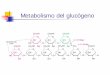
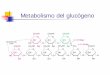
Patologías más destacadas de la alteración del metabolismo del glucógeno.
ALTERACIN DEL METABOLISMO DEL GLUCGENO
Las glucogenosis son trastornos del metabolismo del glucgeno en
los tejidos debido a defectos enzimticos en la va glucoltica o
glucogenoltica. Han sido clasificadas segn la deficiencia enzimtica
y tejido afectado, ya sea en hgado, msculo o en ambos,
describindose 12 formas de presentacin. La incidencia general se ha
estimado en 1: 20 000 a 1:43 000 recin nacidos, siendo la ms comn
la glucogenosis tipo IX y el 80% est representado por las tipo I,
II y IX. La hipoglicemia y la hepatomegalia son los signos y
sntomas presente en todas, pero con una gran heterogeneidad de
presentacin.
Glucogenosis tipo I
La primera glucogenosis fue descrita en 1952 por Cori y Cori, y
es conocida como enfermedad de Von Gierke y se produce por el
dficit de la enzima glucosa-6 fosfatasa, y afectada la
glucogenlisis y neoglucognesis. La principal funcin de la enzima es
proporcionar glucosa en periodos de ayuno y se ubica en hgado y
rin. Est compuesta por 4 subunidades y la subunidad uno est
localizada dentro del retculo endosplasmtico, generando la
glucogenosis tipo I-a. Adicionalmente hay trasportadores para
ingresar y sacar la glucosa-6-fosfato y sus productos del retculo
endoplasmtico. El defecto de la Glucosa -6- fosfato translocasa
(T2) produce la glucogenosis tipo I-b, la deficiencia del
transportados fosfotranslocasa (T2) ocasiona glucogenosis tipo I-c,
y la deficiencia de la glucosa translocasa (T3) que exporta la
glucosa genera la glucogenosis tipo I-d.
Glucogenosis tipo I-a
En 1993 el gen G6PC fue localizado en el cromosoma 17q21 (OMIM
232200). Existe una gran heterogeneidad gentica tnica. Las
mutaciones ms prevalentes en Italia son R83C y Q347K, que
representan al 66.9% de los alelos mutados.
Clnica y diagnstico
Inicialmente los sntomas son causados por la hipoglucemia y no
responde a la administracin de glucagn. Hay temblores,
irritabilidad, hiperventilacin, cianosis, apnea, convulsiones
palidez, sudoracin edema cerebral, coma y la muerte, principalmente
en la maana o despus de alimentarse. En nios mayores tienen cara de
mueca, letargia, alteraciones del sueo, temblor, retardo del
crecimiento, abdomen protuberante por la hepatomegalia y
extremidades delgadas. Puede existir tendencia al sangramiento
nasal, debido a la alteracin plaquetaria. Durante cuadros
infecciosos los sntomas producidos por las hipoglucemias son ms
graves. En edades posteriores hay anemia y raquitismo, problemas de
crecimiento con estatura pequea. Pueden tener diarreas sin
explicacin, al igual que la tipo Ib.
Segn la magnitud de las hipoglicemias se observan crisis de
prdida de conciencia y convulsiones, que es la causa del retardo
mental en los pacientes con mal control metablico. Se han observado
numerosas complicaciones a largo plazo como adenomashepticos que
pueden degenerar en carcinoma hepatocelular, gota, hipertensin
pulmonar.Tambin se ha descrito glomeruloesclerosis segmentaria y
fibrosis intersticial que contribuyen a aumentar la insuficiencia
renal. La hipercolesterolemia, hipertrigliceridemia y disminucin de
las lipoprotenas de alta densidad no se ha asociado a enfermedades
cardiovasculares, pero pueden inducir xantomas o pancreatitis con
elevacin de amilasa, lipasa y tripsina.
