Embed Size (px)
Citation preview

C U R S O S E M B I O L O G I A , B I O Q U Í M I C A , B I O T E C N O L O G I A E E N G E N H A R I A B I O L Ó G I C A
Bioinformática
João [email protected]
Aula T11-T12
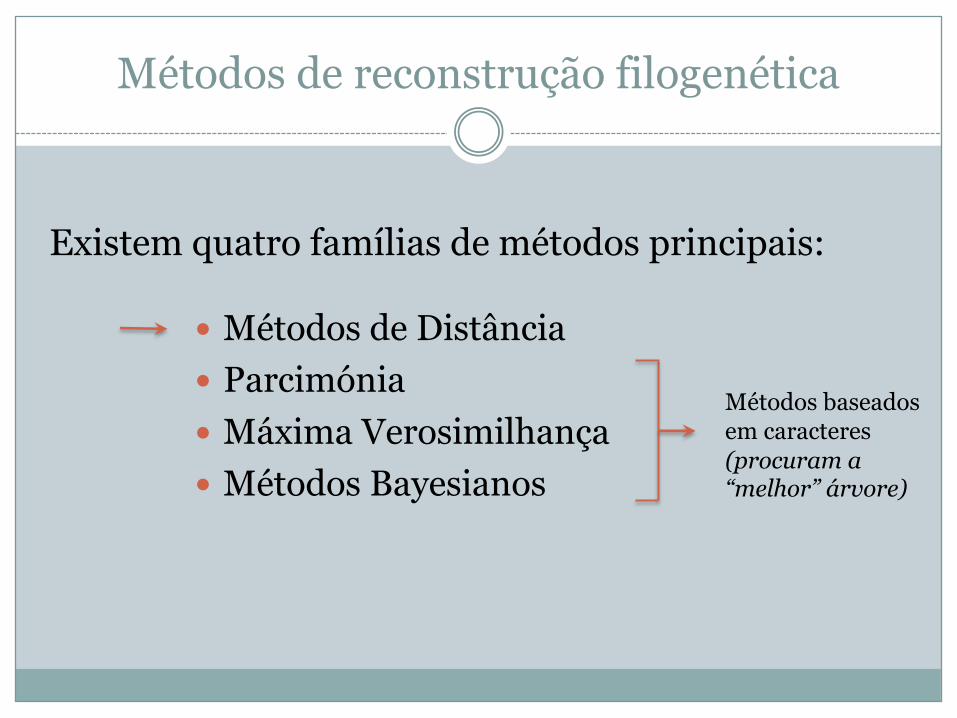
Métodos de reconstrução filogenética
� Métodos de Distância� Parcimónia� Máxima Verosimilhança� Métodos Bayesianos
Existem quatro famílias de métodos principais:
Métodos baseados em caracteres(procuram a “melhor” árvore)

Distâncias evolutivas
Distância evolutiva p:
p = d / l
d = nº de substituições entre 2 sequênciasl = o tamanho de cada uma das sequências comparadas em
aminoácidos ou nucleótidos
Assume a comparação de 2 sequências de cada vez
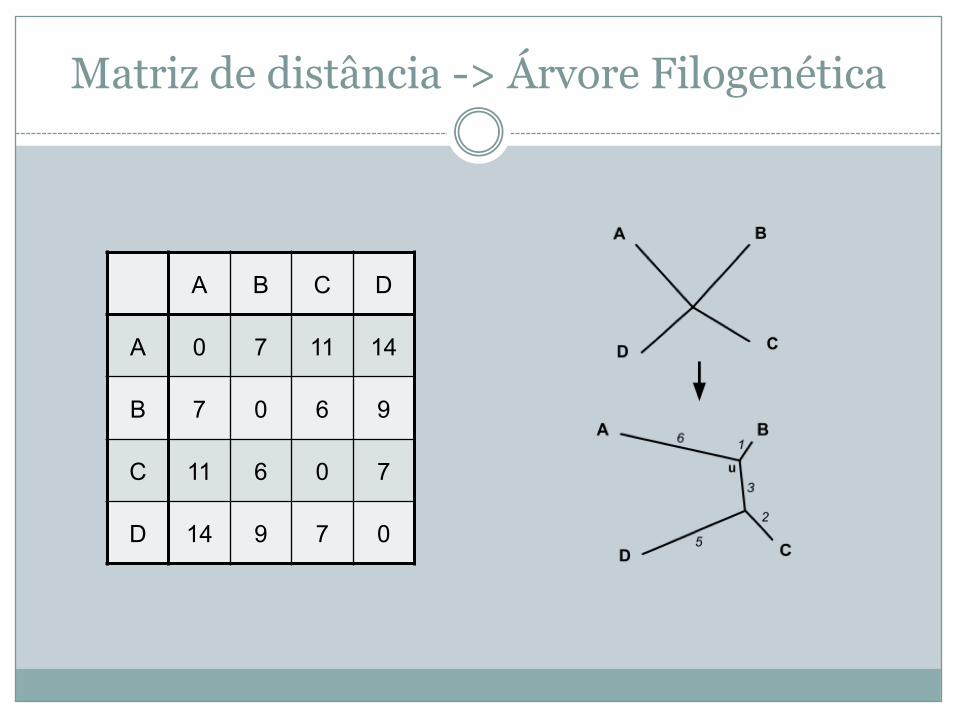
Matriz de distância -> Árvore Filogenética
A B C D
A 0 7 11 14
B 7 0 6 9
C 11 6 0 7
D 14 9 7 0
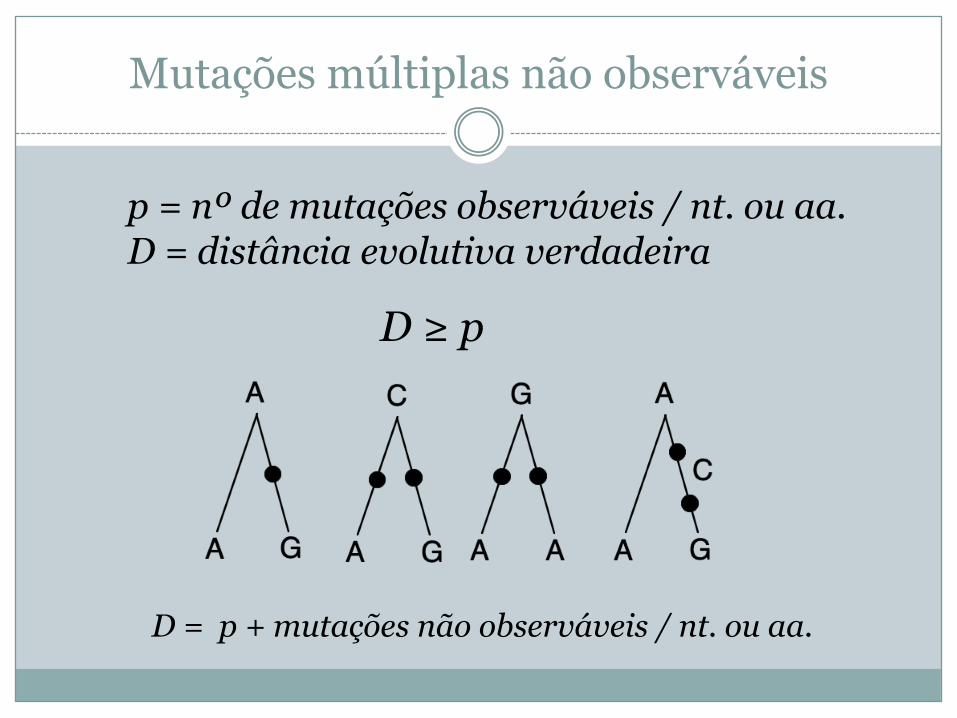
Mutações múltiplas não observáveis
p = nº de mutações observáveis / nt. ou aa. D = distância evolutiva verdadeira
D ≥ p
D = p + mutações não observáveis / nt. ou aa.
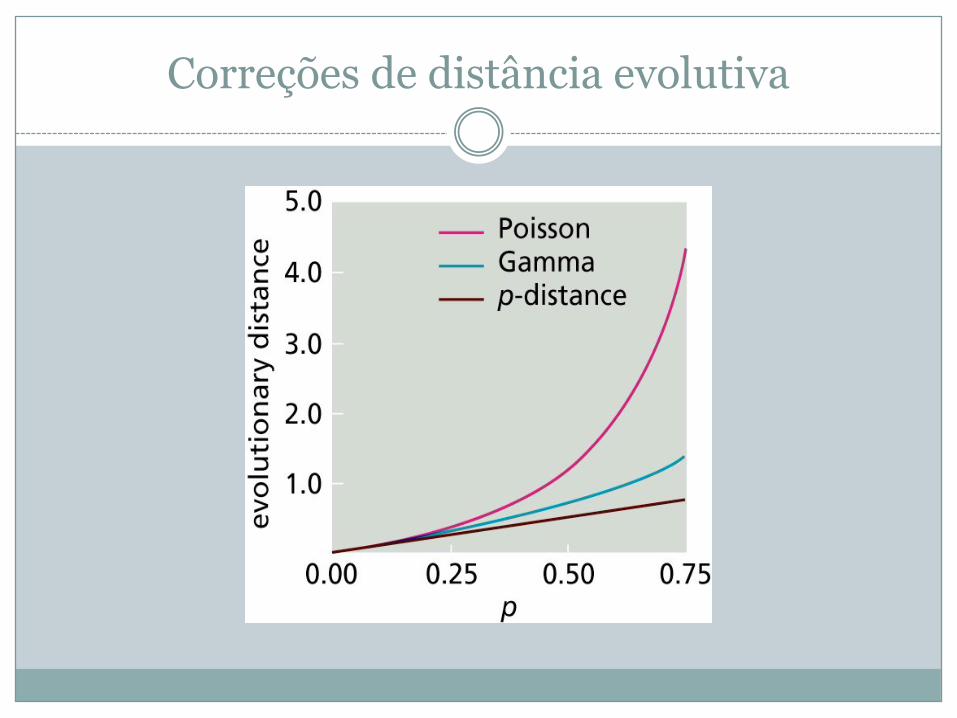
Correções de distância evolutiva
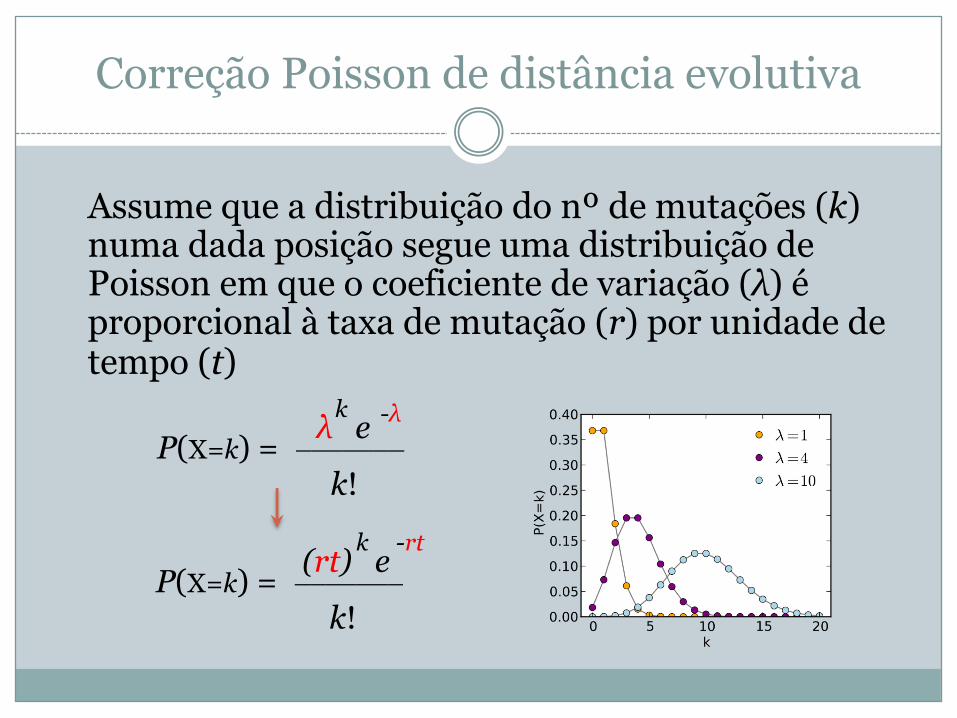
Correção Poisson de distância evolutiva
Assume que a distribuição do nº de mutações (k) numa dada posição segue uma distribuição de Poisson em que o coeficiente de variação (λ) é proporcional à taxa de mutação (r) por unidade de tempo (t)
P(X=k) = _______ λ e
k!
k -λ
P(X=k) = _______ (rt) e
k!
k -rt

