Embed Size (px)
Citation preview

1971
□ CASE REPORT □
Autoimmune Hepatitis Associated with PulmonaryArterial Hypertension
Fumihiko Kaneko 1, Hiroaki Yokomori 1, Kumiko Tahara 1, Tomochika Takeshita 1,Hajime Takeuchi 1, Hide Yoshida 1, Kenta Hoshi 1, Hirobumi Kondo 1,
Makoto Ohbu 2, Tooru Sato 3 and Toshifumi Hibi 3
Abstract
A 46-year-old woman presented with arthralgia. She had a history of fluctuating liver function impairmentfor 6 months. Laboratory investigations revealed elevated liver function test results, positive antinuclear anti-bodies and elevated serum IgG. The histological findings of a liver biopsy were interface hepatitis accompa-nied by plasmocytic infiltration with bridging fibrosis. There was no evidence of cirrhosis on pathological ex-amination and no portal hypertension on endoscopic and radiographic studies. Autoimmune hepatitis was di-agnosed, and treatment with prednisolone improved the liver dysfunction. After 6 months, she complained ofdyspnea. Doppler echocardiography showed a dilated right ventricle, severe tricuspid insufficiency, and sys-tolic pulmonary arterial pressure indicative of pulmonary arterial hypertension. We report this rare case ofautoimmune hepatitis with pulmonary arterial hypertension.
Key words: autoimmune hepatitis, pulmonary arterial hypertension
(Inter Med 47: 1971-1976, 2008)(DOI: 10.2169/internalmedicine.47.1420)
Introduction
Autoimmune hepatitis (AIH) has been shown to be asso-ciated with a number of other autoimmune diseases (1, 2).Patients with AIH may be at increased risk for developingsystemic connective tissue disease (CTD) (3). While AIHand CTD are different entities, the boundary between thetwo disorders can be indistinct (3). Pulmonary arterial hy-pertension (PAH) occurs in a variety of conditions includingCTD and some rheumatic diseases (4). PAH is characterizedby progressive obliteration of small vessels in the pulmonaryvascular bed leading to permanently increased pulmonaryvascular resistance and elevated pulmonary artery pressure,which may result in right heart failure and premature death.However, treatment options for patients with PAH haveevolved to help prolong their survival and improve theirquality of life. Early identification of PAH and its underly-ing or concurrent pathology is important for treatment out-
come (5). We report a rare case of AIH with PAH, whichwas diagnosed from investigating multiple serological mark-ers.
Case Report
A 46-year-old woman was admitted to our hospital with alow-grade fever and arthralgia. The patient had a history offluctuating liver function impairment for 6 months. She de-nied alcohol and drug use. On admission, her temperaturewas 37℃, pulse rate was 70/min, respiration rate was 18/min, and blood pressure was 110/70 mmHg. Physical ex-amination detected no palpable cervical lymph node, butmoderate jaundice was found. Auscultation of the heart andthe lungs were normal. The abdomen was soft with normalbowel sounds. The edge of the liver was detected at 3 cmbelow the right costal margin.Her initial laboratory findings were as follows: Whiteblood cell count (5.40×103/μL; normal range, 3.3-9.0×103)
