Embed Size (px)
Citation preview

Catálisis asimétrica vía enaminas: reacciones nitrosoaldólica y de
Mannich catalizadas por pirrolidinas quirales
Tesis Doctoral Irene Velilla Martínez 2011


CONFORMIDAD DEL DEPARTAMENTO
El Consejo del Departamento de Química Orgánica I
en reunión celebrada el día 25 de octubre de 2011 ha acordado dar la conformidad a
la admisión a trámite de presentación de la Tesis Doctoral titulada: Catálisis
Asimétrica Vía Enaminas: Reacciones Nitrosoaldólica y de Mannich Catalizadas
por Pirrolidinas Quirales dirigida por el/la Dr. Claudio Palomo Nicolau, la Dra.
Antonia Mielgo Vicente y el Dr. Juan Miguel Oyarbide Garmendia y presentada por
Don/ña. Irene Velilla Martínez ante este Departamento.
En Vitoria a 25 de octubre de 2011
Vº Bº DIRECTOR/A DEL DEPARTAMENTO SECRETARIO/A DEL DEPARTAMENTO
Fdo.: Francisco J. Palacios Gambra Fdo.: Ana Mª Ochoa de Retana Mendibil








ACTA DE GRADO DE DOCTOR
ACTA DE DEFENSA DE TESIS DOCTORAL
DOCTORANDO DON/ÑA. Irene Velilla Martínez
TITULO DE LA TESIS: Catálisis asimétrica vía enaminas: reacciones nitrosoaldólica y de
Mannich catalizadas por pirrolidinas quirales
El Tribunal designado por la Subcomisión de Doctorado de la UPV/EHU para calificar la Tesis
Doctoral arriba indicada y reunido en el día de la fecha, una vez efectuada la defensa por el
doctorando y contestadas las objeciones y/o sugerencias que se le han formulado, ha otorgado
por___________________la calificación de: unanimidad ó mayoría
Idioma/s defensa: _______________________________________________________________
En a de de
EL/LA PRESIDENTE/A, EL/LA SECRETARIO/A,
Fdo.: Fdo.:
Dr/a: ____________________ Dr/a: ______________________
VOCAL 1º, VOCAL 2º, VOCAL 3º,
Fdo.: Fdo.: Fdo.:
Dr/a: Dr/a: Dr/a:
EL/LA DOCTORANDO/A,
Fdo.: Irene Velilla Martínez


3
Agradecimientos
Esta Tesis Doctoral ha sido realizada en el Departamento de Química
Orgánica I de la Facultad de Ciencias Químicas de San Sebastián, Universidad
del País Vasco, (U.P.V.-E.H.U.) bajo la dirección del Dr. D. Caludio Palomo
Nicolau, la Dra. Antonia Mielgo Vicente y el Dr. Juan Miguel Oyarbide
Garmendia, a quienes expreso mi más sincero agradecimiento por su
dedicación y esfuerzo durante el transcurso de este trabajo.
La financiación de este trabajo ha provenido de una beca predoctoral de la
Universidad del País Vasco durante el periodo 2007-2011 (PIFA/01/2006/053)
así como de los programas SAIOTEK (Gobierno Vasco) y MICIN (Ministerio de
Ciencia y Tecnología) para la financiación de proyectos de investigación,
entidades a las cuales también deseo agradecer.
Por otro lado, quisiera agradecer a todos los, ya no compañeros, sino
amigos, con los que he compartido lugar de trabajo, proyectos y muchas
alegrías y penas, por hacer más llevadera esta época en muchos sentidos,
especialmente a Silvia, Ixiar y Ángel, que son unas máquinas, y muchos más
(sí, muchos y muchas).
Muchas gracias a mi familia y amigos cercanos, por estar ahí escuchando
estos últimos años mis sermones y penurias, lo que supone un gran desahogo
y apoyo para una doctoranda estresada.
Finalmente, gracias a mi aitá, porque sin él no sería como soy en algunos
aspectos que me gustan, ni abría comenzado esta aventura por el mundo de la
química; sé que estarás orgulloso.


5
“Molecules are everything”. “First from the Iron Age and then the Silicon Age, we are living the Molecular Age”.
(Aunque la silicona sigue estando muy de moda en algunos aspectos…)

6

7
Abreviaturas ác Ácido
AL Ácido de Lewis
Alif Alifático
Ar Aromático
BINAP 2,2'-Bis(difenilfosfino)-1,1'-binaftilo
Bn Bencilo
Boc terc-Butoxicarbonilo
Cat Catalizador
Cbz Benciloxicarbonilo
DCM Diclorometano
DFT Teoría del funcional de la densidad
DIPEA Di-isopropiletilamina
DMAP 4-(dimetilamino)piridina
DMF Dimetilformamida
DMSO Dimetilsulfóxido
dr Relación diastereomérica
E Electrófilo
ee Exceso enantiomérico
EWG Grupo electroatrayente
HOMO Orbital molecular ocupado de mayor energía
HPLC Cromatografía líquida de alta resolución
iPr Isopropilo
Ln Ligando

8
LUMO Orbital molecular no ocupado de menor energía
P.f. Punto de fusión
MS Tamiz molecular/ Espectrometría de masas
Naf Naftilo
nBu Butilo (lineal)
nHex Hexilo (lineal)
NMP N-Metil-2-pirrolidona
Nos Nosilo
nPent Pentilo (lineal)
nPr Propilo (lineal)
Nu Nucleófilo
PCC Clorocromato de piridinio
PG Grupo protector
PMP p-Metoxifenilo
PNBA Ácido p-nitrobenzoico
PNP p-Nitrofenilo
rac Racémico
Rdto. Rendimiento químico
ref. Referencia
RMN Resonancia magnética nuclear
SOMO Siglas de Single Occupied Molecular Orbital
tr Tiempo de retención
t.a. Temperatura ambiente
TBDPS terc-Butildifenilsililo

9
TBS terc-Butildimetilsililo
TEA Trietilamina
THF Tetrahidrofurano
Tf Triflato
TLC Cromatografía de capa fina
TMS Trimetilsililo
Tos Tosilo
TPS Trifenilsililo
TS Estado de transición
pTSA Ácido p-Toluensulfónico
UV Ultravioleta

10

11
1. Introducción……………………………………………………...……………….17
1.1. Quiralidad……………………………………………………...………….…17
1.2. Síntesis asimétrica……………………......………...……………………..21
1.2.1. Utilización de auxiliares quirales…………………………………..22
1.2.2. Biocatálisis………………….………………......…………………...23
1.2.3. Catálisis asimétrica (no enzimática)………..….………...……….24
1.3. Organocatálisis………………...……………………………...…………….26
1.3.1. Desarrollo de la organocatálisis asimétrica……….….………….28
1.3.2. Tipos de activación………………...………………..………..…….31
1.3.2.1. Catálisis no covalente………….……...…………………31
1.3.2.2. Catálisis covalente….……….……...……………………34
1.3.3. Aminocatálisis…..…...…………………………...…………………36
1.3.3.1. Control de la estereoquímica en las reacciones vía
enamina e ion iminio………………...…………………..43
1.3.3.2. Éteres de diarilprolinol como catalizadores en
reacciones vía enamina………………...……………….46
1.4. Objetivos………………...………………………..………………...……….54
2. Reacción nitrosoaldólica………………………………………………………57
2.1. Introducción………………………………………………………………….57
2.2. Reacción nitrosoaldólica catalítica asimétrica…………..……………….65
2.3. Planteamiento 1: reacción N-nitrosoaldólica en ausencia de dadores
de H externos...………………………………..……………………...…….76
2.3.1. Resultados y discusión…………………………..………………..78
2.3.1.1. Viabilidad de la propuesta. Estudio teórico previo……78
2.3.1.2. Viabilidad de la reacción: evaluación experimental
de catalizadores y disolventes………….……………...80
2.3.1.3. Alcance de la reacción…………………………………..82
2.4. Planteamiento 2: reacción O-nitrosoaldólica con ácido de Brønsted
externo…………………………………………….……..………..………...84
2.4.1. Resultados y discusión………………….…………………..……..84
2.4.1.1. Viabilidad de la propuesta. Estudio teórico previo…….84
2.4.1.2. Búsqueda de condiciones de reacción…………………86
2.4.1.3. Alcance de la reacción…………………………………...90

12
2.4.2. Aplicaciones…………………………………………………………92
2.5. Conclusiones………………………………………………………………...95
3. Reacción de Mannich anti-selectiva………...………...……………………..99
3.1. Introducción…………………………………………………………..……...99
3.1.1. Reacción de Mannich asimétrica vía catálsis metálica..………102
3.2. Reacción de Mannich asimétrica vía organocatálisis, precedentes....104
3.2.1. Reacción de Mannich sin-selectiva vía enamina………………104
3.2.2. Reacción de Mannich sin-selectiva con otros catalizadores….109
3.2.3. Reacción de Mannich anti-selectiva vía enamina……………..112
3.2.4. Reacción de Mannich anti-selectiva con otros catalizadores...120
3.2.5. Limitaciones y objetivos………………………….……………….121
3.3. Reacción de Mannich anti-selectiva entre aldehídos y N-tosil-
iminas………………………………………………………………………122
3.3.1. Hipótesis de trabajo y antecedentes…………………………….122
3.3.2. Resultados y discusión……………………………………………124
3.3.2.1. Búsqueda de condiciones de reacción…….….……...124
3.3.2.2. Alcance de la reacción………………………………….128
3.3.2.3. Cálculos computacionales………….………………….130
3.4. Reacción de Mannich anti-selectiva entre aldehídos e iminas
sencillas (bases de Schiff).………………………..……………………..132
3.4.1. Reacción de Mannich con iminas aromáticas………………….133
3.4.2. Reacción de Mannich con iminas propargílicas……..…..…….136
3.4.2.1. Búsqueda de condiciones de reacción…..…....……..139
3.4.2.2. Síntesis del aducto sin-97Ba………………………….144
3.5. Aplicaciones……………………………………………………………….147
3.6. Conclusiones………………………………………………………………149
4. Desarrollo experimental……..………………………………………………..153
4.1. Materiales y métodos generales…………………………………………153
4.2. Síntesis de catalizadores…………………………………………………156
4.2.1. Síntesis de éteres de α,α-dialquilsililo y de α,α-diarilsililo
derivados..…………………………………………………………156
4.2.2. Síntesis de éteres de α,α-dialquilsililo derivados de la
trans-4-hidroxi-L-prolina …..……………………………………..161

13
4.2.3. Espectros de RMN…………………………………….…………..166
4.3. Síntesis de aldehídos…………………………………………….……….172
4.3.1. Síntesis de 17J y 17K…………………………………………….172
4.3.2. Síntesis de 17L y 17M…………………………………………….173
4.4. Procedimientos experimentales del capítulo 2…………………………174
4.4.1. Reacción N-nitrosoaldólica de aldehídos y nitrosobenceno….174
4.4.1.1. Reacción N-nitrosoaldólica: procedimiento general…………175
4.4.1.2. Derivatización del 2-(hidroxifenilamino)heptan-1-ol
con cloruro de benzoilo para análisis de HPLC….…..179
4.4.1.3. Derivatización del 2-(hidroxifenilamino)octan-1-ol
con cloruro de naftoilo para análisis de HPLC….……180
4.4.1.4. Preparación de muestras racémicas………………….180
4.4.1.5. Espectros de 1H y 13C-RMN de algunos
compuestos representativos……..………………….…182
4.4.1.6. Cromatogramas de HPLC……………………………...187
4.4.1.7. Estudios computacionales para la reacción N-
nitrosoaldólica…………………………………………...197
4.4.2. Reacción O-nitrosoaldólica de aldehídos y nitrosobenceno….200
4.4.2.1. Reacción O-nitrosoaldólica: procedimiento general…200
4.4.2.2. Preparación de muestras racémicas…….……………203
4.4.2.3. Preparación del diol 43…………………………………203
4.4.2.4. Procedimiento general para la preparación de 1,2-
aminoalcoholes………………………………………….204
4.4.2.5. Espectros de 1H y 13C-RMN………….………..………209
4.4.2.6. Cromatogramas de HPLC…………..………….………212
4.4.2.7. Estudios computacionales para la reacción O-
nitrosoaldólica……………………...……………………220
4.5. Procedimientos experimentales del capítulo 3……………………...….230
4.5.1. Reacción de Mannich anti-selectiva con N-tosil-iminas……....230
4.5.1.1. Síntesis de N-sulfonil-iminas aromáticas…………..…230
4.5.1.2. Síntesis de la N-Boc-imina 80a………………………..233
4.5.1.3. Reacción de Mannich entre aldehídos y
N-tosil-iminas: procedimiento general..………….……233

14
4.5.1.4. Desprotección y elaboración del aducto 100….…….239
4.5.1.5. Preparación de muestras racémicas………………….241
4.5.1.6. Determinación de la configuración absoluta…………241
4.5.1.7. Espectros de 1H y 13C-RMN………………....………..242
4.5.1.8. Cromatogramas de HPLC ………………..……………250
4.5.1.9. Estudios computacionales para la reacción de
Mannich entre aldehídos y N-sulfonil-iminas………...258
4.5.2. Reacción de Mannich anti-selectiva con iminas sencillas
(bases de Schiff).……………………………………..…………...268
4.5.2.1. Síntesis de iminas N-aromáticas…………..………….268
4.5.2.2. Reacción de Mannich entre aldehídos e iminas N-
aromáticas: procedimiento general………..………….272
4.5.2.3. Espectros de 1H y 13C-RMN………..………………….284
4.5.2.4. Cromatogramas de HPLC.…………..…………..…….298
5. Anexo: publicaciones.…..………………..……..……….……….....………..315

Introducción


Introducción
17
1. Introducción
Desde que Berzelius hiciese la primera distinción entre los compuestos
orgánicos (aquéllos que contienen principalmente carbono) e inorgánicos y
definiera los términos “catálisis” e “isómeros” entre otras aportaciones,1 el
mundo de la química orgánica no ha parado de evolucionar.
Una de las áreas más relevantes de la química orgánica comprende la
síntesis de moléculas. Los compuestos que contienen carbono se denominaron
originalmente orgánicos porque se creía que existían únicamente en los seres
vivos. Sin embargo, pronto se vio que podían prepararse compuestos
orgánicos en el laboratorio a partir de sustancias inertes que contuvieran
carbono. En el año 1828, Friedrich Wöhler, discípulo de Berzelius, consiguió
convertir cianato de plomo en urea por tratamiento con amoniaco acuoso. Así,
una sal inorgánica se convirtió en un producto perteneciente a los seres vivos
(orgánico). A día de hoy se han sintetizado más de diez millones de
compuestos orgánicos y sus aplicaciones son casi ilimitadas.
1.1. Quiralidad
La simetría de formas abunda en la naturaleza. Una esfera, un cubo o
un cono son formas típicas de objetos cotidianos, siendo simétricas y
superponibles a sus imágenes especulares. Sin embargo, muchos otros
objetos, como las manos o pies, no pueden superponerse a sus imágenes
especulares. En geometría, se dice que estos objetos son disimétricos. En
química, esta propiedad se llama quiralidad, y las moléculas que la poseen son
moléculas quirales. Estas moléculas tienen en su estructura al menos un
elemento de asimetría o quiralidad, como un plano, un eje o un centro
estereogénico, que la mayoría de las veces suele ser un carbono con cuatro
sustituyentes diferentes.
1 R. Larsson (Ed.), Perspectives in Catalysis: In Commemoration of J. J. Berzelius, CNK Gleerup, Lund, Sweeden, 1981.

Capítulo 1
18
Figura 1-1: Ilustración de un aminoácido y una mano con su
correspondiente imagen especular.
Como se observa en la Figura 1-1, una mano no es superponible a su
imagen especular. Lo mismo ocurre con el aminoácido mostrado. Lo que
diferencia a las dos moléculas a uno y otro lado del “espejo” imaginario es la
disposición de los grupos en el espacio en torno al carbono estereogénico; son
estereoisómeros. La pareja de estereoisómeros que se relacionan entre sí
como un objeto y su imagen especular no superponible se denominan
enantiómeros y a sus configuraciones R y S, respectivamente, de acuerdo con
las reglas de Cahn-Ingold-Prelog (CIP) internacionalmente aceptadas.2
Los enantiómeros poseen casi todas las propiedades físicas idénticas,
con la excepción de la actividad óptica. Uno de los enantiómeros produce
rotación de la luz polarizada a la derecha (dextrógiro) y el otro rota la luz
polarizada a la izquierda (levógiro). De mucha mayor relevancia es el hecho de
que habitualmente cada uno de los enantiómeros presente unas propiedades
biológicas diferentes. Por ejemplo, el (R)-propranolol se emplea como
anticonceptivo, mientras que su enantiómero, de configuración S, actúa como
antidepresivo (Figura 1-2).
2 Artículos que definieron el sistema de CIP: a) R. S. Cahn, C. K. Ingold, V. Prelog, “Specification of Molecular Chirality” Angew. Chem. Int. Ed. 1966, 5, 385-415. b) V. Prelog, G. Helmchem “Basic Principles of the CIP-System and Proposals for a Revision” Angew. Chem. Int. Ed. 1982, 21, 567-583.

Introducción
19
O NH
OH
O NH
OH
(R)-Propranolol (S)-Propranolol
Figura 1-2
La estructura de las moléculas orgánicas se construye sobre la base
de un esqueleto compuesto de enlaces carbono-carbono, de manera que la
posibilidad de encontrar centros estereogénicos es muy elevada. Basta decir
que más de la mitad de los medicamentos utilizados actualmente son
moléculas quirales.3 Sin embargo, la magnitud de la relación entre la
configuración de una molécula determinada y su actividad biológica no fue
reconocida hasta la década de los 60, cuando se fue testigo de los efectos de
la talidomida (Fig. 1-3). Este fármaco se utilizó como sedante en su forma
racémica (mezcla equimolar de enantiómeros R y S), comprobándose más
tarde que el enantiómero S causa además deformaciones fetales importantes.4
Figura 1-3
Este y otros ejemplos han forzado a la industria, ya no solo
farmacéutica, sino química en general, a precisar más sus objetivos sintéticos;
cuando se trata de compuestos quirales, es de vital importancia lograr su
preparación con una configuración definida (síntesis de compuestos
enantioméricamente puros, EPC). Pero la industria química actualmente se
enfrenta también a otro gran reto: la sostenibilidad de los procesos, incluyendo
el respeto medioambiental. Por ello hoy en día a las reacciones y procesos que
3 R. Mannhod, H. Kubinyi, G. Folkers, Chirality in Drug Research, 2006, Wiley-VCH Vol. 33. 4 a) W. H. De Camp, Chirality, 1989, 1, 2. b) T. Stephens, R. Brynner. Dark Remedy: The Impact of Thalidomide and Its Revival as a Vital Medicine, 2001. Cambridge, MA, Perseus.

Capítulo 1
20
constituyen un protocolo de síntesis se les exige además de buen rendimiento
químico en las reacciones, minimización del gasto de reactivos y/o disolventes,
uso de energía o producción de desechos y la ausencia de sustancias tóxicas o
dañinas en los procesos, conceptos acordes con la Química Verde.5
Los métodos que se utilizan para la obtención de compuestos
enantioméricamente puros (EPC) se pueden clasificar en tres grandes grupos:
- Resolución de mezclas racémicas
- Métodos basados en el “Chiral pool” (fuentes quirales naturales)
- Síntesis asimétrica
La resolución de mezclas racémicas6 se puede considerar como el
procedimiento clásico de obtención de EPC. La estrategia consiste en hacer
reaccionar un compuesto quiral racémico, (rac)-A en la Fig. 1-4, previamente
preparado o aislado de fuentes naturales, con un agente de resolución
homoquiral B*, lo que genera dos diastereómeros separables, (+)-AB* y (-)-
AB*. La separación de éstos y la posterior disociación del producto permite
obtener por separado los enantiómeros (+)-A y (-)-A. El agente de resolución
ideal debe ser recuperable al final del proceso.
Figura 1-4: Resolución de mezclas racémicas.
Una segunda estrategia engloba aquellos métodos en los que se
utilizan materiales de partida quirales y además ópticamente puros, obtenidos
5 Para más información sobre el término “química verde”, ver: R. A. Sheldon, I. Arends, U. Hamefeld, Green Chemisty and Catalysis, 2007, Wiley-VCH. 6 Para una revisión actualizada de los procesos de resolución, ver: N. G. Anderson, Org. Proc. Res. Dep. 2005, 9, 800-813.

Introducción
21
de fuentes naturales (“chiral pool”)7 y sobre los que se llevan a cabo
transformaciones de grupos funcionales presentes en la molécula hasta llegar
al compuesto deseado, pero sin alterar ninguna de las unidades
estereogénicas iniciales. Para este fin se dispone de una gama relativamente
amplia de compuestos naturales que cumplen los requisitos necesarios:
aminoácidos, aminoalcoholes, hidroxiácidos, alcaloides, terpenos o azúcares.
Por ejemplo, siguiendo esta estrategia es posible obtener aminoácidos no
naturales a partir de otros que sí lo son, como se muestra en la secuencia
representada en el Esquema 1-1.8 Como se puede apreciar el sustrato y el
producto mantienen la misma disposición espacial en torno al centro
estereogénico y se han llevado a cabo interconversiones de grupos funcionales
en la molécula sin afectar al carbono estereogénico inicial.
Esquema 1-1: Síntesis de aminoácidos a partir de la (S)-serina.8
Por último, el tercer gran grupo de métodos desarrollados para la
obtención de EPC, y sobre el que se centra la presente Tesis es el de la
síntesis asimétrica.
1.2. Síntesis asimétrica
La síntesis asimétrica9 se puede definir como la obtención
estereoselectiva de compuestos quirales a partir de otros aquirales. La
información quiral (entorno quiral) necesaria para inducir estereoselectividad
7 a) K. C. Nicolau, E. S. Sorensen, Classics in Total Synthesis, 1996, Wiley-VCH. b) S. Hanessian, Pure Appl. Chem. 1993, 65, 1189-1204. c) K. C. Nicolau, S. A. Snyder, Classics in Total Synthesis II, 2003, Wiley-VCH. 8 D. Arnold, J. C. G. Drover, J. C. Vederas, J. Am. Chem. Soc. 1987, 109, 4649-4659. 9 a) R. A. Aitken, S. N. Kilényi, Asymmetric Synthesis, 1992, Chapman & Hall, Cambridge. b) M. Nogradi, Stereoselective Synthesis: A Practical Approach, 1995, Weinheim, New York; VCH.

Capítulo 1
22
puede proceder bien de un auxiliar quiral, de una enzima, o de un catalizador
quiral no enzimático.
1.2.1. Utilización de auxiliares quirales
Un auxiliar quiral10 es un fragmento molecular enantioméricamente
puro que se incorpora covalentemente a un sustrato con objeto de controlar el
curso estereoquímico durante la generación de nuevas unidades
estereogénicas en el sustrato (reacción diastereoselectiva). Una vez llevada a
cabo la transformación del sustrato, una reacción de escisión permite recuperar
el auxiliar quiral, que puede ser reutilizado, con liberación del producto
enantioenriquecido deseado (Fig. 1-5).
Figura 1-5
Si bien el uso de auxiliares quirales entraña sendas etapas de anclaje y
liberación del auxiliar, y por tanto no se ajusta bien a los objetivos de economía
de átomo11 y economía de proceso, es también cierto que la predictibilidad del
transcurso estereoquímico en los procesos mediados por estas estructuras es,
por lo general, grande. Este hecho, unido a su robustez, hace que en algunas
situaciones la eficacia de los auxiliares quirales no haya sido alcanzada por los
métodos catalíticos.
Entre los auxiliares quirales más representativos se encuentran las
sulfinamidas y sulfóxidos,12 auxiliares derivados del alcanfor,13 derivados de
10 Para más información sobre auxiliares quirales, ver: a) J. Seyden-Penne, Chiral Auxiliaries and Ligands in Asymmetric Synthesis, 1995, John Wiley & Sons Ins., New York-Chichester-Brisbane-Toronto-Singapore. b) L. A. Paquette, Handbook of Reagents for Organic Synthesis: Chiral Reagents for Asymmetric Synthesis, 2003, Wiley, New York. c) Y. Gnas, F. Glorius, Synthesis, 2006, 1899-1930. d) D. A. Evans, G. Helmchen, M. Rueping, Asymmetric Synthesis-The Essentials (Part I. Chiral Auxiliaries in Asymmetric Synthesis), 2007, Wiley-VCH. 11 Para más información sobre el término “economía de átomo”, ver: a) B. M. Trost, Science 1991, 254, 1471-1477. b) B. M. Trost, Angew. Chem. Int. Ed. 2005, 34, 259-281. 12 a) M. C. Carreño, Chem. Rev. 1995, 95, 1717-1760. b) M. C. Aversa, A. Barattucci, P. Bonaccorsi, P. Giannetto, Tetrahedron: Asymmetry 1997, 8, 1339-1367.c) J. A. Ellman, Pure

Introducción
23
carbohidratos,14 SAMP/RAMP,15 alcoholes, aminas y aminoalcoholes,16
oxazolidinonas, oxazolinas y oxazolidinas.17 En la Figura 1-6 se muestran
algunos ejemplos de ellos.
OH
Ph
Corey, 1978
O NH
O
BnEvans, 1981
Heathcock-Masamune1981
R
O
OSiMe3
R:H,CH3
NHS
O OOppolzer, 1986
N OMeNH2
Enders, 1976
Ph
OH
Me
Myers, 1995
NHCH3
Figura 1-6: Auxiliares quirales representativos.
1.2.2. Biocatálisis
La biocatálisis,18 también llamada catálisis enzimática, fue el primer
método utilizado para llevar a cabo una reacción asimétrica y obtener así
Appl. Chem. 2003, 75, 39-46. d) I. Fernández, N. Khiar, Chem. Rev. 2003, 103, 3651-3706. e) G. Hanquet, F. Colobert, S. Lanners, G. Solladié, Arkivoc 2003, (vii), 328-401. f) P. Zhou, B. C. Chen, F. A. Davis, Tetrahedron 2004, 60, 8003-8030. 13 a) W. Oppolzer, Tetrahedron 1987, 43, 1969-2004. b) W. Oppolzer, Pure Appl. Chem. 1990, 62, 1241-1250. c) B. H. Kim, D. P. Curran, Tetrahedron 1993, 49, 293-319. 14 a) H. Kunz, K. Ruck, Angew.Chem. Int. Ed. 1993, 32, 336-358. b) H. Kunz, Pure Appl. Chem. 1995, 67, 1627-1635. c) P. G. Hultin, M. A. Earle, M. Sudharshan, Tetrahedron 1997, 53, 14823-14870. d) S. Knauer, B. Kranke, L. Krause, H. Kunz, Curr. Org. Chem. 2004, 8, 1739-1761. 15 a) D. Enders, H. Eichenauer, Angew. Chem. Int. Ed. 1976, 15, 549-551. b) D. Enders, H. Eichenauer, Tetrahedron Lett. 1977, 18, 191-194. c) A. Job, C. F. Janeck, W. Bettray, R. Peters, D. Enders, Tetrahedron 2002, 58, 2253-2329. 16 a) D. J. Ager, I. Prakash, D. R. Schaad, Chem. Rev. 1996, 96, 835-875. b) E. Juaristi, J. L. León-Romo, A. Reyes, J. Escalante, Tetrahedron: Asymmetry 1999, 10, 2441-2491. c) P. Camps, D. Muñoz-Torrero, Curr. Org. Chem. 2004, 8, 1339-1380. 17 a) T. G. Gant, A. I. Meyers, Tetrahedron 1994, 50, 2297-2360. b) D. J. Ager, I. Prakash, D. R. Schaad, Aldrichimica Acta 1997, 30, 3-12. c) D. A. Evans, J. T. Shaw, Actual. Chim. 2003, 35-38. d) A. I. Meyers, J. Org. Chem. 2005, 70, 6137-6151. 18 Para más información, ver: a) M. T. Reetz, Pharmacochemistry Library 2002, 32, 27-37. b) K. Drauz, H. Waldmann (eds), Enzyme Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2002. c) J. W. Saalfrank, W. F. Maier, Angew. Chem. Int. Ed. 2004, 43, 2028-2031. d) M. T. Reetz, B. Brunner, F. Schnerider, C. M. Schulz, M. M. Clouthier, M. Kayser, Angew. Chem. Int. Ed. 2004, 43, 4074-4079. e) H. U. Blazer, E. Schmidth, Asymmetric Catalysis on Industrial Scale, 2004, Wiley. f) N. Zagrebelny, Russ. Chem. Rev. 2005, 74, 285-296. g) C. T. Hou, Handbook of Industrial Biocatalysis, 2005, CRC Press.

Capítulo 1
24
compuestos enantioméricamente puros.19 Mediante la biocatálisis se obtienen
sustancias quirales enantioenriquecidas utilizando para ello enzimas, cultivos
celulares e incluso microorganismos. Las principales ventajas de la biocatálisis
son las suaves condiciones de reacción que requiere (medios fisiológicos), la
utilización de catalizadores compatibles con el medioambiente (enzimas), la
alta actividad catalítica y las elevadas regio- y estereoselectividades que ofrece
para moléculas multifuncionales con mínima necesidad de grupos protectores.
Sin embargo, la alta especificidad de los enzimas y sus problemas de
estabilidad y robustez dificultan su uso generalizado. Ante estas limitaciones,
los químicos sintéticos han desarrollado otros métodos de síntesis
estereoselectiva que complementan los procesos biológicos.
1.2.3. Catálisis asimétrica (no enzimática)
La catálisis es el proceso por el cual una reacción química es acelerada
por la acción de una cantidad relativamente pequeña de una especie química,
llamada catalizador, sin que ésta sea consumida durante la reacción. La
catálisis asimétrica20 hace referencia a la catálisis efectuada por catalizadores
quirales que además de acelerar una reacción inducen la formación preferente
de uno o varios estereoisómeros de todos los posibles. Este proceso implica
una “multiplicación” de la quiralidad y es, por tanto, el método ideal para la
síntesis de compuestos ópticamente puros.
Figura 1-7
19 En 1849 Luis Pasteur observó cómo el microorganismo Penicillium glauca descarboxilaba uno de los isómeros (el D) de una mezcla racémica de tartrato amónico. H.-B. Kagan. In: E. N. Jacobsen, A. Pfaltz, H. Yamamoto (Eds.), Comprehensive Asymmetric Catalysis, Springer-Verlag, Berlin, 1999, Vol. 1, 101-118. 20 a) J. D. Morrison, ed., Asymmetric Synthesis, Vol. 5, Academic Press, New York, 1985. b) B. Bosnich, Asymmetric Catalysis, Martinus Nijhoff, Dordrecht, 1986. c) I. Ojima, ed., Catalytic Asymmetric Synthesis, VCH, Weinheim, 1993. d) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994. e) B. M. Trost, Proc. Nat. Acad. Sci. 2004, 101, 5348-5355. f) K. Mikami, M. Lautens, New Frontiers in Asymmetric Catalysis, Wiley, 2007.

Introducción
25
En función de la naturaleza metálica o no metálica del catalizador, la
catálisis asimétrica puede dividirse en catálisis organometálica21 y
organocatálisis.22 Es en el contexto de esta última donde se desarrolla la
presente Tesis Doctoral.
La catálisis organometálica utiliza como especies catalíticas complejos
metal-ligando orgánico quiral. Existen numerosos complejos metálicos con
actividad catalítica remarcable, si bien en muchas ocasiones no es obvio
conocer la estructura de la especie catalítica debido a que estos complejos
sufren cambios estructurales durante la reacción. Para no incurrir en error, a los
promotores que se adicionan inicialmente a la reacción se les denomina
precursores catalíticos, que suelen ser una sal de un metal de transición y un
ligando quiral orgánico portador de funciones coordinantes que generalmente
contiene átomos de fósforo, nitrógeno, oxígeno o azufre.
Entre los pioneros en este campo cabe destacar a los ganadores del
Premio Nobel de Química en 2001, William R. Knowles,23 Ryoji Noyori24 y Karl
B. Sharpless25 por sus contribuciones a las reacciones asimétricas de
hidrogenación y oxidación vía catálisis organometálica.
En el Esquema 1-2 se muestra uno de los ejemplos más significativos
de Noyori, la hidrogenación de una cetona funcionalizada catalizada por Ru (II),
método que ha hecho posible la síntesis asimétrica a gran escala de un
intermedio común de los antibióticos de tipo carbapenem.26
21 Para revisiones generales sobre catálisis organometálica, ver: a) J-A. Ma, D. Cahard, Angew. Chem. Int. Ed. 2004, 43, 4566-4583. b) Special Issue: “Catalytic Asymmetric Synthesis” Acc. Chem. Res. 2000, 33, 323-440. c) E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Comprehensive Asymmetric Catalysis 1999 (VoIl-III, Sprinter, Berlin). 22 Para revisiones generales sobre organocatálisis, ver: a) H. Pellisser, Tetrahedron 2007, 63, 9267-9274. b) D. W. C. MacMillan, Nature 2008, 455, 304-308. c) S. Bertelsen, K. A. Jørgensen, Chem. Soc. Rev. 2009, 38, 2178-2189. Otros: d) A. Berkessel, H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, 2005. Ver también volúmenes especiales sobre organocatálisis: e) Acc. Chem. Res. 2004, 37(8). f) Adv. Synth. Catal. 2004, 346 (9-10). g) Chem. Rev. 2007, 107. 23 Novel lecture: W. S. Knowles, Angew. Chem. Int. Ed. 2002, 41, 1998-2007. 24 Novel lecture: R. Noyori, Angew. Chem. Int. Ed. 2002, 41, 2008-2022. 25 Novel lecture: K. B. Sharpless, Angew. Chem. Int. Ed. 2002, 41, 2024-2032. 26 a) R. Noyori, Chem. Soc. Rev. 1989, 18, 187-208. b) R. Noyori, H. Takaya, Acc. Chem. Res. 1990, 23, 345-350. c) R. Noyori, CHEMTECH, 1992, 22, 360-367.

Capítulo 1
26
Esquema 1-2: Hidrogenación asimétrica de una cetona funcionalizada catalizada por un complejo quiral de Ru (II), Noyori, 1989.26
Durante muchos años la catálisis organometálica ha resultado ser más
versátil que la biocatálisis. Hubo un tiempo en que se pensó que solamente
mediante el uso de catalizadores que contuvieran metales de transición se
podían obtener los dos posibles enantiómeros de los productos. Más adelante
esta creencia se vino abajo cuando se produjeron nuevos avances
espectaculares en biocatálisis, que ampliaban la versatilidad de los enzimas.
Así empezaba una carrera competitiva entre la catálisis organometálica y la
enzimática por las vías de producción óptimas aplicables en la industria para la
obtención de EPC.18f En este contexto, surge una nueva estrategia que
combina características de ambos tipos de catálisis, la organocatálisis.
1.3. Organocatálisis
Al igual que muchos complejos metálicos y enzimas, las moléculas
orgánicas pueden acelerar reacciones químicas.
El uso de moléculas orgánicas pequeñas (de bajo peso molecular)
como catalizadores quirales es reciente y ofrece posibles alternativas para las
metodologías ya existentes en síntesis asimétrica que requieren catalizadores
metálicos.
El desarrollo de la organocatálisis fue tardío en comparación con la
catálisis organometálica asimétrica, básicamente debido a que ésta última
ofrecía un abanico de posibilidades para todo tipo de reacciones, mientras que
los métodos organocatalíticos se mostraron, en un principio, poco eficaces y de
generalidad limitada. Aunque todavía hoy por hoy la mayoría de las reacciones

Introducción
27
en catálisis asimétrica industrial se apoyan en complejos organometálicos, la
organocatálisis está ganando cada vez más importancia en la química
orgánica, ofreciendo una serie de ventajas frente a los métodos
organometálicos o bioorgánicos.
Muchas reacciones organocatalíticas se pueden llevar a cabo en
condiciones aeróbicas, con disolventes húmedos; de hecho, la presencia de
agua en el medio es beneficiosa para el rendimiento y selectividad de la
reacción en muchos casos. La simplicidad operacional y la robustez de estos
catalizadores que son, además, económicos en la mayoría de los casos, hacen
de la organocatálisis un método atractivo para la síntesis de estructuras
complejas. A estas ventajas hay que añadir la, por lo general, menor toxicidad
de los organocatalizadores y el hecho de que sean usualmente más fáciles de
recuperar mediante tratamientos sencillos post-reacción para su reutilización.
Por todo ello se puede decir que la organocatálisis es más respetuosa con el
medioambiente y se ajusta mejor a los criterios de la Química Verde.
La naturaleza inherentemente “verde” y barata de la organocatálisis
permite augurar que cada vez más métodos industriales en los que se utilizan
catalizadores metálicos sean reemplazados por métodos organocatalíticos.
A modo de resumen, se exponen en la Figura 1-8 las distintas
estrategias para la obtención de EPC:
Figura 1-8: Esquema general de las distintas estrategias para la obtención de EPC.

Capítulo 1
28
1.3.1. Desarrollo de la organocatálisis asimétrica
Para descubrir las raíces históricas de la organocatálisis hemos de
trasladarnos a la época en que algunos químicos intentaban entender e imitar
el funcionamiento de los enzimas mediante el uso de moléculas de bajo peso
molecular, cuando en 1848 Luis Pasteur27 observó que partiendo de una
mezcla racémica de ácido tartárico, el organismo Penicillium glauca
descarboxilaba más rápidamente el enantiómero dextrógiro. Puede decirse que
empezó el interés por las reacciones enantioselectivas. Era el principio de la
catálisis asimétrica.
Aunque el término “organocatálisis” no se implantó hasta el 2001, la
metodología en sí se descubrió casi un siglo antes. Casualmente la primera
reacción organocatalítica fue también una descarboxilación. En 1904,
Mackwald llevó a cabo la descarboxilación de un derivado del ácido malónico
en presencia de brucina para dar el ácido 2-metilbutírico con un 10% de exceso
enantiomérico (Esquema 1-3).28
Esquema 1-3: Primera reacción organocatalítica, Mackwald, 1904.28
Por otro lado, a Breding se le atribuye el mérito de realizar la primera
reacción asimétrica organocatalítica que implicaba la formación de enlaces
carbono-carbono (Esquema 1-4), consistente en la preparación
enantioselectiva de mandelonitrilo mediante la adición de HCN a benzaldehído
en presencia de alcaloides tales como quinina y quinidina como catalizadores.29
Dichos procedimientos destacaron por ser innovadores, pero aún distaban
27 P. I. Dalko, Enantioselective Organocatalysis: Reactions and Experimental Procedures, 2007, Wiley-VCH. 28 W. Mackwald, Ber. Dtsch. Chem. Ges. 1904, 37, 349-354. 29 G. Breding, P. S. Fiske, Biochem. Z. 1912, 46, 7-23.

Introducción
29
mucho de ofrecer resultados aptos para síntesis teniendo en cuenta la baja
enantioselectividad de los productos obtenidos.
Esquema 1-4: Primera reacción organocatalítica asimétrica con formación de enlace C-C,
Breding, 1912.29
A pesar de que las transformaciones catalíticas ganaron importancia
tras la Primera Guerra Mundial, las reacciones asimétricas todavía se
consideraban más bien meramente una curiosidad académica. Este hecho y la
falta de métodos de purificación y análisis fiables para estos compuestos en
esa época dificultaron enormemente el desarrollo de la organocatálisis
asimétrica.
No fue hasta finales de los años 50 cuando se consiguieron niveles de
excesos enantioméricos aceptables desde el punto de vista sintético para una
reacción organocatalítica. Así, Pracejus descubrió que la metil fenil cetena se
podía convertir en (-)-α-fenil metilpropionato de metilo con un 74% de ee
mediante el uso de la O-acetilquinina como catalizador (Esquema 1-5).30
Esquema 1-5: Primera reacción organocatalítica asimétrica con alto exceso enantiomérico,
Pracejus, 1960.30
30 H. Pracejus, Justus Liebigs Ann. Chem. 1960, 634, 9-22.

Capítulo 1
30
En este entorno histórico cabe destacar el descubrimiento de la L-
prolina como catalizador, utilizada por primera vez en los años 70 para la
llamada reacción de Hajos-Parrish-Eder-Sauer-Wiechert, una reacción aldólica
intramolecular que permitió el acceso a intermedios clave para la síntesis de
esteroides (Esquema 1-6).31 Esta reacción, de gran interés sintético y
mecanístico,32 fue el primer ejemplo de aminocatálisis, es decir, de la
organocatálisis promovida por aminas. A pesar de que este extraordinario
resultado fue acogido con entusiasmo por la comunidad científica, este tipo de
activación no se volvió a utilizar hasta pasados varios años.
Esquema 1-6: Anelación de Robinson mediada por la L-Prolina, Hajos-Parrish-Eder-Sauer-Wiechert, 1971.31
A partir de este punto, durante las dos siguientes décadas comenzaron
a descubrirse diversas maneras de llevar a cabo reacciones organocatalíticas
eficientes a la vez que se abrían nuevas vías de investigación dentro de la
organocatálisis. Ejemplos pioneros destacables son la epoxidación de alquenos
catalizada por cetonas quirales, realizada por Shi, Wang y Denmark en 1996,33
o la reacción de Strecker catalizada por una tiourea realizada por Jacobsen dos
años más tarde, 34 resultando ser además esta última la primera organocatálisis
asimétrica por enlace de hidrógeno (ver sección 1.3.2.1). También cabe
destacar la utilización de sales de amonio cuaternarias para la catálisis por
31 a) U. Eder, G. Sauer, R. Wiechert, Angew. Chem. Int. Ed. 1971, 10, 496-497. b) Z. G. Hajos, D. R. Parrish, J. Org. Chem. 1974, 39, 1615-1621. 32 Para la revisión pionera sobre la L-prolina como catalizador: J. Martens, Top. Curr. Chem. 1984, 125, 165-246. 33 a) Y. Tu, Z. Wang, Y. Shi, J. Am. Chem. Soc. 1996, 118, 491-492. b) S. E. Denmark, Z. Wu, C. Crudden, H. Matsuhashi, J. Org. Chem. 1997, 62, 8288-8289. 34 M. Sigman, E. N. Jacobsen, J. Am. Chem. Soc. 1998, 120, 4901-4902.

Introducción
31
transferencia de fase (ver sección 1.3.2.1), cuyos pioneros fueron Lygo, Corey
(1987) y O’Donnell (1989).35
Volviendo a la aminocatálisis, se observa que ésta se estudió
tardíamente con respecto a las demás estrategias organocatalíticas. Tuvieron
que pasar tres décadas desde la reacción de Hajos-Parrish-Eder-Sauer-
Wiechert para que renaciera la catálisis promovida por aminas. Seguidamente,
y antes de exponer con más detalle el funcionamiento y desarrollo de la
aminocatálisis, se detallan los diferentes modos de activación para las
reacciones organocatalíticas.
1.3.2. Tipos de activación
La clasificación más extendida de los organocatalizadores está
relacionada con su mecanismo de reacción durante el ciclo catalítico y su modo
de activación de los sustratos de reacción.36 Así, el catalizador puede activar al
sustrato mediante interacciones débiles (no covalentes), que no alcanzan las 4
kcal/mol, o fuertes, mediante uniones covalentes, en las que la energía de
enlace catalizador-sustrato supera las 15 kcal/mol.
1.3.2.1. Catálisis no covalente
En el primer caso se habla de catálisis no covalente y los catalizadores
activan el nucleófilo o electrófilo mediante interacciones débiles, de tipo par
iónico o de enlace de hidrógeno.
A este grupo pertenece la catálisis por transferencia de fase37 (PTC),
que actúa mediante la formación de un par iónico quiral que genera una
discriminación enantiofacial. Como su nombre indica, las reacciones tienen
lugar en sistemas multifásicos, bien una fase líquida acuosa en contacto con
35 a) M. J. O’Donnell, W. D. Bennett, S. Wu, J. Am. Chem. Soc. 1989, 111, 2353-2355. b) B. Lygo, P. G. Wainwright, Tetrahedron Lett. 1997, 38, 8595-8598. c) E. J. Corey, F. Xu, M. C. Noe, J. Am. Chem. Soc. 1997, 119, 12414-12415. 36 D. W. C. MacMillan, Nature, 2008, 455, 304-308. Existe otra clasificación de los organocatalizadores acorde con su reactividad ácido/base propuesta por List; según ésta, los catalizadores se dividirían en ácidos o bases de Lewis y ácidos o bases de Brønsted, dependiendo de su modo de iniciar el ciclo catalítico: J. Seayad, B. List, Org. Biomol. Chem. 2005, 3, 719-724. 37 Para revisiones sobre catálisis por transferencia de fase, ver: a) T. Hashimoto, K. Maruoka, Chem. Rev. 2007, 107, 5656-5682. b) M. J. O'Donnell, Cat. Rev. 1998, 4, 5-8.

Capítulo 1
32
una orgánica o bien una fase líquida en contacto con una sólida (Fig. 1-9). Éste
es un método atractivo desde el punto de vista medioambiental debido a la
posibilidad de minimizar el uso de disolventes orgánicos.38 Los catalizadores
que han conducido a los mejores resultados en este campo son sales de
amonio cuaternarias derivadas de las cinchonas35 y de binaftilaminas.39
Fase orgánica
(Interfase)
Fase acuosao sólida
Nu-H M+Nu-
M+OH- H2O
[Q+X-]* Cat.
[Nu-Q+]*
MX
EX
ENu*
Nu: nucleófilo, E: electrófilo, M: metal, X: halógeno, Q: base quiral Figura 1-9: Mecanismo de catálisis por transferencia de fase.
Otro tipo de catálisis no covalente que actúa mediante par iónico es
aquélla en la que se encuentran involucradas aminas terciarias quirales que
funcionan como bases de Brønsted.40 En estos casos el catalizador abstrae un
protón del sustrato aumentando su nucleofilia y a la vez aportándole un entorno
quiral a través de la formación de un par iónico (Fig.1-10). Las bases de
Brønsted más generales para reacciones organocatalíticas asimétricas son
aminas terciarias, destacando entre éstas los alcaloides de tipo cinchona,
guanidinas, amidinas e imidazoles.
38 M. Makosza, Pure Appl. Chem. 2000, 72, 1399-1403. 39 R. He, S. Shirakawa, K. Maruoka, J. Am. Chem. Soc. 2009, 131, 16620-16621. 40 Para revisiones sobre catálisis con bases de Brønsted, ver: a) T. Ishikawa, T. Kumamoto, Synthesis 2006, 737-752. b) S. E. Denmark, G. L. Beutner, Angew. Chem. Int. Ed. 2008, 47, 1560-1638. c) C. Palomo, M. Oiarbide, R. López, Chem. Soc. Rev. 2009, 38, 632-653. Para ejemplos, ver: d) T. B. Poulsen, C. Alemparte, S. Saaby, M. Bella, K. A. Jørgensen, Angew. Chem. Int. Ed. 2005, 44, 2896-2899.

Introducción
33
Figura 1-10: Catálisis asimétrica mediante bases de Brønsted quirales.
Por otra parte, otro mecanismo de activación relevante es aquél que
implica la formación de enlaces de H catalizador-sustrato (Fig. 1-11).41 En estos
casos el catalizador, que contiene grupos dadores de hidrógeno, forma uno o
varios enlaces de hidrógeno con el componente electrófilo de la reacción
disminuyendo su densidad electrónica y predisponiéndolo para ser atacado por
un nucleófilo. Según la fortaleza que presente el enlace de hidrógeno y el
número de interacciones que puedan establecerse, podemos encontrarnos con
una activación monodentada o bidentada, e incluso con formación de pares
iónicos cuando haya protonación neta del sustrato por acción del ácido de
Brønsted. Los catalizadores de este tipo más utilizados son las ureas, tioureas,
guanidinas, dioles, ácidos carboxílicos, ácidos fosfóricos y amidas quirales.
Figura 1-11: Mecanismo de activación por enlace de hidrógeno.
Finalmente, comentar que también se incluye en el grupo de catálisis
no covalente aquélla promovida por moléculas quirales de bajo peso molecular,
entre ellas péptidos,42 éteres corona, oligonucleótidos, calixarenos o
ciclodextrinas, que participan en la activación del sustrato mediante la
formación de complejos supramoleculares (host-guest).
41 Para revisiones sobre este tema, ver: a) M. S. Taylor, E. N. Jacobsen, Angew. Chem. Int. Ed. 2006, 45, 1520-1543. b) A. G. Doyle, E. N. Jacobsen, Chem. Rev. 2007, 107, 5713-5743. c) H. Miyabe, Y. Takemoto, Bull. Chem. Soc. Jpn. 2008, 81, 785-795. d) S. J. Connon, Chem. Commun. 2008, 2499-2510. e) X. H. Yu, W. Wang, Chem. Asian J. 2008, 3, 516-532. f) Z. Zhang, P. R. Sheriner, Chem. Soc. Rev. 2009, 38, 1187-1198. g) P. M. Pihko, Hydrogen Bonding in Organic Synthesis. Wiley-VCH, 2009. 42 Para revisiones sobre este tema, ver: a) E. R. Jarvo, S. J. Miller, Tetrahedron 2002, 58, 2481-2495. b) E. A. Colby, S. M. Davie, Y. X. Mennen, S. J. Miller, Chem. Rev. 2007, 107, 5759-5812.

Capítulo 1
34
1.3.2.2. Catálisis covalente
Gran parte de las reacciones organocatalíticas proceden mediante la
formación covalente de un complejo activado catalizador-sustrato. Este modo
de activación implica la existencia de reacciones químicas reversibles para el
anclaje y desanclaje del catalizador al sustrato/producto de forma que se
posibilite la regeneración del catalizador. Los catalizadores más relevantes
dentro de este grupo son los aminoácidos, péptidos, alcaloides y moléculas
sintéticas portadoras de nitrógeno (aminas primarias o secundarias
principalmente).
Las aminas primarias o secundarias quirales (generalmente llamadas
aminocatalizadores) (Fig.1-12) participan en muchos tipos de reacciones
activando los correspondientes sustratos carbonílicos (aldehídos y cetonas)
mediante la formación reversible de un intermedio, principalmente enamina43 o
ion iminio,44 y en menor proporción dienamina45 o catión radical-iminio, también
conocida como catálisis SOMO.46 Estos modos de activación son eficaces para
la α, β y γ funcionalización de aldehídos y cetonas mediante la formación de
enlaces carbono-carbono y carbono-heteroátomo.47
43 Para revisiones sobre catálisis vía enamina, ver: a) B. List, Tetrahedron 2002, 58, 5573-5590. b) B. List, Acc. Chem. Res. 2004, 37, 548-557. c) B. List, Chem. Commun. 2006, 819-824. c) S. Mukherjee, J. W. Yang, S. Hoffmann, B. List, Chem. Rev. 2007, 107, 5471-5569. d) S. Sulzer-Mossé, A. Alexakis, Chem. Commun. 2007, 3123-3135. e) T. Kano, K. Maruoka, Chem. Commun. 2008, 5465-5473. 44 Para revisiones sobre catálisis vía ion iminio, ver: a) G. Lelais, D. W. C. MacMillan, Aldrichimica Acta 2006, 39, 79-87. b) S. B. Tsogoeva, Eur. J. Org. Chem. 2007, 1701-1716. c) D. Almasi, D. A. Alonso, C. Nájera, Tetrahedron: Asymmetry 2007, 18, 299-365. d) A. Erkkilä, I. Majander, P. M. Pihko, Chem. Rev. 2007, 107, 5416-5470. 45 Para ejemplos sobre catálisis vía dienamina, ver: a) S. Bertelsen, M. Marigo, S. Brandes, P. Dinés, K. A. Jørgensen, J. Am. Chem. Soc. 2006, 128, 12973-12980. b) S. Bertelsen, M. Nielsen, K. A. Jørgensen, Angew. Chem. Int. Ed. 2007, 46, 7356-7359. c) G. Bergonzini, S. Vera, P. Melchiorre, Angew. Chem. Int. Ed. 2010, 49, 9685-9688. 46 Para más información sobre este modo de activación, ver: a) T. D. Beeson, A. Mastracchio, J-B. Hong, K. Ashton, D. W. C. MacMillan, Science 2007, 316, 582-585. b) ver ref. 45b. c) P. Renaud, P. Leong, Science, 2008, 322, 55-56. d) P. Melchiorre, Angew. Chem. Int. Ed. 2009, 48, 1360-1363. 47 Muy recientemente se ha descrito otro tipo de activación para la β y ε-funcionalización de dienales, vía trienamina. Para más información, ver: a) Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen, K. A. Jørgensen, J. Am. Chem. Soc. 2011, 133, 5053-5061. b) H. Jiang, B. Gschwend, L. Albrecht, S. G. Hansen, K. A. Jørgensen, Chem. Eur. J. 2011, 17, 9032-9036. c) Z.-J. Jia, Q. Zhou, Q.-Q. Zhou, P.-Q. Chen, Y.-C. Chen, Angew. Chem. Int. Ed. 2011, 50, 8638-8641.

Introducción
35
O
R1
R2
*R2NH NR2*
R1
R2
1) EO
R1
R2
E
O
R1R2
*R2NH NR2*
R1R2
O
R1R2
Nu
Activación vía enamina:
Activación vía ion iminio:
*
1) Nu
*
NR2*
R2
R1
dienamina
NR2*
R1
R2
SOMO
Activaciones vía dienamina:
O
R2
R1
*R2NH
O
R1
R2
1) *R2NH
-H2O 2) H2O
OH
2) H2O
-H2O
2) Ox.
Activaciones vía SOMO:
Figura 1-12: Mecanismos más estudiados de activación por aminocatálisis.
Otros tipos de catalizadores que actúan por interacción covalente con
el sustrato son: los carbenos N-heterocíclicos (A en la Fig.1-13),48 que
funcionan mediante la formación del intermedio de Breslow para sustratos
carbonílicos; algunas aminas terciarias o análogos (B en la Fig.1-13),49 que
promueven la denominada catálisis nucleofílica en reacciones vía iluros;50
piridinas que funcionan vía sales de acilamonio y trialquilfosfinas o
48 Para revisiones sobre este tema, ver: a) D. Enders, O. Niemeier, A. Henseler, Chem. Rev. 2007, 107, 5606-5655. b) N. Marion, S. Diez-González, S. P. Nolan, Angew. Chem. Int. Ed. 2007, 46, 2988-3000. 49 Para revisiones sobre este tema, ver: R. P. Wurz, Chem. Rev. 2007, 107, 5570-5595. 50 Para revisiones sobre este tema, ver: a) S. France, D. J. Guerin, S. J. Miller, T. Lectka, Chem. Rev. 2003, 103, 2985-3012. b) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaws, S. L. Riches, V. K. Aggarwal, Chem. Rev. 2007, 107, 5841-5883. c) M. J. Gaunt, C. C. C. Johanson, Chem. Rev. 2007, 107, 5596-5605.

Capítulo 1
36
trialquilaminas (D en la Fig.1-13),51 que reaccionan con olefinas π-deficientes
para dar lugar a intermedios tipo Baylis-Hillman. Finalmente, mencionar que las
reacciones de oxidación vía oxiranos quirales52 también están incluídas dentro
de este tipo de organocatálisis.
X
X
N
R1
OHAr
*R
intermediode Breslow
A) CarbenosN-heterocíclicos
N*R
R*R*
R1
N R1
O*R
iluros
sales deN-acilamonio
B) Aminas terciariasy análogos
X
R**R R*
R1EWG
intermedios deBaylis-Hillman
D) Trialquilaminasy trialquilfosfinas
Sustratos
X
N
Ar
*R
Catalizador EspecieIntermedia
N*R
R*R*
N*R
C) Piridinas
R1
O
H
Ejemplo:aldehído
XR**R
R*
X: N, PR1
O
H
Ejemplo: aldehídoy alqueno activado
EWG
R1
O
H
Ejemplo:aldehído
R1
O
X
Ejemplo:ácidos oderivados
X-
Figura 1-13: Otros catalizadores y modos de activación en catálisis covalente.
1.3.3. Aminocatálisis
La aminocatálisis englobaría aquellos procesos organocatalíticos
promovidos por aminas. Entre éstos, los que más interés han suscitado son las
51 Para revisiones sobre este tema, ver: a) G. Masson, C. Housseman, J. Zhu, Angew. Chem. Int. Ed. 2007, 46, 4614-4628. b) Y.-L. Shi, M. Shi, Eur. J. Org. Chem. 2007, 2905-2916. c) D. Basavaiah, K. V. Rao, R. J. Reddy, Chem. Soc. Rev. 2007, 36, 1581-1588. d) P. R. Krishna, R. Sachwani, P. S. Reddy, Synlett 2008, 2897-2912. e) V. Declerck, J. Martínez, F. Lamaty, Chem. Rev. 2009, 109, 1-48. 52 Para revisiones sobre este tema, ver: a) M. Frohn, Y. Shi, Synthesis 2000, 1979-2000. b) S. E. Denmark, Z. Wu, Synlett, 1999, 847-859.

Introducción
37
aminas primarias53 y secundarias, que conducen generalmente, por interacción
covalente con un aldehído o cetona, a intermedios enamínicos o imínicos como
especies reactivas nucleofílica y electrofílica respectivamente. La activación vía
enamina (a en Fig. 1-14) produce una mayor densidad electrónica en el
carbono nucleofílico con respecto a la del carbono en α del aldehído o cetona
de partida, mientras que en la activación vía ion iminio (b en Fig. 1-14) se
produce un aumento en la electrofilia del sustrato carbonílico al transformarse
en ion iminio. Así, se puede afirmar que este tipo de catalizadores pueden
actuar de manera similar a los ácidos y bases de Lewis con respecto al tipo de
activación que promueven, es decir, aumentando la electrofilia o nucleofilia del
sustrato de partida correspondiente.
Figura 1-14
En la Figura 1-15 se representan los ciclos catalíticos simplificados
para los procesos de α- y β-funcionalización de compuestos carbonílicos
(aldehídos y cetonas) vía enamina e ion iminio, respectivamente.
En la catálisis mediada por enaminas, el ciclo catalítico, en el sentido
de las agujas del reloj, incluye la formación previa de un intermedio imínico
53 Para revisiones sobre aminas primarias como organocatalizadores, ver: a) F. Peng, Z. Shao, J. Mol. Catal. A: Chem. 2008, 285, 1-13. b) L. W. Xu, Y. Lu, Org. Biomol. Chem. 2008, 6, 2047-2053. c) L. W. Xu, J. Luo, Y. Lu, Chem. Commun. 2009, 1807-1821. Para ejemplos, ver: d) A. Córdova, W. Zou, I. Ibrahem, E. Reyes, M. Engqvist, W. W. Liao, Chem. Commun. 2005, 3586-3588. e) I. Ibrahem, W. Zou, M. Engqvist, Y. Xu, A. Córdova, Chem. Eur. J. 2005, 11, 7024-7029. f) A. Córdova, W. Zou, P. Dziedzic, I. Ibrahem, E. Reyes, Y. Xu, Chem. Eur. J. 2006, 12, 5383-5397.

Capítulo 1
38
entre el compuesto carbonílico y la amina quiral que actúa como catalizador, la
subsiguiente desprotonación de la especie imínica para generar una enamina,
la formación de un enlace C-C ó C-heteroátomo entre la enamina y el
componente electrofílico de la reacción e hidrólisis final del nuevo ion iminio
resultante, con la consecuente liberación del producto de reacción y
recuperación del catalizador.
En la catálisis mediada por ion iminio la especie intermedia en forma de
iminio actúa como aceptor frente a un nucleófilo, formándose una enamina
quiral, la cual, tras protonación e hidrólisis, libera el producto final y el
catalizador.
R*
H2O
RNH
H2O
*
Amina quiral secundaria
H+
E+
-H+
Enamina quiral
Ion iminio
O
R1
R2
N
R1
R2
R R*
N
R1
R2
R R*
N
R1
R2
R R*
E*
O
R1
R2
E
R*
H2O
RNH
H2O
Amina quiral secundaria
Enaminaquiral
Ion iminio
O
R1
R2
N
R1
R2
R R*
Nu-
N
R1
R2
R R*
Nu
N
R1
R2
R R*
Nu
O
R1
R2 Nu
H+
**
*
H+
Ciclo catalítico vía enamina Ciclo catalítico vía ion iminio
Nota: Aunque por claridad se han dibujado flechas de reacción en un único sentido, todas las etapas dibujadasson reversibles.
Figura 1-15: Ciclo catalítico de activación de compuestos carbonílicos saturados y α,β-
insaturados vía enamina e ion iminio respectivamente.
Tras el primer ejemplo de aminocatálisis covalente empleando como
catalizador prolina, (la reacción de Hajos-Parrish-Eder-Sauer descrita en los
años 71-73 antes comentada (Esquema 1-7)), el interés por la prolina como
aminocatalizador renació casi tres décadas después gracias al estudio del
mecanismo de acción de las aldolasas de tipo I (que catalizan la reacción
aldólica vía enamina) realizado por Lerner y Barbas III.54 Éste último reexaminó
la reacción de Hajos-Parrish-Eder-Sauer, esta vez intermolecular, utilizando la
54 J. Warner, R. Lerner, C. F. Barbas III, Science 1995, 270, 1797-1800.

Introducción
39
L-Prolina como catalizador,55 intentando emular el comportamiento de dichos
enzimas. El correspondiente aducto se obtuvo con un rendimiento del 49% y un
74% de exceso enantiomérico.
Me
O
O
Me
Rdto.: 49%74% ee
NH
CO2H
O+
O
MeO
ODMSO
O
O
35 mol %
Cat.:
Esquema 1-7: Reacción de anelación de Robinson intermolecular catalizada por L-Prolina,
Barbas III, 1997.55
El año clave para el renacimiento de la aminocatálisis fue el 2000,
cuando se publicaron dos trabajos pioneros en la catálisis vía enamina e ion
iminio, por Barbas III y List,56 por un lado, y MacMillan57 por otro,
respectivamente.
El primer desarrollo, la reacción aldólica intermolecular (Esquema 1-8),
descrita por Barbas III y List, supuso un hito histórico que afloró el potencial de
la catálisis vía enamina en síntesis.
Esquema 1-8: Reacción aldólica directa catalizada por la L-Prolina, List, Barbas III, 2000.56
Por otro lado, MacMillan describió la reacción de Diels-Alder entre
enales y dienos utilizando como catalizador un nuevo tipo de aminas
secundarias quirales, las imidazolidinonas (1 en este caso) (Esquema 1-9),
dando inicio al desarrollo de la catálisis vía ion iminio. Dicha metodología
conduce a los correspondientes aductos de Diels-Alder (2) con excelente
55 T. Bui, C. F. Barbas III, Tetrehedon Lett. 2000, 41, 6951-6954. 56 B. List, R. A. Lerner, C. F. Barbas III, J. Am. Chem. Soc. 2000, 122, 2395-2396. 57 K. A. Ahrendt, C. J. Bortha, D. W. C. MacMillan, J. Am .Chem. Soc. 2000, 122, 4243-4244.

Capítulo 1
40
selectividad. Fue el propio MacMillan quien, tras este estudio, introdujo el
término organocatálisis, que sustituyó a la hasta entonces nombrada catálisis
libre de metales.
Esquema 1-9: Reacción de Diels-Alder entre enales y dienos vía ion iminio catalizada por la
imidazolidinona 1, MacMillan, 2000.57
A la vista de los excelentes resultados obtenidos, el autor
inmediatamente extendió con éxito esta metodología a las reacciones de
cicloadición 1,3-dipolar58 y de alquilación de Friedel-Crafts.59
Estos ejemplos tan prometedores marcaron el principio de una nueva
etapa en el desarrollo de la organocatálisis, que ha quedado patente en los
abundantes estudios posteriores y los exitosos resultados que han
acompañado.
La prolina presenta ciertas limitaciones como organocatalizador que
impiden extender su uso de una forma universal: principalmente su baja
solubilidad en disolventes orgánicos y la ineficiencia, en términos de
enantioselectividad, en reacciones que involucren electrófilos poco proclives a
formar enlaces de hidrógeno.60 Con objeto de solventar estas limitaciones, se
han diseñado una gran variedad de pirrolidinas quirales derivadas de la L-
prolina que, junto con la propia prolina, han resultado ser muy eficaces en una
gran variedad de reacciones que involucran compuestos carbonílicos.
Aparte de la propia prolina (A en Fig. 1-16) y las imidazolinonas
descritas por MacMillan (B en Fig. 1-16), Hayashi61 y Jørgensen62 desarrollaron
58 W. Jen, J. Wiener, D. W. C. MacMillan, J. Am. Chem. Soc. 2000, 122, 9874-9875. 59 N. Paras, D. W. C. MacMillan, J. Am. Chem. Soc. 2001, 123, 4370-4371. 60 Para una ampliación de este tema, ver: J. Franzén, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18296-18304. 61 Y. Hayashi, H. Gotoh, T. Hayashi, M. Shoji, Angew. Chem. Int. Ed. 2005, 44, 4212-415.

Introducción
41
en 2005 un tipo de pirrolidina (C en Fig. 1-16) que constituyó la tercera clase
general de catalizadores en este campo. El descubrimiento y posterior
desarrollo de los catalizadores de tipo C serán detallados más adelante en este
capítulo.
NH
CO2HNH
Ar
OSiMe3
Ar
NH
NO
R3
R2
R1
A B C Figura 1-16
Desde el año 2000 se han documentado varias aminas secundarias
como organocatalizadores eficaces, en particular derivados estructurales de la
prolina.63 Algunos de los más destacables tanto para la catálisis vía enamina64
como la catálisis vía ion iminio65 se recogen en las Tablas 1-1 y 1-2
respectivamente.
62 a) M. Marigo, T. C. Wabnitz, D. Fielenbach, K. A. Jørgensen, Angew. Chem. Int. Ed. 2005, 44, 794-797. b) M. Marigo, J. Franzén, T. Poulsen, W. Zhuang, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 6964-6965. 63 Para revisiones genereales, ver: a) C. Palomo, A. Mielgo, Angew. Chem. Int. Ed. 2006, 45, 7876-7880. b) A. Mielgo, C. Palomo, Chem. Assian. J. 2008, 922-948. c) A. Lattanzi, Chem. Commun. 2009, 1452-1463. 64 a) J. M. Betancort, C. F. Barbas III, Org. Lett. 2001, 3, 3337-3340. b) J. M. Betancort, K. Sakthivel, R. Thayumanavan, C.F. Barbas III, Tetrahedron Lett. 2001, 42, 4441-4444. c) A. Alexakis, O. Andrey, Org. Lett. 2002, 4, 3611-3614. d) P. Melchiorre, K. A. Jørgensen, J. Org. Chem. 2003, 68, 4151-4157. e) A. J. A. Cobb, D. M. Shaw, S. V. Ley, Synlett 2004, 15, 1831-1834. f) W. Wang, J. Wang, H. Li, L. Liao, Tetrahedron Lett. 2004, 45, 7235-7238. 65 a) Ver ref. 57. b) J. F. Austin, D. W. C. MacMillan, J. Am. Chem. Soc. 2002, 124, 1172-1173. c) N. Halland, P. S. Aburel, K. A. Jørgensen, Angew. Chem. Int. Ed. 2003, 42, 661-665. d) S. Brandau, A. Landa, J. Franzén, K. A. Jørgensen, Angew. Chem. Int. Ed. 2006, 45, 4305-4309. e) K. R. Kundsen, C. E. T. Mitchell, S. V. Ley, Chem. Comm. 2006, 66-68. f) D. Enders, M. H. Bonten, G. Raabe, Synlett 2007, 885-888.

Capítulo 1
42
Tabla 1-1: Catalizadores más representativos derivados de la L-prolina en reacciones vía enamina:
Catalizador Autores pioneros Primeras reacciones
Hajos, Parrish, Eder, Sauer
(1970) List, Barbas (2000)
Reacción aldólica31
Barbas III (2001)
Adición conjugada de aldehídos a nitroalquenos64a
Adición conjugada de
cetonas a alquilidenmalonatos64b
Alexakis (2002)
Adición conjugada de aldehídos y cetonas a
nitroalquenos64c
Jørgensen (2003)
Adición conjugada de
aldehídos a vinilcetonas64d
Ley (2004)
Adición conjugada de
cetonas a nitroalquenos64e
Wang (2004)
Reacción de aminoxilación de aldehídos y cetonas64f
Jørgensen (2005)
Funcionalización de
aldehídos y cetonas62, 64g
Hayashi (2005)
Adición conjugada de
aldehídos a nitroalquenos61

Introducción
43
Tabla 1-2: Catalizadores más representativos utilizados en reacciones vía ion iminio:
Catalizador Autores pioneros Primeras reacciones
MacMillan (2000)
Diels-Alder con enales57
NH
NO
Ph
Me
.HXimidazolidinonas2ª generación
MacMillan (2002)
Adición conjugada de indoles a aldehídos α,β-insaturados65b
Jørgensen (2003)
Adición conjugada de
malonatos a aldehídos α,β-insaturados65c
Jørgensen (2006)
Adición conjugada de
malonatos a aldehídos α,β-insaturados65d
Enders (2007)
Glicosilación conjugada de aldehídos α,β-insaturados65f
NH
N
NN
HN
Ley (2006)
Adición conjugada de malonatos a enonas65e
1.3.3.1. Control de la estereoquímica en las reacciones vía enamina e
ion iminio
El grado de éxito de las estrategias catalíticas que transcurren por
activación tanto vía enamina como vía ion iminio viene condicionado por el
grado de rigidez configuracional y conformacional de las especies intermedias
(enaminas o iones iminio) involucradas en el estado de transición.
Seguidamente se exponen los elementos estereoquímicos más relevantes así
como los elementos de estereocontrol más significativos para cada una de las
estrategias.
En los procesos que transcurren a través de formación de enaminas los
elementos de estereocontrol más relevantes son los siguientes:

Capítulo 1
44
a) Configuración E ó Z del doble enlace enamínico (Fig. 1-17). De
manera general, la enamina E es más estable que la Z.
Figura 1-17: Isómeros configuracionales de enaminas provenientes de aldehídos.
b) Disposición (conformación/rotamería) sin ó anti del sustituyente
pirrolidínico con respecto al doble enlace carbono-carbono de la
enamina de configuración E (Fig. 1-18). Por lo general, para
enaminas provenientes de aldehídos, la disposición anti es más
estable.
N
R
sin
N
anti
R
Figura 1-18: Rotamería sin/anti en enaminas E provenientes de aldehídos.
c) Carácter coordinante o no (presencia o ausencia de dadores de H)
en α al nitrógeno pirrolidínico.
Dependiendo de este último factor, de manera general y asumiendo
que la enamina participa a través de su forma E-anti, existen dos modos de
estereocontrol diferentes con este tipo de catalizadores: aproximación
controlada por enlace de hidrógeno y aproximación dirigida por control estérico.
Ambas estrategias se exponen en la Figura 1-19.66
En el caso del control por enlace de hidrógeno, el catalizador es
portador de un grupo dador de hidrógeno (ácido carboxílico, sulfonamida,
tetrazol, etc.) situado normalmente en la posición α al nitrógeno pirrolidínico, 66 Para la definición de estos modelos, ver ref. 63a y b. Para la configuración y conformación de las enaminas, ver: a) M. B. Schmid, K. Zeitler, R. M. Gschwind, J. Am. Chem. Soc. 2011, 133, 7065-7074. b) M. B. Schmid, K. Zeitler, R. M. Gschwind, Chem. Sci. 2011, 2, 1793-1803.

Introducción
45
que se coordina con el aceptor, aproximándolo por la cara correspondiente a
dicho grupo.67 La L-prolina es un claro ejemplo de este tipo de control y dirige la
aproximación del aceptor por la cara Re de la enamina.
Por otro lado, para ejercer control estérico los catalizadores necesitan
portar grupos voluminosos, situados también normalmente en la posición α al
nitrógeno pirrolidínico, que bloqueen una cara de la enamina, forzando la
aproximación del aceptor por la cara contraria.68 De esta manera funcionan, por
ejemplo, los éteres de diarilsililo derivados del prolinol, que dirigen la
aproximación de los aceptores por la cara Si de la enamina.
De forma general, la configuración de los productos finales por ambas
aproximaciones suele ser opuesta.
Figura 1-19: Justificación de la estereoquímica predominante en reacciones vía enamina según un control por enlace de hidrógeno o por repulsión estérica. Para la definición de estos
modelos, ver ref. 63b y c.
Por otro lado, para las reacciones vía ion iminio, los principales
elementos que influyen en el transcurso estereoquímico de la reacción son
análogos. Al igual que con enaminas quirales, en este caso la configuración del
producto viene determinada en gran medida por la geometría del ion iminio. En
este caso los elementos estereoquímicos relevantes69 serán la geometría E o Z
67 Para una profundización sobre este tema, ver: a) C. Blom, T. Rantanen, I. Sschiffers, L. Zani, Angew. Chem. Int. Ed. 2005, 44, 1758-1763. b) M. Marigo, K. A. Jørgensen. Chem. Commun. 2006, 2001-2011. c) G. Guillena, D. J. Ramón, Tetrahedron: Asymmetry. 2006, 17, 1465-1492. 68 Para una profundización sobre este tema, ver: a) P. Dinér, A. Kjærsgaard, M. L. Alstrup Lie, K. A. Jørgensen. Chem. Eur. J. 2008, 14, 122-127. b) M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen, K. A. Jørgensen. Chem. Commun. 2011, 47, 632-649. 69 Para más detalles sobre cálculos computaciones relativos a la estabilidad del ion iminio cuando se utilizan los éteres de trimetilsililo derivados de diarilprolinol, ver: a) D. Seebach, A. Beck, D. M. Badine, M. Limbach, A. Eschenmoser, A. M. Treasurywala, R. Hobi, Helv. Chim. Acta. 2007, 90, 425-471. b) P. Dinés, M. Nielsen, M. Marigo, K. A. Jørgensen, Angew. Chem.

Capítulo 1
46
del doble enlace imínico C=N y la disposición s-cis (desfavorecida) o s-trans
(favorecida) del sistema diénico. Por lo general, en iones iminio provenientes
de aldehídos se estima que la forma s-trans (E) predomina sobre las demás, tal
como se representa en la Figura 1-20.
RRIon iminio
s-cis (E) Ion iminios-trans (E)
N
R
Ion iminios-trans (Z)
H
HH
NN
Figura 1-20: Geometrías del ion iminio relevantes para la estereoquímica de sus reacciones.
Del mismo modo que en la activación vía enamina, para las reacciones
vía ion iminio existen, de manera general, dos modelos que explican el
estereocontrol: aproximación del nucleófilo (o componente nucleofílico de la
reacción) dirigida por un enlace de hidrógeno y aproximación dirigida por un
control estérico, cuyas representaciones simplificadas se muestran en la Figura
1-21.
Figura 1-21
1.3.3.2. Éteres de diarilprolinol como catalizadores en reacciones vía
enamina
La funcionalización de compuestos carbonílicos enolizables, entre ellos
aldehídos, en diferentes posiciones de la cadena (α, β, γ) es una de las
transformaciones donde la aminocatálisis puede ser aplicada con mayor éxito.
Int. Ed. 2007, 46, 1983-1987. c) D. Seebach, U. Groselj, D. M. Badine, W. B. Schweizer, A. Beck, Helv. Chim. Acta. 2008, 91, 1999-2033. d) I. Ibrahem, P. Hammar, J. Vesely, R. Rios, L. Eriksson, A. Córdova, Adv. Synth. Catal. 2008, 350, 1875-1884. e) U. Groselj, D. Seebach, D. M. Badine, W. B. Schweizer, A. Beck, Helv. Chim. Acta. 2009, 92, 1225-1259.

Introducción
47
En este contexto puede decirse en términos generales que la α-
funcionalización de estos compuestos transcurre vía enamina, la β-
funcionalización vía ion iminio y la γ-funcionalización vía dienamina. La
presente Tesis Doctoral se ha centrado en procesos organocatalíticos que
transcurren mediante activación vía enamina.
Las α-funcionalizaciones que tienen lugar con aminas secundarias de
tipo pirrolidina como catalizadores se pueden dividir en dos grupos
dependiendo de si el nuevo enlace formado es un enlace carbono-carbono o
carbono-heteroátomo (C-N, C-O, C-S, C-Se, C-X, siendo X un halógeno).
Junto con la prolina, los éteres de sililo del diarilprolinol han resultado
ser quizás los aminocatalizadores más versátiles y generales descritos hasta la
fecha, siendo aplicables a una gran variedad de reacciones organocatalíticas
de α, β ó γ-funcionalización de aldehídos y/o cetonas e incluso reacciones
multicomponente. En 2005 los grupos de Hayashi61 y Jørgensen62
independientemente, describieron por primera vez los catalizadores 3 y 4 (Fig.
1-22) pertenecientes a este grupo, los cuales han resultado ser los más
utilizados con posterioridad. A los dos años de su descubrimiento, estas
estructuras, o variantes similares, eran aplicables a más de veinte tipos de
reacciones organocatalíticas diferentes.63a,b
Teniendo en cuenta las anteriores consideraciones, en la Figura 1-22
se presentan las α-funcionalizaciones de aldehídos vía enamina más
representativas promovidas por éteres de aril sililo del prolinol.

Capítulo 1
48
Reaccionesvía enaminapromovidaspor 3 y 4
-Formación de enlaces C-C(Adiciones nucleofílicas)
-Reacción de Mannich
- -Arilaciones
-Adiciones de Michael
- a nitroalquenos
- a vinil cetonas
- a vinil fosfonatos
- a maleimidas
-Formación de enlaces C-X
- -Aminación
- -Hidroxilación
- -Halogenación
- -Sulfenilación
- -Seleniación
- a vinil sulfonas
NH
Ph
OTMS
Ph
NH OTMS
F3C CF3
CF3
CF33 4
Figura 1-22
En este contexto y dentro de las reacciones de formación de enlaces
carbono-carbono se encuentran reacciones tan generales e importantes para la
síntesis orgánica como la reacción de Mannich, la aldólica o diversas adiciones
conjugadas. En el Esquema 1-10 se encuentran recogidas las aportaciones
más representativas de este tipo hasta el momento del inicio de la presente
Tesis doctoral con sus autores pioneros.

Introducción
49
H
N
R1
Adición conjugadaa nitroalquenos
Adición conjugadaa vinilfosfonatos
Adición conjugadaa maleimidas
OHC
R1
P(O)(OEt)2
P(O)(OEt)2
OHC
R1
N
O
O
Ph
Adición conjugadaa vinil cetonas
OHC
R1
O
R2
CórdovaChem. Commun.2007,734-735
AlexakisOrg. Lett.2007, 9,3749-3752 Jørgensen
J. Am. Chem. Soc.2005, 12718296-18304
C
X
X: C,O, N,
R1
OHC
Rto: 79-84%d.r.: 92:8-89:11ee: 94-98%
CO2Et
HNPMP
JørgensenJ. Am. Chem. Soc.2005, 127,18296-18304
Mannich
-Arilación
O
OH
R1
OH
R2
Rto: 65-98%ee: 92-99%
JørgensenAngew. Chem. Int. Ed.2007, 46,5520-5523
OHC NO2
R2
R1
Rto: 52-85%d.r.>94:5ee: 99%
HayashiAngew. Chem.Int. Ed.2005, 44,4212-4215
Rto: 70-91 %ee: 97-99%
Rto: 80-93%ee: 92-95%
Rto: 65-85%ee: 97%
adición nucleofílica
NH
Ph
OTMS
Ph
NH OTMS
F3C CF3
CF3
CF33 4
Catalizadores:
Cat. 3
Cat. 3
Cat. 3
Cat. 3
Cat. 4
Cat. 4
Esquema 1-10: Reacciones de formación de enlaces C-C vía enamina catalizadas por
derivados del prolinol.
Por otro lado, también se han desarrollado varias α–
heterofuncionalizaciones de aldehídos vía enamina mediante el uso de
catalizadores de estructura derivada del prolinol. En el Esquema 1-11 se
recogen las reacciones más representativas de este tipo, desarrolladas con
anterioridad al inicio del presente trabajo de Tesis.

