Embed Size (px)
Citation preview

This article was downloaded by: [Moskow State Univ Bibliote]On: 28 January 2014, At: 06:29Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Journal of Biomolecular Structure and DynamicsPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/tbsd20
Alternatingly twisted β-hairpins and nonglycineresidues in the disallowed II′ region of theRamachandran plotIvan Yu. Torshina, Natalya G. Esipovab & Vladimir G. Tumanyanb
a Department of Chemistry, M.V. Lomonosov State University, 1-73 Leninskie Gory, Moscow,119991, Russian Federation.b V.A. Engelhard Institute of Molecular Biology, Russian Academy of Sciences, Vavilova St.,32, Moscow, 119991, Russian Federation.Published online: 05 Feb 2013.
To cite this article: Ivan Yu. Torshin, Natalya G. Esipova & Vladimir G. Tumanyan (2014) Alternatingly twisted β-hairpins andnonglycine residues in the disallowed II′ region of the Ramachandran plot, Journal of Biomolecular Structure and Dynamics,32:2, 198-208, DOI: 10.1080/07391102.2012.759451
To link to this article: http://dx.doi.org/10.1080/07391102.2012.759451
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) containedin the publications on our platform. However, Taylor & Francis, our agents, and our licensors make norepresentations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of theContent. Any opinions and views expressed in this publication are the opinions and views of the authors, andare not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon andshould be independently verified with primary sources of information. Taylor and Francis shall not be liable forany losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoeveror howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use ofthe Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in anyform to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions

Alternatingly twisted β-hairpins and nonglycine residues in the disallowed II′ region of theRamachandran plot
Ivan Yu. Torshina, Natalya G. Esipovab and Vladimir G. Tumanyanb*aDepartment of Chemistry, M.V. Lomonosov State University, 1-73 Leninskie Gory, Moscow 119991, Russian Federation;bV.A. Engelhard Institute of Molecular Biology, Russian Academy of Sciences, Vavilova St., 32, Moscow 119991, Russian Federation
Communicated by Vsevolod J. Makeev
(Received 18 May 2012; final version received 15 November 2012)
The structure of the SH3 domain of α-spectrin (PDB code 1SHG) features Asn47 in the II′ area of the Ramachandranplot, which as a rule admits only glycine residues, and this phenomenon still awaits its explanation. Here, we undertooka computational study of this particular case by means of molecular dynamics and bioinformatics approaches. We foundthat the region of the SH3 domain in the vicinity of Asn47 remains relatively stable during denaturing molecular dynam-ics simulations of the entire domain and of its parts. This increased stability may be connected with the dynamic hydro-gen bonding that is susceptible to targeted in silico mutations of Arg49. Bioinformatics analysis indicated that Asn47 isin the β-turn of a distinctive structural fragment we called ‘alternatingly twisted β-hairpin.’ Fragments of similar confor-mation are quite abundant in a nonredundant set of PDB chains and are distinguished from ordinary β-hairpins by somesurplus of glycine in their β-turns, lack of certain interpeptide hydrogen bonds, and an increased chirality index. Thus,the disallowed conformation of residues other than glycine is realized in the β-turns of alternatingly twisted β-hairpins.
Keywords: SH3 domain; disallowed conformations; β-turn
Introduction
A Ramachandran plot presents the data on all the dihe-dral angles Φ and Ψ of a polypeptide chain. Originallyproposed four decades ago (Ramachandran & Sasisekha-ran, 1968), this diagram constitutes a valuable instrumentthat allows to detect and deal with abnormalities in thechain tracing that might arise during protein structuredetermination. Apart from the use in structure determina-tion (Bertini, 2003), validation (Lovell, 2003), prediction(Oliva, 1997), and study of conformational changes uponligand binding (Gunasekaran, 2007), Ramachandran plotscan also be used to detect rare conformations of thepolypeptide chain which correspond to functional regions(Hooft, Vriend, Sander, & Abola, 1996).
In a way, a Ramachandran plot represents energeti-cally favorable and unfavorable conformations of indi-vidual residues of a polypeptide chain. The regionsreferred to as ‘disallowed’ correspond to conformationscontaining van der Waals clashes. The latter, in mostcases, energetically ‘prohibit’ such conformations.
Disallowed conformations in Ramachandran plots areoften a result of inconsistencies in the protein structure
determination. Typically, as resolution of diffractionexperiment increases, the number of residues in the‘forbidden’ region decreases. It is interesting that someresidues of this type are unaffected in the course ofstructure refinement. Statistical analyses of the distribu-tion of the residues on the Ramachandran plots (Gun-asekaran, 1996; Pal, 2002; Ramakrishnan, 2007) indicatethat in the regions where only glycine residues areallowed, nonglycine residues certainly occur. Such resi-dues as Ser, Asn, Thr, and Cys have the highest propen-sities to exhibit these conformations while the branchedaliphatic residues are characterized by the lowest propen-sities (Pal, 2002).
One of the disallowed areas is the II′ zone [Φ(60± 30°), Ψ(�120 ± 30°)] which corresponds to the residuesin position ‘L1’ of the type II′ β-turns (Hutchinson &Thornton, 1994). Originally, it was thought that onlyglycine residues can occupy this position of the type II′β-turns and consequently, the II′ region of theRamachandran plot. However, statistical analyses (Gun-asekaran, 1996; Ohage, 1997) indicate that nonglycineresidues occur quite often in the II′ area.
*Corresponding author. Email: [email protected]
Journal of Biomolecular Structure and Dynamics, 2014Vol. 32, No. 2, 198–208, http://dx.doi.org/10.1080/07391102.2012.759451
� 2013 Taylor & Francis
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

The structure of the SH3 domain of α-spectrin featuresa nonglycine residue (Asn47, PDB code 1SHG) in the II′area of the Ramachandran plot. In the experimental studyof the SH3 domain (Vega, Martinez, & Serrano, 2000),the researchers attempted to mutate the residue to othertypes which, supposedly, should have resulted in stabiliza-tion of the protein and removal of the conformationalstrain. However, neither thermodynamic analysis of theN47G and N47A mutants nor X-ray structural analysisindicated any considerable effects of the mutations onthe energetics or conformation of the protein structure.The authors concluded that the energetic definition of theII′ region of the Ramachandran plot should perhaps berevised in structure validation and protein design, at leastfor Ala and Asn residues (Vega et al., 2000).
