Embed Size (px)
Citation preview

MOLEKULARNE METODE Autori:
Søren M. Karst Mads Albertsen Rasmus H. Kirkegaard Morten S. Dueholm Per H. Nielsen
Recenzent: Holger Daims
8.1 UVOD Molekularne metode se u području otpadnih voda mogu koristiti za brzo, pouzdano i jeftino identificiranje relevantnih mikroorganizama. U nekim slučajevima, moguće je i povezati identifikaciju s funkcijom, no iznenađenja su česta. Noviji primjer je to da se sada zna da su određeni nitrifikanti, za koje se vjerovalo da imaju jednostavnu i dobro opisanu metodologiju, puno raznovrsniji nego što se to dosad mislilo (Daims i sur., 2015). Funkcija se tiče i njihove fiziologije (heterotrofi, nitrifikanti, fermentacijski mikroorganizmi itd.) kao i njihove morfologije (nitaste ili jednostanične), što je važno za njihov ukupni učinak u sustavima obrade otpadne vode. Poznavanjem identiteta i funkcije mikroorganizama može biti moguće manipulirati njihovom prisutnošću kako bi se optimizirao rad uređaja, npr. osigurala prisutnost nitrifikanata ili uklonile nitaste vrste koje formiraju pjenu.
Molekularna identifikacija mikroorganizama se obično temelji na genu 16S rRNA. Međutim, u nekim se slučajevima identifikacija umjesto toga radi sekvenciranjem funkcionalnih gena, poput onih koji kodiraju amonijev enzim monooksigenazu (AMO) za mikroorganizme koji oksidiraju amonij (Rotthauwe i sur., 1997; Okano i sur., 2004), budući da oni daju veću
filogenetičku razlučivost, što bi moglo biti korisno za detaljnije studije.
Najčešće korištene metode identifikacije u području otpadnih voda bile su kvantitativna lančana reakcija polimerazom (qPCR) u stvarnom vremenu, stvaranje zbirke klonova i tehnike „uzimanja otiska prstiju“ poput gel elektroforeze u gradijentu denaturirajućeg agensa (DGGE; Muyzer, 1999) ili polimorfizma duljine krajnjih restrikcijskih fragmentata (T-RFLP) (Marsh, 1999; Marzorati i sur., 2008). Međutim, te metode „uzimanja otiska prstiju“ se gotovo više uopće ne koriste budući da ih je često teško koristiti i pružaju manje informacija u usporedbi s metodama koje se temelje na masovnom paralelnom sekvenciranju, te se njihovo daljnje korištenje ne može preporučiti.
Masovno paralelno sekvenciranje može se koristiti za metagenomiku ili metatranskriptomiku, gdje se sekvencira čitava DNA ili eksprimirani geni (mRNA) iz određene zajednice. Ipak, te metode ne smatramo značajnima za većinu čitatelja ove knjige budući da zahtijevaju znatne vještine iz molekularne biologije i bioinformatike.
8

286 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
Umjesto toga, za rutinske analize zajednica mikroorganizama predlaže se masovno sekvenciranje amplikona, koje će i biti detaljnije opisano. Tom se metodom dobiva popis mikroorganizama i procjena njihove relativne brojnosti. Jedna od prvih platformi za sekvenciranje, korištena za masovno sekvenciranje amplikona bila je Roche 454 (često se naziva ‘pirosekvenciranje’). Danas je, međutim, zastarjela (ultimo 2016), a tržištem sekvenciranja amplikona trenutno dominira platforma Illumina. Pomoću platforme Illumina moguće je na brz, lagan i jeftin način analizirati na stotine uzoraka u usporedbi s prethodnim tehnikama.
Identifikacija mikroorganizama se obično provodi uspoređivanjem nepoznatih sljedova s poznatnim referentnim skupom definirane taksonomije. U ovom poglavlju preporučujemo bazu podataka MiDAS (midasfieldguide.org), stručnu bazu podataka namijenjenu specifično za mikroorganizme na području pročišćavanja otpadnih voda. Uključeni su kanonski ili putativni nazivi za najuobičajenije taksone na razini roda i mogu se koristiti kao zajednički vokabular za sve istraživače na predmetnom području radi referiranja na iste organizme. Baza podataka MiDAS također daje sve raspoložive funkcionalne informacije o oko 150 najbrojnijih mikroorganizama prisutnih u danskim uređajima za pročišćavanje otpadnih voda (UPOV), a vjerojatno i širom svijeta (McIlroy i sur., 2015).
U ovom ćemo se poglavlju fokusirati na metode koje se odabiru danas i u idućih nekoliko godina u mikrobiologiji otpadnih voda: ekstrakcija DNA, qPCR i sekvenciranje amplikona.
8.2 EKSTRAKCIJA DNA
8.2.1 Opća razmatranja
Za svaku analizu sastava mikroorganizama pomoću sekvenciranja DNA neophodan je optimiziran i standardiziran protokol za ekstrakciju DNA. Razlog je jednostavna činjenica da se mikroorganizmi jako razlikuju u otpornosti na različite metode razgradnje (lize) (Thomas i sur., 2012; Guillén-Navarro i sur., 2015). Prema tome, mikroorganizmi sa staničnim stjenkama koje je teško lizirati će efektivno izgledati manje brojni ako se koriste protokoli ekstrakcije lošiji od optimalnih (Bollet i sur., 1991; Filippidou i sur., 2015). Nadalje, uzorci aktivnog mulja sadrže razne kemijske tvari zbog kojih neke tehnike, zbog inhibicije, ne uspijevaju (Guo i Zhang, 2013). Dakle, metoda za ekstraciju DNA mora biti robusna kako bi se mogla nositi s izazovima pred koje
je stavlja aktivni mulj. Međutim, unatoč velikom naporu na istraživanju različitih protokola ekstrakcije DNA, nije izgledno da će ikada postojati savršeni protokol. Odstupanja (pristranost) uvedena u ekstrakciju DNA ne mogu se izbjeći, već se tek mogu svesti na minimum (Guo i Zhang, 2013; Albertsen i sur., 2015). Svrha ovog odjeljka je u kratkim crtama prikazati korake uključene u ekstrakciju DNA i dati općenite preporuke pri radu s aktivnim muljem. Uz to, predstavljen je i protokol optimiran za korištenje s aktivnim muljem na temelju protokola koji su razvili Albertsen i sur. (2015).
8.2.2 Uzorkovanje
Od ključne je važnosti da je uzorak reprezentativan za aktivni mulj u bioaeracijskom bazenu uređajaili u reaktoru u laboratorijskom mjerilu. Za velike uređaje se preporučuje da se uzme veći volumen (1 L) iz bazenas dobro izmiješanim sadržajem, potom provede homogenizacija i na kraju uzme poduzorak od 3×2 mL alikvota koji se mogu lako zamrznuti i godinama čuvati na -20 °C dok ih se ne analizira. U idealnom slučaju, čuvaju se biološke kopije kako bi se osiguralo da se može analizirati varijanca uzorka i da je dostupan višak biomase u slučaju da nešto pođe krivo. Važno je vrijeme između uzorkovanja i zamrzavanja svesti na minimum budući da bi promijenjeni uvjeti izvan originalne sredine mogli pogodovati rastu određenih vrsta više od ostalih, zbog čega uzorak ne bi bio prikladan za usporednu analizu (Guo i Zhang, 2013). Uzorkovanje bi po mogućnosti trebalo provoditi prilično često, npr. svaki tjedan, i uzorci bi se trebali čuvati zamrznuti u ‘bio-banci’ za kasnije korištenje. Kako broj uzoraka brzo raste, važno je svaki uzorak jasno označiti i voditi evidenciju s identifikacijskim oznakama uzoraka, identifikacijskim oznakama reaktora/bazena, datumima, srodnim kemijskim mjerenjima, te svim dodatnim informacijama koje bi mogle biti značajneza kasniju analizu mikroorganizama.
8.2.3 Ekstrakcija DNA
Ekstrakcija DNA uključuje nekoliko općih koraka koji se modificiraju i kombiniraju na različite načine u nizu komercijalnih kompleta ovisno o ciljnim organizmima, vrsti sredine, te svrsi ekstrahirane DNA. Zajednički koraci su uništenje i liza stanice, uklanjanje proteina, kemijsko uklanjanje, te eluiranje DNA (slika 8.1).
8.2.3.1 Razgradnja stanice
Razvijene su različite metode za razgradnju stanica kako bi se oslobodila njihova DNA. Neke metode koriste

MOLEKULARNE METODE 287
kemijske tvari za raskidanje stanica, neke koriste enzimsku razgradnju staničnih struktura, a neke koriste fizički stres poput ciklusa zamrzavanja i otapanja ili mehanički stres poput ultrazvuka ili udaranja kuglica (Bollet i sur., 1991; Tsai i Olson, 1991; Zhou i sur., 1996). Razvijeni su standardni kompleti koji koriste kombinacije tih strategija optimizirani za različite vrste stanica i uzoraka. Različita priroda aktivnog mulja predstavlja izazov za nekoliko od tih pristupa zbog različitih vrsti inhibicije (Tullis i Rubin, 1980). Ipak, mehanička razgradnja se pokazala vrlo robusnom i nije pogođena učincima inhibicije (Salonen i sur., 2010; Guo i Zhang, 2013; Albertsen i sur., 2015). Razgradnja stanica se često provodi u otopinama s deterdžentima i surfaktantima koji kasnijim centrifugiranjem podržavaju kidanje i uklanjanje sastavnica stanične membrane kao što su lipidi.
Ako se prilikom razgradnje stanica modificira bilo koji parametar, važno je ispitati učinke na prinos i cjelovitost ekstrahirane DNA. Povećavanjem intenziteta ili trajanja nekih koraka vjerojatno će se povećati prinos do određene razine zasićenja. Međutim, uslijed dužeg trajanja i jačeg intenziteta DNA bi se mogla raspuknuti, čime bi na raspolaganju bio manji broj uzoraka (Bollet i sur., 1991; Bürgmann i sur., 2001).
Slika 8.1 Glavni koraci u ekstrakciji DNA.
8.2.3.2 Inhibiranje aktivnosti nukleaza i uklanjanje proteina
Stanice mikroorganizama posjeduju brojne enzime specijalizirane za razgradnju DNA (nukleaze), zbog čega je neophodno da se iste ukloni ili inhibira čim je prije moguće nakon razgradnje stanice. Uobičajena metoda za uklanjanje aktivnosti nukleaza je dodavanje proteaza, koji su specijalizirane za razgradnju proteina, uključujući nukleaze. Nakon toga proteini se uklanjaju tako da se
poveća koncentracija soli, zbog čega će se proteini taložiti. Talog se kasnije može ukloniti centrifugiranjem, pri čemu će DNA ostati u otopini, a proteini u talogu (Miller i sur., 1999).
8.2.3.3 Pročišćavanje
Zajedno s ekstrahiranom DNA dolazi još je DNA vrsta nukleidne kiseline također prisutne u stanici, a to je RNA. Molekule RNA se često uklanjaju dodavanjem enzima RNaze koji razgrađuje samo RNA, a DNA ostavlja netaknutom (Miller i sur., 1999). Kada se slijede ovi koraci, važno je ukloniti neželjene soli, deterdžente, proteine i ostale reagense korištene u procesu stanične razgradnje. To pročišćavanje se obično provodi taloženjem DNA pomoću etanola, budući da DNA u njemu nije topiva i centrifugiranjem se može istaložiti (Bollet i sur., 1991). DNA se može oprati tako da se supernatant zamijeni novim etanolom. Alternativno, DNA se može adsorbirati na matricu na filtru ili silicijevom gelu što omogućuje pranje te kasnije oslobađanje mijenjanjem koncentracije soli.
8.2.3.4 Eluiranje i pohranjivanje
Nakon što je DNA izolirana od ostalih staničnih sastavnih dijelova i kemikalija korištenih u ekstrakciji, etanol se može uklonitiisparavanjem, a DNA se može otopiti u vodi u kojoj nema DNaze ili u zaštitnoj pufer otopini kao što je pufer Tris-EDTA (TE) (Miller i sur., 1999). Pročišćena DNA se može zamrznuti i pohraniti godinama ili se može nekoliko tjedana čuvati u hladnjaku.
8.2.4 Kvantifikacija i cjelovitost
Ovisno o obradi koja slijedi, važno je kvantificirati DNA i provjeriti da nije previše fragmentirana. Najbolja kvantifikacija se dobiva metodom koja se temelji na fluorescenciji kojom se može razlikovati DNA od RNA, poput one koja se koristi u kompletu Qubit dsDNA. Pomoću takvog kompleta DNA se može točno kvantificirati pri vrlo niskim koncentracijama potrebnima za većinu metoda koja se temelje na sekvenciranju (Singer i sur., 1997). Međutim, popularni sustav nanočestica utemeljen na spektrofotometriji može dati pristojne procjene za više koncentracije DNA (> 20 ng µL-1) kad je DNA jako čista. Spektar zabilježen sustavom nanočestica također daje dodatne informacije o čistoći DNA, budući da omjer između intenziteta pri određenim valnim duljinama ukazuje na vrstu potencijalnih kontaminanata. Ovakva vrsta informacija
Uzorak Uništenje iliza
Uklanjanjeproteina
Uklanjanjekemikalija
Otapanje ipohranjivanje

288 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
može se koristiti za ocjenu jesu li potrebni daljnji koraci pročišćavanja (Wilfinger i sur., 1997).
Veličina DNA može se odrediti pomoću klasične gel elektroforeze (McMaster i Carmichael, 1977) ili novijih, osjetljivijih i obično bržih tehnika provedenih u namjenskim i potpuno automatiziranim sustavima kao što su instrumenti ‘bioanalizator’ i ‘tape-station’ (Panaro i sur., 2000; Padmanaban i sur., 2013). Određivanje veličine oslanja se na činjenicu da će se duži fragmenti DNA kad su izloženi električnoj struji sporije kretati kroz matricu gela nego manji fragmenti. Kad se put koji je ekstrahirana DNA prešla usporedi s putom koji su prešli fragmenti poznatih veličina, moguće je procijeniti raspodjelu dužina fragmenata DNA (McMaster i Carmichael, 1977). Točne procjene dužine i koncentracije molekula DNA su ključne za neke vrste molekularne analize.
8.2.5 Optimizirana ekstrakcija DNA iz aktivnog mulja otpadne vode
Ovaj protokol objašnjava ekstrakciju DNA iz aktivnog mulja iz UPOV-a. Protokol se temelji na kompletu FastDNA Spin za uzorke tla (MP Biomedicals) s određenim modifikacijama, uglavnom u smislu pojednostavljenja i dužeg udaranja kuglica, a objavili su ga Albertsen i sur. (2015).
Kod ekstrakcije DNA ključna je dosljednost, zbog čega ovaj protokol treba doslovce slijediti. Ako odlučite odstupiti od protokola, budite dosljedni u tome, za sve uzorke, za vrijeme trajanja čitavog eksperimenta.
8.2.5.1 Materijali
Za ekstraciju DNA potrebni su sljedeći materijali:
• FastDNA spin kit for soil (MP Biomedicals). • FastPrep-24 (MP Biomedicals). • Mikro-centrifuga (po mogućnosti s hladnjakom). • Kivete (bez DNAze), 1,5 mL. • Kivete Falcon, 15 mL. • Led. • Etanol. • Pipete (raspon 1 µL do 1000 µL). • Nastavci za pipetu bez DNAze (10 µL, 300 µL i 1000
µL). • H2O bez nukleaza (Qiagen). • Trajni marker (otporan na zamrzavanje). • Pisač za oznake (opcionalno).
1 Obično je prihvatljivo 1-4 mg TS.
• Osobna zaštitna sredstva: laboratorijska kuta, zaštitne naočale, rukavice.
8.2.5.2 Ekstrakcija DNA
Ukupno vrijeme potrebno za ekstrakciju DNA iz uzorka je otprilike 4 h.
1. Unos uzorka a. Ciljni volumen: 500 µL. b. Ciljne ukupne krutine (TS): 2 mg. Kritičan korak Nikad nemojte smanjiti broj
okretaja kako biste povećali koncentraciju! 2. Pripremite i označite kivete za čitav tijek rada (po
uzorku): a. 1 × kiveta s matricom za liziranje Lysing Matrix
E (iz kompleta). b. 1 × SPIN™ filtar (iz kompleta). c. 1 × kiveta za hvatanje (iz kompleta). d. 3 × 1,5 mL kiveta bez DNAze. e. 1 × 15 mL Falcon cjevčica.
3. Otopite alikvot uzorka na sobnoj temperaturi i držite ga na ledu do korištenja.
4. U svaku od kiveta Lysing Matrix E dodajte 480 µL pufera natrijevog fosfata: PBS (pH 8,0) i 120 µL pufera MT.
5. Dodajte 250 µL PPS (otopina za taloženje proteina) u jednu kivetu od 1,5 mL za svaki uzorak.
6. Resuspendirajte vezivnu matricu i dodajte 1,0 mL u svaku od Falcon cjevčica od 15 mL.
• Udaranje kuglica 1. Uzorak prije korištenja promiješajte, npr.
vorteksiranjem. 2. Prebacite volumen uzorka od 2 mg TS 1 u kivetu
Lysing Matrix E i dodajte PBS tako da ukupni dodani volumen iznosi 500 µL2.
3. Provedite udaranje kuglica u instrumentu FastPrep-24 a. Vrijeme: 4 × 40 s. b. Brzina: 6 m/s. c. Adapter: Standardan. d. Kritičan korak Ne zaboravite ujednačeno
posložiti kivete. Mogla bi biti potrebna kiveta za ravnotežu.
e. Kritičan korak Između svakog intervala od 40 sekunda, uzorke treba 2 min držati na ledu kako bi se ohladili.
2 Koristite vrh pipete sa širokim otvorom kako bi se izdvojile i velike granule.

