Embed Size (px)
Citation preview

CARACTERIZACION DE
LAS PROTEINAS:
PESO MOLECULAR.
DETERMINACION DE LA
ESTRUCTURA PRIMARIA.

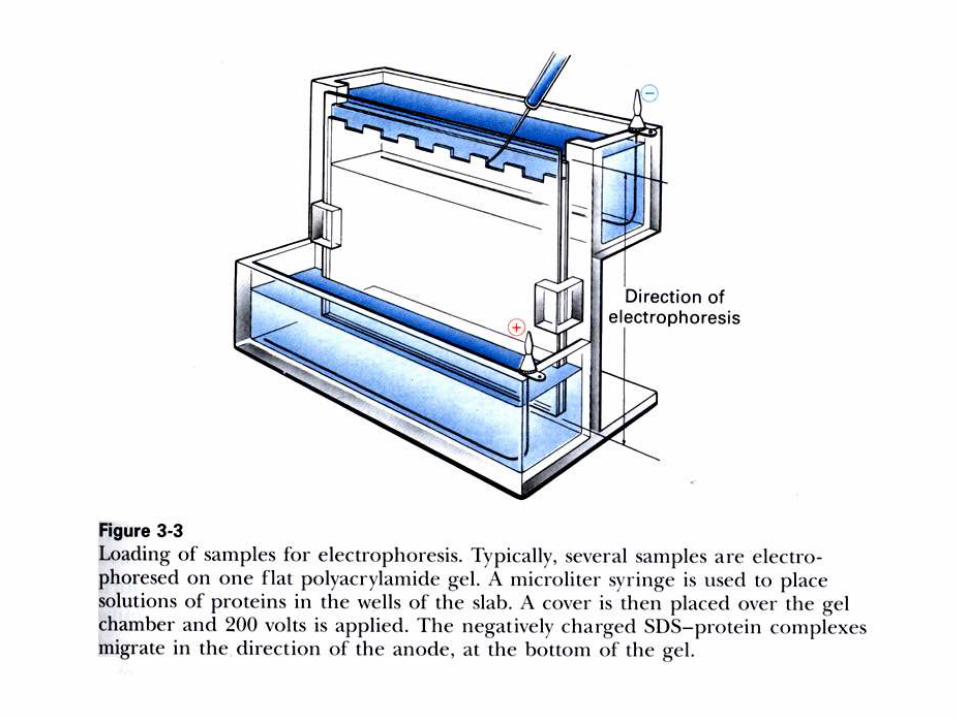
ELECTROFORESIS.
V = E z / f v: velocidad de migración de la molécula en un
campo eléctrico. E: intensidad de campo eléctrico, z: carga neta
de la proteína. f = 6phr es el coeficiente friccional. h (eta) es la
viscosidad del medio y r es el radio de la partícula.
Electroforesis en gel de poliacrilamida: en condiciones nativas
(PAGE) o en presencia de SDS (SDS-PAGE).
Isoelectroenfocado (IEF): Separación por punto isoeléctrico (pI).
Electroforesis bidimensional en gel de poliacrilamida (2D -PAGE):
IEF en una primera dimensión, SDS-PAGE en la segunda.


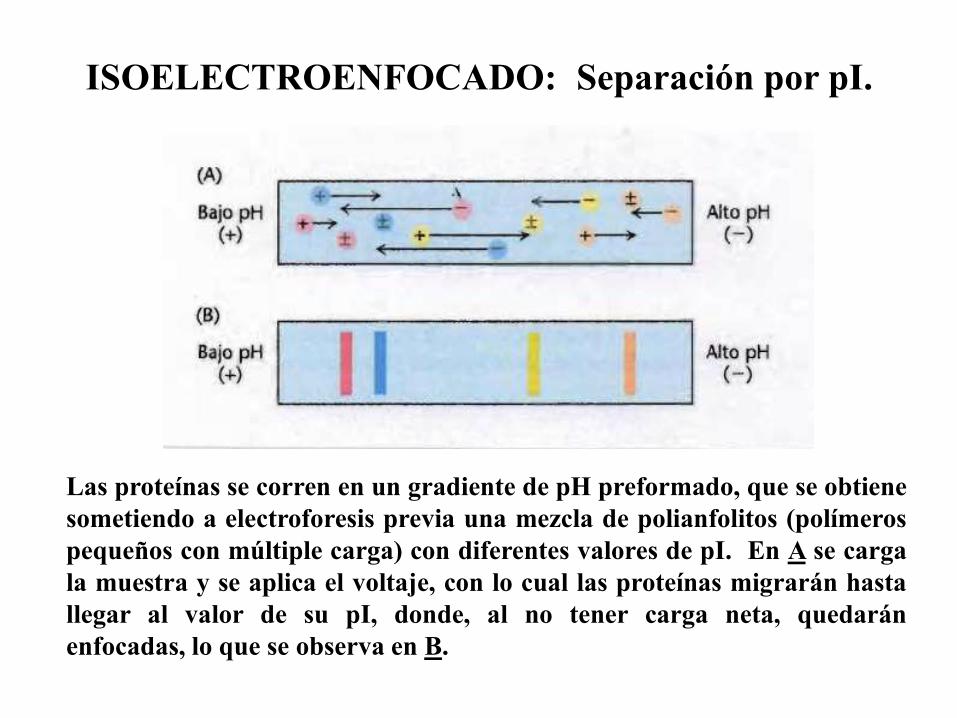
ISOELECTROENFOCADO: Separación por pI.
Las proteínas se corren en un gradiente de pH preformado, que se obtiene
sometiendo a electroforesis previa una mezcla de polianfolitos (polímeros
pequeños con múltiple carga) con diferentes valores de pI. En A se carga
la muestra y se aplica el voltaje, con lo cual las proteínas migrarán hasta
llegar al valor de su pI, donde, al no tener carga neta, quedarán
enfocadas, lo que se observa en B.


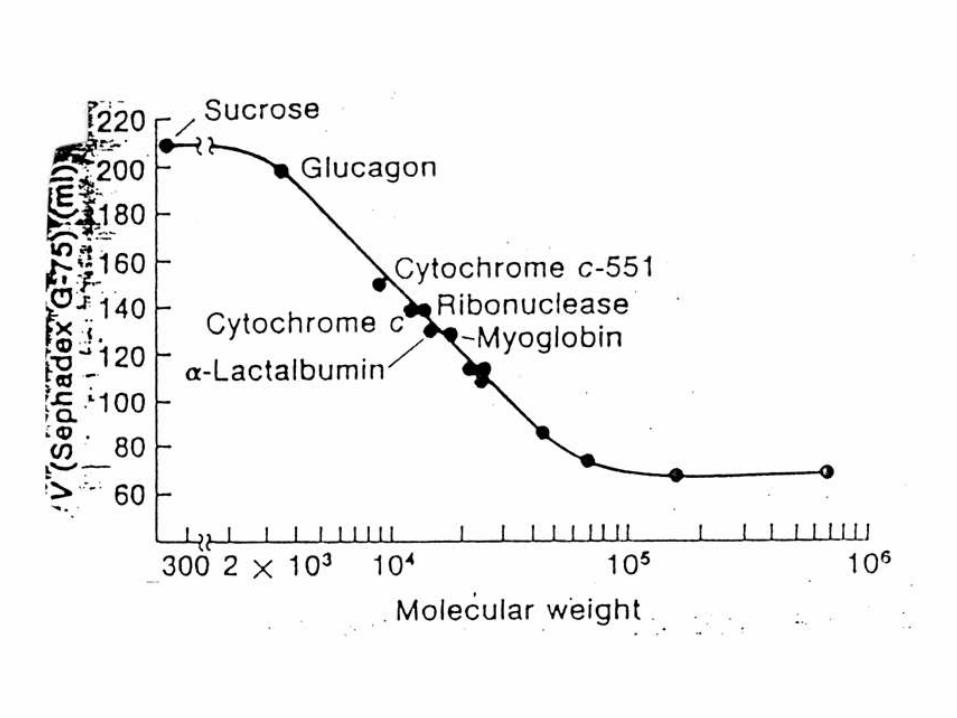
DETERMINACIÓN DEL PESO MOLECULAR DE LAS PROTEINAS.
Mr = masa molecular relativa (por ejemplo, 50,000)
Masa molecular: Por ejemplo 50,000 daltons (50 kDa).
A) Proteína nativa:
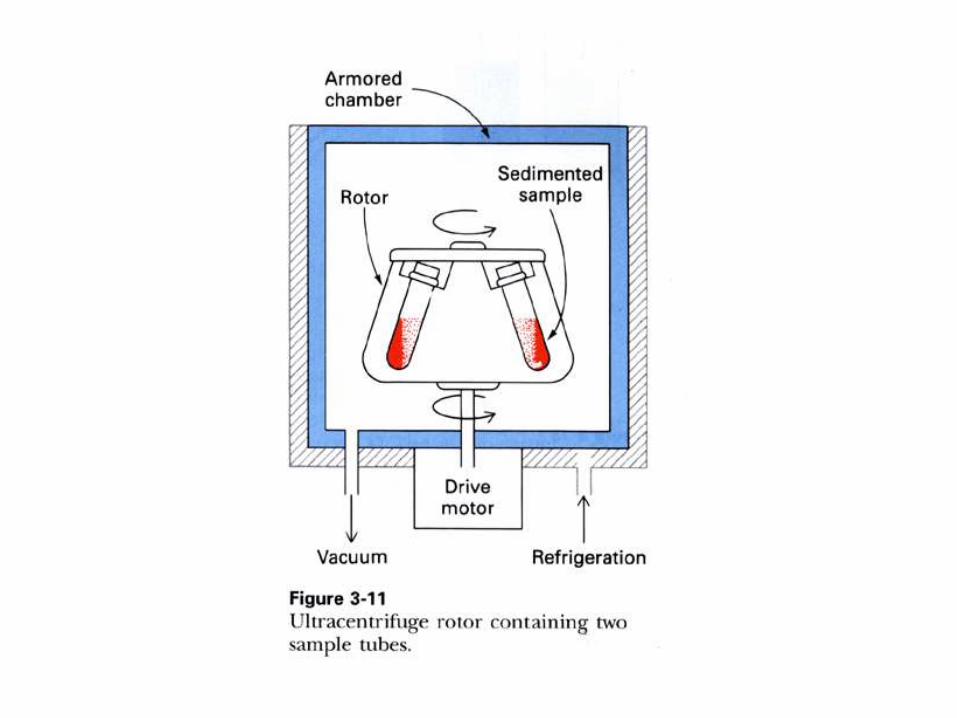
1) Ultracentrifugación.
2) Filtración por gel.
3) Espectrometría de masa.
B) Subunidad:
1) Electroforesis en gel de poliacrilamida en
presencia de dodecil sulfato de sodio (SDS-PAGE).

ULTRACENTRIFUGACION.
Fc= fuerza centrífuga. r = radio del rotor.
w= velocidad angular. w2r = campo centrífugo. = volumen específico parcial
(inversa de la densidad de la proteína).
m’ (masa efectiva) es menor que m (masa real) porque el flúido desplazado
ejerce una fuerza opuesta (1 – r) .
f= coeficiente friccional de la partícula = 6phr , donde h es la viscosidad del
medio y r el radio de la partícula.
v = Fc / f = m(1 – r) w2r / f
s = coeficiente de sedimentación (unidades 10-13 seg = 1 Svedberg).
s = v / w2r = m (1 – r) / f
La velocidad de sedimentación (v) depende en parte de la masa de la
partícula; tambien depende de su forma (una partícula compacta tiene un
valor de f menor que el de una alargada); una partícula densa se mueve
mas rápido que una menos densa porque su valor de (1 – r) es menor; y
depende tambien de la densidad de la solución (r): si r es < 1, se hunde; si es
> 1, flota y si es = 1 no se desplaza.
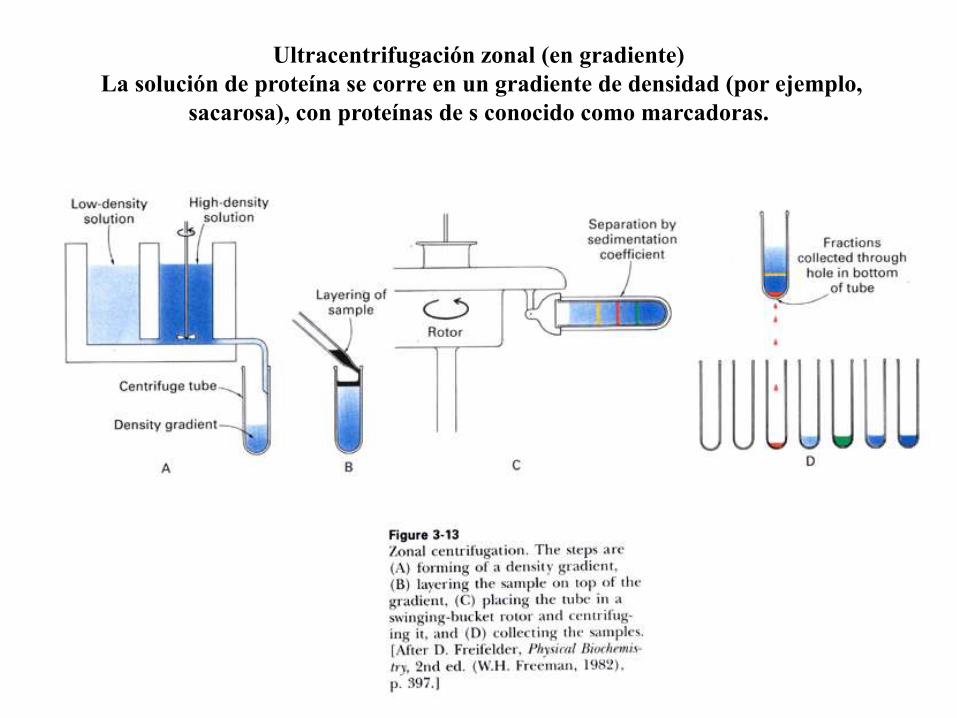
Ultracentrifugación zonal (en gradiente)
La solución de proteína se corre en un gradiente de densidad (por ejemplo,
sacarosa), con proteínas de s conocido como marcadoras.

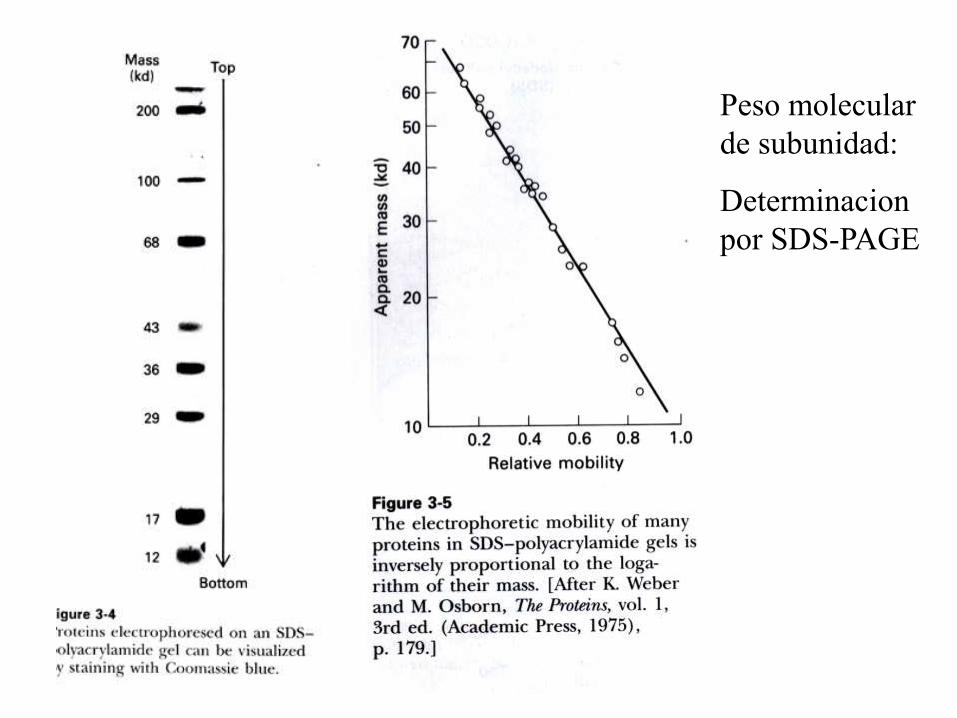
Peso molecular
de subunidad:
Determinacion
por SDS-PAGE

ESPECTROMETRIA DE
MASA: SUS APLICACIONES
EN LA DETERMINACION DE
PESOS MOLECULARES DE
PROTEINAS Y EN LA
SECUENCIACION DE
PEPTIDOS.

