Embed Size (px)
DESCRIPTION
Basico de Cromatografia
Citation preview

CROMATOGRAFÍA
PRINCIPIOS Y APLICACIONES
Clara Pérez González
Blanca Pinedo Martínez
Belén San Román Arenas
Mª del Carmen Fernández Palomar
Elena Jiménez Díaz
Silvia de Juanes Seligmann

1
ÍNDICE
Página 1. CONCEPTOS BÁSICOS DE CROMATOGRAFÍA .................................... 2 2. TIPOS DE CROMATOGRAFÍA ................................................................. 6 3. CROMATOGRAFÍA EN COLUMNA .......................................................... 7 4. CROMATOGRAFÍA PLANA ...................................................................... 8
4.1. Cromatografía en capa fina (TLC) .................................................... 8 4.2. Cromatografía en capa fina de alta resolución (HPTLC) ................. 9 4.3. Cromatografía en papel .................................................................... 9
5. CROMATOGRFÍA DE ALTA RESOLUCIÓN ............................................ 10 5.1. Fundamentos y principios básicos ................................................... 10 5.2. Instrumentación ................................................................................ 13 5.3. Aplicaciones al análisis de alimentos ............................................... 15
6. CROMATOGRAFÍA DE GASES ............................................................... 16 6.1. Fundamentos y principios básicos ................................................... 16 6.2. . Componentes de un cromatógrafo de gases ................................. 17 6.3. Aplicaciones al análisis de alimentos ............................................... 19
7. BIBLIOGRAFÍA ......................................................................................... 21

2
1. CONCEPTOS BÁSICOS DE CROMATOGRAFIA
La cromatografía es una de las técnicas más efectivas para separar e identificar los componentes químicos. El método fue desarrollado por el botánico ruso Mikhail Tswett, en 1906, que utilizó una columna de carbonato cálcico para la separación de pigmentos vegetales, llegando a la conclusión de que la clorofila no era un simple compuesto químico. Las técnicas cromatográficas actuales tienen multitud de formas, la mayoría de las cuales pueden ser automatizadas y están adaptadas para el reparto de gran o pequeña cantidad de sustancias, siendo separadas y purificadas.
�La separación consiste en un proceso de competición donde las moléculas tendrán que elegir en que fase residen (estacionaria, no se mueve a lo largo del proceso de separación, o móvil, líquida o gaseosa)�. Éstas diferencias a la hora de elegir son las que aprovechamos para su separación.
El factor de capacidad de un soluto (K) se define como el cociente del tiempo que el soluto permanece en la fase estacionaria y el que permanece en la fase móvil. En condiciones isocráticas equivale a la relación de moléculas que en un instante de tiempo se encuentran en fase móvil y en fase estacionaria. Es una medida de la retención de los compuestos, siendo función de;
� La composición del relleno de la columna
� La polaridad de la fase móvil
Concentración en fase A −−−−−−−−−−−−−−−−−−−−−−−− = Kd
Concentración en fase B
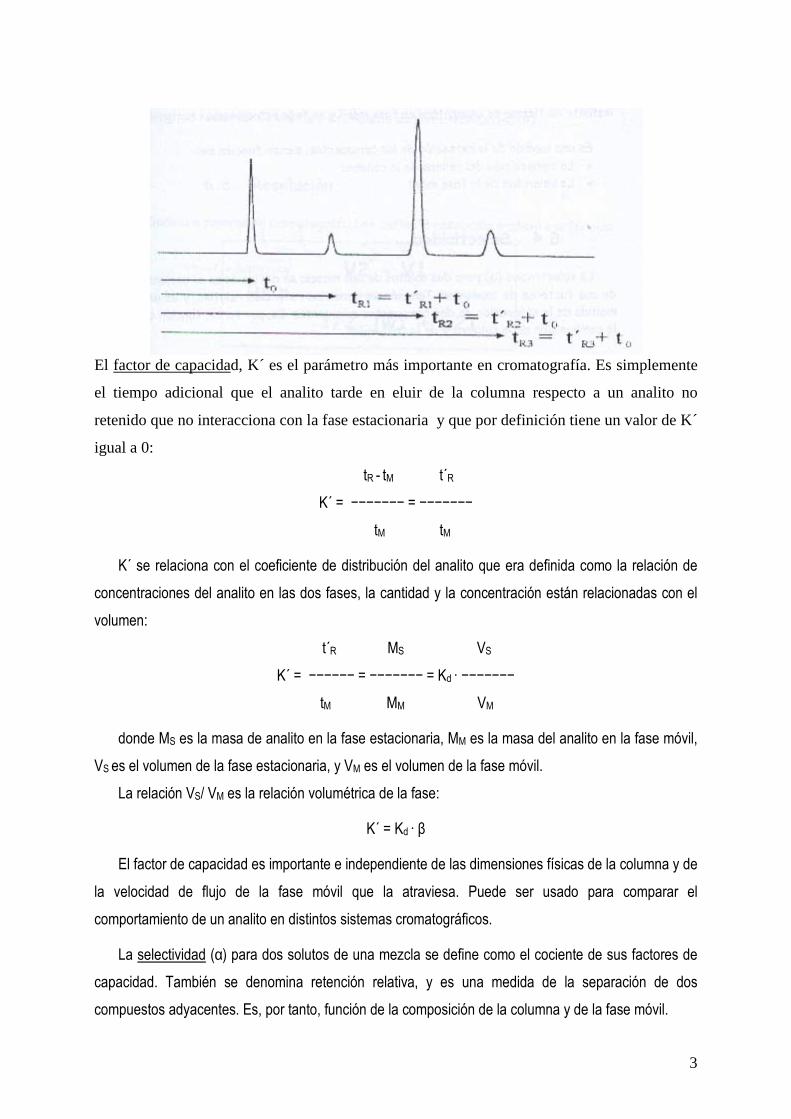
Un cromatograma es la respuesta captada por un detector en función del tiempo de elución o volumen. Consiste en una serie de picos que representan la elución de los analitos individuales. El tiempo de retención tR para cada analito tiene dos componentes. El primero es el tiempo que tarda la molécula de analito en pasar a través de los espacios libres entre la matriz de la fase estacionaria. Este aspecto se refiere al volumen de la columna vacía, V0, y el tiempo que tarde en pasar a través de ella, tM. El valor de tM debe ser el mismo para todos a los analitos y puede ser medido usando un soluto que no interaccione con la fase estacionaria y que simplemente gaste su tiempo de elución en que la fase móvil recorra todo el volumen vacío. El segundo componente es el tiempo en el que el analito es retenido por la fase estacionaria, t´R. Este tiempo es característico de cada analito.
Su valor que es el tiempo de retención viene dado por: t´R = tR - tM
3
El factor de capacidad, K´ es el parámetro más importante en cromatografía. Es simplemente
el tiempo adicional que el analito tarde en eluir de la columna respecto a un analito no
retenido que no interacciona con la fase estacionaria y que por definición tiene un valor de K´
igual a 0:
tR - tM t´R K´ = −−−−−−− = −−−−−−−
tM tM
K´ se relaciona con el coeficiente de distribución del analito que era definida como la relación de concentraciones del analito en las dos fases, la cantidad y la concentración están relacionadas con el volumen:
t´R MS VS K´ = −−−−−− = −−−−−−− = Kd · −−−−−−−
tM MM VM
donde MS es la masa de analito en la fase estacionaria, MM es la masa del analito en la fase móvil, VS es el volumen de la fase estacionaria, y VM es el volumen de la fase móvil.
La relación VS/ VM es la relación volumétrica de la fase:
K´ = Kd · β
El factor de capacidad es importante e independiente de las dimensiones físicas de la columna y de la velocidad de flujo de la fase móvil que la atraviesa. Puede ser usado para comparar el comportamiento de un analito en distintos sistemas cromatográficos.
La selectividad (α) para dos solutos de una mezcla se define como el cociente de sus factores de capacidad. También se denomina retención relativa, y es una medida de la separación de dos compuestos adyacentes. Es, por tanto, función de la composición de la columna y de la fase móvil.

