Embed Size (px)
Citation preview

Therapeutics, Targets, and Chemical Biology
Cotargeting Androgen Receptor and Clusterin DelaysCastrate-Resistant Prostate Cancer Progression byInhibiting Adaptive Stress Response and AR Stability
Hiroaki Matsumoto1, Yoshiaki Yamamoto1, Masaki Shiota1, Hidetoshi Kuruma1, Eliana Beraldi1,Hideyasu Matsuyama2, Amina Zoubeidi1, and Martin Gleave1
AbstractAlthough androgen receptor (AR) pathway inhibitors prolong survival in castrate-resistant prostate cancer
(CRPC), resistance rapidly develops and is often associated with increased stress-activatedmolecular chaperoneslike clusterin (CLU) and continued AR signaling. Because adaptive pathways activated by treatment facilitatedevelopment of acquired resistance, cotargeting the stress response, activated by AR inhibition and mediatedthrough CLU, may create conditional lethality and improve outcomes. Here, we report that CLU is induced by ARantagonism and silencing using MDV3100 and antisense, respectively, to become highly expressed in castrate-andMDV3100-resistant tumors and cell lines. CLU, as well as AKT andmitogen-activated protein kinase (MAPK)signalosomes, increase in response to MDV3100-induced stress. Mechanistically, this stress response iscoordinated by a feed-forward loop involving p90rsk (RPS6KA)-mediated phosphoactivation of YB-1 withsubsequent induction of CLU. CLU inhibition repressed MDV3100-induced activation of AKT and MAPKpathways. In addition, when combined with MDV3100, CLU knockdown accelerated AR degradation andrepressed AR transcriptional activity through mechanisms involving decreased YB-1–regulated expression ofthe AR cochaperone, FKBP52. Cotargeting the AR (with MDV3100) and CLU (with OGX-011) synergisticallyenhanced apoptotic rates over that seen with MDV3100 or OGX-011 monotherapy and delayed CRPC LNCaPtumor and prostate-specific antigen (PSA) progression in vivo. These data indicate that cotargeting adaptivestress pathways activated by AR pathway inhibitors, andmediated through CLU, creates conditional lethality andprovides mechanistic and preclinical proof-of-principle to guide biologically rational combinatorial clinical trialdesign. Cancer Res; 73(16); 5206–17. �2013 AACR.
IntroductionProstate cancer is the second leading cause of cancer deaths
among males in western countries (1). Although early-stagedisease is treated with curative surgery or radiotherapy, themainstay of treatment for locally advanced, recurrent ormetastatic prostate cancer is androgen deprivation therapy(ADT), which reduces serum testosterone to castrate levels andsuppresses androgen receptor (AR) activity. Despite high initialresponse rates after ADT, progression to castrate-resistantprostate cancer (CRPC) occurs within 3 years (2–4). The ARis the principle driver of CRPC (5), and is supported by ADT-activated growth factor signaling pathways (6), survival genes
(7), and cytoprotective chaperone networks (8). Docetaxel (9)was the first therapy to prolong survival in CRPC, stratifyingthe treatment landscape into pre and postchemotherapystates. More recently, the CYP17 inhibitor abiraterone (10)and the AR antagonist MDV3100 (11) have prolonged survivaland are rapidly changing the CRPC treatment landscape.Despite significant responses (11, 12), these novel AR pathwayinhibitors activate redundant survival pathways that adaptive-ly drive treatment resistance and recurrent CRPC progression.Realization of the full potential of AR pathway inhibition willrequire characterization of these stress-activated survivalresponses and rational combinatorial cotargeting strategiesdesigned to abrogate them.
Molecular chaperones play central roles in stress responsesby maintaining protein homeostasis and regulating prosurvi-val signaling and transcriptional networks. Clusterin (CLU), astress-activated chaperone transcriptionally regulated byHSF1 (13), YB-1 (14), and others (15), inhibits stress-inducedapoptosis by suppressing protein aggregation (16), p53-acti-vating stress signals (17), and conformationally altered Bax(17, 18) while enhancing AKT phosphorylation (19) andtransactivation of NF-kB and HSF-1 (13, 20). CLU is expressedin many human cancers (21, 22), including prostate where itincreases following castration to become highly expressed in
Authors' Affiliations: 1The Vancouver Prostate Centre and Department ofUrologic Sciences, University of British Columbia, Vancouver, BritishColumbia, Canada; and 2Department of Urology, Graduate School ofMedicine, Yamaguchi University, Ube, Japan
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author:Martin E Gleave, The Vancouver Prostate Centre,2660 Oak Street, Vancouver, British Columbia V6H 3Z6, Canada. Phone:604-875-4818; Fax: 604-875-5654; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-13-0359
�2013 American Association for Cancer Research.
CancerResearch
Cancer Res; 73(16) August 15, 20135206
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

CRPC (23). CLU overexpression confers treatment resistance(24), whereas CLU inhibition potentiates activity of mostanticancer therapies in many preclinical models (25–27). TheCLU inhibitor, OGX-011 (custirsen, OncoGenex Pharmaceu-ticals), is currently in phase III trials after a randomized phaseII study in CRPC reported 7-month gain in overall survival and50% reduced death rate (HR ¼ 0.50) when combined withdocetaxel (28).Because CLU is induced by treatment stress, including
castration (23, 24), and functions as a key mediator of thestress response (13), we hypothesized that cotargeting the ARand stress-response pathways mediated by CLU may createconditional lethality and improve cancer control. This studywas set out to correlate MDV3100 treatment stress and resis-tance with CLU induction, identify pathways regulating CLUactivation, and define mechanisms by which CLU inhibitionpotentiates anti-AR therapy in CRPC.
Materials and MethodsProstate cancer cell lines and reagentsLNCaP and C4-2 cells were kindly provided by Dr. L.W.K.
Chung (1992, MD Anderson Cancer Center, Houston, Tx) andauthenticated by whole-genome and whole-transcriptomesequencing on Illumina Genome Analyzer IIx in July 2009. PC3cells were purchased from American Type Culture Collection(ATCC) andauthenticatedbyATCC's isoenzymeanalysis. CRPCand MDV3100-resistant tumors and cell lines were generated(Supplementary Fig. S1) as previously described (29). Cyclohex-imide and MG-132 were purchased from Calbiochem, R1881from PerkinElmer, and MDV3100 from Haoyuan ChemexpressCo., Limited. Antibodies used are listed in SupplementaryMaterials and Methods.
CLU siRNA and antisenseCLU and control siRNAs were purchased from Dharma-
con Research, Inc., as previously described (13). Second-generation CLU antisense (OGX-011) and scrambled (ScrB)oligodeoxynucleotides (ODN) with a 20-O-(2-methoxy)ethylmodification were supplied by OncoGenex Pharmaceuticals,whereas AR antisense were supplied by Isis Pharmaceuti-cals; these are described in Supplementary Materials andMethods. Prostate cells were treated with CLU and YB-1siRNA or oligonucleotides, using protocols described pre-viously (13, 14).
Western blot analysis and immunoprecipitationTotal proteins were extracted using radioimmunoprecipita-
tion assay buffer and submitted to Western blot analysis aspreviously described (30). For immunoprecipitation, total pro-teins were precleared with protein-G sepharose (InvitrogenLife Technologies), immunoprecipitatedwith 2mg anti-AR, andWestern blotted as previously described (30).
Quantitative reverse transcription PCRTotal RNA was extracted from cultured cells after 48 hours of
treatment using TRIzol reagent (Invitrogen Life Technologies,Inc.) as previously reported (13). Primers (described in Supple-mentary Materials and Methods) were normalized to b-actin
levels as an internal standard, and the comparative cycle thresh-old (Ct) method was used to calculate relative quantification oftarget mRNAs. Each assay was conducted in triplicate.
ImmunofluorescenceLNCaP cells were grown on coverslips and transfected with
CLU siRNA or ScrB control. Forty-eight hours posttransfection,cells were treated with 10 mmol/LMDV3100� 1 nmol/L R1881for 6 hours. Immunofluorescence was conducted as previouslyreported (30). Photomicrographs were taken at �20 magni-fication using Zeiss Axioplan II fluorescence microscope,followed by analysis with imaging software (Northern Eclipse,Empix Imaging, Inc.).
AR transcription activityLNCaP, CRPC-V16D, and MR49C cells (29) were treated
with OGX-011 or ScrB control, CLU siRNA or Ctr siRNA andthen transfected with PSA-luciferase reporter (�6,100–þ12)or Probasin luciferase reporter along with Renilla plasmid aspreviously described (30). All experiments were carried out intriplicate wells and repeated 5 times using different prepara-tions of plasmids.
Cell proliferation and cell-cycle assaysLNCaP, MR49C, and MR49F cells (Supplementary Fig. S1)
were transfected with CLU siRNA, OGX-011, or ScrB control,and then treated withMDV3100 or dimethyl sulfoxide (DMSO)control 24 hours after transfection. After a time course expo-sure, cell growth was measured by crystal violet assay aspreviously described (30). Detection and quantitation of apo-ptotic cell-cycle populationwere analyzed byflow cytometry aspreviously described (30). Each assay was done in triplicate 3times.
