Embed Size (px)
Citation preview

©2
01
8 D
r. W
alter
F.
de
Aze
ve
do
Jr.
1
000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000111111111110001100000000000000000000001111111111111111111000000001000000000111111111111111111111111000000000000000111111111111111111111111000000000000000011111111111111111111100000000000000001111111111111111111111111000000000011111111111111111111111111111000000001111111111111111111111111111110000000111111111111111111111111111110000000000111111111111111111111111111110000000000000011111111111111111111111111111110000001111111111111111111111111111111111000011111111111111111111111111111111111000001111111111111111111111111111111111100000000011111111111111111111111111111110000000001111111111111111111111111111110000000000001111111111111111111111111110000000000000011111111111111111111111110000000000000000111111111111111111111000000000000000000000000000001111000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000

1. Cristalização. Para
cristalizar uma
macromolécula temos que
levá-la a um estado de
supersaturação, que
favorece a formação de
cristais, como os mostrados
acima. Os cristais de
moléculas biológicas
normalmente apresentam
dimensões inferiores a 1 mm
de comprimento em cada
aresta.
2. Coleta de dados de difração de raios X. Os cristais
apresentam um arranjo ordenado de moléculas, como
uma pilha de tijolos ordenados. Na analogia, cada tijolo
representa uma molécula. As distâncias entre os átomos
são da ordem de 1 Å (0,1 nm ou 10-10 m), usando-se
raios X (com comprimento de onda da odem de Å )
teremos difração.
3. Interpretação do padrão de
difração de raios X. A figura
abaixo é o registro da difração de
raios X de um cristal. Os raios X
interagem com o cristal, o que
produz um padrão de difração. A
análise desta informação
possibilita a resolução de estrutura
3D.
4. Resolução da estrutura.
A partir da análise do
padrão de difração, é
possível gerar mapas de
densidade eletrônica (à
direita). A interpretação de
tais mapas gera a estrutura
3D de molécula.
5. Análise. A partir da estrutura
resolvida, procedemos a análise,
onde relaciona-se a estrutura 3D à
sua função biológica.
Etapas para resolução da
estrutura 3D de
macromoléculas biológicas
por cristalografia
2
Cristalografia

Para cristalizarmos uma macromolécula
biológica é necessário trazê-la a um
estado de supersaturação.
Consideremos uma proteína dissolvida
em um tampão. Para que a proteína seja
levada a formar cristais, é necessário que
as moléculas da proteína sejam trazidas a
uma situação onde as moléculas fiquem
relativamente próximas umas das outras.
Para levar a proteína a à supersaturação,
podemos aumentar sua concentração
(eixo vertical), ou aumentar a
concentração do sal presente na solução
da proteína. O diagrama ao lado ilustra as
diferentes regiões de solubilidade da
proteína.
3
Supersaturação

O processo de cristalização da proteína
normalmente deve ser lento, ou seja,
considerando-se a proteína inicialmente
numa região abaixo da curva de
solubilidade, devemos aumentar a
concentração salina, ou da proteína, de
modo a trazê-la na região de
supersaturação, de forma a propiciar um
arranjo ordenado das moléculas. De uma
forma geral, espera-se que o processo de
cristalização demore horas, dias ou até
meses. A partir o gráfico ao lado, vemos
que na região de supersaturação teremos
as moléculas da proteína próximas umas
das outras, o que, em casos favoráveis,
promoverá o aparecimento dos primeiros
núcleos cristalinos. Esses microcristais
servirão de base para o crescimento de
cristais maiores, adequados para
experimentos de difração de raios X. 4
Supersaturação

Para cristalizarmos proteínas,
normalmente usamos o método de
difusão de vapor. Uma gota de proteína
é colocada sobre uma lamínula. Na gota
adicionamos uma solução contendo sal,
ou outro agente precipitante, como
polietileno glicol (PEG). Colocamos a
lamínula sobre um poço, onde temos a
solução do precipitante. Ao fecharmos o
sistema, ocorrerá difusão de moléculas de
água da gota para o poço, levando a
proteína, em casos favoráveis, a um
estado de supersaturação, que pode levar
à formação dos primeiros núcleos
cristalinos.
5
Supersaturação

