Embed Size (px)
Citation preview

Alignment-free sequence comparison
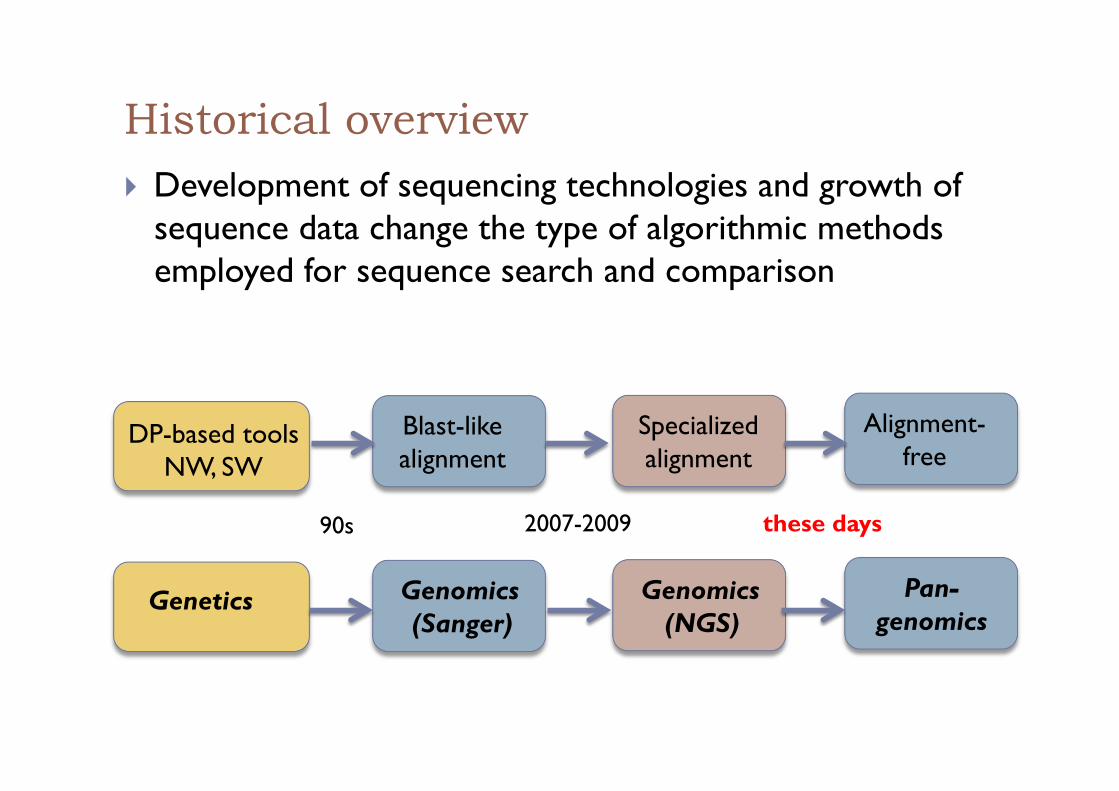
Historical overview ! Development of sequencing technologies and growth of
sequence data change the type of algorithmic methods employed for sequence search and comparison
Genetics
DP-based tools NW, SW
Blast-like alignment
Genomics (Sanger)
Specialized alignment
Genomics (NGS)
Alignment-free
Pan-genomics
2007-2009 90s these days

Alignment-free sequence comparison ! we cannot afford and often don't need alignments
! alignment-free (or composition-based) methods compare sequences by comparing their composition in words/patterns, usually k-mers [Vigna & Almeida 03]
! a sequences is viewed as a bag of words (k-mers), contiguous or spaced, with or without multiplicities
ACGACGAG → {ACG,CGA,GAC,GAG} ACGACGAG → {AC-A,CG-C,GA-G,CG-G}
! Tasks: comparison, search, estimating distance, classification, learning, …

