Embed Size (px)
Citation preview

1
ANHANG I
ZUSAMMENFASSUNG DER MERKMALE DES ARZNEIMITTELS

2
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8. 1. BEZEICHNUNG DES ARZNEIMITTELS Tremfya 100 mg Injektionslösung 2. QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG Jede Fertigspritze enthält 100 mg Guselkumab in 1 ml Lösung. Guselkumab ist ein rein humaner monoklonaler Immunglobulin-G1-Lambda(IgG1λ)-Antikörper (mAk) gegen das Interleukin(IL)-23-Protein, hergestellt durch rekombinante DNA-Technologie in einer CHO-Zelllinie (Chinese-Hamster-Ovary). Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1. 3. DARREICHUNGSFORM Injektionslösung (Injektion) Die Lösung ist klar und farblos bis hellgelb. 4. KLINISCHE ANGABEN 4.1 Anwendungsgebiete Tremfya wird angewendet für die Behandlung erwachsener Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine systemische Therapie in Frage kommen. 4.2 Dosierung und Art der Anwendung Tremfya ist zur Anwendung unter der Leitung und Überwachung eines in der Diagnose und Behandlung der Plaque-Psoriasis erfahrenen Arztes vorgesehen. Dosierung Die empfohlene Dosis für Tremfya beträgt 100 mg als subkutane Injektion in den Wochen 0 und 4, gefolgt von einer Erhaltungsdosis alle 8 Wochen. Bei Patienten, die nach 16 Wochen auf die Behandlung nicht angesprochen haben, sollte ein Absetzen der Behandlung in Erwägung gezogen werden. Ältere Patienten (≥ 65 Jahre) Es ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Bisher liegen nur begrenzte Erfahrungen zu Patienten ≥ 65 Jahre vor. Nieren- oder Leberfunktionsstörung Tremfya wurde bei diesen Patientengruppen nicht untersucht. Es können keine Dosierungsempfehlungen gegeben werden. Bezüglich weiterer Angaben zur Elimination von Guselkumab siehe Abschnitt 5.2.

3
Kinder und Jugendliche Die Sicherheit und Wirksamkeit von Tremfya bei Kindern und Jugendlichen unter 18 Jahren ist bisher noch nicht erwiesen. Es liegen keine Daten vor. Art der Anwendung Subkutane Anwendung. Wenn möglich, sollten Hautbereiche, die von Psoriasis betroffen sind, als Injektionsstelle vermieden werden. Nach angemessener Schulung in subkutaner Injektionstechnik können Patienten Tremfya selbst injizieren, wenn der Arzt dies für angebracht hält. Jedoch soll der Arzt eine entsprechende medizinische Nachbeobachtung der Patienten sicherstellen. Die Patienten sind angewiesen, die vollständige Menge an Tremfya gemäß der Beilage „Hinweise zur Anwendung“, die der Verpackung separat beiliegt, zu injizieren. Für weitere Hinweise zur Anwendung und zu besonderen Vorsichtsmaßnahmen bei der Handhabung, siehe Abschnitt 6.6 sowie die Beilage „Hinweise zur Anwendung“. 4.3 Gegenanzeigen Schwerwiegende Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile. Klinisch relevante aktive Infektionen (z. B. aktive Tuberkulose, siehe Abschnitt 4.4). 4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung Rückverfolgbarkeit Um die Rückverfolgbarkeit bei biologischen Arzneimitteln zu verbessern, sollten Name und Chargenbezeichnung des verabreichten Produkts deutlich protokolliert werden. Infektionen Tremfya kann das Infektionsrisiko erhöhen. Eine Behandlung mit Tremfya sollte bei Patienten mit klinisch relevanten aktiven Infektionen erst nach dem Abklingen oder einer angemessenen Behandlung der Infektion eingeleitet werden. Mit Tremfya behandelte Patienten sollten angewiesen werden, ärztlichen Rat einzuholen, wenn Anzeichen oder Symptome einer klinisch relevanten chronischen oder akuten Infektion auftreten. Wenn ein Patient eine klinisch relevante oder schwerwiegende Infektion entwickelt oder nicht auf die Standardtherapie reagiert, sollte er engmaschig überwacht und Tremfya bis zum Abklingen der Infektion abgesetzt werden. Tuberkulose-Untersuchung vor der Behandlung Vor Beginn der Behandlung mit Tremfya sollten die Patienten auf eine Tuberkulose(TB)-Infektion untersucht werden. Patienten, die Tremfya erhalten, sollten während und nach der Behandlung auf Anzeichen und Symptome einer aktiven TB überwacht werden. Bei Patienten mit latenter oder aktiver TB in der Vorgeschichte, bei denen kein angemessener Behandlungsverlauf bestätigt werden kann, sollte vor dem Einleiten der Behandlung mit Tremfya eine Anti-TB-Therapie in Erwägung gezogen werden. Überempfindlichkeit Im Falle des Auftretens einer schwerwiegenden Überempfindlichkeitsreaktion sollte die Anwendung von Tremfya unverzüglich abgebrochen und eine geeignete Behandlung eingeleitet werden. Impfungen Vor dem Einleiten der Therapie mit Tremfya sollte die Durchführung aller angebrachten Impfungen in Übereinstimmung mit den aktuellen Impfempfehlungen in Erwägung gezogen werden. Bei mit Tremfya behandelten Patienten sollten keine Lebendimpfstoffe angewendet werden. Es liegen keine Daten bezüglich des Ansprechens auf Lebend- oder Totimpfstoffe vor.

4
Vor einer Impfung mit viralen oder bakteriellen Lebendimpfstoffen muss die Behandlung mit Tremfya nach der letzten Gabe für mindestens 12 Wochen ausgesetzt werden und kann frühestens 2 Wochen nach der Impfung wieder aufgenommen werden. Zur weiteren Information und Anleitung zur gleichzeitigen Anwendung von Immunsuppressiva nach der Impfung sollten die verordnenden Ärzte die Zusammenfassung der Merkmale des Arzneimittels (Fachinformation) des spezifischen Impfstoffs heranziehen. 4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen Wechselwirkungen mit CYP450-Substraten In einer Phase-I-Studie an Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis waren die Veränderungen der systemischen Expositionen (Cmax und AUCinf) von Midazolam, S-Warfarin, Omeprazol, Dextromethorphan und Coffein nach einer Einzeldosis von Guselkumab klinisch nicht relevant, was darauf hindeutet, dass Arzneimittelwechselwirkungen zwischen Guselkumab und Substraten unterschiedlicher CYP-Enzyme (CYP3A4, CYP2C9, CYP2C19, CYP2D6 und CYP1A2) unwahrscheinlich sind. Bei gleichzeitiger Anwendung von Guselkumab und CYP450-Substraten ist keine Dosisanpassung erforderlich. Gleichzeitige Immunsuppressiva- oder Phototherapie Die Sicherheit und Wirksamkeit von Tremfya in Kombination mit Immunsuppressiva, einschließlich Biologika oder Phototherapie wurden nicht untersucht. 4.6 Fertilität, Schwangerschaft und Stillzeit Frauen im gebärfähigen Alter Frauen im gebärfähigen Alter sollen während und für mindestens 12 Wochen nach der Behandlung eine zuverlässige Verhütungsmethode anwenden. Schwangerschaft Bisher liegen keine Erfahrungen mit der Anwendung von Guselkumab bei Schwangeren vor. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf Schwangerschaft, embryonale/fetale Entwicklung, Entbindung oder postnatale Entwicklung (siehe Abschnitt 5.3). Aus Vorsichtsgründen soll eine Anwendung von Tremfya während der Schwangerschaft vermieden werden. Stillzeit Es ist nicht bekannt, ob Guselkumab in die Muttermilch übergeht. Da Immunglobuline in die Muttermilch übergehen, kann ein Risiko für das gestillte Kind nicht ausgeschlossen werden. Es muss eine Entscheidung darüber getroffen werden, ob das Stillen während der Behandlung sowie bis zu 12 Wochen nach der letzten Gabe zu unterbrechen ist oder ob die Behandlung mit Tremfya zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der Therapie mit Tremfya für die Frau zu berücksichtigen. Bezüglich Angaben zum Übergang von Guselkumab in die Muttermilch von Tieren (Javaneraffen) siehe Abschnitt 5.3. Fertilität Die Wirkung von Guselkumab auf die Fertilität des Menschen wurde nicht untersucht. Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf die Fertilität (siehe Abschnitt 5.3). 4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen
von Maschinen Tremfya hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.

5
4.8 Nebenwirkungen Zusammenfassung des Sicherheitsprofils Die häufigste Nebenwirkung war eine Infektion der oberen Atemwege. Tabellarische Auflistung der Nebenwirkungen In einer Phase-II- und drei Phase-III-Studien zur Plaque-Psoriasis wurden insgesamt 1.748 Patienten mit Tremfya behandelt. Von diesen hatten 1.393 Psoriasis-Patienten mindestens 6 Monate lang eine Tremfya-Exposition und 728 Patienten hatten eine Exposition über mindestens 1 Jahr (d. h. Behandlung bis einschließlich Woche 48). Die Häufigkeiten der spezifizierten Nebenwirkungen wurden mittels einer gepoolten Analyse bestimmt, in die 823 Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis einbezogen waren, die Tremfya während des placebokontrollierten Zeitraums in zwei Phase-III-Studien erhielten. Die Nebenwirkungen (Tabelle 1) sind nach MedDRA-Systemorganklassen und Häufigkeitskategorien gemäß folgender Konvention eingestuft: sehr häufig (≥ 1/10), häufig (≥ 1/100 bis < 1/10), gelegentlich (≥ 1/1.000 bis < 1/100), selten (≥ 1/10.000 bis < 1/1.000), sehr selten (< 1/10.000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Tabelle 1: Auflistung der Nebenwirkungen in klinischen Studien
Systemorganklasse Häufigkeit Nebenwirkung Infektionen und parasitäre Erkrankungen
Sehr häufig Infektion der oberen Atemwege Häufig Gastroenteritis Häufig Herpes-simplex-Infektionen Häufig Tinea-Infektionen
Erkrankungen des Nervensystems Häufig Kopfschmerzen Erkrankungen des Gastrointestinaltrakts
Häufig Diarrhoe
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig Urtikaria
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig Arthralgie
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig Erythem an der Injektionsstelle Gelegentlich Schmerzen an der Injektionsstelle
Beschreibung ausgewählter Nebenwirkungen Gastroenteritis In zwei klinischen Phase-III-Studien einschließlich des placebokontrollierten Zeitraums trat Gastroenteritis häufiger in der mit Tremfya behandelten Gruppe (1,1 %) als in der Placebo-Gruppe (0,7 %) auf. Gastroenteritis-Nebenwirkungen waren nicht schwerwiegend und hatten bis einschließlich Woche 48 kein Absetzen von Tremfya zur Folge. Reaktionen an der Injektionsstelle In zwei klinischen Phase-III-Studien bis einschließlich Woche 48 waren 0,7 % der Tremfya-Injektionen sowie 0,3 % der Placebo-Injektionen mit Reaktionen an der Injektionsstelle assoziiert. Die Nebenwirkungen Erythem an der Injektionsstelle und Schmerzen an der Injektionsstelle waren alle von leichtem bis mäßigem Schweregrad, waren nicht schwerwiegend und hatten kein Absetzen von Tremfya zur Folge. Immunogenität Die Immunogenität von Tremfya wurde mit Hilfe eines sensitiven und wirkstofftoleranten Immunoassays untersucht. Bei den gepoolten Phase-II- und Phase-III-Analysen bildeten im Verlauf von bis zu 52 Behandlungswochen weniger als 6 % der mit Tremfya behandelten Patienten Anti-Drug-Antikörper. Von den Patienten, die Anti-Drug-Antikörper bildeten, wiesen ca. 7 % als neutralisierend klassifizierte Antikörper auf, was 0,4 % aller mit Tremfya behandelten Patienten entspricht. Anti-Drug-Antikörper waren nicht mit einer geringeren Wirksamkeit oder dem Auftreten von Reaktionen an der Injektionsstelle assoziiert.

