os pontos, para evitar contato entre as amostras. A placa, com a extremidade semeada para baixo, é então mergulhada no solvente selecionado, mantido em cuba fechada (câmara).É importante observar que o nível do solvente deverá ficar abaixo dos pontos de aplicação (Figura 3.2). Durante a corrida do solvente o sistema deve permanecer fechado, para garantir saturação da câmara pelo vapor do solvente. Um pedaço de papel de filtro, colocado na parede interna da câmara, acelera a saturação. A cromatoplaca deverá ser retirada da cuba somente quando a frente do solvente atingir um limite pré-determinado na parte superior da placa (cerca de 1 cm). Após o desenvolvimento as cromatoplacas são secas e as posições dos componentes da amostra visualizadas por um meio adequado. A visualização (revelação) pode ser feita por métodos físicos ou químicos. Placas preparadas com adição de um material fluorescente permitem visualizar facilmente compostos UV-ativos, que aparecem como manchas escuras em fundo fluorescente, se a placa for iluminada com lâmpada de luz ultravioleta (cuidado).

4
Figura 3. (1) Aplicação da amostra na cromatoplaca (2) Eluição de uma placa cromatográfica;
(3) Revelação de uma placa cromatográfica em cuba de iodo Reagentes químicos podem também ser usados. Quase todas as classes de substâncias orgânicas (exceto alcanos e haletos alifáticos) reagem com iodo formando complexos de transferência de carga. Exposição da placa aos vapores do iodo (Figura 3.3) revelam manchas marrom, tanto mais escuras quanto maior o tempo de exposição e a concentração da amostra. A formação desses complexos é reversível (muitas vezes rapidamente), de modo que a posição das manchas escuras deve ser marcada prontamente. A grande vantagem desse método é que, na maioria dos casos, o iodo pode ser eliminado posteriormente por aquecimento da placa, a qual pode ser borrifada, a seguir, com outro revelador (ácido sulfúrico, vanilina sulfúrica, tricloreto de antimônio, permanganato de potássio/ácido sulfúrico, etc.). A literatura química traz uma série de referências a reveladores para cromatografia em camada delgada, variando desde os de aplicação muito específica até os mais gerais. Em geral os métodos químicos de revelação são destrutivos e aplicáveis a separações analíticas, mas não em separações preparativas. Quando as condições experimentais são completamente especificadas é possível identificar uma substância pelo seu valor de Rf. Define-se Rf, como a razão entre a distância
percorrida na placa pela substância e a distância percorrida pelo solvente (medidas no centro da mancha da substância e a frente do solvente, Figura 4b). Contudo, para fins de identificação de uma substância o solvente escolhido para eluição deve carrear o composto a um Rf entre 0,3 e 0,6 e
uma amostra autêntica deve ser aplicada lado a lado na mesma placa, para assegurar igualdade de condições experimentais. Além disso, como muitas substâncias podem ter o mesmo valor de Rf,
assim com podem ter o mesmo ponto de fusão, métodos adicionais devem ser utilizados para identificação inequívoca do composto.
Distância percorrida pela mancha desde a origem (�x)
Rf = ----------------------------------------------------------------------------------- Distância percorrida pelo solvente desde a origem (�y)

5
(b)(a)
Origem
Marca da
frente do
solvente
Componente 1
Componente 2
Mancha
cromatográfica
Origem
Marca da
frente do
solvente
∆x
∆y
Rf = ∆x/∆y
Figura 4. (a) Cromatograma de uma separação em coluna; (b) Determinação de Rf no cromatograma em camada fina
Experimental da Parte A-I – Preparação das placas cromatográficas Experimento demonstrativo
Objetivo: Demonstrar experimentalmente a preparação artesanal de placas para uso na cromatografia em camada delgada. Procedimento 1. As placas limpas e secas, não devem ser tocadas com os dedos. Segure-as pelas bordas. 2. Misture num béquer 30 g de sílica gel com 60 mL de água destilada até obter uma suspensão
homogênea. 3. Mantendo-se as placas em posição horizontal, transferir uma porção adequada da suspensão para
a superfície das placas, espalhando-a uniformemente com o auxílio de um bastão de vidro. 4. Para facilitar a distribuição homogênea da suspensão, manter a placa apoiada sobre a bancada,
erguendo-se alternadamente as extremidades até que visualmente toda a superfície esteja uniforme. A espessura do recobrimento deve ser aproximadamente 0,3 mm.