Locality-sensitive hashing (LSH)
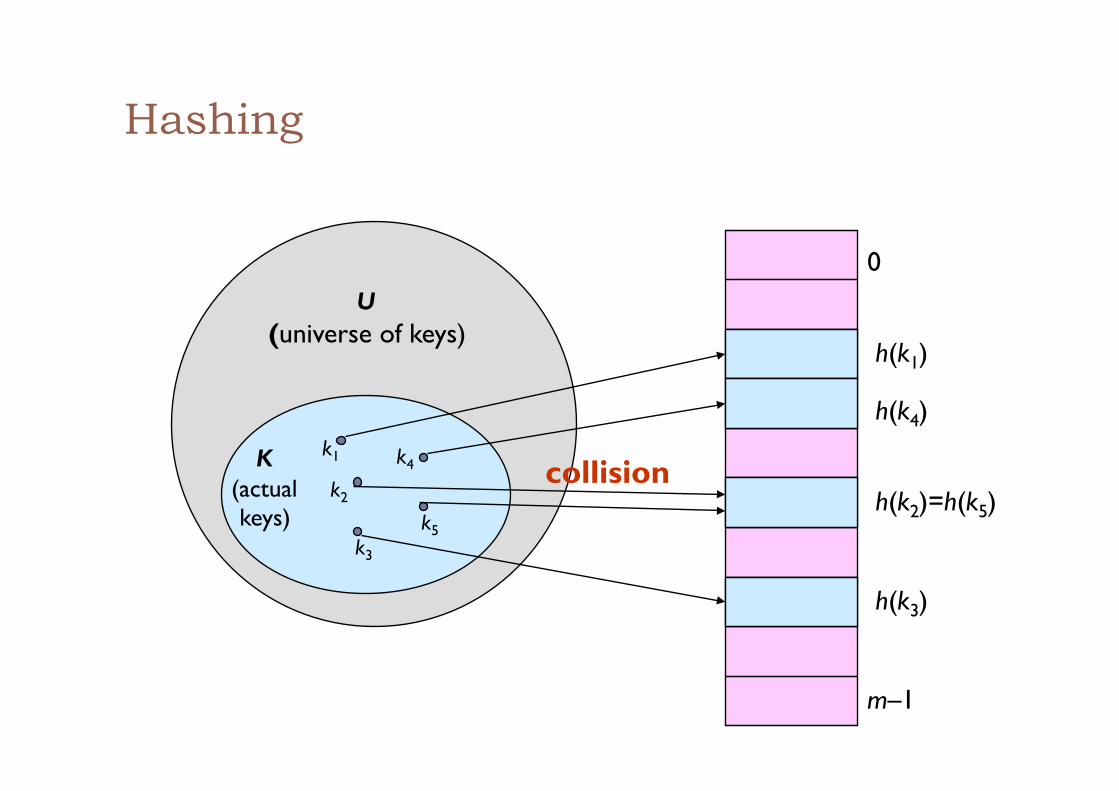
Hashing
0
m–1
h(k1)
h(k4)
h(k2)=h(k5)
h(k3)
U (universe of keys)
K (actual keys)
k1
k2
k3
k5
k4 collision

Locality-sensitive hashing (LSH) ! given objects {x1,…,xn} (points in space of high dimension
d) ! assume a similarity relation Sim(x,y)∈[0,1] ! define a family H of hash functions such that Ph∈H[h(x)=h(y)]=Sim(x,y) ! that is: hash collision captures object similarity

Locality-sensitive hashing (LSH) ! given objects {x1,…,xn} (points in space of high dimension
d) ! assume a similarity relation Sim(x,y)∈[0,1] ! define a family H of hash functions such that Ph∈H[h(x)=h(y)]=Sim(x,y) ! that is: hash collision captures object similarity
! making the collision probability smaller: P[<h1(x),…,hk(x)>=<h1(y),…,hk(y)>]=(Sim(x,y))k

