Embed Size (px)
Citation preview

Universita degli Studi di Perugia
DIPARTIMENTO DI FISICA E GEOLOGIA
Corso di Laurea in Fisica
Tesi di laurea triennale
Dall’entropia termodinamica all’entropiadi entanglement
Candidato:
Tania PitikMatricola 268116
Relatore:
Marta Orselli
Anno Accademico 2016–2017


Indice
1 Entropia in termodinamica 11.1 Entropia in senso macroscopico . . . . . . . . . . . . . . . . . . . 21.2 Entropia in meccanica statistica . . . . . . . . . . . . . . . . . . . 61.3 Entropia e disordine . . . . . . . . . . . . . . . . . . . . . . . . . 9
2 Entropia nella teoria dell’informazione 12
3 L’operatore densita 193.1 Insiemi puri e miscele . . . . . . . . . . . . . . . . . . . . . . . . 193.2 Stati puri . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.3 Descrizione di una miscela statistica . . . . . . . . . . . . . . . . 223.4 Proprieta dell’operatore densita . . . . . . . . . . . . . . . . . . . 233.5 Evoluzione temporale di un insieme statistico . . . . . . . . . . . 253.6 Elementi di matrice dell’operatore densita . . . . . . . . . . . . . 263.7 Matrice densita ridotta . . . . . . . . . . . . . . . . . . . . . . . . 283.8 Postulati della MQ attraverso la matrice densita . . . . . . . . . 30
4 Entanglement 334.1 Sistemi bipartiti . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
4.1.1 Decomposizione di Schmidt . . . . . . . . . . . . . . . . . 344.2 Entropia di Von Neumann . . . . . . . . . . . . . . . . . . . . . . 37
4.2.1 Proprieta dell’entropia di Von Neumann . . . . . . . . . . 374.3 Entanglement di stati puri . . . . . . . . . . . . . . . . . . . . . . 444.4 Stati misti . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454.5 Criteri di separabilita degli stati misti . . . . . . . . . . . . . . . 46
4.5.1 Criterio PPT o della trasposta parziale positiva . . . . . . 464.5.2 Entanglement witness . . . . . . . . . . . . . . . . . . . . 474.5.3 Criterio di riduzione . . . . . . . . . . . . . . . . . . . . . 484.5.4 Criterio di maggiorazione . . . . . . . . . . . . . . . . . . 48
4.6 Misura di entanglement di stati misti . . . . . . . . . . . . . . . . 494.6.1 Entanglement of formation & Concurrence . . . . . . . . 504.6.2 Entanglement distillation . . . . . . . . . . . . . . . . . . 514.6.3 Entanglement cost . . . . . . . . . . . . . . . . . . . . . . 524.6.4 Negativity . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
Bibliografia 58
ii

Introduzione
Entropia . . . . una parola suggestiva, un concetto, uno strumento interpretativoche coinvolge e puo modificare la nostra immagine del mondo. Non e possibiledarne una definizione univoca, perche puo essere presentata sotto vari aspetticon approcci diversi: l’entropia non e una grandezza direttamente misurabile,la sua esistenza non e direttamente suggerita dall’esperienza e non e percepibiledai sensi. Tuttavia e una grandezza fisica fondamentale per capire i fenomeniche avvengono in natura. Il termine “entropia” fu introdotto per la prima voltanel 1865 da Rudolph Clausius, in un testo in cui sintetizza i risultati della allorarecente scienza termodinamica in due lapidarie proposizioni:
• nell’universo l’energia si conserva;
• nell’universo l’entropia tende ad aumentare.
Non ingannino la brevita degli assunti: si tratta della brillante conclusione di unpercorso iniziato il secolo precedente quando si sono costruite le prime macchi-ne termiche, le macchine che utilizzano fonti di calore per ottenere movimento.Intorno alla meta di quel secolo numerosi ricercatori contribuirono, partendo dadiversi punti di vista, a quella che, insieme alla teoria dell’elettromagnetismo,rappresenta il grande risultato della fisica dell’Ottocento: la termodinamica,uno dei grandi capitoli delle scienze naturali. Nata nell’ambito di quest’ultimascienza, nel corso del tempo all’entropia come concetto fu riconosciuto un si-gnificato ben piu vasto, che ha dato luogo a varie estrapolazioni nei campi piusvariati, anche non della fisica, come ad esempio nella teoria dell’informazioneo perfino in ambiti sociologici. Alla base della sua definizione vige la secondalegge della termodinamica, la quale sostiene che in normali condizioni tutti i si-stemi abbandonati a se stessi tendono a divenire disordinati, dispersi e corrottiin relazione diretta al trascorrere del tempo. Ogni cosa vivente e non viventesi consuma, si deteriora e si disintegra. Questa e la sicura fine che tutti gliesseri dovranno affrontare in un modo o nell’altro e, secondo tale legge, questoprocesso inevitabile non ha ritorno.Il proposito di questa tesi e quello di definire e descrivere in maniera il piu pos-sibile rigorosa e quantitativa questa grandezza cosı misteriosa ai molti.Nel primo capitolo viene ripresa la definizione classica di entropia che si incon-tra nei primi corsi di fisica, richiamando alcuni risultati gia noti. Dopodichesi entra brevemente nel campo della meccanica statistica, analizzando con ilsuo formalismo le caratteristiche dei sistemi macroscopici, per arrivare, infine,a evidenziare il legame che sussiste tra l’entropia di un sistema termodinamicoe il grado del suo disordine.Nel secondo capitolo viene fatta una veloce trattazione delle nozioni teoriche che
iii

