Embed Size (px)
Citation preview

1
CARIÓTIPO EABERRAÇÕES
CROMOSSÔMICAS
Profª Ana Luisa Miranda Vilela
2
CARIÓTIPOConjunto de cromossomos típico de uma espécie(número e morfologia).
metacêntrico submetacêntrico acrocêntrico telocêntrico
Classificação dos cromossomos quanto à posição dos centrômeros.

2
3
N° DE CROMOSSOMOS X ESPÉCIESNúmero de cromossomos varia entre as diversasespécies biológicas:
ESPÉCIE N° DE CROMOSSOMOSBrachiaria brizantha (gramínea) 2n = 36
Brachiaria dictyoneura (gramínea) 2n = 42Canis familiaris (cão) 2n = 78
Chrysocyon brachyurus (lobo guará) 2n = 76Equus caballus (cavalo) 2n = 64
Pan troglodytes (chipanzé) 2n = 48Homo sapiens (homem) 2n = 46
4
ANÁLISE DOSCROMOSSOMOS HUMANOS
autossomos não diferencia os sexos
22 pares ~ homens e mulheres
cromossomos sexuaisou alossomos
diferencia os sexos
1 par # homens e mulheres
2n = 23 paresde cromossomos
HOMEM

3
5
EXAME DE CARIÓTIPO
6
CARIÓTIPO HUMANO - MONTAGEMA padronização internacional da nomenclatura doscromossomos engloba uma série de normas:
os cromossomos são identificados pelo tamanho,posição do centrômero e padrão de bandas;são organizados em pares de homólogos, por ordemdecrescente de tamanho.

4
7
CARIÓTIPO HUMANO - BANDAS
O centrômero divideo cromossomo emdois braços:
p = braço curto;q = braço longo.
Nos cromossomosmetacêntricos os doisbraços são de igualtamanho.
8
CARIÓTIPO HUMANO - GRUPOS
A: cromossomos 1 a 3 cromossomos grandes,metacêntricos e submetacêntricos.B: cromossomos 4 e 5 cromossomos grandessubmetacêntricos.C: cromossomos 6 a 12, X cromossomossubmetacêntricos de tamanho médio.
Cromossomo 1:metacêntricoCromossomo 9:
submetacêntrico
p: braço curto
q: braço longo

5
9
CARIÓTIPO HUMANO - GRUPOS
D: cromossomos 13 a 15 cromossomosacrocêntricos.
Cromossomo 14:acrocêntrico
E: cromossomos 16 a 18 cromossomosrelativamente curtos, metacêntricos ousubmetacêntricos.F: cromossomos 19 e 20 cromossomosmetacêntricos pequenos com padrão debandas característico.G: cromossomos 21 e 22, Ycromossomos pequenos acrocêntricos compadrão de bandas característico.
10
CARIÓTIPO HUMANO NORMALMasculino (15 cromossomos no grupo C e 5 no grupo G)
figura abaixo à esquerda.Feminino (16 cromossomos no grupo C e 4 no grupo G)
figura abaixo à direita.

6
11
MÉTODOS DE BANDEAMENTO- HISTÓRICO -
Torbjörn Oskar Caspersson et al.(1968) descobriram quea fluorescência dos cromossomos após coloração comquinacrina mostarda mostra uma seqüência distinta debandas para cada cromossomo.Mais tarde padrões de bandeamento podiam também serproduzidos por coloração Giemsa com técnicas adicionais.Conferência de Paris (1971) padronização e nomenclaturados cromossomos todos os métodos revelaram a mesmaestrutura:
algumas técnicas mostraram alguns segmentos cromossômicosmais claramente, enquanto outras funcionaram melhor comoutros segmentos.
12
MÉTODOS DE BANDEAMENTOOs vários tipos debandas foramdesignados de acordocom a técnica pela qualsão mais claramenterevelados.Métodos disponíveis:1- Bandas Q(quinacrina) bandasfluorescentes visíveisapós coloração comquinacrina mostarda oucompostos similares.

7
13
MÉTODOS DE BANDEAMENTO2- Bandas G(Giemsa)reveladas porcoloração Giemsa etécnicas adicionais
garantem queapenas os segmentoscromossômicos maisprontamentecoráveis captem ocorante idênticasàs bandas Q.
14
BANDEAMENTO G- MASCULINO -

8
15
BANDEAMENTO G- FEMININO -

16
Gene constitutivo (em inglês, house keeping gene):gene expresso continuamente e em todas as células de um organismo. O mesmoque gene de manutenção.