1Departments of Internal Medicine, Kitasato Institute Medical Center Hospital, Kitasato University, Kitamoto, 2Department of Pathology, Schoolof Allied Health Sciences, Kitasato University, Sagamihara and 3Department of Internal Medicine, School of Keio University, TokyoReceived for publication June 17, 2008; Accepted for publication August 4, 2008Correspondence to Dr. Hiroaki Yokomori, [email protected]

Inter Med 47: 1971-1976, 2008 DOI: 10.2169/internalmedicine.47.1420
1972
was within normal range. Red blood cell count was slightlyreduced (3.17×104/μL; normal range, 4.30-5.70×104). Plateletcount (16.7×103/μL; normal range, 140-340×103/μL) waswithin normal range. Total bilirubin (4.2 mg/dL; normalrange: 0.2-1), direct bilirubin (3.2 mg/dL; normal range, 0.0-0.4), aspartate aminotransferase (AST) (159 U/L; normalrange, 5-35), and alanine aminotransferase (ALT) (149 U/L;normal range, 5-45) were elevated. Alkaline phosphatase(ALP) (361 IU/L; normal range: 100-325) was slightly high.Prothrombin time was reduced (48%; normal range, 70-100). Tests for antibodies to Epstein-Barr nuclear antigenand viral capsid antigen (IgG) were both positive. Urinalysiswas normal. Viral serological tests were negative for anti-HCV, anti-hepatitis B core antigen, and anti-hepatitis B sur-face antigen. HCV-RNA was not detected in serum by po-lymerase chain reaction. The concentrations of IgG, IgM,and IgA were 4808 mg/dL (normal range; 870-1,700), 551mg/dL (normal range; 110-410), and 253 mg/dL (normalrange; 46-260), respectively. The test for anti-nuclear anti-bodies was positive, at a titer of 1: 2,560 and showing ho-mogenous pattern. Anti-mitochondria antibody was negative.Human leukocyte antigens (HLA) typing was DR-4, DR14.Abdominal ultrasonography (US), computed tomography(CT) and magnetic resonance imaging (MRI) were con-ducted to investigate the cause of hepatic dysfunction. Thepatient had no splenomegaly. Electronic gastroduodenal en-doscope depicted no esophageal or gastric varices. There
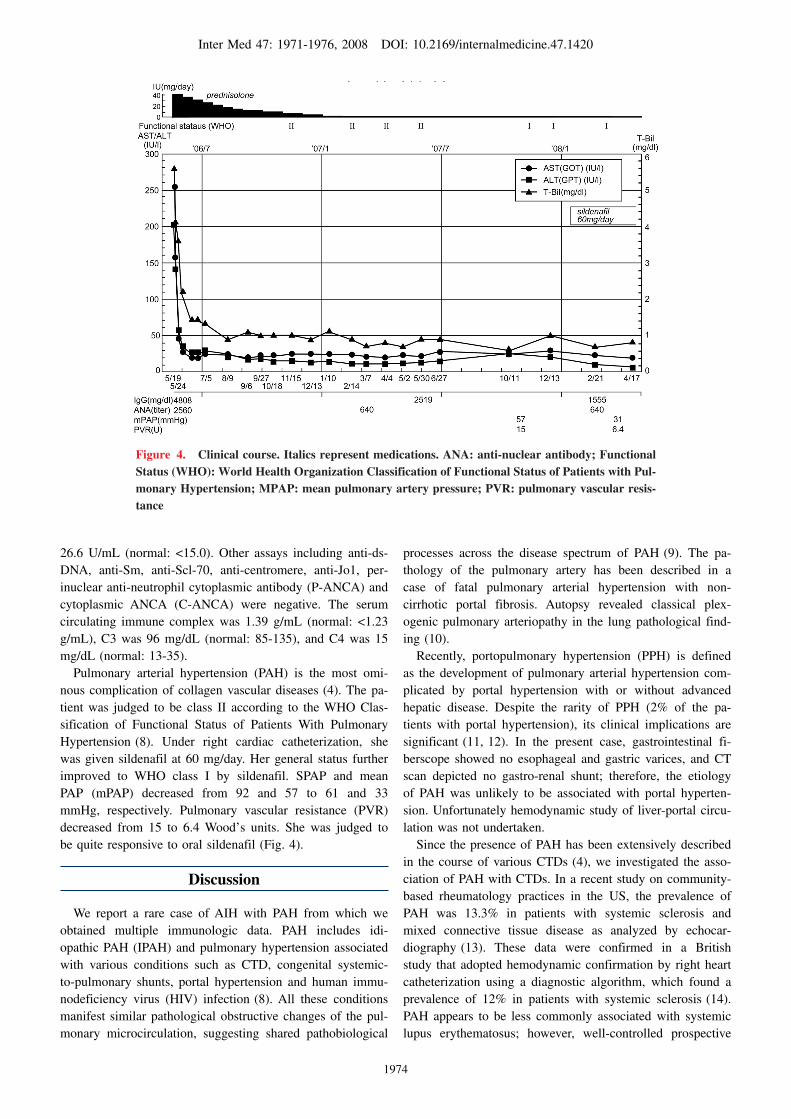
was no evidence of portal hypertension on endoscopic andradiographic studies. Liver biopsy showed moderate to se-vere portal fibrosis with partial nodular formation of paren-chyma, interface hepatitis, and moderate to severe portal in-flammation consisting of lymphocytes and plasma cells. Ro-sette arrangement of hepatocytes was commonly observed inperiportal zones. However, interlobular bile ducts werehardly involved. Inflammation activity and the stage of fi-brosis were classified as A3F3 according to the Inuyamaclassification (6) (Fig. 1). Cirrhosis was not evident. Auto-immune hepatitis was diagnosed provisionally. A final diag-nosis of AIH was established on the basis of a positive anti-nuclear antibody test, high titers of serum globulin, no evi-dence of current hepatitis virus infection, histological find-ings, and a good response to corticosteroid therapy, all ofwhich met the diagnostic criteria for type 1 AIH (7). Thepatient had a pre-treatment AIH score of 26 according to thesystem proposed by the International Autoimmune HepatitisGroup (7).Prednisolone (40 mg/day) was started on the fifth hospital
day, after the diagnosis of AIH. The patient’s elevatedbilirubin levels began to decline after the administration ofsteroids for 3 days, and eventually normalized in one month.Other liver function tests including AST and ALT also de-creased to normal ranges concomitantly. IgG declined gradu-ally and returned to normal range after approximately a year(Fig. 4). She was maintained on prednisolone at 2 mg/day.
Figure 1. Histological findings of liver biopsy. Liver biopsy shows moderate to severe portal in-flammation infiltrated by lymphocytes and plasma cells (a), interface hepatitis (b) (arrow), portal fibrosis with nodular formation (c) and (d) rosette arrangement of hepatocytes (arrow) in peripor-tal zone. (a, b, d: Hematoxylin and Eosin staining; c: Masson-trichrome stain). Magnification for a, b and c: ×100; for d, ×400.

Inter Med 47: 1971-1976, 2008 DOI: 10.2169/internalmedicine.47.1420
1973
After 6 months, she complained of exercise-induced dyspneafor the previous month.CTD is characterized by polyarthralgia, arthritis, lowgrade fever, erythema, sicca symptom, and Raynaud’s phe-nomenon. However, the patient showed none of these char-acteristic symptoms. Echocardiography revealed a dilated,hypertrophied right ventricle and septal flattening in aparasternal short axis view (Fig. 2a). Continuous Dopplerechocardiography revealed a dilated right ventricle, severetricuspid insufficiency and systolic pulmonary arterial pres-sure (sPAP) of at least 95 mmHg, showing evidence of PAH(Fig. 2b). Ventilation/perfusion scan of lungs did not suggest
the presence of thromboembolism. Right heart catheteriza-tion disclosed severe pulmonary arterial hypertension with amean right atrial pressure of 24 mmHg (normal: <6 mmHg),systolic PAP of 95 mmHg (normal: <30 mmHg), mean PAPof 54 mmHg (normal: <14 mmHg), and cardiac output of2.43/min/m2 body surface area (normal: >2.9/min/m2). Im-munological studies showed a positive test for anti-nuclearantibodies at a titer of 1: 640 with homogenous staining pat-tern, while anti-DNA was 14 IU/mL (normal: <6), RAHAtiter was ×80 (normal:<×40), anti-RNP was 25.9 pU/mL(normal: <15), anti-Scl was 6.0 U/mL (normal: <16), anti-SS-A was 109.0 U/mL (normal: <10.0), and anti-SS-B was
Figure 2. Echocardiography. a: A parasternal short axis view reveals a dilated, hypertrophic right ventricle and septal flattening. b: Doppler echocardiography shows a dilated right ventricle, severe tricuspid insufficiency and systolic pulmonary arterial hypertension.
Figure 3. Micrographs of immunohistochemical studies for D2-40 (a) and CD34 (b) in sequential sections. Considering the two stains, D2-40 stains the endothelium of a lymphatic capillary but not the adjacent blood capillary, while CD34 stains the endothelium of portal vein, capillary or arteriole. Comparison of these two photographs shows that the endothelial walls of several vessels are positive for D2-40 (stars and fine arrows in Fig. 3a) but are negative for CD34 (Fig. 3b). These are lymphatic capillaries. On the other hand, several vessels are positive for CD34 (arrowheads and bold arrows in Fig. 3b) but negative for D2-40 (Fig. 3a). These are blood vessels. In this case di-lated lymphatic vessels are observed. P: portal vein. Lym: lymphatic capillary. Black arrowhead denotes portal vein. Black bold arrow denotes blood capillary, arteriole or venule. Black fine arrow denotes lymphatic capillary. Star denotes dilated lymphatic capillaries. Hematoxylin counterstaining.
Inter Med 47: 1971-1976, 2008 DOI: 10.2169/internalmedicine.47.1420
1974
Figure 4. Clinical course. Italics represent medications. ANA: anti-nuclear antibody; Functional Status (WHO): World Health Organization Classification of Functional Status of Patients with Pul-monary Hypertension; MPAP: mean pulmonary artery pressure; PVR: pulmonary vascular resis-tance
26.6 U/mL (normal: <15.0). Other assays including anti-ds-DNA, anti-Sm, anti-Scl-70, anti-centromere, anti-Jo1, per-inuclear anti-neutrophil cytoplasmic antibody (P-ANCA) andcytoplasmic ANCA (C-ANCA) were negative. The serumcirculating immune complex was 1.39 g/mL (normal: <1.23g/mL), C3 was 96 mg/dL (normal: 85-135), and C4 was 15mg/dL (normal: 13-35).Pulmonary arterial hypertension (PAH) is the most omi-
nous complication of collagen vascular diseases (4). The pa-tient was judged to be class II according to the WHO Clas-sification of Functional Status of Patients With PulmonaryHypertension (8). Under right cardiac catheterization, shewas given sildenafil at 60 mg/day. Her general status furtherimproved to WHO class I by sildenafil. SPAP and meanPAP (mPAP) decreased from 92 and 57 to 61 and 33mmHg, respectively. Pulmonary vascular resistance (PVR)decreased from 15 to 6.4 Wood’s units. She was judged tobe quite responsive to oral sildenafil (Fig. 4).
Discussion
We report a rare case of AIH with PAH from which weobtained multiple immunologic data. PAH includes idi-opathic PAH (IPAH) and pulmonary hypertension associatedwith various conditions such as CTD, congenital systemic-to-pulmonary shunts, portal hypertension and human immu-nodeficiency virus (HIV) infection (8). All these conditionsmanifest similar pathological obstructive changes of the pul-monary microcirculation, suggesting shared pathobiological
processes across the disease spectrum of PAH (9). The pa-thology of the pulmonary artery has been described in acase of fatal pulmonary arterial hypertension with non-cirrhotic portal fibrosis. Autopsy revealed classical plex-ogenic pulmonary arteriopathy in the lung pathological find-ing (10).Recently, portopulmonary hypertension (PPH) is definedas the development of pulmonary arterial hypertension com-plicated by portal hypertension with or without advancedhepatic disease. Despite the rarity of PPH (2% of the pa-tients with portal hypertension), its clinical implications aresignificant (11, 12). In the present case, gastrointestinal fi-berscope showed no esophageal and gastric varices, and CTscan depicted no gastro-renal shunt; therefore, the etiologyof PAH was unlikely to be associated with portal hyperten-sion. Unfortunately hemodynamic study of liver-portal circu-lation was not undertaken.Since the presence of PAH has been extensively describedin the course of various CTDs (4), we investigated the asso-ciation of PAH with CTDs. In a recent study on community-based rheumatology practices in the US, the prevalence ofPAH was 13.3% in patients with systemic sclerosis andmixed connective tissue disease as analyzed by echocar-diography (13). These data were confirmed in a Britishstudy that adopted hemodynamic confirmation by right heartcatheterization using a diagnostic algorithm, which found aprevalence of 12% in patients with systemic sclerosis (14).PAH appears to be less commonly associated with systemiclupus erythematosus; however, well-controlled prospective

Inter Med 47: 1971-1976, 2008 DOI: 10.2169/internalmedicine.47.1420
1975
trials are lacking and retrospective studies reported a preva-lence of up to 14% as assessed by echocardiography (15).Patients with AIH may be at an increased risk of developingsystemic CTD (3). Moreover, there appears to be sharedsusceptibility alleles for AIH and CTD in addition to sharedautoantibodies (3). On the other hand, Morrison et al (16)reported an association of type 1 AIH with PAH, althoughthe association is quite rare. Their case series described theclinical and pathologic findings of seven consecutive pa-tients with progressive and fatal pulmonary hypertensionwhich was not explained by predisposing cardiac or pulmo-nary diseases. Bertino et al (17) reported a case of AIH/PAHwith correlated diagnostic and therapeutic implications, andproposed the possibility to classify this condition in the con-text of a more complex “overlap syndrome”.In the liver biopsy, there were many abnormal vessels.
Most vessels in a liver biopsy can be easily identified aslymphatic vessels or blood vessels by a combination of D-240 (lymph vessels) and CD34 (blood vessels) immunostain-ing. D2-40 stains the endothelium of a small lymphatic ves-sel but not that of the adjacent capillary, while CD34 stainsthe endothelium of a portal vein, capillary and arteriole. Us-ing this method, we found that the abnormal vessels in theliver biopsy of the present case were dilated lymphatic ves-sels (Fig. 3). Previous report showed that the number oflymphatics and areas occupied by the lymphatics do not dif-fer significantly with the activity of hepatitis, but differ sig-nificantly in association with the degree of liver fibrosis.These changes are thought to be caused by the disturbanceof microcirculation associated with liver fibrosis and lobulardistortion (18).
As for clinical manifestations, the present patient hadanti-nuclear antigen, anti-SS-A, anti-SS-B, and anti-RNP an-tibodies, but no signs or symptoms suggestive of a systemicautoimmune disease, and did not fulfill the classification cri-teria for defined diseases. Such conditions have been definedas undifferentiated connective tissue diseases (UCTD) (19).The most characteristic symptoms of UCTD are representedby arthritis and arthralgias, Raynaud’s phenomenon andleukopenia, while neurological and kidney involvement isvirtually absent. Eighty percent of these patients have a sin-gle autoantibody specificity, more frequently anti-SS-A andanti-RNP antibodies. Stable UCTD are considered a distinctclinical entity, and therefore it has been proposed to definethose conditions as UCTD. Classification criteria have alsobeen proposed and studies to better define them are underway (19). Patients with signs and symptoms suggestive of asystemic autoimmune disease but not fulfilling the classifi-cation criteria for defined diseases are commonly encoun-tered in clinical practice. Although many aspects of theseconditions have been studied and clarified, there is still noagreement on how best to identify UCTD patients after dis-ease onset. However, such identification is of paramount im-portance, and further analysis is necessary to improve thesensitivity and specificity of the proposed classification cri-teria (19).Therefore, in cases of PAH, thorough immunological andhepatic function studies are always recommended in order toensure an early diagnosis and a prompt treatment for AIH,thus preventing the risk of a rapid progression to severe cir-rhosis and pulmonary hypertension (16).
