Embed Size (px)
Citation preview

5
LA FISIOPATOLOGÍA DE LA HEMOSTASIA:ALGUNOS ASPECTOS SOBRE LA VIDA Y MUERTE DE
LAS PLAQUETAS EN LA CIRCULACION
Dr. Jaime Pereira G. (1)
(1) Profesor Titular, Departamento de Hematología y Oncología.Financiamiento: Fondecyt 1971024, 1990131, 1011010, 8010002, DIPUC 2006/10Correspondencia: [email protected]: 354 3772
RESUMEN
En las últimas dos décadas se ha progresado notablemente en el conocimiento de la fisiología de las plaquetas. Esta nueva información no solamente ha significado cambiar una serie de paradigmas, sino que también ha contribuido a un mejor entendimiento del papel de estas células en enfermedades hemorrágicas y trombóticas. Entre otros aspectos, han surgido nuevos hallazgos con respecto a los procesos de envejecimiento de las plaquetas en la circulación y la descripción de marcadores de edad plaquetaria; mecanismos de remoción desde la circulación y la demostración de que las plaquetas experimentan apoptosis durante su envejecimiento en la sangre. En cuanto a su participación en procesos patológicos, se describió el papel que juegan en la patogenia de los estados de hipercoagulabilidad adquiridos, como los que se observan en pacientes portadores de lupus eritematoso sistémico y en el uso crónico de cocaína.
Finalmente, la demostración de que las plaquetas son capaces de sintetizar y expresar factor tisular funcional, nos lleva a proponer un nuevo modelo de hemostasia basado en células. En éste, las plaquetas constituirían el punto de unión entre hemostasia primaria y secundaria y permitirían formular un modelo más racional para explicar los mecanismos hemostáticos en salud y enfermedad.
INTRODUCCIÓN
Las plaquetas son participantes esenciales en el proceso de hemostasia primaria. Además, aunque las plaquetas carecen de núcleo y podrían ser definidas como “trozos de citoplasma de los megacariocitos”, estas células contienen muchos componentes estructurales, metabólicos y de señalización propios de las células nucleadas. Incluso debido a su participación en diversos procesos fisiológicos y su accesibilidad, han servido como modelo de estudio en biología celular. Las plaquetas, que circulan normalmente en forma inactiva, se adhieren a la pared del vaso dañado, secretan el contenido de sus gránulos e interactúan con otras plaquetas, formando la base del tapón hemostático. Además, las plaquetas participan en la activación del sistema de la coagulación, proveyendo la superficie sobre la cual se ensamblan los complejos de este sistema (1). A la luz de nuestro trabajo experimental y clínico en torno a la fisiopatología plaquetaria, en el presente artículo haremos un recorrido por aquellos aportes más relevantes de nuestro grupo.
EL ENVEJECIMIENTO DE LAS PLAQUETAS EN LA CIRCULACION
Las plaquetas humanas después de ser liberadas por los megacariocitos de la médula ósea, permanecen en la circulación
durante 8 a 10 días, antes de su remoción
por parte de los macrófagos del sistema
reticuloendotelial (SRE) (2). Estudios de
cinética de plaquetas demostraron que
éstas son removidas de la sangre en función
de su edad. Durante su envejecimiento en
la circulación, las plaquetas sufren una
serie de cambios físicos, bioquímicos y
funcionales, que determinan en gran parte
su heterogeneidad. Este fenómeno fue
el primero en ser abordado por nuestro
laboratorio, enfocándonos principalmente
en aquellos cambios que pudieran constituir
marcadores de edad de las plaquetas (3-
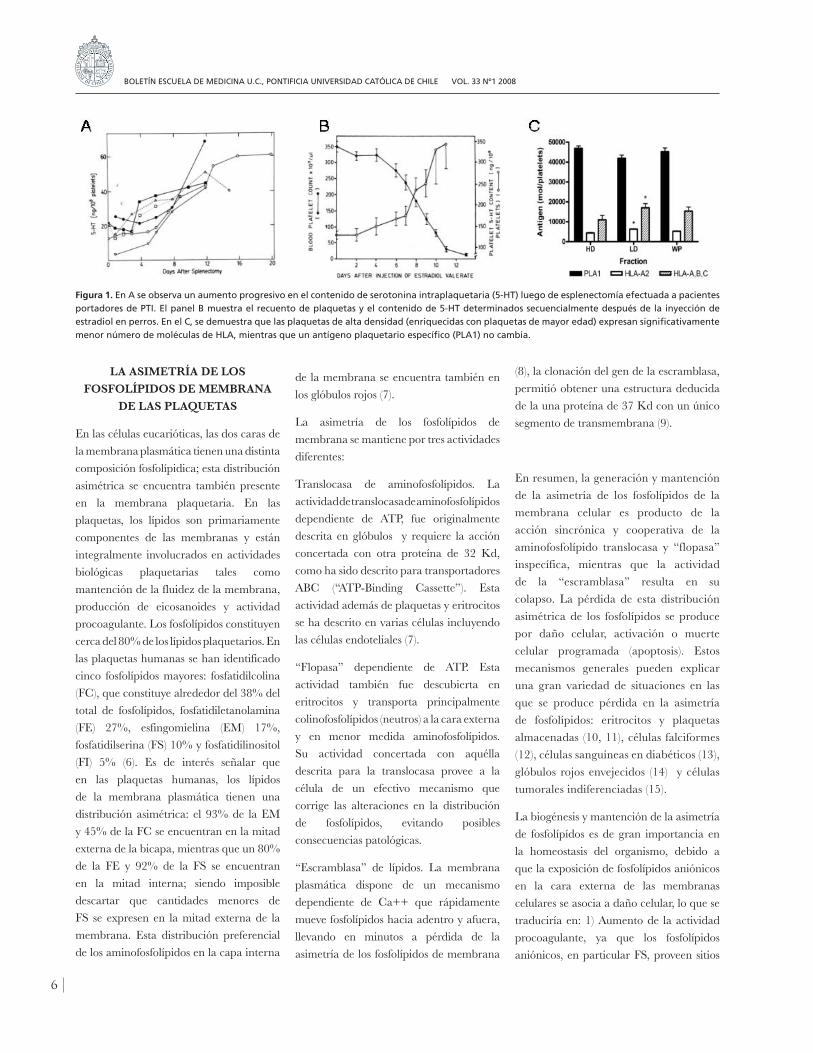
5) (figura 1). Una vez demostrado que
las plaquetas circulantes envejecen, la
siguiente interrogante que surgió fue: ¿Qué
determina, en condiciones fisiológicas, su
remoción desde la circulación al cabo de
8 a 10 días?. Debido a que las plaquetas
normales son retiradas de la circulación en
función de su edad, era lógico suponer que
durante el proceso de envejecimiento se
verificaban alteraciones estructurales y/o
bioquímicas que finalmente determinarían
la remoción al final de su vida útil. Con el
fin de contestar esta pregunta, iniciamos
estudios tendientes a investigar los cambios
plaquetarios durante su envejecimiento in
vivo que pudieran promover su retiro de la
circulación. En este sentido, la investigación
se enfocó en la pérdida de la asimetría de
los fosfolípidos de membrana, como un
mecanismo fundamental.
BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
6
LA ASIMETRÍA DE LOS FOSFOLÍPIDOS DE MEMBRANA
DE LAS PLAQUETAS
En las células eucarióticas, las dos caras de la membrana plasmática tienen una distinta composición fosfolípidica; esta distribución asimétrica se encuentra también presente en la membrana plaquetaria. En las plaquetas, los lípidos son primariamente componentes de las membranas y están integralmente involucrados en actividades biológicas plaquetarias tales como mantención de la fluidez de la membrana, producción de eicosanoides y actividad procoagulante. Los fosfolípidos constituyen cerca del 80% de los lípidos plaquetarios. En las plaquetas humanas se han identificado cinco fosfolípidos mayores: fosfatidilcolina (FC), que constituye alrededor del 38% del total de fosfolípidos, fosfatidiletanolamina (FE) 27%, esfingomielina (EM) 17%, fosfatidilserina (FS) 10% y fosfatidilinositol (FI) 5% (6). Es de interés señalar que en las plaquetas humanas, los lípidos de la membrana plasmática tienen una distribución asimétrica: el 93% de la EM y 45% de la FC se encuentran en la mitad externa de la bicapa, mientras que un 80% de la FE y 92% de la FS se encuentran en la mitad interna; siendo imposible descartar que cantidades menores de FS se expresen en la mitad externa de la membrana. Esta distribución preferencial de los aminofosfolípidos en la capa interna
de la membrana se encuentra también en los glóbulos rojos (7).
La asimetría de los fosfolípidos de membrana se mantiene por tres actividades diferentes:
Translocasa de aminofosfolípidos. La actividad de translocasa de aminofosfolípidos dependiente de ATP, fue originalmente descrita en glóbulos y requiere la acción concertada con otra proteína de 32 Kd, como ha sido descrito para transportadores ABC (“ATP-Binding Cassette”). Esta actividad además de plaquetas y eritrocitos se ha descrito en varias células incluyendo las células endoteliales (7).
