Embed Size (px)
Citation preview

© Laura Condorelli 2012 Pag. 1
CENNI DI TERMODINAMICA Si occupa degli scambi di materia ed energia tra un sistema (porzione di universo in cui si verifica una
trasformazione, separato dall’ambiente esterno mediante una membrana oppure una superficie fisica). Sono
esempi di sistema un cilindro di vetro chiuso, una massa di gas) e l’ambiente (il resto dell’universo). Il
sistema rispetto all’ambiente può essere isolato (non avvengono scambi di alcun tipo), chiuso (vi sono
scambi di energia, ma non di materia), aperto (sono consentiti scambi sia di materia, sia di energia).
Una trasformazione è la variazione subita dal sistema per passare da uno stato di equilibrio iniziale ad uno
finale. In queste trasformazioni cambiano una o più grandezze del sistema (variabili del sistema). Lo stato
del sistema viene definito da variabili intensive (che non dipendono dalle dimensioni, ovvero, temperatura,
pressione, concentrazione) ed estensive (che dipendono dalle dimensioni, come volume, massa, energia
libera, entalpia, entropia).
nessuno scambio energia energia
materia
Sistema isolato Sistema chiuso Sistema aperto
Le trasformazioni termodinamiche possono essere reversibili o irreversibili (non possono essere ripercorse
all’indietro).
Ad ogni sistema si può associare un determinato valore di energia (energia interna U), pari alla somma di
energia cinetica e potenziale, tra loro del tutto interconvertibili (1° principio). L’energia cinetica è dovuta al
movimento dei corpi (traslazione, vibrazione, rotazione) ed è legata alla temperatura
Ec=3/2 KT
L’energia potenziale, invece, dipende dalla posizione reciproca dei costituenti del sistema. Fisicamente ha
maggiore energia potenziale l’acqua presente ad alta quota, rispetto alla stessa al suolo; chimicamente
dipende dalla posizione degli elettroni (hanno maggiore energia gli elettroni più distanti dal nucleo) e dalle
forze di attrazione tra molecole.
Esistono 4 diversi tipi di trasformazione: isoterma (a temperatura costante), isocora (volume costante),
isobara (pressione costante), adiabatica (non consentono lo scambio di calore).
1 principio termodinamica (principio di conservazione delll’energia):
Sono possibili passaggi di energia da una forma all’altra oppure dal sistema
all’ambiente. Il calore è una forma di energia che può passare da un corpo
caldo ad un corpo freddo e non viceversa, esso è equivalente al lavoro (1
caloria= 4,186J)
In un sistema chiuso
∆U= Q-W
L’energia interna (U) è data dalla somma di energia cinetica Ec e potenziale
Ep e si conserva, può essere trasferita (mediante un lavoro o il calore), ma mai
creata. Non è possibile costruire una macchina che produca lavoro senza
consumare energia. Q è il calore scambiato tra ambiente e sistema (>0 se passa
dall’ambiente al sistema, negativo se passa dal sistema all’ambiente) W è il
lavoro compiuto (>0 se passa dal sistema all’ambiente, <=0 se passa

© Laura Condorelli 2012 Pag. 2
dall’ambiente al sistema).
Entalpia:
dal primo principio della termodinamica risulta che
(1) Q=∆U +L con ∆U= U2-U1
In una reazione allo stato gassoso, in cui la pressione si conserva, il lavoro è dato da
(2) L= P∆V con ∆V=V2-V1
Sostituendo nella (1) si ha che Q= (U2-U1)+(PV2-PV1), quindi Q= (U2+PV2)-(U1+PV1)
Si definisce entalpia la funzione H=U+PV
La (1) può quindi essere espressa con la Q=∆H,
che significa che il calore assorbito in una
trasformazione a pressione costante equivale alla
variazione di entalpia di un sistema.
Se ∆H<0 reazione esotermica, aumenta l’energia
cinetica e la temperatura aumenta.
Se ∆H>0 la reazione è endotermica, l’energia
cinetica si trasforma in potenziale e la temperatura
diminuisce.
Tutte le reazioni di combustione sono esotermiche.
Viene definito che l’entalpia standard di riferimento=0 (di elementi non legati, alla temperatura di 25°C,
pressione di 1 atm). L’entalpia di formazione diventa dunque la quantità di calore ceduta o acquistata, a
pressione costante per una mole di sostanza a partire dagli elementi di cui è composta.
Entropia:
Inizialmente si era convinti che qualsiasi reazione esotermica fosse
spontanea, convinzione suffragata dalla constatazione che effettivamente
gli stati tendono verso un minor contenuto di energia potenziale
(immagine, in assenza di energia la pallina sta fissa verso il basso). Vi
sono invero reazioni endotermiche che procedono spontaneamente
(solubilizzazione di alcune sostanze, passaggi di stato, sintesi di NO,
NO2, HI), come ad esempio la fusione del ghiaccio, che pur essendo
endotermica è spontanea. Questo succede ogni volta che una reazione, pur
essendo endotermica, porta ad un aumento del grado di disordine. Si
introduce pertanto un’altra funzione termodinamica
∆S=Q/∆T
Un sistema evolve spontaneamente verso contenuti energetici più bassi (diminuzione entalpia) e verso un
maggior grado di disordine (aumento dell’entropia).
