Embed Size (px)
Citation preview

315
ERRORES INNATOS DEL METABOLISMO DE LOS CICLOS ESPECÍFICOS
5.1. ENFERMEDADES DEL CICLO DE LA UREA (ECU)
Los átomos de nitrógeno contenidos en los alimentos sólo se utilizan con fines biosintéticos,para el crecimiento o la reparación de los tejidos. El exceso de nitrógeno de la dieta debe sereliminado, puesto que no existe ninguna forma de almacenamiento de éste y, además, cons-tituye un sustrato potencialmente tóxico para el ser humano. Para evitar este problema, losmamíferos (animales ureotélicos) han desarrollado una vía de excreción del nitrógeno sobrantemediante la formación de urea (1) (Figura 1). La formación de urea, o ciclo de la urea com-pleto, tiene lugar en el hígado y constituye una vía bioquímica esencial para eliminar el nitró-geno de desecho. Comprende un sistema cíclico de seis reacciones metabólicas, en el quese eliminan dos moles de amonio tóxicas en forma de una molécula de urea (hidrosoluble yno tóxica) en cada vuelta del ciclo (Figura 2). La urea constituye el vehículo más importantepara la excreción del nitrógeno, y su producción aumenta a medida que lo hace la ingesta deproteínas. El amonio también es atrapado por determinados compuestos como el glutamato,el piruvato y el aspartato, y también se utiliza para la síntesis de compuestos que contienennitrógeno (glicina y pirimidinas, incluido el ácido orótico). Se puede producir un bloqueo de es-te ciclo, ya sea por una deficiencia enzimática (carbomilfosfato sintetasa [CPS], ornitintrans-carbamilasa [OTC], N-acetil glutamato sintetasa [NAGS], arginosuccínico sintetasa [AS], argi-nosuccinato liasa [AL] o arginasa [A]), o por depleción de un aminoácido (AA) esencial para elnormal funcionamiento del ciclo, debido a un defecto del transportador, como ocurre en el sín-drome HHH o en la lisinuria con intolerancia a las proteínas (2). Cuando se produce un bloqueode la ureagénesis, van a ocurrir dos hechos: una síntesis inadecuada de urea y, lo que es másimportante, una acumulación de amonio en todas las células del organismo, que es un com-puesto tóxico, fundamentalmente para el cerebro. También se van a acumular compuestosque amortiguan y transportan el exceso de nitrógeno, sobre todo glutamina y alanina.
Puesto que no existe un sistema de eliminación secundario del amonio que sea efectivo,se produce una rápida acumulación de éste y de otros metabolitos precursores que dan
5

lugar a un edema cerebral agudo con un grave compromiso neurológico que puede llegara ser letal. En general, cuanto más próximo sea el defecto enzimático, más severa y re-sistente al tratamiento es la hiperamoniemia (las deficiencias de CPS y OTC son las másseveras), aunque existe una gran variedad, que depende también del grado de deficien-cia enzimática.
El hígado es el único órgano donde el ciclo es completo. Como grupo, los trastornos delciclo de la urea (TCU) son frecuentes, y se estima una incidencia de 1/25.000 recién na-cidos, aunque, dado que se sospecha que existen bastantes casos no diagnosticados, suincidencia real debe de hallarse alrededor de 1/15.000 recién nacidos. Estos trastornosse heredan con carácter autosómico recesivo, salvo la deficiencia de OTC, que tiene unaherencia ligada al cromosoma X (sin embargo, actualmente se están diagnosticando mu-chos casos en mujeres, que podrían corresponder a mutaciones de novo). Su patogeniano es bien conocida y parece deberse al aumento del amonio y de la glutamina, la glicinay la alanina, que originan cambios osmolares (edema cerebral) y que desencadenarían laencefalopatía aguda o crónica, así como la alteración de los neurotransmisores.
Aspectos clínicos
a) Comienzo agudo neonatal (forma clásica): tras un periodo libre de síntomas, a las 24-48 horas del inicio de la alimentación aparece un cuadro neurológico tipo intoxi-
316
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
Figura 1. Relaciones entre las pro-teínas de la dieta, las proteínascorporales y la síntesis de urea. Elnitrógeno de la dieta que no se uti-liza para el crecimiento o la repa-ración tisular debe ser excretado,pues no existe ninguna forma fi-siológica de almacenamiento. Losmamíferos lo eliminan mediantela síntesis y la excreción de urea.Solamente los átomos de nitró-geno contenidos en el amonio(que deriva de diferentes ami-noácidos) y en el aspartato (pro-veniente de la transaminación deloxalacetato y del glutamato) es-tán destinados a la producción deurea, y por ello se denominan áto-mos de nitrógeno de desecho.Cualquier agente farmacológicoque secuestre aminoácidos delpool libre disminuirá los requeri-mientos de síntesis de urea.
cación que evoluciona rápidamente a convulsiones generalizadas. Si el amonio su-pera los 250 µmol/L, sobreviene el coma y el enfermo puede evolucionar hacia elexitus.
b) Forma crónica de presentación tardía: desencadenado por factores ambientales (in-fecciones, alta ingesta proteica, etc.), aparece un cuadro insidioso y potencialmen-te grave que va desde los vómitos y la somnolencia a las convulsiones y el coma,del que se mejora con el ayuno. A menudo se halla una hipertransaminasemia no ex-plicable.
317
EIM DE LOS CICLOS ESPECÍFICOS
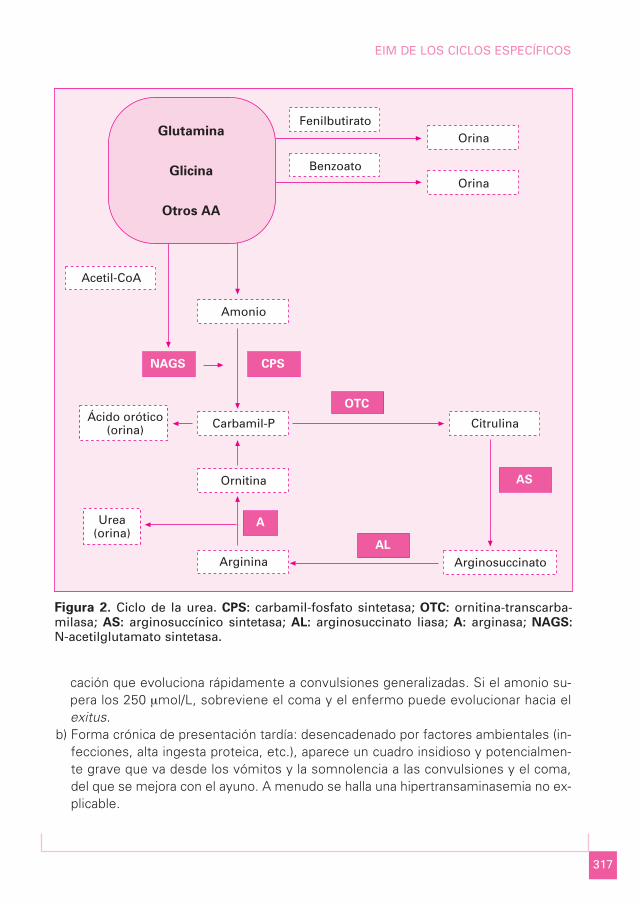
Figura 2. Ciclo de la urea. CPS: carbamil-fosfato sintetasa; OTC: ornitina-transcarba-milasa; AS: arginosuccínico sintetasa; AL: arginosuccinato liasa; A: arginasa; NAGS:
N-acetilglutamato sintetasa.
Acetil-CoA
Carbamil-P
Ornitina
Arginina
Citrulina
Arginosuccinato
Amonio
Benzoato
FenilbutiratoOrina
Orina
Glutamina
Glicina
Otros AA
Ácido orótico(orina)
Urea(orina)
NAGS CPS
OTC
AS
AL
A
En niños mayores, adolescentes y adultos existe una sintomatología neurológica dife-rente (migraña, disartria, ataxia), o bien una sintomatología psiquiátrica (alucinaciones,trastornos del comportamiento).
Esta variabilidad clínica y la existencia de formas paucisintomáticas hace que los TCU pro-bablemente sean infradiagnosticados.
Ante una clínica sugestiva, incluidas las crisis neurológicas repetitivas sin diagnóstico, debesospecharse un TCU, que se diagnostica por la presencia de hiperamoniemia (por encima delos 150 µmol/L durante el periodo neonatal y posteriormente superior a 80 µmol/L), con glu-cemia normal, cetonuria negativa y ácido láctico normal; suele existir alcalosis respiratoria omixta y una moderada afectación hepática en las fases agudas o de descompensación. Eldiagnóstico se realiza mediante la determinación de AA. Los defectos más frecuentes sonlos de la parte alta del ciclo, las deficiencias de OTC y CPS, que cursan con citrulina baja y sediferencian entre sí por la eliminación urinaria de ácido orótico en la deficiencia de OTC (quese debe confirmar con una prueba de sobrecarga con alopurinol). Las deficiencias de enzimascitoplasmáticas se caracterizan por un aumento de la citrulina, la cual ésta mucho más ele-vada (por encima de 250 µmol/L) en las deficiencias de AS (citrulinemia) que en las de AL (aci-duria arginosuccínica) (100-250 µmol/L); la deficiencia de argininemia (A) cursa con citrulina yarginina muy elevadas, e hiperamonemia discreta. La deficiencia de NAGS es el trastorno me-nos frecuente. El diagnóstico definitivo se realiza mediante el estudio genético (Tabla I).
318
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
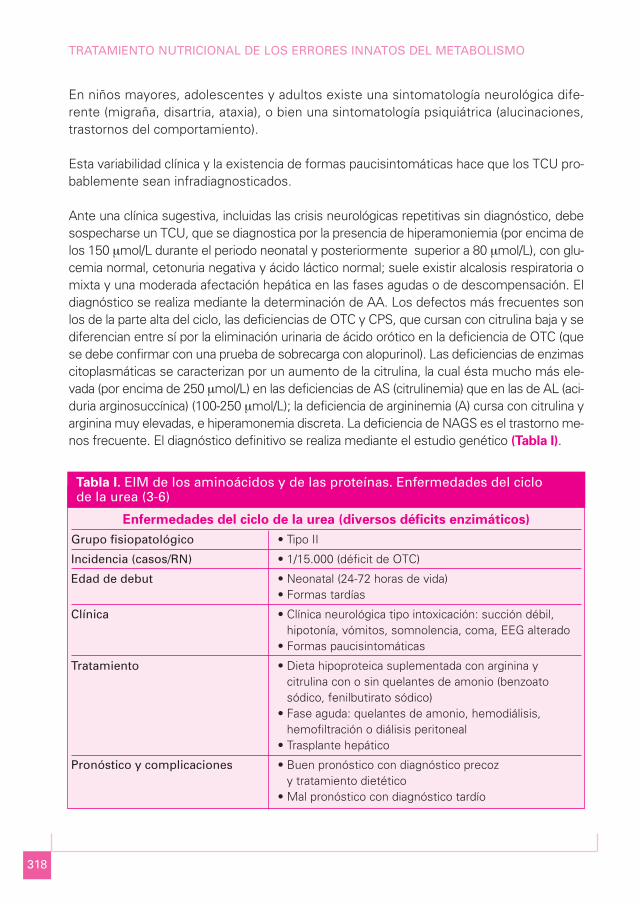
Enfermedades del ciclo de la urea (diversos déficits enzimáticos)
Grupo fisiopatológico • Tipo II
Incidencia (casos/RN) • 1/15.000 (déficit de OTC)
Edad de debut • Neonatal (24-72 horas de vida)• Formas tardías
Clínica • Clínica neurológica tipo intoxicación: succión débil,hipotonía, vómitos, somnolencia, coma, EEG alterado
• Formas paucisintomáticas
Tratamiento • Dieta hipoproteica suplementada con arginina ycitrulina con o sin quelantes de amonio (benzoatosódico, fenilbutirato sódico)
• Fase aguda: quelantes de amonio, hemodiálisis,hemofiltración o diálisis peritoneal
• Trasplante hepático
Pronóstico y complicaciones • Buen pronóstico con diagnóstico precoz y tratamiento dietético
• Mal pronóstico con diagnóstico tardío
Tabla I. EIM de los aminoácidos y de las proteínas. Enfermedades del ciclo de la urea (3-6)