6
Meldung des Verdachts auf Nebenwirkungen Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung über das in Anhang V aufgeführte nationale Meldesystem anzuzeigen. 4.9 Überdosierung In klinischen Studien wurde gesunden Freiwilligen einzelne Guselkumab-Dosen von bis zu 987 mg (10 mg/kg) intravenös und Patienten mit Plaque-Psoriasis einzelne Guselkumab-Dosen von bis zu 300 mg subkutan ohne Auftreten dosislimitierender Toxizität verabreicht. Im Falle einer Überdosierung ist der Patient auf Anzeichen oder Symptome von Nebenwirkungen zu überwachen und unverzüglich eine geeignete symptomatische Behandlung einzuleiten. 5. PHARMAKOLOGISCHE EIGENSCHAFTEN 5.1 Pharmakodynamische Eigenschaften Pharmakotherapeutische Gruppe: Immunsuppressiva, Interleukin-Inhibitoren; ATC-Code: Noch nicht zugewiesen. Wirkmechanismus Guselkumab ist ein humaner monoklonaler IgG1λ-Antikörper (mAk), der mit hoher Spezifität und Affinität selektiv an das Interleukin(IL)-23-Protein bindet. IL-23, ein regulatorisches Zytokin, beeinflusst die Differenzierung, Proliferation und das Überleben bestimmter T-Zellen (z. B. Th17-Zellen und Tc17-Zellen) sowie von unspezifischen lymphoiden Immunzellen (innate lymphoid cells - ILC), die proinflammatorische Effektor-Zytokine (z. B. IL-17A, IL-17F und IL-22) produzieren. Beim Menschen hat sich gezeigt, dass die selektive Blockierung von IL-23 die Produktion dieser Zytokine normalisiert. In der Haut von Patienten mit Plaque-Psoriasis sind die IL-23-Spiegel erhöht. In In-vitro-Modellen wurde gezeigt, dass Guselkumab die Bioaktivität von IL-23 inhibiert, indem es dessen Interaktion mit dem IL-23-Zelloberflächenrezeptor verhindert. Dadurch werden die IL-23 abhängigen Signal-, Aktivierungs- und Zytokinkaskaden unterbrochen. Die klinischen Wirkungen von Guselkumab bei Plaque-Psoriasis beruhen auf der Inhibierung des IL-23-Zytokin-Wegs. Pharmakodynamische Wirkungen In einer Phase-I-Studie führte die Behandlung mit Guselkumab zu einer reduzierten Expression von Genen des IL-23/Th17-Signalwegs und von Psoriasis-assoziierten Genexpressionsprofilen. Das ergaben Analysen der mRNA aus Hautläsionsbiopsien von Patienten mit Plaque-Psoriasis in Woche 12 im Vergleich zu Baseline. In derselben Phase-I-Studie führte die Behandlung mit Guselkumab zu einer Verbesserung histologischer Messgrößen der Psoriasis in Woche 12, u.a. zu einer Reduzierung der Epidermisdicke und der T-Zell-Dichte. Außerdem waren bei mit Guselkumab behandelten Patienten in Phase-II- und Phase-III-Studien reduzierte IL-17A-, IL-17F- und IL-22-Serumspiegel im Vergleich zu Placebo zu beobachten. Diese Ergebnisse decken sich mit dem beobachteten klinischen Nutzen der Guselkumab-Behandlung bei Plaque-Psoriasis.
Klinische Wirksamkeit und Sicherheit Die Wirksamkeit und Sicherheit von Guselkumab wurde in drei randomisierten, doppelblinden, aktiv-kontrollierten Phase-III-Studien an erwachsenen Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die für eine Phototherapie oder systemische Therapie in Frage kamen, beurteilt. VOYAGE 1 und VOYAGE 2 In zwei Studien (VOYAGE 1 und VOYAGE 2) wurde die Wirksamkeit und Sicherheit von Guselkumab vs. Placebo und Adalimumab an 1.829 erwachsenen Patienten untersucht. Patienten, die