Crystal violet assay was used to determine synergism ofcombination therapy. LNCaP cells were treated with 10nmol/L CLU or ScrB siRNA, or 500 nmol/L OGX-011 or ScrBODN, and 1 day later combined with indicated concentra-tion of MDV3100 or DMSO for 48 hours. CalcuSyn softwarewas used to calculate the combination index (CI) at severaleffective doses (CI ¼ 1; additive effect, CI < 1; synergy effect,CI > 1; antagonistic effect).
Protein stability and degradationTo assess the effect of combination treatment on AR protein
stability, LNCaP cells were treated with OGX-011 or ScrB for 48hours followed by RPMIþ5% FBS plus 10 mmol/L cyclohexi-mide and 10 mmol/L MDV3100 for 2 to 6 or 16 hours andWestern blotted using AR and vinculin antibodies.
Animal treatmentMale athymic mice (Harlan Sprague-Dawley, Inc.) were
injected subcutaneoulsy with 1 � 106 LNCaP cells suspendedin Matrigel. When tumors reached 150 mm3, and serumprostate-specific antigen (PSA) was more than 50 ng/mL,mice were castrated. Once tumors progressed to CRPC, micewere randomly assigned to 10 mg/kg MDV3100 daily pluseither 10 mg/kg OGX-011 or ScrB ODN intraperitoneally oncedaily for 7 days and then 3 times per week thereafter. Each
Cotargeting Androgen Receptor and Clusterin for CRPC
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5207
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359
experimental group consisted of 12 mice. Tumor volume andserum PSA were measured as previously described (13). Allanimal procedures were conducted according to the guide-lines of the Canadian Council on Animal Care.
Statistical analysisAll results are expressed as the average � SE. Two-tailed
t tests, one-way ANOVA, or Wilcoxon matched pairs testswere used for statistical analysis. Synergism was calculatedby CalcuSyn software. The differences between single treat-ment and combination treatment was analysis by Freidmantest and done with JMP version 4; �, P < 0.05, ��, P < 0.01, and���, P < 0.001 were considered significant.
ResultsCLU is highly expressed inMDV3100-resistant xenograftsand cell lines
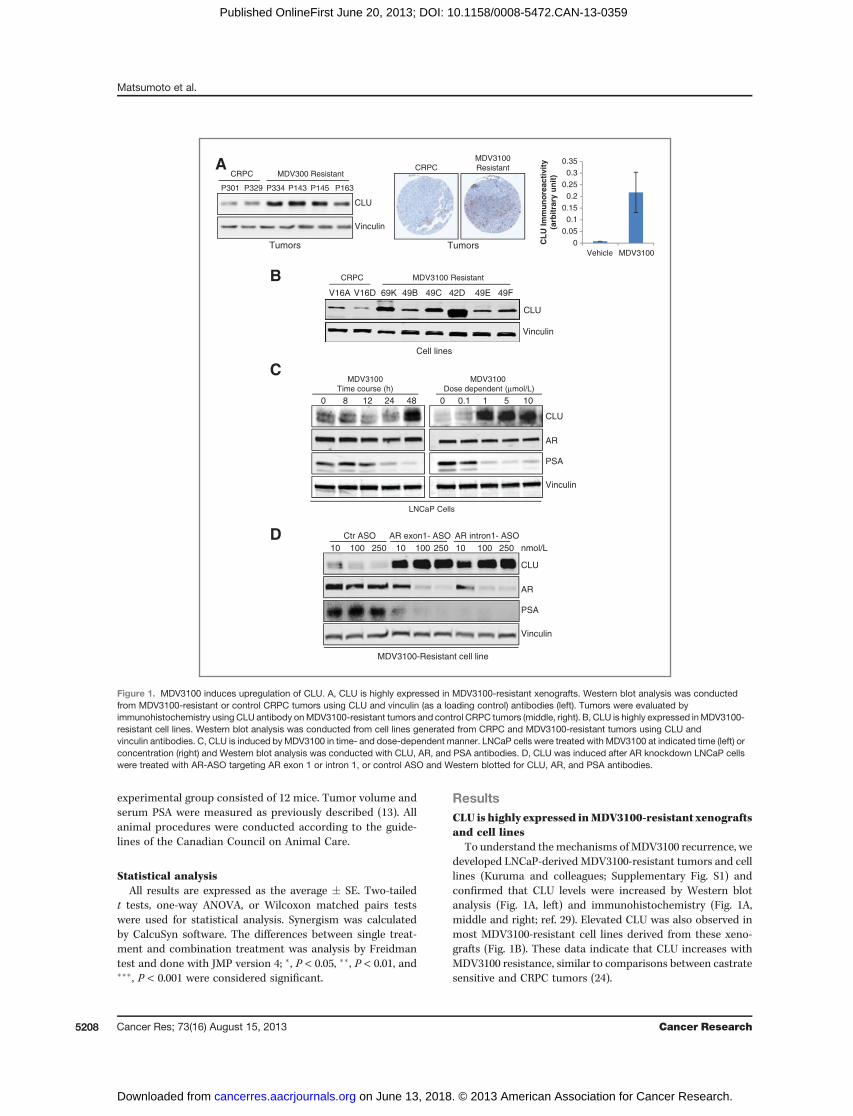
To understand themechanisms of MDV3100 recurrence, wedeveloped LNCaP-derived MDV3100-resistant tumors and celllines (Kuruma and colleagues; Supplementary Fig. S1) andconfirmed that CLU levels were increased by Western blotanalysis (Fig. 1A, left) and immunohistochemistry (Fig. 1A,middle and right; ref. 29). Elevated CLU was also observed inmost MDV3100-resistant cell lines derived from these xeno-grafts (Fig. 1B). These data indicate that CLU increases withMDV3100 resistance, similar to comparisons between castratesensitive and CRPC tumors (24).
P145P143 P163P334P329
MDV300 Resistant CRPC
P301
A
CLU
Vinculin
48241280 10510.10
MDV3100
Time course (h)
MDV3100
Dose dependent (μmol/L)
CLU
AR
PSA
Vinculin
1001025010010 nmol/L25010010250
AR exon1- ASOCtr ASO AR intron1- ASO
CLU
AR
PSA
Vinculin
MDV3100-Resistant cell line
B
D
C
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
MDV3100Vehicle
CL
U Im
mu
no
reac
tivi
ty(a
rbit
rary
un
it)
V16A V16D 69K 49B 49C 42D 49E 49F
CLU
Vinculin
CRPCMDV3100
Resistant
MDV3100 Resistant
Tumors Tumors
Cell lines
CRPC
LNCaP Cells
Figure 1. MDV3100 induces upregulation of CLU. A, CLU is highly expressed in MDV3100-resistant xenografts. Western blot analysis was conductedfrom MDV3100-resistant or control CRPC tumors using CLU and vinculin (as a loading control) antibodies (left). Tumors were evaluated byimmunohistochemistry using CLU antibody onMDV3100-resistant tumors and control CRPC tumors (middle, right). B, CLU is highly expressed inMDV3100-resistant cell lines. Western blot analysis was conducted from cell lines generated from CRPC and MDV3100-resistant tumors using CLU andvinculin antibodies. C, CLU is induced byMDV3100 in time- and dose-dependent manner. LNCaP cells were treated with MDV3100 at indicated time (left) orconcentration (right) and Western blot analysis was conducted with CLU, AR, and PSA antibodies. D, CLU was induced after AR knockdown LNCaP cellswere treated with AR-ASO targeting AR exon 1 or intron 1, or control ASO and Western blotted for CLU, AR, and PSA antibodies.
Matsumoto et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5208
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

AR pathway inhibition induces CLUWe used MDV3100 and AR antisense approaches to assess
effects of AR pathway inhibition on CLU induction inMDV3100-sensitive and -resistant LNCaP cells. Similar tobicalutamide and ADT using charcoal stripped serum (Sup-plementary Fig. S2), MDV3100 induces CLU in both a time-and dose-dependent manner in castrate-sensitive LNCaP(Fig. 1C) and castrate-resistant C4-2 (Supplementary Fig. S3)cells, in parallel with reduced AR activity as indicated bydecreased PSA expression. These data suggest that MDV3100induces activation of CLU, similar to that seen with first-lineADT in castrate-sensitive tumors (24). To determine whetherCLU is also induced as a consequence of AR inhibition even inMDV3100-resistant cells, AR-ASO targeting exon 1 and intron 1of the ARwere used. BothAR-ASOpotently downregulate AR ina dose-dependent and sequence-specific manner in MR49F
cells, with parallel induction of CLU (Fig. 1D). These datasuggest that AR pathway inhibition by ADT, AR antagonism,or antisense knockdown induces CLU in castrate-sensitive and-resistant cells, possibly as an early adaptive response to AR-inhibited stress.