El diagnstico se sospecha al encontrar hipoglicemia,
hiperlactacidemia, cuerpos cetnicos aumentados en sangre y en
orina, hiperlipidemia (principalmente hipertrigliceridemia) e
hiperuricemia. En esta enfermedad la sobrecarga de glucosa puede
ser muy til, puesto que en ayuno hay aumento del cido lctico y
despus de la carga de glucosa este disminuye en la medida que
aumenta la glicemia. En un individuo normal el cido lctico es bajo
inicialmente y aumenta paulatinamente tras la administracin de
glucosa. Se confirma el diagnstico a travs de la medicin de la
actividad enzimtica. El estudio molecular permite realizar
diagnstico y evitar la biopsia heptica.
Tratamiento
El objetivo principal es prevenir la hipoglicemia, evitar la
estimulacin de la neoglucognesis, aumento del glucagn y cido
lctico. La dieta fraccionada impide laaparicin de hipoglucemia; se
sugiere la alimentacin por sonda nasogstrica por goteo continuo o
bolos en lactantes. En nios mayores alimentacin oral fraccionada
diurna, con alimentacin nocturna con ingestin de almidn crudo.
Desde la dcada de los aos 1980 se usa almidn crudo de maz, que
ha permitido prolongar el periodo de ayuno, mantener normoglicemia
hasta 2,5 a 6 horas. No tiene efectividad el mismo si da cocido o
en forma acuosa. La cantidad recomendada es de 1,5 a 2,5 g/kg por
dosis, distribuidos en 3 a 4 veces al da, dejando un 40% para la
noche. Es importante considerarlo dentro del aporte total de energa
diaria (7). Restringir los hidratos de carbono tales como sacarosa
u otros almidones de absorcin rpida, dejando un pequeo margen para
el uso de almidones de absorcin lenta (arroz, trigo, tapioca).
Debido al bloqueo que existe en la transformacin de
glucosa-6-fosfato a glucosa, se prohbe el consumo de frutas
(fructosa, sorbitol) y disminuyendo los lcteos.
La ingesta de energa depender del estado nutricional del
individuo y se utilizan las recomendaciones entregadas por la
recommended dietary intake (RDI 2002).Distribuir un 65% a 75% para
el da y un 25 a 35% nocturno, cantidades excesivas ocasionan
obesidad e hipertrigliceridemia (10). Por la dislipidemia se
aconseja dar un aporte de lpidos del 25% a 30% del total de la
molcula calrica, si hay riesgo de pancreatitis esta restriccin debe
ser mayor.Si hay hiperuricemia se debe tratar con alopurinol (10
mg/kg/da, dividido en 3 dosis) y cuando hay acidosis (bases < 5
mmol/L o bicarbonato < 20mmol/L) usar bicarbonato (1-2
mmol/kg/da en 4 dosis) o citrato de potasio (5-10 mEq cada 8 a 12
hrs). Si con el tratamiento y un buen control de glicemia, persiste
la hipertrigliceridemia sobre10 mmol/L,se deben usar drogas (cido
nicotnico o fibratos), ya que existe el riesgo de pancreatitis o
colelitiasis.
El control de glicemia se realizan diariamente en ayunas y al
final de la noche, en lasprimeras etapas de la dieta o en
adecuaciones nutricionales. Si la glicemia se encuentrabajo 70
mg/dl, se recomienda aumentar el aporte de hidratos de carbono en
un 10% a 15% y reevaluar nuevamente el nivel de glicemia. Un mejor
control metablico y adhesin al tratamiento permiten adems de
normoglicemia, disminuir la lactacidemia, colesterol y triglicridos
y mantener un crecimiento ponderal dentro de rangos de
normalidad.
Glucogenosis tipo Ib
Fue descrita en 1968 y se produce por la deficiencia del
transportador glucosa-6-fosfatotranslocasa (T1), (Figura 3) y el
gen SLC37A4 ha sido clonado en el cromosoma 11q23.
