Embed Size (px)
Citation preview

Cuore
Sede: mediastino antero-inferiore
Rapporti: Sup.→ carena tracheale Inf.→ diaframma Post.→ esofago Ant.→ sterno, lingula
Dimensioni: peso (M/F): 300g ± 50 Asse longitud.: 12 cm “ trasversale: 8 cm spessore delle pareti: Vsn: 15mm Vdx: 5 mm Atri: 2.5 mm
Il peso e le dimensioni del cuore variano quando si debba trovare a fronteggiare un postcarico o un precaricomaggiore: in generale l’↑ postcarico produce almeno inizialmente una ipertrofia concentrica con ↑ spessore dellepareti senza ↑ volume delle cavità cardiache; invece l’ ↑ precarico produce una ipertrofia eccentrica condilatazione della cavità e ↓ spessore delle pareti.
- Ipertensione polmonare e cardiopatia ischemica portano a un raddoppiamento del peso (600g)- Ipertensione arteriosa e stenosi aortica (conc.), insufficienza mitralica e CMPD (ecc.) portano a un ↑di
peso di 2-3 volte → 800 g- Insufficienza aortica (ecc.) e CMPI (conc.) portano a un aumento di oltre 3 volte: 1000 g!
vascolarizzazione: il cuore è vascolarizzato dalle coronarie dx e sn che si dipartono bilateralmente dal bulboaortico a livello dei seni di Valsalva subito al di sopra delle semilunari → una embolizzazione si ha solo perendocardite delle semilunari. I territori di competenza sono:
coronaria sn: dopo un breve tratto si divide in 2 rami:- discendente anteriore sn che irrora la parete anteriore del cuore e i 2/3 anteriori del setto interventricolare e
proseguendo arriva all’ apice e lo abbraccia fino arrivando ad irrorare la parte più anteriore della pareteposteroinferiore.
- circonflessa sn che percorrendo il solco coronario manda un ramo per il margine ottoso e continuaposteriormente. Solo nel 20% dei casi fornisce il ramo discendente posteriore.
Coronaria dx: decorre nel solco coronario e manda rami per le pareti del ventricolo dx e una volta arrivata allacrux fornisce nell’80% dei casi la discendente posteriore che irrora il 1/3 posteriore del setto oltre che unaporzione della parete posteriore del Vsn: in definitiva nell’80% dei casi è questa la coronaria dominante.
IL PERICARDIO consta di un foglietto viscerale o epicardio sottile e trasparente e di uno parietale, più spesso efibroso, che è in continuità col primo e che prende il via dai grossi vasi poco dopo la loro origine e si disponequindi a formare il sacco pericardico.I 2 foglietti sono rivestiti internamente da MESOTELIO.Il pericardio ha funzioni meccaniche:1)di contenimento2)di fissità del cuore attraverso i legamenti sterno-p. e freno-p.3)di scivolamento delle superfici pericardiche l’una sull’altra durante la pulsazione del cuorecome si vedrà i processi patologici possono portare a uno sconvolgimento di queste funzioni.
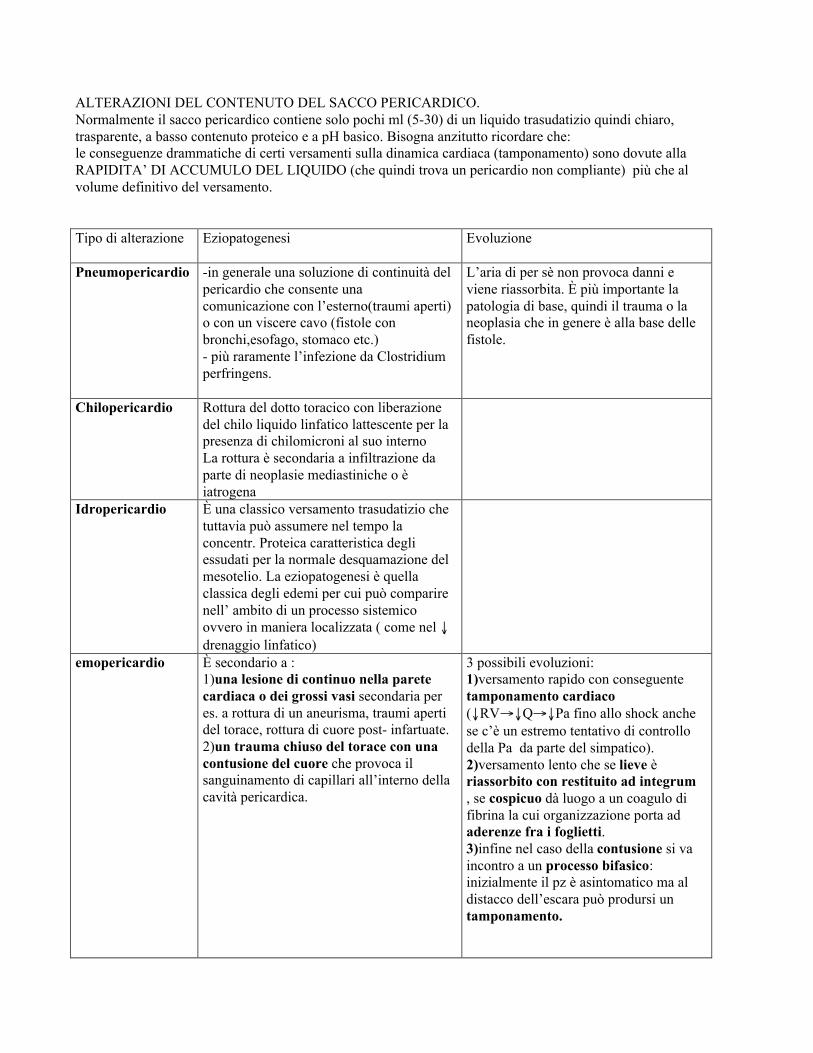
ALTERAZIONI DEL CONTENUTO DEL SACCO PERICARDICO.Normalmente il sacco pericardico contiene solo pochi ml (5-30) di un liquido trasudatizio quindi chiaro,trasparente, a basso contenuto proteico e a pH basico. Bisogna anzitutto ricordare che:le conseguenze drammatiche di certi versamenti sulla dinamica cardiaca (tamponamento) sono dovute allaRAPIDITA’ DI ACCUMULO DEL LIQUIDO (che quindi trova un pericardio non compliante) più che alvolume definitivo del versamento.
Tipo di alterazione Eziopatogenesi Evoluzione
Pneumopericardio -in generale una soluzione di continuità delpericardio che consente unacomunicazione con l’esterno(traumi aperti)o con un viscere cavo (fistole conbronchi,esofago, stomaco etc.)- più raramente l’infezione da Clostridiumperfringens.
L’aria di per sè non provoca danni eviene riassorbita. È più importante lapatologia di base, quindi il trauma o laneoplasia che in genere è alla base dellefistole.
Chilopericardio Rottura del dotto toracico con liberazionedel chilo liquido linfatico lattescente per lapresenza di chilomicroni al suo internoLa rottura è secondaria a infiltrazione daparte di neoplasie mediastiniche o èiatrogena
Idropericardio È una classico versamento trasudatizio chetuttavia può assumere nel tempo laconcentr. Proteica caratteristica degliessudati per la normale desquamazione delmesotelio. La eziopatogenesi è quellaclassica degli edemi per cui può comparirenell’ ambito di un processo sistemicoovvero in maniera localizzata ( come nel ↓drenaggio linfatico)
emopericardio È secondario a :1)una lesione di continuo nella paretecardiaca o dei grossi vasi secondaria peres. a rottura di un aneurisma, traumi apertidel torace, rottura di cuore post- infartuate.2)un trauma chiuso del torace con unacontusione del cuore che provoca ilsanguinamento di capillari all’interno dellacavità pericardica.
3 possibili evoluzioni:1)versamento rapido con conseguentetamponamento cardiaco(↓RV→↓Q→↓Pa fino allo shock anchese c’è un estremo tentativo di controllodella Pa da parte del simpatico).2)versamento lento che se lieve èriassorbito con restituito ad integrum, se cospicuo dà luogo a un coagulo difibrina la cui organizzazione porta adaderenze fra i foglietti.3)infine nel caso della contusione si vaincontro a un processo bifasico:inizialmente il pz è asintomatico ma aldistacco dell’escara può prodursi untamponamento.

PERICARDITIL’infiammazione del pericardio segue la classificazione anatomo-patologica di tutte le sierositi.In base all’essudato infiammatorio avremo le pericarditi:Sierosa→siero-fibrinosa/fibrinosa pura→ siero-fibrino-emorragicain caso di infezione da piogeni la purulenta o siero-fibrino-purulenta.
Tipo Eziopatogenesi Morfologia
Sierosa1)Virali: cox b, adeno, echo negliimmunodepressi; orthomyxo,EBV,VZV.2)collagenopatie3)s. di Dressler _da sensibilizzazionead Ag delle sierose.4)è la presentazione inziale di una p.purulenta.
Liquido con proteine al 2% tende a intorbidirsi e adaccumularsi potendo raggiungere i 1200 ml connotevole ingrandimento dell’ aia cardiaca.Le superfici pericardiche appaiono arrossate, opache inquanto prive di mesotelio e zone con depositi di fibrinadi un certo spessore diventano grigiastre e villose.
Siero-fibrinosa/Fibrinosa
1)reumatica (corpi di Ashoff)2)da radiazioni: in corso ditrattamento per es. della mammella3)infarto subepicardico(epistenocardica)4)uremica5)collagenopatie come il LES.
La pericardite fibrinosa è distinta da spessi depositi difibrina che rendono la superficie grigia, ruvida e opacae che creano:- rumori e dolore da sfregamentosi hanno anche segni sistemici di nfiammazione(febbre).Nella sierofibrinosa compare un fluido denso egiallastronon vi sono i rumori.
Siero-fibrino-purulenta
dovuta alla presenza di microrganismipiogeni che hanno raggiunto il cavopericardico per:- contiguità- via linfatica o ematogena- via traumaticaè più frequente negli immunodepressi.
Essudato siero-fibrino-purulento liquido o cremoso.
Siero-fibrino-emorragica
1)neoplastica2)tubercolare per diffusione diretta(gn dai ln tracheobronch.) i m.infettano il p. con formazioni di noduliche svuotandosi all’interno del p.rilasciano la caseosi colliquata e lacomp. Emorr.3)diatesi emorragiche4)post-chirurgica
Essudato siero-fibrino-emorragico.Sarà possibile riscontrare aspetti peculiari come per es.cellule neoplastiche nell’essudato.

Per quanto riguarda l’evoluzione delle pericarditi si può fare un discorso in generale ovvero: l’evoluzionedipende dalla quantità della fibrina depositata e dalla durata del processo.Infatti se il processo dura poco e la fibrina è poca si ha la digestione della stessa da parte dei macrofagi conrestitutio ad integrum (risoluzione).Se la fibrina è molta come nei processi fibrinosi- o fibrino-purulenti/emorragici inizia il processo diorganizzazione della fibrina che porta alla formazione di vari reliquati di diversa importanza fisiopatologica:1) “placche da lavoro”: ispessimenti fibrosi dell’epicardio2) sottili aderenze fra i due foglietti pericardici3) pericardite adesiva: obliterazione cavità pericardica4) pericardite costrittiva: che può essere anche idiopatica e porta alla formazione di una spessa cotenna
fibrosa con eventuali calcificazioni (concretio cordis) →disfunzione sisto-diastolica ; è un quadro simile allaCMPR
5) mediastino-pericardite adesiva: la estensione del processo infiammatorio anche al mediastino circostanteper es dopo terapia radiante provoca la formazione di aderenze non solo fra i foglietti pericardici ma anche fa ilpericardi e le strutture circostanti come il diaframma e la parete toracica anteriore → ↑ carico di lavoro del cuorecon ipertrofia e sfiancamento: assomiglia a una CMPD.
L’ENDOCARDIO è una membrana trasparente e sottile (il suo spessore è inversamente proporzionale a quellodella parete rivestita): microscopicamente è composta da tre strati:- l’endotelio- uno strato medio a fibre collagene orientate secondo l’asse longitudinale del cuore che ispessendosi può
assumere colore madreperlaceo,- uno strato esterno con fibre diversamente disposte e dotato di vascolarizzazione.
L’endocardio riveste il miocardio, le corde tendinee e i lembi valvolari; questi ultimi sono strutture tipicamenteavascolari costituite in sintesi da un ripiegamento dei dei 2 strati più interni dell’endocardio.
Le endocarditi sono processi infiammatori molto importanti per le loro possibili conseguenze che sono:1)la embolizzazione di trombi generatisi sull’endotelio2)le lesioni a livello valvolare e delle corde tendinee.
Si classificano in base a vari criteri:eziologico: batterica (vegetante o ulcerosa) o asettica (verrucosa o poliposa)anatomo – patologica: in base alla lesioni di cui sopratopografica: parete , valvole o cordeclinico: acuta , cronica , ricorrente

Tipo Eziopatogenesi Morfologia Evoluzione
Reumatica: comparenell’ ambito dellelesioni prodotte incorso di febbrereumatica: a livellocardiaco la patogenesisarebbe dovuta all’azione di Ig anti-streptococco di gruppoA cross-reattive conAg cardiaci:il cheprovoca unapancardite reumatica.La formazione diimmunocomplessi insitu e l’attivazione delcomplementoinnescano la flogosicon la necrosifibrinoide e quindi ilrichiamo di celluleinfiammatorie con laformazione dellelesioni fondamentalidi tipo nodulare e latrombosi che insorgesulle areedisendotelizzatesovrastanti.
Le lesioni macroscopiche sonopiccole escrescenze che sidispongono a filiera lungo i lembivalvolari sulla faccia che che guardail flusso dette verruche.Generalmente è interessata lamitrale con (25%) o senza (65%)interessamento aortico, raramente latricuspide e mai la polmonare. INPARTICOLARE si osservano1)lesioni nodulari dette corpi diAshoff : focolai di necrosifibrinoide circondati da un infiltratolinfocito-macrofagico in cuicompaiono particolari celluleistiocitarie: le c. di Ashoffplurinucleate e le cellule diAnitschkow o “a bruco”. tali lesionisono estese a tutto il cuore nell’ambito di una cosiddetta pancardite2)le verruche che non sono altroche trombi di fibrina cresciuti suaree disendotelizzate.3)le placche di MacCallum sonoispessimenti fibrosi con contorni acarta geografica dell’ endocardioparietale (in genere nell’ atrio sn)secondari a una lesione sub-endocardica tipo Ashoff insortadopo continua sollecitazione dareflusso sistolico.
La febbre reumatica oggi ha unincidenza molto più ridotta .insorge + frequentemente fra i5 e i 15 anni. Si manifesta solonel 3% dei casi di faringitestreptococcica. Si manifestacon una poliartrite migrantedi modesta gravità e con unapancardite: il primo attacco èletale nell’1% dei casi; tuttaviasi ha maggiore suscettibilità ainfezioni ricorrenti conconseguenti recidive.In questo modo nella carditereumatica cronica si hannodanni cumulativi che portanoad alterazioni valvolari come:- ispessimento fibrotico deilembi e possibilecalcificazione.- accorciamento delle cordetedinee.- fusione delle commessure e/odelle corde tendinee- vascolarizzazione dei fogliettiIn genere il tutto si risolve inuna stenosi mitralica(reumatica nel 99% dei casi) oin steno-insufficienza.
Verrucosa
Marantica:è detta ancheterminale in quanto comparein individui fortementedebilitati che abbianodisordini della coagulazione;in particolare si osserva in pz.Affetti da adenocarcinomi delpancreas (la mucina sarebberesponsabile di ino stato diipercoagulabilità) ;anchetraumatismi endocarditipossono costituire un FdR.
La definizione alternativa di e.trombotica abatterica suggeriscela natura della lesioneelementare: si tratta divegetazioni trombotiche nell’ordine di 1-5 mm bassamenteadese alla valvola chegeneralmente (75%) è lamitralica .
La localizzazionemitralica spiega latromboembolia sistemicache colpisce specialmentereni, milza e SNC( qui puòportare a rammollimento oa infarto cerebrale)
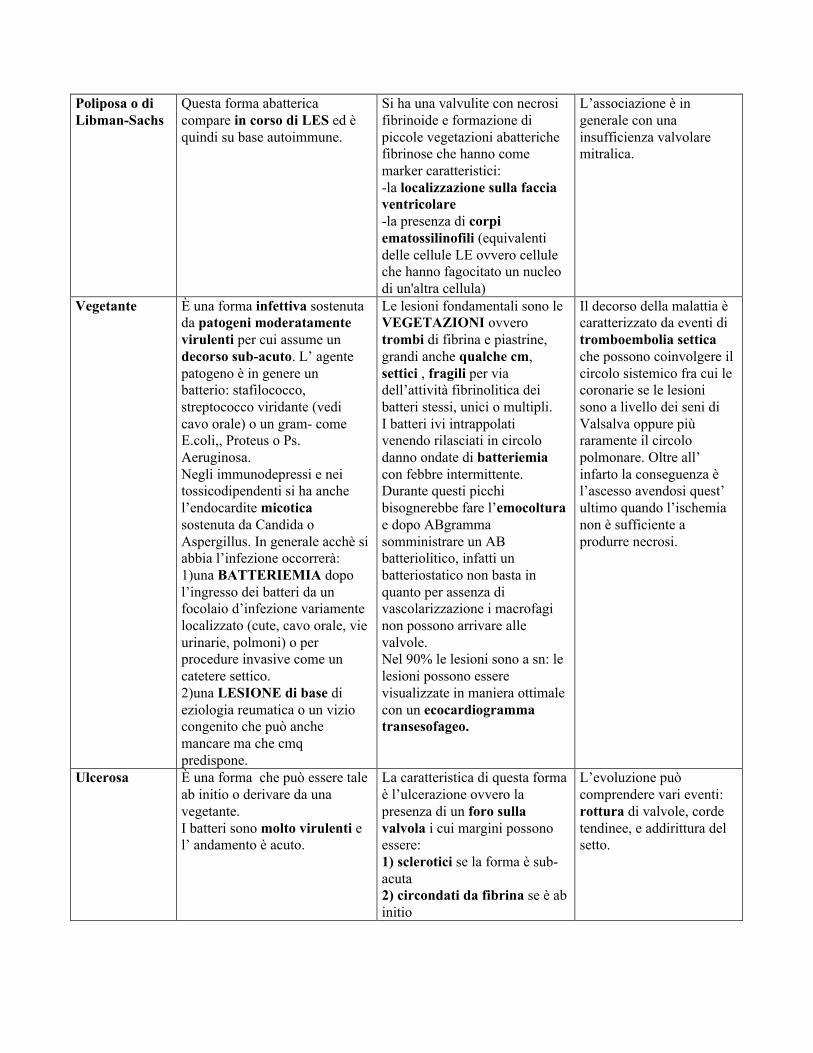
Poliposa o diLibman-Sachs
Questa forma abattericacompare in corso di LES ed èquindi su base autoimmune.
Si ha una valvulite con necrosifibrinoide e formazione dipiccole vegetazioni abatterichefibrinose che hanno comemarker caratteristici:-la localizzazione sulla facciaventricolare-la presenza di corpiematossilinofili (equivalentidelle cellule LE ovvero celluleche hanno fagocitato un nucleodi un'altra cellula)
L’associazione è ingenerale con unainsufficienza valvolaremitralica.
Vegetante È una forma infettiva sostenutada patogeni moderatamentevirulenti per cui assume undecorso sub-acuto. L’ agentepatogeno è in genere unbatterio: stafilococco,streptococco viridante (vedicavo orale) o un gram- comeE.coli,, Proteus o Ps.Aeruginosa.Negli immunodepressi e neitossicodipendenti si ha anchel’endocardite micoticasostenuta da Candida oAspergillus. In generale acchè siabbia l’infezione occorrerà:1)una BATTERIEMIA dopol’ingresso dei batteri da unfocolaio d’infezione variamentelocalizzato (cute, cavo orale, vieurinarie, polmoni) o perprocedure invasive come uncatetere settico.2)una LESIONE di base dieziologia reumatica o un viziocongenito che può anchemancare ma che cmqpredispone.
Le lesioni fondamentali sono leVEGETAZIONI ovverotrombi di fibrina e piastrine,grandi anche qualche cm,settici , fragili per viadell’attività fibrinolitica deibatteri stessi, unici o multipli.I batteri ivi intrappolativenendo rilasciati in circolodanno ondate di batteriemiacon febbre intermittente.Durante questi picchibisognerebbe fare l’emocolturae dopo ABgrammasomministrare un ABbatteriolitico, infatti unbatteriostatico non basta inquanto per assenza divascolarizzazione i macrofaginon possono arrivare allevalvole.Nel 90% le lesioni sono a sn: lelesioni possono esserevisualizzate in maniera ottimalecon un ecocardiogrammatransesofageo.
Il decorso della malattia ècaratterizzato da eventi ditromboembolia setticache possono coinvolgere ilcircolo sistemico fra cui lecoronarie se le lesionisono a livello dei seni diValsalva oppure piùraramente il circolopolmonare. Oltre all’infarto la conseguenza èl’ascesso avendosi quest’ultimo quando l’ischemianon è sufficiente aprodurre necrosi.
Ulcerosa È una forma che può essere taleab initio o derivare da unavegetante.I batteri sono molto virulenti el’ andamento è acuto.
La caratteristica di questa formaè l’ulcerazione ovvero lapresenza di un foro sullavalvola i cui margini possonoessere:1) sclerotici se la forma è sub-acuta2) circondati da fibrina se è abinitio
L’evoluzione puòcomprendere vari eventi:rottura di valvole, cordetendinee, e addirittura delsetto.

ValvulopatieIl difetto valvolare può essere una stenosi o una insufficienza: in genere tuttavia si ha una steno-insufficienza congradi variabili delle due componenti.Una insufficienza funzionale è possibile quando si ha uno sfiancamento dell’ostio per dilatazione della strutturacui è saldato per es. mitralica nella CMPD o altre condizioni dilatative.Semeiologicamente i vizi valvolari si manifestano con soffi sistolici o distolici.La presentazione clinica delle valvulopatie è alquanto variabile e variabili sono gli effetti fisiopatologici diciascun tipo: possono indurre modificazioni non solo a livello miocardico ma anche dei vasi per es. polmonari.Le lesioni più frequenti sono le stenosi aortica e mitralica: insieme coprono i 2/3 delle valvulopatie.L’insufficienza come già accennato può essere provocata da una patologia intrinseca dei lembi valvolari ovveroda una alterazione delle strutture “di sostegno” alla valvola: corde tendinee, mm. papillari, parete cardiaca o delbulbo aortico.
Stenosi aortica : è l’alterazione valvolare più frequente: può essere congenita o acquisita.Fra le forme acquisite la stenosi aortica reumatica (insorgenza in giovane età) è oggi molto meno frequente(10%) e cmq è sempre accompagnata da una lesione della mitralica; si hanno piuttosto forme degenerative condeposizione di sali di calcio che possono avvenire su una valvola aortica normale o bicuspide.La lesione elementare è una sclerosi con calcificazione a livello del punto di inserzione delle cuspidi: questo neimpedisce una corretta apertura e quindi crea stenosi.La fisiopatologia della stenosi aortica prevede una ipertrofia concentrica del ventricolo sn (fino a 800 g nelgiovane; poco più della metà nell’anziano) per far fronte all’↑ postcarico: il quadro a livello miocardico èessenzialmente sovrapponibile a quello della cardiopatia ipertensiva sistemica.Dopo un iniziale compenso si avrà la progressione verso lo scompenso che si manifesta con dilatazione delventricolo conpossibilità di trombosi alla punta.
.Insufficienza aortica: anche in questo caso la distinzione è fra insufficienze organiche (post-endocarditiche,congenite etc.) e funzionali (cardiopatia ipertensiva; aneurisma dissecante tipo A; aneurismaluetico).La fisiopatologia prevede:- ipertrofia eccentrica massiva (fino a 1000 g nel giovane : il cosiddetto cor bovinum ; la metà
nell’anziano).- Lesioni da getto- ↑ P differenziale.
Stenosi mitralica (4→2): la stenosi mitralica può essere l’esito:- di una endocardite (reumatica, di Libman-Sachs, batterica) e allora è in genere prima dei 50 anni: in questo
caso si potranno notare ispessimenti e fusione dei lembi valvolari mitralici.- di una calcificazione dell’ anello mitralico e allora insorge dopo i 60 anni: in qusto caso si noteranno
(anche all’RX) noduli calcifici.
La fisiopatologia della stenosi mitralica è molto significativa; in questo caso tuttavia il ventricolo sn presentacaratteri di normalità tranne lesioni da getto dell’ endocardio dette tasche di Zann; per il resto si osservano:

- placche di McCallum (se la stenosi è reumatica)
- fibrillazione e trombosi auricolare → rischio di tromboembolia sistemica1) per quanto concerne l’atrio sn che è ipertrofico ma soprattutto - pressione dal basso sulla carena tracheale: ↑ ampiezza angolo, tosse, st dilatato muco da compressione del bronco principale sn → infezioni
- dislocazione verso dx dell’ esofago
- dapprima stasi venosa e capillare con edema interstiziale →fibrosi diffusa
- trasudazione alveolare alle basi (rantoli a piccole bolle) → il trasudato è un pabulum per i batteri → broncopolmoniti basali bilaterali2) per quanto concerne il circolo polmonare - ectasia capillare con emorragie per diapedesi → espettorato rugginoso e accumulo di Fe con fibrosi nodulare da emosiderina.
- ipertensione polmonare da rimodellamento delle arterie e delle arteriole → a) ectasia del tronco polmonare con compressione del ricorrente laringeo sn b) cuore polmonare cronico: ipertrofia e secondariamente dilatazione-insuff. Tricuspidale
All’ eo si rileva schiocco di apertura ; rullio diastolico etc.La clinica consiste nella evoluzione verso insufficienza respiratoria e cardiaca oltre che alla possibilità di episodiacuti tromboembolici o di edema polmonare acuto.La terapia consiste nella commissurotomia prima che si stabiliscano lesioni polmonari permanenti.
Insufficienza mitralica:può essere funzionale (CMPD; cardiopatia ipertensiva; miocarditi) oppure organica (post- endocarditica; post-infartuale; ma soprattutto da degenerazione mixomatosa della valvola mitralica).La fisiopatologia è pressochè uguale alla stenosi mitralica: tuttavia la ipertensione in atrio sn non è continua; inoltre si hauna ipertrofia eccentrica (800 g).
PATOLOGIE DEL MIOCARDIO
Quasi tutta la patologia cardiaca è caratterizzata da un interessamento del miocardio.Tuttavia vi sono 3 grossi capitoli in cui l’interessamento il tessuto miocardico è interessato solo secondariamentenell’ ambito del cuore e segnatamente:- cardiopatia ischemica- cardiopatia ipertensiva DX e SN- valvulopatie
Vi sono quindi patologie primitive del miocardio a varia eziopatogenesi che vengono chiamate genericamentecardiomiopatie. Alcuni tuttavia usano questo termine solo per i casi in cui non viene riconosciuta una eziologiaprecisa.
Questa questione è di lana caprina per il clinico perché a dispetto di una grande varietà eziopatogenetica siriconoscono 3 precisi quadri anatomo-patologici e clinici che sono in ordine di frequenza: CMPD (90%) ,CMPI, CMPR se si vuole si può aggiungere un quarto che è la CMPO.

Nelle CMP (nel senso più ampio del termine ovvero “tutte le malattie che nell’ambito del cuoreinteressano primariamente il miocardio”) in generale il danno può essere idiopatico oppure secondarioa una causa nota di tipo:- genetico: distrofie muscolari- tossico: alcol, farmaci con effetto dose dipendente- dismetabolico: emocromatosi- infiltrativo: amiloidosi, sarcoidosi, metastasi carcinomatose o leucemiche- immunologiche: reazione di rigetto e miocarditi
le miocarditi che non a caso abbiamo citato alla fine sono un capitolo per così dire a parte perché:- da una parte possono avere un pattern simile che ricorda la CMPD ( cuore flaccido, dilatato)- dall’ altra sono caratterizzate dal fatto che l’infiltrato infiammatorio precede e non già segue la
necrosi
volendo quindi trattare le miocarditi si può descrivere:
1) la semeiologia anatomo-patologica:- caratteri anatomo-patologici generali:a) cuore dilatato (asse trasverso>8 cm), ma non ipertrofico (peso normale o lievemente
aumentato), pareti sfiancateb) pallido, a carne bollita (perché edematoso),c) di consistenza flaccida,d) possibilità di trovare trombi murali all’ interno delle cavità cardiache.
L’ infiltrato infiammatorio si accompagna a necrosi focali: il quadro è diverso da quello di uninfarto in cui le miofibre dell’ area infartuata sono diffusamente necrotiche.Il tipo di infiltrato varia in base all’ agente eziologico per cui se ne parla nella classificazioneeziologica.
2) la presentazione clinica delle miocarditi è variabile:- si possono avere forme asintomatiche che guariscono senza esiti importanti a- forme sintomatiche con segni sistemici di infiammazione (febbre, etc.) , dolori precordiali, soffio
sistolico da rigurgito mitralico per via della dilatazione cardiaca, segni di iniziale scompenso comedispnea, palpitazioni, facile affaticabilità.
- Forme iperacute che esordiscono con scompenso cardiaco improvviso o addirittura morteimprovvisa.
Infatti le tre complicanze principali e più gravi delle miocarditi sono:a) lo scompenso cardiacob) la tromboemboliac) le aritmie e i blocchi di branca
in un certo senso la miocardite può simulare un IMA.

3) Classificazione eziologica:
Batterica Gram- (pseudomonas,enterobacteriaceae) e piogeni specie inindividui con debole immunità: neonati,bambini, immunodepressi, cachettici,diabetici.Modalità d’arrivo: iatrogena, sepsi(endometrite puerperale), embolo settico
Caratteristici microascessi giallastri confluenti.Restitutio o MCS in relazione alla durata.
Tubercolare TBC miliare Lesioni miliari (granulomi tubercolari)→ l’esito è per lamalattia sistemica più che per la miocardiosclerosi
Virale 1)Enterovirus: COX-B 1-5; echo; polio2) orthomyxo/paramyxo: influenza,orecchioni, morbillo.3) HAV4) CMV,EBV5) HIVLe categorie esposte sono le stesse:immunodepressi, neonati, bambini.
Infiltrato da infezione virale ovvero mononucleatolinfomonocitario.In genere segue ad una infezione delle vie aeree superiori.La diagnosi è sierologica: sieroconversione
Tossiche Un tempo, prima della vaccinazioneantidifterica, la tossina diftericaproduceva danni cardiaci letali in 2settimane!
Degenerazione massiva in seguito al danno tossico coninfiltrato mononucleato.
Parassitaria/Micotica
Rare da noi . Ma si ricordi l’endemia da T.cruzi (agente eziologico della malattia di Chagas) inSudamerica → alta prevalenza in queste aree.Dose-dipendente→Chemioterapici: doxo- e dauno-rubicina;Immunosoppressori: ciclofosfamideMetilxantine,barbiturici,Li.
Necrosi focale da danno tossico (vedi anche CMPD) →infiltrato neutrofilo →mononucleato.La severità dipende dalla dose.
Da farmaci
Dose-indipendente →se c’èipersensibilità a:vari AB (penicilline, tetracicline,streptomicine), α-metildopa
Infiltrato infiammatorio diffuso che evolve in questo senso:Eosinofili → necrosi →eosinofili+neutrofili→ neutrofili.
Da radiazioni Irradiati per neoplasie mediastinicheCollagenopatie
Si tratta di malattie autoimmunitarie come: LES,PAN, sclerodermia, dermatomiosite.
In generale si ha una arteriolite: l’infiltrato èlinfomonocitario:- acuto e diffuso, con eosinofili nel LES- cronico nella sclerodermiale lesioni portano addittivamente amiocardiosclerosi.
Idiopatiche A cellule giganti:Grave forma di miocardite acuta ad eziologia sconosciuta con infiltrato caratteristico(mononucleato + cellule giganti) granulomatoso; la m. di Fiedler è una forma analoga ma senza cellulegiganti

Una patologia particolare è la febbre ruematica:
Epidemiologia: nei paesi industrializzati grazie al miglioramento delle condizioni igienico-sanitarie l’incidenzasi è molto ridotta; tuttavia rimane un problema in paesi sottosviluppati o in sacche di popolazione disagiata neinostri paesi.La malattia colpisce in genere nell’ infanzia (5-15 anni) più raramente oltre.
Eziopatogenesi: la malattia è una manifestazione di ipersensibilità che si verifica dopo faringiti dovute a ceppiparticolari di streptococco β-emolitico di gruppo A. si tratta infatti di una ipersensibilità di II tipo per cuil’organismo produce anticorpi contro antigeni streptococcici che cross-reagiscono con autoantigeni e inparticolare:- Ig anti- proteina M che reagiscono contro il cardiomiocita → lesioni miocardiche- Ig anti- polisaccaride C che reagiscono contro la membrana basale → lesioni a vari livelli
La faringite streptococcica si manifesta con febbre, ↑ VES e leucocitosi; in una piccola % dei casi dopo 3settimane si ha la cosiddetta febbre reumatica con ↑ VES ma senza leucocitosi (→forma abatterica).A questo primo attacco di febbre reumatica possono seguire molti altri in seguito a successive infezioni per cuiviene stimolata la produzione di anticorpi: si va così incontro nel corso della vita a un danno cumulativo che allafine porterà a conseguenze serie.
Per fare diagnosi di febbre reumatica occorre:1) dimostrare una pregressa infezione da streptococco β-emolitico di gruppo A→ sierologia2) dimostrare la presenza di due criteri maggiori o di un criterio maggiore e due minori.I criteri minori sono febbre, artralgia,elevati indici di infiammazione etc.I criteri maggiori sono invece 5:
a) corea di Sydenham: sindrome coreiforme (movimenti rapidi e involontari) dovuta a una infiammazione alivello cerebrale.
b) Eritema anulare
c) Noduli sottocutanei sono reumathoid-like: necrosi fibrinoide circondata da infiltrato infiammatorio
d) Poliartrite migrante: interessa le grosse articolazioni (ginocchio, gomito etc.) e si manifesta con violentaartralgia: si forma nella cavità articolare un essudato ricchissimo di neutrofili che simula un’artrite settica matuttavia il processo si risolve in pochi gg. senza esiti funzionali. Si osservano aree di necrosi fibrinoidecircondate da infiltrato infiammatorio a livello della sinovia.
e) Pancardite reumatica: è la manifestazione più importante (si dice che il RAA “morde il cuore e lambisce le articolazioni”). Interessa tutte 3 le componenti del cuore per cui avremo:
- miocardite reumatica: la patogenesi è riconducibile ad entrambi i tipi di autoanticorpi: gli anti-antigene C provocano a livello interstiziale necrosi fibrinoide con un infiltrato solo inizialmenteaspecifico perché secondariamente compare il tipico granuloma fusato di Ashoff: costituito olter che da linfocitida elementi istiocitari modificati che sono le cellule multinucleate di Ashoff e le cellule di Anitschkow connucleo a bruco. Queste lesioni compaiono in maniera asincrona e durano 4-6 mesi per poi evolvere verso lafibrosi (→oltre un certo periodo si trova solo fibrosi). Questi granulomi insorgono in sede perivasale e possonoportare quindi a una relativa ischemia sia in fase attiva che come esito fibrotico. Fibrosi si produce anche doponecrosi diretta del cardiomiocita mediata da anticorpi anti-proteina M cross-reattivi con il sarcolemma.Gli esiti sul miocardio di per sé possono essere la dilatazione in fase acuta e una progressiva sclerosi in sensocronico.

- la pericardite reumatica che come già visto è una pericardite fibrinosa o siero-fibrinosa: in questo caso visono corpi di Ashoff nel connettivo sottosieroso (sierositi fibrinose reumatiche si possono avere anche acarico di pleure e peritoneo)
- la endocardite reumatica che consta di lesioni diverse dal tipico corpo di Ashoff ovvero:
1) a livello delle cuspidi valvolari (specie mitralica, in subordine aortica) e delle corde tendineefocolai di necrosi fibrinoide ricoperti da vegetazioni verrucose che sono trombi di pastrine e fibrina.Gli esiti dell’interessamento valvolare hanno una particolare importanza fisiopatologica:
stenosi mitralica (99% dei casi di,- nel 65% dei casi è isolata)→ dilatazione atrio sn → fibrillazione→ trombi auricolari→ ipertensione: atriale → vene polmonari (edema polmonare) → arteria polm.(cor pulmonale)
lesioni endocardiche → impianto di batteri → endocardite batterica
2) A livello dell’ endocardio parietale in particolare a livello dell’atrio sn dove si riscontrano lesionisubendocardiche costituite al solito da necrosi fibrinoide con infiltrato infiammatorio: a questelesioni si somma il contributo del rigurgito mitralico per cui si formano tipici ispessimenti fibrosicon contorni a carta geografica detti placche di McCallun.
Infine si ricordi che è possibile anche:- polmonite interstiziale reumatica- vasculite
una glomerulonefrite se la streptococcia è data da particolari ceppi nefritogeni.

AneurismiUn aneurisma è una dilatazione circoscritta di un vaso arterioso, nel caso delle vene si parla infatti di varici.Il calibro e di conseguenza la struttura del vaso colpito è molto variabile potendo variare da un grande vasoelastico come l’aorta, le carotidi o le iliache a un capillare. Tuttavia in generale si parla di aneurismi di grossivasi.La classificazione degli aneurismi è su base:Strutturale per cui si distinguono 3 tipi di lesioni:- aneurisma vero ovvero dilatazione vera e propria della parete del vaso- aneurisma dissecante o dissezione aortica ovvero dilatazione dovuta al passaggio di sangue all’interno della
media con relativo sdoppiamento della stessa.- Aneurisma falso ovvero ematoma post-traumatico che comunicando attraverso un foro nella parete arteriosa
può pulsare per le variazioni di pressione nell’arteria.eziologica;ciascun tipo di aneurisma presenterà lesioni elementari e sede elettiva sue proprie. In grandi linee sipossono individuare 2 grandi capitoli nella patologia aneurismatica:- quello degli aneurismi intracranici- quello degli aneurismi aortici
aneurismi intracranici:a questo livello le arterie predisposte a formare aneurismi sono quelle extraparenchimali che decorrono nellospazio subaracnoideo dove essendo circondate dal liquor non hanno una struttura connettivale che dall’ esternosi opponga allo sfiancamento.Il circolo a questo livello è presente il cosiddetto circolo del Willis che rappresenta una anastomosi fra le arteriedei due lati e fra le anteriori e le posteriori.Le arterie afferenti al cervello sono:
1) la carotide interna la quale dalla base del cranio penetra attraverso la rocca petrosa nella cavità cranicadove, dopo l’attraversamento del seno cavernoso (un seno venoso posto ai lati della sella turcica), dà 3grosse branche:
- la oftalmica- la cerebrale anteriore- la cerebrale media
2) la arteria basilare la quale originatasi per confluenza delle 2 aa. vertebrali alla base del ponte, mandabilateralmente, risalendo lungo il ponte, le cerebellari inferiore e superiore e le aa pontine. Arrivata a livellomesencefalico si divide di nuovo nelle 2 arterie cerebrali posteriori.
Vi sono 3 rami anastomotici che chiudono il circolo del Willis:- La comunicante anteriore, impari e mediana che collega le 2 cerebrali anteriori- Le comunicanti posteriori che, bilateralmente, connettono la cerebrale posteriore alla carotide interna.
Si viene così ad avere un vero e proprio circuito attorno al peduncolo ipofisario che consente una certacompensazione in caso di fenomeni occlusivi o ostruttivi.A questo livello vi sono vari tipi di aneurisma:
- Congenito:Sono aneurismi di natura congenita dovuti quindi a difetti strutturali primitivi nella struttura della paretearteriosa : alterazione della muscolare e della lamina elastica interna.Questi difetti sono focali e ne deriva un tipico aneurisma sacciforme o “a bacca” in quanto solo una parte dellacirconferenza del vaso è interessata; si tratta in genere di lesioni localizzate a livello delle biforcazioni delcircolo del Willis e in particolare nella porzione anteriore, quella “tributaria” della carotide interna.Questi aneurismi sono in genere piccoli (pochi mm) e asintomatici.

Quando sono di dimensioni maggiori (es. 2 cm) possono essere sintomatici per compressione esercitata su variestrutture fra cui i nn. Ottico, oculomotore, abduttore, oppure altri vasi.In ogni caso un aneurisma può rompersi e dare un’ emorragia che può essere: subaracnoidea o in caso diaderenze dell’ aneurisma con la dura o con la pia rispettivamente subdurale o intraparenchimale.Per le conseguenze cliniche di questi eventi si rimanda alla trattazione degli stessi.
- aneurisma aterosclerotico:si tratta stavolta di una dilatazione dovuta a una lesione secondaria della parete vasale l’aterosclerosi appunto:in generale si tratta di un aneurisma di tipo fusiforme perché l’aterosclerosi è diffusa ; la sede elettiva è stavoltaposteriore ovvero la basilare, interessata soprattutto a livello delle biforcazioni ovvero dell’ origine dei sui rami(cerebellari e pontine): la complicanze quindi non sono legate tanto all’ aneurisma quanto ad eventi ischemici acarico del ponte e del cervelletto.Le lesioni elementari sono quelle tipiche dell’ aterosclerosi (vedi aterosclerosi allo stadio III): placche fibrocalcifiche intimali, trombosi sovrapposte etc.
- aneurismi micotici: per la loro eziologia non hanno una sede preferenziale; sono molto piccoli e quindidivengono sintomatici solo per rottura.
aneurismi dell’aorta
aterosclerotico: è l’aneurisma di gran lunga più frequente e rappresenta una complicanza dell’aterosclerosi.Epidemiologia → generalmente oltre i 50 anni e con un rapporto M:F pari a 5:1.La sede elettiva è l’aorta addominale in genere sottorenale ma eventualmente anche soprarenale in questo casol’esordio clinico è più precoce: si ricordi che dall’ aorta derivano:1) arterie segmentali ,intercostali a livello toracico e lombari a livello addominale, le quali mandano rami per il
midollo spinale2) in senso craniocaudale il tripode celiaco, le arterie mesenterica superiore, renali e mesenterica inferiore finoad arrivare alle biforcazione nelle iliache comuni.
La patogenesi consiste nella distruzione delle fibre elastiche, anche oltre la lamina elastica interna → minoreresistenza → dilatazione aneurismatica
Le lesioni elementari consistono:
Nella trombosi all’ interno del fuso aneurismatico per cui a questo livello il lume potra essere ↑(in presenza ditrombosi lieve o assente), costante o ↓ (in questo caso sembra di osservare alla tc un vaso con parete ispessita).
A livello della parete dilatata nella placca fibrocalcifica: la calcificazione è visibile all’ RX e provoca allostesso tempo rigidità e fragilità (guscio d’uovo) della parete.La dilatazione è progressiva ma con velocità variabile: quando è > 5 cm c’è l’indicazione per la chirurgia inquanto è elevato il rischio di rottura.Prima dell’ intervento consistente in un by-pass aorto-bifemorale è importante valutare lo stato funzionale dicarotidi e caronarie ed eventualmente ripristinarlo.
A livello esterno al vaso si hanno lesioni da compressione delle strutture viciniori ovvero:- corpi vertebrali con osteolisi.- uretere di sn con dislocazione (pielografia) e compressione dello stesso di grado: moderato→ stasi→predisposizione ai calcoli.
severo → idronefrosi.- polmone (in caso, più raro, di localizzazione toracica) → atelettasia

COMPLICANZE EMODINAMICHE si hanno a livello di uno o più dei suddetti rami arteriosi a partenzadall’aorta: dette complicanze possono essere:1) croniche nel qual caso sono determinate dall’ estensione dell’aterosclerosi con sovrapposizione anche di
trombosi a livello dei punti d’origine o anche della compressione esercitata dall’aneurisma.2) acute dovute a una tromboembolia.
Queste complicanze sono per così dire sede-dipendenti in quanto sono secondarie alla localizzazioneaddominale e ad un particolare livello dell’aneurisma:
Croniche Acute
Arti inferioriClaudicatio intermittens;Ipotrofia cute, annessi, muscoli dainsufficienza vascolare fino allagangrena
Gangrena
RenaliIpertensione arteriosa →Quadri di Ipoperfusione renale fino alrene grinzo.
Infarto renale
MesentericaSuperiore (sec’è coinvol-gimento dellaregionesoprarenale)
Quadri di ischemia di gravitàprogressiva:- claudicatio meseraica- enterocolite ischemica- ulcera enterocolica → fistola
intestinale
Infarto intestinale
Aneurisma dissecanteNon si tratta tanto di sfiancamento indebolimento e sfiancamento della parete come nell’aneurisma vero quantodi una dissezione della parete aortica da parte del sangue che riesce a penetrarvi attraverso fessure intimali e afarsi strada producendo una progressiva espansione della dissezione stessa.La alterazione di base che consente il prodursi della dissezione è la medionecrosi cistica ovvero una condizionead eziologia multifattoriale caratterizzata da perdite focali di sostanza elastica nella tonaca media, questi spazidefiniti “cistici” contengono sostanza amorfa costituita da mucopolisaccaridi.
I fattori che portano alla medionecrosi cistica sono:
1) la sindrome di Marfan caratterizzata da un difetto primitivo, trasmesso come carattere AD, nella sintesi delle fibre elastiche:si ha un tipico abito marfanoide con statura elevata e lunghi arti con aracnodattilia.I legamenti gialli delle vertebre così come quelli del cristallino sono poco funzionali per cui si hanno cifoscoliosie lussazione del cristallino.A livello cardiaco si hanno dilatazione del bulbo aortico e conseguente insufficienza funzionale nonché prolassomitralico.
2) carenza di Cu in quanto il rame è una componente fondamentale di enzimi implicati nel metabolismo dellefibre elastiche: tale carenza può essere per es. secondaria all’assunzione di farmaci chelanti (vedi penicillamina)in pz. con malattie da accumulo di metalli (vedi m. di Wilson).
2) lesioni di tipo ischemico a livello della media dovute all’ ipertensione, all’arteriosclerosi di vasa vasorum,o a crisi emodinamiche come uno shock.