Capítulo 1
50
Esquema 1-11: Reacciones de formación de enlaces C-heteroátomo vía enamina catalizadas
por derivados del prolinol.
En este contexto podemos añadir que, nuevamente la naturaleza,
donde tienen lugar procesos biosintéticos catalíticos en cascada, ha servido de
fuente de inspiración para el desarrollo de procesos organocatalíticos que
involucran más de una etapa secuencial, en lo que se llaman procesos
multicomponente, tándem (dos o más mecanismos catalíticos involucrados) o
dominó (un solo mecanismo catalítico involucrado).70 La base de estos
70 Para revisiones sobre procesos tándem y/o dominó organocatalíticos, ver: a) J.-C. Wasilke, S. J. Obreg, R. T. Baker, G. C. Bazan, Chem. Rev. 2005, 105, 1001-1020. b) L. F. Tietze, G. Brasche. K. M. Gerike, Domino Reactions in Organic Synthesis, 2006, Wiley-VCH. c) D. Enders, C. Grondal, M. R. M. Hüttl, Angew. Chem. Int. Ed. 2007, 46, 1570-1581. d) A. Dondoni, A. Massi, Angew. Chem. Int. Ed. 2008, 47, 4638-4671. f) P. Melchiorre, M. Marigo, A. Carlone, G. Bartoli, Angew. Chem. Int. Ed. 2008, 47, 6138-6171. g) e) A.-N. Alba, X. Companyo, M. Viciano, R. Ríos, Curr. Org. Chem. 2009, 13, 1432-1474. h) S. Bertelsen, K. A. Jørgensen,

Introducción
51
procesos se halla en la complementariedad de los mecanismos vía enamina e
ion iminio. Así, mediante el uso de un mismo catalizador, de manera secuencial
se pueden llevar a cabo varias reacciones.
Esquema 1-12: Proceso secuencial de dos reacciones basadas en la activación vía
iminio/enamina, respectivamente.
Estos procesos, en los que normalmente participan dos o más
sustratos, permiten la realización “one pot” de varias reacciones sucesivas en
un mismo medio de reacción con la formación simultánea de varios
estereocentros, evitando así las costosas y/o tediosas etapas de purificación de
intermedios y reduciendo la necesidad de protección de grupos funcionales.
Dichas reacciones transcurren mediante activaciones consecutivas enamina-
iminio ó iminio-enamina, teniendo lugar los correspondientes ciclos catalíticos
de forma sucesiva. En este contexto, uno de los ejemplos pioneros y más
elegantes es la reacción en cascada Michael/ Michael/ aldólica descrita por
Enders,71 en la que se forman tres enlaces C-C “one pot” con excelentes regio-
y estereoselectividades (Esquema 1-13) y participan un aldehído lineal, un
nitroalqueno y un aldehído α,β-insaturado.
Chem. Soc. Rev. 2009, 38, 2178-2189. i) F. Santos, M. Sánchez-Roselló, C. del Pozo, Pure Appl. Chem. 2010, 82, 669-677. j) C. Grondal, M. Jeanty, D. Enders, Nature Chemistry 2011, 2, 167-178. Para una distinción entre estos conceptos, ver: j) D. E. Frogg, E. N. do Santos, Coord. Chem. Rev. 2004, 248, 2365-2379. k) Ch. J. Chapman, Ch. G. Frost, Synthesis, 2007, 1-21. 71 D. Enders, M. R. M. Hüttl, C. Grondal, G. Raabe, Nature, 2006, 441, 861-863.

Capítulo 1
52
Esquema 1-13: Ciclo catalítico propuesto para la reacción de triple cascada Michael/ Michael/
aldólica (las flechas pueden considerarse equilibrios), Enders, 2006.71
En el siguiente esquema (1-14) se encuentran recogidas algunas de las
aportaciones más representativas de reacciones dominó partiendo de
compuestos carbonílicos α,β-insaturados (7), siendo procesos en los que se
suceden, nuevamente, mecanismos vía ion iminio-enamina en presencia de
aminas secundarias como catalizadores.
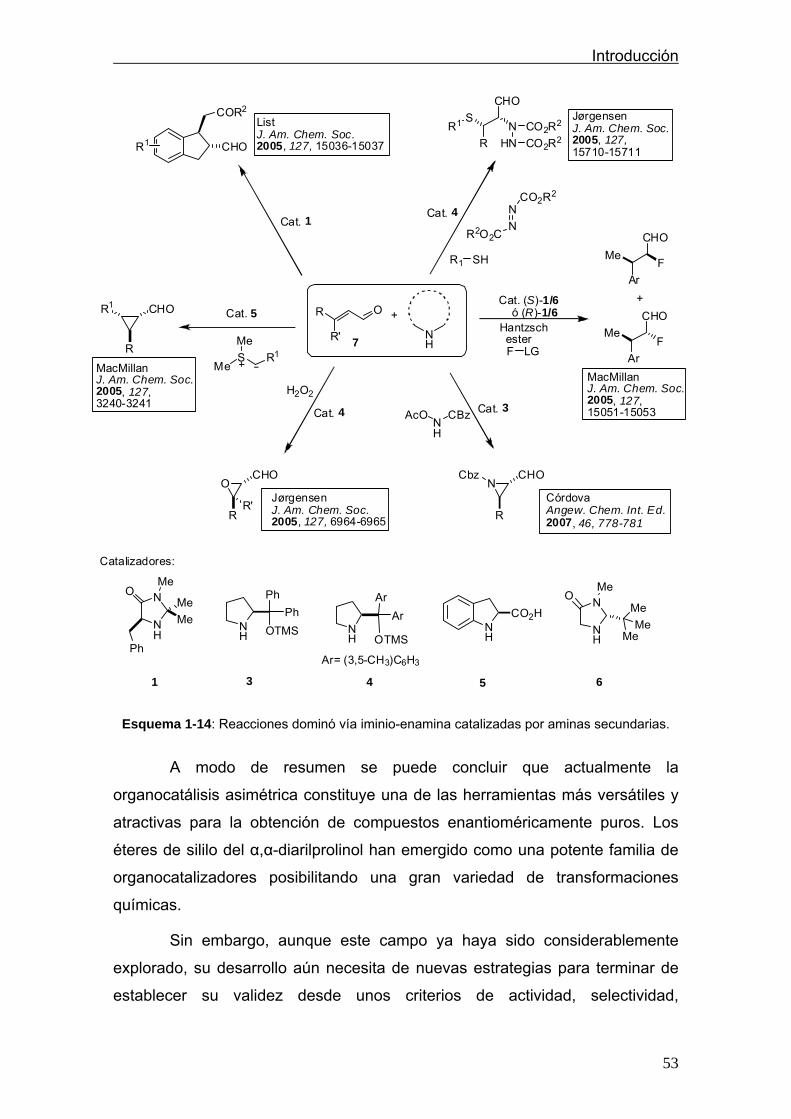
Introducción
53
R O
R' NH
+R1 CHO
R
MeS R1Me
OCHO
RR'
NCbz CHO
R
R1 CHO
COR2
Hantzschester
R1 S
R
CHO
N
HN
CO2R2
CO2R2
R1 SH
R2O2CN
NCO2R2
CHO
Ar
MeF
CHO
Ar
MeF
+
MacMillanJ. Am. Chem. Soc.2005, 127,3240-3241
JørgensenJ. Am. Chem. Soc.2005, 127, 6964-6965
CórdovaAngew. Chem. Int. Ed.2007, 46, 778-781
ListJ. Am. Chem. Soc.2005, 127, 15036-15037
JørgensenJ. Am. Chem. Soc.2005, 127,15710-15711
MacMillanJ. Am. Chem. Soc.2005, 127,15051-15053
NH
CO2HNH
Ar
OTMS
ArNH
Ph
OTMS
Ph
3
NH
NOMe
Me
Me
PhAr= (3,5-CH3)C6H3
NH
NO
Me
Me
MeMe
Catalizadores:
4 51 6
Cat. (S)-1/6ó (R)-1/6
F LG
Cat. 4Cat. 1
Cat. 3Cat. 4
H2O2
AcONH
CBz
Cat. 5
7
Esquema 1-14: Reacciones dominó vía iminio-enamina catalizadas por aminas secundarias.
A modo de resumen se puede concluir que actualmente la
organocatálisis asimétrica constituye una de las herramientas más versátiles y
atractivas para la obtención de compuestos enantioméricamente puros. Los
éteres de sililo del α,α-diarilprolinol han emergido como una potente familia de
organocatalizadores posibilitando una gran variedad de transformaciones
químicas.
Sin embargo, aunque este campo ya haya sido considerablemente
explorado, su desarrollo aún necesita de nuevas estrategias para terminar de
establecer su validez desde unos criterios de actividad, selectividad,

Capítulo 1
54
generalidad, escalado, etc. De esta manera se hace necesario proporcionar
nuevos conocimientos, profundizando en el comportamiento de estos sistemas,
permitiendo así el desarrollo de nuevas metodologías sintéticas cada vez más
sofisticadas, así como su aplicación en la preparación de sistemas cada vez
más complejos.
1.4. Objetivos
La reacción nitrosoaldólica y la reacción de Mannich se encuentran
entre las herramientas más utilizadas para la obtención de compuestos
carbonílicos funcionalizados, más concretamente aminoácidos,
aminoalcoholes, dioles y derivados, que constituyen a su vez estructuras clave
en la síntesis de compuestos naturales o fármacos sintéticos, entre otros.
Al inicio de nuestro trabajo, la reacción nitrosoaldólica estaba poco
desarrollada, mientras que la reacción de Mannich presentaba importantes
limitaciones, aspectos que se detallarán más adelante. La presente Tesis tiene
como objetivo el desarrollo de procedimientos organocatalíticos para llevar a
cabo ambas transformaciones utilizando como sustratos aldehídos y como
catalizadores éteres derivados del prolinol, tanto los comerciales o los ya
descritos, como otros de nuevo diseño (Esquema 1-15).
H
R1
O
H
R1
O
ONH
Ph
H
R1
O
NOH
Ph
+
R
OSiR'3
RNH
ReacciónN-nitrosoaldólica
X
H R2
R1
HNOPG
ReacciónO-nitrosoaldólica
Reacción deMannich
Esquema 1-15
El capítulo 2 recoge los resultados y discusión referentes a las
reacciones N- y O-nitrosoaldólica, incluyendo una previa exposición de los
precedentes y limitaciones de las metodologías existentes.
En el capítulo 3 se hace lo propio respecto a la reacción de Mannich.

Reacción nitrosoaldólica


Reacción Nitrosoaldólica
57
2. Reacción nitrosoaldólica
2.1. Introducción
Las α-oxigenaciones y α-aminaciones de compuestos carbonílicos1 son
procesos de gran importancia para la industria farmacéutica2 y agroquímica
debido a que la mayoría de los compuestos biológicamente activos contienen en
su estructura unidades C-O y C-N. Por esta razón no resulta sorprendente que
exista una gran demanda de métodos enantioselectivos que permitan efectuar
estas transformaciones de modo eficiente, práctico y, cada vez más, respetuoso
con el medioambiente.
En este contexto, los métodos catalíticos asimétricos resultan
interesantes en el área de la síntesis orgánica actual y existen en la bibliografía
varios ejemplos catalíticos promovidos tanto por metales3 como por
organocatalizadores.4 Algunas de estas estrategias se exponen en las Tablas 2-
1 y 2-2.
1 Para revisiones actuales sobre α-aminaciones y α-oxidaciones catalíticas enantioselectivas de compuestos carbonílicos, ver: a) J. M. Janey, Angew. Chem. Int. Ed. 2005, 44, 4292-4300. b) T. Vilaivan, W. Bhanthumnavin, Molecules, 2010, 15, 917-958. c) A. M. R. Smith, K. K. Hii, Chem. Rev. 2011, 111, 1637-1656. 2 S. Caron, R. W. Dugger, S. G. Ruggery, J. A. Ragan, D. H. B. Ripin, Chem. Rev. 2006, 106, 2943-2989. 3 Para ejemplos de α-oxigenaciones enantioselectivas mediante catálisis metálica, ver: a) M. Bougauchi, S. Watanabe, T. Arai, H. Sasai, M. Shibasaki, J. Am. Chem. Soc. 1997, 119, 2329-2330. b) W. Adam, R. T. Fell, V. R. Stegmann, C. R. Saha-Möller, J. Am. Chem. Soc. 1998, 120, 708-714. c) H. Kakei, T. Nemoto, T. Ohsima, M. Shibasaki, Angew. Chem. Int. Ed. 2004, 43, 317-320. d) P. Y. Toullec, C. Bonaccorsi, A. Mezzetti, A. Tongi, Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5810-5817. e) A. M. R. Smith, D. Billen, K. K. Hii, Chem. Commun. 2009, 3925-3927. Para ejemplos de α-aminaciones enantioselectivas mediante catálisis metálica, ver: f) D. A. Evans, S. G. Nelson, J. Am. Chem. Soc. 1997, 119, 6452-6453. g) D. A. Evans, D. S. Johnson, Org. Lett. 1999, 1, 595-598. h) Y. K. Kang, D. Y. Kim, Tetrahedron Lett. 2006, 47, 4565-4568. 4 Para ejemplos de α-oxigenaciones organocatalíticas enantioselectivas, ver: a) M. R. Acocella, O. G. Mancheño, M. Bella, K. A. Jørgensen, J. Org. Chem. 2004, 69, 8165-8167. b) M. Engqvist, J. Casas, H. Sudén, I. Ibrahem, A. Córdova, Tetrahedron Lett. 2005, 46, 2053-2057. c) I. Ibrahem, G.-L. Zhao, H. Sudén, A. Córdova, Tetrahedron Lett. 2006, 47, 4659-4663. d) T. Bekele, M. H. Shah, J. Wolfer, C. J. Abraham, A. Weatherwax, T. Lectka, J. Am. Chem. Soc. 2006, 128, 1810-1811. e) M. P. Sibi, M. Hasegawa, J. Am. Chem. Soc. 2007, 129, 4124-4125. f) F. A. Hernández-Juan, D. M. Cockfield, D. J. Dixon, Tetrahedron Lett. 2007, 48, 1605-1608. g) H. Gotoh, Y. Hayashi, Chem. Commun. 2009, 3083-3085. Para ejemplos de α-aminaciones organocatalíticas enantioselectivas, ver: h) A. Bøgevig, K. Juhl, N. Kumaragurubaran, W. Zhuang, K. A. Jørgensen, Angew. Chem. Int. Ed. 2002, 41, 1790-1793. i) B. List, J. Am. Chem. Soc. 2002, 124, 5656-5657. j) N. Kumaragurubaran, K. Juhl, W. Zhuang, A. Bøgevig, K. A. Jørgensen, J. Am. Chem. Soc. 2002, 124, 6254-6255. k) S. Saaby, M. Bella, K. A. Jørgensen, J. Am. Chem. Soc. 2004, 126, 8120-8121. l) N. S. B. Chowndari, C. F. Barbas III, Org. Lett. 2005, 7, 867-870. m) L. Cheng, L. Liu, D. Wang, Y.-J. Chen, Org. Lett. 2009, 11, 3874-3877.

Capítulo 2
58
Existen múltiples procedimientos de α-oxigenación de compuestos
carbonílicos que emplean diferentes agentes oxidantes, entre los que se
encuentran distintos peróxidos, TEMPO, oxígeno molecular, yodosobenceno,
oxaziridinas o dimetildioxirano. Un ejemplo de oxidación directa organocatalítica
es la α-oxidación de cetonas cíclicas con la oxaziridina 8 catalizada por la
diamina 9, descrita por el grupo de Córdova (Esquema 2-1).4b
NH
O
OH
O
R
ON
Tos
Ph
N
O
9 (30 mol%)
R
Rto.: 27-29%
ee: 34-63%8
Esquema 2-1: α-Oxigenación directa enantioselectiva de cetonas cíclicas con oxaziridinas
catalizada por 9, Córdova, 2005.4b

Reacción Nitrosoaldólica
59
Tabla 2-1: Ejemplos de catalizadores utilizados para α-oxigenaciones enantioselectivas:
Organocatálisis
Catalizadores Reacciones Catalizadores Reacciones
N N
O O
tBu tBu
tBu tBuMn
Cl
-Oxigenación de silil
enol éteres con
dimetildioxirano3b
R1
R2
O
OR3 Cat.
R1
R2
O
-Oxigenación de
-cetoésteres
con oxaziridinas3d
TiClO
ClMeCN NCMe
O
1-Nf
1-Nf
OO
1-Nf
1-Nf
P
PPd
Ph
Ph
OH2
OH2
·2TfO-
-Oxigenación de
-cetoésteres
con
dimetildioxirano3e
NH
N
OMe
N
Me
OH
NH2
NO
TCH2A
NH
CO2H
-Oxigenación de
-cetoésteres
cíclicos con
hidroperóxidos4a
-Ariloxilación de
aldehídos4f
-Oxigenación de
cetonas con
iodosobenceno4b
NH2
NO
BF4Ph
-Aminoxilación de
aldehídos con TEMPO4e
PhPh
OTMS
-Oxibenzoilación de
aldehídos con
peróxido de benzoilo4g
NH
PhPh
OH
-Oxigenación de
aldehídos con
O24c
Catálisis organometálica
2+
*
Por otro lado, a pesar de haberse descrito varios ejemplos de α-
aminaciones catalíticas enantioselectivas, los agentes de aminación utilizados
son menos variados, siendo en casi todos los casos derivados de
azodicarboxilatos. Como ejemplo ilustrativo se muestra en el Esquema 2-2 la α-
aminación enantioselectiva directa de aldehídos catalizada por la pirrolidina 4,
descrita por Jørgensen.5
5 J. Franzén, M. Marigo, D. Fielenbach, T. Wabnitz, A. Kjærgaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18296-18304.
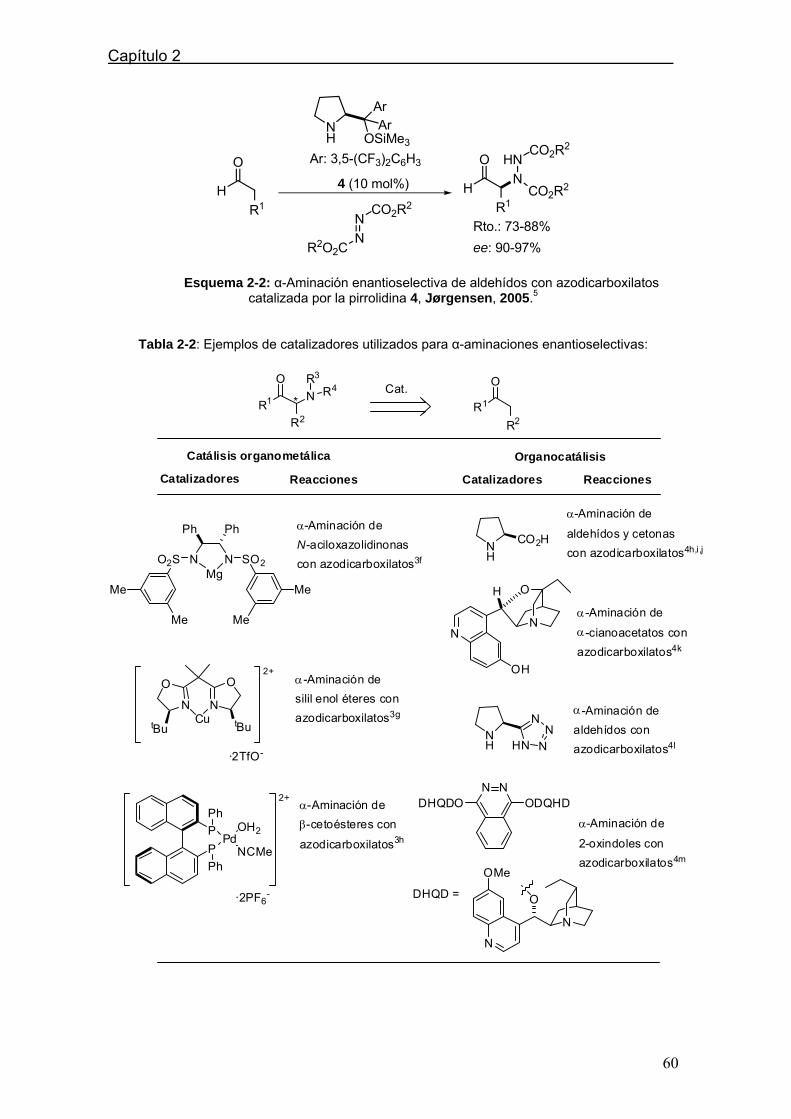
Capítulo 2
60
H
R1
O
NH
R1
O
Rto.: 73-88%
ee: 90-97%
NH
Ar
OSiMe3
Ar
Ar: 3,5-(CF3)2C6H3
4 (10 mol%)
N
NCO2R2
R2O2C
HNCO2R2
CO2R2
Esquema 2-2: α-Aminación enantioselectiva de aldehídos con azodicarboxilatos catalizada por la pirrolidina 4, Jørgensen, 2005.5
Tabla 2-2: Ejemplos de catalizadores utilizados para α-aminaciones enantioselectivas:
Organocatálisis
Catalizadores Reacciones Catalizadores Reacciones
Catálisis organometálica
O2S N N SO2
Me Me
Me MeMg
-Aminación de
silil enol éteres con
azodicarboxilatos3g
R1
R2
O
NCat.
R1
R2
O
O
N N
O
tBu tBuCu
Ph Ph -Aminación de
N-aciloxazolidinonas
con azodicarboxilatos3f
-Aminación de
-cetoésteres con
azodicarboxilatos3h
NH
NN
O
NH
CO2H
-Aminación de
aldehídos y cetonas
con azodicarboxilatos4h,i,j
HN NN
N-Aminación de
aldehídos con
azodicarboxilatos4l
H
OH
-Aminación de
-cianoacetatos con
azodicarboxilatos4k
NNDHQDO ODQHD
N
OMe
N
ODHQD =
-Aminación de
2-oxindoles con
azodicarboxilatos4m
P
PPd
Ph
Ph
OH2
NCMe
·2PF6-
R3
R4
·2TfO-
2+
2+
*

Reacción Nitrosoaldólica
61
Una de las rutas más interesantes para el acceso a compuestos
orgánicos portadores de un enlace C-O o C-N consiste en hacer reaccionar un
carbono nucleofílico con un nitrosocompuesto. Los nitrosocompuestos tienen
una reactividad muy especial,6 ya que tanto el nitrógeno como el oxígeno del
grupo nitroso pueden actuar como electrófilos.
Figura 2-1
Recientemente se han desarrollado varias reacciones asimétricas
catalíticas que utilizan nitrosocompuestos como componentes electrofílicos de la
reacción, entre ellas las reacciones de aminoxilación7 e hidroxiaminación8 de
compuestos carbonílicos enolizables o la reacción de cicloadición [4+2] entre
nitrosocompuestos y dienos, conocida como reacción nitroso Diels-Alder.9 A
pesar del gran potencial de este tipo de reacciones y sus aplicaciones
sintéticas,10 su desarrollo es muy incipiente.11
Una de las transformaciones más importantes en este contexto es la
reacción nitrosoaldólica (NA), que consiste en la reacción entre un
6 Para revisiones sobre propiedades de nitrosocompuestos, ver: a) B. G. Gowenlock, G. B. Richter-Ado, Chem. Rev. 2004, 104, 3315-3340. b) J. Lee, L. Chen, A. H. West, G. B. Richter-Ado, Chem. Rev. 2002, 102, 1019-1065. 7 Primeros ejemplos: a) N. Moriyama, H. Yamamoto, J. Am. Chem. Soc. 2003, 125, 6038-6039. b) N. Moriyama, H. Yamamoto, J. Am. Chem. Soc. 2004, 126, 5360-5361. c) S. P. Brown, M. P. Brochu, C. J. Sinz, D. W. C. MacMillan, J. Am. Chem. Soc. 2003, 125, 10808-10809. d) G. Zhong, Angew. Chem. Int. Ed. 2003, 42, 4247-4250. 8 Primeros ejemplos: a) N. Moriyama, H. Yamamoto, J. Am. Chem. Soc. 2005, 127, 10801081. b)T. Kano, M. Ueda, J. Takai, K. Maruoka, J. Am. Chem. Soc. 2006, 128, 6046-6047. 9 Estudios pioneros: a) Y. Yamamoto, H. Yamamoto, J. Am. Chem. Soc. 2004, 126, 4128-4129. b) Y. Yamamoto, H. Yamamoto, Angew. Chem. Int. Ed. 2005, 44, 7082-7085. 10 Los aductos resultantes de la reacción de aminoxilación, por ejemplo, son fácilmente transformables en 1,2-dioles y derivados, motivos estructurales de gran interés por formar parte de un gran número de productos naturales biológicamente activos y fármacos sintéticos. Para ejemplos recientes, ver: a) P. Jiao, M. Kaswasaki, H. Yamamoto, Angew. Chem. Int. Ed. 2009, 48, 3333-3336. b) L. Yang, R. H. Liu, B. Wang, L. L. Weng, H. Zheng, Tetrahedron Lett. 2009, 50, 2628-2631. c) T. M. Shaikh, A. Sudalai, Tetrahedron: Asymmetry 2009, 20, 2287-2292. d) N. B. Kondekar, P. Kumar, Org. Lett. 2009, 11, 2611-2614. e) M. Lu, D. Zhu, Y. Lu, Y. Hou, B. Tan, G. Zhong, Angew. Chem Int. Ed. 2008, 47, 10187-10191. 11 Para revisiones sobre reacciones con nitrosocompuestos, ver: a) H. Yamamoto, M. Kawasaki, Bull. Chem. Soc. Jpn. 2007, 80, 595-607. b) H. Yamamoto, N. Momiyama, Chem. Commun. 2005, 3514-3525. c) B. Plietker, Tetrahedron: Asymmetry 2005, 16, 3453-3459. d) P. Merino, T. Tejero, Angew. Chem. Int. Ed. 2004, 43, 2995-2997. e) J. M. Janey, Angew. Chem. Int. Ed. 2005, 44, 4292-4300.

Capítulo 2
62
nitrosocompuesto y un compuesto carbonílico enolizable a través de su carbono
en α. Tradicionalmente, el enolato o enamina que actúa como nucleófilo es
formado en una etapa previa, como por ejemplo los enolatos de silicio, y
posteriormente se hace reaccionar con un nitrosocompuesto, pudiendo la
reacción transcurrir según dos posibles caminos: aquél que conduce a
productos aminoxilados (reacción O-Nitrosoaldólica) y aquél que genera
productos oxiaminados (reacción N-Nitrosoaldólica) (Esquema 2-3). Como en
general la transformación implica en ambos casos la formación de un centro
estereogénico (R2 ≠ H), en este tipo de reacciones es relevante no sólo la
regioselectividad, sino también la enantioselectividad del proceso.
R1
R2
O
O-NA
N-NA
R1
R2
O
ONH
Ar
R1
R2
O
NOH
ArN
Ar
O+
aminoxilación
oxiaminación
*
*
Esquema 2-3: Posibles reacciones nitrosoaldólicas entre compuestos carbonílicos y
nitrosoarenos.
A la dificultad del control regio- y estereoquímico de estos procesos hay
que añadir la alta inestabilidad de los nitrosoarenos, compuestos que pueden
encontrarse en forma de monómeros o dímeros en equilibrio (Fig. 2-2). Una
propiedad característica y distintiva de estas formas es su color, ya que éste es
verdoso para la forma monomérica en disolución, color que desparece al
formarse el dímero (ver ref. 6b). De esta manera, el transcurso de las
reacciones de los nitrosoarenos, siempre y cuando el dímero no dé reacción, se
puede monitorizar por la desaparición del color verde, lo que indica que el
nitrosoareno en su forma monomérica se ha consumido completamente.
Figura 2-2: Dimerización de los C-nitrosocompuestos.

Reacción Nitrosoaldólica
63
La primera reacción nitrosoaldólica conocida, en su caso una
oxiaminación, corresponde a la síntesis de una α–hidroxiamino cetona cíclica
llevada a cabo por Lewis y colaboradores en 1972 (Esquema 2-4).12 Estos
autores utilizaron nitrosobenceno como electrófilo y 1-morfolino-1-ciclohexeno
(10) como nucleófilo, y obtuvieron la α–hidroxiamino cetona correspondiente 11
con un 30% de rendimiento. A la vista está que este resultado no despertó
mucho interés entre los químicos sintéticos, ya que este tipo de reacciones no
volvió a estudiarse hasta casi dos décadas después.
Esquema 2-4: Síntesis de la α–hidroxiamino cetona 11 utilizando nitrosobenceno como
electrófilo, Lewis, 1972.12
Posteriormente, en 1992 Oppolzer realizó uno de los estudios más
significativos en este campo, desarrollando α–cloronitroso derivados quirales
(12) para la α-aminación de enolatos proquirales con una alta discriminación
enantiofacial (Esquema 2-5).13
Esquema 2-5: Primera α-aminación con nitrosoderivados con discriminación enantiofacial del
enolato, Opplozer, 1992.13
Más tarde, en 2002, Yamamoto describió la primera reacción
nitrosoaldólica O-selectiva14 de enolatos preformados catalizada por triflato de
trietilsililo (Esquema 2-6). Este trabajo destapó un nuevo y atractivo reto: la
12 J. W. Lewis, P. L. Myers, J. A. Ormerod, J. Chem. Soc., Perkin Trans. 1 1972, 2521-2524. 13 W. Oppolzer, O. Tamura, G. Sundarababu, M. Signer, J. Am. Chem. Soc. 1992, 114, 5900-5902. 14 a) N. Momiyama, H. Yamamoto, Angew. Chem. Int. Ed. 2002, 41, 2986-2988. b) N. Momiyama, H. Yamamoto, Angew. Chem. Int. Ed. 2002, 41, 3313-3316.

Capítulo 2
64
posibilidad de efectuar dos adiciones distintas con un nitrosoareno
(aminoxilación u oxiaminación, ver Esquema 2-3) con el consecuente problema
del control de la regioselectividad que esto suponía. Además, este trabajo
suscitó mucho interés en el mundo de la síntesis orgánica debido a que
descubría una nueva vía para la inserción de un oxígeno en α a un carbonilo.
Esquema 2-6: Primera reacción O-nitrosoaldólica, Yamamoto, 2002.14
De estos estudios parecía deducirse que la regioselectividad (N-adición
frente a O-adición) venía determinada tanto por la naturaleza del enolato (o
enamina) como por la presencia en el medio de reacción de dadores de
hidrógeno y la naturaleza de éstos. Yamamoto, retomando la idea de Lewis de
utlilizar enaminas como dadores, realizó un estudio sobre la reacción
nitrosoaldólica de las mismas en presencia de diferentes ácidos de Brønsted
como aditivos o disolventes. Tras una búsqueda de condiciones óptimas,
observó que en presencia de MeOH, la enamina 10, derivada de la morfolina,
reaccionaba con nitrosobenceno conduciendo al aducto oxiaminado 11,
mientras que la enamina 13 en presencia de ácido acético conducía al aducto
de aminoxilación 14 (Esquema 2-7).15 Estos resultados pioneros parecen indicar
que mediante la selección de la enamina o equivalente de enolato adecuado y
la adición de ácidos de Brønsted específicos puede dirigirse el transcurso de la
reacción por una u otra vía.
Éste fue el principio del control de la regioselectividad en la reacción
nitrosoaldólica, aunque aún no estuviera claro el mecanismo de acción de
dichos dadores de hidrógeno.
15 N. Moriyama, H. Torii, S. Saito, H. Yamamoto, Proc. Natl. Acad. Sci. U.S.A. 2003, 101, 5374-5378.

Reacción Nitrosoaldólica
65
Esquema 2-7: Primeras reacciones nitrosoaldólicas N- y O-regioselectivas, Yamamoto, 2004.15
2.2. Reacción nitrosoaldólica catalítica asimétrica
Las primeras reacciones nitrosoaldólicas realizadas mediante catálisis
asimétrica fueron también descritas por Yamamoto y colaboradores mediante la
utilización de catalizadores ácidos de Lewis.7a,b Así, describieron la reacción de
aminoxilación de enolatos de estaño preformados (15) con nitrosobenceno
utilizando un complejo de plata-BINAP como catalizador (Esquema 2-8), y
obtuvieron el correspondiente producto (16) con excelente enantioselectividad.
Esquema 2-8: Primera reacción nitrosoaldólica catalítica, Yamamoto, 2003.7a,b
Los primeros trabajos de reacciones nitrosoaldólicas asimétricas
organocatalíticas aparecieron coetáneamente. En todos ellos se utiliza L-prolina
como catalizador y nitrosobenceno como electrófilo (Esquema 2-9) y son
aplicables tanto para aldehídos (17) (MacMillan,7c Zhong,7d y Hayashi16), como
para cetonas (18) (Hayashi17 y Córdova18). Éstos no solo resultaron ser los
16 Y. Hayashi, J. Yamaguchi, K. Hibino, M. Shoji, Tetrahedron Lett. 2003, 44, 8293-8296. 17 Y. Hayashi, J. Yamaguchi, J. Sumiya, M. Shoji, Angew. Chem. Int. Ed. 2004, 43, 1112-1115. 18 A. Bøgevig, H. Sudén, A. Córdova. Angew. Chem. Int. Ed. 2004, 43, 1109-1111.

Capítulo 2
66
primeros estudios de la reacción nitrosoaldólica mediante organocatálisis, sino
también las primeras α-oxigenaciones directas de compuestos carbonílicos, sin
la previa preparación de los enolatos o enaminas.19
PhNO
H
O
O
O
ONHPh H
OR
R
ONHPh
NH
COOHee>93%Zhong, McMillan,Hayashi (2003)
ee>95%Hayashi,Córdova (2004)
(R)(R)
Catalizador:
1718
L-prolina
Esquema 2-9: Primeras reacciones nitrosoaldólicas asimétricas organocatalíticas, con L-prolina como catalizador.
En todos los casos la reacción transcurre vía enamina y se obtienen
exclusivamente los aductos aminoxilados. Se postula que esta alta
regioselectividad hacia la O-adición la determina el grupo carboxílico de la
prolina. Existen diversas propuestas mecanísticas a cargo de Houk,20 Córdova21
y Hayashi,22 que proponen estados de transición similares. Aunque no se
descarte la posible participación de los productos de reacción iniciales,23 el
mecanismo mayormente aceptado para el proceso se explica a continuación.
Los tres autores citados consideran que el mecanismo de reacción es
similar al previamente descrito para la reacción aldólica y de Mannich con la L-
prolina como catalizador,24 en el que la prolina actúa como catalizador
bifuncional, activando el sustrato carbonílico vía enamina y el componente
electrofílico mediante enlace de hidrógeno a través del grupo carboxilo. Houk y
colaboradores han llevado a cabo estudios computacionales (DFT) partiendo de
esta idea,20 estudiando como sistema modelo el conformado por la enamina E
19 Para α-oxigenaciones de enol éteres preformados, ver: a) Y. Zhu, Y. Yu, H. Yu, Y. Shi, Tetrahedron Lett. 1998, 39, 7819-7822. b) W. Adam, R. T. Fell, C. R. Saha-Möller, C. G. Zhao, Tetrahedron: Asymmetry 1998, 9, 397-401. c) W. Adam, R. T. Fell, R. T. Stegmann, C. R. Saha-Möller, J. Am. Chem. Soc. 1998, 120, 708-714. d) Ver ref. 14. e) Ver ref. 15. 20 P. H. Cheong, K. N. Houk, J. Am. Chem. Soc. 2004, 126, 13912-13913. 21 A. Córdova, H. Sundén, A. Bøgevig, M. Johansson, F. Himo, Chem. Eur. J. 2004, 10, 3673-3684. 22 Y. Hayashi, J. Yamaguchi, T. Sumiya, K. Hibino, M. Shoji, J. Org. Chem. 2004, 69, 5966-5973. 23 S. P. Mathew, H. Iwamura, D. G. Blackmond, Angew. Chem. Int. Ed. 2004, 43, 3317-3321. 24 S. Bahmanyar, K. N. Houk, Org. Lett. 2003, 5, 1249-1251.

Reacción Nitrosoaldólica
67
derivada de la L-prolina y el propionaldehído (17A), el nitrosobenceno y el
dímero de éste (19). En la Figura 2-3 están representados los tres estados de
transición más plausibles. La estructura de energía más baja resultó ser aquélla
en la que se daba el ataque nucleofílico entre la enamina (E)-anti y el oxígeno
del nitrosobenceno ((R)-O-Anti en Fig. 2-3), que conduce al aducto aminoxilado
R, acorde con los resultados experimentales.
N
O
OPhN
OH
Me
N
O
OHPh N
O
N
O
OH
Me
O
NPh
Me(R)-O-AntiH++= 4.8 kcal/mol
(S)-O-SinH++= 8.3 kcal/mol
(R)-N-AntiH++= 7.1 kcal/mol
Distancia N-H: 1.1Å Distancia N-H: 1.1Å Distancia O-H: 1.7Å
PhNOH
O
MeNH
COOH
17A L-prolina
++ N NPh
O Ph
O
Dímero (19)
+
Estados de transición más estables:
Figura 2-3: Estados de transición de las reacciones de α-aminoxilación y α–
hidroxiaminación con nitrosobenceno de la enamina formada entre la L-prolina y el propanal (17A).20
Los autores explican la selectividad de la reacción por la mayor
basicidad del nitrógeno con respecto al oxígeno, actuando éste último como
electrófilo preferentemente. Esta diferencia de basicidad entre el N y el O se
refleja también en el diferente grado de transferencia de protón que se observa
en los respectivos estados de transición, que se puede deducir por la menor
distancia del enlace N-H en (R)-O-Anti y (S)-O-Sin (1.1Å) frente al enlace O-H
en (R)-N-Anti (1.7Å).
Sin embargo, en el caso de las enaminas provenientes de catalizadores
carentes de grupos dadores de hidrógeno, los cálculos predicen un cambio en la
regioselectividad de la reacción; es decir, para una amina secundaria como
catalizador (20 en Fig. 2-4) carente de grupos dadores de H de ningún tipo, la
N-adición estará favorecida frente a la O-adición (Fig. 2-4).

Capítulo 2
68
Figura 2-4: Estados de transición de α-aminoxilación y α–hidroxiaminación de la enamina
formada entre la dimetilamina (20) y el propanal (17A) con el nitrosobenceno, Houk, 2004.20
Poco más tarde Yamamoto y colaboradores establecieron
experimentalmente la influencia que ejerce sobre la N/O regioselectividad de la
reacción nitrosoaldólica de enaminas el carácter más o menos fuerte del ácido
de Brønsted involucrado. Así, tras un estudio con distintas estructuras quirales
portadoras de hidrógeno, observaron que el ácido 2-(1-naftil) glicólico (21), que
posee grupos ácidos fuertes (en rojo), conducía exclusivamente a los aductos
aminoxilados, mientras que el derivado del TADDOL 22, con dadores de
hidrógeno débiles en su estructura (en azul), conduce a los aductos
oxiaminados, ambos con buenos excesos enantioméricos (Esquema 2-10).8a
Esquema 2-10: Reacciones N-/O-nitrosoaldólicas vía organocatálisis, Yamamoto, 2005.8a
Era de esperar que, de forma similar a la prolina, el ácido carboxílico 21
condujera a los aductos aminoxilados. Por otro lado, los autores proponen que
los grupos hidroxilo presentes en 22, de carácter mucho menos ácido, funcionan
de manera inversa, activando el nitrógeno del nitrosobenceno mediante la
formación de enlace de H con el O del grupo nitroso, para promover la reacción
N-nitrosoaldólica. En este caso, esta activación se debe de nuevo a la diferente
naturaleza del O y el N del nitrosobenceno, teniendo el primero carácter más

Reacción Nitrosoaldólica
69
nucleófilo, lo que conduce a una mayor coordinación que el N con grupos
dadores de H de carácter débil.25
Además de la prolina se han estudiado toda una serie de aminas
secundarias portadoras de un grupo dador de H como catalizadores de la
reacción nitrosoaldólica, y en la Tabla 2-3 se recogen los ejemplos más
significativos. De un examen global de dicha tabla se pueden extraer las
siguientes conclusiones:
-Casi todas las estructuras desarrolladas son derivadas de la L-prolina,
portadoras de grupos acídicos fuertes, como ácidos carboxílicos, triflamidas o
tetrazoles, y promotoras de la O-adición.
-Este grupo de catalizadores conduce al enantiómero R en todos los
casos, al transcurrir la reacción catalizada por prolina mediante aproximación
por enlace de hidrógeno, como se ha explicado anteriormente en el mecanismo
de las reacciones vía enamina.
-Las únicas aminas secundarias que conducen a la N-adición son los
ejemplos de Gong y Maruoka.
-Existen muy pocos organocatalizadores para la obtención de aductos
oxiaminados N-(R) y N-(S) y aminoxilados O-(S) (ver Fig.2-5), presentando
éstos, además, ciertas limitaciones respecto a rendimientos, estereoquímica o
dificultad en la síntesis del catalizador.
-Se han descrito escasos ejemplos para cetonas como dadores, siendo
todos ellos aminoxilaciones y algunos de ellos ofreciendo solamente ejemplos
puntuales con cetonas cíclicas.
Figura 2-5: Posibles aductos de reacción nitrosoaldólica
25 Este punto se explicará ampliamente más adelante, en el apartado de los cálculos teóricos realizados en el contexto de la presente Tesis.

Capítulo 2
70
Tabla 2-3: Aminas secundarias descritas para la reacción nitrosoaldólica:
Catalizador
Autores
Sustrato
Dador
Rto. (%)
Adición (O-/N-) (R/S)
ee (%)
MacMillan, Zhong, Hayashi
Aldehídos26 54-95 >93
Hayashi, Córdova Cetonas cíclicas26
44-96
O-(R) >95
Aldehídos
65-69
98
NH HN N
NN
2004
Yamamoto27 Cetonas
75-97
O-(R) >98
Aldehídos
50-76
>97
Hayashi28 Cetonas
45-76
O-(R) >99
Aldehídos
66-81
>99
Wang29 Cetonas
71-94
O-(R) >97
Gong30
Aldehídos
55-74
N-(R)
46-59
Aldehídos
35-86
>96
NH
CO2HONN
N2007
Pericàs31 Cetonas cíclicas
49-75
O-(R) >96
R1,R2: CPh2OH (2006)32
70-90
N-(S)
>95
R1:NHTf, R2:H
(2008)33
86-96
O-(S)
>96
NH
R1
R2
R1:CO2H, R2:3,4,5-F3C6H2
(2009)34
Maruoka
Aldehídos
69-89
O-(R)
59-88
Zhong35 Aldehídos 74-88 O-(R) 93-99
26 Para aldehídos, ver ref. 7c, 7d, 16. Para cetonas, ver ref. 17, 18. 27 a) Y. Yamamoto, N. Moriyama, H. Yamamoto, J. Am. Chem. Soc. 2004, 126, 5962-5963. b) S. Kumarn, D. M. Shaw, D. A. Longbottom, S. V. Ley, Org. Lett. 2005, 7, 4189-4191. 28 Y. Hayashi, J. Yamaguchi, K. Hibino, T. Sumiya, T. Urushima, M. Shoji, D. Hashizume, H. Koshino, Adv. Synth. Catal. 2004, 346, 1435-1439. 29 W. Wang, J. Wang, H. Li, L. Liao, Tetrahedron Lett. 2004, 45, 7235-7238. 30 H. M. Guo, L. Cheng, L. F. Cun, L. Z. Gong, A. Q. Mi, Y. Z. Jiang, Chem. Commun. 2006, 429-430. 31 D. Font, A. Bastero, S. Salayero, C. Jimeno, M. A. Pericás, Org. Lett. 2007, 9, 1943-1946. 32 T. Kano, M. Ueda, J. Takai, K. Maruoka, J. Am. Chem. Soc. 2006, 128, 6046-6047. 33 T. Kano, A. Yamamoto, K. Maruoka, Tetrahedron Lett. 2008, 49, 5369-5371. 34 T. Kano, A. Yamamoto, F. Shirozu, K. Maruoka, Synthesis 2009, 9, 1557-1563. 35 P. J. Chua, B. Tan, G. Zhong, Green Chem. 2009, 11, 543-547.

Reacción Nitrosoaldólica
71
La reacción nitrosoaldólica catalizada por aminas secundarias también
se encuentra involucrada en varias reacciones catalíticas tándem y/o
multicomponente. Ejemplos destacables son las reacciones O-
nitrosoaldólica/Michael de Yamamoto27a y la Michael/N-nitrosoaldólica de
Dong.36
En el caso de la O-nitrosoaldólica/Michael intramolecular de Yamamoto,
por ejemplo (Esquema 2-11), se obtienen aductos de nitroso Diels-Alder 23
mediante la utilización de un solo organocatalizador, la amina secundaria 24 con
un excelente control tanto de la regio- como de la estereoselectividad.
Esquema 2-11: Reacción tándem O-nitrosoaldólica/Michael intramolecular, Yamamoto, 2004.27a
El único ejemplo de aminoxilación organocatalítica que utiliza un
catalizador que funciona como ácido de Brønsted ha sido descrito por el grupo
de Zhong y consiste en la α–hidroxilación enantioselectiva de compuestos β-
dicarbonílicos con 4-cloronitrosobenceno mediada por el ácido fosfórico quiral
25 (Esquema 2-12). Los correspondientes aductos 26 se aislan con buenos
rendimientos y excesos enantioméricos de moderados a excelentes.37
36 Y.-J. Xu, Q.-Z. Liu, L. Dong, Synlett 2007, 2, 273-277. 37 M. Lu, D. Zhu, Y. Lu, X. Zeng, B. Tan, Z. Xu, G. Zhong, J. Am. Chem. Soc. 2009, 131, 4562-4563.

Capítulo 2
72
Esquema 2-12: α-Hidroxilación de compuestos β–dicarbonílicos en presencia de un ácido de Brønsted, Zhong, 2009.37
Posteriormente al desarrollo de la presente Tesis han sido descritos
algunos ejemplos de reacciones nitrosoaldólicas organocatalíticas y asimétricas,
que se describen a continuación.
Por un lado, Barbas y colaboradores han descrito la reacción de
aminoxilación de oxindoles catalizada por un derivado dimérico de la quinidina
de nuevo diseño (27) (Esquema 2-13).38 Los aductos 28 se obtienen con
buenos rendimientos y enantioselectividades, no estando aún muy claro el
mecanismo de la reacción.
N
R
R1
O PhNO+NR1
O
RONHPh
27 (5-10 mol%)
28
THF, -20 ºC48 h
Rto.: 65-86%ee: 73-96%
N
N
O
OH
N
O
HO
27
Esquema 2-13: α-Aminoxilación de oxindoles con un derivado de la quinidina, Barbas III,
2010.38
38 T. Bui, N. R. Candeias, C. F. Barbas III, J. Am. Chem. Soc. 2010, 132, 5574-5575.

Reacción Nitrosoaldólica
73
Por otro lado, Cheng y Zhu han desarrollado para la aminoxilación de
aldehídos y cetonas con nitrosobenceno el primer organocatalizador iónico (29)
basado en la prolina unida a un grupo imidazolio, utilizando líquidos iónicos
como disolventes (Esquema 2-14).39 El catalizador 29, que se comporta como
un catalizador homogéneo en este medio, conduce a unos resultados
excelentes en cuanto a rendimientos y estereoquímica incluso a temperatura
ambiente.
Esquema 2-14: Aminoxilación de aldehídos y cetonas con 29 en líquidos iónicos, Cheng y Zhu,
2010.39
En lo que respecta a la reacción N-nitrosoaldólica, hasta donde nosotros
sabemos, solamente se ha descrito una nueva versión de esta reacción
mediante organocatálisis. Liu y Chen han llevado a cabo la reacción de
oxiaminación de 2-oxindoles promovida por un alcaloide de la cincona (30),
obteniendo los correspondientes aductos hidroxiaminados (31) con buenos
rendimientos pero excesos enantioméricos moderados (Esquema 2-15).40
Esquema 2-15: Oxiaminación de 2-oxindoles con un derivado de cincona, Liu y Chen, 2010.40
39 X. Ding, W. Tang, C. Zhu, Y. Cheng, Adv. Synth. Catal. 2010, 352, 108-112. 40 T. Zhang, L. Cheng, L. Liu, D. Wang, Y.-J. Chen, Tetrahedron: Asymmetry 2010, 21, 2800-2806.

Capítulo 2
74
Durante el transcurso de nuestro estudio sobre la reacción O-
nitrosoaldólica fueron publicadas reacciones “one pot” o secuenciales en las que
una de las etapas de la secuencia es una reacción de α-aminoxilación con
nitrosoarenos. Un ejemplo representativo descrito por Yamamoto es la adición
de reactivos de Grignard a los aldehídos aminoxilados obtenidos en presencia
del catalizador 24 para obtener 1,2-dioles con excelente diastereoselectividad
(Esquema 2-16).41
Esquema 2-16: Síntesis de 1,2-dioles mediante O-nitrosoaldólica y adición de Grignard,
Yamamoto, 2009.41
Otro ejemplo lo ha desarrollado MacMillan, y consiste en sendos
procesos en cascada vía ion iminio-enamina partiendo de aldehídos α,β-
insaturados y basándose en las imidazolidinonas 32 y 33 y prolina como
catalizadores, en las que la reacción O-nitrosoaldólica constituye la segunda
etapa (Esquema 2-17).42 En todos los casos los productos se obtienen con
buenos rendimientos y diastereo- y enantioselectividad elevadas.
41 P. Jiao, M. Kawasaki, H. Yamamoto, Angew. Chem. Int. Ed. 2009, 48, 3333-3336. 42 B. Simmons, A. M. Walji, D. W. C. MacMillan, Angew. Chem. Int. Ed. 2009, 48, 4349-4353.

Reacción Nitrosoaldólica
75
Ph O
Me
NH
H HtBuO2C
Me
CO2tBu
Me
PhNO
CHCl3/DMSO-20 ºC-t.a.
NaBH4/H2, Pd/C
Ph OH
Me
OH
Ph OH
Me
OH
Hantzsch éster
NH
NO Me
Me
MeMe
NH
NO Me
Me
MeMe
32
32 (10mol%)
L-prolina (30mol%)
32 (10mol%)
D-prolina (30mol%)
A)
Me O
PhNO
CHCl3/DMSO-20 ºC-t.a.NaBH4
Me OH
NCbz
ONHPh
33 (10mol%)
L-prolina (30mol%) TBSON
H
Cbz
33
33 (10mol%)
D-prolina (30mol%)TBSO
Me OH
NCbz
ONHPh
TBSO
Ph
Rto.: 73%ant i/sin 11:1ee: 99%
Rto.: 62%sin/ant i 10:1ee: 99%
Rto.: 74%ant i/sin 17:1ee: 99%
Rto.: 71%sin/anti 14:1ee: 99%
B)
Esquema 2-17: Reacciones “one pot” vía ion iminio-enamina. Paso común: O-nitrosoaldólica
con prolina, MacMillan, 2009.42
Por último, el grupo de Wang ha descrito un método secuencial
asimétrico que comprende una aminoxilación de aldehídos con nitrosobenceno
seguida de una homologación quimioselectiva con diazometano.43 Como
catalizador utiliza L-prolina, para aislar, finalmente, las 3-hidroxi-2-alcanonas 34
con rendimientos moderados y buenos excesos enantioméricos (Esquema 2-
18).
43 L. Yang, R.-H. Liu, B. Wang, L.-L. Weng, H. Zheng, Tetrahedron Lett. 2009, 50, 2628-2631.

Capítulo 2
76
Esquema 2-18: Reacción “one pot”: aminoxilación/homologación con diazometano, Wang,
2009.43
2.3. Planteamiento 1: reacción N-nitrosoaldólica en ausencia
de dadores de H externos
Como se puede concluir de los resultados anteriores, en general la
catálisis vía enamina funciona muy bien en la reacción O-nitrosoaldólica, pero
es mucho menos eficaz o requiere catalizadores más sofisticados para la
reacción N-nitrosoaldólica. Por ejemplo, el único derivado de la prolina descrito
que conduce a esta adición es la L-prolinamida de Gong30 (35), portadora de
dadores de hidrógeno débiles y que cataliza la reacción entre aldehídos y
nitrosobenceno con rendimientos y enantioselectividades moderadas en
tiempos de reacción poco prácticos (Esquema 2-19).
NH
NH HO
Ph
PhO
10 mol%R
O
Me
R
O
Me N PhHO
Rto.: 55-74%ee: 46-59%
Tolueno, -40 ºC2-3 días
(R)35
+ PhNO
Esquema 2-19: Primera reacción N-nitrosoaldólica catalizada por una amina secundaria, Gong,
2005.30
Resultados más satisfactorios en la reacción N-nitrosoaldólica de
aldehídos se obtienen con el catalizador 36 desarrollado por el grupo de
Maruoka, (Esquema 2-20).32 Además de conducir a rendimientos y
enantioselectividades superiores, la carga de catalizador necesaria es menor
que en los casos de los derivados de prolina. Sin embargo, a pesar de conducir
a muy buenos resultados, la preparación de este catalizador requiere ocho
pasos a partir del binaftol y es tediosa.

Reacción Nitrosoaldólica
77
36 (10 mol%)O
R
Rto.: 70-90%ee>95%
THF, 0 ºC1 h
NH
Ph
OH
Ph
OH
PhPh
PhNO+ PhN
O
R
OH
(S)
36
H H
Cat.:
Esquema 2-20: Reacción N-nitrosoaldólica enantioselectiva, Maruoka, 2006.32
A la vista de ello nos propusimos desarrollar un método organocatalítico
práctico para la reacción de oxaminación (N-nitrosoaldólica). Tomamos como
elemento clave para nuestro estudio el empleo de los éteres de diarilprolinol
sobre la base de las siguientes consideraciones:
-De acuerdo al modelo mecanístico de Houk, la reacción N-
nitrosoaldólica está favorecida respecto a la O-nitrosoaldólica en un entorno
desprovisto de dadores de hidrógeno
- La accesibilidad de estos catalizadores y su versatilidad en α–
funcionalización de compuestos carbonílicos.
- La eventual presencia en el medio de dadores de hidrógeno débiles,
provenientes, por un lado, del agua formada durante el ciclo catalítico y, por otro
lado, de los aminoalcoholes resultantes de la reacción.
Además, tal y como se muestra en el estereomodelo de la Figura 2-6,
era de esperar que con estos catalizadores no coordinantes la aproximación del
nitrosobenceno a la enamina se diera por la cara estéricamente menos
congestionada para originar los aductos de configuración (S), complementando
así los métodos basados en la L-prolina y derivados, que conducen
generalmente a los aductos de configuración (R).

Capítulo 2
78
Figura 2-6
2.3.1. Resultados y discusión
En los siguientes apartados se discutirán los resultados obtenidos en la
reacción nitrosoaldólica entre los aldehídos mostrados a continuación y
nitrosobenceno (Esquema 2-21).
Esquema 2-21
2.3.1.1. Viabilidad de la propuesta. Estudio teórico previo
Una vez concebida la idea, en una primera fase se procedió a evaluar
su viabilidad desde una perspectiva teórica. En nuestro grupo de trabajo, el Dr.
Enrique Gómez-Bengoa llevó a cabo un estudio computacional al nivel de teoría
B3LYP/6-31G* de la reacción modelo entre nitrosobenceno, la enamina
derivada del propionaldehído (17A) y la pirrolidina (37) (Fig. 2-7).

Reacción Nitrosoaldólica
79
Primeramente se realizó el estudio sin tener en cuenta posibles enlaces
de hidrógeno con protones del medio, y se comprobó que la N-adición se
encontraba favorecida respecto a la O-adición (Fig. 2-7), resultado acorde con
los estudios previos realizados por Houk.20 Esta diferencia energética de 3.0
kcal/mol entre sendos estados de transición, N-(TSN) frente a O-adición (TSO),
debería ser suficiente para conducir a una buena regioselectividad.
Figura 2-7: Barreras de activación para la reacción entre la enamina del propionaldehído y el nitrosobenceno en los casos de N- u O-adición en ausencia de dadores de hidrógeno. Los
valores de ΔE‡ se dan en kcal/mol y ΔG‡ en paréntesis.
Posteriormente se calcularon los estados de transición de la reacción en
presencia de una molécula de agua, primero, y de una molécula modelo del
producto, la N, N-dimetilhidroxilamina (38) después (Fig. 2-8). De nuevo se
encontró que los estados de transición conducentes a la O-nitrosoaldólica eran
del orden de 3.0 kcal/mol más altos que los correspondientes TS para la
reacción N-nitrosoaldólica. Se presentan en la Figura 2-8 los TS localizados
para ambos casos (B y C), así como el TS para la reacción en ausencia de
dadores de H antes comentada (A). De la comparación de las energías
calculadas se desprende que en los casos B y C, catalizados por dadores de
hidrógeno, las barreras de activación disminuyen considerablemente respecto al
anterior (A) en ausencia de enlaces de hidrógeno. Frente a las 25.6 kcal/mol de
energía libre de activación (ΔG‡) encontradas para ésta, los estados de

Capítulo 2
80
transición estabilizados por una molécula de agua o 38 poseen un ΔG‡ de 20.9 y
20.3 kcal/mol respectivamente.
Adicionalmente se observan unas interacciones débiles entre un H-C
del anillo pirrolidínico y el oxígeno del agua o de 38.44 En el caso del agua,
además, se encontró otra interacción débil entre el oxígeno de ésta y un H-C del
anillo aromático del nitrosobenceno.45
Figura 2-8: Estados de transición computados para la reacción de oxiaminación de la enamina del propionaldehído (17A) y la pirrolidina (37) con nitrosobenceno: A) en ausencia de enlaces de hidrógeno, B) en presencia de agua, y C) en presencia de 38. Los valores de ΔG‡ se dan en kcal/mol.
2.3.1.2. Viabilidad de la reacción: evaluación experimental de
catalizadores y disolventes
Para la exploración experimental inicial se seleccionaron como
catalizadores los éteres de α, α-diarilprolinol representados en la Figura 2-9. Los
dos primeros (3 y 4) son comerciales, mientras que 39 se preparó en pocos
pasos a partir de L-prolina.46
44 Para precedentes sobre la participación de hidrógenos metilénicos del anillo de la pirrolidina en formación de enlaces de hidrógeno intramoleculares, ver: a) S. Bahmanyar, K. N. Houk, H. J. Martin, B. List, J. Am. Chem. Soc. 2003, 125, 2475-2479. Para estudios computacionales sobre formación de enlaces de hidrógeno R3N
+-C-H···O relacionados, ver: b) C. E. Canizzaro, K. N. Houk, J. Am. Chem. Soc. 2002, 124, 7163-7169. 45 Para más información sobre este tipo de interacciones, ver: H. Yamamoto, M. Kawasaki, Bull. Chem. Soc. Jpn. 2007, 80, 595-607. 46 Para más detalles, ver parte experimental.

Reacción Nitrosoaldólica
81
Figura 2-9: Catalizadores preparados para el estudio de la reacción NA.
La evaluación de los catalizadores se efectuó sobre la reacción entre el
butanal (17B) y nitrosobenceno utilizando como disolvente diclorometano. En un
primer ensayo se adicionó el catalizador 3 sobre una disolución de color verde
azulado del nitrosobenceno y el aldehído en diclorometano enfriada a 0 ºC. Es
importante señalar que al adicionar el catalizador se observó una decoloración
casi instantánea de la mezcla de reacción, lo que sugería una transformación
rápida en el producto deseado. Tras reducción del aldehído oxiaminado
intermedio con borohidruro sódico se aisló el correspondiente N-hidroxi-β-
aminoalcohol 40B con 65% de rendimiento y enantioselectividad del 90%, y lo
más importante, con una regioselectividad total (entrada 1 en Tabla 2-4).
Sorprendentemente, en las mismas condiciones, la reacción llevada a cabo en
presencia del catalizador 4 no condujo al producto esperado, ni siquiera
agitando la mezcla durante tiempos de reacción mayores. Por su parte, la
reacción con 39 sí transcurrió, aunque con un rendimiento considerablemente
inferior (Tabla 2-4).
A continuación procedimos a evaluar el comportamiento de la reacción
catalizada por 3 en otros disolventes distintos al diclorometano. Como se puede
apreciar, en todos los casos la regioselectividad fue excelente, obteniéndose
exclusivamente el producto de N-adición. Los mejores resultados, en cuanto a
rendimiento químico y enantioselectividad, se obtuvieron en diclorometano. En
THF o acetonitrilo el rendimiento de la reacción disminuyó sensiblemente, si
bien la enantioselectividad no sufrió variación (entrada 5) o ésta fue muy ligera
(entrada 6). El tolueno resultó no ser un disolvente viable, mientras que la
reacción catalítica puede llevarse a cabo también en agua como medio de
reacción (entrada 7), aunque el rendimiento disminuye notablemente,
presumiblemente debido a la baja solubilidad del nitrosobenceno y el catalizador
en ésta.

Capítulo 2
82
Tabla 2-4: Evaluación de catalizadores y disolventes para la reacción de α-hidroxiaminación de aldehídos con nitrosobenceno:47
H
OCat. (20 mol%) NaBH4, EtOH
40B
Et
HO
N
OH
Ph
PhNO
41B
Et
HO
O
HN
Ph+
Et
CHO
N
OH
Ph
Et
CHO
ONH
Ph
Disolvente, 0 ºC 0 ºC, 30 min17B
Et
Entrada Catalizador Disolvente 40B: 41B[b] Rto. (%)[c] ee (%)[d]
1 3 DCM >99: 1 65 90
2 4 DCM --- --- ---
3 39 DCM >99: 1 32 n.d.
4 3 Tolueno >99: 1 21 n.d.
5 3 THF >99: 1 40 90
6 3 CH3CN >99: 1 50 85
7 3 H2O >99: 1 25[e] 90
[a] Reacciones llevadas a cabo a escala 2 mmol, con 3 equivalentes de butanal (17B), adicionando el catalizador a una disolución del nitrosobenceno y el butanal. [b] Determinada por 1H-RMN del producto crudo. [c] Rendimiento del aminoalcohol 40B aislado tras cromatografía flash en columna. [d] Determinado por análisis de HPLC. [e] Reacción llevada a cabo a 5 ºC. n.d.: no determinado.
2.3.1.3. Alcance de la reacción
Una vez llevada a cabo la optimización de la reacción, se exploró el
alcance de la misma utilizando diversos aldehídos. Como muestran los
resultados obtenidos en la Tabla 2-5, los N-hidroxi-β-aminoacoholes 40 se
aislaron con buenos rendimientos y excelentes enantioselectividades en la
mayoría de los casos. Cabe destacar que a 0 ºC se observó que a mayor
longitud de la cadena del aldehído (Me→Et→Pr→…) el rendimiento era
sensiblemente menor, tendencia que, sin embargo, pudo resolverse al llevar a
cabo la reacción a una temperatura más baja (-20 ºC, entradas 4 y 6). La
reacción también tolera el aldehído ramificado isobutiraldehído (entrada 8).
47 Estudio llevado a cabo en colaboración con la doctora Silvia Vera.

Reacción Nitrosoaldólica
83
Tabla 2-5: Oxiaminación directa enantioselectiva de varios aldehídos con nitrosobenceno:47
Entrada
Aldehído
R
Prod.
Dv.
t
(min)
T
(ºC)
40:41[b]
Rdto. (%)[c]
ee
(%)[d]
1 17A Me 40A DCM 5 20 >99: 1 70 94
2 17B Et 40B DCM 5 0 >99: 1 65 99
3 17C nPr 40C DCM 10 0 >99: 1 60 96
4 17E nPent 40E THF 16 h -20 >99: 1 74 98[e]
5 17F nHex 40F DCM 5 20 >99: 1 42 n.d.
6 17F nHex 40F THF 16 h -20 >99: 1 75 98[e]
7 17G Bn 40G DCM 5 0 >99: 1 70 94
8 17H iPr 40H DCM 30 0 >99: 1 60 99
9 17I 2-MeO-
C6H4CH2 40I DCM 5 20 >99: 1 40 91
[a] Reacciones realizadas con aldehído (3 equiv.) a 0 ºC o temperatura ambiente; se efectuó la adición del catalizador sobre una disolución del adehído y nitrosobenceno. [b] Determinado por 1H-RMN del producto crudo. [c] Rendimiento del aducto 40 después de columna cromatográfica. [d]
Determinado por análisis de HPLC. [e] Determinado por análisis de HPLC de los derivados benzoilados y naftoilados. n.d.: no determinado.
Para determinar la configuración absoluta de los aductos obtenidos se
comparó la actividad óptica del aminoalcohol 40C con el valor descrito para
este compuesto en la bibliografía,32 resultando ser S, tal y como sugería el
estereomodelo más aceptado para este tipo de catalizadores (Figura 2-6).
De esta manera queda demostrada la posibilidad de llevar a cabo la
reacción N-nitrosoaldólica de aldehídos empleando nitrosobenceno con un
organocatalizador comercial y de estructura sencilla (3), y, lo más llamativo e
innovador en este campo, carente de grupos dadores de hidrógeno. Los
correspondientes compuestos oxiaminados resultantes se obtienen con buenos
rendimientos y excelentes regio- y enantioselectividades.

Capítulo 2
84
2.4. Planteamiento 2: reacción O-nitrosoaldólica con ácido de
Brønsted externo
Tras comprobar la viabilidad de la reacción N-nitrosoaldólica con el
empleo de pirrolidinas monofuncionales como catalizadores, nuestro interés viró
entorno a la posibilidad de invertir la regioselectividad del proceso, es decir,
realizar la O-adición utilizando el mismo tipo de catalizadores y un ácido de
Brønsted como cocatalizador (Esquema 2-22). Este aditivo supliría la función de
los grupos acídicos fuertes presentes en los organocatalizadores descritos
hasta el momento para promover la O- frente a la N- adición.
La estrategia, de resultar exitosa, permitiría acceder a los productos
tanto aminoxilados como oxiaminados a partir de un mismo catalizador,
simplemente cambiando las condiciones de reacción.
H
R
O
H
R
O
ONH
Ph
H
R
O
NOH
Ph
PhNO+
Ar
OSiMe3
ArNH
N-nitrosoaldólica
O-nitrosoaldólica
(S)
(S)A-H
Esquema 2-22
2.4.1. Resultados y discusión
2.4.1.1. Viabilidad de la propuesta. Estudio teórico previo
De nuevo, en una primera instancia la viabilidad de la propuesta fue
estudiada computacionalmente por el Dr. Enrique Gómez-Bengoa. En este
caso, y conocidos los buenos resultados obtenidos con el éter derivado del
prolinol 3 en la reacción N-nitrosoaldólica, se realizaron los oportunos cálculos
para la reacción entre la enamina derivada de 3 y propionaldehído con
nitrosobenceno (Fig. 2-10). Como dadores de H se estudiaron moléculas
explícitas de un dador de H débil (agua) y uno fuerte (RCO2H). Se puede
apreciar, como decíamos anteriormente, que la ausencia de dadores de

Reacción Nitrosoaldólica
85
hidrógeno o la presencia de un dador de hidrógeno débil (agua) favorece la N-
adición (ΔG‡ considerablemente menor que la O-adición, diferencia del orden de
3.0 kcal/mol), mientras que en presencia de una o dos moléculas de ácido
benzoico o p-nitrobenzoico se invierte la regioselectividad hacia la O-adición.
Además, de entre los dos ácidos benzoico y p-nitrobenzoico estudiados es este
último ácido el que más favorece la O-adición (3.8 kcal/mol de diferencia frente
a 2.5 kcal/mol de diferencia con benzoico), lo que es una evidencia más de la
correlación entre la fuerza del ácido y la O-selectividad.

Capítulo 2
86
TS N-E-Anti-Si TS O-E-Anti-Si
NMe3SiO
O
NPh
NMe3SiO
N
O
Ph
PhPh
PhPh
H
O
HO
R
ninguno
agua
G‡ = 17.5 (0) G‡ = 21.4 (+3.9)
G‡ = 12.5 (0) G‡ = 15.5 (+3.0)
G‡ = 5.5 (+2.5) G‡ = 3.0 (0)
2 x 4-NO2C6H4CO2H G‡ = 5.1 (+3.8) G‡ = 1.3 (0)
2 x PhCO2H
RO
H
HO O
R
O
RO
H
H
O O
R
NMe3SiO
N
O
PhPhPh
HO
R
NMe3SiO O
NPh
PhPh
H
O-nitrosoaldólicaN-nitrosoaldólicaÁcido de Brønsted
R-OH
R-C-OHO
NMe3SiO O
NPh
PhPh
NMe3SiO
N
O
PhPhPh
2 x
R-C-OHO
4-NO2C6H4CO2H
PhCO2H G‡ = 9.5 (0) G‡ = 9.7 (+0.2)
G‡ = 7.1 (+1.5) G‡ = 5.6 (0)
H
OO
R
H
OO
R
Figura 2-10: Estados de transición N-selectivo y O-selectivo calculados a nivel B3LYP/6-311++G**//B3LYP/6-31G* de teoría en estado de gas.
2.4.1.2. Búsqueda de condiciones de reacción
Con objeto de establecer las condiciones óptimas para la reacción de O-
adición en presencia de ácidos de Brønsted, se eligió la reacción entre el
propanal (17A) y el nitrosobenceno. Así, se procedió a realizar un estudio

Reacción Nitrosoaldólica
87
cambiando factores como la temperatura y la carga del catalizador, además de
la presencia o ausencia de ácido de Brønsted. Fue satisfactorio comprobar que
la reacción, en presencia de 3 y ácido p-nitrobenzoico, transcurrió de forma
efectiva y totalmente regioselectiva tras 2 horas a -20 ºC para generar los O-
nitrosoaldoles. Además, de forma gratamente sorprendente, comprobamos que
para el transcurso de la O-adición era suficiente una carga del catalizador 3 de
2-10 mol%, hecho que contrasta con el 20 mol% necesario en la reacción de N-
adición anteriormente descrita y que puede atribuirse a un efecto co-activante
del ácido de Brønsted.
Tabla 2-6: Búsqueda de condiciones N- versus O-adición:
+
DCM, T, t
NaBH4
EtOH30 min
ONH
OH
Me
Ph NOH
OH
Me
Ph
+
Ph
OSiMe3
Ph
3
NH
aditivo
O- N-
PhNO
3 equiv.
17A 40A41A
H
O
Me
Entrada Cat. 3 (mol%) Aditivo T(ºC) t O-:N-[b]
1 20 --- 0 5min 0:1[c]
2 10 --- -20 2h ---
3 10 Ácido 4-nitrobenzoico
10 mol%
-20 2h 1:0[c]
[a] Reacciones llevadas a cabo a escala 1 mmol, con 3 equivalentes de propanal (17A). [b] Determinado por 1H-RMN del crudo. [c] Conversión total.
Para la optimización de las condiciones de reacción de O-adición se
estudiaron distintos ácidos de Brønsted, aminocatalizadores, y disolventes. En
primer lugar y utilizando 3 como catalizador se comparó la eficacia de los
aditivos. Como indican los resultados recogidos en la Tabla 2-7, de entre estos
ácidos el 4-nitrobenzoico, el ácido de menor pKa de los benzoicos probados
(entradas 1, 2 y 3), era el mejor en términos de reactividad y
enantioselectividad. De la misma manera el ácido cloroacético, el más acídico
de los acéticos probados, también daba mayor rendimiento que el acético
(entradas 4 y 5).

Capítulo 2
88
Tabla 2-7: Estudio de aditivos para la reacción O-nitrosoaldólica:
+2) NaBH4, EtOH
30 min
ONH
OH
Me
Ph(2 mol%)DCM, -20 ºC, 16 hPhNO
3 equiv.17A 41A
H
O
Me
3
NH
PhPh
OTMS1)
Producto O-adición Entrada
Aditivo/pKa
48 Rendimiento[b] ee[c] (%) (%)
1 PhCO2H /4.19 50 >99
2 4-MeOPhCO2H/4.47 22 >99
3 4-NO2PhCO2H/3.44 81 >99
4 CH3CO2H/4.76 35 >99
5 ClCH2CO2H/2.87 52 >99
[a] Reacciones llevadas a cabo a escala 1 mmol en DCM (2 mL) con nitrosobenceno, 3 (2 mol%), ácido de Brønsted (5 mol%) y 3 equiv. de propanal a −20 ºC durante 16 horas. [b] Rendimiento de 41A tras cromatografía de columna flash. [c] Determinado por análisis de HPLC.
Por otra parte, se comparó el comportamiento de los catalizadores 4 y
42 (Esquema 2-23),49 de estructura similar a 3 y que funcionan también según
un mecanismo de estereodiscriminación basado en el control estérico. El
catalizador 4, al igual que ocurría en el caso de la N-adición, resultó ser inactivo
para esta reacción, mientras que el α,α-dihexilprolinol silil éter 42, sintetizado en
nuestro laboratorio de manera análoga a 39, se comportó de manera similar al
catalizador comercial 3.
48 Valores de pKa tomados de: B. G. Tehan, E. J. Lloyd, M. G. Wong, W. R. Pitt, J. G. Montana, D. T. Manallack, E. Gancia, Quant. Struct.-Act. Relat. 2002, 21, 457-472. 49 Para un ejemplo previo de utilización de este catalizador, ver: C. Palomo, A. Landa, A. Mielgo, M. Oiarbide, A. Puente, S. Vera, Angew. Chem. Int. Ed. 2007, 46, 8431-8435.

Reacción Nitrosoaldólica
89
Esquema 2-23
Finalmente se realizó un estudio de disolventes bajo las condiciones de
reacción previamente optimizadas (propanal (17A) en presencia de 3 (2 mol%) y
ácido p-nitrobenzoico (PNBA) (5 mol%)) (Tabla 2-8). Se observó que, además
del diclorometano, el cloroformo y el 1,2-dicloroetano son también efectivos. El
éter dietílico y el THF, así como disolventes más polares como la DMF o
próticos como el metanol no resultaron adecuados. En algunos casos, como en
tolueno y en acetonitrilo, los rendimientos mejoran al aumentar la carga del
catalizador del valor estándar de 2 mol% a 10 mol% (comparar entradas 1 y 9
con 2 y 10).

Capítulo 2
90
Tabla 2-8: Estudio de disolventes para la reacción de α-aminoxilación:
Entrada Disolvente
Rendimiento [b]
(%) ee [c]
(%) 1
Tolueno
22
>99
2
Tolueno
67[d]
>99
3
Et2O
10
n.d.
4
THF
<10
n.d.
5
CHCl3
71
>99
6
DCM
80
>99
7
ClCH2CH2Cl
71
>99
8
MeOH
<10
n.d.
9
CH3CN
41
>99
10
CH3CN
53[d]
>99
11
DMF
<10
n.d.
[a] Reacciones llevadas a cabo a escala 1 mmol en 2 mL de disolvente con nitrosobenceno, propanal (17A, 3 equiv.), catalizador 3 (2 mol%) y PNBA (5 mol%) a −20 ºC durante 16 h. [b] Rendimiento del producto aislado 41A tras cromatografía de columna flash. [c] Determinado por análisis de HPLC. [d] Reacciones llevadas a cabo con catalizador 3 (10 mol%) y PNBA (10 mol%). n.d.: no determinado.
2.4.1.3. Alcance de la reacción
A continuación se procedió a estudiar la generalidad del método
aplicándolo a una selección de aldehídos (Tabla 2-9). Como condiciones
establecidas se utilizaron en todas las reacciones 3 equivalentes de aldehído
con respecto a moles de nitrosobenceno (una disolución 0.5M en
diclorometano), el catalizador (10 mol%) y ácido p-nitrobenzoico (PNBA) (10
mol%) a −20 ºC. Como es habitual para este tipo de reacciones, el transcurso
de la reacción se pudo monitorizar por el cambio de color. Posteriormente, el
crudo de la reacción resultante fue tratado in situ con borohidruro sódico en
etanol para obtener los compuestos aminoxilados 41 como productos de
reacción exclusivos.

Reacción Nitrosoaldólica
91
Tabla 2-9: Reacción catalítica asimétrica de aldehídos con nitrosobenceno promovida por 3:
Entrada
Aldehído
R
Prod.
t (h)
Rto. (%)[b]
ee (%)[c]
1
17A
Me
41A
16
80[d]
>99
2
17C
nPr
41C
2
78
>99
3
17C
nPr
41C
16
71[f]
>99
4
17D
nBu
41D
2
81
>99
5
17G
Bn
41G
5
68
>99
6
17G
Bn
41G
16
68[f]
>99
7
17H
iPr
41H 2
55
>99
8
17H
iPr
41H
3
59[e]
>99
9
17J
BnOCH2
41J
16
45
95
10
17K
BocHN(CH2)4
41K
3
88
>99
11
17L
41L
4
66
>99
[a] Reacciones llevadas a cabo a escala 1 mmol en DCM (2 mL) a -20 ºC utilizando el catalizador 3 (10 mol%), PNBA (10 mol%) y 3 equiv. del correspondiente aldehído (17). [b] Rendimiento de los productos 41 aislados tras cromatografía de columna flash. [c] Determinado por análisis de HPLC tras cromatografía de columna flash. [d] Reacción con 2 mol% de catalizador y 5 mol% de PNBA. [e] Reacción con 20 mol% de catalizador y 20 mol% PNBA. [f] Reacciones con 5 mol% de catalizador y 5 mol% de PNBA.
Como se aprecia en los datos recogidos en la Tabla 2-9, tanto aldehídos
de cadena corta como larga, o con grupos funcionales como éter, carbamato y
olefina son compatibles con la reacción. Incluso el isovaleraldehído, un aldehído
ramificado que es menos reactivo, reacciona para dar el aducto 41H, aunque
con un rendimiento ligeramente inferior (entradas 7 y 8). De manera general se
utilizó una carga de catalizador de 10% para la reacción, pero se observó que
ésta podía disminuirse a un 5% sin comprometer el rendimiento y la
enantioselectividad del proceso (entradas 3 y 6) e incluso a un 2% en el caso
del propionaldehído.

Capítulo 2
92
La configuración absoluta del aducto 41D obtenido de la reacción del
hexanal con nitrosobenceno (entrada 4) se determinó por comparación del valor
de la rotación óptica del diol libre 43 (Esquema 2-24) con el previamente
descrito50 y se confirmó que era S.
Esquema 2-24
2.4.2. Aplicaciones
Debido a su inestabilidad, los aductos de la reacción nitrosoaldólica de
aldehídos no se aislan como tales normalmente, y en su lugar se suelen reducir
y aislar en forma de los correspondientes alcoholes para su estudio. Así y todo,
los aldehídos provenientes de esta reacción pueden funcionar como productos
de partida para otras reacciones típicas de aldehídos. Un ejemplo representativo
de la bibliografía que explica este proceder es la adición de Grignard a
aldehídos aminoxilados descrita por Yamamoto, que ya se ha comentado
anteriormente (p. 74).41
En este sentido, nosotros exploramos una aplicación diferente
consistente en la adición de reactivos de Grignard a las iminas formadas a partir
de los aldehídos aminoxilados. De esta forma se podrían obtener 1,2-
aminoalcoholes, motivos estructurales muy interesantes y frecuentes en
productos naturales y también utilizados como auxiliares quirales.51
Así, por ejemplo, como se muestra en el Esquema 2-25, el α-
aminoxialdehído 44G proveniente de la reacción O-nitrosoaldólica
enantioselectiva con el hidrocinamaldehído (17G) se transformó
cuantitativamente en la imina correspondiente, previa adición de bencidrilamina.
El subsiguiente tratamiento con un magnesiano condujo a los aductos
50 S.-T. Tong, M. A.Brimble, D. Barker, Tetrahedron, 2009, 65, 4801-4807. 51 Para revisiones sobre síntesis asimétrica y aplicaciones de 1,2-aminoalcoholes, ver: a) D. J. Ager, I. Prakash, D. R. Schaad, Chem. Rev. 1996, 96, 835-875. b) H. C. Kolb, K. B. Sharpless, Transition Metals for Organic Synthesis, 1998, M. Beller, C. Bolm Eds.; Willey-VCH: Weinheim; p 243. c) S. C. Bergmeier, Tetrahedron 2000, 56, 2561-2576.
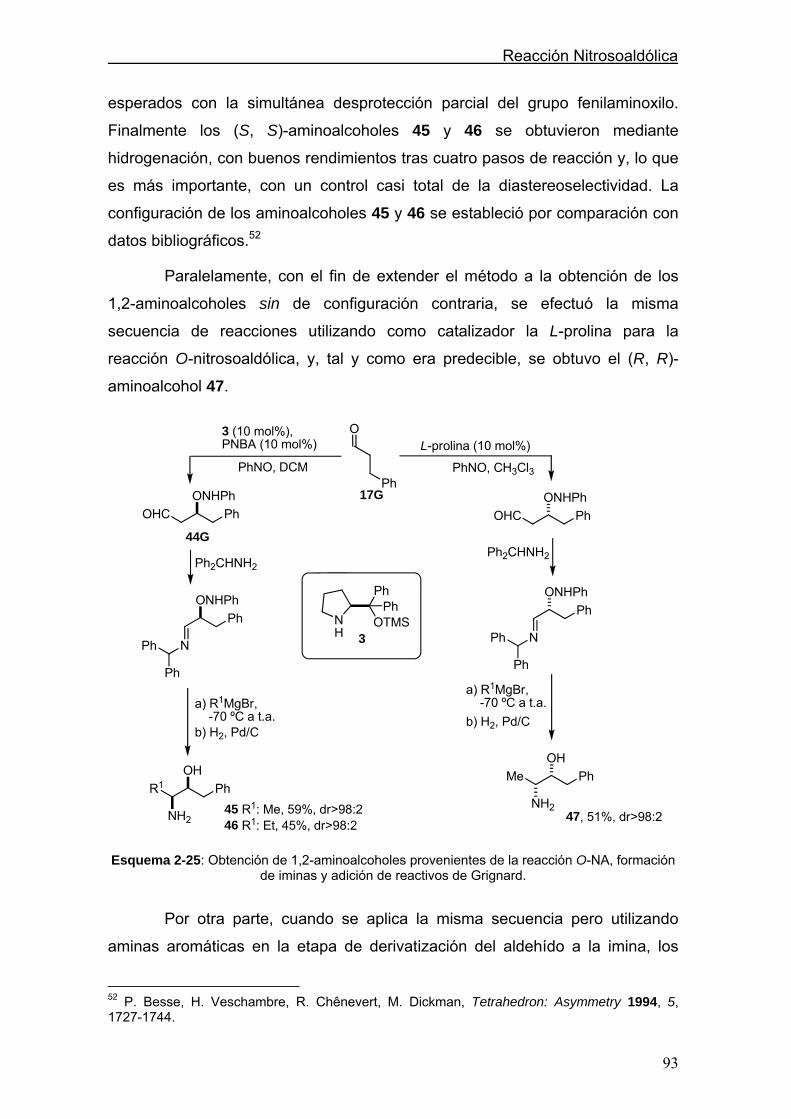
Reacción Nitrosoaldólica
93
esperados con la simultánea desprotección parcial del grupo fenilaminoxilo.
Finalmente los (S, S)-aminoalcoholes 45 y 46 se obtuvieron mediante
hidrogenación, con buenos rendimientos tras cuatro pasos de reacción y, lo que
es más importante, con un control casi total de la diastereoselectividad. La
configuración de los aminoalcoholes 45 y 46 se estableció por comparación con
datos bibliográficos.52
Paralelamente, con el fin de extender el método a la obtención de los
1,2-aminoalcoholes sin de configuración contraria, se efectuó la misma
secuencia de reacciones utilizando como catalizador la L-prolina para la
reacción O-nitrosoaldólica, y, tal y como era predecible, se obtuvo el (R, R)-
aminoalcohol 47.
O
17G
3 (10 mol%),PNBA (10 mol%) L-prolina (10 mol%)
a) R1MgBr,-70 ºC a t.a.
b) H2, Pd/C
45 R1: Me, 59%, dr>98:246 R1: Et, 45%, dr>98:2
47, 51%, dr>98:2
N
Ph
Ph
ONHPh
Ph
Ph2CHNH2
Ph
ONHPh
OHC
44G
R1 Ph
NH2
OH
Ph
ONHPh
OHC
N
Ph
Ph
ONHPh
Ph
Me Ph
NH2
OH
Ph
Ph2CHNH2
a) R1MgBr,-70 ºC a t.a.
b) H2, Pd/C
PhNO, DCM PhNO, CH3Cl3
3
NH
PhPh
OTMS
Esquema 2-25: Obtención de 1,2-aminoalcoholes provenientes de la reacción O-NA, formación de iminas y adición de reactivos de Grignard.
Por otra parte, cuando se aplica la misma secuencia pero utilizando
aminas aromáticas en la etapa de derivatización del aldehído a la imina, los
52 P. Besse, H. Veschambre, R. Chênevert, M. Dickman, Tetrahedron: Asymmetry 1994, 5, 1727-1744.