Correção Poisson de distância evolutiva
P(X=0) = _______ (rt) e 0!
0 -rt
Probabilidade de não ocorrer nenhuma mutação (k = 0) numa dada posição de uma sequência:
= e-rt

Correção Poisson de distância evolutiva
Probabilidade de não ocorrer nenhuma mutação (k = 0) numa dada posição em duas sequências:
P(X=0) ×P(X=0) = e-2rt
d = 2rt
d = distância evolutiva = média do nº de mutações que ocorreram por posição

Correção Poisson de distância evolutiva
p = fração de posições que sofreram mutações1 – p = fração de posições idênticas
e-2rt1 – p = e-d
=
dp = ln(1 – p)
dp = distância Poisson corrigida
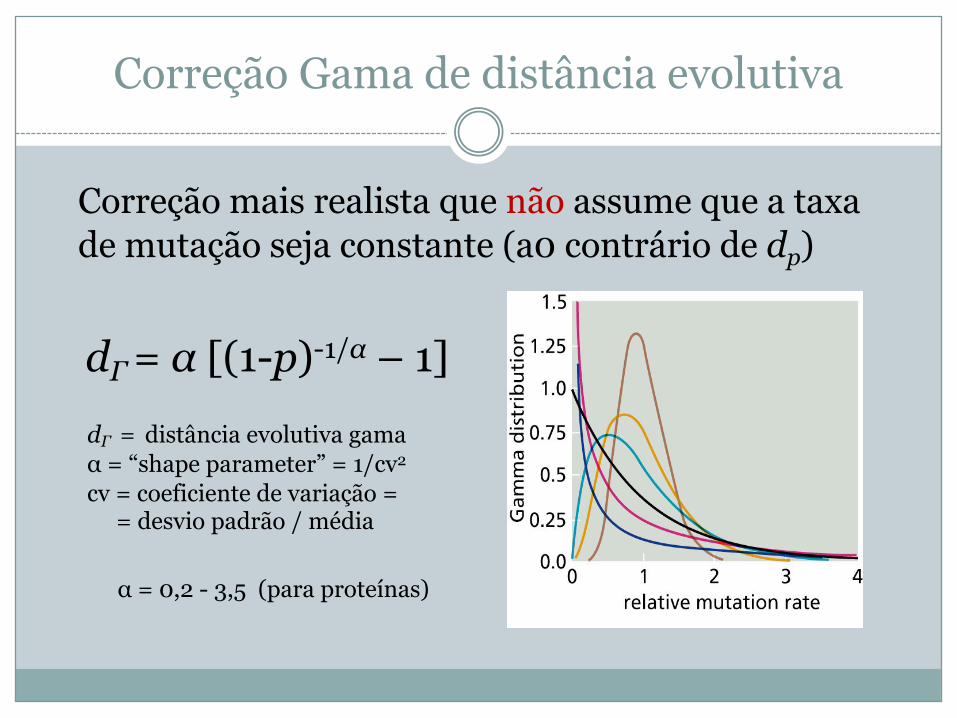
Correção Gama de distância evolutiva
Correção mais realista que não assume que a taxa de mutação seja constante (a0 contrário de dp)
dΓ= α [(1-p)-1/α – 1]dΓ = distância evolutiva gamaα = “shape parameter” = 1/cv2
cv = coeficiente de variação = = desvio padrão / média
α = 0,2 - 3,5 (para proteínas)

Aspetos irrealistas da Correção Gama
� Não há certeza que a distribuição gama seja a correta
� A correção não tem em conta que há uma aparente correlação entre taxas de mutação entre locais diferentes numa sequência
� As taxas de mutação não são constantes ao longo de evolução em diferentes ramos de uma árvore