1
17
MÉTODOS DE BANDEAMENTO
3- Bandas R (reversas)coradas após desnaturaçãocontrolada por aquecimentosituadas entre as bandas Q ouG comportam-se como umnegativo de fotografia.4- Bandas C (heterocromatinaconstitutiva) situadas nasregiões pericentroméricas.5- Bandas T (teloméricas)marcam as regiões teloméricasdos cromossomos. Banda C
18
CITOGENÉTICA MOLECULAR (FISH)FISH hibridação in situ por fluorescência:
utilização de sondas (seqüências de DNA), marcadas comfluorescência ligam-se por complementaridade a seus alvosnos cromossomos ou a seus cromossomos-alvos.possibilidade de se identificar genes, cromossomos ou regiõesespecíficas dos cromossomos.

2
19
INDICAÇÕES DE CARIÓTIPOSuspeita de síndrome cromossômica conhecida.Padrão não reconhecível de duas ou mais malformaçõesprimárias crianças polimalformadas.Recém-nascidos com genitália ambígua definir o sexo.Retardo mental ou atraso de desenvolvimento em criançasdisfórmicas ou com múltiplas malformações.Mulheres com baixa estatura proporcional e amenorréiaprimária.Homens com ginecomastia acentuada e hipogonadismo.Homens com hipertrofia peniana, hipergonadismo e retardomental.Natimortos com malformações ou sem motivo reconhecívelpara a morte fetal.

20
As síndromes de instabilidade cromossômica citadas são de herança autossômicarecessiva.

1
21
ANOMALIAS(ABERRAÇÕES)
CROMOSSÔMICASHUMANAS
22
ANOMALIAS CROMOSSÔMICAS
Constituem uma causa de anormalidades físicas e/oufuncionais, embora existam situações em que não semanifestam clinicamente.Podem ser:
numéricas implicam em acréscimos ou decréscimos nonúmero de autossomos ou de cromossomos sexuais;estruturais podem atingir apenas um gene – mutaçõespontuais – ou resultar em alterações cromossômicas maisalargadas – aberrações cromossômicas (deleção, inversão,translocação etc).
Dependendo do tipo de alteração, podem existir váriasanomalias associadas, que por apresentarem sintomastípicos são preferencialmente designadas de síndromes.

2
23
ABERRAÇÕES CROMOSSÔMICAS1- Numéricas alteração no número decromossomos:
euploidias alteração do conjunto de cromossomos (3n -triploidia; 4n - tetraploidia);aneuploidias alterações no número (falta ou excessode um ou mais de um cromossomo da espécie):
autossômicas (cromossomos não sexuais);alossômicas (cromossomos sexuais).
2- Estruturais alterações na estrutura doscromossomos:
deleção, inversão, translocação, duplicação,isocromossomos, cromossomos dicêntricos, cromossomosem anel.
24
FREQÜÊNCIA DE ANOMALIASCROMOSSÔMICAS EM HUMANOS
In: ANDERSON, P.; GANETZKY, B. An Eletronic Companion to Genetics – 1997.

3
25
ABERRAÇÕESCROMOSSÔMICAS
NUMÉRICAS
26
EUPLOIDIASAlteração do conjunto de cromossomos (3n -Triploidia; 4n - Tetraploidia).

