Embed Size (px)
Citation preview

BIOKEMISKA SEPARATIONSMETODER - EN ÖVERSIKT
Jan-Christer Janson
Institutionen för Kemi
Uppsala Biomedicinska Centrum
Uppsala Universitet
Uppsala

2
INNEHÅLLSFÖRTECKNING
1. INLEDNING ..................................................................................................................... 3
2. ALLMÄNT OM SEPARATION AV MOLEKYLER ...................................................... 4
3. GELMATERIAL FÖR SEPARATIONSÄNDAMÅL ...................................................... 5
3.1. Varför gelmaterial? ..................................................................................................... 5
3.2. Molekylsiktande geler ................................................................................................. 6
3.3. Makroporösa geler ...................................................................................................... 7
4. EXTRAKTION OCH FÖRBEREDANDE SEPARATION ........................................... 7
5. INTERMEDIÄRA OCH HÖGUPPLÖSANDE SEPARATIONSMETODER ................ 9
5.1. Separation efter molekylernas storlek och form ....................................................... 10
5.1.1. Molekylsiktande elektrofores ............................................................................ 10
5.1.2. Molekylsiktande kromatografi: Gelfiltrering ..................................................... 11
5.2. Separation efter molekylernas elektriska laddning ................................................... 13
5.2.1. Elektrofores ........................................................................................................ 13
5.2.2. Isoelektrisk fokusering ....................................................................................... 14
5.2.3. Jonbyteskromatografi ......................................................................................... 17
5.2.4. Kromatofokusering ............................................................................................ 20
5.3. Separation efter molekylernas funktion .................................................................... 21
5.3.1. Affinitetskromatografi av enzymer .................................................................... 21
5.3.2. Immunosorption ................................................................................................. 22
5.3.3. Affinitetskromatografi av glykoproteiner .......................................................... 23
5.3.4. Exempel på affinitetskromatografi av transportproteiner och receptorer .......... 24
5.3.5. Bindning av IgG till Protein A från Staphylococcus aureus ............................. 25
5.4. Hydrofob adsorptionskromatografi ........................................................................... 26
5.5. Kemisorption ("Kovalent kromatografi") ................................................................. 27
5.6. Övriga separationsmetoder ........................................................................................ 28
5.6.1. Metallchelataffinitetskromatografi ..................................................................... 28
6. REFERENSER ................................................................................................................ 29

3
1. INLEDNING
"Den gren av kemin som syftar till ett utforskande av livsprocessernas mekanism - alltså
biokemien - har haft och kommer alltid att ha som en av sina viktigaste uppgifter att ur
biologiskt material isolera ämnen som på ett eller annat sätt spela en väsentlig roll i det
komplexa fenomen som kallas för liv. Detta problem har kommit i förgrunden icke minst
därför, att många av de ämnen det här gäller genom sin molekylstorlek, sin komplicerade
byggnad och sin ömtålighet för vanliga kemiska operationer erbjuder alldeles speciella
problem. Dessutom förekommer många av de viktigaste substanserna i ytterst små
mängder".
Ovanstående citat utgör inledningen till ett föredrag hållet av professor Arne Tiselius
(Nobelpris 1948) i september 1961 (1) och är idag alltjämt lika aktuellt. Tiselius var elev
till professor The Svedberg (Nobelpris 1926) och dessa båda vetenskapsmäns inflytande på
den biokemiska forskningen vid flera svenska universitet kan knappast överskattas. De har
dessutom haft ett avgörande inflytande på utvecklingen av den svenska biokemiskt
inriktade industrin. Utan deras pionjärinsatser skulle vi idag knappast ha ett företag som
GE Healthcare Biosciences AB i Uppsala. Detta företag betyder, med någon överdrift, för
världens biokemister vad COOP eller ICA betyder för Sveriges dagligvarukonsumenter.
Tiselius ägnade hela sin aktiva forskarkarriär åt att utveckla nya separationsmetoder för
biomolekyler, främst äggviteämnen (proteiner) och var en av de ivrigaste förespråkarna för
milda metoder, baserade mer på fysikalisk-kemiska skillnader mellan molekylerna än på de
rent kemiska. Dit hör t.ex. skillnader i storlek och form samt skillnader i elektrisk laddning.
Biokemisk separationsteknik används rutinmässigt av forskare inom de flesta livsveten-
skapliga ämnesområden: Biokemi, molekylärbiologi, mikrobiologi, immunologi, genetik,
farmakognosi etc.. De hämtar sina studieobjekt från levande celler eller deras utsöndrings-
produkter. Förr var djurorgan eller växtmaterial de vanligaste utgångsmaterialen men idag
är det främst rekombinanta proteiner producerade i mikroorganismer som studeras.
Levande celler innehåller utomordentligt komplexa blandningar av tusentals olika
substanser där de enstaka komponenterna sällan förekommer i en procentuell andel över-
stigande 0,01 % av den totala substansmängden. Rekombinanta proteiner däremot når ofta
koncentrationer överstigande flera procent av den totala proteinmängden i utgångsmaterial-
et. Dock måste man komma ihåg att samma typ och antal av värdorganismens proteiner
måste avlägsnas under den följande reningsprocessen oavsett uppnådd expressionsnivå.
Ett typiskt biokemiskt forskningsprojekt har för många forskarstuderande inneburit att
inledningsvis åtskilliga månader ägnats åt att utveckla ett förfarande för renframställning av
det protein vars egenskaper, struktur eller funktion skall studeras. Det kan t.ex. gälla ett
enzym eller ett hormon, ur ett organ, en körtel eller från en bakterie som Escherichia coli,
eller en jästsvamp som Pichia pastoris, i de fall det handlar om ett rekombinant protein.
Ofta har resultatet blivit något eller några få milligram rent protein som sedan undersökts.
Ibland har syftet varit att undersöka proteinets biologiska funktion, ibland att bestämma
dess tredimensionella struktur genom röntgendiffraktion efter kristallisering. Genom att
sålunda studera de enskilda cellkomponenternas egenskaper hoppas man så småningom,
steg för steg, kunna förstå hur den levande cellen fungerar. Många enzymer renas idag för
att användas som verktyg av molekylärbiologer och mikrobiologer, t.ex. vid selektiv ned-

4
brytning och syntes av nukleinsyror. Det är då synnerligen angeläget att dessa inte är
förorenade av andra typer av nukleinsyranedbrytande enzymer, och man driver därför
reningen av sådana enzymer så långt tillgängliga analysmetoder tillåter.
Det är här lämpligt att citera Nobelpristagaren Arthur Kornberg som vid ett seminarium vid
Uppsala Biomedicinska Centrum hösten 1980 yttrade: "There is no point in wasting clean
thinking on dirty enzymes", vilket kan fritt översättas: "Det är bortkastad tid att försöka
tolka resultat som baseras på försök med orena enzymer".
Andra områden där biokemisk separationsteknik används rutinmässigt är inom klinisk
diagnostik, t.ex. vid analys av proteinsammansättningen i blodserum. och inom läke-
medelsindustrin, t.ex. vid renframställning av insulin och humant serumalbumin. Protein-
rening har traditionellt betraktats som ett mycket svårt forskningsområde och anses av
många fortfarande vara mer en konst än en vetenskap. Kanske var det så för några decen-
nier sedan, idag är situationen emellertid en helt annan. Det finns på marknaden ett stort
antal instrument och andra hjälpmedel för renframställning och analys av proteiner och en
mycket rikhaltig litteratur inom området står till biokemisternas förfogande. Med hjälp av
datasökningsmetoder kan man på några minuter få fram en aktuell lista på artiklar som
behandlar rening och isolering av ett speciellt protein, t.ex. ett enzym eller hormon. Saknas
information i litteraturen om det protein man avser isolera, kan ofta värdefull vägledning i
reningsarbetet erhållas från artiklar som beskriver isolering av närbesläktade proteiner.
Vad gäller rekombinanta proteiner kan värdefull information, t.ex. rörande molekylvikten,
primärstrukturen och den teoretiska isoelektriska punkten (det pH där proteinets nettolad-
dning är noll), erhållas ur den till en polypeptidkedja översatta cDNA-sekvensen.
Varför har proteinrening traditionellt betraktats som ett svårt forskningsområde? Den
väsentligaste anledningen, och som redan delvis nämnts i inledningen, är att proteiner dels
är utomordentligt olika till sina egenskaper, dels mycket instabila och känsliga för varia-
tioner i såväl den kemiska som fysikaliska miljön och dels att de har en tendens att binda
sig till andra komponenter såsom nukleinsyror, kolhydrater och lipider. Dessutom binds de
ibland till varandra under de betingelser de tvingas in i under reningsproceduren.
2. ALLMÄNT OM SEPARATION AV MOLEKYLER
Molekyler i blandning kan separeras enligt flera olika huvudprinciper. Inom bioveten-
skaperna, där man arbetar med komplexa blandningar i vattenlösning, kan de flesta
separationsmetoderna inordnas i någon av följande tre kategorier. Inom den första faller
alla förfaranden som baseras på att miljön förändras så att vissa ämnen ej längre kan
existera i lösning utan kristalliserar eller aggregerar. De bildade partiklarna kan sedan
avlägsnas eller tillvaratas genom sedimentation eller filtrering. Hit hör de för alla bio-
kemister välkända s.k. fällningsmetoderna där man stegvis ökar koncentrationen av ett
neutral salt eller ett organiskt lösningsmedel. Till den andra kategorin hör metoder som
utnyttjar skillnader i löslighet hos olika molekyler i de vätskor (lösningsmedel) som ingår i
flytande två-fassystem, och som används i olika motströmsextraktionsprocesser. Den tredje
och sista kategorin innefattar alla metoder som baseras på förutsättningen att två olika
molekyler i lösning reagerar olika med avseende på rörelseriktning och/eller rörelsehastig-

