Embed Size (px)
Citation preview

Uniwersytet Medyczny w Łodzi KATEDRA CHEMII BIOORGANICZNEJ I BIOKOORDYNACYJNEJ
Zakład Chemii Fizycznej i Biokoordynacyjnej
Prof. UM dr hab. ALEKSANDER KUFELNICKI
ĆWICZENIA Z BIOFIZYKI Skrypt dla studentów
Wydziału Farmaceutycznego
ŁÓDŹ 2009

Wydano na zlecenie Senackiej Komisji ds. Wydawnictw Uniwersytetu Medycznego w Łodzi Uchwała z dnia 21.02.2006 r. Recenzja: prof. UŁ dr hab. Adam bald
Uniwersytet Łódzki © Copyright by Uniwersytet Medyczny w Łodzi Łódź 2007 ISBN 83-88940-79-1 Wydanie poprawione Skład komputerowy i opracowanie redakcyjne: Autor

SPIS TREŚCI
1. WSTĘP ................................................................................................................................. 5
2. MIĘDZYNARODOWY UKŁAD JEDNOSTEK SI ............................................................. 6
3. OPRACOWANIE WYNIKÓW POMIARÓW ...................................................................... 9 3.1. Rozkład Gaussa ............................................................................................................... 9 3.2. Rozkład t-Studenta ........................................................................................................ 11 3.3. Błąd bezwzględny i względny ....................................................................................... 12 3.4. Oznaczoność cyfry ........................................................................................................ 14 3.5. Regresja liniowa metodą najmniejszych kwadratów .................................................... 15 3.6. Graficzne przedstawianie wyników pomiarów ............................................................. 17
4. POMIAR NIEKTÓRYCH WIELKOŚCI FIZYCZNYCH .................................................. 19 4.1. Ciśnienie ........................................................................................................................ 19
4.1.1. Zasada pomiaru ciśnienia atmosferycznego za pomocą barometru rtęciowego .... 19 4.1.2. Poprawki wprowadzane przy wyznaczaniu ciśnienia atmosferycznego ................ 21 4.1.3. Typy barometrów ................................................................................................... 23 Ćwiczenie 1 Pomiar ciśnienia atmosferycznego za pomocą barometru rtęciowego .. 24
4.2. Gęstość i cięŜar właściwy .............................................................................................. 26 4.2.1. Pomiar gęstości ciał stałych i cieczy za pomocą piknometru ................................ 26
Ćwiczenie 2 Pomiar gęstości cieczy za pomocą piknometru ..................................... 28 4.3. Lepkość ......................................................................................................................... 29
4.3.1. ZaleŜność lepkości cieczy od temperatury ............................................................. 30 4.3.2. Metody pomiaru współczynnika lepkości .............................................................. 31 Ćwiczenie 3 Badanie wpływu temperatury na lepkość dynamiczną i właściwą
roztworów ............................................................................................. 34 4.4. Napięcie powierzchniowe ............................................................................................. 37
4.4.1. Wygięte błonki powierzchniowe ............................................................................ 38 4.4.2. Zjawisko kapilarne (włoskowatości) ...................................................................... 40 4.4.3. Metody pomiaru napięcia powierzchniowego ....................................................... 41 Ćwiczenie 4 Pomiar napięcia powierzchniowego roztworów kwasu octowego za
pomocą stalagmometru ........................................................................ 45
5. NIEKTÓRE FIZYCZNE METODY BADANIA STRUKTURY MOLEKUŁ .................. 47 5.1. Wielkości addytywne .................................................................................................... 47
5.1.1. Refrakcja molowa .................................................................................................. 47 5.1.2. Parachora ................................................................................................................ 53 Ćwiczenie 5 Pomiar współczynników załamania i wyznaczanie refrakcji molowej
wodnych roztworów mocznika ............................................................. 54 5.2. Czynność optyczna związków ....................................................................................... 57
5.2.1. Zjawisko odbicia światła ........................................................................................ 57 5.2.2. Przejście promieni świetlnych przez kryształy posiadające zdolność podwójnego załamania .......................................................................................................................... 58 5.2.3. Wykorzystanie anizotropii pochłaniania - dichroizm ............................................ 59 Ćwiczenie 6 Wyznaczanie skręcalności właściwej i stęŜenia glukozy .................... 65
6. PRZEWODNICTWO JONOWE ......................................................................................... 67 6.1. Rodzaje przewodników elektryczności ......................................................................... 67 6.2. Ruchliwość jonów ......................................................................................................... 67 6.3. Liczby przenoszenia ...................................................................................................... 69

6.4. Pomiar oporu elektrycznego i przewodnictwa elektrolitów .......................................... 71 6.5. Przewodnictwo właściwe elektrolitów .......................................................................... 73 6.6. Przewodnictwo molowe elektrolitów ............................................................................ 76 6.7. ZaleŜność przewodnictwa elektrolitów od składu roztworu ......................................... 77
Ćwiczenie 7 Pomiar oporu elektrycznego za pomocą mostka prądu przemiennego. 80
7. OGNIWA GALWANICZNE ............................................................................................... 81 7.1. Schemat zapisu ogniw ................................................................................................... 82 7.2. Pomiar siły elektromotorycznej ogniwa ........................................................................ 83 7.3. Termodynamika ogniw galwanicznych......................................................................... 87 7.4. Skoki potencjałów w ogniwach galwanicznych ............................................................ 92 7.5. Ogniwa stęŜeniowe........................................................................................................ 93
Ćwiczenie 8 Kompensator prądu stałego – pomiar SEM ogniwa galwanicznego w róŜnych temperaturach ......................................................................... 95
Ćwiczenie 9 Pomiar SEM ogniwa stęŜeniowego metodą kompensacyjną Poggendorffa........................................................................................ 96

5
1. WSTĘP
Biofizyka jest nauką doświadczalną, która bada właściwości materii oŜywionej, procesy fizyczne zachodzące w organizmach Ŝywych, a takŜe zjawiska fizyczne towarzyszące tym procesom. Poznanie wymienionych zagadnień i wykrywanie ogólnych prawidłowości rządzą-cych przemianami wymaga stosowania doświadczalnych metod badawczych. Biofizyka opiera się na podstawowych działach fizyki, takich jak mechanika klasyczna, termodynamika, elek-tryczność, magnetyzm, drgania i fale, mechanika kwantowa, fizyka jądrowa. Jest nauką ścisłą umoŜliwiającą wyjaśnienie wyników doświadczeń i formułowanie praw ogólnych odnoszą-cych się do właściwości substancji oraz procesów biologicznych zachodzących z ich udzia-łem.
Otrzymywanie i badanie substancji leczniczych, badanie ich przemian w ustrojach Ŝy-wych, a takŜe poza nimi, związane są z przemianami fizycznymi występującymi często w bardzo skomplikowanych układach. Prawidłowe opanowanie szeregu nauk farmaceutycznych wymaga poznania podstaw fizycznych. Dotyczy to w zasadzie wszystkich stosowanych metod doświadczalnych.
Niniejszy skrypt został opracowany w celu przypomnienia znanych i przybliŜenia nowych wybranych pojęć fizycznych studentom Wydziału Farmaceutycznego. Zakres omawianych za-gadnień oraz ilustrujących je ćwiczeń jest dostosowany do obowiązującego programu zajęć. Obejmuje on zasadniczo te dziedziny, które nie są nauczane w ramach nauk pokrewnych, a które są konieczne do naleŜytego opanowania nauk farmaceutycznych podczas dalszych stu-diów.
Pojedyncze doświadczenie, któremu odpowiada jedno ćwiczenie opisane w skrypcie, składać się powinno z następujących etapów:
1) przygotowanie teoretyczne, 2) przygotowanie praktyczne i przeprowadzenie pomiarów, 3) interpretacja wyników.
Pierwszy etap składa się z dwóch części. Jedna z nich to ogólne przygotowanie teoretycz-ne dotyczące przedmiotu badań - część przedmiotowa, inaczej tematyczna. Drugi człon polega na wyborze i poznaniu metody badawczej koniecznej do prawidłowego wykonania pomiarów – jest to część metodyczna.
Część praktyczna wymaga doboru i poznania działania odpowiedniego zestawu przyrzą-dów, pozwalających na uzyskanie obiektywnych i dokładnych wyników. Ta część ćwiczenia moŜe stanowić nawet małą część całości doświadczenia. Niejednokrotnie jednak wykony-wanie pomiarów bywa trudne i pracochłonne.
Interpretacja uzyskanych wyników stanowi uwieńczenie doświadczenia. Opiera się ona na przygotowaniu teoretycznym (przedmiotowym). Wymaga to często uszeregowania wyni-ków, przeliczenia w innych jednostkach lub przedstawienia ich funkcji w postaci wykresów. W przypadku złoŜonych, czasochłonnych obliczeń korzystamy z odpowiednich programów komputerowych dostosowanych do danego ćwiczenia lub programów bardziej ogólnego prze-znaczenia. Z uzyskanych danych ilościowych wynikają wnioski jakościowe dotyczące danego zjawiska lub przemiany fizycznej.
Niniejszy skrypt pt. „Ćwiczenia z biofizyki dla studentów Wydziału Farmaceutycznego” obejmuje tylko grupę ćwiczeń wykonywanych przez studentów kierunku farmacja Uniwersy-tetu Medycznego w Łodzi. Zawiera on przede wszystkim dokładne opisy wykonania ćwiczeń i tylko wybrane podstawy teoretyczne. Wiadomości teoretyczne moŜna uzupełnić na podsta-wie wskazanych lub teŜ innych podręczników.
Autor dziękuje Pani mgr Joannie Gądek-Sobczyńskiej za przygotowanie rysunków oraz współpracę w redagowaniu składu komputerowego skryptu oraz Pani dr Mirosławie Świątek za współpracę w merytorycznej i technicznej korekcie skryptu.

A. Kufelnicki - Ćwiczenia z biofizyki
6
2. MIĘDZYNARODOWY UKŁAD JEDNOSTEK SI
Pomiar, czyli mierzenie, jest to porównywanie dowolnej wielkości z określoną wielkością tego samego rodzaju przyjętą za jednostkę miary. Pomiar moŜe być bezpośredni, w którym poszukiwaną wartość odczytuje się wprost z przyrządu pomiarowego lub pośredni, w którym wartość szukaną uzyskuje się przez pomiar innych wielkości związanych określoną zaleŜno-ścią z wielkością szukaną. Do mierzenia potrzebny jest odpowiedni przyrząd, a do ilościowe-go wyraŜenia wyniku odpowiednia jednostka miary. Zbiór jednostek utworzonych wg okre-ślonych zasad nazywamy układem jednostek. Od 1966 roku obowiązuje w Polsce Międzyna-rodowy Układ Jednostek Miar zwany w skrócie układem SI. Zaletą tego układu jest jego uni-wersalność (stosuje się do wszystkich dziedzin fizyki i techniki) oraz spójność (współczynniki przeliczeniowe równe są jedności). Poza tym układ SI ustala dla określonej wielkości fizycz-nej jedną tylko jednostkę miary, jedną jej nazwę, symbol i wymiar.
Układ SI oparty jest na siedmiu jednostkach podstawowych: metr, kilogram, sekunda, amper, kelwin, mol i kandela oraz dwóch jednostkach uzupełniających: radian i steradian. Jednostki pochodne przyporządkowane róŜnym wielkościom tworzy się w oparciu o odpo-wiednie formuły praw fizyko-chemicznych. KaŜda jednostka podstawowa ma ściśle zdefinio-wany wzorzec. Jednocześnie odpowiednie rozporządzenia dopuszczają przejściowo do stoso-wania jako legalne, wybrane jednostki miar nie naleŜące do układu SI (np. tona, litr, stopień Celsjusza, minuta, godzina).
Jednostki podstawowe: M e t r jest równy drodze, jaką przebywa w próŜni światło w ciągu czasu 1/299792458
sekundy. Dawniejsza definicja: metr jest długością równą 1650763,75 długości fali promie-niowania w próŜni, odpowiadającego przejściu pomiędzy poziomami 2p10 a 5d5 atomu kryp-tonu 86.
K i l o g r a m jest masą międzynarodowego wzorca tej jednostki, przechowywaną w Międzynarodowym Biurze Miar. Masa jednego kg moŜe być odtworzona z błędem nie przekraczającym 2⋅10-9, to znaczy 0,002 mg/kg.
S e k u n d a jest czasem trwania 9192631770 okresów promieniowania, odpowiadające-go przejściu między dwoma nadsubtelnymi poziomami stanu podstawowego atomu cezu 133. Definicja ta pozwala na wyznaczenie sekundy z dokładnością 10-12.
A m p e r jest natęŜeniem prądu elektrycznego nie zmieniającego się, który płynąc w dwóch równoległych przewodach prostoliniowych nieskończenie długich o przekroju okrą-głym znikomo małym, umieszczonych w odległości jednego metra jeden od drugiego w próŜ-ni wywołałby między tymi przewodami siłę równą 2⋅10-7 N na kaŜdy metr długości przewodu. Definicja ta oparta na prawie Ampere'a określa bardzo dokładnie wartość ampera, jednakŜe realizacja tak zdefiniowanego wzorca jest niemoŜliwa. W praktyce dokładny pomiar natęŜenia prądu odbywa się za pomocą tzw. wagi prądowej z błędem względnym 1⋅10-5.
K e l w i n jest jednostką temperatury termodynamicznej w skali, w której temperatura punktu potrójnego wody jest równa dokładnie 273,16 K.
M o l jest miarą liczności materii i oznacza taką jej ilość, która zawiera liczbę cząstek równą liczbie atomów zawartych w masie 0,012 kg czystego nuklidu węgla 12C.
K a n d e l a jest światłością, jaką ma w danym kierunku źródło emitujące monochroma-tyczne promieniowanie o częstości 540·1012 Hz i mające w tym kierunku wydajność energe-tyczną 1/683 W/sr (watów na steradian). Dawniejsza definicja: kandela jest światłością, którą
2. Międzynarodowy układ jednostek SI
7
ma w kierunku prostopadłym pole 1/(6⋅105 m2) powierzchni ciała doskonale czarnego, pro-mieniującego w temperaturze krzepnięcia platyny pod ciśnieniem 101325 Pa (paskali). Jednostki uzupełniające:
R a d i a n jest kątem płaskim o wierzchołku w środku koła, wycinającym z tego koła łuk o długości równej jego promieniowi.
S t e r a d i a n jest kątem bryłowym o wierzchołku w środku kuli, wycinającym z powierzchni tej kuli pole równe kwadratowi jego promienia.
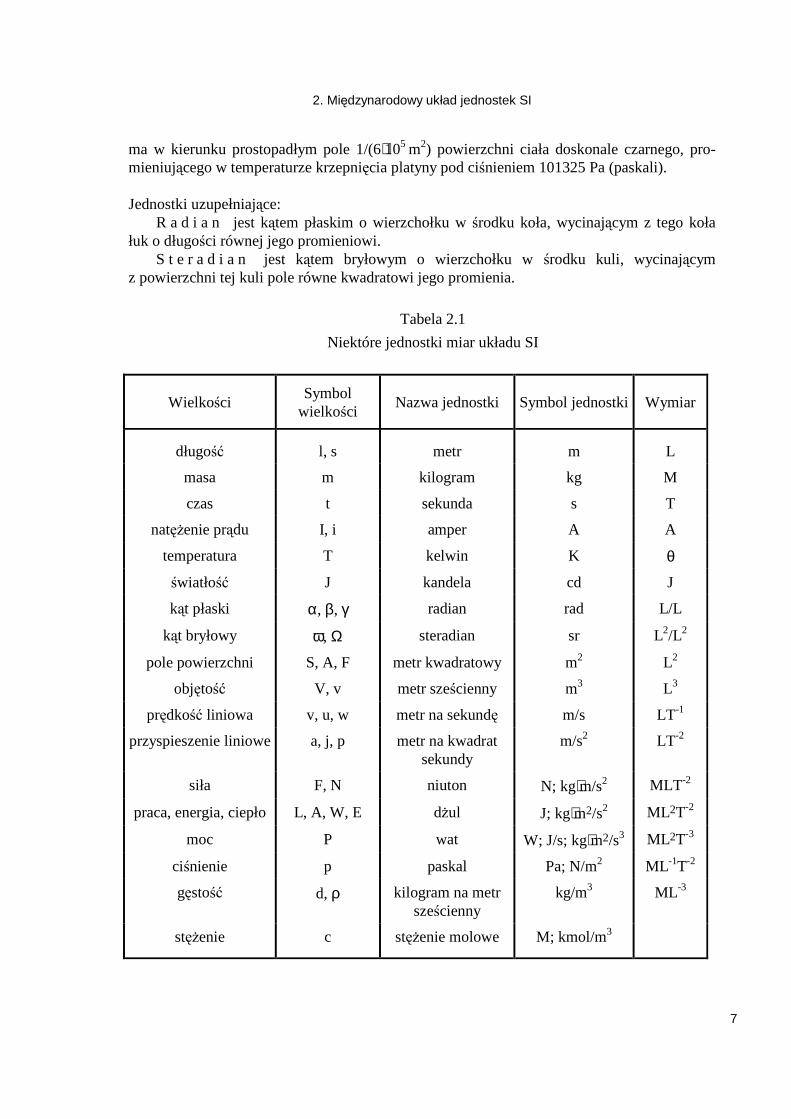
Tabela 2.1
Niektóre jednostki miar układu SI
ciśnienie p paskal Pa; N/m2 ML-1T-2
gęstość d, ρ kilogram na metr sześcienny
kg/m3 ML -3
stęŜenie c stęŜenie molowe M; kmol/m3
Wielkości Symbol
wielkości Nazwa jednostki Symbol jednostki Wymiar
długość l, s metr m L
masa m kilogram kg M
czas t sekunda s T
natęŜenie prądu I, i amper A A
temperatura T kelwin K θ
światłość J kandela cd J
kąt płaski α, β, γ radian rad L/L
kąt bryłowy ω, Ω steradian sr L2/L2
pole powierzchni S, A, F metr kwadratowy m2 L2
objętość V, v metr sześcienny m3 L3
prędkość liniowa v, u, w metr na sekundę m/s LT-1
przyspieszenie liniowe a, j, p metr na kwadrat sekundy
m/s2 LT-2
siła F, N niuton N; kg⋅m/s2 MLT -2
praca, energia, ciepło L, A, W, E dŜul J; kg⋅m2/s2 ML2T-2
moc P wat W; J/s; kg⋅m2/s3 ML2T-3
A. Kufelnicki - Ćwiczenia z biofizyki
8
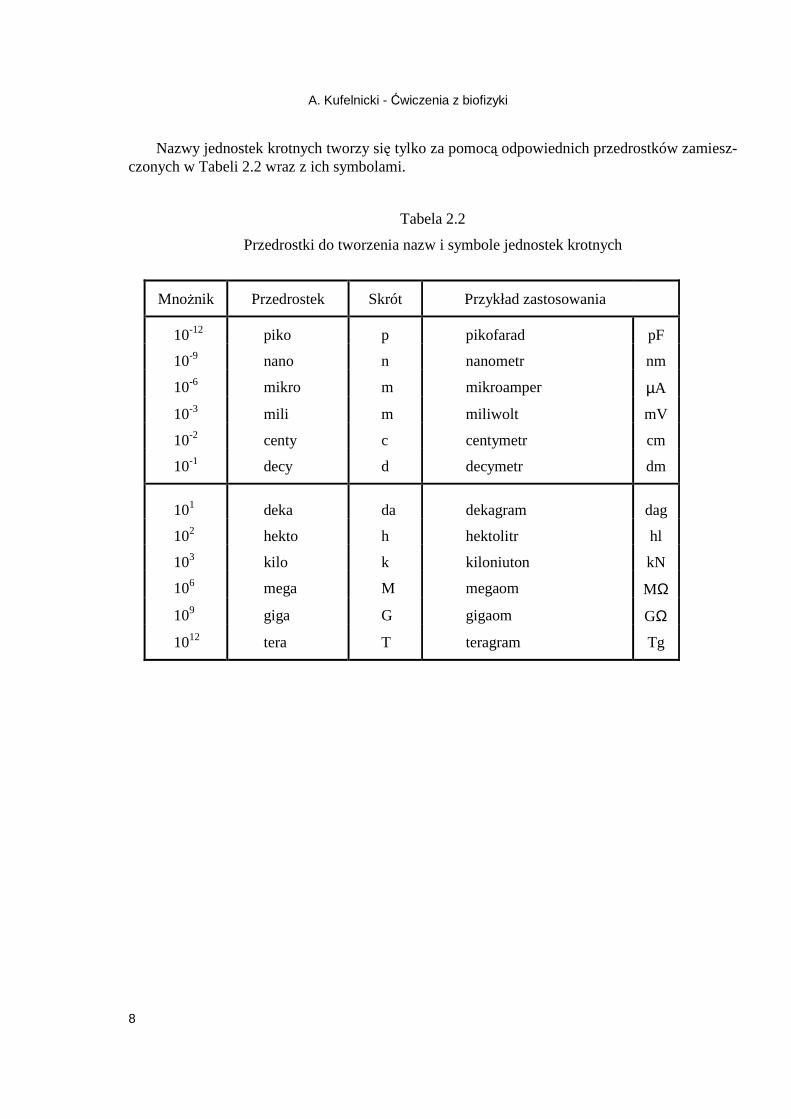
Nazwy jednostek krotnych tworzy się tylko za pomocą odpowiednich przedrostków zamiesz-czonych w Tabeli 2.2 wraz z ich symbolami.
Tabela 2.2
Przedrostki do tworzenia nazw i symbole jednostek krotnych
MnoŜnik Przedrostek Skrót Przykład zastosowania
10-12 piko p pikofarad pF
10-9 nano n nanometr nm
10-6 mikro m mikroamper µA
10-3 mili m miliwolt mV
10-2 centy c centymetr cm
10-1 decy d decymetr dm
101 deka da dekagram dag
102 hekto h hektolitr hl
103 kilo k kiloniuton kN
106 mega M megaom MΩ
109 giga G gigaom GΩ
1012 tera T teragram Tg

9
3. OPRACOWANIE WYNIKÓW POMIARÓW 3.1. Rozkład Gaussa
Podczas pomiarów pewnej wielkości otrzymujemy szereg wyników x1, x2 ,…, xn , z któ-
rych kaŜdy obarczony jest określonym błędem. MoŜe to być b ł ą d p r z y p a d k o w y (bę-dący wynikiem np. przypadkowych zmian temperatury, zmian napięcia w sieci, niedoskonało-ści odczytu itd.) lub b łą d s y s t e m a t y c z n y spowodowany np. złą podziałką przyrzą-du lub nieodpowiednimi warunkami pracy urządzeń pomiarowych.
Zakładając, Ŝe występują tylko błędy przypadkowe (tego rodzajów błędów nie da się cał-kiem wyeliminować) moŜna wykazać, Ŝe do prawdziwej wartości wielkości mierzonej zbliŜa się, wraz z powiększaniem liczby pomiarów, ś r e d n i a a r y t m e t y c z n a :
∑=
=+++=n
ii
n xnn
xxxx
1
21 1... (3.1.1)
Uzyskiwane wyniki osiągają wartości mniejsze lub większe od średniej arytmetycznej. Miarą dokładności pojedynczego pomiaru jest wielkość:
1
)(1
2
−
−=∑=
n
xx
s
n
ii
x (3.1.2)
zwana o d c h y l e n i e m s t a n d a r d o w y m p o j e d y n c z e g o w y n i k u . Wiel-kość n -1 nazywamy l i c z bą s t o p n i s w o b o d y .
Ś r e d n i b łą d k w a d r a t o w y ś r e d n i e j a r y t m e t y c z n e j (zwany teŜ błędem kwadratowym całkowitego pomiaru) oblicza się ze wzoru:
)1(
)(1
2
−
−==∑=
nn
xx
n
ss
n
ii
xx (3.1.3)
Przy bardzo duŜej liczbie pomiarów rozkład wyników doświadczalnych (Rys. 3.1a) staje się symetryczny względem pewnej wartości µ (w przybliŜeniu równej średniej arytmetycznej x ): x≈µ (3.1.4) Odchylenie standardowe sx zbliŜa się do wartości σ zwanej d y s p e r s ją r o z k ł a d u : xs≈σ (3.1.5)
Funkcją analityczną, której odpowiada wówczas rozkład wyników doświadczalnych jest f u n k c j a gę s t oś c i r o z k ł a d u n o r m a l n e g o :
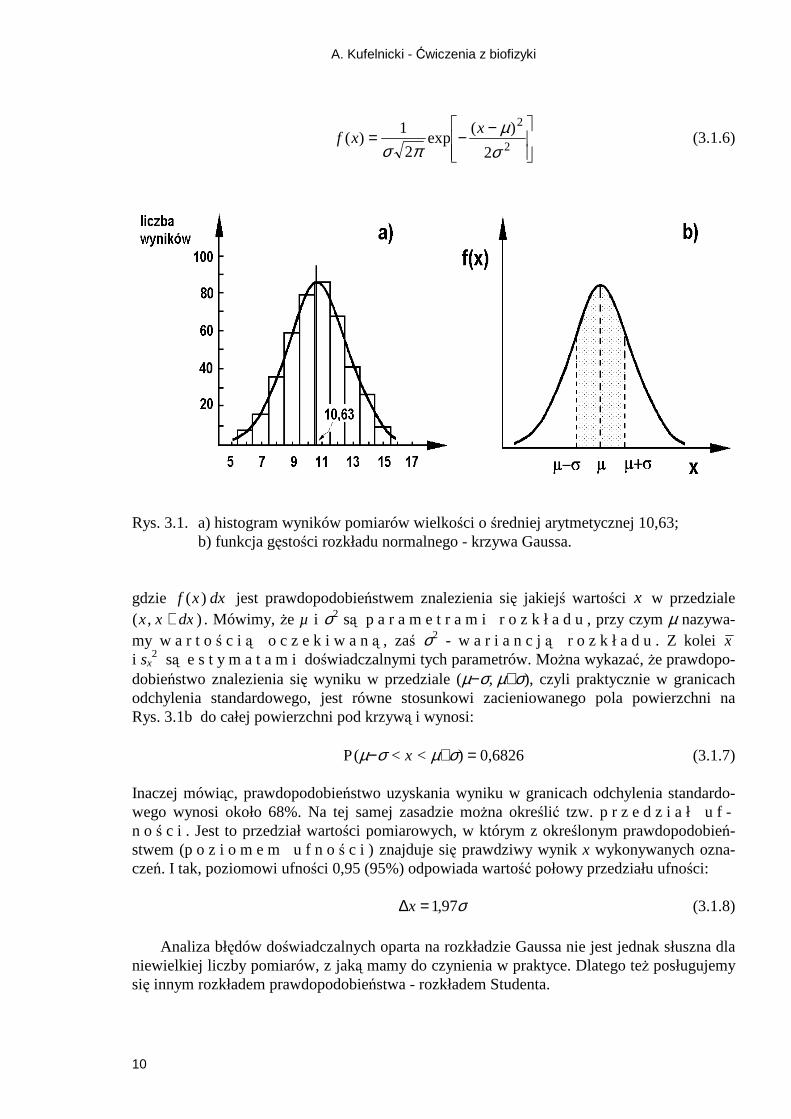
A. Kufelnicki - Ćwiczenia z biofizyki
10
−−=2
2
2
)(exp
2
1)(
σµ
πσx
xf (3.1.6)
Rys. 3.1. a) histogram wyników pomiarów wielkości o średniej arytmetycznej 10,63;
b) funkcja gęstości rozkładu normalnego - krzywa Gaussa. gdzie f x dx( ) jest prawdopodobieństwem znalezienia się jakiejś wartości x w przedziale ( , )x x dx+ . Mówimy, Ŝe µ i σ2 są p a r a m e t r a m i r o z k ł a d u , przy czym µ nazywa-my w a r t oś c i ą o c z e k i w a ną , zaś σ2 - w a r i a n c ją r o z k ł a d u . Z kolei x i sx
2 są e s t y m a t a m i doświadczalnymi tych parametrów. MoŜna wykazać, Ŝe prawdopo-dobieństwo znalezienia się wyniku w przedziale (µ−σ, µ+σ), czyli praktycznie w granicach odchylenia standardowego, jest równe stosunkowi zacieniowanego pola powierzchni na Rys. 3.1b do całej powierzchni pod krzywą i wynosi: P (µ−σ < x < µ+σ) = 0,6826 (3.1.7) Inaczej mówiąc, prawdopodobieństwo uzyskania wyniku w granicach odchylenia standardo-wego wynosi około 68%. Na tej samej zasadzie moŜna określić tzw. p r z e d z i a ł u f -n o ś c i . Jest to przedział wartości pomiarowych, w którym z określonym prawdopodobień-stwem (p o z i o m e m u f n oś c i ) znajduje się prawdziwy wynik x wykonywanych ozna-czeń. I tak, poziomowi ufności 0,95 (95%) odpowiada wartość połowy przedziału ufności: σ97,1=∆x (3.1.8)
Analiza błędów doświadczalnych oparta na rozkładzie Gaussa nie jest jednak słuszna dla niewielkiej liczby pomiarów, z jaką mamy do czynienia w praktyce. Dlatego teŜ posługujemy się innym rozkładem prawdopodobieństwa - rozkładem Studenta.

