Embed Size (px)
Citation preview

Tesi di Dottorato di Ricerca in co-tutela
UNIVERSITÀ DEGLI STUDI DI PARMA Dipartimento di Patologia e Medicina di Laboratorio
Sezione di Microbiologia Dottorato di Ricerca in Microbiologia e Virologia
Coordinatore: Chiar.mo Prof. Carlo Chezzi XVII ciclo
UNIVERSITÀ PARIS XII - VAL DE MARNE U.F.R. di Scienze
Dottorato di Ricerca in “Sciences de la Vie et de la Santé” Disciplina: Biologia Cellulare e Molecolare
Coordinatore: Chiar.mo Prof. Michel Goossens
Aspetti morfologici e funzionali dei proteasomi durante il processo
di differenziamento di cellule muscolari ed in corso di infezione
da citomegalovirus umano in vitro
Dottoranda Dr.ssa Silvia Covan
Commissione
Chiar.mo Prof. Carlo Chezzi Tutore (Università degli Studi di Parma) Chiar.mo Prof. Jean Foucrier Tutore (Università Paris XII – Val de Marne) Dr.ssa Isabelle Saint Girons Esaminatore (Institut Pasteur – Paris) Chiar.ma Prof.ssa Maria Paola Landini Relatore (Università degli Studi di Bologna)

N. attribué par la bibliothèque .........................................................
Thèse de Doctorat en co-tutelle
UNIVERSITÉ DE PARME Département de Pathologie et Médecine de Laboratoire
Section de Microbiologie École Doctorale: Microbiologie et Virologie
Directeur: Pr. Carlo Chezzi XVII cycle
UNIVERSITÉ PARIS XII-VAL DE MARNE U.F.R. de Sciences
École Doctorale: Sciences de la Vie et de la Santé Discipline: Biologie Cellulaire et Moléculaire
Directeur: Pr. Michel Goossens
Présentée et soutenue publiquement
par
Silvia Covan
le 23 Mai 2006
Aspects morphologiques et fonctionnels des protéasomes
au cours de la différenciation des cellules musculaires et pendant
l’infection par le cytomégalovirus humain in vitro
JURY
Pr. Carlo Chezzi Directeur de thèse (Université de Parme) Pr. Jean Foucrier Directeur de thèse (Université Paris XII – Val de Marne) Dr. Isabelle Saint Girons Examinateur (Institut Pasteur – Paris) Pr. Maria Paola Landini Rapporteur (Université de Bologne)

Ai miei genitori
“Casa, il posto da cui non si può fuggire, il posto verso cui ruota la bussola
del cuore…..”
da La spiaggia rubata di Joanne Harris

Indice
INDICE

Indice
-II-
1. RIASSUNTO Pag. 2
2. PRESENTAZIONE DELLA STUDIO Pag. 5
3. INTRODUZIONE
3.1 IL PROTEASOMA Pag. 9
3.1.1 STRUTTURA DEL PROTEASOMA Pag. 9
3.1.2 IL PROTEASOMA 20S E I SUOI COMPLESSI REGOLATORI Pag. 10
3.1.3 IL SISTEMA UBIQUITINA-PROTEASOMA Pag. 12
3.1.4 FUNZIONI DEI PROTEASOMI Pag. 15
3.2 I MODELLI DI STUDIO Pag. 26
3.2.1 IL TESSUTO MUSCOLARE Pag. 26
3.2.2 CITOMEGALOVIRUS Pag. 37
4. OBIETTIVI DELLA RICERCA Pag. 50
5. MATERIALI E METODI
5.1 STUDIO DI LOCALIZZAZIONE INTRACELLULARE DEI PROTEASOMI Pag. 53
5.2 CLONAGGIO IN VETTORI PLASMIDICI Pag. 57
5.3 COLTURE CELLULARI Pag. 66
5.4 IMPIEGO DEI VETTORI PLASMIDICI Pag. 67
5.5 STRATEGIA UTILIZZATA PER LA PRODUZIONE DI VETTORI RETROVIRALI Pag. 73
5.6 PRODUZIONE E IMPIEGO DI PARTICELLE RETROVIRALI (ECOTROPICHE) Pag. 77
5.7 PRODUZIONE DI PARTICELLE RETROVIRALI (ANFOTROPICHE) Pag. 82
5.8 INFEZIONE DI FIBROBLASTI CON UNO STIPITE UMANO DI
CITOMEGALOVIRUS Pag. 84
6. RISULTATI
6.1 PRIMO MODELLO DI STUDIO: CELLULE MUSCOLARI E PROTEASOMI Pag. 88
6.1.1 I PROTEASOMI SONO DISTRIBUITI SECONDO UN PROFILO PSEUDO-SARCOMERICO NEL MUSCOLO SCHELETRICO
Pag. 88
6.1.2 IL PROFILO PSEUDO-SARCOMERICO DI SPECIFICI PROTEASOMI SI MODIFICA IN RAPPORTO ALL’ESTENSIONE DEL SARCOMERO IN VIVO
Pag. 90
6.1.3 UNA SIGNIFICATIVA QUANTITÀ DI PROTEASOMI È ASSOCIATA ALLE MIOFIBRILLE
Pag. 93
6.1.4 NON TUTTI I PROTEASOMI PRESENTI NELLE MIOFIBRILLE SONO ASSOCIATI AD ACTINA
Pag. 102

Indice
-III-
6.1.5 ALLESTIMENTO DI VETTORI DI ESPRESSIONE PER LE SEQUENZE GENICHE DI INTERESSE
Pag. 103
6.1.6 ESPRESSIONE DELLE PROTEINE DI FUSIONE ATTRAVERSO ESPERIMENTI DI TRASFEZIONE
Pag. 108
6.1.7 ALLESTIMENTO DI VETTORI RETROVIRALI PER LE SEQUENZE GENICHE DI INTERESSE
Pag. 116
6.1.8 PROFILO DI DISTRIBUZIONE DI SPECIFICI PROTEASOMI IN POPOLAZIONI DI CELLULE C2.7 CHE ESPRIMONO I COSTRUTTI DI INTERESSE
Pag. 127
6.2 SECONDO MODELLO DI STUDIO: FIBROBLASTI UMANI IN CORSO DI
INFEZIONE DA CITOMEGALOVIRUS IN VITRO E PROTEASOMI Pag. 130
6.2.1 LA FUNZIONE ENZIMATICA DEI PROTEASOMI È RILEVANTE AI FINI DELL’EVOLUZIONE DELL’INFEZIONE PRODUTTIVA DA CITOMEGALOVIRUS IN FIBROBLASTI MRC5
Pag. 130
6.2.2 L’ATTIVITÀ ENZIMATICA DEI PROTEASOMI RISULTA RILEVANTE SOLO IN ALCUNE FASI DEL CICLO CELLULARE, AI FINI DELL’EVOLUZIONE DELL’INFEZIONE PRODUTTIVA DA CITOMEGALOVIRUS IN FIBROBLASTI MRC5
Pag. 132
6.2.3 I PROTEASOMI SONO PRESENTI ANCHE A LIVELLO NUCLEOLARE A TEMPI PRECOCI DOPO L’INFEZIONE DI FIBROBLASTI MRC5 CON CITOMEGALOVIRUS UMANO
Pag. 138
6.2.4 PRODUZIONE DI VETTORI RETROVIRALI RECANTI LE SEQUENZE GENICHE PROTEASOMALI DI INTERESSE E IN GRADO DI INFETTARE CELLULE UMANE, A LORO VOLTA SUSCETTIBILI ALL’INFEZIONE DA CITOMEGALOVIRUS
Pag. 141
7. DISCUSSIONE Pag. 146
8. BIBLIOGRAFIA Pag. 165
9. RINGRAZIAMENTI Pag. 188

1. Riassunto
1. RIASSUNTO

1. Riassunto
-2-
Il lavoro sperimentale svolto ha preso in considerazione due differenti modelli di studio il
cui denominatore comune era rappresentato dai proteasomi. In riferimento al primo
modello, sono stati inizialmente effettuati esperimenti volti ad evidenziare la
localizzazione di specifici proteasomi nell’ambito della struttura sarcomerica nel tessuto
muscolare scheletrico di ratto e di allestire strumenti molecolari idonei allo studio della
distribuzione dei proteasomi durante il processo di differenziamento miogenico. Gli stessi
strumenti molecolari sono stati allestiti durante lo studio del secondo modello
sperimentale, il cui scopo è stato quello di valutare il coinvolgimento di suddetto
complesso multi-enzimatico in corso di infezione produttiva da citomegalovirus umano in
fibroblasti embrionali umani. In particolare, l’espressione di geni precocissimi virali è
stata inizialmente studiata in assenza o in presenza di uno specifico inibitore dell’attività
proteolitica dei proteasomi in monostrati di fibroblasti infettati non sincronizzati.
Successivamente, l’espressione di suddette proteine virali è stata valutata in monostrati di
fibroblasti infettati sincronizzati. Inoltre, è stata studiata la distribuzione intracellulare di
specifici proteasomi nel suddetto modello, allo scopo di verificare eventuali modificazioni
della stessa in corso di infezione produttiva da citomegalovirus umano.
Résumé de la thèse
Deux volets thématiques se rapportant à la biologie des protéasomes ont été abordés au
cours de ce travail de thèse. Le premier a consisté à préciser la localisation des
protéasomes au sein de la structure sarcomérique de muscles striés de rat et d’élaborer
des outils moléculaires appropriés, permettant de suivre la distribution des protéasomes
au cours de la différenciation myogénique. Ces mêmes outils ont également été élaborés
au cours de la deuxième partie du travail, qui a consisté à évaluer la participation de ces
complexes multicatalytiques lors de l’infection de fibroblastes par le cytomegalovirus
humain. L’expression des protéines virales très précoces a été étudiée dans les fibroblastes
synchronisés ou non au cours de l’infection, à la suite de l’utilisation ou non d’un
inhibiteur de la fonction protéolytique des protéasomes. On a aussi étudié la distribution
des protéasomes dans les cellules infectées, afin d’évaluer si celle-ci se modifiait au cours
de l’infection virale.

1. Riassunto
-3-
Thesis summary
Two different topics about proteasomes have been evaluated during this doctoral thesis.
In the first part we have showed the proteasome localization at the level of the sarcomeric
structure of rat skeletal muscle and we have developed useful molecular tools to follow
proteasome distribution during myogenic differentiation. The same tools have been
developed during the second part of this project. The goal of this work was to evaluate the
involvement of this multicatalytic complex in fibroblasts infected by human
cytomegalovirus. Viral immediate-early protein expression was studied using fibroblasts
synchronized or not during viral infection, using an inhibitor of the proteolytic activity of
proteasomes to examine the effects of the lack of this proteasomal function on viral
protein expression. In addition, we investigated the intracellular distribution of
proteasomes in infected cells with the aim to evaluate if proteasome distribution changed
during viral infection.

2. Presentazione dello studio
2. PRESENTAZIONE DELLO STUDIO

2. Presentazione dello studio
-5-
La problematica scientifica prescelta nel quadro di questa tesi si basa su un complesso
lavoro sperimentale, volto a mettere in luce la possibile partecipazione di complessi
molecolari assai rilevanti nell’economia cellulare, i proteasomi, in due condizioni
altamente “dinamiche” per la cellula, ossia il differenziamento muscolare e l’infezione
virale. Se l’intervento dei proteasomi in diverse funzioni regolatorie della cellula in
condizioni fisiologiche è supportato da numerosi dati di letteratura, ancora aperte e del
tutto incomplete rimangono le conoscenze volte a delucidare le modalità di intervento dei
proteasomi in eventi che comportino una cospicua alterazione del normale equilibrio
omeostatico cellulare, provocando, di conseguenza, una decisa e marcata rimodulazione
quali/quantitativa di diverse componenti molecolari.
In riferimento al primo modello di studio, traendo le premesse da dati sperimentali
precedentemente ottenuti dal nostro gruppo di ricerca e da quello del Prof. Jean Foucrier,
sono stati inizialmente effettuati esperimenti volti ad evidenziare la specifica
localizzazione di proteasomi in sezioni di muscolo scheletrico di ratto e in colture di
miofibrille allestite in vitro. I risultati ottenuti hanno evidenziato un profilo di
distribuzione di specifici proteasomi direttamente correlabile al grado di
estensione/contrazione del sarcomero; in particolare, grazie anche al supporto di studi di
immunoelettromicroscopia, è stata appurata la colocalizzazione della subunità α1/p27K
con la regione centrale dei microfilamenti di actina. Tali dati sono stati, inoltre, avvalorati
anche da studi effettuati su preparati di miofibrille precedentemente trattate con
gelsolina, sostanza in grado di rimuovere l’actina.
In una fase successiva della ricerca, sono stati allestiti strumenti molecolari idonei a potere
essere utilizzati, in prospettiva futura, per lo studio della distribuzione dinamica dei
proteasomi in cellule viventi. Per la messa a punto di tali strumenti, alcune sequenze
geniche codificanti per specifiche subunità proteasomali sono state fuse con sequenze
codificanti per proteine fluorescenti naturali, verificandone poi, attraverso differenti
metodi, la funzionalità con esperimenti di trasfezione in cellule CHO e nella linea
cellulare miogenica di topo C2.7; in tal modo, si è potuto verificare che le suddette
proteine di fusione venivano utilizzate per la formazione di nuovi complessi
proteasomali. Sulla base di questi risultati è possibile affermare che l’utilizzo di tali
costrutti per studi su aspetti morfologici e funzionali riguardanti i proteasomi può essere
considerato come rappresentativo del complesso proteasomale vero e proprio, in cui essi
vengono incorporati.

2. Presentazione dello studio
-6-
Le sequenze codificanti per le proteine di fusione sono state successivamente clonate in un
vettore retrovirale, utilizzato poi per eseguire esperimenti di trasfezione della linea
cellulare Bosc-23, allo scopo di consentire la produzione di particelle retrovirali defettive
contenenti i costrutti di interesse e impiegate in seguito per eseguire infezioni della linea
miogenica C2.7. In tal modo, sono state ottenute popolazioni di cellule C2.7 in grado di
esprimere stabilmente le sequenze geniche per le proteine di fusione, che hanno
consentito di dimostrare la presenza delle stesse a livello di proteasomi 20S. In parallelo,
tali costrutti molecolari sono stati valutati anche dal punto di vista della funzione
enzimatica, anch’essa preservata nei complessi proteasomali in cui gli stessi costrutti sono
inclusi. Sono state eseguite quindi osservazioni in microscopia a fluorescenza di
monostrati di cellule C2.7 in grado di esprimere stabilmente i costrutti di interesse; il
segnale di fluorescenza relativo alle proteine di fusione era presente sia nel
compartimento citoplasmatico che in quello nucleare delle suddette cellule, con intensità
decisamente maggiore in quest’ultimo distretto. Inoltre, la fluorescenza riscontrata a
livello del nucleo, in alcuni casi, appariva di tipo granulare e concentrata a livello di
specifiche regioni nucleari; tale distribuzione sembrava accentuarsi nei nuclei dei miotubi,
rispetto a quelli dello stadio mioblastico.
Il secondo modello preso in considerazione per lo studio dei proteasomi ha previsto
l’impiego di cellule fibroblastiche umane (MRC5) per lo studio dell’infezione virale in
vitro da parte di uno stipite umano di citomegalovirus (AD169).
L’espressione dei prodotti maggiori dei geni precocissimi virali IE1 e IE2 è stata
inizialmente studiata in assenza o in presenza di uno specifico inibitore dei proteasomi,
denominato MG132. Il trattamento con MG132 determina una significativa riduzione
dell’espressione dei geni IE già a tempi molto precoci di infezione. L’effetto inibitorio
della sostanza non risulta peraltro della stessa entità per tutte le cellule del monostrato,
suggerendo che l’eventuale inibizione possa esprimersi con differente efficacia
dipendentemente dalla fase del ciclo cellulare in cui l’infezione ha avuto inizio. Le cellule
MRC5 sono state quindi sincronizzate mediante applicazione di uno specifico protocollo
sperimentale. I dati ottenuti hanno dimostrato che, in cellule infettate e non trattate,
l’espressione nucleare delle proteine virali precocissime era buona qualora l’infezione
virale prendesse avvio nell’ambito della fase G1 e decisamente incrementata a livello della
transizione G1/S; essa appariva invece di minore entità in S ed in G2/M. L’intervento
della funzione proteasomale, a sua volta, sembrava maggiormente rilevante in fase G1 ed
alla transizione G1/S.

2. Presentazione dello studio
-7-
Al fine di evidenziare la distribuzione intracellulare di specifiche subunità proteasomali in
corso di infezione virale, è stato effettuato uno studio in microscopia confocale su sezioni
focali di cellule MRC5. I risultati ottenuti hanno evidenziato che i proteasomi si
localizzano principalmente a livello nucleare a tempi precocissimi dopo l’infezione virale,
con accumulo anche nella regione nucleolare di un numero significativo di cellule
infettate; tali esperimenti hanno, inoltre, evidenziato l’esistenza di una colocalizzazione
tra le proteine virali precocissime e specifici proteasomi, spesso presenti con una
distribuzione peculiare in assetto “granulare” e a volte evidenziabili anche in aree peri-
nucleolari.
L’allestimento di strumenti molecolari idonei allo studio dei proteasomi in cellule
suscettibili all’infezione da citomegalovirus ha portato all’elaborazione di particelle
retrovirali defettive recanti le sequenze proteasomali di interesse; successivamente, le
particelle ottenute sono state impiegate per l’infezione di fibroblasti di polmone
embrionale umano immortalizzati (MRC5-hTERT). Sono state eseguite quindi
osservazioni in microscopia a fluorescenza di monostrati di cellule MRC5-hTERT in grado
di esprimere stabilmente i costrutti di interesse; si è, in tal modo, appurato che il segnale
di fluorescenza delle proteine di fusione prescelte era localizzato sia nel compartimento
citoplasmatico che in quello nucleare, ma in quest’ultimo appariva più brillante ed
intenso, con distribuzione spesso granulare; come atteso, il segnale di fluorescenza era
assente a livello del distretto nucleolare. Il confronto del profilo di distribuzione
proteasomale con quello rilevato in cellule MRC5-hTERT non ingegnerizzate ha
consentito di evidenziare che i profili sono sovrapponibili.
I dati ottenuti evidenziano la complessità di rapporti che intercorrono tra proteasomi e
specifici distretti cellulari, quali il nucleo ed il citoplasma, a loro volta articolati in una
serie di compartimenti, ciascuno dei quali con precise collocazioni spaziali e funzionali, e
rendono ragione della complessità di eventi che regolano il differenziamento cellulare,
come anche dei rapporti che intercorrono tra virus e cellula ospite. In particolare, nel caso
del modello di infezione virale preso in considerazione in questo studio, la messa in
evidenza di compartimenti cellulari e di componenti di citomegalovirus che risultino
spazialmente e funzionalmente correlati con il complesso proteasomale, soprattutto a
tempi precocissimi dall’inizio dell’infezione, potrebbe apportare un contributo importante
alla individuazione di quei meccanismi volti ad imprimere una direzione di scelta, tra
ciclo litico e latenza, al rapporto tra citomegalovirus e cellula ospite.

3. Introduzione
3. INTRODUZIONE

3. Introduzione
-9-
a b
c d
Figura 1: Rappresentazione schematica del proteasoma 20S.
3.1 IL PROTEASOMA
I proteasomi sono complessi multi-enzimatici ad elevato peso molecolare (circa 2000kDa
per il complesso con costante di sedimentazione 26S), caratterizzati da una struttura
quaternaria altamente conservata nel corso dell’evoluzione; in particolare, l’aspetto
morfologico dei proteasomi è stato messo in evidenza da diversi studi cristallografici
effettuati in riferimento agli archeobatteri (Löwe J. et al., 1995), ai lieviti (Groll M. et al.,
1997; Groll M. et al., 2000) e, più recentemente, ai mammiferi (Unno M. et al., 2002). I
proteasomi 26S costituiscono il principale sistema di degradazione proteica non
lisosomiale presente nelle cellule eucariote a livello del nucleo, del citoplasma, del reticolo
endoplasmatico liscio e della superficie delle membrane cellulari; la concentrazione di tali
particelle può, inoltre, variare considerevolmente in funzione del tipo di cellula
(Nothwang H.G. et al., 1992; Coux O. et al., 1996; Voges D. et al., 1999). I substrati proteici
da degradare ad opera del proteasoma 26S sono generalmente, anche se non sempre,
preventivamente “etichettati” attraverso il legame covalente con una proteina detta
ubiquitina.
3.1.1 STRUTTURA DEL PROTEASOMA 20S La particella “core”, denominata proteasoma 20S (700kDa), rappresenta la struttura di
base responsabile dell’attività catalitica. Tali particelle sono state identificate per la prima
volta in cellule HeLa mediante l’impiego di tecniche di microscopia elettronica, come
subcomplessi di ribonucleoproteine (RNP) associate alla frazione citoplasmatica di RNA
messaggeri non coinvolti nella traduzione (Schmid H.P. et al., 1984).
Il proteasoma 20S è una struttura a cilindro di 14,8 nm di lunghezza e di 11,3 nm di
diametro, formata da quattro anelli sovrapposti
[Figura 1, pannelli a e b], ciascuno dei quali
costituito da sette subunità che delimitano una
cavità centrale [Figura 1, pannelli c e d]. In
particolare, i due anelli più esterni contengono
subunità di tipo α e quelli più interni subunità
di tipo β; queste ultime racchiudono i siti
catalitici dove i polipeptidi vengono degradati
nelle varie componenti aminoacidiche.

3. Introduzione
-10-
Nell’ambito degli archeobatteri è stata individuata una sola subunità di tipo α e una sola
subunità di tipo β; al contrario, negli eucarioti inferiori, come ad esempio nei lieviti, sono
state evidenziate sette subunità di tipo α e sette di tipo β, ciascuna delle quali codificata da
un gene differente.
Negli eucarioti superiori, più precisamente nei mammiferi, il proteasoma 20S è costituito
da 7 subunità di tipo α e 10 di tipo β. Tre delle subunità β (β1i, β2i e β5i) sono specifiche
del cosiddetto “immunoproteasoma”; in particolare, queste componenti sostituiscono tre
delle subunità definite costitutive (β1, β2 e β5). Le citochine, quali ad esempio IFN-γ e
TNF-α, inducono la sostituzione delle subunità β1, β2 e β5 con le subunità ad esse
strettamente correlate: β1i, β2i e β5i (chiamate anche LMP2, LMP10 e LMP7,
rispettivamente) (Groettrup M. et al., 2001a). Esistono pertanto almeno quattro tipi di
proteasoma 20S: un proteasoma costitutivo contenente le subunità β1, β2 e β5,
l’immunoproteasoma contenente le tre subunità inducibili β1i, β2i e β5i e proteasomi
“ibridi” composti da β1i, β2i e β5 o β1, β2 e β5i. I quattro tipi di proteasoma sono, inoltre,
associati alle subunità β3, β4, β6 e β7. L’attività dell’immunoproteasoma sembra essere
correlata alla produzione dei peptidi antigenici presentati dal complesso maggiore di
istocompatibilità di classe I (Rock K.L. et al, 1994; Groettrup M. et al., 2001b; Kloetzel P.M.,
2001).
3.1.2 IL PROTEASOMA 20S E I SUOI COMPLESSI REGOLATORI
Alle due estremità del proteasoma 20S si possono legare complessi ad attività regolatoria
(complessi PA200, PA28/11S e PA700/19S), che consentono l’apertura della cavità
centrale del proteasoma 20S, attivando in questo modo la funzione proteolitica (Förster A.
et al., 2003) [Figura 2].
Figura 2: Il proteasoma 20S e i suoi complessi regolatori. Ricostruzione tridimensionale delle strutture ottenute in microscopia elettronica (a, b, c: immagini ottenute da “Baumeister W. et al., 1998”; d: immagine ottenuta da “Ortega J. et al., 2005”).

3. Introduzione
-11-
Il complesso regolatore PA700 (o complesso 19S), associato alla particella core 20S, forma
il complesso molecolare di ordine superiore 26S (DeMartino G.N. and Slaughter C.A.,
1999). Questo complesso regolatore è formato da due porzioni denominate “Base” e
“Lid”, ciascuna delle quali è costituita da 8 differenti subunità che variano, in dimensioni,
da 24 a 106kDa. In particolare, la porzione definita “Base”, legata all’anello contenente
subunità di tipo α del proteasoma 20S, è costituita da sei subunità che presentano
un’attività ATPasica a cui è associata la funzione ATP - dipendente del proteasoma 26S,
mentre la porzione “Lid” è più esterna e in alcuni casi può dissociarsi, con conseguente
formazione di un complesso residuo tronco. Quest’ultimo complesso interviene più
frequentemente nella degradazione di substrati non legati a molecole di ubiquitina.
Numerose proteine e fattori ausiliari, quali ad esempio le proteine “chaperone” [per
esempio quelle appartenenti alla famiglia “Heat-shock proteins” (Hsp) 70], cooperano con
il sistema ubiquitina-proteasoma per la rimozione di proteine danneggiate o denaturate
che non possono più essere utilizzate. In particolare, le proteine “chaperone” appartenenti
alla famiglia Hsp70 sono state trovate in associazione al complesso 19S, grazie al legame
che instaurano con alcuni cofattori quale, ad esempio, la proteina BAG-1 (“Bcl2-associated
athano-gene”) (Luders J. et al., 2000; Esser C. et al., 2004). Nello specifico, il complesso
regolatore 19S permette il riconoscimento dei substrati ubiquitinati e di altri potenziali
substrati, consentendo l’apertura della cavità centrale del proteasoma 20S e,
contemporaneamente, la denaturazione dei substrati ubiquitinati e non ubiquitinati. Tali
funzioni richiedono elevati livelli di energia che viene fornita dalle sei subunità della
porzione “Base”, che presentano attività ATPasica.
Nelle cellule dei mammiferi è stato identificato anche un altro complesso attivatore della
funzione peptidasica del proteasoma 20S, che è stato denominato PA28 (alternativamente
11S REG) (Song X. et al., 1996; Hill C.P. et al., 2002). Tale complesso è ATP indipendente ed
è in grado di incrementare di più di cento volte la velocità di idrolisi di alcuni piccoli
peptidi da parte del proteasoma 20S, piuttosto che la degradazione di proteine come, al
contrario, si osserva per il complesso regolatore 19S (Dubiel W. et al., 1992; Ma C.P. et al.,
1992). Il complesso PA28 è costituito da tre subunità omologhe, di circa 28kDa ciascuna,
denominate PA28α, PA28β e PA28γ. Le subunità PA28α e PA28β possono associarsi e
dare luogo a complessi di tipo eteromerico (Ahn K. et al., 1996; Knowlton J.R. et al., 1997;
Zhang Z. et al., 1998; Zhang Z. et al., 1999), mentre la subunità PA28γ porta alla
formazione di complessi di tipo omomerico (Realini C. et al., 1997). Il complesso PA28αβ è
indotto dalla presenza della citochina IFN-γ e da condizioni patologiche quale, ad

3. Introduzione
-12-
esempio, un infezione virale; tale complesso è stato localizzato, in particolare, nel
compartimento citoplasmatico in molti tessuti, come ad esempio nel tessuto cerebrale. Tali
osservazioni suggeriscono che i complessi PA28αβ, associati al proteasoma 20S (in
particolare qualora sia costituito dalle tre subunità inducibili, a formare
l’immunoproteasoma), possano avere un ruolo di spicco nella risposta immunitaria e,
nello specifico, nella produzione di peptidi antigenici di piccole dimensioni, costituiti da
circa 6-8 aminoacidi, successivamente presentati dal complesso maggiore di
istocompatibilità di classe I. I dati di letteratura attualmente disponibili sono contrastanti
per quanto concerne il possibile coinvolgimento dei suddetti complessi nell’assemblaggio
dell’immunoproteasoma e, più in generale, nella presentazione dell’antigene, facendo
supporre piuttosto un loro coinvolgimento limitatamente alla presentazione di specifici
epitopi antigenici (Murata S. et al., 2001). Al contrario, i complessi regolatori PA28,
costituiti da sole subunità di tipo γ (PA28γ), non vengono attivati dalla presenza di
citochine e la loro produzione può venire notevolmente ridotta in corso di infezione
(Masson P. et al., 2001; Khan S. et al., 2001). La localizzazione del complesso PA28γ è
prevalentemente di tipo nucleare; inoltre esso è stato riscontrato in quantità più elevate
nel tessuto cerebrale. È stato ipotizzato un possibile coinvolgimento del complesso PA28γ
nell’inibizione del processo di apoptosi, fatto che giustificherebbe gli elevati livelli del
suddetto complesso regolatore rilevati nel tessuto cerebrale, dal momento che, come è
noto, i neuroni sono caratterizzati da sistemi in grado di impedire l’autodistruzione dei
medesimi (Rechsteiner M. and Hill C.P., 2005). Recentemente sono stati osservati
particolari “ibridi” contenenti un complesso 11S e un complesso 19S associati ad uno
stesso proteasoma 20S, suggerendo che i due complessi regolatori possano avere un ruolo
complementare (Hendil K.B. et al., 1998).
In questi ultimi anni è stato identificato mediante microscopia elettronica un ulteriore
complesso regolatore del proteasoma 20S, denominato PA200 (200KDa). Nonostante i dati
di letteratura siano ancora frammentari, sono state osservate interazioni tra il suddetto
complesso e differenti componenti cellulari coinvolti nei meccanismi di riparazione del
DNA (Ustrell V. et al., 2002; Ortega J. et al., 2005).
3.1.3 IL SISTEMA UBIQUITINA-PROTEASOMA
La cellule eucariote hanno sviluppato diverse strategie per eliminare le proteine mal
assemblate o non più funzionali, oppure semplicemente per diminuire la concentrazione
intracellulare di una certa specie proteica. In particolare, l’interazione covalente tra

3. Introduzione
-13-
ubiquitina, una proteina di 76 aminoacidi (circa 9KDa), ed il gruppo C-terminale di
residui di lisina del substrato, indirizza le molecole a diversi destini, tra cui i più
conosciuti sono la degradazione mediata dal proteasoma 26S, l’endocitosi con
conseguente degradazione del substrato mediata dai lisosomi e la modificazione delle
funzioni proteiche.
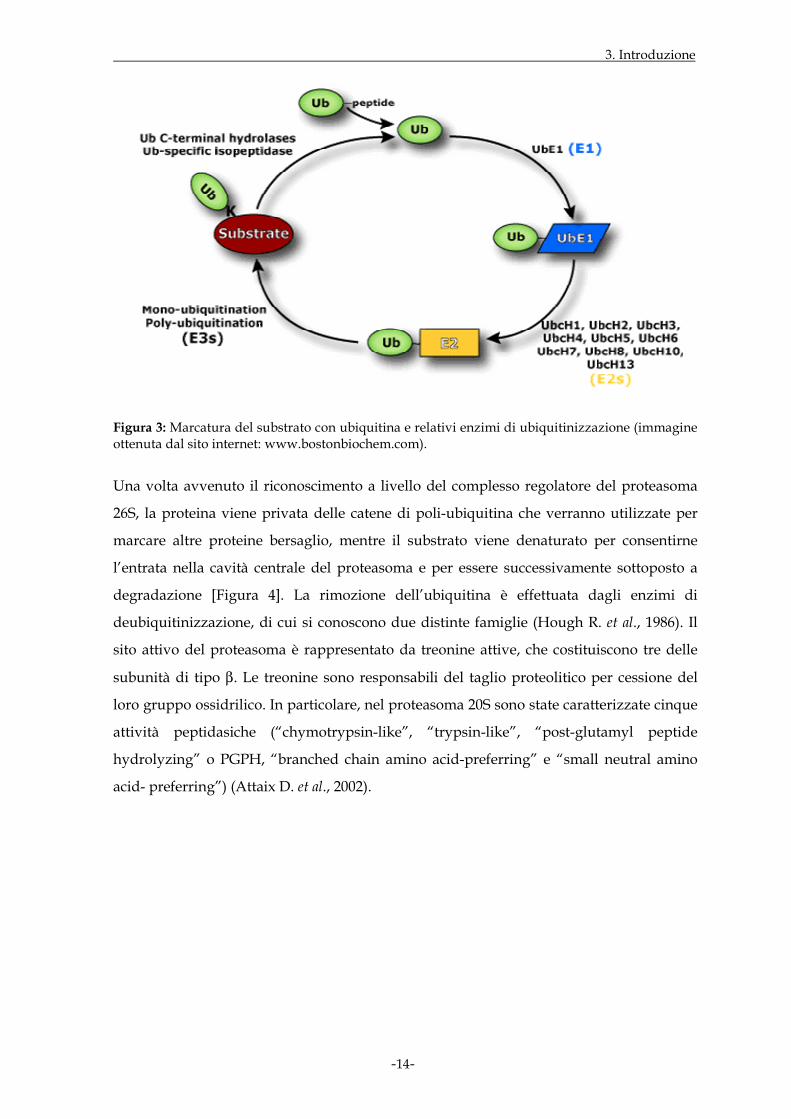
Il processo biochimico inizia con l’attivazione dell’ubiquitina mediante l’enzima E1
(“ubiquitin-activating”) e in presenza di ATP, a cui fa seguito il trasferimento
dell’ubiquitina attivata al residuo cisteinico del sito attivo dell’enzima E2 (“ubiquitin-
conjugating”), con formazione di un tioestere. La fase finale prevede che il complesso E2-
E3 catalizzi, mediante un isopeptide, il legame tra il C-terminale dell’ubiquitina e i residui
di lisina del substrato [Figure 3 e 4]. In molti casi la stessa ubiquitina subisce il processo di
ubiquitinizzazione causando la formazione di catene di poli-ubiquitina dove la lisina
collocata in posizione 48 e il gruppo C-terminale dell’ubiquitina successiva sono connessi
tra di loro.
Esistono due tipologie funzionalmente distinte di E3 (“ubiquitin-protein ligase”): l’enzima
con il dominio “HECT” (esempio NEDD4) che trasferisce l’ubiquitina al substrato
mediante la formazione di un intermedio tioestere covalente (E3-ubiquitina) e l’enzima
con il dominio “RING” (esempio APC/C “anaphase promoting complex/cyclosome” o
SCF “Skp1-Cullin-F-box protein”) che trasferisce le molecole di ubiquitina da E2 al
substrato, agendo come una proteina adattatrice. La varietà di enzimi compresi nella
famiglia E3 fornisce la necessaria specificità funzionale degli stessi nel determinare quale
proteina possa essere modificata (Ciechanover A. et al., 2000; Glickman M.H. and
Ciechanover A., 2001).
La poli-ubiquitinizzazione è una modificazione del substrato strettamente correlata alla
successiva degradazione effettuata tramite l’intervento del proteasoma 26S, mentre la
mono-ubiquitinizzazione è associata a diverse altre funzioni cellulari indipendenti dal
proteasoma come, ad esempio, la regolazione del traffico proteico intracellulare. In
alternativa, sono stati descritti casi in cui la degradazione mediata dal proteasoma 26S è
ubiquitina-indipendente (Murakami Y. et al., 2000).
3. Introduzione
-14-
Figura 3: Marcatura del substrato con ubiquitina e relativi enzimi di ubiquitinizzazione (immagine ottenuta dal sito internet: www.bostonbiochem.com).
Una volta avvenuto il riconoscimento a livello del complesso regolatore del proteasoma
26S, la proteina viene privata delle catene di poli-ubiquitina che verranno utilizzate per
marcare altre proteine bersaglio, mentre il substrato viene denaturato per consentirne
l’entrata nella cavità centrale del proteasoma e per essere successivamente sottoposto a
degradazione [Figura 4]. La rimozione dell’ubiquitina è effettuata dagli enzimi di
deubiquitinizzazione, di cui si conoscono due distinte famiglie (Hough R. et al., 1986). Il
sito attivo del proteasoma è rappresentato da treonine attive, che costituiscono tre delle
subunità di tipo β. Le treonine sono responsabili del taglio proteolitico per cessione del
loro gruppo ossidrilico. In particolare, nel proteasoma 20S sono state caratterizzate cinque
attività peptidasiche (“chymotrypsin-like”, “trypsin-like”, “post-glutamyl peptide
hydrolyzing” o PGPH, “branched chain amino acid-preferring” e “small neutral amino
acid- preferring”) (Attaix D. et al., 2002).

3. Introduzione
-15-
Figura 4: Rappresentazione schematica del processo di degradazione delle proteine mediato dai proteasomi 26S (immagine ottenuta dal sito internet: www.bostonbiochem.com).
3.1.4 FUNZIONI DEI PROTEASOMI
L’interesse rivolto verso i proteasomi, complessi proteici ubiquitari ed altamente
conservati nel corso dell’evoluzione, non risiede nella funzione enzimatica di proteolisi in
sé, in quanto questo sarebbe un modo non esaustivo per definire il ruolo che questi
complessi molecolari assumono nell’ambito dell’economia cellulare.
La proteolisi mediata dai proteasomi è infatti una funzione essenziale non solo per il ruolo
da tempo noto nell’eliminazione di proteine anomale o in sovrannumero, ma anche per il
ruolo che svolge nell’ambito della maturazione di precursori in proteine biologicamente
attive e, negli eucarioti superiori, nel reclutamento di peptidi antigenici per il complesso
maggiore di istocompatibilità di classe I. Il ruolo che i proteasomi hanno nel controllo
dell’omeostasi proteica va inquadrato in un’ottica più ampia, in quanto si applica a
numerosi processi cruciali nell’ambito del metabolismo cellulare. Nello specifico,
l’espletamento dell’attività enzimatica del proteasoma garantisce, in maniera altamente
specifica e regolata, il mantenimento di livelli intracellulari sempre adeguati di numerose
proteine che controllano, a loro volta, processi biologici quali la proliferazione e il
differenziamento (in particolare le cicline ed altri complessi proteici che intervengono
nell’evoluzione del ciclo cellulare), l’apoptosi e la risposta a stimoli o insulti extracellulari,
quali un’infezione virale (Orlowski R.Z., 1999; Grimm L.M. and Osborne B.A., 2000; Mann

3. Introduzione
-16-
C. and Hilt W., 2000; Yew P.R., 2001; Naujokat C. and Hoffmann S., 2002). Nell’ambito dei
molteplici processi in cui è richiesta l’attività enzimatica dei proteasomi, verranno in
questa sezione considerati in maggior dettaglio i meccanismi di controllo che operano a
livello del ciclo cellulare, evento che, come già ricordato, è alla base della proliferazione e
del differenziamento cellulare; su quest’ultimo, in particolare, si impernia uno dei due
modelli presi in considerazione in questo studio, ossia il differenziamento di cellule
muscolari in vitro.
Analogamente, vengono anche riportati esempi di interventi regolatori operati da
proteasomi in situazioni patologiche, quali un‘infezione virale, tenendo presente che il
secondo modello allo studio prende in considerazione l’infezione produttiva da
citomegalovirus umano in sistemi cellulari in vitro. È anche importante sottolineare che un
numero considerevole di dati di letteratura supporta l’intervento da parte del suddetto
virus sul ciclo cellulare, modificandone l’andamento a suo favore. Tali osservazioni
rendono plausibile ipotizzare un concomitante ruolo dei proteasomi nell’ambito del ciclo
replicativo di citomegalovirus (Dittmer D. and Mocarski E.S., 1997; Salvant B.S. et al., 1998;
Sinclair J. et al., 2000; Kalejta R.F. and Shenk T., 2002).
Il ciclo cellulare: punti di regolazione e ruolo dei proteasomi
La progressione del ciclo cellulare è regolata da una serie di fattori geneticamente
caratterizzati, che controllano la divisione cellulare; questi possono a loro volta essere
modulati da fattori endogeni o esogeni, quali i cosiddetti fattori di crescita, che sono in
grado di modificare l’evoluzione del ciclo di divisione. Il ciclo cellulare si articola
abitualmente in quattro fasi: nel corso della prima fase (fase G1) la cellula svolge le sue
normali funzioni metaboliche, si nutre e cresce in volume preparandosi, in un certo senso,
alla fase successiva (fase S), che è quella più impegnativa dal punto di vista energetico. In
quest’ultima fase, viene duplicato il DNA cellulare, così come tutte le altre strutture
cellulari. Terminata la fase S, la cellula intraprende una ulteriore fase di preparazione
(fase G2). Nel loro insieme, le suddette fasi costituiscono l’interfase, che sfocerà nella
divisione cellulare o mitosi (fase M), composta a sua volta da profase, metafase, anafase,
telofase e citochinesi.
Ognuna delle fasi del ciclo cellulare è caratterizzata da numerosi eventi che si susseguono
in maniera estremamente ordinata, grazie ad elaborati meccanismi di regolazione che ne
controllano l’evoluzione. Un ruolo chiave nella regolazione del ciclo cellulare è svolto
dalla fosforilazione e defosforilazione di substrati proteici da parte di specifiche chinasi.

3. Introduzione
-17-
In particolare, nelle cellule somatiche degli eucarioti, le serina-treonina chinasi ciclina-
dipendenti, o cdk, sono deputate a questo ruolo. L’attività di queste chinasi è a sua volta
soggetta ad una serie di complessi meccanismi di controllo. Le subunità catalitiche cdk si
legano a specifiche proteine a funzione regolatoria denominate cicline (A, B, D ed E), per
formare un complesso attivo ciclina-cdk. La formazione, l’attivazione ed il
disassemblamento del complesso ciclina-cdk rappresentano l’evento pilota che guida la
cellula attraverso il ciclo cellulare. Le cicline che intervengono nelle diversi fasi del ciclo
cellulare sono sottoposte serialmente a sintesi e degradazione; in concomitanza, l’attività
delle cdk è regolata positivamente o negativamente, in stretta dipendenza dal progredire
del ciclo cellulare. Tali fluttuazioni di attività delle cdk portano a cambiamenti ciclici nella
fosforilazione di proteine intracellulari, quali appunto le cicline, che danno l’avvio o
regolano l’evoluzione dei maggiori eventi del ciclo cellulare (replicazione del DNA, mitosi
e citochinesi). I cambiamenti ciclici nell’attività delle cdk sono a loro volta regolati dalle
cicline e da piccole proteine (quali ad esempio p16, p15, p18, p19, che appartengono alla
famiglia “INK4 - inhibitors of cdk4” e p21Cip1, p27Kip1, che appartengono, invece, alla
famiglia “Cip/Kip - cdk interacting protein/kinase inhibitory protein”), tutte con funzioni
di inibitori delle chinasi ciclina-dipendenti. Esse formano complessi di natura non
covalente con i dimeri ciclina-cdk, causandone l’inattivazione e rendendo pertanto
possibile un controllo negativo della proliferazione cellulare (Koepp D.M. et al., 1999;
Mann C. and Hilt W., 2000; Yew R.P., 2001).
I complessi ciclina-cdk attivi guidano la cellula da una fase all’altra del ciclo cellulare
attraverso punti di regolazione o “checkpoints”, in cui un particolare corredo di substrati
proteici viene fosforilato. Esistono quattro “checkpoints” nell’ambito del ciclo cellulare, in
particolare in fase G1, G1/S, G2 e M [Figura 5].
La corretta progressione attraverso le varie fasi del ciclo cellulare è dunque dipendente da
una sequenza ordinata di eventi mirati, quali principalmente la replicazione del DNA, la
formazione del fuso mitotico e la divisione nucleare, che sfociano nella divisione cellulare.
Al fine di coordinare l’evoluzione di tali eventi, le cellule eucariote hanno messo a punto
un sistema di regolazione molto raffinato, che è basato, come già menzionato, sull’attività
di regolazione positiva o negativa delle chinasi ciclina-dipendenti. Queste ultime
rappresentano quindi il fulcro attorno al quale ruotano tutti gli “ingranaggi” del ciclo
cellulare, attraverso un processo reversibile di fosforilazione di proteine che intervengono
nella fasi critiche, quali la replicazione del DNA o la condensazione dei cromosomi, e che
risulta direttamente effettuato a livello delle suddette proteine, oppure attraverso

3. Introduzione
-18-
l’attivazione di altre chinasi effettrici (Morgan D.O., 1995; Lees E., 1995; Morgan D.O.,
1997; Nigg E.A., 2001).
Figura 5: Le fasi del ciclo cellulare e relativi punti di regolazione (frecce tratteggiate) (immagine ottenuta dal sito internet: www.users.unimi.it).
Dal momento che i suddetti eventi sono sostanzialmente reversibili, la necessaria
direzionalità delle fasi del ciclo cellulare, ovvero la garanzia di un graduale passaggio,
temporalmente cadenzato, da uno stadio a quello successivo (senza possibilità di ritorno),
deve essere garantito da altre tipologie di meccanismi. In tale ottica, il processo di
degradazione proteica regolata attraverso l’intervento del complesso ubiquitina-
proteasoma rappresenta “la soluzione” mirata ed ideale alla problematica sopra esposta.
In effetti, la rimozione di proteine necessarie in una fase precedente del ciclo cellulare,
mediante rapida degradazione di proteine bersaglio a lento ritmo di sintesi, consente
l’espletamento di un evento irreversibile di “non-ritorno” e garantisce al contempo
l’avanzamento del ciclo verso la fase successiva.
Nell’ambito di questa strategia, il primo bersaglio è rappresentato dalle cicline (ovvero le
subunità che attivano, a loro volta, le cdk), il cui graduale accumulo e rapida
degradazione ad opera del sistema ubiquitina-proteasoma ha un’azione modulante
sull’attività delle cdk stesse, imprimendo il ritmo corretto all’ ”orologio” del ciclo cellulare
(Mann C. and Hilt W., 2000; Yew R.P., 2001).
Analogamente, anche altre proteine che intervengono a differenti livelli di regolazione
sono sottoposte a degradazione proteica mediata dal proteasoma, sempre attraverso una
classica “etichettatura” delle proteine stesse tramite poli-ubiquitinizzazione. Quest’ultima,
come già evidenziato in questa sezione introduttiva, richiede l’azione combinata di enzimi
che legano l’ubiquitina (E2), così come di quelli che operano il legame tra ubiquitina e

3. Introduzione
-19-
substrato proteico da degradare (ubiquitina-ligasi o E3). La specificità della proteolisi
ubiquitina-dipendente è legata proprio all’ubiquitinizzazione del substrato; questo rende
ragione del fatto che gli enzimi ubiquitina-ligasi abbiano un ruolo chiave in molti processi
vitali, tra cui il ciclo cellulare.
In particolare, due complessi enzimatici di tipo E3 svolgono la loro attività nell’ambito del
suddetto processo: il cosiddetto “complesso promotore di anafase” o “ciclosoma”
(“APC/C”) e “SCF” (“Skp1/Cullin/F-box protein”), come di seguito dettagliato.
Per quel che riguarda il ruolo delle cicline, a cominciare dalla fase di “ingresso” nel ciclo
cellulare, è da notare che la sintesi delle cicline D (D1, D2 e D3) è attivata da fattori di
crescita che stimolano il rientro nel ciclo cellulare dalla fase di quiescenza o fase G0. Tali
cicline hanno un tempo di vita media molto breve e i loro livelli diminuiscono
rapidamente quando il fattore di crescita è rimosso. La perdita di ciclina D può innescare
l’uscita della cellula dal ciclo cellulare, con susseguente rientro in fase G0. Le cdk 4 e 6 si
legano con la ciclina di tipo D formando eterodimeri che sono in grado di entrare nel
nucleo, dove vengono fosforilati da una chinasi attivante le cdk. I complessi ciclina D-
cdk4, 6 e, successivamente, ciclina E-cdk2, nell’ambito della transizione G1/S, fosforilano
sequenzialmente la proteina del retinoblastoma (pRb) [Figura 6]. La proteina pRb agisce
durante la fase G1, reprimendo la trascrizione dei geni coinvolti nella regolazione della
progressione del ciclo cellulare verso la fase S. Tale proteina non si lega direttamente al
DNA, ma svolge il suo compito di repressore della trascrizione genica interagendo ed
inibendo l’attività di alcuni fattori di trascrizione, i più studiati dei quali appartengono
alla famiglia dei fattori di trascrizionali E2F (Hiebert S.W. et al., 1992; Helin K. et al., 1993).
I siti di legame per i fattori E2F sono stati identificati nei promotori di molti geni coinvolti
nella regolazione del ciclo cellulare, come quelli per le cicline D1, E ed A e quelli
codificanti per le proteine p107 (famiglia pRb), E2F1, 4 e 5, cdk2 e cdc2 e, infine, per
enzimi coinvolti nella sintesi del DNA come, ad esempio, la timidino-chinasi, la
deidrofolato-reduttasi, la DNA-polimerasi α e cdc6 (Lipinski M.M. and Jacks T., 1999). La
iperfosforilazione di pRb porta ad una riduzione dell’affinità di tale proteina per il fattore
di trascrizione E2F di tipo 1-4, a cui è legata, in cellule quiescenti o nella fase iniziale G1,
nella forma non fosforilata, inducendo il rilascio di E2F e la dissociazione dei complessi di
repressione. Una volta liberate, le proteine E2F attivano la trascrizione dei geni necessari
per la progressione del ciclo cellulare in fase S (Weintraub S.J. et al., 1992; Weintraub S.J. et
al., 1995).

3. Introduzione
-20-
E2F inattivo
trascrizione inibita
E2F attivo
trascrizione attiva
Figura 6: Fattori di regolazione che intervengono nella progressione del ciclo cellulare verso la fase S (immagine ottenuta dal sito internet: www.users.unimi.it). La proteina pRb subisce differenti fosforilazioni durante la progressione del ciclo cellulare
verso la fase S ed ancora verso le fasi G2/M. Molti dei gruppi fosfato vengono invece
rimossi quando la cellula rientra nelle fasi G0/G1. Le forme ipofosforilate predominano in
G0 e nella fase G1 precoce, mentre le forme più fosforilate sono presenti nelle fasi S, G2 ed
M. Ulteriori livelli di fosforilazione possono essere raggiunti dipendentemente dal tipo di
cellula considerata. Il complesso ciclina A-cdk2 si forma durante la fase S e gioca
anch’esso un ruolo importante nella progressione della duplicazione del DNA,
mantenendo la proteina pRb nella forma fosforilata. Il complesso ciclina B-cdk1 è
necessario per rendere possibile l’ingresso in mitosi (si veda paragrafo successivo); i livelli
di ciclina B sono mantenuti bassi durante la fase S dal complesso APC/C, che con la sua
attività di ubiquitina-ligasi (E3) ne causa la degradazione mediata dai proteasomi. Alla
fine della fase S, il complesso ciclina A-cdk2 inattiva il complesso APC/C, fosforilando la
subunità Cdh1 e permettendo, inoltre, l’accumulo di ciclina B e la formazione del
complesso ciclinaB-cdk1, il quale favorisce, a sua volta, la progressione del ciclo cellulare
verso la fase M (Lukas C. et al., 1999). In particolare, l’ingresso in mitosi/meiosi è
consentito solo in presenza del fattore “Maturation Promoting Factor “ (MPF). Questo
fattore è costituito da due tipi di subunità: una ad attività chinasica costitutiva (cdk1, detta
anche cdc2), che fosforila i residui di serina e treonina delle proteine bersaglio ed una
seconda, ciclina B, necessaria per l’attivazione dell’attività chinasica stessa, ma sintetizzata
solo nelle fasi che precedono la mitosi, vale a dire a partire dalla fine della fase S. La
ciclina B, una volta sintetizzata, si coniuga immediatamente alla subunità cdk1
preesistente, inducendo la fosforilazione di molti substrati quali le chinasi, alcune

3. Introduzione
-21-
componenti del citoscheletro, diverse proteine secretorie ed altre proteine che
intervengono nella regolazione del ciclo cellulare. Inoltre, la subunità catalitica cdk1 è
importante per la segregazione del materiale cellulare tra le cellule figlie durante il
processo di divisione cellulare. Data la rilevanza delle funzioni espletate dalla suddetta
subunità è evidente che la sua attività sia finemente regolata, in particolare, attraverso un
processo di fosforilazione a livello di specifici aminoacidi. In effetti, una volta avvenuta la
sintesi di ciclina B, cdk1 viene fosforilata a livello di treonina 161 e tale modificazione
porta all’attivazione delle funzioni di chinasi. D’altra parte, cdk1 può essere fosforilata
anche in posizione treonina 14 e tirosina 15 dall’azione di specifici enzimi, con
conseguente inattivazione del complesso ciclina B-cdk1. Tale inattivazione consente alla
cellula di raggiungere la transizione G2/M, con elevati livelli del complesso ciclina B-cdk1
mantenuto nella forma inattiva. Alcuni sistemi intracellulari hanno il compito di verificare
che tutto sia pronto per la fase successiva di mitosi o meiosi e, una volta che ciò è stato
verificato, tali sistemi attivano la fosfatasi cdc25, che defosforila i residui aminoacidici in
treonina 14 e tirosina 15 di cdk1, attivando il complesso MPF. L’attivazione del complesso
ciclina B-cdk1 porta alla fosforilazione dell’istone H1, con conseguente condensazione
della cromatina, fosforilazione delle lamine e successiva rottura dell’involucro nucleare;
inoltre si osserva anche la concomitante disaggregazione dell’apparato del Golgi e del
reticolo endoplasmatico e, infine, instabilità dei microtubuli con successiva formazione
del fuso mitotico. Una volta completata la fase di mitosi/meiosi, la cellula induce la
degradazione di ciclina B, inattivando cdk1. La degradazione della suddetta proteina
avviene mediante un meccanismo di proteolisi ubiquitina-dipendente, grazie alla
presenza di sequenze aminoacidiche (denominate, nel loro insieme, “destruction box”)
contenute nella regione N-terminale delle cicline. Come già evidenziato, sono state
descritte due tipologie di enzimi ubiquitina-ligasi E3, che sono responsabili della
ubiquitinizzazione delle cicline, vale a dire il complesso APC/C o, in alternativa, il
complesso SCF. In particolare, la proteina APC/C si presenta sotto due forme, definite
sulla base del cosiddetto fattore di specificità Cdc20 o, alternativamente, Cdh1, necessario
per l’attivazione di APC/C. Il complesso APC/C-Cdc20 è attivo esclusivamente durante
la fase di mitosi del ciclo cellulare e la sua funzione è importante per la segregazione dei
cromosomi e per la transizione del ciclo cellulare da metafase ad anafase. In anafase tale
complesso viene sostituito dal complesso APC/C-Cdh1 che rimane attivo fino alla fase
G1/S e la cui funzione è essenziale per la progressione del ciclo cellulare in fase S, in cui
ha inizio la replicazione del DNA cellulare. Inoltre, tale complesso è attivo in cellule

3. Introduzione
-22-
differenziate e in cellule in stato quiescente, risultando importante anche per il
mantenimento delle stesse in fase G0 (Harper J.W. et al., 2002; Peters J.M., 2002). Mentre i
complessi ubiquitina-ligasi di tipo APC/C sono attivi principalmente durante le fasi M e
G1 del ciclo cellulare, quelli di tipo SCF appaiono più versatili, intervenendo a molteplici
stadi del ciclo cellulare e svolgendo diversi ruoli di regolazione, oltre a quello principale
di modulazione dell’attività di cdk (Vodermaier C., 2004) [Figura 7].
Figura 7: Ruolo delle ubiquitina-ligasi APC/C e SCF1 nell’ambito del ciclo cellulare (immagine tratta da “Vodermaier H.C., 2004”).
Altro fattore di rilievo nella regolazione del ciclo cellulare è la proteina p53, che ha il
compito di controllare (specialmente in fase G1) se sono avvenute mutazioni o alterazioni
a carico del DNA. In tali circostanze, i livelli di p53 aumentano, consentendo alla suddetta
proteina di provvedere a riparare il DNA, sfruttando a proprio vantaggio il parallelo
blocco del ciclo cellulare. Se tuttavia il danno a carico del DNA è troppo grave, p53 fa sì
che la cellula vada incontro a morte programmata (apoptosi).

3. Introduzione
-23-
La proteina p53 è un fattore di trascrizione che induce la sintesi ex novo della proteina
p21Cip1 (famiglia “CIP/KIP: cdk interacting protein/kinase inhibitory protein”), in grado
di bloccare l’azione della proteina “proliferating cell nuclear antigen” (PCNA), necessaria
alla formazione della forcella di sostegno richiesta dall’enzima DNA polimerasi durante la
duplicazione del DNA in fase S (El-Deiry W.S. et al., 1994; Luo Y. et al., 1995). Inoltre,
p21Cip1 blocca direttamente i complessi attivati di ciclina D-cdk4 e ciclina D-cdk6 durante
il superamento del punto di restrizione nella fase G1 [Figura 5].
Infezione virale e proteasomi
I proteasomi, il cui intervento è, come già sottolineato, cruciale nella regolazione
dell’omeostasi proteica, vengono sempre più spesso evocati in letteratura anche per il loro
ruolo nell’ambito del ciclo replicativo di numerosi virus che infettano l’uomo. Tale
intervento non è univoco, ma varia dipendentemente dal tipo di virus (es. virus a DNA o
ad RNA, replicazione nucleare o citoplasmatica), come era logico attendersi, dato il ruolo
multifunzionale attribuito ai proteasomi e data la loro collocazione sia a livello
citoplasmatico che nucleare.
Nell’ambito dei virus a DNA, per esempio, è stata messa in evidenza la capacità, da parte
della proteina virale precocissima di Herpes simplex denominata Vmw110, di indurre la
disgregazione dei domìni nucleari ND10 mediante l’attività proteolitica dei proteasomi,
con lo scopo di dirigere l’infezione virale verso il ciclo litico, mentre la mancata
disgregazione dei suddetti domìni consentirebbe un’evoluzione dell’infezione
preferenzialmente verso una condizione di latenza (Everett R.D. et al., 1998). In corso di
infezione virale con papillomavirus umano di tipo 16, è stato osservato che la proteina
virale E7 si lega con alta efficienza alla proteina pRb nella forma ipofosforilata, causando
conseguentemente la rimozione del complesso pRB-E2F e promuovendo, pertanto, la
progressione del ciclo cellulare in fase G1/S. Tuttavia, la proteina E7 è anche in grado di
indurre la degradazione della proteina pRb mediante l’attivazione del proteasoma 26S. In
particolare, è stata osservata una interazione tra una specifica subunità del complesso
regolatore 19S dotata di attività ATPasica e la proteina E7, che funge da tramite tra il
proteasoma 26S e la proteina pRb. L’instaurarsi di questo singolare legame porta alla
degradazione della proteina pRb senza l’intervento del sistema di ubiquitinizzazione.
Parallelamente a quanto appena descritto per la proteina pRb, la proteina E7 può indurre
la degradazione di altre proteine cellulari, quali p130 e p107, che possiedono una elevata
omologia di sequenza con la proteina pRb. Inoltre, è stato osservato che la stessa proteina

3. Introduzione
-24-
E7 può essere sottoposta ad ubiquitinizzazione ed essere successivamente degradata dal
proteasoma 26S (Boyer S.N. et al., 1996; Berezutskaya E. et al., 1997; Jones D.L. and Münger
K., 1997; Reinstein E. et al., 2000; Gonzalez S.L. et al., 2001).
Per quel che riguarda i virus ad RNA, recentemente è stato osservato come la proteina
non strutturale NS3 del virus dell’epatite C (HCV), che svolge un ruolo cruciale nel
meccanismo di rielaborazione delle poliproteine virali, nella replicazione dell’RNA virale
e nella traduzione, sia in grado di legarsi direttamente alla subunità LMP7
dell’immunoproteasoma. Tale legame fa sì che l’attività peptidasica della proteina virale
induca una marcata riduzione della attività del proteasoma. Tale meccanismo andrebbe
ad interferire con il meccanismo di rielaborazione degli antigeni virali per la
presentazione mediata dal complesso maggiore di istocompatibilità di classe I,
proteggendo pertanto il virus HCV dal sistema immunitario dell’organismo ospite e
consentendo una infezione persistente del virus (Khu Y.L. et al., 2004). Similmente, la
proteina virale Tat di HIV-1 e HIV-2 è in grado di inibire l’attività peptidasica del
proteasoma 20S, interferendo con il legame tra il proteasoma 20S e il complesso regolatore
PA28 e non consentendo, pertanto, la formazione di peptidi virali per la presentazione
dell’antigene da parte del complesso maggiore di istocompatibilità di classe I (Seeger M. et
al., 1997). Tuttavia, tale proteina è in grado di incrementare parallelamente la
degradazione di substrati proteici mediata dall’attività del proteasoma 26S.
Dati di letteratura attestano che un intervento del sistema ubiquitina-proteasoma sembra
essere richiesto nell’ambito di eventi cruciali, precoci o tardivi (quali endocitosi e
maturazione-gemmazione), del ciclo replicativo di diverse famiglie virali. Nello specifico,
è stato dimostrato l’intervento dei proteasomi nell’ambito del processo di
internalizzazione del virus influenza, che prevede il passaggio pH-dipendente del virus
attraverso endosomi; l’inibizione della funzione proteolitica mediata da proteasomi
porterebbe al sequestro del virus nel citoplasma, impedendone l’ingresso nel nucleo e la
conseguente evoluzione del suo ciclo replicativo. Tale intervento da parte del sistema
ubiquitina-proteasoma sul processo di endocitosi del virus influenza sembra essere anche
estremamente mirato, in quanto non si verifica per altri virus, non correlati al virus
influenza, quali il virus Semliki Forest (Togaviridae) ed il virus della stomatite vescicolare
(Rhabdoviridae) (Khor R. et al., 2003).
Per quel che riguarda gli eventi tardivi del ciclo di replicazione virale, dati sperimentali
supportano un intervento dei proteasomi nell’ambito dei processi di maturazione e
rilascio dei retrovirus HIV-1 e HIV-2. In particolare, è stata dimostrata un’interazione

3. Introduzione
-25-
funzionale del sistema proteasoma-ubiquitina nel processo di maturazione della
poliproteina Gag, operato dalla proteasi virale. In tale accezione, l’intervento congiunto
dei proteasomi non sembra peraltro interferire con la funzione proteasica virus-specifica;
al contrario, l’assenza della suddetta funzione proteasomale porterebbe ad una ridotta
efficacia del processo di gemmazione, così come ad una significativa diminuzione di
progenie virale infettante (Schubert U. et al., 2000). Analogo intervento da parte dei
proteasomi è stato chiamato in causa per il virus della stomatite vescicolare e per il virus
della rabbia (Rhabdoviridae), le cui proteine di matrice medierebbero il legame con residui
aminoacidici di enzimi di tipo E3 (ubiquitina-ligasi), innescando poi l’attività enzimatica
dei proteasomi, coinvolta nel favorire la gemmazione dei suddetti virus (Harty R.N. et al.,
2001).

3. Introduzione
-26-
3.2 I MODELLI DI STUDIO
3.2.1 IL TESSUTO MUSCOLARE
Caratteristiche generali
Nei vertebrati esistono tre tipi ben distinti di tessuto muscolare: il tessuto muscolare
viscerale, cardiaco e scheletrico. Essi differiscono sia per la struttura delle fibre che li
compongono, che per il modo con cui esse sono organizzate nell’ambito del tessuto;
caratteristica comune a tutte le fibre muscolari è, comunque, la presenza in esse di un
apparato contrattile costituito da filamenti proteici denominati microfilamenti.
- Il tessuto muscolare scheletrico è responsabile del movimento dello scheletro e di organi
come il bulbo oculare e la lingua. I muscoli scheletrici sono spesso definiti muscoli
volontari poiché possono essere controllati dalla volontà.
- Il tessuto muscolare viscerale forma la componente muscolare di strutture quali i vasi
sanguigni, il tratto gastrointestinale, l’utero e la vescica. Poiché questi tipi di muscoli
sono sotto il controllo del sistema nervoso autonomo, essi sono descritti come muscoli
involontari.
- Il tessuto muscolare cardiaco ha caratteristiche funzionali e strutturali intermedie tra quelle
dei due tipi precedenti (muscolo scheletrico involontario), ed è responsabile della
continua e ritmica contrattilità del cuore.
Muscolo scheletrico e sua embriogenesi
Il muscolo scheletrico è costituito da fibre derivanti dall’associazione di cellule molto
allungate, non ramificate, cilindriche, caratterizzate dalla presenza di numerosi nuclei
appiattiti, che risultano disposti quasi ad intervalli regolari e sono localizzati appena sotto
la membrana che avvolge le fibre (sarcolemma).
Durante l’embriogenesi, le cellule mesodermiche dei miotomi si differenziano in
mioblasti, che sono cellule a morfologia allungata, mononucleate e capaci di dividersi per
mitosi. Successivamente, i mioblasti vanno incontro a fusione formando cellule
multinucleate sempre più lunghe, chiamate miotubi. La sintesi delle proteine contrattili
inizia dopo la fusione dei mioblasti; i nuclei dei mioblasti si dispongono dapprima lungo
l’asse centrale del miotubo e, in seguito alla formazione di ulteriori proteine contrattili,
vengono spinti verso la periferia, con formazione della miofibra. Il processo di sviluppo
del muscolo, insieme a quello di innervazione, è quasi del tutto completato alla nascita. La

3. Introduzione
-27-
successiva crescita avviene pertanto per aumento di volume del citoplasma delle cellule
muscolari. Le cellule muscolari mature risultano altamente differenziate e, in seguito a un
eventuale danno muscolare, hanno una limitata capacità di riparazione e rigenerazione;
tuttavia, alcune cellule mononucleate, chiamate cellule satelliti, rimangono localizzate tra
la membrana basale e la membrana citoplasmatica delle fibre muscolari. Le cellule satelliti
sono responsabili dello sviluppo del muscolo; inoltre, quando esso viene danneggiato, tali
cellule vengono indotte a differenziarsi e a fondersi per ricostituire le fibre del muscolo
leso. Queste cellule possono essere coltivate e indotte a differenziarsi anche in vitro (Nag
A.C. and Foster J.D., 1981; Morgan J.E. and Partridge T.A., 2003; Chargé S.B.P. and
Rudnicki M.A., 2004; Dhawan J. and Rando T.A., 2005; Sherwood R.I. and Wagers A.J.,
2006).
Struttura e composizione del muscolo scheletrico
Il muscolo scheletrico è formato da un ventre muscolare, in cui si trovano fasci di fibre
muscolari e due tendini connettivali con cui il muscolo si inserisce sulle ossa. Le fibre
muscolari scheletriche sono allungate e del tutto particolari per struttura e caratteristiche
funzionali. Esse sono riunite in fasci allungati chiamati fascicoli; un delicato tessuto
connettivo (endomisio) occupa gli spazi tra le singole fibre muscolari. I fascicoli sono
circondati da un tessuto connettivo lasso chiamato perimisio. Generalmente, i muscoli
sono composti da molti fascicoli e l’intera massa muscolare è rivestita da un denso
connettivo esterno (epimisio). Grossi vasi ematici e nervi entrano attraverso l’epimisio e si
dividono, per ramificarsi attraverso il muscolo, nel perimisio e nell’endomisio. Ogni fibra
muscolare è delimitata da una membrana (sarcolemma) e al suo interno si trova il
citoplasma (sarcoplasma), contenente numerosi nuclei. Il volume di ogni fibra muscolare
è, tuttavia, quasi interamente occupato dall’apparato contrattile, costituito da numerose
subunità filamentose disposte in modo parallelo nel sarcoplasma (miofibrille), alla cui
struttura è dovuta la striatura trasversa dell’intera fibra muscolare [Figura 8 ].
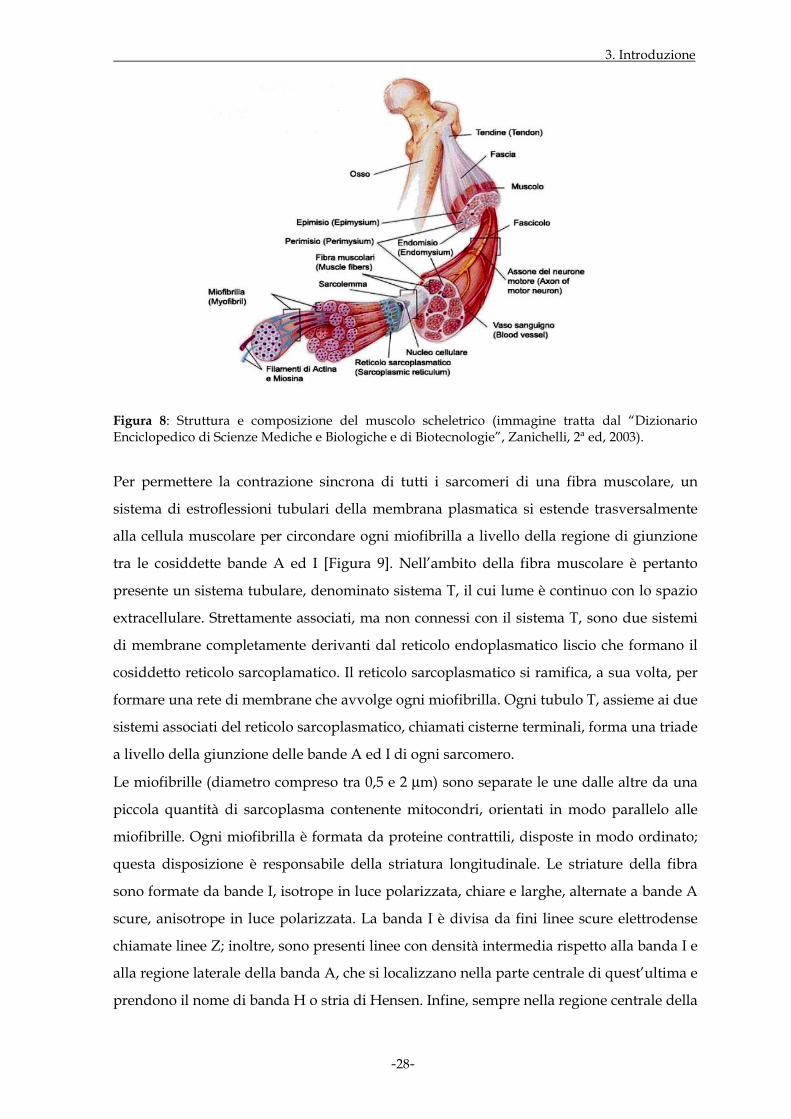
3. Introduzione
-28-
Figura 8: Struttura e composizione del muscolo scheletrico (immagine tratta dal “Dizionario Enciclopedico di Scienze Mediche e Biologiche e di Biotecnologie”, Zanichelli, 2ª ed, 2003).
Per permettere la contrazione sincrona di tutti i sarcomeri di una fibra muscolare, un
sistema di estroflessioni tubulari della membrana plasmatica si estende trasversalmente
alla cellula muscolare per circondare ogni miofibrilla a livello della regione di giunzione
tra le cosiddette bande A ed I [Figura 9]. Nell’ambito della fibra muscolare è pertanto
presente un sistema tubulare, denominato sistema T, il cui lume è continuo con lo spazio
extracellulare. Strettamente associati, ma non connessi con il sistema T, sono due sistemi
di membrane completamente derivanti dal reticolo endoplasmatico liscio che formano il
cosiddetto reticolo sarcoplamatico. Il reticolo sarcoplasmatico si ramifica, a sua volta, per
formare una rete di membrane che avvolge ogni miofibrilla. Ogni tubulo T, assieme ai due
sistemi associati del reticolo sarcoplasmatico, chiamati cisterne terminali, forma una triade
a livello della giunzione delle bande A ed I di ogni sarcomero.
Le miofibrille (diametro compreso tra 0,5 e 2 µm) sono separate le une dalle altre da una
piccola quantità di sarcoplasma contenente mitocondri, orientati in modo parallelo alle
miofibrille. Ogni miofibrilla è formata da proteine contrattili, disposte in modo ordinato;
questa disposizione è responsabile della striatura longitudinale. Le striature della fibra
sono formate da bande I, isotrope in luce polarizzata, chiare e larghe, alternate a bande A
scure, anisotrope in luce polarizzata. La banda I è divisa da fini linee scure elettrodense
chiamate linee Z; inoltre, sono presenti linee con densità intermedia rispetto alla banda I e
alla regione laterale della banda A, che si localizzano nella parte centrale di quest’ultima e
prendono il nome di banda H o stria di Hensen. Infine, sempre nella regione centrale della

3. Introduzione
-29-
banda A, in posizione mediana, si trova la linea M, un tempo definita mesofragma.
Ciascuna miofibrilla è caratterizzata da segmenti uguali, che si ripetono regolarmente e
che prendono il nome di sarcomero. In particolare, il sarcomero (di dimensioni comprese
tra 2,5 e 3 µm) rappresenta l’unità contrattile della miofibrilla ed è compreso tra due linee
Z successive [Figura 9]. Il sarcomero è formato da due tipi di miofilamenti, differenziabili
in filamenti spessi e filamenti sottili.
Figura 9: Rappresentazione schematica del sarcomero (immagine ottenuta dal sito internet: www.accessexcellence.org).
I filamenti spessi, composti principalmente dalla proteina miosina, sono mantenuti tra
loro paralleli poiché sono ancorati a una zona principalmente rappresentata dalla linea M,
mentre i filamenti sottili, composti principalmente dalla proteina actina, sono connessi ad
una zona in cui si trova la linea Z.
La teoria dello “scorrimento dei filamenti” propone che, sotto l’influenza dell’energia
rilasciata dall’ATP, i filamenti spessi e sottili scorrano gli uni sugli altri, determinando
l’accorciamento del sarcomero. La miosina è una proteina ad elevato peso molecolare
(460kDa), caratterizzata da complesse molecole allungate in cui è possibile distinguere
una coda filamentosa e due teste affiancate tra loro, formate da una parte globosa
simmetrica [Figura 10]. Ogni miofilamento è composto da circa 200 molecole di miosina,
organizzate in modo che la “coda” delle molecole sia disposta secondo l’asse del
miofilamento spesso, mentre le “teste”, sostenute dai rispettivi “bracci articolati”,
sporgono ad intervalli regolari dal miofilamento. Le molecole miosiniche sono orientate in
modo contrapposto (coda a coda) rispetto alla metà di ogni miofilamento spesso, in
maniera tale da costituire due strutture speculari comprendenti un centinaio di molecole

3. Introduzione
-30-
ciascuna. In ciascuna metà di ogni miofilamento spesso le singole molecole di miosina
sono regolarmente spostate l’una rispetto all’altra in senso assiale e disposte secondo un
andamento a spirale [Figura 9].
Figura 10: Struttura della molecola di miosina.
I miofilamenti sottili sono costituiti da tre diverse proteine quali actina (43kDa),
tropomiosina (70kDa) e troponina (80kDa); l’actina è costituita a sua volta da molecole
globulari (G-actina) che tendono a legarsi in modo ordinato tra di loro, formando catene
filamentose di F-actina. Ogni miofilamento sottile comprende da 300 a 400 molecole di G-
actina disposte in due lunghe catene di F-actina, a volte organizzate a spirale a formare
una doppia elica. Dal lato in cui i miofilamenti sottili si legano alla linea Z, i due filamenti
di actina si separano e si ancorano alla linea Z in due distinti punti. Il legame è stabilito da
una particolare proteina, denominata α-actinina, intercalata tra le molecole di actina dei
filamenti sottili e le proteine costitutive della linea Z (principalmente desmina e
vimentina). Ogni molecola globulare di actina presenta, in una determinata regione della
sua superficie, un sito di legame ove sono localizzati gruppi chimici specifici capaci di
combinarsi con quelli presenti sulle “teste” di miosina e che risultano sporgenti dai
miofilamenti spessi, in modo da formare “ponti”. Le molecole di actina sono orientate “a
lisca di pesce” nelle due catene di ogni filamento sottile, con un’angolatura opposta nelle
due metà del sarcomero, in modo che i rispettivi “siti di legame” si trovino sempre
allineati con le “teste” miosiniche che pure sporgono dai filamenti spessi, con
un’angolatura opposta, nelle due metà del sarcomero. Il formarsi e il disaggregarsi dei
“ponti” tra le “teste” di miosina ed i siti di legame dell’actina rappresentano momenti
fondamentali nella contrazione del sarcomero.
Nel miofilamento sottile sono presenti, oltre all’actina, altre proteine che svolgono un
ruolo importante nella contrazione muscolare e sono definite accessorie o regolatorie,

3. Introduzione
-31-
quali ad esempio la tropomiosina e la troponina. La tropomiosina è costituita da molecole
a forma di lunghi bastoncelli, composti da due catene polipeptidiche strutturate ad α-
elica; la loro lunghezza è tale da estendersi lungo sette molecole di actina. Le suddette
catene polipetidiche di tropomiosina sono congiunte tra loro alle estremità, in modo da
costituire un filamento continuo; in ogni miofilamento sottile sono presenti due filamenti
di tropomiosina che decorrono nei due solchi (che rappresentano il “passo della spirale”)
esistenti tra le catene di actina. La troponina presenta molecole complesse,
approssimativamente globulari; nel miofilamento sottile ciascuna molecola di troponina è
legata ai filamenti di tropomiosina in vicinanza dei loro punti di giunzione. Ogni
molecola di troponina è composta da tre distinte subunità peptidiche, ciascuna delle quali
presenta differenti caratteristiche. La prima subunità (troponina T) è sede del legame con
la tropomiosina, la seconda subunità (troponina I) ha funzione inibitoria ed è in grado di
legare la G-actina e, infine, la terza subunità (troponina C) ha una elevata affinità per gli
ioni calcio e conferisce alla molecola troponinica il carattere di una calmodulina [Figura
11].
Figura 11: Disposizione delle proteine tropomiosina e troponina nel miofilamento sottile (immagine ottenuta dal sito internet: www.edoc.hu-berlin.de).
Infine, esistono altre proteine del miofilamento sottile che sono in stretto rapporto con le
linee Z, quali per esempio nebulina, proteina filamentosa “gigante” che decorre
parallelamente ai filamenti di actina, regolandone lo stato di polimerizzazione. Per quanto
concerne i miofilamenti spessi (costituiti da miosina), in prossimità della linea M sono
state rinvenute altre proteine, quali la proteina M, la miomesina, la creatina-fosfochinasi
muscolare, la proteina C, la proteina H e la titina (o connectina). Quest’ultima, è la più
grande proteina del sarcomero; essa attraversa tutto lo spazio tra la linea Z e la banda H e
appare estensibile nella regione della banda I, mentre è inestensibile nella regione della
banda A. La sua funzione è quella di stabilizzare la posizione dei miofilamenti nelle

3. Introduzione
-32-
miofibrille e di impedire una sovradistensione del sarcomero durante il processo di
rilasciamento, ovvero di impedire alle linee Z di allontanarsi tra di loro oltre una certa
misura (Clark K.A. et al., 2002).
La disposizione dei miofilamenti nei diversi tratti del sarcomero è tale per cui, in
condizioni di riposo, a livello della banda I vi sono solo miofilamenti sottili, nell’ambito
della banda A vi sono sia miofilamenti sottili che miofilamenti spessi, mentre nella banda
H vi sono solo miofilamenti spessi. Ove sono presenti sia miofilamenti sottili che
miofilamenti spessi, essi sono interposti secondo una simmetria esagonale: ogni
miofilamento spesso è infatti regolarmente delimitato da sei miofilamenti sottili ed ogni
miofilamento sottile è circondato, a sua volta, da tre miofilamenti spessi. L’elevata
estensibilità delle fibre muscolari non esprime una caratteristica meccanica dei
miofilamenti, che infatti hanno una lunghezza costante qualunque sia la lunghezza che
assume il sarcomero, ma è attribuibile, piuttosto, alla capacità dei miofilamenti di
modificare la loro reciproca posizione. Pertanto, quando il sarcomero si allunga o si
accorcia, i miofilamenti sottili scorrono negli spazi esistenti tra essi e quelli spessi, in
maniera tale che solo la lunghezza dei tratti in cui vi è sovrapposizione tra i miofilamenti
dei due tipi venga ad essere modificata. Ne risulta che, quando la fibra muscolare è
contratta, i miofilamenti sottili e quelli spessi sono ampiamente sovrapposti nella zona
“birifrangente” (e la banda H è molto ristretta), mentre quando essa è distesa i
miofilamenti sottili sono parzialmente o totalmente assenti dagli spazi tra quelli spessi (la
banda H è in questo caso più estesa e può occupare anche l’intera banda A) [Figura 12 ].

3. Introduzione
-33-
Figura 12: Rapporti spaziali tra i principali filamenti del sarcomero in condizioni di riposo e di contrazione.
Differenziamento muscolare e proteasomi
Il processo di miogenesi (in particolare, miogenesi murina) di frequente viene utilizzato
come modello per lo studio dei meccanismi di base che riguardano il differenziamento e
la morfogenesi, con particolare attenzione al coinvolgimento di elementi citoscheletrici e
di complessi ad essi associati.
L’uscita dal ciclo cellulare da parte di una cellula mioblastica coincide con l’inizio del
processo di differenziamento ed induce significative alterazioni a carico del profilo delle
proteine cellulari, con comparsa di alcune tipologie ed introduzione di nuove molecole
(Moran J.L. et al., 2002; Tomczak K.K. et al., 2004).; è pertanto possibile affermare che tale
obiettivo venga raggiunto sia mediante riprogrammazione genica ed espressione di
specifici mRNA, che mediante attività proteolitica.
Peraltro, il sistema proteolitico ubiquitina-proteasoma è stato il primo ad essere
caratterizzato in cellule muscolari da Goldberg A.L. e collaboratori (Fagan J.M. et al., 1997)
e sembra giocare un ruolo importante tra i numerosi fattori che intervengono nel controllo
del processo di differenziamento muscolare. Per lo studio del processo di
differenziamento miogenico in vitro, la fase iniziale prevede la coltivazione di mioblasti
proliferanti ed indifferenziati in terreno di coltura in presenza di siero. I fattori di crescita

3. Introduzione
-34-
presenti nel terreno di coltura promuovono la proliferazione dei mioblasti e ne
prevengono il differenziamento. Dopo una prima fase di proliferazione, i mioblasti sono
privati dei fattori di crescita (drastico abbassamento della percentuale di siero nel terreno
di coltura); in questo modo, essi escono definitivamente dal ciclo cellulare ed iniziano ad
esprimere geni muscolo-specifici (differenziamento biochimico). A questo stadio sono
chiamati miociti e, pur essendo ancora mononucleati, sono terminalmente differenziati, in
quanto non possono essere indotti nuovamente a dividersi mediante stimolazione con
fattori di crescita; i miociti mostrano, inoltre, anche una ridotta predisposizione
all’apoptosi. Successivamente, i miociti si fondono per formare grandi sincizi
multinucleati chiamati miotubi (differenziamento cellulare); il differenziamento ha
termine in pochi giorni (Walsh K. and Perlman H., 1997). Dal punto di vista molecolare, il
processo di differenziamento muscolare prevede una serie ordinata di eventi molecolari la
cui realizzazione è fondamentale sia per il raggiungimento dello stato post-mitotico, che
per l’espressione dei geni muscolo-specifici (Kitzmann M. and Fernandez A., 2001).
L’espressione genica muscolo-specifica è attivata dall’azione coordinata di due famiglie di
fattori trascrizionali miogenici: la famiglia bHLM (“basic helix-loop-helix”), che
comprende le molecole MyoD, Myf5, miogenina e MRF4, e la famiglia MEF-2 (“myocyte
enhancer factor 2”) (Weintraub H. et al., 1991; Weintraub H., 1993; Olson E.N. and Klein
W.H., 1994; Kitzmann M. and Fernandez A., 2001). Due componenti della famiglia bHLM,
ossia MyoD e Myf5, sono già presenti nei mioblasti proliferanti e indifferenziati; il loro
ruolo consiste nell’indirizzare il differenziamento delle cellule precursori proliferanti
esclusivamente nella direzione miogenica. I passaggi successivi della miogenesi
richiedono un altro fattore della famiglia bHLM, ossia miogenina. Quest’ultima proteina
coopera con i componenti della famiglia MEF-2 all’attivazione dell’espressione di molti
geni strutturali del muscolo scheletrico. Studi condotti sul differenziamento di mioblasti
in vitro hanno rivelato che l’espressione di miogenina viene indotta 24 ore dopo la
rimozione del siero dal terreno di coltura (Andrés V. and Walsh K., 1996).
Successivamente, queste cellule esprimono l’inibitore delle chinasi ciclina-dipendenti
p21Cip1 ed escono in modo permanente dal ciclo cellulare (Halevy O. et al., 1995). Una
volta che le cellule sono diventate post-mitotiche (circa 36-48 ore dopo la rimozione dei
fattori di crescita), viene indotta l’espressione di proteine miofibrillari, come miosina e
creatinina muscolare. I miociti post-mitotici completano infine il loro differenziamento
mediante fusione in miotubi multinucleati (Lassar A.B. et al., 1994; Lassar A.B. and
Munsterberg A., 1994). È da tempo noto che il prerequisito fondamentale, affinchè venga

3. Introduzione
-35-
indotta l’espressione dei geni muscolo-specifici, è l’uscita definitiva dal ciclo cellulare.
Infatti, ad eccezione della miogenina, la trascrizione dei geni specifici dello stato
differenziato si verifica esclusivamente in cellule post-mitotiche. In particolare, la proteina
pRb, che controlla il differenziamento muscolare, agisce, da un lato, arrestando le cellule
in G0, aumentando l’attività di trascrizione della proteina MyoD e contribuendo
all’accumulo di p21Cip1, e dall’altro, cooperando con MyoD nello stimolare l’attività
trascrizionale della famiglia dei fattori miogenici MEF-2 (David C. and Konieczni S.F.,
1995; Novitch B.G. et al., 1996; Novitch B.G. et al., 1999). Inoltre, è stato recentemente
osservato che i fattori di regolazione del muscolo formano eterodimeri con le cosiddette
proteine E, consentendo l’attivazione del processo di differenziamento miogenico
mediante il loro legame con specifiche sequenze denominate “E box”, presenti a livello
delle regioni regolatorie del promotore dei geni bersaglio del muscolo (Pownall M.E. et al.,
2002).
In particolare, l’attività di trascrizione dei fattori di regolazione del muscolo è
negativamente regolata da una famiglia di proteine denominate “inhibitors of
differentiation” (Id), la cui funzione è quella di inibire il legame dei suddetti fattori con il
DNA. Nello specifico, la proteina MyoD e la proteina E presentano una differente affinità
di legame nei confronti della proteina Id1; la proteina E è infatti caratterizzata da una
maggiore affinità per tale proteina, rispetto alla proteina MyoD. Il legame tra proteina E e
proteina Id1 consente a quest’ultima di inibire l’attività di trascrizione stimolata dalla
proteina MyoD muscolo-specifica. Nei mioblasti la proteina Id1 è presente in elevate
quantità inibendo pertanto la trascrizione dei geni muscolo-specifici, mediata dalla
proteina MyoD, con conseguente assenza delle proteine che intervengono nel processo di
differenziamento muscolare ed inibizione del passaggio da mioblasti proliferanti a
miotubi terminalmente differenziati (Sun X.H. et al., 1991; Jen Y. et al., 1992; Langlands K.
et al., 1997).
Le proteine MyoD e Id1 sono degradate dal sistema ubiquitina-dipendente mediato dal
proteasoma 26S (Abu Hatoum O. et al., 1998; Breitschopf K. et al., 1998; Ciechanover A. et
al., 1999; Floyd Z.E. et al., 2001; Lingbeck J.M. et al., 2003; Fajerman I. et al., 2004). Il legame
tra la proteina MyoD e la proteina Id1, oltre ad inibire l’attività trascrizionale di MyoD, la
rende probabilmente più suscettibili alla degradazione da parte dei proteasomi (Abu
Hatoum O. et al., 1998). Sun L. e collaboratori (Sun L. et al., 2005) hanno recentemente
suggerito che, nel corso del processo di differenziamento miogenico, i livelli cellulari delle
suddette proteine siano strettamente regolati da una combinazione di eventi che

3. Introduzione
-36-
prevedono, da un lato, la sintesi proteica e, dall’altro, la degradazione di tali proteine.
Inoltre, durante il processo di differenziamento muscolare, l’espressione delle proteine
MyoD e Id1 è strettamente associata a quella delle proteine E, che a loro volta sono
degradate attraverso il sistema ubiquitina-dipendente mediato dal proteasoma 26S (Kho
C.J. et al., 1997; Huggins G.S. et al., 1999). Per quanto concerne la localizzazione cellulare
dei proteasomi durante il processo di differenziamento muscolare, in letteratura sono
riportati dati alquanto contrastanti. Nello specifico, alcuni Autori hanno evidenziato una
localizzazione prettamente nucleare (Stauber W.T. et al., 1987), altri esclusivamente
citoplasmatica (Kamura K. et al. ,1988; Beyette J.R. and Mykles D.L., 1992; Kumamoto T. et
al., 2000), altri ancora, sia citoplasmatica che nucleare (Low P. et al., 2000).
Esperimenti di cinetica che si sono avvalsi di un anticorpo monoclonale diretto nei
confronti di una specifica subunità proteica del proteasoma 20S (α1/p27K), hanno inoltre
permesso di mettere in evidenza una distribuzione dinamica di tali complessi durante il
processo di differenziamento di cellule satelliti in vitro. In particolare, in cellule satelliti di
ratto allo stadio di mioblasti, la localizzazione dei proteasomi è risultata inizialmente di
tipo citoplasmatico e, in fasi più tardive, di tipo nucleare. Infine, durante la fase di fusione
dei mioblasti, la loro localizzazione è risultata nuovamente di tipo citoplasmatico, con un
profilo puntiforme, parzialmente sovrapponibile a quello dei microfilamenti di actina e
dei filamenti intermedi di tipo desmina; tale profilo assumeva successivamente una
organizzazione pseudo-sarcomerica in bande, che si sovrapponeva ai microfilamenti di
actina. In particolare, in colture di cellule satelliti di ratto in fase di fusione, i proteasomi
sembrano presentare questo tipo di organizzazione poco prima della formazione di una
struttura sarcomerica matura, messa in evidenza da marcatori di α-actina (Foucrier J. et
al., 1999). Questa organizzazione pseudo-sarcomerica è stata successivamente rilevata
anche in sezioni longitudinali di tessuto muscolare scheletrico e in colture primarie di
cardiomiociti ventricolari di ratto mediante l’impiego di sonde immunologiche dirette nei
confronti di due diverse subunità del proteasoma 20S (α1/p27K e α3/p29K). Inoltre, in
colture primarie di cellule muscolari lisce di aorta di ratto, in cui non si osserva
normalmente la formazione di una struttura sarcomerica, è stata in alcuni rari casi rilevata
la formazione di una struttura pseudo-sarcomerica del proteasoma mediante l’impiego di
sonde immunologiche dirette nei confronti di alcune specifiche subunità del proteasoma
20S (Foucrier J. et al., 2001). Sulla base di questi dati è stato recentemente ipotizzato che la
formazione di una struttura pseudo-sarcomerica da parte di specifici proteasomi possa
svolgere una particolare funzione nell’ambito del sarcomero, mediante l’instaurarsi di un

3. Introduzione
-37-
legame con alcuni degli elementi che controllano il processo di formazione del sarcomero,
rivalutando l’idea secondo la quale i proteasomi non sarebbero componenti proteiche
solubili nel citoplasma, ma bensì complessi molecolari associati a specifiche strutture
cellulari.
In effetti, una ormai sostanziosa mole di dati di letteratura avvalora l’ipotesi secondo la
quale la nozione spaziale, ovvero la specifica distribuzione di una molecola nell’ambito
dei diversi compartimenti cellulari, assume un’importanza analoga a quella della
funzione medesima, che non potrebbe espletarsi al di fuori di quel preciso distretto.
Pertanto, sebbene l’implicazione dei proteasomi nell’ambito della degradazione delle
proteine a livello muscolare sia ben documentata, i dati di letteratura attualmente
disponibili, relativi alla localizzazione dei proteasomi nel corso del processo di
differenziamento, sono ancora piuttosto frammentari e parziali. Lo studio della
localizzazione di specifici proteasomi a livello del tessuto muscolare scheletrico
rappresenta uno degli obiettivi di questo studio.
3.2.2 CITOMEGALOVIRUS
Caratteristiche generali
Il citomegalovirus umano (HCMV) viene ancora oggi considerato uno tra gli agenti
eziologici più importanti nell’indurre anomalie congenite e/o sequele neurologiche
tardive nel bambino, in seguito ad un’infezione primaria intrauterina. In aggiunta a
queste gravi patologie, HCMV costituisce un potenziale problema in altre particolari
condizioni come, ad esempio, in individui con infezione da HIV o in soggetti sottoposti a
trapianto d’organo ed in trattamento terapeutico immunosoppressivo.
Il citomegalovirus umano appartiene alla famiglia Herpesviridae, sottofamiglia
Betaherpesvirinae. HCMV mostra alcune caratteristiche biologiche distintive comuni a tutti
i betaherpesvirus come, ad esempio, uno spiccato tropismo per le ghiandole salivari, uno
spettro d’ospite molto ristretto ed una lenta crescita in cellule in coltura. HCMV si replica
in vitro in cellule fibroblastoidi con corredo cromosomico diploide e provenienti dalla
specie ospite naturale in vivo, mentre le cellule indifferenziate, trasformate o aneuploidi
non sono suscettibili all’infezione. D’altra parte, nell’infezione naturale non solo le cellule
fibroblastoidi, ma anche le cellule epiteliali, muscolari lisce ed endoteliali sono in grado di
sostenere un’infezione produttiva (Sinzger C. et al., 1995).

3. Introduzione
-38-
Il citomegalovirus ha dimensioni variabili (comprese tra 150 e 250 nm) e presenta un
capside a simmetria icosaedrica. Tale virus è, inoltre, rivestito da un involucro lasso,
denominato pericapside, composto principalmente da fosfolipidi e glicoproteine, al di
sotto del quale è presente un tegumento elettrodenso, costituito da materiale fibro-
granulare di natura proteica [Figura 13].
Figura 13: Rappresentazione schematica di citomegalovirus (immagine ottenuta dal sito internet: www.biografix.de).
I virioni presentano uno spiccato pleiomorfismo, dovuto non solo alla variabilità di
spessore del tegumento, ma anche alla possibile presenza di particelle difettive. In
particolare, si è visto che nelle cellule infettate è possibile riscontrare, oltre ai virioni
completi, altri due tipi di particelle virali defettive, chiamate “Dense Bodies” (DB) e “Non
Infectious Envelope Particles” (NIEP) (Sarov I. and Abady I., 1975; Irmiere A. and Gibson
W., 1983; Gibson W., 1996). I virus “DB”, provvisti di un pericapside uguale a quello dei
virioni maturi, sono privi sia di genoma virale, che di struttura capsidica interna. I virus
“NIEP” sono anch’essi privi del genoma, ma provvisti di capside, tegumento e
pericapside virale. Come per tutti i membri della famiglia Herpesviridae, il genoma di
citomegalovirus è costituito da un’unica molecola lineare di DNA a doppia elica, ma di
dimensioni maggiori rispetto agli altri herpesvirus.
nucleocapside
genoma
membrana
glicoproteine
tegumento

3. Introduzione
-39-
Le proteine virali
Nell’ambito delle proteine virali a funzione enzimatica, le più importanti proteine
regolatorie sono rappresentate dai prodotti di due diversi loci genici di HCMV; il locus
IE1/IE2 che codifica per una famiglia di proteine regolatorie denominate “Immediate
Early Modulators” (MIE) e il locus US3 codificante per una proteina immunomodulatoria.
A sua volta, l’attività delle proteine regolatorie virali potrebbe essere modulata da chinasi
(Gallina A. et al., 1999) e fosfatasi (Michelson S. et al., 1996) di origine cellulare, già
presenti nel virione maturo.
Dall’espressione dei geni precocissimi deriva il primo gruppo di proteine virali,
comprendente il complesso antigenico nucleare “Immediate-Early Antigens” (IEA),
composto da almeno due proteine principali: IE1p72 e IE2p86. Queste proteine
precocissime sono caratterizzate dalla presenza di una breve sequenza aminoacidica
comune codificata dall’esone 2 e dall’esone 3 della regione IE. Recentemente è stato
attribuito alla sequenza dell’esone 3 un ruolo di spicco nell’ambito della regolazione della
trascrizione, dell’espressione dei geni virali precoci e nella modulazione delle proteine,
quali ad esempio le cicline, che risultano direttamente coinvolte nella regolazione del ciclo
cellulare (White E.A. and Spector D.H., 2005).
IE2p86 rappresenta la più importante proteina con funzioni regolatorie codificata dal
virus (Spector D.H., 1996; Stenberg R.M., 1996). Il ruolo più rilevante di questa proteina
consiste nell’attivazione dell’espressione sia dei geni β che dei geni γ. Compito di IE2p86
è, inoltre, quello di assicurare che l’espressione dei geni, durante l’infezione, avvenga in
modo sequenziale e temporalmente regolato, oltre che di garantire la repressione
dell’espressione dei due geni α IE1/IE2 e, probabilmente, anche del gene US3, nelle fasi
tardive dell’infezione. I prodotti del gene IE2 funzionano quindi da attivatori
trascrizionali, coadiuvati in questa funzione dai prodotti del gene IE1 che ne promuovono
ulteriormente l’attività. Una funzione inedita è stata recentemente attribuita alla proteina
IE2p86, che risulta coinvolta nella regolazione dell’espressione dei geni che codificano per
proteine che agiscono come mediatori e regolatori dell’immunità innata, quali le citochine.
In corso di infezione virale, le cellule infettate sono in grado di rispondere all’infezione
mediante l’attivazione di citochine e chemochine pro-infiammatorie. In particolare, in
corso di infezione virale le prime citochine ad essere espresse e secrete sono l’interferone β
(IFN-β) e l’interferone α (IFN-α), il cui scopo è quello di bloccare la replicazione del
genoma virale in cellule infettate e nei tessuti circostanti. Inoltre, le cellule infettate
possono produrre chemochine la cui funzione è quella di consentire la rimozione degli

3. Introduzione
-40-
agenti virali mediante il richiamo di leucociti, di macrofagi, di cellule “Natural Killer”
(NK) e di cellule T al sito di infezione, potenziando, da un lato, l’attività citotossica che
caratterizza le cellule NK e T e, dall’altro, bloccando l’entrata degli agenti virali che
riconoscono i recettori per le chemochine.
Numerosi dati di letteratura mettono in evidenza che HCMV regola l’espressione
dell’interferone β (Zhu H. et al., 1998; Browne E.P. et al., 2001; Browne E.P. and Shenk T.,
2003; Taylor R.T. and Bresnahan W.A., 2005), di alcune chemochine, quali per esempio le
chemochine definite come “regulated upon activation normal T cell expressed and
secreted” (RANTES) (Abate D.A. et al., 2004; Browne E.P. et al., 2001; Browne E.P. and
Shenk T., 2003; Zhu H. et al., 1998; Gravel S.P. and Servant M.J., 2005; Schroeder M.B. and
Worthen G.S., 2001), di monociti indotti dall’interferone γ (“monokine induced by
interferon-γ” o “MIG”) (Abate D.A. et al., 2004; Browne E.P. et al., 2001; Browne E.P. and
Shenk T., 2003), delle proteine 1 e 2 di monociti chemiotattici (MCP-1 e -2) (Browne E.P.
and Shenk T., 2003; Hirsch A.J. and Shenk T., 1999), della proteina 1α di macrofagi indotti
dal processo infiammatorio (MIP-1α) (Abate D.A. et al., 2004; Browne E.P. et al., 2001) e,
infine, dell’interleuchina 8 (Browne E.P. and Shenk T., 2003; Compton T. et al., 2003a;
Craigen J.L. et al., 1997; Randolph-Habecker J.R. et al., 2002). L’espressione delle suddette
proteine risulta particolarmente pronunciata quando l’espressione dei geni virali di
HCMV è arrestata, facendo pertanto supporre che una o più proteine virali di nuova
sintesi possano bloccare attivamente l’espressione di questi geni (Browne E.P. et al., 2001;
Hirsch A.J. and Shenk T., 1999; Schroeder M.B. and Worthen G.S., 2001; Zhu H. et al.,
1997). Studi recenti hanno dimostrato che la proteina virale IE2p86 è in grado di impedire
l’induzione dell’interferone β in corso di infezione virale (Taylor R.T. and Bresnahan
W.A., 2005), di intervenire nella soppressione dell’induzione trascrizionale delle citochine
e, parallelamente, di causare un blocco dell’espressione dei geni che codificano per le
citochine e per le chemochine pro-infiammatorie in corso di infezione virale da HCMV
(Taylor R.T. and Bresnahan W.A., 2006).
Per quanto concerne la proteine IE1p72, è da sottolineare che non solo è in grado di
cooperare con la proteina IE2p86, ma anche di influenzare direttamente l’espressione
genica attraverso l’interazione con diversi fattori della trascrizione. IE1p72 è, inoltre,
coinvolta in numerosi processi cellulari quali, ad esempio: regolazione genica,
progressione del ciclo cellulare, trasduzione dei segnali, dispersione dei “PODs” (“PML
oncogenic domains” o “ND10” o “nuclear dots”) e apoptosi (Zhu H. et al., 1995; Ahn J.H.
and Hayward G.S., 1997; Muller S. and Dejean A., 1999; McElroy A.K. et al., 2000).

3. Introduzione
-41-
Sempre nell’ambito dell’espressione dei geni α, sono da evidenziare i diversi prodotti di
“splicing” codificati dal gene US3, che hanno caratteristiche di glicoproteine integrali di
membrana (Jones T.R. et al., 1995; Jones T.R. et al., 1996) e risultano coinvolti nella
diminuita espressione delle molecole del complesso maggiore di istocompatibilità di
classe I, ostacolandone sia il trasporto intracellulare che la maturazione. Il gene US3
rappresenta il primo gene virale, trascritto subito dopo l’inizio dell’infezione, che
contribuisce, insieme a numerose altre strategie attuate da HCMV, all’evasione della
risposta immunitaria (Liu W. et al., 2002).
I prodotti proteici dei geni precoci β includono antigeni virali a localizzazione nucleare
(“EA” o “Early Antigens”), antigeni virali con sede citoplasmatica (“ECA” o “Early
Cytoplasmic Antigens”), distribuiti sia nella regione perinucleare sia diffusamente nel
citoplasma, e, infine, proteine virali di membrana (“EMA” o “Early Membrane
Antigens”). Anche l’enzima DNA-polimerasi virale viene trascritto dai geni β.
Il complesso dei geni tardivi, trascritto dopo la replicazione del DNA virale, codifica per
le proteine strutturali che andranno a comporre il pericapside, il tegumento ed il capside
delle particelle virali.
Il pericapside virale è formato, come già accennato, da un doppio strato lipidico, tipico
delle membrane cellulari, in cui sono inseriti complessi glicoproteici virus-specifici, che
svolgono funzioni di rilievo in diversi processi, quali l’ingresso del virus nella cellula
ospite o, ancora, la risposta immunitaria, in quanto siti di legame per gli anticorpi
neutralizzanti. In particolare, sono due i principali complessi glicoproteici conservati in
tutti i sottogruppi della famiglia Herpesviridae (Britt W.J. and Mach M., 1996). Uno di
questi complessi viene codificato dal gene gB. Il gene gB viene trascritto in un mRNA
codificante per una proteina che, dopo glicosilazione e trasporto nell’apparato del Golgi,
subisce un taglio endoproteolitico che dà luogo ad un dimero della glicoproteina B (gB). Il
secondo complesso glicoproteico è formato dai prodotti di tre geni distinti: gH, gL e gO. Il
genoma di HCMV codifica, inoltre, per numerose proteine con caratteristiche tipiche delle
proteine transmembranarie, come la proteina “integrate membrane protein” (Lehner R. et
al., 1989), che potrebbero quindi rappresentare costituenti minori del pericapside virale ed
essere coinvolte nelle fasi di attacco e di ingresso del virus nella cellula ospite.
Il tegumento, o matrice, è costituito da 25 tipi diversi di proteine fosforilate, come
sottolineato dal prefisso pp (“phosphoprotein”) che le contraddistingue; tali proteine sono
altamente immunogene. Le più rappresentate sono: ppUL83 (pp65), ppUL32 (pp150),
ppUL99 (pp28), ppUL82 (pp71) e ppUL48 (“huge tegument protein”) (Bradshaw P.A. et

3. Introduzione
-42-
al., 1994; Gibson W., 1996). La funzione della maggior parte delle proteine che
compongono il tegumento rimane ignota; molte di queste sembrerebbero comunque
essere coinvolte nella regolazione dell’espressione genica come transattivatori
trascrizionali (Liu B. and Stinski M.F., 1992; Winkler M. et al., 1995; Romanowski M.J. et
al., 1997).
Due proteine del tegumento, la proteina pp150 (“BPP” o “Basic Phosphoprotein”) e la
proteina pp65 (“LMP” o “Lower Matrix Protein”), rappresentano le proteine più
abbondanti durante la replicazione virale. Nel loro insieme, le due proteine costituiscono
il 35% dell’intera massa proteica del virione.
Le proteine pp65 e pp150 sono entrambe bersagli per la fosforilazione attuata dalle chinasi
virioniche. Già nell’ambito della prima ora di infezione la proteina pp65 viene
velocemente traslocata verso il nucleo cellulare grazie a specifici segnali di localizzazione
nucleare. Tali segnali sono costituiti da brevi sequenze di aminoacidi basici, denominate
sequenze di localizzazione nucleare o “NLS”. È stato ipotizzato che queste sequenze
vengono riconosciute, probabilmente a livello citoplasmatico, da proteine con funzione
recettoriale che si legano ad esse trasportandole verso il poro nucleare. In questo processo
le nucleoporine svolgono un ruolo primario, riconoscendo le “NLS” insieme alle proteine
recettoriali a cui sono legate e consentendone il successivo passaggio attraverso il poro
nucleare. La proteina pp65 contiene due “NLS” funzionalmente e strutturalmente distinte,
che risultano localizzate all’estremità carbossi-terminale della proteina; inoltre,
presentano una sequenza addizionale che ne garantisce un efficace trasferimento in sede
nucleare. Il rapido trasporto verso il nucleo, unitamente alla dimostrata attività chinasica
della stessa fosfoproteina, lascia ipotizzare un coinvolgimento della proteina pp65 nella
regolazione della replicazione e dell’espressione genica virale. L’attività chinasica
mostrata dalla proteina pp65 potrebbe essere dovuta alla sua interazione con una chinasi
di derivazione cellulare (Roby C. and Gibson W., 1986; Gallina A. et al., 1999). È
sorprendente il fatto che la proteina pp65 sembri apparentemente superflua per la
replicazione di HCMV in colture cellulari (Schmolke S. et al., 1995), nonostante la sua
abbondanza e il fatto che la presenza di un RNA antisenso per pp65 sia in grado di
bloccare la replicazione del virus (Dal Monte P. et al., 1996). Di particolare rilevanza è la
dimostrazione di un’associazione della proteina pp65 ai cromosomi della cellula infettata
durante la metafase. Il significato di tale associazione resta ancora da indagare, ma
potrebbe essere collegato alla capacità di HCMV di indurre aberrazioni cromosomiche
(Dal Monte P. et al., 1996). Inoltre, tale proteina è in grado di indurre una inibizione

3. Introduzione
-43-
parziale dell’espressione di IFN β e di alcune chemochine in corso di infezione virale,
mentre non sembra essere coinvolta nella regolazione delle citochine pro-infiammatorie, a
differenza di quanto osservato per la proteina IE2p86 (Browne E.P. and Shenk T., 2003;
Abate D.A. et al., 2004; Taylor R.T. and Bresnahan W.A., 2006). La fosfoproteina pp65 è in
grado, inoltre, con un meccanismo per il momento non identificato, di interferire con la
presentazione antigenica delle proteine IE maggiori, denominate pUL122 e pUL123
(Gilbert M.J. et al., 1996).
Un’altra abbondante fosfoproteina del tegumento è la proteina pp71 (“UMP” o “Upper
Matrix Protein”), che costituisce un’importante transattivatore trascrizionale in grado di
attivare l’espressione del locus IE1/IE2 (Liu B. and Stinski M.F., 1992).
Il tegumento contiene, inoltre, numerose proteine funzionalmente non ancora
caratterizzate, come ad esempio la proteina pp28, relativamente abbondante e altamente
immunogena, che si localizza in prossimità della superficie capsidica del virione, e la
proteina pp130, che risulterebbe implicata nell’incapsidamento del genoma virale.
In associazione alle particelle virali purificate sono state osservate diverse proteine di
derivazione cellulare come β2-microglobulina, CD13 (aminopeptidasi N), fosfatasi I,
annexina II e actina; si ipotizza che tali associazioni potrebbero favorire rispettivamente
l’adsorbimento e il trasporto intracellulare del virus durante l’infezione della cellula
ospite (Baldick C.J. and Shenk T., 1996; Varnum S.M. et al., 2004).
Il capside virale è composto da sette tipi di proteine diverse: “MCP” (“Major Capsid
Protein”), che rappresenta il principale componente dei pentoni ed esoni capsidici, “mCP”
(“Minor Capsid Protein”), “mC-BP” (“Minor Capsid Binding Protein”), “SCP” (“Smallest
Capsid Protein”) e, infine, tre distinte proteine che svolgono funzioni diverse nella
costruzione del capside stesso. Di queste ultime tre proteine la più rappresentata è la
proteina “AP” (“Assembly Protein”), che deriva dalla scissione proteolitica del suo
precursore, operata da una proteina virale con attività proteasica, detta assemblina (Welch
A.R. et al., 1991). La proteina “AP” è presente solo nei capsidi virali privi di DNA (virus
“NIEP”) e non nelle particelle virali mature contenenti l’acido nucleico (Robson L. and
Gibson W., 1989); questo depone per un coinvolgimento attivo della stessa proteina nel
processo di incapsidamento del DNA virale.
Il genoma virale
Il genoma di HCMV, che ha una lunghezza compresa tra i 200 e 240 Kb e un peso
molecolare di 150-155x106 daltons, è formato da due sequenze nucleotidiche di lunghezza

3. Introduzione
-44-
diversa, UL e US, fiancheggiate da brevi sequenze di basi ripetute e invertite, indicate
rispettivamente come b (TRL/IRL) e c (IRS/TRS), che consentono l’organizzazione del
genoma in quattro forme isomeriche. Una sequenza ripetuta ma non invertita, definita
sequenza a, si colloca alle estremità della molecola di DNA. La stessa sequenza, ma con
orientamento invertito, si trova localizzata anche nel punto di congiunzione tra le due
sequenze UL ed US. Questa peculiare distribuzione della sequenza a promuove
l’inversione genomica. La sequenza a porta inoltre segnali di regolazione in cis, detti pac-1
e pac-2, altamente conservati in tutti gli herpesvirus e finalizzati al taglio e
all’incapsidamento del genoma virale (McVoy M.A. et al., 1998).
Ciclo di moltiplicazione virale
Lo studio del ciclo replicativo di HCMV è reso complesso dal fatto che tale virus, oltre a
dare luogo ad una infezione produttiva, può rimanere nelle cellule allo stato latente o
indurre in queste un’infezione di tipo persistente. Per quel che riguarda il ciclo litico
[Figura 14], l’attacco alla superficie cellulare è rapido ed efficiente, sia in cellule
permissive che in cellule non permissive, suggerendo una vasta distribuzione dei recettori
cellulari riconosciuti da HCMV. L’interazione tra virus e cellula ospite avviene mediante il
legame degli antirecettori virali a molecole di proteoglicano presenti sulla superficie della
membrana cellulare e a recettori cellulari addizionali (Boyle K.A. and Compton T., 1998;
Compton T. et al., 1993). All’iniziale interazione con i recettori cellulari seguono
l’adsorbimento e la penetrazione del virus che coinvolgono recettori cellulari
abbondantemente rappresentati ma scarsamente caratterizzati.
Figura 14: Il ciclo replicativo di citomegalovirus umano (immagine ottenuta dal sito internet: www.biografix.de).
RE
N
G

3. Introduzione
-45-
Mentre i complessi recettoriali cellulari restano ancora da identificare, gli antirecettori
virali sono rappresentati, con ogni probabilità, dalle glicoproteine gB e gH/gL (Kinzler
E.R. and Compton T., 2005).
La penetrazione del virus è mediata dalla fusione del pericapside virale con la membrana
cellulare, processo che risulta indipendente dall’abbassamento del pH (Compton T. et al.,
1992) e che, probabilmente, coinvolge in prima istanza il complesso glicoproteico virale
gH/gL (Keay S. and Baldwin B., 1991).
Successivamente alla penetrazione nella cellula ospite, il nucleocapside virale si muove
rapidamente verso il nucleo. Il ciclo replicativo di HCMV avviene, infatti,
prevalentemente a livello nucleare, dove il genoma virale circolarizzato, grazie alla
presenza delle strutture palindromiche alle sue estremità, viene trascritto da idonei
enzimi cellulari.
HCMV ha un ciclo replicativo assai lento e induce nelle cellule infettate caratteristiche
modificazioni che portano ad un aumento delle dimensioni cellulari, oltre alla comparsa
di inclusioni nucleari e citoplasmatiche, alle quali il virus deve il suo caratteristico nome.
L’espressione del genoma virale avviene in modo sequenziale e temporalmente regolato.
Sulla base di questo criterio, possono essere identificate tre classi di geni virali: geni
precocissimi (“IE“ o “immediate early“ o α), precoci (“E“ o “early“ o β) e tardivi (“L“ o
“late“ o γ). L’espressione dei geni α avviene immediatamente dopo l’ingresso del virus
nella cellula ospite ed è indipendente dall’espressione di altri geni virali. L’espressione dei
geni β, al contrario, dipende dall’espressione dei geni α, così come dipendente dai geni β è
la successiva espressione dei geni γ. La trascrizione in sequenza temporale dei geni α, dei
geni β e, infine, dei geni γ, avviene ad opera dell’enzima RNA polimerasi II cellulare e di
altri fattori che fanno parte della complessa macchina trascrizionale della cellula ospite,
cooptata dal virus durante l’infezione. I fattori trascrizionali della cellula ospite sono
influenzati da transattivatori codificati dal virus, che modulano l’espressione sia dei geni
virali che dei geni cellulari durante l’infezione. Sempre a livello nucleare, il DNA replicato
viene inserito nei capsidi preformati; in questo modo, le particelle sub-virali risultano
essere di dimensioni tali da non poter abbandonare il distretto nucleare attraverso il
complesso dei pori nucleari. Il capside virale deve quindi attraversare la lamina nucleare
per poter raggiungere la membrana nucleare interna. Questo processo richiede la
depolimerizzazione della lamina nucleare stessa che, molto probabilmente, avviene per
fosforilazione delle proteine che la compongono, in particolare in seguito all’attivazione
di chinasi cellulari reclutate dal virus (Muranyi W. et al., 2002).

3. Introduzione
-46-
Successivamente, gemmando attraverso la membrana nucleare interna verso lo spazio
perinucleare, le particelle sub-virali acquisiscono alcune proteine del tegumento ed un
pericapside primario che, fondendosi con la membrana nucleare esterna, permetterà il
rilascio del nucleocapside nel citoplasma. I nucleocapsidi “nudi” raggiungono in seguito il
reticolo endoplasmatico e, infine, l’apparato del Golgi, acquisendo addizionali proteine
del tegumento e il loro pericapside maturo. I virioni sono quindi trasportati verso la
membrana citoplasmatica in vescicole derivate dall’apparato del Golgi e vengono liberati
all’esterno della cellula ospite per esocitosi (Sanchez V. and Spector D.H., 2002). Il virione
maturo di HCMV, a differenza degli altri virus a DNA, contiene due classi di molecole di
RNA, oltre alla molecola di DNA genomico. Un tipo di RNA forma delle strutture ibride
del tipo RNA-DNA all’interno dell’origine di replicazione “oriLyt“ che potrebbero
facilitare la replicazione dello stesso DNA virale (Prichard M.N. et al., 1998), mentre il
secondo tipo di trascritto sembra essere impacchettato all’interno delle particelle virali
localizzandosi a livello del tegumento e sembra anche essere espresso subito dopo
l’ingresso del virus nella cellula ospite. Le proteine codificate da quest’ultimo tipo di RNA
hanno funzione al momento attuale ancora sconosciuta (Bresnahan W.A. and Shenk T.,
2000).
Effetti dell’infezione da HCMV sulla cellula ospite: alterazioni del ciclo cellulare ed
intervento dei proteasomi
L’impatto che citomegalovirus ha come patogeno sulla salute umana evidenzia di per sé
l’importanza di sviluppare ulteriori conoscenze sulle interazioni con l’ospite, in
particolare a livello cellulare.
È da tempo noto come l’infezione da HCMV induca una complessa serie di modificazioni
nella cellula ospite che si ripercuotono, come è intuitivo, sul metabolismo cellulare, le cui
alterazioni sono in genere profonde, ma anche dipendenti dal tipo di cellula infettata;
come eventi consequenziali dell’infezione sono da menzionare la morte cellulare
(infezione di cellule permissive) o, alternativamente, differenti tipologie di danni cellulari,
che possono condurre a processi di trasformazione o all’instaurarsi di un’infezione virale
di tipo persistente o latente in cellule semi-permissive o non permissive, rispettivamente.
Per quel che riguarda il ciclo litico, è noto come già il legame tra recettori cellulari ed
antirecettori virali inneschi una serie di risposte cellulari simili a quelle da “secondi
messaggeri”, accompagnate anche dall’espressione transiente di mRNA oncogeni, così

3. Introduzione
-47-
come dalla comparsa di ulteriori tipi di messaggeri, analoghi a quelli indotti da
interferone α in cellule non infettate.
In uno scenario così complesso, un ruolo di spicco hanno le interazioni tra HCMV e
l’elaborato sistema che regola il ciclo cellulare. Numerosi dati di letteratura supportano
una netta azione di disturbo, operata da HCMV, sulla normale evoluzione del ciclo
cellulare, con conseguente deregolazione di quest’ultimo (Dittmer D. and Mocarski E.S.,
1997; Salvant B.S. et al., 1998; Sinclair J. et al., 2000; Kalejta R.F. and Shenk T., 2002); ne
rappresenta una chiara evidenza l’induzione di elevati livelli di p53, di complessi attivi
costituiti da cicline E e B e da forme iperfosforilate di Rb (si veda il paragrafo sul ciclo
cellulare di questa sezione introduttiva) (Muganda P. et al., 1994; Jault F.M. et al., 1995).
Tale iperfosforilazione, a sua volta, elimina l’effetto inibitorio di Rb su complessi
transattivatori, che hanno come bersagli principali i geni cellulari che codificano per tutta
una serie di enzimi coinvolti nella replicazione del DNA, alcuni dei quali sono necessari
anche al virus per la replicazione del proprio genoma. In effetti, diversi Autori riportano
un’azione di arresto del ciclo cellulare, in particolare a livello della transizione G1/S, così
come G2/M, indotta da HCMV, con un ruolo prominente attribuito alla proteina
precocissima IEp86 (Bresnahan W.A. et al., 1996; SongY.-J. and Stinski M.F., 2005) e, di
converso, un significativo ritardo nell’espressione dei geni virali precocissimi, qualora
l’infezione venga avviata nell’ambito della fase S (Fortunato E. et al., 2002).
È evidente che i fattori cellulari e virali che intervengono nella deregolazione di
meccanismi a loro volta così complessi, quali appunto il ciclo cellulare, sono senza dubbio
molteplici e sarebbe riduttivo e non corretto attribuire agli uni o agli altri un ruolo da
“protagonista”.
Ciò nonostante, è altrettanto plausibile ipotizzare che il complesso ubiquitina-proteasoma,
che, come già evidenziato in questa sede, ha un ruolo rilevante nel controllo del ciclo
cellulare, sia implicato nella modulazione dei nuovi equilibri che si attuano in corso di
infezione da HCMV.
In particolare, è stato dimostrato che, in modelli in vitro, il blocco dell’espressione dei geni
precocissimi di HCMV, evidenziato nell’ambito della fase S del ciclo cellulare, può essere
rimosso mediante l’uso di inibitori dei proteasomi (Fortunato E. et al., 2002). In questo
contesto, l’ipotesi più accreditata sembrerebbe contemplare la presenza di una proteina
con un tempo di vita media molto breve, presente transientemente in fase S e necessaria al
virus per attivare l’espressione dei geni IE. La presenza di specifici inibitori dei

3. Introduzione
-48-
proteasomi impedirebbe a questi ultimi di rimuovere rapidamente la suddetta proteina
cellulare.
D’altra parte, è stato anche dimostrato che il complesso ubiquitina-proteasoma ha un
ruolo centrale nell’attivazione del fattore di trascrizione NF-kB, che è strettamente
implicato nella patogenesi di molte malattie infiammatorie e in neoplasie, inducendo
l’azione di numerose citochine. Il fattore NF-kB è stato anche indicato, almeno in modelli
cellulari in vitro, come il principale mediatore della stimolazione dell’attività dell’
“enhancer”- promotore dei geni precocissimi di HCMV, cruciale sia nell’ambito del ciclo
litico che per la riattivazione da una condizione di latenza, e potrebbe anche essere,
almeno parzialmente, responsabile dell’immunopatogenesi da HCMV in vivo. Studi
effettuati in presenza di inibitori della funzione proteasomale hanno messo in evidenza
un effetto inibitorio sull’evoluzione dell’infezione da HCMV, in particolare a livello
dell’espressione dei geni precocissimi virali, così come un accumulo citoplasmatico di NF-
kB e relativa deplezione nucleare del suddetto fattore, a cui potrebbe essere, almeno in
parte, imputabile il blocco dell’infezione virale, a causa della mancata attività stimolatoria
sulla trascrizione dei geni IE (Prösch S. et al., 2003).
È anche importante sottolineare altri aspetti rilevanti dell’infezione virale, legati a
specifiche localizzazioni di componenti di HCMV a livello di peculiari compartimenti
nucleari; come già accennato, la corretta collocazione spaziale di complessi molecolari ne
rende anche possibile l’interazione ed il conseguente espletamento della funzione
correlata. In quest’ottica, è interessante evidenziare che una parziale inibizione
nell’espressione del genoma di HCMV potrebbe anche derivare da uno scorretto
posizionamento nucleare. In particolare, è stato osservato che, a tempi precocissimi dopo
l’infezione, una certa quota di DNA del virus infettante si ritrova associata ai domìni
nucleari ND10, il cui numero varia nel corso del ciclo cellulare (ad esempio, diminuisce in
fase S) ed ai quali sono parzialmente associati anche i proteasomi (Anton L.C. et al., 1999;
Rockel T.D. and von Mikecz A., 2002).
A tale proposito e, come già ricordato, è stato messo in evidenza un ruolo di spicco di
questi ultimi complessi nel disassemblamento dei domìni ND10 da parte di una proteina
precocissima di Herpes simplex (Everett R.D. et al., 1998). Anche HCMV svolge un’azione
analoga sugli stessi domìni nucleari, anche se, in quest’ultimo caso, è ancora controversa
l’identificazione del complesso cellulare che medierebbe la disaggregazione dei suddetti
domìni (Nevels M. et al., 2004).

4. Obiettivi della ricerca
4. OBIETTIVI DELLA RICERCA

4. Obiettivi della ricerca
-50-
Il lavoro sperimentale svolto è imperniato sullo studio di complessi molecolari cellulari,
detti proteasomi, in una condizione fisiologica, quale il processo di differenziamento di
cellule muscolari murine in vivo ed in vitro, così come in una condizione patologica,
rappresentata dall’infezione produttiva in vitro di fibroblasti embrionali umani con uno
stipite di riferimento di citomegalovirus; tale studio prende in considerazione sia aspetti
morfologici che funzionali, legati ai suddetti complessi molecolari.
Per quel che riguarda gli aspetti morfologici, in particolare, lo studio effettuato si ispira, in
un certo senso, ad un ormai ricco filone di letteratura, che supporta la nozione secondo
cui esisterebbe un legame imprescindibile tra la funzione espletata da specifici complessi
molecolari e la loro localizzazione all’interno della cellula. In altre parole, la nozione
spaziale assumerebbe un’importanza analoga a quella della funzione stessa, non
espletabile al di fuori di quel preciso distretto.
Sulla base di tali presupposti, è plausibile ritenere che uno degli aspetti più innovativi di
questa ricerca è legato, in particolare, alla progettazione di strumenti molecolari idonei a
permettere l’espressione di costrutti di interesse (recanti le sequenze geniche per
specifiche subunità di proteasoma fuse a quelle per una proteina fluorescente), all’interno
di cellule bersaglio, idonee per lo studio dei due modelli proposti.
La ricerca in oggetto si articola in diverse fasi sperimentali, ognuna delle quali si prefigge
uno specifico obiettivo:
1. Studio della distribuzione di specifici proteasomi in sezioni sottili di muscoli striati
di ratto, così come in miofibrille isolate a partire dagli stessi muscoli, attraverso
l’applicazione di tecniche di immunoelettromicroscopia, microscopia a
fluorescenza e confocale, al fine di approfondire le conoscenze sulla localizzazione
dei suddetti complessi molecolari, individuando il/i possibile/i “partner”/s di
interazione morfologica muscolare, che possa/no verosimilmente rappresentare,
previe ulteriori verifiche, anche il/i più probabile/i candidato/i di interazione a
livello funzionale.
2. Allestimento di vettori di espressione non retrovirali o, alternativamente,
retrovirali, recanti le sequenze geniche per le subunità proteasomali di scelta,
accoppiate a quelle per proteine fluorescenti naturali, per effettuare,
rispettivamente, trasfezioni delle cellule muscolari di interesse, o infezioni di
particolari cellule di impacchettamento. Da queste ultime, è previsto di ottenere

4. Obiettivi della ricerca
-51-
retrovirus defettivi (ma ancora in grado di infettare i modelli allo studio) recanti
l’informazione genica di interesse.
3. Validazione dei suddetti strumenti molecolari, una volta espressi negli idonei
modelli cellulari. Le verifiche saranno effettuate a livello biochimico, al fine di
valutare, da un lato, l’avvenuta incorporazione delle proteine di fusione
nell’ambito dei complessi molecolari proteasomali e, dall’altro, la preservazione
dell’attività enzimatica di tipo proteolitico, da parte di proteasomi che includono i
costrutti in oggetto.
4. Studio dell’espressione di specifici proteasomi durante il processo di
differenziamento dei modelli muscolari prescelti (previa trasfezione/infezione con
i vettori di espressione citati al punto 2) ed analisi dei risultati mediante
microscopia a fluorescenza e/o microscopia confocale.
5. Studio del possibile ruolo di specifici proteasomi in corso di infezione produttiva
da citomegalovirus umano in fibroblasti embrionali umani (modello permissivo),
attraverso l’utilizzo di sostanze (quali MG132) che inibiscono la funzione
proteolitica dei suddetti complessi molecolari, mediante microscopia a
fluorescenza.
6. Studio sulla distribuzione cellulare di specifici proteasomi, con particolare
riguardo a quella nucleare, in corso di infezione produttiva da citomegalovirus
umano in fibroblasti embrionali umani, mediante microscopia confocale.
7. Allestimento di colture cellulari idonee per lo studio in vitro dell’infezione
produttiva da citomegalovirus, che esprimano stabilmente specifiche subunità di
proteasomi e EGFP, applicando tecniche di biologia molecolare analoghe a quelle
menzionate al punto 2 e primo livello di verifica dell’espressione cellulare dei
suddetti costrutti in assenza di infezione virale, mediante analisi in microscopia
confocale.
La messa a punto degli strumenti molecolari da utilizzare nei due modelli di studio
proposti potrà, a sua volta, costituire il punto di partenza per la realizzazione, in
prospettiva futura, di studi di cinetica in cellule viventi, ancora più adeguati a favorire la
comprensione di processi così complessi ed altamente dinamici, quali il differenziamento
muscolare e l’infezione virale.

5. Materiali e Metodi
5. MATERIALI E METODI

5. Materiali e Metodi
-53-
5.1 STUDIO DI LOCALIZZAZIONE INTRACELLULARE DEI PROTEASOMI
5.1.1 ESTRAZIONE DEI MUSCOLI
I muscoli utilizzati per gli studi di localizzazione intracellulare dei proteasomi sono stati
ottenuti da ratti maschi di due mesi e mezzo (razza Wistar; ditta Janvier - Le Genest-St
Isle, Francia). Dopo essere stati sottoposti ad anestesia per somministrazione
intraperitoneale di pentobarbital sodico (dosaggio: 1 ml kg-1 di peso corporeo; ditta
Sanofi), gli animali sono stati sacrificati mediante dislocazione cervicale. Prima di
procedere all’asportazione dei muscoli degli arti inferiori [Soleus (SOL), Extensor Digitorum
Longus (EDL) e Tibialis anteriore], gli animali sono stati lavati con sapone battericida e con
etanolo al 70%.
5.1.2 SEZIONI ISTOLOGICHE
Una volta rimossi, i muscoli sono stati congelati in isopentano freddo ottenuto mediante
l’impiego di azoto liquido e, successivamente, conservati a -80°C fino al momento
dell’uso. Mediante l’impiego di un criostato (Leica CM 3050) sono state preparate sezioni
longitudinali di 10 µm, che sono state fatte aderire su appositi vetrini “Superfrost-plus”
(Labonord). Le sezioni così ottenute sono state lasciate asciugare all’aria per circa 10
minuti e, poi, utilizzate per l’analisi in immunofluorescenza.
5.1.3 ISOLAMENTO DELLE MIOFIBRILLE
Le miofibrille sono state isolate rapidamente a partire da muscoli di ratto, seguendo il
protocollo descritto da Zhukarev V. e collaboratori (Zhukarev V. et al., 1997). In
particolare, i muscoli SOL, EDL e Tibialis anteriore sono stati legati, fissandone le
estremità, allo stantuffo di una siringa di plastica da 50 ml e sono stati immersi per una
notte a 4°C in una soluzione detta di “skinning”, atta a separare le varie componenti
tissutali, [2,5 mM ATP, 170 mM propionato di potassio, 5 mM acido etilen-glico-bis-(2-
aminoetilen) tetraacetico di potassio “K2-EGTA”, 2,5 mM MgCl2, 10 mM imidazolo a
pH=7,0]. Al termine dell’incubazione, il tessuto connettivo e i tendini sono stati rimossi e i
muscoli sono stati disposti in sottili strisce nella soluzione fredda di “skinning”. Le strisce
di muscolo sono state successivamente omogeneizzate in 10 volumi di tampone freddo
denominato “rigor” (10 mM TES, 3 mM MgCl2, 10 mM EGTA a pH=7,1), mediante
l’impiego dell’omogenizzatore Ultra-Turrax T25 a media velocità (3 o 4 impulsi, per 30
secondi ciascuno). La qualità della omogenizzazione è stata monitorata allestendo
preparati a fresco da osservare al microscopio ottico in contrasto di fase. La sospensione,

5. Materiali e Metodi
-54-
ricca di miofibrille, è stata successivamente sottoposta a centrifugazione a bassa velocità
(1.500 x g) per 5 minuti a 4°C ed il sedimento ottenuto è stato lavato per quattro volte con
il tampone “rigor”. Le miofibrille così ottenute sono state raccolte e risospese in tampone
“rigor” addizionato di glicerolo (50%) e poi conservate a -80°C, fino al momento dell’uso.
Per l’allestimento dei preparati microscopici, una goccia della sospensione ottenuta è stata
depositata su vetrini “Superfrost-plus”. I vetrini sono stati quindi incubati a temperatura
ambiente per 10 minuti, per permettere alle miofibrille di aderire alla loro superficie.
5.1.4 ANTICORPI
Per lo studio di localizzazione del proteasoma 20S mediante immunofluorescenza, è stato
impiegato un anticorpo monoclonale non coniugato con fluorocromo in grado di
riconoscere la subunità α1/p27k del proteasoma 20S o, alternativamente, un anticorpo
anti-α1/p27k coniugato con il fluorocromo Alexa-Red (Molecular Probes). Entrambi gli
anticorpi sono stati gentilmente forniti dal Dr. Klaus Scherrer (Institut Jacques Monod-
Parigi, Francia). Entrambi gli anticorpi sono stati impiegati alla diluizione di lavoro di
1:20. Per lo studio della struttura sarcomerica sono stati impiegati un anticorpo
monoclonale anti-titina (clone 9D10) ed un anticorpo anti-miosina (clone MF20),
gentilmente forniti da “Developmental Studies Hybridoma Bank” (University of Iowa,
Stati Uniti); tali anticorpi sono stati impiegati alla diluizione di lavoro di 1:50 e 1:20,
rispettivamente. Inoltre, è stato utilizzato un anticorpo policlonale anti-desmina (Sigma-
Aldrich), alla diluzione di 1:10.
Per la rivelazione dei recettori del reticolo sarcoplasmatico sono stati impiegati anticorpi
monoclonali anti-Serca-1 e anti-Rianodina (ditta ABR), alla diluizione di 1:100. Per la
rivelazione degli anticorpi primari monoclonali sono stati utilizzati anticorpi anti-IgG o
anti-IgM di topo, coniugati con isotiocianato di tetrametilrodamina (TRITC) oppure con
isotiocianato di fluorosceina (FITC) ed utilizzati alla diluizione di 1:50. Per quanto
concerne, invece, gli anticorpi primari policlonali, sono stati utilizzati anticorpi anti-IgG di
coniglio, coniugati con TRITC o FITC (Jackson Immunoresearch), alla diluizione di 1:50.
Per la rivelazione dell’actina in forma filamentosa è stata impiegata falloidina,
direttamente coniugata con TRITC o, alternativamente, con FITC, alla diluizione di 1:100-
1:200.
Tutti gli anticorpi utilizzati, così come falloidina, sono stati diluiti in una soluzione di
siero albumina bovina (BSA) all’1% in tampone salino fosfato (PBS costituito da 137 mM

5. Materiali e Metodi
-55-
NaCl, 2,7 mM KCl, 10 mM NaHPO4, 2 mM KH2PO4, H2O distillata) e Triton-X 100 allo
0,2% in PBS.
5.1.5 IMMUNOFLUORESCENZA INDIRETTA
I preparati microscopici sono stati fissati utilizzando differenti tipi di fissatori (metanolo,
acetone, paraformaldeide al 4% in PBS) o, alternativamente, non sono stati sottoposti a
fissazione. Dopo tre lavaggi di 5 minuti ciascuno in PBS, i siti immunoreattivi non
specifici sono stati saturati con BSA al 6% in PBS per 20 minuti; le cellule sono state
successivamente lavate in PBS, prima di aggiungere l’anticorpo primario.
Per l’immunoreazione, i preparati sono stati incubati in camera umida, con i differenti
anticorpi primari, per 1 ora a temperatura ambiente, oppure per una notte a 4°C. Dopo
cinque lavaggi di 5 minuti ciascuno in PBS, i preparati sono stati incubati con gli anticorpi
secondari specifici e/o con falloidina per 1 ora a temperatura ambiente, sempre in camera
umida. I preparati sono stati poi lavati in PBS (cinque lavaggi da 5 minuti ciascuno) e
montati con 20 µl di liquido di montaggio Mowiol (Calbiochem).
5.1.6 OSSERVAZIONE E MANIPOLAZIONE DELLE IMMAGINI
I preparati sono stati osservati mediante impiego del microscopio Olympus BH-2 a
epifluorescenza, utilizzando un’appropriata combinazione di filtri. Le immagini sono
state acquisite mediante telecamera CMOS-Pro in scala di grigi (Sound Vision, USA) e i
montaggi a colori sono stati ottenuti modificando le immagini acquisite con il
corrispondente canale di “RGB” del programma “Adobe Photoshop Document”. Qualora
le immagini siano state acquisite in contrasto di fase (in particolare come indicatori della
linea Z e della banda A), esse venivano in seguito convertite nel colore blu, in modo tale
da evidenziare le zone scure delle stesse, con particolare riferimento alla linea Z e alla
banda A.
Alcune immagini sono state deconvolute usando il programma “Huygens Essential”
(Scientific Volume Imaging) per Macintosh.
Una parte delle osservazioni di preparati di miofibrille è stata effettuata utilizzando il
microscopio confocale Leica TCS-SP2. Le immagini sono state acquisite in scala di grigi
mediante il programma LCS e successivamente sono state sottoposte allo stesso tipo di
manipolazioni sopra descritto. Le misure dei sarcomeri sono state effettuate direttamente
sullo schermo, dopo che l’immagine relativa alla zona selezionata era stata importata nel
programma “Adobe Illustrator”, impiegando appositi sistemi di misura. La posizione

5. Materiali e Metodi
-56-
delle bande fluorescenti è stata valutata sulla base della localizzazione del segnale più
intenso. Tutte le immagini sono state osservate applicando le stesse condizioni di
calibrazione degli strumenti impiegati. Un riquadro di un micron, in cui un pixel
corrispondeva a 83 nm, è stato usato per calibrare il sistema.
5.1.7 IMMUNOELETTROMICROSCOPIA (METODO DI “PRE-EMBEDDING”)
La reazione di immunoelettromicroscopia è stata effettuata su sezioni sottili congelate e su
preparati di miofibrille non ancora sottoposte a fissazione e a inclusione (metodo di “pre-
embedding”). In particolare, l’anticorpo primario anti-α1/p27k è stato utilizzato su
sezioni sottili congelate e successivamente rivelato mediante un anticorpo secondario
coniugato con perossidasi di rafano (Jackson Immunoreserach). Come substrato
dell’enzima perossidasi è stato impiegato il composto 3,3-diamminobenzidina (DAB)
(Sigma-Aldrich) che, inizialmente incolore, dà poi luogo alla formazione di un prodotto
finale di colore marrone che risulta insolubile in presenza di alcool qualora sia utilizzato
dall’enzima. Successivamente, i preparati sono stati fissati in glutaraldeide al 2% in PBS,
post-fissati con tetrossido di osmio all’1% in acqua distillata e, infine, disidratati
utilizzando concentrazioni crescenti di etanolo. Le sezioni sono state poi incluse in resine
epossidiche (Epon 812). Le sezioni ultrafini sono state osservate mediante microscopio
elettronico a trasmissione (Joel 100 CX II) senza impiego di un mezzo di contrasto o, in
alternativa, con contrasto, ottenuto colorando i preparati con acetato di uranile. Per
quanto concerne i preparati microscopici di miofibrille, anche in questo caso è stato
utilizzato l’anticorpo monoclonale diretto nei confronti della subunità α1/p27k. Tale
anticorpo è stato rivelato utilizzando un anticorpo secondario coniugato con FITC che, a
sua volta, è stato rivelato mediante un anticorpo secondario anti-FITC coniugato con
particelle di oro colloidale (Ultrasmall kit, Aurion). I preparati sono stati sottoposti a
trattamento di intensificazione mediante l’impiego di argento, per circa 20 minuti,
secondo le indicazioni fornite dalla ditta produttrice (Ultrasmall kit, Aurion). Dopo
fissazione in glutaraldeide e post-fissazione con tretrossido di osmio, i campioni sono stati
inclusi in Epon e, infine, esaminati secondo le modalità impiegate per le sezioni di tessuto
congelate.
5.1.8 METODO DI ESTRAZIONE DELL’ACTINA
Al fine di rimuovere selettivamente l’actina dalle miofibrille, è stato impiegato il
composto gelsolina (Sigma-Aldrich) (Funatsu T. et al., 1990; Funatsu T. et al., 1993). Le

5. Materiali e Metodi
-57-
miofibrille adese al vetrino portaoggetti sono state lavate per due volte per 5 minuti
ciascuna con la soluzione fredda “Ca-rigor” (120 mM KCl, 5 mM MgCl2, 0,1 mM CaCl2, 20
mM sale monosodico dell’acido piperazin-1,4-dietansolfonico o “Pipes” a pH=7,0).
Successivamente, è stata aggiunta una soluzione di estrazione (0,2 mg/ml di gelsolina
nella soluzione “Ca-rigor”) per 30-60 minuti in ghiaccio. Le miofibrille sono state lavate
per tre volte con soluzione “Ca-rigor” e successivamente fissate per 20 minuti con
paraformaldeide al 4% in PBS. Una volta terminata la fase di fissazione, le miofibrille sono
state lavate abbondantemente in PBS e processate per la fase di immunofluorescenza
indiretta.
5.2 CLONAGGIO IN VETTORI PLASMIDICI
5.2.1 REAZIONE POLIMERASICA A CATENA PREVIA RETROTRASCRIZIONE (RT-PCR)
Sintesi di cDNA
Le sequenze geniche codificanti per le subunità del proteasoma 20S, α1/p27k e β4/p23k,
sono state ottenute utilizzando il metodo di reazione polimerasica a catena previa
retrotrascrizione (RT-PCR). In particolare, corti oligonucleotidi costituiti da 18
desossitimidine (oligo-dT) sono stati mescolati ad un preparato commerciale di RNA
totale, ottenuto a partire da placenta umana. Gli oligo-dT sono stati utilizzati sia per la
selezione degli mRNA provvisti di code di poli(A) all’estremità 3’, che come sequenze
innesco per consentire all’enzima ricombinante trascrittasi inversa, derivante dal
retrovirus della leucemia murina di Moloney (“Moloney Murine Leukemia Virus” o
“MoMLV”), di sintetizzare cDNA a singolo filamento. Alla miscela di reazione è stato
inoltre aggiunto un inibitore dell’enzima RNAsi. Per la sintesi dei cDNA è stato impiegato
il saggio commerciale “Advantage RT-for-PCR” (Clontech).
Reagenti Volume (µl)
RNA (1 µg/µl) 1
Oligo (dT)18 (20 µM) 1
Acqua distillata senza RNAsi 11,5
La miscela è stata incubata a 72°C per 2 minuti e successivamente conservata in ghiaccio
per almeno 1 minuto. Alla medesima miscela sono stati poi aggiunti i reagenti di seguito
elencati :

5. Materiali e Metodi
-58-
Reagenti Volume (µl)
* Tampone RT 5x 4
dNTP mix (10 mM ciascuno) 1
Inibitore RNase (40 unità/µl) 0,5
Trascrittasi inversa MoMLV (200 unità/µl) 1
* Tampone RT 5x: 250 mM Tris-HCl (pH= 8,3), 375 mM KCl, 15 mM MgCl2
La miscela così ottenuta è stata successivamente incubata a 42°C per 1 ora e la reazione è
stata bloccata mediante riscaldamento a 94°C per 5 minuti. Infine, il preparato è stato
incubato in ghiaccio e diluito con 80 µl di acqua distillata senza RNasi. I frammenti di
cDNA ottenuti sono stati conservati a –80°C fino al momento dell’uso.
Amplificazione del cDNA
I filamenti di cDNA di interesse [α1/p27K (gene PSMA6) e β4/p23K (gene PSMB2)] sono
stati amplificati mediante reazione polimerasica a catena (PCR), impiegando due specifici
oligonucleotidi, di seguito riportati.
PSMA6
senso 5’ ATAAGCTTCCAACATGTCCCGTGGTTCCAGCGC 3’
antisenso 3’ GCGAATTCCGTCTCTCTCTGCTAGAGCAACAAGGT 3’
PSMB2
senso 5’ATAAGCTTCCACCATGGAGTACCTCATCGGTATCCAAGGCCC 3’
antisenso 5’GCGAATTCCGGAGCCCTGTTTGGGGAAGGAAAT 3’
Per la reazione di PCR è stato utilizzato il saggio commerciale “AmpliTaq” (Clontech); di
seguito è riportata la composizione della miscela di reazione impiegata.

5. Materiali e Metodi
-59-
Reagenti Volume (µl)
*Tampone PCR 10x 5
Mix dNTP (10 mM) 1
Sequenza innesco “senso” (10 µM) 1
Sequenza innesco “antisenso” (10 µM) 1
Taq DNA polimerasi (5 unità/µl) 0,4
cDNA 10
Acqua distillata quanto basta (vol. reaz. 50 µl)
* Tampone PCR 10x: 100 mM (pH=8,3), 150 mM KCl, 6 mM MgCl2
L’amplificazione mediante PCR è stata condotta previo utilizzo di un termociclatore
automatico (PerkinElmer), secondo il seguente protocollo: un ciclo di denaturazione
iniziale a 94°C per 2 minuti, 25 cicli di PCR (94°C per 45 secondi, 60°C per 45 secondi e
72°C per 2 minuti) e un ciclo di estensione finale di 7 minuti a 72°C.
5.2.2 CLONAGGIO IN UN VETTORE PLASMIDICO MEDIANTE L’USO DI ENZIMI DI RESTRIZIONE
I frammenti di cDNA a doppio filamento ottenuti nella fase precedente (760 nucleotidi per
il gene PSMA6 e 625 nucleotidi per il gene PSMB2) sono stati inseriti in due vettori
plasmidici commerciali denominati pEGFP-N1 e pDsRed1-N1 (Clontech), in modo che le
sequenze geniche di interesse, introdotte a livello del sito multiplo di clonaggio o
“polylinker” (MCS), fossero espresse come proteine di fusione con la sequenza proteica
per EGFP/DsRed1 all’estremità N-terminale del plasmide. A tal fine, le molecole di cDNA
a doppia elica sono state digerite con gli enzimi di restrizione EcoR I e Hind III, diluiti nel
tampone specifico “NEB 2” [10 mM Tris-HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM
ditiotreitolo (DTT) a pH= 7,9 a 25°C; ditta New England BioLabs], in ragione di 10 unità
per l’enzima Hind III e 20 unità per l’enzima EcoR I. La miscela di reazione è stata
incubata in bagnomaria a 37°C per circa 2 ore. Al termine dell’incubazione, i prodotti sono
stati conservati a -20°C fino al momento dell’uso.
5.2.3 ELETTROFORESI SU GEL DI AGAROSIO
Al fine di verificare la purezza del DNA estratto ed il grado di completezza raggiunto
dalla digestione enzimatica del DNA, nonché separare i frammenti di interesse, sono stati
preparati gel di agarosio a diversa concentrazione, in base alla risoluzione desiderata.

5. Materiali e Metodi
-60-
L’agarosio è stato sciolto alla temperatura di ebollizione in tampone TBE 1x (45 mM Tris-
Borato, 1 mM acido etilen-diammino-tetraacetico o “EDTA”) o, alternativamente in
tampone TAE 1x (40 mM Tris-Acetato e 1 mM EDTA) (Invitrogen). Il gel è stato colato e
lasciato raffreddare nel supporto di un apparato orizzontale da elettroforesi. Una volta
solidificato, il gel è stato ricoperto con tampone di corsa TBE 1x , oppure TAE 1x.
I prodotti della digestione sono stati diluiti in apposito tampone “loading buffer” 10x,
contenente 0,1% sodio-dodecilsolfato (SDS), 50 mM EDTA, 50% glicerolo, 0,25% cianolo di
xilene FF, 0,25% blu di bromofenolo. I campioni sono stati caricati sul gel ed è stata
applicata una differenza di potenziale di 60/120 Volts per un tempo variabile, in funzione
delle dimensioni del frammento di DNA e della concentrazione del gel. Alla fine della
corsa elettroforetica le bande di DNA sono state visualizzate mediante l’aggiunta di
bromuro di etidio (0,5 µg/ml) ( Sigma-Aldrich) che, intercalandosi tra le basi del DNA, ne
permette la visione ai raggi UV (320 nm). Il profilo di migrazione dei diversi frammenti di
DNA e la loro lunghezza sono stati analizzati per confronto con un adatto sistema di
riferimento a peso molecolare noto “SmartLadder” (Eurogentec).
5.2.4 ESTRAZIONE DEL DNA DA GEL DI AGAROSIO
I frammenti di interesse e il DNA plasmidico ottenuti con la digestione enzimatica sono
stati purificati mediante estrazione da banda, dopo corsa elettroforetica su gel di agarosio.
In particolare, per l’estrazione del DNA da gel di agarosio è stato utilizzato il saggio
commerciale “Nucleo Trap” (Clontech). Questo saggio consente di dissolvere le bande di
agarosio contenenti il frammento di interesse in una soluzione di ioduro di sodio a forte
concentrazione salina, permettendo la fissazione del DNA su biglie di vetro. Le biglie
vengono successivamente centrifugate ed il DNA legato è eluito da esse utilizzando
tamponi a bassa forza ionica, secondo le indicazioni fornite dalla ditta produttrice. In
particolare, i campioni sono stati risospesi in 50 µl di tampone TE (10 mM Tris-HCl a
pH=8,2, 1 mM EDTA), al fine di eluire il DNA dalle biglie. I campioni sono stati quindi
incubati a temperatura ambiente per 10 minuti e mescolati brevemente ogni 2 minuti.
Infine, sono stati centrifugati a 4°C per 10 minuti a massima velocità e il surnatante è stato
recuperato e conservato in frigorifero a 4°C.

5. Materiali e Metodi
-61-
5.2.5 VALUTAZIONE DELLA CONCENTRAZIONE DI DNA PLASMIDICO E DEI FRAMMENTI
I frammenti di interesse e il DNA plasmidico sono stati fatti migrare su gel di agarosio
all’1% in TBE 1x e precolorati con bromuro di etidio. L’analisi quantitativa del contenuto
di DNA delle bande è stata effettuata mediante il programma “Gene Tool Syngene”.
5.2.6 LIGAZIONE
Una volta amplificati, purificati e quantificati, i frammenti di cDNA di interesse (PSMA6 e
PSMB2) sono stati utilizzati in reazioni di ligazione che ne hanno permesso il clonaggio
nei vettori plasmidici pEGFP-N1 e pDsRed1-N1. A tale scopo, è stato impiegato l’enzima
DNA-ligasi contenuto nel saggio commerciale “Fast-Link DNA Ligation Kit”(Epicentre).
Al fine di ottenere estremità coesive compatibili, gli amplificati e i plasmidi commerciali
sopra citati sono stati sottoposti a digestione enzimatica, secondo le modalità descritte nel
paragrafo 5.2.2. Al termine della digestione, i prodotti di reazione sono stati utilizzati per
la reazione di ligazione, che è stata eseguita secondo le indicazioni fornite dalla ditta
fornitrice.
5.2.7 TRASFORMAZIONE
I plasmidi ottenuti (pPSMA6-EGFP, pPSMA6-DsRed1, pPSMB2-EGFP e pPSMB2-DsRed1)
sono stati introdotti, mediante un procedimento di trasformazione, in cellule batteriche
modificate di Escherichia coli, contenute nel saggio commerciale “XL1-Blue competent
cells” (Stratagene). In particolare, i batteri competenti conservati a –80°C sono stati
scongelati, avendo cura di mantenerli in ghiaccio; successivamente sono state allestite,
utilizzando appositi tubi di polipropilene “Falcon 2059” (Becton-Dickinson), aliquote, da
50 µl ciascuna, di batteri competenti per ognuno dei quattro plasmidi ricombinanti
ottenuti per ligazione. A ciascuna aliquota sono stati aggiunti 0,85 µl di 2-mercaptoetanolo
1,42 M (concentrazione finale: 25 mM). Le aliquote sono state quindi incubate in ghiaccio
per 10 minuti, mescolandole delicatamente ogni 2 minuti. Infine, ad ogni aliquota, sono
stati aggiunti 8 µl di ciascuna miscela di ligazione. Le cellule sono state incubate in
ghiaccio per 30 minuti. Nel frattempo, il terreno “SOC” è stato preriscaldato a 42°C (si
veda di seguito).

5. Materiali e Metodi
-62-
Terreno SOC Volume (ml)
* Terreno SOB 100
Glucosio (2 M) 1
oppure:
Glucosio (20%) 2
Il terreno è stato filtrato con filtri di porosità pari a 0.22 µm e di volume di 500 ml (Nalge Nunc
International).
Dopo sterilizzazione in autoclave, sono stati aggiunti 10 ml di 1 M MgCl2 e 10 ml di 1 M MgSO4. Il
terreno è stato successivamente filtrato mediante filtri di porosità pari a 0.22 µm e di volume di 500
ml (Nalge Nunc International).
Al termine del periodo di incubazione, le cellule batteriche sono state sottoposte a shock
termico per 45 secondi a 42°C, in modo da alterare la permeabilità della membrana
plasmatica e favorire l’ingresso del DNA trasformante. I batteri sono stati quindi
rapidamente trasferiti in ghiaccio per 2 minuti. A questo punto, ciascuna miscela è stata
posta in agitazione per 1 ora a 37°C, previa aggiunta di 1 ml di terreno “SOC”.
Utilizzando delle biglie di vetro, la sospensione batterica è stata quindi uniformemente
distribuita, in quantità diverse (300 µl, 100 µl e 50 µl), sulla superficie di piastre con
terreno di coltura agarizzato di Luria–Bertani (LB), contenente kanamicina (25 µg/ml)
(Sigma–Aldrich). L’antibiotico di selezione kanamicina consente la crescita dei batteri che
hanno acquisito i plasmidi pEGFP-N1, pDsRed1-N1 e i plasmidi in cui sono state clonate
le sequenze codificanti per le proteine di fusione, dal momento che contengono il gene che
codifica per il fattore di resistenza nei confronti del suddetto antibiotico.
*Terreno SOB Quantità (gr)
Triptone 20
Estratto di lievito 5
NaCl 0,5
Acqua distillata portare a volume finale: 1 litro

5. Materiali e Metodi
-63-
Terreno “Luria-Bertani” (LB) Quantità (gr)
BactoTriptone 10
Estratto di lievito 5
NaCl 10
Bacto agar 20
Acqua distillata Portare a volume finale: 1 litro
Il pH è stato portato a 7,0 con 5 N NaOH. Il terreno è stato successivamente sottoposto a
sterilizzazione in autoclave.
5.2.8 ESTRAZIONE DI DNA PLASMIDICO (“MINI-PREP”)
Prima dell’estrazione del DNA, le colonie batteriche selezionate sono state fatte crescere
per una notte a 37°C, in costante agitazione in 3 ml di terreno liquido LB, addizionato con
50 µg/ml di kanamicina, al fine di favorire la crescita aerobia dei batteri. A partire da
queste sospensioni batteriche è stato possibile preparare delle piccole quantità di DNA
plasmidico (“mini-prep”) ad un livello intermedio di purezza utilizzando il saggio
commerciale “NucleoBond” (Machery-Nagel). A questo scopo, 1,5 ml di coltura batterica
sono stati sedimentati mediante centrifugazione per 30 secondi a 4°C, a velocità massima.
Una volta eliminato il surnatante, il sedimento è stato risospeso in 100 µl di tampone S1
(50 mM Tris-HCl, 10 mM EDTA a pH=8). Per la lisi della parete batterica sono stati
aggiunti 100 µl di tampone S2 (200 mM NaOH, 1% SDS) al sedimento; la sospensione è
stata successivamente mescolata delicatamente per inversione della provetta per un paio
di volte. Dopo incubazione per 5 minuti a temperatura ambiente, sono stati aggiunti 100
µl di tampone S3 (2,60 M acetato di potassio a pH=5,2) e ancora una volta, i campioni sono
stati mescolati per inversione. I campioni sono stati quindi incubati in ghiaccio per 5
minuti. Il lisato batterico è stato centrifugato per 10 minuti a 10.000 x g a 4°C e il
surnatante, contenente il DNA plasmidico, è stato trasferito in una nuova provetta. Il
DNA plasmidico è stato precipitato mediante l’aggiunta di 700 µl di etanolo assoluto. I
campioni sono stati lasciati a temperatura ambiente per 10 minuti e successivamente
centrifugati per 10 minuti a 10.000 x g a 4°C. Dopo un lavaggio con 300 µl di etanolo
freddo al 70% e un’ultima centrifugazione a 10.000 x g per 5 minuti, il surnatante è stato
aspirato e il sedimento, una volta asciugato a temperatura ambiente, è stato risospeso in
100 µl di tampone TE. Gli estratti sono stati conservati in frigorifero a 4°C fino al
momento dell’uso. Poiché non è possibile discriminare le colonie che hanno assunto il
plasmide originale da quelle che lo hanno acquisito ricombinante, dopo l’estrazione del

5. Materiali e Metodi
-64-
DNA, 1 µl di ciascuna “mini-prep” è stato nuovamente sottoposto a digestione enzimatica
con gli enzimi di restrizione EcoR I e Hind III e quindi ciascun campione è stato analizzato
mediante elettroforesi (gel di agarosio 1% in tampone TBE 1x).
5.2.9 ESTRAZIONE DI DNA PLASMIDICO (“MAXI-PREP”)
Una volta verificata la bontà dei plasmidi sulla base dei profili di restrizione, a partire
dalla “minicoltura” batterica di 3 ml è stata allestita una coltura in terreno liquido LB (300
ml) addizionata con 50 µg/ml di kanamicina, al fine di ottenere un quantitativo maggiore
di DNA plasmidico (“maxi-prep”) ad elevata purezza, mediante il saggio commerciale
“NucleoBond Ax 2000” (Machery-Nagel). Questo metodo prevede l’estrazione dell’acido
nucleico per lisi alcalina e la sua successiva purificazione mediante l’uso di colonne
cromatografiche a scambio ionico. Ciascuna coltura batterica è stata successivamente
centrifugata per 15 minuti alla velocità di 5000 x g, allo scopo di ottenere un sedimento di
cellule batteriche. Il sedimento è stato risospeso in 35 ml di tampone S1 (50 mM Tris-HCl,
10 mM EDTA a pH=8,0) addizionato di 100 µg/ml di RNasi A. Alla suddetta sospensione
sono stati aggiunti 35 ml di tampone S2 (200 mM NaOH, 1% SDS) e la miscela è stata
delicatamente mescolata per inversione. La reazione di lisi alcalina è stata lasciata
procedere a temperatura ambiente per una durata non superiore a 5 minuti, trascorsi i
quali sono stati aggiunti 35 ml di tampone S3 (2,60 M di acetato di potassio a pH=5,2), in
grado di far precipitare le membrane e le pareti delle cellule lisate insieme al DNA
genomico e all’RNA ad alto peso molecolare, ad esse associati. Inoltre, il tampone S3 crea
le condizioni appropriate affinché il DNA plasmidico si leghi alla membrana della
colonna cromatografica. I lisati sono stati incubati in ghiaccio per 5 minuti e
successivamente sono stati sottoposti a filtrazione, in modo da allontanare il precipitato
bianco che si forma con l’aggiunta del tampone S3. È importante sottolineare che, prima
del loro utilizzo, le colonne cromatografiche devono essere equilibrate mediante
l’aggiunta di 20 ml di tampone N2 (100 mM Tris, 15% etanolo, 900 mM KCl portato a pH=
6,3 con H3PO4), permettendone lo svuotamento per gravità. I suddetti lisati sono stati
caricati sulle colonne cromatografiche equilibrate per favorire il legame tra DNA e resina,
che risulta localizzata al centro delle colonne. Infine, le colonne sono state lavate per due
volte con 35 ml di tampone N3 (100 mM Tris, 15% etanolo, 1.150 mM KCl a pH= 6,3) in
modo da liberare il DNA dai sali. Il DNA plasmidico è stato eluito dalla colonna mediante
l’aggiunta di 25 ml del tampone di eluizione N5 (100 mM Tris, 15% etanolo, 1 M KCl a

5. Materiali e Metodi
-65-
pH= 6,3). Il DNA eluito è stato precipitato mediante l’aggiunta di 0,7-0,8 volumi di
isopropanolo (corrispondenti a 19 ml), pre-equilibrato a temperatura ambiente e
successivamente centrifugato per 40 minuti a 15.000 x g a 4°C (rotore JA20 Beckman). Una
volta asciugato, il sedimento è stato risospeso in 1 ml di tampone TE. Il DNA plasmidico è
stato nuovamente precipitato mediante l’aggiunta di sodio acetato 0,3 M a pH=5,2 e di 2
volumi di etanolo assoluto. Le provette sono state invertite alcune volte, in modo da
apprezzare la formazione di un addensamento gelatinoso riconducibile al DNA
plasmidico. Infine, i campioni sono stati centrifugati per 20 minuti ad alta velocità (>
15.000 x g) a 4°C e il surnatante è stato rimosso con cura, prestando attenzione a non
perdere il sedimento adeso alle pareti delle provette. Il sedimento è stato lavato
delicatamente con etanolo freddo al 70% e, qualora necessario, è stato nuovamente
centrifugato ad alta velocità per 5 minuti. Una volta rimosso l’etanolo, ogni sedimento è
stato lasciato asciugare a temperatura ambiente. Infine, i preparati di DNA plasmidico
sono stati risospesi in 500 µl di tampone TE e conservati in frigorifero a 4°C fino al
momento dell’uso.
La concentrazione del DNA è stata calcolata a partire da una soluzione diluita 1:100 in
tampone TE, sulla base dei valori di assorbanza rilevati alla lunghezza d’onda di 260 nm
(1 unità di assorbanza a 260 nm per DNA a doppio filamento corrisponde a 50 µg/ml di
DNA).
5.2.10 DETERMINAZIONE DELLA SEQUENZA DI DNA
Al fine di valutare la corretta inserzione dei frammenti al termine di ogni clonaggio, è
stata determinata la sequenza del vettore plasmidico. In particolare, una aliquota dei
plasmidi pPSMA6-EGFP e pPSMB2-EGFP è stata inviata presso la società “Genome
Express“ con sede in Francia, ai fini del loro sequenziamento. Le reazioni di
sequenziamento sono state effettuate per amplificazione mediante PCR e analizzate su
sequenziatore a gel capillare.

5. Materiali e Metodi
-66-
5.3 COLTURE CELLULARI
Nelle diverse fasi sperimentali della ricerca sono state utilizzate le seguenti colture
cellulari :
CHO ( “Chinese Hamster Ovary”): cellule di ovaio di criceto cinese.
Le cellule CHO (Puck T.T. et al., 1958) sono state coltivate a 37°C in atmosfera di ossigeno
e CO2 al 5%, utilizzando, quale terreno di coltura, “Dulbecco’s modified Eagle’s medium”
(D-MEM) contenente 1.000 mg/l di glucosio e addizionato con il 10% di siero di vitello
fetale (SVF) ed antibiotici (streptomicina 10.000 µg/ml e penicillina 10.000 unità/ml).
C2.7: cellule muscolari scheletriche di topo.
Le cellule C2.7 (Pinset C. et al., 1988) sono state coltivate a 37% in atmosfera di ossigeno e
CO2 al 10% in terreno D-MEM contenente 1.000 mg/l di glucosio e addizionato con la
medesima soluzione di antibiotici sopra menzionata ma supplementato con il 20% di SVF.
Il differenziamento della linea cellulare C2.7 è stato indotto sperimentalmente,
modificando le condizioni di mantenimento in coltura: a tale riguardo è stato aggiunto al
terreno di coltura siero di cavallo al 4% e, in alternativa al siero di vitello fetale, lo 0,8% di
un composto sintetico denominato “Ultroser “ (Ciphergen- BioSepra S.A.).
Balb/3T3 clone A31: fibroblasti embrionali di topo.
Le cellule Balb/3T3 (Aaronson S.A. and Todaro G.J., 1968) sono state coltivate a 37°C in
atmosfera di ossigeno e CO2 al 5% utilizzando terreno D-MEM con 4.500 mg/l di glucosio,
addizionato di siero di vitello neonato (10%) e antibiotici (streptomicina 10.000 µg/ml e
penicillina 10.000 unità/ml).
Bosc-23: cellule embrionali di rene umano.
Le cellule Bosc-23 (Pear W.S. et al., 1993) sono state coltivate a 37°C in presenza di CO2 al
5% in terreno D-MEM con 4.500 mg/l di glucosio, addizionato di SVF (10%) ed antibiotici
(streptomicina 10.000 µg/ml e penicillina 10.000 unità/ml).
HEK 293: cellule embrionali di rene umano trasformate con adenovirus di tipo 5.
La linea cellulare HEK 293 (Graham F.L. et al., 1977) è stata coltivata a 37°C in atmosfera
di ossigeno e CO2 al 5%, utilizzando terreno D-MEM con 4.500 mg/l di glucosio,

5. Materiali e Metodi
-67-
addizionato di siero di vitello fetale (10%), sodio piruvato (1 mM), L-glutamina (2 mM) ed
antibiotici (streptomicina 10.000 µg/ml e penicillina 10.000 unità/ml).
MRC5: fibroblasti di polmone embrionale umano.
I fibroblasti MRC5 (Jacobs J.P. et al., 1970) sono stati coltivati in terreno di coltura
“Minimum Essential Medium” (MEM, Invitrogen), modificato con sali di “Earle” ed
addizionato di siero fetale di vitello (10%), sodio piruvato (1%), amminoacidi non
essenziali (1%), L-glutamina (1%) ed antibiotici (penicillina 10.000 unità/ml e
streptomicina 10.000 µg/ml).
MRC5-hTERT: fibroblasti di polmone embrionale umano immortalizzati.
Le cellule MRC5-hTERT, gentilmente fornite dal Dr. Gavin Wilkinson (Section of Infection
and Immunity – University of Wales - College of Medicine – Cardiff, UK) sono state
ottenute per integrazione nel loro genoma del gene che codifica per l’enzima trascrittasi
inversa della telomerasi umana “h-TERT” e sono considerate permissive all’infezione da
citomegalovirus umano (stipite AD169) (McSherry B.P. et al., 2001). Le MRC5-hTERT sono
state coltivate a 37°C in presenza di CO2 al 5%, utilizzando D-MEM con 4500 mg/l di
glucosio, addizionato di siero di vitello fetale (10%), sodio piruvato (1 mM), L-glutamina
(2 mM), amminoacidi non essenziali (1%) ed antibiotici (streptomicina 10.000 µg/ml e
penicillina 10.000 unità/ml).
5.4 IMPIEGO DEI VETTORI PLASMIDICI
5.4.1 TRASFEZIONE DELLE CELLULE CHO E C2.7
I plasmidi pPSMA6-EGFP, pPSMA6-DsRed1, pPSMB2-EGFP, pPSMB2-DsRed1, pEGFP-
N1 e pDsRed1-N1 sono stati impiegati per trasfettare due distinte linee cellulari: le cellule
CHO e le cellule della linea miogenica di topo C2.7.
Il giorno precedente la trasfezione, sono state allestite colture cellulari di CHO e C2.7 in
piastre Petri in polistirene (60 mm di diametro), in ragione di due piastre per ogni linea
cellulare e per ciascuno dei seguenti plasmidi: pPSMA6-EGFP, pPSMA6-DsRed1,
pPSMB2-EGFP, pPSMB2-DsRed1, pEGFP-N1 e pDsRed1-N1 (12 piastre Petri per la linea
cellulare C2.7 e 12 per le cellule CHO). In particolare, sono state allestite colture cellulari
con una densità pari a 7x105 cellule/piastra per cellule CHO e 9x104 cellule/piastra per
cellule C2.7. Per la trasfezione sono state utilizzati monostrati semi-confluenti (25%),

5. Materiali e Metodi
-68-
secondo le indicazioni fornite dal protocollo del saggio commerciale “CalPhos
Mammalian Transfection kit” (Clontech). Per gli esperimenti di trasfezione sono stati
impiegati 20 µg di DNA per ciascun plasmide; in particolare, la soluzione, costituita da
DNA e da CaCl2 (concentrazione finale: 2 M), è stata mescolata con il reagente HBS 2x
(soluzione salina tamponata con HEPES) e quindi incubata per 30 minuti a temperatura
ambiente. In seguito, la soluzione contenente DNA è stata distribuita uniformemente
(goccia a goccia) sul monostrato cellulare.
Dopo 48 ore di incubazione (espressione transitoria) sono stati preparati estratti di proteine
totali da ciascuna linea cellulare e per ogni plasmide utilizzato. Il tappeto cellulare è stato
lavato con tampone PBS e le cellule sono state lisate con 200 µl di tampone “Laemmli” 1x
(50 mM Tris-HCl a pH=6,8, 100 mM DTT, 2% SDS, 25% glicerolo, tracce di blu di
bromofenolo); successivamente le cellule sono state staccate meccanicamente dal
substrato, raccolte in una provetta e conservate fino al momento dell’uso a –20°C.
Con le rimanenti cellule trasfettate sono state allestite subcolture in piastre Petri da 100
mm di diametro, al fine di selezionare le cellule che avevano acquisito stabilmente il DNA
plasmidico (espressione stabile). Per la fase di selezione è stato impiegato l’antibiotico G-418
(Invitrogen) alla concentrazione iniziale di 500 µg/ml e a 750 µg/ml per la successiva fase
di mantenimento. La selezione è durata circa un mese e ha consentito di ottenere i
seguenti cloni cellulari: CHO-EGFP, CHO-PSMA6-EGFP, C2.7-PSMB2-EGFP e C2.7-
EGFP. I cloni così ottenuti sono stati posti in coltura secondo le modalità previste per
ciascuna linea cellulare impiegata (come descritto al paragrafo 5.3) e, dopo 3 giorni di
incubazione, il monostrato cellulare è stato direttamente solubilizzato in tampone
“Laemmli”. Successivamente, gli estratti sono stati raccolti mediante distacco meccanico
dal substrato e scaldati a 100°C per 5 minuti.
Per ciascun estratto, derivante da espressioni transitorie e stabili, è stata valutata la
concentrazione delle proteine totali mediante il saggio commerciale “RC DC protein
assay” (Bio-Rad), basato sul metodo di Lowry.
5.4.2 ELETTROFORESI SU GEL DI POLIACRILAMIDE (“SDS-PAGE”)
Per l’analisi di proteine sottoposte a denaturazione è stato utilizzato il metodo
denominato “SDS-PAGE” che prevede un’analisi elettroforetica in gel di poliacrilamide in
condizioni denaturanti (presenza di SDS). Il detergente SDS è stato aggiunto sia al
tampone di corsa che a quello di diluizione del campione. Gli estratti sono stati
solubilizzati in tampone “Laemmli” e successivamente scaldati a 100°C per 5 minuti.

5. Materiali e Metodi
-69-
Inoltre, per ciascun estratto è stata valutata la concentrazione delle proteine totali secondo
il metodo ”RC DC protein assay” o, alternativamente, il metodo “Bradford” (Bio-Rad).
L’elettroforesi è stata realizzata utilizzando il sistema Bio-Rad modello Mini-Protean III;
questo tipo di elettroforesi sfrutta la combinazione di un gel in cui il campione si
concentra (poliacrilamide al 4%: ”stacking gel”) e di un gel a concentrazione di acrilamide
variabile (poliacrilamide al 12,5%: “resolving gel”) che consente la separazione delle
proteine, in base al loro peso molecolare.
Stacking gel (4%) Volume
* Stacking gel 10x 0,5 ml
30% Acrilamide/Bis 37.5:1 (2,6% C) 0,67 ml
TEMED (N,N,N’,N’-tetrametil-etilenediamina) 97% 5 µl
25% ammonio persolfato (APS) 25 µl
Acqua Milli-Q 3,83 ml * Stacking gel 10x: 0,5 M Tris-HCl (pH=6,8) e 0,4% SDS
Running gel (12,5%) Volume
** Lower gel 4x 2 ml
30% Acrilamide/Bis 37.5:1 (2,6% C) 3,15 ml
TEMED 7,5 µl
25% APS 7,5 µl
Acqua Milli-Q 2,85 ml
** Lower gel 4x : 1,5 M Tris-HCl (pH=8,8) e 0,4% SDS
La migrazione elettroforetica è avvenuta in soluzione elettrolitica contenente 25 mM Tris-
base, 192 mM glicina (pH=8,3) e 0,1% SDS.
In ogni pozzetto preformato nel gel di “stacking” sono state caricate quantità uguali di
proteine per ciascun estratto, fino ad un massimo di 20 µl per ciascun pozzetto; uno dei
pozzetti è stato utilizzato per caricare una miscela di proteine colorate a peso molecolare
noto “SeeBlue Plus 2 Pre-Stained Protein Standard” (Invitrogen).

5. Materiali e Metodi
-70-
5.4.3 WESTERN BLOT
Una volta terminata la migrazione elettroforetica su gel, le proteine sono state trasferite
elettroforeticamente (“elettroblotting”) su un supporto sintetico solido, costituito da una
membrana di fluoruro di polivinilidene (PVDF; Immobilon P., Millipore, Bedford, MA),
mediante il sistema “Novex® Western Transfer Apparatus” (Invitrogen). L’elettroblotting
è stato effettuato assemblando preventivamente i diversi componenti (spugna, carta
assorbente per il blotting, gel, membrana di trasferimento, carta assorbente per il blotting,
spugna); poi i suddetti componenti sono stati immersi in tampone di trasferimento (12
mM Tris-base, 96 mM glicina, 20% metanolo, acqua Milli-Q) e trasferiti
elettroforeticamente a basso voltaggio per una notte a 4°C. Le membrane di PVDF sono
state in seguito saturate per 1 ora a temperatura ambiente con una soluzione contenente
PBS, Tween 20 (0,20%) e latte magro in polvere (4%) (PTL), in modo da impedire il legame
aspecifico degli anticorpi alla superficie della membrana. In seguito, la membrana è stata
incubata con l’anticorpo primario per 1 ora in agitazione a temperatura ambiente. Per
l’analisi delle proteine presenti negli estratti cellulari sono state impiegate sonde
immunologiche dirette nei confronti delle subunità proteasomali α1/p27k (anticorpo
diluito 1:2.000, fornito gentilmente dal Dr. Klaus Scherrer - Institut Jacques Monod-Parigi,
Francia) e β4/p23k (anticorpo diluito 1:500; ditta Affiniti Research), così come anticorpi
contro le proteine fluorescenti GFP (anticorpo diluito 1:500; ditta Abcam) e DsRed
(anticorpo diluito 1:500; ditta Clontech). Dopo l’incubazione con gli anticorpi primari, la
membrana è stata lavata con PTL e successivamente incubata (1 ora, in agitazione a
temperatura ambiente) con l’anticorpo secondario anti-topo [IgG (H+L) purificate da
capra (Bio-Rad)] o, in alternativa, con l’anticorpo anti–coniglio [IgG (H+L) purificate da
capra (Bio-Rad)]; entrambi gli anticorpi erano coniugati con perossidasi di rafano (HRP) e
sono stati diluiti 1:10.000 e 1:5.000, rispettivamente. Una volta terminato il periodo di
incubazione, le membrane sono state lavate per 4 volte con PTL e 1 volta con PBS. Le
proteine sono state valutate mediante chemiluminescenza con il saggio commerciale “BM
Chemiluminescence Blotting Substrate” (Roche Diagnostics) e successiva esposizione
autoradiografica (Kodak X-Omat AR Film). Rispetto ai metodi immunoenzimatici
“classici”, la rivelazione in chemiluminescenza offre il vantaggio di poter riutilizzare le
membrane per successive immunoreazioni (massimo 3); ciò è possibile dal momento che il
prodotto rilevato non è un precipitato colorato fisicamente presente sulla membrana di
PVDF.

5. Materiali e Metodi
-71-
5.4.4 COLORAZIONE CON BLU DI COOMASSIE
Per valutare l’avvenuto trasferimento delle proteine su membrane PVDF dopo
“elettroblotting”, i gel sono stati colorati con il colorante blu di Coomassie. Allo scopo, i
gel sono stati immersi in soluzione acquosa costituita dal colorante Coomassie G250
(0,50%; ditta Bio-Rad), 50% di metanolo e 10% di acido acetico, per 1 ora, a temperatura
ambiente e sotto lenta agitazione. Al termine della colorazione, il colorante è stato raccolto
e conservato per le colorazioni successive, mentre i gel sono stati decolorati per rimuovere
il colore di fondo, con una soluzione costituita da 30% metanolo e 10% acido acetico, per
almeno 3 ore, cambiando la soluzione per 3-4 volte. Una volta decolorati, i gel sono stati
lavati con acqua Milli-Q.
5.4.5 CENTRIFUGAZIONE IN GRADIENTE DI DENSITÀ DI SACCAROSIO
Nella prima fase della ricerca sono stati analizzati estratti citoplasmatici e nucleari ottenuti
dai cloni cellulari CHO-PSMA6-EGFP #1 e #4 e C2.7-PSMB2-EGFP #1 e #3, che
esprimevano stabilmente le proteine di fusione α1/p27k-EGFP e β4/p23k-EGFP. Come
controllo, sono stati impiegati estratti citoplasmatici e nucleari dei cloni cellulari CHO-
EGFP e C2.7-EGFP che esprimevano la sola proteina fluorescente EGFP e cellule CHO e
C2.7 non trasfettate.
Allestimento degli estratti cellulari
Il tappeto cellulare (minimo 1x108 cellule) è stato raccolto mediante l’azione di
tripsina/EDTA, centrifugato a bassa velocità (2.000 x g per 3 minuti), lavato due volte in
PBS freddo e quindi risospeso in 8 volumi di tampone ipotonico (rispetto al volume del
sedimento cellulare), denominato THK-M2 (10 mM Tris-HCl a pH=7,4, 10 mM KCl, 2 mM
MgCl2, 1 mM MnCl2, 5 mM 2-mercaptoetanolo).
Le cellule sono state lisate alla temperatura di 4°C mediante l’impiego di un “dounce” ed
il livello di lisi è stato monitorato attraverso l’allestimento di preparati microscopici a
fresco. Una volta raggiunto il livello di lisi ottimale, l’isotonicità è stata ristabilita con
l’aggiunta di saccarosio 2 M (concentrazione finale: 0,25 M). Il lisato è stato trasferito in
provette Corex (15 ml) e centrifugato a 10.000 x g (rotore JA 20 Beckman), alla
temperatura di 4°C per 30 minuti. Il surnatante è stato raccolto in una nuova provetta
(estratto citoplasmatico) e conservato alla temperatura di 4°C fino al momento dell’uso. Il
sedimento, costituito da materiale nucleare e mitocondriale, è stato invece risospeso in 2
ml del tampone THK (10 mM Tris-HCl a pH=7,4, 50 mM KCl, 5 mM 2-mercaptoetanolo)

5. Materiali e Metodi
-72-
addizionato con il detergente Nonidet P-40 alla concentrazione finale dello 0,1%. La
sospensione è stata incubata a temperatura ambiente per circa 15 minuti, agitata per 10
minuti mediante vortex in camera fredda e, infine, sottoposta a centrifugazione per 45
minuti alla velocità di 12.000 x g (rotore JA 20 Beckman). Il surnatante così ottenuto
(estratto nucleare) è stato raccolto e conservato a 4°C fino al momento dell’uso.
Una volta allestiti gli estratti nucleari e citoplasmatici, è stato preparato per ciascuno di
essi un gradiente esponenziale di saccarosio (5-21%), mediante l’impiego di una pompa
peristaltica.
Proteasoma 20S
Per l’isolamento dei proteasomi 20S sono stati allestiti gradienti esponenziali di saccarosio
(5-21%) contenenti il detergente Sarkosyl (0,2%) (“N-Lauroylsarcosine sodium salt”). La
concentrazione di Sarkosyl impiegata consente di creare condizioni dissocianti alle quali
la struttura del proteasoma 20S è conservata, mentre il complesso di ordine superiore 26S
viene disassemblato.
Composizione chimica dei gradienti esponenziali:
5% (p/p) saccarosio 27,5% (p/p) saccarosio
Saccarosio 7,5 g 41,25 g
THK 10X 14,7 ml (1x) 13,7 ml (1x)
Sarkosyl (10%) 2,94 ml (0,2%) 2,74 ml (0,2%)
H2O portare a volume finale: 147 ml portare a volume finale: 137 ml
2-mercaptoetanolo
(14 M)
51 µl 48 µl
Gli estratti citoplasmatici sono stati diluiti 1:2 con tampone THK addizionato con Sarkosyl
(concentrazione finale 0,2%), mentre agli estratti nucleari è stato aggiunto solo il
detergente Sarkosyl (concentrazione finale 0,2%).
Successivamente, gli estratti sono stati depositati delicatamente sulla superficie dei
gradienti di saccarosio preallestiti, ciascuno dei quali di volume pari a 10 ml in tubi
“Beckman” da 12 ml. I gradienti sono stati quindi sottoposti a centrifugazione alla velocità
di 36.000 rpm, a 4°C, per 14 ore e 30 minuti (rotore SW41 Beckman). Al termine della
centrifugazione, un capillare sottile è stato inserito molto delicatamente (quasi fino al
fondo) in ciascun tubo di gradiente. Mediante l’azione di una pompa peristaltica per ogni
gradiente sono state raccolte in media 70 frazioni di circa 200 µl ciascuna in piastre da 96

5. Materiali e Metodi
-73-
pozzetti con fondo nero. La lettura è stata effettuata mediante un fluorimetro (Thermo-
Electron-LabSystems), utilizzando il filtro 485 nm (eccitazione)/538 nm (emissione). I
valori di intensità di fluorescenza della proteina GFP rilevati per ciascuna frazione sono
stati espressi in unità arbitrarie di fluorescenza e riportati in un grafico. Infine, è stato
effettuato un dosaggio delle proteine, utilizzando il saggio commerciale “Micro BCATM” in
micropiastra (Pierce Biotechnology). Le frazioni ottenute mediante centrifugazione degli
estratti cellulari in gradiente di densità di saccarosio, raggruppate in modo da ottenere 12
frazioni principali per ogni tipologia di cellula presa in considerazione, sono state
analizzate previa elettroforesi e successivo Western Blot, secondo le modalità descritte nei
paragrafi 5.4.2 e 5.4.3.
5.5 STRATEGIA UTILIZZATA PER LA PRODUZIONE DI VETTORI RETROVIRALI
5.5.1 MODIFICAZIONE DEL VETTORE COMMERCIALE PDON-AI
A differenza dei plasmidi di espressione precedentemente descritti, il vettore retrovirale
pDON-AI di 5,6 kb (Takara-Biomedicals) è costituito da sequenze derivanti dal retrovirus
responsabile della leucemia murina di Moloney. In particolare, nel vettore retrovirale il
sito multiplo di clonaggio è localizzato tra le sequenze virali LTR (“long terminal
repeats”); inoltre, nel suddetto vettore è presente il segnale per l’incapsidamento della
progenie virale, definito “psi“ (Ψ). Tuttavia, tali vettori retrovirali risultano defettivi per i
geni virali gag, pol e env, necessari per la produzione di particelle virali. Come in tutti i
plasmidi, sono presenti anche marcatori di selezione, quali i geni che codificano per la
resistenza agli antibiotici ampicillina e neomicina (e del suo analogo G-418).
Rimozione dal vettore pDON-AI del sito di taglio per l’enzima di restrizione Hind III, localizzato
all’esterno delle sequenze LTR
Il vettore retrovirale (20 ng) è stato digerito parzialmente con 2 unità dell’enzima di
restrizione Hind III, diluito nel tampone specifico “NEB 2” (New England BioLabs). La
miscela di reazione è stata incubata in bagnomaria alla temperatura di 37°C e, a tempi
variabili (5, 10, 15, 20 minuti), sono state raccolte aliquote della miscela di reazione. I
prodotti di digestione enzimatica sono stati separati mediante elettroforesi in gel di
agarosio allo 0,8% in tampone TAE ed il frammento, di circa 5,6 kb, è stato in seguito
eluito dal gel e purificato mediante l’impiego del saggio commerciale “NucleoSpin”

5. Materiali e Metodi
-74-
(Stratagene), secondo le modalità fornite dalla ditta produttrice. Successivamente è stata
valutata la concentrazione del suddetto frammento di DNA. Per la rimozione del sito di
taglio dell’enzima di restrizione Hind III, le estremità 3’-OH interne, risultanti dalla
digestione con l’enzima di restrizione Hind III, sono state completate utilizzando l’enzima
DNA polimerasi-frammento di Klenow di E.coli (Roche Diagnostics), in ragione di 2 unità.
Al mezzo di reazione sono stati aggiunti 100 ng del vettore pDONdelta-AI (pDON-AI
modificato) e i nucleotidi trifosfati diluiti nel tampone “NEB 2”, necessari per completare
il doppio filamento di DNA nella porzione mancante. Avvenuta la reazione di
completamento è stato utilizzato l’enzima DNA-ligasi per sigillare le estremità piatte del
vettore mediante il saggio commerciale “Fast-Link DNA Ligation Kit”(Epicentre),
secondo le modalità fornite dalla ditta produttrice.
Una aliquota della miscela è stata impiegata per trasformare cellule batteriche di E.coli
contenute nel saggio commerciale “XL1-Blue competent cells” (Stratagene), secondo il
protocollo descritto nel paragrafo 5.2.7. Dopo un’ora di incubazione a 37°C, la
sospensione batterica è stata seminata mediante biglie di vetro su piastre agarizzate di
terreno di coltura LB, contenente l’antibiotico ampicillina alla concentrazione di 50 µg/ml,
che consente la selezione dei cloni cellulari recanti il plasmide; le piastre sono state
incubate per una notte a 37°C. A partire dai cloni cellulari di interesse si è reso necessario
estrarre e purificare il DNA ricombinante in quantità adeguata per la sua successiva
caratterizzazione secondo il protocollo precedentemente descritto nel paragrafo 5.2.8
mediante l’impiego del saggio commerciale “NucleoBond” (Machery-Nagel). A questo
scopo, sono state trasferite sterilmente singole colonie di E.coli in provette di terreno
liquido LB (3 ml) contenenti ampicillina alla concentrazione di 100 µg/ml. Le provette
sono state poi incubate e mantenute in agitazione per una notte a 37°C, al fine di favorire
la crescita aerobia dei batteri. A partire da queste sospensioni è stato possibile preparare
piccole quantità di DNA plasmidico (“mini-prep”) ad un livello intermedio di purezza.
Le “mini-prep” sono state sottoposte nuovamente a digestione enzimatica per verificarne
la bontà mediante l’impiego degli enzimi di restrizione EcoR I e Hind III, diluiti nel
tampone “NEB 2”, in ragione di 20 unità per EcoR I e di 30 unità per Hind III. La miscela
di reazione è stata incubata in bagnomaria alla temperatura di 37°C per 2 ore e
successivamente impiegata per allestire un gel di agarosio (1%) in tampone TBE.
Una volta verificato che il sito di restrizione per Hind III, localizzato all’esterno delle
sequenze LTR, era stato rimosso (come atteso sono stati ottenuti frammenti di circa 3,8 kb
e 1,8 kb), da una delle “minicolture” batteriche è stata allestita una brodo-coltura batterica

5. Materiali e Metodi
-75-
in 300 ml di LB addizionato di ampicillina (100 µg/ml) al fine di ottenere una maggiore
quantità di DNA plamidico (“maxi-prep”) ad elevata purezza, mediante il saggio
commerciale “NucleoBond Ax 2000” (Machery-Nagel), secondo il protocollo riportato nel
paragrafo 5.2.9. Il plasmide modificato pDONdelta-AI, è stato risospeso in tampone TE e
la concentrazione del DNA è stata calcolata, a partire da una soluzione diluita 1:100 in
tampone TE, sulla base dei valori di assorbanza rilevati alla lunghezza d’onda di 260 nm.
5.5.2 CLONAGGIO IN VETTORI RETROVIRALI
Clonaggio in un vettore retrovirale della sequenza codificante per EGFP
I plasmidi pEGFP-N1 e pDONdelta-AI sono stati sottoposti a doppia digestione con gli
enzimi di restrizione Hind III (20 unità) e Hpa I (10 unità), diluiti in tampone “NEB 4” (20
mM Tris-acetato, 50 mM acetato di potassio, 10 mM acetato di magnesio, 1 mM DTT a
pH= 7,9 a 25°C) (New England BioLabs). In tal modo, dal primo plasmide (pEGFP-N1) è
stato ottenuto l’inserto di circa 900 bp, relativo alla sequenza genica codificante per la
proteina fluorescente (EGFP), mentre dal secondo pDONdelta-AI il vettore di 5,6 kb, in
cui poter inserire le sequenze di interesse. In particolare, mediante i protocolli operativi
precedentemente descritti è stato ottenuto il vettore retrovirale pDONdelta-EGFP (6,5 kb).
Successivamente, è stata verificata la bontà di tale prodotto mediante digestione
enzimatica con gli enzimi di restrizione Not I e Hind III, diluiti nel tampone “NEB 2” in
ragione di 30 unità e di 20 unità, rispettivamente. La miscela di reazione è stata incubata
in bagnomaria alla temperatura di 37°C per 2 ore e successivamente impiegata per
allestire un gel di agarosio (1%) in TBE. Come atteso, sono stati ottenuti per pDONdelta-
EGFP i frammenti di circa 5,7 kb e 779 bp.
Clonaggio in un vettore retrovirale delle sequenze codificanti per α1/p27k-EGFP e β4/p23k-EGFP
Il vettore retrovirale pDONdelta-EGFP è stato utilizzato per la successiva costruzione dei
vettori retrovirali contenenti le sequenze codificanti per le proteine di fusione, dal
momento che tra le sequenze LTR del vettore sopra citato sono presenti i siti di taglio per
gli enzimi di restrizione Hind III e Not I.
Il vettore retrovirale pDONdelta-EGFP e i plasmidi pPSMA6-EGFP e pPSMB2-EGFP sono
stati sottoposti a doppia digestione enzimatica con gli enzimi di restrizione Hind III (10
unità) e Not I (20 unità), diluiti in tampone “NEB 2” e incubati in bagnomaria a 37°C per 2
ore. I frammenti ottenuti dai vettori pPSMA6-EGFP (1,5 kb) e pPSMB2-EGFP (1,3 kb), che

5. Materiali e Metodi
-76-
codificano per le proteine di fusione (α1/p27k-EGFP e β4/p23k-EGFP, rispettivamente),
sono stati estratti e purificati dal gel di agarosio e, successivamente, clonati nel vettore
retrovirale mediante il saggio commerciale “Fast-Link DNA Ligation Kit”(Epicentre).
Seguendo i protocolli precedentemente illustrati e relativi alla preparazione dei vettori di
espressione, sono stati ottenuti i seguenti vettori retrovirali: pDONdelta-PSMA6-EGFP,
pDONdelta-PSMB2-EGFP e pDONdelta-EGFP.
Clonaggio in un vettore retrovirale della sequenza codificante per l’enzima β-galattosidasi
A partire da un plasmide denominato pMonlsLacZ (fornito dal Dr. José Cebrian - Institut
Gustave-Roussy - Instabilité Génétique et Cancer – Villejuif, Francia – dati non
pubblicati), in cui è presente la sequenza che codifica per l’enzima β-galattosidasi, è stato
preparato il corrispondente vettore retrovirale pDONdelta-nlsLacZ, contenente la
sequenza genica codificante per l’enzima β-galattosidasi. In particolare, il vettore
retrovirale pDON-delta-EGFP e il vettore pMonlsLacZ sono stati sottoposti a doppia
digestione enzimatica mediante l’impiego degli enzimi di restrizione BamH II e Not I,
diluiti nel tampone specifico “NEB” per l’enzima BamH II (New England BioLabs). Il
vettore retrovirale così ottenuto (pDONdelta-nlsLacZ) è stato utilizzato come controllo
per le successive fasi di produzione di retrovirus. L’espressione dell’enzima β-
galattosidasi è stata rilevata mediante l’impiego di un composto commerciale inizialmente
incolore, noto come X-gal (5-bromo-4-cloro-3-indol-β-D-galattopiranoside; Sigma-
Aldrich). I monostrati cellulari sono stati dapprima fissati mediante una miscela di
fissazione costituita da formaldeide (1%) e glutaraldeide (0,2%) in PBS e, successivamente,
sottoposti a colorazione per una notte impiegando una soluzione costituita da: 5 mM
ferricianuro di potassio, 5 mM ferrocianuro di potassio, 500 µg/ml X-gal in
dimetilsulfossido e 2 mM MgCl2. La degradazione di X-gal da parte dell’enzima β-
galattosidasi porta alla produzione di un composto blu insolubile, localizzato nel nucleo
delle cellule e facilmente osservabile mediante l’impiego di un microscopio ottico.

5. Materiali e Metodi
-77-
5.6 PRODUZIONE E IMPIEGO DI PARTICELLE RETROVIRALI (ECOTROPICHE)
5.6.1 PRODUZIONE DI PARTICELLE RETROVIRALI DEFETTIVE MEDIANTE L’IMPIEGO DELLE
CELLULE BOSC-23
La linea cellulare denominata Bosc-23 è stata impiegata per esperimenti di trasfezione,
volti ad ottenere la produzione di particelle retrovirali defettive ma in grado di infettare
cellule di origine murina (ecotropiche). Il giorno precedente la trasfezione, sono state
allestite colture di cellule Bosc-23 in tre piastre Petri (35 mm di diametro) per ciascun
plasmide (pDONdelta-PSMA6-EGFP, pDONdelta-PSMB2-EGFP, pDONdelta-EGFP e
pDONdelta-nlsLacZ come controllo), con una densità pari a 1x106 cellule/piastra. Per
incrementare l’efficienza di trasfezione, è stato utilizzato un composto a base di lipidi
(FuGENE-6, Roche Diagnostics). Per la trasfezione è stata impiegata, per ciascun
plasmide, una quantità di DNA pari a 5 µg. Dopo 48 ore dalla trasfezione il surnatante
(terreno di coltura delle cellule Bosc-23, arricchito con progenie virale) è stato raccolto,
chiarificato mediante centrifugazione a 2.000 x g per 3 minuti per la rimozione dei detriti
cellulari e, successivamente, filtrato con filtri del diametro di 0,45 µm. Una aliquota del
surnatante è stata utilizzata per eseguire infezioni della linea cellulare di topo Balb/3T3
per la determinazione del titolo virale e, parallelamente, come controllo di infezione di
cellule di topo, al fine di rendere plausibile l’utilizzo successivo delle particelle retrovirali
per l’infezione della linea miogenica di topo C2.7.
Da un monostrato semiconfluente di Balb/3T3 sono state allestite subcolture in piastre a 6
pozzetti alla concentrazione di 8x104 cellule/piastra. Ciascuna sospensione retrovirale
(PSMA6-EGFP/rv, PSMB2-EGFP/rv, EGFP/rv e nlsLacZ/rv) è stata saggiata a tre diverse
diluizioni (10-1, 10-2, 10-4). Per ridurre le interazioni elettrostatiche tra virus e membrana
cellulare è stata aggiunta al terreno di coltura una sostanza policationica nota come
“polybrene” (“1,5-dimethyl-1,5-diazaundecamethylene polymethobromide”), alla
concentrazione finale di 5 µg/ml (Sigma-Aldrich). Dopo 48 ore dall’infezione è stato
determinato il titolo della sospensione virale sulla base della determinazione quantitativa
dell’infettività volta al rilevamento del numero di cellule che esprimevano la proteina
EGFP (PSMA6-EGFP, PSMB2-EGFP, EGFP) e, alternativamente l’enzima β-galattosidasi
(nlsLacZ).

5. Materiali e Metodi
-78-
5.6.2 INFEZIONE DI COLTURE CELLULARI DELLA LINEA MIOGENICA DI TOPO C2.7 MEDIANTE
RETROVIRUS RICOMBINANTI
Le sospensioni virali PSMA6-EGFP/rv, PSMB2-EGFP/rv, EGFP/rv e nlsLacZ/rv sono
state successivamente utilizzate per infettare la linea cellulare miogenica di topo C2.7. Il
giorno precedente l’infezione sono state allestite colture di cellule C2.7 in tre piastre Petri
(35 mm di diametro), con una densità pari a 4x104 cellule/piastra. Per ridurre le
interazioni elettrostatiche tra virus e membrana cellulare è stato aggiunto “polybrene“ al
terreno di coltura. Dopo 48 ore dall’infezione le cellule sono state tripsinizzate e poste in
coltura in piastre da 100 mm di diametro, in presenza di 750 µg/ml di G-418.
5.6.3 CENTRIFUGAZIONE IN GRADIENTE DI DENSITÀ DI SACCAROSIO
Analogamente a quanto riportato nel paragrafo 5.4.5, per i cloni cellulari CHO-PSMA6-
EGFP #1 e #4 e C2.7-PSMB2-EGFP #1 e #3 sono stati analizzati estratti citoplasmatici e
nucleari ottenuti da popolazioni di cellule C2.7 ingegnerizzate (C2.7-PSMA6-EGFP, C2.7-
PSMB2-EGFP), che esprimevano stabilmente le proteine di fusione α1/p27k-EGFP e
β4/p23k-EGFP, rispettivamente. Come controllo, sono stati analizzati in parallelo gli
estratti ottenuti a partire dalle popolazioni di cellule C2.7 normali ed anche di C2.7-EGFP
che esprimevano la sola proteina fluorescente EGFP. Le frazioni ottenute mediante
centrifugazione degli estratti cellulari in gradiente di densità di saccarosio, raggruppate in
modo da ottenere 12 frazioni principali per ogni tipologia di cellule prese in
considerazione, sono state analizzate previa elettroforesi e successivo Western Blot,
secondo le modalità descritte nei paragrafi 5.4.2 e 5.4.3.
5.6.4 ELETTROFORESI SU GEL DI POLIACRILAMIDE (“NATIVE-PAGE”)
Per l’analisi di proteine non denaturate è stato utilizzato il metodo denominato “NATIVE-
PAGE”, nel cui tampone di corsa e in quello di diluizione del campione non viene aggiunto
SDS. A tal fine sono stati allestiti nuovi estratti citoplasmatici a partire dalle seguenti
popolazione di cellule C2.7: C2.7-EGFP, C2.7-PSMA6-EGFP e C2.7-PSMB2-EGFP, con
metodo analogo a quello descritto nel paragrafo 5.4.5, fatta eccezione per la composizione
del tampone ipotonico THK-M2 a cui è statto aggiunto ATP (5 mM), al fine di preservare
l’attività dei proteasomi 26S.
Le proteine di ciascun estratto citoplasmatico, sono state concentrate mediante il sistema
“UltrafreeR-15 Centrifugal filter device” (Millipore). In sintesi, a ciascun campione è stato
aggiunto ATP (2 mM) e i campioni sono stati in seguito centrifugati a basse temperature
5. Materiali e Metodi
-79-
per 4 ore alla velocità di 2.000 x g, fino a quando tutto il contenuto è stato concentrato.
Complessivamente, il volume iniziale di 3,5 ml è stato ridotto a circa 200 µl per ciascun
campione (PSMA6-EGFP, PSMB2-EGFP e EGFP).
Allestimento di gel di poliacrilamide
Nel frattempo, sono stati allestiti 3 gel di poliacrilamide in condizioni non denaturanti,
utilizzando il sistema “Mini-Protean 3” (Bio-Rad); i suddetti gel avevano uno spessore di
1,5 mm, lunghezza pari a 1,5 cm e a 6,5 cm per lo “stacking” gel e il “resolving” gel
rispettivamente. Di seguito vengono riportate le composizioni dello “stacking” gel e del
“resolving” gel:
Stacking gel (2,5%) Concentrazione finale
30% Bis-Acrilamide (37,5:1 - 2,6% C) 2,5%
* TBE 5x TBE 1x
APS 25% 0,1%
TEMED 0,1%
Resolving gel (4,5%) Concentrazione finale
30% Bis-Acrilamide (37,5:1 - 2,6% C) 4,5%
* TBE 5x TBE 1x
APS 25% 0,04%
TEMED 0,1%
* TBE 1x: 90 mM Tris-Borato, 0,08 mM EDTA
Una volta allestiti i gel, questi sono stati riposti negli appositi apparati per elettroforesi,
ricoperti con tampone TBE freddo e sottoposti a pre-migrazione per 1 ora alla tensione di
50 Volts a 4°C. A ciascun campione sono stati aggiunti 200 µl di TBE freddo contenente 2
mM ATP e blu di bromofenolo. Una volta caricati, i campioni sono stati fatti migrare per
12 ore, a 50 Volts, a 4°C.
Valutazione dell’attività dei proteasomi
Al termine della migrazione, i gel sono stati recuperati e impiegati per misurare l’attività
proteolitica dei proteasomi 20S e 26S (“chymotrypsin-like”), valutando il rilascio del
fluoroforo 7-amino-4 metilcumarin “AMC” (eccitamento 356 nm/emissione 366 nm) dopo

5. Materiali e Metodi
-80-
il taglio del substrato marcato LLVY-AMC (“N-Succinyl-Leu-Leu-Val-Tyr/7-amido-4-
methylcoumarin”, Sigma – Aldrich). I gel sono stati posti in piccole vaschette alle quali
sono stati aggiunti 10 ml di una soluzione contenente 30 mM Tris-HCl a pH=7,5, 5 mM
MgCl2, 10 mM KCl, 0,5 mM DTT, 2 mM ATP e 100 µM del peptide fluorogenico N-Suc-L-
L-V-Y-AMC. Le vaschette sono state poste a 37°C per 1 ora su uno scuotitore a bassa
velocità. Il segnale di fluorescenza è stato osservato esponendo i gel ai raggi UV di un
transilluminatore (360 nm) (Hough R. et al., 1987; Hoffman L. and Rechsteiner M., 1996;
Glickman M.H. et al., 1998; Piccinini M. et al., 2000; Elsasser S. et al., 2005).
Estrazione delle proteine da gel
Le bande a livello delle quali è stata riscontrata l’attività proteolitica dei proteasomi sono
state tagliate mediante un bisturi e raccolte in provette per essere conservate a -40°C, fino
al momento dell’uso. Una volta scongelati, i campioni sono stati frammentati, mediante
l’impiego di un pistone e nuovamente congelati a -40°C (queste operazioni erano atte a
rompere i legami che si formano in seguito a polimerizzazione dei monomeri di
acrilamide). Ai campioni nuovamente scongelati sono stati aggiunti 500 µl di tampone
“Laemmli” senza glicerolo e blu di bromofenolo (50 mM Tri-HCl pH=6,8, 100 mM DTT,
2% SDS). I campioni sono stati incubati a 37°C per una notte. Al fine di concentrare le
proteine è stato utilizzato il protocollo che prevede l’impiego dell’acido tricloroacetico
(TCA). In particolare, a ciascun campione è stata aggiunta, in ragione di una diluizione
1:100, una soluzione di deossicolato (2%) (Sigma-Aldrich). I campioni sono stati mescolati
e lasciati incubare a 4°C per 30 minuti. Successivamente a ciascun campione è stato
aggiunto TCA (100%) diluito 1:10; i campioni sono stati incubati per una notte a 4°C ed
infine centrifugati per 10 minuti a velocità massima (15.000 x g) a 4°C. Il surnatante è stato
delicatamente rimosso per inversione della provetta ed i sedimenti sono stati lavati due
volte con acetone freddo (-20°C) e centrifugati a 15.000 x g per 5 minuti. Una volta
terminati i lavaggi, i sedimenti sono stati asciugati all’aria e risospesi in un minimo
volume di tampone “Laemmli”. La presenza di TCA può far virare il pH del tampone; in
questo caso, sono stati aggiunti pochi µl di NaOH (1N) o, in alternativa, Tris-HCl a
pH=8,5 (1M). Le aliquote dei campioni precipitati con TCA sono state utilizzate per
allestire gel denaturanti (“SDS-PAGE”) da sottoporre ad analisi in Western Blot e,
parallelamente, a colorazione argentica.

5. Materiali e Metodi
-81-
5.6.5 COLORAZIONE ARGENTICA
Al termine della corsa elettroforetica, i gel preparati secondo le modalità descritte nel
paragrafo 5.4.2 sono stati immersi in metanolo al 50% in H2O per 2 ore a temperatura
ambiente, sotto lenta agitazione. La soluzione di fissazione è stata cambiata dopo 1 ora
(per piccoli gel: 500 ml totali, 2 x 250 ml). Nel frattempo è stata preparata la soluzione
argentica come segue:
Soluzione A Soluzione B
0,8 gr AgNO3 in 4 ml di H2O Milli-Q 21 ml NaOH 0,36%
1,4 ml NH4OH 28% (14,8 M)
oppure:
1,53 ml NH4OH 25% (13,4 M)
La soluzione A è stata aggiunta, goccia a goccia, alla soluzione B, mescolando in
continuazione e, infine, sono stati aggiunti 100 ml di acqua Milli-Q; la soluzione deve
presentarsi completamente limpida. Al termine della fase di fissazione in metanolo, i gel
sono stati immersi in questa soluzione per 15 minuti in agitazione e successivamente sono
stati sottoposti a tre lavaggi (10 minuti l’uno) con H2O Milli-Q. La rivelazione è stata
effettuata aggiungendo ai gel una soluzione fresca, costituita da 1 ml di acido citrico
all’1%, 100 µl di formaldeide al 38% e acqua Milli-Q, per un volume totale di 200 ml; i gel
sono stati lasciati in lenta agitazione, fino alla comparsa delle bande. Al comparire delle
bande con l’intensità desiderata, sono stati aggiunti 2 ml di acido acetico (concentrazione
finale: 1%) per bloccare la reazione.
5.6.6 ELETTROFORESI SU GEL DI POLIACRILAMIDE (“SDS-PAGE”)
Per l’analisi delle proteine è stato utilizzato il metodo “SDS-PAGE” descritto nel paragrafo
5.4.2.
Per l’analisi delle proteine presenti negli estratti, sono state impiegate sonde
immunologiche dirette nei confronti delle subunità proteasomali α1/p27k (anticorpo
diluito 1:2.000, fornito gentilmente dal Dr. Klaus Scherrer - Institut Jacques Monod-Parigi,
Francia) e β4/p23k (anticorpo diluito 1:500; ditta Affiniti Research). Inoltre, è stato
utilizzato un anticorpo monoclonale anti-subunità ATPasi, denominata Rpt1, del
complesso regolatore 19S (diluizione 1:2500; ditta Affiniti Research).

5. Materiali e Metodi
-82-
5.6.7 DISTRIBUZIONE DELLE SUBUNITÀ PROTEASOMALI MARCATE CON LA PROTEINA
FLUORESCENTE EGFP
I cloni cellulari CHO-PSMA6-EGFP #1 e #4, C2.7-PSMB2-EGFP #1 e #3, CHO-EGFP e
C2.7-EGFP sono stati posti in coltura, secondo le modalità previste per ciascuna linea
cellulare, come descritto al paragrafo 5.3, allo scopo di osservare la distribuzione delle
subunità proteasomali co-espresse assieme alla proteina fluorescente verde (EGFP) o,
alternativamente, quella della sola proteina fluorescente, impiegata come controllo. A
partire da monostrati cellulari semiconfluenti sono stati preparati vetrini (superficie di 4
cm2) con densità di 1,2x105 cellule/vetrino (cellule CHO) e di 2x104 cellule/vetrino (cellule
C2.7). Il differenziamento della linea cellulare C2.7 è stato indotto sperimentalmente,
modificando le condizioni di mantenimento in coltura, come precedentemente descritto al
paragrafo 5.3. Al fine di studiare la distribuzione delle subunità proteasomali, in
particolare, a differenti stadi del processo di differenziamento miogenico delle cellule
C2.7, queste sono state lavate con PBS e poi fissate con paraformaldeide al 3,7% in PBS per
10 minuti a temperatura ambiente. Dopo tre lavaggi di 5 minuti ciascuno in PBS, le cellule
sono state montate con liquido di montaggio (Immuno-Fluor, NEN) e quindi osservate
mediante microscopio a fluorescenza.
Successivamente, sono state effettuate osservazioni relative alla distribuzione delle
subunità proteasomali marcate con la proteina EGFP e della sola proteina fluorescente
anche nel corso del processo di differenziamento miogenico in popolazioni di cellule C2.7
ottenute dopo infezione con particelle retrovirali (C2.7-PSMA6-EGFP, C2.7-PSMB2-EGFP
e C2.7-EGFP).
5.7 PRODUZIONE DI PARTICELLE RETROVIRALI (ANFOTROPICHE)
5.7.1 PRODUZIONE DI PARTICELLE RETROVIRALI DEFETTIVE MEDIANTE L’IMPIEGO DELLE
CELLULE IN LINEA CONTINUA HEK 293
La produzione di particelle retrovirali defettive infettanti di tipo anfotropico (in grado di
infettare cellule di mammifero) è stata realizzata mediante co-trasfezione della linea
cellulare denominata HEK 293 con i vettori retrovirali recanti i costrutti di interesse
(pDONdelta-PSMA6-EGFP, pDONdelta-PSMB2-EGFP, pDONdelta-EGFP), pVPackG-P
(plasmide contenente i geni retrovirali gag e pol che codificano per le principali proteine
strutturali e funzionali dei retrovirus; ditta Clontech) e pMD2.G (plasmide che contiene il
gene che codifica per la proteina G del pericapside del virus della Stomatite Vescicolare;

5. Materiali e Metodi
-83-
gentilmente fornito dal Dr. Didier Trono – Faculty of Medicine – Department of
Microbiology and Molecular Medicine – University of Geneva, Svizzera) (Soneoka Y. et
al., 1995; Pear W.S. et al., 1997). Ventiquattro ore prima di effettuare la trasfezione, sono
stati allestiti monostrati cellulari di HEK 293 in tre piastre Petri da 60 mm di diametro, con
una densità iniziale di 2,5x106 cellule/piastra. La quantità di cellule è stata scelta in modo
tale che, al momento della trasfezione si avesse una confluenza del 80%. Due ore prima
della trasfezione, il terreno di coltura è stato sostituito con terreno fresco. La trasfezione è
stata effettuata utilizzando il protocollo che prevede l’impiego del CaCl2 (si veda sezione
5.4.1). In particolare, per la trasfezione è stata utilizzata una quantità totale di DNA di 6
µg, distribuita in un rapporto equimolare di 1:2:3 tra i tre vettori (rispettivamente
pMD2.G, pVPackG-P, vettore retrovirale).
Dopo 24 ore dalla trasfezione, il terreno è stata sostituito con terreno fresco e dopo 48 ore
è stato raccolto il primo surnatante. Il secondo surnatante è stato raccolto dopo 72 ore
dalla trasfezione. I surnatanti sono stati centrifugati a 2.000 x g per 3 minuti, per la
rimozione dei detriti cellulari e congelati a –80°C; essi sono stati in seguito utilizzati per
esperimenti di infezione della stessa linea cellulare (HEK 293) al fine di determinare il
titolo virale e, parallelamente, per esperimenti di infezione di cellule MRC5-hTERT. Una
volta rimosso il surnatante contenente le particelle retrovirali, i monostrati di cellule HEK
293 sono stati lavati delicatamente con PBS e poi fissati con paraformaldeide al 3,7% in
PBS per 10 minuti, a temperatura ambiente. L’efficienza di trasfezione è stata valutata
attraverso l’osservazione dei monostrati mediante microscopio a fluorescenza.
5.7.2 INFEZIONE DI FIBROBLASTI DI POLMONE EMBRIONALE UMANO IMMORTALIZZATI
(MRC5-HTERT) CON I RETROVIRUS RECANTI IL COSTRUTTO DI INTERESSE
Le sospensioni virali PSMA6-EGFP/rv, PSMB2-EGFP/rv e EGFP/rv sono state utilizzate
per infettare monostrati di cellule MRC5-hTERT. Il giorno precedente l’infezione, sono
state allestite colture di MRC5-hTERT in piastre Petri (35 mm di diametro), con una
densità pari a 4x104 cellule/piastra. Per ridurre le interazioni elettrostatiche tra virus e
membrana cellulare è stato aggiunto al terreno di coltura il composto “polybrene”. Dopo
alcuni giorni dall’inizio dell’infezione la sostanza selezionante G-418 (500 µg/ml) è stata
aggiunta al terreno di coltura. Al termine della selezione, durata alcune settimane, sono
state ottenute popolazioni di cellule MRC5-hTERT in grado di esprimere in modo stabile
le subunità α1/p27K e β4/p23K del proteasoma 20S marcate con la proteina EGFP

5. Materiali e Metodi
-84-
(MRC5-hTERT-PSMA6-EGFP e MRC5-hTERT-PSMB2-EGFP, rispettivamente), così come
popolazioni di cellule in grado di esprimere la sola proteina EGFP (MRC5-hTERT-EGFP).
5.8 INFEZIONE DI FIBROBLASTI CON UNO STIPITE UMANO DI CITOMEGALOVIRUS
5.8.1 VIRUS
Per eseguire le infezioni, è stato impiegato lo stipite AD169 (ATCC n. VR-538) di
citomegalovirus umano (HCMV), riprodotto in fibroblasti embrionali umani (MRC5) e
titolato, sulla base della determinazione quantitativa dell’infettività, mediante una
reazione di immunofluorescenza, volta al rilevamento del numero di cellule che
esprimevano le proteine precocissime (IE1 e IE2) di HCMV.
5.8.2 INFEZIONE DI FIBROBLASTI EMBRIONALI UMANI (MRC5) CON LO STIPITE AD169 DI
HCMV
Per l’infezione dei monostrati cellulari MRC5 è stato impiegato lo stipite AD169 di HCMV
[titolo: 2,5x106 unità formanti placca (ufp)/ml] alla molteplicità di infezione di 0,5
ufp/cellula. Le cellule sono state fatte crescere su vetrino (“shell-vial”), fino al
raggiungimento della semi-confluenza. Dopo un lavaggio con MEM privo di siero fetale
di vitello, le cellule sono state infettate con l’opportuna diluizione di sospensione virale,
allestita in terreno (MEM) privo di siero e lasciate in incubazione per 60 minuti a 37°C. Al
termine dell’adsorbimento, l’inoculo virale è stato rimosso e sostituito con terreno di
coltura addizionato di siero fetale di vitello al 10%. I monostrati infettati sono stati infine
incubati a 37°C per i tempi prestabiliti.
5.8.3 MONOSTRATI DI CELLULE MRC5 NON SINCRONIZZATE
I monostrati cellulari sono stati fatti crescere su vetrino (“shell vial”) per 48 ore a 37°C, in
terreno addizionato di SVF (10%). Successivamente le cellule MRC5 sono state lavate per
due volte con MEM privo di siero fetale di vitello ed infettate con lo stipite AD169 di
HCMV.
5.8.4 SINCRONIZZAZIONE DEI MONOSTRATI DI CELLULE MRC5
I monostrati cellulari sono stati fatti crescere su vetrino (“shell vial”) per 24 ore a 37°C in
terreno addizionato di SVF (10%). Successivamente le cellule MRC5 sono state lavate per
tre volte con MEM privo di siero fetale di vitello e lasciate in terreno senza siero per 5

5. Materiali e Metodi
-85-
giorni. Al termine di tale periodo, il suddetto terreno è stato sostituito con terreno
addizionato del 10% di SVF per i tempi prestabiliti (9 ore per G1, 18 ore per G1/S, 24 ore
per S, 30 ore per G2/M) (Morel A.P. et al., 2003; Memili E. et al., 2004), prima di infettare le
cellule con lo stipite AD169 di HCMV.
5.8.5 TRATTAMENTO DI MONOSTRATI DI CELLULE MRC5 CON MG132
I monostrati cellulari sincronizzati, così come quelli non sincronizzati, sono stati trattati
con l’inibitore dell’attività dei proteasomi denominato MG132 (Calbiochem). In
particolare, l’inibitore MG132 è stato aggiunto al terreno di coltura alla concentrazione
finale di 0,5 µM (soluzione stock 10 mM in DMSO) per 3 ore prima dell’infezione (Prösch
S. et al., 2003) ed è stato lasciato a contatto con i monostrati cellulari anche durante il
periodo di adsorbimento dell’inoculo virale ed il periodo di infezione prescelto.
5.8.6 ANTICORPI
Per lo studio dell’infezione virale mediante immunofluorescenza, è stato impiegato un
anticorpo monoclonale (Argene–Biosoft) in grado di riconoscere le proteine virali
precocissime IE di HCMV; tale anticorpo è stato utilizzato alla diluizione 1:30 in BSA allo
0,2% in PBS. Per lo studio della localizzazione del proteasoma 20S è stato impiegato un
anticorpo policlonale (gentilmente fornito dal Prof. Dahlmann – Università di Berlino –
Istituto di Biochimica - Berlino, Germania), in grado di riconoscere alcune delle subunità
del proteasoma 20S; l’anticorpo è stato diluito 1:100 in BSA allo 0,2% in PBS. Per la
rivelazione dell’anticorpo primario anti-proteine virali IE è stato utilizzato un anticorpo
anti-IgG murine, coniugato con TRITC (EuroClone), diluito 1:50 in BSA allo 0,2% in PBS,
mentre, per quanto concerne l’anticorpo anti-proteasoma 20S, è stato utilizzato un
anticorpo anti-IgG di coniglio, coniugato con FITC (EuroClone - United Kingdom) e
diluito 1:50 in BSA allo 0,2% in PBS.
5.8.7 IMMUNOFLUORESCENZA INDIRETTA
Le cellule sono state delicatamente lavate con tampone per citoscheletro (CSK), costituito
da 10 mM Pipes (pH=6,9), 100 mM NaCl, 3 mM MgCl2, 300 mM saccarosio, e quindi
permeabilizzate e fissate contemporaneamente, utilizzando una miscela di Triton X-100 al
2,5% in tampone CSK e paraformaldeide all’1% nello stesso tampone, per 20 minuti a
temperatura ambiente. Dopo tre lavaggi di 5 minuti ciascuno in PBS, i siti immunoreattivi

5. Materiali e Metodi
-86-
non specifici sono stati saturati con BSA all’1% in PBS per 15 minuti; le cellule sono state
successivamente lavate con PBS prima di deporre gli anticorpi primari.
Per l’immunoreazione, le cellule sono state incubate con l’anticorpo monoclonale anti-
proteine virali IE e con l’anticorpo policlonale anti-proteasoma 20S, per 1 ora a 37°C, in
camera umida. Dopo tre lavaggi di 5 minuti ciascuno in PBS, le cellule sono state incubate
con gli specifici anticorpi secondari per 45 minuti a 37°C, in camera umida. Le cellule,
sono state lavate in seguito con PBS (tre lavaggi di 5 minuti ciascuno) e montate con
liquido di montaggio (DakoCytomation).
L’immunoreazione è stata osservata mediante microscopio a fluorescenza (Leica) o
mediante microscopio confocale (Zeiss LSM 510 Meta).
5.8.8 ANALISI DELLE CELLULE MRC5 MEDIANTE CITOFLUORIMETRO A FLUSSO
I monostrati cellulari sono stati fatti crescere su piastre da 6 pozzetti (6x105
cellule/pozzetto) per 24 ore a 37°C in terreno MEM addizionato di SVF (10%).
Successivamente le cellule MRC5 sono state lavate per tre volte con MEM privo di SVF e
mantenute in coltura in terreno senza siero per 5 giorni. Terminato questo periodo di
tempo, il terreno privo di siero è stato sostituito con terreno addizionato del 10% di SVF
per i tempi prestabiliti (9 ore per G1, 18 ore per G1/S, 24 ore per S, 30 ore per G2/M). I
tappeti cellulari sono stati raccolti mediante l’azione di tripsina/EDTA, centrifugati a
bassa velocità (1.000 x g per 5 minuti) e risospesi in 50 µl di PBS. I campioni sono stati
conservati a 4°C fino al momento dell’uso. Per l’analisi mediante citofluorimetria a flusso
(FACScan Becton-Dickinson) i campioni sono stati addizionati con 500 µl di tampone
citrato contenente 50 µl di ioduro di propidio (Sigma-Aldrich), 1 mg/ml di ribonucleasi A
(Sigma-Aldrich) e Nonidet P-40 allo 0,1% (Sigma-Aldrich). I campioni sono stati
successivamente incubati al buio a 4°C per almeno 1 ora, prima di essere analizzati
mediante citofluorimetro. Per ciascun campione è stata valutata una quantità minima di
10.000 cellule e i dati ottenuti sono stati analizzati mediante il programma di analisi
“Multicycle Cell Cyle” (Phoenix Flow System, San Diego, CA), basato sul modello
matematico proposto da Dean P.N. e Jett J.H. (Dean P.N. and Jett J.H., 1974).

6. Risultati
6. RISULTATI

6. Risultati
-88-
In questa sezione vengono descritti i risultati ottenuti attraverso lo studio dei due modelli
prescelti, ovvero cellule muscolari di topo, nell’ambito del processo di differenziamento in
vivo e in vitro, e cellule umane durante l’infezione da citomegalovirus in vitro.
Il denominatore comune nell’ambito dei suddetti modelli, così distanti tra loro, è
rappresentato dai proteasomi, con particolare riguardo alla distribuzione degli stessi nel
corso delle due condizioni sopra citate, di cui l’una (differenziamento muscolare)
fisiologica e l’altra (infezione virale) patologica. L’intento di tali studi è stato quello di
mettere in rilievo, a cominciare dagli aspetti morfologici, quello che, come già dettagliato
nella sezione introduttiva, dovrebbe essere un ruolo di spicco dei proteasomi nel controllo
di processi cruciali per la cellula, quale il ciclo cellulare, il cui arresto è fondamentale
nell’ambito del differenziamento e la cui alterazione è una delle strategie vincenti
applicate da citomegalovirus per “sbaragliare” la cellula ospite durante il ciclo litico.
Altro progetto ambizioso a cui si è cercato di dare l’avvio attraverso questo studio
riguarda la messa a punto di strumenti molecolari da potere utilizzare, in un’ottica futura,
per studi sui proteasomi, analoghi a quelli presi in considerazione in questa sede, ma da
effettuare in cellule viventi.
6.1 PRIMO MODELLO DI STUDIO: CELLULE MUSCOLARI E PROTEASOMI
Le osservazioni sperimentali riportate in questa prima parte si riferiscono innanzitutto
allo studio sulla localizzazione di specifici proteasomi nel tessuto muscolare scheletrico di
ratto. Esse sono state effettuate sia in sezioni sottili di muscoli scheletrici, che a partire da
miofibrille isolate dai suddetti muscoli, attraverso l’impiego di tecniche di microscopia
elettronica, microscopia a fluorescenza e microscopia confocale.
Inoltre, attraverso tecniche di ingegneria genetica, sono stati messi a punto e validati
specifici strumenti molecolari atti all’ottenimento di cloni/popolazioni di cellule di topo
in grado di esprimere stabilmente specifiche subunità di proteasomi “marcate” con la
proteina verde naturale EGFP, allo scopo di creare le basi per studi futuri sui proteasomi,
a valenza sia morfologica che funzionale, in cellule muscolari viventi.
6.1.1 I PROTEASOMI SONO DISTRIBUITI SECONDO UN PROFILO PSEUDO-SARCOMERICO NEL
MUSCOLO SCHELETRICO
Precedenti osservazioni ottenute dal nostro gruppo di ricerca e da quello del Prof. Jean
Foucrier, relative al processo di differenziamento di cellule muscolari di topo e di cellule
satelliti di ratto in vitro (De Conto F. et al., 1997; Foucrier J. et al., 1999), hanno messo in

6. Risultati
-89-
evidenza una distribuzione nucleare e/o citoplasmatica di specifici proteasomi,
dipendentemente dalle varie fasi del processo di differenziamento, con particolare
riguardo ad un’organizzazione pseudo-sarcomerica nell’ambito delle tappe finali del
suddetto processo. Queste osservazioni preliminari sono state confermate da studi
successivi, relativi alla localizzazione del proteasoma 20S in sezioni sottili longitudinali
del muscolo scheletrico di ratto e in colture primarie di cardiomiociti ventricolari di ratto,
che hanno anch’essi avvalorato l’esistenza di una caratteristica distribuzione pseudo-
sarcomerica del proteasoma 20S (Foucrier J. et al., 2001).
Alla luce di questi risultati, è sembrato rilevante, oltre che un naturale seguito rispetto a
quanto precedentemente intrapreso, approfondire le conoscenze al riguardo attraverso
l’acquisizione di dati che potessero fornire utili informazioni sulla precisa distribuzione
dei proteasomi nell’ambito del sarcomero, così come sulle possibili interazioni tra questi
ultimi e specifici elementi dell’apparato contrattile muscolare.
Al fine di stabilire con maggiore precisione la localizzazione dei proteasomi nell’ambito
della struttura sarcomerica del muscolo scheletrico, sono state utilizzate sezioni sottili di
muscolo scheletrico di ratto (Extensor Digitorum Longus, EDL); lo studio è stato
inizialmente effettuato attraverso l’impiego di immunoelettromicroscopia. Per quel che
riguarda la tecnica impiegata, l’immunoreazione è stata effettuata prima di sottoporre a
fissazione e ad inclusione i preparati (“pre-embedding”); la localizzazione della subunità
α1/p27k del proteasoma 20S è stata infine rivelata mediante reazione immunoenzimatica
(immunoperossidasi). L’osservazione di tali preparati [Figura 1, pannelli A e B] ha
evidenziato la presenza della subunità α1/p27k, attraverso il rilevamento di precipitati di
3,3-diamminobenzidina (DAB, substrato dell’enzima perossidasi) a livello del sarcomero.
In particolare, sono state individuate due aree principali di positività, concentrate
all’interfaccia tra le bande A ed I, anche se il segnale risultava leggermente diffuso
nell’ambito di tutta la banda I.

6. Risultati
-90-
Figura 1: Localizzazione di α1/p27k in sezioni sottili del muscolo “Extensor Digitorum Longus “ (EDL) di ratto, rilevata mediante immunoelettromicroscopia (tecnica di “pre-embedding”); la reazione immunoenzimatica (immunoperossidasi) è stata rivelata attraverso l’aggiunta del substrato 3,3-diamminobenzidina (DAB); le aree di positività sono evidenziate dalla presenza di precipitati di colore grigio intenso. Per confronto con il controllo (pannello B), il precipitato di DAB, che si osserva nel pannello A è rilevato in associazione alla banda I e, in particolare, all’interfaccia tra le bande A e I (barrette bianche orizzontali). Al contrario, la regione centrale della banda A è priva di precipitati. Barra: 1 µm.
6.1.2 IL PROFILO PSEUDO-SARCOMERICO DI SPECIFICI PROTEASOMI SI MODIFICA IN
RAPPORTO ALL’ESTENSIONE DEL SARCOMERO IN VIVO
Questa serie di esperimenti è stata effettuata su sezioni sottili di muscolo EDL di ratto,
attraverso l’impiego di una tecnica di doppia colorazione in immunofluorescenza, al fine
di individuare i possibili elementi del sarcomero più strettamente associati a specifici
proteasomi. Allo scopo, sono state impiegate sonde immunologiche in grado di
riconoscere la subunità α1/p27K dei proteasomi, assieme a quelle ritenute idonee ad
evidenziare le proteine sarcomeriche di interesse.
In un primo tempo, gli anticorpi anti-α1/p27K sono stati utilizzati in concomitanza a
quelli diretti contro la proteina desmina, che caratterizza i filamenti intermedi del
citoscheletro associati alle linee Z (che delimitano, com’è noto, il sarcomero) [Figura 2,
pannelli A-F].
I risultati ottenuti confermano innanzitutto l’esistenza di una distribuzione spaziale dei
proteasomi, che ricalca quella sarcomerica [Figura 2, pannelli B e E, in rosso]; altro dato
rilevante è che, mentre in alcune delle sezioni analizzate la loro distribuzione si appalesa
in un’unica banda [Figura 2, pannello B, punta di freccia], in altre, essa consta di una
doppia banda [Figura 2, pannello E, freccia]. D’altra parte, il profilo in fluorescenza di
desmina, utilizzata come marcatore della linea Z, è rappresentato da un’unica banda,
come atteso [Figura 2, pannelli A e D, in verde]. Per quel che riguarda lo studio di
colocalizzazione tra α1/p27K (proteasoma) e desmina, realizzato attraverso la
sovrapposizione delle immagini in rosso (proteasoma) ed in verde (desmina) [Figura 2,
pannelli C ed F], esso ha messo in evidenza che, nel caso del profilo dei proteasomi a

6. Risultati
-91-
singola banda [Figura 2, pannello B], la corrispondente immagine di colocalizzazione
[Figura 2, pannello C] mostra i colori rosso (proteasoma) e verde (desmina) ancora
nettamente distinti. In altre parole, questo equivale a dire che i proteasomi sembrano
distribuiti nello spazio tra le linee Z e non sono ad esse associati. Anche nel caso di
distribuzione dei proteasomi a doppia banda [Figura 2, pannello E], l’immagine di
colocalizzazione tra i canali del rosso e del verde [Figura 2, pannello F] evidenzia come le
linee Z sembrino ancora discernibili (conservando quasi integralmente il loro colore
originale), confermando una distribuzione dei proteasomi da esse indipendente. Simili
osservazioni sono state confermate utilizzando diversi metodi di fissazione (metanolo a
freddo, acetone e paraformaldeide), così come in preparati non fissati.
Figura 2: Il profilo di distribuzione di α1/p27k non coincide con quello di desmina. Doppia colorazione in immunofluorescenza: profilo di distribuzione di α1/p27k in associazione a desmina, in sezioni sottili di muscolo EDL di ratto. Pannelli A e D: desmina; pannelli B ed E: α1/p27k; pannelli C ed F: immagini derivanti dalla sovrapposizione di A+B e D+E, rispettivamente. La punta di freccia nel pannello B e la freccia nel pannello E indicano il profilo a banda singola e a doppia banda, rispettivamente, di α1/p27k. Barra: 10 µm.
Il rilevamento di un profilo di distribuzione differenziato dei proteasomi, principalmente
evidenziabile in una singola o in una doppia banda, ha portato ad ipotizzare che queste
differenze potessero essere legate alla lunghezza del sarcomero, che varia
dipendentemente dallo stato di contrazione del muscolo.

6. Risultati
-92-
Per cercare di dirimere il quesito, è stata effettuata una seconda serie di esperimenti di
doppia marcatura in immunofluorescenza su sezioni sottili di muscolo EDL di ratto,
calcolando, in questo caso, la lunghezza media dei sarcomeri su cui è stata effettuata la
suddetta reazione di immunofluorescenza. Quest’ultima è stata eseguita sempre mediante
l’impiego di anticorpi diretti contro la subunità α1/p27K di proteasoma, utilizzati assieme
a sonde immunologiche in grado di riconoscere la proteina titina [Figura 3, pannelli A-C].
In particolare, per quanto concerne quest’ultimo anticorpo, si è scelto di utilizzare il clone
9D10, dal momento che l’epitopo riconosciuto da esso è localizzato a livello di una regione
della molecola di titina la cui distribuzione spaziale nell’ambito del sarcomero risulta
essere più vicina all’interfaccia tra la banda A e la banda I, piuttosto che alla linea Z
(Greaser M.L. et al., 2000). La suddetta regione, riconosciuta dall’anticorpo 9D10, è
denominata “PEVK”, in funzione dei residui aminoacidici da cui è principalmente
costituita (P: prolina; E: acido glutammico; V: valina; K: lisina). Il segmento “PEVK” che,
come già accennato, è localizzato in prossimità della regione centrale della banda I
(porzione intra-sarcomerica), è costituito da un’unica sequenza di 2174 residui
aminoacidici, la cui funzione, nell’ambito della struttura primaria della molecola di titina,
è quella di moderare l’elevato grado di distensione del sarcomero; tale anticorpo è stato
pertanto largamente impiegato quale valido mezzo per studiare la capacità di estensione
dei muscoli scheletrico e cardiaco (Trombitás K. et al., 1998).
I risultati del suddetto esperimento di doppia colorazione in immunofluorescenza hanno
messo in evidenza che sia il profilo di fluorescenza della regione “PEVK” di titina [Figura
3, serie di pannelli A, in verde], che quello di α1/p27K [Figura 3, serie di pannelli B, in
rosso], variano a seconda della lunghezza del sarcomero. In particolare, nei sarcomeri più
corti il profilo di fluorescenza di titina si estrinseca in un’unica banda, in cui si evidenzia a
volte una sorta di micro-fessurazione centrale [Figura 3, pannello A, sarcomero di 1,66
µm; la freccia indica la micro-fessurazione]; le corrispondenti immagini in contrasto di
fase (non riportate in questa sede) hanno permesso di appurare che questa banda si
localizza nelle immediate vicinanze della linea Z. Come atteso e a controprova della
suddetta osservazione, il profilo di distribuzione di α1/p27K nello stesso sarcomero,
anch’esso evidenziabile in un’unica banda [Figura 3, pannello B, sarcomero di 1,66 µm],
non colocalizza con titina [Figura 3, pannello C, sarcomero di 1,66 µm]. In quest’ultimo
caso, l’immagine in contrasto di fase ha permesso di identificare la zona di distribuzione
di α1/p27K a livello della linea M.

6. Risultati
-93-
Nei sarcomeri più lunghi è stata osservata una diversa distribuzione sia di titina che di
α1/p27K, entrambe evidenziabili come doppie bande [Figura 3, pannelli A e B,
rispettivamente; lunghezze dei sarcomeri: 1,81 µm, 2,26 µm, 2,39 µm e 2,69 µm].
Le suddette doppie bande sono non solo sempre più chiaramente discernibili man mano
che il sarcomero considerato aumenta di lunghezza, ma diventano anche piuttosto bene
colocalizzate nei sarcomeri più lunghi, come evidenziato dal colore giallo nel pannello C
(sarcomeri di 2,39 e 2,69 µm). In particolare, e come già più volte ribadito, in condizioni di
muscolo più disteso (sarcomeri più lunghi) la regione “PEVK” di titina è localizzata
all’interfaccia tra le bande A ed I; anche in questo caso (come per quello del sarcomero più
corto), la collocazione spaziale delle suddette bande sarcomeriche è stata confermata
attraverso l’osservazione delle corrispondenti immagini in contrasto di fase (non riportate
in questa sede).
Figura 3: Il profilo di distribuzione di α1/p27k dipende dall’estensione del sarcomero in vivo. Doppia colorazione in immunofluorescenza: profilo di distribuzione di α1/p27k in associazione a titina, in sezioni sottili di muscolo EDL di ratto. I pannelli A e B corrispondono a titina e ad α1/p27k, rispettivamente; nel pannello C sono evidenziate le aree di colocalizzazione (in giallo) tra titina e α1/p27k. Il valore della lunghezza del sarcomero (± ESM) è indicata sotto le corrispondenti immagini delle suddette serie. La doppia banda di titina non è rilevabile nei sarcomeri molto corti (la freccia nella prima immagine della serie di pannelli A mostra la micro-fessurazione nella banda di titina), mentre si vede molto bene man mano che la lunghezza del sarcomero aumenta. La freccia nella serie di pannelli B (immagine con sarcomero di lunghezza di circa 2,39 µm) indica un profilo a doppia banda di α1/p27k. Le punte di freccia a livello delle immagini colocalizzate (serie di pannelli C) indicano la posizione della linea Z, individuata attraverso l’osservazione delle corrispondenti immagini in contrasto di fase. Barra: 10 µm.
6.1.3 UNA SIGNIFICATIVA QUANTITÀ DI PROTEASOMI È ASSOCIATA ALLE MIOFIBRILLE
Le osservazioni sperimentali effettuate nel modello murino in vivo e riportate nei
paragrafi precedenti, sicuramente rafforzano l’ipotesi di una stretta interazione tra
proteasomi e proteine intimamente connesse alla struttura e, verosimilmente, alle funzioni
del sarcomero. In particolare, la distribuzione dei proteasomi concentrata principalmente
A
B
C

6. Risultati
-94-
in una o due bande, a loro volta dislocate alternativamente nella zona centrale, a livello
della linea M (singola banda intra-sarcomerica) in sarcomeri più corti, o tra la banda A e la
banda I (doppia banda intra-sarcomerica) in sarcomeri allungati, suggerisce, anche a
conferma di osservazioni precedenti su modelli di cellule muscolari (De Conto F. et al.,
1997; Foucrier J. et al., 1999; Foucrier J. et al., 2001), così come di altre tipologie cellulari
(Arcangeletti M.C. et al., 1997), una possibile interazione tra proteasomi e filamenti di
actina. Nello specifico, a livello della struttura sarcomerica, in cui lo scorrimento dei
filamenti di actina negli spazi esistenti tra gli stessi ed i filamenti di miosina determina un
accorciamento dei sarcomeri nell’ambito della contrazione muscolare, i proteasomi
sarebbero associati con una regione prossima all’estremità a polarità negativa dei
filamenti di actina, ossia nelle strette vicinanze della zona centrale del sarcomero.
Al fine di confermare ulteriormente l’esistenza di tale interazione, si è passati ad effettuare
un analogo studio in vitro, utilizzando preparati di miofibrille isolate a partire da muscoli
di ratto. In una fase preliminare, sono stati effettuati diversi controlli atti ad appurare
l’adeguatezza dei preparati. Innanzitutto, mediante osservazione al microscopio in
contrasto di fase è stato possibile rilevare, a livello delle singole miofibrille, il classico
profilo striato che caratterizza il tessuto muscolare scheletrico; mediante l’impiego di
sonde immunologiche dirette nei confronti dei principali componenti del sarcomero quali
miosina e titina e, in parallelo, di sostanze quali falloidina, per rilevare la presenza di
actina filamentosa, è stato possibile valutare la bontà dei preparati di miofibrille sulla base
della localizzazione spaziale delle suddette proteine nell’ambito della struttura
sarcomerica.
Inoltre, nei preparati di miofibrille è stata valutata anche l’eventuale presenza di elementi
non prettamente miofibrillari, mediante l’impiego di anticorpi diretti nei confronti della
proteina desmina e dei recettori Serca-1 e rianodina del reticolo sarcoplasmatico. Tali
componenti sono stati rilevati solo in numero esiguo di miofibrille. Infine, la presenza
della subunità di proteasoma α1/p27k, rilevata mediante immunofluorescenza, è stata
accertata in tutte le miofibrille esaminate.
Utilizzando tali preparazioni di miofibrille come modello di studio, è stato possibile
riprodurre innanzitutto le osservazioni effettuate in vivo (sezioni sottili di muscolo), in
particolare quelle relative al rapporto esistente tra il profilo di distribuzione del segnale di
fluorescenza della subunità α1/p27k e la lunghezza del sarcomero [Figure 4 e 5].
Allo scopo di quantificare questo fenomeno, le miofibrille sono state sottoposte sia ad
osservazione mediante microscopio a contrasto di fase, che a reazione di

6. Risultati
-95-
immunofluorescenza, utilizzando un anticorpo monoclonale diretto nei confronti della
subunità α1/p27k dei proteasomi e, simultaneamente, falloidina, che si lega
selettivamente ai filamenti di actina. Per la realizzazione di tale esperimento, sono state
prese in considerazione 44 miofibrille, scelte in modo del tutto casuale; nell’ambito delle
stesse, è stata misurata la lunghezza di 442 sarcomeri, valutando la distanza tra le linee Z
mediante microscopia in contrasto di fase (si veda l’esempio delle quattro porzioni di
miofibrille, con sarcomeri di diversa lunghezza, nelle immagini in contrasto di fase
riportate sopra il grafico di Figura 4). I valori di lunghezza, di tutti i sarcomeri presi in
considerazione, sono riportati lungo l’asse delle ascisse del grafico di Figura 4, mentre
nella parte alta dello stesso grafico compaiono, in parentesi e in corrispondenza di ogni
valore di lunghezza, il numero di sarcomeri analizzati. Attraverso l’analisi dei profili di
fluorescenza di α1/p27k [in rosso nelle quattro tipologie di sarcomeri riportate, in
maniera esemplificativa, sopra il grafico di Figura 4, pannello A], è stata inoltre misurata
sia la distanza tra le bande di proteasomi (α1/p27k) all’interno di un singolo sarcomero
[riportate con simboli blu nel grafico di Figura 4, pannello B], che la distanza tra due
bande proteasomali disposte ai due lati di una linea Z, ossia bande dislocate in due
sarcomeri adiacenti [riportate con simbolo giallo nel suddetto grafico]. Alla misura della
distanza tra bande di α1/p27k intra-sarcomeriche è stato attribuito un valore 0
(approssimando i valori reali di frazioni di micron), quando il profilo di fluorescenza
indicava la presenza di un’unica banda.
Questo risulta apprezzabile, in effetti, sia nelle immagini che nella rappresentazione
grafica di Figura 4, in cui la distanza tra bande intra-sarcomeriche (simbolo in blu) ha
valore 0 per sarcomeri di lunghezza ≤ a circa 2 µm e nei quali il profilo di α1/p27k si
presentava, come già accennato, sottoforma di una sola banda (a livello della linea M),
come evidenziato anche, in maniera esemplificativa, nell’immagine in contrasto di fase e
nel relativo profilo di fluorescenza in rosso nell’ambito del sarcomero che misura 1,95 µm
[Figura 4, pannello A].
Per quel che riguarda queste stesse tipologie di sarcomeri “corti”, le corrispondenti
distanze tra le bande di α1/p27k situate in due sarcomeri adiacenti (ai due lati di una
linea Z) hanno valori diversi dallo 0 (nel grafico, simboli gialli relativi a sarcomeri con
lunghezza ≤ a circa 2 µm). Tali valori risultano essere strettamente correlati alla lunghezza
del sarcomero (si confronti, ad esempio, la misura di bande proteasomali tra sarcomeri
adiacenti, per sarcomeri lunghi 1,7 µm, il cui corrispondente simbolo giallo risulta,
anch’esso, della stessa misura, come desumibile dall’asse delle ordinate destro, o si

6. Risultati
-96-
confronti anche quello di lunghezza 2 µm con relativo simbolo giallo situato in
corrispondenza della stesso valore), come era logico attendersi, dal momento che i
proteasomi appaiono concentrati in un’unica banda nella zona centrale del sarcomero. La
situazione appare decisamente diversa per sarcomeri più estesi (lunghezza ≥ 2,3 µm); in
questo caso, come desumibile dalle immagini in fluorescenza di α1/p27k di Figura 4 e
dalla disposizione dei simboli blu e gialli nella rappresentazione grafica della stessa
Figura, il profilo di distribuzione dei proteasomi passa da una a due bande intra-
sarcomeriche e, di conseguenza, aumenta la distanza tra bande di α1/p27k all’interno del
sarcomero (simboli blu per sarcomeri delle suddette dimensioni). Concomitantemente,
come evidenziato dai rispettivi simboli gialli, diminuisce la distanza tra bande
proteasomali che fiancheggiano la linea Z (situate in due sarcomeri adiacenti).
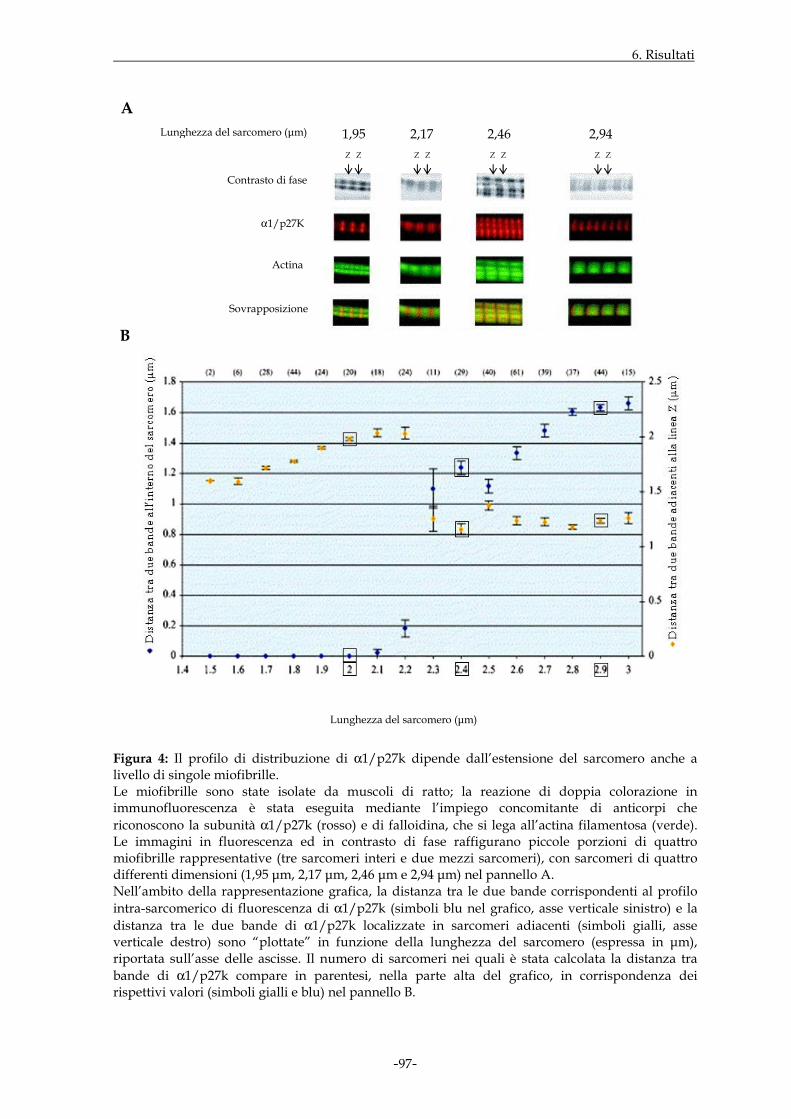
6. Risultati
-97-
Figura 4: Il profilo di distribuzione di α1/p27k dipende dall’estensione del sarcomero anche a livello di singole miofibrille. Le miofibrille sono state isolate da muscoli di ratto; la reazione di doppia colorazione in immunofluorescenza è stata eseguita mediante l’impiego concomitante di anticorpi che riconoscono la subunità α1/p27k (rosso) e di falloidina, che si lega all’actina filamentosa (verde). Le immagini in fluorescenza ed in contrasto di fase raffigurano piccole porzioni di quattro miofibrille rappresentative (tre sarcomeri interi e due mezzi sarcomeri), con sarcomeri di quattro differenti dimensioni (1,95 µm, 2,17 µm, 2,46 µm e 2,94 µm) nel pannello A. Nell’ambito della rappresentazione grafica, la distanza tra le due bande corrispondenti al profilo intra-sarcomerico di fluorescenza di α1/p27k (simboli blu nel grafico, asse verticale sinistro) e la distanza tra le due bande di α1/p27k localizzate in sarcomeri adiacenti (simboli gialli, asse verticale destro) sono “plottate” in funzione della lunghezza del sarcomero (espressa in µm), riportata sull’asse delle ascisse. Il numero di sarcomeri nei quali è stata calcolata la distanza tra bande di α1/p27k compare in parentesi, nella parte alta del grafico, in corrispondenza dei rispettivi valori (simboli gialli e blu) nel pannello B.
A Lunghezza del sarcomero (µm) 1,95 2,17 2,46 2,94
Z Z Z Z Z Z Z Z
Contrasto di fase
α1/p27K
Actina
Sovrapposizione
Lunghezza del sarcomero (µm)
B

6. Risultati
-98-
Questa serie di osservazioni ha portato alla formulazione di un modello interpretativo di
tali evidenze sperimentali, illustrato in Figura 5, in cui sono schematizzate tre tipologie
rappresentative di sarcomeri, di lunghezza pari a 2,9 µm (esteso) in (A), a 2,4 µm
(lunghezza intermedia) in (B) e a 2 µm (“corto”) in (C). A lato vengono anche riportate le
corrispondenti immagini in contrasto di fase, in cui sono visibili le linee Z (evidenziate da
frecce) e la banda A, così come immagini che mostrano il profilo in fluorescenza della
subunità α1/p27k dei proteasomi; in queste ultime, in particolare, la lettera “a” evidenzia
la distanza tra bande di proteasomi intra-sarcomeriche, mentre “b” e “c” quelle tra bande
dislocate in sarcomeri adiacenti. È da sottolineare che le misure effettuate in “b” e in “c”
per tutti i sarcomeri considerati nei preparati di miofibrille, sono risultate sovrapponibili,
a dimostrazione di una distribuzione ripetitiva e regolare delle aree di accumulo (bande)
dei proteasomi.
Nella rappresentazione schematica, le bande proteasomali sono rappresentate in
rosso/arancio, i filamenti di actina in verde, quelli di miosina (delimitanti la banda A) in
azzurro ed infine, le linee Z in nero. Le misure delle distanze tra bande di α1/p27k intra-
sarcomeriche e tra sarcomeri adiacenti sono quelle evidenziate nei riquadri neri,
rispettivamente, di Figura 4, per i sarcomeri di lunghezza corrispondente (valori
sottolineati in ascissa).
È interessante notare che, nel sarcomero di dimensioni intermedie [Figura 5, pannello B]
ed in quello esteso [Figura 5, pannello A], a fronte di aumentate distanze tra bande intra-
sarcomeriche (1,1 e 1,6 µm, rispettivamente), la distanza tra bande di proteasoma di
sarcomeri adiacenti (che fiancheggiano una linea Z) rimangono invariate (circa 1,25 - 1,3
µm). Quest’ultimo valore è risultato paragonabile a quello ottenuto per i filamenti di
actina, la cui lunghezza è stata stimata essere di circa 1,2 µm (dall’estremità a polarità
positiva, situata a livello della linea Z, a quella ad estremità negativa, nella direzione della
banda A). Tale valore si desume misurando la zona di fluorescenza per l’actina (circa 2,4
µm), situata tra le bande H di due sarcomeri adiacenti (la banda H rappresenta la regione
centrale della banda A, sprovvista di filamenti di actina nei sarcomeri più estesi); è
evidente come la misura del suddetto segnale di fluorescenza corrisponda a quella di due
filamenti di actina. L’insieme di queste osservazioni suggerisce, pertanto, che i proteasomi
possano interagire principalmente con la regione centrale dei filamenti di actina.
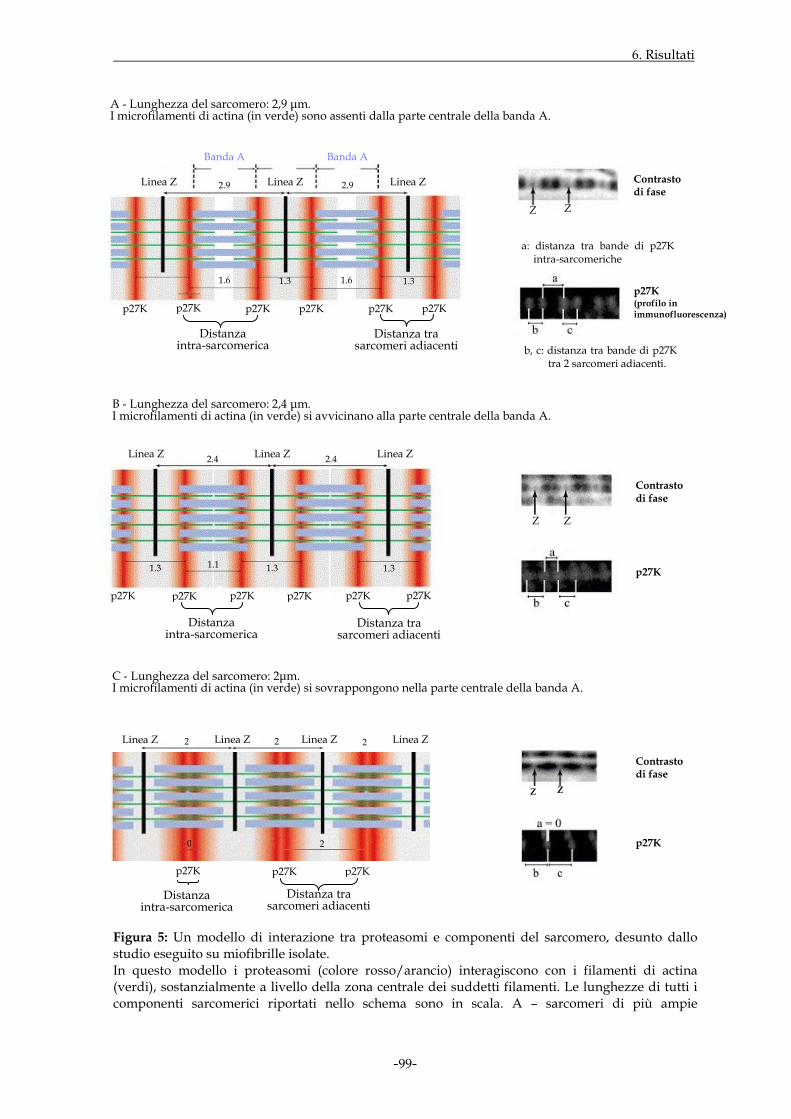
6. Risultati
-99-
Figura 5: Un modello di interazione tra proteasomi e componenti del sarcomero, desunto dallo studio eseguito su miofibrille isolate. In questo modello i proteasomi (colore rosso/arancio) interagiscono con i filamenti di actina (verdi), sostanzialmente a livello della zona centrale dei suddetti filamenti. Le lunghezze di tutti i componenti sarcomerici riportati nello schema sono in scala. A – sarcomeri di più ampie
A - Lunghezza del sarcomero: 2,9 µm. I microfilamenti di actina (in verde) sono assenti dalla parte centrale della banda A.
Linea Z Linea Z Linea Z
Banda A Banda A
2.9 2.9
1.3 1.3 1.6 1.6
Distanza intra-sarcomerica
Distanza tra sarcomeri adiacenti
p27K p27K p27K p27K p27K p27K
Z Z
Contrasto di fase
a: distanza tra bande di p27K intra-sarcomeriche
b, c: distanza tra bande di p27K tra 2 sarcomeri adiacenti.
p27K (profilo in immunofluorescenza)
B - Lunghezza del sarcomero: 2,4 µm. I microfilamenti di actina (in verde) si avvicinano alla parte centrale della banda A.
Linea Z Linea Z Linea Z 2.4 2.4
1.3 1.3 1.3 1.1
p27K p27K p27K p27K p27K p27K
Distanza intra-sarcomerica
Distanza tra sarcomeri adiacenti
Z Z
Contrasto di fase
p27K
C - Lunghezza del sarcomero: 2µm. I microfilamenti di actina (in verde) si sovrappongono nella parte centrale della banda A.
Distanza tra sarcomeri adiacenti
p27K p27K
Linea Z Linea Z Linea Z 2 2 2
0
Linea Z
2
p27K
Distanza intra-sarcomerica
p27K
Z Z
Contrasto di fase

6. Risultati
-100-
dimensioni: la lunghezza del sarcomero (misurata fra due linee Z) e l’ampiezza della banda A sono state desunte da immagini in contrasto di fase (Figura 4 e foto a lato), mentre le caratteristiche di α1/p27k e di actina sono state valutate sulla base dei loro rispettivi profili di fluorescenza (Figura 4 per actina e proteasoma e foto a lato per quest’ultimo). Nelle immagini che riportano il profilo in immunofluorescenza di α1/p27k, (a) indica la distanza tra le bande di α1/p27k all’interno del sarcomero, (b) e (c) la distanza tra le bande di α1/p27k tra due sarcomeri adiacenti (bande che fiancheggiano una delle due linee Z). B – Sarcomeri di dimensioni intermedie: quando il sarcomero si accorcia, la distanza tra le due bande di α1/p27k intra-sarcomeriche diminuisce, mentre la distanza tra bande presenti in sarcomeri adiacenti (che fiancheggiano una linea Z) rimane costante (uguale a quella rilevata in A: 1,3 µm). Questa situazione è rilevabile fino a quando i filamenti di actina si avvicinano senza sovrapporsi (B: sarcomeri lunghi circa 2,4 µm). C – Sarcomero “corto”: quando i filamenti di actina si sovrappongono nell’ambito della regione centrale del sarcomero, solo una banda α1/p27k è distinguibile nella regione mediana del sarcomero stesso.
Gli stessi preparati di miofibrille sono stati in seguito sottoposti ad
immunoelettromicroscopia (metodo “pre-embedding”), per studiare la localizzazione
della subunità α1/p27k nell’ambito del sarcomero, ad un più elevato livello di risoluzione
[Figura 6]. Nello specifico, è stato utilizzato un anticorpo monoclonale anti-α1/p27k;
l’immunoreazione è stata rivelata mediante un anticorpo anti-topo coniugato con
isotiocianato di fluoresceina (FITC). Il segnale veniva in seguito amplificato attraverso
l’utilizzo di un anticorpo anti-FITC, legato a particelle di oro colloidale ed ulteriormente
sottoposto ad amplificazione all’argento. La marcatura mediante particelle di oro
colloidale è stata utilizzata per combinare l’elevata amplificazione del segnale alla natura
discontinua e non diffusibile del prodotto utilizzato per la rivelazione (particelle di oro
colloidale). Questo procedimento è stato utilizzato in alternativa a quello applicato per
l’analisi elettromicroscopica di sezioni sottili di tessuto, mostrata in Figura 1
(immunoperossidasi), proprio al fine di ovviare alla diffusibilità di prodotti, quali i
substrati enzimatici, che potrebbero rendere più difficoltosa l’interpretazione dei risultati
di localizzazione dei complessi molecolari allo studio.
L’osservazione al microscopio elettronico dei preparati di miofibrille, previa marcatura
con oro colloidale e successiva inclusione in resina epossidica, ha chiaramente dimostrato
che la subunità α1/p27k dei proteasomi era presente all’interno della struttura
sarcomerica. Come atteso, nell’ambito dei sarcomeri corti [Figura 6, pannello A] i
proteasomi erano principalmente localizzati (accumulo di particelle d’oro) a livello della
linea M. Nella regione limitrofa alla linea M si potevano osservare dei rigonfiamenti che
apparivano, essi stessi, marcati con le particelle di oro colloidale, a testimonianza del fatto
che la subunità α1/p27k era associata anche a queste protrusioni.

6. Risultati
-101-
Figura 6: Distribuzione sarcomerica della subunità α1/p27k di proteasoma in preparati di miofibrille isolate, mediante immunoelettromicroscopia. Le miofibrille sono state isolate a partire da muscoli di ratto. È stato impiegato un anticorpo monoclonale anti-α1/p27k di proteasoma; l’immunoreazione è stata rivelata mediante un anticorpo secondario coniugato con FITC. Il segnale è stato successivamente amplificato attraverso l’utilizzo di un anticorpo anti-FITC, legato a particelle di oro colloidale. Dopo ulteriore amplificazione argentica, le miofibrille sono state incluse in Epon 812. La punta di freccia indica la posizione della linea Z che in queste due preparazioni non era particolarmente ben visibile. Barra: 1 µm. Pannello A – Sarcomero “corto” (lunghezza approssimativa: 1,5 µm). Il segnale positivo per la subunità α1/p27k dei proteasomi (accumulo di particelle di oro colloidale) è localizzato a livello della linea M. Si nota una sorta di rigonfiamento a livello di questa regione legato, probabilmente, alle protusioni che caratterizzano le estremità a polarità negativa dei filamenti di actina, localizzate a livello della banda A. Pannello B – Sarcomero esteso (lunghezza approssimativa: 2,7 µm). Il segnale è localizzato nell’ambito della banda I del sarcomero. È ravvisabile una possibile organizzazione di α1/p27k in bande multiple all’interno della banda I.
Nelle condizioni sperimentali adottate (l’immunoreazione era volta a mettere in evidenza
esclusivamente i proteasomi, senza concomitante rilevamento di specifiche proteine
sarcomeriche) non è stato possibile determinare con precisione la tipologia di filamento
con cui α1/p27k potesse essere associato. Tuttavia, l’accumulo di particelle d’oro anche a
livello delle protrusioni apprezzabili in vicinanza della linea M evocava l’effetto di
ingombro dovuto alla presenza delle estremità a polarità negativa dei filamenti di actina,
posizionati in direzione della banda A (e sovrapposti a livello della linea M nei sarcomeri
“corti”). Questa ipotesi suggerisce, a sua volta, che l’actina potesse essere implicata
nell’ancoraggio dei proteasomi alla struttura sarcomerica. Nel sarcomero esteso [Figura 6,
pannello B], la subunità α1/p27k dei proteasomi appariva localizzata a livello della banda
I, in associazione con strutture filamentose. In funzione della loro posizione nel sarcomero
e della loro abbondanza, le suddette strutture sono state interpretate come filamenti di
actina. La distribuzione di α1/p27k nella banda I non era uniforme ma localizzata in
distinte sotto-regioni, suggerendo l’esistenza di siti specifici e ripetitivi di attacco a

6. Risultati
-102-
strutture filamentose, non osservabili mediante immunofluorescenza o microscopia
confocale, probabilmente a causa del minore potere risolutivo di questi ultimi metodi.
6.1.4 NON TUTTI I PROTEASOMI PRESENTI NELLE MIOFIBRILLE SONO ASSOCIATI AD ACTINA
L’ipotesi secondo cui esisterebbe una interazione tra filamenti di actina e proteasoma 20S
è stata ulteriormente avvalorata attraverso i risultati ottenuti in esperimenti in cui l’actina
veniva estratta dalle miofibrille. Il metodo utilizzato prevedeva l’uso di un composto
chiamato gelsolina. Nei preparati di miofibrille sottoposti alla suddetta estrazione, è stata
osservata una importante riduzione del segnale di immunofluorescenza relativo alla
subunità α1/p27k dei proteasomi [Figura 7, pannello A], rafforzando l’ipotesi secondo la
quale l’actina rappresenterebbe, in maniera diretta o indiretta, un sito di legame per il
proteasoma.
Tuttavia, è stato osservato che alcuni proteasomi, nonostante il trattamento di rimozione
dell’actina, rimanevano localizzati a livello del sarcomero. Inoltre, tali proteasomi residui
presentavano ancora una organizzazione pseudo-sarcomerica, indipendentemente dalla
presenza dell’actina. Questa osservazione è stata confermata mediante analisi dei
preparati di miofibrille, dopo estrazione dell’actina, in microscopia confocale [Figura 7,
pannelli B e C]; anche tali immagini mettono in evidenza una organizzazione pseudo-
sarcomerica da parte dei proteasomi, in assenza di una apprezzabile organizzazione
sarcomerica dell’actina.
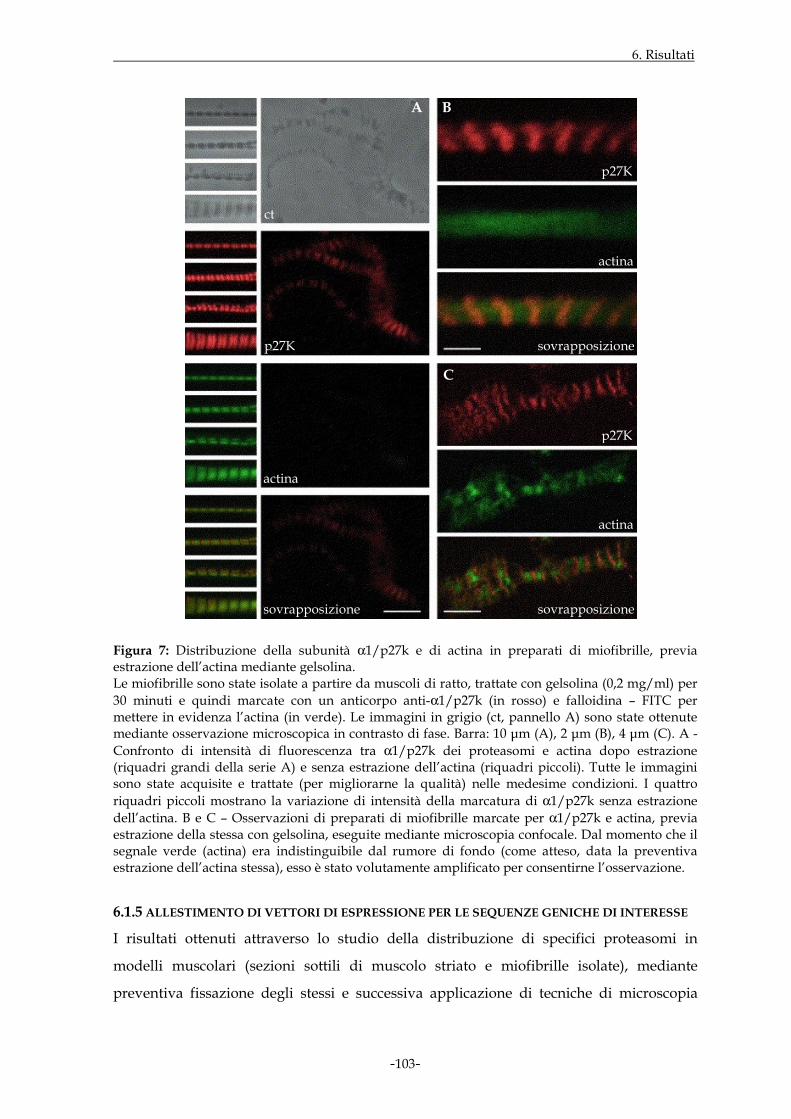
6. Risultati
-103-
Figura 7: Distribuzione della subunità α1/p27k e di actina in preparati di miofibrille, previa estrazione dell’actina mediante gelsolina. Le miofibrille sono state isolate a partire da muscoli di ratto, trattate con gelsolina (0,2 mg/ml) per 30 minuti e quindi marcate con un anticorpo anti-α1/p27k (in rosso) e falloidina – FITC per mettere in evidenza l’actina (in verde). Le immagini in grigio (ct, pannello A) sono state ottenute mediante osservazione microscopica in contrasto di fase. Barra: 10 µm (A), 2 µm (B), 4 µm (C). A - Confronto di intensità di fluorescenza tra α1/p27k dei proteasomi e actina dopo estrazione (riquadri grandi della serie A) e senza estrazione dell’actina (riquadri piccoli). Tutte le immagini sono state acquisite e trattate (per migliorarne la qualità) nelle medesime condizioni. I quattro riquadri piccoli mostrano la variazione di intensità della marcatura di α1/p27k senza estrazione dell’actina. B e C – Osservazioni di preparati di miofibrille marcate per α1/p27k e actina, previa estrazione della stessa con gelsolina, eseguite mediante microscopia confocale. Dal momento che il segnale verde (actina) era indistinguibile dal rumore di fondo (come atteso, data la preventiva estrazione dell’actina stessa), esso è stato volutamente amplificato per consentirne l’osservazione.
6.1.5 ALLESTIMENTO DI VETTORI DI ESPRESSIONE PER LE SEQUENZE GENICHE DI INTERESSE
I risultati ottenuti attraverso lo studio della distribuzione di specifici proteasomi in
modelli muscolari (sezioni sottili di muscolo striato e miofibrille isolate), mediante
preventiva fissazione degli stessi e successiva applicazione di tecniche di microscopia
ct
A B
p27K
actina
sovrapposizione p27K
p27K
actina
sovrapposizione
actina
sovrapposizione
C

6. Risultati
-104-
elettronica, a fluorescenza e confocale, hanno permesso di mettere in evidenza l’esistenza
di una stretta associazione tra specifici proteasomi e componenti di rilievo nell’ambito
della struttura contrattile del muscolo striato (filamenti di actina).
D’altra parte, l’utilizzo di preparati sottoposti a fissazione risulta poco conciliabile con la
possibilità di seguire nel tempo un processo dinamico, quale appunto il differenziamento
muscolare, nella consapevolezza che i procedimenti di fissazione potrebbero, da un lato,
non essere esenti da artefatti legati alla tipologia di fissazione utilizzata e, dall’altro,
mascherare alcuni epitopi bersaglio degli anticorpi utilizzati.
Pertanto, al fine di garantire un ulteriore approfondimento delle conoscenze in questo
ambito, la fase successiva della ricerca è stata focalizzata sulla messa a punto di strumenti
molecolari da potere utilizzare, in un’ottica futura, per studi sui proteasomi analoghi a
quelli presi in considerazione in questa sede, ma da effettuare in cellule viventi.
A tal fine sono stati innanzitutto costruiti vettori di espressione contenenti sequenze
geniche codificanti per specifiche subunità proteasomali (α1/p27k e β4/p23k), accoppiate
a sequenze geniche codificanti per proteine fluorescenti naturali, quali la proteina
fluorescente verde (GFP, in particolare la variante EGFP, di 27kDa) o, alternativamente, la
proteina fluorescente rossa (DsRed, variante DsRed1, di 26kDa). Si fa presente che, sulla
base del nomenclatore attualmente in uso (Coux O. et al., 1994; Baumeister W. et al., 1998),
il gene codificante per la subunità α1/p27k del proteasoma 20S viene chiamato PSMA6,
mentre quello codificante per la subunità del proteasoma 20S β4/p23k è denominato
PSMB2: pertanto, le sequenze di interesse verranno così siglate in questa sede.
Si sottolinea inoltre che, come già dimostrato (Coux O. et al., 1994) e come ulteriormente
evidenziato anche in questa sezione, le subunità proteasomali, quali quelle prese in
considerazione in questo contesto, vengono rapidamente assemblate nell’ambito del
complesso molecolare 20S (e non si ritrovano come subunità libere). Pertanto, gli studi
biochimici e morfologici presentati, basati nella fattispecie sulle subunità proteasomali
α1/p27k e β4/p23k, fanno in realtà riferimento al complesso molecolare 20S.
Per l’allestimento dei suddetti vettori di espressione si è scelto di collocare la proteina
fluorescente prescelta (EGFP o DsRed1) all’estremità C-terminale nella proteina di
fusione. Tale scelta è legata all’esigenza di preservare sia la conformazione che l’attività
enzimatica del proteasoma 20S. In effetti, applicando la suddetta strategia, una volta che
la proteina di fusione (costituita da una delle subunità proteasomali prescelte per lo
studio e dalla proteina fluorescente EGFP o, alternativamente, DsRed1) viene assemblata
nell’ambito del proteasoma 20S, la proteina fluorescente risulta orientata all’esterno del

6. Risultati
-105-
complesso. Tale strategia ha portato a scegliere vettori di espressione, che possiedono un
sito di clonaggio multiplo localizzato a monte delle sequenze che codificano per le
proteine fluorescenti (pEGFP-N1 e pDsRed1-N1).
Tali vettori sono detti di tipo “N”, in quanto la sequenza genica di interesse, in questo
caso quella per una subunità di proteasoma, viene orientata a partire dalle triplette che
codificano per una sua estremità N-terminale, in modo tale che la proteina fluorescente, la
cui sequenza è a valle rispetto alla prima, sia localizzata all’estremità C-terminale.
L’espressione delle suddette sequenze geniche è sotto il controllo del promotore per i geni
precocissimi di citomegalovirus umano (HCMV).
Nella prima fase sperimentale, mRNA provvisti di code di poli(A), contenuti in un
preparato commerciale di RNA totale ottenuto da placenta umana, sono stati
retrotrascritti in cDNA mediante l’impiego di trascrittasi inversa; come molecole innesco
(“primers”) sono stati utilizzate corte sequenze nucleotidiche di timidina (oligo-dT). Le
sequenze codificanti le subunità α1/p27K e β4/p23K sono state successivamente
amplificate mediante reazione polimerasica a catena (PCR), a partire dalla suddetta
miscela di cDNA, attraverso l’impiego di due oligonucleotidi, “primers”, specifici per
ciascuna subunità; in questo modo sono stati ottenuti specifici frammenti di DNA
contenenti le sequenze che codificano per le subunità prescelte. La sequenza innesco
all’estremità 5’ [Figura 8, scritte in blu] presenta il codone di inizio per la traduzione ATG
(in verde) e 5 nucleotidi a monte. Un sito per l’enzima di restrizione Hind III è stato creato
all’estremità 5’ del frammento (sequenza sottolineata in blu), per consentire la successiva
inserzione del medesimo nei vettori di espressione. La sequenza innesco all’estremità 3’
consente di sopprimere il codone di stop TAA e di creare un sito di taglio per l’enzima di
restrizione EcoR I (sequenza sottolineata in nero), al fine di permetterne l’inserzione nel
vettore di espressione e, inoltre, di assicurare la traduzione dei polipeptidi fluorescenti
(EGFP o DsRed1) nella stessa fase di lettura (“in frame”) delle subunità del proteasoma.

6. Risultati
-106-
a) Gene PSMA6 (codifica per la proteina α1/p27K)
- Estremità 5’ ataagcttCCAACATGTCCCGTGGTTCCAGCGCCGGTTTTGACCGCCACATTACCATTTTTTCA----------------
M S R G S S A G F D R H I T I F S ----------------
TattcgaaGGTTGTACAGGGCACCAAGGTCGCGGCCAAAACTGGCGGTGTAATGGTAAAAAAGT----------------
- Estremità 3’
(TAA)
----------------------------------------TGCTCACCTTGTTGCTCTAGCAGAGAGAGACggaattcgc 760
---------------------------------------- A H L V A L A E R D G I
----------------------------------------ACGAGTGGAACAACGAGATCGTCTCTCTCTGccttaagcg
b) Gene PSMB2 (codifica per la proteina β4/p23K)
- Estremità 5’ ataagcttCCAGCATGGAGTACCTCATCGGTATCCAAGGCCCCGACTATGTTCTTGTCGCCTCC----------------
M E Y L I G I Q G P D Y V L V A S ----------------
tattcgaaGGTCGTACCTCATGGAGTAGCCATAGGTTCCGGGGCTGATACAAGAACAGCGGAGG----------------
- Estremità 3’ (TAA)
---------------TTGACAAAAATGGCATCCATGACCTGGATAACATTTCCTTCCCCAAACAGGGCTCCggaattcgc 625
--------------- D K N G I H D L D N I S F P K Q G S G I
---------------AACTGTTTTTACCGTAGGTACTGGACCTATTGTAAAGGAAGGGGTTTGTCCCGAGGccttaagcg
Figura 8: Estremità 5’ e 3’ dei frammenti di PCR attesi (geni PSMA6 e PSMB2). Le lettere maiuscole poste al di sotto dei codoni (in rosso) si riferiscono alle codifiche convenzionali dei corrispondenti aminoacidi derivati da ciascuna tripletta di basi, mentre le lettere minuscole corrispondono ai nucleotidi aggiunti alla sequenza del gene codificante per ognuna delle subunità proteasomali al fine di creare i siti di taglio per gli enzimi di restrizione Hind III e EcoR I. I frammenti ottenuti dopo l’amplificazione mediante PCR constano di 760 nucleotidi per il gene PSMA6 e di 625 nucleotidi per il gene PSMB2.
I prodotti di amplificazione ottenuti mediante PCR e i vettori di espressione sono stati
sottoposti a digestione enzimatica, utilizzando gli enzimi di restrizione Hind III e EcoR I;
in tal modo, i frammenti di 760 bp (PSMA6) e 625 bp (PSMB2) sono stati clonati nei vettori
di espressione pEGFP-N1 e pDsRed1-N1. In Figura 9 è riportato, in particolare, lo schema
che rappresenta l’estremità 3’ del gene PSMA6 e PSMB2, il sito di clonaggio e il codone

6. Risultati
-107-
ATG di inizio della sequenza della proteina fluorescente. Inoltre, questo schema mostra
come la sequenza inserita fosse “in frame” con la sequenza della proteina fluorescente. Il
codone di inizio è evidenziato in verde, la sigla dell’aminoacido corrispondente (M:
metionina) è scritto in verde mentre le codifiche convenzionali degli aminoacidi relativi
alle sequenze che codificano per il sito multiplo di clonaggio sono riportati in rosa.
a) Gene PSMA6 Sal I Sma I
EcoR I Pst I Age I BamH I Nco I
| | | | | | |
--------CTAGCAGAGAGAGACGGAATTCTGCAGTCGACGGTACCGCGGGCCCGGGATCCACCGGTCGCCACCATG--------
-------- L A E R D G I L Q S T V P R A R D P P V A T M --------
--------GATCGTCTCTCTCTGCCTTAAGACGTCAGCTGCCATGGCGCCCGGGCCCTAGGTGGCCAGCGGTGGTAC--------
b) Gene PSMB2
Sal I Sma I
EcoR I Pst I Age I BamH I Nco I
| | | | | | |
--------CCCAAACAGGGCTCCGGAATTCTGCAGTCGACGGTACCGCGGGCCCGGGATCCACCGGTCGCCACCATG--------
-------- P K Q G S G I L Q S T V P R A R D P P V A T M --------
--------GGGTTTGTCCCGAGGCCTTAAGACGTCAGCTGCCATGGCGCCCGGGCCCTAGGTGGCCAGCGGTGGTAC--------
Figura 9: Rappresentazione schematica dell’estremità 3’ della sequenza che codifica per i geni PSMA6 (pannello a) e PSMB2 (pannello b), del sito di clonaggio multiplo e, infine, del codone di inizio “ATG” relativo alla proteina fluorescente.
Nel complesso, sono stati così ottenuti i vettori di espressione di seguito elencati:
pPSMA6-DsRed1, pPSMA6-EGFP, pPSMB2-DsRed1 e pPSMB2-EGFP [Figura 10]. In
particolare, i plasmidi pPSMA6-EGFP e pPSMB2-EGFP sono stati sequenziati al fine di
verificare che, nel corso delle tappe di retrotrascrizione e di PCR (durante le quali sono
frequenti errori di incorporazione dei nucleotidi) non fossero avvenute mutazioni
all’interno delle sequenze codificanti per le subunità proteasomali.

6. Risultati
-108-
Figura 10: Allestimento dei vettori di espressione pPSMA6-DsRed1, pPSMA6-EGFP, pPSMB2-DsRed1 e pPSMB2-EGFP per integrazione delle sequenze codificanti per le subunità α1/p27K e β4/p23K, rispettivamente in vettori di espressione commerciali denominati pDsRed1-N1 e pEGFP-N1.
6.1.6 ESPRESSIONE DELLE PROTEINE DI FUSIONE ATTRAVERSO ESPERIMENTI DI
TRASFEZIONE
Dopo introduzione dei plasmidi di interesse in batteri competenti mediante processo di
trasformazione e successivo ottenimento di adeguate quantità di DNA plasmidico, come
dettagliato nella sezione “Materiali e Metodi”, le sequenze geniche in oggetto sono state
utilizzate per esperimenti di trasfezione di cellule CHO (criceto) e della linea cellulare
miogenica di topo C2.7.
Hind III EcoR I
Hind III (623) EcoR I (630)
Promotore di HCMV
EGFP/DsRed1
Segnale poliA - SV40
f1 ORI
Promotore batterico
Promotore di SV40
Neo/Kan
Segnale poli A HSV TK
ORI
pDsRed1-N1/pEGFP-N1 (4.7 kb)
Hind III
EcoR I
Promotore di HCMV
EGFP/DsRed1
Segnale poli A - SV40
f1 ORI
Promotore di SV40
Neo/Kan
Segnale poli A HSV
ORI
PSMA6/PSMB2
pPSMA6-DsRed1/EGFP
pPSMB2-DsRed1/EGFP
(5.4 kb/5.3 kb)
PSMA6: α1/p27K
oppure
PSMB2: β4/p23K

6. Risultati
-109-
Quale premessa generale a queste tipologie di esperimenti, è bene sottolineare che
l’incorporazione stabile di DNA esogeno nel genoma delle cellule trasfettate rappresenta
un evento raro, che si verifica in una cellula su un numero compreso fra mille e un
milione; inoltre solo circa il 40% di cellule può, di fatto, acquisire il DNA nell’ambito di
una trasfezione, senza peraltro integrarlo nel proprio genoma. In queste cellule, il DNA
esogeno rimane nel nucleo per diversi giorni prima di essere perso, a causa di un insieme
di eventi di degradazione e di diluizione, in quanto il DNA non integrato non è di solito
duplicato, come avviene per i cromosomi della cellula ospite.
Allo scopo di reperire agevolmente la frazione di cellule in cui il DNA plasmidico fosse
stato integrato stabilmente in uno dei cromosomi dell’ospite, si è reso necessario operare
una selezione nei confronti di una proprietà conferita alla cellula da parte di un gene
“marcatore”, presente nei plasmidi utilizzati per la trasfezione. A tal fine, è stato utilizzato
un gene che conferisce uno stato di resistenza cellulare all’antibiotico aminoglicosidico
denominato “G-418” (analogo della neomicina), che è generalmente tossico per le cellule
eucariote. In particolare, il gene marcatore codifica per l’enzima aminoglicoside-
fosfotrasferasi, che permette di degradare l’antibiotico sopra citato. In questo modo, in
presenza del suddetto antibiotico nel terreno di coltura, tutte le cellule che non avevano
integrato il DNA trasfettato sono morte, mentre solo le rare cellule recanti il DNA esogeno
con il marcatore di resistenza sopra menzionato sono rimaste in vita e si sono moltiplicate,
formando cloni. Questi ultimi sono stati selezionati sulla base della emissione di
fluorescenza (per la presenza di una delle proteine fluorescenti co-espresse assieme alle
specifiche subunità di proteasomi), mediante osservazione diretta delle cellule al
microscopio a fluorescenza. La selezione è durata circa un mese e ha consentito di
ottenere i seguenti cloni cellulari: CHO-EGFP e C2.7-EGFP, con espressione della sola
proteina verde fluorescente EGFP in cellule CHO e C2.7, rispettivamente; CHO-PSMA6-
EGFP e C2.7-PSMB2-EGFP, con espressione della proteina di fusione costituita dalla
subunità α1/p27K (PSMA6) di proteasoma e dalla suddetta proteina fluorescente per quel
che riguarda le cellule CHO e della proteina di fusione costituita dalla subunità β4/p23K
(PSMB2) assieme alla proteina fluorescente EGFP per la linea cellulare C2.7.
Successivamente, al fine di verificare la produzione delle proteine di fusione a partire da
cellule CHO e C2.7 (previa trasfezione con i plasmidi ricombinanti), quale risultato di
espressioni transitorie o stabili delle sequenze geniche considerate, sono stati eseguiti
saggi in Western Blot, sugli estratti proteici ottenuti dalle suddette cellule, utilizzando
anticorpi specifici per ognuna delle due subunità di proteasomi di interesse (anti-β4/p23K

6. Risultati
-110-
e anti-α1/p27K), così come anticorpi diretti contro le proteine fluorescenti co-espresse
(anti-GFP e anti-DsRed).
È stato così possibile dimostrare che, quale risultato dell’espressione transitoria delle
sequenze geniche dei plasmidi pPSMB2-EGFP, pPSMB2-DsRed1, pPSMA6-EGFP e
pPSMA6-DsRed1 negli estratti proteici ottenuti da cellule C2.7 dopo trasfezione delle
stesse con i suddetti plasmidi [Figura 11, parte a], erano presenti sia bande corrispondenti
alle proteine endogene β4/p23K (PSMB2), di peso molecolare pari a 23kDa, e α1/p27K
(PSMA6), di 27kDa [Figura 11, pannelli 1 e 2, frecce], che bande corrispondenti alle
relative proteine di fusione β4/p23k-EGFP, di peso molecolare pari a 50kDa, β4/p23k-
DsRed1 di 49kDa, α1/p27k-EGFP di 54kDa e α1/p27k-DsRed1 di 53kDa [Figura 11,
pannelli 1 e 2, punte di freccia]. Come atteso, il segnale positivo per le suddette proteine
di fusione era presente anche quando venivano utilizzati anticorpi specifici per le due
proteine fluorescenti EGFP e DsRed1, co-espresse nell’ambito della chimera [Figura 11,
prima colonna nei pannelli 3 e 4]. D’altra parte, nell’ambito degli estratti proteici ottenuti
da cellule C2.7 trasfettate con i plasmidi pEGFP-N1 e pDsRed1-N1 [Figura 11, colonne 3 e
4, nei pannelli a1 e a2; ultima colonna dei pannelli a3 a4], sono state evidenziate bande
corrispondenti alla proteina endogena β4/p23k [Figura 11, colonne 3 e 4 del pannello a1],
alla proteina endogena α1/p27k [Figura 11, colonne 3 e 4 del pannello a2] e alle proteine
fluorescenti EGFP di 27kDa [Figura 11, asterisco, ultima colonna del pannello a3] e
DsRed1 di 26kDa [Figura 11, asterisco, ultima colonna del pannello a4], quando analizzate
con i rispettivi anticorpi primari. Analoghi risultati sono stati ottenuti utilizzando cellule
CHO (dati non mostrati).
Per quanto riguarda gli estratti proteici derivanti da espressioni stabili delle suddette
sequenze geniche in cellule C2.7 e CHO (integrazione del DNA plasmidico nel genoma
della cellula trasfettata), l’analisi in Western Blot ha consentito di selezionare
ulteriormente i cloni ottenuti (e inizialmente prescelti mediante osservazione al
microscopio a fluorescenza), sulla base della presenza della banda corrispondente alla
proteina di fusione o di quella relativa alla proteina naturale fluorescente (cloni C2.7-
PSMB2-EGFP #1 e #3 e C2.7-EGFP) [Figura 11, parte b, pannelli b1 e b2]; per quel che
riguarda la linea miogenica C2.7, non è stato possibile selezionare cloni C2.7-PSMA6-
EGFP, corrispondenti alla subunità α1/p27k di proteasoma. Di converso, per quanto
attiene alle cellule CHO, sono stati selezionati i cloni CHO-PSMA6-EGFP #1 e #4 e CHO-
EGFP, ma non cloni di tipo CHO-PSMB2-EGFP, relativi alla subunità β4/p23k di
proteasoma (dati non mostrati). Inoltre, non è stato possibile ottenere cloni cellulari in
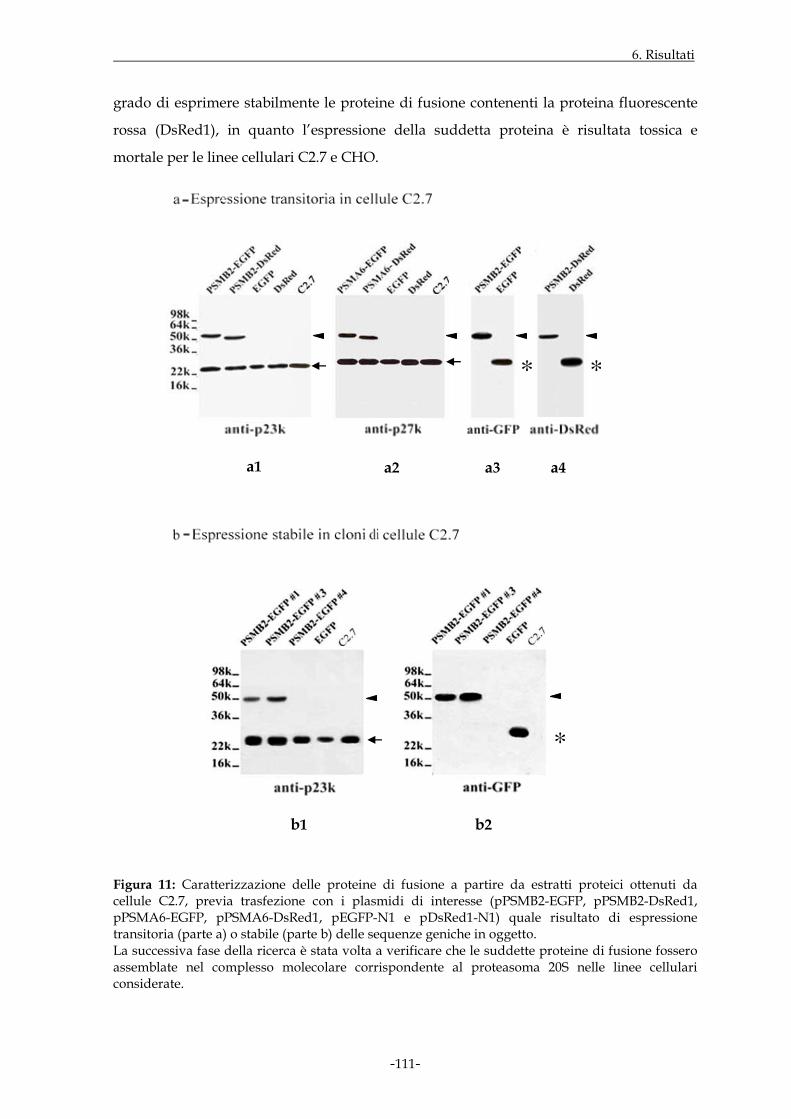
6. Risultati
-111-
* *
*
a1 a2 a3 a4
b1 b2
grado di esprimere stabilmente le proteine di fusione contenenti la proteina fluorescente
rossa (DsRed1), in quanto l’espressione della suddetta proteina è risultata tossica e
mortale per le linee cellulari C2.7 e CHO.
Figura 11: Caratterizzazione delle proteine di fusione a partire da estratti proteici ottenuti da cellule C2.7, previa trasfezione con i plasmidi di interesse (pPSMB2-EGFP, pPSMB2-DsRed1, pPSMA6-EGFP, pPSMA6-DsRed1, pEGFP-N1 e pDsRed1-N1) quale risultato di espressione transitoria (parte a) o stabile (parte b) delle sequenze geniche in oggetto. La successiva fase della ricerca è stata volta a verificare che le suddette proteine di fusione fossero assemblate nel complesso molecolare corrispondente al proteasoma 20S nelle linee cellulari considerate.

6. Risultati
-112-
A tal fine, a partire dai cloni C2.7-PSMB2-EGFP#1 e #3, C2.7-EGFP, CHO-PSMA6-EGFP
#1 e #4 e CHO-EGFP e, in parallelo, dalle stesse tipologie di cellule non sottoposte a
trasfezione, sono stati preparati estratti citoplasmatici e nucleari, che sono stati in seguito
sottoposti a centrifugazione in gradiente di densità di saccarosio, come dettagliato nella
sezione “Materiali e Metodi”, e successiva analisi in Western Blot. Nelle condizioni
sperimentali adottate per questo tipo di gradiente, il picco relativo al proteasoma 20S è
localizzato a tre quarti del gradiente (verso il fondo del gradiente stesso), mentre quello
relativo alle proteine libere (< 5S) si localizza nella parte alta del gradiente.
In Figura 12 e in Figura 13 sono mostrati i risultati dei suddetti esperimenti, relativamente
al clone cellulare C2.7-PSMB2-EGFP #3 (subunità proteasomale β4/p23k) e con
riferimento al gradiente operato sull’estratto citoplasmatico [Figura 12] e nucleare [Figura
13]. In particolare, i pannelli “A” si riferiscono al profilo del gradiente (misura della
fluorescenza dovuta alla presenza della proteina EGFP nell’ambito della proteina di
fusione, tracciati di colore blu), i pannelli “B” alla quantificazione delle proteine ed alla
loro distribuzione nel gradiente (tracciati in blu), mentre i pannelli “C” (parte sinistra)
mostrano i risultati dell’analisi delle frazioni del gradiente mediante Western Blot. Come
già accennato, sono stati parallelamente esaminati estratti citoplasmatici e nucleari
provenienti da lisati di cellule C2.7 non sottoposte a trasfezione [Figure 12 e 13, pannelli A
e B: tracciati di colore rosa; pannello C: parte destra].
Nello specifico, per quel che riguarda i pannelli “A” è possibile osservare un picco di
fluorescenza in prossimità delle frazioni raccolte a circa tre quarti (verso il fondo) del
gradiente, sia nell’estratto citoplasmatico, che in quello nucleare [Figure 12 e 13,
rispettivamente, tracciati in blu]; nell’ambito dell’estratto nucleare, è stato ottenuto un
ulteriore picco (in blu) anche in corrispondenza delle frazioni raccolte verso la parte alta
del gradiente. Per quel che riguarda, invece, il tracciato derivante da cellule C2.7 non
trasfettate (tracciato in rosa), non è stato rilevato alcun picco a livello dell’estratto
citoplasmatico [Figura 12], mentre, anche in questo caso, come per la proteina di fusione, è
stato ottenuto un picco a livello delle frazioni rappresentative della parte alta del
gradiente nell’ambito dell’estratto nucleare [Figura 13].
Inoltre, è stato concomitantemente effettuato un dosaggio delle proteine contenute nelle
diverse frazioni dei gradienti [Figure 12 e 13, pannelli B]; tale dosaggio ha evidenziato
come la maggior parte delle proteine sia concentrata nelle frazioni rappresentative della
parte alta del gradiente.

6. Risultati
-113-
Per quel che riguarda l’analisi mediante Western Blot [Figure 12 e 13, pannelli C],
l’utilizzo di anticorpi anti-β4/p23k e anti-α1/p27k di proteasoma, ha consentito
innanzitutto di evidenziare la presenza di proteine endogene relative alle subunità
proteasomali di 23kDa e 27kDa, con segnale positivo concentrato a livello delle frazioni
corrispondenti a tre quarti del gradiente (verso il fondo), ossia all’altezza del gradiente a
cui sedimenta il proteasoma 20S, sia per quel che riguarda gli estratti citoplasmatici e
nucleari di cellule C2.7 non trasfettate [Figure 12 e 13, pannelli C, parte destra], che per
quanto attiene al clone C2.7-PSMB2-EGFP #3 [Figure 12 e 13, pannelli C, parte sinistra]. In
quest’ultimo caso, come atteso, era inoltre presente un segnale positivo per la proteina di
fusione contenente la subunità β4/p23k di proteasoma (β4/p23k-EGFP, 50kDa), sempre
concentrato alla suddetta altezza del gradiente.
D’altra parte, per quel che riguarda i picchi evidenziati nella parte alta del gradiente,
nell’ambito degli estratti nucleari [Figura 13, pannello A, tracciati in blu e in rosa], essi
non hanno dato luogo ad alcuna risposta al saggio in immunoblotting con anticorpi diretti
contro le due subunità proteasomali di interesse, avvalorando l’ipotesi secondo cui i
suddetti picchi sarebbero verosimilmente riconducibili alla presenza di componenti
fluorescenti aspecifici.
L’utilizzo di un anticorpo anti-GFP ha anch’esso permesso di evidenziare un segnale
positivo all’altezza attesa per la proteina di fusione C2.7-PSMB2-EGFP #3, ossia 50 kDa
(dati non mostrati).
Sulla base di questi risultati, è stato possibile concludere che la proteina di fusione
β4/p23k-EGFP, stabilmente espressa nel clone cellulare C2.7-PSMB2-EGFP #3, era
efficacemente assemblata nel proteasoma 20S, e che il suddetto complesso era presente sia
a livello nucleare che citoplasmatico.
Risultati analoghi a quelli mostrati nelle Figure 12 e 13 sono stati ottenuti utilizzando i
cloni cellulari CHO-PSMA6-EGFP #1 e #4, in questo caso, riguardanti la subunità
α1/p27k di proteasoma (dati non mostrati).
6. Risultati
-114-
Figura 12: Valori di intensità di fluorescenza (pannello A), relativi alla proteina GFP (espressi in unità arbitrarie di fluorescenza), in seguito a lettura al fluorimetro, delle frazioni ottenute dopo centrifugazione in gradiente di densità di saccarosio a partire da estratti citoplasmatici del clone cellulare C2.7-PSMB2-EGFP #3 (in blu) e dalle cellule C2.7 non trasfettate (in rosa). Nel pannello B sono rappresentati in grafico i valori di concentrazione proteica relativi a ciascuna frazione raccolta dopo centrifugazione in gradiente di saccarosio. La maggior parte delle proteine è concentrata nelle frazioni rappresentative la parte alta del gradiente (lato destro del grafico). Profili proteici in Western Blot (pannelli C) di 12 frazioni principali, in cui sono state raggruppate quelle raccolte dopo centrifugazione in gradiente di saccarosio; l’immunoreazione è stata effettuata mediante l’impiego di sonde immunologiche dirette nei confronti delle subunità proteasomali.
B
Clone cellulare C2.7-PSMB2-EGFP #3
Cellule C2.7
Estratto citoplasmatico
Numero frazioni
Numero frazioni
Fondo del gradiente
Unità arbitrarie
di fluorescenza
Concentrazione delle
proteine (m
g/ml)
A
C
6. Risultati
-115-
Figura 13: Valori di intensità di fluorescenza (pannello A), relativi alla proteina GFP (espressi in unità arbitrarie di fluorescenza), in seguito a lettura al fluorimetro, delle frazioni ottenute dopo centrifugazione in gradiente di densità di saccarosio a partire da estratti nucleari del clone cellulare C2.7-PSMB2-EGFP #3 (in blu) e dalle cellule C2.7 non trasfettate (in rosa). Nel pannello B sono rappresentati in grafico i valori di concentrazione proteica relativi a ciascuna frazione raccolta dopo centrifugazione in gradiente di saccarosio. La maggior parte delle proteine è concentrata nelle frazioni rappresentative la parte alta del gradiente (lato destro del grafico). Profili proteici in Western Blot (pannelli C) di 12 frazioni principali, in cui sono state raggruppate quelle raccolte dopo centrifugazione in gradiente di saccarosio; l’immunoreazione è stata effettuata mediante l’impiego di sonde immunologiche dirette nei confronti delle subunità proteasomali.
Unità arbitrarie
di fluorescenza
Numero frazioni
Numero frazioni
Fondo del gradiente
Concentrazione delle
proteine (m
g/ml)
Cellule C2.7
Clone cellulare C2.7-PSMB2-EGFP #3
Estratto nucleare
B
C
A

6. Risultati
-116-
Infine, utilizzando estratti citoplasmatici e nucleari ottenuti a partire dai cloni cellulari
C2.7-EGFP e CHO-EGFP (che esprimono esclusivamente la corrispondente proteina
fluorescente) per esperimenti analoghi a quelli sopra descritti, è stato evidenziato un solo
picco nel profilo dei relativi gradienti, in prossimità della parte alta degli stessi (in cui si
concentrano le proteine < 5S, ossia a più basso peso molecolare). L’analisi in Western Blot,
mediante l’impiego di anticorpi anti-GFP, ha consentito di dimostrare la specificità del
suddetto picco, in quanto corrispondente alla proteina EGFP libera, di peso molecolare
pari a 27kDa. Inoltre, utilizzando anticorpi anti-α1/p27k e anti-β4/p23k nell’ambito della
suddetta analisi, sono stati ottenuti segnali positivi a livello delle frazioni del gradiente
corrispondenti all’altezza del gradiente alla quale si localizza il proteasoma 20S (serie di
risultati non mostrata).
6.1.7 ALLESTIMENTO DI VETTORI RETROVIRALI PER LE SEQUENZE GENICHE DI INTERESSE
Nell’intento di migliorare ulteriormente la strategia per l’ottenimento di strumenti
molecolari fruibili per studi di cinetica in cellule viventi, in particolare allo scopo di
ottenere elevate rese di espressione delle sequenze geniche di interesse, si è
successivamente passati all’allestimento di vettori retrovirali [Figura 14, pannelli A, B e
C], da potere utilizzare per l’infezione di opportuni modelli cellulari.
Le sequenze geniche di interesse, provenienti dai vettori plasmidici precedentemente
descritti, sono state integrate nel vettore commerciale pDON-AI [Figura 14, pannelli B e
C], dopo rimozione del sito di taglio per l’enzima Hind III localizzato all’esterno del sito
multiplo di clonaggio [Figura 14, pannello A], in modo che venissero espresse sotto il
controllo del promotore dei geni precocissimi di HCMV, presente a livello delle sequenze
geniche virali note come LTR (“long terminal repeats”). Nel suddetto vettore retrovirale
sono presenti anche marcatori di selezione, quali i geni che codificano per la resistenza
agli antibiotici ampicillina e neomicina (e del suo analogo G-418) ed il segnale per
l’incapsidamento della progenie virale definito “psi” (Ψ). Tuttavia, tali vettori retrovirali
risultano defettivi per i geni virali gag, pol e env necessari per la produzione di progenie
virale.
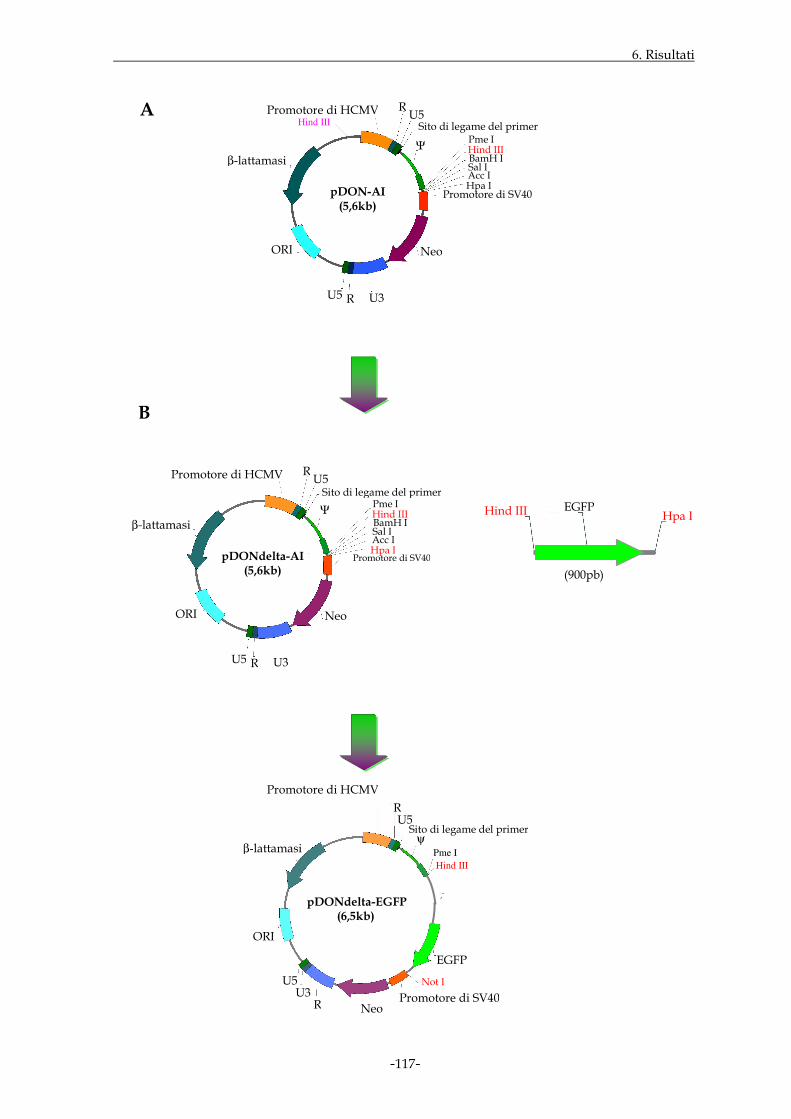
6. Risultati
-117-
pDON-AI (5,6kb)
Promotore di HCMV R U5
Ψ
Promotore di SV40
Neo
U3 R U5
ORI
β-lattamasi
Hind III
Pme I Hind III BamH I Sal I Acc I Hpa I
Sito di legame del primer
pDONdelta-AI (5,6kb)
Promotore di HCMV R U5
Ψ
Promotore di SV40
Neo
U3 R U5
ORI
β-lattamasi
Pme I Hind III BamH I Sal I Acc I Hpa I
Sito di legame del primer
EGFP Hind III Hpa I
(900pb)
pDONdelta-EGFP (6,5kb)
R U5
Sito di legame del primer ψ
Pme I
Hind III
EGFP
Promotore di SV40 Not I
Neo R
U3 U5
ORI
β-lattamasi
Promotore di HCMV
A
B
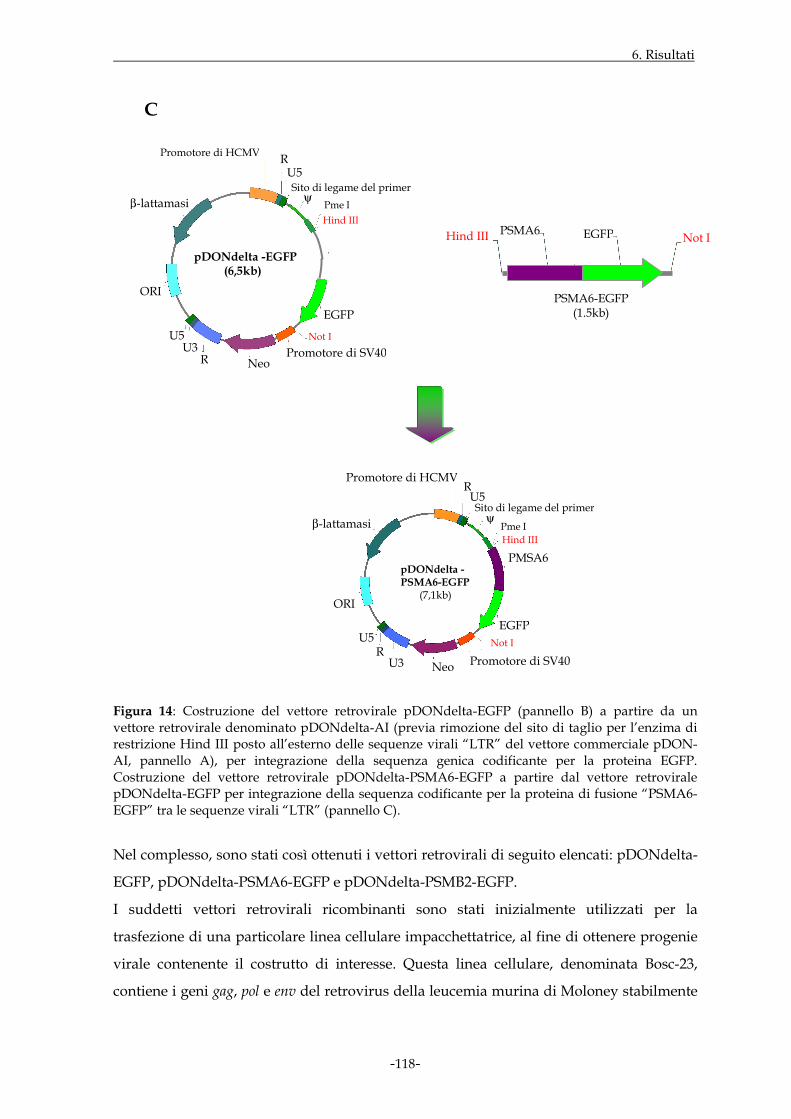
6. Risultati
-118-
Figura 14: Costruzione del vettore retrovirale pDONdelta-EGFP (pannello B) a partire da un vettore retrovirale denominato pDONdelta-AI (previa rimozione del sito di taglio per l’enzima di restrizione Hind III posto all’esterno delle sequenze virali “LTR” del vettore commerciale pDON-AI, pannello A), per integrazione della sequenza genica codificante per la proteina EGFP. Costruzione del vettore retrovirale pDONdelta-PSMA6-EGFP a partire dal vettore retrovirale pDONdelta-EGFP per integrazione della sequenza codificante per la proteina di fusione “PSMA6-EGFP” tra le sequenze virali “LTR” (pannello C).
Nel complesso, sono stati così ottenuti i vettori retrovirali di seguito elencati: pDONdelta-
EGFP, pDONdelta-PSMA6-EGFP e pDONdelta-PSMB2-EGFP.
I suddetti vettori retrovirali ricombinanti sono stati inizialmente utilizzati per la
trasfezione di una particolare linea cellulare impacchettatrice, al fine di ottenere progenie
virale contenente il costrutto di interesse. Questa linea cellulare, denominata Bosc-23,
contiene i geni gag, pol e env del retrovirus della leucemia murina di Moloney stabilmente
C
pDONdelta -EGFP (6,5kb)
R U5 Sito di legame del primer
ψ
Hind III
EGFP
Promotore di SV40 Not I
Neo R
U3 U5
ORI
β-lattamasi
Promotore di HCMV
Pme I
PSMA6-EGFP (1.5kb)
Hind III Not I PSMA6 EGFP
Hind III
Promotore di HCMV
Sito di legame del primer ψ
Pme I β-lattamasi
R
PMSA6
EGFP
Promotore di SV40 Neo U3 R
ORI
U5
pDONdelta -PSMA6-EGFP (7,1kb)
Not I
U5

6. Risultati
-119-
integrati nel proprio genoma. In particolare, il gene env codifica per proteine in grado di
riconoscere solo recettori presenti sulla membrana di cellule di ratto e di topo (tropismo
ecotropico).
Tali cellule impacchettatrici sono in grado di sintetizzare tutte le proteine necessarie per
l’assemblaggio di particelle virali infettanti, fatta eccezione per il segnale di
incapsidamento e per l’RNA virale, che vengono forniti dal DNA del vettore retrovirale
utilizzato per la trasfezione. Le particelle virali sono assemblate nel citoplasma e
gemmano a livello della membrana cellulare, in corrispondenza di zone modificate per
inserimento delle proteine codificate dal gene env. La progenie virale così prodotta può
essere utilizzata per infettare cellule di ratto o di topo; in tali cellule il genoma virale
viene, com’è noto, classicamente integrato nel genoma della nuova cellula ospite, che può
esprimere in modo stabile il nuovo gene introdotto tramite il genoma virale ricombinante
e defettivo, ma non è, ovviamente, in grado di produrre altre particelle virali in quanto il
genoma virale ricombinante manca dei geni necessari per tale processo.
Mediante le cellule impacchettatrici Bosc-23 sono stati prodotti retrovirus nel cui genoma
ricombinante erano presenti i costrutti di interesse. In particolare, sono state ottenute, con
un titolo pari a 4x104 unità formanti colonia (ufc)/ml, le seguenti sospensioni virali:
PSMA6-EGFP/rv (rv: retrovirale), PSMB2-EGFP/rv, EGFP/rv e nlsLacZ/rv (a differenza
delle precedenti, quest’ultima tipologia di particella retrovirale contiene la sequenza
codificante per l’enzima β-galattosidasi).
Tali sospensioni virali sono state utilizzate per eseguire infezioni di colture di cellule
mioblastiche C2.7, al fine di ottenere (previa selezione mediante aggiunta dell’antibiotico
G-418 al terreno di coltura cellulare), popolazioni di cellule in grado di esprimere
stabilmente le proteine di fusione α1/p27k-EGFP e β4/p23k-EGFP o, alternativamente, la
sola proteina fluorescente EGFP o la proteina β-galattosidasi.
Successivamente, a partire dalle popolazioni di mioblasti C2.7-PSMA6-EGFP (per la
subunità proteasomale α1/p27k, accoppiata a EGFP) e C2.7-PSMB2-EGFP (per β4/p23k,
accoppiata a EGFP) [Figure 15 e 16, rispettivamente], sono stati allestiti estratti
citoplasmatici e nucleari per l’analisi dei medesimi in gradiente di densità di saccarosio e,
successivamente, in Western Blot in condizioni sperimentali analoghe a quelle adottate
per gli esperimenti illustrati nelle Figure 12 e 13. Analogamente a quanto osservato in
quel contesto, anche per le popolazioni cellulari C2.7-PSMA6-EGFP e C2.7-PSMB2-EGFP è
stato ottenuto un picco di fluorescenza in prossimità delle frazioni raccolte verso il fondo
(a circa tre quarti) del gradiente, laddove è attesa la sedimentazione del complesso

6. Risultati
-120-
proteasomale 20S, sia nell’estratto citoplasmatico che in quello nucleare derivati dalle
suddette popolazioni di mioblasti [Figure 15 e 16, pannelli A]. Nel caso della popolazione
di mioblasti C2.7-PSMA6-EGFP, è stato tuttavia rilevato un secondo picco nel profilo del
gradiente ottenuto a partire dall’estratto nucleare, in corrispondenza delle frazioni
raccolte verso la parte alta del gradiente [Figura 15 pannello A, parte destra]. Per quel che
riguarda l’analisi mediante Western Blot [Figure 15 e 16, pannelli B], l’impiego di sonde
immunologiche dirette nei confronti delle subunità proteasomali α1/p27k e β4/p23k o,
alternativamente, della proteina fluorescente EGFP [Figure 15 e 16], ha dimostrato la
presenza delle proteine endogene α1/p27k (27kDa) e β4/p23k (23kDa) e delle
corrispondenti proteine di fusione α1/p27k-EGFP (54kDa, Figura 15) e β4/p23k-EGFP
(50kDa, Figura 16) in corrispondenza del picco relativo alle frazioni raccolte a tre quarti
del gradiente, sia per quel che riguarda gli estratti citoplasmatici che quelli nucleari.
D’altra parte, il picco osservato nella parte alta del gradiente relativo all’estratto nucleare
della popolazione C2.7-PSMA6-EGFP, è stato considerato non specifico (assenza di
segnale positivo in Western Blot) e riconducibile, verosimilmente, alla presenza di
componenti fluorescenti non correlate ai costrutti in esame.
L’utilizzo di mioblasti C2.7 di controllo (non sottoposte a infezione con retrovirus defettivi
ricombinanti) ha permesso di avvalorare i risultati ottenuti con le suddette popolazioni
cellulari, mettendo in evidenza, mediante utilizzo di anticorpi anti-subunità di
proteasoma di interesse per l’analisi in Western Blot, un segnale positivo a livello della
regione del gradiente in cui era attesa la presenza del complesso 20S; analogamente, è
stato evidenziato un picco di fluorescenza a livello della parte alta del gradiente
nell’ambito dell’estratto nucleare, attribuibile, anche in questo caso, a fluorescenza
aspecifica (dati non mostrati).
Alla luce di questa serie di risultati è stato possibile affermare che le proteine di fusione,
contenenti le subunità proteasomali α1/p27k e β4/p23k, sono assemblate nel proteasoma
20S e che i suddetti complessi sono presenti sia nel nucleo che nel citoplasma delle
popolazioni di mioblasti C2.7 esaminate.
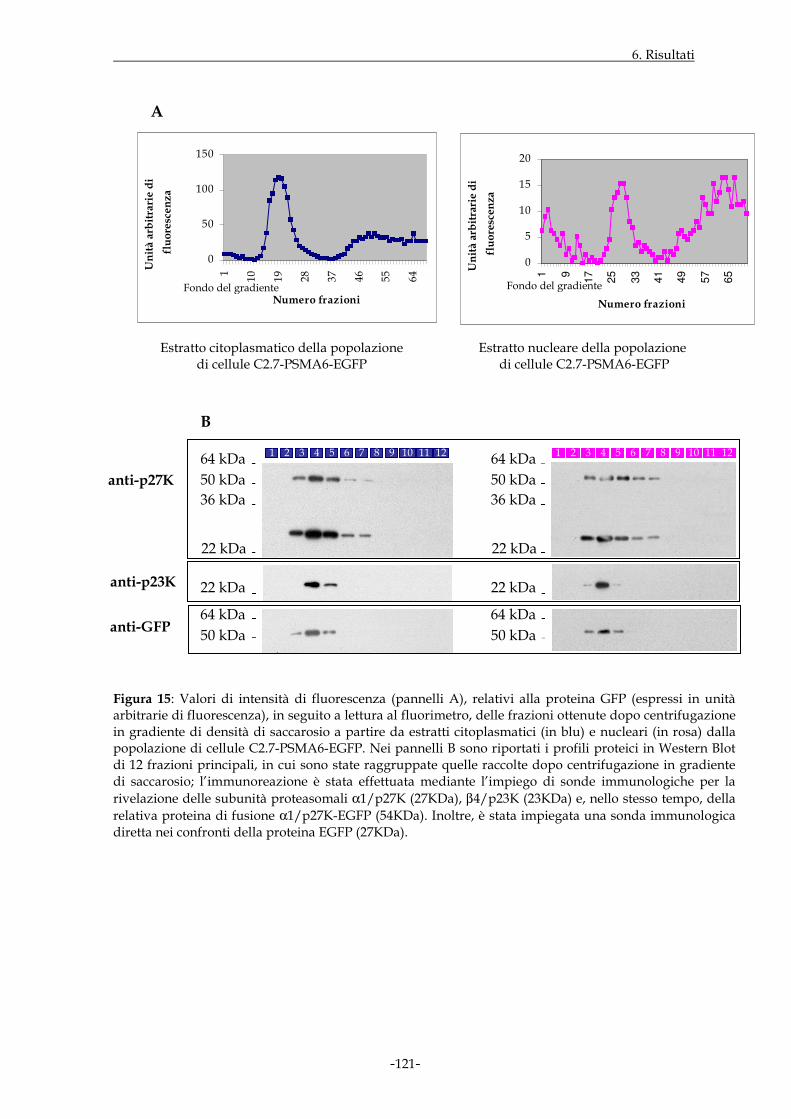
6. Risultati
-121-
Figura 15: Valori di intensità di fluorescenza (pannelli A), relativi alla proteina GFP (espressi in unità arbitrarie di fluorescenza), in seguito a lettura al fluorimetro, delle frazioni ottenute dopo centrifugazione in gradiente di densità di saccarosio a partire da estratti citoplasmatici (in blu) e nucleari (in rosa) dalla popolazione di cellule C2.7-PSMA6-EGFP. Nei pannelli B sono riportati i profili proteici in Western Blot di 12 frazioni principali, in cui sono state raggruppate quelle raccolte dopo centrifugazione in gradiente di saccarosio; l’immunoreazione è stata effettuata mediante l’impiego di sonde immunologiche per la rivelazione delle subunità proteasomali α1/p27K (27KDa), β4/p23K (23KDa) e, nello stesso tempo, della relativa proteina di fusione α1/p27K-EGFP (54KDa). Inoltre, è stata impiegata una sonda immunologica diretta nei confronti della proteina EGFP (27KDa).
anti-p27K
anti-p23K
anti-GFP 64 kDa
22 kDa
22 kDa
50 kDa
64 kDa 50 kDa 36 kDa
8 10 3 5 1 2 4 6 11 12 7 9 10 5 3 1 2 4 6 12 8 11 7 9
22 kDa
22 kDa
50 kDa
64 kDa 50 kDa 36 kDa
64 kDa
0
50
100
150
1 10 19 28 37 46 55 64
Numero frazioni
Unità arbitrarie di
fluorescenza
0
5
10
15
20
1 9
17
25
33
41
49
57
65
Numero frazioni
Unità arbitrarie di
fluorescenza
Estratto citoplasmatico della popolazione di cellule C2.7-PSMA6-EGFP
Estratto nucleare della popolazione di cellule C2.7-PSMA6-EGFP
Fondo del gradiente Fondo del gradiente
A
B
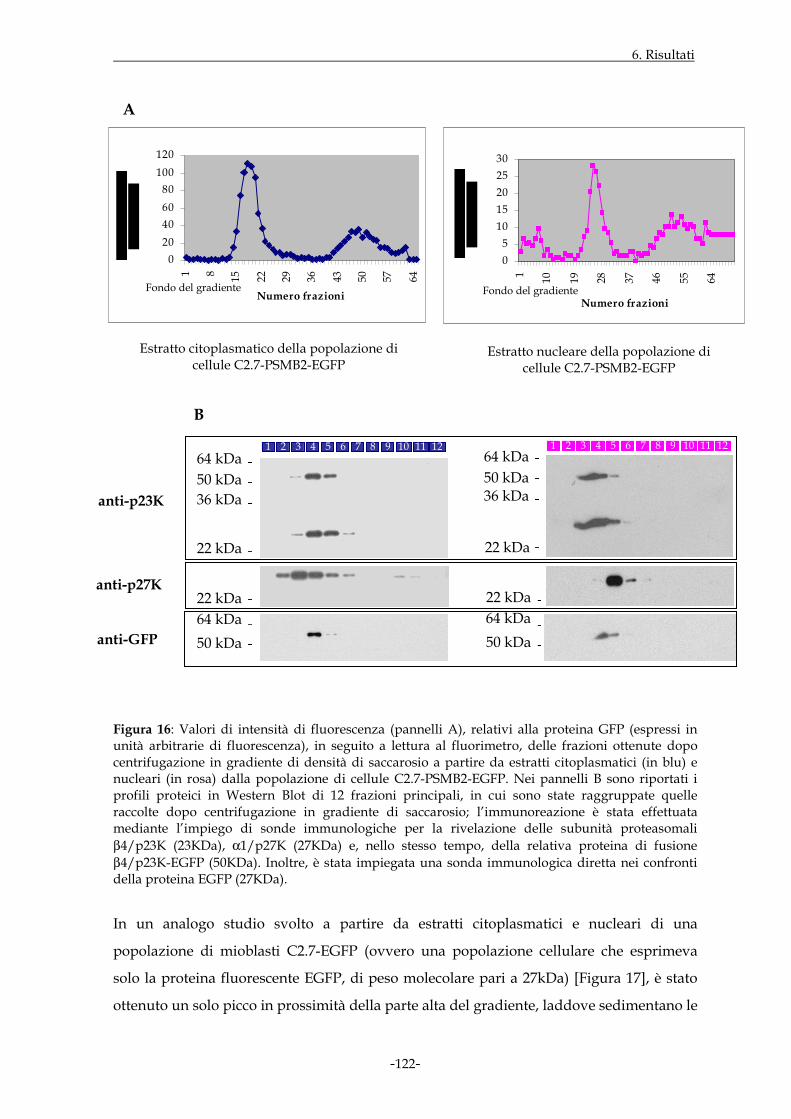
6. Risultati
-122-
anti-p27K
anti-p23K
anti-GFP
36 kDa
22 kDa 22 kDa
50 kDa 50 kDa
8 10 3 5 1 2 4 6 11 12 7 9 64 kDa
36 kDa
22 kDa
10 5 3 1 2 4 6 12 8 11 7 9
22 kDa
64 kDa
64 kDa
50 kDa 50 kDa
64 kDa
05
1015
202530
1 10 19 28 37 46 55 64
Numero frazioni
0
20
4060
80100
120
1 8 15 22 29 36 43 50 57 64
Numero frazioni
Estratto citoplasmatico della popolazione di cellule C2.7-PSMB2-EGFP
Estratto nucleare della popolazione di cellule C2.7-PSMB2-EGFP
Fondo del gradiente Fondo del gradiente
A
B
Figura 16: Valori di intensità di fluorescenza (pannelli A), relativi alla proteina GFP (espressi in unità arbitrarie di fluorescenza), in seguito a lettura al fluorimetro, delle frazioni ottenute dopo centrifugazione in gradiente di densità di saccarosio a partire da estratti citoplasmatici (in blu) e nucleari (in rosa) dalla popolazione di cellule C2.7-PSMB2-EGFP. Nei pannelli B sono riportati i profili proteici in Western Blot di 12 frazioni principali, in cui sono state raggruppate quelle raccolte dopo centrifugazione in gradiente di saccarosio; l’immunoreazione è stata effettuata mediante l’impiego di sonde immunologiche per la rivelazione delle subunità proteasomali β4/p23K (23KDa), α1/p27K (27KDa) e, nello stesso tempo, della relativa proteina di fusione β4/p23K-EGFP (50KDa). Inoltre, è stata impiegata una sonda immunologica diretta nei confronti della proteina EGFP (27KDa).
In un analogo studio svolto a partire da estratti citoplasmatici e nucleari di una
popolazione di mioblasti C2.7-EGFP (ovvero una popolazione cellulare che esprimeva
solo la proteina fluorescente EGFP, di peso molecolare pari a 27kDa) [Figura 17], è stato
ottenuto un solo picco in prossimità della parte alta del gradiente, laddove sedimentano le
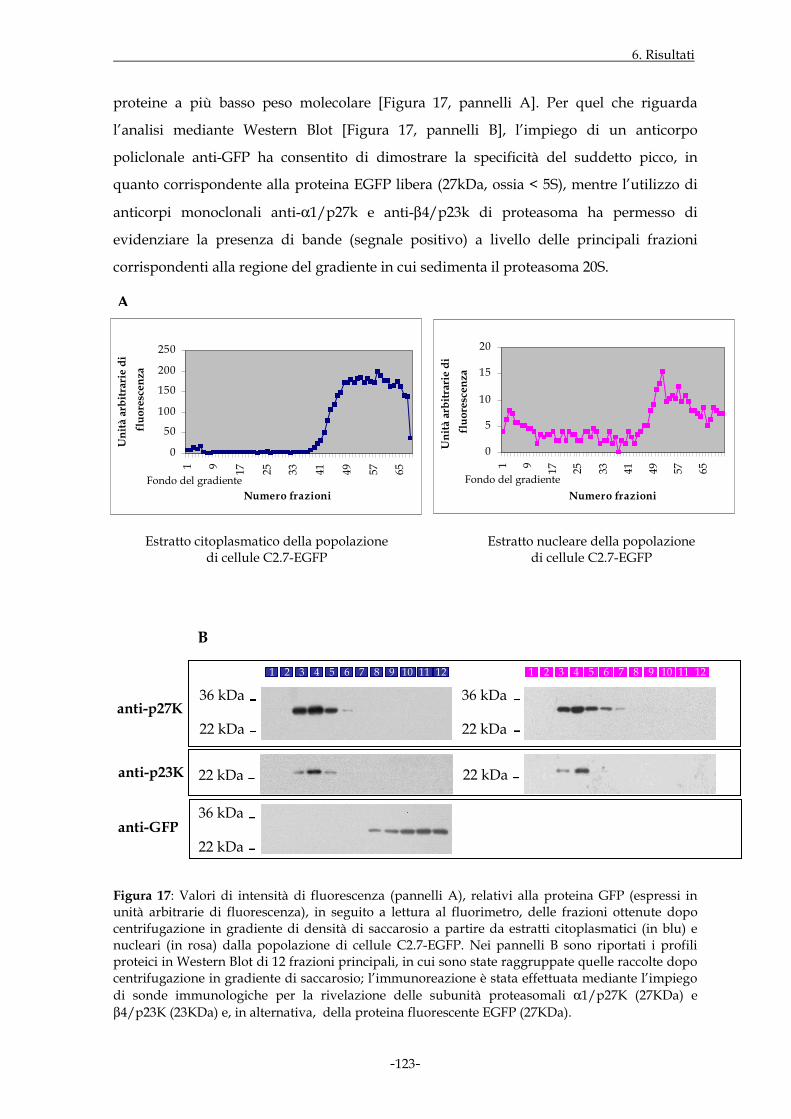
6. Risultati
-123-
anti-p27K
anti-p23K
anti-GFP
36 kDa
22 kDa
22 kDa
8 10 3 5 1 2 4 6 11 12 7 9 10 5 3 1 2 4 6 12 8 11 7 9
36 kDa
22 kDa
36 kDa
22 kDa
22 kDa
0
50
100
150
200
250
1 9 17 25 33 41 49 57 65
Numero frazioni
Unità arbitrarie di
fluorescenza
0
5
10
15
20
1 9 17 25 33 41 49 57 65
Numero frazioni
Unità arbitrarie di
fluorescenza
Estratto citoplasmatico della popolazione di cellule C2.7-EGFP
Estratto nucleare della popolazione di cellule C2.7-EGFP
Fondo del gradiente Fondo del gradiente
A
B
proteine a più basso peso molecolare [Figura 17, pannelli A]. Per quel che riguarda
l’analisi mediante Western Blot [Figura 17, pannelli B], l’impiego di un anticorpo
policlonale anti-GFP ha consentito di dimostrare la specificità del suddetto picco, in
quanto corrispondente alla proteina EGFP libera (27kDa, ossia < 5S), mentre l’utilizzo di
anticorpi monoclonali anti-α1/p27k e anti-β4/p23k di proteasoma ha permesso di
evidenziare la presenza di bande (segnale positivo) a livello delle principali frazioni
corrispondenti alla regione del gradiente in cui sedimenta il proteasoma 20S.
Figura 17: Valori di intensità di fluorescenza (pannelli A), relativi alla proteina GFP (espressi in unità arbitrarie di fluorescenza), in seguito a lettura al fluorimetro, delle frazioni ottenute dopo centrifugazione in gradiente di densità di saccarosio a partire da estratti citoplasmatici (in blu) e nucleari (in rosa) dalla popolazione di cellule C2.7-EGFP. Nei pannelli B sono riportati i profili proteici in Western Blot di 12 frazioni principali, in cui sono state raggruppate quelle raccolte dopo centrifugazione in gradiente di saccarosio; l’immunoreazione è stata effettuata mediante l’impiego di sonde immunologiche per la rivelazione delle subunità proteasomali α1/p27K (27KDa) e β4/p23K (23KDa) e, in alternativa, della proteina fluorescente EGFP (27KDa).
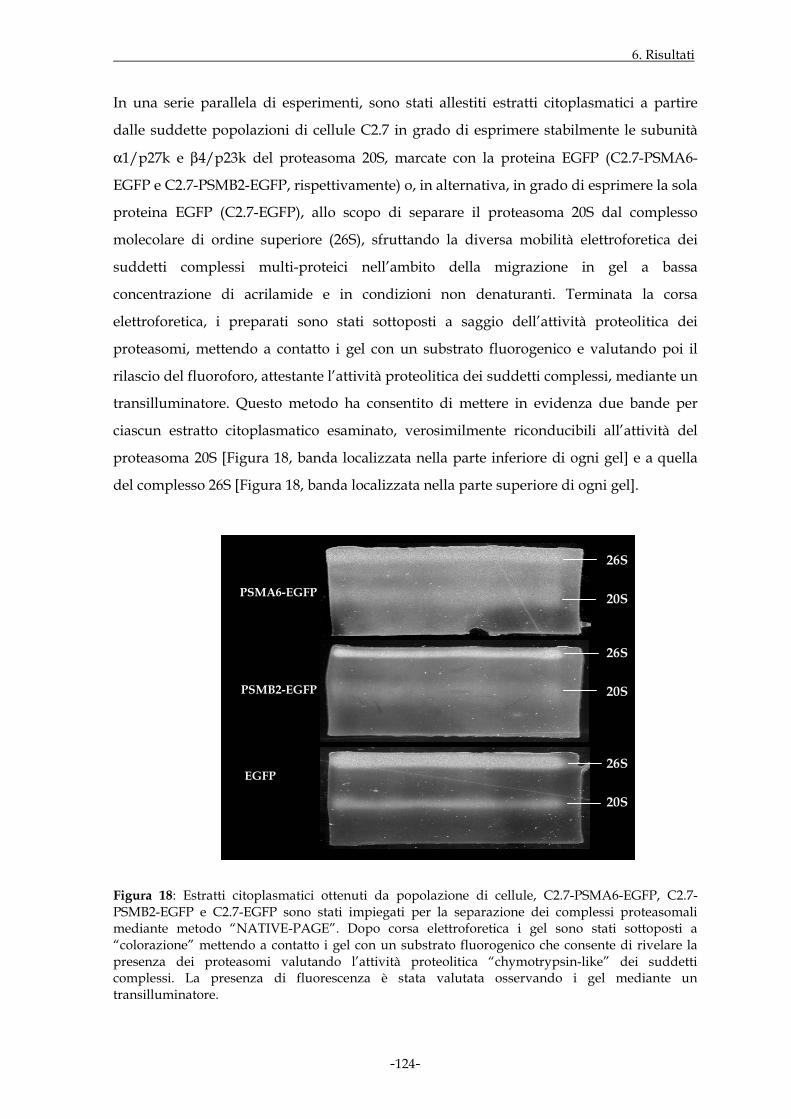
6. Risultati
-124-
In una serie parallela di esperimenti, sono stati allestiti estratti citoplasmatici a partire
dalle suddette popolazioni di cellule C2.7 in grado di esprimere stabilmente le subunità
α1/p27k e β4/p23k del proteasoma 20S, marcate con la proteina EGFP (C2.7-PSMA6-
EGFP e C2.7-PSMB2-EGFP, rispettivamente) o, in alternativa, in grado di esprimere la sola
proteina EGFP (C2.7-EGFP), allo scopo di separare il proteasoma 20S dal complesso
molecolare di ordine superiore (26S), sfruttando la diversa mobilità elettroforetica dei
suddetti complessi multi-proteici nell’ambito della migrazione in gel a bassa
concentrazione di acrilamide e in condizioni non denaturanti. Terminata la corsa
elettroforetica, i preparati sono stati sottoposti a saggio dell’attività proteolitica dei
proteasomi, mettendo a contatto i gel con un substrato fluorogenico e valutando poi il
rilascio del fluoroforo, attestante l’attività proteolitica dei suddetti complessi, mediante un
transilluminatore. Questo metodo ha consentito di mettere in evidenza due bande per
ciascun estratto citoplasmatico esaminato, verosimilmente riconducibili all’attività del
proteasoma 20S [Figura 18, banda localizzata nella parte inferiore di ogni gel] e a quella
del complesso 26S [Figura 18, banda localizzata nella parte superiore di ogni gel].
Figura 18: Estratti citoplasmatici ottenuti da popolazione di cellule, C2.7-PSMA6-EGFP, C2.7-PSMB2-EGFP e C2.7-EGFP sono stati impiegati per la separazione dei complessi proteasomali mediante metodo “NATIVE-PAGE”. Dopo corsa elettroforetica i gel sono stati sottoposti a “colorazione” mettendo a contatto i gel con un substrato fluorogenico che consente di rivelare la presenza dei proteasomi valutando l’attività proteolitica “chymotrypsin-like” dei suddetti complessi. La presenza di fluorescenza è stata valutata osservando i gel mediante un transilluminatore.
PSMA6-EGFP
PSMB2-EGFP
EGFP
26S
20S
26S
20S
26S
20S
EGFP
6. Risultati
-125-
Successivamente, le frazioni proteiche contenute in ciascuna delle due bande fluorescenti,
sono state estratte dai suddetti gel ed utilizzate per una nuova migrazione elettroforetica
delle proteine, in questo caso in condizioni denaturanti (“SDS-PAGE”); al termine della
corsa elettroforetica, i gel sono stati sottoposti a colorazione argentica [Figura 19]. Per
quanto concerne la composizione proteica della banda fluorescente dislocata più in basso
nei gel di Figura 18, riconducibile alla presenza del proteasoma 20S, il profilo
elettroforetico ottenuto in condizioni denaturanti [Figura 19, pannello A] mette in
evidenza bande con peso molecolare compreso tra i 22-36kDa, tipiche del suddetto
complesso molecolare, mentre per quel che riguarda le frazioni proteiche contenute nella
banda fluorescente dislocata più in alto nei gel di Figura 18, riconducibile al proteasoma
26S, il profilo elettroforetico in condizioni denaturanti [Figura 19, pannello B], permette di
apprezzare, da un lato, componenti proteiche corrispondenti al profilo ottenuto in “A”,
tipiche del proteasoma 20S e, dall’altro, bande con peso molecolare più elevato, fino a
circa 98kDa (ossia i pesi molecolari più elevati discernibili con il tipo di gel allestito, 12,5%
poliacrilamide).
Figura 19: Profili proteici corrispondenti a ciascuna banda ottenuta in precedenza utilizzando il metodo “NATIVE-PAGE”. Dopo corsa elettroforetica secondo il protocollo “SDS-PAGE”, i gel sono stati sottoposti a colorazione argentica. Nel pannello A, corrispondente al proteasoma 20S, sono state ottenute bande con peso molecolare compreso tra i 22-36KDa mentre nel pannello B, relativo ai proteasomi 26S, sono state ottenute bande comprese tra i 22-36KDa e da 36KDa fino a circa 98KDa.
EGFP PSMA6EGFP PSMB2EGFP
98 kDa
64 kDa 50 kDa
36 kDa
22 kDa
PROTEASOMA 26S PROTEASOMA 20S
EGFP PSMA6EGFP PSMB2EGFP
98 kDa
50 kDa
36 kDa
22 kDa
64 kDa
A B

6. Risultati
-126-
Le proteine così separate sono state anche elettroforeticamente trasferite su membrane di
fluoruro di polivinilidene ed utilizzate in seguito per analisi attraverso Western Blot
[Figura 20]. Nello specifico, mediante l’impiego sia di sonde immunologiche dirette nei
confronti delle subunità proteasomali α1/p27k e β4/p23k [Figura 20, pannelli A], sia di
anticorpi che riconoscono una subunità ad attività ATPasica del complesso regolatore 19S
[Figura 20, pannelli B], è stato possibile dimostrare, in corrispondenza degli estratti
proteici relativi al proteasoma 20S e 26S (popolazioni cellulari C2.7-PSMA6-EGFP e C2.7-
PSMB2-EGFP) [Figura 20, pannelli A, colonne PSMA6-EGFP 20S e 26S; PSMB2-EGFP 20S
e 26S], la presenza delle proteine endogene α1/p27k (27kDa) e β4/p23k (23kDa) e,
parallelamente, delle corrispondenti proteine di fusione α1/p27k-EGFP (54kDa) e
β4/p23k-EGFP (50kDa). Per quel che riguarda gli estratti proteici derivanti da
popolazioni cellulari che esprimevano solo la proteina fluorescente EGFP, sono stati
evidenziati segnali positivi solo a livello delle subunità proteasomali endogene, di peso
molecolare pari a 27kDa e di 23kDa, come atteso [Figura 20, pannelli A, colonne EGFP
20S; EGFP 26S]. D’altra parte, mediante l’impiego di un anticorpo monoclonale che
riconosce una subunità di peso molecolare pari a 48kDa del complesso 19S [Figura 20,
pannelli B], è stata ottenuta un’unica banda, del peso molecolare atteso, negli estratti
proteici relativi al proteasoma 26S delle popolazioni di cellule C2.7-EGFP, C2.7-PSMA6-
EGFP e C2.7-PSMB2-EGFP.
6. Risultati
-127-
Figura 20: Profili proteici corrispondenti a ciascuna banda ottenuta in precedenza utilizzando il metodo “NATIVE-PAGE”. Dopo corsa elettroforetica secondo il protocollo “SDS-PAGE” i gel sono stati sottoposti ad analisi in Western Blot mediante l’impiego di sonde immunologiche per la rivelazione delle subunità proteasomali α1/p27K di 27KDa, β4/p23K di 23KDa e, nello stesso tempo, delle relative proteine di fusione α1/p27K-EGFP (54KDa), β4/p23K-EGFP (50KDa) (pannelli A). Inoltre, è stata utilizzata una sonda immunologia diretta nei confronti della subunità ATPasica del complesso regolatore 19S denominata Rpt1 di 48KDa (pannelli B).
6.1.8 PROFILO DI DISTRIBUZIONE DI SPECIFICI PROTEASOMI IN POPOLAZIONI DI CELLULE
C2.7 CHE ESPRIMONO I COSTRUTTI DI INTERESSE
La successiva serie di esperimenti è stata incentrata sullo studio dell’espressione delle
proteine di fusione di interesse in cellule muscolari C2.7, in cui i suddetti costrutti erano
stati introdotti tramite infezione con progenie retrovirale defettiva e ricombinante, come
dettagliato precedentemente. Tale linea di cellule mioblastiche può essere coltivata ed
indotta a differenziarsi in vitro, fino al raggiungimento dello stadio di miotubo, ma senza
evolvere fino alla formazione della fibra muscolare con relativa organizzazione
sarcomerica dell’apparato contrattile.
anti-p27K anti-p23K
EGFP 20S
PSMA6-EGFP
20S 26S
EGFP 26S
PSMB2-EGFP
20S 26S
64kDa-
50kDa-
36kDa-
22kDa-
50kDa-
anti-19S anti-19S
EGFP 20S 26S
PSMA6-EGFP
20S 26S
PSMB2-EGFP
20S 26S
64kDa-
36kDa-
22kDa-
A
B

6. Risultati
-128-
I risultati di tali esperimenti sono mostrati in Figura 21, in cui sono rappresentati i profili
di distribuzione del segnale di fluorescenza relativi alle proteine di fusione considerate, in
popolazioni di cellule C2.7-PSMA6-EGFP [subunità proteasomale α1/p27K, pannelli A] e
C2.7-PSMB2-EGFP [Figura 21, subunità proteasomale β4/p23K, pannelli B] nel corso del
processo di differenziamento muscolare [Figura 21, stadio di mioblasti: pannelli a1, a3 e
b1, b3; stadio di miotubi: pannelli a2, a4 e b2, b4; in particolare, a3, a4 e b3, b4
rappresentano immagini a più alto ingrandimento dei corrispondenti stadi di
differenziamento]. Nel pannello C (c1: mioblasti; c2: miotubi) sono mostrate le immagini
ottenute da popolazioni di cellule C2.7 che esprimono solo la proteina fluorescente EGFP.
Come si può notare, sia per le popolazioni di cellule C2.7-PSMA6-EGFP [Figura 21,
pannelli A] che per quelle C2.7-PSMB2-EGFP [Figura 21, pannelli B], il segnale di
fluorescenza era localizzato nel compartimento citoplasmatico, così come in quello
nucleare, ma in quest’ultimo distretto cellulare esso appariva più intenso. Inoltre, la
fluorescenza a livello del nucleo era in alcuni casi distribuita in regioni discrete, sotto
forma di granulazioni, a testimonianza di un particolare accumulo delle corrispondenti
subunità proteasomali nelle suddette regioni. Questa peculiare distribuzione nucleare era
appannaggio di un numero più significativo di nuclei quando le cellule si trovavano allo
stadio di miotubi [Figura 21, pannelli a2, a4 e b2, b4], con particolare riguardo per la
subunità proteasomale β4/p23K [Figura 21, pannelli b2, b4], in accordo con quanto già
osservato da parte del nostro gruppo di ricerca (De Conto F. et al., 2000); inoltre, come
descritto da altri Autori, il segnale fluorescente (Amsterdam A. et al., 1993; Reits E.A.J. et
al., 1997), era sempre assente a livello delle zone nucleolari.
D’altra parte, la distribuzione del segnale di fluorescenza osservato utilizzando la
popolazione di cellule C2.7-EGFP e relativo alla sola proteina EGFP, appariva più spento,
diffuso e omogeneamente distribuito tra il compartimento citoplasmatico e quello
nucleare, sia allo stadio di mioblasti che in quello di miotubi [Figura 21, pannelli C].
Nel complesso il profilo di fluorescenza descritto per le popolazioni di cellule C2.7-
PSMA6-EGFP, C2.7-PSMB2-EGFP e C2.7-EGFP è risultato sovrapponibile a quello
osservato nei cloni cellulari C2.7-PSMB2-EGFP #1 e #3, C2.7-EGFP, CHO-PSMA6-EGFP
#1 e #4 e CHO-EGFP ottenuti nella prima fase della ricerca (risultati non mostrati).
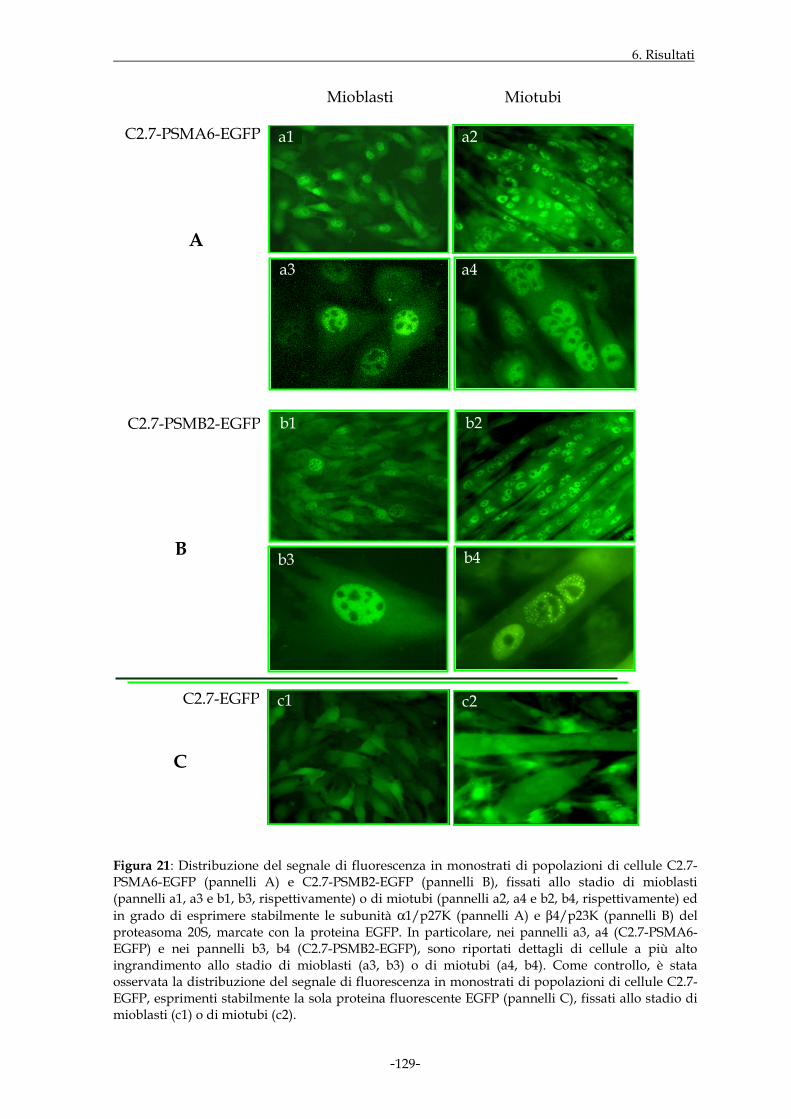
6. Risultati
-129-
a3 a4
Mioblasti
Miotubi
C2.7-PSMA6-EGFP
A
a1 a2
C2.7-EGFP
C
c2 c1
B b3 b4
C2.7-PSMB2-EGFP b1 b2
Figura 21: Distribuzione del segnale di fluorescenza in monostrati di popolazioni di cellule C2.7-PSMA6-EGFP (pannelli A) e C2.7-PSMB2-EGFP (pannelli B), fissati allo stadio di mioblasti (pannelli a1, a3 e b1, b3, rispettivamente) o di miotubi (pannelli a2, a4 e b2, b4, rispettivamente) ed in grado di esprimere stabilmente le subunità α1/p27K (pannelli A) e β4/p23K (pannelli B) del proteasoma 20S, marcate con la proteina EGFP. In particolare, nei pannelli a3, a4 (C2.7-PSMA6-EGFP) e nei pannelli b3, b4 (C2.7-PSMB2-EGFP), sono riportati dettagli di cellule a più alto ingrandimento allo stadio di mioblasti (a3, b3) o di miotubi (a4, b4). Come controllo, è stata osservata la distribuzione del segnale di fluorescenza in monostrati di popolazioni di cellule C2.7-EGFP, esprimenti stabilmente la sola proteina fluorescente EGFP (pannelli C), fissati allo stadio di mioblasti (c1) o di miotubi (c2).

6. Risultati
-130-
6.2 SECONDO MODELLO DI STUDIO: FIBROBLASTI UMANI IN CORSO DI
INFEZIONE DA CITOMEGALOVIRUS IN VITRO E PROTEASOMI
Il secondo modello di studio prescelto, contrariamente al precedente, prevede lo studio di
specifici proteasomi in una condizione patologica, quale quella indotta da un’infezione
virale, nella consapevolezza che, seppure in una situazione molto diversa rispetto al
differenziamento cellulare, anche in questo caso sono comunque previsti cambiamenti
significativi nell’ambito dell’economia cellulare, che prevedono, verosimilmente, una
riprogrammazione genica, con possibile induzione di nuove proteine cellulari e/o la
repressione di altre, in risposta alla suddetta tipologia di “insulto”. D’altra parte,
nell’ambito di questa rimodulazione degli equilibri preesistenti, lo stesso virus applica
strategie atte a cooptare funzioni cellulari a suo proprio beneficio. In questo quadro così
complesso, è verosimile che complessi molecolari di così grande rilevanza per la cellula,
quali i proteasomi, debbano intervenire in corso di infezione virale, come già evidenziato
nella sezione introduttiva.
Nello specifico, quale secondo modello è stato scelto un virus di grande impatto per la
salute umana, quale citomegalovirus (HCMV), durante l’infezione sperimentale di
fibroblasti MRC5 da parte dello stipite umano di riferimento AD169, allo scopo di
studiare aspetti morfologici e funzionali relativi ai proteasomi nell’ambito delle fasi
precoci del ciclo di replicazione virale.
6.2.1 LA FUNZIONE ENZIMATICA DEI PROTEASOMI È RILEVANTE AI FINI DELL’EVOLUZIONE
DELL’INFEZIONE PRODUTTIVA DA CITOMEGALOVIRUS IN FIBROBLASTI MRC5
Sulla base dei suddetti presupposti, la prima fase della ricerca ha avuto come obiettivo
principale quello di verificare, nel modello virus-cellula prescelto, se la funzione
enzimatica dei proteasomi fosse veramente indispensabile per l’evoluzione del ciclo
replicativo litico da HCMV, focalizzando, come già accennato, l’attenzione soprattutto
sulle fasi precoci dell’infezione, in cui vengono espletate le più significative funzioni di
regolazione sull’espressione genica sia virale che cellulare.
L’espressione dei prodotti maggiori dei geni precocissimi virali IE1 e IE2 (IEp72 e IEp86,
rispettivamente) è stata studiata in assenza [Figura 22, pannello “– MG132”], o in
presenza dell’inibitore dei proteasomi denominato MG132 (inibitore reversibile
dell’attività “chymotrypsin-like” del proteasoma 20S) [Figura 22, pannello “+MG132”].
Tale sostanza è stata utilizzata per 3 ore, a concentrazioni non tossiche per la cellula (0,5

6. Risultati
-131-
µM), come pre-trattamento di monostrati di fibroblasti MRC5; i suddetti monostrati
cellulari sono stati successivamente infettati per 2 ore e 30 minuti con lo stipite AD169 di
HCMV, mantenendo MG132 nel terreno di coltura (a causa della rapida reversibilità del
suo effetto inibitorio). Terminato il periodo di infezione, è stato eseguito uno studio sulla
distribuzione nucleare delle proteine precocissime virali IEp72 e IEp86, attraverso una
reazione di immunofluorescenza, mediante l’impiego di una miscela di anticorpi
monoclonali diretta contro epitopi delle suddette proteine di HCMV. I risultati di tale
esperimento dimostrano chiaramente che il trattamento con MG132 determina una
significativa riduzione dell’espressione dei geni precocissimi virali già a tempi molto
precoci di infezione (2 ore e 30 minuti), convalidando l’ipotesi di un intervento rilevante
dei proteasomi nell’ambito del ciclo litico da HCMV [Figura 22, pannello “+ MG132”].
Questi dati hanno anche messo in rilievo che l’effetto inibitorio sull’espressione delle
proteine precocissime virali non è della stessa entità per tutte le cellule del monostrato
(costituito da cellule non sincronizzate), suggerendo che l’inibizione dell’infezione
mediata dal blocco dell’attività proteasomale potesse avere differente efficacia,
dipendentemente dalla fase del ciclo cellulare in cui l’infezione virale ha inizio. Tale
osservazione non è apparsa affatto sorprendente, dal momento che è stata ampiamente
documentata un’azione di “disturbo”, da parte dello stesso virus, sul ciclo cellulare
(Dittmer D. and Mocarski E.S., 1997; Salvant B.S. et al., 1998; Sinclair J. et al., 2000; Kalejta
R.F. and Shenk T., 2002).
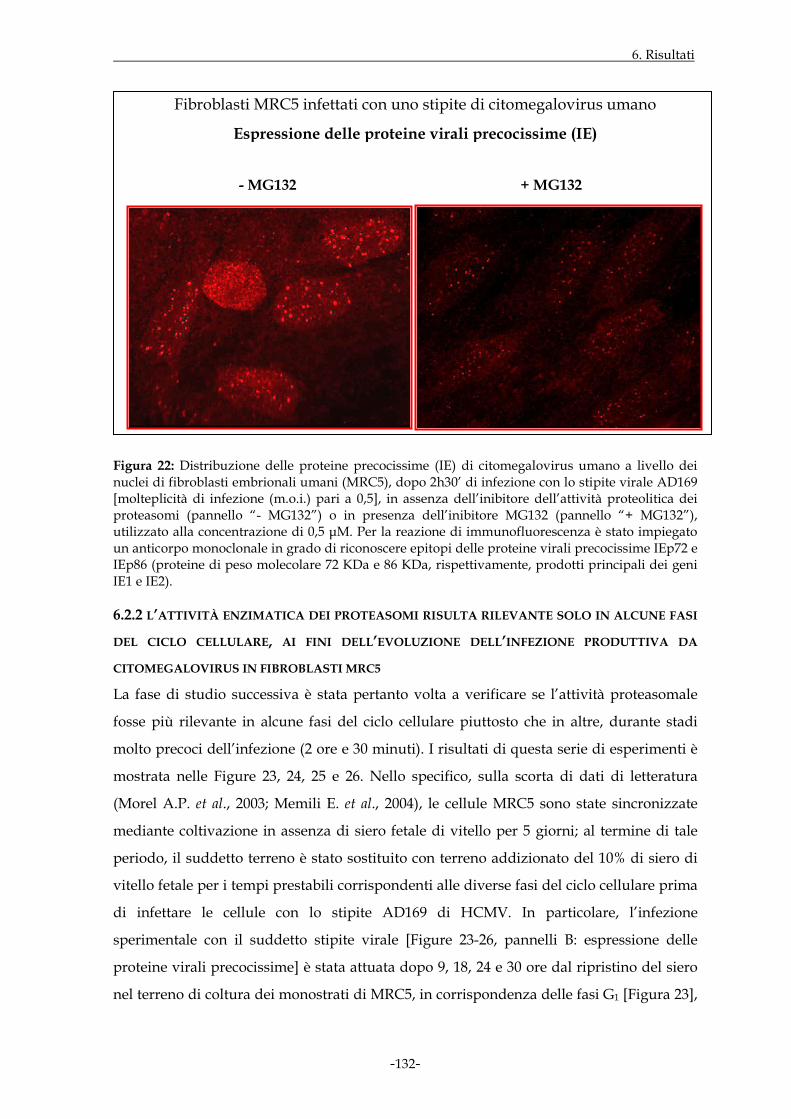
6. Risultati
-132-
Fibroblasti MRC5 infettati con uno stipite di citomegalovirus umano
Espressione delle proteine virali precocissime (IE)
- MG132 + MG132
Figura 22: Distribuzione delle proteine precocissime (IE) di citomegalovirus umano a livello dei nuclei di fibroblasti embrionali umani (MRC5), dopo 2h30’ di infezione con lo stipite virale AD169 [molteplicità di infezione (m.o.i.) pari a 0,5], in assenza dell’inibitore dell’attività proteolitica dei proteasomi (pannello “- MG132”) o in presenza dell’inibitore MG132 (pannello “+ MG132”), utilizzato alla concentrazione di 0,5 µM. Per la reazione di immunofluorescenza è stato impiegato un anticorpo monoclonale in grado di riconoscere epitopi delle proteine virali precocissime IEp72 e IEp86 (proteine di peso molecolare 72 KDa e 86 KDa, rispettivamente, prodotti principali dei geni IE1 e IE2). 6.2.2 L’ATTIVITÀ ENZIMATICA DEI PROTEASOMI RISULTA RILEVANTE SOLO IN ALCUNE FASI
DEL CICLO CELLULARE, AI FINI DELL’EVOLUZIONE DELL’INFEZIONE PRODUTTIVA DA
CITOMEGALOVIRUS IN FIBROBLASTI MRC5
La fase di studio successiva è stata pertanto volta a verificare se l’attività proteasomale
fosse più rilevante in alcune fasi del ciclo cellulare piuttosto che in altre, durante stadi
molto precoci dell’infezione (2 ore e 30 minuti). I risultati di questa serie di esperimenti è
mostrata nelle Figure 23, 24, 25 e 26. Nello specifico, sulla scorta di dati di letteratura
(Morel A.P. et al., 2003; Memili E. et al., 2004), le cellule MRC5 sono state sincronizzate
mediante coltivazione in assenza di siero fetale di vitello per 5 giorni; al termine di tale
periodo, il suddetto terreno è stato sostituito con terreno addizionato del 10% di siero di
vitello fetale per i tempi prestabili corrispondenti alle diverse fasi del ciclo cellulare prima
di infettare le cellule con lo stipite AD169 di HCMV. In particolare, l’infezione
sperimentale con il suddetto stipite virale [Figure 23-26, pannelli B: espressione delle
proteine virali precocissime] è stata attuata dopo 9, 18, 24 e 30 ore dal ripristino del siero
nel terreno di coltura dei monostrati di MRC5, in corrispondenza delle fasi G1 [Figura 23],

6. Risultati
-133-
G1/S [Figura 24], S [Figura 25] e G2/M [Figura 26] del ciclo cellulare, rispettivamente.
L’avvenuta sincronizzazione dei tappeti cellulari nella fase del ciclo cellulare attesa veniva
puntualmente verificata attraverso un’analisi citofluorimetrica del contenuto di DNA in
monostrati di MRC5, allestiti in parallelo a quelli utilizzati per l’infezione e trattati nelle
stesse condizioni sperimentali [Figure 23-26, pannelli A]. Due ulteriori serie di monostrati,
di cui una allestita per lo studio di distribuzione del DNA [Figure 23-26, pannelli C],
l’altra per l’infezione virale [Figure 23-26, pannelli D], venivano anche sottoposte a pre-
trattamento con MG132 per 3 ore; in entrambe le serie, la sostanza veniva costantemente
mantenuta nel terreno di coltura.
I dati ottenuti mostrano innanzitutto che, in cellule infettate e non trattate con MG132
[Figure 23-26, pannelli B: controlli di infezione, in cui i proteasomi sono potenzialmente
attivi], l’espressione nucleare delle proteine virali è buona quando l’infezione inizia in fase
G1 [Figura 23, pannello B], decisamente incrementata quando prende avvio alla
transizione G1/S [Figura 24, pannello B], mentre risulta sensibilmente minore se il ciclo
replicativo virale inizia nelle fasi S [Figura 25, pannello B] e alla transizione G2/M [Figura
26, pannello B]. In cellule trattate con MG132, l’effetto inibitorio sull’espressione dei
prodotti principali dei geni virali precocissimi è evidente se l’infezione parte in G1 [Figura
23, pannello D] , ancora più significativa alla transizione G1/S [Figura 24, pannello D],
scarsa o nulla in S e G2/M [Figure 25 e 26, rispettivamente, pannelli D].
Questa serie di esperimenti, sembra supportare un ruolo di spicco dei proteasomi proprio
durante fasi cruciali del ciclo di replicazione litico di HCMV.
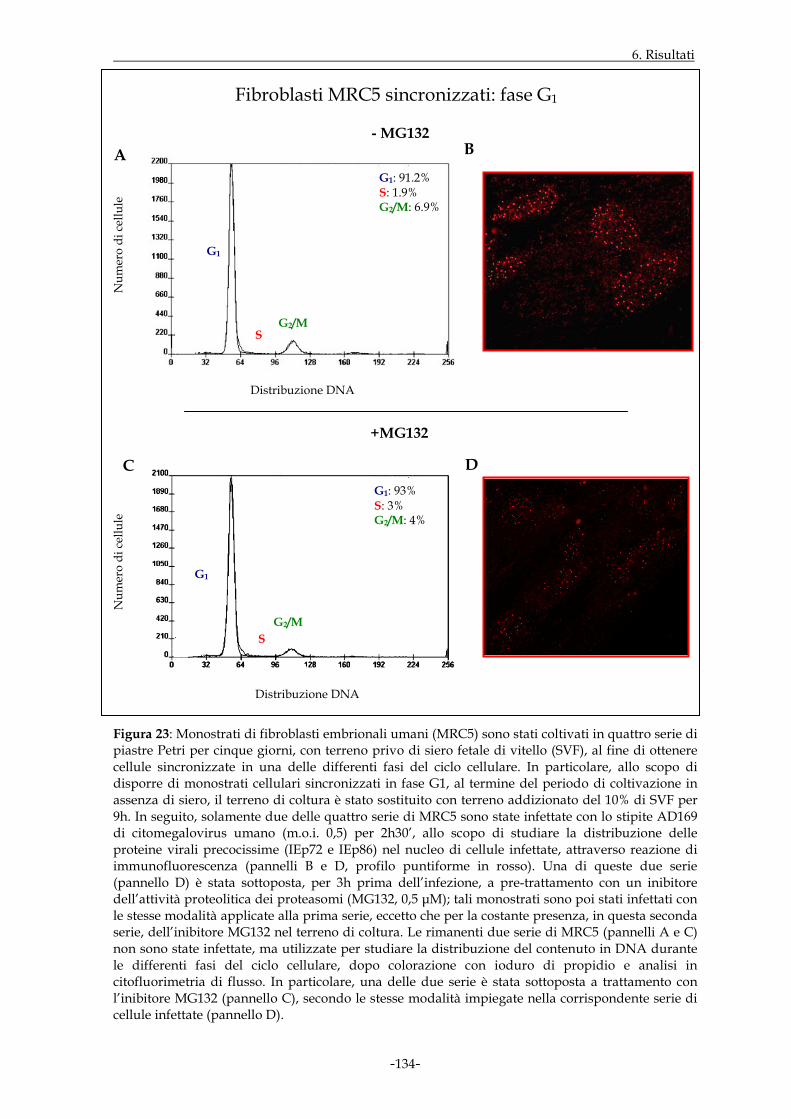
6. Risultati
-134-
Num
ero
di c
ellu
le
G1: 93% S: 3% G2/M: 4%
Distribuzione DNA
G1
S G2/M
C D
A
Distribuzione DNA
Num
ero
di c
ellu
le
G1: 91.2% S: 1.9% G2/M: 6.9%
G1
S G2/M
- MG132 B
+MG132
Fibroblasti MRC5 sincronizzati: fase G1
Figura 23: Monostrati di fibroblasti embrionali umani (MRC5) sono stati coltivati in quattro serie di piastre Petri per cinque giorni, con terreno privo di siero fetale di vitello (SVF), al fine di ottenere cellule sincronizzate in una delle differenti fasi del ciclo cellulare. In particolare, allo scopo di disporre di monostrati cellulari sincronizzati in fase G1, al termine del periodo di coltivazione in assenza di siero, il terreno di coltura è stato sostituito con terreno addizionato del 10% di SVF per 9h. In seguito, solamente due delle quattro serie di MRC5 sono state infettate con lo stipite AD169 di citomegalovirus umano (m.o.i. 0,5) per 2h30’, allo scopo di studiare la distribuzione delle proteine virali precocissime (IEp72 e IEp86) nel nucleo di cellule infettate, attraverso reazione di immunofluorescenza (pannelli B e D, profilo puntiforme in rosso). Una di queste due serie (pannello D) è stata sottoposta, per 3h prima dell’infezione, a pre-trattamento con un inibitore dell’attività proteolitica dei proteasomi (MG132, 0,5 µM); tali monostrati sono poi stati infettati con le stesse modalità applicate alla prima serie, eccetto che per la costante presenza, in questa seconda serie, dell’inibitore MG132 nel terreno di coltura. Le rimanenti due serie di MRC5 (pannelli A e C) non sono state infettate, ma utilizzate per studiare la distribuzione del contenuto in DNA durante le differenti fasi del ciclo cellulare, dopo colorazione con ioduro di propidio e analisi in citofluorimetria di flusso. In particolare, una delle due serie è stata sottoposta a trattamento con l’inibitore MG132 (pannello C), secondo le stesse modalità impiegate nella corrispondente serie di cellule infettate (pannello D).
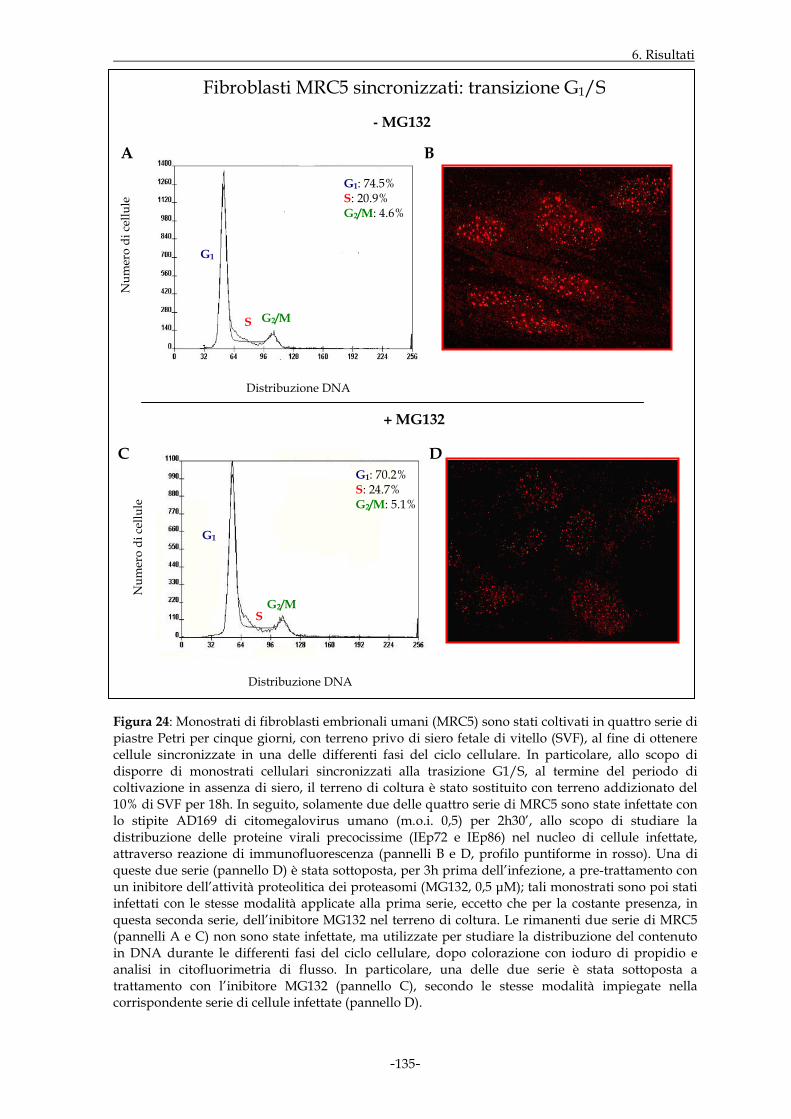
6. Risultati
-135-
Figura 24: Monostrati di fibroblasti embrionali umani (MRC5) sono stati coltivati in quattro serie di piastre Petri per cinque giorni, con terreno privo di siero fetale di vitello (SVF), al fine di ottenere cellule sincronizzate in una delle differenti fasi del ciclo cellulare. In particolare, allo scopo di disporre di monostrati cellulari sincronizzati alla trasizione G1/S, al termine del periodo di coltivazione in assenza di siero, il terreno di coltura è stato sostituito con terreno addizionato del 10% di SVF per 18h. In seguito, solamente due delle quattro serie di MRC5 sono state infettate con lo stipite AD169 di citomegalovirus umano (m.o.i. 0,5) per 2h30’, allo scopo di studiare la distribuzione delle proteine virali precocissime (IEp72 e IEp86) nel nucleo di cellule infettate, attraverso reazione di immunofluorescenza (pannelli B e D, profilo puntiforme in rosso). Una di queste due serie (pannello D) è stata sottoposta, per 3h prima dell’infezione, a pre-trattamento con un inibitore dell’attività proteolitica dei proteasomi (MG132, 0,5 µM); tali monostrati sono poi stati infettati con le stesse modalità applicate alla prima serie, eccetto che per la costante presenza, in questa seconda serie, dell’inibitore MG132 nel terreno di coltura. Le rimanenti due serie di MRC5 (pannelli A e C) non sono state infettate, ma utilizzate per studiare la distribuzione del contenuto in DNA durante le differenti fasi del ciclo cellulare, dopo colorazione con ioduro di propidio e analisi in citofluorimetria di flusso. In particolare, una delle due serie è stata sottoposta a trattamento con l’inibitore MG132 (pannello C), secondo le stesse modalità impiegate nella corrispondente serie di cellule infettate (pannello D).
- MG132
Fibroblasti MRC5 sincronizzati: transizione G1/S
Num
ero
di c
ellu
le
G2/M
S
G1
G1: 70.2% S: 24.7% G2/M: 5.1%
Distribuzione DNA
+ MG132
C D
Distribuzione DNA
Num
ero
di c
ellu
le
G1: 74.5% S: 20.9% G2/M: 4.6%
G1
S G2/M
A B
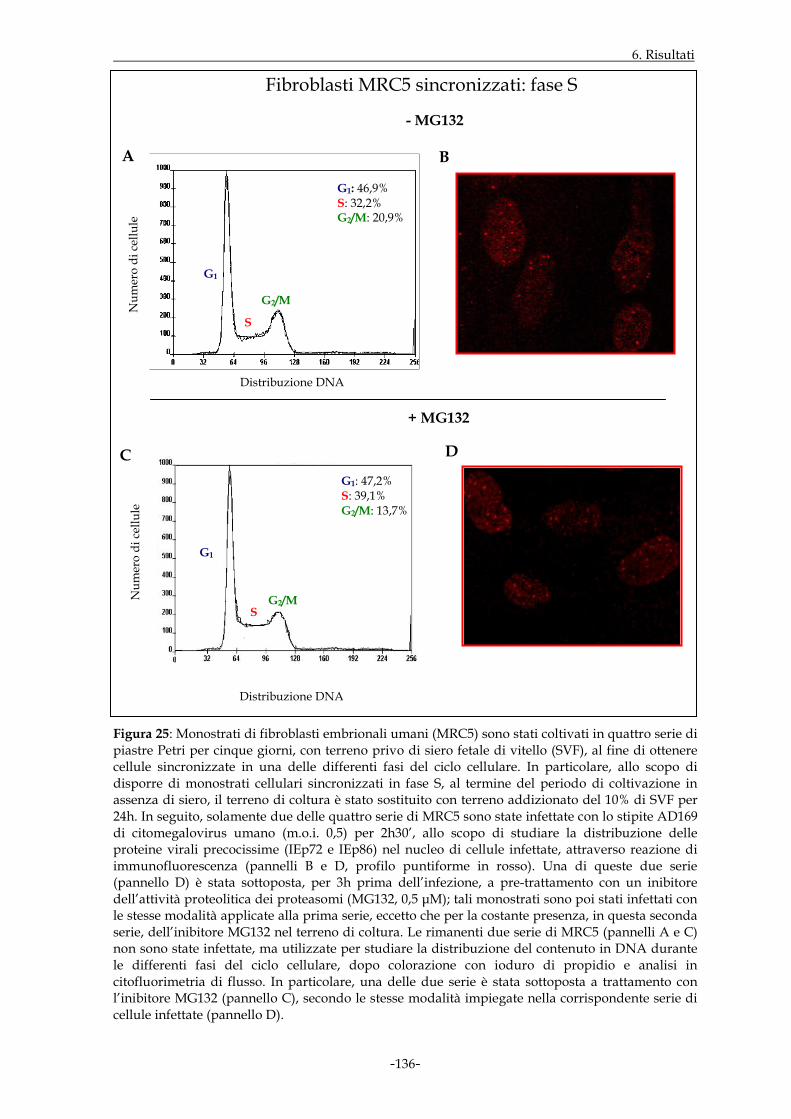
6. Risultati
-136-
+ MG132
Num
ero
di c
ellu
le
G1: 46,9% S: 32,2% G2/M: 20,9%
Distribuzione DNA
G1
S G2/M
Num
ero
di c
ellu
le
G2/M S
G1
G1: 47,2% S: 39,1% G2/M: 13,7%
Distribuzione DNA
C D
Fibroblasti MRC5 sincronizzati: fase S
A B
- MG132
Figura 25: Monostrati di fibroblasti embrionali umani (MRC5) sono stati coltivati in quattro serie di piastre Petri per cinque giorni, con terreno privo di siero fetale di vitello (SVF), al fine di ottenere cellule sincronizzate in una delle differenti fasi del ciclo cellulare. In particolare, allo scopo di disporre di monostrati cellulari sincronizzati in fase S, al termine del periodo di coltivazione in assenza di siero, il terreno di coltura è stato sostituito con terreno addizionato del 10% di SVF per 24h. In seguito, solamente due delle quattro serie di MRC5 sono state infettate con lo stipite AD169 di citomegalovirus umano (m.o.i. 0,5) per 2h30’, allo scopo di studiare la distribuzione delle proteine virali precocissime (IEp72 e IEp86) nel nucleo di cellule infettate, attraverso reazione di immunofluorescenza (pannelli B e D, profilo puntiforme in rosso). Una di queste due serie (pannello D) è stata sottoposta, per 3h prima dell’infezione, a pre-trattamento con un inibitore dell’attività proteolitica dei proteasomi (MG132, 0,5 µM); tali monostrati sono poi stati infettati con le stesse modalità applicate alla prima serie, eccetto che per la costante presenza, in questa seconda serie, dell’inibitore MG132 nel terreno di coltura. Le rimanenti due serie di MRC5 (pannelli A e C) non sono state infettate, ma utilizzate per studiare la distribuzione del contenuto in DNA durante le differenti fasi del ciclo cellulare, dopo colorazione con ioduro di propidio e analisi in citofluorimetria di flusso. In particolare, una delle due serie è stata sottoposta a trattamento con l’inibitore MG132 (pannello C), secondo le stesse modalità impiegate nella corrispondente serie di cellule infettate (pannello D).
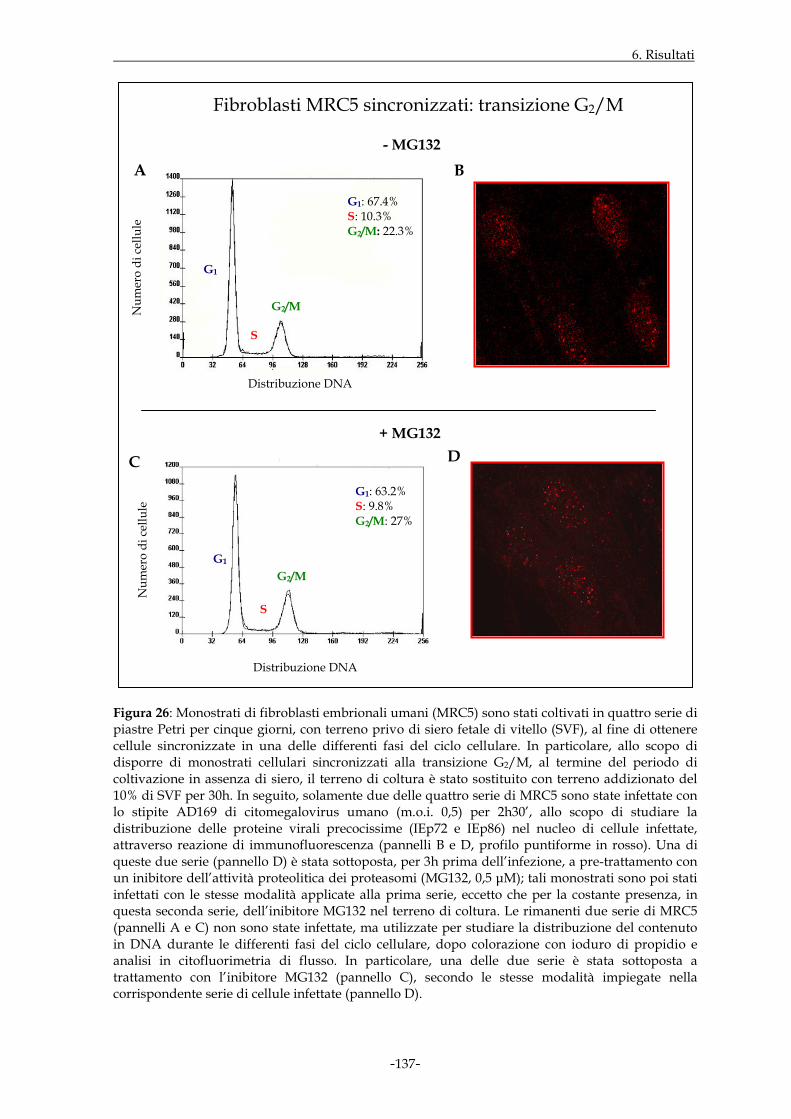
6. Risultati
-137-
Figura 26: Monostrati di fibroblasti embrionali umani (MRC5) sono stati coltivati in quattro serie di piastre Petri per cinque giorni, con terreno privo di siero fetale di vitello (SVF), al fine di ottenere cellule sincronizzate in una delle differenti fasi del ciclo cellulare. In particolare, allo scopo di disporre di monostrati cellulari sincronizzati alla transizione G2/M, al termine del periodo di coltivazione in assenza di siero, il terreno di coltura è stato sostituito con terreno addizionato del 10% di SVF per 30h. In seguito, solamente due delle quattro serie di MRC5 sono state infettate con lo stipite AD169 di citomegalovirus umano (m.o.i. 0,5) per 2h30’, allo scopo di studiare la distribuzione delle proteine virali precocissime (IEp72 e IEp86) nel nucleo di cellule infettate, attraverso reazione di immunofluorescenza (pannelli B e D, profilo puntiforme in rosso). Una di queste due serie (pannello D) è stata sottoposta, per 3h prima dell’infezione, a pre-trattamento con un inibitore dell’attività proteolitica dei proteasomi (MG132, 0,5 µM); tali monostrati sono poi stati infettati con le stesse modalità applicate alla prima serie, eccetto che per la costante presenza, in questa seconda serie, dell’inibitore MG132 nel terreno di coltura. Le rimanenti due serie di MRC5 (pannelli A e C) non sono state infettate, ma utilizzate per studiare la distribuzione del contenuto in DNA durante le differenti fasi del ciclo cellulare, dopo colorazione con ioduro di propidio e analisi in citofluorimetria di flusso. In particolare, una delle due serie è stata sottoposta a trattamento con l’inibitore MG132 (pannello C), secondo le stesse modalità impiegate nella corrispondente serie di cellule infettate (pannello D).
Fibroblasti MRC5 sincronizzati: transizione G2/M
+ MG132
- MG132
Distribuzione DNA
Num
ero
di c
ellu
le G1: 63.2%
S: 9.8% G2/M: 27%
G1
S
G2/M
C D
A B
Num
ero
di c
ellu
le
G1: 67.4% S: 10.3% G2/M: 22.3%
Distribuzione DNA
G1
S
G2/M

6. Risultati
-138-
6.2.3 I PROTEASOMI SONO PRESENTI ANCHE A LIVELLO NUCLEOLARE A TEMPI PRECOCI
DOPO L’INFEZIONE DI FIBROBLASTI MRC5 CON CITOMEGALOVIRUS UMANO
Il successivo quesito scientifico, a cui si è tentato di dare risposta, prende in
considerazione studi di tipo morfologico, atti a stabilire la distribuzione spaziale di
determinati complessi molecolari, potenzialmente in grado di interagire in particolari
condizioni con HCMV. In termini generali, tale filone di studi si basa sul presupposto che
la nozione spaziale sembra essere imprescindibile da quella funzionale, dal momento che
sembra ormai appurato che l’attività di un determinato complesso molecolare non possa
espletarsi se non negli spazi (oltre che nei tempi) corretti.
Pertanto, analogamente a quanto effettuato nell’ambito dello studio del modello
muscolare, è sembrato importante, anche in questo caso, verificare se la distribuzione di
specifici proteasomi potesse subire variazioni in corso di infezione da citomegalovirus,
rispetto ad un profilo previsto in condizioni fisiologiche.
A tale scopo, è stato effettuato uno studio, mediante microscopia confocale, su singole
sezioni focali di cellule MRC5 infettate per 2 ore e 30 minuti con lo stipite AD169 di
HCMV, poi fissate ed utilizzate per reazioni di doppia colorazione in
immunofluorescenza, mediante l’impiego di un anticorpo policlonale diretto contro
diverse subunità del proteasoma 20S ed una miscela di anticorpi monoclonali diretti
contro epitopi di IEp72 e IEp86, prodotti principali dei geni virali precocissimi [Figura 27].
I risultati ottenuti rivelano che i proteasomi si localizzano nel nucleolo di un numero
significativo di cellule infettate (in questa serie di esperimenti le cellule non erano
sincronizzate), a differenza di quanto mostrato nella stessa figura nell’ambito di
fibroblasti MRC5 non infettati (monostrati allestiti in parallelo nell’ambito dello stesso
esperimento); tale osservazione è ulteriormente supportata da quanto descritto in
letteratura per cellule non infettate e appartenenti a diverse tipologie (inclusi i fibroblasti),
in cui i proteasomi sono localizzati nel citoplasma ed in differenti compartimenti nucleari,
ad eccezione del nucleolo (Amsterdam A. et al., 1993; De Conto F. et al., 2000; Reits E.A.J.
et al., 1997; Groothuis T.A.M. and Reits E.A.J., 2005). Inoltre, in questa stessa serie di
esperimenti è stato effettuato uno studio sulla possibile interazione tra proteasomi e
proteine virali precocissime, anch’esso espletato attraverso un’analisi di singole sezioni
focali di nuclei infettati. I dati ottenuti dimostrano l’esistenza di una chiara
colocalizzazione tra le suddette proteine virali e proteasomi, in regioni granulari di
accumulo, a volte dislocate in aree peri-nucleolari. La distribuzione nucleolare del
proteasoma in corso di infezione sperimentale da HCMV, come anche la sua associazione