5
het när de utsätts för ett yttre kraftfält. Till denna kategori hör de flesta moderna biokem-
iska separationsmetoder. De olika krafterna illustreras schematiskt i Fig. 1.
Fig. 1. Krafter som kan utnyttjas för att påverka en molekyl i en separationskammare (utrymmet mellan de
båda horisontella linjerna). ”Impelling force” = framdrivande kraft. ”Retarding force”= bromsande kraft.
”FFF” står för ”Field-Flow Fractionation” en teknik där man kombinerar en axiell framdrivande kraft med en
radiell dito.
Det finns flera sätt att åstadkomma riktad rörelse hos en molekyl. Ett av dessa gäller endast
laddade molekyler och innebär att de placeras i ett elektriskt kraftfält. Beroende på typ och
storlek av molekylernas laddning kommer de att röra sig i en viss riktning med en viss
hastighet. På detta fenomen baseras en separationsteknik som kallas elektrofores. Ett annat
innebär att molekylerna placeras i en flödande vätska som rör sig längs ett stillastående fast
eller flytande ämne. Beroende på molekylernas olika förmåga att fördela sig mellan den
flödande och den stillastående vätskan, eller att binda sig olika starkt till det fasta ämnet
kommer de att transporteras av den flödande vätskan med olika hastighet. På detta fenomen
baseras en teknik som kallas kromatografi. Ett tredje sätt innebär att molekylerna placeras i
ett gravitationsfält, t.ex. i en ultracentrifug. Molekyler med höga molekylvikter kommer här
att sedimentera snabbare än de med låga dito. Metoden har ej använts så mycket för isoler-
ing av proteiner i ren form. En fjärde metod utnyttjar molekylernas egenrörelse, diffusion-
en. Den kallas för dialys och innebär att små molekyler tillåts diffundera genom ett s.k.
semipermeabelt membran, en tunn hinna som innehåller porer vars tvärsnitt är för litet för
att tillåta passage av högmolekylära substanser, t.ex. proteiner. Ett specialfall av dialys är
s.k. ultrafiltrering. Genom att applicera ett hydrostatiskt tryck på ena sidan av det semi-
permeabla membranet åstadkommer man här ett vätskeflöde genom detsamma som
snabbare än diffusionen transporterar små molekyler och salter genom membranet.
Metoden används mest för koncentrering av proteinlösningar. Med speciella membraner
kan man emellertid separera proteiner i storleksklasser.
3. GELMATERIAL FÖR SEPARATIONSÄNDAMÅL
3.1. Varför gelmaterial?
De viktigaste biokemiska separationsmetoderna är elektrofores och kromatografi. Båda kan
betraktas som allmänna begrepp under vilka ett stort antal olika specialtekniker inryms.
Gemensamt för alla (dock med ett par undantag) är att man för att förhindra att redan sepa-
rerade molekyler åter blandas genom oönskade vätskeströmningar, s.k. konvektion, orsak-
ade av skillnader i densitet i olika delar av separationsmediet, eller av virvelbildning, låter

6
separationen äga rum i ett poröst medium, en gel. En gel är en olöslig polymerprodukt vars
förnämsta egenskap är dess förmåga att binda stora mängder lösningsmedel. En bra beteck-
ning är att den sväller i lösningsmedlet. Torrsubstanshalterna i typiska svällda geler ligger
normalt vid 2-10 %. Det viktigaste lösningsmedlet inom biokemin är naturligtvis vatten och
detta leder till att det i första hand är hydrofila (vattenälskande) gelmaterial som kommer
ifråga. Dessa är uppbyggda av polymerer som innehåller polära grupper, t.ex. hydroxyl-
grupper eller amidgrupper.
Varför är nu gelmaterialen så viktiga inom biokemisk separationsteknik? Det finns flera,
vitt skilda orsaker till detta. Den viktigaste är kanske den stora yta. som exponeras inuti ett
gelmaterial och som ger en hög kapacitet per volymsenhet för de reversibla bindningsfeno-
men mellan till gelpolymeren kovalent kopplade, funktionella grupper och de olika mole-
kyler från provblandningen som skall separeras. En annan viktig orsak är den stora volym
av stillastående lösningsmedel, här alltså vatten, som tillåter utnyttjande av ett antal olika
fysikaliska fenomen för separation utan störande inflytande från de svårkontrollerbara
vätskerörelser molekyler utsätts för ute i en fri lösning.
Vid gelelektrofores använder man en homogen gel och de molekyler som skall separeras
befinner sig hela tiden inne i det tredimensionella polymernätverket. Vid kromatografi
använder man en sedimenterad bädd av små gelpartiklar (ofta tillverkade i pärlform och
med diametrar som varierar i storleksintervallet 0,005-0,1 mm) som packas i ett rör av glas,
plast eller stål. Molekylerna som skall separeras transporteras med hjälp av en flödande
vätska som rör sig i det fina kapillärsystemet mellan gelkornen. Separationen åstadkommes
sedan genom att molekylerna genom sin egenrörelse (diffusionen) transporteras in i den
stillastående vätskan i gelkornen, där olika mekanismer för olika kromatografiska tekniker
leder till att vissa molekyler kvarhålles under ett längre tidsintervall än andra innan de åter
kommer att diffundera tillbaka ut i den strömmande vätskan. Beroende på vilken teknik
som tillämpas, används en av två principiellt helt olika geltyper.
3.2. Molekylsiktande geler
Den ena geltypen är uppbyggd av tvärbundna (förnätade), linjära, hydrofila polymerer där
graden av tvärbindning kan varieras och därmed porstorleken i det tredimensionella poly-
mernätverket. Porstorleken hos dessa gelmaterial är av samma storleksordning som de
molekyler som ska separeras vilket leder till molekylsiktningsfenomen som utnyttjas för
separation i storleksklasser. När en sådan geltyp används vid elektrofores kommer större
molekyler att röra sig långsammare genom gelén då de på grund av polymernätverket
tvingas att vandra en längre sträcka än mindre molekyler. Vid kromatografi med sådana
geler kommer tvärtom större molekyler att transporteras snabbare genom kolonnen på
grund av att en mindre delvolym av vätskan i gelkornen är tillgänglig för dem än för
mindre molekyler. Den viktigaste molekylsiktande geltypen för elektrofores av proteiner
och nukleinsyrafragment är uppbyggd av tvärbunden polyakrylamid. Kromatografisk
molekylsiktning brukar kallas gelfiltrering och de viktigaste gelmaterialen är baserade på
tvärbundet dextran och/eller tvärbunden polyakrylamid samt agaros. De förra tillverkas av
GE Healthcare Biosciences i Uppsala under benämningarna Sephadex® och Sephacryl®
samt Sepharose®, Superose®och Superdex®.

7
3.3. Makroporösa geler
Den andra geltypen skiljer sig från den förra främst med avseende på porositet vilket fram-
går av den schematiska illustrationen av de båda geltyperna i Fig. 2. Det inom biokemisk
forskning viktigaste makroporösa gelmaterialet är agaros, en naturligt gelbildande polysac-
karid som framställs ur agar som i sin tur extraherats från vissa marina rödalger. Agaros
används både inom elektrofores och kromatografi i de fall man önskar ett icke siktande
gelmaterial. T.ex. vid olika typer av immunoelektrofores och vid affinitetskromatografi.
Vid agaroskoncentrationer i området 2-4 % erhålles geler vars porositet avsevärt överstiger
diametrarna hos de flesta proteinmolekyler. För de mer högmolekylära nukleinsyrorna är
sådana geler lämpliga för såväl siktande elektrofores som gelfiltrering.
Fig.2. Schematisk framställning av skillnaderna mellan en siktande geltyp (vänstra skissen), uppbyggd av
tvärbundna, linjära polymerer (typ Sephadex), och en makroporös geltyp, här exemplifierad av hur man
tänker sig en agarosgel uppbyggd. (Figuren från Arnott, S. et al., J. Mol. Biol. 90(1974)269-284).
4. EXTRAKTION OCH FÖRBEREDANDE SEPARATION
Ett försök till renframställning av ett protein, t.ex. ett enzym, kan indelas i fyra stadier,
extraktion, förberedande separation (ofta kombinerat med koncentrering), intermediär
separation och avslutande högupplösande separation (i engelskspråkig litteratur används
ofta begreppen ”capturing”, ”purification” och ”polishing” för de tre sistnämnda stadierna).
Vart och ett av dessa stadier kan i sin tur inbegripa flera steg. Extraktionen är helt naturligt
det inledande steget i reningsprocessen och syftar till att frigöra enzymerna från celler eller
cellbeståndsdelar till vilka de uppvisar en starkt varierande bindningstendens. För intra-
cellulära enzymer fordras som regel att cellvägg och cellmembran först sprängs fysikaliskt
eller kemiskt. För animala och vegetabiliska celler utgör detta i allmänhet inget problem.
Animala celler är särskilt lättsönderdelade och oftast räcker det med en passage genom en
köttkvarn, följt av behandling i turmix, d.v.s. ett kärl med roterande knivar i botten.
Det största problemet är närvaron av fettvävnad i utgångsmaterialet och för vissa lipidrika
organ, t.ex. hjärna och lever, framställs ofta acetonpulver, dvs. den restprodukt som erhålles
sedan de homogeniserade vävnaderna befriats från lipider genom extraktion med aceton vid
låg temperatur. Vad beträffar vegetabilisk vävnad, utgör frömaterialen inget större problem
om man undantar de oljerika fröerna. Det finns en rad olika typer av kvarnar i såväl labora-
torie- som storskaleutförande. De oljerika fröerna klarar man ofta genom valsning och

