Embed Size (px)
Citation preview

AMINOÁCIDOS E PROTEÍNASAs proteínas são as macromoléculas mais abundantes nas células vivas. Elas ocorrem em todas as células e em todas as partes das mesmas. As proteínas também ocorrem em grande variedade; milhares de diferentes tipos podem ser encontrados em uma única célula. Ainda mais, as proteínas também exibem uma grande diversidade de funções biológicas. Em um sentido, elas são os instrumentos moleculares através dos quais a informação genética é expressa. As proteínas são cadeias de aminoácidos, cada aminoácido está unido a seus vizinhos por um tipo específico de ligação covalente. O que é mais notável é que as células podem produzir proteínas que têm propriedades e atividades extremamente diferentes, pela reunião dos mesmos 20 aminoácidos em muitas combinações e seqüências diversas. A partir destes blocos de construção distintos, os organismos podem sintetizar produtos largamente diferentes entre si, como as enzimas, hormônios, anticorpos, as proteínas do cristalino do olho, penas de pássaros, teias de aranha, chifres de rinoceronte, proteínas do leite, antibióticos, venenos de fungos e uma grande quantidade de outras substâncias, cada uma delas exibindo atividades biológicas características.

I. AMINOÁCIDOS
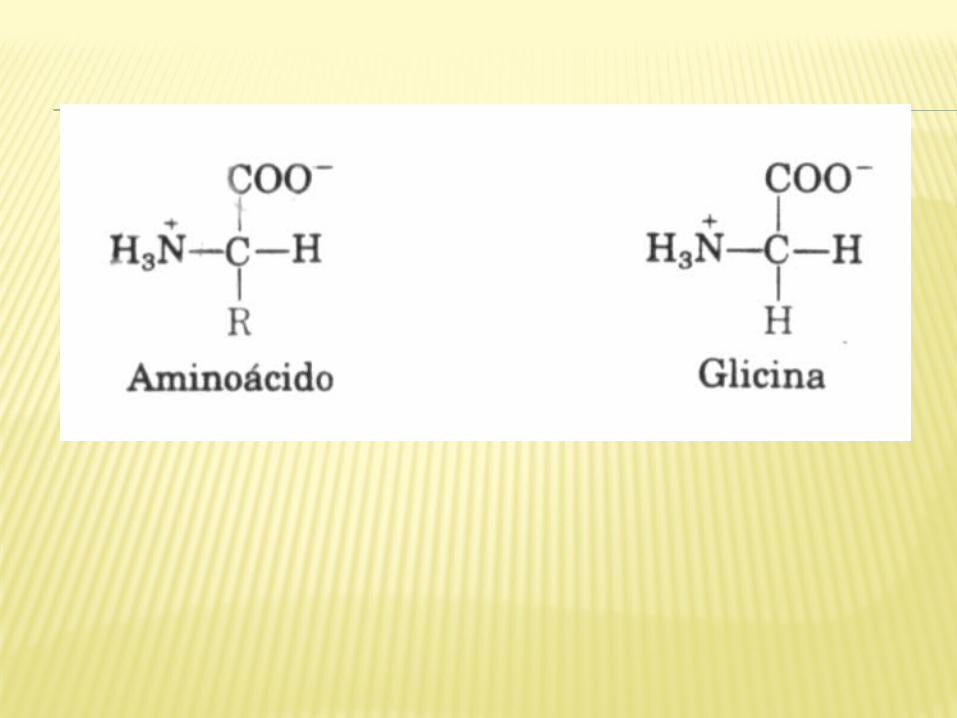
Os aminoácidos têm características estruturais comuns. Todos os 20 aminoácidos encontrados nas proteínas têm um grupo carboxila e um grupo amino ligados ao mesmo átomo de carbono (o carbono α).
Eles diferem um dos outros através de suas cadeias laterais ou grupos R, os quais variam em estrutura, tamanho e carga, e influenciam a solubilidade do aminoácido em água. Os aminoácidos em soluções aquosas estão ionizados e podem agir como ácidos ou bases.
Aproximadamente 300 aminoácidos adicionais foram encontrados nas células e têm uma grande variedade de funções, mas eles nunca aparecem em proteínas. A ornitina e a citrulina merecem uma nota especial, porque são intermediárias importantes na biossíntese da arginina e no ciclo da uréia.

Os peptídeos que ocorrem biologicamente variam muito de tamanho, desde moléculas pequenas contendo dois ou três aminoácidos até grandes macromoléculas contendo milhares de aminoácidos.
Duas moléculas de aminoácidos podem ser unidas covalentemente através de uma ligação amida substituída, chamada ligação peptídica, para formar um dipeptídio. Tal ligação é formada por remoção dos elementos da água de um grupo α-carboxila de um aminoácido e do grupo α-amino de outro.