Fenômeno de salting-in. a)
Macromolécula biológica sem a
presença de íons dissolvidos na
solução. A atração eletrostática entre os
grupos carregados em duas os mais
macromoléculas causa a aglomeração e
precipitação. b) Íons blindam a interação
eletrostática entre as macromoléculas,
aumentando a solubilidade.
Diversos fatores interferem com a solubilidade da proteína, entre eles a concentração
salina. O aumento da solubilidade de uma macromolécula a baixa concentração salina
(<0,5M) é chamada salting-in. Segundo a teoria de Debye-Hückel para soluções
iônicas, um aumento na força iônica reduz a atividade dos íons em solução e aumenta
a solubilidade do composto iônico. Uma forma alternativa de se tratar o fenômeno é
considerar o salting-in como o resultado da competição entre grupos carregados na
superfície da macromolécula e os íons em solução. Na ausência de íons no solvente,
a macromolécula precipita devido à atração de eletrostática entre cargas opostas em
diferentes partes da macromolécula. Se os íons são adicionados à solução, esses
blindam os grupos carregados na macromolécula e aumentam a sua solubilidade.
+
- +
-+
- +
-
+
+
-
-
a) b)
6
Supersaturação

Sequência de eventos para a montagem de uma gota de cristalização: a)
Coloca-se 1-2l da solução da macromolécula biológica sobre a lamínula de vidro.
b) Adiciona-se 1-2l da solução do reservatório à gota com a solução da
macromolécula biológica. c) Ao final temos uma gota (2+2) com a solução de
macromolécula biológica mais a solução do reservatório.
Solução do poço
7
Método da Gota Suspensa (Hanging-drop)

Com o aumento do número de macromoléculas biológicas cristalizadas com
sucesso, tornou-se óbvio que muitas das condições de cristalização se
assemelhavam, ou seja, havia uma concentração de resultados positivos de
cristalização de macromoléculas biológicas usando-se número limitado de
precipitantes, tampões e aditivos. A partir da análise dos resultados positivos de
cristalização, foi possível a proposição de diversos métodos para obtenção de
cristais (Carter & Carter, 1979), onde um número limitado de reagentes eram
testados, usando-se pequenas quantidades da macromolécula biológica, geralmente
por volta de poucos miligramas.
8
0,5mm 0,5mm 0,5mm
Jancarik, J. & Kim, S. -H. (1991) J. Appl. Crystallogr. 24, 409-411.
Matriz Esparsa

A partir da observação dos resultados preliminares dos experimentos cristalização,
era possível determinar que tampão, aditivo e agente precipitante seriam os mais
favoráveis e a partir daí proceder-se a sucessivos melhoramentos até se conseguir
cristais adequados, ou ainda, em casos favoráveis, obter-se cristais adequados já na
primeira tentativa com as condições padrões. Em 1991, A Dra. Jaru Jancarick da
University of California, Berkeley propôs o método da matriz esparsa ( Jancarick &
Kim, 1991), onde diversas condições diferentes são tentadas para se cristalizar a
macromolécula biológica.
Jancarik, J. & Kim, S. -H. (1991) J. Appl. Crystallogr. 24, 409-411.9
0,5mm0,5mm0,5mm
Matriz Esparsa

Parâmetros da matriz de cristalização (Jancarik & Kim, 1991)
Agentes precipitantes
Não-Voláteis Sais Voláteis Mistura
MPD Tartarato de Na,K 2-Propanol Sulfato de NH4 + PEG
PEG 400 Fosfato de NH4 2-Propanol + PEG
PEG 4000 Sulfato de NH4
PEG 8000 Acetato de Na
Sulfato de Li
Formiato de Na
Fosfato de Na,K
Citrato de Na
Formiato de Mg
Faixa de pH: 4,6; 5,6; 6,5; 7,5 e 8,5
Aditivos: Cloreto de Ca, Citrato de Na, Cloreto de Mg, Acetato de NH4, Sulfato de
NH4, Acetato de Mg, Acetato de Zn e Acetato de Ca.
10
Matriz Esparsa

Por tentativa e erro a matriz
multimensional foi simplificada
eliminando-se as condições que podem
ser parcialmente representadas por
resultados de outras condições, a
proposta original apresenta 58 condições.
Comercialmente a empresa Hampton
Research, (USA) simplificou o método
original, e disponibiliza um kit com 50
condições de cristalização.
Comercialmente há outros kits usando-se
como princípio a variação de pH, força
iônica e agentes precipitantes.
Imagem disponível em: <
http://www.hamptonresearch.com/products/ProductDetails.aspx?ci
d=1&sid=17&pid=1 >.
Acesso em: 8 de Abril de 2015.
11
Matriz Esparsa