Capítulo 2
94
productos finales que se obtienen son los 1,2-aminalcoholes N-aril protegidos.
Por ejemplo (Esquema 2-26), el tratamiento del aldehído 44A, proveniente de la
reacción nitrosoaldólica del propanal, con p-anisidina condujo a la imina 48. La
subsiguiente adición de los reactivos de Grignard dio los aductos 49 y 50 con
buenos rendimientos globales y relaciones diastereoméricas del orden de 95:5.
En este caso, la desprotección completa del grupo fenilaminoxilo se produjo
durante la adición del reactivo de Grignard. Esta ruptura del enlace N-O podría
resultar de una transferencia electrónica entre el magnesiano y el aducto
aminoxilado.53
17A
O
Me
3 (10 mol%),PNBA(10 mol%) 4-MeOC6H4NH2
MeR1
OH
NH
49 R1: nPent, 55%, dr: 95:550 R1: Ph, 52%, dr: 94:6
Me
N
ONHPh
48
R1MgBr
Me
ONHPh
OHC
44A
HPhNO
MeO
MeO
Esquema 2-26: Obtención de 1,2-aminoalcoholes provenientes de la reacción O-NA, formación de iminas y adición de reactivos de Grignard.
Es de destacar la elevada estereoselectividad facial observada en la
etapa de adición del correspondiente magnesiano a las iminas derivadas de los
aductos de la reacción O-nitrosoaldólica. El modelo simplificado A de la Figura
2-11 explica el resultado estereoquímico obtenido sobre la base de la formación
de un anillo quelatado de seis miembros, en el que el Mg se coordina tanto al N
imínico como al N del grupo oxiamino. Alternativamente, un quelato análogo
pero de cinco miembros con un sistema O-Mg-N podría también ser invocado
con el mismo resultado estereoquímico.
53 Para ruptura de enlaces N-O mediante reactivos de Grignard, ver: a) O. Wichterle, J. Vogel, Collect. Czech. Chem. Commun. 1949, 14, 209-218. b) T. Q. Dinh, R. W. Armstrong, Tetrahedron Lett. 1996, 37, 1161-1164.

Reacción Nitrosoaldólica
95
R1MgBr
NMg
NO
H
H
R
Ph
PG
R1
R
ONHPh
NHPG
R1MgBr
N
Mg
HN
O
H
H
R
Ph
PG
A B
Figura 2-11: Modelos simplificados para la adición de magnesianos a iminas. PG: Grupo protector
2.5. Conclusiones
A modo de conclusión de este segundo capítulo, se puede afirmar que,
en general, queda demostrada la posibilidad de llevar a cabo las reacciones N-
y O-nitrosoaldólicas directas de aldehídos con nitrosobenceno en presencia de
un organocatalizador, sencillo y comercial. Además, éste es el primer
catalizador utilizado para este fin carente de dadores de hidrógeno de ningún
tipo.
El curso regioquímico de la reacción en uno (O-) y otro (N-) sentido
puede controlarse de manera casi perfecta mediante la adición o no de un dador
de H (ácido de Brønsted) como cocatalizador. Los ejemplos estudiados bajo las
condiciones óptimas conducen en general a los correspondientes aldehídos α-
funcionalizados, que tras reducción son aislados en forma de alcoholes, con
buenos rendimientos y enantioselectividad muy elevada. La configuración del
nuevo estereocentro formado es (S), complementando así los métodos
descritos con la L-prolina y derivados, que por lo general conducen al
enantiómero R.
La posterior elaboración de los aductos de reacción según una
secuencia de formación de imina y adición de un magnesiano habilita una nueva
vía de síntesis organocatalítica de 1,2-aminoalcoholes sencilla, eficaz y de
estereoquímica dirigida y controlada.


Reacción de Mannich anti-selectiva


Reacción de Mannich anti-Selectiva
99
3. Reacción de Mannich anti-selectiva
3.1. Introducción
La reacción clásica de Mannich1 (a en Fig. 3-1) se encuentra entre las
reacciones más interesantes para la formación de enlaces carbono-carbono y
consiste en la condensación de un compuesto carbonílico enolizable con
formaldehído y una amina. El producto de reacción, un compuesto β-amino
carbonílico, se conoce como base de Mannich.2 Por extensión, también se
denomina reacción de Mannich a aquella que utiliza iminas preformadas (b en
Fig. 3-1).
a) Reacción de Mannich “clásica”:
b) Reacción de Mannich a partir de iminas preformadas:
Figura 3-1
Los compuestos β-amino carbonílicos, o bases de Mannich
resultantes de esta transformación, constituyen estructuras muy interesantes y
versátiles desde el punto de vista sintético para la preparación de fármacos o
dianas farmacéuticas,3 productos naturales,4 como alcaloides5 y nucleótidos, y
1 C. Mannich, W. Krosche, Arch. Pharm. 1912, 250, 647-667. 2 Para más detalles, ver: a) B. P. Mundy, M. G. Ellerd, F. G. Favaloro jr., Name Reactions and Reagents in Organic Synthesis, 2005, 2nd ed.; Wiley & Sons: New York. b) T. Laue, A. Plagens, Named Organic Reactions, 2005, 2nd ed.; Wiley & Sons: New York. c) J. J. Li, Name Reactions. A Collection of Detailed Reaction Mechanisms, 2006, 3rd ed.; Springer: New York. d) M.B. Smith, J. March, March´s Advanced Organic Chemistry, Reactions, Mechanisms and Structure, 2007, 6th ed.; Wiley & Sons; New York. 3 Enantioselective Synthesis of β-Amino Acids, E. Juaristi, 1997, Ed.; Wiley-VCH: New York. 4 Para el primer ejemplo de síntesis de productos naturales mediante la reacción de Mannich: a) R. Robinson, J. Chem. Soc. 1917, 762-768. b) E. F. Kleinmann, Comprehensive Organic Synthesis, 1991, B. M. Trost, I. Flemming, Eds.; Pergamon Press: New York, Vol. 2, Chapter 4.1. 5 a) E. Leete, Tetrahedron 1958, 3, 313-314. b) H. Conroy, Tetrahedron Lett. 1960, 1, 34-37. c) E. E. Van Tamelen, R. L. Foltz, J. Am. Chem. Soc. 1960, 82, 2400-2400; d) E. Wenkert, J. Am. Chem. Soc. 1962, 84, 98-102. e) E. Leete, Annu. Rev. Plant Physiology 1967, 18, 179-196. f)

Capítulo 3
100
péptidos con propiedades estructurales únicas.6 En la Figura 3-2 se muestran
dos alcaloides en cuya síntesis está incluida la reacción de Mannich.
Figura 3-2
Dependiendo de la naturaleza del dador (componente carbonílico) y del
aceptor (componente imínico), pueden generarse hasta dos centros
estereogénicos en esta transformación, por lo que el control tanto de la
configuración absoluta (enantioselectividad) como de la relativa
(diastereoselectividad o selectividad sin/anti) es de vital importancia.
Figura 3-3: Posibles estereoisómeros que resultan de la reacción de Mannich.
M. Hesse, Dtsch. Apoth. Ztg. 1968, 108, 1480-1482. g) H. Liebisch, Biosyn. Alkaloids 1969, 101-122. h) K. F. McClure, C. H. Heathcock, J. Am. Chem. Soc. 1989, 111, 1530-1531. i) S. F. Martin, J. Heterocycl. Chem. 1994, 31, 679-686. j) S. F. Martin, Pure Appl. Chem. 1997, 69, 571-576. k) J. C. Lanter, H. Chem, X. Zhang, Z. Sui, Org. Lett. 2005, 7, 5905-5907. l) K. Nagata, K. Nishimura, M. Yokoya, T. Itoh, Heterocycles 2006, 70, 335-344. m) X. Chen, J. Chen, J. Zhu, Synthesis 2006, 23, 4081-4086. n) F. A. Davis, M. Santhanaraman, J. Org. Chem. 2006, 71, 4222-4226. 6 a) T. Hintermann, D. Seebach, Chimia 1997, 50, 244-247. b) D. Seebach, J. L. Matthews, Chem. Commun. 1997, 2015-2022. c) U. Koert, Angew. Chem. Int. Ed. 1997, 36, 1836-1837. d) S. H. Gellman, Acc. Chem. Res. 1998, 31, 173-180.

Reacción de Mannich anti-Selectiva
101
Los primeros métodos estereoselectivos de la reacción de Mannich se
basaban en la utilización de auxiliares quirales7 e implicaban la generación de
los correspondientes enolatos y/o enaminas en una etapa previa e irreversible,
por lo que se conocen como métodos “indirectos” (enolatos o enaminas
preformados). A finales de los 90 fueron documentadas las primeras variantes
asimétricas catalíticas vía enolatos de Si, generados en una etapa previa, y
por lo tanto también indirectos.8
Conviene señalar que el desarrollo de la versión catalítica
enantioselectiva de este proceso,9 basado en catalizadores que funcionan
como ácidos de Lewis, ha resultado complicado debido a que el catalizador
debe ser capaz de activar las iminas por coordinación de las mismas a la vez
que debe ser resistente a una posible inhibición por coordinación con los
productos de reacción, aminas que pueden comportarse como bases de Lewis
fuertes. En este contexto, tanto para la reacción de Mannich como para otras,
inicialmente los catalizadores metálicos fueron los que obtuvieron un mayor
auge.10
7 Para distintos ejemplos, ver: a) R. Kober, K. Papadopoulos, W. Miltz, D. Enders, W. Steglich, H. Reuter, H. Puff, Tetrahedron 1985, 41, 1693-1701. b) D. A. Evans, F. Urpi, T. C. Somers, J. S. Clark, M. T. Bilodeau, J. Am. Chem. Soc. 1990, 112, 8215-8216. c) E. J. Corey, C. P. Decicco, R. C. Newbold, Tetrahedron Lett. 1991, 39, 5287-5290. d) D. Enders, D. Ward, J. Adam, G. Raabe, Angew. Chem. Int. Ed. 1996, 35, 981-984. e) C. Palomo, M. Oiarbide, M. C. Gonzálezs-Rego, A. K. Sharma, J. M. García, A. Landa, A. Linden, Angew. Chem. Int. Ed. 2000, 39, 1063-1065. 8 a) H. Ishitani, M. Ueno, S. Kobayashi, J. Am. Chem. Soc. 1997, 119, 7153-7154. b) D. Ferraris, B. Yong, T. Dudding, T. Leckta, J. Am. Chem. Soc. 1998, 120, 4548-4549. c) A. Fujii, E. Hagiwara, M. Sodeoka, J. Am. Chem. Soc. 1999, 121, 5450-5458. d) H. Ishitani, M. Ueno, S. Kobayashi, J. Am. Chem. Soc. 2000, 122, 8180-8186. 9 Para revisiones sobre reacciones de Mannich catalíticas asimétricas, ver: a) S. Denmark, J.-C. Nicaise, Comprehensive Asymmetric Catalysis, 1999, E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Eds.; Springer: Berlin, Vol. 2, pp 954. b) S. Kobayashi, H. Ishitani, Chem. Rev. 1999, 99, 1069-1094. c) M. Benaglia, M. Cinquini, F. Cozzi, Eur. J. Org. Chem. 2000, 4, 563-572. d) A. Córdova, Accounts of Chemical Research 2004, 37, 102-112. e) S. J. Greco, V. Lacerda, R. B. do Santos, Aldrichimica Acta 2011, 4, 15-24. 10 Para algunos ejemplos pioneros, ver: a) S. Yamasaki, T. Iida, M. Shibasaki, Tetrahedron Lett. 1999, 40, 307-310. b) K. Juhl, N. Gathergood, K. A. Jørgensen, Angew. Chem. Int. Ed. 2001, 40, 2995-2997. c) M. Ueneo, H. Kitagawa, H. Ishitani, S. Yasuda, K. Hanada, S. Kobayashi, Tetrahedron Lett. 2001, 42, 7863-7865. d) S. Kobayashi, R. Matsubara, H. Kitagawa, Org. Lett. 2002, 4, 143-145. e) B. M. Trost, L. M. Terrell, J. Am. Chem. Soc. 2003,125, 338-339. f) D. E. DeMong, R. M. Williams, J. Am. Chem. Soc. 2003, 125, 8561-8565. g) Y. Nakamura, R. Matsubara, H. Kitagawa, S. Kobayashi, K. Kumagai, S. Yasuda, K. Hanada, J. Med. Chem. 2003, 46, 3688-3965. h) H. Liu, S. Cai, J. Xu, J. Pept. Sci. 2006, 12, 337-340.

Capítulo 3
102
3.1.1. Reacción de Mannich asimétrica directa vía catálisis metálica
En contraposición a los métodos “indirectos”, los métodos “directos” se
caracterizan por la no necesidad de generación previa de un enolato o
equivalente en una etapa separada e independiente. En este sentido, la
primera reacción de Mannich catalítica enantioselectiva directa, aunque aún
estuviera lejos de reportar resultados satisfactorios, se llevó a cabo con un
reactivo organometálico. En 1999 Shibasaki y colaboradores, basándose en
sus recientes investigaciones sobre la reacción aldólica directa
enantioselectiva con el complejo LaLi3tris(binaftóxido)11 51 (LLB), describieron
la primera reacción de Mannich asimétrica directa10a (Esquema 3-1),
obteniendo el aducto correspondiente con moderada enantioselectividad y
bajo rendimiento.
Ar
O
RComplejo 51 (30% mol)
MS
Tolueno, 50ºCAr
O
N(C2H5)2R
+
O O
LaO
O
OO
Li Li
Li
Rto.: 61-76%ee: 31-44%
H3CO N(C2H5)2
Complejo 51:
Esquema 3-1: Primera reacción de Mannich directa vía catálisis asimétrica, Shibasaki, 1999.10a
Poco más adelante Jørgensen publicó la primera reacción de Mannich
catalítica enantioselectiva directa eficiente. Basándose en el complejo
organometálico Cu(OTf)2/bisoxazolina 52 este autor describió la reacción entre
11 a) N. Yoshikawa, Y. M. A. Yamada, J. Das, H. Sasai, M. Shibasaki, J. Am. Chem. Soc. 1999, 121, 4168-4178. b) D. Sawada, M. Shibasaki, Angew. Chem. Int. Ed. 2000, 39, 209-213. c) N. Yoshikawa, N. Kumagai, S. Matsunaga, G. Moll, T. Ohshima, T. Suzuki, M. Shibasaki, J. Am. Chem. Soc. 2001, 123, 2466-2467.

Reacción de Mannich anti-Selectiva
103
α-cetoésteres e iminas derivadas del glioxilato de etilo (Esquema 3-2) que
transcurre con buen rendimiento y elevada estereoselectividad.10b
Esquema 3-2: Primera reacción de Mannich directa catalítica enantioselectiva eficiente, Jørgensen, 2001.10b
Posteriormente a estos trabajos otros autores han desarrollado
diversos complejos organometálicos para la reacción de Mannich.12 La Figura
3-4 muestra algunos de los más representativos.13
12 a) Y. Yamashita, M. Ueno, M. Kuriyama, S. Kobayashi, Adv. Synth. Catal. 2002, 344, 929-931. b) D. Ferraris, B. Young, C. Cox, T. Dudding, W. J. III. Drury, L. Ryzhkov, A. E. Taggi, T. Leckta, J. Am. Chem. Soc. 2002, 124, 67-77. c) N. Jaber, F. Carrée, J.-C. Fiaud, J. Collin, Tetrahedron: Asymmetry 2003, 14, 2067-2071. d) S. Matsunaga, N. Kumagai, S. Harada, M. Shibasaki, J. Am. Chem. Soc. 2003, 125, 4712-4713. e) S. Saaby, K. Namaka, M. A. Lie, R. G. Hazell, K. A. Jørgensen, Chem. Eur. J. 2003, 9, 6145-6154. f) N. S. Josephsohn, M. L. Snapper, A. H. Hoveyda, J. Am. Chem. Soc. 2004, 126, 3734-3735. g) Y. Hamashima, N. Sasamoto, D. Hotta, H. Somei, N. Umebayashi, M. Sodeoka, Angew. Chem. Int. Ed. 2005, 44, 1525-1529. h) A. S. González, R. G. Arrayás, J. C. Carretero, Org. Lett. 2006, 8, 2977-2980. i) C. Dubs, Y. Hamashima, N. Sasamoto, T. M. Seidel, S. Suzuki, D. Hashizume, M. Sodeoka, J. Org. Chem. 2008, 73, 5859-5871. 13 a) S. Kobayashi, R. Matsubara, H. Kitagawa, Org. Lett. 2002, 4, 143-145. b) M. Matsunaga, N. Kumagai, N. Harada, M. Shibasaki, J. Am. Chem. Soc. 2003, 125, 4712-4713. c) B. M. Trost, L. M. Terrell, J. Am. Chem. Soc. 2003, 125, 338-339. d) G. A. Cutting, N. E. Stainforth, M. P. John, G. Kociok-Khn, M. Willis, J. Am. Chem. Soc. 2007, 129, 10632-10633.

Capítulo 3
104
O
OH
OH
HO
HO
(Et2Zn)
Shibasaki13b
Trost13c
Me
ON NZn
ArArO
Zn
ArAr
OEt
NH
NH-Naf -Naf
Ph
Cu
Ph
TfO OTf
Kobayashi13a
OO
N NO
Ph Ph
Mg(ClO4)2
Willis13d
Figura 3-4: Otros catalizadores organometálicos descritos para la reacción de Mannich.
3.2. Reacción de Mannich asimétrica vía organocatálisis,
precedentes
En los últimos años se ha estudiado ampliamente la reacción de
Mannich via organocatálisis,14 resultando ser un complemento eficaz a los
métodos basados en metales. De entre los organocatalizadores estudiados,
las aminas secundarias han destacado ampliamente por su eficiencia química
y estereoquímica. En lo que sigue se presenta una relación de estos
procedimientos existentes antes del comienzo de nuestro estudio clasificados
según conduzcan a productos de configuración sin o anti, y obedezcan o no a
un mecanismo vía enamina
3.2.1. Reacción de Mannich sin-selectiva vía enamina
La reacción de Mannich sin-selectiva vía organocatálisis está amplia y
comparativamente más explorada que la anti. La primera reacción de Mannich
directa vía organocatálisis fue descrita por List15 y poco más tarde por el grupo
14 Para revisiones sobre la reacción de Mannich vía organocatálisis, ver: a) A. Ting, S. E. Schaus, Eur. J. Org. Chem. 2007, 5797-5815. b) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg, F. P. J. T. Rutjes, Chem. Soc. Rev. 2008, 37, 29-41. c) P. S. Bhadury, B.-A. Song, Curr. Org. Chem. 2010, 14, 1989-2010. 15 a) B. List, J. Am. Chem. Soc. 2000, 122, 9336-9337. b) B. List, Synlett 2001, 11, 1675-1686. c) B. List, P. Pojarliev, W. T. Biller, H. J. Martin, J. Am. Chem. Soc. 2002, 124, 827-833. d) B. List, Tetrahedron 2002, 58, 5573-5590.

Reacción de Mannich anti-Selectiva
105
de Barbas16 y consiste en el tratamiento de una cetona, un aldehído y p-
metoxianilina en presencia de L-prolina como catalizador, para dar lugar a los
correspondientes compuestos β–amino carbonílicos (Esquema 3-3).
MeR1
O
NH2
OMe
+ +L-prolina
R1: H, OH, Me, OMeR2: PNP, nBu, iBu, iPr, CO2EtRto.: 50-92%, (sin:ant i)>95:5,ee: 70-99%
R2
O
HMe R2
O
R1
HN
OMe
Esquema 3-3: Primera reacción de Mannich sin-selectiva vía organocatálisis, List, 2000, Barbas III, 2001.15, 16
Los mismos autores han propuesto distintos estados de transición
para explicar la estereoquímica de los aductos obtenidos en la reacción (ver
ref. 15a, b, c, d y 16a). Estos modelos postulan la reacción entre la imina (Z) y
la enamina derivada de la cetona y de la prolina según unos estados de
transición de tipo silla y bote, en los cuales se detecta la transferencia del
protón carboxílico de la prolina a los átomos de nitrógeno (a y b en Fig. 3-5).
Posteriormente se propuso un estado de transición que no incluía enlaces de
H internos (c en Fig. 3-5).
Figura 3-5
Más adelante Houk y colaboradores añadieron más luz sobre el
proceso mediante estudios computacionales.17 Acorde con las pruebas
experimentales existentes de que las iminas (E) son mucho más estables que 16 a) W. Notz, K. Sathivel, T. Bui, G. Zhong, C. F. Barbas III, Tetrahedron Lett. 2001, 42, 199-201. b) K. Sathivel, W. Notz, T. Bui, C. F. Barbas III, J. Am Chem. Soc. 2001, 123, 5260-5267. c) A. Córdova, W. Notz, G. Zhong, J. M. Betancort, C. F. Barbas III, J. Am. Chem. Soc. 2002, 124, 1842-1843. d) A. Córdova, S. Watanabe, F. Tanaka, W. Notz, C. F. Barbas III, J. Am. Chem. Soc. 2002, 124, 1866-1867. 17 S. Bahamanyar, K. N. Houk, Org. Lett. 2003, 5, 1249-1251.

Capítulo 3
106
las (Z),18 los modelos de estados de transición estudiados para estas últimas
resultaron mucho menos favorables energéticamente. De entre todos los
estados de transición posibles, el de menor energía de activación resultó ser
el modelo a) representado en la Figura 3-6, que explica satisfactoriamente la
estereoquímica observada experimentalmente. Este mismo estudio aplicado al
propionaldehído (Fig 3-6, b) predice la formación preferente del
diastereoisómero sin.
NO
OMe
N
MeH
PhH
Me Me
O NHPh
NO
OH
N
MeH
PhH
(E), anti, +60º
H Me
O NHPh
MeMe
a)
b)
Figura 3-6: Explicación de la estereoquímica obtenida para la reacción de Mannich con la L-prolina, Houk, 2003.17
A partir de estos primeros trabajos de List y Barbas con la prolina y
sus posteriores versiones,19 incluida la extensión a N-Boc-iminas como
aceptores de Mannich,20 surgieron distintos catalizadores derivados de la
prolina conducentes también a aductos de configuración sin. El propósito
principal a la hora de diseñar estos catalizadores fue mejorar las limitaciones
observadas con la prolina. Por ejemplo, además de la alta carga de
18 H. J. C. Yeh, H. Ziffer, D. M. Jerina, D. R. Boyd, J. Am. Chem. Soc. 1973, 95, 2741-2743. 19 a) Y. Hayashi, W. Tsuboi, I. Ashimine, T. Urushima, M. Shoji, K. Sakai, Angew. Chem. Int. Ed. 2003, 42, 3677-3680. b) W. Notz, S. Watanabe, N. S. Chowdari, G. Zhong, J. M. Betancort, F. Tanaka, C. F. Barbas III, Adv. Synth. Catal. 2004, 346, 1131-1140. c) W. Notz, F. Tanaka, C. F. Barbas III, Acc. Chem. Res. 2004, 37, 580-591. d) A. Córdova, Chem. Eur. J. 2004, 10, 1987-1997. e) Y. Hayashi, T. Urushima, M. Shin, M. Shoji, Tetrahedron, 2005, 61, 11393-11404. f) S. Fustero, D. Jiménez, J. F. Sanz-Cervecera, M. Sánchez-Roselió, E. Esteban, A. Simón-Fuentes, Org. Lett. 2005, 7, 3433-3436. g) B. Westermann, C. Neuhaus, Angew. Chem. Int. Ed, 2005, 44, 4077-4079. h) D. Enders, C. Grondal, M. Vrettou, G. Raabe, Angew. Chem. Int. Ed. 2005, 44, 4079-4083. i) I. Ibrahem, W. Zou, M. Enqvist, Y. Xu, A. Córdova, Chem. Eur. J. 2005, 11, 7024-7029. j) I. Ibrahem, W. Zou, Y. Xu, A. Córdova, Adv. Synth. Catal. 2006, 348, 211-222. 20 a) D. Enders, C. Grondal, M. Vrettou, Synthesis 2006, 21, 3597-3604. b) J. Vesely, R. Rios, I. Ibrahem, A. Córdova, Tetrahedron Lett. 2007, 48, 421-425. c) J. W. Yang, M. Stadtler, B. List, Angew. Chem. Int. Ed. 2007, 46, 609-611.

Reacción de Mannich anti-Selectiva
107
catalizador requerida (20-30 mol%), la reacción de Mannich con la prolina se
tiene que llevar a cabo siempre en disolventes extremadamente polares, como
DMSO o DMF, debido a la baja solubilidad del aminoácido en la mayoría de
disolventes típicos de las reacciones orgánicas, más apolares.
Así, Ley,21 Hayashi22 y Wang23 y más recientemente Carter24 han
identificado otros organocatalizadores de bajo peso molecular derivados de la
L-prolina (Fig. 3-7), que son efectivos en disolventes más apolares como el
dicloromentano, además de requerir menores cargas catalíticas para obtener
resultados similares a los obtenidos con la prolina. Por ejemplo, el catalizador
24 de Ley promueve la reacción entre la dietilcetona y la imina derivada del
glioxilato de etilo y la p-anisidina en diclorometano (Fig. 3-7). Dicho catalizador
presenta en lugar del grupo carboxílico de la prolina una unidad de tetrazol,
con lo que se mejora su solubilidad en disolventes más apolares. Es de
destacar que 1 mol% de este catalizador conduce a resultados similares a los
obtenidos con un 20 mol% de prolina. En el caso del catalizador 56, la
diferencia con la L-prolina es la presencia de un grupo sililoxilo en la posición γ
de la pirrolidina, lo que la hace más reactiva y soluble.
21 A. J. A. Cobb, D. M. Shaw, S. V. Ley, Synlett 2004, 558-560; A. J. A. Cobb, D. M. Shaw, D. A. Longbotton, J. B. Gold, S. V. Ley, Org. Biomol. Chem. 2005, 3, 84-96. Para posteriores contribuciones a la reacción de Mannich con el catalizador 24, ver: a) N. S. Chowdari, M. Ahmad, K. Albertshofer, F. Tanaka, C. F. Barbas III, Org. Lett. 2006, 8, 2839-2842. b) P. Galzerano, D. Agostino, G. Bencivenni, L. Sambri, G. Bartoli, P. Melchiorre, Chem. Eur. J. 2010, 16, 6069-6076. 22 Y. Hayashi, J. Yamaguchi, K. Hibino, T. Sumiya, T. Urushima, M. Shoji, D. Hashizume, H. Koshino, Adv. Synth. Catal. 2004, 346, 1435-1439. 23 W. Wang, J. Wang, H. Li, Tetrahedron Lett. 2004, 45, 7243-746. 24 H. Yang, R. G. Carter, J. Org. Chem. 2009, 74, 2246-2249.

Capítulo 3
108
NH HN N
NN
24
O
Me Me
24 (5 mol%)+
NH
CO2H
TBSO
56Hayashi
NH HN SO2CF3
Wang
57
Entr. 24 (mol%) Rto.(%) (sin:anti) ee(%)
1234
65493770
>95:5>95:5>95:5>95:5
>99>99>99>99
Disolvente
5551
DCM
DCM
MeCNTHF
t(h)
22216
NH
O
NH
SO2R
R: Ph, 53, Ley
R: Me, 54, Ley
R: 4-C12H25C6H4, 55, CarterLey
CO2Et
O
Me
HN
OMe
Me
N
CO2Et
MeO
Ejemplo:
Figura 3-7: Derivados de la L-prolina descritos para la reacción de Mannich sin-selectiva,
Hayashi, 2004,22 Wang, 2004,23 Ley, 2005,21 Carter, 2010.24 Ejemplo de Ley, 2005.
Aparte de la prolina y otras aminas secundarias como las antes
comentadas, también distintos aminoácidos acíclicos (aminas primarias) son
capaces de promover la reacción de Mannich. Por ejemplo, Córdova y
colaboradores25 descubrieron que, sorprendentemente, la serina era capaz de
catalizar la reacción entre la ciclohexanona, 4-nitrobenzaldehído y p-anisidina
en DMSO (Esquema 3-4) con buen control de la diastereo- y
enantioselectividad, obteniendo los correspondientes aductos de Mannich de
configuración sin con buenos rendimientos. En la reacción se observan sólo
trazas de producto de una reacción aldólica, resultados que mejoran los
obtenidos con la prolina en igualdad de condiciones de reacción. Tras un
amplio estudio observaron que casi todos los aminoácidos naturales son
capaces de catalizar la reacción con mayor o menor efectividad.
25 I. Ibrahem, W. Zou, M. Engqvist, Y. Xu, A. Córdova, Chem. Eur. J. 2005, 11, 7024-7029.

Reacción de Mannich anti-Selectiva
109
O OMe
NH2
O
NO2
+ +Cat. (30 mol%)H2O (2 equiv.)
DMSO, t.a.
HNO
OMe
NO2
Cat.:
H2N CO2H
OH
H2N CO2H H2N CO2H H2N CO2H
H2N CO2H
Ph
H2N
HN NN
NH2N CO2H
CO2H
H2N CO2H
Et
H2N CO2H
iPr
H2N CO2H
Et
Rto.: 60-90%(sin:anti): 2:1-6:1ee: 94-99%
Esquema 3-4: Aminas primarias descritas para la reacción de Mannich sin-selectiva, Córdova,
2005.25
3.2.2. Reacción de Mannich sin-selectiva con otros catalizadores
Aparte de los catalizadores de tipo amina secundaria antes
comentados, que actúan por activación del correspondiente sustrato cetónico
o aldehídico vía enamina, se han descrito otros organocatalizadores para la
reacción de Mannich, los cuales actúan bien como bases, bien como ácidos
de Brønsted o bien de forma ambifuncional. Cabe mencionar que gran parte
de los trabajos desarrollados con estos catalizadores involucran el
acetaldehído o derivados del ácido acético y conducen a aductos de Mannich
que poseen un solo estereocentro (ejemplo en Esquema 3-5), y por tanto, no
afrontan el problema del control de la diastereoselectividad sin/anti. Nosotros
nos centraremos de forma principal en aquellos ejemplos que involucran la
formación de dos centros estereogénicos consecutivos y que, por tanto,
pueden conducir preferentemente a aductos de Mannich sin o anti.

Capítulo 3
110
Ph
NHBoc
+
Ph H
NBoc
CO2iPr
5 mol% Cat.
Cat.:
Rto.: 96%ee: 97%
OiPr
OTBS
NH
NH
S
N
N
Me
Bn
O
tBu
HO
tBu tBu
58
59
Esquema 3-5: Reacción de Mannich enantioselectiva entre una N-Boc-imina y el enolato de Si 58 catalizada por la tiourea 59. Jacobsen, 2002.27a
En el caso de catalizadores con carácter de base de Brønsted tiene
lugar una desprotonación del pronucleófilo por parte del catalizador,
formándose un par iónico quiral donde el anión reacciona con la
correspondiente imina, generalmente poseedora de un grupo electroatrayente
en el nitrógeno imínico. Los catalizadores que mayor éxito han reportado
dentro de este grupo son los alcaloides de la cincona26 y sus derivados,
especialmente los portadores de un grupo tiourea27 capaz de activar
simultáneamente el componente imínico.
Un ejemplo representativo de reacción de Mannich sin-selectiva con
este tipo de catalizadores se muestra en el Esquema 3-6. Se trata de la
reacción entre una N-Boc-imina y el β-cetoéster 60 catalizada por 61, descrita
por Takemoto.27b El aducto 62 se obtiene con buen rendimiento y
26 Para ejemplos de reacción de Mannich asimétrica con derivados de cincona como catalizadores, ver: a) T. Poulsen, C. Alemparte, S. Saaby, M. Bella, K. A. Jørgensen, Angew. Chem. Int. Ed. 2005, 44, 2896-2899. b) A. Ting, S. Lou, S. E. Schaus, Org. Lett. 2006, 8, 2003-2006. c) C. M. Bode, A. Ting, S. E. Schaus, Tetrahedron, 2006, 62, 11499-11505. c) J. Song, Y. Wang, L. Deng, J. Am. Chem. Soc. 2006, 128, 6048-6049. f) A. L. Tillman, J. Ye, D. J. Dixon, Chem. Commun, 2006, 1191-1193. g) J. Song, H.-W. Shih, L. Deng, Org. Lett. 2007, 9, 603-606. h) F. Fini, L. Bernardi, R. P. Herrera, D. Pettersen, A. Ricci, V. Sgarzani, Adv. Synth. Catal. 2006, 348, 2043-2046. 27 Para ejemplos de reacción de Mannich asimétrica con derivados de tiourea como catalizadores, ver: a) A. G. Wenzel, E. N. Jacobsen, J. Am. Chem. Soc. 2002, 124, 12964-12965. b) Y. Yamaoka, H. Miyabe, Y. Yasui, Y. Takemoto, Synthesis 2007, 16, 2571-2575. c) J. H. Lee, D. Y. Kim, Synthesis 2010, 11, 1860-1864. d) Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi, K. Nagasawa, Angew. Chem. Int. Ed. 2010, 49, 9254-9257.

Reacción de Mannich anti-Selectiva
111
relativamente buenas relaciones diastereo- y enantioselectivas, aunque no se
pudo extender la reacción a una amplia variedad de sustratos.
NH
NH
NMe2
S
CF3
F3CO
MeO2CN
HPh
Boc
+Ph
ONH
MeO2C
Boc
61
62
Rto.: 89%(sin:anti): 92:8ee: 88%
60
Esquema 3-6: Reacción de Mannich asimétrica entre una N-Boc-imina aromática y 60 catalizada por la tiourea 61. Takemoto, 2006.27b
Otro ejemplo destacable es el descrito por el grupo de Schaus,
utilizando esta vez los catalizadores derivados de la cincona 63 y 64 para
llevar a cabo la reacción de Mannich sin-selectiva entre compuestos 1,3
dicarbonílicos cíclicos y N-acil-iminas26b (Esquema 3-7). Una peculiaridad de
este estudio es que dependiendo del catalizador utilizado (63 o 64) se
obtienen los aductos con similar diastereoselectividad pero enantioselectividad
invertida.
X
O O
Y R1O N
H R2
O
X
O HN
R2
OR1
O
YO
+5mol% Cat.
N
N
HO
H
H
N
H
N
OH
H
Cat.:
ó
Rto.:>90%(sin:anti): 90-99ee: 90-99%
R1:CH3, alilo
R2: Ar, (E)-CH=CH2Ar
63
64
Esquema 3-7: Reacción de Mannich asimétrica entre acil-iminas y compuestos 1,3
dicarbonílicos cíclicos catalizada por los catalizadores 63 y 64. Schaus, 2006.26b
Los organocatalizadores de tipo ácido de Brønsted, por su parte,
actúan protonando la imina, lo que conduce a un ion imínico con su
correspondiente contraión quiral. Este contraión es el responsable de la

Capítulo 3
112
inducción asimétrica durante la etapa de formación del nuevo enlace C-C. Los
catalizadores más comunes de este grupo son los diseñados a partir de
ácidos fosfóricos quirales28 y derivados del TADDOL29 o del BINOL.30
El ejemplo más representativo de este tipo de catalizadores para la
obtención de aductos de Mannich sin es el trabajo de Akiyama y
colaboradores, quienes utilizaron el ácido fosfórico quiral 65 para catalizar la
reacción de Mannich entre la N-o-hidroxifenil-imina 66 y el enolato de silicio
67, obteniendo los correspondientes compuestos β-amino carbonílicos con
excelente rendimiento y buenos excesos diastereo- y enantioméricos
(Esquema 3-8).28a
Esquema 3-8: Reacción de Mannich asimétrica entre la imina 66 y el enolato de Si 67 catalizada por el ácido de Brønsted quiral 65. Akiyama, 2004.28a
3.2.3. Reacción de Mannich anti-selectiva vía enamina
En comparación con los métodos catalíticos que rinden
mayoritariamente aductos de Mannich sin, los métodos disponibles para la
28 Para ejemplos de reacción de Mannich asimétrica con este tipo de catalizadores, ver: a) T. Akiyama, J. Itoh, K. Yokota, K. Fuchibe, Angew. Chem. Int. Ed. 2004, 43, 1566-1568. b) D. Uraguchi, M. Terada, J. Am. Chem. Soc. 2004, 126, 5356-5357. c) M. Yamanaka, J. Itoh, K. Fuchibe, T. Akiyama, J. Am. Chem. Soc. 2007, 129, 6756-6764. d) M. Rueping, E. Sugiono, F. R. Schoepke, Synlett 2007, 9, 1441-1445. e) Q.-X. Guo, H. Liu, C. Guo, S.-W. Luo, Y. Gu, L.-Z. Gong, J. Am. Chem. Soc. 2007, 129, 3790-3791. 29 Para un ejemplo de reacción de Mannich asimétrica con este tipo de catalizadores, ver: T. Akiyama, J. Saitoh, H. Morita, K. Fuchibe, Adv. Synth. Catal. 2005, 347, 1523-1526. 30 Para ejemplos de reacción de Mannich asimétrica con este tipo de catalizadores, ver: a) A. Hasegawa, Y. Naganawa, M. Fushimi, K. Ishihara, H. Yamamoto, Org. Lett. 2006, 8, 3175-3178. b) A. L. Tillman, D. J. Dixon, Org. Biomol. Chem. 2007, 5, 606-609.

Reacción de Mannich anti-Selectiva
113
obtención de aductos anti son más escasos y limitados. Por otro lado, además
de la prolina y de otras aminas coordinantes (bifuncionales) también ciertas
aminas no coordinantes son capaces de catalizar la reacción de Mannich.
Curiosamente, en estos últimos casos el aducto mayoritario obtenido es el
anti.
El primer estudio sobre la reacción de Mannich anti-selectiva vía
organocatálisis fue publicado en 2002. Barbas y Córdova intentaron modificar
la diastereoselectividad del proceso probando distintas pirrolidinas carentes
del grupo carboxilo de la prolina, evitando así el acercamiento por enlace de
hidrógeno y promoviendo, a su vez, el control estérico. Estos autores
descubrieron que la (S)-2-metoximetil pirrolidina (SMP) (68) cataliza
satisfactoriamente la reacción entre aldehídos y N-PMP-iminas derivadas del
glioxilato de etilo.31 La reacción produce mayoritariamente los aductos anti
correspondientes aunque con rendimientos limitados y diastereo- y
enantioselectividades de moderadas a buenas (Esquema 3-9).
PMPN
CO2Et
NH OMe
(20 mol%)
DMSO,t.a., 24-48h
+CO2Et
R
O HNPMP
68 N
R
MeON
CO2Et
H
H
Entr. R Rto.(%) (anti:sin) ee(%)
1 iPrtBuEtnBunPentnHexnCH2CH=CH(CH2)4CH3 trans
234567
52574454786867
95:595:580:2090:1095:595:595:5
82927574767678
PMP
H
O
RH
PMP: p-metoxifenilo
Esquema 3-9: Primera reacción de Mannich anti-selectiva vía organocatálisis, Barbas III y
Córdova, 2002.31
31 A. Córdova, C. F. Barbas III, Tetrahedron Lett. 2002, 43, 7749-7752.

Capítulo 3
114
Hasta el 2005 no volvieron a surgir trabajos sobre la reacción de
Mannich anti-selectiva vía organocatálisis. Fue entonces cuando Jørgensen,32
mediante el uso de la pirrolidina 4 llevó a cabo la reacción de Mannich entre
aldehídos y N-p-metoxifenil(PMP)-iminas derivadas del glioxilato de etilo,
obteniendo una mayor efectividad en términos de rendimiento y
enantioselectividad (Esquema 3-10).
Esquema 3-10: Reacción de Mannich anti-selectiva catalizada por el α,α–diarilprolinol 4,
Jørgensen, 2005.32
El modelo general que explica esta selectividad es el A (Fig. 3-8),
modelo que puede verse reforzado cuando existe un grupo dador de H
estratégicamente colocado en la pirrolidina como en B. (Fig. 3-8).
Los estados de transición propuestos33 para esta reacción
comprenden un ataque por parte de la cara Re de la enamina quiral sobre la
cara Si de la imina, siendo ésta de configuración (E), conduciendo al
correspondiente aducto anti mostrado en la Figura 3-8.
Figura 3-8: Explicación de la estereoquímica obtenida para la reacción de Mannich anti-selectiva catalizada por derivados de la L-prolina que funcionan por boqueo estérico (A) y
bloqueo estérico + control por enlace de hidrógeno (B). PG: grupo protector.
32 J. Franzén, M. Marigo, D Fielenach, T. C. Wabnitz, A. Kjærsgaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18296-18304. 33 a) I. Ibrahem, A. Córdova, Chem. Commun. 2006, 1760-1762. b) S. Mitsumori, H. Zhang, P. H.-Y. Cheong, K. N. Houk, F. Tanaka,C. F. Barbas III, J. Am. Chem. Soc. 2006, 128, 1040-1041.

Reacción de Mannich anti-Selectiva
115
Esta pauta se cumple en la mayoría de los casos, es decir que la L-
prolina y catalizadores derivados que actúan por enlace de H conducen a
aductos sin, mientras que las aminas derivadas del prolinol, que actúan por
impedimento estérico, conducen mayoritariamente a aductos anti.
También en 2005, Maruoka describió una reacción de Mannich anti-
selectiva utilizando como catalizador la amina secundaria derivada del binaftol
69 (Esquema 3-11),34 que ya había sido previamente descrito para la reacción
aldólica por el mismo autor.35 Este catalizador mejora en este caso la
diastereoselectividad del proceso, con valores superiores a 90:10 en todos los
casos con una carga de tan solo 1 mol%.
Esquema 3-11: Reacción de Mannich anti-selectiva catalizada por 69, Maruoka, 2005.34
Con posterioridad se han descrito otros organocatalizadores de tipo
amina secundaria36 para la reacción de Mannich anti-selectiva, de los cuales
los más representativos descritos con anterioridad a nuestro trabajo se
recogen en la Tabla 3-1. Como se observa en ésta, todos los métodos se
restringen al empleo de iminas reactivas, como aquéllas derivadas de
glioxilatos. La extensión del método a iminas menos reactivas se ha mostrado
problemática y tardía.
34 T. Kano, Y. Yamaguchi, O. Tokuda, K. Maruoka, J. Am. Chem. Soc. 2005, 127, 16408-16409. 35 T. Kano, J. Takai, O. Tokuda, K. Maruoka, Angew. Chem. Int. Ed. 2005, 44, 3055-3057. 36 a) Ver ref. 21a y b. b) T. Kano, Y. Hato, K. Maruoka, Tetrahedron Lett. 2006, 47, 8467-8469. c) T. Kano, Y. Hato, A. Yamamoto, K. Maruoka, Tetrahedron 2008, 64, 1197-1203. d) M. Pouliquen, J. Blanchet, M.-C. Lasne, J. Rouden, Org. Lett. 2008, 10, 1029-1032. e) H. Zhang, S. Mitsumori, N. Utsumi, M. Imai, N. García-Delgado, M. Mifsud, K. Albertshofer, P. H.-Y. Cheong, K. N. Houk, F. Tanaka, C. F. Barbas III, J. Am. Chem. Soc. 2008, 130, 875-886.

Capítulo 3
116
Tabla 3-1: Aminas secundarias como catalizadores para la reacción de Mannich anti-selectiva descritas con anterioridad a nuestro trabajo:
O
R2
+
Autor(año) Cat. Rto.(%) (anti:sin) ee(%)
Córdova(2006)33a 45-75 93:7-95:5 97-99
R3Dadores
R1Cat.
NH
Ph
OTMSPh Aldehídos CO2Et
NH
CO2H
Me
Barbas(2006)33b
Aldehídos CO2R 54-92 94:5-98:2 98-99
Maruoka(2006)36b
(2008)36cNH
R2R1HN
Aldehídos,Cetonas
CO2Et 56-99 83:17-95:5*
Blanchet(2008)36d
NH
NHTfAldehídos,Cetonas
CO2Et 56-95 80:20-95:5 60-99
NH
CO2H
Barbas(2008)36e
Aldehídos,Cetonas
CO2R 65-96 90:10-95:5 82-99
anti sin
*Enantiómero mayoritario anti (2S, 3R).
R1: Tf, Nf, R2: H35b
R1: Tf, R2: NHTf35c
90-99
CO2R3R1
R1
HNO
OMe
CO2R3R1
R1
HNO
OMe
+N
R3
MeO
Aparte de estos ejemplos con aminas secundarias, Barbas III y
colaboradores han descrito reacciones de Mannich anti-selectivas utilizando
como catalizadores aminas primarias, como aminoácidos naturales o
derivados (Esquema 3-12).37 Además, las iminas utilizadas en este caso son
aromáticas, siendo éste uno de los pocos ejemplos descritos con iminas poco
reactivas.
37 S. S. V. Ramasastry, H. Zhang, F. Tanaka, C. F. Barbas, J. Am. Chem. Soc. 2007, 129, 288-289.

Reacción de Mannich anti-Selectiva
117
Esquema 3-12: Aminas primarias que catalizan la reacción de Mannich anti-selectiva de tres componentes, Barbas III, 2007.37
Tras el inicio de nuestros estudios en la materia, han sido publicados
diferentes trabajos que describen reacciones de Mannich anti-selectivas
asimétricas mediante organocatálisis.38
Así, en 2008 Melchiorre describió el primer caso de reacción de
Mannich anti-selectiva con N-Boc y N-Cbz-iminas aromáticas. Las iminas se
generan in situ a partir de las correspondientes α-amidosulfonas en presencia
del catalizador 4 y 5 equivalentes de KF como base, con resultados
notables.38a (Esquema 3-13).
Poco más tarde, el mismo grupo extendió el trabajo con diferentes N-
Boc-iminas aromáticas y aldehídos, esta vez en presencia del catalizador 3
(Esquema 3-13).39 Cabe resaltar que los mejores resultados de
diatereoselectividad se obtienen al llevar a cabo la reacción con el aldehído
ramificado (R= iPr).
38 Para estudios de reacción de Mannich anti-selectiva asimétrica con iminas derivadas de glioxilatos paralelos o posteriores a la presente Tesis, ver: a) C. Gianelli, L. Sambri, A. Carlone, G. Bartoli, P. Melchiorre, Angew. Chem. Int. Ed. 2008, 47, 1-4. b) H. Zhang, Y. Chuan, Z. Li, Y. Peng, Adv. Synth. Catal. 2009, 351, 2288-2294. k) J. Pietruszka, R. C. Simon, Chem. Eur. J. 2010, 16, 14534-14544. 39 P. Galzerano, D. Agostino, G. Bencivenni, L. Sambri, G. Bartoli, P. Melchiorre, Chem. Eur. J. 2010, 16, 6069-6076.

Capítulo 3
118
O
R+
NHPG
SO2PhOHC
R
Ar
NHPGKF (5 equiv.)
ArH
CHCl3
NH OTMS
ArAr
Ar: 3,5-(CF3)2C6H3
4 (10 mol%)38a
NH OTMS
PhPh
Catalizador R PG Rto. (%) dr(anti:sin) ee (%)
BocMe
Me
87
CBz 94
92:8
86:14
98
94
BocMe, Bu, Bn 76-94 83:17 76-96
Boc, CBz 60-95 86:14-92:8 84-99iPr3 (10-20 mol%)39
Esquema 3-13: Reacción de Mannich anti-selectiva entre N-Boc y N-Cbz-iminas y aldehídos catalizada por 4 y 3, Melchiorre, 2008 y 2010.38a, 39
Posteriormente Peng obtiene resultados parecidos para la misma
reacción pero con las iminas preformadas y aldehídos ramificados como
dadores, utilizando la pirrolidina bifuncional 70 como catalizador (Esquema 3-
14).40
O
R1+
NHPGOHC
R1Ar
NHPG
ArH
CHCl3
PG: Boc, CBz
Rto.: 70-95%
(anti:sin): 89:11-96:4
ee >98%
NH
HN
HN
OTBDPS
F3C
CF3
S
R270 (10 mol%)
R2
Esquema 3-14: Reacción de Mannich anti-selectiva entre N-Boc y N-Cbz-iminas y aldehídos ramificados catalizada por 70, Peng, 2011.40
Por su parte, el grupo de Maruoka describió en 2009 la reacción de
Mannich anti-selectiva entre aldehídos y N-Boc-iminas aromáticas, incluyendo 40 Y.-M. Chuan, G.-H. Chen, J.-Z. Gao, H. Zhang, Y.-G. Peng, Chem. Commun. 2011, 47, 3260-3262.

Reacción de Mannich anti-Selectiva
119
un primer ejemplo para una N-Boc-imina alifática y utilizando como catalizador
la amina secundaria derivada del binaftol 69 (Esquema 3-15).41
O
R1
+NBoc
OHC
R1
R2
NHBoc1-10 mol% 69
R2H
CHCl30 ºC
Rto.: 66-93%,
(anti:sin): 88:12-96-4
ee >98%
NH
NHTf
69
R2= Ar, ciclohexilo
Esquema 3-15: Reacción de Mannich anti-selectiva entre N-Boc iminas y aldehídos catalizada por 69, Maruoka, 2009.41
Muy recientemente y posteriormente a nuestros trabajos, Hayashi y
colaboradores publican la reacción de Mannich asimétrica enantioselectiva
entre N-tosil-iminas aromáticas y alifáticas formadas in situ y el propanal
catalizada por 442 (Esquema 3-16).
Esquema 3-16: Reacción de Mannich anti-selectiva entre N-tosil iminas aromáticas y alifáticas y el propanal, Hayashi, 2011.42
41 T. Kano, Y. Yamaguchi, K. Maruoka, Angew. Chem. Int. Ed. 2009, 48, 1-4. 42 T. Urushima, H. Ishikawa, Y. Hayashi, Chem. Eur. J. 2011, 17, 8273-8276.

Capítulo 3
120
3.2.4. Reacción de Mannich anti-selectiva con otros catalizadores
También se han descrito reacciones de Mannich anti-selectivas
promovidas por organocatalizadores de tipo ácido o base de Brønsted o
catalizadores ambifuncionales.
Uno de los ejemplos más destacables, descrito por Schaus y
colaboradores, implica la reacción de Mannich entre β-cetoésteres y varias N-
acil-iminas 71 (Esquema 3-17).43 En este caso la reacción es promovida por la
cinconina (derivado de la cincona), que actúa como catalizador ambifuncional
(base de Lewis-ácido de Brønsted quiral).
Esquema 3-17: Reacción de Mannich anti-selectiva catalizada por cinconina, Schaus, 2006.43
Por otro lado, los ácidos de Brønsted que han resultado más exitosos
en reacciones de Mannich anti-selectivas son los derivados del ácido fosfórico
72 y 73, utilizados por Gong para la reacción de tres componentes entre
cetonas cíclicas, anilina y aldehídos aromáticos (Esquema 3-18).44
43 S. Lou, B. M. Taoka, A. Ting, S. E. Schaus, J. Am. Chem. Soc. 2006, 127, 11256-11257. 44 Q.-X. Guo, H. Liu, C. Guo, S.-W. Luo, Y. Gu, L.-Z. Gong, J. Am. Chem. Soc. 2007, 129, 3790-3791.

Reacción de Mannich anti-Selectiva
121
O
OP
OH
O
X
X72, X: Cl73, X: H
X
OO
ArHPhNH2
X
O
Ar
HNPh
+ +
0.5 mol% 72 ó2 mol% 73
Rto.: 67-99%(anti:sin): 77:23-98:2ee: 80-98%
Esquema 3-18: Reacción de Mannich anti-selectiva de tres componentes catalizada por ácidos
de Brønsted quirales, Gong, 2007.44
3.2.5. Limitaciones y objetivos
A pesar de los numerosos estudios, una limitación común a los
métodos descritos con antelación a nuestros trabajos es que solamente los α–
imino ésteres parecen ser lo suficientemente reactivos para dar los aductos de
Mannich de configuración anti con eficiencia tanto química como
estereoquímica. Las únicas excepciones, como se ha visto en las
aportaciones anteriores, se han descrito durante estos dos últimos años,
simultáneamente al transcurso de la presente Tesis Doctoral.
En base a estas consideraciones nos propusimos indagar sobre la
posibilidad de llevar a cabo reacciones de Mannich anti-selectivas con iminas
diferentes a las derivadas del glioxilato y congéneres, con el objeto de ampliar
el potencial de la reacción de Mannich. Más específicamente, el planteamiento
inicial consistió en realizar un estudio de la reacción de Mannich entre
aldehídos e iminas aromáticas con un grupo activante en el nitrógeno,
utilizando diferentes derivados de la pirrolidina como catalizadores.

Capítulo 3
122
3.3. Reacción de Mannich anti-selectiva entre aldehídos y N-
tosil iminas
3.3.1. Hipótesis de trabajo y antecedentes
En los sistemas catalíticos mencionados anteriormente para la
reacción anti-Mannich con aminas como catalizadores (vía enamina) se
propone el estado de transición representado en el modelo A (Fig. 3-9), en el
que se da una sinergia entre la coordinación, o enlace de H, entre un grupo
dador del catalizador y el átomo de nitrógeno imínico y las interacciones
estéricas del grupo voluminoso de la pirrolidina, resultando así un sistema
eficaz en el control de la estereoquímica.
Por otro lado, habiéndose demostrado que el grupo carboxílico de la
prolina (o derivados) actúa como activante de la imina en las reacciones de
Mannich,45 nosotros pensamos que la participación de un ácido de Brønsted
externo podría tener un efecto similar en este sentido, además de dotar de
una mayor flexibilidad a la estructura anteriormente descrita, tal y como se ve
reflejado en el modelo B (Figura 3-9).
Figura 3-9: Estados de transición propuestos para la reacción de anti-Mannich.
La validez de esta propuesta en el contexto de las reacciones de
adición conjugada de aldehídos a nitroalquenos ya había sido demostrada en
nuestro grupo recientemente, mediante el uso de la trans-4-hidroxiprolinamida
45 Para más información sobre el efecto de los ácidos de Brønsted en la activación de iminas, ver: a) Y. Hayasi, M. Urushima, T. Shogi, T. Uchimaru, I. Shiina, Adv. Synth. Catal. 2005, 347, 1595-1604. b) Ver ref. 17. c) Y. Hayashi, T. Okano, T. Itoh, T. Urushima, H. Ishikawa, T. Uchimaru, Angew. Chem. Int. Ed. 2008, 47, 9053-9058. Para la reacción de Mannich catalizada por ácido pipecólico, ver: d) P. H.-Y. Cheong, H. Zhang, R. Thayumanavan, F. Tanaka, K. N. Houk, C. F. Barbas III, Org. Lett. 2006, 8, 811-814.

Reacción de Mannich anti-Selectiva
123
74 (Esquema 3-19).46 Posteriormente otros autores publicaron distintos
estudios con otros catalizadores de estructura parecida, todos ellos
poseedores de este grupo hidroxilo (Esquema 3-19), demostrándose un papel
crucial de dicho grupo en términos de eficiencia de reacción y
estereoselectividad para distintas transformaciones.47
NH
HOO
N
PhPh
NH
HOO
HN NH
HOO
NH
Ph
Ph
OHPh
NH
HOO
N
N
NH
HOO
NH
OH
Me
Me
O
R
+ R1 NO2NO2
O
R
R1
(5-10 mol%)
DCM, 0 ºC, 20 h
Rto.: 70-90%
(sin:anti) > 95:5
ee > 92%
74
75 76
7778
Feng, 200847a
reacción de Biginelli
Nakano/Takeshita, 200947b
adición conjugada de aldehídos a
nitroalquenos, reacción aldólica
Gao, 200947c
adición conjugada de
aldehídos a nitroalquenos
HH
Chen, 200947d
adición conjugada de
aldehídos a nitroalquenos
Esquema 3-19: Adición conjugada de aldehídos a nitroalquenos promovida por 74, Palomo,
2006.46 Otros catalizadores descritos portadores del grupo hidroxilo en la misma posición.
46 C. Palomo, S. Vera, A. Mielgo, E. Gómez-Bengoa, Angew. Chem. Int. Ed. 2006, 45, 5984-5987. 47 a) J. Xing, L. Chang, Z. Hou, D. Shang, X. Liu, X. Feng, Chem. Eur. J. 2008, 14, 3177-3181. b) Y. Okuyama, H. Nakano, M. Watanabe, M. Takeshita, K. Uwai, C. Kabuto, E. Kwon, Tetrahedron Lett. 2009, 50, 193-197. c) C.-Q. Cheng, Z. Bian, Y.-B. He, F.-S. Han, C.-Q. Kang, Z.-L. Ning, L. X. Gao, Tetrahedron: Asymmetry 2009, 20, 1753-1758. d) R. J. Reddy, H.-M. Kuan, T.-Y. Chou, K. Chen, Chem. Eur. J. 2009, 15, 9294-9298.

Capítulo 3
124
3.3.2. Resultados y discusión
Con el fin de establecer la viabilidad del modelo B propuesto
anteriormente (Fig 3-9, p. 122) se seleccionó la reacción de Mannich entre los
aldehídos y las iminas representados en el Esquema 3-20, y los
correspondientes aductos se aislaron en forma de aminoalcoholes tras
reducción in situ con borohidruro sódico.
Esquema 3-20
3.3.2.1. Búsqueda de condiciones de reacción
Para un primer estudio se decidió tomar como modelo la reacción
entre el butanal 17B y la imina 82a en DMF como disolvente, a -60 ºC y en
presencia de un 20% del catalizador 74 (Tabla 3-2, entrada 4), que como se
ha mencionado anteriormente resultó ser efectivo en adiciones conjugadas a
nitroalquenos. Bajo estas condiciones no se observó reacción. Cuando el
mismo experimento se efectuó en presencia de ácido p-nitrobenzoico (PNBA),
que resultó ser el ácido más efectivo de todos los ensayados tal y como se
comentará con posterioridad, se obtuvo el esperado aducto de Mannich con
muy buen rendimiento (88%) y elevada anti-diastereoselectividad (anti:sin
96:4), pero un moderado exceso enantiomérico (75%). A la vista de estos
datos se seleccionaron distintos catalizadores estructuralmente análogos a 74

Reacción de Mannich anti-Selectiva
125
pero que diferían en la presencia o no del grupo hidroxilo en 4 y en la
naturaleza del sustituyente en α al nitrógeno pirrolidínico, con el objeto de
poder así evaluar la influencia de cada uno de estos elementos en el resultado
estereoquímico de la reacción. Los resultados más relevantes se recogen en
la Tabla 3-2.
Tabla 3-2: Evaluación de catalizadores y temperatura para la reacción de Mannich anti-selectiva entre 17B y 82a.
O
Et
+
NTos
Et
NHTos
HO
1) 20 mol% cat.20 mol% PNBA
2) NaBH4EtOH,-40 ºC
DMF
82aMe Me
anti-87Ba
H
17B
Et
NHTos
HO
Mesin-87Ba
+
Entr. Cat. T
(ºC) Conv.[b]
(%) Rto.[c]
(%)
(anti:sin)[b] ee[d] (%)
1 84 -20 n.d.[e] 37 55 : 45 47[f,g]
2 -20 n.d.[e] 70 75 : 25 67[g]
3 -60 >99 29 77 : 23 83[g]
4
74
-60
>99
88
96 : 4
75
5 83 (R: nHex) -20 >99 99 80 : 20 76[f]
6 83 (R: nHex) -20 >99 92 76 : 24 86
7 83 (R: nHex) -60 >99 77 98 : 2 95
8 85 (R: Me) -60 >99 75 93 : 7 92
9
86 (R: nPr) -60 50 43 90 : 10 93
10 4 R:(3,5-CF3)2C6H4 -60 0 0 n.d. n.d.[h]
11 3 (R: Ph) -60 40 15 75 : 25 63
12 42 R: nHex -40 >99 98 78 : 22 67
13
42 R: nHex -60 >99 24 78 : 22 58 [a] Reacciones llevadas a cabo a escala 0.5 mmol en DMF (2 mL) y utilizando los distintos catalizadores (20 mol%), PNBA (20 mol%) y 3 equiv. de 82a. [b] Determinado por espectroscopía 1H-RMN del crudo. [c] Rendimiento del aducto 87Ba aislado tras cromatografía de columna flash. [d] Determinado por análisis de HPLC. [e] n.d.: No determinado. [f] No se usó ácido de Brønsted. [g] Reacciones con 3 equiv. de aldehído. [h] Tiempo de reacción: 3 días.

Capítulo 3
126
Como se observa, mientras que la α-hidroxipirrolidina 84 sola resulta
poco eficaz a -20 ºC (entrada 1 en la tabla), en combinación con 20 mol% de
ácido 4-nitrobenzoico (PNBA) tanto el rendimiento como la diastereo- y
enantioselectividad mejora sensiblemente (entrada 2), lo que nos demuestra la
importancia de la presencia del ácido de Brønsted en la reacción. En estas
condiciones la reacción puede llevarse a cabo incluso a temperaturas bajas (-
60 ºC) con un ligero aumento de la selectividad a expensas de un menor
rendimiento (entrada 3). Como se observa en la tabla, la enantioselectividad
mejora con las pirrolidinas 83, 85 y 86, portadoras todas ellas de grupos más
voluminosos en α que 74, siendo la 83 la óptima, ya que conduce a valores de
diastereoselectividad de 98:2 y exceso enantiomérico de 95% (entrada 7). De
nuevo se comprobó que el ácido juega un papel crucial ya que sin él la
reacción no se da a temperaturas inferiores a -20 ºC (entrada 5). Por último,
para evidenciar la importancia del grupo hidroxilo en la estructura del
catalizador, se evaluaron distintas pirrolidinas (3, 4 y 42) carentes de dicho
grupo (entradas 10-13), de las que 3 y 4 se habían utilizado anteriormente
para esta reacción con iminas derivadas del glioxilato. Como se puede
apreciar, los α,α-diarilprolinoles 3 y 4 se mostraron muy poco o incluso nada
reactivos (entradas 10 y 11), mientras que con 42 se obtienen resultados más
moderados tanto de diastereo- como de enantioselectividad.
Por tanto, en un primer análisis de estos resultados puede concluirse
que los tres elementos, el ácido de Brønsted externo, el sustituyente
voluminoso en α de la pirrolidina y el hidroxilo en la posición γ del anillo son
cruciales para lograr niveles satisfactorios de eficiencia química y
estereoquímica.
Seguidamente se decidió estudiar la influencia de distintos aceptores
para la reacción, para lo cual se sintetizó una serie de iminas derivadas de la
p-metil anilina, portadoras de grupos activantes sulfonilo48 y alcoxicarbonilo en
el átomo de nitrógeno.
48 Para el uso de N-sulfonil-iminas en la reacción de Mannich, ver: a) K. Juhl, N. Gathergood, K. A. Jørgensen, Angew. Chem. Int. Ed. 2001, 40, 2992-2995. b) L. Bernardi, A. S. Gthelf, R. G. Hazell, K. A. Jørgensen, J. Org. Chem. 2003, 68, 2583-2591. c) H. Morimoto, T. Yoshino, T. Yukawa, G. Lu, S. Matsunaga, M. Shibasaki, Angew. Chem. Int. Ed. 2008, 47, 9125-9129. d) J. Hernández Toribio, R. Gómez Arrayas, J. C. Carretero, J. Am. Chem. Soc. 2008, 130, 16150-16151. e) D. Uraguchi, Y. Ueki, T. Ooi, J. Am. Chem. Soc. 2008, 130, 14088-1489.

Reacción de Mannich anti-Selectiva
127
Como indican los resultados obtenidos en la Tabla 3-3, una
comparación de las cuatro iminas seleccionadas para la reacción con butanal
en presencia de un 20 mol% del catalizador 83 y un 20 mol% de ácido
benzoico fue reveladora. Así, ni 79a ni 80a condujeron a los aductos
deseados, mientras que con 81a y 82a se obtuvieron buenos rendimientos,
relaciones anti:sin del orden de 90:10 y enantioselectividades de 90 y 88%
respectivamente. De entre las tres sulfonil-iminas probadas, la única que no
dio la reacción fue 80a, posiblemente debido al impedimento estérico (grupo
protector mayor que 81a y 82a). De entre las dos iminas reactivas 81a y 82a
decidimos estudiar más profundamente la reacción de Mannich con la imina
82a por comodidad de preparación con respecto a la 81a.
Tabla 3-3.: Estudio comparativo del grupo activante de la imina en la reacción con butanal (17B) catalizada por 83:
O
Et
+
+NaBH4
H
17BEtOH
NRNHROH
EtMe
NHROH
EtMe
NH
HOnHex
nHexOTMS
83(20 mol%)
PhCOOH(20 mol%)
DMFanti sin
Me
Entrada R Imina Tª (ºC) Rto. (%) (anti:sin) ee (%)
1 Boc 79a -40/-60 0 n.d. n.d.
2 MesSO2 80a -40/-60 0 n.d. n.d.
3 3-PyrSO2 81a -60 84 91:9 88
4 Tos 82a -60 88 (77) 90:10 (98:2) 90(96)
Nota: los datos en paréntesis son resultados de la reacción con 20 mol% de ácido p-nitrobenzoico. [a] Reacciones llevadas a cabo a escala 0.5 mmol durante 20 h en DMF (2 mL) y 3 equiv. de imina. n.d.: No determinado.
Por otro lado se efectuaron experimentos variando la naturaleza del
ácido benzoico para la reacción entre 17B y 82a. Se utilizaron para ello los
ácidos p-nitrobenzoico (pKa= 3.4 frente a pKa= 4.2 del ácido benzoico) y p-
metoxibenzoico (pKa= 4.5). De manera general se observó que al disminuir la
acidez del ácido se requerían tiempos de reacción más prolongados.
Asimismo también se observaron variaciones leves en los resultados

Capítulo 3
128
estereoquímicos, siendo el ácido p-nitrobenzoico el que mejores resultados
proporcionó (entrada 4, Tabla 3-3). Por ello se seleccionó dicho ácido para los
ensayos posteriores.
3.3.2.2. Alcance de la reacción
Una vez establecida la validez del sistema catalítico formado por la
hidroxipirrolidina 83 y el ácido p-nitrobenzoico, la generalidad de la reacción
de Mannich anti-selectiva se exploró con diferentes aldehídos y N-tosil iminas
aromáticas (Esquema 3-21) bajo las condiciones optimizadas (Tabla 3-4). Los
resultados muestran que la reacción es compatible con diversos aldehídos
alifáticos, tanto lineales como ramificados (entrada 10) así como con N-tosil-
iminas49 con grupos aromáticos de diversa naturaleza. En todos los casos se
obtuvieron buenos rendimientos y excelentes diastereo- y
enantioselectividades. La utilización de relaciones estequiométricas
aldehído:imina 3:1 ó 1:3 no alteró el resultado estereoquímico de la reación
aunque sí generó variaciones moderadas en el rendimiento, siendo éste
generalmente superior al utilizar exceso de aldehído (entradas 1, 2; 3,4 y 6,7).
NH
nHexnHex
OTMS
HO
O
R+
NTos
ArR
Ar
NHTos
HO
1) 83 (20 mol%)20 mol% PNBA
2) NaBH4
-60 ºC, DMF
EtOH
H
anti-8717 82
R1: Me
R1: Et
R1: nPent
R1: iPr
17A
17B
17E
17H
R2/R3: Tos/4-Me-C6H4 82a
R2/R3: Tos/Ph 82b
R2/R3: Tos/4-MeO-C6H4 82c
R2/R3: Tos/3-Me-C6H4 82d
R2/R3: Tos/4-Cl-C6H4 82e
Aldehídos: Iminas:
R
Ar
NHTos
HO
sin-87
+
Esquema 3-21
49 Se ha demostrado que la reacción es igualmente eficiente con N-nosil-iminas, trabajo llevado a cabo por Itziar Otazo en el contexto de su Tesis Doctoral. Ver, también: E. Gómez-Bengoa, M. Maestro, A. Mielgo, I. Otazo, C. Palomo, I. Velilla, Chem. Eur. J. 2010, 16, 5333-5342.

Reacción de Mannich anti-Selectiva
129
Tabla 3-4: Alcance de la reacción de Mannich anti-selectiva con diferentes aldehídos y N-tosil-iminas:
Entrada
Producto
Aldehído:imina
Rto.(%)[b]
(anti:sin)[c]
ee(%)[d]
1 3:1 78 96:4 97
2
87Aa
1:3 68 95:5 97
3 3:1
77(41)[e]
96:4
>99
4
87Ab
1:3 40 n.d. n.d.
5
87Bb
1:3 63 98:2 86
6 3:1 68 94:6 91
7
87Bc
1:3 67 95:5 93
8
87Bd
1:3 60 93:7 91
9
87Ee
3:1 76 87:13 91[f]
10
87Ha
3:1
76
97:3
>99
[a] Reacciones llevadas a cabo a escala 0.5 mmol en DMF (2 mL) y utilizando el catalizador 83 (20 mol %), ácido p-nitrobenzoico (20 mol %). [b] Rendimiento del alcohol 87 aislado tras cromatografía. [c] Determinado por espectroscopía 1H-RMN del crudo. [d] Determinado por análisis de HPLC. [e] Rendimiento entre paréntesis a escala 10 mmol. [f] Tª: -40 ºC. n.d.: no determinado.
Cabe destacar que la reacción se pudo llevar a cabo a una escala de
10 mmol (entrada 3) sin merma en la estereoselectividad, aunque con un
moderado descenso en el rendimiento. Por otra parte conviene señalar que el

Capítulo 3
130
catalizador pudo recuperarse intacto y con un rendimiento del orden de 80%
tras cromatografía de columna flash.
La configuración absoluta del aducto 87Aa50 se estableció como (1S,
2R) por análisis de rayos X (Fig. 3-10). La configuración del resto de aductos
se asignó por analogía y asumiendo un mecanismo de reacción uniforme.
Figura 3-10: Representación del cristal hallado del aducto 87Aa.50
3.3.2.3. Cálculos computacionales
Los resultados obtenidos experimentalmente estarían en concordancia
con el modelo de tres componentes propuesto inicialmente (modelo B en la
Figura 3-9). Con la finalidad de obtener información sobre el mecanismo de
reacción, se llevó a cabo un estudio computacional de tipo DFT, por parte del
Dr. Gómez-Bengoa. A continuación se muestran brevemente los resultados
más relevantes.
50 Los datos cristalográficos del compuesto 87Aa se han depositado en el Cambridge Crystallographic Data Centre, CCDC Nº 726915. Estos datos se pueden obtener online (http://www.ccdc.cam.ac.uk) libre de cargos (o desde el Cambridge Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: (+44)1223-336-033, e-mail: depositccdc.cam.ac.uk).
Me
NHTos
HO
Me87Aa

Reacción de Mannich anti-Selectiva
131
Figura 3-11: Representación de los estados de transición optimizadas con B3LYP/6-31-G* para la reacción entre la imina 88 y la enamina derivada de 17A y la amina 85 ó 89 en
presencia y ausencia de ácido benzoico. Los valores individuales (kcal/mol) a nivel B3LYP/6-311++G** se muestran en cursiva.
Se tomó como modelo la reacción entre el propanal y la imina 88
catalizada por 85 ó 89, asumiendo que la reacción procede mediante un
mecanismo vía enamina. En un primer estudio en ausencia de ácido de
Brønsted externo y presencia de la 4-hidroxipirrolidina 85 como catalizador, se
observó que las energías de activación eran de similar magnitud para los
estados de transición anti o sin, lo que implicaría la formación equimolecular
de diastereómeros (TS1-anti frente a TS1-sin en la Figura 3-11). En un
segundo estudio, en el que se reemplazó el catalizador 85 por 89 (carente de
grupo hidroxilo) y se contempló la participación de un ácido de Brønsted

Capítulo 3
132
externo, la interacción entre el protón del grupo carboxilo y el átomo de
nitrógeno imínico (TS2) contribuye a estabilizar los estados de transición. Sin
embargo, la diferencia de energía entre el TS-anti (10.8 kcal/mol) y sin
(13.0/12.5 kcal/mol) es pequeña como para justificar los niveles de
diastereoselectividad sin/anti observados experimentalmente. Finalmente, la
combinación de las dos funciones, el grupo OH del catalizador y el ácido de
Brønsted externo, condujo a los estados intermedios más estables. De entre
las dos estructuras posibles, TS3-anti y TS3-sin, la diferencia energética era
de 3 kcal/mol a favor de la anti, lo que concuerda perfectamente con los datos
experimentales obtenidos.
3.4. Reacción de Mannich de aldehídos e iminas sencillas
(Bases de Schiff)
Una base de Schiff o azometino (C en Fig. 3-12) es una imina que
contiene un átomo de nitrógeno conectado a un grupo arilo o alquilo, lo que
hace que ésta sea menos electrofílica que las N-tosil-iminas anteriormente
comentadas, y, en consecuencia, menos reactiva en la reacción de Mannich.
Por ello no resulta sorprendente que la reacción de Mannich
organocatalítica y enantioselectiva con estas bases de Schiff esté poco
desarrollada. Es más, en el momento de iniciar nuestro estudio, los escasos
ejemplos existentes eran todos sin-selectivos. Por ello nos propusimos
investigar si nuestro modelo (pirrolidina + ácido de Brønsted) sería lo
suficientemente activo y/o selectivo para poder llevar a cabo reacciones anti-
selectivas con dichos sustratos.
N
EWG
RN
R
EWGN
R
R'
Iminas inherentementereactivas
Ej:
Iminas con grupoactivante en el N
Ej:
Bases de Schiff
A B C
N
EtO2C
PGN
Ar
Boc Ej:N
Ar
Ar
Figura 3-12: Distintas iminas utilizadas para la reacción de Mannich.

Reacción de Mannich anti-Selectiva
133
3.4.1. Reacción de Mannich con iminas N-aromáticas
En base al objetivo anteriormente planteado se seleccionó la reacción
de Mannich entre los aldehídos y las iminas representadas en el Esquema 3-
22 utilizando como sistema catalítico la combinación pirrolidina/ácido de
Brønsted, modelo que había resultado efectivo con las N-tosil-iminas
anteriormente comentadas.
Esquema 3-22
En una primera instancia se seleccionó la reacción entre el propanal
17A y una N-p-metoxifenil-imina aromática (90a) con diferentes
combinaciones de pirrolidinas quirales tanto mono- como bifuncionales y ácido
p-nitrobenzoico a -60 ºC. Satisfactoriamente se observó que la reacción
transcurría en todos los casos tras verificar nuevamente la necesidad de un
ácido de Brønsted.51 El disolvente idóneo en este caso resultó ser el THF
debido a la mejor solubilidad de este tipo de iminas en él. En otros disolventes
comunes, como diclorometano o dietil éter, las iminas resultaron ser
parcialmente solubles o insolubles a bajas temperaturas. En la Tabla 3-5 se
muestran los resultados correspondientes.
51 Se comprobó que en presencia de uno de los catalizadores (3) y en ausencia de ácido de Brønsted la reacción de Mannich no tenía lugar en un rango de temperaturas de 0 a -60 ºC.

Capítulo 3
134
Tabla 3-5: Evaluación de catalizadores para la reacción de Mannich anti-selectiva con N-aril iminas:52
Entrada Cat. dr (anti:sin) ee (%)
1 80:20 96
2 NH OTMS
RR
HO R= nHex, 83
R= Ph, 91
81:19 99
3 81:19 97
4 NH OTMS
RR
R= nHex, 42
R= Ph, 3 79:21 99
5 95:5 98
6 NH OTPS
RR
R= nHex, 92
R= Ph, 39 82:18 99
Como se observa en la tabla, la enantioselectividad fue excelente
(>95%) para todos los casos estudiados, mientras que la selectividad anti:sin
fue del orden de 80:20 con todos los catalizadores evaluados salvo 92, el cual
condujo a un valor remarcablemente mejor de diastereoselectividad (95:5)
(entrada 5 en la Tabla 3-5). De este primer estudio parecen desprenderse las
siguientes conclusiones:
a) La presencia o ausencia del grupo hidroxilo en la posición 4 de
la pirrolidina no parece variar el resultado estereoquímico de la
reacción.
b) La naturaleza alifática/aromática de R del catalizador parece
influir notablemente en la diastereoselectividad del proceso
(entradas 5, 6).
52 Estudio comparativo llevado a cabo por la doctoranda Itziar Otazo en el contexto de su Tesis Doctoral. Conversiones > 80% en todos los casos.

Reacción de Mannich anti-Selectiva
135
c) La naturaleza del grupo OSiR3 en este caso también parece
ser un factor influyente.
Tras comprobar que 92 era el catalizador idóneo, se llevó a cabo la
reacción con una batería de iminas aromáticas y alifáticas provenientes de
distintas anilinas y aldehídos, incluído alguno funcionalizado (17K). Como
puede verse en los datos representados en el Esquema 3-23, los aductos de
Mannich se aislaron en forma de alcoholes con buenos rendimientos,
relaciones diastereoméricas del orden de 95:5 a favor del isómero anti y
excelentes enantioselectividades en todos los casos (Esq. 3-23).

Capítulo 3
136
[a] Reacciones llevadas a cabo a escala 0.5 mmol con 3 equiv. de aldehído 17. El rendimiento de los aductos 93 aislados fue determinado tras cromatografía de columna flash. La relación anti:sin se determinó por espectroscopía 1H-RMN del crudo. La relación enantiomérica se determinó por análisis de HPLC. Esquema 3-23: Alcance de la reacción de Mannich anti-selectiva catalizada por 92 para iminas N-aromáticas.
3.4.2. Reacción de Mannich con iminas propargílicas
Animados por el éxito de esta metodología al emplear iminas
derivadas de anilinas y aldehídos aromáticos o alifáticos, decidimos explorar la
viabilidad del método con un grupo de iminas especiales, las iminas derivadas
de aldehídos porpargílicos.