8
skonsam oljeextraktion, t.ex. med n-hexan. Gröna växtdelar går ej att sönderdela i normala
kvarnar eftersom dessa snabbt kloggar ihop. I laboratorieskala använder man ofta en
turmix. Den numera kanske viktigaste råvarukällan för enzymer är mikroorganismer av
olika slag, t.ex. bakterier, jäst och mögelsvampar. Många enzymer av praktiskt intresse,
t.ex. tvättmedelsenzymerna, är extracellulära, d.v.s. de utsöndras av mikroorganismerna i
det omgivande odlingsmediet och behöver oftast bara koncentreras innan de används. De
flesta mikrobiella enzymer av intresse som verktyg för forskning, t.ex. de s.k. restriktions-
endonukleaserna som används för specifik klyvning av DNA-molekyler inom rekombina-
tions-DNA-teknologin, är emellertid intracellulära och för att komma åt dem måste cell-
väggarna slås sönder. Detta åstadkommes t.ex. med hjälp av u1traljudbehandling eller
genom skakning tillsammans med små glaspärlor. I större skala används ofta modifierade
mjölkhomogenisatorer (t.ex. av fabrikatet APV-Gaulin). Den tillförda energin omvandlas i
samtliga fall huvudsakligen i värme varför det är viktigt att kyla ned suspensionen före och
efter behandlingen så att inte känsliga substanser, t.ex. enzymer, förstörs (denatureras).
Efter extraktionssteget, vilket oftast sker i närvaro av en lämplig buffertsaltlösning,
eventuellt med tillsats av löslighetsbefrämjande ämnen, t.ex. vissa detergenter, vidtar ett
tekniskt viktigt steg, nämligen avskiljandet av de extraherade fasta cellbeståndsdelarna.
Detta sker i laboratorieskala oftast med hjälp av kylda snabbcentrifuger med hastigheter
upp till c:a 25.000 varv per minut. Fibrösa material filtreras oftast genom en silduk,
eventuellt i en s.k. korgcentrifug (liknar en vanlig tvättcentrifug), varefter filtratet befrias
från mindre partiklar genom snabbcentrifugering. I vissa fall är det mycket svårt att uppnå
helt partikelfria extrakt. Det gäller t.ex. för aceton-pulverhomogenat, bakteriehomogenat
och jästautolysat. Sådana extrakt kan i de flesta fall ej direkt användas till kolonnkromato-
grafiska försök utan förbehandling, d.v.s. man sätter in ett eller flera förberedande
fraktioneringssteg. Det klassiska inledande steget vid proteinrening är utfällning med
neutralsalt, särskilt ammoniumsulfat, (NH4)2SO4. Andra proteinfällningsreagens med
vidsträckt användning är etanol samt polymerer som polyetylenglykol och polyetylenimin.
Andra klassiska inledande fraktioneringssteg är värmedenaturering, utfällning genom
tillsats av syra samt utfällning av nukleinsyror med hjälp av spermin, streptomycinsulfat
eller protaminsulfat.
En preparativ teknik för initial provbehandling är adsorption i expanderade partikelbäddar
(”Expanded Bed Adsorption”, ”EBA”). Denna teknik baseras på en ny typ av kolonn och
nya typer av adsorbentmaterial. Tekniken baseras på användning av adsorbentpartiklar av
sfärisk tvärbunden agaros vars densitet höjts genom tillsats av kvartspartiklar eller av
partiklar av en inert metallegering. Adsorbentpartiklarnas storleksfördelning ger upphov till
en stabil, expanderad svävbädd när dessa utsätts för ett homogent linjärt flöde underifrån
genom kolonnen. I den expande-rade bädden kommer de största partiklarna i fördelningen
att anrikas vid kolonnens botten-platta och de minsta att anrikas i den svävande bäddens
toppskikt. Varje adsorbentpartikel finner sin plats i storleksgradienten där det föreligger en
jämvikt mellan dess tendens att sedimentera och att lyftas av vätskeflödet. Det stora
avståndet mellan de svävande partik-larna gör att det är möjligt att använda prover som
innehåller hela celler eller cellfragment. Det lösliga målproteinet i provet kommer att
bindas vid adsorbentpartiklarna under dess transport upp genom den svävande bädden
medan hela celler, cellfragment och icke bundna substanser kommer att tvättas ut genom
kolonnens övre utlopp. När uttvättningen är klar packas bädden och de adsorberade
proteinerna kan frigöras från adsorbentpartiklarna genom att en förträngande

9
buffertsaltlösning pumpas in i kolonnen uppifrån. Detta kan utföras antingen stegvis eller
med hjälp av en koncentrationsgradient. Mer att läsa i ref. (5).
Som sista steg bland de förberedande separationerna har det blivit allt vanligare att använda
avsaltning genom gelfiltrering snarare än dialys. Avsaltning är en sammanfattande term för
den operation där man överför proteiner i en ny miljö med känd sammansättning. Man kan
således byta buffertsaltlösning för att nå lämplig jonstyrka och lämpligt pH som förbered-
else för de intermediära och högupplösande separationsstegen under det sista stadiet av
reningsproceduren. I de fall ändringar i saltkoncentration och/eller pH leder till utfällning
av vissa proteiner i provet, är avsaltning genom dialys att föredra framför gelfiltrering
eftersom de utfällda proteinerna annars fastnar i gelfiltreringskolonnen och hindrar det
normala vätskeflödet.
5. INTERMEDIÄRA OCH HÖGUPPLÖSANDE SEPARATIONSMETODER
De viktigaste reningsstegen utgörs som regel av kromatografiska tekniker. Vid en klassi-
ficering av dessa är det lämpligt att utgå från de egenskaper hos molekylerna som varierar
tillräckligt mycket för att kunna utgöra en bas för deras separation. Alla proteiner är poly-
peptider, uppbyggda av ett tjugotal olika aminosyror. Proteinernas molekylvikter, d.v.s.
summan av de ingående aminosyraresternas molekylmassor, varierar starkt (från c:a
10.000 upp till c:a 1.000.000) liksom de olika proteinernas form. Eftersom aminosyrorna
kan vara såväl positivt som negativt laddade, eller neutrala, kommer antal och typ av
laddade grupper, liksom fördelningen av dessa, på proteinernas yta att variera starkt.
Skillnader i elektrisk laddning och andra egenskaper, t.ex. graden av hydrofobicitet och
biologisk funktion, utgör basen för de separationsmetoder som kommer att beskrivas nedan.
De presenteras översiktligt i Tabell 1.
Tabell 1. SEPARATIONSMETODER INOM BIOKEMIN
Skillnader i egenskaper mellan olika molekyler som kan utnyttjas för separationsändamål.
Molekylstorlek och –form .........
Nettoladdning ............................
Isoelektrisk punkt .......................
Biologisk funktion .....................
Hydrofobicitet ...........................
Innehåll av fri -SH-grupp ..........
Löslighet ...................................
Metall bindande förmåga ..........
Förmåga att bilda charge- .........
transferkomplex
Gelfiltrering, ultrafiltrering, ultracentrifugering,
porgradientgelelektrofores, SDS-gelelektrofores
Jonbyteskromatografi
Zonelektrofores i agarosgel
Isoelektrisk fokusering
Kromatofokusering
Affinitetskromatografi
Affinitetsfördelning
Hydrofob adsorptionskromatografi, omvänd-fas
kromatografi (RPC)
Kemisorption ("kovalent kromatografi")
Utfällning med t.ex. (NH4)2SO4, etanol
eller polyetylenglykol, kristallisering
Metallchelatkromatografi (IMAC),(16)
Charge-transferkomplexkromatografi (17}