Um dos sistema usados para cristalização de proteínas é a placa linbro, mostrada
acima. A placa apresenta 24 poços, que permite testarmos diversas condições de
cristalização. As lamínulas são colocadas sobre cada um dos poços, e vedadas com
graxa de vácuo.
Fonte: http://www.hamptonresearch.com
12
Matriz Esparsa
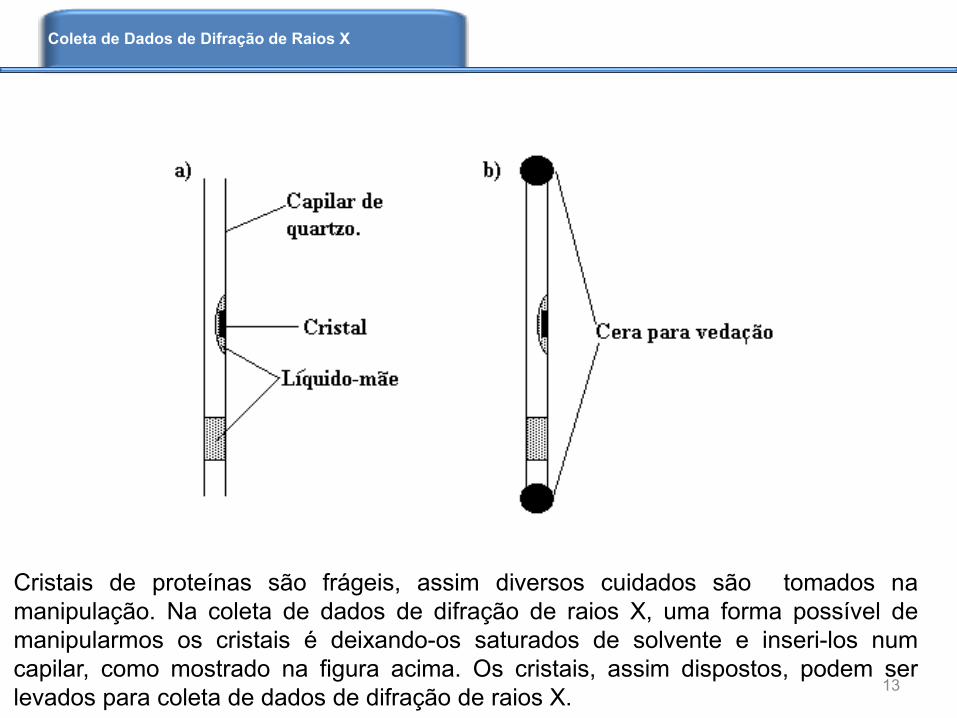
Cristais de proteínas são frágeis, assim diversos cuidados são tomados na
manipulação. Na coleta de dados de difração de raios X, uma forma possível de
manipularmos os cristais é deixando-os saturados de solvente e inseri-los num
capilar, como mostrado na figura acima. Os cristais, assim dispostos, podem ser
levados para coleta de dados de difração de raios X.13
Coleta de Dados de Difração de Raios X

Uma forma alternativa de coletarmos dados é transferir o cristal para uma solução
com protetor criogênico (Polietileno glicol, glicerol entre outros). O cristal é então
exposto a um fluxo de nitrogênio líquido e transferido para uma base metálica
(cabeça goniométrica). O cristal fica pronto para a coleta de dados. As temperaturas
criogênicas minimizam os dados causados pela radiação. 14
Coleta de Dados de Difração de Raios X

Fonte: http://www.hamptonresearch.com
Na figura da esquerda vemos uma base de cobre usada como suporte para o cristal.
À direita temos o cristal inserido num laço. A exposição ao nitrogênio líquido leva à
formação de um filme rígido e transparente que mantém o cristal no laço.15
Coleta de Dados de Difração de Raios X