7
bei der Randomisierung dem Guselkumabarm zugewiesen wurden (N = 825), erhielten 100 mg in den Wochen 0 und 4 sowie anschließend alle 8 Wochen (q8w) bis einschließlich Woche 48 (VOYAGE 1) bzw. Woche 20 (VOYAGE 2). Patienten, die bei der Randomisierung dem Adalimumab-Arm zugewiesen wurden (N = 582), erhielten 80 mg in Woche 0 und 40 mg in Woche 1, gefolgt von 40 mg alle 2 Wochen (q2w) bis einschließlich Woche 48 (VOYAGE 1) bzw. Woche 23 (VOYAGE 2). In beiden Studien erhielten Patienten, die bei der Randomisierung dem Placebo-Arm zugewiesen worden waren (N = 422), 100 mg Guselkumab in den Wochen 16 und 20 sowie anschließend alle 8 Wochen. In VOYAGE 2 wurden Patienten, die bei der Randomisierung in Woche 0 Guselkumab zugewiesen wurden und in Woche 28 eine 90 %ige Verbesserung im Psoriasis Area and Severity Index (PASI) im Vergleich zur Baseline aufwiesen (PASI-90-Responder), rerandomisiert und setzten entweder die Therapie mit Guselkumab q8w fort (Erhaltungstherapie) oder erhielten Placebo (Auslassversuch). PASI-90-Non-Responder aus der Adalimumab-Gruppe erhielten Guselkumab in Woche 28 und 32 sowie anschließend alle 8 Wochen. Alle Patienten wurden bis zu 48 Wochen nach der ersten Verabreichung der Studienmedikation beobachtet. Die Baseline-Krankheitsmerkmale stimmten bei den Studienpopulationen in VOYAGE 1 und 2 überein, mit einem medianen Anteil von 22 % bzw. 24 % der betroffenen Körperoberfläche (Body Surface Area = BSA), einem medianen Baseline-PASI von 19 bei beiden Studien, einem medianen Baseline-Dermatology Life Quality Index (DLQI) von 14 bzw. 14,5, einem Baseline-Investigator Global Assessment (IGA) Score von „schwer“ bei 25 % bzw. 23 % der Patienten sowie Psoriasis-Arthritis in der Vorgeschichte bei 19 % bzw. 18 % der Patienten. Von allen Patienten in VOYAGE 1 und 2 hatten 32 % bzw. 29 % zuvor weder eine konventionelle systemische noch eine Biologika-Therapie erhalten, 54 % bzw. 57 % hatten zuvor eine Phototherapie erhalten, und 62 % bzw. 64 % hatten zuvor eine konventionelle systemische Therapie erhalten. In beiden Studien hatten 21 % zuvor Biologika erhalten, darunter 11 % mindestens ein Anti-Tumornekrosefaktor-α(TNFα)-Biologikum und ca. 10 % ein Anti-IL-12/IL-23-Biologikum. Die Wirksamkeit von Guselkumab wurde im Hinblick auf die gesamte Hautbeteiligung, regionale Hautbeteiligung (Kopfhaut, Hände und Füße und Nägel) sowie Lebensqualität und von Patienten berichtete Ergebnisse (patient reported outcomes = PRO) untersucht. Die ko-primären Endpunkte in VOYAGE 1 und 2 waren die Anteile der Patienten, die einen IGA-Score von „erscheinungsfrei“ oder „nahezu erscheinungsfrei“ (IGA 0/1) sowie ein PASI-90-Ansprechen in Woche 16 vs. Placebo erreichten (siehe Tabelle 2). Gesamte Hautbeteiligung Die Behandlung mit Guselkumab führte zu signifikanten Verbesserungen der Krankheitsaktivität im Vergleich zu Placebo und Adalimumab in Woche 16 sowie im Vergleich zu Adalimumab in Woche 24 und in Woche 48. Die wichtigsten Wirksamkeitsergebnisse für die primären und die wesentlichen sekundären Endpunkte der Studien sind Tabelle 2 zu entnehmen.
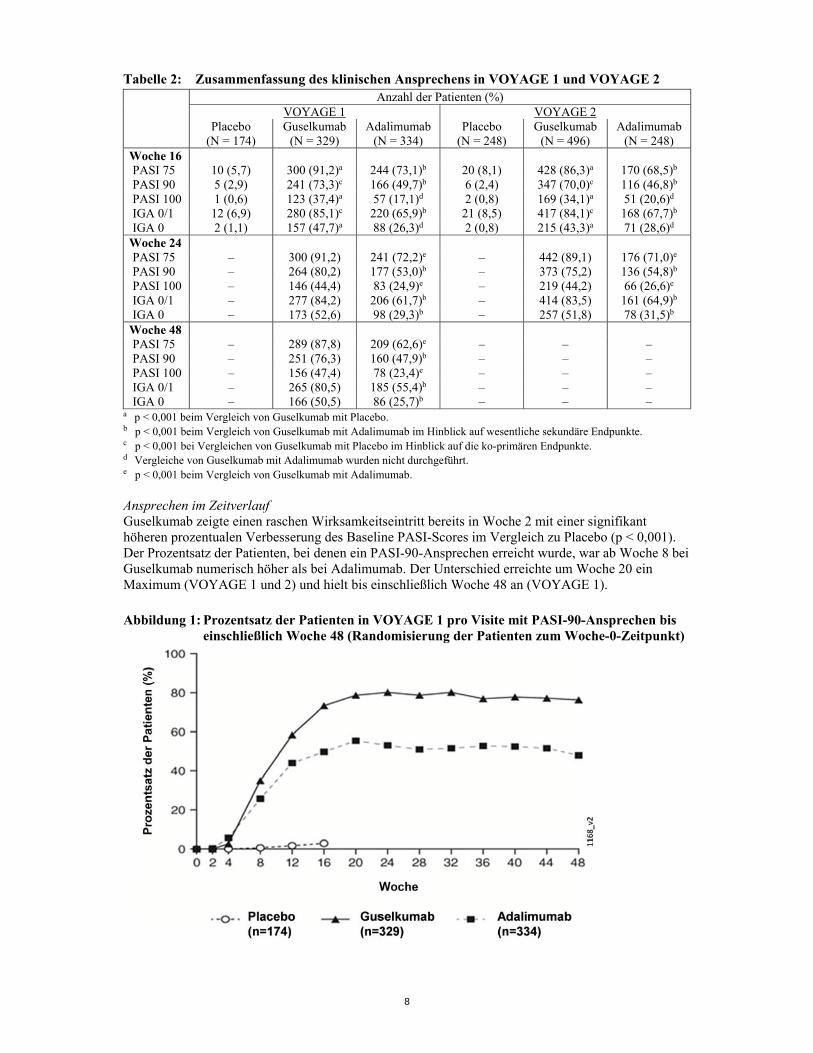
8
Tabelle 2: Zusammenfassung des klinischen Ansprechens in VOYAGE 1 und VOYAGE 2 Anzahl der Patienten (%) VOYAGE 1 VOYAGE 2
Placebo
(N = 174) Guselkumab
(N = 329)Adalimumab
(N = 334)Placebo
(N = 248)Guselkumab
(N = 496) Adalimumab
(N = 248)Woche 16 PASI 75 10 (5,7) 300 (91,2)a 244 (73,1)b 20 (8,1) 428 (86,3)a 170 (68,5)b PASI 90 5 (2,9) 241 (73,3)c 166 (49,7)b 6 (2,4) 347 (70,0)c 116 (46,8)b PASI 100 1 (0,6) 123 (37,4)a 57 (17,1)d 2 (0,8) 169 (34,1)a 51 (20,6)d IGA 0/1 12 (6,9) 280 (85,1)c 220 (65,9)b 21 (8,5) 417 (84,1)c 168 (67,7)b IGA 0 2 (1,1) 157 (47,7)a 88 (26,3)d 2 (0,8) 215 (43,3)a 71 (28,6)d
Woche 24 PASI 75 – 300 (91,2) 241 (72,2)e – 442 (89,1) 176 (71,0)e PASI 90 – 264 (80,2) 177 (53,0)b – 373 (75,2) 136 (54,8)b PASI 100 – 146 (44,4) 83 (24,9)e – 219 (44,2) 66 (26,6)e IGA 0/1 – 277 (84,2) 206 (61,7)b – 414 (83,5) 161 (64,9)b IGA 0 – 173 (52,6) 98 (29,3)b – 257 (51,8) 78 (31,5)b
Woche 48 PASI 75 – 289 (87,8) 209 (62,6)e – – – PASI 90 – 251 (76,3) 160 (47,9)b – – – PASI 100 – 156 (47,4) 78 (23,4)e – – – IGA 0/1 – 265 (80,5) 185 (55,4)b – – – IGA 0 – 166 (50,5) 86 (25,7)b – – –
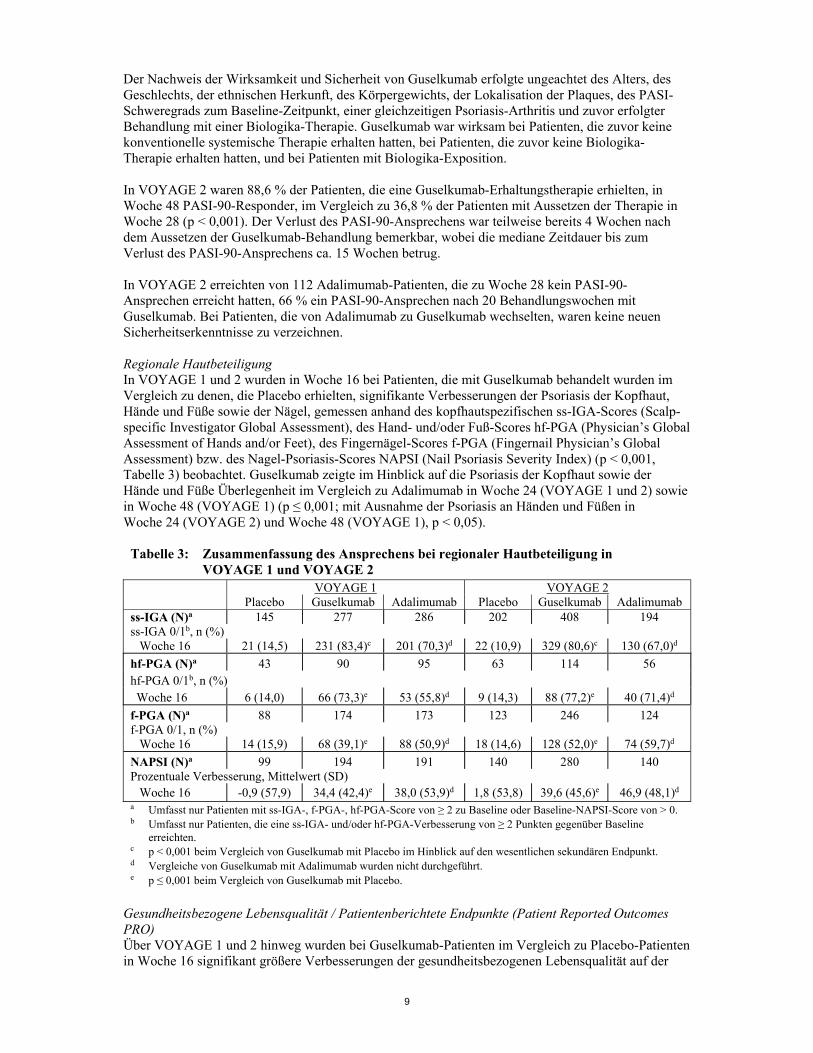
a p < 0,001 beim Vergleich von Guselkumab mit Placebo. b p < 0,001 beim Vergleich von Guselkumab mit Adalimumab im Hinblick auf wesentliche sekundäre Endpunkte. c p < 0,001 bei Vergleichen von Guselkumab mit Placebo im Hinblick auf die ko-primären Endpunkte. d Vergleiche von Guselkumab mit Adalimumab wurden nicht durchgeführt. e p < 0,001 beim Vergleich von Guselkumab mit Adalimumab. Ansprechen im Zeitverlauf Guselkumab zeigte einen raschen Wirksamkeitseintritt bereits in Woche 2 mit einer signifikant höheren prozentualen Verbesserung des Baseline PASI-Scores im Vergleich zu Placebo (p < 0,001). Der Prozentsatz der Patienten, bei denen ein PASI-90-Ansprechen erreicht wurde, war ab Woche 8 bei Guselkumab numerisch höher als bei Adalimumab. Der Unterschied erreichte um Woche 20 ein Maximum (VOYAGE 1 und 2) und hielt bis einschließlich Woche 48 an (VOYAGE 1).
Abbildung 1: Prozentsatz der Patienten in VOYAGE 1 pro Visite mit PASI-90-Ansprechen bis einschließlich Woche 48 (Randomisierung der Patienten zum Woche-0-Zeitpunkt)
9
Der Nachweis der Wirksamkeit und Sicherheit von Guselkumab erfolgte ungeachtet des Alters, des Geschlechts, der ethnischen Herkunft, des Körpergewichts, der Lokalisation der Plaques, des PASI-Schweregrads zum Baseline-Zeitpunkt, einer gleichzeitigen Psoriasis-Arthritis und zuvor erfolgter Behandlung mit einer Biologika-Therapie. Guselkumab war wirksam bei Patienten, die zuvor keine konventionelle systemische Therapie erhalten hatten, bei Patienten, die zuvor keine Biologika-Therapie erhalten hatten, und bei Patienten mit Biologika-Exposition. In VOYAGE 2 waren 88,6 % der Patienten, die eine Guselkumab-Erhaltungstherapie erhielten, in Woche 48 PASI-90-Responder, im Vergleich zu 36,8 % der Patienten mit Aussetzen der Therapie in Woche 28 (p < 0,001). Der Verlust des PASI-90-Ansprechens war teilweise bereits 4 Wochen nach dem Aussetzen der Guselkumab-Behandlung bemerkbar, wobei die mediane Zeitdauer bis zum Verlust des PASI-90-Ansprechens ca. 15 Wochen betrug. In VOYAGE 2 erreichten von 112 Adalimumab-Patienten, die zu Woche 28 kein PASI-90-Ansprechen erreicht hatten, 66 % ein PASI-90-Ansprechen nach 20 Behandlungswochen mit Guselkumab. Bei Patienten, die von Adalimumab zu Guselkumab wechselten, waren keine neuen Sicherheitserkenntnisse zu verzeichnen. Regionale Hautbeteiligung In VOYAGE 1 und 2 wurden in Woche 16 bei Patienten, die mit Guselkumab behandelt wurden im Vergleich zu denen, die Placebo erhielten, signifikante Verbesserungen der Psoriasis der Kopfhaut, Hände und Füße sowie der Nägel, gemessen anhand des kopfhautspezifischen ss-IGA-Scores (Scalp-specific Investigator Global Assessment), des Hand- und/oder Fuß-Scores hf-PGA (Physician’s Global Assessment of Hands and/or Feet), des Fingernägel-Scores f-PGA (Fingernail Physician’s Global Assessment) bzw. des Nagel-Psoriasis-Scores NAPSI (Nail Psoriasis Severity Index) (p < 0,001, Tabelle 3) beobachtet. Guselkumab zeigte im Hinblick auf die Psoriasis der Kopfhaut sowie der Hände und Füße Überlegenheit im Vergleich zu Adalimumab in Woche 24 (VOYAGE 1 und 2) sowie in Woche 48 (VOYAGE 1) (p ≤ 0,001; mit Ausnahme der Psoriasis an Händen und Füßen in Woche 24 (VOYAGE 2) und Woche 48 (VOYAGE 1), p < 0,05).
Tabelle 3: Zusammenfassung des Ansprechens bei regionaler Hautbeteiligung in VOYAGE 1 und VOYAGE 2
VOYAGE 1 VOYAGE 2 Placebo Guselkumab Adalimumab Placebo Guselkumab Adalimumab ss-IGA (N)a 145 277 286 202 408 194 ss-IGA 0/1b, n (%)
Woche 16 21 (14,5) 231 (83,4)c 201 (70,3)d 22 (10,9) 329 (80,6)c 130 (67,0)d
hf-PGA (N)a 43 90 95 63 114 56 hf-PGA 0/1b, n (%) Woche 16 6 (14,0) 66 (73,3)e 53 (55,8)d 9 (14,3) 88 (77,2)e 40 (71,4)d
f-PGA (N)a 88 174 173 123 246 124 f-PGA 0/1, n (%)
Woche 16 14 (15,9) 68 (39,1)e 88 (50,9)d 18 (14,6) 128 (52,0)e 74 (59,7)d
NAPSI (N)a 99 194 191 140 280 140 Prozentuale Verbesserung, Mittelwert (SD)
Woche 16 -0,9 (57,9) 34,4 (42,4)e 38,0 (53,9)d 1,8 (53,8) 39,6 (45,6)e 46,9 (48,1)d a Umfasst nur Patienten mit ss-IGA-, f-PGA-, hf-PGA-Score von ≥ 2 zu Baseline oder Baseline-NAPSI-Score von > 0. b Umfasst nur Patienten, die eine ss-IGA- und/oder hf-PGA-Verbesserung von ≥ 2 Punkten gegenüber Baseline
erreichten. c p < 0,001 beim Vergleich von Guselkumab mit Placebo im Hinblick auf den wesentlichen sekundären Endpunkt. d Vergleiche von Guselkumab mit Adalimumab wurden nicht durchgeführt. e p ≤ 0,001 beim Vergleich von Guselkumab mit Placebo.
Gesundheitsbezogene Lebensqualität / Patientenberichtete Endpunkte (Patient Reported Outcomes PRO) Über VOYAGE 1 und 2 hinweg wurden bei Guselkumab-Patienten im Vergleich zu Placebo-Patienten in Woche 16 signifikant größere Verbesserungen der gesundheitsbezogenen Lebensqualität auf der
10
Basis des Dermatologischen Lebensqualitäts-Index DLQI (Dermatology Life Quality Index) und der von Patienten berichteten Psoriasis-Symptome (Juckreiz, Schmerzen, Brennen, Stechen und Spannen der Haut) und -Anzeichen (Trockenheit, Rissbildung, Schuppenbildung, Abschuppung oder Abschälung der Haut, Rötung und Bluten der Haut) auf der Basis des Psoriasis-Symptom- und -Anzeichen-Tagebuchs PSSD (Psoriasis Symptoms and Signs Diary) beobachtet (Tabelle 4). Anzeichen von Verbesserungen bei den von Patienten berichteten Ergebnissen (PRO) hielten bis einschließlich Woche 24 (VOYAGE 1 und 2) und Woche 48 (VOYAGE 1) an. Tabelle 4: Zusammenfassung der Patientenberichteten Endpunkte (PRO) in VOYAGE 1
und VOYAGE 2 VOYAGE 1 VOYAGE 2
Placebo Guselkumab Adalimumab Placebo Guselkumab AdalimumabDLQI, Patienten mit Baseline-Score
170 322 328 248 495 247
Veränderung gegenüber Baseline, Mittelwert (Standardabweichung) Woche 16 -0,6 (6,4) -11,2 (7,2)c -9,3 (7,8)b -2,6 (6,9) -11,3 (6,8)c -9,7 (6,8)b
PSSD Symptom-Score, Patienten mit Baseline-Score > 0
129 248 273 198 410 200
Symptom-Score = 0, n (%) Woche 16 1 (0,8) 67 (27,0)a 45 (16,5)b 0 112 (27,3)a 30 (15,0)b
PSSD Anzeichen-Score, Patienten mit Baseline-Score > 0
129 248 274 198 411 201
Anzeichen-Score = 0, n (%) Woche 16 0 50 (20,2)a 32 (11,7)b 0 86 (20,9)a 21 (10,4)b
a p < 0,001 beim Vergleich von Guselkumab mit Placebo. b Vergleiche von Guselkumab mit Adalimumab wurden nicht durchgeführt. c p < 0,001 beim Vergleich von Guselkumab mit Placebo im Hinblick auf wesentliche sekundäre Endpunkte.
In VOYAGE 2 zeigten sich bei Patienten mit Guselkumab im Vergleich zu denen mit Placebo in Woche 16 signifikant größere Verbesserungen gegenüber Baseline bei gesundheitsbezogener Lebensqualität, Angst und Depression sowie Arbeitseinschränkung, gemessen anhand verschiedener Fragebögen (36-item Short Form, SF-36, health survey questionaire; Hospital Anxiety and Depression Scale, HADS; Work Limitations Questionnaire, WLQ). Die Verbesserungen im SF-36, HADS und WLQ hielten bei Patienten, die bei der Randomisierung in Woche 28 der Erhaltungstherapie zugewiesen wurden, alle bis einschließlich Woche 48 an. NAVIGATE Die NAVIGATE-Studie untersuchte die Wirksamkeit von Guselkumab bei Patienten mit unzureichendem Ansprechen auf Ustekinumab in Woche 16 (d. h. diejenigen, die kein „erscheinungsfrei“ oder „nahezu erscheinungsfrei“ erreichten, definiert als IGA ≥ 2). Alle Patienten (N = 871) erhielten unverblindet Ustekinumab (45 mg ≤ 100 kg und 90 mg > 100 kg) in Woche 0 und Woche 4. In Woche 16 wurden 268 Patienten mit einem IGA-Score von ≥ 2 mittels Randomisierung entweder der Fortsetzung der Behandlung mit Ustekinumab (N = 133) alle 12 Wochen oder der Einleitung der Behandlung mit Guselkumab (N = 135) in Woche 16, Woche 20 sowie anschließend alle 8 Wochen zugewiesen. Die Baseline-Merkmale der randomisierten Patienten ähnelten den in VOYAGE 1 und 2 beobachteten. Nach der Randomisierung war der primäre Endpunkt die Anzahl der Post-Randomisierungs-Visiten zwischen Woche 12 und 24, bei denen Patienten einen IGA-Score von 0/1 erreichten und eine Verbesserung um ≥ 2 Punkte aufwiesen. Die Patienten wurden in Abständen von vier Wochen untersucht, woraus sich insgesamt vier Visiten ergaben. Unter den Patienten mit unzureichendem Ansprechen auf Ustekinumab zum Randomisierungszeitpunkt war bei den Patienten, die zur Guselkumab-Behandlung wechselten, im Vergleich zu Patienten, welche die Ustekinumab-Behandlung fortsetzten, eine signifikant größere Verbesserung der Wirksamkeit zu beobachten. Zwischen 12 und 24 Wochen nach Randomisierung erreichten Guselkumab-Patienten zweimal so häufig wie Ustekinumab-Patienten einen IGA-Score von 0/1 mit einer Verbesserung um ≥ 2 Punkte (Mittelwerte 1,5 bzw. 0,7 Besuche, p < 0,001). Außerdem erreichte 12 Wochen nach Randomisierung

