Embed Size (px)
Citation preview

Chirality transition in the epoxidation of (�)-a-pinene and successive
hydrolysis studied by Raman optical activity and DFTw
Shi Qiu,zab Guanna Li,zab Peng Liu,ab Changhao Wang,ab Zhaochi Feng*a and
Can Li*a
Received 25th September 2009, Accepted 12th January 2010
First published as an Advance Article on the web 9th February 2010
DOI: 10.1039/b919993d
Characterization of the chirality evolution involved in chemical and biochemical reaction
processes is extremely important to the understanding of the chiral catalysis mechanism. In this
work, the chiral transition from the epoxidation of (�)-a-pinene to a-pinene oxide and successive
hydrolysis to (�)-pinanediol has been studied as an archetype of the asymmetric catalysis by
Raman optical activity (ROA) and the DFT calculation. Minor changes of the absolute
configuration of the chiral products from (�)-a-pinene to (�)-pinanediol lead to the dramatic
variation in ROA spectra indicating that the chirality is delocalized in the whole molecule rather
than only concentrated on the chiral centers. The oxygen atom of a-pinene oxide contributes
strong ROA signals while the two hydroxyl groups of (�)-pinanediol give no apparent
contribution to the chirality in terms of ROA signals. Isolation of the two symmetric anisotropic
invariants shows that the predominant contribution to the ROA signals stems from the electric
dipole–magnetic dipole invariant, and the bond polarizability model is indeed found to be a good
approximation for molecules composed of entirely axially-symmetric bonds in a-pinene oxide and
(�)-pinanediol. This study demonstrates the feasibility of using ROA to sensitively monitor the
variation of the chirality transition during the chiral reactions either in the chemical or biological
system.
Introduction
Direct observation of configurational and conformational
transitions is crucial for understanding the mechanism of
chiral reactions, especially in chemical and biochemical
catalysis. However, these changes are difficult to probe using
conventional methods, although dynamics simulations
sometimes help.1 Chiroptical spectroscopic characterization,
especially vibrational optical activity, including vibrational
circular dichroism (VCD) and Raman optical activity
(ROA), are proven to be an effect way to determine the
configuration, conformation and behavior of chiral
molecules.2–6 In particular, ROA is considered as an ideal
tool for the study of biological molecules in aqueous
solution.7–14
The theoretical background for the ROA phenomenon was
given by Barron and Buckingham in 1971.15 Polavarapu
presented the first complete theoretical study of the CIDs
observed in experimental ROA spectra in 1990,16 using a
static approximation to G0(o) as proposed by Amos.17 The
first correlated gauge-origin independent calculations were
presented by Helgaker et al. using multi-configurational
self-consistent field (MCSCF) wave functions and London
atomic orbitals.18 Bour19 has combined sum-over-states
DFT calculations (including hybrid functionals) of optical
tensors with numerical differentiation with respect to nuclear
displacements. The analytical DFT calculations of ROA
spectra using linear response theory and London atomic
orbitals was presented by Ruud et al.20 A quantum-mechanical
approach for predicting the ROA spectra is essential for
an in-depth interpretation of the experimental spectra,
providing helpful physical insight into the generation of
vibrational optical activity,21–26 and it has been successfully
applied to the studies on small chiral molecules such
as bromochlorofluoromethane,27,28 substituted oxiranes,
biologically significant amino acids and oligopeptides as
primary model molecules for understanding the stereochemical
properties of polypeptides and proteins.29–34
Oxyfunctionalization of cyclic olefins is of industrial and
biological importance due to the possibility of transforming
cheap and readily available substrates to valuable inter-
mediates for fine chemicals and pharmaceutical synthesis.35–43
a-Pinene is a standard molecule frequently used in ROA study.
We chose epoxidation of a-pinene to a-pinene oxide and
a State Key Laboratory of Catalysis, Dalian Institute of ChemicalPhysics, Chinese Academy of Sciences, Dalian, 116023, China.E-mail: [email protected], [email protected];Fax: +86-411-84694447; Tel: +86-411-84379303,+86-411-84379070
bGraduate University of Chinese Academy of Sciences, Beijing,100049, China
w Electronic supplementary information (ESI) available: Synthesisprocedure of a-pinene oxide; introduction of visualization of vibra-tional modes and atomic contribution patterns (ACPs) pictorial; XRDand IR spectra of catalyst ZnAl-[Salen(Mn)]; NMR spectrum ofa-pinene oxide; vibrational modes and ACPs pictorial representation;Mulliken atomic charges distribution; Raman vibrational analysis andanisotropic invariants of the optical activity tensor components. SeeDOI: 10.1039/b919993dz These authors contributed equally to this work.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 | 3005
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online / Journal Homepage / Table of Contents for this issue

subsequent hydrolysis as an archetypal reaction. These reactions
are proper models for assessing the chiral structure changes in
the chemical and biochemical reactions.
Spectroscopic characterization of the absolute configuration
changes is a long standing challenge in chiral catalysis. To our
knowledge, there is no report on the chiral characterization in
the organic synthesis or asymmetric catalysis processes using
ROA spectroscopy. In this work, we studied the chirality
transition in the epoxidation of (�)-a-pinene to a-pinene oxideand the successive hydrolysis to (�)-pinanediol as a typical
reaction in the asymmetric catalysis process. The ROA
differences between the three bicyclo compounds, owing to
the addition of the two new chiral centers and the contribution
from the substituent oxygen atom and hydroxyl groups, are
discussed in detail. Based on the DFT calculations, it is found
that the main contributions to the backscattering ROA
intensities are from the electric dipole–magnetic dipole
symmetric anisotropic invariant while those from the electric
dipole–electric quadrupole invariant are relatively small. This
work also demonstrates that ROA can supply information on
the chiral environment changes and transitions during the
chiral catalytic reactions either in chemical or biological
systems.
Method
Experimental
The chiral transition model is represented by the epoxidation
of (�)-a-pinene and successive hydrolysis to (�)-pinanediol.(�)-a-Pinene and (�)-pinanediol were purchased from
Acros and used without further purification. a-Pinene oxide
was synthesized by the method of Anderson.41 Catalyst
characterization was shown in Fig. S1.w a-Pinene oxide
structure was confirmed by 1H NMR spectrum (in CDCl3)
shown in Fig. S2,w with 83.0% de confirmed by GC-MS.
ROA spectra were recorded on a Chiral RAMAN instru-
ment (BioTools Inc.), which simultaneously provided both
backscattering Raman and scattered circularly polarized ROA
spectra. The instrument utilized a CW 532 nm laser source
(Verdi 2) and a back-thinned CCD detector, which was
optimized for recording spectra in the range 100–2500 cm�1.
Spectral resolution was approximately 7 cm�1. The power at
the laser head output was 600 mW for both (�)-a-pinene anda-pinene oxide and 350 mW for (�)-pinanediol in CCl4solution (4 M). The total exposure time was 18 min for
both (�)-a-pinene and a-pinene oxide, and 137 min for
(�)-pinanediol.As an appropriate experimental quantity, the dimensionless
circular intensity difference (CID) first introduced by Barron15
is defined as
D ¼ IR � IL
IR þ IL
where IR and IL are the scattered intensities in right- and
left-circularly polarized incident light, respectively. Using
the Placzek polarizability approximation, differential CIDs
associated with the four most important scattering geometries
for an isotropic collection of chiral molecules for scattered
circular polarization forms of ROA can be written as:12
Dð0�Þ ¼ 8½45aG0 þ bðG0Þ2 � bðAÞ2�2c½45a2 þ 7bðaÞ2�
Dð180�Þ ¼ 48½bðG0Þ2 þ ð1=3ÞbðAÞ2�2c½45a2 þ 7bðaÞ2�
Dxð90�Þ ¼12½45aG0 þ 7bðG0Þ2 þ bðAÞ2�
c½45a2 þ 7bðaÞ2�
Dzð90�Þ ¼12½bðG0Þ � ð1=3ÞbðAÞ2�
6cbðaÞ2
a and G0 are the isotropic invariants of the polarizability
tensor and the electric dipole–magnetic dipole optical activity
tensor, respectively.
a =1
3aaa =
1
3(axx + ayy + azz),
G0 ¼ 1
3G0aa ¼
1
3ðG0xx þ G0yy þ G0zzÞ;
b(a)2, b(G0)2 and b(A)2 are the anisotropic invariants of the
polarizability tensor and optical activity tensor components.
b(a)2 =1
2(3aabaab � aaaabb),
bðG0Þ2 ¼ 1
2ð3aabG0ab � aaaG0bbÞ;
b(A)2 =1
2oaabeagdAgdb,
Calculations
For the DFT calculation, the program package Gaussian 03
version D 0144 was used. The geometry optimization of
isolated (�)-a-pinene, a-pinene oxide and (�)-pinanediol andthe simulation of the Raman and ROA spectra were
operated using the Becke-three-parameter-Lee–Yang–Parr
(B3LYP)45–47 hybrid functional and the aug-cc-PVDZ basis
set.48–51 We evaluated the relative energy of the two possible
configurations of a-pinene oxide to confirm the abundant one
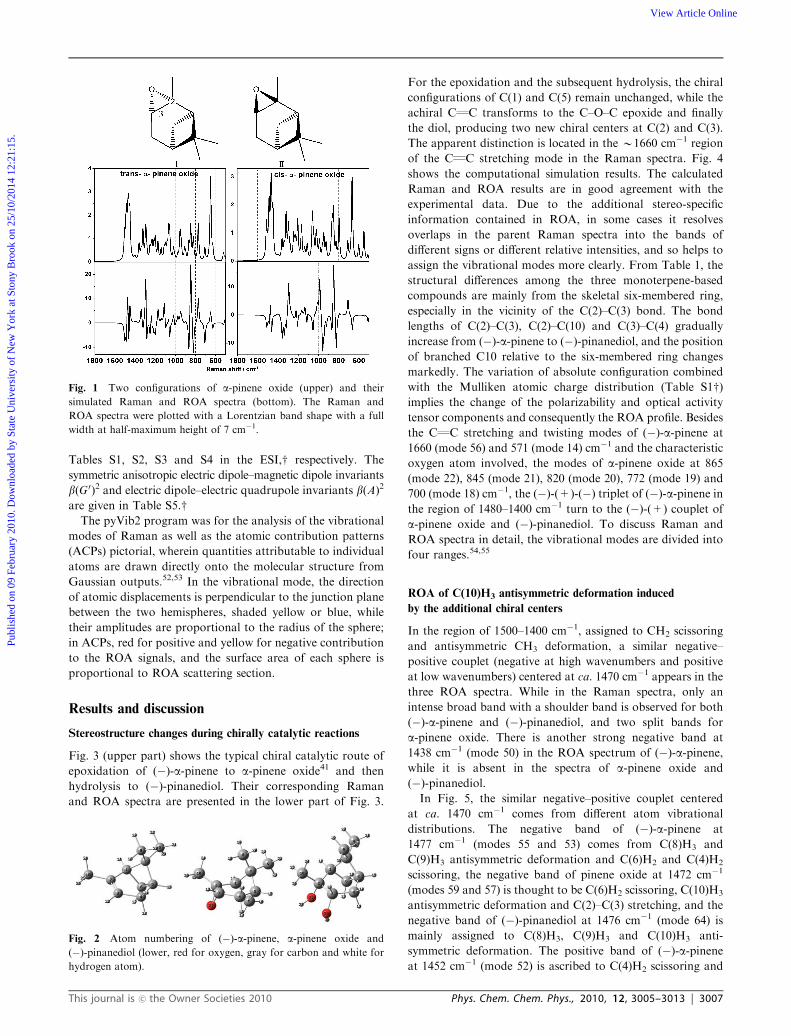
(upper part of Fig. 1). The result shows that configuration I is
more stable than II with an energy difference of 15.75 kJ mol�1,
so the configuration of the epoxide product should be mainly
in configuration I which is consistent with the de% value.
Comparison of the B3LYP/aug-cc-PVDZ ROA spectra of the
two isomers (lower part of Fig. 1) to the experimental ROA
spectrum of a-pinene oxide (middle part of Fig. 3) leads
unambiguously to the conclusion that the trans-isomer is
mainly the configuration of a-pinene oxide deriving from
(�)-a-pinene. For (�)-pinanediol, the two hydroxyl groups
hold cis form attached to C(2) and C(3). The structures and
atom numbering for (�)-a-pinene, trans-a-pinene oxide and
(�)-pinanediol are shown in Fig. 2. The optimized geometry
parameters are listed in Table 1, and the Mulliken atomic
charges distribution and the vibrational modes analysis in
3006 | Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 This journal is �c the Owner Societies 2010
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online
Tables S1, S2, S3 and S4 in the ESI,w respectively. The
symmetric anisotropic electric dipole–magnetic dipole invariants
b(G0)2 and electric dipole–electric quadrupole invariants b(A)2
are given in Table S5.wThe pyVib2 program was for the analysis of the vibrational
modes of Raman as well as the atomic contribution patterns
(ACPs) pictorial, wherein quantities attributable to individual
atoms are drawn directly onto the molecular structure from
Gaussian outputs.52,53 In the vibrational mode, the direction
of atomic displacements is perpendicular to the junction plane
between the two hemispheres, shaded yellow or blue, while
their amplitudes are proportional to the radius of the sphere;
in ACPs, red for positive and yellow for negative contribution
to the ROA signals, and the surface area of each sphere is
proportional to ROA scattering section.
Results and discussion
Stereostructure changes during chirally catalytic reactions
Fig. 3 (upper part) shows the typical chiral catalytic route of
epoxidation of (�)-a-pinene to a-pinene oxide41 and then
hydrolysis to (�)-pinanediol. Their corresponding Raman
and ROA spectra are presented in the lower part of Fig. 3.
For the epoxidation and the subsequent hydrolysis, the chiral
configurations of C(1) and C(5) remain unchanged, while the
achiral CQC transforms to the C–O–C epoxide and finally
the diol, producing two new chiral centers at C(2) and C(3).
The apparent distinction is located in the B1660 cm�1 region
of the CQC stretching mode in the Raman spectra. Fig. 4
shows the computational simulation results. The calculated
Raman and ROA results are in good agreement with the
experimental data. Due to the additional stereo-specific
information contained in ROA, in some cases it resolves
overlaps in the parent Raman spectra into the bands of
different signs or different relative intensities, and so helps to
assign the vibrational modes more clearly. From Table 1, the
structural differences among the three monoterpene-based
compounds are mainly from the skeletal six-membered ring,
especially in the vicinity of the C(2)–C(3) bond. The bond
lengths of C(2)–C(3), C(2)–C(10) and C(3)–C(4) gradually
increase from (�)-a-pinene to (�)-pinanediol, and the position
of branched C10 relative to the six-membered ring changes
markedly. The variation of absolute configuration combined
with the Mulliken atomic charge distribution (Table S1w)implies the change of the polarizability and optical activity
tensor components and consequently the ROA profile. Besides
the CQC stretching and twisting modes of (�)-a-pinene at
1660 (mode 56) and 571 (mode 14) cm�1 and the characteristic
oxygen atom involved, the modes of a-pinene oxide at 865
(mode 22), 845 (mode 21), 820 (mode 20), 772 (mode 19) and
700 (mode 18) cm�1, the (�)-(+)-(�) triplet of (�)-a-pinene inthe region of 1480–1400 cm�1 turn to the (�)-(+) couplet of
a-pinene oxide and (�)-pinanediol. To discuss Raman and
ROA spectra in detail, the vibrational modes are divided into
four ranges.54,55
ROA of C(10)H3 antisymmetric deformation induced
by the additional chiral centers
In the region of 1500–1400 cm�1, assigned to CH2 scissoring
and antisymmetric CH3 deformation, a similar negative–
positive couplet (negative at high wavenumbers and positive
at low wavenumbers) centered at ca. 1470 cm�1 appears in the
three ROA spectra. While in the Raman spectra, only an
intense broad band with a shoulder band is observed for both
(�)-a-pinene and (�)-pinanediol, and two split bands for
a-pinene oxide. There is another strong negative band at
1438 cm�1 (mode 50) in the ROA spectrum of (�)-a-pinene,while it is absent in the spectra of a-pinene oxide and
(�)-pinanediol.In Fig. 5, the similar negative–positive couplet centered
at ca. 1470 cm�1 comes from different atom vibrational
distributions. The negative band of (�)-a-pinene at
1477 cm�1 (modes 55 and 53) comes from C(8)H3 and
C(9)H3 antisymmetric deformation and C(6)H2 and C(4)H2
scissoring, the negative band of pinene oxide at 1472 cm�1
(modes 59 and 57) is thought to be C(6)H2 scissoring, C(10)H3
antisymmetric deformation and C(2)–C(3) stretching, and the
negative band of (�)-pinanediol at 1476 cm�1 (mode 64) is
mainly assigned to C(8)H3, C(9)H3 and C(10)H3 anti-
symmetric deformation. The positive band of (�)-a-pineneat 1452 cm�1 (mode 52) is ascribed to C(4)H2 scissoring and
Fig. 1 Two configurations of a-pinene oxide (upper) and their
simulated Raman and ROA spectra (bottom). The Raman and
ROA spectra were plotted with a Lorentzian band shape with a full
width at half-maximum height of 7 cm�1.