Since physical reasons for occurrence of nonglycineresidues in the II′ region are not entirely clear, in thepresent work, we undertook a theoretical analysis of thebiophysical peculiarities of the SH3 domain of α-spectrinto disclose factors that might determine persistence ofthe disallowed conformation of Asn47. The analysis wasperformed using molecular dynamics simulations,in silico mutagenesis, and bioinfomatics. The point is todiscover global and local factors which allow an energet-ically nonfavorable residue conformation. It is reasonableto hypothesize that the existence of nonglycine residuesin the disallowed region must be balanced by the rest ofthe protein structure supporting the segment under study.In the course of the study, we found the global factor(namely, a peculiar structure which we called an ‘alter-natingly twisted β-hairpin’) that stabilizes nonfavorableconformation of the residue in the β-bend. This conclu-sion is supported by the results of the statistical analysisof similar conformations in a nonredundant subset ofPDB, the relevant amino acid preferences, and putativesequence patterns.
Materials and methods
The protein coordinate files were taken from PDB (Ber-man et al., 2000) using the VAST’s nonredundant set ofthe protein structures (Gibrat, Madej, & Bryant, 1996)(see http://structure.ncbi.nlm.nih.gov/Structure/VAST/vasthelp.html) as of late 2009. The data-set contained2950 protein chains with mutual sequence iden-tity < 25%. Molecular dynamics simulations were per-formed using Energy Calculator for the MacroMolecularStructures (ECMMS) (Torshin, 2004). The package isbased on the universal force field (UFF) force field thatis characterized by an especially high accuracy of thebonding, angular and torsional terms (Rappé & Goddard,1991). The hydrogen atom positions were calculated inaccordance with the geometries specified by the UFFforce field. The denaturing molecular dynamics simula-tions were performed using a modified leapfrog algo-rithm (Cuendet & van Gunsteren, 2007) coupled with
thermostat (400, 600K), without solvent (in order toinduce unfolding), without distance cut-offs, and withoutexplicit hydrogen bonding terms.
Usually, protein molecular dynamics simulations areperformed in solvent environments. Because of the largenumber of water molecules required for full solvation ofa protein macromolecule and computer resource limita-tions, a number of approximate approaches have beenproposed (Davis & McCammon, 1990). In the presentwork, an implicit solvation model is used. The Eisen-berg–McLachlan term is assumed to be proportional tothe atomic solvent accessible surface area (Eisenberg &McLachlan, 1986). Although the accuracy of suchapproximation is uncertain, in the present work, it isused with the sole aim of comparing the conformationalflexibility for different parts of the SH3 domain. Evenwithin quite a short simulation (100 ps), MD allowed usto differentiate between residues with high conforma-tional flexibility (e.g. RMSD greater 5Å) and low flexi-bility (RMSD less than 0.5Å). In this respect, the MDsimulations in the present work serve as an instrument ofbioinformatics rather than a tool for full-scale biophysi-cal modeling of protein unfolding (Daggett, 2002; vander Kamp & Daggett, 2011). Full-scale MD modeling ofthe supertwisted β-hairpin is a separate task beyond thescope of the present paper. In silico protein mutagenesiswas performed using the same script procedure asdescribed previously (Torshin, 2004).
Bioinformatics analyses including the measurementof the Φ, Ψ angles, estimation of the frequency of occur-rence of certain conformations, superposition of proteinfragments, sequence analyses, etc. were performed usingsoftware units developed by I. Yu. T. In particular,hydrogen bonds were detected using INTerGlobular anal-ysis (Torshin, Weber, & Harrison, 2002). The presenceof hydrogen bonds was analyzed in all snapshots duringthe 200 ps simulation run. The hydrogen bonding criteriawere based on geometrical rules [distance H…A<2.5Å;angleD�H…A>90.0° (Torshin et al., 2002)]. In other words,hydrogen bonds were not explicitly modeled. Physically,H-bonds are due to interaction of partial charges andthis, in part, is described by the nonbonding potential.
Surface accessibilities were calculated with SURFaceCalculations (SURFC) (Torshin, 1999) and analysis of thefrequency of conformations was done using the same dec-apeptide algorithm as described previously (Petock, Tor-shin, Weber, & Harrison, 2003) but with different criteriaof similarity. The criteria of the conformation similarityused in the present study were rather strict: RMSD(Cα)< 0.3 Å on all Cα atoms and deviation (Cα)<1.5 Å for anyparticular atom of the decapeptide. The chirality index cal-culations were performed according to the special formula(Pietropaolo, Muccioli, Berardi, & Zannoni, 2008) withtwo modifications: (a) decapeptides and not pentapeptides
Disallowed conformations in polypeptide chain 199
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

were used and (b) the distance cutoff was set to 20Åinstead of 12Å.
Results and discussion
Molecular dynamics of the SH3 domain
Apparently, the entire structure of the SH3 domain of α-spectrin was stable during 100 ps of the denaturing MDsimulations at 400–600K. The data for the 400K simu-lation are presented in Figure 1.
The major events in the structure relaxation occurredbefore 20 ps of the simulation time (Figure 1(A)). Themajor conformational changes occurred in the N-terminalregion of the SH3 domain (residues 10–25) and in region43–45 (Figure 1(D)). These changes (partial unfolding ofsurface accessible regions of the SH3 domain) result inRMSD of side-chain atoms exceeding 1.5Å.
However, the region of interest (Asn47) remained lar-gely unaffected. Although a very small stepwise rise inthe RMSD was observed at 60 ps and at 80 ps, thedynamics after 20 ps were too slow. As shown in theplots of the Φ- and Ψ-angles of the Asn47 residue, theΦ-value of the Asn47 residue oscillated in the range 58–62° with some 20 ps period while the Ψ-value stabilizedat �125° (Figures 1(B,C)). Thus, the conformation ofthe main-chain atoms of this residue was well preservedduring the entire simulation time.
Moreover, as shown with the per-residue RMSD inFigure 1(D), the main chain around residues 46–50 wasone of the least conformationally mobile regions of theSH3 domain structure. It is reasonable to assume that theabnormal Φ, Ψ of the Asn47 residue appear because ofsome peculiarities either of the local environment (resi-dues adjacent in sequence) or global environment (theSH3 domain per se), or where there is some intermediatecase of this residue. Inspection of the structure of theSH3 domain and analysis of the hydrogen bonding sug-gested several possible structural reasons for this stabil-ity. For instance, there is a hydrogen bonding networkformed by residues Lys43, Asp45, Glu48, and Asn50which might contribute to the conformational stability ofthe main chain at residues 46–50. Another example:Val46 is buried inside the hydrophobic core (surfaceaccessibility of 0.0) and thus, might reduce conforma-tional mobility of the main chain. In order to assessplausibility of these and other hypotheses and to find res-idues that might determine conformational stability/mobility of Asn47 and surrounding residues, we under-took a protein-wide alanine scan in silico.