MDV3100-induced CLU is mediated via p90Rsk-YB-1signaling pathway
We next set out to define transcriptional regulation of CLUinduction following MDV3100 treatment stress. We recentlyreported that CLU is upregulated via YB-1 after treatment stress(14); YB-1 is stress-activated via phosphorylation by AKT andp90RSK (a RPS6KA multigene family member; refs. 31, 32),stimulating its nuclear translocation. Although MDV3100 hasbeen reported to activate AKT (33), we found that MDV3100also induces a downstream effector of extracellular signal-
0
0.5
1
1.5
2
2.5
3
3.5
0 h 3 h 12 h 48 hRel
ativ
e C
LU
mR
NA
exp
ress
ion Control siRNA
YB-1 siRNA
C
48241204824120 (h) MDV3100
YB-1
CLU
β-Actin
YB-1 siRNACtr-siRNA
A
p-S6K
RSK
Vinculin
MDV3100
0 24 48 72 (h)
p-RSK
S6K
YB-1
p-YB-1
– + – (LY294002)+
–– (MDV3100)++
CLU
β-Actin
– + – (SL0101)+
–– (MDV3100)++
p-Akt
YB-1
p-YB-1
Akt
p-RSK
YB-1
p-YB-1
RSK
CLU
β-Actin
B
CLU
MDV3100
p-S6
S6K
p-S6
S6K
Figure2. MDV3100-inducedCLU ismediated via p90Rsk-YB-1signaling pathway. A, MDV3100activates AKT and MAPKpathways. LNCaP cells weretreated with 10 mmol/L MDV3100for indicated times and Westernblotted using CLU, p-Rsk/R/Rsk,p-S6K/S6K, and pYB-1/YB-1.Vinculin was used as a loadingcontrol. B, YB-1 knockdowndecreases MDV3100-inducedCLU. LNCaP cells weretransfected with YB-1 or controlsiRNA and treated with 10 mmol/LMDV3100 for indicated time.Proteins were extracted forWestern blot analysis using YB-1and CLU and actin as loadingcontrol (top). Bottom panel showsquantitative reverse transcriptionPCR using CLU-specific primersand GAPDH as internal control. C,MDV3100-induced CLU ismediated via p90Rsk-YB-1signaling pathway. LNCaP cellswere treated with MDV3100, Aktinhibitor (LY294002), or incombination for 24 hours. Proteinswere extracted for Western blotanalysis using CLU, pAkt/Akt, andpYB-1/YB-1 (left). LNCaP cellswere treated with MDV3100,p90Rsk inhibitor (SL0101), or incombination for 24 hours. Proteinswere extracted for Western blotanalyses using indicatedantibodies (right).
Cotargeting Androgen Receptor and Clusterin for CRPC
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5209
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359
regulated kinase (ERK), p90Rsk (Fig. 2A); because bothAKT andERK pathways can activate YB-1 (31, 32), we hypothesized thatMDV3100 induces CLU upregulation via phosphoactivationof YB-1. Figure 2A confirms that MDV3100 increases down-stream effectors of AKT (pS6K) and Erk (p90Rsk), accompaniedby increased phospho-YB-1 levels. YB-1 silencing abrogatesMDV3100-induced CLU expression at both the protein andmRNA levels (Fig. 2B), suggesting that MDV3100-induced CLUis mediated by YB-1. Because YB-1 can be phosphoactivated byboth AKT and p90Rsk (31, 32), we next defined which pathwaymediated MDV3100-induced upregulation of CLU using inhibi-tors ofAKT (LY294002) andp90Rsk (SL0101).MDV3100-inducedupregulation of CLUwas abrogated by p90Rsk (Fig. 2C, left), but
not AKT inhibition (Fig. 2C, right), indicating that p90Rsk-YB-1pathway mediates MDV3100-induced CLU expression.
CLU knockdown synergistically enhances MDV3100-induced inhibition of LNCaP cell growth
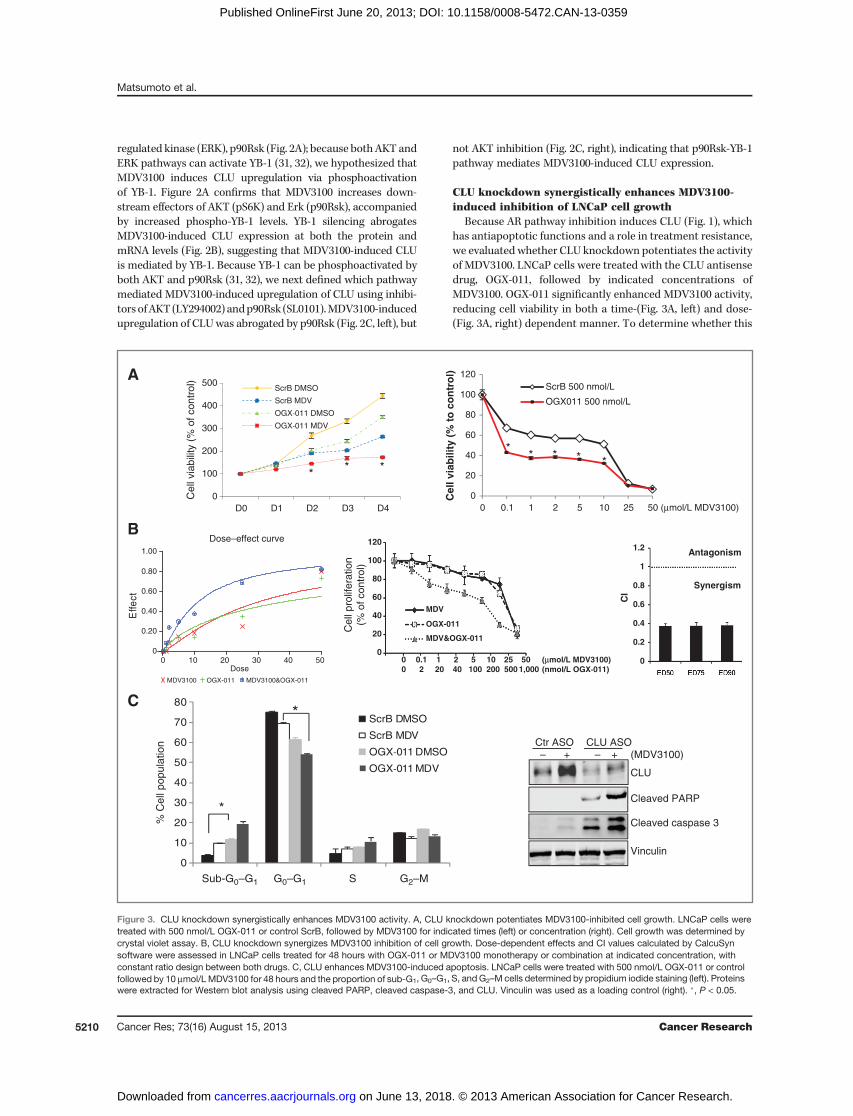
Because AR pathway inhibition induces CLU (Fig. 1), whichhas antiapoptotic functions and a role in treatment resistance,we evaluated whether CLU knockdown potentiates the activityof MDV3100. LNCaP cells were treated with the CLU antisensedrug, OGX-011, followed by indicated concentrations ofMDV3100. OGX-011 significantly enhanced MDV3100 activity,reducing cell viability in both a time-(Fig. 3A, left) and dose-(Fig. 3A, right) dependent manner. To determine whether this
0 10 20 30 40 500
0.20
0.40
0.60
0.80
1.00
Dose
Dose–effect curve
Eff
ect
MDV3100 OGX-011 MDV3100&OGX-011
Synergism
Antagonism
0 0.1 1 2 5 10 25 50 (μmol/L MDV3100)0 2 20 40 100 200 500 1,000 (nmol/L OGX-011)
0
100
200
300
400
500
D4D3D2D1D0
ScrB DMSO
ScrB MDV
OGX-011 DMSO
OGX-011 MDV
Cell
via
bili
ty (
% o
f contr
ol)
A
B
C
CLU
Vinculin
Cleaved PARP
Cleaved caspase 3
Ctr ASO CLU ASO– + – + (MDV3100)
Cell
pro
lifera
tion
(% o
f contr
ol)
0
20
40
60
80
100
120
50 (μmol/L MDV3100)25105210.10
Cel
l via
bili
ty (
% t
o c
on
tro
l)
ScrB 500 nmol/L
OGX011 500 nmol/L
* * * **
0
10
20
30
40
50
60
70
80
G2–MSG0–G1Sub-G0–G1
% C
ell
popula
tion
ScrB DMSO
ScrB MDV
OGX-011 DMSO
OGX-011 MDV
*
* *
** *
CI
120
100
80
60
40
20
0
1.2
1
0.8
0.6
0.4
0.2
0
MDV
OGX-011
MDV&OGX-011
Figure 3. CLU knockdown synergistically enhances MDV3100 activity. A, CLU knockdown potentiates MDV3100-inhibited cell growth. LNCaP cells weretreated with 500 nmol/L OGX-011 or control ScrB, followed by MDV3100 for indicated times (left) or concentration (right). Cell growth was determined bycrystal violet assay. B, CLU knockdown synergizes MDV3100 inhibition of cell growth. Dose-dependent effects and CI values calculated by CalcuSynsoftware were assessed in LNCaP cells treated for 48 hours with OGX-011 or MDV3100 monotherapy or combination at indicated concentration, withconstant ratio design between both drugs. C, CLU enhances MDV3100-induced apoptosis. LNCaP cells were treated with 500 nmol/L OGX-011 or controlfollowed by 10 mmol/LMDV3100 for 48 hours and the proportion of sub-G1, G0–G1, S, and G2–Mcells determined by propidium iodide staining (left). Proteinswere extracted for Western blot analysis using cleaved PARP, cleaved caspase-3, and CLU. Vinculin was used as a loading control (right). �, P < 0.05.