5. Repousa-se a placa em uma superfície plana horizontal e deixa-se secar ao ar durante 15 a 30 minutos, quando o adsorvente adquire uma aparência opaca.
6. Para serem utilizadas, as placas devem ser ativadas por aquecimento em uma estufa a 110 oC durante 30 minutos . Este processo visa eliminar a toda a água adsorvida no suporte.
As placas assim preparadas estão prontas para uso e podem ser guardadas em lugar protegido tomando-se cuidado para não tocar, contaminar ou ferir a camada de adsorvente. Micro-placas, muito úteis na análise rápida de misturas reacionais, podem ser facilmente obtidas por imersão de lâminas de microscópio numa suspensão de 35 g de sílica (para CCD) em cerca de 100 mL de clorofórmio ou tetracloreto de carbono. Duas lâminas, arrumadas face a face, são rapidamente imersas e, com um movimento constante, removidas da suspensão. Após a separação das lâminas, e evaporação do CHCl3 (ou CCl4), umedecem-se as placas com vapor de água para fixar o adsorvente. Obtém-se, assim, duas micro-placas com superfície homogênea de adsorvente em um dos lados, prontas para uso.

6
Parte Experimental A-II. Cromatografia em camada delgada (CCD) na análise de cafeína e N-acetil-p-aminofenol em analgésicos
Objetivo: Investigar a presença de cafeína e N-acetil-p-aminofenol (paracetamol) em analgésicos [Tylenol, Saridon, Dorflex, Paracetamol (Genérico), etc.]. Procedimento 1. Aplicar em pontos separados da placa cromatográfica, a 1 cm da borda inferior e mantendo cerca
de 1 cm de distância entre os pontos de aplicação, uma gotinha (tubos capilares) das soluções de cafeína (1), N-acetil-p-aminofenol (2) e das amostras de analgésicos (soluções 3 a 5).
2. Adicionar uma certa quantidade de acetato de etila à cuba de cromatografia de modo que o mesmo alcance o nível de cerca de 5 mm.
3. Revestir a parede interna da cuba com um pedaço de papel de filtro que vá até o fundo. Feche a cuba e aguardar alguns minutos para que a atmosfera da cuba fique saturada com vapores do solvente de eluição. Eventualmente o nível do líquido deverá ser completado.
4. Introduzir a placa na cuba de modo que o nível do líquido fique abaixo dos pontos de aplicação. Esperar o desenvolvimento do cromatograma mantendo a cuba fechada.
5. Quando a frente do solvente chegar a 1 cm da borda superior da placa, retira-la e marcar a distância percorrida. Deixar a placa secar totalmente ao ar (10 a 15 minutos).
6. Antes da revelação com iodo examinar o cromatograma sob luz UV. Marcar cuidadosamente o contorno das manchas das substâncias UV-ativas, com o auxílio de uma lapiseira ou outro objeto pontiagudo.
7. Para revelação definitiva, inserir a placa em recipiente seco contendo um pouco de cristais de iodo sólido. Fechar devidamente a câmara de iodo e aguardar o surgimento de manchas na placa.
8. Medir (pelo centro da mancha!) as distâncias percorridas pelos componentes de cada amostra analisada.
Discussão 1. Com base no experimento realizado responda as seguintes questões:
(a) Esquematize um desenho do cromatograma e diga quantos componentes foram detectados em cada amostra aplicada?
(b) Determine os Rf das substâncias puras (cafeína e N-acetil-p-aminofenol) e de todos os componentes das soluções de analgésico analisadas (soluções 3 a 5).
(c) Você pode inferir inequivocamente que algumas amostras contêm paracetamol? Ou cafeína? Ou nenhum dos dois? Ou outros constituintes? Explique.
2. Sobre a técnica cromatografia em camada delgada, responda:
(a) Quais os fundamentos básicos? (b) Cite os usos mais consagrados da técnica (importância). (c) Enumere as vantagens e desvantagens da técnica. (d) Que características deve ter um solvente para ser considerado um bom eluente de
desenvolvimento? No caso em questão você considera o acetato de etila um bom solvente de eluição?
(e) Que artifícios podemos utilizar quando suspeitamos que o(s) componente(s) da amostra a ser analisada pode não ser visível a olho nu?
