Embed Size (px)
Citation preview

1
_________________________________________________
SCUOLA DI SPECIALIZZAZIONE
IN GASTROENTEROLOGIA
“CORRELAZIONE TRA POLIMORFISMO DEI
GENI cagA e vacA di HELICOBACTER PYLORI
E VARIETA’ ISTOLOGICA DEL CARCINOMA GASTRICO”
RELATORE
CHIAR.MO PROF. SANTINO MARCHI
CANDIDATA
DR.SSA CARMELA STEFANIA LARDIELLO
ANNO ACCADEMICO 2014/2015

2
INDICE
Considerazioni generali
Helicobacter pylori ..........................................................................5
1.1. Introduzione storica ..................................................................................... 5
1.2. Cenni di epidemiologia e modalità di trasmissione.................7
1.3. Caratteristiche generali del batterio ............................................................... 8
1.4. Caratteri patogenetici particolari e fattori di virulenza ................................ 10
a) Lipopolisaccaridi................................................................................................. 9
b) La citotossina vacuolante e il gene vacA.......................................................... 11
c) CagA e l’isola di patogenicità cag ..................................................................... 16
1.5 Meccanismi pro-infiammatori, immunità e produzione di radicali liberi ...... 20
1.6. Patologie gastrointestinali ............................................................................ 24
a) Gastrite cronica................................................................................................ 24
b) Ulcera peptica .................................................................................................. 27
c) Dispepsia........................................................................................................... 28
d) Malattia da reflusso gastroesofageo e carcinoma esofageo ........................... 29
e) Adenocarcinoma gastrico................................................................................. 29
f) Linfoma MALT gastrico ..................................................................................... 34
1.7. Manifestazioni extra-digestive...................................................................... 36
a) Apparato cardiovascolare ................................................................................ 36
b) Apparato respiratorio ...................................................................................... 38
c) Sistema nervoso ............................................................................................... 39
d) Sistema endocrino............................................................................................ 40

3
e) Manifestazioni ematologiche........................................................................... 41
f) Manifestazioni reumatologiche ........................................................................ 43
g) Cute .................................................................................................................. 44
h) Tessuto osseo….……………………………….……………………………………………………….…45
i) Patologia urologica............................................................................................ 46
j) Patologia ginecologica....................................................................................... 46
k) Infertilità........................................................................................................... 47
1.8. Diagnosi di infezione ..................................................................................... 47
a) Tecniche non invasive ...................................................................................... 49
b) Tecniche invasive ............................................................................................. 51
Infertilità ....................................................................................... 51
2.1. Definizione .................................................................................................... 51
2.2. Infertilità femminile ...................................................................................... 52
2.3. Infertilità maschile: ....................................................................................... 55
2.3.1. Introduzione .............................................................................................. 55
2.3.2. Cause principali.......................................................................................... 63
a) Fattori ambientali............................................................................................ 63
b) Alterazioni anatomiche.................................................................................... 64
c) Difetti genetici .................................................................................................. 65
d) Anomalie dello spermatozoo........................................................................... 68
2.3.3. Ruolo delle infezioni................................................................................... 69
Associazione con Helicobacter pylori……. ............................................................ 70
3. Ormoni regolatori dell’appetito ................................................. 72
3.1. Ghrelina......................................................................................................... 72
3.2. Obestatina..................................................................................................... 75
3.3. Altri ormoni regolatori dell’appetito............................................................. 77

4
3.4. Ormoni dell’appetito ed Helicobacter pylori................................................ 79
3.5. Ormoni dell’appetito e riproduzione ........................................................... 82
Parte sperimentale
4. Scopo della tesi ................................................................................................ 86
5. Risultati ................................................................................................... 88
7. Discussione....................................................................................................... 90
8. Conclusioni .............................................................................................. 93
Bibliografia ........................................................................................................... 94

5
Considerazioni generali
1. Helicobacter pylori
1.1 Introduzione storica
Nel 1982, Barry James Marshall (Kalgoorlie,
30 settembre 1951) e J. Robin Warren
(Adelaide, 11 giugno 1937) si trovarono di
fronte alla scoperta che nel 2005 valse loro il
premio Nobel per la medicina.
I due ricercatori australiani dimostrarono
mediante osservazione di campioni di
mucosa gastrica prelevati da pazienti affetti
da gastrite e ulcera, l’infezione da parte di un microorganismo che inizialmente,
a causa delle analogie morfostrutturali con i batteri del genere Campylobacter e
per il particolare tropismo, fu denominato Campylobacter pyloridis (poi corretto
in Campylobacter pylori). Il termine Helicobacter comparve per la prima volta
nel 1989, proprio per la necessità di una migliore definizione tassonomica di
questo e di altri batteri con analoghe caratteristiche morfologiche (Helicobacter
mustelae, Helicoabacter nemestrinae, Helicobacter felis) ritrovati nella mucosa
gastrica di alcuni animali (furetto, scimmia, cane e gatto). Con pylori si intende,
invece, la principale sede di isolamento del batterio (1).
La scoperta di agenti patogeni all’interno della mucosa gastrica ha, però, radici
più antiche. Alla fine del 1800 Giulio Bizzozero (Varese, 20 marzo 1846 –
Torino, 8 aprile 1901) osservò nello stomaco di alcuni cani e gatti la presenza di
particolari microorganismi a forma di spirale. Il 18 marzo del 1892 Bizzozero
riferì della sua scoperta alla Regia Accademia di Medicina di Torino, e nello
stesso anno descrisse alcune caratteristiche principali del batterio, tra cui la
Figura 1 – I premi Nobel Barry J. Marshall e J.
Robin Warren.

6
presenza dei flagelli e l’interessamento delle
ghiandole del fondo gastrico e del piloro (18).
Tre anni dopo, le tesi di Bizzozero furono
supportate da un medico tedesco, Hugo Solomon,
che descrisse la presenza di batteri spiraliformi
nella mucosa gastrica di cane e la possibilità di
isolare il patogeno e infettare cavie sane.
All’inizio del ventesimo secolo per la prima volta
vennero isolati batteri spiraliformi dalla mucosa
gastrica di pazienti affetti da carcinoma gastrico, merito di un altro ricercatore
tedesco, Krienitz, mentre nel 1906 Balfour li osservò in lesioni gastriche ulcerate
(2).
Tutte queste evidenze andavano nettamente in controtendenza rispetto all’assunto
comune secondo il quale la mucosa gastrica non potesse essere sede di alcuna
infezione batterica, a causa dell’ambiente ritenuto troppo avverso per la
colonizzazione. Questo dogma fu proprio infranto da Marshall e Warren,
mettendo in relazione l’infezione da parte di questi organismi e le varie patologie
del tratto superiore del sistema gastrointestinale, dimostrando con le loro
pubblicazioni il rapporto esistente tra la colonizzazione da parte di questi non
ancora indentificati batteri flagellati e la patogenesi di gastrite ed ulcera. Marshall
e Warren dimostrarono che il batterio rispettava i primi due postulati di Koch (era
presente nella mucosa gastrica del paziente affetto dalla patologia in esame,
ovvero la gastrite, e poteva essere isolato e coltivato) e per soddisfare il terzo e
quarto postulato (ovvero l’instaurarsi di un quadro patologico inoculando l’agente
in un soggetto sano e la possibilità di poter isolare nuovamente il batterio nel
paziente infettato sperimentalmente), lo stesso Marshall utilizzò sé stesso come
cavia (3). Oggi l’Helicobacter pylori è riconosciuto come principale causa delle
affezioni delle alte vie digestive e nel 1994 l’OMS lo ha inserito tra gli agenti
carcinogeni di primo livello, data la sua stretta correlazione con la patologia
neoplastica dello stomaco.
Figura 2 - Giulio Bizzozzero

7
1.2 Epidemiologia e modalità di trasmissione
L’infezione da Helicobacter pylori è un evento estremamente frequente, tanto da
colpire il 50% della popolazione mondiale (paradigmatico è il caso dell’aerea
metropolitana di Mosca in cui l’infezione da Helicobacter pylori ha un tasso di
incidenza dell’88%) (8). Da quanto emerge da numerosi studi, la prevalenza
dipende dall’area geografica, dall’età, dalla razza e dallo status socio-economico.
In particolare il tasso di infezione nelle aree in via di sviluppo è maggiore rispetto
alle regioni più industrializzate: in uno studio condotto nel villaggio di Linqu,
nella provincia di Shandong, in Cina, su 98 bambini esaminati ben il 70% era
positivo per l’infezione da Helicobacter pylori (9). Un esempio, invece, di come
le condizioni socio-economiche incidano sul rischio di infezione proviene dalla
città di Lipsia, in Germania, in cui è stato osservato un aumento della prevalenza
in gruppi di bambini d’età scolare provenienti da famiglie con basso tenore di
vita. Questi sono solo due esempi della molteplicità di studi effettuati a riguardo
(10).
Un altro fattore importante è la familiarità: la presenza di un parente prossimo
affetto da Helicobacter pylori aumenta notevolmente il rischio di infezione
(soprattutto per i bambini) (11), mentre in alcuni studi viene considerata la
possibilità di trasmissione da parte di un coniuge all’altro (12).
Il grafico soprastante (figura 3) (15) riporta un altro interessante dato: nelle
regioni in via di sviluppo c’è una maggiore prevalenza dell’infezione in
popolazioni di giovane età rispetto alle aeree geografiche sviluppate. Come
riportato sul sito www.gastronet.it, questa discrepanza è dovuta al cosiddetto
“effetto coorte”: i
soggetti residenti in
aeree a maggior
tenore di vita hanno
probabilmente vissuto
durante l’infanzia in
condizioni socio-
economiche più basse
rispetto al presente.
Figura 3 - Prevalenza dell'infezione per età di insorgenza

8
Ciò spiega da un lato quanto
sopra detto, dall’altro la
prevalenza più bassa nelle fasce di
età più giovani.
Per quanto riguarda la
trasmissione, nei paesi più
avanzati prevale la trasmissione
orale-orale. In alcuni studi è stato
possibile evidenziare la presenza
del batterio nella saliva di pazienti
affetti mediante metodiche di PCR-assay o isolando il batterio; altri, invece,
hanno dato risultati contrastanti. Particolare attenzione è stata rivolta verso la
placca dentaria: in alcuni studi è stato possibile isolare il batterio dalla placca, ed
è stato anche dimostrato che l’associazione della terapia eradicante con
trattamenti odontoiatrici atti all’eliminazione della placca risulta essere più
efficace rispetto alla sola terapia anti-Helicobacter (13-14). Un’altra possibile via
di trasmissione interumana è la via gastro-orale, da parte di individui positivi
all’Helicobacter pylori e portatori di reflusso gastro-esofageo o con episodi di
rigurgito.
La trasmissione oro-fecale avviene soprattutto in aeree geografiche meno
sviluppate, soprattutto attraverso l’approvvigionamento idrico da fonti
contaminate.
Esistono, inoltre, studi che prendono in considerazione una possibile trasmissione
zoonotica (tramite la saliva, il vomito o le feci di gatti infetti) e iatrogena
(attraverso metodiche endoscopiche) (16).
1.3 Caratteristiche generali del batterio
L’Helicobacter pylori si presenta come un bacillo Gram-negativo, non sporigeno,
flagellato, della lunghezza di 2.25-5 µm e con un diametro trasverso tra 0.5 e 1.2
µm. (1). Caratteristicamente il batterio si presenta ricurvo con conformazione
elicoidale (da cui il nome), ma in culture di lunga data o esposte all’aria il batterio
Figura 4 - Helicobacter pylori visto in microscopia
elettronica

9
può assumere una forma coccoide, aspetto che si verifica in ambienti di crescita
sfavorevoli (4).
Le condizioni ottimali di coltura sono ad una temperatura di 37° e a basso
contenuto di ossigeno (5-15%, il batterio infatti è microaerofilo), su agar-sangue o
cioccolato, formando dopo circa 3-4 giorni (massimo 8 giorni) colonie dalle
dimensioni tra 0,5 e 1 mm. (4-5).
Sotto il profilo biochimico, caratteristica peculiare del batterio è la sua capacità di
scindere l’urea. Proprio per questa proprietà possiamo dividere le specie di
Helicobacter conosciute in due categorie: ureasi positivi e ureasi negativi. Nel
primo gruppo vengono annoverate le specie che colonizzano prevalentemente lo
stomaco (Helicobacter pylori, Helicobacter heilmanii, Helicobacter felis), mentre
nel secondo possiamo inserire alcune specie riscontrabili a livello biliare e
intestinale (Helicobacter canis, Helicobacter epaticus, Helicobacter bilis).
L’ureasi prodotta dalle specie Helicobacter differisce dalle altre ureasi batteriche
per la presenza di due sub-unità, UreA e UreB, anziché tre. I geni deputati alla
produzione dell’enzima sono sette e sono localizzati in un singolo cluster di 6.13
kb (6). L’ureasi è estremamente importante ai fini dell’infezione: la scissione
dell’urea provoca la liberazione di ammonio e bicarbonato che, neutralizzando il
pH acido nelle immediate vicinanze del batterio, assicura la sopravvivenza
nell’avverso ambiente gastrico.
L’adesione del batterio alle cellule epiteliali gastriche avviene tramite più
molecole di adesione. In questo contesto ritroviamo la proteina BabA, il cui target
è l’antigene Lewis B, un normale costituente delle cellule di individui con gruppo
sanguigno 0. Oltre a BabA, sono state riconosciute altre molecole di adesione, tra
cui HpA, AlpA e AlpB, tutte facenti parte della famiglia delle Outer Membrane
Proteins.
La motilità dell’Helicobacter pylori (come anche precedentemente ricordato) è
garantita dalla presenza di flagelli, in numero da 4 a 6. Ogni flagello ha un
diametro di 30 µm, ed è costituito da un filamento interno di circa 12 nm,
circondato a sua volta da una guaina e da una membrana esterna che si continua
con quella della cellula. Nella parte terminale di alcuni flagelli sono presenti dei
bulbi, probabilmente implicati nei processi di fissazione del batterio (7). La spinta

10
dei flagelli unipolari (che producono un peculiare movimento “a cavaturaccioli”),
insieme all’azione lipolitica e proteolitica di alcuni enzimi specifici, fa sì che il
batterio riesca a penetrare nella porzione di muco sovrastante l’epitelio gastrico,
garantendosi un ambiente ideale per la crescita e ancorandosi alla mucosa
mediante adesine distribuite su tutta la superfice batterica (1).
1.4 Caratteri patogenetici particolari e fattori di virulenza.
L’azione patogena dell’Helicobacter pylori è determinata da alcune particolari
caratteristiche del batterio e fattori di virulenza che andiamo ora ad analizzare.
a) Lipopolisaccaride.
Comunemente a molte altre specie batteriche Gram-negative, anche
l’Helicobacter pylori esprime sulla sua superficie un LPS. Questa componente è
indispensabile per l’integrità del batterio e per l’interazione con l’organismo
ospite, ed è imputato nei meccanismi di immunomodulazione e
immunostimolazione (soprattutto tramite la componente lipidica, il lipide A).
L’LPS ha inoltre caratteri di tipo pirogenetico ed endotossico, e mutazioni della
componente saccaridica possono interferire con i normali processi di fagocitosi da
parte delle cellule mononucleate. L’LPS dell’Helicobacter pylori rientra in
maniera limitata nella patogenesi dell’infezione, tuttavia possiede delle
caratteristiche biochimiche uniche. Vari batteri condividono buona parte della
struttura dell’LPS, in particolare la catena polisaccaridica specifica O, il core
oligosaccaridico e la porzione del lipide A. La catena O, composta da 20-40 unità
(può contenere fino a 8 zuccheri), ha un’alta variabilità e possiede specifiche
funzioni di antigenicità e sierospecificità. Il lipide A (che invece ha una struttura
altamente conservata) connette l’LPS alla membrana esterna del batterio, mentre
il core oligosaccaridico (composto invece da una breve catena di zuccheri, da 10 a
15) è essenziale per il legame con i linfociti T attivati. (7)

11
L’LPS dell’Helicobacter pylori ha, invece, una scarsa immunogenicità. Questo è
probabilmente imputabile all’alto contenuto in acidi grassi del lipide A che
presumibilmente diminuisce il potenziale pirogenetico dell’LPS (1). E’ stata
osservata una forte affinità dell’LPS dell’Helicobacter pylori con la laminina,
proteina contenuta sia nella matrice extracellulare che nella membrana basale
delle cellule del tratto gastroenterico, che favorisce l’adesione del batterio
attraverso meccanismi di glicoconiugazione (legame lectin-like) (7). Questo
aspetto è stato osservato soprattutto in pazienti portatori di ulcera peptica.
Nella porzione polisaccaridica sono presenti componenti antigeniche analoghe
agli antigeni Lewis x (Le x) e Lewis y (Le
y), presenti anche (ed in maniera
consistente) come self sulle cellule dell’organismo umano. Le x e Le
y sono
espressi a livello dell’epitelio ghiandolare gastrico, e la dotazione delle stesse
molecole da parte del batterio provoca un
camuffamento di quest’ultimo, sfuggendo
così alla risposta immunologica soprattutto
negli stadi precoci dell’infezione. Gli
antigeni Lewis sono altresì coinvolti
nell’innesco di un processo di
autoimmunità, con produzione di auto-
anticorpi anti-gastrici. La presenza di auto-
anticorpi attivi contro la membrana
luminale dell’epitelio foveolare e contro la membrana canalicolare delle cellule
parietali è stata messa in relazione con l’insorgenza di gastrite, atrofia della
mucosa gastrica e riduzione del pepsinogeno. (20)
b) La citotossina vacuolante e il gene vacA
Nel 1988 uno studio di Leunk et al. osservò che alcuni ceppi dell’allora
conosciuto come Campylobacter pylori producevano una proteina in grado di
indurre vacuolizzazione in diverse linee cellulari (21). I ceppi produttori di questa
proteina sono maggiormente riscontrabili in pazienti con gastrite atrofica e ulcera
peptica, tuttavia sono stati prelevati campioni anche da pazienti asintomatici.
La produzione della citotossina vacuolante avviene a partire dal gene vacA,
presente in tutte i ceppi di Helicobacter pylori, ma la cui espressione è
Figura 5 - Struttura molecolare di VacA

12
strettamente legata alla presenza dell’isola di patogenicità cag. Gli alleli del gene
vacA hanno una lunghezza che va dai 3.864 ai 3.933 nucleotidi, e presentano
differenze considerevoli tra alleli di ceppi diversi. Nel contesto degli alleli sono
state individuate tre sequenze segnale (definite s1a, s1b e s2) e due regioni medie
(m1 e m2), le cui combinazioni (s1a/m1, s1a/m2 e così via) determinano un
polimorfismo tra i vari ceppi secernenti tossine, con livelli di virulenza differenti
(23). E’ stato infatti osservato che ceppi con l’allele m1 risultano essere associati
ad un maggior danno epiteliale rispetto ai ceppi m2 (che sono poco o non
tossigeni), così come i ceppi s1a/m1 hanno virulenza maggiore rispetto a quelli
s1b/m1 (22).
Figura 6 - Struttura del gene vacA e sequenze segnale
Il trascritto del gene vacA è monocistronico e codifica per una pro-tossina di 140
kDa, per un totale di 1300
aminoacidi. Nel passaggio
attraverso la membrana
citoplasmatica del batterio, la
porzione N-terminale della
proteina va incontro a clivaggio,
cosa che avviene anche per il terzo
C-terminale prima dell’esocitosi. A
questo punto si hanno monomeri di
90 kDa circa che, associandosi,
danno origine alla forma
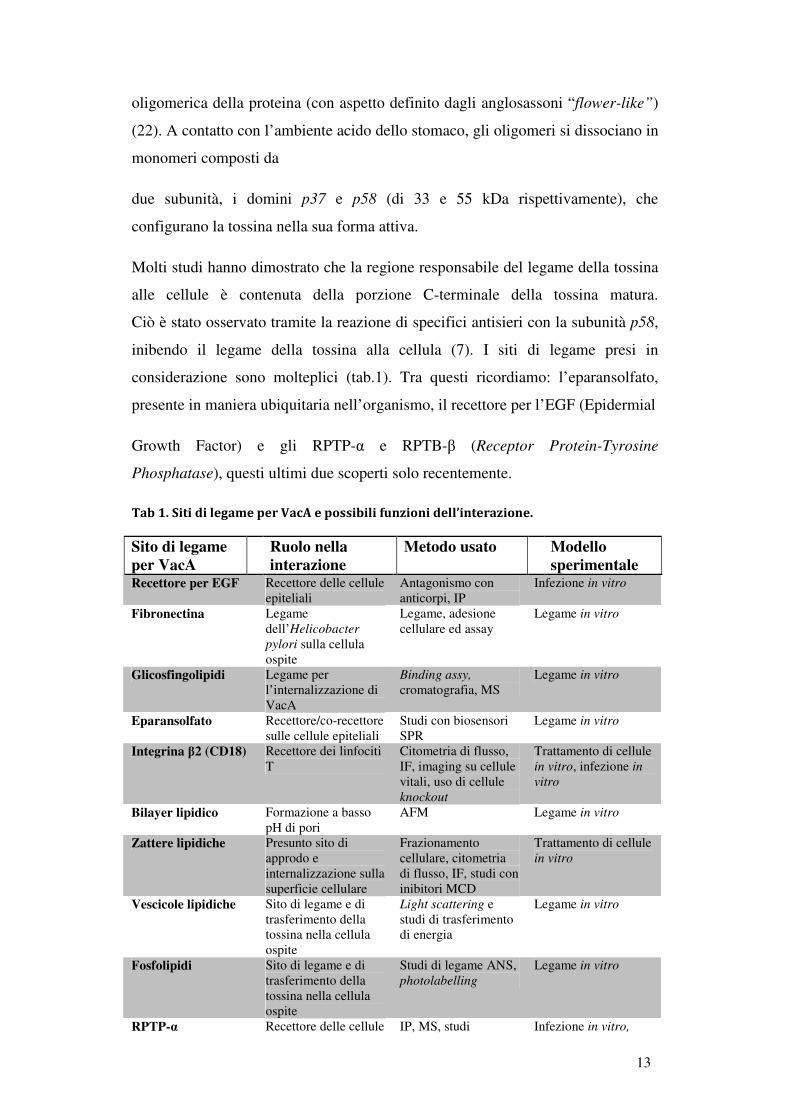
13
oligomerica della proteina (con aspetto definito dagli anglosassoni “flower-like”)
(22). A contatto con l’ambiente acido dello stomaco, gli oligomeri si dissociano in
monomeri composti da
due subunità, i domini p37 e p58 (di 33 e 55 kDa rispettivamente), che
configurano la tossina nella sua forma attiva.
Molti studi hanno dimostrato che la regione responsabile del legame della tossina
alle cellule è contenuta della porzione C-terminale della tossina matura.
Ciò è stato osservato tramite la reazione di specifici antisieri con la subunità p58,
inibendo il legame della tossina alla cellula (7). I siti di legame presi in
considerazione sono molteplici (tab.1). Tra questi ricordiamo: l’eparansolfato,
presente in maniera ubiquitaria nell’organismo, il recettore per l’EGF (Epidermial
Growth Factor) e gli RPTP-α e RPTB-β (Receptor Protein-Tyrosine
Phosphatase), questi ultimi due scoperti solo recentemente.
Tab 1. Siti di legame per VacA e possibili funzioni dell’interazione.
Sito di legame per VacA
Ruolo nella interazione
Metodo usato Modello sperimentale
Recettore per EGF Recettore delle cellule epiteliali
Antagonismo con anticorpi, IP
Infezione in vitro
Fibronectina Legame dell’Helicobacter
pylori sulla cellula ospite
Legame, adesione cellulare ed assay
Legame in vitro
Glicosfingolipidi Legame per l’internalizzazione di VacA
Binding assy,
cromatografia, MS Legame in vitro
Eparansolfato Recettore/co-recettore sulle cellule epiteliali
Studi con biosensori SPR
Legame in vitro
Integrina β2 (CD18) Recettore dei linfociti T
Citometria di flusso, IF, imaging su cellule vitali, uso di cellule knockout
Trattamento di cellule in vitro, infezione in
vitro
Bilayer lipidico Formazione a basso pH di pori
AFM Legame in vitro
Zattere lipidiche Presunto sito di approdo e internalizzazione sulla superficie cellulare
Frazionamento cellulare, citometria di flusso, IF, studi con inibitori MCD
Trattamento di cellule in vitro
Vescicole lipidiche Sito di legame e di trasferimento della tossina nella cellula ospite
Light scattering e studi di trasferimento di energia
Legame in vitro
Fosfolipidi Sito di legame e di trasferimento della tossina nella cellula ospite
Studi di legame ANS, photolabelling
Legame in vitro
RPTP-α Recettore delle cellule IP, MS, studi Infezione in vitro,

14
epiteliali antisense silencing trattamento di cellule in vitro
RPTP-β Recettore delle cellule epiteliali, recettore per la fosforilazione di Git1 (proteina attivatrice della ARF GTPasi)
Citometria di flusso, IF, IP, siRNA, uso di topi e cellule knockout
Legame in vitro, infezione di topi e cellule
RACK-1 Sconosciuto PD, Y2H Legame in vitro Sfingomielina Recettore sulle cellule
epiteliali Siti di legame, ELISA, citometria di flusso, trattamento con sfingomielinasi
Legame e trattamento di cellule in vitro
Abbreviazioni usate: ANS (1-anilina-8-naftalensulfonato), AFM (microscopia a forza atomica),
EGF (fattore di crescita epidermico), ELISA (enzyme-linked immunosorbent assay), IF
(immunofluorescenza), IP (immunoprecipitazione), MCD (metil-β-ciclodestrina), MS
(spettrometria di massa), PD (pull-down), siRNA (silencing RNA knockdown), SPR (risonanza
plasmonica di superficie), Y2H (Yeast-two-hybrid).
L’attività della tossina VacA ha come target due ATPasi, la H+-ATPasi tipo V e
la Na+,K+-ATPasi tipo P. Dopo la formazione di pori a livello della membrana
cellulare, con un aumento della permeabilità della stessa (l’alterazione della
ATPasi tipo P provoca un accumulo di sodio con assorbimento di acqua e
possibile sviluppo di edema intracellulare), avviene l’internalizzazione tramite
endocitosi dei canali anionici attivati. Una volta raggiunti gli endosomi, il legame
tra la tossina e la proteina rab7 qui espressa ostacola le fisiologiche funzioni degli
stessi, mentre l’interazione con l’ATPasi tipo V provoca un’alterazione della
permeabilità, con formazione di un canale selettivo per gli anioni. Ciò provoca un
aumento della concentrazione di H+, con abbassamento del pH e formazione di
una corrente di bicarbonati in entrata. Il vacuolo quindi aumenta di dimensioni,
cosa che si rende irreversibile quando le molecole in entrata vengono protonate
con formazione di NH4+.
Data la semipermeabilità delle membrane vacuolari, le basi andate incontro a
protonazione non possono più diffondere in senso opposto, determinando un
sempre maggiore aumento delle vescicole che occupano l’interno della cellula,
fino alla morte per disgregazione delle membrane.
Gli effetti della tossina VacA, tuttavia, non si esauriscono solo agli aspetti ora
descritti:

15
� Mitocondri: la molecola di VacA esercita a questo proposito un’attività simile a
quella della tossina difterica. La porzione p37 della tossina favorisce l’interazione
tra la membrana endosomiale e la membrana esterna del mitocondrio, laddove il
complesso TOM contribuisce nella formazione di un poro attraverso il quale
avviene l’importazione di VacA. In seguito VacA provoca la formazione di canali
ionici nel contesto della membrana interna mitocondriale, determinando una
iperpolarizzazione della stessa, con inibizione delle funzioni mitocondriali e
dissipazione del potenziale. Un altro fattore precipitante è il clivaggio della poli-
ADP-ribosio polimerasi (PARP), responsabile del rilascio del citocromo c e della
caspasi-3 da parte dei mitocondri. Tutte queste alterazioni conducono al
coinvolgimento di svariati fattori pro-apoptotici, tra cui la proteina Bax (24),
innescando il processo di morte cellulare.
� Cellule immunitarie: uno dei primi studi a riguardo ha evidenziato l’inibizione da
parte della citotossina VacA dei processi di presentazione dell’antigene da parte
dei linfociti T CD4+. VacA, inoltre, pare essere implicata nell’arresto della
maturazione dei fagosomi e l’inibizione dell’espressione della ossido nitrico-
sintetasi (eNOS), probabilmente causata dalla soppressione della kinasi integrino-
dipendente (ILK, Integrin-linked kinase).
La citotossina VacA interferisce con le normali funzioni dei linfociti T, avendo tra
i suoi target CD18. Dopo l’internalizzazione, VacA inibisce la mobilitazione di
Ca2+, provocando l’arresto delle calcineurina, una serina / treonina fosfatasi Ca-
dipendente. con attivazione di NFAT (Nuclear Factor of Activated T cell). La
conseguenza di ciò è la mancata espressione dei geni bersaglio di NFAT, in
particolare il gene di IL-2 e del recettore ad alta affinità per IL-2 (IL-2-R-α).
L’inibizione dei linfociti CD4+ può avvenire anche tramite il recettore CD28.
VacA può, quindi, inibire l’espansione clonale dei linfociti T attivati, manovra di
evasione nei confronti della risposta immune necessaria per la persistenza
dell’infezione.
� Risposta infiammatoria: oltre agli altri fattori di virulenza noti di Helicobacter
pylori, anche VacA è in grado di attivare la risposta infiammatoria. Un esempio è
l’attivazione di MAP-kinasi attraverso l’ATF-2 (Activation Transcription Factor-
2), effetto che sovente si verifica nelle cellule carcinomatose dello stomaco. La
porzione p38 della citotossina, come indicato da modelli sperimentali, stimola la

16
produzione di IL-8 (attraverso l’ATF-2), proteine CREB (cAMP Response
Element Binding) e NFkB (Nuclear transcription Factor kB). Quest’ultimo fattore
viene attivato anche a livello dei linfociti T con effetto pro-infiammatorio. Oltre a
ciò, VacA è in grado di aumentare l’espressione della ciclo-ossigenasi-2 (Cox-2)
da parte di neutrofili e macrofagi, sempre mediante induzione da parte della
componente p33, con incremento della secrezione di prostaglandina E2. Nei
mastociti, invece, è stato osservato un aumento del Ca2+, responsabile diretto di
aumentata chemiotassi e degranulazione (i meccanismi di promozione della
flogosi da parte di Helicobacter pylori sono approfonditi al paragrafo 1.5) (25).
c) CagA e l’isola di patogenicità cag
L’isola di patogenicità cag presente nel genoma dell’Helicobacter pylori, ed in
particolare la proteina CagA, sono state e sono tutt’ora oggetto di studio per la
spiccata virulenza dei ceppi portatori di questi fattori. Il gene cagA (cytotoxic-
associated gene A), sezione dell’isola di patogenicità cag-PAI, è stato rinvenuto
nel 50-70% dei ceppi di Helicobacter pylori isolati in occidente, ed è correlato a
patologie gastrointestinali avanzate.
Figura 8 - Isola di patogenicità cag con rappresentazione schematica dei trascritti
L’isola cag-PAI è una regione di 40 Kb, contenente circa 30 geni che possono
essere presenti in maniera variabile da ceppo a ceppo. La distruzione di uno di
questi geni diminuisce il potenziale di danno del batterio, soprattutto riducendo la
capacità di induzione della secrezione di IL-8 da parte delle cellule dell’epitelio
gastrico. Cag-PAI è divisa in due regioni: la regione a monte cagII, contenente
almeno 14 geni, e la regione a valle cagI, di 16 geni. Il gene cagA è localizzato
nella parte più distale della regione cagI, mentre in alcuni ceppi risulta essere

17
interposto ad un segmento definito IS605 (Insertion Sequence 605) (26). IS605
segna anche il limite tra le due sezioni di cag-Pai, e codifica per due trasponasi
(TnpA e TnpB), enzimi che rientrano nei meccanismi di riorganizzazione del
DNA e responsabili di delezioni o mutazioni.
Cag-PAI pare essere stata acquisita relativamente tardi da parte del batterio,
verosimilmente tramite plasmidi o fagi. In alcuni ceppi batterici compare in tutta
la sua interezza, in altri è deleta o riorganizzata, probabilmente proprio a causa
dell’attività della sequenza IS605 (22).
La secrezione della proteina CagA avviene tramite un apposito apparato di
secrezione di tipo 4, il
T4SS, generalmente
costituito da 11 proteine
definite VirB più la
proteina VirD4, tutte
codificate da cag-PAI. Il
sistema di secrezione 4 con
specifiche funzioni di
virulenza è utilizzato anche
da altri batteri oltre
all’Helicobacter pylori,
nella fattispecie
l’Agrobacterium
tumefaciens (da cui
derivano gran parte della conoscenze su questo tipo di sistema), la Bordetella
pertussis, la Legionella pneumophila e la Brucella suis (probabilmente lo stesso
sistema rientra nei anche meccanismi patogenetici di Rickettsia prowazekii,
Bartonella henselae e Actinobacillus actinomycetemcomitans). Le proteine del
T4SS costituiscono un articolato sistema: un complesso citoplasma/membrana
interna, il canale e un complesso esterno associato al pilo. La prima porzione è
costituita da tre NTPasi, (Virb4, 11 e VirD4) con VirB6 e 8; VirB7, 9 e 10 (il
cosiddetto “core complex”) che danno origine al canale trans-membrana che si
estende dalla membrana interna all’esterna, mentre il complesso esterno collegato
al pilo è costituito dalle componenti VirB2 e 5. VirB3 ha funzione sconosciuta:
Figura 9 - Sistemi di secrezione di tipo IV a confronto:
Agrobacterium tumefaciens (a sinistra) e Helicobacter pylori cag+
(a destra).