Matsumoto et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5210
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359
effect was additive or synergistic, dose-dependent effects withconstant ratio design and CI values were calculated accordingto the Chou and Talalay median effect principal (34). OGX-011synergistically enhances the inhibitory effect of MDV3100 oncell growth in LNCaP (Fig. 3B) as well as C4-2 cells (Supple-mentary Fig. S4, top), but not in AR-negative PC3 cells (Sup-plementary Fig. S4, bottom). Flow cytometric analysis indi-cates that OGX-011 combined withMDV3100 significantly (P <0.001) increases the sub-G1 apoptotic fraction compared withmonotherapy (Fig. 3C, left). In addition, combination OGX-011plus MDV3100 increased caspase-dependent apoptosis, asshown by cleaved PARP and caspase-3 activity (Fig. 3C, right).Collectively, these data indicate that OGX-011 synergisticallyenhances MDV3100-induced apoptosis.
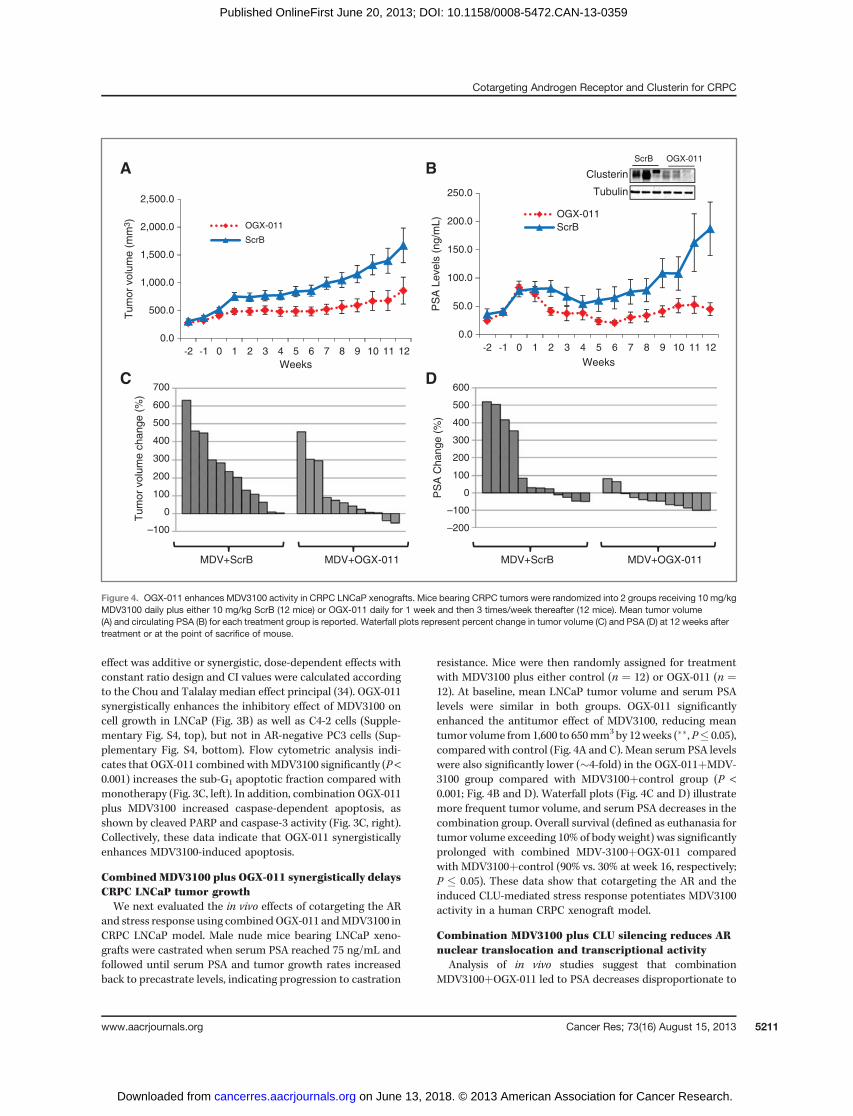
Combined MDV3100 plus OGX-011 synergistically delaysCRPC LNCaP tumor growthWe next evaluated the in vivo effects of cotargeting the AR
and stress response using combinedOGX-011 andMDV3100 inCRPC LNCaP model. Male nude mice bearing LNCaP xeno-grafts were castrated when serum PSA reached 75 ng/mL andfollowed until serum PSA and tumor growth rates increasedback to precastrate levels, indicating progression to castration
resistance. Mice were then randomly assigned for treatmentwith MDV3100 plus either control (n ¼ 12) or OGX-011 (n ¼12). At baseline, mean LNCaP tumor volume and serum PSAlevels were similar in both groups. OGX-011 significantlyenhanced the antitumor effect of MDV3100, reducing meantumor volume from 1,600 to 650mm3 by 12weeks (��, P� 0.05),compared with control (Fig. 4A and C). Mean serum PSA levelswere also significantly lower (�4-fold) in the OGX-011þMDV-3100 group compared with MDV3100þcontrol group (P <0.001; Fig. 4B and D). Waterfall plots (Fig. 4C and D) illustratemore frequent tumor volume, and serum PSA decreases in thecombination group. Overall survival (defined as euthanasia fortumor volume exceeding 10% of body weight) was significantlyprolonged with combined MDV-3100þOGX-011 comparedwith MDV3100þcontrol (90% vs. 30% at week 16, respectively;P � 0.05). These data show that cotargeting the AR and theinduced CLU-mediated stress response potentiates MDV3100activity in a human CRPC xenograft model.
Combination MDV3100 plus CLU silencing reduces ARnuclear translocation and transcriptional activity
Analysis of in vivo studies suggest that combinationMDV3100þOGX-011 led to PSA decreases disproportionate to
0.0
500.0
1,000.0
1,500.0
2,000.0
2,500.0
-2 -1 10 11 129876543210
OGX-011
ScrB
0.0
50.0
100.0
150.0
200.0
250.0
-2 -1 1211109876543210
OGX-011
ScrB
Tum
or
volu
me (
mm
3)
PS
A L
evels
(ng/m
L)
A B
C
Tubulin
OGX-011ScrB
Clusterin
Tum
or
volu
me c
hange (
%)
MDV+ScrB MDV+OGX-011
PS
A C
hange (
%)
MDV+ScrB MDV+OGX-011
WeeksWeeks
D700
600
500
400
300
200
100
0
–100
600
500
400
300
200
100
0
–100
–200
Figure 4. OGX-011 enhances MDV3100 activity in CRPC LNCaP xenografts. Mice bearing CRPC tumors were randomized into 2 groups receiving 10 mg/kgMDV3100 daily plus either 10 mg/kg ScrB (12 mice) or OGX-011 daily for 1 week and then 3 times/week thereafter (12 mice). Mean tumor volume(A) and circulating PSA (B) for each treatment group is reported. Waterfall plots represent percent change in tumor volume (C) and PSA (D) at 12 weeks aftertreatment or at the point of sacrifice of mouse.