Experimentos de cristalização no
espaço normalmente geram cristais
de melhor qualidade para estudos
de difração de raios X. As
condições de microgravidade do
espaço, propiciam um
empacotamento cristalino mais
ordenado, gerando cristais que
difratam à mais alta resolução. A
proteína uropesina (Canduri et al.,
2001) foi cristalizada em condições
de microgravidade na missão STS-
95 do ônibus espacial Discovery (
Disponível em: <
http://www.youtube.com/watch?v=N
9IFiQNY8mE >).
Fonte: http://www.aviationspectator.com/more-aviation-photos?page=405. Crédito: NASA
Cristal de uropepsina .16
Cristalização de Proteínas no Espaço

Uma etapa inicial importante na elucidação da estrutura tridimensional de proteínas, por
meio de cristalografia por difração de raios X, é determinação do conteúdo da cela
unitária. Uma vez tenhamos determinado os parâmetros da cela unitária: a, b, c, alfa,
beta e gama, temos que determinar quantas moléculas temos na unidade assimétrica.
Para isto precisamos, além dos parâmetros da cela unitária, o massa molecular da
proteína cristalizada. O método proposto por Matthews (1968) permite calcular o
número de moléculas por unidade assimétrica. Matthews determinou que para cristais
de proteínas a relação entre volume da cela unitária (Vcell) e a massa molecular da
proteína cristalizada fica na faixa de 1,7 a 3,5 Å3/Da, com a maioria dos valores em
torno de 2,15 Å3/Da. Tal volume é chamado de volume de Matthews (VM), e definido
pela equação abaixo:
onde Z é o número de moléculas na cela unitária, MW a massa molecular da proteína
cristalizada e Vcell o volume cela unitária, que para uma cela unitária qualquer é dada
por:
Vcell = a.b.c [1 + 2 cos cos cos - cos2 - cos2 - cos2]1/2
17
Caracterização de Cristais de Proteína
MWZ
VV cellM
.

A fração de volume ocupada por proteína (Vprotein) é dada por:
A fração de volume de solvente no cristal é dada por:
O Vprotein pode também ser determinado por:
18
Caracterização de Cristais de Proteína
M
proteinV
V23,1
M
solventV
V23,1
1
M
proteinV
V)/cmProteína(g da Específico Volume 3

Principais seções de um artigo de cristalização de proteínas:
1) Clonagem, expressão e purificação da proteína
2) Cristalização
3) Coleta de dados de difração de raios X
4) Outros (teste de atividade, sequenciamento, solução da estrutura e
refinamento cristalográfico parcial.
19
Artigos Científicos Sobre Cristalização de Proteínas

20
Artigos Científicos Sobre Cristalização de Proteínas

The purifed MtCS was concentrated and
dialyzed against 50 mM Tris±HCl buffer pH 7.8
(Hampton Research, USA). The final protein
concentration was about 10 mg.ml-1.
Crystallization was performed by the hanging-
drop vapour-diffusion and sparse-matrix
methods (Jancarik & Kim, 1991) using tissue-
culture multiwell plates with covers (Linbro, ICN
Biomedicals, Inc, USA) at a temperature of 293
K. Each hanging drop was prepared by mixing 1
ml each of protein solution and reservoir
solution and was placed over 700 l reservoir
solution. Initial conditions were screened using
Crystal Screen I and II kits (Hampton Research,
USA).Hexagonal crystals of MtCS. Approximate
dimensions
are 0.30 0.25 0.25 mm.
21
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Dias MV, Ely F, Canduri F, Pereira JH, Frazzon J, Basso LA, Palma MS, de Azevedo WF Jr, Santos DS. Crystallization and
preliminary X-ray crystallographic analysis of chorismate synthase from Mycobacterium tuberculosis. Acta Crystallogr D Biol
Crystallogr. 2004; 60(Pt 11):2003-5.

A data set was collected at a wavelength of 1.427 Å
using a synchrotron-radiation source (Station PCr,
LNLS, Campinas- Brazil). The data set was collected
from a single MtCS crystal using a MAR CCD image-
plate system. The crystal was looped out from the
drop and flash-cooled. The PEG 400 present in the
crystallization conditions served as a cryoprotectant,
X-ray diffraction data were collected at a temperature
of 100 K under a cold nitrogen stream generated and
maintained with an Oxford Cryosystem. The crystal
was rotated through a total of 160o, with a 1o
oscillation range per frame, a crystal-to-detector
distance of 130 mm and an exposure time of 60 s.
Data were processed on a Silicon Graphics Octane2
computer using the programs MOSFLM (Leslie,
1990) and SCALA (CCP4, 1994).
A typical diffraction pattern of the MtCS
crystal with 1 oscillation range. The crystal
diffracts to 2.8 Å resolution.
22
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Dias MV, Ely F, Canduri F, Pereira JH, Frazzon J, Basso LA, Palma MS, de Azevedo WF Jr, Santos DS. Crystallization and
preliminary X-ray crystallographic analysis of chorismate synthase from Mycobacterium tuberculosis. Acta Crystallogr D Biol
Crystallogr. 2004; 60(Pt 11):2003-5.