R H R
H N C H C O H + H N C H C O O
O
1 2
+ 3
-
R H R
H N C H C N C H C O O
O
1 2
+ 3
-
H O2
Três aminoácidos podem ser reunidos por duas ligações peptídicas para formar um tripeptídio, da mesma maneira, os aminoácidos podem ser reunidos para formar tetra e pentapeptídios. Quando um pequeno número de aminoácidos é reunido desta forma a estrutura é chamada de oligopeptídio e quando muitos aminoácidos são reunidos, o produto é chamado de polipeptídio. As proteínas podem ter milhares de unidades de aminoácidos. Embora os termos “proteína” e “polipeptídios” possam ser, algumas vezes, intercambiáveis.
As ligações peptídicas podem ser hidrolisadas por aquecimento tanto com ácido forte quanto com base forte, para liberar os aminoácidos, para liberar os aminoácidos unidos por elas.
As ligações peptídicas podem também ser hidrolisadas por
determinadas enzimas chamadas proteases. Elas são enzimas proteolíticas e são encontradas em todas as células e tecidos, onde elas degradam proteínas que se tornaram desnecessárias ou danificadas, além de ajudarem na digestão dos alimentos protéicos.

Alguns polipeptídios pequenos possuem atividade biológica
Existem muitos oligopeptídios e polipeptídios pequenos que ocorrem naturalmente, possuem atividades biológicas importantes e pronunciadas, e, por isso, exercem seus efeitos em concentrações muito pequenas. Por exemplo, um certo número de hormônios de vertebrados são polipeptídios pequenos. O hormônio insulina contém duas cadeias polipeptídicas, uma com 30 resíduos de aminoácidos e a outra com 21. Outros hormônios polipeptídios são o glucagon, um hormônio pancreático de 29 resíduos que tem ação oposta àquela da insulina, e a corticotrofina, um hormônio com 39 resíduos de aminoácidos, secretado pela hipófise anterior e que estimula o córtex adrenal.

II. PROTEÍNAS
Quase tudo que ocorre nas células envolve uma ou mais proteínas. As proteínas fornecem a estrutura, catalisam as reações celulares e executam milhares de outras tarefas. O papel central ocupado por elas é evidenciado no fato de que a informação genética é, em última instância, expressa como proteínas. Para cada proteína existe um segmento de DNA (um gene) que guarda a informação, especificando sua seqüência de aminoácidos. Em uma célula típica existem milhares de diferentes tipos de proteínas, cada uma delas codificada por um gene e, cada uma delas, executando uma função específica. As proteínas estão entre as macromoléculas biológicas mais abundantes e também são extremamente versáteis em suas funções.

A. Funções biológicas das proteínas
1. Enzimas – o grupo de proteínas mais variado e mais altamente especializado é aqueles cujos componentes exibem atividade catalítica – as enzimas. São catalisadas por enzimas virtualmente todas as reações químicas nas quais participam as biomoléculas orgânicas das células. Muitos milhares de enzimas diferentes, cada uma capaz de catalisar um tipo de reação química diferente, foram descobertos em diferentes organismos.
2. Proteínas transportadoras – proteínas transportadoras existentes no plasma sanguíneo ligam-se a íons ou a moléculas específicas os quais são transportados de um órgão para outro. A hemoglobina dos eritrócitos liga-se ao oxigênio à medida que o sangue atravessa os pulmões, transporta-os até os tecidos periféricos e, aí, libera-o para que possa participar da oxidação dos nutrientes, com concomitante liberação de energia. O plasma sanguíneo também contém lipoproteínas que transportam lipídios do fígado para outros órgãos. Outros tipos de proteínas de transporte estão presentes nas membranas plasmáticas e nas membranas intracelulares de todos os organismos; elas estão aptas a ligarem-se, por exemplo, à glicose, aos aminoácidos ou às outras substâncias e transportá-las através dessas membranas.