Cotargeting Androgen Receptor and Clusterin for CRPC
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5211
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359
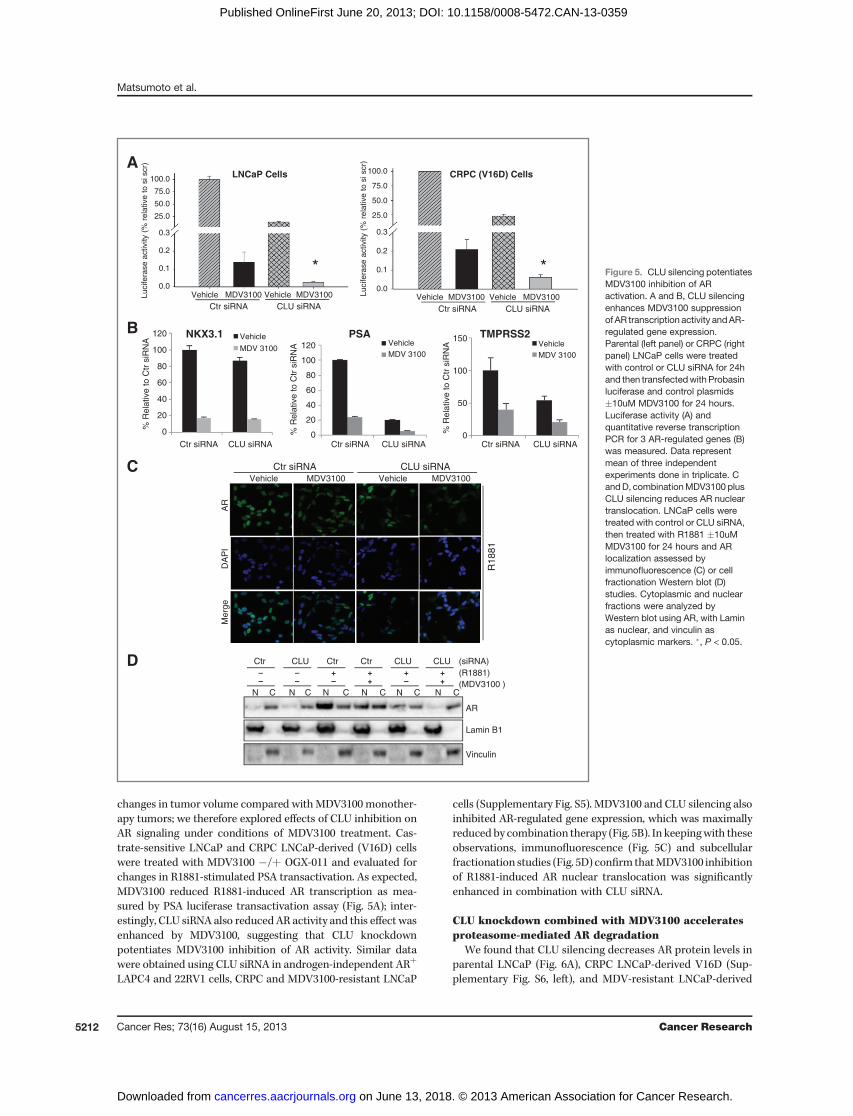
changes in tumor volume compared withMDV3100 monother-apy tumors; we therefore explored effects of CLU inhibition onAR signaling under conditions of MDV3100 treatment. Cas-trate-sensitive LNCaP and CRPC LNCaP-derived (V16D) cellswere treated with MDV3100 �/þ OGX-011 and evaluated forchanges in R1881-stimulated PSA transactivation. As expected,MDV3100 reduced R1881-induced AR transcription as mea-sured by PSA luciferase transactivation assay (Fig. 5A); inter-estingly, CLU siRNA also reduced AR activity and this effect wasenhanced by MDV3100, suggesting that CLU knockdownpotentiates MDV3100 inhibition of AR activity. Similar datawere obtained using CLU siRNA in androgen-independent ARþ
LAPC4 and 22RV1 cells, CRPC and MDV3100-resistant LNCaP
cells (Supplementary Fig. S5). MDV3100 and CLU silencing alsoinhibited AR-regulated gene expression, which was maximallyreduced by combination therapy (Fig. 5B). In keepingwith theseobservations, immunofluorescence (Fig. 5C) and subcellularfractionation studies (Fig. 5D) confirm thatMDV3100 inhibitionof R1881-induced AR nuclear translocation was significantlyenhanced in combination with CLU siRNA.
CLU knockdown combined with MDV3100 acceleratesproteasome-mediated AR degradation
We found that CLU silencing decreases AR protein levels inparental LNCaP (Fig. 6A), CRPC LNCaP-derived V16D (Sup-plementary Fig. S6, left), and MDV-resistant LNCaP-derived
++++–– (R1881)+–+––– (MDV3100 )
N C N C NCNCN
AR
CNC
Lamin B1
Vinculin
Ctr CLU Ctr Ctr CLU CLU (siRNA)
AR
DA
PI
Me
rge
Vehicle MDV3100 Vehicle MDV3100
Ctr siRNA CLU siRNA
R1881
D
C
ALNCaP Cells
**
Vehicle
100.0
75.0
50.0
25.0
Lucifera
se a
ctivity (
% r
ela
tive t
o s
i scr)
Lucifera
se a
ctivity (
% r
ela
tive to s
i scr)
0.3
0.2
0.1
0.0
100.0
75.0
50.0
25.0
0.3
0.2
0.1
0.0MDV3100 Vehicle MDV3100
CLU siRNACtr siRNAVehicle MDV3100 Vehicle MDV3100
CLU siRNACtr siRNA
CRPC (V16D) Cells
B
Ctr siRNA
PSANKX3.1 TMPRSS2
CLU siRNA Ctr siRNA CLU siRNA Ctr siRNA CLU siRNA
Vehicle
MDV 3100Vehicle
MDV 3100
Vehicle
MDV 3100
150
100
50
0
120
100
80
60
40
20
0
120
100
80
60
40
20
0
% R
ela
tive
to
Ctr
siR
NA
% R
ela
tive
to
Ctr
siR
NA
% R
ela
tive
to
Ctr
siR
NA
Figure 5. CLU silencing potentiatesMDV3100 inhibition of ARactivation. A and B, CLU silencingenhances MDV3100 suppressionof AR transcription activity andAR-regulated gene expression.Parental (left panel) or CRPC (rightpanel) LNCaP cells were treatedwith control or CLU siRNA for 24hand then transfectedwith Probasinluciferase and control plasmids�10uM MDV3100 for 24 hours.Luciferase activity (A) andquantitative reverse transcriptionPCR for 3 AR-regulated genes (B)was measured. Data representmean of three independentexperiments done in triplicate. CandD, combinationMDV3100 plusCLU silencing reduces AR nucleartranslocation. LNCaP cells weretreated with control or CLU siRNA,then treated with R1881 �10uMMDV3100 for 24 hours and ARlocalization assessed byimmunofluorescence (C) or cellfractionation Western blot (D)studies. Cytoplasmic and nuclearfractions were analyzed byWestern blot using AR, with Laminas nuclear, and vinculin ascytoplasmic markers. �, P < 0.05.
Matsumoto et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5212
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359
AR
Vinculin
620 16 620 16
ScrB
620 16 620 16
OGX-011
(h, CHX)
MDV3100VehicleMDV3100Vehicle
AR
Vinculin
– + – + (MG132)
+ + + + (MDV3100)
CLUCtr
Hsp90
IP: IgGInput
AR
IP: AR
(siRNA)CLUCtrCLUCtrCLUCtr
(MDV3100)+–+–+–+–+–+–
Acy-Lys AR
Vinculin
AR
CLU
MDV3100Vehicle
Ctr CLU Ctr CLU (siRNA)ScrB OGX ScrB OGX
MDV3100Vehicle
Vinculin
AR
CLU
siRNA
AR
IP:
AR
Ub
ARInput
Vehicle MDV3100
MG132
CLUCtr CLUCtr (siRNA)
CLUCtr CLUCtr (siRNA)
IP: U
b
poly-AR-ub
+ + + + (Ubiquitin)
+ + + + (AR)
Input AR
Ub
Vehicle MDV3100
MG132
B
A
D
C
ScrB OGX-011 ScrB OGX-011
MDV3100DMSO
140
120
100
80
60
40
20
0
140
120
100
80
60
40
20
0
MDV3100DMSO
siRNA Ctr siRNA CLU siRNA Ctr siRNA CLU
AR
mR
NA
levels
(% to S
crB
)
AR
mR
NA
levels
(% to C
tr s
iRN
A)
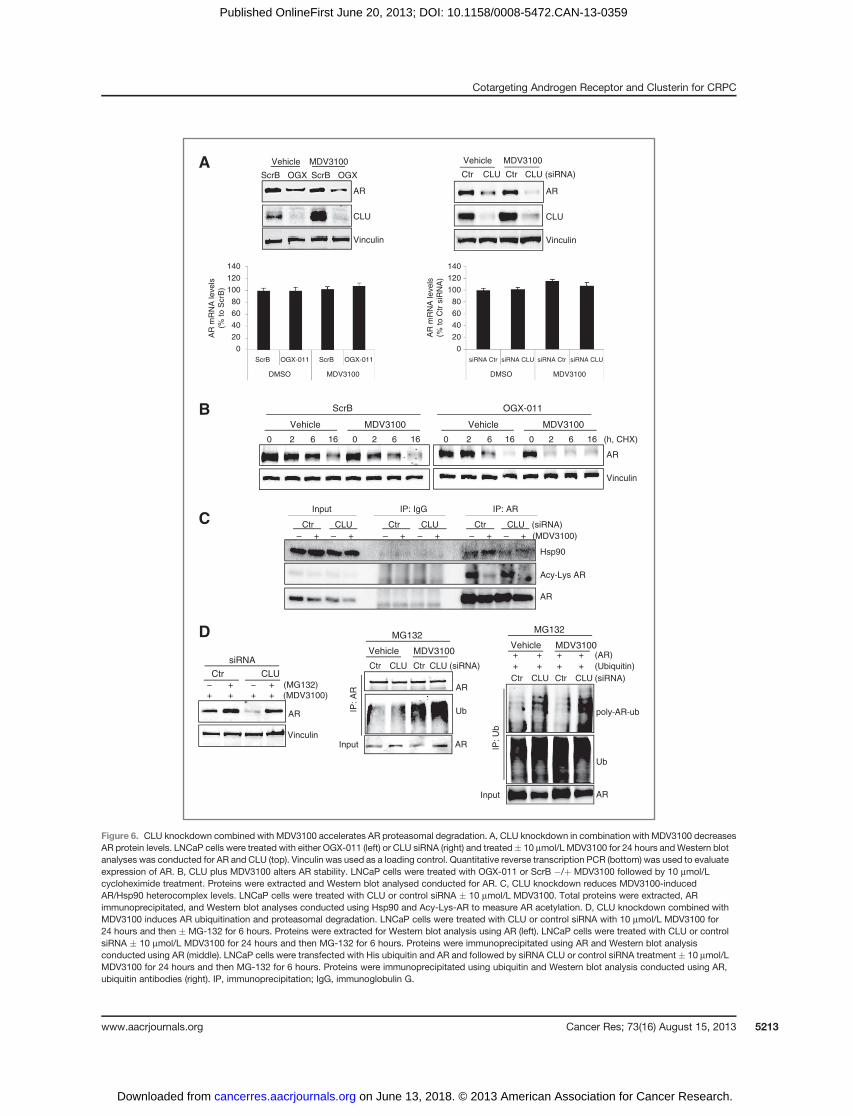
Figure 6. CLU knockdown combined with MDV3100 accelerates AR proteasomal degradation. A, CLU knockdown in combination with MDV3100 decreasesAR protein levels. LNCaP cells were treated with either OGX-011 (left) or CLU siRNA (right) and treated� 10 mmol/L MDV3100 for 24 hours andWestern blotanalyses was conducted for AR and CLU (top). Vinculin was used as a loading control. Quantitative reverse transcription PCR (bottom) was used to evaluateexpression of AR. B, CLU plus MDV3100 alters AR stability. LNCaP cells were treated with OGX-011 or ScrB �/þ MDV3100 followed by 10 mmol/Lcycloheximide treatment. Proteins were extracted and Western blot analysed conducted for AR. C, CLU knockdown reduces MDV3100-inducedAR/Hsp90 heterocomplex levels. LNCaP cells were treated with CLU or control siRNA � 10 mmol/L MDV3100. Total proteins were extracted, ARimmunoprecipitated, and Western blot analyses conducted using Hsp90 and Acy-Lys-AR to measure AR acetylation. D, CLU knockdown combined withMDV3100 induces AR ubiquitination and proteasomal degradation. LNCaP cells were treated with CLU or control siRNA with 10 mmol/L MDV3100 for24 hours and then � MG-132 for 6 hours. Proteins were extracted for Western blot analysis using AR (left). LNCaP cells were treated with CLU or controlsiRNA � 10 mmol/L MDV3100 for 24 hours and then MG-132 for 6 hours. Proteins were immunoprecipitated using AR and Western blot analysisconducted using AR (middle). LNCaP cells were transfected with His ubiquitin and AR and followed by siRNA CLU or control siRNA treatment � 10 mmol/LMDV3100 for 24 hours and then MG-132 for 6 hours. Proteins were immunoprecipitated using ubiquitin and Western blot analysis conducted using AR,ubiquitin antibodies (right). IP, immunoprecipitation; IgG, immunoglobulin G.