3. Opracowanie wyników pomiarów
11
3.2. Rozkład t-Studenta Rozkład Studenta (W.S. Gosseta), zwany teŜ rozkładem t-Studenta, przedstawia zaleŜność:
2
12
1),(
+−
+=
k
k k
tCktS (3.2.1)
gdzie Ck - wielkość zaleŜna wyłącznie od liczby stopni swobody 1−= nk
xx s
xn
s
xt
µµ −=−= (3.2.2)
Rozkład ten jest symetryczny względem osi t = 0, krzywa jest jednak bardziej spłaszczo-
na od krzywej Gaussa. Rozkład t-Studenta dąŜy do rozkładu normalnego dla k → ∞ . Chcąc oszacować przedział ufności, w którym z zadanym prawdopodobieństwem α (po-
ziomem ufności) będzie znajdował się prawdziwy wynik wykonywanych pomiarów, musimy średni błąd kwadratowy średniej arytmetycznej pomnoŜyć przez współczynnik tα,k zaleŜny od tego prawdopodobieństwa i od liczby stopni swobody, czyli: xkxk stxstx ,, αα µ +⟨⟨− (3.2.3)
Wartości t są podane w tablicach. Dla małej liczby pomiarów oszacowany przedział uf-
ności jest znacznie większy od przedziału wynikającego z rozkładu Gaussa, natomiast dla bardzo duŜych k (>30) współczynniki t dąŜą do wartości określonych przez rozkład normalny.
Często w miejsce poziomu ufności uŜywamy pojęcia p o z i o m i s t o t n oś c i : 1-α , np. poziomowi ufności 0,95 odpowiada poziom istotności 0,05.
Przykład
Wyniki pięciu pomiarów pewnej wielkości fizycznej są następujące:
n x xx i − ( ) 6210⋅− xx i
1 1,71 +0,014 196
2 1,73 −0,006 36 3 1,72 +0,004 16 4 1,72 +0,004 16
5 1,74 −0,016 256
x = 1,724 ( )∑ − 2xx i = 520
36
105,09945
10520 −−
⋅=⋅⋅=xs

A. Kufelnicki - Ćwiczenia z biofizyki
12
Połowa przedziału ufności przy α = 0,95 wynosi t0,95;4 ⋅ xs = 2,78 ⋅ 5,099⋅10-3 = 1,418 ⋅10-2, zatem
prawdziwa wartość oznaczonej wielkości zawarta jest w przedziale 1,724 ± 0,014 z prawdopodo-
bieństwem 95%.
3.3. Błąd bezwzględny i względny
Często dysponujemy tylko pojedynczym wynikiem pomiaru danej wielkości, bez moŜli-wości analizy statystycznej rozkładu wyników. Wówczas dokładność wykonanego pomiaru moŜemy ocenić wprowadzając pojęcia błędu bezwzględnego oraz błędu względnego.
B ł ę d e m b e z w z g lę d n y m pomiaru nazywamy wartość bezwzględną róŜnicy pomiędzy wartością zmierzoną xi a wartością prawdziwą x0: 0xxx i −=∆ (3.3.1)
Błąd ten jest liczbą mianowaną, wyraŜoną w jednostkach wielkości mierzonej.
B ł ę d e m w z g lę d n y m pomiaru nazywamy stosunek błędu bezwzględnego do wartości bezwzględnej prawdziwej wartości x0 . Jest to wielkość niemianowana, wyraŜona w procentach:
%100%10000
⋅∆=⋅∆=x
x
x
xxδ (3.3.2)
Na ogół jednak nie znamy prawdziwej wartości wielkości mierzonej, stąd błąd bezwzględny określamy na podstawie dokładności podziałki przyrządu pomiarowego, a błąd względny ob-liczamy dzieląc błąd bezwzględny przez otrzymany wynik. Np. na wadze półtechnicznej otrzymano wynik waŜenia 102,1 g odczytując do 0,1 g. Błąd względny pomiaru, określający dokładność waŜenia, wynosi w tym przypadku (0,1/102,1) ⋅100% ≈ 0,1% .
W wielu eksperymentach obliczana jest wielkość będąca funkcją kilku wielkości pomia-rowych. Np. chcąc wyznaczyć gęstość cieczy musimy zmierzyć jej masę oraz objętość z okre-ślonymi błędami względnymi. Zarówno pomiar masy, jak i objętości wnoszą swój wkład do błędu pomiaru gęstości.
Ogólnie, jeŜeli wielkość u jest funkcją kilku mierzonych parametrów:
u f x y z= ( , , ,...) (3.3.3) korzystamy z m e t o d y r óŜ n i c z k i z u p e ł n e j . RóŜniczką zupełną tej funkcji jest wyraŜenie:
...,...,,...,,...,
+
+
+
= dz
z
fdy
y
fdx
x
fdu
yxzxzy ∂∂
∂∂
∂∂
(3.3.4)
Zastępując du dx dy dz, , , , ... przez skończone przyrosty ∆ ∆ ∆ ∆u x y z, , , , ... otrzymamy:

3. Opracowanie wyników pomiarów
13
...+∆
+∆
+∆
=∆ z
z
fy
y
fx
x
fu
∂∂
∂∂
∂∂
(3.3.5)
Zarówno przyrosty, jak i pochodne mogą mieć wartości ujemne, przyjmuje się zatem ich war-tości bezwzględne:
...+∆+∆+∆=∆ zz
fy
y
fx
x
fu
∂∂
∂∂
∂∂
(3.3.6)
Uzyskuje się w ten sposób błąd bezwzględny wielkości złoŜonej ∆u jako maksymalne odchy-lenie, oparte na załoŜeniu sumowania się błędów cząstkowych. Mówimy, Ŝe ∆u jest m a k -s y m a l n y m b łę d e m w y n i k u. Przykład
W omawianym poprzednio pomiarze gęstości:
V
md =
V
V
mm
VV
V
dm
m
dd ∆∆
1∆∆∆
2−+=+=
∂∂
∂∂
JeŜeli np. m = 138,5 ± 0,1 g , V = 120,0 ± 0,2 cm3 , to:
33 cmg1,15417cmg120,0
138,5 −− ⋅=⋅=d
0,0030,0027560,20,009620,10,00833∆ ≈=⋅−+⋅=d
Zatem: 3cmg0,0031,154 −⋅±=d
Błąd względny pomiaru gęstości wynosi:
%0,2%0,2388%1001,15417
0,002756∆≈≈⋅=
d
d
W przypadku, gdy poszukiwana wielkość jest funkcją typu:
ba yxku = (3.3.7) gdzie k - stały współczynnik, obliczanie błędu maksymalnego wielkości złoŜonej moŜna upro-ścić. Logarytmując obie strony równania:
ybxaku lnlnlnln ++= (3.3.8)

A. Kufelnicki - Ćwiczenia z biofizyki
14
a następnie róŜniczkując logarytmy i zastępując róŜniczki skończonymi przyrostami, otrzyma-my:
yy
bxx
auu ∆+∆=∆
(3.3.9)
Przykład
Wracając do poprzedniego przykładu pomiaru gęstości, otrzymujemy:
%0,2%0,2389120,0
0,21
138,5
0,11
∆ ≈=⋅−+⋅=d
d
a więc wynik niemal identyczny z uzyskanym metodą róŜniczki zupełnej.
3.4. Oznaczoność cyfry
Z omówionym pojęciem błędu względnego, a tym samym z dokładnością pomiaru, wiąŜe się sposób poprawnego zapisywania jego wyniku. Wynik podajemy z odpowiednią liczbą cyfr znaczących. C y f rą z n a c zą c ą liczby przybliŜonej nazywamy kaŜdą róŜną od zera cyfrę rozwinięcia dziesiętnego tej liczby oraz zero, jeśli jest ono zawarte pomiędzy innymi cyframi znaczącymi lub znajduje się na zachowanej pozycji w rozwinięciu dziesiętnym tej liczby.
Zera początkowe oraz niekiedy zera końcowe wchodzące w skład liczby słuŜą do wyzna-czania jej pozycji dziesiętnych i nie są cyframi znaczącymi. Np. liczba 0,0253 zawiera trzy cy-fry znaczące, a liczba 0,00038 tylko dwie. Pojęcia liczby cyfr znaczących nie naleŜy mylić z liczbą cyfr po przecinku, która nie musi wynikać z dokładności pomiaru. W wyniku 36000±100 dwa końcowe zera nie są cyframi znaczącymi, poniewaŜ dokładność wyniku dotyczy tyl-ko trzeciej cyfry znaczącej. Ogólnie przyjmuje się, Ŝe końcowy wynik pomiarów powinien zawierać taką liczbę cyfr znaczących, aby tylko ostatnia z nich była niepewna. W związku z tym duŜe liczby lepiej przedstawiać w tzw. postaci zmiennoprzecinkowej (iloczyn liczby przybliŜonej i odpowiedniej potęgi dziesięciu) - w ostatnim przykładzie: (360±1) ⋅102. W do-kładnych pomiarach podaje się wartość błędu w postaci dwucyfrowej, np. 2,72±0,15.
Przy zaokrąglaniu wielocyfrowej wartości x do rzędu odpowiadającego błędowi pomiaru ∆x , ostatnią cyfrę zachowuje się, jeŜeli następna jest mniejsza od 5. Gdy następna cyfra jest równa 5 lub większa od 5, wówczas ostatnią cyfrę znaczącą podwyŜsza się o 1. Np. 3,5843±0,02 przedstawiamy jako 3,58±0,02, a z kolei 4,279±0,04 zapisujemy poprawnie jako 4,28±0,04.
JeŜeli błąd względny wielkości złoŜonej nie został obliczony, jako ogólną zasadę przyj-mujemy, Ŝe wynik końcowy podajemy z liczbą cyfr znaczących najwyŜej o 1 większą niŜ liczba cyfr znaczących w najmniej dokładnej wielkości cząstkowej wchodzącej do wyniku. Np. wynik obliczenia pewnej wielkości za pomocą danych: p = 546 hPa, T = 298,1 K powi-nien być zapisany z co najwyŜej 4 cyframi znaczącymi.
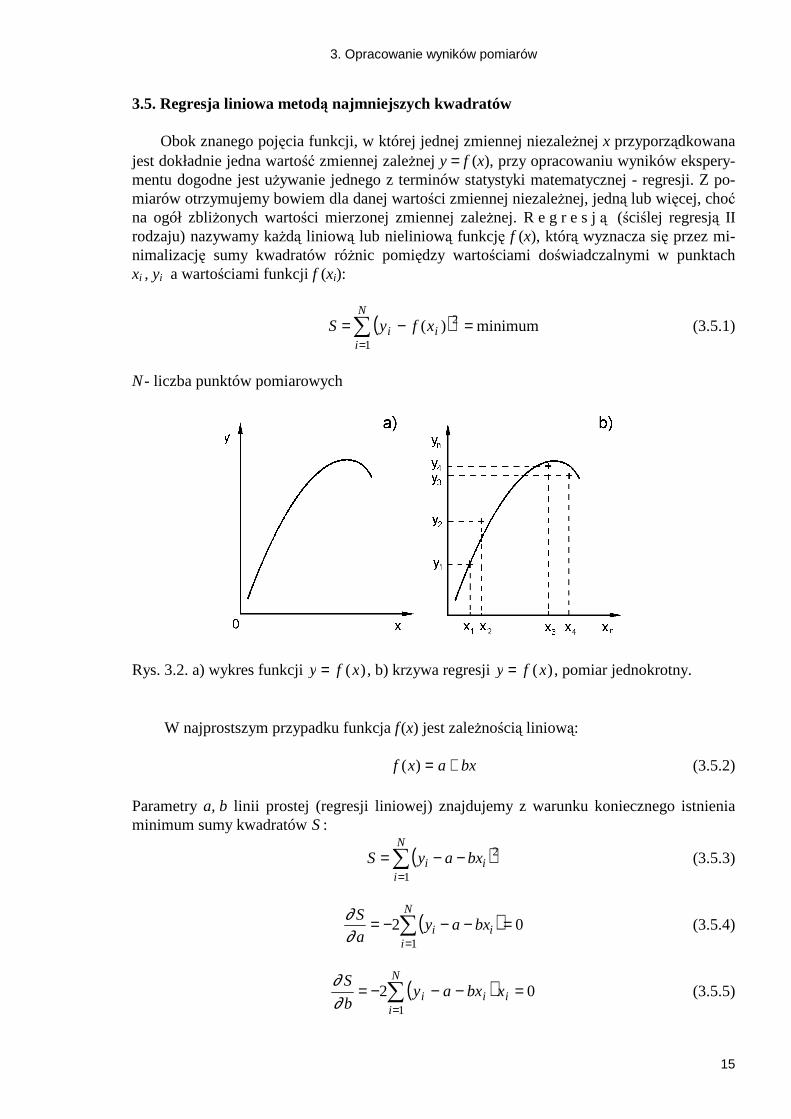
3. Opracowanie wyników pomiarów
15
3.5. Regresja liniowa metodą najmniejszych kwadratów
Obok znanego pojęcia funkcji, w której jednej zmiennej niezaleŜnej x przyporządkowana jest dokładnie jedna wartość zmiennej zaleŜnej y = f (x), przy opracowaniu wyników ekspery-mentu dogodne jest uŜywanie jednego z terminów statystyki matematycznej - regresji. Z po-miarów otrzymujemy bowiem dla danej wartości zmiennej niezaleŜnej, jedną lub więcej, choć na ogół zbliŜonych wartości mierzonej zmiennej zaleŜnej. R e g r e s ją (ściślej regresją II rodzaju) nazywamy kaŜdą liniową lub nieliniową funkcję f (x), którą wyznacza się przez mi-nimalizację sumy kwadratów róŜnic pomiędzy wartościami doświadczalnymi w punktach xi , yi a wartościami funkcji f (xi):
( ) minimum)(1
2 =−=∑=
N
iii xfyS (3.5.1)
N - liczba punktów pomiarowych
Rys. 3.2. a) wykres funkcji y f x= ( ) , b) krzywa regresji y f x= ( ) , pomiar jednokrotny.
W najprostszym przypadku funkcja f (x) jest zaleŜnością liniową:
bxaxf +=)( (3.5.2) Parametry a, b linii prostej (regresji liniowej) znajdujemy z warunku koniecznego istnienia minimum sumy kwadratów S:
( )∑=
−−=N
iii bxayS
1
2 (3.5.3)
( ) 021
=−−−= ∑=
N
iii bxay
a
S
∂∂
(3.5.4)
( ) 021
=−−−= ∑=
N
iiii xbxay
b
S
∂∂
(3.5.5)

A. Kufelnicki - Ćwiczenia z biofizyki
16
Stąd:
( ) 01
=−−∑=
N
iii bxay (3.5.6)
oraz:
( ) 01
=−−∑=
i
N
iii xbxay (3.5.7)
Po przegrupowaniu wyrazów otrzymujemy układ równań liniowych względem parametrów (współczynników) a, b:
∑∑==
=+N
ii
N
ii yxbNa
11
(3.5.8)
∑∑∑===
=+N
iii
N
ii
N
ii yxxbxa
11
2
1
(3.5.9)
Rozwiązując ten układ równań znajdujemy:
∑∑
∑ ∑∑
==
= ==
−
−=
N
ii
N
ii
N
i
N
iii
N
iii
xNx
yxNyx
b
1
22
1
1 11 (3.5.10)
xbyxbyN
aN
ii
N
ii −=
−= ∑∑
== 11
1 (3.5.11)
MoŜna równieŜ wyprowadzić (co nie będzie przedmiotem naszych rozwaŜań) wzory na od-chylenia standardowe parametrów regresji liniowej: sa i sb, a następnie obliczać odpowiednie przedziały ufności. Miarą dopasowania prostej regresji y = a + bx do punktów doświadczal-nych xi , yi jest w s p ó ł c z y n n i k k o r e l a c j i l i n i o w e j , r, określony wzorem:
−
−
−=
∑∑∑∑
∑ ∑∑
====
= ==N
ii
N
ii
N
ii
N
ii
N
i
N
iii
N
iii
yN
yxN
x
yxN
yx
r
1
2
1
2
1
2
1
2
1 11
11
1
(3.5.12)
i przyjmujący wartości z przedziału domkniętego <-1,1>. Moduł r osiąga wartość 1 tylko
wtedy, gdy punkty doświadczalne leŜą dokładnie na prostej (jednak nie równoległej do osi OX). JeŜeli r = 0, mówimy, Ŝe zmienne x i y są nieskorelowane. Dla funkcji liniowych rosną-cych (b > 0) r > 0, dla malejących r < 0.
Współczynnik korelacji moŜemy testować przez obliczenie wartości zmiennej losowej:
3. Opracowanie wyników pomiarów
17
21
2
r
Nrt
−
−= (3.5.13)
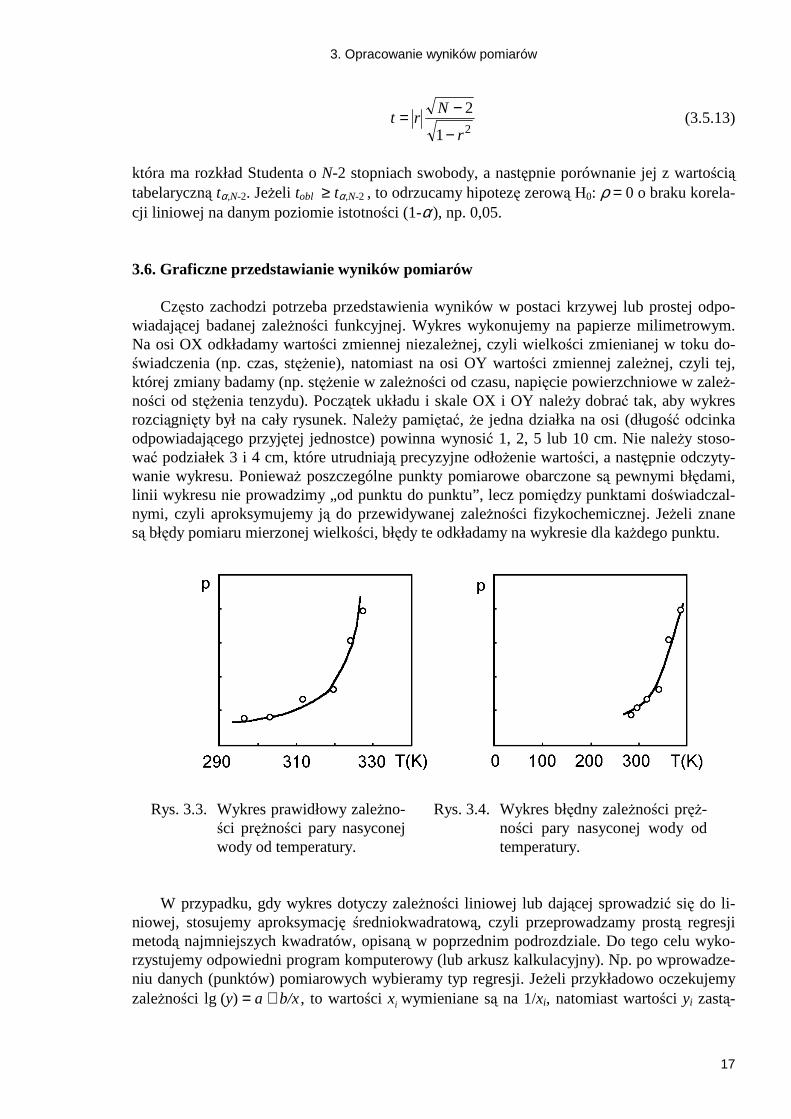
która ma rozkład Studenta o N-2 stopniach swobody, a następnie porównanie jej z wartością tabelaryczną tα,N-2. JeŜeli tobl ≥ tα,N-2 , to odrzucamy hipotezę zerową H0: ρ = 0 o braku korela-cji liniowej na danym poziomie istotności (1-α ), np. 0,05. 3.6. Graficzne przedstawianie wyników pomiarów
Często zachodzi potrzeba przedstawienia wyników w postaci krzywej lub prostej odpo-wiadającej badanej zaleŜności funkcyjnej. Wykres wykonujemy na papierze milimetrowym. Na osi OX odkładamy wartości zmiennej niezaleŜnej, czyli wielkości zmienianej w toku do-świadczenia (np. czas, stęŜenie), natomiast na osi OY wartości zmiennej zaleŜnej, czyli tej, której zmiany badamy (np. stęŜenie w zaleŜności od czasu, napięcie powierzchniowe w zaleŜ-ności od stęŜenia tenzydu). Początek układu i skale OX i OY naleŜy dobrać tak, aby wykres rozciągnięty był na cały rysunek. NaleŜy pamiętać, Ŝe jedna działka na osi (długość odcinka odpowiadającego przyjętej jednostce) powinna wynosić 1, 2, 5 lub 10 cm. Nie naleŜy stoso-wać podziałek 3 i 4 cm, które utrudniają precyzyjne odłoŜenie wartości, a następnie odczyty-wanie wykresu. PoniewaŜ poszczególne punkty pomiarowe obarczone są pewnymi błędami, linii wykresu nie prowadzimy „od punktu do punktu”, lecz pomiędzy punktami doświadczal-nymi, czyli aproksymujemy ją do przewidywanej zaleŜności fizykochemicznej. JeŜeli znane są błędy pomiaru mierzonej wielkości, błędy te odkładamy na wykresie dla kaŜdego punktu.
Rys. 3.3. Wykres prawidłowy zaleŜno-ści pręŜności pary nasyconej wody od temperatury.
Rys. 3.4. Wykres błędny zaleŜności pręŜ-
ności pary nasyconej wody od temperatury.
W przypadku, gdy wykres dotyczy zaleŜności liniowej lub dającej sprowadzić się do li-niowej, stosujemy aproksymację średniokwadratową, czyli przeprowadzamy prostą regresji metodą najmniejszych kwadratów, opisaną w poprzednim podrozdziale. Do tego celu wyko-rzystujemy odpowiedni program komputerowy (lub arkusz kalkulacyjny). Np. po wprowadze-niu danych (punktów) pomiarowych wybieramy typ regresji. JeŜeli przykładowo oczekujemy zaleŜności lg (y) = a + b/x , to wartości xi wymieniane są na 1/xi, natomiast wartości yi zastą-

A. Kufelnicki - Ćwiczenia z biofizyki
18
pione zostają przez lg yi. W dalszej kolejności wybieramy poziom istotności - w typowych opracowaniach 0,05. Otrzymujemy kolejno wartości parametrów a i b, będące podstawą do ewentualnych dalszych obliczeń, średnie odchylenia parametrów regresji sa i sb i połowy przedziałów ufności da oraz db obliczone przy danym poziomie istotności, a takŜe wartość współczynnika korelacji liniowej, r.
Podobne postępowanie pozwala nam porównywać róŜne typy regresji poprzez porówna-nie jakości dopasowania wyników teoretycznych i doświadczalnych, względnie informuje o braku korelacji liniowej przy danym poziomie istotności (inaczej: poprzez testowanie współ-czynnika r nie moŜna odrzucić hipotezy zerowej H0: ρ = 0 o braku korelacji liniowej na da-nym poziomie istotności 1-α ).
Chcąc poprowadzić prostą wystarczy teraz wykorzystać wyniki aproksymacji i obliczyć wartości teoretyczne yi (lub ich funkcji) dla danych lub przekształconych xi , ściśle wg równa-nia prostej najmniejszych kwadratów o parametrach a i b - wzory (3.5.10) i (3.5.11). Aby przeprowadzić prostą regresji, wybieramy dwa dowolne punkty (np. pierwszy i ostatni).
Rys. 3.5. Przeprowadzenie prostej metodą najmniejszych kwadratów dla zaleŜności: lg (y) = a + b/x oraz 1/y = a + bx, b > 0
o - punkty doświadczalne.
JeŜeli dany program stosujący metodę najmniejszych kwadratów nie oblicza wartości rzędnych yi po aproksymacji, to korzystając z równania prostej y = a + b obliczamy wartości rzędnych prostej regresji dla dwóch dowolnych wartości x (moŜliwie odległych od siebie i ła-twych do zaznaczenia na wykresie). Literatura uzupełniająca:
K. DomŜał, E. Gawłowska, „Przykłady zastosowań metod matematycznych i statystycznych w zagadnieniach farmaceutycznych i medycznych”, Akademia Medyczna w Łodzi, 1988. J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 13. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 11.

19
4. POMIAR NIEKTÓRYCH WIELKO ŚCI FIZYCZNYCH 4.1. Ciśnienie
C i ś n i e n i e m nazywamy wielkość fizyczną liczbowo równą sile działającej prosto-padle na jednostkę powierzchni. Jeśli siłę działającą prostopadle do powierzchni S, oznaczy-my przez Fn , to ciśnienie:
S
Fp n= (4.1.1)
W układzie SI jednostką ciśnienia jest N/m2, inaczej paskal (Pa).
C i ś n i e n i e m a t m o s f e r y c z n y m nazywamy stosunek siły wywieranej przez pionowy słup powietrza o wysokości atmosfery do pola powierzchni jego podstawy.
4.1.1. Zasada pomiaru ciśnienia atmosferycznego za pomocą barometru rt ęciowego
Zasadę działania barometru rtęciowego ilustruje doświadczenie z rurką szklaną o długo-ści ok. 1 m, zamkniętą na jednym końcu (Rys. 4.1). Do rurki wlewamy rtęć, zamykamy otwór, odwracamy ją i zanurzamy w naczyniu z rtęcią. Słup rtęci w rurce opada tworząc nad jej po-wierzchnią tzw. próŜnię Torricellego. Wysokość słupa rtęci w rurce liczona w stosunku do poziomu jej swobodnej powierzchni w naczyniu wynosi h. Ciśnienie p wywierane na tym po-ziomie równa się stosunkowi cięŜaru rtęci do pola powierzchni S :
pmg
S= (4.1.2)
Ciśnienie pary rtęci ponad cieczą moŜemy pominąć jako bardzo małe (np. w temp. 20 °C od-powiada ono 0,001 mm Hg). PoniewaŜ masa m równa się iloczynowi gęstości rtęci ρ i objęto-ści V (V = S × h), więc:
pS h g
Sh g= ⋅ ⋅ ⋅ =ρ ρ (4.1.3)
Ciśnienie rtęci wywierane przez słup w rurce całkowicie równowaŜy ciśnienie zewnętrzne wywierane na powierzchnię cieczy w naczyńku (ciśnienie atmosferyczne).
Rys. 4.1. Zasada pomiaru ciśnienia atmos-
ferycznego.

A. Kufelnicki - Ćwiczenia z biofizyki
20
Wartość ciśnienia atmosferycznego wynosi zatem:
ghpatm ρ= (4.1.4)
Wzór (4.1.4) pozwala powiązać podaną poprzednio jednostkę ciśnienia w układzie SI z inny-mi stosowanymi, chociaŜ pozaukładowymi jednostkami, np. atmosferą fizyczną i mm Hg. Ci-śnienie 1 atm fizycznej odpowiada ciśnieniu słupka rtęci o wysokości 760 mm (w skrócie 760 mm Hg) w temp. 0 °C, w tzw. normalnym polu cięŜkości (g = 9,807 m/s2). Podstawiając do wzoru (4.1.4) gęstość rtęci w temp. 0 °C = 13,595 g/cm3 = 13595 kg/m3 otrzymamy: patm = 0,760 m × 13595 kg/m3 × 9,807 m/s2 = 1,0133⋅105 N/m2
(N = kg⋅m/s2) Zatem: 1 atm.fiz = 760 mm Hg (w t = 0 °C, g = 9,807 m/s2) = 1,0133⋅105 Pa = 1013,3 hPa
(1 hPa = 102 Pa).
Spotykaną i tymczasowo uznawaną przez IUPAC jednostką ciśnienia jest teŜ bar. Jeden bar = 105 Pa. Przydatną jednostką pochodną bara jest 1 milibar (mbar = 10-3 bara = 102 Pa), czyli 1 atm.fiz. = 1013,3 mbar.
PoniŜszy przykład ilustruje wykorzystanie ciśnienia atmosferycznego w laboratoryjnej praktyce farmaceutycznej oraz chemicznej. Przykład
W celu rozdzielenia osadu i roztworu, szczególnie w przypadku osadów drobnoziarnistych, często stosuje się sączenie pod zmniejszonym ciśnieniem (Rys. 4.2). Niech ciśnienie atmosferycz-
ne wynosi 750 mm Hg, temperatura 20 °C.
Rys. 4.2. Wykorzystanie ciśnienia atmosferycznego w praktyce laboratoryjnej.
Przed włączeniem pompy na dolne warstwy roztworu oraz na osad działa ciśnienie hydrosta-
tyczne słupa roztworu w sączku, ∆h. Przyjmijmy, Ŝe gęstość roztworu jest bliska gęstości wody ρ
= 103 kg/m3 oraz, Ŝe ∆h = 5 cm = 0,05 m. Ciśnienie atmosferyczne działa z obu stron roztworu - jest takie samo nad powierzchnią cieczy, jak i w kolbie.

4. Pomiar niektórych wielkości fizycznych
21
1) JeŜeli chodzi o otrzymanie klarownego roztworu (np. kropli ocznych) włączamy pompę przy otwartym kranie A, następnie po szybkim odpompowaniu do ciśnienia ok. 20 mm Hg zakrę-camy kran (jeŜeli zauwaŜymy wrzenie roztworu - zwiększamy ciśnienie w kolbie). Na roztwór działa wówczas dodatkowe ciśnienie:
p = 750 − 20 = 730 mm Hg
Jakiej wysokości słupa wody w sączku odpowiada to ciśnienie?
p = ρwody g hwody
ale teŜ: p = ρrtęci g ⋅ 730 mm Hg
czyli: ρwody hwody = ρrtęci ⋅ 730 mm Hg
stąd: m9,931
m0,73013,6 =⋅=wodyh
Widzimy zatem, Ŝe ciśnienie działające na dolne warstwy roztworu wzrasta (9,93+0,05)/0,05 =
9,98/0,05 ≈ 200 razy! Kran A powinien być zamknięty, aby pary roztworu nie opuszczały kolby, przez co zmieniał-by się skład przesączu (przyczyna błędów recepturowych).
2) Przy odsączaniu osadu, kranu A nie musimy zamykać, z tym Ŝe w przypadku stosowania pompy olejowej, pary roztworu powinny być skraplane w tzw. zimnej pułapce (wymraŜaczu), aby zapewnić właściwą eksploatację pompy.
4.1.2. Poprawki wprowadzane przy wyznaczaniu ciśnienia atmosferycznego
PoniewaŜ gęstość rtęci w zakresie zmian temperatury otoczenia jest prawie stała, a zmia-ny przyśpieszenia ziemskiego ze zmianą szerokości geograficznej są niewielkie, ciśnienie moŜna podawać bezpośrednio w mm Hg. Przy dokładniejszych pomiarach musimy jednak pamiętać o wpływie zjawiska rozszerzalności cieplnej rtęci na wysokość słupka rtęci. Dlatego teŜ wysokość słupa rtęci, h, przeliczamy na wysokość h0 w pewnej wybranej temperaturze, np. 0 °C, wprowadzając p o p r a w kę n a z m i a nę w y s o k oś c i s ł u p k a r tę c i z t e m p e r a t u rą .
Obliczmy wysokość słupa rtęci h0 w temperaturze 0 °C, jeśli zmierzona wysokość słupa rtęci w danej temp. t wynosiła h. Z równości ciśnień otrzymamy:
h0 0ρ g = hρg (4.1.5)
gdzie 0ρ - gęstość rtęci w temp. 0 °C, ρ - gęstość rtęci w temperaturze t.
Zatem:

A. Kufelnicki - Ćwiczenia z biofizyki
22
000 ρ
ρρρ
hggh
h == (4.1.6)
Podstawiając:
tγ
ρρ +=1
0 (4.1.7)
(wzór na rozszerzalność objętościową cieczy, gdzie γ - współczynnik rozszerzalności obję-tościowej cieczy; dla rtęci: 0,000182/1 °C ) otrzymamy:
t
hh
γ+=
10 (4.1.8)
PoniewaŜ współczynnik γ jest wielkością bardzo małą, moŜemy zastosować wzór rachunku
przybliŜonego ,011
1
→−≈
+xx
xdla stąd:
)1(0 thh γ−= (4.1.9)
Przy bardzo dokładnych pomiarach musimy takŜe uwzględniać poprawkę na r o z s z e -
r z a l n oś ć c i e p l ną p o d z i a ł k i , na której odczytujemy wysokość słupka rtęci. Odległość dwóch kresek na podziałce jest równa ściśle 1 cm tylko w tej temperaturze, w któ-rej podziałka została sporządzona (zazwyczaj t0 = 0 °C lub 15 °C). W dowolnej temperaturze t odległość dwóch kresek 1 cm odpowiada odległości [1 + α (t − t0)] cm, gdzie α jest współ-czynnikiem rozszerzalności liniowej materiału podziałki, np. dla szkła α = 8⋅10-6/1 °C, dla mosiądzu α = 1,9⋅10-5/1 °C.
Uwzględniając obie dotychczas omawiane poprawki (na rozszerzalność rtęci i podziałki), moŜemy rzeczywistą wysokość słupka rtęci zredukowanego do temperatury 0 °C zapisać wzo-rem: h0 = h [1 + α (t - t0)] (1 - γ t) (4.1.10) Po pomnoŜeniu wyraŜeń w nawiasach i pominięciu wyrazu zawierającego bardzo mały ilo-czyn α×γ, otrzymamy: h0 = h [1 − α t0 − (γ − α)t] (4.1.11) JeŜeli podziałka była cechowana w t0 = 0 °C, to: h0 = h [1− (γ − α)t] (4.1.12) Poprawka h (γ − α) wynosi ok. 1/8 mm Hg na 1 °C. Przykład
Obliczyć wartość poprawki na rozszerzalność rtęci i podziałki mosięŜnej w t = 20 °C, jeŜeli podziałka była cechowana w temp. 0 °C, a odczytana wartość h wynosiła 750 mm Hg. Stosując wzór (4.1.12) otrzymujemy:

4. Pomiar niektórych wielkości fizycznych
23
h0 = h [1 − (γ − α)t] = 750 [1 − (1,8⋅10-4 − 1,9⋅10-5) ⋅20] = 750 (1 − 3,22⋅10-3) = 747,585 mm Hg
Wobec tego wysokość słupa rtęci zredukowana do 0 °C wynosi h0 ≈ 747,6 mm Hg, a wartość po-
prawki h (γ − α) t ≈ 2,4 mm Hg.
W praktyce najczęściej nie obliczamy poprawek, lecz korzystamy z wartości stablicowa-nych dla danego barometru. W bardzo dokładnych pomiarach uwzględniane mogą być takŜe inne poprawki, jak poprawka na obniŜenie menisku rtęci w rurce na skutek zjawiska włosko-watości, na obniŜenie menisku przez ciśnienie pary znajdującej się nad powierzchnią rtęci oraz na zmianę cięŜaru ze zmianą szerokości geograficznej.
4.1.3. Typy barometrów
Do pomiarów ciśnienia atmosferycznego stosujemy zazwyczaj dwa typy barometrów rtę-ciowych (Rys. 4.3) - naczyniowy (Fortina) i lewarowy, a takŜe barometry spręŜynowe (anero-idy lub barografy).
a) Do dokładnych pomiarów ciśnienia uŜywamy b a r o m e t r u n a c z y n i o w e g o , który jest rurką szklaną zatopioną na jednym końcu, napełnioną rtęcią i zanurzoną drugim końcem w zbiorniku z rtęcią. MoŜliwość osiągnięcia duŜej dokładności pomiarów polega na tym, Ŝe poziom rtęci w zbiorniku zmieniamy przez przesuwanie zbiornika w pionie za pomocą odpowiedniej śruby. Poziom ten moŜemy ustawiać dokładnie na stałej wysokości oznaczonej przez dolny koniec ostrzy (ostrza) przytwierdzonych pionowo do pokrywy zbiornika nad powierzchnią rtęci i skierowanych do tej powierzchni. Wymieniony po-ziom jest jednocześnie początkiem podziałki barometru. PołoŜenie górnego menisku rtęci wyznaczamy z dokładnością do 0,1 mm za pomocą noniusza umieszczonego na metalo-wej rurce otaczającej rurkę barometru i przesuwającej się przy obrocie śruby.
b) B a r o m e t r l e w a r o w y jest rurką szklaną o średnicy 8 - 9 mm u dołu rozszerzo-ną (do szerokiego, otwartego naczyńka). Górny koniec jest zatopiony. Przed pomiarem zero w skali barometru ustawiamy na poziomie linii dolnego menisku rtęci. PołoŜenie tej linii na skutek duŜej szerokości naczyńka zmienia się nieznacznie. Barometrów tego typu uŜywamy w pomiarach, w których dokładność do 1 mm jest wystarczająca.
c) Do pomiarów przybliŜonych słuŜą a n e r o i d y , w których miarą ciśnienia jest wiel-kość odkształceń szczelnej, metalowej puszki (pofałdowanej dla większej elastyczności). Z puszki uprzednio wypompowano powietrze. Odkształcenia jej są przenoszone za po-średnictwem układu dźwigni na wskazówkę ukazującą na wyskalowanej tarczy wartość ciśnienia atmosferycznego. W wyniku połączenia aneroidu z urządzeniem samopiszącym powstaje tzw. barograf.