Example: Hamming distance ! objects: bitvectors x∈{0,1}d
! Hamming distance: Ham(x,y) is the nb of unequal corresponding bits, e.g. Ham(00011,10001)=2
! Hamming similarity Sim(x,y)=1-Ham(x,y)/d ! define h(x)=xi randomly sampled bit, i∈[1,d] ! then Ph∈H[h(x)=h(y)]=Sim(x,y) (prove)
! P[<h1(x),…,hk(x)>=<h1(y),…,hk(y)>]=(Sim(x,y))k ! based on this, a practical hash table can be built

Main application: similarity search ! Hash objects of the dataset using chaining ! Given a query x, look through all objects y in bucket h(x),
compute the true similarity dist(x,y) and report those with dist(x,y)<d (filtering)
! MUCH faster than pairwise comparison ! false positives and false negatives
! (d1,d2,p1,p2)-sensitive family of hash functions: ! dist(x,y)<d1 ⇒ P[h(x)=h(y)]>p1 ! dist(x,y)>d2 ⇒ P[h(x)=h(y)]<p2

Gap amplification ! let h(x)=<h1(x),…,hk(x)> and hi's belong to a (d1,d2,p1,p2)-
sensitive family ! by using L distinct hash tables (in OR fashion), we can
construct a (d1,d2, 1-(1-(p1)k)L, 1-(1-(p2)k)L)-sensitive family ! Example: using 4-tuple hash functions and 4 hash tables, a
(0.2,0.6,0.8,0.4)-sensitive family turns to (0.2,0.6,0.8785,0.0985)-sensitive
! the construction can be cascaded to achieve arbitrary large gap
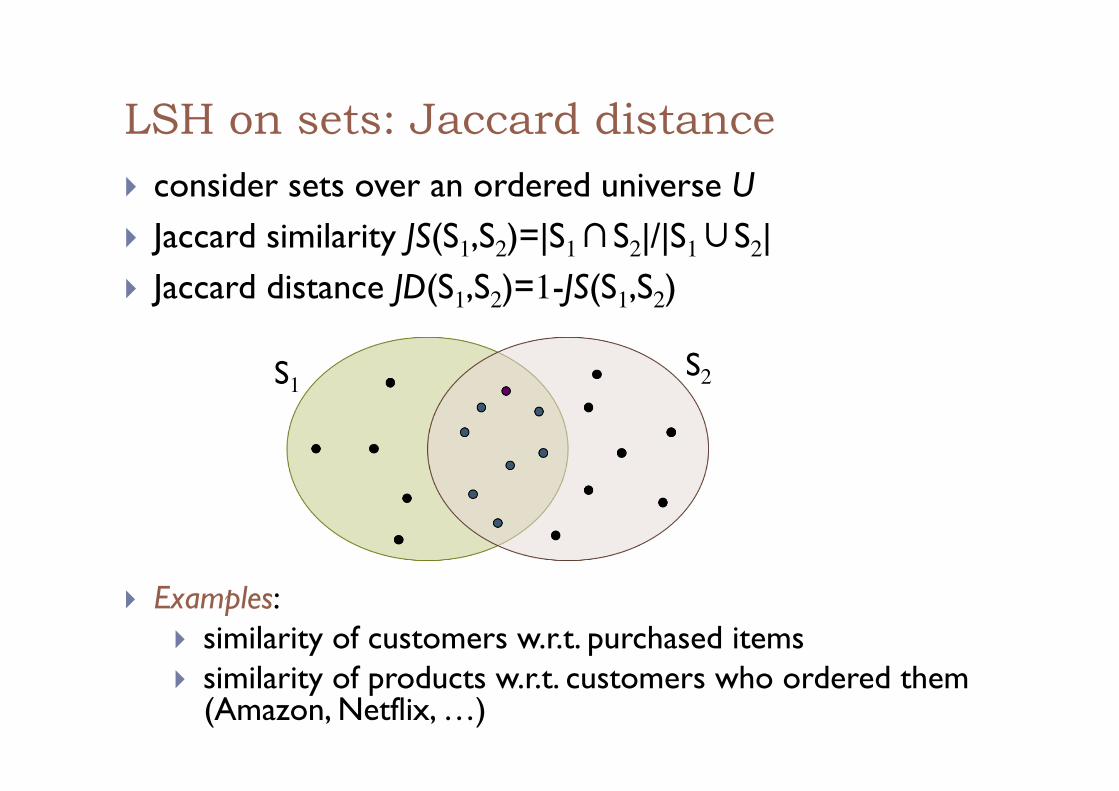
LSH on sets: Jaccard distance ! consider sets over an ordered universe U ! Jaccard similarity JS(S1,S2)=|S1∩S2|/|S1∪S2| ! Jaccard distance JD(S1,S2)=1-JS(S1,S2)
S1 S2
! Examples: ! similarity of customers w.r.t. purchased items ! similarity of products w.r.t. customers who ordered them
(Amazon, Netflix, …)

