Embed Size (px)
Citation preview

A SUBSTITUENT EFFECT IN THE POLYMERIZATION OF lY6-ANHYDRO-2,3,4-TRI-0-
(p-BROMOBENZY L)+D-GLUCOPY RANOSE (2-
A number of 2,3,4-tri-O-substituted 1,6-anhydro sugars have been polymer- ized and copolymerized to give stereoregular homo- and heteropolysaccharides (1). In copolymerization studies, 1,6-anhydro sugars with p-methylbenzyl sub- stituents such as 1.6-anhydro-2.3.4-tri-O-p-methylbenzyl-~-D-galactopyranose (2- 5 ) (G1AnXy3) or 1,6-anhydro-2,3 ,4-tri-O-p-rnethylbenzyl-~-D-galactopyranose (6) have been used as comonomers with perbenzylated anhydro sugars, and it has been shown that the p-methyl group does not significantly affect the prop- agation reaction (5) . Its presence does, however, tend to slow down initiation and also to cause the formation of lower molecular weight polymers by en- hancing transfer or termination reactions (5). A copolymerization of 1,6- anhydro-2,4-di-O-benzyl-3-O-2-butenylQ-D-glucopyranose with lY6-anhydro- 2,3,4-tri-O-benzyl-~-D-glucopyranose (GlAnBn,) demonstrated that the former monomer was more reactive in propagation processes (7). In the present in- vestigation the influence of p-bromo substitution on the polymerization of GlAnBn3 has been investigated.
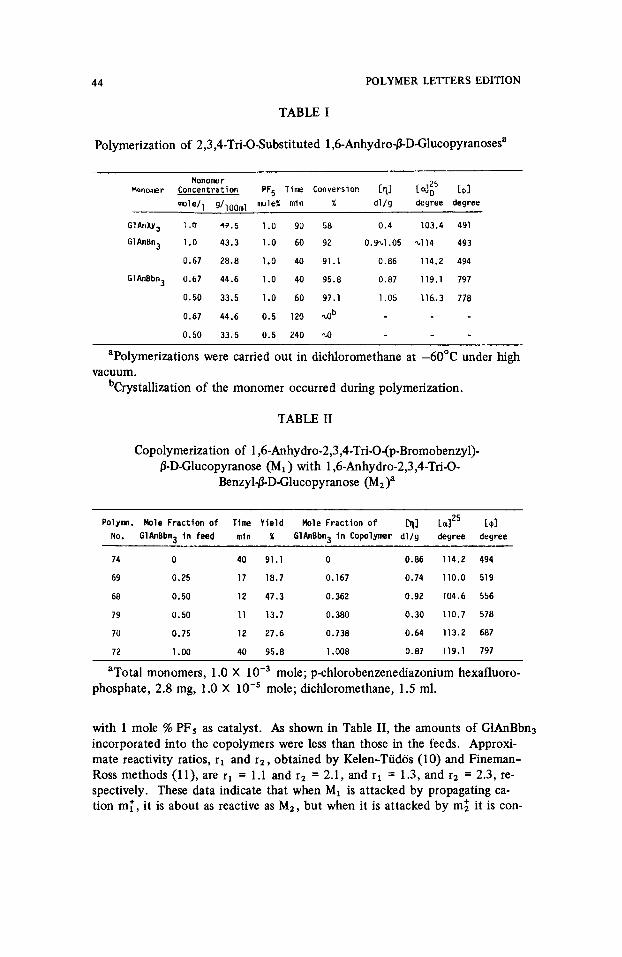
An exact comparison between the polymerization of GlAnBn3 and 1,6- anhydro-2,3,4-tri-O-p-bromobenzyl~-D-glucopyranose (G1AnBbn3) is somewhat difficult since the latter tends to crystallize from the solvent dichloromethane under the usual conditions of polymerization (1 molelliter, concentration at -60°C) (Table I). At a decreased monomer concentration (0.67 mole/liter), however, G1AnBbn3 gave a slightly higher conversion than G1AnBn3, and a polymer of equal viscosity. At even more dilute solution (0.50 mole/liter), the conversion was about the same, and the viscosity about as high as that obtained from G1AnBn3 under optimal conditions. Clearly the results from the two monomers differ very little. There is some suggestion that initiation may be somewhat more efficient with G1AnBbn3 and termination perhaps slightly reduced. Both effects could be caused by the reduced basicity of oxy- gens at C-2, C-3, and C-4 that is the result of p-bromo substitution. Dibenzyl ether has been shown (8) to be of higher basicity than 6,8-dioxabicyclo[3.2.1] - octane (9), the parent skeleton of the 1,6-anhydroglycopyranoses. Therefore, the ether oxygens compete in complexation with the acetal oxygen which must be activated to induce polymerization. Reduced basicity of ether oxygens, due to p-bromo substitution, would result in less competition and favor activa- tion and initiation. Similarly reduced basicity of ether oxygen should reduce chain transfer of cations at these sites and favor somewhat higher molecular weights.
Both effects may be real but appear to be too minor to account for the rather marked difference of the two monomers G1AnBbn3 (MI ) and G1AnBn3 (M,) in copolymerization. The copolymerizations were carried out at 0.67 mole/liter, combined monomer concentration, in dichloromethane at -60°C
Journal of Polymer Science: Polymer Letters Edition, Vol. 19,43-47 (1981) 0 1981 John Wiley & Sons, Inc. CCC 0360-6384/81/0019-0043$01.00
44 POLYMER LETTERS EDITION
TABLE I
Polymerization of 2,3,4-Tri-O-Substituted 1 ,6-Anhydro-P-D-Glucopyranosesa
Mononer kwwr Concentration PF5 Tine Conversion [TI [@I
m o l d l g/loo,l,l nuleX rnin X d l / g degree degree
GIAnXVj 1.0 w.5 1.0 90 58 0.4 103.4 491
GlAnBn3 1.0 43.3 1.0 60 92 0.921.05 1.114 493
0.67 28.8 1.0 40 91.1 0.86 114.2 494
GlAnBbn3 0.67 44.6 1.0 40 95.8 0.87 119.1 797
0.50 33.5 1.0 60 97.1 1.05 116.3 778
0.67 44.6 0.5 120 ab 0.50 33.5 0.5 240 10
aPolymerizations were carried out in dichloromethane at -60°C under high
bCrystallization of the monomer occurred during polymerization. vacuum.
TABLE I1
Copolymerization of 1,6-Anhydr0-2,3,4-Tri-O-(p-Bromobenzyl)- 0-D-Glucopyranose (M, ) with 1,6-Anhydro-2,3 ,4-Tri-O-
Benzylg-D-Glucopyranose (Mz)a
Polym. Mole Frac t ion o f Time Y i e l d Mole Frac t ion of [j] L a p Lo1 No. G1AnBbn3 i n feed min L G1AnBbn3 i n Copolymer d l / g degree degree
74 0 40 91.1 0 0.86 114.2 494
69 0.25 17 18.7 0.167 0.74 110.0 519
68 0.50 12 47.3 0.362 0.92 104.6 556
79 0.50 11 13.7 0.380 0.30 110.7 578
12 27.6 0.738 0.64 113.2 687 70 0.75
72 1 .oo 40 95.8 1.008 0.87 119.1 797
aTotal monomers, 1 .O X mole; p-chlorobenzenediazonium hexafluoro- phosphate, 2.8 mg, 1.0 X lo-’ mole; dichloromethane, 1.5 ml.
with 1 mole % PFS as catalyst. As shown in Table 11, the amounts of G1AnBbn3 incorporated into the copolymers were less than those in the feeds. Approxi- mate reactivity ratios, rl and r z , obtained by Kelen-Tudos (10) and Fineman- Ross methods (ll), are rl = 1.1 and rz = 2.1, and r l = 1.3, and rz = 2.3, re- spectively. These data indicate that when MI is attacked by propagating ca- tion m;, it is about as reactive as M z , but when it is attacked by mf it is con-