A. Kufelnicki - Ćwiczenia z biofizyki
24
Rys. 4.3. Barometry róŜnych typów:
a ) barometr naczyniowy, b) barometr lewarowy, c) aneroid. Ćwiczenie 1
Pomiar ciśnienia atmosferycznego za pomocą barometru rtęciowego Wykonanie pomiarów:
1. Porównać cechy barometru znajdującego się w pracowni z cechami barometrów naczynio-wego i lewarowego.
2. Przesunąć za pomocą śruby podziałkę noniusza tak, aby dolna krawędź podziałki (zero) pokrywała się z górnym meniskiem rtęci. Ustawienie zera najlepiej wykonać zamykając jedno oko - przednia i tylna krawędź rurki powinny pokrywać się na poziomie górnej po-wierzchni menisku.
3. Odczytać całkowite milimetry przy pierwszej kresce pod zerem noniusza. Dziesiętne części milimetra odczytujemy przy tej kresce noniusza, która pokrywa się z jedną z kresek skali głównej.
4. Odczytać temperaturę t na termometrze znajdującym się przy barometrze. Temperatura, w której została sporządzona podziałka wynosi t0 = 0 °C.
5. Obliczyć zredukowaną wysokość słupa rtęci wg wzoru (4.1.12). Porównać uzyskaną war-tość poprawki z wartością zamieszczoną w tabeli przy barometrze - współczynnik rozszerzalności objętościowej rtęci γ = 1,82⋅10-4 - współczynnik rozszerzalności liniowej podziałki mosięŜnej α = 1,9⋅10-5
4. Pomiar niektórych wielkości fizycznych
25
Tabela wyników pomiarów i wielkości obliczonych:
Ciśnienie odczytane,
h
(mm Hg)
Temperatura,
t
(°C)
Temperatura cechowania podziałki,
t0
Zredukowana wysokość słupka rtęci,
h0
(mm Hg)
Poprawka
∆h = h - h0
(mm Hg)

A. Kufelnicki - Ćwiczenia z biofizyki
26
4.2. Gęstość i cięŜar właściwy
G ę s t oś c i ą (inaczej: masą właściwą) nazywamy stosunek masy danego ciała do jego objętości:
ρ = m
V (4.2.1)
Jednostką gęstości w układzie SI jest kg/m3. Często uŜywaną jednostką jest g/cm3 (jednostka z dawniej stosowanego układu CGS).
Gęstość jest wielkością charakterystyczną dla danego rodzaju substancji i stałą przy okre-ślonej temperaturze i danym ciśnieniu. Podobnie jak masa, gęstość jest wielkością niezaleŜną od tego, w jakim miejscu Ziemi ją wyznaczono.
Z tego względu jest ona wielkością wygodniejszą w uŜyciu niŜ c i ę Ŝ a r w ł aś c i -w y . CięŜar właściwy jest równy stosunkowi cięŜaru ciała do jego objętości:
γ = Q
V (4.2.2)
PoniewaŜ cięŜar ciała na Ziemi jest równy iloczynowi jego masy i przyśpieszenia w danym punkcie pola grawitacyjnego Ziemi, Q = m g, więc podstawiając do wzoru (4.2.2) otrzymuje-my związek między cięŜarem właściwym a gęstością:
γ ρ= =mg
Vg (4.2.3)
Jeśli gęstość mierzymy w kg/m3, to cięŜar właściwy ma wymiar N/m3.
Spotykaną jeszcze jednostką cięŜaru właściwego jest kG/m3 (G/cm3). Występujący w niej kilogram cięŜarowy jest równy sile, z jaką Ziemia przyciąga masę 1 kg w danym miejscu (da-nej szerokości geograficznej). Jeśli przyjąć, Ŝe średnia wartość przyśpieszenia ziemskiego wynosi 9,80665 m/s2 ≈ 9,81 m/s2, to:
1 kG(siły) = 1 kg(masy) × 9,81 m/s2 = 9,81 N
Zaletą jednostki kG/m3 jest to, Ŝe gęstość ciała i jego cięŜar właściwy w danym miejscu są liczbowo równe. Niech g będzie średnim przyśpieszeniem ziemskim. Wówczas ciało o gę-stości 1 kg/m3 ma cięŜar właściwy:
1 kg/m3×9,81 m/s2 = 9,81 N/m3 = 1 kG/m3
4.2.1. Pomiar gęstości ciał stałych i cieczy za pomocą piknometru
Jedną z dokładniejszych metod pomiaru gęstości ciał stałych i cieczy jest metoda pikno-metryczna.
Piknometrem nazywamy szklaną kolbkę ze szklanym korkiem dokładnie doszlifowanym. Przez korek przechodzi rurka włoskowata (Rys. 4.4a). Niektóre typy piknometrów mają za-miast korka termometr ze szlifem, rurka włoskowata znajduje się obok (Rys. 4.4c). Przez tę rurkę wylewa się nadmiar cieczy wlanej do piknometru.

4. Pomiar niektórych wielkości fizycznych
27
a) b) c)
Rys. 4.4. Niektóre typy piknometrów.
W innych typach piknometrów (Rys. 4.4b) występuje jedynie termometr ze szlifem speł-niający rolę korka. Aby zmierzyć gęstość ciała stałego, waŜymy najpierw piknometr napełnio-ny wodą (masa m1), po wytarciu bibułą jego zewnętrznych ścian zwilŜonych przez wodę wy-lewającą się przy zamykaniu piknometru. Następnie waŜymy badane ciało (m2), wrzucamy je do piknometru z wodą i po zamknięciu korkiem oraz ponownym osuszeniu waŜymy ponow-nie cały piknometr (m3). JeŜeli masę piknometru pustego oznaczymy przez m0 , to (m1 - m0) stanowi masę wody wypełniającej piknometr przed wrzuceniem badanego ciała, a (m3 - m2 - m0) oznacza masę wody, która pozostała po wrzuceniu ciała stałego. Stąd masa wody wy-pchniętej przez to ciało wynosi:
321231 )()( 00 mmmmmmmm −+=−−−− Objętość wypchniętej wody jest równa objętości badanego ciała, bVV =OH2
. Z definicji gę-
stości (wzór (4.2.1)) otrzymamy więc:
b
mmmmρρ
2
OH
321
2
=−+
(4.2.4)
stąd
OH321
22
ρρ ⋅−+=mmm
mb (4.2.5)
Jak wynika z równania (4.2.5), w piknometrycznych pomiarach gęstości ciała stałego niepo-trzebne jest waŜenie pustego piknometru, m0
. Przy pomiarze gęstości cieczy naleŜy najpierw wyznaczyć masę pustego (suchego) pik-
nometru, m0 . Następnie wyznaczamy masę piknometru wypełnionego wodą destylowaną (m1)
oraz masę piknometru napełnionego cieczą badaną (m2). Znając gęstość wody w temperaturze pomiaru (z tablic) moŜemy określić objętość piknometru:
OH
1
2
0
ρmm
V−
= (4.2.6)
oraz gęstość cieczy:
OH1
222
0
00 ρρmmmm
V
mm−−
=−
= (4.2.7)

A. Kufelnicki - Ćwiczenia z biofizyki
28
Tabela 4.1
Gęstość wody w róŜnych temperaturach
t ( °C) ρH O2 (g/cm3) t ( °C) ρH O2
(g/cm3)
10 0,9997 20 0,9982
11 0,9996 21 0,9980
12 0,9995 22 0,9978
13 0,9994 23 0,9975
14 0,9992 24 0,9973
15 0,9991 25 0,9971
16 0,9989 26 0,9968
17 0,9988 27 0,9965
18 0,9986 28 0,9962
19 0,9984 29 0,9959
Literatura uzupełniająca:
J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 27. Ćwiczenie 2
Pomiar gęstości cieczy za pomocą piknometru
Wykonanie pomiarów: 1. JeŜeli piknometr nie jest suchy, naleŜy wymyć piknometr wodą destylowaną, przepłukać
mieszaniną osuszającą i wysuszyć. 2. Wyznaczyć masę m0 suchego piknometru za pomocą wagi analitycznej (waŜenie wstępne
na wadze technicznej!). 3. Napełnić piknometr wodą destylowaną, starannie wytrzeć bibułą zewnętrzne ścianki pikno-
metru, odczytać temperaturę wody, wyznaczyć masę m1. 4. Wylać wodę z piknometru, starannie osuszyć i zwaŜyć piknometr napełniony cieczą badaną
(masa m2). 5. Obliczyć gęstość badanej cieczy ze wzoru (4.2.7). Tabela wyników pomiarów i wielkości obliczonych:
Temperatura, t ...............°C
Masa piknometru, m0 ............... g
Masa piknometru z wodą, m1 ............... g
Masa piknometru z badaną cieczą, m2 ............... g
Gęstość wody w temp. pomiaru ......... g/cm3
Gęstość badanej cieczy ......... g/cm3

4. Pomiar niektórych wielkości fizycznych
29
4.3. Lepkość
We wszystkich cieczach znajdujących się w ruchu pojawiają się, w wyniku oddziaływań międzycząsteczkowych, siły styczne do kierunku ruchu zwane si ł a m i l e p k oś c i lub t a r c i a w e w nę t r z n e g o . Jeśli wyobrazimy sobie, Ŝe ciecz złoŜona jest z wielu warstw o bardzo małej grubości, to siły tarcia wewnętrznego działają w ten sposób, Ŝe od strony warstwy poruszającej się szybciej działa na warstwę poruszającą się wolniej siła przy-śpieszająca i odwrotnie - od strony warstwy poruszającej się wolniej działa na warstwę szyb-szą siła hamująca.
Rys. 4.5. Siła tarcia wewnętrznego w cieczy.
Zgodnie z równaniem Newtona siła tarcia wewnętrznego F pomiędzy dwiema sąsiednimi warstwami o powierzchni S jest wprost proporcjonalna do pola powierzchni S i gradientu prędkości w kierunku prostopadłym do kierunku ruchu, dv/dx:
dx
dvSF η= (4.3.1)
Gradient (spad) prędkości dv/dx jest wielkością wskazującą, jak szybko zmienia się prędkość przy przechodzeniu z jednej warstwy do drugiej. Współczynnik proporcjonalności, η, zaleŜny od rodzaju cieczy, nazywamy w s p ó ł c z y n n i k i e m l e p k oś c i d y n a m i c z n e j ( w skrócie: w s p ó ł c z y n n i k i e m l e p k oś c i lub l e p k oś c i ą d y n a -m i c z ną ) . JeŜeli siłę F wyrazimy w N, powierzchnię S w m2, prędkość v w m/s, a odle-głość warstw dx w m, to jednostką lepkości dynamicznej będzie N⋅s/m2 (niutonosekunda na metr kwadratowy) lub inaczej Pa⋅s (paskalosekunda). Często spotykaną jednostką z dawnego układu CGS jest puaz (P) lub wygodna jednostka pochodna centypuaz (cP) = 10-2 P. Związki pomiędzy wymienionymi jednostkami: 1 P = 0,1 N⋅s/m2 (4.3.2)
1 cP = 10-3 N⋅s/m2 (4.3.3)
Np. w t = 20 °C lepkość dynamiczna wody wynosi 1,005 cP, rtęci - 1,554 cP, eteru etylowego 0,233 cP, a 93% gliceryny - ok. 400 cP.
Stosunek lepkości dynamicznej, η, do gęstości cieczy, ρ, nazywamy l e p k oś c i ą k i -n e m a t y c z ną (ν = η/ρ) i określamy w m2⋅s-1; w praktyce stosuje się jeszcze jednostkę mm2⋅s-1 zwaną centystokesem (cSt).
Dla większości cieczy, w tym dla czystych rozpuszczalników i bardzo rozcieńczonych roztworów, spełnione jest równanie (4.3.1). Takie ciecze nazywamy idealnie lepkimi lub n i u t o n o w s k i m i . Lepkość ich jest stała w danej temperaturze i przy danym ciśnieniu. W praktyce farmaceutycznej częściej występują jednak substancje ciekłe, które nie spełniają

A. Kufelnicki - Ćwiczenia z biofizyki
30
równania Newtona (nieniutonowskie); naleŜą do nich układy koloidalne, emulsje, ciekłe za-wiesiny, maści, pasty itp.
Miarą róŜnicy lepkości roztworu i rozpuszczalnika jest l e p k oś ć w ł a ś c i w a wy-raŜona wzorem:
100
0wł −=
−= η
ηη
ηηη (4.3.4)
gdzie η i η0 - oznaczają odpowiednio lepkość roztworu i rozpuszczalnika. Cechą charak-terystyczną lepkości właściwej roztworu jest m.in. słabsza niŜ dla lepkości roztworu zaleŜność od temperatury, poniewaŜ w wyraŜeniu η/η0 zwanym l e p k oś c i ą w z g l ę d n ą zarów-no licznik, jak i mianownik maleją wraz ze wzrostem temperatury (choć w róŜnym stopniu).
4.3.1. ZaleŜność lepkości cieczy od temperatury
Lepkość cieczy maleje wraz ze wzrostem temperatury. ZaleŜność tę dla wielu cieczy opi-suje z duŜą dokładnością empiryczne równanie Arrheniusa-Guzmana:
RTElA /e∆=η (4.3.5) lub po logarytmowaniu:
RT
EA l∆
+= lnlnη (4.3.6)
gdzie: A - stała zaleŜna od masy molowej i objętości molowej substancji R - stała gazowa
∆El - energia aktywacji przepływu lepkiego równa najmniejszej nadwyŜce energii w stosunku do wartości średniej, jaką musi posiadać cząsteczka, aby móc poruszać się pomiędzy cząsteczkami sąsiednimi; wartość tę określa się na 1 mol cząsteczek
Równanie (4.3.5) znajduje poglądowe (choć nie tak zadowalające, jak w stosunku do lepkości gazów) uzasadnienie w teorii kinetyczno-molekularnej po wprowadzeniu pojęcia płynności. P ł y n n oś ć jest odwrotnością lepkości:
η
ϕ 1= (4.3.7)
WyraŜa ona tendencję do płynięcia cieczy w odróŜnieniu od lepkości określającej opór prze-ciwko płynięciu. Proces przepływu moŜna porównać do przeciskania się cząsteczek pomiędzy sąsiednimi cząsteczkami. Oznaczmy przez ∆El najmniejszą energię, jaką musi posiadać czą-steczka, aby oderwać się od cząsteczek sąsiednich i móc poruszać się swobodnie pomiędzy warstwami. W danej temperaturze liczba takich cząsteczek jest wprost proporcjonalna do wy-raŜenia wykładniczego określonego znanym z teorii kinetyczno-molekularnej gazów wzorem Boltzmanna. MoŜna więc napisać, Ŝe równieŜ płynność cieczy jest proporcjonalna do tego wyraŜenia:
ϕ ~ RTEl∆−e (4.3.8)

4. Pomiar niektórych wielkości fizycznych
31
Biorąc odwrotność obu stron proporcjonalnej zaleŜności (4.3.8), otrzymamy:
η = 1ϕ ~
RTEl∆e (4.3.9)
lub, zapisując w postaci równania:
RTElA /e∆=η (4.3.10)
Interesujące jest porównanie wartości ∆El z ciepłem parowania cieczy, ∆Epar (∆Epar okre-śla energię konieczną do całkowitego oderwania cząsteczki od cząsteczek sąsiednich). Okazu-je się, Ŝe na ogół energia aktywacji przepływu lepkiego wynosi tylko ok. 1/3 wartości ∆Epar. Wynika to prawdopodobnie z faktu, Ŝe cząsteczka w czasie przepływu warstwowego moŜe wykonywać ruchy w przestrzeni dwuwymiarowej, na co idzie 1/3 tej energii, którą potrzebo-wałaby ta cząsteczka w zjawisku parowania, aby opuścić ciecz i przejść do fazy gazowej – ruch w trzech wymiarach.
4.3.2. Metody pomiaru współczynnika lepkości Omówimy dwa spośród róŜnych typów wiskozymetrów (przyrządów do pomiaru współ-
czynnika lepkości) stosowanych do układów o niezbyt duŜych lepkościach: wiskozymetry Ostwalda i Höpplera. W przypadku maści i past stosuje się róŜnego rodzaju wiskozymetry ro-tacyjne (obrotowe). a) Wiskozymetr Ostwalda
Metoda pomiaru opiera się na prawie Poiseulle'a opisującym laminarny (nie burzliwy) przepływ cieczy przez rurki kapilarne:
η
πl
ptrV
8
4 ∆= (4.3.11)
gdzie V - objętość cieczy, która przepływa w czasie t przez kapilarę o promieniu r i długości l, pod wpływem róŜnicy ciśnień ∆p.
Ze wzoru (4.3.11) wynika, Ŝe współczynnik lepkości wyraŜa równanie:
lV
ptr
8
4 ∆=
πη (4.3.12)
Dla danej kapilary wiskozymetru promień, długość i objętość przepływającej cieczy są niez-mienne (Rys. 4.6), stąd równanie (4.3.12) moŜna zapisać w postaci: pKt∆=η (4.3.13) gdzie K jest stałą. Z kolei wiemy, Ŝe róŜnica ciśnień ∆p jest wytworzona przez ciśnienie hy-drostatyczne słupka cieczy o wysokości równej róŜnicy poziomów w obu ramionach wisko-zymetru, ∆h: hgp ∆=∆ ρ (4.3.14)
gdzie ρ - gęstość cieczy, g - przyśpieszenie ziemskie.

A. Kufelnicki - Ćwiczenia z biofizyki
32
Rys. 4.6. Wiskozymetr kapilarny Ostwalda.
JeŜeli napełniamy wiskozymetr stale taką samą objętością cieczy, to róŜnica ∆p zmienia
się w czasie przepływu cieczy między poziomami (a) i (b) jednakowo dla wszystkich cieczy, a jej średnia wartość (∆pśr) jest stała. Zatem:
ρη tK ′= (4.3.15) gdzie śrhKgK ∆=′ . Dzieląc stronami wyraŜenie typu (4.3.14) dla cieczy badanej i wzorcowej
otrzymamy:
www t
tρρ
ηη = (4.3.16)
Widzimy zatem, Ŝe pomiar lepkości w wiskozymetrze Ostwalda polega na porównaniu
czasu przepływu cieczy pomiędzy dwoma poziomami (a) i (b) dla cieczy badanej i wzorco-wej. Niedogodność w zastosowaniu wiskozymetru Ostwalda, związaną z koniecznością na-pełniania go stale jednakową objętością cieczy badanej i wzorcowej, usunięto w modyfikacji zwanej wiskozymetrem kapilarnym Ubbelohde’a, w którym zostaje ponadto wyeliminowany wpływ napięcia powierzchniowego na wynik pomiaru czasu przepływu. Wiskozymetr Ostwalda (najczęściej w wersji Ubbelohde’a) stosowany jest głównie do pomiaru lepkości strukturalnej, uwarunkowanej strukturą danego ciała. b) Wiskozymetr Höpplera
W wiskozymetrze Höpplera mierzy się szybkość opadania kulki szklanej lub metalowej w badanej cieczy. Na kulkę spadającą pionowo w środowisku lepkim działają 3 siły: 1) siła cięŜkości Q = 4/3π r3ρk g, gdzie r - promień kulki, ρk - gęstość materiału, z którego wy-
konana jest kulka, g - przyśpieszenie ziemskie; 2) siła wyporu (parcie do góry) P = 4/3π r3ρc g , gdzie ρc - gęstość cieczy; 3) siła tarcia wewnętrznego (opór środowiska), która wg prawa Stokesa wynosi F = 6πrηv,
gdzie η - lepkość dynamiczna cieczy, v - prędkość opadania kulki.

4. Pomiar niektórych wielkości fizycznych
33
Siła tarcia wewnętrznego F wzrasta wraz ze wzrostem prędkości kulki, aŜ do chwili, gdy na-stąpi zrównowaŜenie się wszystkich trzech sił : 0=−− PFQ (4.3.17) Wówczas, zgodnie z I zasadą dynamiki, kulka porusza się ruchem jednostajnym ze stałą pręd-kością v. Z równania (4.3.17) PQF −= (4.3.18) i
( )ckgrvr ρρπηπ −= 3
3
46 (4.3.19)
Jeśli prędkość kulki wyrazimy jako stosunek drogi s do czasu t, to wyraŜenie na lepkość przyjmie postać:
( )tsgr
ck ρρη −= 92 2
(4.3.20)
Wzór (4.3.20) stosuje się wówczas, gdy kulka opada w cieczy znajdującej się w naczyniu o tak duŜych wymiarach, Ŝe moŜna pominąć wpływ ścian na ruch kulki.
W wiskozymetrze Höpplera (Rys. 4.7) kulka opada w rurce szklanej o dość małym pro-mieniu wewnętrznym R . Poza tym rura ta posiada niewielkie odchylenie od pionu, skutkiem czego kulka uzyskuje odpowiednie prowadzenie i nie odbija się od ścianek. Stąd teŜ w wyra-Ŝeniu stosowanym praktycznie do obliczania lepkości: ( )tK ck ρρη −= (4.3.21)
stała K - (tzw. stała kulki) róŜni się znacznie od wartości 2r2g/9s wynikającej ze wzoru (4.3.20). Wartość stałej dla danej kulki jest więc wyznaczana doświadczalnie (za pomocą cie-czy o znanej lepkości) i podawana w instrukcji przyrządu.
Rys. 4.7. Wiskozymetr Höpplera.

A. Kufelnicki - Ćwiczenia z biofizyki
34
Wiskozymetr Höpplera umoŜliwia pomiar lepkości cieczy niutonowskich w granicach 10-5 - 103 N⋅s⋅m-2. Rurka, w której opada kulka jest termostatowana płaszczem wodnym. Końce jej zaopatrzone są w nakrętki, które zdejmuje się przy wprowadzaniu lub usuwaniu cieczy i kulki. WzdłuŜ rurki zaznaczony jest stały odcinek drogi (100 mm) i połowa tego dys-tansu (50 mm). Rurka wraz z płaszczem wodnym jest umocowana w statywie, który umoŜli-wia obrót jej w pionie o 180°. Przez odpowiedni obrót rurki, kulkę wprowadza się do góry i mierzy czas jej opadania na wyznaczonej drodze z dokładnością do 0,1 s. Literatura uzupełniająca:
F. Jaroszyk, red., „Biofizyka”, Wydawnictwo Lekarskie PZWL, Warszawa 2002, str. 87. T. W. Hermann, red., „Farmacja fizyczna”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 104. J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 47. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 102. Ćwiczenie 3
Badanie wpływu temperatury na lepkość dynamiczną i właściwą roztworów Wykonanie pomiarów:
1. Włączyć ultratermostat utrzymujący stałą temperaturę w płaszczu wodnym wiskozymetru Höpplera. Termometr kontaktowy w termostacie ustawić na 15 ± 0,5 °C.
2. Przygotować 20% roztwór wodny sacharozy: 20 g cukru + 80 g wody.
3. Napełnić wiskozymetr roztworem sacharozy, a następnie wprowadzić szklaną kulkę o znanej wartości stałej K. Starannie zamknąć rurę pomiarową nakrętką z uszczelką. W przypadku pojawienia się pęcherzyka powietrza odwracamy wiskozymetr tak, aby pęche-rzyk był nad kulką, otwieramy górną nakrętkę i po zniknięciu pęcherzyka ponownie do-pełniamy rurkę pomiarową.
4. Po ustaleniu się temperatury (odczytujemy w termometrze wiskozymetru), obrócić wi-skozymetr o 180° i zmierzyć czas opadania kulki na całym dystansie (100 mm) - trzy-krotnie. Obliczyć średnią arytmetyczną czasów.
5. PodwyŜszyć temperaturę o 1,5 - 3° w stosunku do poprzedniej, np. w serii: 15, 17, 19, 21, 23, 25 °C i ponownie zmierzyć czas opadania jak w punkcie 4.
6. Wyniki pomiarów (czas opadania kulki w sekundach) wpisać do tabeli 1.
7. Lepkość dynamiczną roztworu obliczyć ze wzoru (4.3.21) przyjmując stałą kulki K = 9,90 ⋅10-9 N⋅m⋅kg-1. Gęstość kulki kρ wynosi 2,408 ⋅103 kg/m3. Gęstość roztworu sa-
charozy, cρ , w danej temperaturze odczytać z Tab. 4.2 na końcu rozdziału.
8. Obliczyć lepkość właściwą roztworu ze wzoru (4.3.4), przy czym lepkość wody w danej temperaturze, 0η , odczytać z Tab. 4.3.
9. Narysować wykres zaleŜności log η = f (1/T) i korzystając z programu komputerowego obliczyć współczynnik korelacji liniowej, r, wartość współczynników prostej
4. Pomiar niektórych wielkości fizycznych
35
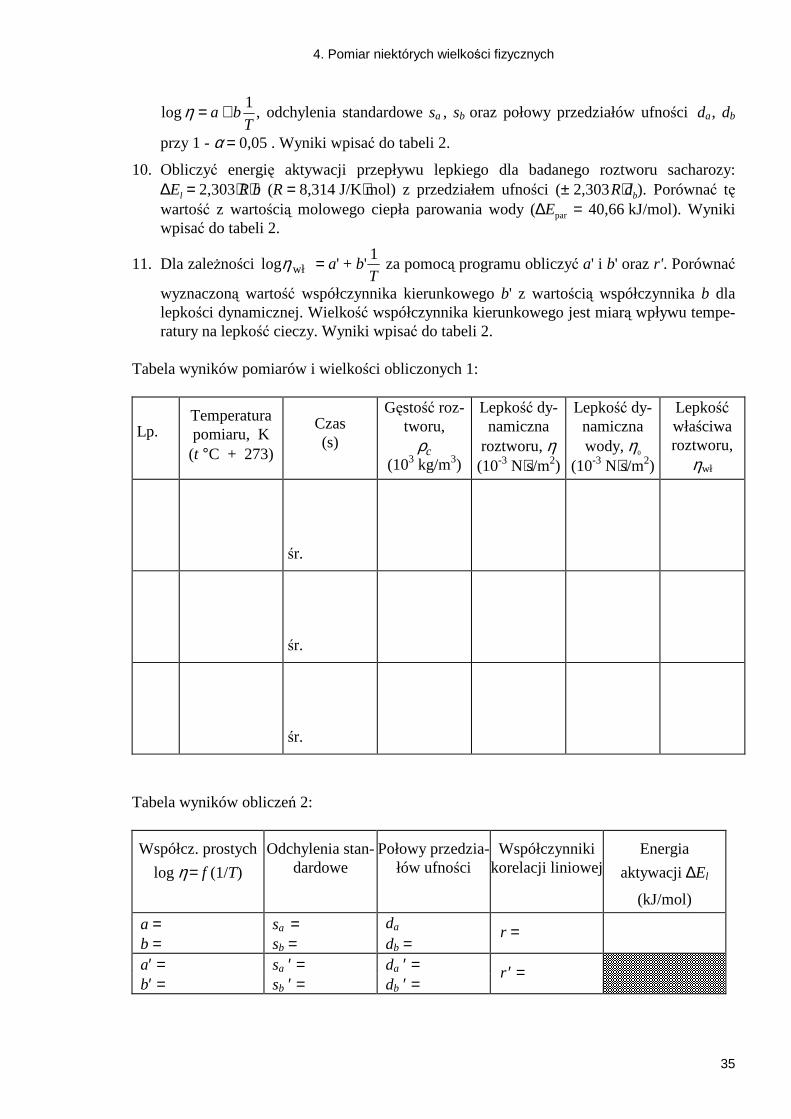
logη = +a bT
1, odchylenia standardowe sa , sb oraz połowy przedziałów ufności da , db
przy 1 - α = 0,05 . Wyniki wpisać do tabeli 2.
10. Obliczyć energię aktywacji przepływu lepkiego dla badanego roztworu sacharozy: ∆El = 2,303⋅R⋅b (R = 8,314 J/K⋅mol) z przedziałem ufności (± 2,303 R⋅db). Porównać tę wartość z wartością molowego ciepła parowania wody (∆Epar = 40,66 kJ/mol). Wyniki wpisać do tabeli 2.
11. Dla zaleŜności włlogη = a' + b'1T
za pomocą programu obliczyć a' i b' oraz r' . Porównać
wyznaczoną wartość współczynnika kierunkowego b' z wartością współczynnika b dla lepkości dynamicznej. Wielkość współczynnika kierunkowego jest miarą wpływu tempe-ratury na lepkość cieczy. Wyniki wpisać do tabeli 2.
Tabela wyników pomiarów i wielkości obliczonych 1:
Lp. Temperatura pomiaru, K (t °C + 273)
Czas (s)
Gęstość roz-tworu,
ρc (103 kg/m3)
Lepkość dy-namiczna
roztworu, η (10-3 N⋅s/m2)
Lepkość dy-namiczna wody, η
0
(10-3 N⋅s/m2)
Lepkość właściwa roztworu,
ηwł
śr.
śr.
śr.
Tabela wyników obliczeń 2:
Współcz. prostych
log η = f (1/T)
Odchylenia stan-dardowe
Połowy przedzia-łów ufności
Współczynniki korelacji liniowej
Energia
aktywacji ∆El
(kJ/mol)
a = sa = da r =
b = sb = db =
a′ = sa ′ = da ′ = r ′ =
b′ = sb ′ = db ′ =
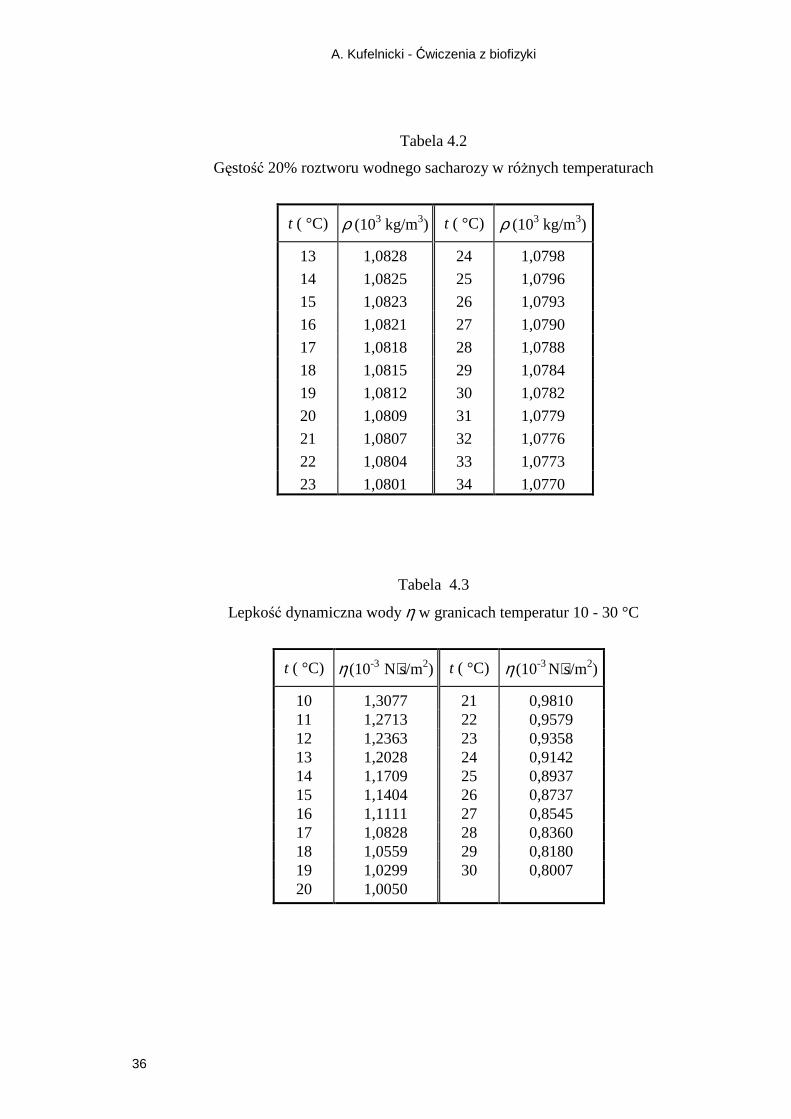
A. Kufelnicki - Ćwiczenia z biofizyki
36
Tabela 4.2
Gęstość 20% roztworu wodnego sacharozy w róŜnych temperaturach
t ( °C) ρ (103 kg/m3) t ( °C) ρ (103 kg/m3)
13 1,0828 24 1,0798
14 1,0825 25 1,0796
15 1,0823 26 1,0793
16 1,0821 27 1,0790
17 1,0818 28 1,0788
18 1,0815 29 1,0784
19 1,0812 30 1,0782
20 1,0809 31 1,0779
21 1,0807 32 1,0776
22 1,0804 33 1,0773
23 1,0801 34 1,0770
Tabela 4.3
Lepkość dynamiczna wody η w granicach temperatur 10 - 30 °C
t ( °C) η (10-3 N⋅s/m2) t ( °C) η (10-3 N⋅s/m2)
10 1,3077 21 0,9810 11 1,2713 22 0,9579 12 1,2363 23 0,9358 13 1,2028 24 0,9142 14 1,1709 25 0,8937 15 1,1404 26 0,8737 16 1,1111 27 0,8545 17 1,0828 28 0,8360 18 1,0559 29 0,8180 19 1,0299 30 0,8007 20 1,0050

4. Pomiar niektórych wielkości fizycznych
37
4.4. Napięcie powierzchniowe
Powierzchniowa warstwa cząsteczek cieczy granicząca z fazą gazową charakteryzuje się odmiennymi właściwościami od pozostałej ich części, znajdującej się w głębi. Cząsteczki znajdujące się na powierzchni podlegają oddziaływaniu innych cząsteczek jedynie od strony wnętrza cieczy. Asymetria ta powoduje wciąganie cząsteczek do wnętrza, co przy stałej obję-tości cieczy prowadzi do rozwinięcia jak najmniejszej powierzchni oraz wytwarza stan me-chanicznego napięcia. Objawia się to szeregiem dostrzegalnych zjawisk, np. powierzchnia cieczy wykazuje właściwości mechanicznej błonki o pewnej spręŜystości; kropla cieczy ma kształt kulisty, a więc o najmniejszej powierzchni, jaką moŜe przyjąć dana objętość. Powie-rzchnia nie jest tworem dwuwymiarowym, ale stanowi warstwę o pewnej skończonej grubo-ści. Zagadnienie struktury tej warstwy, jej grubości, sił i ciśnień w niej panujących naleŜy do bardzo złoŜonych problemów teoretycznych oraz eksperymentalnych.
Zwiększenie powierzchni cieczy wymaga wykonania pracy. Oczywiście, zmniejszenie się powierzchni spowoduje wydzielenie tej pracy (energii). Swobodna powierzchnia cieczy po-siada więc pewien zapas energii zwany energią powierzchniową. Stosunek energii powierzch-niowej do powierzchni danej cieczy nazywa się n a p ię c i e m p o w i e r z c h n i o w y m .
S
Epow=σ (4.4.1)
Wymiarem napięcia powierzchniowego jest J/m2 lub po uproszczeniu - N/m. W przypadku, gdy powierzchnia cieczy jest granicą dwóch faz ciekłych, mówimy o g r a n i c z n y m n a -p i ę c i u m i ę d z y f a z o w y m (lub po prostu o napięciu międzyfazowym).
Jak wiadomo, w przemianach samorzutnych następuje zmniejszenie energii układu. Stąd pochodzi dąŜność do zmniejszenia powierzchni i do zmniejszenia napięcia powierzchniowe-go. Wyrazem tego jest zlewanie się małych kulek rtęci w jedną większą o powierzchni mniej-szej, niŜ suma powierzchni kilku małych kulek.
Właściwości błony powierzchniowej moŜna uwidocznić rozpinając cienką błonkę mydla-ną na ramce drucianej z ruchomą poprzeczką AB , jak na Rys. 4.8.
Rys. 4.8. Właściwości błonki powierzchniowej. Wyobraźmy sobie, Ŝe działając na poprzeczkę AB siłą F stale równą sile zdąŜającej do zmniejszenia powierzchni przesunęliśmy ją na odległość h. Wykonaliśmy pracę W = ∆F⋅h. Uzyskana energia powierzchniowa Epow = W przypada na powierzchnię S = 2⋅ l⋅h (obie strony ramki). Napięcie powierzchniowe wyrazi się więc wzorem:
l
F
hl
hF
22==σ (4.4.2)

A. Kufelnicki - Ćwiczenia z biofizyki
38
W Tabeli 4.5 podano napięcia powierzchniowe kilku cieczy w temperaturze 20 °C. Zwracają uwagę stosunkowo wysokie napięcia powierzchniowe rtęci, szkła i wody. Wysoka wartość napięcia powierzchniowego związana jest z silnymi oddziaływaniami międzyczą-steczkowymi w danej cieczy.
Płyny fizjologiczne ustroju ludzkiego wykazują niŜsze napięcie powierzchniowe niŜ wo-da, poniewaŜ obniŜa je szereg związków: glikogen, kwasy Ŝółciowe, garbniki, lecytyna, estry i tłuszcze. Zmiany napięcia powierzchniowego moczu lub osocza krwi w róŜnych stanach cho-robowych mogą być czynnikami diagnostycznymi.
ObniŜenie napięcia powierzchniowego cieczy powoduje wzrost zdolności zwilŜania po-wierzchni ciał stałych. Fakt ten ma znaczenie przy sporządzaniu płynów owadobójczych, w procesach prania, farbowania oraz w procesach przemysłowych (flotacja).
Tabela 4.5
Napięcia powierzchniowe róŜnych cieczy w temp. 293 K
Ciecz σ (mJ/m2)
woda 72,75
gliceryna 64,00
anilina 42,58
benzen 28,80
kwas octowy 23,50
alkohol etylowy 22,03
alkohol metylowy 22,50
eter dwuetylowy 16,60
rtęć 475
szkło sodowo-wapniowe w 1000 °C 300
4.4.1. Wygięte błonki powierzchniowe
Wygięta błonka powierzchniowa wywiera na ciecz ciśnienie dodatkowe, obok normalnego ciśnienia doznawanego przez ciecz o płaskiej błonie powierzchniowej.
Rys. 4.9. Siły działające na zakrzywionej powierzchni cieczy
a) menisk wypukły, b) menisk wklęsły.