4
27
EUPLOIDIA - TRIPLOIDIAAberração cromossômicamais freqüente (20%) emabortos espontâneos.Retardamento severo docrescimento, letalidadeprecoce (máximo 5 meses).Nascimento de criançascom malformações severasé muito raro.Geralmente causada peladispermia (doisespermatozóides - 1 óvulo)
28
ANEUPLOIDIASAlterações no número (falta ou excesso de um ou maiscromossomos da espécie) erros de não-disjunçãocromossômica nas divisões celulares monossomias,trissomias, tetrassomias, pentassomias.Podem ser:Autossômicas (cromossomos não sexuais):
Síndrome de Down ou Trissomia do 21- 47, XX ou XY + 21Síndrome de Edwards ou Trissomia do 18 - 47, XX ou XY + 18Síndrome de Patau ou Trissomia do 13 - 47, XX ou XY + 13
Alossômicas (cromossomos sexuais):Síndrome de Turner (monossomia) - 45, X0Síndrome de Klinefelter - 47, XXYSíndrome do Triplo X - 47, XXXSíndrome do Duplo Y - 47, XYY
5
29
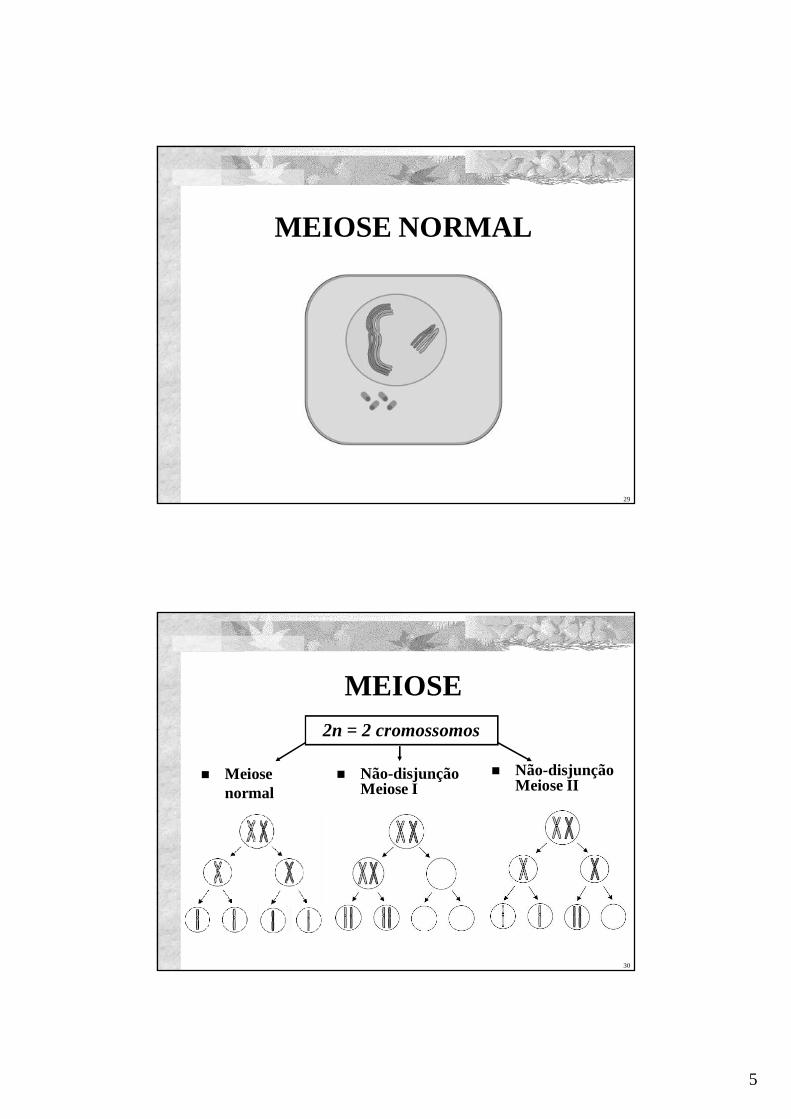
MEIOSE NORMAL
30
MEIOSE
Não-disjunçãoMeiose I
Não-disjunçãoMeiose II
Meiosenormal
2n = 2 cromossomos

6
31
NÃO-DISJUNÇÃO CROMOSSÔMICA
32
IDADE MATERNA X RISCO DENÃO-DISJUNÇÃO CROMOSSÔMICA

7
33
ANEUPLOIDIASAUTOSSÔMICAS
34
TRISSOMIAS

8
35
1- SÍNDROME DE DOWN(TRISSOMIA DO 21)
Trissomia autossômica mais comum:47, XX ou XY + 21.
36
SÍNDROME DE DOWNCaracterísticas típicas: deficiência mental moderada,problemas cardíacos congênitos, além de:
Perfil achatado Orelhas pequenas edismórficas
Olhos com fendaspalpebrais oblíquas(prega epicântica)
Boca aberta com línguagrande, protrusa e sulcada
Encurvamentodos quintos
dígitos(clinodactilia)
Amplo espaço entre oprimeiro e o segundo
dedos dos pés
Prega simiesca