∆S>0 -> reazione spontanea
∆S <0 -> reazione non spontanea
Energia libera
Per combinare le due grandezze entalpia ed entropia viene introdotta da Gibbs-Helmotz la funzione
dell’energia libera ∆G, è l’energia che può essere trasformata in lavoro e corrisponde all’entalpia
diminuita del valore dell’energia che non può essere trasformata in lavoro perché vincolata (T∆S)
∆G=∆H-T∆S
© Laura Condorelli 2012 Pag. 3
Con l’introduzione dell’energia libera si può stabilire con esattezza se una trasformazione è spontanea
∆G<0 -> reazione spontanea
∆G=0 -> reazione all’equilibrio
∆G>0 ->reazione non spontanea
Grandezze termodinamica
Tipo Simbolo Definizione Informazioni sulla reazione
Energia
interna
U Quantità di calore scambiato a
V=K (Qv)
∆U=Q>0 ->reazione esotermica
∆U=Q<0 ->reazione endotermica
Entalpia H Quantità di calore scambiata a
P=K (Qp)
∆H<0 -> reazione esotermica
∆H>0 -> reazione endotermica
Entropia S Quantità di calore scambiato a
T=K
∆S=Qrev/T
indica il grado di disordine
∆S>0 -> maggior grado di disordine e
reazione spontanea
Energia libera G Energia disponibile per compiere
un lavoro
∆G=∆H-T∆S
∆G<0 -> reazione spontanea
∆G=0 -> reazione all’equilibrio
∆G>0 ->reazione non spontanea
In sintesi
1° criterio
Sono spontanee
le reazioni che
avvengono con
diminuzione
dell’energia
potenziale, quindi
∆U>0 (reazioni
esotermiche)
2° criterio
Alcune reazioni
endotermiche sono
spontanee (sintesi di
NO, NO2, HI,
dissoluzione NaCl),
poiché avvengono con
aumento dell’entropia
∆S=Q/T>0
3° criterio
Non conta tanto l’energia termica totale (entalpia), ma solo
quella parte di essa che può essere trasformata in lavoro e
non in calore
Etot = E libera (può produrre lavoro) + E vincolata (non
produce lavoro)
∆H = ∆G +T∆S
∆G=∆H-T∆S <0
2 principio termodinamica: è impossibile che l’unico risultato di una trasformazione sia il passaggio di
calore da un corpo freddo ad uno caldo (Clausius), oppure che produca lavoro a spese del calore sottratto ad
un’unica sorgente (Kelvin). L’entropia totale (numero di stati microscopici di un sistema, cioè i modi in cui
atomi e molecole si distribuiscono in posizione e livello energetico) è invariata se la trasformazione è
reversibile, viceversa aumenta (si ricordi che comunque una trasformazione del tutto reversibile è
irrealizzabile, poiché dovrebbe passare attraverso infiniti stati di equilibrio di P e T e quindi sarebbe troppo
lenta)
© Laura Condorelli 2012 Pag. 4
CINETICA CHIMICA Dalla termodinamica sappiamo che le reazioni sono spontanee se procedono con diminuzione di energia
libera ∆G<0.
In chimica però non avvengono tutte le reazioni termodinamicamente possibili, in quanto alcune avvengono
in tempi talmente lunghi da essere praticamente considerate impossibili.
Ad esempio la reazione
C6H12O6+6O2->6CO2+6H2O
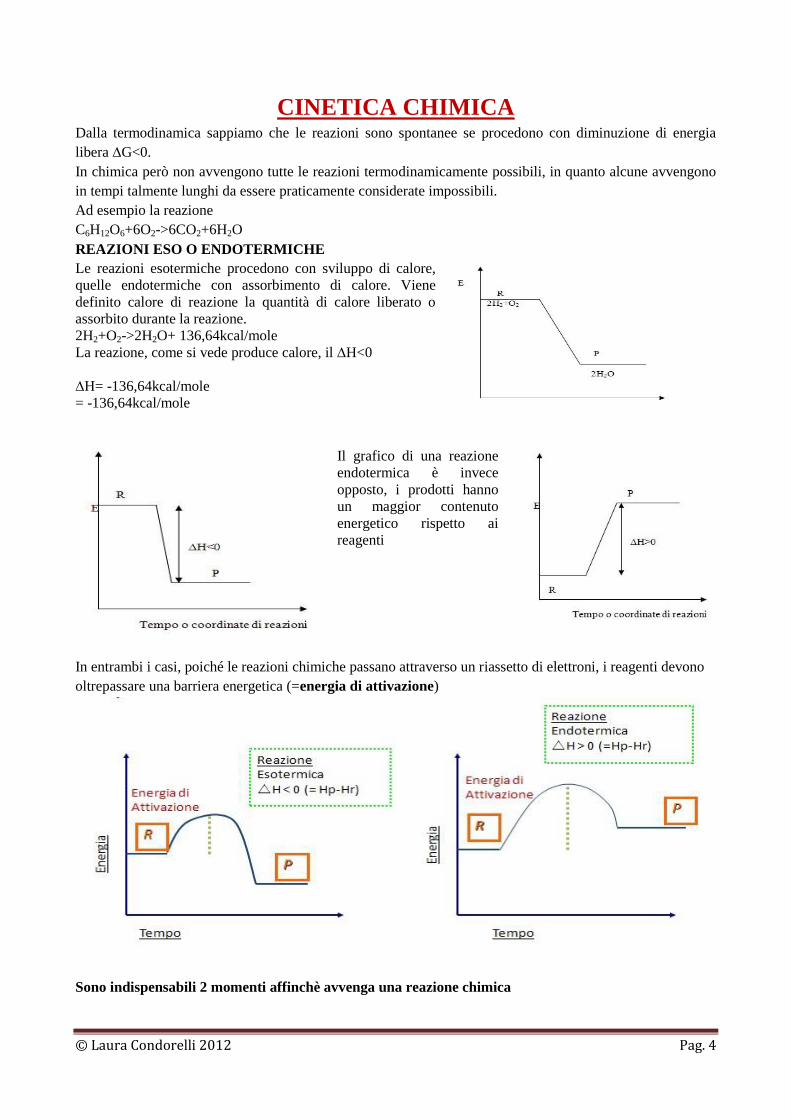
REAZIONI ESO O ENDOTERMICHE
Le reazioni esotermiche procedono con sviluppo di calore,
quelle endotermiche con assorbimento di calore. Viene
definito calore di reazione la quantità di calore liberato o
assorbito durante la reazione.