4. Pomiar niektórych wielkości fizycznych
39
To dodatkowe ciśnienie jest skierowane do dołu w przypadku powierzchni wypukłej (błonka powierzchniowa ciśnie na warstwy cieczy pod nią połoŜone) - Rys. 4.9a, zaś do góry w przypadku powierzchni wklęsłej (błonka powierzchniowa unosi warstwy cieczy dąŜąc do przyjęcia kształtu płaskiego) – Rys. 4.9b.
W celu obliczenia dodatkowego ciśnienia dla przypadku, gdy powierzchnia cieczy stano-wi część powierzchni kuli o promieniu R , weźmy pod uwagę część powierzchni kulistej, ∆S.
Rys. 4.10. Wyznaczanie dodatkowego ci-
śnienia wywieranego przez po-wierzchnię sferyczną.
Siły napięcia powierzchniowego przyłoŜone do obwodu tego segmentu powierzchni są wszę-dzie styczne do powierzchni kulistej. Na element ∆l obwodu działa siła: lF ∆⋅=∆ σ (4.4.3) Składowa tej siły skierowana pionowo w dół wynosi:
ϕsin1 FF ∆=∆ (4.4.4) czyli: ϕσ sin1 lF ∆=∆ (4.4.5)
Na cały segment ∆S działa siła równoległa do promienia krzywizny OC: ϕπσ sin21 rF ⋅= (4.4.6) gdzie r jest promieniem obwodu. Z Rys. 4.10 wynika, Ŝe:
Rr=ϕsin (4.4.7)
gdzie R = OC jest promieniem krzywizny powierzchni, więc po podstawieniu do (4.4.6):
R
rF
2
12πσ ⋅
= (4.4.8)
Dodatkowe ciśnienie wywierane na ciecz otrzymamy po podzieleniu siły F1 przez pole płasz-czyzny ograniczonej przez obwód segmentu:

A. Kufelnicki - Ćwiczenia z biofizyki
40
2
1
r
Fp
π= (4.4.9)
czyli:
R
pσ2= (4.4.10)
Wzór (4.4.10) nosi nazwę w z o r u L a p l a c e ' a dla powierzchni kulistej. Wynika z nie-go, Ŝe dodatkowe ciśnienie wywierane na ciecz przez powierzchnię sferyczną jest wprost pro-porcjonalne do napięcia powierzchniowego i odwrotnie proporcjonalne do promienia krzywi-zny - im bardziej zakrzywiona powierzchnia, (a więc, im mniejsze R), tym większe jest ci-śnienie dodatkowe, p.
4.4.2. Zjawisko kapilarne (włoskowatości)
Wznoszenie się lub opadanie cieczy w cienkich rurkach nazywamy zjawiskiem kapilar-nym, inaczej: włoskowatości. Następuje ono w wyniku powstawania zakrzywionych po-wierzchni. Rodzaj powierzchni zaleŜy od relacji pomiędzy siłami oddziaływań międzyczą-steczkowych - kohezji i adhezji. S i ł y k o h e z j i ( s p ó jn o ś c i ) są to siły występujące pomiędzy cząsteczkami jednego rodzaju, np. cieczy. S i ł y a d h e z j i ( p r z y l e g a -n i a ) są siłami występującymi pomiędzy róŜnymi cząsteczkami. JeŜeli siły adhezji są więk-sze niŜ siły kohezji, to następuje zwilŜanie ścianek rurki kapilarnej i ciecz unosi się do góry (Rys. 4.11a) - w przeciwnym razie poziom cieczy w kapilarze obniŜa się (Rys. 4.11b).
Rys. 4.11. Zjawisko włoskowatości. K ą t e m z w i lŜ a n i a ,ϑ , nazywamy kąt, jaki tworzy styczna do powierzchni cieczy ze ścianką kapilary (powierzchnią zwilŜaną). Od kąta zwilŜania zaleŜy równieŜ np. kształt kropli danej cieczy umieszczonej na płaskiej powierzchni (Rys. 4.12).
a) b)

4. Pomiar niektórych wielkości fizycznych
41
Rys. 4.12. Zachowanie się kropli cieczy dla róŜnych kątów zwilŜania:
ϑ = 0° całkowita zwilŜalność (R = r), cosϑ = 1 ϑ = 180° brak zwilŜalności (−R = r), cosϑ = −1
Ciecz zwilŜająca ścianki kapilary unosi się w niej tworząc menisk wklęsły (Rys. 4.11a) tak wysoko, aŜ dodatkowe ciśnienie wywierane przez zakrzywioną powierzchnię o promieniu R, określone przez równanie Laplace'a (4.4.10), zostanie zrównowaŜone przez ciśnienie hy-drostatyczne słupa cieczy o wysokości h:
ghR
ρσ =2 (4.4.11)
gdzie ρ - gęstość cieczy, g - przyspieszenie ziemskie.
Jeśli przez r oznaczymy promień wewnętrzny rurki, a przez ϑ - kąt zwilŜania, to:
ϑcosr
R = (4.4.12)
a po podstawieniu do (4.4.11):
ghr
ρϑσ =cos2 (4.4.13)
grh ρϑσ cos2= (4.4.14)
Gdy ϑ = 0° (cos ϑ = 1) grh ρσ2= (4.4.15)
4.4.3. Metody pomiaru napięcia powierzchniowego
Spośród wielu metod porównawczych pomiaru napięcia powierzchniowego omówimy tylko trzy: kapilarną, stalagmometryczną i pęcherzykową.

A. Kufelnicki - Ćwiczenia z biofizyki
42
M e t o d a k a p i l a r n a polega na zanurzeniu w badanej cieczy kapilary szklanej i zmierzeniu róŜnicy poziomów cieczy w naczyniu i kapilarze. Ze wzoru (4.4.14) napięcie po-wierzchniowe wynosi więc:
ϑρσ
cos2ghr= (4.4.16)
Przy całkowitej zwilŜalności kąt o0=ϑ (oznacza to, Ŝe promień krzywizny R jest równy promieniowi rurki r, a menisk ma kształt półkuli). Stąd, oczywiście, cosϑ = 1 oraz:
2
ghrρσ = (4.4.17)
Porównanie napięcia powierzchniowego cieczy badanej, σ , z napięciem powierzchniowym wzorca, ′σ , (np. wody) pozwala wyeliminować z równania (4.4.17) wartość g, zaleŜną od miejsca na Ziemi oraz trudny do zmierzenia promień rurki, r:
''' ρ
ρσσ
h
h= (4.4.18)
Jedną z dokładniejszych metod pomiaru napięcia powierzchniowego jest m e t o d a s t a l a g m o m e t r y c z n a polegająca na pomiarze licz-by kropel, w postaci których dana objętość cieczy wypływa z pionowej kapilary. Rysunek 4.13 przedstawia typowy stalagmometr. Kropla urywa się wówczas, gdy jej cięŜar przewaŜy napięcie powierzchniowe utrzymu-jące ją w zawieszeniu. JeŜeli obserwujemy wyciekanie danej objętości cieczy, V, a liczba kropel wynosi n , to wówczas cięŜar kropli równy jest:
ngV
Qρ= (4.4.19)
Siła napięcia powierzchniowego utrzymująca kroplę w zawieszeniu wy-nosi (w przypadku całkowitej zwilŜalności): rF πσ 2⋅= (4.4.20) gdzie r - promień końcówki kapilary. Z porównania sił (4.4.19) i (4.4.20) otrzymamy:
Rys. 4.13. Stalagmometr nr
gV
πρσ
2= (4.4.21)
Znając V, ρ, g, r, moŜna obliczyć σ. Zazwyczaj wykonuje się tym samym stalagmometrem pomiar porównawczy dla cieczy o dokładnie znanym ′σ :

4. Pomiar niektórych wielkości fizycznych
43
nr
gV′
′=′
πρσ
2 (4.4.22)
Dzieląc stronami równania (4.4.21) i (4.4.22) otrzymamy:
nn
ρρ
σσ
′′
=′ (4.4.23)
M e t o d a pę c h e r z y k o w a polega na określeniu nadwyŜki ciśnienia (względem ci-śnienia atmosferycznego) w rurce kapilarnej zanurzonej w badanej cieczy, potrzebnego do oderwania się pęcherzyka powietrza od rurki. Schemat aparatury stosowanej w metodzie pę-cherzykowej przedstawia Rys. 4.14. Ciecz badana znajduje się w termostatowanym naczyniu z otworem dla wyrównania ciśnienia z ciśnieniem zewnętrznym. W cieczy badanej zanurzona jest kapilara o średnicy 0,01 - 0,02 cm na głębokość 0,5 - 1 cm. Kapilara połączona jest z ma-nometrem cieczowym. Do ramienia B wprowadzamy małym strumieniem ciecz manome-tryczną.
Rys. 4.14. Zestaw pomiarowy do wyznaczania napięcia powierzchniowego metodą pęche-
rzykową.
Przy pewnej róŜnicy poziomów, hm , u wylotu kapilary odrywa się pęcherzyk powietrza.
NadwyŜkę ciśnienia w rurce kapilarnej w stosunku do ciśnienia atmosferycznego (tzw. ciśnie-nie kapilarne) określa wówczas róŜnica ciśnień hydrostatycznych: ghhp xxmm )( ρρ −= (4.4.24)

A. Kufelnicki - Ćwiczenia z biofizyki
44
gdzie ρm, ρx - gęstość cieczy manometrycznej i badanej. W chwili odrywania się pęcherzyka parcie wywierane przez powietrze w kapilarze równowaŜy siłę napięcia powierzchniowego działającą na obwodzie rurki, stąd:
rrp πσπ 22 ⋅=⋅ (4.4.25) oraz:
σ = pr2
(4.4.26)
Podobnie, jak w poprzednio opisanych metodach, nie mierzy się bezpośrednio promienia rur-ki, lecz stosuje się pomiar porównawczy. Np. dla wody (znana wartość napięcia powierzch-niowego) spełniona jest zaleŜność:
2
rpww =σ (4.4.27)
Po podzieleniu stronami równań (4.4.26) i (4.4.27) otrzymamy:
ww pp=σ
σ (4.4.28)
Metoda pęcherzykowa sprowadza się zatem do porównania ciśnień kapilarnych cieczy bada-nej (p) i cieczy wzorcowej (pw).
Oprócz metod porównawczych, opartych na wzorcowych cieczach o znanym σ , stoso-wane są takŜe metody bezwzględne, w których napięcie powierzchniowe obliczane jest z od-powiedniego wzoru teoretycznego zawierającego współczynnik korekcyjny. Przykładem ta-kiej metody jest m e t o d a p i e rś c i e n i o w a (Rys. 4.15), w której napięcie powierz-chniowe obliczane jest ze wzoru na maksymalną siłę potrzebną do oderwania pierścienia pla-tynowego od powierzchni cieczy:
σπ RP 4max = (4.4.29)
gdzie R - promień pierścienia (zwykle 0,5 - 1,0 cm). Wartość σ obliczona z tego wzoru obar-czona byłaby duŜym błędem (nawet do ± 30 %). Stąd teŜ zachodzi potrzeba wprowadzenia odpowiedniego współczynnika korekcyjnego (poprawkowego) f:
fR
P⋅=
πσ
4max (4.4.30)
Współczynnik f jest funkcją dwóch parametrów (R/r i R3/V, gdzie r - promień drucika platy-nowego, od 0,1 do 0,5 mm ; V - objętość cieczy uniesionej nad powierzchnią) i znajdujemy go w odpowiednich tablicach. Wartości f zawarte są w granicach 0,75 - 1,05. Płaszczyzna pier-ścienia musi być z duŜą dokładnością zgodna z płaszczyzną powierzchni swobodnej cieczy. Siłę Pmax mierzy się za pomocą wagi torsyjnej (w przeznaczonej do tego celu wersji jest nią tensjometr du Nouy'a) lub przy uŜyciu odpowiednio przygotowanej wagi analitycznej. Wtedy wyznaczana jest maksymalna masa cieczy, M, uniesionej nad powierzchnią; Pmax = Mg. Me-toda pierścieniowa dobrze nadaje się do pomiaru napięcia powierzchniowego: czysta ciecz -

4. Pomiar niektórych wielkości fizycznych
45
gaz, a takŜe do pomiaru napięcia międzyfazowego woda - olej w obecności substancji po-wierzchniowo czynnych.
Rys. 4.15. Pomiar napięcia powierzchniowe-go metodą pierścieniową.
Literatura uzupełniająca:
F. Jaroszyk, red., „Biofizyka”, Wydawnictwo Lekarskie PZWL, Warszawa 2002, str. 100. T. W. Hermann, red., „Farmacja fizyczna”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 99. J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 34. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 10. Ćwiczenie 4
Pomiar napięcia powierzchniowego roztworów kwasu octowego za pomocą stalagmometru
Wykonanie pomiarów:
1. Do czystej zlewki wlać wodę destylowaną i zanurzyć w niej kapilarę stalagmometru. Wciągnąć ciecz powyŜej górnej kreski. Wyjąć stalagmometr z roztworu, zdjąć gruszkę gumową. Wypuszczając ciecz ze stalagmometru rozpocząć liczenie kropel, gdy menisk zrówna się z górną kreską. Przerwać liczenie, gdy menisk osiągnie poziom kreski dolnej. Pomiar powtórzyć trzykrotnie. W tabeli zamieścić wartość średnią (n').
2. Wyznaczyć ilość kropel badanego roztworu kwasu octowego wypływających z tego same-go stalagmometru, w sposób identyczny jak w punkcie pierwszym (wartość średnia n).
3. Wyznaczyć gęstość badanego roztworu kwasu octowego za pomocą piknometru (patrz Ćwiczenie 2).
4. Korzystając z Tabeli 4.6 oraz tabeli gęstości wody w róŜnych temperaturach (Tab. 4.1, Rozdz. 4.2) obliczyć napięcie powierzchniowe badanego roztworu ze wzoru (4.4.23).
A. Kufelnicki - Ćwiczenia z biofizyki
46
Tabela 4.6
ZaleŜność napięcia powierzchniowego H2O od temperatury
°C σ′(mJ/m2) °C σ ′ (mJ/m2)
10 74,22 21 72,59 11 74,07 22 72,44 12 73,93 23 72,28 13 73,78 24 72,13 14 73,64 25 71,97 15 73,49 26 71,82 16 73,34 27 71,66 17 73,19 28 71,50 18 73,05 29 71,35 19 72,90 30 71,18 20 72,75 31 70,38
Tabela wyników pomiarów i wielkości obliczonych:
n n' ρ (103 kg/m3) t ( °C) σ (mJ/m2)

47
5. NIEKTÓRE FIZYCZNE METODY BADANIA STRUKTURY MOLEK UŁ
Budowa cząsteczek materii, w tym materii oŜywionej, warunkuje większość jej właści-
wości fizycznych. Wynika z tego, Ŝe badanie właściwości fizycznych moŜe dostarczyć infor-macji na temat struktury cząsteczek badanej substancji, jej zawartości oraz ewentualnych za-nieczyszczeń. Do najczęściej wykorzystywanych metod naleŜą badania oddziaływania fal elektromagnetycznych z materią, badanie właściwości elektrycznych i magnetycznych. Spo-śród wielkiej liczby róŜnych metod badawczych, tematem tego rozdziału będą tylko badania struktury oraz oznaczanie stęŜeń za pomocą niektórych wielkości addytywnych (np. refrakcji molowej i parachory), a takŜe za pomocą pomiarów skręcalności optycznej. Są to klasyczne, dawno poznane metody, oddające jednak nadal znaczne usługi w badaniach substancji.
5.1. Wielkości addytywne
5.1.1. Refrakcja molowa
Pojęcie refrakcji molowej związane jest ze zjawiskami występującymi przy rozchodzeniu się światła w ośrodkach. W przeźroczystych ośrodkach izotropowych światło rozchodzi się prostoliniowo, równomiernie we wszystkich kierunkach. Takimi ośrodkami są: próŜnia, gazy, prawie wszystkie ciecze, bezpostaciowe przeźroczyste ciała stałe i kryształy układu regularne-go. Prędkość światła zaleŜy od rodzaju ośrodka. Optycznie gęstszym nazywamy ośrodek, w którym prędkość światła jest mniejsza. Najbardziej rzadkim ośrodkiem jest próŜnia. W niej prędkość światła wynosi 2,9979⋅108 m/s i jest niezaleŜna od długości fali promienia świetl-nego.
Promień świetlny padający na płaską granicę dwóch przeźroczystych ośrodków izotro-powych częściowo ulega odbiciu, a częściowo przechodzi do drugiego ośrodka zmieniając swój kierunek, czyli ulega załamaniu. Zjawiska te dla światła monochromatycznego ujmują ilościowo prawa Snelliusa: 1) promienie padający, odbity i załamany leŜą w jednej płaszczyźnie; 2) kąt odbicia jest równy kątowi padania; 3) pomiędzy kątem padania a kątem załamania występuje zaleŜność:
1,22
1
sin
sinn
v
v==
βα
(5.1.1)
gdzie: α - kąt padania (pomiędzy promieniem padającym a prostą prostopadłą do płaszczyzny granicznej ośrodków) β - kąt załamania
v1 i v2 - prędkość światła odpowiednio w ośrodku optycznie rzadszym i gęstszym n2,1 - współczynnik załamania światła ośrodka 2 względem ośrodka 1
Zatem, względny współczynnik załamania światła, n2,1 , jest wielkością niemianowaną, równą stosunkowi prędkości światła w ośrodku rzadszym do prędkości światła w ośrodku gęstszym. JeŜeli ośrodek rzadszy stanowi próŜnia, to wówczas n2,1 charakteryzuje drugi ośrodek i nazy-wa się absolutnym współczynnikiem załamania - nabs (lub po prostu n).
Właściwości optyczne powietrza nieznacznie tylko róŜnią się od właściwości optycznych próŜni. RóŜnica pomiędzy współczynnikiem załamania względem próŜni a względem powie-trza jest niewielka i wynosi ok. 0,03%:
nabs = 1,00027⋅ npow (5.1.2)

A. Kufelnicki - Ćwiczenia z biofizyki
48
Rys. 5.1. Zjawiska załamania i odbicia na granicy dwóch ośrodków.
Gdy promień przechodzi z ośrodka optycznie rzadszego do gęstszego, jak na Rys. 5.1, za-
łamuje się ku prostopadłej: kąt padania jest większy od kąta załamania i n >1. Zwiększenie kąta padania pociąga za sobą zwiększenie kąta załamania. Największy kąt padania w ośrodku rzadszym moŜe wynosić 90°. Wówczas promień padający ślizga się po powierzchni granicz-nej. Kątowi temu odpowiada w ośrodku gęstszym największy kąt załamania, zwany kątem granicznym - βg.
Promienie padające z ośrodka gęstszego na granicę z ośrodkiem rzadszym załamują się od prostopadłej. Wtedy kąt załamania jest większy od kąta padania. JeŜeli kąt padania będzie większy od kąta granicznego, nastąpi zjawisko c a ł k o w i t e g o w e w nę t r z n e g o o d b i c i a - promień świetlny nie opuści ośrodka gęstszego. Pomiędzy promieniem grani-cznym a powierzchnią ośrodka gęstszego występuje przestrzeń, do której nie moŜe przenikać światło z ośrodka rzadszego - jest to przestrzeń ciemna (Rys. 5.1). Granica między przestrze-nią ciemną i jasną, ostro rysująca się w przyrządach optycznych, jest wykorzystywana do po-miaru kąta granicznego, βg. Odpowiada on kątowi padania w ośrodku rzadszym α = 90°, więc:
sin
sin
sin
sin sin,
αβ β β
= = =°
ng g
2 1
90 1 (5.1.3)
ng
2 1
1, sin
=β
(5.1.4)
Ze wzoru (5.1.4) wynika, Ŝe pomiar kąta granicznego umoŜliwia wyznaczanie współczynnika załamania promienia o danej długości fali świetlnej. Wielkość współczynnika załamania zaleŜy od długości fali uŜytego światła i od tempera-tury. Krótsze fale świetlne są silniej załamywane niŜ długie – po załamaniu występuje rozsz-czepienie barw. Z tego powodu do pomiarów współczynnika załamania często stosuje się światło monochromatyczne, przewaŜnie linii D lampy sodowej (λ = 589,3 nm). Jednocześnie pomiar przeprowadza się w stałej, określonej temperaturze. Wzrost temperatury powoduje zmniejszenie współczynnika załamania światła cieczy (ok. 5⋅10-4 na 1 °C); dla ciał stałych zmiana ta moŜe być dodatnia lub ujemna. Dane dotyczące temperatury i długości fali zazna-czamy przy symbolu współczynnika załamania: nD
25 (z prawej u góry temperatura, u dołu ro-dzaj światła).
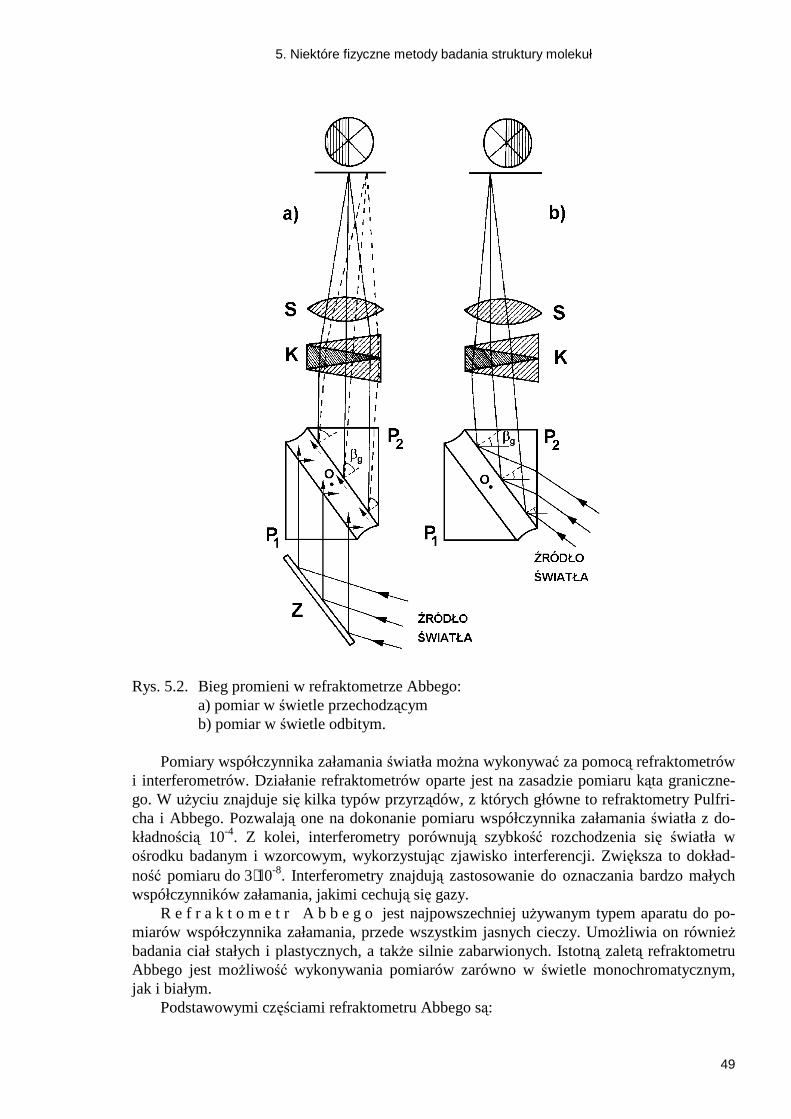
5. Niektóre fizyczne metody badania struktury molekuł
49
Rys. 5.2. Bieg promieni w refraktometrze Abbego: a) pomiar w świetle przechodzącym b) pomiar w świetle odbitym.
Pomiary współczynnika załamania światła moŜna wykonywać za pomocą refraktometrów i interferometrów. Działanie refraktometrów oparte jest na zasadzie pomiaru kąta graniczne-go. W uŜyciu znajduje się kilka typów przyrządów, z których główne to refraktometry Pulfri-cha i Abbego. Pozwalają one na dokonanie pomiaru współczynnika załamania światła z do-kładnością 10-4. Z kolei, interferometry porównują szybkość rozchodzenia się światła w ośrodku badanym i wzorcowym, wykorzystując zjawisko interferencji. Zwiększa to dokład-ność pomiaru do 3⋅10-8. Interferometry znajdują zastosowanie do oznaczania bardzo małych współczynników załamania, jakimi cechują się gazy. R e f r a k t o m e t r A b b e g o jest najpowszechniej uŜywanym typem aparatu do po-miarów współczynnika załamania, przede wszystkim jasnych cieczy. UmoŜliwia on równieŜ badania ciał stałych i plastycznych, a takŜe silnie zabarwionych. Istotną zaletą refraktometru Abbego jest moŜliwość wykonywania pomiarów zarówno w świetle monochromatycznym, jak i białym.
Podstawowymi częściami refraktometru Abbego są:

A. Kufelnicki - Ćwiczenia z biofizyki
50
1) podwójny pryzmat ze szkła o duŜym (1,7) współczynniku załamania światła, 2) luneta z okularem zaopatrzonym w skrzyŜowane nitki pajęcze, 3) skala i układ optyczny do jej odczytywania, umocowana współosiowo z pryzmatem.
RozróŜnia się dwa zasadnicze rodzaje pomiarów – pierwszy, w świetle przechodzącym i drugi, w świetle odbitym. Ciecz badaną zamykamy pomiędzy pryzmatami P1 i P2 (Rys. 5.2) w postaci cienkiej, równoległościennej warstewki. W pierwszej metodzie pomiaru (Rys. 5.2a) promienie odbite od zwierciadła Z padają na pryzmat P1, załamują się w nim i ulegają rozpro-szeniu na jego matowej powierzchni tak, Ŝe w cieczy rozchodzą się w róŜnych kierunkach. Kąty padania na pryzmat P2 zawarte są w granicach 0 - 90°. PoniewaŜ ciecz jest ośrodkiem optycznie rzadszym, więc w pryzmacie P2 światło moŜe rozchodzić się tylko w obrębie kąta granicznego βg . Specjalny układ optyczny, złoŜony z soczewki (S) i kompensatora rozszcze-pienia barwnego (K), kieruje światło do okularu. Przez odpowiedni obrót pryzmatów i wła-ściwe ich oświetlenie kieruje się promienie w ten sposób, aby granica pola ciemnego i jasnego wypadła dokładnie w środku okularu (na skrzyŜowaniu nici pajęczych). Obrót pryzmatów wokół punktu O jest zsynchronizowany ze skalą przyrządu, na której odczytuje się bezpośred-nio wartość współczynnika załamania. Przy pomiarze w świetle odbitym promienie padają na pryzmat P2 (Rys. 5.2b). Do okularu przechodzą tylko te z nich, dla których kąt padania na płaszczyznę zetknięcia z warstewką cieczy jest większy od granicznego, a więc te, które ulegają całkowitemu wewnętrznemu od-biciu. Pozostała część światła załamuje się i ulega rozproszeniu.
Co pewien czas naleŜy sprawdzać ustawienie skali refraktometru. Cechowanie refrakto-metrów przeprowadza się za pomocą wzorcowych cieczy o znanym współczynniku załamania (Tab. 5.1). Najczęściej rolę tę spełnia woda.
Tabela 5.1
Współczynniki załamania cieczy wzorcowych
Ciecz nD20
woda 1,3330 aceton 1,3592 metylocykloheksan 1,4231 toulen 1,4969 benzen 1,5017 chlorobenzen 1,5250 o-nitrotoulen 1,5466 bromoform 1,5973 chinolina 1,6272 1-bromonaftalen 1,6580
W badaniach struktury związków chemicznych wykorzystuje się wielkość zwaną r e -
f r a k c j ą m o l o wą , RM , zdefiniowaną przez H.A. Lorentza i R. Lorenza wzorem (5.1.5):
Rn
n
M
dRM M= −
+⋅ = ⋅
2
2
1
2 m mol3 -1[ ] (5.1.5)
w którym n jest współczynnikiem załamania światła danej (czystej) substancji, M - masą mo-lową, a d - gęstością. WiąŜe się ona głównie ze zjawiskiem p o l a r y z a c j i e l e k t r o -

5. Niektóre fizyczne metody badania struktury molekuł
51
n o w e j w cząsteczce, występującym przy przejściu światła przez materię i jest odpowied-nikiem tzw. polaryzacji molowej:
d
MP
r
rM ⋅
+−
=2
1
εε
(5.1.6)
gdzie: εr - względna przenikalność elektryczna substancji. Polaryzacja elektronowa jest rezultatem zaburzenia rozkładu gęstości elektronowej wokół jąder atomowych. Pod wpływem pola elektrycznego przenoszonego przez falę świetlną indukowany jest w kaŜdym atomie moment dipolowy, proporcjonalny do natęŜenia pola.
Refrakcja molowa jest wielkością charakterystyczną i stałą dla danego związku chemicz-nego, praktycznie nie zaleŜy od temperatury i ciśnienia. ZaleŜy natomiast, za pośrednictwem współczynnika załamania, od długości fali światła (w przypadku światła sodowego stosuje się oznaczenie RD). Najistotniejszą cechą refrakcji molowej jest jej charakter a d d y t y w n y , co oznacza, Ŝe jest ona sumą udziałów refrakcji molowej atomów i wiązań występujących w cząsteczce określonego związku:
RM = Ratomów + Rwiązań (5.1.7)
Ratomów oznacza tu zarówno refrakcję odpowiadającą poszczególnym atomom bez określenia grupy, w której występują, np. C, H, Cl, Br, jak i refrakcję odpowiadającą atomom występu-jącym w danych ugrupowaniach, np. O (w grupie karbonylowej), -NH2 (azot w aminie I rz.) itp. Przez Rwiązań rozumiemy udział pewnych wiązań (nie uwzględnionych w ugrupowaniach) o szczególnej strukturze elektronowej, np. wiązanie podwójne, potrójne, pierścień trójczłono-wy, czteroczłonowy. Układy z takimi wiązaniami wykazują podwyŜszoną polaryzowalność, co prowadzi do zwiększonych wartości refrakcji.
Tabela 5.2 Refrakcje atomowe i wiązań dla linii D światła sodowego
Nazwa RD (cm3mol-1)
C 2,418 H 1,100 O (w grupie karbonylowej) 2,211 O ( w eterach) 1,643 O ( w grupie hydroksylowej) 1,525 N ( w aminie I rz., -NH2 ) 2,322 N ( w aminie II rz., NH ) 2,502 N ( w aminie III rz., N ) 2,840 F 0,997 Cl 5,967 Br 8,865 I 13,900 Wiązanie podwójne 1,733 Wiązanie potrójne 2,398 Pierścień trójczłonowy 0,71 Pierścień czteroczłonowy 0,48