Fig. 2 Atom numbering of (�)-a-pinene, a-pinene oxide and
(�)-pinanediol (lower, red for oxygen, gray for carbon and white for
hydrogen atom).
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 | 3007
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online

C(9)H3 antisymmetric deformation, the positive band of
pinene oxide at 1440 cm�1 (modes 55 and 54) is thought to
be C(4)H2 scissoring, C(8)H3, C(9)H3 and C(10)H3 anti-
symmetric deformation and C(2)–C(3) stretching, and the
positive band of (�)-pinanediol at 1461cm�1 (mode 62) is
ascribed to C(9)H3 and C(10)H3 antisymmetric deformation
and C(4)H2 scissoring modes. The unique sharp negative band
of (�)-a-pinene at 1438 cm�1 (mode 50) is mainly ascribed to
C(4)H2 scissoring and C(8)H3 and C(9)H3 antisymmetric
deformation. Although no apparent similar negative bands
are observed for pinene oxide and (�)-pinanediol, there
exists a negative band in the simulated ROA spectrum of
Table 1 Calculated skeletal bong lengths (A), bond angles (deg) and dihedral angles (deg) for the three molecules (# for distinct variation)
Entry (�)-a-Pinene a-Pinene oxide 2,3-(�)-Pinanediol Note
C1–C2 1.522 1.534 1.539C2–C3 1.343 1.476 1.582 #C2–C10 1.502 1.508 1.531 #C3–C4 1.515 1.526 1.559 #C4–C5 1.541 1.541 1.536C5–C6 1.556 1.555 1.550C5–C7 1.574 1.576 1.569C6–C1 1.562 1.553 1.556C7–C1 1.582 1.582 1.579C7–C8 1.529 1.532 1.533C7–C9 1.536 1.535 1.538C1–C2–C3 116.6 114.0 110.9 #C2–C3–C4 119.9 117.9 114.4 #C3–C4–C5 110.3 111.5 113.6 #C4–C5–C6 108.6 109.1 107.8C5–C6–C1 85.9 86.4 86.4C6–C1–C7 87.1 87.5 87.3C6–C1–C2 107.0 109.7 108.2C1–C7–C5 84.6 84.7 85.0C7–C5–C6 87.6 87.6 87.9C10–C2–C3–C4 �178.5 �150.9 �140.8 #C10–C2–C1–C6 �133.6 �161.5 �178.3 #
Fig. 3 The typical chiral catalytic route of epoxidation of (�)-a-pinene to a-pinene oxide41 and then hydrolysis to (�)-pinanediol (upper) and the
backscattered SCP Raman and ROA spectra of (�)-a-pinene (left part), a-pinene oxide (middle part), and (�)-pinanediol solved in CCl4 (right
part). denotes the chiral centers C(1) and C(5), and denotes the new chiral centers, C(2) and C(3). The numbers label the vibrational modes.
3008 | Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 This journal is �c the Owner Societies 2010
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online

pinene oxide (mode 52), and it is assigned to C(4)H2
scissoring, C(8)H3, C(9)H3, and C(10)H3 antisymmetric
deformation modes.
In this region, C(4)H2 scissoring and C(8)H3, C(9)H3
antisymmetrical deformations dominate the ROA bands of
(�)-a-pinene, and after epoxidation of the CQC bond, the
Fig. 4 The computational simulation of Raman and ROA spectra of (�)-a-pinene (left part), trans-a-pinene oxide (middle part), and
(�)-pinanediol (right part). The Raman and ROA spectra were plotted with a Lorentzian band shape with a full width at half-maximum height
of 7 cm�1.
Fig. 5 Vibrational normal modes (left) and ACPs (right) for (�)-a-pinene (left column), a-pinene oxide (middle column) and (�)-pinanediol(right column) assigned to CH2 scissoring and antisymmetric CH3 deformation (vibrational mode number and the ROA signs are labeled).
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 | 3009
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online

C(10)H3 antisymmetrical deformation shows Raman optical
activity. As the C(10) is adjacent to one of the newly formed
chiral center C(2), it implies that the additional chiral
environment induces the ROA of C(10)H3 antisymmetrical
deformation. It can be also seen from modes 51 and 48 of the
calculated ROA spectrum of (�)-a-pinene, the ACPs
(Fig. S3w) shows that although the two hydrogen atoms of
the C(10)H3 group both have strong ROA contributions, their
signs are opposite, so the total contribution is nearly zero and
the antisymmetrical deformation of C(10)H3 group shows
little ROA in (�)-a-pinene. Overall, the similar negative–
positive couplet in the ROA spectra reflects the chiral
environment of the bicyclic structure.