“Flopasa” dependiente de ATP. Esta actividad también fue descubierta en eritrocitos y transporta principalmente colinofosfolípidos (neutros) a la cara externa y en menor medida aminofosfolípidos. Su actividad concertada con aquélla descrita para la translocasa provee a la célula de un efectivo mecanismo que corrige las alteraciones en la distribución de fosfolípidos, evitando posibles consecuencias patológicas.
“Escramblasa” de lípidos. La membrana plasmática dispone de un mecanismo dependiente de Ca++ que rápidamente mueve fosfolípidos hacia adentro y afuera, llevando en minutos a pérdida de la asimetría de los fosfolípidos de membrana
(8), la clonación del gen de la escramblasa, permitió obtener una estructura deducida de la una proteína de 37 Kd con un único segmento de transmembrana (9).
En resumen, la generación y mantención de la asimetría de los fosfolípidos de la membrana celular es producto de la acción sincrónica y cooperativa de la aminofosfolípido translocasa y “flopasa” inspecífica, mientras que la actividad de la “escramblasa” resulta en su colapso. La pérdida de esta distribución asimétrica de los fosfolípidos se produce por daño celular, activación o muerte celular programada (apoptosis). Estos mecanismos generales pueden explicar una gran variedad de situaciones en las que se produce pérdida en la asimetría de fosfolípidos: eritrocitos y plaquetas almacenadas (10, 11), células falciformes (12), células sanguíneas en diabéticos (13), glóbulos rojos envejecidos (14) y células tumorales indiferenciadas (15).
La biogénesis y mantención de la asimetría de fosfolípídos es de gran importancia en la homeostasis del organismo, debido a que la exposición de fosfolípidos aniónicos en la cara externa de las membranas celulares se asocia a daño celular, lo que se traduciría en: 1) Aumento de la actividad procoagulante, ya que los fosfolípidos aniónicos, en particular FS, proveen sitios
Figura 1. En A se observa un aumento progresivo en el contenido de serotonina intraplaquetaria (5-HT) luego de esplenectomía efectuada a pacientes portadores de PTI. El panel B muestra el recuento de plaquetas y el contenido de 5-HT determinados secuencialmente después de la inyección de estradiol en perros. En el C, se demuestra que las plaquetas de alta densidad (enriquecidas con plaquetas de mayor edad) expresan significativamente menor número de moléculas de HLA, mientras que un antígeno plaquetario específico (PLA1) no cambia.
7
de unión y ensamblaje de los complejos tenasa y protrombinasa, requeridos para la activación del factor X y protrombina respectivamente (7); 2) activación de complemento a través de la vía alterna (16) 3) reconocimiento y remoción de las plaquetas por parte de los macrófagos del SER, que poseen receptores para FS (17, 18).
En ese momento propusimos como hipótesis de trabajo que durante el envejecimiento de las plaquetas en la circulación se produciría pérdida de la asimetría de los fosofolípidos de membrana, lo que determinaría la remoción de las plaquetas de la circulación.
EL ENVEJECIMIENTO DE LAS PLAQUETAS EN LA CIRCULACIÓN
SE ASOCIA A PÉRDIDA DE LA ASIMETRÍA DE LOS
FOSFOLÍPIDOS DE MEMBRANA
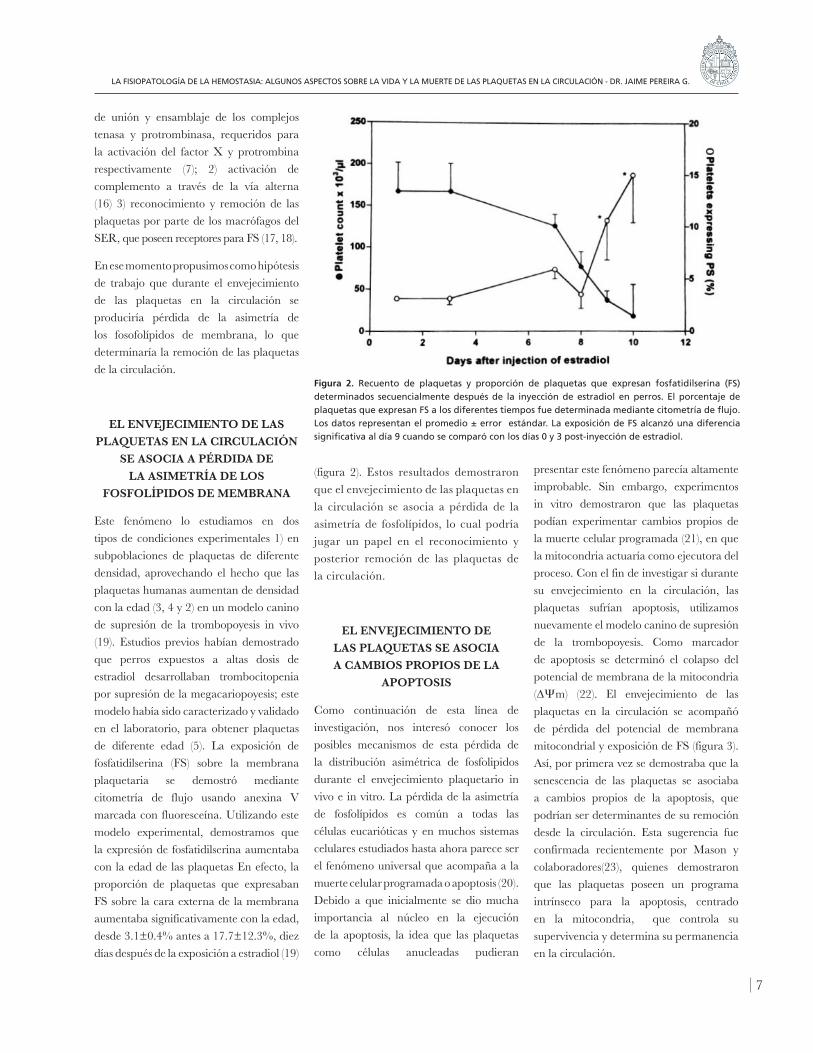
Este fenómeno lo estudiamos en dos tipos de condiciones experimentales 1) en subpoblaciones de plaquetas de diferente densidad, aprovechando el hecho que las plaquetas humanas aumentan de densidad con la edad (3, 4 y 2) en un modelo canino de supresión de la trombopoyesis in vivo (19). Estudios previos habían demostrado que perros expuestos a altas dosis de estradiol desarrollaban trombocitopenia por supresión de la megacariopoyesis; este modelo había sido caracterizado y validado en el laboratorio, para obtener plaquetas de diferente edad (5). La exposición de fosfatidilserina (FS) sobre la membrana plaquetaria se demostró mediante citometría de flujo usando anexina V marcada con fluoresceína. Utilizando este modelo experimental, demostramos que la expresión de fosfatidilserina aumentaba con la edad de las plaquetas En efecto, la proporción de plaquetas que expresaban FS sobre la cara externa de la membrana aumentaba significativamente con la edad, desde 3.1±0.4% antes a 17.7±12.3%, diez días después de la exposición a estradiol (19)
(figura 2). Estos resultados demostraron que el envejecimiento de las plaquetas en la circulación se asocia a pérdida de la asimetría de fosfolípidos, lo cual podría jugar un papel en el reconocimiento y posterior remoción de las plaquetas de la circulación.