MinHash for Jaccard distance ! consider a random permutation π: U → {1,...,|U|} ! for a set S, define h(S)=minx∈S{π(x)} ! then for two sets S1,S2, we have P[h(S1)=h(S2)]=JS(S1,S2)
! Proof: …

MinHash signatures ! consider random permutations π1,..., πk: U → {1,...,|U|} ! permutations are difficult to handle ⇒ replace them by
(random) hash functions π1,..., πk: U → {1,...,N} for a sufficiently large N
! for a set S, define its signature to be sig(S)=<minx∈S{π1(x)},…, minx∈S{πk(x)}> ! NB: signature is easy to compute ! then we have E[#{i|sig(S1)i=sig(S2)i}]/k = JS(S1,S2)

MinHash: toy example S1={0,3}, S2={2}, S3={1,3,4}, S4={0,2,3} π1(x)=(x+1) mod 5π2(x)=(3x+1) mod 5sig(S1)=<1,0>, sig(S2)=<3,2>, sig(S3)=<0,0>, sig(S4)=<1,0>
Then JS(S1,S4) estimated to 1 (true answer 2/3) JS(S1,S3) estimated to 1/2 (true answer 1/4) JS(S3,S4) estimated to 1/2 (true answer 1/5) JS(S1,S2) estimated to 0 (true answer 0)

MinHash for sequences (documents) ! General scenario:
! represent a sequence (text) as a set of k-mers (Q-grams, k-shingles), i.e. tuples of consecutive letters (words) of fixed size
! measure document similarity by Jaccard similarity and apply the MinHash framework
! possibly do gap amplification
! first proposed by Broder (1997) with application to webpage similarity search

Text shingles (k-grams, snippets) The sky is blue and the sun is bright. The sun in the blue sky is bright.
k=1: {and, blue, bright, is, sky, sun, the} {blue, bright, in, is, sky, sun, the}
k=2: {and the, blue and, is blue, is bright, sky is, sun is, the sky, the
sun} {blue sky, in the, is bright, sky is, sun in, the blue, the sun}

MinHash with m min values: example
ACAGTAAC TAAACTAAG
3 10 6 15 13 11 13 11 3 4 6
AC CA AG GT TA AA TA AA AC CT AG
€
π
MinHash={3,6,10} MinHash={3,4,6}
Jaccard index: 4/(2+4+1)=4/7
MinHash similarity: 2/3€
m = 3

Example (DNA sequences) accgcactta cgcattaccg
k=3: {acc, act, cac, ccg, cgc, ctt, gca, tta} {acc, att, cat, ccg, cgc, gca, tac, tta}
! Compare with sequence alignment