18
probabilmente è associata a VirB4, oppure potrebbe rientrare nella composizione
del sistema esterno (27). Nel meccanismo di traslocazione di CagA rilevante
importanza hanno CagF, che lega CagA per la porzione C-terminale, e
VirB5/CagL che si connette insieme a VirB10/CagY alla cellula ospite mediante
interazione con il recettore α5β1.
Una volta iniettata all’interno della cellula ospite, l’oncoproteina CagA subisce
alcune modificazioni che la rendono operativa. In particolare la proteina va
incontro a fosforilazione da parte delle Src tirosin-chinasi (tra l’altro noti fattori
oncogeni) e c-Abl presenti nella cellula ospite. In seguito alla fosforilazione,
CagA interagisce con la tirosin-fosfatasi SHP-2, inducendone l’attività e
determinando modificazioni citoscheletriche (soprattutto tramite fosforilazioni di
proteine leganti l’actina), che allungano le cellule e configurano il cosiddetto
“hummingbird phenotype” (con relativo aumentato rischio di metastasi per
carcinoma gastrico dovuto alla diminuzione dell’inibizione da contatto e la
produzione di uno scattering caratteristico) (27-28).
Figura 10 - Hummingbyrd phenotype ("fenotipo colibrì"): evidente allungamento delle cellule dopo
infezione da Helicobcater pylori cagA-positivo (wild type) rispetto alle cellule non infette (a sinistra) o
esposte a Helicobacter pylori con delezione di cagA (a destra).
L’attività di CagA non si limita solo a quanto sopra descritto, ma lede alla cellula
anche tramite effetti indipendenti dalla fosforilazione. Sono stati descritti ben 11
siti di legame per la CagA non fosforilata, primo fra i quali Grb2, una proteina
adapter che può interagire anche con la CagA fosforilata. Questa interazione si
associa ad un altro fattore, Sos (Son of Sevenless), creando un complesso
CagA/Grb2/Sos che promuove la formazione di GTP da parte di Ras e che avvia
una cascata di eventi responsabili di una incrementata dispersione (scattering) e
proliferazione cellulare (sistema Raf-Mek-Erk).

19
Un altro meccanismo patogenetico non correlato alla fosforilazione è la perdita di
polarità cellulare, con inibizione del sistema Par1b/MARK2 (partitioning-
detective 1/microtubule affinity-regulating kinase 2). Questo sistema è essenziale
per la stabilità dei microtubuli e la polarizzazione del fuso mitotico, con ritardo in
profase/metafase ed errato orientamento dei fusi. Recenti studi hanno dimostrato
che CagA lega anche altre proteine della stessa famiglia (Par1a, Par1c, Par1d),
determinando un’aumentata elongazione delle cellule (29).
Un altro importante effetto di CagA è la stimolazione della produzione di
citochine. La citochina maggiormente chiamata in causa è l’IL-8, secreta dalle
cellule epiteliali gastro-enteriche in risposta all’invasione batterica. I livelli di
questa interleuchina sono stati comparati tra ceppi cagA-positivi e negativi, con
un netto incremento nel primo gruppo (30). Livelli di IL-8 aumentati rispetto a
ceppi CagA-negativi sono stati osservati anche in altri studi: lo studio di Audibert
et al. ha osservato una induzione della produzione di IL-8 nel 58.7% dei batteri da
loro isolati (il gene cagA era presente nel 63% dei pazienti portatori di ulcera
duodenale e nel 56.5% dei soggetti con ulcera gastrica ) (32).
Circa l’oncogenicità di CagA, interessante è l’interazione di quest’ultima con la
proteina ASPP2, un fattore p53 correlato. ASPP2 è una proteina pro-apotpotica,
che esplica il suo compito attraverso l’attivazione del soppressore tumorale p53 e
che normalmente, in seguito a danno sul DNA o stimoli oncogeni, innesca la
cascata della morte cellulare programmata. Uno studio di Butia et al. ha
dimostrato che il legame tra CagA e ASPP2 provoca un sovvertimento funzionale
della proteina, con alterazione del normale meccanismo oncosoppressore di p53
attraverso una degradazione proteosomica di quest’ultima. CagA, quindi, dirotta
le normali funzioni di ASPP2, promuovendo la proliferazione cellulare (33).
Un’altra osservazione sulla correlazione pro-neoplastica di cag-PAI ci giunge da
uno studio sulla metalloproteasi-7 e sulla sua aumentata espressione in seguito
all’infezione da Helicobacter pylori. La MMP-7 fa parte della famiglia degli
enzimi proteolitici zinco-dipendenti, con proprietà pro-neoplastiche e che viene
espressa e secreta dalle cellule epiteliali. I ceppi cag+ provocano una
sovraregolazione di mmp-7, aspetto osservata in condizioni di malignità e pre-
malignità. La cascata che porta a questa condizione risiede in un’aberrazione della

20
catenina p120 a livello nucleare, che rimuove il blocco trascrizionale sul gene
della MMP-7 da parte della proteina “Kaiso” (34).
1.5 Meccanismi pro-infiammatori, immunità e produzione di
radicali liberi
Le modalità attraverso le quali l’Helicobacter pylori esplica la sua attività
patogena sono molteplici e agiscono a vari livelli. Innanzitutto il batterio è in
grado di provocare un danno diretto sulle cellule epiteliali, attraverso l’adesione e
la produzione della citotossina vacuolante. Circa l’attività patogena condotta sia
da VacA che dai prodotti dell’isola di patogenicità cag si è parlato nelle sezioni
precedenti, tuttavia val la pena soffermarsi sul processo di induzione
dell’infiammazione da parte del batterio. Un lavoro di Backert e Naumann ha
inquadrato in maniera molto esaustiva questi meccanismi, suddividendoli in più
punti. Innanzitutto l’infezione da parte dell’Helicobacter pylori provoca una
attivazione immediata di alcuni fattori definiti “precoci” attraverso l’interazione
con specifiche componenti, nelle fasi iniziali soprattutto con le flagelline o l’LPS,
che interagiscono con particolari recettori “Toll-like”. Il Fattore nucleare k-B
(Nf-kB), importante regolatore trascrizionale delle cellule eucariotiche, gioca un
ruolo chiave nella regolazione dell’immunità innata ed adattiva,
nell’infiammazione, e nei meccanismi di sopravvivenza cellulare. Per esempio,
questo fattore è coinvolto nella regolazione della flogosi attraverso l’induzione
alla produzione di citochine, chemochine, fattori di adesione cellulari e altri
effettori. Opinione diffusa è che l’Helicobacter pylori provochi la
sovraregolazione del fattore Nf-kB, soprattutto attraverso i suoi fattori di
virulenza.
AP-1 è un altro fattore di trascrizione implicato nei meccanismi di infiammazione
acuta e cronica. Come Nf-kB è attivato da svariati stimoli, rientrando anche nei
meccanismi di proliferazione, maturazione cellulare e apoptosi. La concomitante
attivazione di Nf-kB è essenziale anche in modalità fisiologiche nella regolazione
dei linfociti T nell’infiammazione cronica, in cui entrambi i fattori promuovono
l’attivazione di geni produttori di citochine.

21
Premesso questo, possiamo suddividere i meccanismi di induzione in base ad
alcune modalità:
1) Attivazione di Nf-kB T4SS dipendente-cagA indipendente. Protagonista è
cagPAI, ed in particolare la porzione implicata nella codifica delle componenti
del sistema T4SS. Probabilmente la porzione del pilo del sistema di trasporto o
l’iniezione di ureasi o peptidoglicano provocano l’attivazione di diversi sistemi
che inducono l’attivazione dell kinasi Nf-kB-induttrice (NIK) e di conseguenza
una sovraespressione di Nf-kB.
2) Attivazione di AP-1 T4SS dipendente-cagA indipendente. Questo meccanismo è
sostenuto da alcune kinasi (JNK-MKK4-PAK1) che contribuiscono all’attivazione
di citochine e chemochine pro-infiammatorie indotte dal sistema di trasporto di
Helicobacter pylori. L’attivazione di AP-1 sembra essere mediata direttamente
dalla kinasi JNK che da il via alla cascata enzimatica che porta all’attivazione di
AP-1.
3) Meccanismi cagA dipendenti. L’iniezione di CagA e la traslocazione di p65
all’interno provocano l’induzione e quindi il rilascio di IL-8., attraverso un
meccanismo guidato da una particolare cascata enzimatica, H-Ras-Raf-MEK.
Quest’ultimo sistema pare intervenga in circa 24 ore. (36).
Per quanto riguarda l’immunità non specifica, l’infezione da Helicobacter pylori
provoca una risposta da parte dei neutrofili, con persistenza di queste cellule
nell’infiammazione cronica. Ciò naturalmente è favorito dall’immissione in
circolo di chemochine, citochine e molecole di adesione che contribuiscono al
reclutamento e all’attivazione dei neutrofili. In particolare uno studio di Svensson
et al. ha evidenziato la sovra-espressione di una particolare molecola di adesione,
la E-selectina, in seguito ad infezione.

22
I ligandi di questa molecola sono Sialyl- e Lewisx, presenti su neutrofili e
monociti; anche lo stesso Helicobacter è in grado di legare l’E-selectina (37).
Figura 11 - Meccanismi pro-infiammatori di Helicobacter pylori
I fagociti mononucleati rivestono una parte importante nelle fasi precoci di
infezione, sia come fonte di mediatori pro-infiammatori sia come cellule
presentanti l’antigene. E’ inoltre possibile osservare un infiltrato di eosinofili, i
quali rilasciano proteine basiche che contribuiscono al danno endoteliale. La
degranulazione degli eosinofili è probabilmente scatenata da immunocomplessi di
IgA e antigeni solubili.

23
L’infiltrato di linfociti T e plasmacellule in corso di gastrite è il testimone
dell’immunità antigene-specifica e della risposta umorale. L’infezione
promuove lo sviluppo di follicoli linfoidi organizzati, che gradualmente
diminuiscono in seguito ad eradicazione. La presenza di cellule accessorie
(macrofagi e cellule dendritiche) facilita i meccanismi di presentazione
dell’antigene. (38) La risposta si avvale anche dei linfociti T, in particolare dei
linfociti CD4+, dei quali è stato osservato un incremento rilevante. La risposta
anticorpale è garantita dalla secrezione prevalentemente di IgG nel torrente
ematico, mentre, a livello locale, si ritrovano principalmente IgA secretorie.
Queste immunoglobuline sembrano non avere un ruolo determinante
nell’eradicazione dell’infezione; malgrado ciò, tra i loro bersagli sono annoverate
anche strutture chiave del batterio, tra i quali i flagelli e le ureasi. Sono immesse
in circolo anche immunoglobuline IgE, queste ultime implicate in reazioni di
liberazione di istamina da parte di mast-cellule e basofili.
Un altro punto interessante è costituito dall’incremento della produzione da parte
dell’Helicobacter pylori di radicali liberi dell’ossigeno (40-41). Sebbene questa
attività sia svolta prevalentemente dai neutrofili, anche il batterio se ne fa
promotore. L’Helicobacter è in grado di generare tali molecole attraverso varie
modalità: si ha, infatti, una produzione passiva di anione superossido O2-
attraverso la catena respiratoria mitocondriale. Inoltre, mediante l’ureasi, l’H.p.
promuove la produzione di altre molecole tossiche, tra cui le cloroammine,
derivate dalla reazione dell’anione clorossido (OCl-) e dell’ammoniaca, scissa
dalle stesse ureasi. CagA, oltre a ciò, favorisce la formazione di radicali liberi
tramite i mitocondri delle cellule dell’epitelio gastrico. Un metodo per ripararsi
dagli stessi radicali, altrimenti lesivi per il batterio, è costituito dalle catalasi e
dalle superossidodismutasi prodotte dallo stesso Helicobacter, che, oltre a
scindere le molecole ossidanti, ne riducono anche la produzione da parte dei
neutrofili (39-40).
Un altro aspetto da tener conto nell’infezione da Helicobacter pylori è l’induzione
di meccanismi di autoimmunità. Numerosi studi, infatti, correlano tale infezione
a numerose patologie a carattere autoimmune, sia a livello gastro-intestinale
(gastrite autoimmune) che extra-intestinale. Le patologie autoimmuni correlate ad

24
Helicobacter pylori saranno trattate in seguito nel contesto delle manifestazioni
extra-digestive dell’infezione.
1.6 Patolologie
gastrointestinali
a) Gastrite cronica
La forma di gastrite cronica da
Helicobacter pylori è sicuramente
la variante di gastrite più comune al
mondo, essendo l’infezione da
Helicobacter presente in circa il
90% dei pazienti con gastrite
cronica antrale (43).
L’evoluzione della gastrite da
Helicobacter pylori può
generalmente seguire due strade:
Gastrite antrale, caratterizzata da
infiammazione limitata all’antro.
Questo pattern è presente con
elevata percentuale in pazienti che sviluppano in seguito ulcera gastrica, a causa
di una elevata attivazione delle cellule secernenti gastrina e susseguente
ipercloridria;
1. Pangastrite e successiva gastrite atrofica multifocale, contraddistinta dal
coinvolgimento del corpo e dell’antro e dalla progressiva distruzione delle cellule
ossintiche e atrofia. Il risultato è una riduzione della secrezione acida, con
ipocloridria o anacloridria, e metaplasia intestinale, con incremento del rischio di
degenerazione neoplastica.
La metaplasia intestinale, come anche sopra ricordato, riveste un ruolo importante
in questa affezione. La classificazione di Filipe e Jass la suddivide in tre stadi
(44):
Figura 12 - Gastrite cronica antrale in maschio di 45 anni.
La colorazione Giemsa mette in risalto l'infezione da
Helicobacter pylori (Brown Medical School -
www.brown.edu)
Figura 13 - Aspetto endoscopico di gastrite da
Helicobacter pylori.

25
Tipo I (completa): cripte prive di alterazioni architetturali, rivestite da cellule
caliciformi mature e cellule assorbenti con un orletto di microvilli ben definito. Le
cellule caliciformi secernono sialomucine e occasionalmente solfomucine. Le
cellule assorbenti non presentano mucine; cellule di Paneth presenti alle basi delle
cripte.
Tipo II (incompleta): cripte con lievi alterazioni architetturali, rivestite da cellule
caliciformi interposte a cellule colonnari mucosecernenti, mentre le cellule
assorbenti sono assenti o rinvenibili in poche unità. Le cellule colonnari
contengono sialomucine e mucine neutre, le caliciformi sialomucine. Cellule di
Paneth rare o assenti;
Tipo III (incompleta): presenza di cripte tortuose e ramificate, con architettura
alterata. Sono presenti prevalentemente cellule colonnari immature secernenti
solfomucine rispetto alle cellule caliciformi contenenti sialomucine e/o
solfomucine. Cellule di Paneth estremamente rare.
Non sono del tutto chiare le modalità patogenetiche che indirizzano la gastrite a
sviluppare l’una o l’altra modalità, tuttavia pare che giochino un ruolo essenziale
alcune citochine, in particolare l’IL1β, che oltre ad essere un potente agente pro-
infiammatorio, induce un decremento della secrezione cloridrica, indirizzando il
quadro verso una pangastrite.
Istologicamente a livello macroscopico non esiste un aspetto endoscopico
caratteristico rispetto agli altri tipi di gastrite. Possono comparire aree di iperemia
e/o ipertrofia, erosioni (in base al grado della gastrite in atto) e, in caso di atrofia,
si osserverà un assottigliamento della mucosa. Microscopicamente, sono visibili
le colonie di Helicobacter pylori che rivestono la mucosa, interessando sia le
regioni antrali sia corpo-fundiche e cardiali. Nel caso sussista una estesa
metaplasia intestinale, le aree di infezione si limitano alle porzioni non
metaplastiche, in particolare a livello del corpo. L’infiltrato infiammatorio,
composto da linfociti e plasmacellule, oltre che da neutrofili nelle fasi attive,
risulta essere preponderante a livello del corpo, con follicoli linfatici iperplastici
(43).
Un altro fattore che riveste notevole importanza è l’ipocloridria. E’ stato
osservato, infatti, che l’ Helicobacter pylori è in grado di provocare ipocloridria

26
già in tre giorni dall’inizio dell’infezione. Questo, naturalmente, facilità la
colonizzazione e promuove l’attivazione di processi pro-infiammatori.
La diminuzione della secrezione acida può essere collegata o a distruzione delle
cellule parietali da parte dell’infiammazione o a un meccanismo di aggressione
condotto dal batterio stesso. Infatti, recentemente è stata dimostrata la capacità
dell’Helicobacter pylori di interferire con una subunità della H+K+ATPasi
(subunità α-catalitica HKα), indispensabile per la secrezione acida. Tale effetto è
realizzato da fattori prodotti dall’isola cagPAI (CagE, CagM, CagL, CagA),
capaci di reprimere la pompa protonica e, di conseguenza, la secrezione acida.
E’ stata evidenziato un ruolo dell’infezione da Helicobacter pylori anche per la
gastrite autoimmune. Infatti, la possibile attivazione di linfociti CD4+ Th1 può
far sì che essi cross-reagiscano con componenti self della mucosa gastrica, come
per esempio con la H+K+ATPasi, e con varie proteine dell’Helicobacter pylori.
Ciò può dare il via ad un processo di autoimmunità gastrica che può avere come
conseguenza finale l’atrofia. In particolare, è stata osservata una stretta omologia
tra le subunità β dell’ureasi batterica e dell’H+K+ATPasi, il ché suggerisceun
possibile mimetismo molecolare tra le componenti batteriche e alcuni
autoantigeni della parete gastrica. Sempre in questi meccanismi sembrano essere
implicati anche gli antigeni Lex e Ley, espressi sia dal lipopolisaccaride batterico
sia a livello della mucosa gastrica (46-47).
Figura 14 - Localizzazioni e interessamento degli strati della parete gastrica in corso di ulcera peptica

27
b) Ulcera peptica
L’ulcera peptica è una patologia estremamente rilevante dal punto di vista sociale:
il 2% della popolazione mondiale è affetta da ulcera attiva, mentre circa il 6-15 %
ha manifestato sintomi riconducibili a essa. Gli uomini sono colpiti con maggiore
frequenza, con un rapporto di 3:1.
Per definizione, si considera come ulcera peptica una lesione focale, con perdita
di sostanza a livello mucoso, che si approfondisce fino alla sottomucosa e supera
la muscularis mucosae. Alla base del fenomeno c’è un difetto dei meccanismi di
difesa della mucosa verso gli agenti dannosi, nella fattispecie l’acido cloridrico e
la pepsina. La localizzazione duodenale è la più frequente, fatta eccezione per le
casistiche giapponesi, in cui la localizzazione gastrica è nettamente preminente
(48). Nel 5-15% dei pazienti, inoltre, è possibile osservare una doppia
localizzazione, sia gastrica che duodenale.
Nella patogenesi dell’ulcera peptica, l’Helicobacter pylori ha un ruolo di primaria
importanza. Ulcere peptiche duodenali si associano sovente a manifestazioni
gastritiche antrali in cui sussiste uno stato di ipergastrinemia, dato che in questo
tipo di gastrite le ghiandole ossintiche risultano essere illese. A questo livello si
verifica un danneggiamento delle cellule D dell’antro, che rimuove l’inibizione
delle cellule G producenti gastrina con conseguente secrezione di acido cloridrico.
A ciò si combina una riduzione di produzione di bicarbonati, probabilmente
dovuta a VacA, che predispone il bulbo duodenale alla metaplasia gastrica, alla
colonizzazione da parte di Helicobacter pylori e al danno epiteliale, rendendo la
mucosa di questo distretto suscettibile all’attacco dei secreti acidi lesivi.
L’Helicobacter pylori rientra in maniera rilevante anche nella patogenesi
dell’ulcera gastrica. In questo caso il primum movens risiede in una alterazione
dello strato di muco che riveste la parete gastrica, con aggressione da parte degli
ioni idrogeno dell’epitelio. L’ulcera gastrica risulta essere, inoltre, maggiormente
correlata ad atrofia e metaplasia, con maggior rischio di trasformazione
neoplastica, probabilmente a causa dell’infiammazione e dell’ipocloridria che
approntano la localizzazione gastrica dell’ulcera. Le sedi di sviluppo più frequenti
sono le aree di transizione fundo-cardiale, antro-pilorica e corpo-antrale.

28
c) Dispepsia
Viene inteso come dispepsia un particolare quadro clinico caratterizzato da dolore
e/o discomfort localizzati ai quadranti addominali superiori (soprattutto in regione
epigastrica) e che persistono nel tempo. La sintomatologia si esacerba in
corrispondenza dei pasti, non si associa ad alterazioni dell’alvo e può avere un
andamento cronico (per almeno 12 settimane negli ultimi 12 mesi).
In termini etiopatologici, la dipsepsia può essere suddivisa in due categorie:
� Dispepsia secondaria, ovvero riconducibile a una nota patologia di base;
� Dispepsia funzionale (o idiopatica), qualora non sia possibile attribuire la causa
del disturbo a nessuna evidente alterazione organica, sistemica o metabolica, e la
cui diagnosi viene effettuata escludendo le cause di dispepsia secondaria. Questo
tipo di dispepsia può essere a sua volta suddivisa in:
� Simil-ulcerosa, presente in pazienti con predominanza di sintomi
quali bruciore retrosternale e dolore epigastrico. Questa tipologia
particolare può essere riconducibile all’infezione da Helicobacter
pylori (è presente una elevata sieroprevalenza dell’infezione in
gruppi di pazienti dispeptici) o a meccanismi di ipersecrezione
acida;
� Simil-motoria, associata prevalentemente a sintomi non dolorosi
(discomfort). La causa risiede probabilmente nel ritardato
svuotamento gastrico, in stati di ectasia dell’antro gastrico o nella
ridotta accomodazione fundica (49).
Le basi fisiopatolgiche della dispepsia indotta da Helicobacter pylori sono
complesse, e esistono numerosi studi a riguardo. L’ipersecrezione acida ha un
ruolo fondamentale: in pazienti con gastrite antrale e ipercloridria, è stata
osservata una riduzione della secrezione di somatostatina, con un conseguente
aumento del rilascio di gastrina e della secrezione acida.
Un altro dato interessante è quello riguardante la ghrelina (vedi seguito): in
diversi studi i livelli sierici dell’ormone risultano essere diminuiti in pazienti
dispeptici e portatori di Helicobacter pylori, specie in coloro che lamentavano

29
sazietà precoce e senso di gonfiore e peso post-prandiale. Da ciò si evince che
l’Helicobacter pylori riveste un ruolo anche nella patogenesi della dispepsia
simil-motoria, e che la determinazione dei livelli sierici di ghrelina può essere in
futuro un utile marker di valutazione per questo disturbo (51).
d) Malattia da reflusso gastroesofageo
L’infezione da Helicobacter pylori non sembra a oggi essere correlata in maniera
evidente con la malattia da reflusso gastro-esofageo (GERD). Dal punto di vista
epidemiologico, non è stata osservata una sostanziale differenza tra pazienti affetti
da GERD e controlli di soggetti non infetti. La questione sulla patogenesi, invece,
sembra essere più spinosa: alcuni studi hanno osservato che l’infezione provoca
una sorta di effetto protettivo nei confronti della patologia da reflusso, a causa
della soppressione delle secrezione acida in pazienti con pangastrite o gastrite
atrofica, escludendo così anche le complicazioni del GERD stesso (con in primis
l’esofago di Barrett e la degenerazione neoplastica). Ciò avverrebbe in maniera
più marcata in pazienti affetti da ceppi CagA-positivi, probabilmente a causa della
maggiore azione pro-infiammatoria che questi ceppi inducono a livello della
mucosa gastrica (49). Tuttavia, in altri studi, è stato osservato che l’eradicazione
dell’infezione non determina né un aggravamento né un miglioramento dei
sintomi in pazienti affetti da GERD e, al contempo, portatori di Helicobacter (50).
Una raccomandazione utile è, comunque, la ricerca dell’infezione in pazienti con
reflusso da lungo tempo trattato con PPI (inibitori della pompa protonica), in
quanto la presenza del batterio, unita all’inibizione della secrezione acida indotta
dal farmaco, possono accelerare il processo di atrofia della mucosa gastrica,
predisponendola allo sviluppo di neoplasie.
e) Adenocarcinoma gastrico
Il carcinoma gastrico è la seconda causa di morte per neoplasia nella popolazione
mondiale, tuttavia negli ultimi cinquant’anni si è assistito ad un declino
dell’incidenza di questa particolare patologia (fatta eccezione per il Giappone, in
cui l’adenocarcinoma assume i caratteri di malattia endemica). In Europa si
verificano annualmente circa 190.000 nuovi casi (23% di tutte le neoplasie), con
un rapporto maschi-femmine di quasi 2:1. L’Europa orientale (34 casi ogni
100.000 abitanti maschi) e meridionale (19 casi per 100.000) sono tra le regioni

30
più colpite (dati dell’Associazione Italiana Ricerca sul Cancro). In Italia
l’incidenza è maggiore nelle regioni centro-settentrionali rispetto al meridione,
con tassi tra 35 e 25 nuovi casi ogni 100.000 abitanti. L’età di insorgenza media è
oltre i 50 anni (sei casi su dieci si sono verificati oltre i 65 anni), raramente sotto i
30.
Per quanto riguarda l’etiopatogenesi, numerosi sono i fattori che possono incidere
sull’insorgenza del carcinoma gastrico. Le abitudini alimentari hanno un ruolo
decisamente importante, in particolare le diete carenti di vegetali o povere in
acido ascorbico e folati. Alcuni fattori genetici, inoltre, sono probabilmente
implicati nella patogenesi del carcinoma gastrico: nell’8% dei casi è stata
osservata una mutazione del gene dell’ E-caderina CDH1, che codifica per una
proteina calcio dipendente di adesione cellulare. Sono state osservate anche
mutazioni della p53 o del gene BRCA-2, e pare essere presente un’aumentata
suscettibilità nei pazienti portatori di HNPCC e con mutazioni germinali dei geni
del mismatch repair. La presenza di displasia, riscontrabile sia in aree di
metaplasia intestinale che in aree non metaplastiche, è un parametro di
valutazione importante, in quanto è stato dimostrato che l’85% delle lesioni con
displasia ad alto grado va incontro a degenerazione maligna in meno di 15 mesi
(43). La gastrite atrofica e la metaplasia intestinale risultano essere protagoniste
nella carcinogenesi, in molti studi considerate una sorta di punto di partenza per lo
sviluppo di un adenocarcinoma di tipo intestinale (52).
In particolare, lo stato di ipocloridria che si instaura nella gastrite atrofica
favorisce la proliferazione di microorganismi capaci di produrre nitriti (agenti
carcinogeni noti) partendo dai nitrati gastrici. Inoltre, c’è una riduzione della
secrezione di acido ascorbico, sostanza con caratteristiche anti-tumorali e che
controbilancia l’eccesso di nitriti.
I meccanismi molecolari attraverso i quali dall’infezione si può passare al
carcinoma sono svariati (fig.15), e contemplano l’effetto tossico diretto sulle
cellule epiteliali, con perdita di polarità e alterazione dell’adesione, lo stress
ossidativo Helicobacter-indotto, gli effetti sulla trasduzione del segnale e
sull’espressione dei geni con turbe nell’equilibrio apoptosi-proliferazione, e le
alterazioni del sistema del DNA mismatch repair (53).