EL ENVEJECIMIENTO DE LAS PLAQUETAS SE ASOCIA A CAMBIOS PROPIOS DE LA
APOPTOSIS
Como continuación de esta línea de investigación, nos interesó conocer los posibles mecanismos de esta pérdida de la distribución asimétrica de fosfolipidos durante el envejecimiento plaquetario in vivo e in vitro. La pérdida de la asimetría de fosfolípidos es común a todas las células eucarióticas y en muchos sistemas celulares estudiados hasta ahora parece ser el fenómeno universal que acompaña a la muerte celular programada o apoptosis (20). Debido a que inicialmente se dio mucha importancia al núcleo en la ejecución de la apoptosis, la idea que las plaquetas como células anucleadas pudieran
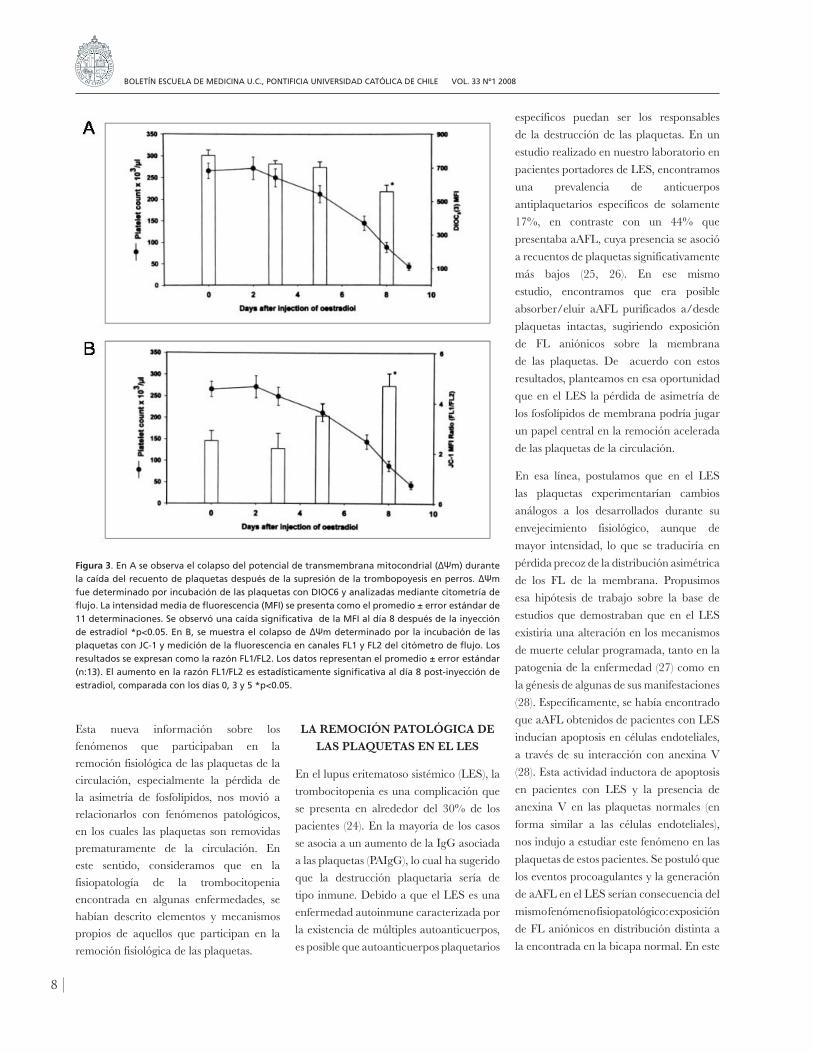
presentar este fenómeno parecía altamente improbable. Sin embargo, experimentos in vitro demostraron que las plaquetas podían experimentar cambios propios de la muerte celular programada (21), en que la mitocondria actuaría como ejecutora del proceso. Con el fin de investigar si durante su envejecimiento en la circulación, las plaquetas sufrían apoptosis, utilizamos nuevamente el modelo canino de supresión de la trombopoyesis. Como marcador de apoptosis se determinó el colapso del potencial de membrana de la mitocondria (∆Ψm) (22). El envejecimiento de las plaquetas en la circulación se acompañó de pérdida del potencial de membrana mitocondrial y exposición de FS (figura 3). Así, por primera vez se demostraba que la senescencia de las plaquetas se asociaba a cambios propios de la apoptosis, que podrían ser determinantes de su remoción desde la circulación. Esta sugerencia fue confirmada recientemente por Mason y colaboradores(23), quienes demostraron que las plaquetas poseen un programa intrínseco para la apoptosis, centrado en la mitocondria, que controla su supervivencia y determina su permanencia en la circulación.
Figura 2. Recuento de plaquetas y proporción de plaquetas que expresan fosfatidilserina (FS) determinados secuencialmente después de la inyección de estradiol en perros. El porcentaje de plaquetas que expresan FS a los diferentes tiempos fue determinada mediante citometría de flujo. Los datos representan el promedio ± error estándar. La exposición de FS alcanzó una diferencia significativa al día 9 cuando se comparó con los días 0 y 3 post-inyección de estradiol.
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.
BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
8
Esta nueva información sobre los fenómenos que participaban en la remoción fisiológica de las plaquetas de la circulación, especialmente la pérdida de la asimetría de fosfolípidos, nos movió a relacionarlos con fenómenos patológicos, en los cuales las plaquetas son removidas prematuramente de la circulación. En este sentido, consideramos que en la fisiopatología de la trombocitopenia encontrada en algunas enfermedades, se habían descrito elementos y mecanismos propios de aquellos que participan en la remoción fisiológica de las plaquetas.
LA REMOCIÓN PATOLÓGICA DE LAS PLAQUETAS EN EL LES
En el lupus eritematoso sistémico (LES), la trombocitopenia es una complicación que se presenta en alrededor del 30% de los pacientes (24). En la mayoría de los casos se asocia a un aumento de la IgG asociada a las plaquetas (PAIgG), lo cual ha sugerido que la destrucción plaquetaria sería de tipo inmune. Debido a que el LES es una enfermedad autoinmune caracterizada por la existencia de múltiples autoanticuerpos, es posible que autoanticuerpos plaquetarios
específicos puedan ser los responsables
de la destrucción de las plaquetas. En un
estudio realizado en nuestro laboratorio en
pacientes portadores de LES, encontramos
una prevalencia de anticuerpos
antiplaquetarios específicos de solamente
17%, en contraste con un 44% que
presentaba aAFL, cuya presencia se asoció
a recuentos de plaquetas significativamente
más bajos (25, 26). En ese mismo
estudio, encontramos que era posible
absorber/eluir aAFL purificados a/desde
plaquetas intactas, sugiriendo exposición
de FL aniónicos sobre la membrana
de las plaquetas. De acuerdo con estos
resultados, planteamos en esa oportunidad
que en el LES la pérdida de asimetría de
los fosfolípidos de membrana podría jugar
un papel central en la remoción acelerada
de las plaquetas de la circulación.
En esa línea, postulamos que en el LES
las plaquetas experimentarían cambios
análogos a los desarrollados durante su
envejecimiento fisiológico, aunque de
mayor intensidad, lo que se traduciría en
pérdida precoz de la distribución asimétrica
de los FL de la membrana. Propusimos
esa hipótesis de trabajo sobre la base de
estudios que demostraban que en el LES
existiría una alteración en los mecanismos
de muerte celular programada, tanto en la
patogenia de la enfermedad (27) como en
la génesis de algunas de sus manifestaciones
(28). Específicamente, se había encontrado
que aAFL obtenidos de pacientes con LES
inducían apoptosis en células endoteliales,
a través de su interacción con anexina V
(28). Esta actividad inductora de apoptosis
en pacientes con LES y la presencia de
anexina V en las plaquetas normales (en
forma similar a las células endoteliales),
nos indujo a estudiar este fenómeno en las
plaquetas de estos pacientes. Se postuló que
los eventos procoagulantes y la generación
de aAFL en el LES serían consecuencia del
mismo fenómeno fisiopatológico: exposición
de FL aniónicos en distribución distinta a
la encontrada en la bicapa normal. En este
Figura 3. En A se observa el colapso del potencial de transmembrana mitocondrial (ΔΨm) durante la caída del recuento de plaquetas después de la supresión de la trombopoyesis en perros. ΔΨm fue determinado por incubación de las plaquetas con DIOC6 y analizadas mediante citometría de flujo. La intensidad media de fluorescencia (MFI) se presenta como el promedio ± error estándar de 11 determinaciones. Se observó una caída significativa de la MFI al día 8 después de la inyección de estradiol *p<0.05. En B, se muestra el colapso de ΔΨm determinado por la incubación de las plaquetas con JC-1 y medición de la fluorescencia en canales FL1 y FL2 del citómetro de flujo. Los resultados se expresan como la razón FL1/FL2. Los datos representan el promedio ± error estándar (n:13). El aumento en la razón FL1/FL2 es estadísticamente significativa al día 8 post-inyección de estradiol, comparada con los días 0, 3 y 5 *p<0.05.

9
n Palquetas Anexina V(+)(%)
MP (nM FS)* ψ(FL1/FL2)
LES activo 16 6.2 + 1.7 0.48 + 0.13 1.1+ 0.2
LES inactivo 14 2.7 + 0.6 0.23 + 0.06 0.9 + 0.2
Controles 30 30 1.9 + 0.3 0.19 + 0.04 0.96 + 0.1
p *** < 0.03 < 0.02 < 0.05
Tabla 1. Marcadores de apoptosis en plaquetas de pacientes portadores de lupus eritematoso
sistémico (LES).
*Micropartículas cuantificadas en plasma mediante actividad procoagulante**Pérdida de potencial de transmembrana mitocondrial determionado por citometría de flujo*** Diferencia entre LES activos y controlespor analisis de variango
sentido, una de las alteraciones celulares que es propia de la apoptosis y que ha sido demostrada en plaquetas, es la generación de micropartículas desde la membrana, probablemente por escisión proteolítica de proteínas del citoesqueleto (21).
En las plaquetas circulantes de pacientes con LES activo, se demostró cambios propios de la apoptosis tales como pérdida de la asimetría de FL, colapso del potencial de membrana de la mitocondria y aumento en la generación de micropartículas (MPs) (29) (Tabla 1).
La pérdida de asimetría de FL y la generación de MPs son fenómenos característicos de las células que experimentan apoptosis; sin embargo, en las plaquetas también pueden presentarse como consecuencia de la activación inducida por distintos agonistas (30). Estas MPs expresan FL cargados negativamente, los cuales proveen una superficie procoagulante para el ensamblaje de los complejos enzimáticos de la coagulación (31, 32); además, existen datos recientes que identifican a las MPs como fuente de factor tisular (FT) funcionalmente activo, el principal iniciador de la coagulación in vivo (33, 34). Desde un punto de vista clínico, las MPs constituyen marcadores patogénicos,
ya que varias condiciones protrombóticas están asociadas a un aumento en el número de MPs circulantes (35-38).
En el LES y otras enfermedades del tejido conectivo se ha descrito circulación de plaquetas activadas y aumento en el número de MPs (39-43). Debido a que la mayoría de los estudios incluyó pacientes con síndrome antifosfolípidos primario o secundario, y con el fin de excluir el efecto de los anticuerpos antifosfolípidos sobre el estado procoagulante, investigamos en pacientes portadores de LES el número y características antigénicas de las MPs circulantes y su capacidad para aumentar la generación de trombina (44).
