Embed Size (px)
Citation preview

Aspects génétiques des épilepsiesI AnS BaulacM BaulacA BriceE Le Guern
Résumé. – Des progrès notables ont été réalisés ces dix dernières années dans le domaine de la génétique desépilepsies tant dans les formes idiopathiques que symptomatiques. L’identification des gènes impliqués dansdes formes familiales d’épilepsie permet peu à peu de mieux en comprendre les mécanismesphysiopathologiques, d’élaborer des modèles expérimentaux de ces maladies et d’envisager de nouvellesstratégies thérapeutiques. Elle rend par ailleurs possible un diagnostic moléculaire direct qui soulève desproblèmes éthiques, en particulier chez des individus asymptomatiques jusque-là.Cet article passe en revue les connaissances actuelles sur les bases génétiques des épilepsies.© 2001 Editions Scientifiques et Médicales Elsevier SAS. Tous droits réservés.
Mots-clés : épilepsies familiales, formes mendéliennes, formes idiopathiques, formes symptomatiques,canaux ioniques, migration neuronale.
Introduction
L’épilepsie est une affection fréquente qui recouvre un ensemblehétérogène de pathologies. La part des facteurs génétiques dansl’étiologie des épilepsies humaines est importante. Les études dejumeaux permettent de l’estimer entre 40 à 60 % [2, 9]. En fait, laparticipation respective des facteurs environnementaux etgénétiques dans le déterminisme d’une épilepsie varie selon lamaladie épileptique considérée. Pour de nombreuses épilepsies, onsoupçonne un mode de transmission complexe. L’épilepsie résultealors de l’action conjointe de facteurs exogènes environnementauxet de gènes (appelés gènes de susceptibilité) qui permettentl’émergence de la maladie. Cependant, même pour les épilepsiesayant une composante génétique forte (les formes monogéniquesd’épilepsies en sont le modèle), les facteurs environnementauxpeuvent également intervenir. Ils pourraient, par exemple, expliquerqu’un individu porteur d’une mutation n’exprime pas la maladie,contrairement à d’autres membres de sa famille porteurs de cettemême mutation (pénétrance incomplète de la maladie), ou que lamaladie épileptique ait une présentation électroclinique ou uneévolution variable chez des individus porteurs de la même mutation(expressivité variable). Enfin, dans les épilepsies les moinsgénétiquement déterminées, dues à des facteurs exogènes acquis(infectieux, toxiques, traumatiques...), des facteurs génétiquespourraient également intervenir, expliquant qu’exposés au mêmefacteur, certains individus développeront ultérieurement uneépilepsie et d’autres non.
La réalisation d’études génétiques dans l’épilepsie afin d’identifierle ou les gènes impliqué(s) est difficile et se heurte à de nombreuxobstacles. Premièrement, la plupart des épilepsies humaines ont unehérédité complexe et nécessitent la mise en œuvre de méthodesparticulières d’analyses dites méthodes non paramétriques (études
Isabelle An : Attachée, unité d’épileptologie.Stéphanie Baulac : Doctorat en Sciences, Inserm U 289.Michel Baulac : Professeur des Universités, praticien hospitalier, chef de service, unité d’épileptologie.Alexis Brice : Professeur des Universités, praticien hospitalier, consultation de génétique et Inserm U 289.Éric Le Guern : Maître de conférences, Inserm U 289.Hôpital de la Salpêtrière, 47, boulevard de l’Hôpital, 75651 Paris cedex 13, France.
de paires de germains, trios, ou études de cas-témoins) quidemandent des séries très larges de patients (souvent plusieurscentaines) difficiles à réunir. Les épilepsies à transmissionmendélienne, qui sont les moins fréquentes, sont théoriquement plusfaciles à étudier génétiquement. Le problème majeur est de disposerde grandes familles où l’on peut étudier de nombreux sujets atteints(au moins 10 pour les épilepsies autosomiques dominantes).Deuxièmement, les difficultés sont également nombreuses à l’étapedu « phénotypage » qui conditionne le succès des analysesgénétiques. Il s’agit principalement de déterminer pour chaqueindividu de la famille étudiée le statut clinique (atteint ou non). Lechoix des critères électrocliniques de départ peut être déterminantet il ne faut pas méconnaître la possibilité de grandes variationsinterindividuelles du phénotype, y compris au sein d’une mêmefamille. La reconstitution de l’histoire clinique de chaque individupeut être problématique, notamment chez les individus les plus âgésde la famille, rendant parfois le statut clinique incertain. Enfin, ilfaut savoir que les phénocopies (patients ayant une présentationclinique identique aux formes génétiques de la maladie maisd’origine non génétique) sont fréquentes dans le domaine del’épilepsie et des convulsions fébriles. En effet, il s’agit d’affectionsfréquentes qui peuvent survenir en dehors de tout contexte familialhéréditaire.Pourtant, malgré ces obstacles, des découvertes fondamentales ontété réalisées ces dix dernières années. Ce sont les épilepsies àtransmission mendélienne aussi bien idiopathiques quesymptomatiques, qui en ont le plus bénéficié.
Épilepsies idiopathiques
Elles sont caractérisées par l’absence de déficit neurologique ouintellectuel, la normalité de la neuro-imagerie et leur âge-dépendance. Depuis longtemps, la forte implication des facteursgénétiques dans les épilepsies idiopathiques a été suspectée(prévalence augmentée d’épilepsie par rapport à la populationgénérale chez les apparentés des malades, taux de concordance plusélevé chez les jumeaux monozygotes que chez les jumeauxdizygotes). La plupart d’entre elles ont une hérédité complexe et
Ency
clop
édie
Méd
ico-
Chi
rurg
ical
e1
7-0
44
-C-7
0 17-044-C-70
Toute référence à cet article doit porter la mention : An I, Baulac S, Baulac M, Brice A et Le Guern E. Aspects génétiques des épilepsies. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés),Neurologie, 17-044-C-70, 2001, 8 p.

sont multifactorielles. Celles qui ont une hérédité mendélienne sontrares mais particulièrement instructives de par les mécanismesfondamentaux qui les sous-tendent.Nous allons détailler celles pour lesquelles des gènes sont identifiés.Les loci et gènes actuellement connus pour les différentes épilepsiesidiopathiques sont répertoriés dans le tableau I qui résume leurscaractéristiques cliniques.
ÉPILEPSIES IDIOPATHIQUESÀ HÉRÉDITÉ MONOGÉNIQUE
¶ Épilepsie frontale nocturne autosomique dominante(«autosomal dominant nocturnal frontal lobe epilepsy»ou ADNFLE)
Ce syndrome familial rare fut décrit pour la première fois en 1994[76, 77]. Il débute le plus souvent dans l’enfance et est caractérisé pardes crises partielles brèves, fréquentes, survenant avec prédilectionpendant le sommeil, le plus souvent en salves. La composantemotrice est prédominante : postures dystoniques soudaines (lanature de ces dystonies paroxystiques nocturnes familiales futlongtemps débattue), pédalage, déambulation. Parfois lasymptomatologie se limite à des réveils nocturnes soudains. Unevocalisation ou une aura de contenu variable précède parfois lesmanifestations motrices. Les généralisations sont possibles maisrares. Des erreurs diagnostiques sont fréquentes avec desmanifestations de parasomnies (somnambulisme ou terreursnocturnes). Les crises persistent souvent à l’âge adulte mais tendentà se raréfier. La sensibilité à la carbamazépine est généralementbonne. Cependant, d’importantes variations intrafamiliales peuventêtre observées tant au plan clinique qu’évolutif. Lorsque lesélectroencéphalogrammes (EEG) percritiques sont interprétables etcontributifs, ils montrent une activité critique frontale et/outemporale.Ce syndrome familial de transmission autosomique dominante etde pénétrance incomplète est sous-tendu par une hétérogénéitégénétique. Un premier locus fut identifié dans une grande familleaustralienne sur le chromosome 20q13.2 [65]. Une mutation dans legène CHRNA4, codant pour la sous-unité alpha-4 du récepteurneuronal nicotinique à l’acétylcholine qui touche le deuxièmedomaine transmembranaire de cette sous-unité, fut identifiée ensuitedans cette famille [86]. D’autres mutations dans cette même sous-unité alpha-4 furent détectées dans une minorité de familles avecADNFLE [34, 72, 85]. Les récepteurs nicotiniques à l’acétylcholine sontdes récepteurs ionotropes hétéropentamériques. Huit gènes codantpour des sous-unités différentes ont été identifiés chez l’homme.Au plan fonctionnel, le second domaine transmembranaire de lasous-unité alpha-4 a un rôle-clef dans la perméabilité ionique ducanal. Il a été démontré in vitro que les mutations décrites affectaientles propriétés du récepteur en réduisant son affinité pourl’acétylcholine et sa perméabilité au calcium [5, 37]. Il semble que lesrécepteurs nicotiniques à l’acétylcholine neuronaux soient presqueexclusivement présynaptiques. Ils pourraient intervenir dans larégulation de la libération de neuromédiateurs et notamment duglutamate. Cependant, on ne sait pas encore par quel mécanismecette altération fonctionnelle du récepteur produit un syndromeépileptique aussi particulier.Un second locus a été identifié sur le chromosome 15q24 dans unefamille unique [66]. Bien que cette région jouxte celle où sont localisésdes gènes codant pour d’autres sous-unités du récepteur nicotiniqueà l’acétylcholine (CHRNA3, CHRNA5 et CHRNB4), aucunemutation dans ces gènes n’a été rapportée à ce jour.Récemment, un troisième locus a été identifié dans la régionpéricentromérique du chromosome 1 [22], et grâce à une stratégiegène-candidat, une équipe italienne et une équipe australienneviennent tout juste d’identifier deux mutations dans le gèneCHRNB2 dans deux familles avec ADNFLE [21, 64]. Ce gène code pourla sous-unité bêta-2 du récepteur nicotinique qui constitue avec lasous-unité alpha-4 le principal récepteur nicotinique neuronal
humain. Cependant, dans la majorité des familles avec ADNFLE, lamaladie n’est associée à aucune de ces sous-unités du récepteurnicotinique à l’acétylcholine [67].