31
Alla base di queste eventi ritroviamo i fattori di virulenza (vedi paragrafo 1.4). La
relazione tra l’infezione da ceppi CagA-positivi e il maggior rischio di
degenerazione neoplastica è supportata da numerosi dati, molti dei quali
provengono dalle regioni dell’Asia orientale. In queste regioni, infatti, esiste
un’alta prevalenza di carcinoma gastrico, con un’elevata percentuale di ceppi
CagA-positivi. Al contrario in Occidente tale percentuale è considerevolmente più
bassa, con una prevalenza del carcinoma gastrico altrettanto minore (54). Questo
evidenzia ancora una volta la stretta correlazione esistente tra il genotipo cag+ e
l’insorgenza di neoplasie.
Dati esaurienti sul rapporto tra Helicobacter pylori e carcinoma gastrico ci
vengono forniti da studi sull’importanza dell’eradicazione dell’infezione nei
pazienti ad alto rischio per tumore allo stomaco. Un trial eseguito in una regione
ad alto rischio della Cina, ad esempio, ha dimostrato che in gruppi affetti da
Helicobacter pylori esisteva una maggiore incidenza di cancro gastrico rispetto ai
controlli (pazienti non infetti), e che l’eradicazione del batterio (sebbene la
regressione della metaplasia intestinale, laddove presente, sia stata osservata solo
in pochi casi) poteva prevenire la comparsa sia di neoplasie de novo in pazienti in
assenza di lesioni precancerose, sia di recidive in soggetti affetti da “hearly
gastric cancer” e trattati con resezione endoscopica.
Per quanto riguarda la metaplasia intestinale, è stato ipotizzato una sorta di “punto
di non ritorno”, oltre il quale, nonostante l’eradicazione del batterio, la
regressione non è più possibile. I meccanismi molecolari alla base di questo
fenomeno, però, non sono chiari (56).
Figura 15 - Meccanismi molecolari di oncogenesi da Helicobacter pylori

32
A questo proposito, tuttavia, alcuni modelli sperimentali hanno dimostrato che
l’eradicazione dell’Helicobacter pylori può provocare una sostanziale
diminuzione delle citochine pro-infiammatorie. In particolare, sono stati dosati i
livelli di IFN-γ prodotta dai linfociti Th-1, evidenziandone una marcata riduzione
in seguito a terapia antibiotica in soggetti con lesioni precancerose. Ciò ha portato
a concludere che l’eradicazione dell’infezione può comportare un’attenuazione
dei processi di flogosi, con parziale regressione delle lesioni precancerose e, di
conseguenza, diminuita incidenza di carcinoma (58).
Tab 2. Classificazione di Borrmann del carcinoma gastrico.
Polipoide Massa a cavolfiore o villosa, scarsa tendenza
ad infiltrare la parete.
Fungoide Massa solida cupoliforme con ulcerazione
centrale. Rappresenta 1/3 dei carcinomi
avanzati
Ulcerativo Forma più comune. Ulcera profonda, fondo
necrotico, margini irregolari e nodosi.
Infiltrante 10% dei tumori avanzati. Non è visibile alcuna
massa, ma è presente un ispessimento della
parete a placca. La variante diffusa viene
definita come “linite plastica”, con stomaco
ridotto di volume e pareti rigide ed ispessite.
Tab 3. Classificazione di Lauren del carcinoma gastrico.
Intestinale Più frequente nei paesi ad alta incidenza, a sede
antro-pilorica con aspetto macroscopico
principalmente fungoide.
Diffuso Carcinoma ad anello con castone
Indefinito Coesistenza di aspetti di carcinoma intestinale
e diffuso

33
Figura 16 - Classificazione TNM del carcinoma gastrico
Interessante è anche il dato proveniente da studi condotti sulla progressione del
carcinoma gastrico in pazienti con ulcera gastrica e infezione da Helicobacter
pylori. L’eradicazione del batterio ha comportato una sostanziale riduzione di
nuovi casi rispetto al controllo (57).
Quanto sopra detto rimanda ad un concetto chiave: lo screening.
La determinazione dell’infezione e la tipizzazione del genotipo CagA+,
soprattutto in aree ad alto rischio, può essere un utile strumento di prevenzione nei
confronti del carcinoma dello stomaco e delle sue condizioni preneoplastiche,
nella fattispecie la gastrite atrofica e la metaplasia intestinale.
Figura 17 - Immagine istologica di linfoma MALT a
basso grado

34
f) Linfoma MALT gastrico
Il linfoma gastrico è una neoplasia di origine linfoide che si localizza
primitivamente allo stomaco. Il linfoma MALT (acronimo di Mucosal Associated
Limphoid Tissue) colpisce gli aggregati linfoidi della mucosa, localizzati
normalmente a livello dell’antro o alla base della mucosa ossintica. Questa
neoplasia vede tra le cause determinanti l’infezione da parte dell’Helicobacter
pylori, con un’associazione che può raggiungere percentuali elevatissime, fino al
98% dei casi osservati (ciò avviene in maniera molto meno frequente per i linfomi
B ad alto grado, in cui l’associazione tumore-Helicobacter è del 25-38 %).
All’esame istologico, il linfoma MALT si presenta macroscopicamente spesso
sotto forma di ulcere superficiali, erosioni, o come pliche mucose ispessite, con
penetrazione generalmente limitata alla mucosa o alla sottomucosa.
Microscopicamente, l’architettura riproduce il MALT normale, con cellule
neoplastiche disposte tra follicoli linfatici pre-esistenti. Con il progredire della
lesione, si ha una colonizzazione ed erosione dei follicoli, fino alla completa
scomparsa. Le cellule linfomatose hanno citoplasma chiaro, con nucleo a contorni
irregolari (43).
Le basi molecolari del linfoma MALT Helicobacter-indotto risiedono nella
cronica stimolazione antigenica effettuata dal batterio, con espansione clonale
delle cellule B. Infatti in corso di infezione da Helicobacter pylori si assiste, sotto
lo stimolo di linfociti T sensibilizzati, all’organizzazione di follicoli linfoidi e
centri germinativi con una zona mantellare ben definita, configurando un sistema
atto a controllare l’infezione. Questo tipo di tessuto, diverso dai normali aggregati
linfoidi sparsi della mucosa gastrica, viene definito come “MALT acquisito”, da
differenziare dal “MALT nativo” che può essere ben osservato nell’ileo distale o
nelle placche di Peyer. L’insorgenza del linfoma si verifica qualora una
popolazione monoclonale si sleghi dal resto della componente MALT, acquisendo
caratteri proliferativi tumorali (59). Finché il tumore resta vincolato alla
stimolazione antigenica, esiste una buona possibilità di regressione dopo
eradicazione dell’Helicobacter pylori, in seguito tale dipendenza svanisce e il
linfoma tende all’evoluzione maligna.

35
Il linfoma MALT Helicobacter-associato possiede alcune caratteristiche
molecolari che probabilmente sono alla base della patogenesi, tra cui delle
traslocazioni cromosomiche caratteristiche. Una di queste è la traslocazione
t(11;18), assente nei linfomi nodali, che provoca una fusione dei geni APJ2-
MALT1, il cui trascritto è probabilmente implicato nella sopravvivenza delle
cellule linfomatose attraverso meccanismi di inibizione dell’apoptosi. Questa
traslocazione pare essere un fattore predittivo di progressione di malattia, in
quanto non è stata riscontrata nei linfomi che regrediscono in seguito ad
eradicazione dell’infezione da Helicobacter pylori. Oltre a ciò, recenti studi hanno
osservato un’aumentata espressione di alcuni geni spesso associati a quadri di
malignità (lamin receptor-1, MDR-1, calgranulina A/Mrp-8), e una iper-
espressione di alcuni markers quali CD1c, CD40, CD44, CD53, CD83, CD86 e
membri della famiglia delle proteine HLA-D (particolarmente sovra-regolate nei
linfomi MALT). In aggiunta, sono state rinvenute metilazioni aberranti del DNA
a livello di vari geni, aspetto che probabilmente riveste un ruolo critico nella
patogenesi della malattia (60). Uno studio di Lehours et al. mette in relazione alla
patogenesi del linfoma MALT gastrico l’espressione degli antigeni Lex e Ley,
presenti nella porzione polisaccaridica del LPS di Helicobacter pylori e
probabilmente implicati nei meccanismi di sensibilizzazione dei linfociti T (61).
L’eradicazione dell’infezione è un provvedimento terapeutico indispensabile per
la remissione del linfoma. Sono stati condotti a riguardo numerosi studi che hanno
mostrato come la terapia anti-Helicobacter provochi una remissione di malattia,
totale o parziale, in pazienti affetti da MALT di basso grado in un’altissima
percentuale di casi (62). Tuttavia esiste una variabilità che è riconducibile a alcuni
fattori predittivi, la cui individuazione permette di capire quali pazienti
effettivamente trarranno beneficio dalla terapia antibiotica. In particolare questi
fattosi sono: il grado di malignità, lo stadio della malattia, la documentata
positività all’infezione, alcuni fattori demografici (età e sesso principalmente,
questi ultimi hanno una valenza limitata) e la durata del follow-up (49).

36
1.6 Manifestazioni extra-digestive
Qui di seguito verranno descritte una serie di patologie provocate da Helicobacter
pylori che esulano dal contesto gastro-intestinale.
a) Apparato cardiovascolare
Per quanto riguarda l’apparato cardiovascolare, da lungo tempo
l’aterosclerosi è stata accostata ad agenti infettivi, e, di conseguenza, la
cardiopatia ischemica è presto divenuta protagonista di studi circa questo tipo di
interazione. Studi sulla possibile compartecipazione infettiva hanno avuto come
protagonisti vari agenti patogeni, tra cui il virus Coxackie B4, il Cytomegalovirus,
l’Herpes simplex e la Chlamidia pneumoniae. Anche l’Helicobacter pylori è stato
chiamato in causa nella patogenesi della cardiopatia ischemica, e tale correlazione
è supportata da varie evidenze epidemiologiche. Il primo a mettere in relazione i
due fattori, Helicobacter e cardiopatia ischemica, è stato uno studio di Mendall et
al. del 1994 (63). Questo lavoro ha evidenziato l’alta prevalenza di infezione in un
campione di pazienti affetti da cardiopatia ischemica, e, soprattutto, in soggetti
infetti dall’età pediatrica. Altri studi in seguito hanno avvalorato questa tesi:
Pellicano et al., tramite misurazione delle IgG anti-Helicobacter pylori in pazienti
ricoverati presso l’Unità di Terapia Intensiva Coronarica dell’ospedale di Novi
Ligure, hanno riscontrato l’infezione nel 77% dei casi osservati (64).
Una review del 2007 curata da Manolakis et al. ha suddiviso gli studi riguardanti
la cardiopatia ischemica Helicobacter-correlata in vari “focus” patogenetici:
1. Lipidi: esistono evidenze sull’alterazione del bilancio delle lipoproteine e dei
trigliceridi in corso di infezione da Helicobacter. Le concentrazioni di HDL in
buona parte dei lavori risultavano essere basse, in maniera del tutto opposta ai
valori di trigliceridi e LDL. Queste rilevazioni sono state effettuate con
concomitante ricerca delle IgG e IgA anti-Helicobacter, e sono stati registrati
reperti significativi anche in campioni di non fumatori e non diabetici, con
aggiustamenti fatti per indice di massa corporea, età e classe sociale.

37
2. Infiammazione: la presenza di un processo infiammatorio cronico in corso di
infezione ha un ruolo decisamente importante (66). Questa ipotesi può essere
spiegata dalla presenza cronica in circolo di alti valori di citochine infiammatorie,
in particolare di IL-6, TNF-α, IL-1b e IL-8, che decrementano considerevolmente
in seguito ad eradicazione. Inoltre la sovra-espressione di molecole di adesione
promossa dall’Helicobacter pylori risulta essere correlabile all’instaurarsi e
all’aggravarsi di quadri di cardiopatia ischemica.
3. Assetto coagulativo: alti valori di fibrinogeno sono stati evidenziati in pazienti
con Helicobacter pylori, con considerevole diminuzione dopo terapia antibiotica
mirata. In questo senso, sono stati riscontrati anche alti livelli di protrombina in
pazienti con gastrite da Helicobacter pylori, confrontati con soggetti sani o con
gastrite Helicobacter-negativa. Inoltre, in modelli murini, è stata riscontrata
un’aumentata aggregazione piastrinica.
4. Omocisteinemia: in pazienti con Helicobacter pylori sono stati osservati alti
livelli sierici di omocisteina associati a riduzione di folati e diminuito
assorbimento di vitamina B12 (situazione importante anche da un punto di vista
neurologico, vedi seguito).
5. Cross-reattività: ipotesi importante è quella circa il mimetismo molecolare tra
antigeni del batterio e proteine omologhe self. Un esempio è dato dal
rinvenimento di anticorpi diretti contro una particolare proteina, hsp65, dei quali è
stato osservato un sostanziale decremento in seguito ad eradicazione
dell’Helicobacter. Inoltre, gli anticorpi anti-CagA pare reagiscano con
componenti citoplasmatiche e nucleari delle cellule muscolari liscie e endoteliali
6. Danno diretto: alcuni evidenze si sono incentrate, infine, sulla ricerca del DNA
batterico nelle placche aterosclerotiche con metodiche di polimerase chain
reaction (65).
I processi infettivi cronici si collocano come fattori di rischio importanti anche per
un’altra patologia altamente debilitante, lo stroke, e la loro identificazione e
terapia può contribuire alla profilassi. In questo caso, però, non è il singolo
batterio ad essere determinante, ma la pluralità dei patogeni a cui un soggetto può
essere esposto, e tra questi non va trascurato anche l’Helicobacter pylori (67).

38
b) Apparato respiratorio
In pazienti con reflusso gastro-esofageo e concomitante infezione da
Helicobacter pylori, qualora il batterio dovesse ritrovarsi all’interno del materiale
refluito, questo potrebbe colonizzare il sistema respiratorio e, potenzialmente,
scatenare un’infiammazione. E’ stata presa in esame una possibile relazione
dell’infezione con alcune patologie polmonari ostruttive croniche (COPD),
osservando la sieroprevalenza dell’infezione mediante titolazione delle IgG anti-
Helicobacter in pazienti affetti da tali patologie, e valutando la FEV1 (Forced
expiratory volume in 1 second) in un gruppo di pazienti Helicobacter-positivi e
negativi. Nel gruppo di soggetti positivi, è stata osservata una riduzione della
FEV1 più marcata rispetto ai pazienti negativi, il che suggerisce un’associazione
quantomeno prognostica con l’infezione (68).
E’ stata ipotizzata anche un’associazione tra l’esposizione al batterio e le
patologie allergiche respiratorie. Infatti secondo la “hygiene hypotesis”,
l’esposizione ai microbi, e in particolare alle infezioni croniche, attraverso lo
sviluppo di un fenotipo immunitario meno suscettibile, pare protegga dalle
manifestazioni allergiche e, nel caso dell’apparato respiratorio, soprattutto
dall’asma bronchiale. Tuttavia, gli studi condotti a riguardo non hanno chiarito a
fondo questa possibile associazione (69). E’ stato condotto anche uno studio sul
possibile ruolo patogenetico dell’Helicobacter pylori con la presenza di
bronchiectasie, tuttavia senza un riscontro positivo (70).
Interessante è, invece, una meta-analisi realizzata da Zuho et al., che metteva in
relazione l’infezione da Helicobacter e il carcinoma polmonare (71). Questa
ipotesi è nata dal presupposto che il polmone condivida le stesse origini
embrionali (endoderma) con le cellule che costituiscono l’apparato digerente.
Zuho et al. hanno selezionato quattro studi caso/controllo, e hanno osservato una
prevalenza dell’infezione da Helicobacter pylori nel 75.5% dei pazienti affetti da
carcinoma e il 57.1% nei controlli. Probabilmente, il meccanismo scatenante
risiede nella up-regulation dell’espressione della gastrina, della quale è
riconosciuta l’attività promuovente la proliferazione cellulare e l’angiogenesi.
Allo stesso tempo anche tossine, fattori di virulenza (CagA soprattutto) e
componenti batteriche possono rendere le cellule bronchiali maggiormente

39
responsive all’azione lesiva di sostanze genotossiche, come il fumo di sigaretta o
alcune componenti dell’inquinamento ambientale (72).
Un’altra patologia neoplastica dell’apparato respiratorio in cui l’Helicobacter
pylori può giocare un ruolo è il carcinoma laringeo. Sempre Zuho et al., in un
lavoro precedente a quello sopra riportato, suggerivano che l’infezione, associata
a reflusso gastro-esofageo e all’indebolimento della mucosa laringea da parte di
tossici ambientali e fumo di sigaretta, potesse essere determinante, seppure in
maniera indiretta, nella patogenesi di questa neoplasia (73)
c) Sistema nervoso
L’Helicobacter pylori potrebbe essere implicato nella patogenesi anche di alcune
patologie neurologiche, prima fra queste la malattia di Alzheimer. Sono stati
valutati i livelli di IgG anti-Helicobacter nel liquido cerebro-spinale di alcuni
pazienti affetti da morbo di Alzheimer, con incremento significativo rispetto ai
controlli, e miglioramento in seguito ad eradicazione (75). I possibili meccanismi
patogenetici sono svariati: la gastrite cronica da Helicobacter pylori, per esempio,
provoca un malassorbimento di vitamina B12 e folati, con difetto nel meccanismo
di metilazione del 5-metil-tetraidrofolato e accumulo di omocisteina. La stessa
omocisteina può dare il via a meccanismi di danno endoteliale che hanno come
risultato disordini atero-trombotici, contribuendo all’insorgere dell’Alzheimer.
Figura 18 - Possibili modalità attraverso le quali l'Helicobacter pylori può contribuire alla patogenesi della
malattia di Alzheimer

40
La stessa omocisteinemia, inoltre, può essere considerata un fattore prognostico di
gravità della demenza. L’Helicobacter pylori, per giunta, promuove l’incremento
dell’aggregazione piastrinica e il rilascio di un grosso quantitativo di citochine
pro-infiammatorie, fattori vasoattivi, eicosanoidi, e proteine di fase acuta, tutte
sostanze coinvolte in disordini di tipo vascolare. L’induzione delle cellule
mononucleate a produrre fattori pro-coagulanti che convertono il fibrinogeno in
fibrina, la formazione di radicali liberi dell’ossigeno (che danneggiano la barriera
endoteliale e promuovono l’adesione leucocitaria) e l’incremento della
produzione di ossido nitrico, sono tutti fattori che possono compartecipare alla
compromissione cerebrale (fig. 18) (76-77).
Le infezioni del tratto gastro-enterico sono state chiamate in causa anche per
un'altra patologia neurologica, il parkinsonismo idiopatico. Tra i possibili
patogeni presi in considerazione come fattori modificanti la prognosi, è stato
quotato anche l’Helicobacter pylori, risultando essere un elemento prognostico
sfavorevole (78). E’ stato osservato che il batterio è in grado di sfruttare l’L-
Dopa per i propri fini di sintesi e colonizzazione della mucosa gastrica (cosa che è
stato possibile valutare in terreni di coltura arricchiti con L-Dopa e noradrenalina,
con accelerazione dei processi di crescita del batterio), con conseguente
alleviamento in seguito a terapia eradicante (79). Inoltre l’infezione da
Helicobacter pylori pare possa contribuire nel peggioramento stesso della
malattia, con passaggio da un parkinsonismo ipercinetico ad un quadro bradi-
ipocinetico (80).
d) Sistema endocrino
Per quanto riguarda la patologia endocrina, studi effettuati negli ultimi
anni hanno preso in considerazione il possibile ruolo patogenetico
dell’Helicobacter pylori nell’insorgenza di patologie tiroidee, in particolare di
natura autoimmune. Questa associazione (posta anche la relazione tra tali auto-
anticorpi e quelli anti-tireoperossidasi e anti-tireoglobulina) è stata maggiormente
osservata in pazienti con infezione da ceppi CagA-positivi, con produzione di
auto-anticorpi monoclonali anti-CagA cross-reagenti con proteine della colloide
tiroidea. Inoltre nel contesto dell’isola di patogenicità cag è stato riscontrato un
gene codificante una perossidasi endogena, con possibile produzione di
immunoglobuline anche verso analoghi self (81). L’associazione Helicobacter

41
pylori-tiroide pare essere anche correlata ad un particolare profilo
immunogenetico legato agli antigeni HLA (nello specifico A1, B8 e
DRB1*0301), dato estrapolato da uno studio su pazienti di giovane età (82). A
rafforzare questa tesi contribuisce il dato della riduzione del titolo degli anticorpi
anti-tiroide in pazienti con infezione da Helicobacter pylori, osservato in seguito
ad eradicazione del batterio (83).
Altra patologia endocrina chiamata in causa tra le manifestazioni extra-digestive è
il diabete mellito. In alcuni studi, tuttavia, non è stata trovata alcuna relazione tra
l’infezione e lo status glicemico (84-85-86). Al contrario, uno studio di Gunji et
al. (2009) esaminava l’associazione tra l’infezione da Helicobacter pylori e
l’insulino-resistenza, dimostrando che “l’infezione da Helicobacter pylori
contribuisce significativamente e indipendentemente nel promuovere l’insulino-
resistenza in una popolazione asintomatica” (87). Wang et al, in uno studio
condotto su 130 pazienti con diabete mellito di tipo II, hanno dimostrato che
l’Helicobacter influisce sui livelli di glicemia giornalieri (88). Un’altra prova
giunge da uno studio di Estraghian et al., in cui su 71 pazienti asintomatici, 43
riportavano infezione da Helicobacter pylori e elevati livelli di insulina sierica a
digiuno, il che sottolinea la possibile relazione tra i due fattori (89). Importante è
anche il dato relativo a un lavoro di So et al (2009), in cui si prendevano in
considerazione gli effetti dell’infezione sulle cellule β pancreatiche su una
popolazione di 288 soggetti. In questo studio, è stato notato come il titolo di
anticorpi anti-Helicobacter pylori, così come il livello di adiponectina e di
leucociti, fossero fattori predittivi indipendenti per iperglicemia e ridotta
sensibilità all’insulina, cosa che contribuisce alla spiegazione dell’alta percentuale
di soggetti affetti da diabete di tipo II (nella fattispecie nella popolazione cinese) e
di come ciò, almeno in questo caso, sia slegato dallo stato di obesità (90).
e) Manifestazioni ematologiche
L’Helicobacter pylori potrebbe rivestire un importante ruolo in una
situazione clinica molto comune, l’anemia ipocromica iposideremica.
Recentemente sono stati condotti degli studi in America Latina (esattamente in
Bolivia, Brasile, Cuba, Messico e Venezuela) senza alcun riscontro
epidemiologico associativo (91). Tuttavia, in queste nazioni la componente
iposideremica delle anemie risulta essere minore rispetto al resto del mondo, e ciò

42
è dovuto anche al fatto che altre cause, tra cui malaria, schistostomiasi, carenze
alimentari e condizioni ereditarie (sferocitosi, talassemia), spesso non vengono
segnalate nel raccordo anamnestico dei soggetti presi in esame, compromettendo i
risultati degli studi (76). Contrariamente a queste osservazioni, infatti, altri studi
hanno portato avanti questa ipotesi: in uno studio condotto su bambini di età
compresa tra 7 e 12 anni, è stata osservata una significativa relazione tra il livello
sierico di ferro, la ferritinemia e il titolo di IgG anti-Helicobacter pylori, la cui
conclusione è che l’infezione da Helicobacter pylori può quantomeno influenzare
l’andamento dell’anemia sideropenica (92). Una meta-analisi di Zhang et al.,
redatta nel 2010, metteva in relazione gli effetti dell’eradicazione del batterio con
l’assetto marziale di pazienti affetti da anemia sideropenica. Sono stati inclusi
nello studio 8 studi per un totale di 800 partecipanti, osservando che, in seguito a
terapia eradicante condotta con successo, in un sottogruppo di pazienti si
osservava un incremento dei livelli ferritina (a un mese e a due mesi dall’inizio
del trattamento). Nelle conclusioni, gli autori di questo studio suggeriscono un
probabile ruolo dell’Helicobacter pylori nell’ostacolare l’assorbimento orale di
ferro e, quindi, nel contribuire allo sviluppo di una iposideremia (93). Oltre a
questo, il possibile sanguinamento occulto in pazienti con erosioni del tratto
gastro-intestinale (specie se affetti da ceppi CagA-positivi) può sortire lo stesso
effetto. In soggetti portatori di infezione cronica da Helicobacter pylori, inoltre,
sono stati riscontrati anticorpi anti-H+,K+-ATPasi, o comunque una auto-
immunità esaltata, e da ciò è possibile supporre un’origine auto-immunitaria
anche per l’anemia sideropenica Helicobacter-indotta (94).
Sempre nel contesto della patologia ematologica, l’Helicobacter pylori è stato
accostato anche alla porpora trombocitopenica idiopatica. In svariati studi è
stata osservato un incremento delle piastrine in seguito a eliminazione del
batterio, in contrapposizione, tuttavia, ad altri studi in cui non è stato riscontrato
alcun beneficio dalla terapia eradicante (citato in 72). Ciò nonostante, Scandellari
et al. suggerivano che il possibile ruolo dell’Helicobacter in questa particolare
patologia potesse essere appannaggio dei ceppi CagA-positivi. In pazienti affetti
da questa variante batterica, l’eradicazione ha sortito un effetto positivo
soprattutto nel gruppo di pazienti con anticorpi anti-CagA. Da ciò i ricercatori
hanno ipotizzato che ci potessero essere analogie tra CagA e alcune componenti
peptidiche piastriniche, e, di conseguenza, un possibile meccanismo di mimetismo

43
molecolare alla base dell’associazione tra questo particolare tipo di porpora e
l’Helicobacter pylori. Concludendo, gli autori di questo studio consigliano lo
screening e l’eradicazione del batterio in pazienti affetti da porpora
trombocitopenica idiopatica, specie se con presenza di anticorpi anti-CagA (95).
f) Manifestazioni reumatologiche
La partecipazione degli agenti infettivi nella patogenesi delle malattie
reumatiche è un punto che si è andato sempre più definendo negli ultimi venti
anni, riconoscendo predisponente l’attività artritogenica di virus e batteri. Non
sempre, però, è possibile isolare i microrganismi responsabili proprio a causa del
sistema immunitario, particolare che, naturalmente, lascia un velo di dubbio sulla
loro reale partecipazione (96).
La documentata attività promotrice dell’autoimmunità da parte dell’Helicobacter
pylori è stata chiamata in causa anche per la patologia reumatica, in particolare
per la Sclerosi Sistemica. Sebbene alcuni studi non abbiano rilevato una
correlazione tra tale patologia, il fenomeno di Raynaud che spesso si accompagna
a essa e l’infezione da Helicobacter pylori (97), altri lavori hanno, invece,
osservato un incrementato tasso di infezione, soprattutto da parte di ceppi CagA-
positivi, in pazienti con Sclerosi Sistemica (98). Alla base dell’associazione,
ritroviamo anche in questo caso il mimetismo molecolare, le reazioni
autoimmunitarie e l’attivazione della risposta cellulo-mediata (99).
Un'altra possibile associazione vagliata è quella con la Sindrome di Sjögren,
tuttavia i risultati di vari studi condotti a riguardo risultano essere contrastanti
(100-101).
Il ruolo dell’Helicobacter pylori nelle malattie reumatiche è stato investigato
anche per le malattie del connettivo. In particolare, uno studio di Kalabay et al.
(2002) poneva particolare attenzione sulla presenza di anticorpi cross-reagenti
tra la proteina hsp60 umana (heat shock protein 60) e la proteina hsp65
micobatterica, evento spesso presente nei disturbi autoimmuni concomitanti ad
infezione da Helicobacter. Alti livelli di anticorpi anti-hsp65 sono stati rilevati nei
pazienti affetti delle patologie sopra citate e Helicobacter-positivi, rispetto al
controllo di pazienti non infetti, suggerendo un possibile ruolo del batterio in tali
patologie autoimmuni (102). Quest’ultimo dato, tuttavia, trova come punto debole

44
la sieroprevalenza, che risulta essere bassa per quanto riguarda patologie quali
Lupus Eritematoso Sistemico, Dermato-polimiosite, Artrite Reumatoide e
Sindrome di Raynaud Primitiva (102-103).
g) Cute
Quello dermatologico è un altro possibile scenario in cui l’Helicobacter
pylori può avere una parte. Le evidenze al momento presenti in letteratura
indirizzano verso un collegamento con alcune patologie, in particolare con
l’orticaria cronica. Uno studio del 2009 di Abdou et al., si è avvalso di biopsie
effettuate tramite gastroscopia in 35 pazienti affetti da questo particolare disturbo,
per verificare l’effettiva incidenza dell’infezione e il suo eventuale collegamento
con l’orticaria cronica. Il 57% di essi erano positivi per l’Helicobacter pylori e
accusavano una sintomatologia più severa rispetto al resto del campione, con
remissione in seguito a terapia antibiotica specifica (104). Sempre sull’efficacia
della terapia eradicante in questa associazione, Akashi et al. hanno condotto uno
studio su 17 pazienti affetti da orticaria cronica e prurigo chronica multiformis,
evidenziando un effettivo miglioramento soprattutto nei soggetti con quest’ultima
variante. Gli autori, in definitiva, suggerivano di prendere in considerazione la
terapia eradicante, soprattutto in casi intrattabili proprio per il possibile ruolo
aggravante dell’Helicobacter pylori (105).
L’acne rosacea (o semplicemente rosacea) è una dermatite cronica che interessa
di solito l'area centrale del volto, e si manifesta con eritema, teleangectasie e
lesioni acneiformi. L’etiopatogenesi è incerta, e proprio a questo riguardo è stata
indagata la possibile causa infettiva, ponendo particolare riguardo per alcuni
patogeni quali Helicobacter pylori, Staphylococcus epidermidis e Chlamidia
pneumoniae. Sebbene alcune osservazioni risultino essere poco convincenti (106),
esistono in letteratura evidenze di un possibile ruolo proprio dell’Helicobacter
pylori. Effettivamente la prevalenza dell’infezione in pazienti affetti da rosacea è
risultata essere significativamente più elevata rispetto ai non infetti, dato rilevato,
ad esempio, in un lavoro effettuato nella città iraniana di Kerman nel 2003, in cui
su 29 individui affetti da rosacea i livelli di IgG anti-Helicobacter apparivano

45
nettamente maggiori rispetto al controllo (107). Oltre a ciò è stato evidenziato un
miglioramento della sintomatologia (sebbene in diversi gradi) in seguito a terapia
eradicante, soprattutto nei soggetti con variante papulo-pustolosa (108).
Altre due patologie dermatologiche sulle quali è possibile trovare in letteratura
delle associazioni con l’Helicobacter pylori sono il lichen planus e la psoriasi.
Per quanto riguarda la prima, è stato condotto uno studio basato su rilevamenti
bioptici endoscopici, che ha0 evidenziato la presenza di DNA batterico sia nelle
lesioni orali sia in quelle gastriche (109). Qayoom e Ahmad, invece, hanno
condotto uno studio sulla prevalenza dell’infezione in soggetti psoriasici del
dipartimento di dermatologia di Srinagar, nel Kashmir, con valori di IgG anti-
Helicobacter sì inscrivibili nei range di aspettativa per la regione in esame, ma
significativamente aumentati nel gruppo in esame rispetto al controllo di pazienti
non infetti (110).
h) Tessuto osseo
In letteratura esistono alcuni lavori che considerano l’osteoporosi come
una possibile manifestazione extra-digestiva dell’infezione da Helicobacter
pylori. Per la verità si tratta di un numero limitato di studi, ma da cui, tuttavia, si
può estrarre qualche dato interessante.
Nel 2005, Figura et al. si avvalsero della misurazione delle concentrazioni di
citochine regolatrici del turn-over osseo (TNF-α, IL-6 etc.) per verificare la
prevalenza di infezione (specie da parte di ceppi CagA-positivi) in pazienti
maschi affetti da osteoporosi, e, eventualmente, dimostrare l’esistenza di una
relazione tra i due fattori. Le differenze tra le concentrazioni dei vari markers,
sebbene differenti tra caso e controllo, non hanno raggiunto valori statisticamente
significativi, tuttavia è emerso che l’Helicobacter pylori può essere considerato
tra i fattori di rischio per l’osteoporosi maschile (111).
Kakehasi et al., in due lavori del 2007 e del 2009, tramite densitometria ossea,
hanno ricercato una possibile via attraverso la quale l’Helicobacter pylori potesse
incidere sull’insorgenza dell’osteoporosi in epoca post-menopausale, e di come la
gastrite cronica agisse sulla densità minerale dell’osso nelle donne, senza tuttavia
trovare una correlazione precisa in entrambi i casi (112-113).