3. Proteínas nutrientes e de armazenamento – as sementes de muitas plantas armazenam proteínas nutrientes necessárias para a germinação e o crescimento do broto. Exemplos particularmente bem estudados são as proteínas das sementes do trigo, milho e arroz. A ovoalbumina, a principal proteína da clara do ovo, e a caseína, a principal proteína do leite, são outros exemplos de proteínas nutrientes. A ferritina encontrada em algumas bactérias e em tecidos animais e vegetais armazena átomos de ferro.
4. Proteínas contráteis ou de motilidade – algumas proteínas
habilitam células e organismos com a capacidade de contraírem-se, de mudarem de forma, ou de se deslocarem no meio ambiente. A actina e a miosina funcionam no sistema contrátil do músculo esquelético e também em muitas células não musculares. A tubulina é a proteína com a qual os microtúbulos são construídos. Os microtúbulos agem de forma concentrada com a proteína dineína nos cílios e flagelos para propelir as células.
a) Proteínas estruturais – muitas proteínas servem como filamentos de suporte, cabos ou lâminas para fornecer proteção ou resistência a estruturas biológicas. O principal componente das cartilagens e dos tendões é a proteína fibrosa colágeno, a qual tem alta resistência à tensão. O couro é quase que colágeno puro. Os ligamentos contêm elastina, uma proteína estrutural capaz de distender-se em duas dimensões. O cabelo, as unhas e as penas consistem principalmente da proteína resistente e insolúvel denominada queratina. O maior componente das fibras da seda e da teia das aranhas é a fibroína. Os ligamentos “em dobradiça” das asas de certos insetos são feitos de resilina, uma proteína que tem propriedades elásticas próximas da perfeição.

5. Proteínas de defesa – muitas proteínas defendem os organismos contra a invasão de outras espécies ou os protegem de ferimentos. As imunoglobulinas ou anticorpos, proteínas especializadas sintetizadas pelos linfócitos dos vertebrados, podem reconhecer e precipitar, ou neutralizar, invasores como bactérias, vírus ou proteínas estranhas oriundas de outras espécies. O fibrinogênio e a trombina são proteínas que participam da coagulação do sangue que previne a perda de sangue quando o sistema vascular é lesado. Venenos de serpente, toxinas bacterianas e proteínas vegetais tóxicas, como a ricina, também parecem ter funções defensivas. Algumas destas proteínas, incluindo o fibrinogênio, a trombina e alguns venenos também são enzimas.
6. Proteínas reguladoras – algumas proteínas ajudam a regular a atividade celular ou fisiológica. Entre elas estão muitos hormônios. Alguns exemplos incluem a insulina, a qual regula o metabolismo dos açúcares e o hormônio do crescimento da hipófise. A resposta celular a muitos sinais hormonais é freqüentemente regulada por uma classe de proteínas que se ligam ao GTP e são chamadas proteínas G (o GTP é estreitamente relacionado ao ATP com a guanina substituindo a adenina). Outras proteínas reguladoras ligam-se ao DNA e regulam a biossíntese de enzimas e das moléculas de RNA envolvidas na divisão celular, tanto em procariotos como em eucariotos.
7. Outras proteínas – existem numerosas outras proteínas cujas funções podem ser
ditas exóticas e de difícil classificação. A monelina, uma proteína de uma planta africana, tem um sabor intensamente doce. Ela está sendo estudada como um adoçante não-tóxico e quase sem calorias para uso humano. O plasma sanguíneo de alguns peixes da Antártica contém proteínas anticoagulantes, as quais protegem do congelamento o sangue destes animais.

Algumas proteínas contêm grupos químicos diferentes dos aminoácidos
Muitas proteínas, como as enzimas ribonuclease e quimotripsina, contêm apenas aminoácidos e nenhum outro grupo químico; elas são consideradas proteínas simples. Entretanto, algumas proteínas contêm componentes químicos em adição aos aminoácidos; elas são chamadas proteínas conjugadas. A parte não-aminoácido de uma proteína conjugada é geralmente chamada de seu grupo prostético. As proteínas conjugadas são classificadas com base na natureza química dos seus grupos prostéticos; por exemplo: lipoproteínas contêm lipídios, glicoproteínas contêm moléculas de açúcares e metaloproteínas contêm um metal específico. Um certo número de proteínas contém mais do que um grupo prostético. Geralmente o grupo prostético desempenha um papel importante na função biológica da proteína.

B. Proteínas conjugadasClasse Grupo prostético Exemplo
Lipoproteínas
Glicoproteínas
Fosfoproteínas
Heme-proteínas
Flavoproteínas
Metaloproteína
Lipídios
Carboidratos
Grupo fosfato
Heme(ferro-porfirina)
Nucleotídeos de flavina
FerroZincoCálcioMolibdênioCobre
Β1-lipoproteína do sangue
ImunoglobulinaG
Caseína do leite
Hemoglobina
Succinato desidrogenase
FerritinaÁlcool desidrogenaseCalmodulinaDinitrogenasePlastocianina

A função de uma proteína depende da sua seqüência de aminoácidos
A bactéria E. coli produz perto de 3.000 proteínas
diferentes. Um ser humano produz de 50.000 a 100.000 proteínas diferentes. Em ambos os casos, cada tipo separado de proteína tem uma estrutura única e esta estrutura confere a ela uma função única. Mais de 1.400 doenças genéticas humanas têm sido identificadas como resultantes da produção de proteínas defeituosas. Talvez um terço dessas proteínas defeituosas assim o são, porque um único aminoácido da seqüência foi mudado; portanto, se a estrutura primária é alterada, a função da proteína também pode mudar.