¶ Convulsions néonatales familiales bénignes(«benign familial neonatal convulsions» ou BFNC)
Ce syndrome est caractérisé par la survenue de crises cloniques uni-ou bilatérales, souvent à bascule, de crises apnéiques, ouéventuellement toniques, au cours du deuxième ou troisième jourde vie d’un nouveau-né normal par ailleurs. Rarement, l’EEGpercritique montre un aspect « thêta pointu alternant » nonspécifique mais évocateur dans le contexte clinique. L’évolution estgénéralement favorable, cependant certains enfants présenterontultérieurement des convulsions fébriles ou développeront uneépilepsie. La forme familiale de ce syndrome (syndrome desconvulsions néonatales familiales bénignes) comporte certainesdifférences par rapport à la forme sporadique (syndrome desconvulsions néonatales bénignes) dans laquelle on n’observe jamaisde crises toniques, où l’aspect EEG « thêta alternant » est plusfréquemment observé, et où le pronostic est meilleur.Il s’agit du premier syndrome épileptique idiopathique dans lequelune liaison génétique fut publiée [44]. Une liaison au chromosome20q [44, 50] puis secondairement au chromosome 8q a étédéterminée [46], mais un troisième locus est suspecté. Des mutationsdans des gènes codant pour des canaux potassium voltage-dépendants, KCNQ2 en 20q (gène majoritairement impliqué) [8, 45, 83]
et KCNQ3 en 8q (rarement impliqué) [11] ont été identifiées. Lescanaux potassium codés par ces deux gènes ont de grandeshomologies de séquences, et sont tous deux exprimés de façonprépondérante dans toutes les régions du cerveau. Ils sontégalement très homologues à KCNQ1, exprimé préférentiellementdans le cœur et l’oreille interne, et qui est impliqué dans deuxsyndromes familiaux : le syndrome du QT long et le syndromecardioauditif de Jervell-Lange-Nielsen [58, 94]. Les canaux codés parKCNQ2 et KCNQ3 sont fonctionnellement liés, ce qui permet decomprendre pourquoi des mutations dans chacun d’eux peuventdonner lieu au même tableau clinique. Ils interviennent dans larepolarisation de la membrane neuronale après une dépolarisation[8, 93]. Les mutations décrites entraînent une perte de fonction ducanal [8, 45, 80]. L’âge-dépendance de ce syndrome pourrait êtreexpliquée par les variations d’expression des canaux potassium aucours de la vie [89].
¶ Syndrome «generalized epilepsy with febrile seizuresplus» (GEFS +)
Dans certaines familles, des convulsions fébriles sont associées à descrises afébriles. C’est le cas dans un nouveau syndrome familialdécrit en 1997 : le syndrome « GEFS + » [75]. Ce syndrome estcaractérisé par un phénotype familial hétérogène dans lequel lesindividus atteints présentent des convulsions fébriles particulières(dites « convulsions fébriles plus ») car elles persistent tardivementau-delà de l’âge de 6 ans (limite supérieure d’âge du syndrome desconvulsions fébriles « classiques »). Elles sont souvent nombreuseschez un individu donné. D’autres membres de la famille peuventprésenter des convulsions fébriles banales, mais l’antécédent deconvulsions fébriles n’est pas constant chez tous les individusatteints. Enfin, des crises afébriles de nature variable sont aussiobservées. Elles étaient décrites, dans la famille princeps, commeétant toutes des crises généralisées (crises tonicocloniques,myocloniques, atoniques, absences) [75]. En fait, d’autres types decrises ont été décrits ultérieurement dans d’autres familles (crisestoniques, hémiconvulsives, temporales ou frontales) [3, 55, 61, 84]. Ledébut de ces crises afébriles est très variable, dans l’enfance, sans ouavec intervalle libre par rapport à la période de convulsions fébriles,ou plus tard à l’âge adulte. Plusieurs types de crises afébrilespeuvent s’observer chez un même individu atteint, donnant lieu àdes tableaux électrocliniques plus ou moins typiques d’épilepsiegénéralisée idiopathique (épilepsie myoclonique juvénile, épilepsie-absence de l’enfant ou de l’adolescent) ou d’épilepsie
17-044-C-70 Aspects génétiques des épilepsies Neurologie
2
myoclonoastatique (ou syndrome de Doose). Parfois le syndromeépileptique est inclassable selon la classification internationale desépilepsies. Un déficit intellectuel est parfois observé [3]. L’évolutionet la pharmacosensibilité sont très variables d’un individu à l’autreau sein de la même famille. L’imagerie cérébrale est normale.