2H2+O2->2H2O+ 136,64kcal/mole
La reazione, come si vede produce calore, il ∆H<0
∆H= -136,64kcal/mole
= -136,64kcal/mole
Il grafico di una reazione
endotermica è invece
opposto, i prodotti hanno
un maggior contenuto
energetico rispetto ai
reagenti
In entrambi i casi, poiché le reazioni chimiche passano attraverso un riassetto di elettroni, i reagenti devono
oltrepassare una barriera energetica (=energia di attivazione)
Sono indispensabili 2 momenti affinchè avvenga una reazione chimica

© Laura Condorelli 2012 Pag. 5
1) Rottura dei legami esistenti (richiede energia)
2) Formazione nuovi legami (libera energia)
Il primo processo (la rottura dei legami) è quello
lento, comunque endoergonico (richiede energia).
Nel caso della reazione 2H2+O2 ->2H2O
In un primo momento si devono rompere i legami
H-H e O=O
La rottura di questi legami richiede energia ed è
proprio l’energia di attivazione. Pertanto nel grafico
dell’andamento energetico il contenuto di energia
sale, sia per le reazioni eso, che in quelle
endotermiche. Questo è anche il motivo per cui a
temperatura ambiente la reazione 2H2+O2 ->2H2O
non avviene.
2H2
O2
La reazione procede attraverso un complesso
attivato in cui si sono rotti i legami dei reagenti, ma
non si sono ancora formati quelli dei prodotti
(l’energia del complesso attivato è pari all’energia di
attivazione).
stato attivato
Alla fine si formano i legami dei prodotti.
2 H2O
Condizioni per ottenere una reazione
Analisi della reazione H2+I2 ->2HI
Le reazioni normalmente avvengono o in soluzione o allo stato gassoso, pertanto le particelle si muovono e
possiedono energia cinetica. E’ proprio l’energia cinetica dei reagenti che può trasformarsi per dar modo alle
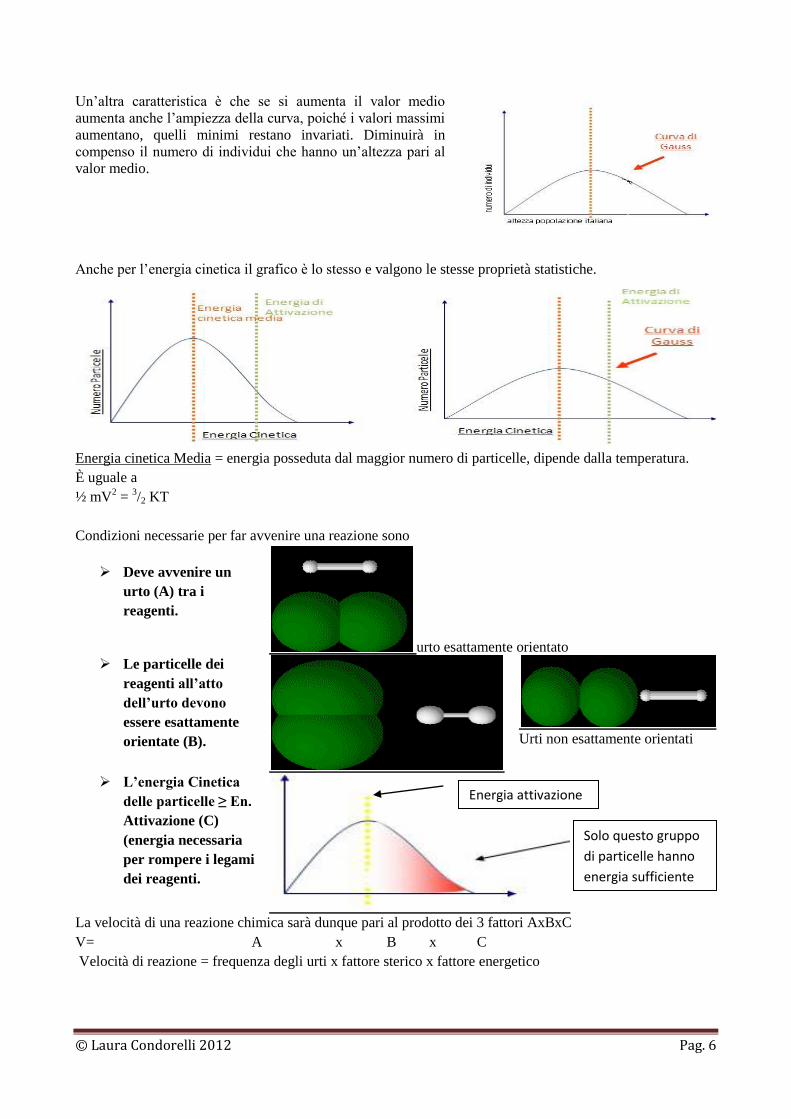
particelle di reagire. In una popolazione di particelle non tutte hanno la stessa energia cinetica. Infatti essa
costituisce un valore medio e viene rappresentata da una curva di Gauss a campana.
Se ad esempio vogliamo misurare l’altezza della popolazione
italiana, vediamo che essa si distribuisce a campana in un
grafico in cui in ordinata figura il numero di individui e in
ascissa l’altezza. Pochi saranno gli individui molto alti o molto
bassi e il maggior numero di individui avrà un’altezza
prossima al valor medio (altezza posseduta dal maggior
numero di individui).
© Laura Condorelli 2012 Pag. 6
Un’altra caratteristica è che se si aumenta il valor medio
aumenta anche l’ampiezza della curva, poiché i valori massimi
aumentano, quelli minimi restano invariati. Diminuirà in
compenso il numero di individui che hanno un’altezza pari al
valor medio.
Anche per l’energia cinetica il grafico è lo stesso e valgono le stesse proprietà statistiche.
Energia cinetica Media = energia posseduta dal maggior numero di particelle, dipende dalla temperatura.
È uguale a
½ mV2 =
3/2 KT
Condizioni necessarie per far avvenire una reazione sono
Deve avvenire un
urto (A) tra i
reagenti.
urto esattamente orientato
Le particelle dei
reagenti all’atto
dell’urto devono
essere esattamente
orientate (B).
Urti non esattamente orientati
L’energia Cinetica
delle particelle ≥ En.
Attivazione (C)
(energia necessaria
per rompere i legami
dei reagenti.