sono alla base della Teoria dell’Informazione per vedere come, anche in questocontesto, si puo richiamare un concetto di entropia analogo a quello classico chefunge da indicatore fondamentale della possibilita di immagazzinare informa-zione in un sistema fisico.Nel terzo capitolo viene introdotto e analizzato il formalismo delle matrici den-sita, utile nell’ambito della meccanica quantistica non solo per trattare sistemistatistici ma anche per la descrizione di sistemi entangled.Nel quarto capitolo, infine, nel caratterizzare i sistemi quantistici bipartiti siaffronta il concetto di entanglement. La necessita di una misura di quest’ulti-mo porta a definire, per gli stati puri, l’entropia di Von Neumann. Viene fattauna rassegna delle proprieta di tale entropia e se ne esaminano brevemente leanalogie con quelle definite precedentemente. In ultima analisi, si affronta ilproblema della separabilita degli stati misti e si esaminano alcune delle misuredi entanglement di quest’ultimi. Per finire, si fa un tentativo di ricollegare lateoria di entanglement alla teoria termodinamica.
iv


Capitolo 1
Entropia in termodinamica
Preliminari
Al fine di avere piu chiari i concetti che saranno presentati in questo capitolovengono qui definite brevemente alcune nozioni di base utili:
• Ogni sistema macroscopico e un sistema termodinamico;
• Uno stato termodinamico e specificato da un insieme di valori di tuttii parametri termodinamici (quantita macroscopiche misurabili come lapressione P, il volumeV e la temperaturaT ) necessari per la descrizionedel sistema.
• Si ha equilibrio termodinamico (meccanico, termico e chimico) quando lostato termodinamico non cambia nel tempo.
• L’equazione di stato e una relazione funzionale tra i parametri termodi-namici per un sistema all’equilibrio. Se tali parametri sono P,V,T essaassume la forma f(P, V, T ) = 0 e definisce una superficie nello spaziotridimensionale P-V-T.
• Una trasformazione termodinamica e un cambiamento di stato. Se lo statoiniziale e uno stato di equilibrio, la trasformazione puo essere indotta soloda cambiamenti delle condizioni esterne al sistema. La trasformazionee quasi statica se la condizione esterna cambia cosı lentamente che adogni istante il sistema e approssimativamente in equilibrio. E reversibilese, riportando indietro nel tempo la condizione esterna, la trasformazioneripercorre indietro la sua storia.
• In termodinamica, una funzione di stato e una grandezza fisica il cui valoredipende solamente dalle condizioni assunte da un sistema all’inizio e allafine di una trasformazione termodinamica, cioe dallo stato iniziale e finale,e non dal particolare percorso seguito durante la trasformazione.
• Una macchina di Carnot e costituita da una qualsiasi sostanza che subiscala trasformazione ciclica costituita da due isoterme e due adiabatiche.
1