Ce syndrome est transmis selon un mode autosomique dominant etla pénétrance est incomplète. Il présente une grande hétérogénéitégénétique. En effet, un premier locus fut identifié sur le chromosome19q13.1, et une mutation dans le gène SCN1B codant pour la sous-unité bêta-1 du canal sodium voltage-dépendant neuronal fut
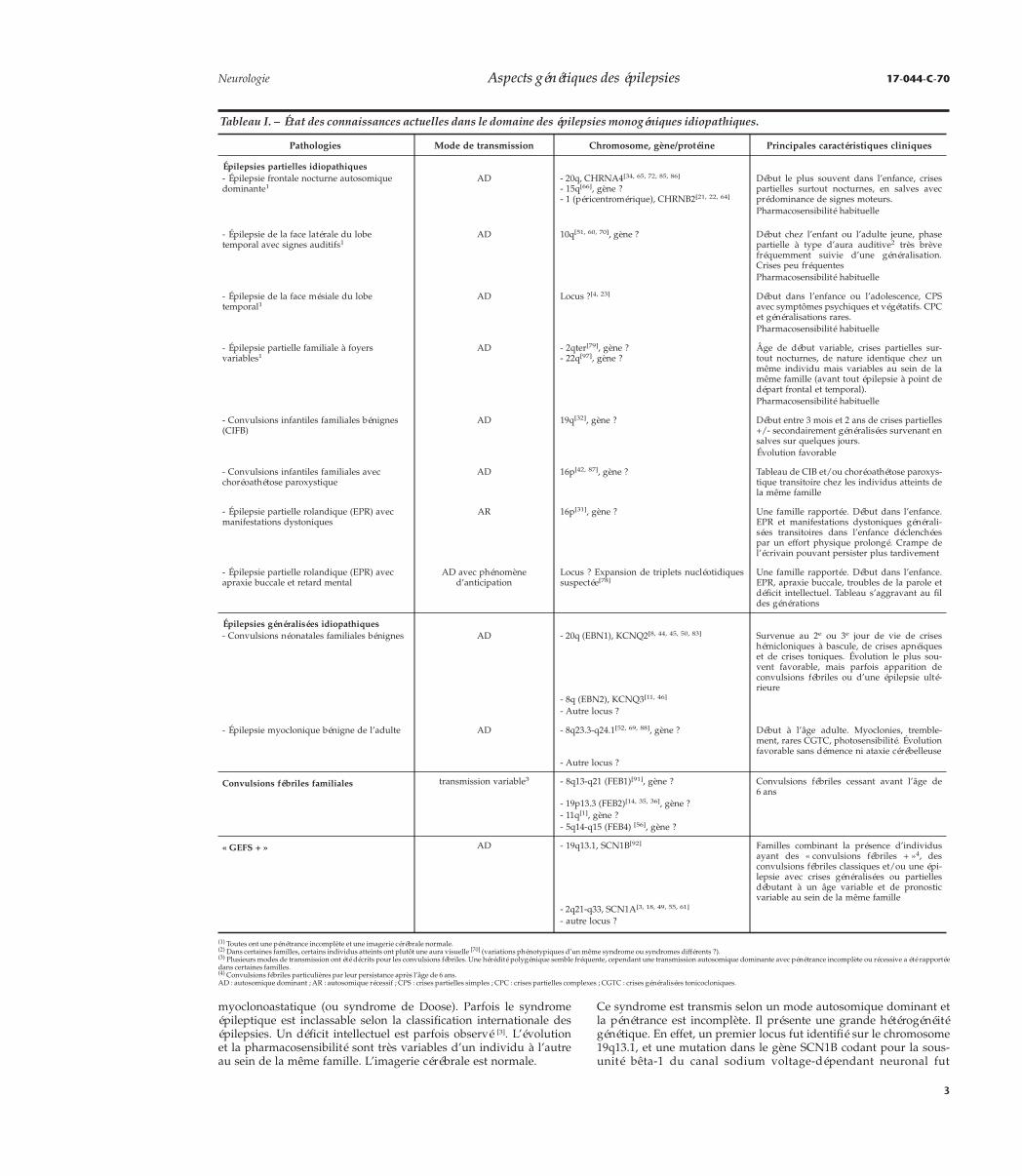
Tableau I. – État des connaissances actuelles dans le domaine des épilepsies monogéniques idiopathiques.
Pathologies Mode de transmission Chromosome, gène/protéine Principales caractéristiques cliniques
Épilepsies partielles idiopathiques- Épilepsie frontale nocturne autosomiquedominante1
AD - 20q, CHRNA4[34, 65, 72, 85, 86]
- 15q[66], gène ?- 1 (péricentromérique), CHRNB2[21, 22, 64]
Début le plus souvent dans l’enfance, crisespartielles surtout nocturnes, en salves avecprédominance de signes moteurs.Pharmacosensibilité habituelle
- Épilepsie de la face latérale du lobetemporal avec signes auditifs1
AD 10q[51, 60, 70], gène ? Début chez l’enfant ou l’adulte jeune, phasepartielle à type d’aura auditive2 très brèvefréquemment suivie d’une généralisation.Crises peu fréquentesPharmacosensibilité habituelle
- Épilepsie de la face mésiale du lobetemporal1
AD Locus ?[4, 23] Début dans l’enfance ou l’adolescence, CPSavec symptômes psychiques et végétatifs. CPCet généralisations rares.Pharmacosensibilité habituelle
- Épilepsie partielle familiale à foyersvariables1
AD - 2qter[79], gène ?- 22q[97], gène ?
Âge de début variable, crises partielles sur-tout nocturnes, de nature identique chez unmême individu mais variables au sein de lamême famille (avant tout épilepsie à point dedépart frontal et temporal).Pharmacosensibilité habituelle
- Convulsions infantiles familiales bénignes(CIFB)
AD 19q[32], gène ? Début entre 3 mois et 2 ans de crises partielles+/- secondairement généralisées survenant ensalves sur quelques jours.Évolution favorable
- Convulsions infantiles familiales avecchoréoathétose paroxystique
AD 16p[42, 87], gène ? Tableau de CIB et/ou choréoathétose paroxys-tique transitoire chez les individus atteints dela même famille
- Épilepsie partielle rolandique (EPR) avecmanifestations dystoniques
AR 16p[31], gène ? Une famille rapportée. Début dans l’enfance.EPR et manifestations dystoniques générali-sées transitoires dans l’enfance déclenchéespar un effort physique prolongé. Crampe del’écrivain pouvant persister plus tardivement
- Épilepsie partielle rolandique (EPR) avecapraxie buccale et retard mental
AD avec phénomèned’anticipation
Locus ? Expansion de triplets nucléotidiquessuspectée[78]
Une famille rapportée. Début dans l’enfance.EPR, apraxie buccale, troubles de la parole etdéficit intellectuel. Tableau s’aggravant au fildes générations
Épilepsies généralisées idiopathiques- Convulsions néonatales familiales bénignes AD - 20q (EBN1), KCNQ2[8, 44, 45, 50, 83] Survenue au 2e ou 3e jour de vie de crises
hémicloniques à bascule, de crises apnéiqueset de crises toniques. Évolution le plus sou-vent favorable, mais parfois apparition deconvulsions fébriles ou d’une épilepsie ulté-rieure
- 8q (EBN2), KCNQ3[11, 46]
- Autre locus ?
- Épilepsie myoclonique bénigne de l’adulte AD - 8q23.3-q24.1[52, 69, 88], gène ? Début à l’âge adulte. Myoclonies, tremble-ment, rares CGTC, photosensibilité. Évolutionfavorable sans démence ni ataxie cérébelleuse
- Autre locus ?
Convulsions fébriles familiales transmission variable3 - 8q13-q21 (FEB1)[91], gène ? Convulsions fébriles cessant avant l’âge de6 ans
- 19p13.3 (FEB2)[14, 35, 36], gène ?- 11q[1], gène ?- 5q14-q15 (FEB4) [56], gène ?
« GEFS + » AD - 19q13.1, SCN1B[92] Familles combinant la présence d’individusayant des « convulsions fébriles + »4, desconvulsions fébriles classiques et/ou une épi-lepsie avec crises généralisées ou partiellesdébutant à un âge variable et de pronosticvariable au sein de la même famille
- 2q21-q33, SCN1A[3, 18, 49, 55, 61]
- autre locus ?
(1) Toutes ont une pénétrance incomplète et une imagerie cérébrale normale.(2) Dans certaines familles, certains individus atteints ont plutôt une aura visuelle [70] (variations phénotypiques d’un même syndrome ou syndromes différents ?).(3) Plusieurs modes de transmission ont été décrits pour les convulsions fébriles. Une hérédité polygénique semble fréquente, cependant une transmission autosomique dominante avec pénétrance incomplète ou récessive a été rapportéedans certaines familles.(4) Convulsions fébriles particulières par leur persistance après l’âge de 6 ans.AD : autosomique dominant ; AR : autosomique récessif ; CPS : crises partielles simples ; CPC : crises partielles complexes ; CGTC : crises généralisées tonicocloniques.
Neurologie Aspects génétiques des épilepsies 17-044-C-70
3

détectée dans une famille [92]. Un second locus en 2q21-q33 sembleplus fréquemment impliqué puisque quatre familles ont étérapportées à ce jour [3, 49, 55, 61]. Deux mutations dans deux famillesfrançaises, situées dans le segment transmembranaire S4 responsablede l’activation du canal, ont été mises en évidence dans le gèneSCN1A qui correspond à ce locus et code pour la sous-unité alpha-1du même canal sodium voltage-dépendant [18].Des analyses fonctionnelles dans les ovocytes de xénope ont montréque les mutations dans les sous-unités alpha-1 et bêta-1 semblentaltérer les propriétés de ce canal. Il faut noter que ce canal sodiumvoltage-dépendant est déjà la cible de bon nombre de moléculesantiépileptiques.Enfin, au moins un troisième locus est suspecté car toutes lesfamilles « GEFS + » ne sont pas liées aux gènes déjà connus.Les mécanismes expliquant qu’une même mutation puisse donnerlieu à des tableaux cliniques aussi variés au sein d’une même famillede « GEFS + » sont actuellement inconnus. Ces dernierscorrespondent-ils à des différences régionales d’expression des gènesimpliqués ? Une autre hypothèse serait que ces gènesinterviendraient à un niveau très général dans le processusd’épileptogenèse, en tant que gènes de susceptibilité ou modulateursdu seuil épileptogène et que d’autres facteurs, génétiques ouenvironnementaux, moduleraient l’expression clinique dusyndrome.