46
Ozdem et al., invece, nel 2007, tramite misurazione dei markers di turn-over
osseo, hanno ricercato l’eventuale esistenza di un rimaneggiamento osseo in corso
di infezione nei bambini, osservando, però, soltanto una diminuzione dei livelli di
vitamina B12 (114).
i) Patologia urologica
Una review di Mohammed Al-Marhoon del 2008, si poneva come quesito
il ruolo dell’Helicobacter pylori nella patologia urologica, raccogliendo le
evidenze a carico sia di patologie neoplastiche sia di altra natura. Per quanto
riguarda il rene, è raccomandata l’eradicazione del batterio, qualora presente, in
pazienti uremici prossimi al trapianto, e, inoltre, è stata osservata un’aumentata
suscettibilità al linfoma renale in soggetti oltre i cinquanta anni di età ed infetti. A
livello della vescica, anche in questo caso la presenza del batterio è stata messa in
relazione con una maggiore incidenza di linfomi, con sostanziale regressione in
caso di eradicazione. La patologia prostatica, invece, è stata accostata
all’Helicobacter pylori soprattutto per il possibile concorso di quest’ultimo nello
sviluppo di prostatiti a carattere cronico. Probabilmente la relazione sta nello
sviluppo di una reazione infiammatoria secondaria ad antigeni sconosciuti, con un
aumento significativo di citochine rilevabili anche nel liquido seminale,
diventando quindi una possibile causa occulta. La patologia neoplastica è ancora
una volta riconducibile all’insorgenza di linfomi, probabilmente in seguito ad
eventi infiammatori cronici con sviluppo di tessuto displastico (115).
j) Patologia ginecologica
Per quel che concerne le patologie di interesse ginecologico, sono presenti
in letteratura alcuni studi circa le implicazioni dell’Helicobacter pylori nella
sindrome dell’ovaio policistico. E’ noto che le infezioni croniche possono
aumentare il rischio di infertilità nelle donne, e la PCOS è una delle cause più
comuni proprio di questa condizione. A questo riguardo è stata osservato un’alta
sieropositività in pazienti portatrici di tale sindrome evidenziata tramite ricerca
delle IgG specifiche (116).
Sono state inoltre prese in esame alcune patologie ostetriche, in particolare
l’iperemesi gravidica e la pre-eclampsia. L’esposizione all’Helicobacter pylori
sembra sia associata ad un incrementato rischio di iperemesi gravidica, sebbene

47
sia presente un’eterogeneità tra risultati di vari studi (117). Per quanto riguarda
invece la pre-eclampsia, è stato osservato un incremento della prevalenza e del
rischio di gestosi nelle pazienti portatrici del batterio (118).
k) Infertilità
Le modalità con cui l’Helicobacter pylori può interferire con la fertilità sia
femminile sia maschile (soprattutto per questa trattazione) verranno esposte nel
capitolo 2 e nella sezione sperimentale.
1.7 Diagnosi di infezione
Di seguito verranno esposte brevemente le metodiche attraverso le quali è
possibile effettuare diagnosi di infezione da Helicobacter pylori. Possiamo
suddividere le tecniche a nostra disposizione in due categorie: metodiche non
invasive e invasive.
a) Tecniche non invasive
I test non invasivi sono fondamentali per il management dei pazienti con
infezione, in quanto di più facile e ripetibile esecuzione e indispensabili nella
strategia “test and treat”.
Quest’ultimo punto prevede di effettuare questo tipo di test in pazienti di età
inferiore a 45 anni, non dispeptici, che non assumono FANS e in assenza di
“sintomi di allarme”. Inoltre tali metodiche sono di estrema utilità nel controllo di
pazienti trattati per valutare l’effettiva eradicazione del batterio.

48
� Analisi sierologica
La ben nota risposta anticorpale all’invasione della mucosa determina la
produzione di IgA e IgM a livello locale (riscontrabili nei succhi gastrici) e IgG
sistemiche. Le tecniche di valutazione degli anticorpi sono molteplici, tuttavia
risultano essere più convenienti quelle su “fase solida”, ovvero basati sul legame
di un antigene su una micropiastra e successiva reazione con gli anticorpi
specifici, presenti nel campione, e anticorpi marcati con molecole fluorescenti o
radioattive.
La metodica ELISA (Enzime Linked ImmunoAdsorbent Assay) , di gran lunga la
più utilizzata, si basa sulla coniugazione dell’anticorpo con enzimi (perossidasi,
fosfatasi alcalina) e sull’aggiunta di coloranti (p-nitrofenilfosfato) per determinare
la presenza di antigeni specifici o anticorpi specifici, con una specificità del 85%
e una sensibilità del 78%. Altre tecniche utili sono il Western Blot, che sfrutta la
separazione delle proteine cellulari tramite elettroforesi, e le reazioni di
agglutinazione con gelatine particolari o sfere di lattice.
L’efficacia dell’indagine sierologica è evidente soprattutto a scopi epidemiologici
o per indagini sui ceppi particolari, quali per esempio i CagA-positivi, oppure per
studi di patogenesi. Per quanto riguarda invece la diagnosi di infezione in atto (ci
danno solo dati sull’avvenuto contatto con il batterio ma non di infezione attiva) o
l’avvenuta eradicazione, queste metodiche sono poco attendibili, in quanto per
avere una significativa riduzione del titolo dell IgG anti-Helicobacter sono
necessari almeno 6 mesi, con una diminuzione media del 25% a 3 mesi e del 50%
a 6 mesi.
� UBT, Urea Breath Test
Analogamente al test all’ureasi, questo sistema sfrutta la capacità del
batterio di produrre questo particolare enzima. Viene utilizzata dell’urea marcata
con 14C o 13C (in Europa si utilizza il 13C), che diffondono attraverso lo strato
mucoso e vengono scisse in anidride carbonica e ammonio. Dopo poco tempo, è
possibile riscontrare l’isotopo marcato nel respiro, in quanto questo diffonde nei

49
vasi della mucosa gastrica. Previa sospensione di terapie a base di PPI, antibiotici
e bismutati almeno una settimana prima del test, viene fatto ingerire il composto
al paziente, determinando poco dopo nell’aria espirata la concentrazione di CO2
tramite apposito scintillatore, che esprime la radioattività in termini percentuali.
La sensibilità e la specificità del test si aggirano intorno al 97% e al 95%. Ad
oggi, il test UBT è considerato il gold standard non invasivo per la
determinazione dell’infezione da Helicobacter pylori.
.
� Determinazione degli antigeni fecali
Un altro strumento a disposizione del clinico per la determinazione
dell’infezione è il test immunoenzimatico su un campione di feci. Sensibilità e
specificità di questa metodica si aggirano intorno al 91% e al 93%, e alcuni studi
(in particolare l’European Helicobacter pylori study group nel 2000 e 2005)
raccomandano l’esecuzione di tale test in concomitanza con l’UBT, per una
diagnosi di certezza e in corso di follow-up.
b) Tecniche invasive
� Valutazione istologica
Le tecniche di diagnosi invasive presuppongono un approccio
endoscopico, tramite il quale è possibile un campionamento seriato della mucosa
e la successiva analisi. Alcuni dati suggeriscono che l’esecuzione di una sola
biopsia, effettuata lungo la piccola curvatura, garantisca la positività del campione
nella quasi totalità dei casi, tuttavia la sensibilità dell’esame può essere
implementata attraverso l’ulteriore campionamento di tessuto a livello dell’antro e
del fondo gastrico, soprattutto in uno stomaco con presenza di metaplasia. Una
volta campionato, il tessuto viene sottoposto a doppia colorazione ematossilina-
eosina, permettendo così la valutazione del grado di flogosi e del relativo
infiltrato infiammatorio. Le colorazioni Giemsa o Genta, inoltre, permettono
l’identificazione del batterio nel campione. Naturalmente, l’endoscopia permette

50
la valutazione della mucosa gastrica, e ciò rende questa metodica estremamente
utile specie in pazienti con gastrite manifesta e/o metaplasia.
� Esame colturale
Sempre grazie all’esame endoscopico è possibile prelevare materiale per
valutare l’infezione da Helicobacter pylori tramite coltura. E’ necessario
effettuare l’esame colturale in breve tempo, a causa della facilità con cui in
ambiente esterno il batterio perde le sue caratteristiche (anche se le biopsie
possono essere conservate per 24h a 4°C in un apposito medium). I terreni di
coltura utilizzati sono basati generalmente sulla formula agar-sangue, con
aggiunta di sostanze quali ciclodestrina, albumina bovina o antibiotici per creare
terreni selettivi. Come già descritto
nelle sezioni precedenti, il batterio
predilige condizioni di micro-aerofilia
(5% O2, 5-10% CO2), a una temperatura
di 37 °C per minimo sette giorni.
La specificità della tecnica è
elevatissima, tuttavia la sensibilità può
essere inficiata da inadeguato
campionamento, esposizione delle
coltura ad aria o operatore-dipendenza.
Anche in questo caso, ai fini di una corretta esecuzione, è necessaria la
sospensione da parte del paziente di terapie a base di PPI da almeno due
settimane.
� Test all’ureasi
La presenza di elevate concentrazioni dell’enzima ureasi al di sotto dello
strato di muco della parete gastrica in corso di infezione da Helicobacter pylori, è
alla base di quest’ultima tecnica che, solitamente, completa l’esame endoscopico.
Prelevato il campione di tessuto, questo viene messo a contatto con una soluzione
contenente urea: il test risulta essere positivo qualora, in presenza dell’enzima e,
quindi, dell’infezione, avvenga una scissione dell’urea, cosa che rende alcalina la
Figura 19 - Test all'ureasi: reperto positivo (a
destra), con caratteristico viraggio della
colorazione, e controllo (a sinistra).

51
soluzione. Ciò viene apprezzato attraverso un indicatore di pH, che evidenzia un
viraggio nella colorazione (figura 19). Questo test ha un’elevata accuratezza
diagnostica, tuttavia la sensibilità dipende dal numero di batteri presenti nella
biopsia e dallo spessore dello strato di muco (esso, infattti, può inficiare l’esame
se in pazienti con carica batterica importante esiste un elevato grado di gastrite,
con assottigliamento dello strato e una bassa rappresentazione di ureasi). Anche in
questo caso sono raccomandate biopsie multiple.
� Indagini molecolari
La PCR (Polymerase Chain Reaction) è sicuramente la metodica
molecolare più utilizzata a scopo di ricerca per le indagini di infezione, soprattutto
per valutare la presenza di Helicobacter pylori nelle biopsie, nel succo gastrico,
nelle feci e in altri distretti. Sono parecchi i loci target di amplificazione utilizzati
per diversi primer, e tra questi ricordiamo il 16s RNA, i loci delle ureasi (A, B e
C), i loci di varie citotossine (vacA e cagA) e quello delle proteine da shock
termico. Inoltre alcune metodiche di amplificazione nucleica possono essere
utilizzate per la ricerca in campo terapeutico, come per esempio le antibiotico-
resistenza dovute a mutazioni puntiformi.
2. Infertilità
2.1 Definizione
Il concetto di infertilità è comunemente sovrapposto a quello di sterilità,
sebbene i due termini sottintendano due significati tecnicamente differenti. In
effetti, effettuando una semplice ricerca su internet è facile incappare in una
moltitudine di definizioni diverse, spesso discordanti o addirittura contraddittorie.
Storicamente, le idee stesse di infertilità e sterilità sono state quasi esclusivamente
relegate al sesso femminile, tuttavia le evidenze accumulatesi nel corso dei

52
decenni hanno contribuito a delineare con maggiore specificità il ruolo della
componente maschile e alla nascita del concetto di “coppia infertile” o “sterilità
di coppia”, inteso come una condizione di impossibilità nel riprodursi non ancora
approfondita clinicamente.
Sfogliando alcuni vecchi testi, nella donna, il termine sterilità sta ad indicare
l’assoluta incapacità riproduttiva, mentre per infertilità è descritta l’inidoneità
della gestante a portare a compimento una gravidanza fino all’epoca di vitalità del
feto. Nell’uomo, il concetto è più articolato, in quanto per uomo infertile viene
inteso il soggetto portatore di una qualche insufficienza del liquido seminale,
inadatto quindi a concepire. Quella di maschio sterile, invece, è una
rappresentazione che contempla l’assoluta mancanza di spermatozoi vivi e/o
validi nell’eiaculato (119).
Ad oggi il Consiglio internazionale per la distribuzione dell’informazione
sull’infertilità (INCIID, International Council on Infertility Information
Dissemination) e la maggior parte degli Autori specificano il concetto di
infertilità come “l’incapacità di concepire dopo un 1 anno di rapporti in assenza
di contraccezione”, presupponendo un sostanziale problema di “performance” a
monte dell’atto della riproduzione (120-121). In questa definizione va inoltre fatta
una distinzione, ovvero tra una infertilità primaria, in cui non è mai stato
possibile il concepimento, ed una secondaria, cioè qualora questa situazione si
verifichi dopo un periodo di documentata fertilità.
2.1 Infertilità femminile

53
L’infertilità femminile è un problema comune, esteso a tutte le latitudini e che
annovera disparate cause, con importanti implicazioni sia di tipo demografico che
in materia di salute pubblica. Una review del 2007 che esaminava studi su 25
popolazioni, con un campione totale di 172.413 donne, indicava una prevalenza di
infertilità del 9%, a fronte di altre stime con dati oscillanti tra il 3% e il 16%.
Basandosi su tale percentuale, Boivin et al. sostengono che ad oggi 72,4 milioni di
donne al mondo sono infertili e 40,5 milioni di esse sono alla ricerca di supporto
medico (esattamente il 56%) (122). Altri studi fissano il dato di prevalenza sul
14% della popolazione femminile mondiale, con valori che possono raggiungere
il 30% nell’Africa Sub-sahariana, il 12% in Europa e il 7.4% negli Stati Uniti.
Uno studio condotto nella provincia di Yazd, in Iran, fissa il dato al 5.3% per le
aree urbane e al 6.8% per le aree rurali, percentuali, quindi, più basse rispetto alle
altre nazioni (123).
Il declino della fertilità femminile (il picco di fertilità si posizione all’età di 23
anni circa) è un evento che si verifica solitamente tra i 32 e i 37 anni d’età per la
progressiva atresia ovarica e la riduzione degli ovociti fecondabili; tuttavia negli
ultimi anni si è assistito ad un netto incremento delle nascite in età superiore ai 30
anni (124).
Il grafico 1 mostra quali sono le cause principali di infertilità femminile, non
tenendo conto però della componente maschile (che risulta essere il 25% di tutte
le cause di infertilità di coppia).
Grafico 1 - Cause principali di infertilità femminile (fonte Stanford University,
www.stanford.edu)

54
Tali percentuali sono abbastanza uniformi in tutto il globo, come testimoniano
studi effettuati in Nigeria (125), Canada (126) o Israele (127-128).
� Infertilità femminile e Helicobacter pylori
Circa il ruolo delle infezioni nell’infertilità femminile si è già accennato
precedentemente; tuttavia il punto da analizzare in questa sede è il possibile
legame tra l’infezione da Helicobacter pylori e questa purtroppo frequente
condizione femminile. I dati in letteratura su questa associazione non sono molti,
tuttavia è possibile ritrovare vari esempi a supporto di questi tesi.
Kurotsuchi et al. incentrarono i loro sforzi sulla plausibilità del nuovo concetto di
“infertilità Helicobacter-correlata” attraverso uno studio retrospettivo su 204
donne. Questo lavoro evidenziò un’aumentata percentuale di sieropositività per
questa infezione nel campione di pazienti con infertilità idiopatica rispetto al
gruppo in cui era riconosciuto almeno un fattore di infertilità; tuttavia in questa
sede non è stato possibile evidenziare un dato epidemiologico significativo (130).
Nel 2010 un’equipe di ricercatori dell’Università degli Studi di Padova ha
pubblicato uno studio che poneva al centro dell’attenzione la presenza di Ig anti-
Helicobacter nel muco cervicale quale causa di infertilità. Sono state arruolate 67
donne con diagnosi di infertilità idiopatica, escludendo turbe ovariche tramite
misurazione ormonale e assenza di patologia tubarica o uterina tramite istero-
salpingografia e ecografia (sia trans-addominale che trans-vaginale). Inoltre sono
state indagate ed escluse infezioni da Chlamydia, Mycoplasma, virus epatitici,
HIV, patogeni del gruppo TORCH ed eventuali cause di infertilità maschile nei
partner. Nelle secrezioni cervicali di pazienti in periodo ovulatorio e positive
all’infezione sono stati individuati alti livelli di IgG anti-Helicobacter. Il muco
cervicale di queste donne è stato fatto interagire con del liquido seminale,
osservando che dopo un’ora soltanto pochi spermatozoi erano stati in grado di
progredire, mentre nel gruppo di controllo (donne non infette) la motilità degli
spermatozoi non è sembrata compromessa (cosa ancora più evidente dopo due
ore). La conclusione cui sono giunti i ricercatori è che la presenza di anticorpi

55
anti-Helicobacter pylori all’interno del muco cervicale possa in qualche modo
interferire con la motilità degli spermatozoi e di conseguenza impedire la
penetrazione di questi ultimi e quindi il concepimento, tuttavia l’esatto
meccanismo attraverso il quale ciò avvenga non è conosciuto. Queste evidenze
fanno sì che l’infezione sia iscritta nelle possibili cause d’infertilità femminile,
essendo assente in questo caso una componente patologica maschile, e fanno dello
screening di questa infezione un’ulteriore utile indagine in caso di infertilità di
coppia senza causa apparente. I titoli di IgG presenti nel muco e nel siero si sono
dimostrati proporzionali, pertanto gli autori dello studio suggeriscono di effettuare
prima uno screening sierico sulle pazienti, seguito dal campionamento del muco
cervicale qualora esse risultassero positive (131).
2.2 Infertilità maschile
2.3.1. Introduzione
Come anche in precedenza anticipato, nel passato, il problema
dell’infertilità vedeva come protagonista esclusiva la donna. Sebbene il problema
sia molto comune, è arduo stabilire quale sia il reale contributo della componente
maschile, cosa soprattutto dovuta alle difficoltà della diagnosi. Il 15% delle
coppie che tentano un primo concepimento ricorrono ad un iter diagnostico per
infertilità a causa dell’insuccesso. L’8% degli uomini in età riproduttiva si avvale
di assistenza medica per problemi legati alla fertilità e di questi l’1-10% riporta
una condizione che compromette il potenziale fecondante, con il varicocele quale
possibile causa più comune (35% circa) (132).
Come anche riportato per l’infertilità femminile, i dati di prevalenza sono diversi
tra le varie regioni del globo, con tassi maggiori nei paesi in via di sviluppo
(anche se da queste zone si hanno pochi dati epidemiologici circa l’infertilità
maschile) (139). Aspetti occupazionali e socio-economici sono stati presi in
considerazione quali fattori di rischio, con particolare attenzione (sebbene i dati

56
non abbiano raggiunto percentuali di elevato interesse statistico) per alcune
categorie di lavoratori, tra cui gli operai delle centrali radio-elettriche, del settore
tipografico e tessile e alcune categorie di impiegati (133). Stando a quanto
riportato da Sherrod e De Coster, non pare esista una differenza della
distribuzione dell’infertilità maschile in popolazioni di razza diversa dello stesso
paese: per esempio negli Stati Uniti non si osserva una significativa diversità
statistica tra soggetti afro-americani e caucasici (134).
Il problema di fondo resta comunque la diagnosi precoce, in quanto ancora oggi
larga parte degli uomini non si sottopone ad indagini di approfondimento.
L’Organizzazione Mondiale della Sanità ha proposto uno schema per la
classificazione diagnostica dell’infertilità maschile basata su un approccio
standardizzato attraverso la valutazione clinica e spermiografica dei pazienti,
tuttavia, molte di queste classificazioni propongono modelli controversi, non
sempre ben visti da parte di alcuni autori (135). Nonostante ciò, le linee guida
della WHO sono state più volte revisionate e aggiornate, fino all’ultima
pubblicazione avvenuta nel 2010.
Di seguito andremo ad analizzare alcuni aspetti di fisiologia della riproduzione
maschile utili nell’inquadramento del problema dell’infertilità.
� Lo spermatozoo.
Schematicamente possiamo suddividere lo spermatozoo in due tratti
principali, la testa e la coda (flagello). La testa è composta da due sezioni: il
nucleo, in cui è contenuta la cromatina aploide andata incontro a fenomeni di
addensamento (cosa possibile grazie alla presenza di particolari tipi di istoni
ricchi in arginina anziché in lisina), e l’acrosoma, struttura necessaria per
l’interazione con la cellula uovo. Quest’ultimo contiene enzimi litici che vengono
liberati al momento della reazione acrosomiale con l’ovulo, aprendo un varco
nella zona pellucida di quest’ultimo necessario per la fecondazione (136).

57
Il flagello degli spermatozoi umani può essere suddiviso in quattro regioni: una
corta porzione di connessione (collo), la porzione centrale, la porzione principale
e la porzione terminale. A livello del collo è presente un centriolo e la placca
basale, mentre nel tratto intermedio è presente la cosiddetta guaina mitocondriale
(ovvero il serbatoio di energia dello spermatozoo) che termina con una struttura
ad anello (annulus). Il tratto principale è costituito essenzialmente dal filamento
assiale rivestito dalle colonne striate, che finisce nel segmento terminale costituito
dalla sola componente microtubulare. L’assonema, o filamento assiale, percorre il
flagello per tutta la sua struttura, costituendo l’apparato motore dello spermatozoo
con la classica struttura 9+2
a doppiette di microtubuli
(suddivisi in sub-unità A e
B) riscontrabile nelle ciglia
e nei flagelli di molte altre
cellule eucariotiche. La
principale costituente dei
microtubuli è la tubulina,
con molecole riarrangiate in
filari a formare dei
protofilamenti. Tra le subunità A e B si protendono dei “bracci” costituiti da
un’ATPasi Ca2+ Mg2+ dipendente, la dineina, in grado di provocare lo
scivolamento delle doppiette di microtubuli l’uno sull’altra e quindi di garantire il
movimento. Le 9 doppiette di microtubuli sono a loro volta connesse tra di loro da
una proteina, la nexina, mentre i microtubuli centrali sono circondati da spirali di
fibre (137).
� Produzione e maturazione delle cellule spermatiche.
Il punto di partenza della produzione dei gameti maschili risiede nel
testicolo. La produzione degli spermatozoi avviene a livello dei tubuli seminiferi
(contenti le cellule del Sertoli) a loro volta circondati da una componente stromale
(in cui sono invece presenti le cellule di Leydig). La produzione degli
spermatozoi è un evento caratterizzato da vari aspetti, che sono la produzione dei
gameti, la loro maturazione e la regolazione ormonale.
Figura 19 - Rappresentazione anatomica del testicolo (Gray's
Anathomy)

58
Il complesso processo di divisione e differenziazione delle cellule germinali segue
un pattern preciso. La durata del ciclo di spermatogenesi (ovvero il primo step di
formazione degli spermatozoi) nell’uomo è in media di 16 giorni, mentre per lo
sviluppo e la differenziazione degli spermatozoi e la produzione di liquido
seminale maturo è necessario un periodo complessivo di circa 64 giorni. Il ciclo
completo di produzione può essere identificato in tre parti: la spermatogenesi
(ovvero la produzione dei gameti), la spermiogenesi (fase differenziativa) e la
spermiazione (ovvero il rilascio dal testicolo) La spermatogenesi inizia con la
divisione delle cellule staminali per mitosi (spermatogoniogenesi) e il
riarrangiamento meiotico delle cellule staminali da tetraploidi (spermatociti) a
aploidi (spermatidi). La meiosi è una fase critica nella gametogenesi, giacchè in
essa avviengono la ricombinazione del materiale genetico, la riduzione dei
cromosomi e lo sviluppo degli spermatidi. La spermiogenesi è il secondo atto del
ciclo, e può anch’esso essere suddiviso in quattro fasi:
1. Fase del Golgi, in cui appare la simmetria cranio-caudale e la bolla acrosomiale;
2. Fase del cappuccio, in cui gli spermatidi appaiono allungati e c’è lo sviluppo
dell’acrosoma che copre per 2/3 la sezione craniale;
3. Fase acrosomiale, in cui continuano la condensazione del nucleo e
l’allungamento della cellula e la maturazione del flagello arriva a completamento;
4. Fase maturativa, il cui principale evento è l’estrusione del citoplasma in eccesso
in quello che viene definito come corpo residuo.
Figura 20- Fasi di maturazione degli spermatozoi

59
La spermiazione è l’ultimo atto e
prevede il rilascio degli spermatozoi
maturi nel lume tubulare. Questo
evento può essere influenzato da
modificazioni ormonali, temperatura e
tossine.
Le cellule del Sertoli hanno un ruolo
importante in questi eventi. Scoperte
nel 1865 da Enrico Sertoli, sono cellule
somatiche localizzate nell’epitelio
germinale, e risultano essere
mitoticamente inattive nell’età adulta.
Il ruolo di queste cellule è molteplice:
contributo nella differenziazione delle
gonadi, repressione prepuberale della
meiosi, nutrizione/protezione delle cellule germinali, regolazione della
proliferazione e del differenziamento, regolazione dell’apoptosi degli
spermatogoni, produzione d’inibina (inibizione ipofisi) e attivina, concentrazione
degli androgeni, trasformazione del testosterone in di-idro-testosterone,
separazione del compartimento basale dal luminale, fagocitosi di corpi
estranei/batteri e della goccia citoplasmatica (corpo residuo), secrezione del fluido
tubulare, produzione del fattore anti-mülleriano (136).
� Controllo ormonale.
Il controllo ormonale della funzione testicolare è guidata dal sistema
ipotalamo-ipofisario. Il rilascio di LH e FSH da parte dalla ghiandola pituitaria
anteriore è garantito dalla produzione di GnRH da parte dell’ipotalamo attraverso
un meccanismo pulsatile.
Il rilascio di GnRH dipende dall’attivazione del recettore GPR54, stimolato dal
peptide kisspeptina. Nell’uomo il maggior ormone che controlla la secrezione di
GnRH è il testosterone, che può inibire la secrezione di gonadotropine con un
feedback negativo sia sull’ipofisi sia sull’ipotalamo (meccanismo anche sostenuto
Figura 21 - Schema ormonale regolatore della
funzione testicolare

60
dall’inibina B prodotta dalle cellule del Sertoli). Il testosterone è inoltre il
principale ormone rilasciato dal testicolo, seguito dal 5α-di-idro-testosterone,
androsterone, androstenedione, 17-idrossi-progesterone, progesterone e
pregnenolone. Il testosterone è sintetizzato dalle cellule di Leydig stimolate
dall’LH e in minima parte dalla corticale surrenalica, agendo come fattore sia
endocrino che paracrino.
Le gonadotropine e il testosterone a livello gonadico rientrano nei meccanismi di
produzione e maturazione delle cellule seminali. FSH e testosterone (quest’ultimo
di primaria importanza per la differenziazione sessuale) sono sicuramente
implicati nella regolazione della spermatogoniogenesi, soprattutto tramite
l’incremento della popolazione degli spermatogoni Ap, cui segue un aumento
delle successive popolazioni cellulari (vedi figura 21). I meccanismi di meiosi,
invece, sembra che siano slegati dal controllo di FSH e testosterone, mentre il
solo FSH pare giochi un ruolo nella condensazione della cromatina durante la
spermiogenesi (non è ancora chiaro quale sia il ruolo del testosterone in questa
fase). Si può concludere quindi che le azioni di LH/testosterone e FSH sono
necessarie per la promozione, il mantenimento e la re-iniziazione della normale
spermatogenesi.
Figura 22 - Organi target del testosterone
Esistono altri regolatori ormonali oltre a quelli già citati. Sono stati ritrovati
recettori di superficie per alcuni fattori di crescita, come TGFα e β, inibina ed
activina, insuline-like growth factor-1 (IGF-1), nerve-growth factor (NGF),

61
fattore di crescita dei fibroblasti (FGF-1) e fattore di crescita epidermico (EGF-1)
(136).
� Maturazione nell’epididimo.
L’epididimo (suddiviso in caput, corpus e cauda) è un dotto di piccolo
diametro e strettamente avvolto che collega i dotti efferenti dal retro di ogni
testicolo al suo dotto deferente. A questo livello avvengono parte della
maturazione e l’acquisizione da parte dello spermatozoo della motilità coordinata
caratteristica. Il complesso assonemale degli spermatozoi testicolari, e quindi la
capacità di movimento, sono attivi solo in alcune cellule per l’attivazione indotta
dall’ATP, tuttavia, solo attraverso il passaggio nell’epididimo, lo status di
maturità dell’assonema è raggiunto, grazie soprattutto all’acquisizione di fattori
promuoventi e alla rimozione di fattori inibitori. Questo discorso include il
cambiamento del pH e delle concentrazioni di Ca2+ e ATP intracellulari, con
anche la modulazione dell’attività dell’adenilato ciclasi e della proteina fosfatasi
PP1γ2 (la cui inibizione sperimentale può provocare infertilità maschile per turbe
della motilità) (138). Anche il bicarbonato prodotto dalle anidrasi carboniche
delle cellule epiteliali epididimali risulta stimolare gli spermatozoi (136).
Il passo successivo della maturazione dello spermatozoo è lo sviluppo della
capacità di interagire con la cellula uovo, iniziando a livello epididimale per poi
completarsi nel tratto genitale femminile. Questa catena di eventi parte dallo
sviluppo dello spermatozoo dell’abilità di compiere la reazione acrosomiale
spontanea, attraverso modificazioni nella composizione dei lipidi di membrana
che permettono alle membrane dell’acrosoma di interagire con l’oocita. La
capacitazione avviene a livello del tratto genitale femminile, e l’interazione
spermatozoo-zona pellucida pare sia garantita da delle glicoproteine presenti
sull’oocita (glicoproteina Zp3). Tuttavia, è stato osservato che una glicoproteina
secreta dall’epididimo, la proteina P34H, è coinvolta attivamente nello sviluppo
della capacità di legare l’uovo, con dimostrazione di casi d’infertilità legata alla
deficitaria interazione spermatozoo-uovo dovuta proprio alla mancanza di questo
peptide (139).