4
K´A KdA t´RA α = ������ = ��������� = ���������
K´B KdB t´RB
Las columnas cromatográficas consisten en un número de zonas adyacentes en cada una de las cuales hay suficiente espacio para que un analito esté en equilibrio entre dos fases. Cada una de estas zonas se conoce con el nombre de plato teórico (de los que hay N en cada columna). La longitud de una columna contiene una altura del plato, H, que tiene unidades de longitud, normalmente en micras. El valor numérico de N y H para cada columna en particular es expresada en referencia a cada analito. La altura del plato está relacionada con la anchura del pico del analito y la distancia que recorre dentro de la columna, X:
σ2 H = ������
X
Donde σ es un estándar de desviación de la banda Gaussiana. Para los picos Gaussianos simétricos la anchura de la base es igual a 4σ y la anchura del pico al punto de inflexión , ωi es igual a 2σ. Por lo tanto el valor de H puede ser calculado desde el cromatograma midiendo la anchura del pico. El número de platos teóricos en la columna entera viene dado por:
L L · X N = ���� = �������
H σ2
Donde L es la longitud de la columna. Si consideramos la posición del pico a X = L con el hecho de que la anchura del pico en su base
es ω, obtenida de las tangentes dibujadas desde los dos puntos con más pendiente del pico, es igual a 4σ, la ecuación anterior se convierte en
16 · L2 N = ��������
ω2
Si en lugar de medir L y ω en unidades de longitud se hace en tiempo, la ecuación quedará:
N = 16 · ( tR / ω ) 2
La nomenclatura refleja el concepto de destilación de la cromatografía de gases; en esta técnica una cierta longitud de la columna cromatográfica es ocupada por un plato teórico (altura equivalente del
5
plato teórico, HEPT). Si dividimos la longitud de la columna por el valor de HEPT, obtendremos el número de platos en la columna.
El número de platos de una columna (eficacia, N) es una medida del ensanchamiento de la banda cromatográfica. Cuando inyectamos un pequeño volumen de analito en una columna forma una banda estrecha en su inicio, pero a medida que el analito migra la banda se va ensanchando. La anchura de pico aumenta con la raíz cuadrada de la longitud que la banda ha recorrido.
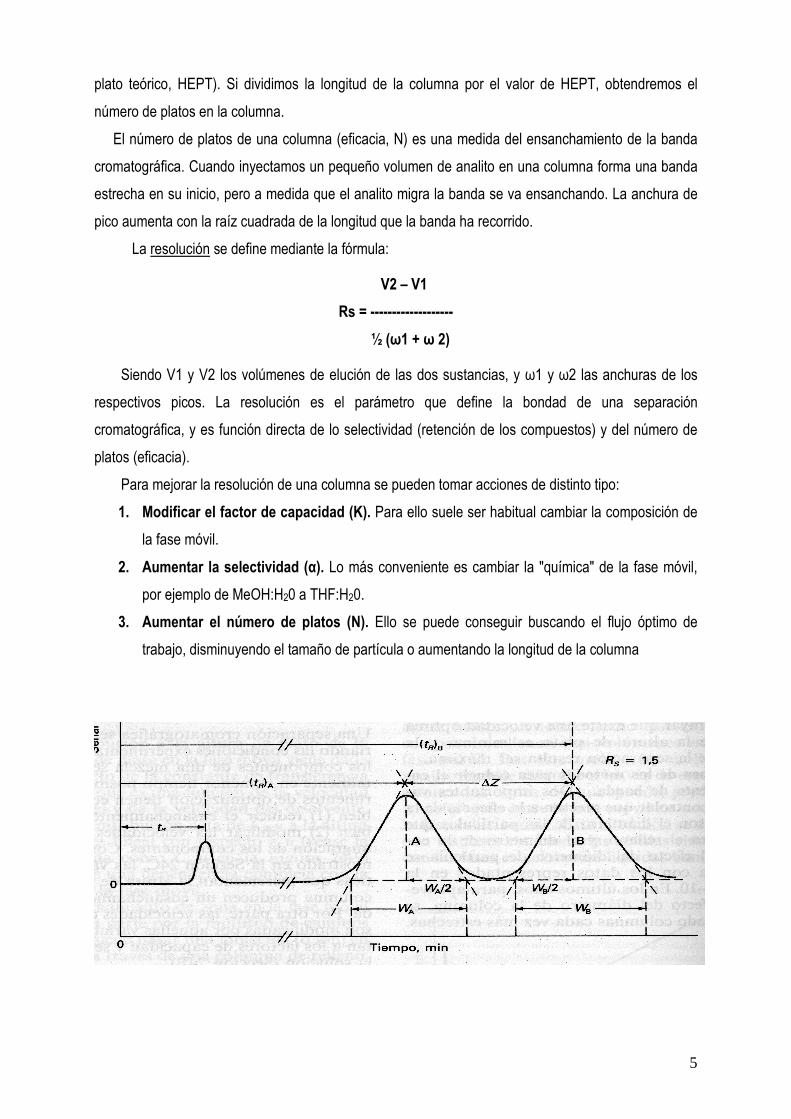
La resolución se define mediante la fórmula:
V2 � V1 Rs = ------------------- ½ (ω1 + ω 2)
Siendo V1 y V2 los volúmenes de elución de las dos sustancias, y ω1 y ω2 las anchuras de los respectivos picos. La resolución es el parámetro que define la bondad de una separación cromatográfica, y es función directa de lo selectividad (retención de los compuestos) y del número de platos (eficacia).
Para mejorar la resolución de una columna se pueden tomar acciones de distinto tipo: 1. Modificar el factor de capacidad (K). Para ello suele ser habitual cambiar la composición de
la fase móvil. 2. Aumentar la selectividad (α). Lo más conveniente es cambiar la "química" de la fase móvil,
por ejemplo de MeOH:H20 a THF:H20. 3. Aumentar el número de platos (N). Ello se puede conseguir buscando el flujo óptimo de
trabajo, disminuyendo el tamaño de partícula o aumentando la longitud de la columna