In pz con medionecrosi cistica quindi potremo trovare una dilatazione del bulbo aortico con o senzainsufficienza aortica: la dissecazione inizia come detto a livello di una fissurazione dell’intima che si puòformare a vario livello cmq sempre nell’aorta prossimale e procede in senso anterogrado o retrogradoslaminando la media che è resa vulnerabile dagli spazi cistici.La classificazione di Stanford prevede 2 tipi di dissezioni:Il tipo A coinvolge il bulbo ± l’arco aortico e l’aorta discendenteIl tipo B coinvolge l’aorta discendente distalmente all’ origine della succlavia sn.
La presentazione della malattia è in giovani adulti.La dissezione aortica di per sé si manifesta con un dolore toracico di tipo anginoso che però non è accompagnatoda alterazioni dell’ECG o da rilascio in circolo di markers di infarto miocardico.
Le complicazioni della dissezione sono:1) la rottura a livello toracico o meno frequentemente retroperitoneale; in caso di dissezione a livello del bulbo
si può avere rottura in cavità pericardica e tamponamento cardiaco.2) analogamente alla dilatazione aneurismatica, anche la dissezione aortica può andare ad interessare l’originedei rami dell’ aorta partendo dalle coronarie* per arrivare, all’ anonima/carotide sn *, e alle intercostali:Al momento in cui si produce dissezione a questi livelli si ha insufficienza vascolare acuta ovvero infarti a
vari livelli (miocardio, cervello etc.) e questa può essere una causa di morte.
La terapia è sia chirurgica sia medica (contro l’ ipertensione).
Aneurisma infiammatorio: è dovuto alla flogosi della parete vasale quindi ovvero ad una arterite. Ha gli stessicaratteri morfologici dell’ aterosclerotico ma l’infiammazione cronica porta qui anche alla formazione diaderenze con gli organi contigui_ vedi aneurismi in corso di P.A.N.
Aneurisma luetico è una delle lesioni della lue terziaria.È secondario a una arterite infettiva a carico dei vasa vasorum dell’aorta.Si osserva un infiltrato linfomonocitario perivascolare (ricorda il carattere granulomatoso della lue) e una endoarteriteobliterante: questo insieme di lesioni si associa a necrosi focali nella media: il quadro generale è detto mesoaortite luetica.A livello dell’ intima si osservano cicatrici “a scrittura cinese” e sovrapposizione di trombi: la sede elettiva dell’ aneurismaè l’aorta prossimale per cui si può avere sfiancamento del bulbo aortico con insufficienza aortica funzionale.L’epidemiologia è quella della lue o meglio della lue terziaria: infatti se la lue è oggi infrequente è ancora più infrequente èil riscontro dello stadio avanzato della malattia la cui progressione può essere bloccata con i farmaci a disposizione. Puòessere ancora riscontrata in popolazioni del terzo mondo.

PREMESSA.
Queste pagine sono state realizzate come schema riassuntivo per fissare nozioni di anatomia patologica e diclinica desunte da più fonti (lezioni, libri, Internet).Si tratta di appunti realizzati da uno studente per preparare il suo esame di anatomia patologica: scritti quindicome promemoria personale e senza alcun intento didattico.
Di conseguenza:
1) Sono estremamente sintetici e danno spesso per scontanti concetti appresi a lezione o sui libri di testo;2) contengono sicuramente errori ortografici e forse anche concettuali; in certi casi la terminologia usata
potrebbe essere scientificamente poco corretta *.
In sintesi questi appunti non vogliono e non possono essere un sostituto del libro di testo consigliato (ilRobbins):possono essere al massimo un punto di vista aggiuntivo con cui confrontarsi e la cui utilità resta peraltro dadimostrare.
Un consiglio di ordine generale è quello di sfogliare gli atlanti del Netter (consultabili in biblioteca) o anche sitiInternet di patologia che riportano fotografie di pezzi anatomici, endoscopie etc. in quanto, vedere le immagini èfondamentale per capire e ricordare una materia come questa.Un altro consiglio è quello di tenere sempre presenti le correlazioni fra patologia e clinica in quanto l’anatomiapatologica non è mai fine a se stessa (come potrebbe sembrare allo studente ancora a digiuno di materiecliniche) ma è la disciplina che in genere conclude un iter diagnostico iniziato da un sintomo o da un segno eche, ponendo una diagnosi certa, orienta verso determinate misure di prevenzione secondaria o di terapia aseconda dei casi.
Infine una considerazione personale: lo studio dell’anatomia patologica è sicuramente molto complesso eimpegnativo ma se condotto in maniera seria vi darà grande soddisfazione perché inizierete ad avere una certapadronanza del sapere medico-chirurgico.
Cordialmente buon lavoro.
* eventuali errori possono essere segnalati all’indirizzo [email protected]
PATOLOGIA DELL’ APPARATO GASTROENTERICO
L’ESOFAGO
Anatomia e fisiologiaL’esofago è un organo cavo che collega il faringe allo stomaco: la sua lunghezza nell’adulto è di circa 25 cm (in genere siindica come punto di repere di una lesione individuata per via endoscopica la distanza dalla arcata dentaria superioreconsiderando che la distanza del cardias è di circa 40 cm).Si estende da C6 a T12. Prossimalmente l’esofago è in continuità con il muscolo crico-faringeo che fa parte delm.costrittore inferiore della faringe,decorre quindi posteriormente alla trachea e al bronco sn dopo la biforcazione a T4:quindi anteriormente ad aorta discendente, dotto toracico e vena azygos (nel suo percorso all’interno del torace si collocanel MEDIASTINO POSTERIORE); attraversato lo iato diaframmatico assume per un breve tratto il rivestimento sieroso

prima di terminare a livello del cardias: si riconoscono quindi 3 parti: cervicale, toracica e addominale: sono 3 anche irestringimenti:-l’UES situato all’inizio presso il m.cricofaringeo-il restringimento dovuto al rapporto con trachea e bronco sn-il LES situato nella porzione addominale (ultimi 4 cm)UES e LES sono sfinteri ovvero zone a maggiore pressione che mantengono chiuso le estremità dell’esofago in condizionibasali.
strutturalmente si riconoscono diverse tonache:
-la mucosa è costituita da un rivestimento epiteliale pavimentoso pluristratificato non cheratinizzato che poggia su unalamina propria di connettivo lasso delimitata all’esterno da una muscularis mucosae costituita da fascetti longitudinali. Alivello del LES si ha la zona giunzionale con passaggio a epitelio colonnare monostratificato di tipo gastrico.-la sottomucosa è un connettivo lasso contenente vasi sanguigni, linfatici, il plesso sottomucoso di Meissner,e infine ilcorpo di ghiandole a secrezione siero-mucosa.
-la muscolare propria contiene cranialmente le fibre striate del crico-faringeo che divengono sempre più rade e commiste afascetti muscolari lisci fino ad esaurirsi a metà esofago, nella metà distale abbiamo unicamente fibrocellule mm. liscedisposte in 2 strati: circolare interno e longitudinale esterno. Distalmente le fibre longitudinali si aprono a ventaglio verso lagrande curvatura formando uno strato che va ad interporsi fra le circolari e le longitudinali a livello gastrico. In questatonaca troviamo il plesso mioenterico di Auerbach.
-esternamente un connettivo avventiziale mantiene in sede l’esofago e gli altri organi adiacenti che quindi sono in intimorapporto con l’esofago. Solo a livello addominale per un breve tratto c’è un rivestimento sieroso. La mancanza di sierosa èmotivo di una maggiore tendenza infiltrativa dei carcinomi esofagei.
L’esofago non è un condotto inerte ma al contrario partecipa al processo della deglutizione che se in apparenza puòsembrare banale in realtà richiede una complessa coordinazione di molti muscoli differenti.
1) in condizioni basali sia l’UES che il LES sono contratti, la pressione del LES eccede di circa 20 cmH2O quellagastrica impedendo così il reflusso gastroesofageo. I meccanismi di continenza del LES sono: la contrazione tonicadella muscolatura, la sua collocazione fra i pilastri diaframmatici, la collocazione della parte più distale in cavitàaddominale.
2) L’atto deglutitivo porta al rilasciamento dell’UES e alla partenza dal faringe di un’ onda peristaltica primaria chesi propaga per tutta la lunghezza dell’esofago in direzione oro-aborale fino al LES che contestualmente si rilascia.Eventuali residui di bolo rimasti in esofago (ma anche un eventuale rigurgito) possono essere spinti in stomaco daun’ onda peristaltica secondaria che si propaga dal punto di distensione al LES. Onde stazionarie non propulsivesono dette peristalsi terziaria e hanno un significato parafisiologico nell’anziano.
FISIOPATOLOGIAI sintomi più comuni della patologia esofagea sono:1) disfagia: deglutizione difficoltosa con senso di arresto retrosternale del bolo ± rigurgito (che differisce dal vomito perchémanca la contrazione dei muscoli addominali).
Il sintomo disfagia può essere dovuto a molte condizioni:- ostruzione intrinseca: stenosi cicatriziale (esofagite IV grado), neoplastica (benigna vs maligna)- compressione ab estrinseco: N da carcinoma polmonare, disfagia lusoria (malformazione congenita dei grossi vasidell’arco aortico che incarcerano l’esofago come il doppio arco aortico o l’anomala origine della succlavia dx a valla dellasucclavia sn.- disfunzioni motorie come l’acalasia.
2) dolore urente/bruciore: tipico della GERD che può o meno sottendere il quadro endoscopico e patologico dell’esofagite da reflusso. “ costrittivo/simil-anginoso: disordini motori (spasmo)3) sanguinamento/ematemesi: lesioni ulcerative.

Anomalie congeniteA) Atresia dell’esofago: consiste nella mancata canalizzazione del tratto digerente a livello esofageo per cui vi saranno duemonconi, superiore e inferiore, uniti da un cordoncino fibroso: si manifesta con oligoidroamnios (il bambino normalmenteingerisce liquido e lo elimina con le urine) e alla nascita con rigurgito del latte poppato, disidratazione e calo ponderale.
B) Fistole esofago-tracheali: uno o entrambi i monconi possono risultare anastomizzati con la trachea e così alla nascita ilbambino aspirerà quello che in realtà dovrebbe deglutire (_ tosse, polmonite ab ingestis) e/o deglutirà di fatto aria mentrepiange (_ meteorismo).
Lesioni stenosantiLe stenosi sono restringimenti fibrotici segmentali che in genere rappresentano l’esito cicatriziale di una lesioneinfiammatoria. Possono aversi per:- guarigione di un’ulcera peptica esofagea (su esofago di Barrett)- esofagite di grado IV, acalasia _ stenosi grave a livello del LES- stenosi da sclerodermiaetc.
Le pliche sono duplicazioni della mucosa esofagea pavimentosa polistratificata che sporgono nel lume dell’ esofagosuperiore.Lesioni analoghe ma rivestite da epitelio cilindrico monostratificato in quanto localizzate alla giunzione squamocolonnaresono i cosiddetti anelli di Schatzky.
L’associazione di pliche + anemia ferro-carenziale + altre lesioni atrofiche delle mucose (glossite, stomatite, acloridria)in donne di età superiore a 40 anni depone per una sindrome carenziale detta di Plummer-Winson che si associa tra l’altroal rischio di sviluppare un carcinoma post-cricoideo.Tale sindrome è distribuita soprattutto nei paesi nordici in relazione a quanto pare a una alimentazione povera di vegetalifreschi e quindi di vitamine del gruppo B e vitamina C.
L’ ernia iatale che in generale è una protrusione dello stomaco in torace attraverso lo iato diaframmatici; può essere di 2tipi:-da scivolamento nel qual caso l’esofago subisce una trazione verso l’alto si trascina dietro una parte del fondo gastrico: inquesto caso il LES è sopra al diaframma: il tutto sarebbe dovuto a una lassità del legamento freno-esofageo chenormalmente tende ad opporsi a questo scivolamento: se il LES diventa incompetente si ha reflusso gastroesofageo. N.B.:questa condizione ha un’altissima prevalenza dopo i 50 anni ma non è una causa frequente di esofagite da reflusso perché ilreflusso deve comunque associarsi a un difetto di clearance.
-paraesofagea in cui una parte di stomaco in genere il fondo si fa strada attraverso lo iato finendo in torace: in questo casoil cardias rimane in sede e le complicazioni sono dovute a strozzamento prodotto a livello dello iato sulla parte di stomacoerniata: si ha compressione venosa con conseguente: edema, aspetto violaceo, erosioni della mucosa, infarcimentiemorragici.
I diverticoli in generale sono delle estroflessioni sacciformi della parete del canale digerente a tutto spessore (tutte le 4tonache); si parla di pseudodiverticoli se l’estroflessione riguarda solamente mucosa e sottomucosa: a livello esofageotroviamo 3 tipi di diverticolo:1) il diverticolo di Zenker è uno pseudodiverticolo che insorge in un locus minoris resistentiae a livello del muscolocricofaringeo.insorge in uno spazio virtuale tra esofago e colonna vertebrale e quindi riempiendosi può comprimerel’esofago.2) il diverticolo da trazione insorge a livello della parte media dell’esofago ed è in genere un esito della retrazionecicatriziale secondaria a una perilinfoadenite.3) il diverticolo epifrenico è un diverticolo da pulsione insorge immediatamente sopra al LES in condizioni di alterazionedella peristalsi e del tono del LES per es. secondariamente ad acalasia:4) un altro diverticolo è quello che può originare in seguito all’asportazione di un leiomioma esofageo nel locus minorisresistentiae che si forma in seguito all’ intervento.

Il diverticolo oltre a manifestarsi con rigurgito può complicarsi per via della stasi del bolo al suo interno con conseguente:alito fetido, diverticolite, peri-diverticolite. Una complicazione iatrogena potrebbe essere una perforazione della sottileparete del diverticolo di Zenker da parte di un “introduttore di tubi” (endoscopista poco attento secondo il prof. Ribacchi)con conseguente pneumomediastino e mediastinite.
le lacerazioni di Mallory-Weiss sono lacerazioni tipiche della parete esofagea: lineari, orientate longitudinalmente,collocate alla giunzione esofago gastrica; la patogenesi sarebbe dovuta almeno negli alcolisti ai conati di vomito ricorrenti.Queste lesioni sono una delle cause di sanguinamento delle alte vie digestive o molto più gravemente andare incontro aperforazione con conseguente mediastinite (s. di Boerehaave)
VARICI ESOFAGEE:in realtà le varici si localizzano a livello dell’esofago distale e della parte prossimale (cardias e fondo) dello stomaco: questevene che fanno parte di un plesso sottomucoso diventano varici ovvero vasi tortuosi e dilatati secondariamenteall’ipertensione portale:l’ipertensione portale può essere dovuta a varie cause come per es. una cirrosi epatica più frequentemente alcolica, la s. diBudd-Chiari consistente nella trombosi delle vene sovraepatiche : in sintesi è classificabile in pre-, intra- e post-epatica.l’ipertensione portale provoca il “reclutamento” di circoli collaterali in corrispondenza delle anastomosi fra sistema portalee vene cave sup. e inf.:1) la vena coronaria stomacica tributaria della porta è in anastomosi con il plesso venoso dell’esofago distale che a sua voltaè tributario dell’ azygos vale a dire della cava superiore.2) il plesso emorroidario.3) il sistema del Retzius : anastomosi fra rami viscerali e rami parietali.4) la vena ombelicale quando ancora pervia con formazione del caput medusae.
In esofagoscopia le varici sono ricoperte da una mucosa andata incontro a leucoplachia per cui appaiono bianco-grigiastre.La complicanza gravissima di queste lesioni è la rottura che porta ad emorragia massiva con ematemesi.Bisogna allora intervenire d’ urgenza con per via endoscopica con un palloncino gonfiabile per frenare l’emorragia: lamortalità ad ogni episodio è circa del 40%!Chirurgicamente si può creare uno shunt porto-cavale che, se da una parte allevia l’ipertensione a livello portale e quindianche delle varici, dall’altra costituisce una via di fuga per il sangue che invece sarebbe destinato al fegato: può risultarneuna encefalopatia porto-sistemica.
DISORDINI DELLA MOTILITA’ ESOFAGEAI disordini della motilità esofagea sono importanti anche dal punto di vista anatomo-patologico perché non sono patologiepuramente funzionali ma al contrario si associano a lesioni organiche caratteristiche.Si dividono dal punto di vista fisiopatologico in due grosse categorie:1) ipocinetici: ipomotilità dell’esofago e incompetenza del LES _ GERD ed esofagite da reflusso2) ipercinetici: ipermotilità del corpo esofageo e ipertono del LES _ acalasia
ACALASIA (= incapacità di rilasciamento).È una condizione caratterizzata funzionalmente (manometria) da un ipertono del LES che si accompagna a una riduzione oassenza della normale peristalsi primaria sostituita da contrazioni sincrone non propulsive del corpo, di durata eampiezza maggiori del normale.Si manifesta clinicamente con disfagia, rigurgito, dolore retrosternale con una prevalenza di 8/100.000.La patogenesi consiste in una neuropatia periferica con perdita di neuroni inibitori a livello del LES e degenerazione fibrevagali a livello del corpo.In genere l’acalasia è idiopatica ma sono descritte forme di acalasia secondaria a neuropatie a causa nota (es. neuropatiadiabetica autonomica): fra queste è particolarmente significativo il morbo di Chagas, una tripanosomiasi (T. cruzi)diffusissima nell’America latina nella quale il parassita distrugge il plesso mioenterico.Dal punto di vista anatomo-patologico si ha dapprima ipertrofia della muscolare tesa a vincere la resistenza offerta dalLES ipertonico.Si va tuttavia verso una fase di scompenso con dolico-megaesofago (esofago allungato e tortuoso, sfiancato).Possono associarsi a questa condizione polmonite ab ingestis, infezioni, carcinoma squamocellulare.Il trattamento è medico (Ca-antagonisti), endoscopico (dilatazione con palloncino o iniezione di botulinica), o chirurgico(esofagomiotomia extramucosa con fundoplicatio antireflusso).

Disordini ipercinetici di tipo puramente funzionale che si manifestano con dolore retrosternale sono per esempio:- l’esofago a schiaccianoci: quadro radiologico di una contrazione peristaltica propagata di ampiezza eccessiva.- lo spasmo esofageo diffuso: contrazione ampia e non propagata di tutto il corpo esofageo con aspetto radiologico di“esofago a cavaturaccioli”.
GERDÈ l’acronimo inglese di malattia da reflusso gastroesofageo.Si tratta di una diagnosi clinica di sindrome caratterizzata da episodi pirosi e rigurgito dovuti a reflusso gastroesofageo.Il golden standard per la diagnosi di GERD è quindi la pH-metria 24h che riesce a correlare il sintomo ad episodi direflusso gastroesofageo oggettivamente dimostrati.
La definizione di GERD che è puramente clinica non va confusa con quella di ESOFAGITE DA REFLUSSO che è inveceendoscopico - patologica.Le due entità condividono la patogenesi per cui sono spesso ma non necessariamente associate (in altre parole possonoesserci GERD senza esofagite così come esofagiti subcliniche).
La patogenesi si compone di episodi reflusso gastroesofageo + deficit di clearance esofagea.
IL REFLUSSO GASTROESOFAGEO, dimostrabile alla pH-metria, può essere di tipo:- acido (pH<4),- basico se biliopancreatico (pH>7) o- misto
il reflusso ACIDO ha come fattore patogenetico fondamentale l’incontinenza del LES che:A) nel 90% dei casi è idiopatica ovvero per cui non è possibile identificare una causa: può trattarsi di un ipotonia basale opiù frequentemente di rilasciamenti transitori inappropriati. Come già detto l’associazione con un difetto di clearance(disordine da ipomotilità) porta alla GERD e/o esofagite.
B) nel 10% dei casi è secondaria:- a patologie organiche: neuropatie, endocrinopatie, collagenopatie come la sclerodermia o esiti di flogosi cronica cheportano a steno-insufficienza del LES, stenosi pilorica potenzialmente neoplastica con impedimento allo svuotamentogastrico, ernia iatale da scivolamento (N.B.: è una condizione parafisiologica fra gli over 50 _ è un fattore predisponentema di certo non sufficiente per lo sviluppo della GERD che altrimenti avrebbe una prevalenza altissima).- a interventi medico-chiururgici: farmaci (Ca-bloccanti, ansiolitici etc.), esofagogastrostomia per un tumore del fondo gastrico (perdita del LES), esofagomiotomia etc.- all’assunzione di determinati alimenti o voluttuari: cioccolato, menta, cibi grassi, alcol, fumo
Per il reflusso BASICO e MISTO devono sussistere oltre a una incontinenza del LES, similmente a quanto accade per ilreflusso acido:
1) alterazione della funzione pilorica che determina reflusso alcalino duodeno-gastrico: lo sfintere pilorico puòessere ancora una volta deficitario per patologia:
- Funzionale: discinesia gastro-piloro-duodenale.- iatrogena: Billroth I : gastro-duodenostomia Billroth II : gastro-digiunostomia, preferita nella resezione del cr gastrico Piloroplastica- Organica: steno-insufficienza in caso di ulcera peptica iuxtapilorica o di suoi esiti.
2a) Per il reflusso BASICO sarà necessaria la acloridria dovuta a compromissione delle ghiandole gastriche secondariaa atrofia della mucosa oppure ad interventi di gastrectomia.- gastrite cronica atrofica- TIPO A- gastrite cronica atrofica del fondo di tipo C secondaria a gastro-enteroanastomosi;- gastrectomia totale con esofago-entero anastomosi
2b) Per il reflusso MISTO si dovrà avere normocloridria visto che per definizione è dovuta a una alternanza di reflussi acidie basici.

Tuttavia E’ UN DEFICIT DI CLEARANCE ESOFAGEA (con conseguente _del tempo di contattofra mucosa e materiale di reflusso) LA CAUSA NECESSARIA PER LA COMPARSA DI GERDe/o ESOFAGITE DA REFLUSSO.Lo dimostra il fatto che in individui sani si hanno anche 50 reflussi/die senza che si sviluppiesofagite _ evidentemente il reflusso non è di per se sufficiente.
Le lesioni sono progressive e si riassumono in 4 gradi che (secondo la classificazione proposta alezione dal prof.) sono i seguenti:
I) endoscopia: mucosa iperemicabiopsia:- scarso infiltrato a livello della mucosa (neutrofili);- iperplasia dello strato basale dell’epitelio ( >20% dello spessore complessivo) +
allungamento delle papille (proiezioni digitiformi della lamina propria).- Microemorragie.
II) endoscopia: chiazze giallastre circondate su mucosa iperemica biopsia: erosioni della mucosa
III) biopsia: perdita di sostanza che supera la muscularis mucosae (lesione ulcerosa)
IV) Stenosi esofagea: a livello del LES si ha una steno-insufficienza che di fatto consente perpetuarsi del reflusso: è diversa dalla stenosi post-ulcerativa (guarigione del IIIgrado) perché è estesa per un tratto maggiore: 4-5 cm vs 1-1,5cm _ anche il trattamento, vista la gravità è diverso: dilatazione con palloncino gonfiabile vs esofagectomia con anastomosi dei monconi (per la stenosi di 4-5 cm).
Oltre alla stenosi che è il risultato della retrazione cicatriziale a livello del segmentoinfiammato si potra avere a livello della mucosa:1) risoluzione immediata (grado I) o in caso di erosione (grado II, III) riepitelizzazione
secondaria alla formazione di un tessuto di granulazione.
2) Rimpiazzamento dell’epitelio squamoso da parte diepitelio colonnare gastrico con o senza ghiandole gastriche tipo cardias o anche corpo-fondoovvero metaplasia gastrica in esofago (una volta chiamata ectopia gastrica in esofago): questo siverifica in prossimità della giunzione squamocolonnare o linea Z.Questa lesione non è ancora il vero esofago di Barrett che si ha quando insorge anche lametaplasia intestinale.In entrambi i casi (metaplasia gastrica e intestinale), sarebbe la mutazione delle condizioniambientali a favorire il differenziamento degli elementi immaturi in cellule colonnari di tipogastrico e intestinale rispettivamente.
Peculiarità dell’ esofago di Barrett:1) Le aree di mucosa gastrica o intestinale in esofago sono rosse e vellutate; si distinguono dalcolore pallido dell’epitelio squamoso e dal colorito marrone della mucosa gastrica.

2) Possono essere in continuità con la mucosa gastrica (completo) tanto da portare a unospostamento verso l’alto della linea Z , un tempo uno spostamento cospicuo della linea Z venivaindicato come “esofago breve” oppure possono essere isole circondate da mucosa esofageaconservata (incompleto).N.B.: vanno differenziate da un esofago di Barrett l’ernia iatale da scivolamento e loscivolamento di mucosa gastrica in esofago a seguito di retrazione cicatriziale: in questi casi c’èuno spostamento meccanico.
3)le aree di mucosa gastrica possono ulcerarsi riproducendo fedelmente in esofago l’ulcera peptica gastrica (ulcera diBarrett).
4) l’esofago di Barrett è una condizione di rischio notevole ( RR = 30-40) per lo sviluppo dell’adenocarcinoma : prima diarrivare all’adenocarcinoma si deve produrre una displasia che può essere di grado:- lieve (nuclei sul versante basale)_ follow up annuale- grave (nuclei in posizione apicale, perdita di polarità etc.) _ follow up semestrale o chirurgia (è una lesioneprecancerosa).La prevenzione secondaria è estremamente importante visto che il Cr. ha un comportamento estremamente subdolopresentandosi spesso in stadio avanzato con una sopravvivenza complessiva a 5 anni del 10-15%.
Quella da reflusso è sicuramente la più frequente e importante, ma esistono anche altritipi di esofagite. Facciamo quindi un breve excursus delle esofagiti:
Sono variamente classificabili:1) in base alle lesioni elementari:sierosa (edema, rubor etc.) _ reflusso I gr.fibrinosa o pseudomembranosa (erosioni epiteliali ricoperte da fibrina) _Candida,reflusso II gr.necrotica o membranosa _ da causticiulcerosa _ virale (HSV, CMV), da reflusso III gr.
2)in base all’ eziologia:1) batterica: invasione della lamina propria e necrosi dell’ epitelio
(erosione): sono rare e associate per lo più a corpi estanei.
2) micotica ovvero da Candida o Aspergillus:la Candida cresce a pH acido = 4 quindi può moltiplicarsi in almeno 3 circostanze:
- nei poppanti come mughetto a livello del cavo orale per via del rigurgito- in associazione a una esofagite da reflusso acido- come opportunista : in pazienti immunodepressi vedi malati di AIDS, panirradiati, pz in trattamento con
antiblastici o in pz trattati con alte dosi di AB.
Sono dimostrabili le ife fungine pas+ dopo brushing o biopsia per via endoscopica delle membrane giallastre chericoprono la mucosa esofagea e che sono costituite da ife proliferanti su aree disepitelizzate. Questo tipo eziologico diesofagite è in genere asintomatico ma è di frequente riscontro in pz sottoposti a endoscopia per altri disturbi.
3) virale ovvero da 2 tipi di herpesvirus che causano lesioni ulcerose responsabili di odinofagia in pazientiimmunedepressi:
- l’HSV produce al solito vescicole che rompendosi lasciano ulcere “a stampo” _ la citologia dimostra degenerazioneballoniforme e nuclei a vetro smerigliato _ terapia con acyclovir.- il CMV produce ulcere serpiginose che biopsate mostrano cellule con tipiche inclusioni nucleari a “occhio di gufo” _terapia con ganciclovir.
4) da radiazione in caso di irradiazione del torace per tumori polmonari, mediastinici (linfomi) o esofageispecialmente se non aggredibili chirurgicamente: la somministrazione frazionata basso dosaggio porta a lesioniprogressivamente più profonde:sierosa _fibrinosa_ulcerosa. La guarigione si ha con riepitelizzazione e stenosi nelcaso in cui non si applichi uno stent.
La sequenza è quindi:condizioni predisponenti(incontinenza LES + deficit diclearance) _ esofagite dareflusso _ lesioni erosive _riparazione con metaplasiagastrica in esofago _ comparsaanche di metaplasia intestinale= esofago di Barrett _displasia lieve _ displasia grave_ adenocarcinoma in situ _adenocarcinoma invasivo.

5) Su base carenziale ovvero la sindrome di Plummer-Winson già vista a proposito delle pliche esofagee a livelloesofageo avremo una esofagite del tratto prossimale caratterizzata da:
- lesioni analoghe a quelle della esofagite da reflusso di I grado e anche leucoplachia- Infiltrato infiammatorio cronico linfomonocitario a livello di lamina propria e sottomucosa con tendenza
verso la sclerosi.- Possibile presenza di membrane.
La malattia di Plummer Winson è importante perché è una condizione di rischio per l’insorgenza di cr a livello orofaringeoed livello esofageo (specie a livello delle membrane per via della stasi del bolo): pertanto le mucositi croniche della malattiadi Plummer-Winson sono da considerarsi lesioni predittive di cr.
6) Chimica da ingestione di acidi o basi forti che è accidentale nel bambino e un tentativo di suicidio nell’adulto: lelesioni si localizzano per lo più a livello dei 3 restringimenti dell’ esofago dove il caustico tende a fermarsi:faringeo, bronchiale, iatale.Gli acidi forti (es. cloridrico) danno necrosi coagulativa mentre le basi forti (es. soda caustica) danno necrosicolliquativa per cui il caustico tende a raggiungere strati sempre più profondi producendo lesioni più gravi.Questi pazienti sono in pericolo di vita (perforazioni esofagee e gastriche, squilibri del Ph, shock etc.) e sesopravvivono possono avere esiti cicatriziali.
NEOPLASIESi distinguono innanzitutto tumori benigni e maligni.
Fra le neoplasie benigne connettivali:- il leiomioma, la più frequente; se non causa disfagia in genere non viene tolta per non generare un locus minoris resistentiae che produce poi un diverticolo.- il lipoma che assume la forma di uno pseudopolipo→ lipoma peduncolato- Il neurinoma o schwannoma che caratteristicamente protrude all’esterno ed allora è classificato come tumore del mediastino o della cavità addominale.- Il rarissimo fibroma.- I polipi fibrovascolari
Fra le neoplasie benigne epiteliali:- i papillomi squamosi sono polipi sessili costituiti da papille con asse connettivale rivestito da un epitelio
squamoso iperplastico: queste lesioni sono associate ad infezioni da parte di particolari sierotipi di HPV(tale infezione è facilitata dalla GERD): questi virus sono in grado di infettare e indurre proliferazionibenigne negli epiteli squamosi a vari livelli (genitali→ conditomi acuminati o piani; cute→ verruche;tratto GI alto e basso→ papilloma). Il marker di infezione è l’anomalia citologica detta koilocitosi (nucleoipercromico circondato da un alone citoplasmatico chiaro). Se il virus ha potenzialità trasformante si puòpassare, attraverso la displasia, al carcinoma ma questo è un evento raro.
I tumori maligni sono rappresentati quasi esclusivamente (95%) da 2 neoplasie di derivazione epiteliale:Il carcinoma squamocellulare e l’adenocarcinoma.Un tempo il primo era nettamente prevalente sul secondo ma oggi l’adenocarcinoma in certe casistiche è arrivato aprevalere.
La eziopatogenesi è basata su diversi fattori e condizioni di rischio:per gran parte ricalca quella del cr gastrico e in effetti la distribuzione geografica è sovrapponibile : Cina , Giappone, Iran,nell’ ambito dell’ Italia Veneto vs Ragusa.
EPIDEMIOLOGIA DEL CR GASTRICOI fattori di rischio fondamentali possono variare:- aree ricche (in genere a bassa incidenza→ alcol e fumo)- vs aree povere (ad alta incidenza→ fattori dietetici e igienici)
l’alcol (soprattutto distillati contenenti nitrosamine) e il fumo sono importanti FdR per entrambi gli istotipi nei paesioccidentali.

Anche una dieta ricca di nitrosamine e povera di vitamine o l’irradiazione di testa e collo correla con _incidenza.
Vi sono poi alcune aree geografiche con una incidenza estremamente elevata; in queste aree la cancerogenesi è stataattribuita a tutta una serie di fattori come:- elevato contenuto di nitriti nell’acqua- carenza di oligoelementi protettivi come Zn e Se- infezione da HPV (che correla scarsa igiene)- conservazione dei cibi inadeguata: affumicatura, salatura _ nitrosamine contaminazione micotica _ tossine- consumo “spiriti caldi” e alimenti molto caldi in generale.
Fra le condizioni di rischio sono importanti:- celiachia- tilosi (cr. epidermoidale)- le condizioni di stasi cronica: acalasia e tutte le stenosi (es. esofagite da caustici) , diverticoli, pliche etc.- le esofagiti croniche (in quanto la flogosi costituisce uno stimolo proliferativo per l’epitelio che
può andare incontro più facilmente a displasia e trasformazione maligna): Plummer-Vinson (cr. epidermoidale nel 1/3 sup.) , esofagite da reflusso ( Barrett _ adenocr. Nel 1/3distale).
in generale il cr. esofageo:1) incide dopo i 50 anni (nelle aree ad alta incidenza tuttavia l’esordio può essere più precoce)2) ha prognosi infausta (sopravvivenza complessiva a 5 anni del 15-20%) perché sintomatica solo in stadio avanzato equesto perché:- l’esofago non ha un rivestimento sieroso _ rapida infiltrazione delle strutture viciniori (trachea, bronco principale sn, aorta, ricorrente laringeo).
- l’ esofago ha una ricca rete linfatica sottomucosa che favorisce la rapida diffusione della malattia in senso sia craniale checaudale con possibile metastatizzazione a vari LN: mediastinici, cervicali e dell’area celiaca.3) la presentazione tipica è con disfagia ingravescente che porta il pz ad anoressia e calo ponderale. Vi può essereanemia sideropenica da stillicidio ematico.Possono poi comparire segni e sintomi di infiltrazione locale: raucedine, singhiozzo, tosse etc.4) in un pz con questi sintomi si fa:endoscopia ± pasto baritato _ diagnosi istologica _ stadiazione (ecoendoscopia, TC spirale etc.).
In ordine di frequenza la sede è 1/3 medio→inferiore→superiore.Il carcinoma (T) può essere:1) in situ→ macroscopicamente non apprezzabile2) in fase precoce: rilevatezza grigio-biancastra → se diagnosticato in questa fase è importante il grading (bene→ perle
cornee; moderatamente; scarsamente differenziato) che correla con la velocità di crescita della neoplasia.3) In fase avanzata, quella in cui purtroppo il cr si rende sintomatico per cui si arriva (tardi) alla diagnosi, si può
presentare in 3 forme:- vegetante ovvero come una lesione polipoide sessile che può presentare aree necrotiche ed emorragiche.- ulcerata: lesione piatta ulcerata a livello del lume e con alta tendenza a infiltrare la parete producendo
fistole in comunicazione con il mediastino (→ mediastinite purulenta) , la trachea (→broncopolmoniti daaspirazione), l’aorta (→emorragia massiva).
- Stenosante: lesione che infiltra la circonferenza della parete producendo ispessimento; irrigidimento;stenosi.
In genere il pz si arriva alla diagnosi per via della disfagia: il pz inconsciamente modifica la dieta e tende a nutrirsi solo conliquidi: anche per questo può divenire cachettico.

FISIOPATOLOGIA GASTRODUODENALELo stomaco è una dilatazione asimmetrica, sacciforme del canale digerente: è quindi un viscere cavo, espandibile fino a 3 lt.Si distinguono:- 2 curvature: grande (margine laterale) e piccola (margine mediale).- 2 incisure: cardiale (fra esofago e fondo sul margine laterale) e angolare (fra corpo e antro sulla piccola curvatura).mediante due piani di taglio, uno orizzontale passante per l’orifizio cardiale, l’altro inclinato passante per l’incisura angolaresi individuano 3 zone:fondo e corpo con una funzione di reservoir e l’antro pilorico con una funzione propulsiva.
3 sono anche gli strati della muscolatura liscia: circolare interno, longitudinale esterno e obliquo intermedio (fibre che sisfioccano dal cardias verso il basso, specie sulla piccola curvatura)
Lo stomaco ha anch’esso 2 innervazioni:1) intrinseca: plessi sottomucoso e mioenterico2) estrinseca: n. vago e nn. splancnici
il vago entra in addome con l’esofago sotto forma di due tronchi ai lati dell’esofago stesso.Visto che il tubo digerente è ruotato verso dx per formare lo stomaco, il ramo dx diventa posteriore e il sn anteriore:entrambi mandano un ramo allo stomaco che decorre lungo la piccola curvatura (nervo di Latarjet anteriore e posteriore) emanda fibre alla rispettiva faccia.Oltre a questo il posteriore manda fibre , attraverso il plesso celiaco, al tenue e alla metà prossimale del colon, mentrel’anteriore manda fibre al fegato attraverso il piccolo omento (legamento gastroepatico).
inoltre le miocellule dello stomaco hanno una loro attività elettrica intrinseca nel senso che presentano delle oscillazionispontanee del potenziale di membrana.Nella zona del fondo il potenziale di riposo coincide praticamente col potenziale soglia per cui a questo livello avremo unacontrazione tonica.Nel resto dello stomaco invece potenziale soglia e potenziale di riposo sono più distanti e quindi l’attività elettrica saràritmica e con contrazioni fasiche.Il ritmo di contrazione massimale è dato da un’area pacemaker situata lungo la grande curvatura al limite fra corpo e fondo.
Lo stomaco prossimale, come già detto, ha una funzione di reservoir.

La sua caratteristica è quella di distendersi in seguito all’arrivo del bolo (rilassamento ricettivo) regolando inoltre il suotono grazie a meccanocettori intraparietali a un livello tale da mantenere stabile la pressione intragastrica (accomodamentogastrico).In questa zona abbiamo quindi una attività tonica di base sulla quale si inseriscono rapide contrazioni fasiche.
Lo stomaco distale ha invece la funzione propulsiva.Lontano dai pasti l’antro la muscolatura antrale è generalmente rilasciata. Vi può essere una attività detta complessomotorio migrante che avrebbe la funzione di rimuovere residui di chimo, muco e cellule esfoliate e che si compone di 3 fasi:quiescenza _ contrazioni irregolari _ contrazioni fasiche propagate in direzione corpo_antro.Dopo i pasti invece si hanno dei complessi di contrazioni a sfintere pilorico chiuso che hanno lo scopo di triturare erimescolare il bolo per consentirne la trasformazione in chimo ( materiale più fluido e predigerito dalla pepsina).
Questa motilità gastrica di base è modulata da molti mediatori umorali (VIP, gastrina, CCK etc.) e neurotrasmettitori (AChetc.).Inoltre esistono dei riflessi a feed-back come: _ contenuto in duodeno _ inibizione stomaco.Il piloro è una valvola, costituita da un ispessimento fibromuscolare ricoperto da pliche mucose longitudinali, che regola laprogressione del contenuto gastrico in duodeno.Lontano dai pasti il piloro è aperto.Dopo i pasti il piloro si contrae in maniera coordinata rispetto all’ antro per favorire il rimescolamento, sminuzzamento efluidificazione cui accennavamo. Vi sono poi dei brevi rilasciamenti che consentono il passaggio solo al materialefluidificato.
Varie condizioni possono alterare la normale motilità gastrica:- la vagotomia _ insufficiente rilasciamento del fondo e svuotamento rapido dei liquidi.- la gastrectomia parziale _ svuotamento rapido dei solidi (manca il piloro).- condizioni neuropatiche che sarebbero alla base della cosiddetta dispepsia funzionale simil-disfunzione motoria:
in questo caso il senso di distensione addominale sarebbe dovuto ad insufficiente rilasciamento ricettivo delreservoir; il senso di sazietà precoce a un’anomala risposta sensoriale alla distensione; la nausea e il vomito a unaeccessiva distensione.
Si ricorda che la dispepsia funzionale è : un dolore o fastidio addominale, cronico o ricorrente, per almeno 3 mesi nell’ultimo anno, senza evidenza clinica, biochimica, endoscopica o ultrasonografica che possa spiegare i sintomi.Tale disordine è classificabile in base al tipo di sintomi in una forma:
- simil-ulcerosa: bruciore- simil-disfunzione motoria:- mista
la mucosa gastrica comprende 2 tipi di epitelio:- epitelio di rivestimento che ricopre le creste e le foveole- epitelio ghiandolare che costituisce il corpo delle ghiandole che si aprono sulle foveole
l’epitelio di rivestimento è uniforme in tutto lo stomaco e secerne muco e bicarbonato che hanno un ruolo protettivo.
L’epitelio ghiandolare comprende invece:- le ghiandole cardiali che secernono essenzialmente muco.- le ghiandole ossintiche situate nel fondo e nel corpo che hanno come elementi peculiari le cellule parietali (HCl e
fattore intrinseco) e le cellule principali (pepsinogeno) ma anche cellule mucose e neuroendocrine (5-HT,istamina, somatostatina, glucagone).
- Le ghiandole piloriche che occupano l’antro e la regione pilorica e che hanno come elementi peculiari le cellule G(gastrina) ma anche cellule mucose neuroendocrini.
La secrezione acida serve a facilitare l’idrolisi peptica delle proteine (fra la pepsina funziona solo a basso pH), sterilizzareil contenuto gastrico, consentire l’assorbimento di sostanze come il Fe (riduzione allo stato ferroso) e il Ca.La secrezione avviene con un complesso meccanismo che prevede lo scambio di cloruro con bicarbonato e di un protonecon uno ione K: il tutto è un processo attivo che richiede energia (si crea un enorme gradiente protonico) e che risulta nellasecrezione netta di HCl.

Un ruolo centrale in tutto il processo è affidato alla pompa protonica che trasporta un protone in cambio di uno ione K. Gliinibitori della pompa protonica agiscono proprio a questo livello producendo un blocco irreversibile dell’enzima.La secrezione è poi modulata da vari mediatori:
1) l’istamina viene prodotta localmente da apposite cellule (ECL) in risposta all’arrivo del cibo _ lega specificirecettori H2 sulle cellule parietali e induce con una cascata cAMP-dipendente una attivazione della pompaprotonica _ gli anti-H2 agiscono a questo livello: è una delle tante vie che _la secrezione gastrica e per questo èmeno efficace dei PPI.
2) La gastrina è prodotta dalle cellule G in risposta a vari stimoli (distensione gastrica, stimolazione chimica da partedei costituenti del cibo, stimolazione vagale) e la secrezione regolata a feedback negativo dal pH ; esercita la suaazione in maniera indiretta ( _ rilascio di istamina) e diretta (legame a recettori specifici sulle cellule parietali).
3) L’ACh è rilasciata dalle terminazioni parasimpatiche e stimola le cellule parietali legando il recettore M3.4) La somatostatina rilasciata dalle cellule D è invece un inibitore della secrezione acida così come di altre
secrezioni sia esocrine che endocrine.5) Le prostaglandine E hanno un importante ruolo citoprotettivo (vedi oltre) e a dosi farmacologiche _ la secrezione
acida.
La secrezione acida può presentarsi alterata in varie condizioni. Fra queste ricordiamo:- la sindrome di Zollinger-Ellison nella quale abbiamo una ipersecrezione acida secondaria a una ipergastrinemia
primitiva: l’ipergastrinemia è determinata da un gastrinoma ovvero una neoplasia producente gastrina che puòessere collocata a livello di pancreas, duodeno o altrove e che può rientrare in una MEN di tipo I (associazionedelle 3 P: pituitaria, paratiroidi, pancreas endocrino).
- la gastrite cronica da HP nella quale potremo avere:1) _secrezione acida se la gastrite è antrale (_somatostatina? _ vedi ulcera peptica duodenale)2) _ secrezione acida se la gastrite è del corpo (atrofia ghiandole ossintiche)
la secrezione di fattore intrinseco è prerogativa delle cellule parietali.Questo fattore serve a legare la vitamina B12 e ne garantisce l’assorbimento per trasporto attivo a livello dell’ileo terminale.Generalmente questo fattore è prodotto in eccesso per cui entro certi limiti la _secrezione non comporta conseguenze.Conseguenze potenzialmente serie possono aversi nel quadro della cosiddetta “anemia perniciosa” una sindrome dacarenza di vitamina B12 dovuta alla gastrite atrofica autoimmune (TIPO A) che si manifesta con anemia megaloblastica,e danni al cordone postero-laterale del midollo spinale.Allo stesso modo interventi di gastrectomia totale annullano l’assorbimento.In queste circostanze è necessario somministrare vitamina B12 per via parenterale.
La secrezione di muco e bicarbonato costituisce un importante mezzo di protezione della mucosa.Entrambi questi fattori sono secreti dall’epitelio di rivestimento: il muco va a formare uno strato di gel che ricopre lasuperficie epiteliale proteggendola dalla pepsina e dalle abrasioni, inoltre questo strato previene blandamente laretrodiffusione protonica e intrappola subito al di sopra della superficie epiteliale il bicarbonato che costituisce un sistematampone.La secrezione di muco e bicarbonato è stimolata dalle prostaglandine che quindi svolgono un importante ruolocitoprotettivo: è per questo che i FANS, inibendo la COX e la sintesi delle PG, sono gastrolesivi.
Il muco, i bicarbonati, la rigenerazione dell’epitelio, le giunzioni strette e la perfusione ematica sono tutti fattoriprotettivi che costituiscono la cosiddetta barriera mucosa gastrica.
STENOSI PILORICAPuò essere di natura:1)congenita ipertrofica: provoca rigurgiti e vomito a getto non biliare nel neonato; si rendono evidenti all’obiettività unaperistalsi visibile e una massa palpabile corrispondente al piloro ipertrofico: curato con la .2) post-infiammatoria in seguito a una gastrite cronica antrale ± ulcera peptica3) neoplastica (piloro, testa del pancreas, linfomi)

LE GASTRITI ACUTELa diagnosi di “gastrite” è eminentemente istologica (la biopsia prevede almeno 2 campioni dal corpo e 2 dall’antro)perché è necessario dimostrare l’infiltrato infiammatorio della mucosa.Infatti si parla più propriamente di gastropatie per indicare quelle condizioni in cui si hanno alterazioni della mucosa senzaun significativo infiltrato infiammatorio.
Le gastriti acute sono dovute ad insulti acuti della barriera mucosa gastrica in una o più delle sue componenti.Clinicamente la gastrite acuta può rimanere asintomatica, produrre una sindrome dispeptica di vario grado (doloreepigastrico, nausea, vomito etc.) con eventuali ematemesi e melena nelle forme emorragiche le quali in certi casi mettono arischio la vita del pz.
Dal punto di vista anatomo-patologico si ha un continuum di lesioni dalla più lieve alla più grave:1) gastrite acuta semplice- rubor (iperemia visibile endoscopicamente che può essere attribuita a gastrite solo dopo dimostrazione istologica diinfiltrato neutrofilo nelle creste gastriche)- tumor (ispessimento pliche gastriche per edema della mucosa ± sottomucosa)le sedi elettive sono le aree non acido secernenti (cardias, antro pilorico e piloro), i bordi delle ulcere e i margini dellegastro-enteroanastomosi (flogosi aspecifica).
2) gastrite acuta emorragicasi distingue dalla precedente in quanto sono presenti microemorragie capillari diffuse e confluenti che danno alla mucosa unaspetto tigrato.Sono presenti microerosioni della mucosa, invisibili a occhio nudo, che tuttavia consentono al sangue di riversarsi nel lumegastrico e di venir perso in quantità anche considerevoli con rischio di shock emorragico.
3) gastrite acuta erosivo-emorragicaall’aspetto diffusamente emorragico della mucosa si accompagnano in questo caso erosioni (perdita di sostanza limitata allamucosa) macroscopicamente evidenti.
4) ulcere acute.Vi sono almeno 2 tipiche ulcere acute gastroduodenali.
Le ulcere di Curling sono associate a tutta una serie di stress acuti: ustioni, chirurgia maggiore, sepsi, ustioni. Lapatogenesi è multifattoriale e prevede ischemia, ipercortisolismo secondario allo stress, rilascio di istamina in corso diustioni etc.
Le ulcere di Cushing sono associate a ipertensione endocranica con conseguente ipergastrinemia e ipersecrezione cloridro-peptica.
Le cause di gastrite sono molte: fisiche (es.radiazioni), chimiche (caustici, antiblastici, alcol, FANS, reflusso duodeno-pancreatico etc.), ischemiche etc.
LE GASTRITI CRONICHEAnche nel caso delle gastriti croniche la diagnosi è istologica ed è definita dalla presenza di un infiltrato infiammatoriomononucleato della lamina propria.Si hanno forme aspecifiche e forme specifiche:
le forme specifiche sono caratterizzate da quadri endoscopico-istologici ben precisi, di raro riscontro in pz che arrivanoall’endoscopia per sintomatologia dispeptica.
quadro endoscopico-istologico DiagnosiNoduli rossastri alla sommità delle pliche (macro);infiltrato linfocitario (micro)
Gastrite varioliforme
Granulomi (micro) DD fra Crohn (ulcere serpiginose in endoscopia),sarcoidosi ( __rapporto CD4/CD8), TBC (Rx torace,coltura)

sarcoidosi ( __rapporto CD4/CD8), TBC (Rx torace,coltura)
Ulcera o placca (macro); iperplasia tessuto linfoide(micro)
Iperplasia linfoide _ follow up
Infiltrato eosinofilo (micro) DD Gastrite eosinofila (ipersensibilità a certi alimenti) vsparassitosi
Le forme aspecifiche sono di riscontro molto più frequente, possono essere del tutto subcliniche ed endoscopicamente (vedigastrite tipo C) inapparenti ma per definizione sono dimostrabili all’esame istologico attraverso il riscontro di un infiltratomononucleato della lamina propria.
In base all’entità dell’infiltrato della lamina propria si distinguono vari “stadi” di malattia:- superficiale se l’infiltrato è limitato alle creste gastriche (porzioni di mucosa frapposti alle fossette).- atrofica se l’infiltrato linfoplasmacellulare si trova anche intorno attorno ai tubuli ghiandolari comprimendoli. Si possonoformare veri e propri follicoli linfatici.- attiva in presenza di neutrofili.
Si hanno poi tutta una serie di lesioni elementari associate alla gastrite come:- aspetti di iperplasia epiteliale: iperplasia compensatoria del colletto, in caso di gastrite tipo A può esserci iperplasia dellecellule G nell’antro.- atrofia ghiandolare + fibrosi della lamina propria: _ghiandole nell’unità di superficie, può interessare corpo-fondo e/oantro.- metaplasia intestinale: consegue evidentemente all’atrofia perché si ha una ipo-acloridria per cui si sviluppa epitelio ditipo intestinale (tipo colon o tipo tenue) con cellule dotate di orletto a spazzola e cellule mucipare.- displasia: comparsa di cellule atipiche per dimensione, orientamento, nucleo etc. Può insorgere su mucosa gastrica ointestinale e essere di grado lieve, moderato o grave (quest’ultima irreversibile) fino ad arrivare al CIS.In presenza di un’area di displasia è possibile che ci sia carcinoma in qualche altro punto.
Si individuano classicamente 3 tipi distinti per eziopatogenesi, estensione, e clinica:
TIPO A come “autoimmune”.Anticorpi anti-cellule parietali e anti-FI titolabili _ risposta infiammatoria autoimmune verso le ghiandole gastricheossintiche (corpo-fundiche) già in giovane età _ atrofia della mucosa corpo-fundica con:
1) ipo/acloridria _ anemia Fe-carenziale (da malassorbimento) e ipergastrinemia secondaria.2) deficit di FI di Castle _ positività al test di Shilling (eliminazione fecale di un carico di vitamina B12 marcata) _
anemia perniciosa: anemia megaloblastica (MCV > 100 µm3) + danni neurologici (degenerazione cordonipostero-laterali del midollo spinale).
1)La gastrite di tipo A non è curabile (nel senso che non c’è guarigione). Come accennato si fa terapia sostitutiva convitamina B12 parenterale.2) Bisogna inoltre attuare un follow-up endoscopico essendo la gastrite atrofica in corso di anemia perniciosa unacondizione ad alto rischio per l’adenocarcinoma gastrico.
TIPO B come “batterica”.È la gastrite da Helicobacter pilori (HP), un batterio spiraliforme G-.Questo batterio è acquisito per via orofecale e colonizza esclusivamente la superficie dell’epitelio gastrico dirivestimento (questo è un punto molto importante come si vedrà nella trattazione dell’ulcera peptica), è interessata inparticolar modo la mucosa antrale (ma non esclusivamente).L’infezione da HP è particolarmente diffusa nella popolazione (> 50% degli over-50).Il batterio possiede vari fattori di virulenza che gli consentono di colonizzare e danneggiare l’epitelio: adesine, enzimimucolitici che gli consentono di farsi strada nel muco, ureasi che gli consente di difendersi dall’acidità gastrica (nicchiaecologica), tossina vacuolizzante etc.Si ha una risposta immune sia umorale che cellulo-mediata che si associa a infiltrato infiammatorio linfoplasmacellularedella lamina propria con danno ulteriore alla mucosa.