62
Il liquido seminale viene quindi immagazzinato nell’epididimo. Gli spermatozoi
accumulati in questa sede sono quiescenti, in un ambiente arricchito dal fluido
epididimale, povero in sodio e ricco in potassio. Sono prodotti inoltre superossido
dismutasi e glutatione perossidasi per proteggere gli spermatozoi dal danno
ossidativo, mentre inibitori dell’acrosina evitano reazioni acrosomiali spontanee.
Un complesso di tight-junctions e fattori d’immunosoppressione proteggono
inoltre il liquido seminale da reazioni auto-antigeniche (136).
� Composizione finale del liquido seminale.
Il liquido seminale è composto di un gruppo eterogeneo di secrezioni, non
solo testicolari ma anche provenienti dalle ghiandole accessorie e dalla prostata.
Sono almeno tre i sistemi che contribuiscono alla composizione dell’eiaculato, i
cui prodotti vengono escreti in successione. La prima componente è quella
prostatica (contenente soprattutto fosfatasi acida, zinco, acido citrico e antigene
prostatico specifico), in secondo luogo c’è la componente spermatica proveniente
da testicoli, epididimo (che secerne anche L-carnitina, glicerofosfocolina e α-
glicosidasi neutra), vasi deferenti e ampolla, e per terzo la vescichette seminali
che secernono con i loro liquidi gran parte del fruttosio rinvenibile e provvedono
ad incrementare il volume del liquido seminale. Le ghiandole bulbo-uretrali di
Cowper secernono una soluzione alcalina (liquido di Cowper) con glicoproteine
che neutralizzano l’acidità del tratto urinario prima dell’eiaculazione (136). Il
liquido prostatico inoltre, come anche il fluido seminale nella sua interezza,
contiene anche un certo numero di ioni (principalmente citrato, cloruro, sodio,
potassio, zinco e magnesio), con concentrazioni che possono variare da soggetto a
soggetto (140).
Nel liquido seminale è possibile rinvenire anche alcuni ormoni. Per esempio,
Abbaticchio et al. hanno osservato la presenza di piccole quantità di ormoni
tiroidei (T3) legate all’albumina che incrementano in caso di flogosi (141), mentre
Brotherton, nel 1990, evidenziò la presenza e misurò le concentrazioni di
cortisolo e transcortina nel liquido seminale e nel liquido amniotico (142). Vari
studi si sono invece concentrati sulla rilevazione delle concentrazioni e sul

63
significato della presenza dell’ormone ghrelina (e obestatina) nel seme, aspetti
che verranno meglio descritti nel capitolo 3 e nella sezione sperimentale.
2.3.2. Cause principali
a) Fattori ambientali
Nella patogenesi dell’infertilità maschile, è opportuno prendere
preventivamente in considerazione alcune situazioni ambientali e tossiche quali
possibili cause. Infatti, alcuni modelli sperimentali hanno dimostrato l’insorgenza
di alterazioni spermatologiche in seguito al contatto con particolari agenti tossici,
come il dibromoetano (soprattutto a livello dell’acrosoma, guaina mitocondriale e
cromatina) (143), il parathion e il malathion, e composti clorurati (a livello di
testa, porzione intermedia e flagello) (144-145). Sostanze tossiche tra cui
idrocarburi, pesticidi, carburanti, olii e altri materiali di derivazione industriale,
possono, quindi, essere importanti fattori di rischio per infertilità.
Anche le radiazioni ionizzanti possono potenzialmente danneggiare gli
spermatozoi, con evidenza di difetti strutturali del nucleo e della cromatina in
modelli murini (146). Un esempio chiaro è lo studio di Bartoov et al. sui
sopravvissuti di Chernobyl e sugli abitanti delle zone circostanti, in cui si è
osservata una diminuzione della motilità degli spermatozoi e difetti ultrastrutturali
(147).
Il fumo di sigaretta è sicuramente un altro importante fattore di rischio, con un
incremento di spermatozoi anomali nei fumatori rispetto ai non fumatori. Un altro
effetto negativo del fumo di sigaretta si ha in associazione al varicocele, con
riduzione della concentrazione e della motilità degli spermatozoi (148-149).
64
b) Alterazioni anatomiche
Varicocele. Per varicocele
s’intende una lesione vascolare
caratterizzata da una dilatazione e
allungamento delle vene del
funicolo spermatico. Colpisce
prevalentemente gli adolescenti, con
un 15% d’incidenza nell’età adulta e
con percentuali sensibilmente più
alte in pazienti con infertilità primaria
e secondaria
Nel 90% dei casi il varicocele si presenta a sinistra, poiché in questa sede la vena
del testicolo s’immette ad angolo retto nella vena renale, a destra, invece, la vena
del testicolo drena direttamente nella vena cava inferiore. Questo provoca un
gradiente di pressione maggiore a sinistra, dando come risultato un più facile
sviluppo della patologia. E’ possibile classificare il varicocele in tre gradi clinici
secondo lo schema proposto da Dubin e Amelar:
1. Allargamento del plesso pampiniforme palpabile solo con manovra di Valsalva,
2. Plesso pampiniforme chiaramente palpabile anche senza manovra di Valsalva,
3. Visibile allargamento del plesso pampiniforme (136).
La diagnosi di certezza si ha solo in seguito ad esame eco-color-doppler che mette
in evidenza la dilatazione dei plessi venosi e la presenza di reflusso anomalo.
Il rapporto tra varicocele e infertilità venne per la prima volta affrontato nel 1965
da McLeod, che osservò alterazioni spermatologiche in pazienti affetti da questa
patologia (151). Oggigiorno quest’associazione è universalmente accettata,
ponendo come base vari possibili scenari patogenetici. In primo luogo
l’ipertermia pare giocare un ruolo fondamentale, poiché il ristagno di sangue
attorno ai testicoli può provocare un innalzamento della temperatura che altera le
normali meccaniche della spermatogenesi. Ciò risiede soprattutto nel fatto che gli
enzimi regolatori della sintesi del DNA e le polimerasi agiscono a una
Figura 23 - Varicocele

65
temperatura ottimale di 33-34° C, con
inibizione a temperature più alte.
Probabilmente, modificazioni della
pressione venosa, presenza di reflusso di
sostanze provenienti dal rene o dalle
ghiandole surrenali con vasocostrizione
cronica testicolare e diminuzione della
funzione, possibili disfunzioni ormonali e
stress ossidativo, possono ulteriormente
contribuire a causar
infertilità (152).
Criptorchidismo.
Per criptorchidismo (da κρυπτός, “occulto”, e ‘όρχις, ”testicolo”) si intende la
mancata discesa del testicolo nello scroto e colpisce il 3% dei neonati.
Solitamente si tratta di un arresto della normale discesa del testicolo, ritrovabile in
un punto del percorso tra il polo inferiore del rene e il sacco scrotale. Nella
maggior parte dei casi, il testicolo occulto si localizza in regione inguinale o
sottofasciale all’interno del canale inguinale (153).
Il criptorchidismo in quei soggetti che ne sono stati affetti si associa ad aumentato
rischio d’infertilità. Questo è dovuto alle modificazioni nelle cellule germinali che
avvengono nel primo anno di vita, epoca della comparsa degli spermatogoni di
tipo A (vedi figura 21), momento critico per la fertilità. Nel testicolo colpito da
quest’anomalia tale passaggio è inibito, e proprio per questo motivo la correzione
del difetto va effettuata entro i primi dodici mesi (154-155).
Con l’aumentare dell’età, la possibilità di divenire infertili incrementa
notevolmente: uno studio del 2007 ha evidenziato come già il 35% dei pazienti
criptorchidi non possieda cellule germinali nel testicolo affetto già a 11 anni
(156). Inoltre spesso è possibile evidenziare anche una degenerazione del testicolo
controlaterale in caso di criptorchidismo monolaterale (155).
Figura 24 - Alterazioni somatiche della sindrome di
Klinefelter

66
c) Difetti genetici
Una parte considerevole dell’etiologia dell’infertilità è rivestita da difetti
genetici (15-30%) e il 5% di tali alterazioni è causata delle anomalie
cromosomiche. Una situazione possibile è l’aneuploidia, ovvero la variazione
della normale quantitativo di cromosomi, condizione presente spesso in pazienti
con azoospermia. Un esempio di patologia cromosomica inerente a questa
situazione è la sindrome di Klinefelter, descritta per la prima volta da Henry
Klinefelter nel 1942. Tale condizione è spesso caratterizzata dalla presenza di un
cariotipo 47 XXY (“non mosaico”), ma possono anche sussistere aneuploidie più
alte, mosaici e anomalie strutturali del cromosoma X. La sindrome di Klinefelter,
oltre a presentare alterazioni fenotipiche caratteristiche (figura 25), determina un
arresto nella produzione di spermatociti primari e di conseguenza oligospermia o
azoospermia. Alcuni pazienti affetti dalla tipologia non mosaico di questa malattia
possiedono comunque spermatozoi nel liquido seminale, negli altri casi la
situazione è più severa, con solo dei residui di spermatogenesi a livello testicolare.
Esistono inoltre altre numerose alterazioni cromosomiche responsabili
d’infertilità, tra cui anche la presenza di traslocazioni derivate da errori in fase di
meiosi. Tra queste sono da ricordare le cosiddette traslocazioni robertsoniane, in
cui due cromosomi acrocentrici (ovvero con il centromero molto vicino al capo
del cromosoma) si fondono a livello del centromero, perdendo così il braccio
corto. In questo caso ci può essere un arresto della produzione delle cellule
germinali con conseguente infertilità. Oltre a queste possono verificarsi
traslocazioni reciproche o inversioni con risultato simile alle precedenti situazioni
descritte (157).
La trisomia 21, o sindrome di Down, è un’altra condizione cromosomica
(autosomica in questo caso) in cui si verifica infertilità per azoospermia o grave
oligospermia.
Importanti in patologie di derivazione cromosomica sono le alterazioni del
cromosoma Y, in quanto in questa sede sono presenti numerosi geni responsabili
del corretto svolgimento dei meccanismi di gametogenesi. Le anomalie possibili
sono molteplici: le microdelezioni del cromosoma Y, per esempio, sono
frequentemente responsabili d’infertilità, con discreta possibilità (15%) di
sviluppare un’azoospermia o una oligospermia (158). Le microdelezioni si

67
sviluppano principalmente sul braccio lungo, con interessamento di una
particolare area, l’azoospermia factor region (AZF, a sua volta costituita da tre
sotto-regioni), la cui delezione provoca un errore critico nello sviluppo dei gameti
(159).
Anche alcuni geni collocati sul cromosoma X possono essere coinvolti in
alterazioni della gametogenesi. Un esempio importante è costituito dalla
sindrome di Morris, detta anche sindrome da insensibilità agli androgeni o
femminilizzazione testicolare, in cui si osserva un’interruzione dello sviluppo
dell’apparato genitale in epoca fetale. Anche se con caratteri sessuali femminili, il
soggetto possiede corredo cromosomico 46 XY. A livello del braccio lungo del
cromosoma X di queste persone è presente una mutazione del gene AR (androgen
receptor), con conseguente insensibilità dei tessuti fetali agli androgeni
normalmente prodotti. I caratteri sessuali esterni, quindi, seguono la linea
femminile, al contrario di quelli interni il cui sviluppo in senso femmineo è stato
interrotto dal fattore anti-mülleriano prodotto dalle cellule del Sertoli. Questa
patologia tuttavia può seguire pattern diversi, dalla completa insensibilità agli
androgeni alla parziale risposta, fino a uno stato più lieve in cui i caratteri esterni
sono maschili (160-161). Essendo gli androgeni implicati nei meccanismi di
meiosi e di spermatogenesi, questi soggetti risultano infertili.
Le mutazioni autosomiche possono giocare un ruolo importante nella patogenesi
dell’infertilità. Per esempio, in pazienti affetti da fibrosi cistica (mutazione del
gene CFTR, cromosoma 7), è possibile osservare atipie dell’apparato genitale,
con atresia congenita dei dotti deferenti che provoca un’ostruzione al deflusso
degli spermatozoi e quindi azoospermia, appunto, ostruttiva (162). La mutazione
del gene SHBG (cromosoma 17), invece, determina un’alterazione di una
glicoproteina legante gli ormoni sessuali con funzioni regolatrici, alterando la
risposta a tali ormoni e quindi la spermatogenesi e la differenziazione sessuale
(163). Questi, tuttavia, sono pochi esempi della moltitudine di alterazioni studiate
e presenti in letteratura (che interessano il gene per il recettore del FSH, il gene
per il recettore degli estrogeni, la deleted azoospermia-like zone DAZL, MTHFR
e altri).
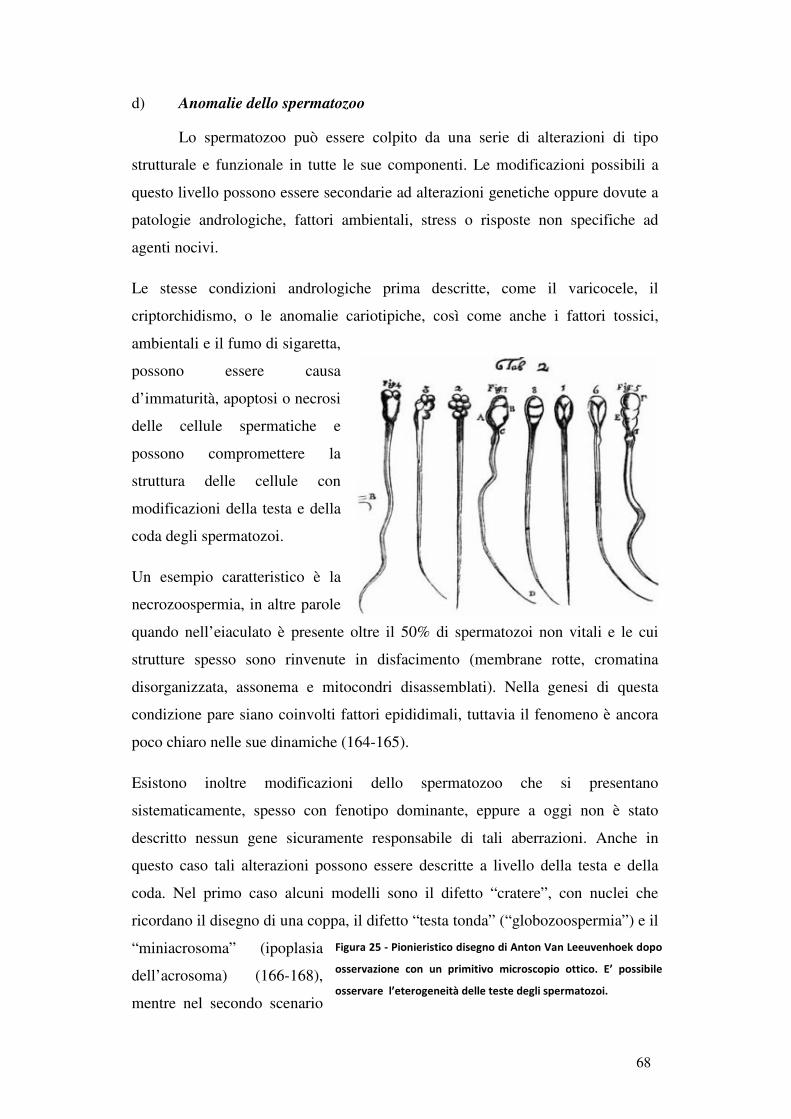
68
d) Anomalie dello spermatozoo
Lo spermatozoo può essere colpito da una serie di alterazioni di tipo
strutturale e funzionale in tutte le sue componenti. Le modificazioni possibili a
questo livello possono essere secondarie ad alterazioni genetiche oppure dovute a
patologie andrologiche, fattori ambientali, stress o risposte non specifiche ad
agenti nocivi.
Le stesse condizioni andrologiche prima descritte, come il varicocele, il
criptorchidismo, o le anomalie cariotipiche, così come anche i fattori tossici,
ambientali e il fumo di sigaretta,
possono essere causa
d’immaturità, apoptosi o necrosi
delle cellule spermatiche e
possono compromettere la
struttura delle cellule con
modificazioni della testa e della
coda degli spermatozoi.
Un esempio caratteristico è la
necrozoospermia, in altre parole
quando nell’eiaculato è presente oltre il 50% di spermatozoi non vitali e le cui
strutture spesso sono rinvenute in disfacimento (membrane rotte, cromatina
disorganizzata, assonema e mitocondri disassemblati). Nella genesi di questa
condizione pare siano coinvolti fattori epididimali, tuttavia il fenomeno è ancora
poco chiaro nelle sue dinamiche (164-165).
Esistono inoltre modificazioni dello spermatozoo che si presentano
sistematicamente, spesso con fenotipo dominante, eppure a oggi non è stato
descritto nessun gene sicuramente responsabile di tali aberrazioni. Anche in
questo caso tali alterazioni possono essere descritte a livello della testa e della
coda. Nel primo caso alcuni modelli sono il difetto “cratere”, con nuclei che
ricordano il disegno di una coppa, il difetto “testa tonda” (“globozoospermia”) e il
“miniacrosoma” (ipoplasia
dell’acrosoma) (166-168),
mentre nel secondo scenario
Figura 25 - Pionieristico disegno di Anton Van Leeuvenhoek dopo
osservazione con un primitivo microscopio ottico. E’ possibile
osservare l’eterogeneità delle teste degli spermatozoi.

69
si possono ritrovare esempi quali la presenza di spermatozoi acefali (169) o
l’assenza di componenti del flagello stesso.
2.3.3. Ruolo delle infezioni
Le infezioni del tratto urogenitale e l’infiammazione che da esse deriva
possono essere un importante fattore etiologico in materia di infertilità. Oltre a
queste, anche infezioni provenienti da altri distretti possono in qualche modo
compromettere la funzione spermatica, e tra queste può aver un ruolo importante
anche l’Helicobacter pylori.
Particolare importanza è rivestita dalle infezioni di tipo batterico, e a questo
proposito, infatti, sono stati dedicati numerosi studi per altrettanto molteplici
patogeni.
I micoplasmi genitali sono micro-organismi molto frequenti sia nei maschi fertili
sia infertili. Questo è particolarmente vero per l’Ureaplasma urealyticum e meno
per il Mycoplasma hominis e il Mycoplasma genitalium. Tali patogeni, sebbene in
maniera non molto chiara, possono essere causa di flogosi del tratto genito-
urinario (uretriti, prostatiti, raramente orchiti), spesso con produzione di specie
reattive dell’ossigeno che possono attaccare gli spermatozoi.
La Neisseria gonorrhoeae è riconosciuta essere causa di uretriti e, anche se
raramente, può provocare infezioni di altre parti dell’apparato genitale, inclusi i
testicoli, con conseguente danno alla capacità fecondante (170).
La Chlamydia trachomatis, batterio distribuito a tutte le latitudini, infetta
entrambi i sessi e ha un impatto maggiore sulle donne rispetto agli uomini. Il
contributo di questo batterio nell’etiologia dell’infertilità è oggetto di discussione,
tuttavia studi epidemiologici e la ricerca di anticorpi anti-LPS della Chlamydia

70
non hanno dato risultati significativi almeno per quanto riguarda la componente
maschile (171-172).
Altri patogeni come le Enterobacteriaceae sono stati considerati potenziali agenti
etiologici. L’Escherichia coli è una delle più comuni cause di epididimo-orchite
non sessualmente trasmessa ed è coinvolta nel 65%-80% delle prostatiti acute.
Cause di prostatiti ed epididimiti possono essere anche batteri quali Klebsiella,
Salmonella, Proteus e Pseudomonas aeruginosa, potendo in questo modo
interferire con la fertilità. Gli enterococchi, invece, possono essere responsabili
di oligospermia e teratospermia (170).
Le infezioni virali possono alla stessa stregua dei batteri giocare un ruolo
importante. DNA di Herpes-virus (in particolare i sierotipi 1 e 7) è stato ritrovato
frequentemente in pazienti infertili. In particolare l’HSV potrebbe essere causa di
leucocitospermia, anche se comunque il reale significato di questa alterazione
risulta poco chiaro in questo contesto (170-173-174-175). Infezioni da papilloma
virus (HPV) sono state riscontrate in biopsie testicolari di uomini azoospermici, e,
quando presente all’interno delle cellule, questo virus può debilitare la motilità
dello spermatozoo e dare astenospermia. Il virus dell’immunodeficienza
acquisita (HIV) è senz’altro responsabile d’infezioni opportunistiche del tratto
genito-urinario potendo essere responsabile d’infertilità; inoltre, la stessa terapia
anti-retrovirale può avere un impatto quasi devastante sull’apparato riproduttivo
maschile (170). Altri virus, come il cytomegalovirus o il virus di Epstein-Barr,
sono stati studiati senza dare risultati convincenti.
Per quanto riguarda i miceti, un possibile ruolo è stato riservato per la Candida
albicans, colonizzando spesso l’uretra e raramente le ghiandole accessorie. La
Candida può essere rinvenuta nel liquido seminale e può compromettere la
fertilizzazione in-vitro, oltre ad avere un effetto inibitore sulla motilità spermatica.
Tra i parassiti il Trichomonas vaginalis può essere causa di uretriti e prostatiti e
può anch’esso inibire la motilità degli spermatozoi (170).
� Associazione con Helicobacter pylori

71
Tra le manifestazioni extradigestive dell’infezione da Helicobacter pylori,
l’infertilità maschile è stata oggetto di alcuni interessanti studi.
Uno dei primi meccanismi ad essere stato preso in considerazione è il mimetismo
molecolare. Le cellule flagellate del liquido seminale, infatti, possiedono strutture
paragonabili ad alcune componenti batteriche e, quindi, possibili bersagli di una
risposta anticorpale. Nel 2002 Figura et al. applicarono questa teoria facendo
reagire spermatozoi umani con un siero iperimmune (creato attraverso
l’immunizzazione di siero di coniglio con componenti del batterio, tra cui VacA,
CagA e HspB - Heat shock protein B) e anticorpi ricavati da fluido follicolare
umano. Effettivamente è stata osservata una cross-reazione tra i sieri e gli
spermatozoi, soprattutto a livello della prima sezione del flagello e del centriolo
(strutture particolarmente ricche in tubulina) e in una piccola parte della zona
equatoriale. E’ stata inoltre osservata un’analogia strutturale tra la flagellina
dell’Helicobacter pylori e la tubulina umana, esempio di quella che potrebbe
essere l’origine di questa interazione e di come essa possa contribuire alla
diminuzione della motilità degli spermatozoi (176).
L’alterazione della qualità dello sperma da parte dell’Helicobacter pylori è stata
oggetto di un altro studio del 2008 di Collodel et al. In questo lavoro è stato
osservato che in soggetti con infertilità idiopatica e infettati dall’Helicobacter
pylori la qualità del liquido seminale dei portatori di ceppi CagA-positivi era
nettamente peggiore rispetto ai soggetti infettati da ceppi CagA-negativi. Nel
primo gruppo, infatti, la motilità e l’indice di fertilità erano diminuiti, così come
la quota di apoptosi e necrosi era aumentata. Queste osservazioni possono essere
ricondotte o a meccanismi di autoimmunità o a eventi legati all’infiammazione.
Alcuni cloni di Helicobacter pylori sono particolarmente efficienti nella
stimolazione della produzione di alcune citochine, in particolare TNF-α e IL-6, sia
a livello locale sia sistemico. Ciò ha portato all’ipotesi che l’alta patogenicità dei
ceppi CagA-positivi, soprattutto per la loro capacità di indurre la produzione di
citochine, può essere legata all’aumentata necrosi e apoptosi rilevata nei campioni
di liquido seminale (177).

72
3. Ormoni regolatori dell’appetito e altri
mediatori
3.1 Ghrelina
Negli ultimi anni sono stati descritti dei nuovi ormoni che rientrano nei
meccanismi di regolazione dell’appetito. Nel 1999 Kojima descrisse per la prima
volta l’ormone peptidico ghrelina, forse il più importante e più studiato ormone di
questa categoria (178).
La struttura dell’ormone è composta di 28 aminoacidi. Particolarità di
quest’ormone è la sua attivazione tramite acilazione della serina in posizione 3 da
parte dell’acido n-octanoico (un acido grasso), aspetto eccezionale per quanto
riguarda gli ormoni. L’acilazione permette alla ghrelina di attraversare la barriera
emato-encefalica. Esistono varianti non acilate della ghrelina che non possiedono
la capacità di legare il recettore GHS-R (vedi seguito), tuttavia possono avere
effetti biologici a livello miocardico (cosa evidenziata soprattutto nelle cavie) e su
alcuni tessuti neoplastici (179). L’acilazione in posizione 3 è indispensabile
affinché si abbia il corretto legame con il recettore (180)
La ghrelina è secreta prevalentemente a livello del fondo gastrico da parte di
cellule che colonizzano le ghiandole ossintiche (cellule X/A like) (181), anche se
altri tessuti possono secernerla sebbene in quantità più limitate (testicolo, rene,
ipofisi, ipotalamo, pancreas, intestino), con concentrazioni che decrementano
progressivamente lungo il tratto
intestinale.
La ghrelina è un ligando endogeno
del recettore secretagogo
dell’ormone della crescita, il
GHS-R (provvisto di due varianti,
1a, completa, e 1b), promuovendo
in maniera spiccata la liberazione
del GH. Il recettore GHS-R fu
scoperto e clonato nel 1996 da
Howard et al. (citato in 182) a Figura 26 – Ricostruzione della struttura dell'ormone
ghrelina

73
livello dell’ipotalamo e dalla ghiandola pituitaria umana e suina, tuttavia in
seguito è stato possibile riscontrarlo in altre strutture cerebrali, come l’ippocampo
e la sostanza nera. Questo particolare recettore è inoltre presente, benché espresso
in maniera minore, anche a livello gastrointestinale (stomaco e pancreas),
ghiandolare (tiroide, surreni, gonadi), a livello miocardico, osseo, linfatico e nel
tessuto adiposo (183-184).
Quali sono effettivamente le funzioni di questo nuovo ormone? Come anche
prima ricordato, il GHS-R è un ormone secretagogo del GH, quindi una delle
funzioni della ghrelina è proprio quella promotrice del rilascio dell’ormone della
crescita. Altra funzione importante è quella oressizzante, ovvero la stimolazione
dell’appetito. E’ stata evidenziata, infatti, la presenza di neuroni rilascianti
ghrelina a livello di vari nuclei dell’ipotalamo (arcuato, dorsale, ventrale,
paraventricolare) con ruolo nell’alimentazione umana. A livello del nucleo
arcuato tali neuroni sono particolarmente espressi, connettendosi ad altre cellule
encefaliche produttrici di neuropeptide Y (NPY) e di proteina agouti-associata
(AgRP), due peptidi oressizzanti. La ghrelina ha inoltre effetto inibitorio sulla
propiomelanocortina, sostanza con effetti anoressizzanti. Un altro sistema è quello
del nucleo paraventricolare, in cui la ghrelina determina un rilascio di ACTH e
cortisolo attraverso la produzione di NPY che inibisce la secrezione di acido γ-
ammino-butirrico (GABA) e promuove la liberazione dell’ormone di rilascio
delle corticotropine (CRH). La ghrelina è, inoltre, antagonista della leptina,
ormone che con i suoi effetti contrasta il rilascio di NPY e AgRP (182).
Il livello di ghrelina plasmatica, proprio a causa della sua attività oressizzante,
aumenta considerevolmente prima dei pasti, riducendo la sua concentrazione dopo
circa un’ora in maniera proporzionale alle calorie ingerite, fino a raggiungere la
quota minima (185). Inoltre la secrezione di ghrelina è provvista di un ritmo
circadiano, con livelli minimi notturni e massimi nella prima mattinata
(rispettivamente all’una di notte e alle nove del mattino).
Importante per la concentrazione della ghrelina è la massa adiposa. I soggetti
affetti da anoressia nervosa tendono ad avere elevati livelli di ghrelina, che
diminuiscono fino a rientrare nei livelli di normalità una volta aumentato il peso
corporeo. Effetto contrario si ha negli individui obesi, in cui i livelli di ghrelina
sono considerevolmente più bassi. L’acil-ghrelina stessa promuove la deposizione

74
di grasso, rientrando anche nella regolazione del metabolismo lipidico. Gli acidi
grassi liberi, inoltre, hanno un effetto soppressivo sul livello plasmatico di
ghrelina (182).
Oltre all’effetto promotore dell’appetito, la ghrelina svolge altre attività. A livello
gastrico stimola la secrezione acida e la motilità del viscere (186), con
promozione della secrezione di gastrina. Inoltre quest’ormone possiede attività
gastro-protettive, funzioni esplicate anche attraverso la produzione di PGE2 e
ossido nitrico da parte degli enzimi ciclo-ossigenasi e ossido nitrico-sintetasi
(COX e NOS). Quest’ultimo effetto ha naturalmente un’azione adiuvante per la
guarigione di eventuali ulcere peptiche (187).
Il metabolismo glucidico è un altro bersaglio della ghrelina, in particolare la
secrezione dell’insulina. Non c’è consenso unanime circa il reale ruolo della
ghrelina a questo livello, infatti, tra gli autori esistono pareri discordanti e sovente
antitetici. Sempre a livello endocrino, si è già accennato circa l’effetto induttore
sulle corticotropine. Alcuni studi hanno osservato, per giunta, diminuiti livelli di
ghrelina sierica in pazienti ipertiroidei (182).
A livello cardiovascolare la ghrelina ha effetti vasodilatatori, con riduzione della
pressione arteriosa media, migliorando anche la funzione di pompa cardiaca
(188).
Effetti della ghrelina possono manifestarsi anche a livello del sistema
immunitario, con il possibile ruolo nei meccanismi di proliferazione e
attivazione delle cellule immunitarie e in alcuni sistemi di regolazione
dell’infiammazione (vedi sezione sperimentale, par. 7).
Altri effetti studiati della ghrelina possono verificarsi a livello del metabolismo
osseo e nei meccanismi del sonno (promuove lo stato di veglia) e della memoria
(182).

75
3.2 Obestatina
Un altro prodotto del gene della ghrelina è l’obestatina, un peptide acido di 23
aminoacidi identificato per la prima volta nello stomaco di ratto. Così come la
ghrelina, l’obestatina pare abbia un ruolo nella regolazione dell’introito
alimentare e gli effetti furono inizialmente osservati nei topi. Nelle cavie, infatti,
l’iniezione intraperitoneale di obestatina sopprimeva l’assunzione di cibo in
maniera tempo e dose-dipendente. Stessa cosa si evidenziava iniettando
l’obestatina per via intra-cerebro-ventricolare. Il trattamento cronico con
obestatina, inoltre, riduceva, sempre nei ratti, il peso corporeo, la secrezione acida
gastrica e la motilità duodenale e digiunale.
E’ stato dimostrato che l’obestatina (prodotta
principalmente dallo stomaco, ma anche da
digiuno, duodeno, colon, pancreas, milza,
ghiandola mammaria e probabilmente a livello
seminale) è un ligando per un recettore
orfano, un G protein-coupled receptor
(GPR39), che possiede caratteristiche simili
al GHSR1a (una delle due varianti del
recettore GHSR) e la cui interazione
porterebbe all’inibizione dell’assunzione di
cibo (189). Questo recettore è infatti coinvolto in meccanismi di regolazione del
peso corporeo, della motilità intestinale, di alcune secrezioni ormonali e della
morte cellulare. Elevati livelli di GPR39 sono stati evidenziati tramite real-time
PCR a livello dell’amigdala, dell’ippocampo, della corteccia uditiva e
dell’ipotalamo, ma anche nel tratto gastro-enterico, fegato, tessuto adiposo e
pancreas. C’è da aggiungere che non tutti gli autori sono concordi sul legame
obestatina-GPR39, tuttavia la questione è ancora fonte di dibattito.
Alcune evidenze sull’attività dell’obestatina ci giungono sempre da modelli
murini. Secondo questi studi il ruolo dell’obestatina sarebbe di antagonista della
ghrelina, con modalità dipendenti dallo stato di nutrizione e dalla secrezione di
GH. Inoltre è stata evidenziata, come anche per la ghrelina, la presenza di un
Figura 27 - Ricostruzione della struttura
dell'obestatina

76
ritmo circadiano pulsatile (190). Al contrario, vari studi tendono a escludere il
ruolo dell’obestatina nella regolazione dell’introito alimentare, non attribuendone
alcuna influenza sull’apporto globale di cibo, perso corporeo, svuotamento
gastrico e motilità duodenale e digiunale.
La concentrazione plasmatica dell’obestatina non sembra essere influenzata
dall’apporto calorico; infatti, i livelli dell’ormone non variano sensibilmente in
seguito all’assunzione di pasti anche molto differenti nella quantità di kilocalorie.
I livelli plasmatici di obestatina risultano inferiori alla media nei soggetti obesi e
in individui sottoposti a gastrectomia (191), tuttavia altri studi suggeriscono
l’opposto. Nei pazienti anoressici, invece, esiste concordanza tra i vari risultati,
concludendo unanimemente che le concentrazioni di obestatina in questi individui
sono maggiori rispetto ai soggetti sani.
Oltre ai presunti effetti anoressizzanti, l’obestatina pare agisca anche ad altri
livelli. E’ stata osservata una inibizione della secrezione insulinica da parte
dell’obestatina esogena sempre in modelli murini (192). Osservazioni circa
possibili influenze dell’obestatina sulla secrezione dell’insulina indotta dal
glucosio hanno dato risultati discordanti. Effetto interessante si ha a livello delle
cellule β pancreatiche, con protezione di queste ultime tramite blocco
dell’apoptosi indotta da citochine. Un lavoro del 2007 ha evidenziato, infatti, una
maggiore incidenza di diabete mellito di tipo 2 e di stati di insulino-resistenza in
pazienti con bassi livelli di obestatina (193). L’importanza dell’obestatina sul
metabolismo glucidico, però, resta a oggi oggetto di dibattito.
Per quanto riguarda il metabolismo lipidico, è stato osservato nelle cavie che
l’obestatina provoca una riduzione del colesterolo sierico e dei livelli di
trigliceridi (182).
L’obestatina, inoltre, pare abbia anche un ruolo nell’inibizione della sete (194),
nella regolazione, alla stessa stregua della ghrelina, del ritmo sonno-veglia (195) e
della memoria (196) e nell’aumento della produzione di enzimi pancreatici (197).