Modelo Jukes-Cantor (JC)
� Nenhum das correções anteriores tem em conta a informação bioquímica (nucleótidos, a.a.)
� Modelo simples que se aplica a sequências nucleotídicas
� Assume que as taxas de mutação são idênticas para todos os locais da sequência e que uma transversão é tão provável como uma transição
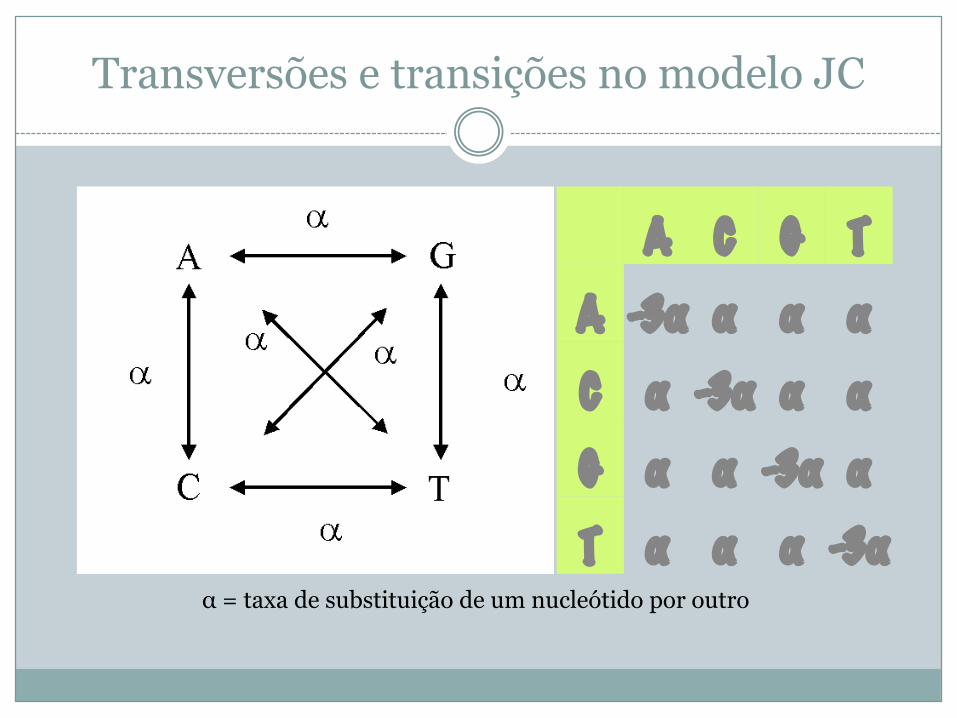
Transversões e transições no modelo JC
α = taxa de substituição de um nucleótido por outro
A C G TA -3α α α αC α -3α α αG α α -3α αT α α α -3α
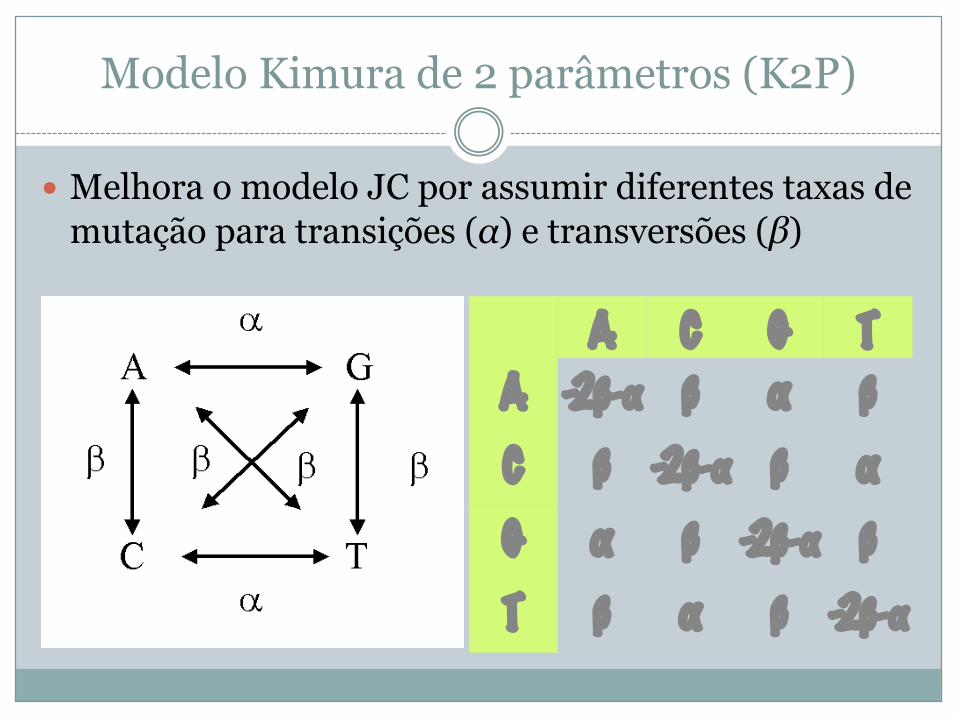
Modelo Kimura de 2 parâmetros (K2P)
� Melhora o modelo JC por assumir diferentes taxas de mutação para transições (α) e transversões (β)
A C G TA -2β-α β α βC β -2β-α β αG α β -2β-α βT β α β -2β-α
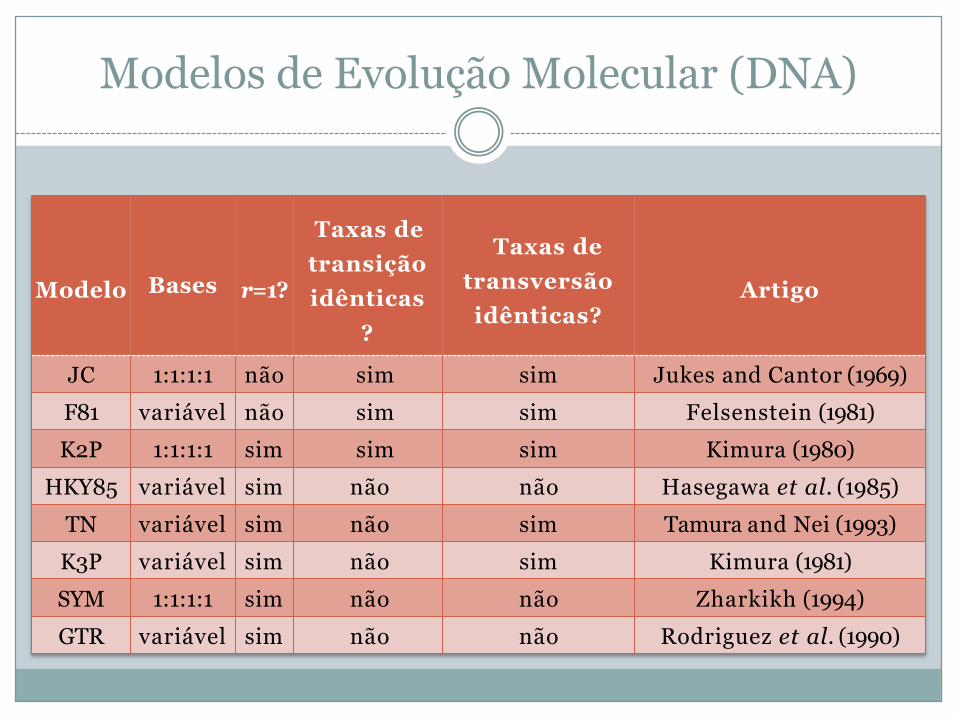
Modelos de Evolução Molecular (DNA)
Modelo Bases r=1?
Taxas de transição idênticas
?
Taxas de transversão idênticas?
Artigo
JC 1:1:1:1 não sim sim Jukes and Cantor (1969)F81 variável não sim sim Felsenstein (1981)K2P 1:1:1:1 sim sim sim Kimura (1980)
HKY85 variável sim não não Hasegawa et al. (1985)TN variável sim não sim Tamura and Nei (1993)K3P variável sim não sim Kimura (1981)SYM 1:1:1:1 sim não não Zharkikh (1994)GTR variável sim não não Rodriguez et al. (1990)
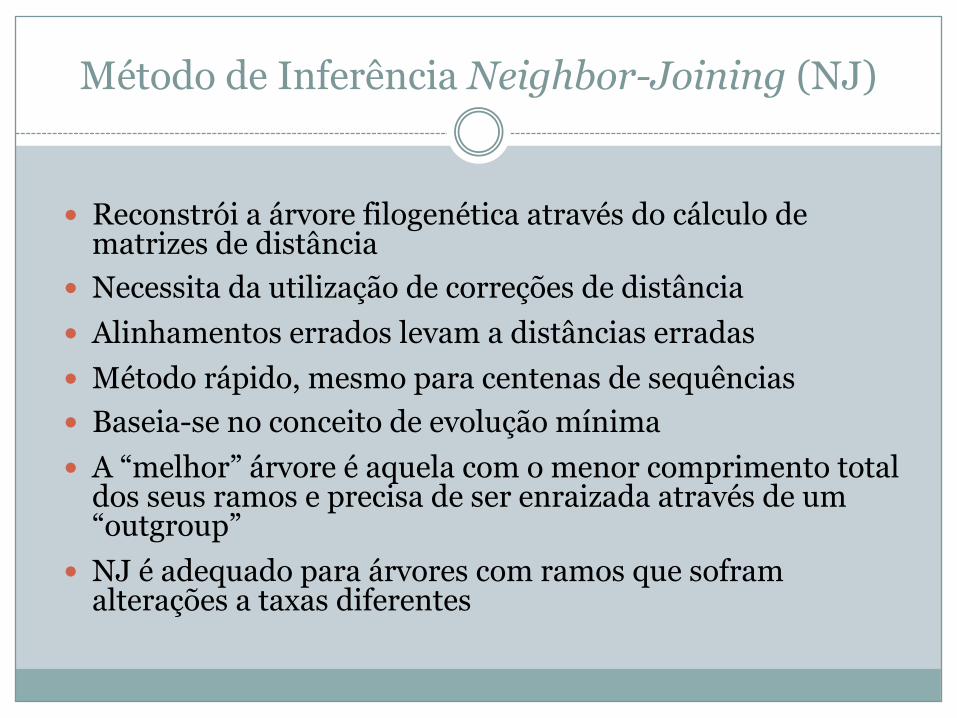
Método de Inferência Neighbor-Joining (NJ)
� Reconstrói a árvore filogenética através do cálculo de matrizes de distância
� Necessita da utilização de correções de distância� Alinhamentos errados levam a distâncias erradas� Método rápido, mesmo para centenas de sequências� Baseia-se no conceito de evolução mínima� A “melhor” árvore é aquela com o menor comprimento total
dos seus ramos e precisa de ser enraizada através de um “outgroup”
� NJ é adequado para árvores com ramos que sofram alterações a taxas diferentes
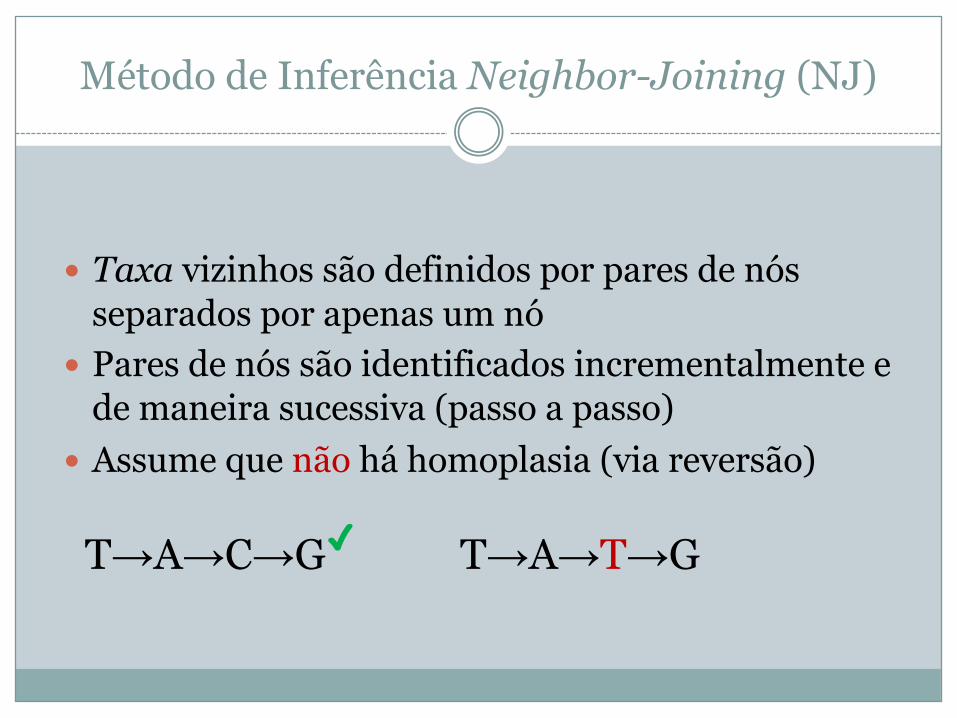
Método de Inferência Neighbor-Joining (NJ)
� Taxa vizinhos são definidos por pares de nós separados por apenas um nó
� Pares de nós são identificados incrementalmente e de maneira sucessiva (passo a passo)
� Assume que não há homoplasia (via reversão)
T→A→C→G✓ T→A→T→G
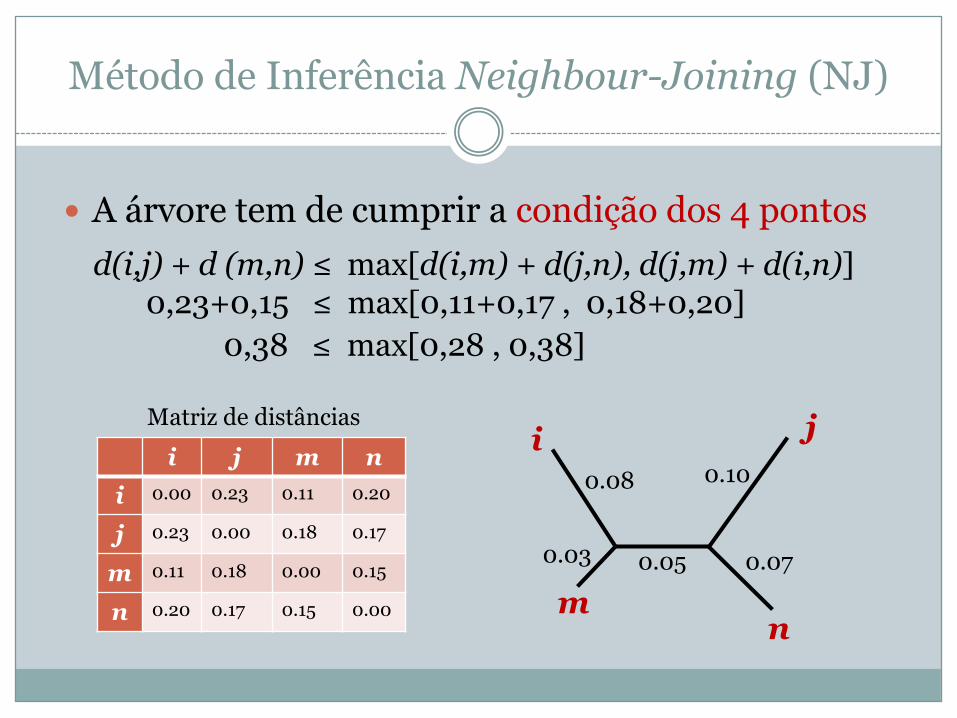
Método de Inferência Neighbour-Joining (NJ)
� A árvore tem de cumprir a condição dos 4 pontosd(i,j) + d (m,n) ≤ max[d(i,m) + d(j,n), d(j,m) + d(i,n)]
i j m ni 0.00 0.23 0.11 0.20
j 0.23 0.00 0.18 0.17
m 0.11 0.18 0.00 0.15
n 0.20 0.17 0.15 0.00
i j
mn
0.03
0.08
0.070.05
0.10
Matriz de distâncias
0,23+0,15 ≤ max[0,11+0,17 , 0,18+0,20] 0,38 ≤ max[0,28 , 0,38]
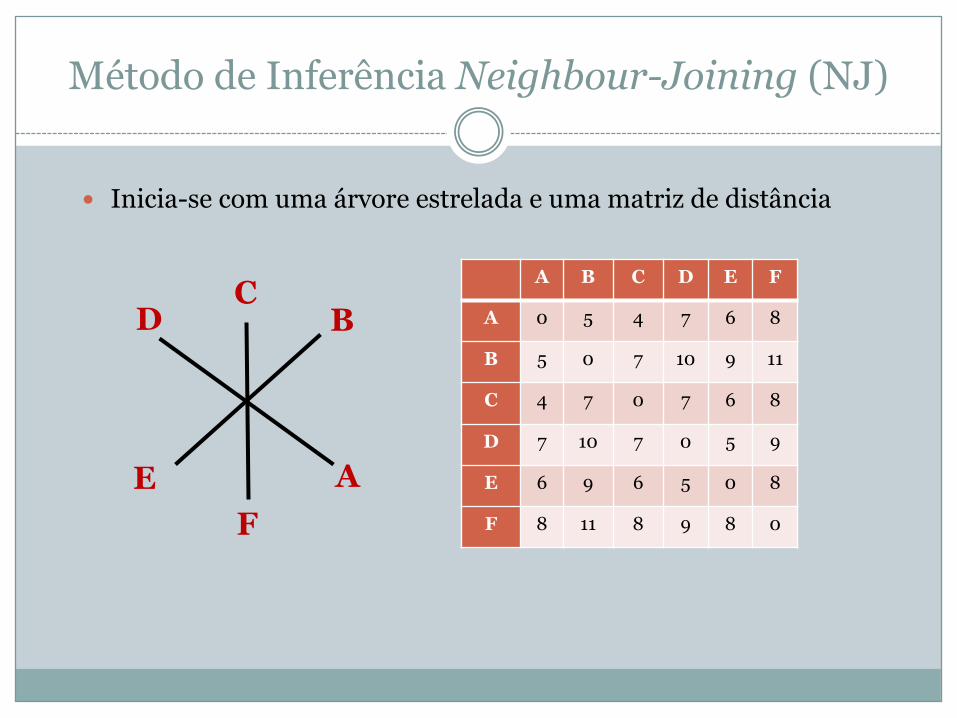
Método de Inferência Neighbour-Joining (NJ)
� Inicia-se com uma árvore estrelada e uma matriz de distância
A B C D E F
A 0 5 4 7 6 8
B 5 0 7 10 9 11
C 4 7 0 7 6 8
D 7 10 7 0 5 9
E 6 9 6 5 0 8
F 8 11 8 9 8 0
A
BC
D
EF
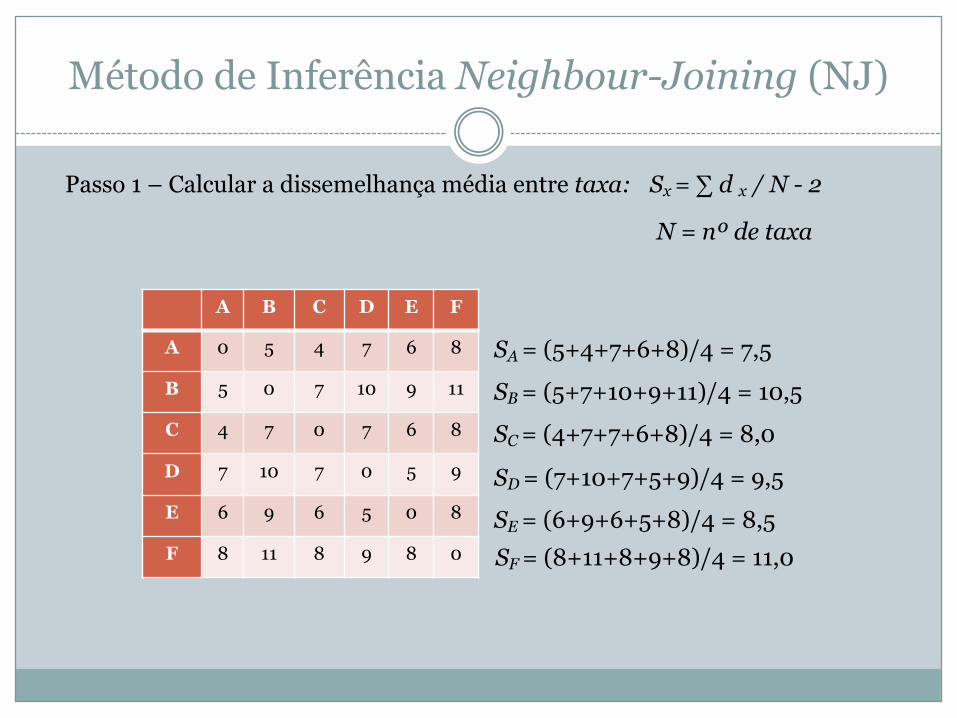
Método de Inferência Neighbour-Joining (NJ)
A B C D E F
A 0 5 4 7 6 8
B 5 0 7 10 9 11
C 4 7 0 7 6 8
D 7 10 7 0 5 9
E 6 9 6 5 0 8
F 8 11 8 9 8 0
Passo 1 – Calcular a dissemelhança média entre taxa: Sx = ∑ d x / N - 2
N = nº de taxa
SA = (5+4+7+6+8)/4 = 7,5
SB = (5+7+10+9+11)/4 = 10,5
SC = (4+7+7+6+8)/4 = 8,0
SD = (7+10+7+5+9)/4 = 9,5
SE = (6+9+6+5+8)/4 = 8,5 SF = (8+11+8+9+8)/4 = 11,0
Método de Inferência Neighbour-Joining (NJ)
dij A B C D E F
A 0 5 4 7 6 8
B 5 0 7 10 9 11
C 4 7 0 7 6 8
D 7 10 7 0 5 9
E 6 9 6 5 0 8
F 8 11 8 9 8 0
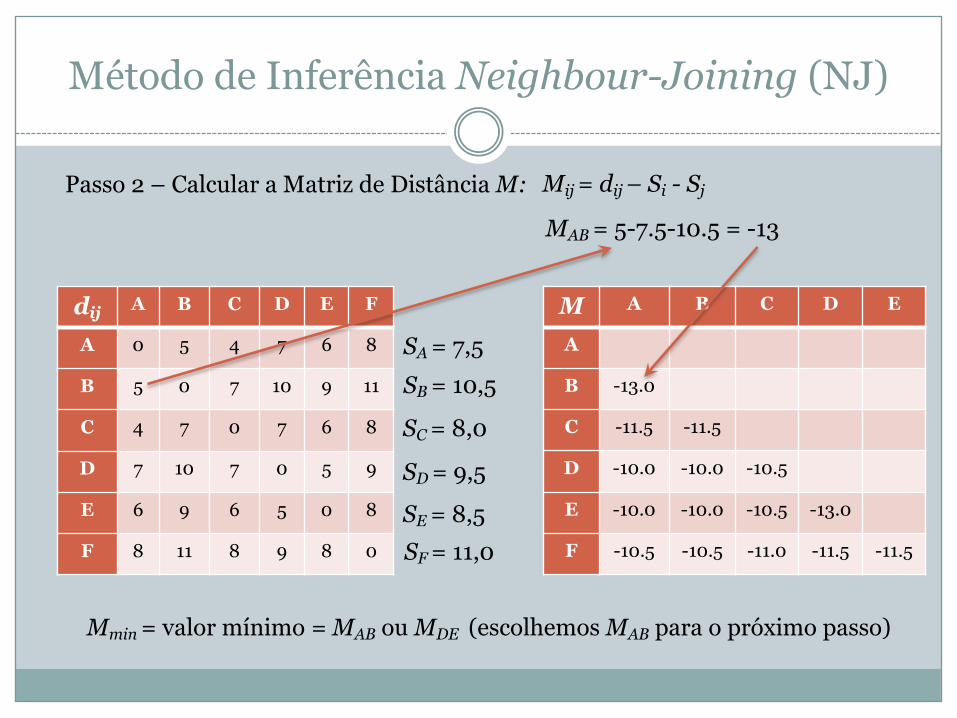
Passo 2 – Calcular a Matriz de Distância M: Mij = dij – Si - Sj
SA = 7,5 SB = 10,5
MAB = 5-7.5-10.5 = -13
M A B C D E
A
B -13.0
C -11.5 -11.5
D -10.0 -10.0 -10.5
E -10.0 -10.0 -10.5 -13.0
F -10.5 -10.5 -11.0 -11.5 -11.5
SC = 8,0
SD = 9,5
SE = 8,5 SF = 11,0
Mmin = valor mínimo = MAB ou MDE (escolhemos MAB para o próximo passo)
Método de Inferência Neighbour-Joining (NJ)
dij A B C D E F
A 0 5 4 7 6 8
B 5 0 7 10 9 11
C 4 7 0 7 6 8
D 7 10 7 0 5 9
E 6 9 6 5 0 8
F 8 11 8 9 8 0
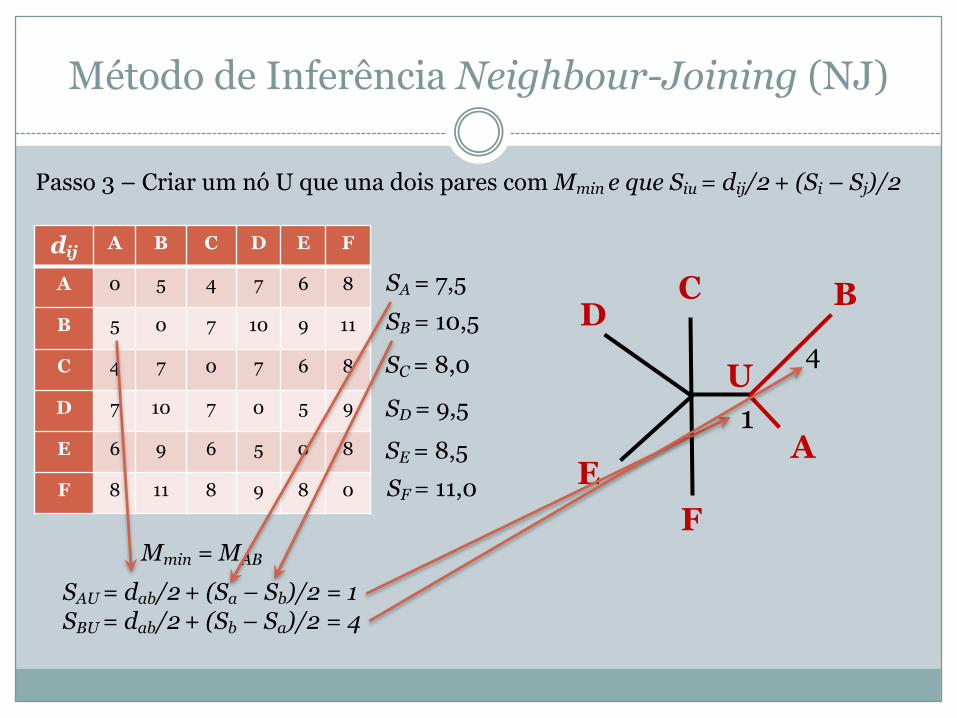
Passo 3 – Criar um nó U que una dois pares com Mmin e que Siu = dij/2 + (Si – Sj)/2
SA = 7,5 SB = 10,5
SC = 8,0
SD = 9,5
SE = 8,5 SF = 11,0
Mmin = MAB
SAU = dab/2 + (Sa – Sb)/2 = 1SBU = dab/2 + (Sb – Sa)/2 = 4
A
BCD
EF
U1
4
Método de Inferência Neighbour-Joining (NJ)
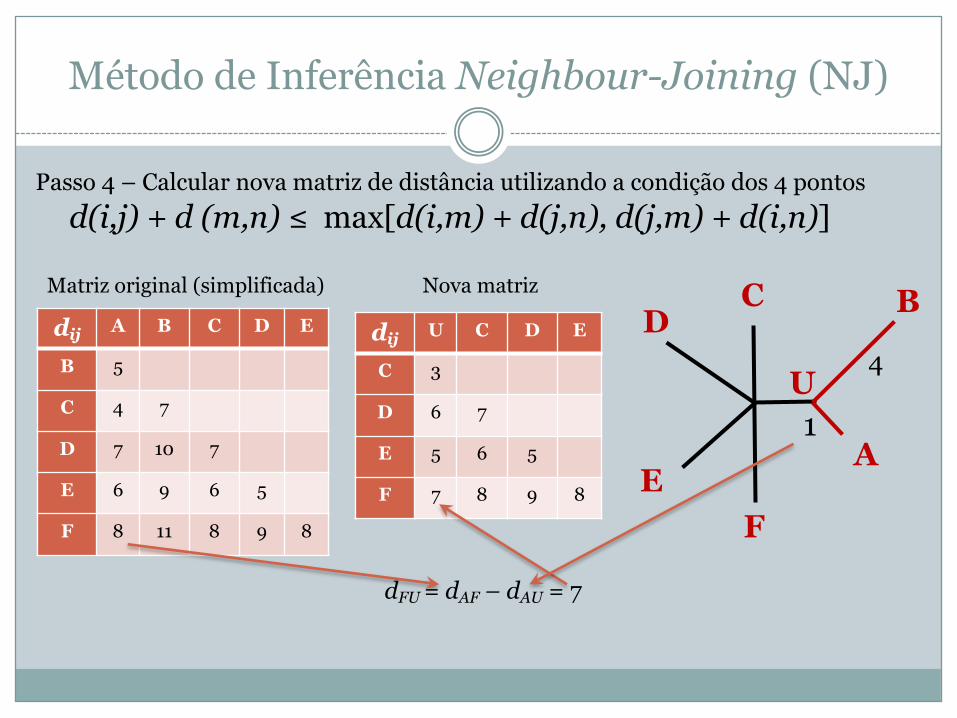
Passo 4 – Calcular nova matriz de distância utilizando a condição dos 4 pontosd(i,j) + d (m,n) ≤ max[d(i,m) + d(j,n), d(j,m) + d(i,n)]
A
BCD
EF
U1
4dij A B C D E
B 5
C 4 7
D 7 10 7
E 6 9 6 5
F 8 11 8 9 8
dij U C D E
C 3
D 6 7
E 5 6 5
F 7 8 9 8
Matriz original (simplificada) Nova matriz
dFU = dAF – dAU = 7
Métodos de reconstrução filogenética
� Métodos de Distância� Parcimónia� Máxima Verosimilhança� Métodos Bayesianos
Existem quatro famílias de métodos principais:
Métodos baseados em caracteres(procuram a “melhor” árvore)

