Embed Size (px)
Citation preview

-1-
Chimie organique 7 :
Substitution électrophile aromatique
La famille des composés aromatiques, fréquemment rencontrés en chimie organique, compte parmi ses membres, le benzène (C6H6), découvert en 1825 par Faraday. A une époque où la chimie quantique et les méthodes spectroscopiques n’existaient pas, la détermination de sa structure a suscité beaucoup d’intérêt et a conduit à de nombreuses propositions, toutes en cohérence avec la formule brute C6H6 :
3-Prismane Benzvalène Benzène Claus Benzène Dewar
Formules de Kékulé :
Proposées en 1865 à une époque où la délocalisation des électrons n’était pas connue Validée par Pauling en 1928 (théorie de la mésomérie)
Les composés aromatiques sont caractérisés par une structure cyclique totalement conjuguée extrêmement stable très difficile à modifier par oxydation ou par addition.
Seules les substitutions électrophiles aromatiques (SEAr) sont relativement faciles à réaliser car elles ne font pas perdre le caractère aromatique à la molécule.
Dans cette réaction (SEAr), le cycle aromatique y joue le rôle de nucléophile et réagit avec un composé électrophile.
Ce chapitre s’intéresse trois aspects :
La notion d’aromaticité,
Les SEAr sur le benzène,
La SEAr sur les cylcles benzéniques déjà substitués.
1. Aromaticité
1.1. Exemples de composés aromatiques
- Dérivés du benzène : présence d’un noyau benzénique
O OH
OH
O
N+
O
ONH2
OH

-2-
- Composés présentant plusieurs noyaux benzéniques accolés :
Naphtalène Phénanthrène Pyrène
- Cycles présentant des hétéroatomes
O N
H
N
Furane Pyrrole Pyridine
- Macrocycles à plusieurs dizaines d’atomes de carbone H H
H
H
H
H
HH
H
H
H
H
HH
HH
H
H
[18]annulène [24]annulène Problématique : Quelle propriété ont en commun les composés aromatiques ? Qu’est-ce qui les caractérise ?
1.2. Stabilité des composés aromatiques
Les composés aromatiques sont extrêmement stables en raison de la délocalisation des électrons sur l’ensemble des atomes du cycle. On constate qu’expérimentalement, il est très difficile de les faire réagir, en particulier lorsque la réaction envisagée supprime la conjugaison au sein du cycle. Pour évaluer la stabilité de la molécule, on détermine l’énergie de résonance, c’est-à-dire l’abaissement énergétique dû à la délocalisation électronique en comparaison avec une molécule fictive les mêmes électrons mais sans délocalisation. a) Evaluation de l’énergie de résonance
Méthode 1 : Enthalpies standard d’hydrogénation
On compare l’enthalpie standard de la réaction d’hydrogénation du benzène et on la compare avec celle du composé fictif analogue au benzène mais sans délocalisation : le cyclohexa-1,3,5-triène.
1. Hydrogénation du cyclohexène : Enthalpie standard d’hydrogénation d’une double liaison : ΔrH°1 = - 120 kJ.mol
-1 (mesure expérimentale)
+ H2 =
2. Hydrogénation de l’hypothétique cyclohexa-1,3,5-triène obtenue en considérant qu’on hydrogène 3 doubles liaisons C=C indépendantes : ΔrH°2 = 3 ΔrH°1 = - 360 kJ.mol
-1 (valeur estimée théoriquement)
+ 3 H2 =

-3-
3. Hydrogénation du benzène : Enthalpie standard de réaction : ΔrH°3 = - 210 kJ.mol-1
(mesure expérimentale)
+ 3 H2 =
4. Report des valeurs sur un axe d’énergie (dans les deux cas, le produit obtenu est le cyclohexane) :
(Benzène)
(Cyclohexatriène)
(Cyclohexane)
E
Energie de résonance : Eres =
La délocalisation des électrons au sein du cycle aromatique est très stabilisante !
Ebenzène << Ecyclohexa-1,3,5-triène
Toute réaction faisant perdre le caractère aromatique à une molécule est très défavorable.
Méthode 2 : Méthode de Hückel
Chaque atome de carbone intervient dans le système π par une orbitale atomique 2p et un électron : le système π du benzène possède donc 6 électrons. Pour assurer la délocalisation, le benzène adopte une structure plane. De cette façon, les orbitales 2p intervenant dans le système π sont parallèles entre elles (condition nécessaire à la délocalisation). Le diagramme des OM π est le suivant :