Reacción de Mannich anti-Selectiva
137
Las unidades de propargilamina son estructuras moleculares muy
versátiles en química orgánica sintética, útiles para la obtención de productos
naturales,53 fármacos54 y/o pesticidas,55 además de estar presentes en
muchos compuestos bioactivos.56 Sin embargo, existen escasas metodologías
para su síntesis; entre ellas destacan la adición de alquinilmetales a iminas57
(Fig. 3-13, ruta a) y las reacciones de 1,2-adición de reactivos alquilmetálicos
a iminas propargílicas58 (Fig. 3-13, ruta b).
NHR1
R3
R2
*
NR1
R3R2
M NR1
R2
Nua b
(Nu: R3M)
Unidad depropargilamina
Figura 3-13
La ruta a generalmente es aplicable solo a iminas no enolizables (R3:
arilo, sec- o terc-alquilo, CO2R’), mientras que los reactivos organometálicos
que se requieren en la ruta b son típicamente dialquilzincs no
53 a) T. Yoon, M. D. Shair, S. J. Danishefsky, G. K. Shulte, J. Org. Chem. 1994, 59, 3752-3754. b) B. Jiang, M. Xu Angew. Chem. Int. Ed. 2004, 43, 2543-2546. c) J. Fleming, J. Du Bois J. Am. Chem. Soc. 2006, 128, 3926-3927. 54 Para ejemplos seleccionados, ver: a) M. Shibasaki, Y. Ishida, G. Iwasaki, T. Iimori, J. Org. Chem. 1987, 52, 3488-3489. b) N. Miyachi, F. Kanda, M. Shibasaki, J. Org. Chem. 1989, 54, 3511-3513. c) A. Hoepping, K. M. Johnson George, J. Flippen-Anderson, A. P. Kozikowski, J. Med. Chem. 2000, 43, 2064-2071. 55 C. Swithenbank, P. J. McNulty, K. L. Viste, J. Agric. Food Chem. 1971, 19, 417-423. 56 a) P. H. Yu, B. Davis, A. A. Boulton, J. Med. Chem. 1992, 35, 3705-3713. b) J. L. Wright, T. F. Gregory, S. P. Kesten, P. A. Boxer, K. A. Serpa, L. T. Meltzer, L. D. Wise, S. A. Espitia, C. S. Konkoy, E. R. Whittemore, R. M. Woodward, J. Med. Chem. 2000, 43, 3408-3419. 57 Para revisiones, ver: a) L. Zani, C. Bolm, Chem. Rev. 2006, 106, 4263-4275. b) B. M. Trost, A. H. Weiss, Adv. Synth. Catal. 2009, 351, 963-983. c) D. Tejedor, S. López-Tosco, F. Cruz-Acosta, G. Méndez-Abt, F- García-Tellado, Angew. Chem. Int. Ed. 2009, 48, 2090-2098. d) G. Blay, A. Monleón, J. R. Pedro, Curr. Org. Chem. 2009, 13, 1498-1539. e) P. de Armas, D. Tejedor, F. García-Tellado, Angew. Chem. Int. Ed. 2010, 49, 1013-1016. Para ejemplos recientes, ver: f) G. Blay, L. Cardona, E. Climent, J. R. Pedro, Angew. Chem. Int. Ed. 2008, 47, 5593-5596. g) Y. Lu, T. C. Johnstone, B. A. Arndtsen, J. Am. Chem. Soc. 2009, 131, 11284-11285. h) J. A. Bishop, S. Lou, S. E. Schaus, Angew. Chem. Int. Ed. 2009, 48, 4337-4404. i) C.-J. Li, Acc. Chem. Res. 2010, 43, 581-590. j) S. Nakamura, M. Ohara, Y. Nakamura, N. Shibata, T. Toru, Chem. Eur. J. 2010, 16, 2360-2362. 58 Para revisiones sobre adiciones 1,2 catalíticas asimétricas a iminas, ver: a) T. Vileivan, W. Bhanthumnavin, Y. Sritana-Anant, Curr. Org. Chem. 2005, 9, 1315-1392. b) G. K. Friestad, A. K. Mathies, Tetrahedron 2007, 63, 2541-2569. c) D. Ferraris, Tetrahedron 2007, 63, 9581-9597. d) K. Yamada, K. Tomioka, Chem. Rev. 2008, 108, 2874-2886. e) R. G. Arrayás, J. C. Carretero, Chem. Soc. Rev. 2009, 38, 1940-1948. f) M. Shimizu, I. Hachiya, I. Mizota, Chem. Commun. 2009, 874-889. g) Y. Mori, J. S. Fossey, M. M. Slater, Chem. Rev. 2011, 4, 2626-2704.

Capítulo 3
138
funcionalizados,59 siendo las dos metodologías mencionadas, dependientes
de reactivos organometálicos como catalizadores.
En vista de la limitación de métodos existentes, resulta atractiva la
posibilidad de obtención de dichas estructuras mediante la reacción de
Mannich con iminas propargílicas, con la consecuente formación adicional de
otro estereocentro (el proveniente del dador cuando R≠ H) en un solo paso
(Fig. 3-14, ruta c).
Figura 3-14
Sin embargo, de entre las numerosas aportaciones para la reacción
de Mannich catalítica enantioselectiva, existe solamente un ejemplo de
Snapper y Hoveyda60 con iminas propargílicas (Esquema 3-24). El método
implica el empleo de enol éteres de sililo y como catalizador una combinación
de una fosfina quiral (94) con acetato de Ag, obteniendo las correspondientes
aminas propargílicas con excesos enantioméricos en el rango de 84-92%. Las
limitaciones y/o inconvenientes de este único ejemplo son: a) dependencia de
un catalizador organometálico, b) formación de un solo centro estereogénico,
c) utilización de enolatos de Si preformados y d) necesidad de iminas con un
sustituyente específico en el N. Los mismos autores extendieron este mismo
estudio a sililoxifuranos como dadores.61
59 a) L. C. Akullian, M. L. Snapper, A. H. Hoveyda, Angew. Chem Int. Ed. 2003, 42, 4244-4247. b) L. C. Akullian, J. R. Porter, J. F. Traverse, M. L. Snapper, A. H. Hoveyda, Adv. Synth. Catal. 2005, 347, 417-425. 60 a) N. S. Josephson, M. L. Snapper, A. H. Hoveyda, J. Am. Chem. Soc. 2004, 126, 3734-3735. b) N. S. Josephson, E. L. Carswell, M. L. Snapper, A. H. Hoveyda, Org. Lett. 2005, 7, 2711-2713. 61 H. Mandai, K. Mandai, M. L. Snapper, A. H. Hoveyda, J. Am. Chem. Soc. 2008, 130, 17961-17969.

Reacción de Mannich anti-Selectiva
139
N
HN
OPPh2 OMe
Me Et
N
R1
MeO
R2
OTMS
+
R1: arilo, alquilo,alquenilo, sililo
94 (1-5 mol%)
1-5 mol% AgOAc, 1.1 equiv. iPrOH,THF húmedo
R2: Ph, Me,OPh
Rto.: 67-93ee: 84-92%
R1
R2
ONH
OMe
Esquema 3-24: Único método de reacción de Mannich con iminas propargílicas vía catálisis asimétrica, Snapper y Hoveyda, 2004.60
Posiblemente, una de las razones que justifiquen la ausencia de
versiones organocatalíticas sea la poca reactividad de las bases de Schiff en
general y la tendencia de las alquinil-iminas a experimentar adiciones 1,4 con
nucleófilos carbonados suaves. Por ejemplo, recientemente Shimizu y
colaboradores han descrito la adición conjugada de ésteres malónicos o β–
ceto ésteres a alquinil-iminas62 para obtener, tras ciclación del intermedio, la 2-
piridona 95 (Esquema 3-25).
Esquema 3-25: Adición 1,4 a iminas propargílicas, Shimizu, 2008.62
3.4.2.1. Búsqueda de condiciones de reacción
De acuerdo con las observaciones anteriores, y en el marco de un
proyecto orientado a la síntesis asimétrica de carbociclos y heterociclos,
actualmente en fase de desarrollo en nuestro laboratorio, decidimos poner a
punto las condiciones óptimas para llevar a cabo la reacción de Mannich
representada en el Esquema 3-26.
62 a) I. Hachiya, Y. Minami, T. Aramaki, M. Shimizu, Eur. J. Org. Chem. 2008, 1411-1417. b) M. Shimizu, I. Hachiya, I. Mizota, Chem. Commun. 2009, 874-889.

Capítulo 3
140
Esquema 3-26
En primer lugar se sintetizaron las N-aril-iminas propargílicas 96a y
96b con el fin de efectuar la reacción de Mannich anti-selectiva con el
propanal 17A utilizando el catalizador 92. No obstante, para efectos
comparativos también se realizó una evaluación de la reacción con otras
pirrolidinas (Esq. 3-27, Tabla 3-6).

Reacción de Mannich anti-Selectiva
141
Esquema 3-27: Catalizadores probados para la reacción de Mannich entre el propanal y las N-aril iminas propargílicas 96a y 96b. PNBA: ácido 4-nitrobenzoico.
Tabla 3-6: Evaluación de catalizadores, disolventes y temperatura para la reacción de Mannich entre 17A y 96a/96b:
Entrada
R
Cat.
Dv.
T(ºC)
Rto.[b](%)
(anti:sin)[c]
ee[d](%)
1 4-ClC6H4
(96a) 3 THF -60 84 84 : 16 >98
2
4 THF -60 63 67 : 33 n.d.[e]
3 39 THF -60 80 77 : 23 99
4 42 THF -60 n.d. 91 : 9 98
5
92 THF -60 71 95 : 5 97
6
92 DCM -40[f] 87 74 : 26 98
7
4-MeOC6H4
(96b) 4 THF -60 72 50 : 50 n.d.
8
3 THF -60 76 83 : 17 98
9
39 THF -60 67 74 : 26 99
10 92 THF -60 85 94 : 6 98]
11
92 DCM -40[f] 73 77 : 23 97
12 92 THF -40
73 88 : 12 97
[a] Reacciones llevadas a cabo a escala 0.5 mmol, con 2 mL de disolvente y utilizando 20 mol% de catalizador, ácido p-nitrobenzoico (20 mol%) y propanal (3 equiv.). [b] Rendimiento de los alcoholes aislados tras cromatografía. [c] Determinado por espectroscopía 1H-RMN del crudo. [d] Determinado por análisis de HPLC. [e] No determinado. [f] Baja solubilidad a temperaturas inferiores con este disolvente.
De nuevo se comprobó la necesidad de la presencia del ácido de
Brønsted externo para el transcurso eficiente de la reacción, por ejemplo, los
diarilprolinoles 3 y 4 sin ácido fueron ineficientes para la reacción de la imina
96a con propanal a bajas temperaturas (Conversión = 0). Sin embargo,
utilizando un 20 mol% de 4 en combinación con un 20 mol% de ácido p-

Capítulo 3
142
nitrobenzoico (PNBA) se obtuvo el aducto deseado con un 63% de
rendimiento y una relación diastereomérica de 67:33 anti:sin, resultados que
mejoraron considerablemente al utilizar 3 en las mismas condiciones
(entradas 1 y 2 en la Tabla 3-6). El catalizador 39, más impedido
estéricamente que 3, también conducía a excelentes relaciones
enantioméricas, pero la relación de diastereómeros resultó algo peor (77:23,
entrada 3) que con el catalizador 3.
Por otro lado, observamos una notable mejoría en la estereoquímica
de la reacción al sustituir los grupos arilo de los catalizadores por alquilos,
concretamente hexilos. Bajo las mismas condiciones de reacción, utilizando
los dialquilprolinoles 42 y 92, las relaciones diastereoméricas mejoraban a
95:5 anti:sin mientras que los excesos enantioméricos seguían siendo
excelentes (entradas 4 y 5). Análogos resultados se obtuvieron con la imina
96b.
En diclorometano las iminas propargílicas 96a y 96b resultaron ser
parcialmente insolubles por debajo de -40 ºC. Si bien a esta temperatura las
reacciones fueron homogéneas, se observó una disminución de la selectividad
anti:sin (entradas 6 y 11 en la Tabla 3-6).
En la Tabla 3-7 se recogen algunos resultados con varias iminas
propargílicas que revelan el potencial de esta reacción. Como se puede
observar, en los cuatro casos se obtuvieron excelentes excesos tanto
diastereo- como enantioméricos, así como buenos rendimientos, siendo éste
más moderado en el caso del hidrocinamaldehído (17G).

Reacción de Mannich anti-Selectiva
143
Tabla 3-7: Estudio de la reacción de Mannich anti-selectiva entre diferentes aldehídos e iminas propargílicas N-aromáticas:
Entrada
Producto
Rto.(%) [b]
(anti:sin)[c]
ee(%)[d]
1
97Aa 71 95:5 97
2
97Ab
85
94:6
98
3
97Ac 62 98:2 98
4 HNHO
OMe
( )4CH3
Ph
97Gc 54 91:9 94
[a] Reacciones llevadas a cabo a escala 0.5 mmol con 3 equiv. de aldehído 17. [b] Rendimiento de los aductos 97 aislados tras cromatografía de columna flash. [c] Determinado por espectroscopía 1H-RMN del crudo. [d] Determinado por análisis de HPLC.
Actualmente en nuestro laboratorio se está estudiando el alcance de
dicha transformación con objeto de incrementar la utilidad de la reaccion de
Mannich anti-selectiva en el contexto de la síntesis de heterociclos
nitrogenados quirales.

Capítulo 3
144
3.4.2.2. Síntesis del aducto sin-97Ab
Con el fin de esclarecer el curso estereoquímico de la reacción, se
llevó a cabo la preparación del aducto sin-97Ab haciendo reaccionar la imina
96b con el propanal en presencia de D-prolina (20 mol%) a 0 ºC en THF
(Esquema 3-28).63 Aunque la relación diastereomérica obtenida fue de un
80:20, se consiguió purificar mediante cromatografía el aducto sin-97Ab para
su caracterización. La comparación sin:anti con respecto al aducto obtenido al
utilizar como catalizador 92 se pudo efectuar por 1H-RMN (Fig. 3-15),
constatando así la configuración absoluta de dicho aducto y del resto por
analogía.64
Esquema 3-28: Síntesis del aducto sin-97Ab
63 Para más información sobre la prolina como catalizador en la reacción de Mannich, ver ref. 15,16,17. 64 Para más información, ver parte experimental.

Reacción de Mannich anti-Selectiva
145
NH
COOH
Me
NHPMP
Ph
HO
Cat: D-prolina
H3
Me
NHPMP
Ph
HOH3
Cat: 92
NH OSiPh3
nHexnHex
= 4.40J2,3= 4.1 Hz
= 4.32J2,3= 7.1 Hz
= 4.40J2,3= 4.1 Hz
= 4.32J2,3= 7.1 Hz
H2
H2
Me
NHPMP
Ph
HOH3
H2
Me
NHPMP
PhHO
H3
H2
4.154.204.254.304.354.404.454.504.55
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
4.154.204.254.304.354.404.454.504.55
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
Figura 3-15: Comparación de señales de 1H-RMN para los aductos sin y anti-97Ab obtenidos,
respectivamente, por la reacción de Mannich catalizada por D-prolina y 92.
Además, se comprobó por cromatografía líquida (HPLC) que el
aducto minoritario obtenido con la D-prolina como catalizador anti-97Ab
AcOEt

Capítulo 3
146
coincidía con el aducto mayoritario obtenido con 92, confirmándose así la
configuración absoluta propuesta (Fig. 3-16).
97Ab-racémico
48.0
65
57.4
53 62.0
40
75.0
26
AU
0.00
0.05
0.10
Minutes
40.00 45.00 50.00 55.00 60.00 65.00 70.00 75.00 80.00 85.00
NHPMP
HO
PhMe
rac-97Ab
Me
NHPMP
Ph
HO
Me
NHPMP
Ph
HO
Me
NHPMP
Ph
HO
Me
NHPMP
Ph
HO

Reacción de Mannich anti-Selectiva
147
Reacción con D-prolina
47.1
40
73.1
18
AU
0.00
0.10
0.20
0.30
0.40
0.50
Minutes
35.00 40.00 45.00 50.00 55.00 60.00 65.00 70.00 75.00 80.00 85.00
Reacción con el catalizador 92
45.9
88
72.0
42
AU
0.00
0.20
0.40
0.60
0.80
Minutes
35.00 40.00 45.00 50.00 55.00 60.00 65.00 70.00 75.00 80.00 85.00
Figura 3-16: Cromatogramas de HPLC para el compuesto racémico 97Ab y sus distintos
isómeros sin y anti mostrados. Para mayor detalle, consultar parte experimental.
3.5. Aplicaciones
Como se decía al principio de este capítulo, los aductos de Mannich
tienen una estructura molecular muy interesante desde el punto de vista
Me
NHPMP
HO
Ph Me
NHPMP
Ph
HO
Me
NHPMP
HO
Ph Me
NHPMP
Ph
HO

Capítulo 3
148
sintético. Los compuestos β-aminocarbonílicos obtenidos pueden ser
fácilmente reducidos a β-aminoalcoholes u oxidados a β-aminoácidos. Por
ejemplo, el β-aminoaldehído 98 (Esquema 3-29) se oxidó mediante
condiciones estándar para dar el derivado 99 con el grupo amino protegido (N-
tosilo), que fue posteriormente desprotegido y convertido en el β-aminoácido
conocido 100. La comparación de los datos espectroscópicos y de rotación
óptica del derivado 99 con los bibliográficos65 confirmó la asignación
estereoquímica realizada con anterioridad.
O
Ph
NHTos
Me
NaHPO4,NaClO2
H2O2,MeOH
O
Ph
NHTos
Me
HO 100 ºC
O
Ph
NH2
Me
HO
98 99 10062%
Rto.: 95%
HCl
Esquema 3-29
Para el caso de los aductos de Mannich provenientes de iminas
sencillas o bases de Schiff se llevaron a cabo diversas manipulaciones
sintéticas que se muestran en el Esquema 3-30. Dichas transformaciones
fueron realizadas por las doctorandas Itziar Otazo y Jacqueline Jiménez.
Esquema 3-30
65 I. Abrahmas, M. Motevalli, A. J. Robinson, P. B. Wyatt, Tetrahedron 1994, 50, 12755-12772.

Reacción de Mannich anti-Selectiva
149
3.6. Conclusiones
A modo de conclusión podemos decir que se han descrito dos nuevos
sistemas catalíticos sencillos y eficientes para la reacción de Mannich anti-
selectiva. Como características principales de los mismos, cabe destacar:
a) Funcionan con iminas poco reactivas.
b) Permiten acceder a productos de configuración anti, con lo cual
ayudan a cubrir las carencias principales dentro de las metodologías
catalíticas y asimétricas de la reacción de Mannich.
Estos logros se han basado en el desarrollo de dos sistemas
catalíticos diferentes. Por un lado, se ha desarrollado un nuevo sistema
catalítico basado en el catalizador bifuncional 83 de diseño propio, eficiente
para la reacción entre aldehídos y N-sulfonil-iminas en combinación con un
ácido de Brønsted externo, que resulta esencial para el transcurso eficiente y
el control de la estereoquímica del proceso.
Por otro lado, se ha encontrado que la combinación de éteres de α,α-
dialquilprolinol en combinación con un ácido de Brønsted es capaz de catalizar
la reacción de Mannich entre aldehídos e iminas sencillas (bases de Schiff),
permitiendo el acceso a aminas propargílicas ópticamente activas entre otras,
las cuales no son fáciles de obtener mediante otros procesos. El método
presentado es eficiente y general, considerando tanto la naturaleza de los
sustituyentes de las iminas y de los aldehídos, siendo apropiado para la
preparación de anti-aminoalcoholes saturados no accesibles vía reacción de
Mannich con iminas derivadas de los correspondientes aldehídos enolizables.
En ambos casos la clave para el estereocontrol eficiente de las
reacciones es el uso de éteres de α,α-dialquilsililo derivados del prolinol como
organocatalizadores, que conducen a resultados considerablemente
superiores a los análogos α,α-diarilsililo.
Los aductos de Mannich, aislados con buenos rendimientos y
excelentes diastereo- y enantioselectividades, pueden tanto oxidarse como
reducirse in situ y desprotegerse para conducir a los correspondientes β–
aminoácidos y β–aminoalcoholes.


Desarrollo experimental


Desarrollo Experimental
153
4. Desarrollo experimental
4.1. Materiales y métodos generales
Reactivos
Los reactivos químicos de partida ordinarios se adquirieron a las casas
comerciales habituales (Aldrich, Acros, Merck, Sigma, Fluka, etc.) y se
utilizaron sin purificación previa salvo cuando se especifique lo contrario. Por
norma general, los catalizadores una vez preparados se conservaron en la
nevera a −30 ºC.
Disolventes
Los disolventes anhidros empleados en las reacciones se trataron y
manipularon mediante técnicas habituales.1 El tetrahidrofurano (THF) y el éter
etílico se pasaron a través de columnas de secado de disolventes It Pure Solv.
La N,N dimetilformamida (DMF) se secó por destilación sobre MgSO4 anhidro y
se almacenó sobre tamiz molecular de 4 Å. El acetonitrilo se secó por
destilación sobre CaH2 justo antes de su utilización. El tolueno se destiló sobre
sodio y se almacenó sobre sodio hilado. Los disolventes utilizados como
eluyentes en cromatografía de columna fueron de calidad técnica, habiendo
sido previamente purificados por destilación.
Condiciones de reacción
Por regla general las reacciones catalíticas se llevaron a cabo en
condiciones anhidras bajo atmósfera de nitrógeno en viales con fondo redondo
y septum. La agitación se realizó mediante la utilización de agitadores
magnéticos.
Cromatografía
El seguimiento de algunas reacciones y de las columnas cromatográficas
se efectuó por cromatografía en capa fina utilizando gel de sílice soportada
sobre placas de aluminio (Merck, kiesekgel 60 F-254). El revelado se realizó 1 D. D. Perrin, D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press: Oxford. 2ª Ed., 1989.

Capítulo 4
154
con luz ultravioleta (λ=254 nm) y/o por calefacción en contacto con disoluciones
de ácido fosfomolíbdico (94 mL de agua, 2.5 g de ácido fosfomolíbdico, 1 g de
nitrato de cerio y amonio y 6 mL de ácido sulfúrico concentrado) o de
permanganato potásico (1% (p/v) KMnO4 en agua).
La purificación de productos mediante cromatografía de columna se realizó
bajo presión, empleando gel de sílice Merck 230-400 mesh (0.040-0.063 mm)
como fase estacionaria y mezclas de disolventes en proporciones adecuadas
(acetato de etilo, hexano, diclorometano, metanol, etc.) como eluyentes.
La cromatografía líquida de alta resolución (HPLC) se realizó en
cromatógrafos Waters 600E con detectores de fotodiodo UV 2996 y 2998. Las
columnas empleadas fueron Chiralpack AD, IA, IB, AD-H, AS-H, IC, y OD-H y
Chiralcell OJ.
Resonancia magnética nuclear (RMN)
Los espectros de 1H-RMN con observación de protones a 200, 300 y 500
MHz fueron registrados en espectrómetros Varian Gemini, Bruker Advance
DXP-300 y Bruker Advance Ultrashield-500 respectivamente. Los espectros de 13C-RMN se registraron en los mismos espectrómetros a 50, 75 y 125 MHz
respectivamente. El disolvente utilizado de manera general fue el CDCl3. Los
valores de los desplazamientos químicos se expresan en unidades de δ (ppm)
respecto a la señal interna del CHCl3 residual δ= 7.26 ppm para el protón y δ=
77.00 ppm para el carbono.
Para la descripción de los espectros se emplearon las siguientes
abreviaturas: m (multiplete), s (singlete), bs (singlete ancho (broad singlet)), d
(doblete), dd (doble doblete), t (triplete), dt (doble triplete) y c (cuadruplete).
Polarímetro
Los valores de rotación óptica de midieron en polarímetros Perkin-Elmer
243B y Jasco P-2000 y se expresan como valores específicos [α], con
indicación del disolvente y la concentración utilizados (g/100 mL).

Desarrollo Experimental
155
Otros
Los puntos de fusión se determinaron en un aparato Büchi SMP-20 y no
están corregidos. Las microdestilaciones se llevaron a cabo en un destilador de
bolas Kügelrohr Büchi GKR-50. La evaporación de los disolventes a presión
reducida se efectuó en rotavapores Büchi R110. Los análisis elementales se
realizaron en un aparato Leo CHNS-923. Los espectros de Masas se
registraron en un espectrómetro de Masas ESI-trampa de iones (Agilent serie
1100 LC/MSD, modelo SL).
Estudios mecanísticos mediante métodos computaciona les
Todos los cálculos se llevaron a cabo con el programa Gaussian 032
utilizando el bifuncional B3LYP.3 Se utilizó la función de base estándar 6-31G*
para describir los átomos de carbono, hidrógeno, oxígeno, nitrógeno, azufre y
silicio. Todos los puntos estacionarios se caracterizaron analizando sus
frecuencias armónicas al mismo nivel de teoría, encontrándose que todos los
reactivos, intermedios y productos poseen Hessianos positivos mientras que
todos los estados de transición (TS) localizados muestran un único autovalor
negativo. Mediante los cálculos IRC (Intrinsic Reaction Coordinate)4 se
verificaron los perfiles de energía que conecta correctamente a cada estado de
transición con los mínimos locales asociados. Las energías de vibración en el
punto cero (ZPVE) también se han calculado al mismo nivel de teoría.
2 M. J. Fisch, G. W. Truncks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Jr. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Perersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian, Inc., Wallingford CT 2004. 3 A. D. J. Becke, Chem. Phys. 1993, 98, 5648. C. Lee, W. Yang, R. G. Parr, Phys. Rev. B. 1998, 37, 785-789. 4 C. González, H. B. J. Schelegel, Phys. Chem. 1990, 94, 5523-5527.

Capítulo 4
156
4.2. Síntesis de catalizadores
Los catalizadores 3 y 4 se adquireron de la casa comercial Sigma-Aldrich.
El resto de catalizadores se prepararon según los procedimientos que se
describen a continuación. Una vez sintetizados, se almacenaron en vial a -30
ºC (nevera) bajo atmósfera de nitrógeno o argón. Cuando se observó la
aparición de un sólido en los catalizadores de aspecto aceitoso, éstos, previo
uso, se basificaron con una disolución saturada acuosa de NaHCO3, se
extrajeron con DCM y se secaron sobre MgSO4.
4.2.1. Síntesis de éteres de α,α-dialquilsililo y de α,α-diarilsililo
derivados
Esquema general:
NH
CO2H
NH
CO2MeMeOHreflujo(cuant.)
toluenoreflujo(95%)
NCO2Me
Bn
THF, t.a.
N
Bn
R
R
OH NH
R
R
OH
4) H2
Pd,C (20% peso)
NH
R
R
OSiR'3
. HCl
BnBr,SOCl2 3) RMgBr
EtOH, t.a.
5) ClSiR'3
base
1)2)
(66-85%)
DIPEA
(73-92%) (64-89%)
1) Preparación del hidrocloruro de (S)-metilpirrolidina-2-carboxilato:5
Se goteó cloruro de tionilo (4.0 mL, 55 mmol) durante 5 minutos sobre una
suspensión de L-prolina (5.76 g, 50 mmol) en MeOH enfriada a 0 ºC y la
mezcla resultante se agitó durante 1 hora a reflujo. Transcurrido este tiempo, el
exceso de cloruro de tionilo y el MeOH se evaporaron a presión reducida
obteniéndose un aceite amarillento que se utilizó en la siguiente etapa sin
previa purificación. Rto: 8.28 g (99%).
5 P. N. Confalone, E. H. Huie, S. S. Ko, G. H. Cole, J. Org. Chem. 1988, 53, 482-487.

Desarrollo Experimental
157
2) Bencilación:6
NH
CO2Me
BnBr (1.1 eq.)
toluenoref lujo, 6 h(95%)· HCl
NCO2Me
Bn
DIPEA (2.5 eq)
Sobre una mezcla de reacción previamente enfriada a 0 ºC del crudo de
reacción obtenido en el paso anterior (8.28 g, ~50 mmol) y diisopropiletilamina
(26.1 mL, 150 mmol) en tolueno (50 mL) se adicionó lentamente bromuro de
bencilo (6.5 mL, 55 mmol). La mezcla de reacción se agitó durante 6 horas a
reflujo. Posteriormente se subió la temperatura de la mezcla a 0 ºC, se vertió
NaHCO3 (sol. sat., 40 mL) y se extrajo con acetato de etilo (2 x 40 mL). La
combinación de las fases orgánicas se secó sobre MgSO4 y el disolvente se
evaporó a presión reducida obteniéndose un aceite marrón. El (S)-metil-1-
bencilpirrolidina-2-carboxilato obtenido se utilizó en la siguiente etapa sin previa
purificación. Rto: 10.30 g (95%).
1H-RMN (300 MHz, CDCl3): δ 7.34, (m, 5H), 5.15 (m, 2H), 4.35 (m, 1H), 3.86-
3.40 (m, 5H), 2.30 -1.80 (m, 4H).
3) Reacción de Grignard:7
Sobre una disolución del producto de la reacción anterior sin previa
purificación (4.38 g, ~20 mmol), en THF (40 mL) enfriada a 0 ºC, se goteó una
disolución del correspondiente bromuro de alquil o aril magnesio (3 equiv.). La
mezcla de reacción se agitó durante 16 horas a temperatura ambiente. Una vez
finalizada la reacción se enfrió la mezcla a 0 ºC y se adicionó una disolución
acuosa saturada de NH4Cl (30 mL). Tras unos minutos de agitación, el líquido
sobrenadante se separó por decantación dejando un precipitado blanco que se
6 Adaptado de: K. Funabashi, M. Jachmann, M. Kanai, M. Shibasaki, Angew. Chem. Int. Ed. 2003, 42, 5489-5492. 7 Adaptado de: K. Soai, H, Hachida, N. Yokota, J. Chem. Soc. Perkin. Trans I 1987, 1909-1914.
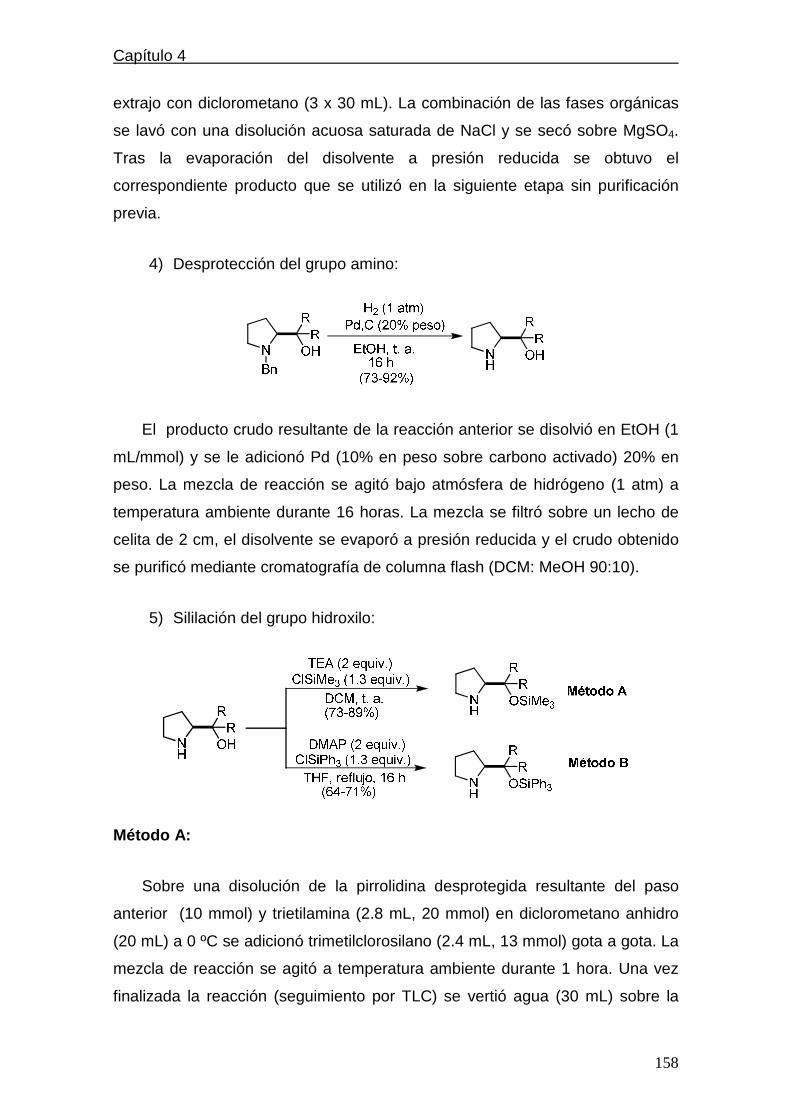
Capítulo 4
158
extrajo con diclorometano (3 x 30 mL). La combinación de las fases orgánicas
se lavó con una disolución acuosa saturada de NaCl y se secó sobre MgSO4.
Tras la evaporación del disolvente a presión reducida se obtuvo el
correspondiente producto que se utilizó en la siguiente etapa sin purificación
previa.
4) Desprotección del grupo amino:
El producto crudo resultante de la reacción anterior se disolvió en EtOH (1
mL/mmol) y se le adicionó Pd (10% en peso sobre carbono activado) 20% en
peso. La mezcla de reacción se agitó bajo atmósfera de hidrógeno (1 atm) a
temperatura ambiente durante 16 horas. La mezcla se filtró sobre un lecho de
celita de 2 cm, el disolvente se evaporó a presión reducida y el crudo obtenido
se purificó mediante cromatografía de columna flash (DCM: MeOH 90:10).
5) Sililación del grupo hidroxilo:
Método A:
Sobre una disolución de la pirrolidina desprotegida resultante del paso
anterior (10 mmol) y trietilamina (2.8 mL, 20 mmol) en diclorometano anhidro
(20 mL) a 0 ºC se adicionó trimetilclorosilano (2.4 mL, 13 mmol) gota a gota. La
mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Una vez
finalizada la reacción (seguimiento por TLC) se vertió agua (30 mL) sobre la

Desarrollo Experimental
159
mezcla y se extrajo con diclorometano (3 x 20 mL). El combinado de las fases
orgánicas se lavó con una disolución acuosa saturada de NaHCO3 (30 mL), la
fase orgánica se secó sobre MgSO4 y el disolvente se evaporó a presión
reducida. El crudo resultante se purificó mediante cromatografía de columna
flash eluyendo con mezclas de DCM:MeOH.
Método B:
Sobre una disolución de la pirrolidina desprotegida resultante del paso
anterior (6 mmol) y DMAP (1.46 g, 12 mmol) en THF anhidro (6 mL) a 0 ºC se
adicionó una disolución de cloruro de trifenilsililo (3.11 g 10.5 mmol) en THF
anhidro (6 mL). La mezcla de reacción se agitó a reflujo durante 16 horas.
Transcurrido este tiempo se vertió agua (25 mL) a la mezcla, se extrajo con
diclorometano (3 x 25 mL) y la combinación de las fases orgánicas se lavó con
una disolución acuosa saturada de NaHCO3 (40 mL). La fase orgánica
resultante se secó sobre MgSO4 y el disolvente se evaporó a presión reducida.
El crudo de reacción se purificó mediante cromatografía de columna flash.
Catalizador 39: ( S)-2-Difenil(trifenilsililoximetil)pirrolidina
El catalizador se preparó a partir del (S)-α,α-difenilprolinol
(20 mmol). La etapa de sililación se efectuó siguiendo el
procedimiento general B. Rendimiento: 75%. Sólido blanco.
P.f.: 147-150 ºC.
[α]D25 = −24.4 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.47-7.41 (m, 8H), 7.39–7.22 (m, 11H), 7.20–7.08
(m, 6H), 3.98 (t, J= 7.6 Hz, 1H), 2.74–2.66 (m, 1H), 2.63–2.49 (m, 1H), 1.88–
1.68 (m, 1H), 1.60–1.38 (m, 3H), 1.30 (t, J= 10.6 Hz, 1H). 13C-RMN (125 MHz, CDCl3) δ: 146.3, 145.1, 136.3, 135.1, 129.3, 129.1, 127.8,
127.4, 127.0, 126.7, 85.0, 65.3, 46.9, 28.0, 25.0.
Anal. calcd . para C35H33NOSi (511.73): C, 82.15; H, 6.50; N, 2.74.
Encontrado: C, 82.38; H, 6.14; N, 2.61.
NH
PhPh
OSiPh3

Capítulo 4
160
Catalizador 42: ( S)-2-((7-Trimetilsililoxi) tridecan-7-il)pirrolidina
El catalizador se preparó a partir de la L-prolina (20
mmol) siguiendo el procedimiento general y utilizando
bromuro de hexilmagnesio (solución 2.0 M en éter dietílico,
30 mL, 60 mmol). La etapa de sililación se efectuó siguiendo el método A. El
rendimiento total tras las cinco etapas fue de 59%. Aceite amarillo.
[α]D25 = −15.6 (c= 1, DCM).
1H-RMN (500 MHz, CDCl3) δ: 3.01-2.98 (m, 1H), 2.88-2.85 (m, 1H), 2.72-2.70
(m, 1H), 1.72-1.42 (m, 8H), 1.36-1.16 (m, 16H), 0.89 (t, J= 7.2 Hz, 3H), 0.11 (s,
9H). 13C-RMN (125 MHz, CDCl3) δ: 79.8, 66.1, 47.2, 38.1, 37.9, 31.9, 31.8, 30.1,
30.0, 26.1, 24.1, 22.7, 22.6, 14.0, 3.0.
Masa exacta calculada para C20H44NOSi (M+H)+: 342.3192, encontrada:
342.3181.
Catalizador Rac-42: Rac-2-((7-Trimetilsililoxi)tridecan-7-il)pirrolidina
El catalizador racémico Rac-42 se preparó siguiendo el
mismo procedimiento experimental general que para el
catalizador quiral 42 pero utilizando la L,D-prolina como
reactivo de partida.
Catalizador 92: ( S)-2-((7-Trifenilsililoxi) tridecan-7-il)pirrolidina
El catalizador se preparó a partir de la L-prolina (20
mmol) siguiendo el procedimiento general y utilizando
bromuro de hexilmagnesio (solución 2.0 M en éter dietílico,
30 mL, 60 mmol). La etapa de sililación se efectuó siguiendo el método B. El
rendimiento total tras las cinco etapas fue de 62%. Aceite amarillo.
[α]D25 = −2.5 (c= 1, DCM).
1H-RMN (500 MHz, CDCl3) δ: 7.73-7.67 (m, 6H), 7.45-7.31 (m, 9H), 3.10-3.03
(m, 1H), 2.89-2.80 (m, 1H), 2.72-2.62 (m, 1H), 1.69-1.52 (m, 4H), 1.44-0.96, m,
20H), 0.82 (t, J= 7.0 Hz, 6H).
NH
nHexnHex
OSiMe3
NH
nHexnHex
OSiPh3
NH
nHexnHex
OTMS

Desarrollo Experimental
161
13C-RMN (125 MHz, CDCl3) δ: 135.6, 129.5, 127.6, 82.5, 66.0, 47.0, 38.4, 37.5,
31.7, 29.8, 26.4, 25.9, 24.1, 23.8, 22.6, 14.1.
Masa exacta calculada para C35H50NOSi (M+H)+: 528.3622, encontrada:
528.3688.
4.2.2. Síntesis de éteres de α,α-dialquilsililo derivados de la trans -4-
hidroxi- L-prolina
Esquema general:
1) Preparación del hidrocloruro de (2S,4R)-metil-4-hidroxipirrolidina-2-
carboxilato:5
Se goteó cloruro de tionilo (4.0 mL, 55 mmol) durante 5 minutos sobre una
suspensión de trans-4-hidroxi-L-prolina (6.55 g, 50 mmol) en MeOH enfriada a
0 ºC y la mezcla resultante se agitó durante 1 hora a reflujo. Transcurrido este
tiempo, el exceso de cloruro de tionilo y el MeOH se evaporaron a presión

Capítulo 4
162
reducida obteniéndose un sólido blanco que se utilizó en la siguiente etapa sin
previa purificación. Rto: 9.05 g (99%).
2) Bencilación:6
Sobre una mezcla de reacción previamente enfriada a 0 ºC del crudo de
reacción obtenido en el paso anterior (9.05 g, ~50 mmol) y diisopropiletilamina
(26.1 mL, 150 mmol) en tolueno (50 mL) se adicionó lentamente bromuro de
bencilo (6.5 mL, 55 mmol). La mezcla de reacción se agitó durante 6 horas a
reflujo. Posteriormente se enfrió la temperatura de la mezcla a 0 ºC, se vertió
NaHCO3 (sol. sat., 40 mL) y se extrajo con acetato de etilo (2 x 40 mL). La
combinación de las fases orgánicas se secó sobre MgSO4 y el disolvente se
evaporó a presión reducida obteniéndose un aceite marrón. El (2S,4R)-metil-1-
bencil-4-hidroxipirrolidina-2-carboxilato obtenido se utilizó en la siguiente etapa
sin previa purificación. Rto: 11.75 g (94%).
3) Reacción de Grignard:7
Sobre una disolución del producto de la reacción anterior sin previa
purificación (4.81 g, ~20 mmol), en THF (40 mL) enfriada a 0 ºC, se goteó una
disolución del correspondiente bromuro de alquil o aril magnesio (3 equiv.). La
mezcla de reacción se agitó durante 16 horas a temperatura ambiente. Una vez
finalizada la reacción se enfrió la mezcla a 0 ºC y se adicionó una disolución
acuosa saturada de NH4Cl (30 mL). Tras unos minutos de agitación, el líquido
sobrenadante se separó por decantación dejando un precipitado blanco que se
extrajo con diclorometano (3 x 30 mL). La combinación de las fases orgánicas

Desarrollo Experimental
163
se lavó con una disolución acuosa saturada de NaCl y se secó sobre MgSO4.
Tras la evaporación del disolvente a presión reducida se obtuvo el
correspondiente producto que se utilizó en la siguiente etapa sin purificación
previa.
4) Protección del grupo hidroxilo de la posición 3 del anillo pirrolidínico:
Sobre una disolución del aducto proveniente de la reacción de Grignard (10
mmol) y DMAP (2.44 g, 20 mmol) en DCM (15 mL) enfriada a 0 ºC y bajo
atmósfera de nitrógeno se adicionó cloroformiato de bencilo (2.85 mL, 20
mmol). La mezcla de reacción se agitó durante 16 horas a temperatura
ambiente. Posteriormente se vertió agua (15 mL) a la mezcla, se extrajo la
mezcla con DCM (3 x 15 mL) y la combinación de las fases orgánicas se lavó
con una disolución acuosa saturada de NaHCO3 (40 mL). El disolvente
orgánico se secó sobre MgSO4 y se evaporó bajo presión reducida. El crudo de
reacción se purificó mediante cromatografía de columna flash eluyendo con
mezclas de hexano:AcOEt.
5) Sililación del grupo hidroxilo:
Sobre una disolución del aducto procedente del paso anterior (10 mmol) y
trietilamina (2.8 mL, 20 mmol) en diclorometano anhidro (20 mL) a 0 ºC se
adicionó trimetilclorosilano (2.4 mL, 13 mmol) gota a gota. La mezcla de
reacción se agitó a temperatura ambiente durante 1 hora. Una vez finalizada la
reacción (seguimiento por TLC) se vertió agua (30 mL) sobre la mezcla y se
extrajo con diclorometano (3 x 20 mL). El combinado de las fases orgánicas se

Capítulo 4
164
lavó con una disolución acuosa saturada de NaHCO3 (30 mL), la fase orgánica
se secó sobre MgSO4 y el disolvente se evaporó a presión reducida. El crudo
resultante se purificó mediante cromatografía de columna flash eluyendo con
mezclas de hexano:AcOEt.
6) Desprotección de los grupos amino e hidroxilo:
El producto crudo resultante de la reacción anterior se disolvió en EtOH (1
mL/mmol) y se le adicionó Pd (10% en peso sobre carbono activado), 20% en
peso. La mezcla de reacción se agitó bajo atmósfera de hidrógeno (1 atm) a
temperatura ambiente durante 16 horas. La mezcla se filtró sobre un lecho de
celita de 2 cm, el disolvente se evaporó a presión reducida y el crudo obtenido
se purificó mediante cromatografía de columna flash eluyendo con mezclas de
DCM:MeOH 90:10.
Catalizador 83: (2 S, 4R)-2-((7-Trimetilsililoxi)tridecan-7-il)pirrolidin-4 -ol 8
El catalizador se preparó a partir de la trans-4-
hidroxi-L-prolina (20 mmol) siguiendo el procedimiento
general y utilizando bromuro de hexilmagnesio (solución
2.0 M en éter dietílico, 30 mL, 60 mmol). El rendimiento total tras las seis
etapas fue de 36%. Aceite amarillo.
[α]D25 = −8.5 (c= 1.6, DCM).
1H-RMN (500 MHz, CDCl3) δ: 4.42-4.32 (m, 1H), 3.29-3.13 (m, 2H), 2.82-2.72
(m, 1H), 2.21 (bs, 1H), 1.75-1.17 (m, 22H), 0.93-0.82 (m, 6H), 0.09 (s, 9H). 13C-RMN (75 MHz, CDCl3) δ: 79.8, 72.9, 63.6, 55.7, 37.8, 37.7, 36.6, 31.9, 31.8,
30.0, 24.2, 23.9, 22.6, 14.0, 3.0.
8 C. Palomo, A. Landa, A. Mielgo, A. Puente, M. Oyarbide, S. Vera, Angew. Chem. Int. Ed., 2007, 46, 8431-8435.
NH
nHexnHex
OSiMe3
HO

Desarrollo Experimental
165
Masa exacta calculada para C20H44NO2Si (M+H)+: 358.3141, encontrada:
358.3125.
Catalizador 85: (2 S, 4R)-2-((2-Trimetilsililoxi) propan-2-il)pirrolidin-4- ol
El catalizador se preparó a partir de la trans-4-
hidroxi-L-prolina (20 mmol) siguiendo el procedimiento
general y utilizando bromuro de metilmagnesio (solución
3.0 M en éter dietílico, 30 mL, 60 mmol). El rendimiento total tras las seis
etapas fue de 60%. Aceite amarillo.
[α]D25 = −3.3 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 4.44-4.35 (m, 1H), 3.24-3.13 (m, 2H), 2.86 (d, J=
12.0 Hz, 1H), 2.34 (bs, 1H), 1.76-1.69 (m, 2H), 1.30 (s, 3H), 1.20 (s, 3H), 0.11
(s, 9H). 13C-NMR (125 MHz) δ: 74.5, 72.6, 67.0, 55.6, 36.7, 28.1, 27.5, 2.4.
Catalizador 86: (2 S, 4R)-((4-trimetilsililoxi) heptan-4-il) pirrolidin-4-o l
El catalizador se preparó a partir de la trans-4-
hidroxi-L-prolina (20 mmol) siguiendo el procedimiento
general y utilizando bromuro de propilmagnesio
previamente preparado según Feringa (nPrBr, Mg (virutas), THF, 45 ºC, 1 h).9
La etapa de sililación se efectuó siguiendo el procedimiento general A. El
rendimiento total tras tres etapas fue de 52%. Aceite amarillo.
[α]D25 = −12.5 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 4.42-4.33 (m, 1H), 3.29 (dd, J= 6.5, 9.8 Hz, 1H),
3.20 (dd, J= 5.0, 12.1 Hz, 1H), 2.77 (d, J= 12.0 Hz, 1H), 1.71-1.19 (m, 11H),
0.89-0.79 (m, 6H), 0.07 (s, 9H). 13C-RMN, 125 MHz) δ: 79.6, 72.5, 63.6, 55.5, 40.3, 40.1, 36.5, 17.5, 17.3, 14.7,
14.6, 2.8.
Masa exacta calculada para C14H32NO2Si (M+H)+: 274.2202, encontrada:
274.2209.
9 R. P. Summeren, S. J. W. Reijmer, B. L. Feringa, A. J. Minnaard, Chem. Commun. 2005, 1387-1389.
NH
MeMe
OSiMe3
HO
NH
nPrnPr
OSiMe3
HO
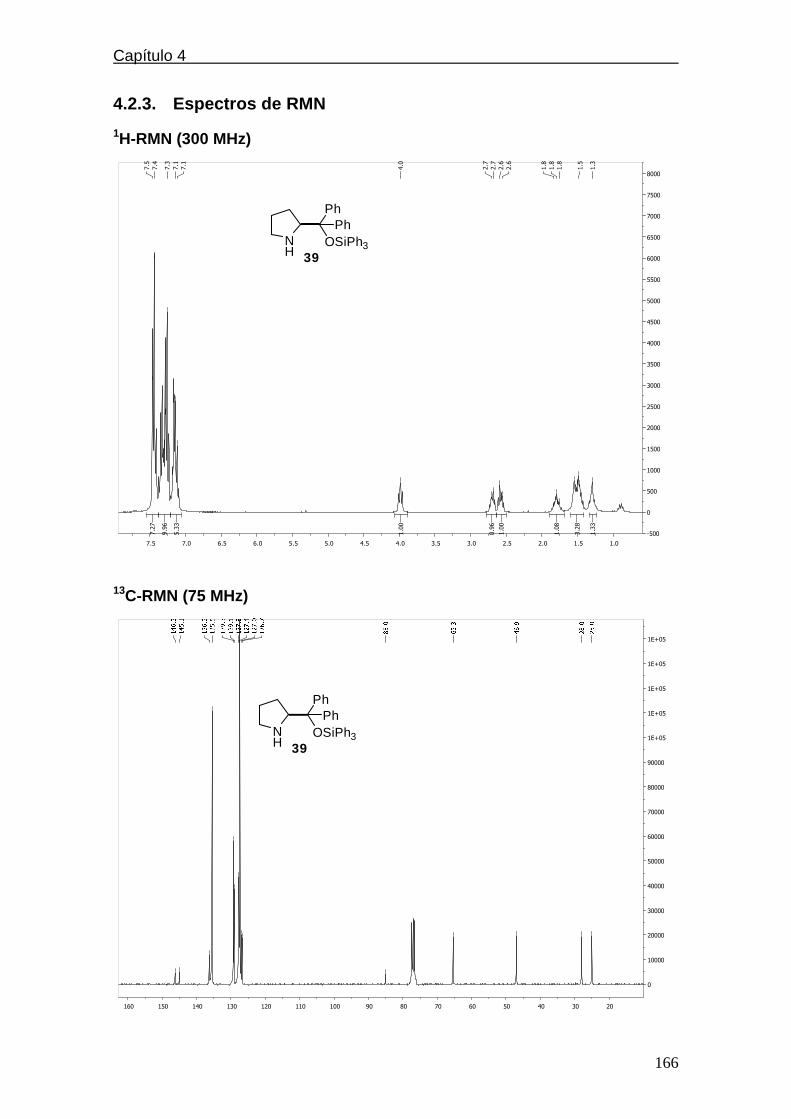
Capítulo 4
166
4.2.3. Espectros de RMN
1H-RMN (300 MHz)
1.01.52.02.53.03.54.04.55.05.56.06.57.07.5
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
1.33
3.28
1.08
1.00
0.96
1.00
5.33
9.96
7.27
1.3
1.5
1.8
1.8
1.8
2.6
2.6
2.7
2.7
4.0
7.1
7.1
7.3
7.4
7.5
13C-RMN (75 MHz)
2030405060708090100110120130140150160
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
1E+05
1E+05
NH
PhPh
OSiPh339
NH
PhPh
OSiPh339

Desarrollo Experimental
167
1H-RMN (300 MHz)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
13C-RMN (75 MHz)
NH
nHexnHex
OSiMe3
42
NH
nHexnHex
OSiMe3
42

Capítulo 4
168
1H-RMN (300 MHz)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
13C-RMN (75 MHz)
-5051015202530354045505560657075808590f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
NH
nHexnHex
OSiMe3
HO
83
NH
nHexnHex
OSiMe3
HO
83

Desarrollo Experimental
169
1H-RMN (300 MHz)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
380
13C-RMN (75 MHz)
05101520253035404550556065707580859095f1 (ppm)
-2000
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
NH
MeMe
OSiMe3
HO
85
NH
MeMe
OSiMe3
HO
85

Capítulo 4
170
1H-RMN (300 MHz)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
13C-RMN (75 MHz)
-5051015202530354045505560657075808590f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
NH
nPrnPr
OSiMe3
HO
86
NH
nPrnPr
OSiMe3
HO
86

Desarrollo Experimental
171
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
13C-RMN (75 MHz)
102030405060708090100110120130140150f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
NH
nHexnHex
OSiPh3
92
NH
nHexnHex
OSiPh3
92

Capítulo 4
172
4.3. Síntesis de aldehídos
4.3.1. Síntesis de 17J y 17K
Procedimiento general:
Los aldehídos 17J y 17K se sintetizaron a partir de los correspondientes
alcoholes mediante oxidación de Swern de acuerdo con el siguiente
procedimiento:10 Una disolución de DMSO (2.27 mL, 32.0 mmol) en
diclorometano (16 mL) se adicionó lentamente sobre una disolución de cloruro
de oxalilo (1.4 mL, 16.0 mmol) en diclorometano (60 mL) previamente enfriada
a −70 ºC. La mezcla resultante se agitó a la misma temperatura durante 5
minutos, tras lo cual se goteó una disolución del alcohol comercial
correspondiente (8.0 mmol) en diclorometano (16 mL) y se agitó durante una
hora adicional a −70 ºC. Posteriormente se goteó trietilamina (6.68 mL, 48
mmol) lentamente y tras dejar que la mezcla resultante alcanzara 0 ºC, se agitó
a dicha temperatura durante una hora. Se vertió agua (40 mL) a la mezcla y se
diluyó con Et2O (400 mL). Tras separar la fase orgánica, ésta se lavó con agua
(2 x 75 mL) y una disolución acuosa saturada de NaCl (2 x 75 mL), se secó con
MgSO4 y se filtró. El disolvente orgánico se evaporó a presión reducida. El
crudo resultante se purificó por cromatografía de columna flash, eluyendo con
mezclas de hexano: acetato de etilo.
10 Adaptado de X. Xiao, S. Antony, G. Kohlhagen, Y. Pommier, M. Cushman, Bioorg. Med. Chem. 2004, 12, 5147-5160.

Desarrollo Experimental
173
3-(Benciloxi)propanal (17J)11
El aldehído 17J se preparó a partir de 3-(benciloxi)-propanol (1.6
mL, 10 mmol) siguiendo el procedimiento general. Líquido incoloro.
Rendimiento: 1.1 g, (69%). Los datos físicos y espectroscópicos
coincidieron con los descritos en la bibliografía.
1H-RMN (300 MHz, CDCl3) δ: 9.82 (1H, s), 7.27-7.42 (5H, m), 4.54 (2H, s),
3.84 (t, J= 6.1 Hz, 2H), 2.72 (t, J= 6.1 Hz, 2H).
6-(Boc-amino)-1-hexanal (17K)10
El aldehído 17K se preparó a partir de 6-(Boc-amino)-1-
hexanol (2.0 mL, 10 mmol) siguiendo el procedimiento general.
Líquido incoloro. Rendimiento: 1.85 g, (86%). Los datos físicos y
espectroscópicos coincidieron con los descritos en la bibliografía.
1H-RMN (300 MHz, CDCl3) δ: 9.76 (t, J= 1.7 Hz,1H), 3.11 (d, J= 6.5 Hz, 2H),
2.43, (dt, J= 1.7, 7.2, 7.3 Hz, 2H), 1.70-1.23 (m, 6H), 1.44 (s, 9H).
4.3.2. Síntesis de 17L y 17M
Procedimiento general:
Los aldehídos 17L y 17M se sintetizaron a partir de los correspondientes
alcoholes mediante oxidación de acuerdo con el siguiente procedimiento:12 Se
adicionó una disolución del correspondiente alcohol comercial (10.0 mmol) en 11 L. Nielsen, K. B. Lindsay, J. Faber, N. C. Nielsen, T. Skrydstrup, J. Org. Chem. 2007, 72, 10035-10044. 12 Adaptado de: M. Rosillo, E. Arnáiz, D. Abdi, J. Blanco-Urgoiti, G. Domíngez, J. Pérez-Castells, Eur. J. Org. Chem. 2008, 3917-3927.
O
NHBoc( ) 5
O
O
Bn

Capítulo 4
174
diclorometano (4 mL) sobre una suspensión de PCC (3.22 g, 15.0 mmol) en
diclorometano (15 mL) a 0 ºC. La mezcla se agitó a temperatura ambiente
hasta completar la reacción (seguimiento por TLC), tras lo cual se diluyó con
Et2O (50 mL) y se filtró a través de un lecho de 2 cm de sílica-gel eluyendo con
Et2O. La disolución resultante se concentró bajo presión reducida a
temperatura ambiente y el residuo así obtenido se utilizó sin posterior
purificación.
5-Hexenal (17L)12
El aldehído 17L se preparó a partir de 5-hexanol (0.96 mL, 8
mmol) siguiendo el procedimiento general. Líquido incoloro.
Rendimiento: 777 mg, (99%). Los datos espectroscópicos de los
productos coincidieron con los de la bibliografía.
1H-RMN (300 MHz, CDCl3) δ: 9.78 (s, 1H), 5.82-5.73 (m, 1H), 5.05-4.95 (m,
2H), 2.44 (t, J= 7.7 Hz, 2H), 2.10-2.08 (m, 2H), 1.76-1.72 (m, 2H).
5-Fenilpentanal (17M)13
El aldehído 17M se preparó a partir de 5-fenilpentanol (1.85 mL,
11 mmol) siguiendo el procedimiento general. El producto crudo se
purificó por cromatografía de columna flash (hexano: AcOEt, 95:5).
Líquido incoloro. Rendimiento: 940 mg, (53%). Los datos espectroscópicos de
los productos coincidieron con los de la bibliografía.
1H-RMN (300 MHz, CDCl3) δ: 9.76 (t, J= 1.8 Hz, 1H), 7.31-7-16 (m, 5H), 2.64 (t,
J= 7.1 Hz, 2H), 2.44 (dt, J= 3.5, 3.5, 4.1 Hz, 2H), 1.72-1.62 (m, 4H).
4.4. Procedimientos experimentales del capítulo 2
4.4.1. Reacción N-nitrosoaldólica de aldehídos y nitrosobenceno
13 A. J. Chalk, S. A. Magennis, J. Org. Chem. 1976, 41, 1206-1209.
O
( ) 3
O
Ph( ) 4

Desarrollo Experimental
175
4.4.1.1. Reacción N-nitrosoaldólica: procedimiento general
Procedimiento general A:
H
O1)
(20 mol%)PhNO, DCM, 0 ºC
2) NaBH4, MeOH30 min.
R
HO
NPh
OHR
NH
OTMS
PhPh
3
40
Sobre una disolución verdosa del correspondiente aldehído (6 mmol) y
nitrosobenceno (2 mmol, 214 mg) en diclorometano (2 mL) a 0 ºC se
adicionaron el catalizador 3 (0.1 mmol, 33 mg) y ácido p-nitrobenzoico (0.1
mmol, 33 mg), observándose casi instantáneamente la decoloración de la
mezcla. Esta disolución se agitó a 0 ºC durante 30 minutos y posteriormente se
adicionaron sucesivamente MeOH (2 mL) y NaBH4 (8 mmol, 302 mg), y tras 30
minutos de agitación se añadió una disolución acuosa saturada de NaCl (5
mL), permitiendo que la mezcla alcanzara la temperatura ambiente. Tras
extraer la mezcla con diclorometano (3 x 5 mL), la combinación de las fases
orgánicas se secó con MgSO4, y el disolvente se evaporó a presión reducida.
El crudo resultante se purificó mediante cromatografía de columna flash para
obtener los aductos 40 puros.
Procedimiento general B:
Sobre una disolución verdosa del correspondiente aldehído (6 mmol) y
nitrosobenceno (214 mg, 2 mmol) en THF (2 mL) a −20 ºC se adicionaron el
catalizador (130 mg, 0.4 mmol). Esta disolución se agitó a −20 ºC durante 16
horas. Posteriormente se adicionó sucesivamente MeOH (2 mL) y NaBH4 (8
mmol) a la misma temperatura, y tras 30 minutos de agitación se añadió una
disolución acuosa saturada de NaCl (6 mL), permitiendo que la mezcla
alcanzara la temperatura ambiente. Tras una previa extracción de la mezcla
con diclorometano (3 x 6 mL), la combinación de las fases orgánicas se secó
con MgSO4, se evaporó el disolvente bajo presión reducida y el crudo
resultante se purificó mediante cromatografía de columna flash para obtener los
aductos 40 puros.

Capítulo 4
176
(S)-2-(Hidroxi(fenil)amino)propan-1-ol (40A) 14
Preparado según el procedimiento general A a partir de
propanal (0.47 mL, 6 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano: AcOEt, 90:10).
Aceite amarillento. Rendimiento: 231 mg (70%). Los datos
espectroscópicos resultaron coincidentes con los existentes en la bibliografía.
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:etanol 90:10, flujo= 1 mL/min, tr: 14.5 min (minoritario) y 15.4 min
(mayoritario).
(S)-2-(Hidroxi(fenil)amino)butan-1-ol (40B)
Preparado según el procedimiento general A a partir de
butanal (0.54 mL, 6 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano: AcOEt, 90:10).
Aceite amarillento. Rendimiento: 236 mg (65%).
[α]D25 = −2.8 (c= 1, DCM).
1H-RMN (500 MHz, CD3OD) δ: 7.19 (t, 2H, J= 7.6 Hz), 7.02 (d, 2H, J= 7.9 Hz),
6.76 (t, 1H, J= 7.6 Hz), 4.28 (t, 1H, J= 5.4 Hz), 3.54-3.52 (m, 1H), 3.46-3.40 (m,
2H), 1.60-1.53 (m, 2H), 0.90 (t, 3H, J= 7.4 Hz).
13C-RMN (75 MHz, CDCl3) δ: 153.1, 128.2, 120.1, 115.4, 68.6, 61.6, 48.7, 48.4,
48.1, 47.8, 47.5, 47.2, 46.9, 19.8, 10.7.
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak IA,
hexano:etanol 98:2, flujo= 0.5 mL/min, tr: 37.8 min (minoritario) y 42.2 min
(mayoritario).
Masa exacta: no determinada.
(S)-2-(Hidroxi(fenil)amino)pentan-1-ol (40C)
Preparado según el procedimiento general A a partir
de pentanal (0.64 mL, 6 mmol). El producto crudo se
purificó por cromatografía de columna flash (hexano:
14 T. Kano, M. Ueda, J. Takai, K. Maruoka, J. Am. Chem. Soc. 2006, 128, 6046-6047.
Me
OH
NPh
HO
OH
NPh
HO
Me
OH
NPh
HO
Me

Desarrollo Experimental
177
AcOEt, 90:10). Aceite amarillento. Rendimiento: 258 mg (66%).
[α]D25 = +8.6 (c= 0.9, DCM).
1H-RMN (500 MHz, CD3OD) δ: 7.19 (t, 2H, J= 7.6 Hz), 7.02 (d, 2H, J= 7.9 Hz),
6.76 (t, 1H, J= 7.5 Hz), 4.28 (t, 1H, J= 5.0 Hz), 3.55-3.34 (m, 3H), 1.38-1.21 (m,
4H), 0.93 (t, 3H, J= 7.3 Hz).
13C-RMN (75 MHz, CDCl3) δ: 153.1, 128.2, 120.0, 115.4, 66.7, 61.8, 29.2, 19.7,
13.2.
Masa calculada para C11H17NO2 (M+H)+: 196.26, encontrada: 197.00.
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:etanol 95:5, flujo= 0.5 mL/min, tr: 10.4 min (minoritario) y 12.7 min
(mayoritario).
(S)-2-(Hidroxi(fenil)amino)heptan-1-ol (40E) 14
Preparado según el procedimiento general B a
partir de heptanal (0.83 mL, 6 mmol). El producto crudo
se purificó por cromatografía de columna flash
(hexano: AcOEt, 90:10). Aceite amarillento.
Rendimiento: 335 mg (75%). Los datos espectroscópicos resultaron
coincidentes con los existentes en la bibliografía.
La pureza enantiomérica se determinó por HPLC en el correspondiente
derivado di-O-benzoilado (ver sección 4.4.1.2.) (Daicel Chiralpak IB,
hexano:etanol 95:5, flujo= 0.5 mL/min, tr: 13.5 min (minoritario) y 15.6 min
(mayoritario).
(S)-2-(Hidroxi(fenil)amino)octan-1-ol (40F) 14
Preparado según el procedimiento general B a
partir de octanal (0.94 mL, 6 mmol). El producto
crudo se purificó por cromatografía de columna flash
(hexano: AcOEt, 95:5). Aceite amarillento.
Rendimiento: 356 mg (75%). Los datos espectroscópicos resultaron
coincidentes con los existentes en la bibliografía.
OH
NPh
HO
Me
OH
NPh
HO
Me

Capítulo 4
178
La pureza enantiomérica se determinó por HPLC en el correspondiente
derivado di-O-naftoilado (ver sección 4.4.1.3.) (Daicel Chiralpak IA, hexano:
etanol 98:2, flujo= 0.5 mL/min, tr: 35.8 min (minoritario) y 39.5 min (mayoritario).
(S)-2-(Hidroxi(fenil)amino)-3-fenilpropan-1-ol (40G) 14
Preparado según el procedimiento general A a partir
de 3-fenilpropanal (0.98 mL, 6 mmol). El producto crudo
se purificó por cromatografía de columna flash (hexano:
AcOEt, 80:20). Aceite amarillento. Rendimiento: 341 mg
(70%). Los datos espectroscópicos resultaron coincidentes con los existentes
en la bibliografía.
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD,
hexano:etanol 60:40, flujo= 0.5 mL/min, tr: 11.6 min (minoritario) y 13.6 min
(mayoritario).
(S)-2-(Hidroxi(fenil)amino)-4-metilpentan-1-ol (40H) 14
Preparado según el procedimiento general A a partir de
isovaleraldehído (0.96 mL, 6 mmol). El producto crudo se
purificó por cromatografía de columna flash (hexano:AcOEt,
95:5). Aceite amarillento. Rendimiento: 176 mg (60%). Los
datos espectroscópicos resultaron coincidentes con los existentes en la
bibliografía. La pureza enantiomérica se determinó por HPLC (Daicel Chiralcell
OJ, hexano:etanol 80:20, flujo= 0.5 mL/min, tr: 12.1 min (mayoritario) y 13.5 min
(minoritario).
(S)-2-(Hidroxi(fenil)amino)-3-(2-metoxifenil)propan-1 -ol (40I)
Preparado según el procedimiento general A a partir de
3-(2-metoxi-fenil)propanal (0.98 mL, 6 mmol). El producto
crudo se purificó por cromatografía de columna flash
(hexano: AcOEt, 80:20). Aceite amarillento. Rendimiento:
219 mg (40%).
[α]D25 : no determinado.
N
HO
OH
Ph
N
HO
OMe OH
Ph
N
HO
Me
OH
Ph
Me

Desarrollo Experimental
179
1H-RMN (200 MHz, CD3OD), δ: 7.20-7.13 (m, 4H), 7.07 (t, 1H, J= 6.6 Hz), 6.94-
6.82 (m, 5H), 3.82 (s, 3H), 3.71-3.65 (m, 1H), 3.64-3.58 (m, 1H) 2.83-2.76 (m,
1H), 2.69-2.60 (m, 1H). 13C-RMN (75 MHz, CD3OD) δ: 157.7, 157.5, 130.7, 129.51, 127.1, 126.8, 120.1,
118.7, 110.2, 72.7, 61.8, 54.5, 28.9.
Masa exacta : no determinada.
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD,
hexano:etanol 80:20, flujo= 0.5 mL/min, tr: 14.2 min (minoritario) y 15.7 min
(mayoritario).
4.4.1.2. Derivatización del 2-(hidroxi(fenil)amino) heptan-1-ol 40E con
cloruro de benzoilo para análisis de HPLC
MeOH
NOH
MeO
N
O
O
O
ClO
TEA
DCM, t.a.
Sobre una disolución de 2-(hidroxi(fenil)amino)heptan-1-ol 40E (223 mg, 1
mmol) en 2 mL de diclorometano anhidro se adicionó trietilamina (0.8 mL, 6
mmol), se goteó cloruro de benzoilo (0.3 mL, 2.6 mmol) a temperatura
ambiente, y tras agitar la mezcla a la misma temperatura durante 1 hora se
vertieron 2 mL de agua. El producto se extrajo con diclorometano (3 x 10 mL) y
la combinación de las fases orgánicas se lavó sucesivamente con una
disolución acuosa saturada de NH4Cl y otra de NaHCO3. La fase orgánica se
secó con MgSO4, se filtró y el disolvente se evaporó bajo presión reducida. El
producto crudo se purificó por cromatografía de columna flash (hexano:AcOEt,
90:10). Sólido amarillento. Rendimiento: 319 mg (74%). Masa calculada para
C27H29NO4 (M+H)+: 432.21, encontrada: 432.10.
Capítulo 4
180
4.4.1.3. Derivatización del 2-(hidroxi(fenil)amino) octan-1-ol 40F con
cloruro de naftoilo para análisis de HPLC
Me OH
NOH
Me O
N
O
O
O
ClO
TEA, DMAP
DCM
Sobre una disolución de 2-(hidroxi(fenil)amino)octan-1-ol 40F (118 mg, 0.5
mmol) en 2 mL de diclorometano anhidro se adicionaron trietilamina (0.42 mL,
3 mmol) y N,N-dimetilaminopiridina (6 mg, 0.05 mmol) y se goteó cloruro de 1-
naftoilo (0.3 mL, 2 mmol) a temperatura ambiente. La mezcla se agitó a la
misma temperatura durante 3 horas y se añadieron 2 mL de agua. El producto
se extrajo con diclorometano (3 x 10 mL) y la combinación de las fases
orgánicas se lavó sucesivamente con una disolución acuosa saturada de NH4Cl
y otra de NaHCO3. La fase orgánica se secó con MgSO4, se filtró y el
disolvente se evaporó bajo presión reducida. El producto crudo se purificó
mediante cromatografía de columna flash (hexano:AcOEt, 90:10) para obtener
el producto deseado como un sólido amarillento. Rendimiento: 237 mg (87%).
Masa calculada para C36H37NO4 (M+H)+: 546.26, encontrada: 546.20.
4.4.1.4. Preparación de muestras racémicas
Procedimiento general A:
Se añadió pirrolidina (0.1 mL, 1.2 mmol) a una disolución del aldehído
correspondiente (12 mmol) y nitrosobenceno (428 mg, 4 mmol) en metanol (12

Desarrollo Experimental
181
mL). La mezcla se agitó durante 30 minutos a temperatura ambiente. Tras
enfriar la mezcla a 0 ºC se adicionó NaBH4 (605 mg, 16 mmol) y ésta se agitó
durante otros 30 minutos a esta temperatura. Posteriormente se añadió una
disolución acuosa saturada de NaCl (15 mL) y la mezcla se extrajo con
diclorometano (3 x 10 mL). La combinación de las fases orgánicas se secó con
MgSO4, se filtró y el disolvente se evaporó bajo presión reducida. El crudo de la
reacción se purificó mediante cromatografía de columna flash (hexano:AcOEt).
Procedimiento general B: 15
Se añadió prolina racémica (92 mg, 0.8 mmol) a una disolución de propanal
(4.8 mL, 0.34 mL) y nitrosobenceno (428 mg, 4 mmol) en DMSO anhidro (8 mL)
a temperatura ambiente. La mezcla se agitó durante 10 minutos a la misma
temperatura. Tras enfriar la mezcla a 0 ºC se adicionaron sucesivamente EtOH
(4 mL) y NaBH4 (605 mg, 16 mmol) y la mezcla resultante se agitó durante
otros 30 minutos a esta temperatura. Posteriormente se vertió a la mezcla una
disolución acuosa saturada de NaCl (15 mL) y el producto se extrajo con
diclorometano (3 x 10 mL). La combinación de las fases orgánicas se secó con
MgSO4, se filtró y el disolvente se evaporó bajo presión reducida. Los crudos
de reacción se purificaron mediante cromatografía de columna flash
(hexano:AcOEt).
15 G. Zhong, Angew. Chem. Int. Ed. 2003, 42, 4247-4250.