Clnica y diagnstico
Es igual a la tipo I-a, pero se agrega la presencia en forma
recurrente de infecciones, neutropenia y disfuncin de los
neutrfilos, predisponindolos a infecciones graves y a enfermedades
inflamatorias intestinales (remedando una enfermedad de Crohn). El
recuento absoluto de neutrfilos es bajo 1.000 clulas por ml.
Presentan fiebre, diarrea y lceras periorales y anales. Hay mayor
prevalencia de tiroiditis o hipotiroidismo. Tambin se ha descrito
leucemia mieloctica aguda. Pueden requerir transplante heptico para
prevenir la aparicin de adenomas y por la hipoglicemia
refractaria.
Tratamiento
Es igual a la tipo I-a. Adicionalmente la terapia de factor de
estimulacin de granulocitos colnico podra restaurar la funcin
mieloide, mejorando el pronstico. Tambin se ha probado la
suplementacin con vitamina E, ya que se ha descrito un aumento de
la apoptosis y de radicales libres provenientes de oxgeno, lo que
podran explicar la neutropenia, observndose excelentes
resultados.
Glucogenosis tipo II
Es la forma ms severa de glucogenosis y su forma clsica, la
enfermedad de Pompe, se produce por el dficit de la enzima -1,4
glucosidasa o maltasa cida. Es una glucogenosis generalizada con
compromiso de corazn, msculo esqueltico, hgado, sistema nervioso
central, rin y leucocitos. La herencia es autosmica recesiva y el
gen GGA ha sido localizado en el cromosoma 17q25.2-q25.3 (OMIM
232300) y se han descrito ms de 300 mutaciones, establecindose una
buena correlacin entre genotipo, actividad residual de la enzima y
gravedad de los sntomas.La incidencia es de 1:40.000 en la poblacin
afroamericano y alemanes, de 1:50.000 en chinos y de 1:146.000 en
la poblacin australiana.
Clnica y diagnstico
Se distinguen 2 formas de presentacin: clsica infantil con
cardiomiopata y una leve de presentacin juvenil o de adulto con
compromiso muscular esqueltico.
Forma clsica infantil
Fue descrita por Pompe en 1933 y se presenta los primeros meses
de vida entre el primer y segundo mes de edad y se asocia a una
actividad nula de la enzima -1,4 glucosidasa. Aparecen problemas
para alimentarse, hay mal incremento ponderal, infecciones
respiratorias, hipotona y pocos movimientos. El corazn est afectado
y la ultrasonografa cardaca muestra cardiomiopata hipertrfica con
engrosamiento de las paredes ventriculares y del septum, lo que
puede producir insuficiencia cardaca. El electrocardiograma muestra
voltajes altos, alteracin en la polarizacin, e intervalos PR corto.
Hay retardo del desarrollo motor y los mayores logros son gatear,
sentarse, no alcanzan posicin vertical y mantenerse de pie.Al
examen clnico se observa problemas para estar en posicin vertical,
no hay control ceflico y los reflejos osteotendineos estn
disminuidos. Otras caractersticas son aumento del tamao de la
lengua, leve hepatomegalia, problemas auditivos por alteraciones
del odo medio, interno y sistema nervios auditivo. Los paciente
fallecen entre los 6 a 8 meses de edad, raramente sobreviven al ao
de vida si no reciben tratamiento.Los exmenes de laboratorio
muestran aumento de la creatinquinasa, que est presente en el 95%
de los casos. Adems est aumentada la aldolasa, lactato
deshidrogenasa, ALT, AST. El diagnstico se establece midiendo la
actividad de la enzima en fibroblastos o msculo, tambin por estudio
molecular.