11
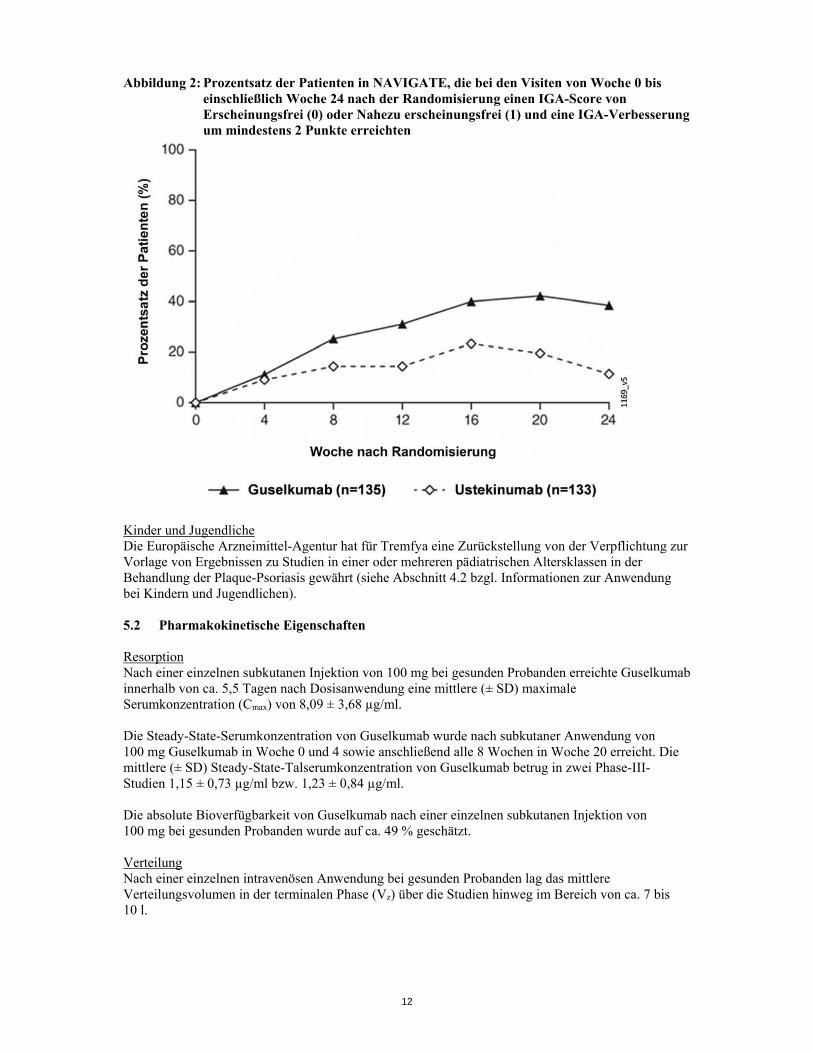
ein höherer Anteil der Guselkumab-Patienten ) im Vergleich zu Ustekinumab-Patienten einen IGA-Score von 0/1 mit einer Verbesserung um ≥ 2 Punkte (31,1 % gegenüber 14,3 %; p = 0,001) und ein PASI-90-Ansprechen (48 % gegenüber 23 %, p < 0,001. Unterschiede hinsichtlich der Ansprechrate zwischen mit Guselkumab und Ustekinumab behandelten Patienten waren bereits 4 Wochen nach Randomisierung bemerkbar (11,1 % bzw. 9,0 %) und erreichten 24 Wochen nach Randomisierung ein Maximum (siehe Abbildung 2). Bei Patienten, die von Ustekinumab zu Guselkumab wechselten, waren keine neuen Sicherheitserkenntnisse zu verzeichnen.
12
Abbildung 2: Prozentsatz der Patienten in NAVIGATE, die bei den Visiten von Woche 0 bis einschließlich Woche 24 nach der Randomisierung einen IGA-Score von Erscheinungsfrei (0) oder Nahezu erscheinungsfrei (1) und eine IGA-Verbesserung um mindestens 2 Punkte erreichten
Kinder und Jugendliche Die Europäische Arzneimittel-Agentur hat für Tremfya eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien in einer oder mehreren pädiatrischen Altersklassen in der Behandlung der Plaque-Psoriasis gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen). 5.2 Pharmakokinetische Eigenschaften Resorption Nach einer einzelnen subkutanen Injektion von 100 mg bei gesunden Probanden erreichte Guselkumab innerhalb von ca. 5,5 Tagen nach Dosisanwendung eine mittlere (± SD) maximale Serumkonzentration (Cmax) von 8,09 ± 3,68 µg/ml. Die Steady-State-Serumkonzentration von Guselkumab wurde nach subkutaner Anwendung von 100 mg Guselkumab in Woche 0 und 4 sowie anschließend alle 8 Wochen in Woche 20 erreicht. Die mittlere (± SD) Steady-State-Talserumkonzentration von Guselkumab betrug in zwei Phase-III-Studien 1,15 ± 0,73 µg/ml bzw. 1,23 ± 0,84 µg/ml. Die absolute Bioverfügbarkeit von Guselkumab nach einer einzelnen subkutanen Injektion von 100 mg bei gesunden Probanden wurde auf ca. 49 % geschätzt. Verteilung Nach einer einzelnen intravenösen Anwendung bei gesunden Probanden lag das mittlere Verteilungsvolumen in der terminalen Phase (Vz) über die Studien hinweg im Bereich von ca. 7 bis 10 l.

