Embed Size (px)
Citation preview

Cuarto encuentro de Final de ResidenciaSocietat Catalana d´Hematologia i HemoteràpiaMarzo 2016 Dra Luz Muñoz
Servicio de LaboratorioCorsorcio Universitario Parc Taulí, Sabadell

SLPC-T
Conjunto de enfermedades que se originan de linfocitos T maduros periféricos.
Entidades poco frecuentes (10-15% de los LNH). Gran dificultad en su diagnóstico.
Tienen unas características biológicas y clínicas muy heterogéneas.
Gran similitud de la célula neoplásica con su originaria normal (en cuanto a fenotipo y a veces morfología).
Ausencia de marcadores fenotípicos y genéticos específicos.

SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS DE FENOTIPO T (SLPC-T) PRIMARIAMENTE LEUCÉMICOS

LEUCEMIA PROLINFOCÍTICA T
Junto con la LLGG-T es el SLPC-T con expresión leucémica más frecuente. Aunque es una enfermedad rara (2% de las leucemias maduras).
CLÍNICA
Clínica agresiva: leucocitosis con linfocitosis rápidamente progresiva (>100.000) con moderada anemia y trombopenia. Se acompaña habitualmente de hepatoesplenomegalia y menos frecuentemente adenopatías y afectación cutánea (20%) sin eritrodermia, y efusiones serosas.
El 10% de los pacientes pueden presentar una forma más indolente con menor linfocitosis y ausencia de organomegalias con estabilidad durante varios meses o años pero con progresión en todos los casos a la forma agresiva.

MORFOLOGÍA EN SANGRE PERIFÉRICA
Existen tres tipos de morfología:
1.Forma clásica: linfocitos de pequeño-moderado tamaño con citoplasma basófilo con “blebs”, con un núcleo irregular con un nucleolo prominente. Más frecuente.
2. Linfocitos son pequeños sin nucleolo visible al microscopio óptico. 20% de los casos.
3. Variante cerebriforme: forma más infrecuente (5%).
Las variedades morfológicas no se asocian ni con la clínica ni con el pronóstico.
LEUCEMIA PROLINFOCÍTICA T
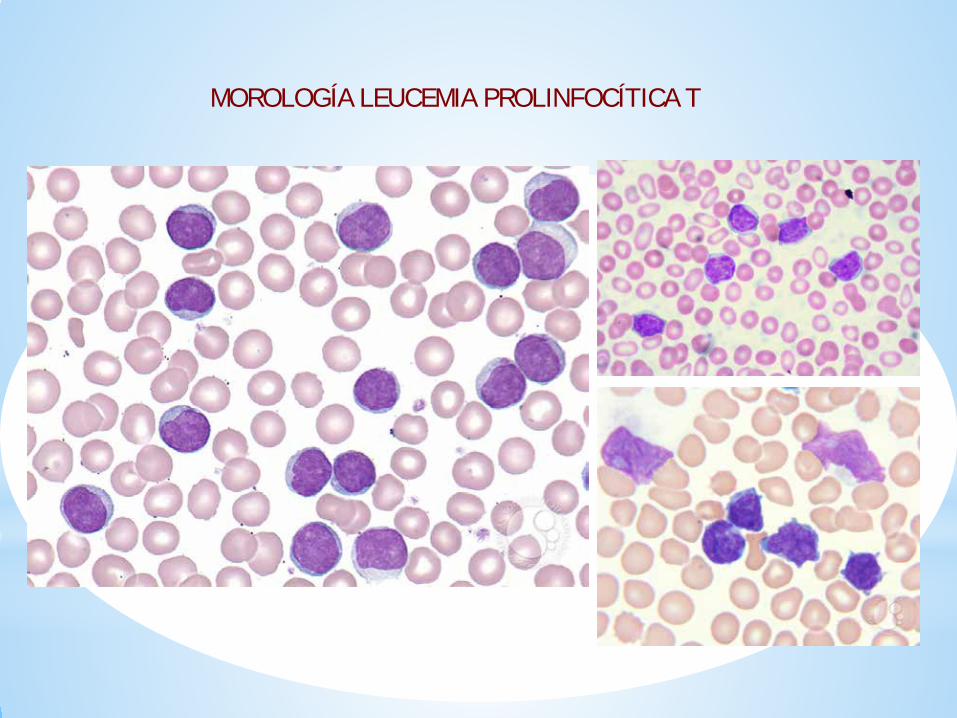
MOROLOGÍA LEUCEMIA PROLINFOCÍTICA T