10
5.1. Separation efter molekylernas storlek och form
Separation av molekyler i storleksklasser kan åstadkommas både genom elektrofores och
genom kromatografi. Den förra tekniken utnyttjas huvudsakligen för analys av proteiner
medan den senare i första hand är en preparativ metod.
5.1.1. Molekylsiktande elektrofores
Vid elektrofores används oftast en homogen polyakrylamidgel som framställts i form av en
platta, ett tunt skikt eller en smal cylinder. Provlösningen med proteinblandningen läggs på
gelén vid dess ena kant och proteinerna får vandra in i gelén som ett tunt band. Separation-
en sker inuti gelén genom att större molekyler kommer att vandra långsammare då de på
grund av polymernätverket tvingas att vandra en längre sträcka än mindre molekyler. Man
brukar säga att molekylerna vandrar med en hastighet som är omvänt proportionell mot
deras molekylvikter. Det senare gäller speciellt i det fall man förbehandlat proteinmole-
kylerna med natriumdodecylsulfat, ett ämne som binder sig till dessa och gör dem starkt
negativt laddade. I engelskspråklig litteratur används ofta benämningen SDS-PAGE för
denna teknik.
Porgradientelektrofores innebär att separationen får äga rum i en polyakrylamidgel vars
siktande egenskaper, d.v.s. porstorleken, förändras i proteinernas vandringsriktning. Olika
storleksklasser av proteinmolekyler kommer att anrikas vid vissa pordiametrar där de inte
har möjlighet att tränga fram med samma hastighet som tidigare. Fördelen med en sådan
porgradientgel är att proteinerna efter infärgning framträder som mycket tunna band, d.v.s.
upplösningsförmågan är mycket hög, och att deras vandringssträcka är korrelerad till
molekylvikten. En kalibreringskurva kan framställas med hjälp av proteiner med kända
molekylvikter. I Fig. 3 (a) och 3 (b) visas principen för gelelektrofores och i Fig. 4 visas ett
exempel på porgradientelektrofores av serum proteiner. Mer att läsa om gelelektrofores
finner Du i referenserna (2) och (6).
Fig.3(a). Principen för gelelektrofores. Vid elektrofores i homogen gel (vänstra skissen) vandrar små
molekyler snabbare än de större, då de i mindre grad bromsas av polymernätverket (de svarta punkterna). Vid
porgradientgelelektrofores (högra figuren) fångas molekylerna upp av polymernätverket vid vissa gelkoncen-
trationer varvid deras vandringshastighet starkt reduceras. Detta leder till smala zoner (hög upplösning).
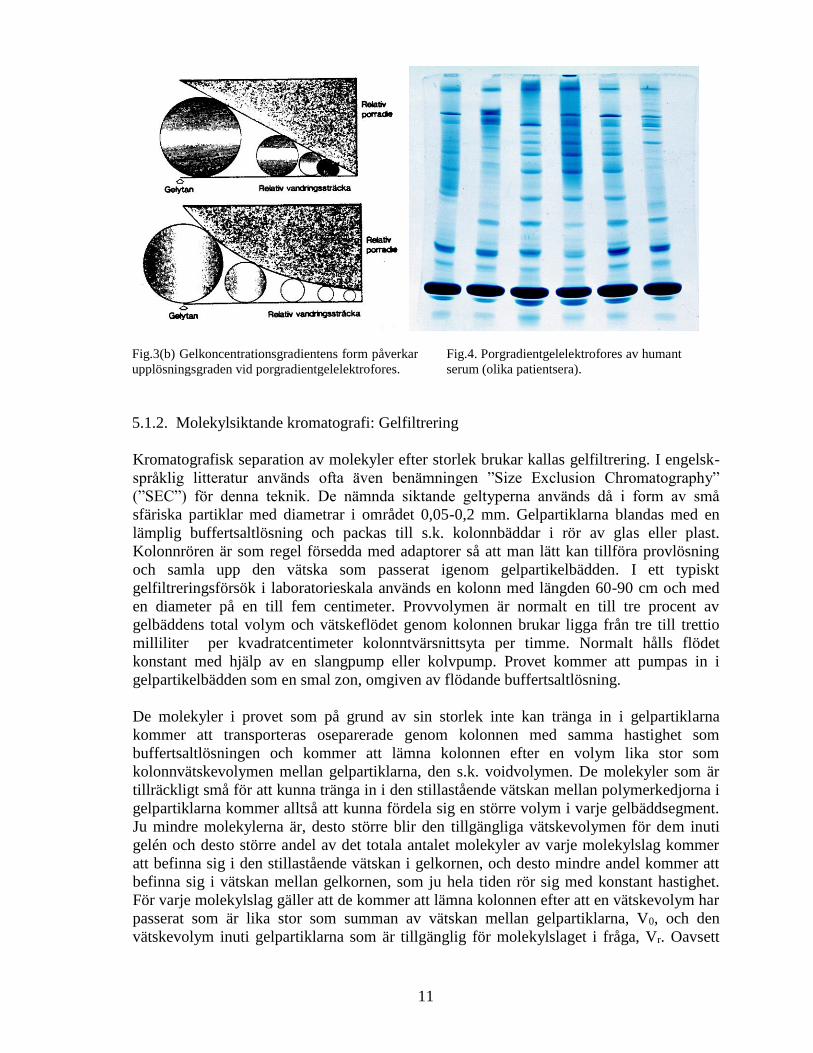
11
Fig.3(b) Gelkoncentrationsgradientens form påverkar
upplösningsgraden vid porgradientgelelektrofores.
Fig.4. Porgradientgelelektrofores av humant
serum (olika patientsera).
5.1.2. Molekylsiktande kromatografi: Gelfiltrering
Kromatografisk separation av molekyler efter storlek brukar kallas gelfiltrering. I engelsk-
språklig litteratur används ofta även benämningen ”Size Exclusion Chromatography”
(”SEC”) för denna teknik. De nämnda siktande geltyperna används då i form av små
sfäriska partiklar med diametrar i området 0,05-0,2 mm. Gelpartiklarna blandas med en
lämplig buffertsaltlösning och packas till s.k. kolonnbäddar i rör av glas eller plast.
Kolonnrören är som regel försedda med adaptorer så att man lätt kan tillföra provlösning
och samla upp den vätska som passerat igenom gelpartikelbädden. I ett typiskt
gelfiltreringsförsök i laboratorieskala används en kolonn med längden 60-90 cm och med
en diameter på en till fem centimeter. Provvolymen är normalt en till tre procent av
gelbäddens total volym och vätskeflödet genom kolonnen brukar ligga från tre till trettio
milliliter per kvadratcentimeter kolonntvärsnittsyta per timme. Normalt hålls flödet
konstant med hjälp av en slangpump eller kolvpump. Provet kommer att pumpas in i
gelpartikelbädden som en smal zon, omgiven av flödande buffertsaltlösning.
De molekyler i provet som på grund av sin storlek inte kan tränga in i gelpartiklarna
kommer att transporteras oseparerade genom kolonnen med samma hastighet som
buffertsaltlösningen och kommer att lämna kolonnen efter en volym lika stor som
kolonnvätskevolymen mellan gelpartiklarna, den s.k. voidvolymen. De molekyler som är
tillräckligt små för att kunna tränga in i den stillastående vätskan mellan polymerkedjorna i
gelpartiklarna kommer alltså att kunna fördela sig en större volym i varje gelbäddsegment.
Ju mindre molekylerna är, desto större blir den tillgängliga vätskevolymen för dem inuti
gelén och desto större andel av det totala antalet molekyler av varje molekylslag kommer
att befinna sig i den stillastående vätskan i gelkornen, och desto mindre andel kommer att
befinna sig i vätskan mellan gelkornen, som ju hela tiden rör sig med konstant hastighet.
För varje molekylslag gäller att de kommer att lämna kolonnen efter att en vätskevolym har
passerat som är lika stor som summan av vätskan mellan gelpartiklarna, V0, och den
vätskevolym inuti gelpartiklarna som är tillgänglig för molekylslaget i fråga, Vr. Oavsett

12
hur små molekylerna är kan de inte lämna kolonnen efter en volym som är större än sum-
man av vätskan mellan gelpartiklarna och den totala vätskevolymen i gelpartiklarna, Vt.
Om en molekyl lämnar kolonnen efter en volym som är större än kolonnens sammanlagda
vätskevolym är det ett tecken på att den på ett eller annat sätt står i bindningsjämvikt med
polymerkedjorna och/eller tvärbindningsbryggorna i gelmaterialet. Tvärbundet dextran,
Sephadex®, har en tendens att svagt binda molekyler med aromatiska grupper. Graden av
bindning kan man påverka genom att variera t.ex. ättiksyrakoncentrationen (3). Fig. 5 visar
principen för gelfiltrering och Fig. 6 en schematisk skiss över en typisk kromatografi
uppställning. Fig. 7 visar separationsmönstret för blodserum efter gelfiltrering på Sephacryl
S-300. Detaljerad information om gelfiltrering erhålls i referenserna (2) och (4).
Fig 5 Principen för gelfiltrering. De små molekylerna (•) tränger in i de molekyl siktande gelpartiklarna och
fördröjs relativt de större molekylerna (O). Pilarna till höger symboliserar vätskeflödet genom kolonnen, vars
"tvärsnitt" avbildats vid försökets start och efter två olika långa tidsintervall därefter.
Prov Buffert Pump Kolonn Fraktionssamlare
Fig.6. Skiss över en typisk apparatuppställning för kromatografi. Vid gelfiltrering med små provmängder
brukar provpåsättningsventilen placeras efter den peristaltiska pumpen, ofta i kombination med en s.k. loop.

13
200 250 300 ml
Fig.7. Gelfiltrering av humant serum på Sephacryl S-300 Superfine. Kolonn: 2.6 x 94 cm, provvolym: 2 ml,
elueringsbetingelser: buffert 0,1 M Tris-HCI pH 8.0 innehållande 0.5M NaCl, flöde: 2.3 ml/cm2/tim.
5.2. Separation efter molekylernas elektriska laddning
De viktigaste separationsmetoderna där man utnyttjar skillnader i storlek och typ av
elektrisk laddning hos molekylerna är elektrofores och jonbyteskromatografi.
5.2.1. Elektrofores
Elektrofores innebär laddade partiklars och molekylers vandring i ett elektriskt fält. Den
elektroforetiska vandringshastigheten är direkt proportionell mot laddningens storlek och
omvänt proportionell mot molekylradien. Ett normalvärde för proteiner är 5-10-5 cm/s vid
fältstyrkan en volt per centimeter. Antal och typ av laddade grupper på proteinernas yta
varierar starkt och har lett till att man talar om sura, neutrala och basiska proteiner, beroen-
de på vid vilket pH-värde proteinerna har sin isoelektriska punkt ,(pI), dvs. det pH-värde
där nettoladdningen är noll och proteinet således ej vandrar i ett elektriskt fält. Fig. 8 visar
hur vandringshastigheten i ett elektriskt fält med styrkan 1 V/cm, mobiliteten, för ett antal
olika serumproteiner ändras med pH-värdet hos den lösning där proteinerna befinner sig.
PROTEINERS ELEKTROFORETISKA MOBILITET SOM FUNKTION AV pH
Fig.8. Mobiliteten för olika serumproteiner som funktion av pH. Skärningspunkten vid mobiliteten noll är
identisk med proteinernas pI.

14
I föregående stycke beskrevs elektrofores i polyakrylamid och den molekylsiktande
egenskapen hos denna geltyp. Elektrofores användes där enbart som ett sätt att "transpor-
tera" molekylerna. Separationen åstadkoms av polymernätverket. I ren elektrofores, där
separationen beror på skillnader i vandringshastighet betingad av skillnader i nettoladdning
mellan olika molekyler, väljer man en icke siktande geltyp, t.ex. agarosgel av låg koncen-
tration (2-4 %). Elektrofores i agarosgel har framför allt blivit en viktig klinisk analys-
metod för proteiner i t.ex. blodserum och ryggmärgsvätska. En intressant utveckling av
metoden innebär att man efter avslutad elektrofores låter de separerade proteinerna
elektroforetiskt vandra, i 90 graders riktning mot den ursprungliga, in i ett agarosgelskikt
som innehåller en lösning av antikroppar mot alla eller vissa proteiner i provet. Alltefter-
som de vandrande proteinerna möter sina antikroppar så kommer de, där koncentrations-
förhållandet är optimalt, att reagera med dessa och bilda en olöslig fällning. Detta inträffar i
koncentrationsgradienten i kanten på den vandrande proteinzonen och leder till att den
successivt minskar i omfattning för att till sist helt försvinna i spetsen på den mer eller
mindre Gausskurveformade linje som utfällningslinjerna bildade längs båda sidorna på den
vandrande proteinzonen. Ytan innanför utfällningslinjen är ett mått på mängden protein i
zonen. Metoden kallas korsad immunoelektrofores och resultatet av ett sådant försök med
blodserum som prov visas i Fig. 9.
En mer detaljerad beskrivning av denna och andra agarosgelelektroforetiska analysmetoder
återfinns i boken "Plasmaproteiner", se (18). Ingående presentationer av elektroforetisk
teori och praktik finner Du i referenserna (2) och (6).
Fig.9. Korsad immunoelektrofores av humant serum i agarosgel innehållande kaninantikroppar mot helt
humanserum. Provet (2u1) sattes på gelén i den lilla runda brunnen till höger i bildens nedre kant.
5.2.2. Isoelektrisk fokusering
En annan viktig elektroforetisk separationsmetod som främst används analytiskt i poly-
akrylamidgel är isoelektrisk fokusering. Metoden bygger på det i början av detta stycke
omnämnda förhållandet att många biomolekyler, främst proteinerna, är amfolyter (d.v.s. de
kan vara både positivt och negativt laddade) med väl definierade isoelektriska punkter,
d.v.s. pH-värden där de ej vandrar i ett elektriskt fält. Vid pH-värden under den isoelektri-
ska punkten är amfolyterna positivt laddade och vandrar mot den negativt laddade