Capítulo 4
182
4.4.1.5. Espectros de 1H y 13C-RMN de algunos compuestos
representativos
1H-RMN (200 MHz)
ppm (f1)0.671.001.331.672.002.332.673.003.333.674.004.334.675.005.335.676.006.336.677.007.337.67
1.00
0.86
1.36
1.32
0.72
1.36
2.82
1H-RMN (200 MHz)
ppm (f1)0.671.001.331.672.002.332.673.003.333.674.004.334.675.005.335.676.006.336.677.007.33
1.13
2.13
1.89
1.77
3.00
Me
OH
NPh
HO
40A, aducto de N-adición
AcOEt AcOEt
Me O
HOHN
Ph
41A, aducto de O-adición

Desarrollo Experimental
183
1H-RMN (200 MHz)
13C-RMN (75 MHz)
OH
NPh
HO
Me
40B
OH
NPh
HO
Me
40B

Capítulo 4
184
1H-RMN (200 MHz)
ppm (f1)0.671.001.331.672.002.332.673.003.333.674.004.334.675.005.335.676.006.336.677.007.337.67
1.00
2.26
0.69
1.32
1.50
2.78
2.31
2.92
13C-RMN (75 MHz)
OH
NPh
HO
Me
40C
OH
NPh
HO
Me
40C
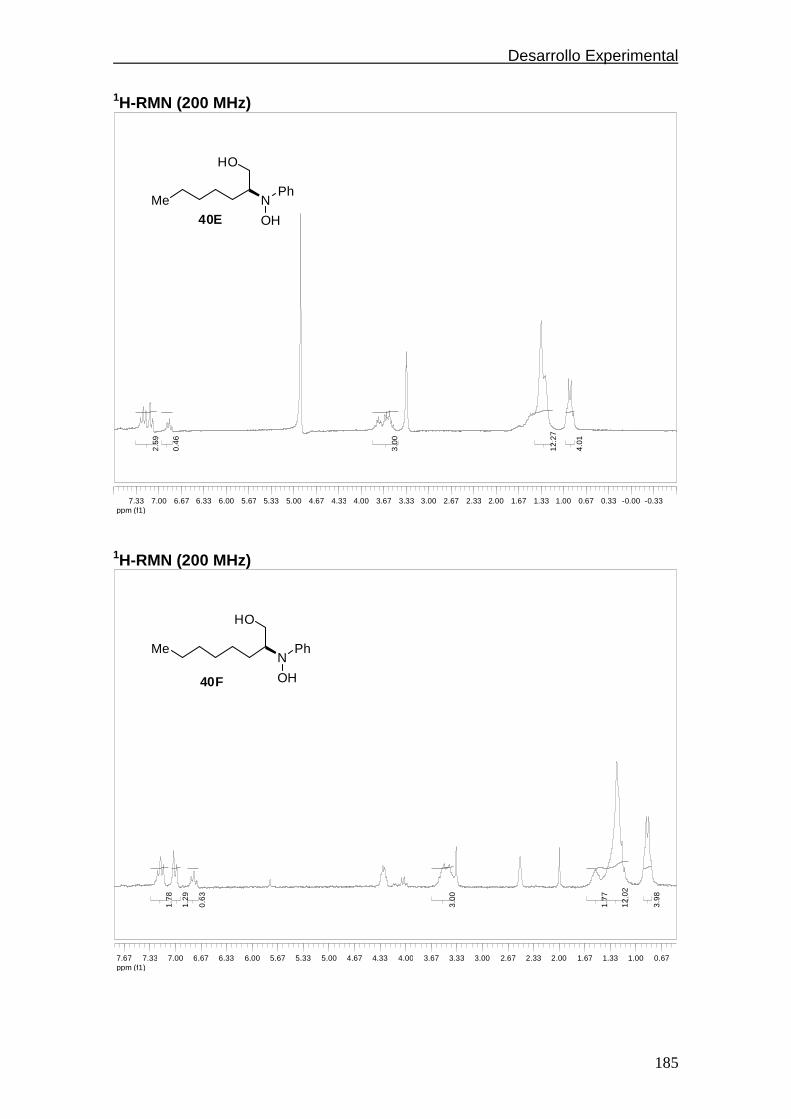
Desarrollo Experimental
185
1H-RMN (200 MHz)
ppm (f1)-0.33-0.000.330.671.001.331.672.002.332.673.003.333.674.004.334.675.005.335.676.006.336.677.007.33
3.00
2.59
0.46
12.2
7
4.01
1H-RMN (200 MHz)
ppm (f1)0.671.001.331.672.002.332.673.003.333.674.004.334.675.005.335.676.006.336.677.007.337.67
3.00
1.77
12.0
2
3.98
0.63
1.29
1.78
OH
NPh
HO
Me
40F
OH
NPh
HO
Me
40E
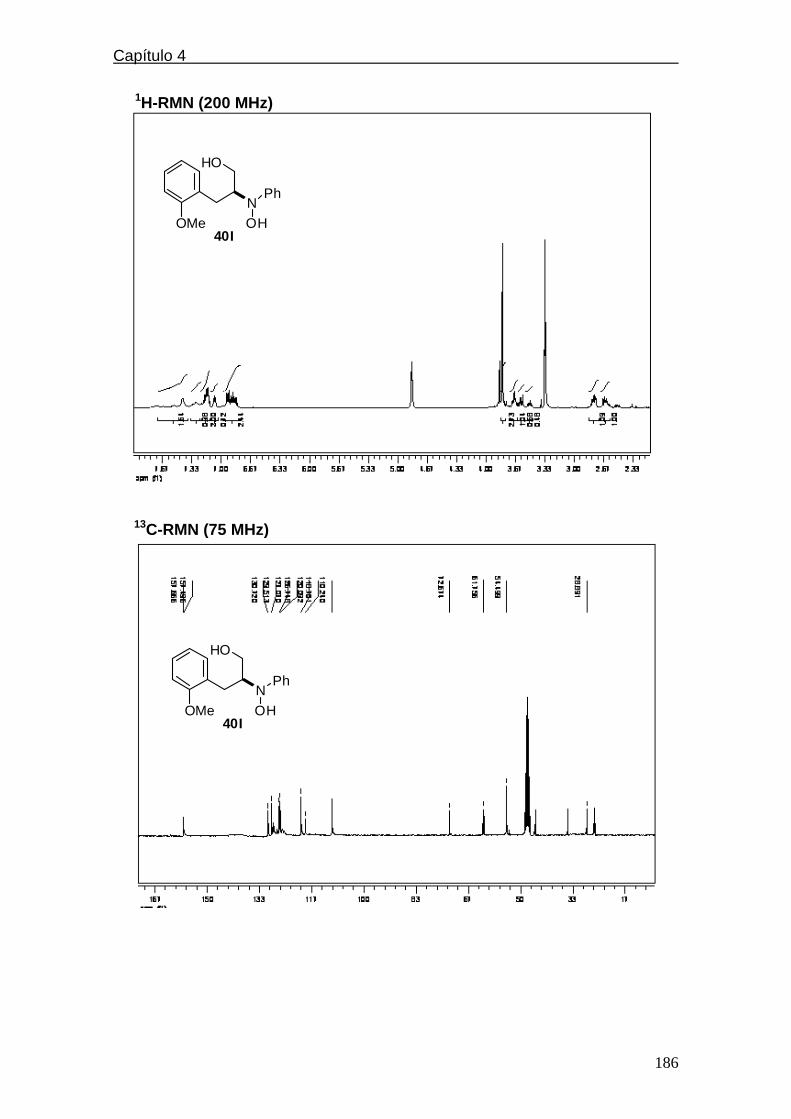
Capítulo 4
186
1H-RMN (200 MHz)
13C-RMN (75 MHz)
N
HO
OMe OH
Ph
40I
N
HO
OMe OH
Ph
40I

Desarrollo Experimental
187
UA
0,00
200,00
400,00
600,00
800,00
1000,00
Minutes20,00 21,00 22,00 23,00 24,00 25,00 26,00 27,00 28,00 29,00
21,9
28
25,6
38
4.4.1.5. Cromatogramas de HPLC
Chiralpak AD-H, 1 mL/min, hexano:etanol 90:10, λ= 210 nm
Chiralpak AD-H, 1ml/min, hexane/ethanol 90:10, λ= 254nm
Me
HO
N
OH
Ph
rac-40A
UA
0,00
200,00
400,00
600,00
Minutes13,80 14,00 14,20 14,40 14,60 14,80 15,00 15,20 15,40 15,60 15,80 16,00 16,20 16,40 16,60 16,80 17,00
14,4
99
15,3
81

Capítulo 4
188
Chiralpak AD-H, 1 mL/min, hexano:etanol 90:10, λ= 210 nm
UA
0,00
100,00
200,00
300,00
400,00
Minutes15,00 16,00 17,00 18,00 19,00 20,00 21,00 22,00 23,00 24,00 25,00 26,00 27,00 28,00
15,9
88
16,6
12

Desarrollo Experimental
189
Enantiómero S
Enantiómero R Enantiómero S
Chiralpak AD-H, 1mL/min, hexano:etanol 90:10, λ= 210 nm
UA
0,00
200,00
400,00
600,00
Minutes13,80 14,00 14,20 14,40 14,60 14,80 15,00 15,20 15,40 15,60 15,80 16,00 16,20 16,40 16,60 16,80 17,00
14,4
99
15,3
81
UA
0,00
500,00
1000,00
Minutes15,00 15,20 15,40 15,60 15,80 16,00 16,20 16,40 16,60 16,80 17,00 17,20 17,40 17,60 17,80 18,00 18,20 18,40
15,9
87
16,6
12
Me
HO
N
OH
Ph
rac-40A

Capítulo 4
190
Chiralpak IA, 0.5 mL/min, hexano:etanol 98:2, λ= 210 nm
HO
N
OH
PhMe
r ac-40B
UA
0,00
100,00
200,00
300,00
Minutes30,00 32,00 34,00 36,00 38,00 40,00 42,00 44,00 46,00 48,00 50,00
37,7
93
42,2
37
HO
N
OH
PhMe
40B
UA
0,00
20,00
40,00
60,00
80,00
Minutes35,00 36,00 37,00 38,00 39,00 40,00 41,00 42,00 43,00 44,00 45,00 46,00 47,00
36,2
82 43,4
80

Desarrollo Experimental
191
Chiralpak AD-H, 0.5 mL/min, hexano:etanol 95:5, λ= 254 nm
UA
0,00
500,00
1000,00
1500,00
2000,00
Minutes10,00 10,20 10,40 10,60 10,80 11,00 11,20 11,40 11,60 11,80 12,00 12,20 12,40 12,60 12,80 13,00 13,20 13,40 13,60 13,80
10,4
00
12,6
81
UA
0,00
500,00
1000,00
1500,00
2000,00
Minutes10,00 10,50 11,00 11,50 12,00 12,50 13,00 13,50 14,00 14,50
10,9
87
13,2
52HO
N
OH
Ph
40C
Me
HO
N
OH
Ph
rac-40C
Me

Capítulo 4
192
Chiralpak IB, 0.5 mL/min, hexano:etanol 95:5, λ= 210 nm
UA
0,00
500,00
1000,00
1500,00
Minutes12,50 13,00 13,50 14,00 14,50 15,00 15,50 16,00 16,50
13,5
55
15,6
37O
N
O
Ph
Me
Ph
O
Ph
O
derivado de 40E
O
N
O
Ph
Me
Ph
O
Ph
O
rac-(derivado de 40E)

Desarrollo Experimental
193
Chiralpak IA, 0.5 mL/min, hexano:etanol 98:2, λ= 241 nm
35.9
80
39.5
75
AU
0.00
1.00
2.00
Minutes
34.00 36.00 38.00 40.00 42.0038
.097
AU
0.00
0.50
1.00
Minutes
32.00 34.00 36.00 38.00 40.00 42.00
O
N
O
PhO
O
Me
r ac-(derivado de 40F)
O
N
O
PhO
O
Me
derivado de 40F

Capítulo 4
194
Chiralpak AD 0.5 mL/min, hexano:etanol 60:40, λ= 210 nm
UA
0,00
500,00
1000,00
Minutes10,50 11,00 11,50 12,00 12,50 13,00 13,50 14,00 14,50
11,6
18
13,6
12
HO
N
OH
Ph
rac-40G
Ph
UA
0,00
200,00
400,00
600,00
800,00
1000,00
Minutes11,00 11,50 12,00 12,50 13,00 13,50 14,00 14,50 15,00 15,50
11,6
83
13,7
50
HO
N
OH
Ph
40G
Ph

Desarrollo Experimental
195
Chiralcell OJ, 0.5 mL/min, hexano:etanol 80:20, λ= 210 nm
UA
0,00
500,00
1000,00
1500,00
Minutes11,00 11,50 12,00 12,50 13,00 13,50 14,00 14,50 15,00
12,1
08
13,5
54
HO
N
OH
Ph
Me
Me
40H
4.5.
Chiralpak AD 0.5 mL/min, hexano:etanol 80:20, λ= 210 nm
UA
0,00
200,00
400,00
600,00
Minutes10,50 11,00 11,50 12,00 12,50 13,00 13,50 14,00 14,50 15,00 15,50 16,00 16,50 17,00
12,1
30
HO
N
OH
Ph
rac-40H
Me
Me

Capítulo 4
196
UA
0,00
100,00
200,00
300,00
Minutes12,50 13,00 13,50 14,00 14,50 15,00 15,50 16,00 16,50 17,00 17,50
14,1
95
15,7
00
UA
0,00
200,00
400,00
600,00
800,00
Minutes13,50 14,00 14,50 15,00 15,50 16,00 16,50 17,00 17,50
14,2
73
16,0
77
HO
N
OH
Ph
40I
MeO

Desarrollo Experimental
197
4.4.1.6. Estudios computacionales para la reacción N-nitrosoaldólica
Coordenadas cartesianas de los puntos estacionarios de la reacción N-
nitrosoaldólica y O-nitrosoaldólica de la enamina derivada del propionaldehído
y la pirrolidina con nitrosobenceno (TSN y TSO respectivamente). TSNwater y
TSN38 corresponden a las mismas reacciones en presencia de agua o de la
N,N-dimetilhidroxilamina 38 (modelo del producto de reacción).
TSN
Atom X Y Z C -2.1844 -1.4007 -0.9030 H -2.1463 -1.2805 -1.9928 N -1.8744 -0.1210 -0.2324 C -0.8867 0.6803 -0.6631 C -0.3234 1.6935 0.1494 H -0.9920 2.1414 0.8820 N 0.4967 0.6456 1.3424 O -0.3836 -0.0136 1.9928 C 1.5349 -0.2124 0.7597 C 2.7406 0.3613 0.3390 H 2.8754 1.4357 0.4029 C 3.7753 -0.4509 -0.1270 H 4.7087 0.0027 -0.4514 C 3.6251 -1.8389 -0.1551 H 4.4361 -2.4700 -0.5087 C 2.4332 -2.4113 0.3013 H 2.3170 -3.4928 0.3017 C 1.3967 -1.6051 0.7651 H 0.4740 -2.0183 1.1575 C -2.9563 0.2745 0.6934 H -2.5378 0.7200 1.5933 H -3.6210 0.9971 0.1930 C -3.6789 -1.0473 0.9603 H -3.1378 -1.6045 1.7326 H -4.7087 -0.8985 1.2981 C -3.5860 -1.7732 -0.3929 H -3.7209 -2.8558 -0.3144 H -4.3515 -1.3906 -1.0785 C 0.6123 2.6957 -0.4850 H 0.0421 3.4928 -0.9777 H 1.2602 2.2322 -1.2356 H 1.2433 3.1595 0.2803 H -0.3305 0.3191 -1.5241 H -1.4353 -2.1478 -0.6204 TSO Atom X Y Z C -0.4571 2.6518 0.4470 C -0.8455 1.4474 1.3120 C -2.1753 1.4451 -0.7426
N
HN
O

Capítulo 4
198
C -2.2244 -0.5289 0.7990 C -3.3029 -2.7471 0.2504 C -2.6244 -1.4731 -0.1832 C -1.7030 2.8725 -0.4270 H -3.2639 1.3687 -0.8437 H -1.4940 3.4443 -1.3359 H -1.9790 -0.8670 1.8015 H -3.1208 -3.5262 -0.4964 H -4.3855 -2.6059 0.3449 H -2.9140 -3.0976 1.2122 H -3.0076 -1.0593 -1.1153 H -2.4728 3.4112 0.1390 H -0.1906 3.5262 1.0479 H 0.4046 2.3867 -0.1749 H -1.3853 1.7529 2.2205 H 0.0127 0.8313 1.6026 H -1.7068 1.0432 -1.6439 N -1.7292 0.6632 0.4362 O -1.1038 -1.9976 -0.6831 N -0.3686 -1.0669 -1.2828 C 0.8539 -0.8970 -0.6433 C 3.4037 -0.4097 0.4817 C 1.7801 -0.0330 -1.2786 C 1.2407 -1.5146 0.5704 C 2.4983 -1.2627 1.1179 C 3.0318 0.2003 -0.7246 H 1.4882 0.4242 -2.2205 H 0.5539 -2.2053 1.0453 H 2.7795 -1.7504 2.0492 H 3.7295 0.8601 -1.2357 H 4.3855 -0.2287 0.9104 TSNwater Atom X Y Z C -2.3122 -1.1026 -1.0991 C 0.8440 2.5932 0.0203 C -0.7602 0.7129 -0.4639 C -0.1457 1.5543 0.4872 C -2.8772 0.3295 0.8010 C -3.9460 -0.7568 0.6208 C -3.8270 -1.1330 -0.8661 H -2.0266 -0.8727 -2.1310 H -1.8566 -2.0509 -0.7994 H 1.4788 2.9103 0.8539 H 0.3210 3.4808 -0.3572 H 1.4878 2.2155 -0.7805 H -0.2231 0.4547 -1.3734 H -0.7804 1.8928 1.3025 H -2.4475 0.3523 1.8015 H -3.2671 1.3273 0.5509 H -3.6920 -1.6223 1.2395 H -4.9421 -0.3991 0.8977 H -4.2556 -2.1126 -1.0951 H -4.3259 -0.3856 -1.4957 N -1.8352 -0.0328 -0.1935 C 1.7044 -0.4614 0.7710 C 2.9342 0.1860 0.5896 C 3.9929 -0.4726 -0.0355
NO
N
H
N
H
N
O
HO
H
HH

Desarrollo Experimental
199
C 3.8418 -1.7916 -0.4668 C 2.6258 -2.4483 -0.2549 C 1.5626 -1.7957 0.3665 H 3.0617 1.1948 0.9674 H 4.9421 0.0404 -0.1683 H 4.6679 -2.3092 -0.9470 H 2.5053 -3.4808 -0.5742 H 0.6291 -2.3163 0.5470 N 0.6735 0.2670 1.5094 O -0.2327 -0.4829 2.0318 H -1.1888 -3.4698 2.1310 H -1.0470 -2.0258 1.6190 O -1.5115 -2.8780 1.4347 TSN38 Atom X Y Z C 0.8514 2.4003 -0.4251 C 0.6271 -1.8388 2.1604 C 0.0607 -0.9648 1.0689 C -0.2040 3.4156 -0.8913 C -1.4327 2.5403 -1.1931 C -1.3869 1.4898 -0.0827 C 0.6270 0.3082 0.8469 H 1.4811 2.0586 -1.2525 H 1.5086 2.7954 0.3590 H -1.9195 0.5743 -0.3299 H -1.7939 1.8947 0.8572 H -1.3240 2.0465 -2.1650 H -2.3711 3.1020 -1.1938 H 0.1383 3.9967 -1.7523 H -1.0173 -1.0530 0.9607 H -0.4345 4.1198 -0.0832 H 0.1671 -1.5913 3.1252 H 1.7105 -1.7198 2.2648 H 0.4095 -2.8906 1.9477 H 1.6796 0.4597 1.0755 N 0.0654 1.2565 0.0898 C 1.8326 -1.6653 -0.9254 C 2.6794 -2.5796 -0.2866 C 4.0257 -2.6531 -0.6459 C 4.5315 -1.8347 -1.6581 C 3.6749 -0.9495 -2.3205 C 2.3311 -0.8688 -1.9637 H 2.2752 -3.2468 0.4668 H 4.0573 -0.3257 -3.1252 H 4.6762 -3.3662 -0.1458 H 1.6370 -0.2160 -2.4809 H 5.5779 -1.9013 -1.9436 N 0.4101 -1.6735 -0.5927 O -0.2924 -1.0374 -1.4682 H -1.9531 -1.4751 -1.1023 O -2.8130 -1.5118 -0.6074 N -3.6881 -2.2997 -1.4439 C -4.0128 -3.4957 -0.6757 H -4.6928 -4.1198 -1.2668 H -4.4883 -3.2636 0.2936 H -3.0931 -4.0569 -0.4913 C -4.8671 -1.4766 -1.6787
N
HN
O
HO
N
H

Capítulo 4
200
H -4.5705 -0.5812 -2.2316 H -5.3655 -1.1673 -0.7427 H -5.5779 -2.0462 -2.2882
4.4.2. Reacción O-nitrosoaldólica de aldehídos con nitrosobenceno
4.4.2.1. Reacción O-nitrosoaldólica: procedimiento general
H
O1)
(20 mol%)PhNO, PNBA, DCM, -20 ºC
2) NaBH4, MeOH30 min.
R
HO
ONHPh
NH
OTMS
PhPh
R
3
41
Sobre una disolución verdosa del correspondiente aldehído (3 mmol) y
nitrosobenceno (107 mg, 1 mmol) en diclorometano (2 mL) a −20 ºC se
adicionaron ácido p-nitrobenzoico (PNBA) (17 mg, 0.1 mmol) y el catalizador 3
(33 mg, 0.1 mmol) y la mezcla se agitó a −20 ºC durante 2 horas
aproximadamente. Posteriormente se adicionaron sucesivamente EtOH (2 mL)
y NaBH4 (302 mg, 8 mmol) y tras 30 minutos de agitación a −20 ºC se vertió
sobre la mezcla una disolución acuosa saturada de NaCl (5 mL), permitiendo
que la mezcla alcanzara temperatura ambiente. El crudo se extrajo con
diclorometano (3 x 5 mL) y la combinación de las fases orgánicas se secó con
MgSO4, se filtró y el disolvente se evaporó bajo presión reducida. El crudo
resultante se purificó mediante cromatografía de columna flash.
(S)-2-(Fenilaminoxi)propan-1-ol (41A) 15
Preparado según el procedimiento general a partir de
propanal (0.22 mL, 3 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:AcOEt, 80:20).
Aceite amarillento. Rendimiento: 133 mg (80%). Los datos
espectroscópicos resultaron coincidentes con los de la bibliografía.
[α]D25= −1.1 (c= 1, CHCl3).
Me ONHPh
HO

Desarrollo Experimental
201
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:etanol 90:10, flujo= 1 mL/min, tr: 14.5 min (mayoritario) y 20.1 min
(minoritario).
(S)-2-(Fenilaminoxi)pentan-1-ol (41C) 15
Preparado según el procedimiento general a partir
de valeraldehído (0.32 mL, 3 mmol). El producto crudo
se purificó por cromatografía de columna flash
(hexano:AcOEt, 85:15). Aceite amarillento. Rendimiento: 152 mg (78%). Los
datos espectroscópicos resultaron coincidentes con los de la bibliografía.
[α]D25= −24.8 (c= 0.9, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:isopropanol 95:5, flujo= 0.8 mL/min, tr: 20.5 min (mayoritario) y 24.5 min
(minoritario).
(S)-2-(Fenilaminoioxi)hexan-1-ol (41D) 15
Preparado según el procedimiento general a
partir de hexanal (0.36 mL, 3 mmol). El producto
crudo se purificó por cromatografía de columna flash
(hexano:AcOEt, 85:15). Aceite amarillento.
Rendimiento: 169 mg (81%). Los datos espectroscópicos resultaron
coincidentes con los de la bibliografía.
[α]D25= −21.1 (c= 1, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:etanol 95:5, flujo= 0.8 mL/min, tr: 19.3 min (mayoritario), y 24.6 min
(minoritario).
(S)-3-Fenil-2-(fenilaminoxi)propan-1-ol (41G)15
Preparado según el procedimiento general a partir de
hidrocinamaldehído (0.39 mL, 3 mmol). El producto crudo se
purificó por cromatografía de columna flash (hexano:AcOEt,
85:15). Aceite amarillento. Rendimiento: 164 mg (68%). Los
datos espectroscópicos resultaron coincidentes con los de la bibliografía.
ONHPh
HO
Me
ONHPh
HO
Me
ONHPh
HO
Ph

Capítulo 4
202
[α]D25= −43.1 (c= 1, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:isopropanol 95:5, flujo= 0.8 mL/min, tr: 40.1 min (mayoritario) y 54.8 min
(minoritario).
(S)-3-Metil-2-(fenilaminoxi)butan-1-ol (41H)15
Preparado según el procedimiento general a partir de
isovaleraldehído (0.32 mL, 3 mmol). El producto crudo se
purificó por cromatografía de columna flash
(hexano:AcOEt, 85:15). Aceite amarillento. Rendimiento:
109 mg (56%). Los datos espectroscópicos resultaron coincidentes con los de
la bibliografía.
[α]D25= −32.6 (c= 1, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:etanol 95:5, flujo= 0.8 mL/min, tr: 13.9 min (mayoritario) y 16.1 min
(minoritario).
(R)-3-(Benciloxi)-2-(fenilaminoxi)propan-1-ol (41J)15
Preparado según el procedimiento general a partir de 3-
(benciloxi)-propanal (0.48 mL, 3 mmol). El producto crudo se
purificó por cromatografía de columna flash (hexano:AcOEt,
80:20). Aceite amarillento. Rendimiento: 122 mg (45%). Los
datos espectroscópicos resultaron coincidentes con los de la bibliografía.
[α]D25= +7.2 (c= 1, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:etanol 95:5, flujo= 1 mL/min, tr: 55.8 min (mayoritario) y 64.0 min
(minoritario).
(S)-terc -Butil 5-(fenilaminoxi)-6-hidroxi-hexilcarbamato (41K)15
Preparado según el procedimiento general a
partir de 6-(Boc-amino)-1-hexanal (645 mg, 3
mmol). El producto crudo se purificó por
ONHPh
HO
Me
Me
ONHPh
HO
OBn
ONHPh
HO
BocHN

Desarrollo Experimental
203
cromatografía de columna flash (hexano:AcOEt, 60:40). Aceite amarillento.
Rendimiento: 306 mg (88%). Los datos espectroscópicos resultaron
coincidentes con los de la bibliografía.
[α]D25= −8.4 (c= 1, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak OD-H,
hexano:isopropanol 92:8, flujo= 1 mL/min, tr: 26.5 min (minoritario) y 30.3 min
(mayoritario).
(S)-2-(Fenilaminoxi)hex-5-en-1-ol (41L) 15
Preparado según el procedimiento general a partir de
hex-5-enal (294 mg, 3 mmol). El producto crudo se purificó
por cromatografía de columna flash (hexano:AcOEt, 80:20).
Aceite amarillento. Rendimiento: 134 mg (66%). Los datos
espectroscópicos resultaron coincidentes con los de la
bibliografía.
[α]D25= −1.4 (c= 1, CHCl3).
La pureza enantiomérica se determinó por HPLC (Daicel Chiralpak AD-H,
hexano:isopropanol 94:6, flujo= 1 mL/min, tr: 15.8 min (mayoritario), y 19.5 min
(minoritario). 4.4.2.2. Preparación de muestras racémicas
Las muestras racémicas se prepararon paralelamente a las
enantioselectivas siguiendo el procedimiento general utilizando el catalizador
rac-42 a 0 ºC.
4.4.2.3. Preparación del diol 43 16
16 Procedimiento adaptado de: a) ver ref.15. Para datos espectroscópicos, ver: b) A. Córdova, H. Sundén, A. Bøgevig, M. Johansson, F. Hilmo, Chem. Eur. J. 2004, 10, 3673-3684. Para datos de rotación óptica, ver: c) S.-T. Tong, M. A.Brimble, D. Barker, Tetrahedron, 2009, 65, 4801-4807.
ONHPh
HO

Capítulo 4
204
El alcohol aminoxilado 41D (0.4 mmol) se hidrogenó en THF (2 mL/mmol)
sobre PtO2 (Catalizador de Adams) (13 mg, 15% en peso) durante 2 horas a
presión atmosférica. La mezcla se filtró sobre un lecho de celita de 2 cm, se
evaporó el disolvente y el crudo resultante se purificó mediante cromatografía
de columna flash (hexano:AcOEt, 60:40) para obtener el diol esperado 43 puro.
Rendimiento: 36 mg (76%). Los datos espectroscópicos resultaron coincidentes
con los de la bibliografía.
[α]D25= −19.5 (c= 0.9, EtOH).
4.4.2.4. Procedimiento general para la preparación de 1,2-aminoalcoholes
17a,g
O
R
1) 3 (10 mol%),PNBA(10 mol%)PhNO, DCM
2) Ph2CHNH2
1) L-prolina(10 mol%),PhNO, CH3Cl3
2) Ph2CHNH2
1) R1MgBr,-70ºC a t.a.
2) H2, Pd/C
3) R1MgBr
4) H2, Pd/C
45 R1: Me46 R1: Et
47
44
R1OH
NH2
MeOH
NH2
N
Ph
Ph
ONHPh
1) 3 (10 mol%),PNBA(10 mol%)PhNO, DCM
2) 4-MeOC6H4NH2
MeR1
OH
HN
49 R1: nPent50 R1: Ph
PMP
Ph
Ph
Ph
Me
NPMP
ONHPh
48
R1MgBr
A
B
C
Método general A:
Sobre una disolución verdosa del correspondiente aldehído (3 mmol) y
nitrosobenceno (107 mg, 1 mmol) en diclorometano (2 mL) a −20 ºC se
adicionaron ácido p-nitrobenzoico (17 mg, 0.1 mmol) y el catalizador 3 (33 mg,
0.1 mmol) y la disolución resultante se agitó a −20 ºC durante 2 horas hasta

Desarrollo Experimental
205
que adquirió un color amarillento. Posteriormente se adicionaron MgSO4 (250
mg) y bencidril amina (0.66 mL, 3 mmol). Se dejó subir la temperatura a 0 ºC y
la mezcla se agitó durante 2 horas a dicha temperatura. Seguidamente se filtró
la disolución, se evaporó el disolvente y el crudo resultante se disolvió en THF
(2 mL). Tras enfriar la disolución a −70 ºC se goteó el reactivo de Grignard
correspondiente (3 M en éter dietílico, 5 mL, 15 mmol), se permitió que la
mezcla alcanzara temperatura ambiente y se agitó durante 16 horas.
Transcurrido este tiempo se añadió a la mezcla una disolución acuosa saturada
de NH4Cl (10 mL), se extrajo con acetato de etilo (2 x 15 mL) y la combinación
de fases orgánicas se lavó con una disolución acuosa saturada de NaCl (15
mL), se secó sobre MgSO4, se filtró y el disolvente se evaporó bajo presión
reducida. El crudo obtenido se purificó por cromatografía de columna flash y se
hidrogenó en EtOH (2 mL/mmol) sobre Pd (sobre carbón, 20% en peso)
durante 48 horas a presión atmosférica. La mezcla se filtró sobre un lecho de
celita de 2 cm, se evaporó el disolvente y el crudo resultante se purificó
mediante tratamiento ácido/base, obteniéndose los 1,2-aminoalcoholes
esperados con relaciones diasteroméricas mayores de 95:5.
Método general B:
Sobre una disolución verdosa del correspondiente aldehído (3 mmol) y
nitrosobenceno (107 mg, 1 mmol) en cloroformo (1 mL) a 0 ºC se adicionó L-
prolina (12 mg, 0.1 mmol) y la disolución resultante se agitó a 0 ºC durante 2
horas hasta que adquirió un color amarillento. Posteriormente se concetró la
disolución bajo presión reducida y se adicionaron 2 mL de diclorometano,
MgSO4 (250 mg) y benzhidril amina (0.66 mL, 3 mmol). La disolución resultante
se agitó durante 2 horas a 0 ºC, después se filtró y el disolvente se evaporó a
presión reducida. El crudo resultante se disolvió en THF (2 mL) y tras enfriar la
disolución a -70 ºC se goteó el reactivo de Grignard correspondiente (3 M en
éter dietílico, 5 mL, 15 mmol) y se dejó que la reacción alcanzara la
temperatura ambiente. La mezcla se mantuvo en agitación durante 16 horas y
a continuación se adicionó una disolución acuosa saturada de NH4Cl (10 mL).
Se extrajo con acetato de etilo (2 x 15 mL) y la combinación de las fases
orgánicas se lavó con una disolución acuosa saturada de NaCl (15 mL), se

Capítulo 4
206
secó sobre MgSO4, se filtró y el disolvente se evaporó bajo presión reducida. El
crudo obtenido se purificó por cromatografía de columna flash y se hidrogenó
en EtOH (2 mL/mmol) sobre Pd (sobre carbón, 20% en peso) durante 48 horas
a presión atmosférica. La mezcla se filtró por un lecho de celita de 2 cm, se
evaporó el disolvente y el crudo resultante se purificó mediante tratamiento
ácido/base, obteniéndose los 1,2-aminoalcoholes esperados con relaciones
diastereoméricas mayores de 95:5.
Método general C:
Sobre una disolución verdosa del correspondiente aldehído (3 mmol) y
nitrosobenceno (107 mg, 1 mmol) en diclorometano (2 mL) a −20 ºC se
adicionaron ácido p-nitrobenzoico (17 mg, 0.1 mmol) y el catalizador 3 (33 mg,
0.1 mmol) y la disolución resultante se agitó a −20 ºC durante 2 horas hasta
que adquirió un color amarillento. Posteriormente se adicionaron MgSO4 (250
mg) y p-anisidina (369 mg, 3 mmol). Seguidamente se dejó subir la temperatura
a 0 ºC y la mezcla se agitó durante 2 horas a dicha temperatura. Se filtró la
disolución, se evaporó el disolvente y el crudo resultante se disolvió en THF (2
mL). Tras enfriar la disolución a −70 ºC se goteó el reactivo de Grignard
correspondiente (3 M en éter dietílico, 5 mL, 15 mmol), se permitió que la
mezcla alcanzara temperatura ambiente y se agitó durante 16 horas a dicha
temperatura. A continuación se adicionó sobre la mezcla una disolución acuosa
saturada de NH4Cl (10 mL), se extrajo con acetato de etilo (2 x 15 mL) y la
combinación de las fases orgánicas se lavó con una disolución acuosa
saturada de NaCl (15 mL), se secó sobre MgSO4, se filtró y el disolvente se
evaporó bajo presión reducida. El crudo obtenido se purificó por cromatografía
de columna flash, obteniéndose los 1,2-aminoalcoholes esperados con
relaciones diasteroméricas del orden de 95:5.
(2S, 3S)-3-Amino-1-fenilbutan-2-ol (45) 17
Preparado según el procedimiento general A a partir de
hidrocinamaldehído (0.39 mL, 3 mmol) y bromuro de metil
17 R. Besse, H. Veschambre, Tetrahedron: Asymmetry 1994, 5, 1727-1744.
Me Ph
NH2
OH

Desarrollo Experimental
207
magnesio (3 M en éter dietílico, 5 mL, 15 mmol). Aceite naranja. Rendimiento:
90 mg (60%).
Los datos espectroscópicos resultaron coincidentes con los de la bibliografía.
[α]D25= −7.8 (c= 0.5, DCM).
(2S, 3S)-3-Amino-1-fenilpentan-2-ol (46)
Preparado según el procedimiento general A a partir de
hidrocinamaldehído (0.39 mL, 3 mmol) y bromuro de etil
magnesio (3 M en éter dietílico, 5 mL, 15 mmol). Aceite
naranja. Rendimiento: 74 mg (45%).
[α]D25= −20.6 (c= 0.5, DCM).
1H-RMN (300 MHz, CDCl3): δ: 7.37-7.16 (m, 5H), 4.99 (bs, 2H), 3.74-3.62 (m,
1H), 2.91-2.75 (m, 2H), 2.71-2.60 (m, 1H), 1.72-1.40 (m, 2H), 0.91 (t, J= 7.5 Hz,
3H). 13C-RMN (125MHz, CDCl3) δ: 138.2, 129.5, 128.4, 126.4, 73.3, 57.2, 40.2, 24.5,
10.0.
Masa exacta : no determinada.
(2R, 3R)-3-Amino-1-fenilbutan-2-ol (47) 17
Preparado según el procedimiento general B a partir de
hidrocinamaldehído (0.39 mL, 3 mmol) y bromuro de metil
magnesio (3 M en éter dietílico, 5 mL, 15 mmol). Aceite
naranja. Rendimiento: 77 mg (51%).
Los datos espectroscópicos resultaron coincidentes con los de la bibliografía.
[α]D25= +20.0 (c= 0.5, DCM).
(2S, 3S)-3-(4-Metoxifenilamino)octan-2-ol (49)
Preparado según el procedimiento general C a partir de
propionaldehído (0.22 mL, 3 mmol) y bromuro de pentil
magnesio (3 M en éter dietílico, 5 mL, 15 mmol). Aceite
naranja. Rendimiento: 155 mg (55%).
[α]D25 (95:5 sin/anti mezcla no separable)= +6.8 (c= 1, DCM).
Et Ph
NH2
OH
Me Ph
NH2
OH
PentMe
NHPMP
OHn

Capítulo 4
208
1H-RMN (300 MHz, CDCl3, diastereoisómero mayoritario sin): δ: 6.80 (d, J= 9.0
Hz, 2H), 6.67 (d, J= 9.0 Hz, 2H), 3.78 (s, 3H), 3.76-3.65 (m, 2H), 3.15-3.04 (m,
1H), 1.74-1.49 (m, 10H), 0.93-0.80 (m, 5H). 13C-RMN (125 MHz, CDCl3, diastereoisómero mayoritario sin) δ: 152.6, 142.8,
128.7, 115.3, 115.0, 69.7, 61.8, 55.8, 32.3, 32.0, 25.6, 22.5, 19.9, 14.0.
Masa exacta calculada para C15H26NO2 (M+H)+: 252.1964, encontrada:
252.1973.
(1S, 2S)-1-Fenil-1-(4-metoxifenilamino)propan-2-ol (50)
Preparado según el procedimiento general C a partir de
propionaldehído (0.22 mL, 3 mmol) y bromuro de fenil magnesio
(3 M en éter dietílico, 5 mL, 15 mmol). Aceite naranja.
Rendimiento: 134 mg (52%).
[α]D25 (94:6 sin/anti mezcla no separable)= +8.2 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3, diastereoisómero mayoritario sin): δ: 7.40-7.26 (m,
5), 6.74 (d, J= 8.9Hz, 2H), 6.59 (d, J= 8.9 Hz, 2H), 4.14, (d, J= 6.3 Hz, 1H), 3.99
(m, 1H), 3.73 (s, 3H), 1.25 (d, J= 6.3 Hz, 3H). 13C-RMN (125 MHz, CDCl3, diastereoisómero mayoritario sin) δ: 152.5, 141.5,
141.3, 128.7, 127.5, 127.1, 115.6, 114.8, 71.7, 66.1, 55.7, 20.0.
Masa exacta : no determinada.
PhMe
NHPMP
OH

Desarrollo Experimental
209
4.4.2.5. Espectros de 1H y 13C-RMN
1H-RMN (300 MHz)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
13C-RMN (125 MHz)
0102030405060708090100110120130140150
-2000
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
Et Ph
NH2
OH
46
Et Ph
NH2
OH
46

Capítulo 4
210
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
13C-RMN (125 MHz)
0102030405060708090100110120130140150160
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
PentMe
NHPMP
OHn
49
PentMe
NHPMP
OHn
49

Desarrollo Experimental
211
1H-RMN (300M Hz)
1.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
13C-RMN (125 MHz)
102030405060708090100110120130140150160170
-2000
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
PhMe
NHPMP
OH
50
PhMe
NHPMP
OH
50

Capítulo 4
212
4.4.2.6. Cromatogramas de HPLC Chiralpak AD-H, 1 mL/min, hexano:etanol 90:10, λ= 210 nm
AU
0,00
0,10
0,20
0,30
0,40
Minutes14,00 15,00 16,00 17,00 18,00 19,00 20,00 21,00 22,00 23,00
14,7
08
19,9
82
AU
0,00
0,50
1,00
1,50
2,00
2,50
3,00
Minutes12,00 13,00 14,00 15,00 16,00 17,00 18,00 19,00 20,00 21,00 22,00 23,00
14,5
08
20,1
59
OH
Me
ONHPh
41A
OH
Me
ONHPh
rac-41A
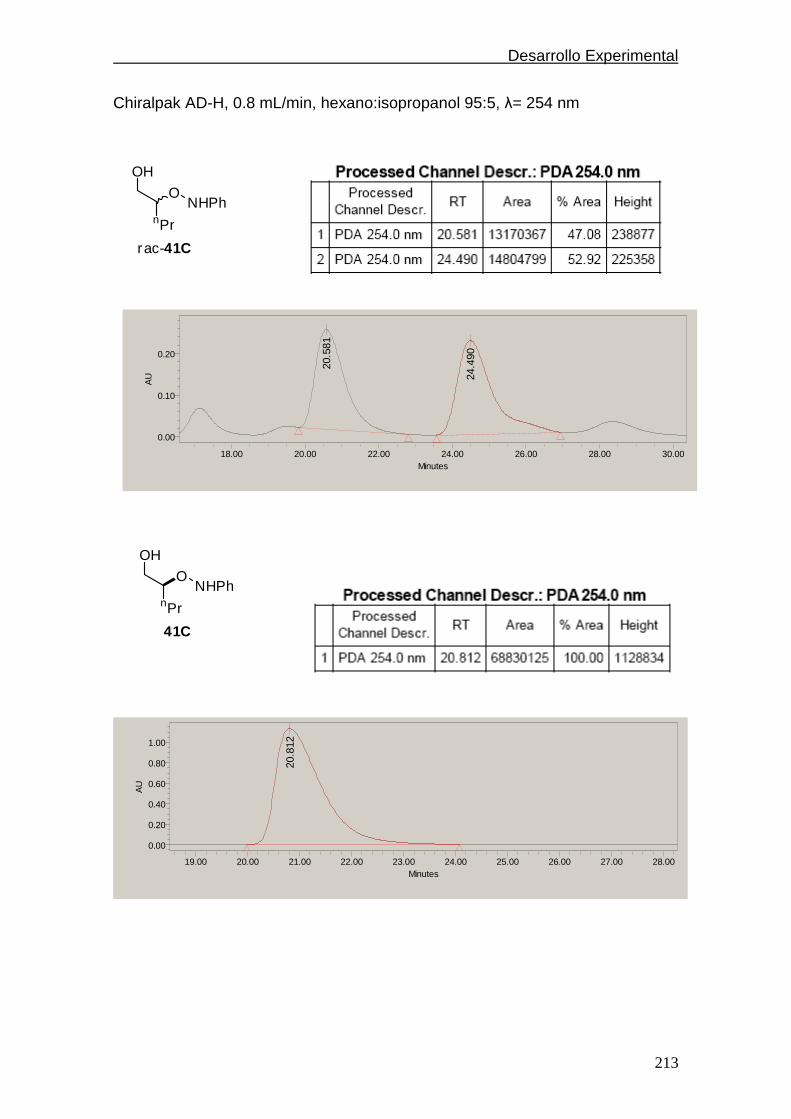
Desarrollo Experimental
213
Chiralpak AD-H, 0.8 mL/min, hexano:isopropanol 95:5, λ= 254 nm
20.8
12
AU
0.00
0.20
0.40
0.60
0.80
1.00
Minutes19.00 20.00 21.00 22.00 23.00 24.00 25.00 26.00 27.00 28.00
OH
nPr
ONHPh
rac-41C
OH
nPr
ONHPh
41C
20.5
81
24.4
90
AU
0.00
0.10
0.20
Minutes18.00 20.00 22.00 24.00 26.00 28.00 30.00

Capítulo 4
214
Chiralpak AD-H, 0.8 mL/min, hexano:isopropanol 95:5, λ= 254 nm
19.3
47
24.6
45
AU
0.00
0.02
0.04
0.06
0.08
Minutes16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00
OH
nBu
ONHPh
rac-41D
OH
nBu
ONHPh
41D
17.8
29
AU
0.00
0.10
0.20
0.30
Minutes16.00 17.00 18.00 19.00 20.00 21.00 22.00 23.00 24.00 25.00 26.00

Desarrollo Experimental
215
Chiralpak AD-H, 0.8 mL/min, hexano:isopropanol 95:5, λ= 210 nm
OH
ONHPh
Ph
41G
OH
ONHPh
Ph
rac-41G
AU
0,00
0,50
1,00
Minutes36,00 38,00 40,00 42,00 44,00 46,00 48,00 50,00 52,00 54,00 56,00 58,00 60,00
40,1
60
54,8
97
AU
0,00
1,00
2,00
Minutes36,00 38,00 40,00 42,00 44,00 46,00 48,00 50,00 52,00 54,00 56,00 58,00
40,0
31

Capítulo 4
216
Chiralpak AD-H, 0.8 mL/min, hexano:isopropanol 95:5, λ= 290 nm
13.9
60
16.1
28
AU
0.00
0.50
1.00
1.50
2.00
Minutes
11.00 12.00 13.00 14.00 15.00 16.00 17.00 18.00 19.00 20.00 21.00
13.8
30
16.2
24
AU
0.00
0.20
0.40
0.60
Minutes
12.00 13.00 14.00 15.00 16.00 17.00 18.00 19.00
OH
ONHPh
Me
rac-41H
Me
OH
ONHPh
Me
41H
Me

Desarrollo Experimental
217
Chiralpak AD-H, 1 mL/min, hexano:isopropanol 95:5, λ= 210 nm
AU
0,00
1,00
2,00
Minutes
45,00 50,00 55,00 60,00 65,00
55,7
85
63,9
77
AU
0,00
0,20
0,40
0,60
Minutes50,00 55,00 60,00 65,00 70,00 75,00
56,3
09
63,5
68
OH
CH2OBn
ONHPh
r ac-41J
OH
CH2OBn
ONHPh
41J

Capítulo 4
218
Chiralpak OD-H, 1 mL/min, hexano:isopropanol 92:8, λ= 210 nm
26.5
33
30.3
42
AU
-0.10
0.00
0.10
0.20
0.30
0.40
Minutes20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00
30.4
09
AU
0.00
0.20
0.40
0.60
Minutes20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00
OH
(CH2)4NHBoc
ONHPh
rac-41K
OH
(CH2)4NHBoc
ONHPh
41K

Desarrollo Experimental
219
Chiralpak AD-H, 1 mL/min, hexano:isopropanol 94:6, λ= 210 nm
AU
0,00
0,20
0,40
0,60
Minutes15,00 20,00 25,00 30,00 35,00
15,8
62
19,5
54
AU
0,00
1,00
2,00
Minutes12,00 14,00 16,00 18,00 20,00 22,00 24,00
15,8
53
OH
ONHPh
rac-41L
OH
ONHPh
41L

Capítulo 4
220
4.4.2.7. Estudios computacionales para la reacción O-nitrosoaldólica
Coordenadas cartesianas de los puntos estacionarios de la reacción N-
nitrosoaldólica y O-nitrosoaldólica de la enamina derivada del propionaldehído
y el catalizador 3 con el nitrosobenceno en presencia de agua (TSN-agua y
TSO-agua ), o dos moléculas de ácido benzoico (TSN-2xPhCO 2H y TSO-
2xPhCO 2H) o dos moléculas de ácido p-nitrobenzoico (TSN-2xp-C6H4CO2H y
TSO-2xp-C6H4CO2H).
TSN-agua Atom X Y Z C -0.4319 0.2964 -1.2471 H -4.7455 4.1423 -1.6409 C -3.8645 0.6918 2.3424 O -4.7483 3.6318 -0.8176 C -2.1595 0.6013 0.4993 H -4.7374 2.6881 -1.1208 C -3.3824 1.1141 0.9766 C -1.5801 2.4356 -1.1255 C -1.3291 2.1544 -2.6061 C -0.2476 1.0542 -2.5884 H -4.9566 0.7603 2.3892 H -0.8602 -0.6950 -1.4028 H -1.8245 -0.3679 0.8481 H -3.6289 2.1428 0.7311 H -2.5552 2.8778 -0.9292 H -0.7969 3.0810 -0.7044 H -2.2551 1.7807 -3.0520 H -1.0064 3.0457 -3.1533 H -0.3336 0.3732 -3.4389 H 0.7469 1.5019 -2.6396 H -3.4557 1.3553 3.1152 H -3.5652 -0.3321 2.5891 N -1.4569 1.0914 -0.5287 C -4.3291 -1.0501 -0.4649 C -4.8292 -1.8857 0.5422 C -4.7485 -3.2716 0.4051 C -4.1929 -3.8366 -0.7455 C -3.7318 -3.0008 -1.7673 C -3.8061 -1.6158 -1.6345 H -5.3013 -1.4471 1.4144 H -5.1395 -3.9119 1.1919 H -4.1420 -4.9166 -0.8552 H -3.3222 -3.4337 -2.6774 H -3.4848 -0.9451 -2.4231 N -4.5047 0.3952 -0.3318 O -4.2929 1.0070 -1.4485 C 0.8953 0.0616 -0.4134 O 0.4711 -0.7580 0.6816 Si 1.1606 -1.9075 1.7099 C 1.5613 -3.5203 0.8186 H 0.6901 -3.8942 0.2684 H 1.8420 -4.2869 1.5528
NMe3SiO O
NPh
PhPh
HO
H

Desarrollo Experimental
221
H 2.3884 -3.4177 0.1096 C 2.6927 -1.2602 2.6046 H 2.5077 -0.2875 3.0734 H 3.5518 -1.1527 1.9345 H 2.9806 -1.9636 3.3967 C -0.2225 -2.2122 2.9540 H -1.1252 -2.5895 2.4589 H -0.4939 -1.2952 3.4899 H 0.0798 -2.9559 3.7016 C 1.9358 -0.7133 -1.2521 C 3.8451 -2.2851 -2.6279 C 1.5473 -1.6231 -2.2486 C 3.3054 -0.6229 -0.9542 C 4.2489 -1.3972 -1.6308 C 2.4883 -2.3940 -2.9321 H 0.5009 -1.7524 -2.5002 H 3.6418 0.0639 -0.1856 H 5.3013 -1.3013 -1.3769 H 2.1551 -3.0853 -3.7016 H 4.5778 -2.8858 -3.1598 C 1.4203 1.4069 0.1233 C 2.2090 3.9424 1.0904 C 2.2440 2.2507 -0.6404 C 1.0027 1.8632 1.3819 C 1.3919 3.1148 1.8609 C 2.6347 3.5029 -0.1630 H 2.6081 1.9245 -1.6090 H 0.3585 1.2340 1.9838 H 1.0515 3.4429 2.8396 H 3.2775 4.1312 -0.7740 H 2.5132 4.9166 1.4634 TSO-agua Atom X Y Z C 0.4856 -0.1204 -2.9011 C 0.1359 0.0140 -1.4049 H 5.2536 -1.9821 -2.8646 C 1.3277 -2.1200 -1.7731 C 1.2348 -1.1100 0.4993 O 4.3537 -1.6235 -2.8712 C 2.5029 -2.0330 2.4740 C 2.3806 -1.8006 0.9892 H 4.2643 -1.1711 -1.9921 C 0.8448 -1.6014 -3.1358 C -2.0464 -1.3252 -1.4053 C -2.0877 -2.3671 -0.4683 C -2.0910 3.5372 -2.4773 C -4.1755 2.3440 -2.3393 C -3.4967 1.2238 -1.8586 C -2.4823 2.7290 1.4629 C -1.4146 2.4178 -1.9892 C -3.4740 3.5080 -2.6544 C -2.1039 1.2349 -1.6754 C -4.1678 0.1267 1.5228 C -1.5748 0.3370 3.1677 C -1.3980 0.0201 -1.0309 C -2.6105 -3.6174 -0.8044 C -3.0634 -2.8277 -3.0330

Capítulo 4
222
C -2.5397 -1.5788 -2.6952 C -3.1019 -3.8540 -2.0882 H 0.8577 -3.0705 -1.4952 H 0.7464 -0.3758 1.1249 H 3.5613 -2.1210 2.7391 H 1.9932 -2.9558 2.7734 H 2.0750 -1.2010 3.0427 H 2.7416 -2.6282 0.3820 H -0.0255 -2.1667 -3.4762 H -0.3239 0.2231 -3.5491 H 1.3564 0.5113 -3.0994 H 0.5470 0.9348 -0.9834 H 2.4100 -2.2397 -1.7705 H -2.1384 0.6977 4.0370 H 1.6390 -1.7051 -3.8788 H -1.4727 3.1545 1.4318 H -2.9831 3.1289 2.3545 H -3.0250 3.0898 0.5838 H -0.5634 0.7550 3.2360 H -1.4933 -0.7527 3.2546 H -4.7578 0.4974 0.6786 H -4.6997 0.4049 2.4419 H -4.1504 -0.9674 1.4665 H -0.3415 2.4934 -1.8548 H -4.0594 0.3262 -1.6274 H -5.2536 2.3018 -2.4705 H -1.5282 4.4356 -2.7153 H -3.9985 4.3808 -3.0338 H -1.7034 -2.1936 0.5297 H -2.5276 -0.7928 -3.4434 H -3.4447 -2.9942 -4.0370 H -2.6335 -4.4069 -0.0576 H -3.5128 -4.8254 -2.3495 N 0.9139 -1.0830 -0.7929 O -1.4055 0.1698 0.3944 Si -2.4217 0.8472 1.5612 C 2.8190 1.9329 0.2854 C 3.6706 1.6677 -1.9796 C 2.7276 3.7922 -1.2787 C 3.4197 1.1094 -0.6981 C 3.3319 2.9846 -2.2555 C 2.4786 3.2530 -0.0148 H 2.6670 1.5329 1.2805 H 2.4727 4.8254 -1.4983 H 3.5386 3.3923 -3.2425 H 2.0261 3.8724 0.7573 H 4.1297 1.0335 -2.7336 N 3.7852 -0.2127 -0.5112 O 3.5722 -0.6277 0.7405
TSN-2xPhCO 2H
Atom X Y Z C -2.0590 0.0655 -1.0720 C -1.2370 -3.7205 2.0780 C -1.5760 -1.8555 0.4240 C -0.7240 -2.7995 1.0060 C -0.3600 -1.5415 -1.7470 C 0.0920 -0.1825 -2.2750

Desarrollo Experimental
223
C -1.1840 0.6815 -2.1950 C -3.8920 -2.6205 -2.0480 C -3.5830 -0.1655 -1.4400 C -6.0600 2.3155 -3.0370 C -3.9260 -3.6765 -2.9600 C -6.1430 1.2535 1.0540 C -7.0590 -1.2555 -0.5080 C -5.4200 -1.5485 2.0940 C -4.2420 1.1545 -1.8950 C -5.5970 3.5455 -2.5680 C -3.7980 2.3995 -1.4230 C -5.3890 1.1385 -2.7050 C -3.5940 -2.1255 -4.7690 C -4.4620 3.5805 -1.7590 C -3.7080 -1.2985 -2.4770 C -3.7750 -3.4355 -4.3250 C -3.5610 -1.0705 -3.8550 H -4.0010 -2.8225 -0.9900 H -4.0690 -4.6915 -2.5980 H -3.4830 -1.9165 -5.8300 H -2.0710 0.6965 -0.1820 H -0.4230 -4.0055 2.7540 H -1.6320 -4.6455 1.6360 H -2.0390 -3.2635 2.6680 H -2.4370 -1.5075 0.9840 H 0.1020 -3.1915 0.4220 H 0.4480 -2.1455 -1.3410 H -0.8830 -2.1215 -2.5200 H 0.8820 0.2215 -1.6380 H 0.4820 -0.2465 -3.2950 H -0.9540 1.7285 -1.9830 H -1.7180 0.6615 -3.1480 H -5.3430 1.7165 1.6440 H -7.0440 1.2325 1.6800 H -6.3460 1.9005 0.1960 H -7.9670 -1.3725 0.0980 H -6.7830 -2.2455 -0.8890 H -7.3140 -0.6255 -1.3650 H -4.6120 -1.1415 2.7130 H -5.1640 -2.5855 1.8460 H -6.3290 -1.5695 2.7080 H -6.1150 4.4645 -2.8300 H -2.9300 2.4655 -0.7770 H -5.7610 0.1935 -3.0870 H -6.9440 2.2685 -3.6680 H -4.0890 4.5285 -1.3810 H -3.8020 -4.2565 -5.0360 H -3.4420 -0.0585 -4.2290 N -1.3370 -1.1775 -0.7010 O -4.1720 -0.5815 -0.2020 Si -5.6840 -0.5045 0.5460 C -0.2960 -0.5785 2.7470 C -0.7110 -1.1045 3.9770 C -1.4150 -0.3055 4.8780 C -1.6910 1.0285 4.5690 C -1.2410 1.5635 3.3560 C -0.5410 0.7705 2.4510 H -0.1580 1.1685 1.5190 H -0.4580 -2.1295 4.2270 H -1.7330 -0.7215 5.8300

Capítulo 4
224
H -2.2310 1.6525 5.2750 H -1.4300 2.6085 3.1230 N 0.4890 -1.4325 1.8760 O 1.0380 -0.7465 0.9250 C 3.2100 -2.9045 -0.4920 C 4.1450 1.5845 -0.9540 C 4.5520 -3.4025 -0.9220 C 4.9800 2.8205 -1.0180 C 7.0110 -4.4355 -1.7680 C 6.4780 5.1725 -1.2050 C 4.6080 -4.6255 -1.6060 C 4.5690 3.8515 -1.8740 C 5.7370 -2.7005 -0.6620 C 6.1450 2.9735 -0.2550 C 6.9610 -3.2175 -1.0860 C 6.8900 4.1485 -0.3500 C 5.8320 -5.1395 -2.0270 C 5.3160 5.0225 -1.9670 H 4.0580 -0.1395 -0.1290 H 2.2630 -1.4315 0.4380 H 3.6800 -5.1545 -1.7950 H 3.6640 3.7105 -2.4560 H 5.7040 -1.7545 -0.1350 H 6.4570 2.1735 0.4070 H 7.8760 -2.6685 -0.8840 H 7.7920 4.2645 0.2440 H 5.8680 -6.0875-2.5560 H 4.9950 5.8195 -2.6330 H 7.9670 -4.8365 -2.0960 H 7.0610 6.0875 -1.2770 O 3.2250 -1.7235 0.1290 O 4.6460 0.6555 -0.1220 O 2.1820 -3.5415 -0.6930 O 3.1150 1.4345 -1.5890 TSO-2xPhCO 2H Atom X Y Z C 1.9525 -0.4825 -0.3140 C 3.3895 -0.2775 0.3130 C 3.8485 1.1865 0.1370 C 3.3985 -0.7415 1.7810 C 0.8095 0.3835 0.2600 C 6.2175 0.1825 -2.2420 C 0.4095 -1.9695 0.8960 C -0.0395 -0.5275 1.1790 C 3.8115 -2.0405 2.1060 C 2.9285 0.0805 2.8180 C 3.7635 -2.5035 3.4220 C 2.8785 -0.3805 4.1340 C 3.2955 -1.6765 4.4430 C 3.4085 1.9765 -0.9380 C 4.8155 1.7335 0.9960 C 5.3215 3.0185 0.7940 C 4.8665 3.7925 -0.2740 C 3.9065 3.2645 -1.1370 C 1.8955 -5.2825 -1.6720 C 1.8985 -2.8915 -0.8670 C 7.0615 -1.0605 0.4660

Desarrollo Experimental
225
C 5.9755 -2.8505 -1.7960 C 1.2415 -4.1255 -0.9820 H 5.2725 -2.9435 -2.6320 H 5.7665 -3.6635 -1.0910 H 2.0755 -0.3165 -1.3870 H 4.1685 -2.6935 1.3180 H 4.0915 -3.5155 3.6470 H 2.5155 0.2785 4.9180 H -0.4055 -2.6205 0.5730 H 0.8885 -2.4255 1.7710 H -1.0995 -0.4035 0.9500 H 0.1125 -0.2865 2.2340 H 0.1895 0.7435 -0.5650 H 1.1905 1.2665 0.7780 H 5.2535 4.7955 -0.4300 H 2.6725 1.6015 -1.6400 H 5.1755 1.1525 1.8380 H 6.0675 3.4135 1.4780 H 3.5405 3.8535 -1.9730 H 3.2595 -2.0345 5.4680 H 2.6125 1.0955 2.6030 H 2.7555 -2.6795 -1.4930 H 0.4265 -4.3565 -0.3050 H 6.9835 -3.0115 -2.1960 H 1.1855 -5.7385 -2.3750 H 2.1895 -6.0575 -0.9530 H 2.7855 -4.9775 -2.2330 H 5.4955 0.1625 -3.0660 H 7.2145 0.0165 -2.6700 H 6.2015 1.1875 -1.8080 H 8.0675 -1.3185 0.1130 H 6.7995 -1.7595 1.2680 H 7.1135 -0.0565 0.9000 N 1.4345 -1.8595 -0.1680 O 4.2275 -1.1385 -0.4680 Si 5.8425 -1.1595 -0.9720 H -1.2935 -2.2055 -1.8840 H -3.3815 -0.5065 -0.6350 C -3.5905 3.9275 -0.6310 C -6.0605 3.7255 0.6550 C -5.5345 4.9815 0.3430 C -5.3545 2.5695 0.3250 C -3.3245 1.4515 -0.6960 C -4.2975 5.0815 -0.3000 C -4.1145 2.6655 -0.3200 H -5.7535 1.5895 0.5620 H -7.0225 3.6475 1.1550 H -3.8875 6.0575 -0.5440 H -2.6295 3.9795 -1.1310 H -6.0875 5.8815 0.6000 O -3.9165 0.3165 -0.3530 O -2.2375 1.5285 -1.2620 C -6.7465 -2.9065 -0.3260 C -4.6685 -4.3055 0.8990 C -4.3705 -3.2825 -0.0110 C -5.9905 -4.6175 1.2060 C -7.0345 -3.9185 0.5920 C -2.9235 -2.9805 -0.3310 C -5.4205 -2.5895 -0.6260 H -7.5545 -2.3655 -0.8120

Capítulo 4
226
H -6.2115 -5.4075 1.9190 H -3.8425 -4.8445 1.3520 H -5.1925 -1.8055 -1.3390 H -8.0675 -4.1635 0.8270 O -2.0515 -3.7895 0.0320 O -2.6715 -1.8905 -0.9890 C -0.3765 -0.1295 -3.2530 C 1.8535 -0.7295 -4.8440 C 0.1565 -1.4315 -3.2730 C 1.2735 -1.7345 -4.0720 C 1.3315 0.5665 -4.8320 C 0.2165 0.8575 -4.0350 H -1.2285 0.1115 -2.6220 H 1.7845 1.3425 -5.4420 H 2.7115 -0.9645 -5.4680 H -0.1985 1.8605 -4.0160 H 1.6445 -2.7525 -4.0980 N -0.4515 -2.4155 -2.4950 O 0.0335 -3.6005 -2.4440 TSN-2xp-C6H4CO2H Atom X Y Z C -2.6735 -0.1065 -1.0140 C -2.0535 -4.0005 2.0500 C -2.2685 -2.0755 0.4440 C -1.4855 -3.0805 1.0070 C -0.9665 -1.7135 -1.6690 C -0.4795 -0.3445 -2.1380 C -1.7505 0.5285 -2.0870 C -4.4865 -2.7455 -2.1320 C -4.1845 -0.3105 -1.4460 C -6.5505 2.2545 -3.0820 C -4.4895 -3.7775 -3.0710 C -6.8625 1.0475 0.9550 C -7.6915 -1.4195 -0.7180 C -6.1915 -1.7795 1.9570 C -4.8095 1.0315 -1.8870 C -6.1025 3.4605 -2.5430 C -4.3815 2.2525 -1.3430 C -5.9095 1.0575 -2.7580 C -4.0765 -2.1825 -4.8240 C -5.0145 3.4525 -1.6700 C -4.2775 -1.4135 -2.5180 C -4.2815 -3.5015 -4.4220 C -4.0745 -1.1515 -3.8830 H -4.6405 -2.9725 -1.0840 H -4.6525 -4.8005 -2.7410 H -3.9215 -1.9475 -5.8740 H -2.7115 0.4995 -0.1080 H -1.2675 -4.3225 2.7420 H -2.4675 -4.9065 1.5860 H -2.8545 -3.5265 2.6260 H -3.1375 -1.7205 0.9860 H -0.6435 -3.4725 0.4470 H -0.1825 -2.3395 -1.2500 H -1.4635 -2.2645 -2.4800 H 0.2865 0.0295 -1.4540 H -0.0455 -0.3825 -3.1410

Desarrollo Experimental
227
H -1.5245 1.5695 -1.8430 H -2.2465 0.5335 -3.0600 H -6.0925 1.4975 1.5920 H -7.7915 1.0075 1.5390 H -7.0305 1.7165 0.1060 H -8.6345 -1.5345 -0.1670 H -7.4045 -2.4075 -1.0960 H -7.8935 -0.7765 -1.5810 H -5.4195 -1.3855 2.6300 H -5.9165 -2.8085 1.6970 H -7.1315 -1.8205 2.5200 H -6.5965 4.3945 -2.7970 H -3.5505 2.2855 -0.6480 H -6.2705 0.1335 -3.1950 H -7.3985 2.2405 -3.7620 H -4.6555 4.3815 -1.2360 H -4.2845 -4.3045 -5.1540 H -3.9365 -0.1315 -4.2260 N -1.9785 -1.3675 -0.6520 O -4.8245 -0.7525 -0.2420 Si -6.3765 -0.6955 0.4250 C -1.0085 -0.8925 2.8580 C -1.4815 -1.4465 4.0560 C -2.2135 -0.6615 4.9460 C -2.4605 0.6835 4.6580 C -1.9575 1.2435 3.4790 C -1.2295 0.4655 2.5840 H -0.8065 0.8835 1.6770 H -1.2515 -2.4805 4.2870 H -2.5765 -1.0955 5.8740 H -3.0235 1.2965 5.3570 H -2.1275 2.2955 3.2640 N -0.2055 -1.7395 2.0090 O 0.3985 -1.0475 1.0970 C 2.5265 -3.2565 -0.2250 C 3.7225 1.0455 -0.6500 C 3.8825 -3.8385 -0.5060 C 4.6085 2.2525 -0.6930 C 6.3295 -5.0005 -1.0700 C 6.1785 4.5225 -0.8370 C 3.9515 -4.9675 -1.3350 C 4.2855 3.2715 -1.5980 C 5.0565 -3.3025 0.0410 C 5.7275 2.3825 0.1400 C 6.2895 -3.8835 -0.2390 C 6.5215 3.5235 0.0720 C 5.1765 -5.5555 -1.6250 C 5.0705 4.4165 -1.6780 H 3.5115 -0.6425 0.2210 H 1.5625 -1.7505 0.6920 H 3.0305 -5.3695 -1.7410 H 3.4145 3.1475 -2.2320 H 5.0065 -2.4345 0.6880 H 5.9705 1.5905 0.8390 H 7.2105 -3.4895 0.1720 H 7.3925 3.6515 0.7030 H 5.2565 -6.4255 -2.2650 H 4.8445 5.2185 -2.3690 N 7.6335 -5.6195 -1.3700 N 7.0155 5.7335 -0.9120

Capítulo 4
228
O 2.5495 -2.1195 0.4630 O 4.1355 0.1275 0.2340 O 1.5025 -3.8075 -0.6040 O 2.7335 0.9215 -1.3500 O 8.6345 -5.1145 -0.8640 O 7.9875 5.8035 -0.1620 O 7.6415 -6.6025 -2.1100 O 6.6895 6.6025 -1.7210 TSO-2xp-C6H4CO2H
Atom X Y Z C 2.4800 -0.8050 -0.0860 C 3.9560 -0.5930 0.4550 C 4.2990 0.9120 0.4790 C 4.1100 -1.2860 1.8230 C 1.3750 -0.0410 0.6840 C 6.5660 0.4530 -2.1680 C 0.9450 -2.4320 0.9220 C 0.2660 -1.0650 0.9970 C 4.5940 -2.6000 1.8900 C 3.7060 -0.6760 3.0230 C 4.6680 -3.2830 3.1050 C 3.7780 -1.3570 4.2390 C 4.2580 -2.6660 4.2870 C 3.7530 1.8020 -0.4610 C 5.2640 1.4140 1.3670 C 5.6580 2.7520 1.3300 C 5.0940 3.6250 0.4000 C 4.1390 3.1420 -0.4950 C 2.8280 -5.4860 -1.7670 C 2.5940 -3.1690 -0.8010 C 7.6550 -1.0890 0.2910 C 6.5590 -2.6220 -2.1550 C 2.0700 -4.4470 -1.0030 H 5.8080 -2.6460 -2.9540 H 6.4540 -3.5410 -1.5670 H 2.5040 -0.5160 -1.1400 H 4.9130 -3.0920 0.9790 H 5.0490 -4.3010 3.1250 H 3.4620 -0.8560 5.1500 H 0.2510 -3.2100 0.5930 H 1.4060 -2.7240 1.8740 H -0.5330 -1.0120 0.2550 H -0.1820 -0.8970 1.9810 H 0.9880 0.7860 0.0870 H 1.7770 0.3870 1.6030 H 5.3940 4.6690 0.3730 H 3.0250 1.4620 -1.1890 H 5.7120 0.7540 2.1020 H 6.4050 3.1100 2.0330 H 3.6930 3.8070 -1.2300 H 4.3160 -3.1950 5.2330 H 3.3540 0.3500 3.0200 H 3.4540 -2.8460 -1.3760 H 1.2540 -4.8000 -0.3850 H 7.5470 -2.6470 -2.6320 H 2.1620 -5.9680 -2.4930 H 3.2040 -6.2730 -1.0990

Desarrollo Experimental
229
H 3.6790 -5.0600 -2.3080 H 5.7850 0.4980 -2.9360 H 7.5360 0.4200 -2.6820 H 6.5240 1.3830 -1.5930 H 8.6560 -1.2100 -0.1440 H 7.4940 -1.9160 0.9910 H 7.6570 -0.1570 0.8650 N 2.0310 -2.2190 -0.0610 O 4.7830 -1.2500 -0.5130 Si 6.3660 -1.0790 -1.0880 H -0.6270 -2.6490 -1.9890 H -3.0470 -1.2180 -1.0120 C -3.5500 3.1710 -0.5840 C -6.2450 2.7130 0.0310 C -5.7180 4.0030 -0.0080 C -5.4020 1.6400 -0.2450 C -3.1140 0.7320 -0.8540 C -4.3800 4.2520 -0.3120 C -4.0530 1.8640 -0.5530 H -5.7820 0.6250 -0.2220 H -7.2910 2.5690 0.2730 H -4.0170 5.2720 -0.3300 H -2.5030 3.3190 -0.8240 N -6.6050 5.1440 0.2820 O -7.7800 4.9000 0.5520 O -6.1160 6.2730 0.2380 O -3.7000 -0.4530 -0.8260 O -1.9280 0.9230 -1.0970 C -6.1440 -3.9090 -0.8780 C -4.0470 -5.0030 0.6180 C -3.7790 -4.0230 -0.3480 C -5.3480 -5.4300 0.8560 C -6.3810 -4.8720 0.1010 C -2.3410 -3.5960 -0.5810 C -4.8360 -3.4870 -1.0960 H -6.9750 -3.5100 -1.4470 H -5.5770 -6.1800 1.6030 H -3.2120 -5.4230 1.1690 H -4.6260 -2.7390 -1.8520 N -7.7610 -5.3170 0.3440 O -1.4390 -4.3150 -0.1240 O -2.1560 -2.5110 -1.2630 O -7.9450 -6.1710 1.2130 O -8.6560 -4.8100 -0.3330 C 0.2520 -0.3880 -3.0790 C 2.5680 -0.7300 -4.6250 C 0.8570 -1.6530 -3.1900 C 2.0190 -1.8260 -3.9640 C 1.9710 0.5310 -4.5260 C 0.8150 0.6920 -3.7520 H -0.6320 -0.2490 -2.4650 H 2.4000 1.3800 -5.0510 H 3.4580 -0.8630 -5.2330 H 0.3430 1.6670 -3.6680 H 2.4500 -2.8160 -4.0580 N 0.2820 -2.7380 -2.5240 O 0.8470 -3.8810 -2.5330

Capítulo 4
230
4.5. Procedimientos experimentales del capítulo 3
4.5.1. Reacción de Mannich anti -selectiva con N-tosil-iminas
4.5.1.1. Síntesis de N-sulfonil-iminas aromáticas 18
Procedimiento general:
Una mezcla del aldehído (7.7 mmol), la sulfonamida correspondiente (7
mmol), MS 4Å en polvo activado19 (2.8 g, 400 mg/mmol) y resina Amberlyst-15
(35 mg/mmol) en tolueno seco (15 mL) se calentó en un tubo sellado,
previamente flameado, a 140 ºC durante 16 horas. La mezcla se filtró a través
de un lecho de 2 cm de celita y se concentró. El sólido amarillo resultante se
disgregó con hexano y se filtró, tras lo cual se cristalizó en hexano:DCM para
dar la sulfonil-imina correspondiente pura.
2,4,6-Trimetil- N-(4-metilbenciliden)bencenosulfonamida (79a) 20
Preparado según el procedimiento general a partir
de p-metilbenzaldehído (0.91 mL, 7.7 mmol) y
mesitilensulfonamida (1,39 g, 7 mmol). Sólido blanco.
Rendimiento: 1.43 g (68%). Los datos físicos y
espectroscópicos resultaron coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.98 (s, 1H), 7.81 (s, J= 8.0 Hz, 2H), 7.33 (d, J=
9.1 Hz, 2H), 6.97 (s, 2H), 2.70 (s, 6H), 2.43 (s, 3H), 2.30 (s, 3H).
18 Adaptado de: J. Esquivias, R. Gómez Arrayás, J. C. Carretero, Angew. Chem. Int. Ed. 2006,
45, 629-633. 19 Calentados a 80ºC en baño de arena durante 2 horas a presión reducida (bomba de vacío 2-4 mbar). 20 M. Ochiai, Y. Kitagawa, J. Org. Chem. 1999, 64, 3181-3189.
NSO2Mes
Me

Desarrollo Experimental
231
4-Metil- N-(4-metibencilideno)bencenosulfonamida (81a) 21
Preparado según el procedimiento general a partir de
p-metilbenzaldehído (0.91 mL, 7.7 mmol) y p-
toluensulfonamida (1,20 g, 7 mmol). Sólido blanco.
Rendimiento: 1.51 g (79%). Los datos físicos y espectroscópicos resultaron
coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.98 (s, 1H), 7.96-7-76 (m, 4H), 7.36-7.26 (m,
4H), 2.43 (s, 6H).
N-(4-Metibencilideno)piridin-2-sulfonamida (82a) 18
Preparado según el procedimiento general a partir
de p-metilbenzaldehído (0.91 mL, 7.7 mmol) y 2-
piridilsulfonamida (1,10 g, 7 mmol). Sólido amarillo.
Rendimiento: 1.09 g (60%). Los datos físicos y
espectroscópicos resultaron coincidentes con los de la
bibliografía.
1H-RMN (300 MHz, CDCl3): δ: 9.27 (s, 1H), 8.77-8-69 (m, 1H), 8.29-8.23 (m,
1H), 8.05-7.94 (m, 2H), 7.69-7.47 (m, 2H), 2.44 (s, 3H).
N-Bencilideno-4-metilbenzenosulfonamida (81b) 21
Preparado según el procedimiento general a partir de
benzaldehído (0.78 mL, 7.7 mmol) y p-toluensulfonamida (1,20 g,
7 mmol). Sólido blanco. Rendimiento: 1.45 g (80%). Los datos
físicos y espectroscópicos resultaron coincidentes con los de la
bibliografía.
1H-RMN (300 MHz, CDCl3): δ: 9.03 (s, 1H), 7.91-7.87 (m, 4H), 7.62-7.42
(m, 3H), 7.34 (d, J= 8.2 Hz, 2H), 2.43 (s, 3H).
21 R. Fan, D. Pu, F. Wen, Y. Ye, X. Wang, J. Org. Chem. 2008, 73, 3623-3625.
NTos
Me
NTos
O2SN
Me
N

Capítulo 4
232
N-(4-Metoxibencilideno)-4-metilbenzenosulfonamida (8 1c)21
Preparado según el procedimiento general a partir de
p-metoxibenzaldehído (0.93 mL, 7.7 mmol) y p-
toluensulfonamida (1,20 g, 7 mmol). Sólido blanco.
Rendimiento: 1.41 g (71%).
1H-RMN (300 MHz, CDCl3): δ: 8.93 (s, 1H), 7.91-7.86 (m, 4H), 7.34-6.97 (m,
4H), 3.87 (s, 3H), 2.43 (s, 3H).
4-Metil- N-(3-metilbencilideno)bencenosulfonamida (81d) 21
Preparado según el procedimiento general a partir de m-
metilbenzaldehído (0.91 mL, 7.7 mmol) y p-toluensulfonamida
(1,20 g, 7 mmol). Sólido blanco. Rendimiento: 1.32 g (69%). Los
datos físicos y espectroscópicos resultaron coincidentes con los
de la bibliografía.
1H-RMN (300 MHz, CDCl3): δ: 8.99 (s, 1H), 7.91-7.88 (m, 2H), 7.74 (s, 1H),
7.61-7.59 (m, 1H), 7.40-7.35 (m, 4H), 2.45 (s, 3H), 2.39 (s, 3H).
N-(4-Clorobencilideno)-4-metilbencenosulfonamida (81 e)21
Preparado según el procedimiento general a partir de p-
clorobenzaldehído (1.08 g, 7.7 mmol) y p-toluensulfonamida
(1,20 g, 7 mmol). Sólido blanco. Rendimiento: 2.94 g (70%).
Los datos físicos y espectroscópicos resultaron coincidentes
con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 9.00 (s, 1H), 7.92-7.85 (m, 4H), 7.48-7.34 (m,
4H), 2.44 (s, 3H).
NTos
MeO
NTos
Me
NTos
Cl

Desarrollo Experimental
233
4.5.1.2. Síntesis de la N-Boc-imina 80a 22
En un matraz previamente secado en el horno (80 ºC) durante 16 horas y
flameado, se adicionó K2CO3 anhidro (4.0 g) y, tras flamear nuevamente se
añadió terc-butil p-tolil(tosil)metilcarbamato (2.63 g, 7 mmol) y THF anhidro (70
mL) bajo atmósfera de nitrógeno. La mezcla resultante se calentó a reflujo
durante 16 horas. Transcurrido este tiempo la mezcla de reacción se filtró sobre
un lecho de celita de 2 cm y se evaporó el disolvente a presión reducida,
manteniendo la atmósfera de nitrógeno. El correspondiente terc-butil p-
metilbencilidencarbamato (80a) se obtuvo como un aceite transparente que se
almacenó posteriormente bajo atmósfera de nitrógeno. Rendimiento: 1.15 g
(75%). Los datos físicos y espectroscópicos resultaron coincidentes con los de
la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.87 (s, 1H), 7.93-7.89 (m, 2H), 7.58-7.42 (m,
2H), 1.43 (s, 3H), 1.59 (s, 9H).
4.5.1.3. Reacción de Mannich entre aldehídos y N-to sil iminas:
procedimiento general
A una disolución de la imina 82 (0.5 mmol, 1 equiv.), ácido p-nitrobenzoico
(17 mg, 0.1 mmol) y el catalizador (0.1 mmol, 20 mol%) en DMF (2 mL) a −60
22 Procedimiento adaptado de: A. G. Wenzel, E. N. Jacobsen, J. Am. Chem. Soc. 2002, 124, 12964-12965. Para más información sobre la preparación de este tipo de iminas, ver Tesis Doctoral de Idoia Múgica Mendiola, Reacción de aza-Henry catalítica y asimétrica en condiciones de transferencia de fase, Universidad del País Vasco, 2009.