Métodos de inferência baseadas em caracteres
� Não calculam distâncias entre duas sequências� Todos os locais de todas as sequências são usados
para procurar a “melhor” árvore� A procura da melhor árvore é feita pelo método
“Hill Climbing” (Parcimónia, Verosimilhança Máxima) ou por Markov Chain Monte Carlo (Métodos Bayesianos)
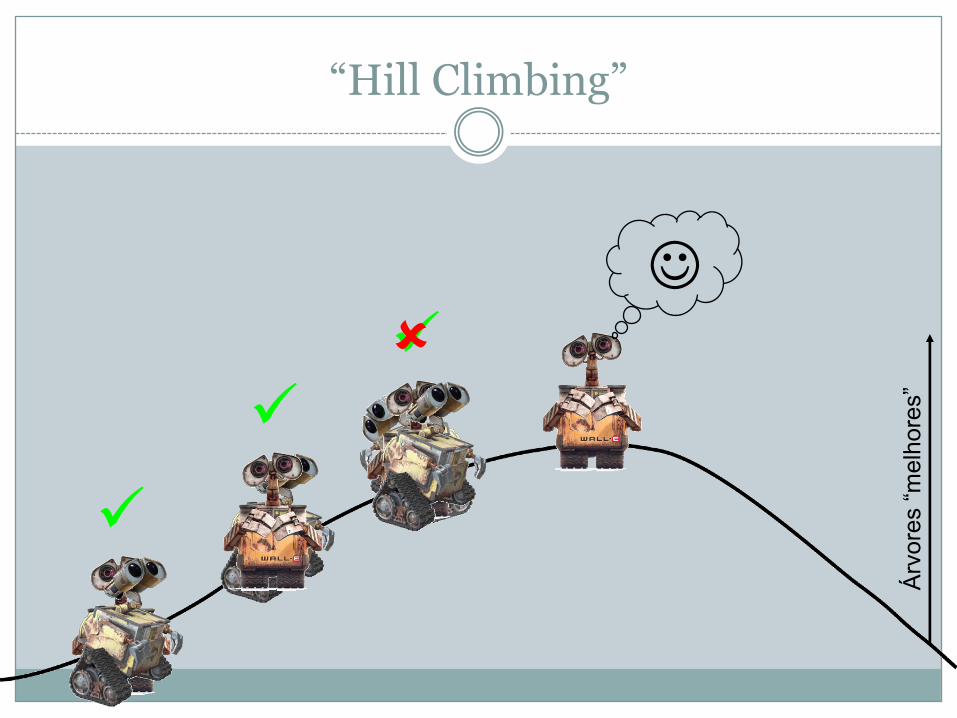
“Hill Climbing”
ü
üüû
J
Árvores“melhores”