Summary of data-collection statistics.
X-ray wavelength (A ° ) 1.427
Space group P6422 or P6222
Unit-cell parameters (Å ) a = 129.74, b = 129.74,
c = 156.77
Highest resolution shell (Å) 2.94–2.8
Asymmetric unit content 2 molecules
Total reflections measured 92610
Number of independent reflection 19341
Completeness (%) 97.9 (97.9)
Rmerge (%) 5.6 (16.5)
Values in parentheses are for the highest resolution shell (2.94–2.8 Å).
23
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Dias MV, Ely F, Canduri F, Pereira JH, Frazzon J, Basso LA, Palma MS, de Azevedo WF Jr, Santos DS. Crystallization and
preliminary X-ray crystallographic analysis of chorismate synthase from Mycobacterium tuberculosis. Acta Crystallogr D Biol
Crystallogr. 2004; 60(Pt 11):2003-5.

Aplicações
1) Determinação do conteúdo da unidade assimétrica da corismato sintase de
Mycobacterium tuberculosis (Dias et al., 2004). A partir dos dados de difração de
raios X foi determinado os parâmetros da cela unitária.
a = 129,74 Å
b = 129,74 Å
c = 156,77 Å
Grupo espacial: P6422 ou P6222
MW = 41,800 kDa
Para cela hexagonal: Vcell = a.b.c.sen ()
Vcell = 129,74. 129,74 . 156,77. sen(120o) = 2.285.290,31 Å3
24
Artigos Científicos Sobre Cristalização de Proteínas

Para determinarmos o conteúdo da unidade assimétrica, calcularemos o volume de
Matthews para diferentes possibilidades de unidade assimétrica e verificaremos qual
valor fica mais próximo da faixa de 1,7 a 3,5 Å3/Da, como segue:
Z VM(Å3/Da)
1 54,67
6 9,11
12 4,56
18 3,04
24 2,28
30 1,82
25
Artigos Científicos Sobre Cristalização de Proteínas
MWZ
VV cellM
.

Os valores dentro da caixa estão na faixa prevista por Matthews, contudo sabemos
que o grupo espacial é hexagonal primitivo, o que significa que temos 6 unidades
assimétricas, assim esperamos um número de moléculas múltiplo de 6, ou seja, 12,
18, 24, 30.., sendo o valor mais provável 24. Assim temos:
Vprotein = 1,23/VM = 0,5395 e Vsolvent = 0,4605
Z VM(Å3/Da)
1 54,67
6 9,11
12 4,56
18 3,04
24 2,28
30 1,82
26
Artigos Científicos Sobre Cristalização de Proteínas

27
Artigos Científicos Sobre Cristalização de Proteínas

C. roseum seeds were ground to a fine powder in a coffee mill. The powder was
stirred with 0.15 M NaCl [1:10(w:v)] at room temperature for 4 h and then centrifuged
at 10 000g for 20 min at 278 K. The resultant supernatant was applied onto a
Sepharose-4B-mannose column (0.5 10 cm) equilibrated with 0.15 M NaCl
containing 5 mMCaCl2 and 5 mMMnCl2. After removing unbound material, the lectin
was eluted with 0.1 M glycine, 0.15 M NaCl pH 2.6. Purified CRL was monitored by
SDS–PAGE as described by Laemmli (1970) and was used to perform further
characterization. N-terminal sequence analysis was performed using an Applied
Biosystems pulsed-liquid phase 477A protein sequencer with a 120A PTH aminoacid
analyzer, following the method described by the manufacturer.
28
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006; 1(62): 235-7.

29
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006; 1(62): 235-7.