A. Kufelnicki - Ćwiczenia z biofizyki
52
Wyznaczając refrakcję molową cząsteczki (przez pomiar n, d i M ) oraz porównując otrzy-mane wyniki z wartościami teoretycznymi obliczonymi na podstawie Tabeli 5.2 dla kilku moŜliwych struktur badanego związku, moŜemy wyciągnąć wnioski odnośnie jego budowy. Zgodność teoretycznej i doświadczalnej wartości refrakcji przemawia za danym wzorem strukturalnym. Przykład
Wyznaczona doświadczalnie wartość refrakcji molowej pewnego związku organicznego o
wzorze sumarycznym C3H6O wynosi RM = 16,97 cm3/mol. Określić, która z dwóch podanych niŜej struktur jest bardziej właściwa:
C O
H3C
H3C
CH2 CH CH2OH
(a) (b) Dla struktury (a) : Dla struktury (b) :
RC ==== 3⋅2,418 ==== 7,254 RC ==== 3⋅2,418 ==== 7,254
RH ==== 6⋅1,100 ==== 6,600 RH ==== 6⋅1,100 ==== 6,600
RO (C=O) ==== 1⋅2,211 ==== 2,211 RO (OH) ==== 1⋅1,525 ==== 1,525
R wiąz. podw. ==== 1⋅1,733 ==== 1,733
R(a) ====16,065
R(b) ====17,112 Jak wynika z porównania obliczonych wartości teoretycznych z wartością doświadczalną, bardziej prawdopodobna jest struktura (b). Refrakcja molowa roztworu, Rroztw, jest sumą iloczynów refrakcji molowych składników (R1 i R2) i ich ułamków molowych w roztworze (x1 i x2 ). Refrakcja molowa roztworu ma więc równieŜ charakter a d d y t y w n y :
Rroztw ==== x1R1 + x2R2 (5.1.8) Jej doświadczalną wartość wyznacza się na podstawie równania:
roztwroztw
roztwroztw d
MxMx
n
nR 2211
2
2
2
1 +⋅
+−= (5.1.9)
gdzie: nroztw i droztw - współczynnik załamania i gęstość roztworu, zmierzone w tej samej tem-peraturze, M1, M2 - masy molowe składników. Pozwala to wyznaczyć refrakcję molową czy-stego składnika, dla którego pomiar gęstości i współczynnika załamania jest technicznie trud-ny. Np. po wyznaczeniu refrakcji molowej roztworu wodnego, Rroztw , obliczamy refrakcję substancji rozpuszczonej, R1:
1
221 x
RxRR roztw −
= ; R2 ==== Rwody (5.1.10)

5. Niektóre fizyczne metody badania struktury molekuł
53
5.1.2. Parachora
Do wielkości addytywnych naleŜy, oprócz refrakcji molowej i objętości molowej, rów-nieŜ wielkość zwana p a r a c h o rą , zdefiniowana przez Sugdena wzorem:
PM
Pc p
= ⋅−
= ⋅ ⋅ ⋅σρ ρ
1 4
kg m s mol1 4 3 -1 2 -1[ ] (5.1.11)
gdzie: M - masa molowa σ - napięcie powierzchniowe cieczy ρc , ρp - gęstości odpowiednio cieczy i jej pary nasyconej.
PoniewaŜ gęstość cieczy jest znacznie większa niŜ gęstość pary, więc w mianowniku
wzoru (5.1.11) moŜemy pominąć wartość ρp . Zatem:
PM
c
= ⋅σρ
1 4
(5.1.12)
Z kolei, zastępując wyraŜenie M/ρc we wzorze (5.1.12) objętością molową V, otrzymuje-my równanie: P V= ⋅σ 1 4 (5.1.13) Parachorę moŜemy więc określić jako iloczyn objętości molowej cieczy i czwartego pierwiastka z jej napięcia powierzchniowego. Parachora jest wielkością niezaleŜną od tempe-ratury i stałą dla danej substancji, mimo Ŝe od temperatury zaleŜą wielkości σ, ρ i V występu-jące w wyraŜeniach (5.1.12) - (5.1.13). Okazuje się, Ŝe parachora danego związku chemicznego jest, podobnie jak refrakcja mo-lowa, sumą udziałów pochodzących od poszczególnych atomów, wiązań i innych elementów strukturalnych (np. pierścieni). Addytywność parachory wyraŜa wzór: P P P Pa w p= + + (5.1.14)
gdzie Pa , Pw i Pp oznaczają odpowiednio udziały atomów, wiązań i pierścieni w parachorze całej cząsteczki. Np. PH = 30,4⋅10-7, PC = 8,5⋅10-7 , PO = 35,6⋅10-7 , P(=) = 41,2⋅10-7 . Wyznaczając doświadczalnie parachorę (przez pomiar napięcia powierzchniowego i gęstości) przy danej masie molowej M, wynikającej ze wzoru sumarycznego oraz porównu-jąc tę wartość parachory z wartością (5.1.14) opartą na danych tablicowych, moŜemy wycią-gnąć wnioski co do poprawności załoŜonego wzoru strukturalnego. Przykład
Napięcie powierzchniowe acetonu w temp. 20 °C wynosi 0,02370 N/m, a jego gęstość w tej
samej temperaturze ρ = 796,0 kg/m3. Obliczyć wartość parachory acetonu i porównać ją z warto-ścią otrzymaną na podstawie udziałów atomowych i strukturalnych. Parachorę doświadczalną obliczamy na podstawie wzoru (5.1.12), przy czym dla acetonu M
= 58,08 g/mol = 58,08⋅10-3 kg/mol:

A. Kufelnicki - Ćwiczenia z biofizyki
54
1213417
3
413
molsmkg10287,0kg/m796,0
N/m)(0,02370kg/mol1058,08 −−−−
⋅=⋅⋅
=P
Z drugiej strony dla cząsteczki acetonu:
C O
H3C
H3C
P = 3⋅PC + 6⋅PH + PO + P(=)
Po podstawieniu danych tablicowych:
12134177 molsmkg10284,71041,2)35,630,468,5(3P −−−− ⋅=⋅++⋅+⋅=
Zgodność wartości P wskazuje na poprawność przyjętej struktury badanego związku che-micznego. Literatura uzupełniająca:
F. Jaroszyk, red., „Biofizyka”, Wydawnictwo Lekarskie PZWL, Warszawa 2002, str. 106. T. W. Hermann, red., „Farmacja fizyczna”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 99, 372. J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 205. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 205. Ćwiczenie 5
Pomiar współczynników załamania i wyznaczanie refrakcji molowej wodnych roztworów
mocznika Wykonanie pomiarów: 1. Odczytać temperaturę pomiaru (termometr przy refraktometrze). 2. Zmierzyć współczynniki załamania wody oraz roztworów mocznika (wzorcowych i bada-
nych). Wyniki zamieścić w tabeli 1. 3. Wyznaczyć gęstość badanych roztworów mocznika w danej temperaturze (ewentualnie od-
czytać z Tabeli na końcu rozdziału). 4. Z wykresu zaleŜności współczynnika załamania od stęŜenia roztworu odczytać stęŜenia
dwóch wybranych roztworów badanych, np. c1 oraz c2; c1 oraz c4 itp. 5. Dla kaŜdego badanego roztworu obliczyć ułamki molowe mocznika i wody, refrakcję mo-
lową roztworu i refrakcję molową mocznika ze wzorów (5.1.9) i (5.1.10) oraz, wykorzystu-jąc Tabelę 4.1 gęstości wody, jej refrakcję molową ze wzoru (5.1.5):

5. Niektóre fizyczne metody badania struktury molekuł
55
mw
mm NN
Nx
+= ;
mw
ww NN
Nx
+=
gdzie:
m
mm M
mN = ;
w
ww M
mN =
N - liczba moli m - masa M - masa molowa (Mw = 18,016 g⋅mol-1; Mm = 60,06 g⋅mol-1 )
Obliczenia liczby moli mocznika i wody najwygodniej jest wykonać dla 1 dm3 roztworu. Wówczas:
N cm =
( )w
mw M
McdN
⋅−⋅=
1000
gdzie: c - stęŜenie roztworu badanego w mol/dm3 d - gęstość roztworu w g/cm3
6. Obliczyć teoretyczną wartość refrakcji molowej mocznika na podstawie Tabeli 5.2. 7. Wyniki pomiarów i wielkości obliczone zamieścić w tabeli 2. Tabela wyników pomiarów 1:
StęŜenie roztworu
Współczynnik załamania (n) w temperaturze ….. °C
(c) w mol/dm3 Pomiar I Pomiar II Pomiar III Średnia 0 1 2 3 4 c1 c2
Tabela wyników pomiarów i wielkości obliczonych 2:
Gęstość badanego roztworu, droztw
Współczynnik załamania, n
Ułamek molowy mocznika, xm
Ułamek molowy wody, xw
Refrakcja molowa wody, Rw
Refrakcja molowa roztworu, Rroztw (dośw.)
Refrakcja molowa mocznika, Rm (dośw.)
Refrakcja molowa mocznika, obliczona z danych Tab. 5.2
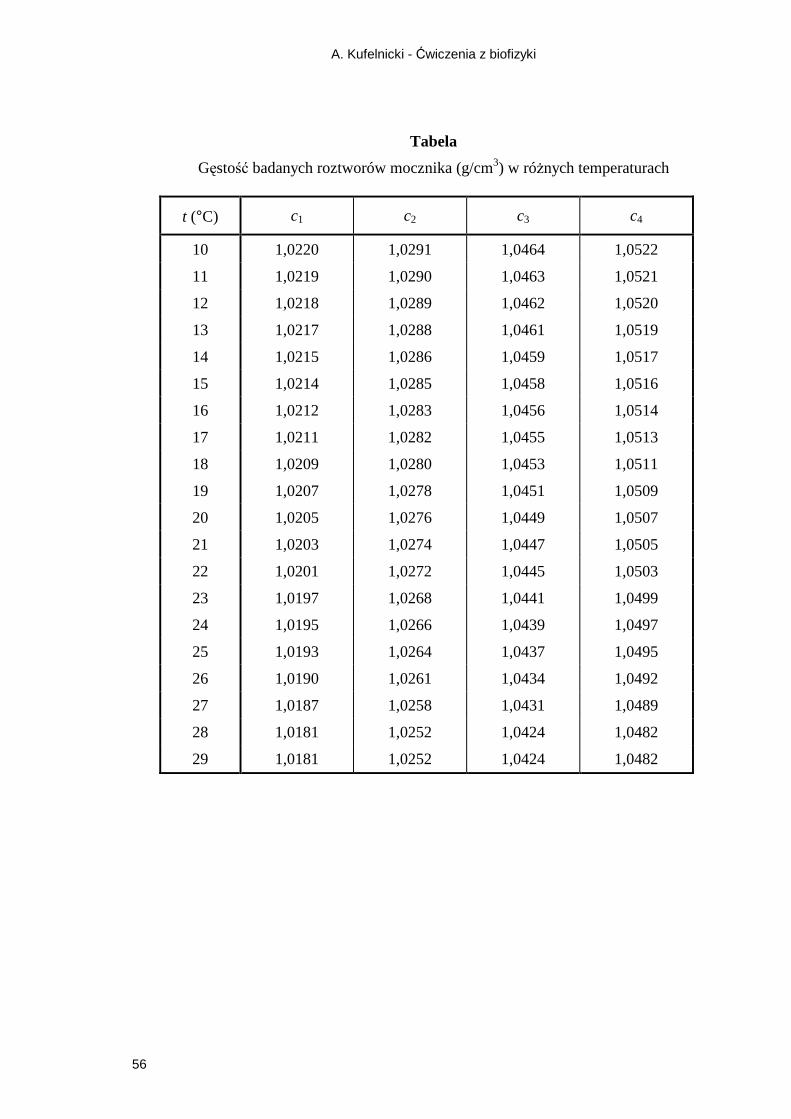
A. Kufelnicki - Ćwiczenia z biofizyki
56
Tabela
Gęstość badanych roztworów mocznika (g/cm3) w róŜnych temperaturach
t (°C) c1 c2 c3 c4
10 1,0220 1,0291 1,0464 1,0522
11 1,0219 1,0290 1,0463 1,0521
12 1,0218 1,0289 1,0462 1,0520
13 1,0217 1,0288 1,0461 1,0519
14 1,0215 1,0286 1,0459 1,0517
15 1,0214 1,0285 1,0458 1,0516
16 1,0212 1,0283 1,0456 1,0514
17 1,0211 1,0282 1,0455 1,0513
18 1,0209 1,0280 1,0453 1,0511
19 1,0207 1,0278 1,0451 1,0509
20 1,0205 1,0276 1,0449 1,0507
21 1,0203 1,0274 1,0447 1,0505
22 1,0201 1,0272 1,0445 1,0503
23 1,0197 1,0268 1,0441 1,0499
24 1,0195 1,0266 1,0439 1,0497
25 1,0193 1,0264 1,0437 1,0495
26 1,0190 1,0261 1,0434 1,0492
27 1,0187 1,0258 1,0431 1,0489
28 1,0181 1,0252 1,0424 1,0482
29 1,0181 1,0252 1,0424 1,0482

5. Niektóre fizyczne metody badania struktury molekuł
57
5.2. Czynność optyczna związków W swobodnej przestrzeni fala elektromagnetyczna jest falą poprzeczną. Nie ma składo-wych podłuŜnych, a drgania wektorów pola elektrycznego,E
r, oraz indukcji magnetycznej,B
r
zachodzą wyłącznie prostopadle do kierunku ruchu fali. Ponadto w fali elektromagnetycznej wektorE
roraz wektor B
rdrgają w płaszczyznach do siebie prostopadłych.
Fale poprzeczne mogą ulegać polaryzacji. Zjawisko polaryzacji polega na uporządkowa-niu drgań względem kierunku ruchu fali. Jeśli drgania kaŜdego z wektorów odbywają się w jednej płaszczyźnie, to mówimy, Ŝe światło jest l i n i o w o ( p ł a s k o ) s p o l a r y z o -w a n e – Rys. 5.3. Płaszczyznę prostopadłą do płaszczyzny drgań wektora E
r nazywamy
p ł a s z c z y z ną p o l a r y z a c j i . Światło rozchodzące się w danym kierunku składa się na ogół z niezaleŜnych ciągów fal elektromagnetycznych, w których płaszczyzny drgań zorientowane są w sposób przypadkowy wokół kierunku ruchu. Takie światło, chociaŜ jest falą poprzeczną, jest niespolaryzowane. Teoria Maxwella i dane doświadczalne (np. zjawisko fotoelektryczne) wykazują, Ŝe za wraŜe-nia świetlne odpowiada tylko wektorE
r, zwany w tym przypadku wektorem świetlnym.
Rys. 5.3. Płasko spolaryzowana fala elektromagnetyczna w próŜni w danej chwili. W celu uzyskania światła liniowo spolaryzowanego stosuje się p o l a r y z a t o r y . Po-laryzator rozdziela światło na dwie składowe spolaryzowane w płaszczyznach do siebie pro-stopadłych, a następnie eliminuje jedną z nich. Najczęściej wykorzystywane, a zarazem naj-prostsze sposoby otrzymania światła spolaryzowanego opierają się na opisanych poniŜej zja-wiskach.
5.2.1. Zjawisko odbicia światła Zasadę zjawiska polaryzacji przez odbicie przedstawia Rys. 5.4. Dla pewnego kąta padania α wiązka odbita i załamana tworzą kąt prosty. Stwierdzono doświadczalnie, Ŝe wówczas wiązka odbita jest całkowicie spolaryzowana w płaszczyźnie padania, natomiast wiązka załamana jest tylko częściowo spolaryzowana. PoniewaŜ:
αββα −== o90i
sin
sin1,2n (5.2.1)
więc:
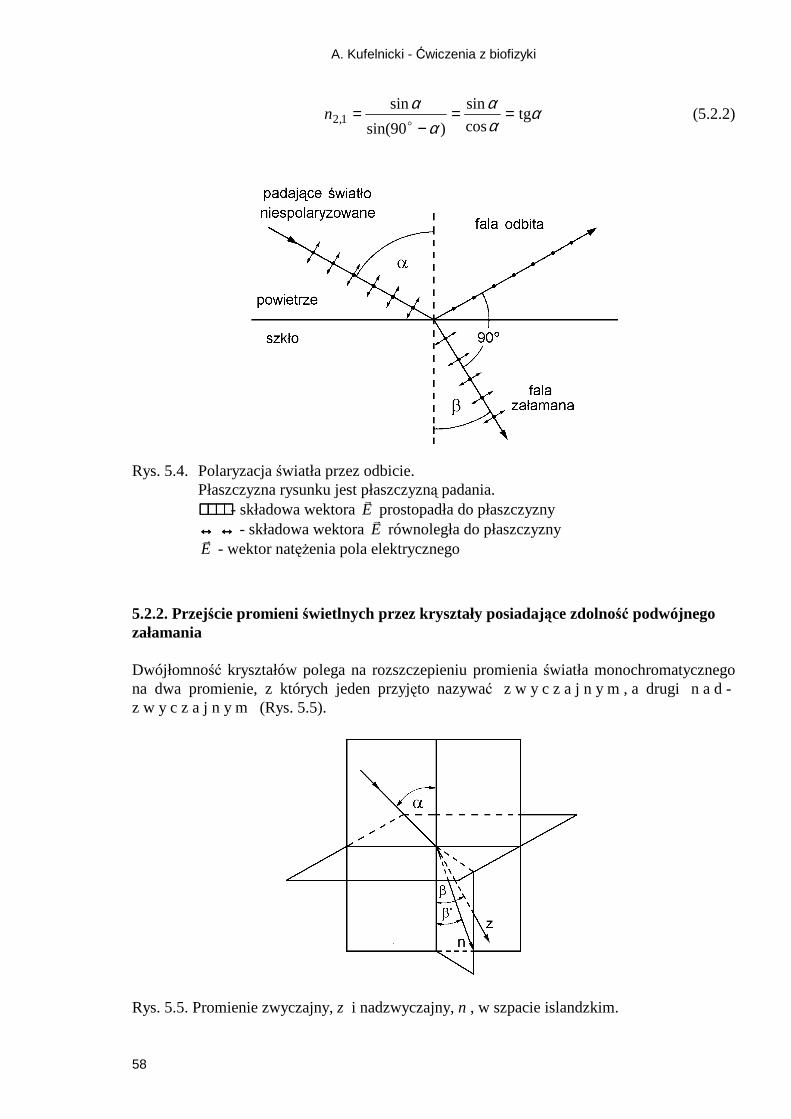
A. Kufelnicki - Ćwiczenia z biofizyki
58
ααα
αα
tgcos
sin
)90sin(
sin1,2 ==
−=
on (5.2.2)
Rys. 5.4. Polaryzacja światła przez odbicie. Płaszczyzna rysunku jest płaszczyzną padania. ⋅⋅⋅⋅ ⋅⋅⋅⋅ ⋅⋅⋅⋅ ⋅⋅⋅⋅ - składowa wektora E
r prostopadła do płaszczyzny
↔↔↔↔ ↔↔↔↔ - składowa wektora Er
równoległa do płaszczyzny E
r - wektor natęŜenia pola elektrycznego
5.2.2. Przejście promieni świetlnych przez kryształy posiadające zdolność podwójnego załamania Dwójłomność kryształów polega na rozszczepieniu promienia światła monochromatycznego na dwa promienie, z których jeden przyjęto nazywać z w y c z a j n y m , a drugi n a d -z w y c z a j n y m (Rys. 5.5).
Rys. 5.5. Promienie zwyczajny, z i nadzwyczajny, n , w szpacie islandzkim.

5. Niektóre fizyczne metody badania struktury molekuł
59
Obydwa promienie są spolaryzowane w płaszczyznach wzajemnie prostopadłych. Głównym kryształem stosowanym w przyrządach polaryzujących jest szpat islandzki (kalcyt, CaCO3). Jednym z polaryzatorów jest pryzmat Nicola (zwany teŜ po prostu nikolem). Stanowi on od-powiednio oszlifowany kryształ szpatu, przecięty w określonej płaszczyźnie i z powrotem sklejony warstewką balsamu kanadyjskiego (Rys. 5.6).
Rys. 5.6. Bieg promieni w pryzmacie Nicola. Balsam kanadyjski jest ośrodkiem optycznie rzadszym dla promienia zwyczajnego niŜ szpat islandzki (współczynnik załamania szpatu islandzkiego dla promienia zwyczajnego, nz = 1,658 jest większy od współczynnika załamania balsamu: 1,542). Promień zwyczajny ulega całkowitemu wewnętrznemu odbiciu od warstwy balsamu ka-nadyjskiego, poniewaŜ pada on na płaszczyznę balsamu pod kątem większym od granicznego. Po odbiciu promień zwyczajny pada na wyczernione ścianki boczne i zostaje pochłonięty, na-tomiast promień nadzwyczajny przechodzi przez warstwę balsamu kanadyjskiego, ulegając tylko nieznacznemu osłabieniu. Omawiane warunki biegu światła w pryzmacie Nicola są spełnione, jeŜeli promienie pa-dające na pryzmat nie tworzą zbyt duŜego kąta z kierunkiem SI, równoległym do krawędzi AC' i A'C (Rys. 5.6), czyli jeśli wi ązka nie jest zbyt rozbieŜna.
5.2.3. Wykorzystanie anizotropii pochłaniania - dichroizm Na tej zasadzie oparte jest działanie polaroidów, np. polaroidu Landa (Rys. 5.7).
Rys. 5.7. Anizotropia pochłaniania światła (dichroizm). Na przeźroczystej błonie znajdują się równolegle ułoŜone względem siebie łańcuchowe cząsteczki polimeru nasyconego atomami jodu. JeŜeli błonę ustawimy prostopadle do wiązki światła niespolaryzowanego, to fale świetlne, dla których kierunki drgań wektora E
r są rów-
noległe do cząsteczek polimeru będą silnie pochłaniane przez atomy jodu, natomiast fale o

A. Kufelnicki - Ćwiczenia z biofizyki
60
drganiach prostopadłych zostaną przepuszczone praktycznie bez zmiany energii. Podobnie, dla wszystkich innych fal świetlnych pochłaniane będą składowe równoległe wektorów E
r, a
przepuszczane tylko składowe prostopadłe. Wiązka światła po przejściu przez polaroid będzie więc spolaryzowana - drgania wektora E
r odbywać się będą w kierunku prostopadłym do łań-
cucha cząsteczek polimeru. C z y n n y m i o p t y c z n i e nazywamy ciała stałe i ciekłe, które skręcają płaszczyznę światła liniowo spolaryzowanego pozostawiając je liniowo spolaryzowanym. JeŜeli skręcenie płaszczyzny polaryzacji nastąpiło w prawo (tzn. zgodnie z ruchem wskazówek zegara patrząc naprzeciw) - mówimy o ciałach prawoskrętnych i oznaczamy je znakiem „+”; w przeciwnym razie mówimy o ciałach lewoskrętnych ze znakiem „-”. Czynność optyczna niektórych ciał stałych jest wynikiem ich budowy krystalicznej i zni-ka po stopieniu lub rozpuszczeniu. Dla cieczy lub substancji rozpuszczonych czynność optyczna jest spowodowana asymetrią budowy cząsteczki - brakiem środka i płaszczyzny sy-metrii. Przyczyną tej asymetrii jest najczęściej obecność róŜnie podstawionego atomu węgla, ale równieŜ np. atomu azotu lub siarki, które tworzą tzw. c e n t r u m c h i r a l n oś c i . Cząsteczka optycznie czynna (chiralna) moŜe występować w dwóch formach e n a n c j o -m e r y c z n y c h mających się do siebie jak przedmiot do odbicia lustrzanego.
C
COOH
HO
H
CH3
a) b)
c)
N
O
H5C2C6H5
CH3
S
OCH3
C6H4COOH
Rys. 5.8. Przykłady związków optycznie czynnych: a) kwas mlekowy, b) aminotlenek, c) sulfotlenek. Takie odmiany enancjomeryczne są ciałami lewo- i prawoskrętnymi. Ich właściwości fi-zyczne i chemiczne są jednakowe z wyjątkiem kierunku skręcania płaszczyzny polaryzacji światła, chociaŜ bezwzględne wartości kątów skręcenia są sobie równe.
Do opisu stereochemii związków chiralnych wprowadzono takŜe oznaczenia D (łac. dextra - prawa) i L (łac. laeva - lewa). Konfiguracje D i L odnosi się umownie do konfiguracji aldehydu glicerynowego prawo- i lewoskrętnego:
aldehyd L-(−−−−)-glicerynowyaldehyd D-(+)-glicerynowy
CHO
H OH
CH2OH
C*
CHO
HO H
CH2OH
C*

5. Niektóre fizyczne metody badania struktury molekuł
61
We wzorach strukturalnych związków D grupę OH zapisuje się po prawej stronie chiralnego atomu węgla (z gwiazdką), a w związkach L - po lewej stronie. Symbole (+) i (−) wiąŜą się z faktem bezwzględnego skręcania płaszczyzny polaryzacji światła spolaryzowanego w prawo lub lewo, natomiast D i L rozróŜniają konfiguracje tego samego związku. Oznacza to, Ŝe związek o konfiguracji np. D moŜe być prawo-, ale teŜ lewo-skrętny, czyli D-(+) lub D-(−).
Występowanie centrów chiralności nie stanowi jednak wystarczającego kryterium czynności optycznej. JeŜeli związek posiada centra chiralności, a cząsteczka jako całość ma płaszczyznę symetrii, albo środek symetrii, to jest on optycznie nieczynny. Przykładem jest kwas mezo-winowy, cząsteczka achiralna mająca płaszczyznę symetrii:
kwas D-(−−−−)-winowy kwas mezo-winowy
COOH
C
C
COOH
HHO
H OH
COOH
COOH
H
H OH
C
C
HO
kwas L-(+)-winowy
*
*
COOH
COOH
OH
H OH
C
C
H
*
**
*
Kwas mezo-winowy jest wobec odmian prawo- i lewoskrętnej d i a s t e r e o i z o m e r e m , a więc w stosunku do cząsteczek tych izomerów, cząsteczki odmiany mezo nie są odbiciami lustrzanymi. Inaczej niŜ dla enancjomerów, właściwości chemiczne i fizyczne diastereoizome-rów są róŜne. Wielkość kąta skręcenia zaleŜy od rodzaju substancji, grubości jej warstwy, temperatury i długości fali uŜytego światła. Zwykle pomiary wykonuje się w temp. 293 K za pomocą linii D światła sodowego. Celem umoŜliwienia porównywania czynności optycznej róŜnych sub-stancji wprowadzono pojęcie s k rę c a l n oś c i w ł a ś c i w e j , [α]. Rodzaj uŜytego światła (λ - długość fali w nm lub nazwę linii widma emisyjnego, np. linia D światła sodowe-go 589,3 nm) i temperaturę (T) zaznacza się po prawej stronie: [α]λ
T . Skręcalność właściwa czystej substancji wyraŜa się wzorem:
[ ]ρ
αα⋅
=l
293D (5.2.3)
α - kąt skręcenia próbki danej substancji optycznie czynnej l - grubość warstwy substancji: dla ciał stałych 1 mm, dla cieczy 1 dm ρ - gęstość substancji optycznie czynnej.
W przypadku roztworów wprowadzamy ich procentowość p ( symbol ρ oznacza teraz gęstość roztworu w g/cm3) lub stęŜenie c wyraŜone w gramach na 100 cm3 roztworu:
[ ] [ ]clpl ⋅
⋅=⋅⋅
⋅= 100 ,
100 293D
293D
ααρ
αα (5.2.4)
Po przekształceniu:
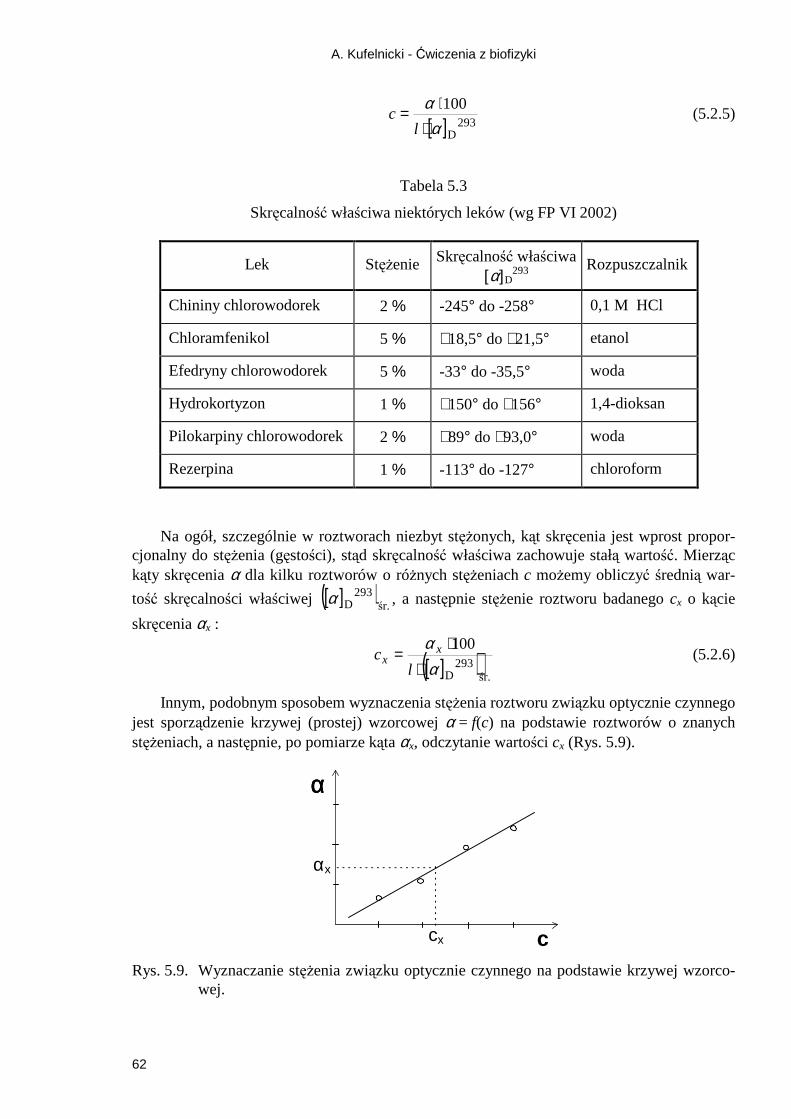
A. Kufelnicki - Ćwiczenia z biofizyki
62
[ ] 293
D
100
αα⋅
⋅=l
c (5.2.5)
Tabela 5.3
Skręcalność właściwa niektórych leków (wg FP VI 2002)
Lek StęŜenie Skręcalność właściwa [α]D
293 Rozpuszczalnik
Chininy chlorowodorek 2 % -245° do -258° 0,1 M HCl
Chloramfenikol 5 % +18,5° do +21,5° etanol
Efedryny chlorowodorek 5 % -33° do -35,5° woda
Hydrokortyzon 1 % +150° do +156° 1,4-dioksan
Pilokarpiny chlorowodorek 2 % +89° do +93,0° woda
Rezerpina 1 % -113° do -127° chloroform
Na ogół, szczególnie w roztworach niezbyt stęŜonych, kąt skręcenia jest wprost propor-cjonalny do stęŜenia (gęstości), stąd skręcalność właściwa zachowuje stałą wartość. Mierząc kąty skręcenia α dla kilku roztworów o róŜnych stęŜeniach c moŜemy obliczyć średnią war-
tość skręcalności właściwej [ ]( )śr.293Dα , a następnie stęŜenie roztworu badanego cx o kącie
skręcenia αx :
[ ]( )śr.293
D
100
αα
⋅
⋅=
lc x
x (5.2.6)
Innym, podobnym sposobem wyznaczenia stęŜenia roztworu związku optycznie czynnego jest sporządzenie krzywej (prostej) wzorcowej α = f(c) na podstawie roztworów o znanych stęŜeniach, a następnie, po pomiarze kąta αx, odczytanie wartości cx (Rys. 5.9).
αααα
αx
ccx
Rys. 5.9. Wyznaczanie stęŜenia związku optycznie czynnego na podstawie krzywej wzorco-wej.