Alanine lscan and Φ/Ψ values of Asn47
We attempted to outline the structural determinants ofthe abnormal Φ, Ψ values of Asn47 by preparing fullatomic models of single mutant-to-alanine mutations of
all residues in the SH3 domain (5–62 using the numera-tion of PDB file 1SHG). The minimization procedureinvolved in the in silico mutagenesis (Torshin, 2004)includes short isothermic runs of the molecular dynam-
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
0 20 40 60 80 100 120
t,ps
rms,
A
phi(ASN47)
0
10
20
30
40
50
60
70
0 20 40 60 80 100 120 140t,ps
phi
psi(ASN47)
-140
-120
-100
-80
-60
-40
-20
00 20 40 60 80 100 120
t,ps
psi
(A)
(C)
(B)
(D)
Figure 1. MD simulation of the SH3 domain (PDB file1SHG) at 400K, no solvent. (A) RMSD plot (main chain), (B)Φ(Asn47) on the simulation time, (C) Ψ(Asn47), and (D) per-residue RMSD of the snapshot at 100 ps.
200 I.Y. Torshin et al.
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

ics. Energy minimization was performed until reaching aplateau of stability after which there was no reduction inenergy values (rule of thumb – a decrease in energy lessthan 1% in 100 minimization steps by conjugategradients).
It is well known that the results of the moleculardynamics considerably depend on the initial conditions(in our case, the velocities). In order to assess the extentof this influence, 20 independent runs of the minimizingprocedure were performed on the native structure usingdifferent sets of initial velocities (randomized Maxwelldistribution) and the dispersion of the Φ and Ψ angles ofAsn47 was estimated. In all of the 20 runs, Φ was within(48…64°) and Ψ (�116…�152°). Accordingly, if amutation leads to a statistically significant effect on thedihedrals of Asn47, the value should lie outside theseerror margins. The mutations that lead to significantchanges in the dihedrals are summarized in Table 1.
The data in Table 1 are divided in two groups: thosethat lead to further distortion of the main chain of Asn47(higher Φ) and those resulting in partial relaxation of thestructure (higher Ψ). The first group shows an apparentprevalence of the Leu-to-Ala mutations. Surface-accessi-ble leucines Leu8, Leu10, and Leu12 form hydrophobicpatches on the protein surface and their disruption leadsto a loss of the optimal packing. Leucines Leu31, Leu33,
and Leu34 as well as residues Val23, Met25, and Val46belong to the compact, densely packed ‘hydrophobiccore’ and their removal because of mutagenesis resultsin destruction of the dense packing and, therefore, itinduces considerable conformational changes of theentire structure of the SH3 domain. Comparison of thedata in the table with conformationally labile residues(Figure 1) indicates that most of the residues thataffected the Φ- and Ψ-angles were of low conformationalmobility. In accordance with the experimental results(Vega et al., 2000), the mutations Asn47Ala andAsn47Gly did not affect the Φ and Ψ values of Asn47.
While the residues whose mutation affected the dihe-drals of Asn47 were scattered throughout the globule(Table 1), most of them were concentrated in region 29–55 which corresponds to three consecutive β-strands(Figure 2). This structure is a compact module and itsformation is likely to be important for the folding of theSH3 domain. There is more information on compactmodules and folding (Viguera, Jimenez, Rico, & Serano,1995). Since most of the residues affecting dihedrals ofAsn47 were located to this segment of the polypeptidechain, we attempted further in silico studies of this frag-ment separated from the rest of the domain. MD simula-tion of the compact module 29–55 indicated that itremained stable during 100 ps of denaturing MD at 400
Table 1. Alanine scan of the SH3 domain and dihedrals of Asn47.
Mutant rACS Φ(°) Ψ(°)
Variants that made Φ (Asn47) higherLeu8Ala 0.08 113 �112Leu10Ala 0.05 114 �113Leu12Ala 0.06 119 �123Arg21Ala 0.1 98 �128Asp29Ala 0.04 96 �125Ile30Ala 0.09 110 �112Leu31Ala 0.0 115 �116Leu33Ala 0.0 115 �114Leu34Ala 0.0 115 �116Trp41Ala 0.03 102 �133Trp42Ala 0.04 101 �132Lys43Ala 0.06 87 �120Arg49Ala 0.07 84 �132Lys59Ala 0.2 87 �134Lys60Ala 0.08 85 �132Variants that made Ψ (Asn47) higherGlu7Ala 0.03 63 �101Glu17Ala 0.07 64 �96Glu22Ala 0.02 63 �98Val23Ala 0.0 67 �105Met25Ala 0.0 50 �105Asn35Ala 0.06 64 �91Asn38Ala 0.09 67 �97Val46Ala 0.0 65 �114
Notes: rACS, relative surface accessibility of the native residue. In accordance with the SURFC parameters used for calculations (23), residues withthe rACS> 0.04 are ‘surface accessible,’ residues with rACS< 0.03 are ‘‘buried”.
Disallowed conformations in polypeptide chain 201
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

or 600K (400K: RMSD stabilized at 1.8 A0after 16 ps of
simulation). However, the N-terminal part of the module(residues 29–40) was significantly more unfolded thanthe C-terminal β-hairpin (RMSD 2.0 A
0vs. 1.4 A
0). Thus,
the most conformationally stable region of the compactmodule was β-hairpin 40–55 and all subsequent simula-tions were performed on this β-hairpin that includesAsn47 (Figure 2(C)).
It is interesting to compare the results of our compu-tational study and the experimental findings (Vigueraet al., 1995) for fragment 40–55. These authors wrotethat they could not detect any nonsequential nuclearoverhauser effects, but this could be a result of the lowconcentration used because of the poor solubility of thepeptide. They marked the similarity of some chemical
shifts in the peptide and in native protein, in spite ofsmall absolute values of the shifts in the peptide. Theconcluding phrase of their paragraph devoted to the 40–55 peptide was: ‘These results indicate that the peptideis adopting, to some extent, a native-like conformation.’
In silico studies of β-hairpin 40–55
Hydrogen bonding network of 40–55
The 200 ps simulation run of the 40–55 β-hairpin at400K has indicated that this fragment of the structurewas also stable on its own, without the support of therest of the domain. Moreover, during the simulation,the Φ and Ψ values of Asn47 still remained within theboundaries corresponding to the II′ region disallowed fornonglycine [Φ (48…64°) and Ψ(�116…�152°)]. Since
Figure 2. The main elements of the structure of the SH3 domain. Residues Asn47 are indicated in a wireframe mode. The figurewas prepared using Rasmol. (A) SH3 domain (PDB file 1SHG); (B) three β-strands of compact module 29–55; (C) ‘alternatinglytwisted’ β-hairpin 40–55.
Table 2. Hydrogen bonding networks of alternatingly twisted hairpin 40–55.