-4-
Energie de résonance :
b) Inertie chimique des composés aromatiques : Contrairement à ce que les formules de Kékulé laissent penser (écriture de doubles liaisons C=C), les cycles aromatiques ne réagissent pas du tout comme les alcènes. Les réactions d’addition sur les alcènes détruiraient les doubles liaisons et donc la conjugaison au sein du cycle… Elles sont donc particulièrement défavorables et sont particulièrement difficiles à réaliser. Les réactions d’addition :
Double liaison C=C Cycle aromatique
Addition de Br2 Très facile Impossible
Epoxydation Facile Impossible
Addition de H2 Très facile
Conditions douces (T et P ambiantes)
Très difficile Conditions très dures
(T et P très fortes)
Expl :
H2, [Ni]
1 bar, 25 °C
H2, [Ni]
100 bar, 200 °C
OH
O
KMnO4 cc
Chauffage
Seules les réactions de substitution sont réalisables facilement car elles n’entraînent pas la disparition du système conjugué cyclique.

-5-
1.3. Propriétés spectroscopiques
- Spectroscopie RMN 1H : H aromatiques très fortement déblindés (δ ~ 7-8 ppm)
- Phénomène de courant de cycle :
Le champ magnétique imposé par l’appareil crée un courant d’électrons au sein du cycle. Ce courant d’électrons est
responsable d’un champ magnétique induit qui s’oppose au champ extérieur
à l’intérieur du cycle.
Les atomes situés à l’intérieur du cycle sont soumis à un champ moins important –
.
Les atomes situés à l’extérieur du cycle sont au contraire soumis à un champ plus important +
: le champ à
appliquer pour atteindre leur valeur normale de résonance est donc moindre. Le déplacement chimique δ
correspondant est par conséquent plus grand.
1.4. Critère d’aromaticité de Hückel
Critère de Hückel : Un composé est aromatique s’il présente :
Sur un cycle plan ou des cycles plans accolés
4p + 2 électrons π (p : entier naturel)
Délocalisés sur l’ensemble des atomes du cycle
Composé Conclusion
N
H
N

-6-
Remarques :
o Limite du décompte des électrons π : L’analyse d’une formule topologique permet, certes, de dire si la condition des 4 p + 2 électrons délocalisés cycliquement est vérifiée ou non, mais elle ne permet pas de dire si la molécule est plane. Par exemple, le [10]annulène vérifie la condition au niveau du décompte des électrons, mais sa structure non plane non prévisible sur une formule topologique ne permet pas, en réalité, une délocalisation sur l’ensemble du cycle. La molécule n’est donc pas aromatique.
o Critère moderne d’aromaticité : Un cycle est aromatique s’il déblinde fortement les atomes d’hydrogène qu’ils portent
et qui sont situés à l’extérieur du cycle.
2. Substitution électrophile aromatique (SEAr) sur le benzène
2.1. Vue d’ensemble
- Equation générale :
o Nucléophile =
o Electrophile =
- Réaction de substitution électrophile aromatique (SEAr) : quatre modifications du cyle au programme :
H remplacé par : Electrophile
Halogénation –H → –X (Atome d’halogène)
Nitration –H → –NO2 (Groupe nitro)
Alkylation de Friedel-Crafts –H → –R (Groupe alkyle)
Acylation de Friedel-Crafts –H → –CO-R (Groupe acyle)
- Nécessité d’une catalyse ! Sans catalyseur, rien ne se produit.

-7-
2.2. Mécanisme général
- Mécanisme : se décompose en trois étapes
Formation du réactif électrophile (différent pour chaque réaction, voir plus loin)
Addition de l’électrophile sur le cycle benzénique = formation d’un ion arénium ou intermédiaire de Wheland
Elimination de l’hydrogène = réaromatisation
Commentaires :
- Profil énergétique (seules les deux dernières étapes sont représentées : formation IdW, puis réaromatisation) :
Conclusion :

-8-
2.3. Réaction de nitration
Réaction de nitration :
Réactif = Acide nitrique HNO3 Catalyseur = Acide nitrique ou sulfurique Electrophile = Ion nitronium NO2+
- Mécanisme :
Formation de l’électrophile (mélange sulfo-nitrique) :
Commentaires :
Addition de l’électrophile sur le cycle benzénique = formation de l’ion arénium
Elimination de l’hydrogène = réaromatisation
- Application de la réaction : Greffage d’une fonction amine −NH2 sur un cycle benzénique
Problème : Impossible de remplacer directement H par un groupe amino –NH2. Solution : Faire d’abord une nitration (remplacer –H par –NO2), puis réduire en groupe amino –NH2.