The lyophilized purified CRL was
dissolved to a concentration of 12 mg ml-1
in 20 mM Tris–HCl pH 8.0 containing 0.5
mM CaCl2 and MnCl2 and used for
crystallization trials. Crystallization
screening by the hanging-drop vapour-
diffusion method was performed in Linbro
plates at 293 K using Hampton Research
Crystal Screens I and II, SaltRx, Index and
PEG/Ion Screens (Hampton Research,
Aliso Viejo, CA, USA). The drops were
composed of equal volumes (2 ml) of
protein solution and reservoir solution and
were equilibrated against 500 ml reservoir
solution. An example of a crystal of CRL is
shown in Fig. 1(a).
30
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006; 1(62): 235-7.

A crystal was transferred to a
cryoprotectant solution consisting of 30%
glycerol in the crystallization reservoir
solution. Data were collected at 1.42 A °
wavelength at a synchrotron-radiation
source (beamline MX1, CPr station,
Laboratório Nacional de LuzSíncrotron–
LNLS, Campinas, Brazil) using a MAR
Research CCD imaging plate at a crystal-
to-detector distance of 70 mm. A set of
100 1 oscillation images was recorded
(an image is shown in Fig. 1b). Diffraction
data were indexed, integrated and scaled
using MOSFLM and SCALA (Collaborative
Computational Project, Number 4, 1994).
31
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006; 1(62): 235-7.

Summary of data-collection statistics for CRL.
X-ray wavelength (A ° ) 1.427
Space group P212121
Unit-cell parameters (A° ) a = 67.82, b = 103.14, c = 122.09
Resolution limits (A ° ) 34.92–1.77
Asymmetric unit content 4 molecules
Total reflections measured 286361
Unique reflections measured 80568
Completeness (%) 97.00 (97.0)
Rmerge (%) 5.4 (32.5)
Values in parentheses are for the highest resolution shell (1.87–1.77 A ° ).
32
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB,Rustiguel JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006; 1(62): 235-7.

Aplicações
1) Determinação do conteúdo da unidade assimétrica da Lectina de Cymbosema
roseum seeds (Cavada et al., 2006). A partir dos dados de difração de raios X
foram determinados os parâmetros da cela unitária.
a = 67,82 Å
b = 103,14 Å
c = 122,09 Å
Grupo espacial: P212121
MW = 25kDa
Vcell = 67,82 . 103,14 . 122,09 = 854.014,03 Å3
33
Artigos Científicos Sobre Cristalização de Proteínas

Para determinarmos o conteúdo da unidade assimétrica, calcularemos o volume de
Matthews para diferentes possibilidades de unidade assimétrica e verificaremos qual
valor fica mais próximo da faixa de 1,7 a 3,5 Å3/Da, como segue:
Z VM(Å3/Da)
1 34,14
2 17,07
3 11,38
4 8,53
5 6,83
6 5,69
7 4,88
8 4,27
Z VM(Å3/Da)
9 3,79
10 3,41
11 3,10
12 2,85
13 2,63
14 2,44
15 2,28
16 2,14
Z VM(Å3/Da)
17 2,01
18 1,90
19 1,80
20 1,71
21 1,63
22 1,55
23 1,49
24 1,42
34
Artigos Científicos Sobre Cristalização de Proteínas
MWZ
VV cellM
.

Os valores dentro das caixas estão na faixa prevista por Matthews, contudo sabemos
que o grupo espacial é ortorrômbico primitivo, o que significa que temos 4 unidades
assimétricas, assim teremos um número de moléculas múltiplo de 4, ou seja, 12, 16 ou
20, sendo o valor mais provável 16. Assim temos:
Vprotein = 1,23/VM = 0,575 e Vsolvent = 0,425
Z VM(Å3/Da)
1 34,14
2 17,07
3 11,38
4 8,53
5 6,83
6 5,69
7 4,88
8 4,27
Z VM(Å3/Da)
9 3,79
10 3,41
11 3,10
12 2,85
13 2,63
14 2,44
15 2,28
16 2,14
Z VM(Å3/Da)
17 2,01
18 1,90
19 1,80
20 1,71
21 1,63
22 1,55
23 1,49
24 1,42
35
Artigos Científicos Sobre Cristalização de Proteínas