Espectrometría de Masa
• La Espectrometría de Masa es una tecnica quepermite determinar la masa de moléculas pormedio de la medida de la relacion masa/carga (m/z).
• La medida de masas moleculares involucra laproduccion, separación y detección de ionesmoleculares en fase gaseosa.
•Cada molécula puede originar iones moleculares
con distinto número de cargas ( y distintas m/z).

Un espectrómetro de masa consiste de tres módulos:
1) Fuente de iones.
2) Analizador de masas.
3) Sistema de detección y adquisición de datos.
Técnicas actuales para trabajo biológico:
1) MALDI-ToF MS (desorción/ionización por laser asistida por
matriz, combinada con análisis de masas por tiempo de vuelo)
2) ES/MS (ionización por electrospray combinada con análisis de
masas por cuadrupolo)

La solución a analizar se pasa a través de una
aguja hipodérmica mantenida a un alto
potencial. Se forma un cono de gotitas muy
finas, altamente cargadas, que irradian de la
punta de la aguja. Se evaporan rápidamente,
pasando las moléculas a analizar a la fase
gaseosa. Si son moléculas grandes se
producen iones con cargas múltiples.
ES/MSGENERACION DE IONES MOLECULARESIONIZACION POR ELECTROSPRAY
(ESI MS)

Aqueous
solution
This ionization method arises in the attempt to couple the LC to MS
The effect of vacuum, temperature and curtain gas causes the formation of
increasingly smaller drops until the molecule is fully desolvated in vacuum.
Sir Geoffrey Taylor (1964). "Disintegration of Water Droplets in an Electric Field". Proc. Roy. Soc. London. Ser. A 280 (1382): 383.
dripping,
burst,
pulsating,
cone-jet
1914, John Zeleny
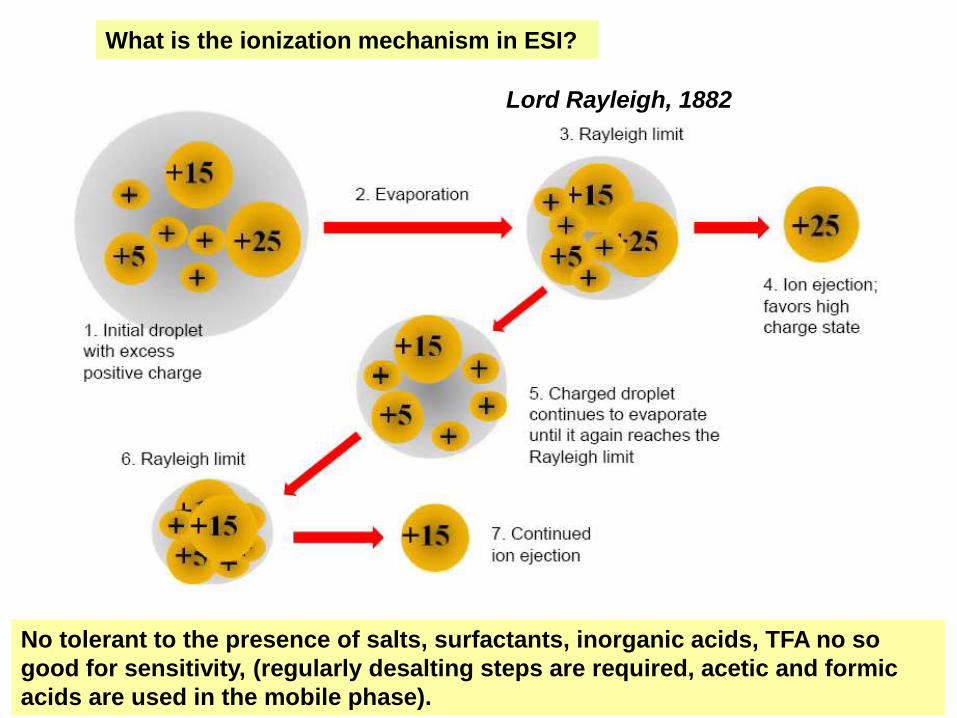
What is the ionization mechanism in ESI?
No tolerant to the presence of salts, surfactants, inorganic acids, TFA no so
good for sensitivity, (regularly desalting steps are required, acetic and formic
acids are used in the mobile phase).
Lord Rayleigh, 1882

ANALISIS DE LA MASA DE LOS IONES
MOLECULARES.
ANALISIS DE LA MASA DE LOS IONES
MOLECULARES
Los iones se dirigen al vacío del espectrómetro de
masa de cuadrupolo a través de un orificio o capilar
calentado. El cuadrupolo consta de cuatro barras
metálicas paralelas que generan un campo eléctrico
oscilatorio. Los iones se separan en este campo:
sólo los de una determinada m/z se dejan pasar a
una determinada amplitud y frecuencia de
oscilación de los potenciales eléctricos aplicados a
las barras, y llegan al detector.
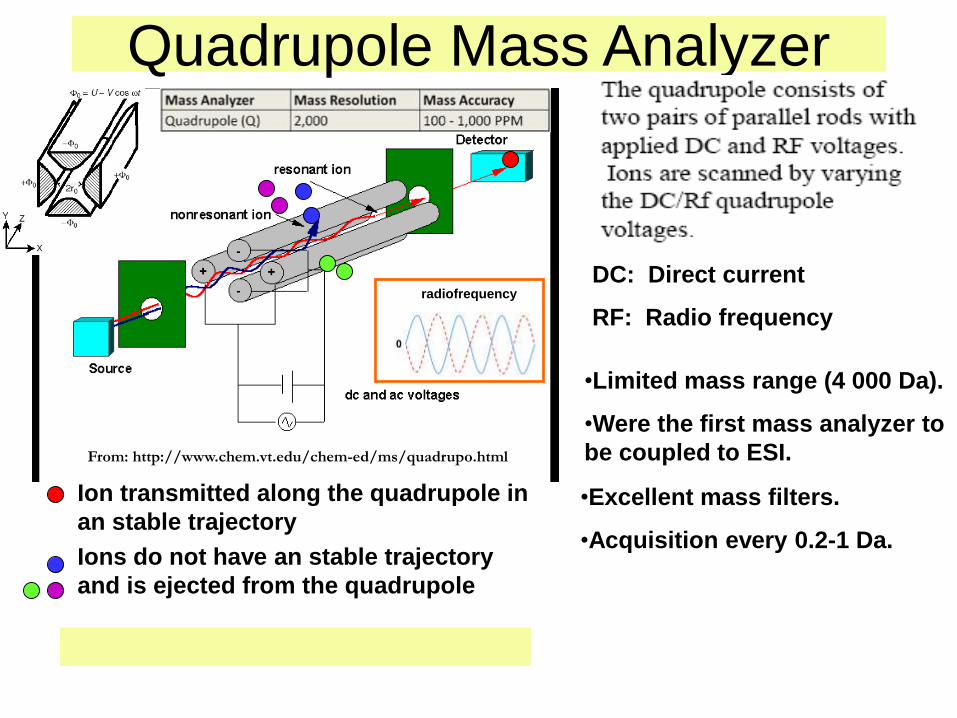
Quadrupole Mass Analyzer
From: http://www.chem.vt.edu/chem-ed/ms/quadrupo.html
Ion transmitted along the quadrupole in
an stable trajectory
Ions do not have an stable trajectory
and is ejected from the quadrupole
•Limited mass range (4 000 Da).
•Were the first mass analyzer to
be coupled to ESI.
•Excellent mass filters.
•Acquisition every 0.2-1 Da.
radiofrequency
DC: Direct current
RF: Radio frequency
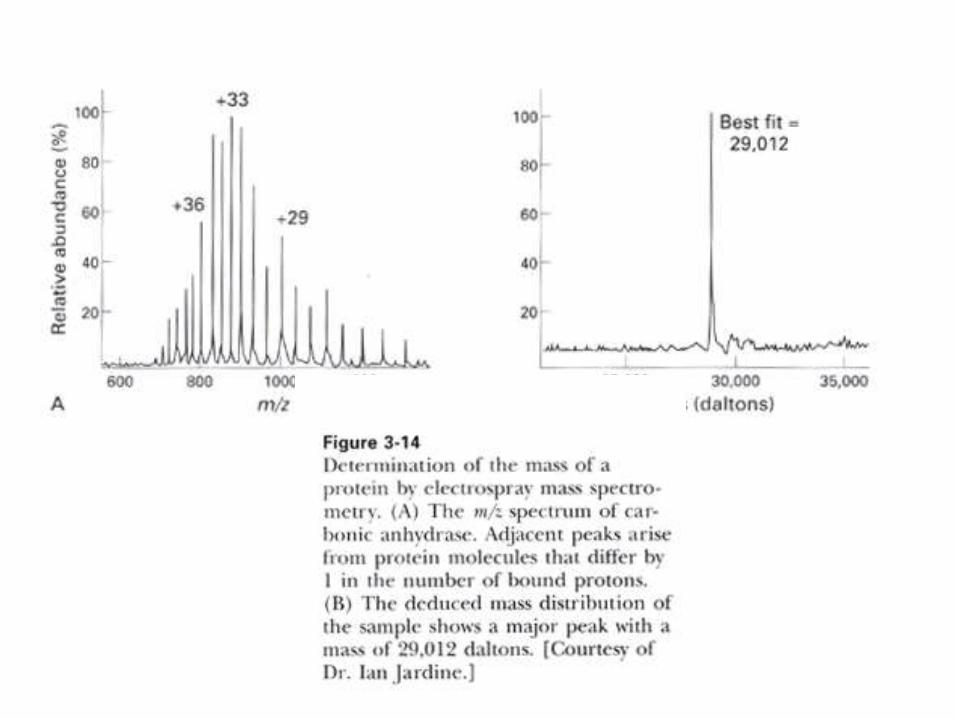