9
37
IDADE MATERNA X SÍNDROME DE DOWN
Freqüências relativas de tipos particulares de alteraçõescromossômicas na síndrome de Down:
Trissomia 21 completa .........................94,0%Trissomia 21/mosaicismo normal.........2,4%Casos de translocação...........................3,3%
38
2- SÍNDROME DE EDWARDS(TRISSOMIA DO 18)

10
39
SÍNDROME DE EDWARDSSegunda trissomia mais comum nos seres humanos:
incidência: 1/6.000-1/8.000 nascimentos.Origem:
trissomia livre: 90-94% dos casos;mosaicismo: 5-10% dos casos;trissomias parciais: incidência muito pequena.
Associada a uma elevada taxa de mortalidade intra-uterina:
estima-se que apenas 2,5% dos conceptos sobrevivam.Mortalidade pós-natal elevada:
55-65% morrem ao redor de 6 meses de idade;apenas 5-10% sobrevivem até o 1° ano;possibilidade de atingir a idade adulta: mosaicismo.
40
SÍNDROME DE EDWARDS
Características típicas:deficiência mental e decrescimento (estatura baixa);anomalias renais e do aparelhoreprodutor;palato alto e estreito (por vezeslábio leporino e palato fendido);defeitos oculares;acentuada malformação cardíaca;além de:

11
41
SÍNDROME DE EDWARDS
Hipertonicidade,esterno e pescoço
curtos, pelveestreita
2° e 5° dedos sesobrepondo ao 3° e 4°
Micrognatia(mandíbula recuada)
Pés viradospara fora ecalcanharsaliente
Crânio alongado na regiãooccipital e orelhas com
implantação baixa
42
SÍNDROME DE EDWARDS
Ocorrência: 1/6.000-1/8.000 nascimentos5-10% sobrevivem durante o 1° ano:
associação com idade materna avançada namaior parte dos casos (85%), o erro é decorrenteda não-disjunção cromossômica durante a meiosematerna (somente 15% são decorrentes da meiosepaterna).

12
43
3- SÍNDROME DE PATAU(TRISSOMIA DO 13)
44
SÍNDROME DE PATAUCaracterísticas típicas:
deficiência mental grave;surdez;anomalias cardíacas;além de:
Punhos cerrados eplantas arqueadas
Polidactilia
Anormalidadesoculares
Lábio leporino e/oupalato fendido

13
45
SÍNDROME DE PATAUA síndrome é caracterizada por anomalias morfológicas emalformação graves do Sistema Nervoso Central (SNC).Em geral há também defeitos cardíacoscongênitos e defeitos urogenitais, gerandoinviabilidade dos afetados.Ocorrência: 1/5.000 88% morrem no 1º mêssó 5% sobrevivem até o 6º mês.Cerca de 20% dos casos resultam de umatranslocação não-balanceada.
Menino com trissomia do 13 aos 7 anos apresentaprejuízos visuais e auditivos significativos sobrevivênciaapós o primeiro ano é rara.
46
4- MOSAICISMO 47, XX OUXY + 8/NORMAL
Trissomia completa do cromossomo 8 é letalgeralmente é mosaicismo.Características:
estrabismo;deficiência mental variável (leve a severa)depende do grau de mosaicismo;crescimento variável;orelhas com lobos alterados;tronco alongado e magro, escápula e esternoanormais;pescoço curto ou alado;mamilos muito afastados;defeitos cardíacos.
Ocorrência: mais de 100 casos relatados.

14
47
5- MOSAICISMO 47, XX OU XY +9/NORMAL
Características:crescimento pré-nataldeficiente;deficiência mental severa;anomalias nas juntas;anomalias cardíacas em 2/3 doscasos.
Maioria morre na fase pós-natal, os que sobrevivemapresentam severa deficiênciamental e motora.
48
ANEUPLOIDIASALOSSÔMICAS

15
49
1- SÍNDROME DE TURNER (45, X0)
50
SÍNDROME DE TURNERCaracterísticas:
desenvolvimento sexualretardado (não apresentamdesenvolvimento das mamasaté os 13 anos e apresentamamenorréia primária ousecundária);geralmente estéreis ou sub-férteis;baixa estatura;tendência à obesidade;pescoço alado;defeitos cardíacos.