6
2. TIPOS DE CROMATOGRAFÍA Existen diferentes criterios de clasificación de la cromatografía, - Por la naturaleza de sus fases:
- Cromatografía líquido - líquido - Cromatografía gas � líquido - Cromatografía líquido � sólido - Cromatografía gas � sólido
- Atendiendo al proceso químico � físico que va a protagonizar el proceso de separación: siendo este el criterio más coherente de clasificación
- Cromatografía de Adsorción (líquido - sólido o cromatografía de fases normales). - Cromatografía de Reparto (o líquido - líquido), se basa en las características de solubilidad
relativa de los solutos entre la fase móvil y una fase estacionaria de un líquido no polar. La fase líquida se impregna a un soporte inerte de sílice o, en el caso de cromatografía de fase invertida, se une químicamente.
- Cromatografía de Intercambio iónico. - Cromatografía de Exclusión.
- En base a la naturaleza del soporte en el que se aloja la fase estacionaria: A. Cromatografía plana: 1. Cromatografía en papel 2. Cromatografía en capa fina (TLC) B. Cromatografía en columna 1. Cromatografía de gases (GC) 2. Cromatografía líquida (LC) a. cromatografía líquido � líquido b. cromatografía sólido � líquido

7
3. CROMATOGRAFÍA EN COLUMNA
Todas las cromatografías denominadas en columna se caracterizan por tener una fase estacionaria que se encuentra dentro de una columna de vidrio de 5 a 30 mm de diámetro por la que se hace pasar una fase móvil líquida o gaseosa que estará en permanente movimiento. Según la afinidad de las moléculas por la fase móvil o la estacionaria, éstas se separaran. Después de cada cromatografía podremos sacar información del cromatograma tanto cualitativa (para identificar los distintos compuestos de la mezcla) como cuantitativa (para poder obtener la cantidad y composición de las sustancias separadas).
Las fases estacionarias pueden ser de materiales muy distintos, existen derivados de dextranos (sefadex), derivados de agarosa, poliacrilamida, esferas de vidrio, sílice, etc. Existen distintos tipos de cromatografía en columna:
3.1. Cromatografía de intercambio iónica: se basa en la afinidad de los iones en solución por los sitios de polaridad opuesta que se encuentran en la fase estacionaria.
3.2. Cromatografía de exclusión: Se basa en la habilidad de materiales de porosidad controlada para separar los componentes de
una mezcla de acuerdo al tamaño y forma de las moléculas.
3.3. Cromatografía de afinidad: se basa en la especificidad de algunas macromoléculas biológicas. Éstas se unen específicamente a la fase estacionaria y para separar dicha macromolécula, bastará con varíar el pH una vez que la columna este limpia y sólo se encuentre la que nos interesa.
3.4. Cromatografía de adsorción: se basa principalmente en las diferencias en la afinidad relativa
de los compuestos por el sólido utilizado como fase estacionaria. Las separaciones obtenidas se determinan casi exclusivamente por interacciones polares, siendo la fase estacionaria más polar que la fase móvil.
3.5. Cromatografía de alta resolución (HPLC). (Ver punto 5) 3.6. Cromatografía de gases. (Ver punto 6)

8
4. CROMATOGRAFÍA PLANA
La mayoría de los principios que se aplican para la cromatografía en columna se aplican también a la cromatografía en superficie plana.
4.1. Cromatografía en capa fina (TLC) Para la cromatografía en capa fina (TLC), la fase estacionaria es una capa de partículas de unos
milímetros de espesor, fijadas sobre un soporte sólido generalmente de aluminio, plástico o vidrio. Después de aplicar el analito cerca de la parte inferior de la placa seca, el disolvente empieza a producir la separación.
La ventaja principal de la TLC es que se analizan simultáneamente la muestra y el patrón, mientras que en la cromatografía en columna las muestras se analizan individualmente. Además, las muestras que son difíciles de separar, se pueden resolver utilizando dos disolventes diferentes por desarrollo de la placa en direcciones perpendiculares.
Otra ventaja es que, si los componentes no se pierden en los vapores que rodean la placa, todos estarán en algún lugar de la misma, mientras que en la cromatografía en columna algunos componentes no eluyen y se pierden. Y a ésto se suma que la placa de TLC se usa una sola vez, por lo que se pueden utilizar condiciones severas de separación.
La mancha en una placa de TLC se caracteriza por la distancia que recorre con relación a la distancia recorrida por la fase móvil.
El grado de retención en cromatografía plana de superficie se expresa como el factor de retardación, o índice de retención Rf.
distancia de desplazamiento del soluto Rf = ������������������ distancia de desplazamiento del disolvente
El �frente� del disolvente es el límite alcanzado por la fase móvil, en éste se mide la distancia en que se ha desplazado éste. El valor de Rf depende de las mismas condiciones experimentales que el valor de K de la cromatografía en columna: la composición de la fase móvil, el tipo de fase estacionaria, la temperatura y el tipo de compuestos separados.
También podemos definir el coeficiente de distribución, K, en términos de la concentración del soluto en la fase móvil, Cl, y en la fase estacionaria, Cs:
K = CS / Cl