77
3.3 Altri ormoni e mediatori
Leptina. La leptina, la cui scoperta risale al 1994, è una proteina costituita
da 167 aminoacidi, codificata dal gene Ob a livello del cromosoma 7. Questo
ormone è prodotto soprattutto a livello del tessuto adiposo bianco, tuttavia può
essere riscontrato anche a livello della mucosa del fondo gastrico, ipofisi, tessuto
muscolare e placenta. La concentrazione plasmatica della leptina è proporzionale
al peso corporeo e quindi al BMI, soprattutto nelle donne (198). Questo pare
essere dovuto alla promozione da parte degli estrogeni della produzione di leptina,
mentre al contrario il testosterone sembra inibirla.
L’attività della leptina si manifesta attraverso l’interazione con il recettore Ob-R
di classe I (componente della famiglia dei recettori delle citochine espresso in vari
organi), determinando una riduzione dell’introito alimentare con dissipazione di
energia (199). Il recettore Ob-R possiede varie isoforme, suddivise in “corte” e
“lunghe”: del primo gruppo fa parte l’isoforma Ob-Re, recettore solubile che lega
la leptina nel siero, mentre nel secondo ritroviamo Ob-Rb, espressa soprattutto a
livello del nucleo arcuato e paraventricolare dell’ipotalamo. Quando la leptina si
lega al suo recettore nel nucleo arcuato provoca un’inibizione della secrezione di
neuropeptide Y con effetto anoressizzante.
A livello pancreatico la leptina promuove la secrezione di glucagone e inibisce il
rilascio d’insulina: è stato osservato che nelle cellule β pancreatiche tale
inibizione avviene attraverso l’attivazione della fosfodiesterasi 3B (PDE3B)
(200). Altre azioni della leptina sono quella lipolitica e tireotropa (aumento del
T3).
Adiponectina. L’adiponectina fu scoperta nel 1995 nel tessuto adiposo
(201), ed è una proteina di 247 aminoacidi con 4 domini e struttura multimerica.
Esistono tre forme di adiponectina prodotta dagli adipociti: LMW formata da tre
monomeri, MMW da sei, e HMW da otto (202). La HMW sembra essere la forma
attiva dell’ormone, con possibili interazioni anche con l’insulina (203). E’ stato

78
osservato in modelli sperimentali che l’insulina stessa e l’insuline-like growth
factor stimolano l’espressione del gene dell’adiponectina.
Esistono due tipi di recettori conosciuti per l’adiponectina, l’adipoR1 e adipoR2,
il primo espresso soprattutto a livello muscolare mentre il secondo a livello
epatico.
L’adiponectina pare abbia un ruolo rilevante nella regolazione della funzione
dell’insulina e nell’omeostasi energetica, essendo infatti ridotta in soggetti con
diabete mellito di tipo II ed obesi (204-205). La concentrazione plasmatica
dell’ormone è inversamente proporzionale al BMI, al grado d’insulino-resistenza,
alla glicemia e alle concentrazioni di insulina e trigliceridi (206). Il meccanismo
d’azione non è del tutto stato chiarito, tuttavia pare che agisca soprattutto a livello
epatico diminuendo la produzione di glucosio e aumentandone il catabolismo a
livello muscolare. L’adiponectina può avere un ruolo anche a livello cardio-
vascolare (con effetti protettivi contro l’ischemia e il danno da riperfusione) e
muscolare (207).
Resistina. La resistina è una proteina ricca in cisteina codificata negli
esseri umani dal gene RETN, scoperta nel 2001 da un team di studiosi
dell’University of Pennsilvania School of Medicine. Il ruolo della resistina è
controverso, ma pare abbia un ruolo di primo piano nello sviluppo dell’obesità e
del diabete mellito di tipo 2. La resistina è una citochina derivata dagli adipociti,
che in modelli murini provoca insulino-resistenza e intolleranza al glucosio e
nell’uomo è iperespressa dal tessuto adiposo (208). A confermare il legame tra
resistina, obesità e diabete mellito di tipo II, uno studio di Stoppan et al. ha
dimostrato come l’espressione del gene della resistina sia ridotta marcatamente in
corso di trattamento con tiazolinedioni, TZD, farmaci anti-diabetici che legano i
recettori PPARγ (Peroxisome proliferator-activated receptors). La resistina,
inoltre, antagonizza l’insulina in modelli sperimentali in vitro; infatti, il
trattamento dei campioni con antisiero anti-resistina provoca un aumento
dell’ingresso di glucosio nelle cellule indotto dall’insulina (209).
Defensine. Le defensine sono piccole proteine costituite da 18-45
aminoacidi con otto residui di cisteina conservati. Le cellule del sistema
immunitario contengono questi peptidi, utili nella difesa contro batteri, funghi e

79
virus. Esistono tre categorie di defensine studiate. Le α-defensine (DEFA1-1B-3-
4-5-6) sono espresse dai neutrofili, linfociti NK e alcuni linfociti T, oltre che dalle
cellule di Paneth per quanto riguarda DEFA5 e 6. Le β-defensine (DEFB1, 2,
103A, 107B, 110, 136) sono secrete da leucociti e cellule epiteliali di varia natura,
come per esempio cellule polmonari e del tratto respiratorio, delle ghiandole
salivari, renali ed esofagee. Le θ-defensine (DEFT1P) sono rare e sono state
ritrovate solo in linfociti di primati.
Alcuni studi sperimentali hanno messo in relazione queste molecole con alcune
patologie, in particolare il diabete. E’ stato osservato in modelli murini che i
livelli di β-defensine (e Toll-like receptors) erano ridotti in ratti diabetici in
confronto a topi omologhi ma sottoposti a terapia insulinica. Questi bassi livelli
potevano essere riportati a normalità somministrando insulina, sottolineando
come il diabete possa diminuire l’efficacia dell’immunità innata (210-211).
Le α-defensine sono incrementate in condizioni infiammatorie croniche e in
alcune patologie neoplastiche. Inoltre, una riduzione delle defensine può
predisporre al morbo di Crohn: è stata infatti osservata una diminuzione dei livelli
di defensine in concomitanza con mutazioni del gene NOD2-CARD15, implicato
nella patogenesi di questa patologia, debilitando i meccanismi di difesa anti-
batterica (212).
3.4 Ormoni dell’appetito e Helicobacter pylori
Numerosi studi hanno messo in relazione l’infezione da Helicobacter
pylori con i livelli sierici di ghrelina e obestatina. In particolare questi ultimi due
ormoni sono secreti dalle cellule X/A presenti a livello delle ghiandole ossintiche,
che vanno incontro a decimazione a causa dell’atrofia in corso di gastrite da
Helicobacter. Questa naturalmente non è una condizione solo attribuibile
all’Helicobacter pylori ma anche ad altre forme di gastrite, come, per esempio, la
gastrite autoimmune.
Una review del 2011 a cura di Nweneka et al. ha raggruppato numerosi studi
riguardanti l’associazione tra ghrelina e Helicobacter per determinare la reale

80
esistenza di questo legame e, se presente, le sue caratteristiche. Gli studi presi in
considerazione prevedevano l’utilizzo di campioni infettati e di controlli sani e la
determinazione dei livelli di ghrelina prima e dopo l’eradicazione del batterio.
Nella grande maggioranza di questi studi è stato evidenziato come i livelli di
ghrelina sierica in pazienti sani fossero significativamente più elevati rispetto ai
pazienti infettati (213). Cindoruk et al., però, puntualizzano in un loro lavoro che
i livelli di ghrelina in pazienti infetti senza gastrite cronica non si discostano
significativamente dal campione, ponendo quindi la gastrite come una condicio
sine qua non (214).
I dati riportati da un lavoro di Nwokolo et al. sostengono che i livelli di ghrelina
dopo l’eradicazione dell’infezione aumentano sensibilmente, fino a una
percentuale massima stimata del 75% (215). La questione sugli effetti benefici
dell’eradicazione, però, è controversa. E’ plausibile che la terapia specifica anti-
Helicobacter provochi un incremento dei livelli sierici di ghrelina, tuttavia dal
momento che l’infezione da Helicobacter pylori causa atrofia a livello della
mucosa, questo concetto risulta essere effettivamente dipendente dalla durata
dell’infezione, dall’entità del danno sulle cellule produttrici di ghrelina e dal
tempo di recupero delle cellule stesse (213). Gli effetti sulle cellule secernenti
l’ormone esercitati dal batterio sono stati studiati tramite misurazione dell’mRNA
della ghrelina a livello gastrico, dimostrando una sensibile diminuzione di
quest’ultimo nei pazienti infetti, così come anche il numero stesso delle cellule
produttrici (216). Il grado stesso di atrofia, quindi, è una variabile importante. E’
stato inoltre osservato che la concentrazione di ghrelina può essere anche messa in
relazione con i livelli di alcuni zimogeni, nella fattispecie il pepsinogeno I e II, le
cui concentrazioni si riducono sensibilmente in corso di gastrite atrofica.
Le discrepanze presenti circa la risposta all’eradicazione dell’infezione possono
anche essere messe in correlazione ai vari ceppi di Helicobacter pylori. Isomoto
et al. hanno infatti dimostrato che la diversità dei ceppi è associata ai livelli di
ghrelina plasmatici e gastrici nell’uomo. Pazienti affetti da ceppi esprimenti VacA
e CagA possedevano livelli di ghrelina sierica nettamente inferiori ai pazienti con
ceppi cagA-negativi (217).

81
Essendo la ghrelina un ormone oressizzante, un suo marcato incremento provoca
anche un aumento dell’appetito con conseguente aumento del peso corporeo
(218).
Uno studio italiano del 2007 ha osservato, contrariamente a quanto finora
descritto, che in 25 pazienti con gastrite atrofica i livelli di ghrelina acilata
risultavano maggiori rispetto al controllo senza tuttavia trovare significative
differenze nel computo totale della ghrelina sierica. Ciò può dipendere da un
aumentato processo di acilazione della ghrelina residua (219).
Sono pochi, contrariamente alla ghrelina, gli studi condotti sul comportamento
dell’obestatina in corso d’infezione da Helicobacter pylori. Uno studio di Sin-
Yuan Gao et al. ha preso in considerazione una popolazione di 100 uomini,
comprendendo anche soggetti con età superiore a 65 anni, con un campione di 50
uomini affetti da gastrite atrofica e 50 controlli. I livelli plasmatici di obestatina,
in accordo con quelli della ghrelina, risultavano essere inferiori nei soggetti con
gastrite. E’ stata inoltre osservata una relazione negativa tra obestatina circolante
e BMI (body mass index), cosa invece non osservata nei pazienti gastritici (220).
L’infezione da Helicobacter può anche alterare il comportamento di altri ormoni
tra quelli descritti in questo capitolo. Tra questi ritroviamo la leptina, la cui
concentrazione e l’espressione del suo mRNA a livello dello stomaco aumenta in
maniera significativa nei soggetti positivi all’Helicobacter, con riduzione in caso
di eradicazione (221). Gli alti livelli di leptina si associano a un aumento della
produzione di IL-1, IL-6 e TNF-α, tipico dell’infezione da Helicobacter, e ciò può
essere ricondotto ad un potenziale ruolo delle leptina nei processi di
infiammazione e risposta immunitaria al batterio. La stessa risposta immunitaria è
associata con l’iperespressione di geni delle adipochine, tra cui anche quelli
dell’adiponectina e delle resistine (222). L’adiponectina e le resistine sono fattori
multi-task e rientrano in molteplici disordini, tra cui anche patologie esofagee, del
piccolo e del grosso intestino. Tuttavia, nell’uomo, bassi livelli di adiponectina
sono stati rinvenuti in soggetti giapponesi di mezza età portatori di gastrite
atrofica da Helicobacter pylori, ma il significato clinico di queste osservazioni
resta ancora poco chiaro (223).

82
E’ stato dimostrato che le cellule epiteliali gastriche sia in vivo sia in vitro
esprimo il peptide β-defensina di tipo I. Al contrario, in seguito ad infezione da
Helicobacter pylori le stesse cellule epiteliali secernono β-defensine di tipo II,
cosa che avviene anche in seguito a stimolazione con IL-1. Dati relativi ad alcuni
studi suggeriscono che tali defensine siano una componente della risposta immune
contro l’Helicobacter pylori (224).
3.5 Ormoni dell’appetito e riproduzione.
Gli ormoni regolatori dell’appetito sono stati per molti aspetti studiati per
quanto riguarda lo sviluppo sessuale e la riproduzione. I punti presi in
considerazione in questi studi sono svariati, ma la maggior parte di essi ha come
protagonista la ghrelina.
Per quel che riguarda le donne, recettori per la ghrelina sono presenti nelle ovaie
di mammiferi e di animali non mammiferi, e probabilmente questi organi sono
target per quest’ormone. Verosimilmente la ghrelina ha un ruolo nella regolazione
della funzione ovarica, con azione inibitoria sulla steroidogenesi ed effetti sullo
sviluppo e maturazione dell’ovocita (studi effettuati su modelli animali) (225).
Uno studio di Messini et al. (2009) ha valutato gli effetti della ghrelina sulla
secrezione delle gonadotropine durante il ciclo mestruale. Secondo tale studio non
esistono modificazioni sostanziali dei livelli di LH e FSH dovuti a un
innalzamento acuto di ghrelina in donne sane, quindi questo ormone esula dalla
regolazione di questo sistema (226). Schöfl et al. hanno invece ricercato il
significato dei bassi livelli di ghrelina riscontrati in donne con sindrome
dell’ovaio policistico (PCOS), non trovando in questo caso una diretta
correlazione dei livelli dell’ormone con LH e FSH ma configurando per la
ghrelina un ruolo nell’insulino-resistenza spesso presente in questa patologia
(227). Panidis et al. nel 2005, invece, osservarono che i livelli di ghrelina erano in
relazione inversamente proporzionale alle concentrazioni di androgeni,
probabilmente a causa del ruolo regolatore della ghrelina nella sintesi degli
steroidi e/o nei loro meccanismi d’azione. E’ anche riportato che le differenti
manifestazioni cliniche e biochimiche della PCOS possono essere anche dovute ai

83
diversi valori della ghrelina (228). Sempre Panidis et al. nel 2003 dimostrarono
ridotti valori di adiponectina in pazienti con policistosi ovarica (229). La
riduzione dell’ormone leptina è stata messo in associazione con alcuni disturbi
della fertilità femminile, tra cui l’amenorrea di origine ipotalamica (230).
Uno studio di El-Eshmawy et al. del 2010 s’incentrava sull’associazione tra
ghrelina e leptina con gli ormoni riproduttivi nel ritardo di crescita e pubertà nei
maschi. Come già prima riportato, la ghrelina rientra nei meccanismi di
secrezione del GnRH con meccanismo inibitorio, mentre la leptina pare abbia un
effetto stimolante. Secondo questo studio l’elevato valore di ghrelina e il ridotto
livello di leptina possono essere implicati nel ritardo di pubertà. Sono stati inoltre
valutati i livelli di FSH, LH e testosterone, ed è stato osservato che mentre la
leptina stimola la produzione di LH e FSH, la ghrelina ha un effetto negativo su
LH e testosterone, soprattutto nel periodo pre-puberale (231). L’associazione
ghrelina-LH è riportata in più studi: in modelli in vitro è stata dimostrata la
riduzione della risposta all’ormone rilasciante LH (LHRH), e per l’appunto ridotta
secrezione di LH in vivo (225).
Come la ghrelina possa rientrare nelle meccaniche della fertilità maschile è
oggetto di svariati studi. L’espressione della ghrelina è stata dimostrata a livello
genitale in roditori e altri mammiferi ed è presente nel testicolo umano,
soprattutto a livello delle cellule di Leydig e del Sertoli. Uno degli effetti della
ghrelina a questi livelli può essere correlato alla steroidogenesi testicolare. In
modelli murini e in vitro, l’incremento della ghrelina inibisce significativamente e
in maniera dose-dipendente il rilascio di testosterone sia hCG- che cAMP-indotto
da parte delle cellule di Leydig con modalità ancora poco chiare. Si può dire,
dunque, che i livelli di testosterone sierico sono inversamente proporzionali
all’espressione della ghrelina da parte delle cellule di Leydig (232–233). Altro
effetto della ghrelina si ha a livello dei tubuli seminiferi. E’ stato osservato che
l’iniezione intratesticolare di ghrelina nei topi provoca un’inibizione
dell’espressione di mRNA prodotto del gene dello Stem Cell Factor (SCF), un
segnale chiave per la produzione delle cellule staminali germinali e probabile
regolatore dello sviluppo delle cellule di Leydig. E’ stato dimostrato che
l’iniezione intratesticolare di ghrelina può anche inibire lo sviluppo delle cellule
di Leydig immature in epoca pre-puberale (234).

84
La ghrelina può essere rinvenuta nel liquido seminale e quest’aspetto e il suo
significato sono oggetto di studio di numerose trattazioni. I livelli di ghrelina nel
liquido seminale sono pari circa al 27% della corrispondente concentrazione
plasmatica di soggetti con normospermia (235.).
I livelli di ghrelina nel liquido seminale potrebbero essere correlati con
l’infertilità, soprattutto come risposta all’infezione da Helicobacter pylori.
Quest’ultima associazione, unita anche alla determinazione dei livelli di
obestatina, è materia della sezione sperimentale di questa tesi.

85
Parte sperimentale

86
4. Scopo della tesi
Lo scopo della presente tesi è di determinare se i tipi istologici di carcinoma
gastrico possono essere associati coi polimorfismi di cagA e di vacA di ceppi di
H. pylori isolati da pazienti gastroresecati per carcinoma gastrico.
Abbiamo visto come la proteina CagA, una volta inoculata nel mucocita, venga
fosforilata e trasformata in un'oncoproteina. Il sito di fosforilazione è costituito
dalla sequenza aminoacidica EPIYA. La regione variale (C'-terminale) di CagA è
polimorfa in quanto comprende una o più ripetizioni contenenti questa sequenza.
L'ipotesi è che maggiore è il numero di ripetizioni, più facilmente i ceppi batterici
che le ospitano possono concorrere allo sviluppo del carcinoma gastrico.
L'altro fattore di virulenza potenzialmente associato allo sviluppo di un carcinoma
gastrico è VacA, la tossina vacuolizzante che mostra anch'essa una variabilità
strutturale nelle sezioni s (da signal sequence) ed m (da middle sequence) della
proteina, cioè la parte iniziale e mediale. E' stato osservato che determinati
polimorfismi genomici di vacA, in particolare s1/m1, appartengano a a ceppi
isolati significativamente più spesso da pazienti con carcinoma gastrico.
Il carcinoma gastrico può essere classificato in due varianti istologiche principali:
il tipo intestinale e il tipo diffuso. Lo sviluppo di carcinoma gastrico tipo
intestinale è preceduto da alterazioni della mucosa gastrica, come atrofia,
metaplasia intestinale e displasia. Il carcinoma gastrico tipo diffuso, invece, non è
preceduto da alterazioni metaplastiche e apparentemente origina de novo dal
colletto iperplastico di ghiandole gastriche attraverso il coinvolgimento di vari
oncogeni e geni oncosoppressori.

87
SEZIONE SPERIMENTALE
Un gruppo di 28 pazienti con carcinoma gastrico non cardiale è stato selezionato
per questo studio; 15 pazienti avevano un carcinoma gastrico varietà intestinale e
13 un carcinoma gastrico varietà diffuso. I ceppi di H. pylori sono stati isolati
mediante coltura su agar selettivo. Da ciascun paziente sono stati isolate fino a 25
colonie di H. pylori. I ceppi venivano subcoltivati e conservati a – 80 °C in brodo
glicerolo fino al momento dell’esame genetico.
Estrazione del DNA e PCR
Un totale di 226 colonie è stato isolato dalla mucosa gastrica resecata. Il DNA di
ciascuna colonia è stato estratto in accordo al Puro gene DNA purificazione KIT
protocol (Gentrasystem Minneapolis, Minn).La concentrazione del DNA era
calcolata allo spettrofotometria. Il tipo strutturale cagA è stato determinato
utilizzando i primer sottoelencati, che comprendono tutta la regione variabile di
cagA.
CagA senso :AACCCTAGTCGGTAATGGGTTAT Cag A antisenso: GTAATTGTCTAGTTTCGC
Per aumentare la specificità della PCR, tutti gli amplificati sono stati ottenuti in
test di amplificazione interna (nested PCR) di ampliconi ottenuti con i seguenti
primer specificy per l’intero gene cagA.
CagA1 :AGCGGTATCAATGGCTAAAGC CagA2:CGTGCTCCATTCTGGATATTG
Gli ampliconi così ottenuti venivano separati elettroforeticamente in un gel di
agarosio e le dimensioni calcolati con 'ausilio di pesi molecolari noti.
Le regioni s ed m di vacA venivano amplificate coi primer sottoriportati

88
Allele Direzione Sequenza bp del prodotto
vacA1 senso 5’-GAAATACAACACACCGC-3’ 201 antisenso 5’-GGCTTGTTTGAGCCCCAG-3’ vacA3 senso 5’GGTCAAAATGCGGTCATGG-3’ 388 antisenso 5’-CATCAGTATTTCGCACCACA-3’ vacA4 senso 5-CCAGGAAACATTGCCGGCAAA-3’ 346 antisenso 5’-CATAACTAGCGCTTGCAC-3’
STATISTICA
I dati sono stati riportati in frequenze assolute e in percentuali. L’associazione tra
variabili considerate sono state valutate col il test del Chi quadro e un valore di p
<0,05 veniva considerato statisticamente significativo. Tutte le analisi sono
effettuate utilizzando in software SPSS v 17 (SPSS INC., Chicago, U.S.A.).
5. Risultati
Associazione tra ceppi cagA positivi e istotipo di carcinoma gastrico
I risultati sono stati riassunti nella tabella 1, riportata di seguito. Dei 226 ceppi
isolati, 214 erano cagA positivi (cagA+) e 12 erano cagA negativi (cagA-). I ceppi
ottenuti da pazienti con carcinoma gastrico tipo intestinale erano 119; quelli
ottenuti da pazienti con carcinoma gastrico diffuso erano 95 (44,3%). Tutti e 12 i
ceppi cagA- sono stati isolati da casi di carcinoma gastrico diffuso (p< 0,001,
Tabella 1).
Associazione tra tipi strutturali cagA e varietà istologica di carcinoma gastrico
Nessun ceppo con un particolare tipo cagA è stato trovato significativamente più
spesso in una delle due varianti di carcinoma gastrico. Ceppi con tipo strutturale
cagA C (no., 4) sono stati identificati soltanto in pazienti con carcinoma gastrico
istotipo intestinale (p=0,063) Il tipo strutturale cagA C corrisponde alla regione
variabile con il più alto numero di ripetizioni EPIYA.

89
Ceppi con tipi strutturali cagA diversi dal tipo C erano distribuiti in modo simile
nelle due varietà di carcinoma gastrico, suggerendo che lo sviluppo di carcinoma
gastrico intestinale o diffuso non dipendeva dal numero di motivi EPIYA presenti
nella regione variabile di cagA.
Dei 15 pazienti con carcinoma gastrico intestinale, 7 erano infettati da ceppi con
un solo tipo cagA, mentre 8 pazienti erano infettati da ceppi con più di un tipo
strutturale.
Tra i 13 pazienti con carcinoma gastrico diffuso, 9 erano colonizzati da ceppi con
un solo tipo cagA e 4 con più di un tipo. Queste differenze non erano
significative.
Questi risultati non dimostrano che il genotipo e il polimorfismo dell'agente
infettante possano influire sulla variante istologica del carcinoma gastrico.
Associazione tra sottotipi vacA e varietà istologica del carcinoma gastrico e tra
tipi strutturali cagA e sottotipi vacA
Il numero totale di ceppi con sottotipo vacA s1/m1 era 205, mentre i ceppi
sottotipo s1/m2 erano 21. Il 43.9% dei ceppi con sottotipo vacA s1/m1 era
associate con carcinoma gastrico intestinale, mentre l’80.9% dei ceppi sottotipo
vacA s1/m2 era isolato da casi di carcinoma gastrico istotipo diffuso (p<0.001).
Ceppi con sottotipo vacA s1/m1 erano distribuiti pressoché uniformemente tra I
diversi tipi strutturali cagA, eccetto il tipo C. L’89.9%, il 91.6% e il 94.2% dei
ceppi con questo sottotipo vacA erano associati con tipi strutturali cagA A(I), A e
B/D, rispettivamente.
Tutti i tipi strutturali cagAerano sottotipi vacA s1/m2.
Ceppi con il sottotipo vacA s1/m2, considerate meno carcinogenetico, erano
identificati per lo più in casi di carcinoma gastrico di tipo intestinale. Tuttavia, è

90
stato anche visto che tutti i ceppi col tipo strutturale cagA C erano del sottotipo
vacA s1/m2 e nessuno era vacA s1/m1 (Tabella 1).
6. Discussione
Associazione tra infezione da ceppi cagA+ e istotipo di carcinoma gastrico
E’ noto che il rischio di sviluppare un carcinoma gastrico associato ad infezione
da H. pylori aumenta ulteriormente se il ceppo colonizzanti esprimono la proteina
CagA (246-248). Il rischio è solo modestamente aumentato tra i soggetti infettati
da ceppi CagA-, rispetto alle persone non infettate. Se però stratifichiamo gli
individui per varietà tumorale, ci accorgiamo che le infezioni da ceppi CagA- non
sono strettamente associati allo sviluppo di una neoplasia gastrica, e possono solo
aumentare il rischio di sviluppare un carcinoma gastrico dell’istotipo diffuso
(247).
Un ulteriore studio ha riportato un’associazione tra l’assenza di cagA nel ceppi
infettanti e sviluppo di carcinoma gastrico diffuso (40).
Nonostante nei nostri pazienti l’infezione da ceppi cagA- fosse associate con un
carcinoma gastrico tipo diffuso, tutti i pazienti alberganti ceppi cagA- ospitavano
anche ceppi cagA+.
Associazione tra tipi strutturali cagA e varietà istologica del carcinoma gastrico
del carcinoma gastrico
In questo studio, abbiamo notato che nessun ceppo con un particolare tipo cagA
era maggiormente prevalente in una delle due varietà istologiche di carcinoma
gastrico, suggerendo che il numero di motivi EPIYA, presenti nella regione

91
variabile di CagA non contribuiva allo sviluppo di uno o dell’altro tipo istologico
di carcinoma gastrico.
Abbiamo anche notato la tendenza dei ceppi con tipo strutturale cagA C (no. = 4)
a concentrarsi nei pazienti con carcinoma gastrico istotipo intestinale (p=0.063); è
probabile che, se il numero dei casi fosse stato più alto, la maggior prevalenza di
ceppi col tipo strutturale cagA C nei pazienti con carcinoma gastrico intestinale
sarebbe stata significativa, in quanto nessun ceppi con tipo cagA C è stato isolato
da pazienti con carcinoma gastrico diffuso.
Associazione tra sottotipi vacA e varietà del carcinoma gastrico e tra tipi
strutturali cagA e sottotipi vacA
In questo studio, abbiamo fatto alcune osservazioni che meritano d’essere
discusse:
• La maggior parte dei ceppi col sottotipo s1/m2, che è considerato
meno carcinogenico, è stata identificate in casi di carcinoma gastrico
istotipo intestinale.
• Tutti i ceppi cagA tipo C erano vacA s1/m2 e nessuno era vacA
s1/m1 (Tabella 1).

92
Tabella 1. Istotipi di carcinoma gastrico, tipi strutturali cagA e sottotipi vacA dei
ceppi isolati.
Case
no.
Carcinom
a
histotype
No. of
colonies
examined
(cagA+and
cagA-)
No. of
colonies
cagAtyp
e A(I)
No. of
colonies
cagAtyp
e A
No. of
colonies
cagAtyp
e B/D
No. of
colonies
cagAtyp
e C
No. of
colonies
vacAtyp
e
s1/m1
No. of
colonies
vacAtyp
e s1/m2
1 Intestinal 6 and 0 0 3 3 0 6 0
4 Intestinal 10 and 0 0 4 4 2 8 2
6 Intestinal 9 and 0 0 4 3 2 7 2
11 Intestinal 4 and 0 4 0 0 0 4 0
12 Intestinal 4 and 0 0 0 4 0 4 0
13 Intestinal 25 and 0 0 25 0 0 25 0
14 Intestinal 5 and 0 5 0 0 0 5 0
15 Intestinal 4 and 0 0 0 4 0 4 0
16 Intestinal 9 and 0 0 3 6 0 9 0
19 Intestinal 8 and 0 0 4 4 0 8 0
21 Intestinal 6 and 0 0 3 3 0 6 0
24 Intestinal 4 and 0 0 4 0 0 4 0
26 Intestinal 4 and 0 0 4 0 0 4 0
27 Intestinal 13 and 0 1 3 9 0 13 0
28 Intestinal 8 and 0 0 3 5 0 8 0
2 Diffuse 4 and 1 0 0 4 0 1 4
3 Diffuse 4 and 0 0 4 0 0 4 0
5 Diffuse 4 and 0 0 4 0 0 4 0
7 Diffuse 4 and 1 0 0 4 0 1 4
8 Diffuse 12 and 3 5 6 1 0 15 0
9 Diffuse 14 and 0 2 3 9 0 14 0
10 Diffuse 6 and 0 0 0 6 0 6 0
17 Diffuse 12 and 4 5 6 1 0 16 0
18 Diffuse 14 and 0 2 4 8 0 14 0
20 Diffuse 5 and 2 0 0 5 0 2 5
22 Diffuse 4 and 1 0 0 4 0 1 4
23 Diffuse 8 and 0 0 8 0 0 8 0
25 Diffuse 4 and 0 0 4 0 0 4 0

93
Queste osservazioni suggeriscono che il ridotto potenziale neoplastico del
sottotipo s1/m2 di vacA potrebbe essere bilanciato dall’elevato potere
carcinogenico di cagA tipo C.
Il carcinoma gastrico è la quarta più comune neoplasia maligna e la sola associate
ad infezione batterica cronica. Gli istotipi intestinale e diffuso del carcinoma
gastrico sono entità differenti e hanno distinti processi carcinogenetici. Il tipo
intestinale di carcinoma gastrico è preceduto da alterazioni della mucosa gastrica,
come atrofia, metaplasia intestinale e displasia
Il carcinoma gastrico varietà diffuse origina su una base di infiammazione cronica
e non è preceduto dalla cosiddetta “cascata di Correa”. Il carcinoma gastrico
diffuso mostra uno sviluppo progressivo e invasivo e il fatto che non è preceduto
da atrofia gastrica e metaplasia intestinale potrebbe ritardare la diagnosi di cancro
e quindi peggiorare la prognosi (249).
7. Conclusioni
In conclusione, questi risultati dimostrano che tutti i pazienti esaminati erano
infettati da almeno un ceppo cagA+, che una considerevole proporzione dei
pazienti era infettata da ceppi con più d’un tipo strutturale cagA e che tutti i
pazienti avevano nel loro stomaco almeno un ceppo con sottotipo vacA s1/m1. Il
polimorfismo del gene cagA non è risultato associato con alcuna varietà istologica
del carcinoma gastrico, mente il polimorfismo di vacA, in particolare il sottotipo
s1/m2, può influire sull’istotipo della neoplasia, dato che l’80,9% dei ceppi con
questo sottotipo è stato isolato da casi di carcinoma gastrico diffuso (p<0.001).