-9-
2.4. Réaction d’halogénation
Réaction de chloration :
Réactif = Dichlore X2 Catalyseur = Acide de Lewis (AlCl3) Electrophile = Ion halogénium X+
- Mécanisme de formation de l’électrophile :
Une fois l’électrophile obtenu, il réagit dans les deux étapes présentées à la page précédente avec le cycle aromatique.
Remarque 1 : Comment éviter la formation de HCl(g) ? Remarque 2 : Pour la réaction de bromation :
2.5. Réaction d’alkylation de Friedel et Crafts
a) A partir d’un halogénoalcane
Réaction d’alkylation :
Réactif = Dérivé chloré R-Cl Catalyseur = Acide de Lewis (AlCl3) Electrophile = Carbocation R+
- Mécanisme de formation de l’électrophile :
Les deux dernières étapes sont identiques à ce qui a été présenté dans le mécanisme général.

-10-
- Problème : Réarrangement possible de carbocations instables : certains carbocations particulièrement instables se réarrangent par migration sur le site adjacent d’un H si le carbocation ainsi obtenu est plus stable :
b) A partir d’un alcool ou d’un alcène
- Il existe d’autres façons de former des carbocations qui peuvent réagir avec le cycle benzénique :
Alcool + acide : Alcène + acide :
2.6. Réaction d’acylation de Friedel et Crafts
a) A partir d’un chlorure d’acyle
Réaction d’acylation :
Réactif = Chlorure d’acyle R-CO-Cl Catalyseur = Acide de Lewis (AlCl3 Electrophile = Ion acylium RCO+
- Mécanisme de formation de l’électrophile :
Les deux dernières étapes sont identiques à ce qui a été présenté dans le mécanisme général.

-11-
Attention ! Complexation par l’oxygène d’AlCl3 : Conséquences :
Hydrolyse acide finale nécessaire pour séparer l’acide de Lewis de la cétone.
Quantité de AlCl3 à introduire : AlCl3 n’étant pas régénéré, il est nécessaire de l’introduire en quantité plus importante que le dérivé aromatique (1,1 éq par exemple).
b) A partir d’un anhydride d’acide
- Autre méthode de formation d’un ion acylium : (La suite du mécanisme est inchangée) c) Application en synthèse : Comment contourner le problème du réarrangement de carbocation ?
Clemmensen a mis au point une méthode de réduction des cétones aromatiques par traitement avec du zinc amalgamé (Zn/Hg) en milieu acide chlorhydrique concentré :
Cl
O O
AlCl3
HCl cc
Zn(Hg)

-12-
3. SEAr sur un cycle déjà substitué
Problématique : Si l’on opère une SEAr sur un cycle déjà substitué par un groupement G, 2 conséquences à envisager :
Le groupe déjà présent accélère-t-il ou ralentit-il la SEAr par rapport au benzène ? (effet cinétique)
Le groupe déjà présent impose-t-il une position privilégiée pour le substituant qui va arriver ? (effet régiosélectif)
3.1. Définitions
- Aspect cinétique : Notion de substituant activant / désactivant :
EE+
k1
E+
k2
G G
E
Si k1 < k2 : le groupe a accéléré la réaction par rapport au cas du benzène : il a rendu le cycle plus nucléophile. Son effet est qualifié d’activant.
Si k1 > k2 : le groupe a ralenti la réaction par rapport au cas du benzène : il a rendu le cycle moins
nucléophile. Son effet est qualifié de désactivant.
- Aspect régiosélectif : Positions du nouveau groupement sur le cycle déjà substitué :
G
3.2. Résultats expérimentaux
- On a obtenu les résultats suivants pour la nitration de différents dérivés aromatiques notés Ph−G
Si évolution gouvernée par le hasard, on doit obtenir :
20 % de produit en position para (1 chance sur 5)
40 % de produit en position méta (2 chances sur 5)
40 % de produit en position ortho (2 chances sur 5) Si ces proportions ne sont pas respectées, le groupe a un effet régiosélectif.