LEUCEMIA PROLINFOCÍTICA T
INMUNOFENOTÍPO
El patrón más frecuente es el del linfocito T maduro naïf:CD4+ CD8-, negatividad para marcadores de inmadurez Tdt. CD1a,con expresión de CD3 débil , CD2+, CD26, CD28 y habitualmenteCD7+.
Menos frecuentemente (25%) pueden ser CD4+ CD8+.En algunos casos el CD7 puede ser negativo (expresión aberrante)
El 80-85% de los casos muestran expresión citoplasmática de TCL1.Este marcador es específico de esta entidad y no se observa enotros SLPC-T.
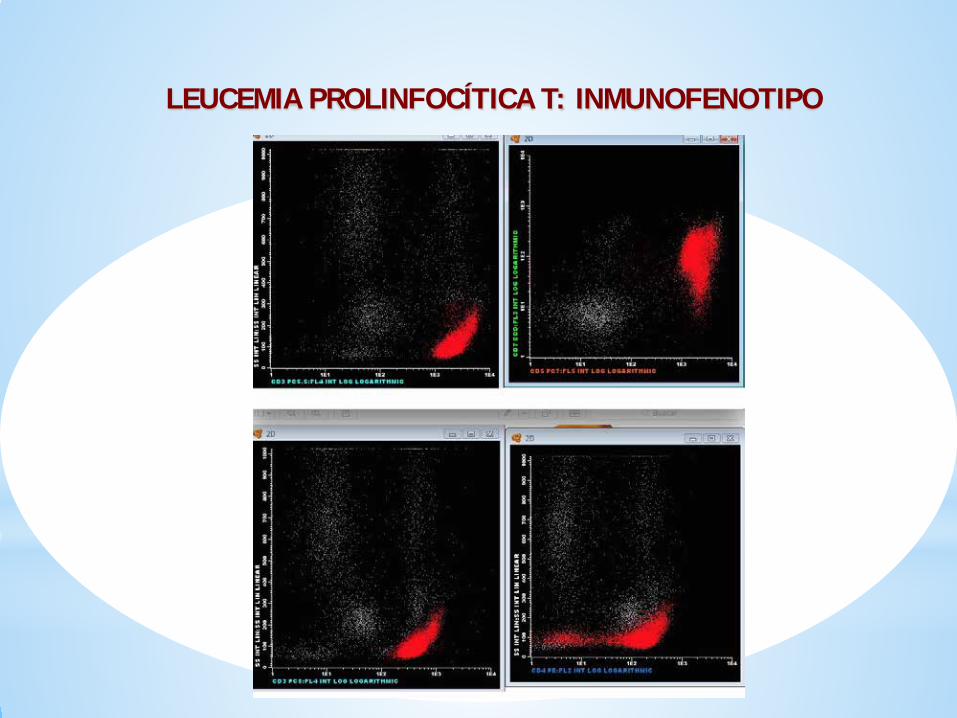
LEUCEMIA PROLINFOCÍTICA T: INMUNOFENOTIPO

LEUCEMIA PROLINFOCÍTICA T
GENÉTICA
Cariotipos complejos con alteraciones recurrentes sobretodo en loscromosomas 14, 8 y 11.
La alteración más frecuente (80%) es la inv 14 (q11;q32). El 10%pueden presentar la t(14,14) (q11; q32).
Importante para el diagnóstico reordenamiento de TCL1 por FISH,en los casos sin alteración citogenética de 14 q32.
Estas alteraciones producen una activación del protoconcogen TCL1(14 q32) con la consecuente sobreexpresión de la proteína tcl1 quese asocia a la malignización de las células.
El estudio mutacional mediante secuenciación ha mostradoalteraciones en la vía de señalización de JAK- STAT (especialmenteen JAK3 y STAT5b). Lo que ofrece posibles opciones terapéuticasmediante inhibidores de estas proteínas.