POLYMER LETTERS EDITION
TABLE 111
45
Physical Properties of 2,4,6-Tri-O-Substituted 1,6-Anhydro-~-D-Glucopyranoses
Moiionie 1- nip Mw L U l o LQ1 r e f .
degree degree degree
25
GlAnXy3 2. 66 474.9 -29.9 142 5
G1AnBn3 2. 90 432.5 -30.8,-31 .5 134.5t1.5 5,12,Pa
GlAnBbn3 2.125 669.2 -21.1 ,-22.5 145.8*5 13. Pa
aP, present paper.
siderably less reactive than Mz . An alternative statement would be that m; is less selective between monomers than is mi , and m t prefers to react with M z .
and since the homopropagation rate of GlAnBbn3 (kl ) kzz 2, & Since - = kZl klZ
is probably not less than that of GlAnBn, (k22) (from a comparison of the in- dividual polymerizations) the cross-propagation reaction kz probably is dis- favored. GlAnBbn, may require more energy to react than does the less mas- sive GlAnBn, and the energy would be available on collision with the more massive cation. Thus there would be little selectivity observed in kl /kl but much more in the ratio kzz/kZ1.
If the cross-propagation reaction is disfavored, it should be possible to co- polymerize GlAnBbn, with a less reactive monomer like 1,6-anhydr0-3,4,6-tri- 0-benzyl-0-D-galactopyranose (GalAnBn, )2 and obtain longer sequences of the second monomer in the copolymer than is possible on copolymerization of GlAnBn, and GalAnBn,.
GlAnBn, and its polymer except in the aromatic region. Aromatic C-1, C-3, and C-4 of the brominated monomer resonate at 137.06, 131.71, and 121.87 ppm, respectively, as single sharp peaks, whereas aromatic C-2 resonances are found at 129.53 and 129.43 ppm. Aromatic C-1 and C-3 resonances of the polymer appear as sharp singlets at 137.76 and 131.77, respectively, but the aromatic C-2 and C-4 differ markedly from the monomer (C-2 at 129.75, 129.39, and 128.84 ppm; C-4 at 121.91, 121.75, and 121.51 ppm). Only one anomeric peak is observable at 97.68 ppm, revealing the high stereoselectivity of the homopolymerization. Copolymers of GlAnBbn, and GlAnBn3 show two aromatic C-1 resonances at 138.94 and 137.80 ppm, with the former cor- responding to the GlAnBn, residue and the latter to the GlAnBbn, unit.
As shown in Table 111, molecular rotations of the three 1,6-anhydrogluco- pyranoses are not very much different from one another. Table 11, however, indicates that although molecular rotations (4) of the polymers of G1AnXy3 and GlAnBn, were identical (491' vs. 493", ref. 5), the GlAnBbn, polymers
13C-NMR spectra of G1AnBbn3 and its polymer are very similar to those of