“Hill Climbing”
� Todas as árvores possíveis (“tree space”) são como se fossem um monte que é preciso escalar
� As “melhores” árvores estão no pico do monte� O programa de inferência é como um robot que está
programado a aceitar apenas instruções para subir e rejeitar instruções para descer
� Este método tem a desvantagem do programa poder encontrar um pico local que poderá não ser o pico mais alto do monte
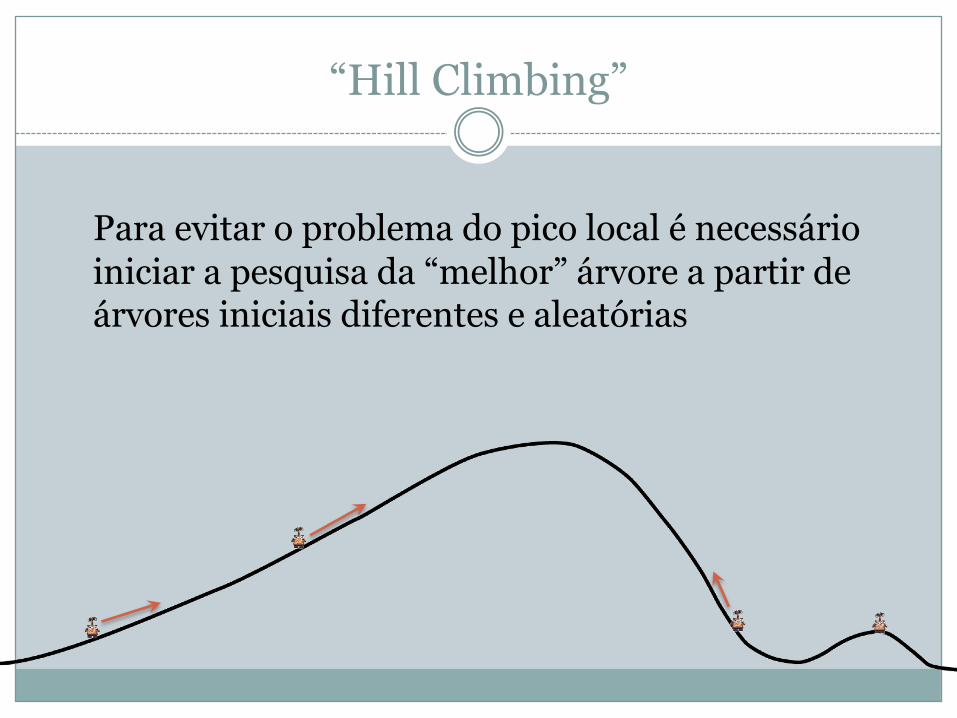
“Hill Climbing”
Para evitar o problema do pico local é necessário iniciar a pesquisa da “melhor” árvore a partir de árvores iniciais diferentes e aleatórias
Métodos de reconstrução filogenética
� Métodos de Distância� Parcimónia� Máxima Verosimilhança� Métodos Bayesianos
Existem quatro famílias de métodos principais:
Métodos baseados em caracteres(procuram a “melhor” árvore)

Métodos de Inferência por Parcimónia
� Procura a melhor árvore por “Hill Climbing”
� O critério de otimização é a parcimónia máxima
� Procura a árvore com o menor score de parcimónia (com a topologia construída com o menor número de alterações de caracteres)
� Método adequado para construir árvores a partir de conjuntos de dados com elevado grau de semelhança e cujos locais são alterados a taxas de mutação diferentes
� Assume que os caracteres são independentes entre si
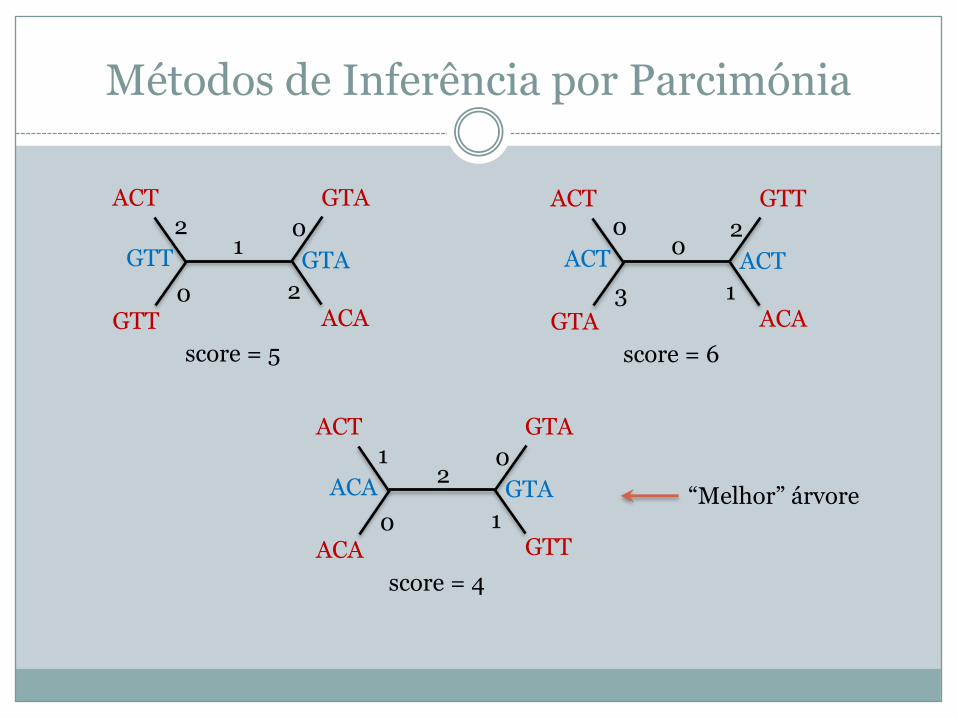
Métodos de Inferência por Parcimónia
GTAACT
GTT ACA
GTT GTA2
0
10
2
score = 5
GTTACT
GTA ACA
ACT ACT0
3
02
1
score = 6
GTAACT
ACA GTT
ACA GTA1
0
20
1
score = 4
“Melhor” árvore
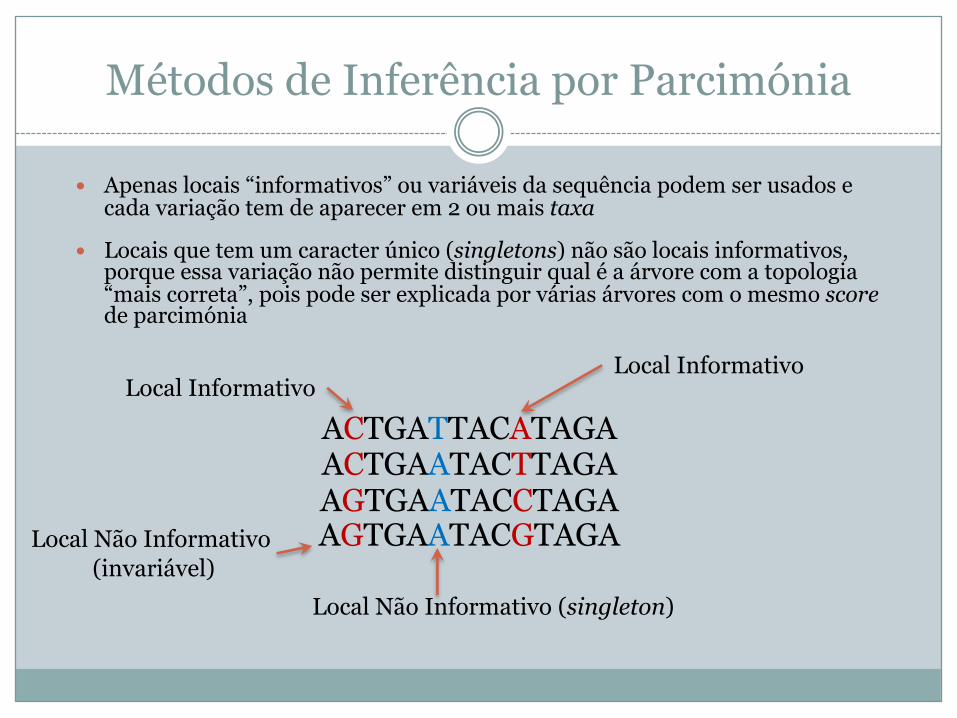
Métodos de Inferência por Parcimónia
� Apenas locais “informativos” ou variáveis da sequência podem ser usados e cada variação tem de aparecer em 2 ou mais taxa
� Locais que tem um caracter único (singletons) não são locais informativos, porque essa variação não permite distinguir qual é a árvore com a topologia “mais correta”, pois pode ser explicada por várias árvores com o mesmo scorede parcimónia
ACTGATTACATAGAACTGAATACTTAGAAGTGAATACCTAGAAGTGAATACGTAGA
Local Informativo
Local Não Informativo (singleton)
Local Informativo
Local Não Informativo(invariável)

Métodos de Inferência por Parcimónia
� Árvores obtidas por métodos de parcimónia máxima só são significativas se os alinhamentos possuírem vários locais informativos
� Se a homoplasia por substituições paralelas ou reversão for alta, as árvores obtidas por este método não são fiáveis
� Parcimónia máxima não é estatisticamente consistente
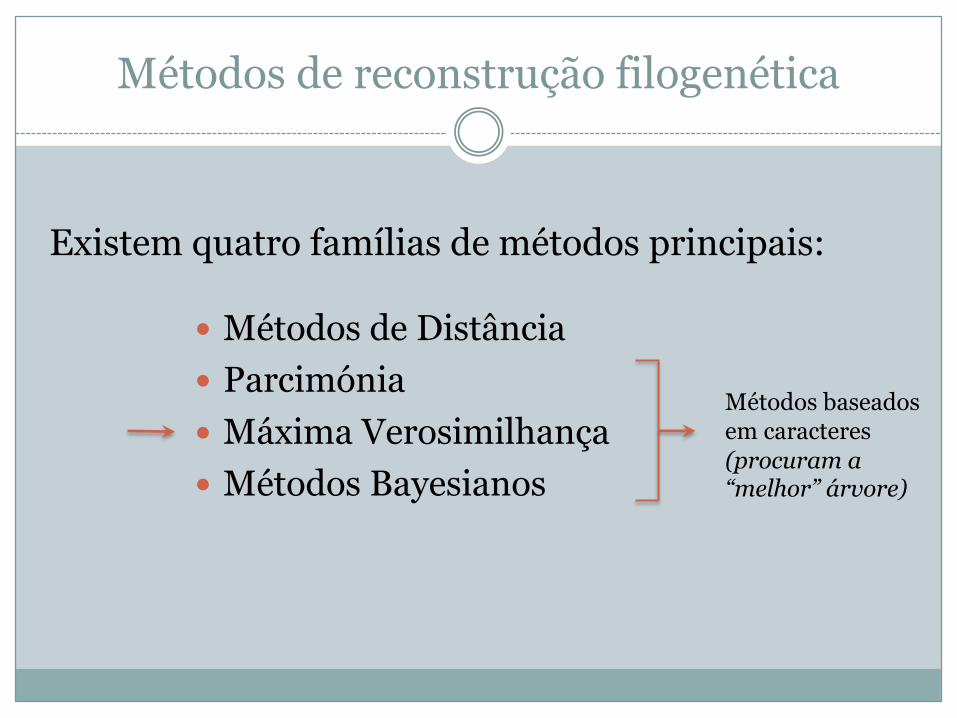
Métodos de reconstrução filogenética
� Métodos de Distância� Parcimónia� Máxima Verosimilhança� Métodos Bayesianos
Existem quatro famílias de métodos principais:
Métodos baseados em caracteres(procuram a “melhor” árvore)

Métodos de Inferência por Verosimilhança Máxima
Verosimilhança Máxima = Maximum Likelihood (ML)
L = p(H|D)
Verosimilhança (L) corresponde a uma probabilidade (p) de uma hipótese (H) explicar um conjunto de dados (D)

Métodos de Inferência por Verosimilhança Máxima
Exemplo de cálculo de L = p(H|D):
� Suponha que está a contar o número de vezes que sai “cara” quando um árbitro atira uma moeda ao ar para saber quem começa um jogo
� Suponha que o árbitro obtém “cara” duas vezes
� Para explicar estes resultados (dados) pode propor duas hipóteses:
Ø Hipótese H1: a moeda é normal: p(H1|cara) = p(H1|coroa) = 0,5
Ø Hipótese H2: a moeda é viciada:p(H2|cara) = 0,8; p(H2|coroa) = 0,2
Qual é a verosimilhança de H1 ou H2?