15
elektroden, katoden. Vid pH-värden över isoelektriska punkten är amfolyterna negativt
laddade och vandrar mot den positiva elektroden, anoden. Om man skapar en stabil pH-
gradient mellan de båda elektroderna, så kommer en amfolyt med en väldefinierad
isoelektrisk punkt, oavsett var den placeras i gradienten, att vandra mot och stanna vid
motsvarande pH-värde. Principen framgår av skissen i Fig. 10.
Fig.10. Principen för isoelektrisk fokusering. I läge A är proteinet negativt laddat och vandrar mot anoden. I
läge B är proteinet positivt laddat och vandrar mot katoden. Vid pI är nettoladdningen noll och proteinzonen
skärps stationärt. Den heldragna linjen visar den stabila pH-gradienten.
Stabila pH-gradienter kan erhållas med hjälp av s.k. bäraramfolyter, dvs. lågmolekylära
substanser med hög buffertkapacitet vid sina isoelektriska punkter. En bra bäraramfolyt-
blandning skall dessutom ha jämn elektrisk ledningsförmåga över hela pH-intervallet.
Avsnitt med låg ledningsförmåga kan ge överhettningsproblem och begränsar den totala
effekt man annars skulle kunna tillföra hela separationskammaren. Avsnitt med hög
ledningsförmåga ger spänningsfall och dålig fokusering. Det finns flera olika fabrikat av
bäraramfolyter på marknaden. De ger i de flesta fall fullt likvärdiga resultat. Skillnader kan
upptäckas först när man pressar betingelserna, t.ex. när de används i mycket tunna gelskikt
och med fältstyrkor upp till 300 volt/cm. Orsaken till att höga fältstyrkor är önskvärda inom
isoelektrisk fokusering beror på att det föreligger ett direkt samband mellan zonbredden hos
ett "fokuserande" protein och den elektriska fältstyrkan. Sambandet framgår av följande
formel:
= mått på zonskärpan, motsvarar c:a 1/4 av proteinets zonbredd
D = diffusionskoefficienten för proteinet
E = fältstyrkan (V/cm)
du/dpH = lutningen hos mobilitetskurvan vid proteinets pI
dpH/dx = lutningen hos pH-gradienten
Bland dessa variabler är D och du/dpH inneboende egenskaper hos det speciella proteinet,
varför endast E och dpH/dx kan varieras experimentellt. En ökning av dpH/dx skärper
visserligen proteinzonen men leder samtidigt till att zoner tillhörande olika proteiner i
prover kommer närmare varandra, varför nettoupplösningen ej påverkas i praktiken. Fig. 11
visar separationsmönstret efter isoelektrisk fokusering av olika muskelproteiner i ett tunt
polyakrylamidgelskikt. Fig. 12 visar ett exempel på 2-dimensionell gelelektrofores där
isoelektrisk fokusering använts i första dimensionen och SDS polyakrylamidgelelektrofores

16
i den andra dimensionen. Konsultera referenserna (2), (6), (7) och (8) för ytterligare
information om isoelektrisk fokusering.
Fig.11. Isoelektrisk fokusering i tunnt polyakrylamidskikt. Prover från vänster: muskelpro-
teiner från gädda, linslektin (isolektiner), muskelproteiner från regnbågsforell och beta-
laktoglobulin. Samma prover återkommer sedan till höger i gelen. Försöksbetingelser:
Bäraramfolyt Pharmalyte 3-10, fältstyrka 300 V/cm.
Fig. 12. 2-dimensionell elektrofores av bagerijästproteiner (Saccharomyces cereviciae) i ett
tunt skikt av polyakrylamidgel. Första dimensionen: IEF i närvaro av urea. Andra dimensi-
onen: SDS-porgradientgelelektrofores. Vänstra gelen färgad med Coomassie Brilliant Blue.
Högra gelen färgad med silver-reagens som ger c:a 10 ggr högre känslighet än färgning
med Coomassie Brilliant Blue.

17
5.2.3. Jonbyteskromatografi
Jonbyteskromatografi är en separationsmetod som bygger på användningen av gelpartiklar
som innehåller kovalent bundna laddade grupper. Dessa laddade grupper är införda genom
en kemisk reaktion i en från början neutral, porös gel. Varje laddad grupp i gelen neutrali-
seras med hjälp av en jon med motsatt laddning, en motjon, som hålls kvar i dess närhet på
grund av elektrostatisk attraktion. Motjonen är alltså ej bunden utan rörlig och kan lätt
bytas ut mot en annan jon med samma laddning. Kloridjonen, Cl-, är en vanlig motjon till
en positivt laddad jonbytargel och natriumjonen. Na+, är en vanlig motjon till en negativt
laddad jonbytare. Positivt laddade jonbytare brukar kallas anjonbytare och negativt laddade
jonbytare katjonbytare. Man brukar dessutom skilja mellan svaga och starka jonbytare. En
svag jonbytare innehåller laddade grupper vars laddning är beroende av den omgivande
lösningens pH-värde. En stark jonbytare är laddad vid alla pH-värden. Exempel på svaga
jonbytare är de som innehåller karboxylgrupper. De blir oladdade, och således oanvändbara
vid pH-värden under c:a 5. Jonbytare innehållande tertiära aminogrupper, t.ex. dietylamino-
etyl (DEAE-) förlorar sin laddning genom protolys vid pH-värden över c:a 8-9. Exempel på
starka jonbytare är de som innehåller sulfonsyragrupper och kvaternära ammoniumgrupper.
Fig. 13 visar exempel på olika typer av jonbytande grupper.
Fig. 13. Exempel på jonbytande grupper i moderna jonbytare för proteiner.
En jonbytare befinner sig i dynamisk jämvikt med jonerna i sin omgivning och är mycket
känslig även för små skillnader i protolyseringsgrad hos olika motjoner. Detta förhållande
insågs mycket tidigt av forskare framförallt i USA och den kanske elegantaste praktiska
tillämpningen av detta fenomen kom i början på 1950-talet i form av aminosyraanalysatorn,

18
en maskin som bygger på användningen av starka katjonbytare och noggrant utprovade
buffertsaltlösningar. Jonbyteskromatografi av proteiner ställer stora krav på gelmaterialets
egenskaper. Proteiner har utvecklats i en vattenmiljö och deras struktur är ofta mycket
känslig för påverkan av hydrofoba grupper. De första användbara jonbytarna för protein-
separation var därför tillverkade av hydrofila gelmaterial baserade på cellulosa, tvärbundet
dextran (Sephadex®) och agaros (t.ex. Sepharose® Fast Flow). Under de senaste
decennierna har emellertid nya jonbytare utvecklats som bygger på syntetiska organiska
polymerer såsom polystyren-divinylbensen. Dessa har högre rigiditet än de ovan nämnda
materialen och kan därför tillverkas med avsevärt mindre partikeldiametrar. Vissa av dessa
har framställts i monodispers pärlform (se Fig. 14). Den hydrofoba polystyrenytan har först
hydrofiliserats och sedan substituerats med laddade grupper. En viktig faktor vid
proteinkromatografi är jonbytarmaterialets porositet. Makroporositet ger hög bind-
ningsförmåga per volymsenhet för proteiner och dessutom snabb jämviktsinställelse, d.v.s.
effektivare separation. Agarosbaserade jonbytare har här stora fördelar framför de som
baseras på siktande geltyper; speciellt för högmolekylära proteiner. Även för nukleinsyror,
oligonukleotider och nukleotider är idag jonbyteskromatografi en ofta använd separations-
metod.
Fig. 14. Monodispersa gelmaterial baserade på polystyren-divinylbensen. Används av GE Healthcare
Biosciences i Uppsala som utgångsmaterial för framställning av olika typer av jonbytare för
proteinseparation.
Det finns två principiellt olika förfaranden vid jonbyteskromatografi. Vid det ena anpassar
man betingelserna med avseende på saltkoncentration och pH så att de komponenter som
skall separeras aldrig helt fastnar på jonbytaren utan alla molekylerna rör sig samtidigt på
samma sätt som beskrevs i föregående kapitel för gelfiltrering. D.v.s. för varje molekylslag
existerar en unik jämvikt mellan bundna och fria molekyler och där proportionen fria
molekyler bestämmer den hastighet med vilken molekylslaget rör sig genom jonbytar-
kolonnen. De molekyler som är för stora för att ens penetrera gelpartiklarna och som ej
visar någon tendens att binda till de laddade grupperna vandrar snabbast och bland dessa
molekyler sker ingen separation alls. De molekyler som helt penetrerar gelpartiklarna (icke
siktande geltyp) men ej binds till de laddade grupperna i gelen vandrar också tillsammans
oseparerade från varandra men långsammare än, och således separerade från, de förra. Alla
övriga molekyler, d.v.s. de som mer eller mindre starkt binder till de laddade grupperna
kommer mer eller mindre fullständigt att separera från varandra under sin vandring genom
jonbytarkolonnen. Ju längre kolonnen är, desto bättre separation får man. Dock blir inte
separationen dubbelt så bra med dubbelt så lång kolonn. Enligt den grundläggande kroma-
tografiteorin blir separationen endast kvadratroten ur 2, d.v.s. c:a 1,4, gånger så bra vid en
fördubbling av kolonnlängden vid den här speciella varianten av jonbyteskromatografi.
Vid den andra jonbyteskromatografiska metoden väljer man betingelser där den kompo-

19
nent man vill renframställa (alternativt, i vissa fall föroreningarna), fastnar på jonbytaren
och inte lossnar förrän man på något sätt förändrat sammansättningen hos den buffert-
saltlösning som provet och jonbytaren från början befann sig i. Genom att ändra t.ex.
saltkoncentrationen eller pH i buffertsaltlösningen stegvis eller kontinuerligt (gradient-
eluering) kommer de olika komponenterna att lossna en efter en och transporteras av den
flödande vätskan genom och ut ur kolonnen. Vid denna typ av kromatografi är man oftast
ej betjänt av långa kolonner, tvärtom får man i de flesta fall bättre separation ju kortare
kolonnerna är. I Fig. 15 visas resultatet av en jonbyteskromatografisk separation av
proteinerna i giftet av kungskobra och i Fig. 16 visas resultatet av jonbyteskromatografisk
renframställning av humant serumalbumin i stor skala. Jonbyteskromatografi, eller oftast
endast adsorption av proteiner till en jonbytargel som blandas med provet och sedan
frånskiljs genom filtrering, har blivit en mycket viktig teknik för renframställning av
proteiner i industriell skala av vissa kliniskt viktiga proteiner, t.ex. serumalbumin,
gammaglobulin och olika blodkoaguleringsfaktorer. Närmare information om detta kan
inhämtas i boken "Plasmaproteiner", se (18). Ytterligare referenser till jonbyteskromato-
grafiska arbeten kan extraheras ur referenserna (2) och (9).
Fraktionsnummer
Fig.15. Jonbyteskromatografi med gradienteluering av kobragift på CM-Sephadex C-50.
Fig.16. Jonbyteskromatografi med stegvis eluering av humant serumalbumin från ett försök med DEAE-
Sepharose CL-6B i stor skala (16 liter gel i en 37 cm diameter och 15 cm hög kolonn, Pharmacia KS370/15)
utgående från 16 liter humanserum.