Features of MinHash ! Simple! easy to compute! ! Can be combined with spaced seeds ! Easy to maintain for growing datasets ! Size (m) does not depend on the size of the dataset ! Suitable for comparing datasets of (roughly) the same size ! Low level of false positives ("accidental similarity")
! Example: fingerprints of 400 32-bit hash values (k=16) are sufficient to discriminate microbial genomes (a few Mb)
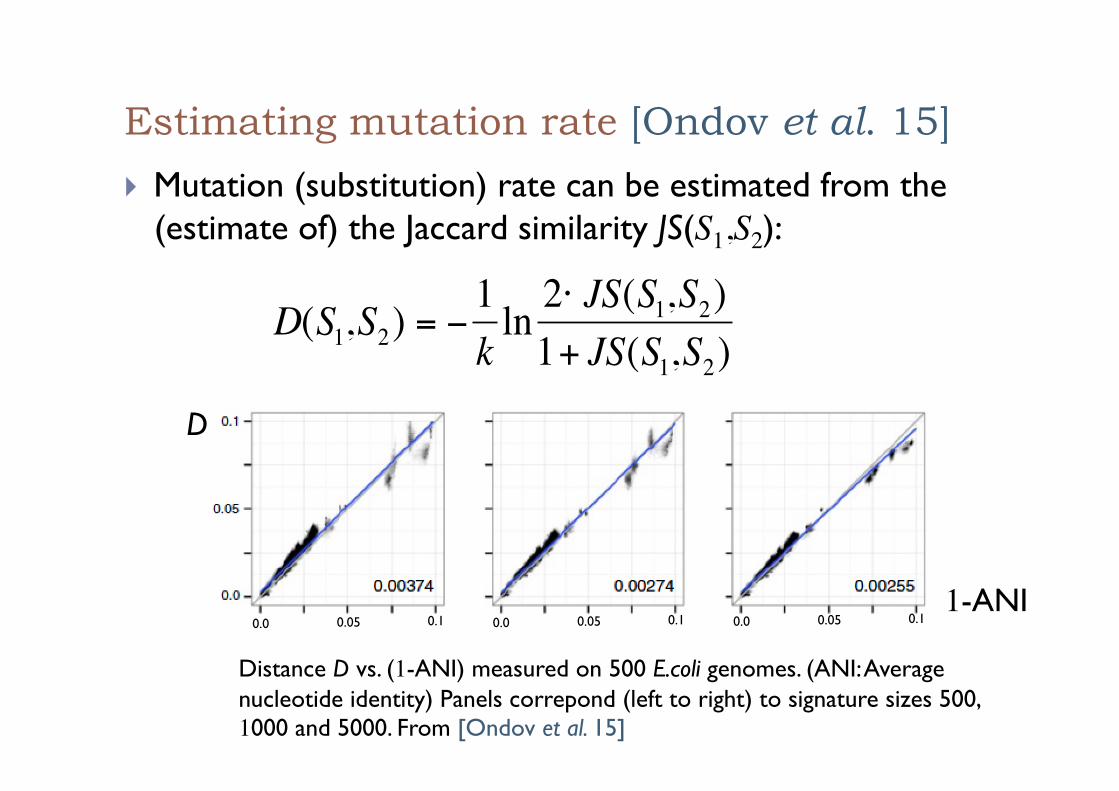
Estimating mutation rate [Ondov et al. 15]
! Mutation (substitution) rate can be estimated from the (estimate of) the Jaccard similarity JS(S1,S2):
€
D(S1,S2) = −1kln 2⋅ JS(S1,S2)1+ JS(S1,S2)
0.0 0.05 0.1 0.0 0.05 0.1 0.0 0.05 0.1
Distance D vs. (1-ANI) measured on 500 E.coli genomes. (ANI: Average nucleotide identity) Panels correpond (left to right) to signature sizes 500, 1000 and 5000. From [Ondov et al. 15]
D
1-ANI

Examples of application [Ondov et al. 15]
! clustering of 54,000 NCBI genomes (entire RefSeq, 618Gbp) in 33 CPU h using 1,000-valued hashes. Resulting hashes: 93MB
! (real-time) matching a sequencing data against a "sketched" database
! scalable clustering of hundreds of metagenomic samples by composition: analysis of 10TB of sequence data using 10,000-valued hashes (k = 21)