3. (Provão 2000) Uma mistura contendo os compostos I, II e III, representados abaixo, foi analisada
pela técnica de cromatografia em camada fina. A mistura foi aplicada em uma cromatoplaca de sílica sem indicador de fluorescência. Após eluição com hexano:éter etílico (4:1) e revelação sob luz ultravioleta (254nm), a cromatoplaca apresentou o seguinte aspecto:

7
O
(I) (II) (III)
Rf = 0,77 Rf = 0,34
Quando a cromatoplaca foi revelada com iodo, foram observadas três manchas. Pode-se afirmar que as manchas obtidas nos Rf 0,34 e 0,77 correspondem, respectivamente, às estruturas: (a) I e II. (b) I e III. (c) II e III. (d) III e I. (d) III e II.
Cromatografia em coluna A cromatografia em coluna é geralmente usada na separação preparativa ou purificação de substâncias orgânicas. A técnica requer uso de tubos de vidro, montados verticalmente, contendo o suporte sólido finamente dividido (fase estacionária), em geral sílica gel ou óxido de alumínio. A substância que se deseja purificar (ou mistura que se quer fracionar) é aplicada no topo da coluna e a eluição da substância efetuada mediante a percolação com solvente adequado. Na parte inferior da coluna recolhe-se um número de frações, nas quais se encontram os componentes da mistura separados. A velocidade com que uma substância trafega pela coluna depende de sua polaridade, da polaridade da fase estacionária e da polaridade do solvente (eluente). Se o composto é mais atraído pela fase estacionária do que pelo solvente, ele migrará lentamente. Se sua afinidade for maior pelo solvente ele migrará mais rapidamente, gastando menos tempo e solvente. O êxito de uma coluna dependerá então da escolha do suporte e solvente adequados. Em alguns casos é possível visualizar a separação dos componentes de uma mistura observando o desenvolvimento de faixas diferentes na coluna ou iluminando-a com luz ultravioleta (a coluna precisa ser de quartzo!). Entretanto, na maioria das vezes, torna-se necessário o acompanhar a separação dos componentes examinando cada fração eluída por cromatografia em camada delgada (ccd). Instruções gerais sobre a técnica de cromatografia em coluna 1. Escolha da fase estacionária e do solvente para uma coluna cromatográfica Os suportes e solventes usados em uma coluna cromatográfica, são normalmente, os mesmos utilizados em CCD. Portanto devem ser escolhidos mediante um exame prévio da amostra por cromatografia em camada delgada. A quantidade de adsorvente utilizada depende da quantidade de amostra a ser separada e do tipo de adsorvente. Misturas pouco complexas, tendo sílica gel como suporte requer, no mínimo, a relação 1:50 amostra/sílica. 2. Empacotamento da coluna As colunas de vidro utilizadas na técnica têm a configuração que lembra uma bureta. Suas dimensões, obviamente, dependem da quantidade da amostra a ser processada. Dois métodos são comumente usados para a introdução do suporte na coluna (empacotamento). No primeiro método o

8
suporte é introduzido seco (Figura 5a,b). Coloca-se uma pequena bucha de algodão na base inferior da coluna para evitar a passagem do suporte sólido pela torneira e a mesma é preenchida até cerca da metade com solvente. Com a ajuda de um funil adaptado na parte superior da coluna introduz-se, lentamente, a fase estacionária que será empacotada com o solvente, tendo-se o cuidado de retirar quaisquer bolhas de ar formadas. O empacotamento poderá ser auxiliado com a introdução e escoamento de novas quantidades de solvente. Nunca deixe a superfície do suporte livre de solvente. No segundo método o suporte é introduzido em suspensão no solvente, portanto, os mesmos cuidados devem ser observados durante a preparação e o empacotamento da coluna.