ÉPILEPSIES IDIOPATHIQUES À HÉRÉDITÉ COMPLEXE
Pour la plupart des épilepsies généralisées idiopathiques (incluantl’épilepsie myoclonique juvénile, l’épilepsie-absence de l’enfant,l’épilepsie-absence de l’adolescent et l’épilepsie à crises grand maldu réveil), c’est un modèle d’hérédité complexe qui semble le plusadapté comme mode de transmission. Ces épilepsies résultent del’interaction de facteurs génétiques (gènes de susceptibilité) et defacteurs environnementaux. Les difficultés rencontrées dans l’étudegénétique de ces épilepsies, que nous avons abordées dansl’introduction, expliquent probablement les résultats contradictoirespubliés : les localisations suggérées par certaines équipes ne sont pasretrouvées par d’autres. Il faut souligner qu’au début de ces étudesil y a toujours un choix conceptuel. Le premier présuppose que pourl’épilepsie, les gènes de susceptibilité sont nombreux et leurs effetsfaibles. Il faut alors des effectifs considérables (au moins100 familles) pour obtenir des liaisons significatives. En corollaire,on peut diminuer le nombre de gènes en cause et renforcer leurseffets (au plan statistique) en homogénéisant le phénotype desfamilles étudiées. L’étape du phénotypage est alors essentielle.L’autre choix est de considérer qu’il existe des gènes de susceptibilitéde l’épilepsie en général (il existerait donc des traits épileptiques) etqu’il existe un « continuum » entre les syndromes épileptiques. Il
est alors possible de grouper des familles avec des épilepsiesdifférentes, ce qui permet d’obtenir des effectifs suffisants enanalyses non paramétriques (analyse dans lesquelles on nedétermine pas le mode de transmission).Une liaison génétique avec les régions 8q [19, 98], 3p [99] et 1p [96] a étérapportée dans des familles comportant une épilepsie généraliséeidiopathique (sans précision syndromique) mais n’a pas étéconfirmée par la suite.Parmi les épilepsies généralisées idiopathiques à hérédité complexe,l’épilepsie myoclonique juvénile a été la plus étudiée au plangénétique. Des résultats contradictoires ont été publiés concernantune liaison au chromosome 6p [17, 29, 30, 47, 74, 95]. Un deuxième locus en15q14 [16] (région comportant notamment le gène codant pour lasous-unité alpha-7 du récepteur cholinergique nicotinique) a étédécrit.Enfin, les convulsions fébriles et l’épilepsie ponctuelle rolandiqueont avant tout une hérédité complexe. Une liaison en 15q futsuggérée pour l’épilepsie ponctuelle rolandique dans une étude [57].
Épilepsies en rapportavec des anomalies héréditairesdu développement cortical (troublesde la migration neuronale)
Elles représentent une cause importante d’épilepsiepharmacorésistante souvent associée à un retard mental. Lescaractéristiques cliniques et génétiques de ces pathologies sontrésumées dans le tableau II. Les mutations dans le gène LIS 1(localisé sur le chromosome 17) codant pour une sous-unité noncatalytique du platelet activating factor (PAF) acétylhydrolase [13, 48] etle gène DCX (localisé sur le chromosome X) codant pour la doublecortine [12, 26] sont les causes les plus fréquentes de lissencéphalie detype 1 (chez l’homme pour DCX). De plus, le gène LIS 1 est contenudans la délétion à l’origine du syndrome de Miller-Dieker [33, 48] etles mutations du gène DCX donnent lieu chez la femme à un tableauclinique moins sévère que chez l’homme (double cortex) [12, 26]. Plusrécemment de rares cas de syndrome du double cortex ont étéégalement rapportés chez des hommes ayant une mutation dans lesgènes LIS 1 ou DCX [25, 68].Les produits de LIS 1 et DCX semblent être impliqués dans lafonction des microtubules et interagissent entre eux. Leur rôle exactdans la migration neuronale au cours du développement cérébralreste à préciser.Un gène a été identifié pour l’hétérotopie nodulaire périventriculairefamiliale. Il s’agit du gène FLN1 qui code pour la filamine 1, une
Tableau II. – Caractéristiques cliniques et génétiques des anomalies héréditaires du développement cortical.
Pathologies Mode de transmission Signes cliniques Gène et localisation
Lissencéphalie de type 1 AD Début précoce, épilepsie pharmacorésis-tante, crises généralisées de types variés,spasmes, retard mental important, symp-tômes variés neurologiques
Délétion ou mutation dans le gène LIS 1(17p13.3) [13, 48]
Lissencéphalie de type 1. Phénotypeobservé chez l’homme
Dominante liée à l’X Gène DCX (Xq22.3-q23) [12, 26]
Syndrome de Miller-Dieker AD Épilepsie généralisée pharmacorésistante,retard mental profond, dysmorphie faciale
Délétion en 17p13.3 emportant le gèneLIS 1 [33, 48]
Hétérotopie en bande ou double cortex Transmission dominante autosomique ouliée à l’X selon le gène impliqué
Épilepsie pharmacorésistante, retard men-tal moins sévère que dans la lissen-céphalie, voire absent
- Chez la femme : gène DCX (Xq22.3-q23)[12, 26]
- Rarement chez l’homme : gène DCX(Xq22.3-q23) ou LIS 1 (17p13.3) [25, 68]
Hétérotopie nodulaire périventriculaire Dominante liée à l’X Létale chez l’embryon masculin. Épilepsiesans retard mental, coagulopathie, canalartériel persistant, anomalies squelettiqueschez la femme
Gène FLN1 (Xq28) codant pour la fila-mine 1 [20]
AD : autosomique dominant ; AR : autosomique récessif.
17-044-C-70 Aspects génétiques des épilepsies Neurologie
4

protéine se liant à l’actine et à d’autres protéines du cytosquelette [20].Les mutations dans ce gène sont létales chez l’homme durant la vieembryonnaire. Chez la femme, une épilepsie pharmacorésistantegénéralement sans retard mental associé, et des anomaliessomatiques diverses sont observées (tableau II).
Épilepsies myocloniques progressiveshéréditaires
Il s’agit d’un groupe de maladies rares ayant en commun certainssignes cliniques (début le plus souvent dans l’enfance oul’adolescence, crises généralisées tonicocloniques, myoclonies,aggravation progressive avec apparition d’une démence et d’uneataxie cérébelleuse), mais dont les étiologies et l’évolution sontvariées. Pendant longtemps, le diagnostic spécifique a reposé sur laréalisation de bilans longs, complexes et coûteux, ne permettant pastoujours d’affirmer l’étiologie précise du syndrome. Plus récemment,le diagnostic génétique est devenu possible pour certaines épilepsiesmyocloniques progressives, simplifiant grandement la démarchediagnostique (tableau III). C’est le cas en particulier de la maladied’Unverricht-Lundborg et de la maladie de Lafora.
MALADIE D’UNVERRICHT-LUNDBORG
Cette maladie de transmission autosomique récessive débuteclassiquement entre 6 et 15 ans. L’évolution est typiquement lente,l’ataxie cérébelleuse et la détérioration intellectuelle classiquementrares, tardives et modérées [24, 59]. En fait, les possibilités actuelles dediagnostic génétique permettent de montrer que des formescliniques moins typiques existent, notamment des formes à débuttardif (jusqu’à l’âge de 32 ans) [28]. La présentation clinique etl’évolution peuvent être aggravées par la prescription dephénytoïne [15]. Les formes balte et méditerranéenne initialementdécrites sont dues à des mutations dans le gène de la cystatine Blocalisé en 21q22.3 [43, 63]. Il existe deux types de mutations qui
conduisent soit à une protéine de structure anormale ayant une pertede fonction, soit à une diminution de la transcription du gène. Ils’agit soit de rares mutations ponctuelles et de délétions dans larégion codante du gène [6, 7, 39, 62], soit majoritairement de l’expansiond’un dodécamère (CCC CGC CCC GCG)n situé en 5’ dans lepromoteur du gène [38, 40, 41, 90]. Tandis qu’à l’état normal ledodécamère existe en deux ou trois copies, les allèles mutéscomportent plus de 30 répétitions de celui-ci. Les premières étudesne montrent pas de corrélation entre la taille de l’expansion et l’âgede début de la maladie [41]. Il semble exister des porteurs deprémutations ayant 12 à 17 répétitions et un phénotype normal, etsusceptibles de transmettre des allèles pathologiques à leurdescendance (instabilité méiotique de l’expansion) [40].Les fréquences respectives des deux types de mutations varientselon l’origine géographique des patients. La forme balte est duegénéralement à la présence d’une mutation ponctuelle sur un desgènes de la cystatine B et à une expansion sur l’autre, plus rarementà une mutation ponctuelle sur les deux gènes. La formeméditerranéenne, dans laquelle l’existence d’une consanguinité estfréquente, est due à une expansion de dodécamère sur chacun desdeux gènes de la cystatine B.La cystatine B, un inhibiteur de cystéine-protéases, paraît impliquéedans la protection contre l’apoptose en inactivant directement ouindirectement les caspases. Cependant, les mécanismes exactsconduisant à la maladie ne sont pas encore connus.
