Embed Size (px)
Citation preview

Cyclodextrins in Polymer Synthesis: Free Radical
Polymerization of b-Cyclodextrin
Complexes of Photosensitive Mesoionic
6-Oxo-1,6-dihydropyrimidin-3-ium-4-olates
in Aqueous Medium
Alexander Theis, Helmut Ritter*
Institute of Organic Chemistry and Macromolecular Chemistry, Universitatsstraße 1, 40225 Dusseldorf, GermanyFax: þ49 211 811478; E-mail: [email protected]
Keywords: copolymerization; cyclodextrin; inclusion chemistry; photosensitive polymers; radical polymerization
Introduction
Mesoionic 6-oxo-1,6-dihydropyrimidin-3-ium-4-olates (1)
are six-membered heterocycles which were synthesized
primarily in 1971.[1,2] Many experiments were performed in
order to investigate 1,4-dipolar cycloadditions with olefins
and alkynes.[3–10] Their pharmacological activities are of
great interest,[11,12] and it was furthermore shown that irra-
diation of mesoionic compounds 1 leads irreversibly to the
structure of bis(b-lactames) 2 as illustrated in Scheme 1.[13]
With the change in molecular structure, changes of
the physical properties, such as color, volume, dipole
moment, and refractive index, can also be expected. For
this reason, it is interesting to prepare polymers containing
mesoionic compounds 1, which can be used as photosensi-
tive materials.[14–16]
Up to now, different approaches toward preparing meso-
ionic polymers have been made using styrene as a poly-
merizable group.[14,17,18] We also prepared new types
of polymers containing mesoionic groups in the main
chain.[16,19] Recently, the preparation of polymerizable
methacrylic derivatives was successfully conducted as
shown in Scheme 2.[15]
The homopolymerization of monomers 7 as well as co-
polymerization with methyl methacrylate (MMA) was
carried out in N,N-dimethylformamide (DMF) solution
at 60 8C with 2,20-azoisobutyronitrile (AIBN) as radical
initiator.[15]
Full Paper: Solid mesoionic 2-[2-(isopropenylcarbonyl-oxy)ethylthio]-1-methyl-6-oxo-3-phenyl-5-propyl-1,6-dihy-dropyrimidin-3-ium-4-olate was complexed in water usingb-cyclodextrin (b-CD) and randomly methylated b-CD,which resulted in polymerizable complexes with 2:1 stoichio-metry. The b-CD complex was characterized using 1H NMR,ROESY NMR and UV spectroscopy. Polymerization of thecomplex prepared from methylatedb-CD led to a photosensi-tive polymer, which precipitated during polymerization andwas nearly free of CD. Polymerization was carried out with awater-soluble redox initiator. In addition, a copolymer withmethyl methacrylate was prepared from the complexes, whichshowed a different mass-dependent distribution in the incor-poration in comparison to a copolymer prepared without CDin organic solvents.
Macromol. Chem. Phys. 2003, 204, 1297–1304 1297
Macromol. Chem. Phys. 2003, 204, No. 10 � WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 1022-1352/2003/1007–1297$17.50þ.50/0

In recent years, a new method for the polymerization
of hydrophobic monomers in aqueous solution came
up.[20–22] It is based on the use of at least stoichiometric
amounts of cyclodextrins, which show the capability of
forming water-soluble inclusion complexes of the mono-
mers. Upon polymerization with a water-soluble radical
initiator, the cyclodextrin (CD) slips off stepwise and
can be used for further complexations, whereby the
resulting polymer is insoluble in water and precipitates.
In contrast to the classical polyreactions in emulsion,[23]
this process can also be applied with solid monomers.[24]
Up to now, this method has been expanded on many other
examples.[25–28]
In order to obtain information about the mechanism, the
kinetics of the polymerization of various monomers[29,30] as
well as the influence of chain-transfer agents[31,32] was
investigated. In addition, examples whereby CDs hinder the
polymerization are known.[33] Furthermore, the influence
of CD on the copolymerization parameters of hydrophobic
monomers was examined.[34,35] In some examples, it was
also shown that the copolymerization of CD-complexed
hydrophobic monomers together with hydrophilic mono-
mers in water could only be performed in good yields by
the use of CDs.[36,37]
Referring to these results, we were encouraged to prepare
CD complexes of high-melting crystalline methacrylic
esters containing mesoionic 6-oxo-1,6-dihydropyrimidin-
3-ium-4-olates in aqueous medium and to investigate their
polymerization behavior.