20
5.2.4. Kromatofokusering
En intressant variant av jonbyteskromatografi är kromatofokusering. Tekniken bygger på
användningen av en speciell jonbytare vars grupper har hög buffertkapacitet över ett brett
pH-intervall. Närvaron av kvaternära ammoniumgrupper garanterar proteinbindnings-
kapacitet även vid höga pH-värden. Eluering sker med pH-gradient. Jonbytaren jämviktas
med en lämplig buffertsaltlösning vid det högre pH-värdet i det valda intervallet. Prov-
lösningen, som innehåller proteiner med isoelektriska punkter inom det valda intervallet är
som regel jämviktad vid samma pH eller i elueringsbuffert. Efter provpåsättningen pumpas
en speciell elueringsbuffert, kallad "Polybuffer", in i kolonnen. Polybufferlösningens pH är
justerat till det lägsta värdet i intervallet. Polybufferlösningen har jämn och hög buffert-
kapacitet över hela det valda intervallet vilket leder till att en jämn och stabil pH-gradient
kommer att utvecklas i den flödande vätskan i kolonnen på grund av att Polybuffer-
lösningen långsamt kommer att titreras av jonbytaren. Proteinerna i provet är vid försökets
början adsorberade som ett smalt band vid kolonnens övre del. I och med att Polybuffer-
lösningen börjar pumpas in i kolonnen sjunker sakta pH-värdet i vätskan kring de
adsorberade proteinerna. När pH-värdet sjunker under den isoelektriska punkten för ett
visst protein lossnar det från jonbytaren och transporteras nedåt i kolonnen med den
flödande Polybufferlösningen. Polybufferlösningens pH i det volymavsnitt där proteinet
befinner sig är emellertid inte konstant utan ändras kontinuerligt genom titrering på grund
av jonbytargruppernas höga buffertkapacitet. Efter en kort stund har pH-värdet passerat
proteinets isoelektriska punkt igen men nu från andra hållet, proteinet blir negativt laddat
och fastnar på jonbytarens positivt laddade grupper. Gradienten rör sig emellertid hela tiden
nedåt i kolonnen och samma procedur upprepas om och om igen. Resultatet blir att varje
protein kommer att anrikas, fokuseras, till en mycket smal zon som kommer att lämna
kolonnens utlopp vid ett pH-värde nära dess isoelektriska punkt. I kolonnen utbildas
således en stabil pH-gradient som hela tiden rör sig framåt, men, vilket har avgörande
betydelse, med en hastighet som är c:a 10 gånger lägre än den hos den flödande vätskan.
Fig. 17 visar schematiskt principen för kromatofokusering och i Fig. 18 visas resultatet av
kromatofokusering av ett älgmuskelproteinextrakt. Mer information om kromatofokusering
kan inhämtas i referenserna (2) och (10).
Fig.14. Den fokuserande effekten vid kromatofokusering beror på att vätskeflodet genom kolonnen (den långa
pilen) är avsevärt högre än den hastighet med vilken pH-gradienten, och således en amfolyt med
isoelektrisk punkt inom gradienten, rör sig genom kolonnen (den korta pilen). Figuren är hämtad ur (10).

21
Fig.18. Kromatofokusering av ett proteinextrakt från älgmuskel. Kolonn: 1 x 45 cm PBE 94, prov: 5 ml,
elueringsbetingelser: startbuffer 0.025M etanolamin-HCI pH 9.4, elueringsbuffert: 0.0075 mmol/pH-enhet/ml
Polybuffer 96, flöde: 20 m1/cm2/timme. (Ur referens (10)).
5.3. Separation efter molekylernas funktion
5.3.1. Affinitetskromatografi av enzymer
Alla proteiner har en specifik biologisk funktion. Den största gruppen är enzymerna,
biokatalysatorerna, som ombesörjer att alla de tusentals olika kemiska reaktionerna i en
levande cell kan fortgå vid de låga fysiologiska temperaturerna. Karakteristiskt för
enzymerna är deras ofta höga specificitet, d.v.s. de katalyserar reaktioner mellan endast
enstaka eller ett fåtal s.k. substrat. På enzymernas yta finns s.k. aktiva ytor där själva
katalysen äger rum. Vid katalystillfället binds substratet till den aktiva ytan, för att
omedelbart efter reaktionen frigöras. Vissa substanser har stor likhet med substratet, men
kan inte omsättas av enzymet. De fortsätter således att vara bundna till den aktiva ytan och
förhindrar substratet att nå fram. Dessa substanser kallas för hämmare eller inhibitorer och
bindningen mellan dem och enzymet kan utnyttjas för separationsändamål under förutsätt-
ning att bindningen är reversibel. En stor grupp enzymer behöver för sin funktion s.k. co-
faktorer. Även co-faktorerna binds vid katalystillfället tillsammans med substratet till
enzymets aktiva yta för att omedelbart därefter frigöras. Det har visat sig att substanser som
liknar co-faktorer också fungerar som hämmare och kan således utnyttjas för separations-
ändamål.
Sättet att utnyttja biologisk parbildning for separationsändamål kallas för affinitetskromato-
grafi och tillgår så att den ena molekylen i paret, i exemplet ovan hämmaren, kemiskt
kopplas till en bärare, oftast en makroporös gelpartikel t.ex. 4 % agaros. Gelpartiklarna
innehållande bundna hämmare packas till en bädd i ett rör av glas eller plast varefter en
provlösning innehållande det enzym man vill renframställa pumpas igenom bädden. Endast
det enzym eller de enzym som hämmas av den bundna molekylen kommer att fastna i
gelbädden, de övriga tvättas ut ur densamma med en lämplig buffertsaltlösning. Genom att
sedan ändra sammansättningen hos buffertsaltlösningen, t.ex. genom att sätta till en
substans som binder till samma ställe på enzymet som hämmaren d.v.s. substratet eller co-
faktorn, eller genom att ändra lösningens pH, kommer enzymet att lossna och kan tas
tillvara från den lösning som lämnar kolonnen. På denna princip bygger ett mycket stort
antal effektiva isoleringsförfaranden för olika enzymer som publicerats sedan tekniken
22
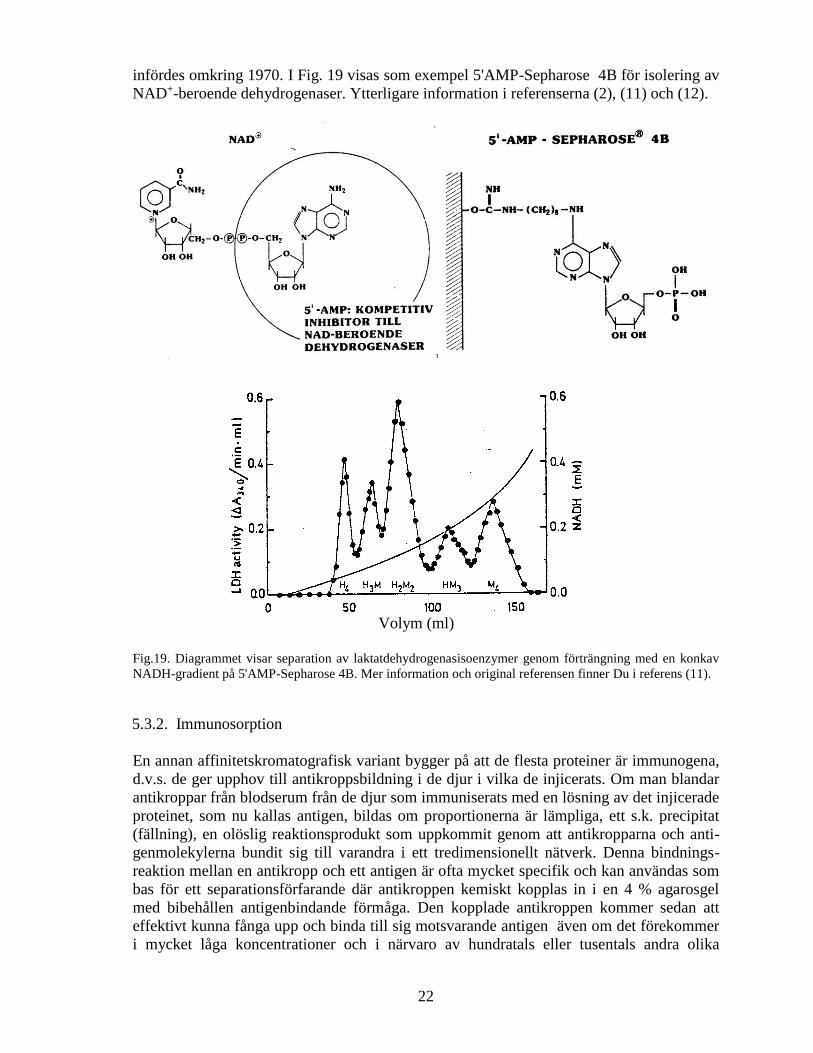
infördes omkring 1970. I Fig. 19 visas som exempel 5'AMP-Sepharose 4B för isolering av
NAD+-beroende dehydrogenaser. Ytterligare information i referenserna (2), (11) och (12).
Volym (ml)
Fig.19. Diagrammet visar separation av laktatdehydrogenasisoenzymer genom förträngning med en konkav
NADH-gradient på 5'AMP-Sepharose 4B. Mer information och original referensen finner Du i referens (11).
5.3.2. Immunosorption
En annan affinitetskromatografisk variant bygger på att de flesta proteiner är immunogena,
d.v.s. de ger upphov till antikroppsbildning i de djur i vilka de injicerats. Om man blandar
antikroppar från blodserum från de djur som immuniserats med en lösning av det injicerade
proteinet, som nu kallas antigen, bildas om proportionerna är lämpliga, ett s.k. precipitat
(fällning), en olöslig reaktionsprodukt som uppkommit genom att antikropparna och anti-
genmolekylerna bundit sig till varandra i ett tredimensionellt nätverk. Denna bindnings-
reaktion mellan en antikropp och ett antigen är ofta mycket specifik och kan användas som
bas för ett separationsförfarande där antikroppen kemiskt kopplas in i en 4 % agarosgel
med bibehållen antigenbindande förmåga. Den kopplade antikroppen kommer sedan att
effektivt kunna fånga upp och binda till sig motsvarande antigen även om det förekommer
i mycket låga koncentrationer och i närvaro av hundratals eller tusentals andra olika