Occurrence (%) Residue 1 Residue 2 Atom1 Atom2 Distance H…A (A0) Angle D–H…A (°)
Static H-bond network100 Asp 40 Trp 42 O N 2.48 125.6100 Trp 42 Val 53 O N 2.37 152.1100 Lys 43 Glu 45 NZ OE2 1.75 164.3100 Val 44 Gly 51 N O 2.50 105.099 Lys 43 Glu 45 NZ OE1 2.27 136.599 ASP 48 Arg 49 OD1 N 2.51 105.7Dynamic H-bonds of Arg4988 Asp 48 Arg 49 OD2 NE 2.45 106.688 Asp 48 Arg 49 OD2 NH2 2.07 130.316 Asp 48 Arg 49 O NH2 2.42 95.135 Val 46 Arg 49 O NH2 2.37 112.321 Val 46 Arg 49 O N 2.31 153.38 Asp 48 Arg 49 O NE 2.49 93.1Dynamic H-bonds of Lys4358 Lys 43 Gln 50 NZ OE1 2.01 152.121 Lys 43 Val 44 NZ O 2.52 131.8Dynamic H-bonds of Gln5067 Ala 47 Gln 50 O NE2 2.23 145.325 Glu 45 Gln 50 OE2 NE2 2.41 95.6
Notes: Occurrence was defined as the number of 1 ps snapshots in which the hydrogen bond was found divided by the total simulation time (200 ps).The donor–acceptor H…A distances and the D�H…A angles are mean values.
202 I.Y. Torshin et al.
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

the context of the whole domain is not significant, thehydrogen bonding is the most likely factor that enhancesthe stability of the β-hairpin. The presence of H-bondswas analyzed in all the snapshots during the 200 ps sim-ulation run. The summary of the occupancy of all thehydrogen bonds detected during > 5% of the simulationtime is presented in Table 2.
The data in Table 2 suggest that the hydrogen bond-ing network of the 40–55 hairpin can be subdivided intostatic and dynamic parts. The static H-bonds (occupancyclose to 100%) stabilize the hairpin structure and includethose formed by the main-chain atoms (W42-V53; V44-G51) and those formed by side chains (Lys43-Glu45;Asp48-Arg49). The dynamic H-bonds were formed bythe side chains of Arg49, Lys43, and Gln50. The side-chain RMSD of Lys43 and Gln50 was <1.0 A
0during the
entire simulation time, thus the dynamic H-bonds formedby these residues rather correspond to oscillation of thestructurally adjacent segments of the chain (i.e. residuesVal44, Glu45, and Ala47). The RMSD of the Arg49 sidechain was > 3.0 A
0which corresponded to oscillation of
the side chain of Arg49 between the side chain of Asp48and the main-chain carboxyl of Val46.
In order to elucidate the structural role of these side-chain interactions in the stabilization of the β-hairpin, weattempted to remove some of these interactions bycharge perturbations and then by using in silico muta-genesis. As the data in the table suggest, the main-chainatoms of at least seven residues, namely 40, 42, 44, 46,47, 48, and 51, were involved in the formation of thehydrogen bonding networks. The charge perturbationconsisted in changing the charge of the main-chain oxy-gen of these residues (from �0.5 to + 0.5) followed by ashort (10 ps) run of MD simulation. In other words, sucha procedure would roughly correspond to a change inthe protonation state of the main-chain oxygen atoms.The results of the charge perturbation analysis are givenin Table 3.
It can be seen that targeted disruption of the hydro-gen bonding network through the charge perturbationhad the greatest impact in the case of the main-chainoxygen atoms of residues Val46, Asn47, and Asp48. Inthese three cases, significant unfolding (RMSD>2.0A)occurred only after 10 ps of simulation. However, onlyin the charge perturbation of Val46.O (carbonyl oxygen),the Φ and Ψ values of Asn47 left the disallowed II′region already after 3 ps of the MD simulation with the
disturbed charge, moved to the allowed region (Φ< 0,Ψ < 0) and stayed there until simulation was terminatedat 10 ps. Therefore, the hydrogen bonds formed by themain chain of Val46 are crucial to sustain the abnormalconformation of the Asn47 residue.
According to the data in Table 2, the main-chain car-bonyl oxygen of Val46 formed interactions with the sidechain and main chain of Arg49. Both of these hydrogenbonds were of low occupancy (<50% of the simulationtime) and oscillation of the Arg49 side chain was biasedtowards interactions with Asp48. The low occupancyduring MD simulation can be explained by the fact thatno explicit hydrogen bonding terms were used (sincethey can introduce unnecessary bias in the simulationresults). Anyway, disruption of the hydrogen bonds withVal46 had a remarkable effect on conformation of theentire β-hairpin and of the abnormal main-chain confor-mation of Asn47 (which was confirmed, in part, by theresults of the in silico mutagenesis, see below).
The analysis of the SH3 domain structures, the sameas those analyzed by Vega et al. (2000), PDB codes1 qkw (N47G), 1 shg (wt), and 1 qkx (N47A), indicatedthat the side-chain atoms of Arg49 formed some hydro-gen bonds in each of the structures but with differentregions of the SH3 domain: with Val46 in 1qkw, withGlu17 in 1shg, and with Thr24 in 1qkx. However, themain-chain H-bonding interactions of Arg49 with Val46were preserved in all of the structures (including the cir-cular permutant PDB 1pwt and the NMR structure1aey). These interactions (Arg49.N…Val46.O andAsp48.N…Val46.O) can compensate the steric hin-drances that arise because of the abnormal main-chainconformation of Asn47/Ala47/Gly47. These steric hin-drances correspond to van der Waals clashes (such asbetween Val46.O and Asn47.Cα, 2.9 A
0; Val46.C′ and
Asn47.Cα, 2.4 A0, etc). Calculations using ECMMS on
the native structure (1 shg) indicated that the destabiliz-ing contribution of all the clashes was only+ 1.5 cal mol�1, whereas the stabilizing effect providedby the hydrogen bonds formed between the main-chainatoms of Val46 and Arg49 was �3.5 cal mol�1.
In silico mutagenesis of 40–55
Mutagenic experiments of any sorts influence mostlyside-chain interactions. Therefore, by using mutagenesis,it is possible to target disruptions of only side-chain but
Table 3. Φ- and Ψ-angles of Asn47 after charge perturbation of the main-chain carbonyl oxygen atoms.
Angle (°) Residue Asp40 Trp42 Val44 Val46 Asn47 Asp48 Gly51
Φ(Asn47) +50 +53 +65 �10 +72 +64 +53Ψ(Asn47) �120 �138 �104 �135 �60 �65 �133
Notes: Each charge perturbation was simulated using at least three different random sets of initial velocities and the data in this table represent medianvalues.
