Embed Size (px)
Citation preview

CHIMICA FARMACEUTICA E TOSSICOLOGICA I
Corso M-Z per Farmacia (Prof. G. Ortar)
Appunti sulla Parte Generale

INTRODUZIONE
Generalità sui farmaci
La chimica farmaceutica si interessa della scoperta, progettazione, identificazione e prepa-razione dei composti biologicamente attivi, dello studio delle loro proprietà, del loro metabolismo, dell’ interpretazione del loro meccanismo d’ azione a livello molecolare, della costruzione delle re-lazioni struttura-attività. E’ una disciplina fondata sulla chimica che coinvolge anche numerosi aspetti delle scienze biologiche, farmacologiche e mediche e che applica i principi di tali scienze alla creazione di conoscenze finalizzate all’ introduzione di nuovi utili farmaci.
I farmaci sono tutte quelle sostanze di natura organica, metallorganica od inorganica, ottenute per via estrattiva, fermentativa o sintetica che vengono impiegate per la diagnosi, la prevenzione e il trattamento delle malattie umane o animali.
Più in dettaglio i farmaci possono essere impiegati per uno dei seguenti scopi: ●Somministrazione di elementi di cui l’ organismo è carente (vitamine, sali, ormoni, idrolizzati
proteici); ●Prevenzione di una infezione (sieri, vaccini); ●Trattamento di una infezione (chemioterapici); ●Blocco temporaneo di una funzione fisiologica (anestetici locali, contraccettivi); ●Correzione di una funzione fisiologica alterata (antiipertensivi, antiaritmici); ●Detossicazione dell’ organismo (antidoti); ●Diagnosi della funzionalità o dello stato di mantenimento di un organo (tests di funzionalità,
mezzi di contrasto). Le principali classi di farmaci sono: ●Farmaci agenti sul sistema nervoso; ●Agenti farmacodinamici (farmaci che agiscono sui processi dinamici dell’ organismo, ad esempio
sistema cardiovascolare, apparato respiratorio e gastrointestinale); ●Chemioterapici; ●Farmaci agenti su funzioni endocrine e su malattie metaboliche (ad esempio antiinfiammatori,
antiartritici, antidiabetici). La grande maggioranza dei farmaci impiegati attualmente è costituita da composti organici e
loro sali. Alcuni sono prodotti dal metaboli-smo di particolari microrganismi e vengono isolati dai brodi di coltura nei quali questi mi-crorganismi crescono (antibiotici); altri sono isolati da organi animali, da parti di piante, da prodotti di queste. La maggior parte dei farmaci però viene ottenuta per sintesi, totale o parziale (questo ultimo termine si riferisce a modificazioni chimiche di sostanze di origine naturale). Con riferimento alla loro origine è mostrata a fianco la distribuzione percentuale dei farmaci essenziali elencati dalla Organiz-zazione Mondiale della Sanità (OMS) (i far-maci essenziali, circa 300, sono i farmaci che soddisfano le esigenze sanitarie della maggior parte della popolazione e che dovrebbero per-ciò essere sempre disponibili in adeguata quantità e nelle opportune forme di dosaggio). Negli ultimi 15 anni sono emerse come impor-tante fonte di nuovi agenti terapeutici le bio-
SIERI 2%
VACCINI 5%
MICROORGANISMI 7%
SINTESI 52%
ANIMALI 6%
MINERALI 8%
SINTESI PARZIALE 11%
PIANTE 8%
ORIGINE DEI FARMACI ESSENZIALI DELL' OMS
1
tecnologie e circa una cinquantina sono i farmaci approvati ottenuti in questo modo. I farmaci, intesi come principi attivi, vengono commercializzati in forme adeguate alla som-
ministrazione (forme farmaceutiche) e nelle quantità necessarie a dare una risposta farmacologica (dose terapeutica). La forma farmaceutica nel suo assieme (principio attivo + ingredienti inerti con funzioni varie, di riempimento, lubrificante, legante, conservante, antiossidante) costituisce il
medicamento. Una forma farmaceutica può contenere più principi attivi (preparato in associazione).
50
50
DEDLIT = Una indicazione piuttosto sommaria del margine di sicurezza di un farmaco
è fornita dal suo indice terapeutico IT, definito come il rapporto tra dose letale nel 50% degli animali da esperimento e dose efficace nel 50% degli individui.
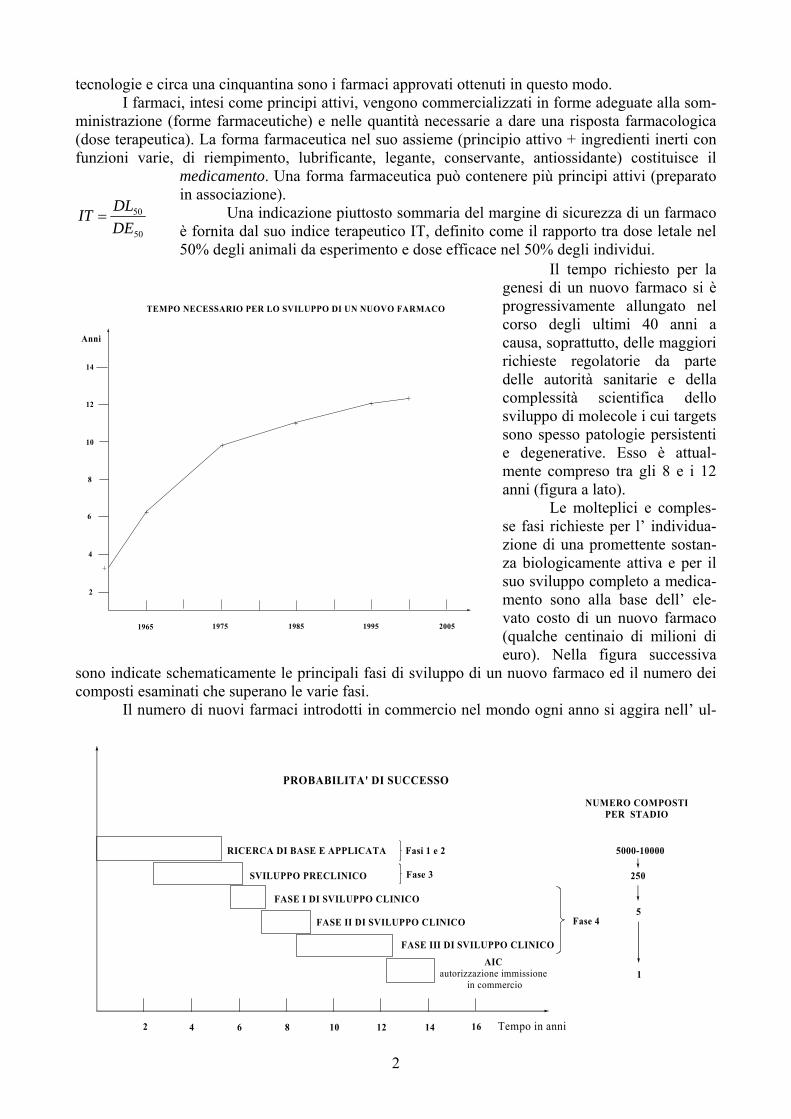
Il tempo richiesto per la genesi di un nuovo farmaco si è progressivamente allungato nel corso degli ultimi 40 anni a causa, soprattutto, delle maggiori richieste regolatorie da parte delle autorità sanitarie e della complessità scientifica dello sviluppo di molecole i cui targets sono spesso patologie persistenti e degenerative. Esso è attual-mente compreso tra gli 8 e i 12 anni (figura a lato). Le molteplici e comples-se fasi richieste per l’ individua-zione di una promettente sostan-za biologicamente attiva e per il suo sviluppo completo a medica-mento sono alla base dell’ ele-vato costo di un nuovo farmaco (qualche centinaio di milioni di euro). Nella figura successiva
sono indicate schematicamente le principali fasi di sviluppo di un nuovo farmaco ed il numero dei composti esaminati che superano le varie fasi.
Anni
TEMPO NECESSARIO PER LO SVILUPPO DI UN NUOVO FARMACO
2
4
6
8
10
12
14
1965 1975 1985 1995 2005
+
+
+
+
++
Il numero di nuovi farmaci introdotti in commercio nel mondo ogni anno si aggira nell’ ul-
2 4 6 8 10 12 14 16
RICERCA DI BASE E APPLICATA
SVILUPPO PRECLINICO
FASE I DI SVILUPPO CLINICO
FASE II DI SVILUPPO CLINICO
FASE III DI SVILUPPO CLINICO
AICautorizzazione immissione
in commercio
Fasi 1 e 2
Fase 3
Fase 4
NUMERO COMPOSTIPER STADIO
5000-10000
250
1
5
PROBABILITA' DI SUCCESSO
Tempo in anni
2

timo quinquennio mediamente attorno ai 30. Il numero di farmaci e di loro associazioni in vendita in Italia (2005) è: ●sostanze singole: 1375 (sali esclusi); ●associazioni: 718. Quello delle specialita' medicinali è: ●confezioni a base di una sola sostanza: 7145; ●a base di piu' sostanze: 1412.
Individuazione di nuovi farmaci e loro sviluppo a medicamenti Di norma, i farmaci di utilità clinica non vengono ‘scoperti’. Ciò che viene invece inizialmente individuato è il cosiddetto ‘lead compound’, o composto guida. Il lead compound è una molecola prototipo che possiede una attività biologica interessante, ma anche quasi sempre caratteristiche indesiderate quali bassa potenza, eccessiva tossicità, problemi di stabilità chimica e/o
metabolica, di assorbimento, eccetera. Tali difetti fanno si che raramente un lead approdi alla approvazione e quindi alla commercializzazione. Esistono, ovviamente, delle eccezioni. Una di queste è rappresentata dal paclitaxel (chiamato precedentemente taxolo), approvato nel 1993, laddove l’ indagine iniziale sull’ estratto della corteccia del Tasso del Pacifico (Taxus brevifolia) che evidenziò una azione citotossica, dovuta al paclitaxel appunto, risale al 1962, mentre la delucidazione della struttura del principio attivo è del 1972.
O
H
OH
OCOCH3
O
O O
CH3COO
H3C
HO
O
OH
OO
NH
Paclitaxel
Le vie più comuni per individuare un lead compound sono le seguenti (da tenere presente che diversi prodotti biologicamente attivi sono stati anche scoperti per caso, ad esempio le penicilline).
a) Miglioramento di farmaci esistenti
b) Screening sistematico
Osservazioni cliniche degli effetti collaterali dei farmacic) Sfruttamento di osservazioni biologiche
Isolamento e sintesi di sostanze naturali
d) Indagini sui meccanismi biochimici e patobiochimici nell' organismo
e) Indagini riguardanti il metabolismo dei farmaci [Sintesi di profarmaci (prodrugs) o di farmaci labili (soft drugs)]
L’ utilizzo di farmaci già noti come modelli (approccio a) per individuare altre molecole attive è una pratica largamente utilizzata a livello industriale. Questo approccio manca di originalità ed i prodotti così ottenuti rientrano nella categoria dei cosiddetti ‘me-too products’ che permettono a più ditte farmaceutiche di accedere, con limitati investimenti, a settori particolarmente remunerativi. Sebbene molto criticata, questa strategia ha anche una sua validità dal punto di vista
3

scientifico, in quanto dalla somma di piccoli e graduali miglioramenti può discendere alla fine una innovazione sostanziale. Lo scrrening sistematico comprende lo screening estensivo e quello casuale (random). Nel primo caso, l’ indagine è condotta su una serie relativamente piccola di molecole chimicamente sofisticate e consiste in un’ ampia varietà di tests biologici e farmacologici. Nel secondo caso, il numero di molecole saggiate è molto ampio, mentre il tipo di attività cercata è unico. Queste due varianti sono state combinate a partire dagli anni '80 come conseguenza della robotizzazione e delle miniaturizzazione delle metodologie di screening (high-throughput screening, screening ad alta capacità) che consentono di valutare simultaneamente centinaia di composti in decine di tests biologici. Il problema di porre la sintesi dei composti al passo con le nuove potenzialità degli screenings è stato in parte risolto mediante la sintesi combinatoriale. L’ idea base della chimica combinatoriale è quella di costruire ampie collezioni di composti contenenti un alto numero di varianti di strutture fondamentali, procedere alla valutazione della loro attività biologica, isolare ed identificare il/i prodotti attivi. La figura seguente illustra il principio della sintesi combinatoriale.
Nella sintesi classica una molecola A viene fatta reagire con una molecola B per dare un prodotto AB il quale, per reazione con C, fornisce il prodotto ABC, e così via. Nella sintesi combinatoriale invece una serie di molecole di tipo A (A1, A2, A3…An) viene fatta reagire con una serie di molecole di tipo B (B1, B2, B3…Bn’) con ottenimento di tutte le possibili combinazioni tra A e B per un totale di nn’ composti. La reazione successiva con una serie di reagenti di tipo C (C1, C2, C3…Cm) porta ad.nn’m composti del tipo ABC, e così via. Il numero dei prodotti totali ottenibili N dipende dal numero di reagenti disponibili in ogni
passaggio. Se ad ogni passaggio si usa sempre lo stesso numero di reagenti n (nell’ esempio 10) ed s è il numero dei passaggi (nell’ esempio 4), N sarà = ns. (nell’ esempio 104).
A B C DAB
A1-n
ABC ABCD
B1-n' AiB i C1-m AiB iCi D1-m' AiB iCiD i
Sintesi classica
Sintesi combinatoriale
++
+ +
+
+ ===
= ==
1 1 1 1 1 1 1
n n' nxn' m nxn'xm m' nxn'xmxm'
10 10210 10 103 10410
Per quanto riguarda l’ approccio c), esso comprende le osservazioni cliniche degli effetti collaterali dei farmaci e l’ isolamento (e l’ eventuale sintesi) di sostanze naturali. La storia della ri-cerca farmaceutica è ricca di esempi in cui gli effetti collaterali osservati durante la sperimentazio-
ne clinica hanno suggerito mani-polazioni della struttura originale allo scopo di produrre nuove mo-lecole nelle quali un effetto se-condario è stato amplificato, fino a diventare quello principale. Uno di tali esempi è costituito dalla sulfanilamide, il precursore di tut-ti i sulfamidici; la sostituzione di un idrogeno dello azoto solfon-ammidico con un eterociclo ha notevolmente aumentato la poten-za antibatterica del capostipite, eliminando nello stesso tempo le azioni secondarie diuretica e ipo-glicemizzante. Tali azioni sono
NH2CH3CONH SO2S
N N
CONHBuNHH3C SO2
OO
H
H Cl
NH2SO2
N
NS
NH2SO2H2N
ETEROCICLONHSO2H2N
Tolbutamide(Ipoglicemizzante)
Sulfanilamide (Antibatterico)Azioni secondarie: Diuretica Ipoglicemizzante Idroclortiazide
(Diuretico)
Acetazolamide (Diuretico)
Sulfamidico(Antibatterico)
4

state invece evidenziate attraverso altri tipi di modifiche che hanno portato alla introduzione in terapia degli ipoglicemizzanti a struttura solfonilureica e dei diuretici solfonammidici. I prodotti di origine naturale, in particolare quelli provenienti dal mondo vegetale, sono la più antica fonte di farmaci. Punto di partenza per l’ isolamento, la delucidazione strutturale e la sintesi di sostanze naturali è l’ osservazione, casuale o derivante da uno screening sistematico, di una attività interessante nell’ estratto di una parte di pianta, di un organo animale, di una coltura batterica o fungina. La ricerca in questo settore può anche essere in qualche modo guidata dalle indicazioni della medicina tradizionale (etnofarmacologia). Gli approcci d) ed e) rappresentano due tipi di strategie razionali per l’ individuazione di nuovi lead. Secondo il modello ‘medico’, le malattie sono la conseguenza della deregolazione di una normale funzione dell’ organismo che può essere definita in termini biochimici ed influenzata da opportuni interventi chimici o fisici. Una volta che il processo biochimico deregolato sia stato individuato e caratterizzato nei dettagli molecolari, il lead può essere rappresentato da una o più sostanze endogene che intervengono nella deregolazione (ad esempio neurotrasmettitori, ormoni, fattori di crescita, citochine, substrati enzimatici vari). Per quanto concerne il punto e), i farmaci subiscono comunemente trasformazioni chimiche nell’ organismo ad opera di sistemi enzimatici. Talvolta, l’ azione farmacologica del farmaco è dovuta in parte o del tutto ad uno o più di questi prodotti di biotrasformazione. Nell’ ambito degli studi sul metabolismo dei farmaci rientra anche la progettazione di profarmaci (prodrugs) e di farmaci labili (soft drugs). I primi sono sostanze inattive che vengono attivate ‘in vivo’, mentre i secondi sono sostanze attive che subiscono ‘in vivo’ una inattivazione metabolica prevedibile e controllata. Una volta che sia stato identificato un nuovo lead, il passo successivo consiste nell’ otti-mizzarne le caratteristiche farmacologiche attraverso opportune modifiche della sua struttura. Gli
obiettivi principali del processo di ottimizzazione sono: aumento della potenza, miglioramento della spe-cificità d’ azione, riduzione della tossicità e miglioramento delle pro-prietà farmacocinetiche del lead. Le modifiche strutturali vengono guidate da criteri di scelta di carat-tere qualitativo e quantitativo. I dettagli di tali strategie verranno esaminati successivamente nel ca-pitolo delle relazioni struttura-atti-vità.
Lead compound
Ottimizzazionedella struttura Ricerca di base
ed applicata
Sostanza attiva adatta adessere sviluppata a medicamento
Elaborazionefarmacocinetica,farmacodinamica
e tossicologica
Elaborazione chimica,chimico-analitica
e galenicaSviluppo esplorativo
(preclinico)
Medicamento
Sperimentazioneclinica
Sviluppo di un metododi produzione industriale
Prodotto prontoper il mercato
Sviluppocompleto
Il processo di ottimizzazio-ne conclude la fase della ricerca di base ed applicata. Ad esso fanno seguito una serie di operazioni di varia natura che costituiscono la fase di sviluppo preclinico (figura a lato). Per elaborazione farmaco-cinetica si intende la definizione dei dati di assorbimento, distribu-zione, metabolismo ed escrezione dei principi attivi ottimizzati; la elaborazione farmacodinamica consiste nel completamento del quadro del loro profilo di attività; l’ elaborazione tossicologica ri-
5

guarda la definizione delle loro tossicità acuta, subacuta e cronica. Nell’ elaborazione chimica viene messa a punto una procedura di preparazione dettagliata, ben riproducibile, il più possibile economica e facilmente trasferibile su scala industriale dei composti ottimizzati mentre nella elaborazione chimico-analitica vengono messi a punto i saggi di identificazione e di determinazione della purezza. La elaborazione galenica consiste nell’ elaborazione dei principi attivi in una o più formulazioni. La sperimentazione clinica condotta sull’ uomo per confermare le proprietà finora definite negli animali da esperimento si articola in 3 fasi (fasi I-III) che differiscono tra loro per il numero dei soggetti coinvolti, la durata ed il grado di approfondimento della sperimentazione. Dopo la registrazione e l’ introduzione in commercio, il farmaco viene ulteriormente controllato in una fase IV (farmacovigilanza).
Classificazione dei farmaci I farmaci possono essere classificati in base a vari criteri: struttura chimica, azione farmacologica, impiego terapeutico, meccanismo d’ azione. La classificazione raccomandata dalla OMS e curata in Italia dal Ministero della Salute è la classificazione ATC (Anatomica-Terapeutica-Chimica). Il sistema base divide i farmaci in 14 Gruppi Anatomici Principali (1° livello) , ognuno dei quali viene poi articolato in Gruppi Terapeutici Principali, Sottogruppi Terapeutici, Sottogruppi Chimico/Terapeutici e Sottogruppi Chimici per un totale di 5 livelli gerarchici come mostrato di seguito. Ad ogni farmaco è associato un codice alfanumerico. I testi di Chimica Farmaceutica impiegano generalmente una classificazione simile a quella ATC, anche se meno capillare.
GRUPPI ANATOMICI PRINCIPALI Antimicrobici generali per uso sistemico Apparato gastrointestinale e metabolismo Dermatologici Farmaci antineoplastici ed immunomodulatori Farmaci antiparassitari, insetticidi e repellenti Organi di senso Preparati ormonali sistemici, esclusi gli ormoni sessuali
Sangue ed organi emopoietici Sistema cardiovascolare Sistema genito-urinario ed ormoni sessuali Sistema muscolo-scheletrico Sistema nervoso centrale Sistema respiratorio Vari
Classificazione ATC
1° livello - Gruppo Anatomico Principale ↓
2° livello - Gruppo Terapeutico Principale ↓
3° livello - Sottogruppo Terapeutico ↓
4° livello - Sottogruppo Chimico/Terapeutico ↓
5° livello - Sottogruppo Chimico
Esempio: Aspirina N02BA01
1° N (Sistema Nervoso) 2° 02 (Analgesici) 3° B (Analgesici non Oppioidi) 4° A (Acido Acetilsalicilico e Derivati) 5° 01 (Acido Acetilsalicilico)
6

Denominazione dei farmaci I farmaci sono caratterizzati da vari tipi di nomi: nomi chimici (costruiti secondo le regole di nomenclatura dell’ IUPAC), nomi brevettati (nomi delle specialità che contengono quel farmaco) e nomi (o denominazioni) comuni. Questi ultimi sono quelli di uso più diffuso, in quanto evitano le difficoltà della nomenclatura chimica, con l’ eccessiva lunghezza dei termini, e la varietà dei nomi brevettati contenenti lo stesso principio attivo. Sono nomi di uso libero facilmente pronunciabili e traducibili nelle varie lingue, raccomandati o proposti dall’OMS e vengono espressi in latino, inglese (INN, international nonproprietary name, denominazione comune internazionale inglese) e francese (DCI, dénomination comune internationale). Dopo traduzione nelle lingue dei vari paesi, essi divengono denominazioni comuni nazionali (ad esempio DC.IT, denominazione comune italiana). L’ OMS raccomanda l’ utilizzo e la protezione delle denominazioni comuni, attraverso la adozione di provvedimenti atti ad evitare la registrazione di specialità con nomi coincidenti o molto simili a quelli delle denominazioni comuni. Per l’ assegnazione di nuove denominazioni comuni, sempre l’ OMS raccomanda di evidenziare l’ analogia esistente con altre sostanze appartenenti allo stesso gruppo terapeutico alle quali già siano state attribuite denominazioni comuni. Ciò può essere realizzato inserendo nel nome sillabe o gruppi di sillabe ben codificate ed ampiamente utilizzate. Alcuni esempi sono riportati qui di seguito. Cardiovascolari ACE-inibitori Benazepril Captopril Cilazapril Enalapril Lisinopril Perindopril Quinapril Ramipril Cardiovascolari β-bloccanti Acebutololo Atenololo Celiprololo Nadololo Pindololo Propranololo Timololo Ipolipemizzanti inibitori della HMG CoA reduttasi Atorvastatina Cerivastatina Fluvastatina Pravastatina Simvastatina Antimicotici imidazolici Bifonazolo Clotrimazolo
Econazolo Fenticonazolo Isoconazolo Ketoconazolo Miconazolo Tioconazolo Antibatterici-Chinoloni Ciprofloxacina Enoxacina Lomefloxacina Norfloxacina Ofloxacina Pefloxacina Rufloxacina Antibatterici-Tetracicline Clortetraciclina Doxiciclina Metaciclina Minociclina Tetraciclina Antibatterici-Penicilline Amoxicillina Ampicillina Bacampicillina Cloxacillina
Meticillina Mezlocillina Piperacillina Ticarcillina Antibatterici-Cefalosporine Cefacloro Cefalexina Cefalotina Cefamandolo Cefazolina Cefoxitina Ceftazidima Cefuroxima Sulfamidici Sulfadiazina Sulfalene Sulfamazone Sulfametoxazolo Sulfametrolo Ansiolitici-Benzodiazepine Bromazepam Diazepam Lorazepam Medazepam Oxazepam Pinazepam
7

MOMENTI D’ AZIONE DEI FARMACI
Introduzione
La maggior parte dei farmaci produce i suoi effetti farmacologici in conseguenza della inte-razione con una specifica macromolecola biologica (proteine o acidi nucleici, soprattutto) al cosiddetto sito d’ azione o biofase (il distretto dell’ organismo dove è localizzato il tessuto bersaglio). Da tenere presente che non necessariamente il sito d’ azione ed il tessuto che manifesta la risposta farmacologica coincidono. L’ azione di un farmaco richiede che esso raggiunga una concentrazione adeguata nel fluido che circonda il tessuto bersaglio. Oltre che nella biofase, il farmaco si distribuisce però in tutta una serie di altri distretti e quindi il farmaco ‘utile’ ai fini della azione terapeutica è solamente una frazione della dose somministrata. La concentrazione raggiunta dal farmaco al sito d’ azione dipende, oltre che dalla dose, anche dall’ entità e dalla velocità con cui si svolge una serie di processi che avvengono successivamente alla sua somministrazione e che costituiscono i vari momenti d’ azione di un farmaco. Tali processi si suddividono tradizionalmente in tre fasi: fase farmaceutica, fase farmacoci-
netica e fase farmacodinamica. La fase farmaceutica com-porta la liberazione del farmaco dal-la forma farmaceutica in cui esso è contenuto, ad esempio la disgrega-zione della compressa e la disag-gregazione dei granuli risultanti, il passaggio in soluzione del principio attivo e la sua diffusione al sito di assorbimento. Lo studio delle rela-zioni che intercorrono tra forma farmaceutica ed attività terapeutica è di pertinenza della Biofarmaceu-tica, una branca della Tecnica Far-maceutica, disciplina che si occupa della trasformazione dei principi at-tivi in forme farmaceutiche adegua-te alla somministrazione (formula-zione).
Effetto
Interazione farmaco-macromolecola biologica
specifica nella biofase
AssorbimentoDistribuzioneMetabolismo Escrezione
Liberazione del farmacodalla forma farmaceuticae passaggio in soluzione
Somministrazione
Forma Farmaceutica
FASE FARMACODINAMICA
FASE FARMACOCINETICA
FASE FARMACEUTICA
La fase farmacocinetica comprende invece il processo di as-sorbimento ed il passaggio in cir-colo, la distribuzione ai vari di-stretti dell’ organismo, biofase in-clusa, il metabolismo e l’ escrezio-ne del farmaco (ADME). La fase farmacodinamica, infine, riguarda il comportamento del farmaco in biofase. La sua azio-ne può anche essere rivolta verso un biosistema estraneo all’ organismo (ad esempio un microrganismo in-vasore), in tal caso si parla di fase chemioterapica.
8

Fase Farmaceutica
Un farmaco può essere somministrato per via topica o per via sistemica. Nel primo caso esso esplica un effetto localizzato al sito a cui è stato somministrato, essendo scarsamente assorbito nel torrente circolatorio. Un tipico esempio di somminstrazione per via topica è quella epidermica. Nel
secondo caso il farmaco esplica un effetto sistemico, cioè su siti lontani da quello di somministrazione, in se-guito a passaggio in rilevante quantità nella circolazione sanguigna. Le principali vie sistemiche di somministra-zione sono quelle enterali (orale, rettale, sublinguale) e parenterali (intravascolare,
e, sottocutanea). E' bene ricordare che in diversi casi farmaci sommi-nistrati topicamente sono capaci di entrare nel torrente circolatorio in quantitativi
sufficienti da provocare effetti sistemici. Di solito tali effetti sono indesiderati. Esistono tuttavia anche esempi di sistemi di rilascio controllato (es. cerotti transdermici) che hanno lo scopo di assicurare la cessione graduale del farmaco alla circolazione sistemica. Viceversa, alcuni farmaci somministrati per via sistemica (ad es. orale) hanno effetti prevalentemente topici (ad es. a livello del tratto gastrointestinale, perchè sono caratterizzati da scarso assorbimento, oppure a livello urinario, perchè, pur essendo bene assorbiti non raggiungono concentrazioni ematiche sufficiente-mente elevate e per un tempo suffcientemente lungo da esercitare un' azione sistemica). Spesso si può scegliere
intramuscolar
la via attraverso la quale somministrare un agente terapeutico e
a della mo-
orale nel coniglio.
EpidermicaPolmonareOculare
Intravascolare (Endovenosa, Endoarteriosa)IntramuscolareSottocutanea
SublingualeOraleRettale
Parenterali
Enterali
Vie Sistemiche
Vie Topiche
Principali Vie di Somministrazione dei Farmaci
quindi ha importanza essenziale la conoscenza delle caratteristiche, dei vantaggi e degli svantaggi delle differenti vie di somministrazione. Così, ad esempio, la via orale è conveniente, sicura ed economica ma richiede la cooperazione del paziente, l’ assorbimento può essere variabile o limitato ed essa può causare irritazione gastrica; la somministrazione intravascolare aggira gli eventuali pro-blemi di assorbimento, produce un effetto rapido, permette un dosaggio esatto ma aumenta il rischio
di effetti indesiderati, non è nor-malmente impiegabile per sostanze non sufficientemente idrosolubili e richiede personale specializzato; la via sottocutanea è adatta per so-spensioni insolubili, ma può pro-durre dolore e necrosi. La forte influenz
HN NH
O O
O
Et
Pentobarbitale
dalità di somministrazione di un farmaco sull' inizio, sulla intensità e sulla durata dell' azione è esem-plificata nella figura accanto che riporta il caso del pentobarbitale (anestetico generale) somministra-to per via endovenosa, intramu-scolare, sottocutanea, rettale ed
9

