Embed Size (px)
Citation preview

Analyse von Gendefekten bei Autoinflammation und Immundefizienz
Prof. Dr. med. Angela Rösen-Wolff
Klinische Forschung Klinik und Poliklinik für Kinder- und Jugendmedizin
Universitätsklinikum Carl Gustav Carus TU Dresden

Systemische autoinflammatorische Erkrankungen
• anscheinend grundlose Entzündungsepisoden
• Fieberepisoden
• Hautbefunde
• häufig Muskel- und Gelenkbeteiligung
• keine Autoantikörper

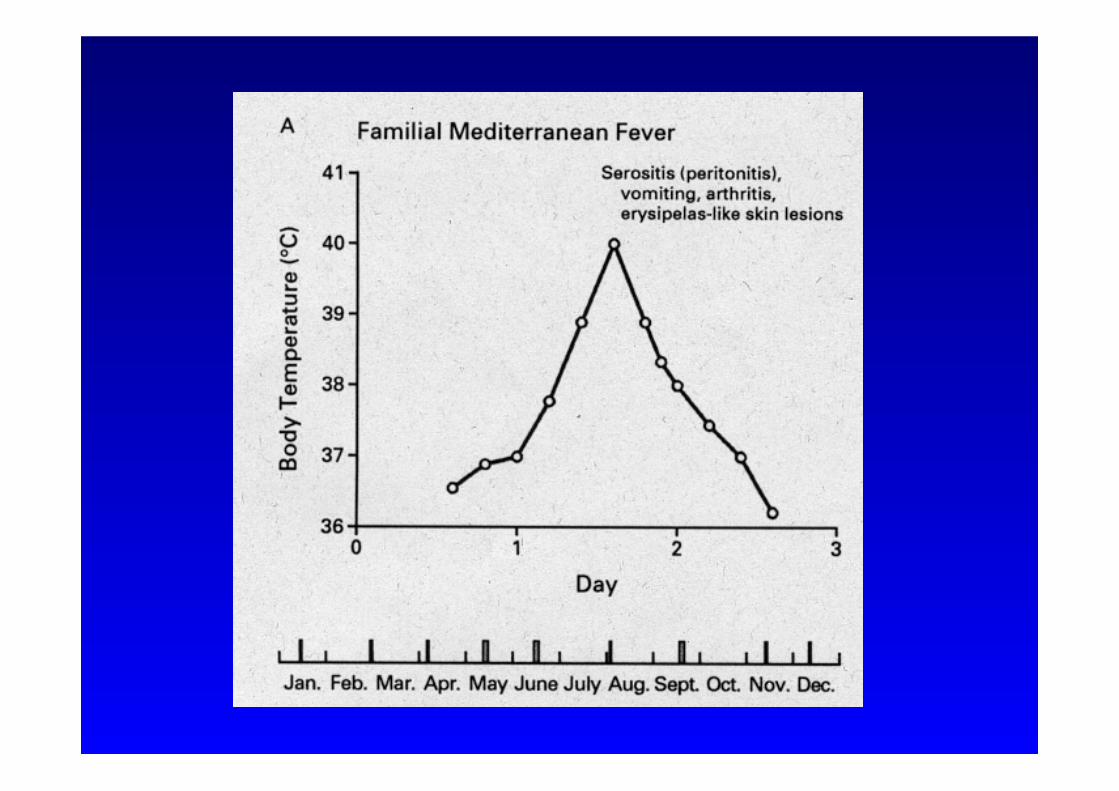
Familiäres Mittelmeerfieber
Erstbeschreibung 1908 durch Janeway und Mosenthal
Symptome: rekurrierende Bauchschmerzen und Fieber
• Kurze Attacken von Serositis (Peritonitis, Pleuritis, Arthritis)
• Beginn der Attacken: <20 Jahre
• Plötzliches Einsetzen der Symptome
• Kurze Dauer: 6 bis 96 Stunden

Familiäres Mittelmeerfieber
Symptome:
95% Bauchschmerzen / akutes Abdomen manchmal milde Beschwerden
75% Monarthritis / Gelenkserguss selten chronische destruierende Arthritis
30% Brustschmerzen / einseitige Pleuritis
1% Pericarditis
Junge männliche Patienten (<20 Jahre): akute Skrotalschwellung

Familiäres Mittelmeerfieber
Genetik und Epidemiologie:
• Autosomal rezessive Erkrankung
• Häufigste periodische Fiebererkrankung (weltweit >10.000 Patienten)
• Menschen mediterraner Herkunft gehäuft betroffen (Sephardische Juden, Araber, Türken, Armenier, seltener Griechen, Italiener)