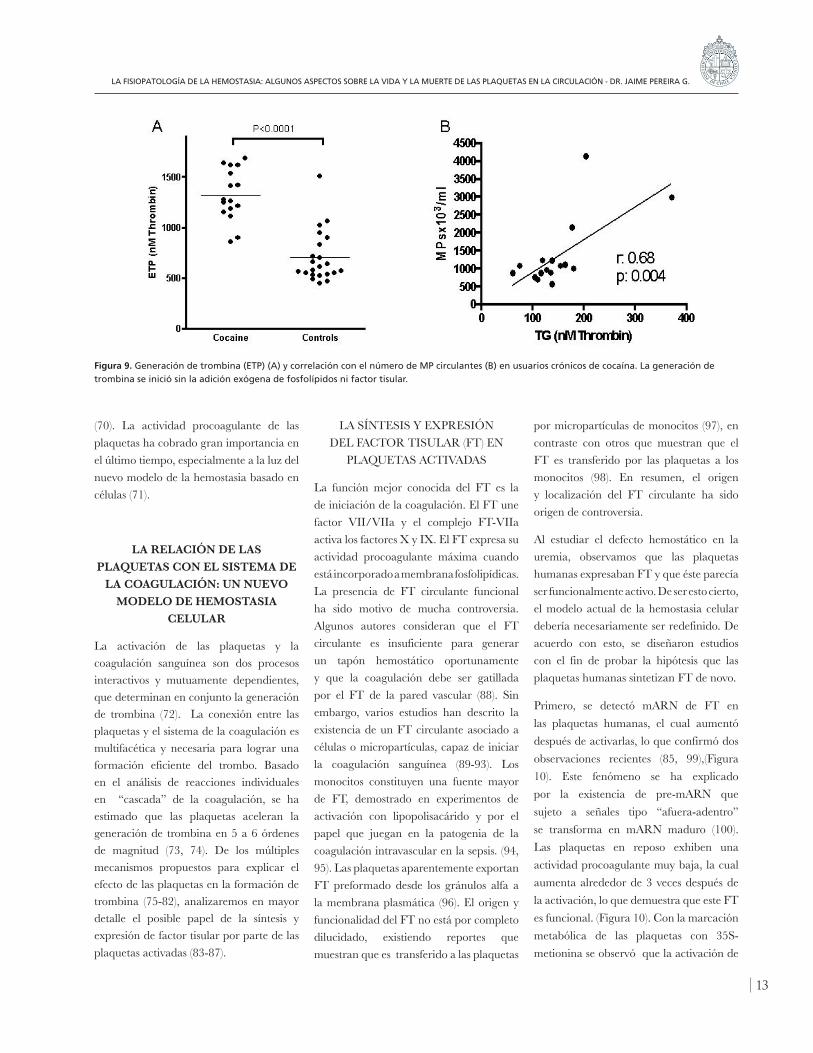
Se cuantificó el número y origen celular de los MPs circulantes y también la generación de trombina (GT) del plasma, medida como el Potencial Endógeno de Trombina (ETP), sin la adición de FL o factor tisular; bajo estas condiciones, la GT depende directamente del número de MPs presentes en el plasma de los pacientes. En la figura 4 se muestran los resultados de la cuantificación de MPs en pacientes y controles. Con respecto al origen celular de las MPs, encontramos que el 76% se originaba en las plaquetas. El ETP fue significativamente mayor en
los pacientes (figura 5) y se correlacionaba en forma directa con las MPs circulantes (figura 6). El aumento en el número de MPs derivadas de plaquetas y su asociación con un aumento del ETP, sugieren que estas MPs pueden jugar un papel importante en el estado protrombótico en esta patología (44).
De acuerdo a los resultados anteriores, parece evidente que las alteraciones plaquetarias en los pacientes con LES no se asocian a trombocitopenia sino más bien a un estado de hipercoagulabilidad adquirido. En este sentido, es importante considerar que los pacientes portadores de LES muestran un riesgo aumentado de presentar trombosis venosas y arteriales (45). Con respecto a las trombosis arteriales, los pacientes con LES tienen mayor predisposición para desarrollar arterioesclerosis acelerada y
Figura 4. Enumeración mediante citometria de flujo de las micropartículas circulantes (MP) en pacientes con LES inactivo o activo (MEX-SLEDAI >0). La enumeración de las MP totales (A) fue realizada usando anexina V-FITC; las MP derivadas de plaquetas (B) se cuantificaron usando doble marca con anexina V-FITC y anti-CD61-PE. Las líneas horizontales muestran la mediana; los cuadrados los rangos intercuartiles y las líneas verticales los valores altos y bajos. Las diferencias entre los grupos se analizaron mediante la prueba de Kruskall-Wallis.
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.

BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
10
enfermedad cardiovascular prematura (45, 46). De hecho, en estos pacientes el riesgo de presentar infarto de miocardio es 50 veces mayor que una población pareada por edad y sexo (47); este mayor riesgo es sólo parcialmente explicado por factores de riesgo clásicos como hipertensión, dislipidemia y diabetes (48). El LES es una enfermedad inflamatoria crónica y la inflamación actualmente se considera que juega un papel fundamental en la patogenia de la ateroesclerosis. Sin
embargo, los mecanismos que participan en el desarrollo de ateroesclerosis prematura en los pacientes con LES, son mayormente desconocidos. En la sangre de pacientes ateroscleróticos con angina inestable o enfermedad coronaria, se han detectado plaquetas activadas (49, 50), sugiriendo que la activación de las plaquetas podría participar en la aterotrombosis. En efecto, se ha demostrado que la infusión de plaquetas activadas exacerba la formación de lesiones ateroescleróticas en ratones deficientes en apolipoproteína E (51), los cuales desarrollan lesiones AE prematuras secundarias a la dislipidemia. Estas observaciones establecieron las bases para una serie de investigaciones, especialmente in vitro y en modelos animales, que demostrarían el papel que juegan las plaquetas en la génesis y progresión de la aterosclerosis.
EL PAPEL DE LAS PLAQUETAS EN LA ATEROESCLEROSIS
Aparte de sus conocidas funciones en los procesos de hemostasia y trombosis, las plaquetas participan directamente en la patogenia de la aterosclerosis y representan un punto de unión entre inflamación y aterogénesis (52, 53).
LA ADHESIÓN DE LAS PLAQUETAS AL ENDOTELIO
Evidencia reciente ha demostrado que la denudación endotelial no es un requisito único para permitir la unión de las plaquetas a la pared arterial. El endotelio previene la reactividad de las plaquetas a través de mecanismos de inhibición tales como la liberación de prostaciclina, óxido nítrico (NO) y expresión de una ecto-ADPasa (CD39). Sin embargo, las células endoteliales dañadas y/o activadas son adhesivas para las plaquetas. Estudios in vitro han demostrado que la unión de las plaquetas al endotelio activado es
dependiente de la glicoproteína (GP) IIb/IIIa, utilizando como proteínas puente el fibrinógeno, fibronectina y FvW (54). Evidencia adicional ha mostrado también la participación de receptores de las células endoteliales tales como ICAM-1 y αvβ3. Estudios in vivo demuestran que bajo condiciones de fuerza de cizalla alta, las plaquetas son capaces de adherirse al endotelio intacto en un proceso que comprende varias etapas. El contacto inicial laxo entre las plaquetas y las células endoteliales está mediado por selectinas presentes en ambas células. La P-selectina es rápidamente expresada por las CE frente a una variedad de estímulos. El contrarreceptor de la P-selectina en las plaquetas sería la glicoproteina GPIb-V-IX, que posteriormente se complementa con la PSGL-1. La unión definitiva de las plaquetas al endotelio se completa por la interacción entre proteínas adhesivas (por ej., fibrinógeno) y la GPIIb/IIIa (Figura 7).
LAS PLAQUETAS UNIDAS AL ENDOTELIO ESTIMULAN LA
INFLAMACIÓN
Durante el proceso de adhesión, las plaquetas se activan y liberan numerosas sustancias proinflamatorias y mitogénicas al micromedioambiente local. Entre las más importantes se destaca a las proteínas adhesivas (fibrinógeno, FvW, trombospondina, P-selectina); factores de crecimiento (PDGF, TGF-β, EGF); quimioquinas (RANTES, FP4); citoquinas (IL-1β, CD40L, β-tromboglobulina) y factores de la coagulación (Factor V, Factor XI, PAI) (55). La acción concertada y regulada de todas estas proteínas promueve una serie de funciones biológicas tales como adhesión celular, quimiotaxis, proliferación celular, coagulación y proteolisis. La IL1-β y el marcador CD40L sobre la superficie de las plaquetas han demostrado ser potentes activadores de las células endoteliales, promoviendo la expresión de moléculas de adhesión (ICAM-1, αvβ3) y la secreción
Figura 6. Correlación entre la capacidad de generar trombina del plasma libre de plaquetas y las MP totales (A) y aquéllas derivadas de plaquetas (B). El análisis se hizo mediante prueba de “correlación por intervalos según Spearman.
Figura 5. Potencial endógeno de trombina (ETP) en pacientes con LES y controles. La generación de trombina se inició sin la adición exógena de fosfolípidos ni factor tisular. La diferencia entre pacientes y controles se analizó con prueba de t de Mann Whitney.

11
de MCP-1, que juega un papel clave en el reclutamiento de monocitos. La acción conjunta de las quimioquinas liberadas por las plaquetas, la presencia de plaquetas activadas y la expresión de moléculas de adhesión sobre las células endoteliales, se traduce finalmente en reclutamiento de neutrófilos y monocitos sobre la superficie endotelial, uno de los fenómenos claves en la iniciación del proceso ateroesclerótico (55).
De acuerdo con las observaciones anteriores, parece evidente que las plaquetas son cruciales en la iniciación, desarrollo y extensión total de las lesiones ateroescleróticas. Sin embargo, gran parte de la evidencia disponible proviene de estudios in vitro o en modelos animales. En este sentido, fue importante encontrar un modelo humano en el que exista daño vascular y ateroesclerosis acelerada, en ausencia de factores de riesgo clásico: es la situación que se observa en los usuarios de cocaína.
EL USO DE COCAÍNA Y DAÑO VASCULAR
Los usuarios crónicos de cocaína tienen un riesgo aumentado de presentar infarto de miocardio, angina, muerte súbita, y accidentes cerebrovasculares, así como
Figura 7. Adhesión de las plaquetas al endotelio. Las células endoteliales activadas expresan p-selectina que constituye un ligando para la GPIb y su receptor (PSGL-1) presentes sobre la superficie de las plaquetas. La interacción inicial es posteriormente reforzada en las plaquetas activadas por la participación de las integrinas (GPIIb/IIIa y αvβ3).
defectos regionales de perfusión cerebral. La patogenia de las lesiones vasculares isquémicas asociadas al uso crónico de cocaína no se ha dilucidado completamente, existiendo varios mecanismos posibles, entre los que destacan: 1) vasoconstricción 2) aumento del consumo de oxígeno y 3) trombogénesis.
Los efectos sobre el tono vasomotor se relacionan a las propiedades simpaticomiméticas de la cocaína; sin embargo, varios estudios concluyen que este efecto no explica completamente la isquemia cerebral y cardíaca. En este sentido, existe evidencia que muestra que la activación de la hemostasia y la trombogénesis asociada participan en la patogenia de estas complicaciones. La demostración de activación de las plaquetas y la evidencia morfológica de formación de trombos arteriales, apoyan el concepto de que en el abuso de cocaína una activación del sistema hemostático, especialmente de sus componentes celulares, juega un papel importante en la isquemia inducida por esta droga. Prácticamente todos estos trabajos estudiaron los efectos de la exposición in vitro de plaquetas a la droga o a infusión e.v. aguda en voluntarios sanos y no se había abordado el sistema hemostático en forma global, considerando sus elementos
celulares, humorales y la interacción entre ellos. En esta línea, en el modelo celular actual de hemostasia se establece que la activación de las plaquetas y la coagulación sanguínea son procesos interactivos y dependientes, que determinan en conjunto la generación de trombina.
LA TROMBOGÉNESIS Y ATEROSCLEROSIS ACELERADA
ASOCIADAS AL USO DE COCAÍNA
Diferentes estudios y casos reportados han demostrado claramente un aumento en la formación de trombos arteriales en usuarios de cocaína (56-58); sin embargo, los estudios in vitro e in vivo sobre sus mecanismos han entregado resultados controvertidos (56, 59-61, 62-64). Minor y cols. (56) en un estudio en pacientes con infarto agudo del miocardio asociado al uso de cocaína, encontraron que un 38% presentaba coronarias sanas, pero en alrededor del 80% había evidencia angiográfica de trombos intracoronarios. A continuación, se discute la evidencia disponible en cuanto al efecto de la cocaína sobre las plaquetas y los vasos sanguíneos, que permite sostener un papel patogénico de la activación del sistema hemostático en la aterotrombosis inducida por cocaína.
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.

BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
12
EL EFECTO DE LA COCAÍNA SOBRE LAS PLAQUETAS
Uno de los primeros estudios que abordó el problema fue el de Eichhorn y cols. (64) que demostró que segmentos de aorta de conejo expuestos in vitro a cocaína presentaban aumento en la producción de tromboxano A2, el cual es un potente agregante plaquetario. La incubación directa de las plaquetas in vitro con cocaína ha mostrado resultados muy variados. Se ha encontrado aumento significativo de la respuesta plaquetaria a ácido araquidónico (62), ausencia de efecto significativo sobre su función o incluso, inhibición de la agregación a diferentes concentraciones de ADP y ácido araquidónico (59).
La expresión de p-selectina (CD62-P), proteína integral de la membrana de los gránulos alfa que se trasloca a la superficie en las plaquetas activadas, se ha utilizado más recientemente como marcador del efecto de la cocaína sobre las plaquetas. Rinder y cols. (61) estudiando voluntarios sanos, demostraron que sólo la exposición crónica -pero no la aguda- a cocaína aumentaba significativamente la expresión de p-selectina sobre la membrana plaquetaria. Usando una aproximación experimental similar, Kugelmass (60) demostró que la incubación in vitro de
plaquetas lavadas con cocaína aumentaba la expresión de p-selectina, el cual no era inhibible por aspirina. Más recientemente, Heesch (65) usando un modelo de administración aguda de cocaína en voluntarios sanos, observó un aumento en la expresión de p-selectina, aumento en la liberación de factor plaquetario 4 y β-tromboglobulina (proteínas de gránulos α) y presencia de microagregados de plaquetas en la circulación. En esta misma línea, Siegel reportó un aumento en el número de eritrocitos y del factor von Willebrand después de exposición a una dosis única de cocaína en usuarios crónicos (66). Zurbano en un modelo en cerdos, demostró que la exposición aguda a cocaína se asociaba a un aumento significativo de la interacción de las plaquetas con estructuras subendoteliales (67).
Algunos estudios que sugieren que la cocaína, bajo ciertas condiciones, puede afectar la función de las plaquetas en forma negativa. En este sentido, Jennings observó que la cocaína producía una inhibición de la agregación plaquetaria inducida por araquidonato, ADP y colágeno, (68); un efecto similar fue reportado por Heesch (65).
Por lo tanto, usando diferentes modelos experimentales y formas de exposición a la droga, es evidente que la cocaína se
asocia a una activación de las plaquetas, especialmente en los estudios de administración a droga in vivo. Debido a la falta de información sobre el estado de activación de las plaquetas en usuarios crónicos de cocaína, sus mecanismos de producción y especialmente su relación con los otros componentes del sistema hemostático, se inició en nuestro laboratorio un estudio con el fin de abordar estos objetivos.
En usuarios crónicos de cocaína se observó un aumento en el número de micropartículas circulantes, de los agregados monocito-plaquetas y de la expresión de p-selectina plaquetaria (figura 8) (69, 70). El aumento en la expresión de estos tres marcadores demuestra activación de las plaquetas in vivo, lo cual podría jugar un papel en el daño vascular prematuro observado en usuarios de cocaína.
Por otra parte, la presencia de plaquetas activadas en la circulación no solamente puede ser importante en la generación y progresión del daño vascular sino también en la generación de un estado protrombótico (figura 9). Esta condición de hipercoagulabilidad es clave en la patogenia de las complicaciones vasculares propias de estos pacientes, tales como infarto de miocardio o defectos de perfusión cerebral
Figura 8. En los pacientes usuarios de cocaína se encontró una activación significativa de las plaquetas circulantes, evidenciada por un aumento de los agregados monocito-plaquetas (A) y de la p-selectina de superficie (B). Las determinaciones se hicieron mediante citometría de flujo de sangre periférica de los pacientes y controles.
13
(70). La actividad procoagulante de las plaquetas ha cobrado gran importancia en el último tiempo, especialmente a la luz del nuevo modelo de la hemostasia basado en células (71).
LA RELACIÓN DE LAS PLAQUETAS CON EL SISTEMA DE
LA COAGULACIÓN: UN NUEVO MODELO DE HEMOSTASIA
CELULAR
La activación de las plaquetas y la coagulación sanguínea son dos procesos interactivos y mutuamente dependientes, que determinan en conjunto la generación de trombina (72). La conexión entre las plaquetas y el sistema de la coagulación es multifacética y necesaria para lograr una formación eficiente del trombo. Basado en el análisis de reacciones individuales en “cascada” de la coagulación, se ha estimado que las plaquetas aceleran la generación de trombina en 5 a 6 órdenes de magnitud (73, 74). De los múltiples mecanismos propuestos para explicar el efecto de las plaquetas en la formación de trombina (75-82), analizaremos en mayor detalle el posible papel de la síntesis y expresión de factor tisular por parte de las plaquetas activadas (83-87).
Figura 9. Generación de trombina (ETP) (A) y correlación con el número de MP circulantes (B) en usuarios crónicos de cocaína. La generación de trombina se inició sin la adición exógena de fosfolípidos ni factor tisular.
LA SÍNTESIS Y EXPRESIÓN DEL FACTOR TISULAR (FT) EN
PLAQUETAS ACTIVADAS
La función mejor conocida del FT es la de iniciación de la coagulación. El FT une factor VII/VIIa y el complejo FT-VIIa activa los factores X y IX. El FT expresa su actividad procoagulante máxima cuando está incorporado a membrana fosfolipídicas. La presencia de FT circulante funcional ha sido motivo de mucha controversia. Algunos autores consideran que el FT circulante es insuficiente para generar un tapón hemostático oportunamente y que la coagulación debe ser gatillada por el FT de la pared vascular (88). Sin embargo, varios estudios han descrito la existencia de un FT circulante asociado a células o micropartículas, capaz de iniciar la coagulación sanguínea (89-93). Los monocitos constituyen una fuente mayor de FT, demostrado en experimentos de activación con lipopolisacárido y por el papel que juegan en la patogenia de la coagulación intravascular en la sepsis. (94, 95). Las plaquetas aparentemente exportan FT preformado desde los gránulos alfa a la membrana plasmática (96). El origen y funcionalidad del FT no está por completo dilucidado, existiendo reportes que muestran que es transferido a las plaquetas
por micropartículas de monocitos (97), en contraste con otros que muestran que el FT es transferido por las plaquetas a los monocitos (98). En resumen, el origen y localización del FT circulante ha sido origen de controversia.
Al estudiar el defecto hemostático en la uremia, observamos que las plaquetas humanas expresaban FT y que éste parecía ser funcionalmente activo. De ser esto cierto, el modelo actual de la hemostasia celular debería necesariamente ser redefinido. De acuerdo con esto, se diseñaron estudios con el fin de probar la hipótesis que las plaquetas humanas sintetizan FT de novo.
Primero, se detectó mARN de FT en
las plaquetas humanas, el cual aumentó
después de activarlas, lo que confirmó dos
observaciones recientes (85, 99),(Figura
10). Este fenómeno se ha explicado
por la existencia de pre-mARN que
sujeto a señales tipo “afuera-adentro”
se transforma en mARN maduro (100).
Las plaquetas en reposo exhiben una
actividad procoagulante muy baja, la cual
aumenta alrededor de 3 veces después de
la activación, lo que demuestra que este FT
es funcional. (Figura 10). Con la marcación
metabólica de las plaquetas con 35S-
metionina se observó que la activación de
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.

BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
14
las plaquetas se acompañó de síntesis de novo de factor tisular (figura 11). La síntesis de proteínas por las plaquetas anucleadas fue sugerida desde hace dos décadas (101), pero sólo recientemente se documentó la síntesis de novo de Bcl-3, interleuquina-1β
y PAI-1 (102-104).
de los complejos de la coagulación sobre su
membrana, pero le demanda una fuente
adicional de FT tisular para gatillar las
reacciones (81). Las plaquetas humanas,
como células sintetizadoras de FT, podrían
constituir estas “células portadoras de FT”
asegurando la localización del FT en el
lugar correcto y en el momento apropiado.
Esta exposición rápida, localización
espacial precisa y depósito progresivo
y ordenado de FT sobre la membrana
plaquetaria, configura un modelo más
racional para explicar los mecanismos
hemostáticos en salud y enfermedad (figura
12). Este concepto resalta el papel único de
las plaquetas para unificar la hemostasia
primaria y secundaria, enfatizando la
autosuficiencia de los componentes
intravasculares para satisfacer todas
las necesidades hemostáticas (87). Por
otra parte, esta nueva concepción de la
hemostasia podría ampliar las posibilidades
de estudio en enfermedades hemorrágicas
y trombóticas, en las que en alrededor del
50% de los casos no es posible demostrar
alteraciones de laboratorio con las pruebas
actualmente disponibles (105).
Figura 10. mARN de factor tisular en plaquetas humanas. RNA de plaquetas se extrajo antes y después de estimular con 5µM de TRAP por 15 minu-tos. En A, amplificación de los transcritos de RNA en plaquetas. En este experimento se detectó mRNA de FT sólo después de la estimulación de las plaquetas (línea 6). En B se muestra la actividad procoagulante de las plaquetas (PCA) después de la estimulación con TRAP. Las plaquetas en reposo y activadas, se incubaron con FX y FVIIa, la generación de FXa se determinó mediante sustrato cromogénico. Se observa ausencia de efecto de la puromicina (inhibidor de la traducción).
Figura 11. Plaquetas con o sin pretratamiento con puromicina se incubaron con [35S]-metionina, y se activaron con TRAP. Las membranas de las plaquetas se inmunoprecipitaron con un anticuerpo anti-FT policlonal. Se observa un aumento significativo de la banda de FT después de activar las plaquetas (línea 3), la cual es reducida por la puromicina (línea 4).
AGRADECIMIENTOS
A Diego Mezzano, mentor, colaborador y amigo, que me contagió su vocación por la investigación. A los estudiantes Iván Palomo, Mónica Soto, Lindi Marie Cotzee y Gino Alfaro que con sus trabajos de tesis generaron muchos de los datos mostrados. A Isabel Pizarro, Patricia Hidalgo, Eduardo Aranda, bioquímicos y tecnólogos médicos del Laboratorio de Hemostasia.
REFERENCIAS
1. PEREIRA J. Estructura, producción, cinética y función de las plaquetas. En: Mezzano D, Pereira J. (Eds). Fisiología de la Sangre. Ediciones Universidad Católica de Chile, Santiago, 1993. Pag. 145-176.
2. GEORGE JN, DALE GL. Platelet kinetics. En: Beutler E, Lichtman MA, Coller BS, Kipps TJ. Hematology, McGraw-Hill, Inc. New York, 1995. Pag. 1202-1205.
3. MARTIN J, TROWBRIDGE A. (Eds). Platelet heterogeneity. Biology and pathology. Springler-Verlag, London 1990.
El paradigma actual del sistema
hemostático basado en células asigna a las
plaquetas un papel central en el ensamblaje

15
Figura 12. Nuevo modelo de la hemostasia basado en células. Se propone que la “TF bearing cell” presente en el subendotelio es fundamental en el inicio de la hemostasia. Sin embargo, el crecimiento y progresión del proceso hemostático, depende de la expresión de FT sobre la superficie de las plaquetas activadas. De esta forma, el proceso modulado en la generación de trombina y el depósito de fibrina sobre la membrana plaquetaria, semejante a la acción de “cemento sobre los ladrillos”, podría construir un tapón hemostático o un trombo.
4. PEREIRA J, CRETNEY C, ASTER RH. Variation of class I HLA antigen expression among platelet density cohorts: a possible index of platelet age? Blood 71: 516-519, 1988.
5. ARANDA E, PIZARRO M, PEREIRA J, MEZZANO D.. Accumulation of platelet 5-hydroxytryptamine by aging platelets: studies in a model of supressed thrombopoiesis in dogs. Thromb Haemostas 71: 488-492, 1994.
6. SCHICK PK. Megakaryocyte and platelet lipids. En: Colman RW, Hirsh J, Marder VJ, Salzman EW. Hemostasis and Thrombosis: Basic principles and clinical
practice. J.B. Lippincott Co., Philadelphia, 1994. Pag. 574-589.
7. ZWAAL RFA, SCHROIT AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89: 1121-1132, 1997.
8. SMEETS EF, COMFURIUS P, BEVERS EM, ZWAAL RFA. Calcium-induced transbilayer scrambling of fluorescent phosholipid analogs in platelets and erythrocytes. Biochim Biophys Acta 1195:281-286, 1994.
9. ZHOU Q, ZHAO J, STOUT JG, LUHM RA, WIEDMER T, SIMS PJ.
Molecular cloning of human plasma membrane phospholipid scramblase. J Biol Chem 272: 18240-18244, 1997.
10. GELDWERTH D, KUYPERS FA, BUTIKOFER P, ALLARY M, LUBIN BH, DEVAUX PF. Transbilayer mobility and distribution of red cell phospholipids during storage. J Clin Invest 93: 92-96, 1993.
11. GAFFET P, BASSÉ F, BIENVENÜE A. Loss of phospholipid asymmetry in human platelet plasma membrane after 1-12 days of storage. Eur J Biochem 222: 1033-1040, 1994.
12. TAIT JF, GIBSON D. Measurement
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.

BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
16
of membrane phospholipid asymmetry in normal and sickle-cell erythrocytes by means of annexin V binding. J Lab Clin Med 123: 741-748, 1994.
13. WILSON MJ, RICHTER-LOWNEY K, DALEKE DL. Hyperglycemia induces a loss of phospholipid asymmetry in human erythrocytes. Biochemistry 32: 11302-11310, 1993.
14. CONNOR J, PAK CH, SCHROIT AJ. Exposure of phosphatidylserine in the outer leaflet of human red blood cells: Relationship to cell density, cell age and clearence by mononuclear cells. J Biol Chem 269: 2399-2404, 1994.
15. UTSUGI T, SCHROIT AJ, CONNOR J, BUCANAN CD, FIDLER IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res 51: 3062-3066, 1991.
16. TEST ST, MITSUYOSHI J. Activation of the alternative pathway of complement by calcium-loaded erythrocytes resulting from loss of membrane phospholipid asymmetry. J Lab Clin Med 130: 169-182, 1997.
17. SAVILL J, FADOK V,HENSON P, HASSLET C. Phagogyte recognition of cells undergoing apoptosis. Immunol Today 14: 131-136, 1993.
18. SCHROIT AJ, MADSEN JW, TANAKA Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membrane. J Biol Chem 260: 5131-5138, 1985.
19. PEREIRA J, PALOMO I, OCQUETEAU M, SOTO M, ARANDA E, MEZZANO D. Platelet aging in vivo is associated with loss of membrane phospholipid asymmetry. Thromb Haemost 1999; 82: 1318-1321.
20. HALE AJ, SMITH CA, et al. Apoptosis: molecular regulation of cell death. Eur J Biochem 236: 1-26, 1996.
21. VANAGS DM, ORRENIUS S, AGUILAR-SANTELISES M. Alterations in Bcl-2/Bax protein levels in platelets form part of an ionomycin-induced process that resembless apoptosis. Br J Haematol 99: 824-831, 1997.
22. PEREIRA J, SOTO M, PALOMO I, OCQUETEAU M, COETZEE LM, ASTUDILLO S, ARANDA E, MEZZANO D. Platelet aging in vivo is associated with activation of apoptotic pathways: studies in a model of suppressed thrombopoiesis in dogs. Throm Haemost 2002; 87: 905-909.
23. MASON K, CARPINELLI MR, FLETCHER JI, et al D. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173-1186.
24. LAURENCE J, NACHMAN R: Hematologic aspects of systemic lupus erythematosus. In Lahita RG Ed.. Systemic lupus erythematosus, New York, John Wiley & Sons Inc, 1987.
25. PEREIRA J, PALOMO I, JACOBELLI S, et al. Anticuerpos antiplaquetarios en pacientes con lupus eritematoso sistémico: asociación con anticuerpos antifosfolípido. X Congreso Chileno de Hematología, Soc Chilena de Hematología, Santiago 1994. Libro de Resúmenes, pág 76.
26. PALOMO I, PEREIRA J, ALARCÓN M, et al. Antiphospholipid antibodies in Chilean patients with systemic lupus erythematosus. J Lab Clin Med. 2002; 140: 336-341.
27. KOTZIN BL. Systemic lupus erythematosus Cell 85: 303-306, 1996.
28. NAKAMURA N, BAN T, YAMAJI K, et al. Localization of the apoptosis-inducing activity of lupus anticoagulant in an annexin V binding antibody subset. J Clin Invest 101: 1951-1959, 1998.
29. PEREIRA J, QUIROGA T,
MASSARDO L, et al. Loss of Platelet
Membrane Phospholipid Asymmetry in
Systemic Lupus Erythematosus: Association
with Disease Activity and Hypercoagulable
State. Blood 2004; 104: 709a.
30. LEYTIN V, ALLEN DJ, MYKHAYLOV
S, et al. Thrombin-triggered platelet
apoptosis. J Thromb Haemost. 2006; 4:
2656-2663.
31.ZWAAL RFA, SCHROIT AJ.
Pathophysiologic implications of
membrane phopholipid asymmetry in
blood cells. Blood 1997; 89: 1121-1132.
32. LEUNG R, GWOZDZ AM, WANG
H, et al. Persistence of procoagulant
surface expression on activated human
platelets: involvement of apoptosis and
aminophospholipid translocase activity. J
Thromb Haemost. 2007; 5: 560-570.
33. MÜLLER I, KLOCKE A, ALEX M,
et al. Intravascular tissue factor initiates
coagulation via circulating microvesicles
and platelets. FASEB J 2003; 17: 476-478.
34. BIRÓ E, STURK-MAQUELIN
KN, VOGEL GMT, et al. Human cell-
derived microparticles promote thrombus
formation in vivo in a tissue factor-
dependent manner. J Thromb Haemost
2003; 1: 2561-2568.
35. WARKENTIN TE, HAYWARD CP,
BOSHKOV LK, et al. Sera from patients
with heparin-induced thrombocytopenia
generate platelet-derived microparticles
with procoagulant activity: an explanation
for the thrombotic complications of
heparin-induced thrombocytopenia. Blood
1994; 84: 3691-3699.
36. COMBES V, SIMON AC, GRAU GE,
et al. In vitro generation of endothelial
microparticles and possible prothrombotic
activity in patients with lupus anticoagulant.
J Clin Invest 1999; 104: 93-102.

17
37. MALLAT Z, BENAMER H, HUGEL B, et al. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000; 101: 841-843.
38. BRETELLE F, SABATIER F, DESPREZ D, et al. Circulating microparticles: a marker of procoagulant state in normal pregnancy and pregnancy complicated by preeclampsia or intrauterine growth restriction. Thromb Haemost 2003; 89: 486-492.
39. NAGAHAMA M, NOMURA S, OZAKI Y, YOSHIMURA C, KAGAWA H, S. Platelet activation markers and soluble adhesion molecules in patients with systemic lupus erythematosus. Autoimmunity 2001; 33: 85-94.
40. JOSEPH JE, HARRISON P, MACKIE IJ, ISENBERG DA, MACHIN SJ. Increased circulating platelet-leukocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol 2001; 115: 451-459.
41. KNIJFF-DUTMAR EAJ, KOERTZ J, NIEUWLAND R, KALSBEEK-BATENBURG EM, VAN DE LAAR MAFJ. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum 2002; 46: 1498-1503.
42. ABID HUSSEIN MN, MEESTERS EW, OSMANOVIC N, ROMIJN FPHTHM, NIEUWLAND R, STURK A. Antigenic characterization of endothelial cell-derived microparticles and their detection ex vivo. J Thromb Haemost 2003; 1: 2434-2443.
43. EKDAHL KN, BENGTSSON AA, ANDERSSON J, et al. Thrombotic disease in systemic lupus erythematosus is associated with maintained systemic platelet activation. Br J Haematol 2004; 125: 74-78.
44. PEREIRA J, ALFARO G,
GOYCOOLEA M, et al. Circulating
platelet-derived microparticles in systemic
lupus erythematosus: association with
increased thrombin generation and
procoagulant state. Thromb Haemost
2006; 95: 94-99.
45. RUIZ-IRASTORZA G,
KHAMASHTA MA, CASTELLINO
G, HUGHES GR. Systemic lupus
erythematosus.Lancet. 2001; 357: 1027-
1032.
46. SALMON JE, ROMAN MJ.
Accelerated atherosclerosis in systemic
lupus erythematosus: implication for patient
management. Curr Opin Rheumatol 2001;
13: 341-344.
47. MANZI S, MEILAHN EN, RAIRIE
JE, et al. Age-specific incidence rates
of myocardial infarction and angina in
women with systemic lupus erythematosus:
comparison with the Framingham Study.
Am J Epidemiol. 1997; 145: 408-15.
48. ESDAILE JM, ABRAHAMOWICZ
M, GRODZICKY T, et al. Traditional
Framingham risk factors fails to fully
account for accelerated atherosclerosis in
systemic lupus erythematosus. Arthritis
Rheum 2001; 44: 2331-2337.
49. FITZGERALD DJ, ROY L, CATELLA
F, FITZGERALD GA. Platelet activation
in unstable coronary disease. N Engl J Med
1986; 315: 983-989.
50. FURMAN MI, BENOIT SE,
BARNARD MR, et al. Increased platelet
reactivity and circulating monocyte-platelet
aggregates in patients with stable coronary
artery disease. J Am Coll Cardiol. 1998;
31: 352-358.
51. HUO Y, SCHOBER A, FORLOW
SB, et al. Circulating activated platelets
exacerbate atherosclerosis in mice
deficient in apolipoprotein E. Nat Med
2003; 9: 61-67.
52. GAWAZ M. Platelets in the onset of atherosclerosis. Blood Cells Mol Dis 2006; 36: 206-210.
53. LINDEMANN S, KRÄMER B, DAUB K, STELLOS K, GAWAZ M. Molecular pathways used by platelets to initiate and accelerate atherogenesis. Curr Opin Lipidol 2007; 18: 566-573.
54. BOMBELI T, SCHWARTZ BR, HARLAN JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa–dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin and GPIbalpha. J Exp Med 1198; 187: 329-339.
55. WEBER C. Platelets and chemokines in atherosclerosis. Partners of a crime. Circ Res 2005; 96: 612-616.
56. MINOR RL JR, SCOTT BD, BROWN DD, WINNIFORD MD. Cocaine-induced myocardial infarction in patients with normal coronary arteries. Ann Intern Med 1991; 115: 797-806.
57. LEE HO, EISENBERG MJ, DREW D, SCHILLER NB. Intraventricular thrombus after cocaine induced myocardial infarction. Am Heart J 1995; 129: 403-405.
58. KARLSSON G, REHMAN J, KALARIA V, BREALL JA. Increased incidence of stent thrombosis in patients with cocaine use. Catheter Cardiovasc Interv 2007; 69: 955-958.
59. HEESCH CM, WILHEM CR, RISTICH J, ADNANE J, BONTEMPO FA, WAGNER WR. Cocaine activates platelets and increases the formation of circulating platelet containing microaggregates in humans. Heart 2000; 83: 688-695.
60. KUGELMASS AD, ODA A, MONAHAN K, CABRAL C, WARE JA. Activation of human platelets by cocaine. Circulation 1993; 88: 876-883.
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.

BOLETÍN ESCUELA DE MEDICINA U.C., PONTIFICIA UNIVERSIDAD CATÓLICA DE CHILE VOL. 33 Nº1 2008
18
61. RINDER HM, AULT KA, JATLOW PI, KOSTEN TR, SMITH BR. Platelet alpha-granule release in cocaine users. Circulation 1994; 90: 1162-1167.