Disallowed conformations in polypeptide chain 203
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14
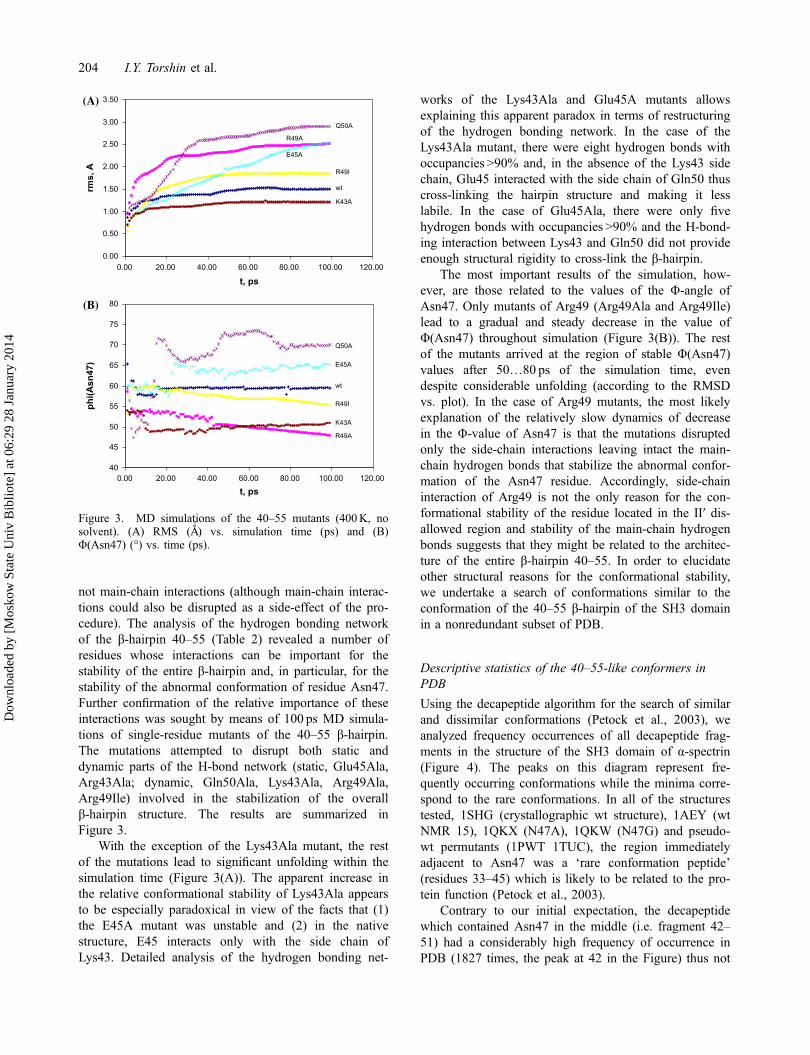
not main-chain interactions (although main-chain interac-tions could also be disrupted as a side-effect of the pro-cedure). The analysis of the hydrogen bonding networkof the β-hairpin 40–55 (Table 2) revealed a number ofresidues whose interactions can be important for thestability of the entire β-hairpin and, in particular, for thestability of the abnormal conformation of residue Asn47.Further confirmation of the relative importance of theseinteractions was sought by means of 100 ps MD simula-tions of single-residue mutants of the 40–55 β-hairpin.The mutations attempted to disrupt both static anddynamic parts of the H-bond network (static, Glu45Ala,Arg43Ala; dynamic, Gln50Ala, Lys43Ala, Arg49Ala,Arg49Ile) involved in the stabilization of the overallβ-hairpin structure. The results are summarized inFigure 3.
With the exception of the Lys43Ala mutant, the restof the mutations lead to significant unfolding within thesimulation time (Figure 3(A)). The apparent increase inthe relative conformational stability of Lys43Ala appearsto be especially paradoxical in view of the facts that (1)the E45A mutant was unstable and (2) in the nativestructure, E45 interacts only with the side chain ofLys43. Detailed analysis of the hydrogen bonding net-
works of the Lys43Ala and Glu45A mutants allowsexplaining this apparent paradox in terms of restructuringof the hydrogen bonding network. In the case of theLys43Ala mutant, there were eight hydrogen bonds withoccupancies >90% and, in the absence of the Lys43 sidechain, Glu45 interacted with the side chain of Gln50 thuscross-linking the hairpin structure and making it lesslabile. In the case of Glu45Ala, there were only fivehydrogen bonds with occupancies >90% and the H-bond-ing interaction between Lys43 and Gln50 did not provideenough structural rigidity to cross-link the β-hairpin.
The most important results of the simulation, how-ever, are those related to the values of the Φ-angle ofAsn47. Only mutants of Arg49 (Arg49Ala and Arg49Ile)lead to a gradual and steady decrease in the value ofΦ(Asn47) throughout simulation (Figure 3(B)). The restof the mutants arrived at the region of stable Φ(Asn47)values after 50…80 ps of the simulation time, evendespite considerable unfolding (according to the RMSDvs. plot). In the case of Arg49 mutants, the most likelyexplanation of the relatively slow dynamics of decreasein the Φ-value of Asn47 is that the mutations disruptedonly the side-chain interactions leaving intact the main-chain hydrogen bonds that stabilize the abnormal confor-mation of the Asn47 residue. Accordingly, side-chaininteraction of Arg49 is not the only reason for the con-formational stability of the residue located in the II′ dis-allowed region and stability of the main-chain hydrogenbonds suggests that they might be related to the architec-ture of the entire β-hairpin 40–55. In order to elucidateother structural reasons for the conformational stability,we undertake a search of conformations similar to theconformation of the 40–55 β-hairpin of the SH3 domainin a nonredundant subset of PDB.
Descriptive statistics of the 40–55-like conformers inPDB
Using the decapeptide algorithm for the search of similarand dissimilar conformations (Petock et al., 2003), weanalyzed frequency occurrences of all decapeptide frag-ments in the structure of the SH3 domain of α-spectrin(Figure 4). The peaks on this diagram represent fre-quently occurring conformations while the minima corre-spond to the rare conformations. In all of the structurestested, 1SHG (crystallographic wt structure), 1AEY (wtNMR 15), 1QKX (N47A), 1QKW (N47G) and pseudo-wt permutants (1PWT 1TUC), the region immediatelyadjacent to Asn47 was a ‘rare conformation peptide’(residues 33–45) which is likely to be related to the pro-tein function (Petock et al., 2003).
Contrary to our initial expectation, the decapeptidewhich contained Asn47 in the middle (i.e. fragment 42–51) had a considerably high frequency of occurrence inPDB (1827 times, the peak at 42 in the Figure) thus not
0.00
0.50
1.00
1.50
2.00
2.50
3.00
3.50
0.00 20.00 40.00 60.00 80.00 100.00 120.00
t, ps
rms,
A
Q50A
R49I
wt
K43A
R49A
E45A
40
45
50
55
60
65
70
75
80
0.00 20.00 40.00 60.00 80.00 100.00 120.00
t, ps
phi(A
sn47
)
Q50A
E45A
wt
R49I
K43A
R49A
(A)
(B)
Figure 3. MD simulations of the 40–55 mutants (400K, nosolvent). (A) RMS (A
0) vs. simulation time (ps) and (B)
Φ(Asn47) (°) vs. time (ps).