La gastrite di tipo B, di per sé, è un problema clinico relativo essendo in genere subclinica.È invece importante per la sua associazione con:
1) ulcera peptica gastrica e duodenale:- fra i pz con ulcera gastrica (e ancor più duodenale) quasi tutti sono HP +.- D’altra parte solo una minoranza di portatori HP sviluppa ulcera: forse c’è virulenza differenziale fra vari ceppi.
2) cr gastrico:- in rapporto alla comparsa di atrofia-displasia: rischio cmq minore rispetto alla gastrite tipo A.
3) linfoma gastrico:- caratteristici linfomi a cellule T che talvolta regrediscono con l’eradicazione di HP.
TIPO C come “chimica”.È associata al reflusso biliopancreatico che si ha in seguito a gastroenteroanastomosi (vedi Billroth II ogastrodigiunoanastomosi). L’antrectomia porta alla diminuzione dello stimolo trofico della gastrina e inoltre il reflussobiliare ha un’azione detergente sul muco gastrico. Si ha quindi una gastrite atrofica corpo-fundica.
ULCERA PEPTICAIn generale è una perdita di sostanza che oltrepassa la muscolaris mucosae.Può essere acuta (sarebbe il grado estremo di una gastrite acuta) o cronica.La sedi sono in ordine di frequenza:
1) duodeno2) stomaco3) esofago (esofagite da reflusso III, ulcera di Barrett)4) digiuno5) diverticolo di Meckel6) anse ileali o colon
qual è l’eziopatogenesi? In ogni caso c’è un attacco acido alla mucosa per cui potremmo dire che è dovuta a una iperaciditàassoluta o relativa (rispetto a una barriera mucosa più debole).
Le cause di iperacidità assoluta sono:1) voluttuari come fumo, alcol, caffè.2) Iperparatiroidismo (ipercalcemia)3) Tensione nervosa4) Stasi ematica a livello gastrico (scompenso di cuore congestizio)5) Vari farmaci: metilxantine, isoniazide etc.6) Ipergastrinemia primitiva ovvero S. di Zollinger-Ellison
A) In generale l’ipersecrezione correla con l’ulcera peptica duodenale _ i pz con ulcera duodenale hanno una secrezioneacida significativamente più alta rispetto ai controlli.Vediamo alcune peculiarità di questo tipo di ulcera.- L’ulcera peptica duodenale è di gran lunga la più frequente (duodenale:gastrica = 1:4).- l’ulcera duodenale ha un’ associazione ancora più forte della gastrica con HP.- la sede tipica dell’ulcera duodenale è il bulbo: prima cioè che il chimo acido venga
tamponato dal secreto alcalino bilio-pancreatico.
I dati di fatto sono l’associazione con ipersecrezione acida gastrica, l’associazione con HP e il fatto che HP cresce solo sumucosa gastrica.

B) le ulcere gastriche insorte in prossimità del piloro (non frequentissime) sonoanch’esse associate a ipersecrezione acida e pertanto a ulcera duodenale.
C) Particolari ulcere da ipersecrezione le ritroviamo nella sindrome di Zollinger-Ellison.In questo caso c’è una ipergastrinemia primitiva dovuta a una neoplasia funzionantedelle cellule G: il gastrinoma.Le cellule G sono elementi neuroendocrini che troviamo nell’antro, nel duodeno e inmisura molto minore nel pancreas.Ciononostante la quasi totalità dei gastrinomi insorge nel pancreas, una piccola quotanel duodeno, rarissimamente nell’antro o nell’ovaio.In 1/3 dei casi si tratta di una MEN I ovvero di una sindrome neuroendocrinaereditaria caratterizzata dall’ interessamento delle “3 P”: pituitaria, paratiroidi,
pancreas endocrino.In questi casi si hanno lesioni multifocali e a carattere benigno (iperplasia o adenoma).
Nel resto dei casi si hanno lesioni solitarie e spesso maligne: come per altre neoplasie endocrine la malignità è indicata solodal riscontro di metastasi o di invasione vascolare.Tuttavia si tenga presente tuttavia che in genere è l’ipergastrinemia più che la metastatizzazione a mettere in pericolo la vitadel pz!
Clinicamente l’ipergastrinemia nel corso della sindrome di Zollinger-Ellison porta a :1) diarrea dovuta a distruzione lipasi pancreatiche, danno alla mucosa intestinale etc.2) formazione di ulcere peptiche multiple, in sedi atipiche ( duodenali oltre il bulbo, addirittura digiunali),
recidivanti.
Diagnosi clinico-laboratoristica:diarrea ± sindrome dispeptica con eventuale episodio di complicazione acuta _ endoscopia: stomaco con mucosa ipertroficae contenente secrezioni nonostante il digiuno _ test biochimici: ipersecrezione acida (_BAO, _BAO/MAO) +ipergastrinemia.
Diagnosi strumentale:- TC, RMN _ in gn testa del pancreas- scintigrafia con somatostatina *- test alla secretina (infusione selettiva intrarteriosa di secretina _ dosaggio gastrina nella vena epatica)
Terapia:- i pz con neoplasia focale sono candidati alla chirurgia (enucleazione se possibile o Whipple) o alla chemio se c’è un carcinoma non operabile.- i pz con MEN I sono candidati alla terapia medica con inibitori della pompa protonica.
CONDIZIONI DI IPERACIDITA’ RELATIVA si hanno in tutti quei casi in cui la barriera mucosa è insufficiente per unmotivo o per l’altro a sostenere l’aggressione acida.
A) La più importante è l’ulcera gastrica che si differenzia dalla duodenale per:- minore incidenza (1:4)- comparsa in età più avanzata e in individui normo/iposecernenti.L’ulcera è associata ad antrite da HP e insorge tipicamente sulla piccola curvatura, su mucosa antrale, spesso incorrispondenza dell’incisura angolare.un dato importantissimo è la necessità di fare diagnosi differenziale fra ulcera peptica gastrica e carcinoma gastricoulcerato.
B) Un altro esempio sono quelle ulcere, peraltro minoritarie, sia gastriche che duodenali, associate all’assunzione cronicadei FANS (riduzione PG che favoriscono il rilascio di muco e bicarbonato).
C) ancora su una gastro-enteroanastomosi se permangono residui di mucosa antrale con conseguente stimolo gastrinicodelle ghiandole ossintiche (gastrite di tipo C sull’anastomosi + attacco acido _ ulcera).
Patogenesi dell’ulceraduodenale.Si è visto che l’ulceraduodenale associata a HP è percosì dire una “gastrite di tipo Bin duodeno”, questa lapatogenesi teorizzata:L’ipersecrezione acida porta ametaplasia gastrica a livello delbulbo _ colonizzazione di HP _“gastrite tipo B in duodeno” _ulcera peptica duodenale.Non è chiaro se l’ipersecrezionesia dovuta a fattori endogeni(es. stress psichico) o siasecondaria a un’antrite da HP(con diminuzione dellasecrezione di somatostatina).

D) ancora l’ulcera peptica associata a diverticolo di Meckel.Il diverticolo di Meckel è un residuo del dotto onfalomesenterico.
Ricorda che a un certo punto dell’embriogenesi esistono 2 cavità, l’amniotica e il sacco vitellino rivestite all’esterno dacellule di derivazione mesodermica (splancnopleura e somatopleura rispettivamente): la cavità amniotica si espanderipiegandosi in posizione cefalica, caudale e ai lati sul sacco vitellino. Questi ripiegamenti, se prendiamo in considerazioneil tronco, si fondono lungo la linea mediana individuando:- la cavità celomatica interna (è la cavità peritoneale rivestita dal mesotelio che deriva dalla splancnopleura ).- l’ intestino primitivo che rimane in comunicazione con il sacco vitellino proprio attraverso il dotto onfalomesenterico, fral’altro l’intestino primitivo ruota attorno all’asse dotto onfalomesenterico-arteria mesenterica superiore;per inciso abbiamo anche l’allantoide, un recesso che parte dall’ultima porzione dell’intestino primitivo , la cloaca, e va afinire a fondo cieco a livello ombelicale (è attorno all’allantoide che si organizzano in origine vasi ombelicali).Dalla sepimentazione della cloaca traggono origine una porzione anteriore che forma le basse vie urinarie e genitali(l’allantoide obliterandosi forma un legamento fibroso che sospende la vescica all’ombelico detto uraco) e una porzioneposteriore che forma il retto.In certi casi si verifica una involuzione incompleta del dotto onfalomesenterico o dell’allantoide (vedi adenocarcinomavescicale).I residui del dotto onfalomesenterico si collocano nell’ileo, 10-90 cm a monte della valvola ileocecale, sul versanteantimesenterico e possono essere:- il diverticolo di Meckel: è il più frequente (1-3 % della popolazione), può contenere varie ectopie (gastrica, colica,pancreas aberrante) _ ulcera peptica intestinale (in presenza di mucosa gastrica), diverticolite/perforazione (addomeacuto appendicite-like), neoplasie su epitelio ectopico.
- un cordoncino fibroso _ volvolo _ occlusione intestinale.- una fistola onfalomesenterica o un seno ombelicale _ scarico di materiale mucoso, purulento o fecaloide all’esterno.- un enterocistoma: cisti per occlusione alle due estremità.
E) un esempio analogo è l’ulcera peptica del colon secondaria a fistola gastro-colica.
CLINICA DELL’ULCERAIn sintesi i due tipi di ulcera più importanti sono la duodenale e la gastrica.
Entrambe si manifestano con una sintomatologia dispeptica : ovvero dolore epigastrico che può essere alleviato dal cibo(ma solo transitoriamente nella gastrica, più a lungo nella duodenale). Sindromi dispeptiche sono diffusissime nellapopolazione e oltre la metà è di tipo funzionale.È improponibile fare una gastroscopia a tutti quelli che lamentano dispepsia. Bisogna valutarne l’opportunità nel singoloindividuo considerando anche la presenza di segni sospetti o di CdR per patologie organiche (età, uso di FANS, HP +).
La EGDscopia evidenzia un’ulcera peptica gastrica o duodenale.1) L’ulcera duodenale è a livello del bulbo (1a parte del duodeno), sulla parete anteriore o posteriore o su entrambe (kissingulcera), è sempre di natura peptica e non serve la biopsia per differenziarla dal CR.2) L’ulcera peptica gastrica classica, quella associata a gastrite da HP, insorge su mucosa antrale, lungo la piccolacurvatura, con massima frequenza a livello dell’angulus.Varianti meno comuni di ulcera gastrica sono l’ulcera iuxtapilorica a eziopatogenesi ipersecretiva e l’ulcera da FANSlocalizzata comunque a livello antrale.Le dimensioni dell’ulcera peptica cronica sono vanno da 0.5 a 4 cm.La forma è ovalare.I margini sono netti e non rilevati, raggiunti dalle pliche mucose.Il fondo è liscio e deterso costituito da un sottile strato di necrosi, su un infiltrato neutrofilo, su un tentativo di riparazioneda parte di tessuto di granulazione che poggia su uno strato già cicatrizzato.Sono presenti vasi trombizzati di colore scuro che vanno attentamente evitati.La profondità (per definizione al di là della mucosa) è variabile.
A livello gastrico è fondamentale fare dd ulcera peptica – ulcera neoplastica.Alcune caratteristiche come le anomalie di sede (es. grande curvatura), dimensioni > 4 cm, forma, la rilevatezza dei marginirispetto alla mucosa circostante, le pliche che non arrivano ai margini dell’ulcera etc. sono suggestive di ulcera maligna.

La diagnosi definitiva è comunque istopatologica e si basa sul prelievo di almeno 6 campioni bioptici e un brushing deimargini (se possibile anche sul fondo).
Le COMPLICAZIONI DELLE ULCERE PEPTICHE sono:A) il sanguinamento _ erosione dei vasi della parete con emorragia delle alte vie digestive (ematemesi, melena). Le ulcerepeptiche sono responsabili di >50% dei casi di sanguinamento massivo delle alte vie digestive (a monte del legamento delTreitz).
In generale il pz con sanguinamento massivo si presenta con ematemesi ± segni di shock emorragico: 1) stabilizzare il pz: infusione liquidi e sangue in vena dietro attento monitoraggio della
pressione arteriosa, della diuresi, del volume di sangue perduto. 2) irrigare con acqua fredda lo stomaco attraverso un tubo nasogastrico che ha effetto di ripulitura e in molti casi terapeutico (emostasi). 3) EGDscopia ha 2 finalità: - diagnostica: caratterizzare il sanguinamento. - terapeutica: tentare l’emostasi per via endoscopica (adrenalina, elettrocauterizzazione, diatermocoagulazione, laser etc.)
in generale se il trattamento medico fallisce o se il pz sta perdendo molto sangue c’è indicazione alla chirurgiad’emergenza che può essere:- nel caso dell’ulcera duodenale la piloroplastica (apertura duodeno)- nel caso dell’ ulcera gastrica la gastrectomia (Billroth II)
B) la stenosi pilorica _ è una stenosi cicatriziale che è aggravata dall’edema e dallo spasmo muscolare. È di fatto unaostruzione e si manifesta con distensione gastrica, vomito (non biliare), calo ponderale, scompensi idroelettrolitici (perditadi acqua, protoni, Cl, K e Na con il vomito, ulteriore perdita di Na bicarbonato a livello renale, conseguenteiperaldosteronismo secondario con perdita ulteriore di K e H: in sintesi bisogna reintegrare acqua, NaCl e KCl).In presenza di una sindrome siffatta è necessario procedere alla DD con stenosi neoplastica (cr. del pancreas, cr. gastrico) ingenere questo è possibile con la gastroscopia.Una opzione è, anche in questo caso, la terapia medica (suzione gastrica mantenuta per diversi giorni) che tuttavia puòessere solo su edema e spasmo.La terapia chirurgica prevede la piloroplastica + vagotomia.
C) la perforazione in genere avviene su ulcere della parete gastrica o duodenale anteriore:questo perché anteriormente non ci sono grossi vasi; grossi vasi, presenti posteriormente, ulcerandosi portano ad emorragiaprima che si sviluppi perforazione. Talvolta si possono avere 2 “kissing ulcers” sul bulbo duodenale, una anteriore, l’altraposteriore: il sanguinamento della posteriore associato alla perforazione dell’anteriore possono portare a emoperitoneo.
il pz ha una peritonite chimica e si presenta con addome acuto ad esordio improvviso e chiari segni di peritonismo:immobilità, posizione rannicchiata, addome ligneo, ileo paralitico.Si ha pneumoperitoneo.La presentazione può essere più sfumata nel caso di formazione di un ascesso subepatico o subfrenico.
NEOPLASIE DELLO STOMACOLungo tutto il tratto gastrointestinale troviamo 2 grosse categorie di tumori:- i tumori epiteliali sono di gran lunga i più frequenti- i tumori stromali, cosiddetti GIST che sono una minoranza
Per polipo gastrico si intende qualunque lesione derivante dalla mucosa e aggettante nel lume dello stomaco. In relazionealla modalità d’impianto si parla di polipi sessili o peduncolati.

Per pseudopolipi si intendono masse aggettanti nel lume ma appartenenti a strati più profondi (vedi i GIST: fibromi, lipomi,leiomiomi e leiomiosarcomi, neurinomi*, neurofibromi) per cui la mucosa gastrica appare sollevata.* vedi s. di von Recklinghausen.
Per quanto riguarda i polipi propriamente detti abbiamo:
1) il p. iperplastico-infiammatorio: è associato a gastrite cronica _ risposta rigenerativa esuberante _ formazioni difrequente riscontro (90%), piccole (<1cm), sessili, generalmente multiple, dello stesso colore della mucosanormale, con evidenza istologica di gastrite.non sono lesioni precancerose ma occorrendo nell’ambito della gastrite possono associarsi ad aree di displasia.
2) Il p. adenomatoso: è una lesione epiteliale displastica.È meno frequente (10%), grande (pochi cm) e generalmente peduncolato, singolo,di colorito rosso vivo esuperficie bernoccoluta.Il significato della displasia è quello di lesione precancerosa: per di più un focus di carcinoma è già presente nel30% dei casi sul polipo e nel 20% sulla mucosa circostante.Questo polipo può essere un reperto endoscopico occasionale oppure per esempio rendersi sintomatico per anemiasideropenica da stillicidio ematico.
3) i p. amartomatosi nella s. di Peutz-Jeghers.
IL CARCINOMA GASTRICOI cancerogeni più importanti sono le nitrosamine responsabili anche del carcinoma esofageo.Fonti di nitrosamine sono il fumo di sigaretta, i distillati (grappa e Calvados), diete ricche di amidi che possono essereterreno di fermentazione batterica.Le nitrosamine potrebbero anche essere sintetizzate endogenamente e questo è favorito: dall’assunzione di nitrati e nitriti(abbondanti nell’acqua di certe aree geografiche, cibi con conservanti), ipocloridria (gastrite atrofica tipo A, B o C),carenza frutta e vegetali freschi e quindi di antiossidanti (vitamine A, C, E, acido gallico).
La prova dell’importanza dell’ambiente è stata dimostrata valutando il calo d’incidenza di cr gastrico che si riscontra neigiapponesi e nei cinesi emigrati negli USA.Negli USA e nei paesi occidentali il forte decremento del carcinoma gastrico è stato possibile grazie alla diffusione delfrigorifero che garantisce la salubrità dei cibi e al miglioramento delle condizioni igieniche con _della gastrite da HP.
La gastrite cronica in generale sembra essere un FdR di rischio per lo sviluppo del cancro gastrico.La flogosi cronica comporta un’ accelerata e protratta proliferazione dell’epitelio che di per sé favorisce la cancerogenesi;inoltre l’atrofia corpo-fundica porta ad ipocloridria e a metaplasia intestinale.Sulla metaplasia intestinale può insorgere displasia lieve, moderata o grave.Macroscopicamente la mucosa displastica o adenomatosa può essere polipoide o piatta.Microscopicamente è caratterizzata da atipie cellulari e alterazioni dell’architettura ghiandolare.Dalla displasia grave si passa al CIS che è un carcinoma di tipo intestinale.Il carcinoma di tipo diffuso si associa anch’esso a gastrite cronica ma insorge de novo senza passare per metaplasiaintestinale e displasia.
La SEDE tipica del carcinoma gastrico è l’antro-piloro sulla piccola curva ovvero la stessa dell’ulcera peptica. In certi casiè difficile definire la sede di insorgenza tanto il tumore è esteso, in altri casi è la regione cardiale.I tumori della regione cardiale oggi vengono considerati come un’entità a sé stante che comprenderebbe anche gliadenocarcinomi dell’esofago distale associati all’esofago di Barrett (GERD = ernia iatale, obesità, sedentarietà, dieta, fumoe alcol).L’incidenza di questa categoria di tumori tende ad aumentare in controtendenza rispetto a quelli dell’antro-piloro.
La MODALITA’ di crescita può essere:1) vegetante: massa sporgente nel lume

2) piatto: tumore infiltrante, difficilmente riconoscibile all’endoscopia o alla radiografia, tipico nella forma diffusa,può portare al quadro della linite plastica (parete dello stomaco rigida e ispessita per infiltrazione estensiva delcarcinoma) che si può avere anche nelle metastasi da cr polmonare o mammario.
3) Ulcerato: forma che pone la necessità di diagnosi differenziale con l’ulcera gastrica benigna che peraltro tende ainsistere sulla stessa zona (antro-piloro sulla piccola curva).
Le TIPOLOGIE ISTOLOGICHE sono essenzialmente 2 secondo Lauren:1) Tipo intestinale: ci si arriva passando per metaplasia intestinale _ displasia (adenoma piatto o polipoide); è
costituito da strutture ghiandolari tipo quelle dell’adenocarcinoma del colon. Cresce con modalità espansiva.2) Tipo diffuso: ci si arriva de novo dalla mucosa gastrica; è costituito da cellule mucipare di tipo gastrico che non
formano ghiandole ma hanno un atteggiamento infiltrativo.una variante del tipo diffuso è quella con cellule a castone nelle quali il nucleo è spinto in periferia dall’accumulodi mucina.
La STADIAZIONE prevede:in base all’estensione della massa tumorale, la “T”:
1) il CIS: intraepiteliale2) il carcinoma in fase precoce (“early gastric cancer”): non oltrepassa la sottomucosa. Se la diagnosi avviene a
questo stadio la sopravvivenza a 5-anni è > 90%!3) il carcinoma in fase avanzata: oltre la sottomucosa.
Per contiguità si può avere infiltrazione del colon trasverso, del pancreas, del piccolo e grande omento.
I ln interessati dalle metastasi per via linfatica sono quelli:1) della piccola curva_ celiaci2) periesofagei (cardias)3) gastroepiploici di sn _ dell’ ilo splenico _ pancreatici supp. di sn _ celiaci4) gastroepiploici di dx _ infrapilorici _ soprapilorici _ pancreatici supp.di dx _ celiaci5) i celiaciè possibile che attraverso il dotto toracico il cr gastrico produca una metastasi linfatica sovraclaveare a sn: il cosiddettolinfonodo di Virchow o segno di Troisier.
Una particolare via di metastatizzazione è quella trans-celomatica:- carcinosi peritoneale- nella donna metastasi bilaterale all’ovaio (tumore di Krukemberg- questo tipo di metastasi è possibile anche per altri cancri addominali).
Per via ematogena sono possibili metastasi a fegato, polmoni, cervello, osso.
Clinicamente il carcinoma gastrico si manifesta quando già è in fase avanzata con una sindrome dispeptica (pesantezzapost-prandiale), quindi una sintomatologia aspecifica.Si può avere quindi anoressia, calo ponderale, vomito (specie per stenosi pilorica), disfagia per i carcinomi del cardias.Il pz può presentare anemia e positività al CEA.il golden standard per la diagnosi è la gastroscopia che consente di ottenere biopsie dalla lesione.Si ricordi a questo proposito che grazie allo screening endoscopico in Giappone nel 30% dei casi il cr viene identificato infase early vs solo il 10% dell’Europa con ovvie ricadute sulla sopravvivenza dei pz.in fase early la sopravvivenza a 5 anni è > 90%in fase avanzata scende di molto.
I CARCINOIDI sono il 2% dei cancri gastrici.Sono del tutto simili a quelli intestinali ai quali si rimanda.
I LINFOMI GASTRICI sono il 3-5% dei cancri gastrici.Possono essere primitivi (MALTomi) oppure metastatici.

Può presentarsi con lo stesso tipo di lesioni del carcinoma: vegetante, infiltrante o ulcerante.L’importanza è quella di tenerle in considerazione nella dd con l’adenocarcinoma gastrico del quale ricalcano lapresentazione clinica.La prognosi varia in base al clone d’origine B vs T e al grading.La terapia è essenzialmente medica.Un tipo di linfoma gastrico è associato all’infezione da HP e può regredire con l’eradicazione del batterio.
I Bezoar sono agglomerati di fibre che si formano all’interno dello stomaco.In genere si tratta di fibre vegetali che non vengono sufficientemente masticate nell’edentulo, un’altra condizione favorenteè la gastrectomia.Questi agglomerati di fibre possono dare ulcere gastriche da decubito oppure possono causare ostruzione intestinale sepassano nel tenue.In gastroscopia è in genere possibile sminuzzare queste formazioni.
La GASTROPATIA IPERTROFICA è un quadro endoscopico con notevole ispessimento delle pliche gastriche. Il quadroendoscopico può essere associato:1) ad ipersecrezione acida nella sindrome di Zollinger-Ellison (secondaria a un gastrinoma) e nella gastropatiaipertrofica-ipersecretoria2) a ipocloridria nella sindrome di Menetriere: in questo caso si ha un iperplasia della mucosa ma a vantaggio dell’epiteliodi rivestimento che si solleva in pliche carnose mentre le ghiandole sono atrofiche. Il risultato è una mucosa fragile che siframmenta facilmente e sanguina _ anemia; produce un’abnorme quantità di muco _ proteino-dispersione con iponchia ededemi periferici; può andare incontro a metaplasia intestinale _ _rischio di adenocarcinoma gastrico.
INTESTINO
Il tenue è un tubo lungo circa 6 che si estende dal piloro al cieco: il duodeno, la prima parte lunga circa 25 cm forma una“c” in sede retroperitoneale e si continua attraverso la flessura duodeno-digiunale nel digiuno corrispondente al primo terzodella restante parte; i due terzi distali sono rappresentati dall’ileo.Il colon è un condotto a lume più ampio e lungo circa 1.5 m ; si divide in ceco, ascendente, traverso, discendente e sigma.Quest’ultimo inizia al margine superiore del cavo pelvico di sn e dopo aver descritto una sorta di “s” è in continuità con ilretto che iniziando a livello della terza vertebra sacrale scende caudalmente per un tratto di circa 15 cm.
Vascolarizzazione:ARTERIE: formano una rete di arcate fittamente anastomizzata nello spessore del mesentere.-la mesenterica superiore irrora un segmento che va dal digiuno (il duodeno spetta al tripode celiaco) alla metà prossimaledel colon trasverso.-La mesenterica inferiore irrora la restante parte fino alla parte prossimale del retto-La parte distale del retto è perfusa dalle arterie emorroidarie media e inferiore che originano rispettivamente dalle arterieipogastrica e pudenda.
VENE: corrispondono grosso modo alle arterie: la mesenterica superiore e la vena lienale che ha ricevuto la mesentericainferiore confluiscono nella vena porta (cui si aggiungono poi anche i rami gastro-duodenali e della colecisti).Vi sono 3 anastomosi porto-cavali:- a livello del retto- a livello delle gastriche brevi- a livello del sistema del Retzius (peritoneo-parietale)
Considerazioni anatomo-funzionali:
Funzione digestiva _ ampia superficie di contatto con il chilo _ villi (mucosa del tenue)

e di assorbimento _ microvilli (enterociti)
Funzione secretiva _ Cellule caliciformi mucipare (cripte e villi) rivolte verso il lume Cellule neuroendocrine (cripte) rivolte alla membrana basale
Funzione immunitaria _ IgA secretorie: linfociti B Linfociti T CD8+ (intraepiteliali) e T CD4+ Noduli linfoidi e placche di Peyer (ileo distale) Cellule M per il trasporto di Ag ai linfociti Cellule di Paneth (fondo delle cripte _ lisozima)
Funzione motoria _ segmentazione e peristalsi _ plesso mioenterico di Auerbach Innervazione estrinseca del SNA
È importante ricordare che giornalmente passano entrano nel canale gastroenterico 9 litri d’acqua (dieta, saliva, succogastrico, succo biliopancreatico, secrezioni intestinali) di questi 8.8 viene riassorbito e 0.2 escreti come feci.Per diarrea si intende l’evacuazione di feci ipoformate e di volume > 200 g/die in almeno 3 scariche/die.Essa si accompagna spesso a sintomi come l’urgenza, il tenesmo (senso fastidioso di evacuazione incompleta),l’incontinenza.La diarrea può essere di vari tipi:
1) Secretoria: diarrea acquosa isotonica col plasma. Si può avere per un’ ipersecrezione di elettroliti e acqua comenel colera o come quando l’epitelio è messo fuori uso da una gastroenterite virale.
2) Osmotica: diarrea acquosa iperosmolare rispetto al plasma. È dovuta a sostanze non assorbibili e osmoticamenteattive all’interno del lume (es. lattulosio, lattosio etc.).
3) Infiammatoria: diarrea purulenta e sanguinolenta di volume variabile (Chron, RCU, colite pseudomembranosaetc.). Per dissenteria si intende l’emissione dolorosa di numerose scariche/die, sanguinolente e di scarso volume.
4) Alterata motilità: feci variabili in volume e consistenza (colon irritabile, neuropatie, carcinoide etc.).
PATOLOGIA INFETTIVALe enterocoliti infettive rappresentano una patologia estremamente frequente e particolarmente gravenei paesi in via di sviluppo dove sono responsabili della metà dei decessi nei bambini al di sotto di 5anni.
I virus colpiscono soprattutto i bambini: i rotavirus colpiscono bambini fra 6-24 mesi e sono estremamente contagiosi, icalicivirus (vedi il Norwalk) colpiscono soprattutto bambini più grandi ma anche adulti.Si osserva un infiltrato linfocitario nella lamina propria e talvolta appiattimento della mucosa: la mucosa secerne ma nonassorbe bene _ diarrea acquosa secretiva.
I batteri possono dare in vario modo sindromi diarroiche nel senso che possono produrre in vari modi un danno allamucosa:- intossicazione: enterotossina stafilococcica.
- tossinfezioni ovvero infezioni intestinali da parte di patogeni che agiscono mediante tossine ad azione:1) secretiva: E. coli enterotossigeno (ETEC _ diarrea del viaggiatore) e V. cholerae.2) citotossica nei confronti degli enterociti con conseguente sindrome dissenterica: Shigella, E. coli enteroemorragico
(EHEC).- invasione e/o distruzione dell’epitelio: E. coli enteroinvasivi (EIEC).
I Protozoi e i parassiti che agiscono a livello intestinale sono estremamente importanti nel terzo mondo.L’Entamoeba histolytica, come suggerisce il nome, è un protozoo ad azione invasiva che produce una sindrome dissentericacon ulcerazioni a livello del colon, potendo per di più diffondere attraverso il sistema portale a fegato generando ascessiepatici.La Giardia lamblia è un parassita non invasivo che disponendosi sulla mucosa intestinale con un meccanismo non chiarito(_superficie assorbente?) provoca una sindrome da malassorbimento.

Sindromi da malassorbimentoLa sindrome da malassorbimento è l’epifenomeno di un deficit delle funzione propria dell’ intestino ovvero assorbire,previa digestione per le macromolecole, le sostanze introdotte con la dieta.Questa funzione si realizza in più tappe successive:
1) digestione intraluminale eseguita dagli enzimi pancreatici di polipeptidi (tripsina etc.), polisaccaridi (amilasi), e lipidiprevia emulsione da parte della bile (lipasi e fosfolipasi)
2) digestione terminale a livello dell’ orletto a spazzola da parte di dipeptidasi, disaccaridasi, e lipasi.
3) assorbimento che si compone di trasporto transcellulare e successivo trasferimento:- al circolo linfatico per chilomicroni (lipoproteine)- al circolo venoso portale per le sostanze idrosolubili.
Fatta questa premessa si possono considerare i molteplici meccanismi eziopatogenetici alla base della sindrome damalassorbimento:
Pochi enzimi: - da insufficienza pancreatica (pancreatite cronica etc.) - secondario a digestione peptica (Zollinger-Ellison) Alteratadigestione poca bile: - disfunzione del circolo enteroepatico a vari livelli: colestasi,intraluminale resezione dell’ileo terminale
troppi batteri: ansa esclusa etc.
alterata deficit congeniti di disaccaridasi: es. intolleranza al lattosiodigestioneterminale varie situazioni acquisite con compromissione dell’orletto a spazzola
riduzione della superficie assorbente: - sindrome dell’intestino corto - sprue celiaca e tropicale - morbo di Crohn
alterato lesione dell’epitelio assorbente: -infezioni e sovracrescita battericaassorbimento deficit congeniti di trasporto: -abetalipoproteinemia
stasi linfatica: linfangectasia congenita o secondaria (Whipple, linfomi, chirurgia etc.)
↓ tempo di transito per iperperistaltismo (vedi s. da carcinoide)Sebbene qualsiasi forma di diarrea determini ovviamente un’alterazione dell’assorbimento dei nutrienti parlare di sindromeda malassorbimento è un po’ più specifico.In questo caso all’inizio c’è un deficit primitivo di tipo cronico dell’assorbimento di uno o più nutrienti dovuto a undeficit delle funzioni di digestione intraluminale, digestione terminale o di assorbimento da parte della mucosa.Ne risulteranno sindromi caratterizzate da:
1) diarrea di tipo osmotico con feci abbondanti e schiumose per la steatorrea.2) manifestazioni di tipo carenziale:- sistemiche: calo ponderale, astenia- gastroenteriche: mucositi- cute: diatesi emorragica (porpora, ecchimosi); ipercheratosi (ipovitaminosi A)
- sangue: anemia (Fe-carenza, deficit di folato e B12), ipoproteinemia ed edemi - osso: osteopenia, ipocalcemia fino alla tetania (Ca, vitamina D etc.) - sistema nervoso: neuropatie da ipovitaminosi A, B12 etc.

Alcune CAUSE DI MALASSORBIMENTO.1) sprue celiaca2) sprue tropicale3) deficit di disaccaridasi4) linfangectasia intestinale5) abetalipoproteinemia
La SPRUE CELIACA e l’ enteropatia da glutine.In soggetti geneticamente predisposti (determinati alleli HLA-DQ), probabilmente anche per l’intervento di altri fattoriambientali (lo dimostra il fatto che c’è concordanza incompleta nei gemelli monozigoti), l’assunzione di alimenti contenentiglutine, una frazione proteica alcol-solubile comprendente la gliadina ed altre proteine dette prolamine, porta a una rispostaautoimmune di tipo cellulare e umorale.Il glutine è contenuto nel grano e in altri cereali come la segale, l’orzo, l’avena.
L’enteropatia consiste nell’infiltrato infiammatorio (linfociti, plasmacellule, macrofagi etc.) della lamina propria conaccorciamento dei villi e iperplasia delle cripte mentre lo spessore complessivo della mucosa rimane invariato (_superficieassorbente).Le lesioni si cocncentrano nel tenue prossimale , duodeno e prima parte del digiuno (perché qui si hanno le maggioriconcentrazioni di Ag !).
Clinicamente l’enteropatia da glutine può:- in genere rendersi sintomatica con sintomi intestinali e extraintestinali (_ sprue celiaca).- essere paucisintomatica: manifestazioni isolate e lievi di malassorbimento.- rimanere del tutto asintomatica.
La diagnosi di enteropatia da glutine si basa su 4 capisaldi:1) clinica: anche se esistono casi asintomatici2) sierologia: anticorpi anti-gliadina e anti-endomisio (o anti-transglutaminasi)3) istologia: dimostrazione dell’enteropatia.4) ex adiuvantibus: regressione dell’enteropatia dopo 1 anno di dieta aglutinata.
È importante diagnosticare e trattare con dieta aglutinata i pz con enteropatia da glutine perchè i pz non trattati hanno unamortalità doppia rispetto alla popolazione generale; questo in ragione del rischio di complicanze come la sprue refrattaria,la digiuno ileite ulcerativa, l’adenocarcinoma del tenue, i linfomi intestinali a cellule T.
La SPRUE TROPICALE o post-infettiva è una sindrome da malassorbimento che si sviluppa in seguito a una sindromediarroica acuta e persiste se non trattata.La malattia viene acquisita in paesi tropicali e la sua patogenesi sarebbe probabilmente una ipersensibilità a certe speciebatteriche che vengono acquisite nel corso di questi soggiorni; questo sarebbe supportato dall’efficacia di una terapiaantibiotica ad ampio spettro.In questo caso l’enteropatia, di grado variabile, è diffusa a tutto il tenue.Un segno spesso presente è l’alterazione in senso megaloblastico degli enterociti secondaria a un deficit di folati e vitaminaB12.
Deficit di disaccaridasiIl deficit riguarda soprattutto la lattasi e può essere talvolta congenito ma più frequentemente acquisito secondariamente adaffezioni del tratto digerente per cui si avrà la comparsa dei sintomi in età differenti.La mancata digestione del lattosio ne impedisce l’assorbimento con conseguente diarrea osmotica in seguito ad assunzionedi alimenti contenenti lattosio: eclatante la sintomatologia nel lattante.
Linfangectasia intestinaleEctasia dei vasi linfatici intramurali con o senza interessamento dei linfatici nello spessore del mesentere.La lesione fondamentale è la dilatazione dei dotti chiliferi che endoscopicamente corrisponde alla comparsa di macchiolinebiancastre (il chilo ha un aspetto lattescente) sulla superficie della mucosa: la ectasia può riguardare anche linfaticiintramurali di calibro maggiore e addirittura i vasi mesenterici.

La eziopatogenesi può essere:1) congenita: anomalia di sviluppo dei linfatici con sindrome da malassorbimento grave e precoce e rapidamente letale.2) secondaria: impedimento allo scarico linfatico in seguito a interventi chirurgici, morbo di Crohn, neoplasie del tenue.
Malattia di WhippleÈ una malattia infettiva provocata da un Actinomicete il quale si ritrova non solo a livello della lamina propria all’ internodi macrofagi pas-positivi, ma anche a livello linfonodale→linfonodi mesenterici ingrossati e linfangectasia intestinale ,della sinovia (artriti) , dell’ encefalo (sintomi neurologici) etc.Oltre a una sindrome cronica da malassorbimento i pz possono presentarsi con manifestazioni aggiuntive da interessamentosistemico.La malattia risponde bene agli antibiotici.
AbetalipoproteinemiaLa mancanza congenita dell’ApoB provoca la scomparsa dal circolo di VLDL, LDL e chilomicroni. Proprio la incapacità dirilasciare sotto forma di chilomicroni i grassi assorbiti, provoca un accumulo di lipidi all’interno dell’enterocita.Vi è pertanto un malassorbimento di lipidi e vitamine liposolubili con diarrea e steatorrea.A livello sistemico si ha iposviluppo, disordini neurologici, retinopatia e acantocitosi.
MALATTIE INFIAMMATORIE INTESTINALISi tratta del morbo di Crohn e della RCU.Entrambe queste patologie sono autoimmuni e a eziologia ignota.La loro prevalenza è simile (50/100.000).Esse però presentano molte differenze: le differenze possono essere importanti nella diagnosi differenziale fra colite diCrohn e RCU.
M. di Crohn Rettocolite ulcerosa
SEDE Potenzialmente qualsiasi punto del canalealimentare dalla bocca all’ano.
Ileo terminale > colon > altre sediInteressamento a salto: zone lese e zone indenni.
Sempre limitata al colon-retto ovvero…
… Parte sempre dal retto-sigma ma puòestendersi prossimalmente , in maniera continua,al colon fino a dare, in certi casi, una pancoliteulcerosa.
LESIONI Interessamento a tutto spessore:
1) infiltrato mononucleato cronicodiffuso reperibile in tutte le tonachema prevalente a carico di mucosa esottomucosa,
sono presenti sparsi granulomi non caseificantiche si ritrovano anche in zone risparmiate, laloro assenza tuttavia non esclude la diagnosi!
Lesioni riscontrabili in ordine di profondità:Ascessi criptici _ piccole ulcere aftoidi _ perconfluenza “ulcere lineari” longitudinali eserpiginose; per estensione in profondità,fissurazioni della parete (_ aspetto endoscopicoe contrastografico a “cobblestone”). Sierosite _ superficie del viscere grigiastra eopacata, iperplasia del tessuto adiposomesenterico, edema e ispessimento fibrotico delmesentere.
interessamento limitato a mucosa esottomucosa:
1) infiltrato mononucleato cronico dellamucosa e ascessi criptici _ mucosa friabile efacilmente sanguinante _ erosioni e ulcere(superficiali, limitate alla parte esterna dellasottomucosa) _ tenesmo rettale, proctorragia,diarrea essudativa: clinicamente la malattia haandamento a pousses (con riacutizzazioni):
possibili esiti:- per rigenerazione esuberante della mucosa si possono avere polipi infiammatori, talvolta ramificati.
- se i margini di due ulcere contigue sono sottominati si può formare un polipo infiammatorio “a ponte”.

mesenterico, edema e ispessimento fibrotico delmesentere.
Correlazioni anatomo-cliniche:1) atrofia mucosa (villi e cripte) _malassorbimento (ricorda che Sali biliari e B12nell’ileo);_ incidenza carcinomi2) edema cronico _ fibrosi _ stenosi post-infiammatoria dell’ileo terminale _ quadrisubocclusivi o occlusivi.
3) Possibilità di :- aderenze- formazione di tramiti fistolosi con l’esterno(le fistole perianali sono un reperto specifico delCrohn) o con un viscere cavo (vescica, vagina).- perforazione _ peritonite diffusa o circoscritta(ascesso).
- l’esito della colite cronica è una atrofiadella mucosa (cripte) e la comparsa didisplasia di vario grado, quindi mucosaadenomatosa in forma piatta o polipoide(_ dd polipo infiammatorio vsneoplastico) che precede il carcinomadel colon-retto per il quale questi pzpresentano un alto RR (20-30), anche inrelazione alla durata e alla estensionedella malattia _ categoria a rischiocandidata a screening endoscopico.
- riparazione con fibrosi della sottomucosa, il resto della parete è indenne (a meno che non si sviluppi come complicanza acuta un megacolon tossico).
Cenni diterapia
- Trattandosi di malattie autoimmuni, peraltro talvolta associate ad altre manifestazioni sistemiche diautoimmunità (eritema nodoso, artriti, iriti, colangite sclerosante), la terapia medica si basa suantinfiammatori e immunosoppressori.- la terapia chirurgica:1) nel Crohn si limita al controllo delle complicanze (stenosi, perforazione, megacolon tossico,fistole) e in ogni caso la recidiva è costante visto che tutto il tubo gastroenterico è colpito.2) nella RCU, oltre alle complicanze (vedi megacolon tossico), la chirurgia (essendo la malattialimitata al colon-retto) può essere pressoché risolutiva con un intervento di procto-colectomia +ricostruzione di una pouch con ileo + anastomosi ileo-anale.
SINDROMI ISCHEMICHE INTESTINALIVascolarizzazione del tratto gastrointestinale.1) tripode celiaco:- a. gastrica sn- a. splenica _ gastriche brevi (fondo), gastroepiploica sn.- a. epatica comune : gastroduodenale _ gastroepiploica dx e pancreatico-duodenale sup. epatica propria _ gastrica dx, cistica, epatiche
2) mesenterica superiore:- pancreatico-duodenale inf.- 15-20 arterie digiuno-ileali- ileocolica- colica dx- colica media
3) mesenterica inferiore- colica sn (anastomizzata con la colica media attraverso l’arcata di Riolano).- 2-3 aa. sigmoidee- emorroidaria superiore
4) arteria ipogastrica- emorroidaria media- pudenda interna _ emorroidaria inferiore

tenue: le arterie digiuno-ileali formano un primo ordine di arcate anastomotiche, da questo sorgono altri ordini di arcate;dall’ultimo ordine di arcate sorgono le arterie rette che si dirigono nel versante antimesenterico dove penetrano nella parete(vedi diverticoli).
colon: il colon riceve sangue dalla mesenterica superiore nella metà prossimale e dalla mesenterica inferiore nella metàdistale. È presente una arteria marginale (di Drummond) ovvero un canale continuo che parte dal ramo colico dellaileocolica e arriva alle sigmoidee ricevendo lungo il suo percorso la colica sn, la colica media e la colica sn.Dalla marginale partono anche in questo caso arterie rette.
ISCHEMIA CRONICAL’aterosclerosi può provocare una stenosi cronica del tripode celiaco e della mesenterica superiore a livello dell’orifizio;più raramente l’albero vascolare mesenterico può essere compromesso in corso di vasculiti.Questo porta a una sindrome caratteristica detta angina meseraica ovvero dolore addominale epigastrico post-prandialeche compare entro 15-30 min e dura almeno un’ora.
Il dolore può essere molto forte e associarsi ad anoressia (il pz evita il cibo per paura del dolore) e calo ponderale.Un soffio è in genere auscultabile in epigastrio.L’arteriografia è diagnostica.La terapia chirurgica consiste nella endoarterectomia o nel bypass aorto-celiaco e aorto-mesenterico: è assolutamenteindicato procedere all’intervento per evitare complicanze letali come l’inanizione o l’infarto intestinale.Nel caso delle vasculiti la terapia è medica.
Talvolta ci possono essere vaghi sintomi addominali accompagnati da diarrea ematica.In questo caso si è prodotta una enterocolite ischemica.In questo caso c’è una ischemia cronica, o anche acuta (es. ipoporfusione intraoperatoria) , che colpisce le tonache piùsensibili all’ischemia ovvero la mucosa e, a seguire, la sottomucosa.In sintesi si va:1) da infarti mucosi (_ erosioni all’apice dei villi)2) a infarti murali (_necrosi mucosa e sottomucosa _ ulcera ischemica).
Queste lesioni sanguinando possono rendere conto dell’ematochezia.Una enterocolite ischemica è importante anche perché entra per esempio con una IBD come il Crohn o una colitepseudomembranosa.Inoltre se la lesione è cronica si va incontro a stenosi ischemica.
In generale le sedi più sensibili all’ischemia sono:- nel tenue: il digiuno e l’ileo distale ovvero i territori più distanti dalle anastomosi della mesenterica superiore con iltripode celiaco e la mesenterica inferiore: non a caso l’infarto mesenterico si ha per occlusione embolica del segmentocritico di Reiner (mesenterica superiore a valle dell’ostio della colica media).- nel crasso: la flessura splenica ovvero il punto corrispondente all’anastomosi mesenterica superiore _ inferiore
INSUFFICIENZA MESENTERICA ACUTA:l’insufficienza vascolare mesenterica è suddivisibile dal punto di vista patogenetico come segue:
1) 50% - tromboembolia arteriosa (fibrillazione atriale, trombi murali)2) 25% - trombosi dell’arteria mesenterica superiore (complicanza dell’aterosclerosi, il pz ha una storia di angina
meseraica)3) 20% - malattia nonocclusiva: in caso di shock per la bassa pressione di perfusione e la vasocostrizione splancnica
l’intestino è uno degli organi meno per fusi. Questi pz hanno un’alto tasso di mortalità dovuto naturalmente anchealla malattia di base.
4) 5% - trombosi venosa (condizioni di ipercoagulabilità, ipertensione portale)
l’entità della lesione dipende sia dal grado di ischemia (funzionalità del circolo collaterale, livello dell’occlusione, durata)sia dalla riperfusione (liberazione di radicali liberi).Il pz con infarto si presenta in genere con un dolore addominale fortissimo e diffuso, possibile evidenza di sanguinamento(vomito, diarrea), leucocitosi.Obiettività estremamente povera rispetto al dolore.