Cotargeting Androgen Receptor and Clusterin for CRPC
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5213
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

cells (Supplementary Fig. S6, right) as well as in LAPC4 and22RV1 cells (Supplementary Fig. S7); these changes are seen atthe protein, but not mRNA levels (Fig. 6A, bottom) andenhanced by MDV3100, suggesting that CLU knockdown incombination with MDV3100 alters AR protein stability. Theeffect of CLU silencing�/þMDV3100 onARbiosynthesis usingcycloheximide was next evaluated. Both MDV3100 and CLUsiRNA induced rapid andprofound decrease of AR 6 hours aftercyclohexamide, whereas this effect was seen after only 2 hourswhenMDV3100 was combined with CLU knockdown (Fig. 6B).These data suggest that CLU silencing and MDV3100, bothdecrease AR protein turnover, but this effect is potentiatedwhen combined.
Protein ubiquitination and acetylation are important deter-minants of AR transcriptional activation (35, 36). AR forms aheterodimer complex with Hsp90 to provide stability forligand-unbound AR; without Hsp90 binding, the AR is ubiqui-tinated and degraded by the proteasome (37, 38). We foundthat MDV3100 increases AR-Hsp90 heterocomplex levels (Fig.6C), consistent with prior reports thatMDV3100 sequesters ARin the cytoplasm (11), with decreased AR acetylation (Fig. 6C)and AR transcriptional activity (Fig. 5A). Interestingly, CLUknockdown profoundly decreases AR/Hsp90 heterocomplexand AR acetylation levels (Fig. 6C). These data are consistentwith a view that the MDV3100-AR-Hsp90 heterocomplexbecomes more vulnerable to degradation under conditions ofCLU silencing. The proteasome inhibitor, MG132, abrogatedAR degradation under conditions of CLU knockdown plusMDV3100 (Fig. 6D, left) with increased ubiquitinated AR levels(Fig. 6D,middle and right). Collectively, these data suggest thatwhen CLU knockdown is combined with MDV3100, the AR isdeacetylated and ubiquitinated with accelerated proteasome-mediated degradation.
CLU knockdown decreases levels of the molecularcochaperone FKBP52
The preceding data indicate thatMDV3100-sequestered, AR-Hsp90 cytoplasmicheterocomplexes,when combinedwithCLUknockdown, are destabilized and lead to AR ubiquitination anddegradation with reduced AR nuclear translocation and activ-ity. One explanation is that CLU inhibition may lower Hsp90levels through its regulation of HSF-1 activity (13); however,MDV3100 did not induce HSF-1 activity (Supplementary Fig.S8), and combination therapy did not lower the levels of theHSF-1–regulated gene, Hsp90 (Fig. 6C, input). These observa-tions, along with data in Fig. 2, implicate YB-1, rather than HSF-1, as the key stress-activated transcription factor mediatingMDV3100 increases of CLU. Because Hsp90 cooperates withother cochaperones to stabilize client proteins, we initially usedan unbiased approach to identify AR-Hsp90 cochaperonesregulated by CLU. Gene profiling analysis from LNCaP andPC-3 cells treatedwith control versus CLU siRNA indicated thatCLU expression correlated with the Hsp90 cochaperoneFKBP52 (Hsp56) and that CLU knockdown reduces FKBP52,but not FKBP51 or Hsp90, protein levels while abrogatingMDV3100-induced phosphorylation of YB-1, pAKT, and p90Rsk(Fig. 7A). To define how FKBP52 levels decrease under condi-tions of MDV3100-induced stress and CLU knockdown, we
mined public databases and found that YB-1 binds to FKBP52DNA with a high stringency of 12 in chromatin immunopre-cipitation (ChIP) on ChIP analysis (39). Western blotting con-firmed that YB-1 knockdown decreases FKBP52 expressionlevels (Fig. 7B). To ascertain the role of FKBP52 in AR stabilityunder conditions of CLU silencing, FKBP52 was overexpressedafter CLU knockdown and AR expression was evaluated. Figure7C shows that FKBP52 partially rescues AR from degradationinduced by CLU knockdown. FKBP52 overexpression alsopartially restores PSA expression, indicating increased ARactivity when FKBP52 levels are restored under conditions ofCLU knockdown. These data suggest that CLU regulates thestability of the AR–cochaperone complex by affecting FKBP52levels. Although FKBP52 rescue can partially reverse CLUknockdown effects on AR stability in the absence of MDV3100,AR levels were not rescued when MDV3100 was present (Fig.7C). These data define anMDV3100-induced feed-forward loopinvolving pYB-1, p90rsk, CLU, and the AR cochaperone FKBP52that collectively support AR stability, nuclear translocation, andactivation that may be cotargeted using OGX-011.
DiscussionMany strategies used to kill cancer cells induce stress and
redundant survival responses that promote survival andemergence of treatment resistance, which is the underlyingbasis for most cancer deaths. This therapeutic resistanceresults from the Darwinian interplay of innate and adaptivesurvival pathways activated by selective pressures of treat-ment. In prostate cancer, castration induces apoptosis andclinical responses in most patients but also triggers pro-gression to CRPC (2, 3). Similarly, newer AR pathwayinhibitors like abiraterone and MDV3100 prolong survivalbut resistance develops in many initial responders (11,12, 40). Disease progression frequently correlates with risingPSA levels, indicating continued AR signaling and highlight-ing need for additional therapies targeting the molecularbasis of treatment-resistant CRPC. Defining interactionsbetween the AR and redundant survival pathways will buildnew combinatorial strategies that control progression andimprove outcomes.
Persistent AR signaling in CRPC can occur via AR amplifi-cation or mutations, as well as altered levels of AR coactivatorsor cochaperones (41–44). Another more dynamic mechanisminvolves reciprocal feedback regulation between AR and phos-phoinositide 3-kinase (PI3K) pathways whereby AR inhibitionactivates AKT signaling by reducing the levels of the AKTphosphatase PHLPP, and PI3K inhibition activates AR signal-ing by relieving feedback inhibition of HER kinases; inhibitionof one activates the other, thereby enhancing survival (33).These insights guide design of combinatorial regimens cotar-geting the AR pathway and AKT or Hsp chaperones Hsp90 andHsp27 (45).