Fase Farmacocinetica
Una volta che il farmaco somministrato si sia liberato dalla forma farmaceutica in cui è
-
Assorbimento
Con il termine di assorbimento imento del principio attivo dal sito di ssorbimento al torrente circolatorio. Un assorbimento adeguato costituisce il presupposto per una
interaz
ento è
ane assorbenti
contenuto e si sia reso disponibile per l’ assorbimento mediante solubilizzazione si verifica una serie di eventi schematicamente illustrati nella figura seguente. Un farmaco si muove nell’ orga-nismo secondo due modalità: per trasferimento di massa mediante la circolazione sanguigna e per
mico-fisiche del farmaco non hanno grande influenza, le caratteristiche diffusionali differiscono marcatamente tra i singoli farmaci. E’ in particolare il movimento tra i vari compartimenti (per compartimento si intende un distretto dell' organismo omogeneo dal punto di vista farmacocinetico, ad es. tratto gastrointestinale, sangue, tessuto adiposo), che generalmente comporta la penetrazione di barriere idrofobiche, a determinare dove e per quanto tempo un farmaco sarà presente nell’ orga-nismo.
trasferimento diffusionale. Mentre nel trasferimento mediante il flusso sanguigno le proprietà chi
si indica il trasfer
aione significativa con il sito attivo e quindi per la produzione dell' effetto terapeutico.
Un farmaco, per abbandonare il sito di assorbimento, deve attraversare una complessa barriera lipidica rappresentata da una o più membrane biologiche. Il processo di assorbimassente nel caso di somministrazione intravascolare. Nella Tabella successiva sono indicate le membrane assorbenti per le varie vie di somministrazione.
Vie di somministrazione e membr
Vie Membrane
Orale Sublinguale
I Sottocutan uscolare
Mucose del tratto gastroenterico Mu le
Endotelio dei fatici Mucosa d spiratorio
Rettale ntravascolare
ea, IntramInalatoria
Epidermica
cosa sublinguaMucosa rettale
Nessuna capillari vascolari e lin
el tratto reEpitelio cheratinizzato e annessi cutanei
LiberoLegato
SITO D' AZIONEBiopolimero bersaglio
LiberoLegato
DEPOSITITISSUTALI
BIOTRASFORMAZIONE
ESCREZIONEASSORBIMENTO
Metaboliti
Farmaco libero
Farmaco legato
CIRCOLAZIONE SISTEMICA
10

Il passaggio attraverso le membrane biologiche è anch processi di distribuzione, metabolismo ed escrezione poichè, in gener cellule e non t
tea cs. Uno dei param
nfinissime particelle che si scioglieranno poi rapidamente a causa dell’ eleva Un parametro farmacocinetico legato al grado di assorbimento e d
per esercitare il suo effetto sistemico. Essa dipende non solo dalla rmul
te), una quota di esso
e implicato nei successiviale, i farmaci passano attraverso le
ra cellula e cellula. Quindi le membrane biologiche rappresentano una barriera comune ai movimenti dei farmaci. Sono perciò molto importanti i fattori chimico-fisici che influenzano il pas-saggio dei farmaci attraverso le membrane e che saranno esaminati più in dettaglio successiva-mente. Oltre ad essi, influiscono sul processo anche altri fattori quali la concentrazione del farmaco, il flusso sanguigno che irrora il sito di assorbimento ed infine l' area della superficie assorbente.
I farmaci somministrati in soluzione acquosa vengono assorbiti più rapidamente di quelli somministrati in sospensione o in forma solida. Per quelli somministrati in forma solida, la velocità
di dissoluzione può essere il fattore limitante nel loro assorbimento. La legge di Noyes-Whitney esprime la dipendenza della velocità di dissoluzione da una se-rie di parametri: D = coefficiente di dissoluzione; A = area superficiale totale delle particelle di farmaco; cs = solubilità del farmaco; c = concentrazione del farmaco al tempo t; h = spessore dello strato di diffusione, cioè del sottile film alla superficie della particella solida con una concentrazione del farmaco pari
etri sui quali agire per aumentare la velocità di passaggio in soluzione è l’ area superficiale complessiva A. Per rendere tale area elevata si può somministrare il farmaco in forma micronizzata (cioè polverizzata mediante l’ impiego di un mulino a getto d’ aria) oppure facendo in modo che finissime particelle di farmaco si generino nei pressi del sito di assorbimento. Ad esempio,
l’ aspirina è un acido de-bole ed è relativamente insolubile nel contenuto acido dello stomaco. La aggiunta di una sostanza tampone (idrossido di magnesio ed alluminio glicinato basico) crea uno strato di diffusione con un pH superiore a quello dello stomaco che è in grado di solubiliz-zare il farmaco sotto for-ma di sale. In un secon-do tempo, venendo a contatto per diffusione dissociato sotto forma di to valore di A. i metabolizzazione presi-
stemica (cioè precedente all’ ingresso nella circolazione generale) di un farmaco è la sua biodisponibilità, definita come la quota di farmaco che è in grado di raggiungere intatta la circola-zione sistemica ed è disponibile
stazionario adiacen
con il pH più basso dello stomaco, l’ aspirina precipiterà come acido i
fo azione ma anche da variazioni nell’ attività di enzimi farmaco-metabolizzanti. Se consideriamo ad esempio un farmaco somministrato per via orale che viene assorbito nel
tratto gastrointestinale, esso deve, prima di raggiungere la circolazione sistemica, entrare in contatto con la mucosa intestinale e poi attraversare il fegato mediante la circolazione portale. Se il farmaco viene in parte metabolizzato nell’ intestino e/o nel fegato (e ciò avviene comunemen
verrà persa prima del raggiungimento della circolazione generale, anche nel caso di un assorbimento completo. Con l’ espressione “effetto di primo passaggio” si intende la diminuzione di biodisponibilità dovuta alla degradazione metabolica di un farmaco assunto per via orale nel corso del primo passaggio nell’ intestino e nel fegato.
Si parla di biodisponibilità assoluta (indicata con F, fraction escaping metabolism) quando viene misurata la quantità totale di farmaco che raggiunge la circolazione sistemica. Essa viene
Granulo di aspirina AHcon sostanza tampone
Strato didiffusione
pH 5-6
Succo gastricopH 1-3
Microprecipitatodi AH
CO2H
OCOCH3
AH =
Ridissoluzione
Strato didiffusione
Membrana del trattogastroenterico
TorrentecircolatorioA-
hts=
ccDAq )( −
11
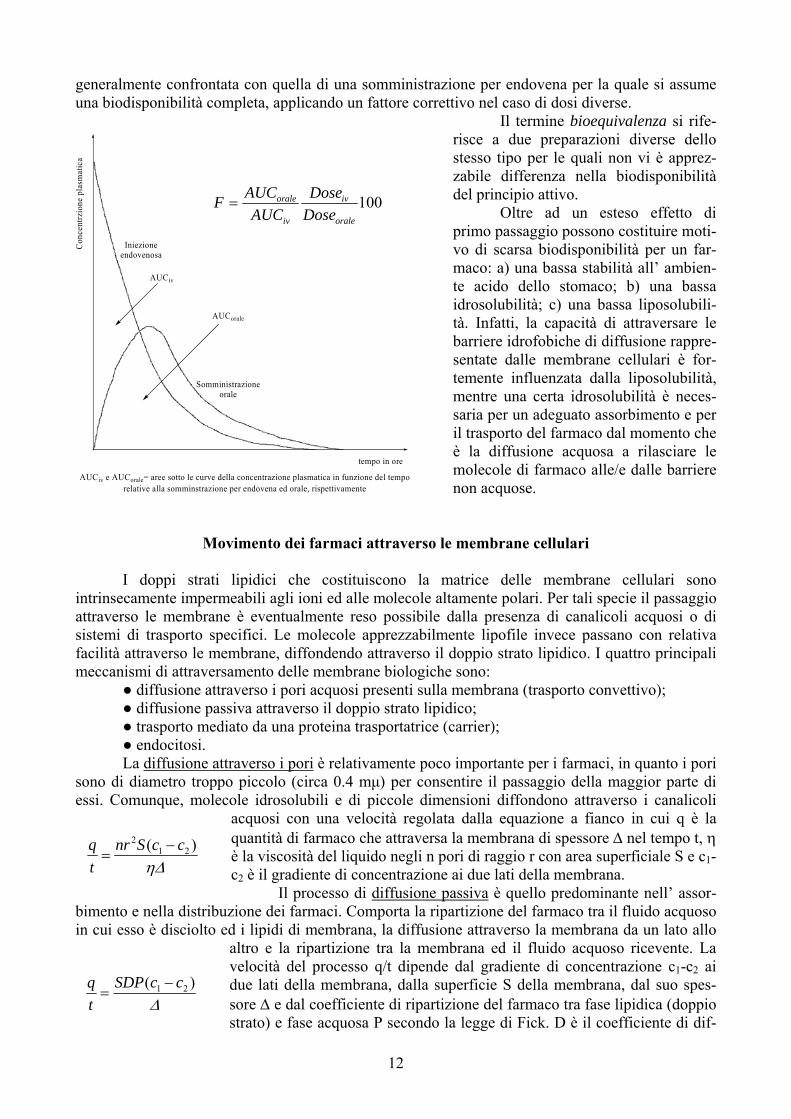
generalmente confrontata con quella di una somministrazione per endovena per la quale si assume una biodisponibilità completa, applicando un fattore correttivo nel caso di dosi diverse.
isponibilità del prin
tabilità all’ ambien-te acid
e I doppi strati lipidici che costituiscono la m membrane cellulari sono
trinsecamente impermeabili agli ioni ed alle molecole altamente polari. Per tali specie il passaggio ttraverso le membrane è eventualmente reso possibile dalla presenza di canalicoli acquosi o di
sistemi di trasporto ssano con relativa cilità attraverso le membrane, diffondendo attraverso il doppio strato lipidico. I quattro principali eccan
Il termine bioequivalenza si rife-risce a due preparazioni diverse dello stesso tipo per le quali non vi è apprez-zabile differenza nella biod
Iniezioneendovenosa
Somministrazioneorale
AUCiv
AUCorale
AUCiv e AUCorale= aree sotto le curve della concentrazione plasmatica in funzione del temporelative alla somminstrazione per endovena ed orale, rispettivamente
tempo in ore
Con
cent
rzio
ne p
lasm
atic
a
cipio attivo. Oltre ad un esteso effetto di
primo passaggio possono costituire moti-vo di scarsa biodisponibilità per un far-maco: a) una bassa s
o dello stomaco; b) una bassa idrosolubilità; c) una bassa liposolubili-tà. Infatti, la capacità di attraversare le barriere idrofobiche di diffusione rappre-sentate dalle membrane cellulari è for-temente influenzata dalla liposolubilità, mentre una certa idrosolubilità è neces-saria per un adeguato assorbimento e per il trasporto del farmaco dal momento che è la diffusione acquosa a rilasciare le molecole di farmaco alle/e dalle barriere non acquose.
membrane cellulari
atrice delle
Movimento dei farmaci attraverso l
ina
specifici. Le molecole apprezzabilmente lipofile invece pafam ismi di attraversamento delle membrane biologiche sono: ● diffusione attraverso i pori acquosi presenti sulla membrana (trasporto convettivo); ● diffusione passiva attraverso il doppio strato lipidico; ● trasporto mediato da una proteina trasportatrice (carrier); ● endocitosi. La diffusione attraverso i pori è relativamente poco importante per i farmaci, in quanto i pori
arte di i diffondono attraverso i canalicoli
quazione a fianco in cui q è la quantità di farmaco che attraversa la membrana di spessore ∆ nel tempo t, η
sono di diametro troppo piccolo (circa 0.4 mµ) per consentire il passaggio della maggior pessi. Comunque, molecole idrosolubili e di piccole dimension
acquosi con una velocità regolata dalla e
è la viscosità del liquido negli n pori di raggio r con area superficiale S e c1-c2 è il gradiente di concentrazione ai due lati della membrana.
Il processo di diffusione passiva è quello predominante nell’ assor-ione dei farmaci. Comporta la ripartizione del farmaco tra il fluido acquoso
d i lipidi di membrana, la diffusione attraverso la membrana da un lato allo altro e la ripartizione tra la membrana ed il fluido acquoso ricevente. La velocità del processo q/t dipende dal gradiente di concentrazion
bimento e nella distribuzin cui esso è disciolto e
e c1-c2 ai due lati della membrana, dalla superficie S della membrana, dal suo spes-sore ∆ e dal coefficiente di ripartizione del farmaco tra fase lipidica (doppio strato) e fase acquosa P secondo la legge di Fick. D è il coefficiente di dif-
∆η)( 21 ccSnr
tq
=2 −
∆t=
)( 21 ccSDPq −
100orale
iv
iv
orale
DoseDose
AUCAUCF =
12

fusione, costituisce una misura della mobilità delle molecole all’ in-terno del doppio strato lipidico e varia solo leggermente tra i vari far-maci. P è il rapporto all’ equilibrio tra le concentrazioni del farmaco in fase lipidica ed acquosa. Per la determinazione sperimentale di P si impiega come fase lipidica n-ottanolo o CHCl3 ed un tampone fosfato
a pH 7.4 come fase acquosa. Come si evidenzia dalla legge di Fick, vi è una stretta correlazione tra liposolubilità di un farmaco e permeabilità della membrana ad esso. Per questo motivo la liposolu-bilità costituisce uno dei fattori più importanti per le caratteristiche farmacocinetiche di un farmaco e numerose proprietà quali il grado e la velocità di assorbimento nel tratto gastrointestinale, la penetrazione nei tessuti e nel cervello ed il grado di eliminazione renale possono essere predette dalla conoscenza della liposolubilità del farmaco. Un esempio del ruolo di P è illustrato nella figura successiva in cui si osserva una soddisfacente correlazione lineare tra % di farmaco assorbito dal colon di ratto e relativo coefficiente di ripartizione CHCl3/H2O per una serie di barbiturici.
Sebbene i farmaci di elevata lipofilia e quindi con un elevato coefficiente di ripartizione siano nella condizione di venire facilmente assorbiti, è tuttavia indispensabile che essi abbiano anche un certo grado di solubilità in acqua, dal momento che i fluidi biologici sono per loro natura acquosi. Quindi da un punto di vista pratico è necessario che i farmaci
[ ][ ] acquosafase
lipidicafase
farmacofarmaco
P
=
H2OCHCl3
Allobarbitale..
....
...
% Assorbimento
Esetale
SecobarbitalePentobarbitale
CiclobarbitaleButetale
AprobarbitaleFenobarbitale
Barbitale
604020
1
5
10
50
400PO
OOR'R
NHHN
Esetale
Secobarbitale
Pentobarbitale
Ciclobarbitale
Butetale
Allobarbitale
Aprobarbitale
Fenobarbitale
Barbitale
R'R
[ ][ ]
[ ][ ]+
−
+=+=BH
BpKpHAHApKpH aa lg ;lg assumono le due forme indicate a fianco per un
acido debole AH
possiedano un corretto rapport
le note equazioni di Henderson-Hasselbach che
o tra lipofilia ed idrofilia. Si ha generalmente un buon assorbimento per valori di log P compresi tra -2 e +4.
Un fattore complicante il processo di diffusione passiva è che molti farmaci sono acidi o basi deboli e quindi esistono nelle forme non ionizzata ed ionizzata in equilibrio tra loro. Il rapporto
tra esse dipende dai valori di pK e di pH secondo
A- + H+ e per una base debole B BH+, rispettivamente (pKa è riferito in quest' ultimo caso all' acido coniugato BH+). In anno una liposolubilità molto bassa e non sono er diffusione passiva, mentre le specie neutre AH
ile da perm re una rapida permeazione.
entrambi i casi le specie ionizzate BH+ e A- hvirtualmente in grado di permeare le membrane pe B per molti farmaci sono sufficientemente lipof ette
13
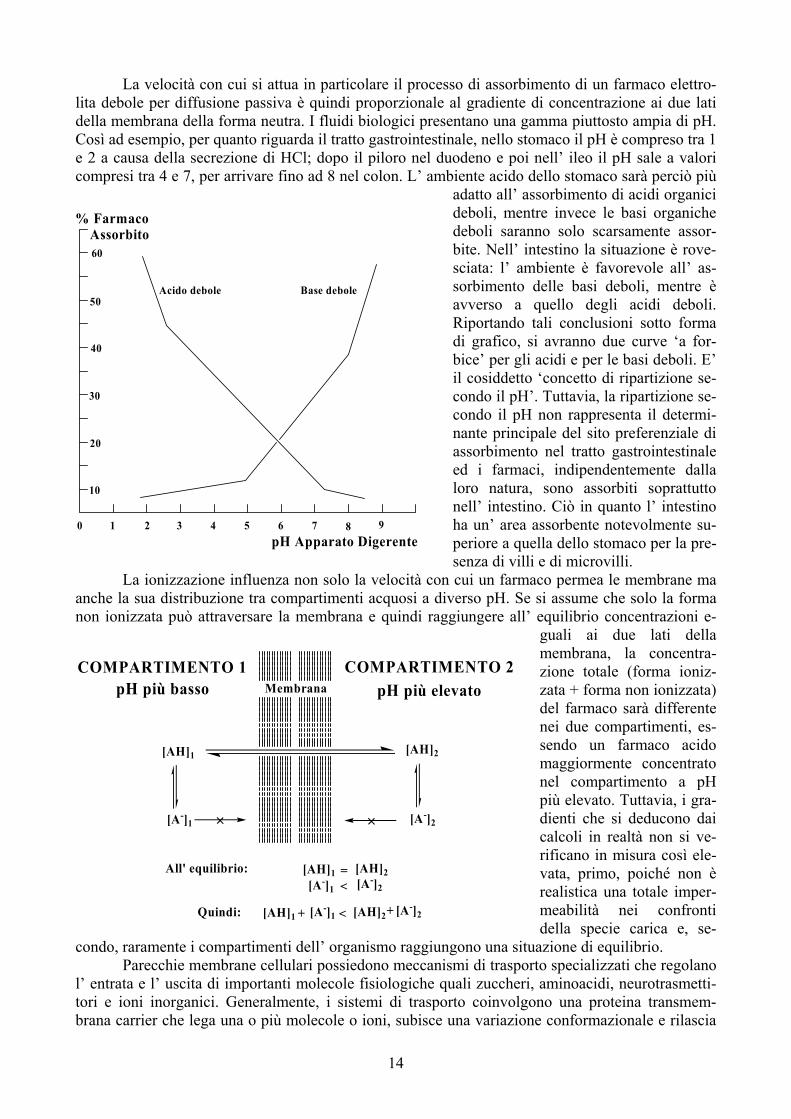
La velocità con cui si attua in particolare il processo di assorbimento di un farmaco elettro-lita debole per diffusione passiva è quindi proporzionale al gradiente di concentrazione ai due lati della membrana della forma neutra. I fluidi biologici presentano una gamma piuttosto ampia di pH. Così ad
n anche la sua distribuzione tra compartimenti acquosi a divnon ionizzata può attraversare la membrana e quindi ragg ntrazioni e-
a Parecchie membrane cellulari possiedono meccanismi di trasportl’ entrata e l’ uscita di importanti molecole fisiologiche quali zuccheri,
smem-brana carrier che lega una o più molecole o ioni, subisce una variazione conformazionale e rilascia
esempio, per quanto riguarda il tratto gastrointestinale, nello stomaco il pH è compreso tra 1 e 2 a causa della secrezione di HCl; dopo il piloro nel duodeno e poi nell’ ileo il pH sale a valori compresi tra 4 e 7, per arrivare fino ad 8 nel colon. L’ ambiente acido dello stomaco sarà perciò più
adatto all’ assorbimento di acidi organici deboli, mentre invece le basi organiche deboli saranno solo scarsamente assor-bite. Nell’ intestino la situazione è rove-sciata: l’ ambiente è favorevole all’ as-sorbimento delle basi deboli, mentre è avverso a quello degli acidi deboli. Riportando tali conclusioni sotto forma di grafico, si avranno due curve ‘a for-bice’ per gli acidi e per le basi deboli. E’ il cosiddetto ‘concetto di ripartizione se-condo il pH’. Tuttavia, la ripartizione se-condo il pH non rappresenta il determi-nante principale del sito preferenziale di assorbimento nel tratto gastrointestinale ed i farmaci, indipendentemente dalla loro natura, sono assorbiti soprattutto nell’ intestino. Ciò in quanto l’ intestino ha un’ area assorbente notevolmente su-periore a quella dello stomaco per la pre-senza di villi e di microvilli. cui un farmaco permea le membrane ma erso pH. Se si assume che solo la forma iungere all’ equilibrio conce
La ionizzazione influenza non solo la velocità co
guali ai due lati della membrana, la concentra-zione totale (forma ioniz-zata + forma non ionizzata) del farmaco sarà differente nei due compartimenti, es-sendo un farmaco acido maggiormente concentrato nel compartimento a pH più elevato. Tuttavia, i gra-dienti che si deducono dai calcoli in realtà non si ve-rificano in misura così ele-vata, primo, poiché non è realistica una totale imper-meabilità nei confronti della specie carica e, se-zione di equilibrio. o specializzati che regolano aminoacidi, neurotrasmetti-
tori e ioni inorganici. Generalmente, i sistemi di trasporto coinvolgono una proteina tran
Base deboleAcido debole
pH Apparato Digerente9876543210
60
50
40
30
20
10
[A-]1
[A-]2[A-]1
[AH]2[AH]1
[A-]2
[AH]2[AH]1
[A-]2[A-]1 [AH]2[AH]1
% Farmaco Assorbito
<All' equilibrio: =
pH più elevatopH più basso
Quindi: <+
MembranaCOMPARTIMENTO 2COMPARTIMENTO 1
+
++
condo, raramente i compartimenti dell’ organismo raggiungono una situ
14

le specie sull’ altro lato della membrana. Tali sistemi possono operare passivamente, senza dispendio di energia; in tal caso essi semplicemente facilitano il processo nella direzione del
gradiente di concentrazione od elettrochimico e per ciò si parla di diffusione facilitata. Inizialmente il sito di legame per la specie trasportata si trova sul lato esterno della membrana. Una volta che la specie si sia legata, ha luogo una modificazione conformazionale che permette alla specie di attraversare la membrana. La proteina trasportatrice rilascia la specie all’ interno della cellula e recupera la conformazione originaria. Il processo può avvenire nei due sensi e la direzione del trasporto dipende dai gradienti di concentrazione o elettrochimici. Altri sistemi di trasporto sono invece accoppiati ad una fonte di energia (ATP o gradienti ionici). In questo caso il trasporto può avvenire contro gradiente di concentrazione o elettrochimico (trasporto attivo). Tra i vari meccanismi fisiologici di trasporto attivo vi sono quelli coinvolti nella secrezione di ioni H+ nel succo gastrico, nella captazione di iodio da parte della tiroide,
P
P
P
2K+
2K+
2K+
2K+
2K+
P
P
3Na+
3Na+
3Na+
nel riassorbimento di glucosio e di aminoacidi dall’ urina al sangue e la Na+/K+ATPasi, la cosiddetta pompa del so-dio, che trasporta da un lato all’ altro delle membrane cellulari ioni Na+ e K+, sfruttando l’ idrolisi dell’ ATP come fonte di energia. La Na+/K+ATPasi è in grado di as-sumere due conformazioni diverse a seconda che essa sia fosforilata o no, con diversa affinità per i due ioni. Il sito
di legame per gli ioni è rivolto verso l' interno della cellula in una conformazione e verso l’ esterno nell’ altra. Un modello di funzionamento della pompa del sodio è mostrato nella figura seguente.
3Na+3Na+
ATPESTERNOCELLULA
Membranacellulare
Diffusione facilitata
INTERNOCELLULA
Na+/K+ATPasi Inversione
Fosforilazione
Rilascio
LegameDefosforilazione
Inversione
Rilascio
Modello di Funzionamento della Na+/K+ ATPasi
Legame
15

Il legame dell’ Na+ all’ interno della cellula attiva la fosforilazione della pompa. Il processo
eccanismo di
ro la velocità del trasporto cresce in proporzione di-
modifica la sua conformazione, cosicchè il sito di legame per gli ioni è rivolto ora verso l’ esterno della cellula. La nuova conformazione ha bassa affinità per lo ione Na+ che viene quindi rilasciato, mentre ha alta affinità per lo ione K+ che si lega al posto dell’ Na+. Il legame con lo ione K+ promuove la defosforilazione della pompa. La pompa defosforilata non è stabile nella conformazione con il sito di legame per gli ioni rivolto verso l’ esterno e si riconverte in quella iniziale che ha bassa affinità per lo ione K+ che viene ceduto all’ interno della cellula.
Esigenza comune per l’ esistenza di un mtrasporto di un farmaco mediato da un carrier è che vi sia una certa somiglianza strutturale tra il farmaco ed un qualche sub-strato fisiologico che attraversa la membrana cellulare serven-dosi di tale sistema. Così, ad esempio, l’ antineoplastico meto-tressato utilizza il sistema di trasporto attivo dell’ acido folico. Da un punto di vista farmacocinetico, vi sono comunque solo pochi distretti dell’ organismo nei quali il trasporto di farmaci mediato da carriers è importante: tubuli renali, tratto biliare, intestinale.
A differenza della diffusione passiva in cui
CO H
barriera ematoencefalica, tratto gast
retta con il gradiente di concentrazione, nel trasporto mediato da carriers i siti del trasportatore si saturano ad alte concentrazioni di farmaco e la velocità non aumenta ulteriormente oltre tale punto. Il termine endocitosi è riassuntivo delle due
Metotressato: R = NH2; R1 = CH3Acido folico: R = OH; R1 = H
N
N N
N N
NH
O
CO2H
2
R1H2N
R
denominazioni di fagocitosi e di pinocitosi con cui si indicavano in precedenza l’ inglobamento, ri-spettivamente, di particelle solide e di soluti all’ in-terno della massa citoplasmatica. L’ endocitosi comporta l’ invaginazione di una parte della mem-brana che ingloba il farmaco, la coalescenza delle due facce della membrana, la separazione di una vescicola che migra all’ interno della cellula dove viene poi degradata dagli enzimi lisosomiali. Questo meccanismo appare essere importante per il trasporto di certe macromolecole come l’ insulina che passa in tal modo la barriera ematoencefalica.
Distribuzione
Una volta che il farmaco abbia raggiunto il circolo, esso si distribuirà nei quattro principali ompar
seguente possibilità di ingresso nell’ SNC di farmaci normalmente non permeanti.
c timenti acquosi dell’ organismo: fluido extracellulare (che comprende il plasma, il fluido interstiziale e la linfa), fluido intracellulare (che è la somma dei fluidi contenuti in tutte le cellule dell’ organismo) e fluido transcellulare (che comprende i fluidi cerebrospinale, intraoculare, perito-neale, pleurico e sinoviale e le secrezioni digestive). Per entrare nei compartimenti transcellulari da quelli extracellulari, un farmaco deve attraversare una barriera cellulare. Un esempio par-ticolarmente importante tra esse è la barriera ematoencefalica. La barriera ematoencefalica è una barriera a permeabilità selettiva per la diffusione passiva di sostanze dal torrente circolatorio alle varie regioni del sistema nervoso centrale (SNC). Non è completamente definita anatomicamente e non è assoluta. La principale caratteristica strutturale che sta alla base della sua esistenza è la stretta adesione degli astrociti alla membrana basale dei capillari. L’ SNC è inaccessibile a parecchi far-maci ad azione sistemica con insufficiente liposolubilità. Tuttavia, nel caso di infiammazione in for-ma di meningite si può avere una interruzione dell’ integrità della barriera ematoencefalica con con-
16