13
Biotransformation Der exakte Stoffwechselweg von Guselkumab wurde nicht charakterisiert. Es ist zu erwarten, dass Guselkumab als humaner IgG-mAk in der gleichen Weise wie endogenes IgG über katabolische Wege zu kleinen Peptiden und Aminosäuren abgebaut wird. Elimination Die mittlere systemische Clearance (CL) nach einer einzelnen intravenösen Anwendung bei gesunden Probanden lag über die Studien hinweg im Bereich von 0,288 bis 0,479 l/Tag. Die mittlere Halbwertszeit (T1/2) von Guselkumab betrug über die Studien hinweg bei gesunden Probanden ca. 17 Tage und bei Patienten mit Plaque-Psoriasis ca. 15 bis 18 Tage. Linearität/Nicht-Linearität Die systemische Guselkumab-Exposition (Cmax und AUC) nahm nach einer einzelnen subkutanen Injektion über einen Dosisbereich von 10 mg bis 300 mg hinweg bei gesunden Probanden und Patienten mit Plaque-Psoriasis in annähernd dosisproportionaler Weise zu. Ältere Patienten Es wurden keine spezifischen Studien mit älteren Patienten durchgeführt. Von den 1.384 Plaque-Psoriasis-Patienten mit Guselkumab-Exposition, die in die populationsspezifische Analyse der Pharmakokinetik eingingen, waren 70 Patienten im Alter von mindestens 65 Jahren, darunter 4 Patienten im Alter von mindestens 75 Jahren. Populationsspezifische Analysen der Pharmakokinetik zeigten, dass es bei Patienten im Alter von ≥ 65 Jahren im Vergleich zu Patienten im Alter von < 65 Jahren keine offensichtlichen Veränderungen des CL/F-Schätzwerts gab, was darauf hindeutet, dass keine Dosisanpassung für ältere Patienten erforderlich ist. Patienten mit Nieren- oder Leberfunktionsstörung Es wurde keine spezifische Studie zur Ermittlung der Auswirkungen von Nieren- oder Leberfunktionsstörungen auf die Pharmakokinetik von Guselkumab durchgeführt. Die Elimination von unverändertem Guselkumab, einem IgG-mAk, über die Nieren ist vermutlich gering und von geringer Bedeutung; desgleichen wird nicht erwartet, dass Leberfunktionsstörungen die Guselkumab-Clearance beeinflussen, da die Elimination von IgG-mAk hauptsächlich über den intrazellulären Katabolismus erfolgt. 5.3 Präklinische Daten zur Sicherheit Basierend auf den konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Reproduktions- sowie prä- und postnataler Entwicklungstoxizität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. In Studien zur Toxizität bei wiederholter Gabe an Javaneraffen war Guselkumab bei intravenöser und subkutaner Anwendung gut verträglich. Eine wöchentliche subkutane Dosis von 50 mg/kg bei Affen ergab Expositionswerte (AUC) und Cmax-Werte von mindestens dem 49-fachen bzw. > 200-fachen der im Rahmen der klinischen PK-Studie am Menschen gemessenen Werte. Außerdem wurden bei der Durchführung der Studien zur Toxizität bei wiederholter Gabe und in einer zielgerichteten pharmakologischen Studie zur Herz-Kreislauf-Sicherheit an Javaneraffen keine unerwünschten immuntoxischen Wirkungen oder pharmakologischen Wirkungen auf die Herz-Kreislauf-Sicherheit beobachtet. Bei histopathologischen Untersuchungen wurden keine präneoplastische Veränderungen beobachtet, weder bei Tieren, die bis zu 24 Wochen behandelt wurden noch nach einer anschließenden 12-wöchigen Erholungsphase, während der das Arzneimittel im Serum feststellbar war. Es wurden keine Studien zur Mutagenität oder zum kanzerogenen Potential von Guselkumab durchgeführt. Bei einer postnatalen Messung an Tag 28 war Guselkumab in der Muttermilch von Javaneraffen nicht nachweisbar.

14
6. PHARMAZEUTISCHE ANGABEN 6.1 Liste der sonstigen Bestandteile Histidin Histidinmonohydrochlorid-Monohydrat Polysorbat 80 Sucrose Wasser für Injektionszwecke 6.2 Inkompatibilitäten Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden. 6.3 Dauer der Haltbarkeit 2 Jahre 6.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung Im Kühlschrank lagern (2°C – 8°C). Nicht einfrieren. Die Fertigspritze im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. 6.5 Art und Inhalt des Behältnisses 1 ml Lösung in einer Fertigspritze aus Glas mit einer daran befestigten Nadel und einer Nadelhülle, versehen mit einem automatischem Nadelschutz. Guselkumab ist in einer Packung mit 1 Fertigspritze erhältlich. 6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur
Handhabung Nach der Entnahme von Tremfya aus dem Kühlschrank ist die Fertigspritze im Umkarton zu belassen und bis zur Injektion 30 Minuten zu warten, damit die Fertigspritze Raumtemperatur erreicht. Die Fertigspritze darf nicht geschüttelt werden. Vor Gebrauch der Fertigspritze empfiehlt es sich, diese visuell zu prüfen. Die Lösung sollte klar und farblos bis hellgelb sein und kann wenige kleine weiße oder durchsichtige Partikel enthalten. Tremfya darf nicht verwendet werden, wenn die Lösung trübe oder verfärbt ist oder große Partikel enthält. Jeder Packung Tremfya liegt eine separate Beilage mit dem Titel „Hinweise zur Anwendung“ bei, in welcher die Vorbereitung und Anwendung der Fertigspritze ausführlich beschrieben sind. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen. 7. INHABER DER ZULASSUNG Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgien

15
8. ZULASSUNGSNUMMER(N) EU/1/17/1234/001 9. DATUM DER ERTEILUNG DER ZULASSUNG/VERLÄNGERUNG DER
ZULASSUNG Datum der Erteilung der Zulassung: 10. STAND DER INFORMATION Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ verfügbar.

16
ANHANG II
A. HERSTELLER DES WIRKSTOFFS BIOLOGISCHEN
URSPRUNGS UND HERSTELLER, DER FÜR DIE CHARGENFREIGABE VERANTWORTLICH IST
B. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE
ABGABE UND DEN GEBRAUCH
C. SONSTIGE BEDINGUNGEN UND AUFLAGEN DER GENEHMIGUNG FÜR DAS INVERKEHRBRINGEN
D. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE
SICHERE UND WIRKSAME ANWENDUNG DES ARZNEIMITTELS

17
A. HERSTELLER DES WIRKSTOFFS BIOLOGISCHEN URSPRUNGS UND HERSTELLER, DER FÜR DIE CHARGENFREIGABE VERANTWORTLICH IST
Name und Anschrift der Hersteller des Wirkstoffs biologischen Ursprungs Biogen Inc. (BIIB) 5000 Davis Drive Research Triangle Park NC27709 USA Janssen Sciences Ireland UC Barnahely Ringaskiddy Co. Cork Irland Name und Anschrift des Herstellers, der für die Chargenfreigabe verantwortlich ist Janssen Biologics B.V. Einsteinweg 101 2333 CB Leiden Niederlande B. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE ABGABE UND DEN
GEBRAUCH Arzneimittel auf eingeschränkte ärztliche Verschreibung (siehe Anhang I: Zusammenfassung der Merkmale des Arzneimittels, Abschnitt 4.2). C. SONSTIGE BEDINGUNGEN UND AUFLAGEN DER GENEHMIGUNG FÜR DAS
INVERKEHRBRINGEN Regelmäßig aktualisierte Unbedenklichkeitsberichte Die Anforderungen an die Einreichung von regelmäßig aktualisierten Unbedenklichkeitsberichten für dieses Arzneimittel sind in der nach Artikel 107 c Absatz 7 der Richtlinie 2001/83/EG vorgesehenen und im europäischen Internetportal für Arzneimittel veröffentlichten Liste der in der Union festgelegten Stichtage (EURD-Liste) - und allen künftigen Aktualisierungen - festgelegt. Der Inhaber der Genehmigung für das Inverkehrbringen legt den ersten regelmäßig aktualisierten Unbedenklichkeitsbericht für dieses Arzneimittel innerhalb von 6 Monaten nach der Zulassung vor. D. BEDINGUNGEN ODER EINSCHRÄNKUNGEN FÜR DIE SICHERE UND
WIRKSAME ANWENDUNG DES ARZNEIMITTELS Risikomanagement-Plan (RMP)
Der Inhaber der Genehmigung für das Inverkehrbringen führt die notwendigen, im vereinbarten RMP beschriebenen und in Modul 1.8.2 der Zulassung dargelegten Pharmakovigilanzaktivitäten und Maßnahmen sowie alle künftigen vereinbarten Aktualisierungen des RMP durch. Ein aktualisierter RMP ist einzureichen:
nach Aufforderung durch die Europäische Arzneimittel-Agentur;

18
jedes Mal wenn das Risikomanagement-System geändert wird, insbesondere infolge neuer eingegangener Informationen, die zu einer wesentlichen Änderung des Nutzen-Risiko-Verhältnisses führen können oder infolge des Erreichens eines wichtigen Meilensteins (in Bezug auf Pharmakovigilanz oder Risikominimierung).

