Embed Size (px)
Citation preview

Antimigrañosos &
AntigotososEquipo 11
Paula Ipiña ÁlvarezManuel Alejandro Jiménez Pérez
Oscar Antonio Ledesma OrtaCarlos Andrés Leija Hernández
Alán Giresse Lozano AlanísBrandon Yair Lozano Valdez
Salvador Armando Luna Flores

Antimigrañosos

Migraña Tiene diversas causas como:• La dieta• El embarazo • Estrés• Fiebre• Origen neuronal

La migraña es un fenómeno neuronal de hiperexcitabilidad paroxística que
implica a las neuronas del sistema trigémino-vascular mediado por
anomalías en los canales iónicos responsables de los potenciales de
acción neuronales, disfunciones en la producción de energía o del control
tónico que ejercen los sistemas neuronales aminérgicos (serotonina,
adrenalina, dopamina).

Migraña sin aura (migraña común) Migraña con aura (clásica):
◦ Fotofobia, hiperacusia, poliuria y diarrea.
El aura también puede presentarse sin cefalea subsiguiente.
Manifestaciones variables

Tratamiento con Agonistas 5-HT• Triptános.
• Derivados del hongo de centeno (Ergot y sus alcaloides).
• Fármacos que actúen en 5-HT 1D. *Depende de la fisiopatología de la migraña

Clasificación

Sumatriptán Vía subcutánea.
Rizatriptán Mejor efecto para el ataque inmediato.
Zolmitriptán Mayor afinidad. Profármaco 5-HT 1D y 1B.
NO COMBINAR con IMAO, porque es su lugar de metabolización.
Triptános

Son eficaces en el tratamiento agudo de la migraña (con o sin aura).
NO debe de prescribirse junto con derivados de Cornezuelo de Centeno.
Naritriptán
• No se metaboliza por MAO (CYP3A4).
• V ½ MÁS LARGA.
Eleptriptán
• Su metabolito activo le da un efecto más prolongado

Alcaloides de Cornezuelo de Centeno Antagonistas de la Serotonina
Ergotamina (Vasoselectivo) Ketanserina
Ergonovina*
Bloquean α1 y producen vasoconstricción
Tienen receptores:• Adrenérgicos
• Dopaminérgicos • Serotoninérgicos c

Si se presenta crisis migrañosas más de 3 veces al
mes.
Se debe comenzar lo más pronto
posible.
Tip
os
de t
rata
mie
nto
Profiláctico
Farmacológico
No Farmacológico
Abortivo
Farmacológico
No Farmacológico

Es un cuidado paliativo, no alivia la migraña, disminuye su frecuencia de aparición.
Es un tratamiento diario y prolongado (6 meses aprox.)
Profiláctico farmacológico
Familia Fármaco
B-bloqueadores Propanolol
Inhibidores de canales de Ca++
Nicardipina y Nimodipina
Antidepresivos Antidepresivos tricíclicos
Antiepilépticos Valproato y Topiramato
AINES Aspirina y Naproxeno
Antiserotoninérgicos MetisergidaSaltar explicación