Forma juvenil o adulto
Se presenta esencialmente como una miopata de severidad variable
que semeja una distrofia muscular de cinturas o de Duchenne, pero
que afecta ambos sexos. Hay debilidad de predominio cervical y de
cinturas pelviana y escapular, elevacin de creatinfosfoquinasa
(CPK) y electromiografa mioptica. El compromiso cardaco es
espordico. La enfermedad de Pompe en adulto ha sido descrita como
una patologa progresiva y que los sntomas aparecen en etapa
escolar. Un estudio hecho en 52 pacientes mostr deterioro motor
sustancial, diferentes grados de discapacidad que pueden requerir
uso de silla de ruedas. Hay problemas respiratorios y necesitar
oxigenoterapia nocturna. Durante este estudio fallecieron 4 casos
de edades de 44 a 68 aos, indicando que el tratamiento debe ser
usado en estos pacientes al igual que en la forma clsica
infantil.Se confirma el diagnstico por determinacin de la enzima en
cultivo de fibroblastos. El uso de sustratos artificiales como
4-metilumbeliferil--D-gluocopiranoside, permite hacer el diagnstico
diferencial entre la forma clsica infantil y las de presentacin
tarda, ya que detecta hasta el 1 a 2% de la enzima residual (20).
El diagnstico prenatal es posible por medicin de la actividad de la
-1,4 glucosidasa en cultivo de fibroblastos de lquido amnitico.
Tratamiento
Hasta el ao 2006 esta enfermedad no tena tratamiento aprobado y
en la actualidad se comenz a usar un tratamiento de reemplazo
enzimtico (TRE), que consiste en entregar la enzima recombinada
rhGAA (-glucosidasa ). El tratamiento en un comienzo se focaliz en
la forma clsica infantil, pero se ha ampliado a todas las formas de
presentacin con buen resultado. Estudios multicntricos han descrito
que dosis de 20 mg/kg cada semana, disminuyen la cantidad de
glucgeno en clulas endoteliales, vasos sanguneos del msculo liso,
en clulas de perineurium y Schwann de nervios perifricos. A largo
plazo se ha visto que mejoran las funciones motoras y
respiratorias. No se han observado efectos colaterales, slo se
menciona una respuesta inmunolgica despus de 3 meses de tratamiento
al desarrollar anticuerpos antirhGAA.
No obstante es necesarios estudios de seguimiento a largo plazo
que permitan comprender el potencial que tiene la TRE y con ello
poder delinear las recomendaciones para su uso en el tratamiento de
esta enfermedad. La dieta con ingesta alta en protenas y baja en
hidratos de carbono es beneficiosa en adultos.
Glucogenosis III
Se produce por el dficit de la enzima amilo 1-6-glucosidasa o
enzima desramificadora, tiene 2 sitios activos independientes
(oligo-1,4-1,4 glucantransferasa y amilo-1,6 glucosidasa. El gen se
ha localizado en el cromosoma 1p21 (OMIM 232400) y las mutaciones
ms frecuentes son R864X, R1228X, Y1510X. La incidencia estimada en
Europa es de 1: 83.000 recin nacidos y de 1: 100.000 para
Norteamrica.Se han descrito diversas variantes debido a que la
enzima tiene una subunidad transferasa que transfiere 3 residuos de
glucosa de una cadena a otra. En la glucogenosis tipo III-a existe
dficit de transferasa y de glucosidasa tanto en hgado como en
msculo y representa al 80%. La glucogenosis tipo III-b es una forma
exclusivamente heptica y equivale al 15% del total. Otras formas
poco frecuentes son la tipo III-c donde hay prdida selectiva de
actividad de la glucosidasa y la glucogenosis tipo III-d que
presenta dficit de transferasa en hgado y msculo.
Clnica y diagnstico
Se caracteriza por hepatomegalia e hipoglucemia, estatura baja,
dislipidemia y en algunos casos leve retardo mental. Los sntomas
musculares pueden comenzar junto con los hepticos, pero los
hepticos mejoran con la edad y siempre desaparecen en la
pubertad.Adultos con la tipo III-a presentan fatiga progresiva con
agotamiento de msculos distales, alteraciones cardacas que van
desde la hipertrofia ventricular a una evidente cardiomegalia. Un
bajo porcentaje presenta slo alteraciones musculares sin otro
sntoma.Dentro de las alteraciones bioqumicas los marcadores son
hipoglucemia por ayuno prolongado, aumento de creatinaquinasa (CK),
aspartato transaminasa, alanina transaminasa. La dislipidemia,
especialmente la hipertrigliceridemia se incrementa con la edad y
est asociada principalmente a un aumento de la beta oxidacin de
grasas inducido por la hipoglucemia. Frecuentemente hay fibrosis
periportal y algunas veces cirrosis micronodular progresiva.