23
proteiner. Högsta tänkbara specificitet erhålles med s.k. monoklonala antikroppar som är
riktade mot endast en s.k. antigen determinant på proteinets yta. En antigen determinant
utgörs av ett begränsat avsnitt av proteinytans strukturmönster och ett och samma protein
kan således innehålla ett stort antal olika antigena determinanter. Från klassisk immunodif-
fusionsanalys vet man att olika proteiner kan ha gemensamma antigena determinanter,
emellertid finns det alltid unika sådana för varje protein och monoklonala antikroppar mot
dessa kan användas för konstruktion av mycket specifika isoleringsförfaranden. Det finns
idag metoder för framställning av monoklonala antikroppar med hjälp av s.k. hybridom,
d.v.s. hybridcellinjer framställda genom sammansmältning av antikroppsbildande celler
(mjältceller) med tumörceller (myelom). Dessa hybridom kan, till skillnad från normala
antikroppsproducerande celler, hållas vid liv under mycket lång tid och man kan använda
denna teknik för framställning av monoklonala (d.v.s. från en enda celltyp härledda)
antikroppar i stor skala. Det bör tilläggas att det framför allt är hybridom från vit mus man
arbetar med. Den starka bindningen mellan en antikropp och dess antigen är en förutsätt-
ning för att man skall kunna isolera de proteiner som förekommer i koncentrationer under
c:a 10-8 M, men utgör samtidigt ett problem vid försöken att lösgöra proteinet från den
kopplade antikroppen. Det enda sättet går ut på att förändra antikroppens och/eller det till
antikroppen adsorberade proteinets rymdstruktur så att förutsättningarna för adsorptionen
upphör och proteinet lossnar. Bland de strukturförändrande substanser som prövats med
varierande framgång hör syror (d.v.s. pH-sänkning till c:a 3), urinämne, guanidinium-
hydroklorid samt chaotropa joner t.ex. SCN-, J-, ClO4-, Cl3CCOO- och F3CCOO-. Det är
viktigt att omedelbart efter det att proteinet lossnat återföra detsamma och den kopplade
antikroppen i den ursprungliga fysiologiska miljön. Det finns annars risk för att både
proteinet och antikroppen ej kan återställas i sin ursprungliga form. De blir "irreversibelt
denaturerade", och förlorar sin biologiska funktion. Återförandet kan ske antingen genom
dialys eller, snabbare, genom gelfiltrering. Den kopplade antikroppen förs snabbt över i
"rätt" miljö genom tvättning av gelbädden med fysiologisk buffertsaltlösning. Referens
(13) ger mer information om denna teknik.
5.3.3. Affinitetskromatografi av glykoproteiner
Vissa proteiner, s.k. glykoproteiner, bär kovalent bundet kolhydrat (oftast heterologa
oligosackarider) på ytan. Med speciell teknik, en variant av affinitetskromatografi, baserad
på användningen av till agarosgelpärlor kovalent kopplade, kolhydratbindande proteiner,
s.k. lektiner, kan man effektivt separera glykoproteiner från proteiner som saknar kolhydrat.
Genom att koppla lektiner med olika specificitet erhålles adsorbent som dessutom separerar
olika glykoproteiner från varandra under förutsättning att de innehåller olika sockerarter i
sina kolhydratdelar. Lektinbaserade adsorbent har en mycket tilltalande egenskap, nämligen
den för det adsorberade glykoproteinet mycket skonsamma desorptionsmetoden. Proteinet
lösgörs nämligen från lektinet genom tillsats av en lösning av den sockerart som ingår i
proteinets kolhydratdel och som binder till lektinet. Principen framgår av innehållet i Fig.
20. Metoden är så skonsam att man med framgång använt den för separation av olika
levande celler. Således kan T- och B-lymfocyter separeras på en agarosgel innehållande
kopplat lektin från vinbergssnäckan, He1ix pomatia. Detta lektin är specifikt mot kolhydrat
innehållande N-acetyl--D-galaktosamin. Det mest kända och använda lektinet är
Concanavalin A från jackböna, Canavalia ensiformis. Det binder till bl.a. mannos, en
sockerart som finns i många glykoproteiner t.ex. i blodplasma. Concanavalin A kopplat till
agarospärlor har därför blivit ett mycket populärt separationshjälpmedel för de forskare

24
som studerar blodplasmaglykoproteiner. Referenserna (2), (11) och (14) ger ytterligare
information om lektiner och lektinadsorbent.
Fig.20. Förenklad framställning av principen för affinitetskromatografi av glykoproteiner med hjälp av lektin
kopplat till agarosgel. Det på bilden kopplade lektinet binder proteiner innehållande galaktos (G), övriga
glykoproteiner (t.ex. mannosinnehållande (M)) och proteiner som saknar kolhydrat, binder ej och kan tvättas
bort. Det adsorberade glykoproteinet kan sedan förträngas med hjälp av överskott galaktos.
5.3.4. Exempel på affinitetskromatografi av transportproteiner och receptorer
En del proteiner har specifik transportfunktion, t.ex. för hormoner och vissa vitaminer.
Andra fungerar som receptorer för hormoner på ytan av vissa celler. Genom att koppla
vitamin B12 till agarosgelpärlor (se Fig. 21) får man ett adsorbent som kan fånga upp det
vitamin B12-transporterande proteinet i blodplasma, transcobalamin II
Fig.21. Strukturen för det vitamin B12-adsorbent som användes vid den i Tabell 3 redovisade reningen av
transcobalamin II.
Koncentrationen av detta protein i plasma är mycket låg, man har uppskattat att det finns en
molekyl transcobalamin II per två miljoner serumalbuminmolekyler. Talesättet att hitta en
nål i en höstack är mycket passande i detta fall. Alla försök att renframställa detta protein
med konventionella metoder hade misslyckats. Det var först i och med utvecklingen av den
affinitetskromatografiska tekniken i början på 1970-talet som detta blev möjligt. I Tabell 2
visas en uppställning av de olika stegen i reningen av transcobalamin II. Det framgår klart

25
att affinitetskromatografi steget är direkt avgörande för isoleringen av detta protein.
Tabell 2. Rening av transcobalamin II frän human plasma
RENINGSSTEG VOLYM (ml) PROTEIN (mg)
Human plasma 1.400.000 98.000.000
Cohn fraktion III 372.000 5.280.000
CM-Sephadex® C-50 19.700 760.000
Vitamin B12-kopplad
Sepharose® 4B 62 25
DEAE-cellulosa 61 11
3,3’-diaminodipropylamin-
kopplad Sepharose 4B 82 10
Sephadex G-150 9 6
Ett annat anmärkningsvärt exempel på användningen av affinitetskromatografi för att lösa
ett svårt reningsproblem är isolering av insulinreceptorn från fettcellplasmamembraner i
råttlever. Insulinkoncentrationen i blod är mycket låg, c:a 10-10 M, och det krävs därför en
hög bindningskonstant för komplexet mellan hormonet och dess receptor. Receptorkoncen-
trationen är också mycket låg, man har uppskattat att det endast finns c:a tiotusen receptor-
molekyler per fettcell, varför en hög bindningskonstant underlättar möjligheten att med
hjälp av ett adsorbent bestående av agarosgelpärlor innehållande kovalent bundet insulin
specifikt fånga upp de fåtaliga receptormolekylerna i ett detergentextrakt av råttleverfett-
cellmembraner. Receptorerna är normalt bundna till membranytan men kan överföras i
löslig form med hjälp av ett icke-joniskt detergent. Den på så sätt lösliggjorda insulin-
receptorn visar samma bindningsegenskaper till insulin som den intakta receptorn. Sedan
receptordetergentkomplexet adsorberats till det kopplade insulinet och övriga substanser i
provet tvättats ut ur get bädden kunde receptorn lösgöras från insulinet genom en kombina-
tion av pH-sänkning och tillsats av 4,5 M urinämne. Reningsfaktorn i detta steg blev c:a
8000 och totalt för hela proceduren från leverhomogenat till den renade receptorn blev
reningsfaktorn c:a 250 000.
5.3.5. Bindning av IgG till Protein A från Staphylococcus aureus
Ett mycket speciellt fall av bindningsbenägenhet som fått stor praktisk användning är här
värt att beskrivas. Bundet till cellväggen hos bakterien Staphylococcus aureus finns ett
protein som har egenskapen binda till immunglobuliner av typ G, d.v.s. IgG, de proteiner
man normalt förknippar med begreppet antikroppar. Bakterieproteinet brukar kallas Protein
A och dess bindning till antikropparna beror ej på en vanlig immunkemisk reaktion mellan
de senare och ett antigen. Protein A binder nämligen till den s.k. Fc-delen av antikroppen
och lämnar den antigenbindande s.k. Fab-delen fri. Genom att kovalent binda Protein A till
agarosgelpärlor får man ett specifikt adsorbens för IgG-molekyler och således ett utomor-
dentligt bekvämt separationshjälpmedel för alla de forskare som önskar isolera IgG från
olika serumprover. Komplexet mellan den bundna Protein A-molekylen och antikroppen
spjälkas normalt genom sänkning av pH till c:a 3,0. Efter uttvättning justeras pH omedel-
bart till det fysiologiska för att undvika irreversibel denaturering av proteinerna. Fig. 22
visar principen för rening av IgG med Protein A Sepharose. Se även referens (2) och (11)