Fármaco: Beta-bloqueadores.Ejemplo: Propanolol.Indicaciones: Migrañas secundarias a enfermedades tratadas con estos medicamentos.Contraindicaciones: No administrar en pacientes asmáticos, con depresión o diabetes.Efectos adversos: Disfunción eréctil, broncoconstricción, bradicardia, fatiga, alteraciones del sueño.
Fármaco: Bloqueadores de canales de Ca++.
Ejemplo: Nicardipina y Nimodipina.
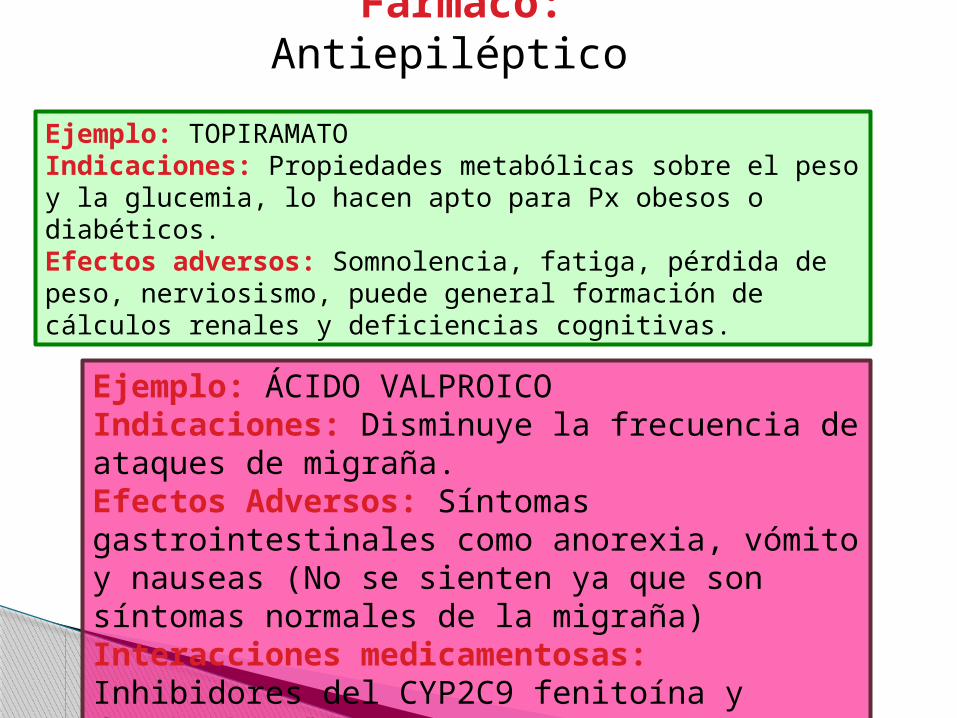
Ejemplo: ÁCIDO VALPROICO Indicaciones: Disminuye la frecuencia de ataques de migraña.Efectos Adversos: Síntomas gastrointestinales como anorexia, vómito y nauseas (No se sienten ya que son síntomas normales de la migraña)Interacciones medicamentosas: Inhibidores del CYP2C9 fenitoína y fenobarbital.
Ejemplo: TOPIRAMATO Indicaciones: Propiedades metabólicas sobre el peso y la glucemia, lo hacen apto para Px obesos o diabéticos.Efectos adversos: Somnolencia, fatiga, pérdida de peso, nerviosismo, puede general formación de cálculos renales y deficiencias cognitivas.
Fármaco: Antiepiléptico

Fármaco: AINESEjemplo: NAPROXENO Y ASPIRINAUso terapeutico: Efectivo en tratamiento profiláctico perimenstrual de migraña.
Fármaco: AntiserotoninérgicosEjemplo: MetisergidaEfectos adversos: MUCHOS efectos secundarios

Inyecciones pericraneales de Botox. Ejercicios de relajación (música, masajes, sonidos
relajantes, aromaterapía).
Profiláctico no farmacológico.

Abortivo farmacológico.
Agonistas de los receptores 5-HT
Triptános• Primera generación• Segunda generación
Alcaloides del Cornezuelo
ErgotaminaErgovina
BromocriptinaLSD

Vía de administración
Alcanzar CM
Parenteral 12 min
Intramuscular 60 min
Oral 90 min
Agonista de receptores 5-HT 1B y 1D afinidad más baja por F, A y E, además de receptores adrenérgicos, dopaminérgicos y muscarínicos. Semivida en plasma 2 horas.
Alivia la cefalea, náuseas, fotofobia y fonofobia.
Sumatriptán
Primera
generaci
ón

Efectos adversos 20-40% cefalea de intensidad moderada o grave tras la primera dosis, es necesaria una segunda dosis (Velázquez).◦ Los pacientes rara vez sugieren algo más que ardor en el sitio de
inyección. Vía oral; astenia, fatiga, rubor, sensación de presión, tirantez o dolor
en el tórax, cuello o quijada.
Pacientes con antecedentes de arteriopatía coronaria isquémica o vasoespástica, apoplejía, enfermedad vascular cerebral, HTA u otras enfermedades cardiovasculares.
Efectos Adversos
Contraindicaciones

Nueva generació
n
Fárm
ac
o
• Naratriptán• Zolmitriptán• Rizatriptán• Eleptriptán• Frovatriptán• Amlotriptán• Avitriptán
Ventajas principalmente FARMACOCINÉTICAS
Mayor biodisponibilidad.Semivida plasmática más
larga.Absorción más rápida.Mayor lipofilia.
• Provocan vasoconstricción de arterias coronarias.
• Su uso para tratar la migraña está contraindicado. Su uso es en tratamiento de
enfermedades cerebrovasculares, enfermedades isquémicas cardiacas e hipertensión no controlada.

El primero en descubrirse fue la Ergotamina, seguido de Ergonovina.
Sintéticos: Bromocriptina (disminuye la secreción de prolactina) y LSD.
Alcaloides del cornezuelo.