El diagnstico se hace midiendo la actividad de la enzima en
hgado, msculo, corazn eritrocitos y cultivo de fibroblastos. Se
puede hacer estudio de mutaciones que es menos invasivo.
Tratamiento
El tratamiento es nutricional y su objetivo es evitar las
hipoglicemias. Se da una dieta fraccionada, con hidratos de carbono
complejos, de preferencia almidn crudo. A diferencia de la
glucogenosis tipo Ia, se permite el consumo de lcteos y frutas, ya
que la va de la glucosa -6-fosfato est normal, por tanto la
galactosa y fructosa pueden ser transformadas en glucosa. La
alimentacin nocturna por sonda y el almidn crudo ayudan a mejorar
el crecimiento y disminuye el tamao del hgado.
Debido a que la neo glucognesis funciona regularmente y es la va
de obtencin rpida de glucosa, se recomienda aportar mayor cantidad
de protenas. La composicin de la dieta para lactantes y nios es de
un 55% a 60% de hidratos de carbono, 15% a 20% de protenas y los
lpidos para completar aporte de caloras. En nios mayores o adultos
puede ser 20-25% protenas, 50-55% hidratos de carbono complejos, y
25% de lpidos. Es importante proporcionar el 3% como cidos grasos
esenciales de la familia de los 6 y un 1% de los 3.
Glucogenosis tipo IV
Fue descrita por Andersen en 1956 y se produce por la
deficiencia de la enzima amilo-1,4 a 1,6-transglucosidasa o enzima
ramificadota (Figura 3), su herencia es autosmica recesiva y el gen
ha sido codificado en el cromosoma 3p12 (OMIM 232500). Las
mutaciones R515C, F257L, R524X, R515H se asocian a la forma
heptica, las mutaciones Y329S, R524Q, IV5+2T>C, L224P a la forma
no progresiva juvenil.
Clnica y diagnstico
Clnicamente es extremadamente heterognea, debido a la existencia
de isoformas. La forma clsica heptica precozmente progresa a una
cirrosis e insuficiencia heptica, seguida de fallecimiento a causa
de la falla heptica o de complicaciones derivadas de la hipertensin
portal asociada. Presentan miopata perifrica, con o sin
cardiomiopata, neuropata y cirrosis heptica. La presentacin
muscular est dividida en 4 grupos segn presentacin clnica: forma
perinatal (fetal) con hidrops fetal y polihidroamnio, artrogriposis
en extremidades; forma congnita: hipotona, hiporreflexia,
cardiomiopata; forma infantil neuromuscular: miopata, o cardiopata,
intolerancia al ejercicio; forma adulta: distrofia muscular,
dificultad progresiva para caminar, debilidad de extremidades
proximales, mayor en brazos que piernas, tetraparesia piramidal,
neuropata perifrica.
El diagnstico se establece demostrando depsitos de glucgeno
anormal en hgado y eventualmente msculo, aunque hay casos
publicados sin depsito patolgico muscular. Se confirma mediante
medicin de actividad enzimtica en hgado, eritrocitos, y
fibroblastos. Es posible identificar heterocigotos y realizar
diagnstico prenatal.
Tratamiento
No hay tratamiento especfico disponible y los numerosos intentos
teraputicos usandocorticoesteroides, dietas alta en protenas o baja
en hidratos de carbono, o la administracin de alfa-1,4-glucosidasa
y alfa-1,6-glucosidasa no han tenido xito. Hay publicaciones que
destacan una mayor sobrevida con un control diettico estricto de la
hipoglicemia de ayuno.El trasplante heptico ha sido beneficioso no
slo por los problemas hepticos sino que tambin por los
musculares.