C. Existem quatro níveis na arquitetura das proteínas 1. Estrutura primária – inclui todas as ligações covalentes
entre os aminoácidos que compõem uma proteína e é definida pela seqüência dos aminoácidos unidos por ligações peptídicas e pela localização das pontes dissulfeto. O arranjo espacial relativo dos aminoácidos não é especificado.
2. Estrutura secundária – refere-se aos arranjos regulares e
recorrentes no espaço de resíduos de aminoácidos adjacentes em uma cadeia polipeptídica.
3. Estrutura terciária – refere-se ao relacionamento espacial
entre todos os aminoácidos em um polipeptídio. 4. Estrutura quaternária – especifica a relação espacial dos
polipeptídios, ou subunidades, no interior de uma dada proteína.


D. As proteínas perdem a estrutura e a função quando desnaturadas A maneira de demonstrar a importância da estrutura específica das proteínas para a função
biológica que exercem é alterar esta estrutura e determinar o efeito que isto causa nesta função. Uma alteração extrema é a perda total da sua estrutura tridimensional, um processo chamado desnaturação. Este é o processo familiar que ocorre quando um ovo é cozido. A clara do ovo, a qual contém a proteína solúvel albumina do ovo, ou ovoalbumina, coagula pelo aquecimento para formar uma substância branca e sólida. Esta substância não redissolverá, quando resfriada, para reproduzir a solução límpida de proteína que era, antes do aquecimento, a clara do ovo original. O aquecimento da albumina do ovo produziu, portanto, uma mudança irreversível. Este efeito do calor ocorre em, virtualmente, em todas as proteínas globulares, independentemente do seu tamanho ou da sua função biológica, embora, a temperatura precisa na qual o processo ocorre possa variar e o seu efeito nem sempre seja irreversível. Algumas proteínas globulares desnaturadas pelo calor, extremos de pH, ou reagentes desnaturantes, recuperarão a sua estrutura nativa e sua atividade biológica, um processo chamado renaturação. Quando estes agentes são retirados da solução em que as proteínas se encontram, esta mesma solução retorna às condições nas quais a conformação protéica nativa é possível e estável. A mudança na estrutura produzida pela desnaturação é quase invariavelmente associada à perda de função. Isto é uma conseqüência esperada do princípio de que a estrutura tridimensional específica das proteínas é crítica para o exercício de suas funções.
As proteínas podem ser desnaturadas não somente pelo aquecimento, mas também por valores extremos de pH, por alguns solventes orgânicos miscíveis com a água, como o etanol e a acetona, por algumas substâncias em solução como a uréia, ou por exposição da proteína a substâncias detergentes.

PROTEÍNAS GLOBULARES Com o arranjo desses elementos estruturais fundamentais em
diferentes combinações, é possível construir uma grande diversidade de proteínas, as quais serão capazes de desempenhar uma variedade de funções especializadas.
I. HEMEPROTEÍNAS GLOBULARES
Hemeproteínas são um grupo especializado de proteínas, as quais contém heme como grupo prostético firmemente ligado. O papel do grupo heme é determinado pelo ambiente criado pela estrutura tridimensional da proteína. Por exemplo, o grupo heme de citocromo funciona como um carreador de elétrons, sendo alternadamente oxidado e reduzido. Em contraste, o grupo heme da enzima catalase é parte do sítio ativo da enzima, a qual catalisa a quebra do peróxido de hidrogênio. Na hemoglobina e na mioglobina, as duas hemeproteínas mais abundantes em humanos, o grupo heme serve para ligar, de forma reversível, o oxigênio.

A. A estrutura do heme O heme é um complexo entre a protoporfirina IX e
o íon ferroso (Fe2+)

B. Estrutura e função da mioglobina A mioglobina, uma hemeproteína presente no coração e no músculo
esquelético, funciona tanto como um reservatório de oxigênio quanto como um carreador de oxigênio, que aumenta a velocidade de transporte de oxigênio dentro da célula muscular. A mioglobina consiste em uma única cadeia polipeptídica, a qual é estruturalmente similar a uma das cadeias polipeptídicas individuais que constituem as subunidades da molécula da hemoglobina.
C. Estrutura e função da hemoglobina A hemoglobina é encontrada exclusivamente nos eritrócitos, onde sua
principal função e transportar oxigênio dos pulmões até os capilares dos tecidos. A hemoglobina A, a principal hemoglobina em adultos, é composta por quatro cadeias polipeptídicas – duas cadeias alfa (α) e duas cadeias beta (β) – mantidas unidas por meio de ligações não–covalentes. Cada subunidade contém segmentos de estrutura em hélice α , onde se liga o grupo heme, de forma similar ao da mioglobina. A molécula tetramérica da hemoglobina, no entanto, é estrutural e funcionalmente mais complexa do que a mioglobina. Por exemplo, a hemoglobina pode transportar CO2 dos tecidos até os pulmões e pode carregar quatro moléculas de O2 dos pulmões às células dos tecidos do corpo.