Il pz va incontro a ileo paralitico (distensione addominale, dolorabilità)
L’infarto intestinale arterioso si differenzia da quello venoso solo per alcuni particolari come:1) limiti più netti2) stasi meno marcata ma cmq presente
qualitativamente si hanno gli stessi fenomeni la differenza è solo quantitativa per alcuni di essi.
Le lesioni fondamentali sono:
1) edema della parete dovuto alla stasi che cmq è presente anche nell’ infarto arterioso per la _ vis a tergo.Il trasudato si accumula anche nel lume che è dilatato se la peristalsi è bloccata (ileo paralitico).
2) colore bluastro dovuto alla desaturazione dell Hb.3) Infarcimento emorragico progressivo dovuto a rottura dei vasi (infarto rosso)_anche in questo caso il
sangue si riversa attraverso le erosioni della mucosa nel lume per cui l’epifenomeno sarà diarrea ematica (sec’è iperperistaltismo).
4) Sierosite fibrinosa con estensione anche al peritoneo parietale (addome acuto) _ perforazione con peritonitefecaloide, pneumoperitoneo etc.
5) La proliferazione e l’ingresso in circolo di batteri può portare a shock settico.
La prognosi in caso di infarto intestinale dipende dal momento della diagnosi e dall’eziopatogenesi:precoce (mortalità molto bassa) vs tardiva (molto alta).
--------------------------------
intussuscezione, volvolo, ernie.
Vediamo ora 3 patologie di interesse chirurgico che possono essere pericolose per 2 tipi di complicazione:1) occlusione intestinale2) strangolamentocome premessa generale queste 3 patologie interessano tratti di intestino che sono dotati di una certa libertà dimovimento in quanto sono, fisiologicamente o patologicamente (per anomalie congenite), dotate di un meso.
INTUSSUSCEZIONE:1) è nei 4/5 dei casi una patologia pediatrica: in questo caso è primitiva ovvero su base funzionale: lo spasmodi un segmento intestinale ne provoca la invaginazione nel successivo.
2) Negli adulti è una patologia gn secondaria ovvero dovuta a una massa polipoide di varia natura che spintadalla peristalsi si trascina dietro il tratto di tenue da cui origina.Più spesso per questioni di diametro è interessato il colon ascendente con invaginazioni ileo-coliche (l’ileo èinvaginato nel colon ascendente).nella intussuscezione si ha una trazione del mesentere _ conseguente strozzamento e rischio di infarto:l’intervento di riduzione è chirurgico anche se è possibile risolverlo con un clisma di acqua sotto pressione.
VOLVOLO: è la torsione di un ansa di intestino generalmente sull’asse del meso che la sostiene conconseguente occlusione e strozzamento.Un volvolo può potenzialmente insorgere a livello di:

TENUENei neonati si ha un volvolo del tenue da mancata fissazione del mesentere per cui un lungo tratto di intestinodal duodeno alla metà del colon trasverso rimane sospeso al sottile “istmo duodeno colico” nel quale passa lamesenterica superiore.
Nell’ adulto i volvoli del tenue sono dovuti a condizioni predisponenti come:- briglie aderenziali fra le branche di un’ ansa (alla base dell’ansa) oppure fra l’ansa e la parete addominale.- raramente da un tumore in una branca che provoca torsione per gravità oppure dalla persistenza del dotto vitellino.
CIECOse è dotato di meso può torcersi:- secondo un asse longitudinale oppure- in senso orario secondo un asse obliquo in basso e internamente _ va a finire in ipocondrio sn.
SIGMAÈ la sede più frequente (70%)condizioni favorenti sono1) un dolicocolon che può essere in parte congenito in parte secondario ad abitudini alimentari o unasindrome dell’intestino irritabile;2) una briglia aderenziale che avvicina alla base due segmenti del sigma.
Condizioni scatenanti sono sforzi come quelli della defecazione e del parto (quindi stipsi e gravidanza sonocondizioni di rischio).L’ andamento è cronico-ricorrente con crisi che si risolvono da sole anche se occasionalmente si potrannoprodurre lesioni irreversibili.
ERNIE
L’ernia intestinale in generale è costituita da un’ ansa:
- che si impegna in un recesso intraddominale costituito da briglie aderenziali (ernia interna ) o
- avvolta in un sacco erniario, costituito da peritoneo parietale, in un varco che si crea a livello di un locusminoris resistentiae della parete addominale (ernia esterna).
L’ernia può andare incontro a varie complicazioni come:- irriducibilità o incarceramento- intasamento/occlusione intestinale- strangolamento o strozzamento
Fra le ernie esterne le più frequenti sono:- inguinali: dirette o indirette (congenite o acquisite)- crurali (attraverso la lacuna vasorum)- ombelicali e della linea alba- postoperatorieetc.
I diverticoli sono estroflessioni circoscritte della parete degli organi cavi: la parete può essere composta:1) da tutti gli strati → diverticoli congeniti (Meckel) e da trazione2) da tutti gli strati assottigliati → diverticoli da cedimento (vedi sclerodermia)3) solo da mucosa e sottomucosa → diverticoli da pulsione o pseudodiverticoli (es. Zenker)

i diverticoli gastrici sono rari possono essere congeniti (a livello cardiale) o acquisiti a livello pilorico.
I diverticoli duodenali sono di due tipi:- diverticoli congeniti a livello peri-ampollare per cui si può avere compressione del coledoco.- diverticoli acquisiti: pseudodiverticoli da pulsione a monte di una stenosi peptica del bulbo duodenale.
I diverticoli digiuno-ileali sono infrequenti (~1/1000) si tratta di diverticoli da pulsione che originano nei “loci minorisresistentiae” ovvero nei punti in cui i rami delle arterie rette penetrano nella parete intestinale. Pur essendo a bocca stretta ilmateriale non ristagna perché a questo livello è liquido; solo in caso di diverticoli plurimi si può avere una sindrome damalassorbimento da iperproliferazione batterica: questo è qualcosa di simile alla sindrome dell’ansa esclusa ovvero lacomplicazione di un intervento chirurgico , volto a diminuire la superficie di assorbimento intestinale, che viene praticatonei casi di obesità mostruosa.
I diverticoli colici sono i più frequenti: non colpiscono il retto ma il sigma e tutto il colon restante con frequenzadecrescente procedendo in senso prossimale.Anche in questo caso si tratta di diverticoli da pulsione che originano a livello delle tenie antimesenteriche nei punti diingresso delle arterie rette. Hanno una bocca estremamente stretta e un diametro di 0.5-1 cm: il materiale anche per lamaggiore consistenza tende a ristagnare e a disidratarsi formando i cosiddetti coproliti.È una patologia in ↑ che colpisce soprattutto dopo i 50 anni (prevalenza di ~50% a 60 anni)La patogenesi è dovuta:
1) alla presenza di loci minoris resistentiae2) all’ipertensione intraluminale alla dieta carnea e povera di fibre: che porta a una riduzione del volume fecale e
alla comparsa di una attività di segmentazione spastica del sigma.La patologia può essere asintomatica ed allora si parla di diverticolosi ; se invece diventa sintomatica si parla di malattiadiverticolare che può estrinsecarsi semplicemente con una sintomatologia tipo intestino irritabile (dolore addominale, alvoalternante etc.) oppure con complicazioni quali:
- diverticolite: infiammazione della mucosa- peridiverticolite: flogosi del grasso pericolico- peritonite circoscritta (ascesso pericolico) o anche diffusapresentazione appendicite-like ma a dx: dolore, dolorabilità, difesa, febbre, leucocitosi etc.
- stenosi: flogosi cronica con ispessimento fibrotica della parete del sigma a tutto spessore- fistole: con gli organi cavi viciniori (colon-ileali o colon-vescicali) secondariamente alla
formazione di aderenze.- sanguinamento _ il 40% dei sanguinamenti inferiori è attribuibile a malattia diverticolare !Si noti come l’ematochezia sia attribuibile in un altro 20 % a lesioni angiodisplastiche (cieco e colon dx), in un 10% acoliti, in un 10% a lesioni neoplastiche (benigne o maligne), 20% indeterminate e miscellanea.Solo in 1/10 dei casi si tratta quindi di neoplasia!TUMORI INTESTINALI (tenue e colon)
Polipi non neoplasticiIperplastici (colon)Infiammatori (colon)Linfoidi (colon)Amartomatosi: giovanili (sporadico e poliposi) e s. di Peutz-Jeghers
Lesioni epiteliali neoplasticheadenomaadenocarcinomacarcinoide intestinalecarcinoma anale
Lesioni mesenchimali o GISTLipomi

AngiomiNeurinomiLeiomioma
Linfomi
------------------------------------
il tenue è incredibilmente poco colpito da patologia neoplastica (solo il 3-5% dei tumori intestinali).
TUMORI BENIGNI.1) adenomi : si tratta di lesioni polipoidi sessili o peduncolate,
unici o multipli. A differenza della loro controparte colica non sono considerati precancerosi.
2) GIST tra cui:- lipomi.- leiomioma.- neurinomi _ se multipli possono rientrare nella s. di von Recklinghausen.- emangiomi: capillare (gomitolo di capillari), cavernoso (grosse lacune a pareti sottili rivestite da endotelio).
3) polipi amartomatosi:in generale per polipo amartomatoso si intende una malformazione della mucosa: in sintesi le cellule sono normali (nonc’è displasia epiteliale) ma vanno a formare una struttura abnorme.La sindrome di Peutz-Jeghers è una patologia a trasmissione AD caratterizzata dall’associazione poliposi (presenza dipolipi amartomatosi peduncolati lungo tutto il tratto gastro-intestinale ma particolarmente a livello del tenue) +pigmentazione mucocutanea attorno agli orifizi.
Clinicamente le neoplasie benigne del tenue possono presentarsi per:- occlusione intestinale: per effetto massa, volvoli (masse extraluminali), intussuscezioni (masse polipoidi)- emorragia: emorragia intestinale se intraluminale (anemia sideropenica) oppure emoperitoneo se extraluminale.TUMORI MALIGNI DEL TENUE.Sono, come abbiamo detto, rari.Comprendono:
1) adenocarcinoma: un’adenocarcinoma del tenue cresce in genere in forma vegetante (polipoide) o stenosante (adanello). La presentazione clinica è simile a quella degli adenomi: per sanguinamento o per occlusione.La diffusione dell’adenocarcinoma avviene per contiguità (mesentere, anse contigue), per via linfatica, per viaematogena al fegato.
2) Carcinoide intesstinale: i carcinoidi intestinali e bronchiali appartengono al folto gruppo delle neoplasie diderivazione neuroendocrina che comprende anche il cr. midollare della tiroide, feocromocitomi e paragangliomi, leneoplasie endocrine del pancreas funzionanti e non, etc.i carcinoidi derivano da cellule endocrine associate alla mucosa intestinale e bronchiale.Si tratta di noduli giallastri che insorgono in ordine di frequenza a livello di:- appendice- tenue (soprattutto ileo)- retto- stomaco- colonistologicamente si tratta di cordoni di cellule monomorfe con nucleo rotondo e regolare.Tecniche immunoistochimiche sono usate per la diagnosi di natura di tumori neuroendocrini.
Il comportamento biologico del tumore è indolente:

quando è piccolo il tumore è asintomatico _ crescendo può manifestarsi per occlusione o sanguinamento _ inoltre sihanno metastasi ai linfonodi regionali e per via ematogena al fegato: in questo caso, una minoranza di pz presenta ilquadro classico della sindrome da carcinoide ovvero una sindrome paraneoplastica da iperincrezione di 5-idrossitriptamina (5-HT) - per gli amici serotonina -.La serotonina è catabolizzata principalmente dal fegato in acido 5-idrossindolacetico (5-HIAA) e questo ha dueimplicazioni importanti:1) il dosaggio del 5-HIAA nel sangue o nelle urine fa fare diagnosi.2) è necessario che vi siano metastasi epatiche (_ epatomegalia nodulare) affinché entri nel circolo sistemico una quantità di serotonina tale da produrre la sindrome che consiste in:
- flushing cutaneo e cianosi- diarrea da iperperistaltismo- asma bronchiale- fibrosi endocardica particolarmente a dx _ vizi alle valvole tricuspide e polmonare.
3) linfomi gastrointestinali (in generale): il tratto gastroent. è la prima sede per insorgenza di linfomi extranodali ovviamente a partenza dal tessuto linfoide associato alle mucose. Hanno vari gradi di malignità; in genere si tratta di linfomi a cellule B, raramente dei più aggressivi linfomi a cellule T. Possono essere sporadici o associati a varie condi- zioni predisponenti fra cui: - gastrite cronica da HP - sprue celiaca : provoca i linfomi T - origine mediterranea: plasmocitosi della mucosa intestinale _ m. immunoproliferativa del tenue
4) Leiomiosarcomi
I POLIPI DEL COLON
Premessa:“Polipo” è un termine generico per indicare una massa che aggetta nel lume , descrittivamente un polipo può esseresessile (ovvero a larga base d’impianto) o peduncolato (ovvero dotato di una testa sostenuta da un peduncolofibrovascolare).In genere si chiama polipo una massa che tragga comunque origine dalla mucosa.Lesioni aggettanti che prendono origine da altre tonache come un leiomioma, un lipoma o un neurinoma sono dettepseudopolipi.
1) Il polipo non è necessariamente di natura neoplastica _ è importante conoscere i vari tipi di polipo per poter fareuna diagnosi differenziale.
2) è particolarmente importante identificare la natura di un polipo perché:a) quasi sempre (97%) il carcinoma colorettale insorge su un polipo adenomatoso (vedi oltre).b) è possibile, visto che lesioni adenomatose (benigne) impiegano parecchi anni prima di trasformarsi in carcinoma, fare
profilassi secondaria per il carcinoma del colon-retto.
Fra i polipi non neoplastici del colon ricordiamo:
- i p. amartomatosi ovvero lesioni malformative della mucosa che possono presentarsi come:a) polipo giovanile: gn a livello del retto, ha una testa grande(anche 3 cm) e
lobulata nonchè un lungo peduncolo: può sanguinare e la testa essere espulsa con l’atto della defecazione _ la lesione è del tutto benigna.
b) poliposi giovanile: presenza di polipi amartomatosi multipli (malattia su base AD) che si associa ad _R di carcinoma.c) sindrome di Peutz-Jeghers: malattia AD: presenza di polipi amartomatosi multipli dallo
stomaco in giù (specie nel tenue) + macchie melanosiche a livello degli orifizi. _ si associa a un _rischio di carcinomi extraintestinali (pancreas, mammella, ovaio, utero).

- i p. infiammatori (allungati e ramificati o a ponte _ vedi colite di Crohn, RCU)
- i p. linfoidi benigni: epitelio sollevato da tessuto linfoide intramucoso benigno.
- ma soprattutto i p. iperplastici:Questi ultimi hanno un’altissima prevalenza (50%) nei soggetti > 60 anni.Sono polipi piccoli, sessili, lisci e bruni, per lo più a livello sigmo-rettale, microscopicamente costituiti da cripte profonde elarghe con epitelio affollato per cui si formano ripiegamenti simil papillar nel lume.Rappresentano il 90% di tutti i polipi del colon, non sono una condizione di rischio per il CCR.
I polipi neoplastici sono i p. adenomatosi.Si tratta di polipi costituiti da un epitelio proliferante displastico o adenomatoso (cellule alte, ipercromatiche, disordinate)che si solleva formando polipi di tipo sessile o peduncolato.La displasia può essere lieve, moderata, grave (CIS).
Dal punto di vista microscopico i polipi adenomatosi sono classificabili come:- tubulari (> 90%): componente tubulare > 70%- tubulo-villosi (<10%): componente tubulare del 50-70%- villosi (< 1%): componente papillare > tubulare
Gli adenomi tubulari si presentano:- se piccoli: sessili e lisci- se grandi: peduncolati con una testa lobulata; il peduncolo è un asse fibrovascolare che appartiene alla sottomucosa e che è in genere ricoperto da mucosa normale anche se talvolta può essere ricoperto da mucosa displastica e allora la radicalità della polipectomia può essere compromessa.
Gli adenomi villosi sono lesioni sessili, vellutate o a cavolfiore.- possono anche essere lesioni piatte, non polipoidi.- sono facilmente sanguinanti.- mancano del peduncolo che rappresenta una specie di tampone all’espansione del tumore e tendono quindi a infiltrare più rapidamente la muscolare propria.
Gli adenomi misti tubulo-villosi hanno caratteristiche intermedie e possono essere sessili o peduncolati.
I polipi adenomatosi si ritrovano più o meno con la stessa frequenza degli iperplastici (~50%) nella popolazioneultrasessantenne; in termini assoluti però, se in un paziente trovo 10 polipi epiteliali 9 saranno iperplastici, 1adenomatoso.
I polipi adenomatosi sono estremamente importanti perché, come già accennato, si calcola che il 97% dei CCR insorga daun carcinoma. In pratica l’adenoma sarebbe una delle tappe di un processo multistep che porta alla trasformazione epitelionormale _ CCR.Si calcola che la trasformazione adenoma _ CCR richieda mediamente 10 anni.La sequenzialità adenoma _carcinoma è supportata da molteplici evidenze.
I polipi si distribuiscono per quasi la metà nel retto-sigma (50%), vengono quindi il segmento ascendente e traverso (15%ciascuno) e il discendente (10%).
La probabilità di avere o sviluppare un carcinoma è correlata:1) alla presenza della componente villosa (che si associa più frequentemente a displasia severa)2) alla dimensione dei polipi adenomatosi3) al numero dei polipi adenomatosi

A questo proposito vi sono alcune sindromi ereditarie che si associano a un gran numero di polipi adenomatosi (poliposi delcolon) con aumentato rischio di sviluppare cancro già in età giovanile.
La più importante è la FAP o poliposi familiare adenomatosa una sindrome con una frequenza di 1/10.000 nascite,consistente nella presenza di almeno 100 (ma possono essere migliaia) polipi nel colon-retto.La malattia è AD a penetranza incompleta.È associata a certezza (rischio del 100%) di sviluppare cancro del colon per cui si procede alla colectomia profilattica e alloscreening sui familiari (rettosigmoidoscopia biennale fino ai 40 aa per vedere se compaiono i polipi _ la malattia è apenetranza incompleta!).
Altre sindromi sono:la sindrome di Gardner associata fra l’altro a osteomi della mandibola, lipomi, fibromi, cisti epidermoidali.
la sindrome di Turcot associata fra l’altro a neoplasie cerebrali (gliomi).
la sindrome di Lynch o HNPCC che consiste nel riscontro di CCR in almeno 3 membri della stessa famiglia dei qualialmeno 2 devono essere genitore/figlio e almeno 1 deve aver sviluppato il CCR prima dei 50 anni. Questa non è unapoliposi, la displasia dalla quale insorge il carcinoma è piatta.
Abbiamo già parlato delle poliposi amartomatose nelle quali i polipi si distribuiscono soprattutto nel tenue e che sonoassociate a un rischio anche se non alto come nelle precedenti sindromi di carcinomi: sono la sindrome di Peutz-Jeghers e lapoliposi giovanile.
IL CARCINOMA DEL COLON-RETTOle CdR sono:
- l’età (>50 anni)- I polipi adenomatosi
- Le sindromi familiari, soprattutto la FAP e la HNPCC (tuttavia questi casi sono una minoranza).- Le IBDs in particolare la RCU di lunga data e molto estesa (RR=20) e in pz con Chron (RR=5).
i FdR sono soprattutto legati al tenore di vita delle nazioni occidentali:- dieta ricca di grassi saturi di origine animale _ _turnover acidi biliari _ _acidi che arrivano in colon e vengono quideconiugati e riassorbiti _ gli acidi biliari deconiugati sono lesivi per la mucosa.- dieta povera di fibre _ diluiscono eventuali cancerogeni e regolarizzando l’alvo ne consentono un più rapidoallontanamento.- vita sedentaria, obesità _ stipsi- cibi fritti e alla brace- alcol, fumo
il principale fattore protettivo è una dieta equilibrata, ricca di fibre, vitamine e oligoelementi e povera di grassi.
Paradigmatico della cancerogenesi ambientale è il caso della popolazione giapponese emigrata negli USA. Dopo duegenerazioni, per il cambiamento delle abitudini alimentari si nota una _del cr. gastrico (associato al consumo di carboidratinon raffinati e cibi conservati con metodi tradizionali etc.) e un _del cr. colorettale associato alla dieta americana ricca dicarboidrati raffinati poveri di fibra, eccesso di grassi insaturi di origine animale etc.
Clinica
1) generalmente i polipisono asintomatici.
2) talvolta possonomanifestarsi perchésanguinano con:anemia ferro-carenziale (_sangueocculto nelle feci) oematochezia.
3) possono essere unreperto incidentale (onon) nel corso diclisma a doppiocontrasto oendoscopia.
L’endoscopia (retto-sigmoidoscopia o meglioancora la pancolonscopia) è unaprocedura a doppia finalità:
1) diagnostica2) curativa
all’endoscopia infatti siriescono ad asportare i polipipeduncolati e piccoli polipisessili.Talvolta se il polipo è sessile alarga base d’impianto o se ipolipi sono molti potrebbeessere preferibile la resezionechirurgica del segmentointestinale interessato.
Una volta asportato il polipoviene esaminato dal patologo

SEDEIl cancro si distribuisce in ordine di frequenza a:1) retto _ pur essendo lungo solo 15 cm è la sede del 45%2) sigma _ 25%3) cieco e colon ascendente _ 18%4) colon discendente _ 5%5) colon trasverso _ 9%
ISTOLOGIA
- Tubulare- cellule a castone: il muco disseca i tessuti colici rendendo la neoplasia più aggressiva- Indifferenziato a grandi cellule- indifferenziato a piccole cellule (tipo microcitoma polmonare, può secernere)
STADIAZIONE
La stadiazione (più patologica che clinica) più seguita dai chirurghi è quella di Dukes-Astler-Coller, più semplice rispettoal TNM che viene riportato fra parentesi, a ciascuno stadio corrisponde una prognosi diversa:
A: entro la sottomucosa (T1)
B1: entro la muscolare propria (T2) B2: raggiunge e supera la sierosa (T3) _ _C1: T1-2, N+ C2: T3, N +
D: metastasi a distanza (Tx, Nx, M1)
ProfilassiIl problema dello screening sulla popolazione generale si pone perché il colon è una patologia socialmente importante (è la3a causa di morte per cancro in entrambi i sessi) , con un lungo periodo “prodromico” (fase di adenoma che dura 10-15anni) e quindi potenzialmente controllabile con un intervento di screening.I metodi di screening sono diversi:- alcuni economici e non invasivi ma poco sensibili o specifici come il sangue occulto nelle feci.- Altri metodi sono al contrario costosi e invasivi ma più sensibili, specifici o addirittura curativi come la colonscopia. Viste anche le limitate risorse della Sanità Pubblica si deve fare un’attenta analisi dell’efficacia e dell’efficienza di questiscreening.
È sicuramente opportuno controllare con un Follow up endoscopico pazienti ad alto rischio come in caso di:1) pregresso adenoma o CCR (con periodicità variabile in base a vari fattori)2) portatori di RCU o colite di Crohn: iniziare a 10 anni dall’esordio dei sintomi3) membri di famiglie HNPCC: dai 25 anni in poi.4) membri di famiglie FAP: dall’adolescenza ai 40 anni (basta la rettosigmoidoscopia).

Il fegato è un organo parenchimatoso collocato nel quadrante superiore dx dell’addome, esso svolge molteplici funzioni.
Dal punto di vista anatomico si può dire che:- il peso è di 1,2-1,5 Kg- il fegato è “servito” in entrata da:1) vena porta e2) arteria epaticache provvedono rispettivamente a 2/3 (65%) e a 1/3 (33%) dell’ apporto ematico.In uscita troviamo:3) le vene sovraepatiche che confluiscono con la cava inferiore e4) i dotti epatici dx e sn che confluiscono nel dotto epatico comune e che sono deputati al drenaggio del secreto biliare.
- Il parenchima epatico può essere suddiviso in più parti:1) 2 lobi , dx e sn, individuati da un piano passante per la cava e la colecisti,2) 8 segmenti che sono unità irrorate con una circolazione di tipo terminale che consente la loro resecabilità
chirurgica. Essi sono:- i laterali superiore e inferiore nella zona laterale (al di là del legamento falciforme).- il caudato e il quadrato al di qua del legamento falciforme nel lobo anatomico sn.- i 2 anteriori (superiore e inferiore)- i 2 posteriori (superiore e inferiore)
- Microscopicamente l’unità anatomica del fegato è il lobulo epatico il quale consiste di un aggregato di lamine diepatociti disposte radialmente (travate di Remak) e di una lamina limitante esterna o piatto limitante e infinecircondato da tessuto connettivo che si organizza nello SPAZIO PORTALE dove decorrono i rami:
1) dell’a. epatica2) della vena porta3) delle vie biliari intraepatiche
il sangue che irrora il lobulo è misto e quindi relativamente povero di O2.Esso fluisce all’ interno dei sinusoidi, capillari a lume ampio e ampiamente fenestrati che decorrono fra le lamine diepatociti per cui ciascun epatocita verrà ad avere un polo vascolare al quale si affaccia sul sinusoide; la direzione delflusso è centripeta ovvero dallo spazio portale al centro del lobulo dove il sangue venoso viene drenato dalla venacentrolobulare tributaria delle vene sovraepatiche: è ovvio che gli epatociti pericentrolobulari siano i più suscettibiliall’ipossiemia in quanto vengono in contatto con sangue già deossigenato alla periferia del lobulo.Si può quindi individuare l’unità funzionale o metabolica del fegato ovvero l’acino triangolare o di Rappaportsuddivisibile in 3 zone: 1, 2 e 3 procedendo dal piatto limitante alla vena centrolobulare.Le zone dell’acino di Rappaport (periportale, intermedia, pericentrolobulare) sono importanti perché ci sono gradientidi attività enzimatiche e differente suscettibilità a vari tipi di insulti.
Fra il sinusoide e il polo vascolare dell’epatocita si trova lo spazio di Disse.In questo spazio troviamo:

1) cellule istiocitiche dette cellule del Kuppfer che appartengono al SRE, tali cellule hanno la funzione di filtro per ilmateriale estraneo, di riserva del Fe (ferritina _ emosiderina), di difesa in corso di danno epatico.
2) cellule di Ito che fungono da deposito per la vitamina A, nel corso di danno epatico possono andare incontro adifferenziamento in elementi miofibroblastici e rendersi responsabili della fibrosi (vedi oltre).
L’epatocitaAl versante vascolare l’epatocita possiede microvilli che servono ad ottimizzare i processi di assorbimento e inparticolare quello della bilirubina indiretta (non coniugata): questi villi sono assenti dopo le epatiti virali per cui si haiperbilirubinemia post-epatitica.
La bile (o meglio la pre-bile visto che il secreto subisce modificazioni nella composizione lungo le vie biliari) è secretadall’epatocita al polo biliare: anche in questo caso si hanno microvilli che servono per ottimizzare la secrezione dellabile che comprende fra l’altro bilirubina indiretta; la scomparsa di questi microvilli che si ha per es. in caso di terapiaanticoncezionale provoca un ittero epatico da _ bilirubina indiretta.I canalicoli biliari non hanno una parete propria bensì sono costituiti dalla giustapposizione di due epatociti con laformazione di un lume di 1-2 micron; nel caso di necrosi epatocitaria la continuità del sistema canalicolare vieneinterrotta e la bile passa nei sinusoidi per cui si ha ittero.Alla periferia del lobulo si trovano canali rivestiti da elementi endoteliali (canali di Hering); nello spazio portaletroviamo invece i dotti biliari interlobulari delimitati da epitelio cubico maturo.
Per quanto riguarda il sistema di scarico linfatico si hanno linfonodi:1) sovraepatici2) ilari
per quanto riguarda i quadri degenerativi dell’ epatocita osservabili in corso di epatopatie abbiamo:
1) degenerazione torbida: edema cellulare (ipossia, tossine, infezioni virali) con _ volume citoplasmatico; questo siassocia a un _ volume e di peso al taglio quando è di grado particolarmente grave si parla di degenerazioneballoniforme (epatiti): il rigonfiamento porta a schiacciamento dei sinusoidi con conseguente lieve ipertensioneportale ma soprattutto con danno ipossico per la _ perfusione del lobulo.
2) A livello citoplasmatico può osservarsi dapprima un viraggio degli epatociti verso la eosinofilia, a livelloperinucleare possono quindi comparire formazioni eosinofile dette corpi di Mallory : questi aggregati di proteinepresenti in cellule sparse preludono alla necrosi e si osservano in genere nella cirrosi alcolica.
Formazioni esonofile analoghe al Mallory sono i corpi di Councilman i quali tuttavia sono:- residui di cellule andate incontro ad apoptosi e non a necrosi.- di forma rotondeggiante e non irregolare.- Localizzate ai margini delle travate di Remak in quanto sono espulse per essere riassorbite- Si ritrovano nelle epatiti virali.
3) la degenerazione grassa o steatosi è caratterizzata dall’ accumulo di lipidi intracitoplasmatici in forma:- microvescicolare: vacuoli che non dislocano il nucleo è dovuta a un difetto della _-ossidazione mitocondriale degli
acidi grassi_ gravidanza e malattia alcolica- macrovescicolare: grande vacuolo che disloca il nucleo è dovuta a meccanismi che portano ad _sintesi di
trigliceridi nel fegato_ diabetici, obesi, alcolisti., dislipidemia.
4) vi sono anche altri tipi di degenerazione come la degenerazione schiumosa in corso di colestasi
la morte cellulare o necrosi può avvenire per lisi oppure per apoptosi con la formazione dei suddetti corpi di Councilman.La localizzazione e l’estensione della necrosi è varia potendo essere pericentrolobulare specie per danno ischemico otossico-farmacologico, più raramente periportale o intermedia;La piecemeal necrosis è data dal coinvolgimento della lamina limitante esterna.

La necrosi a ponte è data dalla confluenza di zone di necrosi fra spazio portale e vena centrolobulare.Queste lesioni istologiche sono importanti nella stadiazione dell’epatite cronica.
L’infiammazione del parenchima epatico altrimenti detta epatite è costituita dall’ infiltrazione di celluledell’infiammazione acuta o cronica, la quale infiltrazione può essere conseguente (danno tossico) o determinante (dannoimmuno-mediato come nell’ HB) la necrosi.
La rigenerazione è un aspetto successivo al danno epatico: si presenta con mitosi degli epatociti e un relativoscompaginamento delle travate; si osserva inoltre proliferazione dei dotti biliari a livello portale: condizione necessaria èl’integrità delle fibre reticolari che costituiscono lo stroma del lobulo.
La fibrosi rappresenta l’evoluzione cui si va incontro in conseguenza del danno epatico: può essere presente a livellocentrolobulare , portale, così come parenchimale a livello dell’interstizio che nel lobulo è lo spazio di Disse.La fibrosi è una lesione irreversibile e tuttavia statica, non evolutiva.Viceversa la cirrosi è una forma progressiva con la formazione di ponti connettivali che delimitano noduli di epatocitirigeneranti.
Ittero e colestasiLa bilirubina è il catabolita dell’eme che a livello epatico viene captato, coniugato con il glucuronato e secreto entrando cosia far parte della bile (_ escrezione con le feci cui conferisce il colore brunastro).La bilirubina non coniugata o indiretta è relativamente insolubile e viaggia nel sangue legata all’ albumina → non hasignificativa escrezione renale, passa la BEE ancora immatura del neonato potendo provocare in tutti i casi di ↑ della quotalibera (l’eritroblastosi fetale da incompatibilità Rh o ABO è la forma classica) gravi danni neurologici (kernicterus).L’eritroblastosi fetale che può portare a kernittero non va confusa con l’ittero fisiologico del neonato, una iperbilirubinemiaindiretta dovuta al fatto che fino alla 2a settimana i meccanismi di coniugazione ed escrezione della bilirubina non sonoancora del tutto efficienti. I bambini allattati al seno hanno una maggiore tendenza a sviluppare questo ittero fisiologicoperché il latte materno contiene _-glicuronidasi che scindono i glicuronidi della bilirubina a livello intestinale favorendone ilriassorbimento.
La bilirubina coniugata o diretta è invece un pigmento solubile, non tossico, che può essere escreto in caso di↑concentrazione sierica (ittero ostruttivo) anche per via renale.
La concentrazione normale di bilirubina è inferiore a 1 mg/dl; fra 1 e 4 si ha subittero in cui è possibile rilevare lacolorazione giallastra delle sclere e oltre 4 si ha ittero franco (colore giallastro della cute).Questi valori si riferiscono ai valori di bilirubina nell’ insieme: tuttavia ad aumentare può essere soprattutto la bilirubinadiretta ovvero la indiretta a seconda del meccanismo che ha portato all’ittero.
FISIOPATOLOGIA DELL’ITTERO- eccessiva produzione di bilirubina → condizioni di eritropoiesi inefficace e/o emolisi- ↓ captazione → ittero post-epatitico (carenza di villi al polo vascolare),- alterata coniugazione epatica→ varie sindromi su base ereditaria come :
1) Crigler-Najjar _ con deficit totale (tipo I: incompatibile con la vita _ kernittero letale) o solo parziale (tipo II: sviluppo normale ma rischio di kernittero)
2) Gilbert: molto frequente (5% della popolazione) e quindi benigna: episodi di subittero in relazione a stress fisici- alterata secrezione della bilirubina coniugata al polo canalicolare→ sindromi di Dubi Johnson (caratteristiche inclusioni intracellulari di pigmento) e di Rotor ad andamento assolutamente benigno.- Nel quadro più ampio della colestasi ostruttiva (ritenzione bilirubina e Sali biliari).
la colestasi è per definizione la ritenzione dei costituenti fondamentali della bile: si ricordi a questo proposito come, a parte iprodotti di detossificazione (metaboliti escreti in forma coniugata fra cui la bilirubina diretta), i veri costituenti funzionali(emulsificazione dei lipidi ingeriti) siano i sali biliari e i fosfolipidi.La colestasi si divide fondamentalmente in due tipi:
1) il primo, seppure raro, è quello epatocellulare _ difetti congeniti nell’escrezione di Sali biliari e fosfolipidi.L’unica lesione è in questo caso la degenerazione schiumosa dell’epatocita.

2) il secondo è quello ostruttivo _ in questo caso l’ostruzione può essere secondaria a svariati processi patologici edefinibile in base alla sede come intraepatica oppure extraepatica. In entrambi la via biliare a monte della stenosipresenta:
- ipertensione _ distensione, proliferazione dei dotti biliari- stasi _ edema degli spazi portali, concrezioni di pigmento nei dotti, degenerazione schiumosa degli epatociti.È importante notare come la colestasi ostruttiva porti con il passare del tempo a lesioni parenchimali (es. necrosiepatociti e formazione di laghi di bile) che evolvono in fibrosi e cirrosi.
La stasi biliare ha come manifestazioni cliniche:- il prurito dato dal passaggio in circolo dei sali biliari- talvolta gli xantomi cutanei dovuti all’iperlipidemia secondaria a ritenzione di colesterolo- l’ elevazione dei marker di lesione delle vie biliari come la fosfatasi alcalina- una sindrome da malassorbimento specie riguardo a nutrienti liposolubili come alcune vitamine.
L’insufficienza epatica è l’esito terminale delle epatopatie: in generale il fegato ha un ampia riserva funzionale per cui deveessere perduto almeno l’80-90% del parenchima. La insufficienza epatica può sottendere 3 diversi quadri eziopatogenetici-morfologici:
- necrosi epatica massiva (atrofia giallo-bruna o fegato a noce moscata) come nel caso di epatite acuta fulminante odi avvelenamento da A. phalloides, da CCl4, da vari farmaci (acetaminofene, alotano, isoniazide).
- Malattia epatica cronica- Disfunzioni epatiche in assenza di necrosi: es. s. di Reye
Le funzioni dell’epatocita sono moltissime, il fegato è una vera e propria centrale biochimica:1) metabolismo dei carboidrati: glicogeno, gluconeogenesi etc.2) metabolismo dei lipidi : trigliceridi, colesterolo, lipoproteine, chetoni.3) metabolismo dell’eme: formazione di bilirubina e sua coniugazione4) metabolismo dell’azoto: formazione di urea5) detossificazione: farmaci, tossici, ormoni.6) sintesi di plasmaproteine: albumina, fattori della coagulazione, complemento, etc.
L’insufficienza epatica comporta varie alterazioni ovviamente dovute a ↓ produzione di fattori di sintesi epatica e ↓biotrasormazione di molecole anche endogene per cui avremoDa una parte: ipoalbuminemia, coagulopatia, ipocomplemetemia.Dall’altra: ↓clearance di estrogeni (ipogonadismo e ginecomastia, spider nevi), mercaptani (fetor hepaticus), ammoniaca ealtri tossici (encefalopatia epatica).
LA CIRROSI EPATICAPer “cirrosi epatica” si intende una fibrosi epatica diffusa, irreversibile e progressiva con formazione di noduli diparenchima rigenerante delimitati da tralci fibrosi.I noduli possono essere:micro (< 3 mm) oppure macro (> 3 mm fino a diversi cm)
la cirrosi è il risultato di insulti cronici al parenchima epatico.dal punto di vista patogenetico è caratterizzata da una deposizione, abnorme, di collagene tipo I e III che normalmente è presente solonegli spazi portali e in sede centrolobulare anche a livello lobulare; le cellule responsabili sarebbero le cellule di Ito che subiscono unametamorfosi assumendo un fenotipo miofibroblastico.I determinanti che producono la fibrosi sono le citochine rilasciate in corso di infiammazione come il TNF-α e il TGF-β; la distruzionedella matrice extracellulare etc.
la cirrosi epatica è un problema sanitario rilevante: 26.000 nuovi casi/anno.In Italia il 90% delle cirrosi è di origine virale o alcolica:
- Da virus B (HBsAg) con eventuale virus D (anti-Delta)- Da virus C (anticorpi anti-C o HCV-RNA)- Da alcol (consumo di >40g/die -20 per la donna- per almeno 10 anni)
Nel Nord Italia sono più frequenti le forme alcoliche, nel resto del paese quelle infettive.

Oggi la mortalità per cirrosi è diminuita: ciononostante questa è una patologia che per le sue complicazioni ha grossericadute sulla qualità della vita e inoltre è _l’incidenza del Cr. Epatocellulare.
Nella restante parte dei casi:- malattie delle vie biliari (cirrosi biliare)- emocromatosi ereditaria- morbo di Wilson- deficit di _1-antitripsina
il cirrotico attraversa una prima fase di compenso nella quale la sintomatologia èmolto scarsa.Si può avere astenia nelle forme virali, febbricola in quelle autoimmuni o prurito inquelle colestatiche.I segni di malattia sono sovrapponibili a quelli di una epatite cronica: epatomegalia emodesta alterazione degli enzimi.La DD epatite – cirrosi (si noti che la prima è conseguenza della seconda) può quindirichiedere l’esecuzione di una biopsia epatica.
Sintomi e segni ben più significativi si hanno quando si passa alla fase di scompenso:Si avranno allora tutta una serie di segni e sintomi caratteristici.
1) secondari all’insufficienza epatica2) secondari alla ipertensione portale
L’ ipertensione portale in corso di cirrosi è dovuta:a) all’↑R intraepatiche dovuto sia allo sconvolgimento dell’ architettura sia alla
capillarizzazione (scomparsa fenestrature per via della deposizione di collagenenello spazio di Disse) e vasocostrizione dei sinusoidi (da parte delle cellule di Itoa fenotipo miofibroblastico).
b) All’ iperafflusso di sangue al circolo portale in seguito a una vasodilatazionearteriolare viscerale _ ne può conseguire un circolo ipercinetico.
In ogni caso l’ipertensione portale si associa a 4 principali conseguenze:1) Ascite _ raccolta di liquido di almeno 500 ml nel cavo peritoneale (talvolta molto
di più con il classico addome “batraciano”): al sinusoide epatico si ha uncospicuo passaggio di liquido nello spazio di Disse (ricorda che le proteine fannofatica a passare direttamente in circolo per la capillarizzazione del sinusoide _
vengono trattenute nello spazio di Disse e provocano trasudazione di liquido) _ Questo liquido costituisce un grossoquantitativo di linfa che eccede la capacità di drenaggio del dotto toracico per cui si ha filtrazione in cavità peritonealeattraverso la capsula epatica.Meccanismi additivi sono la filtrazione dal microcircolo intestinale (ipertensione portale) e l’iperaldosteronismosecondario.
2) circoli collaterali _ caput medusae (vena ombelicale), varici esofago-gastriche (gastriche brevi ex splenica e venacoronaria stomacica), plesso emorroidario (mesenterica inferiore) , sistema del Retzius (collaterali nel retroperitoneo).
3) shunt porto-sistemici _ encefalopatia porto-sistemica.
4) splenomegalia congestizia _ talvolta c’è ipersplenismo secondario
PATOLOGIA INFIAMMATORIA DEL FEGATO ovvero LE EPATITI
La cirrosi epatica, una delleprime dieci cause di morte almondo, rappresental’evoluzione di patologie adeziologia varia per cui sidistinguono le cirrosi:
- alcolica- biliare- post-epatite virale- da sovraccarico di Fe
(emocromatosigenetica)
- da sovraccarico di Cu(morbo di Wilson)
- altre cause rare
Dal punto di vista istologicouna volta che la cirrosi si èsviluppata è difficile risalireall’eziologia.
Si noti cmq come la cirrosi siauna forma di fibrosi:1) diffusa2) progressiva ed
irreversibile3) caratterizzata da noduli di
parenchima micro o macrodelimitati da tralci fibrosiporto-portali; porto-centrolobulari;
con complessivosconvolgimento quindi dell’architettura parenchimale maanche di quella vascolaredovuta agli shunt fra spaziportali e vena centrolobulare
Oltre al coinvolgimento dellafunzionalità del parenchimaepatico la cirrosi è la principalecausa intraepatica diipertensione portale:si noti infatti come questacondizione possa essere ancheprovocata da cause:
- pre-epatiche:trombosi, stenosiportale, splenomegaliamassiva (i.p. daiperafflusso)
- post-epatica:scompenso dx severo;pericardite costrittiva;s. di Budd-Chiari

Il reperto di flogosi del parenchima epatico è di per sé molto aspecifico nel senso che molte cause possono esserneresponsabili: d’altra parte l’infiammazione può essere sia causa (forme autoimmuni, ipersensibilità a farmaci, epatite B) checonseguenza del danno al parenchima epatico (es. epatite A, tossicità diretta dei farmaci etc.).Se il reperto di epatite è per definizione anatomo-patologico, nella pratica clinica la diagnosi di epatite è per lo più clinico –laboratoristica:La presentazione clinica può essere eclatante nelle forme acute itteriche o acute fulminanti, e d’altro canto può essereassente o insidiosa nelle forme acute anitteriche o nell’epatite cronica. Importante è l’anamnesi: sia familiare (precedenti)che personale (alcol, farmaci, FdR per epatiti virali etc.).
Il laboratorio offre:- innanzitutto una diagnosi biochimica di epatopatia e/o colestasi- in secondo luogo test per caratterizzare l’epatopatia di tipo sierologico (epatiti virali ma non solo), biochimico, etc.
A parte i reperti autoptici, l’anatomia patologica è in vivo un importante strumento diagnostico : la biopsia epatica puòinfatti essere importante per la conferma diagnostica e il follow- up delle epatiti croniche che, come tali, evolvono nel tempoa cirrosi.
In base all’eziologia si riconoscono epatiti:- infettive- autoimmuni- da farmaci- in corso di disordini ereditari etc.
in base al decorso clinico si riconoscono epatiti:- fulminanti- acute- croniche.
EPATITI INFETTIVESi tratta:- principalmente di epatiti virali ovvero dovute a virus epatotropi, molto più raramente, nei pazienti immunodepressi si puòavere coinvolgimento del fegato in corso di infezioni virali sistemiche in particolare da parte delle herpesviridae (HSV,CMV, EBV) o da rosolia, adenovirus, enterovirus.- secondariamente di epatiti batteriche e parassitarie.
LE EPATITI VIRALIVi sono essenzialmente 5 tipi di virus epatotropi:
HAV e HEV sono virus a trasmissione oro-fecale (acque inquinate, cibi non lavati e/o non cotti etc.).L’infezione , che è unicamente acuta, può restare asintomatica (epatite acuta anitterica), oppure con un periodo dincubazione che arriva a 6 settimane, può dare epatite acuta, oppure ancora epatite fulminante (raramente l’HAV; altamortalità -20%- invece per le infezioni da HEV in gravidanza).Il superamento dell’infezione lascia uno stato di immunità segnalato da anticorpi che sono anche diagnostici (IgM _ IgG).
HBV, HCV e HDV sono virus a trasmissione parenterale: apparente (trasfusioni di sangue o emoderivati infetti,condivisioni di siringhe fra tossicodipendenti) o inapparente (verticale madre-figlio, sessuale).Danno un’ infezione che anche stavolta risulta dal punto di vista clinico asintomatica, epatite acuta o fulminante.
In questo caso la guarigione non è scontata.Se l’infezione da virus B non cronicizza quasi mai (ad eccezione di individui immunodeficenti potremmo dire nell’1% deltotale; il 90% solo se l’infezione è perinatale) l’infezione da virus C cronicizza nel 90% dei casi.Il virus D non ha un comportamento suo proprio ma segue, essendo difettivo, quello del virus B.
Quindi l’infezione cronica potrà avere come controparte clinica:- lo status di portatore sano- l’epatite cronica

è l’epatite cronica che evolve con una certa frequenza in cirrosi.
L’HBV ha un’incubazione variabile da 1-6 mesi.I markers di malattia sono:- l’antigene virale solubile o HBsAg che in ogni caso segnala un’infezione in atto.- gli antigeni virali “core” e “pre-core”, HBcAg e HBeAg, il primo solo intracellulare, il secondo presente in circolo, che assieme all’HBV-DNA sono markers di replicazione virale in atto (NB. il virus B integrato non replica).- gli anticorpi anti-HBsAg che sono indice di guarigione
in sintesi sono particolarmente importanti l’HBsAg che indica infezione in atto e l’anti-HBsAg che indica guarigione.L’epatite cronica B evolve a cirrosi e comporta il rischio di HCC.
L’HDV segue la storia naturale dell’HBV.In caso di coinfezione il comportamento biologico è generalmente sovrapponibile a una semplice epatite B.
In caso di superinfezione ovvero di infezione da virus D che avviene su un soggetto con infezione cronica da HBV si hauna più spesso epatite D acuta severa, talvolta fulminante.Molto spesso (90%) l’infezione da virus D cronicizza e si ha una più rapida progressione a cirrosi rispetto all’epatite cronicada virus B.Si tenga presente che nel corso di infezione da HDV, l’HBV è sempre presente ma generalmente inattivo (non c’è replica diHBV).Il marker di uso comune per HDV è l’anti-HD totale.
L’HCV è un problema sanitario importante per la sua alta prevalenza nella popolazione generale (1-5% a seconda delle areegeografiche!).HCV è a trasmissione parenterale: la modalità più efficiente è quella apparente(rara la trasmissione verticale e tramiterapporti sessuali è molto meno efficiente) :- prima del 1990 (anno di introduzione dello screening dei donatori per HCV) era molto frequente la trasmissione per viatrasfusionale.- Oggi la via di trasmissione più frequente è la condivisione delle siringhe fra tossicodipendenti; - --- rimane possibile latrasmissione in corso di traumatismi come interventi chirurgici, odontoiatrici, piercing, tatuaggi, taglio della barba dal barbiere, se non vengono rispettate le norme igieniche di base.
L’infezione da virus C presenta una incubazione mediamente di 2-3 mesi.L’infezione acuta provoca una epatite anitterica, raramente itterica, rarissimamente forme fulminanti _ può passareinosservata.Nell’85% degli individui l’infezione cronicizza mentre un 15% guarisce.L’HCV è responsabile del 70% di casi di epatite cronica nei paesi industrializzati.L’epatite cronica ha un decorso asintomatico o paucisintomatico: essa rappresenta un problema in quanto porta in un tempopiù o meno lungo a cirrosi (vedi):questo avviene oltre i 10 anni dal contagio, in genere fra 20-30 anni.Un’ accelerazione del processo si può avere in caso di alcolismo, immunosoppressione, coinfezione con altri virus comeHBV e HDV.È ovvio che l’epatite cronica è un problema rilevante in base all’aspettativa di vita del pz.Inoltre la cirrosi da HCV è una importante condizione di rischio per l’HCC.
I marker per l’HCV sono due:- gli anti-HCV che compaiono entro qualche settimana e indicano in genere infezione in atto, raramente infezione guarita come gli anti-HBsAg.- l’HCV-RNA che implica replica virale in atto.
L’epatite acuta anitterica può essere scoperta a posteriori con un esame sierologico.