ROA distributions among similar vibrational modes
In the 1400–1300 cm�1 region, Raman bands are mainly from
the vibrations of CH2 wagging and symmetric CH3 deformation.
In Fig. 6, the positive band of (�)-a-pinene at 1379 cm�1
(mode 46) in the ROA spectra is ascribed to C(10)H3
symmetric deformation. Its counterpart for a-pinene oxide is
at 1396 cm�1 (mode 49) and is nearly absent for (�)-pinanediol(mode 56). The relative position of C(10)H3 in the chiral
environment gradually transforms, and the ROA changes
although they have similar Raman vibrational modes.
A similar situation is also found for the positive band at
1270 cm�1 of (�)-a-pinene (mode 41), the band at 1283 cm�1
of a-pinene oxide (mode 44) and the band at 1224 cm�1 of
(�)-pinanediol (modes 49) involving the C(1)H and C(5)H
wagging, C(4)H2 twisting and C(7)–C(5) stretching modes
(Fig. 7). The relatively large shift in the band of (�)-pinanediolis thought to be the involvement of the OH wagging and
C(6)H wagging vibrations. Although they all show one
positive band, their ROA signals from the atom contributions
are not the same. The predominant contribution to the
positive ROA signal is from C(7) for (�)-a-pinene and the
two hydrogen atoms linked to C(4) make almost exactly
opposite contribution. For a-pinene oxide, the hydrogen atom
linked to C(3) and one of the hydrogen atoms linked to C(4)
make comparable contributions to ROA with that of C(7).
While for (�)-pinanediol, C(5), C(2), the hydrogen atom
linked to C(3) and one of the hydrogen atoms linked to C(6)
make relatively large positive contributions and the hydrogen
atom linked to C(5) make strong negative contribution
although the contribution of C(7) is still positive.
Apparently, the similarity of the vibrational modes comes
from their similar bicyclic structure, however, the optical
activity tensor derivatives with respect to the normal coordinate
vary in the slightly different asymmetric environments. Thus
the atomic contributions to ROA differ obviously and produce
ROA signals with the same or opposite signs. Some other
vibrational modes alike are shown in Fig. S4a–d.w
ROA induced by substituted oxygen atom and diol
In the region 1200–600 cm�1, all the three monoterpene-based
compounds show very characteristic strong ROA. For the
a-pinene oxide, the substituent oxygen atom makes a large
contribution to the ROA signals in this region. In Fig. 8, it is
easy to observe positive or negative effect from the oxygen
atom, which is in good agreement with the sign of related
ROA bands for a-pinene oxide. For example, the strongly
negative band at 865 cm�1 (mode 22) is ascribed to
C(2)–O–C(3) ring deformation. The ACPs suggest that the
substituted oxygen atom contributes negatively to the ROA
intensity. Similarly, in the next ring deformation and H
bending mode at 845 cm�1 (mode 21), the oxygen atom has
a positive contribution to the whole ROA intensity which is
consistent with the observed strong positive band. The negative
contribution from the oxygen atom in the skeletal deformation
and branched CH3 wagging mode at 820 cm�1 (mode 20) is
also in accordance with the strong negative band of the
experimental spectrum. The same correlation is observed for
the positive band at 772 cm�1 (mode 19) assigned to the ring
deformation, and other modes at 1144 (mode 36), 1066 (mode 32),
1045 (mode 31), 700 (mode 18) and 572 cm�1 (mode 15)
as well. The excellent correlation of the contributions of
substituted oxygen atom with the sign of ROA signals is
distinct to a-pinene oxide.
Fig. 6 Vibrational normal mode (left), ACPs (middle), b(G0)2 and
b(A)2/3 invariants (right) for (�)-a-pinene mode 46 (upper), a-pineneoxide mode 49 (middle), and (�)-pinanediol mode 56 (lower)
(the position of atom C(10) is labeled).
Fig. 7 Vibrational normal mode (left), ACPs (middle), b(G0)2 and
b(A)2/3 invariants (right) for (�)-a-pinene mode 41 (upper), a-pineneoxide mode 44 (middle), and (�)-pinanediol mode 49 (lower) (the
position of atoms C(2), C(3), C(4), C(5), C(6) and C(7) is labeled).
3010 | Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 This journal is �c the Owner Societies 2010
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online

In contrast, the contributions from the two hydroxyl groups
of the diol are relatively small and have opposite signs in some
bands (Fig. S5w). For example, the contributions from the two
oxygen atoms of the diol to the positive band at 1136 cm�1
(mode 41) are relatively small and opposite in amplitude.
Similarly, the contributions from the two hydrogen atoms of
the diol to the positive band at 1052 cm�1 (mode 37) are also
relatively small and opposite, although the amplitude is not
the same. Thus, the correlation between the hydroxyl group
contributions and ROA signals is not as direct as that of the
oxygen atom in the a-pinene oxide. This can be explained in
terms of the rigid structure of the newly formed three-member
ring C(2)–O–C(3) in the asymmetric environment of a-pineneoxide, whereas the small ROA activity of hydroxyl group in
the chiral environment of (�)-pinanediol in the region of
1400–600 cm�1 and the cancelling effect from the two hydroxyl
groups adjacent to chiral carbon atoms of opposite configuration
(C(2) takes R configuration and C(3) takes S configuratrion).
Although the ROA signals below 600 cm�1 of a-pineneoxide and (�)-pinanediol are relatively weak, there is still a
significant band for (�)-a-pinene, i.e. a negative band at
566 cm�1 (mode 14) corresponding to the CQC twisting which
is absent for a-pinene oxide and (�)-pinanediol.
Delocalization of chirality represented by ROA
Fig. 7 shows that the atomic contributions to the ROA signals
are different while the vibrational modes are similar (as
discussed above and also in Fig. S4a–dw). It may be inferred
that ROA signals are very sensitive to minor changes of the
chiral environment, even if the vibrational modes seem alike,
as the bicyclic structure has little change. The two new formed
chiral centers from the epoxidation of (�)-a-pinene change theoptical activity not only of the adjacent groups, but also of the
whole chiral molecules, especially the bicyclic structure.
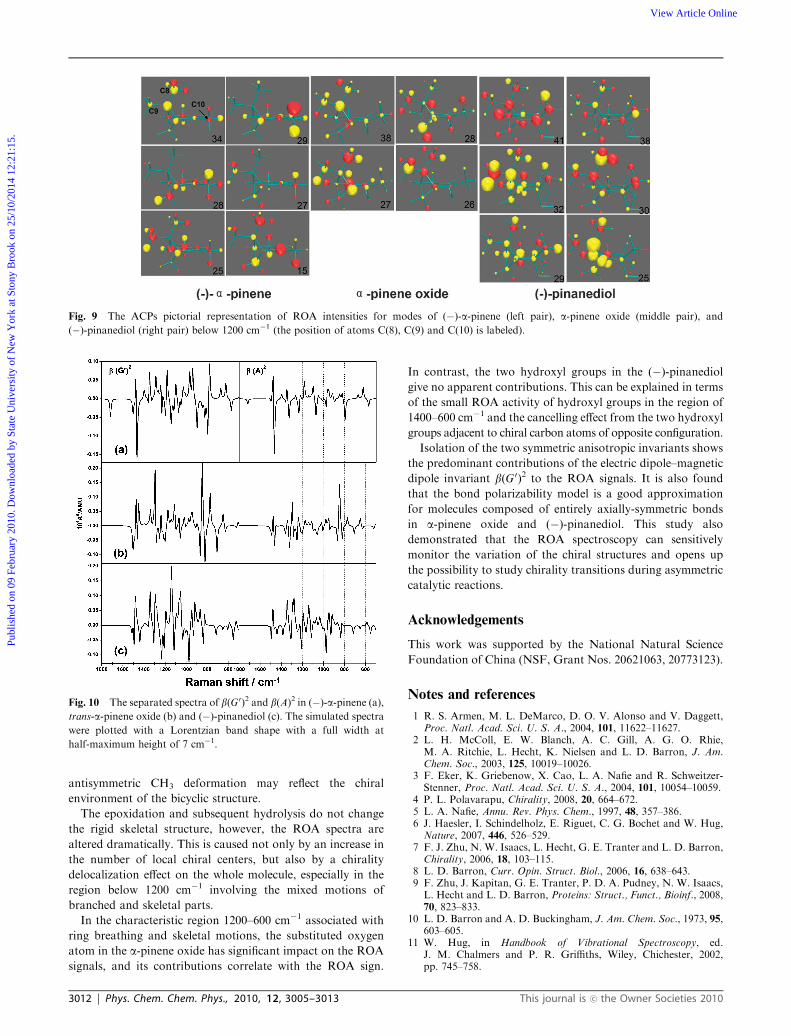
Furthermore, in Fig. 9, the groups not linked directly to the
chiral centers, i.e. C(8)H3, C(9)H3 and C(10)H3 in (�)-a-pinene, and C(8)H3 and C(9)H3 both in a-pinene oxide and
(�)-pinanediol, show medium or strong ROA activity in some
modes, especially in the region below 1200 cm�1 involving the
mixed motions of branched and skeletal parts. The C(8)H3,
C(9)H3 and C(10)H3 groups are away from the local chiral
centers C(1) and C(5) in (�)-a-pinene, and after epoxidation,
C(10)H3 is adjacent to the new-born chiral center C(2), yet the
C(8)H3 and C(9)H3 groups are still distant from the four chiral
centers C(1), C(2), C(3) and C(5) in a-pinene oxide and
(�)-pinanediol. Similar situations are also observed for the
modes 55 of (�)-a-pinene and 62 in (�)-pinanediol in Fig. 5.
These phenomena are most likely attributable to the
delocalization of chirality in the whole molecule rather than
just concentrated on the chiral centers themselves.
Contribution of electric dipole–magnetic dipole to ROA
The differential Raman backscattering intensity for the SCP
form of ROA contains the symmetric anisotropic electric
dipole–magnetic dipole invariants b(G0)2 and electric dipole–
electric quadrupole invariants b(A)2. Both the seperated
optical acitivity tensor invariants of the three bicyclic compounds
are evaluated in Fig. 10 (and the isolated invariants in Fig. S6
and Table S5w) and the relative ratio of b(G0)2 and b(A)2 in
some modes are shown in Fig. 6 and 7. In the region
1800–500 cm�1, the contributions of the b(A)2 are generally
much smaller than those of the b(G0)2, so the symmetric
anisotropic electric dipole–magnetic dipole invariant b(G0)2
plays a predominant role in the generation of ROA signals.
This phenomenon is also found in other small chiral molecules,
e.g. (S)-methyloxirane, (S)-glycidol and (+)-trans-pinane.56,57
The exceptions are only for few cases, for example in Fig. S6,wthe mode 42 in (�)-a-pinene, the mode 45 in a-pinene oxide, themodes 61, 59, 54, 51 and 48 in (�)-pinanediol mainly ascribed
to C–H wagging. However, their ROA signals are very weak.
For both of a-pinene oxide and (�)-pinanediol, b(G0)2 and
b(A)2 are nearly equal in many modes, yet not the case in
a-pinene, for which even the signs of b(G0)2 and b(A)2 are
opposite. In the bond polarizability model of ROA,13 if all the
bonds are axially-symmetric, the b(A)2 contribution will be
exactly 1/3 that of the b(G0)2. Since the bonds are all axially
symmetric in the a-pinene oxide and (�)-pinanediol (but
perhaps with some distortion in the oxide), but not in
a-pinene, this approximation should hold much better for
the oxide and diol. Such studies show that it is indeed found
to be a good approximation for molecules composed of
entirely axially-symmetric bonds, then the speed of ab initio
ROA calculations could be significantly increased, thereby
facilitating the application to some quite large systems, e.g. all
the bonds in oligo and polysaccharides are axially symmetric.
Conclusions
For the first time, the chirality transition in the epoxidation of
(�)-a-pinene to a-pinene oxide and subsequent hydrolysis to
(�)-pinanediol as an archetype of the asymmetric catalysis has
been studied by ROA and DFT calculations.
After epoxidation, the rigid bicyclic structure of (�)-a-pinene persists and two new chiral centers are born. The new
chiral centers induce the ROA signals of adjacent C(10)H3
antisymmetric deformation. The similar ROA couplet in the
region of 1480–1400 cm�1 associated with CH2 scissoring and
Fig. 8 The ACPs pictorial representation of ROA intensities of
a-pinene oxide (the position of atom O(27) is labeled).
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 | 3011
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online
antisymmetric CH3 deformation may reflect the chiral
environment of the bicyclic structure.
The epoxidation and subsequent hydrolysis do not change
the rigid skeletal structure, however, the ROA spectra are
altered dramatically. This is caused not only by an increase in
the number of local chiral centers, but also by a chirality
delocalization effect on the whole molecule, especially in the
region below 1200 cm�1 involving the mixed motions of
branched and skeletal parts.
In the characteristic region 1200–600 cm�1 associated with
ring breathing and skeletal motions, the substituted oxygen
atom in the a-pinene oxide has significant impact on the ROA
signals, and its contributions correlate with the ROA sign.
In contrast, the two hydroxyl groups in the (�)-pinanediolgive no apparent contributions. This can be explained in terms
of the small ROA activity of hydroxyl groups in the region of
1400–600 cm�1 and the cancelling effect from the two hydroxyl
groups adjacent to chiral carbon atoms of opposite configuration.
Isolation of the two symmetric anisotropic invariants shows
the predominant contributions of the electric dipole–magnetic
dipole invariant b(G0)2 to the ROA signals. It is also found
that the bond polarizability model is a good approximation
for molecules composed of entirely axially-symmetric bonds
in a-pinene oxide and (�)-pinanediol. This study also
demonstrated that the ROA spectroscopy can sensitively
monitor the variation of the chiral structures and opens up
the possibility to study chirality transitions during asymmetric
catalytic reactions.
Acknowledgements
This work was supported by the National Natural Science
Foundation of China (NSF, Grant Nos. 20621063, 20773123).
Notes and references
1 R. S. Armen, M. L. DeMarco, D. O. V. Alonso and V. Daggett,Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11622–11627.
2 L. H. McColl, E. W. Blanch, A. C. Gill, A. G. O. Rhie,M. A. Ritchie, L. Hecht, K. Nielsen and L. D. Barron, J. Am.Chem. Soc., 2003, 125, 10019–10026.
3 F. Eker, K. Griebenow, X. Cao, L. A. Nafie and R. Schweitzer-Stenner, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10054–10059.
4 P. L. Polavarapu, Chirality, 2008, 20, 664–672.5 L. A. Nafie, Annu. Rev. Phys. Chem., 1997, 48, 357–386.6 J. Haesler, I. Schindelholz, E. Riguet, C. G. Bochet and W. Hug,Nature, 2007, 446, 526–529.
7 F. J. Zhu, N. W. Isaacs, L. Hecht, G. E. Tranter and L. D. Barron,Chirality, 2006, 18, 103–115.
8 L. D. Barron, Curr. Opin. Struct. Biol., 2006, 16, 638–643.9 F. Zhu, J. Kapitan, G. E. Tranter, P. D. A. Pudney, N. W. Isaacs,L. Hecht and L. D. Barron, Proteins: Struct., Funct., Bioinf., 2008,70, 823–833.
10 L. D. Barron and A. D. Buckingham, J. Am. Chem. Soc., 1973, 95,603–605.
11 W. Hug, in Handbook of Vibrational Spectroscopy, ed.J. M. Chalmers and P. R. Griffiths, Wiley, Chichester, 2002,pp. 745–758.
Fig. 9 The ACPs pictorial representation of ROA intensities for modes of (�)-a-pinene (left pair), a-pinene oxide (middle pair), and
(�)-pinanediol (right pair) below 1200 cm�1 (the position of atoms C(8), C(9) and C(10) is labeled).
Fig. 10 The separated spectra of b(G0)2 and b(A)2 in (�)-a-pinene (a),trans-a-pinene oxide (b) and (�)-pinanediol (c). The simulated spectra
were plotted with a Lorentzian band shape with a full width at
half-maximum height of 7 cm�1.
3012 | Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 This journal is �c the Owner Societies 2010
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online

12 L. D. Barron, L. Hecht, I. H. McColl and E. W. Blanch, Mol.Phys., 2004, 102, 731–744.
13 L. D. Barron, in Molecular Light Scattering and Optical Activity,Cambridge University Press, Cambridge, 2004.
14 F. J. Zhu, N. W. Isaacs, L. Hecht and L. D. Barron, Structure,2005, 13, 1409–1419.
15 L. D. Barron and A. D. Buckingham, Mol. Phys., 1971, 20,1111–1119.
16 P. L. Polavarapu, J. Phys. Chem., 1990, 94, 8106–8112.17 R. D. Amos, Chem. Phys. Lett., 1982, 87, 23–26.18 T. Helgaker, K. Ruud, K. L. Bak, P. Jørgensen and J. Olsen,
Faraday Discuss., 1994, 99, 165–180.19 P. Bour, J. Comput. Chem., 2001, 22, 426–435.20 K. Ruud, T. Helgaker and P. Bour, J. Phys. Chem. A, 2002, 106,
7448–7455.21 Z. Deng, P. L. Polavarapu, S. J. Ford, L. Hecht, L. D. Barron,
C. S. Ewig and K. Jalkanen, J. Phys. Chem., 1996, 100, 2025–2034.22 J. Kapitan, V. Baumruk and P. J. Bour, J. Am. Chem. Soc., 2006,
128, 2438–2443.23 J. Kapitan, V. Baumruk, V. Kopecky, R. Pohl, Jr. and P. Bour,
J. Am. Chem. Soc., 2006, 128, 13451–13462.24 P. K. Bose, L. D. Barron and P. L. Polavarapu, Chem. Phys. Lett.,
1989, 155, 423–429.25 R. D. Amos, Chem. Phys. Lett., 1986, 124, 376–381.26 M. Pecul and K. Ruud, Int. J. Quantum Chem., 2005, 104, 816–829.27 P. L. Polavarapu, Angew. Chem., Int. Ed., 2002, 41, 4544–4546.28 J. Costante, L. Hecht, P. L. Polavarapu, A. Collet and
L. D. Barron, Angew. Chem., Int. Ed. Engl., 1997, 36, 885–887.29 P. L. Polavarapu, L. Hecht and L. D. Barron, J. Phys. Chem.,
1993, 97, 1793–1799.30 L. D. Barron, A. R. Gargaro, L. Hecht and P. L. Polavarapu,
Spectrochim. Acta, Part A, 1991, 47, 1001–1016.31 J. Kapitan, F. J. Zhu, L. Hecht, J. Gardiner, D. Seebach and
L. D. Barron, Angew. Chem., Int. Ed., 2008, 47, 6392–6394.32 G. S. Yu, T. B. Freedman, L. A. Nafie, Z. Y. Deng and
P. L. Polavarapu, J. Phys. Chem., 1995, 99, 835–843.33 M. Pecul, E. Larnparska, C. Cappelli, L. Frediani and K. Ruud,
J. Phys. Chem. A, 2006, 110, 2807–2815.34 M. Pecul, Chem. Phys. Lett., 2006, 427, 166–176.35 A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107,
2411–2502.36 R. N. Moore, C. Golumbic and G. S. Fisher, J. Am. Chem. Soc.,
1956, 78, 1173–1176.37 J. M. Encinar, F. J. Beltran and J. M. Frades, J. Chem. Technol.
Biotechnol., 1994, 61, 359–365.38 E. F. Murphy, T. Mallat and A. Baiker, Catal. Today, 2000, 57,
115–126.39 P. Gallezot, Catal. Today, 2007, 121, 76–91.
40 J. L. F. Monteiro and C. O. Veloso, Top. Catal., 2004, 27, 169–180.41 S. Bhattacharjee, T. J. Dines and J. A. Anderson, J. Catal., 2004,
225, 398–407.42 B. S. Lane and K. J. Burgess, J. Am. Chem. Soc., 2001, 123,
2933–2934.43 B. S. Lane, M. Vogt, V. J. DeRose and K. Burgess, J. Am. Chem.
Soc., 2002, 124, 11946–11954.44 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,
M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr.,T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam,S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi,G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada,M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida,T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li,J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev,A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala,K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg,V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain,O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari,J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford,J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham,C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill,B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez andJ. A. Pople, GAUSSIAN 03 (Revision D.01), Gaussian, Inc.,Wallingford, CT, 2004.
45 A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652.46 A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38,
3098–3100.47 C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter,
1988, 37, 785–789.48 T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023.49 R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys.,
1992, 96, 6796–6806.50 D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98,
1358–1371.51 D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100,
2975–2988.52 W. Hug, Chem. Phys., 2001, 264, 53–69.53 M. Zerara, J. Comput. Chem., 2008, 29, 306–311.54 T. Brocki, M. Moskovits and B. Bosnich, J. Am. Chem. Soc., 1980,
102, 495–500.55 G. S. Yu, T. B. Freedman and L. A. Nafie, J. Raman Spectrosc.,
1995, 26, 733–743.56 D. Che and L. A. Nafie, Chem. Phys. Lett., 1992, 189, 35–42.57 S. Luber, C. Herrmann and M. Reiher, J. Phys. Chem. B, 2008,
112, 2218–2232.
This journal is �c the Owner Societies 2010 Phys. Chem. Chem. Phys., 2010, 12, 3005–3013 | 3013
Publ
ishe
d on
09
Febr
uary
201
0. D
ownl
oade
d by
Sta
te U
nive
rsity
of
New
Yor
k at
Sto
ny B
rook
on
25/1
0/20
14 1
2:21
:15.
View Article Online





