In ognuno dei compartimenti acquosi, le molecole di farmaco esistono sia in forma libera che in forma legata con componenti compartimentali; inoltre, i farmaci che sono acidi o basi deboli
coefficiente di ripartizione P). parente di distribuzione di un farmaco Vd è definito come il
farmaco Q alla stessa
o o, poiché esse sono di dimensioni troppo grandi er att
di entrare facilmente nelle cellule a causa
co di farmaci relativamente liposolubili che passano facilmente attraverso le mem-
l valore del volume di distribuzione è superiore a quello totale della
Escrezione
I farmaci vengono eliminati dall’ odificati o come metaboliti. Gli organi scretori generalmente eliminano più facilmente i composti polari che le sostanze caratterizzate da
sono soggetti anche all’ equilibrio di dissociazione. In definitiva, il profilo di distribuzione di un farmaco tra i vari compartimenti dipende dai seguenti fattori: ●permeabilità tra le barriere tissutali; ●legame con i componenti compartimentali; ●ripartizione in funzione del pH; ●ripartizione tra fasi lipidiche e fasi acquose (
Il volume apvolume di fluido richiesto per contenere la quantità totale di
pd C
QV = concentrazione di quella presente nel plasma Cp. In altri termini è il volume che il farmaco occuperebbe se la quantità totale di esso nell’ organismo fosse in soluzione ad una concentrazione uguale a quella plasmatica. Il volume del plasma è ~ 0.05 lt/kg di peso corporeo. Farmaci con Vd intorno a no sostanze confinate nel compartimento plasmatictale valore s p raversare con facilità la parete dei capillari oppure, più spesso, a causa di un
forte legame con le proteine plasmatiche. Numerosi farmaci sono presenti nel plasma principal-mente in forma legata con le proteine plasmatiche (in particolare albumina per i farmaci di natura acida e α1-glicoproteina acida per i farmaci di natura basica). Alle normali concentrazioni terapeu-tiche, i siti di legame su tali proteine sono lontani dalla saturazione per cui la concentrazione di farmaco legato varia in proporzione diretta con la concentrazione di farmaco libero. La relativa non specificità dei siti di legame per numerosi tipi di farmaci fa si che si possa avere competizione tra essi. La somministrazione di un farmaco B può cioè ridurre il legame con le proteine plasmatiche di un farmaco A, somministrato in precedenza e quindi aumentarne la concentrazione della forma libera, con conseguenze sull’ intensità della sua azione. Il volume extracellulare totale è ~ 0.2 lt/kg e questo è approssimativamente il volume appa-rente di distribuzione per farmaci che non sono in gradodella loro bassa liposolubilità e che quindi sono distribuiti prevalentemente nel compartimento extracellulare. I fluidi totali dell’ organismo si aggirano attorno ai 0.55 lt/kg. Questo volume di distribuzio-ne è caratteristibrane cellulari, non si legano ad alcun costituente tissutale e si distribuiscono quindi in maniera uni-forme nell’ acqua corporea. Infine, se un farmaco si lega selettivamente ad un qualche costituente tissutale al di fuori del compartimento plasmatico, iacqua corporea dell’ organismo.
organismo imm
eelevata liposolubilità. Il rene è l’ organo più importante per l’ eliminazione dei farmaci. Le sostanze lipofile non sono eliminate facilmente dai reni. Di conseguenza, molti farmaci lipofili sono metabolizzati a prodotti più polari i quali sono poi eliminati con le urine. L’ escrezione renale implica tre processi: filtrazione glomerulare, secrezione e riassorbimento tubulari. Il primo ed il terzo sono normalmente dei processi di diffusione passiva, mentre il secondo è un processo di trasporto mediato da carriers. La quota di farmaco che entra nel lume tubulare per filtrazione dipende dall’ entità del legame con le proteine plasmatiche, poiché solo la frazione di farmaco libera viene filtrata. I principi che governano il riassorbimento dei farmaci dal filtrato glomerulare sono gli stessi che sono stati esaminati per l’ assorbimento e la distribuzione. Farmaci con elevato coefficiente di ripartizione sono facilmente riassorbiti. Il riassorbimento di acidi e basi deboli dipende dal pH dell’ urina tubulare (4.5-8): gli acidi deboli sono scarsamente riassorbiti nel caso di
17

urine alcaline, così come le basi deboli in urine acide. Le cellule dei tubuli prossimali trasportano attivamente numerose sostanze dal plasma all’urina tubulare. Oltre a meccanismi di trasporto attivo specifici, esistono due sistemi di trasporto relativamente aspecifici, uno per i cationi organici, l’ al-tro per gli anioni organici, utilizzabili da numerosi farmaci.
Alcuni farmaci e metaboliti di farmaci che si formano nel fegato sono secreti nella bile. Tali sostanze hanno in genere un peso molecolare superiore a 500, mentre i composti con peso moleco-lare co
Metabolismo
I farmaci subiscono nell’ organism rente una serie di modificazioni, preva-ntemente enzimatiche, con formazione di uno o (generalmente) più prodotti di biotrasformazione etab
, sesso, razza), fattori farmacodinamici (dose, frequenza e via di somministra-
nervoso e nel asma
iò ne favorisce l’ eliminazione. Il metabolismo inoltre porta di solito ad una attenuazione o
l metabolismo dei rmac
i oppure modificano quelli esistenti
Reazioni di Ossidazione
tipi principali: quelle microsomiali e quelle non microsomiali. Le prime ono catalizzate da sistemi enzimatici presenti nel reticolo endoplasmatico liscio, soprattutto, ma
mpreso tra 300 e 500 si ripartiscono tra bile ed urine. Il trasporto nella bile è mediato da car-riers aspecifici. Essi sono poi escreti con le feci o riassorbiti dall’ intestino (in tal caso si instaura un circolo enteroepatico). L’ escrezione polmonare è importante esclusivamente per le sostanze gasso-se o altamente volatili. Infine, i farmaci possono essere eliminati anche con il latte materno o il sudore.
o in misura diffe
le(m oliti). Solo in pochi casi, il farmaco viene eliminato completamente inalterato. Già nel tratto gastrointestinale, i farmaci possono essere modificati per l’ azione idrolitica dell’ ambiente acido dello stomaco. Di gran lunga più importanti sono però i processi di trasformazione enzimatica in vari organi e tessuti. Il metabolismo di un farmaco può essere influenzato da vari fattori fra cui: fattori genetici, fattori fisiologici (etàzione) e fattori ambientali (interazioni con altri farmaci ed altre sostanze in genere). La sede principale del metabolismo dei farmaci è il fegato. Sistemi enzimatici farmaco-metabolizzanti sono presenti anche nell’ intestino, nei reni, nei polmoni, nel tessutopl . I prodotti di biotrasformazione sono generalmente più polari e meno lipofili dei loro proge-nitori e cad una perdita completa di attività. Meno frequentemente, può tuttavia verificarsi la formazione di metaboliti attivi, alcune volte con lo stesso tipo di attività del farmaco di orgine, altre volte invece con una attività diversa, potenzialmente utile oppure fonte di effetti indesiderati. Le reazioni metaboliche sono suddivise in due categorie principali: reazioni della fase I e reazioni della fase II. Nella pagina seguente è mostrato lo schema generale defa i e la localizzazione subcellulare degli enzimi coinvolti. Le reazioni di fase I comprendono reazioni di ossidazione, riduzione ed idrolisi ed introducono nella molecola di farmaco nuovi gruppi funzionalcon il risultato di convertire il farmaco in un metabolita più polare che può essere più o meno attivo del progenitore, possedere un’ attività diversa, oppure essere completamente inattivo. Le reazioni della fase II sono invece reazioni in cui il farmaco di origine (o un suo metabolita della fase I) è condensato con un substrato endogeno (coniugante) di natura glicidica, lipidica o aminoacidica per dare i cosiddetti coniugati. Essi sono, con qualche eccezione, meno attivi rispetto ai loro precursori, oppure, più spesso, completamente inattivi. Anche le reazioni della fase II normalmente portano ad una diminuzione della liposolubilità. Le reazioni della fase I introducono spesso nel farmaco gruppi funzionali che servono come punti di attacco per i sistemi coniuganti della fase II. Perciò, la maggior parte dei farmaci viene coinvolta sequenzialmente nelle due fasi.
Reazioni della Fase I
Ve ne sono di dues
18

19
one degli Enzimi Coinvolti
non esclusivamente degli epatociti. Sono indicati come enzimi microsomiali in quanto, a seguito di omogeneizzazione, il reticolo endoplasmatico liscio viene demolito in piccoli frammenti che si richiudono in vescicole chiamate microsomi che possono poi essere isolati per centrifugazione differenziale dell’ omogenato tissutale nella frazione microsomiale.
Schema Generale del Metabolismo dei Farmaci e Localizzazi
Reazioni Metaboliche Localizzazione Enzimi
Reazioni della Fase I
e , S-Ossidazione
lazione
ldeidi
one zoriduzione
deidi
i Esteri rolisi di Ammidi
II (Coniugazioni)
Ossidazioni C-OssidazionNN, O, S-DealchiDeaminazione Ossidazione di Alcoli Ossidazione di A Riduzioni NitroriduziARiduzione di Al Idrolisi Idrolisi dId
Reazioni della Fase
olfoconiugazione urica
Glicuronazione SConiugazione ippMercapturazione Acetilazione Metilazione
Microsomi Microsomi
Mitocondri Citosol
icrosomi, Citosol icrosomi
icrosomi, Citosol icrosomi, Citosol
icrosomi
Citosol Mitocondri
Gli enzimi micros
zimi che catalizzano l’ inserzione di uno dei due atomi dell’ O2 nella m
-O2CH2CH2C CH2CH2CO2-
H3C
CH3CH3
CH3
NN
N N
Fe
Microsomi Microsomi, Microsomi, Citosol MMCitosol MM
M
Microsomi, Citosol Mitocondri, Citosol Citosol
omiali sono delle monoossigenasi, en-
Ferroprotoporfirina IX
olecola del farmaco AH che viene ossidato ad AOH mentre l’ altro atomo di ossigeno viene ridotto ad H2O. La prin-cipale monoossigenasi farmaco-metabolizzante è il citocromo P-450 (CYP450). I citocromi sono una ampia categoria di pro-teine enzimatiche coinvolte in reazioni ossidoriduttive che pos-siedono come cofattore l’ eme, cioè il sistema ferro-protopor-firinico. Sono quindi eme proteine. Ne esistono di tre classi principali: a, b, c individuate in base alla natura delle catene laterali dell’ eme. Il citocromo P-450 appartiene alla classe b, come la mioglobina e l’ emoglobina ed il suo sistema eme è la

ferroprotoporfirina IX. Il ferro oscilla tra gli stati di ossidazione +2 e +3. Delle sei posizioni di coordinazione del ferro(II) quattro sono impegnate con l’ anello porfirinico, il quinto ligando è un residuo di cisteina dell’ apoenzima, mentre il sesto è un ligando facilmente scambiabile (probabil-mente H2O). Il nome del’ enzima deriva dal fatto che il complesso della forma ferrosa con l’ ossido di carbonio forma un composto rosa (pink) che presenta un massimo di assorbimento a ~ 450 nm. Il sistema P-450 comprende un’ ampia famiglia (superfamiglia) di enzimi con diverse specificità, di-visa in famiglie ed in sottofamiglie. Questi enzimi vengono descritti con la sigla CYP seguita da un numero arabo (famiglia), da una lettera (sottofamiglia) e da un altro numero arabo (gene individua-le). Le famiglie coinvolte nel metabolismo dei farmaci sono CYP1-4.
L’ ossidazione ad opera del citocromo P-450 richiede ossigeno molecolare, NADPH che trasferisce due elettroni all’ ossigeno ed una flavoproteina (NADPH-P-450 reduttasi) che assicura la cession
combina con la forma os-
nossidato AOH con rigenerazione del citocromo nello stato iniziale. Il don
NADPH
NADP+
FAD
P-450-AHO2- 2H+
H2O
AOH
Fe3+.O22-
e al P-450 di un elettrone per volta. Il ciclo catalitico del citocromo P-450 è illustrato nella figura seguente.
Il substrato AH si
sidata del citocromo P-450 (Fe3+) che viene suc-cessivamente ridotta alla forma Fe2+ dalla flavo-proteina. Il complesso ri-dotto lega O2 per dare un complesso ternario. Un secondo elettrone conver-te l' ossigeno in radicale O2
-. Il risultante comples-so attivato ed altamente instabile si dissocia con formazione di H2O e di un complesso ossene fer-ro perferrile–farmaco (FeV=O)3+.AH. Quest’ ul-timo astrae un atomo di idrogeno dal substrato azione produce il farmaco atore finale di elettroni è
NADPH. AH+ NADPH + H
P-450-AH
Fe2+.O2
P-450-AH
P-450-AH
Fe2+
P-450-AH
Fe3+
P-450
(Fe=O)3+.AH
Fe2+.O2-
AH
P-450
Fe3+
-2e-
1e-
1e-
O2FADH.
FADH2
per formare una coppia di radicali liberi A.·(FeIVOH)3+. La loro ricombi
+ + O2 → AOH + NADP+ + H2O
20
H
H
RH3C
H3C
O
ON
NN
NO
O-POO P
NH2
N
N
NN
OHHO
O O
OHHOHHOHHOHHHH
FAD
2H+, 2e-
H
FADH2
R
H
H3C
H3C
O
ONH
NN
NHH3C
H3C
O
ON
NN
N
H+, 2e-CONH2
NO-
O P
O
PO
O-
NH2
N
N
NN
OPO32-HO
O O
OHHO
O O
NADPH
R
HHCONH2
N
NADP+
.
FADH.
FLAVIN ADENIN DINUCLEOTIDE
+
NICOTINAMIDE ADENINDINUCLEOTIDE FOSFATO
O-
Sitoreattivo
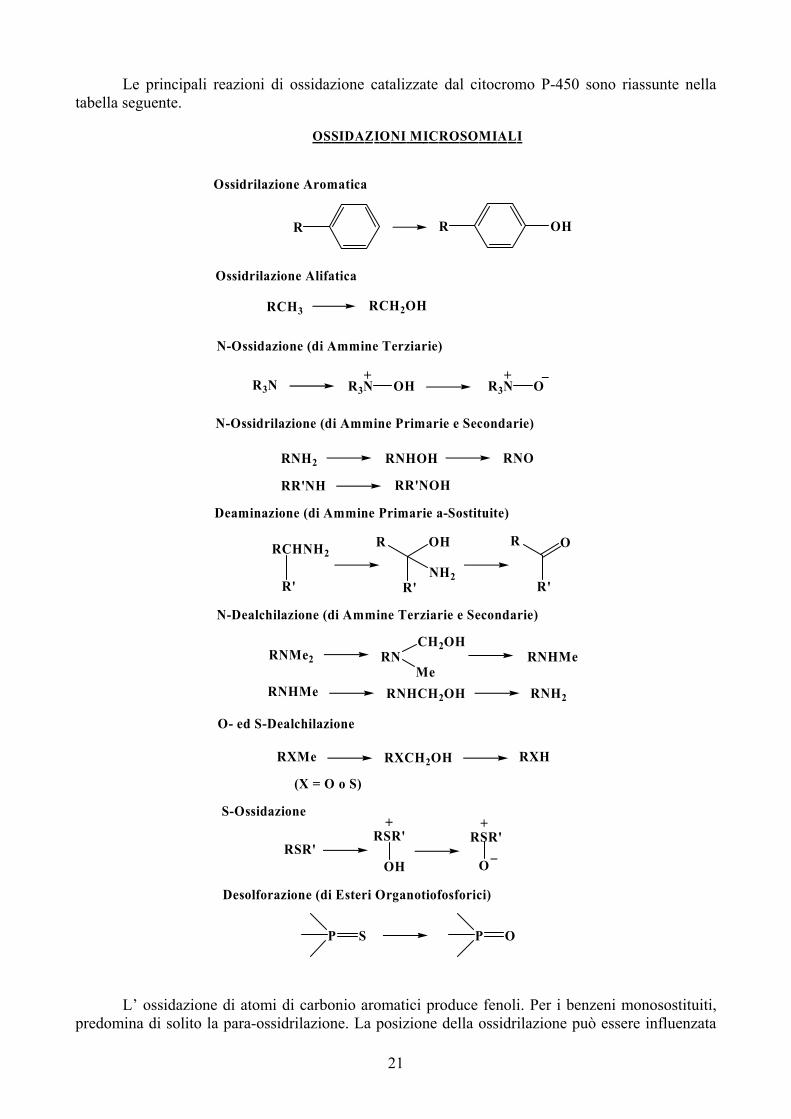
Le principali reazioni di ossidazione catalizzate dal citocromo P-450 sono riassunte nella tabella seguente.
OR3NOHR3NR3N
NH2
OH
R'
R O
R'
RRCHNH2
OPSP
O
RSR'
OH
RSR'RSR'
RXHRXCH2OHRXMe
OHRR
RNH2RNHCH2OHRNHMe
RNHMeMe
CH2OHRNRNMe2
RCH2OHRCH3
RR'NOHRR'NH
RNORNHOHRNH2
_++
_
++
(X = O o S)
S-Ossidazione
O- ed S-Dealchilazione
N-Dealchilazione (di Ammine Terziarie e Secondarie)
Deaminazione (di Ammine Primarie a-Sostituite)
N-Ossidrilazione (di Ammine Primarie e Secondarie)
N-Ossidazione (di Ammine Terziarie)
Ossidrilazione Alifatica
Ossidrilazione Aromatica
Desolforazione (di Esteri Organotiofosforici)
OSSIDAZIONI MICROSOMIALI
R'
L’ ossidazione di atomi di carbonio aromatici produce fenoli. Per i benzeni monosostituiti, predomina di solito la para-ossidrilazione. La posizione della ossidrilazione può essere influenzata
21
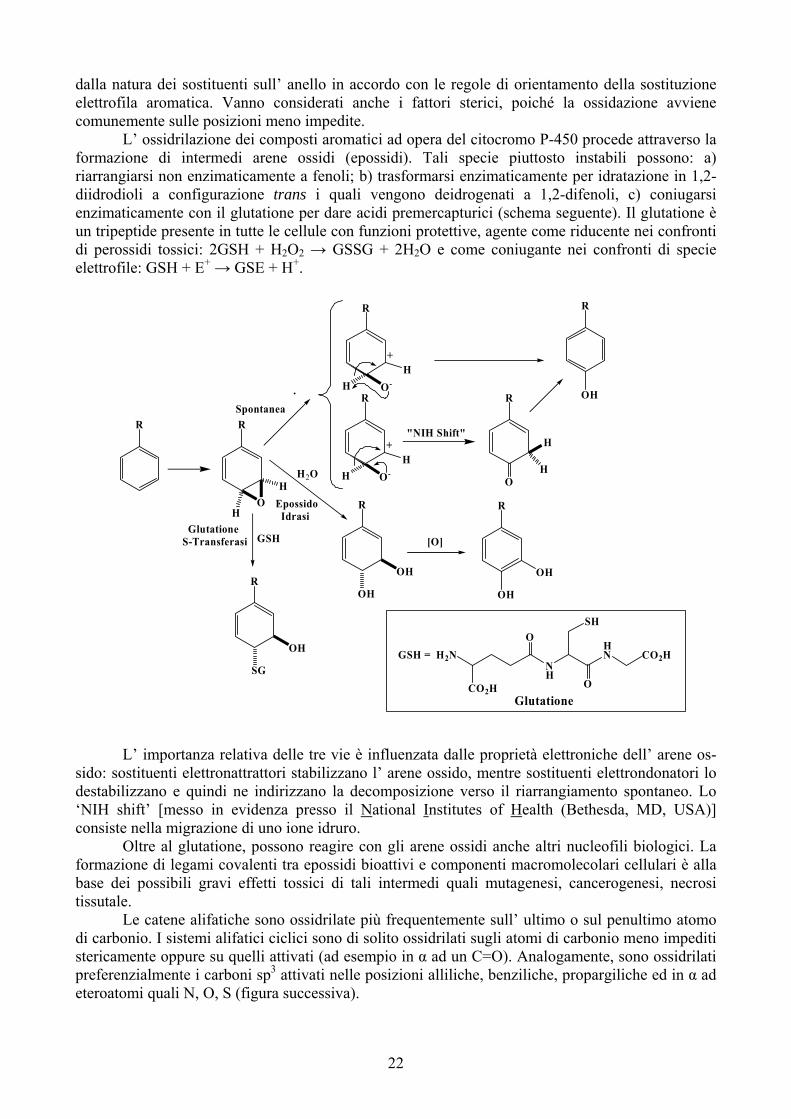
dalla natura dei sostituenti sull’ anello in accordo con le regole di orientamento della sostituzione elettrofila aromatica. Vanno considerati anche i fattori sterici, poiché la ossidazione avviene comunemente sulle posizioni meno impedite. L’ ossidrilazione dei composti aromatici ad opera del citocromo P-450 procede attraverso la formazione di intermedi arene ossidi (epossidi). Tali specie piuttosto instabili possono: a) riarrangiarsi non enzimaticamente a fenoli; b) trasformarsi enzimaticamente per idratazione in 1,2-diidrodioli a configurazione trans i quali vengono deidrogenati a 1,2-difenoli, c) coniugarsi enzimaticamente con il glutatione per dare acidi premercapturici (schema seguente). Il glutatione è un tripeptide presente in tutte le cellule con funzioni protettive, agente come riducente nei confronti di perossidi tossici: 2GSH + H2O2 → GSSG + 2H2O e come coniugante nei confronti di specie elettrofile: GSH + E+ → GSE + H+.
[O]
OH
SG
R
H
H
H2N
CO2H O
OSH
CO2HNN
+H
H O-
R
OH
R
OH
OH
R
OH
OH
R
H
H
O
R
+H
H O-
R
H
HO
RRSpontanea
GSH GlutationeS-Transferasi
GSH =
"NIH Shift"
Epossido Idrasi
Glutatione
H2O
L’ importanza relativa delle tre vie è influenzata dalle proprietà elettroniche dell’ arene os-sido: sostituenti elettronattrattori stabilizzano l’ arene ossido, mentre sostituenti elettrondonatori lo destabilizzano e quindi ne indirizzano la decomposizione verso il riarrangiamento spontaneo. Lo ‘NIH shift’ [messo in evidenza presso il National Institutes of Health (Bethesda, MD, USA)] consiste nella migrazione di uno ione idruro.
Oltre al glutatione, possono reagire con gli arene ossidi anche altri nucleofili biologici. La formazione di legami covalenti tra epossidi bioattivi e componenti macromolecolari cellulari è alla base dei possibili gravi effetti tossici di tali intermedi quali mutagenesi, cancerogenesi, necrosi tissutale. Le catene alifatiche sono ossidrilate più frequentemente sull’ ultimo o sul penultimo atomo di carbonio. I sistemi alifatici ciclici sono di solito ossidrilati sugli atomi di carbonio meno impediti stericamente oppure su quelli attivati (ad esempio in α ad un C=O). Analogamente, sono ossidrilati preferenzialmente i carboni sp3 attivati nelle posizioni alliliche, benziliche, propargiliche ed in α ad eteroatomi quali N, O, S (figura successiva).
22

R CH2 CH3
R CH2 CH2OH
CH3R CH3OHOH
R R
OH
R ROH
RX R
X
OH
R XH
O
H+
X = O, NR', S
Per quanto riguarda la N-dealchilazione, i gruppi alchilici vengono rimossi ossidativamente uno alla volta ad iniziare dal gruppo più piccolo. In genere, le ammine terziarie sono dealchilate a secondarie più rapidamente delle secondarie a primarie; si verifica pertanto un sensibile accumulo di ammine secondarie e primarie come metaboliti e spesso esse contribuiscono all’ azione farmaco-logica della sostanza progenitrice. Anche nel caso delle O- ed S-dealchilazioni, la velocità della reazione è funzione della lunghezza della catena alifatica che viene rimossa: un aumento della lunghezza diminusce la velocità.
La maggior parte dei prodotti di N-ossidazione è bioinattivata a prodotti pola-ri che sono facilmente escre-ti con le urine. In alcuni casi, tuttavia, la N-ossidazione può generare metaboliti reat-tivi e tossici. Ad esempio, le idrossilammine derivanti da ammine aromatiche possono
evolvere in urine acide a ioni nitrenio i quali reagiscono covalentemente con nucleofili biologici, comportandosi quindi da cancerogeni.
NH2 NHOH NH+
NH+NHH
NuNH2Nu
O H+
eccetera
Nu-
-H2O
Nelle frazioni mitocondriali e citosoliche sono presenti altre ossidasi che catalizzano le reazioni indicate a fianco.
RCO2HRCHO
RCHORCH=NHRCH2NH2
RR'CORR'CHOH
RCHORCH2OH
Acido 6-tiourico6-Mercaptopurina
Ossidazione di Purine
Deaminazione Ossidativa
Ossidazione di Aldeidi
Ossidazione di Alcoli
OSSIDAZIONI NON MICROSOMIALI
N
N N
N
H
SH
N
N N
N
H
SH
OHOH
L’ alcool deidrogenasi è un enzima localizzato nella frazione solubile degli omogenati tissutali (citosol) che ossida la maggior parte degli alcoli primari ad aldeidi; solo alcuni alcoli secondari sono convertiti a chetoni, mentre altri alcoli secondari e tutti quelli terziari sono escreti inalterati oppure come metaboliti coniugati. Le aldeidi, sia quelle prodotte per os-sidazione degli alcoli primari che quelle ot-tenute per deamminazione ossidativa delle ammine (vedi successivamente), sono ossi-date ad acidi da una serie di enzimi quali al-deide deidrogenasi, aldeide ossidasi, xantina ossidasi.
23