204 I.Y. Torshin et al.
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

comprising a decapeptide of a rare conformation. Theseconformations were found in �1100 protein chains withmutual sequence similarity <25%. Conformations similarto the entire β-hairpin 40–55 were much less frequent(only < 400 times in PDB). However, the core of theconformation which we refer to here as an ‘alternatinglytwisted hairpin’ (i.e. the 42–51 decapeptide) is,apparently, a fragment that commonly occurs in proteinstructures. The subsequent bioinformatic analyses wererestricted to the conformation of the 42–51 decapeptide.
The abnormal values of the Φ- and Ψ-angles appearto be an inherent feature of the alternatingly twisted hair-pin conformation. Thus, 85% (1552/1827) conformationssimilar to 42–51 contained at least one residue withabnormal Φ>0 at positions +7, +8 or +9 (which corre-spond to residues 47, 48, and 49 of the SH3 domain ofα-spectrin, PDB 1shg numeration). Defining the II′ disal-lowed region as Φ(60 ± 30°), Ψ(�120 ± 30°), residues at+7, +8, and +9 were located in the II′ region in 36%(658/1827) of the conformations similar to the 42–51decapeptide. Positional analysis of the amino acids inthese 658 conformations indicated that glycines com-prised >95% of the residues in the II′ regions at all posi-
tions of the decapeptides with the exception of positions+7 and +8 (which correspond to Asn47 and Asp48 ofthe SH3 domain, respectively). At these two positions,only 67% of the residues were glycines and the rest werehydrophilic residues Asp, Asn, Glu, Lys, Arg, Ser, andGln (the order corresponds to the frequency ofoccurrence, most frequent first). Only a few residues atpositions +7 and +8 were hydrophobic.
The results of the positional analysis of amino acidsin the II′-regions we obtained clearly indicate preferencesfor certain residue types at positions +7 and +8. Theydiffer somewhat from the earlier data of Hutchinson andThornton (1994), who studied β-turns and the II′-turn inparticular. For instance, in their Table 3 (Hutchinson &Thornton, 1994) ‘positional potential’ of glycines washigher at the i+1 position of the β-turn (position +7 inour case) and at i+3 but lower at the i+2 position. Inour alternatingly twisted β-hairpins sample, glycines aremore frequent at positions +7 and +8 and considerablyless frequent at +9 (Table 4 here). The bias againsthydrophobic residues at positions +7, +8, and +9 in ourcase is even more prominent than in the previous work(Hutchinson & Thornton 1994). Thus, in alternatinglytwisted β-hairpins, the chances to find a hydrophobic res-idue at these positions are 5–8 times lower than at theother positions (P(χ2-test) < 0.005). We suppose thatthese differences are inherent in alternatingly twisted β-hairpins. At the same time, the statistics could be influ-enced by the considerable expansion of PDB since 1994.
In attempt to deduce possible sequence patterns thatmight correspond to the alternatingly twisted β-haripinconformation, the analysis of amino acid preferences wasperformed on the entire set of the alternatingly twisted β-hairpin conformations (i.e. the 1827 conformations werefound in PDB to be similar to decapeptide 42–51). Theresults are summarized in Table 4.
The results of the amino acid analysis of the alternat-ingly twisted β-hairpin conformations suggest severalconclusions. First, there was apparent prevalence of gly-
Table 4. Amino acid preferences per position of alternatingly twisted β-hairpin.
Pos. G D N E K A R S Q L I V T F Y M H C
+2 58 63 45 67 81 106 54 73 56 178 154 235 98 119 89 21 34 25+3 87 29 31 92 77 111 57 109 55 181 199 240 148 112 112 31 24 39+4 67 50 33 109 82 128 92 64 43 159 182 243 114 144 159 41 44 21+5 39 180 108 132 137 77 121 120 84 113 146 170 128 46 45 39 49 52+6 97 133 97 112 116 80 75 140 54 109 121 169 156 75 82 24 70 22+7 418 220 207 180 127 110 63 102 69 30 17 27 58 28 24 17 28 8+8 512 226 200 94 127 55 73 136 52 42 16 21 124 31 24 8 33 15+9 277 64 102 140 234 75 147 76 105 78 54 126 96 59 60 25 63 18+10 38 20 44 121 123 89 93 107 64 194 146 225 170 81 73 40 26 42+11 56 22 40 81 99 82 63 81 43 204 213 257 115 126 173 35 47 24
Notes:+2 corresponds to residue 42 of the SH3 domain of α-spectrin, +7 to residue 47, etc. Numbers in cells are the total number of occurrences ofthe amino acid type at a specific position in the 40–55 alternatingly twisted β-hairpin.
Figure 4. Occurrence of the decapeptide conformations of theSH3 domain in a nonredundant PDB subset. Abscissa, residuenumber (1 corresponds to the decapeptide 1–10 of SH3, etc);ordinate, number of hits.
Disallowed conformations in polypeptide chain 205
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

cine residues in at least one of the three positions +7, +8and +9 which correspond to the β-turn of the hairpin.Indeed, 76% (1253/1649) of all glycines that occurred atall positions in 1827 decapeptides occurred at these threepositions. The presence of a glycine in at least one posi-tion of the β-turn makes the structure more flexible thusallowing the β-hairpin to twist during folding of thiscompact module. In particular, the β-hairpin can twistinto a conformation that belongs to the disallowed II′region. What forces could twist the hairpin? Main-chainH-bonds between the strands pull the strands togetherwhile van der Waals interactions of the side chains resistexcessive closing of the two strands. Therefore, twistingof the hairpin is one of the ways to saturate van derWaals contacts of side chains without loosing the main-chain H-bonds that stabilize the entire β-hairpin struc-ture. Second, the residues at the three positions +7, +8,and +9 were either glycines or hydrophilic residues butrarely hydrophobic: hydrophobic residues comprised<14% of the residues at position +7,<15% at position+8, and <18% at position +9.
In this connection, it is noteworthy that while no sta-bilizing effects were found for the Asn47Ala and theAsn47Gly mutations, the Asp48Gly substitution pro-duced a significant stabilizing effect on the SH3 domain(Martinez, Pisabarro, & Serano, 1998). The alternatinglytwisted β-hairpins concept allows an explanation of thisexperimental fact. As evident from Table 4, in alternat-ingly twisted β-hairpins, it is the glycines that are mostpreferred at the +8 position. The Asp48Gly mutation pro-duced by Martinez et al. (1998) corresponds to placingthe type of residue preferred for alternatingly twistedβ-hairpins – the ‘flexible’ glycine – into position +8. Theglycine at this position relaxes the conformational restric-tions that a β-turn imposes on the whole β-hairpin. As aresult, the alternatingly twisted β-hairpin becomes morecompact, stabilizing the entire structure of the SH3domain.