L’epatite acuta itterica presenta 4 fasi:- incubazione: variabile in base al virus- preitterica: sintomi prodromici come febbre, nausea e vomito, malessere generale, anoressia.- itterica (occlusione canalicoli biliari): iperbilirubinemia diretta, prurito etc.- convalescenzal’infezione cronica che si ha con i virus a trasmissione parenterale può:- restare del tutto inapparente _ non c’è _transaminasi.- epatite cronica _ _transaminasi ed evidenza di infiammazione alla biopsia. Clinicamente può rimanere asintomatica o manifestarsi con sintomi aspecifici tipo astenia, segni clinici altrettanto vaghi; talvolta si possono avere manifestazioni da ipersensibilità di III tipo: glomerulonefrite, vasculiti etc.MorfologiaLe lesioni elementari dell’epatite acuta sono:- i quadri degenerativi dell’epatocita:
epatociti a vetro smerigliato per l’HBV (particelle di HBsAg),degenerazione balloniforme (frustoli di citoplasma in una cellula gigante),necrosi (lisi della cellula con richiamo di macrofagi) eapoptosi (citoplasma ridotto e intensamente eosinofilo, carioressi),
degenerazione grassa, degenerazione schiumosa.- infiltrato infiammatorio (+ iperplasia delle cellule di Kupffer) sia nello spazio portale che all’interno del lobulo.
Le lesioni elementari dell’epatite cronica sono di grado variabile e vanno conosciute perché servono per fare uno staging(stadio di avanzamento) e un grading (velocità di progressione) di questa malattia cronico-degenerativa.
1) infiltrato infiammatorio per lo più mononucleato a livello degli spazi portali che può organizzarsi in aggregati linfoidi(epatite cronica persistente). La penetrazione dell’infiltrato nel murarium (lamina limitante esterna) va a costituire la“piecemeal necrosis” - necrosi a boccone, come se il lobulo fosse stato morso- e configura una epatite cronica attiva.Quando la necrosi e l’infiltrato arrivano alla vena centrolobulare si ha la “necrosi a ponte”.
Col procedere della necrosi si ha formazione di tralci fibrosi periportali e a ponte (porto-centrolobulari).
D’altra parte si ha la comparsa di noduli rigenerativi.In poche parole in un tempo variabile ma comunque nell’ordine di 10-30 aa si arriva a cirrosi epatica.
EPATITE ACUTA FULMINANTEÈ un quadro clinico di insufficienza epatica fulminante che corrisponde a un quadro anatomo-patologico di necrosi epaticamassiva.Vi sono in genere 2 tipi di cause:- virale- tossica (vedi epatiti da farmaci)morfologicamente si ha il quadro dell’atrofia giallo-bruna: il fegato è incredibilmente piccolo (da 1.5 kg passa a 500 g).Talvolta la necrosi è talmente acuta che non c’è tempo per l’insediamento di un infiltrato infiammatorio.Questi pazienti hanno ovviamente alta mortalità.
EPATITI BATTERICHEI batteri possono colpire il fegato in vari modi:- in corso di sepsi per una flogosi mediata dalle endotossine circolanti- con infezione vera e propria del parenchima epatico: microascessi da stafilococco, febbre tifoide…- in corso di colangite ascendente (vedi patologia delle vie biliari).
l’infezione del fegato da batteri piogeni (stafilococco) ma anche da parassiti (primo fra tutti E. hystolitica) tende a produrreascessi epatici.Gli ascessi possono essere piccoli e multipli (via ematogena o biliare ascendente) oppure unici e grandi (traumi, contiguità).

Gli ascessi possono rompersi in cavità addominale provocando peritonite acuta, o talvolta nel cavo pleurico conconseguente empiema.Gli ascessi epatici provocano febbre e dolore in ipocondrio dx.Quelli grandi, ovviamente, richiedono drenaggio chirurgico che va eseguito prima che sia troppo tardi.
EPATITI AUTOIMMUNILe epatiti autoimmuni sono quadri clinico-morfologici ampiamente sovrapponibili a quelli dell’epatite virale ma con alcunepeculiarità:- incidenza in giovani donne- associazione con altre manifestazioni autoimmuni: artriti, rash, Sjogren, RCU etc.- sierologia positiva per autoanticorpi di vario tipo.
EPATITI DA FARMACILa diagnosi differenziale di qualsiasi forma di epatopatia dovrebbe sempre prendere in considerazione l’esposizione adagenti tossici.La patogenesi può essere di 2 tipi:- tossicità diretta dose-dipendente (acetaminofene, tetracicline, antineoplastici, CCl4)- reazione idiosincrasica dose-indipendente (alotano, allopurinolo etc.)
i quadri clinici e morfologici sono i più vari:- steatosi- epatite acuta- epatite cronica- colestasi
EPATOPATIA ALCOLICAL’etilismo cronico porta a danno epatico che può presentarsi in 3 quadri:- steatosi epatica- epatite alcolica- cirrosi
la steatosi che consegue all’ingestione di alcol è inizialmente microvescicolare, in seguito per esposizione cronica divienemacrovescicolare (grosso vacuolo che sposta alla periferia il nucleo – come in un adipocita). Il processo inizia dalla venacentrolobulare ma può estendersi a tutto il lobulo; a lungo andare inoltre si osserva fibrosi sempre a partenza centrolobulare.C’è epatomegalia (fino a 4-6 kg) e aspetto giallastro, molle e untuoso.Clinicamente è una situazione spesso asintomatica: ci possono essere lievi alterazioni degli indici di epatopatia e dicolestasi.L’astensione dall’alcol e una dieta adeguata possono far regredire la malattia.
L’epatite alcolica è un quadro acuto che si produce in genere per ingestione di grosse quantità di superalcolici.Si tratta di una epatite che può andare da una forma anitterica a una forma fulminante.I sintomi sono quelli tipici di un’epatite acuta: malessere, anoressia, calo ponderale, dolore in ipocondrio dx e lieveepatomegalia. Si ha poi ittero, _enzimi epatici, leucocitosi neutrofila.È una patologia grave: la letalità ad ogni attacco non è trascurabile (10-20%)Con l’astensione dall’alcol e una dieta adeguata l’epatite lentamente , ma non sempre, può guarire.Dal punto di vista morfologico il fegato può essere di volume normale o aumentato.Microscopicamente si notano lesioni come:- degenerazione epatociti: degenerazione balloniforme, corpi di Mallory (accumuli eosinofili di citocheratine in posizioneperinucleare), necrosi segnalata dall’arrivo di infiltrato di neutrofili e macrofagi.- fibrosi
La cirrosi alcolica è un quadro ad evoluzione insidiosa che rappresenta l’esito di una steatosi di lunga data o di attacchiripetuti di epatite alcolica.La fibrosi progressiva e irreversibile porta inizialmente a un aspetto micronodulare; con il passare del tempo quindi a unaspetto macronodulare.il fegato si coarta, la superficie è scura e bernoccoluta, con aree giallastre dovute all’accumulo di bile.

Il cirrotico alcolico ha tutte le complicanze della cirrosi consistenti nell’ipertensione portale, nel precario stato di compensodella funzione epatica con tutto ciò che questo comporta, nel rischio di HCC.
QUADRI DEGENERATIVI SU BASE EREDITARIA
Emocromatosi genetica:l’emocromatosi è in generale un accumulo di Fe a livello sistemico e può essere primitiva (emocromatosi genetica) osecondaria ad altre condizioni particolari che comportano uno sbilanciamento nel metabolismo del Fe (trasfusioni ripetute,eritropoiesi inefficace etc.).l’emocromatosi genetica è una malattia ereditaria a trasmissione AR con una frequenza di omozigosi nella popolazione di1/400.la malattia è caratterizzata da eccessivo assorbimento di Fe per alterazione di una proteina (HFE) che influenzal’assorbimento del Fe in maniera ancora ignota,ne consegue accumulo del Fe in forma di ferritina e emosiderina nel SRE ma anche in altre cellule:
1) fegato _ epatomegalia, superficie nodulare, colorito rosso mattone. Microscopicamente si osserva l’accumulo di ferrointraepatocitario con il blu di Prussia. Nel tempo si sviluppa cirrosi micronodulare.2) Pancreas _ deposito parenchimale con iperigmentazione e atrofia delle cellule acinari e insulari con conseguente diabetemellito.3) Cute _ colorazione grigiastra dovuta alla combinazione del pigmento emosiderinico + quello melaninico4) Cuore _ cardiomiopatia con dilatazione e aspetto marrone.5) Ghiandole endocrine _ colorazione brunastra.6) Testicoli _ atrofia (da _ gonadotropine).7) Articolazioni _ sinoviti.
Il quadro dell’emocromatosi si manifesta quando si accumula una quantità sufficiente di Fe (si può arrivare oltre i 50 g vs i6 gm massimi nell’individuo sano).L’accumulo del Fe è favorito nell’uomo rispetto alla donna ed è tale da produrre sintomi solo oltre i 40 anni.In sintesi in un paziente adulto con una pigmentazione caratteristica della cute si possono riscontrare alterazioni di tipoendocrino (diabete mellito, ipogonadismo), cardiopatia, cirrosi epatica. I pazienti hanno forte rischio (RR=200) disviluppare HCC.
Fondamentali sono la saturazione della transferrina (proteina di trasporto) e la ferritinemia (proteina di stoccaggio).Si può valutare la _ceruloplasmina, il Fe epatico (su tessuto secco), o con la PCR dimostrare la mutazione genetica allabase della malattia.La terapia è attraverso i salassi finchè l’accumulo di Fe non regredisce.Molti sintomi migliorano grazie a questa misura.
Malattia di Wilson:La malattia di Wilson è trasmessa con modalità AR e l’omozigosi ha una frequenza nella popolazione di 1/25.000.In questo caso l’accumulo di rame è dovuto a una insufficiente escrezione epatica: tra l’altro il fegato sintetizza anche laceruloplasmina, una metallotioneina che trasporta il rame a livello del plasma e che lega oltre il 90% del pool di rame; laceruloplasmina senescente viene ricaptata dal fegato, degradata ed escreta con la bile; in tal modo il pool del rame vienemantenuto in equilibrio.
Il gene implicato nella malattia di Wilson è una ATPasi coinvolta nell’escrezione del rame al polo canalicolaredell’epatocita.il rame che arriva all’epatocita in queste condizioni è in eccesso rispetto a quello mobilizzato con la ceruloplasmina _ ilsoggetto omozigote presenta nei primi 5 anni di vita un accumulo di rame negli epatociti _ in seguito il rame passa incircolo: a questo punto compaiono segni di accumulo in altri organi e aumenta l’escrezione urinaria.
Il fegato è quindi l’organo primitivamente coinvolto: presenta quadri di steatosi, epatite cronica, cirrosi. Il rame èevidenziabile con colorazioni speciali come l’orceina e la sua concentrazione chimicamente quantificabile.Quando il rame dal fegato passa in circolo, si hanno lesioni a livello di:- eritrociti _ emolisi

- occhio _ anello di Kayser-Fleischer ovvero sottile deposito di rame (colore verdastro) attorno al limbus (donde il nome alternativo di sindrome epato-lenticolare).- SNC _ degenerazione neuronale soprattutto a livello dei nuclei della base _ spasticità, tremore, atetosi etc.
la terapia con penicillamina, farmaco chelante il rame, consente di arrestare la degenerazione.
Deficit di _1-antitripsinaL’_1-antitripsina è un inibitore delle proteasi, in particolare delle elastasi dei neutrofili, sintetizzato a livello epatico erilasciato in circolo.Un particolare allele, l’allele Z, allo stato omozigote ovvero PiZZ, è associato a livelli sierici di antitripsina estremamentebassi che sono responsabili della comparsa di enfisema polmonare panacinoso.I bassi livelli sono dovuti alla ritenzione nel reticolo endoplasmatico rugoso dell’epatocita della proteina mutante: in generequesta proteina non si accumula perché sono attivi meccanismi di degradazione da parte del reticolo endoplasmatico stesso.Una piccola percentuale di individui tuttavia non smaltisce adeguatamente la proteina aberrante che si accumula, soprattuttonell’omozigote, all’interno dell’epatocita in forma di granuli fortemente PAS+ (glicoproteine).In sintesi si ha una epatopatia (epatite cronica, cirrosi) con presentazione variabile:- neonatale- nell’adolescenza- nell’adultoEPATITE NEONATALENel neonato, accanto a un ittero fisiologico, vi sono itteri patologici.Una iperbilirubinemia coniugata (DD dalle malattie di tipo emolitico) di lunga durata detta colestasi neonatale è imputabilefondamentalmente a 2 condizioni:- l’atresia delle vie biliari extraepatiche- le epatiti neonatale
è importante fare diagnosi differenziale fra queste 2 entità in quanto:La prima è una malformazione congenita che richiede un approccio terapeutico di tipo chirurgico.Le seconde sono un gruppo estremamente eterogeneo che comprende moltissime opzioni diagnostiche (spesso la causarimane sconosciuta e la diagnosi è di epatite neonatale idiopatica) e che in genere richiede un approccio medico.
MALATTIE DELLE VIE BILIARI INTRAEPATICHE
CIRROSI BILIARE SECONDARIAAbbiamo detto che la ritenzione della bile o colestasi può essere epatocellulare o, molto più frequentemente, ostruttiva.Nella colestasi ostruttiva l’ostruzione può essere a qualsiasi livello della via biliare e quindi dal canalicolo biliare (vedi peresempio nelle epatiti) fino all’ampolla di Vater,l’ostruzione può essere congenita o acquisita.Cause congenite sono:- l’atresia biliare- la vanishing bile duct syndrome (S. di Alagille)- la cisti del coledoco- la fibrosi cisticaCause acquisite sono:- la calcolosi del coledoco- neoplasie delle vie biliari e della testa del pancreas- stenosi post-chirurgiche
è importante dire che l’ostruzione cronica delle vie biliari extraepatiche porta a un complesso di lesioni che rappresentano lacirrosi biliare secondaria.Volendo procedere in ordine logico avremo dilatazione di tutti i dotti biliari a monte, il fegato è iperconsistente everdastro.a livello dello spazio portale si hanno: proliferazione e dilatazione dei dotti biliari, edema, infiltrato infiammatorio, fibrosi.L’infiammazione può essere severa in caso di colangite ascendente.

a livello lobulare si hanno trombi di bile, degenerazione schiumosa epatociti e cellule di Kupffer, laghi di bile, fibrosi;quando si arriva alla cirrosi vi sono noduli rigenerativi.
CIRROSI BILIARE PRIMITIVAÈ una patologia autoimmunitaria caratterizzata dalla distruzione dei dotti biliari intraepatici di medio calibro da partedi un infiltrato infiammatorio in forma di granulomi.L’esito è una cicatrice che sostituisce il dotto con ovvia colestasi a monte.Il paziente tipo è una donna di mezza età con sintomi (prurito ma senza ittero) e segni (xantomi, xantelasmi, ALP, _GT)riconducibili a una condizione di colestasi.Possono essere presenti altri disordini autoimmuni: Sjogren, sclerodermia, tiroidite, AR, Gn membranosa, malattia celiaca.Sono di frequente riscontro anticorpi anti-mitocondrio (AMA).La cirrosi è l’esito di questo quadro infiammatorio-ostruttivo.
COLANGITE SCLEROSANTEÈ una patologia ad eziopatogenesi incerta.La sua forte associazione con la RCU fa pensare a fenomeni di autoimmunità, tossici etc.in ogni caso colpisce giovani adulti con un’incidenza doppia nel maschio.La lesione elementare è una flogosi segmentaria e plurifocale dell’albero biliare intra e extraepatico: l’infiltratoinfiammatorio mononucleato viene poi sostituito da una sclerosi a bulbo di cipolla che restringe fortemente il lume _ fra lestenosi i dotti si dilatano _ la via biliare assume un aspetto “a corona di rosario”.Anche in questo caso l’esito è la cirrosi.
MALFORMAZIONI DELL’ALBERO BILIARE INTRAEPATICO
1) complessi di Von Meyenburg: microamartomi costituiti da duttuli biliari immersi in uno stroma fibroso. Sonoclinicamente silenti ma possono simulare radiologicamente un fegato con metastasi multiple.
2) Malattia policistica del fegato: cisti epatiche multiple rivestite da epitelio biliare cubico ma senza comunicazionecon l’albero biliare per cui contengono un liquido sieroso paglierino. Questa patologia si associa al renepolicistico dell’adulto (AD).Può essere causa di dolore addominale.in altri casi si possono trovare cisti epatiche solitarie.
3) Fibrosi epatica congenita: gli spazi portali sono sostituiti da spessi fasci di collagene con dotti biliari alterati: laconseguenza fondamentale è l’ipertensione portale.È fortemente associata al rene policistico del bambino (AR).
4) Malattia di Caroli: dilatazioni multiple sacculari dei dotti intraepatici che provocano ristagno di bile con colelitiasi intraepatica, colangite ascendente, ascessi epatici. Si associa anch’essa a fibrosi portale.
Sia la fibrosi epatica congenita che la malattia di Caroli presentano un rischio di colangiocarcinoma.
5) vanishing bile duct syndrome: in un fegato peraltro normale mancano i dotti biliari a livello portale. Questipazienti vanno incontro a cirrosi biliare secondaria e insufficienza epatica. Nella sindrome di Alagille si associauna caratteristica facies con anomalie scheletriche e cardiovascolari.
PATOLOGIA VASCOLARE DEL FEGATOPremessa: il flusso ematico dipende per 1/3 dall’a. epatica e per 2/3 dalla vena porta.
L’occlusione del ramo principale dell’arteria epatica non comporta necrosi dell’organo per la presenza di collateraliarteriosi e ovviamente della vena porta. Un infarto epatico si ha raramente per occlusione di rami intraepatici dell’arteria.

L’occlusione della vena porta può essere dovuta a tromboflebite della vena porta (pileflebite) o a una ostruzione di altranatura, le conseguenze sono soprattutto congestizie a carico del territorio drenato (splenomegalia, varici esofagee,congestione intestinale).L’occlusione di un ramo intraepatico della vena porta per esempio dovuta a una neoplasia, non provoca necrosi ischemicama solo un’area di atrofia parenchimale.A livello dei sinusoidi l’ostruzione al flusso ematico si può avere in corso di:- cirrosi epatica: ricorda la capillarizzazione e vasocostrizione dei sinusoidi.- anemia falciforme- CID- eclampsia- microtrombi neoplastici
a livello delle vene centrolobulari si può avere la cosiddetta malattia veno-occlusiva ovvero una fibrosipericentrolobulare che occlude l’efflusso epatico e provoca congestione del lobulo.
a livello delle vene sovraepatiche si può avere una trombosi vuoi idiopatica vuoi in associazione a determinate condizionipredisponenti come la policitemia vera, la gravidanza, il postpartum, i carcinomi intraddominali etc.ne risulta la sindrome di Budd-Chiari ovvero una ipertensione portale post-epatica con congestione a monte:epatomegalia, dolore addominale, ascite.Se la congestione è acuta la mortalità è alta, se cronica molto meno.
Infine sono caratteristici le lesioni del fegato in corso di scompenso cardiaco.Lo scompenso cardiaco sn può portare a ipoperfusione con necrosi centrolobulare.Lo scompenso congestizio invece può portare invece a necrosi emorragica centrolobulare, in questo caso il cuore assumeun tipico aspetto marezzato a “noce moscata”.L’evoluzione è a fibrosi pericentrolobulare che un tempo veniva detta “cirrosi cardiaca”.
NEOPLASIE DEL FEGATONel parenchima epatico possono essere presenti masse di varia natura che possono essere scoperte anche incidentalmente,per esempio con un’ eco-addome.Le masse possono essere benigne o maligne.
Fra le masse benigne abbiamo:- cisti solitarie
- emangiomi cavernosi: noduli costituiti da ampie lacune vascolari rivestite da endotelio, non pungere! Alla TC con mdc il contrast enhancement è caratteristico: tardivo e persistente.
- iperplasia nodosa focale: nodulo privo di capsula ma ben demarcato contenente una cicatrice stellata centrale, è una formazione pseudotumorale si tratta infatti di una sorta di amartoma più che di neoplasia: sono presenti abbozzi di lobuli epatici.
- iperplasia nodulare rigenerativa: noduli multipli di epatociti in iperplasia e ipertrofia rigenerativa, gn dopo condizioni di ipoperfusione epatica.
- adenomi epatocellulari: sono noduli grigiastri, generalmente sottocapsulari, ben demarcati, costituiti da cordoni di cellulesimili agli epatociti ma senza alcuna tendenza alla formazione di pseudolobuli (dd con l’iperplasia nodosa). Sono associati aterapia anticoncezionale orale.Sono importanti soprattutto perché presentano una ricca vascolarizzazione arteriosa e sono a rischio di rottura conematomi intraepatici o emoperitoneo per cui c’è l’indicazione alla rimozione chirurgica.
A livello epatico possiamo trovare vari tipi di cancro, in ordine di frequenza abbiamo:1) cancri secondari _ si ricordi infatti che il fegato è il principale organo “filtro” assieme al polmone: in particolare il fegato è la sede elettiva per i carcinomi del tratto gastrointestinale (stomaco, pancreas, colon) ma qualsiasi neoplasia maligna può dare metastasi epatiche. Le

metastasi appaiono come noduli multipli nel parenchima epatico: questo può portare a epatomegalia massiva con una funzione epatica sorprendentemente ben conservata.
2) cancri primitivi _ HCC, colangiocarcinoma, neoplasie maligne rare
Fra i cancri primitivi rari ricordiamo l’angiosarcoma epatico che è un cancro strettamente relato all’esposizione a clorurodi vinile monomero, e l’epatoblastoma ovvero un tumore a cellule embrionarie.
Il carcinoma epatocellulare o HCC, ha una epidemiologia molto particolare.
A) Vi sono aree ad alta incidenza (fino al 40% dei casi di cancro!) in paesi a basso tenore socio-economico. In questi paesil’incidenza è nella prima età adulta e non è relata alla cirrosi bensì:
1) all’endemia da HBV: in questi paesi la trasmissione è verticale ovvero avviene da madre a figlio (parto,allattamento etc.); il neonato non è ancora immunologicamente preparato e l’infezione cronicizza. Il genoma diHBV viene integrato a livello dell’epatocita e qui svolge un azione trasformante per cui in assenza di cirrosi, nellaprima età adulta si sviluppa un HCC.
2) Alla contaminazione dei cibi con aflatossine che sono agenti mutageni prodotti da certe muffe, accanto a unasuscettibilità geneticamente determinata alle aflatossine (deficit di detossificazione epatica).
B) Nei paesi occidentali ad elevato tenore socio-economico l’incidenza è molto più bassa e l’HCC è un carcinomarelativamente raro. Nei nostri paesi l’HCC insorge quasi sempre come complicanza della cirrosi epatica (in quanto ifenomeni flogistici e necrotici costituirebbero uno stimolo rigenerativo persistente che favorisce la comparsa dellaneoplasia) e quindi in età decisamente più avanzata (generalmente dopo i 60 aa).Si ricordi che le principali cause di cirrosi sono: l’epatopatia alcolica, l’epatite cronica C, l’emocromatosi etc.
L’HCC può presentare una crescita:- unifocale: un grosso nodulo- multifocale: tanti noduli- infiltrante: crescita diffusala lesione può essere più o meno differenziata: si tratta di cordoni di grosse cellule poligonali disposte in cordoni chepossono produrre bile.Il tumore non ha una spiccata tendenza alla metastatizzazione (che può interessare LN, osso, polmone) ma piuttosto infiltralocalmente (capsula, diaframma, vena porta etc.).Il paziente come detto è generalmente cirrotico per cui la presentazione clinica può essere estremamente insidiosa. Esami discreening possono essere utili per una diagnosi precoce.Un marker per l’HCC è l’AFP che tuttavia non è del tutto sensibile e specifica.In sintesi questi pazienti hanno una patologia di base che è la cirrosi la quale viene complicata dal carcinoma epatocellulare:la chirurgia resettiva è indicata solo per lesioni unifocali, intraepatiche e con una cirrosi in stadio A (vedi la stadiazioneclinica della cirrosi).Alternative sono la palliazione con chemioembolizzazione, alcolizzazione, radiofrequenza.In genere questi pazienti muoiono nel giro di pochi mesi per insufficienza epatica, sanguinamento da varici esofagee,cachessia, rottura del tumore con emorragia fatale.
Una prognosi decisamente migliore è quella di una variante detta carcinoma fibrolamellare;questo insorge nel giovane adulto su un fegato non cirrotico e si presenta come un nodulo lobulato per la presenza di densifasci fibrosi.In questo caso è potenzialmente praticabile una chirurgia curativa.
LE VIE BILIARIAnatomia:A) vie biliari intraepatiche:1) canalicolo biliare : 1-2µm, formato per giustapposizione degli epatociti2) canale di Hering: alla periferia del lobulo, rivestito da cellule epiteliali piatte.3) dotto interlobulare e varie generazioni dei dotti biliari: nello spazio portale, inizia il rivestimento epiteliale cubico.
B) vie biliari extraepatiche:

le vie biliari extraepatiche (compreso il cistico e la cistifellea) constano di un4) dotti epatici destro e sn5) dotto epatico comune _ coledoco dopo aver ricevuto il dotto cistico (dotto d’efflusso della colecisti).6) ampolla di Vater nei 2/3 dei casi per congiunzione con il Wirsung.
NB: esiste una discreta variabilità delle vie biliari extrapancreatiche per cui il chirurgo deve sempre prestare moltaattenzione a quello che sta legando o tagliando in questa regione.
Strutturalmente i grossi dotti biliari e la cistifellea sono costituiti da:- una tonaca fibromuscolare esterna- una tonaca mucosa interna: questa è costituita da un epitelio di rivestimento cilindrico con orletto a spazzola (concentrazione della bile) e contiene ghiandole tubulari mucosecernenti (in definitiva, a parte i villi, la mucosa ricorda quella intestinale). Tenere presente questa struttura aiuta a capire vari tipi di patologia come: a) le neoplasie epiteliali _ adenomi e adenocarcinomi b) l’idrope della colecisti
La cistifellea è un sacchettino a forma di pera, lungo 9 cm, con una capacità di 50ml: si distinguono un fondo, un corpoe un collo che si continua nel dotto cistico.Alterazioni congenite della colecisti possono essere:- la agenesia- la duplicazione- la c. bilobata- la c. intraepatica- la c. a cappello frigio (introflessione del fondo)
La colecisti ha due funzioni:1) concentrare la bile epatica2) contrarsi in risposta alla CCK (rilasciata dalle cellule “I” in seguito all’arrivo di grassi e protidi in duodeno) la
quale fra l’altro stimola la secrezione di zimogeni pancreatici e il rilasciamento dello sfintere d’Oddi.
La bile è una soluzione acquosa contenente lipidi, pigmenti, proteine, elettroliti.particolarmente importante ai fini della funzione digestiva è la componente lipidica nella quale riconosciamo:
- Sali biliari: sono Sali di acidi biliari. Gli acidi biliari sono steroli anfifilici sintetizzati nel fegato a partire dal colesterolo. Per la verità nella bile sono presenti 2 tipi di Sali: a) primari cioè sintetizzati come tali nel fegato e b) secondari cioè derivati dei sali primari attraverso la trasformazione batterica intestinale.
i sali vengono escreti dal fegato in forma coniugata con glicina o con taurina.I sali biliari sono soggetti al cosiddetto circolo entero-epatico per cui esiste un pool in continuo ricircolo fral’intestino e l’epatocita.Il 90% dei Sali biliari rientra in circolo dal tenue: i glicocolici dal digiuno e ileo prossimale per trasportopassivo, i taurocolici dall’ileo distale per trasporto attivo.Il restante 10% passa nel colon dove avviene può avvenire la trasformazione sale primario _ sale secondario e unulteriore riassorbimento.Solo una piccolissima quota di sali biliari viene escreta con le feci e una stessa quantità è sintetizzata “de novo” apartire dal colesterolo per mantenere il pool in equilibrio.
- la lecitina o fosfatidilcolina
- il colesterolo non esterificato
lecitina, sali biliari e colesterolo formano micelle che hanno la funzione di emulsionare i lipidi introdotti con la dieta perconsentirne la digestione.Il colesterolo non esterificato in acqua tende spontaneamente a precipitare in forma di cristalli:sono i sali biliari e la lecitina a mantenerlo in soluzione.

La LITIASI COLESTERINICA è dovuta proprio a un difetto nella solubilizzazione del colesterolo.
I fattori causali sono:1) la sovrasaturazione in colesterolo della bile: possiamo immaginare che i Sali biliari e i fosfolipidi abbiano nel lorocomplesso una capacità legante il colesterolo così come l’Hb con l’O2.Questa capacità legante può non bastare:
- per _ del colesterolo _ obesità, dislipidemie, stimolo estrogenico (nel sesso femminile), farmaci che _l’escrezionebiliare del colesterolo come il clofibrato, dieta povera di fibre/_tempo di transito (_ eccesso dell’acido biliaresecondario desossicolico per _metabolismo batterico che strappa colesterolo alle membrane cellulari-evidentemente a carico delle cellule delle vie biliari).
- per difetto di sali biliari e/o fosfolipidi.
2) la presenza di fattori pronucleanti: muco, proteine di flogosi
3) la stasi biliare: colecisti più grande e con difetto di svuotamento, vagotomia tronculare, nutrizione parenterale.
i calcoli di colesterolo possono essere puri (10% del totale) o misti (75% del totale); quelli misti contengono anchebilirubina e Ca.
la LITIASI PIGMENTARIA rappresenta il 15% del totale.Essa comprende:
1) i calcoli neri: contenenti oltre a Sali inorganici di calcio il bilirubinato di Ca . Essi si formano solo all’internodella colecisti e si associano a condizioni di aumentata eritrocateresi o emolisi (protesi valvolari, anemieemolitiche croniche) o anche a epatopatia cronica.
2) I calcoli marroni hanno una composizione mista ovvero bilirubinato di Ca, palmitato di Ca e colesterolo (<25%).Si formano unicamente nelle vie biliari e sono particolarmente associati a infezioni e parassitosi biliari eparticolarmente frequenti nei paesi in via di sviluppo. Questi calcoli, insorgendo nella via biliare, recidivano dopocolecistectomia.
Bisogna ricordare che a differenza dei calcoli renali solo una piccola quota (10%) dei calcoli biliari è radiopaca (inquanto contenente una quantità adeguata di Ca).Di questi la maggioranza (75%) sono calcoli pigmentari.
La prevalenza della colelitiasi è molto alta, in Italia è del 20% per le donne e del 10% per i maschi. L’incidenza annua èattorno al 4 per mille.
Lo sviluppo di sintomi in 10-20 anni avviene solo nel 15-30% degli individui affetti da litiasi.La colecistectomia non è indicata in via profilattica.
la colelitiasi può rendersi sintomatica:1) con la colica biliare:
si tratta di un dolore non realmente a colica ma continuo della durata di 30minuti-4 ore , di intensità variabile che recede spontaneamente e gradualmente ma si possono somministrare antispastici. Spesso insorge di notte, è localizzato al quadrante addominale superiore dx e può irradiarsi alla scapola (punto di Boas) e alla spalla omolaterali, può associarsi a ittero ostruttivo se il calcolo è impegnato nel coledoco.
2) con varie complicanze:- la colecistite cronica dovuta allo stimolo irritativo cronico dei calcoli, decorre asintomatica e porta a una sclerosi
della colecisti.Talvolta è possibile osservare all’ecografia o alla diretta dell’addome una colecisti calcifica, la cosiddetta colecistia porcellana, che rappresenta un esito di colecistite cronica e che consiglia una colecistectomia a scopo profilatticoper la sua associazione con il carcinoma della colecisti.

Altre volte nel quadro di una colelitiasi da colesterolo questo stesso colesterolo può accumularsi nella parete dellacolecisti in forma diffusa (colecisti a fragola) o localizzata (polipi di colesterolo).Infine si possono riscontrare dei polipi che rientrano nel quadro della adenomiosi diffusa, una condizione diiperplasia della tonaca muscolare con la presenza anche di formazioni ghiandolari esuberanti.
- La colecistite acuta litiasica ovvero una flogosi acuta della colecisti dovuta all’ostruzione del dotto cistico da partedi un calcolo (l’ostruzione porta di per se a una colecistite chimica per l’accumulo di lisolecitina
altre rare volte si può avere una colecistite acuta alitiasica ovvero da altre cause come stenosi neoplastica, invasionebatterica primitiva o occlusione dell’arteria cistica.
- un esito dovuto alla prolungata ostruzione della colecisti è l’idrope dellacolecisti nella quale mentre si ha il riassorbimento della bile, si produce unaccumulo di muco con distensione progressiva della colecisti fino araggiungere dimensioni ragguardevoli.
- la evoluzione della colecistite acuta verso la suppurazione portaall’empiema della colecisti con accentuazione di tutti i sintomi e segni sialocali (dolore) che sistemici (febbre, leucocitosi). Il rischio di perforazione èelevato e il pz va subito inviato all’intervento con adeguata coperturaantibiotica.
- La perforazione può essere:localizzata _ ascesso pericolecistico (spazio sottoepatico e pericolico),
libera in peritoneo _ peritonite diffusauna fistola in un organo cavo come il duodeno _ si associa all’aerobilia (reperto radiografico) e può rendersisintomatica con ileo da calcolo (fistola cisto-enterica) o con steatorrea (cisto-colica).
- La migrazione nel dotto cistico con compressione del dotto epatico comune provoca colestasi, colangite acuta,emobilia (ovvero emorragia all’interno della via biliare principale per erosione della parete della via biliare).
Si ricordi che talvolta attacchi ricorrenti tipo colica possono essere attribuibili a una disfunzione motoria della colecisti inassenza di calcoli. Si tratta della cosiddetta colecistopatia alitiasica.
TERAPIALa colica biliare si tratta con antispastici e analgesici ad azione centrale o FANS (diclofenac).
La calcolosi della colecisti è una condizione ad alta prevalenza, l’approccio terapeutico deve quindi essere estremamenterazionale (non si possono operare tutti i casi di colelitiasi!).
Se asintomatica, la calcolosi non presenta indicazioni al trattamento se non in giovani diabetici (rischio di colecistiteenfisematosa da coliformi o clostridi) o donne giovani che vogliano avere gravidanze.
Se sintomatica (con coliche biliari ripetute ) vi sono 3 opzioni:- nel caso di calcoli radiotrasparenti, del diametro inferiore a 1.5 cm, con colecisti funzionante (la capacità di
concentrazione della bile si verifica tramite colecistografia orale) c’è indicazione alla terapia con acidi biliari chesomministrati per un anno riescono a solubilizzare il calcolo: tale terapia può essere vanificata da insuccesso orecidiva
- nel caso di un grosso calcolo sempre radiotrasparente ma di diametro > 2cm si può ricorrere alla litotrissiaextracorporea con somministrazione di acidi biliari per aiutare a sciogliere i frammenti.
- Nel caso di calcoli che non rispondano ai precedenti requisiti quindi calcoli calcificati, in caso di colecisti nonfunzionante, in caso di insuccesso della terapia medica e/o litotriptica si ricorre alla colecistectomia (in genere è laterapia d’elezione in quanto sicuramente risolutiva, gli altri 2 approcci sono possibili ma in genere non effettuati).
Se complicata si ricorre alla chirurgia in urgenza per via laparotomica (es. colecistite suppurativa) o in elezione per viageneralmente laparoscopica (colecistite acuta).
Presentazione: febbree leucocitosineutrofila, dolorecontinuoall’ipocondrio dx,colecisti dolorabile(segno di Murphy),nausea, vomito.Ecotomografia:colecisti distesa(>9cm) e a paretiispessite (>4mm),Murphyultrasonografico.

CALCOLOSI DELLE VIE BILIARI (coledocolitiasi)La presenza di calcoli nelle vie biliari può essere classificata in base alla sede:
1) calcoli migrati dalla colecisti (1/6 dei pz con calcolosi della colecisti ha coinvolgimento della via biliare) _ è il casopiù frequente 70%.
2) calcoli nel coledoco con colecisti alitiasica3) calcoli nelle vie biliari intra- ed extraepatiche4) calcoli del coledoco dopo colecistectomia _ si tratta di calcoli marroni.
La presenza di calcoli nel coledoco può rendersi responsabile di:
1) colica biliare accompagnata da ittero ostruttivo.
2) colangite batterica _ l’ostruzione coledocica provoca stasi _ proliferazione di eventuali batteri che avevanocolonizzato la via biliare _ infezione con diffusione retrograda dei batteri al fegato e in circolo (colangiteascendente) _
triade di Charcot: dolore, ittero ostruttivo, stato tossinfettivo
Sono alterati: bilirubina, transaminasi, fosfatasi alcalina, _-GT, eventualmente le amilasi e lipasi.Nella forma suppurativa, la più grave, il pz entra in letargia e stato di shock.La diagnostica è ecotomografica (dilatazione via biliare, visualizzazione pancreas).La colangiografia è controindicata in fase acuta.Si può ricorrere alla sfinterotomia endoscopica per decomprimere l’albero biliare.
3) pancreatite acuta _ incuneamento del calcolo nell’ampolla _ _pressione nel sistema duttale pancreatico.
L’ittero ostruttivo senza colica può essere dovuto a molte altre cause che vanno escluse:- stenosi neoplastica (a carico delle vie biliari o della testa del pancreas)- stenosi cicatriziale (per esempio calcolo incarcerato)- cirrosi biliare primitiva- colangite sclerosante etc.
Nel neonato l’ittero colestatico può essere dovuto essenzialmente a 2 sindromi:- le epatiti neonatali- l’atresia biliare.Quest’ultima è l’ assenza o della distruzione (per cause non del tutto conosciute come infezioni, autoimmunità, ischemiaetc.) dei dotti biliari extraepatici nel neonato (incidenza 1/10.000).Si tratta di una condizione estremamente drammatica che porta rapidamente a cirrosi biliare secondaria e morte delbambino.In base all’ estensione dell’interessamento duttale la malattia può essere correggibile chirurgicamente oppure no.Sfortunatamente questa ultima eventualità è la più frequente per cui l’unica opzione terapeutica è il trapianto di fegato edotti biliari.
NEOPLASIE DELLE VIE BILIARIIl più importante tumore delle vie biliari è il colangiocarcinoma.
Istologicamente è un adenocarcinoma scirroso ovvero costituito da tubuli ad epitelio cuboidale (tipo dotti biliari) immersiin un abbondante connettivo denso che le conferisce un colore grigiastro e una consistenza estremamente solida.
Può insorgere nei dotti intraepatici o extraepatici.
A livello intraepatico la clinica del colangiocarcinoma ricalca quella del HCC: il colangiocarcinoma però :- è decisamente più raro (9:1)

- non si associa a cirrosi- non produce né bile né _-fetoproteina- rispetto all’HCC (infiltrazione locale), ha una tendenza più spiccata alla metastatizzazione ai linfonodi e a distanza
fattori di rischio specifici sarebbero condizioni di stasi e/o di flogosi dell’albero biliare:- parassitosi (Clonorchis, Ascaris)- colangite sclerosante (quadro di flogosi segmentaria di tutto l’albero con dotti a corona di rosario)- particolari tossici
A livello extraepatico questi tumori possono insorgere:1) a livello di uno dei dotti epatici2) a livello della confluenza dei dotti epatici/dotto epatico comune3) a livello del coledocola zona a più alta insorgenza è quella peri-ilare (2): in questa sede esso prende il nome di tumore di Klatskin.
Il comportamento biologico del tumore consiste in una crescita piuttosto lenta, lungo i dotti con metastatizzazione ailinfonodi ilari e meno frequentemente con metastasi a distanza.
La presentazione può essere:- modesto dolore addominale- epatomegalia- ittero ostruttivo (bilirubina diretta, bilirubinuria, prurito) _ NB solo se è ostruita la via biliare comune, l’ostruzione
di un solo dotto epatico non provoca ittero.- markers di colestasi (AP, _-GT).- segno di Courvoisier _ se si ha ostruzione a valle della confluenza col cistico.
Nel caso di ostruzione complicata da colangite acuta vi sarà la classica triade di CharcotLa clinica e il laboratorio chiariscono un quadro di ittero colestatico (se c’è stenosi a valle della confluenza dotto epaticodx-sn) o di colestasi senza ittero (se è interessato solo il dotto epatico dx o sn) _ c’è colestasi _ quindi è opportuno fare unostudio per immagini della via biliare:
- l’ecotomografia _ dilatazione della via biliare, presenza di colelitiasi- la ERCP nel caso si sospetti un’ostruzione bassa- la THC nel caso si sospetti un’ostruzione alta (es. Klatskin)- la colangio-RMN consente di visualizzare la via biliare e di valutare i rapporti di una eventuale neoplasia con le
strutture circostanti.Si noti come la ERCP è una metodica polivalente in quanto può servire a:
- eseguire manovre decompressive come la sfinterotomia o l’inserimento di un drenaggio biliare interno- visualizzare i dotti biliari e/o pancreatici- prelevare campioni da analizzare (brushing ; aspirato)
la terapia segue un ben preciso algoritmo.La resezione chirurgica del tumore (con eventuali parti di fegato) viene eseguita solo dopo un iter ben preciso (ecografialaparoscopica, ecografia intraoperatoria) che abbia stabilito la resecabilità stessa del tumore: in genere sono lesioni piccole edistali.Se il tumore, come spesso accade, non è resecabile si effettuano interventi palliativi di:
- drenaggio biliare per es con ERCP o THC- radioterapia esterna o brachiterapia
CARCINOMA DELLA COLECISTIÈ il carcinoma delle vie biliari più frequente, al 5° posto fra i tumori dell’apparato digerente: nel complesso tuttavia èrelativamente raro.
La neoplasia compare in età avanzata (> 50 anni) e soprattutto nel sesso femminile (4:1).FdR possono trovarsi nell’ambito di certe industrie (gomma, derivati del petrolio)

in generale ciò che si associa al tumore (come per l’HCC e per molti altri) è la flogosi cronica, ovvero la colecistite cronica,che comporta un continuo stimolo rigenerativo e proliferativo per l’epitelio.In altre parole le lesioni predisponenti sono:- colecistite cronica litiasica: nei paesi ricchi come il nostro la flogosi cronica della colecisti è fortemente associata alla colelitiasi (e qui ritorna anche il discorso dell’età e del sesso femminile). Vi sono particolari lesioni, in corso di colecistite cronica, che sembrano avere una certa associazione con la comparsa di carcinoma come la colecisti a porcellana, i polipi della colecisti, l’adenomiosi della colecisti.- colecistite cronica alitiasica: nei paesi poveri una causa molto importante di colecistite cronica, accanto alla litiasi che è molto meno comune, sono le infezioni croniche della cistifellea per es. da S. typhi o da parassiti.
In genere il tumore è un adenocarcinoma:può essere del tipo scirroso per cui tende a infiltrare la parete e le strutture contigue come il fegato oppure può essere di tipovegetante per cui protrude all’interno spesso sanguinante o ulcerato con minore tendenza alla diffusione per contiguità e pervia linfatica.
la diagnosi precoce di carcinoma della colecisti è estremamente difficile.1) perché la clinica non offre un quadro molto specifico: dolore, calo ponderale, eventuale ittero ostruttivo ,
epatomegalia etc.2) Perché spesso i primi sintomi sono tardivi.
Il paziente quindi è in genere inoperabile e riceve unicamente terapie palliative con una sopravvivenza a 5 annibassissima.
NEOPLASIE DELL’AMPOLLAL’ampolla di Vater può essere sede di adenomi (ovvero epitelio con displasia di variogrado) che sembra diano poi origine ad adenocarcinomi.Sia l’adenoma che l’adenocarcinoma dell’ampolla possono presentarsi con itterocolestatico (colorazione giallastra di cute e mucose, urine scure, prurito, feciipocoliche) in maniera del tutto simile a quanto avviene per il carcinoma della testadel pancreas, anche se qui la neoplasia è più precocemente ostruttiva vista la
localizzazione strategica.si può naturalmente avere dolore epigastrico (di tipo pancreatitico dopo i pasti)Si può avere il segno di Courvoisier.Un reperto meno frequente è il sanguinamento gastrointestinale: infatti questo carcinoma può affacciarsi sul lumeduodenale e sanguinare.Il sanguinamento può aver portato ad anemia ferro-carenziale.
La diagnosi si fa all’ERCP:nel 75% dei casi il tumore si presenta sul lume duodenale (in questo caso ci si potrebbe porre l’interrogativo - che tuttaviaha un senso solo a fini speculativi e non pratici- se il tumore sia insorto dalla papilla o dall’ampolla) e si può prendere subitola biopsia, altrimenti dopo sfinterotomia bisogna aspettare una decina di gg prima di prendere la biopsia per la possibilità diartefatti.L’esame con mezzo di contrasto dimostra la massa stenosante.
La terapia chirurgica radicale consiste nella operazione di Whipple, la stessa eseguita per tumori resecabili della testa delpancreas _ la differenza rispetto al carcinoma della testa del pancreas è che il 75% delle lesioni è resecabile con una buonasopravvivenza (50%) a 5-anni.In caso di neoplasia non resecabile si fa terapia palliativa mediante posizionamento di endoprotesi.
Bisogna dire che:1) una certa quota di tumoriviene trovata incidentalmentedopo colecistectomie eseguiteper colelitiasi _ in questi casi lacolecistectomia può salvare lavita del paziente.2) La colecistectomia a scopoprofilattico non ha invece alcunvalore perché i rischidell’operazione superano ilbeneficio.