5. Niektóre fizyczne metody badania struktury molekuł
63
Skręcalność właściwa niektórych substancji zmienia się jednak wraz ze zmianą stęŜenia. Polarymetryczne oznaczanie stęŜeń roztworów takich substancji zwykle wykonuje się tylko przez porównanie kątów skręcenia dla roztworów o bardzo zbliŜonych stęŜeniach.
Do pomiaru kąta skręcenia płaszczyzny polaryzacji stosowane są p o l a r y m e t r y . Za-sada działania polarymetrów polega na uŜyciu dwóch nikoli lub polaroidów. Zadaniem jed-nego z nich jest spolaryzowanie wiązki świetlnej i dlatego nazywa się on p o l a r y z a t o -r e m . Drugi słuŜy do obserwowania i ustalenia połoŜenia płaszczyzny polaryzacji po przej-ściu przez substancję badaną i nazywa się a n a l i z a t o r e m .
Rys. 5.10. Zasada działania polarymetru. PołoŜenie polaryzatora jest stałe, a analizatora zmienne - posiada on urządzenie do pomiaru kąta, o jaki obrócono jego płaszczyznę polaryzacji. NatęŜenie światła przepuszczonego przez analizator zaleŜy od kąta obrotu analizatora i zgodnie z p r a w e m M a l u s a wynosi: I I= ⋅0
2cosα (5.2.7) gdzie: I - natęŜenie światła przepuszczonego przez analizator I0 - natęŜenie światła przepuszczonego przez polaryzator α - kąt obrotu analizatora. Wartości I mają więc swoje maksima w połoŜeniach analizatora 0° i 180°, a przy 90° i 270° są równe zeru. Do badania zjawiska czynności optycznej roztworów stosujemy polarymetry róŜnych ty-pów. Zasadniczo zbudowane są one z soczewki kolimacyjnej S, polaryzatora P, rurki do na-pełniania cieczą badaną R i analizatora A . Dla zwiększenia dokładności odczytów polaryme-try posiadają zazwyczaj jeszcze pryzmat półcieniowy PP.
Rys. 5.11. Schemat polarymetru. Pryzmat polaryzatora i pryzmat półcieniowy polaryzują światło w płaszczyznach tworzą-cych z sobą pewien, niewielki kąt. Ustawienie analizatora tak, aby jego płaszczyzna polaryza-cji była prostopadła do płaszczyzny polaryzacji promieni przechodzących przez nikol polary-zatora powoduje zaciemnienie odpowiedniej połowy pola widzenia (Rys. 5.12a). Zmieniając

A. Kufelnicki - Ćwiczenia z biofizyki
64
połoŜenie analizatora moŜna otrzymać równe zaciemnienie obu połówek pola widzenia (Rys. 5.12b) w momencie, gdy płaszczyzna polaryzacji analizatora utworzy jednakowe kąty z płaszczyznami polaryzacji polaryzatora i pryzmatu półcieniowego. Dalszy obrót analizatora powoduje zaciemnienie tej połówki pola widzenia, która poprzednio była jasna (Rys. 5.12c). PołoŜenie analizatora, przy którym obie połówki pola widzenia są równo zaciemnione, jest połoŜeniem zerowym.
Rys. 5.12. Obraz obserwowany w okularze polarymetru dwupolowego. Jeszcze lepsze moŜliwości odczytu zapewnia polarymetr trójpolowy. Przyrząd wyregulo-wany jest w ten sposób, Ŝe bez cieczy optycznie czynnej zerowa kreska podziałki kątowej powinna pokrywać się z zerem podziałki noniusza. Wówczas trzy części pola widzenia w okularze są jednakowo jasno oświetlone (Rys. 5.13b). W przypadku, gdy do aparatu wstawi-my rurkę polarymetryczną wypełnioną prawoskrętną substancją optycznie czynną, „+”, środ-kowa część pola widzenia ulegnie zaciemnieniu (Rys. 5.13a). JeŜeli obraz w okularze jest przeciwny (Rys. 5.13c), oznacza to substancja, Ŝe substancja optycznie czynna jest lewoskręt-na, „-”. Pomiar polega na takim ustawieniu analizatora, aby uzyskać pierwotną jasność wszystkich trzech części pola widzenia. Wartość kąta, o jaką badana substancja skręciła płaszczyznę światła spolaryzowanego odczytuje się na tarczy za pomocą lupy i noniusza z do-kładnością do 0,05°.
Rys. 5.13. Obraz ·obserwowany w okularze polarymetru trójpolowego. S a c h a r y m e t r i a . Do oznaczania stęŜeń cukrów najczęściej stosuje się metodę po-larymetryczną. Sacharymetry przemysłowe posiadają skalę wycechowaną w jednostkach sa-charymetrycznych, a nie kątowych. 100° S tej skali odpowiada roztworowi, który zawiera 26,000 g cukru w temperaturze 20 °C i objętości 100 cm3, badanemu w rurce polarymetrycz-nej o długości 200 mm. Stopnie sacharymetryczne przelicza się na kątowe zgodnie z równa-niem:
1° S = 0,34613° Literatura uzupełniająca:
F. Jaroszyk, red., „Biofizyka”, Wydawnictwo Lekarskie PZWL, Warszawa 2002, str. 23. T. W. Hermann, red., „Farmacja fizyczna”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 375.
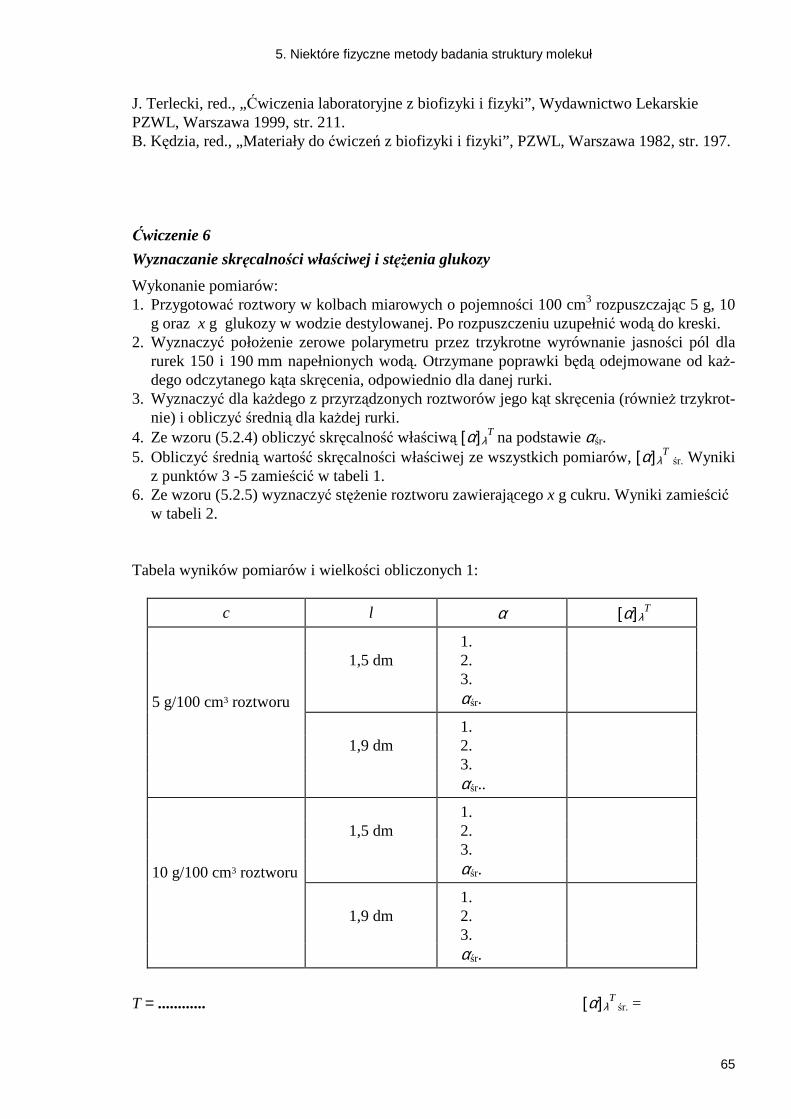
5. Niektóre fizyczne metody badania struktury molekuł
65
J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 211. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 197. Ćwiczenie 6
Wyznaczanie skręcalności właściwej i stęŜenia glukozy
Wykonanie pomiarów: 1. Przygotować roztwory w kolbach miarowych o pojemności 100 cm3 rozpuszczając 5 g, 10
g oraz x g glukozy w wodzie destylowanej. Po rozpuszczeniu uzupełnić wodą do kreski. 2. Wyznaczyć połoŜenie zerowe polarymetru przez trzykrotne wyrównanie jasności pól dla
rurek 150 i 190 mm napełnionych wodą. Otrzymane poprawki będą odejmowane od kaŜ-dego odczytanego kąta skręcenia, odpowiednio dla danej rurki.
3. Wyznaczyć dla kaŜdego z przyrządzonych roztworów jego kąt skręcenia (równieŜ trzykrot-nie) i obliczyć średnią dla kaŜdej rurki.
4. Ze wzoru (5.2.4) obliczyć skręcalność właściwą [α]λT na podstawie αśr.
5. Obliczyć średnią wartość skręcalności właściwej ze wszystkich pomiarów, [α]λT śr. Wyniki
z punktów 3 -5 zamieścić w tabeli 1. 6. Ze wzoru (5.2.5) wyznaczyć stęŜenie roztworu zawierającego x g cukru. Wyniki zamieścić
w tabeli 2. Tabela wyników pomiarów i wielkości obliczonych 1:
c l α [α]λT
1.
1,5 dm 2. 3.
5 g/100 cm3 roztworu αśr.
1.
1,9 dm 2. 3. αśr..
1.
1,5 dm 2. 3.
10 g/100 cm3 roztworu αśr.
1.
1,9 dm 2. 3. αśr.
T = ............ [α]λT śr. =
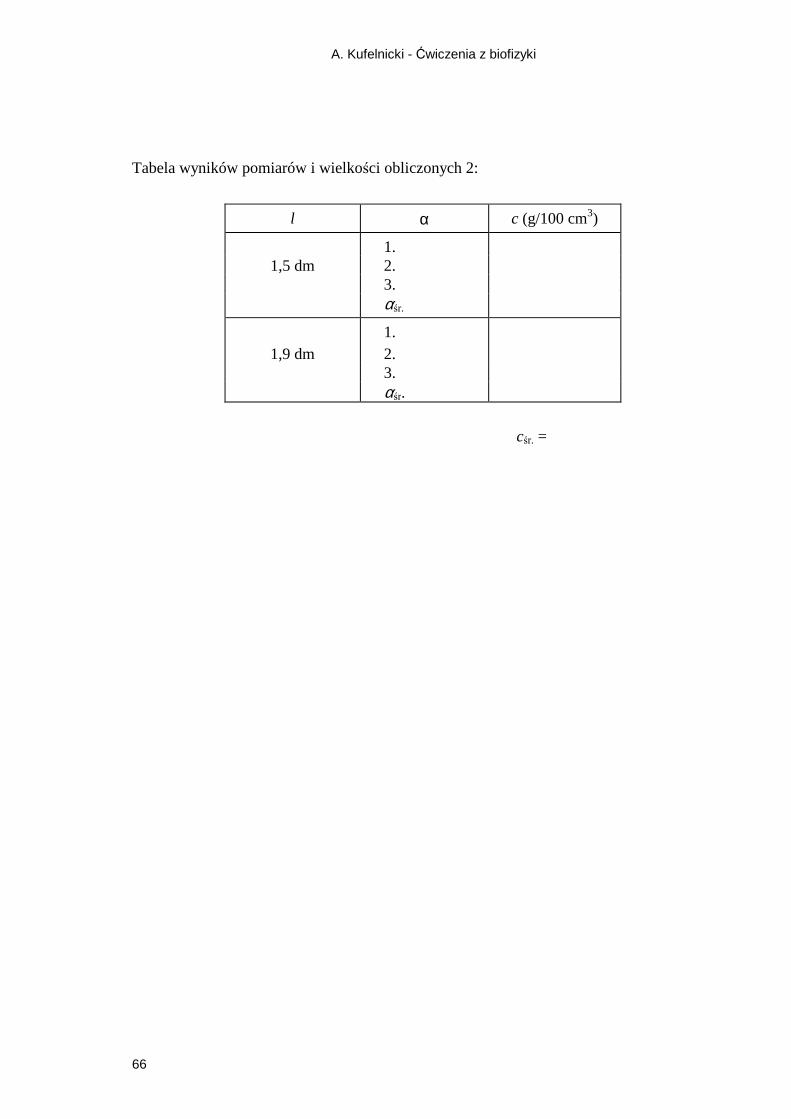
A. Kufelnicki - Ćwiczenia z biofizyki
66
Tabela wyników pomiarów i wielkości obliczonych 2:
l α c (g/100 cm3)
1.
1,5 dm 2. 3. αśr.
1.
1,9 dm 2. 3. αśr.
cśr. =

67
6. PRZEWODNICTWO JONOWE
6.1. Rodzaje przewodników elektryczności
W zaleŜności od rodzaju nośników ładunku wyróŜniamy następujące rodzaje przewodni-ków elektryczności: P r z e w o d n i k i I r o d z a j u ( e l e k t r o n o w e ) - nośnikami ładunku są elektrony; przepływ prądu nie powoduje efektów w przewodniku, gdyŜ elektrony są nierozróŜnialne. Dzięki temu, równieŜ przepływ prądu przez takie przewodniki jest bardzo szybki. Przewodni-kami I rodzaju są metale i ich stopy, grafit, niektóre tlenki, siarczki i węgliki metali. Przewod-nictwo maleje wraz ze wzrostem temperatury, a wzrasta w niskich temperaturach - w bardzo niskich temperaturach występuje nawet zjawisko nadprzewodnictwa. P r z e w o d n i k i I I r o d z a j u ( j o n o w e ) - nośnikami ładunku są jony, obserwuje się charakterystyczne skutki przepływu prądu przez roztwory elektrolitów, polegające na zmianach chemicznych i zmianach stęŜeń. Przewodnikami drugiego rodzaju są roztwory soli, kwasów i zasad, sole stopione lub w stanie stałym. Ich przewodnictwo na ogół rośnie wraz ze wzrostem temperatury. Właściwości przewodników II rodzaju, równieŜ występujących w układach biologicznych, będą głównym przedmiotem rozwaŜań niniejszego rozdziału. P ó ł p r z e w o d n i k i ( p r z e w o d n i k i e l e k t r o n o w e i d z i u r o w e ) - no-śnikami ładunku są elektrony i dodatnio naładowane dziury elektronowe. Tego rodzaju prze-wodniki znajdują szerokie zastosowanie w elektronice (tranzystory, obwody scalone i inne). 6.2. Ruchliwość jonów
Wartość wektora n a tę Ŝ e n i a p o l a e l e k t r y c z n e g o ,rE , w danym punkcie
pola elektrostatycznego jest równa stosunkowi siły Fr
działającej na mały, dodatni ładunek q umieszczony w tym punkcie pola do wartości tego ładunku (Rys. 6.1). ZastrzeŜenie dotyczące niewielkiej wartości ładunku q (a takŜe jego bardzo małych rozmiarów geometrycznych) wią-Ŝe się z tym, Ŝe pole utworzone przez wspomniany ładunek, zwany często ładunkiem „prób-nym”, nie powinno zakłócać badanego pola elektrycznego. Linie sił pola dla ładunku dodat-niego mają zwrot zgodny z kierunkiem sił działających na ten ładunek.
l+
F E= Fq
+
_
Rys. 6.1. Jednorodne pole elektrostatyczne.
MoŜna takŜe wykazać, Ŝe natęŜenie pola elektrycznego w danym punkcie jest gradientem (inaczej: spadem) potencjału pola elektrycznego w tym punkcie. JeŜeli do roztworu zanurzy-my dwie płaskie elektrody i przyłoŜymy do nich stałe napięcie zewnętrzne, U, to wytworzymy między nimi w przybliŜeniu jednorodne pole elektryczne (o stałej wartości natęŜenia pola we wszystkich punktach i równoległych liniach sił). W tym przypadku gradient potencjału upraszcza się do wyraŜenia:

A. Kufelnicki - Ćwiczenia z biofizyki
68
EU
l= (6.2.1)
gdzie: E - natęŜenie pola elektrycznego U - napięcie między elektrodami l - odległość między elektrodami
Na jony oraz inne cząstki obdarzone ładunkiem pole elektryczne oddziałuje siłą kulom-bowską F, która jest wprost proporcjonalna do natęŜenia pola i ładunku q:
F E qU
lq
U
lz e= = = (6.2.2)
gdzie: z - wartościowość jonu e - ładunek elektronu (tzw. ładunek elementarny = 1,60⋅10-19 C).
Pod wpływem siły F jon o masie m uzyskuje przyspieszenie proporcjonalne do tej siły (a = F / m) . Wraz ze wzrostem prędkości jonu rośnie takŜe opór ośrodka zwany siłą tarcia we-wnętrznego, F ′. Siła tarcia wewnętrznego skierowana jest przeciwnie do kierunku ruchu i dla sferycznego jonu wynosi, zgodnie z równaniem Stokesa:
′ =F r v6π η (6.2.3)
gdzie: r - promień cząstki kulistej v - prędkość η - lepkość ośrodka zaleŜna od takich czynników, jak: rodzaj ośrodka, stęŜenie
rozpuszczonej w nim substancji, temperatura i ciśnienie.
Początkowo przyspieszony ruch jonów bardzo szybko przechodzi w jednostajny (od mo-mentu, gdy siła kulombowska F zostanie zrównowaŜona przez siłę tarcia wewnętrznego, F ′). Prędkość tego ruchu, dla kaŜdego jonu w danym środowisku, zaleŜy od natęŜenia pola elek-trycznego. Z równań (6.2.2) i (6.2.3) oraz warunku F = F ′ wynika, Ŝe:
vE q
r=
6π η (6.2.4)
Chcąc porównywać szybkości ruchu róŜnych jonów w polu elektrycznym, naleŜy znor-
malizować warunki. Porównuje się więc prędkości jonów w polu elektrycznym o natęŜeniu jednostkowym (1 V/m). Prędkość poruszania się jonu pod wpływem pola elektrycznego o na-tęŜeniu jednostkowym nazywamy r u c h l i w oś c i ą j o n u , u:
uv
E= (6.2.5)
Wstawiając v z równania (6.2.4) do równania (6.2.5) otrzymujemy zaleŜność:
uq
r=
6π η (6.2.6)
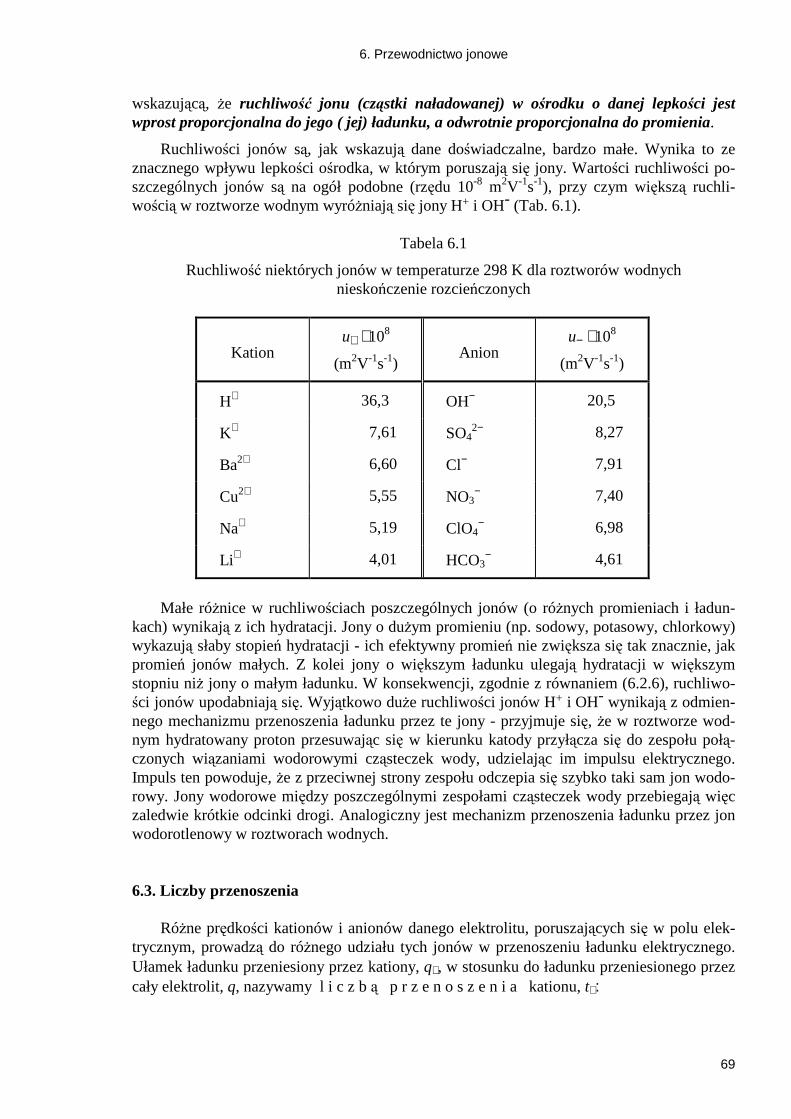
6. Przewodnictwo jonowe
69
wskazującą, Ŝe ruchliwość jonu (cząstki naładowanej) w ośrodku o danej lepkości jest wprost proporcjonalna do jego ( jej) ładunku, a odwrotnie proporcjonalna do promienia.
Ruchliwości jonów są, jak wskazują dane doświadczalne, bardzo małe. Wynika to ze znacznego wpływu lepkości ośrodka, w którym poruszają się jony. Wartości ruchliwości po-szczególnych jonów są na ogół podobne (rzędu 10-8 m2V-1s-1), przy czym większą ruchli-wością w roztworze wodnym wyróŜniają się jony H+ i OH- (Tab. 6.1).
Tabela 6.1
Ruchliwość niektórych jonów w temperaturze 298 K dla roztworów wodnych nieskończenie rozcieńczonych
Kation u+ ⋅ 108
(m2V-1s-1) Anion
u− ⋅ 108
(m2V-1s-1)
H+ 36,3 OH− 20,5
K+ 7,61 SO42− 8,27
Ba2+ 6,60 Cl− 7,91
Cu2+ 5,55 NO3− 7,40
Na+ 5,19 ClO4− 6,98
Li + 4,01 HCO3− 4,61
Małe róŜnice w ruchliwościach poszczególnych jonów (o róŜnych promieniach i ładun-
kach) wynikają z ich hydratacji. Jony o duŜym promieniu (np. sodowy, potasowy, chlorkowy) wykazują słaby stopień hydratacji - ich efektywny promień nie zwiększa się tak znacznie, jak promień jonów małych. Z kolei jony o większym ładunku ulegają hydratacji w większym stopniu niŜ jony o małym ładunku. W konsekwencji, zgodnie z równaniem (6.2.6), ruchliwo-ści jonów upodabniają się. Wyjątkowo duŜe ruchliwości jonów H+ i OH- wynikają z odmien-nego mechanizmu przenoszenia ładunku przez te jony - przyjmuje się, Ŝe w roztworze wod-nym hydratowany proton przesuwając się w kierunku katody przyłącza się do zespołu połą-czonych wiązaniami wodorowymi cząsteczek wody, udzielając im impulsu elektrycznego. Impuls ten powoduje, Ŝe z przeciwnej strony zespołu odczepia się szybko taki sam jon wodo-rowy. Jony wodorowe między poszczególnymi zespołami cząsteczek wody przebiegają więc zaledwie krótkie odcinki drogi. Analogiczny jest mechanizm przenoszenia ładunku przez jon wodorotlenowy w roztworach wodnych. 6.3. Liczby przenoszenia
RóŜne prędkości kationów i anionów danego elektrolitu, poruszających się w polu elek-trycznym, prowadzą do róŜnego udziału tych jonów w przenoszeniu ładunku elektrycznego. Ułamek ładunku przeniesiony przez kationy, q+, w stosunku do ładunku przeniesionego przez cały elektrolit, q, nazywamy l i c z bą p r z e n o s z e n i a kationu, t+:

A. Kufelnicki - Ćwiczenia z biofizyki
70
tq
q++= (6.3.1)
oraz analogicznie dla anionu:
tq
q−−= (6.3.2)
PoniewaŜ q+ + q− = q
więc: t+ + t− = 1 (6.3.3)
Liczby przenoszenia jonów związane są z ich ruchliwościami. W przypadku elektrolitu syme-trycznego (kation i anion o tej samej wartościowości):
tu
u u++
+ −=
+ t
u
u u−−
+ −=
+ (6.3.4)
W przypadku ogólnym, gdy roztwór zawiera większą liczbę róŜnych rodzajów jonów o róŜ-nych wartościowościach zi:
tc z u
c z uii i i
i i i
=∑
(6.3.5)
gdzie ci - stęŜenie molowe jonów danego rodzaju.
Ruchliwości jonów w danym rozpuszczalniku zaleŜą od stęŜenia roztworu i od jego tem-peratury. Od tych samych parametrów, choć w mniejszym stopniu, zaleŜą liczby przenoszenia (Tab. 6.2). Następuje tu częściowa kompensacja wpływu obu zmiennych. Dla elektrolitu sy-metrycznego ze wzrostem temperatury liczby przenoszenia dąŜą do wartości 0,5.
Tabela 6.2
Liczby przenoszenia niektórych kationów w roztworach wodnych (temp. 298 K)
Elektrolit t+
0,1 n 0,01 n nieskończenie
rozcieńczony
HCl 0,831 0,825 0,821
CH3COONa 0,559 0,554 0,551
KNO3 0,510 0,508 0,507
KCl 0,490 0,490 0,491
LiCl 0,317 0,329 0,336
AgNO3 0,468 0,465 0,464
Na2SO4 0,383 0,385 0,386
NaleŜy podkreślić, Ŝe liczby przenoszenia danych kationów i anionów odnoszą się wy-
łącznie do określonego elektrolitu. W innym elektrolicie liczba przenoszenia tego samego jo-nu jest najczęściej inna, a wynika to z róŜnego udziału jonu przeciwnego znaku w przenosze-

6. Przewodnictwo jonowe
71
niu ładunku. Na przykład liczba przenoszenia kationu Na+ w 0,1 n octanie sodu wynosi 0,559, ale w 0,1 n Na2SO4 tylko 0,383. 6.4. Pomiar oporu elektrycznego i przewodnictwa elektrolitów
P r z e w o d n i c t w e m e l e k t r y c z n y m (λ) lub przewodnością elektryczną na-zywamy odwrotność oporu elektrycznego, λ = 1/R . Jednostką przewodnictwa jest om-1 (Ω-1), zwany simensem (S). Przewodnictwo elektrolitu jest zaleŜne od stęŜenia i rodzaju jonów oraz od wymiarów geometrycznych warstwy roztworu zawartego między elektrodami. Naczyńka stosowane do pomiarów przewodnictwa zawierają elektrody platynowe, zazwyczaj pokryte elektrolitycznie wydzieloną czernią platynową (inaczej: platyną w rozproszeniu atomowym) w celu zwiększenia powierzchni czynnej elektrod oraz zmniejszenia efektów polaryzacyjnych. Przykładowe rozwiązania budowy konduktometrycznych naczyniek pomiarowych przedsta-wia Rys. 6.2. W środkowym naczyńku doprowadzenie do elektrod zapewniają warstwy rtęci.
.
Rys. 6.2. Naczyńka do pomiarów przewodnictwa elektrycznego.
Pomiary przewodnictwa elektrolitów powinny być wykonywane w naczyńkach termo-statowanych, poniewaŜ zmiana temperatury o 1 °C zmienia przewodnictwo elektrolitu średnio o 2 – 3%. Rozpuszczalnik uŜywany do sporządzania roztworów musi być dokładnie oczysz-czony, szczególnie wtedy, gdy badane są roztwory bardzo rozcieńczone. Najczęściej stosowa-nym rozpuszczalnikiem jest woda, której przewodnictwo wzrasta nawet z powodu śladowych zanieczyszczeń.
Dokładne pomiary oporu elektrolitu przeprowadza się w układzie, który stanowi m o -s t e k W h e a t s t o n e ’ a w m o d y f i k a c j i K o h l r a u s c h a (Rys. 6.3). Do koń-cówek AB przykładamy napięcie przemienne i symetryczne. Rz jest oporem o wartości znanej, Rx jest oporem badanym. W gałęzi CD umieszczamy przyrząd zerowy pokazujący przepływ małego prądu (np. mikroamperomierz). Końcówka D tej gałęzi styka się z opornikiem AB i moŜe być wzdłuŜ niego przesuwana. W chwili, gdy w gałęzi CD prąd przestaje płynąć, poten-cjał punktu C równy jest potencjałowi punktu D. Spadek napięcia na oporze Rz musi być wte-dy równy spadkowi napięcia na oporze AD, a spadek napięcia na oporze Rx równy jest spad-kowi napięcia na oporze DB. Zapiszemy to w postaci równań:
2AD1 IRIRz ⋅=⋅ (6.4.1)

A. Kufelnicki - Ćwiczenia z biofizyki
72
R I R Ix ⋅ = ⋅1 2DB (6.4.2)
Rys.6.3. Schemat mostka Wheatstone’a – Kohlrauscha.
Dzieląc stronami równania (6.4.1) i (6.4.2), a następnie odwracając otrzymamy:
R
R
R
Rx
z= DB
AD (6.4.3)
Stąd:
AD
DB
R
RRR zx = (6.4.4)
Oznacza to, Ŝe nieznany opór Rx moŜemy zmierzyć przez porównanie go ze znanym opo-rem Rz.
W praktycznie stosowanych mostkach, stosunek RDB/RAD (tzw. p o d s t a w a m o s t -k a ) jest wielkością stałą, zwaną „mnoŜnikiem” lub „dzielnikiem” mostka. Wartość ta wyno-si 1, 10, 100 lub 1000. JeŜeli np. mnoŜnik równa się 1, to RDB = RAD ; jeŜeli 10, to RDB = 10 RAD itp. W przypadku dzielnika liczba 10 oznacza: RDB = 1/10 RAD, liczba 100: RDB = 1/100 RAD itd. Opornikiem Rz jest opornica dekadowa zwana dekadą porównawczą. Opór ten moŜna zmieniać skokowo o wielokrotności 1000 Ω, 100 Ω, 10 Ω, 1 Ω i 0,1 Ω.
W przypadku pomiaru oporu elektrolitu napięcie zasilające mostek nie moŜe być napię-ciem stałym. PrzyłoŜenie napięcia stałego spowodowałoby wystąpienie zjawiska elektrolizy, a tym samym polaryzację elektrod. Powstająca siła elektromotoryczna polaryzacji zmniejszały-by napięcie zasilające, co prowadziłoby do zmniejszania natęŜenia prądu I1. UniemoŜliwiłoby to precyzyjny pomiar oporu Rx. Na ogół zatem mostek Wheatstone’a – Kohlrauscha (w odróŜ-nieniu od samego mostka Kohlrauscha) zasilany jest z generatora drgań niegasnących napię-ciem przemiennym o częstotliwości 1000 – 3000 Hz. Jako wskaźnik w gałęzi CD moŜna sto-sować wówczas słuchawkę telefoniczną, miernik prądu zmiennego lub miernik prądu stałego z prostownikiem i wzmacniaczem.
Zastosowanie prądu przemiennego pociąga za sobą jednak szereg zjawisk ubocznych, jak pojawienie się oporów indukcyjnych i pojemnościowych zaleŜnych od częstotliwości prądu. Aby natęŜenie prądu w gałęzi CD spadło do zera, proporcja Rx/Rz = RDB/RAD musi być speł-niona takŜe dla oporów indukcyjnych i pojemnościowych. W praktyce nigdy nie udaje się spełnić tego warunku, wobec czego za punkt kompensacji przyjmuje się takie połoŜenie styku D, które odpowiada minimum natęŜenia prądu. W dokładniejszych pomiarach do odgałęzienia

6. Przewodnictwo jonowe
73
mostka AC włącza się równolegle kondensator o zmiennej pojemności (Rys. 6.3). Pozwala to na skompensowanie pojemności układu pomiarowego.
Seryjnie produkowane przyrządy do pomiaru przewodnictwa, tzw. konduktometry, są obecnie przewaŜnie przyrządami mikroprocesorowymi, cechowanymi bezpośrednio w jed-nostkach przewodnictwa właściwego (patrz Rozdział 6.5): mS⋅cm-1 lub µS⋅cm-1. Zasadę dzia-łania konduktometru przedstawia Rys. 6.4.
Wartość oporności naczyńka pomiarowego Rx określa natęŜenie prądu płynącego przez stały (dla danego zakresu pomiarowego) opór R. Prowadzi to do odpowiedniego spadku na-pięcia na końcach tego oporu. Wartość spadku napięcia pokazuje odpowiedno wyskalowany miernik M. Zakres pomiarowy przewodnictwa właściwego w konduktometrach mieści się w określonych granicach, np. od 0 do 2000 mS⋅cm-1 w 6 podzakresach. Źródłem napięcia zmiennego jest oscylator o częstotliwości np. 80 Hz i 3 kHz (w zaleŜności od podzakresu po-miarowego).
Rys. 6.4. Zasada działania konduktometru: M - woltomierz tranzystorowy (wyskalowany w simensach) Rx - opór badany R - opór wymienny.
6.5. Przewodnictwo właściwe elektrolitów
Przewodnictwo właściwe (κ – kappa) jest odwrotnością oporu właściwego, κ = 1/ρ. Po-niewaŜ opór elektryczny:
Rl
S= ρ (6.5.1)
gdzie: l - odległość między elektrodami S - pole przekroju poprzecznego warstwy roztworu między elektrodami więc:
λ ρ κ= = ⋅ =11
RS
l
S
l (6.5.2)
Z równania (6.5.2):
κ λ λ= =l
Sk (6.5.3)
gdzie l/S = k nazywamy s t a łą k o n d u k t o m e t r y c z ną naczyńka. Wymiarem k jest m-1 lub cm-1. Na podstawie równania (6.5.3) moŜemy powiedzieć, Ŝe p r z e w o d n i c t w o w ł a ś c i w e danego elektrolitu jest przewodnictwem elektrycznym warstwy tego elektrolitu