Métodos de Inferência por Verosimilhança Máxima
L = p(H|D) em que D = «sair duas vezes “cara”» depende da hipótese:
� Hipótese H1: L(H1|D) = 0,5 x 0,5 = 0,25
� Hipótese H2: L(H2|D) = 0,8 x 0,8 = 0,64
Ø Neste caso, a Hipótese H2 parece ser a mais verosímil.
Ø Porém, se atirássemos a mesma moeda 20 vezes ao ar e se obtivéssemos “cara” 8 vezes, a resposta seria a mesma?
Qual é a verosimilhança de H1 ou H2?

Métodos de Inferência por Verosimilhança Máxima
� Há várias combinações possíveis para obter o mesmo conjunto de dados Cara (C )= 8x, Coroa (R) = 12x
Combinação 1: CRCCC RRRCR CRRRC RRRCR ouCombinação 2: CCCCC CCCRR RRRRR RRRRR ... ou ainda mais 125.968 combinações possíveis.
� Para calcular o número de combinações possíveis que poderiam explicar os mesmos dados e assim calcular as respetivas probabilidades:
n!nCk = ______________k!(n-k)!
n = 20, k = 8
=20 × 19 × 18 ... 2 × 1_____
(8×7× ... ×1) (12×11×... ×1) = 125.970

Métodos de Inferência por Verosimilhança Máxima
L = p(H|D) em que D = «sair oito vezes “cara” em 20 tentativas»:
LH1 = p(H1|D) = 125.970 × (0,5)8 × (0,5)12 = 0,120134353638
LH2 = p(H2|D) = 125.970 × (0,8)8 × (0,2)12 = 0,0000865659248443
• Probabilidades tão baixas causam problemas em bioinformática devido ao conhecido problema de “underflow” na conversão de números decimais em números binários e vice-versa, o que causa erros de arredondamento
• Assim, prefere-se converter estes números a logaritmos naturais:LH1 = ln(0,120134353638) = -2,119
LH2 = ln(0,0000865659248443) = -9,355
Neste caso, H1 é mais verosímil por ter um valor de verosimilhança maior
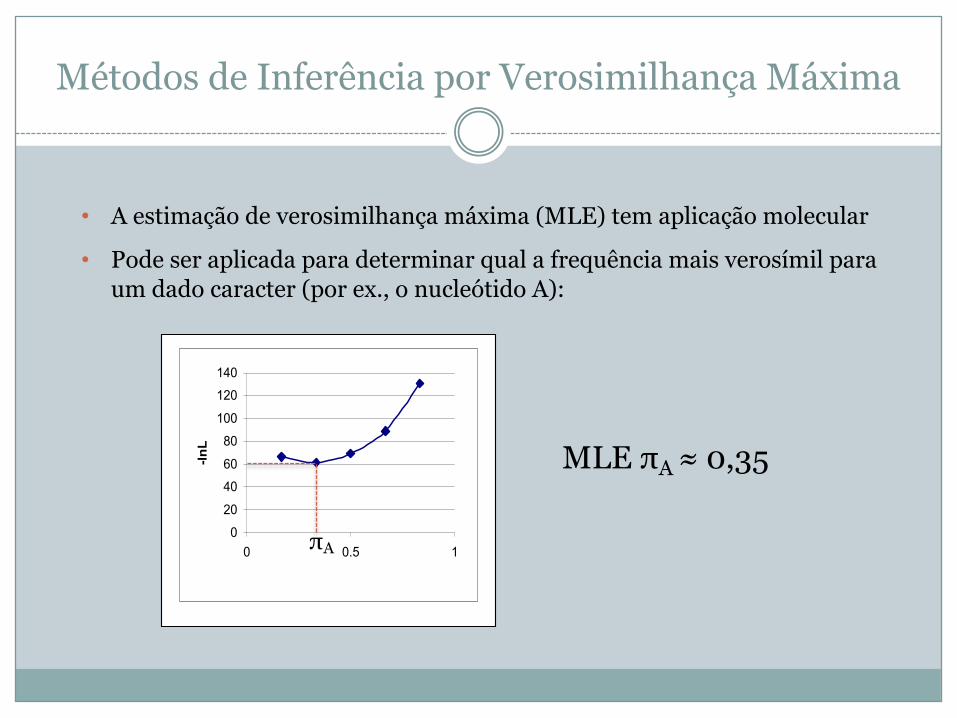
Métodos de Inferência por Verosimilhança Máxima
• A estimação de verosimilhança máxima (MLE) tem aplicação molecular
• Pode ser aplicada para determinar qual a frequência mais verosímil para um dado caracter (por ex., o nucleótido A):
MLE πA ≈ 0,35
020406080100120140
0 0.5 1
p values
-lnL
πA
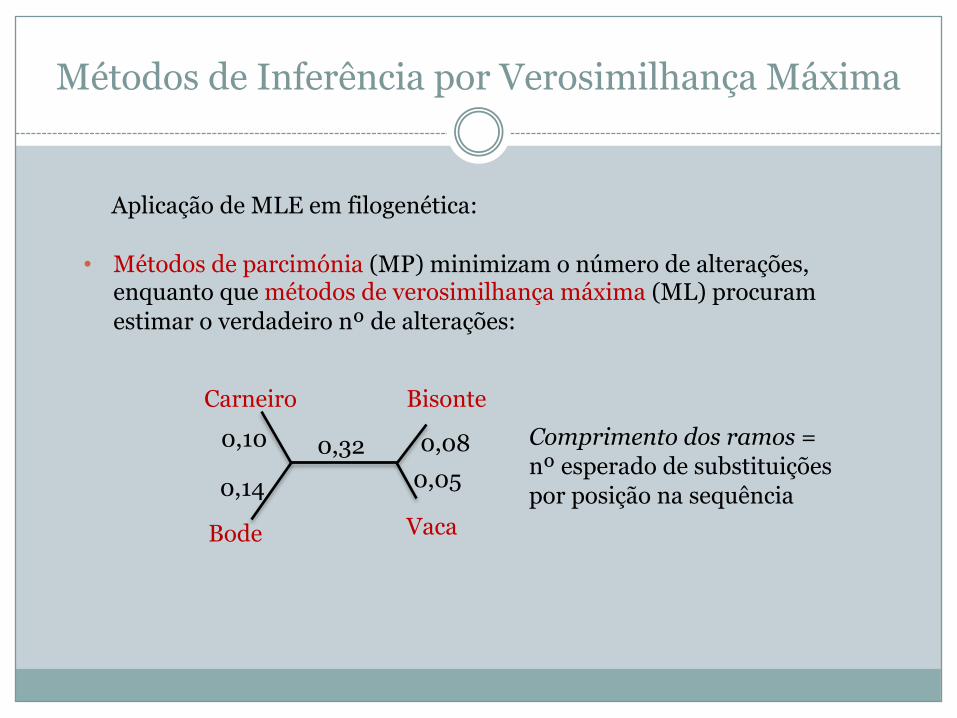
Métodos de Inferência por Verosimilhança Máxima
• Métodos de parcimónia (MP) minimizam o número de alterações, enquanto que métodos de verosimilhança máxima (ML) procuram estimar o verdadeiro nº de alterações:
Aplicação de MLE em filogenética:
Carneiro
Bode Vaca
Bisonte0,10
0,14
0,32 0,080,05
Comprimento dos ramos =nº esperado de substituições por posição na sequência
Métodos de Inferência por Verosimilhança Máxima
• MP não têm em conta a informação dada de como certos locais na sequência evoluem, enquanto que ML assume que há mecanismos comuns de evolução cuja informação pode ser obtida de outros locais
• ML assume, porém, que estes locais evoluem independentemente
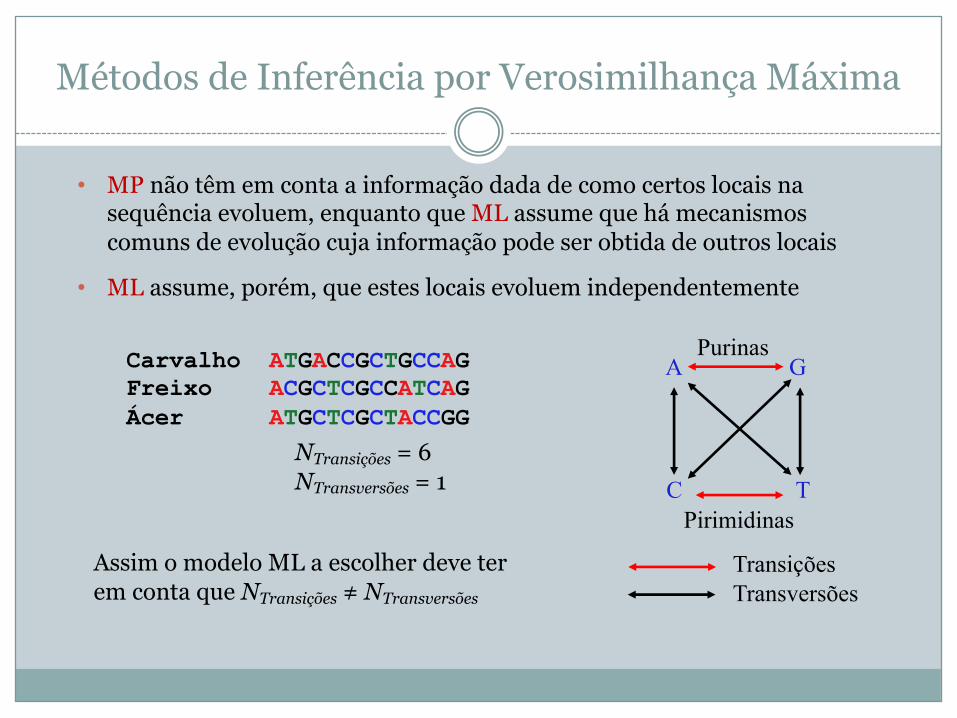
Carvalho ATGACCGCTGCCAG Freixo ACGCTCGCCATCAGÁcer ATGCTCGCTACCGG
TransiçõesTransversões
A G
C TPirimidinas
Purinas
NTransições = 6NTransversões = 1
Assim o modelo ML a escolher deve ter em conta que NTransições ≠ NTransversões