Familiäres Mittelmeerfieber
Molekulargenetik:
• Kurzer Arm Chromosom 16
• MEFV, pyrin, marenostrin
• 781 Aminosäuren, 86 kD
• >30 Mutationen beschrieben
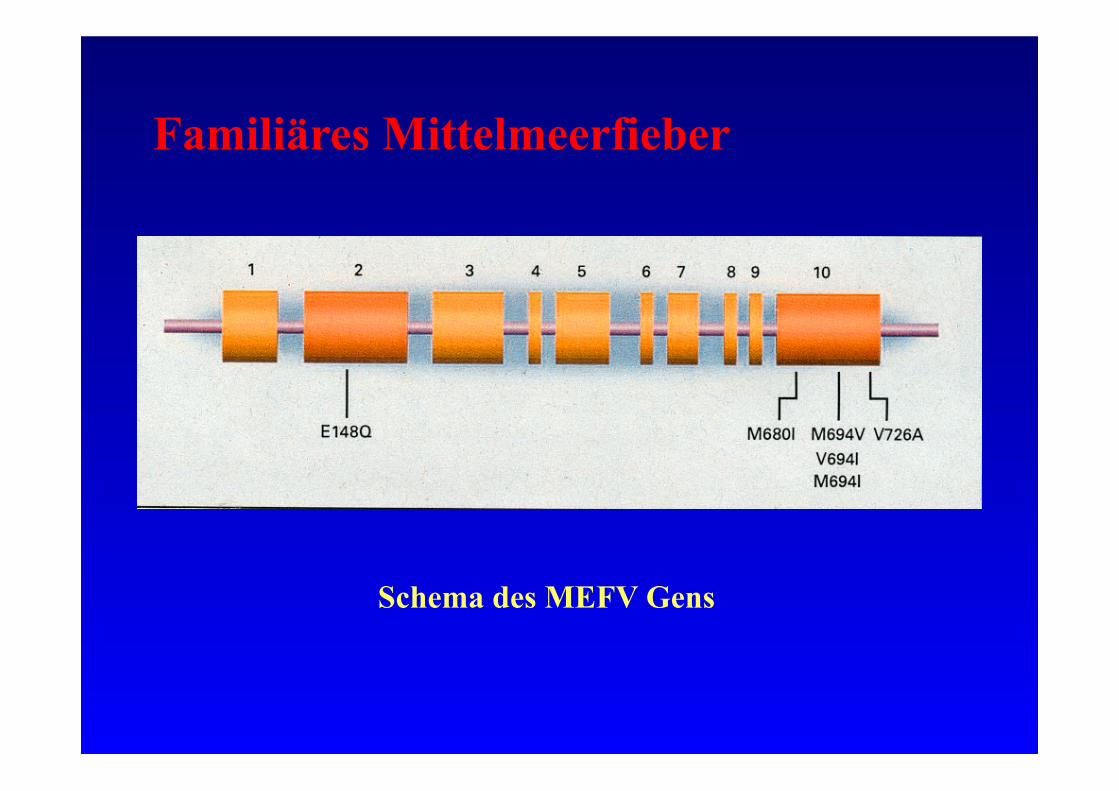
Familiäres Mittelmeerfieber
Schema des MEFV Gens

Familiäres Mittelmeerfieber
Pathogenese:
• Expression des Pyrin in myeloischen Zellen • Während Differenzierung hochreguliert • Expression wird durch Interferon-γ und TNF-α stimuliert • Funktion: antiinflammatorisch / interagiert mit Caspase-1 (pro-inflammatorisch?)

Familiäres Mittelmeerfieber
Therapie:
• Mittel der Wahl: Colchicin • Wirksamkeit in 2 kontrollierten klinischen Studie bestätigt • Dosierung: 1 mg/Tag (Steigerung möglich) • Verhindert bei 60% der Patienten die Fieberschübe • Bei 20-30% wesentliche Reduktion der Fieberschübe • Compliance sehr wichtig!!! • Absetzen der Medikation kann Fieberschub auslösen • Nebenwirkungen: Diarrhoe Myopathie, Neuropathie, Leukopenie (selten)
Im akuten Schub keine Wirkung von Colchicin Zur Schmerztherapie: Diclofenac

Familiäres Mittelmeerfieber
Prognose:
• abhängig von der Ausbildung einer Amyloidose • ohne Amyloidose normale Lebenserwartung
• vor der Colchicin Ära: Amyloidose bei 60% der Patienten (>40 Jahre)
• selbst wenn Colchicin die Fieberattacken nicht reduziert:
Amyloidose kann verhindert werden!!!