D. A ligação do oxigênio à mioglobina e a hemoglobina
A mioglobina pode ligar somente uma molécula
de oxigênio (O2), porque contém apenas um grupo heme. Em contraste, a hemoglobina pode ligar quatro moléculas de oxigênio – uma para cada um de seus quatro grupos heme. O grau de saturação (Y) desses sítios de ligação ao oxigênio em todas as moléculas de mioglobina ou hemoglobina pode variar de zero (quando todos os sítios estão vazios) a 100% (quando todos os sítios estão preenchidos).

1. Curva de dissociação do oxigênio. Uma curva da saturação (Y) medida em diferentes pressões parciais de oxigênio (pO2) é chamada de curva de dissociação do oxigênio. As curvas para a mioglobina e para a hemoglobina apresentam diferenças importantes. Esse gráfico ilustra que a mioglobina tem maior afinidade pelo oxigênio do que a hemoglobina. A pressão parcial de oxigênio necessária para obter metade da saturação dos sítios de ligação (P50) é de aproximadamente 1 mm Hg para a mioglobina e 26 mm Hg para a hemoglobina. (Quanto maior a afinidade pelo oxigênio [isto é, quanto mais fortemente liga o oxigênio], menor a P50).

a. Mioglobina. A curva de dissociação do oxigênio da mioglobina possui uma forma hiperbólica. Isso reflete o fato de que a mioglobina liga-se reversivelmente a apenas uma molécula de oxigênio. Assim, a mioglobina oxigenada (MbO2) e a desoxigenada (Mb) estão em um equilíbrio simples: Mb + O2 ↔ MbO2
O equilíbrio é desviado para a direita ou para a esquerda à medida que o oxigênio é adicionado ou removido do sistema. (a mioglobina tem a função de ligar o oxigênio liberado pela hemoglobina nas baixas pO2 encontradas no músculo. A mioglobina, por sua vez, liberará o oxigênio dentro da célula muscular em resposta à demanda de oxigênio)
b. Hemoglobina. A curva de dissociação do oxigênio da hemoglobina tem forma sigmoidal, indicando que as subunidades cooperem na ligação do oxigênio. A ligação cooperativa do oxigênio às quatro subunidades da hemoglobina significa que a ligação de uma molécula de oxigênio a um dos grupos heme aumenta a afinidade pelo oxigênio dos grupos heme restantes na mesma molécula de hemoglobina. Esse efeito é denominado interação heme-heme. Embora seja mais difícil para a primeira molécula de oxigênio ligar-se à hemoglobina, a ligação subseqüente de oxigênio ocorre com alta afinidade, como demonstrado pela curva rapidamente ascendente na região de 20 a 30 mm Hg.

E. Outras hemoglobinas É importante lembrar que a hemoglobina A humana (HbA) é apenas um membro de uma
família funcional e estruturalmente relacionada de proteínas, as hemoglobinas. Algumas hemoglobinas, como HbF, normalmente são sintetizadas somente durante o desenvolvimento fetal, enquanto outras, como a HbA2, são sintetizadas no adulto, embora em baixos níveis, se comparados à HbA. A HbA pode também ser modificada pela adição covalente de uma hexose. Por exemplo, a adição de glicose forma um derivado glicosilado da hemoglobina, a HbA1C.
1. Hemoglobina fetal. Nas primeiras semanas após a concepção, o feto começa a sintetizar a HbF na medula óssea em desenvolvimento. A HbF é a principal hemoglobina encontrada no feto e no recém-nascido, perfazendo cerca de 60% da hemoglobina total dos eritrócitos durante os últimos meses da vida fetal. A síntese da HbA inicia na medula óssea por volta do oitavo mês de gravidez e substitui gradualmente a HbF. Em condições fisiológicas, a HbF possui uma afinidade maior pelo oxigênio do que a HbA, pois a HbF liga-se apenas fracamente ao 2,3-BPG. Uma vez que o 2,3-BPG serve para reduzir a afinidade da hemoglobina pelo oxigênio, a interação mais fraca entre 2,3-BPG e HbF resulta em uma maior afinidade da HbF pelo oxigênio, quando comparada com a HbA. A maior afinidade da HbF pelo oxigênio facilita a transferência do oxigênio da circulação materna para os eritrócitos do feto através da placenta.
2. Hemoglobina A2 (HbA2). A HbA2 é um componente menor da hemoglobina normal do adulto, surgindo inicialmente cerca de 12 semanas após o nascimento e finalmente constituído cerca de 2% da hemoglobina total.
3. Hemoglobina A1C. Sob condições fisiológicas, a HbA é glicosada de forma lenta e não-enzimática; a extensão da glicosilação depende da concentração plasmática de uma determinada hexose. Quantidades aumentadas de HbA1C são encontradas nos eritrócitos de pacientes com diabetes melito, pois a HbA desses pacientes está em contato com maiores concentrações de glicose durante os 120 dias de vida de seus eritrócitos.