Figura 5. Colunas cromatográficas. (a) Solvente adicionado à coluna;
(b) suporte sólido sendo introduzido na coluna. 3. Introdução da mistura de compostos a ser separada; eluição dos compostos e monitoramento da coluna Quando a coluna estiver preparada deixa-se escoar o solvente até que reste uma diminuta quantidade de solvente na superfície (evitar a todo custo que a coluna seque!). Introduzir, na sequência, uma solução concentrada (no solvente de eluição) do material que se deseja separar, com a ajuda de uma pipeta de Pasteur, tendo cuidado para não perturbar a superfície do adsorvente. Se a mistura for líquida ela poderá ser introduzida diretamente na coluna. Uma metodologia alternativa envolve a impregnação da amostra em pequena quantidade do adsorvente, com o auxílio de um solvente, seguida de evaporação e introdução do pó resultante (farofa) na coluna. Esse procedimento é particularmente útil quando pelo menos um dos constituintes da amostra se dissolve apenas em solventes muitos polares. Inicialmente deixa-se eluir a amostra com um pouco de solvente até quase a secura (evitando-se que a coluna seque!). Introduz-se uma nova quantidade de solvente e a operação repetida. A coluna é, então, preenchida com o solvente, e iniciada a eluição propriamente dita. À medida que as frações vão sendo recolhidas na base da coluna (usando vários Erlenmeyers numerados) torna-se necessário adicionar mais solvente (Figura 6), cuja polaridade eventualmente pode ser modificada mediante mistura com outro solvente polar. Faça um monitoramento das frações recolhidas por CCD, para verificar se todos os componentes da amostra foram eluídos e quais as frações podem ser combinadas, pois um componente individual pode estar presente em várias frações contíguas. Algumas frações podem conter mais de um componente, essas frações não devem ser reunidas com as que contém apenas um componente. Posto que as frações recolhidas encontram-se diluídas, é necessário evapora-las para obter o componente puro. Em alguns casos, a evaporação do solvente é indispensável, mesmo antes da análise cromatográfica.

9
Figura 6. Coluna cromatográfica acoplada a um funil de separação
para manter o nível do solvente na superfície superior.
Cromatografia Rápida (flash-column chromatography, FC)1 Uma técnica muito utilizada no fracionamento de uma mistura de compostos é a cromatografia rápida ou cromatografia sob baixa pressão. A técnica, que se vale de colunas curtas e eluição acelerada sob pressão, efetua a separação de componentes com diferenças de Rf ≥ 0,05 em tempo relativamente reduzido (1-3h). Pressões mais elevadas permitem a separação de maneira mais rápida (10-15min), entretanto, a resolução é apenas moderada (∆Rf ≥ 0,15). O aparato necessário para a técnica consiste em uma coluna com diâmetro apropriado, equipada com reservatório de solvente e válvula de controle de pressão (Figura 7). A pressão necessária é fornecida por cilindro de nitrogênio ou argônio, e a quantidade de suporte a ser utilizada normalmente calculada multiplicando-se a quantidade de material que se deseja separar por fator de 20-60. A coluna empacotada deverá ter cerca de 10 a 15 cm. A escolha do eluente é feita da mesma forma que numa coluna normal (CCD), recomendando-se o eluente capaz de carrear o componente desejado da mistura a um Rf = 0,35. A coluna é preenchida com adsorvente (sílica-gel 60) e o solvente de eluição forçado sob pressão até que a coluna esteja empacotada (livre de bolhas de gás). Adiciona-se, então, a amostra no topo da coluna (com as precauções mencionadas na cromatografia de coluna convencional) e procede-se a eluição sob pressão, a uma velocidade controlada (≈ 2 pol/min medida pelo nível do eluente). Uma separação típica por esta técnica é mostrada na Figura 8. Por essa técnica também se recomenda não deixar a coluna secar.
1 Still, W. C.; Kahn, M.; Mitra, A.; Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. J. Org. Chem., 1978, 43, 2923.

10
Figura 7. Coluna para cromatografia rápida.
Figura 8. Separação típica em cromatografia rápida.