Hyper-IgD Syndrom
1984 - Erkennung als separate Entität vorher als Variante des Still-Syndroms beschrieben
Symptome:
• Rekurrierende Fieberschübe • Beginn <1 Jahr • Schüttelfrost • Steiler Fieberanstieg • Dauer: 4-6 Tage • Provokation durch: Impfung, Trauma, OP, Stress
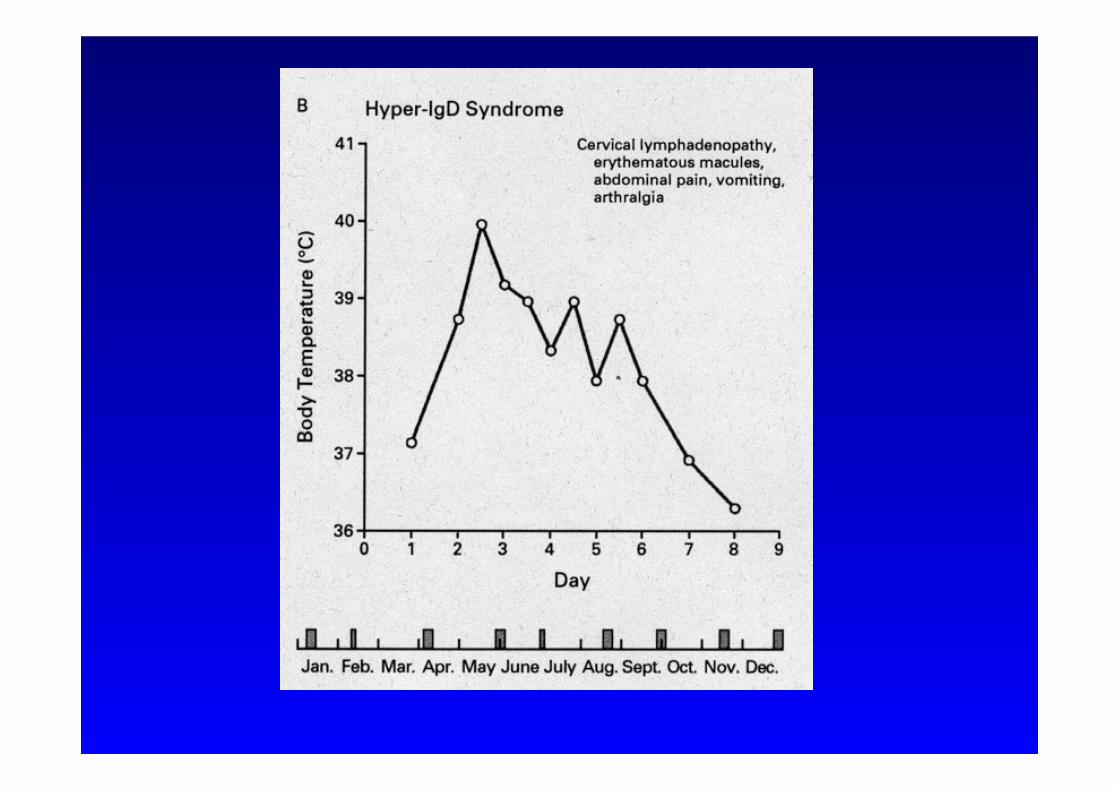