La velocità di una reazione chimica sarà dunque pari al prodotto dei 3 fattori AxBxC
V= A x B x C
Velocità di reazione = frequenza degli urti x fattore sterico x fattore energetico
Solo questo gruppo
di particelle hanno
energia sufficiente
Energia attivazione
© Laura Condorelli 2012 Pag. 7
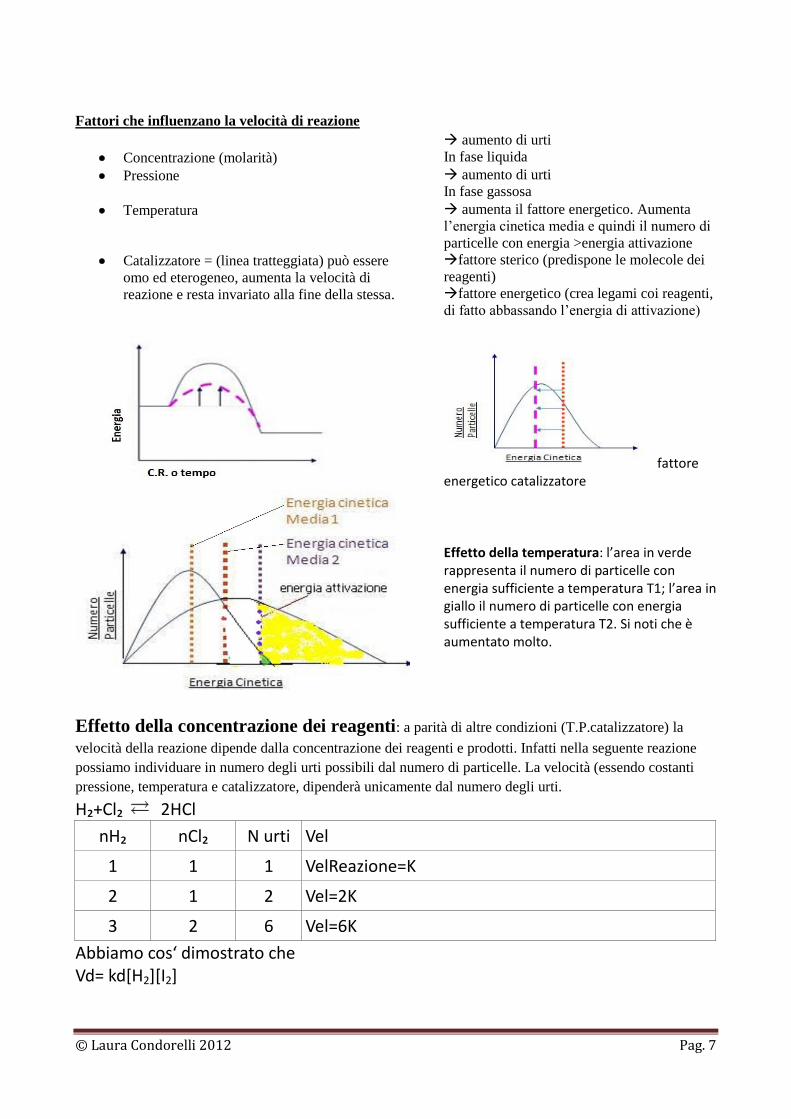
Fattori che influenzano la velocità di reazione
Concentrazione (molarità)
aumento di urti
In fase liquida
Pressione
aumento di urti
In fase gassosa
Temperatura aumenta il fattore energetico. Aumenta
l’energia cinetica media e quindi il numero di
particelle con energia >energia attivazione
Catalizzatore = (linea tratteggiata) può essere
omo ed eterogeneo, aumenta la velocità di
reazione e resta invariato alla fine della stessa.
fattore sterico (predispone le molecole dei
reagenti)
fattore energetico (crea legami coi reagenti,
di fatto abbassando l’energia di attivazione)
fattore
energetico catalizzatore
Effetto della temperatura: l’area in verde rappresenta il numero di particelle con energia sufficiente a temperatura T1; l’area in giallo il numero di particelle con energia sufficiente a temperatura T2. Si noti che è aumentato molto.
Effetto della concentrazione dei reagenti: a parità di altre condizioni (T.P.catalizzatore) la
velocità della reazione dipende dalla concentrazione dei reagenti e prodotti. Infatti nella seguente reazione
possiamo individuare in numero degli urti possibili dal numero di particelle. La velocità (essendo costanti
pressione, temperatura e catalizzatore, dipenderà unicamente dal numero degli urti.
H₂+Cl₂ 2HCl
nH₂ nCl₂ N urti Vel
1 1 1 VelReazione=K
2 1 2 Vel=2K
3 2 6 Vel=6K
Abbiamo cos‘ dimostrato che Vd= kd[H2][I2]

© Laura Condorelli 2012 Pag. 8
Reazioni Reversibili ed equilibri chimici Avvengono nei 2 sensi mA + nB pC + qD .
Dove A,B,C,D sono le specie chimiche di reagenti e
prodotti: m, n, p, q sono i coefficienti stechiometrici.
La velocità di reazione diretta (da reagenti a
prodotti) diminuisce nel tempo, poiché nel tempo i
reagenti si trasformano in prodotti e la loro
concentrazione diminuisce. La velocità inversa (da
prodotti a reagenti) aumenta, poiché all’inizio ho
solo reagenti e la velocità inversa è nulla, ma col
tempo i prodotti si formano e allora cominciano
anche a ri-trasformarsi in reagenti. Si arriva
all’equilibrio chimico, caratterizzato dal fatto che i
reagenti si trasformano in prodotti con la stessa
velocità in cui i prodotti si trasformano in reagenti.
Andamento nel tempo della velocità di reazione
Vd = Vi
Kd [A]m
∙ [B]n = Ki [C]
p ∙ [D]
q
E quindi un equilibrio dinamico, che può essere rappresentato da una costante.