46 POLYMER LETTERS EDITION
exhibited greatly enhanced molecular rotations (797", 778"). The abnormally high molecular rotations of the latter polymers might be ascribed to the higher refractive index induced by the p-bromo substitution, which in the case of the monomer is cancelled out because the benzyl group on C3 is antiparallel to those on C2 and C4 ('C4 conformation). While molecular rotations of the GlAnXy,-GlAnBn, copolymers stayed constant (481O-499") irrespective of the composition of the copolymers (9, the GlAnBbn3-GlAnBn3 copolymer sys- tem gave gradually increased molecular rotations with increase in the mole fraction of GlAnBbn3 in the copolymer (Table 11).
dro sugars may have at least two possible applications. The difference in reac- tivity compared to benzylated anhydro sugars may permit the formation of copolymers of wider range of compositions and sequence distributions, and the higher melting points may in some cases make the monomers more tract- able experimentally, especially when the benzylated monomers are syrupy or low melting.
The use of p-bromobenzyl substituents instead of benzyl substituents on anhy-
Experimental
NMR spectra were measured with a Varian XL-100 spectrometer in deuterio- chloroform with tetramethylsilane (TMS) as the internal standard. Chemical shifts are expressed in parts per million (ppm) downfield from the TMS reso- nance. Optical rotations were determined in chloroform at 25°C in a Perkin- Elmer model 141 polarimeter with a jacketed 1-dm cell. Viscosities were meas- ured in a Cannon-Ubbelohde semimicro viscometer at 25OC. Mole fractions of GlAnBbn, in copolymers and in recovered monomers were determined from bromine analyses performed by Micro-Analysis, Inc. (Wilmington, DE 19808).
Monomers 1.6-Anhydr0-2,3,4-tri-O-p-bromobenzyl-~-D-glucopyranose (GIAnBbn, ) was
synthesized according to the method previously described (9) except that p- bromobenzylation was performed with powdered potassium hydroxide in tol- uene instead of sodium hydride in N,N-dimethylformamide. The synthesis of 1,6-anhydro-2,3,4-tri-O-benzyl-/3-D-glucopyranose (GlAnBn, ) was described previously.
GlAnBbn,: mp 125.5-126S°C [lit. 124.0-1250°C (13)] ; [a] 'D" -22S0(cl,CHC13) [lit. -21 .1"(cl,CHCl3) (13)]
GlAnBn, : mp 89.5-90.0"C [lit. 89.5-90.5"C (12)] ; [a] -31.5" (cl,CHC13) [lit. -30.8" (5,12)]
Polymerization Total quantity of monomer in each polymerization was 1.0 X lo-, mole.
Polymerization and copolymerization were carried out under high vacuum at -60°C in anhydrous dichloromethane with PF, generated in situ by pyrolysis