Hyper-IgD Syndrom
• Begleitsymptome:
• zervikale Lymphadenopatie • Bauchschmerzen mit Erbrechen • Diarrhoe
• Hepatosplenomegalie • Kopfschmerzen • Arthralgien • Erythem • Petechien • Purpura
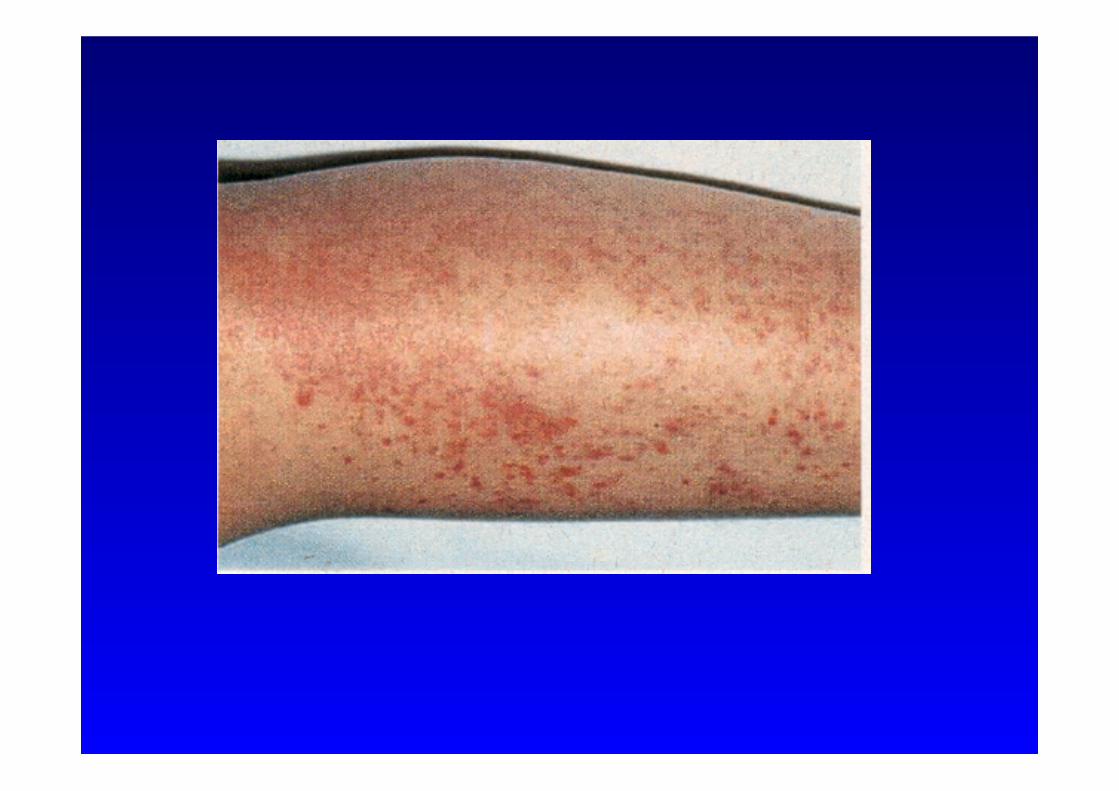

Hyper-IgD Syndrom
Paraklinik:
• IgD >100 IU/ml (Ausnahmen bei sehr jungen Patienten) • IgA Spiegel erhöht (>80% der Patienten) • Leukocytose • CRP erhöht • Serum Amyloid A erhöht
• Urin: Mevalonsäure erhöht (im Fieberschub)

Hyper-IgD Syndrom
Genetik und Epidemiologie:
• Vererbung autosomal rezessiv • weltweit ca. 200 Fälle bekannt • bevorzugt weiße Westeuropäer betroffen • 60% Holländer oder Franzosen
• Lokalisation: langer Arm Chromosom 12
• Mevalonatkinase-Gen: Hot-Spot V377I reduziert Aktivität des Enzyms

Hyper-IgD Syndrom
Pathogenese:
Exakte Pathogenese bislang nicht aufgeklärt!
• Mevalonatkinase: Schlüsselenzym der Cholesterinsynthese • <15% der normalen Aktivität • Serum Cholesterinwerte leicht erniedrigt
• <1% der Patienten: komplette Defizienz Mevalonacidurie seltene Stoffwechselerkrankung - Entwicklungsverzögerung - Hypotonie, Ataxie, Myopathie - Katarakt

Hyper-IgD Syndrom
Therapie:
• Keine einheitliche Therapie empfohlen • Anekdotische Therapieerfolge mit: Steroiden Immunglobulinen i.m. Colchicin Cyclosporin Anakinra Prognose:
• Lebenslange Fieberschübe • Reduzierung mit zunehmendem Lebensalter • selten Amyloidose • Bei Arthritiden: Gelenksdestruktion sehr selten