GENERACION DE IONES MOLECULARESPseudocristales mixtos de una matriz (p.ej.: ácido 3,5-
dimetoxi-4-hidroxicinámico). Se los irradia con un pulso
de laser. La sublimación rápida de los cristales lleva a la
formación de iones moleculares protonados en fase gaseosa.
MALDI-ToF MS
ANALISIS DE LA MASA DE LOS IONES MOLECULARES.Se aceleran a una energía cinética de aprox. 25 KeV y se los
deja volar a través de una distancia fija sin campo eléctrico.
Teniendo energía cinética idéntica, los iones chicos se
mueven más rápido que los grandes y llegan antes al
detector. M/z se determina a partir del tiempo de vuelo,
comparando con standards.
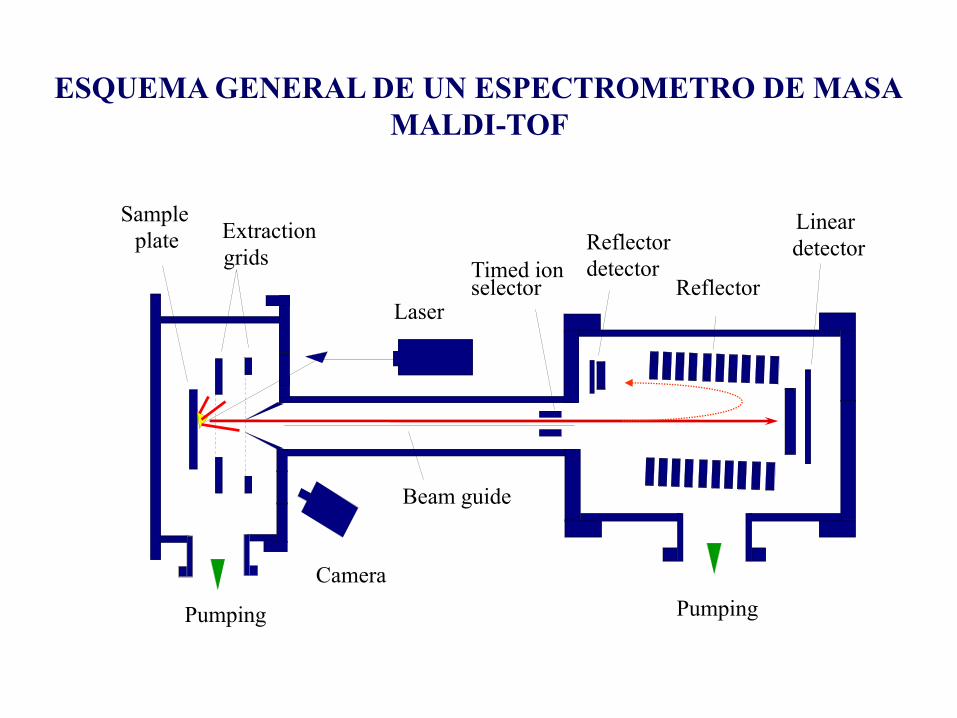
Camera
Laser
Sample
plate
Pumping Pumping
Beam guide
Timed ion selector Reflector
Linear
detectorExtraction
gridsReflector
detector
ESQUEMA GENERAL DE UN ESPECTROMETRO DE MASA
MALDI-TOF
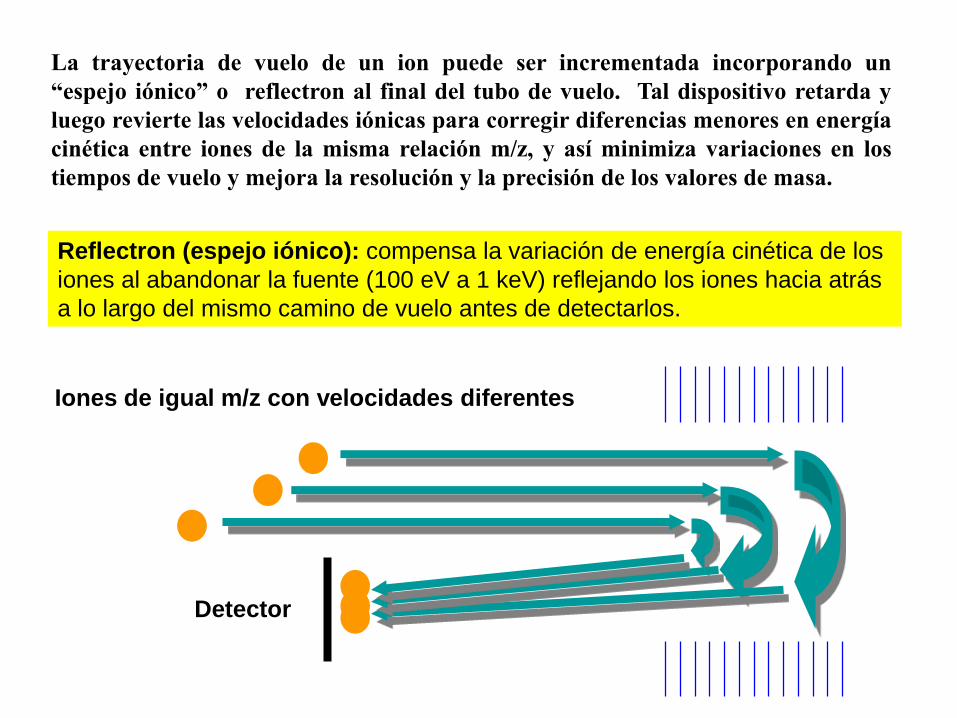
Detector
Iones de igual m/z con velocidades diferentes
Reflectron (espejo iónico): compensa la variación de energía cinética de los
iones al abandonar la fuente (100 eV a 1 keV) reflejando los iones hacia atrás
a lo largo del mismo camino de vuelo antes de detectarlos.
La trayectoria de vuelo de un ion puede ser incrementada incorporando un
“espejo iónico” o reflectron al final del tubo de vuelo. Tal dispositivo retarda y
luego revierte las velocidades iónicas para corregir diferencias menores en energía
cinética entre iones de la misma relación m/z, y así minimiza variaciones en los
tiempos de vuelo y mejora la resolución y la precisión de los valores de masa.
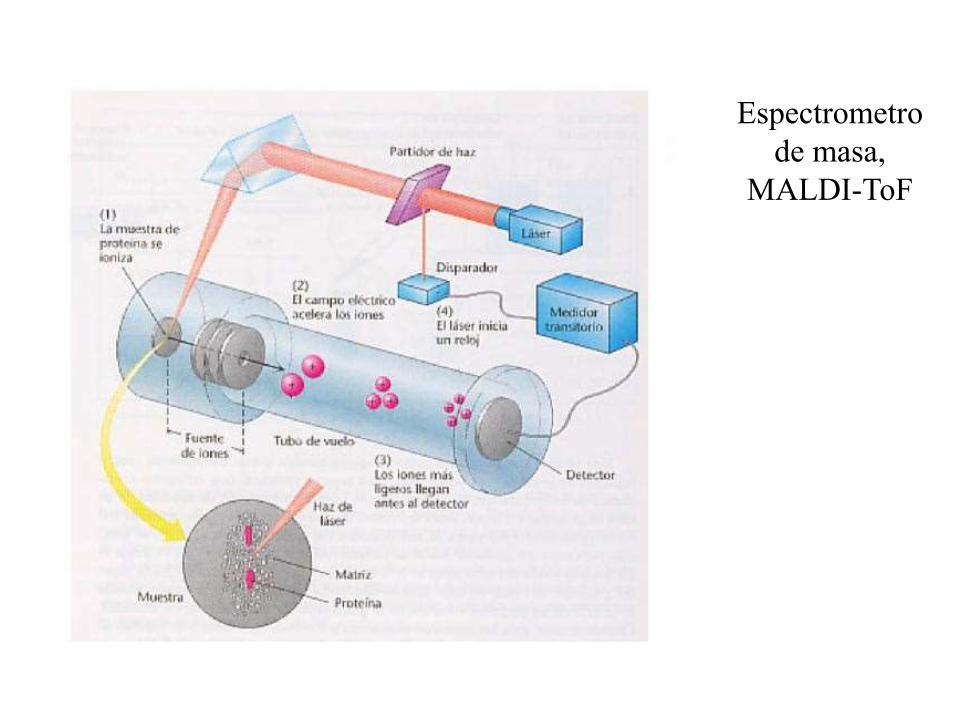
Espectrometro
de masa,
MALDI-ToF
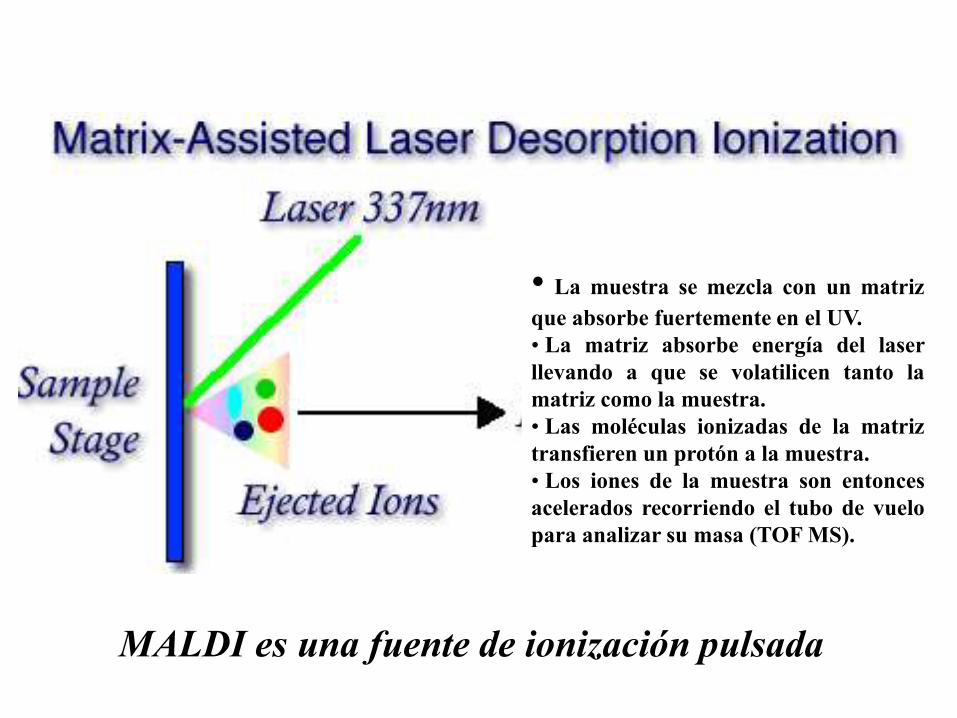
MALDI es una fuente de ionización pulsada
• La muestra se mezcla con un matriz
que absorbe fuertemente en el UV.
• La matriz absorbe energía del laser
llevando a que se volatilicen tanto la
matriz como la muestra.
• Las moléculas ionizadas de la matriz
transfieren un protón a la muestra.
• Los iones de la muestra son entonces
acelerados recorriendo el tubo de vuelo
para analizar su masa (TOF MS).
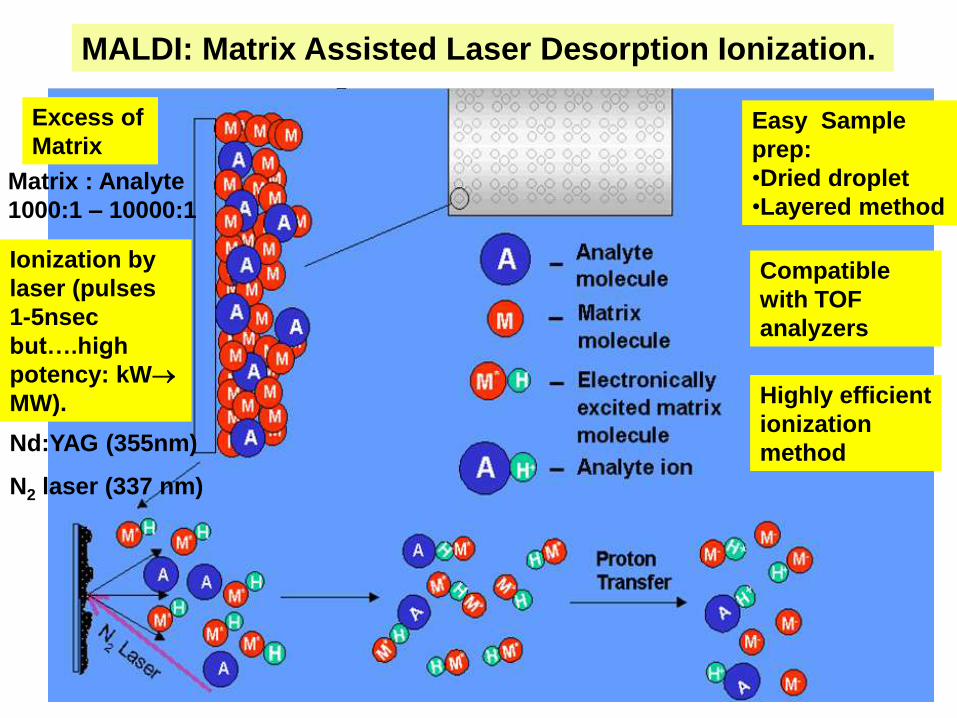
MALDI: Matrix Assisted Laser Desorption Ionization.
Matrix : Analyte
1000:1 – 10000:1
Excess of
MatrixEasy Sample
prep:
•Dried droplet
•Layered method
Nd:YAG (355nm)
N2 laser (337 nm)
Ionization by
laser (pulses
1-5nsec
but….high
potency: kW
MW).
Compatible
with TOF
analyzers
Highly efficient
ionization
method
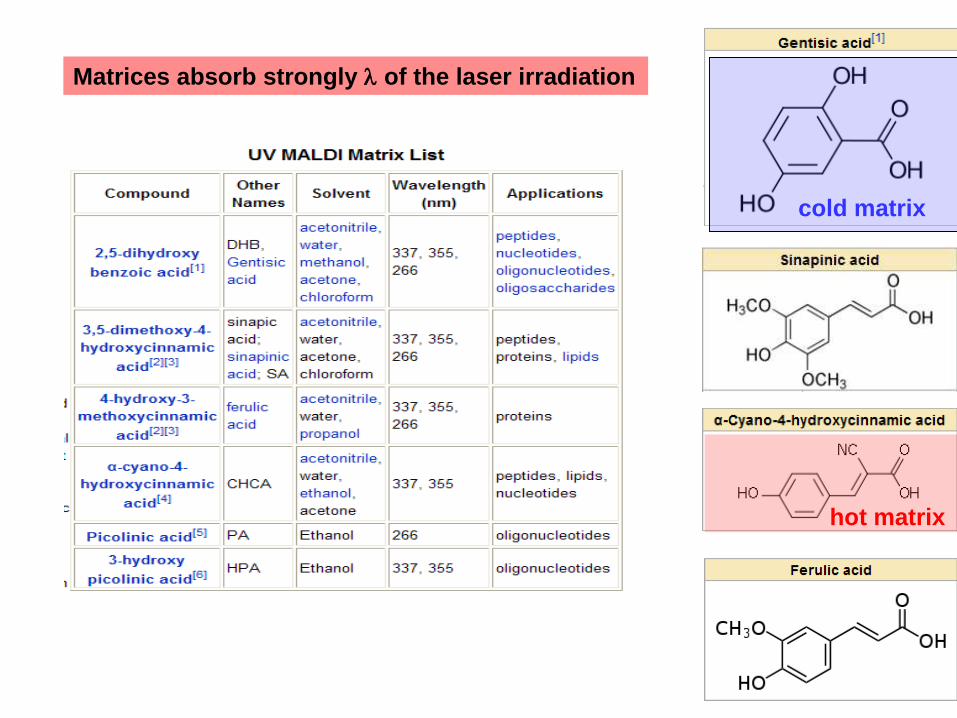
Matrices absorb strongly l of the laser irradiation
cold matrix
hot matrix

La relación masa a carga de un ion es proporcional al cuadrado
de su tiempo de vuelo.
t = Tiempo de vuelo
L = Longitud recorrida por los iones
dentro del analizador
m = Masa
K = Energía cinética del ion
z = Número de cargas en el ion
2
22
L
Kt
z
m

MALDI-MS
1. Los espectros de MALDI-MS son muy simples, en general sólo
se observan iones con una sola carga: (M+H)+ o (M-H)-.
2. Es un método de ionización muy eficiente, y tiene alta
sensibilidad para el análisis de biomoléculas y biopolímeros.
3. Es ideal para el análisis de mezclas de péptidos trípticos para la
identificación de proteínas por PMF, peptide mass fingerprinting.
4. Es un análisis de high throughput (permite el análisis de un
número grande de muestras en corto tiempo).
5. Es compatible con el analizador de masa de tiempo de vuelo.
6. La intensided del laser puede inducir fragmentaciones que
permiten obtener información estructural (secuencia).

SECUENCIACION
DE PEPTIDOS Y
PROTEINAS

¿Cuales son lo objetivos de la secuenciación de péptidos?
• Comparar estructuras primarias de diferentes proteínas paraestablecer relaciones evolutivas (actualmente, se hace mayormenteusando secuencias traducidas de secuencias de nucleótidos).
• Obtener una secuencia de péptido de una proteína desconocidaproveniente de un organismo cuyo genoma no está secuenciado; estopermitirá la síntesis de oligonuclótidos degenerados para identificary clonar el gen que la codifica.
•Confirmar la identidad de una proteína sospechada a partir de unexperimento de PMF.
•Identificar modificaciones post-traduccionales.
.

SECUENCIACION DE PROTEINAS Y PEPTIDOS.
Para secuenciar una proteína siempre se debe partir de la proteína
purificada. La purificación puede consistir en obtener la proteína
pura en un tubo, a través de los métodos de purificación ya
mencionados, o, aún si no está completamente homogénea, como
una banda discreta en un gel de SDS-PAGE.
Para secuenciar una proteína siempre se la debe fragmentar para
obtener péptidos de menor tamaño, secuenciables por distintos
procedimientos.
Tanto para secuenciar por el método de Edman como por
espectrometría de masa, debe comenzarse con estos tres pasos:
1) Desnaturalizar, con urea por ejemplo, y reducir los puentes
disulfuro.
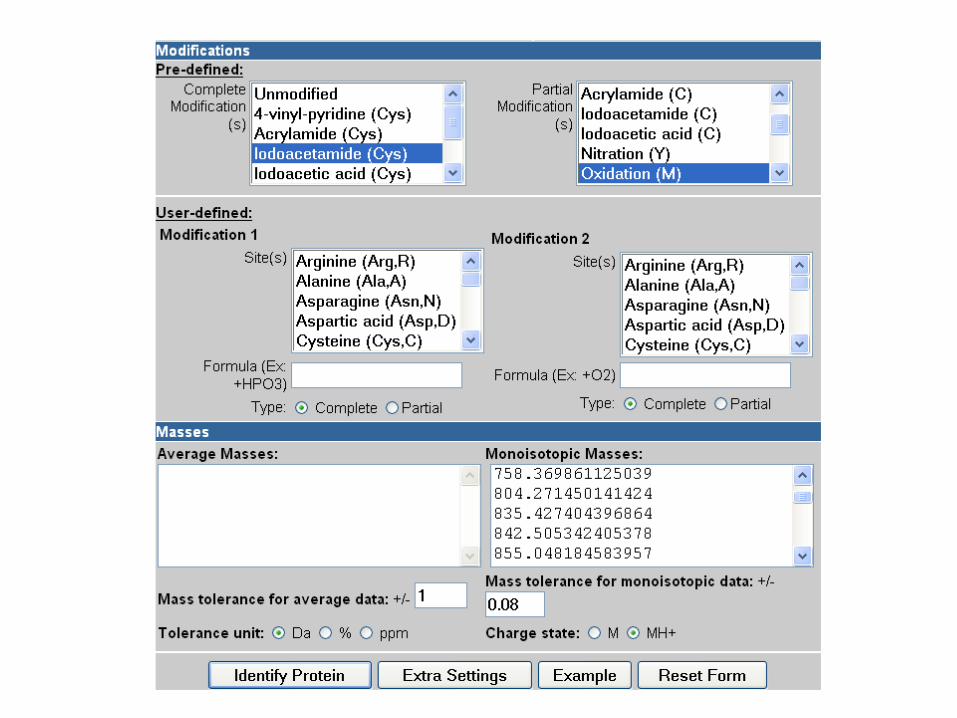
2) Bloquear los grupos –SH, con iodoacetamida por ejemplo.
3) Fragmentar la proteína en péptidos mas pequeños, usando dos
métodos diferentes (por ejemplo, digestiones con tripsina y BrCN)
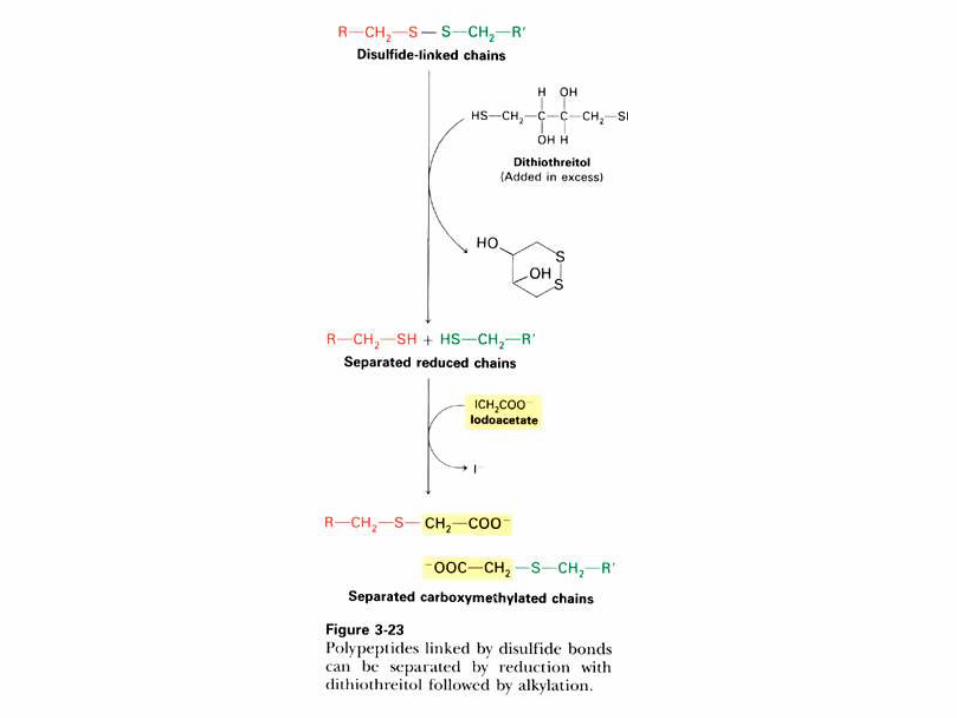
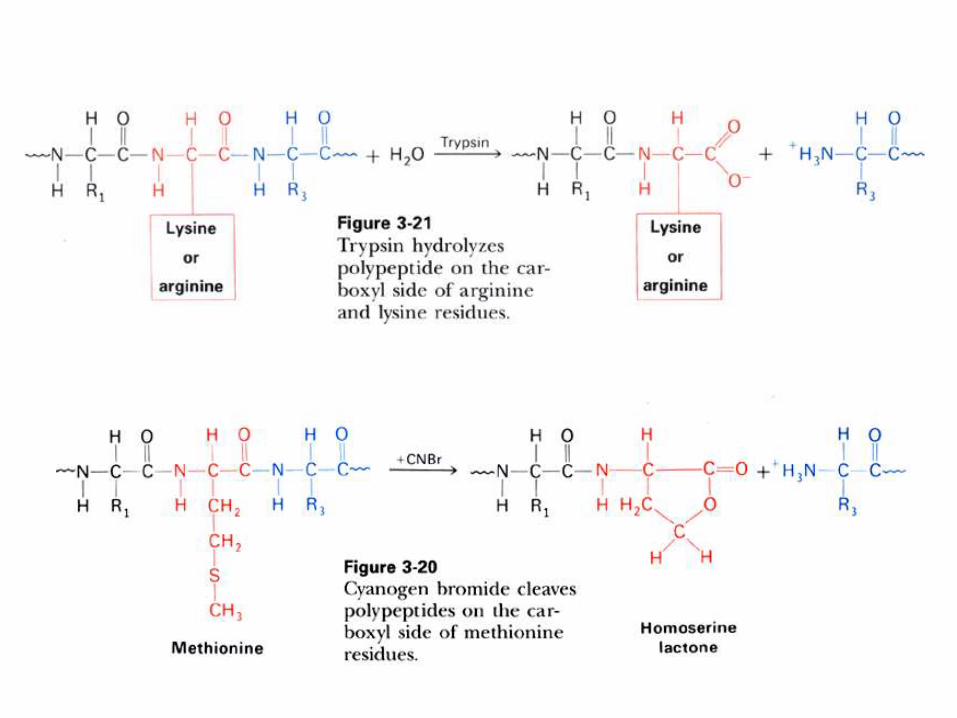