L’ ossidazione ad opera dell’ alcool deidrogenasi è la principale via di metabolizzazione del-l’ etanolo ad acetaldeide (~ 2/3), che viene poi a sua volta ossidata ad acido acetico dall’ aldeide de-idrogenasi; il rimanente ~ 1/3 viene invece ossidato da un sistema enzimatico microsomiale (MEOS, microsomal ethanol oxidizing system). Il metanolo viene ossidato piuttosto lentamente a formaldei-de dall’ alcool deidrogenasi (velocità ~ 1/6 rispetto a quella dell’ EtOH) e ciò costituisce la base dell’ impiego dell’ EtOH nel trattamento delle intossicazioni acute da MeOH. L’ EtOH deprime ul-teriormente la velocità della ossidazione dell’ MeOH a CH2O (metabolita tossico, così come l’ aci-do formico che si forma per successiva ossidazione della formaldeide), agendo come substrato com-petitivo per l’ enzima. Le monoammino ossidasi (MAO) e le diammino ossidasi (DAO) catalizzano la deammina-zione ossidativa delle ammine ad aldeidi. Le MAO sono enzimi mitocondriali esistenti in due forme, A e B, ed i loro substrati sono le monoammine primarie prive di sostituenti sul carbonio in α RCH2NH2. Le ammine secondarie vengono ossidate dalle MAO a patto che il sostituente sia un gruppo metilico RCH2NHMe. Fisiologicamente, esse intervengono nella regolazione della degra-dazione metabolica nei tessuti neurali delle catecolammine e della serotonina, ammine biogene con funzione di neurotrasmettitori; inoltre svolgono un ruolo cruciale nella inattivazione delle monoam-mine circolanti e di quelle presenti negli alimenti (tiramina) o prodotte da essi nel tratto gastointe-stinale.
NH2
HO
HO
NHMeOH
HO
HO
OHNH2
HO
HO
Dopamina
Epinefrina oAdrenalina
Norepinefrina oNoradrenalina
CATECOLAMMINE
HO NH2
NH
NH2
NH
HO
NH2
Serotonina
Triptamina
Tiramina
Le diammine del tipo H2N(CH2)nNH2 con n < 6 non sono attaccate dalle MAO. Se la distanza intramolecolare tra i due gruppi NH2 cresce, la velocità di ossidazione da parte delle MAO
aumenta. Le DAO (enzimi citosolici presenti nei reni, nell’ intestino, nel fegato, nei polmoni e nel tessuto ner-voso) attaccano tutte le diammine e l’ istamina, forman-do, come le MAO, aldeidi. Esse svolgono un importante ruolo di regolazione degli effetti biologici dell’ istamina e delle poliamine biogene quali putrescina
H2N(CH2)4NH2, spermina H2N(CH2)3NH(CH2)4NH(CH2)3NH2 e spermidina H2N(CH2)3NH (CH2)4NH2.
N
NH
NH2 N
NH
CHO
Istamina
Numerosi derivati purinici vengono ossidati ad analoghi dell’ acido urico ad opera della xan-tina ossidasi, un enzima citosolico che interviene fisiologicamente negli stadi finali del catabolismo dei nucleotidi derivanti dalla degradazione degli acidi nucleici. Se le basi puriniche libere che si for-mano per idrolisi dai nucleotidi non vengono recuperate e riutilizzate, esse sono trasformate in aci-do urico che viene escreto nelle urine (schema pagina successiva). Reazioni di Riduzione Oltre a sistemi ossidativi, i microsomi epatici contengono sistemi enzimatici che catalizzano la riduzione di azo- e nitrocomposti ad ammine primarie: RN=NR’ → RNH2 + R’NH2 e RNO2 →
24
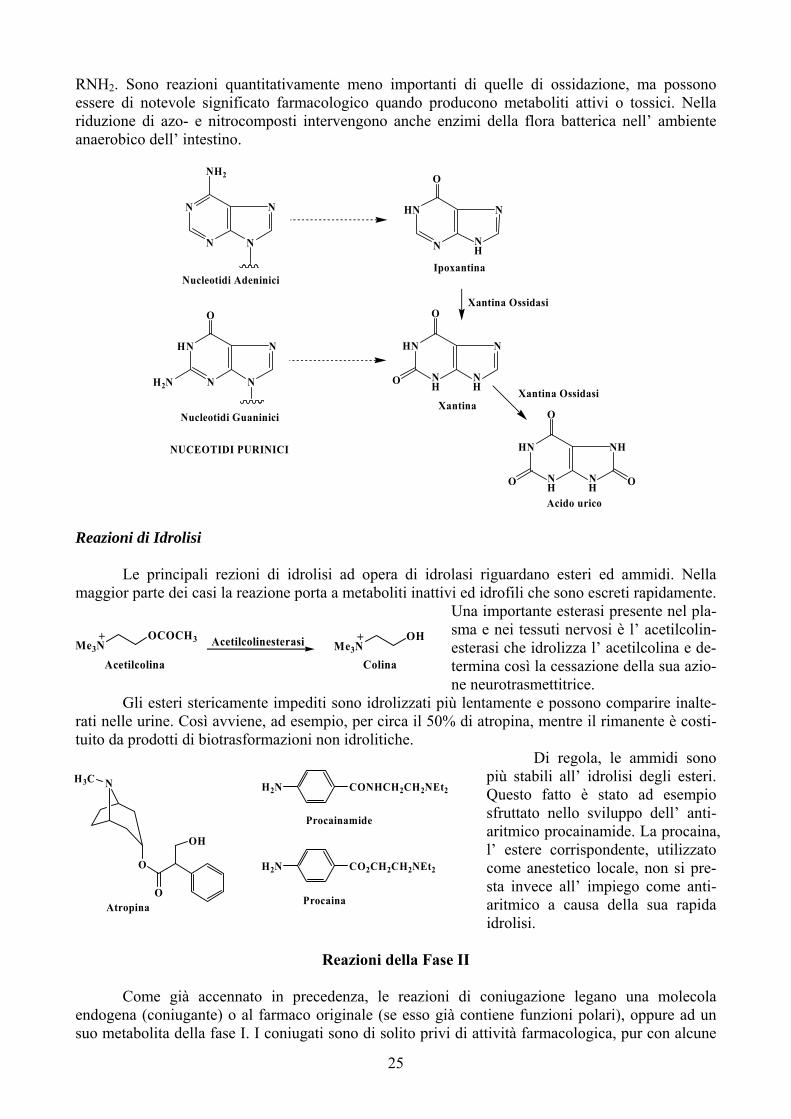
RNH2. Sono reazioni quantitativamente meno importanti di quelle di ossidazione, ma possono essere di notevole significato farmacologico quando producono metaboliti attivi o tossici. Nella riduzione di azo- e nitrocomposti intervengono anche enzimi della flora batterica nell’ ambiente anaerobico dell’ intestino.
O
O
O NH
NH
NH
HN
O
O
NH
N
NH
HN
O
NH
N
N
HN
H
H2N
O
N
N
N
N
NH2
N
N
N
N
Xantina Ossidasi
Xantina Ossidasi
Acido urico
Xantina
Ipoxantina
Nucleotidi Guaninici
Nucleotidi Adeninici
NUCEOTIDI PURINICI
Reazioni di Idrolisi Le principali rezioni di idrolisi ad opera di idrolasi riguardano esteri ed ammidi. Nella maggior parte dei casi la reazione porta a metaboliti inattivi ed idrofili che sono escreti rapidamente.
Una importante esterasi presente nel pla-sma e nei tessuti nervosi è l’ acetilcolin-esterasi che idrolizza l’ acetilcolina e de-termina così la cessazione della sua azio-ne neurotrasmettitrice.
Me3NOCOCH3+
Me3NOH+
Acetilcolina
Acetilcolinesterasi
Colina
Gli esteri stericamente impediti sono idrolizzati più lentamente e possono comparire inalte-rati nelle urine. Così avviene, ad esempio, per circa il 50% di atropina, mentre il rimanente è costi-tuito da prodotti di biotrasformazioni non idrolitiche.
Di regola, le ammidi sono più stabili all’ idrolisi degli esteri. Questo fatto è stato ad esempio sfruttato nello sviluppo dell’ anti-aritmico procainamide. La procaina, l’ estere corrispondente, utilizzato come anestetico locale, non si pre-sta invece all’ impiego come anti-aritmico a causa della sua rapida idrolisi.
NH3C
O
O
OH
Atropina
H2N CONHCH2CH2NEt2
H2N CO2CH2CH2NEt2
Procainamide
Procaina
Reazioni della Fase II
Come già accennato in precedenza, le reazioni di coniugazione legano una molecola endogena (coniugante) o al farmaco originale (se esso già contiene funzioni polari), oppure ad un suo metabolita della fase I. I coniugati sono di solito privi di attività farmacologica, pur con alcune
25
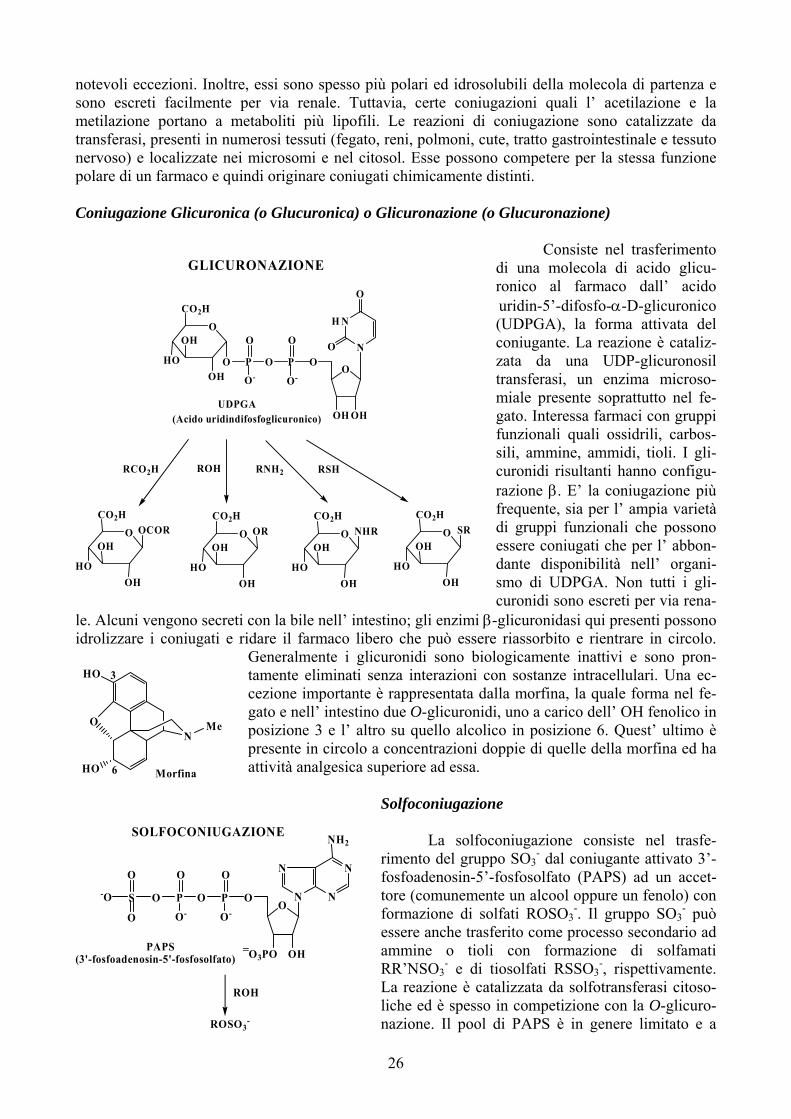
notevoli eccezioni. Inoltre, essi sono spesso più polari ed idrosolubili della molecola di partenza e sono escreti facilmente per via renale. Tuttavia, certe coniugazioni quali l’ acetilazione e la metilazione portano a metaboliti più lipofili. Le reazioni di coniugazione sono catalizzate da transferasi, presenti in numerosi tessuti (fegato, reni, polmoni, cute, tratto gastrointestinale e tessuto nervoso) e localizzate nei microsomi e nel citosol. Esse possono competere per la stessa funzione polare di un farmaco e quindi originare coniugati chimicamente distinti. Coniugazione Glicuronica (o Glucuronica) o Glicuronazione (o Glucuronazione)
Consiste nel trasferimento di una molecola di acido glicu-ronico al farmaco dall’ acido uridin-5’-difosfo-α-D-glicuronico (UDPGA), la forma attivata del coniugante. La reazione è cataliz-zata da una UDP-glicuronosil transferasi, un enzima microso-miale presente soprattutto nel fe-gato. Interessa farmaci con gruppi funzionali quali ossidrili, carbos-sili, ammine, ammidi, tioli. I gli-curonidi risultanti hanno configu-razione β. E’ la coniugazione più frequente, sia per l’ ampia varietà di gruppi funzionali che possono essere coniugati che per l’ abbon-dante disponibilità nell’ organi-smo di UDPGA. Non tutti i gli-curonidi sono escreti per via rena-
le. Alcuni vengono secreti con la bile nell’ intestino; gli enzimi β-glicuronidasi qui presenti possono idrolizzare i coniugati e ridare il farmaco libero che può essere riassorbito e rientrare in circolo.
Generalmente i glicuronidi sono biologicamente inattivi e sono pron-tamente eliminati senza interazioni con sostanze intracellulari. Una ec-cezione importante è rappresentata dalla morfina, la quale forma nel fe-gato e nell’ intestino due O-glicuronidi, uno a carico dell’ OH fenolico in posizione 3 e l’ altro su quello alcolico in posizione 6. Quest’ ultimo è presente in circolo a concentrazioni doppie di quelle della morfina ed ha attività analgesica superiore ad essa.
(Acido uridindifosfoglicuronico)UDPGA
O-O-
HOOH
OH
CO2H
O
O
O
P O
O
P
OHOH
OO
H
O
O
N
N
RSHRNH2ROHRCO2H
SR
HOOH
OH
CO2H
ONHR
HOOH
OH
CO2H
OOR
HOOH
OH
CO2H
OOCOR
HOOH
OH
CO2H
O
GLICURONAZIONE
O
HO
HO
NMe
3
6 Morfina
Solfoconiugazione
-O
O
O
S O
O
P O
O
P
O
O
NH2
ROSO
RO
3PO OH
ON
N
N
N
3-
H
=(3'-fosfoadenosin-5'-fosfosolfato)
PAPS
SOLFOCONIUGAZIONE
O- O-
La solfoconiugazione consiste nel trasfe-- al coniugante attivato 3’-
ato (P PS) ad un accet-ol op nolo) con O
rimento del gruppo SO3 dfosfoadenosin-5’-fosfosolf Atore (comunemente un alco pure un feformazione di solfati ROSessere anche trasferito comammine o tioli con foRR’NSO3
- e di tiosolfati La reazione è catalizzata dliche ed è spesso in compenazione. Il pool di PAPS
3-. Il gruppo SO3
- può e processo secondario ad rmazione di solfamati RSSO3
-, rispettivamente. a solfotransferasi citoso-tizione con la O-glicuro-è in genere limitato e a
26
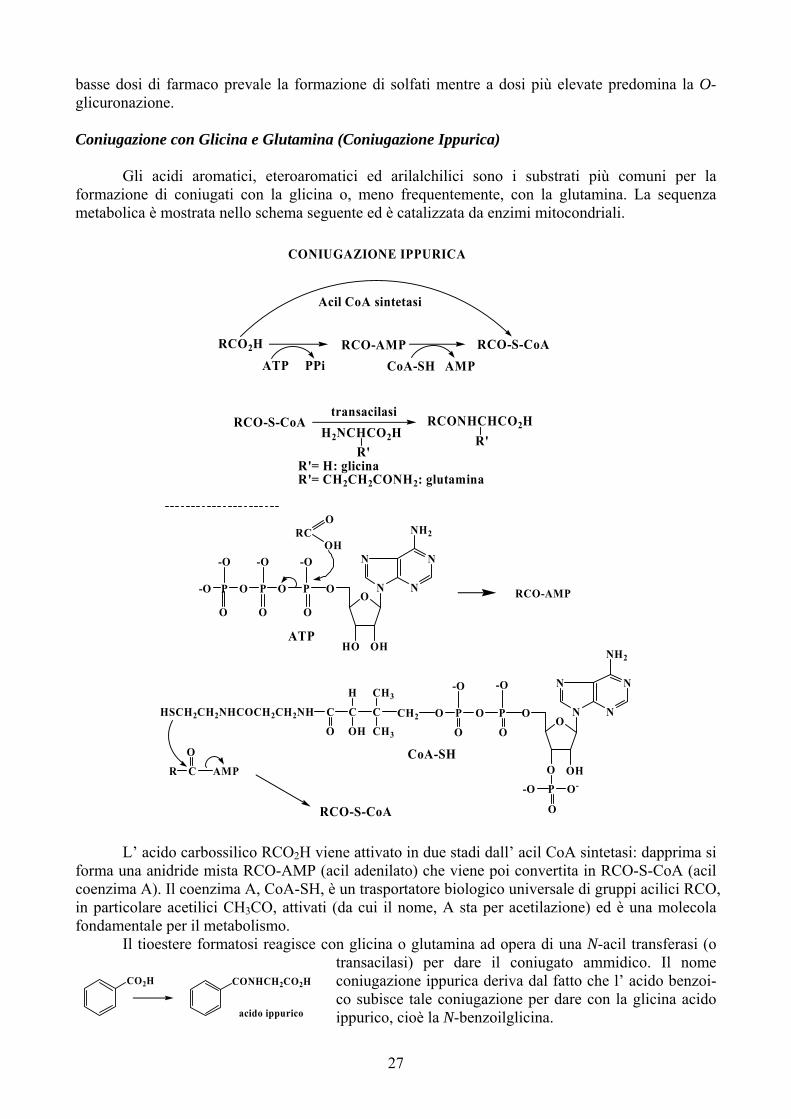
basse dosi di farmaco prevale la formazione di solfati mentre a dosi più elevate predomina la O-glicuronazione. Coniugazione con Glicina e Glutamina (Coniugazione Ippurica) Gli acidi aromatici, eteroaromatici ed arilalchilici sono i substrati più comuni per la formazione di coniugati con la glicina o, meno frequentemente, con la glutamina. La sequenza metabolica è mostrata nello schema seguente ed è catalizzata da enzimi mitocondriali.
O
CR AMP
RCO-AMP
R'= H: glicinaR'= CH2CH2CONH2: glutamina
R'H2NCHCO2H
transacilasi
R'RCONHCHCO2HRCO-S-CoA
Acil CoA sintetasi
RCO-S-CoAAMPCoA-SHPPiATP
RCO2H
CONIUGAZIONE IPPURICA
OH
O O
-O-O
-O P O P O
O
-O
P O
HO OH
O
NH2
N
N
N
N
RCO-S-CoA
CoA-SH
HSCH2CH2NHCOCH2CH2NH
O
C
OH
H
C
CH3
CH3
C CH2
O
-O
O P O
O
-O
P
O
O-P
O
O
OH
O
NH2
N
N
N
N
RC
RCO-AMP
O
-O
ATP
L’ acido carbossilico RCO2H viene attivato in due stadi dall’ acil CoA sintetasi: dapprima si forma una anidride mista RCO-AMP (acil adenilato) che viene poi convertita in RCO-S-CoA (acil coenzima A). Il coenzima A, CoA-SH, è un trasportatore biologico universale di gruppi acilici RCO, in particolare acetilici CH3CO, attivati (da cui il nome, A sta per acetilazione) ed è una molecola fondamentale per il metabolismo. Il tioestere formatosi reagisce con glicina o glutamina ad opera di una N-acil transferasi (o
transacilasi) per dare il coniugato ammidico. Il nome coniugazione ippurica deriva dal fatto che l’ acido benzoi-co subisce tale coniugazione per dare con la glicina acido ippurico, cioè la N-benzoilglicina.
CO2H CONHCH2CO2H
acido ippurico
27
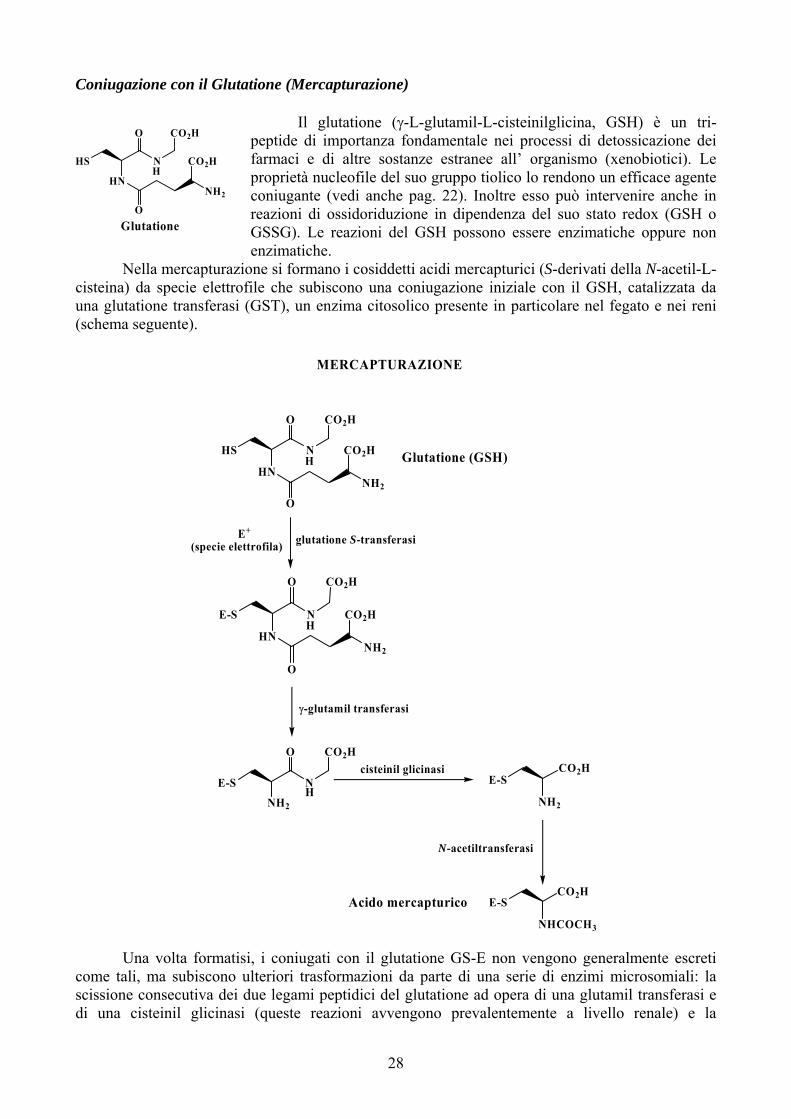
Coniugazione con il Glutatione (Mercapturazione)
Il glutatione (γ-L-glutamil-L-cisteinilglicina, GSH) è un tri-peptide di importanza fondamentale nei processi di detossicazione dei farmaci e di altre sostanze estranee all’ organismo (xenobiotici). Le proprietà nucleofile del suo gruppo tiolico lo rendono un efficace agente coniugante (vedi anche pag. 22). Inoltre esso può intervenire anche in reazioni di ossidoriduzione in dipendenza del suo stato redox (GSH o GSSG). Le reazioni del GSH possono essere enzimatiche oppure non enzimatiche.
H
CO2H
HS N
O
NH2
CO2H
O
HN
Glutatione
Nella mercapturazione si formano i cosiddetti acidi mercapturici (S-derivati della N-acetil-L-cisteina) da specie elettrofile che subiscono una coniugazione iniziale con il GSH, catalizzata da una glutatione transferasi (GST), un enzima citosolico presente in particolare nel fegato e nei reni (schema seguente).
(specie elettrofila)
HN
O
CO2H
NH2
O
N
CO2H
E-SH
Acido mercapturicoNHCOCH3
E-SCO2H
NH2
E-SCO2Hcisteinil glicinasi
NH2
E-S
CO2H
NH
O
E+
H
CO2H
HS N
O
NH2
CO2H
O
HN
MERCAPTURAZIONE
Glutatione (GSH)
N-acetiltransferasi
γ-glutamil transferasi
glutatione S-transferasi
Una volta formatisi, i coniugati con il glutatione GS-E non vengono generalmente escreti come tali, ma subiscono ulteriori trasformazioni da parte di una serie di enzimi microsomiali: la scissione consecutiva dei due legami peptidici del glutatione ad opera di una glutamil transferasi e di una cisteinil glicinasi (queste reazioni avvengono prevalentemente a livello renale) e la
28
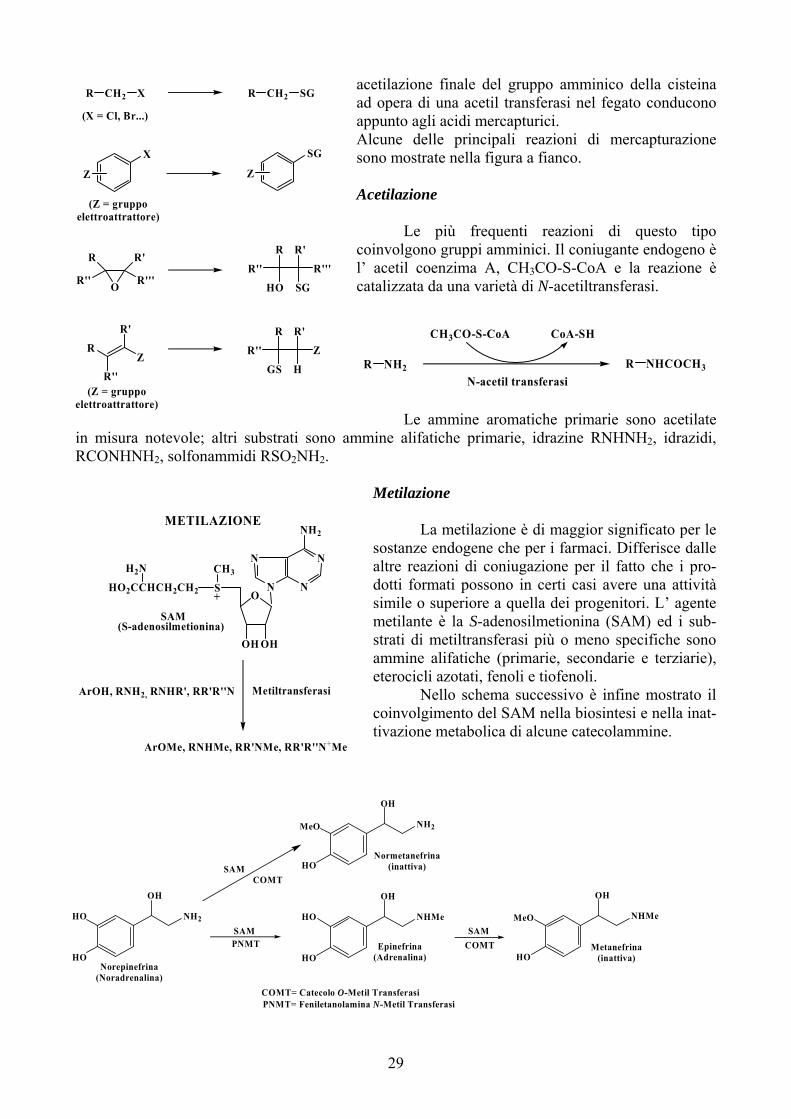
acetilazione finale del gruppo amminico della cisteina ad opera di una acetil transferasi nel fegato conducono appunto agli acidi mercapturici. Alcune delle principali reazioni di mercapturazione sono mostrate nella figura a fianco. Acetilazione Le più frequenti reazioni di questo tipo coinvolgono gruppi amminici. Il coniugante endogeno è l’ acetil coenzima A, CH3CO-S-CoA e la reazione è catalizzata da una varietà di N-acetiltransferasi.
Le ammine aromatiche primarie sono acetilate in misura notevole; altri substrati sono ammine alifatiche primarie, idrazine RNHNH2, idrazidi, RCONHNH2, solfonammidi RSO2NH2.
R CH2 X R CH2 SG
(X = Cl, Br...)
X
Z
SG
Z
(Z = gruppo elettroattrattore)
OR''
R'
R'''R''
HO
R R'
R'''
SG
R
Z
R'
R
R''
R''
R R'
Z
HGS
(Z = gruppo elettroattrattore)
CoA-SHCH3CO-S-CoA
NHCOCH3RNH2RN-acetil transferasi
Metilazione
HO2CCHCH2CH2 S
OH OH
O
NH2
N
N
N
N
ArOMe, RNHMe, RR'NMe, RR'R''N+Me
ArOH, RNH2, RNHR', RR'R''N
(S-adenosilmetionina)
+
Metiltransferasi
SAM
METILAZIONE
H2N CH3
La metilazione è di maggior significato per le sostanze endogene che per i farmaci. Differisce dalle altre reazioni di coniugazione per il fatto che i pro-dotti formati possono in certi casi avere una attività simile o superiore a quella dei progenitori. L’ agente metilante è la S-adenosilmetionina (SAM) ed i sub-strati di metiltransferasi più o meno specifiche sono ammine alifatiche (primarie, secondarie e terziarie), eterocicli azotati, fenoli e tiofenoli. Nello schema successivo è infine mostrato il coinvolgimento del SAM nella biosintesi e nella inat-tivazione metabolica di alcune catecolammine.
MeO
OH
NHMe
HO
MeO
OH
NH2
HO
OH
NHMe
HO
HO
OH
NH2
HO
HO
Metanefrina(inattiva)
Epinefrina(Adrenalina)
Normetanefrina(inattiva)
Norepinefrina(Noradrenalina)
PNMT= Feniletanolamina N-Metil TransferasiCOMT= Catecolo O-Metil Transferasi
PNMT COMT
COMT
SAM SAM
SAM
29

Fase Farmacodinamica
Introduzione
La fase farmacodinamica descrive il comportamento del farmaco al sito d’ azione (o biofase), quel distretto dell’ organismo cioè in cui il farmaco interagisce con un bersaglio biologico specifico e da tale interazione scaturisce una variazione di un qualche processo o di una qualche funzione fisiologica ed in ciò consiste l’ effetto farmacologico del farmaco. I farmaci quindi agiscono influenzando (potenziando o deprimendo a seconda dei casi) funzioni normali e preesistenti dei tessuti e degli organi, non generando nuove funzioni. L’ effetto di un farmaco è sempre espresso in termini relativi, in relazione alle condizioni fisiologiche al momento della somministrazione. Le macromolecole biologiche bersaglio dei farmaci sono principalmente proteine, di cinque tipi: ● enzimi; ● canali ionici; ● sistemi trasportatori; ● proteine strutturali; ● recettori.
La sola eccezione importante alle proteine come bersagli sono gli acidi nucleici (DNA). Tra i componenti del sistema biologico ed i farmaci possono avere luogo numerose altre interazioni che sono state esaminate nel capitolo Farmacocinetica, ad esempio il legame con l’ albumina plasmatica. Queste interazioni hanno conseguenze secondarie per l’ azione del farmaco, quali la durata e la ve-locità dell’ azione.
Una minoranza di farmaci agisce extracellularmente su componenti non cellulari dell’ orga-nismo. In questo caso non sono coinvolte interazioni con biopolimeri. Esempi al riguardo sono gli antiacidi che neutralizzano gli ioni H+ del succo gastrico, gli agenti chelanti che legano metalli pe-santi tossici ed i diuretici e purganti osmotici che agiscono in virtù delle loro proprietà colligative, inducendo per osmosi appropriate modificazioni nella distribuzione di acqua. Enzimi Numerosi farmaci agiscono su sistemi enzimatici, attivandoli o, più spesso, inibendoli. Fre-quentemente il farmaco si comporta da analogo del substrato enzimatico ed agisce come inibitore competitivo, o reversibilmente o irreversibilmente. Rimandando al corso di Biochimica per una di-scussione completa dell’ argomento, gli schemi cinetici relativi alle interazioni substrato-enzima ed inibitore-enzima sono i seguenti:
a) un enzima E si combina con il substrato S per dare il complesso reversibile S.E il quale può evolvere nel prodotto P, rigenerando l’ enzima libero; b) nel caso di un inibitore reversibile e competitivo (cioè in grado di competere con il substrato per il sito attivo dell’ enzima) si ha la formazione di un complesso non produttivo I.E; c) se l’ inibizione è non competitiva (se cioè l’ inibitore si lega ad un sito enzimatico diverso da quello del substrato) si ha la formazione dei complessi inattivi I.E e I.S.E, poiché I è in grado di legarsi sia all’ enzima libero che al complesso S.E; d) un inibitore che si lega soltanto al complesso S.E è un inibitore acompetitivo (o incompetitivo); e) nel caso, infine, di inibizione irreversibile di tipo competitivo, alla formazione del complesso I.E fa se-
S + E S.E P + E
E
I
I.E I.S.ES
S.ES
I
I + E I.E
I + E I.E I E
30

guito generalmente quella di un addotto covalente tra enzima ed inibitore. Una serie di esempi di enzimi bersaglio di farmaci correnti è la seguente:
Enzima bersaglio Impiego clinico
Anidrasi carbonica DNA polimerasi virale 3C proteasi del Rhinovirus Xantina ossidasi Trombina Cicloossigenasi PBP ACE Dopa decarbossilasi β-lattamasi Na+/K+ ATPasi Trascrittasi inversa dell’ HIV Testosterone 5α-reduttasi Timidilato sintasi HMG-CoA reduttasi Diidrofolato reduttasi DNA girasi H+/K+ ATPasi
Glaucoma Herpes Raffreddore Gotta Malattie cardiovascolari Infiammazione Infezioni batteriche Ipertensione Morbo di Parkinson Resistenza batterica Malattie cardiovascolari AIDS Iperplasia prostatica Cancro Ipercolesterolemia Cancro, infezioni batteriche Infezioni batteriche Ulcera
Canali ionici Alcuni tipi di canali ionici sono collegati ad un recettore e si aprono solo quando viene attivato il recettore. Altri tipi invece possono essere bersagli diretti di farmaci. L’ argomento verrà precisato successivamente, nel paragrafo sui recettori accoppiati a canali ionici. Sistemi trasportatori Esempi di molecole carrier e di loro inibitori sono:
Carrier Inibitore
Sistema di ricaptazione 1 della noradrenalina Cotrasportatore di Na+/K+/2Cl-
Na+/K+ATPasi
Antidepressivi triciclici, Cocaina Diuretici dell’ ansa Glicosidi cardiaci
Proteine strutturali Un esempio importante è la tubulina, costituente dei microtubuli, in particolare del fuso mitotico, con cui interagiscono gli Alcaloidi della Vinca ed i Taxani. Recettori
I recettori sono elementi sensoriali di natura proteica, localizzati sulla membrana cellulare o all' interno della cellula, facenti parte del sistema di comunicazione che coordina le funzioni di tutte le differenti cellule dell' organismo. I messaggeri chimici che assicurano le comunicazioni tra le cellule sono ormoni, neurotrasmettitori, fattori di crescita, citochine. Alcuni di essi attraversano la membrana cellulare e portano il messaggio direttamente all’ interno della cellula, interagendo con recettori intracellulari; altri invece interagiscono con recettori localizzati sulla membrana cellulare ed il segnale che essi contengono viene trasferito all’ interno della cellula mediante vari meccanismi.
31

I recettori sono in altri termini proteine in grado di attivare determinate funzioni biochimiche all’ in-terno di una cellula a seguito dell’ interazione con molecole endogene di varia natura con funzione di messaggeri chimici (mediatori fisiologici) ed, eventualmente, con farmaci (mediatori esogeni); il termine ligando recettoriale viene impiegato per indicare sia i mediatori endogeni che quelli esogeni. Quasi tutti i farmaci agiscono su recettori per i quali sono noti i ligandi endogeni (ad esempio i recettori dell’ acetilcolina, della noradrenalina, dell’ istamina, della serotonina, della dopamina, dell’ insulina, degli ormoni steroidici). Vi sono però anche esempi di recettori per farmaci per i quali non sono stati ancora identificati i mediatori endogeni (recettori orfani). Il termine recettore viene talvolta impiegato in senso estensivo per indicare qualsiasi biopolimero bersaglio di un farmaco, per cui sono considerati ‘recettori’ anche gli acidi nucleici, gli enzimi e le proteine trasportatrici e strutturali. Tuttavia, è preferibile riservare il termine recettore alle proteine regolatrici che trasmettono informazioni alle cellule a seguito di interazione con messaggeri chimici.
Famiglie Recettoriali Sulla base della struttura molecolare e della natura dei meccanismi di trasduzione (meccanismi di segnalazione transmembrana coinvolgenti il legame del ligando con il recettore e la trasmissione del segnale all’ interno della cellula ad opera di una o più proteine ‘effettori’) si possono distinguere quattro tipi o superfamiglie di recettori: ● Recettori accoppiati ad un canale ionico (ionotropi); ● Recettori accoppiati alle proteine G (metabotropi); ● Recettori accoppiati ad una chinasi (recettori con attività enzimatica intrinseca); ● Recettori intracellulari (attivatori della trascrizione). I recettori delle prime tre categorie sono tutti proteine di membrana, mentre quelli del quarto tipo sono proteine citosoliche o intranucleari. Recettori Ionotropi I recettori accoppiati ad un canale ionico rappresentano i recettori di diversi neu-rotrasmettitori (recettori nicotinici dell’ acetilcolina, di aminoacidi eccitatori come aspartato e gluta-mato e di aminoacidi inibitori come il γ-aminobutirrato e la glicina). Il sito di legame del ligando ed il canale ionico fanno parte di uno stesso complesso proteico e l’ apertura del canale avviene come risposta diretta al legame ligando-recettore. L’ azione del neurotrasmettitore si svolge in una frazione di millisecondi e decade entro pochi millisecondi.
LIGANDOMembrana cellulare
Interno cellula
Esterno cellula
CANALE IONICO
RECETTORE
EFFETTI CELLULARI
IPERPOLARIZZAZIONE O DEPOLARIZZAZIONE
RECETTORI ACCOPPIATI AD UN CANALE IONICO (IONOTROPI)
IONI
32

I neurotrasmettitori eccitatori come l’ acetilcolina inducono l’ apertura di canali cationici, in particolare del sodio e del potassio. Il risultato immediato è l’ afflusso di ioni Na+ nella cellula con conseguente depolarizzazione e generazione di un potenziale d’ azione. L’ attivazione dei recettori di neurotrasmettitori inibitori invece induce l’ apertura di canali anionici (Cl-) con iperpolarizza-zione della cellula. I canali ionici collegati direttamente (come nel caso appena esaminato) oppure indiretta-mente (come nel caso di certi tipi di recettori accoppiati alle proteine G, vedi successivamente) ad un recettore e che si aprono solo in presenza di un ligando sono indicati come canali ionici regolati da un ligando (ligand-gated ion channels). Altri canali ionici sono invece regolati da differenze di potenziale (voltage-gated ion channels). Il più semplice tipo di interazione di questi canali con i farmaci consiste nel blocco fisico del canale (ad esempio, nel caso degli anestetici locali). Recettori Metabotropi
Questa famiglia di recettori comprende recettori di vari ormoni e trasmettitori lenti quali i recettori muscarinici dell’ acetilcolina, i recettori adrenergici, dopaminergici ed oppiacei. L' attiva-zione del recettore da parte di un ligando produce la attivazione di un ‘trasduttore’ il quale, a sua volta, provoca l’ attivazione (o l’ inattivazione) di un enzima (oppure l’ apertura, o la chiusura, di un canale ionico). L’ attivazione dell’ enzima si concretizza nella produzione di un ‘secondo messaggero’ (il primo messaggero è il ligando recettoriale) il quale interviene nella regolazione di specifiche funzioni cellulari (figura successiva). La scala dei tempi è dell' ordine dei secondi. Meccanismi di questo genere danno luogo ad una amplificazione del segnale iniziale, poiché un singolo complesso ligando-recettore può attivare numerose molecole di proteine G, ognuna delle quali, a sua volta, può rimanere associata all’ enzima abbastanza a lungo da provocare la produzione di numerosissime molecole di secondo messaggero.
EFFETTI CELLULARI
ALTROFOSFORILAZIONE DI PROTEINE
RILASCIO DI Ca
PROTEINA G(TRASDUTTORE)
SECONDO MESSAGGERO (es. cAMP)
ENZIMA(AMPLIFICATORE)es. Adenilato Ciclasi
LIGANDOMembranacellulare
Interno cellula
Esterno cellula
RECETTORE
RECETTORI ACCOPPIATI ALLE PROTEINE G (METABOTROPI)
Il trasduttore è comunemente rappresentato dalle proteine G. Le proteine G (così chiamate per la loro interazione con i nucleotidi guaninici GTP e GDP) consistono di tre subunità ancorate alla membrana plasmatica, α, β e γ. I nucleotidi guaninici sono legati alla subunità α.
33

Nello stato a riposo, la proteina G esiste come trimero indissociato con GDP che occupa il sito sulla subunità α. Quando il recettore viene attivato da un ligando, avviene un cambiamento conformazionale nel recettore che lo porta ad associarsi alla proteina G. L’ associazione recettore-proteina G provoca la sostituzione di GDP con GTP che, a sua volta, induce la dissociazione del trimero con rilascio delle subunità α-GTP e β,γ. La subunità α (ma anche il dimero β,γ) può diffondere lungo la membrana ed associarsi con vari enzimi, ad es. adenilato ciclasi, ma anche canali ionici, causandone la attivazione o l’ inattivazione (o la apertura o la chiusura). Si conoscono proteine G stimolatrici e proteine G inibitrici. L’ attivazione dell’ adenilato ciclasi stimola la formazione di AMP ci-clico. Il processo termina con l' idrolisi del GTP a GDP ad opera della subunità α dotata di attività GTPasica. La risultante subunità α-GDP si dissocia dall’ enzima bersaglio e si riunisce al dimero β,γ, ripristinando il trimero e completando così il ciclo.
GDP
GTPcAMP
ATP
GTP
GTP GDP
GDP
GDP
GTP
γβ ACα
γβ ACα
γβ AC
α
INTERNO
ESTERNO
Proteina G
L = LigandoR = Recettore
AC = Adenilato Ciclasi
ACα γβL
ACα γβ
L
ACα γβ
ACα γβLR
R
R
R
R
R
R
N
NHN
N
OOP
HO OH
O-O
OPO-
-OO
PO-
-OO
N
NHN
N
OOP
HO OH
O-O
OPO-
OO
GTP GDP
O O
NH2 NH2
Oltre al sistema adenilato ciclasi/cAMP, altri effettori accoppiati alle proteine G sono il sistema fosfolipasi C/inositolo fosfato e certi canali ionici (in quest’ ultimo caso non sono coinvolti secondi messageri e la proteina G interagisce direttamente con il canale).
Recettori Accoppiati Ad Una Chinasi
I recettori con attività enzimatica intrinseca incorporano un dominio intracellulare con attività di proteina chinasi (in genere una tirosina chinasi). Comprendono i recettori dell’ insulina e di varie citochine e fattori di crescita. Sono coinvolti principalmente in eventi di controllo della crescita e della differenziazione cellulari ed, indirettamente, di regolazione della trascrizione genica.
EFFETTI CELLULARI
FOSFORILAZIONE DI PROTEINE
ENZIMA (CHINASI)
LIGANDOMembrana cellulare
Interno cellula
Esterno cellula
RECETTORE
RECETTORI ACCOPPIATI AD UNA CHINASI
In dettaglio, il legame del ligando porta alla dimerizzazione di una coppia di recettori. L’ as-sociazione dei due domini proteici
34

con attività enzimatica determina l’ autofosforilazione dei residui di tirosina delle chinasi. I residui autofosforilati si legano al dominio SH2 di varie proteine che poi attivano altre proteine, ad esempio le proteine Ras che funzionano come le proteine G. L’ attivazione delle proteine Ras attiva a sua volta una serie di chinasi a cascata, l’ ultima delle quali fosforila un fattore di trascrizione che dà inizio all’ espressione genica. Recettori intracellulari (attivatori della trascrizione) La regolazione recettore-mediata della trascrizione del DNA è caratteristica degli ormoni steroidei e tiroidei. Alcuni di tali recettori sono localizzati nel citoplasma, altri nel nucleo ed i loro ligandi sono tutti composti lipofili in grado di attraversare facilmente la membrana cellulare. A se-guito del legame, ad esempio di una molecola di steroide, il recettore dimerizza. Il dimero si lega a sequenze specifiche del DNA note come elementi di risposta all’ormone (HRE, hormone-responsi-ve elements) che sono localizzate circa 200 coppie di basi a monte dei geni la cui espressione viene regolata dall’ ormone. Altre molecole che agiscono in maniera simile sono la vitamina D e l’ acido retinoico.
Nucleo
DNA
EFFETTI CELLULARI
SINTESIDI PROTEINE
SINTESIDI mRNA
Membranacellulare
Interno cellula
Esterno cellula
RECETTORE
LIGANDO
RECETTORI INTRACELLULARI ATTIVATORI DELLA TRASCRIZIONE
35

Agonisti ed Antagonisti Un agonista recettoriale è un ligando in grado di attivare un recettore inducendo una risposta biologica intrinseca. Un farmaco che si comporta da agonista simula l’ azione del mediatore natu-rale (ligando endogeno, agonista fisiologico) e ne riproduce gli effetti. Un antagonista recettoriale è invece un ligando che si lega ad un recettore senza attivarlo. Quindi un antagonista può bloccare la capacità di un agonista di indurre una risposta.
Ad esempio, il neurotrasmettitore acetilcolina CH3COOCH2CH2N(CH3)3
+ agisce su due sottotipi di re-cettori indicati come recettore muscarinico e recettore ni-cotinico poiché i due gruppi di risposte che l’ acetilcolina produce attivandoli possono essere riprodotti da due al-caloidi: muscarina e nicotina, rispettivamente. L’ attiva-zione dei recettori muscarinici
da parte dell’ acetilcolina o della muscarina provoca diminuzione della frequenza e della forza contrattile cardiache, vasodi-latazione periferica, restringimento della pupilla (miosi), aumento delle secrezioni ghiandolari, con-trazione dei tratti gastrointestinale ed urinario. Un antagonista muscarinico produce effetti opposti: diminuzione della secrezione salivare e del succo gastrico, diminuzione della peristalsi intestinale, dilatazione della pupilla. Farmaci con quest’ ultimo tipo di attività sono di potenziale utilità nel trat-tamento dell’ ulcera, in oftalmologia, come antispastici.
O
NMe+
OHH3C
(+)-2S,4R,5S-muscarina
N
N
CH3
H
(-)-S-nicotina
Indipendentemente dalla natura specifica del recettore R, l’ interazione di un ligando L con esso può essere descritta dallo schema seguente:
L + R L.R Effetto Alla formazione reversibile di un complesso ligando-recettore fa seguito, nel caso di un agonista, l’attivazione di un meccanismo di trasduzione (apertura di un canale ionico, formazione di un secondo messaggero, eccetera) che si traduce in una risposta biologica. Nel caso di un antagonista, i meccanismi di trasduzione non si attivano e l’ effetto osservato è la conseguenza del blocco recettoriale. Gli antagonisti possono essere classificati in base alle loro cinetiche di interazione con il recettore in antagonisti reversibili ed irreversibili. L’ antagonismo irreversibile si verifica non solo con antagonisti che formano legami covalenti con il recettore (evento piuttosto occasionale nelle interazioni farmaco-recettore, mentre è più frequente in quelle farmaco-enzima e farmaco-DNA) ma anche con antagonisti che formano legami non covalenti, ma che si dissociano molto lentamente dal recettore). In base invece al sito di legame, gli antagonisti si suddividono in antagonisti competitivi e non competitivi. I primi interagiscono con lo stesso sito di legame dell’ agonista e competono con esso nel processo di interazione. I secondi interagiscono con un sito distinto da quello di legame dell’ agonista, ma attraverso determinati meccanismi prevengono egualmente l’ attivazione del recettore da parte dell’ agonista.
Le varie categorie di antagonisti hanno effetti diversi sulle curve dose-risposta di un agonista. E
Emax
lg A
Se l’effetto di un agonista, espresso come percentuale della risposta massima, viene riportato in funzione della sua concentrazione, la curva asume la forma di una sigmoide su scala semilogaritmica. Nel caso di antagonisti competitivi e reversibili, la loro aggiunta provoca uno spostamento parallelo verso destra delle curve dose-risposta dell’ agonista. Si parla in questo caso di antagonismo sormontabile. Se in-vece si ha una diminuzione della risposta massima dell’ ago-nista con o senza spostamento delle curve verso destra, l’ an-
36
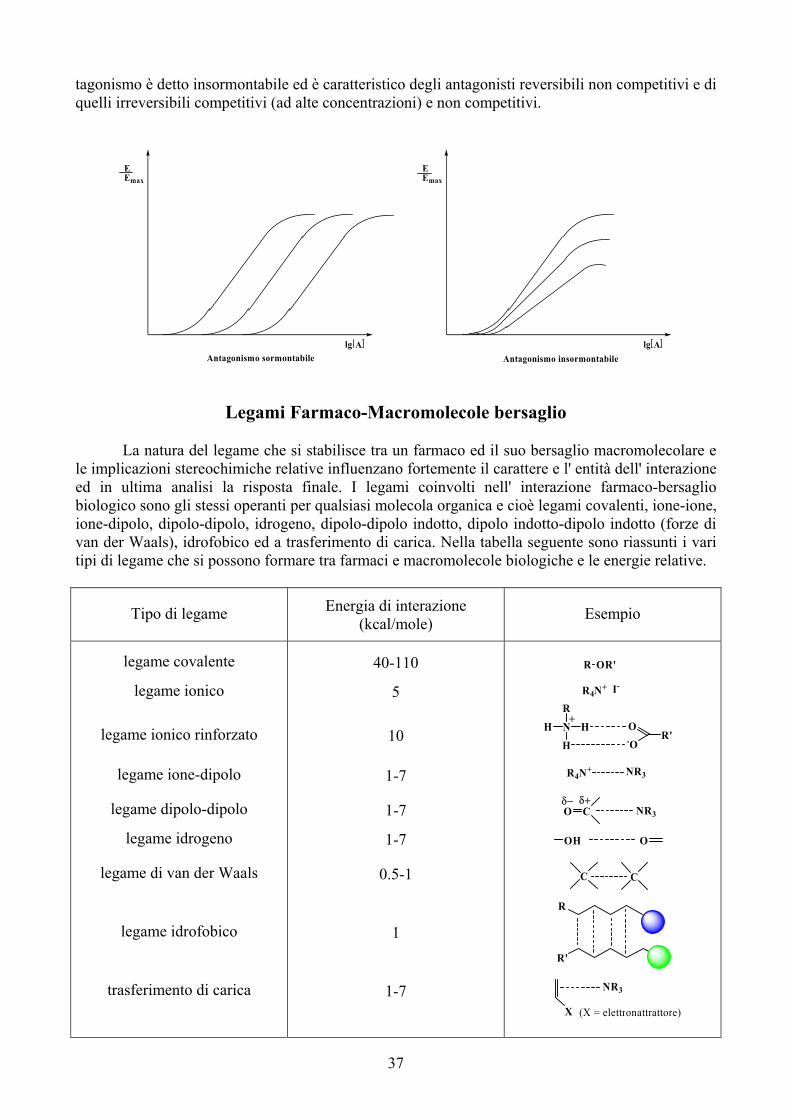
tagonismo è detto insormontabile ed è caratteristico degli antagonisti reversibili non competitivi e di quelli irreversibili competitivi (ad alte concentrazioni) e non competitivi.
EEmax
lg A
EEmax
lg AAntagonismo sormontabile Antagonismo insormontabile
Legami Farmaco-Macromolecole bersaglio La natura del legame che si stabilisce tra un farmaco ed il suo bersaglio macromolecolare e le implicazioni stereochimiche relative influenzano fortemente il carattere e l' entità dell' interazione ed in ultima analisi la risposta finale. I legami coinvolti nell' interazione farmaco-bersaglio biologico sono gli stessi operanti per qualsiasi molecola organica e cioè legami covalenti, ione-ione, ione-dipolo, dipolo-dipolo, idrogeno, dipolo-dipolo indotto, dipolo indotto-dipolo indotto (forze di van der Waals), idrofobico ed a trasferimento di carica. Nella tabella seguente sono riassunti i vari tipi di legame che si possono formare tra farmaci e macromolecole biologiche e le energie relative.
Tipo di legame Energia di interazione (kcal/mole) Esempio
legame covalente
legame ionico
legame ionico rinforzato
legame ione-dipolo
legame dipolo-dipolo
legame idrogeno
legame di van der Waals
legame idrofobico
trasferimento di carica
40-110
5
10
1-7
1-7
1-7
0.5-1 1
1-7
R OR'
N
R
HH
H
+ O-O
R'
I-R4N+
O Cδ+δ−
NR3
OH O
R
R'
CC
R4N+ NR3
NR3
X (X = elettronattrattore)
37
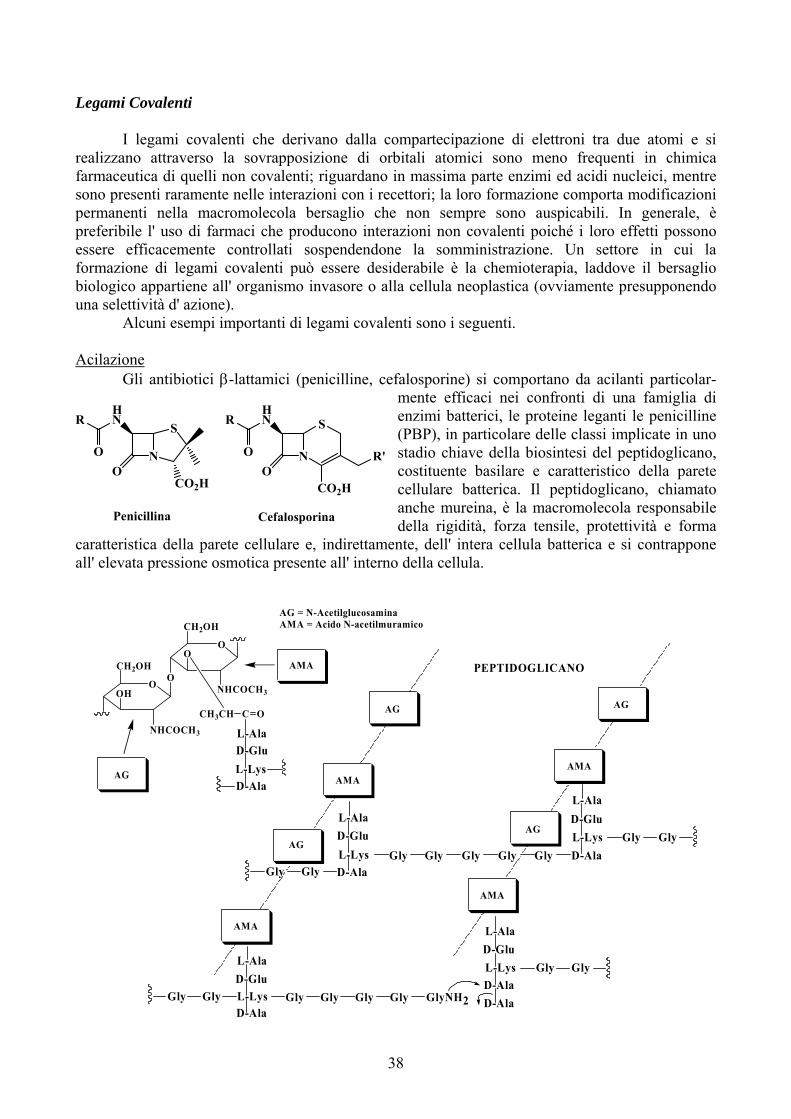
Legami Covalenti I legami covalenti che derivano dalla compartecipazione di elettroni tra due atomi e si realizzano attraverso la sovrapposizione di orbitali atomici sono meno frequenti in chimica farmaceutica di quelli non covalenti; riguardano in massima parte enzimi ed acidi nucleici, mentre sono presenti raramente nelle interazioni con i recettori; la loro formazione comporta modificazioni permanenti nella macromolecola bersaglio che non sempre sono auspicabili. In generale, è preferibile l' uso di farmaci che producono interazioni non covalenti poiché i loro effetti possono essere efficacemente controllati sospendendone la somministrazione. Un settore in cui la formazione di legami covalenti può essere desiderabile è la chemioterapia, laddove il bersaglio biologico appartiene all' organismo invasore o alla cellula neoplastica (ovviamente presupponendo una selettività d' azione). Alcuni esempi importanti di legami covalenti sono i seguenti. Acilazione Gli antibiotici β-lattamici (penicilline, cefalosporine) si comportano da acilanti particolar-
mente efficaci nei confronti di una famiglia di enzimi batterici, le proteine leganti le penicilline (PBP), in particolare delle classi implicate in uno stadio chiave della biosintesi del peptidoglicano, costituente basilare e caratteristico della parete cellulare batterica. Il peptidoglicano, chiamato anche mureina, è la macromolecola responsabile della rigidità, forza tensile, protettività e forma
caratteristica della parete cellulare e, indirettamente, dell' intera cellula batterica e si contrappone all' elevata pressione osmotica presente all' interno della cellula.
N
S
O
HNR
O
CO2H
Penicillina
NO
HNR
O
S
CO2H
R'
Cefalosporina
OH
NHCOCH3
CH2OH
O
O
ONHCOCH3
CH2OH
O
OCCH3CH
AG L-Lys
L-Ala
D-Glu
Gly GlyL-Lys
L-Ala
D-AlaGly GlyGly Gly Gly D-Ala
L-LysD-GluL-Ala
Gly Gly
D-Glu
D-Ala
PEPTIDOGLICANO
D-AlaL-LysD-GluL-Ala
D-Ala
Gly Gly
L-LysD-GluL-Ala
D-AlaGly GlyGly Gly GlyNH2Gly Gly
AG = N-AcetilglucosaminaAMA = Acido N-acetilmuramico
AMA
AG
AG AG
AG
AMA
AMA
AMA
AMA
38

Il peptidoglicano (figura precedente) è un complesso eteropolimero costituito da una parte glicidica e da una peptidica. La componente glicidica è una catena lineare di n unità di un disaccaride costituito da N-acetilglucosamina ed acido N-acetilmuramico uniti tra loro mediante legame glicosidico tra il carbonio 1 della N-acetilglucosamina ed il carbonio 4 dell' acido N-acetilmuramico. Questa componente è sostanzialmente identica in tutti i batteri. La componente peptidica della mureina è formata da un tetrapeptide (la composizione più comune è L-Ala-D-Glu-L-Lys-D-Ala) unito alla porzione glicidica da un legame ammidico tra il carbossile dell' acido N-acetilmuramico e l' aminogruppo del primo aminoacido del tetrapeptide. Nel loro insieme, queste due componenti costituiscono l' unità strutturale fondamentale del peptidoglicano. Esso tuttavia non sarebbe un polimero molto robusto e non conferirebbe resistenza alla parete cellulare se non si avessero anche dei legami crociati tra il tetrapeptide di una unità e quello di una unità adiacente. Esiste un' alta variabilità nella natura del ponte tra le unità. Nello S. aureus il ponte è formato da cinque glicine. Nella formazione del legame crociato, il gruppo amminico terminale ad una estremità della catena pentaglicinica attacca il legame peptidico tra due residui di D-alanina terminali di un' altra unità peptidica. Il processo è catalizzato dalle PBP con attività transpeptidasica che agiscono formando un legame covalente con la penultima D-alanina del peptide per attacco nucleofilo di un residuo serinico presente al sito attivo dell' enzima sul carbonile ammidico tra le due D-Ala. L' intermedio acil enzima reagisce poi con il gruppo amminico della glicina terminale formando il legame trasversale e rigenerando l' enzima (Figura successiva).
CH2OH NHGly OC
CH2OH
CO2-
OC
NH
OC
CH2O
H2N Gly
Enzima
D-Ala
D-Ala
Enzima
D-Ala
D-Ala
D-Ala
Enzima
H
N
NH
H
Me
CO2H
H
O
H
Me
Me
MeS
H
O
H
CO2H
N
N
R
O
O
H
Peptidoglicano
Penicillina
Residuo D-Ala-D-Ala
Analogia strutturale tra una penicillina ed il residuo D-Ala-D-Ala del pentapeptide
39
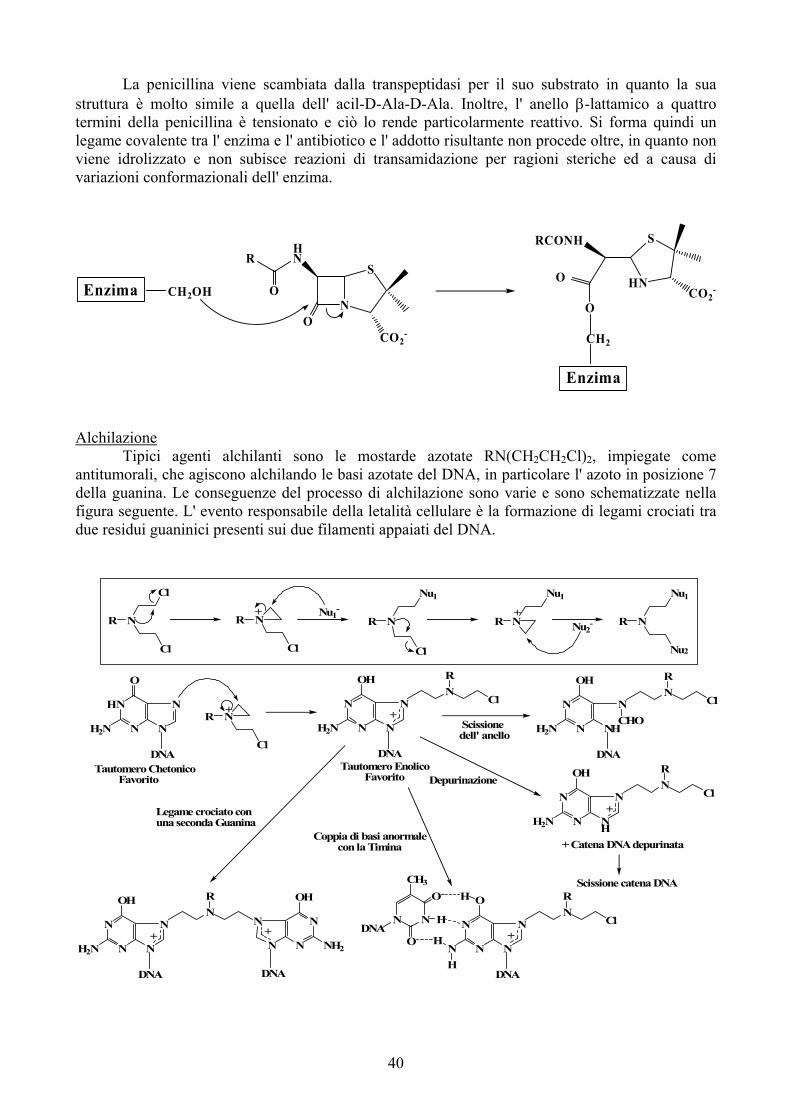
La penicillina viene scambiata dalla transpeptidasi per il suo substrato in quanto la sua struttura è molto simile a quella dell' acil-D-Ala-D-Ala. Inoltre, l' anello β-lattamico a quattro termini della penicillina è tensionato e ciò lo rende particolarmente reattivo. Si forma quindi un legame covalente tra l' enzima e l' antibiotico e l' addotto risultante non procede oltre, in quanto non viene idrolizzato e non subisce reazioni di transamidazione per ragioni steriche ed a causa di variazioni conformazionali dell' enzima.
CH2
O
O
RCONH
CH2OH
Enzima
EnzimaN
SHN
OCO2
-
R
O HN
S
CO2-
Alchilazione Tipici agenti alchilanti sono le mostarde azotate RN(CH2CH2Cl)2, impiegate come antitumorali, che agiscono alchilando le basi azotate del DNA, in particolare l' azoto in posizione 7 della guanina. Le conseguenze del processo di alchilazione sono varie e sono schematizzate nella figura seguente. L' evento responsabile della letalità cellulare è la formazione di legami crociati tra due residui guaninici presenti sui due filamenti appaiati del DNA.
DNA
H
H
H
O
N N
N
N
N Cl
RN
DNA
CH3
H
O
NN
H
H
Nu1
NR Nu2-
Cl
Cl
NR
Cl
Nu1
NRNu1
-
Cl
NR
Nu
Nu1
NR
Cl
RN
OH
H2N N
N
N
N
N NH2
N
OH
N
N
RN
DNA
OH
H2N N
N
N
N
Cl
RN
DNA
OH
H2N N
N
N
N
Cl
NR
DNA
H
H2N
O
N
N
N
N Cl
RN
CHO
OH
H2N N
N
N
N
DNA
++
Legame crociato conuna seconda Guanina
Coppia di basi anormale con la Timina
Depurinazione
Scissionedell' anello
2
Scissione catena DNA
+
+ +
DNA
+ Catena DNA depurinata
++
Tautomero Chetonico Favorito
Tautomero Enolico Favorito
O +
40

Fosforilazione I composti organofosforici sono inibitori dell' acetilcolinesterasi, dell' enzima deputato alla idrolisi e quindi all' inattivazione dell' acetilcolina.
R'YR'Y
X
ORY P HOH2C ORY P
OH2CAcetilcolinesterasi
R, R' = Alchile o ArileY, Y' = O, talvolta SX = Gruppo uscente
Due dei tre esempi riportati si riferiscono a farmaci le cui azioni farmacologiche sono dovute alla loro capacità di inibire irreversibilmente un enzima, modificando covalentemente un gruppo funzionale enzimatico essenziale per la sua attività catalitica. Inibitori irreversibili di questo tipo sono indicati con il termine di marcatori per affinità (affinity labels). Un marcatore per affinità è quindi un reagente diretto al sito attivo dell' enzima e che possiede un gruppo chimico reattivo di natura elettrofila in grado di formare un addotto irreversibile con l' enzima bersaglio. Lo schema cinetico è quello già visto per una inbizione irreversibile competitiva.
EII EI + E
Un meccanismo inibitorio diverso dal precedente, anche se comunemente di tipo irreversibile anch' esso, è l' inibizione basata sul meccanismo o inibizione suicida. Un inibitore suicida è una sostanza che non possiede inizialmente le caratteristiche di inibitore, viene trattata dall' enzima come un substrato e, una volta legatasi al sito attivo dell' enzima, viene trasformata dall' enzima stesso in una specie reattiva (inibitore) che si lega fortemente all' enzima prima del rilascio dal sito attivo. L' enzima quindi produce il proprio inbitore a partire da un composto inattivo e commette con ciò suicidio. I tipi di inibizione che si possono verificare sono di vario tipo. La varietà 'classica' e più frequente di inibitori suicidi comprende intermedi reattivi con proprietà elettrofile che si combinano covalentemente con l' enzima e producono una inibizione irreversibile. Lo schema cinetico di tali inibitori è il seguente, dove S' = pseudosubstrato, IR = intermedio reattivo e P = prodotto della reazione enzimatica.
EIRS' ES' + E EIR
P + E
k1 k2
k3
k4
k-1 inattivaz.normale
Mentre i marcatori per affinità possono, a causa della loro reattività intrinseca reagire con enzimi (o altre biomolecole) diversi da quello bersaglio, gli inibitori suicidi sono in linea di principio attivati solamente dall' enzima bersaglio, con il conseguente vantaggio di minori effetti indesiderati. La strategia più seguita nella progettazione degli inibitori suicidi consiste nella generazione, catalizzata dall' enzima, di specie elettrofile che reagiscono poi covalentemente con gruppi nucleo-fili presenti al sito attivo dell' enzima.Gli schemi seguenti illustrano alcuni dei principali tipi di rea-zione che hanno luogo nell' attivazione del substrato e nella successiva inattivazione dell' enzima.
41
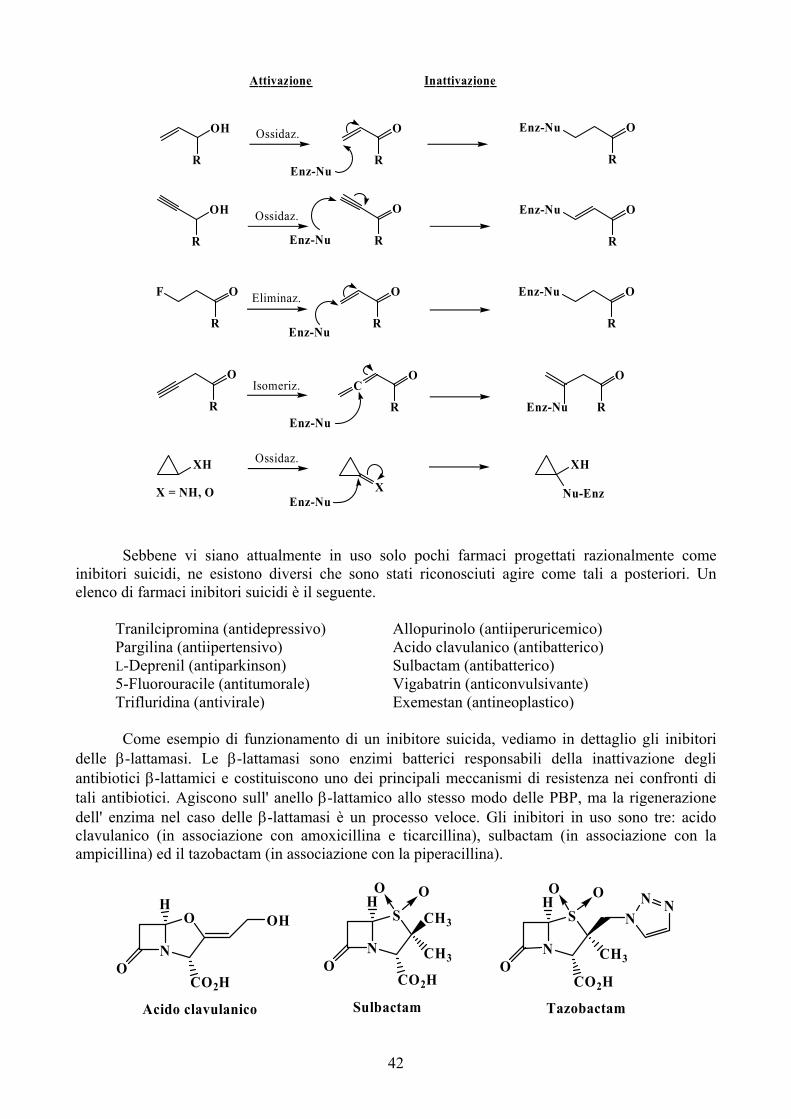
OH
R
O
R
O
R
Enz-Nu
OH
R
O
R
O
R
Enz-Nu
Enz-Nu
Enz-Nu
O
R
F O
R
O
R
Enz-Nu
O
R
O
R
CO
REnz-Nu
XH
X Nu-Enz
XH
Enz-Nu
Enz-Nu
Enz-Nu
Attivazione Inattivazione
Ossidaz.
Ossidaz.
Eliminaz.
Ossidaz.
Isomeriz.
X = NH, O
Sebbene vi siano attualmente in uso solo pochi farmaci progettati razionalmente come inibitori suicidi, ne esistono diversi che sono stati riconosciuti agire come tali a posteriori. Un elenco di farmaci inibitori suicidi è il seguente.
Tranilcipromina (antidepressivo) Pargilina (antiipertensivo) L-Deprenil (antiparkinson) 5-Fluorouracile (antitumorale) Trifluridina (antivirale)
Allopurinolo (antiiperuricemico) Acido clavulanico (antibatterico) Sulbactam (antibatterico) Vigabatrin (anticonvulsivante) Exemestan (antineoplastico)
Come esempio di funzionamento di un inibitore suicida, vediamo in dettaglio gli inibitori delle β-lattamasi. Le β-lattamasi sono enzimi batterici responsabili della inattivazione degli antibiotici β-lattamici e costituiscono uno dei principali meccanismi di resistenza nei confronti di tali antibiotici. Agiscono sull' anello β-lattamico allo stesso modo delle PBP, ma la rigenerazione dell' enzima nel caso delle β-lattamasi è un processo veloce. Gli inibitori in uso sono tre: acido clavulanico (in associazione con amoxicillina e ticarcillina), sulbactam (in associazione con la ampicillina) ed il tazobactam (in associazione con la piperacillina).
OH
N
O
OCO2H
HOO
N
S
O
CH3
CH3
CO2H
H H
CO2H
CH3O
S
N
NN N
OO
Acido clavulanico Sulbactam Tazobactam
42
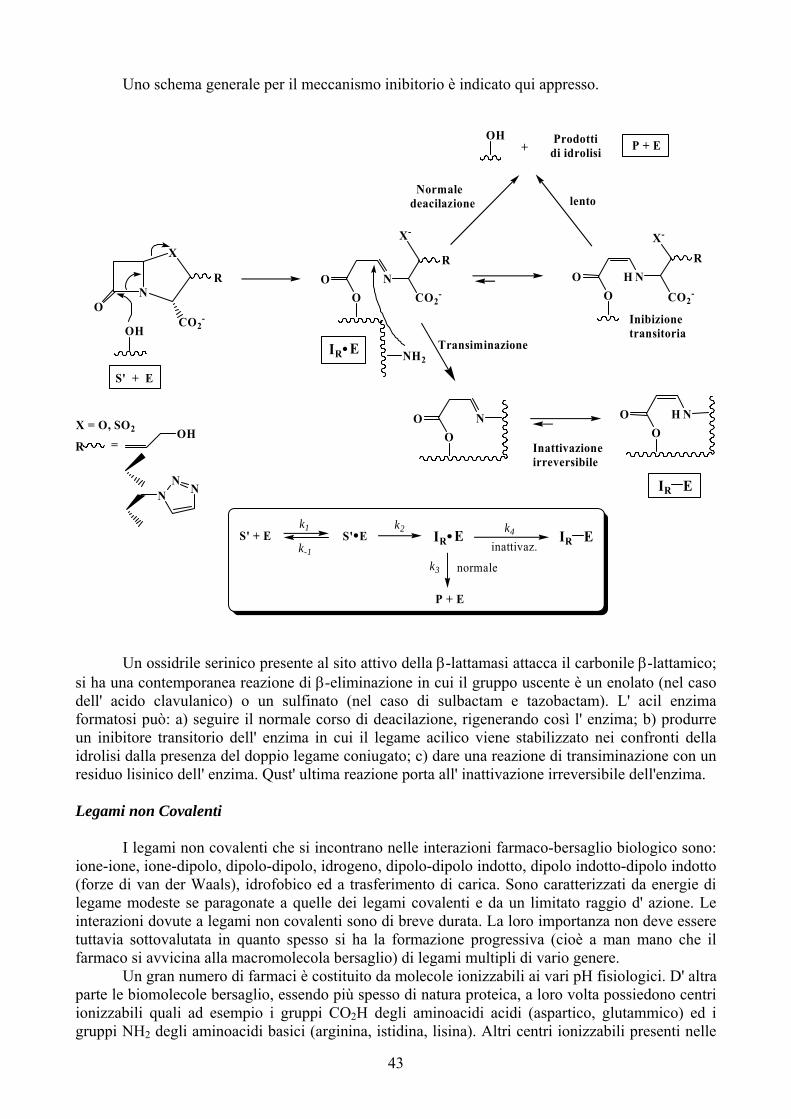
Uno schema generale per il meccanismo inibitorio è indicato qui appresso.
OHR
HO
O N
R
O
X-
CO2-
N
ON
NH2
HR
OO
X-
CO2-
N
OH
R
CO2-
O
X
N
OH
=X = O, SO2
Transiminazione
S' + E
Inibizionetransitoria
Inattivazioneirreversibile
Prodottidi idrolisi+
Normaledeacilazione lento
EIR
S' ES' + E
EIR
P + E
k1 k2
k3
k4
k-1 inattivaz.
normale
N NN
O
O
P + E
EIR EIR
Un ossidrile serinico presente al sito attivo della β-lattamasi attacca il carbonile β-lattamico; si ha una contemporanea reazione di β-eliminazione in cui il gruppo uscente è un enolato (nel caso dell' acido clavulanico) o un sulfinato (nel caso di sulbactam e tazobactam). L' acil enzima formatosi può: a) seguire il normale corso di deacilazione, rigenerando così l' enzima; b) produrre un inibitore transitorio dell' enzima in cui il legame acilico viene stabilizzato nei confronti della idrolisi dalla presenza del doppio legame coniugato; c) dare una reazione di transiminazione con un residuo lisinico dell' enzima. Qust' ultima reazione porta all' inattivazione irreversibile dell'enzima. Legami non Covalenti I legami non covalenti che si incontrano nelle interazioni farmaco-bersaglio biologico sono: ione-ione, ione-dipolo, dipolo-dipolo, idrogeno, dipolo-dipolo indotto, dipolo indotto-dipolo indotto (forze di van der Waals), idrofobico ed a trasferimento di carica. Sono caratterizzati da energie di legame modeste se paragonate a quelle dei legami covalenti e da un limitato raggio d' azione. Le interazioni dovute a legami non covalenti sono di breve durata. La loro importanza non deve essere tuttavia sottovalutata in quanto spesso si ha la formazione progressiva (cioè a man mano che il farmaco si avvicina alla macromolecola bersaglio) di legami multipli di vario genere. Un gran numero di farmaci è costituito da molecole ionizzabili ai vari pH fisiologici. D' altra parte le biomolecole bersaglio, essendo più spesso di natura proteica, a loro volta possiedono centri ionizzabili quali ad esempio i gruppi CO2H degli aminoacidi acidi (aspartico, glutammico) ed i gruppi NH2 degli aminoacidi basici (arginina, istidina, lisina). Altri centri ionizzabili presenti nelle
43

macromolecole biologiche sono i residui fosforici degli acidi nucleici, i centri carichi dei fosfolipidi e dei polisaccaridi. Pertanto le interazioni tra ioni di segno opposto sono molto frequenti. Anche le molecole neutre possono avere gruppi in cui si hanno separazioni di cariche parziali con formazione di dipoli permanenti, come risultato della differenza di elettronegatività in particolare tra il carbonio ed atomi quali azoto, ossigeno, alogeni. Chetoni, esteri, eteri, ammidi, nitrili sono tutti gruppi contenenti dipoli responsabili potenzialmente di interazioni dipolari con il bersaglio biologico. Il legame idrogeno, caso particolare di interazione dipolo-dipolo, consiste in una interazione elettrostatica tra una coppia di elettroni non condivisi legati ad un eteroatomo (N, O, S) ed un idrogeno elettron-deficiente di gruppi quali NH, OH, SH e rappresenta il meccanismo fondamentale per accrescere la solubilità di un farmaco polare non elettrolita. Le forze di van der Waals sono le forme di attrazione tra gli atomi più universale. Esse inter-vengono ogni qualvolta due atomi di molecole diverse si avvicinano sufficientemente. Sono anche indicate come forze di dispersione di London e sono basate sulla induzione di asimmetria nella nube elettronica di un atomo da parte del nucleo di un atomo vicino. Si verifica una mutua polarizzazione delle nuvole elettroniche con formazione di dipoli momentanei complementari. La energia in gioco è piuttosto debole e decresce molto rapidamente con la distanza tra le molecole; ad esempio per una coppia di gruppi CH2 assomma a ~ 0.7 Kcal/mole. Tuttavia, quando le porzioni non polari interagenti sono sufficientemente estese e di appropriata configurazione, le forze di van der Waals possono contribuire significativamente alla stabilizzazione del complesso farmaco-biomolecola. Gli anelli aromatici e le catene alifatiche lineari o poco ramificate soddisfano a questi requisiti. Il legame idrofobico rappresenta una delle più importanti interazioni implicate nella conservazione della struttura terziaria delle proteine. Il termine legame non è in realtà appropriato, dal momento che l' interazione è sempre del tipo van der Waals, ma deriva da un guadagno di entropia del solvente acquoso ed più corretto perciò parlare di effetto idrofobico. Le porzioni apolari di una molecola presente in soluzione acquosa sono circondate da un mantello idratante estremamente ordinato (strutture a gabbia) a bassa entropia che non è compensata a sufficienza dall' energia (fattore entalpico) associata alle deboli interazioni di tipo dipolo-dipolo indotto tra l' acqua e le porzioni apolari (si ricordi che la variazione di energia libera per un processo è data da ∆G = ∆H – T ∆S, dove ∆H è la variazione di entalpia, ∆S è la variazione di entropia e T è la temperatura assoluta. Per un processo spontaneo ∆G < 0). Di conseguenza, le
parti apolari di due molecole diverse tendono ad aggregarsi spontaneamente tra loro in modo da diminuire il numero delle strutture a gabbia attorno ad esse ed accrescere quindi lo stato di disordine del sistema con aumento del fattore entropico (l' aumento di entropia del solvente è maggiore in valore assoluto della diminuzione di entropia del soluto) che compensa ampiamente la variazione sfavorevole di entalpia. La forza motrice che determina quindi l' instaurarsi delle interazioni di van der Waals tra due porzioni apolari di due molecole (in particolare di un farmaco e di una biomolecola) in soluzione acquosa è l' au-mento di entropia del solvente. I complessi a trasferimeno di carica si originano tra molecole elettron-ricche che agiscono da donatrici (D) e molecole
elettron-povere che si comportano da accettrici (A). Il termine 'trasferimento di carica' si riferisce ad
Catena apolaredel farmaco
Catena apolaredel biopolimero
molecolad' acqua
44

una successione di eventi tra una molecola D ed una molecola A che può andare dalle deboli interazioni di van der Waals fino alla formazione di una coppia ionica. In altri termini, un complesso a trasferimento di carica è un ibrido tra due strutture limiti, una in cui le molecole interagiscono tramite le forze di dispersione e l' altra in cui D ed A sono legate da un legame ionico.
D + A D.....A Dδ+ Aδ− D+ A-
Tipiche molecole donatrici sono gli eterociclici π elettron-ricchi pirrolo, furano e tiofene (contengono 6 elettroni π distribuiti su 5 atomi), i composti aromatici con sostituenti elettron-donatori ed i composti con doppietti elettronici non condivisi; molecole accettrici sono invece ad esempio sistemi eterociclici π elettron-poveri quali gli anelli purinico e pirimidinico e composti aromatici con sostituenti elettron-attrattori.
Stereochimica ed Attività
I processi che costituiscono la farmacocinetica e la farmacodinamica di un farmaco comprendono una serie continua di interazioni con biopolimeri dotati spesso di grande specificità strutturale. E' evidente quindi che gli aspetti stereochimici di tali interazioni rivestono un ruolo di grande importanza ai fini delle caratteristiche farmacologiche di un farmaco. Vi è una chiara tendenza negli ultimi 10-15 anni, stimolata da raccomandazioni al riguardo da parte degli enti sanitari di controllo sui farmaci, verso lo sviluppo di agenti stereoisomericamente (in particolare enantomericamente) puri. Richiami di Stereochimica Organica Stereoisomeri sono isomeri che differiscono soltanto per il modo con cui gli atomi che li costituiscono sono disposti nello spazio. Essi possono essere classificati in base all' entità delle barriere energetiche che ne limitano la interconversione ed in base a criteri di simmetria. Sulla base di criteri di interconvertibilità, gli stereoisomeri possono essere distinti in isomeri configurazionali, interconvertibili per rottura di un legame covalente, ed in isomeri conformazionali, interconvertibili per rotazione attorno ad un legame singolo. La facilità di interconversione è carat-teristica di quasi tutti gli isomeri conformazionali, mentre gli isomeri configurazionali l' intercon-versione implica la rottura di un legame covalente, per la quale rottura la barriera energetica è molto alta. Sulla base di criteri di simmetria gli stereoisomeri sono distinguibili in enantiomeri (isomeri ottici) ed in diastereoisomeri, a seconda che le coppie di stereoisomeri siano o meno immagini speculari (non sovrapponibili). La non sovrapponibilità delle immagini speculari è definita chiralità e gli atomi della molecola responsabili della chiralità sono indicati come centri chirali. Mentre due diastereoisomeri hanno proprietà fisiche e chimiche differenti, due enantiomeri hanno identiche proprietà chimiche e fisiche ad eccezione del comportamento verso reagenti chirali e del senso di rotazione del piano della luce polarizzata, rispettivamente. Sebbene la maggior parte delle molecole che esistono come coppie di enantiomeri contenga almeno un atomo di carbonio asimmetrico (cioè legato a quattro sostituenti diversi), la presenza di atomi di carbonio asimmetrici non è condizione né necessaria né sufficiente per l' esistenza di enantiomeri. La diastereoisomeria si origina dall' esistenza in una molecola di due o più centri chirali, oppure dalla ristretta rotazione attorno ad un legame (isomeria geometrica o cis-trans). Nel primo caso, se n sono i centri chirali, il numero massimo di stereoisomeri è 2n. Le coppie di enantiomeri sono 2n/2 ed ogni enantiomero di una coppia è in relazione di diastereoisomeria con tutti i componenti delle altre coppie. Nel secondo caso, le due categorie più importanti sono quelle derivanti dalla presenza di doppi legami e di cicli. L' isomeria geometrica può essere o meno accompagnata da quella ottica.
45

Enantiomeria ed Attività In linea generale ed indipendentemente dallo stadio (o da gli stadi) in cui è operante la selettività, si verifica, come risultato più frequente, che due enantiomeri mostrano risposte quantitativamente differenti, cioè uno dei due enantiomeri è più potente del' altro. L' enantiomero più potente è denominato eutomero, quello meno potente distomero. Il rapporto tra la potenza dello eutomero e quella del distomero è definito rapporto eudismico. Esso aumenta all' aumentare della potenza dell' eutomero (regola di Pfeiffer). Altre possibilità sono che uno solo dei due enantiomeri sia attivo (l' enantiomero inattivo potrebbe tuttavia contribuire agli effetti collaterali), che i due enantiomeri posseggano qualitativamente e quantitativamente la stessa attività e che i due enantiomeri mostrino attività di tipo differente. Due enantiomeri possono perfino produrre effetti opposti, cioè comportarsi come una coppia agonista-antgonista. In quest' ultimo caso però le potenze sono diverse. Tornando al caso più spesso osservato, e cioè stesso tipo di attività ma diversa potenza per i due enantiomeri, tale risultato è la conseguenza di processi selettivi che possono avvenire sia nel corso della fase farmacocinetica che di quella farmacodinamica. Per quanto concerne le possibilità di selezione di uno dei due enantiomeri durante la fase farmacocinetica, si possono fare le seguenti generalizzazioni. Selettività di membrana: in genere, i processi di difusione passiva non sono stereoselettivi, mentre lo sono quelli che coinvolgono proteine trasportatrici; Biopolimeri non specifici: le due principali proteine sieriche coinvolte in legami non specifici con pressoché tutti i farmaci sono l' albumina e l' α1-glicoproteina acida. Entrambe mostrano affinità diverse per gli enentiomeri di numerosi farmaci; Captazioni stereoselettive: Sono state osservate captazioni selettive nei confronti di alcuni farmaci da parte del tessuto adiposo e di alcuni organi quali cuore, cervello, polmoni; Escrezione selettiva: La filtrazione glomerulare ed il riassorbimento tubulare (processi passivi) non sono in grado di discriminare tra due enantiomeri. La selettività eventualmente osservata durante l' escrezione renale può quindi essere la conseguenza o di un legame preferenziale con le proteine plasmatiche, oppure di una secrezione tubulare stereoselettiva; Metabolismo selettivo: Numerosi enzimi farmaco-metabolizzanti interagiscono preferen-zialmente, anche se non esclusivamente, con uno dei due enantiomeri; inoltre essi possono anche produrre preferibilmente uno dei due possibili enantiomeri a partire da un precursore non chirale. Le reazioni metaboliche dal punto di vista stereochimico possono presentare: - stereoselettività di substrato, quando gli enantiomeri di un substrato chirale sono metabolizzati con velocità differenti; - stereoselettività di prodotto, quando in un substrato non chirale viene creato un centro chirale ed uno dei due enantiomeri è prodotto a preferenza; - stereoselettività di substrato-prodotto in cui viene creato, preferibilmente a carico di uno dei due enantiomeri, un ulteriore centro chirale in una molecola già chirale e uno dei due possibili diastereoisomeri viene prodotto a preferenza.
C Cachir.
C Cachir.
conf. R
conf. S*
*
no biotrasformaz.
C Cconf. R
*
C Cconf. R
*
*
*
conf. R
conf. S
preferenz.
DiastereoisomeripossibiliX
X
46
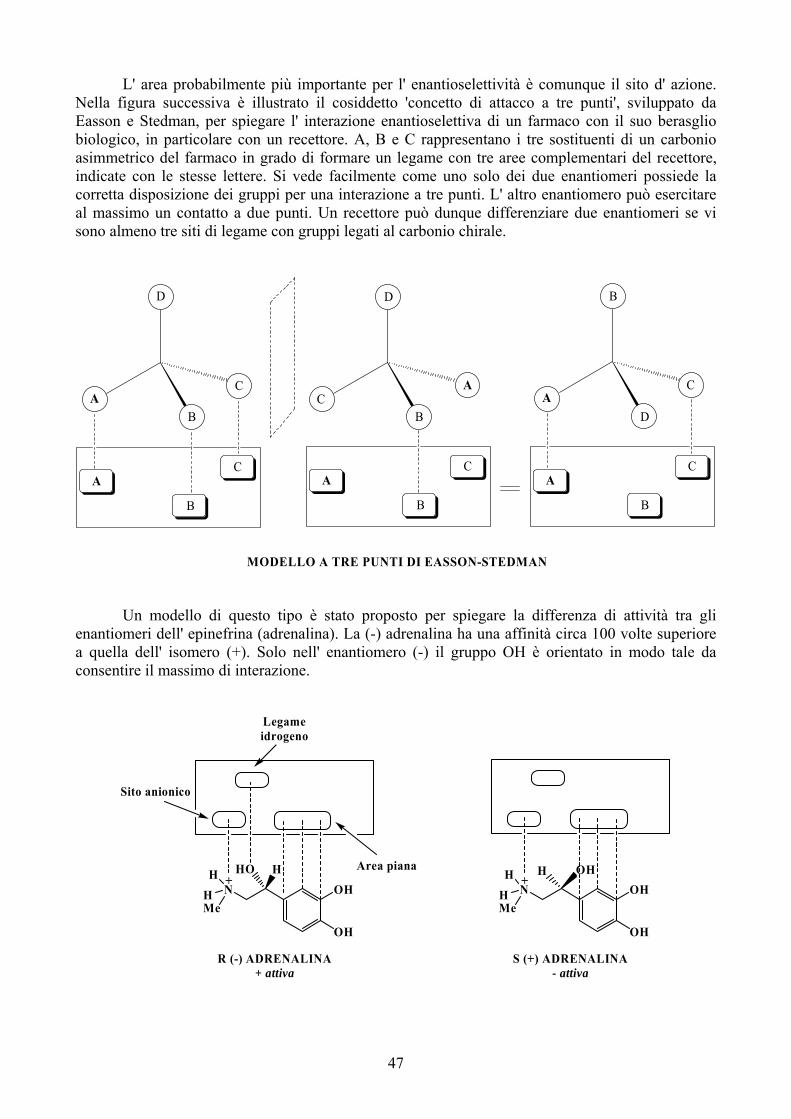
L' area probabilmente più importante per l' enantioselettività è comunque il sito d' azione. Nella figura successiva è illustrato il cosiddetto 'concetto di attacco a tre punti', sviluppato da Easson e Stedman, per spiegare l' interazione enantioselettiva di un farmaco con il suo berasglio biologico, in particolare con un recettore. A, B e C rappresentano i tre sostituenti di un carbonio asimmetrico del farmaco in grado di formare un legame con tre aree complementari del recettore, indicate con le stesse lettere. Si vede facilmente come uno solo dei due enantiomeri possiede la corretta disposizione dei gruppi per una interazione a tre punti. L' altro enantiomero può esercitare al massimo un contatto a due punti. Un recettore può dunque differenziare due enantiomeri se vi sono almeno tre siti di legame con gruppi legati al carbonio chirale.
AAA
A
B
BD
CB D
D
CC
A
B
CA
B
CA
B
C
MODELLO A TRE PUNTI DI EASSON-STEDMAN Un modello di questo tipo è stato proposto per spiegare la differenza di attività tra gli enantiomeri dell' epinefrina (adrenalina). La (-) adrenalina ha una affinità circa 100 volte superiore a quella dell' isomero (+). Solo nell' enantiomero (-) il gruppo OH è orientato in modo tale da consentire il massimo di interazione.
N OH
OH
HH
Me
HO H+
N OH
OH
HH
Me
H OH+
Sito anionico
Legameidrogeno
Area piana
R (-) ADRENALINA+ attiva
S (+) ADRENALINA- attiva
47
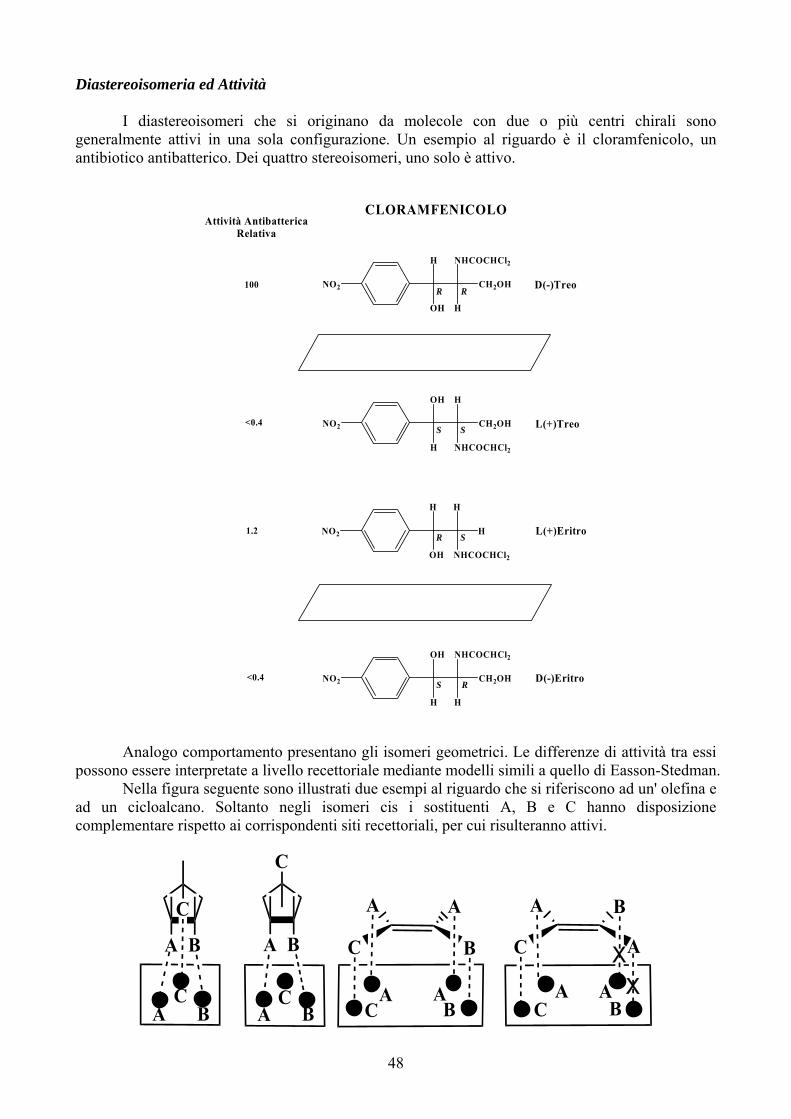
Diastereoisomeria ed Attività I diastereoisomeri che si originano da molecole con due o più centri chirali sono generalmente attivi in una sola configurazione. Un esempio al riguardo è il cloramfenicolo, un antibiotico antibatterico. Dei quattro stereoisomeri, uno solo è attivo.
S RCH2OH
NHCOCHCl2
H
OH
H
NO2
SRH
H
NHCOCHCl2
H
OH
NO2
SSCH2OH
H
NHCOCHCl2
OH
H
NO2
RRCH2OH
NHCOCHCl2
H
H
OH
NO2
CLORAMFENICOLOAttività Antibatterica
Relativa
<0.4
1.2
D(-)Eritro
L(+)Eritro
<0.4 L(+)Treo
100 D(-)Treo
Analogo comportamento presentano gli isomeri geometrici. Le differenze di attività tra essi possono essere interpretate a livello recettoriale mediante modelli simili a quello di Easson-Stedman. Nella figura seguente sono illustrati due esempi al riguardo che si riferiscono ad un' olefina e ad un cicloalcano. Soltanto negli isomeri cis i sostituenti A, B e C hanno disposizione complementare rispetto ai corrispondenti siti recettoriali, per cui risulteranno attivi.
A B
C
A B
C
C B
A A
C A
A B
A BBCC
AA
CA
BA A
C BX
X
48

Conformazione ed Attività La conformazione di una molecola denota i differenti possibili arrangiamenti che gli atomi costituenti la molecola possono assumere per rotazione attorno ad uno o più legami semplici. La conformazione con la quale un farmaco interagisce con il proprio bersaglio biologico (conformazione attiva) spesso non coincide con la conformazione più stabile in assoluto. E' generalmente accettato che ognuna delle conformazioni più stabili può essere quella che interagisce con il biopolimero. E' ragionevole ritenere comunque che tutte le conformazioni al di sopra di una certa energia debbano pagare un prezzo energetico troppo alto per adattare la loro conformazione al biopolimero. Informazioni sulle caratteristiche conformazionali di una molecola possono essere ottenute mediante principalmente tre metodologie: (a) la cristallografia ai raggi X permette di individuare la conformazione di una sostanza allo stato solido con precisione. Tale conformazione può tuttavia essere anche molto diversa da quella preferita a livello di interazione biologica; (b) la risonanza magnetica nucleare (NMR) permette di studiare la conformazione di un composto in soluzione; questo è un vantaggio rispetto alla difrattometria ai raggi X, specie se il solvente utilizzato ha caratteristiche che si avvicinano all' ambiente fisiologico; (c) i metodi teorici coinvolgono calcoli nei quali sono presi in considerazione vari parametri molecolari quali angoli e lunghezze di legame e distribuzione elettronica. In ogni caso tutti questi metodi di analisi conformazionale forniscono informazioni sulle conformazioni preferite di una molecola in assenza di bersaglio biologico il quale può in una certa misura imporre una conformazione di legame alla sostanza. E' chiaro che il metodo definitivo per risolvere il problema della caratterizzazione della conformazione attiva sarebbe quello di studiare la conformazione all' atto dell' interazione. Tuttavia, ciò è attualmente possibile solo per quei bersagli biologici per i quali è nota la struttura del sito attivo e nei quali è possibile studiare direttamente il complesso con il ligando: in pratica enzimi ed acidi nucleici. Un metodo indiretto, adottato da diversi decenni, consiste nella progettazione di analoghi a flessibilità molecolare ridotta che rappresentano un congelamento di possibili conformeri della mo-lecola originale. Per ridurre la libertà conformazionale di una molecola è necessario introdurre in essa degli elementi strutturali aggiuntivi che da una parte la irrigidiscono ma dall' altra possono mo-dificare le sue caratteristiche farmacocinetiche e farmacodinamiche. Per questo motivo, è opportuno che la riduzione di flessibilità venga effettuata con le minime variazioni strutturali possibili. Un esempio ormai classico di applicazione di tale procedura riguarda l' identificazione della
conformazione attiva dell' acetilcolina. Allo stato solido, in soluzione ed allo stato gasso-so la conformazione più stabile, dettata non dall' ingombro sterico tra i due sostituenti ma dalla interazione ione-dipolo tra il gruppo ammonico quaternario ed il carbonile estereo, appare quella sinclinale (gauche). I calcoli teorici indicano anche che la differenza ener-getica tra le varie conformazioni possibili è piccola. Molte evidenze, accumulate nel cor-so dei numerosissimi studi dedicati a questo neurotrasmettitore, indicavano che la confor-mazione attiva a livello del recettore musca-rinico era, probabilmente, quella trans (anti-periplanare). Il problema è stato risolto con
la applicazione della metodologia della restrizione conformazionale. Sono stati sintetizzati e saggia-ti gli isomeri (+)-cis e -trans dell' analogo ciclopropanico 2-acetossiciclopropiltrimetilammonio
H
H OCOCH3
NMe3
HH
OCOCH3
H H
NMe3
HH
+ +
sinclinale antiperiplanare
Me3N+
H
OCOCH3
H
Me3N+
H
H
OCOCH3
(+)cis-2-acetossiciclopropil-trimetilammonio ioduro
(+) trans-2-acetossiciclopropil-trimetilammonio ioduro
I- I-
49

ioduro. Dei due isomeri, solamente quello trans, corrispondente alla conformaziona antiperiplanare è equipotente con l' acetilcolina, mentre l' isomero cis è del tutto inattivo.
Relazioni Struttura-Attività Con il termine relazioni struttura-attività (SAR) si intendono le relazioni tra la struttura chimica e la attività farmacologica di una serie di composti. Esse vengono costruite nel corso del processo di ottimizzazione di un composto guida attraverso modificazioni molecolari del prototipo e sono a loro volta utilizzate per la progettazione di nuovi analoghi e per una comprensione dettagliata del meccanismo d' azione e della topografia della biomolecola bersaglio. Possono essere di carattere qualitativo e di carattere quantitativo (QSAR). In quest' ultimo caso, si utilizzano per la loro costruzione parametri chimico-fisici e trattazioni matematiche. Un passaggio fondamentale negli studi SAR è rappresentato dalla individuazione del farmacoforo. Per farmacoforo si intende l' insieme delle caratteristiche strutturali di una molecola biologicamente attiva che sono necessarie per l' interazione con il biopolimero bersaglio e quindi per l' attività. Il bersaglio biologico riconosce l' arrangiamento di certi gruppi della molecola nello spazio tridimensionale e le loro caratteristiche chimico-fisiche. Alcune modificazioni vengono realizzate al fine di esplorare gli effetti secondari della molecola (vedi Farmacocinetica, pg. 4). Dopo anni di studi SAR si sono messe in evidenza come produttive alcune strategie standard di modificazione molecolare utilizzabili per l' esplorazione primaria delle relazioni SAR a partire da un nuovo lead compound con una attività di qualsiasi tipo. Tali strategie comprendono: -dissociazione (o semplificazione) molecolare; -associazione (o complicazione) molecolare; -processi speciali: preparazione di profarmaci, costruzione di serie omologhe, sostituzioni bioisosteriche.
O
N
Me
O
NMe2
Me
ClO
N
N
Cl
Me
O
N
N
Cl
OMe
N
N
Y
Me
Cl
O
N
N
O
Me
N
N
Cl
S
MeO
N
N
OOMe
Cl N
SN
Me
Cl
O
N
N
Me
Cl
O
N
N
Me
Cl
O
N
N
R2
R1
Me
Cl
O
N
N
ESPLORAZIONE PRIMARIA DELLE SAR SUL DIAZEPAM
50

Il cosiddetto approccio globale consiste nell' applicare in maniera sistematica tutte le strategie suddette, individuando così sulla carta un numero molto alto di variazioni strutturali. La ricchezza di questa procedura è illustrata nella figura precedente, laddove essa è applicata al noto ansiolitico diazepam. Alcune regole possono aiutare a codificare con precisione l' uso delle varie strategie e ad aumentarne l' efficacia: -regola 1: dare priorità ad analoghi che derivano dal prototipo attraverso piccole variazioni; -regola 2: utilizzare prima possibile i dati biochimici; -regola 3: utilizzare i dati strutturali; -regola4: scegliere opportunamente i sostituenti sugli anelli aromatici; -regola 5: dare la preferenza ad analoghi la cui sintesi sia più semplice e meno costosa; -regola 6: eliminare i centri chirali; Dissociazione (o Semplificazione) molecolare Consiste nella sintesi di analoghi più semplici del prototipo. Il fatto che il farmacoforo sia in genere limitato ad alcuni elementi strutturali di una molecola che può essere più o meno complessa fa sì che si possa procedere spesso ad una semplificazione della molecola, sfrondandola degli elementi che non appartengono al farmacoforo (molecular pruning). Uno degli esempi più noti di semplificazione molecolare che, partendo da un prodotto naturale complesso, arriva via via alla creazione di molecole sempre più semplici ma ancora farmacologicamente attive è rappresentato dalla morfina e suoi congeneri.
Me
Me
N
HO
MeNEtO2C
MeN
HO
O
MeN
HO
HO
Me
Me
N
Me
COEt
Meperidina
Fenazocina(Benzomorfano)
E
Levorfanolo(Morfinano)
Morfina
SEMPLIFICAZIONE MOLECOLARE
Metadone
D
C
B
A
La morfina è un analgesico narcotico, riduce cioè la sensibilità al dolore e produce sedazione, diminuzione dell' attività mentale ed induzione al sonno. Possiede i seguenti effetti collaterali: depressione respiratoria, costipazione, nausea, vomito, euforia. Per l' azione euforizzante la morfina è uno stupefacente; già dopo breve tempo produce dipendenza fisica e psichica ed assuefazione. Partendo dalla morfina, viene inizialmente rimosso l' anello furanico D con ottenimento dei morfinani (ad es. levorfanolo). Per eliminazione anche dell' anello C si ha la serie dei benzomorfani (ad es. fenazocina). La più semplice modificazione della morfina può essere osservata nella mepe-ridina. Infine, perfino nella molecola del metadone si riconoscono i resti dell' anello piperidinico (E).
51

Associazione (o Complicazione) Molecolare Consiste nella sintesi di analoghi più complessi del composto guida. I processi di complicazione molecolare si possono distinguere in processi di: - replicazione molecolare; - ibridazione molecolare; - addizione molecolare. La replicazione molecolare consiste nell' associazione di unità identiche mediante formazione di legami covalenti (raddoppiamento molecolare, triplicazione molecolare, etc.):
nA A An-1
es: 2A A A3A A A A
Il caso di gran lunga più frequente è rappresentato dal raddoppiamento molecolare. L' ibridazione molecolare consiste invece nella associazione di unità diverse, sempre mediante formazione di legami covalenti:
A + B A B L' addizione molecolare consiste infine nell' associazione tra due unità differenti mediante interazioni non covalenti:
A + B A B
Il raddoppiamento e l' ibridazione molecolari riguardano la costruzione dei cosidetti "twin drugs", farmaci che derivano appunto dalla combinazione covalente di due porzioni farmacofore identiche o diverse. Essi possono essere legati direttamente fra loro, separati da uno spaziatore oppure addirittura sovrapposti parzialmente.
A BA
A
A A A B
A+ o A A B
A A B
o
o
o
I twin drugs possono rigenerare in vivo i costituenti (questo si verifica soprattutto per gli ibridi molecolari) oppure no (questo invece vale per entrambi i tipi di twin drugs). Nel primo caso si tratta di profarmaci reciproci (i due principi attivi si rendono profarmaci a vicenda); il twin drug è di per sé inattivo, si scinde in vivo (in genere attraverso un processo di idrolisi) e rigenera i due componenti attivi. Lo scopo del twin drug è quello di migliorare le proprietà farmaceutiche e farmacocinetiche. Un profarmaco reciproco avrà un' alta probabilità di successo a patto che esso venga ben assorbito, entrambi i componenti siano rilasciati contemporaneamente e quantitativa-mente dopo assorbimento ed ingresso in circolo, l' effetto massimo si verifichi con un rapporto tra essi di ~ 1:1 (in altre parole non ci deve essere una differenza troppo grande tra le concentrazioni attive dei due componenti).
52
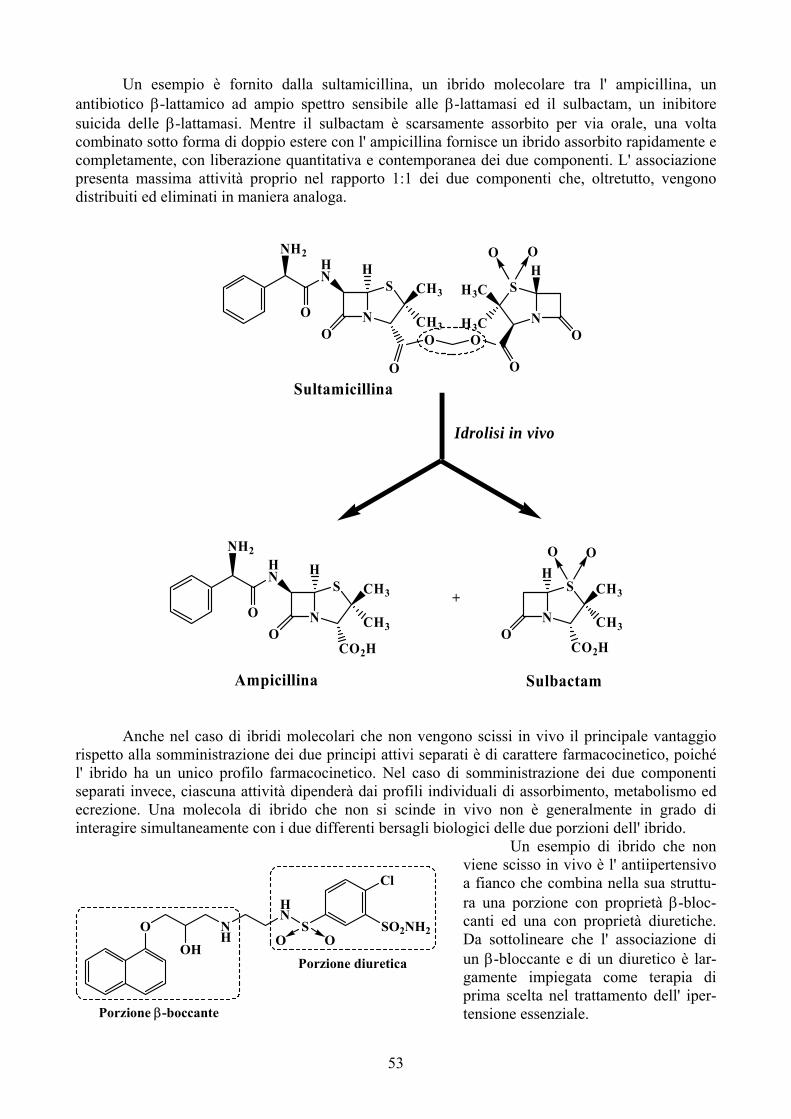
Un esempio è fornito dalla sultamicillina, un ibrido molecolare tra l' ampicillina, un antibiotico β-lattamico ad ampio spettro sensibile alle β-lattamasi ed il sulbactam, un inibitore suicida delle β-lattamasi. Mentre il sulbactam è scarsamente assorbito per via orale, una volta combinato sotto forma di doppio estere con l' ampicillina fornisce un ibrido assorbito rapidamente e completamente, con liberazione quantitativa e contemporanea dei due componenti. L' associazione presenta massima attività proprio nel rapporto 1:1 dei due componenti che, oltretutto, vengono distribuiti ed eliminati in maniera analoga.
NH2HN H
CO2H
CH3
CH3
O
S
N N
S
O
CH3
CH3
CO2H
H
+
O
O
HH3C
H3C
S
N
O
NH2HN H
CH3
CH3
O
S
NOO
O
O
O O
O O
Sultamicillina
Ampicillina Sulbactam
Idrolisi in vivo
Anche nel caso di ibridi molecolari che non vengono scissi in vivo il principale vantaggio rispetto alla somministrazione dei due principi attivi separati è di carattere farmacocinetico, poiché l' ibrido ha un unico profilo farmacocinetico. Nel caso di somministrazione dei due componenti separati invece, ciascuna attività dipenderà dai profili individuali di assorbimento, metabolismo ed ecrezione. Una molecola di ibrido che non si scinde in vivo non è generalmente in grado di interagire simultaneamente con i due differenti bersagli biologici delle due porzioni dell' ibrido.
Un esempio di ibrido che non viene scisso in vivo è l' antiipertensivo a fianco che combina nella sua struttu-ra una porzione con proprietà β-bloc-canti ed una con proprietà diuretiche. Da sottolineare che l' associazione di un β-bloccante e di un diuretico è lar-gamente impiegata come terapia di prima scelta nel trattamento dell' iper-tensione essenziale.
O
OH
NH
HN
S
Cl
SO2NH2O O
Porzione β-boccante
Porzione diuretica
53
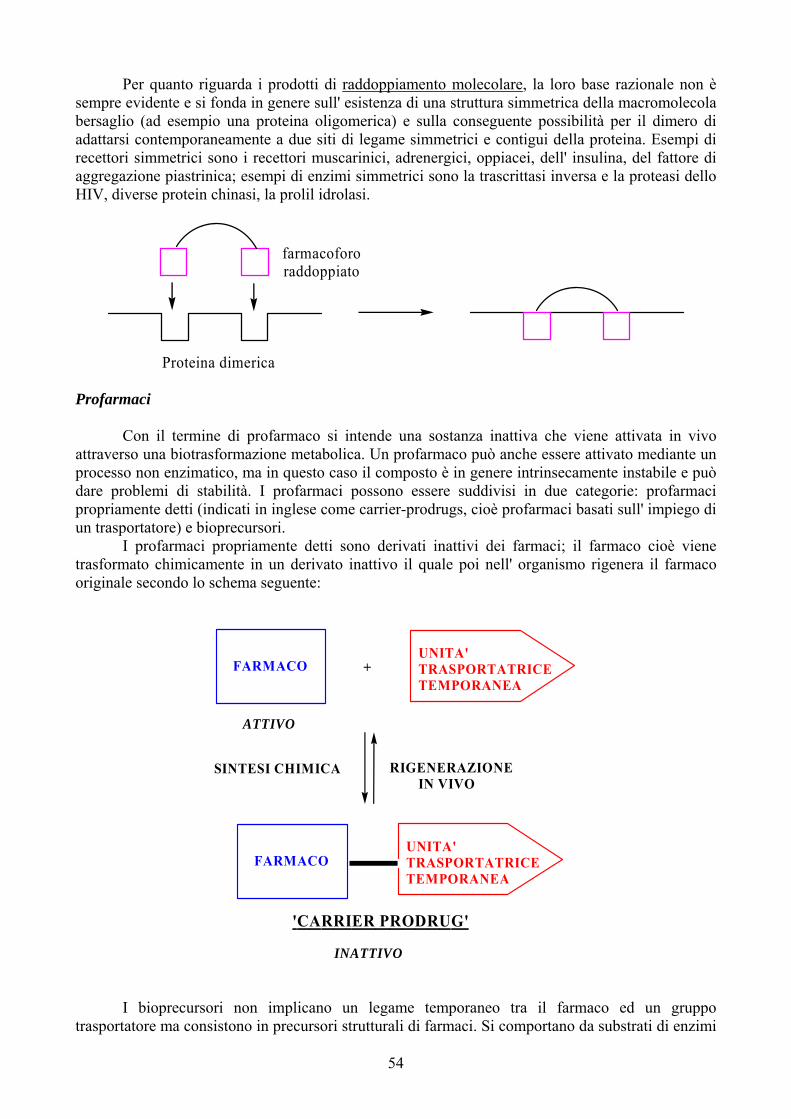
Per quanto riguarda i prodotti di raddoppiamento molecolare, la loro base razionale non è sempre evidente e si fonda in genere sull' esistenza di una struttura simmetrica della macromolecola bersaglio (ad esempio una proteina oligomerica) e sulla conseguente possibilità per il dimero di adattarsi contemporaneamente a due siti di legame simmetrici e contigui della proteina. Esempi di recettori simmetrici sono i recettori muscarinici, adrenergici, oppiacei, dell' insulina, del fattore di aggregazione piastrinica; esempi di enzimi simmetrici sono la trascrittasi inversa e la proteasi dello HIV, diverse protein chinasi, la prolil idrolasi.
Proteina dimerica
farmacofororaddoppiato
Profarmaci Con il termine di profarmaco si intende una sostanza inattiva che viene attivata in vivo attraverso una biotrasformazione metabolica. Un profarmaco può anche essere attivato mediante un processo non enzimatico, ma in questo caso il composto è in genere intrinsecamente instabile e può dare problemi di stabilità. I profarmaci possono essere suddivisi in due categorie: profarmaci propriamente detti (indicati in inglese come carrier-prodrugs, cioè profarmaci basati sull' impiego di un trasportatore) e bioprecursori. I profarmaci propriamente detti sono derivati inattivi dei farmaci; il farmaco cioè viene trasformato chimicamente in un derivato inattivo il quale poi nell' organismo rigenera il farmaco originale secondo lo schema seguente:
UNITA'TRASPORTATRICETEMPORANEA
FARMACO
'CARRIER PRODRUG'
RIGENERAZIONE IN VIVO
SINTESI CHIMICA
FARMACO +
INATTIVO
ATTIVO
UNITA'TRASPORTATRICETEMPORANEA
I bioprecursori non implicano un legame temporaneo tra il farmaco ed un gruppo trasportatore ma consistono in precursori strutturali di farmaci. Si comportano da substrati di enzimi
54

delle categorie ossidasi, reduttasi, chinasi, soprattutto e generano in vivo il metabolita attivo. Esempi di bioprecursori che subiscono una attivazione ossidativa sono il proguanile, la ciclofosfamide e la procarbazina; esempi di bioprecursori che subiscono invece una attivazione riduttiva sono la sulfasalazina e la mitomicina; esempi infine di bioprecursori che subiscono una attivazione fosforilativa sono tutti gli analoghi nucleosidici ad attività antivirale e antineoplastica. Un esempio storico di bioprecursore è rappresentato dal Prontosil rosso, un colorante azoico che viene convertito in vivo a sulfanilamide, l' effettivo responsabile dell' attività antibatterica; tale scoperta aprì la strada nella seconda metà degli anni trenta allo sviluppo dei sulfamidici.
H2N
NH2
N N SO2NH2
SO2NH2
H2N
NH2
H2N
NH2Prontosil rosso
Sulfanilamide
Nel quadro seguente sono riassunti le principali finalità dei profarmaci.
a) miglioramento delle caratteristiche farmacocinetiche (assorbimento e distribuzione)
b) miglioramento della stabilità metabolica
c) miglioramento dell' accettabilità
d) mascheramento degli effetti collaterali e della tossicità
Aumento sitospecificitàAumento biodisponibilitàAumento durata d' azione
Vediamo alcuni esempi di realizzazioni di profarmaci. La trasformazione di un farmaco in un suo derivato più lipofilo può avere come obiettivi un miglioramento dell' assorbimento (con conseguente aumento della biodisponibilità) ed un aumento della durata d' azione. L' ampicillina che possiede sia una funzione amminica che una funzione carbossilica è zwitterionica e quindi viene assorbita incompletamente nel tratto gastrointestinale. Il suo ampio spettro d' azione associato ad una quota non trascurabile del farmaco non assorbito nel tratto intestinale è responsabile dell' elevata incidenza di diarrea (8-10%). La bacampicillina, un doppio estere inattivo dell' ampicillina è invece bene assorbita e, una volta entrata in circolo, viene rapidamente e quantitativamente idrolizzata per ridare l' ampicillina. L' utilizzo di un doppio estere è dettato dal fatto che nelle penicilline l' intorno del gruppo carbossilico è altamente ingombrato e quindi esteri alifatici semplici non sono sufficientemente labili.
55
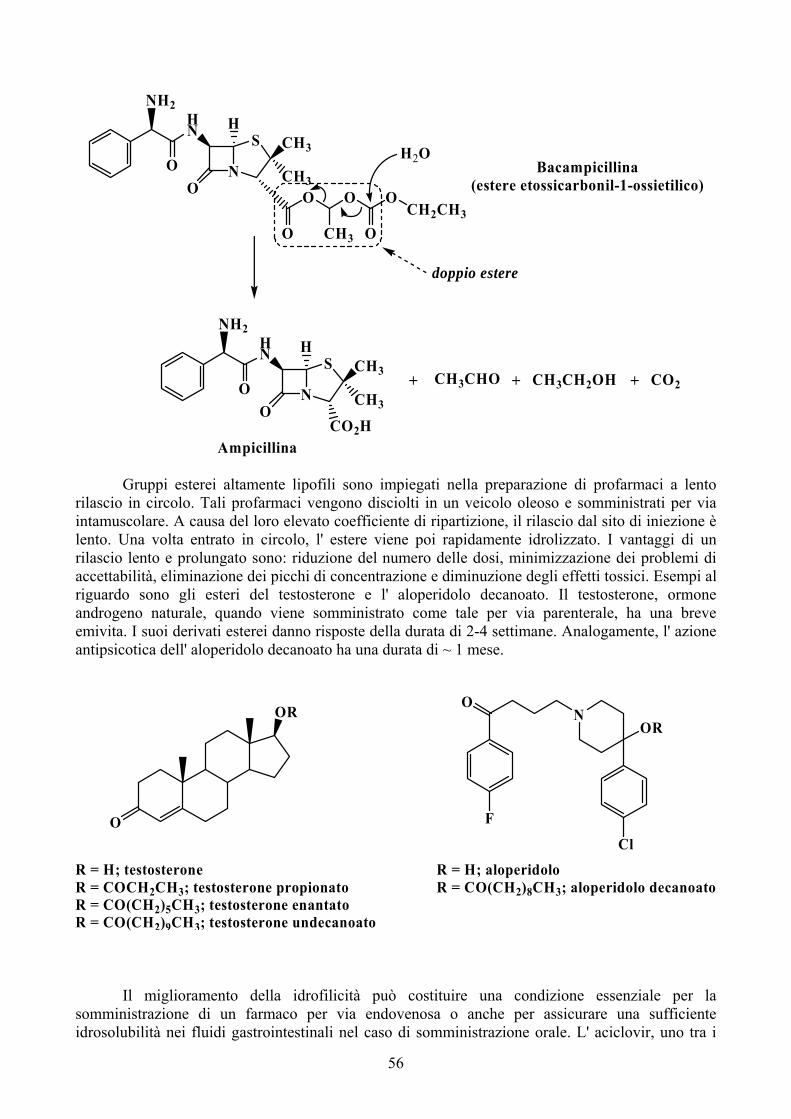
HNH2
O
N H
CH3
CH3
O
S
N
HNH2
O
N H
CO2H
CH3
CH3
O
S
N
O O OCH2CH3
O CH3 O
H2O
+ CH3CHO +CH3CH2OH CO2+
Ampicillina
Bacampicillina(estere etossicarbonil-1-ossietilico)
doppio estere
Gruppi esterei altamente lipofili sono impiegati nella preparazione di profarmaci a lento rilascio in circolo. Tali profarmaci vengono disciolti in un veicolo oleoso e somministrati per via intamuscolare. A causa del loro elevato coefficiente di ripartizione, il rilascio dal sito di iniezione è lento. Una volta entrato in circolo, l' estere viene poi rapidamente idrolizzato. I vantaggi di un rilascio lento e prolungato sono: riduzione del numero delle dosi, minimizzazione dei problemi di accettabilità, eliminazione dei picchi di concentrazione e diminuzione degli effetti tossici. Esempi al riguardo sono gli esteri del testosterone e l' aloperidolo decanoato. Il testosterone, ormone androgeno naturale, quando viene somministrato come tale per via parenterale, ha una breve emivita. I suoi derivati esterei danno risposte della durata di 2-4 settimane. Analogamente, l' azione antipsicotica dell' aloperidolo decanoato ha una durata di ~ 1 mese.
OR
O
R = H; testosteroneR = COCH2CH3; testosterone propionatoR = CO(CH2)5CH3; testosterone enantatoR = CO(CH2)9CH3; testosterone undecanoato
F
ON
OR
ClR = H; aloperidoloR = CO(CH2)8CH3; aloperidolo decanoato
Il miglioramento della idrofilicità può costituire una condizione essenziale per la somministrazione di un farmaco per via endovenosa o anche per assicurare una sufficiente idrosolubilità nei fluidi gastrointestinali nel caso di somministrazione orale. L' aciclovir, uno tra i
56
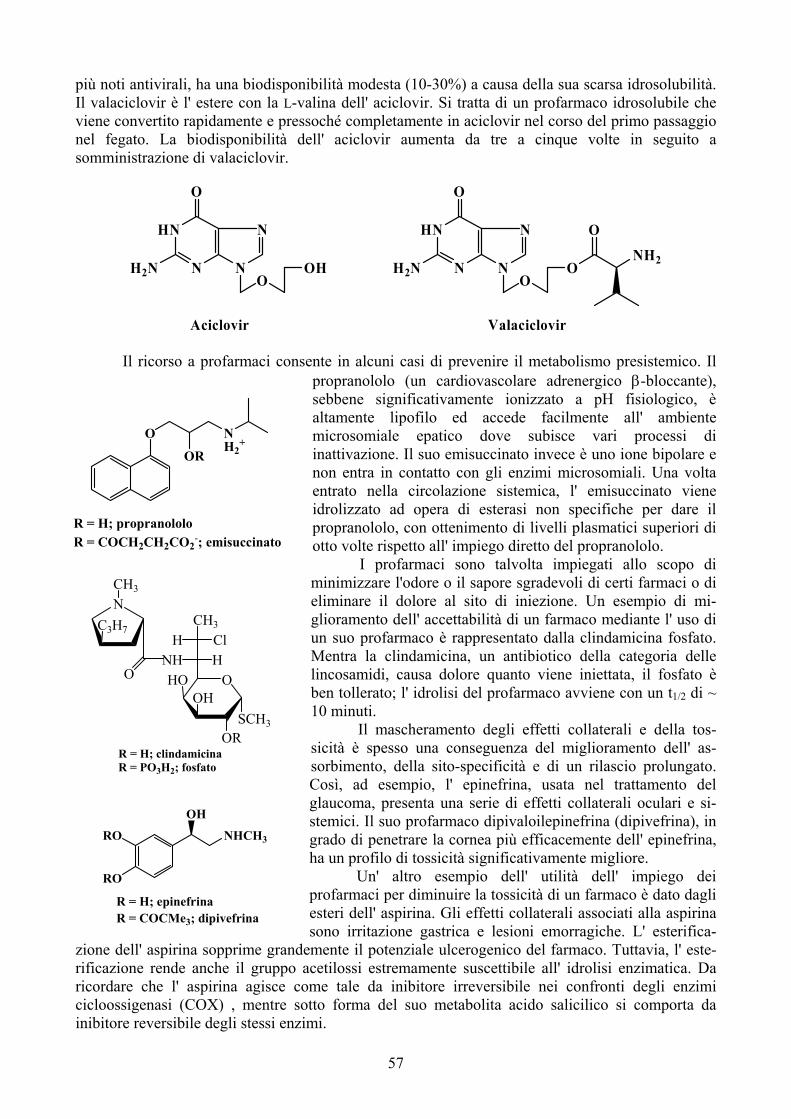
più noti antivirali, ha una biodisponibilità modesta (10-30%) a causa della sua scarsa idrosolubilità. Il valaciclovir è l' estere con la L-valina dell' aciclovir. Si tratta di un profarmaco idrosolubile che viene convertito rapidamente e pressoché completamente in aciclovir nel corso del primo passaggio nel fegato. La biodisponibilità dell' aciclovir aumenta da tre a cinque volte in seguito a somministrazione di valaciclovir.
HN
N N
N
O
OOHH2N
Aciclovir
HN
N N
N
O
OOH2N
NH2
O
Valaciclovir
Il ricorso a profarmaci consente in alcuni casi di prevenire il metabolismo presistemico. Il propranololo (un cardiovascolare adrenergico β-bloccante), sebbene significativamente ionizzato a pH fisiologico, è altamente lipofilo ed accede facilmente all' ambiente microsomiale epatico dove subisce vari processi di inattivazione. Il suo emisuccinato invece è uno ione bipolare e non entra in contatto con gli enzimi microsomiali. Una volta entrato nella circolazione sistemica, l' emisuccinato viene idrolizzato ad opera di esterasi non specifiche per dare il propranololo, con ottenimento di livelli plasmatici superiori di otto volte rispetto all' impiego diretto del propranololo.
O
OR
NH2
+
R = H; propranololoR = COCH2CH2CO2
-; emisuccinato
I profarmaci sono talvolta impiegati allo scopo di minimizzare l'odore o il sapore sgradevoli di certi farmaci o di eliminare il dolore al sito di iniezione. Un esempio di mi-glioramento dell' accettabilità di un farmaco mediante l' uso di un suo profarmaco è rappresentato dalla clindamicina fosfato. Mentra la clindamicina, un antibiotico della categoria delle lincosamidi, causa dolore quanto viene iniettata, il fosfato è ben tollerato; l' idrolisi del profarmaco avviene con un t1/2 di ~ 10 minuti.
O
C3H7
CH3
NCH3
ClHHNH
HOOH
SCH3OR
O
R = H; clindamicinaR = PO3H2; fosfato
Il mascheramento degli effetti collaterali e della tos-sicità è spesso una conseguenza del miglioramento dell' as-sorbimento, della sito-specificità e di un rilascio prolungato. Così, ad esempio, l' epinefrina, usata nel trattamento del glaucoma, presenta una serie di effetti collaterali oculari e si-stemici. Il suo profarmaco dipivaloilepinefrina (dipivefrina), in grado di penetrare la cornea più efficacemente dell' epinefrina, ha un profilo di tossicità significativamente migliore. Un' altro esempio dell' utilità dell' impiego dei profarmaci per diminuire la tossicità di un farmaco è dato dagli esteri dell' aspirina. Gli effetti collaterali associati alla aspirina sono irritazione gastrica e lesioni emorragiche. L' esterifica-
zione dell' aspirina sopprime grandemente il potenziale ulcerogenico del farmaco. Tuttavia, l' este-rificazione rende anche il gruppo acetilossi estremamente suscettibile all' idrolisi enzimatica. Da ricordare che l' aspirina agisce come tale da inibitore irreversibile nei confronti degli enzimi cicloossigenasi (COX) , mentre sotto forma del suo metabolita acido salicilico si comporta da inibitore reversibile degli stessi enzimi.
NHCH3
OH
RO
RO
R = H; epinefrinaR = COCMe3; dipivefrina
57

Omologia Lineare e Ciclica Consiste nell' esplorazione di una serie omologa, cioè una serie di composti che differiscono tra loro per una unità costante, generalmente un gruppo CH2. I casi più frequenti in chimica farmaceutica sono quelli dei derivati monoalchilati, dei composti ciclometilenici e dei composti
bifunzionalizzati polimetilenici. Il risultato che si osserva più frequentemente in una serie omolga è un aumento, regolare o irregolare dell' attività con l' aumentare del nu-mero di gruppi CH2 fino ad un mas-simo, seguito poi da una diminu-zione. Questo andamento è interpre-tato comunemente come conseguen-za di un coefficiente di ripartizione ottimale associato alla massima fa-cilità di attraversamento delle mem-brane biologiche. Il successivo calo di attività è attribuito alla insuffi-ciente idrosolubiltà che rende pro-blematico il trasporto del farmaco. Un esempio al riguardo è costituito da composti anticoliner-
gici a struttura di sali d' ammonio quaternari di dialchilammino etil esteri dell' acido benzilico.
R X R CH2X R CH2CH2X , eccetera
derivati monoalchilati
(CH2)n X (CH2)n+1 X
composti ciclometilenici
X (CH2)n Y X (CH2)n+1 Y
composti bifunzionali polimetilenici
R2
R3
R1
CO2CH2CH2N
OH
R1 = R2 = Me
(Anticolinergici)
+
Attività
H Me Et nPr nBu R3
L' attività cresce con il crescere della lunghezza della catena alifatica R3, riducendosi già dopo C2. Oltre ad influenzare la farmacocinetica di un farmaco, la lunghezza di una catena alifatica è in grado di influenzarne la farmacodinamica nel caso in cui la catena sia coinvolta nell' interazione con la macromolecola bersaglio.
Un caso interessante che il-lustra come allungamenti progressi-vi di catene alifatiche possano por-tare a variazioni nel meccanismo di azione è quello dei derivati poli-metilene-bisammonici con struttura Me3N+-(CH2)n-+NMe3 ad azione ganglioplegica (blocco della tra-smissione nervosa mediata dai recet-tori nicotinici a livello gangliare) e
I I I I I I I I I I I I I1 2 3 4 5 6 7 8 9 10 11 12 13
Attivitàganglioplegica
Attivitàcurarizzante
curarizzante (blocco a livello invece
58

della p
N
CO2Et
Et
CO2Et
NEt
Meperidina Etoeptazina
lacca motrice). A partire da n = 1 si ha un incre-
loomologia è fornito
ostituzioni Bioisosteriche
Le sostituzioni bioisosteriche sono modificazioni in cui un atomo o un gruppo di atomi della
stato originariamente introdotto da Langmuir nel 1919 per spiegare
iderate ad esempio N2 e CO (14 elettroni), CO2 ed N2O (22
Proprietà CO2 N2O
mento del primo tipo di attività che raggiunge il mas-simo per n = 6 (esametonio bromuro). L' attività gan-glioplegica quindi diminuisce fino a scomparire mentre compare l' attività curarizzante che raggiunge l' apice per n = 10 (decametonio ioduro). Un esempio infine di cicdalla copia meperidina-etoeptazina (analgesici morfino-simili).
S molecola prototipo sono sostituiti con atomi o gruppi con caratteristiche steriche ed elettroniche approssimativamente simili con il risultato di ottenere analoghi in grado di interagire con lo stesso sistema biologico del prototipo , comportandosi da agonisti o da antagonisti. Il termine bioisosteri si applica sia agli atomi o gruppi con caratteristiche simili che alle molecole ottenute mediante sostituzioni bioisosteriche. Esso costituisce un' estensione al campo dei composti biologicamente attivi del concetto di isosteria. Il concetto di isosteria èperché alcune specie chimiche presentassero proprietà chimico-fisiche simili tra loro. Tale fatto venne da Langmuir attribuito alla identità nel numero complessivo degli elettroni di tali specie che furono indicate col termine di isosteri. Coppie di isosteri vennero conselettroni), N3
- e NCO- (21 elettroni) e CH2N2 e CH2CO (22 elettroni). Il confronto delle proprietà chimico-fisiche di CO2 ed N2O è una evidente dimostrazione delle similitudini tra isosteri.
Carica elettronica complessiva
atm)
) 0 °C
°C)
22 44,01
1
31,9 0,0506
22 44,02
1
35,4 0,0506
Peso molecolare Viscosità (20°, 1 Pressione critica (atm) Temperatura critica (°CConducibilità termica a 10Indice di rifrazione del liquido (0Costante dielettrica del liquido (0 °C) Solubilità in acqua (0 °C) Solubilità in alcool (15 °C)
48x10-6
77
1,190 1,582 1,780 3,13
48x10-6
75
1,193 1,598 1,305 3,25
Il concetto di isosteria è stato successivamente elaborato da Grimm (1924) con la sua regola dello spostamento degli idruri con cui costruire serie di isosteri. Colonne di isosteri si ottengono agiungendo un atomo di idrogeno ad un elemento di una riga del sistema periodico e scalando di un posto verso destra l' aggruppamento così ottenuto ed indicato con il termine di pseudoatomo. La tabella seguente è limitata agli elementi biologicamente interessanti.
59
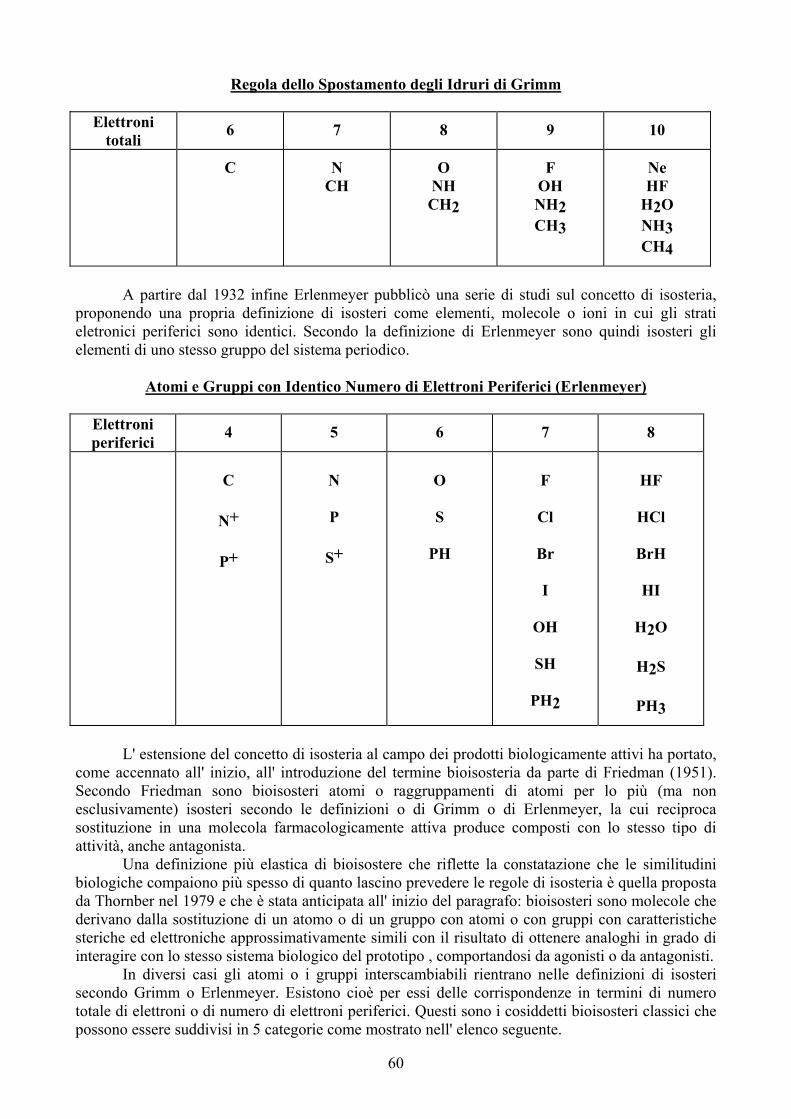
Regola dello Spostamento degli Idruri di Grimm
Elettroni 6 7 8 9 10
totali C N
CH O
NH F
OH Ne
HCH2 NH2 CH3
HF 2O
NH3 CH4
A partire dal 1932 infine Erlenmeyer pubblicò una serie di studi sul concetto di isosteria,
Atomi e Gruppi con Identico Numero di Elettroni Periferici (Erlenmeyer)
proponendo una propria definizione di isosteri come elementi, molecole o ioni in cui gli strati eletronici periferici sono identici. Secondo la definizione di Erlenmeyer sono quindi isosteri gli elementi di uno stesso gruppo del sistema periodico.
Elettroni 4 5 6 7 8
periferici
C
N
P
N
P
S
O
PH
Cl
Br
OH
SH
PH2
HF
HCl
BrH
HI
H
H
PH3
+ +
+
S
F
I
2O 2S
L' estensione del concetto di isosteria al campo dei prodotti biologicamente attivi ha portato, ome a
elastica di bioisostere che riflette la constatazione che le similitudini
possono essere suddivisi in 5 categorie come mostrato nell' elenco seguente.
c ccennato all' inizio, all' introduzione del termine bioisosteria da parte di Friedman (1951). Secondo Friedman sono bioisosteri atomi o raggruppamenti di atomi per lo più (ma non esclusivamente) isosteri secondo le definizioni o di Grimm o di Erlenmeyer, la cui reciproca sostituzione in una molecola farmacologicamente attiva produce composti con lo stesso tipo di attività, anche antagonista. Una definizione piùbiologiche compaiono più spesso di quanto lascino prevedere le regole di isosteria è quella proposta da Thornber nel 1979 e che è stata anticipata all' inizio del paragrafo: bioisosteri sono molecole che derivano dalla sostituzione di un atomo o di un gruppo con atomi o con gruppi con caratteristiche steriche ed elettroniche approssimativamente simili con il risultato di ottenere analoghi in grado di interagire con lo stesso sistema biologico del prototipo , comportandosi da agonisti o da antagonisti. In diversi casi gli atomi o i gruppi interscambiabili rientrano nelle definizioni di isosteri secondo Grimm o Erlenmeyer. Esistono cioè per essi delle corrispondenze in termini di numero totale di elettroni o di numero di elettroni periferici. Questi sono i cosiddetti bioisosteri classici che
60

BIOISOSTERI CLASSICI
1. Atomi e Gruppi Monova
2 3
alenti
valenti
i Tetrasostituiti
ari
-
lenti 4. Atom
-F, -OH, -NH2, -CH3 l, -SH, -PH , -SiH -C
-Cl, -Br, -I
ruppi Biv2. Atomi e G -O-, -S-, -NH-, -CH2- 3. Atomi e Gruppi Tri -N=, -P=, -CH=
=N+=, =P+=, =C= 5. Equivalenti anul -CH=CH-, -S-, -O-, -NH
=, -CH= -N
Il significato della quinta categoria è illustrato dalla seguente serie di sistemi anulari
S
ONH
NN
S
NN
N
NN
N
N
Essi possono essere scambiati reciprocam te in una molecola con mantenimento probabile del tipo di attività.
o di Erlenmeyer. Le somiglianze tra essi sono più vaghe e spesso il loro
I NON CLASSICI
en
I bioisosteri non classici invece sono serie di gruppi che non rientrano nelle definizioni di isosteri di Grimm accostamento deriva dalla constatazione che in vari tipi di molecole il loro interscambio ha prodotto serie di composti con lo stesso tipo di attività.
BIOISOSTER
1. Gruppi Interscambiab
O-, -SO2- O H, -PO(OR)OH
NHR
erti - Modelli Chiusi
ili 2. Modelli Ap
-Cl, -CF3 -C-CO2H, -S 3-CO2H, -SO2-NO2, -SO2CH3, -COCH3
61

Relazioni Quantitative Struttura-Attività' (QSAR)
L' attività di un composto è, com' è noto, influenzata in maniera determinante dalle sue proprietà chimico-fisiche. Lo scopo delle QSAR è di individuare delle relazioni con valore
predittivo tra le proprietà biologiche del composto ed una serie di parametri che esprimono quantitativamente certe proprietà chimico-fisiche (ad es. parametri elettronici, di ripartizione, sterici). Nei metodi 2D QSAR l' attività è espressa in funzione di parametri chimico-fisici all' interno di un set di composti strutturalmente omogenei, di composti cioè con una struttura base comune e
o di Hansch
L' approccio matematico più pop ansch. Esso considera gli aspetti chimico-fisici sia del trasporto e della distribuzione del farmaco
e originale ipotizza che in un
π;
che differiscono tra loro per la natura, la posizione ed il numero dei sostituenti presenti. Le variazioni di attività sono conseguenza delle variazioni dei parametri considerati, variazioni a loro volta legate alle caratteristiche dei sostituenti.
Metod
olare è il metodo di H che dell' interazione di esso con il recettore. Nella sua formulaziondato gruppo di principi attivi con strutture analoghe ed agenti mediante lo stesso meccanismo giochino un ruolo principale tre parametri chimico-fisici che quantificano le proprietà elettroniche, di lipofilia e steriche dei sostituenti: - la costante di sostituente di Hammett σ; - la costante di sostituente di lipofilia- il parametro sterico di Taft Es. a) Effetti Elettronici Sono espressi dalla costante di sostituente di Hammett σ. La costante σ è stata introdotta in
li anni '40 da Hammett per quantificare l' influenza sulla reattività chimica di chimica organica negun composto aromatico dei sostituenti presenti sull' anello stesso. Compare nelle equazioni di Hammett: lg(kx/ko) = ρσ, in cui k può essere sia una costante di velocità che una costante di equilibrio; kx si riferisce al composto con il sostituente X, ko al composto senza sostituenti. Il valore di σ dipende dalle proprietà elettroniche e dalla posizione del singolo sostituente ( così, σmeta e σpara per lo stesso sostituente non sono generalmente uguali) e rappresenta una misura della capacità del sostituente X di cedere o attrarre elettroni. Più elettronattrattore è un sostituente, maggiormente positivo è il valore di σ (rispetto ad H che è posto pari a 0). La costante σ è additiva, cioè sostituenti multipli esercitano un' influenza complessiva pari alla somma dei singoli sostituenti. La seconda costante presente nelle equazioni di Hammett, costante ρ, chiamata costante di reazione, dipende invece dal tipo di reazione ed indica la sensibilità della reazione agli effetti elettronici dei sostituenti. Reazioni che sono favorite da una elevata densità elettronica nello stato di transizione hanno valori negativi di ρ mentre, al contrario, posseggono valori positivi di ρ reazioni che sono agevolate da una bassa densità elettronica. b) Effetti di Lipofilia Si è già detto dell' importanza del coefficiente di ripartizione P ai fini della attività (pg. 13).
coefficiente di ripartizione tra n-ottanolo ed acqua di una serie di derivati, ha
PX - lg PH = lg (PX/PH)
Hansch, studiando il evidenziato, in analogia con quanto fatto da Hammett per il parametro σ, un parametro π (costante di sostituente di lipofilia) definito come:
π = lg
62

dove PX è il coefficiente di ripar ostituente X e PH è quello del omposto non sostituito. Come nel caso della costante di sostituente di Hammett, anche π può
tizione del composto con il scessere positiva o negativa ed è additiva, cioè sostituenti multipli esercitano un' influenza complessiva pari alla somma dei singoli sostituenti. c) Effetti Sterici Un altro importante parametro chimico-fisico è il parametro sterico di Taft. Taft ha definito
ico Es allo stesso modo in cui Hammett ha quantizzato gli effetti elettronici dei
Es = lg (kX/kH)
in cui kX e kH sono le costanti di veloc ida degli esteri metilici di a-cidi acetici onosostituiti XCH2CO2Me e dell' acetato di metile CH3CO2Me, rispettivamente. Taft usò questa
lg(1/C) = -aπ + bπ + ρσ + cEs + d
dove C è la concentrazione mol efinito effetto biologico mentre , b, c e d sono dei coefficienti che possono essere ricavati mediante metodi matematici (analisi di
bassa, mentre il segno negativo per il termine π riflette il fatto che esiste un valore ttimal
Nella scelta dei sostituen una volta stabiliti i segni e le randezze dei vari coefficienti, diagrammi quali quello di Craig (pagina seguente) che esprime mbin
ridotte. Sono stati successivamente impiegati parametri diversi da π,
parametro) + e
il parametro stersostituenti e Hansch quelli di lipofilia:
ità di idrolisi ac
mreazione di riferimento dal momento che la sua velocità è controllata da soli fattori sterici. I valori di Es sono tutti negativi. I parametri elettronici, di lipofilia e sterici descritti sono stati inseriti da Hansch nella seguente equazione:
2
are di farmaco che provoca un ben d
aregressione lineare multipla), misurando le concentrazioni C per una serie di sostanze strutturalmente correlate. Una volta che tali coefficienti siano stati ottenuti, l' equazione può essere utilizzata per predire i valori di C e quindi l' attività di composti non saggiati o non ancora sintetizzati. Il reciproco della concentrazione C riflette il fatto che una maggiore potenza è associata ad una dose più 2
o e ai fini dell' attività del coefficiente di ripartizione P. La curva che descrive l' effetto del lg P sulla risposta biologica, espressa come lg (1/C), è infatti una parabola e la sua espressione è:
lg (1/C) = - k (lg P)2 + k' lg P + k''
ti da inserire possono essere utili,gco azioni di coppie π-σ. I sostituenti si distribuiscono in 4 quadranti identificati dalle coppie: +σ, -π; +σ, +π; -σ, +π; -σ, -π. L' equazione ρ-σ-π di Hansch è stata applicata con successo in centinaia di casi sia nella sua forma completa che in versioniσ ed Es per descrivere le proprietà globali della molecola ed il contributo dei singoli sostituenti. La forma più generale dell' equazione impiegata nell' analisi di Hansch è quindi: lg (1/C) = a (parametro idrofobico) + b (parametro elettronico) + c (parametro sterico) + d (altro
63

Diagramma σ-π di Craig per sostituenti in para
Metodo di Free-Wilson
Non molto dopo che Hansch propose il suo approccio, Free e Wilson riportarono un metodo matematico per l' ottimizzazione dei sostituenti di una struttura fondamentale basato sulla assunzione che la risposta biologica è la somma dei contributi indipendenti dei sostituenti e della struttura fondamentale. In tal modo, se si assegna ai possibili sostituenti un certo valore di contributo all' attività biologica, si possono predire i composti con la massima attività. Naturalmente ciò funziona solo se i contributi dei sostituenti sono additivi. Nel metodo di Free-Wilson si prescinde dalle proprietà chimico-fisiche dei vari sostituenti e si attribuisce a ciascuno di essi un certo contributo di attività che è indipendente dall' effetto dei sostituenti nelle altre posizioni. Si potrà così scrivere che l' attività [espressa come lg (1/C)] del composto i-esimo di una serie è:
lg (1/C) = Σ aiXi + µo
dove ai è il contributo del sostituente i-esimo, Xi è il sostituente i-esimo con un valore di 1 se presente e di 0 se assente e µo è l' attività della struttura base. Anche in questo caso, l' analisi di regressione è impiegata per determinare ai e µo. Questo metodo ha trovato applicazione soprattutto nelle fasi iniziali della ricerca di relazioni quantitative struttura-attività quando siano disponibili già diversi analoghi dei quali non si conoscono i dati chimico-fisici.
64

Metodo di Topliss Topliss ha sviluppato nel 1972 una guida all' uso dei principi di Hansch non matematica, non statistica e non computerizzata. Si tratta di un metodo empirico nel quale ciascun composto è testato prima di pianificare un analogo successivo e confrontato in termini di proprietà chimico-fisiche con analoghi già provati. Questo metodo è soprattutto utile quando la sintesi di un gran numero di composti è difficoltosa. Costituisce un approccio manuale per l' efficiente ottimizzazione di un composto guida, minimizzando il numero dei composti da sintetizzare. L' approccio è basato soprattutto sui valori di σ e π. Vediamo un esempio riferito ad un ipotetico composto aromatico. Sono disponibili anche schemi operativi per sistemi non-aromatici. Supponiamo di avere un composto di natura aromatica e non sostituito sull' anello benzenico con una attività interessante che giustifichi la ricerca di possibili derivati più attivi. Poichè molti sistemi sono +π dipendenti, cioè la loro attività aumenta con l' aumentare della lipofilia, una buona scelta per il primo analogo è rappresentata dal 4-Cl derivato poichè esso possiede un buon π (0.71) ed una media capacità elettronattrattrice (σ = 0.23). Una volta preparato e testato il derivato, si presenterà uno dei casi seguenti: il 4-Cl derivato è più attivo (P), egualmente attivo (E) oppure meno attivo (M) del composto non sostituito. Se esso è più potente (P), ciò può esere attribuito ad un effetto +π, ad un effetto +σ, oppure ad entrambi. Si può allora sintetizzare il 3,4-dicloro derivato per il quale si ha un valore di π = 1.25 ed un valore di σ = 0.52 e testarlo. Nel caso di un ulteriore aumento di attività, la determinazione dell' importanza relativa degli effetti +π e +σ può essere effettuata selezionando successivamente il 4-PhS analogo (π = 2.32, σ = 0.18) oppure il 3-CF3, 4-NO2 analogo (π = 0.60, σ = 1.21). A questo punto si può costruire uno schema e preparare altri analoghi.
4-H
4-Cl
3,4-Cl2
M E P
3-Cl o 4-OMe
4-Me o 4-NO2
M E P
4-SPho 3-CF3 , 4-NO2
4-CF3 P
4-NO2
o 4-Et M E P
Se invece il 3,4-dicloro derivato si fosse dimostrato meno potente del 4-Cl analogo, la causa poteva essere attribuita o ad un superamento dei valori ottimali di π e σ oppure ad un effetto sterico sfavorevole. Quest' ultima ipotesi può essere verificata provando il 4-CF3 analogo (π = 0.88; σ = 0.54), che non ha un sostituente in 3 ma possiede un alto valore di σ ed un valore intermedio di π. Se questo analogo è più potente del 4-Cl, si possono sintetizzare il 4-NO2 (π = -0.28; σ = 0.78) op-pure il 4-Et (π = 1.02; σ = -0.15) allo scopo di determinare l' importanza di π e σ, rispettivamente. Se il 4-Cl analogo è meno potente del composto guida, si deve dedurre o che vi è un problema sterico alla posizione 4 oppure che l' aumento di potenza dipende da valori -π o -σ. Si può sintetizzare il 3-Cl analogo (π = 0.71, σ = 0.37) per verificare la prima ipotesi ed il 4-MeO derivato
65

(π = -0.04; σ = -0.27) per valutare le conseguenze dell' introduzione di un sostituente -σ. Nel caso di un aumento di attività, si possono provare altri sostituenti con valori di π e/o di σ più negativi. Se infine il 4-Cl derivato è equipotente con il composto guida, ciò può essere la conseguenza di un bilanciamento tra un favorevole effetto +π ed uno sfavorevole effetto -σ, o viceversa. Nel primo caso, il 4-Me analogo (π = 0.56; σ = -0.17) dovrebbe mostrare una maggiore potenza, nel secondo caso dovrebbe essere invece il 4-NO2 (π = -0.28; σ = 0.78) ad essere più attivo.
3D QSAR Le QSAR tridimensionali si applicano in genere ad una serie di composti con maggiore varietà strutturale rispetto alle QSAR bidimensionali. Ciò deriva dal fatto che le molecole sono descritte da proprietà calcolate direttamente dalle loro strutture tridimensionali mentre nelle 2D QSAR le proprietà vengono stimate da costanti di sostituente misurate su molecole strutturalmente correlate tra loro. Quindi una 3D QSAR può essere più generale di una 2D QSAR. Il metodo più usato nelle 3D QSAR è l' analisi comparativa del campo molecolare (CoMFA). L' analisi CoMFA inizia con la scelta di un gruppo di molecole (training set), anche molto diverse tra loro, per le quali si hanno a disposizione i dati biologici. Le molecole vengono quindi allineate sulla base di studi di modellistica molecolare rivolti all' individuazione della conformazione farmacofora e del farmacoforo per ciascuna molecola. Nella figura successiva è mostrata la sovrapposizione di una serie di farmaci agenti come antagonisti sul recettore 5-HT1A
Metitepina (verde), Spiperone (celeste), Propranololo (rosa), Buspirone (viola). Il contorno nero rappresenta l' ipotetico volume della cavità recettoriale
S
SCH3
N
NCH3
R-Metitepina
F
O
N
NH
N
O
Spiperone
O NH
OH
Propranololo
N
N N
NN
O
O
Buspirone
Una volta che le molecole siano state allineate sulla base di caratteristiche molecolari comuni in modo da occupare lo stesso volume di spazio, esse vengono circondate da una griglia tridimensionale che si estende per circa 4 Å attorno a tutte le molecole (figura a lato). La spaziatura dei nodi della griglia è di solito di 1-2 Å. In ogni punto della griglia vengono quindi calcolati i valori delle intensità dei campi di interazione intermolecolare che circondano ogni molecola. Tali campi sono valutati mediante piccoli gruppi o atomi sonda con proprietà steriche ed elettrostatiche specifiche alle intersezioni di una griglia 3D che racchiude le molecole. Vengono
S
SCH3
N
N
CH3
66

calcolati campi sterici ed elettrostatici mentre nel CoMFA standard non sono presi in considerazioni quelli idrofobici. Come in tutti gli studi QSAR, anche nel CoMFA si costruisce una tavola di dati in cui l' attività biologica di ciascun composto è contenuta nella prima colonna mentre in tutte le altre sono contenuti i parametri strutturali e cioè le intensità delle interazioni steriche ed elettrostatiche del composto con le sonde.
Composto Attività S001 S002...S998 E001...E998 C1 C2 Cn
Attività = a + b (S001) + c(S002) + ...m(S998) + n(E001) + ...z(E998) Si tratta poi di trovare una equazione che correli l' attività biologica di ciascuna molecola con i campi esercitati da essa su qualsiasi atomo che la circondi. La difficoltà matematica consiste nel dovere risolvere una equazione con migliaia di coefficienti metre le misure biologiche sono poche decine. La procedura impiegata è quella della regressione multivariata PLS. L' equazione viene in genere visualizzata come mappa di contorno tridimensionale che rappresenta regioni nello spazio attorno alla molecola in cui si verificano interazioni steriche o elettrostatiche positive o negative con il recettore (Figura successiva). I contributi sterici ed elettrostatici son espressi con contorni separati così come i contributi positivi e quelli negativi. I contorni sterici positivi mostrano regioni dello spazio che, se occupate da gruppi del farmaco, aumentano la potenza del composto; mentre i contorni negativi la diminuiscono. Per quanto riguarda invece i contorni elettronici, vi saranno zone in cui i gruppi con carica parziale di un certo segno determinano una interazione favorevole con il recettore, per cui in quella zona vanno evitati gruppi con carica parziale opposta.
I poliedri verdi e gialli identificano zone in cui una maggiore interazione sterica aumenta e diminuisce, rispettivamente, l' affinità per il recettore. I poliedri rossi identificano zone in cui un potenziale elettrostatico più negativo migliora l' affinità; quelli blu individuano regioni in cui al contrario l' affinità migliora con un potenziale più positivo.
67



















![valutaz rischio testai.ppt [modalità compatibilità] - iss.it rischio Testai.1212139345.pdf · La valutazione tossicologica L’obbiettivo è quello di identificare i possibili rischi](https://img.pdfslide.tips/doc/110x75/5a7927267f8b9adb5a8e08d3/valutaz-rischio-modalit-compatibilit-issit-rischio-testai1212139345pdfla.jpg)