TNF-α Rezeptor assoziiertes periodisches Fiebersyndrom (TRAPS)
Erstbeschreibung 1982 in einer großen Irischen Familie zunächst Hibernian Fever genannt
Symptome:
• Rekurrierende Fieberschübe • Myalgien • Erythem • Bauchschmerzen, Diarrhoe, Obstipation, Erbrechen • Konjunktivitis • Dauer: > 1 Woche
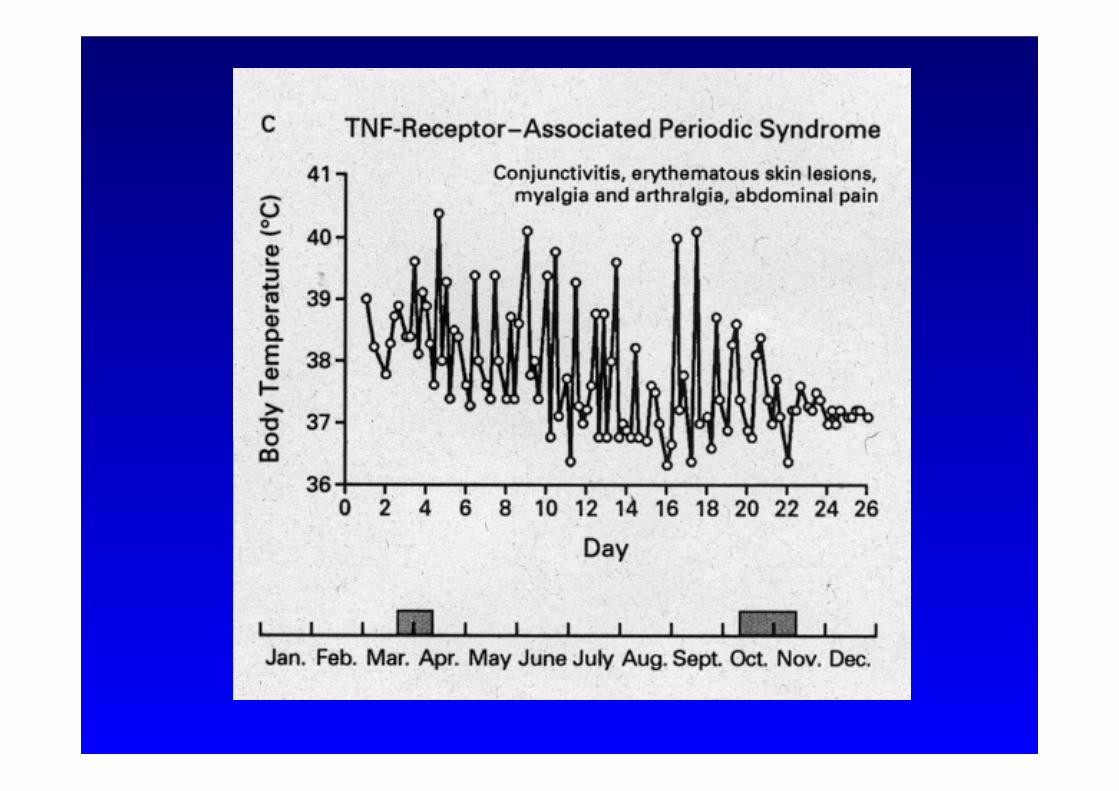

TNF-α Rezeptor assoziiertes periodisches Fiebersyndrom (TRAPS)
Paraklinik:
• Neutrophilie • CRP erhöht • IgA erhöht • IgD erhöht <100 IU/ml
• Löslicher TNF-α Rezeptor erniedrigt

TNF-α Rezeptor assoziiertes periodisches Fiebersyndrom (TRAPS)
Genetik und Epidemiologie:
• Autosomal dominante Vererbung • mehr als 50 Familien beschrieben • verschiedene ethnische Gruppe
• Kurzer Arm Chromosom 12 • TNF-α Rezeptor 1 (TNFR1; TNFRSF1A)