LEUCEMIA PROLINFOCÍTICA T
PRONÓSTICO Y TRATAMEINTO
Curso agresivo con supervivencia inferior a 1 año.
Pueden haber formas indolentes (10%) con un curso mas establedurante meses o años pero con progresión siempre a las formasagresivas.
Refractariedad al tratamiento quimioterápico convencional.
Mejores respuestas con alemtuzumab (80% de RC) y consolidacióncon trasplante de médula ósea.Otros esquemas pueden ser alemtuzumab y FCM o alemtuzumab ydeoxycoformicina.

LEUCÉMIA DE LINFOCITOS T GRANDES GRANULARES
Expansión clonal de linfocitos T maduros citotóxicos. Su morfología ysu immunofenotipo es igual al de la célula normal por lo que sudiagnóstico con frecuencia es difícil. Espectro variado: procesosreactivos/benignos transitorios o crónicos, procesos neoplásicosindolentes, procesos neoplásicos más sintomáticos.
CLÍNICA
Los pacientes suelen estar asintomáticos (1/3)
Las manifestaciones clínicas más típicas son las citopenias(especialmente la neutropenia) y las consecuentes infeccionesbacterianas de repetición, anemia y trombopenia. Esplenomegalia(40% de los casos) y muy poco frecuente adenopatías, síntomas B,etc.Esta entidad se puede asociar a diferentes situaciones comoenfermedades autoinmunes (artritis reumatoide), síndromesmielodisplásicos, linfomas, etc.

MORFOLOGÍA
La linfocitosis es variable y aunque no existe un consenso comocriterio diagnóstico se requiere >2x109 /L (según la OMS), perosegún otros grupos se requiere >1x109 /L por un período >6 meses o> 25% de linfocitos grandes granulares.
La célula predominante es un linfocito de tamaño moderado conamplio citoplasma con granulación azurófila. La morfología esidéntica al linfocito T normal.
Médula ósea
Importante en el diagnóstico especialmente en casos con mínimalinfocitosis. Infiltración característica nodular o intersticial variable peroen general no muy importante. La neutropenia es por acción de lascitoquinas o anticuerpos antineutrófilo más que por infiltraciónmedular.
LEUCEMIA DE LINFOCITOS T GRANDES GRANULARES

MORFOLOGÍA DE LA LEUCEMIA DE LINFOCITOS T GRANDES GRANULARES

INMUNOFENOTIPO
El patrón más característico es:CD3+, TCR αβ+, CD8+ CD4- (80% de los casos)
Más infrecuentemente pueden expresar: CD4+ CD8-, CD4- CD8- ,CD4+ CD8+.
Genética
No existe una alteración citogenética típica.
Reordenamiento de TCR, obligatorio para el diagnóstico.
LEUCÉMIA DE LINFOCITOS T GRANDES GRANULARES

PRONÓSTICO Y TRATAMIENTO
Los casos asintomáticos no deben tratarse.
Solo requerirán tratamiento los casos con citopenias severas(neutropenia con infecciones de repetición) o bien en los que seproduce un importante incremento de la linfocitosis conesplenomegalia.
El tratamiento de elección son inmunosupresores como metotrexato,ciclofosfamida ,ciclosporina A. Consiguen mejoría de las citopenias perono la remisión completa de la enfermedad.Los casos con gran carga tumoral pueden tratarse con alemtuzumab.
El curso suele ser crónico son supervivencias superiores a 10 años y suevolución a linfoma agresivo es rara.
LEUCEMIA DE LINFOCITOS T GRANDES GRANULARES

LEUCEMIA / LINFOMA T DEL ADULTO
EPIDEMIOLOGÍA
Neoplasia causada por la infección del retrovirus humano HTLV-1.
Esta infección tiene una distribución geográfica con zonasendémicas como Japón, Caribe y algunas zonas de África central. Enel resto del mundo se han descrito también casos esporádicos, através de la inmigración de pacientes de las zonas endémicas.
En España con mayor frecuencia se han detectado pacientesinmigrantes de centroamérica (Chile y Brasil) o la región central oeste de África. Entre los años 1989 y 2012 se han registrado 229casos de infección por HTLV-1, de estos 17 pacientes desarrollaronuna leucemia/linfoma T del adulto.
Para el diagnóstico es imprescindible la seropositividad de HTLV-1.