Manejo
Estrategias para el abordaje terapéutico de estos pacientes
Restringir la ingesta de proteínas
El objetivo de esta restricción es disminuir las necesidades de excretar nitrógeno. La to-lerancia proteica dependerá del grado de déficit enzimático y de la edad del niño. Comosiempre, se debe buscar el techo de tolerancia proteica de cada paciente (aquel que per-mita un adecuado crecimiento sin desestabilización metabólica).
Tras el nacimiento, el objetivo es estabilizar al niño en una ingesta de proteínas de1,5 g/kg/día (3), empezando con cantidades de 0,5-0,7 g/kg/día, que se irán aumentandogradualmente (incrementos progresivos no mayores del 10% cada vez). Los requeri-mientos proteicos de los recién nacidos cambian durante los primeros meses, con un pe-riodo inicial de “luna de miel” en que toleran una alta ingesta de proteínas; a partir de losseis meses, los requerimientos por peso corporal disminuyen. Para confirmar que la in-gesta proteica es adecuada, además de los parámetros clínicos (crecimiento, signos dedesnutrición, etc.), se utilizan parámetros bioquímicos que incluyen las determinacionesplasmáticas de amonio, aminoácidos esenciales, glutamina, hemoglobina, hematocrito,albúmina, prealbúmina, transferrina, ferritina y proteínas totales. En la Tabla II se expo-nen los objetivos bioquímicos para un adecuado control (7).
319
EIM DE LOS CICLOS ESPECÍFICOS
• Amonio plasmático <40 µmol/L (otros autores: <80 µmol/L)
• Glutamina plasmática <1.000 µmol/L
• Niveles plasmáticos de alanina, glicina, lisina Normales, salvo en la deficienciay arginina de arginasa
• Concentraciones de aminoácidos En el límite bajo de la normalidad
• Excreción de orotato urinario Normal (<3 µmol/mmol de creatinina)
• Concentración de proteínas plasmáticas (albúmina, Normalprealbúmina, transferrina y proteínas totales)
Estos objetivos a veces no son alcanzables y el tratamiento debe ser siempre individualizado.De todas estas investigaciones de laboratorio, el amonio plasmático y la cuantificación de aminoácidosresultan muy útiles, aunque los niveles de amonio están sujetos a numerosos factores (ingesta deproteínas, hora de la última comida, etc.), así como a los artefactos de recogida de la muestra; por ello,puede no ser un buen índice de control a largo plazo, y se utiliza la glutamina plasmática como una guía másfiable.Unos niveles elevados de glutamina y amonio pueden reflejar que el amonio corporal está elevado por unaelevada ingesta proteica, pero también se debe considerar la posibilidad de una ingesta de proteínas ycalorías insuficiente.La excreción urinaria de orotato resulta útil en el déficit de OTC.
Tabla II. Objetivos bioquímicos para un control óptimo de las ECU

Aunque es necesario alcanzar el mínimo proteico recomendado, en las formas más se-veras de déficit enzimático la gran restricción proteica puede no cubrir el mínimo protei-co indispensable. En otros casos, los pacientes no ingieren sus necesidades proteicas,pues sufren aversión a las proteínas. En ambos grupos, una determinada cantidad de pro-teínas (entre un 25% y un 50%) puede ser reemplazada por una mezcla comercial deaminoácidos esenciales que se añaden a la fórmula o, en el caso de los niños mayores,se ingieren como bebida o pasta. Se empieza con una cantidad de 0,2-0,5 g/kg/día de pro-teínas, hasta alcanzar un máximo de 0,7 g/kg/día dividido en 2-3 comidas. De esta ma-nera, las necesidades de aminoácidos esenciales pueden alcanzarse, con la ventaja de lamenor densidad de nitrógeno que éstos poseen; además, al limitar la ingesta de los ami-noácidos no esenciales, el nitrógeno sobrante se utiliza para sintetizar éstos, gracias a locual se consigue el objetivo de reducir el flujo de nitrógeno a través del ciclo de la urea.Cuando los pacientes están clínicamente estables, incluso aquellos con formas severas,existe cierta flexibilidad para proporcionar proteínas adicionales sin provocar un aumen-to de los niveles de amonio y de glutamina. Esto no es posible en aquellos con un malcontrol, pues su estatus metabólico se deterioraría rápidamente.
Aunque una importante restricción proteica puede dar lugar a un déficit de aminoácidosesenciales, probablemente existe un mayor riesgo de deficiencia de micronutrientes, enespecial de hierro y zinc. Por ello es necesario utilizar suplementos de vitaminas y mine-rales.
También es importante asegurar un aporte calórico adecuado, para evitar la movilizaciónendógena de proteínas y conseguir un ahorro nitrogenado máximo. Ello se consigue conalimentos de muy bajo contenido proteico. La limitada cantidad de proteínas que se pue-den aportar nunca debe ser considerada como fuente calórica, sino exclusivamente ana-bólica. Proporcionar más energía de la necesaria no va a reducir aún más el catabolismoproteico, y lo único que se conseguirá es un sobrepeso.
Emplear fármacos que utilicen vías alternativas para la excreción de nitrógeno
Se trata de compuestos que se conjugan con los aminoácidos y se excretan rápidamen-te. El motivo de su uso es que el nitrógeno se elimina como compuestos diferentes a laurea, con lo que disminuye la cantidad de los que entran en el ciclo de la urea. Se utilizanel benzoato sódico (250 mg/kg/día cada 6-8 horas), que se conjuga con la glicina formandohipurato, el cual se excreta rápidamente. Por cada mol de benzoato se excreta un mol denitrógeno. Los principales efectos secundarios son náuseas, vómitos e irritabilidad.
El fenilbutirato sódico (Ammonaps®) se oxida en el hígado a fenilacetato, y éste se con-juga con la glutamina formando fenilacetilglutamina; por cada mol de fenilbutirato se ex-cretan dos moles de nitrógeno. La dosis es de 200-600 mg/kg/día, en función de si seutiliza en combinación con el benzoato o solo. La excreción de hipurato y fenilacetilglu-
320
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO

tamina aumenta la pérdida urinaria de potasio, lo que puede originar hipopotasemia y al-calosis metabólica. Una sobredosificación de estos compuestos puede dar lugar a sínto-mas que imitan una crisis hiperamoniémica, tales como agitación, confusión, e hiper-ventilación (8).
Reemplazar los nutrientes deficitarios
La arginina es un aminoácido no esencial, pues se sintetiza en el ciclo de la urea. Entodos los pacientes con trastornos del ciclo de la urea, excepto en el caso del défi-cit de arginasa, la arginina se convierte en semiesencial o esencial a causa del blo-queo metabólico, por lo que es necesario suplementarlo. Las dosis empleadas osci-lan entre los 100 y los 170 mg/kg/día, aunque los pacientes con citrulinemia y aciduriaarginosuccínica tienen unos requerimientos mayores (400-700 mg/kg/día) a causa dela importante pérdida de ornitina a través de la orina, que debe ser restablecida. Losniveles plasmáticos de arginina deben mantenerse entre 50 y 200 µmol/L. Los pre-parados comerciales habituales son en forma de clorhidrato (orales o ev) y hay quevigilar la tendencia a la acidosis hiperclorémica. Puede aumentar los niveles de ci-trulina y ácido arginosuccínico que, al eliminarse por la orina, arrastran nitrógeno conellos.
En ocasiones, en variantes muy graves de deficiencia de CPS y OTC puede sustituirse laarginina por citrulina a dosis de 170 mg/kg/día, con la cual se consigue además la elimi-nación suplementaria de un nitrógeno procedente del aspartato.
Restauración del ciclo de la urea
En la deficiencia de NAGS se produce una alteración de la síntesis de N-acetilglutamato,que actúa como cofactor de la enzima carbamilfosfato sintasa I (CPS I). El ácido carglú-mico (Carbaglu®) activa la CPS I en ausencia de su cofactor endógeno (9) y restituye elciclo. El tratamiento permite una mayor tolerancia proteica.
Otros tratamientos
• Citrato: reduce la elevación postprandial de amonio y podría desempeñar un papel im-portante en el control de la aciduria arginosuccínica al corregir el déficit de aspartato,pero su efecto a largo plazo es desconocido.
• Carnitina: durante las crisis de descompensación hiperamoniémicas se produce una de-pleción de carnitina por consumo, por lo que se recomienda su empleo a 100 mg/kg/día.En las fases de estabilidad clínica también es útil la administración de carnitina a dosisde 30-50 mg/kg/día, en función del déficit plasmático.
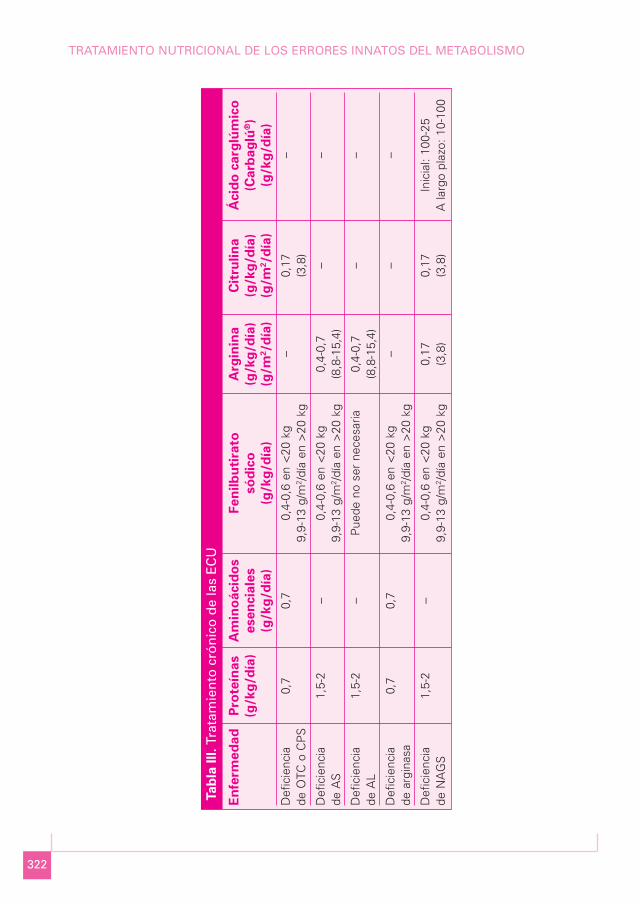
En la Tabla III se resume el tratamiento de las ECU.
321
EIM DE LOS CICLOS ESPECÍFICOS
322
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
En
ferm
ed
ad
Pro
teín
as
Am
ino
ácid
os
Fen
ilb
uti
rato
Arg
inin
aC
itru
lin
aÁ
cid
o c
arg
lúm
ico
(g/k
g/d
ía)
esen
cia
les
só
dic
o(g
/kg
/día
)(g
/kg
/día
)(C
arb
ag
lú®)
(g/k
g/d
ía)
(g/k
g/d
ía)
(g/m
2/d
ía)
(g/m
2/d
ía)
(g/k
g/d
ía)
Def
icie
ncia
0,7
0,7
0,4-
0,6
en <
20 k
g–
0,17
–de
OTC
o C
PS
9,9-
13 g
/m2 /
día
en >
20 k
g(3
,8)
Def
icie
ncia
1,5-
2–
0,4-
0,6
en <
20 k
g0,
4-0,
7–
–de
AS
9,9-
13 g
/m2 /
día
en >
20 k
g(8
,8-1
5,4)
Def
icie
ncia
1,5-
2–
Pue
de n
o se
r ne
cesa
ria0,
4-0,
7–
–de
AL
(8,8
-15,
4)
Def
icie
ncia
0,7
0,7
0,4-
0,6
en <
20 k
g–
––
de a
rgin
asa
9,9-
13 g
/m2 /
día
en >
20 k
g
Def
icie
ncia
1,5-
2–
0,4-
0,6
en <
20 k
g0,
170,
17In
icia
l: 10
0-25
de N
AG
S9,
9-13
g/m
2 /dí
a en
>20
kg
(3,8
)(3
,8)
A la
rgo
plaz
o: 1
0-10
0
Tab
la III.
Trat
amie
nto
cró
nic
o d
e la
s E
CU

Tratamiento de urgencia del coma hiperamoniémico inicial
y de las descompensaciones agudas
El coma hiperamoniémico es una emergencia médica, por lo que se debe instaurar untratamiento inmediato y agresivo en un intento de prevenir o minimizar el daño cere-bral (10, 11). Unos niveles plasmáticos de amonio de 100-200 µmol/L ya se puedenasociar con síntomas clínicos de letargia, confusión y vómitos, y niveles mayores des-embocan en coma. En los déficits de OTC (la más frecuente de las ECU), más de la mi-tad de los pacientes que sobreviven presentan un daño neurológico severo, y los ni-veles de amonio plasmático al diagnóstico constituyen el único factor de pronósticoneurológico (12, 13).
Fases del tratamiento (4-16)
• Establecer vía aérea: ventilación asistida (hiperventilación), pues estos pacientes pre-sentan inicialmente una alcalosis respiratoria.
• Perfusión intravenosa: conseguir una buena hidratación del paciente (vigilar el posibleedema cerebral) y aportar calorías no proteicas con soluciones glucosadas (8-10 mg/kg/mi-nuto) y emulsiones lipídicas (Intralipid® o Lipofundina® a 1-2 g/kg) para alcanzar aportescalóricos superiores a 80 kcal/kg/día, y minimizar así la proteólisis endógena.
• Supresión total del aporte proteico durante 24-48 horas. Si el paciente presenta buenatolerancia enteral, se iniciará alimentación por sonda nasogástrica o transpilórica conmódulos de carbohidratos y lípidos. El aporte será complementario al endovenoso.
• Eliminación del amonio acumulado: el método más rápido es la diálisis y, dentro de és-ta, los sistemas más rápidos son la oxigenación con membrana extracorpórea conec-tada a una máquina de hemodiálisis (ECMO/HD), y la hemofiltración (14). La exangui-notransfusión no es efectiva. La diálisis debe suspenderse cuando los niveles de amoniocaigan por debajo de 200 µmol/L.
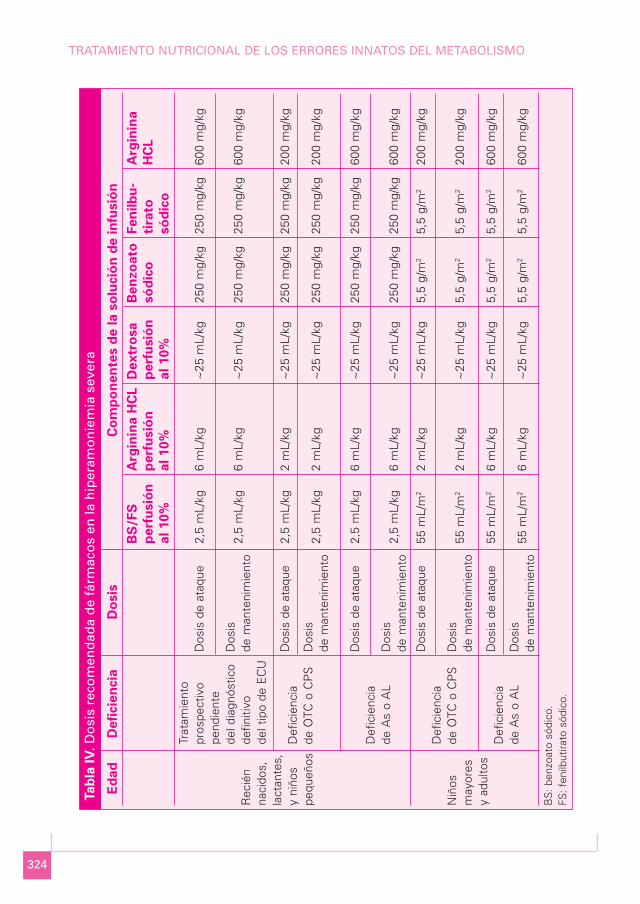
• En segunda línea de tratamiento están los fármacos que utilizan las vías alternativas deeliminación del amonio. En la Tabla IV se detallan las dosis de ataque y de manteni-miento durante la fase previa al diagnóstico definitivo, según el defecto enzimático delque se trate (14). La dosis de arginina se ha aumentado respecto a los protocolos pre-vios (250-500 mg/kg), debido a que parece que su infusión rápida ejerce un impacto po-sitivo en la deficiencia de AS y AL, y es relativamente segura en los pacientes con dé-ficit de OTC, CPS y NAGS. Los fármacos quelantes de amonio se diluyen en solucionesglucosadas, ya que 1 g de benzoato sódico contiene 160 mg de sodio, y un gramo defenilbutirato sódico incluye 147 mg de sodio, por lo que no es necesario sodio adicio-nal. Sin embargo, se debe incorporar potasio por la depleción secundaria que se pro-duce con el empleo de estos fármacos. Las dosis de mantenimiento ev se mantendránhasta que se pase a la vía oral, una vez que el paciente se haya estabilizado (niveles deamonio de 100-200 µmol/L) y haya reiniciado la alimentación. Estos fármacos se pue-den emplear como de primera línea, si los niveles de amonio no son muy elevados o
323
EIM DE LOS CICLOS ESPECÍFICOS
324
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
Ed
ad
Defi
cie
ncia
Do
sis
Co
mp
on
en
tes d
e la s
olu
ció
n d
e in
fusió
n
BS
/FS
Arg
inin
a H
CL
Dextr
osa
Ben
zo
ato
Fen
ilb
u-
Arg
inin
a
perf
usió
np
erf
usió
np
erf
usió
nsó
dic
oti
rato
H
CL
al 10%
al 10%
al 10%
só
dic
o
Trat
amie
nto
pros
pect
ivo
Dos
is d
e at
aque
2,5
mL/
kg6
mL/
kg~
25 m
L/kg
250
mg/
kg25
0 m
g/kg
600
mg/
kgpe
ndie
nte
del d
iagn
óstic
o D
osis
2,5
mL/
kg6
mL/
kg~
25 m
L/kg
250
mg/
kg25
0 m
g/kg
600
mg/
kgR
ecié
nde
finiti
vo
de m
ante
nim
ient
ona
cido
s,de
l tip
o de
EC
Ula
ctan
tes,
y ni
ños
Def
icie
ncia
Dos
is d
e at
aque
2,5
mL/
kg2
mL/
kg~
25 m
L/kg
250
mg/
kg25
0 m
g/kg
200
mg/
kg
pequ
eños
de O
TC o
CP
SD
osis
2,5
mL/
kg2
mL/
kg~
25 m
L/kg
250
mg/
kg25
0 m
g/kg
200
mg/
kgde
man
teni
mie
nto
Dos
is d
e at
aque
2,5
mL/
kg6
mL/
kg~
25 m
L/kg
250
mg/
kg25
0 m
g/kg
600
mg/
kgD
efic
ienc
iade
As
o A
LD
osis
2,5
mL/
kg6
mL/
kg~
25 m
L/kg
250
mg/
kg25
0 m
g/kg
600
mg/
kgde
man
teni
mie
nto
Dos
is d
e at
aque
55 m
L/m
22
mL/
kg~
25 m
L/kg
5,5
g/m
25,
5 g/
m2
200
mg/
kgD
efic
ienc
iaN
iños
de O
TC o
CP
SD
osis
55 m
L/m
22
mL/
kg~
25 m
L/kg
5,5
g/m
25,
5 g/
m2
200
mg/
kgm
ayor
esde
man
teni
mie
nto
y ad
ulto
sD
osis
de
ataq
ue55
mL/
m2
6 m
L/kg
~25
mL/
kg5,
5 g/
m2
5,5
g/m
260
0 m
g/kg
Def
icie
ncia
de A
s o
AL
Dos
is55
mL/
m2
6 m
L/kg
~25
mL/
kg5,
5 g/
m2
5,5
g/m
260
0 m
g/kg
de m
ante
nim
ient
oB
S: b
enzo
ato
sódi
co.
FS: f
enilb
utira
to s
ódic
o.
Tab
la IV
.D
osi
s re
com
end
ada
de
fárm
aco
s en
la h
iper
amo
nie
mia
sev
era

no se dispone de métodos dialíticos inmediatos. También se pueden utilizar combina-dos con la diálisis, pues los efectos se suman.
En ocasiones se utiliza la citrulina por vía enteral (150-200 mg/kg/día), para los RN condeficiencias de OTC y CPS, pues, al combinarse con el aspartato, aumenta el aclara-miento del nitrógeno. No debe administrarse en los defectos de AS y AL, ya que con-tienen cantidades excesivas de ésta, si bien la citrulina no es tóxica.
• Reintroducción de las proteínas una vez se han estabilizado las concentraciones de amo-nio plasmático (por debajo de 100 µmol/L), empezando por dosis de 0,25-0,5 g/kg/día.
• Recordar que no se debe emplear el ácido valproico para el tratamiento de las convul-siones, puesto que disminuye la actividad del ciclo de la urea, lo que agrava la hipera-moniemia.
Tratamiento de la hiperamoniemia intercurrente
Cualquier situación de estrés, como un proceso infeccioso, el ayuno prolongado, una ci-rugía, etc., puede precipitar una crisis de hiperamoniemia. Es importante que los padresestén informados e instruidos sobre lo que deben hacer en estos casos de riesgo, o an-te la sospecha de descompensación.
Será necesario disminuir el aporte proteico a la mitad (a veces suspenderlo), se iniciaráun régimen de emergencia (con polímeros de glucosa y/o soluciones azucaradas), y semantendrá el tratamiento farmacológico. Si el paciente no tolera los líquidos orales o suestado general empeora, se recomienda el ingreso hospitalario.
Otros tratamientos
Trasplante hepático
Se debe considerar en cualquier paciente que no pueda cumplir las restricciones dieté-ticas necesarias, o que sufra episodios recurrentes de hiperamoniemia pese a seguir unadecuado tratamiento médico. En el subgrupo de pacientes con formas severas de de-ficiencia de OTC y CPS está indicada una evaluación temprana para iniciar un programade trasplante, ya que la historia natural de estas enfermedades se caracteriza por un di-fícil control. En cambio, a medida que el niño crece, los afectados de citrulinemia y de-ficiencia de arginosuccinato liasa tienen una mayor tolerancia a las proteínas y presen-tan una disminución de la frecuencia de las crisis de hiperamoniemia. Tras el trasplantehepático, los niveles de amonio se controlan con rapidez, y ya no es necesaria la res-tricción proteica o el uso de medicaciones. Dado que no se corrigen los defectos enzi-máticos del ciclo de la urea en el resto del cuerpo (intestino, riñón), la deficiencia de labiosíntesis de arginina persiste, por lo que puede ser necesaria la suplementación (15).
325
EIM DE LOS CICLOS ESPECÍFICOS

Terapia génica
Actualmente, aún se halla en estudio con vectores adenovirales.
Dietas tipo
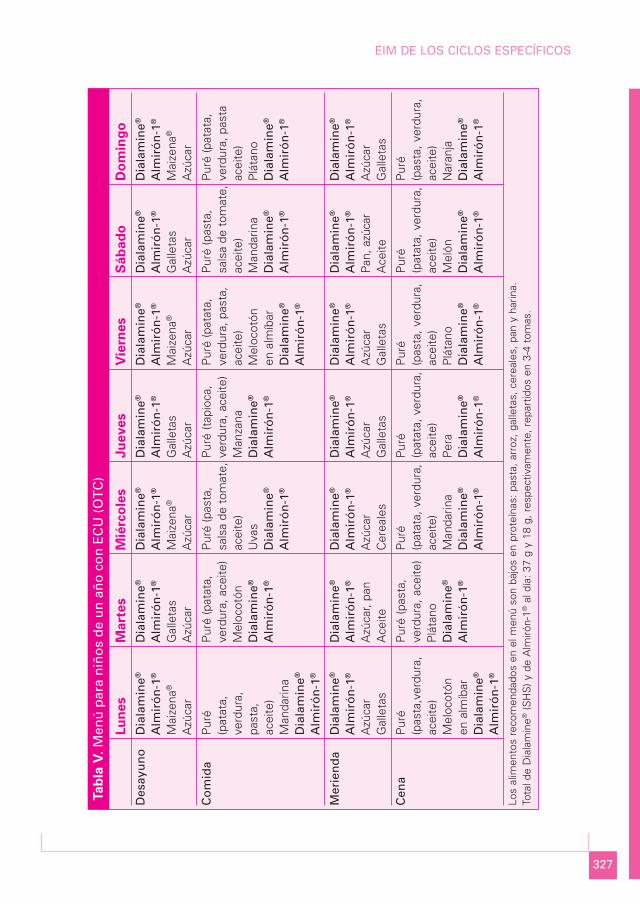
MENÚ PARA NIÑOS DE UN AÑO CON ECU (OTC) (Tabla V)
Media semanal
Componente Cantidad Porcentaje
Proteína bruta (g) 16,5 6%Lípidos totales (g) 47 38%Carbohidratos (g) 158 56%Energía (kcal) 1.121
Proteína total 16,5 gEquivalente proteico 9,25 gProteína natural: 7,25 g• Almirón-1® 1,92 g• Fruta-verdura 5,33 gCálculo proteico 1,5 g/kg
Menú semanal
a) Lunes:
• Desayuno: 10 g de Dialamine®; 5 g de Almirón-1®; 10 g de Maizena®; 5 g de azúcar.• Comida: triturado con 100 g de patata, 20 g de cebolla, 25 g de zanahoria, 50 g de
judías verdes, 15 g de pasta y 20 mL de aceite de oliva; 50 g de mandarina; 4 g deAlmirón-1®; 9 g de Dialamine®.
• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 5 g de azúcar; 15 g de galletas.• Cena: triturado con 50 g de pasta, 50 g de calabacín, 50 g de zanahoria y 15 mL de
aceite de oliva; 50 g de melocotón en almíbar; 5 g de Almirón-1®; 9 g de Dialamine®.
b) Martes:
• Desayuno: 10 g de Dialamine®; 5 g de Almirón-1®; 15 g de galletas; 5 g de azúcar.• Comida: triturado con 100 g de patata, 50 g de zanahoria, 50 g de judías verdes
y 20 mL de aceite de oliva; 50 g de melocotón; 4 g de Almirón-1®; 9 g de Diala-mine®.
• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 5 g de azúcar; 20 g de pan; 5 mLde aceite de oliva.
326
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
327
EIM DE LOS CICLOS ESPECÍFICOS
Lu
nes
Mart
es
Mié
rco
les
Ju
eves
Vie
rnes
Sáb
ad
oD
om
ing
o
Des
ayu
no
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®
Mai
zena
®G
alle
tas
Mai
zena
®G
alle
tas
Mai
zena
®G
alle
tas
Mai
zena
®
Azú
car
Azú
car
Azú
car
Azú
car
Azú
car
Azú
car
Azú
car
Co
mid
aP
uré
Pur
é (p
atat
a,P
uré
(pas
ta,
Pur
é (t
apio
ca,
Pur
é (p
atat
a,P
uré
(pas
ta,
Pur
é (p
atat
a,(p
atat
a,ve
rdur
a, a
ceite
)sa
lsa
de t
omat
e,ve
rdur
a, a
ceite
)ve
rdur
a, p
asta
,sa
lsa
de t
omat
e,ve
rdur
a, p
asta
verd
ura,
Mel
ocot
ónac
eite
)M
anza
naac
eite
)ac
eite
)ac
eite
)pa
sta,
Dia
lam
ine®
Uva
sD
iala
min
e®M
eloc
otón
M
anda
rina
Plá
tano
acei
te)
Alm
iró
n-1
®D
iala
min
e®A
lmir
ón
-1®
en a
lmíb
arD
iala
min
e®D
iala
min
e®
Man
darin
aA
lmir
ón
-1®
Dia
lam
ine®
Alm
iró
n-1
®A
lmir
ón
-1®
Dia
lam
ine®
Alm
iró
n-1
®
Alm
iró
n-1
®
Mer
ien
da
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®
Azú
car
Azú
car,
pan
Azú
car
Azú
car
Azú
car
Pan
, azú
car
Azú
car
Gal
leta
sA
ceite
Cer
eale
sG
alle
tas
Gal
leta
sA
ceite
Gal
leta
s
Cen
aP
uré
Pur
é (p
asta
,P
uré
Pur
éP
uré
Pur
éP
uré
(pas
ta,v
erdu
ra,
verd
ura,
ace
ite)
(pat
ata,
ver
dura
,(p
atat
a, v
erdu
ra,
(pas
ta, v
erdu
ra,
(pat
ata,
ver
dura
,(p
asta
, ver
dura
,ac
eite
)P
láta
noac
eite
)ac
eite
)ac
eite
)ac
eite
)ac
eite
)M
eloc
otón
Dia
lam
ine®
Man
darin
aP
era
Plá
tano
Mel
ónN
aran
jaen
alm
íbar
Alm
iró
n-1
®D
iala
min
e®D
iala
min
e®D
iala
min
e®D
iala
min
e®D
iala
min
e®
Dia
lam
ine®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®A
lmir
ón
-1®
Alm
iró
n-1
®
Alm
iró
n-1
®
Los
alim
ento
s re
com
enda
dos
en e
l men
ú so
n ba
jos
en p
rote
ínas
: pas
ta, a
rroz
, gal
leta
s, c
erea
les,
pan
y h
arin
a.To
tal d
e D
iala
min
e®(S
HS
) y d
e A
lmiró
n-1®
al d
ía: 3
7 g
y 18
g, r
espe
ctiv
amen
te, r
epar
tidos
en
3-4
tom
as.
Tab
la V
.M
enú
par
a n
iño
s d
e u
n a
ño
co
n E
CU
(O
TC
)

• Cena: triturado con 50 g de pasta, 25 g de tomate, 25 g de zanahoria y 15 mL deaceite de oliva; 50 g de plátano; 5 g de Almirón-1®; 9 g de Dialamine®.
c) Miércoles:
• Desayuno: 10 g de Dialamine®; 5 g de Almirón-1®; 10 g de Maizena®; 5 g de azúcar.
• Comida: triturado con 50 g de pasta, 50 g de salsa de tomate y 20 mL de aceite deoliva; 50 g de uvas; 4 g de Almirón-1®; 9 g de Dialamine®.
• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 5 g de azúcar; 20 g de cereales.• Cena: triturado con 100 g de patata, 50 g de calabacín, 25 g de judías verdes y 15 mL
de aceite de oliva; 50 g de mandarina; 5 g de Almirón-1®; 9 g de Dialamine®.
d) Jueves:
• Desayuno: 10 g de Dialamine®; 4 g de Almirón-1®; 20 g de galletas; 5 g de azúcar.
• Comida: triturado con 50 g de tapioca, 15 g de cebolla, 25 g de puerro, 15 g de tomate y 20 mL de aceite de oliva; 50 g de manzana; 4 g de Almirón-1®; 9 g deDialamine®.
• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 5 g de azúcar; 20 g de galletas.• Cena: triturado con 100 g de patata, 20 g de acelgas, 50 g de judías verdes y 15 mL
de aceite de oliva; 50 g de pera; 5 g de Almirón-1®; 9 g de Dialamine®.
e) Viernes:
• Desayuno: 10 g de Dialamine®; 5 g de Almirón-1®; 10 g de Maizena®; 5 g de azúcar.
• Comida: triturado con 50 g de patata, 25 g de cebolla, 25 g de coliflor, 25 g de za-nahoria, 20 g de pasta y 20 mL de aceite de oliva; 50 g de melocotón en almíbar; 4 gde Almirón-1®; 9 g de Dialamine®.
• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 5 g de azúcar; 20 g de galletas.• Cena: triturado con 50 g de pasta, 25 g de tomate, 25 g de champiñón y 15 mL de
aceite de oliva; 50 g de plátano; 5 g de Almirón-1®; 9 g de Dialamine®.
f) Sábado:
• Desayuno: 10 g de Dialamine®; 5 g de Almirón-1®; 20 g de galletas; 5 g de azúcar.• Comida: triturado con 50 g de pasta, 30 g de salsa de tomate y 20 mL de aceite de
oliva; 50 g de mandarina; 4 g de Almirón-1®; 9 g de Dialamine®.• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 20 g de pan; 5 g de azúcar; 5 mL
de aceite de oliva.
328
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO

• Cena: triturado con 100 g de patata, 100 g de calabacín y 15 mL de aceite de oliva;75 g de melón; 5 g de Almirón-1®; 9 g de Dialamine®.
g) Domingo:
• Desayuno: 10 g de Dialamine®; 5 g de Almirón-1®; 10 g de Maizena®; 5 g de azúcar.• Comida: triturado con 100 g de patata, 25 g de puerro, 25 g de zanahoria, 10 g de pas-
ta y 20 mL de aceite de oliva; 50 g de plátano; 4 g de Almirón-1®; 9 g de Dialamine®.• Merienda: 9 g de Dialamine®; 4 g de Almirón-1®; 5 g de azúcar; 20 g de galletas.• Cena: triturado con 50 g de pasta, 15 g de judías verdes, 15 g de zanahoria y 15 mL
de aceite de oliva; 50 g de naranja; 5 g de Almirón-1®; 9 g de Dialamine®.
Los alimentos recomendados en todos los menús son bajos en proteínas: pasta, arroz,galletas, cereales, pan y harina.
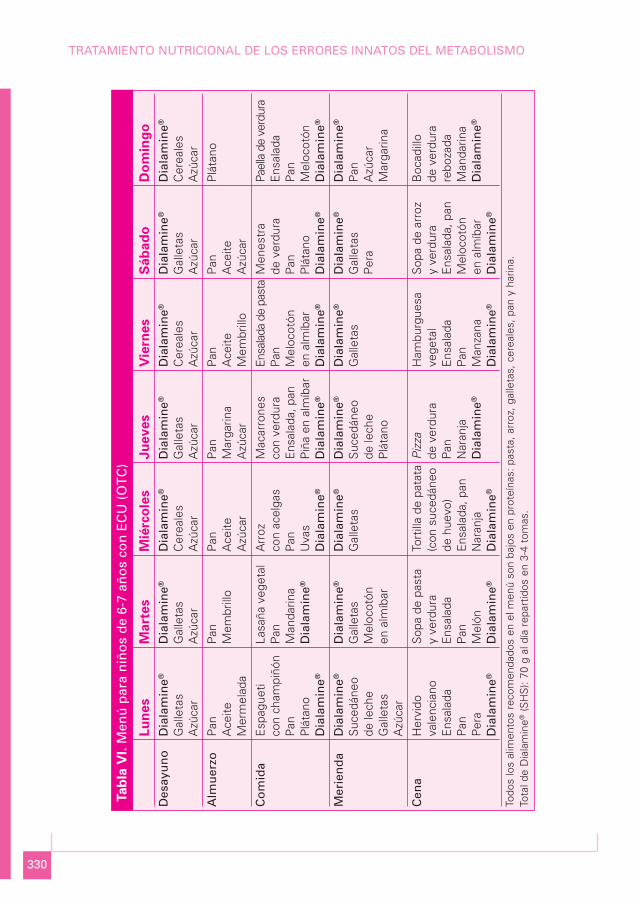
MENÚ PARA NIÑOS DE 6-7 AÑOS CON ECU (OTC) (Tabla VI)
Media semanal
Componente Cantidad Porcentaje
Proteína bruta (g) 25,4 6%Lípidos totales (g) 60,5 31%Carbohidratos (g) 282,6 64%Energía (kcal) 1.777
Proteína total 25,4 gEquivalente proteico 17, 5 gProteína natural 7, 9 gCálculo proteico 1 g/kg
Menú semanal
a) Lunes:
• Desayuno: 20 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 50 g de pan; 10 mL de aceite de oliva; 20 g de mermelada.• Comida: espagueti con champiñón (60 g de pasta, 25 g de champiñón, 30 g de salsa de
tomate y 20 mL de aceite de oliva); 15 g de pan; 100 g de plátano; 15 g de Dialamine®.• Merienda: 15 g de Dialamine®; 200 mL de sucedáneo de leche; 10 g de azúcar; 20 g
de galletas.• Cena: hervido valenciano (75 g de patata, 50 g de judías verdes y 20 mL de aceite
de oliva); ensalada (50 g de lechuga y 10 g de aceitunas); 15 g de pan; 100 g de pe-ra; 20 g de Dialamine®.
329
EIM DE LOS CICLOS ESPECÍFICOS
330
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
Lu
nes
Mart
es
Mié
rco
les
Ju
eves
Vie
rnes
Sáb
ad
oD
om
ing
o
Des
ayu
no
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Gal
leta
sG
alle
tas
Cer
eale
sG
alle
tas
Cer
eale
sG
alle
tas
Cer
eale
sA
zúca
rA
zúca
rA
zúca
rA
zúca
rA
zúca
rA
zúca
rA
zúca
r
Alm
uer
zoP
anP
anP
anP
anP
anP
anP
láta
noA
ceite
Mem
brill
oA
ceite
Mar
garin
aA
ceite
A
ceite
Mer
mel
ada
Azú
car
Azú
car
Mem
brill
oA
zúca
r
Co
mid
aE
spag
ueti
Lasa
ña v
eget
alA
rroz
Mac
arro
nes
Ensa
lada
de
past
aM
enes
tra
Pael
la d
e ve
rdur
aco
n ch
ampi
ñón
Pan
con
acel
gas
con
verd
ura
Pan
de v
erdu
raE
nsal
ada
Pan
Man
darin
aP
anE
nsal
ada,
pan
Mel
ocot
ónP
anP
anP
láta
noD
iala
min
e®U
vas
Piñ
a en
alm
íbar
en a
lmíb
arP
láta
noM
eloc
otón
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Mer
ien
da
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Suc
edán
eoG
alle
tas
Gal
leta
sS
uced
áneo
Gal
leta
sG
alle
tas
Pan
de le
che
Mel
ocot
ónde
lech
eP
era
Azú
car
Gal
leta
sen
alm
íbar
Plá
tano
Mar
garin
aA
zúca
r
Cen
aH
ervi
doS
opa
de p
asta
Tort
illa
de p
atat
aPi
zza
Ham
burg
uesa
Sop
a de
arr
ozB
ocad
illo
vale
ncia
noy
verd
ura
(con
suc
edán
eode
ver
dura
vege
tal
y ve
rdur
ade
ver
dura
Ens
alad
aE
nsal
ada
de h
uevo
)P
anE
nsal
ada
Ens
alad
a, p
anre
boza
daP
anP
anE
nsal
ada,
pan
Nar
anja
Pan
Mel
ocot
ónM
anda
rina
Per
aM
elón
Nar
anja
Dia
lam
ine®
Man
zana
en a
lmíb
arD
iala
min
e®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Todo
s lo
s al
imen
tos
reco
men
dado
s en
el m
enú
son
bajo
s en
pro
teín
as: p
asta
, arr
oz, g
alle
tas,
cer
eale
s, p
an y
har
ina.
Tota
l de
Dia
lam
ine®
(SH
S):
70 g
al d
ía r
epar
tidos
en
3-4
tom
as.
Tab
la V
I.M
enú
par
a n
iño
s d
e 6-
7 añ
os
con
EC
U (
OT
C)

b) Martes:
• Desayuno: 20 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 50 g de pan; 30 g de membrillo.• Comida: lasaña vegetal (60 g de pasta, 25 g de berenjena, 25 g de calabacín, 15 g de
cebolla, 30 g de salsa de tomate y 20 mL de aceite de oliva); 15 g de pan; 100 g demandarina; 15 g de Dialamine®.
• Merienda: 15 g de Dialamine®; 100 g de melocotón en almíbar; 15 g de ga-lletas.
• Cena: sopa de pasta y verdura (30 g de pasta, 50 g de patata, 20 g de judíasverdes, 60 g de zanahoria y 20 mL de aceite de oliva); ensalada (50 g delechuga y 10 g de aceitunas); 15 g de pan; 100 g de melón; 20 g de Diala-mine®.
c) Miércoles:
• Desayuno: 20 g de Dialamine®; 20 g de cereales; 10 g de azúcar.• Media mañana: 50 g de pan; 10 mL de aceite de oliva; 10 g de azúcar.• Comida: arroz con acelgas (70 g de arroz, 50 g de acelgas, 15 g de salsa de to-
mate y 20 mL de aceite de oliva); 15 g de pan; 75 g de uvas; 15 g de Diala-mine®.
• Merienda: 15 g de Dialamine®; 20 g de galletas.• Cena: tortilla de patata (100 g de patata, 50 g de cebolla, 20 g de sucedáneo de hue-
vo y 20 mL de aceite de oliva); ensalada con 50 g de tomate; 15 g de pan; 100 g denaranja; 20 g de Dialamine®.
d) Jueves:
• Desayuno: 20 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 50 g de pan; 15 g de margarina; 10 g de azúcar.• Comida: macarrones (60 g de pasta, 50 g de calabacín, 50 g de salsa de tomate y
20 mL de aceite de oliva); ensalada con 30 g de lechuga; 15 g de pan; 100 g de piñaen almíbar; 15 g de Dialamine®.
• Merienda: 15 g de Dialamine®; 200 mL de sucedáneo de leche; 100 g de plátano.• Cena: pizza (100 g de harina, 20 g de champiñón, 30 g de cebolla, 30 g de salsa de
tomate, 15 g de aceitunas y 30 mL de aceite de oliva); 100 g de naranja; 20 g de Dia-lamine®.
e) Viernes:
• Desayuno: 20 g de Dialamine®; 20 g de cereales; 10 g de azúcar.• Media mañana: 50 g de pan; 10 g de aceite; 30 g de membrillo.
331
EIM DE LOS CICLOS ESPECÍFICOS

• Comida: ensalada de pasta (60 g de pasta, 30 g de zanahoria, 50 g de tomate;30 g de maíz y 20 mL de aceite de oliva); 15 g de pan; 100 g de melocotón enalmíbar; 15 g de Dialamine®.
• Merienda: 15 g de Dialamine®; 20 g de galletas.• Cena: hamburguesa vegetal (50 g de patata, 20 g de nabo, 25 g de zanahoria, 20 g de
acelgas, 15 g de champiñón, 10 g de sucedáneo de huevo y 20 mL de aceite de oliva);ensalada con 50 g de lechuga; 15 g de pan; 100 g de manzana; 20 g de Dialamine®.
f) Sábado:
• Desayuno: 20 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 50 g de pan; 10 mL de aceite de oliva; 10 g de azúcar.• Comida: menestra (75 g de patata, 20 g de alcachofa, 50 g de judías verdes, 20 g de
cebolla, 30 g de salsa de tomate y 20 mL de aceite de oliva); 15 g de pan; 100 g deplátano; 15 g de Dialamine®.
• Merienda: 15 g de Dialamine®; 15 g de galletas; 100 g de pera.• Cena: sopa de arroz y verdura (40 g de arroz, 30 g de zanahoria, 30 g de calabacín,
15 g de champiñón y 20 mL de aceite de oliva); ensalada con 50 g de tomate; 15 gde pan; 100 g de melocotón en almíbar; 20 g de Dialamine®.
g) Domingo:
• Desayuno: 20 g de Dialamine®; 20 g de cereales; 10 g de azúcar.• Media mañana: 100 g de plátano.• Comida: paella de verduras (100 g de arroz, 20 g de coliflor, 40 g de nabo, 25 g
de judías verdes, 25 g de zanahoria, 15 g de tomate y 20 mL de aceite de oliva);ensalada con 25 g de lechuga; 15 g de pan; 100 g de melocotón; 15 g de Diala-mine®.
• Merienda: 15 g de Dialamine®; 50 g de pan; 5 g de azúcar; 10 g de margarina.• Cena: bocadillo de verdura rebozada (70 g de pan, 75 g de berenjena, 75 g de cala-
bacín, 15 g de sucedáneo de huevo y 20 mL de aceite de oliva); 100 g de mandari-na; 20 g de Dialamine®.
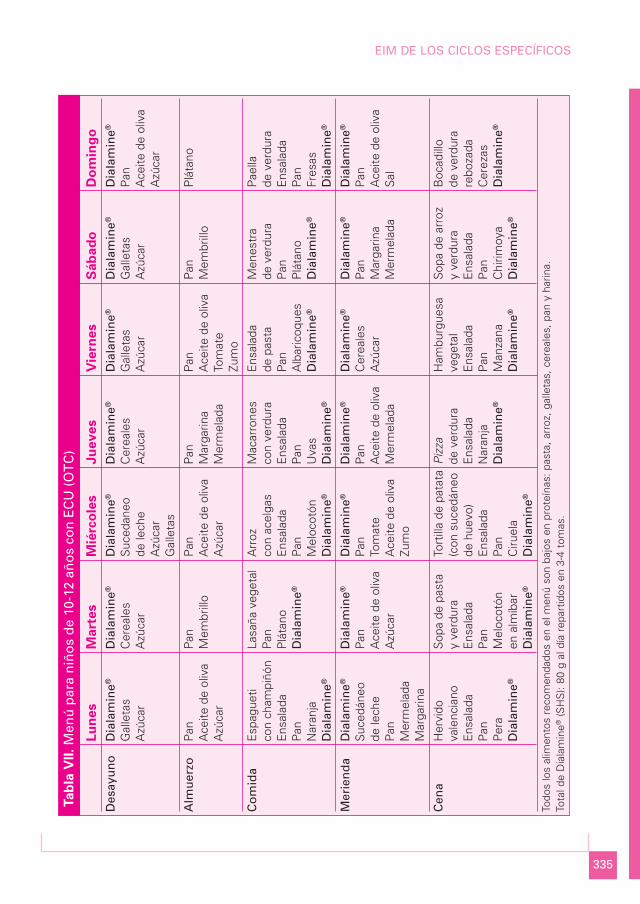
MENÚ PARA NIÑOS DE 10-12 AÑOS CON ECU (OTC) (Tabla VII)
Media semanal
Componente Cantidad Porcentaje
Proteína bruta (g) 42 7%Lípidos totales (g) 80,5 31%Carbohidratos (g) 370,3 62%Energía (kcal) 2.373
332
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO

Proteína total 42,0 gEquivalente proteico 31,5 gProteína natural 10,5 gCálculo proteico 1 g/kg
Menú semanal
a) Lunes:
• Desayuno: 35 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 60 g de pan; 15 mL de aceite de oliva; 10 g de azúcar.• Comida: espagueti con champiñón (100 g de pasta, 50 g de champiñón, 50 g de ca-
labacín y 20 mL de aceite de oliva); ensalada con 50 g de lechuga; 15 g de pan; 100 gde naranja; 28 g de Dialamine®.
• Merienda: 28 g de Dialamine®; 200 mL de sucedáneo de leche; 40 g de pan; 20 g demermelada; 20 g de margarina.
• Cena: hervido valenciano (100 g de patata, 50 g de judías verdes, 50 g de cebolla y20 mL de aceite de oliva); ensalada con 50 g de tomate; 15 g de pan; 100 g de pe-ra; 35 g de Dialamine®.
b) Martes:
• Desayuno: 35 g de Dialamine®; 20 g de cereales; 10 g de azúcar.• Media mañana: 60 g de pan; 40 g de membrillo.• Comida: lasaña vegetal (100 g de pasta, 50 g de berenjena, 50 g de calabacín, 25 g
de cebolla, 50 g de salsa de tomate y 20 mL de aceite de oliva); 15 g de pan; 100 gde plátano; 28 g de Dialamine®.
• Merienda: 25 g de Dialamine®; 40 g de pan; 10 mL de aceite de oliva; 10 g deazúcar.
• Cena: sopa de pasta y verdura (50 g de pasta, 30 g de patata, 30 g de judías verdes,30 g de zanahoria y 20 mL de aceite de oliva); ensalada con 50 g de tomate; 15 g depan; 100 g de melocotón almíbar; 35 g de Dialamine®.
c) Miércoles:
• Desayuno: 35 g de Dialamine®; 200 mL de sucedáneo de leche; 10 g de azúcar; 20 gde galletas.
• Media mañana: 60 g de pan; 15 mL de aceite de oliva; 10 g de azúcar.• Comida: arroz con acelgas (100 g de arroz, 75 g de acelgas, 30 g de salsa de toma-
te y 20 mL de aceite de oliva); ensalada con 50 g de lechuga; 15 g de pan; 100 g demelocotón; 28 g de Dialamine®.
• Merienda: 28 g de Dialamine®; 50 g de pan; 50 g de tomate; 10 g de aceite; 200 mLde zumo.
333
EIM DE LOS CICLOS ESPECÍFICOS

• Cena: tortilla de patata (100 g de patata, 100 g de cebolla, 20 g de sucedáneo de hue-vo y 20 mL de aceite de oliva); ensalada (50 g de lechuga y 15 g de aceitunas); 15 gde pan; 100 g de ciruela; 35 g de Dialamine®.
d) Jueves:
• Desayuno: 35 g de Dialamine®; 20 g de cereales; 10 g de azúcar.• Media mañana: 60 g de pan; 15 g de margarina; 20 g de mermelada.• Comida: macarrones (100 g de pasta, 50 g de calabacín, 75 g de salsa de tomate y
20 mL de aceite de oliva); ensalada con 50 g de lechuga; 15 g de pan; 75 g de uvas;28 g de Dialamine®.
• Merienda: 28 g de Dialamine®; 40 g de pan; 10 mL de aceite de oliva; 20 g de mer-melada.
• Cena: pizza (100 g de harina, 40 g de champiñón, 30 g de pimiento, 50 g de salsa detomate y 10 g de aceitunas); ensalada (50 g de lechuga y 30 mL de aceite de oliva);100 g de naranja; 35 g de Dialamine®.
e) Viernes:
• Desayuno: 35 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 60 g de pan; 10 mL de aceite de oliva; 50 g de tomate; 200 mL de
zumo.• Comida: ensalada de pasta (100 g de pasta, 50 g de zanahoria, 50 g de tomate, 30 g
de maíz, 25 g de lechuga y 20 mL de aceite de oliva); 15 g de pan; 100 g de albari-coque; 28 g de Dialamine®.
• Merienda: 28 g de Dialamine®; 20 g de cereales; 10 g de azúcar.• Cena: hamburguesa vegetal (100 g de patata, 30 g de nabo, 25 g de berenjena, 20 g
de acelgas, 15 g de champiñón, 20 g de sucedáneo de huevo y 20 mL de aceite deoliva); ensalada con 50 g de lechuga; 15 g de pan; 100 g de manzana; 35 g de Diala-mine®.
f) Sábado:
• Desayuno: 35 g de Dialamine®; 20 g de galletas; 10 g de azúcar.• Media mañana: 60 g de pan; 40 g de membrillo.• Comida: menestra (75 g de patata, 20 g de alcachofa, 50 g de judías verdes, 20 g de
cebolla, 20 g de pimiento, 50 g de salsa tomate y 20 mL de aceite de oliva); 15 g depan; 100 g de plátano; 28 g de Dialamine®.
• Merienda: 28 g de Dialamine®; 40 g de pan; 15 g de margarina; 20 g de mermelada.• Cena: sopa de arroz y verdura (70 g de arroz, 50 g de zanahoria, 50 g de calabacín,
30 g de judías verdes y 20 mL de aceite de oliva); ensalada (50 g de tomate y 10 gde maíz); 15 g de pan; 100 g de chirimoya; 35 g de Dialamine®.
334
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
335
EIM DE LOS CICLOS ESPECÍFICOS
Lu
nes
Mart
es
Mié
rco
les
Ju
eves
Vie
rnes
Sáb
ad
oD
om
ing
o
Des
ayu
no
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Gal
leta
sC
erea
les
Suc
edan
eoC
erea
les
Gal
leta
sG
alle
tas
Pan
Azú
car
Azú
car
de le
che
Azú
car
Azú
car
Azú
car
Ace
ite d
e ol
iva
Azú
car
Azú
car
Gal
leta
s
Alm
uer
zoP
anP
anP
anP
anP
anP
anP
láta
noA
ceite
de
oliv
aM
embr
illo
Ace
ite d
e ol
iva
Mar
garin
aA
ceite
de
oliv
aM
embr
illo
Azú
car
Azú
car
Mer
mel
ada
Tom
ate
Zum
o
Co
mid
aE
spag
ueti
Lasa
ña v
eget
alA
rroz
Mac
arro
nes
Ens
alad
aM
enes
tra
Pae
llaco
n ch
ampi
ñón
Pan
con
acel
gas
con
verd
ura
de p
asta
de v
erdu
rade
ver
dura
Ens
alad
aP
láta
noE
nsal
ada
Ens
alad
aP
anP
anE
nsal
ada
Pan
Dia
lam
ine®
Pan
Pan
Alb
aric
oque
sP
láta
noP
anN
aran
jaM
eloc
otón
Uva
sD
iala
min
e®D
iala
min
e®Fr
esas
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Mer
ien
da
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Dia
lam
ine®
Suc
edán
eoP
anP
anP
anC
erea
les
Pan
Pan
de le
che
Ace
ite d
e ol
iva
Tom
ate
Ace
ite d
e ol
iva
Azú
car
Mar
garin
aA
ceite
de
oliv
aP
anA
zúca
rA
ceite
de
oliv
aM
erm
elad
aM
erm
elad
aS
alM
erm
elad
aZu
mo
Mar
garin
a
Cen
aH
ervi
doS
opa
de p
asta
Tort
illa
de p
atat
aPi
zza
Ham
burg
uesa
Sop
a de
arr
ozB
ocad
illo
vale
ncia
noy
verd
ura
(con
suc
edán
eode
ver
dura
vege
tal
y ve
rdur
ade
ver
dura
Ens
alad
aE
nsal
ada
de h
uevo
)E
nsal
ada
Ens
alad
aE
nsal
ada
rebo
zada
Pan
Pan
Ens
alad
aN
aran
jaP
anP
anC
erez
asP
era
Mel
ocot
ónP
anD
iala
min
e®M
anza
naC
hirim
oya
Dia
lam
ine®
Dia
lam
ine®
en a
lmíb
arC
iruel
aD
iala
min
e®D
iala
min
e®
Dia
lam
ine®
Dia
lam
ine®
Todo
s lo
s al
imen
tos
reco
men
dado
s en
el m
enú
son
bajo
s en
pro
teín
as: p
asta
, arr
oz, g
alle
tas,
cer
eale
s, p
an y
har
ina.
Tota
l de
Dia
lam
ine®
(SH
S):
80 g
al d
ía r
epar
tidos
en
3-4
tom
as.
Tab
la V
II.
Men
ú p
ara
niñ
os
de
10-1
2 añ
os
con
EC
U (
OT
C)

g) Domingo:
• Desayuno: 35 g de Dialamine®; 40 g de pan; 15 mL de aceite de oliva; 10 g deazúcar.
• Media mañana: 100 g de plátano.• Comida: paella de verduras (100 g de arroz, 50 g de coliflor, 25 g de alcachofa,
50 g de judías verdes, 15 g de salsa tomate y 20 mL de aceite de oliva); ensalada(25 g de lechuga y 15 g de aceitunas); 20 g de pan; 100 g de fresas; 28 g de Dia-lamine®.
• Merienda: 28 g de Dialamine®; 40 g de pan; 15 mL de aceite de oliva; sal.• Cena: bocadillo de verdura rebozada (100 g de pan, 75 g de berenjena, 75 g de ca-
labacín, 15 g de sucedáneo de huevo, 50 g de tomate y 20 mL de aceite de oliva);100 g de cerezas; 35 g de Dialamine®.
Total de Dialamine® al día: 80 g repartidos en 3-4 tomas.
Pautas de seguimiento de las enfermedades del ciclo de la urea
Debut
• Normalmente, el debut se produce durante los primeros días de vida, y su presenta-ción es aguda y grave.
• El tratamiento y los controles iniciales dependen de la gravedad del cuadro:
- Se deben estabilizar las constantes.- Se utilizarán depuradores de amonio: fenilbutirato, benzoato sódico, diálisis perito-
neal o hemofiltración.- Se ha de asegurar un aporte calórico adecuado, mediante soluciones glucosadas y li-
pídicas ev y/u orales.
En todos los casos
• Eliminar las proteínas naturales de la dieta durante 24-48 horas (valorar el momento desu reintroducción según los niveles de amonio).
• Iniciar arginina a 100-200 mg/kg (excepto en la acidemia argininsuccínica).• Iniciar tratamiento con ácido carglúmico (Carbaglu®) en la deficiencia de NAGS. Dosis
inicial: 100-250 mg/día. A largo plazo: 10-100 mg/día. Se debe ajustar la dosis según elpaciente.
• Facilitar el contacto con la Asociación de Padres de ECM de la comunidad.• Controles: amonio, carnitina, aminoácidos (del ciclo de la urea y un aminoácido esen-
cial), y parámetros nutricionales (vitamina B12).
336
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO

Controles ambulatorios
La determinación de amonio sólo puede realizarse en sangre total. Cada control suponeuna venopunción.
a) Durante los dos primeros meses:
• Controlar el amonio y los aminoácidos cada 7-15 días.• Control por el Servicio de Nutrición cada 7-15 días.
b) De los dos a los seis meses:
• Controlar el amonio y los aminoácidos cada 30 días.• Control por Nutrición cada 30 días.
c) De los seis a los doce meses:
• Controlar el amonio y los aminoácidos cada tres meses.• Control por Nutrición cada tres meses, con especial vigilancia en el momento de la
introducción de alimentos.
d) Del primer al segundo año:
• Controlar el amonio y los aminoácidos cada tres meses.• Control por Nutrición cada tres meses.
e) A partir de los dos años:
• Controlar el amonio y los aminoácidos cada 4-6 meses.• Control por Nutrición cada 4-6 meses.
Otros controles sugeridos
• Control por Neurología, si se producen secuelas tras el debut.• Control de las funciones hepática y renal.• Control de la osteoporosis: densitometría a los diez años.• Control psicológico.
Hoja de control
Para un mejor seguimiento del niño, se puede cumplimentar una hoja de control comola que se muestra a continuación:
337
EIM DE LOS CICLOS ESPECÍFICOS
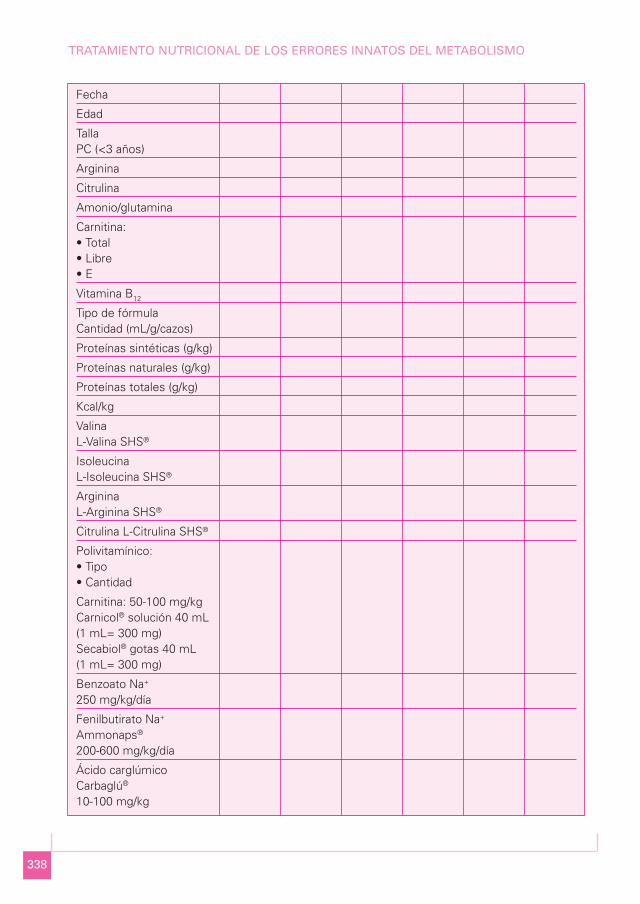
Fecha
Edad
Talla PC (<3 años)
Arginina
Citrulina
Amonio/glutamina
Carnitina:• Total• Libre• E
Vitamina B12
Tipo de fórmulaCantidad (mL/g/cazos)
Proteínas sintéticas (g/kg)
Proteínas naturales (g/kg)
Proteínas totales (g/kg)
Kcal/kg
ValinaL-Valina SHS®
IsoleucinaL-Isoleucina SHS®
ArgininaL-Arginina SHS®
Citrulina L-Citrulina SHS®
Polivitamínico:• Tipo• Cantidad
Carnitina: 50-100 mg/kgCarnicol® solución 40 mL(1 mL= 300 mg)Secabiol® gotas 40 mL(1 mL= 300 mg)
Benzoato Na+
250 mg/kg/día
Fenilbutirato Na+
Ammonaps®
200-600 mg/kg/día
Ácido carglúmicoCarbaglú®
10-100 mg/kg
338
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO

Parámetros de control
Se deben solicitar controles de aminoácidos en sangre, de amonio y de carnitina.
Parámetros bioquímicos
Para hallarse dentro de la normalidad, los valores de amonio deben ser inferiores a 80 µmol/Ldurante el primer mes, e inferiores a 60 µmol/L a partir de entonces.
Los niveles de glutamina deben ser inferiores a 800-1.000 µmol/L, los de arginina deben hallar-se por encima de los 100 µmol/L y, finalmente, los de citrulina deben oscilar entre 13 y 33 µmol/L.
Dispensación
El tipo de dispensación varía según la comunidad autónoma y puede consistir en diver-sos productos:
• L-Arginina SHS®/L-Citrulina SHS®, bote de 100 g: se puede obtener en farmacia hospi-talaria, en oficinas de farmacia con receta o como nutrición enteral domiciliaria. Esteproducto está financiado por el Sistema Nacional de Salud.
• Essential Amino Acid Mix® y Dialamine®: se obtienen en forma de nutrición enteral do-miciliaria, en farmacia hospitalaria o en oficinas de farmacia, y también reciben finan-ciación del Sistema Nacional de Salud.
• Módulos nutricionales: se pueden obtener en farmacia hospitalaria, en oficinas de far-macia con receta o como nutrición enteral domiciliaria; también están financiados.
• Productos especiales bajos en proteínas: se pueden adquirir en farmacias, tiendas dedietética o en las grandes superficies, pero no reciben ningún tipo de financiación.
• Benzoato Na+: el benzoato de sodio se obtiene con receta verde como fórmula magis-tral, a través de la farmacia hospitalaria y encapsulado a cargo de la familia. La finan-ciación se realiza mediante la petición del facultativo al inspector médico.
• Fenilbutirato: se obtiene mediante dispensación hospitalaria, con receta blanca o en far-macia hospitalaria, y no recibe financiación del Sistema Nacional de Salud.
Posología del fenilbutirato y de la arginina
Nombre Dosis Envase
Fenilbutirato: 200-600 mg/kg Polvo: 266 gAmmonaps® Comprimidos: 500 g
L-Arginina SHS®:Arginina clorhidrato 200-600 mg/kg Bote de 100 g, oralArginine Veyron® 200-600 mg/kg Ampollas de 1 g/5 mL ev
Arginina aspartato: Potenciator® 200-600 mg/kg Ampollas bebibles de 1 y 5 g vo
1 g de arginina clorhidrato= 1,66 g de arginina aspartato
339
EIM DE LOS CICLOS ESPECÍFICOS

Preparación
• Arginina 20 mg/mL: 100 mL de H2O + 2 g de arginina.• Citrulina 20 mg/mL: 100 mL de H2O + 2 g de citrulina.• Benzoato Na+ (250 mg/mL): 100 mL H2O + 31,25 g de benzoato.
Tiempo de permanencia en la nevera: 14 días.
Fórmulas especiales (100 g)
Fórmula AA Kcal Edad
Essential Amino Acid Mix® (SHS) 94,5 316 Niños menores de un año
Dialamine® (SHS) 30 360 Niños mayores de tres años
340
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
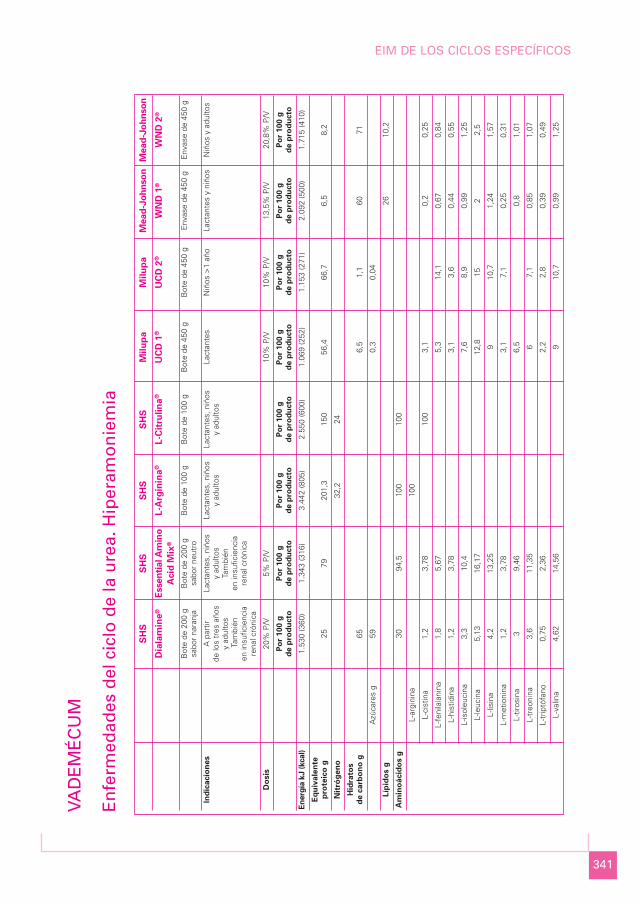
341
EIM DE LOS CICLOS ESPECÍFICOS
VAD
EM
ÉC
UM
En
ferm
edad
es d
el c
iclo
de
la u
rea.
Hip
eram
on
iem
ia
SH
SS
HS
SH
SS
HS
Milu
pa
Milu
pa
Mead
-Jo
hn
so
nM
ead
-Jo
hn
so
n
Dia
lam
ine
®E
ssen
tial A
min
oL-A
rgin
ina
®L-C
itru
lin
a®
UC
D 1
®U
CD
2®
WN
D 1
®W
ND
2®
Acid
Mix
®
Bot
e de
200
gB
ote
de 2
00 g
Bot
e de
100
gB
ote
de 1
00 g
Bot
e de
450
gB
ote
de 4
50 g
Env
ase
de 4
50 g
Env
ase
de 4
50 g
sabo
r na
ranj
asa
bor
neut
ro
Ind
icacio
nes
A p
artir
La
ctan
tes,
niñ
osLa
ctan
tes,
niñ
osLa
ctan
tes,
niñ
osLa
ctan
tes
Niñ
os >
1 añ
oLa
ctan
tes
y ni
ños
Niñ
os y
adu
ltos
de lo
s tr
es a
ños
y ad
ulto
sy
adul
tos
y ad
ulto
sy
adul
tos
Tam
bién
Ta
mbi
énen
insu
ficie
ncia
en in
sufic
ienc
iare
nal c
róni
care
nal c
róni
ca
Do
sis
20%
P/V
5%
P/V
10
% P
/V10
% P
/V13
,5%
P/V
20,8
% P
/V
Po
r 100 g
Po
r 100 g
Po
r 100 g
Po
r 100 g
Po
r 100 g
Po
r 100 g
Po
r 100 g
Po
r 100 g
de p
rod
ucto
de p
rod
ucto
de p
rod
ucto
de p
rod
ucto
de p
rod
ucto
de p
rod
ucto
de p
rod
ucto
de p
rod
ucto
En
erg
ía k
J (
kcal)
1.53
0 (3
60)
1.34
3 (3
16)
3.44
2 (8
05)
2.55
0 (6
00)
1.06
9 (2
52)
1.15
3 (2
71)
2.09
2 (5
00)
1.71
5 (4
10)
Eq
uiv
ale
nte
pro
teic
o g
2579
201,
315
056
,466
,76,
58,
2
Nit
róg
en
o32
,224
Hid
rato
s
de c
arb
on
o g
656,
51,
160
71
Azú
care
s g
590,
30,
04
Líp
ido
s g
2610
,2
Am
ino
ácid
os g
3094
,510
010
0
L-ar
gini
na10
0
L-ci
stin
a1,
23,
7810
03,
10,
20,
25
L-fe
nila
lani
na1,
85,
675,
314
,10,
670,
84
L-hi
stid
ina
1,2
3,78
3,1
3,6
0,44
0,55
L-is
oleu
cina
3,3
10,4
7,6
8,9
0,99
1,25
L-le
ucin
a5,
1316
,17
12,8
152
2,5
L-lis
ina
4,2
13,2
59
10,7
1,24
1,57
L-m
etio
nina
1,2
3,78
3,1
7,1
0,25
0,31
L-tir
osin
a3
9,46
6,5
0,8
1,01
L-tr
eoni
na3,
611
,35
67,
10,
851,
07
L-tr
iptó
fano
0,75
2,36
2,2
2,8
0,39
0,49
L-va
lina
4,62
14,5
69
10,7
0,99
1,25
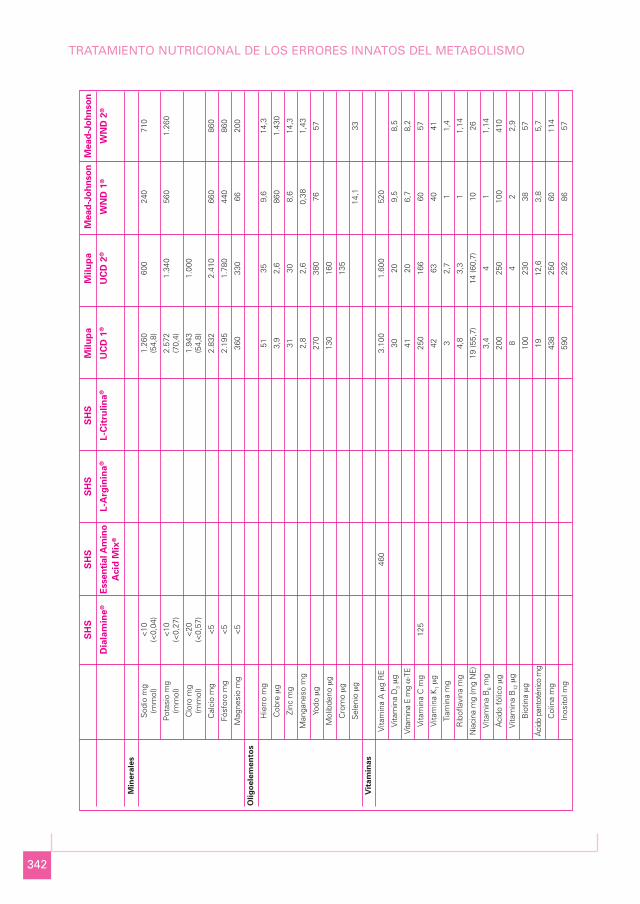
342
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
SH
SS
HS
SH
SS
HS
Milu
pa
Milu
pa
Mead
-Jo
hn
so
nM
ead
-Jo
hn
so
n
Dia
lam
ine
®E
ssen
tial A
min
oL-A
rgin
ina
®L-C
itru
lin
a®
UC
D 1
®U
CD
2®
WN
D 1
®W
ND
2®
Acid
Mix
®
Min
era
les
Sod
io m
g<
101.
260
600
240
710
(mm
ol)
(<0,
04)
(54,
8)
Pot
asio
mg
<10
2.57
21.
340
560
1.26
0(m
mol
)(<
0,27
)(7
0,4)
Clo
ro m
g<
201.
943
1.00
0(m
mol
)(<
0,57
)(5
4,8)
Cal
cio
mg
<5
2.83
22.
410
660
860
Fósf
oro
mg
<5
2.19
51.
780
440
860
Mag
nesi
o m
g<
536
033
066
200
Olig
oele
men
tos
Hie
rro
mg
5135
9,6
14,3
Cob
re µ
g3,
92,
686
01.
430
Zinc
mg
3130
8,6
14,3
Man
gane
so m
g2,
82,
60,
381,
43
Yodo
µg
270
380
7657
Mol
ibde
no µ
g13
016
0
Cro
mo
µg
135
Sel
enio
µg
14,1
33
Vit
am
inas
Vita
min
a A
µg
RE
460
3.10
01.
600
520
Vita
min
a D
3µ
g30
209,
58,
5
Vita
min
a E
mg α
-TE
4120
6,7
8,2
Vita
min
a C
mg
125
250
166
6057
Vita
min
a K
1µ
g42
6340
41
Tiam
ina
mg
32,
71
1,4
Rib
ofla
vina
mg
4,8
3,3
11,
14
Nia
cina
mg
(mg
NE)
19 (5
5,7)
14 (6
0,7)
1026
Vita
min
a B
6m
g3,
44
11,
14
Áci
do f
ólic
o µ
g20
025
010
041
0
Vita
min
a B
12µ
g8
42
2,9
Bio
tina
µg
100
230
3857
Áci
do p
anto
téni
co m
g19
12,6
3,8
5,7
Col
ina
mg
438
250
6011
4
Inos
itol m
g59
029
286
57
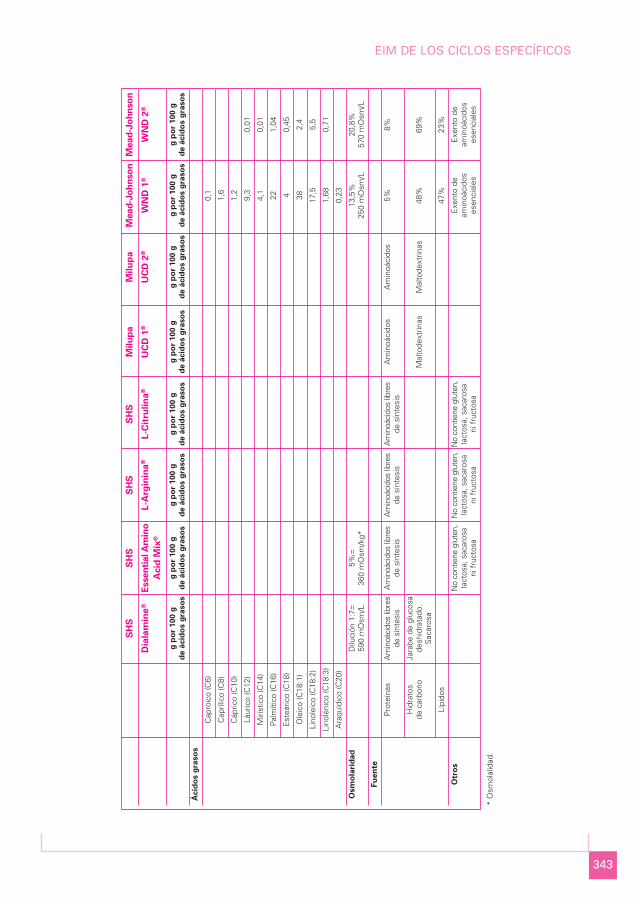
343
EIM DE LOS CICLOS ESPECÍFICOS
SH
SS
HS
SH
SS
HS
Milu
pa
Milu
pa
Mead
-Jo
hn
so
nM
ead
-Jo
hn
so
n
Dia
lam
ine
®E
ssen
tial A
min
oL-A
rgin
ina
®L-C
itru
lin
a®
UC
D 1
®U
CD
2®
WN
D 1
®W
ND
2®
Acid
Mix
®
g p
or
100 g
g p
or
100 g
g p
or
100 g
g p
or
100 g
g p
or
100 g
g p
or
100 g
g p
or
100 g
g p
or
100 g
de á
cid
os g
raso
sd
e á
cid
os g
raso
sd
e á
cid
os g
raso
sd
e á
cid
os g
raso
sd
e á
cid
os g
raso
sd
e á
cid
os g
raso
sd
e á
cid
os g
raso
sd
e á
cid
os g
raso
s
Ácid
os g
raso
s
Cap
roic
o (C
6)0,
1
Cap
rílic
o (C
8)1,
6
Cáp
rico
(C10
)1,
2
Láur
ico
(C12
)9,
30,
01
Mirí
stic
o (C
14)
4,1
0,01
Pal
míti
co (C
16)
221,
04
Est
eáric
o (C
18)
40,
45
Ole
ico
(C18
:1)
382,
4
Lino
leic
o (C
18:2
)17
,55,
5
Lino
léni
co (C
18:3
)1,
680,
71
Ara
quíd
ico
(C20
)0,
23
Osm
ola
rid
ad
Dilu
ción
1:7
=5%
=
13,5
%20
,8%
590
mO
sm/L
360
mO
sm/k
g*25
0 m
Osm
/L57
0 m
Osm
/L
Fu
en
te
Pro
teín
asA
min
oáci
dos
libre
sA
min
oáci
dos
libre
sA
min
oáci
dos
libre
sA
min
oáci
dos
libre
sA
min
oáci
dos
Am
inoá
cido
s5%
8%de
sín
tesi
sde
sín
tesi
sde
sín
tesi
sde
sín
tesi
s
Hid
rato
sJa
rabe
de
gluc
osa
de c
arbo
node
shid
rata
do.
Mal
tode
xtrin
asM
alto
dext
rinas
48%
69%
Sac
aros
a
Lípi
dos
47%
23%
Otr
os
No
cont
iene
glu
ten,
No
cont
iene
glu
ten,
No
cont
iene
glu
ten,
Exe
nto
deE
xent
o de
lact
osa,
sac
aros
a la
ctos
a, s
acar
osa
lact
osa,
sac
aros
a am
inoá
cido
sam
inoá
cido
sni
fru
ctos
ani
fru
ctos
ani
fru
ctos
aes
enci
ales
esen
cial
es
* O
smol
alid
ad.

344
TRATAMIENTO NUTRICIONAL DE LOS ERRORES INNATOS DEL METABOLISMO
BIBLIOGRAFÍA
1. Brusilow S, Maestri N. Urea cycle disorders: Diagnosis, pathophysiology, and therapy.Advances in Pediatrics 1996; 43: 127-70.
2. Summar M, Tuchman. Proceedings of a consensus conference for the managementof patients with urea cycle disorders. J Pediatr 2001; 138 (Suppl.): 6-10.
3. Leonard JV. The nutritional management of urea cycle disorders. J Pediatr 2001; 138(Suppl.): 40-5.
4. Pintos G, Briones MP, Marchante C et al. Protocolo para el diagnóstico, tratamiento y se-guimiento de los trastornos del ciclo de la urea. An Esp Pediatr 1997; (Suppl. 89): 1-8.
5. Sanjurjo P, Montejo M, García MA, Pintos G. Errores innatos del ciclo de la urea. Ac-tualidad Nutricional 1993; 24: 16-21.
6. Ruiz M, Santana C, Trujillo R, Sánchez-Valverde F, Dalmau J. Aproximación al trata-miento nutricional de los errores innatos del metabolismo (V). Acta Pediatr Esp 2002;60: 677-84.
7. Berry GT, Steiner RD. Long-term management of patients with urea cycle disorders.J Pediatr 2001; 138 (Suppl.): 56-61.
8. Batshaw M, McArthur R, Tuchman M. Alternative pathway therapy for urea cycle di-sorders: Twenty years later. J Pediatr 2001; 138 (Suppl.): 46-55.
9. Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, Tuchman M.Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamyl-glutamate. J Pediatr 2004 Oct; 145 (4): 552-4.
10. Ogier DE, Baulny H. Management and emergency treatments of neonates with a sus-picion of inborn of metabolism. Semin Neonatol 2002; 7: 17-26.
11. Saudubray JM, Nassogne MC, de Lonlay P, Touati G. Clinical approach to inheriteddisorders in neonates: An overview. Semin Neonatol 2002; 7: 3-15.
12. Maestri N, Clissold D, Brusilow S. Neonatal onset ornithine transcarbamylase defi-ciency: A retrospective analysis. J Pediatr 1999; 134: 268-72.
13. Nicolaides P, Liebsch D, Dale N et al. Neurologic outcome of patients with ornithinecarbamoyltransferase deficiency. Arch Dis Child 2002; 86: 54-6.
14. Summar M. Current strategies for the management of neonatal urea cycle disorders.J Pediatr 2001; 138 (Suppl.): 30-9.
15. Lee B, Goss J. Long-term correction of urea cycle disorders. J Pediatr 2001; 138(Suppl.): 62-71.
16. Prietsch V, Lindner M, Zschocke J, Nyhan WL, Hoffmann GF. Emergency manage-ment of inherited metabolic diseases. J Inher Metabol Dis 2002; 25: 531-46.

345
EIM DE LOS CICLOS ESPECÍFICOS
CONCLUSIÓN
“Individualmente, los EIM son poco frecuentes, pero en conjunto son muy numerosos.”Esta aseveración es posiblemente el gran reto de los EIM. Sin ningún género de duda, setrata de un grupo de enfermedades que se están empezando a comprender y que se diag-nostican cada vez con mayor frecuencia. Probablemente, durante los próximos años seirá comprobando cómo diversos cuadros clínicos que hasta el momento no se habían cla-sificado se explican en sus aspectos etiológicos y clínicos a raíz del descubrimiento desus fundamentos metabólicos.
A la espera de la terapia génica que se aplicará en el futuro, en muchos casos el trata-miento de estas enfermedades se ha de basar en la dieta. En el momento actual, la for-mación de equipos de pediatras y dietistas especializados en el metabolismo, con la co-laboración de otros profesionales en ciertas ocasiones, constituye la mejor alternativa parael diagnóstico y el seguimiento de estos pacientes.
Este libro, como todo proyecto interdisciplinario, ha supuesto un reto apasionante paralos autores, que han tenido que responder a la necesidad de alimentar a los pacientesmetabólicos, teniendo en cuenta además, en muchas ocasiones, que esta dieta era te-rapéutica.
La presente obra acumula muchas horas de trabajo de pediatras, dietistas y otros profe-sionales que han tenido a su cargo a este tipo de pacientes. Al mostrar esta experiencia,se pretende facilitar la toma de decisiones clínicas y nutricionales a aquellos compañerosque, en cualquier parte del mundo, se van a enfrentar a situaciones similares.


BIBLIOGRAFÍA GENERALRECOMENDADA
• Nyhan WL, Barshop BA, Ozand PT. Atlas of Metabolic Diseases, 2nd ed. Hodder ArnoldPublication 2005.
• Hoffmann GF, Nyhan WL, Zschoke J, Khaler SG, Mayatepek E. Inherited metabolic dis-eases. Philadelphia: E. Lippincot Williams&Wilkins 2002.
• Saudubray JM, Ogier de Baulny H, Charpentier C. Clinical approach to inherited meta-bolic diseases. In: Fernandes J, Saudubray JM, Van den Berghe G (eds.). Inborn Meta-bolic Diseases. Diagnosis and Treatment, 4th ed. Springer Medicine Verlag 2006.
• Pampols T. Cromosomas, genes y mutaciones: bases bioquímicas y moleculares de lasenfermedades genéticas En: Pampols T (coord.). Del cromosoma al gen. Las anoma-lías cromosómicas y las enfermedades metabólicas hereditarias, dos modelos para-digmáticos de enfermedad genética. Barcelona: Instituto de Ediciones de la Diputación1995: 39-74.
• Sanjurjo P, Baldellou A (eds). Diagnóstico y tratamiento de las enfermedades metabó-licas hereditarias, 2ª ed. Madrid: Ergon SA 2006.
• Sanjurjo P, Baldellou A, Aladamiz L. Enfermedades congénitas del metabolismo: basesdiagnósticas para el pediatra. Madrid: Ergon 2003.
• Schaw V, Lawson M (eds.). Clinical Paediatric Dietetics, 2nd ed. Oxford: Blackwell Scien-tific Publishing 2002.
• Blau N, Hoffmann GF, Leonard J, Clarke JTR (eds.). Physician’s Guide to the Treatmentand Follow-Up of Metabolic Diseases, 1st ed. Springer 2005.
• Scriver CR, Blau N, Durán M, Blaskovics ME, Gibson KM. Physician’s Guide to the La-boratory Diagnosis of Metabolic Diseases, 2nd ed. Springer 2004.
• Clarke JTR. A Clinical Guide to Inherited Metabolic Diseases, 3rd ed. Cambridge Uni-versity Press 2006.
• Fernandes J, Saudubray JM, van den Berghe G, Walter J. Springer Medicine Verlag2006.
347




















![Transtornos Del Ciclo d Ela Urea[1]](https://img.pdfslide.tips/doc/110x75/5572139c497959fc0b92a5bf/transtornos-del-ciclo-d-ela-urea1.jpg)