19
ANHANG III
ETIKETTIERUNG UND PACKUNGSBEILAGE

20
A. ETIKETTIERUNG

21
ANGABEN AUF DER ÄUSSEREN UMHÜLLUNG TEXT FÜR DEN UMKARTON DER FERTIGSPRITZE (100 mg) 1. BEZEICHNUNG DES ARZNEIMITTELS Tremfya 100 mg Injektionslösung Guselkumab 2. WIRKSTOFF(E) Jede Fertigspritze enthält 100 mg Guselkumab in 1 ml. 3. SONSTIGE BESTANDTEILE Sonstige Bestandteile: Histidin, Histidinmonohydrochlorid-Monohydrat, Polysorbat 80, Sucrose, Wasser für Injektionszwecke. 4. DARREICHUNGSFORM UND INHALT Injektionslösung 1 Fertigspritze 5. HINWEISE ZUR UND ART(EN) DER ANWENDUNG Nicht schütteln. Subkutane Anwendung. Packungsbeilage beachten. 6. WARNHINWEIS, DASS DAS ARZNEIMITTEL FÜR KINDER UNZUGÄNGLICH
AUFZUBEWAHREN IST Arzneimittel für Kinder unzugänglich aufbewahren. 7. WEITERE WARNHINWEISE, FALLS ERFORDERLICH 8. VERFALLDATUM verwendbar bis 9. BESONDERE VORSICHTSMASSNAHMEN FÜR DIE AUFBEWAHRUNG Im Kühlschrank lagern. Nicht einfrieren.

22
Die Fertigspritze im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. 10. GEGEBENENFALLS BESONDERE VORSICHTSMASSNAHMEN FÜR DIE
BESEITIGUNG VON NICHT VERWENDETEM ARZNEIMITTEL ODER DAVON STAMMENDEN ABFALLMATERIALIEN
11. NAME UND ANSCHRIFT DES PHARMAZEUTISCHEN UNTERNEHMERS Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgien 12. ZULASSUNGSNUMMER(N) EU/1/17/1234/001 13. CHARGENBEZEICHNUNG Ch.-B. 14. VERKAUFSABGRENZUNG 15. HINWEISE FÜR DEN GEBRAUCH 16. ANGABEN IN BLINDENSCHRIFT tremfya 100 mg 17. INDIVIDUELLES ERKENNUNGSMERKMAL – 2D-BARCODE 2D-Barcode mit individuellem Erkennungsmerkmal. 18. INDIVIDUELLES ERKENNUNGSMERKMAL – VOM MENSCHEN LESBARES
FORMAT PC: SN: NN:

23
MINDESTANGABEN AUF KLEINEN BEHÄLTNISSEN TEXT AUF DEM ETIKETT DER FERTIGSPRITZE (100 mg) 1. BEZEICHNUNG DES ARZNEIMITTELS SOWIE ART(EN) DER ANWENDUNG Tremfya 100 mg Injektion Guselkumab s.c. 2. HINWEISE ZUR ANWENDUNG 3. VERFALLDATUM EXP 4. CHARGENBEZEICHNUNG Lot 5. INHALT NACH GEWICHT, VOLUMEN ODER EINHEITEN 1 ml 6. WEITERE ANGABEN

24
B. PACKUNGSBEILAGE

25
Gebrauchsinformation: Information für Anwender
Tremfya 100 mg Injektionslösung in einer Fertigspritze Guselkumab
Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Sie können dabei helfen, indem Sie jede auftretende Nebenwirkung melden. Hinweise zur Meldung von Nebenwirkungen, siehe Ende Abschnitt 4. Lesen Sie die gesamte Packungsbeilage sorgfältig durch, bevor Sie mit der Anwendung dieses Arzneimittels beginnen, denn sie enthält wichtige Informationen. - Heben Sie die Packungsbeilage auf. Vielleicht möchten Sie diese später nochmals lesen. - Wenn Sie weitere Fragen haben, wenden Sie sich an Ihren Arzt, Apotheker oder das
medizinische Fachpersonal. - Dieses Arzneimittel wurde Ihnen persönlich verschrieben. Geben Sie es nicht an Dritte weiter.
Es kann anderen Menschen schaden, auch wenn diese die gleichen Beschwerden haben wie Sie. - Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker oder das
medizinische Fachpersonal. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Siehe Abschnitt 4.
Was in dieser Packungsbeilage steht 1. Was ist Tremfya und wofür wird es angewendet? 2. Was sollten Sie vor der Anwendung von Tremfya beachten? 3. Wie ist Tremfya anzuwenden? 4. Welche Nebenwirkungen sind möglich? 5. Wie ist Tremfya aufzubewahren? 6. Inhalt der Packung und weitere Informationen 1. Was ist Tremfya und wofür wird es angewendet? Tremfya enthält den Wirkstoff Guselkumab, bei dem es sich um einen als „monoklonaler Antikörper“ bezeichneten Proteintyp handelt. Dieses Arzneimittel wirkt, indem es die Aktivität eines Proteins namens IL-23, das bei Personen mit Psoriasis in erhöhter Konzentration vorliegt, blockiert. Tremfya wird für die Behandlung von Erwachsenen mit mittelschwerer bis schwerer Plaque-Psoriasis verwendet, einer entzündlichen Erkrankung, die sich auf die Haut und die Nägel auswirkt. Tremfya kann den Zustand der Haut und das Aussehen der Nägel verbessern und Symptome, wie Schuppenbildung, Abschuppung, Abschälung, Juckreiz, Schmerzen und Brennen der Haut, reduzieren. 2. Was sollten Sie vor der Anwendung von Tremfya beachten? Tremfya darf nicht angewendet werden, wenn Sie allergisch gegen Guselkumab oder einen der in Abschnitt 6. genannten sonstigen
Bestandteile dieses Arzneimittels sind. Wenn Sie vermuten, allergisch zu sein, fragen Sie vor der Anwendung von Tremfya Ihren Arzt um Rat.
wenn Sie eine aktive Infektion, einschließlich einer aktiven Tuberkulose, haben.

26
Warnhinweise und Vorsichtsmaßnahmen Bitte sprechen Sie mit Ihrem Arzt, Apotheker oder dem medizinischen Fachpersonal, bevor Sie Tremfya anwenden, wenn bei Ihnen eine Infektion behandelt wird. wenn Sie eine anhaltende oder immer wiederkehrende Infektion haben. wenn Sie Tuberkulose haben oder engen Kontakt mit jemandem hatten, der Tuberkulose hat. wenn Sie glauben, dass Sie eine Infektion haben oder Symptome einer Infektion aufweisen
(siehe unten unter „Achten Sie auf Infektionen und allergische Reaktionen“). wenn Sie kürzlich geimpft wurden oder wenn bei Ihnen während der Behandlung mit Tremfya
eine Impfung fällig wird. Wenn Sie nicht sicher sind, ob einer der oben genannten Punkte auf Sie zutrifft, sprechen Sie mit Ihrem Arzt, Apotheker oder dem medizinischen Fachpersonal, bevor Sie Tremfya anwenden. Achten Sie auf Infektionen und allergische Reaktionen Durch Tremfya können Ihre Abwehrkräfte gegen Infektionen verringert und deshalb Ihre Infektionsanfälligkeit und das Risiko für allergische Reaktionen erhöht sein. Informieren Sie Ihren Arzt oder suchen Sie sofort einen Arzt auf, wenn Sie Anzeichen einer Infektion oder einer allergischen Reaktion bemerken, während Sie Tremfya anwenden. Im Folgenden sind derartige Anzeichen aufgeführt. Infektionen - Fieber oder grippeähnliche Symptome - blutiger Auswurf (Schleim) - Muskelschmerzen - Gewichtsverlust - Husten - Durchfall oder Magenschmerzen - Kurzatmigkeit - warme, rote oder schmerzende Haut
oder Wunden am Körper, bei denen es sich nicht um Ihre Psoriasis handelt
- Brennen beim Wasserlassen oder häufigeres Wasserlassen als gewöhnlich
Allergische Reaktionen - Atem- oder Schluckbeschwerden - Schwellung von Gesicht, Lippen, Zunge oder Rachen - starkes Hautjucken mit rotem Ausschlag oder erhabenen Beulen. Kinder und Jugendliche Tremfya wird für Kinder und Jugendliche unter 18 Jahren nicht empfohlen, da es in dieser Altersgruppe nicht untersucht wurde. Anwendung von Tremfya zusammen mit anderen Arzneimitteln Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel anwenden, kürzlich andere Arzneimittel angewendet haben oder
beabsichtigen, andere Arzneimittel anzuwenden. wenn Sie kürzlich geimpft wurden oder bei Ihnen eine Impfung fällig ist. Während der
Anwendung von Tremfya dürfen Sie bestimmte Arten von Impfstoffen (Lebendimpfstoffe) nicht erhalten.
Schwangerschaft und Stillzeit Tremfya darf während der Schwangerschaft nicht angewendet werden. Die Wirkung dieses
Arzneimittels bei Schwangeren ist nicht bekannt. Wenn Sie eine Frau im gebärfähigen Alter sind, wird Ihnen geraten, eine Schwangerschaft zu vermeiden, und Sie müssen während der Anwendung von Tremfya sowie für den Zeitraum von mindestens 12 Wochen nach der letzten Dosis von Tremfya eine zuverlässige Verhütungsmethode anwenden. Sprechen Sie mit Ihrem Arzt, wenn Sie schwanger sind oder wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden.
Sprechen Sie mit Ihrem Arzt, wenn Sie stillen oder vorhaben, zu stillen. Sie und Ihr Arzt müssen zwischen dem Stillen und der Anwendung von Tremfya entscheiden.