IL PANCREASIl pancreas è una ghiandola a secrezione sia esocrina che endocrina annessa all’apparato digerente.
Morfologia:il pancreas è un organo relativamente piccolo (circa 100g di peso) , di forma allungata (15 x 3 x 2), di colore giallo-roseo, diconsistenza relativamente dura (che non cambia molto, quindi, in caso di neoplasia o pancreatite cronica).
Rapporti:il pancreas è disposto trasversalmente a livello di L1 in sede retroperitoneale, contrae rapporto posteriormente con:
1) colonna vertebrale2) cava inferiore3) aorta4) vena e arteria mesenterica superiore (rapporto stretto: vedi l’uncino a livello della testa).5) rene sn
anteriormente il pancreas è in rapporto con la radice del mesocolon traverso e si affaccia sulla retrocavità degli epiploon.il pancreas si divide in 3 porzioni:
- la testa è disposta nella C duodenale, accoglie al suo interno il 1/3 distale del coledoco che poi confluisce colWirsung nella papilla di Vater.
- il corpo separato dalla testa tramite un istmo individuabile dal solco lasciato dai vasi mesenterici superiori.- La coda è l’estremità sn del pancreas che contrae rapporto con l’ilo splenico.
Struttura:Il pancreas ha un architettura lobulare. il singolo lobulo è costituito da:
- acini pancreatici: globosi, con lume ristretto e pareti formate da cellule piramidali con orientamento funzionale(nucleo, RE, Golgi, granuli di zimogeno).
- dotti escretori intralobulari: piccoli, dal lume ristretto, rivestiti da un epitelio cubico monostratificatoresponsabile della secrezione di bicarbonato e con citoplasma scarsamente colorabile, la porzione iniziale èaggettante nel lume dell’acino con le cosiddette cellule centroacinose.
- Isolotti di Langerhans: sono aggregati di cellule endocrine di tipo _ (glucagone) , _ (insulina), _ (somatostatina).Sono concentrate soprattutto nel corpo e nella coda.
- vasi intralobulari e fibre nervose.
fra un lobulo e l’altro abbiamo travate connettivali nelle quali ritroviamo:- dotti interlobulari: lume più ampio, rivestiti da epitelio cilindrico.- dotti collettori: sono i dotti di calibro maggiore denominati di Wirsung e Santorini, hanno una mucosa con
epitelio cilindrico di rivestimento dotato di orletto a spazzola e ghiandole tubulari mucosecernenti _ (correlarequesta struttura con la patologia neoplastica !); la mucosa poggia su una tonaca fibromuscolare.
- Fibre nervose- Vasi interlobulari

Vascolarizzazione:il pancreas , schematizzando, è irrorato da arterie che derivano:
1) dal tripode celiaco:- arteria lienale lungo il margine superiore _ rami penetranti a livello di corpo e coda che formano piccole arcate
anastomotiche.- Arteria gastroepatica _ gastroduodenale _ pancreatico-duodenale superiore2) dall’arteria mesenterica superiore:- arteria pancreatica inferiore (omologa della lienale)- arteria pancreatico-duodenale inferiore
le arterie pancreatico-duodenali superiore e inferiore si biforcano entrambe e quindi si anastomizzano a pieno canaleformando due arcate anteriore e posteriore per la testa del pancreas e il duodeno.
Il pancreas è tributario della vena porta che origina dietro la testa per confluenza delle vene mesenterica superiore e lienale(che ha ricevuto la mesenterica inferiore).Vi sono vene dirette alla lienale, alla mesenterica o alla porta stessa.
Il linfatici si dirigono ai LN:- dell’ilo splenico- del margine superiore del pancreas (3-8 ln satelliti della vena splenica)- pilorici ed epatici (nello spessore del piccolo omento)
- mesenterici
FISIOLOGIA DEL PANCREASIl pancreas esocrino ha fondamentalmente 2 funzioni:
1) secrezione di zimogeni _ le cellule piramidali degli acini secernono almeno 10 idrolasi differenti (tripsina,chimotripsina, elastasi, amilasi, lipasi, fosfolipasi) tali enzimi sono immagazzinati in appositi granuli in forma diproenzimi.La secrezione è controllata da:- pancreozimina-colecistochinina (CCK) rilasciata dalle cellule I del duodeno in risposta all’ arrivo di peptoni(polipeptidi grossolanamente digeriti)- gastrina- stimolazione vagaleFisiologicamente, i proenzimi vengono attivati solo dopo il loro arrivo in duodeno dalla enterochinasi presentenell’orletto a spazzola dell’enterocita.Esistono diversi meccanismi di difesa contro l’attivazione intrapancreatica degli zimogeni:
1) la compartimentazione (separazione dagli enzimi lisosomiali)2) la presenza di inibitori specifici (antitripsina)3) la coesione dell’epitelio duttale (ostacolo alla retrodiffusione nell’interstizio)4) la pressione dello sfintere d’Oddi ( barriera antireflusso)5) il flusso del succo pancreatico (rimozione di enzimi attivati)
nella pancreatite si ha l’indebita attivazione enzimatica intrapancreatica è responsabile di tutta una serie di lesioni locali e sistemiche.
È importante ricordare che gli enzimi pancreatici (amilasi ma ancor più specifiche – ricorda le ghiandole salivari - le lipasi) sono dosabili nel sangue costituendo un importante marker di patologia pancreatica.
2) secrezione di un succo alcalino ricco in bicarbonato _ questa secrezione sierosa alcalina serve a tamponarel’acidità del chimo garantendo un pH ottimale per l’attività degli enzimi pancreatici.la secrezione avviene ad opera delle cellule dei dotti intralobulari ed è stimolata dalla secretina, un polipeptiderilasciato dalle cellule S della mucosa duodenale in risposta alla _pH.

EMBRIOGENESI e ANOMALIE CONGENITE DEL PANCREASIl pancreas all’ inizio è rappresentato da due gemme endodermiche che si dipartono, una dorsale e una ventrale, dal tubodigerente.L’ abbozzo dorsale dà origine alla parte superiore della testa, al corpo e alla coda: esso ha un suo dotto escretore.L’abbozzo ventrale dà origine solo alla parte inferiore della testa ma esso è connesso anche alla via biliare che ha originedalla stessa gemma di endoderma: anche questo abbozzo ha un suo dotto escretore.
L’abbozzo ventrale ruota, verso dx e posteriormente, attorno all’intestino primitivo finchè non arriva in contatto conl’abbozzo dorsale nella sua posizione definitiva (considera anche che l’abbozzo dorsale è venuto a trovarsi a sndell’intestino per la rotazione della C duodenale).Venuti in contatto i due abbozzi si fondono e si formano 2 dotti escretori:
1) un grosso dotto detto principale o di Wirsung che nel 70% dei casi si fonde con il coledoco prima di terminarenella papilla di Vater.
2) un piccolo dotto detto accessorio o di Santorini che drena nella papilla minore.
Detto questo passiamo alle principali alterazioni congenite:
PANCREAS DIVISUMnel 5-10% della popolazione i due dotti non si fondono e pertanto il dotto di Santorini rimane a drenare la maggior parte delparenchima pancreatico (derivato dall’abbozzo dorsale) attraverso la papilla minore mentre il pancreatico ventrale drenasolo una piccola porzione di pancreas e riceve il coledoco.Questa è una condizione di rischio per pancreatiti croniche ricorrenti.Inoltre richiede al chirurgo una notevole attenzione quando opera nella zona periampollare.
PANCREAS ANULARESe gli abbozzi pancreatici si fondono in maniera anomala può derivarne la formazione di un pancreas che si avvolto amanicotto attorno al duodeno: se questo manicotto è completo e abbastanza stretto, si ha una stenosi duodenale.Questa comporta:1) dilatazione gastrica e del bulbo duodenale (segno della doppia bolla in radiologia)2) sintomatologia consistente in vomito post-prandiale3) ulcera duodenale da ristagno della secrezione gastrica4) eventuale ipertrofia della tonaca muscolare a monte della stenosi
PANCREAS ECTOPICOQualsiasi formazione di parenchima pancreatico in sede anomala.Le sedi in ordine di frequenza sono:1) duodeno2) stomaco3) digiuno4) diverticolo di Meckel5) ileo
il tessuto pancreatico può trovarsi :a) nella sottomucosab) nella muscolare: assume l’aspetto di uno pseudopolipo e può presentare una ombelicatura che è l’orifizio del dotto
escretorec) nella sierosa (cresce all’esterno come uno schwannoma)d) in tutti gli strati
possono restare asintomatici oppure infiammarsi e dare dolore, sanguinare e infine essere sede di tumori pancreatici.

PANCREATITE ACUTALa pancreatite acuta consiste in una flogosi acuta del tessuto pancreatico con estensione anche al tessuto peripancreatico chepuò presentarsi dal punto di vista anatomo-patologico e clinico in maniera differente.
- una forma di pancreatite è quella edematosa in cui si hanno solo 2 lesioni:a) edema infiammatorio del pancreas e dei tessuti peripancreatici che è dovuto alla risposta flogistica più che a una
vera e propria autodigestione.b) Focolai di steatonecrosi ovvero digestione del tessuto adiposo (lipasi) con saponificazione del grasso. Il grasso
trasudato può andare a formare un versamento peritoneale a brodo di pollo. Rappresenta la forma più comune, definita da un punto di vista clinico p. acuta lieve in
quanto recede in 1-2 settimane.
- L’altra forma ben più grave è la necrotico-emorragica nella quale si hanno:a) aree di necrosi-emorragica per digestione del parenchima (fosfolipasi) e dei vasi (elastasi).b) Ancora steatonecrosi.Le lesioni quindi sono molto più serie, si hanno complicazioni locali ma anche sistemiche (vedi oltre), dal punto divista clinico si parla di p. acuta severa.
L’eziologia della pancreatite è molto varia, tuttavia 2 cause sono responsabili dell’ 80% dei casi ovvero la colelitiasi el’alcolismo.Fra le cause “minori” di pancreatite individuiamo diversi tipi: - metaboliche: iperlipidemia, ipercalcemia
- tossiche: diuretici tiazidici e dell’ansa, azatioprina, sulfamidici etc.- infettive: parotite, Coxsackie- traumatiche: traumi addominali, chirurgia (es. interventi sul pancreas), ERCP- ischemiche: emboli, trombosi, shock, vasculiti etc.
Nel 10-20% dei casi infine la pancreatite è idiopatica (a causa ignota).
Patogenesila forma edematosa non richiede l’attivazione degli enzimi, in generale solo la lipasi è già attiva: per questo lasteatonecrosi è presente già in questa forma.
Sia la coledocolitiasi che l’alcolismo possono determinare l’ostruzione duttale: la prima se un calcolo si incuneanell’ampolla di Vater, il secondo per le concrezioni che possono prodursi nei dotti e per la papillite indotta da alcol.Se poi si considera che un pasto abbondante o l’assunzione stessa di alcol può indurre una ipersecrezione abbiamo:ostruzione + ipersecrezione _ ipertensione duttale.
L’ipertensione duttale può dare l’accumulo interstiziale di liquido ricco di enzimi, di questi la lipasi è già attivata per cui sipuò avere la steatonecrosi. Inoltre i leucociti presenti nell’interstizio possono iniziare una risposta infiammatoria con_permeabilità vascolare ed essudazione.Si produce quindi la forma edematosa.
La forma necrotico-emorragica necessita invece l’attivazione enzimatica da parte della tripsina della pro-fosfolipasi (lafosfolipasi produce la necrosi) e della pro-elastasi (l’elastasi provoca le emorragie).
Ci si chiede come possa avvenire l’attivazione degli enzimi e quindi la forma necrotico-emorragica: questa attivazionerichiede che le idrolasi lisosomiali vengano in contatto con i proenzimi, ciò può avvenire :
1) per lesioni della cellula acinare (es. da tossici, virus, traumi, ischemia anche conseguente alla forma edematosa).2) per alterato trasporto intracellulare: si è visto che in molte forme di pancreatite sperimentalmente indotte si ha co-
localizzazione dei proenzimi e dell’enzima lisosomiale catepsina B.Comunque si realizzi, l’attivazione intrapancreatica dei proenzimi provoca autodigestione del viscere.
CLINICA:dolore epigastrico di intensità progressiva fino a un massimo che si mantiene per ore o giorni.Il dolore è a barra, può essere irradiato al dorso a cintura oppure può essere diffuso.

Il carattere è continuo con picchi di intensità ed è di tipo trafittivo.Il pz assume una posizione antalgica con le ginocchia contro il tronco.Il dolore è dovuto a varie componenti come l’ipertensione a livello pancreatico, l’ irritazione nervosa, una eventualeperitonite chimica.
Il quadro della pancreatite severa è molto più drammatico:in questi casi la pancreatite:
- in genere è necrotizzante- può comportare tutta una serie di complicanze locali e sistemiche- ha prognosi peggiore- va trattata in maniera energica
le complicanze sistemiche sono:1) CID2) Shock eventualmente di tipo settico (per sovrainfezione batterica, vedi oltre)3) ARDS4) insufficienza renale5) iperglicemia (_glucagone e _insulina)6) ipocalcemia
le complicanze locali sono:1) pseudocisti pancreatiche2) ascessi = infezione delle aree necrotiche3) versamento pleurico a sn4) ittero ostruttivo da compressione del coledoco: tuttavia l’ittero ostruttivo può essere già presente nelle forme da
coledocolitiasi.5) Versamento peritoneale (abbiamo detto “a brodo di pollo”)6) peritonite
indici di laboratorio alterati:- _amilasi sierica (x 5 o più)_ ma questo non è un indice del tutto sensibile (es. pancreatite da iperlipidemia) e
specifico (.- _amilasi urinaria- _lipasemia (dopo 72h)- ipocalcemia- indici di infiammazione: leucocitosi, CRP, VES
- le pseudocisti pancreatiche sono raccolte di liquido ricco di enzimi pancreatici contenute da una parete fibrosa, priva dirivestimento epiteliale, si differenziano quindi dalle cisti che hanno una parete rivestita da epitelio.Le pseudocisti possono originare:
a) dalla organizzazione di tessuto fibroso attorno a una raccolta liquida peripancreatica in seguito a pancreatite.b) da una cisti da ritenzione (dovuta a ostruzione duttale) che infine perde il rivestimento epiteliale.
Esse si accrescono tramite microtraumi _ coagulo _ lisi _ _pressione osmotica _ richiamo di liquidi: possono raggiungeredimensioni ragguardevoli (specie nella pancreatite acuta).
Clinicamente le pseudocisti sono asintomatiche oppure responsabili della persistenza o della ricomparsa del doloreepigastrico dopo trattamento della pancreatite acuta.All’ esame obiettivo può aversi una massa palpabile e dolorabile.
Le pseudocisti entrano in DD con altre lesioni cistiche del pancreas come:
1) Le cisti vere sono cavità rivestite da epitelio e ripiene di un liquido mucoso o sieroso.Sono di natura congenita e si accrescono per secrezione e sfaldamento epiteliale.Cisti multiple pancreatiche possono comparire nell’ambito di patologie ereditarie come il rene policistico o la malattia diVHL.
2) Le neoplasie cistiche del pancreas (vedi oltre)

LA PANCREATITE CRONICALa pancreatite cronica è un processo infiammatorio cronico che coinvolge inizialmente i dotti e secondariamente ilparenchima sia esocrino che endocrino.
La pancreatite cronica per definizione è caratterizzata da una condizione favorente che persiste nel tempo e che porta,attraverso riacutizzazioni successive, a un quadro di degenerazione caratteristica con:
1) lesioni duttali: stenosi dei dotti, ostruzioni dei dotti per concrezioni di proteine e sali di calcio, alterazionidell’epitelio duttale (in senso atrofico, iperplasico, metaplasico), formazione di pseudocisti.
2) lesioni parenchimale: atrofia degli acini e _volume, sclerosi con _consistenza, relativo risparmio delle insule(almeno inizialmente).
Abbiamo detto che c’è una causa che persiste nel tempo: ma di che tipo?- la causa fondamentale nel mondo occidentale è l’etilismo cronico:possiamo ora precisare che
l’etilista cronico non ha pancreatite acuta ma piuttosto una pancreatite cronica che va incontro ariacutizzazioni.L’alcol provoca un danno tossico delle cellule acinari; inattivazione della litostatina, una proteina solubilizzante il Ca(vedi anche fumo di sigaretta); stimolo secretivo + papillite.
- in secondo luogo possiamo avere ostruzioni duttali per esempio esiti di una pregressa pancreatiteacuta (vedi cause), il pancreas divisum, neoplasie etc.
- in terzo luogo può esserci una flogosi autoimmune dei dotti un po’ come avviene per i dotti biliari(possibile associazione con colangite sclerosante, cirrosi biliare primitiva o altre m. autoimmuni).
- infine ci possono essere alterazioni genetiche come nella pancreatite ereditaria (carente inattivazione della tripsina) o nella fibrosi cistica.
La pancreatite cronica quindi può essere la sequela di una pancreatite acuta o può essere cronica ab initio.Nella pancreatite cronica si ritrovano le stesse lesioni ma distribuite a lungo nel tempo della pancreatite acuta.
Storia clinica:in genere si compone:
1) di una prima fase (5 anni) nella quale il pz sperimenta ripetute riacutizzazioni.
2) Di una seconda fase nella quale il pz sperimenta la riduzione delle crisi dolorose e l’inizio dei segni tipici dellainsufficienza pancreatica esocrina e/o endocrina:
- Steatorrea (malassorbimento lipidi)- creatorrea (malassorbimento proteine)- carenza di vitamine liposolubili (raramente)- intolleranza al glucosio _ diabete mellito
obiettività:1) epigastralgia e dolorabilità2) subittero, febbricola, versamento pleurico sn
le associazioni più frequenti sono con:- cisti da ritenzione _ pseudocisti- ulcera peptica da ridotto tamponamento- cirrosi epatica (negli alcolisti)- colelitiasi (associazione molto meno importante rispetto quella con pancreatite acuta)- patologie autoimmuni- CdR per l’adenocarcinoma
La diagnosi di pancreatite cronica viene posta su varie basi:

1) laboratoristica:- _amilasi, lipasi in fase di riacutizzazione- _ indici di colestasi- curva da carico di glucosio- test al glucagone- test di tolleranza all’insulina- escrezione fecale di grassi (normale <7g/24 h)- dosaggio attività enzimi pancreatici nelle feci- pancreolauryl test- dosaggio HCO3 nell’aspirato duodenale (_)
2) diagnostica per immagini:- US: ecostruttura, dilatazione duttale, calcificazioni, cisti, biopsia ecoguidata di lesioni sospette _ è anche trans-
duodenale _ importante per le biopsie.- ERCP: è il gold standard della diagnostica; conformazione del Wirsung ( a catena di rosario ; uniformemente
dilatato in caso di pancreatite cronica ostruttiva; compresso ab estrinseco nella autoimmune) , deformazione dottisecondari, cisti comunicanti.
- TC: dd pancreatite settoriale/cancro- Colangio - RMN: esame sovrapponibile alla ERCP.
TERAPIAMEDICA:
- terapia antinfiammatoria con corticosteroidi nella forma autoimmune.- Sospensione di alcol e fumo (profilassi recidive)- Dieta a base di cibi facilmente digeribili- Somministrazione ai pasti di preparati contenenti enzimi pancreatici: la potenziale in attivazione da parte
dell’HCl-pepsina richiede l’uso di preparati gastroprotetti o di farmaci antisecretivi- Terapia insulinica in caso di diabete; biguanidi se insulino-resistente.
ENDOSCOPICA:- sfinterotomia in caso di pancreatite cronica ostruttiva- sfinterotomia post-litotrissia extracorporea sui concrementi duttali.
CHIRURGIA:- trattamento delle complicanze come le cisti- pancreaticodigiunostomia
duodeno-cefalopancreasectomia
TUMORI CISTICI DEL PANCREASInsorgono in genere su corpo-coda (raramente danno ittero ostruttivo) _ presentazione per : massa palpabile, doloreepigastrico, reperto TC.Si dividono in Cistoadenomi e Cistoadenocarcinomi.
CISTOADENOMA SIEROSOÈ in genere microcistico, ovvero presenta cisti < 2 cm e aspetto spugnoso al taglio.Le cisti sono piene di un liquido sieroso demarcate da un epitelio cuboidale ricco di glicogeno senza potenzialitàmaligna (il cistoadenocarcinoma sieroso è rarissimo) _ se l’escissione è troppo pericolosa, si può lasciare in situ senzarischio.Si associa ad _CEA.Il cistoadenoma sieroso macrocistico è rarissimo.
CISTOADENOMA MUCOSOSi presenta come macrocistico ovvero presenta grandi cisti (2-20 cm) che spesso producono un aspetto “settato”all’imaging.Le cisti sono piene di un fluido vischioso rivestite da epitelio alto e con cellule caliciformi spesso in forma papillare.Evolve nel tempo a cistoadenocarcinoma _ è d’obbligo l’escissione del tumore.

È associato ad _ CA 15.3.
CISTOADENOCARCINOMA MUCOSOInsorge dal cistoadenoma mucoso.Le cellule sono maligne, polistratificate, invasive localmente e a distanza.
ADENOCARCINOMA DEL PANCREASSi tratta di una neoplasia a partenza dalla mucosa dei dotti pancreatici.Strutturalmente si tratta di abbozzi di tubuli ghiandolari formati da cellule cilindriche in un denso stroma fibroso (reazionedesmoplastica) ; vi sono poi varianti rare come l’adenosquamosa, quella a cellule acinari etc.
Il comportamento biologico dell’adenocarcinoma è estremamente aggressivo con spiccata tendenza ad infiltrare localmentee a metastatizzare ai linfonodi loco-regionali e a distanza.La presentazione clinica (e quindi la diagnosi) è in genere tardiva e infatti la resezione del tumore è possibile solo nel 10%dei casi.La prognosi è quindi decisamente infausta.
Da un punto di vista puramente didattico cerchiamo di tracciare la figura del “malato tipo”, in altre parole un individuo chepresenti FdR e quadro clinico suggestivi di adenocarcinoma del pancreas.Innanzitutto il malato tipo potrà arrivare all’attenzione medica per sintomi:
1) precoci in quanto possono comparire con malattia in stadio ancora operabile:- dolore post-prandiale tipo pancreatite: si ha per ostruzione del Wirsung ma è un reperto aspecifico specie quando preesiste pancreatite cronica (vedi oltre).- ittero ostruttivo: un piccolo carcinoma della testa, se collocato al punto giusto, può bastare a dare un quadro (piuttosto eclatante) di ittero ostruttivo _ per questo l’ittero è l’unico segno che, in una
certa percentuale dei casi, consente una diagnosi precoce ovvero la possibilità di resezionechirurgica.A questo proposito ricordiamo che i carcinomi sono così distribuiti: ~ 65% testa, ~ 20% corpo,~15%
coda. L’ostruzione del coledoco può essere dovuta a: - una coledocolitiasi: questa si accompagna acutamente a una colica biliare ma cronicamente può dare flogosi del dotto con una reazione di sclerosi che porta a ostruzione cronica benigna. - una neoplasia fra le seguenti: tumore della testa del pancreas, ampulloma, colangiocarcinoma del coledoco. Esiste allora un segno clinico che, ove presente, consente di fare diagnosi differenziale fra coledocolitiasi e neoplasia pancreatica: è il reperto di colecisti distesa e palpabile (segno di Courvoisier). Il principio di Courvoisier-Terrier recita infatti che la colecisti distesa e palpabile è quasi invariabilmente associata a una neoplasia ostruente la via biliare (in genere un carcinoma della testa del pancreas). Questo è dovuto al fatto che i calcoli del coledoco sono in genere provenienti dalla colecisti _ quindi abbiamo una colelitiasi _ quindi abbiamo una colecistite cronica _ quindi la colecisti è sclerotica e inestensibile in seguito a ostruzione. NB: l’ittero ostruttivo è il classico segno di presentazione dei carcinomi della testa, questo solo in una minoranza casi consente una diagnosi precoce, il più delle volte, per l’estrema aggressività del tumore la chirurgia con intenti curativi è comunque esclusa.
2) tardivi costituiscono la presentazione tipica dei tumori del corpo-coda:- dolore dorsale severo e continuo da infiltrazione dei plessi nervosi nel retroperitoneo.- cachessia- tromboflebite migrante (s. di Trousseau)- diabete mellito

il paziente si presenta a noi con questo quadro clinico; a questo punto chiediamoci: quali sono gli elementi anamnestici cheavvalorano l’ipotesi diagnostica “carcinoma del pancreas”? In altre parole: quali sono i FdR e le CdR per il carcinoma delpancreas?1) l’età : > 50 aa.2) il sesso maschile : M/F = 23) il fumo di sigaretta e l’etilismo cronico _ in altre parole la pancreatite cronica (vedi anche la forma ereditaria) . Quiritorna il discorso delle flogosi croniche che in vari organi favoriscono l’insorgenza delle neoplasie.Abbiamo a proposito della pancreatite cronica che fra le lesioni compaiono alterazioni dell’epitelio duttale (atrofia,metaplasia, iperplasia, displasia).4) la dieta ricca di lipidi (come per il cr. del colon)
A questo punto si procede con gli accertamenti diagnostici:
l’imaging: ci dà una diagnosi di massa pancreatica ed eventualmente dati su lesioni ripetitive ai linfonodi o al fegato.- eco-addome _ vie biliari intraepatiche e extraepatiche, metastasi epatiche- eco-endoscopia- ERCP possibilità di brushing e citologia per fare dd da una pancreatite cronica- TC spirale con m.d.c. _ T, N, M
la biopsia: è indispensabile per fare diagnosi di natura
il laboratorio: possibile _CA19.9 e CEA: come al solito non tanto per la diagnosi quanto per il follow-up.
La diagnostica per immagini è importante per la stadiazione clinica del tumore.A questo proposito è importante ricordare i criteri di operabilità che sono:1) per la T: tumore che non infiltri vasi mesenterici, vena porta o arteria epatica.2) N0: i linfonodi sono i peripancreatici, distribuiti lungo i vasi del pancreas (vena splenica, ilo splenico, mesentericasuperiore, sottopilorici e soprapilorici, del tripode celiaco e della mesenterica superiore).3) M0: il primo filtro che si incontra è il fegato dopodiché qualsiasi organo può essere interessato (polmone, osso etc.)
Come accennavamo la prognosi dell’adenocarcinoma duttale del pancreas è estremamente infausta.

PREMESSA.Queste pagine sono state realizzate come schema riassuntivo per fissare sulla carta alcune nozioni di anatomia patologica edi clinica prese da più fonti (lezioni, libri e talvolta anche Internet).Si tratta di appunti realizzati da uno studente per prepararsi all’esame di anatomia patologica : scritti quindi per un usopersonale e senza alcun intento “didattico”.
Di conseguenza:
3) Sono estremamente sintetici e danno spesso per scontanti concetti appresi a lezione o sul libro di testo;4) contengono sicuramente errori ortografici e concettuali *.5) Non contengono disegni e immagini che sono utilissime per capire le lesioni anatomo-
patologiche e per le quali si consiglia di ricorrere anche agli atlanti del Netter (consultabili in biblioteca).
In sintesi questi appunti non vogliono e non possono essere un sostituto del libro di testo:possono essere al massimo un punto di vista in più (la cui utilità è tutta da dimostrare) con cui confrontarsi.
Per concludere con una considerazione personale: lo studio dell’anatomia patologica è sicuramente molto complesso ma secondotto in maniera seria vi darà alla fine grande soddisfazione perché avrete l’impressione di cominciare a“padroneggiare” la Medicina.
Cordialmente buon lavoro, l’autore.
* eventuali errori possono essere segnalati all’indirizzo [email protected]
Patologia polmonare - indice
- Anatomia normale- patologie congenite- bronchiettasie e atelettasie- mucoviscidosi- polmoniti- TBC- pneumoconiosi e sarcoidosi- BPCO: enfisema polmonare e bronchite cronica- patologie del circolo polmonare: ipertensione; iperemia; embolia polmonare- patologia del cavo pleurico (versamenti, pneumotorace)- neoplasie bronco-polmonari
Anatomia normale dell’ apparato respiratorioLe vie aeree sono costituite procedendo distalmente da :- cavità nasali- faringe- laringe- trachea- bronchi- parenchima polmonare corrispondente agli alveoli, dove avvengono gli scambi gassosi

le vie aeree sono rivestite da mucosa costituita da epitelio cilindrico monostratificato ciliato (solo a livello dell’ orofaringe edel laringofaringe si tratta di epitelio pavimentoso pluristratificato) e da lamina propria contenente ghiandole siero-mucoseanaloghe alle salivari (→vedi cr.adenoido-cistico e mucoepidermoide): la funzione è quella di intrappolare particelleinspirate nello strato di muco che mosso dalle ciglia come un nastro trasportatore le convoglia verso l’esterno.Non esiste muscolaris mucosae né sottomucosa; più esternamente la parete è composta da una componente cartilaginea emuscolare che serve a variare il calibro delle vie aeree.A livello alveolare la struttura è semplificata al massimo essendo costituita da un epitelio pavimentoso monostratificatosostenuto da una trame di fibre elastiche che assicurano la compliance del polmone.
Trachea:La trachea è un condotto che collega la laringe ai bronchi principali: inizia a C6 e termina a T4: decorre quindi in parte nelcollo e in parte nel toracein posizione mediana: in entrambe le regioni contrae rapporto con importanti strutture quali:- la tiroide- il fascio vascolonervoso del collo- l’esofago (posteriormente)- la vena brachiocefalica sn (anteriormente)- a sn con la pleura sn, l’aorta e i vasi che da essa originano- a dx con la pleura dx, la vena azygos e la cava superiore (che d’altro canto deriva dalla confluenza dei tronchi
brachiocefalici)- a livello della carena contrae rapporto con l’atrio sn.- Naturalmente si hanno rapporti con i vari linfonodi: tracheobronchiali e paratracheali (vedi oltre).
è costituita da circa 15 anelli cartilaginei, incompleti posteriormente, che la mantengono beante.Arrivati alla carena si ha l’origine dei due bronchi principali che presentano così come i due polmoni alcune differenzefraloro: vediamo di dedurle con un ragionamento sequenziale:
1) il cuore occupa prevalentemente l’emitorace sn2) il polmone sn è di minori dimensioni → il bronco sn è di minor calibro (10 vs 15 mm)3) l’ilo polmonare di sn è più lontano dalla trachea che è in posizione mediana → il bronco di sn deve essere più
lungo (5 vs 2.5 cm) e più angolato (45 vs 25).
per quanto riguarda i polmoni sono suddivisi da incisioni dette scissure in lobi:- a dx abbiamo 3 lobi: superiore, medio e inferiore demarcati dalle scissure principale e secondaria- a sn abbiamo solo 2 lobi : superiore e inferiore , il superiore includendo la porzione omologa del lobo medio di dx detta
lingulaciascun lobo è fornito da un bronco lobare, e da arterie e vene lobari proprie:gli schemi secondo cui i bronchi principali dx e sn si suddividono sono i seguenti:
bronco principale dx → lobare superiore + intermedio → lobare medio + inferiore
bronco principale sn → lobare superiore → della divisione superiore + lingulare inferiore
ciascun lobo è poi diviso in segmenti areati da bronchi segmentari (3a generazione) e forniti di una propria arteria matuttavia sprovvisti di una propria vena poiché il drenaggio venoso è garantito da una rete intersegmentaria.I segmenti sono pensabili come delle formazioni piramidali con la base verso la pleura e l’apice verso l’ilo: la suddivisionedel polmone in segmenti apparentemente complessa è in realtà abbastanza semplice da ricordare:
Nel Superiore abbiamo : apicale, anteriore e posterioreNel medio (dx) e lingula (sn) abbiamo rispettivamente: anteriore+posteriore e superiore+inferioreNell’ inferiore abbiamo anteriore+laterale+posteriore e un segmento aggiuntivo a dx (il polmone è più grande) dettomediale.
Istologicamente nei bronchi notiamo:

1) mucosa costituita da epitelio, membrana basale e lamina propria con ghiandole acinose ramificate a secrezionesieromucosa: nell’ epitelio si ritrovano vari tipi cellulari:
- cellule cilindriche ciliate: il movimentociliare è bloccato da anestesia e nel fumatore cronico→ ristagno secrezioni- cellule con microvilli- cellule di rimpiazzo- cellule mucose- cellule a secr. Sierosa- cellule di Kultchinsky→ sono cellule del sistema apud
2) anelli e placche cartilaginee
si noti tuttavia come distalmente ( a livello di bronchiolo terminale ovvero di lobulo) si abbia scomparsa delle cartilagini,tarnsizione aepitelio cubico e comunque scomparsa di muco e microvilli.
Per quanto concerne l’anatomia macroscopica del polmone abbiamo che:
1) il polmone di DX è di dimensioni e di peso maggiore del sn: 400 vs 350g2) la capacità anatomica polmonare ovvero il volume massimo che i polmoni possono contenere è all’ incirca 3.5 lt e si
compone di: volume residuo (dopo una espirazione massimale) + capacità vitale (inspirazione massimale).3) Il colore è rosso scuro nel feto a termine, rosso vivo nel neonato, rosa nell’adulto e progressivamente antracotico4) La consistenza è fisiologicamente molle, può essere aumentata in caso di fibrosi5) Alla palpazione si produce un crepitio dovuto al passaggio di aria fra gli alveoli: il crepitio è ↑ in caso di enfisema e ↓
in caso di atelettasia o polmonite.6) la superficie del polmone è ricoperta dalla pleura, una sierosa costituita da un rivestimento mesoteliale che poggia suun piano fibroelastico in continuazione col parenchima polmonare: la pleura è fisiologicamente liscia, trasparente, lucida;reperti patologici possono essere una pleura opaca e ruvida per la deposizione di fibrina oppure grigiastra e ispessita.
Il lobulo polmonare è l’unità strutturale dell’ organo: è rifornito dal bronchiolo terminale il quale dà origine a bronchiolirespiratori la particolarità dei quali è di avere una parete parzialmente costituita da alveoli e i quali terminano in vari sacchialveolari a fondo cieco: gli alveoli costituiscono il parenchima polmonare e il livello a cui avvengono gli scambi gassosi: lamembrana di scambio è costituita da almeno 5 strati:- epitelio alveolare- m. basale- interstizio dotato di ff. elastiche- m. basale- endotelio capillare
l’epitelio alveolare è costituito dagli pneumociti:- in gran parte del tipo I: cellule pavimentose che consentono gli scambi- in minor misura di tipo II: cellule cubiche sporgenti nel lume secernenti una lipoproteina ad attivita tensioattiva detta
surfattante- all’ interno dell’ epitelio si trovano macrofagi alveolari e qualche neutrofilo: è importante che a questo livello siano
presenti fattori ad attivita antiproteasica come l’α1-antitripsina: un deficit genetico o acquisito (vedi enfisemi) provocala digestione delle fibre elastiche da parte delle proteasi rilasciate dai leucociti.
I linfatici del polmone:
Nel polmone vi sono 2 reti linfatiche fra loro anastomizzate:- profonda che scarica in senso centripeto all’ilo- periferica che scrica ai vasi linfatici subpleurici i quali poi confluiscono all’ilo
dalla periferia al centro incontriamo i ln:intrapolmonari → ilari → tracheobronchiali inferiori (carenali) e tracheobronchiali superiori* → paratracheali
*a sn c’è anche un linfonodo dell’ arco aortico che si colloca nella cosiddetta finestra aorto-polmonare a dx i linfonodi tracheobronchiali superiori ricevono afferenze dall’ ilo di sn per cui si veda la stadiazione dell’ N nel crbroncogeno.

PATOLOGIE CONGENITE
AGENESIA-IPOPLASIA POLMONARE: può essere:-secondaria a una massa occupante spazio all’ interno del torace come un’ ernia diaframmatica che porta a dislocazione divisceri addominali e che è in genere localizzata a sn.- primitiva ovvero dovuta a un mancato sviluppo degli abbozzi dell’ albero respiratorio che comincia a formarsi a partire
dalla quarta settimana; nel qual caso può essere:1)bilaterale (ovviamente incompatibile con la vita) con presenza di un abbozzo tracheale o di trachea fino alla biforcazionecon o senza abbozzi dei bronchi principali; nel “migliore” dei casi ci sono bronchi con un poco di tessuto polmonare e con ivasi polmonari: il tutto dipende dal momento in cui l’organogenesi si arresta.2)monolaterale nel qual caso può essere: A)completa se manca del tutto un bronco principale B)con abbozzo di bronco a fondo cieco C)con bronco principale e abbozzo di parenchima polmonare immaturo che può assumere la tipologia di cisti unica, cistimultiple o parenchima di tipo fetale non aereato. In questi casi si hanno sequele infiammatorie dovute al fatto che il ristagnodi muco che porta a infezione e bronchite purulenta circoscritta; le conseguenze possono essere:-espettorato purulento-emoftoe-mediastinite-pneumotorace spontaneo secondario-ascesso che si sviluppa nell’abbozzo di parenchima (ricorda gli anaerobi)
per l’agenesia lobare valgono gli stessi discorsi: no bronco, abbozzo di bronco,bronco con abbozzo di parenchima.
BRONCO SOVRANNUMERARIOPuò essere presente un bronco sovrannumerario che partendo dalla trachea in genere sul lato dx può:-terminare a fondo cieco (abbozzo)-terminare in un abbozzo di parenchima-contrarre un’ anastomosi con un bronco segmentartio del lobo superiorementre in quest’ultimo caso è asintomatico nei primi due porta alle stesse conseguenze già viste per l’agenesia polmonaremonolaterale.
SEQUESTRO POLMONAREÈ una porzione di parenchima che non comunica con l’albero bronchiale e che presenta una vascolarizzazioneanomala da parte di un ramo dell’ aorta discendente. Può essere:Extralobare: nel qual caso è un vero e proprio polmone in miniatura, con una sua pleura, localizzato in generesull’emidiaframma sn. Il parenchima può assumere gli aspetti già visti nell’ipoplasia di tipo C. il sangue refluo va allaazygos: in definitiva il sequestro extralobare è uno shunt inserito in parallelo al circolo sistemico.Intralobare: in questo caso il parenchima interessato è all’interno di un lobo polmonare in genere inferiore sn, puòpresentare un certo grado di aereazione per via dei fori di Kohn; il drenaggio da parte delle vene polmonari che èconservato.