Bonus: Bloom filters

Bloom filters: generalities ! Bloom (1970) ! generalizes the bitmap representation of sets ! supports INSERT and LOOKUP ! LOOKUP only checks for the presence, no satellite data ! produces false positives (with low probability) ! cannot iterate over the elements of the set ! DELETE is not supported (in the basic variant) ! very space efficient ! Example: forbidden passwords

Bloom filter: how it works ! U : universe of possible
elements ! K : subset of elements,
|K| = n ! m : size of allocated bit
array ! define d hash functions h1,…,hd: U → {0,…,m-1}
! INSERT(k): set hi(k) ← 1 for all i ! LOOKUP(k): check hi(k)=1 for all i
! false positives but no false negatives

Bloom filters: analysis ! P[specific bit of filter is 0] =
! P[false positive] =
! Optimal number d of hash functions: ! Therefore, for the optimal number of hash functions,
P[false positive] =
! E.g. with 10 bits per element, P[false positive] is less than 1% ! To insure the FP rate ε: m≈1.44⋅n⋅log2(1/ε)
€
(1−1/m)dn ≈ e−dn /m ≡ p
€
(1− p)d = (1− e−dn /m )d
€
d = ln(2)⋅ (m /n)
€
2− ln(2)⋅(m / n ) ≈ 0.6185m / n

Dependence on the nb of hash functs
m/n = 8
Opt d = 8 ln 2 = 5.45... n elements m bits d hash functions

Bloom filter: properties ! For the optimal number of hash function, about a half of
the bits is 1 [show]! The Bloom filter for the union is the OR of the Bloom
filters ! Is similar true for the intersection? [explain] ! If a Bloom filter is sparse, it is easy to halve its size ! Various generalizations exist, e.g. counting Bloom filters
(support counting and deletion)

Bloom filters: applications ! Bloom filters are very easy to implement ! Used e.g. for
! spell-checkers (in early UNIX-systems) ! unsuitable passwords ! "approximate" unsuitable passwords (Manber&Wu 1994) ! filtering in databases ! …
! Sometimes it is possible to store the set of false positives in a separate data structure








![Havo A 5.1 Stijgen en dalen. Intervallen a-8 ≤ x < 3 [ -8, 3 › b4 < x ≤ 4½ ‹ 4, 4½ ] c5,1 ≤ x ≤ 7,3 [ 5,1 ; 7,3 ] d3 < x ≤ π ‹ 3, π ] -83 l l ○● 44½4½](https://img.pdfslide.tips/doc/110x75/5551a0ed4979591f3c8b55c3/havo-a-51-stijgen-en-dalen-intervallen-a-8-x-3-8-3-b4-x-4-4-4-c51-x-73-51-73-d3-x-3-83-l-l-444.jpg)




![`GRIV:W$TE=ER$S;'Z5BT@$TEJ X$KT @«J« qttrfile.siam2web.com/krittasilph/files[document]/201777_44678.pdf · §”π” ‡ªÑ“À¡“¬°“√ªØ ‘√Ÿª°“√» ÷°…“„π∑»«√√…∑](https://img.pdfslide.tips/doc/110x75/5fd7ea74d9336545b20a1209/grivwteersz5bttej-xkt-j-document20177744678pdf-aa-aaoeaoeaoea.jpg)





![`GRIV:W$TE=ER$S;'Z5BT@$TEJ X$KT @«J« qttrfile.siam2web.com/krittasilph/files[document]/201777_44636.pdf · §”π” ‡ªÑ“À¡“¬°“√ªØ ‘√Ÿª°“√» ÷°…“„π∑»«√√…∑](https://img.pdfslide.tips/doc/110x75/5fd7ede5f301a37fdc7fe143/grivwteersz5bttej-xkt-j-document20177744636pdf-aa-aaoeaoeaoea.jpg)








![+ã HêeT dü+e‘·‡s¡ l X¯è+π>] l »>∑∑](https://img.pdfslide.tips/doc/110x75/5e7a46ec2f3be374b40f2853/-het-deaas-l-x-l-aa.jpg)