TNF-α Rezeptor assoziiertes periodisches Fiebersyndrom
(TRAPS)
Pathogenese:
• Aktivierung des TNFR1 induziert Rezeptor-Shedding • Abspalten des Rezeptors von der Zellmembran • Löslicher Rezeptor fungiert als Antagonist
• Mutationen führen zu reduziertem Rezeptor-Shedding • Überexpression in der Zellmembran • Kontinuierlicher Stimulierung durch TNF-α • Unkontrollierte Entzündung
• Reduzierte Apoptose aktivierter Zellen
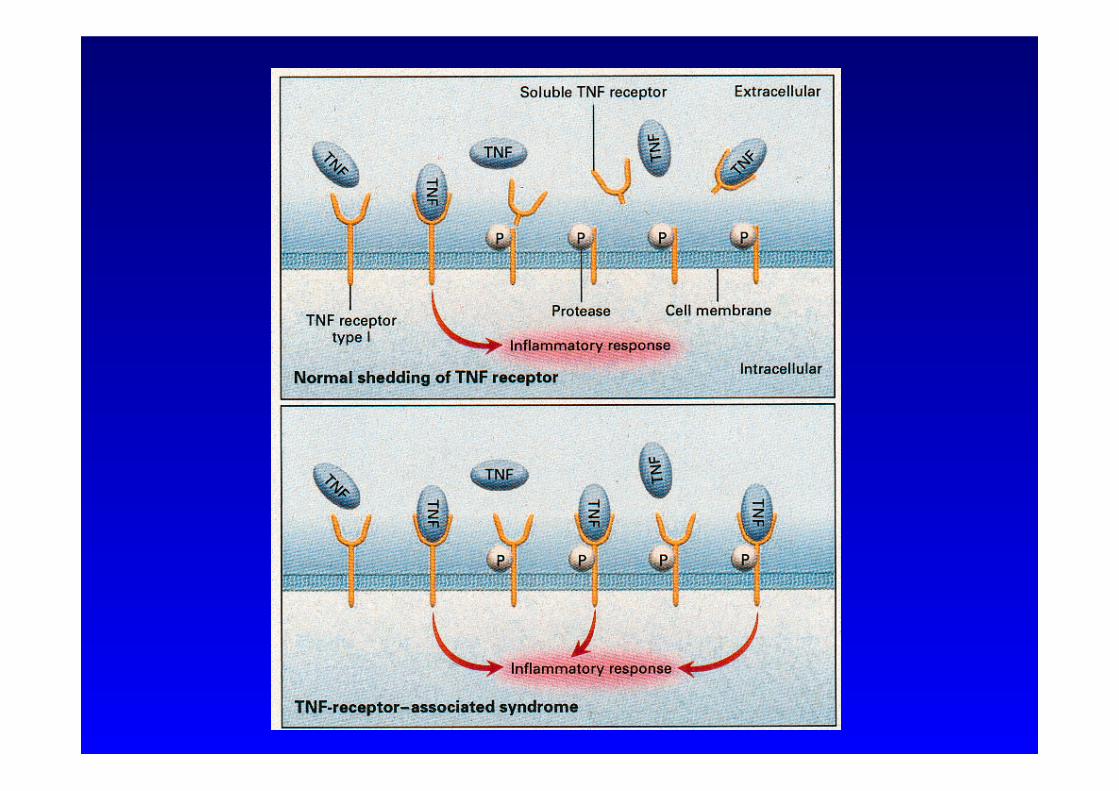

TNF-α Rezeptor assoziiertes periodisches Fiebersyndrom
(TRAPS)
Therapie:
• Ethanercept (rekombinanter löslicher TNFR2; konjugiert an Fc-Fragment des humanen IgG) bindet freies und membrangebundenes TNFα 25 mg 2 x pro Woche

TNF-α Rezeptor assoziiertes periodisches Fiebersyndrom
(TRAPS)
Prognose:
• wird von der Ausbildung einer Amyloidose bestimmt • 25% entwickeln Amyloidose • Nierenversagen (Proteinurie als initiale Manifestation) • Leberversagen

Periodisches Fieber ohne Fieber
• monosymptomatische Nierenamyloidose bei einer TNFR1 mutations-tragenden ansonsten unauffälligen Schwester eines TRAPS Patienten (Kallinich et al., Ann Rheumatol Dis 2005, epub ahead of print)
• Periodisches Auftreten einer Psychose ohne Fieber bei nachgewiesener TRAPS induzierender TNFR1 Mutation (Hurst et al., J Clin Rheumatol 11, 329-330, 2005)

PAPA Syndrom: Pyogene sterile Arthritis, Pyoderma gangraenosum, Akne Mutationen im CD2BP1 Gen (Chromosom 15) Blau Syndrom: granulomatöse Arthritis Uveitis Exanthem Mutationen im CARD15 Gen (Chromosom 16) ebenfalls bei M. Crohn
PFAPA: Periodisches Fieber, aphthöse Stomatitis, Pharyngitis, cervikale Lymphadenitis keine familiäre Häufung (Gen unbekannt)

Cryopyrin-assoziierte periodische Fiebersyndrome (CAPS):
Familiäre Kälteurtikaria: autosomal dominant Beginn im ersten Lebensjahr Kälte induziertes nicht-juckendes Exanthem Schüttelfrost, Fieber Arthralgien, Myalgien, Kopfschmerzen Amyloidose (selten) Therapie: Kälteexposition meiden Muckle-Wells Syndrom: s.o. Kälte triggert Symptome nicht sensoneurale Taubheit Therapie: keine CINCA-Syndrom: Chronic Infantile Neurological Cutaneous
and Articular Syndrome - chronische aseptische Meningitis - Urtikaria - Arthritis Therapie: Nicht-steroidale Antiphlogistika, Steroide,
Mutationen im CIAS1 Gen (Chromosom 1)