CISTI CONGENITE POLMONARIUna cisti è una struttura cavitaria con una parete che in genere riproduce quella della struttura da cui prende origine. Le cisti possono essere distinte in base al punto d’origine in:Paramediastiniche: cisti singole di origine molto prossimale, possono essere enucleate dal tessuto polmonareIntraparenchimali: cisti broncogene spesso multiple, non enucleabiliQueste cisti broncogene sono costituite da epitelio cilindrico da gh. Sieromucose, tessuto muscolare liscio e cartilagine: ilcontenuto è mucoso.Talvolta tuttavia possono presentare un epitelio pavimentoso polistratificato come conseguenza di metaplasia squamosa.In sede più distale si va verso le cisti piccole e numerose che sostituiscono un intero segmento, lobo o polmone: ilcosiddetto polmone policistico.Il rivestimento delle cisti bronchiolari è cubico monostratificato e manca di gh.mucose per cui il contenuto è sieroso: ladifferenza con il quadro anatomo patologico della bronchiectasia cistica consiste nella mancata formazione di un alberobronchiale completo.Infine le cisti alveolari sono piccole cavità multiple (talvolta anche secondarie) prive di un rivestimento cubicomonostratificato e con parete connettivale.Le complicanze delle cisti possono essere:-assenti-ipertensione e cuore polmonare in caso di interessamento massivo-infezione:suppurazione→broncopolmonite-rottura:pneumotorace spontaneo secondario-emorragia:emoftoe o emottisi in base vaso usurato
ALTERAZIONI DEL CALIBROFistole tracheo-esofagee:1)se associata ad atresia dell’esofago avvero a un’ agenesia focale (moncone esofageo superiore e/o inferioreanastomizzati alla trachea) si presenta da subito: il bambino ha tosse con espettorazione del latte ad ogni poppata, se c’ècomunicazione fra trachea e moncone inferiore si ha aerofagia quando il bambino piange,il bambino non acquista peso e sidisidrata progressivamente.2)se non associata ad atresia si presenta come un canalicolo che mette in comunicazione un esofago ben canalizzato con latrachea; in questo caso la malformazione può essere scoperta in età adulta nel pz con una storia di broncopolmonitiricorrenti.
Malformazioni della trachea:Stenosi da membrane o da riduzione distrettuale del diametro degli anelli
Tracheomalacia congenita da agenesia di alcuni anelli cartilaginei: collasso in fase inspiratoria
Alterazioni del calibro dei bronchi: sono le broncostenosi e le bronchiectasie per le quali va fatto un discorso a parteessendo importanti patologie acquisite
BRONCOSTENOSILa broncostenosi può essere dovuta a:1) un corpo estraneo2) una neoplasia che cresce all’interno del bronco: in genere benigna o a basso grado di malignità3) una flogosi della parete bronchiale4) una patologia che dall’esterno comprime il bronco come: una neoplasia mediastinica o una linfoadenite tubercolare
che può produrre stenosi attraverso vegetazione (di tessuto di granulazione nel lume bronchiale), compressione ocicatrizzazione. In genere si verifica in un punto di biforcazione bronchiale nel cui angolo è collocato un linfonodo:questa condizione anatomica si verifica per il bronco lobare medio di dx per cui si parla di sindrome del lobo medio(atelettasia e bronchiectasie secondarie a stenosi).
Le conseguenze della broncostenosi possono essere:

1) se la stenosi è lieve, nessuna2)se la stenosi è severa iperinflazione dinamica (meccanismo a valvola) e bronchiectasie secondarie (ristagnosecrezioni)*3)in caso di ostruzione completa atelettasia
*Da notare che il ristagno del muco provoca una complicanze infettive per cui si potrà avere una broncopolmonite focalericorrente secondaria alla lesione anatomica.
BRONCHIECTASIELa dilatazione del bronco può essere:1)congenita pura: molto rara2)acquisita su base congenita: meiopragia connettivale congenita, s. di Kartagener, mucoviscidosi3)secondaria a broncostenosi (ostruttiva o compressiva) o cmq associata a infiammazione
la dilatazione può essere di vario tipo:cilindrica: l bronchi/oli sono uniformemente dilatatati e arrivano fin sotto la pleura: questa tipologia è in genere congenita ecolpisce più frequentemente i lobi inferiorisacciforme: il bronchiolo presenta una dilatazione a forma di clava, in genere hanno eziologia infettiva (oggi tendono ascomparire) e si localizzano prevalentemente agli apici e in questa localizz. Sono “secche”: non muco ma emoftoe.Cistiche: sono dilatazioni bronchiali multiple, a grappolo, differenziabili dal polmone policistico in quanto qui i bronchi sidilatano verso la periferia.La patogenesi della bronchiectasia è complessa:
In grassetto sono evidenziati i tre fattori tra loro interdipendenti che danno inizio alla lesione:L’ostruzione non precede necessariamente gli altri due: per es. un ristagno di muco come nella mucoviscidosi o nella s. diKartagener può esso stesso ostruire il bronco oppure la bronchiectasia può svilupparsi per infezione da parte dimicrorganismi particolarmente virulenti senza che sussistano precedenti alterazioni della clearance del muco.
In corsivo sono indicate la sintomatologia: tosse produttiva con escreato purulento e fetido, emoftoe; nonché le principalicomplicazioni: l’ascesso cerebrale da embolo micotico, l’interessamento pleurico con pneumotorace spontaneo secondario ol’empiema, l’amiloidosi sistemica e non indicato il cuore polmonare cronico in caso di patologia massiva.
Sono sottolineate le lesioni elementari a livello della bronchiectasia:A livello della mucosa vi sono aree di desquamazione, ulcerazioni, metaplasia squamosa.A livello della parete è presente un infiltrato infiammatorio variamente composto (neutrofili→monociti)Vi è in fine una estensione del processo infiammatorio alle aree circostanti di parenchima con conseguente fibrosiperibronchiectasica.
ATELETTASIAIl termine indica il collassamento di una parte del parenchima polmonare per cui quella porzione avrà un contenuto d’ariabasso o nullo.Vi sono fondamentalmente 2 tipi di atelettasia che si distinguono dal punto di vista eziopatogenetico e non solo:
Atelettasia da ostruzione Atelettasia da compressione
1)ostruzione
ulcerazioni
Debolezza parete
2)Ristagno muco 3)Suppurazione-flogosi
Escreato
purulento
al mattino
(vomica)
emoftoeDilatazione datrazione/distensione
Metaplasiasquamosa
Ascesso cerebr. (emboli micotici)
Fibrosi peribronchiale
Possibile atelettasia
Coinvolgimentopleurico
Amiloidosi sistemica post-infiammatoria
Pneumotorace sp.
Pleurite,empiema

Eziologia Qualsiasi causa porti ad un’ ostruzionecompleta di un bronco: corpo estraneo,neoplasia , tappo mucoso, sindrome del lobomedio.
Qualsiasi causa possa determinare unacompressione dall’esterno: versamentopleurico(in genere idrotorace nei cardiopatici),Neoplasia es.mesotelioma pl. , pneumotorace.
Patogenesi edevoluzione
L’aria alveolare viene riassorbita conconseguente collassamento degli alveoli→si hauna continua trazione sulle vie aeree da partedel parenchima circostante aerato per cuiavremo:-un aumento relativo della perfusione delparenchima atelettasico (shunt) che però ècontrastato da meccanismi di compenso.-l’accumulo di trasudato porta a edemaalveolare (polmone umido) che favorendol’infezione predispone a broncopolmonitepost-atelettasica; cronicamente poi si hal’evoluzione a fibrosi (carnificazionepolmonare): quindi se non si interviene intempo si hanno lesioni irreversibili.- la tendenza alla bronchiectasie post-atelettasiche di tipo cilindrico.
Nel caso di una compressione estrinseca delparenchima non c’è un intrappolamentodell’aria con conseguente riassorbimento masolo una ↓contenuto d’aria (il polmonegalleggia e al tatto è crepitante) secondaria alrelativo collassamento degli spazi aerei.
Anche in questo caso essendo la V della zonamolto ridotta si avrà un mismatching delrapporto V/Q (shunt)
La lesione è completamente reversibilequando venga eliminata la causa che producecompressione
Reperti patologici La zona atelettasica si presentaDepressa rispetto alle circostantiDi colorito bluastro (sangue venoso che nonviene ossigenato)C’è spostamento del mediastinoomolateralmente all’atelettasia
In questo caso il polmone( lo pneumotorace locomprime nel suo insieme) si presentacollassato e raccolto verso l’ilo; il colorito èugualmente tendente al bluastro ma lospostamento mediastinico è controlaterale.
Zone di atelettasia possono formarsi anche in caso di lesioni polmonari fibrotiche che impediscono una regolare espansionedi zone di parenchima (a. da contrazione).
Un tipo particolare di atelettasia è quella fetale che si associa alla SINDROME DA DISTRESS RESPIRATORIO DELNEONATO:L’insufficienza respiratoria nel neonato può essere dovuta a varie cause: immaturità cerebrale o dei polmoni, inalazione dicoaguli o di liquido amniotico durante il parto, blocco dei centri della respirazione in caso di somministrazione di anesteticialla madre.L a patologia di gran lunga più frequente come causa di insufficienza respiratoria è la cosiddetta MALATTIA AMEMBRANE IALINE:Si manifesta clinicamente subito dopo la nascita con cianosi ingravescente:_La malattia è dovuta alla carenza di surfattante, una sosatnza lipoproteica prodotta dagli pneumociti di tipo II, ad azionetensioattiva: il surfattante infatti1)abbassa la tensione superficiale degli alveoli opponendosi al collasso delle pareti alveolari2)stabilizza le dimensioni alveolari consentendo un adattamento della tensione in funzione del volume alveolare
Carenza disurfattante
↑ tensionealveolare atelettasia
MismatchingV/Q (shunt)
Ipossiemia,ipercapnia eacidosi
Danno endotelialee agli pneumociti
Formazione dellemembrane ialine.
↓ diffusibilità (alveoli)
Ulteriore ostruzione (bronchioli)

In pratica mentre dopo il primo respiro i polmoni di un neonato normale trattengono il 40% dell’aria, nel neonato affetto daquesta malattia i polmoni si svuotano ad ogni espirazione rendendo il lavoro respiratorio troppo gravoso.La carenza di surfattante può essere dovuta a:1)una nascita prematura in quanto la produzione di surfattante avviene in modo significativo solo dopo la 35a settimana(in tutto sono 38 a partire dall’ovulazione).2)la nascita da madre diabetica in quanto l’iperinsulinemia riduce la sintesi di corticosteroidi che stimolano la sintesi delsurfattante3)il parto cesareo in quanto la manca lo stress del parto che stimola la secrezione di corticosteroidi
come profilassi nelle situazioni a rischio si fa:-analisi dei fosfolipidi nel liquido amniotico-somministrazione prenatale di corticosteroidi-somministrazione di surfattante al momento della nascita-somministrazione di ossigeno e ventilazione assistita
esiste anche un'altra sindrome da distress respiratorioARDS: SINDROME DA DISTRESS RESPIRATORIO ACUTO
Patogenesi:
Questi eventi possono avvenire anche in più organi producendo la cosiddetta disfunzione d’organo multipla.Le conseguenza è danno alla parete alveolare con edema alveolare e riduzione dei livelli di surfactante e atelettasia deglialveoli. In definitiva il volume polmonare a una data p risulta significativamente ridotto (↓compliance).
Anatomia patologica:1)vasodilatazione e permeabilizzazione capillari.2)infiltrato neutrofilo.3)necrosi pneumociti di tipo I che desquamano nel lume alveolare con denudamento della membrana basale (DAD).
4)formazione di membrane ialine di fibrina negli alveoli.5)microtrombi a livello arteriolo-capillare.
Innesco:
Un eventoprecipitante (es.sepsi) provoca ilrilascio di citochinequali TNF-α e IL-1
Amplificazione:
Cellule effettrici,soprattutto, ineutrofili arrivanosul posto e vengonoattivati
Danno:
Il rilascio di radicalidell’ossigeno e diproteasi provocadanno cellulare

FIBROSI CISTICA o MUCOVISCIDOSI
Si tratta della patologia genetica letale più comune nella razza bianca con alta incidenza soprattutto nel Nord Europa 1/5000nati vivi.È una patologia a trasmissione autosomica recessiva che potremmo definire ad espressività variabile incui il difetto genetico risiede in una proteina trasportatrice del Cl; l’alterazione funzionale di questaproteina negli omozigoti provoca:1) un ↓ assorbimento di NaCl a livello dei dotti escretori delle ghiandole sudoripare per cui si ha un sudore ad elevata
concentrazione salina che gn consente di fare diagnosi.2) Un ↑ riassorbimento di NaCl e acqua a livello delle ghiandole mucipare di vari organi → ne consegue la presenza di
un muco particolarmente vischioso e tenace che quindi tende ad accumularsi invece di essere espulso (suggestivo è aquesto riguardo il nome della malattia).
Le lesioni nella mucoviscidosi possono essere varie:
1) nel 5-10% a causa di un muco particolarmente denso si sviluppa dopo la nascita un quadro di ostruzione intestinaledetto ileo da meconio (vedi la definizione di meconio) che è rapidamente letale.
2) Nell’ 85-90% dei casi si ha la comparsa di fibrosi cistica del pancreas ovvero:Occlusione duttale che porta a una pancreatite cronica per cui il pancreas presenterà:- dilatazioni cistiche piene di mucine eosinofile.- fibrosi con ↓ volume e ↑ consistenzaoltre a un possibile interessamento delle insule con comparsa di diabete, si avrà una sindrome da malassorbimento conconseguenti deficit nutrizionali (es. malassorbimento lipidi e vitamine liposolubili) e feci abbondanti, putride, schiumose.
3) si potrà avere anche ostruzione delle vie biliari con conseguente cirrosi biliare in una piccola percentuale di casi.
4) in tutti i casi si ha interessamento polmonare caratterizzato da:accumulo di muco vischioso → ostruzione e difetto di clearance → sovrapposizione di infezioni batteriche conbronchiectasie, broncopolmoniti, ascessi polmonari: i patogeni più comuni sono S. aureus e P. aeruginosa.
La sopravvivenza è variabile: con varie terapie di supporto è possibile arrivare all’età adulta (vita media attuale attorno ai30 anni).

PolmonitiSono processi infiammatori a carico del parenchima polmonare nei quali l’essudato infiammatorio può raccogliersi a livelloalveolare (alveoliti) o solo interstiziale.Si possono classificare in termini di:
Eziologia:Infettive: batteriche , da micoplasma, actinomicosi, tubercolosi, Virali (orthomyxo e paramyxovirus; VZV;HSV e CMV) micotiche (Aspergillus e Candida) protozoi (P.carinii)da cause chimiche: uremia, pneumoconiosida cause fisiche: radiazioni ionizzanti
distribuzione delle lesioni:lobare: sono interessati nella loro totalità uno o più lobilobulare o broncopolmonite: sono interessati parecchi lobuli distribuiti “a pelle di leopardo”
andamento:acutosubacuto-cronico: le polmoniti virali hanno lenta risoluzione, bisogna escludere in caso di polmonite recidivante localizzatasempre nella stessa sede una broncopolmonite da stenosi ceh può avere alla base un carcinoma broncogeno.
Lesioni fondamentali:
Sierosa: liquido sieroso essudatizio in cavità alveolare. Si differenzia dall’edema alveolare perché è localizzato enongeneralizzato e inoltre perché contiene qualche neutrofilo (non troppi altrimenti sarebbe purulenta!).Si tratta di un processo virale o del primo step verso essudati “più consistenti”.Evolve per restitutio ad integrum.
Siero-fibrinosa: la permeabilità è maggiore e passa in alveolo una buona quantità di fibrina; si presenta come tale in seguitoad uremia o a radiazioni oppure e uno step verso la purulenta. L’evoluzione di una fibrinosa pura è in generel’oganizzazione delle “membrane ialine” di fibrina da parte di un tessuto di granulazione, infatti la relativa carenza dienzimi proteolitici che digeriscano la fibrina porta all’ inizio del processo di organizzazione entro la 7-10 giornata.
Siero-fibrino-purulenta: è la tipica polmonite batterica da piogeni. L’elemento distintivo è l’essudato ricco di neutrofili.L’evoluzione è in genere per lisi della fibrina (che può essere più o meno abbondante a seconda del batterio patogeno) erestitutio ad integrum: la lisi è possibile in quanto vi sono molti enzimi proteolitici.Tuttavia in caso di batteri particolarmente virulenti secernenti tossine necrotizzanti come lo stafilococco e se c’è uno scarsoapporto ematico (↓pH, ↓arrivo di Ig) l’evoluzione può essere quella della necrosi colliquativa con formazione di un ascessopolmonare.
Siero-emorragica: in genere si distingeue da una emorragia pura in quanto è evidente che non c’è solo sangue maessudato+sangue. Può essere dovuta a radiazioni, agenti virali etc. in base alla quantita di fibrina può evolvere con restitutioad integrum o carnificazione. Possibile anche la sovrapposizione batterica.

Interstiziale: è in genere di eziologia virale e infatti si osserva un infiltrato mononucleato (linfociti) a livello dei setti cehsecondariamente da anche origine a una alveolite sierosa. L’alveolite sierosa guarisce come già detto per restitutio adintegrum. La infiammazione interstiziale è ad evoluzione lenta per cui può guarire con restitutio ma anche con una fibrosiinterstiziale.Inoltre può essere un buon terreno per l’attecchimento di batteri per cui si avranno polmonite interstiziale+purulenta.
Polmoniti battericheNella maggior parte dei casi si tratta dello pneumococco, tuttavia sono possibili agenti eziologici anche S. pyogenes, lostafilococco, nonche gram – come legionella, pseudomonas, enterobacteriaceae come Klebsiella o E.coli, o ancora anaerobidel cavo orale, o mycoplasma il quale dà una polmonite atipica.
La presentazione può essere quella di una polmonite lobare o di una brocopolmonite
Polmonite lobareIn genere è data dallo pneumococco, molto meno frequentemente da altre specie. L’estensione lobare oggi è poco frequenteperché l’uso degli antibiotici limita l’estensione del processo solo a una porzione del lobo; infatti l’estensione lobare sembradovuta sia alla suscettibilità dell’ospite sia alla particolare capacità di diffusione del patogeno.Fra gli 80 ceppi di di pneumococco differenziati in base alla capsula polisaccaridica danno solitamente polmonite i ceppi 1,2e 3.In generale l’infezione insorge in condizioni di ↓ efficienza delle difese dell’ albero respiratorio (per es. infezioni virali oraffreddamenti che ↓ la clearance mucociliare) o dell’ organismo in generale (pz immunodepressi).La polmonite lobare da pneumococco essendo la più frequente è anche definita “tipica”: ha un esordio acuto e una duratabreve, si presenta con febbre alta e continua e guarisce in circa 8 gg.
Si sviluppa in 4 fasi:1) congestione: il lobo è congesto, presenta un consolidamento (graduale scomparsa del crepitio) omogeneo dovuto
all’essudato ancora sieroso , è quindi aumentato di peso e di volume tanto che possono osservarsi le impronte dellecoste. Per diffusione dal parenchima sottostante si ha una pleurite siero-fibrinosa circoscritta al lobo interessato; lapresentazione clinica sarà quindi con una febbre alta e continua, tosse dovuta all’interessamento di una zonariflessogena come la pleura, dolore pleurico da sfregamento quando compare la fibrina,dispnea e cianosi dovuta allaperdita di funzione di un lobo.
2) epatizzazione rossa : presenta le caratteristiche della prima fase ma in misura maggiore: il crepitio è del tuttoscomparso perché gli alveoli sono pieni di essudato che diventa sempre più purulento, l’aspetto rosso del polmone èdovuto all’iperemia e allo stravaso di eritrociti in alveolo per diapedesi; sulla superficie pleurica si evidenzia lacomponente fibrinosa con opacamento prima e formazione di una vera e propria membrana poi.
3) Epatizzazione grigia: l’essudato è francamente purulento e dà: 1)compressione dei setti per cui senza l’iperemia ilpolmone assume un colorito grigiastro; 2) tosse produttiva.
4) Risoluzione: la lisi dei macrofagi e dei neutrofili libera proteasi che digeriscono la fibrina per cui si ha fluidificazione etosse produttiva, anche gli enzimi che arrivano per via ematica in seguito al ripristino della perfusione contribuiscono alprocesso che quindi si conclude con restitutio ad integrum in circa 8 gg, prima cioè che la fibrina possa andare incontroad organizzazione.
Un tipo di polmonite lobare più rara, frequente più che altro come infezione ospedaliera nei pz con ventilazione assistita onei diabetici è quella da Klebsiella che ha una evoluzione ascessuale.
Le complicanze della polmonite possono essere:-mancata lisi della fibrina ed organizzazione della stessa che porta a carnificazione-evoluzione ad ascesso nel caso di patogeni particolarmente virulenti come lo pneumococco di tipo 3 e la klebsiella-interessamento del cavo pleurico con un empiema-disseminazione per batteriemia con infiammazioni purulente a vari livelli: endocardio, reni, milza, encefalo, articolazionietc.
Polmonite lobulare o broncopolmoniteÈ la variante anatomo-patologica di più frequente riscontro ed è costituita da focolai lobulari multipli che se confluisconopossono essere mal distinguibili da una lobare.

Colpisce soprattutto nell’infanzia e nella vecchiaia e in quest’ultimo caso è in relazione all’ immunodepressione, acondizioni di stasi a livello polmonare che creano il terreno per l’attecchimento dei batteri, ad infezioni virali checompromettono i meccanismi di difesa delle vie respiratorie.Da non dimenticare l’associazione con condizioni di broncoostruzione che danno broncopolmoniti recidivanti sempre nellastessa zona: l’ostruzione potrebbe essere dovuta a un carcinoma broncogeno.
Differisce dalla lobare solo in quanto a distribuzione delle lesioni: infatti si tratta di focolai di infiammazione purulentache danno anch’essi pleurite siero-fibrinosa (e quindi dolore) se ubicati in sede subpleurica, disposti in generebilateralmente e per lo più nelle zone più declivi (basi in ortostatismo e docce costovertebrali in clinostatismo).L’evoluzione è in genere per restitutio ad integrum ma possono verificarsi le stesse complicanze della lobare.
Inoltre le broncopolmoniti sono più varie in quanto ad eziologia: stafilo, strepto, haemophilus, enterobacteriaceae (coli eklebsiella), pseudomonas, legionella etc. tutti questi danno in genere polmonite lobulare e occasionalmente lobare.
Ascesso polmonareÈ una processo necrotizzante a carico del parenchima polmonare la cui eziologia può essere varia: in genere si tratta dipatogeni fortemente virulenti come certi streptococchi, lo stafilococco, la klebsiella o anaerobi del cavo orale.La modalità di arrivo al polmone può essere varia in quanto può trattarsi di:1)evoluzione ascessuale di una polmonite preesistente (vedi polmoniti da strepto tipo 3 o klebsiella; polmoniti micotiche)2)arrivo del batterio per via ematogena: vedi stafilococco per es. nei tossicodipendenti con endocardite al cuore dx che dàemboli settici.3)aspirazione di corpi estranei (per es bolo) per cui si ha polmonite ab ingestis con evoluzione ascessuale: in questo caso peril decorso delle vie aeree la sede è frequentemente dx.4)associazione con processi che interessano i bronchi come le bronchiectasie o una broncoostruzione da parte di unaneoplasia.5)diffusione per contiguità6)causa non identificabile per cui l’ascesso è criptogenetico.
L’ascesso è una cavità contenente pus (fetido nel caso degli anaerobi) la parete è costituita dal parenchima circostante coninfiltrato infiammatorio di macrofagi e neutrofili e una reazione connettivale che tende a limitare il processo: se questareazione connettivale manca significa che il processo è in fase di espansione ed evolverà in gangrena.Le dimensioni sono importanti perché mentre ascessi svuotati del ∅< 4cm possono guarire spontaneamente per cicatricequando il ∅> 4cm la guarigione è più difficile per cui può essere indicato il trattamento chirurgico.Quando la parete dell’ascesso arriva a contatto con la parete di un bronco di medio calibro può aprirsi nel bronco e svuotarsiparzialmente formando un livello idro-aereo (che peraltro può essere gia presente nel caso di batteri ceh producano gas).Le complicazioni dell’ ascesso sono più o meno le stesse della polmonite: pleurite fibrinosa circoscritta, batteriemia, sepsi(la cicatrice è la modalità di guarigione) tuttavia l’ascesso può dare due complicazioni caratteristiche:-nel caso in cui si apra nel cavo pleurico dà una pleurite purulenta diffusa o empiema.-nel caso in cui si apra anche in un bronco dà uno pneumotorace.
Una forma particolare di infezione ad evoluzione ascessuale è l’actinomicosi prodotta dall’actinomices un gram + a strutturafilamentosa preferenzialmente anaerobio che può trovarsi nel cavo orale in associazione con le carie dentarie.Questi microrganismi crescono raggomitolati in colonie giallastre dette “grani di zolfo”.Pertanto la lesione fondamentale è un microascesso di pochi mm contenente il grano di zolfo all’interno di una zona dinecrosi circondata dal parenchima sano con infiltrato infiammatorio.Questi microascessi per coalescenza danno cavità nodulari più ampie che possono formare tragitti fistolosi e scaricareall’esterno il loro contenuto.Le localizzazioni dell’actinomicosi possono essere:-cervico-facciale: gengiva e mandibola-raramente polmonare dove può essere primitiva o secondaria.-intestinale: colie che può dare peritonite-uterina: specialmente iatrogena
Polmoniti micoticheCome noto le micosi profonde colpiscono in genere solo individui con deboli difese immunitarie per cui sono daconsiderare opportunistiche. Una forma è quella membranosa a livello di organi cavi come la bocca, l’esofago etc. per cui ilmicete cersce su sree disepitelizzate formando assieme a neutrofili eresidui necrotici una membrana.

A livello di organi parenchimatosi invece la lesione può essere un nodulo o un ascesso.Il nodulo è una reazione infiammatoria in cui un infiltrato di neutrofili ed eosinofili circoscrive le ife. L’ascesso è del tutto simile a quello batterico solo che all’interno vi saranno ife al posto dei batteri e nell’infiltrato periascessuale troveremo una componente di eosinofili: rappresenta l’evoluzione necrotica del nodulo.
I miceti più importanti sono Candida e AspergillusLa candidosi e l’aspergillosi sono affezioni infiammatorie nodulari (non propriamente granulomatose in quanto l’infiltratonel granuloma comprende caratteristicamente cellule giganti) del parenchima polmonare che può evolvere ad ascesso.Aspergillus può anche dare l’aspergilloma che consiste in un ammasso di ife ceh cresce in una cavità preformata come unacaverna tubercolotica, una cisti, un ascesso, una bronchiectasia.All’interno della cavità si forma una palla di ife che è di poco inferiore al diametro della cavità stessa per cui c’è uno spaziovuoto fra le due: la terapia in questo caso è solo chirurgica.
Polmonite atipicaPer polmonite atipica si intende una polmonite a decorso più prolungato e a localizzazione prettamente interstiziale per cuisi avrà un infiltrato mononucleato a livello dei setti alveolari che può corrispondere ad una radiopacità ma che non siaccompagna a tosse produttiva nè a segni di consolidamento polmonare all’esame obiettivo.In realtà, visto che si è pur sempre in presenza di una flogosi, un essudato a livello alveolare può essere presente ed è dinatura sierosa o siero-emorragica in relazione all’effetto citopatico sugli pneumociti.
Gli agenti patogeni sono fondamentalmente alcuni virus, il M. pneumoniae, le Chlamidiae, Pneumocystis carinii etc.
Il Mycoplasma pneumoniae è l’agente più frequentemente responsabile di polmonite atipica con le solite caratteristiche diquesto tipo di presentazione. Le lesioni sono distribuite anche a livello tracheo-bronchiale e pleurico. Inoltre può colpite sitiextrapolmonari come il miocardio, il snc (mielite e meningite), la cute attorno agli orifizi (eritema polimorfo)
Le polmoniti virali possono essere provocate essenzialmente in 3 condizioni fondamentali:
1)Da adenovirus, orthomyxovirus (influenzale) e paramyxovirus (RSV,parainfluenzale ) in qualsiasi tipo di soggetto: si tratta di virus che colpiscono in genere le vie respiratorie superiori e i grossi bronchi:i virus influenzali infatti danno una tracheobronchite con tosse secca.gli adeno e il RSV invece sono responsabili di riacutizzazioni nel bronchitico cronico e di bronchioliti nella prima infanzia.A livello polmonare si ha come detto l’infiltrato interstiziale che sarà bilaterale e che potrà essere accompagnato da unessudato sieroso-emorragico con una componente fibrinosa più o meno accentuata in relazione all’effetto citopatico delvirus sugli pneumociti. La risoluzione si ha in 3-4 settimane per restitutio ad integrum a meno che non si sovrapponga unapolmonite batterica che in questo caso può facilmente evolvere ad ascesso.
È importante la vaccinazione anti-influenzale soprattutto in soggetti che presentano un deficit di funzionalità polmonarecome i portatori di enfisema o di scompenso cardiaco: in questi soggetti la sovrapposizione di una infezione influenzaleprecipiterà la situazione già precaria.
2)da VZV o da virus del morbillo che possono indurre polmonite in soggetti non immuni o in soggetti immunodepressi(vedi anche lo zoster). Tali virus producono una polmonite che può associarsi al caratteristico rash cutaneo; un markerdifferenziale sono le cellule giganti multinucleate a livello del setto.
3)da CMV o HSV si tratta in questo caso di infezioni opportunistiche in pz. immunodepressi come malati di AIDS, soggettitrapiantati etc. i markers di queste lesioni sono le cellule con inclusioni nucleari.
Pneumocystis colpisce caratteristicamente come opportunista pz con AIDS; oltre alla componente interstiziale c’è unacomponente alveolare ricca in proteine.
La clamidia psittaci è responsabile della ornitosi o psittacosi una zoonosi trasmessa dai pappagalli per cui sono a rischioallevatori, venditori e proprietari. La caratteristica è l’assenza quasi completa di componente essudatizia alveolare mentre

l’infiltrato interstiziale è particolarmente abbondante per cui si avrà un ostacolo più pronunciato agli scambi alveolari conconseguente dispnea grave.Polmonite da radiazioniCompare in soggetti irradiati al torace per neoplasia con dosi di almeno 1500-2000 rad.La gravità della lesione è tuttavia dovuta oltre che alla dose complessiva:-al frazionamento della dose-agli intervalli fra le dosi-alla sensibilità dell’individuole lesioni ovviamente saranno generalizzate a tutte le strutture e colpiranno in particolare le cellule più radiosensibili ovverogli epiteli, i mesoteli e gli endoteli e quindi si avrà dalla periferia verso il centro:-pleurite siero-fibrinosa con tosse secca , dolore e che evolve con la formazione di aderenze.-alveolite siero-fibrinosa con la formazione di membrane ialine ed evoluzione cicatriziale.-frammentazione delle fibre elastiche a livello settale-danno agli endoteli con trombosi ed emorragie-lesione dell’epitelio bronchiale che precocemente ripara con metaplasia mucosa.
L’evoluzione come detto in precedenza è per sclerosi.Inoltre si ricordi che per il rilascio di istamina dalle zone irradiate si può avere una lesione acuta a livello gastrico del tipogastrite acuta erosivo-emorragica/ulcera acuta da stress.
Polmonite uremicaÈ una patologia che si sviluppa in corso di uremia cronica, una condizione oggi meno frequente per via dell’emodialisi.la metilguanidina si accumula in corso di uremia e porta a sierositi siero-fibrinose; prima di tutto a pericardite ma anche apleurite e alveolite. L’evoluzione nel caso di polmonite uremica non trattata è verso la sclerosi.Bisogna inoltre ricordare che per le condizioni generali del pz. che può anche presentare scompenso cardiaco si ha unrischio di sviluppare anche polmoniti batteriche.
La tubercolosiLa TBC è oggi stata debellata nei paesi occidentali grazie alla disponibilità di farmaci ma cmq resta un problema per pzimmunodepressi e per gli immigrati.Inoltre è importante da conoscere perché si può porre il problema di fare DD (terapie diverse !!!) fra:1) nodulo tubercolare o neoplastico2) tubercolosi miliare o collagenopatia

Un tempo l’ agente eziologico poteva esser anche il M. bovis che dava la forma intestinale ma oggi questa è da noiscomparsa grazie alla pastorizzazione del latte per cui l’ agente eziologico è rimasto il M. tubercolosis.Questo è un batterio gram + , ma si colora con la Ziehl-Nielsen, a crescita lenta (24h per la replicazione; 48 gg per averecolonie!) e cresce solo in ambiente:- Aerobio e a pH neutro o debolmente basico ♦ non cresce nella caseosi ma può farlo sulle pareti di una caverna aperta o
nella zona periferica di un granuloma caseificato.- fra 35 e 41 gradi ♦ non cresce all’ esterno
il contagio quindi avviene direttamente per via respiratoria da parte di un pz che abbia caverne tubercolari aperte.
la patogenesi della TBC non è tanto una tossicità del batterio quanto una reazione di ipersensibilità dell’ ospite.Le componenti strutturali in particolare i lipidi di rivestimento (richiamo di monociti e loro trasformazione in celluleepitelioidi e cellule di Langhans), le proteine (vedi test alla tubercolina che è una tipica ipersensibilità di tipo ritardato) e ipolisaccaridi portano a una risposta immune cellulare con frormazione di granulomi di 2 mm di diametro i quali inglobano imicobatteri e possono presentare vari stadi evolutivi:1) solo macrofagi e micobatteri2) caseosi e cellule giganti di Langhans, vallo di cellule epitelioidi, strato esterno linfoplasmacellulare : questo è il classico
granuloma tubercolare ma la caseosi può essere assente e allora si pone il problema della DD con granulomi noncaseosici del tutto simili come quelli da sarcoidosi e berilliosi.
Il granuloma evolve verso la fibrosi con eventuale calcificazione sovrapposta; le lesioni più evidenti sono i noduli cheoriginano dalla confluenza di più granulomi e arrivano a dimensioni di 2-3 cm.Vi possono essere noduli che presentano tutti e tre gli stadi del processo ovvero caseosi, sclerosi e calcificazione.
La diagnostica comprende come detto l’intradermoreazione (comparsa di un pomfo a 48 h dall’ inoculo che deve essere didiametro > 1cm per avere la positività) la cui positività documenta un contatto col bacillo di Koch: la tecnica che indaga lapositività alla tubercolina nel corso degli anni è la Mantoux.Infatti un pz positivo può:- negativizzarsi dopo qualche anno dall’infezione primaria perché l’organismo è riuscito ad eliminare il bacillo- rimanere positiva per tutta la vita se il batterio rimane “murato vivo” e viene sporadicamente rilasciato in circolo- negativizzarsi se c’è una immunosoppressione o una linfocitolisi da virosi come un morbillo o se c’è una diffusione per
via ematogena del batterio come una forma miliare.- Negativizzarsi in corso di sarcoidosi che è quindi visto anche il pattern granulomatoso una candidata alla DD.La TBC primaria può presentarsi come tipica o atipica.
La TBC primaria tipica è caratterizzata dalla “triade di Gohn” che comprende:1) focolaio primario tipico ovvero una lesione che normalmente si ritrova al lobo medio di dx la quale:- misura ca 2 cm di →- è in sede sottopleuricail decorso della lesione può variare dalla restitutio ad integrum alla caseificazione in relazione al rapporto fra virulenza delbacillo e risposta dell’ospite.2) linfangite tubercolare: costituita da granulomi disposti lungo il decorso dei linfatici afferenti all’ilo dal focolaio
primario.3) linfoadenite satellite: si tratta del coinvolgimento dei linfonodi satelliti con lesioni capsulari caseose che per
estensione producono una perilinfoadenite: entrambe le lesioni evolvono con fibrosi ± calcificazioni : un tempo lafibrosi dovuta a linfoadenite del linfonodo adiacente al bronco lobare medio produceva la sindrome del lobo medio.
Dal punto di vista clinico una TBC primaria tipica può essere asintomatica; presentarsi con una febbricolaaspecifica; presentarsi con pleurodinia in caso di interessamento per contiguità della pleura.
La TBC primaria atipica è un quadro di maggiore gravità e di maggiore estensione rispetto alla atipica e si verifica in pzcon debole risposta immune ed esposizione ad alte cariche del bacillo. Il tipo di lesione è lo stesso, cambia solo l’estensionedel processo: l’interessamento sia parenchimale che linfonodale è infatti massivo:1) a livello parenchimale si hanno focolai caseosi multipli che si configurano come una broncopolmonite tubercolare: in
questo caso l’evoluzione a caverna primaria (pareti non fibrotiche) con drenaggio nell’ albero bronchiale dellacaseosi porta alla diffusione del processo, specie alle basi, per aspirazione.
2) A livello linfonodale si ha il coinvolgimento di più linfonodi eventualmente bilateralmente; si manifesta con unasindrome mediastinica ed evolve con una fibrodsi che può mimare una neoplasia ilare.

La tbc allergica comprende un insieme di entità anatomo-patologiche che sono di possibile riscontro in individuigeneralmente giovani con una spiccata reattività al bacillo di Koch i quali infatti hanno una positività molto accentuata,con reazione ulcero-necrotica alla tubercolina. Si tratta di flogosi di tipo siero-fibrinoso con scarso infiltratolinfomonocitario che in questi individui accompagnano il complesso primario e possono riguardare:1) il parenchima circostante al focolaio primario ♦ flogosi perifocale: alone sfumato attorno al focolaio ♦ durata
relativamente breve (2-3 mesi) e restitutio ad integrum2) un intero lobo (è una lobite) ♦ epitubercolosi ♦ la durata è lunga (5-8 mesi) e si ha conseguentemente una fibrosi del
lobo interessato3-4) la pleura delle scissure o la pleura parietale ♦ scissurite o pleurite ♦ può evolvere con restitutio ad integrum o con
aderenze.
La tbc post-primaria deriva dalla deriva da una reinfezione che in genere deriva da una riattivazione endogena perparticolari condizioni di immunodepressione dell’ ospite o più molto raramente può essere esogena.Il carattere delle lesioni che si producono può essere:- prevalentemente produttivo ovvero con scarsa componente di caseosi- prevalentemente essudativo ovvero con una franca evoluzione verso la caseosila riattivazione non avviene in un alta percentuale dei casi ma tuttavia se compare è caraterizzata da lesioni ben più serie diquelle della tubercolosi primariale forme prevalentemente produttive sono:
1) TUBERCOLOSI NODULARE DELL’ APICE (sopraclavicolare): se i batteri che diffondono non sono molti si haattecchimento solo nelle aree più favorevoli ovvero bilateralmente agli apici polmonari dove c’è una alta PAO2 , quisi osservano pochi piccoli noduli che si sviluppano in sede subpleurica. Questi noduli hanno la caratteristicacomposizione e hanno generalmente una evoluzione verso la fibrosi ± calcificazione± caseosi residua; più raramentel’evoluzione è però verso la caverna secondaria con cercine fibroso ben evidente e broncopolmonite tubercolareprodotta per diffusione endobronchiale; gli esiti della fibrosi apicale possono essere:
- l’enfisema bolloso- la bronchiectasia/broncostenosi
2) TUBERCOLOSI MILIARE: in relazione al numero di bacilli rilasciati e alla reattività dell’ospite si hanno formeprogressivamente più disseminate.Il bacillo dal focolaio in genere linfonodale può diffondere per via linfoematogena o per via ematogena: questo lo portaprimariamente nel piccolo (polmone) o nel grande (sistemico) circolo: questa distinzione è però fittizia perché se la caricache diffonde è adeguata si ha interessamento generalizzato.La riattivazione può verificarsi in condizioni particolari come: gravidanza, virosi che danno linfocitolisi come il morbillo,malattie debilitanti di varia natura: in genere quindi prevale prima dei 30 e dopo i 60 anni.
La tbc miliare si suddivide dal punto di vista clinico in acuta e subacuta/cronica.La TBC miliare acuta è caratterizzata da un elevatissimo numero di granulomi tubercolari di colore giallastro (similiappunto a grani di miglio) localizzati non solo in tutto il parenchima polmonare ma anche a livello sistemico : fegato, milza,midollo osseo, retina, e vari altri organi. La manifestazione clinica è una febbricola e in seconda istanza con una dispneadovuta alla invasione del parenchima polmonare con evoluzione verso la fibrosi ; naturalmente sono presenti anche i segnidell’ interessamento di altri organi come le meningi.A livello polmonare il quadro è simile da quello della sarcoidosi per la quale va ovviamente fatta DD prima di intraprendereuna terapia.La DD con una miliare primaria (che si può avere in pz immunodepressi con TBC primaria ) è possibile grazie alla assenzadi una linfoadenite in atto.La TBC miliare subacuta/cronica è caratterizzata da un numero molto inferiore di granulomi che si concentrano nelcampo medio polmonare e dalla maggiore durata: quindi oltre alla febbricola aspecifica non si avrà tanto dispnea(l’interessamnto polmonare è circoscritto) quanto (per la possibile evoluzione tisiogena) emoftoe.Un'altra entità che è piuttosto radiologica è il tubercoloma ovvero una massa radio-opaca dai contorni dentellati frequentenei campi medio-superiore specialmente a dx che mima pertanto un cr. polmonare e deve entrare con esso in DD.La natura anatomo patologica può essere varia (fibrosi a cipolla, agglomerato di granulomi, caseosi disidratata, cavernaoccupata da un coagulo) possono essere presenti calcificazioni.

Le forme prevalentemente essudative sono come sottolineato in precedenza date da una evoluzione verso la caseosi. Si haquindi una alveolite siero-fibrino-caseosa con una reazione infiammatoria che tende a circoscrivere la caseosi: la evoluzioneverso la caseosi è dovuta a una scarsa resistenza dell’ ospite.
1) INFILTRATO SOTTOCLAVEARE DI ASMAN-REDEKER: è un nodulo di pochi cm in sede sottoclaveare (campomedio); l’evoluzione è tisiogena per cui il nodulo tende a svuotarsi in un bronco. In genere questa forma è dovuta areinfezione esogena.
2) INFILTRATI PRECOCI MULTIPLI: è la stessa lesione soltanto con presentazione multipla : può mimare un crmetastatico al polmone: è più frequente che la reinfezione sia stata endogena
3) LOBITE TUBERCOLARE: stesse lesioni elementari concentrate in 1 lobo in genere il superiore dx con gli spaziinternodulari interessate da una flogosi aspecifica: i noduli tendono a svuotarsi e a confluire in una grande caverna
4) BRONCOPOLMONITE/ POLMONITE TUBERCOLARE: interessamento di ampie zone in seguito a diffusione pervia ematogena con consolidamento pneumonico o broncopneumonico (vedi anche la TBC primaria atipica).
In sintesi si ha :Infezione primaria ♦ è in genere tipica per cui si presenta classicamente con focolaio primario (+ granulomi fanno 1nodulo < 2 cm), subpleurico, al lobo medio; linfangite e linfoadenite satellite: sintomatologia assente o vaga febbricola, almassimo pleurodinia.*Raramente (in pz debilitati) si ha la presentazione atipica con noduli multipli > 2 cm per cui si può avere un quadro dibroncopolmonite o polmonite tubercolare primaria; l’evoluzione è essudativa e tisiogena: si formano caverne primarie;inoltre si ha una linfoadenopatia ilare massiva.**Al contrario soggetti particolarmente reattivi alla tubercolina e quindi al bacillo hanno una TBC allergica che va al di ladel focolaio infettivo e può dare: flogosi perifocale, epitubercolosi (lobite), pleurite e scissurite.
Infezione secondaria ♦ può avvenire per varie vie e in base alla risposta dell’ ospite sarà a carattere:
Produttivo se c’è scarsa caseosi:- tubercolosi nodulare dell’apice: noduli sottopleurici bilateralmente in regione apicale- tubercolosi miliare: acuta (disseminata♦ fibrosi polmonare e lesioni sistemiche) o cronica (possibile evoluzione a
microcaverne)- tubercoloma: massa fibrotica a margini sfrangiati che simula un carcinoma (specie zona medio-alta a dx)
essudativo se c’è molta caseosi e allora avremo una malattia a livello polmonare con estensione più o meno ampia edevoluzione tisiogena.- infiltrato sottoclaveare ♦ nodulo monolaterale pieno di caseosi al campo medio- infliltrati precoci multipli- infiltrato precoce gigante (lobite)- broncopolmonite tubercolarele modalità d’infezione sono le solite per queste forme: la presentazione differente è dovuta:-alla carica e alla virulenza batterica-alla immunità dell’ ospite in quanto una risposta troppo forte provoca un pattern localizzato ed essudativomentre una risposta debole provoca scarsa essudazione e diffusione sistemica.

Asbestosi: amianto è un termine che comprende vari cristalli di silicati. Le due varietà di amianto sono:1)il serpentino:fibre morbide e flessibili 2)l’anfibolo: fibre rigide e diritte, è meno diffuso ma è associato al mesotelioma. La minoresolubilità e la sua rigidità ne influenzano negativamente la clearance.È stato usato in vari settori per le sue proprietà di isolante termico.Il tempo necessario per la comparsa di lesioni significative varia con l’esposizione e la suscettibilità individuale si va daqualche mese a 20 anni: in media 5-10 anni.Le fibre più lunghe non raggiungono gli alveoli ma possono penetrare attraverso la parete dei bronchioli nel connettivoperibronchiale: sono queste le fibre maggiormente responsabili della fibrosi interstiziale.
I macrofagi tentano la fagocitosi delle fibre ma vanno incontro a necrosi e rilascio degli enzimi lisosomiali. Si instaura unarisposta infiammatoria cronica che porta a fibrosi polmonare diffusa (pattern reticolare) la quale parte dai bronchioli e sidiffonde agli alveoli adiacenti; l’inizio delle lesioni è nei lobi inferiori: il reperto caratteristico sono i numerosi corpi dell’asbesto ovvero fibre di amianto rivestite da manicotti ferro-proteici: corpi dell’asbesto efibre d’asbesto possono essererilevati nell’ espettorato, nel parenchima polmonare in aree normali, di fibrosi o all’interno di un carcinoma, nelle feci graziealla deglutizione del muco: il che spiega anche l’arrivo di fibre a livello del digerente dove si potrà avere un mesoteliomaperitoneale.In fase avanzata si ha coalescenza delle lesioni, con distorsione del parenchima e ed evoluzione in polmone a favo d’api.Altre conseguenze dell’esposizione all’asbesto sono:
- PLACCHE PLEURICHE:ispessimenti fibro-calcifici della pleura parietale molto frequenti a prevalentelocalizzazione diaframmatica e senza aderenza fra i due foglietti; la fibrosi polmonare alle basi può dare una lesionesimile in quanto si associa ad ispessimento fibroso dei foglietti pleurici che divengono aderenti (pachipleurite cronica).
- Versamenti pleurici sierosi o siero-emorragici.- Mesotelioma: associato all’ anfibolo ma non al fumo- carcinoma broncogeno: i lavoratori dell’amianto hanno un RR=5 che sale addirittura a 55 se sono anche fumatori;
oltre al potere cancerogeno intrinseco l’amianto ha anche la capacità di adsorbire altri cancerogeni come quelli delfumo.il carcinoma polmonare in questi individui è frequentemente un cancro cicatrice ovvero un adenocarcinoma.
*si ricordi la fibrosi da polmonite da radiazioni: in quel caso c’è frammentazione delle fibre elastiche in questo caso no.

Antracosi: interessa i lavoratori del carbone già estratto.
-l’antracosi asintomatica è una pigmentazione distribuita vista l’opera dei macrofagi, in corrispondenza della rete linfaticadei polmoni compreso il malt e i linfonodi bronchiali: nel complesso il pigmento si concentra all’ilo.
-la CWP (pnemuconiosi dei lavoratori carbone) semplice è caratterizzata da una fibrosi incipiente con opacità reticolariseguita con il protrarsi dell’ esposizione dalla comparsa di noduli nell’ordine del mm.
-la CWP complicata si sviluppa in una % minoritaria degli esposti ma è una condizione progressiva anche in caso dicessazione dell’esposizione: si tratta di una FMP (fibrosi massiva progressiva) ovvero di una forma nodulare sclerosante.Noduli nell’ordine del cm compaiono nella metà superiore del polmone e possono confluire fino ad occupare un lobo intero.
-la sindrome di Caplan è caratterizzata dall’associazione artrite reumatoide+FMP.
Si ricordi che l’antracosi è associata con la bronchite cronica e l’enfisema centrolobulare.
Silicosi: la pneumoconiosi più frequente è provocata dall’inalazione di silice cristallina (es. quarzo),molto meno efficaci sono i silicati che si ritrovano in forma amorfa (es. talco): sono a rischio i minatori,i lavoratori del cemento e della ceramica.L’azione dei cristalli è dovuta alla formazione di legami idrogeno con le molecole delle membranecellulari da parte dei gruppi SiOH di superficie nonché dei radicali liberi che si producono quando icristalli vengono meccanicamente “rotti”. Le particelle inalate attivano quindi quei macrofagi chesopravvivono con inizio dell’ infiammazione e della fibrogenesi:una volta che la silice è in sede ilprocesso patologico continua anche se il pz si allontana dal luogo di lavoro.
Anche in questo caso si avrà il riscontro di opacità ai lobi superiori dapprima finemente nodulari: istologicamente nodulo asfoglia di cipolla (eventualmente associate a strie calcifiche a guscio d’uovo nei linfonodi ilari) indi per coalescenzasempre più grandi fino a una FMP (in corso della quale si avrà una palese sintomatologia): la disposizione delle lesioni ailobi superiori sarebbe dovuta al fatto che qui il drenaggio linfatico è meno efficiente; quella parailare delle lesioni tardive alfatto che a lungo andare si accumula in questa sede molta silice.
L’evoluzione del nodulo silicotico può essere per cavitazione come nel caso della silico-antracosi (lavoratori delle minieredi carbone dove c’è sia carbone che silice) o della sindrome di Kaplan in quest’ultima la presenza di una collagenopatia dibase esacerba la gravità della silicosi.
La kaolinosi è una variante di silicosi che può colpire i lavoratori del kaolino (un silicato di alluminio usato per es.nell’industria della ceramica): la patologia è del tutto simile ma è caratterizzata da una evoluzione più lenta verso la sclerosi.
Una pneumoconiosi simile ma più rara rispetto alla silicosi è la siderosi: anch’essa ha un evoluzione sclerotica ed èpotenzialmente cancerogena.