6. Risultati
-139-
con i prodotti maggiori dei geni virali precocissimi, sottolinea di nuovo un ruolo di spicco
di questi complessi molecolari cellulari nell’ambito del ciclo litico da HCMV. La
dislocazione dei proteasomi in aree nucleari non occupate in condizioni fisiologiche, quali
i nucleoli, indica inoltre come questi compartimenti nucleari rappresentino,
verosimilmente, siti di interesse nell’ambito dell’infezione virale.
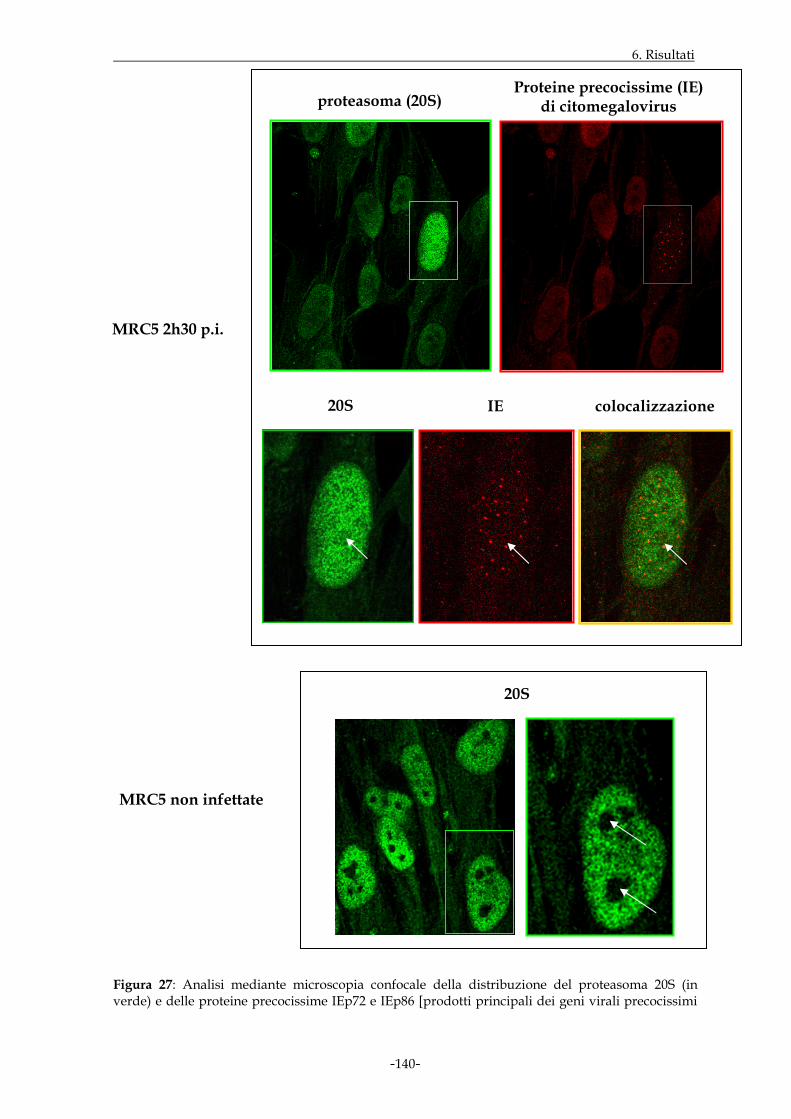
6. Risultati
-140-
Figura 27: Analisi mediante microscopia confocale della distribuzione del proteasoma 20S (in verde) e delle proteine precocissime IEp72 e IEp86 [prodotti principali dei geni virali precocissimi
MRC5 2h30 p.i.
proteasoma (20S) Proteine precocissime (IE)
di citomegalovirus
20S IE colocalizzazione
20S
MRC5 non infettate

6. Risultati
-141-
(IE1 e IE2) di citomegalovirus umano (in rosso), in monostrati di fibroblasti embrionali umani MRC5, dopo 2h30’ di infezione con lo stipite virale AD169 (m.o.i. = 0,5)]. Il profilo di distribuzione proteasomale in cellule MRC5, in corso di infezione virale, è messo a confronto con quello osservato nella stessa tipologia di cellula in condizioni fisiologiche (pannelli “MRC5 non infettate”). Le figure mostrano un solo piano focale, che passa attraverso la regione centrale dei nucleoli (indicati dalle frecce a livello dell’immagini a più alto ingrandimento, sia nell’ambito di MRC5 in corso di infezione, che nelle stesse cellule non infettate). La reazione di immunofluorescenza è stata eseguita mediante l’impiego di un anticorpo policlonale diretto nei confronti di diverse subunità del complesso proteasomale 20S e di una miscela di anticorpi monoclonali che riconoscono epitopi delle proteine virali precocissime IEp72 e IEp86 di citomegalovirus. Lo studio di colocalizzazione mostra la presenza di diverse regioni di sovrapposizione tra il proteasoma 20S e le proteine virali (profilo puntiforme in arancio).
6.2.4 PRODUZIONE DI VETTORI RETROVIRALI RECANTI LE SEQUENZE GENICHE
PROTEASOMALI DI INTERESSE ED IN GRADO DI INFETTARE CELLULE UMANE, A LORO VOLTA
SUSCETTIBILI ALL’INFEZIONE DA CITOMEGALOVIRUS
Quale ultimo, ambizioso “tassello” di questo progetto ed in analogia a quanto prodotto
nell’ambito del modello muscolare, ci si è proposti di allestire strumenti molecolari che
potessero risultare idonei, in prospettiva futura, allo studio dei proteasomi in modelli
cellulari permissivi per l’infezione con HCMV.
Nel tentativo di aggirare alcuni ostacoli tecnici legati alla presenza di fattori di inibizione,
prodotti nelle linee cellulari di origine murina, che causano, a loro volta, una
considerevole riduzione del titolo di retrovirus ricombinanti, l’approccio sperimentale si è
ispirato a quanto di recente è comparso in letteratura, riguardo alla produzione di
particelle retrovirali ricombinanti, ovvero all’utilizzo di linee cellulari di cane o umane; in
particolare, è stato introdotto un sistema (Soneoka Y. et al., 1995; Pear W.S. et al., 1997) che
prevede una co-trasfezione di cellule denominate HEK 293 (cellule embrionali di rene
umano).
Nello specifico, la linea cellulare HEK 293 è stata trasfettata contemporaneamente con tre
vettori di espressione: un plasmide recante i geni gag-pol del retrovirus della leucemia
murina (MoMLV), un plasmide recante il gene VSV-G del virus della stomatite vescicolare
ed infine, i vettori retrovirali ricombinanti allestiti nell’ambito di questo progetto
(pDONdelta-PSMA6-EGFP, pDONdelta-PSMB2-EGFP), in cui sono presenti le sequenze
proteasomali di interesse in associazione a quella per la proteina fluorescente EGFP o, in
alternativa, la sola sequenza relativa alla proteina EGFP (pDONdelta-EGFP). Tale sistema
ha consentito la produzione di particelle retrovirali con titoli pari a 1x10x5 ufc/ml. .
La scelta del modello cellulare permissivo all’infezione da citomegalovirus umano è
caduta sulla linea cellulare MRC5-hTERT; in particolare, si tratta di fibroblasti di polmone
embrionale umano immortalizzati, del tutto analoghi, quanto a recettività all’infezione da

6. Risultati
-142-
HCMV, alle corrispondenti cellule non immortalizzate (MRC5) (McSherry B.P. et al.,
2001). Il vantaggio strategico è stato quello di potere utilizzare MRC5-hTERT per ottenere
popolazioni di cellule che esprimessero le proteine di fusione di interesse, potendole
conservare, potenzialmente, per periodi di tempo illimitati e superando così l’ostacolo
legato al potenziale di vita finito dei fibroblasti MRC5.
La linea cellulare MRC5-hTERT è stata infettata con le sospensioni virali PSMA6-EGFP/rv
e PSMB2-EGFP/rv, al fine di selezionare e ottenere popolazioni di cellule MRC5-hTERT
che esprimessero stabilmente le proteine di fusione costituite dalle sequenze proteasomali
oggetto di questo studio e, al contempo, la proteina EGFP. Come controllo, sono state
ottenute anche popolazioni di cellule MRC5-hTERT in grado di esprimere la sola proteina
fluorescente verde EGFP.
L’analisi mediante microscopia confocale delle suddette popolazioni cellulari [Figura 28,
pannelli A], a confronto con la distribuzione dei proteasomi ottenuta nelle stesse cellule
(non ingegnerizzate con le proteine di fusione sopra menzionate), attraverso una reazione
di immunofluorescenza con anticorpi policlonali diretti contro diverse subunità del
complesso 20S [Figura 28, pannelli B], mostra, in entrambi i casi, una distribuzione sia
citoplasmatica che nucleare dei proteasomi, anche se nettamente a favore del distretto
nucleare, spesso accentrata in regioni di accumulo che si estrinsecano sotto forma di
granuli brillanti. In entrambi i casi (cellule ingegnerizzate e non), il nucleolo risulta
escluso dalla distribuzione proteasomale nucleare, come chiaramente dimostrato dalle
immagini di Figura 28, corrispondenti ad una singola sezione focale, che passa attraverso
la parte centrale dei nucleoli.
Questa ultima serie di dati, sebbene preliminare, sembra attestare, in assenza di infezione
da citomegalovirus, la bontà degli strumenti molecolari messi a punto e suggerisce quindi
il potenziale impiego delle suddette popolazioni cellulari per studi futuri di cinetica,
nell’ambito del ciclo replicativo litico da HCMV in cellule viventi.
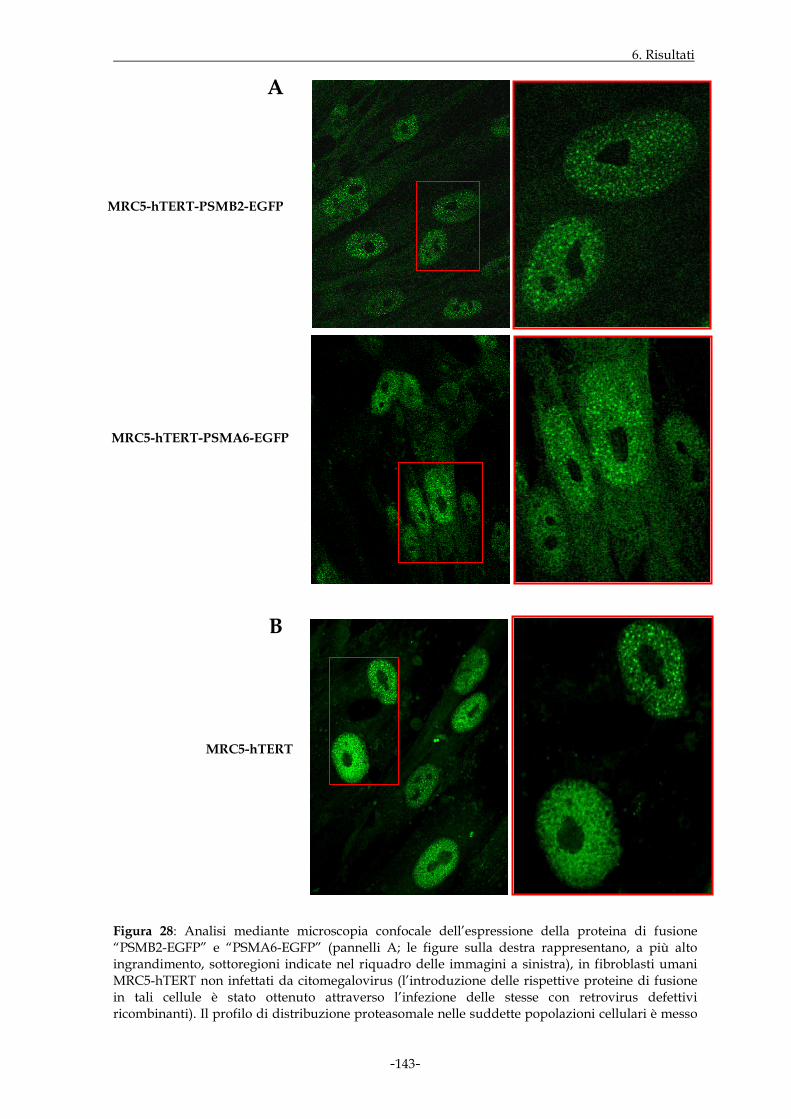
6. Risultati
-143-
MRC5-hTERT-PSMA6-EGFP
MRC5-hTERT-PSMB2-EGFP
A
MRC5-hTERT
B
Figura 28: Analisi mediante microscopia confocale dell’espressione della proteina di fusione “PSMB2-EGFP” e “PSMA6-EGFP” (pannelli A; le figure sulla destra rappresentano, a più alto ingrandimento, sottoregioni indicate nel riquadro delle immagini a sinistra), in fibroblasti umani MRC5-hTERT non infettati da citomegalovirus (l’introduzione delle rispettive proteine di fusione in tali cellule è stato ottenuto attraverso l’infezione delle stesse con retrovirus defettivi ricombinanti). Il profilo di distribuzione proteasomale nelle suddette popolazioni cellulari è messo

6. Risultati
-144-
a confronto con quello osservato nella stessa tipologia di cellula non ingegnerizzata per l’espressione delle proteine di fusione in oggetto (pannelli B). Le figure mostrano un solo piano focale, che passa attraverso la parte centrale dei nucleoli. Pannelli B: la reazione in immunofluorescenza è stata eseguita mediante l’impiego di un anticorpo policlonale diretto nei confronti di diverse subunità del complesso proteasomale 20S. Anche in questo caso, la figura sulla destra rappresenta, a più alto ingrandimento, una sottoregione indicata nel riquadro dell’immagine a sinistra.

7. Discussione
7. DISCUSSIONE

7. Discussione
-146-
Il lavoro sperimentale oggetto di questo studio nasce da un’interessante problematica
scientifica volta a mettere in luce la partecipazione dei proteasomi, complessi molecolari
di grande rilievo nell’ambito dell’economia cellulare, nel controllo di processi cruciali per
la cellula, non solo in condizioni fisiologiche, ma anche in corso di infezione virale.
Numerosi dati di letteratura depongono per una diretta partecipazione dei proteasomi
nella regolazione di diversi eventi cellulari, quali la trascrizione, l’apoptosi e la corretta
progressione del ciclo cellulare (Orlowski R.Z., 1999; Grimm L.M. and Osborne B.A., 2000;
Mann C. and Hilt W., 2000, Yew P.R., 2001; Naujokat C. and Hoffmann S., 2002); in
particolare, tali complessi molecolari agirebbero modulando e scandendo il “turnover”
proteico, attraverso una selettiva e puntuale ricognizione delle proteine marcate con
molecole di ubiquitina. I proteasomi sarebbero, inoltre, direttamente implicati in processi
di attivazione/repressione di specifici fattori di trascrizione, come anche nella produzione
di peptidi antigenici in grado di associarsi al complesso maggiore di istocompatibilità di
tipo I (Rock K.L. et al., 1994; Groettrup M. et al., 2001b; Kloetzel P.M., 2001). Il processo di
degradazione proteica ubiquitina-dipendente non rende comunque ragione del complesso
ruolo svolto dai proteasomi; tali particelle, inizialmente osservate come subcomplessi di
ribonucleoproteine non tradotte (Schmid H.P. et al., 1984), sembrano, infatti, essere
implicate in importanti funzioni regolatorie per la cellula, non solo in termini di
catabolismo inteso in senso stretto, ma piuttosto di omeostasi proteica, la cui realizzazione
è di fondamentale rilievo soprattutto nel corso di situazioni altamente “dinamiche”, quali,
ad esempio, il differenziamento cellulare e l’infezione virale (Fagan J.M. et al., 1997;
Kitzmann M. and Fernandez A., 2001; Everett R.D. et al., 1998; Gonzalez S.L. et al., 2001;
Berezutskaya E. et al., 1997; Boyer S.N. et al., 1996; Jones D.L. and Münger K., 1997;
Reinstein E. et al., 2000; Kalejta R.F. and Shenk T., 2002). In diretto riferimento a
quest’ultima condizione, in realtà, rimangono ancora aperte e del tutto incomplete le
conoscenze volte a delucidare le modalità di intervento dei proteasomi in eventi che
comportano una cospicua alterazione del normale equilibrio omeostatico cellulare,
provocando, di conseguenza, una decisa e marcata rimodulazione quali/quantitativa di
diverse componenti molecolari.
In correlazione alla complessa problematica scientifica sopra enunciata, ossia allo scopo di
cercare di appurare ulteriormente l’eventuale partecipazione di specifici proteasomi in
eventi ritenuti cruciali per la cellula, nell’ambito di tale lavoro sperimentale sono stati
impiegati due distinti modelli di riferimento, rappresentati, rispettivamente, da cellule
muscolari di ratto, impiegate allo scopo di effettuare studi sul differenziamento cellulare,

7. Discussione
-147-
e da cellule fibroblastiche umane, utilizzate, invece, per lo studio dell’infezione virale in
vitro da parte di uno stipite umano di citomegalovirus.
In riferimento al primo modello di studio proposto, alcuni dati ottenuti in precedenza dal
nostro gruppo di ricerca (De Conto F. et al., 1997), come anche dal gruppo di ricerca
diretto dal Prof. Jean Foucrier (Foucrier J. et al., 1999), avevano consentito di appurare la
presenza di specifici proteasomi (20S) in cellule muscolari di topo (mioblasti) e in colture
primarie di cellule satelliti di ratto coltivate in vitro. Tali complessi molecolari erano stati
evidenziati impiegando, allo scopo, sonde immunologiche dirette nei confronti di
specifiche subunità proteasomali; inoltre, le osservazioni morfologiche ottenute erano
state successivamente avvalorate dall’esito di studi biochimici che avevano consentito di
mettere in luce l’assenza di subunità proteasomali libere; pertanto, la localizzazione
evidenziata per le proteine esaminate poteva riferirsi direttamente a quella delle
corrispondenti particelle proteasomali includenti tali subunità. L’analisi dei risultati
ottenuti aveva, in particolare, consentito di evidenziare una caratteristica e peculiare
distribuzione dei proteasomi, sia a livello del nucleo che del citoplasma, con una
localizzazione strettamente correlabile allo specifico stadio di differenziamento muscolare
raggiunto ed al tipo di subunità proteasomale esaminata, partendo in ogni caso dallo
stadio iniziale di riferimento, rappresentato da cellule mioblastiche, per giungere poi alla
valutazione di uno stadio più avanzato di differenziamento, rappresentato dalla fusione
delle stesse cellule in miotubi. Nello specifico, tali studi avevano evidenziato che, se nei
mioblasti di topo la localizzazione di specifici proteasomi appare di tipo prevalentemente
citoplasmatico, seppure con un eventuale marcaggio transiente della regione nucleare,
durante la fase di fusione dei mioblasti la distribuzione di specifiche subunità
proteasomali torna inizialmente ad essere appannaggio del nucleo, per poi stabilizzarsi
definitivamente nel citoplasma, dove si esprime con un caratteristico profilo puntiforme e
dove risulta parzialmente sovrapponibile, in particolare, alla distribuzione dei
microfilamenti di actina (De Conto F. et al., 1997; Foucrier J. et al., 1999).
Nei miotubi di colture primarie di cellule satelliti di ratto (queste ultime in grado di
raggiungere uno stadio più elevato di differenziamento muscolare), i proteasomi si
dispongono secondo una caratteristica distribuzione che “mima” l’assetto sarcomerico,
ossia presentano un’organizzazione che precorre la formazione di una struttura
sarcomerica ben definita (Foucrier J. et al., 1999). Tale organizzazione proteasomale è stata
successivamente osservata anche in sezioni longitudinali di tessuto muscolare scheletrico

7. Discussione
-148-
e in colture primarie di cardiomiociti ventricolari di ratto. Infine, tali dati sono stati
ulteriormente avvalorati dal rilevamento di un’analoga distribuzione del profilo proteico
proteasomale in cellule muscolari lisce dell’aorta di ratto, nonostante, in quest’ultima
tipologia di cellule, non si arrivi, se non raramente, ad osservare la formazione di una
struttura sarcomerica completamente funzionale, dal momento che la miogenesi si arresta
in una fase assai precoce (Foucrier J. et al., 2001).
La localizzazione intracellulare dei proteasomi costituisce, a tutt’oggi, un interessante e
vivace elemento di discussione e di dibattito, dal momento che tali particelle sono state
rinvenute sia a livello nucleare che citoplasmatico in diversi modelli cellulari (Rivett A.J. et
al., 1998) e, in entrambi i casi, erano presenti quale componente molecolare libera (Reits
E.A.J. et al., 1997; Groothuis T.A.M. and Reits E.A.J., 2005), oppure direttamente associati a
specifici elementi cellulari, in particolar modo a filamenti del citoscheletro (Olink-Coux M.
et al., 1994; Arcangeletti M.C. et al., 2000; Arcangeletti M.C. et al., 1997). In aggiunta a tali
considerazioni, questi ed altri studi hanno posto l’accento sul fatto che la localizzazione
dei proteasomi non sembrerebbe essere di natura statica, bensì sono state evidenziate
diverse possibilità di scambio e di veicolazione di tali complessi tra i compartimenti
nucleare e citoplasmatico, rispettivamente (Reits E.A.J. et al., 1997; De Conto F. et al., 1997,
Foucrier J. et al., 1999; Groothuis T.A.M. and Reits E.A.J., 2005).
Nello specifico, l’implicazione dei proteasomi in corso di differenziamento cellulare è
stata, ad esempio, avvalorata da diversi studi che ne hanno messo in luce il possibile ruolo
svolto non solo nel corso della miogenesi, ma anche in altri modelli cellulari di
riferimento. Così, ad esempio, alcuni Autori hanno valutato il ruolo svolto da tali
complessi molecolari nell’elaborazione di molecole segnale nel corso della morfogenesi
del tessuto osseo, soprattutto in riferimento a particolari categorie di proteine prodotte
dagli osteoblasti; in tale studio, è stata evidenziata l’importanza dell’attività proteasomale
che, in concomitanza e in diretta associazione allo svolgersi di altre reazioni enzimatiche
parallele, consentirebbe una costante azione di controllo sulla genesi tissutale (Zhao M. et
al., 2004). Altri dati di letteratura emersi recentemente hanno posto, invece, l’accento
sull’interazione che intercorre tra la proteina “proline-rich homeodomain protein/Hex”
(PHR), che sembrerebbe svolgere un ruolo di rilievo nel controllo della proliferazione e
del differenziamento cellulare reprimendo la trascrizione, e specifici complessi
proteasomali. Tale associazione si è resa funzionale, in particolar modo, in cellule
ematopoietiche, dove la degradazione proteica mediata dai proteasomi favorirebbe la
produzione di molecole di PHR modificate strutturalmente rispetto alla loro morfologia

7. Discussione
-149-
originaria; tali modificazioni non comprometterebbero in alcun modo la capacità di PHR
di legarsi al DNA e consentirebbero una modulazione delle funzioni codificanti
soprattutto in corso di differenziamento cellulare (Bess K.L. et al., 2003).
L’interpretazione del possibile ruolo svolto dai proteasomi in corso di differenziamento
muscolare non può prescindere, peraltro, dall’esistenza di un rigoroso e assai complesso
programma di regolazione dell’espressione genica, nell’ambito del quale risultano
direttamente coinvolti diversi fattori di regolazione miogenica, afferenti alla famiglia
bHLM quale per esempio la molecola MyoD, che funge da attivatore trascrizionale in
grado di interagire direttamente con il DNA cellulare (Davis R.L. et al., 1987), e, in una
fase più tardiva, altre molecole, tra le quali miogenina.
La fibra muscolare scheletrica matura è, inoltre, contraddistinta da un’organizzazione
strutturale assai complessa, derivante dall’accumulo progressivo di specifici elementi
citoscheletrici, dal momento che le miofibrille, che sono in grado di rimanere ancorate
nella loro posizione grazie alla presenza di una ricca rete di fibre di desmina, appaiono
costituite quasi integralmente da elementi contrattili, rappresentati principalmente dalle
proteine actina e miosina.
In letteratura sono state riportate alcune indicazioni relative alla capacità, mostrata da
numerosi enzimi, di interagire direttamente con il citoscheletro, specialmente nel tessuto
muscolare; tali interazioni, che sarebbero generalmente di natura sito-specifica,
soprattutto per quanto concerne funzioni enzimatiche deputate ad attività di tipo
metabolico (Suarez R.K., 2003), sono state evidenziate in misura maggiore in corso di
degradazione della struttura della fibra muscolare; d’altra parte, rimangono a tutt’oggi
ancora carenti, o comunque in parte confuse e discordanti tra loro, le nozioni circa la
specifica localizzazione dei proteasomi nel muscolo scheletrico maturo.
Vale la pena sottolineare, a tale proposito, che esiste ormai un nutrito filone di letteratura
che avvalora la nozione di spazio, anche nell’ambito degli studi di funzioni cellulari, sia in
condizioni fisiologiche che patologiche. In altre parole, il nucleo ed il citoplasma sono
articolati in una serie di compartimenti con precise collocazioni spaziali e funzionali, nei
quali l’aggregazione delle molecole deputate ad una specifica funzione si verifica in
maniera estremamente dinamica in risposta a specifici segnali di attivazione. Di
conseguenza, la nozione spaziale assumerebbe un’importanza analoga a quella della
funzione stessa, che non potrebbe espletarsi al di fuori di quel preciso distretto.
Sulla base di tali premesse, la ricerca in oggetto si è posta l’obiettivo iniziale di
approfondire i risultati precedentemente ottenuti dal nostro gruppo e da quello del Prof.

7. Discussione
-150-
Jean Foucrier, in particolar modo andando a valutare, in maniera più puntuale, la
localizzazione di specifici proteasomi nell’ambito della struttura sarcomerica del muscolo
scheletrico di ratto.
Un secondo, rilevante aspetto che, in maniera innovativa, ci si è proposti di sviluppare
nello studio condotto, riguarda la messa a punto di strumenti molecolari atti a permettere
l’espressione transiente o stabile di specifici proteasomi, accoppiata a quella di molecole
fluorescenti, nei modelli cellulari prescelti, nella consapevolezza del loro potenziale
utilizzo, in un’ottica futura, per studi morfologici, così come a valenza funzionale, su
cellule viventi.
L’impiego di marcatori diretti nei confronti di alcune subunità del complesso
proteasomale 20S, denominate α1/p27K e α3/p29K, nei modelli di cellule muscolari di
ratto utilizzati, ha consentito di evidenziare la distribuzione intracellulare dei
corrispondenti complessi proteasomali e di valutare se questa potesse, in qualche modo,
essere “regolata” dalla presenza di alcune componenti costitutive facenti parte della
struttura sarcomerica delle miofibrille. A diretto sostegno di tale ipotesi sperimentale e
come già accennato precedentemente, esistono numerosi dati di letteratura secondo i
quali i proteasomi non devono essere considerati come complessi proteici solubili, liberi
nel citoplasma, ma bensì associati a specifiche strutture cellulari (Arcangeletti M.C. et al.,
1997; Arcangeletti M.C. et al., 2000; Rivett A.J. et al., 2001); diversi Autori sembrano
concordi nell’affermare che, fatta eccezione per la subunità α5, tutte le altre subunità
proteiche proteasomali sono sempre state rilevate in associazione al complesso 20S
(Schmidtke G. et al., 1997; Jorgensen L. and Hendil K.B., 1999).
In particolare, per quanto attiene a questa prima parte dello studio, incentrata sulla
localizzazione proteasomale ed effettuata su sezioni sottili di muscolo scheletrico di ratto
(Bassaglia Y. et al., 2005) essa ha preso in considerazione, quali elementi strutturali di
riferimento per individuare l’organizzazione della struttura sarcomerica, le proteine:
titina, desmina e miosina. Relativamente a titina, che è stata evidenziata impiegando
un’anticorpo “anti-PEVK”, in grado di riconoscere la porzione di molecola adiacente alla
zona intermedia della banda I, è stato osservato che, in sarcomeri in stato di contrazione,
titina appare localizzata a livello della linea Z; d’altra parte, in sarcomeri in stato di
estensione, il profilo della proteina evolve ad una doppia banda, presente in prossimità
dell’interfaccia tra le bande A ed I. Parzialmente diversificato, rispetto a quello di titina,
appare il profilo di distribuzione dei proteasomi, malgrado anch’esso, come quello della
suddetta proteina sarcomerica, sia direttamente correlabile al grado di

7. Discussione
-151-
estensione/contrazione del sarcomero, suggerendo, pertanto, un’associazione stabile di
tali complessi molecolari a determinate componenti strutturali del sarcomero. Più
precisamente, il segnale di fluorescenza relativo alla subunità α1/p27K è stato osservato
sottoforma di una singola banda a livello della linea M in sarcomeri in stato di
contrazione, mentre, in sarcomeri in stato intermedio di estensione, sono state evidenziate
due bande proteasomali distinte, localizzate in prossimità della banda A; in condizioni di
massima estensione della fibra muscolare si è, infine, resa evidente una colocalizzazione
con la regione PEVK della molecola di titina. Sembra evidente potere concludere che,
malgrado tali dati supportino la nozione di localizzazione dei proteasomi a livello della
banda I, in condizioni di sarcomero disteso, evidenziata dalla sovrapposizione con titina,
è poco probabile che quest’ultima rappresenti il sito di legame ed, eventualmente, di
interazione molecolare con i proteasomi, dal momento che i profili di distribuzione di
questi due complessi proteici (proteasomi e titina) si sfasano in condizioni di sarcomero
contratto (linee M e Z, rispettivamente).
Gli esperimenti effettuati hanno, inoltre, permesso di escludere un’interazione dei
proteasomi con desmina, che risulta localizzata a livello della linea Z, come anche con
miosina, presente, invece, nella banda A. Per quanto concerne miosina, è importante
sottolineare che la sua localizzazione nell’ambito della struttura sarcomerica non varia
durante la contrazione e l’allungamento del sarcomero, a differenza di quanto riscontrato
invece per i proteasomi.
Le fasi successive della ricerca, supportate anche da studi di immunoelettromicroscopia,
hanno consentito di appurare la colocalizzazione della subunità α1/p27K con i
microfilamenti di actina; più precisamente, in condizioni di riposo, la subunità α1/p27K si
localizza a livello delle due estremità a polarità negativa dei microfilamenti di actina
presenti nel sarcomero, assumendo, pertanto, un assetto a due bande distinte; al contrario,
quando il sarcomero è in stato di contrazione, le estremità negative dei filamenti di actina
si sovrappongono in prossimità della linea M, dando ragione del fatto che la subunità
α1/p27K del proteasoma, ad esse ancorata, si appaia sottoforma di una singola banda.
Le osservazioni sopra riportate sono state successivamente confermate mediante
l’impiego di preparati di miofibrille allestiti in vitro a partire da muscoli di ratto, previa
rimozione della membrana plasmatica e sulla base di uno specifico protocollo operativo,
in grado di preservare le componenti costitutive delle miofibrille (Zhukarev V. et al.,
1997). Dato rilevante, in tali condizioni sperimentali, è che, oltre ai componenti costitutivi
del sarcomero, anche la subunità α1/p27K del proteasoma 20S continuava ad essere

7. Discussione
-152-
presente, suggerendo, pertanto, che i proteasomi siano stabilmente associati a componenti
della struttura sarcomerica (Bassaglia Y. et al. 2005). La messa in evidenza di tale
interazione rafforza ulteriormente quanto osservato in sezioni sottili di muscoli scheletrici
di ratto, del resto più direttamente riconducibili alla situazione riscontrabile in vivo, e
consente anche di escludere la possibile implicazione di eventi artefattuali, imputabili alla
particolare procedura sperimentale adottata per l’isolamento delle miofibrille.
I preparati di miofibrille sono stati, inoltre, utilizzati per definire meglio, sulla base di
rigorose valutazioni di tipo matematico, la localizzazione delle bande relative alla
subunità α1/p27K. I dati ottenuti hanno consentito di affermare che la subunità α1/p27K
è, in realtà, localizzata in corrispondenza della regione centrale dei microfilamenti
nell’ambito di un sarcomero, piuttosto che in corrispondenza della loro estremità
negativa. Tali dati sono stati supportati anche da studi effettuati su preparati di miofibrille
precedentemente trattati con gelsolina, sostanza in grado di rimuovere l’actina; i risultati
ottenuti hanno consentito di rilevare una cospicua diminuzione del segnale di
fluorescenza corrispondente alla subunità proteasomale oggetto di studio, ad ulteriore
sostegno dell’ipotesi relativa alla partecipazione di actina e, probabilmente, anche di altri
elementi citoscheletrici ad essa strettamente correlati (es. nebulina), nel determinare la
localizzazione di specifici proteasomi a livello della struttura sarcomerica.
L’associazione di specifiche subunità proteasomali con actina potrebbe rendere ragione
del fatto che tali particelle siano direttamente implicate nell’omeostasi proteica muscolare,
senza peraltro essere causa di un iniziale disassemblamento della struttura miofibrillare
(Solomon V. and Goldberg A.L., 1996; Eble D.M. et al., 1999). Del resto, non è possibile
escludere anche l’eventuale partecipazione di altri meccanismi proteolitici cellulari di tipo
non-lisosomiale, quale, ad esempio, la degradazione proteica calcio-dipendente mediata
da calpaina, che potrebbero concorrere, in concomitanza con i proteasomi, ad una corretta
morfogenesi del tessuto muscolare (Belcastro A.N. et al., 1998; Williams A.B. et al., 1999).
Tuttavia, il significato della colocalizzazione e di una eventuale associazione molecolare
tra proteasomi e microfilamenti, sistema citoscheletrico altamente dinamico ed implicato
non solo nello svolgimento di funzioni strutturali all’interno della cellula, ma anche
responsabile della specifica localizzazione citoplasmatica dei ribosomi e, pertanto,
dell’apparato di sintesi proteica (Hesketh J., 1994; Percipalle P. et al., 2001; Klyachko N.L.
et al., 2003), rimane a tutt’oggi oggetto di un’ampia gamma di interpretazioni che
evidenziano, comunque, l’importanza di effettuare ulteriori indagini sperimentali allo
scopo di approfondire, in primo luogo, la valenza funzionale dell’interazione riscontrata,

7. Discussione
-153-
soprattutto a livello molecolare. A tale riguardo, è importante sottolineare di nuovo, in
questa sede, come l’uscita dal ciclo cellulare da parte delle cellule mioblastiche coincida
con l’inizio del processo di differenziamento ed induca significative alterazioni a carico
del profilo delle proteine cellulari, comportando inoltre la comparsa di nuove molecole
(Moran J.L. et al., 2002; Tomczak K.K. et al., 2004); è pertanto ragionevole supporre che tale
stadio venga raggiunto non solo tramite un’intensa attività proteolitica, effettuata
principalmente da sistemi di tipo non-lisosomiale, tra cui, principalmente, i proteasomi,
ma anche mediante una cospicua riprogrammazione genica, allo scopo di promuovere la
sintesi di specifici mRNA richiesti dal processo di differenziamento cellulare in atto.
Come già anticipato, in una fase successiva della ricerca ci si è proposti di allestire
strumenti molecolari idonei, prospetticamente, allo studio della distribuzione dinamica
dei proteasomi durante il processo di differenziamento muscolare in cellule viventi, anche
ovviando alle possibili aberrazioni a volte indotte dall’impiego di preparati fissati.
Allo scopo, alcune sequenze geniche codificanti per specifiche subunità proteasomali sono
state fuse con sequenze codificanti per proteine fluorescenti naturali, quali la proteina
fluorescente verde (GFP, variante EGFP) o, alternativamente, la proteina fluorescente
rossa (DsRed, variante DsRed1).
L’utilizzo della proteina GFP rappresenta uno dei sistemi più diffusi per marcare proteine
cellulari, dal momento che i costrutti ottenuti possono essere espressi come proteine di
fusione, dopo opportuna trasfezione delle stesse nel modello cellulare oggetto di studio
(Zhang J. et al., 2002; Miyawaki A. et al., 2003). L’osservazione della fluorescenza emessa
dalla proteina GFP tende a riflettere, di conseguenza, la localizzazione intracellulare della
proteina presa in esame e può anche fornire importanti informazioni addizionali
riguardanti, ad esempio, i livelli di espressione del gene che l’ha codificata. Inoltre, i
cambiamenti di localizzazione intracellulare del segnale di fluorescenza riscontrato
possono essere considerati un importante indicatore della mobilità dei corrispondenti
complessi proteici marcati, sia qualora questi risultino strettamente confinati all’interno di
specifici compartimenti cellulari, come ad esempio il nucleo, sia qualora risultino in grado
di essere veicolati tra differenti compartimenti cellulari.
Alcuni interessanti dati di letteratura (Reits E.A.J. et al., 1997), ottenuti previo marcaggio
della subunità β5i dell’immunoproteasoma 20S con la proteina fluorescente GFP, al fine di
valutare se nel modello cellulare esaminato i complessi proteasomali contenenti la
proteina di fusione occupassero distretti cellulari ben determinati o, in alternativa, fossero
in grado di diffondere liberamente all’interno del compartimento citoplasmatico e/o

7. Discussione
-154-
nucleare, hanno evidenziato che tale tipologia di proteasomi è in grado di diffondere
lentamente, con un flusso unidirezionale, dal compartimento citoplasmatico verso quello
nucleare, mentre, durante la divisione cellulare, nel momento in cui la membrana
nucleare si disaggrega, i proteasomi sono liberi di diffondere nel compartimento
citoplasmatico, senza incontrare barriere selettive di alcun tipo. Una volta che la divisione
cellulare ha avuto termine, in seguito a ricostituzione delle membrane nucleari, si assiste
nuovamente ad un rallentamento del processo di diffusione dei proteasomi tra i diversi
compartimenti cellulari.
Un altro lavoro sperimentale, strettamente correlato al precedente, si riferisce invece allo
studio della localizzazione del complesso proteasomale 20S in cellule di lievito,
utilizzando, quali elementi di riferimento, le subunità α, o, in alternativa, componenti del
complesso regolatore 19S (Enenkel C. et al., 1998; Wilkinson C.R. et al., 1998). Inoltre, sono
anche stati effettuati studi atti ad approfondire le conoscenze riguardanti lo svolgimento
dei complessi meccanismi di ubiquitinizzazione delle proteine, marcando direttamente le
molecole di ubiquitina con la proteina GFP e valutandone le successive interazioni con i
complessi proteasomali (Varshavsky A., 2000; Qian S.B. et al., 2002).
Per consentire l’allestimento ottimale degli strumenti molecolari sopra menzionati, sono
state selezionate, quale oggetto di studio, le subunità α1/p27k e β4/p23k del proteasoma
20S, dal momento che tali proteine erano già state oggetto di studi di localizzazione dei
proteasomi (De Conto F. et al., 1997; Foucrier J. et al., 1999; De Conto F. et al., 2000;
Foucrier J. et al., 2001; Bassaglia Y. et al., 2005). Una volta sintetizzate le sequenze relative
alle subunità proteasomali sopra menzionate, queste sono state clonate in opportuni
vettori di espressione contenenti anche le sequenze codificanti per la proteina fluorescente
GFP o, alternativamente, per DsRed. Le fasi successive della ricerca hanno avuto, come
scopo iniziale, quello di verificare, attraverso differenti metodi impiegati in parallelo, la
bontà e la funzionalità delle proteine di fusione ottenute, eseguendo, allo scopo,
esperimenti di trasfezione in due distinti modelli cellulari di riferimento (cellule CHO e
linea mioblastica C2.7) ed ottenendo cellule che esprimevano transitoriamente o
stabilmente le proteine di fusione di interesse. Nell’uno e nell’altro caso, è stato analizzato
il profilo proteico di estratti ottenuti dalle suddette cellule, allo scopo di verificare
l’effettiva presenza dei prodotti proteici attesi.
In una fase successiva della ricerca, a partire da estratti citoplasmatici e nucleari, ottenuti
da specifici cloni cellulari (selezionati in quanto rappresentativi dell’espressione stabile
delle suddette proteine di fusione), è stato possibile dimostrare, utilizzando metodi di tipo

7. Discussione
-155-
biochimico, che le proteine di fusione α1/p27k-EGFP e β4/p23k-EGFP venivano
immediatamente utilizzate per la formazione di nuovi complessi proteasomali e, pertanto,
non sussistevano a livello intracellulare quali subunità molecolari libere.
Al fine di ottenere strumenti molecolari più “duttili” ed a maggiore concentrazione
rispetto a quelli utilizzati per i precedenti esperimenti di trasfezione, le sequenze
codificanti per le proteine di fusione α1/p27k-EGFP e β4/p23k-EGFP e, alternativamente,
la sequenza codificante per la sola proteina EGFP sono state clonate in un vettore
commerciale retrovirale, realizzato a partire da sequenze del virus della leucemia murina
di Moloney. I retrovirus offrono l’indubbio vantaggio di poter trasportare quantità
considerevoli di DNA eterologo e di garantirne un’espressione a lungo termine, in virtù
della loro capacità di integrarsi nel genoma della cellula ospite.
Inizialmente i vettori retrovirali ottenuti sono stati utilizzati per eseguire esperimenti di
trasfezione della linea cellulare Bosc-23, allo scopo di consentire la produzione di
particelle retrovirali defettive, contenenti i costrutti di interesse (PSMA6-EGFP/rv,
PSMB2-EGFP/rv, EGFP/rv e nlsLacZ/rv); tali particelle virali sono in grado di infettare
esclusivamente cellule di ratto e di topo (spettro d’ospite ecotropico).
Le sospensioni virali PSMA6-EGFP/rv, PSMB2-EGFP/rv e EGFP/rv sono state in seguito
utilizzate per eseguire infezioni della linea cellulare miogenica di topo C2.7; dopo
opportuna selezione, sono state ottenute popolazioni di cellule C2.7 in grado di esprimere
stabilmente le sequenze geniche codificanti per le proteine di fusione α1/p27k-EGFP e
β4/p23k-EGFP o, alternativamente, per la sola proteina EGFP. A partire da queste
popolazioni sono stati ottenuti estratti proteici, impiegati poi al fine di dimostrare la
presenza delle proteine di fusione in stato integrato a livello di proteasomi 20S.
In parallelo, tali costrutti molecolari sono stati valutati anche dal punto di vista della
funzione enzimatica, che può essere espletata previa incorporazione delle suddette
proteine di fusione (trasportate dai vettori retrovirali) nei complessi proteasomali 20S e
26S; anche questa serie di esperimenti ha dato risultati più che soddisfacenti,
confermando la presenza di attività proteolitica nei complessi proteasomali che
includevano le proteine di fusione in oggetto, nell’ambito dei modelli cellulari prescelti.
Come già più volte sottolineato, lo svolgimento delle procedure sperimentali molto
complesse sopra descritte, è stato finalizzato alla messa a punto di strumenti molecolari
propedeutici ad effettuare osservazioni circa la distribuzione delle subunità proteasomali
nei modelli cellulari prescelti in situazioni dinamiche, quali appunto il differenziamento
muscolare.

7. Discussione
-156-
Al di là di un futuro e più ottimale utilizzo di tali strumenti in cellule viventi, a
completamento di questa fase iniziale dello studio sono state impiegate le cellule
muscolari C2.7, esaminate sia allo stadio di mioblasti che di miotubi e preventivamente
fissate. I risultati ottenuti hanno permesso di evidenziare che il segnale di fluorescenza
relativo alle proteine di fusione era presente sia nel compartimento citoplasmatico che in
quello nucleare, con intensità decisamente maggiore in quest’ultimo distretto. Inoltre, la
fluorescenza riscontrata a livello del nucleo, in alcuni casi, appariva di tipo granulare e
concentrata a livello di regioni nucleari evocanti il profilo di distribuzione di specifici
domìni, quali i PML o i siti di “splicing” dei trascritti; tale distribuzione sembrava
accentuarsi nei nuclei dei miotubi, rispetto a quelli dello stadio mioblastico. D’altra parte,
in accordo con quanto osservato precedentemente da alcuni Autori (Amsterdam A. et al.,
1993; Reits E.A.J. et al., 1997; De Conto F. et al., 2000), il segnale di fluorescenza
proteasomale era invece assente a livello delle zone nucleolari.
Complessivamente, tali risultati hanno consentito, innanzitutto, di apportare nuovi
elementi di conoscenza relativi alla distribuzione spaziale di specifici proteasomi
nell’ambito del sarcomero di cellule muscolari striate, suggerendo anche i più probabili
“partners” di interazione molecolare. Inoltre, attraverso la messa a punto di idonei
strumenti molecolari, essi costituiscono una premessa importante per cercare di
approfondire ulteriormente, attraverso studi di dinamica, le conoscenze circa il ruolo
svolto dai proteasomi in corso di differenziamento cellulare.
Il secondo modello preso in considerazione per lo studio dei proteasomi prevede
l’impiego di cellule fibroblastiche umane, utilizzate per lo studio dell’infezione virale in
vitro da parte di uno stipite umano di citomegalovirus.
Cellule che si trovano in una condizione patologica, quale quella indotta da un’infezione
virale, possono essere considerate, in analogia al precedente modello, come ugualmente
assoggettate ad una profonda riprogrammazione dell’espressione genica, in questo caso
virus-indotta, che si verifica, peraltro, in concomitanza ad importanti variazioni di assetto
morfologico della cellula, con possibili ripercussioni sulla struttura e sulla funzionalità dei
diversi compartimenti cellulari.
Come evidenziato nella sezione introduttiva, esistono diversi dati di letteratura che
depongono per un ruolo importante svolto dai proteasomi in corso di infezione virale, sia
in termini di una corretta evoluzione del ciclo replicativo virale, che di possibile
potenziamento delle difese dell’ospite nei confronti dell’infezione stessa (Everett R.D. et

7. Discussione
-157-
al., 1998; Boyer S.N. et al., 1996; Berezutskaya E. et al., 1997; Jones D.L. and Münger K.,
1997; Reinstein E. et al., 2000; Gonzalez S.L. et al., 2001; Khu Y.L. et al., 2004).
In riferimento al virus Herpes simplex di tipo 1 (HSV 1) è, ad esempio, stato appurato che la
proteina virale ICP27 è in grado di interagire con l’enzima RNA polimerasi di tipo II, allo
scopo di cooptare l’enzima stesso per favorire gli eventi connessi alla trascrizione dei geni
virali. Il trattamento di cellule con inibitori della funzione proteasomale in corso di
infezione sperimentale con HSV 1 causerebbe un incremento di particolari isoforme della
suddetta proteina virale, incapaci di interagire nuovamente con l’enzima cellulare,
causando, pertanto, un netto decremento della sintesi di proteine virali tardive, come
anche dei titoli della progenie virale neoformata. Nello specifico, gli Autori sostengono
che il ruolo dei proteasomi sarebbe rilevante soprattutto nelle fasi tardive dell’infezione
virale, quando cioè i livelli di trascrizione e di sintesi di DNA virale appaiono
notevolmente pronunciati (Dai-Ju J.Q. et al., 2006).
Un’altro lavoro sperimentale recentemente emerso (Diaz-Griffero F. et al., 2006), evidenzia
l’importanza di una rapida degradazione di fattori di restrizione specie-specifica nei
confronti di particelle retrovirali; tali molecole sarebbero altamente differenziate nei
vertebrati e presenti, generalmente, a livello citoplasmatico. Per importanza,
spiccherebbero le proteine TRIM5alpha e TRIMCyp, la cui emivita non è peraltro
superiore a 50 minuti; i corretti livelli intracellulari di tali molecole verrebbero mantenuti
grazie alla concomitante funzionalità di rapidi processi di sintesi proteica e alla successiva
degradazione di tali proteine mediata dai proteasomi.
Per quel che riguarda la ricerca in oggetto, il modello virus-cellula prescelto è
rappresentato, nello specifico, dallo stipite AD169 di citomegalovirus umano (HCMV)
nell’ambito del ciclo replicativo litico di fibroblasti polmonari umani (MRC5); in
particolare, tale studio è stato finalizzato alla comprensione del ruolo svolto dai
proteasomi nel corso delle fasi precocissime dell’infezione virale, essendo questo il
momento in cui vengono espletate le più significative funzioni di regolazione
sull’espressione genica, sia di tipo virale che cellulare.
Allo scopo, l’espressione dei prodotti maggiori dei geni precocissimi virali IE1 e IE2 è
stata inizialmente studiata in assenza o in presenza di uno specifico inibitore dei
proteasomi, denominato MG132; tale sostanza è stata utilizzata in concentrazioni non
tossiche per la cellula, pretrattando i monostrati di fibroblasti, che venivano poi infettati
per diversi tempi, prima di essere sottoposti a reazioni di immunofluorescenza volte ad
individuare le proteine virali. Si è, in tal modo, potuto osservare che il trattamento con

7. Discussione
-158-
MG132 determina una significativa riduzione dell’espressione dei geni precocissimi IE già
a tempi molto precoci di infezione (2 ore 30 minuti), convalidando pertanto l’ipotesi di un
intervento rilevante dei proteasomi nel modulare il ciclo replicativo litico di HCMV.
I risultati di tali esperimenti hanno anche messo in rilievo come l’effetto inibitorio della
sostanza sull’espressione delle proteine precocissime virali non sia della stessa entità per
tutte le cellule del monostrato (costituito, peraltro, da cellule non sincronizzate tra loro),
suggerendo perciò che un’eventuale inibizione dell’infezione virale, mediata dal blocco
dell’attività proteasomale, potesse esprimersi con differente efficacia, dipendentemente
dalla fase del ciclo cellulare in cui l’infezione virale aveva avuto inizio. Tale osservazione
non è apparsa irrilevante, dal momento che è nota e ampiamente documentata l’azione di
alterazione della normale evoluzione del ciclo cellulare, indotta da HCMV (Lu M. and
Shenk T., 1996; Dittmer D. and Mocarski E.S, 1997; Salvant B.S. et al., 1998; Sinclair J. et al.,
2000; Kalejta R.F. and Shenk T., 2002); in particolare, il virus è in grado di arrestarne
l’evoluzione a livello della transizione G1/S, condizione che si rivela maggiormente
favorevole alla sua replicazione. Verosimilmente, è possibile affermare che quest’ultima
fase del ciclo cellulare si rivela estremamente vantaggiosa per il virus, dal momento la
cellula risulta provvista degli enzimi necessari per la replicazione del genoma cellulare
che verranno, pertanto, cooptati dal virus, inducendo, in parallelo, l’inibizione della
sintesi di DNA cellulare.
La fase di studio successiva è stata pertanto focalizzata a verificare se l’attività
proteasomale potesse essere rilevante in specifiche fasi del ciclo cellulare piuttosto che in
altre, sempre nel corso di stadi molto precoci dell’infezione virale.
Allo scopo, le cellule MRC5 sono state sincronizzate mediante applicazione di un
protocollo sperimentale che prevedeva la sottrazione del siero nel terreno di
mantenimento delle colture cellulari (Morel A.P. et al., 2003; Memili E. et al., 2004); tale
condizione sperimentale veniva mantenuta per uno specifico arco temporale, in seguito al
quale il siero veniva ripristinato e le cellule venivano successivamente infettate con
citomegalovirus, a diversi tempi dopo il ripristino stesso. I tempi di infezione prescelti
coincidevano, in particolare, con l’inizio delle diverse fasi del ciclo cellulare (G1, G1/S, S,
G2/M), come puntualmente confermato mediante analisi citofluorimetrica delle colture
cellulari.
In una serie parallela di esperimenti, condotti con identiche modalità sperimentali, i
monostrati di cellule MRC5 venivano anche sottoposti a pretrattamento con MG132 per 3
ore prima dell’infezione; la sostanza veniva poi costantemente mantenuta nel terreno di

7. Discussione
-159-
coltura (data la rapida reversibilità del suo effetto), per l’intera durata dell’infezione
stessa. I dati ottenuti hanno dimostrato che, in cellule infettate e non trattate con MG132,
l’espressione nucleare delle proteine virali precocissime (IE1 e IE2) era decisamente
incrementata qualora l’infezione virale prendesse avvio nell’ambito della transizione
G1/S, come era logico attendersi per quanto precedentemente descritto a proposito
dell’azione interferente operata dal virus proprio a questo stadio. Il rilevamento delle
suddette proteine virali risultava di entità relativamente minore qualora il ciclo replicativo
virale iniziasse nella fase G1 e decisamente più esiguo in fase S ed alla transizione G2/M;
tali osservazioni sono in accordo con quanto riportato in letteratura (Fortunato E. et al.,
2002). L’intervento della funzione proteasomale nell’ambito dell’evoluzione del ciclo
replicativo di HCMV sembrava essere, a sua volta, maggiormente rilevante in fase G1 ed
alla transizione G1/S, rispetto agli altri stadi del ciclo replicativo considerati, come
dimostrato dalla serie di dati ottenuti in presenza dell’inibitore MG132.
Al fine di evidenziare la distribuzione intracellulare di specifiche subunità proteasomali,
relativamente a quella di proteine precocissime di HCMV, è stato successivamente
effettuato uno studio in microscopia confocale su singole sezioni focali di cellule MRC5,
precedentemente infettate per due ore e mezzo con citomegalovirus e poi fissate e
sottoposte ad una reazione di immunofluorescenza, impiegando sonde dirette nei
confronti di alcune subunità del complesso proteasomale 20S e dei prodotti principali dei
geni virali precocissimi.
I risultati ottenuti hanno evidenziato che i proteasomi si localizzano principalmente a
livello nucleare a tempi precocissimi dopo l’infezione virale, con accumulo anche nella
regione nucleolare di un numero significativo di cellule infettate, a differenza di quanto
riportato in letteratura per diverse tipologie di cellule in condizioni fisiologiche, in cui i
proteasomi sembrano invece essere localizzati nel citoplasma ed in differenti
compartimenti nucleari, ad eccezione del nucleolo (Reits E.A.J. et al., 1997; Groothuis
T.A.M. and Reits E.A.J., 2005). Tuttavia, in letteratura sono anche stati riportati esempi
particolari in cui viene invece descritta una precipua localizzazione nucleolare dei
proteasomi, come ad esempio, in condizioni di aumento della sintesi della proteina Myc
(Arabi A. et al., 2003), o qualora si verifichi un’alterata localizzazione delle proteine
nucleari integrate nei complessi PML (Mattson K. et al., 2001).
Per quel che riguarda, nello specifico, i risultati conseguiti in questo studio, l’analisi al
microscopio confocale di singole sezioni di nuclei di cellule MRC5 infettate con HCMV ha
consentito di dimostrare non solo la presenza di proteasomi a livello nucleolare, ma anche

7. Discussione
-160-
l’esistenza di una chiara colocalizzazione tra le proteine virali precocissime considerate ed
i proteasomi, spesso evidenziabili con una distribuzione peculiare in assetto “granulare”,
probabilmente riconducibile ad un accumulo di diversi complessi proteasomali che, a
volte, risultavano presenti anche in aree peri-nucleolari.
La distribuzione nucleolare dei proteasomi in corso di infezione sperimentale da HCMV,
come anche la loro associazione con i prodotti maggiori dei geni precocissimi, sottolinea e
pone nuovamente l’accento sul ruolo di spicco di questi complessi molecolari nell’ambito
del ciclo litico di HCMV. La dislocazione dei proteasomi in aree nucleari, quali i nucleoli,
non occupate da tali complessi in condizioni cellulari fisiologiche, indica chiaramente
come tali distretti cellulari rappresentino importanti siti in cui, presumibilmente, si
svolgono funzioni rilevanti per il virus nell’ambito del suo ciclo replicativo.
Per quanto riguarda l’associazione di proteine di HCMV a domìni funzionali nucleari è
stato dimostrato, in particolare, che la fosfoproteina pp65, il maggiore componente del
tegumento di HCMV possiede segnali di localizzazione nucleare, che permettono la sua
traslocazione veloce verso il nucleo. L’accentuato tropismo nucleare di pp65 a tempi
precocissimi dopo l’infezione, unitamente ad una dimostrata attività protein-chinasica, la
rendono candidata ideale quale fattore di regolazione dell’espressione genica virale
(Gallina A. et al., 1999). Dati pubblicati dal nostro gruppo hanno messo inoltre in evidenza
un significativo accumulo della proteina pp65 a livello nucleolare, visibile già entro la
prima ora di infezione per il virus “parentale”, come anche dopo venti ore, per quello
neoformato (Arcangeletti M.C. et al., 2003).
È in effetti noto come il nucleolo costituisca un compartimento nucleare altamente
specializzato, sede dei geni per l’RNA ribosomiale, di fattori di trascrizione e maturazione
del suddetto RNA, come anche di complessi molecolari adibiti al trasporto dello stesso
verso le sedi citoplasmatiche, attraverso proteine “navetta”, che possono traslocare da e
verso il nucleolo; in esso si distinguono tre regioni, dotate di diversa specializzazione
funzionale (Strouboulis J. and Wolffe A.P., 1996). Dati di letteratura descrivono un suo
coinvolgimento nella maturazione e nel trasporto di alcuni mRNA (Schneiter R. et al.,
1995) e tRNA (Bertrand E. et al., 1998), e in numerose altre funzioni, come la stessa
regolazione del ciclo cellulare (Visintin R. and Amos A., 2000; Carmo-Fonseca M. et al.,
2000). È stato inoltre ipotizzato che, oltre a svolgere tali funzioni, il nucleolo possa
rappresentare uno dei siti di elezione per la regolazione della trascrizione virale (il
distretto perinucleolare è, peraltro sede di molti fattori di trascrizione cellulari), per
promuovere la replicazione virale o per alterare il ciclo cellulare (Hiscox J.A., 2002).

7. Discussione
-161-
Inoltre, anche per la proteina precocissima IEp72 (prodotto principale del gene
precocissimo IE1 di HCMV), è stata messa in evidenza una colocalizzazione con il
nucleolo che, in questo caso e contrariamente a quanto evidenziato per la proteina pp65,
non è appannaggio della totalità delle cellule del monostrato e si estrinseca attraverso la
localizzazione di addensamenti granulari di tale proteina all’interno dei nucleoli
(Arcangeletti M.C. et al., 2003).
IEp72 riveste notevole importanza nel bilancio dell’infezione da HCMV, essendo uno dei
componenti virali di spicco nel regolare la trascrizione (sia in senso positivo che negativo)
dei geni posizionati a valle rispetto a quelli precocissimi; a tale proposito, è stato
ipotizzato che questa proteina possa interagire con l’enzima RNA polimerasi II cellulare,
oltre che con promotori virali.
La sua localizzazione in regioni discrete del nucleo (che precede la successiva diffusione a
sempre più vaste regioni nucleari), verosimilmente in stretta associazione spaziale con i
proteasomi, come dimostrano i dati ottenuti in questo studio, evoca un profilo di
distribuzione paragonabile a quello di domìni nucleari, come i fattori di trascrizione
cellulari o quelli di “splicing” di RNA pre-messaggeri, o anche altre regioni specializzate,
come i “PML nuclear bodies”. È stata infatti descritta la capacità, da parte di questo
prodotto del gene IE1, di disassemblare tali “PML” durante il ciclo litico (Kelly C. et al.,
1995; Ahn J.H. and Hayward G.S., 1997; Wilkinson G.W. et al., 1998).
Il ruolo dei PML non è ancora del tutto chiaro, anche se essi sembrano avere numerose
funzioni come soppressori della crescita, mediatori dell’apoptosi, soppressori tumorali,
oltre ad avere un coinvolgimento nella regolazione del ciclo cellulare (Ruggero D. et al.,
2000). La capacità di specifiche proteine di virus a DNA di provocare una
riorganizzazione o la distruzione di questi domìni nucleari, suggerisce un possibile ruolo
dei “PML bodies” nell’ambito dei meccanismi cellulari di difesa nei confronti delle
infezioni virali (Parkinson J. and Everett R.D., 2000; Adamson A.L. and Kenney S., 2001;
Florin L. et al., 2002).
È interessante inoltre sottolineare come la disgregazione di questi peculiari compartimenti
nucleari da parte di una proteina precocissima di Herpes simplex sembrerebbe cruciale per
dirigere l’infezione virale verso il ciclo litico, mentre la mancata disgregazione avvierebbe
l’infezione preferenzialmente verso una condizione di latenza (Everett R.D. et al., 1998).
In linea con gli esperimenti effettuati in precedenza in relazione al modello di
differenziamento muscolare, l’attenzione è stata successivamente focalizzata

7. Discussione
-162-
all’allestimento di strumenti molecolari idonei allo studio dei proteasomi in cellule
suscettibili all’infezione da HCMV.
Allo scopo, sono state inizialmente prodotte delle particelle retrovirali defettive di tipo
anfotropico (in grado di infettare cellule di mammifero), recanti le sequenze proteasomali
di interesse, oltre a quella per EGFP; successivamente, le particelle retrovirali così ottenute
sono state impiegate per eseguire esperimenti di infezione di fibroblasti di polmone
embrionale umano immortalizzati, denominati MRC5-hTERT (McSherry B.P. et al., 2001).
L’obiettivo principale di tali esperimenti è consistito nel mettere a punto modelli
sperimentali di derivazione cellulare umana che fossero, da un lato, permissivi
all’infezione da HCMV, e, dall’altro, che consentissero l’espressione stabile delle proteine
di fusione di interesse, ovviando, al contempo, all’esiguo potenziale di vita dei fibroblasti
MRC5, a numero di passaggi limitato.
In tale ottica sperimentale, sono state eseguite osservazioni preliminari in microscopia a
fluorescenza di monostrati di cellule MRC5-hTERT in grado di esprimere stabilmente i
costrutti di interesse, previa fissazione degli stessi; si è, in tal modo, appurato che il
segnale di fluorescenza delle proteine di fusione α1/p27k-EGFP e β4/p23k-EGFP era
localizzato sia nel compartimento citoplasmatico che in quello nucleare, ma in
quest’ultimo appariva più brillante ed intenso, con distribuzione spesso granulare; come
atteso, il segnale di fluorescenza era assente a livello del distretto nucleolare.
Inoltre, l’osservazione più rilevante di quest’ultima serie di esperimenti preliminari è
emersa dal confronto del profilo di distribuzione proteasomale ottenuto con i suddetti
fibroblasti, che esprimevano stabilmente le sequenze di interesse, con quello rilevato in
cellule MRC5-hTERT non ingegnerizzate e preventivamente sottoposte a reazione di
immunofluorescenza, mediante l’impiego di una sonda immunologica diretta nei
confronti di specifiche subunità del complesso proteasomale 20S; i profili di distribuzione
dei proteasomi sono infatti risultati sovrapponibili in entrambi i casi.
I risultati sopra descritti costituiscono, allo stato attuale, una fase del tutto preliminare
nell’ambito delle varie tappe previste per tale ricerca; in un’ottica futura, l’impiego di
popolazioni di cellule MRC5-hTERT in grado di esprimere stabilmente le sequenze
proteasomali prescelte potrebbe costituire un’utile strumento per lo studio della
localizzazione intracellulare dei proteasomi in studi di cinetica in cellule viventi, al fine di
approfondire le conoscenze sul ruolo svolto da tali complessi molecolari in corso di
infezione virale, studiandone, in maniera dettagliata, le possibili interazioni con
strutture/compartimenti nucleari funzionalmente rilevanti.

7. Discussione
-163-
I dati ottenuti in quest’ultima parte dello studio confermano in maniera significativa la
complessità dei meccanismi che regolano i rapporti tra virus e cellula ospite. In
particolare, la messa in evidenza di compartimenti cellulari e di componenti di
citomegalovirus che risultino spazialmente e funzionalmente correlati con il complesso
proteasomale, soprattutto a tempi precocissimi dall’inizio dell’infezione, potrebbe
apportare un contributo importante alla individuazione di quei meccanismi volti ad
imprimere una direzione di scelta, tra ciclo litico e latenza, al rapporto tra citomegalovirus
e cellula ospite.

8. Bibliografia
8. BIBLIOGRAFIA

8. Bibliografia
-165-
1. Aaronson S.A. and Todaro G.J. (1968). Development of 3T3-like lines from Balb-c
mouse embryo cultures: transformation susceptibility to SV40. J. Cell Physiol., 72: 141-148.
2. Abate D.A., Watanabe S. and Mocarski E.S. (2004). Major human cytomegalovirus
structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the
interferon reponse. J. Virol., 78: 10995-11006.
3. Abu Hatoum O., Gross-Mesilaty S., Breitschopf K., Hoffman A., Gonen H.,
Ciechanover A. and Bengal E. (1998). Degradation of myogenic transcription factor MyoD
by the ubiquitin pathway in vivo and in vitro: regulation by specific DNA binding. Mol.
Cell Biol., 18: 5670-5677.
4. Adamson A.L. and Kenney S. (2001). Epstein-Barr virus immediate-early protein
BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol., 75:
2388-2399.
5. Ahn J.H. and Hayward G.S. (1997). The major immediate-early proteins IE1 and
IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies
at very early times in infected permissive cells. J. Virol., 71: 4599-4613.
6. Ahn K., Erlander M., Leturcq D., Peterson P.A., Fruh K. and Yang Y. (1996). In vivo
characterization of the proteasome regulator PA28. J. Biol. Chem., 27: 18237-18242.
7. Amsterdam A., Pitzer F. and Baumeister W. (1993). Changes in intracellular
localization of proteasomes in immortalized ovarian granulosa cells during mitosis
associated with a role in cell cycle control. P.N.A.S. U. S.A., 90: 99-103.
8. Andrés V. and Walsh K. (1996). Myogenin expression, cell cycle withdrawal and
phenotypic differentiation are temporally separable events that precede cell fusion upon
mygenesis. J. Cell Biol., 132: 657-666.
9. Anton L.C., Schubert U., Bacik I., Princiotta M.F., Wearsch P.A., Gibbs J., Day P.M.,
Realini C., Rechsteiner M.C., Bennink J.R. and Yewdell J.W. (1999). Intracellular
localization of proteasomal degradation of a viral antigen. J. Cell Biol., 146: 113-124.
10. Arabi A., Rustum C., Hallberg E. and Wright A.P.H. (2003). Accumulation of c-
Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J.
Cell Science, 116: 1707-1717.
11. Arcangeletti M.C., De Conto F., Ferraglia F., Pinardi F., Gatti R., Orlandini G.,
Calderaro A., Motta F., Medici M.C., Martinelli M., Valcavi P., Razin S.V. Chezzi C. and
Dettori G. (2003). Human cytomegalovirus proteins PP65 and IEP72 are targeted to

8. Bibliografia
-166-
distinct compartments in nuclei and nuclear matrices of infected human embryo
fibroblasts. J. Cell. Biochem., 90: 1056-1067.
12. Arcangeletti M.C., De Conto F., Sutterlin R., Pinardi F., Missorini S., Geraud G.,
Aebi U., Chezzi C. and Scherrer K. (2000). Specific types of prosomes distribute
differentially between intermediate and actin filaments in epithelial, fibroblastic and
muscle cells. Eur. J. Cell Biol., 79: 423-437.
13. Arcangeletti M.C., Pinardi F., Missorini S., De Conto F., Conti G., Portincasa P.,
Scherrer K. and Chezzi C. (1997). Modification of cytoskeleton and prosome networks in
relation to protein synthesis in influenza A virus-infected LLC-MK2 cells. Virus Res., 51:
19-34.
14. Attaix D., Cobaret L. and Taillandier D. (2002). The ubiquitin-proteasome-
dependent proteolytic pathway in muscle wasting: from molecular biology to therapeutic
strategies. Riv. Italiana Nutr. Parent. Entrale, 1: 1-6.
15. Baldick C.J. Jr and Shenk T. (1996). Proteins associated with purified human
cytomegalovirus particles. J. Virol., 70: 6097-6105.
16. Bassaglia Y., Cebrian J., Covan S., Garcia M. and Foucrier J. (2005). Proteasomes
are tightly associated to myofibrils in mature skeletal muscle. Exp. Cell Res., 302: 221-232.
17. Baumeister W., Walz J., Zuhl F. and Seemuller E. (1998). The proteasome:
paradigm of a self-compartmentalizing protease. Cell, 92: 367-380.
18. Belcastro A.N., Shewchuk L.D. and Raj D.A. (1998). Exercise-induced muscle
injury: a calpain hypothesis. Mol. Cell Biochem., 179: 135-145.
19. Berezutskaya E., Yu B., Morozov A., Raychaudhuri P. and Bagchi S. (1997).
Differential regulation of the pocket domains of the retinoblastoma family proteins by the
HPV 16 E7 oncoprotein. Cell Growth Differ., 8: 1277-1286.
20. Bertrand E., Houser-Scott F., Kendall A., Singer R.H. and Engelke D.R. (1998).
Nucleolar localization of early tRNA processing. Genes Dev., 12: 2463-2468.
21. Bess K.L., Swingler T.E., Rivett A.J., Gaston K. and Jayaraman P.S. (2003). The
transcriptional repressor protein PRH intercats with the proteasome. Biochem. J., 374: 667-
675.
22. Beyette J.R. and Mykles D.L. (1992). Immunocytochemical localization of the
multicatalytic proteinase (proteasome) in crustacean striated muscles. Muscle Nerve, 15:
1023-1035.

8. Bibliografia
-167-
23. Boyer S.N., Wazer D.E. and Band V. (1996). E7 protein of human papilloma virus-
16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome
pathway. Cancer Res., 56: 4620-4624.
24. Boyle K.A. and Compton T. (1998). Receptor-binding properties of a soluble form
of human cytomegalovirus glycoprotein B. J. Virol., 72: 1826-1833.
25. Bradshaw P.A., Duran-Guarino M.R., Perkins S., Rowe J.I., Fernandez J., Fry K.E.,
Reyes G.R., Young L. and Foung S.K.H. (1994). Localization of antigenic sites on human
cytomegalovirus virion structural proteins encoded by UL48 and UL56. Virology, 205:
321-328.
26. Breitschopf K., Bengal E., Ziv T., Admon A. and Ciechanover A. (1998). A novel
site ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential
for conjugation and degradation of the protein. EMBO J., 17: 5964-5973.
27. Bresnahan W.A. and Shenk T. (2000). A subset of viral transcripts packaged within
viral particles. Science, 288: 2373-2376.
28. Bresnahan W.A., Boldogh I., Thompson E.A. and Albrecht T. (1996). Human
cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in
late G1. Virology, 224: 150-160.
29. Britt W.J. and Mach M. (1996). Human cytomegalovirus glycoproteins.
Intervirology, 39: 401-412.
30. Brown E.P. and Shenk T. (2003). Human cytomegalovirus UL83-coded pp65 virion
protein inhibits antiviral gene expression in infected cells. P.N.A.S. U.S.A., 100: 11439-
11444.
31. Brown E.P., Wing B., Coleman D. and Shenk T. (2001). Altered cellular mRNA
levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of
antiviral mRNAs. J. Virol., 75: 12319-12330.
32. Carmo-Fonseca M., Mendes-Soares L. and Campos I. (2000). To be or not to be in
the nucleolus. Nat. Cell Biol., 2: E107-E112.
33. Chargé S.B.P. and Rudnicki M.A. (2004). Cellular and molecular regulation of
muscle regeneration. Physiol. Rev., 84: 209-238.
34. Ciechanover A., Breitschopf K., Hatoum O.A. and Bengal E. (1999). Degradation of
MyoD by the ubiquitin pathway: regulation by specific DNA-binding and identification
of a novel site for ubiquitination. Mol. Biol. Re., 26: 59-64.

8. Bibliografia
-168-
35. Ciechanover A., Orian A. and Schwartz A.L. (2000). Ubiquitin-mediated
proteolysis: Biological regulation via destruction. BioEssays, 22: 442-451.
36. Clark K.A., McElhinny A.S., Beckerle M.C. and Gregorio C.C. (2002). Striated
muscle cytoarchitecture: an intricate web of form and function. Annu. Rev. Cell Dev. Biol.,
18: 637-706.
37. Compton T., Kurt-Jones E.A., Boehme K.W., Belko J., Latz E., Golenbock D.T. and
Finberg R.W. (2003). Human cytomegalovirus activates inflammatory cytokine responses
via CD14 and Toll-like receptor 2. J. Virol., 77: 4588-4596.
38. Compton T., Nepomuceno R.R. and Nowlin D.M. (1992). Human cytomegalovirus
penetrates host cells by pH-independent fusion at the cell surface heparan sulfate.
Virology, 191: 387-395.
39. Compton T., Nowlin D.M. and Cooper N.R. (1993). Initiation of human
cytomegalovirus infection requires initial interaction with cell surface heparan sulfate.
Virology, 193: 834-841.
40. Coux O., Nothwang H.G., Silva Pereira I., Recillas Targa F., Bey F. and Scherrer K.
(1994). Phylogenic relationships of the amino acid sequences of prosome (proteasome,
MCP). Mol. Gen. Genet., 245: 769-780.
41. Coux O., Tanaka K. and Goldberg A.L. (1996). Structure and functions of the 20S
and 26S proteasomes. Annu. Rev. Biochem., 65: 801-847.
42. Craigen J.L., Yong K.L., Jordan N.J., MacCormac L.P., Westwick J., Akbar A.N. and
Grundy J.E. (1997). Human cytomegalovirus infection up-regulates interleukin-8 gene
expression and stimulates neutrophil transendothelial migration. Immunology, 92: 138-
145.
43. Dai-Ju J.Q., Li L., Johnson L.A. and Sandri-Goldin R.M. (2006). ICP27 interacts with
the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes
simples virus 1 transcription sites, where it undergoes proteasomal degradation during
infection. J. Virol., 80: 3567-3581.
44. Dal Monte P., Bessia C., Ripalti A., Landini M.P., Topilko A., Plachter B., Virelizier
J.L. and Michelson S. (1996). Stably-expressed antisense RNA to cytomegalovirus UL83
inhibits viral replication. J. Virol., 70: 2086-2094.
45. David C.L. and Konieczny S.F. (1995). Transcription factor families: muscling in on
the myogenic program. FASEB J., 9: 1595-1604.

8. Bibliografia
-169-
46. Davis R.L., Weintraub H. And Lassar A.B. (1987). Expression of a single
transfected cDNAconverts fibroblasts to myoblasts. Cell, 51: 987-1000.
47. De Conto F., Missorini S., Arcangeletti M.C., Pinardi F., Montarras D., Pinset C.,
Vassy J., Geraud G., Chezzi C. and Scherrer K. (1997). Prosome cytodistribution relative to
desmin and actin filaments in dividing C2.7 myblasts and during myotube formation in
vitro. Exp. Cell Res., 233: 99-117.
48. De Conto F., Pilotti E., Razin S. V., Ferraglia F., Geraud G., Arcangeletti M.C. and
Scherrer K. (2000). In mouse myoblasts nuclear prosomes are associated with the nuclear
matrix and accumulate preferentially in the perinucleolar areas. J. Cell Science, 113: 2399-
2407.
49. Dean P.N. and Jett J.H. (1974). Mathematical analysis of DNA distributions
derived from flow microfluorometry. J. Cell. Biol., 60: 523-527.
50. DeMartino G.N. and Slaughter C.A. (1999). The proteasome, a novel protease
regulated by multiple mechanisms. J. Biol. Chem., 274: 22123-22126.
51. Dhawan J. and Rando T.A. (2005). Stem cells in postnatal myogenesis: molecular
mechanism of satellite cell quiescence activation and replenishment. Trend in Cell Biol.,
15: 666-673.
52. Diaz-Griffero F., Javanbakht H., Song B., Welikala S., Stremlau M. and Sodroski J.
(2006). Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5.
Virology, in corso di stampa (doi:10.1016/j.virol.2005.12.040).
53. Dittmer D. and Mocarski E.S. (1997). Human cytomegalovirus infection inhibits
G1/S transition. J. Virol., 71: 1629-1634.
54. Dubiel W., Pratt G., Ferrell K. and Rechsteiner M. (1992). Purification of an 11 S
regulator of the multicatalytic protease, J. Biol. Chem., 267: 22369–22377.
55. Eble D.M., Spragia M.L., Ferguson A.G. and Samarel A.M. (1999). Sarcomeric
myosin heavy chain is degraded by the proteasome. Cell Tissue Res., 296: 541-548.
56. El-Deiry W.S., Harper J.W., O’Connor P.M., Velculescu V.E., Canman C.E.,
Jackman J., Pietenpol J.A., Burrell M., Hill D.E., Wang Y., Wiman K.G., Mercer W.E.,
Kastan M.B., Kohn K.W., Elledge S.J., Kinzler K.W. and Vogelstein B. (1994). WAF1/CIP1
is induced in p53-mediated G1 arrest and apoptosis. Cancer Res., 54: 1169-1174.
57. Elsasser S., Schmidt M. and Finley D. (2005). Characterization of the proteasome
using native gel electrophoresis. Methods in Enzimology, 398: 353-363.

8. Bibliografia
-170-
58. Enenkel C., Lehmann A. and Kloetzel P.M. (1998). Subcellular distribution of
proteasomes implicates a major location of protein degradation in the nuclear envelope-
ER network in yeast. EMBO J., 17: 6144-6154.
59. Esser C., Alberti S. and Hohfeld J. (2004). Cooperation of molecular chaperones
with the ubiquitin/proteasome system. Biochim. Biophys. Acta, 1695: 171-188.
60. Everett R.D., Freemont P., Saitoh H., Dasso M., Orr A., Kathoria M. and Parkinson
J. (1998). The disruption of ND10 during Herpes simplex virus infection correlates with the
Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol., 72: 6581-
6591.
61. Fagan J.M., Waxman L. and Goldberg A.L. (1987). Skeletal muscle and liver
contain soluble ATP+ ubiquitin-dependent proteolytic system. J. Biochem., 243: 335-343.
62. Fajerman I., Schwartz A.L. and Ciechanover A. (2004). Degradation of the Id2
developmental regulator: targeting via N-terminal ubiquitination. Biochem. Biophys. Rev.
Commun., 314: 505-512.
63. Florin L., Schafer F., Sotlar K., Streeck R.E. and Sapp M. (2002). Reorganization of
nuclear domain 10 induced by papillomavirus capsid protein L2. Virology, 295: 97-107.
64. Floyd Z.E., Trausch-Azar J.S., Reinstein E., Ciechanover A. and Schwartz A.L.
(2001). The nuclear ubiquitin-proteasome system degrades MyoD. J. Biol. Chem., 276:
22468-22475.
65. Förster A., Whitby F.G. and Hill C.P. (2003). The pore of activated 20S proteasomes
has an ordered 7-fold symmetric conformation. EMBO J., 22: 4356-4364.
66. Fortunato E.A., Sanchez V., Yen J.Y. and Spector D.H. (2002). Infection of cells with
human cytomegalovirus during S phase results in a blockade to immediate-early gene
expression that can be overcome by inhibition of the proteasome. J. Virol., 76: 5369-5379.
67. Foucrier J., Bassaglia Y., Grand M.C., Rothen B., Perriard J.C. and Scherrer K.
(2001). Prosomes form sarcomere-like banding patterns in skeletal, cardiac and smooth
muscle cells. Exp. Cell Res., 266: 193-200.
68. Foucrier J., Grand M.C., De Conto F., Bassaglia Y., Geraud G., Scherrer K. and
Martelly I. (1999). Dynamic distribution and formation of a para-sarcomeric banding
pattern of prosomes during myogenic differentiation of satellite cells in vitro. J. Cell
Science, 112: 989-1001.

8. Bibliografia
-171-
69. Funatsu T., Higuchi H. and Ishiwata S. (1990). Elastic filaments in skeletal muscle
revealed by selective removal of thin filaments with plasma gelsolin. J. Cell Biol., 110: 53-
62.
70. Funatsu T., Kono E., Higuchi H., Rimura S., Ishiwata S., Yoshioka T., Maruyama K.
and Tsukita S. (1993). Elastic filaments in situ in cardiac muscle: deep-etch replica analysis
in combination with selective removal of actin and myosin filaments. J. Cell Biol., 120: 711-
724.
71. Gallina A., Simoncini L., Garbelli S., Percivalle E., Pedrali-Noy G., Lee K.S.,
Erikson R.L., Plachter B., Gerna G. and Milanesi G. (1999). Polo-like kinase 1 as a target for
human cytomegalovirus pp65 lower matrix protein. J. Virol., 73: 1468-1478.
72. Gibson W. (1996). Structure and assembly of the virion. Intervirology, 39: 389-400.
73. Gilbert M.J., Riddell S.R., Plachter B. and Greenberg P.D. (1996). Cytomegalovirus
selectively blocks antigen processing and presentation of its immediate-early gene
product. Nature, 383: 720-722.
74. Glickman M.H. and Ciechanover A. (2001). The ubiquitin-proteasome proteolytic
pathway: destruction for the sake of construction. Physiol Rev., 82: 373-428.
75. Glickman M.H., Rubin D.M., Fried V.A. and Finley D. (1998). The regulatory
particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol., 18: 3149-3162.
76. Gonzales S.L., Stremlau M., He X., Basile J.R. and Munger K. (2001). Degradation
of the Retinoblastoma Tumor Suppressor by the Human Papillomavirus type 16 E7
oncoprotein is important for functional inactivation and is separable from proteasomal
degradation of E7. J. Virol., 75: 7583-7591.
77. Graham F.L., Smiley J., Russell W.C. and Nairn R. (1977). Characteristics of a
human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol., 36:
59-74.
78. Gravel S.P. and Servant M.J. (2005). Roles of an Ikappa B kinase-related pathway
in human cytomegalovirus-infected vascular smooth muscle cells: a molecular link in
pathogen-induced proatheroscleortic conditions. J. Biol. Chem., 280: 7477-7486.
79. Greaser M.L., Wang S.M., Berri M., Mozdziak P. and Kumazawa Y. (2000).
Sequence and mechanical implications of titin’s PEVK region. Adv. Exp. Med. Biol., 481:
53-63.

8. Bibliografia
-172-
80. Grimm L.M. and Osborne B.A. (2000). The role of the proteasome in apoptosis. In:
Hilt W. and Wolf D.H., editors. Proteasomes: The world of regulatory proteolysis. Gorge-
town, Texas: Landes Bioscience, 315-325.
81. Groettrup M., Khan S., Schwarz K. and Schmidtke G. (2001a). Interferon-gamma
inducible exchanges of 20S proteasome active site subunits: why? Biochimie, 83: 367-372.
82. Groettrup M., van den Broek M., Schwarz K., Macagno A., Khan S., de Giuli R. and
Schmidtke G. (2001b). Structural plasticity of the proteasome and its function in antigen
processing. Crit. Rev. Immunol., 21: 339-358.
83. Groll M., Bajorek M., Kohler A., Moroder L., Rubin D.M., Huber R., Glickman
M.H. and Finley D. (2000). A gated channel into the proteasome core particle. Nat. Struct.
Biol., 7: 1062-1067.
84. Groll M., Ditzel L., Lowe J., Stock D., Bochtler M., Bartunik H.D. and Huber R.
(1997). Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature, 386: 463-471.
85. Groothuis T.A.M. and Reits E.A.J. (2005). Monitoring the distribution and
dynamics of proteasomes in living cells. Methods in Enzymology, 398: 549-563.
86. Halevy O., Novitch B.G., Spicer D.B., Skapek S.X., Rhee J., Hannon G.J., Beach D.
and Lassar A.B. (1995). Correlation of terminal cell cycle arrest of skeletal muscle with
induction of p21 by MyoD. Science, 17: 1018-1021.
87. Harper J.W., Burton J.L. and Solomon M.J. (2002). The anaphase-promoting
complex: It’s not just for mitosis any more. Genes Dev., 16: 2179-2206.
88. Harty R.N., Brown M.E., McGettigan J.P., Wang G., Jayakar H.R., Huibregtse J.M.,
Whitt M.A. and Schnell M.J. (2001). Rhabdoviruses and the cellular ubiquitin-proteasome
system: a budding interaction. J Virol., 75: 10623-9.
89. Helin K., Harlow E. and Fattaey A.R. (1993). Inhibition of E2F-1 transactivation by
direct binding of the retinoblastoma protein. Mol. Cell Biol., 13: 6501-6508.
90. Hendil K.B., Khan S. and Tanaka K. (1998). Simultaneous binding of PA28 and
PA700 activators to 20 S proteasomes. Biochem. J., 332: 749-754.
91. Hesketh J. (1994). Translation and the cytoskeleton: a mechanism for targeted
protein synthesis. Mol. Biol. Rep., 19: 233-243.
92. Hiebert S.W., Chellappan S.P., Horowitz J.M. and Nevins J.R. (1992). The
interaction of pRb with E2F inhibits the transcriptional activity of E2F. Genes Dev., 6: 177-
185.

8. Bibliografia
-173-
93. Hill C.P., Masters E.I. and Whitby F.G. (2002). The 11S regulators of 20S
proteasome activity. Current Top. Microbiol. Immunol., 268: 73-89.
94. Hirsch A.J. and Shenk T. (1999). Human cytomegalovirus inhibits transcription of
the CC chemokine MCP-1 gene. J. Virol., 73: 404-410.
95. Hiscox J.A. (2002). The nucleolus: a gateway to viral infection? Arch. Virol., 147:
1077-1089.
96. Hoffman L. and Rechsteiner M. (1996). Nucleotidase activities of the 26S
proteasome and its regulatory complex. J. Biol. Chem., 271: 32538-32545.
97. Hough R., Pratt G. and Rechsteiner M. (1986). Ubiquitin-lysozyme conjugates. J.
Biol. Chem., 261: 2400-2408.
98. Hough R., Pratt G. and Rechsteiner M. (1987). Purification of two high molecular
weight proteases from rabbit reticulocyte lysate. J. Biol. Chem., 262: 8303-8313.
99. Huggins G.S., Chin M. T., Siringa N. E.S., Lee S.-L., Haber E. and Lee M.E. (1999).
Characterization of the mUBC9-binding sites required for E2A protein degradation. J. Biol
Chem., 274: 28690-6.
100. Irmiere A. and Gibson W. (1983). Isolation and characterization of a noninfectious
virion-like particle released from cells infected with human strains of cytomegalovirus.
Virology, 130: 118-133.
101. Jacobs J.P., Jones C.M. and Baille J.P. (1970). Characteristics of a human diploid cell
designated MRC-5. Nature, 227: 168-170.
102. Jault F.M., Jault J.M., Ruchti F., Fortunato E.A., Clark C., Corbeil J., Richman D.D.
and Spector D.H. (1995). Cytomegalovirus infection induces high levels of cyclins,
phosphorylated RB, and p53, leading to cell cycle arrest. J. Virol., 69: 6697-6704.
103. Jen Y., Weintraub H. and Benezra R. (1992). Overexpression of Id protein inhibits
the muscle differentiation program: in vivo association of Id with E2A proteins. Genes
Dev., 6: 1466-1479.
104. Jones D.L. and Münger K. (1997). Analysis of the p53-mediated G1 growth arrest
pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J. Virol.,
71: 2905-2912.
105. Jones T.R., Hanson L.K., Sun L., Slater J.S., Stenberg R.M. and Campbell A.E.
(1995). Multiple independent loci within the human cytomegalovirus unique short region

8. Bibliografia
-174-
down-regulate expression of major histocompatibility complex class I heavy chains. J.
Virol., 69: 4830-4841.
106. Jones T.R., Wiertz E. J., Sun L., Fish K.N., Nelson J.A. and Ploegh H.L. (1996).
Human cytomegalovirus US3 impairs transport and maturation of major
histocompatibility complex complex class I heavy chains. P.N.A.S. U.S.A., 93: 11327-11333.
107. Jorgensen L. and Hendil K.B. (1999). Proteasome subunit zeta, a putative
ribonuclease, is also found as a free monomer. Mol. Biol. Rep., 26: 119-123.
108. Kalejta R.F. and Shenk T. (2002). Manipulation of the cell cycle by human
cytomegalovirus. Front Biosci., 7: D295-306.
109. Kamura K., Ishiura S., Nonaka I. and Sugita H. (1988). Localization of ingensin in
rat central nervous system and skeletal muscle. J. Neurosci. Res., 20: 473-478.
110. Keay S. and Baldwin B. (1991). Anti-idiotype antibodies that mimic gp86 of human
cytomegalovirus inhibit viral fusion but not attachment. J. Virol., 65: 5124-5128.
111. Kelly C., Van Driel R. and Wilkinson G.W. (1995). Disruption of PML-associated
nuclear bodies during human cytomegalovirus infection. J. Gen. Virol., 76: 2887-2893.
112. Khan S., van den Broek M., Schwarz K., de Giuli R., Diener P.A. and Groettrup M.
(2001). Immunoproteasomes largely replace constitutive proteasomes during an antiviral
and antibacterial imune response in the liver. J. Immunology, 167: 6859-6868.
113. Kho C.J. , Huggins G. S., Endege W.O., Hsieh C.-M., Lee M.-E. and Haber E.
(1997). Degradation of E2A Proteins through a Ubiquitin-conjugating Enzyme, UbcE2A. J.
Biol. Chem., 272: 3845-3851.
114. Khor R., McElroy L.J. and Whittaker G.R. (2003). The ubiquitin-vacuolar protein
sorting system is selectively required during entry of influenza virus into host cells.
Traffic, 4: 857-68.
115. Khu Y.L., Tan Y.J., Lim S.G., Hong W. and Goh P.Y. (2004). Hepatitis C virus non-
structural protein NS3 interacts with LMP7, a component of the immunoproteasome, and
affects its proteasome activity. Biochem. J., 38: 401-409.
116. Kinzler E.R. and Compton T. (2005). Characterization of Human Cytomegalovirus
glycoprotein-induced cell-cell fusion. J. Virol., 79: 7827-7837.
117. Kitzmann M. and Fernandez A. (2001). Crosstalk between cell cycle regulators and
the myogenic factor MyoD in skeletal myoblasts. Cell. Mol. Life Sciences, 58: 571-579.

8. Bibliografia
-175-
118. Kloetzel P.M. (2001). Antigen processing by the proteasome. Nat. Rev. Mol. Cell
Biol., 2: 179-187.
119. Klyachko N.L., Kulikova A.L. and Erokhina M.A. (2003). Plant polysome binding
to the actin cytoskeleton as a target for physiological regulation. Cell Biol. Int., 27: 217-218.
120. Knowlton J.R., Johnston S.C., Whitby F.G., Realini C., Zhang Z. and Hill C.P.
(1997). Structure of the proteasome activator REGalpha (PA28alpha). Nature, 11: 639-43.
121. Koepp D.M., Harper W.J. and Elledge S.J. (1999). How the cyclin became a cyclin:
regulated proteolysis in the cell cycle. Cell, 97: 431-434.
122. Kumamoto T., Fujimoto S., Ito T., Horinouchi H., Ueyama H. and Tsuda T. (2000).
Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta
Neuropathol. (Berl.), 100: 595-602.
123. Langlands K., Yin X., Anand G. and Prochownik E.V. (1997). Differential
interactions of Id proteins with basic helix-loop-helix transcriptio factors. J. Biol. Chem.,
272: 19785-19793.
124. Lassar A.B. and Munsterberg A. (1994). Wiring diagrams: regulatory circuits and
the control of skeletal myogenesis. Curr. Opin. Cell Biol., 6: 432-442.
125. Lassar A.B., Skapek S.X. and Novitch B. (1994). Regulatory mechanisms that
coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell
Biol., 6: 788-794.
126. Lees E. (1995). Cyclin dependent kinase regulation. Curr. Opin. Cell Biol., 7: 773-
780.
127. Lehner R., Meyer H. and Mach M. (1989). Identification and characterization of a
human cytomegalovirus gene coding for a membrane protein that is conserved among
human herpesvirus. J. Virol., 63: 3792-3800.
128. Lingbeck J.M., Trausc-Azar J.S., Ciechanover A. and Scwartz A.L. (2003).
Determinants of nuclear and cytoplasmic ubiquitin-mediated degradation of MyoD. J.
Biol. Chem., 278: 1817-1823.
129. Lipinski M.M. and Jacks T. (1999). The retinoblastoma gene family in
differentiation and development. Oncogene, 18: 7873-7882.
130. Liu B. and Stinski M.F. (1992). Human cytomegalovirus contains a tegument
protein that enhances transcription from promoters with upstream ATF and AP-1 cis-
acting elements. J. Virol., 66: 4434-4444.

8. Bibliografia
-176-
131. Liu W., Zhao Y. and Biegalke B.J. (2002). Analysis of human cytomegalovirus US3
gene products. Virology, 301: 32-42.
132. Low P., Hastings R.A., Dawson S.P., Sass M., Billett M.A., Mayer R.J. and Reynolds
S.E. (2000). Localisation of 26S proteasomes with different subunit composition in insect
muscles undergoing programmed cell death. Cell Death Differ., 7: 1210-1217.
133. Löwe J., Stock D., Jap B., Zwickl P., Baumeister W. and Huber R. (1995). Crystal
structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution.
Science, 268: 533-539.
134. Lu M. and Shenk T. (1996). Human cytomegalovirus infection inhibits cell cycle
progression at multiple points, including the transition from G1 to S. J. Virol., 70: 8850-
8857.
135. Luders J., Demand J. and Hoehfeld J. (2000). The ubiquitin-related BAG-1 provides
a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol.
Chem., 275: 4613-4617.
136. Lukas C., Sorensen C.S., Kramer E., Santoni-Rugiu E., Lindeneg C., Peters J.M.,
Bartek J. and Lukas J. (1999). Accumulation of cyclin B1 requires E2F and cyclin-A-
dependent rearrangement of the anaphase-promoting complex. Nature, 401: 815-818.
137. Luo Y., Hurwitz J. and Massague J. (1995). Cell cycle inhibition by independent
CDK and PCNA binding domains in p21Cip1. Nature, 375: 159-161.
138. Ma C.P., Slaughter C.A. and DeMartino G.N. (1992). Identification, purification,
and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J.
Biol. Chem., 267: 10515–10523
139. Mann C. and Hilt W. (2000). The ubiquitin-proteasome system in cell cycle control.
In: Hilt W. and Wolf D.H., editors. Proteasomes: The world of regulatory proteolysis.
Gorge-town, Texas: Landes Bioscience, 264-301.
140. Masson P., Andersson O., Petersen U.M. and Young P. (2001). Identification and
characterization of a Drosophila nuclear proteasome regulator. A homolog of human 11S
REGg (PA28g). J. Biol. Chem., 276: 1383-1390.
141. Mattsson K., Pokrovskaja K., Kiss C., Klein G. and Szekely L. (2001). Proteins
associated with the promyelocytic leukemia gene product (PML)-containing nuclear body
move to the nucleolus upon inhibition of proteasome-dependent protein degradation.
P.N.A.S. U.S.A., 98: 1012-1017.

8. Bibliografia
-177-
142. McElroy A.K., Dwarakanath R.S. and Spector D.H. (2000). Dysregulation of cyclin
E gene expression in human cytomegalovirus-infected cells requires viral early gene
expression and is associated with changes in the Rb-related protein p130. J. Virol., 74:
4192-4206.
143. McSherry B.P., Jones C. J., Skinner J.W., Kipling D. and Wilkinson G.W.G. (2001).
Human telomerase reverse transcriptase-immortalized MRC-5 and HCA2 human
fibroblasts are fully permissive for human cytomegalovirus. J. Gen. Virol., 82: 855-863.
144. McVoy M.A., Nixon D.E., Adler S.P. and Mocarski E.S. (1998). Sequences within
the herpesvirus-conserved pac1 and pac2 motifs are required for cleavage and packaging
of the murine cytomegalovirus genome. J. Virol., 72: 48-56.
145. Memili E., Behboodi E., Overton S.A., Kenney A.M., O’Coin M., Zahedi A.,
Rowitch D.H. and Echelard Y. (2004). Synchronization of goat fibroblast cells at quiescent
stage and determination of their transition from G0 to G1 by detection of cyclin D1
mRNA. Cloning Stem Cell, 6: 58-66.
146. Michelson S., Turowski P., Picard L., Goris J., Landini M.P., Topilko A., Hemmings
B., Bessia C., Garcia A. and Virelizier J.L. (1996). Human cytomegalovirus carries
serine/threonine protein phosphatases PP1 and host-cell derived PP2A. J. Virol., 70: 1415-
1423.
147. Miyawaki A., Sawano A. and Kogure T. (2003). Lighting up cells: Labelling
proteins with fluorophores. Nat. Cell Biol., Suppl.: S 1-7.
148. Moran J.L., Li Y., Hill A.A., Mounts W.M. and Miller C.P. (2002). Gene expression
changes during mouse skeletal myoblast differentiation revealed by transcriptional
profiling. Physiol. Genomics, 10: 103-111.
149. Morel A.P., Sentis S., Bianchin C., Le Romancer M., Jonard L., Rostan M.C.,
Rimokh R. and Corbo L. (2003). BTG2 antiproliferative protein interacts with the human
CCR4 complex existing in vivo in three cell cycle-regulated forms. J. Cell Science, 116:
2929-2936.
150. Morgan D.O. (1995). Principles of CDK regulation. Nature, 374: 131-134.
151. Morgan D.O. (1997). Cyclin-dependent kinases: engines, clocks, and
microprocessors. Annu. Rev. Cell Dev. Biol., 13: 261291.
152. Morgan J.E. and Partridge T.A. (2003). Muscle satellite cells. Int. J. Biochem. Cell
Biol., 35: 1151-1156.

8. Bibliografia
-178-
153. Muganda P., Mendoza O., Hernandez J. and Qian Q. (1994). Human
cytomegalovirus elevates levels of the cellular protein p53 in infected fibroblasts. J. Virol.,
68: 8028-8034.
154. Muller S. and Dejean A. (1999). Viral immediate-early proteins abrogate the
modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body
disruption J. Virol., 73: 5137-5143.
155. Murakami Y., Matsufuji S., Hayashi S.I., Tanahashi N. and Tanaka K. (2000).
Degradation of ODC by the 26S proteasome. Biochem. Biophys. Rev. Commun., 267: 1-6.
156. Muranyi W., Haas J., Wagner M., Krohne G. and Koszinowski. (2002).
Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science,
297: 854-857.
157. Murata S., Udono H., Tanahashi N., Hamada N., Watanabe K., Adachi K., Yamano
T., Yui K., Kobayashi N., Kasahara M., Tanaka K. and Chiba T. (2001).
Immunoproteasome assembly and antigen presentation in mice lacking both PA28a and
PA28b. EMBO J., 20: 5898-5907.
158. Nag A.C. and Foster J.D. (1981). Myogenesis in adult mammalian skeletal muscle
in vitro. J. Anat., 132: 1-18.
159. Naujokat C. and Hoffmann S. (2002). Role and function of the 26S proteasome in
proliferation and apoptosis. Laboratory Investigation, 82: 965-980.
160. Nevels M., Brune W. and Shenk T. (2004). SUMOylation of the Human
Cytomegalovirus 72-Kilodalton IE1 protein facilitates expression of the 86-kilodalton IE2
protein and promotes viral replication. J. Virol., 78: 7803-7812.
161. Nigg E.A. (2001). Mitotic kinases as regulators of cell division and its check-points.
Nat. Rev. Mol. Cell Biol., 2: 21-32.
162. Nothwang H.G., Coux O., Bey F. and Scherrer K. (1992). Prosomes and their
multicatalytic proteinase activity. Eur. J. Biochem., 207: 621-630.
163. Novitch B.G., Mulligan G.J., Jacks T. and Lassar A.B. (1996). Skeletal muscle cells
lackin the retinoblastoma protein display defects in muscle gene expression and
accumulate in S and G2 phase of the cell cycle. J. Cell Biol., 135: 441-456.
164. Novitch B.G., Spicer D.B., Kim P.S., Cheung W.L. and Lassar A.B. (1999). pRb is
required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal
muscle differentiation. Curr. Biol., 6: 449-459.

8. Bibliografia
-179-
165. Olink-Coux M., Arcangeletti M.C., Pinardi F., Minisini R., Huesca M., Chezzi C.
and Scherrer K. (1994). Cytolocation of prosome antigens on intermediate filament
subnetworks of cytokeratin, vimentin and desmin type. J. Cell Science, 107: 353-366.
166. Olson E.N. and Klein W.H. (1994). BHLH factors in muscle development:
deadlines and commitments, what to leave in and what to leave out. Genes Dev., 8: 1-8.
167. Orlowski R.Z. (1999). The role of the ubiquitin-proteasome pathway in apoptosis.
Cell Death Diff., 6: 303-313.
168. Ortega J., Heymann J.B., Kajava A.V., Ustrell V., Rechsteiner M. and Steven A.C.
(2005). The axial channel of the 20S proteasome opens upon binding of the PA200
activator. J. Mol. Biol., 346: 1221-1227.
169. Parkinson J. and Everett R.D. (2000). Alphaherpesvirus proteins related to herpes
simplex viru type 1 ICPO affect cellular structures and proteins. J. Virol., 74: 10006-10017.
170. Pear W.S., Nolan G.P., Scott M.L. and Baltimore B. (1993). Production of high-titer
helper-free retroviruses by transient transfection. P.N.A.S. U.S.A., 90: 8392-8396.
171. Pear W.S., Scott M.L., Nolan G.P. (1997). Generation of High-Titer, Helper-Free
Retroviruses by Transiente Transfection. In Methods in Molecular Medecine: Gene
Therapy Protocols. P.D. Robbins (ed.) Humana Press, Totawa, New Jersey.
172. Percipalle P., Zhao J., Pope B., Weeds A., Lindberg U. and Daneholt B. (2001).
Actin bound to the heterogeneous nuclear ribonucleoprotein hrp36 is associated with
Balbiani ring mRNA from the gene to polysomes. J. Cell Biol., 153: 229-236.
173. Peters J.M. (2002). The anaphase-promoting complex: proteolysis in mitosis and
beyond. Mol. Cell, 9: 931-943.
174. Piccinini M., Tazartes O., Mostert M., Musso A., DeMarchi M. and Rinaudo M.T.
(2000). Structural and functional characterization of 20S and 26S proteasomes from bovine
brain. Mol. Brain Res., 7: 103-114.
175. Pinset C., Montarres D., Chenevert J., Minty A., Barton P., Laurent C. and Gros F.
(1988). Control of myogenesis in the mouse myogenic C2 cell line by medium composition
and by insulin: characterization of permissive and inducible C2 myoblasts.
Differentiation, 38: 28-34.
176. Pownall M.E., Gustafsson M.K. and Emerson C.P. Jr (2002). Myogenic regulatory
factors and the specification of muscle progenitors in vertebrate embryos. Annu. Rev. Cell
Dev. Biol., 18: 747-783.

8. Bibliografia
-180-
177. Prichard M.N., Jairath S., Penfold M.E., St. Jeor S., Bohlman M.C. and Pari G.S.
(1998). Identification of persistent RNA-DNA hybrid structures within the origin of
replication of human cytomegalovirus. J. Virol., 72: 6997-7004.
178. Pröch S., Prienner C., Hoflich C., Liebenthal C., Babel N., Kruger D.H. and Volk
H.D. (2003). Proteasome inhibotors: a novel tool to suppress human cytomegalovirus
replication and virus-induced immune modulation. Antiviral Therapy, 8: 555-567.
179. Puck T.T., Cieiura S.J. and Robinson A. (1958). Genetics of somatic mammalian
cells III. Long-term cultivation of euploid cells from human and animal subjects. J. Exp.
Med., 108: 945-956.
180. Qian S.B., Ott D.E., Schubert U., Bennink J.R. and Yewdell J.W. (2002). Fusion
proteins with COOH-terminal ubiquitin are stable and maintain dual functionality in vivo.
J. Biol. Chem., 277: 38818-38826.
181. Randolph-Habecker J.R., Rahill B., Torok-Storb B., Vieira J., Kolattukudy P.E.,
Rovin B.H. and Sedmak D.D. (2002). The expression of the cytomegalovirus chemokine
receptor homolog US28 sequesters biologically active CC chemokines and alters IL-8
production. Cytokine, 19: 37-46.
182. Realini C. , Jensen C.C. , Zhang Z., Johnston S.C., Knowlton J.R., Hill C.P. and
Rechsteiner M. (1997). Characterization of Recombinant REGα, REGβ, and REGγ
Proteasome Activators. J. Biol. Chem., 272: 25483-25492.
183. Rechsteiner M. and Hill C.P. (2005). Mobilizing the proteolytic machine: cell
biological roles of proteasome activators and inhibitors. Trends in Cell Biol., 15: 27-33.
184. Reinstein E., Scheffner M., Oren M., Ciechanover A. and Schwartz A. (2000).
Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-protesome
system: targeting via ubiquitination of the N-terminal residue. Oncogene, 19: 5944-5950.
185. Reits E.A. J., Benham A.M., Plougastel B., Neefjes J. and Trowsdale J. (1997).
Dynamics of proteasome distribution in living cells. EMBO J., 16: 6087-6094.
186. Rivett A.J. (1998). Intracellular distribution of proteasomes. Curr. Opin. Immunol.,
10: 110-114.
187. Rivett A.J., Bose S., Brooks P. and Broadfoot K.I. (2001). Regulation of proteasome
complex by gamma-inteferon and phosphorylation. Biochimie, 83: 363-366.
188. Robson L. and Gibson W. (1989). Primate cytomegalovirus assembly protein:
genome location and nucleotide sequence. J. Virol., 63: 669-676.

8. Bibliografia
-181-
189. Roby C. and Gibson W. (1986). Characterization of phosphoprotein and protein
kinase activity of virions, non-infectious enveloped particles, and dense bodies of human
cytomegalovirus. J. Virol., 59: 714-727.
190. Rock K.L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D. and
Goldberg A.L. (1994). Inhibitors of the proteasome block the degradation of most cell
protein and the generation of peptides presented on MHC class I molecules. Cell, 78: 761-
771.
191. Rockel T.D. and von Mikecz A. (2002). Proteasome-dependent processing of
nuclear proteins is correlated with their subnuclear localization. J. Structural Biol., 140:
189-199.
192. Romanowski M.J., Garrido-Guerrero E. and Shenk T. (1997). pIRS1 and pTRS1 are
present in human cytomegalovirus virions. J. Virol., 71: 5703-5705.
193. Ruggero D., Wang Z.G. and Pandolfi P.P. (2000). The puzzling multiple lives of
PML and its role in the genesis of cancer. Bioessays, 22: 827-835.
194. Salvant B.S., Fortunato E.A. and Spector D.H. (1998). Cell cycle dysregulation by
human cytomegalovirus: influence of the cell cycle phase at the time of infection and
effects on cyclin transcription. J. Virol., 72: 3729-3741.
195. Sanchez V. and Spector D.H. (2002). CMV makes a timely exit. Science, 297: 778-
779.
196. Sarov I. and Abady I. (1975). The morphogenesis of human cytomegalovirus.
Isolation and polypeptide characterization of cytomegalovirus and dense bodies.
Virology, 66: 464-473.
197. Schmid H.P., Akhayat O., Martines de Sa C., Puvion F., Koehler K. and Scherrer K.
(1984). The prosome: an ubiquitous morphologically distinct RNP particle RNP associated
with repressed mRNPs and containing specific ScRNA and a characteristic set of proteins.
EMBO J., 3: 29-34.
198. Schmidtke G., Schmidt M. and Kloetzel P.M. (1997). Maturation of mammalian 20S
proteasome: purification and characterization of 13S and 16S proteasome precursor
complexes, J. Mol. Biol., 268: 95-106.
199. Schmolke S., Keren P., Drescher P., Jahn G. and Plachter B. (1995). The dominant
phosphoprotein pp65 (ppUL83) of human cytomegalovirus is dispensable for growth in
cell culture. J. Virol., 69: 5959-5968.

8. Bibliografia
-182-
200. Schneiter R., Kadowaki T. And Tartakoff A.M. (1995). mRNA transport in yeast:
time to reinvestigate the functions of the nucleolus. Mol. Biol. Cell, 6: 357-370.
201. Schroeder M.B. and Worthen G.S. (2001). Viral regulation of RANTES expression
during human cytomegalovirus infection of endothelial cells. J.Virol., 75: 3383-3390.
202. Schubert U., Ott DE, Chertova E.N., Welker R., Tessmer U., Princiotta M.F.,
Bennink J.R., Krausslich H.G. and Yewdell J.W. (2000). Proteasome inhibition interferes
with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. P.N.A.S.
U.S.A., 97: 13057-13062.
203. Seeger M., Ferrell K., Frank R. and Dubiel W. (1997). HIV-1 Tat inhibits the 20S
proteasome and its 11S regulator-mediated activation. J. Biol. Chem., 272: 8145-8148.
204. Sherwood R.I. and Wagers A.J. (2006). Harnessing the potential of myogenic
satellite cells. Trends in Mol. Med., in corso di stampa (doi: 10.1016/j.molmed.2006.03.002)
205. Sinclair J., Baillie J., Bryant L. and Caswell R. (2000). Human cytomegalovirus
mediates cell cycle progression through G(1) into early S phase in terminally
differentiated cells. J. Gen. Virol., 81: 1553-1565.
206. Sinzger C., Grefte A., Plachter B., Gouw A.S., The T.H. and Jahn G. (1995).
Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of
human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol., 76:
741-50.
207. Solomon V. and Goldberg A.L. (1996). Importance of the ATP-ubiquitin-
proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit
muscle extracts. J. Biol. Chem., 271: 26690-26697.
208. Soneoka Y., Cannon P.M., Ramsdale E.E., Griffiths J.C., Romano G., Kingsman
S.M. and Kingsman A.J. (1995). A transient three-plasmid expression system for the
production of high titer retroviral vectors. Nucleic Acids Research, 23: 628-633.
209. Song X. Mott J.D., von Kampen J., Pramanik B., Tanaka K., Slaughter C.A. and
DeMartino G.N. (1996). A model for the quaternary structure of the proteasome activator
PA28. J. Biol Chem., 271: 26410-26417.
210. Song Y.-J. and Stinski M.F. (2005). Inhibition of cell division by the Human
Cytomegalovirus IE86 protein: role of the p53 pathway or cyclin-dependent kinase
1/Cyclin B1. J. Virol., 79: 2597-2603.

8. Bibliografia
-183-
211. Spector D.H. (1996). Activation and regulation of human cytomegalovirus early
genes. Intervirology, 39: 361-377.
212. Stauber W.T., Fritz V.K., Maltin C.A. and Dahlmann B. (1987). Localization of a
multi-catalytic, high-molecular mass proteinase in the nuclei of muscle cells. J.
Histochem., 19: 594-597.
213. Stenberg R.M. (1996). The human cytomegalovirus major immediate-early gene.
Intervirology, 39: 343-349.
214. Strouboulis J. and Wolffe A.P. (1996). Functional compartmentalization of the
nucleus. J. Cell Science, 109: 1991-2000.
215. Suarez R.K. (2003). Shaken and stirred: muscle structure and metabolism. J. Exp.
Biol., 206: 2021-2029.
216. Sun L., Trausch-Azar J.S., Ciechanover A. and Schwartz A.L. (2005). Ubiquitin-
proteasome-mediated degradation, intracellular localization, and protein synthesis of
MyoD and Id1 during muscle differentiation. J. Biol. Chem., 280: 26448-26456.
217. Sun X.H., Copeland N.G., Jenkins N.A. and Baltimore D. (1991). Id proteins Id1
and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol.
Cell Biol., 11: 5603-5611.
218. Taylor R.T. and Bresnahan W.A. (2005). Human Cytomegalovirus immediate-early
2 gene expression blocks virus-induced beta interferon production. J. Virol., 79: 3873-3877.
219. Taylor R.T. and Bresnahan W.A. (2006). Human Cytomegalovirus immediate-early
2 protein IE86 blocks virus-induced chemokine expression. J. Virol., 80: 920-928.
220. Tomczak K.K., Marinescu V.D., Ramoni M.F., Sanoudou D., Montanaro F., Han
M., Kunkel L.M., Kohane I.S. and Beggs A.H. (2004). Expression profilig and identification
of novel genes involved in myogenic differentiation. FASEB J., 18: 403-405.
221. Trombitás K., Greaser M., French G. and Granzier H (1998). PEVK extension of
Human soleus muscle titin revealed by immunolabelling with the anti-titin antibody
9D10. J. Struct. Biol., 122: 188-196.
222. Unno M., Mizushima T., Morimoto Y., Tomisugi Y., Tanaka K., Yasuoka N. and
Tsukihara T. (2002). The Structure of the Mammalian 20S Proteasome at 2.75 Å
Resolution. Structure (Camb.), 10: 609-618.
223. Ustrell V., Hoffman L., Pratt G. and Rechsteiner M. (2002). PA200, a nuclear
proteasome activator involved in DNA repair. EMBO J., 13: 3516-3525.

8. Bibliografia
-184-
224. Varnum S.M., Streblow D.N., Monroe M.E., Smith P., Auberry K.J., Pasã- Tolic L.,
Wang D., Camp II D.G., Rodland K., Wiley S., Britt W., Shenk T., Smith R.D. and Nelson
J.A. (2004). Identification of proteins in Human Cytomegalovirus (HCMV) particles: the
HCMV proteome. J. Virol., 78: 10960-10966.
225. Varshavsky A. (2000). Ubiquitin fusion technique and its descendants. Methods in
Enzymology, 327: 578-593.
226. Visintin R. and Amon A. (2000). The nucleolus: the magician’s hat for cell cycle
tricks. Curr. Op. Cell Biol., 12: 372-77.
227. Vodermaier H.C. (2004). APC/C and SCF: controlling each other and the cell cycle.
Current. Biol., 14: R787-796.
228. Voges D., Zwickl P. and Baumeister W. (1999). The 26S proteasome: a molecular
machine designed for controlled proteolysis. Annual Rev. Biochem., 68: 1015-1068.
229. Walsh K. and Perlman H. (1997). Cell cycle exit upon myogenic differentiation.
Curr. Opin. Genet. Dev., 7: 597-602.
230. Weintraub H. (1993). The MyoD family and myogenesis: redundancy, networks,
and thresholds. Cell, 75: 1241-1244.
231. Weintraub H., Davis R., Tapscott S., Thayer M., Krause M., Benezra R., Blackwell
T.K., Turner D., Rupp R., Hollenberg S., Zhuang Y. and Lassar A. (1991). The MyoD gene
family: nodal point during specification of muscle cell lineage. Science, 251: 761-766.
232. Weintraub S.J., Chow K.N.B., Luo R.X., Zhang S.H., He S. and Dean D.C. (1995).
Mechanism of active transcriptional repression by the retinoblastoma protein. Nature,
375: 812-816.
233. Weintraub S.J., Pater C.A. and Dean D.C. (1992). Retinoblastoma protein switches
the E2F site from positive to negative element. Nature, 358: 259-261.
234. Welch A.R., Woods A.S., McNally L.M., Cotter R.J. and Gibson W. (1991). A
herpesvirus maturational proteinase, assemblin: identification of its gene, putative active
site domain, and cleavage site. P.N.A.S. U.S.A., 88: 10792-10796.
235. White E.A. and Spector D.H. (2005). Exon 3 of the Huma Cytomegalovirus major
immediate-early region is required for efficient viral gene expressione and for cellular
cyclin modulation. J. Virol., 79: 7438-7452.

8. Bibliografia
-185-
236. Wilkinson C.R., Wallace M., Morphew M., Perry P., Allshire R., Javerzat J.P.,
McIntosh J.R. and Gordon C. (1998). Localization of the 26S proteasome during mitosis
and meiosis in fission yeast. EMBO J., 17: 6465-6476.
237. Wilkinson G.W., Kelly C., Sinclair J.H. and Rickards C. (1998). Disruption of PML-
associated nuclear bodies mediated by the human cytomegalovirus major immediate
early gene product. J. Gen. Virol., 79: 1233-1245.
238. Williams A.B., Decourten-Myers G.M., Fischer J.E., Luo G., Sun X. and Hasselgren
P.O. (1999). Sepsis stimulates release of myofilaments in skeletal muscle by a calcium-
dependent mechanism. FASEB J., 13: 1435-1443.
239. Winkler M., Schmolke S., Plachter B. and Stamminger T. (1995). The pUL69 protein
of human cytomegalovirus (HCMV), a homologue of the herpes simplex virus ICP27, is
contained within the tegument of virions and activates the major immediate early
enhancer of HCMV in synergy with the tegument protein pp71 (ppUL82). Scand. J. Infect.
Dis. Suppl., 99: 8-9.
240. Yew P.R. (2001). Ubiquitin-mediated proteolysis of vertebrate G1- and S-phase
regulators. J. Cell Physiol., 187: 1-10.
241. Zhang J., Campbell R.E., Ting A.Y. and Tsien R.Y. (2002). Creating new fluorescent
probes for cell biology. Nat. Rev. Mol. Cell Biol., 3: 906-918.
242. Zhang Z., Clawson A. and Rechsteiner M. (1998). The proteasome activator 11 S
regulator or PA28. Contribution by both alpha and beta subunits to proteasome
activation. J. Biol. Chem., 13: 30660-30668.
243. Zhang Z., Krutchinsky A., Endicott S., Realini C., Rechsteiner M. and Standing
K.G. (1999). Proteasome activator 11S REG or PA28: recombinant REG alpha/REG beta
hetero-oligomers are heptamers. Biochem., 27: 5651-5658.
244. Zhao M., Qiao M., Harris S.E., Oyajobi B.O., Mundy G.R. and Chen D. (2004).
Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. J. Biol.
Chem., 279: 12854-12859.
245. Zhu H., Cong J.P. and Shenk T. (1997). Use of differential display analysis to assess
the effect of human citomegalovirus infection on the accumulation of cellular RNAs:
induction of interferon-responsive RNAs. P.N.A.S. U.S.A., 94: 13985-13990.
246. Zhu H., Cong J.P., Mamtora G., Gingeras T. and Shenk T. (1998). Cellular gene
expression altered by human cytomegalovirus: global monitoring with oligonucleotide
arrays. P.N.A.S. U.S.A., 95: 14470-14475.

8. Bibliografia
-186-
247. Zhu H., Shen Y. and Shenk T. (1995). Human cytomegalovirus IE1 and IE2
proteins block apoptosis. J. Virol., 69: 7960-7970.
248. Zhukarev V., Ranger J.M., Ranger J.W., Goldman Y.E. and Shuman H. (1997).
Distribution and orientation of rhodamine-phalloidin bound to thin filaments in skeletal
and cardiac myofibrils. Cell Motil. Cytoskeleton, 37: 363-377.

9. Ringraziamenti
9. RINGRAZIAMENTI

9. Ringraziamenti
-188-
Vorrei ringraziare, innanzitutto, il prof. Carlo Chezzi, Direttore del Dipartimento di
Patologia e Medicina di Laboratorio – Università degli Studi di Parma e il prof. Giuseppe
Dettori, Responsabile della Sezione di Microbiologia del Dipartimento di Patologia e
Medicina di Laboratorio – Università degli Studi di Parma, che mi hanno permesso di
frequentare la Sezione di Microbiologia per lo svolgimento del Dottorato di Ricerca.
Desidero, inoltre, ringraziare il prof. Carlo Chezzi per avermi dato l’opportunità di
svolgere questo lavoro di tesi sotto la sua direzione nel gruppo di ricerca da lui
coordinato. A tal proposito, un sentito ringraziamento va alla dr.ssa Maria Cristina
Arcangeletti e alla dr.ssa Flora De Conto per il prezioso aiuto e per gli insegnamenti
inerenti il citomegalovirus umano. Inoltre, vorrei ringraziare la mia collega e compagna di
“infezioni”, la dr.ssa Federica Motta, per la simpatia e per il sostegno in campo
informatico.
Ringrazio tutti gli amici, i colleghi e il personale tecnico, il personale laureato strutturato, i
docenti e il personale amministrativo del laboratorio per avermi trasmesso la passione e le
conoscenze relative alla microbiologia e alla virologia e per avermi aiutato e sostenuto con
il loro affetto durante questi lunghi anni trascorsi presso la Sezione di Microbiologia.
Per quanto concerne il mio periodo di permanenza presso il laboratoire CRRET
(Croissance cellulaire, la Réparation et la Régénération Tissulaires) - Université Paris XII -
Val de Marne, vorrei ringraziare il prof. Denis Barritault e il prof. Jean Pierre Caruelle, che
si sono susseguiti alla direzione del laboratorio CRRET, per avermi consentito di svolgere
parte del Dottorato di Ricerca nell’ambito di una tesi in co-tutela Italia-Francia presso il
loro laboratorio. A tal fine, desidero ringraziare il prof. Jean Foucrier per avermi dato
l’opportunità di svolgere il Dottorato di Ricerca sotto la sua direzione. Inoltre, ringrazio il
prof. Yann Bassaglia per la sua disponibilità e per avermi trasmesso le conoscenze relative
alla tematica del muscolo.
Ringrazio con tutto il cuore gli studenti, il personale tecnico, i docenti, i ricercatori e il
personale amministrativo che ho avuto modo di incontrare durante i miei due anni di
permanenza presso il laboratoire CRRET per la simpatia, per il supporto tecnico-
scientifico e per finire per l’incoraggiamento linguistico che mi ha permesso di
apprendere e, nello stesso tempo di apprezzare, una nuova lingua straniera…..il francese!
Un ringraziamento speciale al dr. José Cebrian, con cui ho avuto la fortuna e il privilegio
di lavorare presso il laboratoire CRRET, per la competenza e la professionalità con cui mi
ha guidato durante questi anni di dottorato consentendomi di approfondire l’aspetto
inerente la biologia molecolare. Inoltre, desidero ringraziarlo, per i suoi preziosi consigli e

9. Ringraziamenti
-189-
per aver sopportato con grande pazienza e simpatia le mie “crisi” informatiche con il
sistema Macintosh.
Vorrei ringraziare, infine, la mia grande famiglia che mi ha sempre sostenuta e circondata
di affetto dandomi così la forza di superare anche i momenti più difficili.
Ricorderò sempre tutti con affetto e riconoscenza.

9. Remerciements
-190-
Je voudrais tout d’abord remercier le professeur Carlo Chezzi, Directeur du Département
de Pathologie et de Médecine de Laboratoire - Université de Parme ainsi que le Professeur
Giuseppe Dettori, Responsable de la Section de Microbiologie du Département de
Pathologie et de Médecine de Laboratoire - Université de Parme lesquels m’ont permis de
suivre les cours à la Section de Microbiologie pour la préparation de mon Doctorat en
Recherche. Mes plus sincères remerciements et ma plus grande reconnaissance à
monsieur le professeur Carlo Chezzi qui m’a donné l’opportunité de préparer mon
Doctorat en Recherche sous sa direction dans le groupe qu’il a lui-même coordonné. A ce
propos, j’aimerais remercier Mme Maria Cristina Arcangeletti et Mlle Flora De Conto
pour leur aide précieuse et pour leur enseignement concernant le cytomégalovirus. Par
ailleurs, je voudrais remercier ma collègue et compagne “d’infections”, madame Federica
Motta, pour sa sympathie ainsi que pour son soutien dans le domaine informatique. Je
remercie tous les amis, colleguès et tous les membres du laboratoire, qui m’ont transmis la
passion et les connaissances relatives à la microbiologie et à la virologie et m’ont aidée et
soutenue par leur affection au cours de ces longues années passées à la Section de
Microbiologie. En ce qui concerne ma période de permanence au laboratoire CRRET
(Croissance cellulaire, la Réparation et la Régénération Tissulaires) - Université Paris XII -
Val de Marne je voudrais remercier le professeur Denis Barritault et le professeur Jean
Pierre Caruelle qui se sont succédé à la direction du laboratoire CRRET et qui m’ont
permis de préparer une partie de mon Doctorat dans le cadre d’une thèse en co-tutelle
Italie-France dans leur laboratoire. Mes plus sincères remerciements et ma plus grande
reconnaissance à monsieur le professeur Jean Foucrier qui m’a donné l’opportunité de
préparer mon Doctorat en Recherche sous sa Direction. Je remercie également le
professeur Yann Bassaglia de sa disponibilité et de m’avoir transmis les connaissances
dans le domaine du muscle. Je remercie de tout cœur les étudiants, le personnel
technique, les enseignants, les chercheurs et le personnel administratif que j’ai eu
l’occasion de rencontrer durant mes deux années de permanance au Laboratoire CRRET
pour leur sympathie, le support technico-scientifique et pour finir pour l’encouragement
linguistique grâce auquel j’ai pu apprendre et en même temps apprécier une nouvelle
langue......le Français! Un remerciement spécial à monsieur le docteur José Cebrian avec
qui j’ai en la chance et le privilège de travailler au laboratoire CRRET et dont la
compétence et la professionalité m’ont guidée durant ces années de doctorat me
permettant d’approfondir l’aspect inhérent à la biologie moléculaire. Je le remercie
également pour ses précieux conseils et les nombreuses discussions scientifique et, en

9. Remerciements
-191-
outre, d’avoir supporté avec beaucoup de patience et de sympathie mes “crises”
informatiques avec le système Macintosh.
Merci, enfin, à ma grande famille qui m’a toujours soutenue et entourée d’affection en me
donnant la force de surmonter les moments les plus difficiles.
Je garde de tous un souvenir affectieux ainsi que de la reconnaissance.