SECUENCIACION DE PROTEINAS Y
PEPTIDOS.
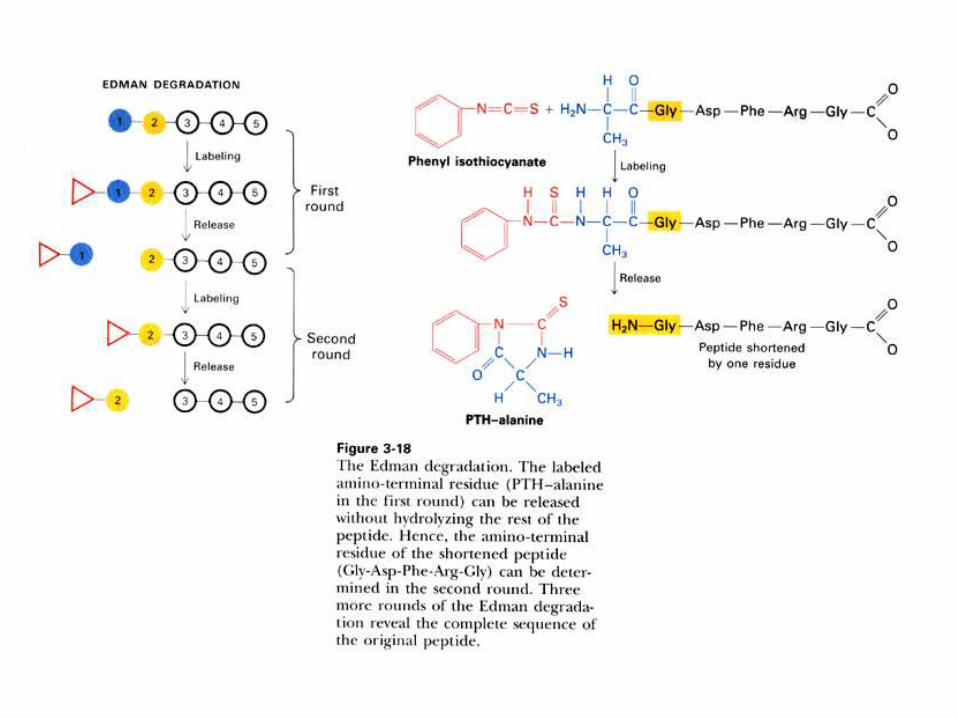
1) Método de Edman:
Previa separación de los péptidos por
HPLC en fase reversa, secuenciarlos
químicamente por reacción con el reactivo
de Edman (isotiocianato de fenilo), en un
secuenciador automático.

Pasos a seguir para secuenciar una proteína pura
por el método de Edman.
1) Desnaturalizar, con urea por ejemplo, y reducir los puentes disulfuro.
2) Bloquear los grupos –SH, con iodoacetamida por ejemplo.
3) Fragmentar la proteína en péptidos mas pequeños, usando dos
métodos diferentes (por ejemplo, digestiones con tripsina y BrCN)
4) Separar los péptidos obtenidos por el primer método por HPLC en fase
reversa (RP-HPLC).
5) Tratar cada péptido purificado con el reactivo de Edman, isotiocianato
de fenilo, en un secuenciador automático.
6) Identificar los residuos de aminoácidos por RP-HPLC de los derivados
de feniltiohidantoína (PTH-aminoácido) on line en el secuenciador.
7) Hacer lo mismo con los péptidos obtenidos por el segundo método de
ruptura de la proteína.
8) Armar la secuencia por superposición de los péptidos obtenidos por
ambos métodos.
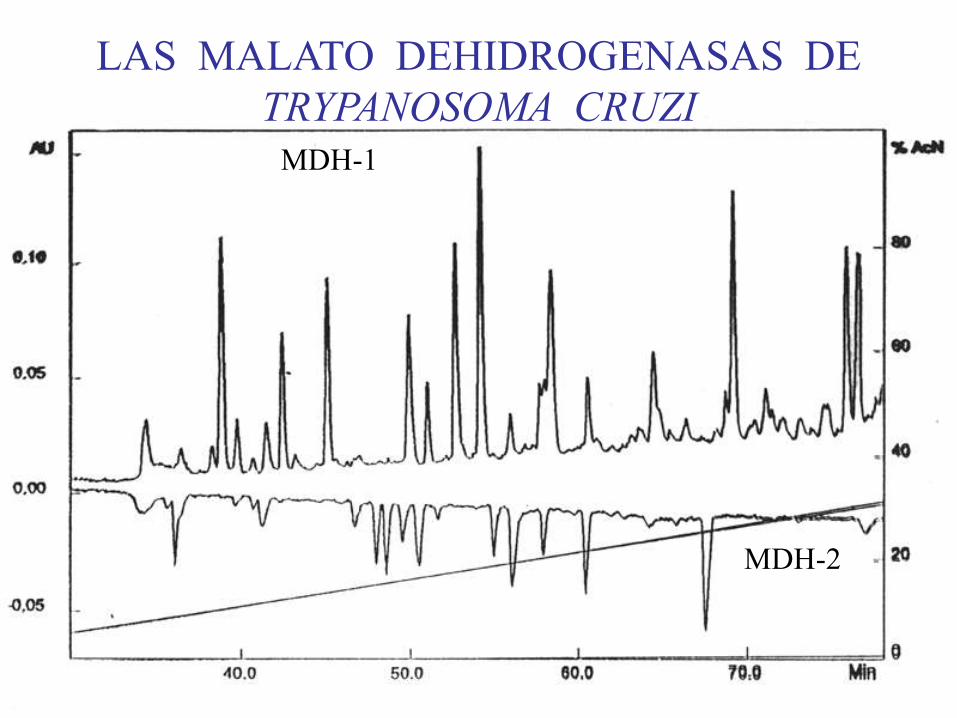
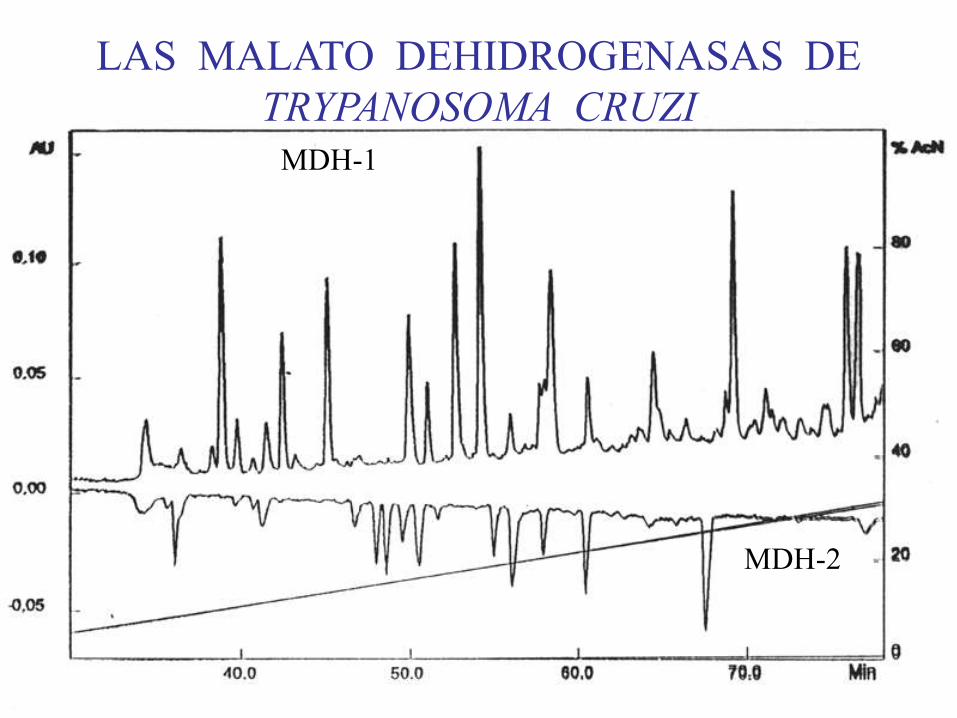
MDH-1
MDH-2
LAS MALATO DEHIDROGENASAS DE
TRYPANOSOMA CRUZI

El Procise Protein Sequencing System de Applied Biosystems
Está compuesto por 4 módulos integrados: el Secuenciador propiamente dicho, el Microgradient Delivery
System, el detector de UV de longitud de onda variable, y la computadora equipada con Procise control
software y SequencePro software. El sistema controla la entrega precisa de hasta 12 solventes y reactivos
diferentes. Incluye el cartucho de reacción, donde se obtiene el primer derivado, el frasco de conversión
donde se lo transforma en el PTH-derivado, que luego se transfiere a la columna de HPLC en fase reversa,
donde se separan por el gradiente de solvente, se lo detecta en el detector UV/VIS por su absorbancia y se lo
identifica por el tiempo de retención en la columna. La calibración se hace en base a una corrida con los
PTH derivados de los 20 aminoácidos proteicos, además en algún caso de un derivado correspondiente a una
modificación post-traduccional que se quiere detectar.
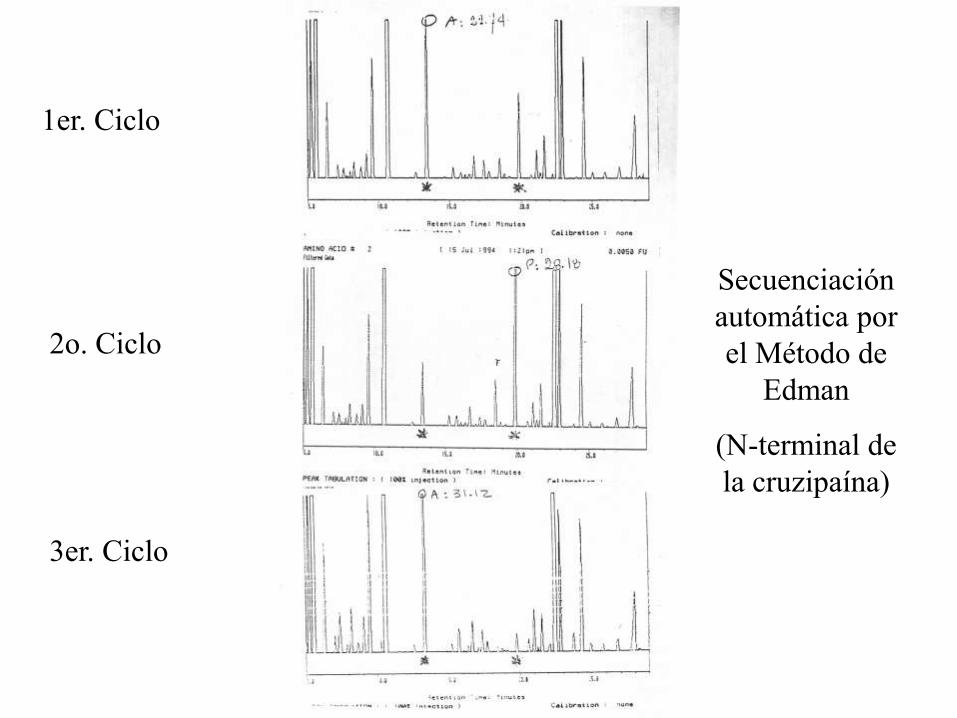
1er.Ciclo
2
3er.
DEGRADACI
ON DE
EDMANREACCION DEL
RESIDUO N-
TERMINAL CON
ISOTIOCIANATO
DE FENILO
1er. Ciclo
2o. Ciclo
3er. Ciclo
Secuenciación
automática por
el Método de
Edman
(N-terminal de
la cruzipaína)


2) Espectrometría de masa:
Puede utilizarse para secuenciar péptidos o para identificar
proteínas.
2a) Secuenciación: Seleccionar un péptido determinado por
MS y fragmentarlo, obteniendo su secuencia a partir de las
masas de los fragmentos resultantes (CID, fragmentación en
cámara de colisión, o PSD, decaimiento después de la fuente de
iones)
2b) Identificación: Sin separar los péptidos, determinar la
masa de cierto número de los mismos por MS e identificar a la
proteína en los bancos de datos, por comparación con las masas
de digeridos virtuales de todas las proteínas ya secuenciadas. Se
conoce como peptide mass fingerprinting (PMF). Es óptimo
cuando el genoma del organismo de origen está completamente
secuenciado.
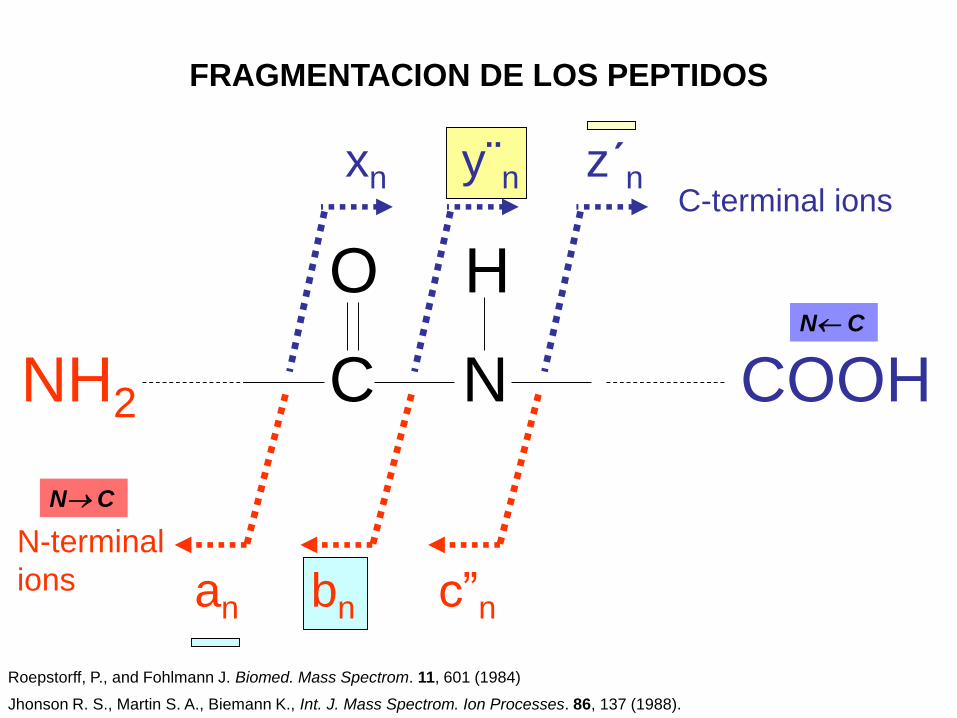
FRAGMENTACION DE LOS PEPTIDOS
C
O
N
H
COOHNH2
y¨n z´nxnC-terminal ions
bnan c”n
N-terminal
ions
Roepstorff, P., and Fohlmann J. Biomed. Mass Spectrom. 11, 601 (1984)
Jhonson R. S., Martin S. A., Biemann K., Int. J. Mass Spectrom. Ion Processes. 86, 137 (1988).
N C
N C