Capítulo 4
234
ºC se añadió el aldehído (1.5 mmol, 3 equiv.) y la disolución resultante se agitó
a −60 ºC durante 24 horas. Posteriormente, se adicionaron a esta temperatura
EtOH (1 mL) y NaBH4 (4.5 mmol, 8equiv.), y tras agitar la mezcla durante 30
min a −40 ºC se adicionó a dicha temperatura una disolución acuosa saturada
de NaCl (2 mL) y se permitió que la mezcla alcanzara temperatura ambiente.
Tras extraer con Et2O (3 x 4 mL), la combinación de las fases orgánicas se lavó
con una disolución acuosa saturada de NaCl (3 x 15 mL), se secó con MgSO4,
se filtró y se evaporó el disolvente bajo presión reducida. El crudo resultante se
purificó mediante cromatografía de columna flash. La diastereoselectividad del
proceso se determinó por análisis de 1H-NMR del crudo.
N-[(1S,2R)-3-Hidroxi-2-metil-1-p-tolilpropil]-4-metilbenceno sulfonamida
(87Aa)
Se siguió el procedimiento general partiendo
de propanal (0.11 mL, 1.5 mmol) y N-tosil-tolilimina
(137 mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:AcOEt 80:20) y se disgregó con Et2O.
Sólido blanco. Rendimiento: 130 mg (78%).
P.f.: 104-106 ºC.
[α]D25 = −68.2 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.53 (d, J= 8.3 Hz, 2H), 7.13 (d, J= 8.0 Hz, 2H),
6.97 (d, J= 7.9 Hz, 2H), 6.84 (d, J= 8.1 Hz, 2H), 5.65 (d, J= 7.5 Hz, 1H), 4.17 (t,
J= 8.1 Hz, 1H), 3.97 (dd, J= 11.0 Hz, 1H), 3.58 (dd, J= 4.5, 11.2 Hz, 1H), 2.39
(s, 3H), 2.30 (s, 3H), 2.02-1.89 (m, 1H), 0.81 (d, J= 7.0 Hz, 3H). 13C-RMN (125 MHz, CDCl3) δ: 142.8, 137.5, 137.0, 136.8, 129.27, 128.9, 127.1,
126.9, 65.0, 61.4, 40.9, 21.4, 21.0, 14.5.
Masa exacta: no determinada.
Cristalografía de rayos X (ver sección 4.5.1.6.).
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 97:3, flujo= 1 mL/min, tr: 126.7 min
(minoritario) y 137.5 min (mayoritario)).
Me
NHTos
HO
Me

Desarrollo Experimental
235
N-[(1S,2R)-3-Hidroxi-2-metil-1-fenilpropil]-4-metilbencenosu lfonamida
(87Ab)
Se siguió el procedimiento general partiendo de
propanal (0.11 mL, 1.5 mmol) y N-tosil-fenilimina (129 mg,
0.5 mmol). El producto crudo se purificó por cromatografía
de columna flash (hexano:AcOEt 80:20) y se disgregó con Et2O. Sólido blanco.
Rendimiento: 123 mg (77%).
P.f.: 127-130 ºC.
[α]D25 = −82.2 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.52 (d, J= 8.3 Hz, 2H), 7.18-7.08 (m, 5H), 7.00-
6.96 (m, 2H), 5.81 (s, 1H), 4.22 (m, 1H), 3.96 (dd, J= 3.1, 11.2 Hz, 1H), 3.59
(dd, J= 4.6, 11.2 Hz, 1H), 2.38 (s, 3H), 1.98 (m, 1H), 0.82 (d, J= 7.0 Hz, 3H).
13C-RMN (125 MHz, CDCl3): δ 142.9, 139.9, 137.3, 129.6, 129.2, 128.2, 127.1,
126.4, 64.9, 61.5, 40.9, 21.4, 14.5.
Masa exacta calculada para C17H21NO3S (M+H-OH)+: 302.1215, encontrada:
302.1252.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IA, hexano:isopropanol 95:5, flujo= 0.8 mL/min, tr: 36.7 min
(minoritario) y 43.7 min (mayoritario)).
N-[(1S,2R)-2-(Hidroximetil)-1- p-tolilbutil]-4-metilbencenosulfonamida
(87Ba)
Se siguió el procedimiento general partiendo
de butanal (0.14 mL, 1.5 mmol) y N-tosil-tolilimina
(137 mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:AcOEt
80:20). Aceite amarillo. Rendimiento: 134 mg (77%).
[α]D25 = −19.8 (c= 0.5, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.54 (d, J= 8.3 Hz, 2H), 7.13 (d, J= 8.2 Hz, 2H),
6.97 (d, J= 7.9 Hz, 2H), 6.87 (d, J= 8.1 Hz, 2H), 5.81 (d, J= 7.9 Hz, 1H), 4.35 (t,
J= 7.8 Hz, 1H), 3.93 (d, J= 11.1 Hz, 1H), 3.72-3.63 (m, 1H), 2.39 (s, 3H), 2.30
(s, 3H), 1.66-1.53 (m, 1H), 1.44-1.18 (m, 2H), 0.88 (t, J= 7.4 Hz, 3H).
Me
NHTos
HO
Et
NHTos
HO
Me

Capítulo 4
236
13C-RMN (125 MHz, CDCl3): δ 142.8, 137.7, 137.4, 136.8, 129.2, 128.9, 127.1,
126.6, 61.2, 60.0, 47.6, 29.7, 21.4, 21.0, 11.6.
Masa exacta calculada para C19H25NO3S (M+H-OH)+: 330.1528, encontrada:
330.1522.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 95:5, flujo= 0.8 mL/min, tr: 51.5
min (minoritario) y 61.8 min (mayoritario)).
N-[(1S,2R)-2-(Hidroximetil)-1fenilbutil]-4-metilbenzenosulfo namida (87Bb)
Se siguió el procedimiento general partiendo de
butanal (0.14 mL, 1.5 mmol) y N-tosil-fenilimina (129 mg,
0.5 mmol). El producto crudo se purificó por cromatografía
de columna flash (hexano:AcOEt 80:20) y se disgregó con
Et2O. Sólido blanco. Rendimiento: 122 mg (68%).
P.f.: 123-125 ºC.
[α]D25 = +72.7 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.53 (d, J= 8.3 Hz, 2H), 7.17-7.08 (m, 5H), 7.02-
6.99 (m, 2H), 5.95 (d, J= 8.07 Hz, 1H), 4.42 (t, J= 7.7 Hz, 1H), 3.91 (m, 1H),
3.38 (m, 1H), 2.38 (s, 3H), 1.69-1.59 (m, 1H), 1.49-1.21 (m, 2H), 0.89 (t, J= 7.4
Hz, 3H). 13C-RMN (125 MHz, CDCl3): δ 142.8, 140.4, 137.7, 129.2, 128.3, 127.0, 126.7,
61.3, 60.2, 47.5, 21.4, 21.1, 11.6.
Masa exacta calculada para C18H23NO3S (M+H-OH)+: 316.1371, encontrada:
316.1411.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IA, hexano:isopropanol 95:5, flujo= 0.75 mL/min, tr: 51.3 min
(minoritario) y 57.7 min (mayoritario)).
N-[(1S,2R)-2-(Hidroximetil)-1-(4-metoxifenil)butil]-4-metilb enceno
sulfonamida (87Bc)
Se siguió el procedimiento general partiendo
de butanal (0.14 mL, 1.5 mmol) y N-tosil-4-
Et
NHTos
HO
Et
NHTos
HO
OMe

Desarrollo Experimental
237
metoxifenilimina (145 mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:AcOEt 80:20) y se disgregó con Et2O.
Sólido blanco. Rendimiento: 114 mg (63%).
P.f.: 117-121 ºC.
[α]D25= +58.4 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.53 (d, J= 8.3 Hz, 2H), 7.13 (d, J= 8.0 Hz, 2H),
6.91 (d, J= 8.6 Hz, 2H), 6.68 (d, J= 8.7 Hz, 2H), 5.97 (d, J= 7.8 Hz, 1H), 4.33 (t,
J= 7.7 Hz, 1H), 3.94 (d, J= 11.2 Hz, 1H), 3.77 (s, 3H), 3.68 (d, J= 8.0 Hz, 1H),
2.38 (s, 3H), 2.29 (s, 1H), 1.67-1.55 (m, 1H), 1.44-1.16 (m, 2H), 0.87 (t, J= 7.4
Hz, 3H). 13C-RMN (125 MHz, CDCl3): δ 158.7, 142.8, 137.7, 132.5, 129.2, 127.9, 127.1,
113.6, 61.3, 59.8, 55.2, 47.6, 21.4, 21.0, 11.6.
Masa exacta calculada para C19H25NO4S (M+H-C7H8NO2S)+: 193.1229,
encontrada: 193.1260.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IA, hexano:isopropanol 95:5, flujo= 0.75 mL/min, tr: 79.6 min
(minoritario) y 92.3 min (mayoritario)).
N-[(1S,2R)-2-(Hidroximetil)-1- m-tolilbutil]-4-metilbenzenosulfonamida
(87Bd)
Se siguió el procedimiento general partiendo de
butanal (0.14 mL, 1.5 mmol) y N-tosil-3-metilfenilimina
(137 mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:AcOEt 80:20).
Aceite amarillo. Rendimiento: 104 mg (60%).
[α]D25 = −39.8 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.51 (d, J= 8.3 Hz, 2H), 7.13-7.01 (m, 3H), 6.94
(d, J= 7.5 Hz, 1H), 6.83 (d, J= 7.6 Hz, 1H), 6.68 (s, 1H), 6.10 (d, J= 8.1 Hz, 1H),
4.33 (t, J= 7.9 Hz, 1H), 3.93 (d, J= 11.1 Hz, 1H), 3.68 (d, J= 10.7 Hz, 1H), 2.51
(bs, 1H), 2.37(s, 3H), 2.17(s, 3H), 1.70-1.57 (m, 1H), 1.47-1.20 (m, 2H), 0.88 (t,
J= 7.4 Hz, 3H).
13C-RMN (125 MHz, CDCl3): δ: 142.7, 140.1, 137.7, 129.1, 128.2, 127.7, 127.1,
123.8, 61.2, 60.2, 47.4, 21.4, 21.2, 21.0, 11.6.
Et
NHTos
HO
Me

Capítulo 4
238
Masa exacta calculada para C19H25NO3S (M+H-OH)+: 330.1528, encontrada:
330.1522.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IA, hexano:isopropanol 98:2, flujo= 0.7 mL/min, tr: 91.1 min
(minoritario) y 98.8 min (mayoritario)).
N-[(1S,2R)-1-(4-Clorofenil)-2-(hidroximetil)heptil]-4-
metilbenzenosulfonamida (87Ee)
Se siguió el procedimiento general partiendo de
heptanal (0.21 mL, 1.5 mmol) y N-tosil-4-
clorofenilimina (147 mg, 0.5 mmol). El producto crudo
se purificó por cromatografía de columna flash
(hexano:AcOEt 80:20) y se disgregó con Et2O. Sólido
blanco. Rendimiento: 161 mg (76%).
P.f.: 121-124 ºC.
[α]D25 = −58.9 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.54 (d, J= 8.3 Hz, 2H), 7.13 (m, 4H), 7.01 (d, J=
8.5 Hz, 2H), 6.39 (d, J= 7.4 Hz, 1H), 4.40 (t, J= 7.0 Hz, 1H), 3.81 (d, J= 11.2 Hz,
1H), 3.61 (m, 1H), 2.40 (s, 3H), 2.31 (m, 1H), 1.65 (m, 1H) 1.40-1.05 (m, 8H),
0.86 (t, J= 7.0 Hz, 3H). 13C-RMN (125 MHz, CDCl3) δ: 143.1, 139.2, 137.7, 132.8, 129.3, 128.3, 127.0,
61.9, 60.1, 45.6, 31.8, 28.2, 26.7, 22.4, 21.4, 14.0.
Masa exacta : no determinada.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 95:5, flujo= 0.8 mL/min, tr: 31.7
min (minoritario) y 49.1 min (mayoritario)).
N-[(1S,2R)-2-(Hidroximetil)-3-metil-1- p-tolilbutil]-4-
metilbenzenosulfonamida (87Ha)
Se siguió el procedimiento general partiendo
de isovaleraldehído (0.16 mL, 1.5 mmol) y N-tosil-
tolilimina (137 mg, 0.5 mmol). El producto crudo se
purificó por cromatografía de columna flash
NHTos
HO
Cl
Me
NHTos
HO
MeMeMe

Desarrollo Experimental
239
(hexano:AcOEt 80:20) y se disgregó con Et2O. Sólido blanco. Rendimiento: 134
mg (76%).
P.f.: 145-148 ºC.
[α]D25 = −70.8 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3) δ: 7.50 (d, J= 8.2 Hz, 2H), 7.08 (d, J= 8.1 Hz, 2H),
6.97-6.90 (m, 4H), 6.70-6.55 (bs, 1H), 4.60 (d, J= 6.8 Hz, 1H), 3.82-3.68 (m,
2H), 2.36 (s, 3H), 2.28 (s, 3H), 1.80-1.65 (m, 1H), 1.46-1.36 (m, 1H), 1.34-1.24
(bs, 1H), 0.92 (dd, J= 6.7, 20.5 Hz, 6H). 13C-RMN (125 MHz, CDCl3): δ 142.6, 138.0, 137.7, 136.4, 129.0, 128.7, 127.0,
126.9, 60.3, 59.6, 53.5, 51.9, 25.7, 21.3, 20.9, 18.8.
Masa exacta : no determinada.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IA, hexano:isopropanol 95:5, flujo= 0.5 mL/min, tr: 50.1 min
(minoritario) y 54.4 min (mayoritario)).
4.5.1.4. Desprotección y elaboración del aducto 100
Ácido (2 R,3S)-2-metil-3-(fenil)-3-(4-metilfenilsulfonamido)-pro panoico (99)
A una disolución de N-tosilfenilimina 81b (0.5 mmol, 1
equiv.), ácido p-nitrobenzoico (17 mg, 0.1 mmol) y el
catalizador 83 (0.1 mmol, 20 mol%) en DMF (2 mL) a −60
ºC se añadió propanal (0.11 mL, 3 equiv.) y la disolución resultante se agitó a
esta temperatura durante 24 horas. Posteriormente se adicionaron MeOH (2.5
Me
NHTos
HO
O

Capítulo 4
240
mL), KH2PO4 (570 mg, 4.15 mmol) y NaClO2 (338 mg, 3.15 mmol) y se
permitió que la temperatura de la mezcla subiera a 0 ºC. Tras la adición de
H2O2 (solución 35%, 4.5 mL) la mezcla se agitó durante 2 horas a temperatura
ambiente.23 Seguidamente se ajustó el pH a 3 mediante la adición de HCl 1 M y
se añadió lentamente una disolución acuosa saturada de Na2SO3 (10 mL). La
mezcla resultante se extrajo con Et2O (3 x 10 mL), la combinación de fases
orgánicas se lavó con una disolución acuosa saturada de NaCl (4 x 15 mL), se
secó sobre MgSO4, y se filtró. El crudo se concentró bajo presión reducida y se
purificó por cromatografía de columna flash (DCM:MeOH 98:2), obteniéndose
el derivado protegido como un sólido blanco. Rendimiento: 100 mg (62%). Los
datos espectroscópicos y físicos resultaron coincidentes con los previamente
descritos.24
P.f.: 128-130 ºC.
[α]D25 = −25.8 (c= 1, AcOEt).
1H-RMN (300 MHz, CDCl3) δ: 7.54-7.43 (m, 2H), 7.18-6.97 (m, 7H), 6.12 (bs,
1H), 4.55-4.46 (m, 1H), 2.98-2.81 (m, 1H), 2.30 (s, 3H), 1.19 (d, J= 7.0 Hz, 3H).
Ácido (2 R,3S)-3-amino-3-fenil-2-metilpropanoico (100)
Una suspensión del aducto 99 (100 mg, 0.31 mmol)
en HCl concentrado se agitó a 100ºC durante 24
horas.25 La mezcla resultante se concentró bajo presión
reducida para dar el aminoácido desprotegido 100.
Rendimiento: 52 mg (95%). Los datos espectroscópicos y físicos resultaron
coincidentes con los previamente descritos.26 1H-RMN (300 MHz, CDCl3) δ: 7.37-7.29 (m, 5H), 4.20 (d, J= 7.1 Hz, 1H), 2.76-
2.70 (m, 1H), 0.91 (d, J= 7.2 Hz, 3H).
23 Procedimiento de oxidación adaptado de: S. Brandau, A. Landa, J. Franzén, M. Marigo, K. A. Jørgensen, Angew. Chem. Int. Ed. 2006, 45, 4305-4309. 24 I. Abrahams, M. Motevalli, A. J. Robinson, P. B. Wyatt, Tetrahedron 1994, 50, 12755-12772. 25 Procedimiento de desprotección adaptado de: D. Uraguchi, Y. Ueki, T. Ooi, J. Am. Chem. Soc. 2008, 130, 14088-14089. 26 M. Shindo, K. Itoh, C. Tsuchiya, K. Shishido, Org. Lett. 2002, 4, 3119-3121.
Me
NH2
HO
O

Desarrollo Experimental
241
4.5.1.5. Preparación de muestras racémicas
Las muestras racémicas se prepararon paralelamente a las
enantioselectivas siguiendo el procedimiento general y utilizando el catalizador
rac-42 a 0 ºC o temperatura ambiente.
4.5.1.6. Determinación de la configuración absoluta
La configuración absoluta de los aductos se estableció inequívocamente
mediante el análisis de rayos-X de un cristal de (2R,3S) 87Aa.27
(2R,3S) 87Aa
27 Los datos cristalográficos del compuesto 87Aa se han depositado en el Cambridge Crystallographic Data Centre, CCDC Nº 726915. Estos datos se pueden obtener online (http://www.ccdc.cam.ac.uk) libre de cargos (o desde el Cambridge Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: (+44)1223-336-033, e-mail: depositccdc.cam.ac.uk). Igualmente se encuentran recogidos en el S.I. del artículo: E. Gómez-Bengoa, M. Maestro, A. Mielgo, I. Otazo, C. Palomo, I. Velilla, Chem. Eur. J. 2010, 16, 5333-5342.
Me
NHTos
HO
Me

Capítulo 4
242
4.5.1.7. Espectros de 1H y 13C-RMN
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
13C-RMN (125 MHz)
102030405060708090100110120130140150f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
Me
NHTos
Me
HO
87Aa
Me
NHTos
Me
HO
87Aa
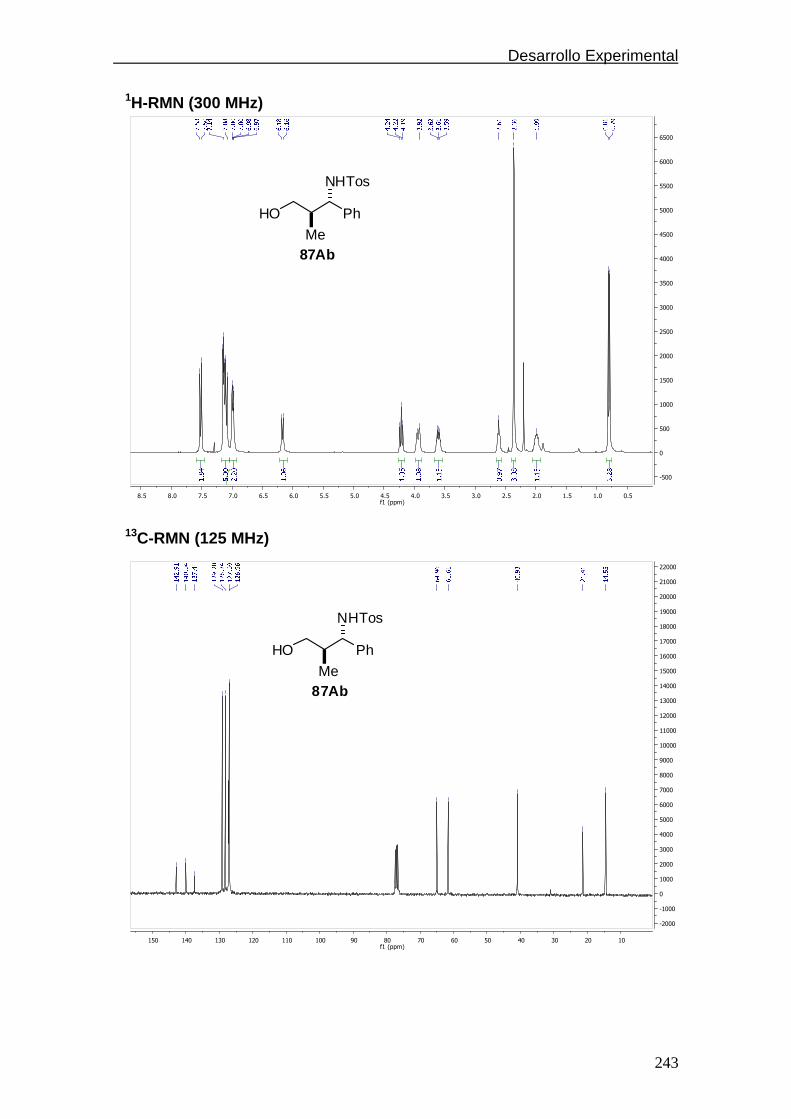
Desarrollo Experimental
243
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
13C-RMN (125 MHz)
102030405060708090100110120130140150f1 (ppm)
-2000
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000
20000
21000
22000
Me
NHTos
PhHO
87Ab
Me
NHTos
PhHO
87Ab

Capítulo 4
244
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
13C-RMN (125 MHz)
102030405060708090100110120130140150f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
1E+05
1E+05
2E+05
2E+05
OH
Et
NHTos
Me87Ba
OH
Et
NHTos
Me87Ba

Desarrollo Experimental
245
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
13C-RMN (125 MHz)
0102030405060708090100110120130140150f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
Et
NHTos
PhHO
87Bb
Et
NHTos
PhHO
87Bb

Capítulo 4
246
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
8500
9000
3.11
2.35
0.98
1.02
2.83
0.90
2.79
0.98
0.94
0.91
1.91
1.88
1.83
1.75
0.84
0.87
0.89
1.20
1.24
1.28
1.34
1.60
2.29
2.38
3.66
3.69
3.77
3.92
3.95
4.31
4.33
4.36
5.96
5.98
6.66
6.69
7.11
7.14
7.51
7.54
13C-RMN (125 MHz)
0102030405060708090100110120130140150160170f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
11.62
21.03
21.41
47.62
55.23
59.77
61.32
113.65
127.10
127.91
129.18
132.53
137.75
142.78
158.66
OH
Et
NHTos
OMe87Bc
OH
Et
NHTos
OMe87Bc
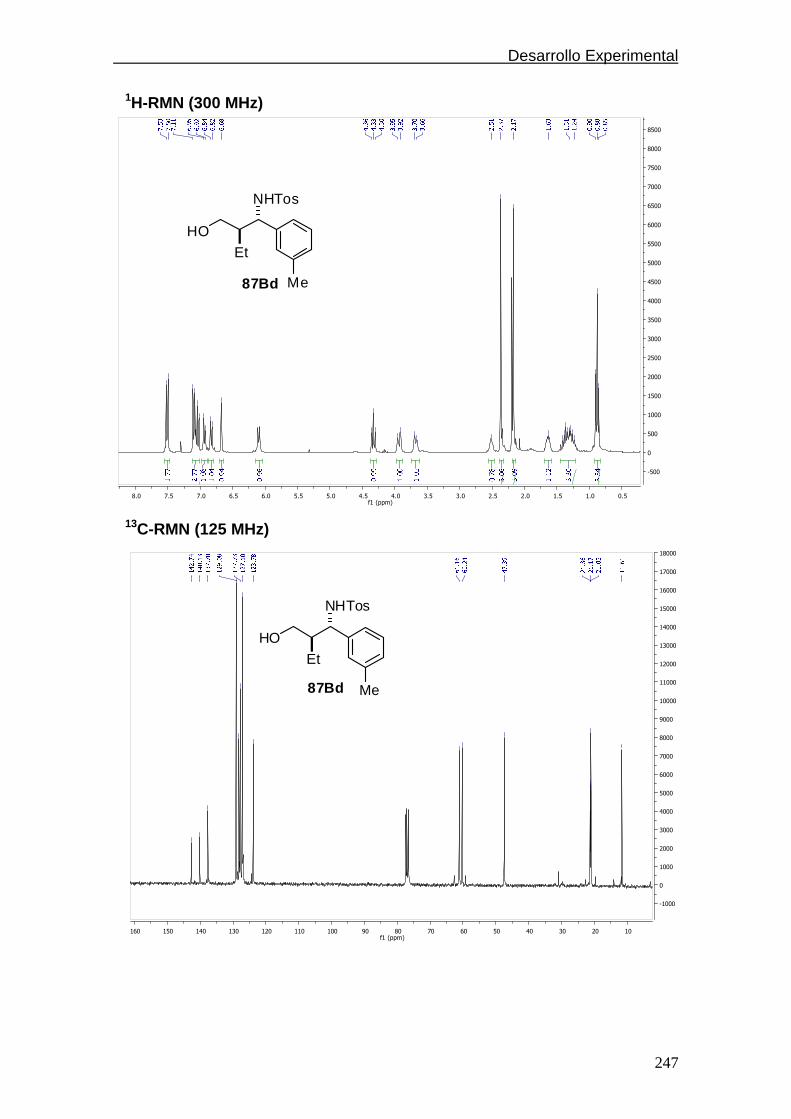
Desarrollo Experimental
247
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
8500
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
Et
NHTos
HO
Me87Bd
Et
NHTos
HO
Me87Bd

Capítulo 4
248
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
13C-RMN (125 MHz)
102030405060708090100110120130140150f1 (ppm)
-2000
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000
20000
NHTos
HO
Cl
Me87Ee
NHTos
HO
Cl
Me87Ee

Desarrollo Experimental
249
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
0
500
1000
1500
2000
2500
3000
3500
4000
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
1E+05
1E+05
2E+05
2E+05
2E+05
2E+05
2E+05
OH NHTos
Me
87Ha
MeMe
OH NHTos
MeMeMe
87Ha

Capítulo 4
250
4.5.1.8. Cromatogramas de HPLC Chiralpack IA, 1 mL/min, hexano:isopropanol 97:3, λ= 210 nm
AU
0,00
0,10
0,20
0,30
0,40
Minutes118,00 120,00 122,00 124,00 126,00 128,00 130,00 132,00 134,00 136,00 138,00 140,00 142,00 144,00 146,00 148,00 150,00
125,
733
136,
858
AU
0,00
0,20
0,40
0,60
Minutes
120,00 122,00 124,00 126,00 128,00 130,00 132,00 134,00 136,00 138,00 140,00 142,00 144,00 146,00 148,00 150,00 152,00 154,00
126,
820
137,
596
rac-87Aa
OH NHTos
MeMe
OH NHTos
Me87Aa
Me

Desarrollo Experimental
251
Chiralpack IA, 0.8 mL/min, hexano:isopropanol 95:5, λ= 210 nm
AU
0,00
0,10
0,20
0,30
0,40
Minutes34,00 35,00 36,00 37,00 38,00 39,00 40,00 41,00 42,00 43,00 44,00 45,00 46,00 47,00 48,00
36,6
73
43,6
56
AU
0,00
0,05
0,10
0,15
0,20
Minutes28,00 30,00 32,00 34,00 36,00 38,00 40,00 42,00 44,00
40,1
28
rac-87Ab
OH NHTos
Me
OH NHTos
Me87Ab

Capítulo 4
252
Chiralpack AD-H, 0.8 mL/min, hexano:isopropanol 95:5, λ= 210 nm
51.0
01
60.8
17AU
0.00
0.02
Minutes
46.00 48.00 50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00 66.00 68.00
51.4
62
60.2
28
AU
0.00
1.00
Minutes
48.00 50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00 66.00 68.00
rac-87Ba
OH NHTos
EtMe
OH NHTos
EtMe
87Ba

Desarrollo Experimental
253
Chiralpack IA, 0.75 mL/min, hexano:isopropanol 95:5, λ= 245 nm
51.2
95
57.6
82
AU
0.00
0.05
Minutes
50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00 66.00
52.3
18 57.1
91
AU
0.00
0.05
0.10
Minutes
50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00 66.00
rac-87Bb
OH NHTos
Et
OH NHTos
87BbEt
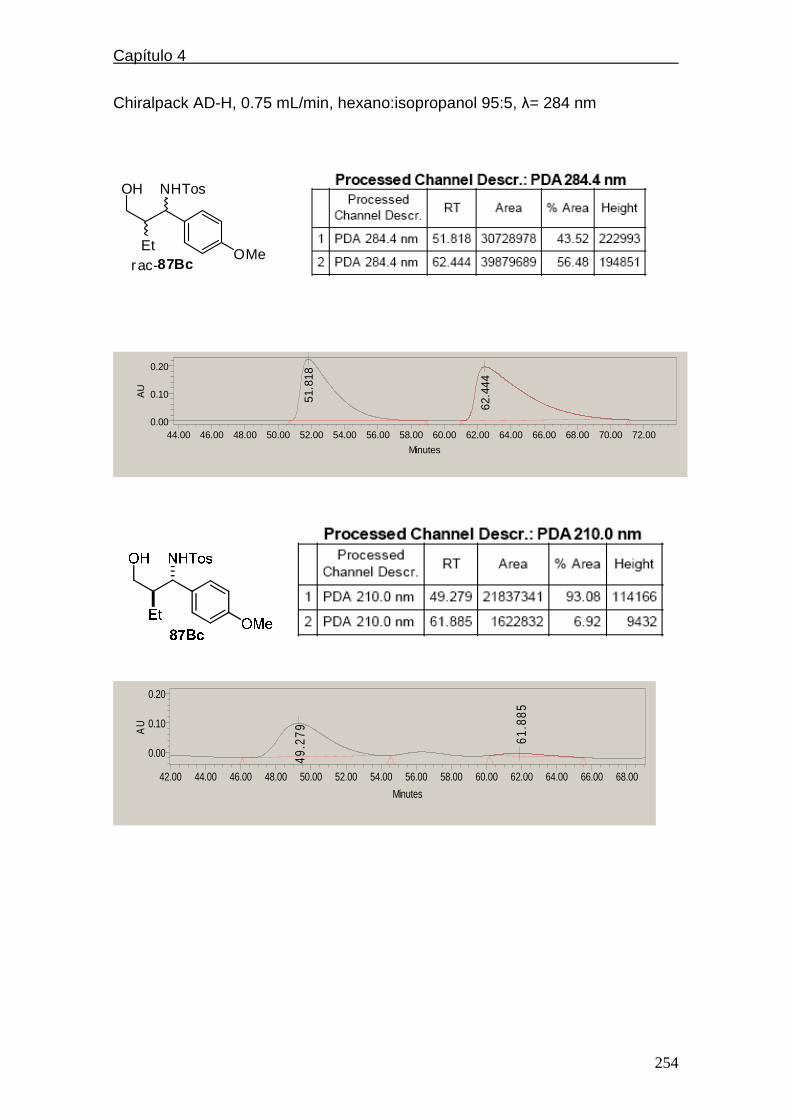
Capítulo 4
254
Chiralpack AD-H, 0.75 mL/min, hexano:isopropanol 95:5, λ= 284 nm
51.8
18
62.4
44
AU
0.00
0.10
0.20
Minutes
44.00 46.00 48.00 50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00 66.00 68.00 70.00 72.00
49.2
79 61.8
85
AU
0.00
0.10
0.20
Minutes
42.00 44.00 46.00 48.00 50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00 66.00 68.00
rac-87Bc
OH NHTos
EtOMe

Desarrollo Experimental
255
Chiralpack IA, 0.7 mL/min, hexano:isopropanol 98:2, λ= 210 nm
91.1
50
98.8
28AU
0.00
0.10
0.20
Minutes
86.00 88.00 90.00 92.00 94.00 96.00 98.00 100.00 102.00 104.00 106.00 108.00
95.3
58
105.
240
AU
-0.10
0.00
Minutes
95.00 100.00 105.00 110.00 115.00 120.00 125.00 130.00
rac-87Bd
OH NHTos
Et
Me
OH NHTos
Et
Me87Bd

Capítulo 4
256
Chiralpack AD-H, 0.8 mL/min, hexano:isopropanol 95:5, λ= 254 nm
31.6
37
50.4
50AU
0.000
0.010
Minutes
30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00 52.00 54.00
31.7
31
49.1
03
AU
0.00
0.05
Minutes
28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00 52.00 54.00
rac-87Ee
NHTos
HO
Cl
Me3
87Ee
NHTos
HO
Cl
Me3

Desarrollo Experimental
257
Chiralpack IA, 0.5 mL/min, hexano:isopropanol 95:5, λ= 210 nm
50.1
16
54.4
36
AU
0.00
0.50
1.00
Minutes
47.00 48.00 49.00 50.00 51.00 52.00 53.00 54.00 55.00 56.00 57.00
54.2
79
AU
0.00
2.00
Minutes
40.00 42.00 44.00 46.00 48.00 50.00 52.00 54.00 56.00 58.00 60.00 62.00 64.00
rac-87Ha
NHTos
HO
MeMe Me
NHTos
HO
MeMe Me
87Ha

Capítulo 4
258
4.5.1.9. Estudios computacionales para la reacción de Mannich entre
aldehídos y N-sulfonil-iminas
Coordenadas cartesianas de los puntos estacionarios de la reacción de
Mannich de las enaminas derivadas del propionaldehído y los catalizadores 85
y 89 con la N-sulfonil-imina 88 en presencia o ausencia de ácido benzoico.
TS1-anti
Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z
--------------------------------------------------------------------------------------------- 1 6 0 -0.421647 0.984110 2.978414 2 6 0 -1.407638 -0.219037 2.909169 3 6 0 -1.880193 -0.351581 1.440067 4 6 0 -0.801022 1.809793 1.735378 5 6 0 -0.909395 0.787025 -0.524934 6 6 0 -0.011179 1.681314 -1.172857 7 6 0 -0.110286 1.729269 -2.689949 8 6 0 -3.421632 -0.373039 1.204241 9 6 0 -4.126048 0.906316 1.678073 10 6 0 -3.995631 -1.612632 1.909424 11 1 0 -2.251260 -0.051803 3.586149 12 1 0 -0.595843 1.570694 3.886354 13 1 0 -0.887655 -1.123152 3.229835 14 1 0 -1.479247 -1.263776 0.996660 15 1 0 -1.657008 2.465835 1.938086 16 1 0 0.029598 2.408948 1.370528 17 1 0 -1.238864 -0.097691 -1.058857 18 1 0 0.071984 2.666315 -0.718683 19 1 0 0.740982 2.270875 -3.113797 20 1 0 -1.018992 2.244983 -3.026229 21 1 0 -0.114664 0.717884 -3.112017 22 1 0 -5.206254 0.823500 1.517537 23 1 0 -3.769541 1.779972 1.124087 24 1 0 -3.970776 1.081553 2.747618 25 1 0 -5.067617 -1.696253 1.708423 26 1 0 -3.859055 -1.562140 2.994385 27 1 0 -3.504257 -2.518208 1.540041 28 1 0 1.204155 0.279555 2.114963 29 7 0 -1.209266 0.790677 0.753379 30 8 0 0.935638 0.629069 3.003786 31 6 0 2.566572 2.125023 -0.540813 32 6 0 4.168071 4.419307 -0.227737 33 6 0 3.144560 2.756235 -1.652555 34 6 0 2.822514 2.648570 0.737242 35 6 0 3.611192 3.788059 0.887893 36 6 0 3.937700 3.894472 -1.499414 37 6 0 3.423441 -2.801636 -1.451574 38 6 0 5.059285 -3.095043 0.800367 39 6 0 3.003548 -2.345129 -0.202609 40 6 0 4.675621 -3.409837 -1.569219 41 6 0 5.492750 -3.554255 -0.447497
NTMSO
N
Ph
OHPhO2S

Desarrollo Experimental
259
42 6 0 3.811559 -2.488688 0.928964 43 6 0 1.715432 0.902298 -0.737201 44 1 0 1.850050 0.458566 -1.727231 45 1 0 2.993919 2.338741 -2.644902 46 1 0 2.418487 2.147328 1.611213 47 1 0 3.802120 4.178395 1.884325 48 1 0 4.382700 4.362864 -2.373547 49 1 0 5.693825 -3.212756 1.674631 50 1 0 5.009059 -3.773289 -2.537648 51 1 0 4.786661 5.304384 -0.104619 52 1 0 3.459790 -2.136301 1.893169 53 1 0 2.766133 -2.689480 -2.307093 54 1 0 6.466574 -4.027427 -0.542283 55 7 0 1.712482 0.021440 0.328543 56 8 0 0.753541 -2.150443 1.146991 57 8 0 0.717573 -1.685122 -1.352288 58 16 0 1.397786 -1.542482 -0.037203 59 8 0 -3.566777 -0.532558 -0.209252 60 14 0 -4.814007 -0.356134 -1.322044 61 6 0 -4.179528 -1.243522 -2.853917 62 1 0 -4.909907 -1.195402 -3.670995 63 1 0 -3.978187 -2.299839 -2.643601 64 1 0 -3.246054 -0.797175 -3.215501 65 6 0 -5.113038 1.468678 -1.717452 66 1 0 -5.818027 1.566718 -2.552897 67 1 0 -4.182124 1.966980 -2.013767 68 1 0 -5.532001 2.021589 -0.869129 69 6 0 -6.431916 -1.140089 -0.736484 70 1 0 -7.193595 -1.049361 -1.521569 71 1 0 -6.834748 -0.657071 0.161506 72 1 0 -6.309707 -2.207230 -0.518750 TS1-sin Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ------------------------------------------------------------------------ 1 6 0 -0.303949 -2.613473 0.830290 2 6 0 1.060389 -2.596045 1.548567 3 6 0 1.818653 -1.379792 0.957080 4 6 0 0.084290 -2.128270 -0.570988 5 6 0 1.021059 0.182592 -0.777131 6 6 0 0.107363 0.734675 -1.712382 7 6 0 0.621406 1.909836 -2.531113 8 6 0 3.331334 -1.598997 0.656973 9 6 0 3.571704 -2.653766 -0.432291 10 6 0 4.034973 -1.986531 1.967505 11 1 0 3.877186 -1.212720 2.726068 12 1 0 -0.725699 -3.626520 0.788461 13 1 0 1.595974 -3.531214 1.351108 14 1 0 0.926793 -2.504379 2.628398 15 1 0 1.761327 -0.522264 1.632069 16 1 0 0.627569 -2.920197 -1.103565 17 1 0 -0.764198 -1.806167 -1.171463 18 1 0 1.753434 0.845035 -0.325670 19 1 0 -0.457293 0.019490 -2.307370 20 1 0 -0.210823 2.428655 -3.019434 21 1 0 1.307594 1.581025 -3.321733 22 1 0 1.152692 2.636166 -1.905481
NTMSO
N
Ph
O
HSO2Ph

Capítulo 4
260
23 1 0 4.645589 -2.780535 -0.603780 24 1 0 3.108385 -2.354396 -1.376945 25 1 0 3.174052 -3.631828 -0.143202 26 1 0 5.111243 -2.086232 1.801414 27 1 0 3.665428 -2.939255 2.359854 28 1 0 -1.561324 -1.058223 0.865724 29 7 0 1.021564 -1.031394 -0.258443 30 8 0 -1.197939 -1.742851 1.491124 31 6 0 -5.853881 0.254970 -0.759503 32 6 0 -4.706360 -0.423407 -0.351213 33 6 0 -7.060090 -0.743251 1.084641 34 6 0 -4.717015 -1.265802 0.764789 35 6 0 -5.901542 -1.417133 1.483297 36 6 0 -7.035865 0.089716 -0.034446 37 6 0 -0.743296 2.190296 0.501538 38 6 0 0.368313 3.803670 2.523593 39 6 0 -0.462154 1.654099 1.768496 40 6 0 -0.479684 3.548383 0.274023 41 6 0 0.074421 4.349822 1.274519 42 6 0 0.093159 2.454878 2.766034 43 6 0 -1.338132 1.359122 -0.601460 44 1 0 -1.751808 1.976767 -1.405609 45 1 0 -7.980806 -0.868645 1.648423 46 1 0 -3.812919 -1.781096 1.074028 47 1 0 -5.920747 -2.063892 2.356310 48 1 0 -7.936081 0.612844 -0.345664 49 1 0 0.795781 4.424926 3.306226 50 1 0 -0.688267 0.612495 1.973010 51 1 0 -0.725797 3.985945 -0.690161 52 1 0 0.265525 5.401734 1.078504 53 1 0 0.300117 2.026166 3.743521 54 1 0 -5.808820 0.897873 -1.631936 55 7 0 -2.129152 0.314893 -0.194443 56 8 0 -2.800055 -1.595971 -1.754159 57 8 0 -3.469134 0.761936 -2.380862 58 16 0 -3.204354 -0.234628 -1.319589 59 8 0 3.807926 -0.316618 0.234365 60 14 0 5.191559 0.249990 -0.540454 61 6 0 5.122761 -0.103752 -2.394835 62 1 0 4.187745 0.262748 -2.834982 63 1 0 5.201511 -1.172732 -2.621420 64 1 0 5.948937 0.402772 -2.909919 65 6 0 6.782879 -0.474804 0.178064 66 1 0 6.866897 -1.556754 0.022336 67 1 0 6.869503 -0.280709 1.253265 68 1 0 7.650131 -0.011389 -0.309745 69 6 0 5.139154 2.106100 -0.236764 70 1 0 4.222160 2.552628 -0.638513 71 1 0 5.988926 2.608851 -0.714587 72 1 0 5.175326 2.331938 0.834977 TS2-anti Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ------------------------------------------------------------------------ 1 6 0 -0.678669 -2.875889 -0.636874 2 6 0 -3.837615 -0.846060 -1.458981 3 6 0 -2.020777 -2.728114 -1.355865

Desarrollo Experimental
261
4 6 0 -2.027737 1.601109 2.857951 5 6 0 -2.329692 -1.221360 -1.255490 6 6 0 -0.840960 -1.955795 0.568468 7 6 0 -4.026848 0.630375 -1.864463 8 6 0 -1.979869 0.205704 0.790391 9 6 0 -4.402236 -1.704801 -2.610480 10 6 0 -1.326446 0.592208 1.971625 11 6 0 -6.079019 0.952202 1.068729 12 6 0 -6.148586 -2.025936 1.865156 13 6 0 -7.435895 -1.098474 -0.787826 14 1 0 -4.439959 -2.761234 -2.335650 15 1 0 -3.792818 -1.599549 -3.515184 16 1 0 -5.085413 0.821564 -2.073458 17 1 0 -3.458724 0.844713 -2.774817 18 1 0 -7.015093 1.121773 1.616273 19 1 0 -1.352055 -2.459604 1.401071 20 1 0 -5.418609 -1.379785 -2.852118 21 1 0 -1.746200 -0.691904 -2.016273 22 1 0 -0.454876 -3.904023 -0.339754 23 1 0 0.109131 -1.559588 0.919182 24 1 0 -2.692359 0.895235 0.362568 25 1 0 -2.796813 -3.300194 -0.832762 26 1 0 -1.986968 -3.062004 -2.395579 27 1 0 -3.696338 1.345887 -1.111953 28 1 0 -0.766333 -0.174230 2.499865 29 1 0 -1.320353 2.101510 3.526608 30 1 0 -2.789118 1.126592 3.492081 31 1 0 -2.531883 2.373575 2.263136 32 1 0 -7.417655 -0.391900 -1.625799 33 1 0 -8.395394 -0.964253 -0.271862 34 1 0 -7.424095 -2.112580 -1.203290 35 1 0 -5.317217 -1.894939 2.567176 36 1 0 -6.128804 -3.065290 1.517806 37 1 0 -7.082281 -1.876086 2.421141 38 1 0 -6.031708 1.693878 0.263572 39 1 0 -5.253031 1.155910 1.760490 40 7 0 -1.723820 -0.871762 0.070553 41 8 0 -4.514252 -1.173994 -0.246789 42 14 0 -6.010386 -0.823229 0.423599 43 6 0 1.207340 3.866423 -2.162144 44 6 0 2.137022 4.792839 -2.638652 45 6 0 3.427707 4.380972 -2.973794 46 6 0 3.793267 3.038433 -2.838545 47 6 0 0.355259 1.614257 1.153182 48 6 0 1.151951 1.740128 2.406184 49 6 0 1.153005 2.968060 3.085636 50 6 0 1.929209 0.685629 2.913267 51 6 0 2.687754 0.865036 4.067475 52 6 0 2.687919 2.094044 4.734528 53 6 0 1.918177 3.146228 4.239746 54 6 0 1.585797 2.530072 -2.029387 55 6 0 2.873244 2.103247 -2.367644 56 1 0 3.283664 2.227983 5.633543 57 1 0 0.564631 3.795382 2.696079 58 1 0 1.934676 -0.276699 2.410060 59 1 0 3.283261 0.039569 4.448432 60 1 0 1.912796 4.107330 4.747124 61 1 0 4.796957 2.718175 -3.104400 62 1 0 4.149273 5.104442 -3.343890 63 1 0 3.145179 1.058276 -2.262395
NTMSO
N
Ph
PhO 2S O OH

Capítulo 4
262
64 1 0 0.195023 4.161646 -1.908597 65 1 0 1.849906 5.834857 -2.751120 66 1 0 -0.245100 2.495121 0.920046 67 7 0 0.924948 0.948951 0.117044 68 8 0 0.530642 0.126762 -2.241848 69 8 0 -0.918763 2.029151 -1.365781 70 16 0 0.390498 1.332976 -1.405548 71 6 0 6.751088 -4.071199 -0.560217 72 6 0 6.615446 -2.790740 -1.101962 73 6 0 5.488252 -2.023912 -0.810605 74 6 0 4.489145 -2.537042 0.027402 75 6 0 4.630030 -3.821460 0.569175 76 6 0 5.756678 -4.585885 0.276016 77 6 0 3.263630 -1.753778 0.369948 78 1 0 3.844945 -4.199046 1.215978 79 1 0 5.374588 -1.028599 -1.225185 80 1 0 7.388641 -2.390162 -1.752080 81 1 0 5.861294 -5.581864 0.697958 82 1 0 7.630630 -4.667441 -0.789376 83 1 0 2.360007 -0.074197 0.026668 84 8 0 3.227554 -0.546758 -0.189934 85 8 0 2.387612 -2.190254 1.109407 86 1 0 0.136366 -2.509382 -1.268258 TS2-sin Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ------------------------------------------------------------------------ 1 6 0 -1.177816 0.777927 -3.541164 2 6 0 -3.367041 1.374942 -0.430084 3 6 0 -2.364214 1.449398 -2.845909 4 6 0 -1.780108 -3.609995 0.182798 5 6 0 -2.132284 1.132980 -1.357685 6 6 0 -0.997433 -0.531495 -2.770480 7 6 0 -2.968894 1.476145 1.057395 8 6 0 -1.704595 -1.210670 -0.520023 9 6 0 -3.996926 2.727471 -0.825121 10 6 0 -1.080575 -2.473777 -0.538637 11 6 0 -5.322973 -1.207142 1.621643 12 6 0 -6.463010 -1.370633 -1.241098 13 6 0 -6.913771 1.161491 0.470945 14 1 0 -4.781966 2.995097 -0.112052 15 1 0 -4.443195 2.678846 -1.821094 16 1 0 -3.247955 3.527504 -0.813653 17 1 0 -3.841102 1.765596 1.654408 18 1 0 -2.198029 2.242382 1.184295 19 1 0 -3.312036 1.004469 -3.172028 20 1 0 -1.344904 0.602262 -4.607515 21 1 0 -2.572839 0.549493 1.474865 22 1 0 -2.403644 2.524903 -3.032229 23 1 0 -1.319347 1.762839 -0.979823 24 1 0 -1.560707 -1.357908 -3.221834 25 1 0 0.051383 -0.813455 -2.691945 26 1 0 -2.295247 -0.955699 0.349000 27 1 0 -0.595067 -2.769012 -1.465418 28 1 0 -1.080075 -4.424644 0.403163 29 1 0 -2.585779 -4.038115 -0.427917 30 1 0 -2.217675 -3.282282 1.132569
O
O
H
NTMSO
N
Ph
SO2Ph

Desarrollo Experimental
263
31 1 0 -7.380292 -1.840080 -0.864984 32 1 0 -4.630214 -2.033397 1.422707 33 1 0 -6.243958 -1.643444 2.029370 34 1 0 -4.880869 -0.582641 2.405942 35 1 0 -6.536671 1.822610 1.259862 36 1 0 -7.857224 0.732703 0.832946 37 1 0 -7.146988 1.779200 -0.403701 38 1 0 -5.767576 -2.171344 -1.517446 39 1 0 -6.717795 -0.822143 -2.155145 40 7 0 -1.586131 -0.260690 -1.430094 41 8 0 -4.298127 0.323201 -0.681770 42 14 0 -5.704700 -0.230020 0.049515 43 6 0 -0.322187 -0.312383 3.652966 44 6 0 -0.140771 -2.665744 4.151152 45 6 0 0.199175 -2.914386 2.820100 46 6 0 0.280231 -1.867763 1.889168 47 6 0 4.818785 -0.771161 -0.872774 48 6 0 5.930340 -3.205607 -0.045443 49 6 0 4.060723 -1.944463 -0.887090 50 6 0 6.140809 -0.825072 -0.432294 51 6 0 6.695836 -2.038184 -0.016776 52 6 0 4.605922 -3.164292 -0.482828 53 6 0 0.680150 -2.179468 0.485246 54 6 0 -0.404223 -1.362694 4.572128 55 6 0 0.017661 -0.558809 2.323780 56 1 0 -0.192519 -3.490402 4.857242 57 1 0 -0.666664 -1.164493 5.608012 58 1 0 6.740864 0.080824 -0.419584 59 1 0 0.419843 -3.930420 2.503028 60 1 0 0.075063 0.267038 1.621522 61 1 0 -0.519359 0.707577 3.973010 62 1 0 0.887971 -3.239245 0.321116 63 1 0 7.727473 -2.074286 0.323142 64 1 0 6.365483 -4.151329 0.266109 65 1 0 4.000830 -4.063135 -0.532144 66 1 0 4.377201 0.161582 -1.204777 67 7 0 1.500017 -1.294570 -0.141664 68 8 0 1.954773 -3.284384 -1.751161 69 8 0 2.282681 -0.876898 -2.513032 70 16 0 2.348236 -1.891016 -1.442305 71 6 0 3.742131 3.667010 0.546791 72 6 0 1.582742 4.764944 0.581812 73 6 0 3.606901 6.075066 0.727536 74 6 0 2.211830 6.002752 0.689680 75 6 0 2.343226 3.590679 0.507978 76 6 0 4.369696 4.906892 0.657120 77 6 0 1.627689 2.283257 0.392640 78 1 0 1.944341 0.363738 0.181306 79 1 0 4.324741 2.754189 0.493932 80 1 0 5.454474 4.962829 0.688125 81 1 0 1.617453 6.910809 0.745333 82 1 0 4.098838 7.040604 0.812539 83 1 0 0.501106 4.684401 0.553355 84 8 0 2.442135 1.240271 0.309096 85 8 0 0.401639 2.201853 0.390873 86 1 0 -0.277503 1.393710 -3.440502

Capítulo 4
264
TS2’-sin Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ----------------------------------------------------------------------- 1 6 0 3.749092 0.954525 -2.533169 2 6 0 4.415088 -1.367170 0.326614 3 6 0 4.308732 -0.404575 -2.081319 4 6 0 -0.078505 1.513451 1.685605 5 6 0 3.516273 -0.778200 -0.802383 6 6 0 3.369884 1.625050 -1.210703 7 6 0 3.615028 -1.665705 1.603329 8 6 0 1.717887 0.519043 0.277460 9 6 0 5.038266 -2.673335 -0.200145 10 6 0 0.783077 1.573864 0.434838 11 6 0 5.720927 0.578896 3.349269 12 6 0 7.742554 1.115195 1.084895 13 6 0 7.480472 -1.720408 2.283851 14 1 0 5.653976 -3.137995 0.575253 15 1 0 5.673540 -2.487699 -1.070543 16 1 0 4.258345 -3.388476 -0.485333 17 1 0 4.250187 -2.185746 2.328752 18 1 0 2.754965 -2.310890 1.392509 19 1 0 5.368993 -0.303507 -1.835703 20 1 0 4.471337 1.542976 -3.106850 21 1 0 3.254669 -0.747581 2.073797 22 1 0 4.203492 -1.172907 -2.851150 23 1 0 2.722532 -1.495227 -1.031616 24 1 0 4.251903 2.027543 -0.698585 25 1 0 2.625856 2.414731 -1.312438 26 1 0 1.427286 -0.445732 0.682909 27 1 0 1.140508 2.571638 0.187759 28 1 0 -0.946896 2.170555 1.602598 29 1 0 0.492826 1.821958 2.570515 30 1 0 -0.449606 0.497996 1.851060 31 1 0 8.518204 1.389993 1.810151 32 1 0 5.138057 1.474458 3.103479 33 1 0 6.480936 0.873283 4.084460 34 1 0 5.047886 -0.132188 3.840675 35 1 0 6.827564 -2.480617 2.728539 36 1 0 8.255297 -1.486818 3.025369 37 1 0 7.978925 -2.170371 1.417623 38 1 0 7.220783 2.034458 0.794082 39 1 0 8.243796 0.720609 0.193583 40 7 0 2.834635 0.498417 -0.428712 41 8 0 5.438089 -0.398791 0.575743 42 14 0 6.547737 -0.145078 1.812625 43 6 0 -2.538913 4.387378 -1.026793 44 6 0 -0.756618 4.648559 -2.631549 45 6 0 -0.301078 3.368763 -2.320040 46 6 0 -0.956884 2.579761 -1.360955 47 6 0 -2.908037 -3.012387 -0.823047 48 6 0 -3.351574 -2.882206 -3.585901 49 6 0 -2.095500 -2.228454 -1.641261 50 6 0 -3.955113 -3.733403 -1.400110 51 6 0 -4.178269 -3.666542 -2.776538 52 6 0 -2.302637 -2.159158 -3.020631 53 6 0 -0.415842 1.209900 -1.082427 54 6 0 -1.880180 5.163832 -1.981767 55 6 0 -2.086135 3.104339 -0.715708
O
OH
NTMSO
N PhPhO2S

Desarrollo Experimental
265
56 1 0 -0.240959 5.238301 -3.385195 57 1 0 -2.240248 6.161107 -2.220810 58 1 0 -4.592683 -4.350797 -0.773142 59 1 0 0.564729 2.965589 -2.842714 60 1 0 -2.608742 2.522224 0.035289 61 1 0 -3.414526 4.778778 -0.515755 62 1 0 0.348733 0.934134 -1.813075 63 1 0 -4.994335 -4.229743 -3.221509 64 1 0 -3.521950 -2.836853 -4.658033 65 1 0 -1.643550 -1.556205 -3.636446 66 1 0 -2.712462 -3.053747 0.242348 67 7 0 -1.329648 0.221991 -0.795662 68 8 0 0.409380 -1.376044 -1.869182 69 8 0 -0.486180 -1.893411 0.427333 70 16 0 -0.733621 -1.310196 -0.915420 71 6 0 -6.239135 -0.906430 1.438857 72 6 0 -6.089371 0.916575 3.028877 73 6 0 -7.991576 -0.572393 3.072741 74 6 0 -7.305695 0.531080 3.587532 75 6 0 -5.549765 0.200255 1.952886 76 6 0 -7.457231 -1.289079 1.999249 77 6 0 -4.243897 0.653673 1.381574 78 1 0 -2.924640 0.188768 0.025421 79 1 0 -5.815487 -1.454760 0.604494 80 1 0 -7.991285 -2.146641 1.598383 81 1 0 -7.720490 1.089372 4.422627 82 1 0 -8.941440 -0.873094 3.507680 83 1 0 -5.538347 1.770581 3.408999 84 8 0 -3.829081 -0.099370 0.364394 85 8 0 -3.637911 1.628030 1.808944 86 1 0 2.850703 0.818413 -3.145503 TS3-anti Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ----------------------------------------------------------------------- 1 6 0 -0.891422 -3.230974 0.647471 2 6 0 -3.619492 -1.286276 -1.253540 3 6 0 -2.199208 -3.183979 -0.140929 4 6 0 -2.214770 2.101375 2.556271 5 6 0 -2.245958 -1.739589 -0.671755 6 6 0 -0.873245 -1.865446 1.349078 7 6 0 -3.486145 -0.032423 -2.139854 8 6 0 -2.033676 0.234247 0.906922 9 6 0 -4.158484 -2.426650 -2.142127 10 6 0 -1.458369 0.913066 1.997868 11 1 0 -5.048697 -2.088936 -2.680519 12 1 0 -4.431342 -3.298564 -1.542764 13 1 0 -3.412544 -2.730258 -2.885057 14 1 0 -4.463083 0.225366 -2.564563 15 1 0 -2.804156 -0.240117 -2.970435 16 1 0 0.984835 -3.058791 0.112807 17 1 0 -3.057042 -3.370725 0.515656 18 1 0 -0.883248 -4.042134 1.392075 19 1 0 -3.087594 0.843180 -1.627142 20 1 0 -2.187881 -3.924502 -0.942117 21 1 0 -1.493655 -1.630316 -1.459733 22 1 0 -1.317623 -1.925992 2.351007
NTMSO
N
Ph
PhO 2S
O H
O
O
H

Capítulo 4
266
23 1 0 0.137598 -1.466223 1.430215 24 1 0 -2.725040 0.772169 0.275101 25 1 0 -0.956862 0.299501 2.741256 26 1 0 -1.553882 2.745822 3.146206 27 1 0 -3.035028 1.793979 3.218945 28 1 0 -2.647740 2.708798 1.751840 29 7 0 -1.718658 -0.985213 0.511384 30 8 0 0.156232 -3.386505 -0.287702 31 6 0 2.227045 4.534619 -2.907907 32 6 0 3.380862 3.947266 -3.430346 33 6 0 2.608019 1.765673 -2.720003 34 6 0 0.281845 1.759476 1.146783 35 6 0 1.093041 1.898837 2.389560 36 6 0 2.642411 2.267379 4.709484 37 6 0 1.120006 3.138582 3.046882 38 6 0 1.857758 0.842973 2.913366 39 6 0 2.622782 1.028067 4.062113 40 6 0 1.889768 3.323158 4.196923 41 6 0 1.259342 3.744874 -2.285218 42 6 0 3.568810 2.565641 -3.337358 43 6 0 1.461762 2.366808 -2.193754 44 1 0 3.241957 2.406753 5.605024 45 1 0 0.551260 3.971244 2.640754 46 1 0 1.852820 -0.125637 2.423176 47 1 0 3.208418 0.200707 4.454082 48 1 0 1.902951 4.293437 4.686283 49 1 0 4.462429 2.108319 -3.753259 50 1 0 4.131716 4.564919 -3.915874 51 1 0 2.737375 0.691571 -2.649278 52 1 0 0.347072 4.178849 -1.890465 53 1 0 -0.278464 2.655675 0.879325 54 1 0 2.075490 5.607489 -2.990035 55 7 0 0.825479 1.039596 0.131373 56 8 0 0.174295 0.073310 -2.126471 57 8 0 -1.012967 2.172778 -1.299397 58 16 0 0.215319 1.345669 -1.386240 59 6 0 3.293113 -1.524784 0.306537 60 6 0 4.606764 -2.159397 -0.003942 61 6 0 7.045455 -3.410726 -0.546817 62 6 0 4.823671 -3.483968 0.398526 63 6 0 5.619108 -1.462452 -0.678508 64 6 0 6.834586 -2.088975 -0.947427 65 6 0 6.038644 -4.107750 0.126419 66 1 0 2.263273 0.117678 0.066731 67 1 0 4.029553 -4.005904 0.921975 68 1 0 5.445253 -0.436386 -0.983011 69 1 0 7.618335 -1.546924 -1.469580 70 1 0 6.202051 -5.135693 0.438165 71 1 0 7.994006 -3.897212 -0.758953 72 8 0 2.405161 -2.118085 0.924971 73 8 0 3.184862 -0.279251 -0.121632 74 8 0 -4.502870 -1.091620 -0.145168 75 14 0 -6.011975 -0.386442 0.055680 76 6 0 -7.242783 -0.872202 -1.295164 77 1 0 -6.962966 -0.485669 -2.281969 78 1 0 -8.233402 -0.463757 -1.056626 79 1 0 -7.346658 -1.960134 -1.377547 80 6 0 -6.602661 -1.045306 1.718055 81 1 0 -5.891142 -0.802534 2.515646 82 1 0 -6.715605 -2.135316 1.694643

Desarrollo Experimental
267
83 1 0 -7.572800 -0.613821 1.993931 84 6 0 -5.877706 1.496896 0.128592 85 1 0 -5.501393 1.924357 -0.807234 86 1 0 -5.206076 1.816750 0.934173 87 1 0 -6.860600 1.944530 0.324153 TS3-sin Center Atomic Atomic Coordinates (Angstroms) Number Number Type X Y Z ----------------------------------------------------------------------- 1 6 0 -1.130318 1.879960 -2.827597 2 6 0 -3.626048 1.397429 0.084342 3 6 0 -2.465292 2.181652 -2.127770 4 6 0 -1.799250 -3.433889 -0.598090 5 6 0 -2.314106 1.514918 -0.752024 6 6 0 -0.824304 0.440338 -2.376349 7 6 0 -3.334442 1.127144 1.575640 8 6 0 -1.763634 -0.942387 -0.571188 9 6 0 -4.367883 2.747864 -0.000023 10 6 0 -1.080273 -2.130624 -0.895990 11 1 0 -5.214099 2.751704 0.693059 12 1 0 -4.750382 2.922999 -1.008200 13 1 0 -3.706954 3.578512 0.272441 14 1 0 -4.274424 1.073444 2.135531 15 1 0 -2.750216 1.954671 1.992835 16 1 0 0.246508 2.585493 -1.611253 17 1 0 -3.303650 1.721027 -2.664890 18 1 0 -1.230010 1.929535 -3.916003 19 1 0 -2.775612 0.209123 1.762993 20 1 0 -2.625342 3.258976 -2.063910 21 1 0 -1.604822 2.100891 -0.155365 22 1 0 -1.167229 -0.295944 -3.112826 23 1 0 0.242923 0.290361 -2.206902 24 1 0 -2.467698 -0.981909 0.248510 25 1 0 -0.511220 -2.140435 -1.821236 26 1 0 -1.097677 -4.276519 -0.605457 27 1 0 -2.568603 -3.654904 -1.349388 28 1 0 -2.287994 -3.413009 0.383491 29 7 0 -1.611530 0.247230 -1.128743 30 8 0 -0.124466 2.812746 -2.487028 31 6 0 -0.452468 -0.614925 3.608970 32 6 0 -0.607199 -3.015395 3.802750 33 6 0 -0.183535 -3.143975 2.478478 34 6 0 0.110321 -2.011856 1.704676 35 6 0 4.927847 -1.129681 -0.621652 36 6 0 5.729271 -3.804170 -0.389884 37 6 0 4.049060 -2.169528 -0.935943 38 6 0 6.213346 -1.441120 -0.180495 39 6 0 6.613964 -2.774727 -0.062999 40 6 0 4.438870 -3.505607 -0.829485 41 6 0 0.600091 -2.192547 0.307637 42 6 0 -0.741578 -1.750009 4.373034 43 6 0 -0.031707 -0.741786 2.286440 44 1 0 6.044350 -4.840981 -0.309280 45 1 0 7.618550 -3.010794 0.277814 46 1 0 3.741526 -4.289158 -1.105413 47 1 0 4.607462 -0.098939 -0.723710 48 1 0 6.905824 -0.640703 0.065350
O
O
H
NTMSO
N
Ph
O
H
SO2Ph

Capítulo 4
268
49 1 0 0.698104 -3.237752 0.007482 50 1 0 -0.062340 -4.133428 2.045070 51 1 0 0.177329 0.143637 1.694575 52 1 0 -0.552268 0.374683 4.047476 53 1 0 -0.823587 -3.905116 4.388187 54 1 0 -1.065624 -1.646369 5.405132 55 7 0 1.566055 -1.341810 -0.116155 56 8 0 1.826463 -3.033589 -2.071796 57 8 0 2.477796 -0.583430 -2.346584 58 16 0 2.382638 -1.786952 -1.501239 59 6 0 1.996024 2.068973 0.469233 60 6 0 2.764032 3.324923 0.705186 61 6 0 4.146015 5.715230 1.133365 62 6 0 2.176065 4.549585 0.360698 63 6 0 4.048736 3.303024 1.266234 64 6 0 4.734559 4.496836 1.481431 65 6 0 2.867201 5.740214 0.570639 66 1 0 2.136521 0.130245 0.467480 67 1 0 1.183538 4.549483 -0.077713 68 1 0 4.496086 2.351495 1.531316 69 1 0 5.729106 4.477875 1.918681 70 1 0 2.411093 6.687003 0.295058 71 1 0 4.684506 6.644754 1.298881 72 8 0 0.831348 2.070470 0.060091 73 8 0 2.660018 0.964093 0.756492 74 8 0 -4.422735 0.376833 -0.512226 75 14 0 -5.828678 -0.445331 -0.097923 76 6 0 -7.185451 0.693256 0.562646 77 1 0 -6.930209 1.142083 1.529714 78 1 0 -8.107813 0.116301 0.708667 79 1 0 -7.413215 1.506153 -0.136059 80 6 0 -6.380483 -1.233476 -1.714551 81 1 0 -5.593191 -1.872283 -2.130822 82 1 0 -6.621573 -0.472569 -2.465632 83 1 0 -7.272410 -1.855057 -1.568449 84 6 0 -5.477049 -1.789223 1.183365 85 1 0 -4.717308 -2.495161 0.827650 86 1 0 -6.388776 -2.366342 1.384649 87 1 0 -5.131250 -1.380661 2.139570
4.5.2. Reacción de Mannich con iminas sencillas (ba ses de Schiff)
4.5.2.1. Síntesis de iminas N-aromáticas
Una mezcla del aldehído (10 mmol), amina (10 mmol) y MgSO4 (1 g, 100
mg/mmol) en diclorometano seco (50 mL) se agitó a temperatura ambiente
durante 16 horas. La mezcla se filtró sobre celita y se evaporó el disolvente
bajo presión reducida para dar la imina correspondiente.

Desarrollo Experimental
269
N-(4-Metilbenzcilideno)anilina (90a) 28a
Se siguió el procedimiento general a partir de
p-metilbenzaldehído (1.25 mL, 10 mmol) y p-
anisidina (1.23 g, 10 mmol). El producto crudo se
trituró con mezclas de hexano:DCM. Sólido
blanco. Rendimiento: 1.62 g (72%). Los datos
físicos y espectroscópicos resultaron coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.45 (s, 1H), 7.83 (d, J= 8.5 Hz, 2H), 7.46-7.22
(m, 4H), 6.96-6.90 (m, 2H), 3.84 (s, 3H), 2.43 (s, 1H).
N-(4-Metilbenzcilideno)anilina (90b) 28b
Se siguió el procedimiento general a partir de p-
metilbenzaldehído (1.25 mL, 10 mmol) y anilina (0.91 mL,
10 mmol). El producto crudo se trituró con mezclas de
hexano:DCM. Sólido blanco. Rendimiento: 1.58 g (81%).
Los datos físicos y espectroscópicos resultaron coincidentes con los de la
bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.42 (s, 1H), 7.80, (d, J= 8.0 Hz, 2H), 7.43-7.19
(m, 7H), 2.42 (s, 1H).
28 a) H. Neuvonen, K. Neuvonen, F. Fulop, J. Org. Chem. 2006, 71, 3143-3148. b) Ch.-F. Chen, Z.-G. Zheng, B.-Z. Yan, J.-H. Xu, Tetrahedron. Lett. 1994, 35, 9221-9224. c) Z. Wang, Q. Ding, X. He, J. Wu, Org. & Biom. Chem. 2009, 7, 863-865. d) S. Failla, P. Finocchiaro, M. Latronico, Phosp. Sulf. Sil. Rel. Elem. 1995, 101, 261-266. e) E. M. Smith, S. K. Sorota, M. Hyunjin, B. A. McKittrick, T. L. Nechuta, C. Bennett, C. Knutson, D. A. Burnett, J. Kieselgof, Z. Tan, D. Rindgen, T. Bridal, X. Zhou, Y.-P. Jia, Z. Dong, D. Mullins, X. Zhang, T. Priestley, C. C. Correll, D. Tulshian, M. Czarniecki, W. J. Greenlee, Bioorg. & Med. Chem. Lett. 2010, 20, 4602-4606. f) M. Shimizu, T. Nishi, A. Yamamoto, Synlett 2003, 1469-1437. g) I. Hachiya, K. Ogura, M. Shimizu, Org. Lett. 2002, 4, 2755-2757. J. A. Townes, M. A. Evans, J. Queffelec, S. J. Taylor, J. P. Morken, Org. Lett. 2002, 4, 2537-2540.
NPh
Me
Me
N
OMe

Capítulo 4
270
N-(4-Metilbenzcilidenoamino)benzonitrilo (90c) 2828a
Se siguió el procedimiento general a partir de p-
metilbenzaldehído (1.25 mL, 10 mmol) y p-
cianoanilina (1.18 g, 10 mmol). El producto crudo se
trituró con mezclas de hexano:DCM. Sólido blanco.
Rendimiento: 1.56 g (71%). Los datos físicos y
espectroscópicos resultaron coincidentes con los de la bibliografía.
1H-RMN (300 MHz, CDCl3): δ: 8.38 (s, 1H), 7.76 (dd, J= 8.1, 24.6 Hz, 4H), 7.34-
7.22 (m, 4H), 2.46 (s, 3H).
4-Fluoro- N-(4-metilbencilideno)anilina (90d) 28c
Se siguió el procedimiento general a partir de p-
metilbenzaldehído (1.25 mL, 10 mmol) y p-fluoroanilina
(0.95 mL, 10 mmol). El producto crudo se trituró con
mezclas de hexano:DCM. Sólido blanco. Rendimiento:
1.62 g (76%). Los datos físicos y espectroscópicos
resultaron coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.41 (s, 1H), 7.78 (d, J=7.9 Hz, 2H), 7.30-7.03
(m, 6H), 2.42 (s, 1H).
N-(Ciclohexilmetileno)-4-fluoroanilina (90e) 28d
Se siguió el procedimiento general a partir de
ciclohexanocarboxaldehído (1.20 mL, 10 mmol) y p-
fluoroanilina (0.95 mL, 10 mmol). El disolvente orgánico se
evaporó bajo presión reducida a temperatura ambiente
bajo atmósfera de nitrógeno. Sólido blanco. Rendimiento
cuantitativo. Los datos físicos y espectroscópicos resultaron coincidentes con
los de la bibliografía.
1H-RMN (300 MHz, CDCl3): δ: 7.74 (d, J= 4.9 Hz, 1H), 7.05-7.00 (m, 4H), 2.46-
2.33 (m, 1H), 2.01-1.25 (m, 10H).
N
Me
CN
N
Me
F
N
F

Desarrollo Experimental
271
N-(2-Metilpropilideno)anilina (90g) 2828e
Se siguió el procedimiento general a partir de
isobutiraldehído (0.91 mL, 10 mmol) y anilina (0.91 mL, 10
mmol). El disolvente orgánico se evaporó bajo presión reducida
a temperatura ambiente bajo atmósfera de nitrógeno. Sólido
blanco. Rendimiento cuantitativo. Los datos físicos y espectroscópicos
resultaron coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 7.73 (d, J= 4.9 Hz, 1H), 7.23-7.17 (m, 3H), 7.05
(d, J= 7.2 Hz, 2H), 2.73-2.59 (m, 1H), 1.24 (d, J= 6.9 Hz, 6H).
4-Fluoro- N-(4-metoxibencilideno)anilina (90h) 28a
Se siguió el procedimiento general a partir de
p-metoxibenzaldehído (1.36 mg, 10 mmol) y p-
fluoroanilina (0.95 mL, 10 mmol). El producto
crudo se trituró con mezclas de hexano:DCM.
Sólido blanco. Rendimiento: 1.60 g (70%). Los
datos físicos y espectroscópicos resultaron coincidentes con los de la
bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 8.37 (s, 1H), 7.86-7.81 (m,2H), 7.21-7-14 (m,
2H), 7.10-7.03 (m, 2H), 6.99 (d, J= 8.8 Hz, 2H), 3.88 (s, 3H).
4-Cloro- N-(3-fenilprop-2-inilideno)anilina (96a) 28f
Se siguió el procedimiento general a partir de
fenilpropargilaldehído (1.22 mL, 10 mmol) y p-
cloroanilina (1.28 g, 10 mmol). Sirope naranja.
Rendimiento cuantitativo. Los datos físicos y
espectroscópicos resultaron coincidentes con los de la
bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 7.92 (s, 1H), 7.60 (dd, J= 1.7, 8.9 Hz, 2H), 7.43-
7.32 (m, 5H), 7.17-7.11 (m, 2H).
N
N
MeO
F
N
Ph
Cl

Capítulo 4
272
N-3-Fenilprop-2-inil-(4-metoxilideno)anilina (96b) 28g
Se siguió el procedimiento general a partir de
fenilpropargilaldehído (1.22 mL, 10 mmol) y p-
anisidina (1.23 g, 10 mmol). Sirope naranja.
Rendimiento cuantitativo. Los datos físicos y
espectroscópicos resultaron coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 7.95 (s, 1H), 7.59 (dd, J= 2.0, 7.6 Hz, 2H), 7.41-
7.36 (m, 3H), 7.24 (d, J= 9.8 Hz, 2H), 6.92 (d, J= 8.9 Hz, 2H), 3.83 (s, 3H).
4-Metoxi- N-(oct-2-inilideno)anilina (96c) 28h
Se siguió el procedimiento general a partir de 2-
octinal (1.43 mL, 10 mmol) y p-anisidina (1.23 g, 10
mmol). Sirope naranja. Rendimiento cuantitativo. Los
datos físicos y espectroscópicos resultaron
coincidentes con los de la bibliografía. 1H-RMN (300 MHz, CDCl3): δ: 7.70 (s, 1H), 7.16 (d, J= 8.9 Hz, 2H), 6.88 (d, J=
8.9 Hz, 2H), 3.81 (s, 3H), 2.43 (t, J= 7.1 Hz, 2H), 1.67-1.57 (m, 2H), 1.47-1.28
(m, 4H), 0.92 (t, J= 7.0 Hz, 3H).
4.5.2.2. Reacción de Mannich entre aldehídos e imin as N-aromáticas:
procedimiento general
Procedimiento general A:
A una disolución de la imina (0.5 mmol, 1 equiv.), ácido p-nitrobenzoico (17
mg, 0.1 mmol) y el catalizador (0.1 mmol, 20 mol%) en THF seco (1.5 mL) a
−40 ºC ó −60 ºC se añadió el correspondiente aldehído (1.5 mmol, 3 equiv.). La
disolución resultante se agitó a la misma temperatura durante 24 horas, tras lo
N
Ph
OMe
N
OMe
( ) 4

Desarrollo Experimental
273
cual se adicionaron a la misma temperatura EtOH (1 mL) y NaBH4 (4.5 mmol, 8
equiv.) y la mezcla resultante se agitó durante 30 minutos a −40 ºC.
Seguidamente se adicionó una disolución acuosa saturada de NaCl (2 mL), y
se permitió que la mezcla alcanzara temperatura ambiente. Tras extraer con
DCM (3 x 4 mL), la combinación de las fases orgánicas se lavó con una
disolución acuosa saturada de NaCl y se secó con MgSO4, se filtró y se
evaporó el disolvente bajo presión reducida. El crudo resultante se purificó
mediante cromatografía de columna flash. La diastereoselectividad del proceso
se determinó por análisis de 1H-RMN del crudo.
Procedimiento general B:
A una disolución de la anilina correspondiente (0.55 mmol, 1.1 equiv.) en
THF seco a temperatura ambiente, se adicionaron el aldehído correspondiente
(0.5 mmol, 1 equiv.) y ácido p-nitrobenzoico (17 mg, 0.1 mmol) y la mezcla se
agitó durante 30 minutos. Posteriormente se enfrió la mezcla a −40 ºC y se
adicionaron el catalizador (0.1 mmol, 20 mol%) y el correspondiente aldehído
(1.5 mmol, 3 equiv.). La disolución resultante se agitó a la misma temperatura
durante 24 horas, tras lo cual se añadieron EtOH (1 mL) y NaBH4 (4.5 mmol, 8
equiv.) y la mezcla resultante se agitó durante 30 minutos a −40 ºC.
Transcurrido este tiemplo se adicionaron una disolución acuosa saturada de
NaCl (2 mL), y se permitió que la mezcla alcanzara temperatura ambiente. El
tratamiento posterior fue similar al del procedimiento general A.

Capítulo 4
274
Procedimiento general C:
+
a)
THF, 0 ºC
b) NaBH4, EtOH,
NH
COOH
NPMP
Me
NPMP
PhPh
Rto.: 80%(sin:anti): 80:20ee (sin): 99%HOH
Me
O
96b17A sin-97Ab(mayoritario)
A una disolución de la imina 96b (0.5 mmol, 1 equiv.) y D-prolina (12 mg,
0.1 mmol) en THF seco (1.5 mL) a 0 ºC se añadió el propionaldehído (17A)
(1.5 mmol, 3 equiv.). La disolución resultante se agitó a la misma temperatura
durante 20 horas. Seguidamente se añadieron EtOH (1 mL) y NaBH4 (4.5
mmol, 8 equiv.) a esta temperatura, y tras agitar la mezcla durante 30 minutos
se vertió sobre la misma una disolución acuosa saturada de NaCl (2 mL) y se
permitió que alcanzara temperatura ambiente. El tratamiento posterior fue
similar al del procedimiento general A.
(2R,3S)-3-(Fenilamino)-2-metil-3- p-tolilpropan-1-ol (93Ab)
Preparado según el procedimiento general A a
−40 ºC partiendo de propanal (0.11 mL, 1.5 mmol) y
N-(4-metilbenciliden) anilina 90b (98 mg, 0.5 mmol).
El producto crudo se purificó por cromatografía de
columna flash (hexano:acetato de etilo 80:20). Sólido blanco. Rendimiento: 122
mg (96%).
P.f.: 85-89 ºC.
[α]D20 = −49.2 (c= 0.5, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.2 (d, J= 8.0 Hz, 2H), 7.15-7.08 (m, 4H), 6.67 (t,
J= 7.3 Hz, 1H), 6.59 (d, J= 7.6 Hz, 2H), 4.34 (d, J= 7.0 Hz, 1H), 3.79-3.67 (m,
2H), 2.34 (s, 3H), 2.16-2.06 (m, 1H), 0.94 (d, J= 7.0 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 147.4, 139.2, 136.6, 129.2, 129.1, 126.9, 117.7,
114.1, 66.7, 62.7, 41.4, 21.1, 14.7.
Anal. Calcd para C17H21NO: C, 79.96%; H, 8.29%; N, 5.49%. Encontrado: C,
79.77%; H, 8.02%; N, 5.41%.
Me
NHPh
HO
Me

Desarrollo Experimental
275
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 95:5, flujo= 0.5 mL/min, tr: 28.5
min (minoritario) y 39.3 min (mayoritario)).
4-((1S,2R)-3-Hidroxi-2-metil-1- p-tolilpropilamino)benzonitrilo (93Ac)
Preparado según el procedimiento general A
a −60 ºC partiendo de propanal (0.11 mL, 1.5 mmol)
y 4-(4-metilbencilidenamino)benzonitrilo 90c (110
mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 70:30). Sólido blanco. Rendimiento: 117 mg (84%).
P.f.: 135-139 ºC.
[α]D20 = −61.5 (c= 0.65, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.33 (d, J= 8.8 Hz, 2H), 7.23-7.15 (m, 4H), 6.49
(d, J= 8.9 Hz, 2H), 5.89 (s, 1H), 4.42 (t, J= 6.0 Hz, 1H), 3.81-3.75 (m, 1H), 3.68-
3.62 (m, 1H), 2.36 (s, 3H), 2.11 (bs, 1H), 2.11-2.06 (m, 1H), 1.65 (t, J= 4.6 Hz,
1H), 1.09 (d, J= 7.1 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 151.1, 138.2, 137.0, 133.5, 129.3, 126.8, 120.7,
112.8, 97.9, 65.1, 61.7, 41.1, 21.1, 14.8.
Anal. Calcd para C18H20N2O: C, 77.11%; H, 7.19%; N, 9.99%. Encontrado: C,
76.92%; H, 6.96%; N, 9.91%.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IC, hexano:isopropanol:etanol 95:4:1, flujo= 0.8 mL/min, tr:
57.3 min (minoritario) y 70.3 min (mayoritario)).
(2R,3S)-3-(4-Fluorofenilamino)-2-metil-3- p-tolilpropan-1-ol (93Ad)
Preparado según el procedimiento general A
a −40 ºC partiendo de propanal (0.11 mL, 1.5 mmol)
y 4-fluoro-N-(4-metilbenzilideno) anilina 90d (107
mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 85:15). Aceite amarillo. Rendimiento: 115 mg (84%).
[α]D20 = −16.9 (c= 0.35, DCM).
Me
HN
HO
Me
CN
Me
HN
HO
Me
F

Capítulo 4
276
1H-RMN (300 MHz, CDCl3): δ 7.16 (q, J= 8.2, 8.2, 8.1 Hz, 4H), 6.83-6.77 (m,
2H), 6.54-6.49 (m, 2H), 4.24 (d, J= 7.3 Hz, 1H), 3.80-3.67 (m, 2H), 2.35 (s, 3H),
2.12-2,06 (m, 1H), 0.91 (d, J= 7.0 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 157.6, 154.5, 143.7, 139.0, 136.7, 129.2, 126.9,
115.6, 115.3, 115.2, 66.9, 63.9, 41.3, 21.1, 14.7.
Masa exacta calculada para C17H20NOF (M, H+): 273.1529; encontrada:
273.1524.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 95:5, flujo= 0.5 mL/min, tr: 29.2
min (minoritario) y 35.9 min (mayoritario)).
(2R,3R)-3-Ciclohexil-3-(4-fluorofenilamino)-2-metilpropan -1-ol (93Ae)
Preparado según el procedimiento general A a −40
ºC partiendo de propanal (0.11 mL, 1.5 mmol) y N-
(ciclohexilmetileno)-4-fluoroanilina 90e (103 mg, 0.5
mmol). El producto crudo se purificó por cromatografía de
columna flash (hexano:acetato de etilo 80:20). Aceite
amarillo. Rendimiento: 58 mg (44%).
[α]D20 = −32.6 (c= 0.5, DCM).
1H-RMN (300 MHz, CDCl3): δ 6.88 (t, J= 8.7 Hz, 2H), 6.62-6.57 (m, 2H), 3.70
(d, J= 4.9 Hz, 2H), 3.14-3.10 (m, 1H), 1.94-1.56 (m, 7H), 1.30-0.96 (m, 5H),
1.03 (d, J= 6.9 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 157.0, 153.9, 115.8, 115.6, 113.9, 113.8, 66.7,
63.2, 41.4, 37.7, 31.5, 27.1, 26.5, 26.4, 15.5.
Masa exacta calculada para C16H24NOF (M, H+): 265.1842; encontrada:
265.1839.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 95:5, flujo= 0.5 mL/min, tr: 30.2
min (minoritario) y 41.4 min (mayoritario)).
Me
HN
HO
F

Desarrollo Experimental
277
(2R,3R,E)-5-Fenil-3-(4-fluorofenilamino)-2-metilpent-4-en-1 -ol (93Af)
Preparado según el procedimiento general B
partiendo de propanal (0.11 mL, 1.5 mmol), 4-
fluoroanilina (47µL, 0.5 mmol) y cinamaldehído (63 µL,
0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 85:15). Aceite amarillo. Rendimiento: 75 mg (53%).
[α]D20 = −60.0 (c= 0.65, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.39-7.26 (m, 5H), 6.93-6.87 (m, 2H), 6.67-6.64
(m, 2H), 6.56 (d, J= 15.9 Hz, 1H), 6.1 (dd, J= 7.2, 15.9 Hz, 1H), 3.97 (t, J= 7.0
Hz, 1H), 3.83 (dd, J= 4.3, 10.9 Hz, 1H), 3.74 (dd, J= 6.9, 10.9 Hz, 1H), 2.07-
1.99 (m, 1H), 1.06 (d, J= 7.0 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 157.8, 154.6, 143.6, 136.7, 135.5, 131.8, 129.6,
128.6, 127.6, 126.4, 115.8, 115.5, 115.4, 66.8, 61.2, 40.1, 14.0.
Masa exacta calculada para C18H20NOF (M, H+): 285.1529; encontrada:
285.1526.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AS-H, hexano:isopropanol 97:3, flujo= 0.6 mL/min, tr: 40.9
min (mayoritario) y 45.1 min (minoritario)).
((2R,3R)-2,4-Dimetil-3-(fenilamino)pentan-1-ol (93Ag)
Preparado según el procedimiento general A a −40 ºC
partiendo de propanal a (0.11 mL, 1.5 mmol) y 4-fluoro-N-
(2-metilpropilideno) anilina 90g (83 mg, 0.5 mmol). El
producto crudo se purificó por cromatografía de columna
flash (hexano:acetato de etilo 90:10). Aceite amarillo. Rendimiento: 50 mg
(48%).
[α]D20 = +16.4 (c= 0.25, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.18 (t, J= 7.9 Hz, 2H), 6.71-6.66 (m, 3H), 3.71
(d, J= 4.9 Hz, 2H), 3.24 (dd, J= 4.3, 8.4 Hz, 1H), 2.11-2.00 (m, 1H), 1.88-1.78
(m, 1H), 1.05 (d, J= 6.9 Hz, 3H), 0.97 (d, J= 2.3 Hz, 3H), 0.94 (d, J= 2.4 Hz,
3H).
Me
HN
HO
F
Ph
Me
HN
HO
Me
Me

Capítulo 4
278
13C-RMN (75 MHz, CDCl3): δ 149.6, 129.4, 117.1, 113.2, 66.8, 62.2, 38.7, 30.7,
21.0, 16.2, 15.5.
Masa exacta calculada para C13H22NO (M, H+): 208.1701; encontrada:
208.1705.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 97:3, flujo= 0.5 mL/min, tr: 25.7
min (mayoritario) y 40.4 min (minoritario)).
(R)-2-((S)-(4-Fluorofenilamino)(4-metoxifenil)metil) heptan- 1-ol (93Eh)
Preparado según el procedimiento general A a −40
ºC partiendo de heptanal (0.21 mL, 1.5 mmol) y 4-
flouro-N-(4-metoxibenzilideno) anilina 90h (115 mg, 0.5
mmol). El producto crudo se purificó por cromatografía
de columna flash (hexano:acetato de etilo 96:4). Aceite amarillo. Rendimiento:
83 mg (48%).
[α]D20 = −0.8 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3): 7.23 (d, J= 8.6 Hz, 2H), 6.88 (d, J= 8.7 Hz, 2H),
6.81 (t, J= 8.8 Hz, 2H), 6.49-6.45 (m, 2H), 4.38 (d, J= 6.0 Hz, 1H), 3.82 (s, 3H),
3.82-3.77 (m, 1H), 3.69 (dd, J= 5.4, 11.0 Hz, 1H), 1.90-1.81 (m, 1H), 1.52-1.22
(m, 8H), 0.90 (t, J= 6.7 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 158.5, 157.3, 154.2, 143.9, 134.5, 127.9, 115.6,
115.3, 114.7, 114.6, 113.9, 63.6, 61.9, 55.2, 46.4, 32.0, 28.7, 27.0, 22.5, 14.0.
Masa exacta calculada para C21H29NO2F (M, H+): 346.2182; encontrada:
346.2185.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IB, hexano:isopropanol 98:2, flujo= 0.5 mL/min, tr: 29.6 min
(minoritario) y 34.4 min (mayoritario)).
HN
HO
F
( )4OMe

Desarrollo Experimental
279
terc -Butil(5 R,6S)-6-(4-fluorofenilamino)-5-(hidroximetil)-6- p-
tolilhexilcarbamato (93Kd)
Preparado según el procedimiento general A a
−40 ºC partiendo de 6-N-Boc-aminohexanal (323 mg,
1.5 mmol) y 4-fouoro-N-(4-metilbenciliden) anilina 90d
(107 mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 80:20). Aceite amarillo. Rendimiento: 110 mg
(51%).
[α]D22 = +5.5 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.14 (dd, J= 8.0, 22.2 Hz, 4H), 6.75 (t, J= 8.8 Hz,
2H), 6.41 (dd, J= 4.4, 9.0 Hz, 2H), 4.37 (d, J= 5.1 Hz, 1H), 3.72-3.58 (m, 2H),
3.28-2.96 (m, 2H), 2.30 (s, 3H), 1.80-1.56 (m, 2H), 1.48-1.24 (m, 5H), 1.44 (s,
9H). 13C-RMN (75 MHz, CDCl3): δ 157.0, 156.4, 153.9,144.0, 139.5, 136.4, 135.7,
129.2, 126.8, 115.6, 115.3, 114.0, 79.4, 62.3, 46.4, 39.6, 30.4, 28.4, 28.1, 23.9,
21.0.
Masa exacta calculada para C25H36N2O3F (M, H+): 431.2710; encontrada:
431.2727.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IC, hexano:isopropanol 95:5, flujo= 0.5 mL/min, tr: 35.9 min
(minoritario) y 41.2 min (mayoritario)).
(R)-5-Fenil-2-(( S)- (4-fluorofenilamino)(4-metoxifenil)metil)- penta n-1-ol (93Mh)
Preparado según el procedimiento general A a
−40 ºC partiendo de 5-fenilpentanal (243 mg, 1.5
mmol) y 4-fouoro-N-(4-metoxibenzilideno) anilina 90h
(115 mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 90:10). Aceite amarillo. Rendimiento: 124 mg (63%).
[α]D20 = +3.6 (c= 0.7, DCM).
HN
HO
Me
F
NHBoc
( )3
HN
HO
F
Ph OMe

Capítulo 4
280
1H-RMN (300 MHz, CDCl3): 7.32-7-15 (m, 7H), 6.87 (d, J= 8.7 Hz, 2H), 6.81 (t,
2H, J= 8.8 Hz, 2H), 6.48-6.43 (m, 2H), 4.38 (d, J= 6.0 Hz, 1H), 3.82 (s, 3H),
3.82-3.77 (m, 1H), 3.68 (dd, J= 5.3, 11.0 Hz, 1H), 2.64-2.58 (m, 1H), 1.90-1.85
(m, 1H), 1.75-1.48 (m, 5H).
13C-RMN (75 MHz, CDCl3): δ 158.6, 157.3, 143.8, 142.2, 134.3, 128.3, 127.8,
125.8, 115.6, 115.3, 114.6, 114.5, 113.9, 63.5, 61.7, 55.2, 46.3, 36.0, 29.1,
28.4.
Masa exacta calculada para C25H28NO2F (M, H+): 393.2121; encontrada:
393.2104. La pureza enantiomérica del diastereoisómero mayoritario se
determinó por HPLC (Daicel Chiralpak IB, hexano:isopropanol 95:5, flujo= 0.5
mL/min, tr: 41.7 min (mayoritario) y 46.4 min (minoritario)).
(2R,3S)-3-(4-Clorofenilamino)-5-fenil-2-metilpent-4-in-1- ol (97Aa)
Preparado según el procedimiento general A a −60
ºC partiendo de propanal (0.11 mL, 1.5 mmol) y 4-cloro-
N-(3-fenilprop-2-inilideno) anilina 96a (120 mg, 0.5
mmol). El producto crudo se purificó por cromatografía
de columna flash (hexano:acetato de etilo 80:20).
Sirope amarillo. Rendimiento: 106 mg (71%).
[α]D22 = −113.1 (c= 0.35, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.42-7.37 (m, 2H), 7.34-7.28 (m, 3H), 7.20 (d, J=
8.9 Hz, 2H), 6.76 (d, J= 8.9 Hz, 2H), 4.48 (d, J= 6.0 Hz, 1H), 3.89 (dd, J= 4.3,
10.9 Hz, 1H), 3.79 (dd, J= 7.4, 10.9 Hz, 1H), 2.26-2.17 (m, 1H), 1.19 (d, J= 6.9
Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 145.3, 131.7, 129.1, 128.3, 123.5, 122.7, 115.8,
87.7, 84.7, 66.1, 50.2, 39.9, 13.1.
Masa exacta calculada para C18H19NOCl (M, H+): 300.1155; encontrada:
300.1168.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 97:3, flujo= 0.6 mL/min, tr: 43.3
min (minoritario) y 54.6 min (mayoritario)).
Me
HN
HO
Cl
Ph

Desarrollo Experimental
281
(2R,3S)-5-Fenil-2-metil-3-(4-metoxifenilamino)-pent-4-in- 1-ol (97Ab)
Preparado según el procedimiento general A a −60
ºC partiendo de propanal (0.11 mL, 1.5 mmol) y N-(3-
fenilprop-2-inilideno)-4-metoxianilina 96b (118 mg,
0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 80:20). Sirope amarillo. Rendimiento: 133 mg (90%).
[α]D22 = −135.5 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.36-7.32 (m, 2H), 7.29-7.24 (m, 3H), 6.83-6.79
(m, 4H), 4.28 (d, J= 7.2 Hz, 1H), 3.85-3.72(m, 2H), 3.77 (s, 3H), 2.22-2.12 (m,
1H), 1.14 (d, J= 6.9 Hz, 3H).
13C-RMN (75 MHz, CDCl3): δ 153.7, 140.4, 131.7, 128.2, 128.1, 117.4, 116.8,
88.6, 84.6, 67.2, 55.7, 53.1, 40.2, 13.6.
Masa exacta calculada para C19H22NO2 (M, H+): 296.1651; encontrada:
296.1665.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 97:3, flujo= 0.6 mL/min, tr: 62.4
min (minoritario) y 75.3 min (mayoritario)).
(2R,3S)-5-Fenil-2-metil-3-(4-metoxifenylamino)-pent-4-in- 1-ol (sin -97Ab)
Preparado según el procedimiento general C
partiendo de propanal (0.11 mL, 1.5 mmol) y N-(3-
fenilprop-2-inilideno)-4-metoxianilina 96b (118 mg,
0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 80:20). Sirope amarillo. Rendimiento: 118 mg (80%). Tras la cromatografía
flash en columna el aducto sin puro se purificó por placa preparativa de silica
gel eluyendo con hexano:Et2O:AcOEt 5:4:1.
[α]D22 = +87.8 (c= 0.5, DCM).
1H-RMN (300 MHz, CDCl3): δ 7.36 (dd, J= 3.1, 6.8 Hz, 2H), 7.29-7.25 (m, 3H),
6.84-6.77 (m, 4H), 4.36 (d, J= 4.2 Hz, 1H), 3.98-3-91 (m, 1H), 3.83-3.78 (m,
1H), 3.76 (s, 3H), 2.32-2.20 (m, 1H), 1.15, (d, J= 7.0 Hz, 3H).
Me
HN
HO
OMe
Ph
Me
HN
HO
OMe
Ph

Capítulo 4
282
13C-NMR (75 MHz, CDCl3): δ 153.4, 140.8, 131.7, 128.2, 128.1, 116.8, 115.9,
114.8, 88.4, 84.7, 66.2, 55.7, 52.5, 39.6, 13.6.
Masa exacta : no determinada.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AD-H, hexano:isopropanol 97:3, flujo= 0.6 mL/min, tr: 47.14
min (mayoritario) y 57.45 min (minoritario)).
(2R,3S)-2-Metil-3-(4-metoxifenilamino)-dec-4-in-1-ol (97A c)
Preparado según el procedimiento general A a
−60 ºC partiendo de propanal (0.11 mL, 1.5 mmol) y
4-metoxi-N-(oct-2-iniliden) anilina 96c (115 mg, 0.5
mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
etilo 80:20). Aceite amarillo. Rendimiento: 89 mg
(62%).
[α]D22 = −110.7 (c= 0.5, DCM).
1H-RMN (300 MHz, CDCl3): δ 6.82-6.73 (m, 4H), 4.02 (d, J= 7.1 Hz, 1H), 3.76
(s, 3H), 3.76-3.64 (m, 2H), 2.16-2.10 (m, 2H), 2.09-1.98 (m, 1H), 1.49-1.39 (m,
2H), 1.33-1.22 (m, 4H), 1.04 (d, J= 6.9 Hz, 3H), 0.90-0.83 (m, 3H). 13C-RMN (75 MHz, CDCl3): δ 153.5, 140.6, 117.3, 114.7, 85.0, 79.1, 67.3, 55.7,
52.7, 40.1, 30.9, 28.4, 22.1, 18.6, 13.9, 13.4.
Masa exacta calculada para C18H28NO2 (M, H+): 290.2120; encontrada:
290.2115.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak IC, hexano:isopropanol 90:10, flujo= 0.5 mL/min, tr: 25.1 min
(mayoritario) y 32.9 min (minoritario)).
(2R,3S)-2-Bencil-3-(4-metoxifenilamino)dec-4-in-1-ol (97G c)
Preparado según el procedimiento general A a
−60 ºC partiendo de hidrocinamaldehído (0.20 mL, 1.5
mmol) y 4-metoxi-N-(oct-2-iniliden) anilina 96c (115
mg, 0.5 mmol). El producto crudo se purificó por
cromatografía de columna flash (hexano:acetato de
Me
HN
HO
OMe
( )4
HN
HO
OMe
( )4Ph

Desarrollo Experimental
283
etilo 90:10). Aceite amarillo. Rendimiento: 98 mg (54%).
[α]D22 = −67.6 (c= 1, DCM).
1H-RMN (300 MHz, CDCl3): δ .7.36-7-22 (m, 5H), 6.85-6.80 (m, 2H), 6.72-6.67
(m, 2H), 4.20 (d, J= 6.3 Hz, 1H), 3.93-3.85 (m, 1H), 3.79 (s, 1H), 3.71 (dd, J=
7.0, 11.1 Hz, 1H), 3.10 (dd, J= 5.7, 13.7 Hz, 1H), 2.69 (dd, J= 8.7, 13.7 Hz),
2.27-2.15 (m, 3H), 1.56-1.46 (m, 2H), 1.40-1.26 (m, 4H), 0.91 (t, J= 7.1 Hz, 3H). 13C-RMN (75 MHz, CDCl3): δ 153.2, 140.7, 129.2, 128.4,126.2, 116.8, 114.6,
85.4, 79.5, 64.0, 55.7, 50.9, 46.9, 34.6, 31.0, 28.5, 22.1, 18.6, 13.9.
Masa exacta calculada para C24H32NO2 (M, H+): 366.2433; encontrada:
366.2447.
La pureza enantiomérica del diastereómero mayoritario se determinó por HPLC
(Daicel Chiralpak AS-H, hexano:isopropanol 95:5, flujo= 0.5 mL/min, tr: 29.5
min (minoritario) y 34.2 min (mayoritario)).