A. Kufelnicki - Ćwiczenia z biofizyki
74
o długości jednostkowej i jednostkowym polu przekroju poprzecznego. Wymiarem prze-wodnictwa właściwego jest om-1⋅m-1 (S⋅m-1) lub om-1⋅cm-1 (S⋅cm-1).
Bezpośrednie ustalenie stosunku l/S dla kaŜdego naczyńka byłoby często niemoŜliwe, a przynajmniej bardzo uciąŜliwe i obarczone duŜym błędem. Dlatego przy wyznaczaniu stałej konduktometrycznej postępuje się inaczej. Np. wykorzystuje się stablicowane, dokładnie usta-lone przewodnictwa właściwe roztworów KCl dla róŜnych stęŜeń i temperatur (Tab. 6.3 ). Roztwory te traktuje się jako wzorcowe. Gdy wypełnimy roztworem wzorcowym dowolne naczyńko elektrolityczne i znajdziemy jego przewodnictwo, to z równania (6.5.3) obliczymy stałą k:
KCl
KCl
λκ
=k (6.5.4)
a stąd dla roztworu badanego KCl
KCl
λκ
λκ = . Pomiar przewodnictwa właściwego ma zatem cha-
rakter porównawczy. Ponadto naleŜy pamiętać, Ŝe symbol λKCl we wzorze (6.5.4) dotyczy tyl-ko przewodnictwa jonów K+ i Cl− i dlatego λKCl odpowiada dokładnie wartości λKCl - λH2O. JeŜeli jednak roztwór został sporządzony w dostatecznie czystym rozpuszczalniku (wodzie destylowanej), to λH2O << λKCl i poprawkę λH2O moŜna zaniedbać.
Tabela 6.3
Przewodnictwa właściwe wodnych roztworów chlorku potasu w om-1⋅cm-1
Temp. ( °C)
c (mol⋅dm-3)
0,01 0,1 1 ⋅10-3 ⋅10-2 ⋅10-1
15 1,145 1,047 0,9298
16 1,171 1,071 0,9439
17 1,198 1,095 0,9630
18 1,225 1,119 0,9822
19 1,251 1,143 1,0014
20 1,278 1,167 1,0207
21 1,305 1,191 1,0400
22 1,332 1,215 1,0594
23 1,359 1,239 1,0789
24 1,386 1,264 1,0984
25 1,413 1,288 1,1180
Przykład
Opór naczyńka zmierzony dla 0,01 M roztworu KCl w temp. 20 °C ma wartość 593 Ω. Ob-liczyć stałą konduktometryczną. Ile wynosi przewodnictwo właściwe 0,01 M roztworu NaCl w tej
samej temperaturze, jeŜeli jego opór elektryczny równa się 708 Ω? Korzystając z danych Tab. 6.3 moŜemy obliczyć:
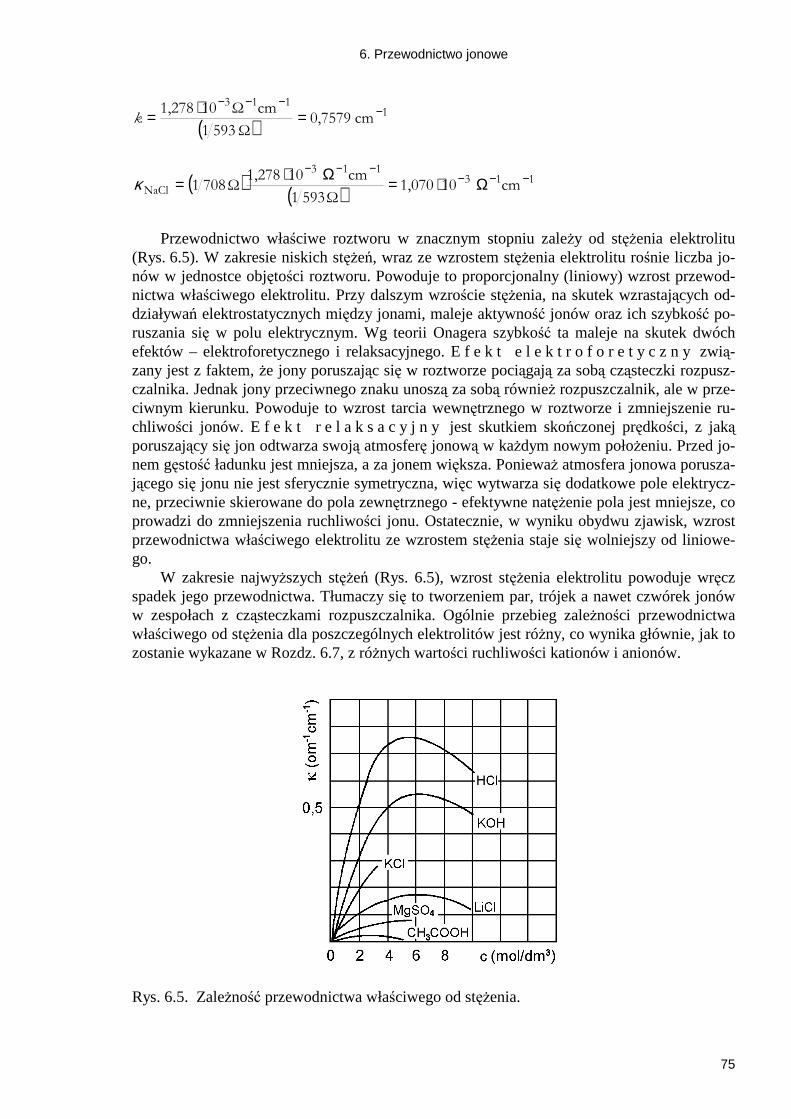
6. Przewodnictwo jonowe
75
( )1
113
cm0,7579Ω5931
cmΩ101,278 −−−−
=⋅=k
( ) ( )113
113
NaCl cm101,070Ω5931
cm101,278Ω7081 −−−
−−−Ω⋅=Ω⋅=κ
Przewodnictwo właściwe roztworu w znacznym stopniu zaleŜy od stęŜenia elektrolitu
(Rys. 6.5). W zakresie niskich stęŜeń, wraz ze wzrostem stęŜenia elektrolitu rośnie liczba jo-nów w jednostce objętości roztworu. Powoduje to proporcjonalny (liniowy) wzrost przewod-nictwa właściwego elektrolitu. Przy dalszym wzroście stęŜenia, na skutek wzrastających od-działywań elektrostatycznych między jonami, maleje aktywność jonów oraz ich szybkość po-ruszania się w polu elektrycznym. Wg teorii Onagera szybkość ta maleje na skutek dwóch efektów – elektroforetycznego i relaksacyjnego. E f ek t e l e k t r o f o r e t y c z n y zwią-zany jest z faktem, Ŝe jony poruszając się w roztworze pociągają za sobą cząsteczki rozpusz-czalnika. Jednak jony przeciwnego znaku unoszą za sobą równieŜ rozpuszczalnik, ale w prze-ciwnym kierunku. Powoduje to wzrost tarcia wewnętrznego w roztworze i zmniejszenie ru-chliwości jonów. E f e k t r e l a k s a c y j n y jest skutkiem skończonej prędkości, z jaką poruszający się jon odtwarza swoją atmosferę jonową w kaŜdym nowym połoŜeniu. Przed jo-nem gęstość ładunku jest mniejsza, a za jonem większa. PoniewaŜ atmosfera jonowa porusza-jącego się jonu nie jest sferycznie symetryczna, więc wytwarza się dodatkowe pole elektrycz-ne, przeciwnie skierowane do pola zewnętrznego - efektywne natęŜenie pola jest mniejsze, co prowadzi do zmniejszenia ruchliwości jonu. Ostatecznie, w wyniku obydwu zjawisk, wzrost przewodnictwa właściwego elektrolitu ze wzrostem stęŜenia staje się wolniejszy od liniowe-go.
W zakresie najwyŜszych stęŜeń (Rys. 6.5), wzrost stęŜenia elektrolitu powoduje wręcz spadek jego przewodnictwa. Tłumaczy się to tworzeniem par, trójek a nawet czwórek jonów w zespołach z cząsteczkami rozpuszczalnika. Ogólnie przebieg zaleŜności przewodnictwa właściwego od stęŜenia dla poszczególnych elektrolitów jest róŜny, co wynika głównie, jak to zostanie wykazane w Rozdz. 6.7, z róŜnych wartości ruchliwości kationów i anionów.
Rys. 6.5. ZaleŜność przewodnictwa właściwego od stęŜenia.

A. Kufelnicki - Ćwiczenia z biofizyki
76
6.6. Przewodnictwo molowe elektrolitów
P r z e w o d n i c t w e m m o l o w y m elektrolitu (Λ) nazywamy przewodnictwo, ja-kie wykazywałaby warstwa elektrolitu o grubości jednostkowej i objętości tak duŜej, by za-wierała jeden mol danego elektrolitu. Oznaczając tę objętość przez V mamy w myśl definicji:
Λ = κ⋅V (6.6.1) lub
Λ = κc (6.6.2)
gdzie c – stęŜenie molowe.
Wymiarem przewodnictwa molowego jest: m2 om-1 mol-1 lub cm2 om-1 mol-1.
Przykład
Ile wynosi objętość zawierająca 1 mol roztworu o stęŜeniu 0,1 mol/dm3? Obliczyć przewodnic-
two molowe 0,1 molowego roztworu KCl, jeŜeli jego przewodnictwo właściwe w temp. 20 °C
wynosi 1,167⋅10-3 om-1cm-1?
molm0,01moldm10molcm10000cm1000mol0,1
1
dmmol0,1
11 333
33======
cV
1123113 molomcm11,67molcm10000cmom101,167 −−−−− =×⋅== VΛ κ
Przewodnictwo molowe wodnych roztworów wszystkich elektrolitów zaleŜy w określo-
nym stopniu od stęŜenia i rośnie wraz z jego rozcieńczeniem. Na podstawie przebiegu zaleŜ-ności od pierwiastka ze stęŜenia molowego, Λ = f ( c ), w zakresie niskich stęŜeń, moŜna po-dzielić ogół elektrolitów na dwie grupy: elektrolity mocne, dla których przewodnictwo molo-we maleje proporcjonalnie do c , oraz elektrolity słabe, dla których ta zaleŜność jest krzywo-liniowa (Rys. 6.6).
P r z e w o d n i c t w e m g r a n i c z n y m elektrolitu (Λ0) nazywamy graniczną war-tość, do jakiej dąŜy przewodnictwo molowe, gdy stęŜenie elektrolitu dąŜy do zera. Wielkość ta jest miarą zdolności przenoszenia ładunku elektrycznego przez 1 mol substancji roz-puszczonej i jest wielkością charakterystyczną dla danego elektrolitu.
Przewodnictwo graniczne elektrolitów mocnych, Λ0, moŜna wyznaczyć metodą ekstrapo-lacji, czyli metodą przedłuŜenia poza zakres pomiarowy wykazanej przez Kohlrauscha zaleŜ-ności liniowej dla roztworów rozcieńczonych: Λ Λ= −0 a c (6.6.3) (gdzie a - stała empiryczna, c – stęŜenie molowe) w kierunku zerowego stęŜenia roztworu, czyli do przecięcia się z osią rzędnych, jak to przedstawiono na Rys. 6.6.
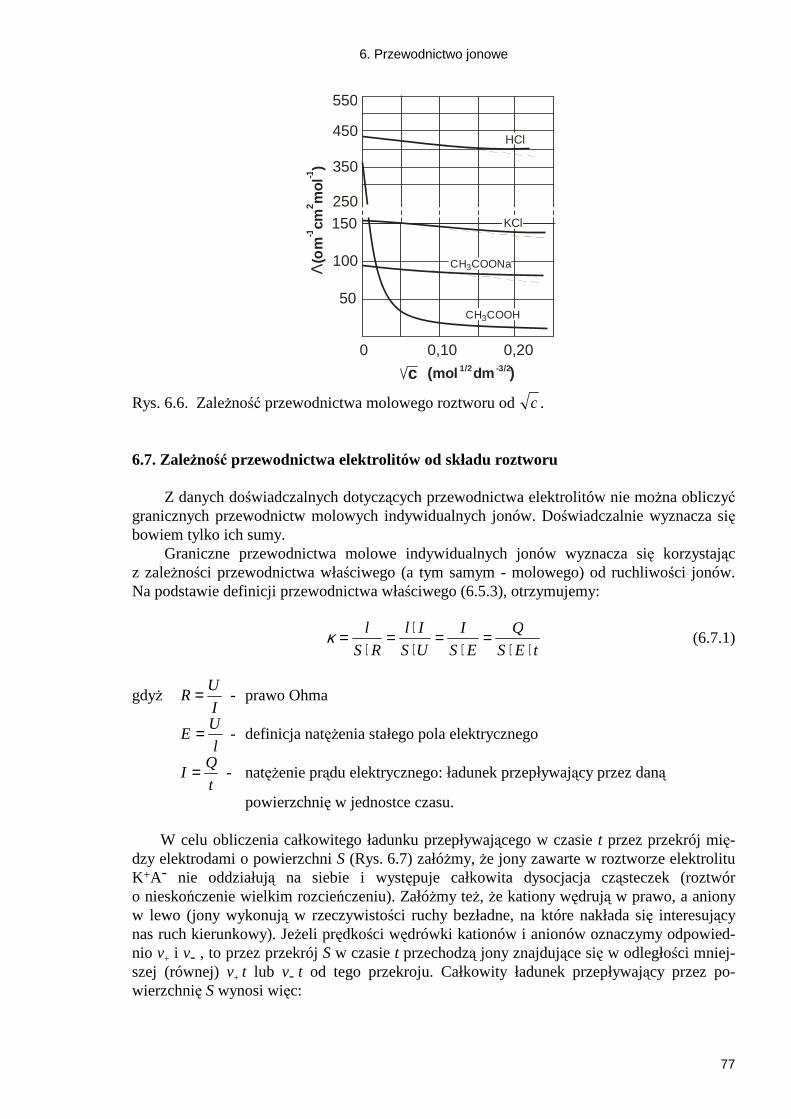
6. Przewodnictwo jonowe
77
0 0,10 0,20
50
100
150250
350
450
550
Λ(o
mcm
mol
)-1
2-1
HCl
KCl
CH COOH3
CH COONa3
c ( )mol dm1/2 -3/2
Rys. 6.6. ZaleŜność przewodnictwa molowego roztworu od c . 6.7. ZaleŜność przewodnictwa elektrolitów od składu roztworu
Z danych doświadczalnych dotyczących przewodnictwa elektrolitów nie moŜna obliczyć granicznych przewodnictw molowych indywidualnych jonów. Doświadczalnie wyznacza się bowiem tylko ich sumy.
Graniczne przewodnictwa molowe indywidualnych jonów wyznacza się korzystając z zaleŜności przewodnictwa właściwego (a tym samym - molowego) od ruchliwości jonów. Na podstawie definicji przewodnictwa właściwego (6.5.3), otrzymujemy:
κ =⋅
= ⋅⋅
=⋅
=⋅ ⋅
l
S R
l I
S U
I
S E
Q
S E t (6.7.1)
gdyŜ RU
I= - prawo Ohma
EU
l= - definicja natęŜenia stałego pola elektrycznego
IQ
t= - natęŜenie prądu elektrycznego: ładunek przepływający przez daną
powierzchnię w jednostce czasu.
W celu obliczenia całkowitego ładunku przepływającego w czasie t przez przekrój mię-dzy elektrodami o powierzchni S (Rys. 6.7) załóŜmy, Ŝe jony zawarte w roztworze elektrolitu K+A- nie oddziałują na siebie i występuje całkowita dysocjacja cząsteczek (roztwór o nieskończenie wielkim rozcieńczeniu). ZałóŜmy teŜ, Ŝe kationy wędrują w prawo, a aniony w lewo (jony wykonują w rzeczywistości ruchy bezładne, na które nakłada się interesujący nas ruch kierunkowy). JeŜeli prędkości wędrówki kationów i anionów oznaczymy odpowied-nio v+ i v- , to przez przekrój S w czasie t przechodzą jony znajdujące się w odległości mniej-szej (równej) v+ t lub v- t od tego przekroju. Całkowity ładunek przepływający przez po-wierzchnię S wynosi więc:

A. Kufelnicki - Ćwiczenia z biofizyki
78
Q ne S t v v= ++ −( ) (6.7.2) gdzie n - liczba ładunków elementarnych w jednostce objętości.
Rys. 6.7. Schemat ułatwiający obliczenie ładunku przepływającego przez dowolną powierz-
chnię S między elektrodami. Po podstawieniu wyraŜenia (6.7.2) do równania (6.7.1) otrzymamy: κ = ++ −ne u u( ) (6.7.3) Przewodnictwo właściwe roztworu elektrolitu o nieskończenie wielkim rozcieńczeniu jest wprost proporcjonalne do liczby ładunków elementarnych w jednostce objętości, ale rów-nieŜ do sumy ruchliwości kationów i anionów tego elektrolitu.
Z dość dobrym przybliŜeniem wniosek ten znajduje potwierdzenie eksperymentalne rów-nieŜ dla roztworów elektrolitów o niskim, ale określonym, skończonym stęŜeniu. Jest to wi-doczne na Rys. 6.5 w zestawieniu z danymi Tab. 6.1. ZaleŜność przewodnictwa właściwego od stęŜenia roztworu jest w przybliŜeniu liniowa dla bardzo niskich stęŜeń, a nachylenie jest większe dla elektrolitów o większej sumie ruchliwości kationów i anionów.
Gdy ładunek przenoszony jest przez 1 mol jonów kaŜdego znaku, n = N (liczba Avoga-dra), otrzymujemy wyraŜenie na przewodnictwo molowe: Λ = ++ −F u u( ) (6.7.4) gdzie F = ne - stała Faraday′a.
Dla podkreślenia, Ŝe wyprowadzone zaleŜności otrzymano dla roztworów nieskończenie rozcieńczonych, ruchliwości jonów oznaczamy: u0+ i u0− , zatem: )( 000 −+ +=Λ uuF (6.7.5)
Z równania (6.7.5) wynika, Ŝe graniczne przewodnictwa molowe indywidualnych jonów moŜna wyznaczyć z zaleŜności: ++ = 00 uFl i −− = 00 uFl (6.7.6)
Wartości l0 moŜna takŜe wyznaczyć mierząc Λ0 całego elektrolitu oraz liczbę przenosze-
nia jednego z jonów. PoniewaŜ:
−+
++ +
=00
00 uu
ut i
−+
−− +
=00
00 uu
ut (6.7.7)

6. Przewodnictwo jonowe
79
więc mnoŜąc licznik i mianownik w tych wyraŜeniach przez stałą F otrzymujemy:
0
00 Λ
= ++
lt i
0
00 Λ= −
−l
t (6.7.8)
stąd: 000 Λ= ++ tl 000 Λ= −− tl (6.7.9)
W Tabeli 6.4 zwraca uwagę wyjątkowo duŜe przewodnictwo graniczne jonów H+ oraz OH-, co wiąŜe się z ich wyjątkowo duŜymi ruchliwościami (patrz Rozdz. 6.3, Tab. 6.2).
Tabela 6.4
Graniczne przewodnictwa molowe niektórych jonów w roztworach wodnych (temp. 298 K)
Kation l 0+ Anion l 0- (om-1cm2mol-1) (om-1cm2mol-1)
H+ 349,82 OH− 198,6
Na+ 50,10 Cl− 76,35
K+ 73,50 NO3− 71,46
Ca2+ 119,0 ClO4− 67,4
Mg2+ 106,2 SO42− 160,0
Ba2+ 127,2 CO32− 138,6
Z równań (6.7.4) i (6.7.5) wynika takŜe teoretyczne uzasadnienie postulatu (wzoru) Arr-heniusa dla elektrolitów spełniających prawo Ohma, bowiem w przypadku słabego elektrolitu niecałkowicie zdysocjowanego, równanie (6.7.4) dla roztworu 1-molowego przyjmuje postać: Λ = ++ −α F u u( ) (6.7.10) gdzie α - stopień dysocjacji.
Zakładając, Ŝe dla rozcieńczonego roztworu przy niskim stopniu dysocjacji u+ ≈ u0+ oraz u- ≈ u0− , po podzieleniu stronami przez równanie (6.7.5) otrzymujemy wzór Arrheniusa:
0Λ
Λ=α (6.7.11)
Literatura uzupełniająca: F. Jaroszyk, red., „Biofizyka”, Wydawnictwo Lekarskie PZWL, Warszawa 2002, str. 198. T. W. Hermann, red., „Farmacja fizyczna”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 262. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 149.

A. Kufelnicki - Ćwiczenia z biofizyki
80
Ćwiczenie 7
Pomiar oporu elektrycznego za pomocą mostka prądu przemiennego 1. Włączyć układ pomiarowy do sieci.
2. Dokonać pomiaru oporu elektrycznego wody destylowanej: ustawić przełącznik (x) na 100 i przełącznik (:) na 1. Podstawa mostka wynosi
100 : 1 = 100, elektrodę pomiarową kilkakrotnie przepłukać wodą destylowaną, napełnić naczyńko pomiarowe wodą destylowaną i zanurzyć w nim elektrodę pomia-
rową, uruchomić mostek przyciskiem (B), a następnie wcisnąć przycisk (G 0,1). Przy pomocy
pięciu dekad porównawczych znaleźć punkt kompensacji mostka przełączając kolejno dekady, od najwyŜszej do najniŜszej, aŜ mikroamperomierz wskaŜe minimum natęŜe-nia prądu elektrycznego. Wyłączyć (G 0,1),
wcisnąć przycisk (G) i powtórnie dokonać kompensacji mostka, wartość zmierzonego oporu elektrycznego wynosi:
R = (wartość odczytana na 5-ciu dekadach) · (podstawa mostka)
wynik umieścić w tabeli pomiarów i wyników.
3. Zmierzyć opór elektryczny 0,01 M roztworu KCl, pamiętając o dokładnym przepłukaniu elektrody przed pomiarem, najpierw wodą destylowaną, a następnie badanym roztworem. Podstawę mostka dobrać tak, aby uzyskać jak największą dokładność pomiaru, tzn. war-tość oporu moŜna zacząć odczytywać od najwyŜszej dekady (oznaczonej 1000).
4. Obliczyć stałą konduktometryczną naczyńka korzystając z wartości przewodnictwa wła-ściwego 0,01 M roztworu KCl w danej temperaturze (Tabela 6.3).
5. Wynik zamieścić w tabeli.
6. Zmierzyć opór elektryczny pozostałych roztworów, podobnie jak w punkcie 3.
7. Dla wody destylowanej i wodociągowej obliczyć tylko przewodnictwo (λ) i przewodnic-two właściwe (κ). Obliczyć przewodnictwo (λ), przewodnictwo właściwe (κ) i przewod-nictwo molowe (Λ) badanych roztworów NaCl i CH3COOH. Dla kwasu odjąć λH2O wody destylowanej.
8. Wyniki obliczeń zamieścić w tabeli. Tabela wyników pomiarów i wielkości obliczonych:
Roztwór c
(mol/dm3)
R
(Ω)
λ
(S)
κ
(S·cm-1)
Λ
(S·cm2 mol-1)
Temp. pomiaru = κKCl (0,01 M) = k =

6. Przewodnictwo jonowe
81

81
7. OGNIWA GALWANICZNE
Na powierzchni metalu, zanurzonego w roztworze elektrolitu zawierającym jony tego me-talu, zachodzą dwa przeciwstawne procesy:
a) odrywanie się kationów metalu z siatki krystalicznej i przechodzenie ich do roztworu, b) osadzanie się kationów na powierzchni metalu.
JeŜeli na przykład początkowa szybkość pierwszego procesu jest większa - metal ładuje się ujemnie, natomiast roztwór dodatnio (Rys. 7.1). Z kolei, gdy szybciej zachodzi wiązanie kationów z roztworu na powierzchni metalu - metal ładuje się dodatnio, a roztwór ujemnie. Na granicy faz metal-roztwór powstaje p o d w ó j n a w a r st w a e l e k t r y c z n a . W obydwu przypadkach wytwarza się więc róŜnica potencjałów pomiędzy metalem a roztworem, która na zasadzie oddziaływań elektrostatycznych hamuje proces szybszy i przyspiesza wol-niejszy, aŜ do wyrównania się szybkości. Ustala się stan równowagi dynamicznej między jo-nami przechodzącymi od metalu, przez warstwę podwójną do roztworu i od roztworu do me-talu.
Rys.7.1. Powstawanie róŜnicy potencjału na granicy metal – roztwór.
Metal wraz z otaczającym go roztworem elektrolitu nazywamy e l e k t r o dą lub p ó ł o g n i w e m . Utworzoną, w wyniku opisanych zjawisk na granicy faz, róŜnicę poten-cjałów pomiędzy metalem a roztworem nazywamy p o t e n c j a ł e m N e r n s ta lub p o t e n c j a ł e m e l e k t r o d y (π).
Układ dwóch elektrod (półogniw), których roztwory elektrodowe połączone są w odpo-wiedni sposób nazywamy o g n i w e m g a l w a n i c z n y m . Połączenie to moŜe być zre-alizowane poprzez wspólny elektrolit, przegrodę porowatą lub tzw. klucz elektrolityczny. Je-Ŝeli z kolei obwód zewnętrzny ogniwa zamkniemy przewodnikiem, to stwierdzimy w nim przepływ prądu elektrycznego. Elektrony w tym obwodzie zewnętrznym płyną od elektrody naładowanej ujemnie do elektrody bardziej dodatniej. W roztworze elektrolitu, czyli obwo-dzie wewnętrznym, nośnikiem ładunku elektrycznego są jony. Na granicy zetknięcia metalu z roztworem zachodzi przekazywanie ładunku poprzez reakcje chemiczne utleniania i redukcji. Elektrodę, na której zachodzi utlenianie nazywamy a n o dą ; k a t o dą jest elektroda, na której zachodzi redukcja. Określenia „anoda” i „katoda” dotyczą jednak konkretnego ogniwa i konkretnego procesu rozładowania bądź ładowania. Łatwo sobie wyobrazić, Ŝe ta sama elek-troda, która w jednym ogniwie jest np. anodą, moŜe być w innym ogniwie (w stosunku do in-nej elektrody) katodą.
W ogniwie galwanicznym zachodzi bezpośrednia zamiana energii chemicznej na energię elektryczną. ZaleŜnie od rodzaju procesów prowadzących do powstania energii elektrycznej, dzielimy ogniwa galwaniczne na c h e m i c z n e i s tę Ŝ e n i o w e . W pierwszych ener-

A. Kufelnicki - Ćwiczenia z biofizyki
82
gia elektryczna powstaje w wyniku sumarycznie efektywnej reakcji utleniania-redukcji, w drugich zaś na skutek wyrównywania się stęŜeń roztworów elektrodowych – co prawda reak-cje utleniania i redukcji zachodzą przy poszczególnych elektrodach, ale sumarycznie Ŝaden z tych procesów nie zachodzi. 7.1. Schemat zapisu ogniw
Sposób zapisywania ogniw galwanicznych omówimy na przykładzie o g n i w a D a -n i e l l a zbudowanego z elektrody cynkowej i elektrody miedziowej (Rys. 7.2). Elektrodę cynkową stanowi cynk metaliczny w roztworze siarczanu cynku, elektrodę miedziową - meta-liczna miedź w roztworze siarczanu miedzi. Roztwory stykają się poprzez klucz elektrolitycz-ny lub przez porowatą przegrodę, która zapobiega ich mieszaniu się, pozwala jednak na prze-noszenie ładunku. W obwodzie zewnętrznym moŜemy włączyć odbiornik energii, na przykład Ŝarówkę, jak na Rys. 7.2.
Rys.7.2. Ogniwo Daniella.
Klucz elektrolityczny stanowi łącznik napełniony stęŜonym roztworem soli o zbliŜonej ruchliwości kationu i anionu, jak np. NH4NO3, KCl, KNO3 . Przy zamkniętym obwodzie ze-wnętrznym na elektrodzie cynkowej samorzutnie przebiega proces utleniania cynku, a jony cynku przechodzą do roztworu: Zn Zn2+ + 2e anoda (−) (7.1.1) Na elektrodzie miedziowej następuje proces redukcji jonów miedziowych: Cu2+ + 2e Cu katoda (+) (7.1.2) Samorzutna reakcja sumaryczna przebiega zatem w kierunku: Zn + Cu2+ Zn2+ + Cu (7.1.3) W obwodzie zewnętrznym elektrony płyną od ujemnie naładowanej blaszki cynkowej (anody) do elektrody miedziowej (katody), gdzie z kolei zobojętniają dopływające z roztworu kationy miedzi. Na katodzie osadza się miedź metaliczna.
W 1953 r. na konferencji IUPAC (Międzynarodowa Unia Chemii Czystej i Stosowanej) w Sztokholmie przyjęto następujący schemat zapisu ogniw: anodę zapisuje się po lewej stro-nie schematu, katodę po prawej. Tak więc ogniwo Daniella zapiszemy schematem:

7. Ogniwa galwaniczne
83
Zn ZnSO4 (c1) CuSO4 (c2) Cu (7.1.4)
gdzie | oznacza granicę faz ciało stałe - roztwór || oznacza połączenie roztworów elektrodowych poprzez klucz elektrolityczny (z praktycznym wyeliminowaniem tzw. potencjału dyfuzyjnego na styku roztworów) c1 i c2 - stęŜenie roztworów odpowiednich elektrolitów .
Połączenie roztworów elektrodowych poprzez przegrodę porowatą oznaczamy pionową linią kreskowaną ( ).
PowyŜszy zapis ogniwa jest zgodny z kierunkiem reakcji przebiegającej w ogniwie samo-rzutnie, a więc kierunkiem reakcji, dla której zmiana entalpii swobodnej jest mniejsza od zera (∆G < 0). Według Konwencji Sztokholmskiej s i ł a e l e k t r om o t o r y c z n a ogniwa (w skrócie SEM) jest równa róŜnicy, przy otwartym ogniwie, między potencjałem elektry-cznym przewodnika metalicznego przyłączonego do elektrody prawej a potencjałem przewo-dnika z tego samego materiału dołączonego do elektrody lewej:
SEM = πp − πl (7.1.5) Dla ogniwa Daniella zapisanego schematem (7.1.4): SEM > 0.
Określenie jednej elektrody jako prawej, a drugiej jako lewej ma sens tylko w odniesieniu do ogniwa, które zostało w dany sposób zapisane. Zapisanie ogniwa w przeciwnym kierunku zmienia znak przypisanej mu SEM na przeciwny. 7.2. Pomiar siły elektromotorycznej ogniwa
Jak wynika z definicji SEM ogniwa podanej w Konwencji Sztokholmskiej, pomiaru siły elektromotorycznej musimy dokonywać przy otwartym ogniwie (bez przepływu prądu przez ogniwo). Warunku tego nie zapewnia pomiar zwykłym woltomierzem. Wówczas mierzone napięcie jest mniejsze od siły elektromotorycznej o spadek napięcia na oporze wewnętrznym ogniwa, Rw . Wynika to bowiem z II prawa Kirchhoffa dla obwodu zamkniętego ze źródłem SEM - Rys. 7.3. Mówi ono, Ŝe w dowolnym zamkniętym obwodzie suma wszystkich spad-ków napięć i sił elektromotorycznych w tym obwodzie jest równa zeru - obowiązuje przy tym umowa, Ŝe przy „obchodzeniu” obwodu w określonym kierunku spadki napięcia na oporach wynoszą -IR, gdy kierunek ten jest zgodny z kierunkiem przepływu prądu, a odwrotnie: +IR. Znaki SEM są dodatnie, gdy źródło SEM podwyŜsza potencjał w kierunku ruchu - na Rys. 7.3, gdy „przechodzimy” przez źródło od „−„ do”+”.
Rys.7.3 Obwód zamknięty ze źródłem
SEM.

A. Kufelnicki - Ćwiczenia z biofizyki
84
W naszym przypadku napięcie mierzymy woltomierzem o oporze Rz, zatem
E − IRw − IRz = 0 (7.2.1) i
E = IRw + IRz (7.2.2) PoniewaŜ U = IRz, więc: U = E − IRw (7.2.3)
Z równania (7.2.3) wynika, Ŝe wartość U dąŜy do E, gdy natęŜenie prądu płynącego przez obwód dąŜy do zera. Praktycznie moŜna to osiągnąć przez zastosowanie woltomierza o znacznym oporze Rz , bowiem z równania (7.2.1):
zw RR
I+
= E (7.2.4)
więc dla Rz >>Rw
EE ≈+
=zw
z
RR
RU (7.2.5)
PowyŜsze przybliŜenie moŜe jednak nie być wystarczające przy dokładnych pomiarach
SEM, stąd najczęściej pomiaru siły elektromotorycznej dokonuje się m e t o dą k o m -p e n s a c y j ną zaproponowaną przez Poggendorffa z uŜyciem odpowiednich kompensato-rów oraz przy pomocy m i l i w o l t o m i e r z y t r a n z y s t o r o w y ch .
Rys. 7.4. Schemat układu kompensacyjnego do pomiaru siły elektromotorycznej ogniwa me-
todą Poggendorffa.
W metodzie kompensacyjnej brak przepływu prądu przez ogniwo w momencie pomiaru uzyskuje się drogą dokładnego zrównowaŜenia (skompensowania) SEM badanego ogniwa przez róŜnicę potencjałów przyłoŜoną z zewnątrz. Rys. 7.4 wyjaśnia zasadę metody kompen-sacyjnej Poggendorffa. Źródło prądu stałego (najczęściej akumulator lub zasilacz) o SEM większej od SEM badanych ogniw połączone jest z drutem oporowym AB o jednakowym przekroju na całej długości. Przed właściwym pomiarem wykonuje się kompensację ogniwa o stałej i znanej sile elektromotorycznej (tzw. normalnego ogniwa Westona). Elektroda dodatnia ogniwa połączona jest z dodatnim biegunem akumulatora. Przesuwając ruchomy styk S, tak dobieramy jego połoŜenie, aby galwanometr G nie wykazywał przepływu prądu przez obwód

7. Ogniwa galwaniczne
85
boczny (prąd I2 = 0). Obwód zamykamy na krótką chwilę za pomocą przerywacza K, co za-bezpiecza ogniwo przed polaryzacją elektrod przy długotrwałym przepływie prądu. Po kom-pensacji, przez obwód główny płynie prąd I = I1, a spadek napięcia na odcinku AS drutu opo-rowego jest równy sile elektromotorycznej ogniwa normalnego (jest to teraz ogniwo otwarte). II prawo Kirchhoffa dla obwodu AEnGSA zapiszemy w postaci:
EEEE n − IRAS = 0 (7.2.6) Stąd
EEEE n = IRAS (7.2.7) Wykorzystując zaleŜność oporu przewodnika od jego długości i przekroju poprzecznego,
otrzymamy:
S
lIn
ASρ=E (7.2.8)
gdzie: ρ - opór właściwy drutu, lAS - długość odcinka AS, S - przekrój poprzeczny drutu. Następnie w miejsce ogniwa normalnego włączamy ogniwo badane i analogicznie znaj-
dujemy punkt kompensacji S′. Równanie (7.2.8) zapiszemy wówczas w postaci:
S
lIx
SA ′= ρE (7.2.9)
gdzie prąd I jest, podobnie jak poprzednio, określony przez SEM akumulatora (zasilacza) i opór obwodu głównego. Dzieląc stronami równania (7.2.8) i (7.2.9) otrzymamy:
SA
AS
′=
ll
x
n
E
E (7.2.10)
i
AS
SAll
nx′= EE (7.2.11)
Stosowane w praktyce przyrządy do pomiaru SEM metodą kompensacyjną noszą nazwę
k o m p e n s a t o r ó w p rą d u s t a ł e g o lub k o m p e n s a t o r ó w t e c h n i c z -n y c h . Ich budowa (oparta na tej samej zasadzie) umoŜliwia uzyskiwanie dokładniejszych wyników. W przyrządach tych (Rys. 7.5) kompensacja SEM ogniwa normalnego odbywa się poprzez zmianę napięcia zasilającego za pomocą dzielnika napięcia (potencjometru oporowe-go o ciągłych zmianach oporu), natomiast opór, na którym spadek potencjału kompensuje SEM ogniwa normalnego jest stały. Z kolei kompensacja SEM ogniwa badanego odbywa się za pomocą potencjometrów skokowych (tzw. dekad), wyskalowanych bezpośrednio w wol-tach (miliwoltach). Szczegółowy opis pomiarów jest następujący:
Kompensacja SEM ogniwa normalnego, pozycja En (przycisk klawiszowy En wciśnięty).
Przełączniki P1 i P2 łączą ogniwo Westona z obwodem głównym AR2R3...R4R1. Poprzez dobór oporu R1 (regulacja ciągła) doprowadzamy do kompensacji SEM tego ogniwa przez spadek napięcia na oporze R4 (galwanometr G nie wykazuje przepływu prądu). Jedyny prąd, jaki pły-nie w obwodzie bocznym z ogniwem normalnym, ale tylko przez opór R4, to prąd I, określony przez napięcie zasilacza A i opór obwodu głównego, a więc z II prawa Kirchhoffa:

A. Kufelnicki - Ćwiczenia z biofizyki
86
04 =− IRnE
a stąd:
4IRn =E
Rys. 7.5. Uproszczony schemat kompensatora prądu stałego (niskooporowego) – zaznaczone są tylko dwie dekady oporowe, R2 i R3.
Kompensacja SEM ogniwa badanego, pozycja X1 lub X2 (przycisk klawiszowy X1 lub X2 wci-śnięty).
Przełączniki P1 i P2 łączą teraz ogniwo badane z takimi częściami oporów R2, R3..., których suma wynosi R. Przy odpowiednim doborze R przez galwanometr G nie płynie prąd, zatem:
0=− IRxE
IRx =E
PoniewaŜ natęŜenie prądu I w obwodzie głównym jest równe natęŜeniu prądu ustalonemu w pierwszej części pomiaru, więc:
4R
R
n
x =E
E
a stąd:
4R
Rnx EE =
Opory dekadowe R2, R3,... składające się na wartość R są wyskalowane w mV (×100 mV, ×10 mV, ×1 mV,...). Odczytujemy więc wartość mierzonej SEM ogniwa badanego.
Metoda kompensacyjna nie moŜe być stosowana do pomiarów SEM ogniw o oporze we-wnętrznym większym niŜ ∼105 Ω ze względu na zbyt niskie prądy niepełnej kompensacji (ustalenie punktu kompensacji byłoby praktycznie niemoŜliwe). Tymczasem wiele ogniw, jak
I I

7. Ogniwa galwaniczne
87
np. ogniwa zawierające elektrodę szklaną lub rozcieńczone roztwory elektrolitów w rozpusz-czalnikach organicznych, ma opory znacznie większe. Przy pomiarze SEM takich ogniw sto-sowane są potencjometry (kompensatory), w których następuje wzmocnienie prądów niepeł-nej kompensacji oraz miliwoltomierze oparte na układach tranzystorowych i scalonych. 7.3. Termodynamika ogniw galwanicznych
JeŜeli proces przebiegający w ogniwie podczas przepływu nieskończenie małego prądu jest odwracalny w sensie termodynamicznym, to takie ogniwo nazywamy ogniwem o d -w r a c a l n y m . Oznacza to, Ŝe w wszystkie procesy spowodowane przepływem nieskończe-nie małego ładunku w jednym kierunku powinny być dokładnie „odwracane” przez przepływ takiego samego ładunku w kierunku przeciwnym. Przykłady ogniw odwracalnych:
a) niektóre ogniwa bez połączeń ciekłych (mające wspólny roztwór dla obu półogniw), np.
O g n i w o H a r n e d a : PtH2 (p)HCl(c)AgClAg
Rys. 7.6. Ogniwo Harneda.
reakcja samorzutna: 12
H2 + AgCl H+ + Cl- + Ag
reakcja wymuszona: H+ + Cl- + Ag12
H2 + AgCl
O g n i w o W e s t o n a :
12,5% Cd(Hg)nasyc. CdSO4⋅8/3 H2OHg2SO4, Hg

A. Kufelnicki - Ćwiczenia z biofizyki
88
Rys. 7.7. Nasycone ogniwo Westona.
reakcja samorzutna: Cd + Hg2SO4 + 8/3 H2O CdSO4⋅8/3 H2O + 2 Hg
reakcja wymuszona: CdSO4⋅8/3 H2O + 2 Hg Cd + Hg2SO4 + 8/3 H2O
Normalne (nasycone) ogniwo Westona stosowane jest jako ogniwo wzorcowe ze względu na stałą i odtwarzalną siłę elektromotoryczną oraz bardzo mały współczynnik temperaturowy SEM (Tab. 7.1) .
Tab. 7.1
ZaleŜność siły elektromotorycznej ogniwa Westona od temperatury
t (°C) SEM (mV) t (°C) SEM (mV)
15 1018,5 22 1018,2
16 1018,4 23 1018,2
17 1018,4 24 1018,1
18 1018,4 25 1018,1
19 1018,3 26 1018,0
20 1018,3 27 1018,0
21 1018,2
Wymienione cechy ogniwa Westona są zachowane pod warunkiem, Ŝe przez ogniwo nie płyną prądy większe niŜ 10-6 A. Dlatego teŜ ogniwo Westona moŜe słuŜyć jako wzo-rzec SEM tylko w bezprądowych metodach pomiaru SEM, a nie powinno być źródłem prądu. Przepływ większych prądów przez to ogniwo powoduje znaczne zmiany, w wyniku których układy elektrodowe bardzo powoli powracają do stanu równowagi;
b) niektóre ogniwa z połączeniem ciekłym, jeŜeli został wyeliminowany potencjał dyfuzyjny,
np. o g n i w o D a n i e l l a z kluczem elektrolitycznym (w przybliŜeniu odwracalne);

7. Ogniwa galwaniczne
89
c) ogniwa regenerowalne (a k u m u l a t o r y )
Akumulator magazynuje w procesie ładowania energię elektryczną i oddaje ją ponownie, bez większych strat, przy rozładowaniu. KaŜde ogniwo odwracalne (np. ogniwo Daniella) moŜe w zasadzie spełniać rolę akumulatora. W praktyce wymagane są jednak: − odpowiednio duŜa pojemność − moŜliwość pobierania duŜego prądu − odpowiednia wydajność prądowa i energetyczna.
Np. w akumulatorze ołowiowym (kwasowym)
PbPbSO4, H2SO4PbO2,Pb
podczas wyładowania zachodzą reakcje:
biegun ujemny Pb + SO4 2- PbSO4 + 2 e-
biegun dodatni PbO2 + 4 H+ + SO4 2- + 2 e- PbSO4 + 2 H2O
Sumarycznie:
Pb + PbO2 + 2 H2SO4 wyładowanie
ładowanie 2 PbSO4 + 2 H2O
Dla reakcji zachodzącej w ogniwie odwracalnym spełnione jest równanie na zmianę en-
talpii swobodnej w warunkach izotermiczno-izobarycznych: ∆G = WuŜ = Wodwr + pdV (7.3.1) Jak moŜna wykazać, jest ona równa maksymalnej pracy uŜytecznej (maksymalnej pracy w procesie odwracalnym pomniejszonej o pracę zmiany objętości). JeŜeli rozwaŜanym układem jest ogniwo, to podczas przepływu prądu o nieskończenie małym natęŜeniu zostaje wymie-niona z otoczeniem praca elektryczna (Wel) i objętościowa (-pdV): Wodwr = Wel + Wobj = Wel − pdV (7.3.2) Wstawiając Wodwr = Wel − pdV z równania (7.3.2) do równania (7.3.1) otrzymujemy: ∆G = WuŜ = Wel (7.3.3)
Praca prądu elektrycznego jest więc równa zmianie entalpii swobodnej w reakcji odwra-calnej zachodzącej w ogniwie. Tym samym praca ta jest równa maksymalnej pracy uŜytecz-nej, jaką moŜe wykonać ogniwo. Z drugiej strony wiadomo, Ŝe praca przeniesienia ładunku Q między elektrodami, dla których stała róŜnica potencjałów wynosi EEEE , jest równa:
Wel = -EEEE Q (7.3.4) Znak (-) wynika z umowy na temat znakowania pracy wykonywanej przez układ. Praca wyko-nywana nad układem jest dodatnia, wykonywana przez układ – ujemna. Połączenie równań (7.3.3) i (7.3.4) prowadzi do wzoru: ∆G = -EEEE Q (7.3.5)

A. Kufelnicki - Ćwiczenia z biofizyki
90
JeŜeli ładunek, który przepłynął przez obwód ogniwa wynosi zF, gdzie z jest liczbą elektro-nów wymienianych w danej reakcji biegnącej w ogniwie, zaś F - stałą Faraday′a, to:
∆G = - zFEEEE (7.3.6)
Wzór (7.3.6) przedstawia waŜną zaleŜność między zmianą entalpii swobodnej w reakcji zachodzącej w ogniwie odwracalnym a siłą elektromotoryczną tego ogniwa i jest punktem wyjścia do wyprowadzenia innych zaleŜności termodynamicznych. Wynika z niego np. zwią-zek pomiędzy siłą elektromotoryczną ogniwa i aktywnościami reagujących substancji. Korzy-stając z podstaw termodynamiki, zmianę entalpii swobodnej dla reakcji odwracalnej:
a A + b B c C + d D (7.3.7) przebiegającej w ogniwie galwanicznym zapiszemy w postaci:
ba
dc
aa
aaRTGG
BA
DCln+∆=∆ o (7.3.8)
gdzie: ∆G° - standardowa zmiana entalpii swobodnej aA, aB ... - aktywności reagujących substancji. Po zastosowaniu wyraŜenia (7.3.6) otrzymamy z kolei:
ba
dc
aa
aaRTFzFz
BA
DCln+−=− oEE (7.3.9)
czyli
ba
dc
aa
aa
zF
RT
BA
DCln−= oEE (7.3.10)
Równanie (7.3.10) nosi nazwę r ó w n a n i a N e r n s t a . EEEE ° oznacza standardową siłę elektromotoryczną. Jest ona równa SEM ogniwa, w którym aktywności wszystkich reagentów są równe jedności. Wielkość ta jest charakterystyczna dla danego ogniwa i określona w danej temperaturze. Standardowa siła elektromotoryczna związana jest ze stałą równowagi reakcji (7.3.7). PoniewaŜ ∆G° = -zFEEEE ° , ale teŜ ∆G° = - RT ln Ka , więc:
aKzF
RTln=oE (7.3.11)
gdzie ba
dc
aaa
aaK
BA
DC= , kreski nad aktywnościami oznaczają ich wartości w stanie równo-
wagi. Gdy reakcja zachodząca w ogniwie galwanicznym osiąga stan równowagi, ogniwo prze-staje wytwarzać siłę elektromotoryczną (EEEE = 0, gdyŜ ∆G = 0). Wówczas strzałki reakcji od-wracalnej w równaniu (7.3.7) naleŜy zastąpić strzałkami równowagowymi z jednym grotem ( ).

7. Ogniwa galwaniczne
91
Przykład Ogniwo Daniella moŜe w określonych warunkach pracować w sposób odwracalny. Prak-
tycznie oznacza to włączenie do obwodu zewnętrznego na krótki okres czasu. W tym przypadku ogólną reakcję (7.3.7) zapiszemy w postaci:
Zn + Cu2+ Zn2+ + Cu
W drodze umowy przyjęto, Ŝe aktywność czystych ciał stałych (a takŜe czystych cieczy) jest
równa jedności: aZn = aCu = 1. Stąd równanie Nernsta dla ogniwa Daniella w temperaturze T ma postać:
+
+−=
2
2
Cu
Znlna
a
zF
RToEE
Stałą równowagi reakcji moŜemy obliczyć ze wzoru (7.3.11) przekształconego do postaci:
RT
FzK
2,303log
oE
=
Podstawiając EEEE ° = 1,100 V (standardowa SEM ogniwa Daniella w temperaturze 298 K, odczytana
z tablic) otrzymamy:
3737,2K298KmolJ8,314 2,303
V1,100C 965002log
11≈=
⋅⋅⋅⋅= −−K
zatem:
37
K298Cu
Zn 102+
2+
=
=
=Ta
aK
Tak wysoka stała równowagi oznacza, Ŝe reakcja zachodząca w ogniwie Daniella przebiegłaby praktycznie całkowicie w prawo, gdyby było ono w dłuŜszym okresie czasu zamknięte obwodem zewnętrznym.
Wzór (7.3.6) umoŜliwia takŜe wyznaczenie zmiany entalpii i entropii reakcji biegnącej
w ogniwie drogą pomiaru SEM i współczynnika temperaturowego SEM bez konieczności wykonywania skomplikowanych pomiarów kalorymetrycznych, a mianowicie korzystając z tego wzoru i równania Gibbsa - Helmholtza (patrz: piśmiennictwo na końcu rozdziału) otrzy-mamy:
−−=∆ T
TzFH
p∂∂ E
E (7.3.12)

A. Kufelnicki - Ćwiczenia z biofizyki
92
oraz pT
zFS
=∆
∂∂ E
(7.3.13)
Współczynnik temperaturowy siły elektromotorycznej ( )pT∂∂E wyznaczamy przez wykona-
nie pomiarów SEM w kilku temperaturach. W niezbyt szerokim zakresie temperatur EEEE jest li-niową funkcją temperatury, określoną równaniem linii prostej, które wynika z przekształcenia wzoru (7.3.12):
TTzF
H
p
⋅
+∆−=
∂∂ E
E (7.3.14)
Wartość ∆H obliczona z równania (7.3.12) nie jest równa ciepłu wymienianemu między
otoczeniem i ogniwem w czasie jego pracy, poniewaŜ jak juŜ wspomniano, ogniwo poza pra-cą objętościową wymienia z otoczeniem równieŜ pracę elektryczną. Przy odwracalnej pracy ogniwa w stałej temperaturze ciepło wymieniane jest równe STQodwr ∆= , natomiast ∆H
z równania (7.3.12) byłoby równe ciepłu reakcji, gdyby została ona przeprowadzona poza ogniwem (w ten sposób by jedyną pracą wymienioną była praca objętościowa). 7.4. Skoki potencjałów w ogniwach galwanicznych
Na siłę elektromotoryczną ogniwa składa się szereg skoków (róŜnic) potencjałów, z których najistotniejsze są: opisane juŜ poprzednio skoki potencjałów na granicach faz me-tal/roztwór elektrodowy, zwane potencjałami elektrod (półogniw) oraz tzw. p o t e n c j a ł d y f u z y j n y występujący na granicy zetknięcia dwóch roztworów elektrolitów. JeŜeli jed-na z elektrod ma wyŜszy potencjał, πK, a więc jest katodą, a druga niŜszy, πA (jest anodą), to: dπππ +−= AKE (7.4.1)
Potencjałem dyfuzyjnym nazywamy skok potencjału wytwarzający się na granicy ze-
tknięcia dwóch roztworów elektrolitów o róŜnym składzie (na tzw. połączeniach ciekłych) w wyniku zachodzącego procesu dyfuzji. Dla uproszczenia weźmy pod uwagę styk (ciekłe połą-czenie) dwóch roztworów tego samego elektrolitu róŜniących się stęŜeniem, np. dwóch roz-tworów kwasu solnego:
HCl(c1) HCl(c2)
JeŜeli c1 > c2, co pociąga za sobą a1 > a2, to jony z roztworu 1 będą migrowały do roz-tworu 2. Wobec róŜnej ruchliwości jonów elektrolitu (jony wodorowe mają znacznie większą ruchliwość niŜ jony chlorkowe) kationy wodorowe będą wyprzedzały aniony chlorkowe. Wskutek tego od strony roztworu o mniejszym stęŜeniu powstanie warstwa o przewadze ła-dunku dodatniego; za nią w kierunku roztworu bardziej stęŜonego uformuje się druga warstwa z przewagą jonów ujemnych. Wytworzy się pole elektryczne pomiędzy warstwami - jego siły będą hamowały jony poruszające się szybciej (kationy), a przyspieszały jony powolniejsze (aniony). Po krótkim czasie (praktycznie natychmiast po zetknięciu się roztworów) osiągnięty zostanie stan stacjonarny, charakteryzujący się określonym skokiem potencjału na styku obu faz ciekłych, zwanym potencjałem dyfuzyjnym.

7. Ogniwa galwaniczne
93
W przypadku wyŜej opisanym, czyli gdy po obu stronach ciekłego połączenia występują roztwory tego samego elektrolitu dysocjującego na jony jednowartościowe, przy załoŜeniu niewielkiej róŜnicy stęŜeń obu roztworów, potencjał dyfuzyjny moŜna obliczyć za pomocą w z o r u H e n d e r s o n a , który upraszcza się do postaci:
πd t tRT
F
a
a= −+ −( ) ln 1
2
(7.4.2)
lub po uwzględnieniu związku między liczbami przenoszenia i ruchliwościami:
πd
u u
u u
RT
F
a
a= −
++ −
+ −
ln 1
2
(7.4.3)
gdzie: t+ i t- - liczby przenoszenia kationu i anionu u+ i u- - ruchliwość kationu i anionu a1 i a2 - aktywności elektrolitu po obu stronach ciekłego połączenia.
Jak wynika z równań (7.4.2) i (7.4.3), potencjał dyfuzyjny jest tym większy, im większy jest stosunek aktywności obydwu roztworów oraz im większa jest róŜnica liczb przenoszenia jonów (ruchliwości jonów) danego elektrolitu. W szczególnym przypadku, gdy t+ = t- = 0,5 potencjał dyfuzyjny równa się zeru. Warunek ten spełnia w przybliŜeniu np. KCl w roztwo-rach wodnych (tK+ = 0,49; tCl- = 0,51).
Potencjały dyfuzyjne zwykle osiągają wartości rzędu kilku lub kilkunastu miliwoltów, a w przypadku zetknięcia roztworów półogniw, z których jedno zawiera jony wodorowe lub wodorotlenowe, potencjały dyfuzyjne mogą osiągać znacznie większe wartości.
W celu istotnego zmniejszenia lub wyeliminowania potencjału dyfuzyjnego stosowany jest k l u c z e l e k t r o l i t y c z n y , zwany teŜ mostkiem solnym. Jest to zwykle U-rurka wypełniona nasyconym roztworem KCl, KNO3 lub NH4NO3, za pośrednictwem której łączy się roztwory półogniw badanego ogniwa. Działanie klucza elektrolitycznego polega na tym, Ŝe na granicy dwóch roztworów, z których jeden ma znaczne stęŜenie (np. roztwór KCl w kluczu), a drugi jest roztworem rozcieńczonym, potencjał dyfuzyjny jest prawie wyłącznie wynikiem dyfuzji jonów elektrolitu stęŜonego. Jeśli zatem jony elektrolitu wypełniającego klucz mają bliskie siebie ruchliwości, to potencjał dyfuzyjny dąŜy do zera. NiezaleŜnie od te-go, potencjały dyfuzyjne powstające na obu końcach klucza są równe co do wartości bez-względnej, a róŜne co do znaku, jeśli ł ączone za pomocą klucza roztwory są rozcieńczone. W konsekwencji obydwa potencjały dyfuzyjne kompensują się. 7.5. Ogniwa stęŜeniowe
O g n i w a s tę Ŝ e n i o w e zbudowane są z dwóch półogniw róŜniących się tylko stę-Ŝeniami elektrolitu, np.
AgAgNO3 (c1)NH4NO3 nasyc.AgNO3 (c2)Ag
c2 > c1
Źródłem SEM jest w tych ogniwach praca przeniesienia ładunku z roztworu o wyŜszym
stęŜeniu (wyŜszym potencjale termodynamicznym) do roztworu o niŜszym stęŜeniu (niŜszym potencjale termodynamicznym). W zaleŜności od sposobu połączenia roztworów elektrodo-

A. Kufelnicki - Ćwiczenia z biofizyki
94
wych rozróŜniamy ogniwa stęŜeniowe bez przenoszenia oraz ogniwa stęŜeniowe z przenosze-niem.
O g n i w a s tę Ŝ e n i o w e z p r z e n o s z e n i e m charakteryzują się obecnością granic (kontaktów) między roztworami, jakie powstają np. przy zastosowaniu przegrody porowatej lub klucza elektrolitycznego. Na granicach roztworów występują potencjały dyfu-zyjne.
O g n i w o s tę Ŝ e n i o w e b e z p r z e n o s z e n i a to ogniwo nie zawierające styku róŜnych roztworów. MoŜna to osiągnąć przez połączenie szeregowe dwóch ogniw che-micznych wypełnionych jednym elektrolitem, zbudowanych z elektrod, z których jedna jest odwracalna względem kationu, a druga względem anionu tego elektrolitu. UŜyjmy np. dwóch ogniw Harneda:
Pt, H2(p)HClAgCl, Ag Wówczas ogniwo stęŜeniowe powstałe po zwarciu ze sobą elektrod chlorosrebrnych zapisze-my jako:
Pt, H2(p)HCl(c1)AgCl, Ag - Ag, AgClHCl(c2)H2(p), Pt
c2 > c1 Wynikiem pracy powyŜszego ogniwa jest zniknięcie pewnej liczby moli HCl w ogniwie Har-neda o wyŜszym stęŜeniu, a pojawienie się takiej samej liczby moli w drugim ogniwie.
Wracając do ogniwa podanego na wstępie, które jest ogniwem stęŜeniowym z prze-noszeniem, naleŜy przypomnieć, Ŝe przy zastosowaniu odpowiedniego klucza elektrolitycz-nego zamiast przegrody porowatej, zostaje ostatecznie prawie całkowicie wyeliminowany po-tencjał dyfuzyjny i otrzymuje się wartości SEM ogniwa, takie jak w ogniwach bez przenosze-nia. W omawianym przykładzie na elektrodach zachodzą reakcje: anoda: Ag = Ag+(1) + e (7.5.1) katoda: Ag+(2) + e = Ag (7.5.2) a proces sumaryczny sprowadza się do zapisu Ag+
(2) = Ag+(1) (7.5.3)
Zgodnie z równaniem Nernsta (7.3.10) siła elektromotoryczna tego ogniwa pracującego
w sposób odwracalny wynosi:
(1)Ag
(2)+Ag
(2)+Ag
+Ag
+
(1) lnlna
a
F
RTa
a
F
RT =−=E (7.5.4)
poniewaŜ standardowa siła elektromotoryczna EEEE ° = 0, jako róŜnica standardowych potencja-łów dwóch identycznych elektrod.
Potencjał wyŜszy uzyska półogniwo o większym stęŜeniu jonów Ag+. Podczas pracy og-niwa elektroda ujemna (o niŜszym stęŜeniu jonów srebra) oddaje jony Ag+ do roztworu, do którego z kolei poprzez klucz elektrolityczny dopływają aniony NO3
- - cały elektrolit musi

7. Ogniwa galwaniczne
95
być elektrycznie obojętny. Na elektrodzie dodatniej osadzają się jony srebra z roztworu oraz odpływa do klucza równa ilość jonów azotanowych. W rezultacie roztwór o mniejszym stęŜe-niu zwiększa swoje stęŜenie, a roztwór o większym stęŜeniu zmniejsza je - zachodzi wyrów-nywanie stęŜeń. Literatura uzupełniająca:
F. Jaroszyk, red., „Biofizyka”, Wydawnictwo Lekarskie PZWL, Warszawa 2002, str. 193. T. W. Hermann, red., „Farmacja fizyczna”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 273. J. Terlecki, red., „Ćwiczenia laboratoryjne z biofizyki i fizyki”, Wydawnictwo Lekarskie PZWL, Warszawa 1999, str. 166. B. Kędzia, red., „Materiały do ćwiczeń z biofizyki i fizyki”, PZWL, Warszawa 1982, str. 155. Ćwiczenie 8
Kompensator prądu stałego – pomiar SEM ogniwa galwanicznego w róŜnych temperatu-
rach W ćwiczeniu zestawiamy ogniwo:
Pt | Fe(CN)63-, Fe(CN)6
4- || MnO4-, 8 H+, Mn2+ | Pt
złoŜone z dwóch półogniw redoks:
Pt | Fe(CN)63-, Fe(CN)6
4- , π° = 0,356 V
Pt | MnO4-, 8 H+/Mn2+ , π° = 1,50 V przy aH+ = 1
Na sumaryczną reakcję:
MnO4- + 5 Fe(CN)6
4- + 8 H+ Mn2+ + 5 Fe(CN)63- + 4 H2O
składają się reakcje:
utleniania 5 Fe(CN)64- 5 Fe(CN)6
3- + 5 e-
i redukcji MnO4- + 8 H+ + 5 e- Mn2+ + 4 H2O Wykonanie pomiarów:
1. Włączyć ultratermostat i nastawić termometr kontaktowy na temperaturę 293 K. 2. Do kolby miarowej o pojemności 100 cm3 odpipetować 10 cm3 roztworu nadmanganianu
potasu o stęŜeniu 0,01 mol/dm3. Dodać 10 cm3 stęŜonego roztworu H2SO4 (7,5 mol/dm3) oraz 1 cm3 roztworu chlorku manganu(II) o stęŜeniu 0,1 mol/dm3. Dopełnić wodą destylo-waną do kreski. Zawartość kolby przelać do butelki oznaczonej (+). Umieścić elektrodę platynową oraz termometr w roztworze elektrodowym.

A. Kufelnicki - Ćwiczenia z biofizyki
96
3. Do kolby miarowej na 100 cm3 odpipetować po 1 cm3 0,1 M roztworów heksacyjanoŜela-zianu(II) potasu oraz heksacyjanoŜelazianu(III) potasu. Dopełnić wodą destylowaną do kre-ski. Zawartość kolby przelać do butelki oznaczonej (−). Umieścić elektrodę platynową oraz termometr w roztworze elektrodowym.
4. Połączyć roztwory elektrodowe sporządzonych półogniw kluczem elektrolitycznym. 5. Wstawić badane ogniwo do termostatu i połączyć je przewodami elektrycznymi z odpo-
wiednimi zaciskami kompensatora. 6. Zmierzyć i zanotować siłę elektromotoryczną badanego ogniwa w temp. 293 K po ustale-
niu się temperatury w roztworach elektrodowych. 7. Dokonać pomiarów SEM badanego ogniwa w kilku innych temperaturach z przedziału
293 - 323 K, np. co 5 K . Wyniki pomiarów zestawić w tabeli. 8. Sporządzić wykres zaleŜności EEEE = f (T). Obliczyć metodą najmniejszych kwadratów, korzy-
stając z programu komputerowego, współczynniki prostej typu y = A + B x odpowiadającej równaniu (7.3.14).
AH
zF= − ∆
; pT
B
=
∂∂ E
9. Na podstawie wyznaczonych współczynników A i B obliczyć SEM ogniwa w temperaturze 298 K, a następnie z równania (7.3.6) zmianę entalpii swobodnej G∆ w temp. 298 K oraz odpowiednio z równań (7.3.12) i (7.3.13) zmianę entalpii, H∆ i entropii, S∆ dla reakcji przebiegającej w badanym ogniwie.
Tabela wyników pomiarów:
A = B =
298G∆ =
298H∆ =
298S∆ = S∆ =
Ćwiczenie 9
Pomiar SEM ogniwa stęŜeniowego metodą kompensacyjną Poggendorffa
Zestawiamy następujące ogniwo stęŜeniowe:
AgAgCl nasyc., 0,1 M KCl || AgNO3 (0,1 M)Ag
(I )
Na podstawie znajomości siły elektromotorycznej tego ogniwa stęŜeniowego moŜemy wyzna-czyć iloczyn rozpuszczalności soli trudnorozpuszczalnej AgCl.
T (K) EEEE (V)

7. Ogniwa galwaniczne
97
W roztworze elektrodowym lewego półogniwa aktywność jonów Ag+ (a1) jest określona przez iloczyn rozpuszczalności AgCl: Kir = a1⋅aCl-, przy czym aktywność jonów chlorkowych wynika prawie wyłącznie ze stęŜenia KCl. Korzystając z równania (7.5.4) i podstawiając k = 2,303 RT/F oraz a1 = Kir/aCl- otrzymamy:
irK
aak
2Cllog−
=E (7.5.5)
Znając aktywności jonów chlorkowych w 0,1 M KCl (aCl- = 0,0769) oraz jonów srebro-wych 0,1 M AgNO3 (a2 = 0,0734) w temp. 298 K, moŜna wyraŜenie na SEM badanego ogniwa zapisać w postaci:
AgCl
0734,00769,0log
irKk
⋅=E (7.5.6)
stąd
log Kir = log 0,0769 + log 0,0734 − k
E (7.5.7)
Aby zmniejszyć błąd względny pomiaru, nie mierzymy bezpośrednio SEM ogniwa (I ), lecz dwóch ogniw:
HgHg2Cl2, KCl nasyc.KNO3 nasyc. AgNO3 (0,1 M)Ag
(II )
HgHg2Cl2, KCl nasyc.KNO3 nasyc.AgCl nasyc. , 0,1 M KClAg
(III )
z których kaŜde jest połączone szeregowo z ogniwem Westona.
Odejmując od SEM ogniwa (II ) połączonego z ogniwem Westona, SEM ogniwa (III ) po-łączonego z ogniwem Westona, otrzymamy wartość równą SEM naszego zasadniczego og-niwa pomiarowego (I ): IIIIIIIIIII EEEEEEEE −=+−+== )()( nnx (7.5.8)
Wykonanie pomiarów:
1. Po wyjęciu klucza elektrolitycznego z nasyconego roztworu KNO3 opłukać go wodą desty-lowaną oraz osuszyć bibułą. Ramię oznaczone symbolem (Ag+) zanurzyć do roztworu 0,1 M AgNO3, w którym umieszczona jest elektroda srebrowa. Drugie ramię klucza zanu-rzyć do roztworu elektrodowego elektrody kalomelowej. W ten sposób otrzymaliśmy ogniwo o schemacie (II ).
2. Zestawić układ pomiarowy Poggendorffa wg Rys. 7.4. 3. Ogniwo pomiarowe (II ) połączyć szeregowo z ogniwem Westona i włączyć do obwodu
pomiarowego. Znaleźć punkt kompensacji, l1 . Uwaga: obwód główny zamykać tylko na czas pomiaru, obwód boczny zamykać tylko na okres nie dłuŜszy niŜ 3 - 4 s.
4. W miejscu ogniwa (II ) włączyć ogniwo przedstawione schematem (III ) szeregowo z ogni-wem Westona, jak w punkcie 3. Pamiętać o opłukaniu i osuszeniu końcówki klucza elek-trolitycznego. Znaleźć punkt kompensacji l2 .
5. Znaleźć punkt kompensacji dla ogniwa Westona, l3 . 6. Powtórzyć czynności wymienione w punktach 1 - 5 jeszcze dwukrotnie.

A. Kufelnicki - Ćwiczenia z biofizyki
98
7. Końcówki klucza elektrolitycznego opłukać wodą destylowaną i zanurzyć do nasyconego roztworu KNO3.
8. Obliczyć EEEE x wg równania
3
21l
llnx
−= EE
dla kaŜdej trójki liczb: l1, l2 i l3. Następnie obliczyć wartość średnią EEEE x. 9. Z równania (7.5.7) obliczyć wartość Kir AgCl . Tabela wyników pomiarów i wielkości obliczonych:
l1
(cm)
l2
(cm)
l3
(cm)
EEEE x
(mV) Kir AgCl
Temperatura =