Sia data una generica reazione in cui A e B reagiscono tra loro per dare C e D. Siano m, n, p, q i relativi
coefficienti stechiometrici. Man mano che la reazione procede A e B diminuiscono, C e D aumentano,
diminuisce così la velocità della reazione diretta (da reagenti a prodotti) e aumenta la velocità della reazione
inversa (da prodotti a reagenti). All’equilibrio si avrà che Vd=Vi.
m A+ n B ↔ p C+q D
vd= Kd ∙ [A]m ∙ [B]
n vi= Ki ∙ [C]
p ∙ [D]
q
All’equilibrio vd=vi => Kd ∙ [A]m ∙ [B]
n = Ki ∙ [C]
p ∙ [D]
q
Kd/Ki= (= Ke) =
All’equilibrio si avrà:
Kc=
dove
[ ]= molarità
[ A ]=
Kn=
dove
n= n° moli
Kx=
dove
X= fraz. molare=n/ntot
XA =
Kp=
dove
p= X∙ pressione totale
pA= XA ∙ Ptot
In un solo caso tutte le K hanno lo stesso valore, ovvero quando m+n=p+q. Il valore della costante indica il
verso della reazione, se K>1 la reazione è spostata a destra verso i prodotti, se K<1, al contrario, si avranno
più reagenti che prodotti.

© Laura Condorelli 2012 Pag. 9
Principio di Le Chatelier
Se dall’esterno apporto una modifica ad un sistema in equilibrio il sistema reagisce in modo da annullare gli
effetti prodotti dal cambiamento stesso
Se aumento la temperatura il sistema la abbassa, ovvero nelle reazioni esotermiche l’equilibrio si
sposta verso i reagenti, in quelle endotermiche verso i prodotti (viceversa se abbasso la temperatura)
ΔH>0 (reazioni endotermiche)
2H2O +calore 2H2+ O2
Dim temp eq a sinistra
Aum temp ed a destra
ΔH<0 (reazioni esotermiche)
2NO2 N2O4+calore
Dim temp eq a destra
Aum temp ed a sinistra
Se aumento la pressione l’equilibrio si sposta nella direzione in cui trovo un minor numero di moli
Reazioni che procedono con aumento del numero di
moli
2H2O 2H2+ O2
2moli/P< 3 moli/P>
Dim P eq a destra
Aum P ed a sinistra
Reazioni che procedono con diminuzione del
numero di moli
2NO2 N2O4+calore
2 moli/P> 1mole/P<
Dim P eq a sinistra
Aum P ed a destra
Se diminuisco la concentrazione di uno o più reagenti l’equilibrio si sposta verso i reagenti
(viveversa se l’aumento)
Se diminuisco la concentrazione di uno o più prodotti l’equilibrio si sposta verso i prodotti
(viceversa se l’aumento)
L’uso del catalizzatore non ha alcun effetto sull’equilibrio
Processo Haber (sintesi dell’ammoniaca)
(esempio di applicazione del principio di le Chatelier)
• È una reazione di sintesi
N2+3H2 -->2NH3 + calore
• Procede con diminuzione del numero di moli ed è una reazione esotermica
• Va quindi eseguita a bassa temperatura e ad alta pressione
EQUILIBRI IN SOLUZIONE
1 DISSOCIAZIONE DELL’ACQUA E PH
L’acqua in soluzione acquosa si dissocia pochissimo, secondo la reazione
H2O ↔ H+ + OH -
In realtà, poiché lo ione H+ non resiste in acqua, combinandosi subito con una molecola di H2O a dare H3O
+, la reazione dovrebbe essere scritta così
2 H2O ↔ H3O+ + OH - Noi continueremo a scriverla in modo semplificato,
intendendo con H+ lo ione H3O+
Ke =
Anche per la dissociazione dell’acqua, dunque, è possibile trovare una costante di concentrazione. Dal momento che l’acqua è poco dissociata si può inglobare il suo valore nella K

© Laura Condorelli 2012 Pag. 10
Ke ∙ [ H2O ] = KW= [H+] ∙ [OH -] = 1∙ 10 -14 La costante dell’acqua (Kw) o costante del prodotto ionico dell’acqua vale 1 ∙ 10 – 14 ed è costante
[H+] =[ OH -]
[H+]2 = 1∙ 10 -14 [H+] = 1∙ 10 -7
[OH-]2 = 1∙ 10 -14 [OH-] = 1∙ 10 -7
Inoltre le concentrazioni di [H+] e di [ OH -] devono necessariamente essere uguali , quindi .
pH = - log [H+] pOH= -log[OH-] pH + pOH = 14
In una soluzione neutra le concentrazioni di [H+] =[ OH
-] e [H+
] = 1∙ 10 -7
e[OH-] = 1∙ 10
-7 .
Si introduce la funzione di pH= logaritmo negativo della concentrazione di ioni H+
, come di pOH ==
logaritmo negativo della concentrazione di ioni OH -.
In una soluzione neutra pH = 7 e p OH = 7. E evidente che pH +pOH = 14 (sempre).
Soluzione neutra
[H+] = 1∙ 10 -7 [OH-] = 1∙ 10 -7 pH=7 OH=7
Soluzione acida
10-14
Soluzione basica
10-14
[H+] ∙[ OH -] = 1∙ 10 -14 pH+ pOH = 14
2.SOLUZIONI ACIDE, ACIDI FORTI E DEBOLI
H2O ↔ H+ + OH- Alla reazione di dissociazione dell’acqua si deve aggiungere la reazione di dissociazione acida
HA ↔ H+ + A- In una soluzione acida, infatti, il soluto è un acido che si dissocia in soluzione acquosa.
Ka =
Come per tutte le reazioni di equilibrio, anche in questo caso si può definire una costante di dissociazione acida. Se Ka>1, allora l’acido si troverà prevalentemente in forma dissociata (o ionica) e si dirà forte, se Ka<1 allora prevale la forma indissociata o molecolare e l’acido si dirà debole.