16
51
SÍNDROME DE TURNER
Ocorrência: 1/2.000 nascimentos do sexofeminino.
52
2- SÍNDROME DE KLINEFELTER (47,XXY) ou 48, XXXY ou 49, XXXXY

17
53
SÍNDROME DE KLINEFELTERCaracterísticas:
homens altos e magros, commembros superiores e inferioresalongados;hipogonadismo (evidente apóspuberdade);caracteres sexuais secundáriossubdesenvolvidos; sub-férteis;desenvolvimento de seios;problemas comportamentais,incluindo irritabilidade, agitação,hiperatividade;QI normal (XXY).
Ocorrência: 1/500 nascimentosdo sexo masculino.
A Síndrome caracteriza-se pela presença do cariótipo 47, XXY ou em mosaicos.
54
48, XXXY 49, XXXXYCaracterísticas: quanto maior for a aneuploidia maissevera será a deficiência mental e a feminilização.
Indivíduos XXY XXXY XXXY XXXXY

18
55
3- TRISSOMIA DO X (47, XXX) ou48, XXXX ou 49, XXXXX
56
TRISSOMIA DO XCaracterísticas:
mulheres com genitálianormal e leve retardamentomental;deficiência de crescimentopré-natal;baixa estatura (XXXXX).
Ocorrência: 1/1000nascimentos do sexofeminino quase todos oscasos resultam de erros nameiose materna.
XXXXX

19
57
4- SÍNDROME DO DUPLO Y (XYY)
58
SÍNDROME DO DUPLO YÉ um dos cariótipos mais freqüentemente observadosdespertou grande interesse após observar-se que aproporção era bem maior entre os detentos de uma prisãode segurança máxima, sobretudo entre os mais altos, doque na população em geral.

20
59
SÍNDROME DO DUPLO YCaracterísticas:
maioria fenotipicamente normal;estatura muito elevada;problemas comportamentais como distração,hiperatividade e crises de fúria na infância e início daadolescência.
60
ABERRAÇÕESCROMOSSÔMICAS
ESTRUTURAIS

21
61
ALGUNS TIPOS DE REARRANJOSCROMOSSÔMICOS
62
1 – DELEÇÃO OU DEFICIÊNCIAPerda de um segmento do cromossomo.
Deleção terminal dobraço curto
Deleção terminal dobraço longo
Deleção do braçolongo

22
63
DELEÇÃO OU DEFICIÊNCIAMonossomia parcial.Resulta de:
quebras casuais e perda do segmentoacêntrico;crossing-over desigual;segregação anormal de translocação ouinversão equilibrada.
64

23
65
SÍNDROME DE WOLF-HIRSCHHORNSWH 46, XX ou XY, del(4)(p16.3)Características típicas:
microcefalia,anomalias faciais hipertelorismo(distância grande entre as paredesmediais das órbitas), glabelaproeminente, nariz largo e/ou em bico,micrognatia, comissuras labiaisinclinadas para baixo, pavilhõesauriculares displásicos,retardo mental e de crescimento,cardiopatia congênita,anomalias urogenitais.
Glabelaproeminente
66
SÍNDROME DE WOLF-HIRSCHHORNAs deleções submicroscópicaspodem ser detectadas através detécnicas de citogenética molecularhibridização in situ comfluorescência – FISH, por exemplo.

24
67
SÍNDROME DE CRI DU CHATTambém chamada síndrome do “Miado de gato”.46, XX ou XY, del(5)(p14) deleção no braço curtodo cromossomo 5).
68
SÍNDROME DE CRI DU CHAT
Características típicas:choro típico que lembra o miado de gatos.peso baixo ao nascer, crescimento lento;deficiência mental;hipotonia muscular;microcefalia;pavilhão auditivo dismórfico;pregas epicântricas;hipertelorismo;malformações dos membros.
Freqüência: 1:50.000 nascimentos.

25
69
Neuropatia Hereditária Sensível àCompressão (HNPP)
Decorrente de deleção de umsegmento do cromossomo 17(17p11.2-p12), onde se localiza ogene PMP22.Herança autossômica dominanteinício na adolescência ou na faseadulta jovem.Caracterizada por paralisia(geralmente não-severa) de pressãoreincidente (sensorial e motora) emum único nervo compressão físicareal do nervo pode ou não estarpresente alguns indivíduos sãoassintomáticos.
70
2- INVERSÃO
Quando um segmento do cromossomo originadode duas quebras sofre rotação de 180° e éressoldado.Quando não envolve o centrômero é denominadaparacêntrica (quebras e inversão ocorrem nomesmo braço cromossômico); quando envolve,pericêntrica.