MALADIE DE LAFORA
Cette maladie de transmission autosomique récessive débute entre10 et 18 ans. Elle est caractérisée par une aggravation neurologiquerapide avec détérioration intellectuelle précoce, et est fatale au boutd’une dizaine d’années d’évolution. Des crises d’épilepsie focalesoccipitales sont fréquentes dans cette maladie [71].Jusqu’à récemment, le diagnostic reposait sur la recherched’inclusions intracellulaires particulières par biopsie de peau (corpsde Lafora) [10]. Le diagnostic génétique est à présent possible. Le gène
Tableau III. – Épilepsies myocloniques progressives héréditaires.
Maladie Mode de transmission Locus, gène, protéine
Maladie d’Unverricht-Lundborg AR 21q, EMP1, cystatine B
Maladie de Lafora AR 6q, EMP2A, laforineAutres locus ?
Céroïdo-lipofuschinoses- Forme infantile précoce, forme infantile tardive et variante de laforme juvénile, toutes trois avec dépôts cytoplasmiques granulairesosmiophiles
AR 1p32 (CLN1), palmitoyl thioestérase 1 (lysosomale)
- Forme infantile tardive classique AR 11p15 (CLN2), tripeptidyl peptidase I (lysosomale)- Variante de la forme infantile tardive AR 15q21-23 (CLN6), gène ?- Variante finlandaise de la forme infantile tardive AR 13q21-q32 (CLN5), nouvelle protéine membranaire de fonction inconnue- Variante turque de la forme infantile tardive AR (CLN7), localisation ? Gène ?- Forme juvénile AR 16p12 (CLN3), nouvelle protéine membranaire intervenant dans la régu-
lation du pH lysosomal- Maladie de Kufs (forme adulte) AD (CLN4), localisation ? Gène ?
MERRF (myoclonus épilepsy ragged-red tibers syndrome) Transmission maternelle Mutations variables dans le génome mitochondrial (8344 ARNt la plusfréquente)
Sialidoses AR - 20q, protéine stabilisatrice du complexe alpha-neuraminidase et bêta-galactosidase- 6p, alpha-neuraminidase
Maladie de Gaucher (forme juvénile)(1) AR 1q, bêta-glucocérébrosidase
Certaines gangliosidoses AR Variables selon le type
Atrophie dentato-rubro-pallido-luysienne(1)
AD 12p, atrophine
Maladie de Huntington (forme juvénile)(1) AD 4q, huntingtine
Mutation dans le gène de la neuroserpine(1) AD 3q26, neuroserpine (une famille publiée)
Forme familiale de la maladie d’Alzheimer(1) AD 14q, préséniline 1
(1) Certaines formes cliniques de ces maladies peuvent donner lieu à un tableau d’épilepsie myoclonique progressive.AD : autosomique dominant ; AR : autosomique récessif ; ARNt : acide ribonucléique de transfert.
Neurologie Aspects génétiques des épilepsies 17-044-C-70
5

impliqué est localisé en 6q23-25 [73, 81]. Il s’agit du gène de la laforine[53, 82], une protéine tyrosine-phosphatase qui inhibe l’action destyrosine-kinases. Cette enzyme pourrait être impliquée dans lemétabolisme du glycogène. Des délétions et mutations ponctuelleshomozygotes dans la région codante du gène ont été démontréesdans les familles atteintes [53, 82].Un second locus pourrait être impliqué dans la maladie de Lafora[27, 54].Les autres affections génétiques susceptibles de se manifester parune épilepsie myoclonique progressive sont indiquées dans letableau III.
Maladies de transmissionmendélienne et anomalieschromosomiques pouvant comporterune épilepsie parmi les signescliniques
De nombreuses maladies héréditaires du système nerveux peuventcomporter une épilepsie dans leur tableau clinique souventcomplexe. Les principales sont regroupées dans le tableau IV.Certaines anomalies chromosomiques peuvent également comporter,avec une fréquence variable, une épilepsie. Les principales sont :
– la trisomie 21 (syndrome de Down) ;
– le syndrome d’Angelman (monosomie partielle 15q11) ;
– la trisomie 12p ;
– le syndrome de Wolf-Hirschhorn (monosomie partielle 4p) ;
– le syndrome de Klinefelter (XXY) ;
– le chromosome 20 en anneau.Enfin, diverses maladies métaboliques héréditaires peuvents’accompagner d’une épilepsie (aminoacidopathies, maladies ducycle de l’urée, du métabolisme des purines...).
Conclusion
Si l’analyse génétique de syndromes épileptiques bien définis au départa permis la découverte de gènes impliqués dans certains d’entre eux, lagénétique a inversement permis d’individualiser de nouveauxsyndromes non répertoriés dans la classification internationale desépilepsies et syndromes épileptiques ou d’enrichir le spectre cliniqued’entités déjà décrites.
Plus les connaissances avancent dans le domaine, plus l’importance dela composante génétique se confirme, mais plus les bases génétiques del’épilepsie semblent complexes : transmission polygénique oumendélienne, hétérogénéité allélique (même gène impliqué dans unsyndrome mais types de mutations différents) et/ou hétérogénéitégénétique (implication de gènes différents), pénétrance incomplète,expressivité variable.
L’identification des premiers gènes responsables démontre que lesmécanismes sous-jacents donnant lieu à une épilepsie ne sont pasunivoques. Cependant, à côté de gènes impliqués dans des anomaliesmorphologiques cérébrales, l’importance des canaux ioniques dansl’épileptogenèse s’affirme à travers l’étude des épilepsies idiopathiquesfamiliales (épilepsie frontale nocturne autosomique dominante et lerécepteur nicotinique à l’acétylcholine, convulsions néonatalesfamiliales bénignes et les canaux potassium voltage-dépendants,« GEFS + » et canal sodium voltage-dépendant). Aujourd’hui, on esttenté de faire de ces épilepsies des « canalopathies ». Il est intéressant deconstater que dans un même syndrome, différentes sous-unités quiparticipent à la formation d’un canal peuvent être altérées. Néanmoins,le mécanisme par lequel l’altération de canaux ioniques dontl’expression cérébrale semble diffuse peut conduire à des phénotypesaussi différents reste à élucider.
La plupart des découvertes génétiques ont bénéficié aux épilepsies àhérédité monogénique qui sont pourtant les plus rares. Toutefois, il estprévisible que ces découvertes pourront avoir des retombées pour lacompréhension des épilepsies à hérédité complexe.
Tableau IV. – Principaux exemples de maladies héréditaires comportant une épilepsie dans le phénotype.
Maladie Mode de transmission Locus, gène, protéine
Sclérose tubéreuse de Bourneville AD 9q34, TSC1, tubérine16p13.3, TSC2, hamartine
Neurofibromatose de type 1 AD 17q11.2, NF1, neurofibromine
Cavernomatose cérébrale familiale AD 7q, gène KRIT17p3q
Syndrome de Rett Dominant lié à l’X Xq28, gène MECP2
Certaines gangliosidoses AR Variables selon le type
MELAS (mitochondrial, myopathy, encephalopathy, lactic acidosis andstroke)
Transmission maternelle Mutations variables dans le génome mitochondrial (3243 ARNt laplus fréquente)
Syndrome de l’X fragile Dominant lié à l’X(1) Xq27.3, FMR1, FMR2
(1) Avec des particularités liées au type de mutation en cause : existence des femmes transmettrices ayant un retard mental et d’hommes normaux transmetteurs.AD : autosomique dominant ; AR : autosomique récessif ; ARNt : acide ribonucléique de transfert.
17-044-C-70 Aspects génétiques des épilepsies Neurologie
6

Références[1] Anderson VE, Rich SS, Wilcox KJ, Ahrens MJ, Weber JL,
Dubovsky J. Gene maping studies in febrile convulsions.Epilepsia 1995 ; 36 (suppl 3) : S215
[2] Annegers JF, Rocca WA, Hauser WA. Causes of epilepsy:contributions of the Rochester epidemiology project.Mayo Clin Proc 1996 ; 71 : 570-575
[3] Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M,Prudhomme JF, Baulac M et al. A second locus for familialgeneralized epilepsy with febrile seizures plus maps tochromosome 2q21-q23. Am J Hum Genet 1999 ; 65 :1078-1085
[4] BerkovicSF,McIntoshA,HowellRA,MitchellA,SheffieldLJ,Hopper JL. Familial temporal lobe epilepsy: a commondisorder identified in twins.Ann Neurol1996 ;40 :227-235
[5] Bertrand S, Weiland S, Berkovic SF, Steinlein OK, BertrandD. Properties of neuronal nicotinic acetylcholine receptormutants fromhumans suffering fromautosomaldominantnocturnal frontal lobe epilepsy. Br J Pharmacol 1998 ; 125 :751-760
[6] Bespalova IN, Adkins S, Pranzarelli M, Burmeister M. Novelcystatin B mutation and diagnostic PCR assay in anUnverricht-Lundborg progressive myoclonus epilepsypatient. Am J Med Genet 1997 ; 74 : 467-471
[7] Bespalova IN, Pranzatelli M, Burmeister M. G to C transver-sion at a splice acceptor site causes exon skipping in thecystatin B gene. Mutat Res 1997 ; 382 : 67-74
[8] BiervertC,SchroederBC,KubischC,BerkovicSF,ProppingP, Jentsch TJ et al. A potassium channel mutation in neona-tal human epilepsy. Science 1998 ; 279 : 403-406
[9] Bird TD. Epilepsy. In : King RA, Rotter JI, Motulsky AG eds.The genetic basis of common diseases. Oxford : OxfordUniversity Press, 1992 : 732-752
[10] Carpenter S, Karpati G. Sweat gland duct cells in Laforadisease: diagnosis by skin biopsy. Neurology 1981 ; 31 :1564-1568
[11] Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJet al. A pore mutation in a novel KQT-like potassiumchannel gene in an idiopathic epilepsy family. Nat Genet1998 ; 18 : 53-55
[12] Des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A,Carrie A, et al. A novel CNS gene required for neuronalmigration and involved in X-linked subcortial laminarheterotopia and lissencephaly syndrome. Cell 1998 ; 92 :51-61
[13] Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissen-cephaly. A humain brain malformation associated withdeletion of the LIS1 gene located at chromosome 17p13.JAMA 1993 ; 270 : 2838-2842
[14] Dubovsky J, Weber JL, Orr HT, Rich SS, Gil-Nagel A, Ander-son VE et al. A second gene for familial febrile convulsionsmaps on chromosome 19p. Am J Hum Genet 1996 ; 59(suppl 1) : A223
[15] Eldridge R, Iivanainen M, Stern R, Koerber T, Wilder BJ.Baltic myoclonus epilepsy: hereditary disorder of child-hood made worse by phenytoin. Lancet 1983 ; 2 : 838-842
[16] Elmslie FV, Rees M, Williamson MP, Keer M, Kjeldsen MJ,Pang KA et al. Genetic mapping of a major susceptibilitylocus for juvenilemyoclonicepilepsyonchromosome15q.Hum Mol Genet 1997 ; 6 : 1329-1334
[17] Elmslie FV, Williamson MP, Rees M, Kerr M, Kjeldsen MJ,Pang KA et al. Linkage analysis of juvenile myoclonic epi-lepsy and microsatellite loci spanning 61 cM of humanchromosome 6p in 19 nuclear pedigrees provides no evi-dence for a susceptibility locus in this region. Am J HumGenet 1996 ; 59 : 653-663
[18] Escayg A, Mac Donald BT, Meisler MH, Baulac S, HuberfeldG, An-Gourfinkel I et al. Mutations of SCN1A, encoding aneuronal sodium channel, in two families with GEFS+2.Nat Genet 2000 ; 24 : 343-345
[19] Fong CG, Shah PU, Gee MN, Serratosa JM, Castroviejo IP,Khan S et al. Childhood absence epilepsy with tonic-clonicseizures and electroencephalogram 3-4 Hz spike andmultispike-slow wave complexes linkage to chromosome8q24. Am J Hum Genet 1998 ; 63 : 1117-1129
[20] Fox JW, Lamperti E, Eksioglu YZ, Hong SE, Feng Y, GrahamDA et al. Mutations in filamin 1 prevent migration of cere-bralcorticalneurons inhumanperiventricularheterotopia.Neuron 1998 ; 21 : 1315-1325
[21] Fusco MD, Becchetti A, Patrignani A, Annesi G, Gambar-della A, Quattrone A et al. The nicotinic receptor beta2subunit is mutant in nocturnal frontal lobe epilepsy. NatGenet 2000 ; 26 : 275-276
[22] Gambardella A, Annesi G, De Fusco M, Patrignani A,Aguglia U, Annesi F et al. A new locus for autosomal domi-nant nocturnal frontal lobe epilepsy maps to chromo-mose 1. Neurology 2000 ; 55 : 1467-1471
[23] Gambardella A, Messina D, Le Piane E, Oliveri RL, Annesi G,Zappia M et al. Familial temporal lobe epilepsy: autosomaldominant inheritance in a large pedigree from SouthernItaly. Epilep Res 2000 ; 38 : 127-132
[24] Genton P, Michelucci R, Tassinari CA, Roger J. The RamsayHunt syndrome revisited: Mediterranean myoclonusversus mitochondrial encephalomyopathy with ragged-red fibers and Baltic myoclonus. Acta Neurol Scand 1990 ;81 : 8-15
[25] Gleeson JG.Classical lissencephalyanddoublecortex(sub-cortical band heterotopia): LIS1 and doublecortin. CurrOpin Neurol 2000 ; 13 : 121-125
[26] Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S,Scheffer IE et al. Doublecortine, a brain specific genemutated in human X-linked lissencephaly and doublecortex syndrome encodes a putative signaling protein. Cell1998 ; 92 : 63-72
[27] Gomez-Garre P, Anta B, Castro-Gago M, Lindhout D, Tas-sinariCA,MichelucciRetal.Reductionof theLaforadiseasecandidate gene region to a 2 cM interval in chromosome6q24 and evidence for genetic heterogeneity. Eur J HumGenet 1998 ; 6 : 152
[28] Gouider R, Ibrahim S, Fredj M, Gargouri A, Saidi H, Ouez-zani R et al. Unverricht-Lundborg disease: clinical and elec-trophysiologic study of 19 Maghreb families. Rev Neurol1998 ; 154 : 503-507
[29] Greenberg DA, Delgado-Escueta AV, Widelitz H, SparkesRS, Treiman L, Maldonado HM et al. Juvenile myoclonicepilepsy (JME) may be linked to the BF and HLA loci onhumanchromosome6.AmJMedGenet1988;31:185-192
[30] Greenberg DA, Durner M, Resor S, Rosenbaum D, ShinnarS. The genetics of idiopathic generalized epilepsies of ado-lescent onset: difference between juvenile myoclonic epi-lepsy and epilepsy with random grand mal and with awa-kening grand mal. Neurology 1995 ; 45 : 942-946
[31] Guerrini R, Bonanni P, Nardocci N, Parmeggianni L, Picci-rilli M, De Fusco M et al. Autosomal recessive rolandic epi-lepsy with paroxysmal exercise-induced dystonia and wri-ter’s cramp: delineation of the syndrome and genemapping to chromosome 16p12-11. 2. Ann Neurol 1999 ;45 : 344-352
[32] Guipponi M, Rivier F, Vigevano F, Beck C, Crespel A,Echenne B et al. Linkage mapping of benign familial infan-tile convulsions to chromosome 19q. Hum Mol Genet1997 ; 6 : 473-477
[33] Hattori M, Adachl H, Tsujlmoto M, Aral H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brainplatelet-activating factor. Nature 1994 ; 370 : 216-218
[34] HiroseS, IwataH,AkiyoshiH,KobayashiK, ItoM,WadaKetal .AnovelmutationofCHRNA4responsible for autosomaldominantnocturnal frontal lobeepilepsy.Neurology1999;53 : 1749-1753
[35] Johnson EW, Dubovsky J, Rich SS, O’Donovan CA, Orr HT,Anderson VE et al. Evidence for a novel gene for familialfebrile convulsions, FEB2, linked to chromosome 19p in anextended family from the Midwest. Hum Mol Genet 1998 ;7 : 63-67
[36] Kugler SL, Stenroos ES, Mandelbaum DE, Lehner T, McKoyVV, Prossick T et al. Hereditary febrile seizures: phenotypeandevidence for a chromosome19p locus.Am J Med Genet1998 ; 79 : 354-361
[37] Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J.Mutation causing autosomal dominant nocturnal frontallobe epilepsy alters Ca2+ permeability, conductance, andgating of human alpha4beta2 nicotinic acetylcholinereceptors. J Neurosci 1997 ; 17 : 9035-9047
[38] Lafreniere RG, Rochefort DL, Chretien N, Rommens JM,Cochius JI, Kalviainen R et al. Unstable insertion in the5’flanking region of the cystatin B gene is the mostcommonmutation inprogressivemyoclonusepilepsy type1, EPM1. Nat Genet 1997 ; 15 : 298-302
[39] Lalioti MD, Mirotsou M, Buresi C, Peitsch MC, Rossier C,Ouazzani R et al. Identification of mutations in cystatin B,the gene responsible for the Unverricht-Lundborg type ofprogressive myoclonus epilepsy (EPM1). Am J Hum Genet1997 ; 60 : 342-351
[40] Lalioti MD, Scott HS, Buresi C, Rossier C, Bottani A, MorrisMAetal.Dodecamerrepeatexpansion incystatinBgene inprogressive myoclonus epilepsy. Nature 1997 ; 386 :847-851
[41] Lalioti MD, Scott HS, Genton P, Grid D, Ouazzani R,M’Rabet A et al. A PCR amplification method reveals insta-bility of the dodecamer repeat in progressive myoclonusepilepsy (EPM1) and no correlation between the size of therepeat and age at onset. Am J Hum Genet 1998 ; 62 :842-847
[42] Lee WL, Tay A, Ong HT, Goh LM, Monaco AP, SzepetowskiP.Associationof infantileconvulsionswithparoxysmaldys-kinesias (ICCA syndrome): confirmation of linkage tohuman chromosome 16p12-q12 in a Chinese family. HumGenet 1998 ; 103 : 608-612
[43] LehesjokiAE,KoskiniemiM,SistonenP,Miao J,Hastbacka J,NorioRet al. Localizationof agene forprogressivemyoclo-nusepilepsy tochromosome21q22.ProcNatl AcadSciUSA1991 ; 88 : 3696-3699
[44] Leppert M, Anderson VE, Quattlebaum T, Stauffer D,O’Connell P, Nakamura Y et al. Benign familial neonatalconvulsions linked to genetic markers on chromosome 20.Nature 1989 ; 337 : 647-648
[45] Lerche H, Bievert C, Alekov AK, Schleithoff L, Lindner M,Klingler W et al. A reduced K+ current due to a novel muta-tion in KCNQ2 causes neonatal convulsions. Ann Neurol1999 ; 46 : 305-312
[46] Lewis TB, Leach RJ, Ward K, O’Connell P, Ryan SG. Geneticheterogeneity in benign familial neonatal convulsions:identification of a new locus on chromosome 8q. Am J HumGenet 1993 ; 53 : 670-675
[47] Liu AW, Delgado-Escueta AV, Serratosa JM, Alonso ME,Medina MT, Gee MN et al. Juvenile myoclonic epilepsylocus in chromosome 6p21. 2-p11: linkage to convulsionsand electroencephalography trait. Am J Hum Genet 1995 ;57 : 368-381
[48] Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R,Ledbetter DH. Point mutations and an intragenic deletionin LIS1, the lissencephaly causative gene in isolated lissen-cephaly sequence and Miller-Dieker syndrome. Hum MolGenet 1997 ; 6 : 157-164
[49] Lopes-Cendes I, Scheffer IE, Berkovic SF, Rousseau M,AndermannE,RouleauGA.Anewlocus forgeneralizedepi-lepsy with febrile seizures plus maps to chromosome 2. AmJ Hum Genet 2000 ; 66 : 698-701
[50] Malafosse A, Leboyer M, Dulac O, Navelet Y, Plouin P, BeckC et al. Confirmation of linkage of benign familial neonatalconvulsions toD20S19andD20S20.HumGenet1992;89:54-58
[51] Mautner VF, Lindenau M, Gottesleben A, Goetze G, KluweL.Supportingevidenceofagene forpartial epilepyon10q.Neurogenetics 2000 ; 3 : 31-34
[52] Mikami M, Yasuda T, Terao A, Nakamura M, Ueno SI,Tanabe H et al. Localization of a gene for benign adultfamilial myoclonic epilepsy to chromosome 8q23. 3-q24.Am J Hum Genet 1999 ; 65 : 745-751
[53] Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S,Mungall AJ et al. Mutations in a gene encoding a novelprotein tyrosine phosphatase cause progressive myoclo-nus epilepsy. Nat Genet 1998 ; 20 : 171-174
[54] Minassian BA, Sainz J, Serratosa JM, Gee M, Sakamoto LM,BohlegaSetal.Genetic locusheterogeneity inLafora’spro-gressive myoclonus epilepsy. Ann Neurol 1999 ; 45 :262-265
[55] Moulard B, Guipponi M, Chaigne D, Mouthon D, Buresi C,Malafosse A. Identification of a new locus for generalizedepilepsywith febrile seizuresplus (GEFS+)onchromosome2q24-q33. Am J Hum Genet 1999 ; 65 : 1396-1400
[56] Nakayama J, Hamano K, Iwasaki N, Nakahara S, HorigomeY, Saitoh H et al. Significant evidence for linkage of febrileseizures to chromosome 5q14-q15. Hum Mol Genet 2000 ;9 : 87-91
[57] Neubauer BA, Fiedler B, Himmelein B, Kampfer F, LasskerU, Schwabe G et al. Centrotemporal spikes in families withrolandic epilepsy: linkage to chromosome 15q14. Neuro-logy 1998 ; 51 : 1608-1612
[58] Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C,Barhanin J et al. A novel mutation in the potassium channelgene KVLQT1 causes the Jervell and Lange-Nielsen cardio-auditory syndrome. Nat Genet 1997 ; 15 : 186-189
[59] Norio R, Koskiniemi M. Progressive myoclonus epilepsy:genetic and nosological aspects with special reference to107 Finnish patients. Clin Genet 1979 ; 15 : 382-398
[60] Ottman R, Risch N, Hauser WA, Pedley TA, Lee JH, Barker-Cummings C et al. Localization of a gene for partial epi-lepsy to chromosome 10q. Nat Genet 1995 ; 10 : 56-60
[61] Peiffer A, Thompson J, Charlier C, Otterud B, Varvil T,Pappas C et al. A locus for febrile seizures (FEB3) maps tochromosome 2q23-24. Ann Neurol 1999 ; 46 : 671-678
[62] Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virta-neva K, Miao J et al. Mutations in the gene encoding cysta-tin B in progressive myoclonus epilepsy. Science 1996 ;271 : 1731-1734
[63] Pennacchio LA, Myers RM. Isolation and characterizationof the mouse cystatin B gene. Genome Res 1996 ; 6 :1103-1109
Neurologie Aspects génétiques des épilepsies 17-044-C-70
7

[64] Phillips HA, Favre I, Kirkpatrick M, Zuberi SM, Goudie D,Heron SE et al. CHRNB2 is the second acetylcholine recep-tor subunit associated with autosomal dominant noctur-nal frontal lobe epilepsy. Am J Hum Genet 2001 ; 68 :225-231
[65] Phillips HA, Scheffer IE, Berkovic SF, Hollway GE, Suther-land GR, Mulley JC. Localization for a gene for autosomaldominant nocturnal frontal lobe epilepsy to chromosome20q13. 2. Nat Genet 1995 ; 10 : 117-118
[66] Phillips HA, Scheffer IE, Crossland KM, Bhatia KP, Fish DR,Marsden CD et al. Autosomal dominant nocturnal frontal-lobe epilepsy: genetic heterogeneity and evidence for asecond locus at 15q24. Am J Hum Genet 1998 ; 63 :1108-1116
[67] Picard F, Baulac S, Kahane P, Hirsch E, Sebastianelli R,Thomas P et al. Dominant partial epilepsies. A clinical elec-trophysiological and genetic study of 19 European fami-lies. Brain 2000 ; 123 : 1247-1262
[68] Pilz DT, Kuc J, Matsumoto N, Bodurtha J, Bernadi B, Tassi-nari CA et al. Subcortical band heterotopia in rare affectedmales can be causes by missense mutations in DCX (XLIS)or LIS1. Hum Mol Genet 1999 ; 8 : 1757-1760
[69] Plaster NM, Uyama E, Uchino M, Ikeda T, Flanigan KM,Kondo I et al. Genetic localization of the familial adult myo-clonic epilepsy (FAME) gene to chromosome 8q24. Neuro-logy 1999 ; 53 : 1180-1183
[70] Poza JJ, Saenz A, Martinez-Gil A, Cheron N, Cobo AM,Urtasun M et al. Autosomal dominant lateral temporal epi-lepsy: clinical and genetic study of a large Basque pedigreelinkedtochromosome10q.AnnNeurol1999;45:182-188
[71] Roger J, Genton P, Bureau M. Progressive myoclonus epi-lepsies. In : Dam M, Gram L eds. Comprehensive epilepto-logy. New York : Raven Press, 1990 : 215-231
[72] Saenz A, Galan J, Caloustian C, Lorenzo F, Marquez C,Rodriguez N et al. Autosomal dominant nocturnal frontallobe epilepsy in a Spanish family with a Ser252Phe muta-tion in the CHRNA4 gene. Arch Neurol 1999 ; 56 :1004-1009
[73] Sainz J, Minassian BA, Serratosa JM, Gee MN, SakamatoLM, Iranmanesh R et al. Lafora progressive myoclonus epi-lepsy: narrowing the chromosome 6q24 locus by recom-binations and homozygosities. Am J Hum Genet 1997 ; 61 :1205-1209
[74] Sander T, Bockenkamp B, Hildmann T, Blasczyk R, Kretz R,Wienker TF et al. Refined mapping of the epilepsy suscep-tibility locus EJM1onchromosome6. Neurology 1997 ;49 :842-847
[75] Scheffer IE, Berkovic SF. Generalized epilepsy with febrileseizures plus: a genetic disorder with heterogeneous clini-cal phenotypes. Brain 1997 ; 120 : 479-490
[76] Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, MarsdenCD, Andermann F et al. Autosomal dominant frontal epi-lepsy misdiagnosed as sleep disorder. Lancet 1994 ; 343 :515-517
[77] Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, MarsdenCD, Andermann E et al. Autosomal dominant nocturnalfrontal epilepsy: a distinctive clinical disorder. Brain 1995 ;118 : 61-73
[78] Scheffer IE, Jones L, Pozzebon M, Howell RA, Saling MM,Berkovic SF. Autosomal dominant rolandic epilepsy andspeech dyspraxia: a new syndrome with anticipation. AnnNeurol 1995 ; 38 : 633-642
[79] Scheffer IE, PhillipsHA,O’BrienCE,SalingMM,Wrennal JA,Wallace RH et al. Familial partial epilepsy with variable foci:anewpartial epilepsy syndromewithsuggestionof linkageto chromosome 2. Ann Neurol 1998 ; 44 : 890-899
[80] SchroederBC,KubischC,SteinV, JentschTJ.Moderate lossof function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+
channels causes epilepsy. Nature 1998 ; 396 : 687-690[81] Serratosa JM, Delgado-Escueta AV, Posada I, Shih S, Drury
I, Berciano J et al. The gene for progressive myoclonus epi-lepsy of the Lafora type maps to chromosome 6q. Hum MolGenet 1995 ; 4 : 1657-1663
[82] Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, DeBernabeBV,LinahoutDetal.Anovelproteintyrosinephos-phatase gene is mutated in progressive myoclonus epi-lepsy of the Lafora type (EPM2). Hum Mol Genet 1999 ; 8 :345-352
[83] Singh NA, Charlier C, Stauffer D, Dupont BR, Leach RJ,Melis R et al. A novel potassium channel gene, KCNQ2, ismutated in an inherited epilepsy of newborns. Nat Genet1998 ; 18 : 25-29
[84] Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalisedepilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol 1999 ; 45 :75-81
[85] Steinlein OK, Magnusson A, Stoodt J, Bertrand S, WeilandS, Berkovic SF et al. An insertion mutation of the CHRNA4gene in a family with autosomal dominant nocturnalfrontal lobe epilepsy. Hum Mol Genet 1997 ; 6 : 943-947
[86] Steinlein OK, Mulley JC, Propping P, Wallace RH, PhillipsHA, Sutherland GR et al. A missense mutation in the neu-ronal nicotinic acetylcholine receptor alpha 4 subunit isassociated with autosomal dominant nocturnal frontallobe epilepsy. Nat Genet 1995 ; 11 : 201-203
[87] Szepetowski P, Rochette J, Berquin P, Piussan C, LathropGM, Monaco AP. Familial infantile convulsions and paro-xysmal choreoathetosis: a new neurological syndromelinked to the pericentromeric region of human chromo-some 16. Am J Hum Genet 1997 ; 61 : 889-898
[88] Terada K, Ikeda A, Mima T, Kimura K, Nagahama Y,Kamioka Y et al. Familial cortical myoclonic tremor as aunique form of cortical reflex myoclonus. Mov Disord1997 ; 12 : 370-377
[89] Tinel N, Lauritzen I, Choabe C, Lazdunski M, Borsotto M.The KCNQ2 potassium channel: splice variants, functionaland developmental expression. Brain localization andcomparison with KCNQ3. FEBS Lett 1998 ; 438 : 171-176
[90] Virtaneva K, D’Amato E, Miao J, Koskiniemi M, Norio R,Avanzini G et al. Unstable minisatellite expansion causingrecessively inheritedmyoclonusepilepsy,EPM1.NatGenet1997 ; 15 : 393-396
[91] WallaceRH,BerkovicSF,Howell RA, SutherlandGR,MulleyJC. Suggestion of a major gene for familial convulsionsmapping to 8q13-21. J Med Genet 1996 ; 33 : 308-312
[92] Wallace RH, Wang DW, Singh R, Scheffer IE, George AL,Phillips HA et al. Febrile seizures and generalized epilepsyassociated with a mutation in the Na+-channel beta1subunit gene SCN1B. Nat Genet 1998 ; 19 : 366-370
[93] Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS etal. KCNQ2 and KCNQ3 potassium channel subunits:molecular correlates of the M-channel. Science 1998 ; 282 :1890-1893
[94] Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM,Vanraay TJ et al. Positional cloning of a novel potassiumchannel gene: KVLQT1 mutations cause cardiac arrhy-thmias. Nat Genet 1996 ; 12 : 17-23
[95] Weissbecker KA, Durner M, Janz D, Scaramelli A, SparkesRS, Spence VA. Confirmation of linkage between juvenilemyoclonic epilepsy locus and the HLA region of chromo-some 6. Am J Med Genet 1991 ; 38 : 32-36
[96] Westling B, Weissbecker KA, Serratosa JM, Jara-Prado A,Alsonso ME, Cordova S et al. Evidence for linkage of juve-nile myoclonic epilepsy with absence to chromosome 1p.Am J Hum Genet 1996 ; 59 (suppl 1) : A241
[97] XiongL, LabudaM,LiDS,HudsonTJ,DesbiensR,PatryGetal . Mapping of a gene determining familial partial epilepsywith variable foci to chromosome 22q11-q12. Am J HumGenet 1999 ; 65 : 1698-1710
[98] Zara F, Bianchi A, Avanzini G, Di Donato S, Castellotti B,Patel PI et al. Mapping of genes predisposing to idiopathicgeneralized epilepsy. Hum Mol Genet 1995 ; 4 : 1201-1207
[99] Zara F, Labuda M, Garofalo PG, Durisotti C, Bianchi A,Castellotti B et al. Unusual EEG pattern linked to chromo-some 3p in a family with idiopathic generalized epilepsy.Neurology 1998 ; 51 : 493-498
17-044-C-70 Aspects génétiques des épilepsies Neurologie
8





