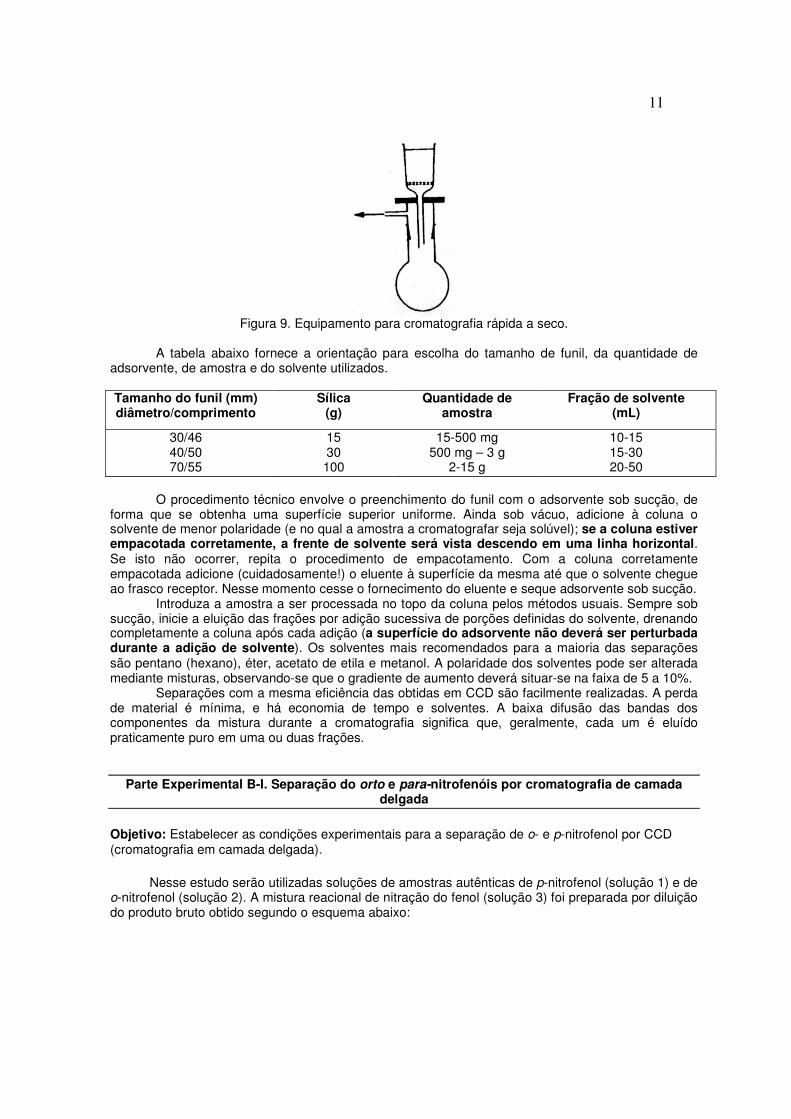
A cromatografia rápida é um bom método para a separação de componentes de uma mistura de complexidade moderada. Entretanto, a necessidade de pressão e aparelhagem sofisticada (colunas com diâmetros variáveis, válvula para controle de pressão, reservatório de solvente, cilindro de gases) constituem fatores limitantes da técnica. Cromatografica rápida a seco (dry-column flash chromatografy)2 Um método alternativo à cromatografia rápida (FC) é a cromatografia rápida a seco. A técnica combina as vantagens de aparelhagem e operação simples. Um fator de distinção entre ambas é o uso de sucção em lugar da pressão positiva. Um segundo fator de diferenciação é que os solventes de eluição são adicionados em volumes pré-determinados e a coluna drenada até a secura, antes da adição de uma nova adição do eluente. A aparelhagem usada constitui-se apenas de um funil cilíndrico sinterizado, adaptado a um balão com saída para trompa d’água (Figura 9).
2 Harwood, L. M.; “Dry-Column” Flash chromatography. Aldrichimica Acta, 1985, 18, 25; (b) Yau, E. K.; Coward,
J. K.; Filtering-Column Chromatography - A Fast, Convenient Chromatographic Method - Aldrichimica Acta, 1988, 21, 106.
11
Figura 9. Equipamento para cromatografia rápida a seco.
A tabela abaixo fornece a orientação para escolha do tamanho de funil, da quantidade de
adsorvente, de amostra e do solvente utilizados. Tamanho do funil (mm) diâmetro/comprimento
Sílica (g)
Quantidade de amostra
Fração de solvente (mL)
30/46 15 15-500 mg 10-15 40/50 30 500 mg – 3 g 15-30 70/55 100 2-15 g 20-50
O procedimento técnico envolve o preenchimento do funil com o adsorvente sob sucção, de
forma que se obtenha uma superfície superior uniforme. Ainda sob vácuo, adicione à coluna o solvente de menor polaridade (e no qual a amostra a cromatografar seja solúvel); se a coluna estiver empacotada corretamente, a frente de solvente será vista descendo em uma linha horizontal. Se isto não ocorrer, repita o procedimento de empacotamento. Com a coluna corretamente empacotada adicione (cuidadosamente!) o eluente à superfície da mesma até que o solvente chegue ao frasco receptor. Nesse momento cesse o fornecimento do eluente e seque adsorvente sob sucção.
Introduza a amostra a ser processada no topo da coluna pelos métodos usuais. Sempre sob sucção, inicie a eluição das frações por adição sucessiva de porções definidas do solvente, drenando completamente a coluna após cada adição (a superfície do adsorvente não deverá ser perturbada durante a adição de solvente). Os solventes mais recomendados para a maioria das separações são pentano (hexano), éter, acetato de etila e metanol. A polaridade dos solventes pode ser alterada mediante misturas, observando-se que o gradiente de aumento deverá situar-se na faixa de 5 a 10%.
Separações com a mesma eficiência das obtidas em CCD são facilmente realizadas. A perda de material é mínima, e há economia de tempo e solventes. A baixa difusão das bandas dos componentes da mistura durante a cromatografia significa que, geralmente, cada um é eluído praticamente puro em uma ou duas frações.
Parte Experimental B-I. Separação do orto e para-nitrofenóis por cromatografia de camada delgada
Objetivo: Estabelecer as condições experimentais para a separação de o- e p-nitrofenol por CCD (cromatografia em camada delgada).
Nesse estudo serão utilizadas soluções de amostras autênticas de p-nitrofenol (solução 1) e de o-nitrofenol (solução 2). A mistura reacional de nitração do fenol (solução 3) foi preparada por diluição do produto bruto obtido segundo o esquema abaixo:

12
OH
HNO3-H2O
OH
NO2
OH
NO2
+
Procedimento
1. Preparar três câmaras cromatográficas com alguns mL dos solventes em ensaio (por ex. diclorometano, acetato de etila, hexano, metanol, puros ou com polaridades ajustadas por misturas). Colocar meia folha de papel de filtro encostada à parede das câmaras para acelerar a saturação das mesmas com o vapor do solvente.
2. Preparar três micro-placas com sílica gel para CCD (confira métodos descritos na parte teórica sobre CCD). Com um tubo capilar, aplicar em cada micro-placa, devidamente espaçadas entre si e a cerca de 10 mm da margem, as soluções autênticas de p-nitrofenol e o-nitrofenol, e a solução da mistura reacional da nitração do fenol. As manchas resultantes não devem atingir diâmetros superiores a 3 mm.
3. Marcar na placa o ponto até onde o solvente deve atingir - cerca de 10 mm da borda superior. 4. Mergulhar cuidadosamente cada micro-placa em cada solvente escolhido, de modo que os
pontos de aplicação das amostras fiquem acima do nível do solvente. Aguardar o desenvolvimento dos cromatogramas. Atingido o ponto final, remover as placas.
5. Observar os resultados, estabelecendo qual solvente efetuou melhor separação (as manchas devem ser visíveis!) Para fins de registro, desenhar as cromatoplacas.
Discussão
1. Com base nos resultados escolha o melhor solvente de desenvolvimento (melhor separação). 2. Determine os valores de Rf de cada componente, obtidos na melhor separação.
3. Você pode inferir inequivocamente que além do o- e p-nitrofenol, existe um outro componente na solução 3? Em caso afirmativo, que impureza (contaminante) poderia estar presente na solução reacional? Explique.
4. Qual (ais) dos eluentes testados você recomendaria para eluir a coluna cromatográfica? Justifique.
PARTE EXPERIMENTAL B-II. Separação da mistura de o- e p-nitrofenol por cromatografia em coluna
Procedimento
1. Preparação da coluna
• Colocar um pequeno chumaço de algodão hidrófilo na base inferior da coluna (use bastão de tamanho adequado). Encher a coluna até cerca de 1/4 com o primeiro eluente da série. Testar se a torneira está estanque.
• Pesar aproximadamente 20 g de sílica-gel em béquer de 100 mL; adicionar o primeiro eluente em quantidade bastante para formar uma papa homogênea; misturar bem removendo as bolhas de ar.

13
• Abrir a coluna de modo a gotejar o solvente em um Erlenmeyer durante o processo de empacotamento.
• Com a ajuda de bastão de vidro deitar, de modo contínuo, a suspensão de sílica na coluna (o solvente recolhido no Erlenmeyer deve ser utilizado novamente para carrear todo adsorvente para a coluna). Se a adição da suspensão de adsorvente à coluna for interrompida por algum tempo, o mesmo tende a sedimentar-se, formando zonas estratificadas. Uma maneira de evitar esta situação é agitar o solvente na coluna com uma vareta de vidro entre duas adições sucessivas.
• Deixar gotejar o solvente 5-10 minutos após o enchimento da coluna para facilitar a sedimentação da sílica. Fechar a torneira quando o solvente apenas cobrir a coluna de sílica.
• Opcionalmente colocar uma rodela de papel de filtro, ou fina camada de algodão, no topo da coluna para proteger a superfície da sílica (recomendável após introdução da amostra a fracionar! ).
2. Preparação e aplicação da amostra
• Com o auxílio de pipeta ou seringa introduzir, lentamente, no topo da coluna cerca de 2 mL da solução de nitrofenóis.
• Deixar que a solução a cromatografar penetre na coluna mediante controle da torneira. Com pequenas quantidades de solvente carrear toda a amostra para dentro da coluna.
3. Eluição dos componentes da amostra
• Encher a coluna com o solvente (ou mistura de solventes) menos polar, tomando extrema precaução para não perturbar a superfície do adsorvente (a rodela de papel de filtro ou camada de algodão são úteis nesse estágio!).
• Recolher as frações eluídas em Erlenmayers de 50 mL, previamente numerados. O volume de cada fração deve, em média, ser de aproximadamente 1/10 do volume da coluna. Vigiar constantemente o volume de solvente na coluna, a qual deve jamais secar!
• Monitorar a separação e pureza de cada fração por cromatografia em camada delgada (ccd). A eluição deverá ser continuada até a saída total de todos os componentes.
• As frações contendo cada componente puro deverão ser reunidas em balões apropriados e o eluente evaporado com auxílio de roto-evaporador.
Discussão
1. Baseado em argumentos estruturais, comente sobre separação do o-nitrofenol e p-nitrofenol. Que outras diferenças nas propriedades físicas desses dois isômeros podem ser explicadas usando-se os mesmos argumentos? (Propriedades do o-nitrofenol e p-nitrofenol descritas no Index Merck, cópia anexa).
2. Compare os obtidos na CCD com os tempos de retenção observados em cromatografia em fase gasosa (CG, cromatogramas anexos).
3. (Provão 2001) Um químico deseja separar os compostos abaixo por cromatografia em coluna, utilizando sílica como adsorvente.
CH2CH3 CH2CH3
N
CH2CH3
(X) (Y) (Z) Para tal, percolou a coluna seqüencialmente com os solventes I, II e III, que formam uma série eluotrópica. A primeira fração continha o composto X, a segunda fração, o composto Y, e a última

14
fração, o composto Z. Qual dos sistemas de solventes abaixo serve como fase líquida para justificar a ordem de eluição encontrada? Solvente I Solvente II Solvente III (A) hexano acetonitrila tolueno (B) hexano tolueno CH2Cl2 (C) tolueno hexano acetonitrila (D) acetonitrila CH2Cl2 hexano (E) CH2Cl2 tolueno hexano
1. (Provão 2001) A cromatografia líquida de alta eficiência (CLAE) é um dos métodos
cromatográficos mais modernos utilizados em análise (CLAE analítica) e separação/purificação de misturas (CLAE preparativa). Abaixo são dados os cromatogramas X, Y e Z de uma mistura de compostos presentes em analgésicos: aspirina (A), cafeína (B), fenacetina (C) e paracetamol (D), utilizando três fases móveis diferentes, no modo isocrático, em uma mesma coluna.
Avaliando esses cromatogramas, responda às perguntas abaixo. (a) Qual a fase móvel mais apropriada para ser utilizada em escala preparativa, e a fase móvel mais
adequada para utilização em escala analítica, considerando um grande número de amostras a serem analisadas? Justifique sua resposta.
(b) Qual o tipo de coluna (fase reversa ou fase normal) utilizada nestes três experimentos? Justifique sua resposta.
(c) Sabendo-se que o composto mais polar elui primeiro, qual o composto de maior tempo de retenção? Justifique sua resposta.
DISPOSIÇÃO DOS RESÍDUOS E INSUMOS � Alguns resíduos aquosos poderão ser descartados na pia com água corrente. O demais resíduos
líquidos e sólidos (solventes, mistura de solventes, sílica, etc.) deverão ser acondicionados em frascos apropriados, segundo orientação do instrutor.
� Os produtos sólidos e líquidos deverão ser reservados, após a purificação, para utilização como
insumos em práticas posteriores.





