9
Detección y cuantificación por TLC La detección y localización de las manchas en la placa de TLC se hace observando los cambios
en la fluorescencia, o absorción en el U.V., de un indicador que se incorporó a la fase estacionaria. Toda la placa fluoresce bajo luz UV cuando no hay solutos presentes. Los lugares en los que reside el soluto aparecen como manchas oscuras. También podemos observar el color de los compuestos después de reaccionar con reactivos específicos para grupos o metales, rociados sobre la placa, como la fluorescamina para aminas.
Existen instrumentos para determinar directamente la fluorescencia y/o color de las manchas en la placa. Los instrumentos deben ser capaces de cuantificar la medida sobre el área total de la mancha.
4.2.Cromatografía en capa fina de alta resolución (HPTLC)
Como las separaciones en columna y en TLC se basan en el mismo fenómeno, las mejoras incorporadas a la cromatografía líquida también se aplican a la TLC.
Con HPTLC se puede obtener una separación equivalente con menores distancias de migración, lo que significa que se requiere menos tiempo para análisis equivalentes.
Una desventaja es que el tamaño de la muestra para HPTLC debe ser significativamente menor (en el rango de nanogramos) que para la TLC convencional (en el rango de los microgramos). En general, el HPTLC requiere más equipamiento y más experiencia que el TLC convencional.
Los materiales que se usan para la fase estacionaria de la cromatografía en capa fina pueden ser los mismos que los de relleno de la columna: alúmina, sílice, alquilsilanos enlazados y celulosa. Sin embargo, aunque se use el mismo material para el relleno, el comportamiento de un determinado soluto puede no ser exactamente comparable. La principal razón de ésto es que los solutos y los disolventes en TLC interaccionan con superficies secas que inicialmente no estaban en equilibrio con el disolvente, mientras que en una columna la fase estacionaria está constantemente en contacto con el disolvente (fase móvil).
4.3. Cromatografía en papel
En este caso, el soporte y fase estacionaria es papel. El papel que normalmente se utiliza es de
celulosa. La celulosa es muy polar en el agua y se vuelve electronegativa. El papel tiene propiedades de intercambio iónico débiles, así como de adsorción.
La mayoría de las propiedades varían de papel a papel, lo que provoca variaciones en el Rf. Actualmente existen papeles modificados con resinas cambiadores de iones, alúmina, etc.
El aparato que se usa consta: de un soporte para el papel, un recipiente para el disolvente y una cámara hermética para desarrollar el cromatograma. La cámara hermética es necesaria para evitar la
10
evaporación de los disolventes volátiles debido a la gran superficie del papel expuesta. Antes de sumergir el papel en el disolvente de elución se aplica la muestra en forma de punto diminuto con cualquier objeto que pueda transferir un pequeño volumen.
El papel debe estar en equilibrio con los vapores del disolvente antes de empezar el desarrollo. La cromatografía en papel se ha aplicado con buen éxito a problemas de química orgánica e
inorgánica y de bioquímica.
5. Cromatografía de alta resolución (HPLC)
5.1 Fundamentos y principios básicos
El proceso cromatográfico puede ser considerado como: un conjunto de fuerzas que compiten de manera selectiva por un compuesto (analito o soluto) para, por una parte, fijarlo al relleno de la columna o fase estacionaria, o llevarlo disuelto en los líquidos que fluyen a través de la columna o fase móvil. Los distintos tipos de fuerzas entre fase estacionaria y solutos definen los distintos tipos de cromatografía.
TIPOS DE CROMATOGRAFÍA LÍQUIDA DE ALTA RESOLUCIÓN (HPLC)
Denominación Proceso cromatográfico
Fuerzas implicadas Nombre alternativo Español Genérica
Adsorción - reparto De adsorción Líquido-sólido CLS HPLSC Partición De partición Líquido-líquido CLL HPLLC
De intercambio iónico. CIE HPIEC
Iónicas
De par iónico
De supresión iónica
____
CPI HPIPC
Penetrabilidad en poros
De exclusión
Cromatografía de geles (filtración o permeación)
CEM
HPSEC
En la cromatografía de reparto se utilizan fases estacionarias polares (sílice, alúmina,
hidroxiapatito) y fases móviles apolares (ciclohexano, isooctano, tetracloruro de carbono, etc.), aplicándose a la separación de solutos apolares. El origen de la retención es, por tanto, la interacción de los grupos polares de los analitos con los grupos polares de la superficie del relleno.
En la de partición la fase estacionaria es líquida, es necesario ligarla o embeberla sobre partículas inertes (generalmente de sílice) para que permanezca fija en la columna. Hoy en día los grupos funcionales se fijan mediante enlace químico a las partículas de relleno, lo que permite a la fase estacionaria resistir las altas presiones aplicadas en HPLC.