27
Verkehrstüchtigkeit und Fähigkeit zum Bedienen von Maschinen Es ist unwahrscheinlich, dass Tremfya einen Einfluss auf Ihre Verkehrstüchtigkeit und Ihre Fähigkeit zum Bedienen von Maschinen hat. 3. Wie ist Tremfya anzuwenden? Wenden Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt, Apotheker oder dem medizinischen Fachpersonal an. Fragen Sie bei Ihrem Arzt, Apotheker oder dem medizinischen Fachpersonal nach, wenn Sie sich nicht sicher sind. Menge und Dauer der Anwendung von Tremfya Ihr Arzt wird entscheiden, wie lange Sie Tremfya benötigen. Die Dosis beträgt 100 mg (der Inhalt von 1 Fertigspritze) und wird unter die Haut injiziert
(subkutane Injektion). Diese Injektion erfolgt möglicherweise durch Ihren Arzt oder das medizinische Fachpersonal.
Nach der ersten Dosis werden Sie die nächste Dosis 4 Wochen später erhalten und danach alle 8 Wochen.
Zu Beginn wird Ihr Arzt oder das medizinische Fachpersonal Tremfya injizieren. Sie können jedoch auch zusammen mit Ihrem Arzt entscheiden, sich Tremfya selbst zu injizieren. In diesem Fall werden Sie ausreichend darin geschult, wie Sie sich Tremfya richtig injizieren müssen. Sprechen Sie mit Ihrem Arzt oder dem medizinischen Fachpersonal, wenn Sie Fragen dazu haben, wie Sie sich selbst eine Injektion verabreichen sollen. Es ist wichtig, dass Sie erst von Ihrem Arzt oder vom medizinischen Fachpersonal geschult wurden, bevor Sie versuchen, sich die Injektion selbst zu verabreichen. Vor der Anwendung von Tremfya lesen Sie bitte sorgfältig die ausführliche Anleitung „Hinweise zur Anwendung“, die dem Karton separat beiliegt. Wenn Sie eine größere Menge von Tremfya angewendet haben, als Sie sollten Wenn Sie mehr Tremfya erhalten haben, als Sie sollten, oder wenn die Dosis früher als verordnet angewendet wurde, informieren Sie Ihren Arzt. Wenn Sie die Anwendung von Tremfya vergessen haben Wenn Sie vergessen haben, eine Dosis Tremfya zu injizieren, informieren Sie Ihren Arzt. Wenn Sie die Anwendung von Tremfya abbrechen Sie sollten die Anwendung von Tremfya nicht abbrechen, ohne vorher mit Ihrem Arzt gesprochen zu haben. Wenn Sie die Behandlung abbrechen, können die Psoriasis-Symptome wieder auftreten. 4. Welche Nebenwirkungen sind möglich? Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen. Die folgenden Nebenwirkungen sind alle leicht bis mittelschwer. Wenn sich eine dieser Nebenwirkungen zu einer schweren Nebenwirkung entwickelt, informieren Sie sofort Ihren Arzt, Apotheker oder das medizinische Fachpersonal. Manche Nebenwirkungen sind sehr häufig (kann mehr als 1 von 10 Behandelten betreffen): - Infektionen der oberen Atemwege Manche Nebenwirkungen sind häufig (kann bis zu 1 von 10 Behandelten betreffen): - Kopfschmerzen

28
- Gelenkschmerzen (Arthralgie) - Durchfall - Magen-Darm-Grippe (Gastroenteritis) - Rötung an der Injektionsstelle - Nesselausschlag - Pilzinfektion der Haut, beispielsweise zwischen den Zehen (z. B. Fußpilz) - Herpes-simplex-Infektionen Manche Nebenwirkungen treten gelegentlich auf (kann bis zu 1 von 100 Behandelten betreffen): - Schmerzen an der Injektionsstelle Meldung von Nebenwirkungen Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker oder das medizinische Fachpersonal. Dies gilt auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem* anzeigen. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden. 5. Wie ist Tremfya aufzubewahren? Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf. Sie dürfen dieses Arzneimittel nach dem auf dem Spritzenetikett nach „EXP“ und auf dem Umkarton nach „verwendbar bis“ angegebenen Verfalldatum nicht mehr verwenden. Das Verfalldatum bezieht sich auf den letzten Tag des angegebenen Monats. Die Fertigspritze im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Im Kühlschrank lagern (2°C – 8°C). Nicht einfrieren. Nicht schütteln. Sie dürfen dieses Arzneimittel nicht verwenden, wenn Sie bemerken, dass das Arzneimittel trübe oder verfärbt ist oder große Teilchen enthält. Nehmen Sie den Umkarton vor Gebrauch aus dem Kühlschrank, belassen Sie die Fertigspritze im Umkarton und lassen Sie sie Zimmertemperatur erreichen, indem Sie 30 Minuten warten. Dieses Arzneimittel ist nur für die einmalige Anwendung bestimmt. Entsorgen Sie Arzneimittel nicht im Abwasser oder Haushaltsabfall. Fragen Sie Ihren Apotheker, wie das Arzneimittel zu entsorgen ist, wenn Sie es nicht mehr verwenden. Sie tragen damit zum Schutz der Umwelt bei. 6. Inhalt der Packung und weitere Informationen Was Tremfya enthält - Der Wirkstoff ist: Guselkumab. Jede Fertigspritze enthält 100 mg Guselkumab in 1 ml Lösung. - Die sonstige Bestandteile sind: Histidin, Histidinmonohydrochlorid-Monohydrat, Polysorbat 80,
Sucrose und Wasser für Injektionszwecke. Wie Tremfya aussieht und Inhalt der Packung Injektionslösung (Injektion). Tremfya ist eine klare, farblose bis hellgelbe Lösung. Sie wird in einem Umkarton geliefert, der eine 1-ml-Fertigspritze aus Glas enthält.

29
Pharmazeutischer Unternehmer Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgien Hersteller Janssen Biologics B.V. Einsteinweg 101 NL-2333 CB Leiden Niederlande Falls Sie weitere Informationen über das Arzneimittel wünschen, setzen Sie sich bitte mit dem örtlichen Vertreter des pharmazeutischen Unternehmers in Verbindung. België/Belgique/Belgien Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Tel/Tél: +32 14 64 94 11
Lietuva UAB "JOHNSON & JOHNSON" Geležinio Vilko g. 18A LT-08104 Vilnius Tel: +370 5 278 68 88
България „Джонсън & Джонсън България” ЕООД ж.к. Младост 4 Бизнес Парк София, сграда 4 София 1766 Тел.: +359 2 489 94 00
Luxembourg/Luxemburg Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Belgique/Belgien Tél/Tel: +32 14 64 94 11
Česká republika Janssen-Cilag s.r.o. Karla Engliše 3201/06 CZ-150 00 Praha 5 - Smíchov Tel.: +420 227 012 227
Magyarország Janssen-Cilag Kft. Nagyenyed u. 8-14 H-Budapest, 1123 Tel.: +36 1 884 2858
Danmark Janssen-Cilag A/S Bregnerødvej 133 DK-3460 Birkerød Tlf: +45 45 94 82 82
Malta AM MANGION LTD. Mangion Building, Triq Ġdida fi Triq Valletta MT-Ħal-Luqa LQA 6000 Tel: +356 2397 6000
Deutschland Janssen-Cilag GmbH Johnson & Johnson Platz 1 D-41470 Neuss Tel: +49 2137 955 955
Nederland Janssen-Cilag B.V. Graaf Engelbertlaan 75 NL-4837 DS Breda Tel: +31 76 711 1111
Eesti UAB "JOHNSON & JOHNSON" Eesti filiaal Lõõtsa 2 EE-11415 Tallinn Tel: +372 617 7410
Norge Janssen-Cilag AS Postboks 144 NO-1325-Lysaker Tlf: +47 24 12 65 00

30
Ελλάδα Janssen-Cilag Φαρμακευτική Α.Ε.Β.Ε. Λεωφόρος Ειρήνης 56 GR-151 21 Πεύκη, Αθήνα Tηλ: +30 210 80 90 000
Österreich Janssen-Cilag Pharma GmbH Vorgartenstraße 206B A-1020 Wien Tel: +43 1 610 300
España Janssen-Cilag, S.A. Paseo de las Doce Estrellas, 5-7 E-28042 Madrid Tel: +34 91 722 81 00
Polska Janssen-Cilag Polska Sp. z o.o. ul. Iłżecka 24 PL-02-135 Warszawa Tel.: +48 22 237 60 00
France Janssen-Cilag 1, rue Camille Desmoulins, TSA 91003 F-92787 Issy Les Moulineaux, Cedex 9 Tél: 0 800 25 50 75 / +33 1 55 00 40 03
Portugal Janssen-Cilag Farmacêutica, Lda. Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835
Hrvatska Johnson & Johnson S.E. d.o.o. Oreškovićeva 6h 10010 Zagreb Tel: +385 1 6610 700
România Johnson & Johnson România SRL Str. Tipografilor nr. 11-15 Clădirea S-Park, Corp B3-B4, Etaj 3 013714 Bucureşti, ROMÂNIA Tel: +40 21 207 1800
Ireland Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG United Kingdom Tel: +44 1 494 567 444
Slovenija Johnson & Johnson d.o.o. Šmartinska cesta 53 SI-1000 Ljubljana Tel: +386 1 401 18 30
Ísland Janssen-Cilag AB c/o Vistor hf. Hörgatúni 2 IS-210 Garðabær Sími: +354 535 7000
Slovenská republika Johnson & Johnson, s.r.o. CBC III, Karadžičova 12 SK-821 08 Bratislava Tel: +421 232 408 400
Italia Janssen-Cilag SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI Tel: +39 02 2510 1
Suomi/Finland Janssen-Cilag Oy Vaisalantie/Vaisalavägen 2 FI-02130 Espoo/Esbo Puh/Tel: +358 207 531 300
Κύπρος Βαρνάβας Χατζηπαναγής Λτδ Λεωφόρος Γιάννου Κρανιδιώτη 226 Λατσιά CY-2234 Λευκωσία Τηλ: +357 22 207 700
Sverige Janssen-Cilag AB Box 4042 SE-16904 Solna Tel: +46 8 626 50 00

31
Latvija UAB "JOHNSON & JOHNSON" filiāle Latvijā Mūkusalas iela 101 Rīga, LV-1004 Tel: +371 678 93561
United Kingdom Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG - UK Tel: +44 1 494 567 444
Diese Packungsbeilage wurde zuletzt überarbeitet im {MM.JJJJ}. Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur http://www.ema.europa.eu/ verfügbar.