Métodos de Inferência por Verosimilhança Máxima
• ML estima p(D|A) = a probabilidade p de observarmos os dados D tendo em conta a árvore A; ao passo que:
• Métodos Bayesianos estimam p(A|D) = a probabilidade pde obtermos a árvore A tendo em conta os dados D
• ML é um método probabilístico para encontrar a árvore mais verosímil e, assim, determinar qual a hipótese de sequências ancestrais (não observáveis) mais provável
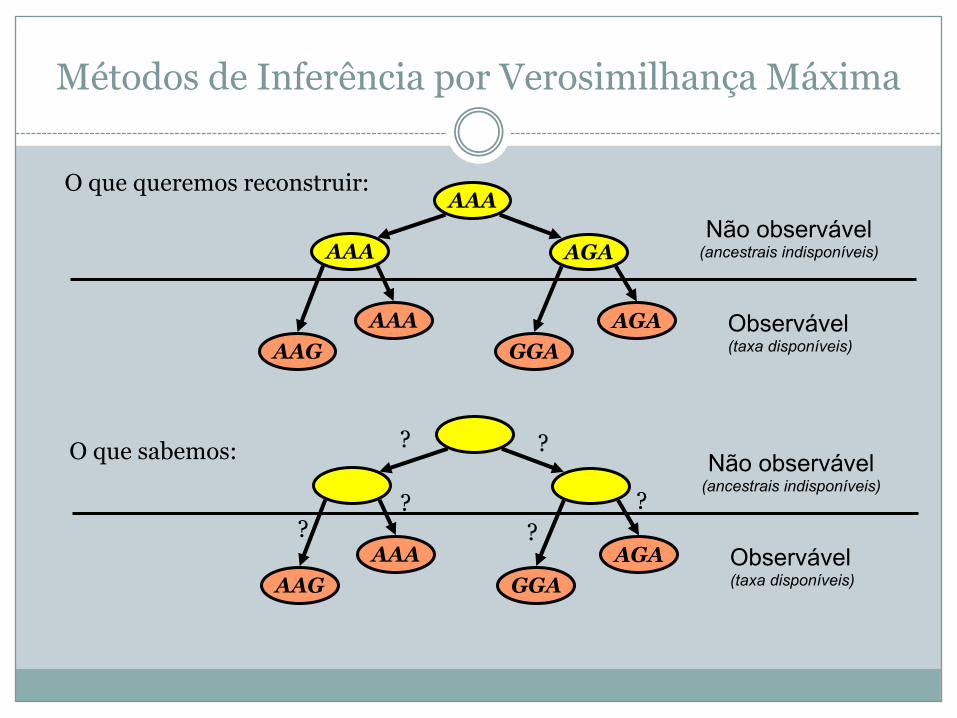
Métodos de Inferência por Verosimilhança Máxima
AGAGGA
AAAAAG
AAA AGA
AAA
Observável(taxa disponíveis)
Não observável(ancestrais indisponíveis)
O que queremos reconstruir:
AGAGGA
AAAAAG
Observável(taxa disponíveis)
Não observável(ancestrais indisponíveis)
O que sabemos: ? ?
??
??

Métodos de Inferência por Verosimilhança Máxima
Para estimar por ML (MLE) é necessário:
• um conjunto de observações (x1, x2, ..., xn) independentes e com distribuição idêntica, mas com uma distribuição de dados com uma função de densidades de probabilidade, f0(x), desconhecida
• um conjunto de parâmetros ! = {!1, !2, ... , !k}, de modo a que:
• f0 = f (xi |!)
• embora os dados possam ter distribuição normal, binomial (ex., resultado de atirar uma moeda ao ar), Poisson, etc.
Objetivo:
• obter o MLE que maximize o valor de verosimilhança do conjunto de parâmetros !, L(!| x1, x2, ..., xn)

Métodos de Inferência por Verosimilhança Máxima
Uma vez que se assume que as observações (x1, x2, ..., xn) são independentes:
• f0 = f (xi |!) = f (x1, x2, ..., xn |!) = "n f (xi | !)i=1
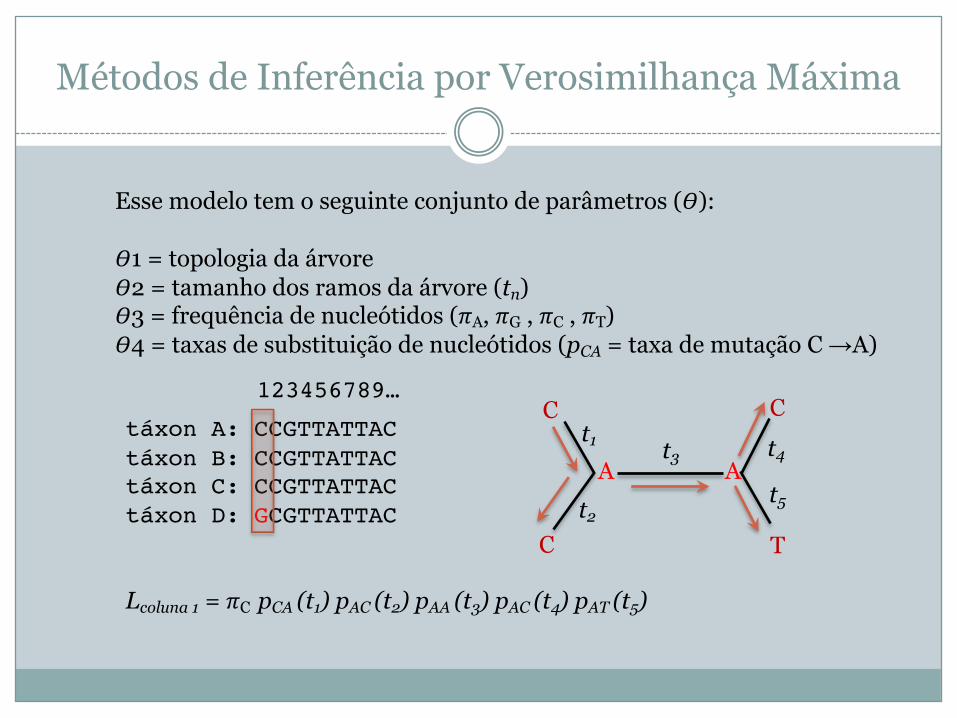
Como aplicar estes conceitos a filogenia? Suponhamos que que queremos estudar as seguintes sequências:
táxon A: CCGTTATTACtáxon B: CCGTTATTACtáxon C: CCGTTATTACtáxon D: GCGTTATTAC
Modelo probabilístico para a determinar qual a hipótese que melhor explique a evolução de uma sequência ancestral para as 4 sequências dos taxa atuais
Métodos de Inferência por Verosimilhança Máxima
táxon A: CCGTTATTACtáxon B: CCGTTATTACtáxon C: CCGTTATTACtáxon D: GCGTTATTAC
Esse modelo tem o seguinte conjunto de parâmetros (!):
!1 = topologia da árvore!2 = tamanho dos ramos da árvore (tn)!3 = frequência de nucleótidos ("A, "G , "C , "T)!4 = taxas de substituição de nucleótidos (pCA = taxa de mutação C →A)
C
C T
Ct1
A At2
t3 t4
t5
Lcoluna 1 = "C pCA (t1) pAC (t2) pAA (t3) pAC (t4) pAT (t5)
123456789…
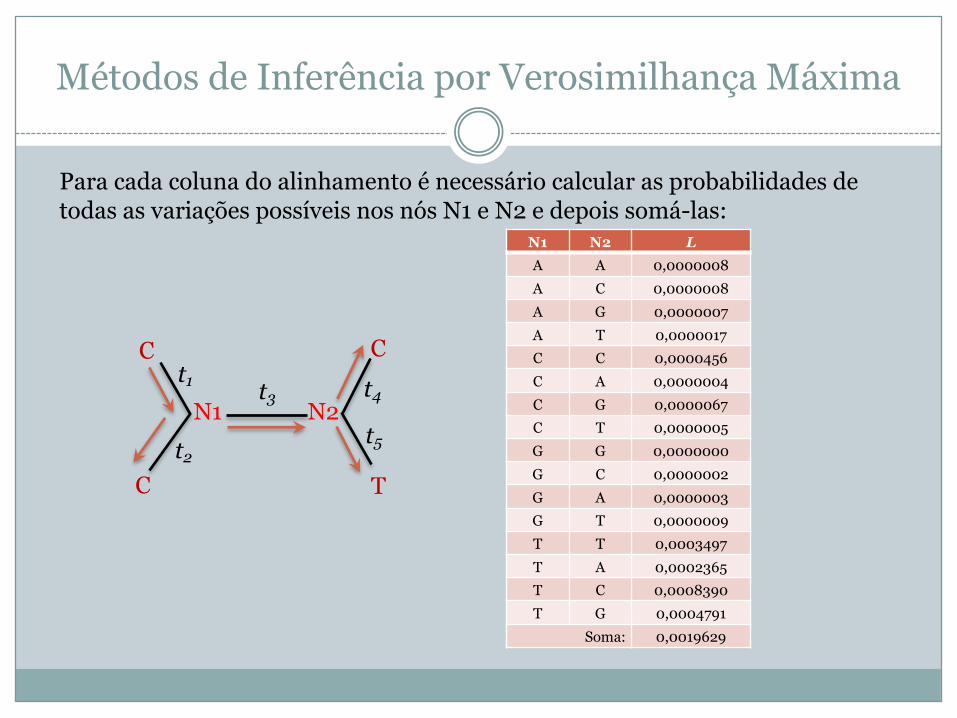
Métodos de Inferência por Verosimilhança Máxima
C
C T
Ct1
N1 N2t2
t3 t4
t5
Para cada coluna do alinhamento é necessário calcular as probabilidades de todas as variações possíveis nos nós N1 e N2 e depois somá-las:
N1 N2 LA A 0,0000008A C 0,0000008A G 0,0000007A T 0,0000017C C 0,0000456C A 0,0000004C G 0,0000067C T 0,0000005G G 0,0000000G C 0,0000002G A 0,0000003G T 0,0000009T T 0,0003497T A 0,0002365T C 0,0008390T G 0,0004791
Soma: 0,0019629

Métodos de Inferência por Verosimilhança Máxima
Mas como os computadores não são bons para fazer cálculos com números tão pequenos, aqueles valores são transformados em logaritmos naturais:
L = L1 + L2 + ... + Ln = !n f (xi | ") = !n Lii=1 i=1
Para cada coluna do alinhamento é necessário calcular as probabilidades de todas as variações possíveis nos nós N1 e N2 e depois somá-las:
ln(L) = ln(L1) + ln(L2) + ... + ln(Ln) = !n f (xi | ") = !n ln(Li)i=1 i=1
Estes valores são chamados em inglês: log-likelihood

Métodos de Inferência por Verosimilhança Máxima
• Os métodos de inferência por ML são considerados dos mais fiáveis
• Necessitam, em geral, de outros programas para definir qual o modelo a usar para um dado conjunto de sequências (DNA: Jukes-Cantor, Kimura-2, etc...; Proteína: Dayhoff, JTT, WAG, etc.)
• Em phylogeny.fr, o modelo que usarão será o WAG (Whelan and Goldman, 2001), uma vez que este é o modelo que é considerado o mais fiável atualmente para filogenia de sequências de aminoácidos
• Porém, são computacionalmente caros
• Por isso, não escalam bem para conjuntos grandes de sequências sendo impossível de procurar a melhor árvore por teste exaustivo de todas as árvores possíveis quando se tem mais de 10 taxa.