MOLEKULARNE METODE 289
• Taloženje proteina i vezivanje DNA uz matricu 1. 10 min Centrifugirajte uzorke na > 10,000 ×g, po
mogućnosti na 4 °C. 2. Nakon centrifugiranja, prebacite supernatante u
kivete od 1,5 mL s PPS-om i ručno protresite kivete 10 puta.
3. Kritičan korak Kivete držite na ledu sve dok se svi uzorci ne obrade.
4. 5 min centrifugirajte kivete na 14.000 ×g kako bi se peleti istaložili.
5. Prebacite supernatant u kivetu od 15 mL sa suspenzijom matrice za vezivanje.
6. Preokrenite je rukom na 2 min kako bi se DNA mogla vezati na matricu.
7. Stavite kivetu u stalak na 3-5 min (ili dok se tekućina ne izbistri) kako bi se matrica na silicijevom gelu mogla istaložiti.
8. Uklonite i bacite do 2 ×750 µL supernatanta, pri čemu treba paziti da se ne dira matrica za vezivanje.
9. Resuspendirajte matricu za vezivanje u preostaloj količini supernatanta.
• Pranje i eluiranje DNA 1. Prebacite otprilike 750 µL mješavine u SPIN™ filtar
i onda 1 min centrifugirajte na 14.000 ×g.3 2. Ispraznite kivetu za hvatanje. 3. Kritičan korak Osigurajte da je etanol dodan
koncentriranom SEWS-M. 4. Dodajte 500 µL pripremljenog SEWS-M i nježno
resuspendirajte granule pomoću sile tekućine iz vrha pipete – ili miješanjem vrhom pipete.
5. 1 min centrifugirajte na 14.000 ×g. 6. Ispraznite kivetu za hvatanje i ponovno je
upotrijebite. 7. 2 min centrifugirajte na 14.000 × g kako bi se
‘osušila’ matrica preostale otopine za pranje. 8. Bacite kivetu za hvatanje i zamijenite je novom
kivetom.4 9. Ostavite SPIN™ filtar 5 min da se osuši na sobnoj
temperaturi s otvorenim poklopcem. 10. Nježno resuspendirajte matricu za vezivanje (iznad
SPIN filtra) u 60 µL DES. Vrhom pipete miješajte matricu dok ne postane tekuća. Pazite da ne uništite filtar.
11. Centrifugirajte 1 min na 14.000 × g kako bi se eluirana DNA dovela u čistu kivetu za hvatanje. Bacite SPIN filtar.
12. Kivetu valjano označite ili isprintanom naljepnicom ili markerom otpornim na zamrzavanje.
3 Ako imate više od 750 µL uzorka, ovaj korak biste trebali ponoviti.
13. Pauza Pohranite DNA na -20 °C za kratkotrajnu pohranu i na -80 °C za dugotrajnu pohranu.
8.3 KVANTITATIVNI PCR (qPCR) U STVARNOM VREMENU
8.3.1 Opća razmatranja
Premda su tehnike masovnog paralelnog sekvenciranja kao što su metagenom i sekvenciranje amplikona (vidi odjeljak 8.4) revolucionizirale način na koji ispitujemo zajednice mikroorganizama, kvantitativna lančana reakcija polimerazom u stvarnom vremenu (qPCR) je i dalje najosjetljivija tehnika za kvantificiranje specifičnih vrsta DNA. Uz to, u optimalnim uvjetima ona omogućuje otkrivanje jedne tražene sekvence unutar analiziranog uzorka, premda se takvi uvjeti rijetko postižu s uzorcima iz okoliša zbog prisustva tvari koje inhibiraju PCR. Nadalje, qPCR se može koristiti za pretvaranje podataka o relativnoj brojnosti dobivenih sekvenciranjem amplikona u apsolutne količine, premda se to rijetko zahtijeva. U sustavima pročišćavanja otpadne vode, qPCR se može koristiti za procjenu ukupne brojnosti bakterija (Horz i sur., 2005) ili za kvantificiranje bakterija koje pripadaju specifičnim taksonomskim skupinama (Matsuda i sur., 2007) pomoću početnica (primera) koje ciljaju na nevarijabilne odnosno varijabilne regije gena rRNA (16S ili 23S). Može se koristiti i za procjenu brojnosti bakterija koje pripadaju specifičnim funkcionalnim skupinama, kao što su nitrifikanti ili bakterije koje akumuliraju polifosfate (PAO), pomoću početnica koje ciljaju na ključne funkcionalne gene (Ge i sur., 2015). qPCR je koristan alat i za određivanje sudbine pojedinačnih bioaugmentacijskih sojeva. To se može postići pomoću početnica koje ciljaju jedinstvene genomske regije (Dueholm i sur., 2015). qPCR se nadalje može koristiti za praćenje širenja gena za otpornost na antibiotike (Volkmann i sur., 2004) i infektivnih virusa (Kitajima i sur., 2014). Kombiniranjem qPCR s reverznom transkripcijom (RT-qPCR) može se kvantificirati aktivnost (transkripcija) specifičnih gena (Nolan i sur., 2006), ali time se ovdje nećemo baviti. qPCR je dorada klasične PCR tehnike (vidi odjeljak 8.4.3) (Saiki i sur., 1985) u kojoj se produkti PCR otkrivaju nakon svakog ciklusa PCR (slika 8.2). Tehnika se oslanja na činjenicu da se u tipičnoj PCR, ciljni slijed amplificira otprilike dvostruko za svaki ciklus PCR, dok jedan ili više reagensa ne postanu ograničavajući (Kubista i sur.,
4 Nova kiveta je kiveta u kojoj treba pohraniti uzorak pa se pobrinite da bude označenaprikladno.
290 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
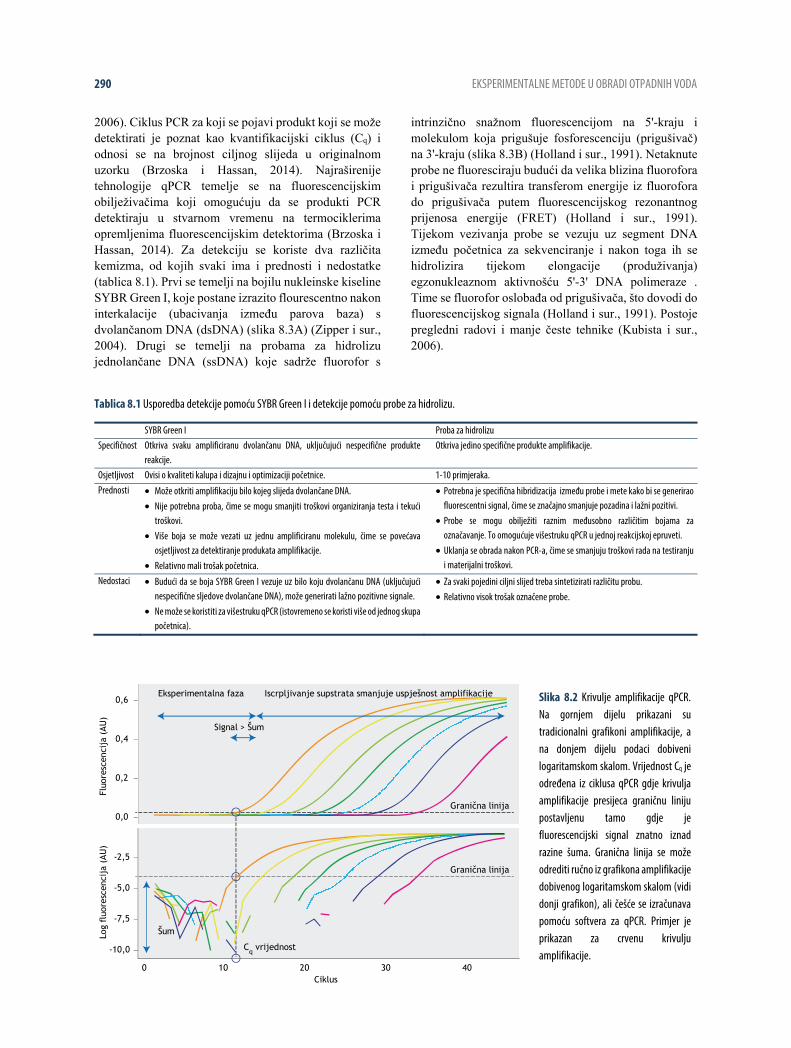
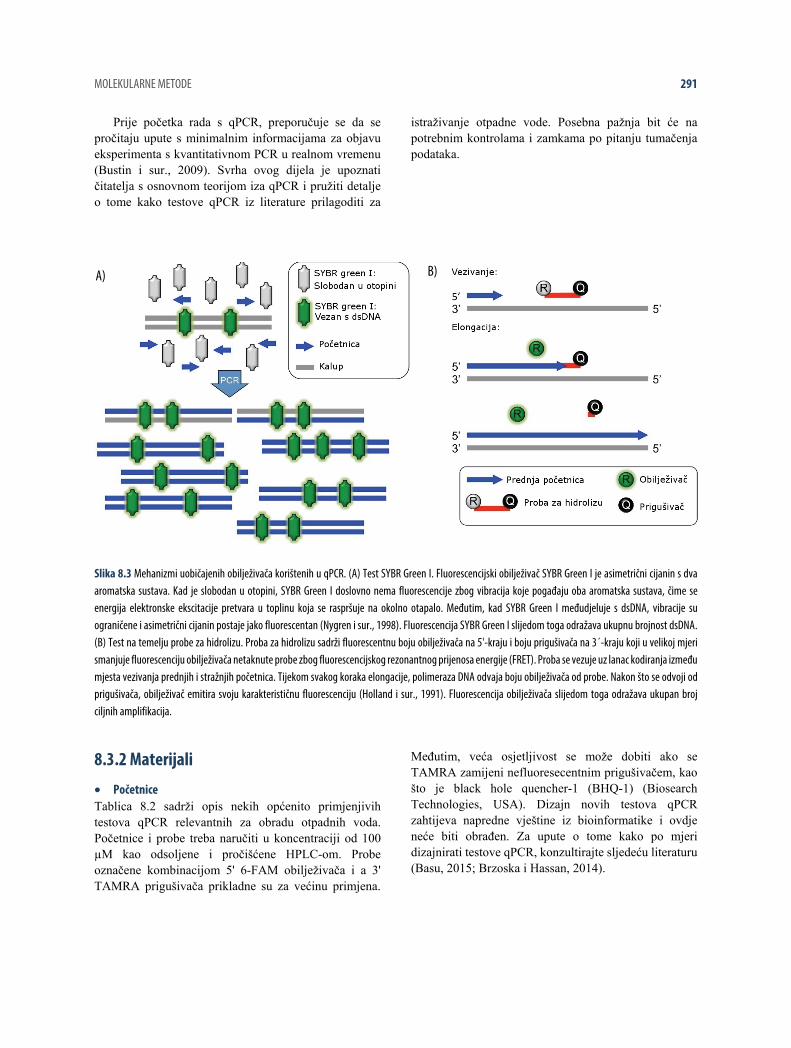
2006). Ciklus PCR za koji se pojavi produkt koji se može detektirati je poznat kao kvantifikacijski ciklus (Cq) i odnosi se na brojnost ciljnog slijeda u originalnom uzorku (Brzoska i Hassan, 2014). Najraširenije tehnologije qPCR temelje se na fluorescencijskim obilježivačima koji omogućuju da se produkti PCR detektiraju u stvarnom vremenu na termociklerima opremljenima fluorescencijskim detektorima (Brzoska i Hassan, 2014). Za detekciju se koriste dva različita kemizma, od kojih svaki ima i prednosti i nedostatke (tablica 8.1). Prvi se temelji na bojilu nukleinske kiseline SYBR Green I, koje postane izrazito flourescentno nakon interkalacije (ubacivanja između parova baza) s dvolančanom DNA (dsDNA) (slika 8.3A) (Zipper i sur., 2004). Drugi se temelji na probama za hidrolizu jednolančane DNA (ssDNA) koje sadrže fluorofor s
intrinzično snažnom fluorescencijom na 5'-kraju i molekulom koja prigušuje fosforescenciju (prigušivač) na 3'-kraju (slika 8.3B) (Holland i sur., 1991). Netaknute probe ne fluoresciraju budući da velika blizina fluorofora i prigušivača rezultira transferom energije iz fluorofora do prigušivača putem fluorescencijskog rezonantnog prijenosa energije (FRET) (Holland i sur., 1991). Tijekom vezivanja probe se vezuju uz segment DNA između početnica za sekvenciranje i nakon toga ih se hidrolizira tijekom elongacije (produživanja) egzonukleaznom aktivnošću 5'-3' DNA polimeraze . Time se fluorofor oslobađa od prigušivača, što dovodi do fluorescencijskog signala (Holland i sur., 1991). Postoje pregledni radovi i manje česte tehnike (Kubista i sur., 2006).
Tablica 8.1 Usporedba detekcije pomoću SYBR Green I i detekcije pomoću probe za hidrolizu.
SYBR Green I Proba za hidrolizuSpecifičnost Otkriva svaku amplificiranu dvolančanu DNA, uključujući nespecifične produkte
reakcije. Otkriva jedino specifične produkte amplifikacije.
Osjetljivost Ovisi o kvaliteti kalupa i dizajnu i optimizaciji početnice. 1-10 primjeraka.Prednosti • Može otkriti amplifikaciju bilo kojeg slijeda dvolančane DNA.
• Nije potrebna proba, čime se mogu smanjiti troškovi organiziranja testa i tekući troškovi.
• Više boja se može vezati uz jednu amplificiranu molekulu, čime se povećava osjetljivost za detektiranje produkata amplifikacije.
• Relativno mali trošak početnica.
• Potrebna je specifična hibridizacija između probe i mete kako bi se generirao fluorescentni signal, čime se značajno smanjuje pozadina i lažni pozitivi.
• Probe se mogu obilježiti raznim međusobno različitim bojama za označavanje. To omogućuje višestruku qPCR u jednoj reakcijskoj epruveti.
• Uklanja se obrada nakon PCR-a, čime se smanjuju troškovi rada na testiranju i materijalni troškovi.
Nedostaci • Budući da se boja SYBR Green I vezuje uz bilo koju dvolančanu DNA (uključujući nespecifične sljedove dvolančane DNA), može generirati lažno pozitivne signale.
• Ne može se koristiti za višestruku qPCR (istovremeno se koristi više od jednog skupa početnica).
• Za svaki pojedini ciljni slijed treba sintetizirati različitu probu. • Relativno visok trošak označene probe.
0,6
0,4
0,2
0,0
-2,5
-5,0
-7,5
-10,0
Fluo
resc
enci
ja (
AU
)Lo
g fl
uore
scen
cija
(A
U)
Šum
Signal > Šum
Eksperimentalna faza Iscrpljivanje supstrata smanjuje uspješnost amplifikacije
Granična linija
Granična linija
20Ciklus
Cq vrijednost
30100 40
Slika 8.2 Krivulje amplifikacije qPCR. Na gornjem dijelu prikazani su tradicionalni grafikoni amplifikacije, a na donjem dijelu podaci dobiveni logaritamskom skalom. Vrijednost Cq je određena iz ciklusa qPCR gdje krivulja amplifikacije presijeca graničnu liniju postavljenu tamo gdje je fluorescencijski signal znatno iznad razine šuma. Granična linija se može odrediti ručno iz grafikona amplifikacije dobivenog logaritamskom skalom (vidi donji grafikon), ali češće se izračunava pomoću softvera za qPCR. Primjer je prikazan za crvenu krivulju amplifikacije.
MOLEKULARNE METODE 291
Prije početka rada s qPCR, preporučuje se da se pročitaju upute s minimalnim informacijama za objavu eksperimenta s kvantitativnom PCR u realnom vremenu (Bustin i sur., 2009). Svrha ovog dijela je upoznati čitatelja s osnovnom teorijom iza qPCR i pružiti detalje o tome kako testove qPCR iz literature prilagoditi za
istraživanje otpadne vode. Posebna pažnja bit će na potrebnim kontrolama i zamkama po pitanju tumačenja podataka.
Slika 8.3 Mehanizmi uobičajenih obilježivača korištenih u qPCR. (A) Test SYBR Green I. Fluorescencijski obilježivač SYBR Green I je asimetrični cijanin s dva aromatska sustava. Kad je slobodan u otopini, SYBR Green I doslovno nema fluorescencije zbog vibracija koje pogađaju oba aromatska sustava, čime se energija elektronske ekscitacije pretvara u toplinu koja se raspršuje na okolno otapalo. Međutim, kad SYBR Green I međudjeluje s dsDNA, vibracije su ograničene i asimetrični cijanin postaje jako fluorescentan (Nygren i sur., 1998). Fluorescencija SYBR Green I slijedom toga odražava ukupnu brojnost dsDNA. (B) Test na temelju probe za hidrolizu. Proba za hidrolizu sadrži fluorescentnu boju obilježivača na 5'-kraju i boju prigušivača na 3´-kraju koji u velikoj mjeri smanjuje fluorescenciju obilježivača netaknute probe zbog fluorescencijskog rezonantnog prijenosa energije (FRET). Proba se vezuje uz lanac kodiranja između mjesta vezivanja prednjih i stražnjih početnica. Tijekom svakog koraka elongacije, polimeraza DNA odvaja boju obilježivača od probe. Nakon što se odvoji od prigušivača, obilježivač emitira svoju karakterističnu fluorescenciju (Holland i sur., 1991). Fluorescencija obilježivača slijedom toga odražava ukupan broj ciljnih amplifikacija.
8.3.2 Materijali
• Početnice Tablica 8.2 sadrži opis nekih općenito primjenjivih testova qPCR relevantnih za obradu otpadnih voda. Početnice i probe treba naručiti u koncentraciji od 100 µM kao odsoljene i pročišćene HPLC-om. Probe označene kombinacijom 5' 6-FAM obilježivača i a 3' TAMRA prigušivača prikladne su za većinu primjena.
Međutim, veća osjetljivost se može dobiti ako se TAMRA zamijeni nefluoresecentnim prigušivačem, kao što je black hole quencher-1 (BHQ-1) (Biosearch Technologies, USA). Dizajn novih testova qPCR zahtijeva napredne vještine iz bioinformatike i ovdje neće biti obrađen. Za upute o tome kako po mjeri dizajnirati testove qPCR, konzultirajte sljedeću literaturu (Basu, 2015; Brzoska i Hassan, 2014).
A) B)
292 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
Tablica 8.2 Primjeri općenito primjenjivih testova qPCR za obradu otpadne vode.
Meta i sredstvo1 Opis Referenca Većina bakterija (HP) Dizajniran je i evaluiran jedinstveni test qPCR za amplifikaciju gena 16S rRNA iz područja bakterija. Test
omogućuje kvantifikaciju ukupne brojnosti bakterija unutar uzorka. (Nadkarni i sur., 2002)
Većina arheja (HP) Evaluiran je obuhvat većeg broja početnica za gen 16S rRNA iz područja Archaea i bakterija. Specifične početnice omogućuju da se u zajednicama mikroorganizama iz različitih staništa odredi relativna brojnost arheja i bakterija.
(Wang and Qian, 2009)
Većina gljiva (HP) Jedinstveni test qPCR za amplifikaciju gena 18S rRNA u gljivama dizajniran i evaluiran je in silico i in vitro. Test omogućuje kvantificiranje ukupne brojnosti gljiva unutar uzorka.
(Liu i sur., 2012)
Pojedinačni sojevi (HP) Općenita tehnika za izradu specifičnih testova qPCR je prezentirana i korištena za dizajn testa qPCR za bioaugmentacijski soj Pseudomonas monteilii SB3074. Test je kasnije korišten za ocjenu postojanosti soja u aktivnom mulju.
(Dueholm i sur., 2015)
Nitrifikacija (HP) Test qPCR namijenjen za podjedinicu alfa gena za monooksigenazu amonija (amoA), koji je ključni enzim u oksidaciji amonija od strane bakterija koje oksidiraju amonij (AOB), je razvijen i korišten za procjenu veličine populacije AOB u uzorcima tla.
(Okano i sur., 2004)
Redukcija nitrata (SG) Testovi qPCR namijenjeni za uz membranu vezane (narG) i periplazmatske (napA) protobakterijske reduktaze nitrata su razvijeni i korišteni za određivanje njihove relativne brojnosti u različitim sredinama.
(Bru i sur., 2007)
Anaerobna oksidacija amonija (ANAMMOX) (SG)
Test qPCR koji specifično cilja gen 16S rRNA svih poznatih ANAMMOX bakterija dizajniran i korišten je za određivanje njihove brojnosti u močvarnim tlima.
(Humbert i sur., 2012)
Razgradnja aromatskih ugljikovodika (HP)
Razvijen je test qPCR koji cilja bssA, koji kodira α-podjedinicu benzilsukcinatne sintaze, glavnog enzima povezanog s anaerobnom razgradnjom toluena i ksilena. Test je korišten za proučavanje toga kako ispuštanja mješavine benzina i etanola iz propusnih podzemnih spremnika djeluje na prirođene bakterije koje degradiraju toluen.
(Beller i sur., 2002)
Otpornost na antibiotike (HP)
Dizajnirani su testovi qPCR koji ciljaju gene vezane uz otpornost na antibiotike vanA, ampC i mecA, koji su povezani s enterokokima otpornima na vankomicin (VRE), Enterobactreiaceae otporne na β-laktamske antibiotike odnosno Staphylococcus aureus (MRSA) otporne na meticilin. Testovi su korišteni za otkrivanje gena otpornosti u komunalnim i otpadnim vodama bolnica.
(Volkmann i sur., 2004)
Patogeni virusi koji se prenose vodom (HP)
Testovi qPCR su korišteni za određivanje relativne brojnosti 11 različitih virusa na ulazu i izlazu iz dva uređaja za pročišćavanje otpadne vode.
(Kitajima i sur., 2014)
1 SG: SYBR Green I; HP: Proba za hidrolizu.
• Termocikler s analizom u stvarnom vremenu Termocikleri s analizom u stvarnom vremenu mogu se nabaviti od velikog niza dobavljača, uključujući Agilent Technologies (SAD), Applied Biosystems Inc. (SAD), Bio-Rad (SAD); Eppendorf International (Njemačka) te Roche Applied Science (Švicarska). Prijavljeno je dobro iskustvo korištenja sustava Agilent Mx3005P qPCR tvrtke Agilent Technologies.
• Reagensi za qPCR Dostupan je čitav niz komercijalnih kompleta za qPCR. Imamo dobro iskustvo s III Ultra-Fast SYBR Green qPCR Master Mix (Agilent Technologies) za testove s SYBR Green I i EXPRESS qPCR Supermix (Life Technologies, SAD) za testove s probama za hidrolizu.
• Oprema za mjerenje koncentracije DNA Autori preporučuju korištenje Qubit fluorometra (Life Technologies, SAD) ili slične, na probama utemeljene tehnike za određivanje koncentracija DNA. Može se koristiti i NanoDrop spectrofotometar (Thermo Scientific, SAD), ali ta tehnika je osjetljivija na nečistoće u uzorku (vidi odjeljak 8.2.4).
8.3.3 Metode
• Priprema standarda za qPCR Apsolutna koncentracija tražene sekvence DNA se određuje uspoređivanjem Cq vrijednosti uzorka sa serijom razrjeđenja standarda s poznatim koncentracijama tražene sekvence (amplikon) (slika 8.4).

MOLEKULARNE METODE 293
Slika 8.4 Procjena učinkovitosti amplifikacije iz nagiba pravca linearne regresije serije razrjeđenja standarada.
Standardi se mogu napraviti iz genomske DNA, plazmida ili produkata PCR. Premda se produkti PCR tražene sekvence lako dobiju PCR-om pomoću qPCR početnica, ti produkti često dovode do loših serija razrjeđenja standarda, budući da je zbog male veličine teško napraviti ponovljive otopine. Za rutinske qPCR testove, autori stoga preporučuju korištenje lineariziranih plazmida koji sadrže amplikon. Linearizacija je važna budući da kružna struktura plazmidne DNA može potisnuti amplifikaciju PCR (Hou i sur., 2010). Linearizirani standardi plazmidne qPCR se lako naprave kako je opisano u nastavku. Ako nije navedeno drugačije, komplet i enzime treba koristiti u skladu s preporukama proizvođača.
1. Amplificirajte ciljni slijed pomoću početnica qPCR i standardne Taq-polimeraze.
2. Klonirajte produkt PCR u pCR4-TOPO plazmid pomoću kompleta za kloniranje TOPO TA za sekvenciranje i stanica E. coli One Shot TOP10 (Life Technologies, SAD).
3. Inokulirajte 10 mL LB medija koji sadrži 50 µg mL-1 kanamicina s pozitivnim klonom i uzgojite kulturu preko noći (37 °C, 200 rpm).
4. Pročistite plazmide iz kulture pomoću kompleta QIAprep Spin Miniprep Kit (Qiagen, SAD).
5. Linearizirajte plazmide pomoću FastDigest ScaI ili FastDigest SspI (Thermo Scientific, SAD).
6. Pročistite linearizirane plazmide pomoću kompleta za suspenziju QIAEX II (Qiagen, SAD).
7. Odredite koncentraciju DNA pomoću fluorometra Qubit ili NanoDrop spektrofotometra.
8. Izračunajte molekularnu težinu lineariziranog plazmida s umetkom koristeći donju jednadžbu.
MW = konačna veličina plazmida u bp × 607.4 g mol-1
9. Izračunajte brojnost ciljnog slijeda u uzorku pomoću donje jednadžbe.
Kopije ciljnog slijeda po µL = koncentracija u ng µL-1× 10-9 × 6.022 × 1023 / MW
10. Razrijedite zalihu amplikona u 109 kopija po µL pomoću 10 mM tris pufera, pH 8,5.
11. Napravite seriju desetorostrukog razrjeđenja u rasponu od 108 do 101 kopija po µL s 10 mM tris pufera, pH 8,5. Nakon svakog razrjeđenja koristite novi vrh pipete i vorteksirajte uzorak.
12. Prebacite 100 µL standarda u niz od 8 epruveta od 200 µL za PCR i do korištenja ih čuvajte na -18 °C.
• Priprema uzorka 1. Pročistite DNA od uzoraka kako je opisano u
‘Ekstrakcija DNA’ u odjeljku 8.2.5. 2. Odredite koncentraciju DNA pomoću fluorometra
Qubit ili NanoDrop spektrofotometra.
• Organiziranje qPCR 1. Pripremite qPCR glavnu mješavinu prema protokolu
koji dolazi uz komplet za qPCR. Glavna mješavina se obično priprema tako da bude dovoljna za 5 µL uzorka.
2. Stavite glavnu mješavinu na qPCR mikropločicu. 3. Centrifugirajte mikroploču na 2,200 ×g 5 min. 4. Dodajte duplikate serije s razrijeđenim standardom u
prve dvije kolone qPCR pločice. 5. Dodajte duplikate uzorka na qPCR ploču. 6. Dodajte niže opisane kontrole na qPCR pločicu.
a. NTC (Nema kontrole kalupa/slijepa proba): NTC se umjesto s DNA kalupom priprema s vodom u kojoj nema DNA. Služi kao opća kontrola za vanjsku kontaminaciju nukleinske kiseline. Kad se koristi boja SYBR Green, služi i kao važna kontrola za formiranje dimera početnica.
b. NAC (Nema kontrole amplifikacije): NAC se priprema bez DNA polimeraze. Funkcionira kao kontrola za pozadinsku fluorescenciju koja nije funkcija PCR. Takvu fluorescenciju obično uzrokuje korištenje djelomično razgrađenih proba za hidrolizu. Slijedom toga, NAC nije potreban u testovima sa SYBR Green.
c. Kontrole razrijeđenog uzorka: Koriste se kako bi se odredilo sadrži li uzorak inhibitore PCR. To je
Cq
vrij
edno
st
log kopije/µL
16
20
24
28
32
2 3 4 5 6
y = 3,546 x + 38,60R2 = 0,998Učinkovitost = 10^(-1/-3,546)-1 = 91,4%

294 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
slučaj ako razrijeđeni uzorak daje znatno veći broj kopija od uzorka nakon korekcije za faktor razrjeđenja.
d. Uzorci kojima je dodan amplikon: Odabranim uzorcima se dodaje poznata visoka koncentracija amplikona i služe kao kontrole za prisutnost inhibitora PCR-a.
7. Centrifugirajte mikroploču na 2.200 ×g 5 min. 8. Provedite qPCR u skladu s protokolom isporučenim
s kompletom za qPCR. Prilagodite temperaturu vezivanja i vrijeme elongacije u skladu s opisom testa.
9. Ako se koristi test SYBR Green, završite proces qPCR s analizom krivulje topljenja. Time se može identificirati formiranje dimera početnica i proizvodnja nespecifičnih produkata. Oboje se mogu promatrati kao dodatni maksimumi na grafikonima prvih derivativa krivulja topljenja. Dimeri početnica imaju znatno niže temperature topljenja od ciljnog amplikona.
10. Kada se novi qPCR test koristi prvi put, uvijek je dobro validirati test. Kako biste to napravili, pročistite proizvedeni produkt PCR-a i pošaljite mali alikvot i bilo prednju ili stražnju početnicu firmi koja provodi sekvenciranje Sangerovom metodom. Na temelju podataka sekvenciranja potvrdite da je amplificirani produkt doista ciljni slijed. Pročišćeni proizvod bi se mogao analizirati i na agarnom gelu. Trebalo bi promatrati jednu prugu u predviđenoj duljini.
8.3.4 Obrada podataka
• Određivanje broja kopija uzorka Većina termociklera s analizom u stvarnom vremenu može automatski obraditi podatke i za svaki uzorak dati broj kopija. Broj kopija se može izračunati i tako da se Cq vrijednosti uzorka ručno usporede s vrijednostima standardnih serija razrjeđenja. Kako bi se to napravilo, na grafikonu prikažite Cq vrijednosti serije razrjeđenja standarda u odnosu na logaritam (log10) brojnosti ciljnog slijeda i potom napravite linearnu regresiju. Dobivena jednadžba se kasnije može koristiti za određivanje brojnosti željene sekvence iz vrijednosti Cq uzoraka (slika 8.4).
• Ocjena učinkovitosti PCR-a Učinkovitost PCR-a opisuje kako amplifikacija odstupa od idealne situacije, pri čemu se koncentracija amplikona udvostručuje nakon svakog ciklusa PCR-a. Učinkovitosti PCR-a manje od 90 % mogu ukazivati na dizajn PCR početnice/probe manji od optimalnog, prisutnost inhibitora PCR-a ili neispravno pipetiranje uzorka ili
reagensa, dok do učinkovitosti iznad 100 % uvijek dolazi zbog neispravnog pipetiranja. Kao smjernica, predloženo je da bi za okolišne uzorke učinkovitost PCR-a trebala biti između 80 i 115 % (Zhang i Fang, 2006).
Učinkovitost PCR-a se može odrediti iz nagiba pravca linearne regresije gore opisanih serija razrjeđenja standarda (jedn. 8.1, Rasmussen, 2001). Izračunata učinkovitost pretpostavlja da svi standardi i uzorci imaju istu učinkovitost amplifikacije (Souazé i sur., 1996). To se potvrđuje korištenjem ranije opisanih kontrola razrijeđenog uzorka ili uzorka kojemu je dodan amplikon.
Učinkovitost = 10 -1slope‒ 1
8.3.5. Rezultat i tumačenje podataka
Konačni rezultat analize qPCR je popis brojnosti tražene sekvence za svaki uzorak. Međutim, pri analiziranju podataka na umu treba imati neke važne aspekte koji će znatno djelovati na konačne zaključke (Kim i sur., 2013).
• Ekstrakcija nukleinskih kiselina je pristrana Uzorci iz sustava za pročišćavanje otpadne vode sadrže raznolike mikroorganizme koji pokazuju priličnu varijaciju u arhitekturi svojih staničnih stjenki (Saunders i sur., 2015). Neke od njih je lako razgraditi, dok je s drugima to teže. Slijedom toga, odabir postupka ekstrakcije nukleinske kiseline uvodi znatnu pristranost (Albertsen i sur., 2015). U skladu s time, može biti vrlo teško među studijama usporediti apsolutnu kvantifikaciju. Protokol ekstracije DNA opisan u odjeljku 8.2.5.2 daje rezultate usporedive s rezultatima kvantitativne FISH analize za uzorke iz sustava za pročišćavanje otpadne vode.
• Kvaliteta kalupa DNA Uzorci iz okoliša, poput onih iz sustava za pročišćavanje otpadne vode, često sadrže spojeve koji štetno djeluju na amplifikaciju PCR (Bessetti, 2007). To mogu biti humimske kiseline, teški metali, polisaharidi, fenolni spojevi ili urea. Takvi inhbitori se mogu ukloniti tako da se uzorak obrađuje pomoću adsorbenata, kemijskim pranjem ili pročišćavanjem gelom (Schriewer i sur., 2011). Međutim, važno je uvijek empirijski ocijeniti uklanjanje inhibitornih spojeva, kako je opisano u odjeljku 8.3.3 (Stults i sur., 2001).
• Specifičnost testova qPCR širokog spektra Testovi qPCR korišteni u obradi otpadne vode često ciljaju skupine mikroorganizama, a ne pojedinačne vrste
8.1

MOLEKULARNE METODE 295
ili sojeve. Slijedom toga, testovi koriste generičke početnice i probe dizajnirane na temelju poznate degenerativnosti ciljnog slijeda. Međutim, poznata degenerativnost ne mora uvijek odražavati ono što se vidi u prirodi, što dovodi do precjenjivanja ili podcjenjivanja kocnentracije tražene sekvence. Problem predstavlja i korištenje jako iskvarenih početnica i proba. Ako je zajednica mikroorganizama jako obogaćena specifičnim organizmima, savršene odgovarajuće početnice za te organizme će se brzo potrošiti, dok će početnice za malobrojne organizme biti prisutne duže vrijeme. Amplifikacija će slijedom toga biti pristrana prema malobrojnom organizmu, što će rezultirati podcjenjivanjem brojnosti ciljnog slijeda. Na kraju, mogu postojati razlike u učinkovitosti amplifikacije za svaki organizam zbog varijacije u njihovom sadržaju GC (Kim i sur., 2013).
• Amplifikacija izvanstanične DNA (eDNA) Biološki procesi poput obrade otpadne vode se oslanjaju na aktivnu populaciju mikroorganizama. Međutim, qPCR ne može razlikovati između DNA koja potječe iz aktivnih bakterija i izvanstanične DNA (eDNA) koja potječe od mrtvih i razgrađenih stanica. Budući da uzorci iz sustava za pročišćavanje otpadne vode sadrže značajne količine eDNA, to može dovesti do odstupanja u podacima (Dominiak i sur., 2011). Zbog toga treba paziti kad se donose zaključci o aktivnosti na temelju rezultata qPCR.
• Varijacija u broju kopija gena Genomi mikroorganizama sadrže veliku varijaciju u broju kopija metabolički važnih gena kao što je 16S rRNA gen (Větrovský i Baldrian, 2013). To može dovesti do pristrane kvantifikacije specifičnog broja bakerija ako se ne zna broj kopija. Uz to, broj čitavih genoma po stanici može varirati ovisno o stanju rasta bakterija (Ludwig i Schleifer, 2000). Ako treba istražiti relativnu brojnost specifične vrste bakterija, preporučuje se da se koriste podaci o aplikonu 16S rRNA (vidi odjeljak 8.4).
8.3.6 Rješavanje problema • Uzorak sadrži inhibitore PCR-a Učinci inhibitora PCR-a mogu se spriječiti na tri načina. Najjednostavnija opcija je razrjeđivanje uzorka. Inhibitori PCR-a su učinkoviti tek iznad određene koncentracije. Međutim, razrjeđivanjem uzorka će se smanjiti i signal, što će dovesti do manje osjetljivog testa. Druga opcija je dodatno pročišćavati DNA. To zahtijeva koncentrirani uzorak, budući da se tijekom pročišćavanja uvijek gubi materijal. Pročišćavanje uzorka u velikom mjerilu može se provesti na PCR pločama s 96 jažica
pomoću kompleta za pročišćavanje na temelju magnetnih kuglica. Treće i konačno rješenje je pročistiti DNA iz originalnog uzorka pomoću drugog kompleta za pročišćavanje koji je optimiran za dani inhibitor.
• Dizajn početnice ili probe nije optimalan Nikada ne bi trebalo koristiti test qPCR koji se temelji na lošim početnicama i probama. Umjesto toga, dizajnirajte i evaluirajte nove početnice i skup proba. Smjernice daje nekoliko autora (Basu, 2015; Brzoska i Hassan, 2014).
• Neispravno pipetiranje uzorka i reagensa Neispravna kalibracija pipeta štetna je za qPCR. Zbog toga se preporučuje da se čuva namjenski komplet pipeta za qPCR koje se redovno kontroliraju. Preporučuje se i korištenje pipete za višestruko raspršivanje budući da pojednostavljuje baratanje uzorcima. Na kraju, može biti dobra ideja da prije provedbe qPCR provjerite svoje tehnike pipetiranja.
8.3.7 Primjer
Pospješena razgradnja određenih onečišćujućih tvari može se postići tako da se aktivnom mulju u UPOV-ima dodaju katabolički značajni sojevi bakterija (El Fantroussi i Agathos, 2005). To je poznato kao bioaugmentacija. Uspješna bioaugmentacija zahtijeva da dodani sojevi imaju mogućnost napredovati u novoj sredini (Thompson i sur., 2005). qPCR se može koristiti za evaluaciju postojanosti sojeva bioaugmentacije in situ. qPCR test specifičan za određeni soj može se razviti na temelju jedinstvenih genomskih sljedova u sojevima bioaugmentacije. Brojnost sojeva se kasnije može odrediti pomoću DNA ekstrahirane iz aktivnog mulja u različitim trenucima nakon dodavanja bioaugmentacijskog soja (Dueholm i sur., 2015). Ovdje ćemo pokazati primjer kako je qPCR korišten za ocjenu postojanosti bioaugmentacijskih sojeva Pseudomonas monteilii SB3078 i SB3101, koji se koriste za razgradnju aromatskih ugljikovodika (Dueholm i sur., 2014; 2015).
8.3.7.1 Uzorci
Bioaugmentacijski soj P. monteilii SB3078 ili SB3101 je ubačen u 100 mL svježeg aktivnog mulja dobivenog iz UPOV-a Aalborg East u količini od 1 % na temelju broja stanica. Grubo je procijenjeno da i suspendirane tvari (SS) = 1 g L-1 (aktivni mulj) i OD600 nm = 1 (čista kultura Pseudomonas) odgovaraju otprilike 109 stanica mL-1 kako je ranije pokazano (Frølund i sur., 1996). Dodan je benzen do koncentracije 10 µg mL-1. Tikvice za uzgoj kulture su zatvorene pomoću zatvarača od butilne gume i inkubirane su 4 dana na 25 °C, 150 rpm. Tikvice su

296 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
otvarane svakih 12 h na 30 min kako bi isparili preostali tragovi benzena i istaložile čestice mulja. Potom je uklonjeno 50 mL efluenta i zamijenjeno s 50 mL primarno istaložene otpadne vode, čime se simuliralo hidrauličko vrijeme zadržavanja od 24 h. Uzeti su uzorci za ekstrakciju DNA i ponovno je dodan bezen do 10 µg mL-1. Tikvice su potom zatvorene i nastavilo se s inkubacijama (Dueholm i sur., 2015). DNA je u osnovi ekstrahirana kako je opisano u odjeljku 8.2.
8.3.7.2 Organiziranje qPCR 1. Pripremite glavnu mješavinu za qPCR kako je
opisano u nastavku. Početnice i probe ciljaju i na SB3078 i na SB3101.
Stavka Konačna koncentracija
Po reakciji (20μL)
100 × reakcije
EXPRESS qPCR Supermix
1 × 10 μL 1.000 μL
ROX (25 μM) 50 nM 0,04 μL 4 μL Prednja početnica (100 μM)
500 nM 0,10 μL 10 μL
Stražnja početnica (100 μM)
500 nM 0,10 μL 10 μL
Proba za hidrolizu (100 μM)
200 nM 0,04 μL 4 μL
DEPC voda - 4,72 μL 472 μL Alikvot - 15 μL -
2. Stavite glavne mješavine na qPCR mikroploču i
dodajte 5 µL uzoraka i kontrole (vidi gore). 3. Centrifugirajte mikroploču 5 min na 2.200 ×g. 4. Provedite qPCR prema sljedećem programu:
- 50 °C 2 min (UDG inkubacija). - 95 °C 2 min. - 45 ciklusa od:
- 95 °C 15 s. - 60 °C 1 min.
8.3.7.3 Rezultati
Počeli smo ocjenjivanjem učinkovitosti amplifikacije. Cq vrijednosti serije razrjeđenja standarda su prikazane grafički u odnosu na logaritam broja kopija, nakon čega je napravljena linearna regresija. Time je dobivena sljedeća jednadžba:
y = -3,349 · x + 39,80; R2 = 1
Potom je učinkovitost izračunata iz nagiba pravca kao:
Učinkovitost = 10 (-1/slope) ‒1 = 10(-1/-3.349) ‒1 = 98,9 %
To je unutar prihvatljivog raspona za uzorke iz okoliša od 80-115 % (Zhang i Fang, 2006).
Zatim smo ocjenjivali kontrole. NTC i NAC se nisu amplificirali unutar 45 ciklusa. Time je potvrđeno da u reagensima nisu bili prisutni kontaminanti koji se mogu amplificirati, odnosno da su probe bile stabilne. Analiziran je i uzorak koji je sadržavao DNA ekstrahiranu iz neobrađene otpadne vode. Niti ova kontrola se nije amplificirala, čime je potvrđena specifičnost qPCR testa. Na kraju, istraživali smo amplifikaciju nekoliko razrijeđenih uzoraka. Oni su proizveli rezultate slične nerazrijeđenim uzorcima, čime je potvrđeno da inhibitori nemaju značajniji učinak. Bacili smo pogled i na eksperimentalne podatke (slika 8.5).
Slika 8.5 Postojanost P. monteilii SB3078 i SB3101 u aktivnom mulju tretiranom kao u SBR sustavu. Relativna brojnost bioaugmentacijskih sojeva određena je pomoću qPCR testa specifičnog za predmetni soj. Modelirani podaci predstavljaju teoretsko smanjivanje broja stanica koje bi se opazilo da nema neto rasta i da su sve bioaugmentacijske stanice izvan flokula. Volumen koji zauzima kruti materijal je izračunat na temelju indeksa volumena razrijeđenog mulja, a te informacije su zajedno s hidrauličkim vremenom zadržavanja korištene za izračun brzine kojom se ispiru planktonske stanice.
qPCR test je pokazao da se oko 90 % dodanih bioaugmentacijskih sojeva izgubilo unutar prva 24 h. Tome je vjerojatno bilo tako zbog uklanjanja stanica koje nisu vezane u flokule s efluentom, budući da je zamjenjivan svježom otpadnom vodom svakih 12 h. Preostale bioaugmentacijske stanice su se stabilizirale unutar mulja i mogle su preživjeti čitav eksperiment (4 d). Podaci su nadalje pokazali da je SB3078 postojaniji od SB3101 u aktivnom mulju.
SB8078
SB3101
Model
Sojevi
Rela
tivn
a br
ojno
st (
%)
0,00
0,25
0,50
0,75
1,00
1,25
12 24 36 48 60 72 80 960
Vrijeme (h)

MOLEKULARNE METODE 297
8.4 SEKVENCIRANJE AMPLIKONA
8.4.1 Opća razmatranja
Prvi korak u pokušaju razumijevanja kako bakterije u aktivnom mulju djeluju na rad uređaja za obradu otpadne vode je dobiti pregled zajednice bakterija. To uključuje identificiranje bakterija, njihove brojnosti i saznanja o
tome što one rade. Napredak u sekvenciranju DNA je omogućio da se bakterije identificiraju velikom razlučivošću i propusnošću čitanjem gena 16S ribosomske RNA (rRNA) bakterija i njihovim korištenjem kao ‘otisaka prstiju’.
Taj se pristup naziv sekvenciranje amplikona 16S rRNA i sastoji se od niza koraka prikazanih na slici 8.6.
Slika 8.6 Prikaz osnovnih koraka u analizi zajednica mikroorganizama pomoću sekvenciranja amplikona 16S rRNA.
Prvi korak je ‘ekstrakcija DNA’, pri čemu se ekstrahira i pročišćuje genomska DNA iz svih bakterija u uzorku (vidi odjeljak 8.2). Nakon toga, geni 16S rRNA se amplificiraju i pripremaju za sekvenciranje ‘lančanom reakcijom polimerazom (PCR)’. Amplikoni gena 16S rRNA se potom očitavaju, odnosno ‘DNA se sekvencira’, pomoću suvremenih instrumenta za sekvenciranje. Rezultat su sljedovi svih gena 16S rRNA u uzorku. Ti sljedovi se potom grupiraju u skupine koje predstavljaju vrstu i broji se relativni broj gena 16S rRNA koji pripadaju svakoj skupini. Svaka skupina vrste se identificira kroz ‘Taksonomsku klasifikaciju’ uspoređivanjem reprezentativnog slijeda svake skupine s bazom podataka poznatih bakterija. Rezultat je tablica s nazivom svake vrste u uzorku i njihova relativna brojnost. Ta tablica je temelj za vizualiziranje i analiziranje zajednice bakterija. Naziv vrste se može koristiti i kao poveznica s funkcionalnim informacijama koje se nalaze u literaturi ili javnim bazama podataka kao što su MiDAS, midasfieldguide.org (McIlroy i sur.,
2015), npr. ako se za neku od identificiranih vrsta zna da formiraju pjenu ili da su nitrifikanti.
U odjeljcima 8.4.2 do 8.4.7 će se detaljno opisati svaki korak pristupa sekvenciranja amplikona 16S rRNA. Opisi se temelje na sekvenciranju amplikona 16S rRNA pomoću platforme Illumina za sekvenciranje. Međutim, osnovna ideja je ista za sve platforme sekvenciranja.
8.4.2 Gen 16S rRNA kao filogenetski genski biljeg
Filogenetski genski biljeg kodira esencijalnu funkciju koju dijele svi organizmi koji će biti ciljani i koji nisu izloženi lateralnom prijenosu gena. Uz to, genski biljeg mora imati i evolucijski jako očuvane položaje kao i jako varijabilne položaje u svojem slijedu nukleotida. Očuvani dijelovi omogućuju ciljanje gena pomoću PCR-a i potrebni su za ispravnu filogenetsku analizu, dok nam

298 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
varijabilni dijelovi omogućuju da razlikujemo različite organizme i istražimo njihovu povezanost (filogenija).
Ribosomski geni su odabrani za filogenetsku analizu budući da su ih Woese i Fox 1977. godine koristili da prikažu podjelu živih bića u tri odvojene domene (Woese i Fox, 1977; Pace i sur., 2012). Danas je gen 16S rRNA daleko najčešće korišten filogenetski genski biljeg u ekološkim studijama raznolikosti bakterija.
Gen 16S rRNA kodira dio RNA koji čini funkcionalni dio bakterijskog ribosoma. Ribosomi su
tvornice proteina svih oblika staničnog života koji su se razvili rano u evoluciji. Očuvane regije su ključne za ispravnu strukturu i funkciju ribosoma, što znači da većina mutacija u tim regijama evolucijski ne odabire. Varijabilne regije imaju veću mogućnost promjene i mutacije su puno češće (Madigan i Martinko, 2006). Prema tome, gen 16S rRNA sadrži nekoliko očuvanih otoka među kojima se nalaze varijabilne regije, nazvane varijabilne regije od 1 do 9 (V1 do V9) (Ashelford i sur., 2005); vidi sliku 8.7.
Slika 8.7 Varijabilnost u sastavu slijeda unutar gena 16S rRNA (prilagođeno iz Ashelford i sur., 2005).
Navedeno čini gen 16S rRNA izvrsnim genskim biljegom budući da može biti ciljan u cijelosti ili u dijelovima, što daje određenu tehničku fleksibilnost. Gen 16S rRNA se kao genski biljeg koristi mnogo godina, a stečena saznanja su sakupljena u opširne baze podataka koje se koriste za uspoređivanje podatka o genu 16S rRNA i određivanje filogenetske pripadnosti novih gena 16S rRNA te njihovo pripisivanje nekoj skupini unutar taksonomije bakterija. Obično je moguće odrediti taksonomiju bakterija do razine vrsta pomoću njihovih sljedova gena 16S rRNA. Rasprostranjenost i razlučivost gena 16S rRNA, zajedno s resurisma baza podataka, ga čine preferiranim genskim biljegom za analizu zajednice bakterija.
Za analizu zajednice bakterija koriste se i ostali genski biljezi ali u manjoj mjeri, često kako bi se dobila veća filogenetska razlučivost, npr. do razine soja. Za te genske biljege je uobičajeno da budu više varijabilni u sastavu slijeda, čime se dobiva veća filogenetska razlučivost. Međutim, to ujedno znači da oni ciljaju samo
specifične podskupine bakterija. Primjeri takvih genskih markera su amoA za organizme koji oksidiraju amonij
(AOO) i mcrA u metanogenima; vidi i odjeljak 8.3 o qPCR-u. Načela za analiziranje tih ostalih genskih markera su slična genu 16S rRNA, premda geni koji kodiraju proteine kao što je amoA mogu biti analizirani na razini slijeda aminokiselina. Za razlučivost soja potrebni su sljedovi nukleotida, ali moraju biti poredani prema kodonu na temelju slijeda aminokiselina u proteinu (Juretschko i sur., 2000).
8.4.3 Amplifikacija PCR-om
Prvi korak u sekvenciranju amplikona 16S rRNA je amplificiranje gena 16S rRNA pomoću PCR-a.
8.4.3.1 Reakcija PCR
PCR se koristi za selektivnu amplifikaciju gena 16S RNA iz pozadinske genomske DNA, tako da postoji dovoljno materijala koji je praktično analizirati pomoću DNA sekvenatora (slika 8.8).
Pros
ječn
a fr
ekve
ncij
a na
jčeš
ćih
osta
taka
kod
50
otvo
ra b
aze
0,5
0,6V1
V2
V3
V4 V5
V6
V7
V8
V9
0,7
0,8
0,9
1,0
100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 15000
Pozicija baze u genu 16S rRNA

MOLEKULARNE METODE 299
Slika 8.8 Osnovni koraci u amplifikaciji PCR-om i sekvenciranju gena 16S rRNA. Sivo predstavlja genomsku DNA, a svijetlo plavo predstavlja gen 16S rRNA. Obojani dijelovi adaptera i početnica predstavljaju različite funkcionalne sljedove, koji su opisani u odjeljku 8.4.11. Strelice označuju smjer elongacije/sekvenciranja.
PCR koristi termostabilni enzim polimerazu za kopiranje DNA. Polimeraza kopira DNA pomoću komadića sintetičke DNA (15-25 parova baza: bp), koji se zovu početnice (primeri), kao početne točke za amplifikaicju. Kad se analiziraju geni 16S rRNA, početnice su dizajnirane da selektivno odgovaraju nevarijabilnim dijelovima gena 16S rRNA, što omogućuje selektivnu amplifikaciju i ostalih dijelova DNA između njih. Za vrijeme PCR-a, kako bi se omogućila amplifikacija, koriste se ciklusi grijanja i hlađenja. Prvo se dsDNA denaturira grijanjem, kojim se dva lanca međusobno razdvajaju. Kao drugo, reakcija se hladi kako bi se početnice mogle vezati uz svoja ciljna mjesta na DNA. Kao treće, temperatura se ponovno povećava kako bi se dobila optimalna temperatura za aktivnost polimeraze, koja produžavanjem početnice počinje kopirati gen 16S rRNA. Jednim ciklusom se stvaraju dvije kopije gena 16S rRNA iz jednog originala, a same kopije mogu funkcionirati kao kalupi. Ciklus se ponavlja 25-35 puta za vrijeme PCR-a, što rezultira eksponencijalnom amplifikacijom gena 16S rRNA. Kopirani genski produkt iz amplifikacije PCR-om se zove amplikon, otkuda potječe i naziv sekvenciranje amplikona 16S rRNA. Pored DNA, početnica i
polimeraze, reakcija sadrži i nukleotide koji su ugrađeni u novu DNA zajedno s puferima i ostalim dodacima (soli Mg2+ itd.) koji osiguravaju optimalne uvjete za vezivanje početnica i aktivnost polimeraze. Načela na kojima se temelji PCR detaljno su obradili Green i Sambrook (2012).
Druga važna uloga PCR-a je omogućiti sekvenciranje amplikona 16S rRNA. To se postiže pomoću adaptera za sekvenciranje pričvršćenog na kraj početnica korištenih u PCR-u. Adapter se pričvršćuje na sve proizvedene amplikone 16S rRNA. Adapter se sastoji od sintetičkog komadića DNA (oko 50 bp) koji ima različite ‘aktivne’ komponente (vidi odjeljak 8.4.11). Te komponente omogućuju da naprava za sekvenciranje uhvati amplikone 16S rRNA, počne ih čitati i prepozna iz kojeg je uzorka potekao svaki amplikon 16S rRNA, što je poznato kao šifriranje ili indeksiranje. Ideja je istom šifrom označiti sve amplikone 16S rRNA iz istog uzorka. To omogućuje da se velik broj različitih uzoraka zajedno pomiješa (multipleksiranje) i istovremeno očitava na DNA sekvenatoru, a da se ipak kasnije mogu razdvojiti podaci iz pojedinačnih uzoraka (demultipleksiranje) (Illumina Inc., 2015; Caporaso i sur., 2010).

300 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
Konačni produkt PCR-a se zove zbirka sekvenciranja amplikona 16S rRNA. Kod pripreme zbirki sekvenciranja amplikona 16S rRNA mogu se koristiti različite strategije, ali glavna ideja je ista. Gore opisana strategija koristi jedan korak za amplificiranje i pričvršćivanje adaptera, a ostale strategije koriste dva odvojena PCR-a. Te strategije imaju prednosti i nedostatke u pogledu troška, vremena i potreba sekvenciranja. Trenutno je izvedivo sekvencirati jedino dugačke fragmente gena 16S rRNA opisanom strategijom PCR od jednog koraka. To uključuje fragmet V1-3 koji se obično koristi u aktivnom mulju (Albertsen i sur., 2015).
8.4.3.2 Odstupanja kod PCR-a
U PCR se mogu uvesti različite vrste sustavnih odstupanja, što će djelovati na konačno opaženu strukturu zajednice. Odstupanje zbog početnica je jedno od najznačajnijih i bit će obrađeno u idućem stavku (Albertsen i sur., 2015). Odstupanje PCR-a je odstupanje do kojeg dovode stohastička zbivanja u prvim ciklusima PCR-a gdje je u replikaciju uključeno relativno malo molekula ili jednostavno varijacije u postupanju s reagensom/uzorkom (pipetiranje, pozicija u termocikleru itd.). Do odstupanja pri odabiru PCR-a dolazi zbog promjenjive učinkovitosti amplifikacije koju uzrokuju fizička svojstva slijeda nukleotida gena 16S rRNA (Polz i sur., 1998; Kennedy i sur., 2014). Kako bi se smanjio utjecaj odstupanja PCR-a i odabira, provode se replike PCR reakcija, broj PCR ciklusa se drži na minimumu, a količina kalupa DNA bi trebala biti oko 10 ng. U većini standardnih protokola sekvenciranja amplikona ti parametri su optimirani.
8.4.3.3 Odabir početnica
Kako je gore spomenuto, skup početnica odgovara nevarijabilnim dijelovima gena 16S rRNA. Međutim, ne može se izbjeći određena varijabilnost u ‘evolucijski očuvanom’ nevarijabilnom dijelu gena 16S rRNA. Prema tome, početnice će nekim bakterijama odgovarati više nego drugima, a nekima uopće neće odgovarati (Klindworth i sur., 2013). Time se u čitavu analizu uvodi znatno odstupanje zbog početnica i važno je toga biti svjestan.
Kad se analizira gen 16S rRNA, bilo bi idealno sekvencirati čitav gen (otprilike 1,600 bp) budući da se time dobiva maksimalna filogenetska razlučivost. Međutim, zbog ograničenja u tehnologiji sekvenciranja Illumina, trenutno se mogu sekvencirati jedino fragmenti do 550 bp gena 16S rRNA.
Kao posljedica odstupanja zbog početnica i ograničenja u dužini očitanja, dizajnirani su brojni skupovi početnica koji ciljaju različite varijabilne regije bakterijskog gena 16S rRNA. Najčešće korišteni skupovi početnica ciljaju regije V1-3, V4 i V3-4 (Albertsen i sur., 2015). Skupovi početnica imaju različita odstupanja, te pri odabiru skupa početnica treba voditi računa o nekoliko stvari.
a. Skup početnica trebao bi imati najmanje moguće odstupanje prema bakterijama u uzorcku koje vas najviše zanimaju. I premda je moguće dobiti ideju o odstupanju početnica putem analize in silico (Klindworth i sur., 2013), uvijek se preporučuje testirati početnice sekvenciranjem.
b. Skup početnica bi trebao biti isti kao u radovima s kojima želite usporediti svoje rezultate. Baza podataka MiDAS, u kojoj se nastoje sažeti sva trenutna znanja o važnim bakterijama u aktivnom mulju temelji se na početnicama koje ciljaju regiju V1-3. Tome je tako zbog dobre razlučivosti i velike pokrivenosti bakterija koje su odgovorne za procese od interesa u zajednici aktivnog mulja (Albertsen i sur., 2015).
Za uzorke iz specijaliziranih sustava s aktivnim muljem može biti dobra ideja testirati ostale skupove početnica. Na primjer, skup početnica V1-3 ne cilja dobro na najčešće ANAMMOX bakterije i bolje je koristiti skup početnica V4 (Laureni i sur., 2015; Gilbert i sur., 2014). Također je moguće dizajnirati nove početnice, ali to se obično ne preporučuje, budući da to zahtijeva stručna znanja o ekosustavima i filogeniji mikroorganizama kao i puno vremena provedenog u laboratoriju na optimizaciji i validaciji.
8.4.4 Sekvenciranje DNA
8.4.4.1 Platforma sekvenciranja
Nakon što se pripreme zbirke amplikona 16S rRNA, postoji niz različitih opcija za sekvenciranje DNA. Međutim, svaka metoda koristi jasno različite strategije za sekvenciranje, što znači da su proizašli podaci prikladni za različite svrhe i potrebe. Vezano uz sekvenciranje amplikona 16S rRNA, najvažniji kriteriji su dužina sekvenciranja (> 200 bp), kvaliteta sekvenciranja (< 1 % pogrešaka), prinos podataka (> 10.000 očitanja po uzorku), trajanje, trošak, te lakoća izrade zbirke. Kada treba postići kompromis između tih kriterija, trenutno, početkom 2016. godine, najprikladnijom se čini metoda platforme Illumina MiSeq. Illumina MiSeq omogućuje analizu do 400 zbirki

MOLEKULARNE METODE 301
amplikona 16S rRNA (50.000 očitanja po uzorku) u jednom sekvenciranju (56 h). MiSeq trenutno može sekvencirati 301 bp sa svakog kraja amplikona 16S rRNA. To se zove i sekvenciranje uparenih krajeva (paired-end, u daljnjem tekstu PE), a svaki od dva slijeda od 301 bp se zove ‘očitanje’. Tijekom obrade podataka dva očitanja se spajaju na preklapajućim krajevima kako bi se dobila maksimalna dužina od oko 550 bp. Za specijaliziranije korištenje koje zahtijeva veću dužinu očitanja ili kraće vrijeme procesa prikladnije su druge platforme, npr. Pacbio RS II (Pacific Biosciences) ili Ion Proton System (Thermo Fisher Scientific Inc), a uskoro i MinION (Oxford Nanopore Technologies). Valja imati na umu da za različite platforme sekvenciranja treba koristiti različite protokole pripreme zbirke.
8.4.4.2 Dubina sekvenciranja
Kad se provodi sekvenciranje amplikona, važno je imati grubu procjenu potrebne dubine sekvenciranja (broj očitanja po uzorku) kako bi se odgovorilo na pitanja postavljena kroz eksperimentalni dizajn. Za opću analizu zajednice bakterija koja cilja na V1-3 gena 16S rRNA u aktivnom mulju, rutinski se koristi 50.000 sirovih PE očitanja po uzorku. To se radi iz razloga što se često uspoređuju prilično slične zajednice i obično je važno dobiti robusne procjene brojnosti pojedinačnih članova zajednice. Pravilo je da ne bude manje od 100 očitanja bakterija od interesa. Ispod 100 očitanja konačni rezultati postanu vrlo nesigurni zbog biološke i tehničke varijacije (Albertsen i sur., 2015). Ako je potrebna veća razlučivost, najbolja opcija je uključiti više bioloških replika. Međutim, ako to nije opcija, može se koristiti i duže sekvenciranje. Aktivni mulj sadrži na tisuće različitih bakterija, a najvažnijih 100 od njih čini više od 70 % ukupnog broja zajednice (Saunders i sur., 2015). U
prosjeku svaka od tih 100 vrsta čini > 0,5 % ukupne zajednice. Kako bi se dobilo > 100 očitanja iz svake od tih vrsti, potrebno je najmanje 20.000 bioinformatički obrađenih očitanja po uzorku ili > 30.000 neobrađenih PE očitanja, ovisno o kvaliteti sekvenciranja. Treba napomenuti da trošak sekvenciranja obično nije najskuplji dio analize, zbog čega se često preferira, kako bi se osiguralo da je sekvencirano dovoljno očitanja.
8.4.5 Bioinformatička obrada
8.4.5.1 Dostupni softver
Rigorozna standardna procedura za obradu podataka sekvenciranja 16S rRNA još uvijek nije definirana i vjerojatno još neko vrijeme i neće biti budući da se to polje brzo razvija. Ipak, opća ideja ostaje ista i prikazana je na slici 8.9.
Mnoge istraživačke skupine obrađuju podatke prilagođenim workflow računalnim alatima uz upotrebu programiranja, a neke su od toga napravile softverske pakete koji mogu funkcionirati gotovo u potpunosti automatski. Najpopularniji su QIIME (Caporaso i sur., 2010), Mothur (Schloss i sur., 2009) i UPARSE (Edgar, 2013). Njihove postavke i temeljne pretpostavke se razlikuju, također unutar verzija, što će dovesti do nešto drugačijih rezultata čak i za podatke istog slijeda. Zbog toga ne bi trebalo uspoređivati rezultate iz različitih softverskih paketa i verzija. Općenito, savjetuje se da se analiziraju svi podaci iz nekog eksperimenta pomoću jednog softverskog paketa odjednom i da se čitava analiza ponovi ako su neki podaci dodani ili su se postavke promijenile. U nastavku slijede neka opća zapažanja o bioinformatičkoj obradi.
Slika 8.9 Osnovni koraci u bioinformatičkoj obradi podataka o 16S rRNA iz platforme Illumina MiSeq. Boje označuju sljedove koji su > 97 % identični. Slova A i B označuju uzorak iz kojeg dolazi slijed.

MOLEKULARNE METODE 302
8.4.5.2 Neobrađeni podaci
Iz sekvenatora Illumina MiSeq se dobiva par datoteka za svaki uzorak. Svaki par datoteka sadrži sve Read1 uparenih čitanja u prvoj datoteci i sve Read2 u drugoj datoteci. Sve datoteke su datoteke fastq (.fastq ili .fq), odnosno jednostavne tekstovne datoteke sa specifičnim pravilima za navođenje informacija o slijedu (slika 8.10).
Svako očitanje u datoteci fastq zauzima četiri reda: i) ID (identifikacijska oznaka) konkretnog očitanja koja počinje s ‘@’, ii) Neobrađena slova slijeda konkretnog očitanja, iii) ‘+’ koji radi kao opisno polje, ali najčešće je prazno te iv) Phred bodovanje kvalitete označeno slovima ASCII za svaku od baza u očitanju (Cock i sur., 2010).
Slika 8.10 Primjer datoteke fastq s tri očitanja. Očitanja su skraćena radi lakše čitljivosti.
8.4.5.3 Bodovanje kvalitete i filtriranje
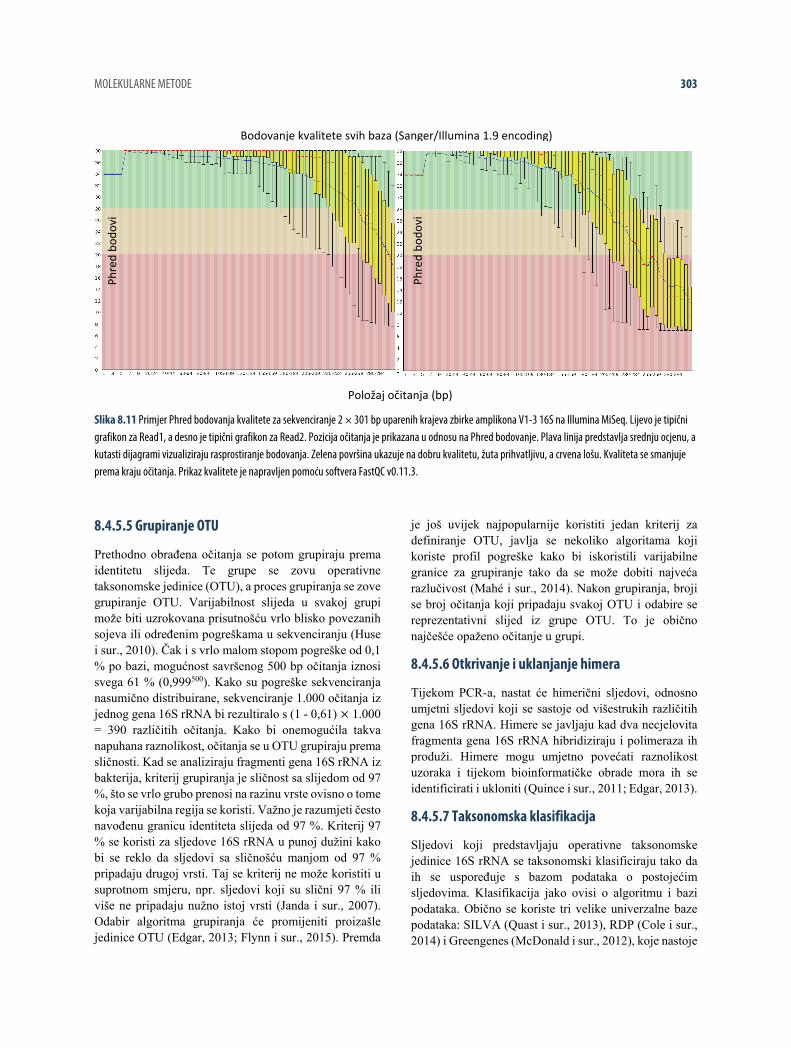
Phred bodovanje kvalitete je mjera vjerojatnosti da određena baza sadrži pogrešku (Q10 = 10 %, Q20 = 1 % i Q30 = 0,1 %) (Cock i sur., 2010). Za sekvenciranje amplikona 16S rRNA preferira se Q20 i više. Kvaliteta se može ocijeniti tako da se na grafikonu prikažu Phred bodovi za kvalitetu i stopa pogreške internog standarda. Za sekvenciranje 2 × 301 bp uparenih krajeva kvalitetnih zbirki amplikona na MiSeq, prosječna kvaliteta je obično iznad Q30 za prvih 250 bp od Read1 i 200 bp od Read2, nakon čega se kvaliteta očitanja obično pogoršava (slika 8.11). Ako je kvaliteta većine očitanja ispod Q20, možda je tijekom sekvenciranja nešto pošlo krivo. Podaci loše kvalitete se uklanjanju skraćivanjem i filtriranjem. Ako dugački komadi očitanja imaju ispod Q20, komadi se često skraćuju od 3’ kraja. Kad se sekvenciraju amplikoni V1-3 16S rRNA, očitanja < 275 bp nakon skraćivanja se odbacuju budući da nisu prikladna za spajanje.
8.4.5.4 Spajanje očitanja uparenih krajeva
Nakon filtriranja prema kvaliteti, uparena očitanja u preostalim kvalitetnim podacima se spajaju kako bi se stvorio jedan kontinuirani slijed gena 16S rRNA dužine 250-550 bp ovisno o ciljnim varijabilnim regijama. Nakon filtriranja i spajanja obično preostane oko 70 % očitanja, ovisno o kvaliteti specifičnog sekvenciranja. Međutim, ključno je biti pažljiv kod spajanja budući da se ovdje mogu unijeti velike greške. Dužina varijabilnih regija se razlikuje među vrstama, npr. regija V1-3 gena 16S rRNA može biti između 425 i 525 bp. Ako je kvaliteta očitanja, 2 × 300 bp uparenih krajeva bi se mogla skratiti na 2 × 245 bp (490 bp), što je prekratko za spajanje parova očitanja koji potječu iz vrsta s velikim fragmentima V1-3. Parovi očitanja koji se ne spoje se uvijek odbace, čime se uvodi odstupanje. To se može spriječiti tako da se prije spajanja kako je gore navedeno odbace sva očitanja < 275 bp.
MOLEKULARNE METODE 303
Slika 8.11 Primjer Phred bodovanja kvalitete za sekvenciranje 2 × 301 bp uparenih krajeva zbirke amplikona V1-3 16S na Illumina MiSeq. Lijevo je tipični grafikon za Read1, a desno je tipični grafikon za Read2. Pozicija očitanja je prikazana u odnosu na Phred bodovanje. Plava linija predstavlja srednju ocjenu, a kutasti dijagrami vizualiziraju rasprostiranje bodovanja. Zelena površina ukazuje na dobru kvalitetu, žuta prihvatljivu, a crvena lošu. Kvaliteta se smanjuje prema kraju očitanja. Prikaz kvalitete je napravljen pomoću softvera FastQC v0.11.3.
8.4.5.5 Grupiranje OTU
Prethodno obrađena očitanja se potom grupiraju prema identitetu slijeda. Te grupe se zovu operativne taksonomske jedinice (OTU), a proces grupiranja se zove grupiranje OTU. Varijabilnost slijeda u svakoj grupi može biti uzrokovana prisutnošću vrlo blisko povezanih sojeva ili određenim pogreškama u sekvenciranju (Huse i sur., 2010). Čak i s vrlo malom stopom pogreške od 0,1 % po bazi, mogućnost savršenog 500 bp očitanja iznosi svega 61 % (0,999500). Kako su pogreške sekvenciranja nasumično distribuirane, sekvenciranje 1.000 očitanja iz jednog gena 16S rRNA bi rezultiralo s (1 - 0,61) × 1.000 = 390 različitih očitanja. Kako bi onemogućila takva napuhana raznolikost, očitanja se u OTU grupiraju prema sličnosti. Kad se analiziraju fragmenti gena 16S rRNA iz bakterija, kriterij grupiranja je sličnost sa slijedom od 97 %, što se vrlo grubo prenosi na razinu vrste ovisno o tome koja varijabilna regija se koristi. Važno je razumjeti često navođenu granicu identiteta slijeda od 97 %. Kriterij 97 % se koristi za sljedove 16S rRNA u punoj dužini kako bi se reklo da sljedovi sa sličnošću manjom od 97 % pripadaju drugoj vrsti. Taj se kriterij ne može koristiti u suprotnom smjeru, npr. sljedovi koji su slični 97 % ili više ne pripadaju nužno istoj vrsti (Janda i sur., 2007). Odabir algoritma grupiranja će promijeniti proizašle jedinice OTU (Edgar, 2013; Flynn i sur., 2015). Premda
je još uvijek najpopularnije koristiti jedan kriterij za definiranje OTU, javlja se nekoliko algoritama koji koriste profil pogreške kako bi iskoristili varijabilne granice za grupiranje tako da se može dobiti najveća razlučivost (Mahé i sur., 2014). Nakon grupiranja, broji se broj očitanja koji pripadaju svakoj OTU i odabire se reprezentativni slijed iz grupe OTU. To je obično najčešće opaženo očitanje u grupi.
8.4.5.6 Otkrivanje i uklanjanje himera
Tijekom PCR-a, nastat će himerični sljedovi, odnosno umjetni sljedovi koji se sastoje od višestrukih različitih gena 16S rRNA. Himere se javljaju kad dva necjelovita fragmenta gena 16S rRNA hibridiziraju i polimeraza ih produži. Himere mogu umjetno povećati raznolikost uzoraka i tijekom bioinformatičke obrade mora ih se identificirati i ukloniti (Quince i sur., 2011; Edgar, 2013).
8.4.5.7 Taksonomska klasifikacija
Sljedovi koji predstavljaju operativne taksonomske jedinice 16S rRNA se taksonomski klasificiraju tako da ih se uspoređuje s bazom podataka o postojećim sljedovima. Klasifikacija jako ovisi o algoritmu i bazi podataka. Obično se koriste tri velike univerzalne baze podataka: SILVA (Quast i sur., 2013), RDP (Cole i sur., 2014) i Greengenes (McDonald i sur., 2012), koje nastoje
Bodovanje kvalitete svih baza (Sanger/Illumina 1.9 encoding)
Položaj očitanja (bp)
Phre
d bo
dovi
Phre
d bo
dovi