Se limita su uso a Px con jaquecas frecuentes y moderadas o poco frecuentes muy intensas.
Tanto Ergotamina como Ergonovina van conjugadas con cafeína en forma de tabletas orales y supositorios.
Presentación Dosis
Tabletas sublinguales 2mg por intervalos de 30min hasta alcanzar los 6 mg en 24h o 10mg en 1 semana
Aerosol nasal .5mg por narina cada 15 min hasta completar un total de 2mg.
Solución Inyectable 1mg inicial y una segunda aplicación parar lograr 2mg IV o 3mg IM ó SC

Vía oral 10% presentan náuseas y vómitos. Que aumentan al doble en vía parenteral.
Debilidad en piernas y dolores musculares a veces intensos.
Ardor o parestesias en pies y manos.
• No usar en pacientes embarazadas, causa aborto.• En caso de sobredosis se trata con anticoagulantes,
dextrano de bajo peso molecular y vasodilatadores. • No administrar si no hasta 24 hrs antes o tras un
triptano u otro fármaco vasoconstrictor.
Efectos Adversos
Contraindicaciones

Mecanismo de acción
La importancia de la disfunción vascular y neurológica en la jaqueca sigue siendo controversial
Implica la capacidad de los receptores 5-HT1B/1D para provocar contracción de los vasos sanguíneos intracraneales. Una serie de acontecimientos desconocidos provoca la dilatación anormal de las anastomosis arterio-venosas de la cabeza generando hipoxia e isquemia cerebral.
En base a esto el fármaco efectivo contra jaqueca cerraría las desviaciones y restablecería la circulación cerebral.
Los receptores 5-HT1B y 5-HT1D modulan la liberación de neurotransmisor a partir de las terminales neuronales.
Los agonistas de los receptores 5-HT1 pueden bloquear la liberación de neuropeptidos proinflamatorios.
Triptanos
1era Hipótesis
2nda Hipótesis

Sumatriptán
Zolmitriptán Naratriptán Rizatriptán
Vía subcutánea (concentración
plasmática máxima)
12 min(Biodisponibilida
d 97%)- - -
Vía oral (concentración
plasmática máxima)
1 o 2 hrs (biodisponibilida
d 14 a 17%)
1.5 a 2 hrs (biodisponibilida
d 40%)
2 a 3 hrs (biodisponibilida
d de 70%)
1 a 1.5 hrs (biodisponibilidad
45%)*Existe una
presentación oral que se absorbe más
lentamente
Semivida de eliminación 1 o 2 hrs De 2 a 3 hrs 6 hrs -
Vía metabólica
Se metabólica por medio de la
MAO-A, sus metabolitos se
excretan en orina
Se convierte a un metabolito N-desmetilado
activo
50% de la dosis se excreta inalterada en la orina, 30% se
extrema como producto de la
oxidación
Desanimación oxidativa por
medio de la MAO-A
Farmacocinética

Ergotamina• Debido a su gran metabolismo de primer paso, la
administración oral genera una una concentración sistémica indetectable
• Después de su administración vía rectal su biodisponibilidad es mayor
• Se metabólica en hígado por vías desconocidas• 90% de los metabolitos se excretan por bilis• Provoca vasoconstricción que dura 24hrs o más
Ergonovina y metilergonovina• Se absorben rápidamente por vía oral,
alcanzando su concentración plasmática máxima a los 60 a 90 minutos
• La semiárida de la metilergonovina en el plasma varía de 30 min a 2hrs
Farmacocinética

Agonistas de los receptores 5-HT. Son selectivos en los receptores 5-
HT subfamilia 1 subtipo B y D. Y sus efectos están limitados a
esta familia de receptores y tienen poco afinidad o nula hacia otros receptores.
Capaces de disminuir las nauseas y vomito de la migraña.
Son mas selectivos que los alcaloides del Ergot.
Causa constricción de los vasos sanguíneos intracraneales.
Efectos Farmacológicos

Eficaces en el tratamiento agudo de la migraña con o sin aura, pero no en su profilaxis
Se debe iniciar el tratamiento tan pronto comience la migraña.
Indicaciones terapéuticas

Con base al mecanismo de acción del sumatriptán y otros agonistas 5-HT1B/1D probablemente la acción de los alcaloides del cornezuelo de centeno a nivel de los receptores 5-HT1B/1D es mediar sus efectos inmediatos contra la jaqueca.
Alcaloides de Cornezuelo
Mecanismo de acción

Por sus efectos variados y complejos sus usos terapéuticos son limitados.
Es un agonista-antagonista parcial de los receptores adrenérgicos, dopaminergicos y serotoninérgicos.
Se utiliza para el tratamiento de la migraña pero por sus múltiples efectos es difícil determinar el mecanismo de acción.
Probablemente los receptores 5-HT de la subfamilia 1 subtipo B y D median la acción antimigrañosa.
Ergotaimina y sus alcaloides
Efectos Farmacológicos

Metisergida (derivado del ergot) es antagonista de el receptor 5-HT y se utiliza como tratamiento profiláctico de la cefalea magañosa.
Su uso se restringe a pacientes con ataques de migraña moderados y frecuentes o graves pero poco frecuentes.
Se debe administrar tan pronto sea posible después de iniciar una cefalea.
Indicaciones terapéuticas

Antigotosos
Tratamiento de la gota aguda. Se considera tratamiento de segunda línea.
Colchicina

Tiene efectos antimitóticos, interfiriendo con la formación de micro túbulos y el huso.
Torna mas rígidas las membranas y disminuye la secreción de factores quimotácticos.
Inhibe la liberación de células cebadas de gránulos que contienen histamina, la secreción de insulina y el movimiento de gránulos de melanina en melanóforos.
Mecanismo de Acción

La temperatura corporal
La sensibilidad a depresores centrales
Deprime el centro respiratorio
Mejora la respuesta a fármacos simpaticomiméticos.
Contrae vasos sanguíneos, e induce hipertensión.

Se absorbe con rapidez. Concentración plasmática max. : ½ a 2 hrs. 50% unido a proteínas plasmáticas. Su metabolismo incluye desacetilación
hepática. 10 a 20% se excreta en orina. La t ½ es de 9 hrs.
Farmacocinética & Metabolismo

Gastropatía hemorrágica
.
Efectos tóxicos
Primeros signos de toxicidad inminente
Nauseas, vómitos, diarrea, dolor
abdominal
Intoxicación Aguda
Proteinuria, hematuria y
necrosis tubularIntoxicación Grave

Por V.I. da complicaciones importantes de la medula ósea y renales.
Uso prolongado: Agranulocitosis, miopatía proximal,
azoospermia y nefropatía gotosa. Toxicidad se acompaña de:
Supresión de la medula ósea, leucocitosis, trombocitopenia.

Gota aguda En 2/3 de los pacientes es eficaz si se
administra dentro de las 24 horas iníciales. Después de 12 hrs remite el dolor, la
tumefacción el enrojecimiento y desaparecen por completo a las 48 a 72 hrs.
Aplicaciones terapéuticas
Dosis oral es de: 0.6 mg cada hora por un total de 3 dosis.
No debe repetirse por más de 7 días.

Se usa en el Tx de la hiperuricemia de pacientes con gota.
Alopurinol
Mecanismo de Acción• Inhibidor competitivo de xantinooxidasa*
concentración de ácido úrico• Se convierte en oxipurinol (metabolito
activo).• Disuelve los tofos.• + Colchicina en 1er mes.• Aumenta excreción de xantinas.• Evita formación de cálculos y neuropatías.

Se absorbe con relativa rapidez. Vía oral. Concentraciones maxima en plasma: 60 a 90
min. 20% se excreta en heces en 48-72 hrs. 10 - 30% se excreta sin modificar por orina. El resto se metaboliza en oxipurolol.
Farmacocinética
• T ½ del alopurinol es de 1 a 2 hrs y la de oxipurinol es de 18 a 30 hrs.
• Ninguno de los compuestos se une a proteínas del plasma.

Aumenta la t ½ del Probencid e incrementa su efecto uricosúrico.
Inhibe la inactivación enzimática de la mercaptopurina por oxidasa de xantina.
Puede interferir con la inactivación hepática de la warfarina.
El riesgo de supresión de la medula ósea es mayor cuando se administra con fármacos citotóxicos.
Interacciones farmacológicas
Hiperuricemia primaria de la gota y secundaria a policitemia verdadera.
Metaplasia mieloide. Lisis tumoral aguda.
Aplicaciones terapéuticas

Contraindicado: en Px con efectos adversos o reacciones de hipersensibilidad al medicamento.
Objetivo: reducir las concentraciones plasmáticas de ac. úrico a menos de 6 mg/dl.
Dosis inicial de 100 mg, se aumenta en intervalos de 100 mg a intervalos semanales.
Es útil en pacientes con Sd. De Lesch – Nyhan.

Reacciones de hipersensibilidad.
Erupción pruriginosa, eritematosa o maculo-papular.
Necro lisis epidérmica toxica (rara vez).
Leucopenia. Eosinofília. Hepatomegalia.
Efectos Adversos

FebuxostatMecanismo de
AcciónEs un inhibidor no purinico de la xantina oxidasa, el febuxostat forma un complejo estable con la enzima tanto reducida como oxidada e inhibe la función catalítica en los dos estados.

Se absorbe con rapidez y alcanza concentraciones plasmáticas en 1 y 1.5 horas después de la dosis.
El hidróxido de magnesio y el de aluminio retrasan la absorción una hora , al igual que la comida.
Metabolizado en forma extensa mediante oxidación por CYP1A2, 2C8, 2C9 y conjugación con enzimas que no son las CYP a través de las UGT 1-A1, 1-A3, 1-A9, 2-B7.
Tiene una semivida de 5/8 horas.
Eliminado por vía hepática y renal.
Absorción, Distribución y Eliminación

Autorizado para los pacientes hiperúricos con crisis de gota, pero no se recomienda para tratar la hiperuricemia asintomática.
40 mg de Febuxostat al día disminuyen el acido úrico sérico a concentraciones similares que 300 mg de alopurinol al día.
Se debe iniciar el tratamiento con 40 mg al día e incrementarse la dosis si la concentración sérica de acido úrico elegida como objetivo no se alcanza al cabo de dos semanas.
Aplicaciones terapéuticas

Los mas frecuentes fueron las anomalías de la función hepática, nausea, artralgias y exantema.
El tratamiento profiláctico concomitante con un NSAID o Colchicina suele ser necesario.
Hubo una tasa de infarto del miocardio y apoplejía numéricamente mas alta en pacientes con Febuxostat que con Alopurinol.
Efectos Adversos

No se debe utilizar en los pacientes que reciben Azatioprina , Mercatopurina o Teofilina.
Interacciones farmacológicas

RASBURICASE• Es una uratooxidasa recombinante que cataliza la
oxidación enzimática de acido úrico en el metabolito soluble e inactivo alantoina.
• Indicado para el tratamiento inicial de las concentraciones altas de acido úrico en los pacientes pediátricos con leucemia, linfoma y neoplasias malignas solidas que están recibiendo tratamiento antineoplásico con la expectativa de que produzcan lisis del tumor e hiperuricemia importante.

Producido por la cepa Saccharomyces Cerevisiae pero se dificulta su eficacia por la producción de anticuerpos contra el fármaco.
Rasburicase se ha relacionado con hemolisis en los pacientes con deficiencia de la enzima glucosa-6-fosfato deshidrogenasa (G6PD),metahemoglobinemia, insuficiencia renal aguda y anafilaxia.
Produce degradación enzimática de acido úrico en muestras de sangre y se necesita un manejo especial para evitar las concentraciones falsamente bajas de acido úrico en pacientes que reciben el fármaco.
Efectos Adversos
*Vomito, fiebre, nausea, cefalea, dolor abdominal, estreñimiento, diarrea y mucositis.

Excreción de ácido úrico.Transportador bidireccional urato-anión orgánico/inorgánico.
Los fármacos uricosúricos compiten con el urato por el transportador del borde en cepillo inhibiendo su resorción.
◦ Dosis bajas Disminuye la excreción ◦ Dosis altas Incremento de la excreción
Uricosúricos

Derivado del ácido benzoico.
Altamente liposoluble.
Para el tratamiento de la gota.
Probenecid


Efectos
Adversos Comunes
• 2% presenta irritación gastrointestinal. 2-4% reacciones alérgicas.
• Contraindicado en pacientes con antecedentes de úlcera péptica.
• Ineficiente en pacientes con insuficiencia renal.
• Sobredosis de Probenecid, origina estimulación del SNC, convulsiones y muerte.
• Inhibe la secrecion renal de los metabolitos glucuronidos inactivos de AINES, aumentando sus concentraciones en plasma.

Uso
Terapéutico
• Vía oral, dosis inicial de 250 mg dos veces al día, a la semana se incrementa a 500-1000mg dos veces al día.
• El tratamiento aumenta el urato en la orina, por lo que se deben consumir muchos líquidos, para evitar formación de cálculos renales.
• Contraindicado en pacientes con gota, o producción excesiva de ácido úrico.
• Se usa junto con la Colchicina para evitar precipitar un ataque de gota.
• El Probenecid, se creo con el fin de retardar la excreción de penicilina, y aun se usa para esto.

Se utiliza en Europa. Inhibidor activo y reversible del intercambiador
de urato-anión en el túbulo proximal. Activo en pacientes con insuficiencia renal, y
pacientes alérgicos. Benzbromarona + Alopurinol = disminuyen las
concentraciones séricas de ácido úrico.
Benzbromarona

Por vía oral alcanza su concentración máxima plasmática a las 4 hrs.
Se excreta por la bilis. Es eficaz en una sola dosis diaria de 40 a 80
mg. Es mas potente que la mayoría de los
uricosúricos.