94
Questi dati indicano che la variabilità genomica dei principali fattori di virulenza
di H. pylori può contribuire a determinare il tipo istologico del cancro. Infine,
questo studio suggerisce che, per una migliore comprensione del ruolo che hanno
i principali determinanti di virulenza nelle malattie associate ad infezione, è
necessario esaminare diverse colonie per paziente e non a limitarsi ad una,
considerata rappresentativa del clone infettante
8.Bibliografia
1. M. La Placa – Principi di microbiologia medica. Società editrice Esculapio. 2005.
2. D. Janulaitytė-Günther, T. Günther1, A. Pavilonis, L. Kupčinskas. What Bizzozero
never could imagine – Helicobacter pylori today and tomorrow. 2003.
3. Richard J. Linch. Helicobacter pylori and ulcers. Milestones. Milestones in
investigative patology. 2005.
4. M. Moroni, R. Esposito, F. de Lalla. Malattie infettive. Elsevier Masson. 2008.
5. Ellin, D. Segal, S. Falkow, L. Tompkins. Helicobacter pylori attachment to gastric cells
induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins.
Proc. Natl. Acad. Sci. USA. 1996.
6. M. Clyne, B. Drumm. The urease enzyme of helicobacter pylori does not function as an
adhesin. Infection and Immunity. 1996
7. Mobley HLT, Mendz GL, Hazell SL. Helicobacter pylori: Physiology and Genetics.
Asm press. 2001.
8. German SV, Zykova IE, Modestova AV, Ermakov NV. Epidemiological
characteristics of Helicobacter pylori infection in Moscow. 2011.
9. Ma JL, You WC, Gail MH, et al. Helicobacter pylori infection and mode of
transmission in a population at high risk of stomach cancer. Int J Epidemiol. 1998.
10. Bauer S, Krumbiegel P, Richter M, Richter T, Röder S, Rolle-Kampczyk U,
Herbarth O. Influence of sociodemographic factors on Helicobacter pylori prevalence
variability among schoolchildren in Leipzig, Germany. A long-term follow-up study. Cent
Eur J Public Health. 2011.
11. M. Kivi, A.L.V. Johansson, M.Reilly, Y. Tindberg. Helicobacter pylori status in family
members as risk factors for infection in children. Epidemiol. Infect. 2005.

95
12. G. I. Perez-Perez, S.S. Witkin, M.D. Decker, M.J. Blaser. Seroprevalence of
Helicobacter pylori Infection in Couples. Journal of clinical microbiology. 1991.
13. J Pytko-Polonczyk, S J Konturek, E Karczewska, W Bielański, A Kaczmarczyk-
Stachowska. Oral cavity as permanent reservoir of Helicobacter pylori and potential
source of reinfection. Journal of physiology and pharmacology - an official journal of the
Polish Physiological Society. 1996.
14. S. Zaric, B. Boijc, Lj. Janckovic, B. Dapcevic, B.Popovic, S.Cakic, J.Milasin.
Periodontal therapy improves gastric Helicobacter pylori eradication. Journal of dental
research. 2009.
15. R.P.H. Logan, M.M. Walker. Epidemiology and diagnosis of Helicobacter pylori
infection.
16. L.Morris Brown. Helicobacter pylori: Epidemiology and routes of transmission.
Epidemiologic Reviews. 2000.
17. Biselli R, Fortini M, Matricardi PM, Stroffolini T, D'Amelio R. Incidence of
Helicobacter pylori infection in a cohort of Italian military students .Infection. 1999.
18. G. Pareti, C. Cerletti, G. de Gaetano. How old is Helicobacter pylori? The Lancet.
2002.
19. S. O. Hyne, S. Teneberg, N. Roche, T. Wadstro¨m. Glycoconjugate binding of gastric
and enterohepatic Helicobacter spp. Infection and Immunity. 2003
20. G Faller, H Steininger, J Kränzlein, H Maul, T Kerkau, J Hensen, E G Hahn, T
Kirchner. Antigastric autoantibodies in Helicobacter pylori infection: implications of
histological and clinical parameters of gastritis. Gut. 1997
21. R . D. Leunk. , T. Johnson, C. David, G. Kraft, R. Morgan. Cytotoxic activity in
broth-culture filtrates of Campylobacter pylori. J. Med. Microbiology. 1988.
22. J.C. Atherton. H. pylori virulence factor. British medical bulletin. 1998.
23. J. C. Atherton, P. M. Sharp, T. L. Cover, G. Gonzalez-Valencia, R. M. Peek Jr. S. A.
Thompson, C. J. Hawkey, M. J. Blaser. Vacuolating cytotoxin (vacA) alleles of
Helicobacter pylori comprise two geographically widespread types, m1 and m2, and
have evolved through limited recombination. Curr microbiology. 1999.
24. A. Galmiche, J. Rassow. Targeting of Helicobacter pylori vacA to mitochondria. Gut
microbes. 2010.
25. S. Backert, N. Tegtmeye. The versatility of the Helicobacter pylori vacuolating
cytotoxin vacA in signal transduction and molecular crosstalk. Toxins 2010, 2, 69-92.

96
26. S. Maeda, H. Yoshida, T. Ikenoue, K. Ogura, F. Kanai, N. Kato, Y. Shiratori, M.
Omata. Structure of cag pathogenicity island in Japanese Helicobacter pylori isolates.
Gut 1999;44:336–341.
27. L. Terradot, G. Waksma. Architecture of the Helicobacter pylori Cag-type IV secretion
system. FEBS Journal. 2011. 1213–1222.
28. H. Higashi, R. Tsutsumi, A. Fujita, S. Yamazaki, M. Asaka, T. Azuma, M.
Hatakeyama. Biological activity of the Helicobacter pylori virulence factor CagA is
determined by variation in the tyrosine phosphorylation sites. PNAS. 2002. 14428–
14433.
29. S. Backert, N. Tegtmeyer, M. Selbach. The versatility of Helicobacter pylori CagA
effector protein functions: the master key hypothesis. Helicobacter. 2010. ISSN 1523-
5378.
30. J.E. Crabtree, S.M. Farmery, I.J.D. Lindley, N. Figura, P. Peichl, D.S. Tompkin.
CagA/cytotoxic strains of Helicobacter pylont and interleukin-8 in gastric epithelial cell
lines. J. Clin. Pathol. 1994. 47:945-950.
31. Y. Yamaoka, M. Kita, T. Kodama, N. Sawai, J. Imanishi. Helicobacter pylori cagA
gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology
1996; 110:1744–1752
32. C. Audiberta, B. Janviera, B. Grignona, L. Salaüna, C. Burucoaa, J.C. Lecronb, J.L.
Fauchèrea. Correlation between IL-8 induction, cagA status and vacA genotypes in 153
French Helicobacter pylori isolates. Res. Microbiol. 2000. 191–200.
33. L. Butia,, E. Spoonera, A. G. Van der Veena, R. Rappuoli, A. Covacci, H. L.
Ploegha. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-
stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. PNAS. 2011.
34. S.R. Ogden, L.E. Wrobleswski, C. Weydig, J. Romero-Gallo, D.P. O’Brien, D.A.
Israel, U.S. Krishna, B. Fingleton, A. B. Reynolds, S. Wessler, R, M. Peek jr. p120
and Kaiso regulate Helicobacter pylori-induced expression of matrix metalloprotease-7.
Molecular Biology of the Cell. 2008. 4110-4121.
35. S. Manxhuka-Kerliu, S. Telaku, E. Devolli-Disha, A. Ahmetaj, V. Sahatçiu-Meka, A.
Kerliu, S. Loxha, B. Shahini, G. Gashi, A. Podrimaj. Helicobacter pylori gastritis
Updated Sydney Classification applied in our material. Contributions, Sec. Biol. Med.
Sci., MASA, XXX, 1, p. 45–60 (2009) - ISSN 0351–3254.
36. S. Backert, M. Nauman. What a disorder: proinflammatory signaling pathways induced
by Helicobacter pylori. Trends in Microbiology Vol.18 No.11.
Doi:10.1016/j.tim.2010.08.003.

97
37. H. Svensson, M. Hansson, J. Kilhamn, S. Backert , M. Quiding-Järbrink. Selective
upregulation of endothelial E-selectin in response to Helicobacter pylori-induced
gastritis. Infect Immun. 2009 Jul;77(7):3109-16. Epub 2009 May 4.
38. K. Bodger, J. E. Crabtre. Helicobacter pylori and gastric inflammation. British Medical
Bulletin 1998;54 (No. 1): 139-150
39. N.Figura, C. Gonnelli, C. Lenzi, D. Vaira. Patogenesi dell’infezione da Helicobacter
pylori. L’infezione da Helicobacter pylori, diagnosi e terapia. 2006.
40. O. Handa, Y. Naito, T. Yoshikawa. Redox biology and gastric carcinogenesis: the role
of Helicobacter pylori. Redox Rep. 2011;16(1):1-7.
41. M. Mashimo, M. Nishikawa, K. Higuchi, M. Hirose, Q. Wei, A. Haque, E. Sasaki, M.
Shiba, K. Tominaga, T. Watanabe, Y. Fujiwara, T. Arakawa, M. Inoue. Production
of reactive oxygen species in peripheral blood is increased in individuals
with Helicobacter pylori infection and decreased after its eradication. Helicobacter. 2006
Aug;11(4):266-71.
42. M. Stolte, A. Meining. Update Sydney System: classification and grading of gastritis as
the basis of diagnosis and treatment. Review. Can J Gastroenterology, vol 15, No 9,
September 2001.
43. G. M. Mariuzzi – Anatomia Patologica. Piccin 2007.
44. M.L. Caruso, R. Fiocca. Gastriti croniche, linee guida e criteri diagnostici minimi.
Gruppo italiano patologi dell’apparato digerente. Pathologica, 1997; 89: 592-597.
45. A. Saha, C. E Hammond, C. Beeson, R. M Peek Jr, and A. J. Smolka. Helicobacter
pylori represses proton pump expression and inhibits acid secretion in human gastric
mucosa. Gut. 2010 July; 59(7): 874–881. doi:10.1136/gut.2009.194795.
46. M. M. D’Elios, B. J. Appelmelk, A. Amedei, M. P. Bergman, G. Del Prete. Gastric
autoimmunity: the role of Helicobacter pylori and molecular mimicry. TRENDS in
Molecular Medicine Vol.10 No.7 July 2004 (da aggiungere nella sezione gastrite)
47. F. Presotto, B. Sabini, A. Cecchetto, M. Plebani, F. De Lazzari, B. Pedini, C.
Betterle. Helicobacter pylori infection and gastric autoimmune diseases: is there a link?
Helicobacter. Volume 8, number, 2003
48. R. Dionigi. Chirurgia. Masson. 2006.
49. C. Ricci, D. Vaira. Helicobacter pylori e malattie gastroduodenali. L’infezione da
Helicobacter pylori, patogenesi, diagnosi e terapia. 2006.
50. B. Qian, S. Ma, L. Shang, J. Qian, G. Zhang. Effects of Helicobacter pylori eradication
on gastroesophageal reflux disease. Helicobacter ISSN 1523-5378. 2011.

98
51. H. Suzuki, J. Matsuzaki, T. Hibi. What Is the Difference Between Helicobacter pylori
Associated Dyspepsia and Functional Dyspepsia? J Neurogastroenterol Motil, Vol. 17
No. 2 April, 2011. doi:10.5056/jnm.2010.17.2.124
52. N. Uemura, H. Okamoto, O.Yamamoto, O. Matsumura, H. Yamaguchi, I.
Yamakido, I. Taniyama, A. Sasaki, R. J. Schlemper. Helicobacter pylori infection and
the development of gastric cancer. The New England journal of medicine. Vol. 345, No.
11. September 13, 2001.
53. M. Asaka, A. R. Sepulveda, T. Sugiyama, D. Y. Graham. Gastric Cancer.
Helicobacter pylori, fisiology and genetics. Chapter 40. Asm press. 2001.
54. T. Matysiak-Budnika, F. Me´graudb. Helicobacter pylori infection and gastric cancer.
European Journal Of Cancer 42 (2006) 708 –716.
55. A. Venkateshwari, D. Krishnaveni1, S. Venugopal1, P. Shashikumar, A. Vidyasagar,
A. Jyothy. Helicobacter pylori infection in relation to gastric cancer progression. Indian
Journal of Cancer. 2011. Doi: 10.4103/0019-509X.75840, Pmid: 21248438.
56. B.Chun-Yu Wong, S. Kum Lam, W. Man Wong, J. Shun Chen, T. Ting Zheng, R. E.
Feng, K. Chuen Lai, W. Hsing Cheng Hu, S. Tsan Yuen, S. Yi Leung, D. Yee Tak
Fong, J. Ho, C. Kong Ching, J. Shi Chen. Helicobacter pylori Eradication to prevent
gastric cancer in a high-risk region of China. A randomized controlled trial. JAMA,
January 14, 2004—Vol 291, No. 2.
57. S. Take, M. Mizuno, K. Ishiki, Y. Nagahara, T. Yoshida, K. Yokota, K. Oguma, H.
Okada, Y. Shiratori. The effect of eradicating Helicobacter pylori on the development of
gastric cancer in patients with peptic ulcer disease. The American Journal of
Gastroenterology 100, 1037-1042 (May 2005) | doi:10.1111/j.1572-0241.2005.41384.
58. J.Romero-Gallo, E. J Harris, U. Krishna, M. K. Washington, G. I. Perez-Perez, R.
M. Peek Jr. Effect of Helicobacter pylori eradication on gastric carcinogenesis. Lab
Invest. 2008 March ; 88(3): 328–336. doi:10.1038/labinvest.3700719.
59. S. R. Owens, L. B. Smith. Molecular aspects of H. pylori -related MALT lymphoma –
Review article. Pathology Research International. Volume 2011, Article ID 193149,
doi:10.4061/2011/19314.
60. H. Suzuki, Y. Saito, T. Hib. Helicobacter pylori and gastric Mucosa-associated
Lymphoid Tissue (MALT) lymphoma: updated review of clinical outcomes and the
molecular pathogenesis. Gut and Liver, Vol. 3, No. 2, June 2009, pp. 81-87.
61. P. Lehours, Z. Zheng, A. Skoglund, F. Me´graud, L. Engstrand. Is there a link
between the lipopolysaccharide of Helicobacter pylori gastric MALT lymphoma

99
associated strains and lymphoma pathogenesis? PlosOne. October 2009, volume 4, issue
10, e7297.
62. A. Stathis, C. Chini, F. Bertoni, I. Proserpio, C. Capella, L. Mazzucchelli, E.
Pedrinis, F. Cavalli, G. Pinotti, E. Zucca. Long-term outcome following Helicobacter
pylori eradication in a retrospective study of 105 patients with localized gastric marginal
zone B-cell lymphoma of MALT Ann Oncol. 2009 Jun;20(6):1086-93. Epub 2009 Feb 4.
63. M. A. Mendall, P. M. Goggin, N. Molineaux, J. Levy, T. Toosy, D. Strachan, A. J.
Camm, T. C. Northfield. Relation of Helicobacter pylori infection and coronary heart
disease. Br Heart - 1994;71:437-439
64. R. Pellicano, M.G. Mazzarello, S. Morelloni. Acute myocardial infarction and
Helicobacter pylori seropositivity. Int J Clin Lab Res 1999;29:141.
65. A. Manolakis, A. N. Kapsoritakis, S. P. Potamiano. A review of the postulated
mechanisms concerning the association of Helicobacter pylori with ischemic heart
disease. Helicobacter 12: 287–297. 2007.
66. G.S. Tamer, I. Tengiz, E. Ercan, C. Duman, E. Alioglu, U.O. Turk. Helicobacter
pylori seropositivity in patients with acute coronary syndromes. Dig Dis Sci. 2009
Jun;54(6):1253-6. Epub 2008 Sep 4.
67. F. Palm, C. Urbanek, A. Grau. Infection, its treatment and the risk for stroke. Curr
Vasc Pharmacol. 2009 Apr;7(2):146-52.
68. M. Gencer, E. Ceylan, F. Yıldız Zeyrek, N. Aksoy. Helicobacter pylori
Seroprevalence in Patients with Chronic Obstructive Pulmonary Disease and Its Relation
to Pulmonary Function Tests. Respiration 2007;74:170–175.
69. D. Fullerton, J. R. Britton, S. A. Lewis, I. D. Pavord, T. M. McKeever, A. W.
Fogarty. Helicobacter pylori and lung function, asthma, atopy and allergic disease. A
population-based cross-sectional study in adults. International Journal of Epidemiology
2009; 38:419 – 426 doi: 10.1093/ije/dyn348.
70. J. Angrilla, N. Sa´nchezb, C. Agustı´a, J.Ma. Guilemanyc, R. Miqueld, J. Gomeze,
A. Torresa. Does Helicobacter pylori have a pathogenic role in bronchiectasis?
Respiratory Medicine (2006) 100, 1202–1207.
71. W.L. Zhuo, B. Zhu, Z.L. Xiang, X.L. Zhuo, L. Cai, Z. T. Chena. Assessment of the
relationship between Helicobacter pyloriand and lung cancer: A Meta-analysis. Archives
of Medical Research 40 (2009) 406e410.
72. N. Figura, F. Franceschi, A. Santucci, G. Bernardini, G. Gasbarrini, A. Gasbarrini.
Extragastric manifestations of Helicobacter pylori infection. Helicobacter. 15. 2010,
ISSN 1523-5378.

100
73. X. L. Zhuo, Y. Wang, W. L. Zhuo. Possible association of Helicobacter pylori infection
with laryngeal cancer risk: an evidence-based meta-analysis. Arch Med Res
2008;39:625e628.
74. U. R. M. Bohr, B. Annibale, F. Franceschi, D. Roccarina, A. Gasbarrini. Extragastric
manifestations of Helicobacter pylori infection – Other Helicobacters. Helicobacter. 12.
2007 (Suppl. 1): 45–53. ISSN 1523-5378.
75. J. Kountouras, M. Boziki, E. Gavalas, Ch. Zavos1, G. Deretzi1, N. Grigoriadis, M.
Tsolaki, D. Chatzopoulos, P. Katsinelos, D. Tzilves, A. Zabouri1, I. Michailidou.
Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s Disease.
International Journal of Neuroscience. 2009, Vol. 119, No. 6 , Pages 765-777,
doi:10.1080/00207450902782083.
76. M. Malaguarnera, R. Bella,, G. Alagona, R. Ferri, A. Carnemolla, G. Pennisi.
Helicobacter pylori and Alzheimer’s disease: a possible link. European Journal of
Internal Medicine 15 (2004) 381– 386.
77. J. Kountouras, E. Gavalas, C. Zavos, C. Stergiopoulos, D. Chatzopoulos, N.
Kapetanakis, D. Gisakis. Alzheimer’s disease and Helicobacter pylori infection:
Defective immune regulation and apoptosis as proposed common links. Medical
Hypotheses (2007) 68, 378–388
78. A. Charlett, R. J.. Dobbs, S. M. Dobbs, C. Weller, M. A.A. Ibrahim, T. Dew, R.
Sherwood, N. L. Oxlade, J. M. Plant, J. Bowthorpe, A. J. Lawson, A. Curry, D. W.
Peterson, I. T. Bjarnason. Blood profile holds clues to role of infection in a premonitory
state for idiopathic parkinsonism and of gastrointestinal infection in established disease.
Gut Pathogens 2009, 1:20 doi:10.1186/1757-4749-1-20
79. M. Lyte. Microbial endocrinology as a basis for improved L-DOPA bioavailability in
Parkinson's patients treated for Helicobacter pylori. Med Hypotheses. 2010
May;74(5):895-7. Epub 2009 Dec 3.
80. R. J. Dobbs, S. M. Dobbs, C. Weller, A. Charlett, I. T. Bjarnason, A. Curry, D. S.
Ellis, M. A. A. Ibrahim, M. V. McCrossan, J. O’Donohue, R. J. Owen, N. L. Oxlade,
A. B. Price, J. D. Sanderson, M. Sudhanva, J. Williams. Helicobacter Hypothesis for
Idiopathic Parkinsonism: Before and Beyond. 2009. Helicobacter ISSN 1523-5378.
81. N. Figura, G. Di Cairano, F. Lorè, E. Guarino, A. Gragnoli, D. Cataldo, R.
Giannace, D. Vaira, L. Bianciardi, S. Kristodhullu, C. Lenzi, V. Torricelli, G.
Orlandini, C. Gennari. The infection by Helicobacter pylori strains expressing CagA is
highly prevalent in women with autoimmune thyroid disorders. J Physiol Pharmacol.
1999 Dec;50(5):817-26.

101
82. D. Larizza, V. Calcaterra, M. Martinetti, R. Negrini, A. De Silvestri, M. Cisternino,
A.M. Iannone, E. Solcia. H. Pylori infection and autoimmune thyroid disease in young
patients: the disadvantage of carrying the hla-drb1*0301 allele. Journal of Clinical
Endocrinology & Metabolism. First published November 1, 2005 as doi:10.1210/jc.2005-
1272
83. G. Bertalot, G. Montresor, M. Tampieri, A. Spasiano, M. Pedroni, B. Milanesi, M.
Favret, N. Manca, R. Negrini. Decrease in thyroid autoantibodies after eradication of
Helicobacter pylori infection. Clin Endocrinol (Oxf). 2004 Nov;61(5):650-2.
84. I. Ciortescu, C. Sfarti, M. Stan, M. Graur, C. Stanciu. Prevalence of Helicobacter
pylori infection in patients with diabetes mellitus. Rev Med Chir Soc Med Nat Iasi
2009;113:1048–55.
85. I. Krause, J.M. Anaya, A. Fraser, O. Barzilai, M. Ram, V. Abad, A. Arango, J.
García, Y. Shoenfeld. Anti-infectious antibodies and autoimmune-associated
autoantibodies in patients with type I diabetes mellitus and their close family members.
Ann N Y Acad Sci. 2009 Sep;1173:633-9.
86. Lutsey PL, Pankow JS, Bertoni AG, Szklo M, Folsom AR. Serological evidence of
infections and Type 2 diabetes: the Multi-Ethnic Study of Atherosclerosis. Diabet Med
2009;26:149–52.
87. T. Gunji, N. Matsuhashi, H. Sato, K. Fujibayashi, M. Okumura, N. Sasabe, A.
Urabe. Helicobacter pylori infection significantly increases insulin resistance in the
asymptomatic Japanese population. Helicobacter 2009;14:144–50.
88. S.Z. Wang, Y.N. Shi, J. Zhao, Z.D. Wang. Effects of Helicobacter pylori on blood
glucose fluctuation in type 2 diabetic patients. Zhonghua Yi Xue Za Zhi 2009;89:958–61.
89. A. Eshraghian, S.A. Hashemi, A. Hamidian Jahromi,H. Eshraghian, S.M.
Masoompour, M.A. Davarpanah. Helicobacter pylori infection as a risk factor for
insulin resistance. Dig Dis Sci 2009;54:1966–70.
90. W.Y. So, P.C. Tong, G.T. Ko, R.C. Ma, R. Ozaki, A.P. Kong. Low plasma adiponectin
level, white blood cell count and Helicobacter pylori titre independently predict
abnormal pancreatic betacell function. Diabetes Res Clin Pract 2009;86:89–95.
91. I.S. Santos, J. Boccio, L. Davidsson, M. Hernandez-Triana, E. Huanca-Sardinas, M.
Janjetic, Helicobacter pylori is not associated with anaemia in Latin America: results
from Argentina, Brazil, Bolivia, Cuba, Mexico and Venezuela. Public Health Nutr
2009;12:1862–70.

102
92. M. Hoseinzadeh, A. Khosravi, K. Sayemiri, M. Hossein Rasoli, A. Mohaveri. The
antibody titers to Helicobacter pylori in 7 - 12 year old iron deficiency anemic children,
in Ilam. J Res Med Sci. 2010 Nov-Dec; 15(6): 324–330.
93. Z.F. Zhang, N. Yang, G. Zhao, L. Zhu, Y. Zhu, L.X. Wang. Effect of Helicobacter
pylori eradication on iron deficiency. Chin Med J 2010;123(14):1924-1930.
94. R. Rad, R.M. Schmid, C. Prinz. Helicobacter pylori, iron deficiency, and gastric
autoimmunity. Blood 2006;107:4969–70.
95. R. Scandellari, E. Allemand, S. Vettore, M. Plebani, M.L. Randi, F. Fabris. Platelet
response to Helicobacter pylori eradication therapy in adult chronic idiopathic
thrombocytopenic purpura seems to be related to the presence of anticytotoxin-
associated gene A antibodies. Blood Coagul Fibrinolysis. 2009 Mar;20(2):108-13.
96. H. Amital, M. Govoni, R. Maya, P.L. Meroni, B. Ori, Y. Shoenfeld, A. Tincani, F.
Trotta, P. Sarzi-Puttini, F. Atzeni. Role of infectious agents in systemic rheumatic
diseases. Clin Exp Rheumatol. 2008 Jan-Feb;26(1 Suppl 48):S27-32.
97. A. Sulli, B. Seriolo, V. Savarino, M. Cutolo. Lack of correlation between gastric
Helicobacter pylori infection and primary or secondary Raynaud's phenomenon in
patients with systemic sclerosis. J Rheumatol. 2000 Jul;27(7):1820-1.
98. S. Danes, A. Zoli, F. Cremonini, A. Gasbarrini. High prevalence of Helicobacter pylori
type I virulent strains in patients with systemic sclerosis. J Rheumatol. 2000
Jun;27(6):1568-9.
99. M. Radic, D. Martinovic, J. Radic. Helicobacter pylori infection and systemic
sclerosis–is there a link? Joint Bone Spine. 2011 Jul;78(4):337-40. Epub 2010 Dec 8.
100. N. Giordano, N. Figura, M. Senesi, E. Battisti, F. Palumbo, P. Nardi, C. Gennari.
Helicobacter pylori infection and primary Sjögren's syndrome: a possible association
and new therapeutic approach. Current Therapeutic Research.Volume 56, Issue 12,
December 1995, Pages 1265-1269
101. E. Theander, I. Nilsson, R. Manthorpe, L.T. Jacobsson, T. Wadström.
Seroprevalence of Helicobacter pylori in primary Sjögren's syndrome. Clin Exp
Rheumatol. 2001 Nov-Dec;19(6):633-8.
102. L. Kalabay, B. Fekete, L. Czirják, L. Horváth, M.R. Daha, A. Veres, G. Fónyad, A.
Horváth, A. Viczián, M. Singh, I. Hoffer, G. Füst, L. Romics, Z. Prohászka.
Helicobacter pylori infection in connective tissue disorders is associated with high levels
of antibodies to mycobacterial hsp65 but not to human hsp60. Helicobacter. 2002
Aug;7(4):250-6.

103
103. Y. Showji, R. Nozawa, K. Sato, H. Suzuki. Seroprevalence of Helicobacter pylori
infection in patients with connective tissue diseases. Microbiol Immunol.
1996;40(7):499-503.
104. A. G. Abdou, E.I. Elshayeb, A.G. Farag, N.F. Elnaidany. Helicobacter pylori
infection in patients with chronic urticaria: correlation with pathologic findings in
gastric biopsies. Int J Dermatol. 2009 May;48(5):464-9.
105. R. Akashi, N. Ishiguro, S. Shimizu, M. Kawashima. Clinical study of the relationship
between Helicobacter pylori and chronic urticaria and prurigo chronica multiformis:
Effectiveness of eradication therapy for Helicobacter pylori. J Dermatol. 2011
Aug;38(8):761-6. doi: 10.1111/j.1346-8138.2010.01106.x. Epub 2010 Nov 22.
106. E. Lazaridou, C. Giannopoulou, C. Fotiadou, E. Vakirlis, A. Trigoni, D. Ioannides.
The potential role of microorganisms in the development of rosacea. JDDG; 2011 • 9:21–
25 DOI: 10.1111/j.1610-0387.2010.07513.
107. S. Zandi, S. Shamsadini, M. J. Zahedi, M. Hyatbaksh. Helicobacter pylori and
rosacea. East Mediterr Health J. 2003 Jan-Mar;9(1-2):167-71.
108. D. Boixeda de Miquel, M. Vázquez Romero, E. Vázquez Sequeiros, J. R. Foruny
Olcina, P. Boixeda de Miquel, A. López San Román, S. Alemán Villanueva, C.
Martín de Argila de Prados. Effect of Helicobacter pylori eradication therapy in
rosacea patient. Revista Española De Enfermedades Digestivas Copyright © 2006 Arán
Ediciones, S. L. 1130-0108/2006/98/7/501-509.
109. E. A. Attia, N. S. Abdel Fattah, H. M. Abdella. Upper gastrointestinal findings and
detection of Helicobacter pylori in patients with oral lichen planus. Clin Exp Dermatol.
2010 Jun;35(4):355-60. Epub 2009 Jul 29.
110. S. Qayoom, Q.M. Ahmad. Psoriasis and Helicobacter pylori. Indian journal of
dermatology, venereology and leprology. Year : 2003 , volume : 69, issue : 2.
111. N. Figura, L. Gennari, D. Merlotti, C. Lenzi, S. Campagna, B. Franci, B. Lucani, L.
Trabalzini, L. Bianciardi, C. Gonnelli, A. Santucci, A. Nut. Prevalence of
Helicobacter pylori infection in male patients with osteoporosis and controls. Dig Dis
Sci. 2005 May;50(5):847-52.
112. A. M. Kakehasi, C. M, Mendes, L. G. Coelho, L. P. Castro, A. J. Barbosa . The
presence of Helicobacter pylori in postmenopausal women is not a factor to the decrease
of bone mineral density. Arq Gastroenterol. 2007 Jul-Sep;44(3):266-70.
113. A .M. Kakehasi, C. B. Rodrigues, A. V. Carvalho, A. J. Barbosa. Chronic gastritis
and bone mineral density in women. Dig Dis Sci. 2009 Apr;54(4):819-24. Epub 2008
Aug 7.

104
114. S. Ozdem, M. Akcam, A. Yilmaz, M. Gultekin, R. Artan. Biochemical markers of
bone metabolism in children with Helicobacter pylori infection. Dig Dis Sci. 2007
Apr;52(4):967-72. Epub 2007 Feb 22.
115. M. S. Al-Marhoon. Is there a role for Helicobacter pylori infection in urological
diseases? Urology Journal Vol 5 No 3 Summer 2008.
116. I. Yavasoglu, M. Kucuk, B. Cildag, E. Arslan, M. Gok, S. Kafkas. A novel association
between polycystic ovary syndrome and Helicobacter pylori. Am J Med Sci. 2009
Sep;338(3):174-7.
117. I. Sandven, M. Abdelnoor, B.I. Nesheim, K. K. Melby. Helicobacter pylori infection
and hyperemesis gravidarum: a systematic review and meta-analysis of case-control
studies. Acta Obstet Gynecol Scand. 2009;88(11):1190-200.
118. H. Aksoy, A. Ozkan, F. Aktas, B. Borekci. Helicobacter pylori seropositivity and its
relationship with serum malondialdehyde and lipid profile in preeclampsia. J Clin Lab
Anal. 2009;23(4):219-22.
119. G. Pescetto, L. De Cecco, D. Pecorari. Manuale di clinica ostetrica e ginecologica.
Società editrice Universo, Roma. 1981.
120. M. Rochon. Sterility and infertility: two concepts. Cah Que Demogr. 1986 Apr;15(1):27-
56.
121. C. Niederberger, G. F. Joyce, M. Wise, R. B. Meacham. Male Infertility. Chapter 14.
Urologic Diseases in America. NIH Publication No. 07-5512. 2007.
122. J. Boivin, L. Bunting, J. A.Collins, K. G .Nygren. International estimates of infertility
prevalence and treatment-seeking: potential need and demand for infertility medical
care. Human Reproduction Vol.22, No.6 pp. 1506–1512, 2007
123. A. Aflatoonian, S. M. Seyedhassani, N. Tabibnejad. The epidemiological and
etiological aspects of infertility in Yazd province of Iran. Iranian Journal of Reproductive
Medicine Vol.7. No.3. pp: 117-122, Summer 2009
124. M. Fox. Age and infertility: the biological clock: fact or fiction? Jacksonville Medicine /
May, 2000.
125. J. O. Sule, P. Erigbali, L. Eruom. Prevalence of infertility in women in a southwestern
nigerian community. African Journal of Biomedical Research, Vol. 11 (2008); 225 – 227
ISSN 1119 – 5096.
126. S. Norris. Reproductive infertility: prevalence, causes, trends and treatments. Parliament
research branch, Library of Parliament. In Brief, January 2001. PRB 00-32E.