Capítulo 4
284
4.5.2.3. Espectros de 1H y 13C-RMN 1H-RMN (300 MHz)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
13C-RMN (125 MHz)
0102030405060708090100110120130140150160f1 (ppm)
-400
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
Me
NHPh
HO
Me93Ab
Me
NHPh
HO
Me93Ab

Desarrollo Experimental
285
1H-RMN (300 MHz)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
Me
HN
HO
Me
CN
93Ac
Me
HN
HO
Me
CN
93Ac

Capítulo 4
286
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-2000
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
Me
HN
HO
Me
F
93Ad
Me
HN
HO
Me
F
93Ad

Desarrollo Experimental
287
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
13C-RMN (125 MHz)
0102030405060708090100110120130140150160f1 (ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
Me
HN
HO
F
93Ae
Me
HN
HO
F
93Ae

Capítulo 4
288
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-5000
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
65000
Me
HN
HO
F
Ph
93Af
Me
HN
HO
F
Ph
93Af

Desarrollo Experimental
289
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
13C-RMN (125 MHz)
102030405060708090100110120130140150f1 (ppm)
-5000
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
65000
70000
75000
Me
HN
HO
Me
Me
93Ag
Me
HN
HO
Me
Me
93Ag

Capítulo 4
290
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
0
500
1000
1500
2000
2500
3000
3500
4000
4500
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
PMP
HN
HO
F
( )4
93Eh
PMP
HN
HO
F
( )4
93Eh

Desarrollo Experimental
291
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
1E+05
1E+05
2E+05
2E+05
2E+05
2E+05
HN
HO
Me
F
NHBoc
93Kd
HN
HO
Me
F
NHBoc
93Kd
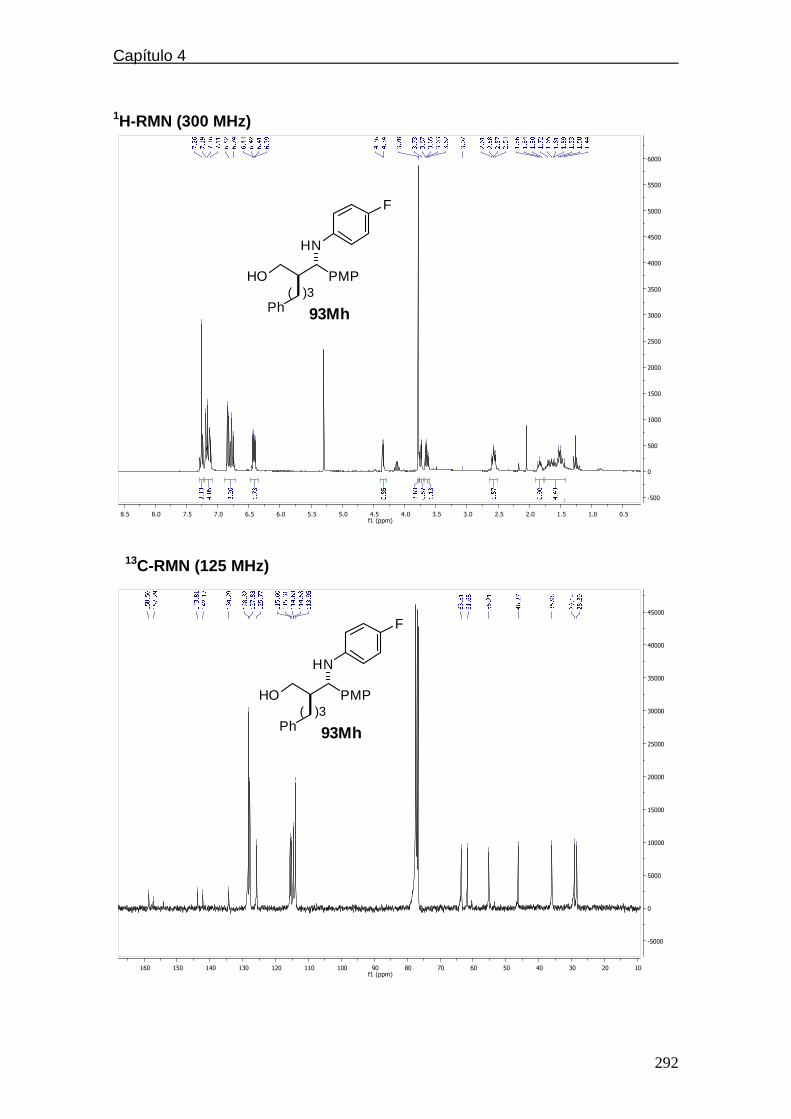
Capítulo 4
292
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-5000
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
( )3PMP
HN
HO
F
Ph 93Mh
( )3PMP
HN
HO
F
Ph 93Mh

Desarrollo Experimental
293
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
13C-RMN (125 MHz)
0102030405060708090100110120130140150f1 (ppm)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
Me
HN
HO
Cl
Ph97Aa
Me
HN
HO
Cl
Ph97Aa
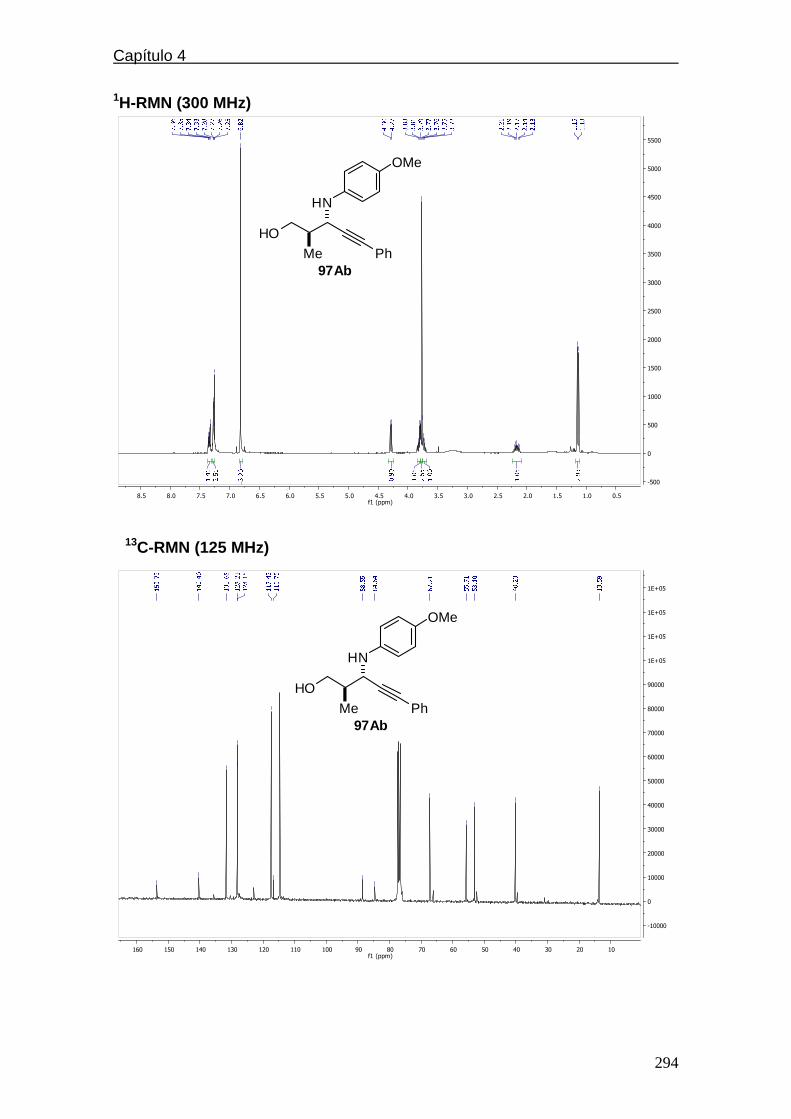
Capítulo 4
294
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
1E+05
Me
HN
HO
OMe
Ph97Ab
Me
HN
HO
OMe
Ph97Ab

Desarrollo Experimental
295
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
3600
3800
4000
13C-RMN (125 MHz)
0102030405060708090100110120130140150160170f1 (ppm)
-4000
-2000
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
22000
24000
26000
28000
30000
32000
34000
Me
HN
HO
OMe
Ph
sin-97Ab
Me
HN
HO
OMe
Ph
sin-97Ab

Capítulo 4
296
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
13C-RMN (125MHz)
102030405060708090100110120130140150160f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
Me
HN
HO
OMe
( )497Ac
Me
HN
HO
OMe
( )497Ac

Desarrollo Experimental
297
1H-RMN (300 MHz)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5f1 (ppm)
0
500
1000
1500
2000
2500
3000
3500
13C-RMN (125 MHz)
102030405060708090100110120130140150160f1 (ppm)
-10000
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
1E+05
1E+05
1E+05
1E+05
1E+05
2E+05
2E+05
2E+05
2E+05
2E+05
HN
HO
OMe
( )4Ph
97Gc
HN
HO
OMe
( )4Ph
97Gc

Capítulo 4
298
4.5.2.4. Cromatogramas de HPLC
Chiralpack AD-H, 0.5 mL/min, hexano:isopropanol 95:5, λ= 280 nm
25.3
47
36.2
60
AU
0.00
0.10
0.20
Minutes
22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00
28.5
56
39.3
13
AU
0.00
0.20
0.40
Minutes
24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00
NHPh
HO
MeMe
r ac-93Ab
Me
NHPh
HO
Me93Ab

Desarrollo Experimental
299
Chiralpack IC, 0.8 mL/min, hexano:isopropanol 95:4:1, λ= 254 nm
52.5
90
65.8
88
AU
0.00
0.05
0.10
Minutes
40.00 45.00 50.00 55.00 60.00 65.00 70.00 75.00 80.00 85.00 90.00
57.3
17
70.3
13
AU
0.00
0.20
Minutes
45.00 50.00 55.00 60.00 65.00 70.00 75.00
Me
HN
HO
Me
CN
93Ac
HN
HO
Me
CN
Me
rac-93Ac

Capítulo 4
300
Chiralpack AD-H, 0.5 mL/min, hexano:isopropanol 95:5, λ= 254 nm
28.8
26
35.9
15
AU
0.00
0.50
1.00
Minutes
24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00
29.2
48
35.9
13
AU
0.00
1.00
2.00
Minutes
24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00
Me
HN
HO
Me
F
93Ad
HN
HO
Me
F
Me
rac-93Ad

Desarrollo Experimental
301
Chiralpack AD-H, 0.5 mL/min, hexano:isopropanol 95:5, λ= 254 nm
29.6
55
41.3
40
AU
0.00
0.20
0.40
0.60
0.80
Minutes
20.00 25.00 30.00 35.00 40.00 45.00 50.00 55.00
30.1
74
41.4
43
AU
0.00
0.50
1.00
Minutes
25.00 30.00 35.00 40.00 45.00 50.00 55.00 60.00
HN
HO
F
Me
rac-93Ae
Me
HN
HO
F
93Ae

Capítulo 4
302
Chiralpack AS-H, 0.6 mL/min, hexano:isopropanol 97:3, λ= 254 nm
41.9
84
45.8
93
AU
0.00
0.20
0.40
Minutes
30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00 52.00 54.00 56.00 58.00
40.8
92
45.0
89AU
0.00
0.20
0.40
0.60
Minutes
32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00 52.00 54.00
HN
HO
F
Ph
Me
r ac-93Af
Me
HN
HO
F
Ph
93Af

Desarrollo Experimental
303
Chiralpack AD-H, 0.5 mL/min, hexano:isopropanol 97:3, λ= 254 nm
AU
0,00
0,10
0,20
0,30
0,40
0,50
Minutes15,00 20,00 25,00 30,00 35,00 40,00 45,00 50,00
25,9
27
40,4
04
AU
0,00
0,20
0,40
0,60
0,80
Minutes18,00 20,00 22,00 24,00 26,00 28,00 30,00 32,00 34,00 36,00 38,00 40,00 42,00 44,00
25,7
52
40,3
68
HN
HO
Me
Me
Merac-93Ag
Me
HN
HO
Me
Me
93Ag

Capítulo 4
304
Chiralpack IB, 0.5 mL/min, hexano:isopropanol 98:2, λ= 210 nm
29.6
08
34.3
66
AU
0.00
0.20
0.40
0.60
0.80
1.00
Minutes
26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00
29.6
20
33.9
97
AU
0.00
0.20
0.40
0.60
0.80
1.00
Minutes
24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00
PMP
HN
HO
F
93Eh
( )4
PMP
HN
HO
F
rac-93Eh
( )4

Desarrollo Experimental
305
Chiralpack IC, 0.5 mL/min, hexano:isopropanol 95:5, λ= 210 nm
36
.532
42.1
58
AU
0.00
0.05
0.10
Minutes
26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00
35.8
82
41.2
78
AU
0.00
0.10
0.20
Minutes
28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00
rac-93Kd
HN
HO
Me
F
NHBoc
HN
HO
Me
F
NHBoc93Kd

Capítulo 4
306
Chiralpack IB, 0.5 mL/min, hexano:isopropanol 95:5, λ= 254 nm
41.7
38
46.4
42
AU
0.000
0.001
0.002
0.003
0.004
Minutes
36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00
42.5
27
47.2
17AU
0.000
0.005
0.010
Minutes
41.00 42.00 43.00 44.00 45.00 46.00 47.00 48.00
PMP
HN
HO
F
Ph( )3
rac-93Mh
PMP
HN
HO
F
Ph( )3
93Mh
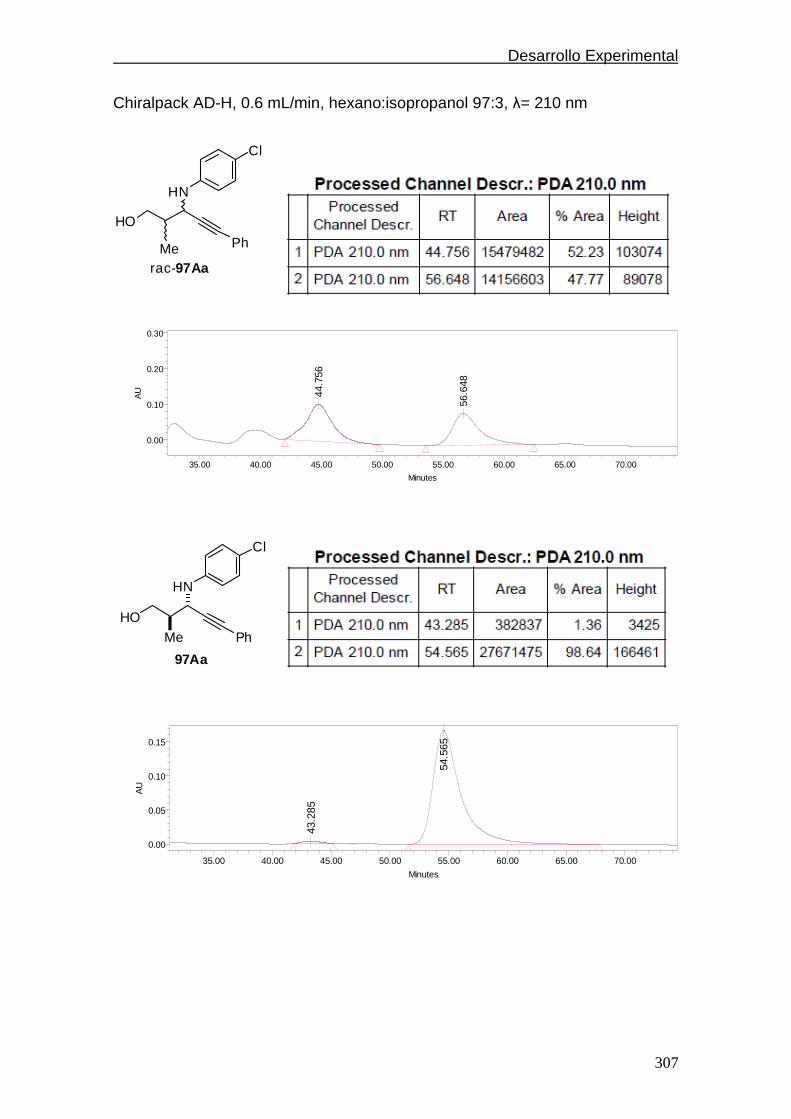
Desarrollo Experimental
307
Chiralpack AD-H, 0.6 mL/min, hexano:isopropanol 97:3, λ= 210 nm
44.7
56
56.6
48
AU
0.00
0.10
0.20
0.30
Minutes
35.00 40.00 45.00 50.00 55.00 60.00 65.00 70.00
43.2
85
54.5
65
AU
0.00
0.05
0.10
0.15
Minutes
35.00 40.00 45.00 50.00 55.00 60.00 65.00 70.00
Me
HN
HO
Cl
Ph
97Aa
HN
HO
Cl
PhMerac-97Aa
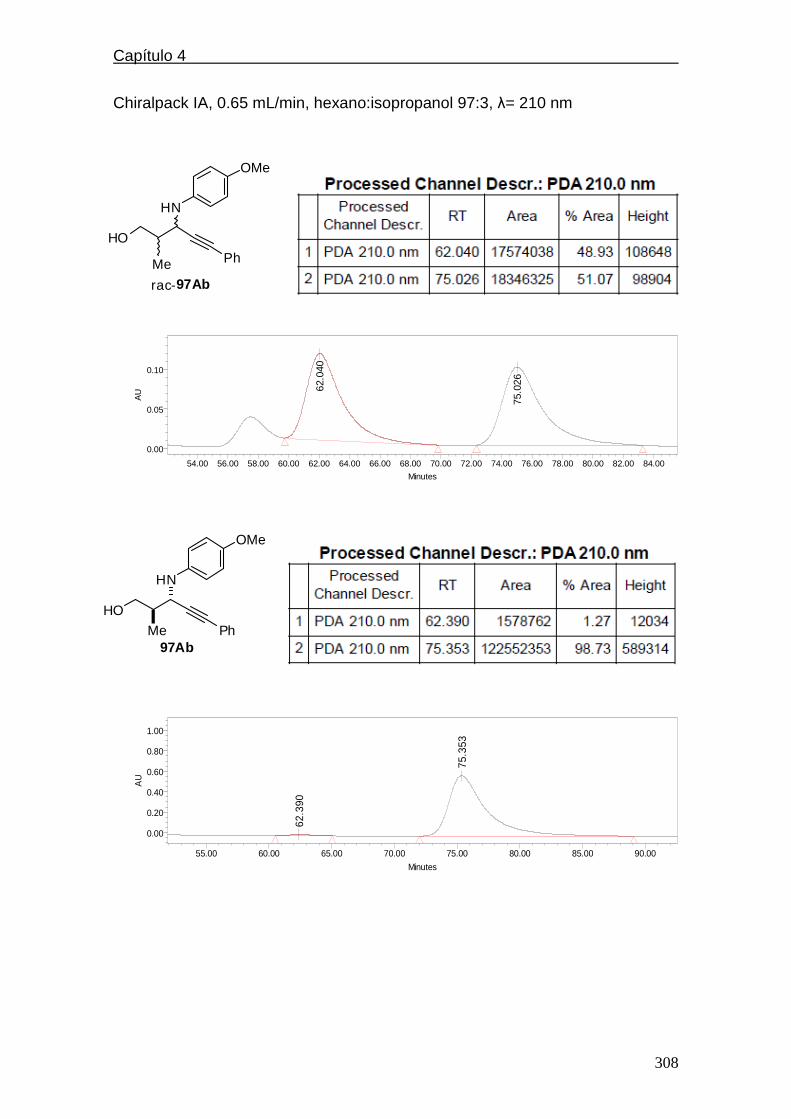
Capítulo 4
308
Chiralpack IA, 0.65 mL/min, hexano:isopropanol 97:3, λ= 210 nm
62.0
40
75.0
26
AU
0.00
0.05
0.10
Minutes
54.00 56.00 58.00 60.00 62.00 64.00 66.00 68.00 70.00 72.00 74.00 76.00 78.00 80.00 82.00 84.00
62.3
90
75.3
53
AU
0.00
0.20
0.40
0.60
0.80
1.00
Minutes
55.00 60.00 65.00 70.00 75.00 80.00 85.00 90.00
HN
HO
OMe
PhMe
rac-97Ab
Me
HN
HO
OMe
Ph97Ab

Desarrollo Experimental
309
Chiralpack IA, 0.65 mL/min, hexano:isopropanol 97:3, λ= 210 nm
Diastereoisómeros sin
48.0
65
57.4
53 62.0
40
75.0
26
AU
0.00
0.05
0.10
Minutes
40.00 45.00 50.00 55.00 60.00 65.00 70.00 75.00 80.00 85.00
47.1
40
73.1
18
AU
0.00
0.10
0.20
0.30
0.40
0.50
Minutes
35.00 40.00 45.00 50.00 55.00 60.00 65.00 70.00 75.00 80.00 85.00
Me
HN
HO
OMe
Phsin-97Ab
HN
HO
OMe
PhMe
rac-97Ab

Capítulo 4
310
Chiralpack IC, 0.5 mL/min, hexano:isopropanol 90:10, λ= 210 nm
25.1
24
32.9
38
AU
0.00
0.20
0.40
0.60
0.80
Minutes
18.00 20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00
24.5
13
32.1
65
AU
0.00
0.50
1.00
1.50
Minutes
16.00 18.00 20.00 22.00 24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00
Me
HN
HO
OMe
( )497Ac
Me
HN
HO
OMe
( )4
rac-97Ac
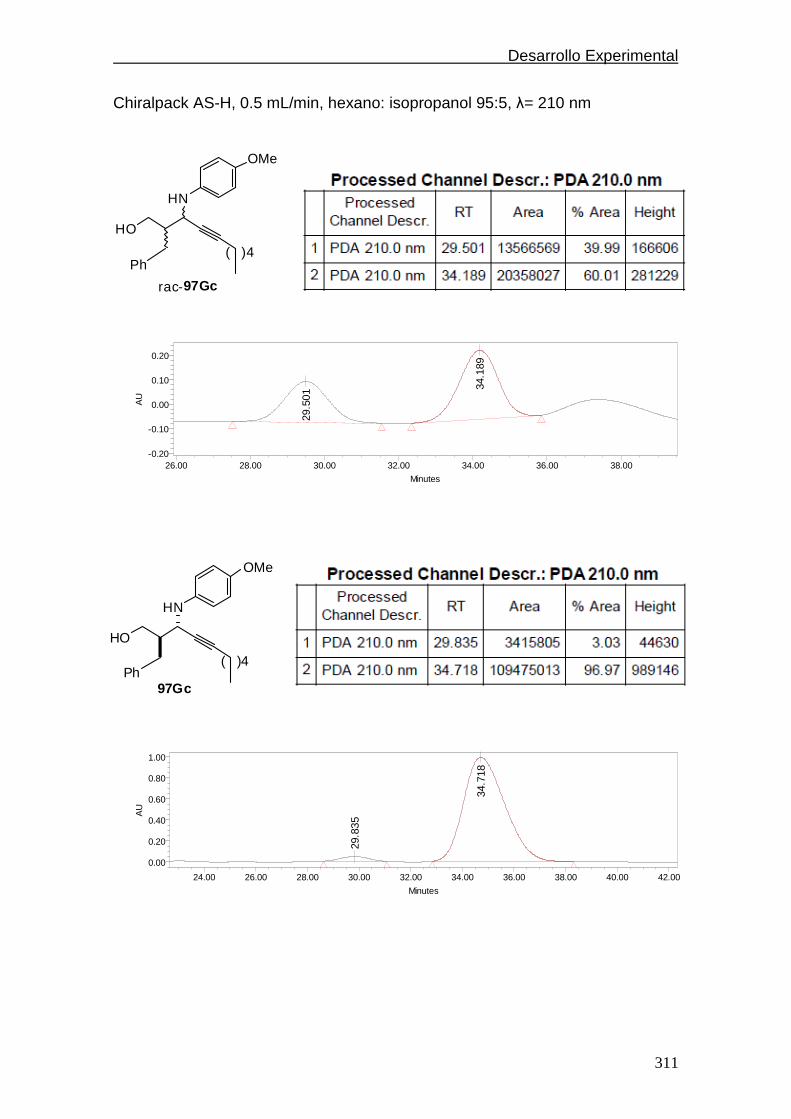
Desarrollo Experimental
311
Chiralpack AS-H, 0.5 mL/min, hexano: isopropanol 95:5, λ= 210 nm
29.5
01
34.1
89
AU
-0.20
-0.10
0.00
0.10
0.20
Minutes
26.00 28.00 30.00 32.00 34.00 36.00 38.00
29.8
35
34.7
18
AU
0.00
0.20
0.40
0.60
0.80
1.00
Minutes
24.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00
HN
HO
OMe
( )4Ph
rac-97Gc
HN
HO
OMe
( )4Ph
97Gc


Anexo: publicaciones


315
5. Anexo: publicaciones Parte de los resultados obtenidos durante la elaboración de la presente Tesis
Doctoral han sido objeto de las siguientes publicaciones:
“Regio- and Enantioselective Direct Oxyamination Reaction of Aldehydes
Catalyzed by α,α-Diphenylprolinol Trimethylsilyl Ether”.
C. Palomo, S. Vera, I. Velilla, Antonia Mielgo, E. Gómez-Bengoa
Angew. Chem. Int. Ed. 2007, 42, 8054 – 8056.
“A 4-Hydroxypyrrolidine-Catalyzed Mannich Reaction of Aldehydes: Control of
anti-Selectivity by Hydrogen Bonding Assisted by Brønsted Acids”.
E. Gómez-Bengoa, M. Maestro, A. Mielgo, I. Otazo, C. Palomo, I. Velilla
Chem. Eur. J. 2010, 16, 5333 – 5342.
“Brønsted Acid Assisted Regio- and Enantioselective Direct O-Nitroso Aldol
Reaction Catalyzed by S-(-)-α,α-Diphenylprolinol Trimethylsilyl Ether”.
C. Palomo, I. Velilla, A. Mielgo, E. Gómez-Bengoa
Chem. Eur. J. 2010, 16, 7496 – 7502.


OrganocatalysisDOI: 10.1002/anie.200703001
Regio- and Enantioselective Direct Oxyamination Reaction ofAldehydes Catalyzed by a,a-Diphenylprolinol Trimethylsilyl Ether**Claudio Palomo,* Silvia Vera, Irene Velilla, Antonia Mielgo, and Enrique G�mez-Bengoa
The nitroso function is recognized as a unique source toprepare nitrogen- and oxygen-containing molecules, andvarious catalytic asymmetric reactions of nitroso com-pounds[1] such as aminoxylation,[2] oxyamination,[3] and ni-troso Diels–Alder reactions[4] have recently been developedwhich exploit their unique properties. Owing to the highreactivity of nitroso derivatives toward nucleophiles, control-ling the regioselectivity of either the nitrogen or oxygen topreferentially react with the nucleophile is a challenge offundamental importance. Investigations of the reaction ofnitrosobenzene with a silyl or metal enolate have revealedthat the O versus N selectivity is dependent on the nature ofthe enolate and the presence or absence of a Lewis acidcatalyst.[5] Yamamoto and Momiyama reported the reactionbetween preformed enamines and nitrosobenzene in thepresence of catalytic amounts of glycolic acid to preferentiallyafford the O-nitroso aldol product, while in the presence ofTADDOL (a,a,a’,a’-tetraaryl-2,2-dimethyl-1,3-dioxolan-4,5-dimethanol) the a-amino derivatives are exclusively obtained(Scheme 1).[6]
Organocatalyzed reactions of nitrosobenzene and car-bonyl compounds with proline and its derivatives as catalyststo give a-oxygenated compounds as the major products have
also been actively investigated.[2d–o,7] In sharp contrast, onlytwo contributions on organocatalyzed direct nitrosoaldolreactions of carbonyl compounds that take place preferen-tially at the nitrogen have been reported.[2a,8] The first byMaruoka and co-workers[3a] described the asymmetric oxy-amination reaction of aldehydes with nitrosobenzene cata-lyzed by the novel binaphthyl-based axially chiral secondaryamine 1 (Figure 1). Although the preparation of the catalyst
requires several steps, high yields and excellent enantioselec-tivities were achieved. The second report implied the directnitrosoaldol reaction of a-branched aldehydes in the presenceof the l-prolinamide derivative 2 (Figure 1) as catalyst.[3b] Inthis case, the catalyst is structurally simple, but the reportedenantioselectivities were modest. Here, we present ourfinding that prolinol ether derivatives are capable of effi-ciently promoting the oxyamination reaction of aldehydeswith nitrosobenzene.
A structural feature of catalysts 1 and 2 for the directoxyamination of aldehydes is the presence of a weak hydro-gen-bond donor (OH group), which apparently coordinatesthe oxygen atom of nitrosobenzene to facilitate the oxy-amination pathway.[9] We hypothesized that the a-oxyamina-tion of aldehydes could be promoted by catalysts which lackhydrogen-bond donors. As the reaction results in the pro-duction of hydroxylamine, either the product itself or watergenerated from formation enamine could activate the oxy-amination reaction through hydrogen-bond coordination tothe oxygen of nitrosobenzene. In a first instance and owing tothe recent emergence of a,a-diarylprolinol ether derivativesas fairly general organocatalysts,[10] we considered thatderivative 3[11] (Figure 1) could be a reasonable candidate toevaluate the above assumption. Concordant with our expect-ations, product 6b (Table 1, entry 3), obtained from thereaction of butanal with nitrosobenzene promoted by 3 inmethylene chloride as solvent, was produced after subsequentreduction of the resulting oxyaminated intermediate, in goodyield and, most remarkably, with essentially perfect regiose-
Scheme 1. Possible products from the nitroso aldol reaction.
Figure 1. Developed organocatalysts for the N-nitroso aldol reaction ofaldehydes (1 and 2) and a complementary alternative proposal (3).
[*] Prof. Dr. C. Palomo, S. Vera, I. Velilla, Dr. A. Mielgo,Dr. E. G/mez-BengoaDepartamento de Qu3mica Org4nica IFacultad de Qu3micaUniversidad del Pa3s Vasco. Apdo. 107220080 San Sebasti4n (Spain)Fax: (+34)943-015-270E-mail: [email protected]
[**] This work was financially supported by The University of the BasqueCountry (UPV/EHU) and the Ministerio de Educaci/n y Ciencia(MEC, Spain). Predoctoral grants to S.V. (MEC) and to I. Velilla(UPV/EHU) are also acknowledged. We also thank the SGI/IZO-SGIker UPV/EHU for allocation of computational resources.
Supporting information for this article is available on the WWWunder http://www.angewandte.org or from the author.
Communications
8054 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2007, 46, 8054 –8056

lectivity and very high enantioselectivity. Screening of othersolvents for this reaction revealed that THF and acetonitrileare also efficient, although methylene chloride is somewhatsuperior.
Results with other aldehydes are shown in Table 2. Thereactions were carried out by dropwise addition of catalyst 3to a greenish solution of nitrosobenzene and the aldehyde inmethylene chloride at 0 8C or room temperature, whereby analmost instantaneous decoloration was observed after theaddition (Table 2, entries 1–3, 7–10). In general, the oxy-amination reaction proceeded fast and the subsequentreduction with sodium borohydride provided the correspond-ing N-hydroxy b-aminoalcohols 6 in good yields over the twosteps and with excellent enantioselectivities. Long-chainaldehydes afford at room temperature the expected adductsin somewhat lower yields (Table 2, entry 7). In these cases, thebest results are obtained by carrying out the reaction at
�20 8C overnight (Table 2, entries 4–6) in either methylenechloride or THF.
The absolute configuration of the resulted adduct in thereaction of pentanal with nitrosobenzene was determined bycomparison of the value for the optical rotation with thatpreviously described[3a] and was found to be S. This result is inaccordance with the approach of the nitrosobenzene by theSi face of the enamine as shown in Figure 2.
To gain more insight into our assumption, we computed bydensity functional theory (DFT) the different transition statesfor the formation of the C�N and C�O bonds in the reactionbetween propionaldehyde and nitrosobenzene in the absenceof any added hydrogen-bonding source (non-functionalizedpyrrolidine catalyst) and in the presence of water or N,N-dimethylhydroxylamine (8 ; mimicking product 6 or itsprecursor).[12] We found that the free activation energy DG�
for the non-catalyzed oxyamination is 25.6 kcalmol�1 (Fig-ure 3A). Water or N,N-dimethylhydroxylamine (8) catalyzethe reaction with similar efficiency, reducing the activationbarrier to 20.9 kcalmol�1 for water (Figure 3B) and 20.3 kcalmol�1 for 8 (Figure 3C).
In agreement with the experimentally encountered regio-selectivity of over 99:1 in favor of the formation of 6, the
computed DG� values for the non-catalyzed and catalyzed aminoxyla-tion are in all cases over 3.0 kcalmol�1 higher than those of the oxy-amination.
In summary, we have demon-strated that the direct and regiose-lective oxyamination reaction ofaldehydes with nitrosobenzene canbe successfully achieved in the pres-ence of a structurally simple andreadily accessible organocatalystsuch as a,a-diphenylprolinol trime-thylsilyl ether to afford the oxy-aminated compounds in good yieldsand with excellent regio- and enan-tioselectivities.
Experimental SectionGeneral procedure for the oxyaminationof aldehydes with nitrosobenzene: Thecatalyst (0.4 mmol, 20 mol%) was added
Table 1: Solvent screening in the oxyamination reaction of butanal withnitrosobenzene.[a]
Entry Solvent 6b/7b Yield [%][b] ee [%][c]
1 toluene >99:1 21 n.d.[d]
2 THF >99:1 40 903 CH2Cl2 >99:1 65 904 CH3CN >99:1 50 855 H2O >99:1 25[e] 90
[a] Reactions were conducted with aldehyde (3 equiv) at 0 8C, withaddition of the catalyst to a solution of the aldehyde and nitrosobenzene.[b] Isolated yield of product 6b after column chromatography. [c] Deter-mined by HPLC analysis. See Supporting Information for further details.[d] Not determined. [e] Nitrosobenzene shows poor solubility in water.
Table 2: Enantioselective oxyamination of various aldehydes with nitrosobenzene.[a]
Entry 6,7 R Solvent t [min] T [8C] 6/7[b] Yield [%][c] ee [%][d]
1 a Me CH2Cl2 5 RT >99:1 70 942 b Et CH2Cl2 5 0 >99:1 65 993 c nPr CH2Cl2 10 0 >99:1 60 964 c nPr CH2Cl2 30 �20 >99:1 66 985 d n-Pent THF 16 h �20 >99:1 74 98[e]
6 e n-Hex THF 16 h �20 >99:1 75 98[e]
7 e n-Hex CH2Cl2 5 RT >99:1 42 n.d.[f ]
8 f Bn CH2Cl2 5 0 >99:1 70 949 g 2-MeOC6H4CH2 CH2Cl2 5 RT >99:1 40 91
10 h iPr CH2Cl2 30 0 >99:1 60 99
[a] Reactions were conducted with aldehyde (3 equiv) at 0 8C or RT, with addition of the catalyst to asolution of the aldehyde and nitrosobenzene. See Experimental Section for the experimental procedureat �20 8C. [b] Determined by 1H NMR spectroscopic analysis of the crude mixture. [c] Isolated yield ofproduct 6 after column chromatography. [d] Determined by HPLC analysis. See Supporting Informationfor further details. [e] Determined by HPLC analysis of the benzoyl and naphthoyl derivatives. SeeSupporting Information for further details. [f ] Not determined.
Figure 2. The approach of nitrosobenzene from the Si face of theenamine.
AngewandteChemie
8055Angew. Chem. Int. Ed. 2007, 46, 8054 –8056 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org

to a solution of the aldehyde (6 mmol, 3 equiv) in THF (1 mL) at�20 8C. A solution of nitrosobenzene (2 mmol, 1 equiv) in THF(1 mL) was then added dropwise to the reaction mixture, and theresulting solution was stirred at �20 8C for 16 h. EtOH (2 mL) andNaBH4 (8 mmol) were successively added at the same temperature,and after stirring for 30 min the reaction was quenched with saturatedNaCl (3 mL) and allowed to reach room temperature. Afterextraction with CH2Cl2 (3 B 4 mL), the combined organic phaseswere dried over MgSO4 and concentrated under reduced pressure,and the residue was purified over silica gel by flash columnchromatography to afford the expected adducts.
Received: July 5, 2007Revised: July 18, 2007Published online: September 12, 2007
.Keywords: aldehydes · enantioselectivity · organocatalysis ·oxyamination · regioselectivity
[1] For reviews on nitroso compounds, see: a) P. Zuman, P. Shap,Chem. Rev. 1994, 94, 1621 – 1641; b) L. Soghyuk, C. Li, H. W.Ann, Chem. Rev. 2002, 102, 1019 – 1066; c) H. Yamamoto, M.Kawasaki, Bull. Chem. Soc. Jpn. 2007, 80, 595 – 607. For a reviewon catalytic, enantioselective a-aminations and oxygenations,see: d) J. M. Janey,Angew. Chem. 2005, 117, 4364 – 4372;Angew.Chem. Int. Ed. 2005, 44, 4292 – 4300.
[2] For reviews, see: a) P. Merino, T. Tejero, Angew. Chem. 2004,116, 3055 – 3058; Angew. Chem. Int. Ed. 2004, 43, 2995 – 2997;b) B. Plietker, Tetrahedron: Asymmetry 2005, 16, 3453 – 3459;c) H. Yamamoto, N. Momiyama, Chem. Commun. 2005, 3514 –3525. For l-proline as catalyst, see: d) Z. Zhong, Angew. Chem.2003, 115, 4379 – 4382; Angew. Chem. Int. Ed. 2003, 42, 4247 –4250; e) S. P. Brown, M. P. Brochu, C. J. Sinz, D. W. C. MacMil-lan, J. Am. Chem. Soc. 2003, 125, 10808 – 10809; f) Y. Hayashi, J.Yamaguchi, T. Sumiya, K. Hibino, M. J. Shoji, J. Org. Chem.2004, 69, 5966 – 5973; g) K. Huang, Z.-Z. Huang, Y.-L. Li, J. Org.
Chem. 2006, 71, 8320 – 8323; h) A. CGrdova, H. SundHn, A.Bøgevig, M. Johansson, F. Himo, Chem. Eur. J. 2004, 10, 3673 –3684. For a proline-based tetrazole derivative, see: i) N.Momiyama, H. Torii, S. Saito, H. Yamamoto, Proc. Natl. Acad.Sci. USA 2004, 101, 5374 – 5378; j) S. Kumarn, D. M. Shaw, D. A.Longbottom, S. V. Ley, Org. Lett. 2005, 7, 4189 – 4191; k) Y.Yamamoto, N. Momiyama, H. Yamamoto, J. Am. Chem. Soc.2004, 126, 5962 – 5963; l) S. G. Kin, T.-H. Park, Tetrahedron Lett.2006, 47, 9067 – 9071. For trans-4-tert-butyldimethylsilyloxy-l-proline, see: m) Y. Hayashi, J. Yamaguchi, K. Hibino, T. Sumiya,T. Urushima, M. Shoji, D. Hashizume, H. Koshino, Adv. Synth.Catal. 2004, 346, 1435 – 1439. For a polymer-supported prolinederivative, see: n) D. Font, A. Bastero, S. Salayero, C. Jimeno,M. A. PericLs, Org. Lett. 2007, 9, 1943 – 1946. For a pyrrolidinesulfonamide catalyst, see: o) W. Wang, J. Wang, H. Li, L. Liao,Tetrahedron Lett. 2004, 45, 7235 – 7238.
[3] a) T. Kano, M. Ueda, J. Takai, K. Maruoka, J. Am. Chem. Soc.2006, 128, 6046 – 6047; b) H.-M. Guo, L. Cheng, L.-F. Cun, L.-Z.Ghong, A.-Q. Mi, Y.-Z. Jiang, Chem. Commun. 2006, 429 – 431.
[4] a) Y. Yamamoto, H. Yamamoto, J. Am. Chem. Soc. 2004, 126,4128 – 4129; b) Y. Yamamoto, H. Yamamoto, Angew. Chem.2005, 117, 1508 – 1511; Angew. Chem. Int. Ed. 2005, 44, 7082 –7085.
[5] a) N.Momiyama, H. Yamamoto,Angew. Chem. 2002, 114, 3459 –3461; Angew. Chem. Int. Ed. 2002, 41, 2986 – 2988; b) N.Momiyama, H. Yamamoto, Org. Lett. 2002, 4, 3579 – 3582;c) N. Momiyama, H. Yamamoto, J. Am. Chem. Soc. 2003, 125,6038 – 6039.
[6] N. Momiyama, H. Yamamoto, J. Am. Chem. Soc. 2005, 127,1080 – 1081.
[7] For the origins of selectivities in proline-catalyzed a-amino-xylations, see: a) P. H.-Y. Chong, K. N. Houk, J. Am. Chem. Soc.2004, 126, 13912 – 13913; b) S. P. Mathew, H. Iwamura, D. G.Blackmond, Angew. Chem. 2004, 116, 3379 – 3383; Angew.Chem. Int. Ed. 2004, 43, 3317 – 3321.
[8] For reviews on organocatalytic enantioselective a-heterofunc-tionalisation of carbonyl compounds, see: a) G. Guillena, D. J.RamGn, Tetrahedron: Asymmetry 2006, 17, 1465 – 1492; b) M.Marigo, K. A. Jørgensen, Chem. Commun. 2006, 2001 – 2011.
[9] It has also been reported recently that a pyrrolidine-basedtetrazole catalyst also promotes the oxyamination. In thisinstance, lower levels of regio- and enantioselectivities, areproduced; see Ref. [2l] for details.
[10] a) C. Palomo, A. Mielgo, Angew. Chem. 2006, 118, 8042 – 8046;Angew. Chem. Int. Ed. 2006, 45, 7876 – 7880. For a recent reviewon asymmetric aminocatalysis, see: b) B. List, Chem. Commun.2006, 819 – 824, and references therein.
[11] Y. Hayashi, H. Gotog, T. Hayashi, M. Shoji,Angew. Chem. 2005,117, 4284 – 4287; Angew. Chem. Int. Ed. 2005, 44, 4212 – 4215.
[12] All the calculations were performed at the standard 6-31G* basisset (see Supporting Information for details). Further studies athigher level involving the actual catalyst 3, as suggested by onereferee, are currently underway.
Figure 3. Computed transition states for the oxyamination reaction ofpropionaldehyde and nitrosobenzene: A) in the absence of any hydro-gen-bonding source, B) in the presence of water, and C) in thepresence of 8. DG� values are given in kcalmol�1.
Communications
8056 www.angewandte.org � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2007, 46, 8054 –8056

DOI: 10.1002/chem.200903537
A 4-Hydroxypyrrolidine-Catalyzed Mannich Reaction of Aldehydes: Controlof anti-Selectivity by Hydrogen Bonding Assisted by Brønsted Acids
Enrique G�mez-Bengoa,[a] Miguel Maestro,[b] Antonia Mielgo,[a] Itziar Otazo,[a]
Claudio Palomo,*[a] and Irene Velilla[a]
Dedicated to Professor Josep M. Rib� on the occasion of his 70th birthday
Introduction
Inspired by the efficiency, elegance, and selectivity of enzy-matic catalysis, the design of organic molecules capable ofthe efficient and enantioselective promotion of carbon–carbon or carbon–heteroatom bond processes is a formida-ble challenge that is currently receiving considerable atten-tion. In this context, several examples of chiral Brønstedacid[1] and chiral Brønsted base[2] promoted organocatalytictransformations are known. More significantly, moleculesthat combine a site with Brønsted base character and anoth-
er site with hydrogen-bond donor ability (ambifunctionalcatalysts) that are capable of activating concurrently boththe donor and acceptor components have emerged as abetter alternative to achieve high levels of reaction enantio-selection.[3] Besides the thiourea/amine-based catalysts,[4] rel-evant examples in the above context are some secondaryamines that activate the donor aldehyde or ketone throughenamine[5] formation and the electrophile through hydro-gen-bond interactions (Scheme 1a). Prototypical catalysts ofthis type are proline 1,[5a,6] the analogues 2[7] and 3,[6d, 8] andsome prolylamides[9] in which the hydrogen-bond donor islocated in the substituent at the a-position of the pyrrolidinenitrogen. A related family of amine catalysts works onlythrough steric control (Scheme 1b). These molecules acti-vate the donor via enamine formation and possess a sterical-ly hindered group that directs the approach of the electro-phile from the less-hindered enamine face and thereby con-trols the stereoselectivity of the reaction. In this secondgroup, the most representative small organic molecules areMacMillan�s imidazolidinones[10] and a,a-diarylprolinolethers[11] (4 in Scheme 1), which have been shown to be verygeneral for a broad range of reactions. However, catalysts ofthis latter family with an additional hydrogen-bond donorthat could also activate the pronucleophile concurrently(Scheme 1c) are quite scarce despite the fact that they
Abstract: An anti-selective Mannichreaction of aldehydes with N-sulfonylimines has been developed by using a4-hydroxypyrrolidine in combinationwith an external Brønsted acid. Thecatalyst design is based on three ele-ments: the a-substituent of the pyrroli-dine, the 4-hydroxy group, and theBrønsted acid, the combination ofwhich is essential for high chemicaland stereochemical efficiency. The re-
action works with aromatic aldehyde-derived imines, which have rarely beenemployed in previously reported enam-ine-based anti-Mannich reactions. Ad-ditionally, both N-tosyl and N-nosyl
imines can be successfully used and theMannich adducts can be easily reducedor oxidized, and after N-deprotectionthe corresponding b-amino acids andb-amino alcohols can be obtained withgood yields. The results also show thatthis ternary catalytic system may bepractical in other enamine-based reac-tions.
Keywords: asymmetric catalysis ·Brønsted acids · enamine activa-tion · hydrogen bonds · Mannich re-action
[a] Dr. E. G�mez-Bengoa,+ Dr. A. Mielgo, I. Otazo, Prof. Dr. C. Palomo,I. VelillaDepartmento de Qu�mica Org�nica IUniversidad del Pa�s Vasco, Manuel Lardizabal 3.20018-San Sebasti�n (Spain)Fax: (+34) 943015270E-mail : [email protected]
[b] Dr. M. MaestroDepartmento de Qu�mica FundamentalFacultad de Ciencias, Universidad da CoruÇaCampus Zapateira s/n, 15071 A CoruÇa (Spain)
[+] Computational study.
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.200903537.
Chem. Eur. J. 2010, 16, 5333 – 5342 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 5333
FULL PAPER

would be, in principle, more active as a result of this syner-gic action.
Herein we report a new simple family of amine catalysts,trans-4-hydroxy-2-dialkylsilyl pyrrolidines, that fulfils thesestructural requirements and we demonstrate their capabilityof promoting anti-selective Mannich reactions with veryhigh diastereo- and enantioselectivity.
Results and Discussion
Background and working plan : Previously we introducedtrans-4-hydroxyprolylamide 8 as an efficient catalyst for con-jugate addition reactions of aldehydes to nitroalkenes.[12] In
a subsequent related investigation by Feng[13] it has beenshown that catalyst 9 is capable of promoting the Biginellireaction with good to moderate levels of enantioselectivity,and more recently, Nakano/Takeshit,[14] Gao,[15] and Chen[16]
independently revealed catalysts 10, 11, and 12, respectively,to be effective for the conjugate addition of aldehydes to ni-troalkenes, with the former being also efficient in aldol reac-tions. In all these cases, it was assumed that the hydroxygroup of each catalyst played an important role in reactionefficiency and stereoselectivity.
In our initial studies on this subject, the addition reactionof a survey of enolizable aldehydes to either b-alkyl or b-aryl-substituted nitroalkenes promoted by catalyst 8 was ob-served to proceed with excellent syn/anti ratios, typicallygreater than 95:5, and with very high enantiomeric excess(ee) values for the major syn diastereomer (>92 % ee ;Scheme 2). The optimum results were achieved when the re-
actions were carried out in CH2Cl2 at room temperature inthe presence of 10 % of catalyst 8 for b-branched aldehydesand 5 mol % for linear-chain aldehydes at 0 8C. Under theseconditions, the enantioselectivities obtained were above90 % for essentially all substrates that were explored. Eventhe more challenging b-alkyl-substituted nitroalkenes gaveadducts with diastereomeric ratios greater than 99:1 andenantioselectivities of up to 99 %, results that were attribut-ed to a cooperative effect between the hydroxy group andthe substituent of the catalyst according to our proposal(Scheme 1c).
Given these results, it was considered to be instructive toevaluate the scope of this concept and the Mannich reactionwas elected for that purpose. Not only does this transforma-tion establish one carbon–carbon bond and up to two ste-reogenic centers in a single synthetic operation, it also playsa pivotal strategic role in the synthesis of numerous nitro-gen-containing compounds.[17] As a result, both the activa-tion of the reaction and control of its stereochemical out-come have been and continue to be the subject of intensiveresearch activity.[18] However, despite the recent progressmade in the area, methods based on direct approaches lead-ing to anti-a,b-disubstituted b-amino carbonyl compoundsare still demanding. To date the most common practice toaccess anti-b-amino a-substituted carbonyl compounds is thealkylation of their corresponding a-unsubstituted deriva-tives, which can be readily obtained by several catalyticenantioselective procedures.[19]
Early formulations and challenges : At the outset of our in-vestigation, however, it was not clear which kind of imineswould be best suited for the Mannich reaction. Whilstimines derived from aromatic aldehydes are effective in the
Scheme 1. Prototypical organocatalysts for enamine and iminium ionbased reactions and their mode of action for enamine reactions. a) Hy-drogen-bond approach. b) Steric-control approach. c) Proposed model.
Scheme 2. Catalytic conjugate addition of aldehydes to nitroalkenes andrepresentative examples.
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 5333 – 53425334

reactions promoted by proline and its congeners to give synadducts with high enantioselectivity, they are rarely em-ployed in the reported anti-selective Mannich reactions.[18, 20]
The first example of an anti-selective Mannich reaction byenamine catalysis was described in 2002 by Barbas III[21] byusing (S)-2-methoxymethylpyrrolidine (13, Scheme 3) and
glyoxylate imines. The identification by Jørgensen[22a] of thediaryl prolinol silyl ether 4 a led to significant improvementsin terms of yields and enantioselectivity. Maruo-ka[23]reported the first bifunctional catalyst for the same re-action, the axially chiral amino sulfonamide 14. Although itspreparation involves tedious multistep synthesis, the catalysthas the ability of activating concurrently the aldehyde viaenamine formation and the Mannich acceptor through hy-drogen bonding. Later, Barbas III and Tanaka showed thatthe installation of a carboxyl group at the b-position of thepyrrolidine nitrogen (catalyst 15[24] and 16[25]) suffices forhigh enantio- and anti-diastereocontrol of this reaction. Pyr-rolidines 17[26] and 18[27] that work through the same modelhave also been reported by Blanchet and Maruoka, inde-pendently. More recently, the thiourea-based catalyst 19 thatcombines structural features of catalysts 13 and 18 has been
reported by Peng for the same reaction.[28] Accordingly, themain limitation of all these contributions is that only a-imino esters appear to be sufficiently reactive to give Man-nich adducts with both chemical and stereochemical effi-ciency.[29] One solution has been recently presented by Mar-uoka who has revealed that N-Boc (tert-butoxycarbonyl)imines from arylaldehydes may be utilized in the Mannichreaction by using chiral sulfonamide 14.[30] On the otherhand, catalyst 4 b, which is more readily available, seems tobe less active since long reaction times (4–7 days) areneeded for the reaction with this kind of imines to occurwith good yields. In this instance, glyoxylate-derived iminesare, once again, the best substrates to give the Mannich ad-ducts with high anti-selectivity and enantioselectivity.[31]
In most of the above catalytic systems, a transition-statemodel A (Scheme 4) is invoked in which a hydrogen-bond
interaction with the imine nitrogen is postulated to be thekey organizational element in a two-component arrange-ment in analogy with our proposal for conjugate additions.Apparently, glyoxylate imines have the appropriate bondlengths and angles to fit properly in this model.[32] We rea-soned that in the presence of an external Brønsted acid amore fluxional model could be provided in which otherimines could be accommodated in a three-component ar-rangement, such as model B (Scheme 4).
To evaluate this assumption, N-sulfonyl imines[33] werechosen because of the poor coordinating ability of theoxygen atoms toward hydrogen bonding that would ensurean effective imine activation.
Catalyst screening and conditions : The validity of the abovehypothesis soon became clear. Whilst hydroxypyrrolidine 20alone (Table 1, entry 1) was quite inefficient in promotingthe reaction of butyraldehyde 22 (R=Et) with N-tosylimine23 a at �20 8C, in combination with a 20 mol% of p-nitro-benzoic acid (PNBA), higher yield and diastereo- and enan-tioselectivity were attained (Table 1, entry 2). Decreasingthe temperature to �608C (Table 1, entry 3) provided ahigher ee, although with similar diastereoselectivity and alower yield. By using catalyst 8, a further improvement wasachieved albeit still insufficient providing the amino alcohol29 a, after in situ reduction of the intermediate Mannich
Scheme 3. Representative chiral pyrrolidines for anti-Mannich reactionswith glyoxylate derived imines. a) Catalysts working through steric con-trol. b) Catalysts working through hydrogen-bond control.
Scheme 4. Features of the proposed models for anti-Mannich reactions.
Chem. Eur. J. 2010, 16, 5333 – 5342 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 5335
FULL PAPERA 4-Hydroxy Pyrrolidine-Catalyzed Mannich Reaction of Aldehydes

adduct, with a good diastereomeric ratio and moderate ee(Table 1, entry 4).
Subsequent efforts to increase stereoselectivity by increas-ing the steric bulk of the a-substituent of the pyrrolidinering resulted in the development of 4-hydroxy derivatives 5,6, and 7 (Scheme 5).[34] Concordant with our expectations,the aminoalcohol product 29 a obtained from the reaction ofaldehyde 22 (R=Et) and imine 23 a promoted by catalyst 5in the presence of PNBA was indeed formed after reductionof the intermediate aldehyde with an excellent high diaste-
reomeric ratio (93:7) and92 % ee (Table 1, entry 5). Cata-lyst 6 enabled further stereo-chemical improvement, and cat-alyst 7 proved to be the bestgiving the anti-Mannich adduct29 a in 77 % yield and with a re-markably high diastereomericratio (98:2) and 95 % ee (en-tries 6, 7). The less acidic ben-zoic acid is also effective underthe same conditions but alonger reaction time is neededfor reaction completion, whilstp-methoxybenzoic acid leads tolower yield after 20 h. Addi-tionally, decreasing or increas-ing the amount of PNBA to 10or 40 %, respectively, whilemaintaining the catalyst loadingat 20 % affords similar enantio-and diastereoselectivity, but sig-nificantly lower yields for thesame reaction time (50 and58 % yield, respectively). Asthe results in Table 1 show, inthe absence of PNBA (entry 8),
the reaction only proceeded at �20 8C, and low diastereo-and enantioselectivity were obtained. No variations wereobserved with the aid of PNBA in this instance (Table 1,entry 9), thus suggesting that a low reaction temperature isalso needed for achieving good results and that the acid co-catalyst is necessary not only for reactivity,[35] but also forstereocontrol. To further prove these observations catalysts4 a and 4 b, which work well when glyoxylate imines are em-ployed,[11] were evaluated. Whilst catalyst 4 a in combinationwith PNBA was totally ineffective under the conditions ex-amined, catalyst 4 b afforded a 15 % yield and poor diaster-eo- and enantioselectivity (Table 1, entries 10, 11).
To further demonstrate the influence of the hydroxygroup in catalyst 7, the reaction of butyraldehyde with imine23 a was conducted under the same conditions in the pres-ence of the analogous catalyst 21 lacking the hydroxyl group(Table 1, entries 12 and 13). These data show that all threeelements, the external Brønsted acid, the a-substituent inthe pyrrolidine, and the 4-hydroxy group of the ring are es-sential for high chemical and stereochemical efficiency.
Scope of the Mannich reaction : The generality of the enan-tioselective Mannich reaction catalyzed by 7 was then ex-plored with different aldehydes and N-p-toluensulfonylimines under the optimized conditions (Table 2). The resultsshow that the reaction is quite general with respect the alde-hyde donor and imine acceptor. N-Tosyl imines derivedfrom aromatic aldehydes with electron-poor and -rich sub-stituents can be employed leading to the anti adducts in upto 78 % yield, 97:3 diastereomeric ratio, and 99 % ee. Initial-
Table 1. Catalyst screening for the Mannich reaction of butyraldehyde 22 (R =Et) with imine 23a in DMF asthe solvent to give 32a.[a]
Entry Cat. T[8C]
Conv. [%][b] Yield[%][c]
d.r.[b] ee[%][d]
1 20 �20 nd[e] 37 55:45 47[f,g]
2 20 �20 nd [e] 70 75:25 67[g]
3 20 �60 >99 29 77:23 83[g]
4 8 �60 >99 88 96:4 75
5 5 (R2 =Me) �60 >99 75 93:7 926 6 (R2 =nPr) �60 50 43 90:10 937 7 (R2 =nHex) �60 >99 77 98:2 958 7 (R2 =nHex) �20 >99 99 80:20 76[f]
9 7 (R2 =nHex) �20 >99 92 76:24 86
10 4 a (R2 =3,5-(F3C)2C6H3) �60 0 0 – –[h]
11 4 b (R2 =Ph) �60 40 15 75:25 6312 21 (R2 =nHex) �40 >99 98 78:22 6713 21 (R2 =nHex) �60 >99 24 78:22 58
[a] Reactions performed at 0.5 mmol scale in DMF (2 mL) and by using catalysts 4a, 4 b, 5, 6, 7, and 21(20 mol %), PNBA (20 mol %), and imine 23a (3 equiv). Data related to 32 a after in situ reduction of 26 a. Re-action time 20 h. [b] Determined by 1H NMR spectroscopy on the crude material. [c] Isolated yield of 32 aafter flash column chromatography. [d] Determined by HPLC analysis; see the Supporting Information for de-tails. [e] nd: not determined. [f] No Brønsted acid was used. [g] Reactions conducted by using 3 equiv of alde-hyde. [h] Reaction time 3 days.
Scheme 5. Mannich reaction between aldehydes 22 and N-sulfonyl-imines23–25 catalyzed by 5, 6, 7, 20, and 21.
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 5333 – 53425336
C. Palomo et al.

ly, reactions were performed in a 3:1 molar ratio of imine/al-dehyde. However changing the stoichiometry to 3:1 alde-hyde/imine led to significantly higher yields without appar-ent changes in the stereoselectivity.
Given the utility of the nitrobenzenesulfonyl group asboth activating and protecting group of the amine func-tion,[36] we next extended the method to nosyl imines. Asthe results in Table 3 show, the Mannich reaction also pro-ceeded efficiently with o-nosyl- and p-nosyl-protectedimines. In these cases, both benzoic acid and p-nitrobenzoicacid can be used, with the latter slightly superior in terms ofyield.
The assigned absolute configuration of the major productswas established by a single-crystal X-ray analysis of 29 a andby assuming an uniform reaction mechanism.[37, 38] In addi-tion, the b-amino aldehyde 26 c upon oxidation under stan-dard conditions provided the N-tosyl-protected derivative37, which then was converted into the known b-amino acid38 (Scheme 6).[39]
Complementing this protocol, the N-nosyl derivatives canalso be easily deprotected[40] under the conditions shown inScheme 7 to afford the free aminoalcohols. For example,treatment of 31 c with tiophenol and potassium carbonate inacetonitrile and some drops of DMSO afforded amino alco-hol 39 in 80 % yield.
Mechanistic insights : The above results suggest that the ini-tially invoked three-component arrangement for the Man-nich reaction to proceed with anti selectivity is indeed oper-ating. To get a better understanding of this activation mech-anism and provide a rationale for the stereochemical out-come of the reaction, some quantum calculations were car-ried out.
On the basis of enamine formation,[41] we assumed the re-action to proceed through an enamine mechanism[38] rather
Table 2. Catalytic asymmetric Mannich reactions of aldehydes 22 with N-tosyl imines 23 promoted by 7.[a]
Entry Product T[8C]
Yield[%][b]
d.r.[c] ee[%][d]
1 29 a �60 68 (78) 96:4 97
2 29 c �60 40 (77) 96:4 >99
3 32 a �60 77 98:2 95
4 32 b �60 63 98:2 86
5 32 c �60 67 (68) 94:6 91
6 32 e �60 60 93:7 91
7 34 d �40 – (76) 87:13 91
8 35 a �60 – (76) 97:3 >99
[a] Reactions performed at 0.5 mmol scale in DMF (2 mL) and by usingcatalyst 7 (20 mol %), p-nitrobenzoic acid (20 mol %), aldehydes 22, andimines 23 (3 equiv). [b] Isolated yield of aminoalcohols 29, 32, and 34after flash column chromatography. Yields in brackets were obtained byusing a ratio of aldehyde/imine of 3:1. [c] Determined by 1H NMR spec-troscopy on the crude material. [d] Determined by HPLC analysis afterflash chromatography; see the Supporting Information for details.
Table 3. Catalytic asymmetric Mannich reactions of aldehydes 22 with N-nosyl imines 24 and 25 promoted by 7.[a]
Entry Product T [8C] Yield[%][b]
d.r.[c] ee[%][d]
1 30a �60 – (61) 93:7 93
2 31c �60 – (59)[e] 91:9 97
3 33a �60 – (71) 93:7 91
4 36b �60 – (51)[e] 99:1 >99
[a] Reactions performed at 0.5 mmol scale in DMF (2 mL) and by usingcatalyst 7 (20 mol %), p-nitrobenzoic acid (20 mol %), aldehydes 22(3 equiv), and imines 24 and 25. [b] Isolated yield of aminoalcohols afterflash column chromatography. Yields in brackets were obtained by usinga ratio of aldehyde/imine of 3:1. [c] Determined by 1H NMR spectrosco-py on the crude material. [d] Determined by HPLC analysis after flashchromatography; see the Supporting Information for details. [e] Benzoicacid (20 mol %) was used as cocatalyst.
Scheme 6. Oxidation and deprotection of Mannich adducts.
Scheme 7. Deprotection of the N-nosyl adducts.
Chem. Eur. J. 2010, 16, 5333 – 5342 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 5337
FULL PAPERA 4-Hydroxy Pyrrolidine-Catalyzed Mannich Reaction of Aldehydes

than an enol intermediate.[42] Accordingly, computationalDFT[43] data based on the model shown in Scheme 8 indicat-ed that the activation of the iminic N by the pyrrolidine OH
group is of similar magnitude for anti or syn transitionstates, anticipating an almost equal ratio of diastereomers(TS1-anti vs TS1-syn, Scheme 8). Better activation of theiminic nitrogen was obtained by replacing the intramolecu-lar hydrogen bonding by an intermolecular hydrogen-bondinteraction with the aid of an external Brønsted acid (TS2,Scheme 8). However, the anti stereoselectivity is still pre-dicted to be low. In fact, TS2-syn and TS2’-syn staggeredconformations equally contribute to the formation of thesyn isomer in this case. Remarkably, however, the combina-tion of both tools, the catalyst hydroxy group and the exter-nal Brønsted acid, led to a model that correctly predicts theresults obtained, differentiating TS3-anti and TS3-syn by3 kcal mol�1 (Scheme 8).
Conclusion
In summary, we have reported a highly efficient catalyticsystem for the anti-selective Mannich reaction of aldehydeswith N-sulfonyl imines. This catalyst system combines anamino group to activate the aldehyde donor substrate, and ahydroxy group together with an external Brønsted acid toactivate the imine acceptor component, while controllingthe stereochemistry of the process. The Mannich adductscan be easily reduced or oxidized and then deprotected togive the corresponding b-amino acids and b-amino alcohols.Specifically, the synthetic utility of the present anti-selective
Mannich reaction has been significantly expanded by theutilization of imines other than those derived from glyoxy-late. The results also show that this ternary system may beof utility in the development of other enamine-based reac-tions.
Experimental Section
General methods : All reactions were carried out under a nitrogen atmos-phere in flame-dried glassware with efficient magnetic stirring. Methyl-ene chloride (CH2Cl2) was distilled from CaH2, and toluene was dried inthe presence of sodium metal. Ethyl acetate and DMF were used as re-agent grade. Purification of reaction products was carried out by flashcolumn chromatography by using silica gel 60 (0.040–0.063 mm, 230–400mesh). Analytical TLC was performed on 0.25 mm silica gel 60-F plates.Visualization was accomplished with UV light and a solution obtained byadmixing in 470 mL of water ammonium molybdate (21 g), cerium sul-phate (1 g), and concentrated sulphuric acid (31 mL), followed by heat-ing. Melting points were measured with a Buchi SMP-20 melting pointapparatus and are uncorrected. 1H and 13C NMR spectra were recordedon Bruker Advance-300 and are reported in ppm from internal tetrame-thylsilane (TMS). Analytical HPLC was performed on waters-600E con-troller chromatographs, equipped with 2996 and 2998 photodiode arrayUV detector, by using Daicel Chiralpak AD-H, IC and IA columns. Op-tical rotations were recorded on a Jasco P-2000 polarimeter. MS spectrawere recorded on an ESI-ion trap mass spectrometer (Agilent 1100 seriesLC/MSD, SL model). Diarylprolinol trimethylsilyl ether catalysts 4a and4b were purchased from Aldrich and used without further purification.
General procedure for the Mannich reaction : The aldehyde (1.5 mmol,3 equiv) was added to a solution of the imine (0.5 mmol, 1 equiv), theacid (0.1 mmol, 20 mol %), and the catalyst (0.1 mmol, 20 mol %) inDMF (2 mL) at �60 8C. The resulting solution was stirred at �60 8C for24 h. EtOH (1 mL) and NaBH4 (4.5 mmol, 8 equiv) were successivelyadded at the same temperature, and after stirring for 30 min at �40 8C,the reaction was quenched with brine (2 mL) and allowed to reach roomtemperature. After extraction with Et2O (3 � 4 mL), the combined organ-ic phases were washed with brine and dried over MgSO4, concentratedunder reduced pressure, and purified over silica gel by flash column chro-matography to afford the expected adducts. The diastereoselectivity ofthe process was determined by 1H NMR spectroscopic analysis on thecrude material.
N-[(1S,2R)-3-Hydroxy-2-methyl-1-p-tolylpropyl]-4-methylbenzenesulfo-namide (29 a): Prepared according to the general procedure by startingfrom propanal (0.11 mL, 1.5 mmol) and N-tosyl-tolylimine (23a) (137 mg,0.5 mmol). The crude material was purified by flash column chromatog-raphy on silica gel (eluting with hexane/ethyl acetate 80:20) and then dis-agregated with Et2O to give the title compound as a white solid. Yield:78% (130 mg); m.p. 104–06 8C; [a]20
D =�68.2 (c=1 in CH2Cl2); 1H NMR(300 MHz, CDCl3): d=7.53 (d, J =8.3 Hz, 2H), 7.13 (d, J =8.0 Hz, 2H),6.97 (d, J =7.9 Hz, 2 H), 6.84 (d, J=8.1 Hz, 2H), 5.65 (d, J=7.5 Hz, 1H),4.17 (t, J =8.0 Hz, 1 H), 3.97 (dd, J=11.0, 11.0 Hz, 1H), 3.58 (dd, J =4.5,11.2 Hz, 1H), 2.39 (s, 3H), 2.30 (s, 3H), 2.02–1.89 (m, 1 H), 0.81 ppm (d,J =7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): d =142.8, 137.5, 137.0,136.8, 129.27, 128.9, 127.1, 126.9, 65.0, 61.4, 40.9, 21.4, 21.0, 14.5 ppm; theenantiomeric purity of the major diastereomer was determined by HPLCanalysis (Daicel Chiralpak AD-H, hexane/2-propanol 97:3): flow rate=
1 mL min�1; retention times: 126.7 (minor), 137.5 min (major).
N-[(1S,2R)-3-Hydroxy-2-methyl-1-phenylpropyl]-4-methylbenzenesulfo-namide (29 c): Prepared according to the general procedure by startingfrom propanal (0.11 mL, 1.5 mmol) and N-tosyl-phenylimine (23c)(129 mg, 0.5 mmol). The crude material was purified by flash columnchromatography on silica gel (eluting with hexane/ethyl acetate 80:20)and then disgregated with Et2O to give the title compound as a whitesolid. Yield: 77% (123 mg); m.p. 127–130 8C.; [a]20
D =�82.2 (c =1 inCH2Cl2); 1H NMR (300 MHz, CDCl3): d= 7.52 (d, J =8.3 Hz, 2H), 7.18–
Scheme 8. Representation of the computed transition structures opti-mized at B3LYP/6-31-G*. Single-point values [Kcal mol�1] at the B3LYP/6-311++ G** level are shown in italics.
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 5333 – 53425338
C. Palomo et al.