II. HEMOGLOBINOPATIAS As hemoglobinopatias têm sido tradicionalmente
definidas como uma família de distúrbios causados pela produção de uma molécula de hemoglobina estruturalmente anormal, pela síntese de quantidades insuficientes de hemoglobina normal ou, raramente, por ambas. A anemia falciforme (HbS), a doença da hemoglobina C (HbC) e a síndrome da talassemia são hemoglobinopatias representativas, que podem ter conseqüências clínicas graves. As duas primeiras condições resultam da produção de hemoglobinas com seqüências alteradas de aminoácidos, enquanto as talassemias são devidas a uma produção diminuída de hemoglobina normal.

A. Anemia falciforme (doença da hemoglobina S) A anemia falciforme (também denominada doença falciforme) é uma
doença genética do sangue causada pela alteração de um único nucleotídeo (mutação pontual) no gene da cadeia de β-globina. A anemia falciforme é uma doença homozigota recessiva. Ela ocorre em indivíduos que herdaram dois genes mutantes (um de cada um dos pais) que codificam a síntese das cadeias β das moléculas de globina. A presença da doença não se manifesta no bebê, até que uma quantidade suficiente de HbF seja substituída pela HbS, quando os eritrócitos começam a assumir a morfologia falciforme. A anemia falciforme caracteriza-se por episódios dolorosos (crises) que ocorrem durante toda a vida, por uma anemia hemolítica crônica e pelo aumento da suscetibilidade a infecções, que começam em geral no início da infância. Outros sintomas incluem síndrome aguda do peito, acidentes vasculares cerebrais e disfunção esplênica e renal. A vida média de um eritrócito homozigótico para HbS é de cerca de 20 dias, comparada com os 120 dias para células normais. Os heterozigotos, representando um em cada dez afro-americanos, possuem um gene normal e um gene falciforme. Os eritrócitos desses heterozigotos contêm ambas, HbS e HbA. Esses indivíduos possuem o traço falciforme. Em geral eles não apresentam sintomas clínicos e podem ter uma expectativa de vida normal.

1. As alterações falciformes nos eritrócitos causam anóxia tecidual. Em condições de baixa pressão de oxigênio, a HbS polimeriza dentro dos eritrócitos, primeiro formando um gel, subseqüentemente se agregando em uma rede de polímeros fibrosos que distorce as células, produzindo eritrócitos rígidos e deformados. Essas células falciformes freqüentemente bloqueiam o fluxo sangüíneo nos capilares de pequeno diâmetro. Essa interrupção no suprimento de oxigênio leva à anóxia localizada (privação de oxigênio) no tecido, causando dor e, eventualmente, a morte (infarto) das células na vizinhança da área do bloqueio.
2. Possível vantagem seletiva do estado heterozigoto. A elevada
freqüência do gene HbS entre os africanos, apesar de seus efeitos lesivos no estado homozigoto, sugere que exista uma vantagem seletiva para indivíduos heterozigotos. Por exemplo, os heterozigotos para o gene da anemia falciforme são menos suscetíveis à malária, causada pelo parasito Plasmodium falciparum. Esse organismo desenvolve parte obrigatória de seu ciclo vital no eritrócito. Uma vez que, em indivíduos heterozigotos, assim como nos homozigotos para HbS, essas células apresentam um tempo de vida menor do que o normal, o parasito não pode completar seu estágio de desenvolvimento intracelular. Esse fato pode oferecer uma vantagem seletiva aos heterozigotos que vivem em regiões onde a malária é uma das maiores causas da morte.