105
127. J. Farhi, A. Ben-Haroush. Distribution of causes of infertility in patients attending
primary fertility clinics in Israel. Isr Med Assoc J. 2011 Jan;13(1):51-4.
128. J. O. Schorge, J. I. Schaffer, L. M. Halvorson, B. L. Hoffman, K. D. Bradshaw, F. G.
Cunningham. Williams’ gynecology. McGraw-Hill Companies. ISBN 978-0-07-147257-
9. MHID 0-07-147257-6. 2008.
129. S. Kurotsuchi, H. Ando, A. Iwase, Y. Ishida, N. Hamajima, F. Kikkawa. The
plausibility of Helicobacter pylori-related infertility in Japan. Fertil Steril. 2008
Sep;90(3):866-8. Epub 2007 Oct 1.
130. G. Ambrosini, A. Andrisani, C. Fiore, D. Fabbrini, D. D’antona, E. Ragazzi, M.
Plebani, D. Amanini. Anti-Helicobacter pylori antibodies in cervical mucus: a new
cause of infertility. Eur J Obstet Gynecol Reprod Biol. 2011 Apr;155(2):157-60. Epub
2010 Dec 28.
131. S. C. Esteves, A. Agarwal. Novel Concepts in Male Infertility. International Braz J Urol.
Vol. 37 (1): 5-15, January - February, 2011 doi: 10.1590/S1677-55382011000700002
132. E. Buiatti, A. Barchielli, M. Geddes, L. Nastasi, D. Kriebel, M. Franchini, G.
Scarselli. Risk factors in male infertility: a case-control study. Arch Environ Health.
1984 Jul-Aug;39(4):266-70.
133. R. J. Leke, J. A. Oduma, S. Bassol-Mayagoitia, A. M. Bacha, K. M. Grigor. Regional
and geographical variations in infertility: effects of environmental, cultural, and
socioeconomic factors. Environmental Health Perspectives Supplements. 101 (Suppl. 2):
73-80 (1993)
134.R. A. Sherrod, J. DeCoster. Male infertility: an exploratory comparison of African -
american and white men.. J Cult Divers. 2011 Spring;18(1):29-35.
135. D. S. Irvine. Epidemiology and aetiology of male infertility. Human Reproduction
Volume 13 Supplement 1998. From humrep.oxfordjournals.org by guest on August 24,
2011.
136. E. Nieschlag, H. M. Nehre, S. Nieschlag. Andrology, male reproduction, health and
dysfunction. 3rd Edition. Springer-Verlag Berlin Heidelberg 2010. ISBN: 978-3-540-
78354-1. DOI: 10.1007/978-3-540-78355-8.
137. J. G. Grundzinskas, J. L. Yovich. Gametes, the spermatozoon. Cambridge reviews in
human reproduction. Cambridge University Press, 1995. ISBN 052147491.
138. J. S. Tash, G. E. Bracho. Regulation of sperm motility: emerging evidence for a major
role for protein phosphatase. Journal of Andrology. Vol 15, No 6. December 1994.
15(6):505-9.

106
139. F. Boué, R. Sullivan. Cases of human infertility are associated with the absence of P34H
an epididymal sperm antigen. Biol Reprod. 1996 May;54(5):1018-24.
140. J. P. Kavanagh. Sodium, potassium, calcium, magnesium, zinc, citrate and chloride of
human prostatic and seminal fluid. J. Reprod. Fertil. (1985) 75,35-41.
141. G. Abbaticchio, R. Giorgino, F. M. Gentile, A. Cassano, F. Gattuccio, G. Orlando,
A. Iannì. Hormones in the seminal fluid. The transport proteins of the thyroid hormones.
Acta Eur Fertil. 1981 Dec;12(4):307-11.
142. J. Brotherton. Cortisol and transcortin in human seminal plasma and amniotic fluid as
estimated by modern specific assays. Andrologia, Volume 22, Issue 3, 1990.
DOI: 10.1111/j.1439-0272.1990.tb01966.
143. F. Hrudka, A. H. ElJack. The effect of ethylene dibromide on differentiation of the
acrosome, nucleus, and transient nuclear appendages in ram spermatids. J Ultrastruct
Res. 1979 May;67(2):135-51.
144. E. Bustos-Obregón, O. Díaz. Ultrastructure of mouse teratozoospermia induced by
parathion. Asian J Androl. 1999 Jun;1(1-2):79-80.
145. H. R. Contreras, E. Bustos-Obregón. Morphological alterations in mouse testis by a
single dose of malathion. J Exp Zool. 1999 Aug 1;284(3):355-9.
146. B. L. Sailer, L. K. Jost, K. R. Erickson, M. A. Tajiran, D. P. Evenson. Effects of X-
irradiation on mouse testicular cells and sperm chromatin structure. Environ Mol
Mutagen. 1995;25(1):23-30.
147. B. Bartoov, N. Zabludovsky, F. Eltes, V. V. Smirnov, V. I. Grischenko, A. Fischbein.
Semen Quality of Workers Exposed to Ionizing Radiation in Decontamination Work after
the Chernobyl Nuclear Reactor Accident. Int J Occup Environ Health. 1997 Jul;3(3):198-
203.
148. G. Collodel, S. Capitani, A. Pammolli, V. Giannerini, M. Geminiani, E. Moretti.
Semen quality of male idiopathic infertile smokers and nonsmokers: an ultrastructural
study. J Androl. 2010 Mar-Apr;31(2):108-13. Epub 2009 Sep 10.
149. G. Collodel, S. Capitani, F. Iacoponi, M. G. Federico, N. A. Pascarelli, E. Moretti.
Retrospective assessment of potential negative synergistic effects of varicocele and
tobacco use on ultrastructural sperm morphology. Urology. 2009 Oct;74(4):794-9. Epub
2009 Aug 5.
150. J. P. Jarow. Effects of varicocele on male fertility. Hum Reprod Update. 2001 Jan-
Feb;7(1):59-64.

107
151. J. MacLeod. Seminal cytology in the presence of varicocele. Fertil Steril. 1965 Nov-
Dec;16(6):735-57.
152. C. K. Naughton, A. K. Nangia, A. Agarwal. Varicocele and male fertility: part II.
Pathofisiology of varicocele in male infertility. Human Reproduction Update. Vol 7, No
5, 473-482. 2001.
153. J. C. Trussell, P. A. Lee. The relationship of cryptorchidism to fertility. Curr Urol Rep.
2004 Apr;5(2):142-8.
154. F. Hadziselimovic, B. Hoecht. Testicular histology related to fertility outcome and
postpubertal hormone status in cryptorchidism. Klin Padiatr. 2008 Sep-Oct;220(5):302-
7. Epub 2008 Apr 9.
155. F. Hadziselimovic, B. Herzog. The importance of both an early orchidopexy and germ
cell maturation for fertility. Lancet. 2001 Oct 6;358(9288):1156-7.
156. D. Cortes, J. Thorup, E. Hogdall, B. Norgaard-Pedersen, B. L. Petersen, C. Hogdall.
The relation of germ cells per tubule in testes, serum inhibin B and FSH in cryptorchid
boys. Pediatr Surg Int. 2007 Feb;23(2):163-9. Epub 2006 Dec 14.
157. A. Ferlin, F. Raicu, V. Gatta, D. Zuccarello, G. Palka, C. Foresta. Male infertility:
role of genetic background. Reprod Biomed Online. 2007 Jun;14(6):734-45.
158. C. Foresta, A. Ferlin, E. Moro, P. Marin, A. Rossi, C. Scandellari. Microdeletion of
chromosome Y in male infertility: role of the DAZ gene. Ann Ital Med Int. 2001 Apr-
Jun;16(2):82-92.
159. P. H. Vogt. AZF deletions and Y chromosomal haplogroups: history and update based
on sequence. Hum Reprod Update. 2005 Jul-Aug;11(4):319-36. Epub 2005 May 12.
160. J. Jääskeläinen. Molecular biology of androgen insensitivity. Mol Cell Endocrinol. 2011
Aug 17.
161. B. Gottlieb, L. K. Beitel M. A. Trifiro. Androgen Insensitivity Syndrome. Source Gene
Reviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-.1999 Mar 24
[updated 2007 May 24].
162. M. Chillón, T. Casals, B. Mercier, L. Bassas, W. Lissens, S Silber, M. C. Romey, J.
Ruiz-Romero, C. Verlingue, M. Claustres, V. Nunes, C. Férec, X. Estivill. Mutations
in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl
J Med 1995; 332:1475-1480June 1, 1995
163. L. Lazaros, N. Xita, A. Kaponis, K. Zikopoulos, N. Sofikitis, I. Georgiou. Evidence
for association of sex hormone-binding globulin and androgen receptor genes with
semen quality. Andrologia. 2008 Jun;40(3):186-91.

108
164. H. E..Chemes, V. Y. Rawe. Sperm pathology: a step beyond descriptive morphology.
Origin, characterization and fertility potential of abnormal sperm phenotypes in infertile
men. Human Reproduction Update, Vol.9, No.5 pp. 405±428, 2003
165. E. Moretti, B. Baccetti, G. Scapigliati, G. Collodel. Transmission electron microscopy,
immunocytochemical and fluorescence in situ hybridisation studies in a case of 100%
necrozoospermia: case report. Andrologia. 2006 Dec;38(6):233-8.
166. B. Baccetti, A. G. Burrini, G. Collodel, A. R. Magnano, P. Piomboni, T. Renieri, C.
Sensini. Crater defect in human spermatozoa. Gamete Res. 1989 Mar;22(3):249-55.
167. A. H. D. M. Dam, I. Feenstra, J. R. Westphal, L. Ramos, R. J. T. van Golde, J. A. M.
Kremer. Globozoospermia revisited. Human Reproduction Update, Vol.13, No.1 pp. 63–
75, 2007
168. B. Baccetti, A. G. Burrini, G. Collodel, P. Piomboni, T. Renieri. A "miniacrosome"
sperm defect causing infertility in two brothers. J Androl. 1991 Mar-Apr;12(2):104-11.
169. B. Baccetti, A. G. Burrini, G. Collodel, A. R. Magnano, P. Piomboni, T. Renieri, C.
Sensini. Morphogenesis of the decapitated and decaudated sperm defect in two brothers.
Gamete Res. 1989 Jun;23(2):181-8.
170. D. Pellati, I. Mylonakis, G. Bertoloni, C. Fiore, A. Andrisani, G. Ambrosini, D.
Armanini. Genital tract infections and infertility. European Journal of Obstetrics &
Gynecology and Reproductive Biology 140 (2008) 3–
11doi:10.1016/j.ejogrb.2008.03.009.
171. I. Günyeli, F. Abike, I. Dünder, C. Aslan, O. L. Tapısız, O. Temizkan, A. Payaslı, E.
Erdemoğlu. Chlamydia, Mycoplasma and Ureaplasma infections in infertile couples and
effects of these infections on fertility. Arch Gynecol Obstet. 2011 Feb;283(2):379-85.
Epub 2010 Oct 27.
172. W. Eggert-Krause, M. Weltin, T. Strowitzki. Are chlamydial lipopolisaccaride-
directed antibodies in seminal plasma or serum clinically significant during investigation
of male infertility? Urology, 77 (5), 2001. 0090-4295/11/$36.00
doi:10.1016/j.urology.2010.11.014
173. E. Neofytou, G. Sourvinos, M. Asmarianaki, D. A. Spandidos, A. Makrigiannakis.
Prevalence of human herpes virus types 1–7 in the semen of men attending an infertility
clinic and correlation with semen parameters. Fertility and Sterility Vol. 91, No. 6, June
2009. doi:10.1016/j.fertnstert.2008.03.074.
174. N. Kapranos, E. Petrakou, C. Anastasiadou, D. Kotronias. Detection of herpes
simplex virus, cytomegalovirus, and Epstein-Barr virus in the semen of men attending an
infertility clinic. Fertility and sterility, Vol. 79, suppl. 3, june 2003.

109
175. W. Krause, F. Hubstreit, W. Slenzka. Are viral infections cause of leukocytospermia?
Andrologia, 34, 87-90 (2002).
176. N. Figura, P. Piomboni, A. Ponzetto, L. Gambera, C. Lenzi, D. Vaira, C. Peris, M
.R. Lotano, L. Gennari, L. Bianciardi, T. renieri, P. E. Valesin, S. Capitani, E.
Moretti, R. Colapinto, B. Baccetti, C. Gennari. Helicobacter pylori infection and
infertility. European journal of gastroenterology & epatology. 2002. Vol.14. No 6. 0954-
691 x
177. G. Collodel, E. Moretti, M. S. Campagna, S. Capitani, C. Lenzi, N. Figura. Infection
by CagA-positive Helicobacter pylori strains may contribute to alter the sperm quality of
men with fertility disorders and increas the systemic levels of TNF-α. Dig. Dis. Sci.
(2010) 55:94-100. Doi 10.1007/s10620-008-0704-1.
178. M. Kojima, H. Hosoda, Y. Date, M. Nakazato, H. Matsuo, K. Kangawa. Ghrelin is a
growth-hormone-releasing acylated peptide from stomach. Nature. 1999 Dec
9;402(6762):656-60.
179. M. Korbonits, A. P. Goldstone, M. Gueorguiev, A. B. Grossman. Ghrelin--a hormone
with multiple functions. Front Neuroendocrinol. 2004 Apr;25(1):27-68.
180. H. Hosoda, M. Kojima, H. Matsuo, K. Kangawa. Ghrelin and des-acyl ghrelin: two
major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res
Commun. 2000 Dec 29;279(3):909-13.
181. H. Ariyasu, K. Takaya, T. Tagami, Y. Ogawa, K. Hosoda, T. Akamizu, M. Suda, T.
Koh, K. Natsui, S. Toyooka, G. Shirakami, T. Usui, A. Shimatsu, K. Doi, H. Hosoda,
M. Kojima, K. Kangawa, K. Nakao. Stomach is a major source of circulating ghrelin,
and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J
Clin Endocrinol Metab. 2001 Oct;86(10):4753-8.
182. C. Y. Chen, A. Asakawa, M. Fujimiya, S. D. Lee, A. Inui. Ghrelin gene products and
the regulation of food intake and gut motility. Pharmacological Reviews. 61:430–481,
2009. 0031-6997/09/6104-430–481$20.00
183. S. Gnanapavan, B. Kola, S. A, Bustin, D. G. Morris, P. McGee, P. Fairclough, S.
Bhattacharya, R. Carpenter, A. B. Grossman, M. Korbonits. The tissue distribution
of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin
Endocrinol Metab. 2002 Jun;87(6):2988.
184. M. Kojima, H. Hosoda, K. Kangawa. Purification and distribution of ghrelin: the
natural endogenous ligand for the growth hormone secretagogue receptor. Horm Res.
2001;56 Suppl 1:93-7.

110
185. D. E. Cummings, J. Q. Purnell, R. S. Fravo, K. Schmidova, B. E. Wisse, D. S.
Weigle. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in
humans. Diabetes. 2001 Aug;50(8):1714-9.
186. Y. Masuda, T. Tanaka, N. Inomata, N. Ohnuma, S. Tanaka. Z. Itoh, H. Hosoda, M.
Kojia, K. Kangawa. Ghrelin stimulates gastric acid secretion and motility in rats.
Biochem Biophys Res Commun. 2000 Oct 5;276(3):905-8.
187. A. Szlachcic, T. Brzozowski, J. Majka, R. Pajdo, P. C. Konturek, M. Pawlik, S.
Kwiecien, D. Drozdowicz, W. Bielanski, S. J. Konturek, W. W. Pawlik. Involvement
of orexigenic peptides in the mechanism of gastric mucosal integrity and healing of
chronic gastric ulcers. Curr Pharm Des. 2010;16(10):1214-23.
188. N. Nagaya, M. Kijima, M. Uematsu, M. Yamagishi, H. Hososda, H. Oya, Y.
Hayashi, K. Kangawa. Hemodynamic and hormonal effects of human ghrelin in healthy
volunteers. Am J Physiol Regul Integr Comp Physiol. 2001 May;280(5):R1483-7.
189. J. V. Zhang, R. Pei-Gen, O. Avsian-Kretchmer, L. Ching-Wei, R. Rauch, C. Klein,
A. J. W. Hsueh. Obestatin, Peptide Encoded by the Ghrelin Gene, Opposes Ghrelin’s
Effects on Food Intake. Science. 2005 Nov 11;310(5750):996-9.
190. P. Zizzari, R. Longchamps, J. Epelbaum, M. T. Bluet-Pajot. Obestatin partially
affects ghrelin stimulation of food intake and growth hormone secretion in rodents.
Endocrinology. 2007 Apr;148(4):1648-53. Epub 2007 Jan 4.
191. M. S. Huda, B. H. Durham, S. P. Wong, T. M. Dovey, P. McCulloch, D. Kerrigan, J.
H. Pinkney, W. D. Fraser, J. P. WildingLack of an acute effect of ghrelin on markers of
bone turnover in healthy controls and post-gastrectomy subjects.2007 - Bone 41:406–
413.
192. A. J. Ren, Z. F. Guo, Y. K. Wang, L. K. Wang, W. Z. Wang, L. Lin, X. Zheng, W. J.
Yuan. Inhibitory effect of obestatin on glucose-induced insulin secretion in rats.
Biochem Biophys Res Commun 369:969–972. 2008.
193. X Qi, G. Yang, J. Liu, K. Li, Y. Tang, H. Liou, G. Boden. Circulating obestatin levels
in normal subjects and in patients with impaired glucose regulation and type 2 diabetes
mellitus. Clin Endocrinol (Oxf). 2007 Apr;66(4):593-7.
194. W. K. Samson, M. M. White, C. Price, A. V. Ferguson. Obestatin acts in brain to
inhibit thirst. Am J Physiol Regul Integr Comp Physiol. 2007 Jan;292(1):R637-43. Epub
2006 Aug 24.
195. E. Szentirmai, J. M. Krueger. Obestatin alters sleep in rats. Neurosci Lett. 2006 Aug
14;404(1-2):222-6. Epub 2006 Jun 23.

111
196. V. P. Carlini,H. B. Schiöth, S. R. Debarioglio. Obestatin improves memory
performance and causes anxiolytic effects in rats. Biochem Biophys Res Commun. 2007
Jan 26;352(4):907-12. Epub 2006 Dec 4.
197. M. Kapica, M. Zabielska, I. Puzio, A. Jankowska, I. Kato, A. Kuwahara, R.
Zabielski. Obestatin stimulates the secretion of pancreatic juice enzymes through a vagal
pathway in anaesthetized rats - preliminary results. J Physiol Pharmacol. 2007 Aug;58
Suppl 3:123-30.
198. R. V. Consindine, J. F. Caro. Leptin and the regulation of body weight. Int J Biochem
Cell Biol. 1997 Nov;29(11):1255-72.
199. L. A. Tartaglia, M. Dembski, X. Weng, N. Deng, J. Culpepper, R. Devos, G. J.
Richards, L. A. Campfield, F. T. Clark, J. Deeds, C. Muir, S. Sanker, A. Moriarty,
K. J. Moore, J. S. Smutko, G. G. Mays, E. A. Wool, C. A. Monroe, R. I. Tepper.
Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995 Dec
29;83(7):1263-71.
200. A. Z. Zhao, K. E. Bornfeldt, J. A. Beavo. Leptin inhibits insulin secretion by activation
of phosphodiesterase 3B. J Clin Invest. 1998 Sep 1;102(5):869-73.
201. P. E. Scherer, S. Williams, M. Fogliano, G. Baldini, H. F. Lodish. A novel serum
protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995 Nov
10;270(45):26746-9.
202. H. Waki, T. Yamauchi, J. Kamon, Y. Ito, S. Uchida, S. Kita, K. Hara, Y, Hada, G.
Vasseur, P. Froguel, S. Kimura, R. Nagai, T. Kadowaki. Impaired multimerization of
human adiponectin mutants associated with diabetes. Molecular structure and multimer
formation of adiponectin. J Biol Chem. 2003 Oct 10;278(41):40352-63. Epub 2003 Jul
23.
203. U. B. Pajvani, M. Hawkins, T. P. Combs, M. W. Rajala, T. Doebber, J. P. Berger, J.
A. Wagner, M. Wu, A. Knopps, A. H. Xiang, K. M. Utzschneider, S. E. Kahn, J. M.
Olefsky, T. A. Buchanan,P. E. Scherer. Complex distribution, not absolute amount of
adiponectin, correlates with thiazolidinedione-mediated improvement in insulin
sensitivity. J Biol Chem. 2004 Mar 26;279(13):12152-62. Epub 2003 Dec 29.
204. A. H. Berg, T. P. Combs, P. E. Scherer. ACRP30/adiponectin: an adipokine regulating
glucose and lipid metabolism. Trends Endocrinol Metab. 2002 Mar;13(2):84-9.
205. P. J. Havel. Control of energy homeostasis and insulin action by adipocyte hormones:
leptin, acylation stimulating protein, and adiponectin. Curr Opin Lipidol. 2002
Feb;13(1):51-9.

112
206. K. Hotta, T. Funahashi, Y. Arita, M. Takahashi, M. Matsuda, Y. Okamoto, H.
Iwahashi, H. Kuriyama, N. Ouchi, K. Maeda, M. Nishida, S. Kihara, N. Sakai, T.
Nakajima, K. Hasegawa, M. Muraguchi, Y. Ohmoto, T. Nakamura, S. Yamashita,
T. Hanafusa, Y. Matsuzawa. Plasma concentrations of a novel, adipose-specific
protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000
Jun;20(6):1595-9.
207. N. Ouchi, S. KiharA, Y. Arita, K. Maeda, H. Kuriyama, Y. Okamoto, K. Hotta, M.
Nishida, M. Takahashi, T. Nakamura, S. Yamashita, T. Funahashi, Y. Matsuzawa.
Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein
adiponectin. Circulation. 1999 Dec 21-28;100(25):2473-6.
208. C. L. McTernan, P. G. McTernan, A. L. Harte, P. L. Levick, A. H. Barnett, S.
Kumar F. Resistin, central obesity, and type 2 diabetes. The Lancet. Volume 359, Issue
9300, 5 January 2002, Pages 46-47. doi:10.1016/S0140-6736(02)07281-1.
209. C. M. Steppan, S. T. Bailey, S. Bhat, E .J. Brown, R. R. Banerjee, C. M. Wright, H.
R. Patel, R. S. Ahima, M. A. Lazar. The hormone resistin links obesity to diabetes.
Nature. 2001 Jan 18;409(6818):307-12.
210. O. Froy, A. Hananel, Z. Madar. Differential effect of insulin treatment on decreased
levels of beta-defensins and Toll-like receptors in diabetic rats. Molecular Immunology.
Volume 44, Issue 5, February 2007, Pages 796-802
211. M. Barnea, Z. Madar, O. Froy Glucose and insulin are needed for optimal defensin
expression in human cell lines. Biochemical and Biophysical Research Communications.
Volume 367, Issue 2, 7 March 2008, Pages 452-456
212. J. Wehkamp, J. Harder, M. Weichenthal, M. Schwab, E Schäffeler, M Schlee, K. R.
Herrlinger, A. Stallmach, F. Noack, P. Fritz, J. M. Schröder, C. L. Bevins, K.
Fellermann, E. F. Stange. NOD2 (CARD15) mutations in Crohn’s disease are
associated with diminished mucosal α-defensin expression. Gut 2004;53:1658-1664
doi:10.1136/gut.2003.032805
213. C. V. Nweneka, A. M. Prentice. Helicobacter pylori infection and circulating ghrelin
levels – A sistematic review. BMC Gastroenterol. 2011 Jan 26;11:7.
214. M. Cindoruk, I. Yetkin, S. M. Deger, T. Karakan, E. Kan, S. Unal. Influence of H.
pylori on plasma ghrelin in patients without atrophic gastritis. World Journal of
Gastroenterology 2007, 13(10):1595-1598.
215. C. U. Nwokolo, D. A. Freshwater, P. O'Hare, H. S. Randeva. Plasma ghrelin
following cure of Helicobacter pylori. Gut 2003; 52: 637-640.

113
216. A. Tatsuguchi, K. Miyake, K. Gudis, S. Futagami, T. Tsukui, K. Wada, T. Kishida,
Y. Fukuda, Y. Sugisaki, C. Sakamoto. Effect of Helicobacter pylori infection on ghrelin
expression in human gastric mucosa. Am J Gastroenterol. 2004 Nov;99(11):2121-7.
217. H. Isomoto, Y. Nishi, K. Ohnita, Y. Mizuta, S. Kohno, H. Ueno, M. Nakazato M. The
relationship between plasma and gastric ghrelin levels and strain diversity in
helicobacter pylori virulence. American Journal of Gastroenterology 2005, 100(6):1425-
1427.
218. H. Osawa. Ghrelin and Helicobacter pylori infection. World J Gastroenterol 2008
November 7; 14(41): 6327-6333
219. D. Campana, F. Nori, U. Pagotto, R. De Iasio, A. M. Morselli-Labate, R. Pasquali,
R. Corinaldesi, P. Tomassetti. Plasma acylated ghrelin levels are higher in patients
with chronic atrophic gastritis. Clinical Endocrinology Volume 67, Issue 5, pages 761–
766, November 2007.
220. G. Xin-Yuan, K. Hong-Yu, L. Xiao-Min, M. Zhi-Bin, N. Hao-Jie, G. Hong. Plasma
obestatin levels in men with chronic atrophic gastritis. Peptides. Volume 29, Issue 10,
October 2008, Pages 1749-1754. doi:10.1016/j.peptides.2008.05.027
221. T. Azuma, H. Suto, Y. Ito, M. Ohtani, M. Dojo, M. Kuriyama, T. Kato. Gastric leptin
and Helicobacter pylori infection. Gut. 2001 Sep;49(3):324-9.
222. M. J. Blaser, J. C. Atherton. Helicobacter pylori persistence: biology and disease. J
Clin Invest. 2004;113(3):321–333.
223. E. K. Tiaka, a. C. Manolakis, A. N. Kapsoritakis, S. P. Potamianos. The implication
of adiponectin and resistin in gastrointestinal diseases. Cytokine & Growth Factor
Reviews 22 (2011) 109–119.
224. D. A. O’Neill, S. P. Cole, E. Martin-Porter, M. P. Housley, L. Liu, T. Ganz, M. F.
Kagnoff. Regulation of human -defensins by gastric epithelial cells in response to
infection with helicobacter pylori or stimulation with interleukin-1. Infection and
Immunity, September 2000, p. 5412-5415, Vol. 68, No. 9. 0019-9567/00/$04.00+0.
225. J. Dupont, V. Maillard, S. Coyral-Castel, C- Ramè, P. Froment. Ghrelin in female
and male reproduction. Int J Pept. 2010;2010. pii: 158102. Epub 2010 Mar 14.
226. C. I. Messino, K. Dafopoulos, N. Chalvatzas, P. Georgoulias, I. E. Messinis. Effect of
ghrelin on gonadotrophin secretion in women during the menstrual cycle. Human
Reproduction, Vol.24, No.4 pp. 976–981, 2009
227. C. Schöfl, R. Horn, T. Schill, H. W. Schlösser, M. J. Müller, G. Brabant. Circulating
ghrelin levels in patients with polycystic ovary syndrome. The Journal of Clinical
Endocrinology & Metabolism 87(10):4607–4610.

114
228. D. Panidis, D. Farmakiotis, G. Koliakos, D. Rousso, A. Koutris, I. Katsikis, C.
Asteriadis, V. Karayannis, E. Diamanti-Kandarakis. Comparative study of plasma
ghrelin levels in women with polycystic ovary syndrome, in hyperandrogenic women and
in normal controls. Human Reproduction Vol.20, No.8 pp. 2127–2132, 2005
doi:10.1093/humrep/dei055.
229. D. Panidis, A. Kourtis, D. Farmakiotis, T. Mouslech, D. Rousso, G. Koliakos. Serum
adiponectin levels in women with polycystic ovary syndrome. Hum. Reprod. (2003) 18
(9): 1790-1796. doi: 10.1093/humrep/deg353.
230. S. Moschos, J. L. Chan, C. S. Mantzoros. Leptin and reproduction: a review. Fertility
and Sterility. Volume 77, Issue 3, March 2002, Pages 433-444. doi:10.1016/S0015-
0282(01)03010-2.
231. M .M. El-Eshmawy, J. A. Abdel Aal, A. K. El Hawary. Association of ghrelin and
leptin with reproductive hormones in constitutional delay of growth and puberty. Reprod
Biol Endocrinol. 2010 Dec 22;8:153.
232. M. Tena-Sempere, M. L. Barreiro, L. C. González. Novel expression and functional
role of ghrelin in rat testis. Endocrinology. 2002;143(2):717–725.
233. T. Ishikawa, H. Fujioka, T. Ishimura, A. Taneaka, M. Fujisawa. Ghrelin Expression
in Human Testis and Serum Testosterone Level. Journal of Andrology, Vol. 28, No. 2,
March/April 2007
234. M. L. Barreiro, M. Tena-Sempere. Ghrelin and reproduction: a novel signal linking
energy status and fertility? Molecular and Cellular Endocrinology. 2004;226(1-2):1–9.
235. D. Panidis, D. G. Goulis, I Katsikis, G. Koliakos, N. A. Geogopuolos, E. Diamanti-
Kandarakis. Serum and seminal plasma ghrelin levels in men with normospermia and
dyspermia. Gynecol Endocrinol. 2008 Jun;24(6):320-5.
236. World Health Organization. WHO laboratory manual for the examination of human
semen and semen-cervical mucus interaction. 4th ed. Cambridge University Press, 1999.
237. World Health Organization. WHO laboratory manual for the examination of human
semen and semen-cervical mucus interaction. Cambridge University Press, 1992.
238. J. Kawashima, S. Ohno, T. Sakurada, H. Takabayashi, M. Kudo, S. Ro et al.
Circulating acylated ghrelin level decrease in accordance with the extent of atrophic
gastritis. J. Gastroenterol 2009, 44:1046-54.
239. E. Moretti, G. Collodel, F. Iacoponi, M. Germiniani, A. N. Pascarelli, S. Campagna
et al. Detection of obestatin in seminal plasma and its relationship with ghrelin and
semen parameters. Fertil Steril 2011, 95:2303-9.

115
240. P. L. Jeffery, A. C. Herinton, L. K. Chopin. Expression and action of the growth
hormone releasing peptide and its receptor in prostate cancer lines. J. Endocrinology.
2002, 72:R7-11
241. P. Cassoni, C. Ghé, T. Marrocco, E. Tarabra, E. Allia, F. Catapano. Expression of
ghrelin and biological activity of the specific receptors for ghrelin and des-acylated
ghrelin in human prostate neoplasm and related cell lines. Eur J Endocrinol 2004,
150:173-184.
242. D. Radelman, L. A. Welniak, D. Traub, W. J. Murphy. Neurendocrine hormones such
as growth hormone and prolactin are integral members of the immunological cytokine
network. Cell Immunol 2008; 252: 111-21.
243. D. Traub. Novel concepts between the neuroendocrine and immune system: the ghrelin
immunoregolatory network. Vitam Hor 2008, 77:325-46
244. A. Kheradmand, M. Alizarei, P. Asadian, E. Alavi Rafiei, S. Joorabi. Antioxidant
enzyme activity and MDA level in rat testis following chronic administration of ghrelin.
Andrologia 2009, 41:335-40.
245. D. Basso, F. Navaglia, L. Brigato, F. Di Mario, M. Rugge, M. Plebani. Helicobacter
pylori non-cytotoxic genotype enhances mucosal gastrin and mast cell trypatase. J. Clin
Pathol 1999, 52:210-14.
246. Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. cag, a
pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-
associated virulence factors. Proc Natl Acad Sci U S A 1996; 93:14648-14653.
247. Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in
people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997;
40:297-301.
248. Azuma T, Ohtani M, Yamazaki Y, Higashi H, Hatakeyama M. Meta-analysis of the
relationship between CagA seropositivity and gastric cancer. Gastroenterology 2004;
249. Correa P. Gastric cancer: overview. Gastroenterol Clin North Am 2013; 42:211-217





