CLÍNICAExisten 4 formas de presentación:
1.Forma aguda (más frecuente): marcada leucocitosis,linfoadenopatias generalizadas, hepatoesplenomegalia, rash cutáneo,hipercalcemia con lesiones óseas líticas. Clínica grave y aguda.
2.Variante linfomatosa: clínica similar a la aguda pero sin tantaexpresión en sangre periférica, presencia de adenopatías generalizadasmenor afectación ósea.
3.Variante crónica: afectación cutánea con rash exfoliativo, menorexpresión en sangre periférica y sin hipercalcemia.
4.Variante smoldering: afectación cutánea o pulmonar, con cifra deleucocitos normal y menos del 5% de linfocitos neoplásicos circulantes.
El 25 % de las formas crónicas evolucionan a agudas tras un periodo(meses o años).
LEUCEMIA / LINFOMA T DEL ADULTO

LEUCEMIA / LINFOMA T DEL ADULTO
MORFOLOGÍA

LEUCEMIA / LINFOMA T DEL ADULTO
INMUNOFENOTIPO

GENÉTICA
Con frecuencia hay alteraciones citogenéticas múltiples queafectan mas frecuentemente a 14q y 3p.
Frecuente alteraciones en tumores supresores como p53 op14/p16. Importancia la detección de la mutación de p53 para eltratamiento.
Se detecta también integración monoclonal de HTLV-1 en lascélulas neoplásicas y reordenamiento clonal del receptor T.
LEUCEMIA / LINFOMA T DEL ADULTO

TRATAMIENTO Y PRONÓSTICO
Son factores pronósticos el subtipo clínico, la edad, la LDH y losniveles de calcio.Las formas aguda y linfomatosa tienen mal pronóstico con unasupervivencia que va de semanas a algo más de 1 año. Con frecuenciala causa de muerte son infecciones oportunistas.
La formas crónica y smoldering tienen mejor pronóstico con mayorsupervivencia aunque algunos casos se transforman a formas agudas.
El tratamiento incluye zidovudina, interferón y quimioterapia seguidode alotrasplante.
Los pacientes con mutación de p53 no responderían a zidovudina ypodrían tratarse con alentuzumab y consolidación con trasplante.
LEUCEMIA / LINFOMA T DEL ADULTO

SÍNDROME DE SÉZARY
Se define como la tríada:
1. Afectación cutánea en forma de eritrodermia
2. Adenopatías, generalmente
3. Presencia de linfocitos T neoplásicos (células de Sézary) en sangre periférica, ganglio o piel.
Para la definición de síndrome de Sézary se requiere junto con la
eritrodermia uno o más de estos datos:
1. Cifra absoluta de células de Sézary (>1.000/ mm3) o un 5% de la fórmula leucocitaria.
2. Alteración CD4/CD8 > 10, fenotipo aberrante (pérdida de expresión de antígenos T).
3. Demostración clonalidad T mediante biología molecular.

CLÍNICA
Afecta a adultos especialmente a partir de 60 años.
Afectación cutánea en forma de eritrodermia generalizada yadenopatías. Prurito, alopecia, hiperqueratosis palma y plantar.
Histología cutanea
Similar a la de la micosis fungoide: infiltración por célulascerebriformes con epidermotropismo y presencia de microabceos dePautrier. Con al progresión tumoral la infiltración en la dermis sehace mas difusa y puede perderse el epidermotropismo.
SÍNDROME DE SÉZARY

MORFOLOGÍA
Existen dos variantes de la célula de Sézary la variante grande y lavariante pequeña.
La variante pequeña (célula de Lutzner) es muy difícil de reconocermicroscopio óptico es un linfocito de pequeño tamaño, con el núcleoirregular (es la variante mas frecuente).
La variante grande, es un linfocito de mas tamaño similar a unmonocito, con un núcleo grande irregular con pliegues o surcos. Enocasiones pueden observarse alguna célula polilobular, similar a la de laleucemia/linfoma T del adulto.
En el mismo paciente suelen coexistir a la vez las dos variantesmorfológicas en número variable.
No suele haber afectación medular.
SÍNDROME DE SÉZARY

Inmunofenotipo
CD4+, CD8-, CD3+, TCRβ, CD5, expresión débil o nula de CD7 o de otros marcadores T : CD5, CD2, CD3. Característicamente son CD25 – (a diferencia de la leucemia/linfoma T del adulto).- Recientemente se ha detectado expresión aberrante de CD158 en estos pacientes.
GenéticaCariotipos complejos sin ninguna alteración recurrente o específica.Reordenamiento de TCR.
SÍNDROME DE SÉZARY

PRONÓSTICO Y TRATAMIENTO
La mayoría de pacientes requieren tratamiento sistémico:diferentes estrategias interferón, bexaroteno, inhibidores deHDAC, fotoaféresis extracorpórea, alemtuzumab,poliquimioterapia (CHOP) y TPH.
La supervivencia a los 5 años es del 25%.
SÍNDROME DE SÉZARY

CONCLUSIONES
Los SLP-T maduros son un grupo heterogéneo de enfermedades conuna patogenia, clínica y pronóstico muy diferente, pero quecomparten características morfológicas, fenotípicas y genéticas entreellos e incluso con las célules T normales, lo que dificultaenormemente el diagnóstico de estas entidades.
Es imprescindible la integración de toda la información clínica,morfológica, fenotípica y genética para el correcto diagnóstico.
En los casos con afectación leucémica primaria el exhaustivo estudiomorfológico, inmunofenotípico y genético de la sangre periférica esnecesario para el correcto diagnóstico.
El avance en los conocimientos patogenéticos moleculares de estasenfermedades nos ayudará en su diagnóstico así como en conseguirnuevos tratamientos diana que aumenten la supervivencia.

CASO CLÍNICO

Hombre de 56 años ingresa por shock séptico de origen respiratorio.
Antecedentes patológicosHiperreactividad bronquial.Dislipemia
Historia hematológica
Diagnosticado previamente de neutropenia crónica idiopática (6 añosantes) con cifra de neutrófilos < 0,5x109/L.El paciente había presentado varios episodios infecciosos graves: sepsispor celulitis necrotizante, infección grave con absceso muscular porPseudomona Aeruginosa y varios ingresos por infecciones respiratorias.
Aspirado medular: disminución de la serie granulopoyética sin otrasalteraciones.

NEUTROPENIA CRÓNICA IDIOPÁTICA
Entidad poco frecuente y difícil de diagnosticar. Más frecuente en mujeres.
Leucopenia con neutropenia de intensidad variable.
Poco frecuente la clínica infecciosa grave.
Respuesta a G-CSF.

Enfermedad actual
Ingresa por shock séptico de foco respiratorio.Hipertensión pulmonar grave con disfunción ventricular derecha.
Analítica: leucocitos 2.5x109/L (3% neutrófilos < 0,1x109 /L) (92%linfocitos).
Morfología de sangre periférica: 37% de linfocitos grandesgranulares (0.9x109/L)
Aspirado medular: celularidad abundante, presencia de las 3 serieshematopoyéticas, sin rasgos dismórficos significativos. Presencia deun 19 % de linfocitos maduros, polimorfos algunos congranulaciones citoplasmáticas.
Cariotipo medula ósea: normal.

92% linfocitos (37% linfocitos grandes granulares , cifra absoluta: 0.9x109/L)

INMUNOFENOTIPO
Reordenamiento TCR: clonal

ClínicaNeutropenia grave y sintomáticaHipertensión pulmonar grave idiopática
Sangre periférica<1x109/L linfocitos grandes granularesFenotipo CD8+ con alguna aberración fenotípicaReordenamiento TCR clonal
Aspirado medul.larInfiltración intersticial por 20% de linfocitos grandes granulares.

DIAGNÓSTICO
LEUCEMIA DE LINFOCITOS T GRANDES GRANULARES
v Clínica: citopenias, esplenomegalia,
> 2.000 linfocitos grandes granularesv Linfocitosis:
< 1.000 linfocitos grandes granulares en sp necesario población T aberrante por fenotipo y reordenamiento TCR
v Presencia de una población T citotóxica en médula ósea.
Algoritmo diagnóstico

Evolución
Inició tratamiento con metotrexate junto con tratamientopara la hipertensión pulmonar.
Después de varios meses y por refractariedad altratamiento (persistencia de la neutropenia y de la HTP) secambió a ciclofosfamida con buena respuesta(normalización del hemograma, no clínica infecciosa ymejoría de la hipertensión pulmonar.
El paciente ha mantenido esta respuesta durante variosaños hasta que hace unos meses ha vuelto ha aparecer laneutropenia con la presencia de la población T citotóxicaaberrante y empeoramiento de la clínica pulmonar.





