Como determinar a fiabilidade de uma árvore?
• Testar e comparar árvores (re-)construídas por métodos de inferência diferentes - teste de congruência de inferência filogenética
• Reamostrar os dados (alinhamentos) e substituí-los por outros (por duplicação ou omissão) e verificar se obtemos consistentemente a mesma árvore ou, pelo menos, alguns nós dessa árvore – bootstrapping (simula o que aconteceria se tivéssemos dados adicionais)
• Subamostrar os dados (por omissão) sem substituí-los por outros de modo a criar um conjunto mais pequeno de dados e verificar se obtemos consistentemente a mesma árvore ou, pelo menos, alguns nós dessa árvore – jacknifing (simula o que aconteceria se tivéssemos menos dados)
• Embora bootstrapping seja o método mais usado, Anisimova & Gascuel(2006) demonstraram que o método aLRT (approximate Likelihood Ratio Test) consegue substituir bootstrapping usando um algoritmo mais rápido comparando a verosimilhança da melhor e da 2º melhor topologia à volta do ramo a ser testado
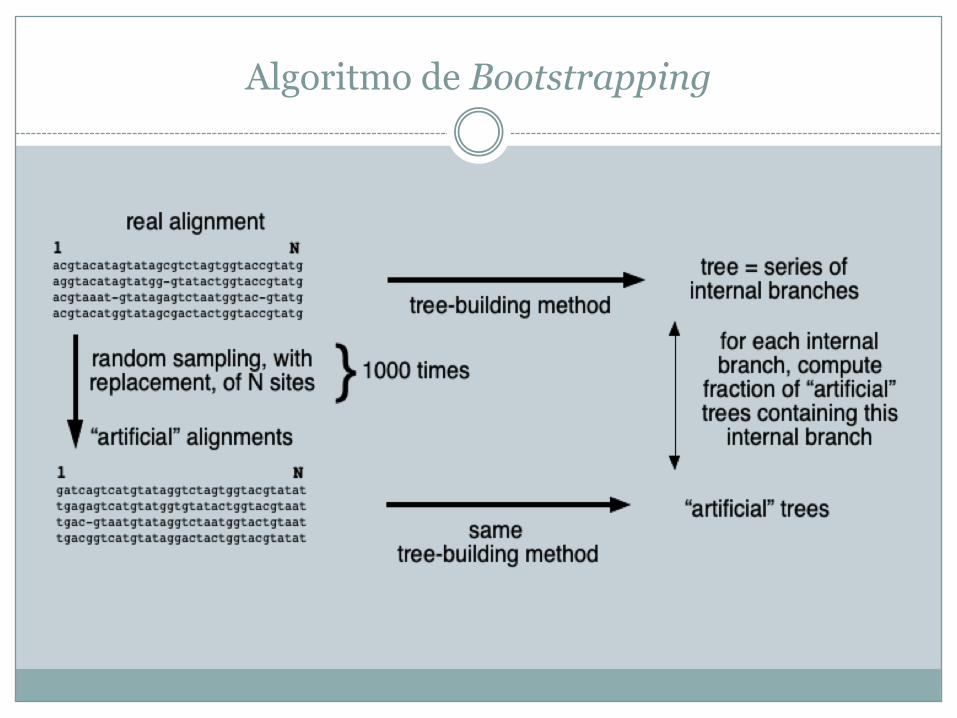
Algoritmo de Bootstrapping
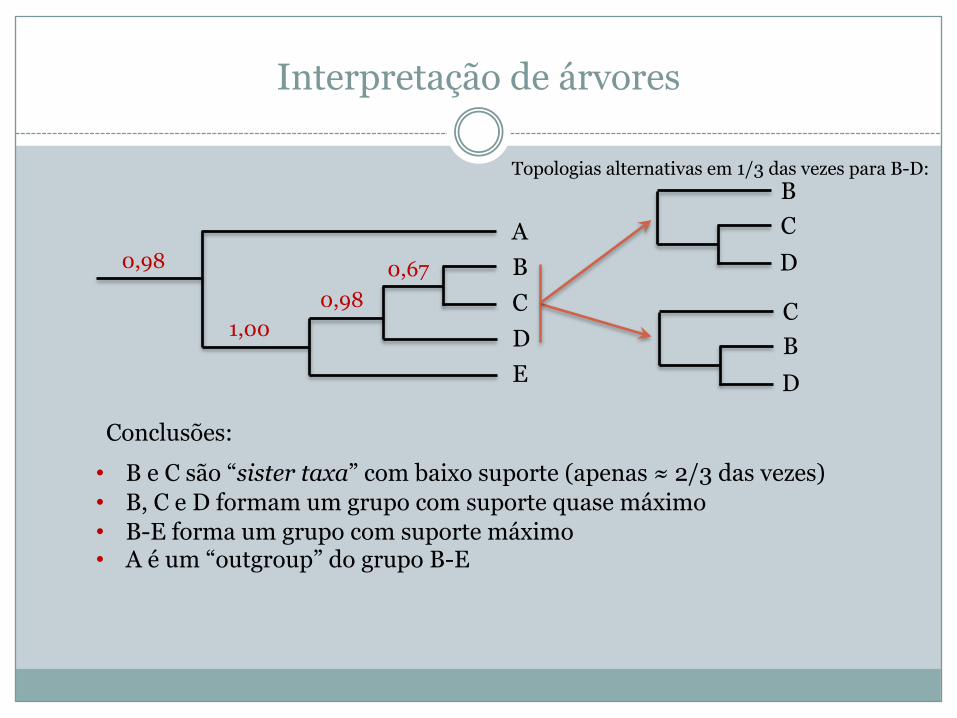
Interpretação de árvores
ABCDE
0,98 0,670,98
1,00
• B e C são “sister taxa” com baixo suporte (apenas ≈ 2/3 das vezes)• B, C e D formam um grupo com suporte quase máximo• B-E forma um grupo com suporte máximo• A é um “outgroup” do grupo B-E
Conclusões:
CBD
BCD
Topologias alternativas em 1/3 das vezes para B-D:
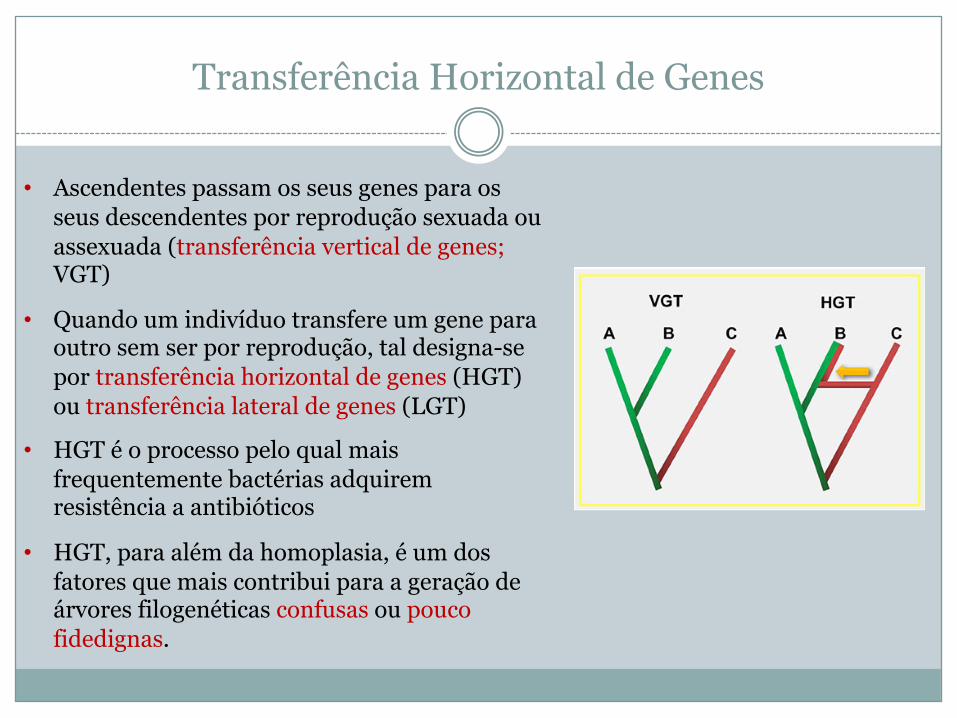
Transferência Horizontal de Genes
• Ascendentes passam os seus genes para os seus descendentes por reprodução sexuada ou assexuada (transferência vertical de genes; VGT)
• Quando um indivíduo transfere um gene para outro sem ser por reprodução, tal designa-se por transferência horizontal de genes (HGT) ou transferência lateral de genes (LGT)
• HGT é o processo pelo qual mais frequentemente bactérias adquirem resistência a antibióticos
• HGT, para além da homoplasia, é um dos fatores que mais contribui para a geração de árvores filogenéticas confusas ou pouco fidedignas.
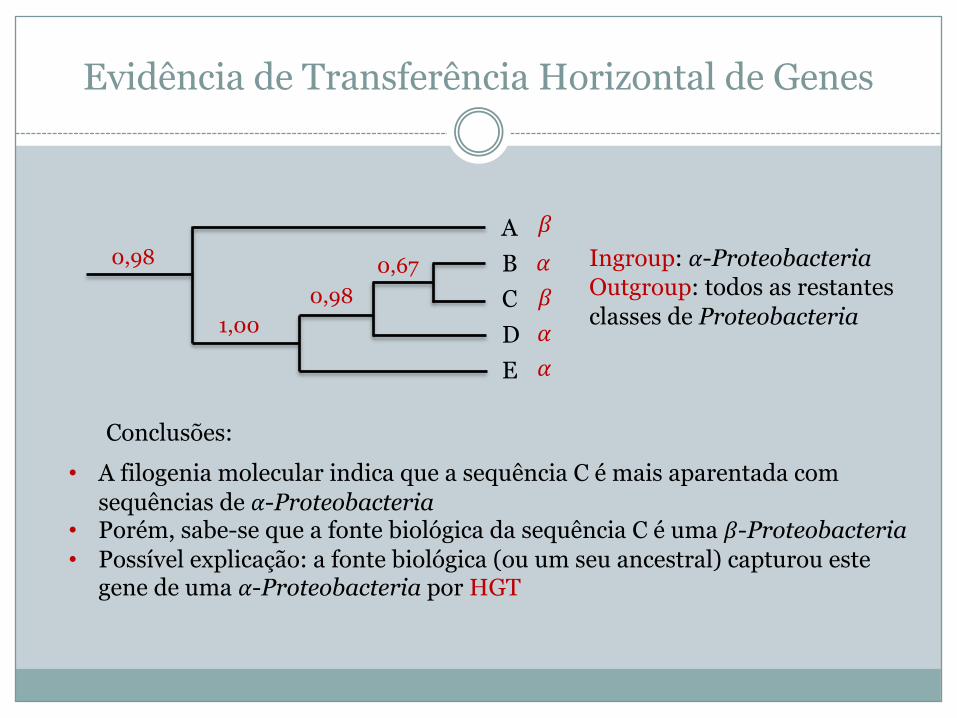
Evidência de Transferência Horizontal de Genes
ABCDE
0,98 0,670,98
1,00
• A filogenia molecular indica que a sequência C é mais aparentada com sequências de !-Proteobacteria
• Porém, sabe-se que a fonte biológica da sequência C é uma "-Proteobacteria• Possível explicação: a fonte biológica (ou um seu ancestral) capturou este
gene de uma !-Proteobacteria por HGT
Conclusões:
Ingroup: !-ProteobacteriaOutgroup: todos as restantes classes de Proteobacteria
!!
!"
"

Aula T12
� RNA� Proteína
ou a sistematização da função de produtos génicos:
Ontologia Génica

Ontologia Génica (GO)
Iniciativa bioinformática (geneontology.org) para definir termos GO, únicos, e com nº de acesso específicos em três domínios diferentes:
� Componente celular (biologia celular)local onde os produtos génicos atuam
� Função molecular / bioquímica (biologia molecular)actividade dos produtos génicos
� Processo Biológico (biologia de sistemas)vias ou processos biológicos compostos por produtosgénicos com diferentes funções moleculares e componentes celulares
Ontologia Génica (GO)
Iniciativa bioinformática (geneontology.org) para definir termos GO, únicos, e com nº de acesso específicos em três domínios diferentes:

Termo GO
� Vem da designação “GO term” em inglês� Em que GO = Gene Ontology� O termo GO é único� O termo GO tem sempre um nº de acesso com
formato: GO:nnnnnnn� Permite sistematizar funções moleculares,
processos biológicos e componentes celulares

Ontologia Génica
� Componente celular (biologia celular)
� Função molecular / bioquímica (biologia molecular)
� Processo Biológico (biologia de sistemas)
Iniciativa bioinformática (geneontology.org) para definir termos em trêsdomínios diferentes:

Biologia molecular de um gene
� Função bioquímica / molecular de um gene e respectivo produto (RNA e / ou proteína) (por ex., catálise enzimática)
� Ligação a outras moléculas
Exs: lactase, acetil-CoA carboxilase, ligação a DNA

Ontologia Génica
� Componente celular (biologia celular)
� Função molecular / bioquímica (biologia molecular)
� Processo Biológico (biologia de sistemas)

Processos biológicos
� Metabolismo das purinas� Metabolismo de glícidos� Locomoção� Fototactismo / Fototaxia� Fotossíntese� Respiração� Etc.
Conjunto de eventos moleculares com princípio e fim bem definidos:

Exemplo de termo GO
� glycerol-3-phosphate dehydrogenase [NAD(P)+] activity
� GO term: GO:0047952

A base de dados Gene Ontology é hierárquica
Inferred Tree View da base de dados GO:

Utilização de termos GO no Annotathon
� Escolher a função molecular e / ou o processo biológico em que participa a proteína codificada pela ORF selecionada
� Selecionar o termo GO mais específico dentro das hipóteses dadas pelo Annotathon (procurar esse termo na Inferred Tree View da base de dados GO)
� explicar a vossa decisão nas Conclusões gerais da anotação

Como selecionar o termo GO?
Por exemplo, o Annotathon só oferece a possibilidade de escolher entre atividade de oxidorredutase e atividade catalítica, logo escolher a primeira hipótese por ser menos abrangente que a segunda hipótese: