Embed Size (px)
Citation preview

BIOEQUIVALENCIA
- Métodos, requerimientos y exenciones
- Diseño y evaluación
-Interpretación de resultados
INES FUENTES N

Bioequivalencia
� Medicos, mismos productos:� Falta de respuesta
terapéutica
� Buena respuesta
� Toxicidad
Diferencias debidas a respuestas farmacocinéticas y/o farmacodinámicas diferentes entre individuos.
� Factores farmacodinámicos:
� Diferencias en sensibilidad del receptor
� Edad, tolerancia, interacciones y
� Factores patofisiológicos desconocidos.

Bioequivalencia
� Para realizar los estudios de bioequivalencia ydemostrar, que un producto de prueba (genérico) es igual que un innovador en la biodisponibilidad del fármaco (velocidad y extensión), se necesitan técnicas estadísticassensibles (detecten diferencias no atribuibles a la variabilidad individual.
� Requerimientos para los estudios:
� Estudios clínicos bien controlados (BPC)
� Los productos presentan un indice terapéutico angosto que requieren dosificación cuidadosa y monitoreo del paciente
� Falta de bioequivalencia con presencia de reacciones adversas.

Bioequivalencia
� Requerimientos para los estudios:� Evidencia fisicoquímica
� El fármaco presenta poca solubilidad en agua (menor de 5 mg/mL)
� La velocidad de disolución de uno o mas productos es lenta (menos de 50% en 30 minutos probados con algún método compendial)
� El tamaño de partícula y/o área superficial es crítico al determinar su biodisponibilidad
� Fármaco presenta formas polimórficas, complejos, solvatos que se disuelven muy poco y afectan la absorción
� Relación de excipientes al fármaco activo es alta (5 a 1)

Bioequivalencia
� Evidencia farmacocinética� El fármaco se absorbe en un
sitio específico del tracto GI.
� El grado de absorción del fármaco es pobre
� Existe rápido metabolismo del fármaco en la pared intestinal o en el hígado durante la absorción.
� El fármaco se metaboliza y excreta rápoidamente de tal manera que se requiere rápida disolución y absorción para el efecto
� El fármaco es inestable en porciones específicas del tracto GI. Y se requieren formulaciones especiales
� El fármaco tiene cinética dosis dependiente en el intervalo terapéutico.

Exenciones en Bioequivalencia
� Si el producto tiene alguno de los siguientes criterios estará exento de estos estudios:� El producto farmacéutico está
en solución (admon. Iv.)
� Preparación tópica (crema, aceite, gel, efecto terapéutico local)
� Formas de dosificación oral que se pretende no se absorba (antiácido, medio radiopaco)
� Productos que se inhalan (gas o vapor)
� Soluciones orales, elixires, jarabes, etc

Diseño y evaluación� Diseño, trabajo de equipo
� Protocolo
� Técnicas estadísticas (transformación logarítmica de los datos, efecto de secuencia y consideraciones especiales)
� Antes del estudio el Comité de ética, clínico, revisará el protocolo bajo las normas establecidas.
� El estudio normalmente se realiza en sujetos sanos, voluntarios, masculinos
� El número de sujetos dependeráde la variabilidad interindividual. Existen criterios de inclusión y exclusión para admitirlos.
� Los sujetos generalmente están en ayunas 10 a 12 horas antes del estudio (noche) y continúan de 2 a 4 h después.
� En los estudios los productos tanto el de pruebe como el de referencia tienen:� Mismo fármaco activo
� Misma forma de dosificación (libaración inmediata o liberación controlada)
� Misma vía de admon.
� Dosis simple o dosis múltiple
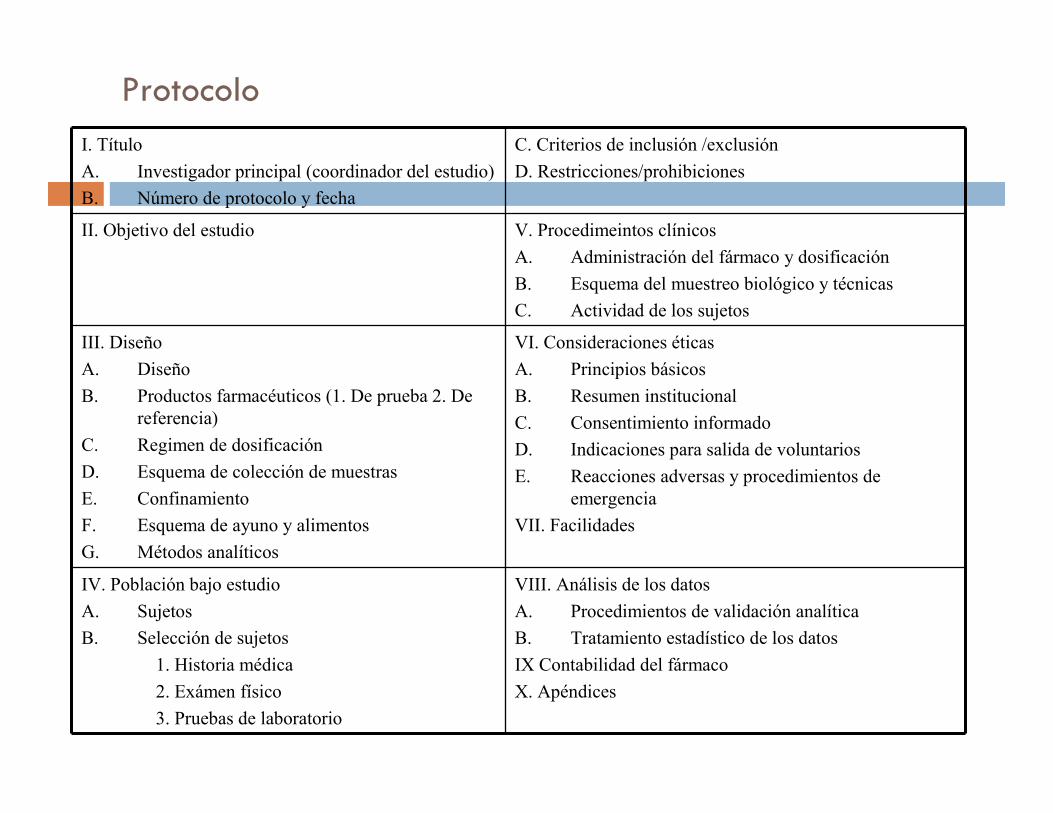
Protocolo
I. Título
A. Investigador principal (coordinador del estudio)
B. Número de protocolo y fecha
C. Criterios de inclusión /exclusión
D. Restricciones/prohibiciones
II. Objetivo del estudio V. Procedimeintos clínicos
A. Administración del fármaco y dosificación
B. Esquema del muestreo biológico y técnicas
C. Actividad de los sujetos
III. Diseño
A. Diseño
B. Productos farmacéuticos (1. De prueba 2. De
referencia)
C. Regimen de dosificación
D. Esquema de colección de muestras
E. Confinamiento
F. Esquema de ayuno y alimentos
G. Métodos analíticos
VI. Consideraciones éticas
A. Principios básicos
B. Resumen institucional
C. Consentimiento informado
D. Indicaciones para salida de voluntarios
E. Reacciones adversas y procedimientos de
emergencia
VII. Facilidades
IV. Población bajo estudio
A. Sujetos
B. Selección de sujetos
1. Historia médica
2. Exámen físico
3. Pruebas de laboratorio
VIII. Análisis de los datos
A. Procedimientos de validación analítica
B. Tratamiento estadístico de los datos
IX Contabilidad del fármaco
X. Apéndices

Bioequivalencia
� Diseño cruzado
� Factores para el número de sujetos: (mínimo 12-24)
� De la diferencia mínima que se pretende detectar en las formulaciones
� Del nivel de significancia
estadíatica, α,
� De la potencia del ensayo (1-
β ) , de la probabilidad de detectar la diferenciua si es que existe
� De la varianza residual
� Consideraciones éticas
� Voluntarios entre 18 –55 años, de preferencia masculinos, (ambos sexos en el caso de no afectar la reproducción) no fumadores.
� Considerar el polimorfismo genético.

Bioequivalencia
� Administración de los tratamientos:� Para disminuir al máximo la
variabilidad inter e intraindividual se estandarizan las condiciones: Secuencia y periodo adoptados, en condiciones de ayuno a la misma hora e idéntica ingesta de líquidos
� Se evitarán las comidas y bebidas que puedan interaccionar con la función renal, hepática
� Obtención y manipulación de las muestras biológicas� Tipo, sangre total, plasma,
suero u orina.
� Procedimiento de obtención, conservación, y horario de tomas.
� Etiquetadas adecuadamente: Voluntario, tiempo de toma, periodo de administración y tratamiento
� Tiempos delimitan las fases de las curvas de niveles plasmáticos (4, 3, 5)
� Diseños cruzados, periodo de lavado.

Estudios de bioequivalencia� Métodos analíticos
� Validación en las condiones experimentales del estudio.� Recuperación y precisión
� Especificidad
� Linealidad
� Sensibilidad
Preparación curva estándar
Controles, concentraciones y frecuencia de uso
LCM
LDM
Tolerancia
Estabilidad
Exactitud
� Parámetros a comparar� ABC0-t, ABC0-∞, Cmax,
tmax
� (Qet. Qe∞ y dQ/dt.)
� (T1/2, TMR)

Diseño cruzado
� Cada sujeto recibe el producto de prueba y el de referencia una sola vez
� Cada sujeto es su propio control
� La variación sujeto a sujeto se reduce. (variaciones debido a secuencia, periodo y tratamiento).(Tratamiento son formulaciones, periodo es el intervalo de tiempo del estudio, secuencia es el número de ordenes de tratamiento)
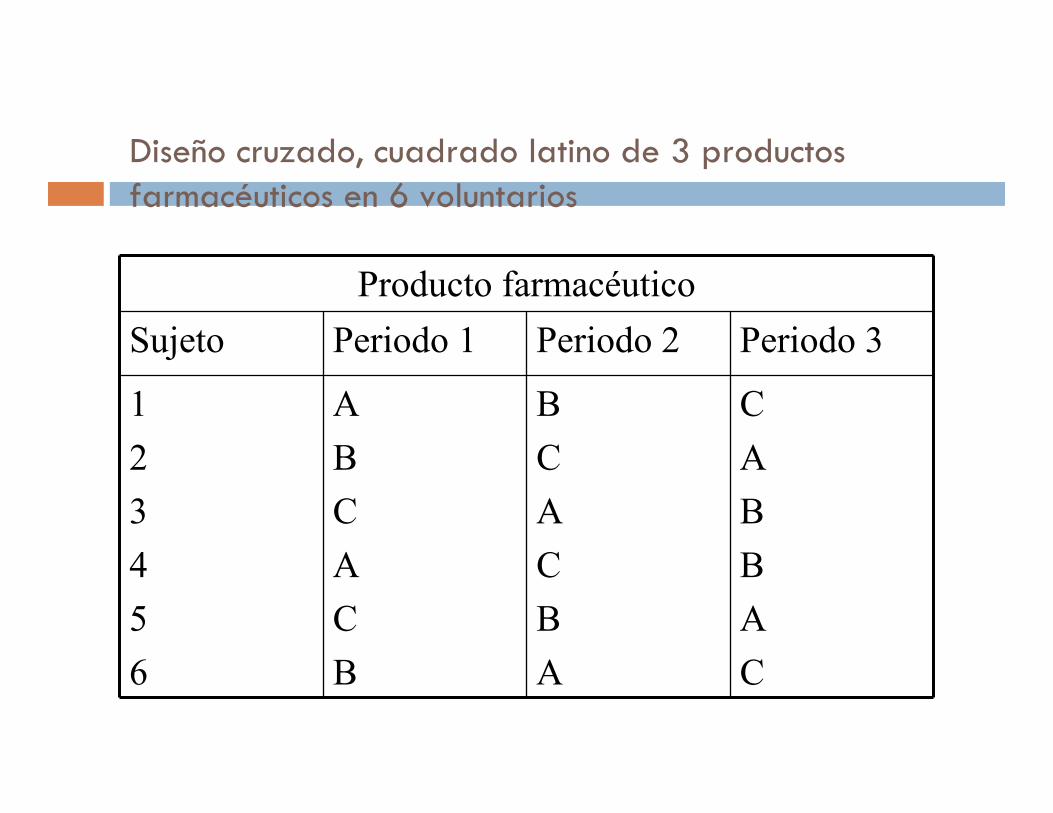
Diseño cruzado, cuadrado latino de 3 productos farmacéuticos en 6 voluntarios
Producto farmacéutico
Sujeto Periodo 1 Periodo 2 Periodo 3
1
2
3
4
5
6
A
B
C
A
C
B
B
C
A
C
B
A
C
A
B
B
A
C
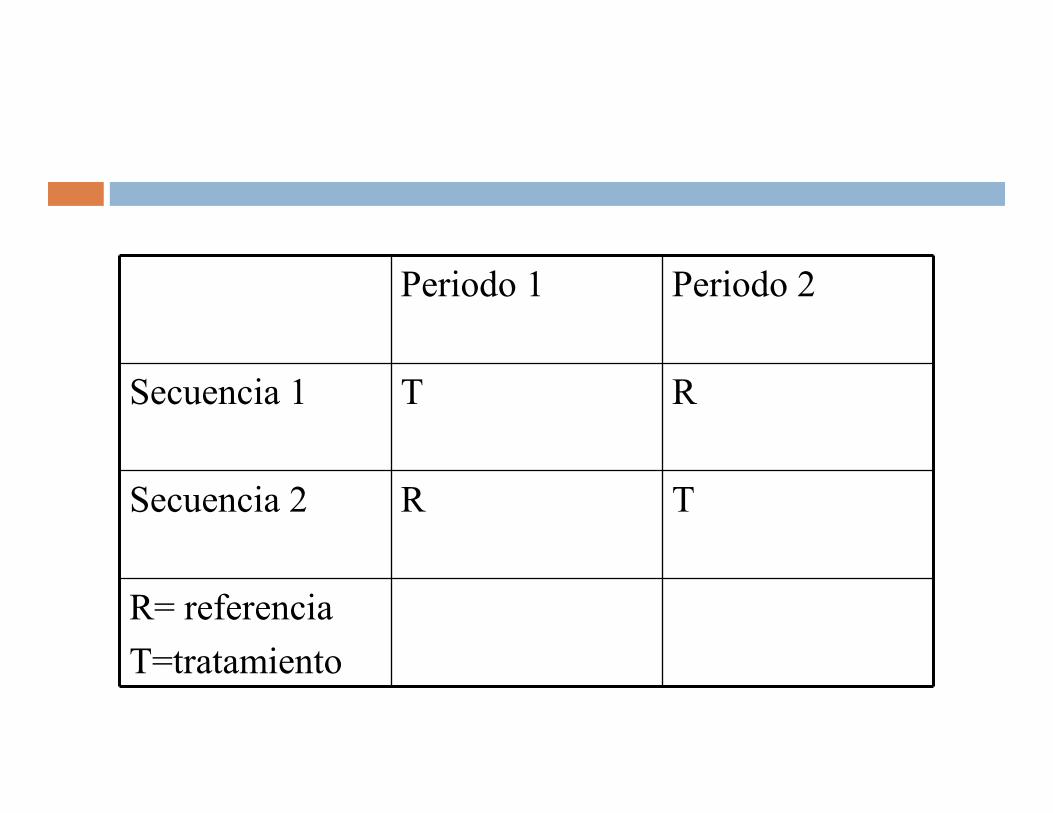
Periodo 1 Periodo 2
Secuencia 1 T R
Secuencia 2 R T
R= referencia
T=tratamiento
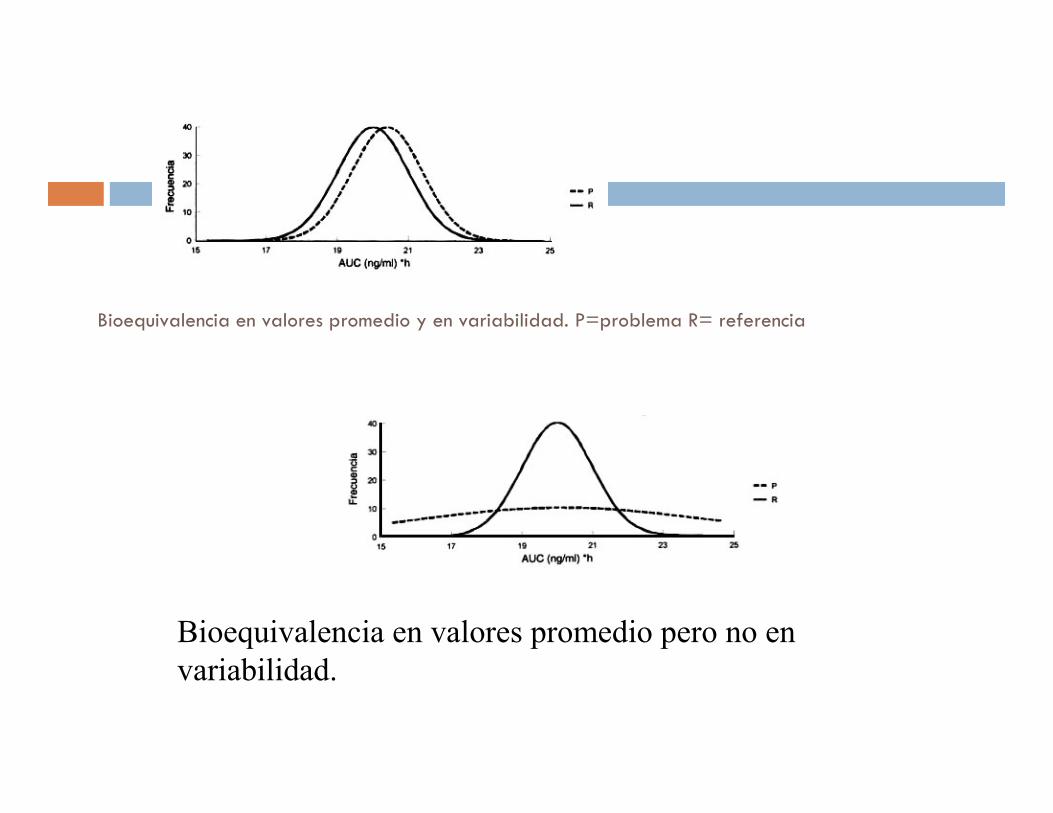
Bioequivalencia en valores promedio y en variabilidad. P=problema R= referencia
Bioequivalencia en valores promedio pero no en
variabilidad.
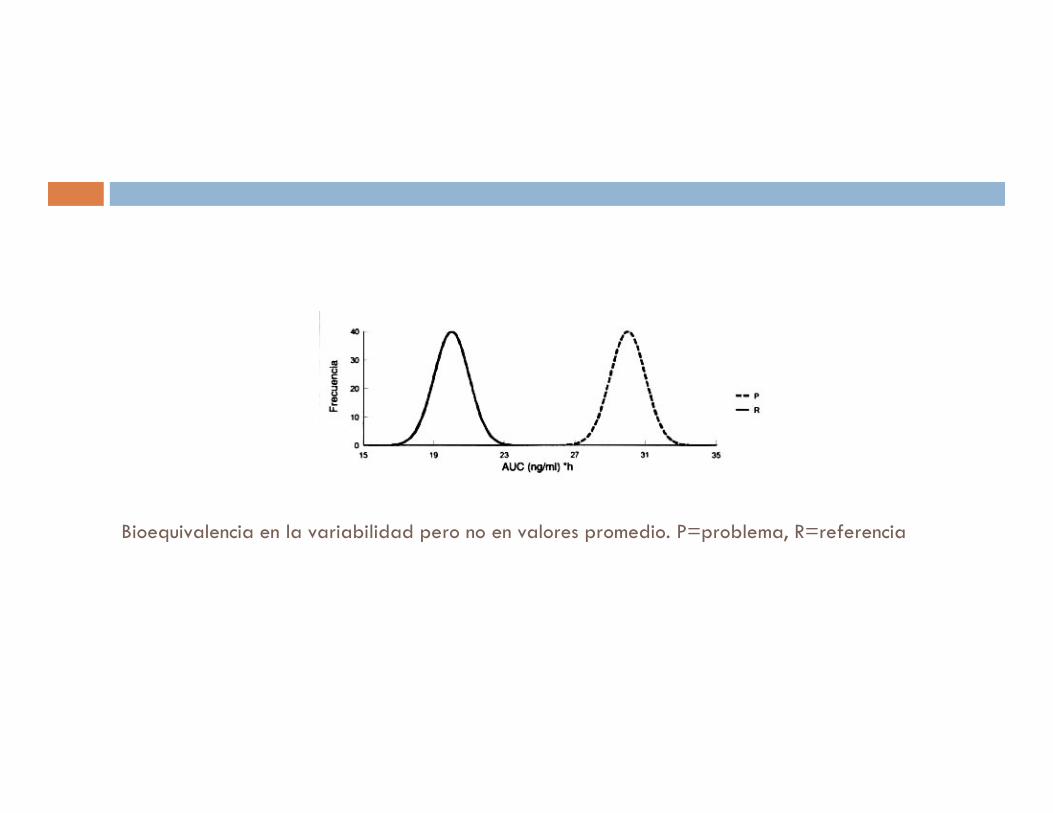
Bioequivalencia en la variabilidad pero no en valores promedio. P=problema, R=referencia
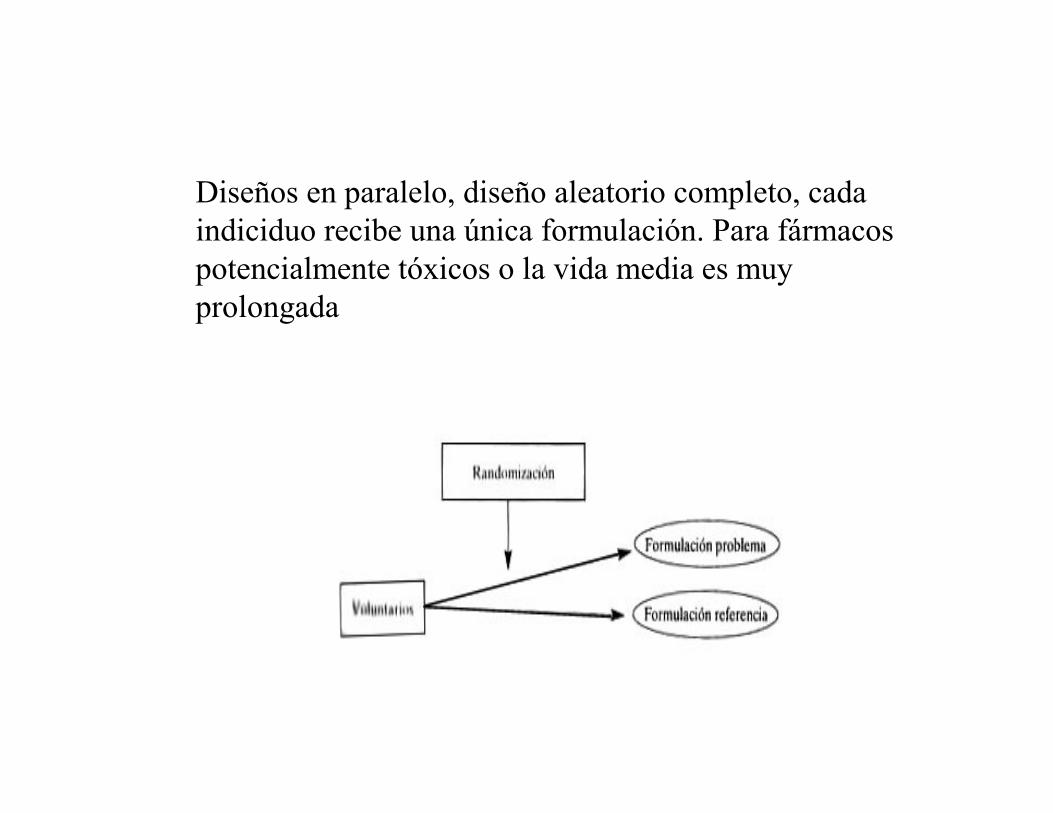
Diseños en paralelo, diseño aleatorio completo, cada
indiciduo recibe una única formulación. Para fármacos
potencialmente tóxicos o la vida media es muy
prolongada

Análisis de varianza
� Procedimiento estadístico usado para probar las diferencias entre y dentro de los tratamientos en grupos control
� Un producto bioequivalente no producirá diferencias significativas en los parámetros farmacocinéticos estudiados.
� Puede evaluar la variabilidad en sujetos, grupos del tratamiento, periodo del estudio, formulaciones, etc.
� Poder de la prueba (probabilidad de que el ANOVA sea válido) es 0.80, alfa= 0.2 con nivel de significancia de 0.05.
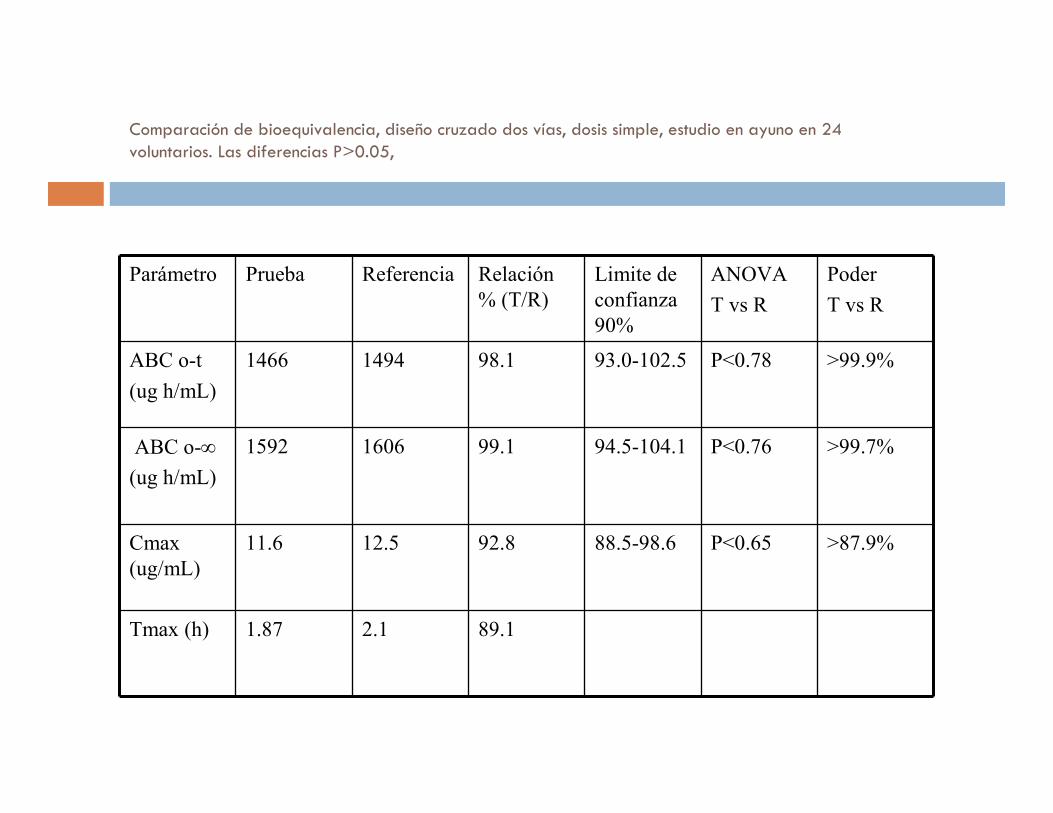
Comparación de bioequivalencia, diseño cruzado dos vías, dosis simple, estudio en ayuno en 24 voluntarios. Las diferencias P>0.05,
Parámetro Prueba Referencia Relación
% (T/R)
Limite de
confianza
90%
ANOVA
T vs R
Poder
T vs R
ABC o-t
(ug h/mL)
1466 1494 98.1 93.0-102.5 P<0.78 >99.9%
ABC o-∞
(ug h/mL)
1592 1606 99.1 94.5-104.1 P<0.76 >99.7%
Cmax
(ug/mL)
11.6 12.5 92.8 88.5-98.6 P<0.65 >87.9%
Tmax (h) 1.87 2.1 89.1
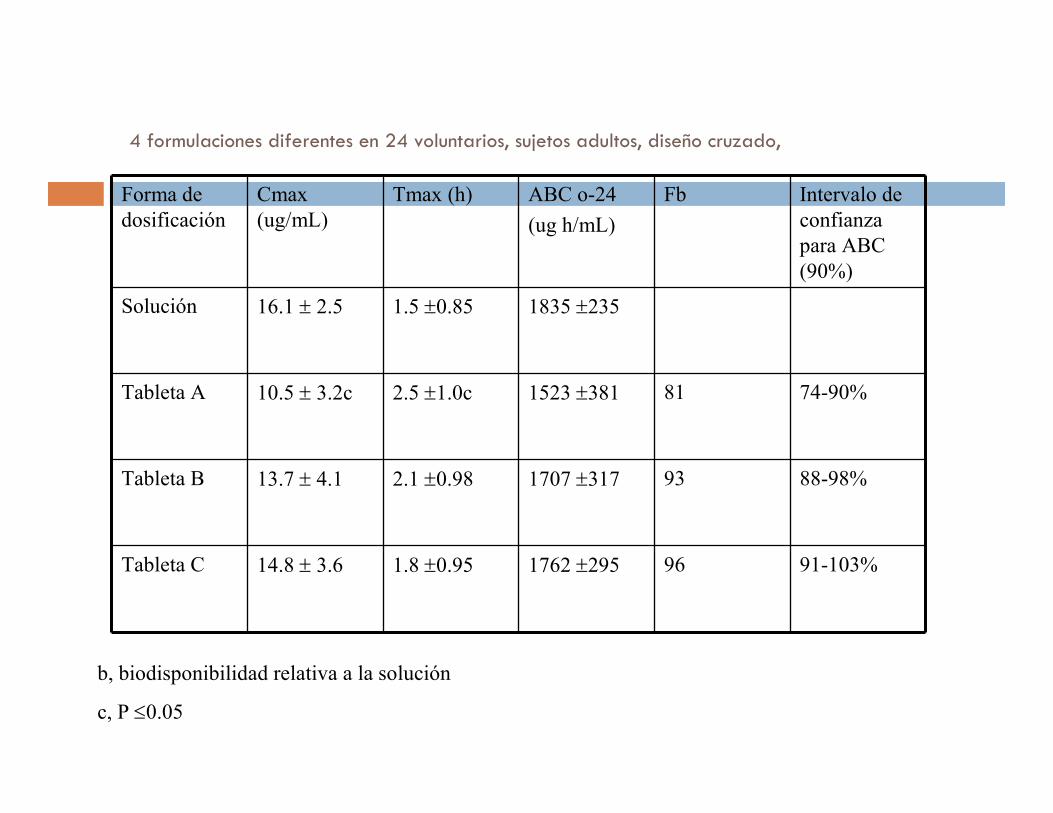
4 formulaciones diferentes en 24 voluntarios, sujetos adultos, diseño cruzado,
Forma de
dosificación
Cmax
(ug/mL)
Tmax (h) ABC o-24
(ug h/mL)
Fb Intervalo de
confianza
para ABC
(90%)
Solución 16.1 ± 2.5 1.5 ±0.85 1835 ±235
Tableta A 10.5 ± 3.2c 2.5 ±1.0c 1523 ±381 81 74-90%
Tableta B 13.7 ± 4.1 2.1 ±0.98 1707 ±317 93 88-98%
Tableta C 14.8 ± 3.6 1.8 ±0.95 1762 ±295 96 91-103%
b, biodisponibilidad relativa a la solución
c, P ≤0.05
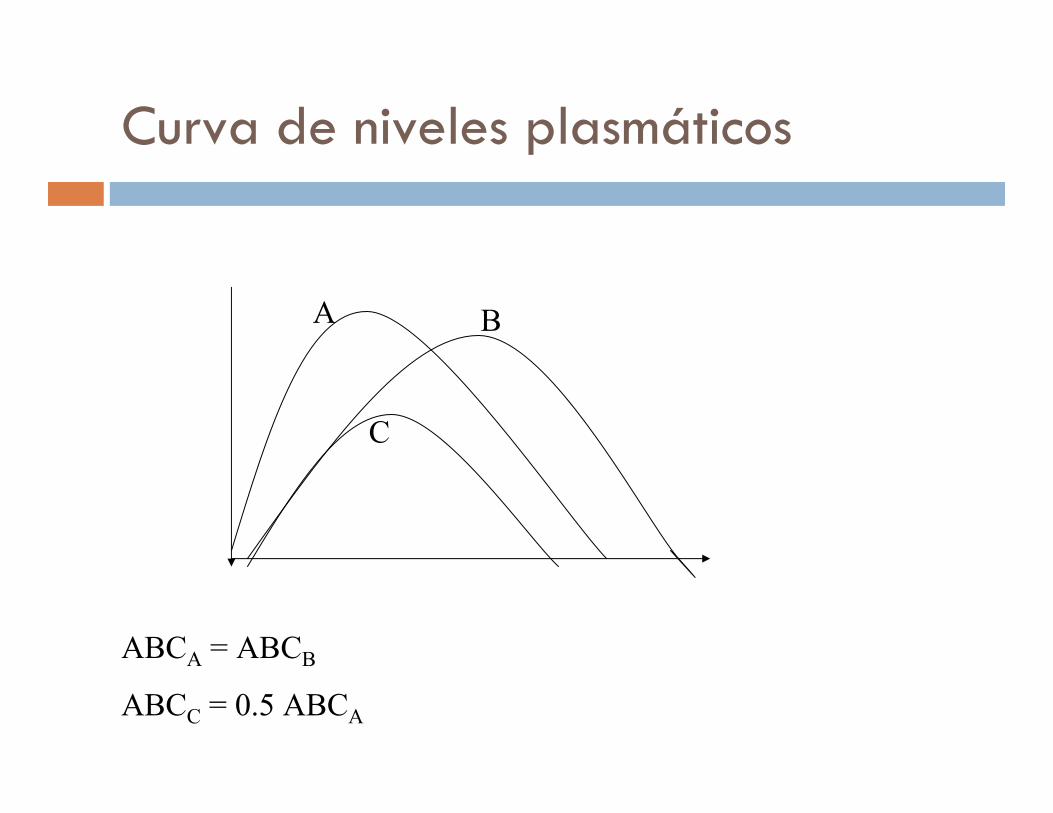
Curva de niveles plasmáticos
ABCA = ABCB
ABCC = 0.5 ABCA
A B
C
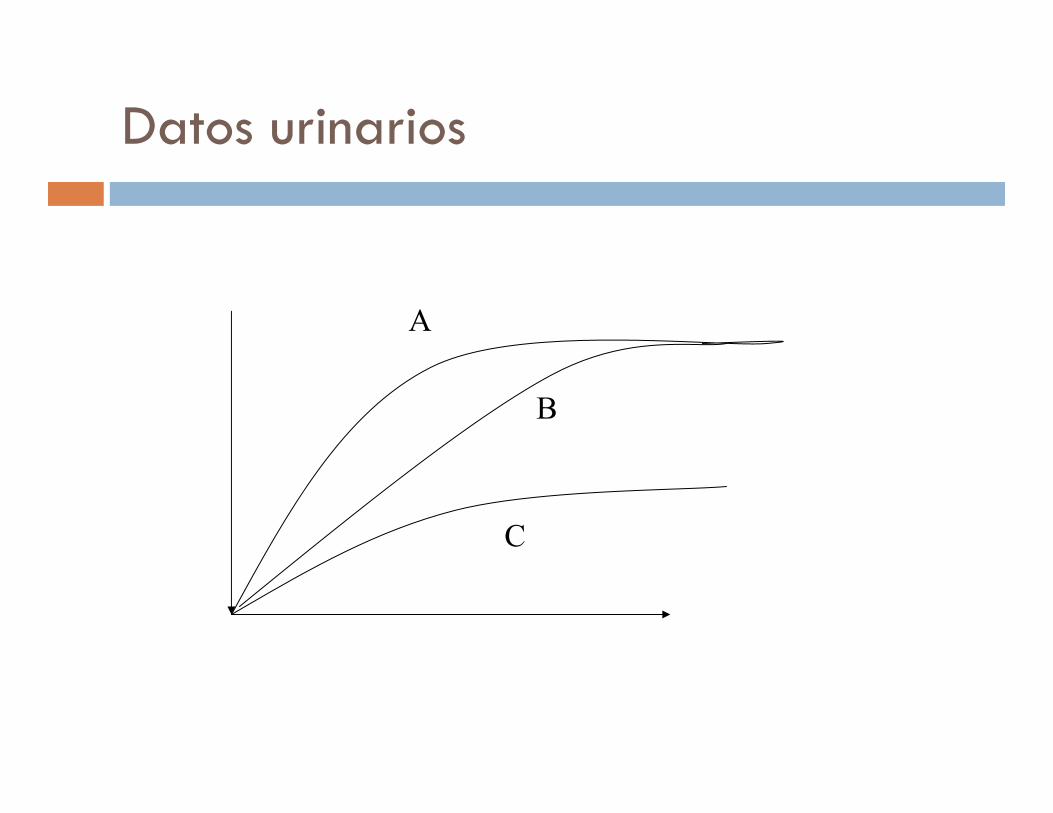
Datos urinarios
A
B
C
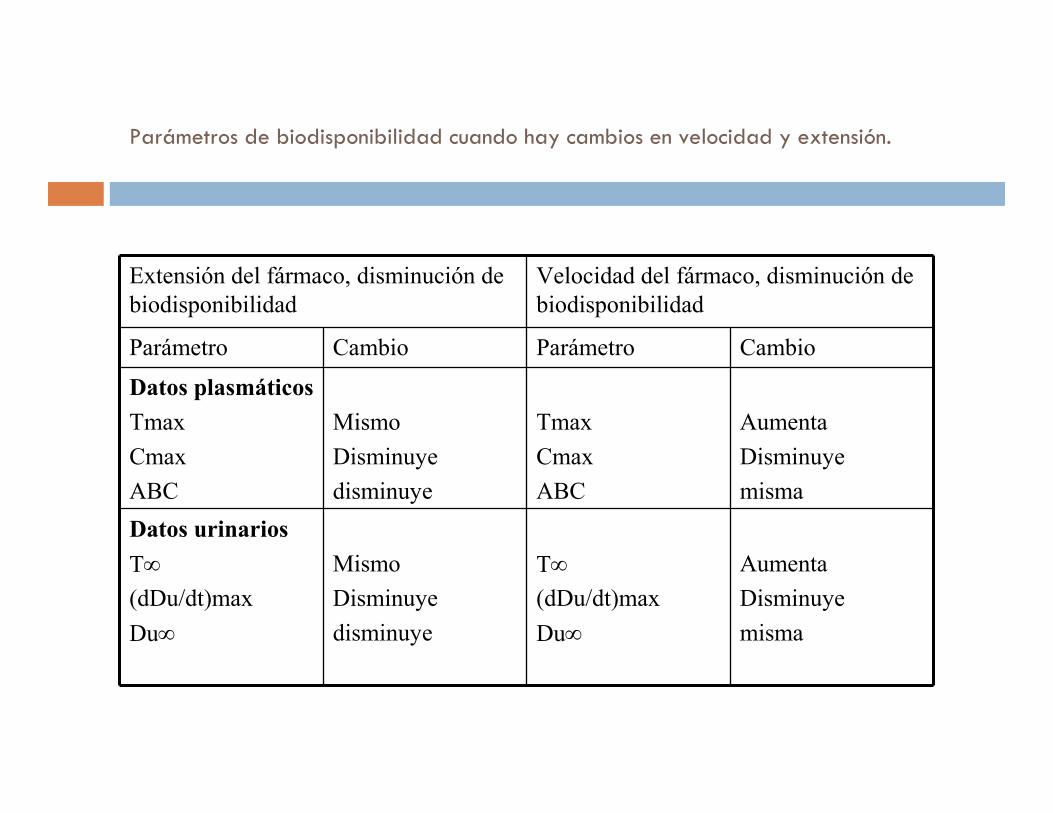
Parámetros de biodisponibilidad cuando hay cambios en velocidad y extensión.
Extensión del fármaco, disminución de
biodisponibilidad
Velocidad del fármaco, disminución de
biodisponibilidad
Parámetro Cambio Parámetro Cambio
Datos plasmáticos
Tmax
Cmax
ABC
Mismo
Disminuye
disminuye
Tmax
Cmax
ABC
Aumenta
Disminuye
misma
Datos urinarios
T∞
(dDu/dt)max
Du∞
Mismo
Disminuye
disminuye
T∞
(dDu/dt)max
Du∞
Aumenta
Disminuye
misma









![Bioequivalencia PDF[1]](https://img.pdfslide.tips/doc/110x75/5571fd8249795991699941fe/bioequivalencia-pdf1.jpg)


