26
för närmare information. Det bör nämnas att ett liknande protein från streptokocker, Protein
G, också har fått vidsträckt användning för samma ändamål.
Steg 1. Provpåsättning
Steg 2. Adsorption av IgG och uttvättnmg av
icke adsorberade proteiner (t.ex. IgA, IgD och
övriga serumproteiner).
Steg 3. Desorption av IgG genom pH-sänkning
till ca 3, t.ex. med 1M ättiksyra. IgG överförs
omedelbart i fysiologisk miljö genom gelfiltrer-
ing.
Fig.22. Principen för isolering av IgG från andra immunglobuliner (och övriga serumproteiner) genom
adsorption till Protein A Sepharose®.
5.4. Hydrofob adsorptionskromatografi
Vissa proteiner exponerar på sin yta mer eller mindre lättillgängliga hydrofoba ytor eller
klyftor. Denna skillnad i hydrofobicitet mellan olika proteiner kan utnyttjas för separation
genom s.k. hydrofob adsorptionskromatografi. Tekniken baseras på att man inför hydrofoba
grupper t.ex. fenylgrupper eller oktylgrupper i den normalt helt hydrofila agarosgelen.
Adsorption av proteiner till dessa grupper underlättas av hög saltkoncentration och vid elu-
eringen av de adsorberade proteinerna brukar man använda en kombination av kontinuerligt
minskande saltkoncentration och en på samma sätt ökande koncentration av etylenglykol.
Fig. 23. Principen för s.k. hydrofob adsorption (”Hydrophobic Interaction Chromatography”, HIC). Den
välkända formeln för ändringen av den fria energin för en isotermisk process kan här användas för att förklara
varför adsorptionen sker spontant. För att erhålla den nödvändiga minskningen av Gibbs fria energi måste
systemets entropi öka. Detta sker genom att ordnat vatten kring de hydrofoba ytorna på adsorbentet och på
proteinet övergår till att bli oordnat i och med att adsorptionen äger rum.

27
Fig. 24. Elueringsdiagrammet från ett försök att rena -amylas från ett kornmjölsextrakt genom kromatografi
på Phenyl Sepharose. I referens (15) finner Du mer data om denna teknik.
5.5. Kemisorption ("Kovalent kromatografi")
En grupp proteiner innehåller fria sulfhydrylgrupper (tiol-grupper) härrörande från
aminosyran cystein. Denna egenskap kan utnyttjas för separation genom s.k. kovalent
kromatografi där -SH-innehållande proteiner lätt kan avskiljas från övriga proteiner i ett
prov. Tekniken bygger på att till agarosgelpärlor kovalent bundna tiolgrupper överförs till
aktiverade disulfider genom reaktion med föreningen dipyridyldisulfid. Den till agaros-
gelen bundna, aktiverade, blandade disulfiden kommer så snart den kommer i kontakt med
en molekyl innehållande en fri tiolgrupp att omedelbart reagera med denna genom tiol-
disulfidutbyte. Den tidigare fria, tiolhaltiga molekylen kommer därvid att ersätta den
aktiverade, lämnande gruppen och kovalent bindas till gelen via en disulfidbrygga. Övriga,
icke tiolhaltiga, molekyler i provet kan nu tvättas ut ur gelbädden varefter den bundna
molekylen kan elueras genom ett nytt tiol-disulfidutbyte. Detta åstadkommes genom tillsats
av lågmolekylära, enkla tioler som t.ex. merkaptoetanol. Tioladsorbentet kan sedan åter
aktiveras med hjälp av dipyridyldisulfid. Reaktionen mellan en tiol och den aktiverade,
blandade disulfiden går praktiskt taget stökiometriskt eftersom den frigjorda pyridyltiolen
omedelbart omlagras till en tiopyridon. Den senare har ett karakteristiskt absorptionsmaxi-
mum vid 343 nm varför det är lätt att kvantitativt mäta mängden inkopplad tiol. Bland de
proteiner som med framgång renats med denna teknik kan nämnas ureas från jackböna och
papain från papayafrukter. Fig. 25 visar principen för kemisorption (kovalent kromatogra-
fi). Ytterligare information och fler referenser kan inhämtas ur referenserna (2) och (11) .
He's The world expert on isolation of Axolotl toe-nail proteins!

28
Fig.25. Principen för kemisorption ("kovalent kromatografi"). Reaktionsförlöppet kommenteras i texten.
5.6. Övriga separationsmetoder
5.6.1. Metallchelataffinitetskromatografi
I engelskt språkbruk kallas denna metod ”Immobilized metal ion affinity chromatography”
eller IMAC. Den bygger på egenskapen hos vissa proteiner att binda till sig vissa
övergångsmetalljoner såsom Cu2+, Zn2+ och Ni2+. Man har visat att det är på proteinets yta
exponerade imidazolgrupper tillhörande aminosyran histidin som i första hand är ansvariga
för denna bindning. Bindningsförmågan bibehålls när metalljonen delvis kelateras, t.ex. till
en till en agarosgel bunden iminodiättiksyragrupp (IDA) (se Fig. 25). På detta faktum
bygger metoden att rena histidinhaltiga proteiner med hjälp av IMAC. I de allra flesta fall
fungerar IDA-substituerade geler mycket bra på grund av att man kan påverka
bindningsstyrkan genom att välja olika metalljoner. Således binder kopparjoner starkare än
zink- och nickeljoner. Som en tumregel prövar man först zinkjonmättade IDA-geler och
prövar sedan kopparjonen om bindningen är för svag.
Hög selektivitet kan ibland erhållas med nickeljonmättade geler. Ett speciellt fall av stor
betydelse är den upptäckt som Eric Hochuli och medarbetare vid Hoffman LaRoche gjorde
i mitten på 80-talet, nämligen att Ni2+-joner bundna till immobiliserade nitrilotriacetat-
grupper (NTA) (se Fig. 25 och läs referens 19), ej band ”normala” histidinhaltiga proteiner
men däremot rekombinanta proteiner som försetts med en s.k. ”His-tag”, dvs. En N- eller
C-terminalt fuserad oligo-His-peptid med 6-8 His-rester. Denna upptäckt gjorde det möjligt
att mycket effektivt, direkt från råextrakt av rekombinanta colibakterier, isolera de
rekombinanta proteinerna i mycket ren form. Det bör dock sägas att metoden inte fungerar i
samtliga system men bör alltid prövas som ett alternativ till traditionella reningsförfaranden
om förutsättningarna i övrigt är tillhanda.
Det bör även sägas att det inte alltid är nödvändigt att använda NTA-geler för att isolera
His-taggade proteiner. Ni2+-mättade IDA-geler (t.ex. Chelating Sepharose FF och andra)
fungerar lika bra i de flesta fall. För mer läsning rekommenderas kapitlet om IMAC av
Lennart Kågedal i referens (2).

29
Fig. 25 Strukturerna för immobiliserade IDA- och NTA-grupper.
6. REFERENSER
1. A. Tiselius, "Kemisk analys och biologisk struktur". Föredrag vid presidiets nedläggande den 29
sept. 1961, Kungl. Vetenskaps Societetens i Uppsala årsbok 1961.
2. J.-C. Janson, ed. "Protein Purification, Principles, High Resolution Methods and Applications", 3:rd
edition, John Wiley & Sons Inc., New York, 2011, pp. 1-517
3. J.-C. Janson, "Adsorption Phenomena on Sephadex", J. Chromatography, 28 (1967) 12-20.
4. Gel Filtration: Principles and Methods. Handbok från GE Healthcare Biosciences AB.
5. Expanded Bed Adsorption Handbook. Handbok från GE Healthcare Biosciences AB.
6. R. Westermeier, “Electrophoresis in Practice”, VCH Weinheim, 1993, 1-277.
7. P.G. Righetti and J.W. Drysdale, "Isoelectric focusing", paper back i serien "Laboratory Techniques in
Biochemistry and Molecular Biology (T.S. Work and E. Work), Vol. 5, part 2, North Holland,
Amsterdam 1976, 341-590.
8. P.G. Righetti, E. Gianazza and K. Ek, "New Developments in Isoelectric Focusing, J. Chromatography
184:4 (1980) 415-456.
9. Ion Exchange Chromatography: Principles and Methods. Handbok från GE Healthcare Biosciences
AB.
10. Chromatofocusing with PBE. Handbok från GE Healthcare Biosciences AB.
11. Affinity Chromatography: Principles and Methods. Handbok från GE Healthcare Biosciences AB.
12. C.R. Lowe, "An Introduction to Affinity Chromatography", paperback i serien "Laboratory Techniques
in Biochemistry and Molecular Biology (T.S. Work and E. Work), Vol 7, part 2, North Holland,

30
Amsterdam 1979, 269-522.
13. T. Kristiansen, "Matrix bound antigens and antibodies", i "Affinity Chromatography" (0. Hoffman-
Ostenhof et al.), Pergamon Press, Oxford 1978, 191-206.
14. P. Vretblad, "Lektiner - proteiner på frammarsch inom cellbiologin". Kemisk Tidskrift (1977), 36-38.
15. Hydrophobic Interaction Chromatography: Principles and Methods. Handbok från GE Healthcare
Biosciences AB.
16. J. Porath, "Metal chelate affinity Chromatography, a new approach to protein fractionation", Nature,
Vol 258, No 5536 (1975) 598-599.
17. J. Porath, "Explorations into the field of charge-transfer adsorption", J. Chromatography 159 (1978)
13-24.
18. B. Blombäck och L.A. Hansson, "Plasmaproteiner", AB Kabi och AWE/Gebers, Stockholm 1976, 1-
385.
19. E. Hochuli, W. Bannwart, H. Döbeli, R. Gentz and D. Stüber, Biotechnology Nov. 1988, 1321-1325.





