11
Hay dos posibilidades en la cromatografía de partición: cromatografía en fase normal (cuando la fase estacionaria es polar, por ejemplo si contiene grupos ciano, diol, amino o dimetilamino) y cromatografía en fase inversa (cuando la fase estacionaria es apolar, por ejemplo con grupos C8 ó C18). La cromatografía de partición en fase inversa es la más utilizada de todas las técnicas HPLC, pues las fases móviles polares permiten separar una amplia variedad de compuestos de interés bioquímico, farmacológico o químico. En fase inversa la retención esta basada en una atracción primaria entre la fase estacionaria y la región no polar del analito. El orden de elución es de hidrofílico a hidrofóbico, de polar a no polar. Para aumentar la retención de un compuesto, es necesario aumentar la polaridad de la fase móvil. La fase estacionaria más común es C18. Las fases móviles más comunes son; acetonitrilo, metanol, tetrahidrofurano, etc. con agua como componente de la fase móvil.
En cromatografía de intercambio iónico la fase móvil compite con la estacionaria por los solutos mediante fuerzas iónicas. La limitación es que el soluto sea iónico (un anión o un catión). Se trata de una técnica muy útil para separar iones inorgánicos, proteínas, etc. Mediante supresión iónica se separan compuestos iónicos, suprimiendo (o reduciendo) su estado iónico en solución a través del control del pH, de manera que quedan más retenidos en rellenos de fase reversa. Se trata, por tanto, de adaptar la cromatografía de partición en fase reversa a la separación de mezclas que contienen uno o varios compuestos ionizables. Funciona bien para ácidos débiles y bases débiles. Otro tipo es la cromatografía de pares iónicos que se forma al pasar una fase móvil polar que contiene un contraión de carga contraria a la de los solutos, se suele añadir a la fase móvil un catión o un anión hidrofóbico, que interacciona con el analito formando el par iónico.
La cromatografía de exclusión molecular separa los solutos en función de su tamaño molecular. El principal inconveniente es que se trata de un método de baja resolución. No obstante, es muy útil para la separación de proteínas y otras moléculas de alto peso molecular. Es importante que no haya interacción entre la fase estacionaria y la muestra.
Las reglas básicas en HPLC son cuatro: 1. Fase móvil y estacionaria han de ser de polaridad opuesta. 2. Todos los solutos han de ser solubles en la fase móvil (es decir, polaridad similar). 3. Todo soluto que entra en la columna ha de salir de ella (propagación). 4. Y, a ser posible, con los distintos solutos separados (migración diferencial).
La elución de los componentes de una muestra comprende el lavado de una parte de la muestra disuelta en la fase móvil a través de una columna de fase estacionaria por agregado de disolvente fresco. La porción de muestra se introduce por la parte superior de la columna (tiempo t0), los componentes se distribuyen por sí mismos entre las dos fases según la naturaleza química de éstos

12
con respecto a las fases móvil y estacionaria. Adiciones posteriores del disolvente llevan a las moléculas de soluto hacia abajo en la columna, hasta que salen de ella en una tiempo determinado (tx).
La velocidad a la cual un soluto migra depende de la fracción de tiempo que ha necesitado para salir de la columna, tiempo que es detectado por un detector. Esta velocidad será pequeña para solutos fuertemente retenidos por la fase estacionaria, y será grande para solutos que tengan mayor afinidad por la fase móvil.
El detector se coloca en el extremo final de la columna, y la señal se transforma en una gráfica en función del tiempo denominada cromatograma. El cromatograma discurre horizontalmente paralelo a la escala de tiempos (línea de base), zona que informa de las condiciones generales del sistema en ausencia de solutos. Luego se observa un brusco incremento sobre la línea base, seguido de un descenso más o menos simétrico que enlaza con la continuación de la misma (pico cromatográfico). Idealmente el pico tiene apenas anchura: es estrecho, casi una línea vertical. Pero muchas veces presenta una anchura apreciable, tomando el aspecto habitual de una gaussiana. También se presenta una primera banda peculiar ascendente-descendente (frente del disolvente o frente de inyección), no siempre observable, y que suele hacerse coincidir con el tiempo muerto del sistema o tiempo necesario para que un fluido sin retención atraviese físicamente la columna, tubos, etc. del sistema.
Las moléculas de un mismo soluto, pese a hallarse sometido a las mismas fuerzas, no eluyen todas al mismo tiempo, sino obedeciendo a un modelo gaussiano.
Los picos sobre el eje de los tiempos se emplean para identificar tanto cualitativa (posición en el eje) como cuantitativamente (área bajo los picos) los componentes de la muestra.
La cromatografía cuantitativa se basa en una comparación de la altura o del área del pico de un analito con el de uno o más estándares. Ambos parámetros varían linealmente con la concentración. La cromatografía cualitativa se basa en la comparación de la posición de los picos (tiempos determinados de retención) con cromatogramas estándares.

13
Ejemplo de una determinación de la composición de aminoácidos de una proteína derivatizando los aminoácidos (resultantes de la hidrólisis ácida de la proteína) con un reactivo fluorogénico.
La efectividad de la columna cromatográfica para la separación de solutos depende de las
velocidades relativas a las cuales las especies son eluídas. La eficiencia máxima para la cromatografía líquida ocurre a muy bajas velocidades de flujo. Se puede incrementar la eficiencia de la columna si se disminuye el tamaño de la partícula del empaque de la columna, se reduce la viscosidad de la fase móvil o se incrementa la temperatura. En una primera etapa, la cromatografía de líquidos se llevaba a cabo en columnas de vidrio con diámetros de 1-5 cm y longitudes de 50-500 cm, cuyas partículas de la fase estacionaria no superaban las 150-200 µm de diámetro a fin de asegurar unos caudales razonables. Incluso así, los tiempos de separación eran largos, podían llevar varias horas.
La cromatografía líquida de alta eficacia o HPLC (High Performance Liquid Chromatography) es el método más sofisticado y moderno de la cromatografía líquida, que utiliza partículas de fase estacionaria para el interior de la columna con diámetros muy pequeños para aumentar la eficiencia, en torno a 3-10 µm. Así, se ha conseguido reducir el tamaño de la columna a 10-30 cm de largo y 4-10 mm de diámetro en la HPLC, y el tiempo necesario para la separación.
5.2 Instrumentación Todo cromatógrafo de HPLC dispone de, al menos, cinco módulos: bomba o sistema de bombeo,
inyector (manual o automático), columna, detector y sistema de visualización de resultados. Hay otros módulos complementarios, a veces necesarios para la aplicación: horno de columnas, un
segundo detector y un colector de fracciones. Existen también una serie de pequeños accesorios: soportes del inyector y de columnas,
reservorios o botellas especiales para la fase móvil, restrictores de presión a la salida del detector, filtros intermedios, etc.