26
71
INVERSÃO PARACÊNTRICA DOBRAÇO CURTO
Quebras e inversãoocorrem no braço curtodo cromossomo.
72
INVERSÃO PARACÊNTRICA DOBRAÇO LONGO
Quebras e inversãoocorrem no braço longodo cromossomo.
27
73
74
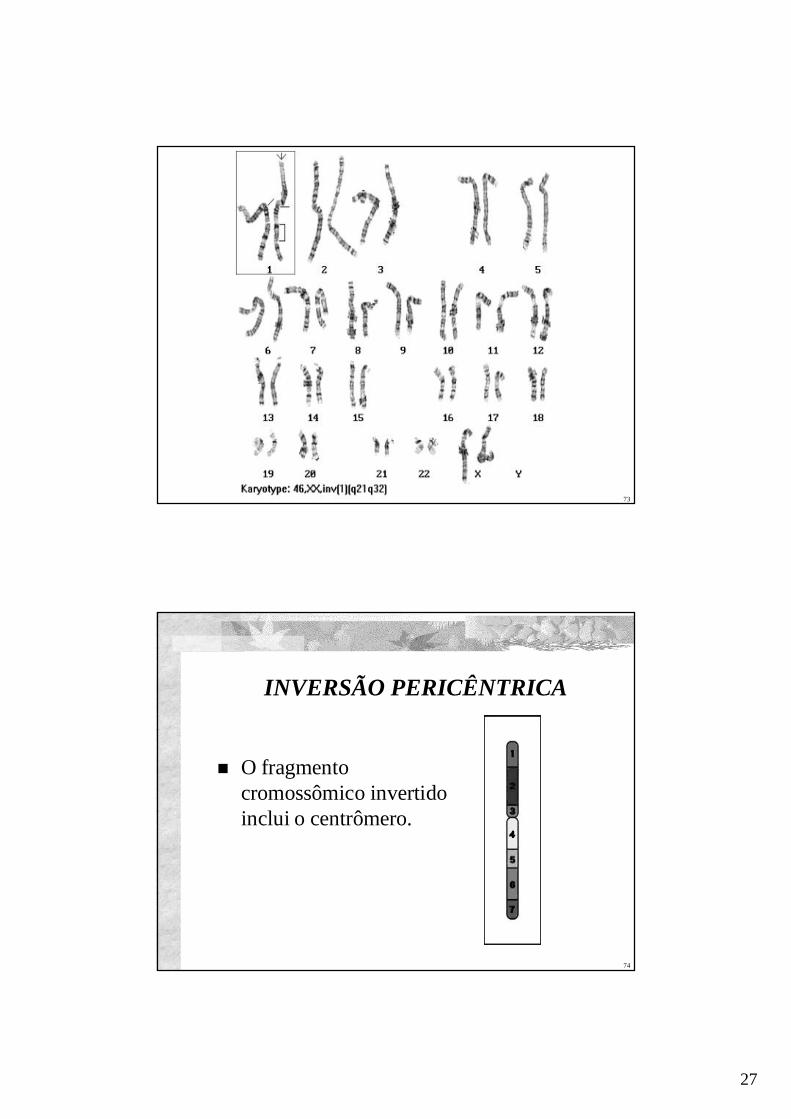
INVERSÃO PERICÊNTRICA
O fragmentocromossômico invertidoinclui o centrômero.

28
75
76
3- TRANSLOCAÇÃO
Troca de segmentos entre cromossomosnão homólogos:
simples;recíproca;robertsoniana.

29
77
TRANSLOCAÇÃO SIMPLES
Quando o segmento perdidopor deleção de umcromossomo é inserido emoutro cromossomo.
78
TRANSLOCAÇÃO RECÍPROCA
Resultam de quebra decromossomos nãohomólogos, com trocasrecíprocas de segmentossoltos.

79
Balanceada: quando não há excesso de material cromossômico.

1
80
Leucemia Mielóide Crônica (LMC)Decorrente de translocação recíproca entre os cromossomos9 e 22 t(9; 22) (q34; q11).
81
Leucemia Mielóide Crônica (LMC)Novo cromossomo formado:
cromossomo Philadelphiaprimeira alteração
cromossômica a serrelacionada ao câncerhumano:
proto-oncogene abl(localizado no braço longo docromossomo 9) é translocadopara o braço longo docromossomo 22 no gene bcr
encurtamento do braçolongo do cromossomo 22.

