Embed Size (px)
DESCRIPTION
Cinética
Citation preview

Tema 1
Métodos de determinación de mecanismos de reacción. Métodos de determinación de mecanismos de reacción.
Efecto de la estructura molecular en la reactividad.

Repaso de conceptos de cinética aprendidos en la
asignatura:
Fundamentos de Química (primer curso del Grado)Fundamentos de Química (primer curso del Grado)
Algunas transparencias de la introducción están basadas
en transparencias de los Drs. Luis Rodríguez y Esteve Fàbregues








Obtención ecuación de velocidad de una reacción compuesta de varias etapas elementales
Teoria del estado Estacionario:
La concentración de los intermedios
permanece constante a lo largo de
la reacción
Reacción global: A + B + D E + F
A + B Ck1
k-1
C + D E + Fk2
v = k1[A][B]-k-1[C]
v = k2[C][D]
k1[A][B] = k2[C][D] + k-1[C]
formación de C destrucción de C
[C] =
k1[A][B]
k2[D] + k-1
Ejemplo:
Mecanismo:
“rds”: rate determining step, la que
Si k2[D] » k-1 v = k1[A][B] la 1ª etapa es la rds
formación de C destrucción de C
v = k2[C][D] = k2
k1[A][B]
k2[D] + k-1
[D]
Si k2[D] « k-1 v = [A][B][D] la 2ª etapa es la rdsk2k1
k-1
“rds”: rate determining step, la que
supone pasar por el punto más alto
en la coordenada energética de la
reacción
E
ECR
CR




Ecuación de Eyring
k = eKBT
h
–∆G‡/RT
Ecuación de Eyring:
e . e–∆H‡/RT ∆S‡/RKBT
hk =
Ecuación de Arrhenius:
k = A e-Ea/RT
Haciendo logaritmos,
Ln k = ln [Ae (-Ea/RT)] = ln A - Ea/RT
Entalpía de activación Entropía de activación

Deducción ecuación de Eyring
Teoría del estado de transición
A + B X‡
i) Reactivos en equilibrio con el complejo activado
ii) Todos los complejos activados evolucionan hacia los productos
a una velocidad fija
K‡ =[X‡]
[A][B]
K T
X‡ C
15
v = [X‡]KBT
h
[X‡] = K‡[A][B]
v = K‡[A][B]KBT
h
∆G‡ = -RT lnK‡ ; K‡ = e –∆G‡/RT
k = eKBT
h
–∆G‡/RT
Equación de Eyring
v = [X‡]KBT
h
e . e–∆H‡/RT ∆S‡/RKBT
hk =

Teoría del estado de transición. Perfil de reacción
El Estado de transicion (ET o TS) es el estado de mayor energía de los reactivos en su camino de reacción
hacia los productos. La estructura específica de los reactivos en el ET se llama complejo activado.
Los datos cinéticos dan información sobre la composición del complejo activado o el TS para el paso
determinante de la velocidad de la reacción, pero no dan información sobre la estructura del intermedio,
(ej. SN1 i SN2)
Perfil de reacción: diagrama bidimensional de la energía potencial respecto a la coordenada de Perfil de reacción: diagrama bidimensional de la energía potencial respecto a la coordenada de
reacción, que representa la evolución energética de la reacción (evolución de reactivos a productos)
La coordenada de reacción representa el progreso de la reacción en el transcurso de una modificación
paulatina de reactivos a productos (diversos parámetros se pueden representar en la coordenada de
reacción, per ejemplo una distancia de enlace para un enlace que se forma o se rompe)

Ejemplos de perfiles de reacción para una reacción de una sola etapa y para una de dos etapas
Teoria del estado de transición. Perfil de reacción

Se pueden relacionar con sus análogos termodinámicos.
Reflejan la estructura del complejo activado o estado de transición.
Entalpía de activación
Por definición: ∆∆∆∆H≠ = Hº (complejo activado) – Hº (reactivos)
Asociada a cambios energéticos de la reacción
(reorganización de enlaces, rotura o formación, efectos electrónicos)
Parámetros de activación
18
(reorganización de enlaces, rotura o formación, efectos electrónicos)
Se determina a partir de la pendiente de la recta que resulta de representar
ln k/T vs 1/ T en la expresión derivada de la ec. d’Eyring (dividir todo por T y aplicar ln)
ln k/T = ∆∆∆∆S≠ / R + ln kB/h – (∆∆∆∆H≠/R)1/T La pendiente es - ∆∆∆∆H≠/R
También se puede estimar por cálculos teóricos.