14
Las columnas de HPLC estas hechas de acero inoxidable, con un diámetro interno de 2-5 mm y una longitud variable de 10 a 30 cm, dependiendo del diámetro de las micropartículas que contiene la columna (fase estacionaria), que puede ser de 3-10µm. Como cierre de las columnas se utilizan placas filtrantes de acero que no dejen escapar las micropartículas de la columna. En muchos casos se utiliza una precolumna para eliminar contaminantes y partículas de polvo.
La mayor parte de los instrumentos comerciales modernos están equipados con calentadores de columna, que controlan la temperatura desde la cercana al ambiente hasta 150 ºC. Manteniendo la temperatura constante se obtienen mejores cromatogramas.
Existe una gran variedad de materiales de relleno de las columnas según las distintas separaciones. Básicamente, las micropartículas utilizadas están compuestas de sílice, alúmina o resina. Pueden ser de dos tipos:
-Partículas enteramente porosas, de forma irregular y diámetro 3-10µm. -Partículas esféricas de superficie porosa, con diámetros de 30-60µm y un núcleo imposible de
atravesar. Son necesarias bombas de presión de varios centenares de atmósferas para conseguir
velocidades de flujo razonables con micropartículas de 3-10µm debido a la resistencia que ofrecen. Como consecuencia, el equipo de HPLC es más costoso y elaborado que el de otros tipos de cromatografía. Las elevadas presiones que generan las bombas no constituyen un peligro de explosión, puesto que los líquidos no son muy compresibles.
Se dispone de varios reservorios de disolvente que pueden contener cada uno más de 500 ml. Es necesario que estos reservorios vayan acompañados de desgasificadores para eliminar los gases del disolvente, pues producen burbujas en la columna interfiriendo en los resultados.
Para una buena elección de la fase móvil o disolvente, hay que tener en cuenta que la interacción con la fase estacionaria (micropartículas) debe ser óptima y la separación debe ser lo más rápida posible. Los parámetros que determinan el tipo de disolvente a utilizar son: viscosidad, transparencia al UV, punto de ebullición, índice de refracción, inercia frente a la muestra, resistencia a la corrosión, toxicidad, precio y mantenimiento.
El sistema de inyección de muestra que más se emplea se basa en "loops de muestra", que son bucles o válvulas dosificadoras que permiten una aplicación cuantitativa y reproducible a presión elevada. Proporcionan una selección del tamaño de la muestra que va desde 5 a 500µl. La muestra se pasa primero al bucle sin presión, para luego pasar a la columna accionando el flujo del disolvente con una llave de varias vías o válvulas de membrana.
La muestra no debe contener sólidos para que no se atasque la columna, si es necesario deberá filtrarse por un filtro de membrana o precolumna. Lo mejor es disolver la muestra en el eluyente que se

15
vaya a emplear en la separación. El volumen inyectado de muestra debe ser lo más pequeño posible, para que los resultados aparezcan más nítidos.
Para HPLC se emplean distintos detectores dependiendo de la naturaleza de la muestra. -Detectores selectivos: responden a una propiedad del soluto en disolución. Son los detectores
ultravioleta/visible (UV/Vis), detector de fluorescencia y detector electroquímico. El más utilizado es el UV/Vis, (también puede clasificarse como detector universal) sobre todo el
espectrofotómetro, que registra sustancias que absorben la radiación ultravioleta o visible. Los hay muy sensibles, son relativamente independientes de las oscilaciones de temperatura y se pueden utilizar en la elución en gradiente.
-Detectores universales: responden cuando una propiedad de la fase móvil es cambiada por la presencia de un soluto. Son el detector del índice de refracción(RI) y el detector de conductividad.
El más utilizado es el detector RI, que registran todas aquellas sustancias que presenten un índice de refracción distinto al de la fase móvil pura. La señal será tanto mayor cuanto mayor sea la diferencia en el índice de refracción. Es necesario un control estricto de la temperatura y no se pueden utilizar para separaciones por elución en gradiente.
5.3 Aplicaciones al análisis de alimentos La HPLC permite muy buenas separaciones e identificaciones de sustancias o grupos de
sustancias en un tiempo corto, tanto cualitativa como cuantitativamente. Como condición, es imprescindible que la muestra sea soluble en un disolvente, al ser la fase móvil un líquido.
Es especialmente adecuada para compuestos poco volátiles , termolábiles e iónicos. Algunas aplicaciones incluyen sustancias tan diversas como aminoácidos, proteínas, ácidos nucléicos, hidrocarburos, carbohidratos, fármacos, terpenos, plaguicidas, antibióticos, esteroides, especies organometálicas y una gran variedad de sustancias inorgánicas.
Ejemplos de utilización de HPLC: - Detección y cuantificación de cafeína y teobromina que son alcaloides muy importantes en el café, té y el cacao, especialmente por su acción estimulante. El contenido de estos compuestos se utiliza para evaluar su calidad. (Detección UV) - Determinación de azúcares. La analítica de carbohidratos no resulta fácil pues presenta acumulación de grupos funcionales iguales, los grupos hidroxilo, y además posee gran número de esteroisómeros. Tras una dilución los azúcares se separan cromatográficamente por medio de HPLC en una fase amínica. (Detección RI) - Determinación cuantitativa de polisacáridos como el almidón, las dextrinas, los dextranos, el glucógeno, la inulina, la celulosa, las hemicelulosas, las pectinas, así como sustancias mucilaginosas de algas marinas y las gomas vegetales.

16
- Detección de ciclomato en jugos de fruta. - Separación de ésteres de ácidos grasos por fase inversa y con argentización de columnas (Silicato de Al con Ag). - Separación de Triglicéridos por la longitud de la cadena y el grado de insaturación por fase inversa y detector de dispersión luminosa, para el estudio de aceites y grasas adulteradas. - Determinación del contenido de Vit A y sus precursores ( y β caroteno) en margarina y aceites de hígado por fase inversa. - Determinación del contenido en Vit D en leche en polvo y cereales por fase normal.
6. CROMATOGRAFÍA DE GASES (GC)
6.1. Fundamentos y principios básicos
Este tipo de cromatografía es una técnica que proporciona información tanto cuantitativa como cualitativa, válida para la separación de casi todo tipo de compuestos, siempre que tengan diferente volatilidad. La fase móvil es un gas inerte que arrastrará los compuestos inyectados a una velocidad de migración que dependerá de la naturaleza de éstos, de la naturaleza y velocidad de la fase móvil, de la naturaleza de la fase estacionaria y de la temperatura, sin interaccionar con los compuestos.
El resultado será un cromatograma donde quedará reflejado el tiempo que ha tardado cada compuesto en salir (tiempo de retención bruto). Podemos encontrar dos tipos de GC atendiendo a la naturaleza de la fase móvil:
! Cromatografía gas-líquido. La fase móvil es un gas y la fase estacionaria es un líquido con alto punto de ebullición (para que no se volatilice), de naturaleza inerte y que proporcione películas uniformes. Este líquido es inmovilizado por impregnación o por enlace sobre un soporte inerte que puede ser simplemente la pared de la columna. La naturaleza de la fase estacionaria nos determinará el orden de salida de los compuestos:
Apolar: primero saldrán los compuestos más pequeños, en orden de volatilidad creciente. A igual nº de carbonos, los compuestos con insaturaciones saldrán antes. Ej: en una serie homóloga de ác. grasos C12 - C14 - C16 - C18...
Si hay insaturaciones C (18:3) � C (18:2) � C (18:1)... Polar: si hay insaturaciones, éstas reaccionarán por fuerzas de Van der Waals, así pues
en una serie homóloga los compuestos saldrán en orden de tamaño creciente, pero a igual nº de carbonos los saturados saldrán antes, luego los monoinsaturados, etc.
El parámetro implicado es el coeficiente de reparto K, pues la separación de los analitos depende de las diferentes fracciones de tiempo en las que se mantienen disueltos en la fase estacionaria liquida.

17
Los componentes más solubles se retienen durante más tiempo, dado que pasan menos tiempo viajando con el gas portador.
! Cromatografía gas-sólido. La fase estacionaria es un sólido poroso (grafito o gel de sílice o
alúmina generalmente) y la fase móvil es un gas. La retención de los analitos se debe al equilibrio proporcionado por la adsorción y desorción sobre la superficie del sólido. Este tipo de GC es muy efectivo para análisis de mezclas de gases o de compuestos con bajo punto de ebullición. El parámetro implicado es el coeficiente de adsorción.
La cromatografía de gases es el método de separación elegido para analizar tanto compuestos orgánicos como gases, pues ambos grupos poseen presiones de vapor suficientemente altas como para ser arrastrados en la fase móvil gaseosa. Lo que limita el uso de la GC es la descomposición térmica.
La GC requiere muestras volátiles, por lo que los compuestos que no lo son (azúcares, ácidos grasos, aminoácidos...) tienen que ser derivatizados, lo que significa transformar químicamente grupos químicos que originan puntos de presión bajas (punto de ebullición alto) en compuestos más volátiles (en azúcares bloqueo de OH con cloruro de sílice trimetilado, en ácidos grasos se producen sus ésteres metílicos, etc.). La derivatización también puede ser empleada cuando los componentes reaccionan para dar grupos que proporcionan una fuerte respuesta en el detector empleado (adición de cloruros cuando se usa un detector de captura electrónica, etc.).
6.2. Componentes de un cromatógrafo de gases
Desde un punto de vista funcional, un equipo de GC está compuesto por tres módulos específicos: un inyector, una columna, un detector y un horno.
• Gas portador: constituye la fase móvil. Suele ser Helio, Nitrógeno o Hidrógeno, proveniente de un cilindro bajo presión (200 � 250 atmósferas en el interior de la botella. Saldrá a 1 � 3 atmósferas gracias a tres manoreductores) o de un generador. Este gas debe estar libre de trazas de hidrocarburos, vapor de agua y oxígeno, ya que pueden reducir la sensibilidad de algunos detectores y actúan como impurezas. La viscosidad y velocidad de este gas tienen influencias sobre la dispersión de los compuestos en la fase estacionaria y sobre la difusión en la fase móvil.
• Inyector: Es la puerta de entrada de la muestra en el cromatógrafo. Además tiene la función de vaporizar y arrastrar la muestra mezclada con el gas portador a la cabeza de la columna. Para volatilizar las muestras de una forma rápida y de modo que todas las partículas inicien el proceso en el mismo punto, la temperatura del inyector debe ser muy elevada, unos 100 ºC mayor que la de la columna. Además para minimizar la difusión de la muestra debe ser un

18
espacio lo más reducido posible. La muestra es introducida en el equipo con una microjeringa de la que existen numerosos modelos adaptados a los diversos inyectores y columnas. Para controlar mejor la reproducibilidad de las inyecciones suele hacerse uso de inyectores automáticos. Las características de los inyectores así como la de los modos de inyección dependen del tipo de columna que se utilice.
Encontramos: Inyectores de vaporización directa Inyectores con o sin división (splitt o splittless)
Inyectores a temperatura programable Inyección directa en la columna
• Recinto termostatizado: (también denominado horno). Es una cámara calentada de volumen suficiente y fácil acceso para instalar la columna. Permite una subida rápida y controlada de la temperatura, y además se mantiene muy estable. Mediante cambios de temperatura en esta cámara, se consigue alcanzar la temperatura óptima para cada compuesto o fracción que queramos separar.
• Columnas: existen dos tipos de columnas, las empaquetadas y las capilares, ambas con diferente eficacia. " Empaquetadas: tienen un diámetro interno de entre 2 y 4 mm, y una longitud de entre 1 y
3 m. Se construyen utilizando como armazón un tubo de acero o de vidrio. Contienen un soporte poroso e inerte, con contorno esférico que permiten la unión o impregnación de la fase estacionaria.
" Capilares: su diámetro interno varía entre 100 y 500 µm (conocidas como micro y
megabore respectivamente) y su longitud es del orden de los 100 m. Suelen estar revestidas por un polímero estable térmicamente, lo que permite enrollarlas en un soporte metálico circular. La fase estacionaria recubre la pared interna con un espesor controlado
de unos 0.05 a 5 µm. Las columnas capilares se clasifican en función del método de
inmovilización de la fase estacionaria. Actualmente las columnas capilares están sustituyendo a las empaquetadas ya que
permiten un mayor nivel de resolución.
• Detectores: los hay universales (sensibles a prácticamente todos los compuestos eluidos) y específicos (sensibles a un tipo particular de molécula). También hay detectores que sólo conducen a valores de tiempos de retención, y otros que proporcionan información también de la estructura de los compuestos detectados. Todos los detectores dan una respuesta en función de la concentración molar o másica de la disolución en el gas portador. También es posible conectar varios detectores en serie. Algunos de los principales detectores empleados son:

19
1. Detector de conductividad térmica (TCD) 2. Detector de ionización de llama (FID) 3. Detector termoiónico (NPD) 4. Detector de captura de electrones (ECD) 5. Detector de fotoionización (PID) 6. Detector de emisión atómica 7. Detector de masas 8. Detector infrarrojo o ultravioleta
Los detectores del 1 al 4 no dan información alguna sobre la naturaleza de los compuestos eluidos, como mucho son selectivos, lo que conlleva el problema de que cuando los picos del cromatograma son cercanos, pueden confundirse compuestos. El resto de detectores aportan información estructural.
6.3. Aplicaciones al análisis de alimentos
- Muy importante en el análisis de la materia grasa porque informa simultáneamente de las propiedades físicas y características nutricionales. Los ácidos grasos se hacen más volátiles por esterificación de sus grupos carboxílicos con metanol. El orden de elución de los ácidos grasos es función de la naturaleza de la fase estacionaria de la columna. Si fuese apolar, saldrán primero los más volátiles; si tiene instauraciones cuanto más insaturado más pequeño es el ácido graso. Si la fase es polar, pueden presentarse fenómenos de interacción si hay dobles enlaces, actúan como dipolos y van a quedar retenidos.
- Otra de las aplicaciones más importantes es el análisis de glúcidos digestibles. La cromatografía de gases permite identificar y cuantificar simultáneamente las pentosas,

20
hexosas, los di, tri y oligosacáridos. Se recomienda para la caracterización de las moléculas glucídicas, tras su hidrólisis en monómeros y transformación en compuestos volátiles. Los azúcares se transforman en compuestos volátiles mediante un tratamiento de acetilación o de sililación (éste es el método más corriente) de sus hidroxilos. La sililación consiste en transformar los monosacáridos en trimetilsililos (TMS) en los que los grupos OH han sido convertidos en O-Si-(CH3)3 . El donador más poderoso de grupos sililos es el trifluoroacetamina (BSTFA) al que se le añade un catalizador, el trimetilclorosilano (TMCS), para obtener una derivación cuantitativa y rápida.
- En la identificación y cuantificación de los esteroles. El principio consiste en fraccionar el insaponificable (vitaminas liposolubles y esteroles que se encuentran libres) por cromatografía de capa fina (TLC) y después analizar la fracción de los esteroles por cromatografía de gases.
- Identificación y cuantificación de glúcidos indigestibles. - En la identificación y determinación de alcoholes. Los alcoholes presentes en disolución
acuosa se separan por cromatografía de gases utilizando Parapack (fase separativa) que es un polímero poroso adecuado para la separación de sustancias volátiles de bajo peso molecular. Las sustancias se eluyen de acuerdo con sus puntos de ebullición.

21
7. Bibliografía Skoog, D.A., West, D.M., Holler F.J. � Fundamentos de Química Analítica� Ed. Reverté, S.A. (Barcelona) 1997. Rubinson, J.F., Rubinson, K.A., �Química analítica contemporánea� Pearson educación. 2000. Pecsok, R.L., Shields, L.D. "Métodos modernos de análisis químico" Ed. LIMUSA, S.A. 1983. Rubinson, K.A., Rubinson, J.F. "Análisis instrumental" Pearson educación, S.A. 2001. Skoog, D.A., Leary, J.J. “Análisis instrumental” McGraw-Hill (Madrid) 1994.
Adrian, J.,Potus, J., Poiffait, A., Dauvillier, P., �Análisis nutricional de los alimentos� Ed. Acribia, S.A. Matissek, Schnepel, Steiner, �Análisis de los alimentos. Fundamentos, métodos y aplicaciones�. Ed. Acribia S.A. 1992. Kirk, R.S., Sawyer, R., Egan, H., �Composición y Análisis de los Alimentos de Pearson� 2ª ed 1996. Rouessac, F. y Rouessac A. � Análisis Químico: Métodos y técnicas instrumentales modernas� Editorial Mc Grawn Hill Interamericana de España, S. A. U. 2000. Robards, K., Hadad, P.R., Jacksin, P.E., �Principles and practice of modern chromatographic methods� Academic press (London)1994 http://www.uib.es/depart/dqu/dquo/pau/Cromatograf%92a/chrom10/chrom/GC/concept/main.htm http://www.redhucyt.oas.org/RLQ/tutoriales/cromatografia/Gas.htm http://ntri.tamuk.edu/hplc/mtamez.html http://www.netaccess.on.ca/~dbc/cic_hamilton/liquid.html


![Cromatografia Gasosa - 01 - CTGAS-ER VIRTUALead2.ctgas.com.br/a_rquivos/inspecao_sistemas_de_gas/Cromatografia/... · pigmentos pigmentos separados Cromatografia = kroma [cor] + graph](https://img.pdfslide.tips/doc/110x75/5c1615aa09d3f252588c0d5d/cromatografia-gasosa-01-ctgas-er-pigmentos-pigmentos-separados-cromatografia.jpg)


