References
1. Krawitt EL. Autoimmune hepatitis. N Engl J Med 354: 54-66,2006.
2. Read AE, Sherlock S, Harrison CV. Active ’juvenile’ cirrhosisconsidered as part of a systemic disease and the effect of corti-costeroid therapy. Gut 4: 378-393, 1963.
3. West M, Jasin HE, Medhekar S. The development of connectivetissue diseases in patients with autoimmune hepatitis: a case se-ries. Semin Arthritis Rheum 35: 344-348, 2006.
4. Distler O, Pignone A. Pulmonary arterial hypertension and rheu-matic diseases―from diagnosis to treatment. Rheumatology (Ox-ford) 45(Suppl 4): iv22-iv25, 2006.
5. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary ar-terial hypertension. N Engl J Med 351: 1425-1436, 2004.
6. Ichida F, Tsuji T, Omata M, et al. New Inuyama classification;new criteria for histological assessment of chronic hepatitis. IntHepatol Commun 6: 112-119, 1996.
7. Johnson PJ, McFarlane IG. Meeting report: International Autoim-mune Hepatitis Group. Hepatology 18: 998-1005, 1993.
8. Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differentialassessment of pulmonary arterial hypertension. J Am Coll Cardiol43 (Suppl 12): 40S-47S, 2004.
9. Galié N, Torbicki A, Barst R, et al. Task Force. Guidelines on di-agnosis and treatment of pulmonary arterial hypertension. TheTask Force on Diagnosis and Treatment of Pulmonary ArterialHypertension of the European Society of Cardiology. Eur Heart J25: 2243-2278, 2004.
10. Goenka MK, Mehta SK, Malik AK, Kumar YR, Kochhar R. Fatalpulmonary arterial hypertension complicating noncirrhotic portalfibrosis. Am J Gastroenterol 87: 1203-1205, 1992.
11. Mandell MS, Groves BM. Pulmonary hypertension in chronicliver disease. Clin Chest Med 17: 17-33, 1996.
12. Tam NL, He XS. Clinical management of portopulmonary hyper-tension. Hepatobiliary Pancreat Dis Int 6: 464-469, 2007.
13. Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hyper-tension in subjects with connective tissue disease at the secondaryhealth care level of community-based rheumatologists (the UN-COVER study). Arthritis Rheum 52: 2125-2132, 2005.
14. Mukerjee D, St George D, Coleiro B, et al. Prevalence and out-come in systemic sclerosis associated pulmonary arterial hyperten-sion: application of a registry approach. Ann Rheum Dis 62:1088-1093, 2003.
15. Johnson SR, Gladman DD, Urowitz MB, Ibanez D, Granton JT.Pulmonary hypertension in systemic lupus. Lupus 13: 506-509,2004.
16. Morrison EB, Gaffney FA, Eigenbrodt EH, Reynolds RC, BujaLM. Severe pulmonary hypertension associated with macronodular(postnecrotic) cirrhosis and autoimmune phenomena. Am J Med69: 513-519, 1980.
17. Bertino G, Ardiri AM, Boemi PM, Ierna D, Pulvirenti D, Neri S.A case of overlap syndrome: autoimmune idiopathic hepatitis/pul-monary idiopathic hypertension. Clin Ter 158: 313-315, 2007.

Inter Med 47: 1971-1976, 2008 DOI: 10.2169/internalmedicine.47.1420
1976
18. Yamauchi Y, Michitaka K, Onji M. Morphometric analysis of lym-phatic and blood vessels in human chronic viral liver diseases. AmJ Pathol 153: 1131-1137, 1998.
19. Mosca M, Tani C, Bombardieri S. Undifferentiated connective tis-sue diseases (UCTD): a new frontier for rheumatology. Best PractRes Clin Rheumatol 21: 1011-1023, 2007.
Ⓒ 2008 The Japanese Society of Internal Medicinehttp://www.naika.or.jp/imindex.html





