B. Talassemias As talassemias são doenças hemolíticas hereditárias, nas quais ocorre um desequilíbrio na síntese das
cadeias de globina. Como um grupo, são os distúrbios mais comuns de um único gene em humanos. Nas talassemias, a síntese de uma cadeia α ou β é defeituosa. A talassemia pode ser causada por uma série de mutações, incluindo deleção de genes, ou ainda substituições ou deleções de um ou vários nucleotídeos no DNA.
1. β-Talassemias. Nessas doenças, a síntese das cadeias β está diminuída ou ausente, enquanto a
síntese das cadeias α está normal. As cadeias de α-globina não podem formar tetrâmeros estáveis e, portanto, precipitam, causando a morte prematura das células inicialmente destinadas a tornarem-se eritrócitos maduros. Assim sendo, indivíduos com defeitos no gene da cadeia β apresentam o traço de β-talassemia (β-talassemia menor), se tiverem apenas um dos genes defeituoso, ou β-talassemia maior, se ambos os genes para a cadeia de β-globina forem defeituosos. Uma vez que o gene da β-globina não é expresso até o final da gestação, as manifestações físicas das β-talassemias só aparecem após o nascimento. Os indivíduos com β-talassemia menor produzem algumas cadeias β, e geralmente não necessitam tratamento específico. Entretanto, as crianças que nascem com β-talassemia maior possuem o triste destino de parecerem saudáveis ao nascimento, tornando-se gravemente anêmicos, em geral durante o primeiro ou segundo ano de vida. Esses pacientes requerem transfusões de sangue regulares.
2. α-Talassemias. Esses são defeitos nos quais a síntese das cadeias de α-globina está diminuída ou
ausente. Uma vez que o genoma de cada indivíduo contém quatro cópias do gene de α-globina (dois em cada cromossomo 16), existem muitos níveis de deficiência de cadeias α. Se um dos quatro genes é defeituoso, o indivíduo é chamado de portador silencioso da α-talassemia, pois não apresenta qualquer das manifestações físicas da doença. Se dois genes da α-globina são defeituosos, o indivíduo é denominado como possuidor do traço de α-talassemia. Se três genes da cadeia α estão defeituosos, o indivíduo tem a doença da hemoglobina H (HbH), uma anemia hemolítica leve a moderadamente grave. Se todos os quatro genes de α-globina são defeituosos, ocorrem hidropisia fetal e morte do feto, pois as cadeias α são necessárias para a síntese da HbF.

PROTEÍNAS FIBROSASO colágeno e a elastina são exemplos de proteínas fibrosas bem caracterizadas, que apresentam função estrutural no organismo. Por exemplo, o colágeno e a elastina são componentes da pele, do tecido conectivo, da parede dos vasos sangüíneos e da córnea e da esclera do olho. Cada proteína fibrosa apresenta propriedades mecânicas especiais, resultado de sua estrutura única, a qual é obtida pela combinação de aminoácidos específicos em elementos regulares de estrutura secundária. Isso está em contraste com as proteínas globulares, cuja forma é o resultado de interações complexas entre elementos estruturais secundários, terciários e, às vezes, quaternários.

I. COLÁGENO O colágeno é a proteína mais abundante no corpo humano. Uma típica
molécula de colágeno é longa, com uma estrutura rígida, na qual três cadeias polipeptídicas (referidas como “cadeias α”) estão torcidas ema em volta da outra, de forma semelhante a uma corda de tripla hélice. Apesar dessas moléculas serem encontradas em todo o corpo, seus tipos e sua organização são ditados pelo papel estrutural que o colágeno desempenha em cada órgão em particular. Em alguns tecidos, o colágeno pode estar disperso como um gel que dá suporte à estrutura, como na matriz extracelular ou no humor vítreo. Em outros tecidos, o colágeno pode estar torcido firmemente em fibras paralelas, o que confere grande resistência, como nos tendões. Na córnea, o colágeno está empilhado de forma a transmitir a luz com o mínimo de dispersão. O colágeno dos ossos ocorre como fibras arranjadas em ângulo tal, umas em relação às outras, que lhe permite apresentar alta resistência mecânica em qualquer direção.
A super família das proteínas de colágeno inclui mais de 20 tipos de
colágeno, assim como outras proteínas que apresentam domínios semelhantes ao colágeno.

A. Biossíntese do colágeno Os polipeptídeos precursores da molécula do colágeno são formados
nos fibroblastos (ou nas células relacionadas osteoblastos dos ossos e condroblastos da cartilagem), e são secretados na matriz extracelular.
1. Hidroxilação. As cadeias pró-α são processadas por diversos
passos enzimáticos dentro do lúmen do RER, enquanto os polipeptídeos ainda estão sendo sintetizados. Essas reações de hidroxilação requerem oxigênio molecular e o agente redutor vitamina C, sem o qual as enzimas hidroxilantes, são incapazes de funcionar. No caso da deficiência de ácido ascórbico (e, dessa forma, ausência de hidroxilação da prolina e da lisina), as fibras do colágeno não podem estabelecer ligações cruzadas, diminuindo enormemente a resistência á tensão nas fibras reunidas. Uma doença resultante dessa deficiência é o escorbuto. Pacientes com deficiência de ácido ascórbico também apresentam hematomas nos membros, como resultado do extravasamento subcutâneo de sangue (fragilidade capilar).