Using the data in the table, it was possible to formu-late a number of sequence patterns that could distinguishbetween alternatingly twisted β-hairpins and other con-formations. Such patterns were not very sensitive(detected less than a half of the 1827 hairpins) and hadlow specificity (false positive rate of 25% when testedon random sequences). However, application of thesepatterns to the entire set of decapeptides from PDByielded statistically significant results. For example, thepattern [ADEIKLNSTVY] – [ADEKNRSG] – [DAK-NSTG] – [RKESTVWG] – [AEFGIKLSTV] (corre-sponding to positions +6...+10) was 1.6-fold more likelyto occur in an alternatingly twisted β-haripin than in anyother conformational fragment (OR= 1.6, 95% CI 1.4–1.8, χ2 p < 10�10).
Analysis of the SCOP classes of 1100 protein chainsthat contained the 1827 alternatingly twisted β-hairpins
indicated that conformations similar to the alternatinglytwisted β-hairpin of the SH3 domain is a commonphenomenon: it occurs in at least 30% of the proteinstructures in the nonredundant set and in various kindsof the protein architectures (Table 5).
Chirality index analysis of alternatingly twistedβ-hairpins
A chirality index is the base of a methodology fordescription of the secondary structure of proteins (Pietro-paolo et al., 2008). Roughly, the chirality index describesthe twist of the protein backbone and from the values ofthe index along the sequence, it is possible to recognizemany structural motifs of the protein. Chirality calcula-tions are based on assigning a chirality parameter to thebackbones of short amino acid sequences (pentapeptides)and as such, the chirality index is not a good measure ofthe overall hairpin topology (decapeptides or greater). Inorder to evaluate the extent of twisting in various ele-ments of the secondary structure including alternatinglytwisted β-hairpins, we performed calculations of the chi-rality index on various sets of decapeptides. The distribu-tions of the chirality index values are presented inFigure 5.
Figure 5 compares distributions of the chirality indexof decapeptides that correspond to β-hairpin, alternating-ly twisted β-hairpin, and α-helices. The left-handed poly-proline helices were too short to be given in the same
Table 5. Structural classification of structural domains in 1100protein chains that contained alternatingly twisted β-hairpins.
SCOP classificationNumberof folds
Number of domainrepresentatives
A ‘all alpha’ 27 65B ‘all beta’ 178 527C ‘a/b’ 120 302D ‘a + b’ 184 464E ‘multi-domain’ 21 48F ‘membrane’ 11 31G ‘small’ 39 105
Total 580 1542
Figure 5. Distributions of the chirality index values indifferent elements of the secondary structure.
206 I.Y. Torshin et al.
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

figure that shows α- and β-types of the structure. Thevalue of the chirality index for poly-proline helices ofmore than four residues (e.g. 1JMQ 50-58) varied from0.06 to 0.11 (the right side of the Figure). On the whole,the results indicate that the chirality index is a goodshort-range measure of the backbone twist. The analysisrevealed that the distribution of the chirality index of thealternatingly twisted β-hairpin (Figure 5) differs from thatof other kinds of secondary structure elements. In particu-lar, alternatingly twisted β-hairpins had a considerableprevalence of positive values of the chirality index. More-over, the decapeptides with the chirality index rangingfrom �0.01 to +0.015 were 11 times more likely to corre-spond to an alternatingly twisted β-hairpin than to anyother kind of the secondary structure (OR= 11.04; 95%9.25–14.2; χ2 p < 10�20). Therefore, the fragment calledhere as an ‘alternatingly twisted β-hairpin’ (including the40–55 fragment of the SH3 domain) has chiral propertiesdistinct from other types of the secondary structure.
Hydrogen bonding patterns of alternatingly twistedhairpins
Analysis of the similarity of both the conformation(Figure 4) and chirality index (Figure 5) suggests thatalternatingly twisted β-hairpins are indeed geometricallydifferent from conventional β-hairpins. Analysis of theamino acid contents (Table 4) suggests that the highproportion of glycines in β-turns leads to greater flexibil-ity and additional twisting of the hairpin. In order toelucidate further differences of the alternatingly twisted
hairpins, we compared hydrogen bonding patterns inconventional β-hairpins (which have not violations of theΦ and Ψ angles) and in the set of 1552 alternatinglytwisted hairpins (that contained at least one residue atpositions + 7, +8, or +9 with Φ>0). For each β-hairpin,hydrogen bonds were found and the frequencies ofoccurrence (occupancy) of the chain hydrogen bondbetween all pairs of residues were calculated. The resultsare presented in Figure 6 which shows only those main-chain hydrogen bonds that occurred in at least 20% ofall studied hairpins (20% occupancy).
On average, each β-hairpin in the set had from 5 to 8main-chain hydrogen bonds. This was the case of boththe conventional and alternatingly twisted hairpins. How-ever, the hydrogen bonding patterns were distinctly dif-ferent. While in the both types of hairpins, the hydrogenbonding (+3, +12), (+4, +11), (+5, +10), and (+6, +9)occurred with comparable frequencies, the alternatinglytwisted β-hairpins lacked the hydrogen bonding withoccupancies >20%. At the same time, there was a muchgreater amount of hydrogen bonding with occu-pancy >20% in the β-turn of the conventional β-hairpin.This observation suggests that conventional β-hairpinsare much more rigid and more uniform in their structure.In part, this conclusion is supported by the difference inthe half-width (the peak width at half-height) of the twopeaks in the chirality index distributions: the super-twisted hairpin apparently had a greater half-width thanconventional ones (Figure 5). Another differencebetween the two types of β-hairpins is that in the case ofa conventional one, the residue in position +5 apparentlyserves as a ‘hub’ for hydrogen bonds to positions +7, +8,+9, and +10. At the same time, in an alternatinglytwisted hairpin, the residue in position +5 forms hydro-gen bonds mostly to positions +9 and +10. A hydrogenbond between positions +6 and +8 appears to be a uniquefeature of alternatingly twisted β-hairpins.
Conclusions
The II′ region of the Ramachandran plot (Φ > 0, Ψ< 0) isdisallowed for nonglycine residues because of steric con-siderations. Interestingly, while most of the residueslocated in the II′ region are glycines, nonglycine residuesdo occur in the II′ region rather often. In order to investi-gate possible biophysical underpinnings of the occurrenceof the nonglycine residue types, we undertook a study of aparticular case (residue Asn47 of the SH3 domain of α-spectrin located within β-hairpin 40–55) followed by abioinformatic analysis of the occurrence of similar struc-tural fragments in PDB. Molecular dynamics of the SH3domain, alanine scanning, and other in silico studies of β-hairpin 40–55 of the SH3 domain (including analysis ofthe static and dynamic parts of the hydrogen bonding net-work and in silico mutagenesis) indicated that stability of
Figure 6. Hydrogen bonding (main chain) in conventionaland alternatingly twisted hairpins. The ‘occurrences’ of eachhydrogen bond are shown as percentages. Right, the hydrogenbonding within the set of 1552 alternatingly twisted hairpinswith Φ>0. Left, hydrogen bond patterns in the set of 1500regular hairpins.