Experimental Part
Materials and Methods
Mesoionic 2-[2-(isopropenylcarbonyloxy)ethylthio]-1-methyl-6-oxo-3-phenyl-5-propyl-1,6-dihydropyrimidin-3-ium-4-olatewas prepared according to the literature.[15] b-CD was receivedfrom Fluka. Randomly methylated b-CD (Me-b-CD) with adegree of methylation of 1.8 was purchased from Wacker. 1HNMR spectra and ROESY NMR spectra were recorded on aBruker AC200 or AM400 at room temperature. The deuteratedsolvent was used as lock and internal standard for the d scalerelative to TMS. Infrared spectra were measured using a NicoletFT-IR spectrometer 5DXC (DTGS detector) or 5SXB (MCTdetector). UV-vis spectra were taken on a Zeiss diode-arrayspectrometer MCS 320 and MCS 340. Differential scanningcalorimetry (DSC) measurements were performed on a PerkinElmer DSC 7.Gelpermeation chromatography (GPC) measure-ments were measured on a PSS installation with chloroform aseluent. 150 ml were injected on a column arrangement of PSSSDV 5m, 100, 1 000, and 10 000 A porosity. A TSP UV 2000UV-vis detector, and a Shodex differential refractometer wereused as detectors. The data were evaluated using PSS-WinGPC6.20. The response factors for copolymer evaluation werecalculated from the signals of the homopolymers.
Preparation of b-CD Complex 9a
1 g (0.88 mmol) b-CD 8a was dissolved in 45 ml of warmdemineralized water and cooled to room temperature. Then0.341 g (0.88 mmol) mesoionic 2-[2-(isopropenylcarbonyl-oxy)ethylthio]-1-methyl-6-oxo-3-phenyl-5-propyl-1,6-dihydro-pyrimidin-3-ium-4-olate (7a) was added and stirred for 5 d
Scheme 1. Intramolecular photocyclization.
Scheme 2. Synthesis of methacryl-substituted mesoions.
1298 A. Theis, H. Ritter

at room temperature. The obtained insoluble precipitationwas filtered and the filtrate was lyophilized to give 0.75 g(29% relative to the guest molecule) of complex 9a as ayellow powder. Water solubility: 17 mg in 1 ml of water atroom temperature.
1H NMR (400 MHz, D2O): d¼ 0.88 (t, 3H, mesoion.-propyl-CH3), 1.44 (tq, 2H, mesoion.-propyl-CH2–CH2–CH3),1.81 (s, 3H, mesoion.-methacryl-CH3), 2.32 (t, 2H, mesoion.-propyl-CH2–CH2–CH3), 3.35 (t, 2H, mesoion.-S–CH2), 3.56(dd, 14H, CD-4-H), 3.63 (dd, 14H, CD-2-H), 3.79–3.89 (m,42H, CD-5,6,60-H), 3.91 (dd, 14H, CD-3-H), 3.93 (s, 3H,mesoion.-N–CH3), 4.36 (t, 2H, mesoion.-O–CH2), 5.04 (d, 14H,CD-1-H), 5.67 (s, 1H, mesoion.-methacryl-CH2), 5.96 (s, 1H,mesoion.-methacryl-CH2), 7.30 (d, 2H, mesoion.-aryl-ortho-CH), 7.58–7.60 (m, 3H, mesoion.-aryl-meta-þ para-CH).
IR (KBr): 3387 (O–H), 2927 (aliph. C–H), 1596 (ar. C C),further signals at 1695, 1635, 1317, 1330, 1298, 1252, 1200,1156, 1079, 1031, 947, 860, 756, 707 cm�1.
UV (H2O): lmax/nm (log e)¼ 226 (4.42), 267 (3.90),343 (3.53).
Preparation of Me-b-CD Complex 9b
4 g (3.00 mmol) Me-b-CD 8b was dissolved in 25 ml ofdemineralized water. Then 0.776 g (2.00 mmol) mesoionic 7awere added and stirred for 5 d at room temperature. Theobtained insoluble precipitation was filtered and the filtratecontaining the complex was used for further reactions withoutisolation. 5 ml of the solution were lyophilized for character-ization. Yield of complex 9b relative to the complete batch:0.87 g (71% relative to guest molecule) as a yellow powderof. Water solubility: � 800 mg in 1 ml of water at roomtemperature.