62. TOGNA G, TEMPESTA E, TOGNA AR, DOLCI N, CEBO B, CAPRINO L. Platelet responsiveness and biosynthesis of thromboxane and prostacyclin in response to in vitro cocaine treatment. Haemostasis. 1985;15(2):100-107.
63. KUGELMASS AD, SHANNON RP, YEO EL, WARE JA. Intravenous cocaine induced platelet activation in the conscious dog. Circulation 1995; 91: 1336-1340.
64. EICHHORN EJ, DEMIAN SE, ALVAREZ LG. Cocaine induced alterations in prostaglandin production in rabbit aorta. J Am Coll Cardiol 1992; 19: 696-703.
65. HEESCH CM, STEINER M, HERNÁNDEZ JA, et al. Effects of cocaine on human platelet aggregation in vitro. J Toxicol Clin Toxicol 1996; 34: 673-684.
66. SIEGEL AJ, SHOLAR MB, MENDELSON JH, et al. Cocaine-induced erythrocytosis and increase in von Willebrand factor: evidence for drug-related blood doping and prothrombotic effects. Arch Intern Med 1999; 159: 1925-1929.
67. ZURBANO MJ, HERAS M, TIGOL M, et al. Cocaine administration enhances platelet reactivity to subendothelial components: studies in a pig model. Eur j Clin Invest 1997; 27: 116-120.
68.JENNINGS LK, WHITE MM, SAUER CM, MAUER AM, ROBERTSON JT. Cocaine-induced platelet defects. Stroke 1993; 24: 1352-1359.
69. PEREIRA J, ALFARO G, MASSARDO T, et al. Elevated Levels of Cell-Derived Microparticles with Procoagulant Activity in Cocaine Abusers: Association with Increased Thrombin Generation and a
Prothrombotic State. Blood 2005; 106: 2629a.
70. MASSARDO T, JAIMOVICH R, PALLAVICINI J, QUINTANA JC, PANES O, HIDALGO P, MEZZANO D, SÁEZ C, PEREIRA J. Defectos de perfusión cerebral se asocian a activación del sistema hemostático en usuarios crónicos de cocaína. XXIX Congreso Chileno de Medicina Interna, Santiago, Ocubre 2007.
71. HOFFMAN M, MONROE DM. A cell-based model of hemostasis. Thromb Haemost 2001; 85: 958-965.
72. VANSCHOONBEEK K, FEIJGE MAH, VAN KAMPEN RJW, KENIS H, HEMKER HC, GIESEN PLA, HEEMSKERK JWM. Initiating and potentiating role of platelets in tissue-factor-induced thrombin generation in the presence of plasma: subject-dependent variations in thrombogram characteristics. J Thromb Haemost 2003; 2: 476-484.
73. WALSH PN. Role of platelets in blood coagulation. En: Hemostasis and Thrombosis: Basic principles and clinical practice. 5a Edición, RW Colman, VJ Marder, AW Clowes, JN George, SZ Goldhaber. (eds). JB Lippincott, Philadelphia, 2006: 605-616.
74. BAGLIA FA, WALSH PN. Prothrombin is the cofactor for the binding of factor XI to the platelet surface and for platelet-mediated factor XI activation by thrombin. Biochemistry 1998; 17: 2271-2281.
75. BEVERS EM, COMFURIUS P, VAN RIJN JMLM, HEMKER C, ZWAAL RFA. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur J Biochem 1982; 122: 429-436.
76. SIMS PJ, FAIONI EM, WIEDMER T, SHATTIL SJ. Complement C5b-9 cause release of membrane vesicles from the
platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 1988; 263: 18205-18212.
77. SIMS PJ, WIEDMER T, ESMON CT, WEISS HJ, SHATTIL SJ. Assembly of the platelet prothrombinase complex is linked to visiculation on the platelet plasma membrane. Studies in Scott syndrome: an isolated defect in platelet procoagulant activity. J Biol Chem 1989; 264: 17049-17057.
78. NESHEIM ME, FURMANIAK-KAZMIERCZAK E, HENING C, COT G. On the existence of platelet receptors for factor V(a) and factor VIII(a). Thromb Haemostas 1993; 70: 80-86.
79. OSTERUD B, RAPAPORT SI, LAVINE KK. Factor V activity of platelets: evidence for an activated factor V molecule and for a platelet activator. Blood 1977; 49: 819-834.
80. NEUENSCHWANDER P, JETY J. A comparison of phospholipid and platelets in the activation of human factor VIII by thrombin and factor Xa, and in the activation of factor X. Blood 1988; 72: 1761-1770.
81. HOFFMAN M, MONROE DM. A cell-based model of hemostasis. Thromb Haemost 2001; 85: 958-965.
82. SOLUM NO. Procoagulant expression in platelets and defects leading to clinical disorders. Arterioscler Thromb Vasc Biol 1999; 19: 2841-2846.
83. SIDDIQUI FA, DESAI H, AMIRKHOSRAVI A, et al. The presence and release of tissue factor from human platelets. Platelets 2002; 13: 247-253.
84. LÖSCHE W, SCHOLZ T, TEMMIER U, OBERLE V, CLAUS RA. Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets 2004; 15: 109-115.

19
85. CAMERA M, FRIGERIO M, TOSCHI V, et al. Platelet activation induces cell-surface immunoreactive tissue factor expression, which is modulated differently by antiplatelets drugs. Arterioscler Thromb Vasc Biol 2003; 23: 1690-1696.
86. WEYRICH A, SCHWARTZ H, TOLLEY ND, ZIMMERMAN GA, MACKMAN N. pre-mRNA splicing modulates the thrombogenecity of platelets. Blood 2006; 108: 40a.
87. PANES O, MATUS V, SÁEZ CG, QUIROGA T, PEREIRA J, MEZZANO D. Human platelets synthesize and express functional tissue factor. Blood 2007; 109; 5242-5250.
88. DAY SM, REEVE JL, PEDERSEN B, et al. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105: 192-198.
89. GIESEN PLA, RAUCH U, BOHRMANN B, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci USA. 1999;96:2311-2315.
90. BERCKMANS RJ, NIEUWLAND R, BÖING AN, et al. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost. 2001;85:639-646.
91. MÜLLER I, KLOCKE A, ALEX M, KOTZSCH M, et al. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17:476-478.
92. CHOU J, MACKMAN N, MERRILL-SKOLOFF G, et al. Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation
during thrombus propagation. Blood. 2004;104:3190-3197.
93. BOGDANOV VY, BALASUBRAMANIAN V, HATHCOCK J, et al. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med. 2003;9:458-462.
94. BROUSSAS M, CORNILLET-LEFÈBVRE P, POTRON G, NGUYÊN P. Adenosine inhibits tissue factor expression by LPS-stimulated human monocytes: involvement of the A3 adenosine receptor. Thromb Haemost. 2002;88:123-130.
95. WARR T, RAO LVM, RAPAPORT SI. Disseminated intravascular coagulation in rabbits induced by administration of endotoxin of tissue factor: effect of anti-tissue factor antibodies and measurement of plasma extrinsic pathway inhibitor activity. Blood. 1990; 75:1481-1489.
96. ZILLMANN A, LUTHER T, MÜLLER I, et al. Platelet-associated tissue factor contributes to the collagen-triggered activation of blood coagulation. Biochem Biophys Res Comm. 2001; 281; 603-609.
97. FALATI S, LIU Q, GROSS P, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197:1585-1598.
98. LÖSCHE W, SCHOLZ T, TEMMLER U, OBERLE V, CLAUS RA. Platelet-derived microvesicles transfer tissue factor to monocytes but not to neutrophils. Platelets. 2004;15:109-115.
99. SCHWERTZ H, TOLLEY ND, FOULKS JM, et al. Signal-dependent splicing of tissue factor pre-mRNA
modulates the thrombogenicity of human platelets J Exp Med. 2006; 203: 2433-2440.
100. DENIS MM, TOLLEY ND, BUNTING M et al. Escaping the nuclear confines: signal-dependent pre mRNA splicing in anucleate platelets. Cell 2005; 122: 379-391.
101. KIEFFER N, GUICHARD J, FARCET JP, VAINCHENKER W, BRETON-GORIUS J. Biosynthesis of major platelet proteins in human blood platelets. Eur J Biochem. 1987;164:189-195.
102. WEYRICH AS, DIXON DA, PABLA R, et al. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA. 1998;95:5556-5561.
103. LINDEMANN S, TOLLEY ND, DIXON DA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485-490.
104. BROGREN H, KARLSSON L, ANDERSSON M, et al. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. 2004;104:3943-3948.
105. QUIROGA T, GOYCOOLEA M, PANES O, ARANDA E, MARTÍNEZ C, BELMONT S, MUÑOZ B, ZÚÑIGA P, PEREIRA J, MEZZANO D. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica 2007;92:357-365
LA FISIOPATOLOGÍA DE LA HEMOSTASIA: ALGUNOS ASPECTOS SOBRE LA VIDA Y LA MUERTE DE LAS PLAQUETAS EN LA CIRCULACIÓN - DR. JAIME PEREIRA G.





