CIAS1 kodiert für Cryopyrin:
• Aminoterminale Pyrin-Domäne
• Carboxyterminale leucinreiche Domäne (LRR)
• Zentrale Nukleotidbindungsstelle gehört zur NACHT Unterfamilie der
NTPasen - reguliert die Aktivierung von NFkB
Mutationen in NACHT Domäne zerstören putative Nukleotidbindungstelle
Expression von CIAS1 hauptsächlich in Leukozyten und Chondrozyten
Dramatische Anstiege von IL-1b, IL-6, TNFalpha, IL-3 und IL-5 bei Patienten
CIAS1 Pyrin-Domäne interagiert mit Apoptoseprotein ASC
- Interaktion zwischen ASC und Caspase 1 (IL-1 converting enzyme)
- erhöhte IL-1b Produktion (IL-1Ra ebenfalls erhöht)
potentieller Ansatz für Therapie
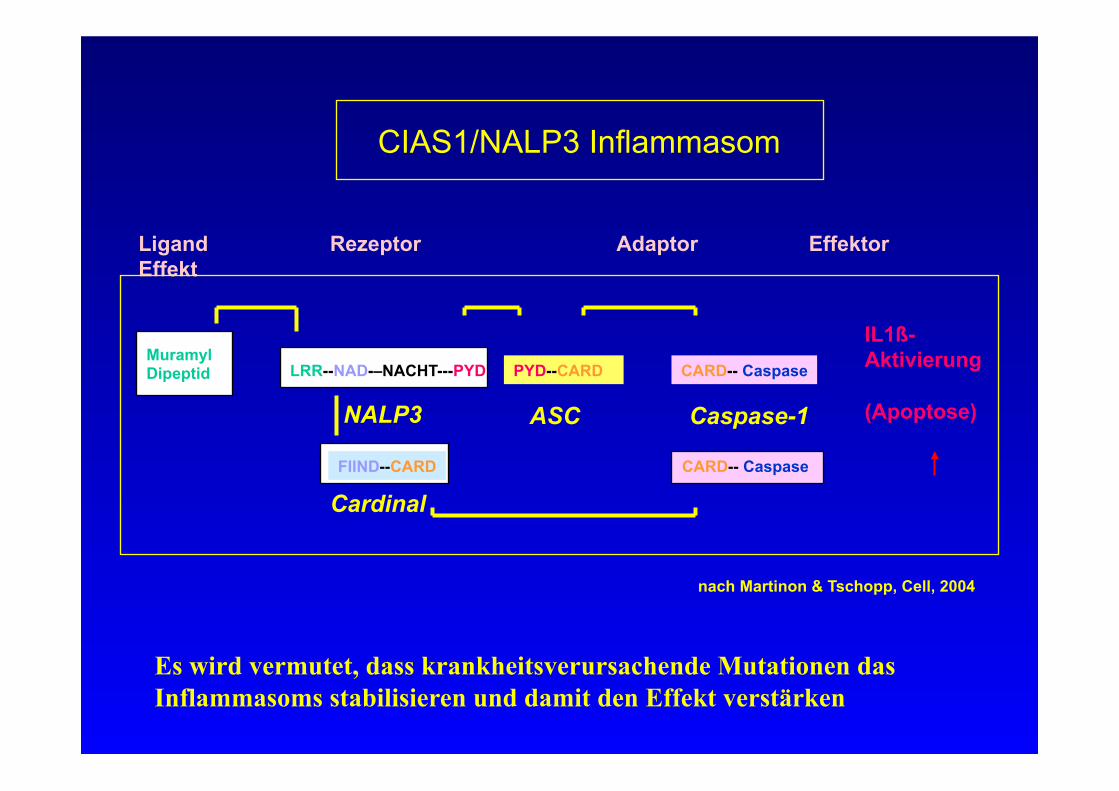
PYD--CARD
CIAS1/NALP3 Inflammasom
Ligand Rezeptor Adaptor Effektor Effekt
Muramyl Dipeptid LRR--NAD-–NACHT---PYD
NALP3
CARD-- Caspase
ASC Caspase-1
IL1ß- Aktivierung
(Apoptose)
CARD-- Caspase FIIND--CARD
Cardinal
nach Martinon & Tschopp, Cell, 2004
Es wird vermutet, dass krankheitsverursachende Mutationen das Inflammasoms stabilisieren und damit den Effekt verstärken
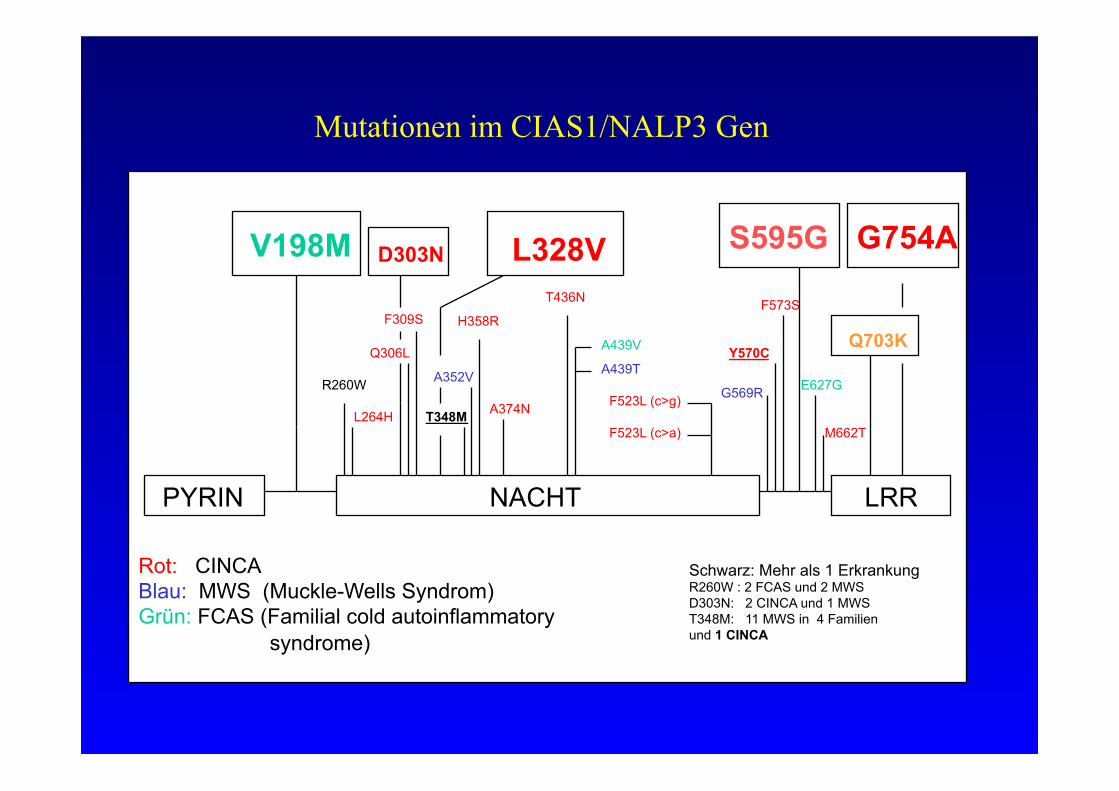
PYRIN NACHT LRR
V198M
R260W
L264H
D303N
Q306L
F309S
T348M
A352V
H358R
A374N
T436N
A439V
A439T
F523L (c>g)
F523L (c>a)
G569R
Y570C
E627G
M662T
F573S
Rot: CINCA Blau: MWS (Muckle-Wells Syndrom) Grün: FCAS (Familial cold autoinflammatory syndrome)
Schwarz: Mehr als 1 Erkrankung R260W : 2 FCAS und 2 MWS D303N: 2 CINCA und 1 MWS T348M: 11 MWS in 4 Familien und 1 CINCA
Mutationen im CIAS1/NALP3 Gen
S595G
Q703K
L328V G754A