1H NMR (400 MHz, D2O): d¼ 0.89 (t, 3H, mesoion.-propyl-CH3), 1.44 (tq, 2H, mesoion.-propyl-CH2–CH2–CH3), 1.82 (s,3H, mesoion.-methacryl-CH3), 2.32 (t, 2H, mesoion.-propyl-CH2–CH2–CH3), 3.92 (s, 3H, mesoion.-N–CH3), 4.36 (t, 2H,mesoion.-O–CH2), 5.67 (s, 1H, mesoion.-methacryl-CH2), 5.96(s, mesoion.-1H, mesoion.-methacryl-CH2), 7.29 (m, 2H,mesoion.-aryl-ortho-CH), 7.55–7.59 (m, 3H, mesoion.-aryl-meta-þ para-CH). Due to the statistical methylation, the signalsof 8b are not listed, the resonance signal of mesoion.-S–CH2 isoverlapped by the Me-b-CD signals and could not be separated.Integration of the signals of host and guest resulted in a 2:1complex stoichiometry.
IR (KBr): 3427 (O–H), 2931, 2840 (aliph. C–H), furthersignals at 1689, 1640, 1455, 1386, 1325, 1253, 1194, 1157,1085, 1046, 968, 859, 757, 704 cm�1.
UV (H2O): l/nm (log e)¼ 221 (4.31), 269 (3.95), 336(3.44).
Radical Homopolymerization of Complex 9b
(a) 5 ml of complex 9b were taken. (b) For comparison, a sus-pension of 0.116 g (0.3 mmol) of 7a in 5 ml of demineralizedwater was prepared. The solutions were carefully degassedwith nitrogen; 2.695 mg potassium peroxodisulfate and1.018 mg sodium hydrogensulfite (10 mmol each) were added,and the samples were heated to 60 8C for 1 h with stirring. Then,the precipitated polymer was separated by filtration, washed
with water and dried in vacuum to give (a) 60 mg (52%) and (b)110 mg (95%) of a yellow solid.
1H NMR (200 MHz, CDCl3): d¼ 0.75–0.98 (m, syndiot.methacryl-CH3þ propyl-CH3), 1.00–1.15 (m, heterot. meth-acryl-CH3), 1.15–1.55 (m, isot. methacryl-CH3þ isot. main-chain-CH2þ propyl-CH2–CH2–CH3), 1.55–2.00 (m, isot.þsyndiot. main-chain-CH2), 2.16 (m, propyl-CH2–CH2–CH3),2.74 (m, S–CH2), 3.34 (m, N–CH3), 4.07 (m, O–CH2), 7.16 (m,aryl-ortho-CH), 7.32–7.59 (m, aryl-metaþ para-CH).
IR (KBr): 2960, 2933, 2873 (aliph. C–H), 1729 (C O), 1639(mesoion. C O), 1594 (aryl C C), further signals at 1686,1491, 1455, 1254, 1150, 1112, 1044, 968, 755, 708, 695 cm�1.
GPC (trichloromethane, polystyrene standards): (a) conver-sion: 55%; Mn¼ 3 000; Mw¼ 3 800; D¼ 1.3, and (b) conver-sion: 14%; Mn¼ 2 500; Mw¼ 11 000; D¼ 4.5
The given conversions represent the polymer fractionin the measured solid. These values have to be multi-plied with the yield of solid in order to obtain overallconversions.
Radical Copolymerization of Complexed 7a with ComplexedMMA 11
(a) 0.687 g (0.516 mmol) of 8b were dissolved in 5 ml ofdemineralized water. Then 0.133 g (0.344 mmol) mesoionic 7awere added and the complex was prepared analogously to 9b. Itwas used without previous isolation (the solution containedapprox. 0.258 mmol of the complex).
(b) 13.31 g (10.0 mmol) 8b were dissolved in 35 ml ofdemineralized water. Then 1.00 g (10.0 mmol) MMA 11 wasadded and the suspension was stirred for 1 h at room tempe-rature. For the first 30 min, the mixture was periodically sonica-ted. The dissolved complex 12 (1:1 host/guest stoichiometry)was used without previous isolation.
(c) 5 ml of each solution derived from (a) and (b) werecombined and degassed with nitrogen for 30 min. 19.32 mg(71.5 mmol) potassium peroxodisulfate and 7.44 mg(71.5 mmol) sodium hydrogensulfite were added and the mix-ture was heated with stirring for 1 h at 60 8C. Then the pre-cipitated polymer was separated by filtration, washed withwater and dried in vacuum to give 0.12 g (50%) of a yellowsolid.
(d) 0.100 g (0.258 mmol) mesoionic 7a, 0.142 g (1.42 mmol)MMA 11 and 8.26 mg (50.3 mmol) AIBN were dissolved in1 ml of dry DMF. The mixture was degassed with nitrogen for30 min and polymerized for 24 h at 60 8C. The dissolvedpolymer was precipitated in a mixture of 50 ml methanol and50 ml water and dried under vacuum. For further purification,the product was dissolved in 2 ml methylene chloride, preci-pitated in 50 ml diethyl ether and again dried under vacuum togive (d) 0.13 g (54%) of a yellow solid.