ACIDI FORTI ACIDI DEBOLI
BINARI
HCl, HBr, HI
H2S, HF, HCN
TERNARI nO≥ nH +2
H2SO4, HClO3, HNO3, HClO4 nO<nH+2
H2CO3, H3BO3,H3PO3, H3PO4, HNO2, H2SO3,
HClO, HClO2
Tutti gli acidi organici
RCOOH
pH= - log Ma [H+] = Ma Se ho il pH e cerco [H+] [H+] = 10 -pH
pH= -log (M= molarità)
[H+ ] = [H+] = 10 -pH

© Laura Condorelli 2012 Pag. 11
3.SOLUZIONI BASICHE, BASI FORTI E DEBOLI
H2O ↔ H+ + OH- Alla reazione di dissociazione dell’acqua si deve aggiungere la reazione di dissociazione basica
BOH ↔ B+ + OH- In una soluzione basica, infatti, il soluto è un idrossido che si dissocia in soluzione acquosa (basi di Arrhenius).
Kb =
Come per tutte le reazioni di equilibrio, anche in questo caso si può definire una costante di dissociazione basica. Se Kb>1, allora la base si troverà prevalentemente in forma dissociata (o ionica) e si dirà forte, se Kb<1 allora prevale la forma indissociata o molecolare e la base si dirà debole.
BASI FORTI BASI DEBOLI
Tutti gli idrossidi degli elementi dei gruppi 1A e 2A
ad eccezione di Be(OH)2
Tutti gli altri, compreso NH4OH (NH3)
pOH= - log Mb [OH-] = Cb Se ho pOH e cerco [OH-] [OH-]= 10 -pOH
pOH= -log (M= molarità)
[OH-] = [OH-]= 10 -pOH
Sono definiti anche pKa= -log Ka e pKb= -log Kb. Pka e pKb hanno un valore tanto maggiore quanto più gli
acidi e le basi sono deboli.
Definizione di acido e base (Arrhenius) Acide sono tutte quelle sostanze che in soluzione acquosa
si dissociano liberando ioni H+.
Possono essere monoprotici, come HCl, oppure di protici,
come H2SO4, o triprotici, come H3PO3.
HCl↔H+ +Cl
-
H2SO4↔ H+ +HSO4
-↔2 H
+ + SO4
- -
H3PO3↔ H+ +H2PO3
-↔ 2H
+ +HPO3
- -↔3H
++HPO
3-
-
La prima reazione di dissociazione acida è sempre la
più forte, prenderemo in considerazione solo quella.
Basi sono tutte quelle sostanze che in soluzione
acquosa liberano ioni OH-.
Anche in questo caso le basi possono essere
mono, bi o anche pentavalenti.
NaOH↔Na+ + OH
–
Ca(OH)2↔CaOH+ + OH
–↔Ca
++ + 2OH
–
Bi(OH)5↔Bi(OH)4+ + OH
–↔Bi (OH)3
++ +
2OH ––
↔Bi (OH)2+++
+ 3OH ––
↔Bi
OH++++
+ 4OH –↔Bi
5+ + 5OH
–
Anche in questo caso si prenderà in esame
solo la prima reazione di dissociazione
basica.
© Laura Condorelli 2012 Pag. 12
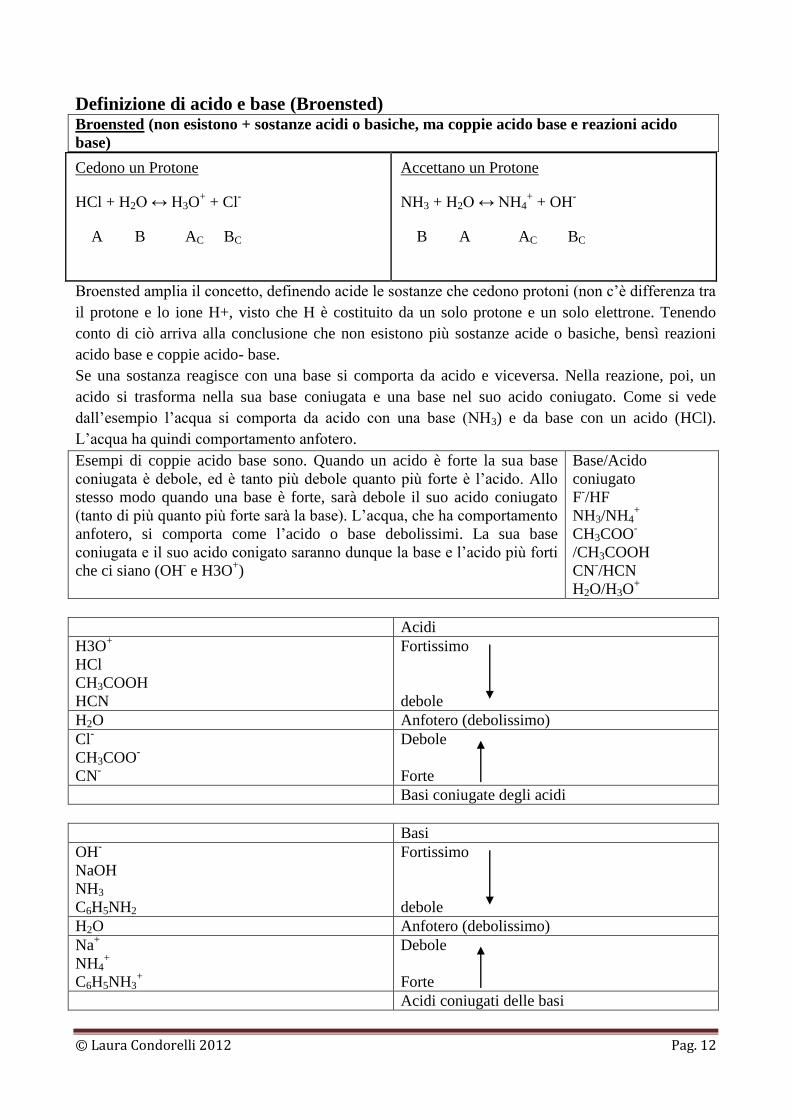
Definizione di acido e base (Broensted) Broensted (non esistono + sostanze acidi o basiche, ma coppie acido base e reazioni acido
base)
Cedono un Protone
HCl + H2O ↔ H3O+ + Cl
-
A B AC BC
Accettano un Protone
NH3 + H2O ↔ NH4+ + OH
-
B A AC BC
Broensted amplia il concetto, definendo acide le sostanze che cedono protoni (non c’è differenza tra
il protone e lo ione H+, visto che H è costituito da un solo protone e un solo elettrone. Tenendo
conto di ciò arriva alla conclusione che non esistono più sostanze acide o basiche, bensì reazioni
acido base e coppie acido- base.