2
82
Leucemia Mielóide Crônica (LMC)Gene quimérico bcr/abl serve como marcador deLMC:
mRNA híbrido bcr/abl, de 8,5 kb proteína híbrida de210 Kd, com atividade de tirosinaquinase:
similar à proteína codificada pelo gene c-abl, de localizaçãoexclusivamente nuclear, porém mais ativa e com localizaçãocitoplasmática.
83
Linfoma de Burkitt
Leucemia Linfóide (ouLinfoblástica) Aguda LLA.Segundo O.M.S. neoplasiamaligna pertencente ao grupo doslinfomas de células B maduras.Resultado de translocaçãocromossômica envolvendo o geneMyc t (8;14), t(8;2), t(8;22).

3
84
Linfoma de BurkittTerceira neoplasia mais freqüente em menores de15 anos:
acomete o sistema hematopoético, apresentandocomportamento biológico agressivo;2/3 das crianças e adolescentes diagnosticadosapresentam doença localmente disseminada oumetastática.
Linfoma de Burkitt: aspectocaracterístico de “céuestrelado”.
85
TRANSLOCAÇÃO ROBERTSONIANA
Envolve dois cromossomosacrocêntricos que se fundempróximos à região docentrômero com perda dosbraços curtos.

86
Translocações Robertsonianas equilibradas:Quando o conjunto cromossômico possui o complemento normal deinformações.Todas as informações genéticas estão presentes, mas acondicionadasde modo diferente.

1
87
88
4- DUPLICAÇÃO
Quando um segmento deum cromossomoapresenta-se duplicado.Resulta de:
crossing-over desigual;segregação anormal nameiose em um portador detranslocação ou inversão.

2
89
90
Neuropatia de Charcot-Marie-Tooth tipo 1A (CMT1A)
Forma mais freqüente de CTM grupo heterogêneo dedoenças genéticas que afetam nervos periféricosmotores e sensoriais, com diferentes padrões de herança,evoluções clínicas e característicaseletroneuromiográficas.Decorrente de duplicação de um segmento docromossomo 17, onde se localiza o gene PMP22dup(17)(p11.2-p12) mesma região deletada naNeuropatia Hereditária Sensível à Compressão (HNPP).Herança autossômica dominante.Pacientes apresentam velocidade de condução nervosamotora diminuída.

3
91
Neuropatia de Charcot-Marie-Tooth tipo 1A (CMT1A)
Diagnóstico através de exames moleculares.
92
5- ISOCROMOSSOMOSResultam de:
erro na divisão do centrômeroque ao invés de separar ascromátides, separa os braçosdo cromossomo;translocação entre cromátides-irmãs de um mesmocromossomo, próximas aocentrômero.

4
93
ISOCROMOSSOMOS
São cromossomos queapresentam deficiência totalde um dos braços eduplicação completa dooutro.
94
6- CROMOSSOMOS DICÊNTRICOSFusão de 2 segmentos cromossômicos, extremidade aextremidade, cada um com um centrômero, com perdados fragmentos acêntricos.Tendência de se quebrar na anáfase.

5
95
Síndrome de EdwardsTambém decorrente de trissomias do cromossomo 18.Duplicações:
maior importância do braço longo na manifestação dossintomas:
associação entre apresentação clínica de diversas característicasfenotípicas e envolvimento combinado de uma porção proximal eoutra mais distal dup(18)(q11 q12.1) combinada comdup(18)(q21 qter);deficiência mental mais severa associada à duplicação maisproximal do braço longo dup(18)(q12.3 q21.1);duplicações quase completas pouca ou nenhuma característicada síndrome.

96
Cromossomos dicêntricos: são cromossomos que apresentam dois centrômeros.Os cromossomos dicêntricos tendem a quebrar-se na anáfase, se os doiscentrômeros estiverem próximos; se um centrômero for inativado, umcromossomo dicêntrico pode ser estável.

1
97
7- CROMOSSOMOS EM ANELQuando ocorrem deleções nas extremidades deum cromossomo (telômeros) e posterior fusão dopedaço mediano que se curva, formando um anel.
98
CROMOSSOMOS EM ANEL
As deleções terminaisnos dois braços de umcromossomo podem darorigem a umcromossomo em anel, seas extremidades livresfraturadas se soldarem.

2
99





