1H NMR (400 MHz, CDCl3): d¼ 0.55–2.08 (m, methacryl-CH3 þ propyl-CH3 þmain-chain-CH2 þ propyl-CH2–CH2–CH3), 2.15 (m, propyl-CH2–CH2–CH3), 2.73 (m, S–CH2), 3.31(s, N–CH3), 3.55 (s, O–CH3), 4.04 (m, O–CH2), 7.13 (d, aryl-ortho-CH), 7.37–7.59 (m, aryl-meta-þ para-CH).
IR (KBr): 2993, 2951 (aliph. C–H), 1731 (C O), 1649(mesoion. C O), further signals at 1482, 1449, 1387, 1245,1194, 1151, 1072, 989, 843, 754, 708 cm�1.
Cyclodextrins in Polymer Synthesis: Free Radical Polymerization . . . 1299

GPC (trichloromethane, polystyrene standard): (c) Mn¼8500, Mw¼ 20 000, D¼ 2.4, and (d) Mn¼ 13 000, Mw¼31 000, D¼ 2.3.
DSC: Tg¼ 96 8C.
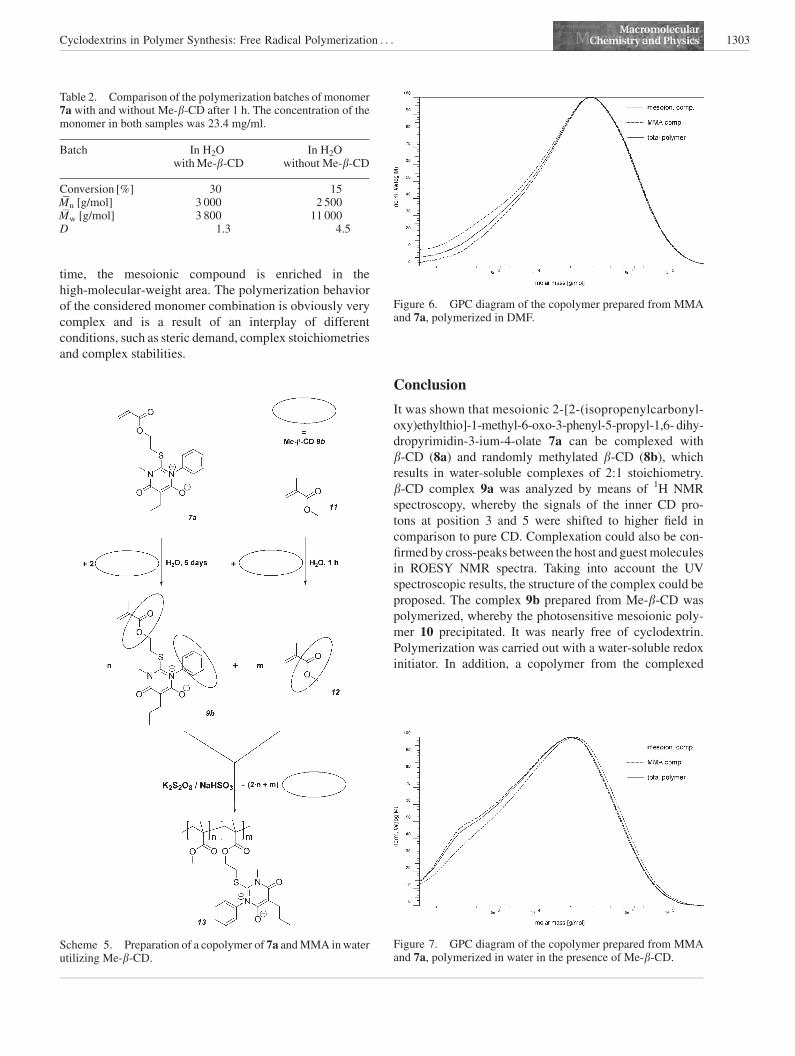
Results and Discussion
In order to prepare complexes, monomers 7a, 7d, and 7fwere stirred in equimolar amounts in saturated solutions of
native b-CD 8a (Scheme 3). After 5 d, the insoluble mono-
mer residuals were removed by filtration. The solution of
complex 9a showed a yellow color caused by the
complexed yellow monomer 7a whereas the solutions of
monomers 7d and 7f showed only minor coloration. The
solutions were lyophilized and the solid residuals were
analyzed by 1H NMR spectroscopy in D2O. The solution of
7a showed signals of b-CD and monomer in a ratio of 2:1,
the other solutions showed only negligible amounts of
monomer, indicating that no complexation occurred. This
can be explained by the fact that the monomer must be at
least sparingly soluble in water, so that monomer and b-CD
can come in contact. Monomer 7a bears the fewest non-
polar substituents, so it should show the highest water
solubility and therefore the highest complexation kinetics.