Entropía de activación
Por definición: ∆∆∆∆S≠ = Sº (complejo activado) – Sº (reactivos)
Puede ser > 0 o < 0 (a diferencia de la entalpia de activación que es siempre > 0)
Relacionada con el grado de orden del complejo activado respecto a los reactivos
Cuanto mayor sea el aumento del orden menor probabilidad de que se de la reacción.
∆∆∆∆S≠ < 0 TS más ordenado (menor entropía) que los reactivos
Parámetros de activación
19
∆∆∆∆S≠ < 0 TS más ordenado (menor entropía) que los reactivos
Reacción retardada o de baja probabilidad
∆∆∆∆S≠ > 0 TS menos ordenado (mayor entropía) que reactivos
Reacción acelerada o de probabilidad elevada
Se determina a partir de la ordenada de origen de la recta que resulta de representar
ln k/T vs 1/ T en la expresión derivada de la ec. d’Eyring (dividir todo por T i aplicar ln)
ln k/T = ∆∆∆∆S≠ /R + ln kB/h – (∆∆∆∆H≠/R)1/T ordenada es ∆∆∆∆S≠ /R + ln kB/h

∆∆∆∆H≠ = 15.5 Kcal/mol
∆∆∆∆S≠ = - 34 eu
Parámetros de activación
1) Dimerización del ciclopentadieno en la fase gas:
2) Descomposición del 1,1’-diazobutano en fase gas:
∆∆∆∆H≠ = 52 Kcal/mol
∆∆∆∆S≠ = + 19 eu
Disminución del grado de libertad traslacional, rotacional y vibracional en (A) comporta disminución de entropía en ET (DS≠ < 0)
Aumento de grados de libertad traslacional, rotacional y vibracionales en (B) comporta aumento de entropía en ET (DS≠ > 0)
∆G≠ = ∆ H≠ - T ∆ S≠

Permite relacionar las estructuras de los ET con las de los intermedios, reactivos y productos.
Si dos estados (por ej. un ET y un intermedio) se producen consecutivamente durante
la reacción y tienen energías similares, su interconversión implica solamente una
pequeña reorganización de la estructura molecular.
De forma más sencilla quiere decir que:
Postulado de Hammond
21
Un paso de reacción muy exotérmico presentará un ET similar a los reactivos
(early or reactant-like) (caso 1 diapositiva siguiente)
Un paso de reacción muy endotérmico presentará un ET similar a productos
(late or product-like) (caso 3 diapositiva siguiente)
Si en el perfil de reacció el ET es de energía mucho más elevada que reactivos y
productos, ni unos ni otros son buenos modelos de l’ET (caso 2 diapositiva siguiente)

Postulado de Hammond

Postulado de Hammond
23
El ET de la e.d.v es de tipo product-like (similar al complejo intermedio σσσσ)





Control cinético/Control termodinámico
CH(CH2)2CH3
H3C
O
CH3C(CH2)3CH3
O
H2C
O
(CH2)3CH3
CH(CH ) CH
HO
H C
OH
B B
Enolato termodinámico Enolato cinético
1
2 3
Formación de enolatos a partir de cetonas asimétricas
28
Con base fuerte y voluminosa, en disolvente aprótico se forma mayoritariamente el
enolato cinético 3.
Con la base más débil y en disolvente prótico, el enolato mayoritario es el
termodinámicamente más estable 2.
Disolventes próticos favorecen los equilibrios.
CH(CH2)2CH3
H3C
H2C
(CH2)3CH3

Control cinético/Control termodinámico
k = eKBT
h
–∆G‡/RT
Ecuación de Eyring:
Control Cinético
ET
Control Termodinámico
Estado de transición
(ET)
Intermedio
Intermedio ∆G‡A
∆G‡B
Energía
PB
h
e . e–∆H‡/RT ∆S‡/RKBT
hk =
Producto
cinético
Producto
de partida
Intermedio
Producto
termodinámico
PA
∆G0A
∆G0B
Coordenada de reacción