36
Artigos Científicos Sobre Cristalização de Proteínas

The pheA gene (Rv3838c) encoding prephenate dehydratase from M. tuberculosis was
amplified by the polymerase chain reaction (PCR) from genomic DNA. The forward (5’-
TGCATATGGTGCGTATCGCTTACCTCGGTCC- 3’) and reverse (5’-
ACAAGCTTTCATGCTTGCGCCCCCTGGTCG- 3’) synthetic oligonucleotide primers
were based on the amino-terminal coding and carboxyterminal non-coding strands of
the pheA gene (Cole et al., 1998) containing 50 NdeI and 30 HindIII restriction sites,
respectively. The PCR product was cloned into pET-23a(+) expression vector
(Novagen) and the recombinant plasmid was sequenced to confirm the identity of the
cloned DNA fragment and to ensure that no mutations had been introduced by the
PCR amplification step. ...
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. 2006; F62: 357-60.
37
Artigos Científicos Sobre Cristalização de Proteínas

The supernatant was loaded onto a Q-
Sepharose Fast Flow (2.6 8.2 cm) anion-
exchange column (GE Healthcare) and
fractionated using a 0.0–0.5 M NaCl linear
gradient. The fractions were pooled and
ammonium sulfate was added to a final
concentration of 0.6 M; the mixture was
then loaded onto a HiLoad 16/10 Phenyl
Sepharose HP hydrophobic interaction
column (GE Healthcare). The active
fractions were loaded onto a Mono Q HR
16/10 anion-exchange column (GE
Healthcare) and eluted using a 0.0–0.5 M
NaCl linear gradient.
38
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. 2006; F62: 357-60.

Crystallization trials were initially
performed by the hanging-drop vapour-
diffusion method at 292 K. Hampton
Crystal Screen and Crystal Screen 2 kits
(Hampton Research) were used to
determine the initial crystallization
conditions. Hanging drops were prepared
by mixing 1 ml of a solution containing 10
mg ml1 recombinant protein in 50 mM
Tris–HCl pH 7.8 and 1 ml reservoir
solution. Crystals were obtained with a
reservoir solution containing 0.1 M
HEPES pH 7.5, 28%(v/v) PEG 400, 0.2 M
calcium chloride.
39
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. 2006; F62: 357-60.

The data set for recombinant M.
tuberculosis prephenate dehydratase was
collected at a wavelength of 1.438 Å using
a synchrotronradiation source (Station
MX1, LNLS, Campinas) and a MAR CCD
detector. The crystal was flash-frozen at
100 K in liquid nitrogen. The oscillation
range used was 0.8, the crystal-to-
detector distance was 150 mm and the
exposure time was 90 s. The crystal
diffracted to 3.2 Å resolution. All data were
processed and scaled using the programs
MOSFLM and SCALA from the CCP4
program suite (Collaborative
Computational Project, Number 4, 1994).
40
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. 2006; F62: 357-60.

Summary of data-collection statistics for M. tuberculosis prephenate dehydratase
X-ray wavelength (A ° ) 1.438
Temperature (K) 100
Resolution range (A° ) 65.94–3.20 (3.37–3.20)
Total/unique reflections 71611/23215
Space group I222 or I212121
Matthews coefficient (Å3 Da-1) 2.7
Unit-cell parameters
a (Å) 98.26
b (Å ) 133.22
c (Å ) 225.01
Mosaicity () 0.43
Data completeness (%) 94.4 (97.2)
Average I/(I) 5.7 (1.5)
Multiplicity 3.1 (3.0)
Rmerge(%) 0.120 (0.434)
41
Artigos Científicos Sobre Cristalização de Proteínas
Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. 2006; F62: 357-60.

Fonte: Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst
Commun. F62, 357-360, 2006
42
Artigos Científicos Sobre Cristalização de Proteínas
Aplicações
1) Determinação do conteúdo da unidade assimétrica da MtPD. A partir dos dados de
difração de raios X foi determinado que os parâmetros da cela unitária.
a = 98,26 Å
b = 133,22 Å
c = 225,01 Å
Grupo espacial: I222 ou I212121
MW = 33,6 kDa
Vcell = 98,26 . 133,22 . 225,01 = 2.945.425,3 Å3

Para determinarmos o conteúdo da unidade assimétrica, calcularemos o volume de
Matthews para diferentes possibilidades de unidade assimétrica e verificaremos qual
valor fica mais próximo da faixa de 1,7 a 3,5 Å3/Da, como segue:
Z VM(Å3/Da)
1 87,66
8 10,96
16 5,48
24 3,65
32 2,74
40 2,19
48 1,83
43
Artigos Científicos Sobre Cristalização de Proteínas
MWZ
VV cellM
.