FRAGMENTACION DE LOS PEPTIDOS
Un péptido cargado puede ser fragmentado en dos trozos de tres maneras, que
pueden producir un par de iones a y x, un par de iones b e y, o un par de iones c
y z.
Teóricamente una fragmentación puede tener lugar en cualquier lugar de un
péptido, y se espera un espectro que contenga todos los posibles picos de iones.
En la práctica, debido a la fortaleza irregular de las ligaduras en las diferentes
posiciones, los diferentes iones aparecen con distintas frecuencias.
Los mas abundantes son los iones y, los cuales a menudo forman la serie
completa en un espectro. Los siguientes en frecuencia son los iones a y b, de
los cuales muchos no se observan. Los iones c, x y z se presentan mucho menos
frecuentemente. Adicionalmente, estos iones pueden formar nuevos iones por
pérdida de agua o de amonio.
La introducción de un grupo sulfónico en el N-terminal de un péptido tríptico
favorece la fragmentación que da lugar a los iones y, y suprime a los de la serie
b, dando un espectro simplificado, que facilita la determinación de la secuencia.
SECUENCIACION POR ESPECTROMETRIA DE MASA
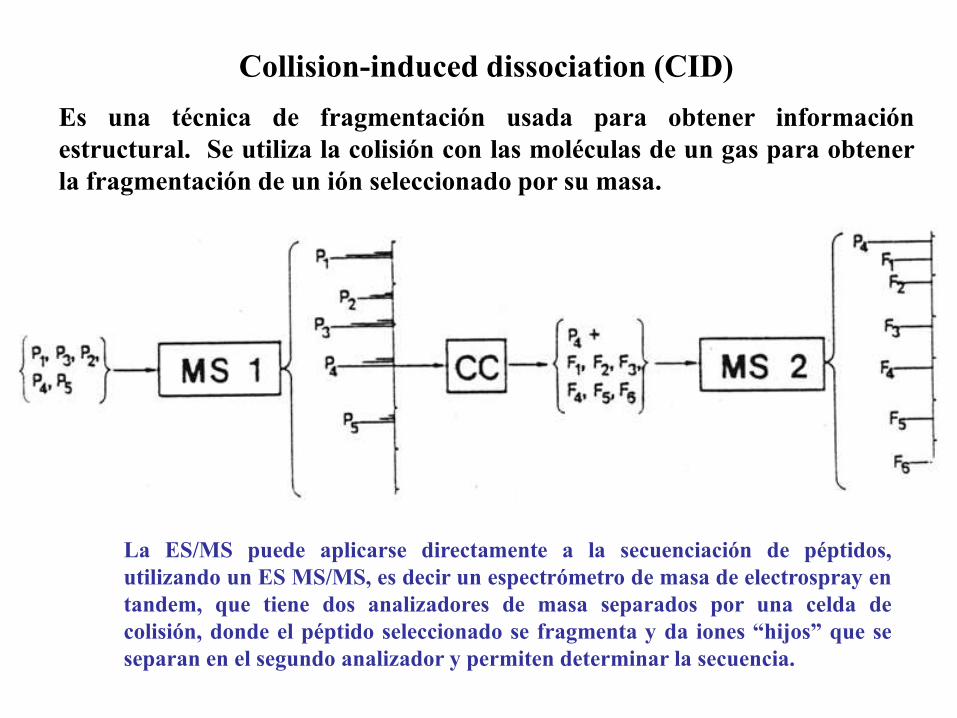
La ES/MS puede aplicarse directamente a la secuenciación de péptidos,
utilizando un ES MS/MS, es decir un espectrómetro de masa de electrospray en
tandem, que tiene dos analizadores de masa separados por una celda de
colisión, donde el péptido seleccionado se fragmenta y da iones “hijos” que se
separan en el segundo analizador y permiten determinar la secuencia.
Collision-induced dissociation (CID)
Es una técnica de fragmentación usada para obtener información
estructural. Se utiliza la colisión con las moléculas de un gas para obtener
la fragmentación de un ión seleccionado por su masa.
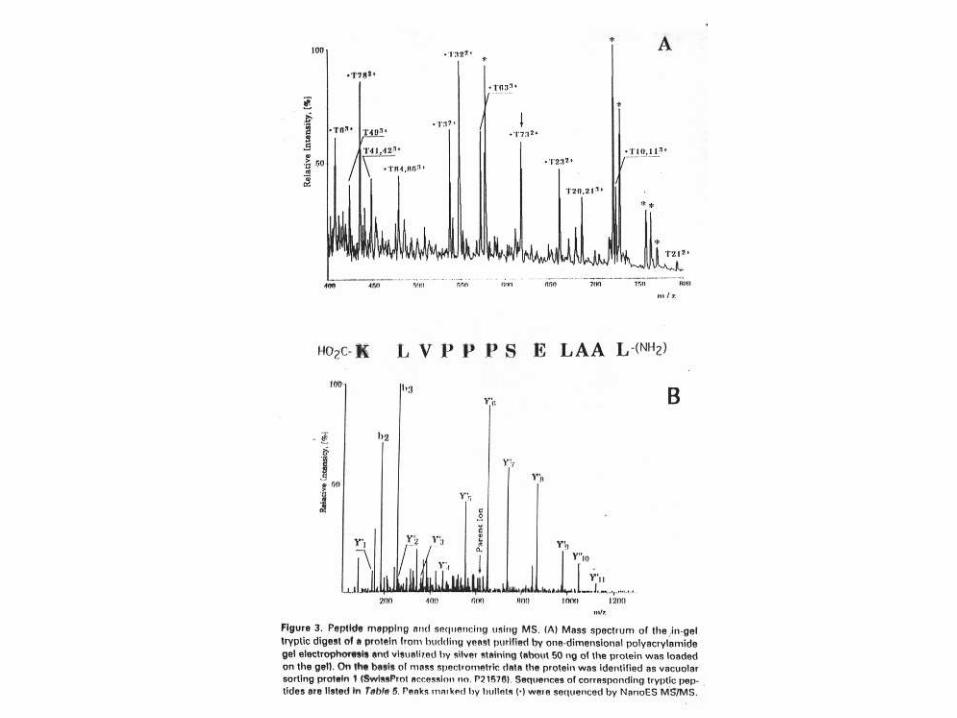

Es una técnica de fragmentación usada con
MALDI-ToF MS para obtener información
estructural, típicamente a partir de péptidos
menores que 2 kDa. El decaimiento de un ion
precursor seleccionado por su masa tiene
lugar después que el ión deja la fuente de
iones, en el tubo de vuelo, y los fragmentos
iónicos son separados por el reflectrón.
Post-source decay (PSD)

1. PSD se refiere a un método de detección y medida
de las masas de fragmentos de iones que se forman
a partir de un ión precursor seleccionado.
2. Los fragmentos de iones se forman principalmente
por descomposición unimolecular después que los
iones precursores están completamente acelerados
(después que dejan la fuente de iones, de ahí el
nombre de post-source decay)
3. Los fragmentos de iones son separados y
detectados en el reflectrón.
Fundamento de Post Source Decay
(PSD)
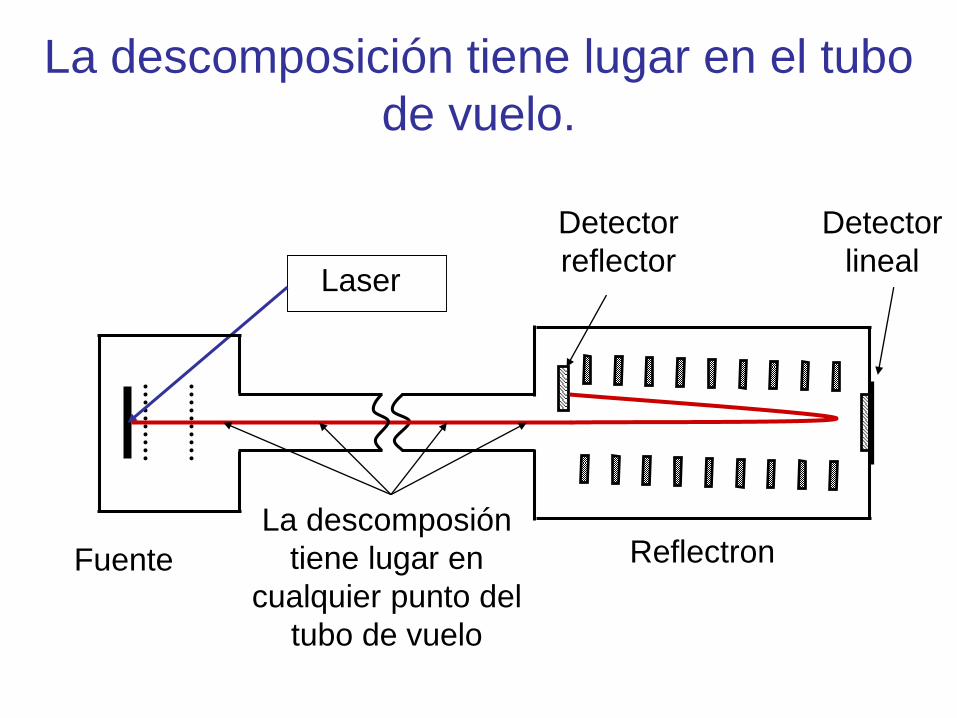
Laser
ReflectronFuente
Detector
lineal
Detector
reflector
La descomposión
tiene lugar en
cualquier punto del
tubo de vuelo
La descomposición tiene lugar en el tubo
de vuelo.
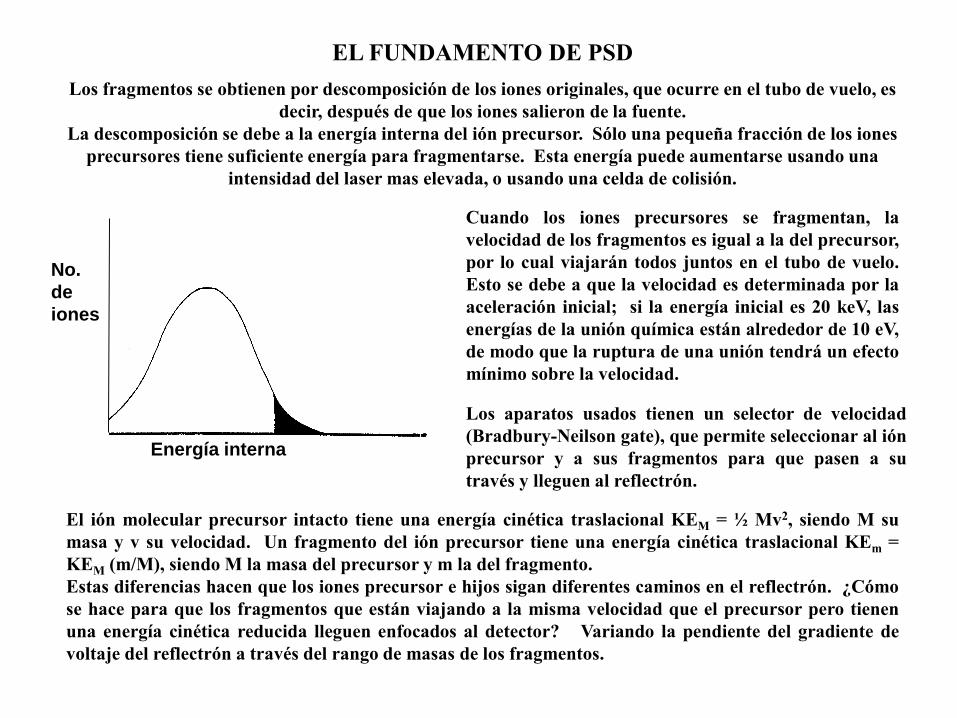
No.
de
iones
Energía interna
Los fragmentos se obtienen por descomposición de los iones originales, que ocurre en el tubo de vuelo, es
decir, después de que los iones salieron de la fuente.
La descomposición se debe a la energía interna del ión precursor. Sólo una pequeña fracción de los iones
precursores tiene suficiente energía para fragmentarse. Esta energía puede aumentarse usando una
intensidad del laser mas elevada, o usando una celda de colisión.
EL FUNDAMENTO DE PSD
Cuando los iones precursores se fragmentan, la
velocidad de los fragmentos es igual a la del precursor,
por lo cual viajarán todos juntos en el tubo de vuelo.
Esto se debe a que la velocidad es determinada por la
aceleración inicial; si la energía inicial es 20 keV, las
energías de la unión química están alrededor de 10 eV,
de modo que la ruptura de una unión tendrá un efecto
mínimo sobre la velocidad.
Los aparatos usados tienen un selector de velocidad
(Bradbury-Neilson gate), que permite seleccionar al ión
precursor y a sus fragmentos para que pasen a su
través y lleguen al reflectrón.
El ión molecular precursor intacto tiene una energía cinética traslacional KEM = ½ Mv2, siendo M su
masa y v su velocidad. Un fragmento del ión precursor tiene una energía cinética traslacional KEm =
KEM (m/M), siendo M la masa del precursor y m la del fragmento.
Estas diferencias hacen que los iones precursor e hijos sigan diferentes caminos en el reflectrón. ¿Cómo
se hace para que los fragmentos que están viajando a la misma velocidad que el precursor pero tienen
una energía cinética reducida lleguen enfocados al detector? Variando la pendiente del gradiente de
voltaje del reflectrón a través del rango de masas de los fragmentos.
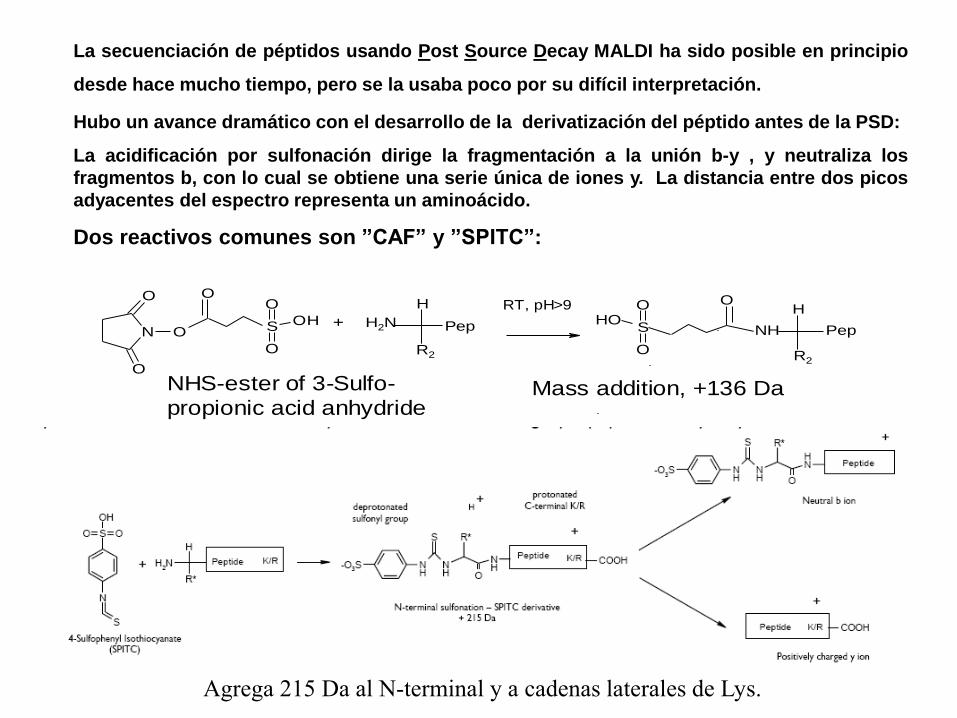
La secuenciación de péptidos usando Post Source Decay MALDI ha sido posible en principio
desde hace mucho tiempo, pero se la usaba poco por su difícil interpretación.
Hubo un avance dramático con el desarrollo de la derivatización del péptido antes de la PSD:
La acidificación por sulfonación dirige la fragmentación a la unión b-y , y neutraliza los
fragmentos b, con lo cual se obtiene una serie única de iones y. La distancia entre dos picos
adyacentes del espectro representa un aminoácido.
Dos reactivos comunes son ”CAF” y ”SPITC”:
N O
O
O
O
H2N
H
Pep
R2
+H
R2
Pep
O
NHS
O
OHO
S OH
O
O RT, pH>9
NHS-ester of 3-Sulfo-
propionic acid anhydrideMass addition, +136 Da
Agrega 215 Da al N-terminal y a cadenas laterales de Lys.