304 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
obuhvatiti sve trenutno poznate mikrobe. Međutim, zbog njihovog širokog opsega, nisu prikladni za specifične ekosustave. Baza podataka MiDAS je stručno organizirana verzija baze podataka SILVA, koja sadrži nazive na razini roda za većinu njarsprostranjenijih vrsta ekosustava aktivnog mulja (McIlroy i sur., 2015). Nazivi su važni budući da daju poveznicu s drugim studijama i literaturom gdje se mogu pronaći funkcionalne informacije.
Algoritam korišten za usporedbu dobivenih operativnih taksonomskih jedinica s bazom podataka može koristiti različite strategije za klasifikaciju. Neki od uobičajenih algoritama koriste različite verzije pristupa najnižeg zajedničkog pretka (LCA) (Pruesse i sur., 2012). Taj pristup uzima u obzir to da neki slijed OTU može biti sličan ne samo jednom slijedu u bazi podataka. Ako je tako, algoritam operativnoj taksonomskoj jedinici dodjeljuje najnižu dijeljenu taksonomiju između tih sljedova baze podataka.
8.4.5.8 Tablica OTU
Konačni rezultat bioinformatičke obrade je tablica operativnih taksonomskih jedinica. Redovi tablice predstavljaju različite OTU, a stupci predstavljaju svaki uzorak u analizi. Polja tablice prikazuju brojevne vrijednosti za određene OTU u određenim uzorcima. Svaka OTU ima i taksonomsku klasifikaciju. Klasifikacija je često razgraničen (odvojen zarezom ili sl.) tekstualni niz koji sadrži klasifikaciju na svakoj taksonomskoj razini (carstvo, koljeno, razred, red, porodica, rod i vrsta). Ako na određenoj razini nedostaje klasifikacija, to znači da na tim razinama nije bila moguća robusna klasifikacija. Uz to, dobiva se i datoteka u fasta formatu koji sadrži sljedove DNA referentnih operativnih taksonomskih jedinica.
8.4.6 Analiza podataka
8.4.6.1 Definiranje cilja analize podataka
Teoretske mogućnosti u sklopu analize podataka amplikona 16S rRNA su mnogobrojne. Međutim, opseg analize koji je moguće postići je definiran eksperimentalnim dizajnom i varijabilnošću unutar generiranih podatka. Zbog toga se preporučuje da se provede predstudija u svrhu određivanja varijacije unutar vrste uzoraka koji će se proučavati. Sekvenciranjem određenog broja bioloških kopija moguće je donijeti utemeljene odluke o eksperimentalnom dizajnu i kopijama potrebnima da se odgovori na postavljena pitanja.
U nastavku će se prikazati kratki primjeri različitih vrsta analize podataka s poveznicama na primjere istraživanja koje mogu služiti kao daljnja inspiracija. Za konkretnije i podacima potaknute primjere preporučuje se da se konzultira online dokumentacija QIIME (Carporaso i sur., 2010), Mothur (Schloss i sur., 2009), PhyloSeq (McMurdie i Holmes, 2013), vegan (Oksanen i sur., 2015) i ampvis (Albertsen i sur., 2015).
8.4.6.2 Validacija podataka i kontrola ispravnosti
Prije započinjanja s glavnom analizom, preporučljivo jenapraviti malu istraživačku analizu kako bi se validirali podaci i otkrile moguće pogreške u obradi. To je naročito važno ako su sekvenciranje i bioinformatička obrada povjereni vanjskim izvršiteljima. Tijek rada koji se temelji na amplikonu ima brojne korake u kojima može doći do pogrešaka. To su obično pomiješani uzorci, unakrsna kontaminacija, kriva bioinformatička obrada ili loša kvaliteta sekvenciranja. Često ih se može lako otkriti tako da se dobije objektivan pregled podataka s općim statističkim podacima i jednostavnim preglednim grafikonima.
Slika 8.12 Evaluacija sekvenciranja pomoću krivulja razrijeđenja. Crvena linija ukazuje na to da svako novo očitanje rezultira identifikacijom nove OTU. Zelena linija ukazuje na to da se otkrivanje novih OTU smanjuje intenzitetom sekvenciranja. Prema tome, zeleni uzorak je dovoljno uzorkovan.
Za sve uzorke bi trebalo proučiti broj neobrađenih očitanja prije i nakon bioinformatičke obrade. Ako uzorak općenito ima malo očitanja, to bi moglo ukazivati da je nešto pošlo krivo kod pripreme zbirke. Ako se tijekom bioinformatičke obrade izgubi mnogo očitanja, to bi moglo ukazivati na lošu kvalitetu podataka. Pomiješani uzorci se mogu otkriti tako da se naprave grafikoni analize glavnih sastavnica (Principal
Broj
OTU
Intenzitet sekvenciranja

MOLEKULARNE METODE 305
component analysis - PCA) na temelju broja OTU svih uzoraka. Na grafikonima PCA, uzorci sa sličnim zajednicama mikroorganizama će se grupirati zajedno. Prema tome, koristite zdrav razum i vizualnu kontrolu grupiraju li se uzorci prema očekivanju. Na primjer, grupiraju li se kopije zajedno? Nakon što je provjerena ispravnost podataka, preporučljivo je da se provjeri je li broj sekvenciranih očitanja bio dovoljan da se obuhvati opažena raznolikost u uzorcima. To se radi analizom razrijeđenja , pri čemu se generira krivulja koja prikazuje broj identificiranih operativnih taksonomskih jedinica na različitim dubinama sekvenciranja kroz poduzorkovanje. Krivulja bi se trebala spljoštiti kao funkcija rastuće dubine sekvenciranja (slika 8.12), što ukazuje da je većina raznolikosti obuhvaćena (Schloss i Handelsman, 2005).
8.4.6.3 Zajednice ili pojedinačne vrste?
Jedinica ili perspektiva za analizu se može u grubo podijeliti u dvije kategorije: zajednice i pojedinačne vrste. U analizi koja se temelji na zajednici, parametar za usporedbu je to postoji li sveobuhvatna razlika u strukturi zajednice ili raznolikost među uzorcima, dok se iz perspektive vrsta nastoji razumjeti uloga i učinak pojedinačnih vrsta u sustavu.
• Perspektiva zajednice Analiza koja se temelji na zajednici može se podijeliti na analizu alfa-raznolikosti (unutar uzoraka) ili beta-
raznolikosti (među uzorcima). Analize alfa-raznolikosti se često koriste da se istraži ima li određena obrada učinak na broj različitih opaženih vrsta (naziva se i bogatstvo) ili na jednolikost u brojnosti vrsta u uzorku. Kako bi se usporedili uzorci, svaki uzorak se izražava u jednakovrijednim jedinicama, kojima se onda može napraviti usporedba među svim uzorcima (Magurran, 2004; Lozupone i Knight, 2008). Analize beta-raznolikosti se koriste za usporedbu zajedničke raznolikosti među uzorcima, bilo kao broj zajedničkih vrsta bilo kao količina zajedničke filogenetske raznolikosti (Lozupone i Knight, 2008).
• Perspektiva vrste I premda se na podacima o amplikonu 16S rRNA mogu napraviti mnoge izrazito složene statističke analize, kod goleme većine analiza se jednostavno radi o identificiranju najprisutnijih bakterija i njihovom povezivanju s funkcionalnim informacijama, npr. kako bi se utvrdila brojnost nitrifikanata ili nitastih bakterija u uzorku (slika 8.13). Ako se taksonomija MiDAS (McIlroy i sur., 2015) koristi za taksonomski zadatak, MiDAS Field Guide (midasfieldguide.org) se može koristiti da se ručno potraže funkcionalne informacije o pojedinim vrstama povezanima s aktivnim muljem. Alternativno, ampvis R paket (Albertsen i sur., 2015) se može koristiti da se podaci o amplikonu 16S rRNA izravno povežu s funkcionalnim informacijama u MiDAS Field Guide.
Slika 8.13Kombiniranje brojnosti vrsta s funkcionalnim informacijama je polazna točka za većinu analiza. Slika je napravljena pomoću ampvis R package (Albertsen i sur., 2015), kojim se nazivi roda izravno povezuju s funcionalnim informacijama u MiDAS Field Guide (www.midasfieldguide.org).

306 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
8.4.6.4 Identificiranje glavnih i kratkotrajnih vrsta Kad se istražuje određen sustav, često je od interesa identificirati vrste koje su važne za procese u sustavu i one koje bi mogle izazvati probleme. Polazna točka za tu analizu je identificirati glavne i kratkotrajne vrste (Grime, 1998; Gibson i sur., 1999). Za ovu analizu su potrebni višestruki uzorci i mogla bi se provesti ili u jednom uređaju za obradu otpadne vode (vremenski niz) ili u više različitih uređaja za obradu otpadne vode.
Tradicionalna definicija glavnih vrsta je da su to one vrste koje su prisutne u svim istraživanim uzorcima (Saunders i sur., 2015). Međutim, zbog velike osjetljivosti metode sekvenciranja amplikona 16S rRNA, u glavne vrste se ovakvom definicijom ubraja i velik broj jako malobrojnih vrsta. Te vrste su prisutne u svim uzorcima, ali po svoj prilici ne pridonose znatno glavnom procesu u ekosustavu. Prema tome, često se koristi praktičnija definicija glavnih vrsta zajednice, kojom se određuje da je bakterija brojna ako je, u osam od deset uzoraka, uključena u skupinu brojnih vrsta koje čine 80 % zajednice (Saunders i sur., 2015). U sustavima s visokim stupnjem inokuluma iz influenta, moglo bi biti potrebno i istražiti dolaze li glavni organizmi jednostavno s influentom ili aktivno rastu. To se realno može procijeniti ako se analizira bakterijski sastav u influentu i aktivnom mulju i kombinira s masenom bilancom sustava (Saunders i sur., 2015).
8.4.6.5 Istraživačka analiza pomoću multivarijatne statistike.
Skupovi podataka generirani sekvenciranjem amplikona 16S rRNA mogu biti izuzetno veliki tako da čak s deset uzoraka (svaki s tisućama vrsta) postaje teško dobiti pregled podataka i identificirati zanimljive obrasce. Zbog toga se često koriste različite inačice metoda ordinacije, npr. PCA. Te metode se mogu koristiti za vizualno grupiranje uzoraka u skladu s time koliko su slične njihove zajednice mikroorganizama i za identificiranje koje vrste su odgovorne za opaženo grupiranje uzoraka (slika 8.14).
Istraživački grafikoni, gdje su uzorci obojani različitim okolišnim varijablama, često su dobro mjesto za početak prije korištenja formalnog testiranja statističke hipoteze. Međutim, specifičan odabir metode ordinacije i transformacije podataka značajno ovise o postavljenom pitanju, broju uzoraka i distribuciji brojnosti. Zbog toga se preporučuje osvrt na specifičnu literaturu kako bi se razumjele primjene različitih metoda (Legendre i Gallagher, 2001; Ramette, 2007; Zuur i sur., 2007).
Slika 8.14 Istraživačka analiza pomoću PCA. Uzorci se grupiraju prema pet uređaja za obradu otpadne vode u Danskoj. Grafikon je napravljen pomoću ampvis R paketa (Albertsen i sur., 2015).
8.4.6.6 Korelacijska analiza
Druga mogućnost s podacima o amplikonu 16S rRNA je istražiti korelacije među različitim vrstama ili između vrsta i okolišnih faktora. Su-pojavljuju li se neke vrste, postoje li temeljne vrste koje su potrebne za funkcioniranje ekosustava ili je li brojnost specifičnih vrsta u korelaciji s okolišnim faktorima poput temperature? Zbog broja mogućih korelacija, rezultati se često predstavljaju u mreži grafova koji prikazuju pozitivne ili negativne korelacije između vrsta i okolišnih varijabli (Faust i sur., 2012; 2015). Međutim, u korelacijskoj analizi podataka o amplikonu 16S rRNA postoje mnoge zamke i odabiri koje treba napraviti. Na primjer, korelacijska analiza se često radi na podacima iz vremenskih nizova i korelacije bi mogle vremenski kasniti. Iznenadno povećanje koncentracije amonija moglo bi rezultirati povećanjem nitrifikanata u razdoblju od nekoliko tjedana.
8.4.6.7 Učinak obrada na pojedinačne vrste
Kako bi se statistički ispitao utjecaj specifičnih obrada na pojedinačne vrste, razvijen je niz različitih statističkih metoda koje u obzir uzimaju prirodu podataka o amplikonu na temelju brojanja. Većina metoda je modificirana ili izravno uvezena iz statističkih okvira razvijenih za analizu podataka ekspresije gena (mRNAseq) (Robinson i sur., 2010; Love i sur., 2014).
PC2
(13,
6%)
-6
-4
-2
0
2
4
-2 0 2
PC1 (15,6%)
4 6
Hjoerring
Ejby
Aalborg West
Aalborg East
Egaa

MOLEKULARNE METODE 307
8.4.7 Opća opažanja
8.4.7.1 Relativna analiza
Važno je napomenuti da je analiza zajednice mikroorganizama pomoću sekvenciranja amplikona 16S rRNA relativna analiza. To znači da se brojnost očitanja koja predstavljaju pojedinačne vrste navodi kao postotak ukupnih. Prema tome, nema veza između postotka i apsolutnog broja stanica. Prema potrebi, takva veza se može uspostaviti pomoću qPCR (vidi odjeljak 8.3).
8.4.7.2 Odstupanje kod broja kopija
I dok je gen 16S rRNA univerzalan među bakterijama, različite bakterije mogu u svojem genomu imati od jedne do više od petnaest kopija gena 16S rRNA (Farrelly i sur., 1995; Angly i sur., 2014). Prema tome, da su prisutne u jednakom broju stanica, vrsta bakterije s deset kopija gena 16S rRNA bi djelovala deset puta brojnija nego vrsta s jednim genom 16S rRNA. Uz to, neke bakterije imaju više od jedne kopije svojeg genoma, što će dovesti do dodatnog odstupanja kod procjene brojnosti (Mendell i sur., 2008; Pecoraro i sur., 2011). Prema tome, umjesto da se na postotke dobivene iz podataka o amplikonu 16S rRNA spominje kao ‘brojnosti’, trebalo bi ih spominjati kao ‘očitane brojnosti’ kako bi se implicirala inherentna odstupanja.
8.4.7.3 Odstupanja kod početnica
Kao što je već spomenuto, odabir početnice i uvjeta PCR-a također ima velik učinak na opaženu zajednicu mikroorganizama. ‘Univerzalne’ početnice su dizajnirane usporedbom slijeda nevarijabilnih regija gena 16S rRNA iz tisuća različitih bakterija, nakon čega se konsenzusom napravio slijed koji cilja većinu bakterija. Međutim, nije moguće dizajnirati početnice koje jednako dobro ciljaju sve bakterije. Uz to, odstupanja su često specifična za određenu taksonomsku kategoriju, što znači da su čitave taksonomske skupine pod-zastupljene ili čak u cijelosti zanemarene. Mnogi od novih kandidata iz koljena koji se otkrivaju pomoću metoda neovisnih o početnicama, kao što je metagenomika, pokazuju velike devijacije u nevarijabilnim lokacijama početnica ili čak umetanja velikih sljedova u sam gen 16S rRNA, što je glavni razlog zašto su nam desetljećima izmicali pažnji (Brown i sur., 2015).
8.4.7.4 Standardizacija
Bilo je mnogo pokušaja da se gore opisana odstupanja uklone, ali sa složenim sustavom kakav je aktivni mulj to vjerojatno nikad neće uspjeti. Nadalje, vrlo je teško
potvrditi jesu li odstupanja uklonjena, budući da je često teško provesti kvalitetne kontrole. Na prvi pogled bi se reklo da to potkopava metodu analize. No, to samo postavlja određena ograničenja o tome na koja se pitanja može odgovoriti i kako bi trebalo dizajnirati eksperimente.
Glavna poruka je ta da ako su odstupanja ista za sve analizirane uzorke, sekvenciranje amplikona 16S rRNA je vrlo snažno sredstvo za relativne usporedbe i opažanja prisutnosti ili odsutnosti specifičnih bakterija. Kako bi se osiguralo da su sva odstupanja ista, uzorci u jednom eksperimentu moraju se tijekom čitave obrade tretirati na sasvim jednak način. Poželjno je da ih se prikupi i pohrani, DNA se ekstrahira, pripreme se zbirke sekvenciranja i podaci se obrade na sasvim jednak način.
8.4.7.5 Utjecaj metode
Unatoč određenim ograničenjima, sekvenciranje amplikona 16S rRNA je danas jedan od najvažnijih alata u ekologiji mikroorganizama. Glavni razlozi su razlučivost i broj podataka koje omogućuje, gdje su prethodne tehnike bile puno inferiornije. 2010. godine, klonske zbirke i DGGE su još uvijek bile standardne tehnike nadopunjene FISH-om za in situ identifikaciju i kvantifikaciju. Velika studija u to doba bi uključivala 100-1.000 sljedova 16S rRNA podijeljenih u 10 uzoraka. Danas se, za isti trošak, u jednom tjednu mogu analizirati na stotine uzoraka s tisućama očitanja po uzorku. Međutim, to ujedno postavlja nove zahtjeve za vještinu ekologa koji se bave mikroorganizmima kako bi mogli baratati iznimno složenim eksperimentima i golemom količinom podataka.
8.4.8 Protokol: Illumina zbirke V1-3 amplikona 16S rRNA
Ovaj protokol opisuje kako napraviti Illumina zbirke sekvenciranja amplikona 16S rRNA varijabilnih regija 1-3 bakterijskog gena 16S rRNA. Biblioteke su prikladne za sekvenciranje na Illumina MiSeq pomoću kompleta reagensa sa 600 ciklusa. Ukupno vrijeme potrebno za provedbu ovog protokola je nekih 10 h.
8.4.8.1 Aparatura
• ND-1000 spektrofotometar (Thermo Scientific) ili slični UV-vis spektrofotometri za mjerenje koncentracije DNA i procjenu čistoće DNA.
• Korisnički priručnik Nanodrop User Manual (NanoDrop Technologies Inc. 2007).

308 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
• Qubit 2.0 fluorometar (Life Technologies), Infinite M1000 PRO (Tecan) ili sličan fluorometar za precizno mjerenje koncentracije DNA pomoću boja koje se vezuju uz DNA.
• Upute za korištenje testa Qubit (Thermo Scientific 2015a; 2015b).
• Standardni PCR termocikler s grijanim poklopcem. • Magnetno postolje za pločice s 96 jažica korištenih
za pročišćavanje DNA, npr. MagneSphere Technology Magnetic Separation Stand (Promega) ili Magnetic Stand-96 (AM10027, Ambion).
• Sustav Tapestation 2200 (Agilent) za gel elektroforezu u svrhu provjere kvalitete zbirke sekvenciranja. Alternativno koristite kovencionalnu opremu za gel elektroforezu (Green i Sambrook, 2012).
• Priručnici Tapestation 2200 (Agilent Technologies, 2012; 2013; 2015).
• Pipete (raspon od 1 µL do 1.000 µL).
8.4.8.2 Materijali
Centrifuga za PCR pločicu • Nastavci bez DNAze (10 µL, 300 µL i 1.000 µL). • Epruvete bez DNAze (1,5 mL). • Prozirne epruvete tankih stjenki bez DNAze za PCR
(500 µL). • PCR pločice s 96 jažica (#82006-664, VWR). • PCR poklopci u nizu. • Mikroploče OptiPlate-96 Black (Perkin Elmer). • H2O bez nukleaze (Qiagen). • Fluorescentne boje koje se vezuju uz DNA, npr.
komplet Qubit dsDNA HS assay kit (Life Technologies), komplet širokog raspona Quant-iT dsDNA assay kit (Life Technologies), komplet visoke osjetljivosti Quant-iT dsDNA assay kit (Life Technologies).
• Komplet Platinum Taq DNA Polymerase High Fidelity kit (Life Technologies).
• dNTP mješavina. • Šifrom označene mješavine adaptera gena V1-3 16S
rRNA (5 µM svakog prednjeg i stražnjeg adaptera), vidi odjeljak 8.4.11.
• Agencourt AMPure XP (Beckman Coulter). • Etanol, 99 %.+ • D1000 Screentape (Agilent) i Genomic DNA
Screentapes (Agilent). Alternativno koristite reagense za konvencionalnu gel elektroforezu (Green i Sambrook, 2012).
8.4.8.3. Protokol
• Kontrola kvalitete i razrjeđivanje DNA uzorka (2,5 h) U ovom se odjeljku kontrolira kvaliteta ekstrahirane genomske DNA i DNA se razrjeđuje do koncentracije prikladne za PCR. Više informacija u odjeljku 8.4.9 Tumačenje i rješavanje problema.
1. Fluorescencijsko mjerenje koncentracije DNA a. Koristite test Qubit dsDNA BR ili test širokog
raspona Quant-iT dsDNA u skladu s protokolom koji je preporučio proizvođač.
b. Koristite 2 µL svakog uzorka po reakciji. c. Provedite jedno mjerenje po uzorku.
2. UV-vis kontrola kvalitete (opcionalno) a. Koristite Nanodrop1000 u skladu s protokolom
koji je preporučio proizvođač (Nanodrop Technologies Inc., 2007).
b. Za velike šarže uzorka promislite o mjerenju podskupa uzoraka (npr. 8 od 96).
c. Pokrenite i postavite očitanje instrumenta na nulu s istim puferom u kojemu su uzorci eluirani.
d. Koristite 2 µL uzorka po mjerenju. e. Provedite jedno mjerenje po uzorku.
3. Gel elektroforeza (opcionalno) a. Koristite Tapestation 2200 u skladu s protokolom
koji je preporučio proizvođač. b. Koristite Genomic Screentapes s referentnom
skalom DNA. c. Provedite jedno mjerenje po uzorku. d. Za velike šarže uzorka promislite o mjerenju
podskupa uzoraka (npr. 7 uzoraka od 96 + 1 skala).
4. Razrjeđivanje uzorka a. Na temelju fluorescencijskog mjerenja
koncentracije DNA, izračunajte koliko je vode bez nukleaze potrebno za svaki od uzoraka da ih se razrijedi do 5 ng µL-1. Korištena formula glasi:
Vsample·Csample
Cfinal‒Vsample =
VH2O → 5µL·Csample
5 ngµL
‒5µL=VH2O
b. Prebacite 5 µL uzorka u praznu jažicu na PCR
pločici s 96 jažica. c. Razrijedite uzorak s izračunatom količinom vode
bez nukleaze. Kritični korak Ako je koncentracija uzorka 5 ng
µL-1 ili manje, uzorak koristite nerazrijeđen ili bacite uzorak. Ako razrijeđivanje zahtijeva vodu
8.2

MOLEKULARNE METODE 309
s > 150 µL nukleaze, moglo bi biti potrebno prethodno razrjeđenje.
d. Ponovite za sve uzorke. e. Zatvorite pločicu s PCR poklopcima u nizu. Pauza Razrijeđeni uzorci se mogu čuvati na -20 °C
najmanje mjesec dana.
• PCR zbirke (2,0 h) Ovaj se dio tiče pripreme za zbirke sekvenciranja pomoću amplifikacije PCR-om. Ulazni kalup je genomska DNA (5 ng µL-1), a izlaz su amplikoni V1-3 16S rRNA veličine otprilike 614 bp.
1. Priprema a. Reakcija PCR zbirke se provodi dvaput za svaki
uzorak. b. Ne zaboravite uključiti negativnu kontrolu (H2O
bez nukleaze) i pozitivnu kontrolu (DNA zajednice mikroorganizama za koju se zna da se amplificira pomoću 16S rRNA PCR).
c. Zabilježite koji V1-3 adapteri s jedinstvenim šiframa su dodijeljeni kojim uzorcima.
2. Pomiješajte PCR reakciju a. Pripremite glavnu mješavinu za (uzorci +
kontrole) × 2 + 3. Svaki višak glavne mješavine će nadoknaditi gubitak tijekom pipetiranja.
b. Dodajte reagense zadanim redom kako bi se proizvela glavna mješavina.
c. Prebacite 13 µL glavne mješavine u jažice nove PCR pločice s 96 jažica.
d. Dodajte 10 µL dodijeljene šifrirane mješavine adaptera V1-3 16S rRNA (1 µM) u svaku jažicu i pipetu povlačite gore-dolje 10 puta kako bi se izmiješao sadržaj. Konačna koncentracija adaptera je 400 nM.
Kritičan korak Velik rizik da će se pobrkati uzorci i adapteri. Budite pažljivi.
e. Dodajte 2 µL kalupa DNA (ukupno 10 ng DNA) i pomiješajte tako da pipetu povlačite gore-dolje. Konačni volumen je 25 µL.
Kritičan korak Velik rizik da će se pobrkati uzorci. f. Zatvorite PCR pločicu s 96 jažica s PCR
poklopcima u nizu. g. Zavrtite PCR pločicu s 96 jažica kako bi se
reakcijska mješavina PCR istaložila na dno pločice.
Reagensi Konačna konc. u 25 μL
reakciji
Volumen (μL) za 1 r×n
Voda bez nukleaze - 7,65×10 pufer Platinum High Fidelity ×1 2,5dNTP (5 mM) 400 uM 2MgSO4 (50 mM) 1,5 mM 0,75Platinum Taq DNA Polymerase High Fidelity (5 U μL-1)
0,02 U μL-1 0,1
Ukupni volumen 13
3. Pokrenite PCR inkubaciju a. Programirajte termocikler sa sljedećim
programom:
Korak Temperatura Trajanje
Denaturacija 95 °C 2 min
30 ciklusa
Denaturacija 95 °C 20 s
Vezivanje 56 °C 30 s
Amplifikacija 72 °C 60 s
Amplifikacija 72 °C 5 min
Pohrana 4 °C Neograničeno
b. Nakon reakcije PCR, ponovno promiješajte
mješavinu za reakciju PCR. c. Uklonite PCR poklopce u nizu i objedinite
dvostruke reakcije PCR za svaki pojedinačni uzorak. Konačni volumen je 50 µL.
d. Nakon PCR-a, uzorci se sada spominju kao ‘zbirke sekvenciranja’ ili ‘kratke zbirke’. Pauza Zbirke se mogu čuvati na -20 °C najmanje mjesec dana.
• Čišćenje zbirke (2,0 h) Ovaj dio uključuje čišćenje PCR reakcija. Svrha je ukloniti preostale reagense i moguće kratke (< 200 bp) nespecifične produkte PCR-a. Rezultat su čiste zbirke sekvenciranja koje se sastoje jedino od amplikona 16S rRNA (oko 614 bp).
1. Priprema a. Nježno protresite Agencourt AMPure XP bočicu
kako biste resuspendirali kuglice, uklonite potreban volumen 40 µL kuglica × [n(uzorci)+3] i ostavite da se uravnoteži na sobnoj temperaturi.