Therapiestudien
2 Patienten (Mutation R260W) Interleukin-1 Rezeptor Antagonist (Anakinra, Kineret, Amgen) Dosierung: 100 mg / Tag s.c.
Effekte: • Entzündung kam innerhalb von Stunden zum Stillstand • Serum Amyloid A Konzentrationen sanken auf Normalwerte • Proteinurie kam zum Stillstand • Therapieerfolg seit Jahren konstant (Hawkins et al. 2003, NEJM 348; 2583-2584)
Therapie wird nun auch bei CINCA und Familiärer Kälteurtikaria eingesetzt (Goldbach-Mansky et al. 2006, NEJM 355; 581-592)

Systemische autoinflammatorische Erkrankungen
• anscheinend grundlose Entzündungsepisoden
• Fieberepisoden
• (Hautbefunde)
• häufig Muskel- und Gelenkbeteiligung
• keine Autoantikörper
Diagnosestellung: Molekulargenetik

Warum Molekulargenetik bei Immundefekten?
• Kombination mit funktionellen Tests bzw. Phänotypisierung
• Genetische Beratung
• Pränatale Diagnostik bei weiterem Kinderwunsch
• Überträgerinnen bei X-chromosomal vererbten Erkrankungen
• Untersuchung weiterer Familienmitglieder





