Glucogenosis tipo V
La glucogenosis tipo V o enfermedad de McArdle (31) se produce
por el dficit de miofosforilasa (Figura 4), el gen PYGM ha sido
localizado en el cromosoma 11q13 (OMIM 608455). Se han descrito
isoformas de esta enzima en hgado y corazn pero estn normales en
esta patologa. Es la glucogenosis del msculo esqueltico y miopatias
ms frecuente, con una prevalencia de 1:100.000 en USA.
Clnica y diagnstico
Presentan intolerancia a la contraccin muscular esttica e
isomtrica (levantar peso o empuar la mano) y al ejercicio dinmico
(subir escaleras, correr, caminar rpido) causando cansancio,
fatigabilidad, mialgias de intensidad variable, calambres, sensacin
de rigidez o de aumento de volumen muscular, debilidad,
sensibilidad a la palpacin muscular.Estos sntomas se alivian despus
del reposo de manera que el paciente aprende a evitarlos, adecuando
sus actividades diarias y reduciendo la actividad fsica. Cualquier
forma de malestar desencadenado por el ejercicio y aliviado con
reposo se debe sospechar una deficiencia de fosforilasa.
Los sntomas pueden variar segn la edad, existiendo tres fases de
sntomas: en infancia y adolescencia el nico sntoma puede ser la
fatigabilidad fcil. Hacia los 20 aos pueden aparecer los calambres
y debilidad con el ejercicio, a menudo asociados a mioglobinuria
transitoria. La mioglobinuria puede ser tan severa que cause
insuficiencia renal. Finalmente en la adultez se desarrolla
debilidad y franca atrofia muscular. Estas fases son extremadamente
variables en intensidad y pueden no estar presentes en todos los
pacientes.La mayora de los casos se diagnostican en la adultez; en
la infancia las molestias son inespecficas como cansancio y
fatigabilidad, de modo que estos nios son catalogados a menudo como
nios flojos o sedentarios.
La anamnesis es fundamental para el diagnstico. El examen entre
los episodios sintomticos es, en general, completamente normal. En
algunos casos la mioglobinuria puede ser evidenciada despus de
ejercicio. Los calambres pueden ser reproducidos sometiendo al
paciente a ejercicio isqumico, usando un manguito de presin. Esto
produce acortamiento y rigidez del msculo, generando una verdadera
contractura con calambre que puede ser muy dolorosa y que puede
persistir una vez que la circulacin es restablecida.Despus de esta
prueba no se observa el aumento de 2 a 5 veces sobre el valor basal
de lactato y piruvato que se produce en un individuo normal. Esto
puede ser medido en sangre venosa de la zona isqumica y es
caracterstico del dficit de miofosforilasa aunque no patognomnico,
pues adems se ve en la ausencia de otras enzimas de la va
glucoltica.La histoqumica muestra ausencia de fosforilasa muscular,
y el exceso de glucgeno sospechado mediante tincin de PAS es
confirmado por microscopia electrnica. El diagnstico debe ser
confirmado por la biopsia muscular y por estudio molecular en el
gen PYGL.
Tratamiento
La mayora de los pacientes aprende a restringir su actividad
fsica. El uso de dietas ricas en protenas o grasas no mejora la
tolerancia al ejercicio. Estudios recientes han observado que al
proporcionar una dieta rica en hidratos de carbonos complejos
(almidn) se mejora la tolerancia al ejercicio y aumenta la
capacidad oxidativa, si se comparar con el uso de una dieta rica en
protenas. Basndose en estos resultados, se recomienda una dieta muy
parecida a la que se entrega para prevenir la arteriosclerosis. La
suplementacin con creatina, D-ribosa o vitamina B6 no ha tenido
resultados positivos.