310 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
b. Pripremite svježu otopinu 80 %-tnog etanola tako da 20 mL etanola (99 %) prebacite u greiner (ili falcon) kivetu (50 mL) i dodate 5 mL vode bez nukleaze. Izmiješajte tako da preokrenete epruvetu.
2. Zbirke povežite s kuglicama a. Prebacite 40 µL otopine kuglice po jažici u novu
PCR pločicu s 96 jažica što odgovara broju uzoraka.
b. Dodajte 50 µL zbirke svakoj jažici s kuglicama i promiješajte tako da pipetu povlačite gore-dolje 10 puta.
Kritičan korak Važno je da omjer kuglice i uzorka iznosi 4:5. Ako zbog nekog razloga ima manje/više od 50-µL uzorka, u skladu s time treba korigirati volumen kuglica.
c. Inkubirajte 5 min na sobnoj temperaturi. 3. Operite uvezane zbirke
a. Postavite PCR pločicu s 96 jažica na magnetno postolje i pričekajte dok tekućina ne postane bistra (2-4 min). Svi kasniji koraci se provode s pločicom na magnetnom postolju.
b. Pomoću pipete uklonite i bacite koliko je najviše moguće tekućine.
Kritičan korak Pazite da ne prebacite kuglice (smeđe granule).
c. Operite kuglice-granule s 200 µL etanola (80 %) tako da etanol nježno pipetom raspodijelite preko kuglica. Pustite ga da se odmori 30 s i potom uklonite tekućinu.
d. Ponovite gornji korak (3c). e. Pobrinite se da nakon pranja ne ostane višak
tekućine. Ako ga ima, uklonite ga pipetom od 10 µL.
f. Ostavite granule da se osuše 7 min. Kritičan korak Izbjegavajte prekomjerno sušenje
grijanjem ili duga sušenja budući da će to otežati eluiranje DNA.
4. Eluirajte zbirku a. Uklonite PCR pločicu s 96 jažica sa sasušenim
kuglicama-granulama s magnetnog postolja. b. Dodajte 33 µL vode bez nukleaze i promiješajte
tako da pipetu povlačite gore-dolje 10 puta kako bi se kuglice resuspendirale.
c. Inkubirajte 2 min na sobnoj temperaturi. d. Vratite PCR pločicu s 96 jažica na magnetno
postolje i pričekajte dok se tekućina ne izbistri (1-2 min).
e. Prebacite 30 µL tekućine u praznu jažicu u novoj pločici s 96 jažica.
Kritičan korak Pazite da ne prebacite kuglice.
Pauza Pročišćene zbirke se mogu čuvati na -20 °C najmanje 6 mjeseci (vidi niži odjeljak Pohrana i transport).
• Kontrola kvalitete zbirke (1,5 h) Ovaj dio se tiče mjerenja koncentracije DNA pročišćenih zbirki i kasnije kontrole kvalitete pomoću gel elektroforeze. Potrebno je potvrditi da je u zbirkama prisutan jedino ciljni amplikon 16S rRNA.
1. Fluorescencijsko mjerenje koncentracije DNA a. Koristite test visoke osjetljivosti Qubit dsDNA ili
test visoke osjetljivosti Quant-iT dsDNA u skladu s protokolom koji je preporučio proizvođač.
b. Koristite 2 µL svakog uzorka po reakciji. c. Provedite jedno mjerenje po uzorku.
2. Gel elektroforeza a. Koristite Tapestation 2200 u skladu s protokolom
koji je preporučio proizvođač. b. Koristite D1000 Screentapes s referentnom
skalom DNA. c. Provedite jedno mjerenje po uzorku. d. Za velike šarže uzorka razmotrite mjerenje
podskupa uzoraka (npr. 15 od 96). U analizu uvijek uključite negativnu i pozitivnu kontrolu.
• Objedinjavanje zbirki (2,0 h) Ovaj se dio tiče objedinjavanja svih zbirki. Svrha je dobiti konačni uzorak koji sadrži ekvimolarne koncentracije (isti broj molekula) svake zbirke. Volumeni se izračunavaju iz koncentracija DNA gore dobivenih zbirki.
1. Izračunajte potrebni volumen svakog uzorka a. Zbirke s koncentracijom manjom od 1 ng µL-1
treba isključiti (ili ih izostavite ili ponovno provedite PCR).
b. Otkrijte uzorak s najmanjom koncentracijom i tu koncentraciju pomnožite s 15 µL (npr. 1 ng µL-1 × 15 µL = 15 ng). To je količina zbirke koja se želi od svake zbirke.
c. Izračunajte volumene potrebne da se dobije ista količina zbirke za svaku od ostalih zbirki.
d. Ako su za neke zbirke potrebni volumeni manji od 1 µL, razmotrite razrjeđivanje zbirke i ponovno izračunajte potrebne volumene.
2. Objedinite zbirke a. Koristite novu epruvetu (1,5 mL). b. U epruvetu prebacite izračunati volumen svakog
uzorka. c. Dobro promiješajte nakon što su dodani svi
uzorci.

MOLEKULARNE METODE 311
3. Fluorescencijsko mjerenje koncentracije DNA a. Koristite test visoke osjetljivosti Qubit dsDNA u
skladu s protokolom koji je preporučio proizvođač.
b. Koristite 2 µL fonda zbirki po reakciji. c. Mjerenje provedite tri puta. d. Izračunajte prosječnu koncentraciju. e. Na temelju koncentracije, izračunajte
nanomolarnu koncentraciju pomoću sljedeće formule.
cnM=cng µL⁄ ·1,000,000
µLL
650 g mol⁄
bp · 614 bp
f. Ako je koncentracija manja od 4 nM, koncentrirajte uzorak pomoću AMPure kuglica za pročišćavanje (vidi raniji dio o čišćenju zbirke u ovom poglavlju). Pobrinite se da koristite omjer kuglica i uzorka od 4:5 (npr. ako imate 100 µL iz fonda uzoraka trebate koristiti 80 µL AMP otopine kuglica). Morate izračunati koliko je vode bez nukleaze potrebno za eluiranje kako bi se dobila koncentracija od 4 nM ili više. Predvidite i gubitak do 50 % produkta. Dakle, ako je potreban 2× koncentrat, tada bi 100 µL fonda zbirki koje se unose trebalo eluirati u 25 µL vode bez nukleaze). Ponovite mjerenje koncentracije DNA koncentriranog fonda.
• Pohrana i transport Ovaj dio se tiče pohrane zbirki i fonda zbirki.
1. Ako se planira pohrana fonda zbirki, nemojte ga razrijediti. DNA bolje podnosi pohranu ako je se čuva u koncentriranom obliku (> 5 ng µL-1).
2. Za kratkotrajnu pohranu ili transport (< 14 d), pročišćena DNA u zbirci se može čuvati na sobnoj temperaturi.
3. Za srednjoročnu pohranu (< 12 mjeseci), zbirke se mogu čuvati na -20 °C.
4. Za dugotrajnu pohranu (> 12 mjeseci), zbirke bi trebalo čuvati na -80 °C.
8.4.9 Tumačenje i rješavanje problema
8.4.9.1 Kontrola kvalitete i razrjeđivanje DNA uzorka
Vezano uz ulaznu DNA u obzir treba uzeti tri stvari : (i) količinu, (ii) kvalitetu i (iii) potencijalne kontaminante. Te karakteristike se istražuju pomoću fluorescencijskog testa koncentracije DNA, UV-vis spektrofotometrije i gel elektroforeze.
Preporučena količina DNA zajednice mikroorganizama za PCR iznosi 1-100 ng ukupne DNA, pri čemu se često koristi 10 ng (otprilike 2×106 stanica). Ako se koristi više DNA, tada se povećava rizik od amplificiranja slučajnih dijelova DNA i može doći do inhibicije reakcije PCR-a. Slučajni amplikoni su problematični budući da rezultiraju smanjenim prinosom podataka iz sekvenciranja i lošom kvalitetom. Niska koncentracija DNA povećava rizik od neuspjeha PCR-a i uvodi varijancu za malobrojne članove zajednice (Kennedy i sur., 2014). UV-vis spektrofotometrija se može koristiti da se izmjeri koncentracija DNA, ali procjena može biti nesigurna budući da na nju djeluje kontaminacija reagensima, prisutnost nukleotida i RNA. Metode utemeljene na fluorescenciji su uvijek bolje, a UV-vis spektrofotometriju bi trebalo koristiti jedino kao rezervu (Li i sur., 2014).
Kvaliteta DNA je važna budući da djeluje na efektivnu količinu DNA dostupnu za reakciju PCR. Ako je DNA teško degradirana (većina DNA < 5.000 bp), povećava se rizik da će se genski biljezi raspasti, čime postaju nedostupni za PCR (Beers i sur., 2006; Wilson i sur., 1997).
Ne-DNA kontaminanti se odnose na kemijske tvari ili organske molekule unesene iz uzorka (npr. huminske kiseline i složeni šećeri) ili za vrijeme ekstrakcije DNA (tj. SDS, alkoholi, denaturirajuće soli), a koje mogu inhibirati reakciju PCR, čime se smanjuje učinkovitost (Wilson i sur., 1997). UV-vis spektar čiste DNA ima vrlo svojstvenu krivulju. Anomalije u ovoj krivulji su znak kontaminacije i često ih se testira tako da se gleda omjer absorbancije na 260 nm i 280 nm (A260/280) i omjer na 260 nm i 230 nm (A260/230). A260/280 čiste DNA je obično oko 1,8, ali ako se jako razlikuje (npr. ± 0,4), tada bi to moglo ukazivati na proteinsku kontaminaciju ili rezidualne reagense kao što su alkoholi iz pribora za ekstrakciju. A260/230 čiste DNA je obično između 2,0 i 2,2, a ako se jako razlikuje to bi moglo ukazivati na rezidualne ugljikohidrate ili reagense iz ekstrakcije DNA. UV-vis spektrofotometrija je vrlo osjetljiva metoda i vrsta pufera i pH mogu rezultate učiniti nejasnima (Thermo Scientific, 2015).
DNA kontaminanti mogu biti DNA iz živih oblika koji nisu od interesa kad se ciljaju bakterije, tj. eukariotska DNA iz gljiva ili biljaka. Prisutnost kontaminirajuće DNA smanjuje efektivnu količinu ciljne DNA u uzorku i smanjuje učinkovitost PCR (Tebbe i sur., 1993). To se ne može mjeriti unaprijed, ali mogle bi se dobiti indicije tako da se prije ekstrakcije DNA
8.3

312 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
vizualno pregleda biomasa (npr. ima li vidljivog biljnog materijala?).
Ako postoji sumnja na kontaminaciju, dobra ideja je provesti testni PCR s nekoliko uzoraka kako bi se vidjelo je li u tome problem. PCR često djeluje unatoč kontaminaciji, ali ako PCR ne uspije, ponekad rješenje može biti dodatno pročišćavanje. Ne zaboravite koristiti metodu čišćenja koja može obuhvatiti veliku molekularnu genomsku DNA (> 10.000 bp), npr. kuglice Agencourt AMPure XP. Mnogi na koloni zasnovani kompleti za pročišćavanje su dizajnirani za DNA < 10.000 bp, pa će prema tome ukloniti genomsku DNA.
8.4.9.2 PCR zbirke
Kad se provodi PCR, sastav reakcijske smjese i postavke inkubacije će utjecati na opažen sastav zajednice u konačnim podacima. U istraživačkom okruženju, uobičajeno je optimirati uvjete PCR-a kako bi se povećala količina proizvedenog ciljnog amplikona i smanjila količina neželjenih, slučajnih produkata PCR-a. Međutim, za sekvenciranje amplikona 16S rRNA, postavke PCR-a bi trebalo držati istima za sve uzorke koje treba usporediti. Kako bi se osigurala konzistentnost, preporučuje se da se koriste standardne postavke iz testiranih protokola. Cjelovitosti radi, u nastavku slijedi kratak prikaz onoga što se obično mijenja tijekom optimizacije PCR-a.
Koncentracija Mg2+ i temperatura vezivanja će djelovati na to koliko su početnice specifične kad pronalaze svoje mete. Ako su prestroge, opazit će se negativno odstupanje prema određenim vrstama. Ako nisu dovoljno stroge, postojat će povećan rizik od amplificiranja slučajnih dijelova DNA. Broj ciklusa PCR određuje koliko se produkta proizvodi i koliko puta prividno neuspjeli PCR mogu uspjeti ako se jednostavno poveća broj ciklusa. Međutim, prijavljeno je i da je povećanim brojem ciklusa PCR-a uvedena greška osjetljivosti PCR-a (Polz i sur., 1998; Kennedy i sur., 2014). Isto tako, ako se provede previše ciklusa i potroše se početnice i ostali reagensi, postoji povećan rizik od nastanka himeričnih produkata (Qiu i sur., 2001).
8.4.9.3 Čišćenje zbirke
Čišćenjem zbirke uklanjaju se ostaci reagensa (početnice, nukleotidi, polimeraza itd.) kao i mali (< 200 bp), slučajni komadići DNA. Svi ti kontaminanti mogu omesti sekvenciranje DNA, bilo tako da se smanji kvaliteta podataka ili tako da sekvenciranje doživi potpun neuspjeh.
Metoda pročišćavanja korištena u protokolu se temelji na taloženju DNA na male, magnetne plastične kuglice koje se zovu SPRI (od Solid Phase Reversible Immobilization) kuglice. Princip metode nije dobro opisan u literaturi (DeAngelis i sur., 1995), no različiti blogovi predstavljaju neke hipoteze (Hadfield, 2012). Zbog njezinog kemijskog sastava, DNA ima ukupan negativni naboj i lako se topi u vodi, gdje elektrostatski međudjeluje s polarnim molekulama vode. Kako bi se DNA istaložila iz otopine, dodaje se sol NaCl. Nastaju ioni Na+, koji štite negativne naboje DNA, zbog čega je manje topiva, čime se potiče taloženje. PEG koji svojom koncentracijom smanjuje udio vode i povećava efektivnu koncentraciju Na+ (engl. crowding) drastično povećava učinak zaštite. Kad se DNA taloži, ona preferira površinu SPRI kuglica. Ta sklonost je suprotna predviđanju budući da je površina kuglica negativno nabijena, zbog čega bi trebala odbijati DNA u negativno nabijenom ili zaštićenom stanju. O ovoj se temi puno raspravlja i nema jasnog objašnjenja, ali neki smatraju da u interakciji između DNA i SPRI kuglica posreduje sloj vode ili iona Na+ koji oblažu SPRI kuglice. Prema tome, miješanje otopljene DNA i kuglica u otopini koja sadrži kemikalije koje smanjuju udio vode će rezultirati time da se DNA taloži na kuglice. Otopina se može ukloniti, a kuglice s DNA se mogu oprati pomoću 80 %-tnog etanola. Sastav otopine za pranje je optimiran kako bi se otopili mali kontaminanti kao što su sol, nukleotidi itd., ali osigurava da se DNA neće ponovno otopiti. Nakon pranja, DNA se ponovno otapa u vodi bez nukleaze ili puferu i spremna je za korištenje.
Male molekule DNA će prije ostati u otopini nego veće molekule DNA kad se doda određena količina tvari koje smanjuju udio vode. To znači da se mali dijelovi DNA mogu ukloniti dodavanjem ispravne količine takve otopine. U ovom protokolu, za 50 µL uzorak koristi se 40 µL otopine Agencourt AMPure XP (kuglice + „crowding“ otopina). Omjer kuglica i uzorka od 4:5 pogoduje vezivanju DNA > 200 bp uz kuglice, a manji dijelovi ostaju u otopini i kasnije se uklanjaju. Treba paziti kad se odmjerava uzorak ili otopina s kuglicama. Ako je omjer kuglice/uzorak < 0,5, uklanja se i amplikon 16S rRNA. Ako je omjer > 1,0, tada se kontaminacija neće ukloniti kako treba.
Kod sušenja preostalog etanola iz kuglica nakon pranja, pazite da ne pretjerate. Nemojte koristiti toplinu ili ih predugo sušiti. Ako kuglica-granula postane presuha (previše pukotina u granuli), bit će teško ponovno otopiti DNA i produkt će se izgubiti.

MOLEKULARNE METODE 313
Umjesto kuglica mogu se koristiti druge metode pročišćavanja kao što su metode zasnovane na koloni, npr. QIAquick PCR Purification Kit (Qiagen). Opća načela pročišćavanja su slična.
8.4.9.4 Kontrola kvalitete zbirke
Koncentracija DNA se mjeri kako bi se pružio temelj za objedinjavanje zbirki. Mjerenje koncentracije ne treba biti jako precizno (± 20 % je prikladno), tako da se provodi samo jedno mjerenje.
Gel elektroforeza se provodi kako bi se osiguralo da je uklonjena sva kontaminirajuća DNA (početnice i nasumično amplificirana DNA) i da je prisutan samo ciljni 16S rRNA amplikon (614 bp). Općenito, provodi se samo za podskup svih uzoraka budući da je gel elektroforeza skupa i oduzima puno vremena, a nasumični podskup bi trebao dati naznaku uspjeha pročišćavanja. Kad odabirete uzorke za podskup, uvijek treba uključiti negativne i pozitivne uzorke. Isto tako, na temelju mjerenja koncentracije, uključite uzorke koji imaju nisku koncentraciju kako bi se vidjelo je li prisutan produkt amplikona V1-3 16S rRNA. Ako produkt amplikona V1-3 16S rRNA nije prisutan, tada ponovite PCR na tim uzorcima.
Produkt amplikona V1-3 16S rRNA ima prosječnu veličinu od 614 bp, no među različitim vrstama veličina može varirati za čak ± 100 bp. Za zajednice bakterija s mnogim različitim vrstama to se obično manifestira kao široki maksmimum s vršnim maksimumom od oko 614 bp kad se koristi elektroforeza Tapestation. Ako se analizira zajednica sa svega nekoliko članova, na rezultatu elektroforeze Tapestation se mogu pokazati višestruki maksimumi između 500 i 700 bp.
Ne bi trebalo ostati dijelova DNA manjih od 300 bp. Ako ostanu, trebalo bi izmijeniti korak pročišćavanja i ponoviti ga. Ako kontaminant čini < 1 % ukupne DNA u uzorku, sekvenciranje bi i dalje trebalo dati koristan rezultat i ponavljanje pročišćavanja se može izbjeći. Rijeko se može vidjeti nepoznata kontaminacija veća od 700 bp. Ako je prisutna, pokušajte ponoviti PCR. Ako je i dalje prisutna, svejedno pokušajte sekvencirati zbirku. Kontaminant će se moći isfiltrirati pomoću bioinformatike.
Pozitivni uzorak bi trebao pokazati jasan produkt od oko 614 bp. Negativni uzorak ne bi trebao pokazati nikakav produkt ili slab produkt koji je < 1 ‰ ukupne DNA ostalih uzoraka. Često je teško u potpunosti izbjeći kontaminaciju, ali prihvatljiva je sve dok je razina kontaminacije relativno niska. Ako negativna kontrola
ima relativno visoku razinu kontaminacije, vjerojatno je da su svi uzorci kontaminirani. Izmijenite strukturu PCR i ponovite PCR reakcije za sve uzorke, po mogućnosti s novim šaržama reagensa.
8.4.9.5 Objedinjavanje zbirki
Ekvimolarno objedinjavanje zbirki se provodi kako bi se osiguralo da se iz svakog uzorka dobiju jednake količine podataka tijekom sekvenciranja. Pogreške u objedinjavanju će imati izravni učinak na količinu dobivenih podataka. Ako se kod objedinjavanja naprave pogreške, ako je moguće, krenite ispočetka.
8.4.9.6 Kontrola kvalitete i razrjeđivanje fonda
Mjerenje koncentracije fonda zbirki je jako važno budući da se ono koristi za određivanje koliko zbirke treba staviti na sekvenator. Ako se koncentracija izmjeri višom nego što zaista jest, iz sekvenciranja će se dobiti manje podataka. Ako je izmjerena koncentracija niža nego što zaista jest, sekvenator se može preopteretiti, što rezultira lošom kvalitetom podataka ili blokiranjem rada sekvenatora.
Zbog toga bi fond zbirki trebalo mjeriti tri puta i uprosječiti koncentracije. Pobrinite se da prije korištenja fluorometar kalibrirate referentnim uzorcima za koje se zna da nisu kontaminirani ili degradirani.
8.4.9.7 Pohranjivanje
DNA bi trebalo pohraniti u ultra-čistoj vodi ili TE puferu (pH 7-8). Bilo je prijava o kvarenju DNA nakon dugotrajne pohrane u ultra-čistoj vodi, čemu je vjerojatno uzrok krhka ravnoteža pH, budući da nema nikakvog pufera. Čista, otopljena DNA je vrlo stabilna čak i na sobnoj temperaturi i može se na kratko vrijeme sigurno pohraniti ili transportirati pri sobnoj temperaturi. Za srednjoročnu ili dugoročnu pohranu, preporučuje se zamrzavanje DNA na -20 ili -80 °C (Smith i Monn, 2005). Izbjegavajte ponavljanje ciklusa zamrzavanja i otapanja DNA budući da to degradira DNA. Ako će se uzorak koristiti više puta, pripremite alikvote.
8.4.10 Protokol: Illumina V1-3 16S sekvenciranje amplikona
Ovaj protokol opisuje važne dodatke protokolu kad se priprema sekvenciranje zbirki amplikona Illumina V1-3 16S rRNA na Illumina MiSeq. Za pojedinosti o koracima koji slijede standardnu proceduru, čitatelja upućujemo na korisnički priručnik MiSeq System User Guide (Illumina