Disallowed conformations in polypeptide chain 207
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14

the hairpin was maintained despite the steric hindrancebecause of the dynamic hydrogen bonding network whichcan be disrupted by targeted mutations of the Arg49 resi-due. In the course of computations, we delineated a mini-mal segment included residues 40–55 that retains itsproperty to stabilize the disallowed conformation ofAsn47 in the β-turn. Bioinformatic analyses indicated thatconformations similar to β-hairpin 40–55 occur quite oftenin PDB, and are characterized by a higher concentration ofglycine residues in their β-turns. The presence of glycinesmakes the entire β-hairpin structure more flexible to thetwisting effects of cooperation between the main-chainhydrogen bonding and packing. We call this fragment asan ‘alternatingly twisted β-hairpin’ and demonstrate thatsuch fragments tend to have chiral properties and prefer-ences in hydrogen bonding patterns distinct from conven-tional β-hairpins without violations of the Φ and Ψ angles.Thus, an alternatingly twisted β-hairpin may be treated asa global factor supporting the existence of a nonglycineresidue in the II′ region.
Acknowledgements
This work was supported by a grant from the ‘Molecular andCellular Biology’ Program of the Presidium RAS and by StateContract of Russian Ministry of Science and Education(07.514.11.4006).
ReferencesBerman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat,
T. N., Weissig, H., … Boume, P. E. (2000). The proteindata bank. Nucleic Acids Research, 28, 235–242.
Bertini, I. (2003). A use of Ramachandran potentials in proteinsolution structure determinations. Journal of BiomolecularNMR, 26, 355–366.
Cuendet, M. A., & van Gunsteren, W. F. (2007). On the calcu-lation of velocity-dependent properties in molecular dynam-ics simulations using the leapfrog integration algorithm.Journal of Chemical Physics, 127, 184102–184110.
Daggett, V. (2002). Molecular dynamics simulations of the pro-tein unfolding/folding reaction. Accounts in ChemicalResearch, 35, 422–429.
Davis, M. E., & McCammon, J. A. (1990). Electrostatics inbiomolecular structure and dynamics. Chemical Reviews,90, 509–521.
Eisenberg, D., & McLachlan, A. D. (1986). Solvation energyin protein folding and binding. Nature, 319, 199–203.
Gibrat, J. F., Madej, T., & Bryant, S. H. (1996). Surprisingsimilarities in structure comparison. Current Opinion inStructural Biology, 6, 377–385.
Gunasekaran, K. (1996). Disallowed Ramachandran conforma-tions of amino acid residues in protein structures. Journalof Molecular Biology, 264, 191–198.
Gunasekaran, K. (2007). How different are structurally flexibleand rigid binding sites? Sequence and structural featuresdiscriminating proteins that do and do not undergo confor-mational change upon ligand binding. Journal of MolecularBiology, 365, 257–273.
Hooft, R. W., Vriend, G., Sander, C., & Abola, E. E. (1996).Errors in protein structure. Nature, 381, 272–272.
Hutchinson, E. G., & Thornton, J. M. (1994). A revised set ofpotentials for β-turn formation in proteins. Protein Science,3, 2207–2216.
Lovell, S. C. (2003). Structure validation by Cα geometry: u, ψand Cβ deviation. Proteins: Structure, Function, and Bioin-formatics, 50, 437–450.
Martinez, J. C., Pisabarro, M. T., & Serano, L. (1998). Obliga-tory steps in protein folding and the conformational diver-sity of the transition state. Nature Molecular Biology, 5,721–729.
Ohage, E. C. (1997). β-Turn propensities as paradigms for theanalysis of structural motifs to engineer protein stability.Protein Science, 6, 233–241.
Oliva, B. (1997). An automated classification of the structureof protein loops. Journal of Molecular Biology, 266, 814–830.
Pal, D. (2002). On residues in the disallowed region of theRamachandran map. Biopolymers, 63, 195–206.
Petock, J. M., Torshin, I. Y., Weber, I. T., & Harrison, R.W. (2003). Analysis of protein structures reveals regionsof rare backbone conformation at functional sites. Pro-teins: Structure, Function, and Bioinformatics, 53, 872–879.
Pietropaolo, A., Muccioli, L., Berardi, R., & Zannoni,C. (2008). A chirality index for investigating proteinsecondary structures and their time evolution. Proteins:Structure, Function, and Bioinformatics, 70, 667–677.
Ramachandran, G. N., & Sasisekharan, V. (1968). Conforma-tion of polypeptides and proteins. Advances in ProteinChemistry, 23, 283–438.
Ramakrishnan, C. (2007). Structural compromise of disallowedconformations in peptide and protein structures. Proteinand Peptide Letters, 14, 672–682.
Rappé, A. K., & Goddard, W. A. (1991). Charge equilibrationfor molecular-dynamics simulation. The Journal of PhysicalChemistry, 95, 3358–3363.
Torshin, I. (1999). Activating oligomerization as intermediatelevel of signal transduction: Analysis of protein–proteincontacts and active sites in several glycolytic enzymes.Frontiers in Bioscience Journal, 4, D557–570.
Torshin, I. Y. (2004). Computed energetics of nucleotides inspatial ribozyme structures: An accurate identification offunctional regions from structure. Scientific World Journal,4, 228–247.
Torshin, I. Y., Weber, I. T., & Harrison, R. W. (2002). Geomet-ric criteria of hydrogen bonds in proteins and identificationof ‘bifurcated’ hydrogen bonds. Protein EngineeringDesign and Selection, 15, 359–363.
van der Kamp, M. W., & Daggett, V. (2011). Moleculardynamics as an approach to study prion protein misfoldingand the effect of pathogenic mutations. Topics in CurrentChemistry, 305, 169–197.
Vega, M. C., Martinez, J. C., & Serrano, L. (2000). Thermody-namic and structural characterization of Asn and Ala resi-dues in the disallowed II′ region of the Ramachandran plot.Protein Science, 9, 2322–2328.
Viguera, A. R., Jimenez, M. A., Rico, M., & Serano,L. (1995). Conformational analysis of peptides correspond-ing to β-hairpins that represent the entire sequence of α-spectrin SH3 domain. Journal of Molecular Biology, 255,507–521.
208 I.Y. Torshin et al.
Dow
nloa
ded
by [
Mos
kow
Sta
te U
niv
Bib
liote
] at
06:
29 2
8 Ja
nuar
y 20
14