Control cinético/Control termodinámico
Coordenada de reacción

Principio de Curtin-Hammett
∆G‡A ∆G‡
B
- En una situación de equilibrio rápido entre dos
moléculas A y B que pueden reaccionar para dar
dos productos distintos: PA y PB
- La proporción entre los dos productos no depende
de la proporción inicial entre las moléculas A y B,
sino exclusivamente del ∆G‡ entre los Estados de
Transición que lleven a esos productos.
Energía
G‡B - G‡
A
- - - - - - - - - - - - - - - - - - - - - - -
- - - - - - - -
K1, K-1 >> KA, KB
KA KBK1
K-1
[PA]
[PB]=
d[PA]/dt
d[PB]/dt=
kA[A]
kB[B]
kA[B]
kBKeq[B]= =
kA
kBKeq
=
Transición que lleven a esos productos.
Coordenada de reacción
K = K B T
e -∆G‡/RT------h
Ecuación Eyring:
Ecuación Equilibrio:
K eq = [B]
[A]------
***
*
* ****
***
K eq = e –∆G0eq/RT

Efecto cinético de isótopo
Efecto Cinético de Isótopo:
- Variación de la velocidad de una reacción al substituir un determinado átomo de uno de los
reactivos por uno de sus isótopos.
- El caso más habitual es sustituir Hidrógeno por Deuterio.
Efecto Cinético de Isótopo Primario:
- En una reacción elemental: Al cambiar hidrógeno por
deuterio, un átomo más pesado, la velocidad de la
reacción disminuye: kH > kD
- A temperatura ambiente:E
32
kH
kD máximo
< 7
- Origen: la energía del nivel vibracional más
bajo de un enlace, es menor al aumentar la
masa de los átomos implicados.
Eo = ½ hνpunto cero:
niveles
vibracionales
permitidos
r
En = (n + ½) hν , n = 0, 1, 2, …
Llei de Hook: ν = 1
2π
f
m
2 <mínimo

∆G‡ < ∆G‡ H D
kH > kD
- En una reacción elemental donde el enlace C-H
o C-D se rompe: en el estado de transición (ET)
ya no existe energía vibracional (Evibr)∆G‡
D∆G‡
Hproducto
ET
R-HR-D
- Energía del nivel vibracional mas bajo:
enlace C-H > enlace C-D.
E
r
Efecto cinético de isótopo
1) - En una reacción compleja, se observará Efecto Cinético Isótopo Primario cuando el enlace
C-H en el que se ha sustituido el H por D se rompa antes o durante la rds.
2) – El valor del Efecto Cinético Isótopo Primario indica la situacíón del ET en la coordenada de
reacción de la etapa donde se rompe el enlace:
- ET semejante al reactivo o al producto: enlace
C-H (D) poco roto, o muy formado en la nueva
molécula
- El ET mantiene las diferencias de Evibr:
Efecto Cinético Isótopo pequeño
- ET simétrico en la coordenada de reacción:
antiguo enlace C-H (D) medio roto, nuevo
enlace C-H (D) medio formado
- El ET no mantiene las diferencias de Evibr :
Efecto Cinético Isótopo cercano a 7

El efecto cinético teórico se puede calcular considerando que las diferencias en las energías
de activación corresponden a las diferencias en los puntos cero de los enlaces C-H / C-D
(aproximación)
A modo de ejemplo podéis ver la tabla siguiente que contiene los valores calculados
para diferentes parejas de isotopos:
Efecto cinético de isótopo
34
Nuclis H /D H / T 12C/13C 12C/14C 14N/15N 16O/18O 32S/34S
klleuger/kpesat ~ 7 ~ 13 1.04 1.07 1.03 1.02 1.01

Efecto cinético de isótopo
- Ocurre cuando el H sustituido por D está unido a un C que cambia de hibridación en el
transcurso de la reacción antes de la rds (H o D no están implicados directamente en la reacción)
Efecto cinético de isótopo secundario
- La diferencia E entre los niveles vibracionales
más bajos de los enlaces C-H y C-D es menor.
Efecto Cinético de isótopo
Secundario normal:
kH
kD
> 1Efecto Cinético de isótopo
Secundario inverso:
kH
kD< 1
- La diferencia E entre los niveles vibracionales
más bajos de los enlaces C-H y C-D es mayor.
Al pasar de C sp3 a C sp2 (enlace
C_H (D) más corto)
Al pasar de C sp2 a C sp3 (enlace
C-H (D) más largo)
(1,3 – 1) (0,7 – 1)
35
∆G‡ > ∆G‡ D H
kD < kH
sp3C-H
C-D
sp2C-HC-D
∆G‡ < ∆G‡ D H
kH < kD
sp3
sp2
C-H
C-D
C-HC-D