The 1H NMR spectrum of the complex (Figure 1) shows
significant shifts of the CD protons in position 3 and 5
to higher field in comparison to pure b-CD. These protons
are directed into the cavity of b-CD (see Figure 2), thus
a shifted signal is an indicator for a different environment
inside the cavity and therefore an evidence for successful
complexation.
Table 1 shows the strong shift of the 1H NMR resonance
of inner proton 3-H in comparison to the other protons,
which are shifted only irreducibly. For pure b-CD 8a, the
signal of the inner proton 5 is overlaid by the signals of the
protons 6, 60. In complex 9a, the signal is strongly shifted to
higher field so that it is separated from the other signals.
ROESY NMR experiments show a cross-peak between the
ortho protons of the guest’s phenyl group and the CD proton
in position 3 as well as the meta/para protons of the guest
and the CD protons in position 5 and 6. Therefore, the wider
side of the CD is preferably orientated towards the meso-
ionic function. Also, methacrylic CH2 and –CH3 protons
show cross-peaks to the inner CD protons in position 3, 5,
and 6. The other aliphatic protons show minor cross-peaks
to different inner and outer CD protons. Thus, complexation
could be confirmed, but the exact structure of the 2:1
complex in aqueous solution could not be resolved.
The UV spectrum of the complexed monomer 9a in
aqueous solution shows a strong hypsochromic shift of
the longest wavelength transition from 380 nm (dichloro-
methane without CD) to 340 nm (Figure 3). This is a result
of the enlarged energy difference between the high polar
ground state and the less polar excited state of the chromo-
phore caused by a more polar environment. The same mono-
mer 7a in ethanol shows the highest wavelength transition
at 345 nm, which indicates an even more polar adjacency of
the mesoionic function of complexed monomer 9a in com-
parison to the dissolved monomer 7a in ethanol. This is a
strong hint for sandwich-like complexation, whereby the
mesoionic function is not included into the cavity of CD and
thus surrounded by highly polar water molecules.
The water solubility of complex 9a is 17 mg/ml, which
corresponds to 2.5 mg/ml of the guest molecule 7a. For a
radical polymerization, this concentration is very low. In
order to improve the water solubility of the mesoionic
monomers, complex 9b with Me-b-CD 8b was prepared.
The preparation of complex 9b was done in analogy to
complex 9a, but due to the higher water solubility, less
amounts of water were necessary. Here, the 1H NMR
resonance signals of the inner Me-b-CD protons in complex
9b were also shifted in comparison to the signals of pure
Me-b-CD 8b, however, due to the statistical methylation,
a complete assignment of the signals could not be done.
Complex 9b has a solubility of 800 mg in 1 ml of water,
which is about fifty times higher than that of complex 9a. A
clear deep-yellow highly viscous solution was obtained.
For radical polymerization, complex 9b was used in the
concentration of 160 mg/ml as received, which corresponds
to 23.4 mg/ml of the guest molecule. At this concentration,
the viscosity is comparable to that of DMF, which is nor-
mally used as a solvent for radical polymerizations of meso-
ionic monomers. However, it must be considered that
polymerizations in DMF need much higher monomer
concentrations of 200–250 mg/ml to be successful.[15] The
polymerizationofcomplex9bwascarriedoutwith3.3mol-%
of a redox initiator, which is composed of potassium
peroxodisulfate and sodium hydrogensulfite (Scheme 4).
The NMR and IR spectra of the processed product clearly
show that polymer 10 is nearly free of Me-b-CD, which
shows that the Me-b-CD is slipped off from the monomer
during polymerization.Scheme 3. Preparation of the complexes.
1300 A. Theis, H. Ritter

In Figure 4, a distinct bimodal distribution is visible.
Besides the polymer with a mass distribution from 1 000
to 10 000 g/mol, an oligomeric fraction of nearly the
same total area with chain lengths from 1 to 4 could be
found.
In order to verify the influence of Me-b-CD, a sample of
monomer 7a under the same conditions but in the absence
of CD was polymerized and characterized by means of GPC
(Figure 5).
The conversions and GPC results are listed in Table 2. The
conversion values consider only the polymer fraction in the
precipitated product referring to the amount of monomer at
the beginning.