•El digerido tríptico se analiza por PMF.
•Para evitar la sulfonación de los e-aminos de Lys se pueden bloquear las Lys por
reacción con imidazol, que agrega 68 Da/Lys. Esto es específico, y no afecta al N-
terminal. Si no se lo hace, se observarán después de la sulfonación sólo los péptidos
con Arg. Luego se hace la sulfonación con el reactivo.
•La mezcla de péptidos modificados se analiza de nuevo por MALDI.
•Se buscan las masas que representan péptidos con Arg o Lys terminal, o sea los que
aumentaron su masa en 136 o 204 Da.
•Se pasa el aparato de MALDI a PSD y se selecciona esa masa de péptido.
•Se hace el análisis de PSD, que lleva aproximadamente 2 minutos en un TOF/TOF
MALDI.
Procedimiento experimental
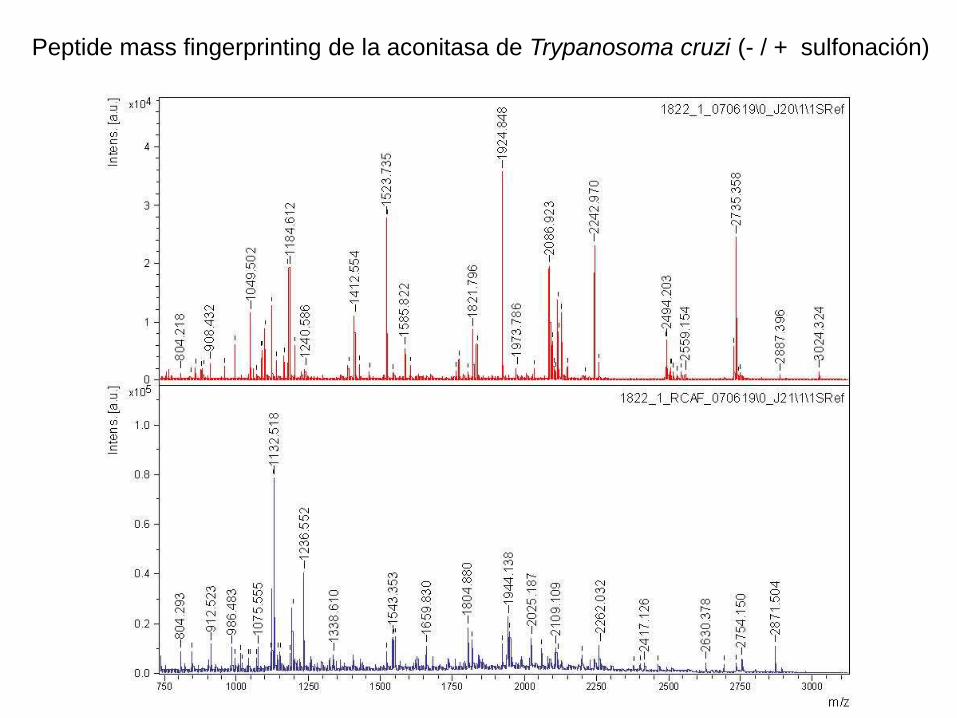
Peptide mass fingerprinting de la aconitasa de Trypanosoma cruzi (- / + sulfonación)
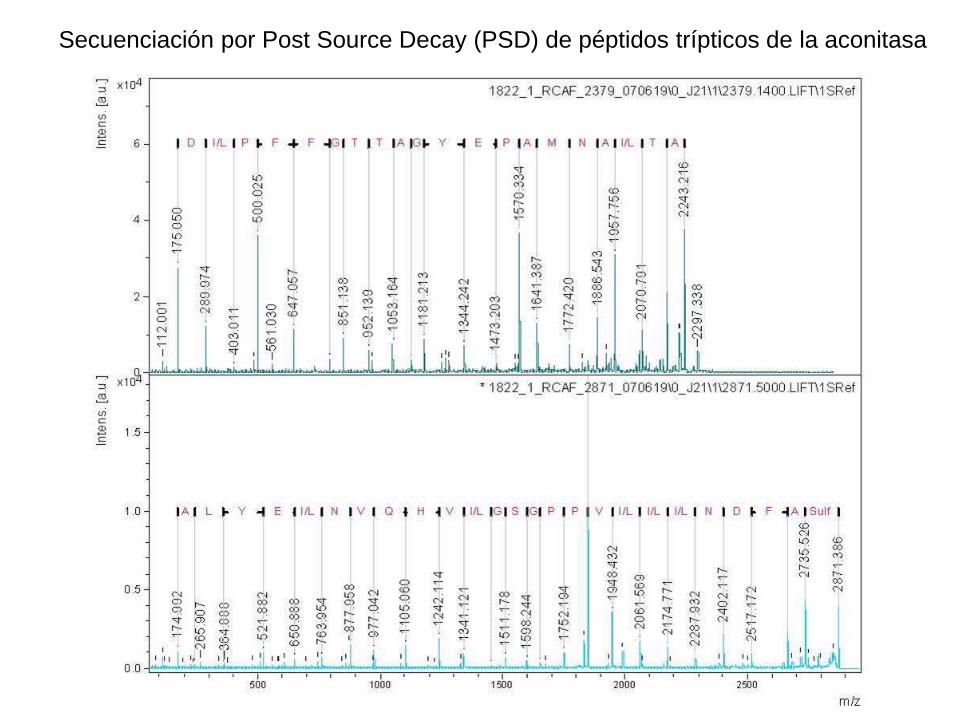
Secuenciación por Post Source Decay (PSD) de péptidos trípticos de la aconitasa

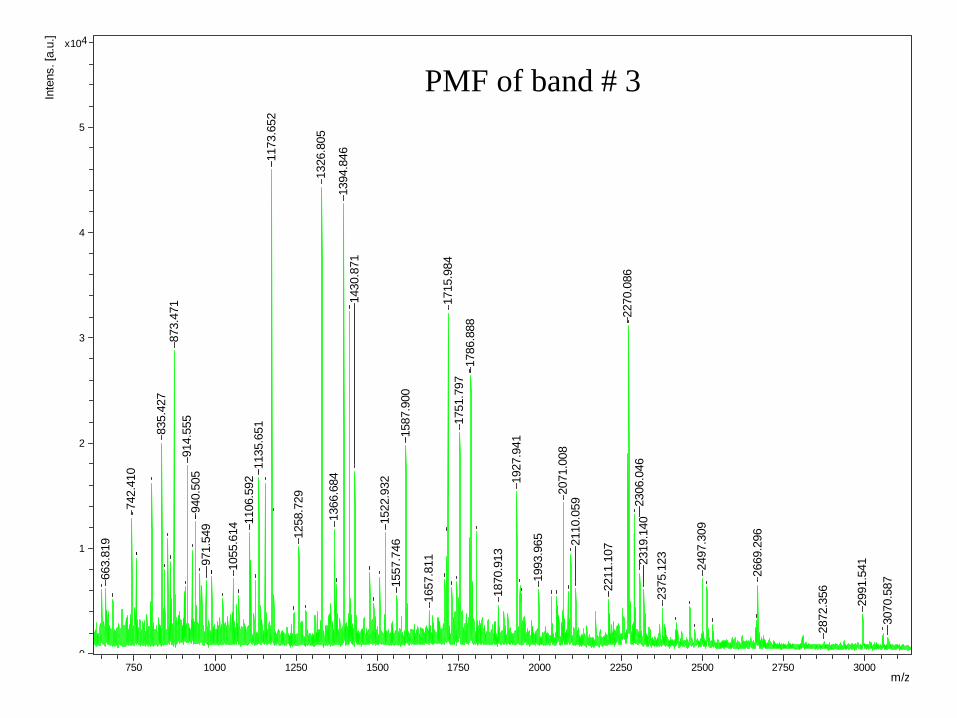
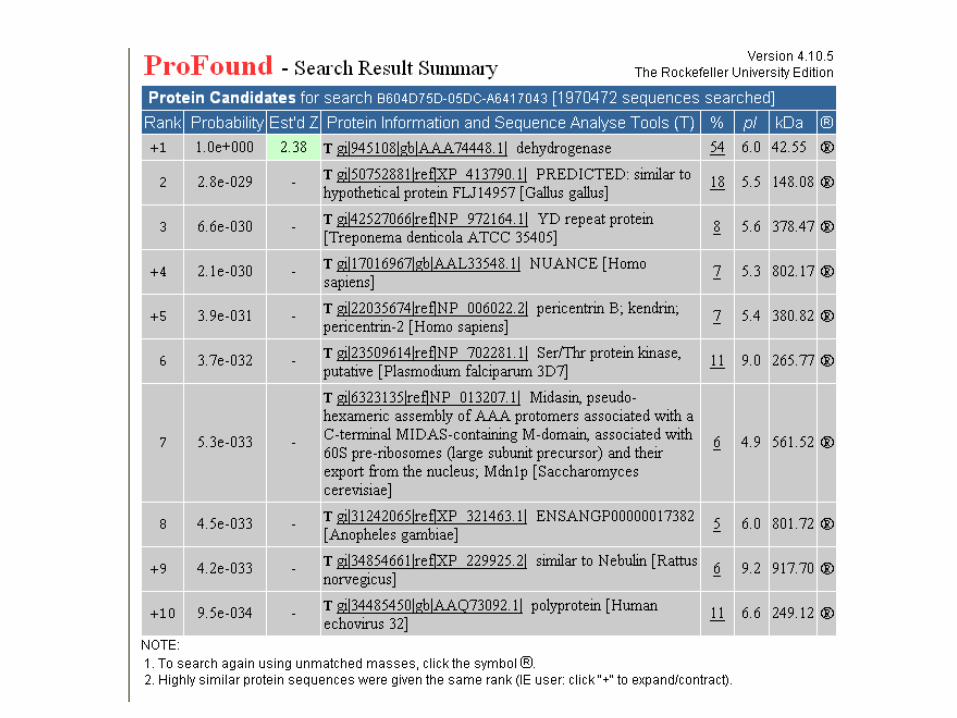
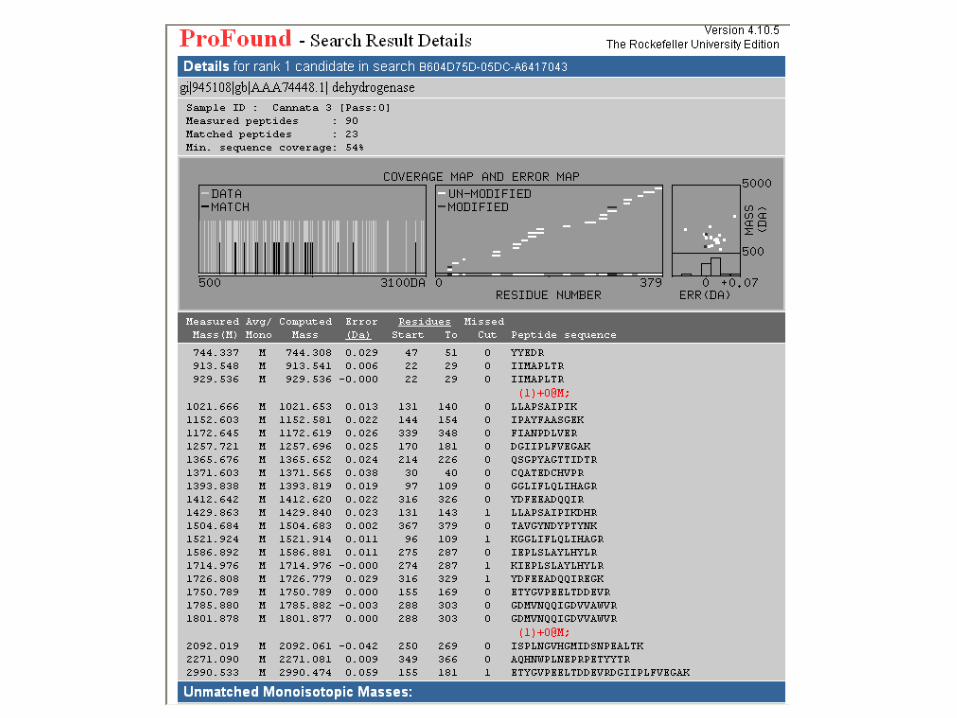
IDENTIFICACIÓN DE PROTEÍNAS POR PEPTIDE
MASS FINGERPRINTING (PMF)
• La muestra reducida y alquilada (en un fragmento de gel o mancha) se digiere
con una proteasa (tripsina?).
•La mezcla de péptidos resultante se analiza por MALDI-ToF-MS.
•La lista de masas de péptidos se utiliza para escanear un digerido in-silica de
una base de datos de secuencias por Internet.
•El resultado de la búsqueda se presenta como probability scores y otras
medidas de confianza.
•La homogeneidad de la muestra y la precisión de la medida son factores muy
importantes para una interpretación segura de los resultados.
1326.8
05
1173.6
52
1394.8
46
1715.9
84
873.4
71
1786.8
88
835.4
27
1751.7
97
1587.9
00
1430.8
71
914.5
55
1135.6
51
2270.0
86
1927.9
41
2071.0
08
742.4
10
1366.6
84
1522.9
32
1106.5
92
1258.7
29
940.5
05
971.5
49
1055.6
14
2306.0
46
2497.3
09
1870.9
13
2110.0
59
1993.9
65
663.8
19
1557.7
46
1657.8
11
2211.1
07
2669.2
96
2319.1
40
2375.1
23
2991.5
41
3070.5
87
2872.3
56
0
1
2
3
4
5
4x10In
tens. [a
.u.]
750 1000 1250 1500 1750 2000 2250 2500 2750 3000m/z
PMF of band # 3
MDH-1
MDH-2
LAS MALATO DEHIDROGENASAS DE
TRYPANOSOMA CRUZI
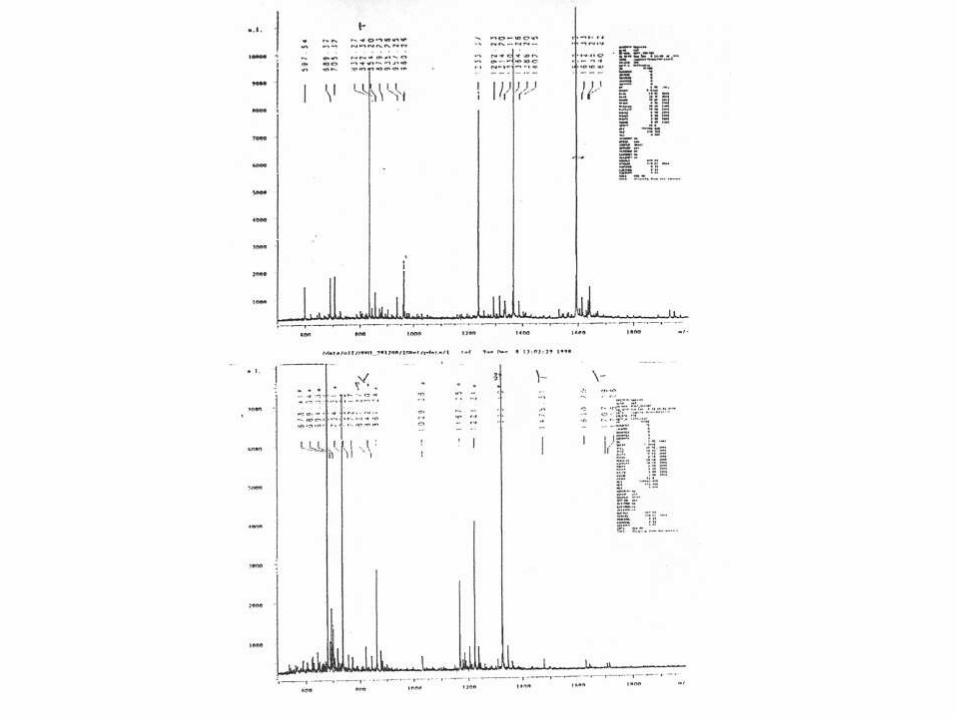
MDH-1
MDH-2

EDMAN


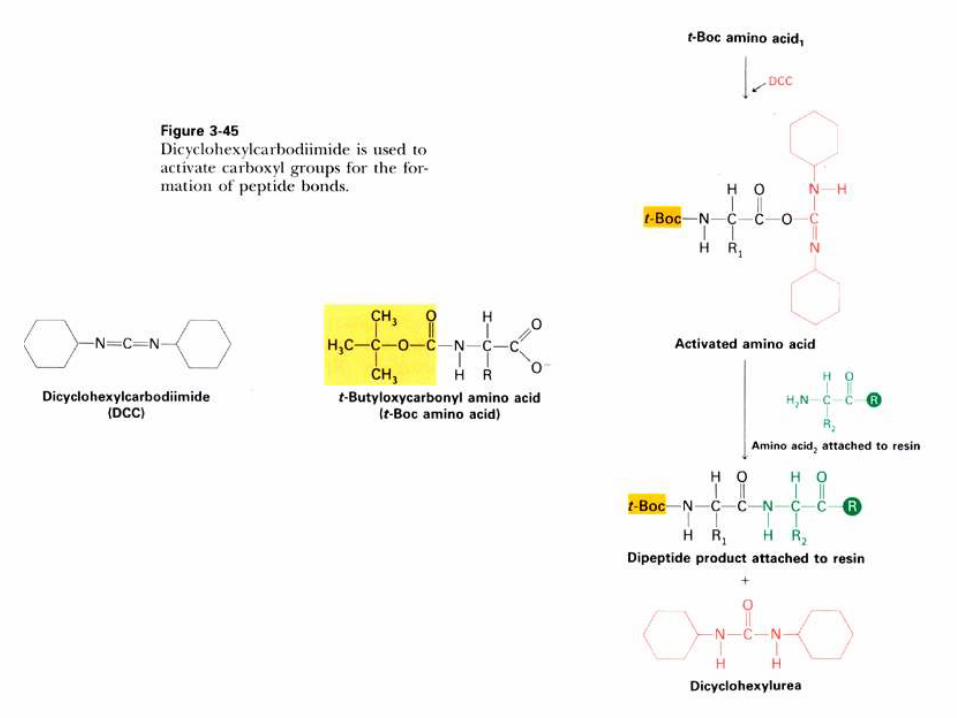
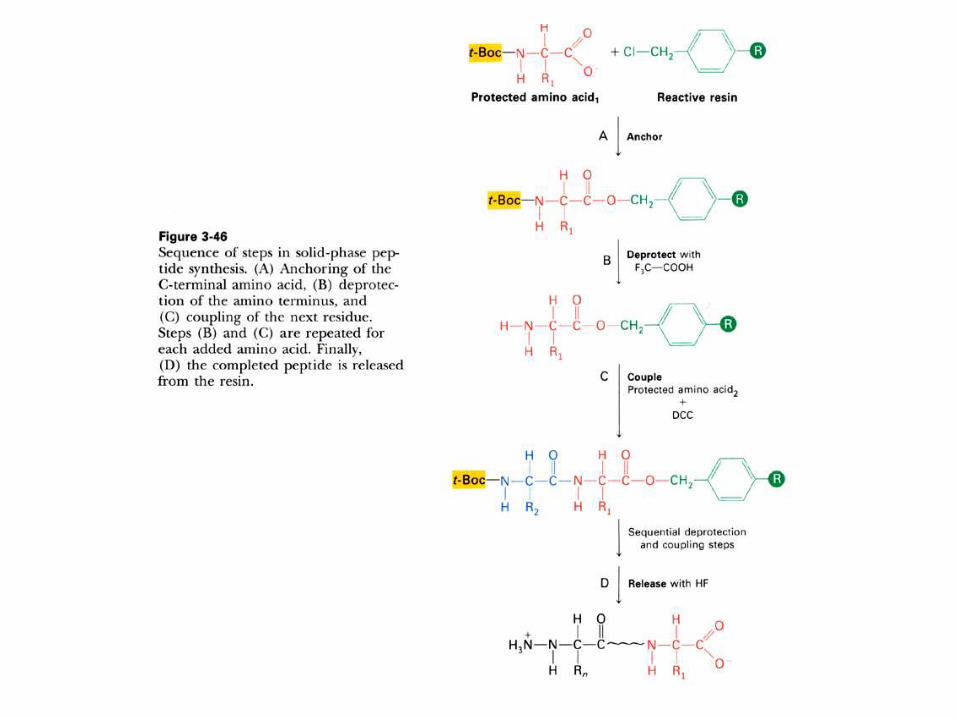
SINTESIS QUIMICA DE PEPTIDOS
La sintesis quimica de peptidos es muy importante actualmente, pues
permite a) estudiar el plegamiento de regiones individuales de una
proteína; b) aislar, usandolos como ligandos para cromatografía de
afinidad, a receptores y otras proteínas, c) usarlos como drogas,
p.ej.: la 1-desamino-8-D-Arg-vasopresina, hormona antidiurética sin
efectos sobre la presión sanguinea; d) para generar anticuerpos
específicos.
La sintesis se realiza en fase sólida, aplicando el método
desarrollado por Merrifield, que le permitió sintetizar interferon (155
residuos) y ribonucleasa (124 residuos), ambos activos
Los aminoácidos que se introducen deben estar bloqueados en los
sitios en que no debe producirse reacción, y luego desbloquearlos
para continuar la etapa siguiente. Se efectúa con el residuo C-
terminal ligado a una resina, lo que permite eliminar facilmente
subproductos.

INTRODUCCION
A LA
PROTEOMICA

GENOMA Y PROTEOMA
GENOME: “GENes and ChromosOMEs” (Winkler,
1920): Conjunto completo de los cromosomas y sus
genes.
PROTEOME: Conjunto completo de las proteínas de
un organismo (el conjunto de proteínas codificadas
por un genoma, Wilkins y Williams, 1994; Wasinger et
al., 1995).

La secuenciación de los genomas se hizo posible con la
introducción de los métodos de secuenciación del ADN, a partir
del establecimiento del método utilizando dideoxynucleótidos
por Frederick Sanger, en 1975, que le permitió secuenciar el
genoma completo del bacteriófago X174, con 5,375 pares de
bases, en 1977. La primera secuenciación completa del genoma
de un organismo de vida libre fué la de Haemophilus influenzae
(1.8 Mb), en 1995. El Human Genome Project comenzó en
1990 y se completó en 2003; la estimación del número total de
genes en el genoma humano se estima entre 20,000 y 25,000. Al
2 de Enero de 2018 la Kyoto Encyclopedia of Genes and
Genomes (KEGG) registraba ya 429 genomas eucarióticos
completos, además de 4521 genomas de bacterias y 263 de
Archaea.

DETERMINACIÓN DE LA FUNCIÓN DE LOS GENES
Determinación de las características químicas, bioquímicas y
biológicas de las proteínas.
1) Estructura molecular.
2) Modificaciones post-traduccionales.
3) Capacidad de unir ligandos.
4) Velocidad de síntesis y degradación. Concentracion
intracelular.
5) Especificidad de tejido, célula y organela.
6) Especificidad de sexo.
7) Especificidad de estadío de desarrollo embrionario, post natal
y de envejecimiento.
Se estima en 252 el número de tipos celulares diferentes que se encuentran
en el hombre. Todas esas células tienen el mismo DNA, pero todas
ellas presentan diferencias cuali y cuantitativas en sus proteínas. El
DNA almacena información; las proteínas hacen todo lo demás.
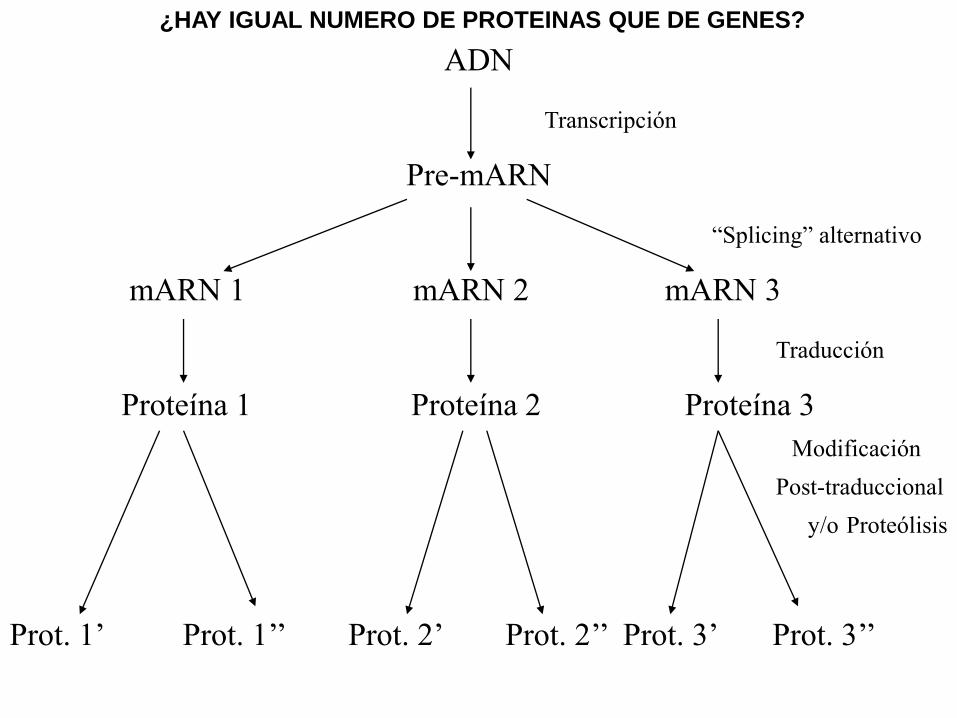
ADN
Transcripción
Pre-mARN
“Splicing” alternativo
mARN 1 mARN 2 mARN 3
Traducción
Proteína 1 Proteína 2 Proteína 3
Modificación
Post-traduccional
y/o Proteólisis
Prot. 1’ Prot. 1’’ Prot. 2’ Prot. 2’’ Prot. 3’ Prot. 3’’
¿HAY IGUAL NUMERO DE PROTEINAS QUE DE GENES?

Para la funcionalidad adecuada de una proteína se
requiere mucho mas que la integridad del gen
estructural que la codifica. Si este está mutado, y el
producto es inactivo, naturalmente la proteína no será
funcional. Pero puede suceder algo parecido si el gen
estructural está intacto, pero hay mutaciones en otros
genes que participan en lo que realmente le interesa a la
célula, que es la proteína perfectamente plegada, con las
modificaciones post-traduccionales requeridas y
localizada en el compartimento subcelular correcto.
LA NATURALEZA POLIGENICA DE LAS PROTEINAS.
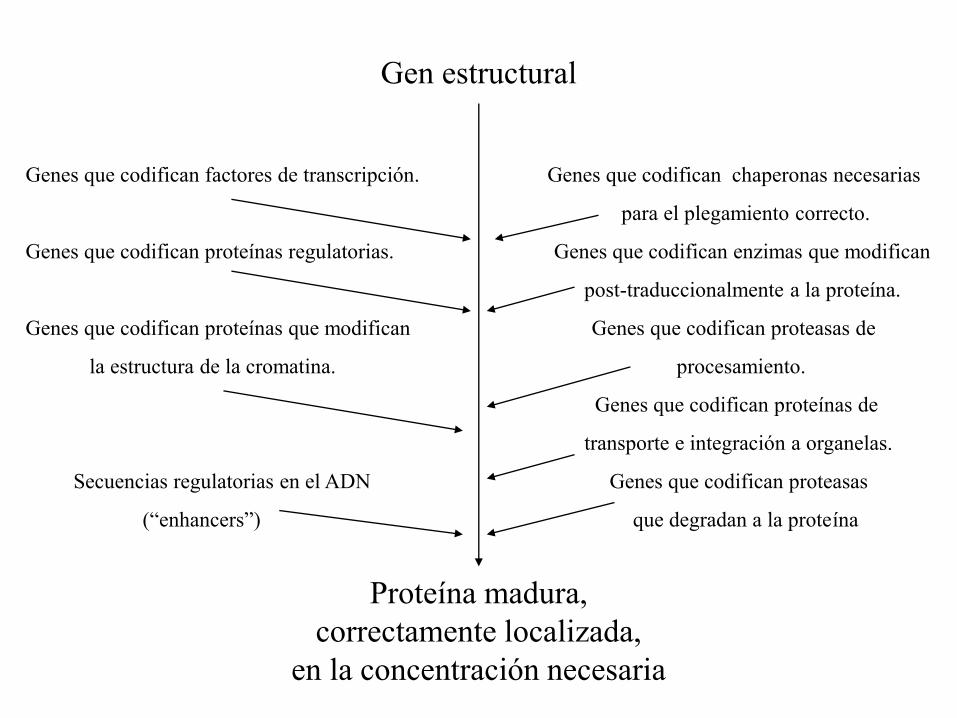
Gen estructural
Genes que codifican factores de transcripción. Genes que codifican chaperonas necesarias
para el plegamiento correcto.
Genes que codifican proteínas regulatorias. Genes que codifican enzimas que modifican
post-traduccionalmente a la proteína.
Genes que codifican proteínas que modifican Genes que codifican proteasas de
la estructura de la cromatina. procesamiento.
Genes que codifican proteínas de
transporte e integración a organelas.
Secuencias regulatorias en el ADN Genes que codifican proteasas
(“enhancers”) que degradan a la proteína
Proteína madura,
correctamente localizada,
en la concentración necesaria

DESAFIOS TECNICOS DE LA PROTEOMICA.
1) Si suponemos alrededor de 22,000 genes en el genoma humano, y
que cada gen da origen a entre 3 y 6 o más proteínas, el número total
de proteínas en un organismo superior puede variar entre 50,000 y
500,000.
2) Las proteínas tienen una heterogeneidad mucho mayor que los
ácidos nucleicos. La composición y secuencia de aminoácidos de una
proteína determina: a) su masa molecular; b) su carga total a
diferentes valores de pH; c) su hidrofobicidad; d) su forma, es decir
el plegamiento de la proteína nativa. Esto a su vez determina la
solubilidad de las proteínas en diferentes solventes, su
comportamiento en los métodos de separación y purificación, y su vida
media in vitro. La diversidad de propiedades fisicoquímicas es
muchísimo mayor que la de los ácidos nucleicos.
3) Los valores de pI de las proteínas varían entre menos de 3 y más de
12, con dos grandes agrupamientos, uno alrededor de 5.5 y el otro
alrededor de 8.5. Los ácidos nucleicos tienen un rango estrecho de pI,
en la zona ácida.

4) Los ácidos nucleicos son muy solubles en soluciones acuosas, en
tanto que muchas proteínas son muy hidrofóbicas, y por ello insolubles
en agua. Se estima que más del 15 % de las proteínas totales nunca
entrarán en un gel de 2D-PAGE por su extrema hidrofobicidad.
5) La masa molecular de las proteínas varía entre unos pocos miles y
varios millones de Daltons, y su tamaño no puede predecirse con
certeza a partir de la secuencia de aminoácidos derivada de la
secuencia de DNA, por las modificaciones post-traduccionales.
6) La concentración de las proteínas varía dentro de un rango muy
amplio, probablemente mayor que el rango de los mRNAs. La
diferencia en concentración de proteínas abundantes y raras en los
flúidos corporales es mayor que 12 órdenes de magnitud.