32
Hinweise zur Anwendung Tremfya
Fertigspritze
Wichtig Wenn Ihr Arzt entscheidet, dass Sie sich selbst oder eine Pflegeperson Ihnen die Tremfya-Injektionen zuhause geben können, sollten Sie vor der ersten Injektion über die richtige Art und Weise der Vorbereitung und Injektion der Tremfya-Fertigspritze geschult werden. Bitte lesen Sie diese Hinweise vor der ersten Anwendung der Tremfya-Fertigspritze sowie bei jeder neuen Anwendung. Eventuell liegen neue Informationen vor. Diese Anleitung ersetzt nicht das Gespräch mit Ihrem Arzt über Ihren Gesundheitszustand oder Ihre Behandlung. Bitte lesen Sie vor dem Beginn Ihrer Injektion auch die Packungsbeilage sorgfältig durch und besprechen Sie alle eventuellen Fragen mit Ihrem Arzt oder dem medizinischen Fachpersonal. Die Tremfya-Fertigspritze ist für Injektionen unter die Haut und nicht in Muskeln oder Venen vorgesehen. Nach der Injektion wird die Nadel in den Spritzenkörper eingezogen und rastet dort ein.
Angaben zur Lagerung Im Kühlschrank lagern (2°C – 8°C). Nicht einfrieren. Tremfya und alle Arzneimittel für Kinder unzugänglich aufbewahren. Die Fertigspritze niemals schütteln.
Einmalige Anwendung

33
Die Fertigspritze auf einen Blick Vor der Injektion
Kolben Halten Sie niemals den Kolben fest oder ziehen Sie niemals daran.
Sicherheitsschutz
Fingerauflage Spritzenkörper Fassen Sie den Spritzenkörper unter der Fingerauflage an. Sichtfenster
Nadelhülle Entfernen Sie die Nadelhülle erst, wenn Sie für die Tremfya-Injektion bereit sind (siehe Schritt 2).

34
Nach der Injektion
Sie benötigen folgende Materialien: • 1 Alkoholtupfer • 1 Wattebausch oder Mulltupfer
• 1 Heftpflaster • 1 durchstichsicheren Behälter (siehe Schritt 3)
Kolben rastet ein.
Der Sicherheitsschutz wird aktiviert.
Die Nadel zieht sich in den Spritzenkörper.

35
Überprüfen Sie den Umkarton Nehmen Sie den Umkarton mit der Fertigspritze aus dem Kühlschrank. Belassen Sie die Fertigspritze im Umkarton und lassen Sie diesen vor Gebrauch auf einer ebenen Fläche für mindestens 30 Minuten bei Raumtemperatur stehen. Erwärmen Sie die Fertigspritze nicht auf andere Weise. Überprüfen Sie das Verfalldatum („verwendbar bis“) auf dem Umkarton. Verwenden Sie das Arzneimittel nicht mehr nach Ablauf des Verfalldatums. Verwenden Sie das Arzneimittel nicht, wenn die Perforationen des Umkartons beschädigt sind. Fragen Sie bei Ihrem Arzt oder Apotheker nach einem Ersatz.
Injektionsstelle wählen Für die Injektion stehen die folgenden Bereiche zur Auswahl: • Vorderseite der Oberschenkel (empfohlen) • Unterer Bauch Nicht im Umkreis von 5 Zentimetern um den Bauchnabel injizieren. • Rückseite der Oberarme (wenn eine Pflegeperson die Injektion vornimmt). Nicht an Stellen injizieren, an denen die Haut empfindlich, rot, schuppig oder hart ist oder Blutergüsse aufweist. Nicht in Bereichen injizieren, die Narben oder Dehnungsstreifen aufweisen.
1. Bereiten Sie sich für Ihre Injektion vor
30 MIN.

36
Reinigen Sie die Injektionsstelle Waschen Sie die Hände sorgfältig mit Seife und warmem Wasser. Wischen Sie die gewählte Injektionsstelle mit einem Alkoholtupfer ab und lassen Sie diese trocknen. Berühren Sie die gereinigte Injektionsstelle nicht, fächeln Sie keine Luft darauf und pusten Sie auch nicht darauf.
Überprüfen Sie die Flüssigkeit Entnehmen Sie die Fertigspritze aus dem Umkarton. Überprüfen Sie die Flüssigkeit im Sichtfenster. Sie muss klar bis leicht gelblich sein und kann winzige weiße oder durchsichtige Teilchen enthalten. Außerdem können eine oder mehrere Luftblasen sichtbar sein. Das ist normal. Nehmen Sie keine Injektion vor, wenn die Flüssigkeit trübe oder verfärbt ist oder große Teilchen aufweist. Wenn Sie sich nicht sicher sind, fragen Sie bei Ihrem Arzt oder Apotheker nach einem Ersatz.

37
Nadelhülle entfernen Fassen Sie die Spritze am Spritzenkörper und ziehen Sie die Nadelhülle gerade ab. Das Erscheinen eines Flüssigkeitstropfens ist normal. Injizieren Sie innerhalb von 5 Minuten nach dem Entfernen der Nadelhülle. Setzen Sie die Nadelhülle nicht wieder auf, da dies die Nadel beschädigen kann. Berühren Sie nicht die Nadel und berühren Sie mit der Nadel auch keine Oberflächen. Verwenden Sie die Tremfya-Fertigspritze nicht, wenn sie fallen gelassen wurde. Fragen Sie bei Ihrem Arzt oder Apotheker nach einem Ersatz.
Positionieren Sie Ihre Finger und führen Sie die Nadel ein Platzieren Sie Daumen, Zeige- und Mittelfinger direkt unter der Fingerauflage, wie dargestellt. Berühren Sie nicht den Kolben und den Bereich oberhalb der Fingerauflage, da dadurch der Nadelschutz aktiviert werden kann. Erfassen Sie mit der anderen Hand eine Hautfalte an der Injektionsstelle. Positionieren Sie die Spritze in einem Winkel von ungefähr 45 Grad auf der Haut. Es ist wichtig, dass Sie eine so große Hautfalte erfassen, dass die Injektion unter die Haut und nicht in den Muskel verabreicht wird. Stechen Sie die Nadel mit einer raschen, pfeilwurfähnlichen Bewegung ein.
2. Nehmen Sie die Injektion von Tremfya mit Hilfe der Fertigspritze vor
38
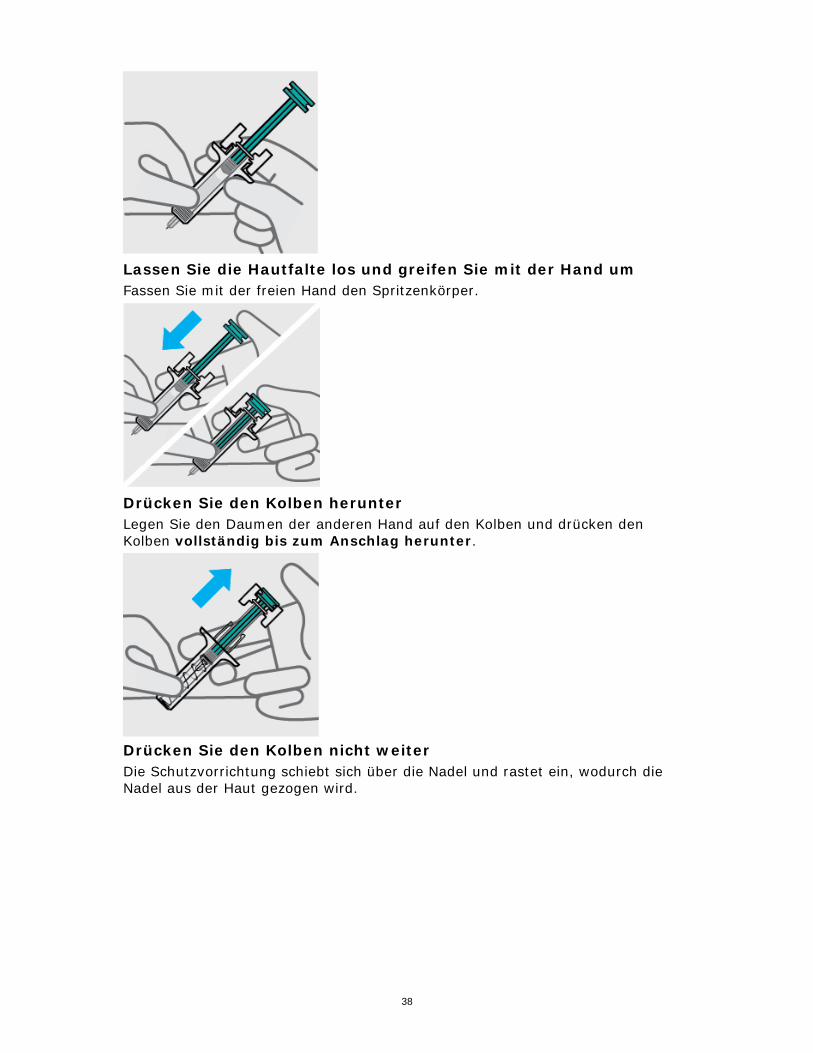
Lassen Sie die Hautfalte los und greifen Sie mit der Hand um Fassen Sie mit der freien Hand den Spritzenkörper.
Drücken Sie den Kolben herunter Legen Sie den Daumen der anderen Hand auf den Kolben und drücken den Kolben vollständig bis zum Anschlag herunter.
Drücken Sie den Kolben nicht weiter Die Schutzvorrichtung schiebt sich über die Nadel und rastet ein, wodurch die Nadel aus der Haut gezogen wird.

39
Entsorgen Sie die gebrauchte Fertigspritze Entsorgen Sie die gebrauchte Fertigspritze unmittelbar nach Gebrauch in einen durchstichsicheren Behälter. Stellen Sie sicher, dass der volle Abfallbehälter gemäß den Anweisungen des behandelnden Arztes oder des medizinischen Fachpersonals entsorgt wird.
Überprüfen Sie die Injektionsstelle An der Injektionsstelle kann eine geringe Menge Blut oder Flüssigkeit sichtbar sein. Üben Sie mit einem Wattebausch oder Mulltupfer Druck auf die Haut aus, bis eine etwaige Blutung vollständig gestillt ist. Reiben Sie nicht an der Injektionsstelle. Versehen Sie die Injektionsstelle bei Bedarf mit einem Pflaster. Damit ist die Injektion abgeschlossen!
Benötigen Sie Hilfe? Wenden Sie sich mit etwaigen Fragen an Ihren Arzt. Wenn Sie weitere Unterstützung benötigen oder eine Rückmeldung geben möchten, verwenden Sie bitte die Kontaktdaten Ihres zuständigen örtlichen Vertreters in der Packungsbeilage oder besuchen Sie www.janssen.com.
3. Nach Ihrer Injektion





