According to the GPC results of the polymer prepared
from the non-complexed monomer 7a, 85% of 7a was unre-
acted after 1 h. Additionally, the molecular mass distribution
of the polymer is bimodal and the polystyrene of analogous
molecular weight reaches values up to 200 000 g/mol. In this
sample, initiation obviously takes place with a low amount
of dissolved monomer. Then, the resulting oligomers form
aggregates, possibly stabilized by the ionic chain ends. At
the same time, conversion is much lower because, as men-
tioned above, the effective concentration of the monomer
molecule in solution is very low.
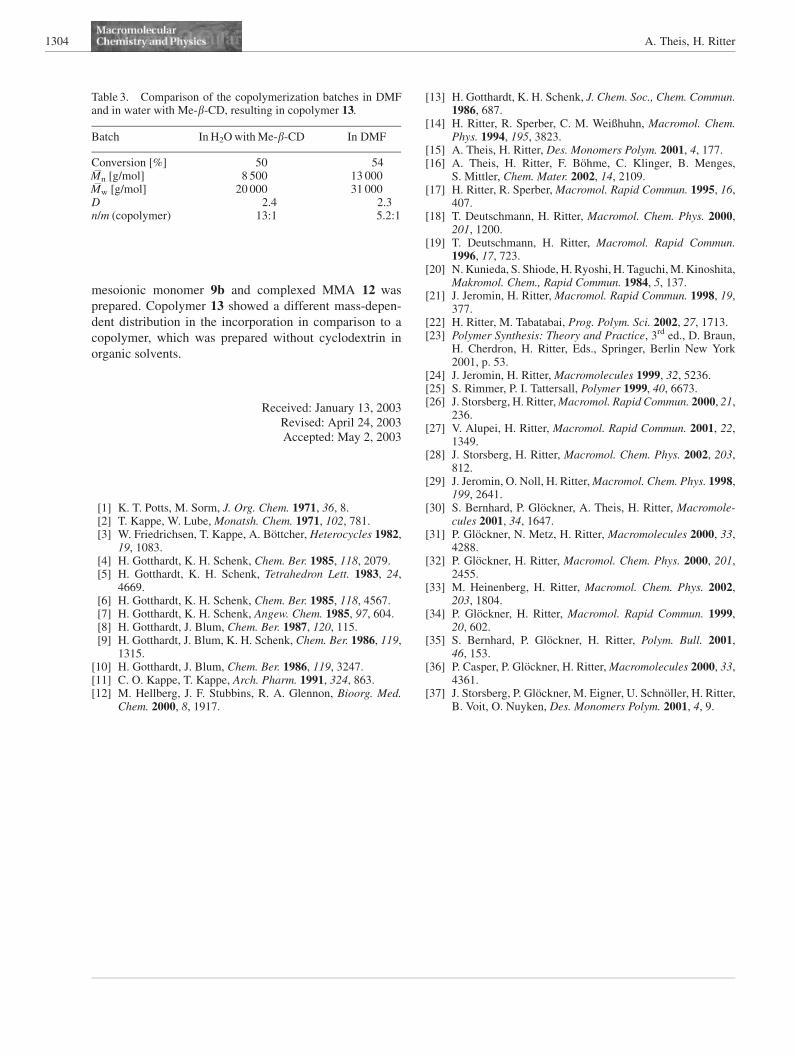
Besides the synthesis of homopolymers, also copolymers
were prepared. On the one hand, copolymers of mesoionic
monomers with complexed liquid standard monomers
are also of technical interest,[15] and on the other hand,
Table 1. 1H NMR spectroscopic shifts of the CD protons ofcomplex 9a.
Sample 1-H 2-H 3-H 4-H
Pure CD 8a 5.049 3.627 3.947 3.564Complex 9a 5.042 3.628 3.912 3.558Difference 0.007 0.001 0.035 0.006
Figure 1. 400 MHz 1H NMR spectrum of b-CD 8a and complex 9a between b-CD and 7ain D2O.
Figure 2. Schematic representation of the b-cyclodextrin torus.
Cyclodextrins in Polymer Synthesis: Free Radical Polymerization . . . 1301

copolymerization can also give information about the
mechanism of the polymerization of complexed monomers.
Due to its comparable double bond reactivity, MMA 11was
used as comonomer. A different tendency of polymeriza-
tion therefore can only be expected due to different
solubility or different steric demand, i.e. a different
diffusion coefficient.