-13-
- Conclusion : La présence d’un groupe sur le cycle rend la SEAr régiosélective !
- On constate trois situations = règles de Holleman :
Exemples Effet cinétique Régiosélectivité
G = électrodonneur -OH -CH3
-tBu (= -C(CH3)3) Activant Ortho et Para
G = électroattracteur -NO2
-CF3 -CO-OEt
Désactivant Méta
G = halogène -Cl -Br -I
Désactivant Ortho et Para
3.3. Cadre théorique de l’étude
- Le but de l’étude qui suit est de retrouver les règles de Holleman. Ces règles permettent de prévoir le composé majoritaire et l’effet cinétique d’un substituant G déjà présent sur une nouvelle SEAr.
Comment conduire le raisonnement pour retrouver les règles de Holleman ?
1. Argument 1 : La SEAr est supposée sous contrôle cinétique : le produit majoritaire est le produit le plus
facilement/vite formé.
2. Argument 2 : L’étape qui contrôle la vitesse et la sélectivité est la formation de l’ion arénium car :
a. Aspect régiosélectif : c’est au cours de cette étape que se fixe le nouveau substituant sur le cycle.
b. Aspect cinétique : c’est l’étape supposée cinétiquement déterminante
Trouver le produit majoritaire revient donc à identifier l’ion arénium formé le plus vite par cette étape.
3. Argument 3 : L’état de transition de l’étape de formation de l’ion arénium est inconnu : son niveau
d’énergie ne peut être estimé qu’au moyen du postulat de Hammond :
Etat de transition tardif (voir 2.2) Postulat de Hammond : l’état de transition a une structure proche de celle de l’ion arénium.
En pratique :
a) On écrit les différents ions arénium possibles b) On identifie celui qui est le plus stabilisé (donc le plus bas en énergie). c) Le postulat de Hammond permet d’affirmer que l’état de transition qui lui est associé est le plus
bas en énergie. d) Ce processus est celui associé à l’énergie d’activation la plus basse, donc processus le plus rapide.
Conclusion :
Le produit majoritaire provient du chemin réactionnel passant par l’intermédiaire de Wheland le plus stabilisé.
Attention ! Erreur fréquente ! On étudie l’influence du groupe déjà présent sur le cycle et du nouveau groupe introduit.
-14-
O
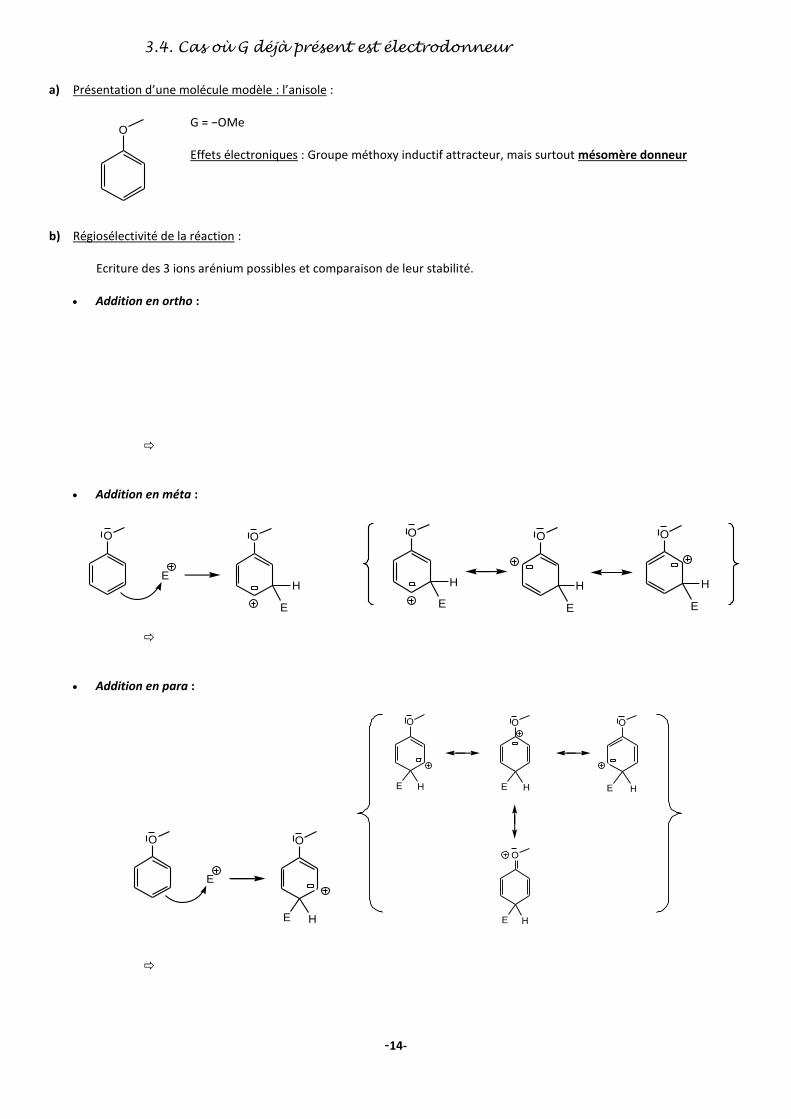
3.4. Cas où G déjà présent est électrodonneur
a) Présentation d’une molécule modèle : l’anisole :
G = −OMe Effets électroniques : Groupe méthoxy inductif attracteur, mais surtout mésomère donneur
b) Régiosélectivité de la réaction : Ecriture des 3 ions arénium possibles et comparaison de leur stabilité.
Addition en ortho :
Addition en méta :
O
E
O
H
E
O O O
H
E
H
E
H
E
Addition en para :
O
E
O
E H
O OO
O
E H E H E H
E H