314 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
Inc. 2014b). Za izvođenje protokola potrebno je otprilike 3,5 h, no za sekvenciranje je potrebno 56 h.
8.4.10.1 Aparatura
Za izvođenje protokola potrebna je sljedeća aparatura:
• MiSeq (Illumina). • Mikropipete (raspona od 1 uL do 1.000 uL). • Korisnički priručnik MiSeq System User Guide
(Illumina Inc. 2014b). • Illumina Experimental Manager v1.9 (illumina.com).
8.4.10.2 Reagensi
Za izvođenje protokola potrebni su sljedeći reagensi:
• Led. • Komplet reagensa MiSeq Reagent kit v3, 600 ciklusa
(Illumina). Uključuje kazetu s reagensima i HT1 pufer.
• 2 M NaOH, (čistoća: molecular grade) • Početnice za sekvenciranje (Read1, Read2 i Index),
vidi odjeljak 8.4.11. • Nastavci bez DNAze (10 µL, 300 µL i 1.000 µL). • Epruvete bez DNAze (1,5 mL). • Voda bez nukleaze (Qiagen). • Kontrolna zbirka PhiX control library v3, 10 nM
(Illumina). • Etanol, 70 % (čistoća: molecular grade). • Laboratorijske maramice, bez dlačica. • Maramice za čišćenje mikroskopskih leća.
8.4.10.3 Protokol
• Pripremiti MiSeq (2,0 h) U ovom se dijelu opisuje otapanje reagensa te priprema instrumenta MiSeq i datoteke Sample sheet.
1. Pranje instrumenta a. U skladu s preporukama u Korisničkom vodiču.
2. Ponovno pokretnite MiSeq kako bi se ponovno podesila memorija. a. Pod Manage Instrument u kontrolnom softveru
MiSeq pritisnite Reboot i pričekajte (to bi moglo potrajati 10 min).
b. Kontrolni softver MiSeq će početi pokretati instrument nakon ponovnog pokretanja. Kad je to gotovo, pojavit će se Control interface.
3. Otapanje reagensa a. Stavite kazetu s reagensima i HT1 pufer 1 h u
vodenu kupelj na sobnoj temperaturi. Nakon otapanja čuvajte na 4 °C do korištenja.
Pauza Otopljena kazeta s reagensima MiSeq se može čuvati tjedan dana na 4 °C.
b. Pregled kazete s reagensima: preokrenite kazetu deset puta, pregledajte ima li taloga, a onda kucnite kazetom o stol kako bi se uklonili mjehurići.
c. Otopite početnice za sekvenciranje (Read1, Read2 i Index) i 2 M NaOH na sobnoj temperaturi. Početnice nakon otapanja stavite na led, a 2 M NaOH ostavite na sobnoj temperaturi do korištenja.
4. Pripremite obrazac Sample Sheet a. Otvorite predložak MiSeq ‘SampleSheet.csv’ u
Notepad++ ili nekom drugom jednostavnom programu za uređivanje teksta.
Kritičan korak ‘SampleSheet.csv’ je tekstna datoteka s vrijednostima odvojenim zarezom (.csv), ali je ne bi trebalo otvarati u programu Microsoft Excel! Excel bi mogao pokvariti formatiranje datoteke.
b. Promijenite informacije o projektu i uzorku: [Header] Investigator (Istraživač), Project name (Naziv projekta), Experiment Name (Naziv eksperimenta), Date (Datum); [Data] Sample_ID (Uzorak_ID), Sample_Name (Uzorak_Naziv), index i index2.
Kritičan korak Važne informacije su: [Header] Chemistry, [Reads] i [Data] kolone index i index2. Te informacije imaju značajan utjecaj na to kako se izvodi proces. Sve ostale informacije se mogu kasnije promijeniti ako su pogrešne.
c. Nakon ispunjavanja obrasca Sample Sheet, provjerite cjelovitost ‘SampleSheet.csv’ tako da ga prebacite u Illumina ‘Experimental Manager’. Ako se može prebaciti, tada bi trebao biti kompatibilan s MiSeq.
d. Pomoću USB memorije prebacite ‘SampleSheet.csv’ u MiSeq.
• Pripremiti zbirke za sekvenciranje (1,0 h) U ovom se dijelu opisuje denaturacija i razrjeđivanje zbirki za sekvenciranje. 1. Pregled uzorka
a. Kontrolna zbirka: PhiX control library v3, 10 nM. b. Fond zbirki (do 400 uzoraka), > 4 nM. c. Slijedite sljedeće korake i za kontrolnu zbirku
PhiX i za fond zbirki. 2. Otopite zbirke i čuvajte ih na ledu. 3. Vodom bez nukleaze razrijedite zbirke za
sekvenciranje do 4 nM. 4. Pripremite 0,1 M otopine NaOH.
a. 475 µL DNA H2O i 25 µL 2 M NaOH 5. Denaturirajte zbirke za sekvenciranje

MOLEKULARNE METODE 315
a. Pomiješajte 5 µL zbirke + 5 µL 0,1 M NaOH. Konačna koncentracija zbirke je 2 nM.
b. Pipetu povlačite gore-dolje 10 deset puta kako biste promiješali sadržaj.
c. Inkubirajte 5 min na sobnoj temperaturi. 6. Razrijedite denaturirane zbirke (2 nM) do 20 pM.
a. Pomiješajte 10 µL denaturirane zbirke s 990 prethodno rashlađenog HT1. Koncentracija je 20 pM.
7. Pomiješajte zbirku PhiX (20 pM) s fondom zbirki (20 pM) tako da čine 20 % odnosno 80 % konačne mješavine. a. Pomiješajte 120 µL zbirke PhiX s 480 µL fonda
zbirke. b. Mješavinu PhiX i fonda stavite na led do
korištenja. Pauza Razrijeđene zbirke se mogu čuvati na -20 °C
do mjesec dana. Duža razdoblja pohrane bi mogla dovesti do smanjene koncentracije, a slijedom toga i do smanjenog sekvenciranja.
• Postavite uzorak i početnice na kazetu s reagensima 1. Dodavanje početnica na kazetu s reagensima.
a. Odredište početnica: Read1 = well 12. Index = well 13. Read2 = well 14. Kritičan korak Brojčano označavanje jažica na
kazeti s reagensima može biti zbunjujuće. Uzmite si vremena kako biste se uvjerili da se koriste prave jažice.
b. Za svaku početnicu: Vrhom pipete od 1.000 µL probušite foliju koja prekriva ciljnu jažicu i nakon toga prebacite 100 µL sadržaja jažice u epruvetu. Dodajte 3,4 µL predmetne početnice i dobro promiješajte. Otopinu prebacite natrag u jažicu iz koje je potekla i promiješajte. Ponovite za sve početnice.
2. Dodavanje uzorka u kazetu s reagensima a. Vrhom probijte jažicu br. 17 i dodajte mješavinu
600 µL PhiX/fond.
• Sekvenciranje (0,5 + 56 h) U ovom se dijelu opisuje pokretanje procesa sekvenciranja. 1. Na sučelju kontrolnog softvera MiSeq pritisnite
Sequence i slijedite upute u Korisničkom priručniku MiSeq System User Guide za pripremu/punjenje ćelije, punjenje kazete s reagensima, referiranje na obrazac Sample Sheet i pokretanje procesa sekvenciranja.
2. Tijekom sekvenciranja napredak se može pratiti otvaranjem mape procesa u softveru Sequence analysis Viewer i promatranjem procesa.
8.4.10.4. Tumačenje i rješavanje problema
• Pripremiti MiSeq i metapodatke Važno je osigurati da je prije sekvenciranja opran MiSeq. Unakrsna kontaminacija između procesa može biti problem. Studije su pokazale da dolazi do prenošenja uzoraka iz jednog procesa u drugi. Obično prenošenjee neće djelovati na analizu amplikona 16S iz aktivnog mulja, zbog čega je između pranja prihvatljiv zadan Post Run Wash. Ipak, za osjetljive uzorke provedite jedno ili dva Maintenance Washes između procesa i/ili posebno pranje linije mulja. Ta pranja su detaljnija i preostala kontaminacija se razrjeđuje. U protivnom slijedite upute o pranju u korisničkom priručniku MiSeq System user guide.
Slika 8.15 Primjer SampleSheet. Datoteka SampleSheet.csv je otvorena u Notepad++. Nazivi odjeljaka su označeni [ ].
Ponovno pokretanje MiSeq pomaže ponovno podesiti računalo instrumenta, zbog čega je manja vjerojatnost da će se blokirati tijekom sekvenciranja.
Pripazite kad otapate reagense MiSeq. Kvaliteta reagensa djeluje na kvalitetu sekvenciranja, naročito na 3’-krajevima očitanja. Iskustvo pokazuje da sekvenciranje i dalje može uspjeti s reagensima koji su čuvani 24 h na sobnoj temperaturi, ali za to nema nikakvog jamstva.

316 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
Datoteka ‘SampleSheet.csv’ kaže instrumentu MiSeq kako bi se trebalo odvijati sekvenciranje, kako demultipleksirati uzorke i neke osnovne metapodatke povezane s uzorcima. Obrazac s metapodacima se ne može pripremiti pomoću Illumina Experiment Manager kad se koriste adapters i šifre koji nisu kupljeni od Illumine. Datoteka ‘SampleSheet.csv’ mora biti pripremljena u programu za uređivanje teksta kao što je Notepad++. Pronađite predložak online ili napravite vlastiti iz Illumina Experiment Manager. Više informacija o SampleSheet.csv pronađite kod Illumina Inc. (2013).
Objašnjenje ključnih informacija u datoteci ‘SampleSheet.csv’:
Chemistry, Amplicon Postavka amplikona omogućuje korištenje dva indeksa (index1 i index2) [Reads] 301 301 Daje naredbu MiSeq-u da provede sekvenciranje uparenih krajeva, pri čemu je svako očitanje dugačko 300 bp. Sample_ID, Sample_Name, index, index2, Description Kolona Sample_ID: Identificikacijska oznaka (ID) vaših uzoraka. Kolona Sample_Name: Naziv uzoraka. Tako će se zvati izlazni podaci. Kolona Index: Šifra prvog očitanja vaših uzoraka. Utipkajte slijed šifre. Prisutnost i dužina šifre daje sekvenatoru do znanja da želite sekvencirati šifru. Kolona Index2: Slično onome gore. Kolona Description: Bilješke o uzorcima.
• Pripremiti zbirke za sekvenciranje U ovom koraku se zbirke za sekvenciranje (kontrolna zbirka PhiX i fond zbirki) denaturiraju visokim pH (NaOH) kako bi se dobili amplikoni zbirke u jednolančanom obliku. Amplikoni zbirke moraju biti jednolančani kako bi ih MiSeq mogao uhvatiti.
pH je vrlo važan. I previsok i prenizak pH će spriječiti hvatanje amplikona zbirke. Po mogućnosti koristite 2 M NaOH čistoće molecular grade.
Nakon denaturacije, zbirke su razrijeđene. Koncentracija je iznimno važna budući da ona izravno određuje količinu proizvedenih podataka. 20 pM proizvodi otprilike 18-25 milijuna očitanja. Ako je koncentracija niža ili viša, rezultat će biti proporcionalno
niži ili viši. Izvan opsega od 2-25 pM postoji opasnost da će se proces sekvenciranja sasvim blokirati.
Kontrolna zbirka PhiX se koristi za procjenu stope pogreške i kao pomoć u kalibriranju instrumenta za vrijeme sekvenciranja. To je naročito važno za fondove zbirki male složenosti. Mala složenost znači da analizirani sljedovi imaju sličan sastav slijeda. Kako gen 16S rRNA ima očuvane regije, to je slučaj sa zbirkama amplikona 16S.
• Postaviti uzorak i početnice na kazetu s reagensima Dodavanje početnica za sekvenciranje u MiSeq kazetu s reagensima je potrebno kad se sekvenciraju zbirke pripremljene s adapterima koji nisu kupljeni od Illumine. Početnice pokreću očitavanje Read1, Read2 i Index1. Proces neće uspjeti ako one nisu prisutne.
• Sekvenciranje Nakon što sekvenciranje započne, prvi statistički podaci će se pojaviti nakon otprilike 4 h. Rezultat po uzorku se može dobiti nakon 32 h, a proces će završiti za 56 h.
Gustoća klastera otkriva procijenjeni rezultat procesa. Prosječni proces pripremljen prema gornjim protokolima će proizvesti između 700 i 1.000 k mm-2, čime se dobiva 17-25 milijuna PE očitanja.
Klaster PF otkriva koliko podataka ispunjava osnovne zahtjeve interne kvalitete. Kod sekvenciranja zbirki amplikona V1-3 16S rRNA, vrijednost od > 90 % je standard. To bi se moglo malo promijeniti tijekom sekvenciranja.
% ≥ Q30 otkriva broj baza u čitavom procesu za koje se očekuje da će imati ocjenu kvalitete iznad Q30. To se jako mijenja tijekom sekvenciranja. Za read1 prosjek je obično > 70 %, a za Read2 prosjek je obično > 60 %.
Stopa pogreške objašnjava stvarnu izmjerenu stopu pogreške u sekvenciranju kontrolne zbirke PhiX. Tijekom sekvenciranja MiSeq prepoznaje amplikone zbirke iz PhiX i uspoređuje ih s referentnim PhiX genomom kako bi se otkrile i izmjerile pogreške u sekvenciranju. Jednostavnim rječnikom rečeno, ocjena kvalitete je teoretska kvaliteta gdje je stopa pogreške empirijska kvaliteta.
Usklađeni statistički podaci objašnjavaju koliki dio čitavog procesa predstavlja kontrolna zbirka PhiX. Ako se slijedio gornji protokol, trebala bi biti blizu 20 %.

MOLEKULARNE METODE 317
8.4.11 Dizajn Illumina 16S adaptera za sekvenciranje amplikona
U ovom se dijelu opisuje dizajn adaptera/početnice i funkcije različitih dijelova.
Za pripremu zbirki amplikona 16S rRNA koriste se tzv. adapteri. Oni dolaze u parovima koji se sastoje od prednjeg i stražnjeg adaptera. Svaki adapter se sastoji od dijela s adapterom i dijela s početnicom. Tijekom PCR-a zbirke, dijelovi prednjeg i stražnjeg adaptera s
početnicom se koriste za specifično amplificiranje varijabilne regije od 1 do 3 (V1-3) gena 16S rRNA (slika 8.7). Konačni amplikoni zbirke sadrže sljedove V1-3 16S rRNA i dijelove s adapterom. Početnice su preuzete iz Projekta ljudskog mikrobioma (HMP, 2010) i zovu se 27F i 534R. Dijelovi s adapterom su preuzeti od Caporasa i sur. (2011; 2012) i od Illumina Inc. (2014a).
Tijekom sekvenciranja, početnice za sekvenciranje se pričvršćuju uz adaptere i pokreću sekvenciranje (slika 8.16).
Slika 8.16 Konceptualni prikaz oligonukleotida korištenih u sekvenciranju amplikona 16S. Adapteri se uvode za vrijeme PCR-a, a ostatak oligonukleotida se koristi za sekvenciranje. Obojani dijelovi adaptera/početnica predstavljaju različite funkcionalne sljedove.
Ukupno se očitavaju četiri slijeda: Read1 i Read2, koji zahvaćaju dio gena V1-3 16S rRNA i čine sljedove očitanja uparenih krajeva, te index i index2, koji čine šifrirani dio koji se koristi za identificiranje iz kojeg uzorka potječe svaki amplikon zbirke. Šifrirani dio se obrađuje izravno na MiSeq i nije dio izlaznih podataka.
Adapteri i početnice su sintetizirani DNA oligonukleotidi koji se mogu naručiti od bilo koje velike kompanije koja proizvodi reagense. Sljedovi pojedinih
DNA oligonukleotida mogu se pronaći na slici 8.17. Različiti dijelovi oligonukleotida imaju nazive i namjensku ulogu u pripremi i/ili sekvenciranju zbirke. Dio adaptera s indeksom (NNNNNNNN) je različit za svaki uzorak koji će se sekvencirati. Na primjer, ako će se sekvencirati 96 uzoraka, tada će obično biti 8 prednjih adaptera i 12 stražnjih adaptera, od kojih svaki ima jedinstveni indeks. Prema tome, može se dobiti ukupno 96 jedinstvenih kombinacija prednjih i stražnjih adaptera.
16S rRNA genV1-3 regija
PCR
Genom DNA
Sekvenciranje
Read1 Index
Index2 Read2
Prednji adapter:Stražnji adapter:Read1 početnica:Index početnica:Index2 početnica:Read2 početnica:

318 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
Slika 8.17 Adapteri i početnice su sintetizirani DNA oligonukleotidi. Prikazani su sljedovi pojedinih DNA oligonukleotida. Različiti dijelovi oligonukleotida imaju nazive i namjensku ulogu u pripremi i/ili sekvenciranju zbirke. Dio adaptera s indeksom (NNNNNNNN) je različit za svaki uzorak koji će se sekvencirati.
8.5 OSTALE METODE Fluorescencijska in situ hibridizacija (FISH) je neovisna metoda za vizualiziranje mikroorganizama pomoću fluorescentno označenih oligonukleotidnih proba koje ciljaju na 16S rRNA. FISH je sama po sebi vrlo snažna metoda za analizu zajednice mikroorganizama, ali je isto tako izvrsna prateća tehnika za potvrdu rezultata iz sekvenciranja amplikona. FISH je detaljno opisan u 7. poglavlju.
Postoje napredne tehnike utemeljene na sekvenciranju, kao što su metagenomika, metatranskriptomika i
metaproteomika, koje omogućuju o kulturi neovisnu analizu funkcija u zajednicama mikroorganizama. Istraživačka zajednica ih počinje primjenjivati na sustave aktivnog mulja. Međutim, te tehnike su složene i neće biti dovoljno razvijene kako bi bile pogodne za raširenu primjenu u predvidivoj budućnosti. Zbog toga će opis tih tehnika biti ograničen na prikaz u osnovnim crtama.
Metagenomika, ili okolišna genomika (Wooley i sur., 2010), je izučavanje čitave DNA zajednice dobivene izravno iz uzoraka iz okoliša. Tijek rada je prikazan na slici 8.18.
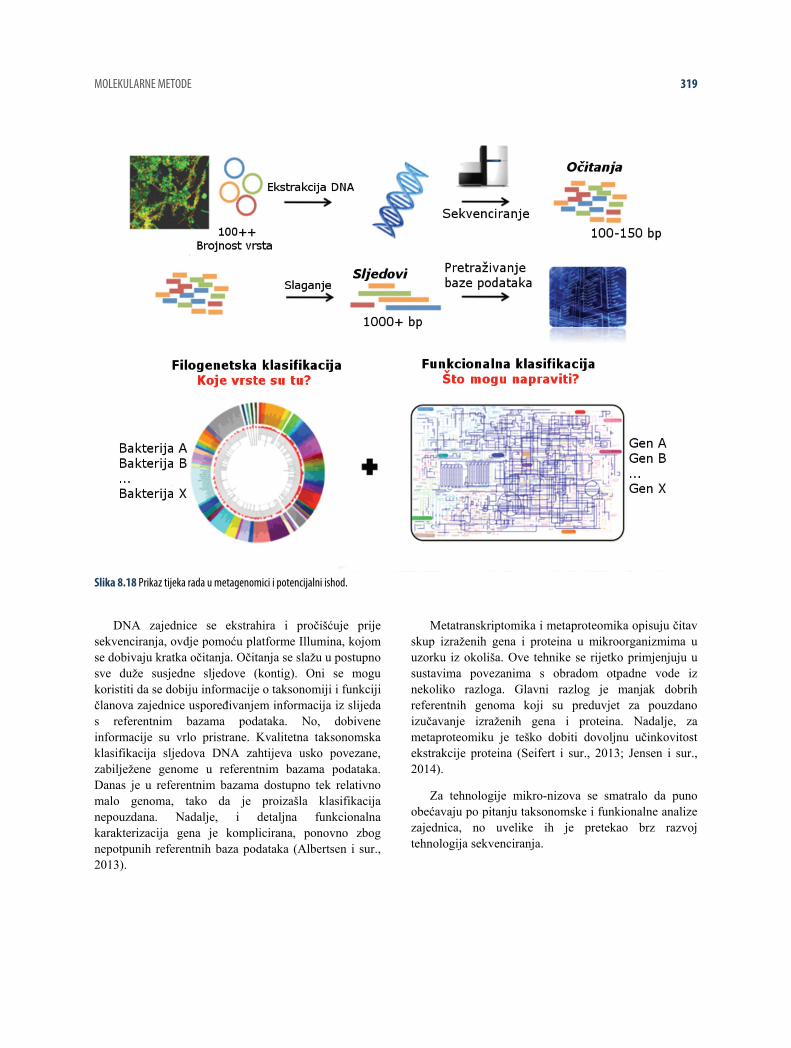
MOLEKULARNE METODE 319
Slika 8.18 Prikaz tijeka rada u metagenomici i potencijalni ishod.
DNA zajednice se ekstrahira i pročišćuje prije sekvenciranja, ovdje pomoću platforme Illumina, kojom se dobivaju kratka očitanja. Očitanja se slažu u postupno sve duže susjedne sljedove (kontig). Oni se mogu koristiti da se dobiju informacije o taksonomiji i funkciji članova zajednice uspoređivanjem informacija iz slijeda s referentnim bazama podataka. No, dobivene informacije su vrlo pristrane. Kvalitetna taksonomska klasifikacija sljedova DNA zahtijeva usko povezane, zabilježene genome u referentnim bazama podataka. Danas je u referentnim bazama dostupno tek relativno malo genoma, tako da je proizašla klasifikacija nepouzdana. Nadalje, i detaljna funkcionalna karakterizacija gena je komplicirana, ponovno zbog nepotpunih referentnih baza podataka (Albertsen i sur., 2013).
Metatranskriptomika i metaproteomika opisuju čitav skup izraženih gena i proteina u mikroorganizmima u uzorku iz okoliša. Ove tehnike se rijetko primjenjuju u sustavima povezanima s obradom otpadne vode iz nekoliko razloga. Glavni razlog je manjak dobrih referentnih genoma koji su preduvjet za pouzdano izučavanje izraženih gena i proteina. Nadalje, za metaproteomiku je teško dobiti dovoljnu učinkovitost ekstrakcije proteina (Seifert i sur., 2013; Jensen i sur., 2014).
Za tehnologije mikro-nizova se smatralo da puno obećavaju po pitanju taksonomske i funkionalne analize zajednica, no uvelike ih je pretekao brz razvoj tehnologija sekvenciranja.