Efecto cinético de isótopo

Ecuación de Hammett
Aproximación donde se comparan cambios en ∆G≠ como resultado de cambios en el
medio o en la estructura de los reactivos (diferentes sustituyentes sobre un anillo
aromático) para dos reacciones similares.
También se pueden comparar cambios en ∆G≠ con cambios en ∆Gº para la misma
reacción o entre reacciones diferentes
Efectos cinéticos electrónicos. Relaciones lineales de energía libre
37
reacción o entre reacciones diferentes
Si les variaciones en el medio o en la estructura son suficientemente pequeñas, se
encuentran relaciones lineales que describen las sensibilidades relativas entre la
reacción en estudio y la reacción de referencia delante de un cambio particular.
Linear free energy relationship (LFER) ∆∆∆∆∆∆∆∆G’ = (∆∆∆∆∆∆∆∆G). cte

Efectos cinéticos electrónicos. Relaciones lineales de energía libre
Correlation of acid dissociation constants of benzoic acids with rates of basic hydrolysis of ethyl benzoates
Advanced Organic Chemistry, Part A: Structure and Mechanisms (Ed. A. Carey, R. J. Sundberg), 2007. Springer

Ecuación de Hammet
Ecuación de Hammett:
- Origen: Hammett constata que pueden
correlacionarse las elucidades de hidrólisis alcalina de
los benzoatos de etilo con las constantes de
disociación de los ácidos benzoicos con (L. P.
Hammett J. Am. Chem. Soc. 1937, 59, 96).
log = σ ρσ ρσ ρσ ρk
ko
p-NO2
m-NO2
p-Cl
p-Me
p-OMe m-NH2
Hlog
k/k
o
2.0
0.8
0.0
-0.4
39
p-NH2
-0.8 -0.4 0 0.4 0.8 1.2
-1.2
-2.0
log K/Ko
La Ecuación de Hammett solo puede aplicarse
en reacciones donde el centro reactivo está
unido a un anillo aromático que pueda estar
diferentemente sustituido (X).

Esta relación lineal indica que el cambio en la energía libre de activación en la
hidrólisis de los benzoatos de etilo a causa de la introducción de una serie de
sustituyentes, es directamente proporcional al cambio en la energía libre de
ionización causado por la misma serie de sustituyentes en los ácidos benzoicos.
La ec. de Hammett para datos de equilibrio y de velocidad:
Efectos cinéticos electrónicos. Relaciones lineales de energía libre
40
log KK0
= σρ
log kk0
= σρ

Tal y como se ha definido, el valor de σσσσ refleja el efecto del sustituyente en la
energía libre de la ionización de los ácidos benzoicos. Después se extrapola este
efecto a las energías libres de activación de las reacciones en estudio.
El efecto de cada sustituyente resulta de la combinación de diversos factores
(inductivos, de campo, resonantes)
Efectos cinéticos electrónicos. Ecuación de Hammett
41
Efectos inductivos: transmisión de los enlaces dipolares a través del esqueleto de
enlaces sigma por polarización sucesiva de cada enlace. Disminuyen con la distancia.
Efectos resonantes: polarización y redistribución de carga a través de un sistema π
conjugado. Se mantienen con la distancia.
Efectos de campo: interacción electrostática a través del espacio.