Se una sostanza reagisce con una base si comporta da acido e viceversa. Nella reazione, poi, un
acido si trasforma nella sua base coniugata e una base nel suo acido coniugato. Come si vede
dall’esempio l’acqua si comporta da acido con una base (NH3) e da base con un acido (HCl).
L’acqua ha quindi comportamento anfotero.
Esempi di coppie acido base sono. Quando un acido è forte la sua base
coniugata è debole, ed è tanto più debole quanto più forte è l’acido. Allo
stesso modo quando una base è forte, sarà debole il suo acido coniugato
(tanto di più quanto più forte sarà la base). L’acqua, che ha comportamento
anfotero, si comporta come l’acido o base debolissimi. La sua base
coniugata e il suo acido conigato saranno dunque la base e l’acido più forti
che ci siano (OH- e H3O
+)
Base/Acido
coniugato
F-/HF
NH3/NH4+
CH3COO-
/CH3COOH
CN-/HCN
H2O/H3O+
Acidi
H3O+
HCl
CH3COOH
HCN
Fortissimo
debole
H2O Anfotero (debolissimo)
Cl-
CH3COO-
CN-
Debole
Forte
Basi coniugate degli acidi
Basi
OH-
NaOH
NH3
C6H5NH2
Fortissimo
debole
H2O Anfotero (debolissimo)
Na+
NH4+
C6H5NH3+
Debole
Forte
Acidi coniugati delle basi
© Laura Condorelli 2012 Pag. 13
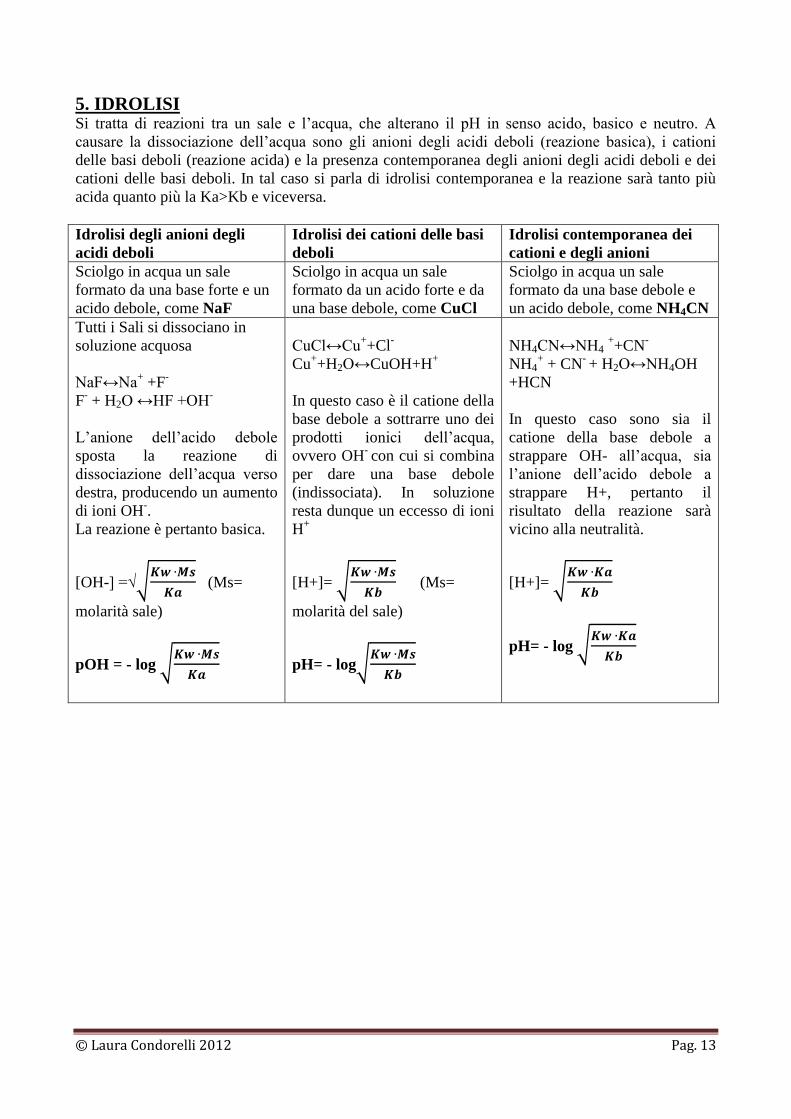
5. IDROLISI Si tratta di reazioni tra un sale e l’acqua, che alterano il pH in senso acido, basico e neutro. A
causare la dissociazione dell’acqua sono gli anioni degli acidi deboli (reazione basica), i cationi
delle basi deboli (reazione acida) e la presenza contemporanea degli anioni degli acidi deboli e dei
cationi delle basi deboli. In tal caso si parla di idrolisi contemporanea e la reazione sarà tanto più
acida quanto più la Ka>Kb e viceversa.
Idrolisi degli anioni degli
acidi deboli
Idrolisi dei cationi delle basi
deboli
Idrolisi contemporanea dei
cationi e degli anioni
Sciolgo in acqua un sale
formato da una base forte e un
acido debole, come NaF
Sciolgo in acqua un sale
formato da un acido forte e da
una base debole, come CuCl
Sciolgo in acqua un sale
formato da una base debole e
un acido debole, come NH4CN
Tutti i Sali si dissociano in
soluzione acquosa
NaF↔Na+ +F
-
F- + H2O ↔HF +OH
-
L’anione dell’acido debole
sposta la reazione di
dissociazione dell’acqua verso
destra, producendo un aumento
di ioni OH-.