Complex 12 between MMA and Me-b-CD was pre-
pared from a dispersion of MMA and an aqueous solution
of Me-b-CD via stirring and ultrasonic treatment. The solu-
tions of complexes 9b and 12 were combined in a ratio
5.5:1 and polymerized with 5.2 mol-% of the redox ini-
tiator (Scheme 5). For comparison, samples of the non-
complexed monomers in DMF as the solvent with 3 mol-%
of AIBN as the initiator were also polymerized.
Both copolymers were analyzed by means of GPC. Due
to the UVabsorbance of the mesoionic monomer 7a, which
is not overlapping with the absorbance of methyl meth-
acrylate 11, both compounds of the copolymer could be
detected separately by using UVand RI detectors. The GPC
diagrams calculated according to the copolymer evaluation
are displayed in Figure 6 and Figure 7.
In the sample, which was polymerized in DMF (Figure 6)
the mesoionic compound is preferably incorporated in the
low-molecular-weight area. The GPC curve of the polymer,
which was prepared with Me-b-CD in water (Figure 7)
shows a completely different feature. The difference in the
ratio in common is lower and the low-molecular-weight area
is enriched with incorporated MMA.
Table 3 shows, that the conversion and polydispersity of
both samples is nearly identical, but the molecular weight of
the copolymer prepared with Me-b-CD in water is
somewhat lower and less amounts of the mesoionic
compounds are incorporated. Nevertheless this method of
copolymerization represents a good alternative to the
standard methods because no organic solvents are needed,
and the classical emulsion polymerization cannot be used
due to the high melting solid monomer.
The influence of CD on the kinetics and molecular
weight is strongly dependent on the monomer. Accordingly,
Me-b-CD-complexed acrylates show different copolymer-
ization parameters in comparison to the free monomers in
organic solvents, which was explained by different complex
stabilities.[33,34]
In the case of the copolymerization of 9b and 12,
the MMA component is built in preferably. At the same
Figure 3. UV spectra of monomer 7a and complex 9a in differ-ent solvents.
Scheme 4. Polymerization of complex 9b.
Figure 4. GPC diagram of 10, polymerized with 3.3 mol-%redox initiator in water by the use of Me-b-CD.
Figure 5. GPC diagram of 10, polymerized with and withoutcyclodextrin (polymer fraction).
1302 A. Theis, H. Ritter
time, the mesoionic compound is enriched in the
high-molecular-weight area. The polymerization behavior
of the considered monomer combination is obviously very
complex and is a result of an interplay of different
conditions, such as steric demand, complex stoichiometries
and complex stabilities.
Conclusion
It was shown that mesoionic 2-[2-(isopropenylcarbonyl-
oxy)ethylthio]-1-methyl-6-oxo-3-phenyl-5-propyl-1,6- dihy-
dropyrimidin-3-ium-4-olate 7a can be complexed with
b-CD (8a) and randomly methylated b-CD (8b), which
results in water-soluble complexes of 2:1 stoichiometry.
b-CD complex 9a was analyzed by means of 1H NMR
spectroscopy, whereby the signals of the inner CD pro-
tons at position 3 and 5 were shifted to higher field in
comparison to pure CD. Complexation could also be con-
firmed by cross-peaks between the host and guest molecules
in ROESY NMR spectra. Taking into account the UV
spectroscopic results, the structure of the complex could be
proposed. The complex 9b prepared from Me-b-CD was
polymerized, whereby the photosensitive mesoionic poly-
mer 10 precipitated. It was nearly free of cyclodextrin.
Polymerization was carried out with a water-soluble redox
initiator. In addition, a copolymer from the complexed
Table 2. Comparison of the polymerization batches of monomer7a with and without Me-b-CD after 1 h. The concentration of themonomer in both samples was 23.4 mg/ml.
Batch In H2Owith Me-b-CD
In H2Owithout Me-b-CD
Conversion [%] 30 15Mn [g/mol] 3 000 2 500Mw [g/mol] 3 800 11 000D 1.3 4.5
Scheme 5. Preparation of a copolymer of 7a and MMA in waterutilizing Me-b-CD.
Figure 6. GPC diagram of the copolymer prepared from MMAand 7a, polymerized in DMF.
Figure 7. GPC diagram of the copolymer prepared from MMAand 7a, polymerized in water in the presence of Me-b-CD.
Cyclodextrins in Polymer Synthesis: Free Radical Polymerization . . . 1303
mesoionic monomer 9b and complexed MMA 12 was
prepared. Copolymer 13 showed a different mass-depen-
dent distribution in the incorporation in comparison to a
copolymer, which was prepared without cyclodextrin in
organic solvents.