LAS HERRAMIENTAS PARA EL ESTUDIO DEL
PROTEOMA
La técnica clásica, y hasta hace unos pocos años, la
principal utilizada, es la separación de las proteínas por
electroforesis bidimensional en geles de poliacrilamida, seguida de
identificación de dichas proteínas por secuenciación o, en general
actualmente, por espectrometría de masa, después de hidrólisis
enzimatica parcial.
Actualmente lo mas utilizado para estudios amplios de
proteómica son otras combinaciones de técnicas, por ejemplo
cromatografía de alta resolución (HPLC) combinada con
espectrometría de masa, o electroforesis capilar tambien combinada
con espectrometría de masa. Este tipo de técnicas permite la
identificación de proteínas en mezclas complejas, previa hidrólisis
enzimática parcial.
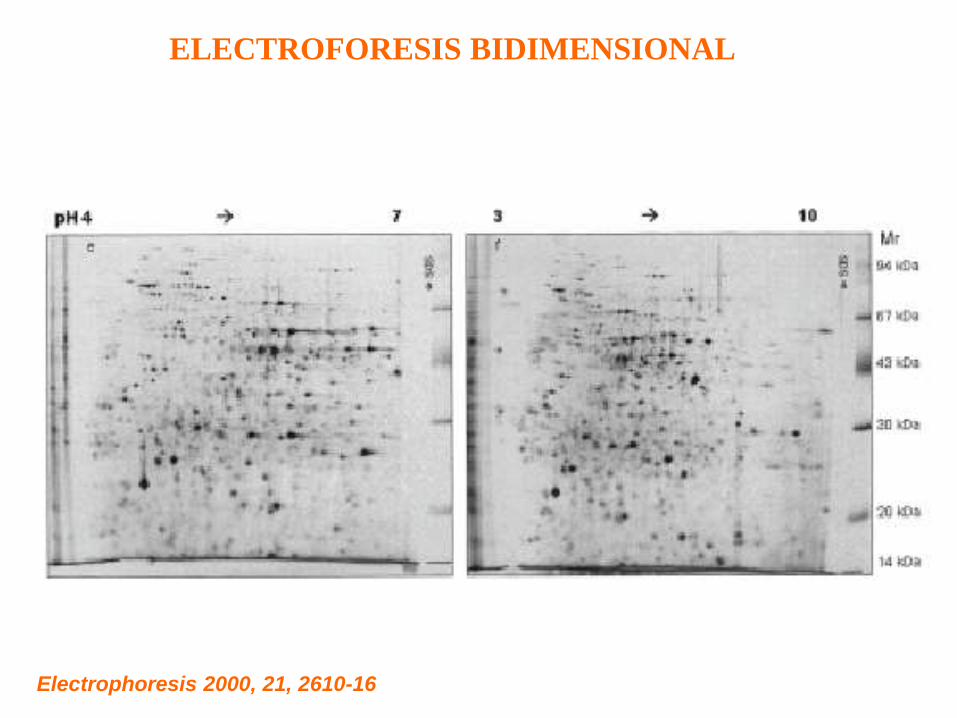
ELECTROFORESIS BIDIMENSIONAL
Electrophoresis 2000, 21, 2610-16

VENTAJAS Y DESVENTAJAS DE LA ELECTROFORESIS BIDIMENSIONAL
EN EL ESTUDIO DEL PROTEOMA.
Ventajas:
Excelente resolución: pueden identificarse a veces hasta 10,000 proteínas
en un solo gel. Es una técnica capaz de analizar simultáneamente miles
de proteínas.
Desventajas:
1)Pueden ser necesarios varios geles, corridos en diferentes condiciones
de rango de pH y de concentración de acrilamida, para resolver las
mezclas muy complejas.
2) Es difícil detectar proteínas muy grandes o muy chicas, muy ácidas o
muy básicas, o proteínas de membrana (muy hidrofóbicas).
3) Se detectan sólo las proteínas de alta y mediana abundancia en el
material en estudio. En cualquier célula, sus proteínas pueden variar en
concentración en seis órdenes de magnitud. Para ver proteínas
minoritarias, es necesario en general purificar la organela en que se
encuentran y trabajar con ese material. Tambien se puede trabajar previa
marcación radioactiva, para aumentar la sensibilidad de detección.
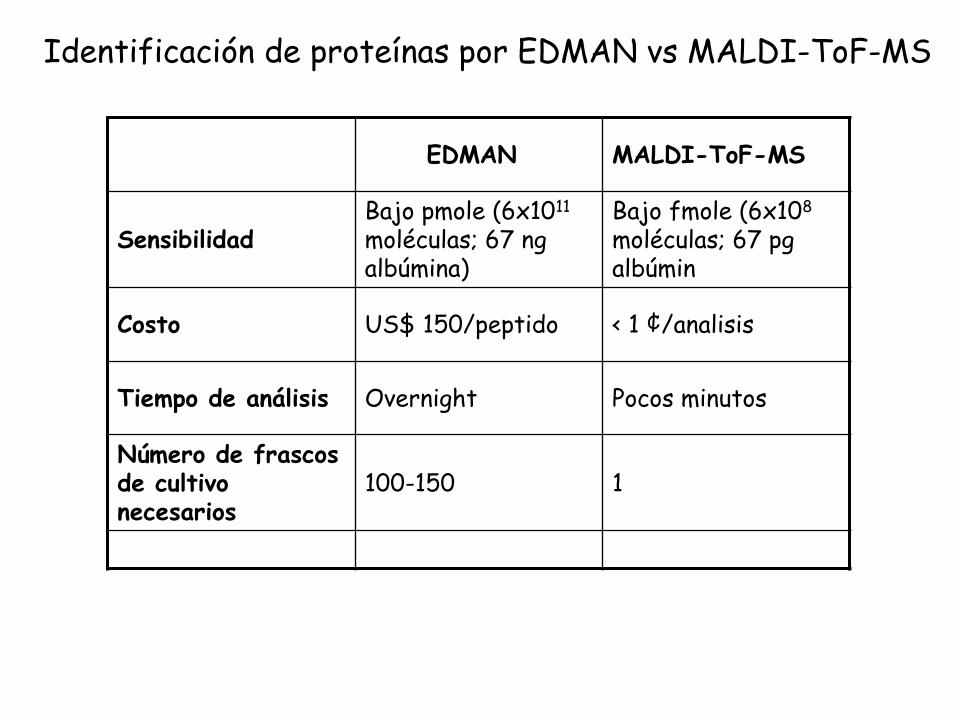
EDMAN MALDI-ToF-MS
SensibilidadBajo pmole (6x1011
moléculas; 67 ng albúmina)
Bajo fmole (6x108
moléculas; 67 pg albúmin
Costo US$ 150/peptido < 1 ¢/analisis
Tiempo de análisis Overnight Pocos minutos
Número de frascos de cultivo necesarios
100-150 1
Identificación de proteínas por EDMAN vs MALDI-ToF-MS

Electroforesis diferencial en gel (DIGE).
Se marcan las dos muestras de proteína que se quieren comparar
con dos colorantes diferentes, reactivos con el amino, y diseñados
para mantener la misma movilidad relativa.
Se mezclan las dos muestras de proteínas marcadas y se las corre
en un mismo gel bidimensional.
Después de la corrida se crean dos imágenes de fluorescencia del
gel que se superponen para identificar diferencias en el patrón.
Esta técnica hace innecesario comparar geles diferentes.
Está disponible comercialmente.
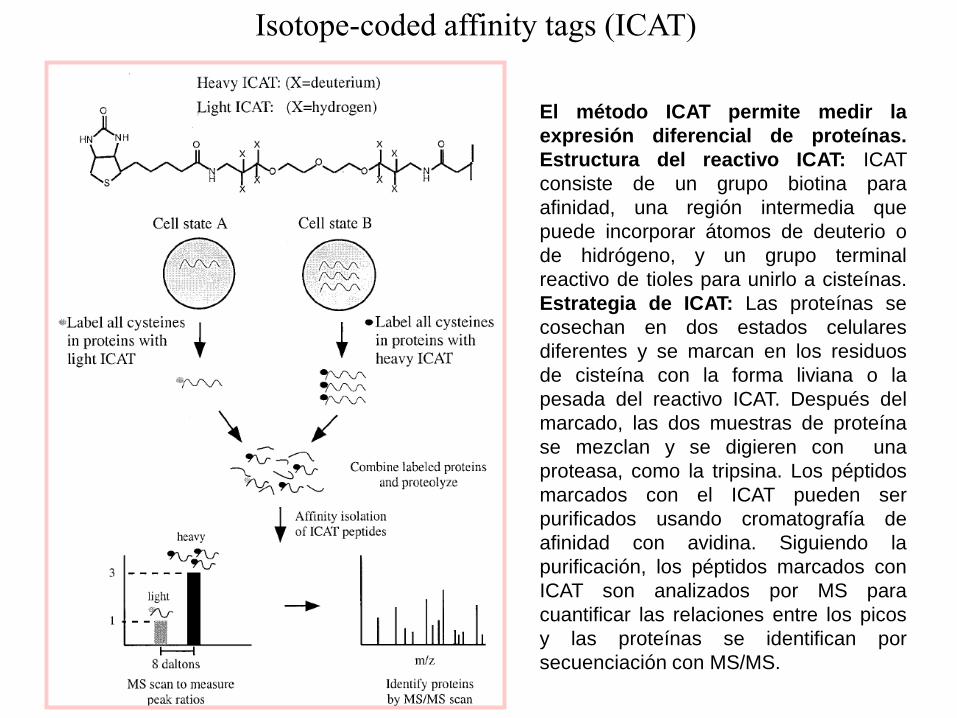
El método ICAT permite medir la
expresión diferencial de proteínas.
Estructura del reactivo ICAT: ICAT
consiste de un grupo biotina para
afinidad, una región intermedia que
puede incorporar átomos de deuterio o
de hidrógeno, y un grupo terminal
reactivo de tioles para unirlo a cisteínas.
Estrategia de ICAT: Las proteínas se
cosechan en dos estados celulares
diferentes y se marcan en los residuos
de cisteína con la forma liviana o la
pesada del reactivo ICAT. Después del
marcado, las dos muestras de proteína
se mezclan y se digieren con una
proteasa, como la tripsina. Los péptidos
marcados con el ICAT pueden ser
purificados usando cromatografía de
afinidad con avidina. Siguiendo la
purificación, los péptidos marcados con
ICAT son analizados por MS para
cuantificar las relaciones entre los picos
y las proteínas se identifican por
secuenciación con MS/MS.
Isotope-coded affinity tags (ICAT)

Si bien la idea última detrás de los estudios de
proteómica es llegar algún día al conocimiento completo
de las proteínas de un organismo, lo mas frecuente
actualmente es hacer proteómicas parciales: la
proteómica de un tipo celular determinado; la proteómica
de una organela aislada; la proteómica de las proteínas
blanco de una determinada modificación post-
traduccional; las diferencias en la expresión de proteínas
antes y después de la diferenciación de un estadío a otro
en un organismo que tiene un ciclo de vida complejo; las
diferencias en la expresión de proteínas de una misma
célula antes y después de un determinado tratamiento.
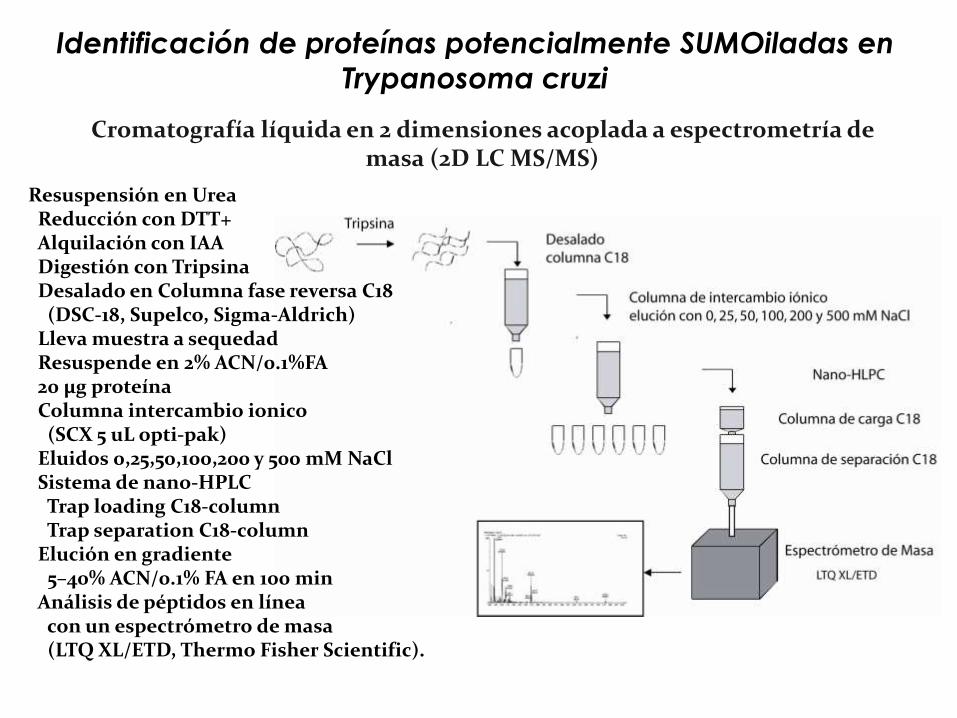
Cromatografía líquida en 2 dimensiones acoplada a espectrometría de masa (2D LC MS/MS)
Identificación de proteínas potencialmente SUMOiladas en
Trypanosoma cruzi
Resuspensión en UreaReducción con DTT+ Alquilación con IAADigestión con TripsinaDesalado en Columna fase reversa C18
(DSC-18, Supelco, Sigma-Aldrich)Lleva muestra a sequedadResuspende en 2% ACN/0.1%FA 20 μg proteínaColumna intercambio ionico
(SCX 5 uL opti-pak)Eluidos 0,25,50,100,200 y 500 mM NaClSistema de nano-HPLCTrap loading C18-columnTrap separation C18-column
Elución en gradiente 5–40% ACN/0.1% FA en 100 min
Análisis de péptidos en línea con un espectrómetro de masa(LTQ XL/ETD, Thermo Fisher Scientific).
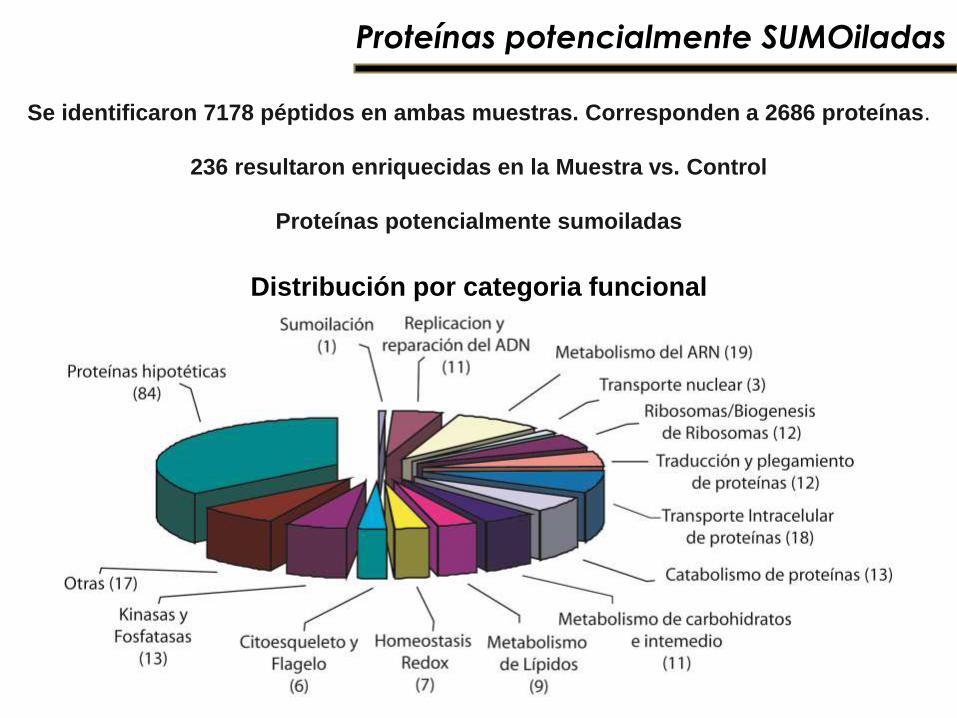
Proteínas potencialmente SUMOiladas
Distribución por categoria funcional
Se identificaron 7178 péptidos en ambas muestras. Corresponden a 2686 proteínas.
236 resultaron enriquecidas en la Muestra vs. Control
Proteínas potencialmente sumoiladas
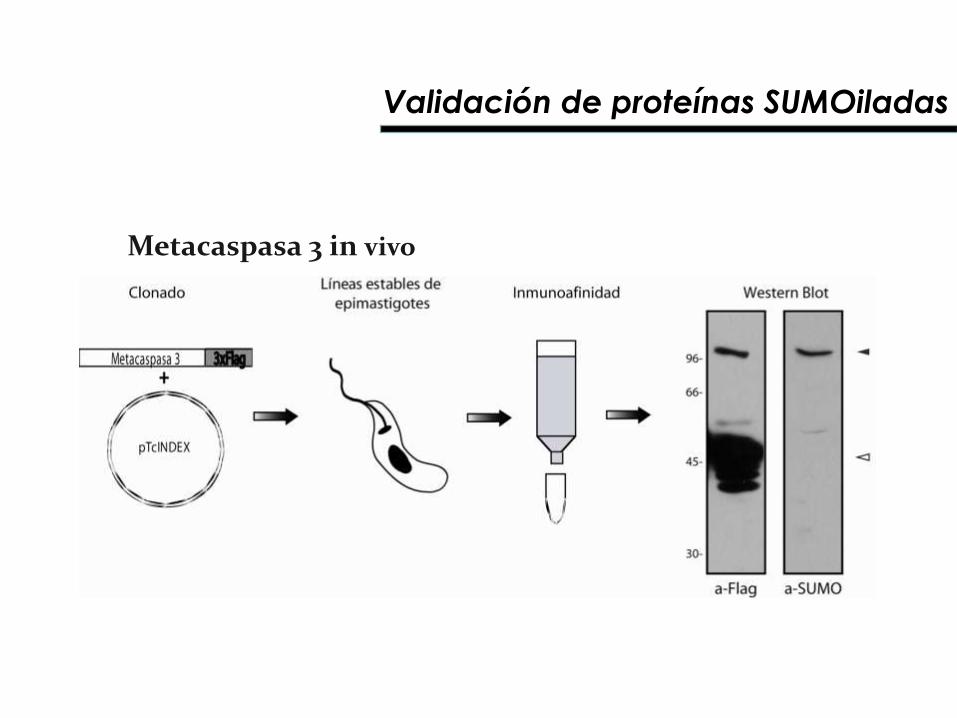
Metacaspasa 3 in vivo
Validación de proteínas SUMOiladas
















![[aminoacidos peptidos]](https://img.pdfslide.tips/doc/110x75/5571f35249795947648dd668/aminoacidos-peptidos.jpg)