Glucogenosis tipo VI
La glucogenosis tipo VI o enfermedad de Hers, es una forma poco
frecuente, y la enzima defectuosa es la fosforilasa heptica, el gen
PYGL se encuentra en el cromosoma 14q21- q22 (OMIM 232700) y se han
identificado varias mutaciones.
Clnica y diagnstico
Se presenta con hepatomegalia y abdomen prominente y una
tendencia moderada a la hipoglicemia en ayuno. El crecimiento del
hgado disminuye lentamente y desaparece en la pubertad. El bazo y
riones estn de tamao normal. Tiene leve retardo del crecimiento y
su desarrollo mental es normal.
En los exmenes de laboratorio se encuentra aumento en suero de
transaminasas, triglicridos y colesterol. El diagnstico se confirma
por estudio enzimtico por biopsia heptica, pero algunos casos
pueden tener una actividad enzimtica elevada, por ello es necesario
realizar estudio molecular para identificar las mutaciones.
Tratamiento
No necesitan tratamiento especfico, excepto si se vuelven
sintomticos durante cuadros infecciosos o ayunos prolongados. La
alimentacin debe ser fraccionada para prevenir ayunos y evitar las
hipoglicemias especialmente en los ms jvenes. Es importante
proporcionar una comida en la noche, antes de acostarse, rica en
protenas y almidn, evitar ayunos prolongados nocturnos. Las grasas
poliinsaturadas son beneficiosas para evitar la
hipercolesterolemia. El retraso del crecimiento inicial se
normaliza posteriormente, llegando a tener estura normal y mnima
hepatomegalia en etapa de adulto. Evitar el alcohol. La esperanza
de vida es normal.
Glucogenosis tipo VII
Tarui describi esta enfermedad en 1965 y se produce por una
deficiencia de la actividad de la fosfofructoquinasa muscular
(Figura 4), (OMIM 610681), el gen ha sido localizado en el
cromosoma 12q13.3 y se han identificado 20 mutaciones. La
fosfofructoquinasa es una enzima tetramrica compuesta por tres
tipos de subunidades: muscular (M), heptica(L) y plaquetaria (P) de
tamao molecular parecido y que se expresan variablemente en
diferentes tejidos
Clnica y diagnstico
Se divide en dos formas de presentacin: grave infantil que
manifiestan insuficiencia respiratoria y la del adulto con
intolerancia al ejercicio, calambres, fatigabilidad, En ambas
formas aparece rabdomilisis y mioglobinuria, frecuentemente
asociado a anemia hemoltica e hiperuricemia (41). El
electrocardiograma muestra un bajo voltaje, taquicardia ectpica
supraventricular, ensanchamiento e insuficiencia de la vlvula
mitral, agrandamiento del atrium izquierdo, hipertrofia ventricular
izquierda y disfuncin sistlica.Las manifestaciones neurolgicas
progresan a convulsiones parciales complejas, visin doble, parlisis
facial, bradidiadocoquinesias, debilidad distal de extremidades
superiores y aumento de los calambres.Los glbulos rojos pueden ser
usados para llegar al diagnstico bioqumico, pero para establecer un
diagnstico certero se recomienda biopsia muscular con
estudioinmunohistoqumico y estudio molecular.
Tratamiento
Las opciones de tratamiento son semejantes a la enfermedad de Mc
Ardle.
Glucogenosis tipo IX
Se produce por la deficiencia de la fosforilasa quinasa (Figura
4), haloenzima compuesta por 4 subunidades (, , , ). La isoforma
est ubicada en el gen PHKA1 (msculo) localizada en el cromosoma
Xq12-q12 y el gen PHKA2 (hgado) en el cromosoma Xp22.2-p22.21 (OMIM
30600). La subunidad en el gen PHKB ubicado en el cromosoma 16q12-
q13. La isoforma tiene 2 sitios activos, los cuales han sido
ubicados en el gen PHKG1 (cromosoma 7p11.2) y gen PHKG2 (cromosoma
16p12.1-p11.2) y se han encontrado 12 mutaciones. La ltima
subunidad o calmodulin, inhibe la actividad de la fosforilasa
quinasa con una disminucin de calcio inico y se han descrito tres
genes independientes: CALM1, CALM2, CALM3. Todas las formas son de
herencia autosmica recesiva, excepto la isoforma .