Received: January 13, 2003Revised: April 24, 2003Accepted: May 2, 2003
[1] K. T. Potts, M. Sorm, J. Org. Chem. 1971, 36, 8.[2] T. Kappe, W. Lube, Monatsh. Chem. 1971, 102, 781.[3] W. Friedrichsen, T. Kappe, A. Bottcher, Heterocycles 1982,
19, 1083.[4] H. Gotthardt, K. H. Schenk, Chem. Ber. 1985, 118, 2079.[5] H. Gotthardt, K. H. Schenk, Tetrahedron Lett. 1983, 24,
4669.[6] H. Gotthardt, K. H. Schenk, Chem. Ber. 1985, 118, 4567.[7] H. Gotthardt, K. H. Schenk, Angew. Chem. 1985, 97, 604.[8] H. Gotthardt, J. Blum, Chem. Ber. 1987, 120, 115.[9] H. Gotthardt, J. Blum, K. H. Schenk, Chem. Ber. 1986, 119,
1315.[10] H. Gotthardt, J. Blum, Chem. Ber. 1986, 119, 3247.[11] C. O. Kappe, T. Kappe, Arch. Pharm. 1991, 324, 863.[12] M. Hellberg, J. F. Stubbins, R. A. Glennon, Bioorg. Med.
Chem. 2000, 8, 1917.
[13] H. Gotthardt, K. H. Schenk, J. Chem. Soc., Chem. Commun.1986, 687.
[14] H. Ritter, R. Sperber, C. M. Weißhuhn, Macromol. Chem.Phys. 1994, 195, 3823.
[15] A. Theis, H. Ritter, Des. Monomers Polym. 2001, 4, 177.[16] A. Theis, H. Ritter, F. Bohme, C. Klinger, B. Menges,
S. Mittler, Chem. Mater. 2002, 14, 2109.[17] H. Ritter, R. Sperber, Macromol. Rapid Commun. 1995, 16,
407.[18] T. Deutschmann, H. Ritter, Macromol. Chem. Phys. 2000,
201, 1200.[19] T. Deutschmann, H. Ritter, Macromol. Rapid Commun.
1996, 17, 723.[20] N. Kunieda, S. Shiode, H. Ryoshi, H. Taguchi, M. Kinoshita,
Makromol. Chem., Rapid Commun. 1984, 5, 137.[21] J. Jeromin, H. Ritter, Macromol. Rapid Commun. 1998, 19,
377.[22] H. Ritter, M. Tabatabai, Prog. Polym. Sci. 2002, 27, 1713.[23] Polymer Synthesis: Theory and Practice, 3rd ed., D. Braun,
H. Cherdron, H. Ritter, Eds., Springer, Berlin New York2001, p. 53.
[24] J. Jeromin, H. Ritter, Macromolecules 1999, 32, 5236.[25] S. Rimmer, P. I. Tattersall, Polymer 1999, 40, 6673.[26] J. Storsberg, H. Ritter,Macromol. Rapid Commun. 2000, 21,
236.[27] V. Alupei, H. Ritter, Macromol. Rapid Commun. 2001, 22,
1349.[28] J. Storsberg, H. Ritter, Macromol. Chem. Phys. 2002, 203,
812.[29] J. Jeromin, O. Noll, H. Ritter, Macromol. Chem. Phys. 1998,
199, 2641.[30] S. Bernhard, P. Glockner, A. Theis, H. Ritter, Macromole-
cules 2001, 34, 1647.[31] P. Glockner, N. Metz, H. Ritter, Macromolecules 2000, 33,
4288.[32] P. Glockner, H. Ritter, Macromol. Chem. Phys. 2000, 201,
2455.[33] M. Heinenberg, H. Ritter, Macromol. Chem. Phys. 2002,
203, 1804.[34] P. Glockner, H. Ritter, Macromol. Rapid Commun. 1999,
20, 602.[35] S. Bernhard, P. Glockner, H. Ritter, Polym. Bull. 2001,
46, 153.[36] P. Casper, P. Glockner, H. Ritter, Macromolecules 2000, 33,
4361.[37] J. Storsberg, P. Glockner, M. Eigner, U. Schnoller, H. Ritter,
B. Voit, O. Nuyken, Des. Monomers Polym. 2001, 4, 9.
Table 3. Comparison of the copolymerization batches in DMFand in water with Me-b-CD, resulting in copolymer 13.
Batch In H2O with Me-b-CD In DMF
Conversion [%] 50 54Mn [g/mol] 8 500 13 000Mw [g/mol] 20 000 31 000D 2.4 2.3n/m (copolymer) 13:1 5.2:1
1304 A. Theis, H. Ritter