Because AR pathway inhibition is known to induce stressand CLU with reciprocal pathway activation of AKT (34),targeting the stress response activated by AR pathway inhibi-tors is another combinatorial strategy. This study defines feed-forward links between stress-induced YB-1 to CLU activationin parallel with enhanced AKT and mitogen-activated protein
Matsumoto et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5214
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

kinase (MAPK) signaling, which collectively support AR sta-bility and activity during MDV3100 treatment. YB-1 and CLUare both stress-activated survival chaperones functionallyassociated with cancer treatment resistance (14). Under stressconditions, YB-1 is phosphoactivated by AKT (31) and p90RSK(32), stimulating its nuclear translocation and binding to targetpromoters. Our findings identify p90RSK as the predominantpathway increasing CLU expression after MDV3100 treatment,whereas YB-1 knockdown abrogates MDV3100-induced CLU.We previously reported that YB-1 transcriptionally enhancesCLU following treatment stress and CLU functions as a keymediator of paclitaxel resistance in prostate cancer (14).Together, these data suggest that MDV3100-induced CLUoccurs via p90RSK and YB-1 signaling pathway.This stress-activated antiapoptotic function for CLU can lead
to resistance to many anticancer therapies. The CLU antisenseinhibitor, OGX-011, enhances cancer cell death in combinationwith therapeutic stressors in many preclinical cancer models(15). Phase III trials evaluating combined docetaxel plus OGX-011 are underway in men with metastatic CRPC after random-ized phase II studies reported a survival benefit when OGX-011was added to docetaxel (15). Although CLU inhibition canenhance castration and delay time toCRPC in castrate-sensitivexenografts (24, 46), MDV3100 now enables investigation of ARpathway and CLU inhibition in CRPC models. In this study, weshow that CLU is induced by MDV3100 and AR knockdown inMDV-sensitive and -resistantLNCaP cells, respectively, and that
CLU remained highly expressed in most MDV3100-resistantxenografts and cell lines. Mechanistically, MDV3100-inducedcross-talk activationofAKTandMAPKpathwayswas repressedwhen combined with OGX-011, and cotargeting AR and CLUsynergistically enhanced apoptotic rates over monotherapy.Unexpectedly, we found that AR ubiquitination and protea-some-mediated degradation rates were accelerated whenMDV3100 was combined with CLU knockdown. Stress activa-tion of YB-1 and MAPK was blunted with combined therapy,resulting indecreasedYB-1–regulated levels of FKBP52 (Hsp56),which led to AR ubiquitination, proteasomal degradation anddecreased transcriptional activity beyond that observed withMDV3100 monotherapy.
These results highlight a role for CLU in supporting YB-1–mediated expression of other molecular chaperones undercontext-dependent stress conditions, analogous to CLU-enh-anced HSF-1–mediated transactivation of Hsp70 and Hsp27after Hsp90 inhibition (13). HSF-1 and YB-1 orchestrate expres-sion of other molecular chaperones involved in processes offolding, trafficking, and transcriptional activation of the AR. Inthe absence of ligand, AR is predominately cytoplasmic, main-tained in an inactive, but highly responsive state by a largedynamic heterocomplex composed of Hsp90 and Hsp70, andcochaperones like FKBP52. These Hsp-AR cochaperones sup-port AR stability and activation (47, 48). Compared with thefirst-generation AR antagonist bicalutamide, which leads to ARdissociation from its Hsp heterocomplex and does not inhibit
A
YB-1
FKBP52
(h) MDV310048241204824120
YB-1 siRNACtr siRNAB
Vinculin
C
CLUCtr CLUCtr (siRNA)
FKBP52
Vinculin
FKBP51
Akt
p-Akt
YB-1
p-YB-1
Hsp90
p-RSK1
MDV3100
CLU
AR
PSA
CLU
FKBP52
Flag
Vinculin
NT CLUCtr NT CLUCtr (siRNA)
Empty vector FKBP52
NT CLUCtr NT CLUCtr
Empty vector FKBP52
MDV3100Vehicle
Figure 7. CLU knockdown decreases FKBP52 levels. A, CLU knockdown decreases MDV3100-induced AKT and MAPK activation as well as FKBP52 levels.LNCaP cells were transfected with CLU or control siRNA and treated with 10 mmol/L MDV3100 for indicated times. Proteins were extracted forWestern blot analysis using indicated antibodies with actin used as loading control. B, YB-1 knockdown decreases FKBP52 expression. LNCaP cells weretransfected with YB-1 or control siRNA and treated with 10 mmol/L MDV3100 for indicated times. Proteins were extracted for Western blot analysisusing indicated antibodies. C, FKBP52 partially rescues AR degradation after CLU knockdown. LNCaP cells were transfected with FKBP52 Flag-taggedconstruct or empty vector followed by CLU knockdown. Forty-eight hours posttransfection, cells were treated with 10 mmol/L MDV3100 or vehicle for24 hours. Proteins were extracted for Western blot analysis using indicated antibodies.
Cotargeting Androgen Receptor and Clusterin for CRPC
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5215
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

AR nuclear transport, MDV3100-bound AR remains cyto-plasmic and complexed with Hsp90, which may increase ARsusceptibility to degradation under conditions of stress andCLU suppression. MDV3100-induced changes in AR acetyla-tion and/or conformation may render it dependent to othercofactors in addition to FKBP52.
In summary, we define a stress-induced feed-forward loopinvolving MDV3100-induced AKT and p90rsk, phosphoactiva-tion of YB-1, and increased expression of CLU. CLU knockdownabrogates MDV3100-induced activation of both AKT andp90rsk, defining another mechanism by which CLU inhibitionpotentiates anti-AR therapy.Hence, cotargeting adaptive stresspathways activated by AR pathway inhibitors, and mediatedthrough CLU, potentiate anti-AR activity by decreasing ARexpression levels and activity, as well as activation of cross-talksignaling pathways induced by MDV3100. These results pro-vide mechanistic and preclinical proof-of-principle to supportcombinatorial clinical studies with MDV3100 and OGX-011.
Disclosure of Potential Conflicts of InterestM.E. Gleave is employed as an advisor, has commercial research grant and
ownership interest (including patents) inOncoGenex, and is a consultant/advisory
board member of Teva and Astellas. No potential conflicts of interest weredisclosed by the other authors.
Authors' ContributionsConception and design: H. Matsumoto, H. Kuruma, A. Zoubeidi, M.E. GleaveDevelopment of methodology: H. Matsumoto, H. Kuruma, M.E. GleaveAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): H. Matsumoto, Y. Yamamoto, M. Shiota, H. Kuruma,M.E. GleaveAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis):H. Matsumoto, Y. Yamamoto, M. Shiota, M.E. GleaveWriting, review, and/or revision of the manuscript: H. Matsumoto,H. Matsuyama, A. Zoubeidi, M.E. GleaveAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): Y. Yamamoto, H. Kuruma, E. BeraldiStudy supervision: H. Kuruma, E. Beraldi, M.E. Gleave
AcknowledgmentsThe authors thankMary Bowden, Virginia Yago, and Igor Moskalev for in vivo
studies and Dr. Susan Ettinger for expression profiling assistance.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received February 3, 2013; revised May 10, 2013; accepted May 30, 2013;published OnlineFirst June 20, 2013.
References1. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the
impact of eliminating socioeconomic and racial disparities on prema-ture cancer deaths. CA Cancer J Clin 2011;61:212–36.
2. GleaveME, Goldenberg SL, Chin JL, Warner J, Saad F, Klotz LH, et al.Randomized comparative study of 3 versus 8-month neoadjuvanthormonal therapy before radical prostatectomy: biochemical andpathological effects. J Urol 2001;166:500–6.
3. Gleave M, Goldenberg SL, Bruchovsky N, Rennie P. Intermittentandrogen suppression for prostate cancer: rationale and clinical expe-rience. Prostate Cancer Prostatic Dis 1998;1:289–96.
4. Bruchovsky N, Sadar M, Akakura K, Goldenberg SL, Matsuoka K,Rennie PS. Characterization of 5 a-reducatase gene expression instroma and epithelium of human prostate. J Steroid BiochemMol Biol1996;59:397–404.
5. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al.Molecular determinants of resistance to antiandrogen therapy. NatMed 2004;10:33–9.
6. MiyakeH,NelsonC, Rennie PS,GleaveME.Overexpression of insulin-like growth factor binding protein-5 helps accelerate progression toandrogen-independence in the human prostate LNCaP tumor modelthrough activation of phosphatidylinositol 30-kinase pathway. Endo-crinology 2000;141:2257–65.
7. Miyake H, Tolcher A, Gleave ME. Antisense Bcl-2 oligodeoxynu-cleotides inhibit progression to androgen-independence aftercastration in the Shionogi tumor model. Cancer Res 1999;59:4030–4.
8. Rocchi P, So A, Kojima S, Signaevsky M, Beraldi E, Fazli L, et al. Heatshock protein 27 increases after androgen ablation and plays acytoprotective role in hormone-refractory prostate cancer. CancerRes 2004;64:6595–602.
9. Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones JA, TaplinME, et al. Docetaxel and estramustine compared with mitoxantroneand prednisone for advanced refractory prostate cancer. N Engl JMed2004;351:1513–20.
10. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al.Abiraterone and increased survival in metastatic prostate cancer.N Engl J Med 2011;364:1995–2005.
11. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Devel-opment of a second-generation antiandrogen for treatment ofadvanced prostate cancer. Science 2009;324:787–90.
12. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al.Antitumour activity of MDV3100 in castration-resistant prostate can-cer: a phase 1-2 study. Lancet 2010;375:1437–46.
13. Lamoureux F, Thomas C, Yin MJ, Kuruma H, Beraldi E, Fazli L, et al.Clusterin inhibition using OGX-011 synergistically enhances Hsp90inhibitor activity by suppressing the heat shock response in castrateresistant prostate cancer. Cancer Res 2011;71:5838–49.
14. Shiota M, Zoubeidi A, Kumano M, Beraldi E, Naito S, Nelson CC, et al.Clusterin is a critical downstream mediator of stress-induced YB-1transactivation in prostate cancer. Mol Cancer Res 2011;9:1755–6.
15. Zoubeidi A, GleaveM. Small heat shock proteins in cancer therapy andprognosis. Int J Biochem Cell Biol 2012;44:1646–56.
16. Poon S, Rybchyn MS, Easterbrook-Smith SB, Carver JA, PankhurstGJ, Wilson MR. Mildly acidic pH activates the extracellular molecularchaperone clusterin. J Biol Chem 2002;277:39532–40.
17. Trougakos IP, Lourda M, Antonelou MH, Kletsas D, Gorgoulis VG,Papassideri IS, et al. Intracellular clusterin inhibits mitochondrial apo-ptosis by suppressing p53-activating stress signals and stabilizing thecytosolic Ku70-Bax protein complex. Clin Cancer Res 2009;15:48–59.
18. Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R,Wang CY. Clusterininhibits apoptosis by interacting with activated Bax. Nat Cell Biol2005;7:909–15.
19. Ammar H, Closset JL. Clusterin activates survival through thephosphatidylinositol 3-kinase/Akt pathway. J Biol Chem 2008;283:12851–61.
20. Zoubeidi A, Ettinger S, Beraldi E, Hadaschik B, Zardan A, Klomp LW,et al. Clusterin facilitates COMMD1 and I-kappaB degradation toenhance NF-kappaB activity in prostate cancer cells. Mol Cancer Res2010;8:119–30.
21. Kruger S, Ola V, Fischer D, Feller AC, Friedrich M. Prognostic signif-icance of clusterin immunoreactivity in breast cancer. Neoplasma2007;54:46–50.
22. Zhang S, Zhang D, Zhu Y, Guo H, Zhao X, Sun B. Clusterin expressionand univariate analysis of overall survival in human breast cancer.Technol Cancer Res Treat 2006;5:573–8.
23. July LV, Akbari M, Zellweger T, Jones EC, Goldenberg SL, Gleave ME.Clusterin expression is significantly enhanced in prostate cancer cellsfollowing androgen withdrawal therapy. Prostate 2002;50:179–88.
24. Miyake H, Nelson C, Rennie PS, Gleave ME. Testosterone-repressedprostate message-2 is an antiapoptotic gene involved in progression
Matsumoto et al.
Cancer Res; 73(16) August 15, 2013 Cancer Research5216
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

to androgen independence in prostate cancer. Cancer Res 2000;60:170–6.
25. Miyake H, Hara I, GleaveME. Antisense oligodeoxynucleotide therapytargeting clusterin gene for prostate cancer: vancouver experiencefrom discovery to clinic. Int J Urol 2005;12:785–94.
26. Gleave M, Miyake H. Use of antisense oligonucleotides targeting thecytoprotective gene, clusterin, to enhance androgen- and chemo-sensitivity in prostate cancer. World J Urol 2005;23:38–46.
27. Zoubeidi A, Chi K, Gleave M. Targeting the cytoprotective chaperone,clusterin, for treatment of advanced cancer. Clin Cancer Res 2010;16:1088–93.
28. Chi KN, Hotte SJ, Yu EY, Tu D, Eigl BJ, Tannock I, et al. Randomizedphase II study of docetaxel and prednisonewith or without OGX-011 inpatients with metastatic castration-resistant prostate cancer. J ClinOncol 2010;28:4247–54.
29. Kuruma H, Matsumoto H, Shiota M, Bishop JL, Lamoureux F, ThomasC, et al. A novel anti-androgen, Compound 30, suppresses castration-resistant andMDV3100-resistant prostate cancer growth in vitro and invivo. Mol Cancer Ther 2013;12:567–76
30. Zoubeidi A, Zardan A, Beraldi E, Fazli L, Sowery R, Rennie P, et al.Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res2007;67:10455–65.
31. Evdokimova V, Ruzanov P, Anglesio MS, Sorokin AV, Ovchinnikov LP,Buckley J, et al. Akt-mediated YB-1 phosphorylation activates trans-lation of silent mRNA species. Mol Cell Biol 2006;26:277–92.
32. Stratford AL, Fry CJ, Desilets C, Davies AH, Cho YY, Li Y, et al. Y-boxbinding protein-1 serine 102 is a downstream target of p90 ribosomal S6kinase in basal-likebreast cancer cells. BreastCancerRes 2008;10:R99.
33. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chan-darlapaty S, et al. Reciprocal feedback regulation of PI3K and andro-gen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell2011;19:575–86.
34. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships:the combined effects of multiple drugs or enzyme inhibitors. AdvEnzyme Regul 1984;22:27–55.
35. Nawaz Z, O'Malley BW. Urban renewal in the nucleus: is proteinturnover by proteasomes absolutely required for nuclear receptor-regulated transcription? Mol Endocrinol 2004;18:493–9.
36. Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, et al.Cyclic, proteasome-mediated turnover of unliganded and ligandedERalpha on responsive promoters is an integral feature of estrogensignaling. Mol Cell 2003;11:695–707.
37. Solit DB, Scher HI, Rosen N. Hsp90 as a therapeutic target in prostatecancer. Semin Oncol 2003;30:709–16.
38. VanajaDK,Mitchell SH, Toft DO, YoungCY. Effect of geldanamycin onandrogen receptor function and stability. Cell Stress Chaperones2002;7:55–64.
39. Finkbeiner MR, Astanehe A, To K, Fotovati A, Davies AH, Zhao Y, et al.Profiling YB-1 target genes uncovers a new mechanism for METreceptor regulation in normal and malignant human mammary cells.Oncogene 2009;28:1421–31.
40. Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgendeprivation therapy: progress in understanding mechanisms of resis-tance and optimizing androgen depletion. Nat Clin Pract Urol 2009;6:76–85.
41. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicingof a novel androgen receptor exon generates a constitutively activeandrogen receptor that mediates prostate cancer therapy resistance.Cancer Res 2008;68:5469–77.
42. GuoZ,YangX,Sun F, JiangR, LinnDE,ChenH, et al. A novel androgenreceptor splice variant is up-regulated during prostate cancer pro-gression and promotes androgen depletion-resistant growth. CancerRes 2009;69:2305–13.
43. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA,et al. Castration resistance in human prostate cancer is conferred by afrequently occurring androgen receptor splice variant. J Clin Invest2010;120:2715–30.
44. Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al.Expression of androgen receptor splice variants in prostate cancerbone metastases is associated with castration-resistance and shortsurvival. PLoS ONE 2011;6:e19059.
45. Loriot Y, Zoubeidi A, Gleave ME. Targeted therapies in metastaticcastration-resistant prostate cancer: beyond the androgen receptor.Urol Clin North Am 2012;39:517–31.
46. Miyake H, Nelson C, Rennie PS, Gleave ME. Acquisition of chemore-sistant phenotype by overexpression of the antiapoptotic gene tes-tosterone-repressed prostatemessage-2 in prostate cancer xenograftmodels. Cancer Res 2000;60:2547–54.
47. Cheung-Flynn J, Prapapanich V, Cox MB, Riggs DL, Suarez-Quian C,Smith DF. Physiological role for the cochaperone FKBP52 in androgenreceptor signaling. Mol Endocrinol 2005;19:1654–66.
48. Yang Z,Wolf IM, ChenH, Periyasamy S, Chen Z, YongW, et al. FK506-binding protein 52 is essential to uterine reproductive physiologycontrolled by the progesterone receptor A isoform. Mol Endocrinol2006;20:2682–94.
Cotargeting Androgen Receptor and Clusterin for CRPC
www.aacrjournals.org Cancer Res; 73(16) August 15, 2013 5217
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359

2013;73:5206-5217. Published OnlineFirst June 20, 2013.Cancer Res Hiroaki Matsumoto, Yoshiaki Yamamoto, Masaki Shiota, et al. Adaptive Stress Response and AR StabilityCastrate-Resistant Prostate Cancer Progression by Inhibiting Cotargeting Androgen Receptor and Clusterin Delays
Updated version
10.1158/0008-5472.CAN-13-0359doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/06/18/0008-5472.CAN-13-0359.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/73/16/5206.full#ref-list-1
This article cites 48 articles, 18 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/73/16/5206.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/16/5206To request permission to re-use all or part of this article, use this link
on June 13, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 20, 2013; DOI: 10.1158/0008-5472.CAN-13-0359





