7.08 (m, 5 H), 7.00–6.96 (m, 2H), 5.81 (s, 1H), 4.22 (m, 1 H), 3.96 (dd, J=
3.1, 11.2 Hz, 1 H), 3.59 (dd, J=4.6, 11,2 Hz, 1H), 2.38 (s, 3H), 1.98 (m,1H), 0.82 ppm (d, J =7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): d=
142.9, 139.9, 137.3, 129.6, 129.2, 128.2, 127.1, 126.4, 64.9, 61.5, 40.9, 21.4,14.5 ppm; the enantiomeric purity of the major diastereoisomer was de-termined by HPLC analysis (Daicel Chiralpak IA, hexane/2-propanol95:5): flow rate= 0.8 mL min�1; retention times: 36.7 (minor), 43.7 min(major).
N-[(1S,2R)-2-(Hydroxymethyl)-1-p-tolylbutyl]-4-methylbenzenesulfona-mide (32 a): Prepared according to the general procedure by startingfrom butanal (0.14 mL, 1.5 mmol) and N-tosyl-tolylimine (23 a) (137 mg,0.5 mmol). The crude material was purified by flash column chromatog-raphy on silica gel (eluting with hexane/ethyl acetate 80:20) to give thetitle compound as a yellow oil. Yield: 77 % (134 mg); [a]20
D =�19.8 (c=
0.5 in CH2Cl2); 1H NMR (300 MHz, CDCl3): d =7.54 (d, J=8.3 Hz, 2H),7.13 (d, J =8.2 Hz, 2 H), 6.97 (d, J=7.9 Hz, 2H), 6.87 (d, J=8.1 Hz, 2H),5.81 (d, J= 7.9 Hz, 1H), 4.35 (t, J =7.8, Hz, 1 H), 3.93 (d, J =11.1 Hz,1H), 3.72–3.63 (m, 1H), 2.39 (s, 3H), 2.30 (s, 3H), 1.66–1.53 (m, 1H),1.44–1.18 (m, 2H), 0.88 ppm (t, J =7.4, 7.4 Hz, 3 H); 13C NMR (125 MHz,CDCl3): d=142.8, 137.7, 137.4, 136.8, 129.2, 128.9, 127.1, 126.6, 61.2, 60.0,47.6, 29.7, 21.4, 21.0, 11.6 ppm; the enantiomeric purity of the major dia-stereomer was determined by HPLC analysis (Daicel Chiralpak AD-H,hexane/2-propanol 95:5): flow rate=0.8 mL min�1; retention times: 51.5(minor), 61.8 min (major).
N-[(1S,2R)-2-(Hydroxymethyl)-1-(4-methoxyphenyl)butyl]-4-methylben-zene sulfonamide (32 b): Prepared according to the general procedure bystarting from butanal (0.14 mL, 1.5 mmol) and N-tosyl-4-methoxyphenyli-mine (23 b) (145 mg, 0.5 mmol). The crude material was purified by flashcolumn chromatography on silica gel (eluting with hexane/ethyl acetate80:20) and then disgregated with Et2O to give the title compound as awhite solid. Yield: 63 % (114 mg); m.p. 117–121 8C; [a]20
D =�54.8 (c =1 inCH2Cl2); 1H NMR (300 MHz, CDCl3): d=7.53 (d, J =8.3 Hz, 2 H), 7.13(d, J =8.0 Hz, 2 H), 6.91 (d, J =8.6 Hz, 2 H), 6.68 (d, J =8.7 Hz, 2 H), 5.97(d, J =7.8 Hz, 1H), 4.33 (t, J =7.7, 7.7 Hz, 1 H), 3.94 (d, J =11.19 Hz,1H), 3.77 (s, 3 H), 3.68 (d, J =8.0 Hz, 1 H), 2.38 (s, 3 H), 2.29 (s, 1 H),1.67–1.55 (m, 1 H), 1.44–1.16 (m, 2 H), 0.87 ppm (t, J =7.4, 7.4 Hz, 3H);13C NMR (125 MHz, CDCl3): d =158.7, 142.8, 137.7, 132.5, 129.2, 127.9,127.1, 113.6, 61.3, 59.8, 55.2, 47.6, 21.4, 21.0, 11.6 ppm; the enantiomericpurity of the major diastereomer was determined by HPLC analysis(Daicel Chiralpak IA, hexane/2-propanol 95:5): flow rate=
0.75 mL min�1; retention times: 79.6 (minor), 92.3 min (major).
N-[(1S,2R)-2-(Hydroxymethyl)-1-phenylbutyl]-4-methylbenzenesulfona-mide (32 c): Prepared according to the general procedure by startingfrom butanal (0.14 mL, 1.5 mmol) and N-tosyl-phenylimine (23c)(129 mg, 0.5 mmol). The crude material was purified by flash columnchromatography on silica gel (eluting with hexane/ethyl acetate 80:20)and then disgregated with Et2O to give the compound as a white solid.Yield: 68% (122 mg); m.p. 123–125 8C; [a]20
D =�72.7 (c =1 in CH2Cl2);1H NMR (300 MHz, CDCl3): d =7.53 (d, J =8.3 Hz, 2H), 7.17–7.08 (m,5H), 7.02–6.99 (m, 2H), 5.95 (d, J=8.07 Hz, 1H), 4.42 (t, J= 7.7, 7.7 Hz,1H), 3.91 (m, 1 H), 3.38 (m, 1 H), 2.38 (s, 3 H), 1.69–1.59 (m, 1 H), 1.49–1.21 (m, 2H), 0.89 ppm (t, J=7.4, 7.4 Hz, 3 H); 13C NMR (125 MHz,CDCl3): d =142.8, 140.4, 137.7, 129.2, 128.3, 127.0, 126.7, 61.3, 60.2, 47.5,21.4, 21.1, 11.6 ppm; the enantiomeric purity of the major diastereomerwas determined by HPLC analysis (Daicel Chiralpak IA, hexane/2-prop-anol 95:5): flow rate=0.75 mL min�1; retention times: 51.3 (minor),57.7 min (major).
N-[(1S,2R)-2-(Hydroxymethyl)-1-m-tolylbutyl]-4-methylbenzenesulfona-mide (32 e): Prepared according to the general procedure by startingfrom butanal (0.14 mL, 1.5 mmol) and N-tosyl-3-methylphenylimine(23e) (137 mg, 0.5 mmol). The crude material was purified by flashcolumn chromatography on silica gel (eluting with hexane/ethyl acetate80:20) to give the title compound as a yellow oil. Yield: 60% (104 mg);[a]20
D =�39.8 (c=1 in CH2Cl2); 1H NMR (300 MHz, CDCl3): d=7.51 (d,J =8.3 Hz, 2 H), 7.13–7.01 (m, 3H), 6.94 (d, J=7.5 Hz, 1 H), 6.83 (d, J=
7.6 Hz, 1 H), 6.68 (s, 1H), 6.10 (d, J=8.1 Hz, 1 H), 4.33 (t, J =7.9, 7.9 Hz,1H), 3.93 (d, J =11.1 Hz, 1H), 3.68 (d, J =10.7 Hz, 1H), 2.51 (br s, 1 H),2.37 ACHTUNGTRENNUNG(s, 3 H), 2.17 ACHTUNGTRENNUNG(s, 3 H), 1.70–1.57 (m, 1H), 1.47–1.20 (m, 2H), 0.88 ppm
(t, J =7.4, 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3): d= 142.7, 140.1,137.7, 129.1, 128.2, 127.7, 127.1, 123.8, 61.2, 60.2, 47.4, 21.4, 21.2, 21.0,11.6 ppm; the enantiomeric purity of the major diastereomer was deter-mined by HPLC analysis (Daicel Chiralpak IA, hexane/2-propanol 98:2):flow rate =0.7 mL min�1; retention times: 91.1 (minor), and 98.8 min(major).
N-[(1S,2R)-1-(4-Chlorophenyl)-2-(hydroxymethyl)heptyl]-4-methylben-zene sulfonamide (34 d): Prepared according to the general procedure bystarting from heptanal (0.21 mL, 1.5 mmol) and N-tosyl-4-chlorophenyli-mine (23 d) (147 mg, 0.5 mmol). The crude material was purified by flashcolumn chromatography on silica gel (eluting with hexane/ethyl acetate80:20) and then disgregated with Et2O to give the compound as a whitesolid. Yield: 76 % (161 mg); m.p. 120–124 8C; [a]20
D =�58.9 (c=1 inCH2Cl2); 1H NMR (300 MHz, CDCl3): d=7.54 (d, J =8.3 Hz, 2 H), 7.13(m, 4H), 7.01 (d, J=8.5 Hz, 2H), 6.39 (d, J =7.4 Hz, 1 H), 4.40 (t, J=
7.0 Hz, 1H), 3.81 (d, J=11.2 Hz, 1H), 3.61 (m, 1H), 2.40 (s, 3H), 2.31(m, 1 H), 1.65 (m, 1H) 1.40–1.05 (m, 8H), 0.86 ppm (t, J= 7.0 Hz, 3H);13C NMR (125 MHz, CDCl3): d =143.1, 139.2, 137.7, 132.8, 129.3, 128.3,127.0, 61.9, 60.1, 45.6, 31.8, 28.2, 26.7, 22.4, 21.4, 14.0 ppm; the enantio-meric purity of the major diastereomer was determined by HPLC analy-sis (Daicel Chiralpak AD-H, hexane/2-propanol 95:5): flow rate=
0.8 mL min�1; retention times: 31.7 (minor), 49.1 min (major).
N-[(1S,2R)-2-(Hydroxymethyl)-3-methyl-1-p-tolylbutyl]-4-methylben-zene sulfonamide (35 a): Prepared according to the general procedure bystarting from isovaleraldehyde (0.16 mL, 1.5 mmol) and N-tosyl-tolyli-mine (23a) (137 mg, 0.5 mmol). The crude material was purified by flashcolumn chromatography on silica gel (eluting with hexane/ethyl acetate80:20) and then disgregated with Et2O to give the title compound as awhite solid. Yield: 76 % (134 mg); m.p. 145–148 8C; [a]20
D =�70.8 (c =1 inCH2Cl2); 1H NMR (300 MHz, CDCl3): d=7.50 (d, J =8.2 Hz, 2 H), 7.08(d, J =8.1 Hz, 2 H), 6.97–6.90 (m, 4H), 6.70–6.55 (br s, 1 H), 4.60 (d, J=
6.8 Hz, 1H), 3.82–3.68 (m, 2 H), 2.36 (s, 3 H), 2.28 (s, 3H), 1.80–1.65 (m,1H), 1.46–1.36 (m, 1 H), 1.34–1.24 (br s, 1H), 0.92 ppm (dd, J =6.7,20.5 Hz, 6 H); 13C NMR (125 MHz, CDCl3): d =142.6, 138.0, 137.7, 136.4,129.0, 128.7, 127.0, 126.9, 60.3, 59.6, 53.5, 51.9, 25.7, 21.3, 20.9, 18.8 ppm;the enantiomeric purity of the major diastereomer was determined byHPLC analysis (Daicel Chiralpak IA, hexane/2-propanol 95:5): flowrate=0.5 mL min�1; retention times: 50.1 (minor), 54.4 min (major).
N-[(1S,2R)-3-Hydroxy-2-methyl-1-p-tolylpropyl]-2-nitrobenzenesulfona-mide (30 a): Prepared according to the general procedure by startingfrom propanal (0.12 mL, 1.5 mmol) and N-o-nosyl-p-methyl-phenylimine(24a) (152 mg, 0.5 mmol). The crude material was purified by flashcolumn chromatography on silica gel (eluting with hexane/ethyl acetate80:20) to give the title compound as a yellowish oil and as a diastereo-meric mixture syn/anti in a ratio of 7:93. Yield: 61 % (111 mg); spectro-scopic data of the major anti diastereomer: 1H NMR (300 MHz, CDCl3):d=7.76–7.67 (m, 2 H), 7.62–7.53 (m, 1H), 7.48–7.40 (m, 1 H) 6.98–6.83(m, 4H), 6.62 (d, J=8.7 Hz, 1 H), 4.49 (t, J =8.3 Hz, 1 H), 4.00—3.88 (m,1H), 3.74–3.62 (m, 1 H), 2.22 (s, 3H), 2.08–1.90 (m, 2H), 0.95 ppm (d, J=
7.0, 3 H); 13C NMR (125 MHz, CDCl3): d=137.2, 136.2, 134.7, 132.5,132.2, 130.8, 130.6, 128.9, 126.8, 124.8, 64.5, 62.2, 40.7, 20.9, 14.6 ppm; theenantiomeric purity of the major diastereomer was determined by HPLCanalysis (Daicel Chiralpak IC, hexane/2-propanol 70:30): flow rate=
0.5 mL min�1; retention times: 108.5 (minor), 116.5 min (major).
N-[(1S,2R)-3-Hydroxy-2-methyl-1-phenylpropyl]-4-nitrobenzenesulfona-mide (31 c): Prepared according to the general procedure by startingfrom propanal (0.12 mL, 1.5 mmol) and N-p-nosyl-phenylimine (25c)(145 mg, 0.5 mmol). The crude material was purified by flash columnchromatography on silica gel (eluting with hexane/ethyl acetate 80:20) togive the title compound as a white solid and as a diastereomeric mixturesyn/anti in a ratio of 9:91. Yield: 59% (90 mg); spectroscopic data of themajor anti diastereomer: 1H NMR (300 MHz, CDCl3): d=8.12–8.05 (m,2H), 7.75–7.69 (m, 2 H), 7.19–7.10 (m, 3H), 7.04–6.98 (m, 2 H), 4.43 (d,J =7.7 Hz, 1 H), 3.93–3.83 (m, 1H), 3.70–3.62 (m, 1H), 2.05–1.95 (m,1H), 0.91 ppm (d, J =7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): d=
149.5, 146.7, 139.3, 128.4, 128.1, 127.7, 127.1, 123.6, 65.4, 63.0, 40.3,14.7 ppm; the enantiomeric purity of the major diastereomer was deter-mined by HPLC analysis (Daicel Chiralpak IC, hexane/2-propanol
Chem. Eur. J. 2010, 16, 5333 – 5342 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 5339
FULL PAPERA 4-Hydroxy Pyrrolidine-Catalyzed Mannich Reaction of Aldehydes

80:20): flow rate =0.5 mL min�1; retention times: 34.4 (minor), 42.8 min(major).
N-[(1S,2R)-2-(Hydroxymethyl)-1-p-tolylbutyl]-2-nitrobenzenesulfona-mide (33 a): Prepared according to the general procedure by startingfrom butanal (0.2 mL, 1.5 mmol) and N-o-nosyl-p-methyl-phenylimine(24a) (152 mg, 0.5 mmol). The crude material was purified by flashcolumn chromatography on silica gel (eluting with hexane/ethyl acetate80:20) to give the title compound as a yellowish oil and as a diastereo-meric mixture syn/anti in a ratio of 7:93. Yield: 71 % (134 mg); spectro-scopic data of the major anti diastereomer: 1H NMR (300 MHz, CDCl3):d=7.71 (dd, J =7.9, 25.2 Hz, 2H), 7.55 (t, J =7.7 Hz, 1 H), 7.40 (t, J=
7.7 Hz, 1H), 6.99 (d, J= 7.9 Hz, 2 H), 6.95 (d, J =13.5 Hz, 1H), 6.87 (d,J =7.8 Hz, 2 H), 4.72–4.64 (m, 1H), 3.98–3.82 (m, 1H), 3.80–3.69 (m,1H), 1.86–1.79 (m, 1 H), 1.72–1.61 ACHTUNGTRENNUNG(m, 1 H), 1.56 (s, 3H), 1.54–1.43 (m,2H), 0.97 ppm (t, J =7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3): d=
137.4, 137.0, 135.4, 132.8, 132.5, 131.1, 129.2, 127.2, 125.1, 61.7, 61.3, 47.6,21.7, 21.3, 12.0 ppm; the enantiomeric purity of the major diastereomerwas determined by HPLC analysis (Daicel Chiralpak IC, hexane/2-propa-nol 80:20): flow rate=0.5 mL min�1; retention times: 112.2 (minor),123.3 min (major).
N-[(1S,2R)-2-(Hydroxymethyl)-1-(4-methoxyphenyl)-3-methylbutyl]-4-nitrobenzene sulfonamide (36 b): Prepared according to the general pro-cedure by starting from isovaleraldehyde (0.16 mL, 1.5 mmol) and N-p-nosyl-4-methoxy-phenylimine (25 b) (160 mg, 0.5 mmol). The crude mate-rial was purified by flash column chromatography on silica gel (elutingwith hexane/ethyl acetate 80:20) to give the title compound as a whitesolid and as a diastereomeric mixture syn/anti in a ratio of 1:99. Yield:52% (106 mg); spectroscopic data of the major anti diastereomer: 1HNMR (300 MHz, CDCl3): d =8.12–8.02 (m, 2H), 7.74–7.64 (m, 2H),6.99–6.87 (m, 2H), 6.68–6.56 (m, 2H), 4.81–4.74 (m, 1 H), 3.87–3.78 (m,2H), 3.73 (s, 3H), 1.90–177 (m, 1 H), 1.47–1.35 (m, 1H), 1.06 (d, J =
7.8 Hz, 3H), 0.94 ppm (d, J =6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3):d=158.9, 149.2, 147.1, 131.7, 128.1, 123.5, 113.6, 60.7, 60.0, 55.3, 51.6,25.6, 21.2, 18.8 ppm; the enantiomeric purity of the major diastereomerwas determined by HPLC analysis (Daicel Chiralpak IC, hexane/2-propa-nol 90:10): flow rate=0.8 mL min�1; retention times: 57.8 (minor),65.6 min (major).
Deprotection and elaboration of the Mannich adducts
Preparation of 38 : Propanal (1.5 mmol, 3 equiv) was added to a solutionof N-tosyl phenylimine (23c) (0.5 mmol, 1 equiv), p-nitrobenzoic acid(0.1 mmol, 20 mol %), and catalyst 7 (0.1 mmol, 20 mol %) in DMF(2 mL) at �60 8C. The resulting solution was stirred at �60 8C for 24 h.Then MeOH (2.5 mL), KH2PO4 (570 mg, 4.15 mmol), and NaClO2
(338 mg, 3.15 mmol) were added. The reaction was allowed to reach 0 8Cand after the injection of H2O2 (35 % solution, 4.5 mL), the mixture waswarmed up to RT and stirred for 2 h. The pH was adjusted to 3 by the ad-dition of 1m HCl and saturated Na2SO3 solution (10 mL) was added. Theresulting mixture was extracted with Et2O (3 � 10 mL), the combined or-ganic layers were washed with sat. NaCl solution (4 � 15 mL) and driedover MgSO4. The organic layer was concentrated in vacuo to give thetitle crude compound, which was then purified by flash column chroma-tography on silica gel (eluent: CH2Cl2/MeOH 98:2). This yielded the pro-tected derivative 37 as a white solid. Yield: 62% (100 mg); spectroscopicand physical properties were in agreement with those previously repor-ted.[39c] Deprotection of the tosyl group was carried out by treatment of36 with HCl at 100 8C; this yielded compound 38 in 95% yield.[39]
Preparation of 39 : K2CO3 (277 mg, 2 mmol), PhSH (0.26 mL, 2.5 mmol),and DMSO (2 mol %) were added to a solution of Mannich adduct 31 c(diastereomeric ratio (d.r.)= 91:9, 143 mg, 0.5 mmol) in anhydrousCH3CN (1 mL) under a N2 atmosphere. The mixture was stirred at roomtemperature for 24 h. The solvents were evaporated under reduced pres-sure and then CH2Cl2 (5 mL) and NaOH (1 m, 5 mL) were added to theresulting residue. The organic phase was separated, dried, and evaporatedunder reduced pressure. The crude product was purified by flash columnchromatography (CH2Cl2 to CH2Cl2/NH3 (7 n MeOH solution) 90:10) togive a colorless oil. Yield: 80 % (66 mg); spectroscopic data of the majoranti diastereomer: 1H NMR (300 MHz, CDCl3): d=7.40–7.21 (m, 5H),3.80–3.58 (m, 3 H), 2.58 (br s, 2 H), 2.08–1.92 (m, 1H), 0.65 ppm (d, J=
6.9 Hz, 3 H); 13C NMR (125 MHz, CDCl3): d =128.9, 127.5, 127.05, 126.5,69.1, 40.2, 29.7, 15.1 ppm.
Acknowledgements
This work was financially supported by The University of the BasqueCountry (UPV/EHU), and Ministerio de Educaci�n y Ciencia (MEC,Grant CTQ2007-68095-C02, Spain). Predoctoral grants to I.O. (from GV)and I.V. (from UPV/EHU) are also acknowledged. We also thankSGIker (UPV/EHU) for NMR, HRMS, and computational facilities.
[1] For selected reviews, see: a) T. Akiyama, Chem. Rev. 2007, 107,5744 – 5758; b) T. Akiyama, J. Itoh. , K. Fuchibe, Adv. Synth. Catal.2006, 348, 999 – 1010; for binaphtol-derived phosphoric acids, see:c) M. Terada, Chem. Commun. 2008, 4097 – 4112; for small mole-cules as H-bond donors in asymmetric catalysis, see: d) A. G. Doyle,E. N. Jacobsen, Chem. Rev. 2007, 107, 5713 –5743.
[2] For a review on chiral Brønsted bases, see: C. Palomo, M. Oiarbide,R. L�pez, Chem. Soc. Rev. 2009, 38, 632 – 653.
[3] For the concept of bifunctional catalysis, see: a) G. J. Rowlands, Tet-rahedron 2001, 57, 1865 –1882; b) J.-A. Ma, D. Cahard, Angew.Chem. 2004, 116, 4666 – 4683; Angew. Chem. Int. Ed. 2004, 43, 4566 –4583; c) S. Matsunaga, M. Shibasaki, Bull. Chem. Soc. Jpn. 2008, 81,60– 75.
[4] For reviews, see: a) S. J. Connon, Synlett 2009, 354 – 376; b) H.Miyabe, Y. Takemoto, Bull. Chem. Soc. Jpn. 2008, 81, 785 –795;c) S. J. Connon, Chem. Commun. 2008, 2499 –2510; d) Y. Takemoto,H. Miyabe, Chimia 2007, 61, 269 – 275; e) S. J. Connon, Chem. Eur.J. 2006, 12, 5418.
[5] For a recent review on enamine catalysis, see: a) A. Erkkil�, I.Majander, P. M. Phiko, Chem. Rev. 2007, 107, 5416 –5470; for lead-ing references on organocatalysis, see: b) C. F. Barbas III, Angew.Chem. 2008, 120, 44 –50; Angew. Chem. Int. Ed. 2008, 47, 42 –47;c) A. Dondoni, A. Massi, Angew. Chem. 2008, 120, 4716 –4739;Angew. Chem. Int. Ed. 2008, 47, 4638 – 4660; d) D. W. C. MacMillan,Nature 2008, 455, 304 – 308; e) Enantioselective Organocatalysis: Re-actions and Experimental Procedures (Ed.: P. I. Dalko), Wiley-VCH,Weinheim, 2007; f) P. Kocovsky, A. V. Malkov, Tetrahedron 2006, 62,255 (thematic issue on Organocatalysis in Organic Synthesis nos. 2 –3); g) A. Berkessel, H. Grçger, Asymmetric Organocatalysis: FromBiomimetic Concepts to Applications in Asymmetric Synthesis,Wiley-VCH, Weinheim, 2005.
[6] Pionering works: a) Z. J. Hajos, D. R. Parrish, J. Org. Chem. 1974,39, 1615 –1621; b) U. Eder, G. Sauer, R. Wietcher, Angew. Chem.1971, 83, 492 – 493; Angew. Chem. Int. Ed. Engl. 1971, 10, 496 –497;c) B. List, A. Lerner, C. F. Barbas III, J. Am. Chem. Soc. 2000, 122,2395 – 2396; d) W. Nutz, B. List, J. Am. Chem. Soc. 2000, 122, 7386 –7387; e) K. Sakthivel, W. Nutz, T. Bui, C. F. Barbas III, J. Am.Chem. Soc. 2001, 123, 5260 –5267.
[7] For the first independent and almost simultaneous organocatalyticapplication, see: a) H. Torii, M. Nakadai, K. Ishihara, S. Saito, H.Yamamoto, Angew. Chem. 2004, 116, 2017 –2020; Angew. Chem. Int.Ed. 2004, 43, 1983 – 1986; b) A. Hartikka, P. I. Arvidsson, Tetrahe-dron: Asymmetry 2004, 15, 1831 – 1834; c) A. J. A. Cobb, D. M.Shaw, S. V. Ley, Synlett 2004, 558 –560; for recent reviews on the useof this catalyst, see: d) D. A. Longbottom, V. Franckevicius, S.Kumarn, A. J. Oelke, V. Wascholowski, S. V. Ley, Aldrichimica Acta2008, 41, 3 –11; e) D. A. Longbottom, V. Franckevicius, S. V. Ley,Chimia 2007, 61, 247 –256; f) M. Limbach, Chem. Biodiversity 2006,3, 119 –133.
[8] a) S. Saito, M. Nakadai, H. Yamamoto, Synlett 2001, 1245 –1248;b) M. Nakadai, S. Saito, H. Yammamoto, Tetrahedron 2000, 56,8167 – 8177.
[9] For a recent review on prolylamide bifunctional organocatalystsworking through hydrogen-bonding interactions, see: X. Liu, L. Lin,X. Feng, Chem. Commun. 2009, 6145 –6148.
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 5333 – 53425340
C. Palomo et al.

[10] G. Lelais, D. W. C. MacMillan, Aldrichimica Acta 2006, 39, 79– 87.[11] For the first reports on the use of catalysts 4a and 4b, see: a) M.
Marigo, T. C. Wabnitz, D. Fielenbach, K. A. Jørgensen, Angew.Chem. 2005, 117, 804 –807; Angew. Chem. Int. Ed. 2005, 44, 794 –797; b) J. Franzn, M. Marigo, D. Fielenbach, T. C. Wabnitz, A.Kjærsgaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18 296 –18304; c) Y. Hayashi, H. Gotoh, T. Hayashi, M. Shoji, Angew.Chem. 2005, 117, 4284 –4287; Angew. Chem. Int. Ed. 2005, 44, 4212 –4215; for reviews, see: d) C. Palomo, A. Mielgo, Angew. Chem.2006, 118, 8042 – 8046; Angew. Chem. Int. Ed. 2006, 45, 7876 –7880;e) A. Mielgo, C. Palomo, Chem. Asian J. 2008, 3, 922 – 948.
[12] C. Palomo, S. Vera, A. Mielgo, E. G�mez-Bengoa, Angew. Chem.2006, 118, 6130 –6133; Angew. Chem. Int. Ed. 2006, 45, 5984 –5987.
[13] J. Xing, L. Chang, Z. Hou, D. Shang, X. Liu, X. Feng, Chem. Eur. J.2008, 14, 3177 –3181.
[14] Y. Okuyama, H. Nakano, M. Watanabe, M. Takeshita, K. Uwai, C.Kabuto, E. Kwon, Tetrahedron Lett. 2009, 50, 193 – 197.
[15] C.-Q. Cheng, Z. Bian, Y.-B. He, F.-S. Han, C.-Q. Kang, Z.-L. Ning,L. X. Gao, Tetrahedron: Asymmetry 2009, 20, 1753 – 1758.
[16] R. J. Reddy, H.-M. Kuan, T.-Y. Chou, K. Chen, Chem. Eur. J. 2009,15, 9294 –9298.
[17] For detailed information, see, for example: Enantioselective Synthe-sis of b-Amino Acids (Eds.: E. Juaristi, V. A. Soloshonok), Wiley,New York, 2005.
[18] For reviews on Mannich reactions, see: a) M. Arend, B. Wester-mann, N. Risch, Angew. Chem. 1998, 110, 1096 – 1122; Angew.Chem. Int. Ed. 1998, 37, 1044 – 1070; b) J. M. M. Verkade, L. J. C.van Hemert, P. J. L. Quaedflieg, F. P. J. T. Rutjes, Chem. Soc. Rev.2008, 37, 29– 41; c) A. Ting, S. E. Schaus, Eur. J. Org. Chem. 2007,5797 – 5815; d) M. M. B. Marques, Angew. Chem. 2006, 118, 356 –360; Angew. Chem. Int. Ed. 2006, 45, 348 –352; e) J. A. Ma, Angew.Chem. 2003, 115, 4426 – 4435; Angew. Chem. Int. Ed. 2003, 42, 4290 –4299; f) A. C�rdova, Acc. Chem. Res. 2004, 37, 102 – 112; g) M. Shi-basaki, S. Matsunaga, J. Organomet. Chem. 2006, 691, 2089 – 2100.
[19] a) H. Estermann, D. Seebach, Helv. Chim. Acta 1988, 71, 1824 –1839; for more information, see: b) Houben–Weyl, StereoselectiveSynthesis, Vol. 2 (Eds.: G. Helmchen, R. W. Hoffman, J. Mulzer, E.Schaumann), Thieme, Stuttgart, 1996.
[20] For pioneering works, see: a) B. List, J. Am. Chem. Soc. 2000, 122,9336 – 9337; b) B. List, P. Pojarliev, W. T. Biller, H. J. Mart�n, J. Am.Chem. Soc. 2002, 124, 827 –833; c) A. C�rdova, W. Notz, G. Zhong,J. M. Betancort, C. F. Barbas III, J. Am. Chem. Soc. 2002, 124, 1842 –1843; for selected recent examples on this subject, see: d) Y. Haya-shi, W. Tsuboi, I. Ashime, T. Urushima, M. Shoji, K. Sakai, Angew.Chem. 2003, 115, 3805 – 3808; Angew. Chem. Int. Ed. 2003, 42, 3677 –3680; e) N. Utsumi, H. Zhang, F. Tanaka, C. F. Barbas III, Angew.Chem. 2007, 119, 1910 – 1912; Angew. Chem. Int. Ed. 2007, 46, 1878 –1880; f) E. Alza, C. Rodriguez-Escrich, S. Sayalero, A. Bastero,M. A. Perics, Chem. Eur. J. 2009, 15, 10167 –10172; g) H. Yang,R. G. Carter, J. Org. Chem. 2009, 74, 2246 –2249; for leading refer-ences to anti adducts, see: h) S. S. V. Ramasastry, H. Zhang, F.Tanaka, C. F. Barbas III, J. Am. Chem. Soc. 2007, 129, 288 –289;i) H. Zhang, S. S. V. Ramasastry, F. Tanaka, C. F. Barbas III, Adv.Synth. Catal. 2008, 350, 791 – 796; j) B. T. Hahn, R. Frçhlich, K.Harms, F. Glorius, Angew. Chem. 2008, 120, 10134 –10137; Angew.Chem. Int. Ed. 2008, 47, 9985 – 9988.
[21] a) A. C�rdova, C. F. Barbas III, Tetrahedron Lett. 2002, 43, 7749 –7752; b) W. Notz, F. Tanaka, S. Watanabe, N. S. Chowdari, J. M.Turner, R. Thayumanaran, C. F. Barbas III, J. Org. Chem. 2003, 68,9624 – 9634.
[22] a) J. Franzn, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærs-gaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18 296 – 18304;for the use of catalyst 4 b in the same reaction, see: b) I. Ibrahem,A. C�rdova, Chem. Commun. 2006, 1760 – 1762.
[23] T. Kano, Y. Yamaguchi, O. Tokuda, K. Maruoka, J. Am. Chem. Soc.2005, 127, 16408 –16409.
[24] S. Mitsumori, H. Zhang, P. H.-Y. Cheong, K. N. Houk, F. Tanaka,C. F. Barbas III, J. Am. Chem. Soc. 2006, 128, 1040 –1041.
[25] H. Zhang, S. Mitsumori, N. Utsumi, M. Imai, N. Garc�a-Delgado, M.Mifsud, K. Albertshofer, P. H.-Y. Cheong, K. N. Houk, F. Tanaka,C. F. Barbas III, J. Am. Chem. Soc. 2008, 130, 875 – 886.
[26] T. Kano, Y. Hato, K. Maruoka, Tetrahedron Lett. 2006, 47, 8467 –8469.
[27] a) T. Kano, Y. Hato, A. Yamamoto, K. Maruoka, Tetrahedron 2008,64, 1197 – 1203; b) M. Pouliquen, J. Balnchet, M.-C. Lasne, J.Rouden, Org. Lett. 2008, 10, 1029 – 1032.
[28] H. Zhang, Y. Chuan, Z. Li, Y. Peng, Adv. Synth. Catal. 2009, 351,2288 – 2294.
[29] For an example that utilizes fluorinated aldimines, see: S. Fustero, F.Mojarrad, M. D. Prez Carri�n, J. F. Sanz-Cervera, J. L. AceÇa, Eur.J. Org. Chem. 2009, 5208 – 5214.
[30] a) T. Kano, Y. Yamaguchi, K. Maruoka, Angew. Chem. 2009, 121,8828 – 8830; Angew. Chem. Int. Ed. 2009, 48, 1838 – 1840; b) T. Kano,Y. Yamaguchik Maruoka, Chem. Eur. J. 2009, 15, 6678 –6687; for thefirst use of N-Boc imines or a-amidosulfone precursors in an asym-metric Mannich reaction, see: c) A. M. Kanazawa, J.-N. Denis, A. E.Greene, J. Org. Chem. 1994, 59, 1238 –1240; d) C. Palomo, M. Oiar-bide, M. C. Gonz�lez-Rego, A. K. Sharma, J. M. Garc�a, C. Landa,A. Linden, Angew. Chem. 2000, 112, 1105 – 1107; Angew. Chem. Int.Ed. 2000, 39, 1063 – 1065; e) C. Palomo, M. Oiarbide, A. Landa,M. C. Gonz�lez-Rego, J. M. Garc�a, A. Gonz�lez, J. M. Odriozola,M. Martin-Pastor, A. Linden, J. Am. Chem. Soc. 2002, 124, 8637 –8643; for pioneering work by using this kind of imines in a syn-selec-tive enamine-based enantioselective Mannich reaction, see: f) J. W.Wang, M. Stadtler, B. List, Angew. Chem. 2007, 119, 615 –617;Angew. Chem. Int. Ed. 2007, 46, 609 –611; for further examples onthe use of N-acyl imines in enantioselective Mannich reactions, see:g) D. Dziedzic, P. Schyman, M. Kullberg, A. C�rdova, Chem. Eur. J.2009, 15, 4044 –4048.
[31] G. Cainelli, L. Sambri, A. Carlone, G. Bartoli, P. Melchiorre, Angew.Chem. 2008, 120, 8828 – 8830; Angew. Chem. Int. Ed. 2008, 47, 8700 –8702.
[32] This argument has been invoked for catalyst 16-promoted Mannichreactions of imines of arylaldehydes wherein poor stereoselectivitiesare observed. For more information on this subject, see refer-ence [25].
[33] For leading references on the use of N-sulfonylimines in Mannichreactions, see: a) K. Juhl, N. Gathergood, K. A. Jørgensen, Angew.Chem. 2001, 113, 3083 – 3085; Angew. Chem. Int. Ed. 2001, 40, 2995 –2997; b) L. Bernardi, A. S. Gthelf, R. G. Hazell, K. A. Jørgensen, J.Org. Chem. 2003, 68, 2583 –2591; c) H. Morimoto, T. Yoshino, T.Yukawa, G. Lu, S. Matsunaga, M. Shibasaki, Angew. Chem. 2008,120, 9265 – 9269; Angew. Chem. Int. Ed. 2008, 47, 9125 – 9129; d) J.Hern�ndez-Toribio, R. G�mez Arrayas, J. C. Carretero, J. Am.Chem. Soc. 2008, 130, 16150 –16151; e) D. Uraguchi, Y. Ueki, T.Ooi, J. Am. Chem. Soc. 2008, 130, 14088 –14089; f) E. A. Tiong, J. L.Gleason, Org. Lett. 2009, 11, 1725 – 1728; g) L. Cheng, L. Liu, H. Jia,D. Wang, Y.-J. Cheng, J. Org. Chem. 2009, 74, 4650 – 4653; h) L.Cheng, X. Han, H. Huang, M. W. Wong, Y. Lu, Chem. Commun.2007, 4143 –4145; i) X.-F. Xiong, Z. J. Jia, X. Du, K. Jiang, T.-Y. Liu,Y.-C. Chen, Chem. Commun. 2009, 6994 – 6996.
[34] These pyrrolidines are readily accessible from the commerciallyavailable and inexpensive (S)-trans-4-hydroxyproline in few routinesteps, an additional aspect of the model that is of practical interest.For a review on trans-4-hydroxy-l-prolines, see: P. Remuzon, Tetra-hedron 1996, 52, 13803 –13835; also see the Supporting Informationfor details.
[35] It has been recently found that an acetaldehyde-based Mannich re-action with aromatic aldehyde-derived benzoyl imines promoted bya diarylprolinol silyl ether is activated by the aid of a Brønsted acid.For details, see: a) Y. Hayashi, T. Okano, T. Itoh, T. Urishima, H.Ishikawa, T. Uchimaru, Angew. Chem. 2008, 120, 9193 –9198;Angew. Chem. Int. Ed. 2008, 47, 9053 –9058; for pioneering workson enamine-based Mannich reactions of acetaldehyde, see: b) J. W.Yang, C. Chandler, M. Stadler, D. Kampen, B. List, Nature 2008,452, 453 – 455; c) C. Chandler, P. Golzerano, A. Michrowska, B. List,
Chem. Eur. J. 2010, 16, 5333 – 5342 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 5341
FULL PAPERA 4-Hydroxy Pyrrolidine-Catalyzed Mannich Reaction of Aldehydes

Angew. Chem. 2009, 121, 2012 –2014; Angew. Chem. Int. Ed. 2009,48, 1978 –1980.
[36] T. Kan, T. Fukuyama, Chem. Commun. 2004, 353 – 359.[37] CCDC-726915 contains the supplementary crystallographic data for
the structural analysis of 29 a. These data can be obtained free ofcharge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
[38] For the origin of the stereoselectivity in organocatalyzed reactionswith a trimethylsilyl-protected diarylprolinol, see: P. Dinr, A. Ka-jæsgaard, M. A. Lie, K. A. Jørgensen, Chem. Eur. J. 2008, 14, 122 –127.
[39] a) D. Uraguchi, Y. Ueki, T. Ooi, J. Am. Chem. Soc. 2008, 130,14088 – 14089; b) F. A. Davis, G. V. Reddy, C.-H. Liang, TetrahedronLett. 1997, 38, 5139 – 5142; c) I. Abrahams, M. Motevalli, A. J. Rob-inson, P. B. Wyatt, Tetrahedron 1994, 50, 12755 –12772.
[40] T. Fukuyama, M. Cheung, C.-K. Jow, Y. Hidai, T. Kan, TetrahedronLett. 1997, 38, 5831 –5834.
[41] For the characterization of enamine intermediates by mass spec-trometry, see: a) W. Schrader, P. P. Handayani, J. Zhou, B. List,Angew. Chem. 2009, 121, 1491 –1494; Angew. Chem. Int. Ed. 2009,48, 1463 –1466. Also, see: b) I. Fleischer, A. Pfaltz, Chem. Eur. J.2010, 16, 95 –99; for characterization of enamines by 1H NMR spec-troscopy and/or X-ray analysis, see: c) T. J. Peelen, Y. Chi, S. H.Gellman, J. Am. Chem. Soc. 2005, 127, 11598 – 11599; d) D. S. See-bach, D. M. Badine, W. B. Schweizer, A. K. Beck, I. Krossing, P.Klose, Y. Hayashi, T. Uchimaru, Helv. Chim. Acta 2009, 92, 1225 –1259.
[42] C. Teck Wong, Tetrahedron 2009, 65, 7491 – 7497.[43] See the Supporting Information for further details.
Received: December 23, 2009Published online: March 22, 2010
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 5333 – 53425342
C. Palomo et al.

DOI: 10.1002/chem.201000376
Brønsted Acid Assisted Regio- and Enantioselective Direct O-Nitroso AldolReaction Catalysed by a,a-Diphenylprolinol Trimethylsilyl Ether
Antonia Mielgo, Irene Velilla, Enrique G�mez-Bengoa, and Claudio Palomo*[a]
Dedicated to Professor Saverio Florio on the occasion of his 70th birthday
Introduction
The asymmetric O-nitroso aldol reaction is an importanttool for the preparation of enantioenriched a-hydroxy car-bonyl compounds.[1] In this reaction two important issuesare the enantioselectivity and regioselectivity—attackthrough the nitrogen atom (oxyamination reaction) oroxygen atom (aminoxylation reaction).[2] First insights byYamamoto et al. on the reaction of nitrosobenzene with silylor metal enolates revealed that the regioselectivity of theprocess is dependent on the nature of the enolate and thepresence or absence of a Lewis acid catalyst.[3] Later, thesame author reported that in the presence of sub-stoichio-metric amounts of glycolic acid the reaction of nitrosoben-zene with pre-formed enamines preferentially afforded O-nitroso aldol products (aminoxylation). Conversely, in thepresence of a,a,a’,a’-tetraaryl-2,2-dimethyl-1,3-dioxolan-4,5-dimethanol (TADDOL), the a-amino derivatives were ex-clusively obtained (oxyamination).[4] Some examples ofhighly enantioselective nitroso aldol reactions have alsobeen realised through in situ generation of the reactive en-
amine by using proline (1) and secondary amines 2–6(Scheme 1) in sub-stoichiometric quantities.[5] A commonstructural feature of all these catalysts is the presence of a
Brønsted acid functionality as key element for controllingregioselectivity.[6] Thus, catalysts with strong acidic function-alities, such as carboxylic acids or sulfonamides (1–4 inScheme 1A), predominantly give a-oxygenated products,whereas those that contain less acidic functionalities,namely, hydroxy groups (5, 6 in Scheme 1B), afford mainlya-oxyaminated compounds.
Keywords: amino alcohols · enam-ine activation · aldehydes · nitrosoaldol reaction · organocatalysis
Abstract: In the presence of p-nitrobenzoic acid, the O-nitroso aldol reaction ofnitrosobenzene with enolisable aldehydes may be promoted by commercially avail-able a,a-diphenylprolinol trimethylsilyl ether. The reaction proceeds with goodyields and essentially complete enantioselectivity, with catalyst loadings in the 5–10 mol % range. The resulting a-oxyaldehyde adducts may be transformed in situinto a-oxyimines, which provide 1,2-amino alcohols upon treatment with Grignardreagents, in good overall yield (45–59 %) and with typical diastereomeric ratios�95:5.
[a] Dr. A. Mielgo, I. Velilla, Dr. E. G�mez-Bengoa,+ Prof. Dr. C. PalomoDepartamento de Qu�mica Org�nica I, Universidad del Pa�s VascoManuel Lardizabal 3, 20018 San Sebasti�n (Spain)Fax: (+34) 943015270E-mail : [email protected]
[+] Computational study.
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.201000376.
Scheme 1. Organocatalysts for the nitroso aldol reaction: A) Catalysts foraminoxylation, B) Catalysts for oxyamination. TMS= trimethylsilyl,TBS = tert-butyldimethylsilyl.
� 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 7496 – 75027496

a,a-Diarylprolinol silyl ethers, such as 7, have proven tobe very effective chiral amine catalysts in a number of orga-nocatalytic transformations.[7,8] In contrast to proline and itscongeners, these catalysts work through steric control[9] andvery high levels of enantioselectivity may be achieved. Thisconcept has not been applied to the O-nitroso aldol reaction(Scheme 2) as yet. Herein, we report the first O-nitrosoaldol reaction catalysed by commercially available a,a-di-phenylprolinol trimethylsilyl ether (7), for the formation ofa-aminoxylated products with extremely high regio- andenantiocontrol.
Results and Discussion
Background, catalyst screening and conditions : Previously,we reported that the reaction of aldehydes with nitrosoben-zene may be promoted by catalyst 7 to produce a-oxyami-nated products as single regioisomers.[10] We envisaged thatin the presence of an external Brønsted acid a switch in re-giocontrol might occur in the above reaction, as a result of aprotonation of the nitrogen atom of nitrosobenzene.[11] Thequestion was to establish whether O-nitroso aldol adductswould be produced as sole reaction products to render theprocedure operationally simple and practical. In this way,generation of both a-amino and a-hydroxy carbonyl com-pounds from a common chiral source should be made feasi-ble, which would be useful from a practical viewpoint. Theresulting O-nitroso aldol adducts could be easily trans-formed in situ into 1,2-diols and related compounds, whichare of considerable interest because these structural moiet-ies are widely found in biologically active natural productsand synthetic pharmaceuticals.[12]
From the outset we realised that the search for reactionconditions under which the oxyamination would be slow ortotally suppressed would be central for the success of thisapproach. In our initial studies, it was observed that the re-action of nitrosobenzene with freshly distilled enolisable al-dehydes proceeds efficiently in CH2Cl2 at temperatures�0 8C (typically in the range 0 8C–RT), or in THF at �20 8Cfor long-chain aldehydes. In all cases 20 mol % of catalyst 7was required for the reaction to proceed and the resultingN-nitroso aldol products were obtained in 60–75 % yield,
with 91–99 % enantiomeric excess (ee).[13] We were pleasedto find that in CH2Cl2 at �20 8C a catalyst loading between2 and 10 mol % sufficed for the O-nitroso aldol reaction toproceed with the assistance of a Brønsted acid, without ap-preciable formation of N-nitroso aldol products (Scheme 3).
For example, compound 12 a, obtained from the reaction ofpropanal with nitrosobenzene (11) in the presence of cata-lyst 7 (2 mol %) and benzoic acid (5 mol %), followed by insitu reduction with NaBH4, was formed with essentiallycomplete regio- and enantioselectivity (Table 1, entry 1).
When this reaction was conducted in the absence of acid, noa-oxyaminated nor a-aminoxylated adducts were detectedby 1H NMR spectroscopy. Other Brønsted acids[14] were ex-amined and it was found that PNBA (Table 1, entry 3) wassuperior in terms of both chemical yield and enantioselectiv-ity, whereas the less acidic p-methoxybenzoic acid (Table 1,entry 2) provided the product in lower yield. Similarly, themore acidic chloroacetic acid provided better yield thanacetic acid (Table 1, entries 4 and 5).
Scheme 2. Catalytic nitroso aldol reaction promoted by secondary aminecatalysts 1–4 and 7.
Scheme 3. O-Nitroso aldol reaction between aldehydes 10 and nitroso-benzene (11), catalysed by 7. PNBA= p-nitrobenzoic acid, Boc= tert-bu-tyloxycarbonyl, Bn=benzyl.
Table 1. Brønsted acid screening for the O-nitroso aldol reaction ofpropanal (10a) with nitrosobenzene (11) promoted by catalysts 7–9.[a]
Entry Catalyst Additive Yield of12 a[b] [%]
ee[c]
[%]
1 PhCO2H 50 >992 4-MeOC6H4CO2H 22 >993 4-NO2C6H4CO2H 81 >994 CH3CO2H 35 >995 ClCH2CO2H 52 >99
6 4-NO2C6H4CO2H <5 nd[d]
7 4-NO2C6H4CO2H 70 >99
[a] Reactions performed on a 1 mmol scale in CH2Cl2 (2 mL) with cata-lyst (2 mol %), Brønsted acid (5 mol %) and 10a (3 equiv) at �20 8C for16 h. [b] Isolated yield after flash column chromatography. [c] Deter-mined by HPLC analysis after flash chromatography; see the SupportingInformation for details. [d] Not determind.
Chem. Eur. J. 2010, 16, 7496 – 7502 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 7497
FULL PAPER

For comparison, other catalysts that work through stericcontrol (Table 1, entries 6 and 7) were tested. Catalyst 8,[7a, b]
which has proven to be very effective in diverse enamine-based reactions,[8,15] did not lead to the expected adducts,whereas the a,a-dihexylprolinol silyl ether 9[16] behaved sim-ilarly to the commercially available catalyst 7.
Screening of other solvents for this reaction (Table 2) re-vealed that CHCl3 and 1,2-dichloroethane were also effec-tive. Diethyl ether and THF, or more polar solvents such asmethanol and DMF, were not appropriate. In acetonitrileand toluene the yield was modest but could be improved byincreasing the catalyst loading to 10 mol % (Table 2, en-tries 1 versus 2 and 9 versus 10).
Results obtained from the reaction of 11 with other alde-hydes are shown in Table 3. The reactions were carried outby addition of the aldehyde 10 a–h (3 equiv) to a solution ofthe catalyst (10 mol %), PNBA (10 mol %) and 11 in di-
chloromethane at �20 8C. The reaction was easily monitoredby complete disappearance of the initial green colour. Theresulting adducts were reduced in situ by treatment withsodium borohydride in ethanol to afford the aminoxylatedcompounds 12 as one regioisomer.[17] Both, short- and long-chain aldehydes, as well as functionalised aldehydes, are tol-erated. Even the less reactive isovaleraldehyde was convert-ed to 12 e, although afforded the expected adduct in some-what lower yield (Table 3, entries 7 and 8). The reactions offreshly distilled hydrocinnamaldehyde with 11 conductedunder these conditions, but in the absence of PNBA, afford-ed neither N- nor O-nitroso aldol products.[13] In general, wehave employed 10 mol% of catalyst for the O-nitroso aldolreaction, although it can be reduced to 5 mol % without det-riment to the yield or ee value (Table 3, entries 3 and 6).With the exception of propanal, the use of 2 mol % of cata-lyst was not effective.
The absolute configuration of the adduct 12 b from the re-action of 10 b with 11 was determined by comparison of thevalue of the optical rotation of the free diol with that previ-ously described and was found to be S.[5f] This approach pro-vides (S)-aminoxylated aldehydes from the S-prolinol silylether 7, thus complements the results obtained with S-pro-line and its congeners, which afford the R enantiomer.[18]
Synthetic applications : The a-oxyaldehyde adducts are gen-erally unstable and must be isolated as the corresponding al-cohols. Nonetheless, they may undergo reactions typical ofaldehydes,[12] such as Grignard additions to produce 1,2-dis-ubstituted diols with remarkably high diastereoselectivity.[12a]
We now show that 1,2-aminoalcohols, common structuralmotifs in naturally occurring and synthetic molecules,[19] mayalso be produced with equal efficiency by Grignard additionto the corresponding imines.
As illustrated in Scheme 4, the nitroso aldol reaction fromhydrocinnamaldehyde (10 d) with catalyst 7 provided thecorresponding aldehyde adduct 12 d, which could be trans-formed in situ to the imine 13. At this stage, Grignard addi-tion[20] afforded the expected adducts with concomitant par-tial cleavage of the phenylaminoxy group.[12a] Subsequenthydrogenation provided the expected (S,S)-amino alcohols14 and 15 in good yields over four steps and, most remarka-bly, with essentially perfect diastereocontrol.[21] As predict-ed, the same sequence of reactions and the use of l-prolinefor the nitroso aldol addition[5b] led to the production of(R,R)-amino alcohol 16. N-Aryl imines[22] can also beformed and treated with Grignard reagents. For instance,following the same sequence, the aldehyde adduct from thereaction of propanal with 11 afforded imine 17 after treat-ment with p-anisidine. Subsequent reaction with Grignardreagents provided adducts 18 and 19 in good overall yieldsand with diastereomeric ratios of 95:5.[23] In this case, com-plete deprotection of the phenylaminoxy group occurredduring the Grignard addition, thus the final hydrogenationwas no longer necessary and, as a result, the protocol wasrendered compatible with aryl Grignard reagents. The sim-plified model A explains the observed stereochemical out-
Table 2. Solvent screening for the aminoxylation reaction of propanal(10a) with nitrosobenzene (11) promoted by catalyst 7.[a]
Entry Solvent Yield of 12a[b] [%] ee[c] [%]
1 toluene 22 >992 toluene 67[d] >993 Et2O 10 nd[e]
4 THF <10 nd[e]
5 CHCl3 71 >996 CH2Cl2 80 >997 ClCH2CH2Cl 71 >998 MeOH <10 nd[e]
9 CH3CN 41 >9910 CH3CN 53[d] >9911 DMF <10 nd[e]
[a] Reactions conducted with 10a (3 equiv), 7 (2 mol %) and PNBA(5 mol %) at �20 8C for 16 h. [b] Isolated yield after column chromatog-raphy. [c] Determined by HPLC analysis; see the Supporting Informationfor further details. [d] Reactions conducted with catalyst 7 (10 mol %)and PNBA (10 mol %). [e] Not determined.
Table 3. Catalytic asymmetric O-nitroso aldol reactions of aldehydes 10with nitrosobenzene (11) promoted by catalyst 7.[a]
Entry R Product t [h] Yield[b] [%] ee[c] [%]
1 Me 12a 16 80[d] >992 nPr 12b 2 78 >993 nPr 12b 16 71[e] >994 nBu 12c 2 81 >995 CH2Ph 12d 5 68 >996 CH2Ph 12d 16 68[e] >997 iPr 12e 2 55 >998 iPr 12e 3 59[f] >999 BnOCH2 12 f 16 45 9510 BocHN ACHTUNGTRENNUNG(CH2)4 12g 3 88 >99
11 12h 4 66 >99
[a] Reactions performed on a 1 mmol scale in CH2Cl2 (2 mL) at �20 8Cwith 7 (10 mol %), PNBA (10 mol %) and 10 (3 equiv). [b] Isolated yieldafter flash column chromatography. [c] Determined by HPLC analysisafter flash chromatography; see the Supporting Information for details.[d] Reaction performed with catalyst 3 (2 mol %) and PNBA (5 mol %).[e] Reaction conducted with catalyst 7 (5 mol %). [f] Reaction performedwith catalyst 7 (20 mol %) and PNBA (20 mol %).
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 7496 – 75027498
C. Palomo et al.

come of the Grignard addition,although a five-membered-che-late model should not be ex-cluded.[20]
Mechanistic insights : To firmlyestablish the origin of regio-and enantioselectivity in thepresent catalytic system, wecomputed the different transi-
tion states for the formation of the C�N and C�O bonds inthe reaction between propionaldehyde and nitrosobenzene,promoted by catalyst 7 in the presence of hydrogen-bonddonors, by density functional theory (DFT). On the basis ofevidence for enamine formation,[24] we assumed that the re-action proceeds through an enamine mechanism[6,9] (ratherthan via an enol intermediate)[25] and that the activation ofthe oxygen and nitrogen atoms of 11 takes place by hydro-gen bonding, with participation of two molecules of acid.[26]
In each case, the activation energies were computed by thedifference between the transition-state energy and theenergy of the corresponding pre-transition-state complexes.After an extensive conformational search, we found that thelowest-energy transition states (TS-N and TS-O) involve theattack of 11 from the opposite face to the bulky�C(Ph)2OSiMe3 directing group (in accordance with previ-ous observations on related enamine-based reactions).[9]
Also the enamine double bond is in the E configuration andanti to the silyl group. The energy data clearly point to a hy-drogen-bond reduction of the Gibbs free energy of activa-tion (DG�) in the presence of PNBA, from 21.4 (absence ofcatalyst) to 1.3 kcal mol�1. More interestingly, the N-selectiv-ity computed in the absence of catalyst, or in the presence
of the weak hydrogen-bond-donor water, is reversed to aclear O-selective addition for the acid species (benzoic acidand PNBA), see Figure 1.
Conclusion
We have demonstrated that commercially available a,a-di-phenylprolinol trimethylsilyl ether (7) in the presence of aBrønsted acid is a very effective and general catalyst for thereaction of aldehydes with nitrosobenzene to afford O-ni-troso aldol adducts with both high enantio- and regioselec-tivity. Because S enantiomers are produced, this procedurecomplements the (S)-proline (1)-catalysed reaction, in whichR enantiomers are formed, and provides a new entry toenantiomerically pure (S,S)- and (R,R)-1,2-aminoalcohols.
Experimental Section
General methods : All reactions were carried out under a nitrogen atmos-phere in flame-dried glassware, with efficient magnetic stirring. CH2Cl2
was distilled from CaH2. Toluene, THF and Et2O were dried in the pres-ence of sodium metal. Reagent grade methanol, ethyl acetate and DMFwere used. Purification of reaction products was carried out by flashcolumn chromatography on silica gel 60 (0.040–0.063 mm, 230–400mesh). Analytical TLC was performed on 0.25 mm silica gel 60-F plates.Visualisation was accomplished with UV light and by dipping the devel-oped plate into a solution of cerium ammonium molybdate (ammoniummolybdate (21 g), cerium sulfate (1 g), concentrated sulfuric acid(31 mL), water (470 mL)), followed by heating. Melting points were mea-sured with a B�chi SMP-20 melting point apparatus and are uncorrected.1H and 13C NMR spectra were recorded on a Bruker Advance-500 (or300) spectrometer and chemical shifts (d) are reported in ppm relative tointernal tetramethylsilane (TMS). Analytical HPLC was performed onWaters-600E, Waters-2996 and Hewlett–Packard series 1050 chromato-graphs equipped with diode array UV detectors and Daicel ChiralpakAD-H and OD-H columns. Optical rotations were recorded on a JascoP-2000 polarimeter. MS spectra were recorded on an ESI ion-trap massspectrometer (Agilent 1100 series LC/MSD, SL model). Prolinol trime-thylsilyl ether catalysts 7 and 8 were purchased from Aldrich and usedwithout further purification. Catalyst 9 was prepared by a previously de-scribed procedure.[15]
Scheme 4. 1,2-Amino alcohols from a nitroso aldol reaction, imine forma-tion and Grignard addition sequence.
Figure 1. N- and O-selective transition states calculated at the B3LYP/6-311++G**//B3LYP/6-31G* level of theory in the gas phase.
Chem. Eur. J. 2010, 16, 7496 – 7502 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 7499
FULL PAPERRegio- and Enantioselective O-Nitroso Aldol Reaction

General procedure for the aminoxylation reaction : Nitrosobenzene(1 mmol, 1 equiv) and the freshly distilled aldehyde[27] (3 mmol, 3 equiv)were successively added to a solution of the catalyst (0.1 mmol,10 mol %) and PNBA (0.1 mmol, 10 mol %) in CH2Cl2 (2 mL) at �20 8C.The resulting green solution was stirred at �20 8C until the colour turnedyellow. EtOH (2 mL) and NaBH4 (8 mmol) were added successively at�20 8C. After stirring for 30 min, the reaction was quenched with a satu-rated aqueous solution of NaCl (3 mL) and allowed to reach room tem-perature. After extraction with CH2Cl2 (3 � 4 mL), the combined organicphases were dried over anhydrous MgSO4, concentrated under reducedpressure and purified by flash column chromatography on silica gel toafford the expected adducts. The regioselectivity of the process was de-termined by 1H NMR spectroscopy analysis of the crude products andwas found to be >99:1 in all cases.
(S)-2-(Phenylaminooxy)propan-1-ol (12a): Prepared according to thegeneral procedure from propanal (0.22 mL, 3 mmol). The crude materialwas purified by column chromatography on silica gel (hexane/ethyl ace-tate 80:20) to give 12a as a yellow oil (80 %, 133 mg). Spectroscopic dataare in agreement with published data for the R enantiomer.[5a] The enan-tiomeric excess (>99%) was determined by HPLC analysis (Daicel Chir-alpak AD-H, hexane/ethanol 90:10, flow rate= 1 mL min�1; retentiontimes: 14.5 min (major), 20.1 min (minor)). [a]25
D =�1.1 (c =1, CHCl3).
(S)-2-(Phenylaminooxy)pentan-1-ol (12b): Prepared according to thegeneral procedure from pentanal (0.32 mL, 3 mmol). The crude materialwas purified by column chromatography on silica gel (hexane/ethyl ace-tate 85:15) to give 12 b as a yellow oil (78 %, 152 mg). Spectroscopic dataare in agreement with published data for the R enantiomer.[5a] The enan-tiomeric excess (>99%) was determined by HPLC analysis (Daicel Chir-alpak AD-H, hexane/isopropanol 95:5, flow rate =0.8 mL min�1; reten-tion times: 20.5 min (major), 24.5 min (minor)). [a]25
D =�24.8 (c =0.9,CHCl3).
(S)-2-(Phenylaminooxy)hexan-1-ol (12c): Prepared according to the gen-eral procedure from hexanal (0.36 mL, 3 mmol). The crude material waspurified by column chromatography on silica gel (hexane/ethyl acetate85:15) to give 12 c as a yellow oil (81 %, 169 mg). Spectroscopic data arein agreement with published data for the R enantiomer.[5a] The enantio-meric excess (>99%) was determined by HPLC analysis (Daicel Chiral-pak AD-H, hexane/ethanol 95:5, flow rate=0.8 mL min�1; retentiontimes: 19.3 min (major) and 24.6 min (minor)). [a]25
D =�21.1 (c =1,CHCl3).
(S)-3-Phenyl-2-(phenylaminooxy)propan-1-ol (12d): Prepared accordingto the general procedure from hydrocinnamaldehyde (0.39 mL, 3 mmol).The crude material was purified by column chromatography on silica gel(hexane/ethyl acetate 85:15) to give 12 d as an orange oil (68 %, 164 mg).Spectroscopic data are in agreement with published data for the R enan-tiomer.[5a] The enantiomeric excess (>99%) was determined by HPLCanalysis (Daicel Chiralpak AD-H, hexane/isopropanol 95:5, flow rate=
0.8 mL min�1; retention times: 40.1 min (major) and 54.8 min (minor)).[a]25
D =�43.1 (c =1, CHCl3).
(S)-3-Methyl-2-(phenylaminooxy)butan-1-ol (12e): Prepared according tothe general procedure from isovaleraldehyde (0.32 mL, 3 mmol). Thecrude material was purified by column chromatography on silica gel(hexane/ethyl acetate 85:15) to give 12e as a yellow oil (56 %, 109 mg).Spectroscopic data are in agreement with published data for the R enan-tiomer.[5a] The enantiomeric excess (>99%) was determined by HPLCanalysis (Daicel Chiralpak AD-H, hexane/ethanol 95:5, flow rate=
0.8 mL min�1; retention times: 13.9 min (major) and 16.1 min (minor)).[a]25
D =�32.6 (c =1, CHCl3).
(R)-3-(Benzyloxy)-2-(phenylaminooxy)propan-1-ol (12 f): Prepared ac-cording to the general procedure from 3-(benzyloxy)propanal (0.48 mL,3 mmol). The crude material was purified by column chromatography onsilica gel (hexane/ethyl acetate 80:20) to give 12 f as an orange oil (45 %,122 mg). Spectroscopic data are in agreement with published data for theR enantiomer.[5a] The enantiomeric excess (95 %) was determined byHPLC analysis (Daicel Chiralpak AD-H, hexane/ethanol 95:5, flowrate=1 mL min�1; retention times: 55.8 min (major) and 64.0 min(minor)). [a]25
D =++7.2 (c=1, CHCl3).
(S)-tert-Butyl-6-hydroxy-5-(phenylaminooxy)hexylcarbamate (12g): Pre-pared according to the general procedure from 6-(tert-butoxycarbonyla-mino)-1-hexanal (645 mg, 3 mmol). The crude material was purified bycolumn chromatography on silica gel (hexane/ethyl acetate 60:40) to give12g as an orange oil (88 %, 306 mg). Spectroscopic data are in agreementwith published data for the R enantiomer.[28] The enantiomeric excessACHTUNGTRENNUNG(>99%) was determined by HPLC analysis (Daicel Chiralpak OD-H,hexane/isopropanol 92:8, flow rate =1 mL min�1; retention times:26.5 min (minor) and 30.3 min (major)). [a]25
D =�8.4 (c= 1, CHCl3).
(S)-2-(Phenylaminooxy)hex-5-en-1-ol (12h): Prepared according to thegeneral procedure from hexen-5-al (294 mg, 3 mmol). The crude materialwas purified by column chromatography on silica gel (hexane/ethyl ace-tate 80:20) to give 12 h as a yellow oil (66 %, 134 mg). Spectroscopic dataare in agreement with published data for the R enantiomer.[28] The enan-tiomeric excess (>99%) was determined by HPLC analysis (Daicel Chir-alpak AD-H, hexane/isopropanol 94:6, flow rate=1 mL min�1; retentiontimes: 15.8 min (major) and 19.5 min (minor)). [a]25
D =�1.4 (c =1,CHCl3).
General procedure for the preparation of 1,2-amino alcohols: Method A :Nitrosobenzene (1 mmol, 1 equiv) and aldehyde (3 mmol, 3 equiv) wereadded successively to a solution of the catalyst (0.1 mmol, 10 mol %) andPNBA (0.1 mmol, 10 mol %) in CH2Cl2 (2 mL) at �20 8C. The resultinggreen solution was stirred at �20 8C until the colour turned yellow. Anhy-drous MgSO4 (0.250 g) and benzhydryl amine (3 mmol, 3 equiv) wereadded to this solution. The reaction was allowed to reach 0 8C and themixture was stirred at this temperature for 2 h before the solution was fil-tered and the solvent evaporated. The residue was dissolved in THF(2 mL) and the solution was cooled to �60 8C. The correspondingGrignard reagent (3 m in diethyl ether, 15 mmol, 15 equiv) was addeddropwise at this temperature; the mixture was allowed to reach roomtemperature and was stirred overnight. The reaction was quenched witha saturated aqueous solution of NH4Cl (10 mL), extracted with ethyl ace-tate (2 � 15 mL), washed with brine, dried over anhydrous MgSO4 andevaporated. The residue was purified by flash column chromatographyon silica gel and then hydrogenated in EtOH (2 mL mmol�1) over Pd/C(20 % w/w) for 48 h at atmospheric pressure. The mixture was filteredthrough Celite, evaporated and purified by an acid–base workup. Thisyielded the expected 1,2-aminoalcohols as almost single diastereomers.
General method B : Nitrosobenzene (1 mmol, 1 equiv) and aldehyde(3 mmol, 3 equiv) were added successively to l-proline (0.1 mmol,10 mol %) in CHCl3 (1 mL) at 0 8C and the resulting green solution wasstirred at 0 8C until the colour turned yellow. The solution was then con-centrated and CH2Cl2 (2 mL), anhydrous MgSO4 (0.250 g) and benzhy-dryl amine (3 mmol, 3 equiv) were added. The reaction was stirred at0 8C for 2 h before the solution was filtered and evaporated. The residuewas dissolved in THF (2 mL) and cooled to �60 8C. The correspondingGrignard reagent (3 m in diethyl ether, 15 mmol, 15 equiv) was addeddropwise at this temperature, the mixture was allowed to reach roomtemperature and was stirred overnight. The reaction was quenched witha saturated aqueous solution of NH4Cl (10 mL), extracted with ethyl ace-tate (2 � 15 mL), washed with brine, dried over anhydrous MgSO4 andevaporated. The residue was purified by flash column chromatographyon silica gel and then hydrogenated in EtOH (2 mL mmol�1) over Pd/C(20 % w/w) for 48 h at atmospheric pressure. The mixture was filteredthrough Celite, evaporated and purified by an acid–base workup. Thisyielded the expected 1,2-aminoalcohols as almost single diastereomers.
General method C : Nitrosobenzene (1 mmol, 1 equiv) and aldehyde(3 mmol, 3 equiv) were added successively to a solution of the catalyst(0.1 mmol, 10 mol %) and PNBA (0.1 mmol, 10 mol %) in CH2Cl2 (2 mL)at �20 8C and the resulting green solution was stirred at �20 8C until thecolour turned yellow. Anhydrous MgSO4 (0.250 g) and p-anisidine(3 mmol, 3 equiv) were added to this solution. The reaction was allowedto reach 0 8C and the mixture was stirred at this temperature for 2 hbefore the solution was filtered and evaporated. The residue was dis-solved in THF (2 mL) and cooled to �60 8C. The corresponding Grignardreagent (3 m in diethyl ether, 15 mmol, 15 equiv) was added dropwise atthis temperature and the solution was allowed to reach room tempera-ture and stirred overnight. The reaction was quenched with a saturated
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 7496 – 75027500
C. Palomo et al.

aqueous solution of NH4Cl (10 mL), extracted with ethyl acetate (2 �15 mL), washed with brine, dried over anhydrous MgSO4 and evaporated.The residue was purified by flash column chromatography on silica gel.ACHTUNGTRENNUNG(2S, 3S)-3-Amino-1-phenylbutan-2-ol (14): Prepared according to thegeneral method A from hydrocinnamaldehyde (0.39 mL, 3 mmol) andmethyl magnesium bromide. Compound 14 was obtained as an orange oil(60 %, 90 mg). Spectroscopic data are in agreement with publisheddata.[28] [a]25
D =�17.8 (c =0.5, CH2Cl2).ACHTUNGTRENNUNG(2S, 3S)-3-Amino-1-phenylpentan-2-ol (15): Prepared according to thegeneral method A from hydrocinnamaldehyde (0.39 mL, 3 mmol) andethyl magnesium bromide. Compound 15 was isolated as an orange oil(45 %, 74 mg). [a]25
D =�20.6 (c=0.5, CH2Cl2); 1H NMR (300 MHz,CDCl3): d=0.91 (t, J=7.5 Hz, 3 H), 1.72–1.40 (m, 2 H), 1.72–1.56 (m,1H), 2.71–2.60 (m, 1 H), 2.91–2.75 (m, 2H), 3.74–3.62 (m, 1H), 4.99 (s,2H), 7.37–7.16 ppm (m, 5 H); 13C NMR (125 MHz, CDCl3): d =10.0, 24.5,40.2, 57.2, 73.3, 126.4, 128.4, 129.5, 138.2 ppm.ACHTUNGTRENNUNG(2R, 3R)-3-Amino-1-phenylbutan-2-ol (16): Prepared according to thegeneral method B from hydrocinnamaldehyde (0.39 mL, 3 mmol) andmethyl magnesium bromide. Compound 16 was isolated as an orange oil(51 %, 77 mg). Spectroscopic data are in agreement with publisheddata.[28] [a]25
D =++20.0 (c =0.5, CH2Cl2).ACHTUNGTRENNUNG(2S, 3S)-3-(4-Methoxyphenylamino)octan-2-ol (18): Prepared accordingto the general method C from propionaldehyde (0.22 mL, 3 mmol) andpentyl magnesium bromide. Compound 18 was obtained as an orange oil(55 %, syn/anti 95:5, 155 mg). [a]25
D (95:5 syn/anti non-separable mix-ture) =++6.8 (c =1, CH2Cl2); 1H NMR (300 MHz, CDCl3, major syn dia-stereomer): d=0.93–0.80 (m, 5 H), 1.49–1.74 (m, 10H), 3.15–4.04 (m,1H), 3.76–3.65 (m, 2H), 3.78 (s, 3 H), 6.67 (d, J =9.0 Hz, 2H), 6.80 ppm(d, J =9.0 Hz, 2 H); 13C NMR (125 MHz, CDCl3, major syn diastereo-mer): d=14.0, 19.9, 22.5, 25.6, 32.0, 32.3, 55.8, 61.8, 69.7, 115.0, 115.3,128.7, 142.8, 152.6 ppm.ACHTUNGTRENNUNG(1S,2S)-1-(4-Methoxyphenylamino)-1-phenylpropan-2-ol (19): Preparedaccording to the general method C from propionaldehyde (0.22 mL,3 mmol) and phenyl magnesium bromide. Compound 19 was obtained asan orange oil (52 %, syn/anti 94:6, 134 mg). [a]25
D (94:6 syn/anti non-sepa-rable mixture)=++8.2 (c= 1, CH2Cl2); 1H NMR (300 MHz, CDCl3, majorsyn diastereomer): d =1.25 (d, J =6.3 Hz, 3H), 3.73 (s, 3 H), 3.99 (m,1H), 4.14 (d, J =6.3 Hz, 1H), 6.59 (d, J =8.9 Hz, 2 H), 6.74 (d, J =8.9 Hz,2H), 7.40–7.26 ppm (m, 5H); 13C NMR (125 MHz, CDCl3, major syn dia-stereomer): d =20.0, 55.7, 66.1, 71.7, 114.8, 115.6, 127.1, 127.5, 128.7,141.3, 141.5, 152.5 ppm.
Acknowledgements
This work was financially supported by The University of the BasqueCountry (UPV/EHU), Basque Government (SAIOTEK s-pe09un40) andMinisterio de Ciencia e Innovaci�n (MICIN, Grant CTQ2007-68095-C02,Spain). A predoctoral grant to I.V. (from UPV/EHU) is also acknowl-edged. We also thank SGIker (UPV/EHU) for NMR, HRMS and com-putational facilities.
[1] For reviews on nitroso compounds, see: a) P. Zuman, P. Shap, Chem.Rev. 1994, 94, 1621 –1641; b) L. Soghyuk, C. Li, H. W. Ann, Chem.Rev. 2002, 102, 1019 –1066; c) Y. Yamamoto, M. Kawasaki, Bull.Chem. Soc. Jpn. 2007, 80, 595 – 607. For a review on catalytic, enan-tioselective a-aminations and oxygenations, see: d) J. M. Janey,Angew. Chem. 2005, 117, 4364 –4372; Angew. Chem. Int. Ed. 2005,44, 4292 –4300.
[2] For reviews, see: a) P. Merino, T. Tejero, Angew. Chem. 2004, 116,3055 – 3058; Angew. Chem. Int. Ed. 2004, 43, 2995 – 2997; b) B.Plietker, Tetrahedron: Asymmetry 2005, 16, 3453 –3459; c) H. Yama-moto, N. Nomiyama, Chem. Commun. 2005, 3514 – 3525.
[3] a) N. Momiyama, H. Yamamoto, Angew. Chem. 2002, 114, 3459 –3461; Angew. Chem. Int. Ed. 2002, 41, 3313; b) N. Momiyama, H.
Yamamoto, Org. Lett. 2002, 4, 3579 –3582; c) N. Momiyama, H. Ya-mamoto, J. Am. Chem. Soc. 2003, 125, 6038 –6039.
[4] N. Momiyama, H. Yamamoto, J. Am. Chem. Soc. 2005, 127, 1080 –1081.
[5] For examples of l-proline (1) as a catalyst, see: a) Z. Zhong, Angew.Chem. 2003, 115, 4379 – 4382; Angew. Chem. Int. Ed. 2003, 42, 4247 –4250; b) S. P. Brown, M. P. Brochu, C. J. Sinz, D. W. C. MacMillan, J.Am. Chem. Soc. 2003, 125, 10808 –10809; c) Y. Hayashi, J. Yamagu-chi, K. Hibino, M. Shoji, Tetrahedron Lett. 2003, 44, 8293 –8296;d) Y. Hayashi, J. Yamaguchi, T. Sumiya, K. Hibino, M. J. Shoji, J.Org. Chem. 2004, 69, 5966 –5973; e) Y. Hayashi, J. Yamaguchi, T.Sumiya, M. Shoji, Angew. Chem. 2004, 116, 1132 – 1135; Angew.Chem. Int. Ed. 2004, 43, 1112 – 1115; f) A. C�rdova, H. Sund�n, A.Bøgevig, M. Johansson, F. Hilmo, Chem. Eur. J. 2004, 10, 3673 –3684; g) S.-G. Kim, T.-H. Park, Tetrahedron Lett. 2006, 47, 9067 –9071; h) K. Huang, Z.-Z. Huang, Y.-L. Li, J. Org. Chem. 2006, 71,8320 – 8323. For 2 as catalyst, see: i) N. Momiyama, H. Torii, S. Saito,H. Yamamoto, Proc. Natl. Acad. Sci. USA 2004, 101, 5374 –5378;j) S. Kumarn, D. M. Shaw, D. A. Longbottom, S. V. Ley, Org. Lett.2005, 7, 4189 –4191; k) Y. Yamamoto, N. Momiyama, H. Yamamoto,J. Am. Chem. Soc. 2004, 126, 5962 – 5963. For catalyst 3, see: l) W.Wang, J. Wang, H. Li, L. Liao, Tetrahedron Lett. 2004, 45, 7235 –7238. For catalyst 4, see: m) Y. Hayashi, J. Yamaguchi, K. Hibino, T.Sumiya, T. Urushima, M. Shoji, D. Hashizume, H. Koshino, Adv.Synth. Catal. 2004, 346, 1435 – 1439. For a polymer-supported pro-line-derivative, see: n) D. Font, A. Bastero, S. Salayero, C. Jimeno,M. A. Perics, Org. Lett. 2007, 9, 1943 – 1946. For catalyst 5, see:o) H.-M. Guo, L. Cheng, L.-F. Cun, L.-Z. Ghong, A.-Q. Mi, Y.-Z.Jiang, Chem. Commun. 2006, 429 – 431. For catalyst 6, see: p) T.Kano, M. Ueda, J. Takai, K. Maruoka, J. Am. Chem. Soc. 2006, 128,6046 – 6047.
[6] For DFT studies on the enamine-based nitroso aldol reaction cata-lysed by chiral Brønsted acids, see: a) M. Akakura, M. Kawasaki, H.Yamamoto, Eur. J. Org. Chem. 2008, 4245 – 4249; catalysed by pro-line (1): b) P. H.-Y. Cheong, K. N. Houk, J. Am. Chem. Soc. 2004,126, 13912 – 13913.
[7] For the first reports describing these catalysts, see: a) M. Marigo,T. C. Wabnitz, D. Fielenbach, K. A. Jørgensen, Angew. Chem. 2005,117, 804 –807; Angew. Chem. Int. Ed. 2005, 44, 794 – 797; b) J. Fran-z�n, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard, K. A.Jørgensen, J. Am. Chem. Soc. 2005, 127, 18 296 – 18304; c) Y. Haya-shi, H. Gotoh, T. Hayashi, M. Shoji, Angew. Chem. 2005, 117, 4284 –4287; Angew. Chem. Int. Ed. 2005, 44, 4212 –4215.
[8] For reviews, see: a) C. Palomo, A. Mielgo, Angew. Chem. 2006, 118,8042 – 8046; Angew. Chem. Int. Ed. 2006, 45, 7876 –7880; b) A.Mielgo, C. Palomo, Chem. Asian J. 2008, 3, 922 – 948.
[9] For the mechanism of a-heterofunctionalisation of aldehydes pro-moted by 7 via enamine, see: P. Din�r, A. Kærsgaard, M. A. Lie,K. A. Jørgensen, Chem. Eur. J. 2008, 14, 122 – 127.
[10] C. Palomo, S. Vera, I. Velilla, A. Mielgo, E. G�mez-Bengoa, Angew.Chem. 2007, 119, 8200 – 8202; Angew. Chem. Int. Ed. 2007, 46, 8054 –8056.
[11] For a chiral Brønsted acid catalysed direct reaction of nitrosoben-zene with b-dicarbonyl compounds, see: M. Lu, D. Zhu, Y. Lu, X.Zeng, B. Tan, Z. Xu, G. Zhong, J. Am. Chem. Soc. 2009, 131, 4562 –4563.
[12] For recent representative examples, see: a) P. Jiao, M. Kawasaki, H.Yamamoto, Angew. Chem. 2009, 121, 3383 –3386; Angew. Chem. Int.Ed. 2009, 48, 3333 – 3336; b) L. Yang, R.-H. Liu, B. Wang, L. L.Weng, H. Zheng, Tetrahedron Lett. 2009, 50, 2628 –2631; c) T. M.Shaikh, A. Sudalai, Tetrahedron: Asymmetry 2009, 20, 2287 – 2292;d) N. B. Kondekar, P. Kumar, Org. Lett. 2009, 11, 2611 – 2614; e) G.Zhong, Y. Yu, Org. Lett. 2004, 6, 1637 –1639; f) M. Lu, D. Zhu, Y.Lu, Y. Hou, B. Tan, G. Zhong, Angew. Chem. 2008, 120, 10341 –10345; Angew. Chem. Int. Ed. 2008, 47, 10 187 –10 191; g) S. Kumarn,A. J. Oelke, D. M. Shaw, D. A. Longbottom, S. V. Ley, Org. Biomol.Chem. 2007, 5, 2678 – 2689.
[13] For more details see reference [10] and the Supporting Information.
Chem. Eur. J. 2010, 16, 7496 – 7502 � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 7501
FULL PAPERRegio- and Enantioselective O-Nitroso Aldol Reaction

[14] pKa values (water) for the employed Brønsted acids: 4-methoxyben-zoic acid, 4.47; benzoic acid, 4.19; 4-nitrobenzoic acid, 3.44; aceticacid, 4.76; chloroacetic acid, 2.87; see: B. G. Tehan, E. J. Lloyd,M. G. Wong, W. R. Pitt, J. G. Montana, D. T. Manallack, E. Gancia,Quant. Struct.-Act. Relat. 2002, 21, 457 – 472, and references therein.
[15] For reviews on a-heterofunctionalisation of carbonyl compounds,see: a) G. Guillena, D. J. Ram�n, Tetrahedron: Asymmetry 2006, 17,1465 – 1492; b) M. Marigo, K. A. Jørgensen, Chem. Commun. 2006,2001 – 2011; for a general review on enantioselective enamine-basedreactions, see: c) S. Mukherjee, J. W. Yang, S. Hoffmann, B. List,Chem. Rev. 2007, 107, 5471 –5569.
[16] C. Palomo, A. Landa, A. Mielgo, M. Oiarbide, A. Puente, S. Vera,Angew. Chem. 2007, 119, 8583 – 8587; Angew. Chem. Int. Ed. 2007,46, 8431 –8435.
[17] The absence of N-nitroso aldol products is easily established by300 MHz NMR spectroscopy because, for each aldehyde, the signalscorresponding to CHN(OH)Ph appear further upfield than those ofCHONHPh. For more details see reference [10] and the SupportingInformation.
[18] For an enantioselective switch in the nitroso aldol reaction by usingbinaphthyl-based chiral secondary amines with identical axial chiral-ity, see: a) T. Kano, A. Yamamoto, K. Maruoka, Tetrahedron Lett.2008, 49, 5369 – 5371; b) T. Kano, A. Yamamoto, F. Shirozu, K. Mar-uoka, Synthesis 2009, 1557 – 1563.
[19] S. C. Bergmeier, Tetrahedron 2000, 56, 2561 –2576.[20] For Grignard additions to N-benzyl and N-benzhydryl imines de-
rived from a-oxyaldehydes, see: a) T. Franz, M. Hein, U. Veith, V.Jger, Angew. Chem. 1994, 106, 1308 –1311; Angew. Chem. Int. Ed.Engl. 1994, 33, 1298 –1301; b) U. Vieth, S. Leurs, V. Jger, Chem.Commun. 1996, 329 – 330.
[21] The configuration of aminoalcohols 14 and 15 was established bycomparison with reported data; see: P. Besse, H. Veschambre, R.
ChÞnevert, M. Dickman, Tetrahedron: Asymmetry 1994, 5, 1727 –1744; see the Supporting Information for details.
[22] K. Imioka, I. Inoue, M. Shindo, K. Koga, Tetrahedron Lett. 1991, 32,3095 – 3098.
[23] For N-dearylation methods of 1,2-amino alcohols, see: a) G. Bartoli,M. Bosco, A. Carlone, M. Locatelli, M. Massaccesi, P. Melchiorre,L. Sambri, Org. Lett. 2004, 6, 2173 – 2176; b) G. E. Keck, A. P.Truong, Org. Lett. 2002, 4, 3131 –3134.
[24] For the characterisation of enamine intermediates by mass spec-trometry, see: a) W. Schrader, P. P. Handayani, J. Zhou, B. List,Angew. Chem. 2009, 121, 1491 –1494; Angew. Chem. Int. Ed. 2009,48, 1463 –1466; also, see: b) I. Fleischer, A. Pfaltz, Chem. Eur. J.2010, 16, 95 –99; for characterisation of enamines by 1H NMR spec-troscopy and/or X-ray crystallographic analysis, see: c) T. J. Peelen,Y. Chi, S. H. Gellman, J. Am. Chem. Soc. 2005, 127, 11598 –11599;d) D. S. Seebach, D. M. Badine, W. B. Schweizer, A. K. Beck, I.Krossing, P. Klose, Y. Hayashi, T. Uchimaru, Helv. Chim. Acta 2009,92, 1225 –1259.
[25] a) For the a-heterofunctionalisation of aldehydes promoted by 7through an enol mechanism, see: C. Teck Wong, Tetrahedron 2009,65, 7491 –7497; b) for the nitrosoaldol reaction via an enol mecha-nism, see: C. T. Wong, Tetrahedron Lett. 2009, 50, 811 – 813.
[26] The results from one molecule of acid did not fit well with the ex-perimental observations. For details, see the Supporting Information.Also see reference [6a].
[27] No traces of any acid should be present in the reaction to avoid theO-nitroso aldol reaction completely.
[28] R. Besse, H. Veschambre, Tetrahedron: Asymmetry 1994, 5, 1727 –1744.
Received: February 11, 2010Published online: April 30, 2010
www.chemeurj.org � 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2010, 16, 7496 – 75027502
C. Palomo et al.





