MOLEKULARNE METODE 320
Reference Agilent Technologies (2012). Agilent 2200 TapeStation User
Manual edition 8. Manual Part number G2966-90001. Agilent Technologies (2013). Agilent D1000 ScreenTape System
Quick Guide edition 10. Manual Part number G2964-90032 Rev. B.
Agilent Technologies (2015). Agilent Genomic DNA ScreenTape System Quick Guide edition 6. Manual Part number G2964-90040 Rev. D.
Albertsen, M, Karst, S.M., Ziegler, A.S., Kirkegaard R.H., Nielsen P.H. (2015). Back to Basics – The influence of DNA extraction and primer choice on phylogenetic analysis of activated sludge communities. PLoS ONE 10:e0132783.
Albertsen M., Saunders, A.M., Nielsen K.L. and Nielsen P.H. (2013): Metagenomes obtained by "deep sequencing" - what do they tell about the EBPR communities? Wat. Sci. Tech., 68: 1959-1968.
Angly, F.E., Dennis, P.G., Skarshewski, A., Vanwonterghem, I., Hugenholtz, P., and Tyson, G.W. (2014). CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome, 2, 11.
Ashelford, K.E., Chuzhanova, N.A., Fry, J.C., Jones, A. J., and Weightman, A.J. (2005). At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 71: 7724-7736.
Basu, C. (2015). PCR primer design (New York: Humana Pr). Beers, E.H. Van, Joosse, S.A., Ligtenberg, M.J., Fles, R.,
Hogervorst, F.B.L., Verhoef, S., and Nederlof, P.M. (2006). A multiplex PCR predictor for aCGH success of FFPE samples. British J. Cancer 94: 333-337.
Beller, H.R., Kane, S.R., Legler, T.C., and Alvarez, P.J.J. (2002). A real-time polymerase chain reaction method for monitoring anaerobic, hydrocarbon-degrading bacteria based on a catabolic gene. Environ. Sci. & Technol. 36: 3977–3984.
Bessetti, J. (2007). An introduction to PCR inhibitors. J. Microbiol. Meth. 28: 159-167.
Bollet C, Gevaudan M.J., de Lamballerie X., Zandotti C., de Micco P. 1991. A simple method for the isolation of chromosomal DNA from Gram positive or acid-fast bacteria. Nucleic Acids Research 19:1955.
Brown, C.T., Hug, L.A, Thomas, B.C., Sharon, I., Castelle, C.J., Singh, A., … Banfield, J. F. (2015). Unusual biology across a group comprising more than 15 % of domain Bacteria. Nature, 523: 208-211.
Bru, D., Sarr, A., and Philippot, L. (2007). Relative abundances of proteobacterial membrane-bound and periplasmic nitrate reductases in selected environments. Appl. Environ. Microbiol. 73: 5971-5974.
Brzoska, A.J., and Hassan, K.A. (2014). Quantitative PCR for detection of mRNA and gDNA in environmental isolates. In Environmental Microbiology, I.T. Paulsen, and A.J. Holmes, eds. (Totowa, NJ: Humana Press), pp. 25-42.
Bürgmann H, Pesaro M, Widmer F, Zeyer J. (2001). A strategy for optimizing quality and quantity of DNA extracted from soil. J Microbiol. Methods 45: 7-20.
Bustin, S.A., Benes, V., Garson, J.A., Hellemans, J., Huggett, J., Kubista, M., Mueller, R., Nolan, T., Pfaffl, M.W., Shipley, G.L., i sur. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55: 611-622.
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., … Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 7: 335-6.
Caporaso, J.G., Lauber, C.L., Walters, W.A, Berg-Lyons, D., Huntley, J., Fierer, N., … Knight, R. (2012). Ultra-high-
throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME Journal 6: 1621-1624.
Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-Lyons, D., Lozupone, C.A., Turnbaugh, P.J., … Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108. Suppl 1, 4516-4522.
Cole, J.R., Wang, Q., Fish, J. a., Chai, B., McGarrell, D.M., Sun, Y., … Tiedje, J.M. (2014). Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Research, 42: 633–642.
Cock, P.J.A., Fields, C.J., Goto, N., Heuer, M.L., and Rice, P.M. (2010). The Sanger FASTQ file format for sequences with quality scores, and the Solexa / Illumina FASTQ variants, Nucleic Acids Research 38: 1767-1771.
Daims, H., Lebedeva, E.V., Pjevac, P., Han, P., Herbold, C., Albertsen, M., … and Wagner, M. (2015). Complete nitrification by Nitrospira bacteria. Nature Doi: 10.1038/nature16461.
DeAngelis M.M., Wang D.G., Hawkins T.L. (1995). Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Research 23: 4742-4743.
Dominiak, D.M., Nielsen, J.L., and Nielsen, P.H. (2011). Extracellular DNA is abundant and important for microcolony strength in mixed microbial biofilms. Environ. Microbiol. 13: 710-721.
Dueholm, M.S., Albertsen, M., D’Imperio, S., Tale, V.P., Lewis, D., Nielsen, P.H., and Nielsen, J.L. (2014). Complete genome sequences of Pseudomonas monteilii SB3078 and SB3101, two benzene-, toluene-, and ethylbenzene-degrading bacteria used for bioaugmentation. Genome Announc. 2: 524-14.
Dueholm, M.S., Marques, I.G., Karst, S.M., D’Imperio, S., Tale, V.P., Lewis, D., Nielsen, P.H., and Nielsen, J.L. (2015). Survival and activity of individual bioaugmentation strains. Biores. Technol. 186: 192-199.
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10: 996-8.
El Fantroussi, S., and Agathos, S.N. (2005). Is bioaugmentation a feasible strategy for pollutant removal and site remediation? Curr Opin Microbiol 8: 268-275.
Farrelly, V., Rainey, F.A, and Stackebrandt, E. (1995). Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl. Environ. Microbiol. 67: 2798-2801.
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nature Reviews. Microbiology, 10, 538–50.
Faust, K., Lahti, L., Gonze, D., de Vos, W. M., and Raes, J. (2015). Metagenomics meets time series analysis: unraveling microbial community dynamics. Current Opin Microbiol., 25: 56-66.
Filippidou, S., Junier, T., Wunderlin, T., Lo, C-C., Li, P-E., Chain, P.S., Junier, P. (2015). Under-detection of endospore-forming Firmicutes in metagenomic data. Comput. and Structural Biotechnol. J. 13: 299-306.
Fisher, S., Barry, A., Abreu, J. (2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biology, 12, R1 doi:10.1186/gb-2011-12-1-r1
Flynn, J. M., Brown, E. a., Chain, F. J. J., MacIsaac, H. J., and Cristescu, M. E. (2015). Toward accurate molecular identification of species in complex environmental samples: testing the performance of sequence filtering and clustering methods. Ecol. Evol., 5: 2252-2266.
Frølund, B., Palmgren, R., Keiding, K., and Nielsen, P.H. (1996). Extraction of extracellular polymers from activated sludge using a cation exchange resin. Wat. Res. 30: 1749-1758.

MOLEKULARNE METODE 321
Ge, S., Wang, S., Yang, X., Qiu, S., Li, B., and Peng, Y. (2015). Detection of nitrifiers and evaluation of partial nitrification for wastewater treatment: A review. Chemosphere, 140: 85-98.
Gibson, D.J., Ely, J.S., Collins, S.L. (1999). The core-satellite species hypothesis provides a theoretical basis for Grime’s classification of dominant, subordinate, and transient species. J. Ecol. 87: 1064-1067.
Gilbert, E.M. ,S. Agrawal, S.M. Karst, H. Horn, P.H. Nielsen, S. Lackner (2014). Low temperature partial nitritation/anammox in a moving bed biofilm reactor treating low strength wastewater. Environ. Sci. Technol., 48: 8784-8792.
Green, M.R., Sambrook, J. (2012). Molecular Cloning: A Laboratory Manual (Fourth Edition). Cold Spring Harbor Laboratory Press. Cold Spring Harbor, New York.
Grime, J. (1998). Benefits of plant diversity to ecosystems: immediate, filter and founder effects. J. Ecol. 86: 902-910.
Guillén-Navarro, K., Herrera-López, D., López-Chávez, M.Y., Cancino-Gómez, M., Reyes-Reyes, A.L. (2015). Assessment of methods to recover DNA from bacteria, fungi and archaea in complex environmental samples. Folia Microbiol. 60: 551-558.
Guo, F., Zhang, T. (2013). Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl. Microbiol. Biotechnol. 97: 4607-4616.
Holland, P.M., Abramson, R.D., Watson, R., and Gelfand, D.H. (1991). Detection of specific polymerase chain reaction product by utilizing the 5’----3’ exonuclease activity of Thermus aquaticus DNA polymerase. PNAS 88: 7276-7280.
Horz, H.P., Vianna, M.E., Gomes, B.P.F.A., and Conrads, G. (2005). Evaluation of universal probes and primer sets for assessing total bacterial load in clinical samples: General implications and practical use in endodontic antimicrobial therapy. J. Clin. Microbiol. 43: 5332-5337.
Hou, Y., Zhang, H., Miranda, L., and Lin, S. (2010). Serious overestimation in quantitative pcr by circular (supercoiled) plasmid standard: Microalgal pcna as the model gene. PLoS ONE 5, e9545.
Humbert, S., Zopfi, J., and Tarnawski, S.-E. (2012). Abundance of anammox bacteria in different wetland soils. Environ. Microbiol. Rep. 4: 484-490.
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12: 1889-1898.
Illumina Inc. (2013) MiSeq Sample Sheet, Quick Reference Guide, Part # 15028392 Rev. J
Illumina Inc. (2014a). Illumina Customer Sequence Letter. Oligonucleotide sequences © 2007-2013 Illumina, Inc. All rights reserved. Derivative works created by Illumina customers are authorized for use with Illumina instruments and products only. All other uses are strictly prohibited.
Illumina Inc. (2014b) MiSeq System User Guide, Part # 15027617 Rev. O
Illumina, Inc. (2015). An Introduction to Next-Generation Sequencing Technology, www.illumina.com
Janda, J. M., and Abbott, S. L. (2007). Minireview: 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 45: 2761-2764.
Jensen S.H., A. Stensballe, P.H. Nielsen, F.-A. Herbst (2014). Metaproteomics: Evaluation of protein extraction from activated sludge. Proteomics 14(21-22), 2535-2539.
Hadfield, J. (2012). How do SPRI beads work? http://core-genomics.blogspot.dk/2012/04/how-do-spri-beads-work.html
Human Microbiome Project (HMP) (2010). Jumpstart Consortium Human Microbiome Project Data Generation Working Group (2010). 16S 454 Sequencing Protocol HMP Consortium. Version 4.2.2.
Juretschko, S., Purkhold, U., Pommerening-ro, A., Schmid, M. C., Koops, H., and Wagner, M. (2000). Phylogeny of all
recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: Implications for molecular diversity surveys. Appl. Environ. Microbiol. 66, 5368–5382.
Kim, J., Lim, J., and Lee, C. (2013). Quantitative real-time PCR approaches for microbial community studies in wastewater treatment systems: Applications and considerations. Biotechnology Advances 31: 1358-1373.
Kitajima, M., Iker, B.C., Pepper, I.L., and Gerba, C.P. (2014). Relative abundance and treatment reduction of viruses during wastewater treatment processes identification of potential viral indicators. Sci. Total Environ. 488-489: 290-296.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., and Glöckner, F. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research, 41, e1.
Kubista, M., Andrade, J.M., Bengtsson, M., Forootan, A., Jonák, J., Lind, K., Sindelka, R., Sjöback, R., Sjögreen, B., Strömbom, L., i sur. (2006). The real-time polymerase chain reaction. Mol. Aspects Medicine 27: 95-125.
Laureni, M., Weissbrodt, D.G., Szivák, I., Robin, O., Nielsen, J.L., Morgenroth, E., Joss, A. (2015). Activity and growth of anammox biomass on aerobically pre-treated municipal wastewater. Wat. Res., 80: 325-336.
Legendre, P., and Gallagher, E. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia, 129: 271-280.
Li, X., Wu, Y., Zhang, L., Cao, Y., Li, Y., Li, J., Wu, G. (2014). Comparison of three common DNA concentration measurement methods. Analytical Biochemistry, 451: 18-24.
Liu, C.M., Kachur, S., Dwan, M.G., Abraham, A.G., Aziz, M., Hsueh, P.-R., Huang, Y.-T., Busch, J.D., Lamit, L.J., Gehring, C.A., i sur. (2012). FungiQuant: A broad-coverage fungal quantitative real-time PCR assay. BMC Microbiol. 12: 255.
Love, M.I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15: 550.
Lozupone, C.A., and Knight, R. (2008). Species divergence and the measurement of microbial diversity. FEMS Microbiol. Rev., 32: 557-578.
Ludwig, W., and Schleifer, K.-H. (2000). How quantitative is quantitative PCR with respect to cell counts? Syst. Appl. Microbiol. 23: 556–562.
Madigan, M.T., Martinko, J.M. (2006). Brock Biology of Microorganisms, 11th ed. Pearson Education International. ISBN 0131968939.
Magurran, A. E. (2004). Measuring biological diversity. Oxford, UK: Blackwell Publishing.
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2014). Swarm: robust and fast clustering method for amplicon-based studies. PeerJ, 1-12.
Marsh, T.L., 1999. Terminal restriction fragment length polymorphism (T-RFLP): an emerging method for characterizing diversity among homologous populations of amplification products. Curr. Opin. Microbiol. 2: 323-327.
Marzorati, M., Wittebolle, T., Boon, N., Daffonchio, D., Verstraete, W. (2008). How to get more out of molecular fingerprints: Practical tools for microbial ecology. Environ. Microbiol. 10: 1571-1581.
Matsuda, K., Tsuji, H., Asahara, T., Kado, Y., and Nomoto, K. (2007). Sensitive quantitative detection of commensal bacteria by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 73: 32-39.
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., … Hugenholtz, P. (2012). An improved Greengenes taxonomy with explicit ranks for

322 EKSPERIMENTALNE METODE U OBRADI OTPADNIH VODA
ecological and evolutionary analyses of bacteria and archaea. ISME Journal, 6: 610-618.
McIlroy, S.J., Saunders, A.M., Albertsen, M., Nierychlo, M., McIlroy, B., Hansen A.A., Karst, S.M., Nielsen, J.L., Nielsen, P.H. (2015). MiDAS: the field guide to the microbes of activated sludge. Database bav062. doi: 10.1093/database/bav062.
McMaster, G.K., Carmichael, G.G. (1977). Analysis of single- and double-stranded nucleic acids on polyacrylamide and agarose gels by using glyoxal and acridine orange. PNAS 74: 4835-4838.
McMurdie, P.J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One, 8, e61217.
Mendell, J.E., Clements, K.D., Choat, J.H., and Angert, E.R. (2008). Extreme polyploidy in a large bacterium. PNAS 105: 6730-6734.
Miller, D.N., Bryant, J.E., Madsen, E.L., Ghiorse, W.C. (1999). Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 65: 4715-4724.
Muyzer, G. (1999).DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 2: 317-322.
Nadkarni, M.A., Martin, F.E., Jacques, N.A., and Hunter, N. (2002). Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148: 257-266.
Nanodrop Technologies, Inc. (2007). ND-1000 Spectrophotometer V3.3 User’s manual, rev.3.
Nolan, T., Hands, R.E., and Bustin, S.A. (2006). Quantification of mRNA using real-time RT-PCR. Nature Protocols 1: 1559-1582.
Nygren, J., Svanvik, N., and Kubista, M. (1998). The interactions between the fluorescent dye thiazole orange and DNA. Biopolymers 46: 39-51.
Okano, Y., Hristova, K.R., Leutenegger, C.M., Jackson, L.E., Denison, R.F., Gebreyesus, B., Lebauer, D., and Scow, K.M. (2004). Application of real-time PCR to study effects of ammonium on population size of ammonia-oxidizing bacteria in soil. Appl. Environ. Microbiol. 70: 1008-1016.
Oksanen, J., Blanchet, G. F., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., … Wagner, H. (2015). Vegan: Community Ecology Package. http://r-forge.r-project.org/projects/vegan/.
Pace, N. R., Sapp, J., and Goldenfeld, N. (2012). Phylogeny and beyond: Scientific, historical, and conceptual significance of the first tree of life. PNAS, 109: 1011-1018
Padmanaban, A., Inche, A., Gassmann, M., Salowsky, R.. 2013. High-Throughput DNA Sample QC Using the Agilent 2200 Tapestation System. J. Biomol. Techn. JBT 24:S41.
Panaro, N.J., Yuen, P.K., Sakazume, T., Fortina, P., Kricka, L.J., Wilding, P. 2000. Evaluation of DNA fragment sizing and quantification by the Agilent 2100 bioanalyzer. Clinical Chemistry 46: 1851-1853.
Pecoraro, V., Zerulla, K., Lange, C., and Soppa, J. (2011). Quantification of ploidy in proteobacteria revealed the existence of monoploid, (mero-)oligoploid and polyploid species. PLoS ONE, 6. e16392. doi:10.1371/journal.pone.0016392.
Polz, M.F., and Cavanaugh, C.M. (1998). Bias in template-to-product ratios in multitemplate PCR, Appl Environ Microbiol. 64(10): 3724-3730.
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28(14): 1823-1829.
Qiu, X., Wu, L., Huang, H., Donel, P. E. M. C., Palumbo, A. V, Tiedje, J. M., and Zhou, J. (2001). Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S
rRNA gene-based cloning, Appl Environ Microbiol. 67(2): 880-887.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., … Glöckner, F. O. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41. doi:10.1093/nar/gks1219
Quince, C., Lanzen, A., Davenport, R. J., and Turnbaugh, P. J. (2011). Removing noise from pyrosequenced amplicons. BMC Bioinformatics, 12: 38.
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes, Bioinformatics 28(14): 1823-1829.
Ramette, A. (2007). Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol., 62: 142-60.
Rasmussen, R. (2001). Quantification on the LightCycler. In Rapid Cycle Real-Time PCR, P.D. med S. Meuer, P.D.C. Wittwer, and D.K.-I. Nakagawara, eds. (Springer Berlin Heidelberg), pp. 21-34.
Robinson, M.D., McCarthy, D.J., and Smyth, G.K. (2010). EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26: 139-40.
Rotthauwe, J.H., Witzel, K.P., Liesack, W., (1997). The ammonia monooxygenase structural gene amoA as a functional marker: Molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl Environ Microbiol. 63: 4704-4712.
Saiki, R.K., Scharf, S., Faloona, F., Mullis, K.B., Horn, G.T., Erlich, H.A., and Arnheim, N. (1985). Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230: 1350-1354.
Salonen, A., Nikkilä, J., Jalanka-Tuovinen, J., Immonen, O., Rajilić-Stojanović, M., Kekkonen, R. a., Palva, A., de Vos, W.M. (2010). Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiological Methods 81: 127-34.
Saunders, A.M., Albertsen, M., Vollertsen, J., and Nielsen, P.H. (2015). The activated sludge ecosystem contains a core community of abundant organisms. ISME J.,doi:10.1038/ismej.2015.117
Schloss, P.D. and Handelsman, J. (2005). Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biol. 6: 229.
Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., … Weber, C.F. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol., 75: 7537-41.
Schriewer, A., Wehlmann, A., and Wuertz, S. (2011). Improving qPCR efficiency in environmental samples by selective removal of humic acids with DAX-8. J. Microbiol. Methods 85: 16-21.
Seifert, J.F.-A. Herbst, P.H. Nielsen, F.J. Planes, M. Ferrer, M. von Bergen (2013) Bioinformatic progress and applications in metaproteogenomics for bridging the gap between genomic sequences and metabolic functions in microbial communities. Proteomics 13: 2786-2804. doi: 10.1002/pmic.201200566.
Singer, V.L., Jones, L.J., Yue, S.T., Haugland, R.P. (1997). Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Analytical Biochemistry 249: 228-238.
Smith, S., Morin, P.A. (2005). Optimal storage conditions for highly dilute DNA samples: a role for trehalose as a preserving agent. J. Forensic Sci., 50: 1101-1108.
Souazé, F., Ntodou-Thomé, A., Tran, C.Y., Rostène, W., and Forgez, P. (1996). Quantitative RT-PCR: limits and accuracy. BioTechniques 21: 280-285.
Stults, J.R., Snoeyenbos-West, O., Methe, B., Lovley, D.R., and Chandler, D.P. (2001). Application of the 5 fluorogenic

MOLEKULARNE METODE 323
exonuclease assay (taqman) for quantitative ribosomal DNA and rRNA analysis in sediments. Appl. Environ. Microbiol. 67: 2781-2789.
Tebbe, C.C., and Vahjen, W. (1993). Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and a yeast. Appl. Environ. Microbiol 59(8): 2657-2665.
Thermo Scientific (2015). Assessment of Nucleic Acid Purity, T042‐TECHNICAL BULLETIN, T042 Rev 1/11.
Thermo Scientific (2015a), Qubit dsDNA HS Assay Kits – User Guide, MAN0002326, P32851, Revision: B.0
Thermo Scientific (2015b), Qubit dsDNA BR Assay Kits – User Guide, MAN0002325, P32850, Revision: A.0
Thomas, T., Gilbert, J., Meyer, F. (2012). Metagenomics - a guide from sampling to data analysis. Microb. Inform. Exp. 2:3. 10.1186/2042-5783-2-3.
Thompson, I.P., Van Der Gast, C.J., Ciric, L., and Singer, A.C. (2005). Bioaugmentation for bioremediation: the challenge of strain selection. Environ. Microbiol .7: 909-915.
Tsai, Y., Olson, B. (1991). Rapid method for direct extraction of DNA from soil and sediments. Appl. Environ. Microbiol. 57: 1070-1074.
Tullis, R.H., Rubin, H. (1980). Calcium protects DNase I from proteinase K: a new method for the removal of contaminating RNase from DNase I. Analyt. Biochem.107: 260-264.
Větrovský, T., and Baldrian, P. (2013). The variability of the 16S rRNAS gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 8. e57923. doi:10.1371/journal.pone.0057923.
Volkmann, H., Schwartz, T., Bischoff, P., Kirchen, S., and Obst, U. (2004). Detection of clinically relevant antibiotic-resistance
genes in municipal wastewater using real-time PCR (TaqMan). J. Microbiol. Methods 56: 277-286.
Wang, Y., and Qian, P.-Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16s ribosomal DNA amplicons in metagenomic studies. PLoS ONE 4, e7401. doi:10.1371/journal.pone.0007401.
Wilfinger, W.W., Mackey, K., Chomczynski, P. (1997). Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. BioTechniques 22: 474-476.
Wilson, I.G. (1997). Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol., 63(10): 3741-3751.
Woese, C.R., and Fox, G.E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. PNAS, 74: 5088-90.
Wooley, J.C., Godzik, A., Friedberg, I. (2010). A primer on metagenomics. PLoS Comput. Biol. 6(2): e1000667. doi:10.1371/journal.
Zhang, T., and Fang, H.H.P. (2006). Applications of real-time polymerase chain reaction for quantification of microorganisms in environmental samples. Appl.Microbiol. Biotechnol. 70: 281-289.
Zhou, J., Bruns, M.A., Tiedje, J.M. (1996). DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62: 316-322.
Zipper, H., Brunner, H., Bernhagen, J., and Vitzthum, F. (2004). Investigations on DNA intercalation and surface binding by SYBR Green I, its structure determination and methodological implications. Nucl. Acids Res. 32: e103–e103.
Zuur, A.F., Ieno, E.N. and Smith, G.M. (2007) Analysing Ecological Data. Springer, New York.





