Os valores dentro das caixas estão na faixa prevista por Matthews, contudo sabemos
que o grupo espacial é ortorrômbico com centragem I, o que significa que temos 16
unidades assimétricas, assim teremos um número de moléculas múltiplo de 16, ou
seja, 16, 32,…, sendo o valor mais provável 32. Assim temos:
Vprotein = 1,23/VM = 0,4489 e Vsolvent = 0,5511
Z VM(Å3/Da)
1 87,66
8 10,96
16 5,48
24 3,65
32 2,74
40 2,19
48 1,83
44
Artigos Científicos Sobre Cristalização de Proteínas

Artigo indicado
Segue um artigo de revisão sobre cristalografia de proteínas:
Canduri F, de Azevedo WF. Protein crystallography in drug discovery. Curr Drug
Targets. 2008; 9(12):1048-53.
45
Material Adicional (Artigo Indicado)

Canduri F, de Azevedo WF. Protein crystallography in drug discovery. Curr Drug Targets. 2008; 9(12):1048-53.
Cavada BS, Marinho ES, Souza EP, Benevides RG, Delatorre P, Souza LA, Nascimento KS, Sampaio AH, Moreno FB, Rustiguel
JK, Canduri F, de Azevedo WF Jr, Debray H. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006; 1(62): 235-7.
Dias MV, Ely F, Canduri F, Pereira JH, Frazzon J, Basso LA, Palma MS, de Azevedo WF Jr, Santos DS. Crystallization and
preliminary X-ray crystallographic analysis of chorismate synthase from Mycobacterium tuberculosis. Acta Crystallogr D Biol
Crystallogr. 2004; 60(Pt 11):2003-5.
Drenth, J. (1994). Principles of Protein X-ray Crystallography. New York: Springer-Verlag.
Moreno FBMB, Delatorre P, Freitas BT, Rocha BAM, Souza EP, Facó F, Canduri F, Cardoso ALH, Freire VN, Lima Filho JL,
Sampaio AH, Calvete JJ, De Azevedo Jr. WF, Cavada BS. Crystallization and preliminary X-ray diffraction analysis of the lectin
from Canavalia gladiata seed. Acta Cryst. 2004; D60: 1493-5.
Moreno FBMB, Martil DE, Cavada BS, de Azevedo Jr. WF. Crystallization and preliminary X-ray diffraction analysis of an anti-H(O)
lectin from Lotus tetragonolobus seeds. Acta Cryst. 2006; F62: 680-3.
Rhodes, G. (2000). Crystallography Made Crystal Clear. 2nd ed.San Diego: Academic Press.
Stout, G. H. & Jensen, L. H. (1989). X-Ray Structure Determination. A Practical Guide. 2nd ed. New York: John Wiley & Sons.
Vivan AL, Dias MVB, Schneider CZ, de Azevedo Jr. WF, Basso LA, Santos DS. Acta Crystallogr Sect F Struct Biol Cryst Commun.
2006; F62: 357-60.
Última atualização em: 30 de agosto de 2018.
46
Referências

Calcule o volume de Matthews (VM), a fração de volume ocupada por proteína (Vprotein),
a fração de volume de solvente no cristal (Vsolvent) e o número de moléculas de
proteína na cela unitária para os cristais descritos nos seguintes artigos:
1) Moreno FBMB, Martil DE, Cavada BS, de Azevedo Jr. WF. Crystallization and
preliminary X-ray diffraction analysis of an anti-H(O) lectin from Lotus
tetragonolobus seeds. Acta Cryst. 2006; F62: 680-3.
2) Moreno FBMB, Delatorre P, Freitas BT, Rocha BAM, Souza EP, Facó F, Canduri F,
Cardoso ALH, Freire VN, Lima Filho JL, Sampaio AH, Calvete JJ, De Azevedo Jr.
WF, Cavada BS. Crystallization and preliminary X-ray diffraction analysis of the
lectin from Canavalia gladiata seed. Acta Cryst. 2004; D60: 1493-5.
Indique o desenvolvimento do cálculo, como mostrado na aula de hoje, onde é
calculado o VM para diversos valores de Z e selecionado aquele que se encontra
dentro da faixa esperada de VM de 1,7 a 3,5 Å3/Da.
Data da entrega: 14 de setembro de 2018. 47
Trabalho





