B. Degradação do colágeno
Colágenos normais são moléculas altamente estáveis, tendo meias-vidas que podem chegar a muitos meses. Entretanto, o tecido conectivo é muito dinâmico e está constantemente sendo remodelado, muitas vezes em resposta ao crescimento ou a lesão do tecido. A quebra das fibrilas do colágeno é dependente da ação proteolítica de colagenases.
C. Doenças do colágeno Defeitos em qualquer um dos muitos passos da síntese das fibras
do colágeno podem resultar em doenças genéticas envolvendo a incapacidade do colágeno em formar fibras e, dessa forma, prover os tecidos com a necessária resistência à tensão normalmente promovida pelo colágeno. Mais de 1.000 mutações foram identificadas em 22 genes que codificam 12 tipos de colágeno. A seguir, são fornecidos alguns exemplos de doenças resultantes da síntese defeituosa do colágeno.

1. Síndrome de Ehlers-Danlos (EDS). Essa doença é constituída por um grupo heterogêneo de desordens generalizadas do tecido conectivo, resultantes de erros inatos do metabolismo das moléculas de fibrilas de colágeno. O colágeno contendo cadeias mutantes não é secretado, e é degradado ou acumulado em grande quantidade nos compartimentos intracelulares. Uma vez que o colágeno do tipo III é um importante componente das artérias, podem ocorrer problemas vasculares letais. (Embora o colágeno do tipo III seja apenas um componente menor das fibrilas do colágeno da pele, por razões desconhecidas os pacientes com EDS também apresentam defeitos nas fibrilas de colágeno do tipo I. Isso resulta em pele elástica e articulações frouxas)

II. ELASTINA Em contraste com o colágeno, o qual forma fibras que apresentam alta
resistência à tensão, as da elastina é uma proteína do tecido conectivo com propriedades semelhantes às da borracha. Fibras elásticas compostas de microfibrilas de elastina e de glicoproteínas são encontradas no pulmões, na parede de grandes artérias e nos ligamentos elásticos. Elas podem ser distendidas em várias vezes o seu comprimento normal, mas retornam ao formato original quando a força de tensão é relaxada.
A. Papel da α1- antitripsina na degradação da elastina 1. α1-antitripsina. O sangue e outros fluidos corporais contêm uma
proteína, a α1-antitripsina (α1-AT, também comumente chamada de α1-antiproteinase), que inibe diversas enzimas proteolíticas (também chamadas de proteases ou proteinases), as quais hidrolisam ou destroem proteínas. A α1-AT tem o importante papel fisiológico de inibir a elastase de neutrófilos – uma potente protease que é liberada no espaço extracelular e degrada a elastina da parede alveolar, assim como outras proteínas estruturais em vários tecidos. A maioria da α1-AT encontrada no plasma é sintetizada e secretada pelo fígado. O restante é sintetizado em diversos tecidos, incluindo monócitos e macrófagos alveolares, os quais podem ser importantes na prevenção da lesão tecidual local pela elastase.

2. Papel da α1-AT nos pulmões. No pulmão normal, os alvéolos são expostos cronicamente a baixos níveis de elastase de neutrófilos, a qual é liberada a partir de neutrófilos ativos ou em degeneração. Essa atividade proteolítica pode destruir a elastina na parede alveolar se não for contraposta pela ação inibitória da α1-AT, o mais importante inibidor da elastase dos neutrófilos. Uma vez que o tecido pulmonar não pode se regenerar, o enfisema resulta da destruição do tecido conectivo da parede alveolar.
3. Enfisema resultante da deficiência de α1-At. São conhecidas várias mutações no gene α1-AT que causam a deficiência dessa proteína, mas uma única mutação na base purina (GAG AAG, resultando na substituição de uma lisina por glutamato na posição 342 da proteína) é, clinicamente, a mais encontrada. Um indivíduo deve herdar dois alelos α1-AT anormais para correr o risco de desenvolver enfisema. Em um indivíduo heterozigoto, com um gene normal e um gene defeituoso, os níveis de α1-AT são suficientes para proteger os alvéolos da lesão. Fumar causa a oxidação e a subseqüente inativação desse resíduo de metionina, diminuindo assim a eficiência do inibidor em neutralizar a elastase. Fumantes com deficiência de α1-AT, portanto, apresentam maior taxa de lesão pulmonar e menor taxa de sobrevivência, comparados com indivíduos não-fumantes portadores da deficiência)