-15-
Conclusion : Profils énergétiques avec groupe G électrodonneur déjà présent
Ep
CR
Conclusion : c) Vitesse de réaction : d) Extension aux groupes inductifs donneurs :

-16-
NOO
3.5. Cas où G déjà présent est électroattracteur
a) Présentation de la molécule modèle : le nitrobenzène :
G = −NO2
Effets électroniques : groupe nitro inductif attracteur et mésomère attracteur
b) Vitesse de réaction : c) Régiosélectivité de la réaction :
Sous contrôle cinétique, on forme majoritairement l’isomère obtenu par passage par l’IdW le plus bas en énergie On compare donc les énergies des ions arénium issus des trois additions possibles
Addition en ortho :
NO
E
NO
E
H
O O
NO
E
H
NO
E
H
NO
E
H
O O O
Addition en méta :
NO
E
NO
H
E
O O
NO
NO
NO
H
E
H
E
H
E
O O O
Addition en para :
NO
E
NO
E H
O O
NO
NO
NO
E H E H E H
O O O

-17-
Cl
Conclusion : Profils énergétiques avec groupe G électroattracteur déjà présent
Ep
CR
3.6. Cas où G déjà présent est un halogène
a) Cas du chlorobenzène :
G = −Cl
Effets électroniques : Deux effets antagonistes : mésomère donneur et inductif attracteur. Les deux effets s’expriment.
b) Vitesse de réaction : C’est l’effet attracteur qui prédomine : les halogènes sont désactivants. En attirant les électrons, ils diminuent la densité électronique sur le cycle. Le cycle aromatique est donc moins nucléophile que le benzène. La réaction est plus lente qu’avec le benzène. c) Régiosélectivité de la réaction :
C’est l’effet mésomère donneur qui prédomine : les halogènes sont ortho/para orienteurs. Lorsque le cycle a perdu temporairement son caractère aromatique (ion arénium), l’atome d’halogène est capable de de
délocaliser un doublet non liant pour stabiliser les ions arénium ortho et para (lacune au pied de l’halogène) par rapport à l’ion arénium méta.
Les ions arénium ortho et para sont donc plus vite formés. Expl : ion arénium ortho
ClE
H
ClE
H
ClE
H
ClE
H

-18-
NO2
Br
OH
4. Stratégie en synthèse organique
4.1. Cas où le cycle est déjà disubstitué
- Il arrive que l’on réalise une SEAr sur un benzène disubstitué, et que les deux orientations prévues par les règles de Holleman pour les deux substituants en place concordent : on parle de « synergie des orientations » Expl :
- Parfois, par contre, les orientations ne concordent pas : Expl :
Comment trancher ?
4.2. Importance de l’ordre des réactions
- Lorsqu’on veut synthétiser à partir du benzène un produit disubstitué, il faut réfléchir à l’ordre des réactions permettant cette synthèse.
Expl : on désire synthétiser :
Br
O

-19-
4.3. Problème de la polyalkylation
- Toute réaction d’alkylation d’un cycle aromatique pose des problèmes de polyalkylation :
4.4. Inversion d’orientation
- Dans le cas où l’on désire réaliser une disubstitution du benzène, on peut parfois modifier le 1e substituant du cycle afin
d’inverser l’orientation de la 2e SEAr
Expl : on désire synthétiser :
NO2
O





