POLYMER LETTERS EDITION 47
of p-chlorobenzenediazonium hexafluorophosphate. Polymerization was termi- nated at -60°C by adding cold methanol. Polymer was precipitated by pour- ing chloroform solution into petroleum ether. Because of the very high crys- tallizability of GlAnBbn3 in petroleum ether, reprecipitation was repeated several times to remove the unreacted monomer completely. The polymer was isolated by freeze-drying from benzene and proved to be free of the monomer by gel permeation chromatography (styragel 200 A, toluene) and 'H-NMR. The combined supernatant solution was concentrated on a flash evaporator to dryness, further dried under vacuum, and subjected to bromine analysis.
This research was completed with financial support from the National Insti- tute of General Medical Science, National Institutes of Health, U.S. Public Health Service, research grant G.M. 06168.
References
(1) C. Schuerch, Acc. Chem. Res., 6, 184-191 (1973). (2) J. W.-P. Lin and C. Schuerch, Macromolecules, 6, 320-324 (1973). (3) K. Kobayashi and C. Schuerch, J. Polym. Sci. Polym. Chem. Ed., 15,
(4) W. H. Lindenberger and C. Schuerch, J. Polym. Sci. Polym. Chem. Ed.,
(5) H. Ito and C. Schuerch, J. Polym. Sci. Polym. Chem. Ed., 16, 2217-
(6) H. Ito, V. Marousek, and C. Schuerch, J. Polym. Sci. Polym. Chem.
(7) H. Ito and C. Schuerch, J. Am. Chem. SOC., 101, 5797-5806 (1979). (8) W. Gordy and S. C. Stanford, J. Chem. Phys., 9, 204-214 (1941). (9) M. Okada, H. Sumitomo, and Y. Hibino, Polym. J., 6, 256-263 (1974).
(10) J. Kelen and F. Tudos, J. Macromol. Sci. Chem., A9, 1-27 (1975). (11) M. Fineman and S. D. Ross, J. Polym. Sci., 5, 259-262 (1950). (12) E. R. Ruckel and C. Schuerch, J. Org. Chem., 31, 2233-2239 (1966). (13) R. Eby and H. Ito, Carbohydr. Res., to appear.
913-926 (1977).
11, 1225-1235 (1973).
2224 (1978).
Ed., 17, 1299-1307 (1979).
Hiroshi Ito Conrad Schuerch
Department of Chemistry State University of New York College of Environmental Science and Forestry Syracuse, New York 13210
Received September 11, 1980 Accepted September 30, 1980








![Supplementary Materials - Royal Society of Chemistry · Supplementary Materials Imidazo[1,5-a]pyridin-3-ylidenes as π-Accepting Carbene Ligands: Substituent Effects on Properties](https://img.pdfslide.tips/doc/110x75/5ec0ffb8f8271e7b336e6711/supplementary-materials-royal-society-of-supplementary-materials-imidazo15-apyridin-3-ylidenes.jpg)










![Synthesis of an organic-soluble -conjugated [3]rotaxane ... · s1 Supporting ... rotaxane via rotation of glucopyranose units in permethylated -cyclodextrin Jun Terao*, Yohei Konoshima,](https://img.pdfslide.tips/doc/110x75/5b2a88127f8b9a853a8b4f83/synthesis-of-an-organic-soluble-conjugated-3rotaxane-s1-supporting-.jpg)

![Nov 23 2018.ppt [Kompatibilitätsmodus] - Limes-Institut-Bonn · Figure 11-5 The anomeric monosaccharides -D-glucopyranose and -D-glucopyranose, drawn as both Haworth projections](https://img.pdfslide.tips/doc/110x75/5f7f813523e6cd4b030ea89a/nov-23-2018ppt-kompatibilittsmodus-limes-institut-bonn-figure-11-5-the-anomeric.jpg)