Este complejo enzimtico es activador especfico de la fosforilasa
quinasa y una llave de control en transporte de glucosa desde el
glucgeno. La complejidad de esta enzima, lleva a una variabilidad
en los sntomas clnicos.
Clnica y diagnstico
La isoforma PHKA2 se caracteriza por hipoglicemias,
hepatomegalia, enfermedad heptica crnica, retardo del crecimiento,
y del desarrollo motor y mental. Manifiestan hipercolesterolemia,
hipertrigliceridemia, e hiperquetonemia por un ayuno prolongado.
Los sntomas disminuyen con la edad. En la PHKA1 se produce
intolerancia al ejercicio, calambres, mialgias, decaimiento y
mioglobinuria. En la subunidad los sntomas son menos graves, y
presentan hepatomegalia, hipoglicemia slo despus de un ayuno
prolongado. La isoforma PHKG2 presenta hepatomegalia, hipoglicemias
recurrentes a ayunos, respuesta alterada de glucagn, fibrosis y
desarrollan cirrosis o adenomas.El diagnstico se hace por biopsia
heptica o muscular, pero se establece el diagnstico diferencial
entre las diferentes formas de presentacin por estudio de
mutaciones.
Tratamiento
Esta dirigido a prevenir las hipoglicemias, el uso de
alimentacin enteral, con ingesta alta en hidratos de carbono,
almidn crudo, fraccionado, y alimentacin nocturna por sonda
nasogstrica permite un crecimiento en parmetros de normalidad y
buena respuesta clnica.
Glucogenosis tipo XI
Es el defecto congnito del transportador de glucosa de los
hepatocitos y clulas tubulares renales con prdida funcional (Figura
4). Se conoce como sndrome de Fanconi-Bickel. Es de herencia
autosmica recesiva, el gen SLC2A2 o GLUT-2 localizado en el
cromosoma 3q26.1-26.3 (OMIM 227810) y se han descrito 23
mutaciones, de las cuales 12 son de Turqua.
La hipoglicemia en ayuno se produce por la alteracin del
transporte de la glucosa desde el hepatocito al torrente sanguneo,
resultando aumento de la glucosa intracelular porque la
neoglucognesis inhibe la degradacin de glucgeno y ste se acumula en
el hepatocito y clulas tubulares renales. La prdida de glucosa por
el rin contribuye a la hipoglucemia. Se produce galactosuria y
galactosemia por este defecto, se puede confundir con una
galactosemia clsica.
Clnica y diagnstico
Generalmente durante el primer ao de vida hay detencin del
crecimiento, osteopenia y posteriormente raquitismo, hepatomegalia,
nefropata de Fanconi.Los exmenes de laboratorio muestran
hipoglicemia y cetonemia en ayuno e hiperglicemia e
hipergalactosemia en estados post absortivos, hipercolesterolemia e
hipertrigliceridemia hipofosfatemia, aminoaciduria generalizada,
fosfaturia, galactosuria, glucosuria y proteinuria.El diagnstico se
realiza por biopsia heptica que muestra acumulacin de glucgeno y
esteatosis. Por la intolerancia a la galactosa, pueden ser
diagnosticados por pesquisa neonatal para galactosemia.
Tratamiento
El tratamiento est dirigido a estabilizar la glicemia y
compensar las prdidas renales de agua, electrolitos y vitamina D
(1,25-dihidroxicolecalciferol ). Restringir la galactosa e indicar
una dieta como para una diabetes mellitus, con fraccionamiento de
comidas e hidratos de carbono complejos como almidn crudo. Como no
est alterado el metabolismo de la fructosa se puede usar como
alternativa, ya que mejora el transporte intestinal de monosacridos
y su absorcin intestinal.