La ec. de Hammett se aplica solamente a sustituyentes en posiciones meta y para,
ya que en estas se puede suponer que los efectos son puramente electrónicos,
mientras que los sustituyentes en orto presentaran una combinación de efectos
electrónicos y efectos estéricos
Efectos cinéticos electrónicos. Ecuación de Hammett
log kk
= σρ
42
σ > 0 para sustituyentes más electronegativos que H
σ< 0 para sustituyentes menos electronegativos que H
σm = σI ; σp = σI + σR (I = inductiva; R = resonante)
log k0
= σρ

Ejemplos correlaciones tipo Hammet aplicadas a otras reacciones:
-1) Ionización de fenoles:
ρ = 2.26
Ecuación de Hammet
- Valores de ρ > 0: reacciones en las que hasta la etapa
determinante de la velocidad de reacción (rds) tiene lugar un
-Sustituyentes electrón-atrayentes: valores de σ > 0
-Sustituyentes electrón-donadores: valores de σ < 0
p-
NO
2
m-NO2
p-Cl
p-Me
p-OMe
p-CN
H
0.8 0.4 0 0.4 0.
8
1.0
1
0
2
3
4
5
6
7
8
0.6 0.2 0.2 0.6
m-Mem-OMe
p-Br
m-Brm-Cl
m-CO2Et
p-CO2Et
σ +-
m-CN
ρ = -4.54
- 2) Solvólisis de cloruros de fenildimetilcarbinol:
- Valores de 0: reacciones en las que hasta la etapa
determinante de la velocidad de reacción (rds) tiene lugar un
desarrollo neto de carga negativa (o desaparición de positiva).
- Valores de ρ < 0: reacciones en las que hasta la etapa rds
tiene lugar un desarrollo neto de carga positiva (o
desaparición de negativa).

44

Efectos cinéticos electrónicos. Ecuación de Hammett
Advanced Organic Chemistry, Part A: Structure and Mechanisms (Ed. A. Carey, R. J. Sundberg), 2007. Springer

El parámetro ρρρρ representa la sensibilidad relativa de la reacción al cambio
electrónico, es decir, a la presencia de diferentes sustituyentes.
Es importante en investigación mecanística, ya que su signo y su
magnitud permiten hacer deducciones sobre el desarrollo de carga
en el centro reactivo en el estado de transición.
Efectos cinéticos electrónicos. Ecuación de Hammett
46
ρ > 0 si la reacción es acelerada por sustituyentes electroatrayentes
(en el paso de reactivos a ET se crea carga negativa o se destruye
carga positiva)
ρ < 0 si la reacción es acelerada por sustituyentes electrodadores
(en el paso de reactivos a ET se crea carga positiva o se destruye
carga negativa)

Efectos cinéticos electrónicos. Ecuación de Hammett
Advanced Organic Chemistry, Part A: Structure and Mechanisms (Ed. A. Carey, R. J. Sundberg), 2007. Springer

Efectos cinéticos electrónicos. Ecuación de Hammett

Parámetros σσσσ+ y σσσσ- de Hammett:
σ+ se utiliza para reacciones en las que hay una interacción resonante directa entre
un sustituyente electrodador y un centro reactivo catiónico.
σ- se utiliza para reacciones en las que hay una interacción resonante directa entre un
sustituyente electroatrayente y un centro reactivo aniónico.
En estos casos, los valores habituales de σ no se ajustarían a la recta
Efectos cinéticos electrónicos. Ecuación de Hammett
49
Direct resonance interaction with cationic center
Direct resonance interaction with anionic center

Efectos cinéticos electrónicos. Ecuación de Hammett
Curvas típicas de reacciones que cambian la etapa determinante de la velocidad según los sustituyentes

Efectos cinéticos electrónicos. Ecuación de Hammett
Gráficas típicas de reacciones que cambian de mecanismo en función de los sustituyentes
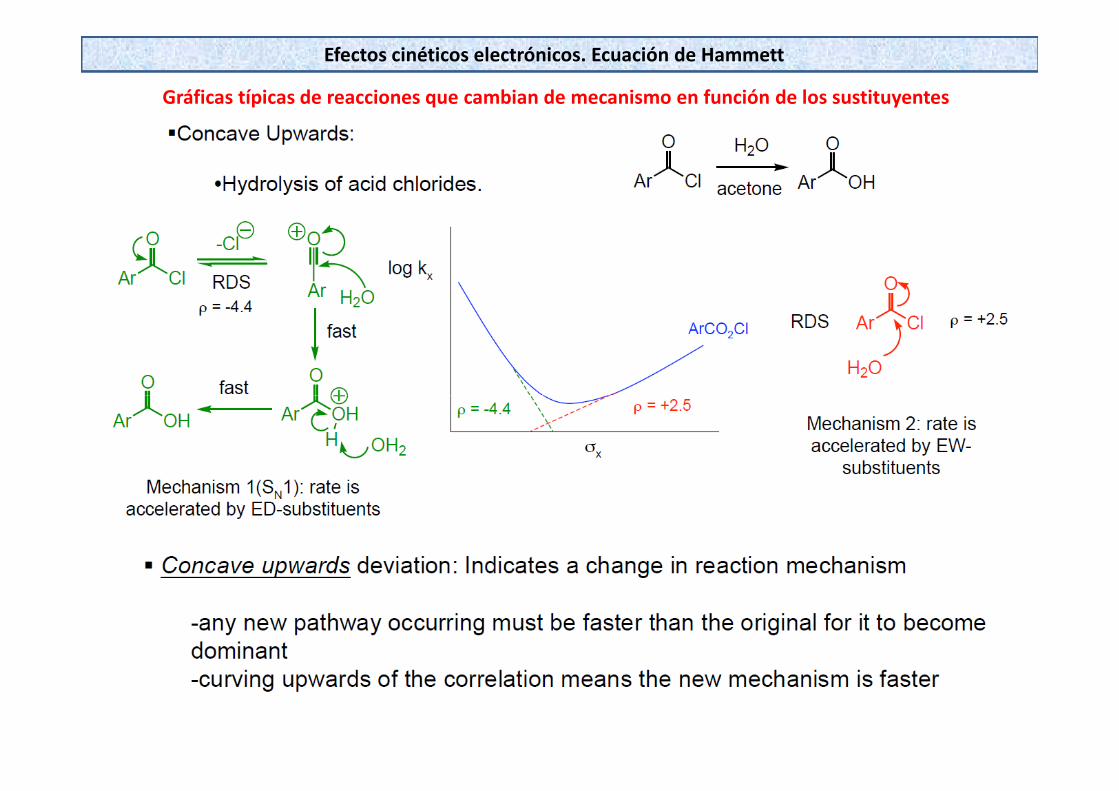
Efectos cinéticos electrónicos. Ecuación de Hammett
Gráficas típicas de reacciones que cambian de mecanismo en función de los sustituyentes

Ejemplos en investigación mecanística :
Hidròlisis bàsica de ésteres
(cambios en la etapa determinante de velocidad)
(se obtienen dos tramos rectos de pendientes diferentes)
Solvólisis de tosilatos bencílicos sustituidos
Efectos cinéticos electrónicos. Ecuación de Hammett
53
Solvólisis de tosilatos bencílicos sustituidos
(cambios de mecanismo: SN1 i SN2)
(se obtienen dos tramos rectos de pendientes diferentes)
Acetòlisis de tosilatos de ββββ-ariletilo
(cambios de mecanismo: SN2 i SN con asistencia anquimérica)
(se obtienen dos tramos rectos de pendente diferente)

Análisis de productos:
a) estructura de los productos de reacción
b) marcaje isotópico y/o experimentos de cruce
Métodos no cinéticos en el estudio de mecanismos de reacción
54
c) datos estereoquímicos

Marcaje isotópico
Proporciona una manera de conocer en que punto del producto final acaben los
átomos del producto de partida.
Una manera conveniente de determinar si el marcaje isotópico es apropiado para
resolver un problema mecanístico particular es numerar todos los átomos del
Métodos no cinéticos en el estudio de mecanismos de reacción
55
resolver un problema mecanístico particular es numerar todos los átomos del
reactivo y determinar si los diferentes mecanismos a considerar llevan a diferentes
conectividades en el producto final.
Hay que tener en cuenta las dificultades de síntesis de los reactivos marcados
isotópicamente, la asequibilidad de los isótopos requeridos y como llevar a cabo la
localización del marcaje en el producto final.

Isótopos más utilizados en marcaje: deuterio, tritio, carbono-13, carbono-14
Localización de D per EM o bien por ausencia de señal en RMN
Tritio y carbono-14 son radioactivos y se pueden detectar en base a medidas de
Métodos no cinéticos en el estudio de mecanismos de reacción
56
radioactividad, pero se requiere proceso degradativo para localizar en que punto
concreto es encuentra el marcaje. Puede resultar largo y laborioso.
El carboni-13 se puede localizar por RMN (tiene momento magnético nuclear),
dará una señal de mayor intensidad en la posición marcada. No se requiere
proceso degradativo

Técnica útil cuando se formen intermedios de reacción más simétricos que los
reactivos. Ejemplo de preparación de anilinas a partir de clorobencenos y amiduro
potásicos.
Ejemplo de aplicación del marcaje isotópico a la transposición de Claisen de éteres
Métodos no cinéticos en el estudio de mecanismos de reacción
57
alilarílicos. Mecanismo inter o intramolecular?
Experimentos de cruce para distinguir mecanismos inter- e intramoleculares en
transposiciones unimoleculares (reactivos doblemente marcados o sustratos
marcados de manera diferente, experimentos de incorporación). Ejemplos de
transposición de Fries de ésteres fenólicos y transposiciones aniónicas 1,2.

Métodos no cinéticos en el estudio de mecanismos de reacción
Marcaje isotópico
Propuesta de mecanismo para la isomerización del Z-1-fenilbutadieno al E-dieno
correspondiente en medio ácido:
Experimento de marcaje isotópico: uso de D+ en D2O
Mecanimo postulado:

Métodos no cinéticos en el estudio de mecanismos de reacción
Marcaje isotópico
SNAr via Bencino. Marcaje radioactivo con 14C
Degradación controlada: 50% marcaje en C1 I 50% en los dos C orto idénditoc

Métodos no cinéticos en el estudio de mecanismos de reacción
Experimentos de cruce
Isomerización de
sulfuros alílicos iniciada
por luz
Experimento de cruce: seguido por espectroscopía de masas
1 1 2
Propuesta de mecanismo: reacc. radicalaria en cadena, iniciada por ArS·

Datos estereoquímicos
a) Obtener información mecanística en procesos de estereoquímica establecida
b) Establecer la estereoquímica de nuevos procesos
Estudios de racemización en reacciones con sustratos quirales para determinar
Métodos no cinéticos en el estudio de mecanismos de reacción
61
Estudios de racemización en reacciones con sustratos quirales para determinar
formación de intermedios aquirales (Ej. SN1)
Correlación de configuraciones: determinación de la configuración absoluta
desconocida de una molécula determinada mediante su conversión en otros
compuestos de configuración conocida. Las reacciones implicadas en esta
transformación han de ser estereoquímicamente definidas (hay que saber si
transcurren con retención o inversión de configuración).

Intermedios de reacción
Corresponden a mínimos locales en el perfil de reacciones multietapa,
tienen vida media corta
es decir, si el intermedio se acumula y evoluciona lentamente hacia Si
Métodos no cinéticos en el estudio de mecanismos de reacción
Reactivos Intermedio Productosk1 k2
K1 > K2
62
productos, existe la posibilidad de detectarlo y/o aislarlo. Si
Cualquier intermedio real aislado de una reacción interrumpida ha de dar los
productos de reacción cuando se sitúa en las condiciones originales.
Si las dos k1 y k2 tienen valores grandes, no se podrán aislar, pero quizá se podrán
detectar espectroscópicamente (UV-Vis, IR, RMN, EPR, …) o atrapar.
Intermedios comunes: carbaniones, carbocationes, radicales, carbenos, nitrenos,
arinos
K1 > K2
![Ciencias Sociais Tema 2 v2 - moodle03.s3.amazonaws.commoodle03.s3.amazonaws.com/UNIFICADO/[10001]CIENCIAS_SOCIAIS/[4]Tema... · 5 Ciências Sociais | Tema 2 2014 de elementos culturais,](https://img.pdfslide.tips/doc/110x75/5bda65e409d3f2e2478c9386/ciencias-sociais-tema-2-v2-moodle03s3-10001cienciassociais4tema.jpg)















![1-ESO Estudiante v2[1]](https://img.pdfslide.tips/doc/110x75/577cd9331a28ab9e78a2f897/1-eso-estudiante-v21.jpg)









![I [1] Tema Inv V2](https://img.pdfslide.tips/doc/110x75/55cf8c545503462b138b7cfc/i-1-tema-inv-v2.jpg)
![Claudelina machuca v2[1]](https://img.pdfslide.tips/doc/110x75/58a6e1b91a28abef698b6811/claudelina-machuca-v21.jpg)