Berilliosi:l’esposizione a questo metallo usato per fabbricare alcune leghe metalliche provoca unapatologia acuta e una cronica in una piccola % di soggetti esposti, a quanto pare geneticamentepredisposti;
La forma acuta è una polmonite interstiziale simile a quella virale.
La forma cronica è una granulomatosi non caseosa (cellule giganti, vallo epitelioide, linfomonociti efibrosi) a livello polmonare ma anche ilare e sistemico che va differenziata dalla sarcoidosi con lamisurazione del berillio tissutale.le lesioni granulomatose portano nel tempo a una fibrosi dall’aspettofinemente nodulare.
Sarcoidosi:È un malattia infiammatoria a carattere granulomatoso non caseoso, ad eziologia ignota e con interessamentosistemico ma in prevalenza a polmone (90%) , linfonodi ilari (90%), occhi (25%) e cute (25%); meno frequenti altre sedi.Nelle varie sedi si formano granulomi compatti non caseosi (cellule istiocitarie: giganti tipo Langhans ed epitelioidi ,linfociti) che nella forma cronica evolvono in fibrosi.
Autoimmunità: La malattia compare in genere prima dei 40 anni e vista la oligoclonalità dei linfociti T che la sostengonosarebbe dovuta a uno o più Ag permanenti ma sconosciuti. Di certo la patogenesi si basa su una abnorme risposta dei TH1 iquali sono in grado una volta attivati di replicarsi a livello tissutale (IL-2) nonchè di richiamare i monociti dal circolo eattivarli per la formazione del granuloma. La risposta Th1 è tanto forte che il rapporto CD4+/CD8+ aumenta di cinque volte.
Clinica: \In genere la diagnosi avviene nel pz. sintomatico e a questo proposito la sarcoidosi può presentarsi in formainsidiosa con maggiore probabilità di cronicizzazione o meno frequentemente in forma subacuta accompagnata in questocaso da segni sistemici di infiammazione come febbre, astenia, calo ponderale etc.
Polmoni: l’interessamento è molto frequente; solo nel 50% si hanno lesioni permanenti e solo nel 10 la fibrosi progressiva. Isintomi sono dispnea e tosse non produttiva.
Linfonodi: la linfoadenopatia ilare ma anche sistemica è altrettanto frequente: i l. sono alla palpazione ingrossati, nondolenti, non aderenti, di consistenza duro-elastica.Vi sono 3 pattern radiologici: tipo I (solo l.adenopatia; in genere forma subacuta), II (opacità parenchimali+adenopatia), III(solo parenchima); II e III corrispondono in genere alla forma cronica.
Occhio: si possono avere uveiti che portano anche a cecità nonché congiuntiviti e nel caso di interessamento delle gh.Lacrimali una cheratocongiuntivite secca; dopo quelle polmonari sono le sequele più serie per importanza e frequenza.
Cute: sono varie le lesioni riscontrate: eritema nodoso, placche, rash maculopapulare.
Diagnosi:Non c’e un reperto patognomonico di sarcoidosi; la diagnosi si basa su una serie di reperti non specifici in varieindagini: anamnesi, esame obiettivo, biopsia, test ematici (linfocitopenia, ↑ACE), BAL (↑linfociti e rapporto CD4/CD8),test di funzionalità polmonare, scintigrafia con Ga-67.
BPCO (bronco-pneumopatie cronico-ostruttive).Il termine si riferisce a un gruppo di patologie polmonari che presentano un pattern fisiopatologico cronico o ricorrente ditipo ostruttivo ovvero una riduzione dei flussi e del FEV1 durante un’ espirazione forzata.Le patologie che rientrano in tale definizione sono:1) enfisema2) bronchite cronica

FUMO
L’ENFISEMA POLMONARE consiste in una abnorme dilatazione di spazi aerei distali al bronchioloterminale associata a distruzione di strutture settali e in assenza di segni di fibrosi.Nel centroacinoso sono dilatate le strutture intralobulari più prossimali (bronchioli respiratori)non gli alveoli più distali e periferici: è il tipo di gran lunga prevalente (95%), si associa al fumo ealla bronchite cronica; la localizzazione è prevalentemente apicale .nel panacinoso ogni acino è coinvolto nella sua totalità anche nelle porzioni più distali; si associa aun deficit di α1-antitripsina un inibitore enzimatico rilasciato in circolo dal fegato (menoimportante il contributo di neutrofili e monociti) che inibisce le elastasi rilasciate dai neutrofili.lalocalizzazione è più evidente alle basi.
Vi sono poi altri tipi di enfisema:1)parasettale che coinvolge solo le parti distali degli acini lungo i setti interlobulari; e associato adaree di fibrosi, atelettasia o cicatrizzazione2)irregolare in quanto non ha una localizzazione precisa nel contesto dell’ acino. È associato a fenomeni di cicatrizzazione3) L’iperinflazione differisce dall’enfisema in quanto non comporta la rottura dei setti alveolari.può essere di natura:
- ostruttiva che si ha per una ostruzione a valvola in un bronco o per la presenza di una via di ventilazione collateralein caso di ostruzione completa.
- Compensatoria come quella che segue a lobectomia o pneumectomia.4)e.senile che corrisponde a una situazione fisiologica legata all’invecchiamento caratterizzata da dilatazione dei dottialveolari e restrizione degli alveoli.5)e.bolloso che corrisponde a una lesione anatomo patologica aspecifica in quanto a eziologia consistente nella formazionedi bolle subpleuriche ovvero cavità di almeno 1 cm di ∅.6)e.interstiziale corrispondente alla penetrazione di aria nello stroma connettivale del polmone, nel mediastino e/o nelsottocutaneo può essere dovuta a rottura di una via aerea o a traumi del torace.
PATOGENESILa patogenesi dell’enfisema consiste in una perturbazione del normale equilibrio elastasi-antielastasi: come già detto l’elastasi più importante è quella liberata dai neutrofili che ha comespecifico inbitore l’α-1 antitripsina. Anche i macrofagi tuttavia possono dare il loro contributoall’attività elastasica e questo sembra importante soprattutto per l’enfisema centrolobulare che correlacon il fumo di sigaretta.
Il fumo di sigaretta infatti ha diversi effetti dannosi:
Potenziale ossidante
azione chemotatticadiretta e indiretta (IL-8)
Stress
ossidativo
diretto e
indiretto
↓ attività
anti-
proteasica↑Rilascio dielastasi dai PMNe dei macrofagi
↑ attività
elastasica
ENFISEMA
Attività pro-infiammatoria ingenerale
InfiammazioneDelle vie aeree
BRONCHITECRONICA eBRONCHIOLITE

Come si vede il fumo è il fattore eziologico principale sia dell’enfisema centrolobulare sia dellabronchite cronica: si può affermare che queste due patologie siano sovrapposte nel senso che nel pz.fumatore coesistono in vari gradi 2 componenti: bronchitica ed enfisematosa.Altri cofattori sono gas inquinanti come il biossido di zolfo e per la bronchite le infezioni.
Per quanto riguarda l’e. panacinoso è ovvio il ruolo del fumo come fattore scatenante in una situazionecongenita di deficit di α1-antitripsina.la malattia si sviluppa clinicamente solo in pochi individui fra iportatori della mutazione non fumatori (la mutazione più frequente , Pizz, è puntiforme e porta a unaproteina inattiva che polimerizza facilmente).
La BRONCHITE CRONICA è clinicamente diagnosticata con il riscontro di tosse persistenteproduttiva per un periodo di almeno 3 mesi per almeno 2 anni di seguito. Vi sono 3 tipi dibronchite cronica:1)la b.c. semplice così detta in quanto non complicata da una significativa ostruzione2)la b.c. asmatica così detta in quanto costellata da episodi di broncospasmo.si differenzia dall’asma inquanto non vi sono fra i broncospasmi periodi asintomatici.3)la b.c. ostruttiva così detta in quanto complicata da una significativa ostruzione al flusso; è tipica deiforti fumatori.Il pz allora è anche dispnoico.
Eziopatogenesi: il principale fattore eziologico come già visto è il fumo (1 pacchetto al giorno peralmeno 20 anni), la malattia è riscontrata molto raramente nei non fumatori. Tuttavia un cofattore importante sono le infezioni tracheobronchiali che si inscrivono comeesacerbazioni di un quadro che in genere è stabile e lentamente progressivo.
La tosse si aggrava e l’espettorato diventa abbondante, denso e purulento; la dispnea si aggravaanch’essa con peggioramento dei parametri respiratori.Le infezioni possono essere batteriche o virali.Queste esacerbazioni possono mettere in serio pericolo la vita del pz.; devono essere escluse altrepatologie che possono intervenire in un quadro di bronchite cronica (es. pneumotorace oinsuff.cardiaca)
Anatomia patologica: in generale l’infiammazione porta a un restringimento delle vie aeree. Iperplasia cellule caliciformi→iperproduzione muco Edema e infiltrato infiammatorio mucoso e sottomucosoBronchioli iperplasia muscolatura lisci(∅<2−3mm) Fibrosi (TGF-β)
Le lesioni bronchiolari, almeno inizialmente, sono le principali cause di ostruzione
Le INFEZIONIBatteriche o viraliprovocanoesacerbazioni
Inibizionemeccanismi di difesa(ciglia e macrofagialv.)

Bronchi : la lesione fondamentale è l’ipertofia delle gh. Mucosecernenti sottomucose Questa lesione corrisponde a un ↑indice di Reid ovvero del rapporto Spessore strato ghiandolare/spessore parete (epit.-cartil.); questo che fisiol. È attorno a 0.44 arriva in genere oltre 0.50.le lesioni bronchiali sono responsabili della tosse produttiva.
FISIOPATOLOGIAPer definizione si ha una ostruzione al flusso specialmente in espirazione forzata con ↓FEV1.Le cause della ↑resistenza al flusso delle vie aeree sono diverse:
iperinflazioneUn’altra conseguenza della perdita di ritorno elastico polmonare, visto che esso serve a controbilanciare il ritornoelastico del torace, è l’ ↑CFR (iperinflazione).Una quota dinamica di iperinflazione sarebbe dovuta alla difficoltà espiratoria, essa va ad accentuare ladispnea in quanto l’espansione polmonare tende ad appiattire il diaframma rendendo meno efficacel’azione di questo muscolo. In definitiva è ostacolata la ventilazione.
Alterazione degli scambi gassosiInizialmente si ha con la bronchiolite una ↓V che porta a un ↓rapporto V/Q con lieve ipossemia.Con l’evoluzione enfisematosa si ha anche una ↓Q per cui il rapporto tende a normalizzarsi ma ↑ lospazio morto effettivo (come dire che è troppo basso il rapporto sup./vol. delle cavità enfisematose)
La risposta del pz. è diversa probabilmente in relazione a una diversa sensibilità dei sistemi nervosi per l’ omeostasi dei gasematici o anche alla resistenza delle vie aeree che è notevolmente aumentata se prevale la bronchite rispetto all’enfisema.1)Pz. che riescono a iperventilare, a costo di una dispnea cronica, ottengono una PCO2 normale, e una PaO2 su livellinormali nonostante l’↑ PAO2- PaO2 (dipende nell’enfisema da ↑Vd/Vt =spazio morto)Questi pz sono detti “soffiatori rosa”(vivono per respirare): il soffiare consente di prolungare l’espirazione diminuendo ilcollassamento dei bronchioli che come visto hanno una maggiore collassabilità nel polmone enfisematoso.2)I pz che invece non iperventilano presentano invece ipossiemia severa con conseguente cianosi e ipercapnia.Questi pzsono detti “aringhe blu”(respirano per sopravvivere)
circolazione polmonarevari meccanismi contribuiscono all’insorgenza di IPERTENSIONE POLMONARE:
Restringimento intrinsecodel lume (bronchiolite)
↓ ritorno elastico delparenchima *(↑ compliance: enfisema)
↓ trazione elastica radialesulle vie aeree ovvero↑ collassabilità
↑ resistenza al flusso↑ lavororespiratorioe dispnea
↓∅ aa. e a.ole polmonaridovute a vasocostr. e ispess.secondario all’ipossia

Adattamenti neuroumorali
Ipossia e ipercapnia croniche portano a una attivazione simpatica che riproduce gli adattamentineuroumorali che agiscono anche nell’ insufficienza cardiaca: rilascio di norepinefrina e attivazione delmeccanismo RAA che portano a un sovraccarico di lavoro per il miocardio.
cachessiaa lungo andare si ha un generale deperimento dell’organismo dovuto sia all’ aumentato dispendioenergetico per il lavoro di respirazione sia ad aumentati livelli di TNF-α in circolo.CLINICALa malattia insorge in generale in soggetti fumatori nella quinta decade:Inizia con una tosse produttiva specialmente mattutina; sono presenti con frequenza non troppo altaesacerbazioni periodiche.Col progredire della patologia le esacerbazioni diventano sempre più frequenti, si sviluppa la dispneaprogressiva, gli scambi gassosi sono alterati (possono aversi eritrocitosi e cianosi secondarieall’ipossiemia) la tolleranza all’esercizio è sempre più ridotta.Alla fine si arriva a seri squilibri dei gas ematici che si accompagnano a cuore polmonare, sovraccaricodi liquidi ed edemi; il calo ponderale (cachessia) è un indice prognostico negativo.
ESAME OBIETTIVONon offre reperti specificiInizialmente si possono trovare solo sibili in e.f. e un ↑ tempo di e.f.Segni di iperinflazione (es ↑∅ anteroposteriore del torace, riduzione aia e scomparsa toni cardiaci)Sono presenti in caso di enfisema .Atteggiamenti caratteristici possono essere la postura “a tripode” e il soffiare in espirazione.Infine possono essere presenti i segni di cuore polmonare e scompenso dx:Turgore delle giugulari, edemi, epatomegalia da stasi.
I reperti radiografici non presentano alterazioni significative se la malattia è lieve.L’enfisema è segnalato da una ipertrasparenza localizzata ai lobi sup.per il panacinoso e a quelli inf.Per il centroacinoso; gli emidiaframmi sono appiattiti (↑l’angolo sterno-diaframmatico);un repertomolto specifico sono le bolle.
Di importanza fondamentale per la diagnosi è il riscontro fisiopatologico di ostruzione al flusso d’aria:-FEV1: è diminuita in vario grado tanto che una stadiazione può essere fatta sulla base della sua entitàrispetto ai valori attesi. Inoltre è possibile verificare la reversibilità dell’ostruzione se c’è un ↑FEV1 dialmeno 200 ml dopo somministrazione di un broncodilatatore.-curva flusso-volume: flussi diminuiti a qualsiasi valore di volume e curva “shiftata” verso valori divolume più alti del normale.-misurazione del volume polmonare che è ↑ a causa dell’ iperinflazione.I livelli di α1-AT dovrebbero essere valutati in pz :Non fumatori con ostruzione al flusso,
Perdita di capillari alveolarisecondaria all’enfisema
Iperviscosità dovuta a eritrocitosisecondaria all’ipossiemia
Ipertensionepolmonare
Cuorepolmonarecronico

Affetti da bronchiectasie , da cirrosi epatica, da enfisema prematuro e a localizzazione basilare.Individui con anamnesi familiare positiva.
Trattamento:1)cessazione dell’ abhtudine al fumo2)broncodilatatori: - agonisti β-adrenergici a breve o a lunga azione
- anticolinergici- teofillina
3)glucocorticoidi4)somministrazione di α1-AT5)ossigenoterapia
profilassi per l’influenza e lo pneumococco consigliata.
Gli enfisemi polmonariGli enfisemi sono classificabili in base alla sede come:
Interstiziale : accumulo di aria nel connettivo lasso interstiziale che può prodursi:- in traumi del torace →associazione con pneumotorace.- rottura di una via area dovuta ad un forte ↑ della pressione intrabronchiale: manovra di Valsalva durante parto,
defecazione, sollevamento pesi.- Incidente in pz in ventilazione assistita con pressione positiva.In questi casi l’aria attraversol’interstizio arriva nel mediastino e risale fin nel collo e nella faccia insinuandosi nel tessuto
sottocutaneo per cui alla palpazione si avrà un crepitio analogo a quello della gangrena gassosa.
Alveolari: ↑ volume degli spazi alveolari a valle del bronchiolo terminale con o senza rottura dei setti.Senza rottura dei setti (è più propriamente detto iperinflazione):1) enfisema vicario ovvero sovradistensione compensatoria del restante parenchima polmonare quando vi sia unaatelettasia o una lobectomia.2) enfisema a valvola: stenosi di una via aerea per cui in espirazione una certa quantita d’aria rimane intrappolata a valle.

3) enfisema senile: ↑ volumetrico del polmone che in età senile è dovuto a una ↓ delle fibre elastiche e a modificazionidella gabbia toracica (↑ ∅ anteroposteriore) cui il polmone si adatta.
Con rottura dei setti: ecco finalmente il vero e proprio enfisema polmonare.Seguendo la prassi fin qui adottata iniziamo dai tipi meno significativi per lasciare poi spazio ai 2 enfisemi più importanti:centrolobulare e panlobulare. Avremo allora:3 tipi di enfisema tutti associati a fenomeni di fibrosi/cicatrizzazione: sono quindi focali e asintomatici e si differenzianoper la sede delle lesioni in:- parasettale: rottura dei setti a livello della periferia del lobulo, dirimpetto al setto interlobulare.- Irregolare: rottura dei setti senza una sede precisa.- Bolloso: formazione di bolle con ∅>1 cm in sede subpleurica e preferenzialmente all’ apice, in associazione con la
cicatrizzazione che segue alla TBC dell’ apice. È importante perché può dare come manifestazione clinica unopneumotorace.

Panlobulare CentrolobulareEziologia Deficit congenito di α1-
antitripsina, un fattore adattività anti-elastasica che èprodotto dal fegato. Gliindividui che ammalano sonosoprattutto gli omozigoti PiZZ.
abitudine al fumo e/o l’esposizionead altri agenti irritanti (polveri).
Patogenesi Il difetto generalizzato di α1-antitripsina provoca unaeccessiva degradazione dellefibre elastiche particolarmentegrave e precoce se il soggetto èpure un fumatore.Il danno è panlobulare eprevalentemente localizzato alle basipolmonari.
Il fumo di sigaretta è un agente conproprietà ossidanti (↓ attivitàantitripsina), e pro-infiammatorie(arrivo neutrofili e macrofagi conrilascio di elastasi) dirette eindirette.
Il danno è centrolobulare (bronchiolo respiratoriocollassante e alveoli contigui per digestione eiperinflazione) e prevalentemente medio-apicale.
Patologieassociate
Epatopatia caratterizzata dall’accumulo di α1-antitripsina nelRER degli epatociti → granulipas+: si possono avere in etàprecoce epatite, cirrosi, ↑incidenza di cr epatocellularenon necessariamente associatoa cirrosi.
Bronchite cronica con la qualecondivide la eziologia.Bronchiolite: flogosi, edema,metaplasia mucosa, collassabilitàdel bronchiolo respiratorio →ostruzioneBronchite: iperplasia delle gh.Sieromucose → ↑ indice di Reid(0.44→+ di 0.50) coniperproduzione di muco e tosseproduttiva.
Epidemiologia
È più infrequente; ad insorgenzaprecoce < 40 anni; ereditarietà.
È il tipo + frequente e compare dopo i50 anni
Caratterimacroscopici
Sono sovrapponibili. Il polmone sarà:- ↑ di volume → impronte costali; riduzione aia di ottusità
cardiaca.- di colorito pallido- di consistenza ↓ con ↑ crepitio- ipertrasparente alla radiografia con aree ancor più trasparenti
corrispondenti a bolle (specialmente nel panlobulare)
Fisiopatologia:Distruzione del parenchima polmonare che dà luogo a un quadro ostruttivo →

→ in base alla entità dell’ ostruzione (più grave se c’è anche bronchite cronica) e alla sensibilità dei centri respiratori siavranno 2 tipi di pz.:1) lo sbuffatore rosa il quale a costo di un grande sforzo riesce a ventilare per cui avrà pressioni parziali dei gas ematici
pressoche normali: l’atto del “soffiare” consente attraverso un ↑ della pressione endobronchiale di prolungarel’espirazione.
2) L’aringa blu cosiddetto perché non riesce a sostenere una ventilazione adeguata e per questo avrà una ipercapnia euna ipossiemia con conseguente poliglobulia.
Questo per quanto riguarda i parametri respiratori, tuttavia vi saranno alterazioni anche a carico:a) della gabbia toracica → torace a cilindro con orizzontalizzazione delle coste, appiattimento del diaframma, ipertrofia
dei muscoli respiratorib) del circolo polmonare → ↑ resistenze polmonari per rimodellamento del circolo dovuto all’ipossia ma anche
distruzione del letto capillare e poliglobulia → ipertensione polmonarec) del cuore dx → cuore polmonare cronico dovuto all’ ipertensione secondaria all’enfisema con tutte le implicazioni che
questo comporta: fase di compenso con ipertrofia concentrica → fase di scompenso con dilatazione, insufficienzatricuspidale, dilatazione atrio sn e auricola con fibrillazione e poliglobulia che producono trombosi auricolare e rischiodi embolia polmonare.
d) del circolo venoso sistemico → stasi venosa con turgore delle giugulari, fegato da stasi, edemi alle zone declivi, ascite,ulcera peptica dovuta alla stasi, trombosi degli arti inferiori con rischio di embolia polmonare.
Ipertensione polmonareDovendo stabilire le cause di ipertensione polmonare si può considerare che:
Pp - Pad = RQ ovvero: Pp= RQ + Pad
Allora si potra avere ipertensione polmonare per:1) ↑Pad (in sè per sè poco influente)2) ↑Q3) ↑R
Alcune patologie a carico del cuore sn (stenoinsufficienza mitralica, insufficienza ventricolare sn) ma anche gli shunt sn-dx in genere su base congenita provocano una condizione di iperemia polmonare , passiva nel primo caso (↑Pad) , attivanel secondo (↑Q), che a lungo andare produce ugualmente ipertensione polmonare sostenuta da un ↑R con lesioni dasovraccarico pressorio del ventricolo sn. In questo caso poichè preesisteva una patologia cardiaca non è corretto parlare dicuore polmonare.
Dunque il CPC è dovuto a patologie non cardiache che inducono in vari modi un ↑R polmonari con conseguenteipertensione polmonare e lesioni a carico del cuore dx. tali patologie sono:
1)malattie a carico dei vasi polmonari :- embolia polmonare ricorrente- ipertensione polmonare primitiva- vasculiti/collagenopatie2) malattie a carico del parenchima e/o delle vie respiratorie:- BPCO : distruzione setti, ipossia- Malattie intersiziali ad evoluzione in fibrosi e sclerosi (polmoniti interstiziali, tubercolosi, sarcoidosi, pneumoconiosi):
ipossia ?- Infiltrazione carcinomatosa:- Pneumectomia- Ostruzione bronchiale3) malattie extrapolmonari che riducono la ventilazione polmonare:- cifoscoliosi- obesità marcata (s. di Pickwick)- malattie neuromuscolari
meccanismi patogenetici implicati nelle forme suddette sono:

- la distruzione del parenchima che provoca una ↓ delle resistenze in parallelo e quindi un↑ R : vedi enfisema, malattie ad evoluzione sclerosante come la silicosi, cavitaria come la TBC , la pneumectomia, l’infiltrazione neoplastica.- la organizzazione dei microemboli multipli nell’embolia polmonare ricorrente.- Modificazioni nei vasi del piccolo circolo su base idiopatica (arteriopatia plessiforme) nella ipertensione polmonare
primitiva.- il rimodellamento della parete dei vasi polmonari indotto dall’ ipossia alveolare e dall’ipossiemia : questo provoca
una vasocostrizione che ha il ruolo fisiologico di riequilibrare il rapporto V/Q in aree poco ventilate: l’ipossia alveolaresi può avere per ridotta tensione di O2 nell’aria inspirata (es. alle alte quote) o per difettosa ventilazione alveolare e inquesto caso si hanno tutta una serie di patologie extrapolmonari e polmonari ostruttive come l’asma, la fibrosicistica, l’enfisema , la bronchite cronica e restrittive fibrosi polmonare di varia natura.
A lungo andare si possono produrre lesioni permanenti a carico della parete specialmente a livello dellearterie muscolari e delle arteriole con 40<∅<300 µm con iperplasia della media e fibrosi sottointimaleche comportano riduzione del calibro e a anche a livello delle aa elastiche (∅>1mm) a livello dellequali compare ateromatosi: in definitiva le lesioni sono analoghe a quelle dell’ipertensione sistemica.Queste modificazioni sono valide per l’ipertensione polmonare in generale e una volta che avvenutesono irreversibili per cui è inutile la rimozione ove possibile della causa che le ha prodotte (es.stenosimitralica).
Polmone da stasi o iperemia polmonare
L’iperemia a livello polmonare si può produrre acutamente in genere per un’ insufficienza acuta del cuore sn o in corso diipertensione endocranica o cronica se la causa è cronica come può essere l’ insufficienza cronica del ventricolo sn, i vizimitralici, gli shunt sn-dx.
Il polmone iperemico ha dei caratteri particolari che sono facilmente intuibili:-è congesto, di colorito rosso-bluastro, ↑ di peso, con crepitio ↓.-in caso di iperemia cronica è di consistenza ↑ per via della fibrosi (edema molle →edema duro)
a livello microscopico si notano altri particolari:
-i capillari sono dilatati ed è presente edema interstiziale per cui i setti alveolari presentano un ↑ di spessore di 3-5 volte(0.3→1.3-1.5 µm) eventualmente con sovrapposizione di fibrosi all’edema. Questo provoca un ostacolo agli scambi(blocco alveolo capillare) per cui si ha cianosi, dispnea e come compenso una poliglobulia che ↑la viscosità ematica epredispone alla trombosi.
-l’edema per motivi di pressione idrostatica è più pronunciato alle basi e qui si ritroverà anche negli alveoli (rantoli);l’edema alveolare è un ottimo terreno per i batteri e predispone alle broncopolmoniti batteriche.
-a livello alveolare si ritrovano anche eritrociti che stanno a testimoniare emorragie per diapedesi o per rottura vera epropria dei capillari. Tali eritrociti vengono fagocitati dai macrofagi alveolari che diventano carichi di granuli diemosiderina e vengono detti “cellule da vizio cardiaco”; conferiscono all’espettorato striature rugginose o in alternativaprendono la via dei linfatici. Lungo questa via cosi come accade nelle pneumoconiosi si deposita col tempo l’emosiderinache ha un potere sclerogeno tale da produrre noduli siderotici.
Nel caso per es. di insufficienza ventricolare sn a breve termine prevarrà la congestione polmonare aggravata da un ↑ delRV al cuore sn come durante lo sforzo o in posizione di decubito (dispnea da sforzo e ortopnea); a lungo termine invece siavranno modificazioni a carico dei vasi a resistenza (arteriole e piccole arterie muscolari) che allevieranno la congestione alivello dei setti e la relativa sintomatologia ma produrranno ipertensione polmonare.
Embolia polmonare e cuore polmonare L’embolia polmonare è una patologia estremamente frequente.

Vi sono embolie di varia natura:1)tromboembolia: è sicuramente la più frequente ed è dovuta al distacco di trombi formatisi in genere a livello delle venedegli arti inferiori pianta del piede, della gamba, la femorale, la iliaca esterna e anche i vasi del bacino: raramente letromboembolie possono derivare dalla safena (varici), dal cuore dx, o addirittura dal cuore sn per shunt sn-dx.la formazione del trombo avviene notoriamente su un endotelio danneggiato: per esempio le infiammazioni delle veneprovocano trombi per cui si parla di tromboflebiti: questi trombi sono a larga base d’impianto per la lesione endoteliale cheli sottende e l’infiammazione si associa a dolore, edema, rubor: sono pertanto trombosi a basso rischio di embolia e cmq benriconoscibili.Diverso il discorso per le flebotrombosi le quali possono insorgere in varie circostanze riconducibili comunque alla triade diVirchow ovvero1) danno endoteliale: per es. interventi chirurgici o incannulamento2)alterazione del flusso : stasi dovuta a immobilità troppo prolungata (per es.lunghi viaggi in aereo o pz allettati che vannomobilizzati il prima possibile per ridurre questo rischio) o anche alla gravidanza.3)ipercoagulabilità (vedi gravidanza, neoplasie, iperviscosità).La flebotrombosi è più insidiosa in quanto non da sintomi ne segni apprezzabili (è spesso profonda), la base d’impianto deltrombo è più piccola e quindi si stacca più facilmente.2) e. gassosa si verifica se non vengono rispettate le tappe di decompressione durante la risalità da una immersione4) e. grassosa: per passaggio di frammenti di tessuto adiposo dopo fratture ossee o anche liposuzione5) 4) e. da midollo osseo5) e. da liquido amniotico6) e. da cotone
Patologia pleurica:Versamenti pleuriciSi parla di versamento pleurico quando si accumula una quantità di liquido di almeno 30 volte il

normale. I versamenti massivi possono avere pesanti ripercussioni sui volumi polmonari in quantoproducono compressione e collassamento delle vie aeree.La eziopatogenesi del versamento ricalca quella generale degli edemi (↑p idrostatica; ↓π;↑permeabilità;↓drenaggio linfatico).
Una prima distinzione è quella che può essere ottenuta dopo toracentesi, analizzando il fluido:Se la [Proteine] è almeno 50% di quella del sieroSe la [LDH] è almeno il 60% di quella del sieroSe la [LDH] è almeno i 2/3 del limite massimo di normalità del siero
Se almeno uno di questi 3 criteri è soddisfatto siamo in presenza di un essudato.
GENERALMENTE GLI ESSUDATI SONO DOVUTI A UN FATTORE LOCALE CHEINFLUENZA L’EQUILIBRIO FORMAZIONE-DRENAGGIO DEL FLUIDO INTRAPLEURICO.
Questo fattore può essere:1) una pleurite (che per definizione porterà alla formazione di un essudato) la cui tipologia ed
eziologia (infezioni varie, infarto polmonare secondario a embolia diffusa,collagenopatie evasculiti, uremia, radiazioni, farmaci etc.) può essere varia,
2) una neoplasia primitiva (mesotelioma correlato all’esposizione all’asbesto) o assai piùfrequentemente metastatica (cr. Polmonare, mammario e linfoma le più frequenti), ma anche
3) patologie gastrointestinali come una perforazione esofagea, una patologia pancreatica, unascesso subfrenico.
4) la rottura del dotto toracico (chilotorace) secondaria a traumi o a tumori mediastinici(controllare i linfonodi),
5) o la rottura di un vaso sanguigno (emotorace) come in caso di aneurismi aortici o traumi,6) cause iatrogene come interventi chirurgici di bypass etc.
Allora quando si è in presenza di un essudato occorre l’analisi dell’essudato.
Aspetto: lattescente (chilo o essudato sieroso torbido?); ematico (essudato siero-emorragico o emorragia vera e propria?); purulento (empiema da non confondere con il versamento metapneumonico)
Composizione chimica: glucosio (↓in caso di tumore in cavità pleurica o infezione batterica) Lipidi (↑ in caso di chilotorace: sopranatante di aspetto cremoso) Amilasi (↑ in caso di patologia esofagea o pancreatica) pH
HT: se è almeno il 50% di quello del sangue periferico si è in presenza di emotorace
conta dei leucociti: ↑linfociti: vedi tubercolosi eosinofilia: vedi farmaci esame colturale
citologia ed esami con anticorpi monoclonali per la ricerca di neoplasie

GENERALMENTE I TRASUDATI SONO DOVUTI FATTORI SISTEMICI OVVERO A UNAPATOLOGIA CHE NON INTERESSA primitivamente L’EQUILIBRIO FILTRAZIONE-RIASSORBIMENTO A LIVELLO pleurico.
Insufficienza Vsn L’edema interstiziale si scarica in parte in cavità pleurica,bilateralmente. Il trattamento è quello diuretico.
Cirrosi epatica Il meccanismo patogenetico principale non è la ↓π ma il passaggio diliquido ascitico attraverso piccoli fori del diaframma in cavitàpleurica; la localizzazione è prevalentemente dx.
Sindrome nefrosica Edemi generalizzati dovuti alla ↓π e al sovraccarico di liquidiEmbolia polmonare Si accompagna a dispnea: il versamento è trasudatizio se l’embolia
interessa un grosso ramo della a. polmonare (ipertensione),essudatizio se post-infartuale.
PNEUMOTORACELo pneumotorace può essere classificato come:Spontaneo: suddiviso in idiopatico (rottura di bolle subpleuriche in pz relativamente giovani senzanessuna apparente patologia polmonare preesistente) e secondario a patologie polmonari in genereBPCO, e in questo caso per la ridotta riserva polmonare lo p. è un evento più pericoloso.
Vedere differenza fra bolle e blebs!
Traumatico: può prodursi in seguito a traumi sia chiusi (es.frattura costale) che aperti (es.ferita daarma da taglio) del torace.
Pneumotorace iperteso: è uno p. con pressione intrapleurica positiva; può aversi per la presenza di unmeccanismo a valvola o per es. in corso di respirazione assistita o di manovre di rianimazione. Questop. è un emergenza, un emitorace si presenta espanso e il polmone omolaterale collassato conspostamento del mediastino controlateralmente. Oltre al deficit ventilatorio del polmone collassato siha una ↓RV per l’↑ pressione intratoracica con conseguente ipossemia e caduta della Q che possonoessere fatali. L’inserimento di un ago a livello del II spazio intercostale serve a riportare la p. pleuricaalla P atmosferica finchè non viene praticata la toracostomia.
Carcinoma polmonareQuesta definizione comprende le varie neoplasie maligne che originano dalle cellule dell’ epitelio delle vierespiratorie(bronchi bronchioli e alveoli).A ben vedere però la maggior parte di questi tumori insorge nella parte più prossimale dell’albero bronchiale: probabilmentequesto è dovuto al fatto che i cancerogeni che arrivano al polmone per via aerea si accumulano maggiormente a livelloprossimale (quantità che si arresta qui + quantità trasportata per via mucociliare).Bronchi I-III ordine: 60%Bronchi IV-VII ordine: 30%Cr. bronchiolo-alveolare: 5-10% →cancerogeni volatili e per via ematogena (tipo nitrosamine)
CANCEROGENESI ed EPIDEMIOLOGIAI principali fattori di rischio per il cr. Polmonare sono:

1)il fumo: l’azione cancerogena del fumo di sigaretta è provata in diversi modi:statisticamente verificata la correlazione con consumo giornaliero di sigarette, durata dell’abitudine, tendenza ad aspirare:il RR per un fumatore accanito di lunga data arriva a 60-70; l’80% dei pz è un fumatore.Morfologicamente è dimostrata l’alta prevalenza nei fumatori di lesioni elementari prodotte dal fumo:- ridotta attività ciliare- lesioni predittive di vario tipo: metaplasia squamosa, iperplasia dello strato basale.- lesioni precancerose di tipo displasico ovvero contenenti cellule atipiche (displasia lieve, moderata, grave e CIS)Sperimentalmente i dati ricavati sugli animali dimostrano comunque il potere cancerogeno del fumo.
2)l’esposizione a cancerogeni presenti nell’ambiente o sul luogo di lavoro fra questi: -l’esposizione a radiazioni o a sostanze radioattive come l’uranio o il radon -l’asbesto (RR=5) particolarmente se associato al fumo (RR=55) -molte altre sostanze come arsenico (insetticidi), cromo (cromatori), nichel, carbone, Fe etc.
3)la presenza di una patologia pregressa per il cosiddetto “cancro- cicatrice” che in genere è un adenocarcinoma
4)in certi casi una predisposizione genetica: probabilmente legata a una particolare attività di alcuni sistemi enzimatici(alcuni agenti sono pro-cancerogeni) o alla trasmissione di mutazioni per geni oncosoppressori.
ANATOMIA PATOLOGICAL’OMS classifica il cr polmonare in 4 tipi che in ordine di frequenza sono:1)Adenocarcinoma (compreso il cr bronchioloalveolare) ,2)cr squamocellulare o epidermoidale,3)cr a piccole cellule o microcitoma4)cr a grandi cellule (anaplastiche)
vi è poi una classificazione di utilità clinica che individua 2 categorie:1)cr a piccole cellule (è molto aggressivo, metastatizza molto presto, e viene in genere diagnosticato troppo tardi per lachirurgia; tuttavia risponde meglio degli altri alla chemioterapia→è a rapida crescita: la sopravvivenza è la più bassa)2)cr non a piccole cellule (può essere diagnosticato anche in fase precoce e quindi trattabile con la chirurgia, tuttaviarisponde peggio del microcitoma alla chemioterapia):
Anatomia patologica Eziopatogenesi Clinica

La sede è prevalentemente medio-parenchimale.Si tratta di lesionicontenenti elementi ghiandoarimucosecernenti variamenteorganizzati (tubuli, papille etc.)
È la varietà conl’associazione piùdebole (75%) alfumo: quellaprevalente nei nonfumatori e nelledonne. Puòinsorgere su lesionicicatriziali.
Crescita e metastatizzazionerelativamente lente ma anchemetastasi ematogena.In certi casi si può intervenirechirurgicamente: lobectomia +aportazione dei linfonodi ilari.
Adenocarcinoma(fino al 35%)
Il carcinoma bronchiolo-alveolare èper definizione periferico* si presentasolitamente con lesioni plurifocalicostituite da cellule cubico-cilindriche con o senza mucine chevanno a rivestire i setti alveolari e siorganizzano in formazioni papillari
Nelle pecoreinsorge su baseinfettiva unalesione del tuttosimile. Non è statoprovato nell’uomoniente del genere.
Non è a crescita veloce né a precocemetastatizzazione: il problema è lapossibile multifocalità e la resistenzaalla terapia (vedi non microcitomi).
Crepidermoidale(35%)
La sede è in genere ilare (grossibronchi) ma può essere medio-parenchimale e allora tende anecrotizzarsi centralmente (crascesso). Vari i gradi didifferenziamento: i più differenziati(epidermoidali) producono perlecornee e spine.
È strettamenteassociato al fumo.
Crescita lenta e metastatizzazione piùtardiva prevalentemente linfonodale(caratteristica di tutti gliepidermoidali).
Cr a piccolecellule (20%)
Ha localizzazione ilare. Le lesionisono costituite da nidi di piccolecellule con scarso citoplasma di formaovale, fusata o poligonale (linfocito-simili tanto che un tempo eradiagnosticato come linfosarcoma):questi sollevano la mucosa che rimaneliscia, non ulcerata. Vi possono esserecellule con granuli ormono-secretrici(vedi oltre).
È il piùstrettamenteassociato al fumo(99%): vedi ancheassociazione conl’uranio (lavoratoriin miniera).
La crescita è molto rapida cosi comeprecoce la metastatizzazione per vialinfatica (cresce nella tonaca propria)ed ematogena →.Alla diagnosi ègeneralmente inoperabile: è indicatala biopsia alla cresta iliaca!!!Talvolta può essere “misto”(+ epid. O+adeno) e allora la prognosi èmigliore.
Cr a grandicellule (10%)
È un cr. medio parenchimale (apalla) o ilare (vegetante): le cellulesono grandi e anaplastiche nellevarietà giganti, chiare e fusate.
È strettamenteassociato al fumo
Accrescimento rapido e infiltranteverso le strutture ilo-mediastiniche.metastatizzazioneprecoce.
*N.B.: periferico indica la localizzazione alle vie aeree più distali e quindi, in generale, sottopleurica: ricorda tuttavia che unsiffatto tumore può ritrovarsi anche in corrispondenza dell’ilo visto che anche lì c’è la pleura viscerale.
CLINICALe manifestazioni cliniche del carcinoma polmonare sono molte e derivano dalla crescita locale, dalla infiltrazione dellestrutture viciniori, dalla disseminazione linfatica ed ematogena, e da sindromi paraneoplastiche.In generale solo una piccola percentuale dei casi (5-10%) viene diagnosticata per es. attraverso una radiografia diroutine, allo stato ancora asintomatico.
CRESCITA LOCALELa crescita endobronchiale può dare tosse, emoftoe (ulcerazione), può produrre una broncostenosi o un’ ostruzionecompleta con ciò che ne consegue:dispnea, broncopolmonite ricorrente post-atelettasica (segno estremamenteimportante), atelettasia oppure enfisema a valvola.La crescita verso l’esterno del bronco può produrre dispnea su base restrittiva, tosse e dolore pleurico e versamento incaso di interessamento di questa struttura, evoluzione ascessuale e relativi sintomi.
CRESCITA DIRETTA ALLE STRUTTURE VICINIORI

Il carcinoma può infiltrare direttamente le strutture adiacenti (parete toracica, mediastino,diaframma) o provocarecompressione in seguito a metastasi linfonodali (es. esofago con disfagia); possono essere interessati:-il nervo frenico con paralisi dell’emidiframma e conseguente dispnea-il nervo ricorrente laringeo con raucedine e voce bitonale.-per i tumori dell’apice polmonare anche molto piccoli:a)il plesso brachiale (radici c8-t1-t2) con conseguente s. di Pancoast (dolore a collo,spalla,braccio e amiotrofia della manoomolateralmente) e/ob)i nervi simpatici con comparsa della sindrome di Horner che porta attraverso una perdita dell’ azione del simpatico alivello della meta omolaterale della faccia a:- miosi- anidrosi- enoftalmo e ptosi palpebrale
-la vena cava superiore con conseguente sindrome (edema a mantellina etc.)-il pericardio e persino il miocardio
T0TX
Ilare Medio-parenchimale SottopleuricoT1 No interessamento bronco princ. ∅<3cm No interessamento pleura visceraleT2 Interessamento bronco princ. A
>2cm dalla carena/ atelettasiapolmonare
∅>3cm Interessamento pleura viscerale
T3 Interassamento br. Principale a<2 cm dalla carena/atelettasia diun intero polmone
Interessamento pleura parietale estrutture parietali viciniori: paretetoracica, diaframma, pericardio.
T4 Interessamento carena Interessamento organi mediastinici: venacava, trachea, esofago, miocardio, corpivertebrali oppure Versamento pleuricomaligno
ESTENSIONE PER VIA LINFATICAIl carcinoma può accrescersi anche per via linfatica rertrograda ovvero centrifuga infiltrando i linfatici sempre piùperiferici: questo porta a una linfangite carcinomatosa con difetto di drenaggio linfatico, edema interstiziale eidrotorace.Per quanto riguarda la via anterograda ovvero centripeta bisogna ricordare il sistema di drenaggio della linfa: questaprocede attraversando i linfonodi polmonari→ bronchiali→ tracheobronchiali inferiori → tracheobronchialisuperiori→ tracheali.Tutti i lobi di dx scaricano ai linfonodi omolaterali; tuttavia lo stesso non avviene per il polmone sn: infatti itracheobronchiali inferiori di sn che raccolgono la linfa dal lobo inferiore e in parte dalla lingula scaricano aitracheobronchiali superiori di dx!!!Pertanto un tumore al lobo inferiore sn e meno frequentemente alla lingula, può metastatizzare ai linfonodi mediastinici (tbsup e paratracheali controlatererali→N3)
Stadiazione dell’N: presenza di metastasi ai linfonodi loco-regionaliN0: non evidenziabiliN1: bronchiali omolateraliN2: tracheobronchiali inferiori (carenali) o ai mediastinici omolateraliN3: metastasi ai ln controlaterali o ai linfonodi sovraclaveari
DIFFUSIONE PER VIA EMATOGENATutti gli organi possono essere interessati dalle metastasi per via ematica→ il filtro è in questo caso il microcircolosistemico: con particolare frequenza saranno interessati:- gh. Surrenali → un nodulo surrenalico può essere un adenoma non secernente oppure una metastasi: allora per
assegnare la M1 bisogna fare DD.- Encefalo → quasi sempre una neoplasia cerebrale è metastatica

- osso- fegato
la stadiazione dell’M è alquanto semplice:M0: non evidenza di metastasi a distanzaM1: evidenza di metastasi a distanza
SINDROMI PARANEOPLASTICHEAlcune sono dovute al rilascio di fattori umorali da parte del carcinoma (in particolare il microcitoma*, ma anchel’epidermoidale** e l’adenocarcinoma***) con le conseguenze che un rilascio eccessivo e incontrollato di questi fattoriprovoca:1) *ADH → iposmolarità liquidi corporei2) *ACTH → Cushing3) **PTH → ipercalcemia4) calcitonina→ ipocalcemia5) ***gonadotropine → ginecomastia6) serotonina → s. da carcinoide
vi sono anche s. paraneoplastiche non associate al rilascio di fattori umorali da parte del tumore ma cmq secondarie ad essocome:1) s. miastenica di Lambert-Eaton: una reazione autoimmune2) dita a bacchetta di tamburo3) neuropatie periferiche4) Acanthosis nigricans: patologia dermatologica
In generale la diagnostica di lesioni polmonari si basa su:- semeiotica classica- esami strumentali- esame citologico su: espettorato, campione “broncoscopico”, agobiopsia.
CARCINOIDI o TUMORI A BASSO GRADO DI MALIGNITA’I carcinoidi rientrano in un gruppo più ampio di tumori derivati da elementi cellulari del cosiddetto sistema neuroendocrinodiffuso. Questi elementi epiteliali specializzati sono presenti un po’ in tutte le mucose e a livello bronchiale assumono ilnome di cellule di Kulchitsky.Si ritiene in base ad elementi morfofunzionali che almeno 3 tipi di neoplasie possano derivare da queste cellule ovvero:1) lesioni benigne dette tumorlets2) cr a basso grado di malignità o carcinoidi3) il microcitoma
il carcinoide rappresenta meno del 5% di tutti i tumori polmonari ma oltre il 90% delle lesioni che venivano chiamate“adenomi bronchiali” piuttosto impropriamente perché in realtà sono, anche se a basso grado, dei carcinomi. L’altro 10% èrappresentato da due neoplasie che traggono origine dalle gh. Sieromucose bronchiali che sono del tutto analoghi aneoplasie delle gh. Salivari:- cr. adenoido-cistico- cr. mucoepidermoide
L’epidemiologia è particolare perché:- insorge in età tipicamente giovanile : 20-30 anni! E senza prdilezione di sesso: F/M=1- non mostra relazione causale con fattori ambientali come il fumo di sigaretta (l’opposto del suo “cugino maligno”: il
microcitoma)- può rientrare nel quadro di una MEN
in genere i carcinoidi sono tumori non aggressivi con cellule dai nuclei rotondi organizzate in nidi separati da un sottilestroma fibroso: le dimensioni sono limitate ma per via della modalità di crescita almeno parzialmente vegentante (massa chesolleva la mucosa formando un polipo sessile) si hanno sintomi come tosse persistente, emottisi (per ulcerazione dellamucosa), alterazioni del drenaggio con broncopolmoniti ricorrenti.Solo una minoranza dei casi può avere un comportamento più aggressivo con metastasi a distanza.

Anche la classica s. da carcinoide dovuta a una iperproduzione di serotonina è in realtà presente solo in una minoranza deicasi ( con riferimento a questo argomento vedi anche alle voci: cardiomiopatia obliterativa e carcinoidi intestinali).
Altri tumori sono:1) l’amartoma: ammasso di tessuti fisiologici (cartilagine, connettivo mixoide, tessuto muscolare, adiposo, linfoide,
epiteliale) ma disordinatamente disposti: sono a lenta crescita→ 1 mm/anno →spesso<4 cm (coin lesion→difficileinterpretazione → DD); l’unico problema è quello di operare se la crescita è endobronchiale (ma è più frequentementeintraparenchimale, rarissimamente intrapleurico) o se è rapida e il pz è giovane→al solito vale l’analisirischio/beneficio. È una lesione frequente (1/400)
2) condroma : è a lentissima crescita ma endobronchiale→ va asportato.3) fibroma




![[eBook ITA] Project Management](https://img.pdfslide.tips/doc/110x75/557201324979599169a0fcc8/ebook-ita-project-management.jpg)
![[eBook - ITA] [Arredamento] MANUALE-Arredare](https://img.pdfslide.tips/doc/110x75/5571f90e49795991698eb0f8/ebook-ita-arredamento-manuale-arredare.jpg)

![[eBook Ita] Manuale Ita Pratico Di Java](https://img.pdfslide.tips/doc/110x75/557213f8497959fc0b937032/ebook-ita-manuale-ita-pratico-di-java-55cb7b9e618ac.jpg)






![[eBook - ITA] Disegnare Fumetti](https://img.pdfslide.tips/doc/110x75/5695d1771a28ab9b0296a6b7/ebook-ita-disegnare-fumetti.jpg)

![[eBook ITA] Pernigotti Leggere i Geroglifici.pdf](https://img.pdfslide.tips/doc/110x75/563db85b550346aa9a92f0b7/ebook-ita-pernigotti-leggere-i-geroglificipdf.jpg)
![Dante Alighieri - Rime [eBook ITA]](https://img.pdfslide.tips/doc/110x75/557202844979599169a3ab5b/dante-alighieri-rime-ebook-ita.jpg)


![[EBook ITA] - Diritto - Formulario Diritto Amministrativo](https://img.pdfslide.tips/doc/110x75/568bd9041a28ab2034a57be3/ebook-ita-diritto-formulario-diritto-amministrativo.jpg)
![[Med ITA] Anatomia - Muscoli (Dall'Edi-Ermes)](https://img.pdfslide.tips/doc/110x75/55cf882655034664618dd96e/med-ita-anatomia-muscoli-dalledi-ermes.jpg)
![[eBook - Ita] Blender Manuale](https://img.pdfslide.tips/doc/110x75/557210cd497959fc0b8db181/ebook-ita-blender-manuale.jpg)
![Giovanni Verga - Novelle Rusticane [eBook ITA]](https://img.pdfslide.tips/doc/110x75/55cf9db9550346d033aee7a8/giovanni-verga-novelle-rusticane-ebook-ita.jpg)
![[eBook - Ita - ECONOMIA] Finanza Aziendale](https://img.pdfslide.tips/doc/110x75/5572027e4979599169a39e86/ebook-ita-economia-finanza-aziendale.jpg)
![[eBook Ita] La magia satanica](https://img.pdfslide.tips/doc/110x75/55cf9a6f550346d033a1b637/ebook-ita-la-magia-satanica.jpg)
![Anonimo - Il Novellino [eBook ITA]](https://img.pdfslide.tips/doc/110x75/54205dd97bef0ab4128b457b/anonimo-il-novellino-ebook-ita.jpg)

![[eBook ITA] Benson_AmiciziadiCristo](https://img.pdfslide.tips/doc/110x75/577cdbdf1a28ab9e78a94ed3/ebook-ita-bensonamiciziadicristo.jpg)
![[ebook ita] corso_sui_cristalli](https://img.pdfslide.tips/doc/110x75/577dabbd1a28ab223f8ce2a0/ebook-ita-corsosuicristalli.jpg)
![[EBOOK ITA] S_Tommaso_dAquino_De_malo](https://img.pdfslide.tips/doc/110x75/5571f97749795991698fa450/ebook-ita-stommasodaquinodemalo.jpg)


![[eBook ITA] Grammatica Spagnola](https://img.pdfslide.tips/doc/110x75/577cdd3d1a28ab9e78ac9282/ebook-ita-grammatica-spagnola.jpg)