La reazione è pertanto basica.
[OH-] =√
(Ms=
molarità sale)
pOH = - log
CuCl↔Cu++Cl
-
Cu++H2O↔CuOH+H
+
In questo caso è il catione della
base debole a sottrarre uno dei
prodotti ionici dell’acqua,
ovvero OH- con cui si combina
per dare una base debole
(indissociata). In soluzione
resta dunque un eccesso di ioni
H+
[H+]=
(Ms=
molarità del sale)
pH= - log
NH4CN↔NH4 ++CN
-
NH4+ + CN
- + H2O↔NH4OH
+HCN
In questo caso sono sia il
catione della base debole a
strappare OH- all’acqua, sia
l’anione dell’acido debole a
strappare H+, pertanto il
risultato della reazione sarà
vicino alla neutralità.
[H+]=
pH= - log
© Laura Condorelli 2012 Pag. 14
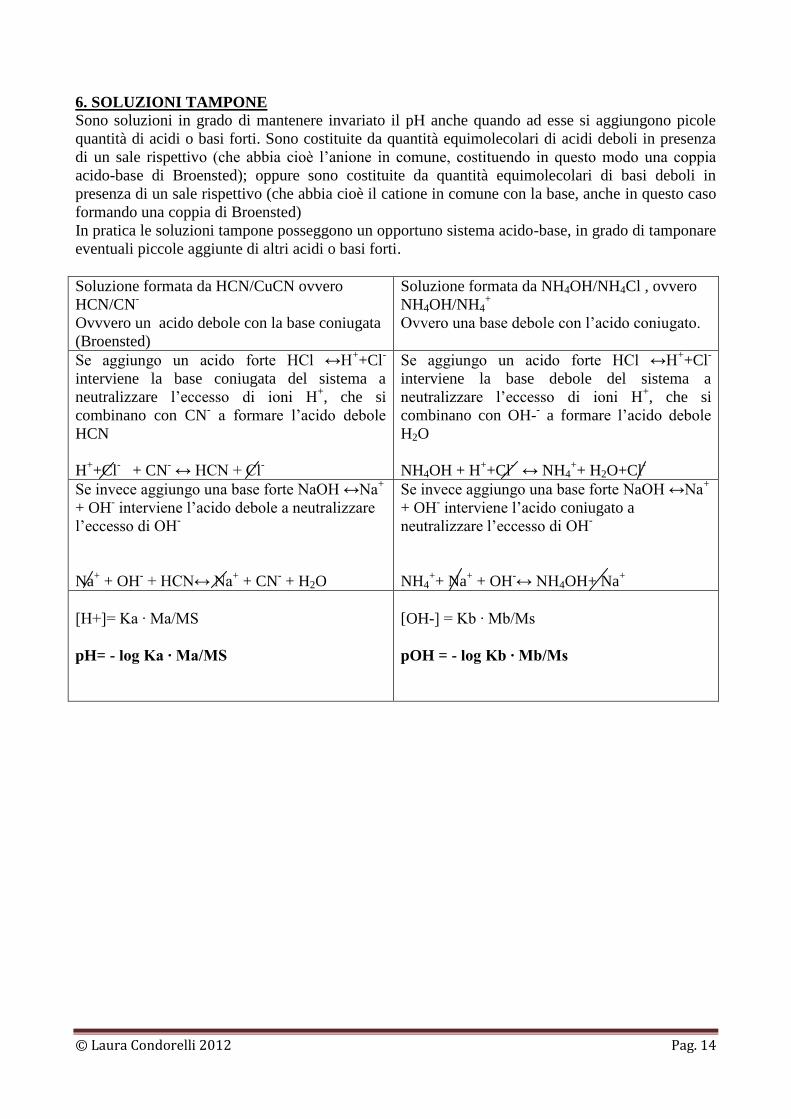
6. SOLUZIONI TAMPONE
Sono soluzioni in grado di mantenere invariato il pH anche quando ad esse si aggiungono picole
quantità di acidi o basi forti. Sono costituite da quantità equimolecolari di acidi deboli in presenza
di un sale rispettivo (che abbia cioè l’anione in comune, costituendo in questo modo una coppia
acido-base di Broensted); oppure sono costituite da quantità equimolecolari di basi deboli in
presenza di un sale rispettivo (che abbia cioè il catione in comune con la base, anche in questo caso
formando una coppia di Broensted)
In pratica le soluzioni tampone posseggono un opportuno sistema acido-base, in grado di tamponare
eventuali piccole aggiunte di altri acidi o basi forti.
Soluzione formata da HCN/CuCN ovvero
HCN/CN-
Ovvvero un acido debole con la base coniugata
(Broensted)
Soluzione formata da NH4OH/NH4Cl , ovvero
NH4OH/NH4+
Ovvero una base debole con l’acido coniugato.
Se aggiungo un acido forte HCl ↔H++Cl
-
interviene la base coniugata del sistema a
neutralizzare l’eccesso di ioni H+, che si
combinano con CN- a formare l’acido debole
HCN
H++Cl
- + CN
- ↔ HCN + Cl
-
Se aggiungo un acido forte HCl ↔H++Cl
-
interviene la base debole del sistema a
neutralizzare l’eccesso di ioni H+, che si
combinano con OH-- a formare l’acido debole
H2O
NH4OH + H++Cl
- ↔ NH4
++ H2O+Cl
-
Se invece aggiungo una base forte NaOH ↔Na+
+ OH- interviene l’acido debole a neutralizzare
l’eccesso di OH-
Na+ + OH
- + HCN↔ Na
+ + CN
- + H2O
Se invece aggiungo una base forte NaOH ↔Na+
+ OH- interviene l’acido coniugato a
neutralizzare l’eccesso di OH-
NH4++ Na
+ + OH
-↔ NH4OH+ Na
+
[H+]= Ka ∙ Ma/MS
pH= - log Ka ∙ Ma/MS
[OH-] = Kb ∙ Mb/Ms
pOH = - log Kb ∙ Mb/Ms





