1.1 Entropia in senso macroscopico
La funzione di stato entropia (o “calore non utilizzabile”) fu introdotta perfornire una misura dell’utilizzo del calore come fonte di lavoro e trovare unarisposta alla degradazione dell’energia e all’irreversibilita delle trasformazioni.L’entropia e una grandezza scalare che rende conto della propensione di un cor-po o di un sistema fisico, durante processi chimici e termodinamici, a scambiareo trasformare energia in un certo modo piuttosto che in altri. Nella fattispecie,si puo dire che un sistema fisico tende a modificarsi o interagire con altri sistemiin modo da aumentare la propria entropia, o quantomeno a non farla diminuire.Per definire questa grandezza fisica, occorre far riferimento al seguente teorema.
Teorema di Clausius: In ogni trasformazione ciclica, durante la quale latemperatura e definita, vale la seguente disuguaglianza:∮
dQ
T≤ 0
dove l’integrale e calcolato su un ciclo della trasformazione. Se la trasformazionee reversibile allora vale l’uguaglianza.
Dimostrazione. Dividiamo la trasformazione ciclica in questione in n passi infi-nitesimi, in ciascuno dei quali la temperatura possa essere considerata costante.Si immagini, ad ogni passo, di portare il sistema a contatto con serbatoi dicalore che si trovano rispettivamente alle temperature T1, T2, . . . Tn. Sia Qi laquantita di calore, proveniente dal serbatoio Ti, assorbita dal sistema durante ilpasso i -esimo. Costruiamo un insieme di n macchine di Carnot (C1, C2, . . . Cn)tali che Ci:
1. operi tra Ti e T0 (T0 > Ti, per ogni i);
2. assorba una quantita di calore Qi0 da T0;
3. riversi in Ti una quantita di calore Qi.
Per la definizione della scala di temperatura, abbiamo:
Qi(0)
Qi=T0
Ti
Il risultato netto di questo ciclo e che una quantita di calore
Q0 =
n∑i=1
Q(0)i = T0
n∑i=1
(QiTi
)viene assorbita dal serbatoio T0 e convertita interamente in lavoro senza altrieffetti. Per la seconda legge questo e impossibile a meno che Q0 ≤ 0, quindi
n∑i=1
(QiTi
)≤ 0
Questo dimostra la prima parte del teorema.Se la trasformazione e reversibile allora la invertiamo. Attraverso i medesimi
2

ragionamenti, arriviamo alla stessa disuguaglianza eccetto il fatto che ora Qi hail segno cambiato:
−n∑i=1
(QiTi
)≤ 0
Combinando questa disuguaglianza con la precedente otteniamo:
n∑i=1
(QiTi
)= 0
L’equazione si generalizza al caso in cui avvengano scambi di calore infinitamentepiccoli δQ tra il sistema e la sorgente di calore a temperatura T∮
dQ
T≤ 0
valida in ogni caso. In particolare, nel caso di una trasformazione reversibile siha ∮
dQ
T= 0
Corollario
Per una trasformazione reversibile l’integrale∫dQ
T
e indipendente dal cammino della trasformazione e dipende soltanto dagli statiiniziale e finale.
Dimostrazione. Sia A lo stato iniziale e B lo stato finale. Indichiamo con I e IIdue cammini arbitrari e reversibili che connettono A con B e sia II ’ il camminoinverso di II.Il teorema di Clausius implica che∫
I
dQ
T+
∫II′
dQ
T= 0
Ma ∫II′
dQ
T= −
∫II
dQ
T
Quindi ∫I
dQ
T=
∫II
dQ
T
3
Figura 1.1: Generica trasformazione ciclica nel piano di Clapeyron
Questo corollario ci consente di definire una funzione di stato, l’entropia S,come segue. Si sceglie uno stato arbitrario fisso O come riferimento. L’entropiaS (A) per ogni stato A e definita come
S(A) ≡∫ A
O
dQ
T
in cui il cammino di integrazione e un qualsiasi cammino reversibile congiungenteO con A. In tal modo l’entropia e definita solo a meno di una costante additiva.In ogni caso e completamente definita la differenza di entropia tra due stati:
S(B)− S(A) =
∫ B
A
dQ
T
in cui il cammino di integrazione e un qualsiasi cammino reversibile congiun-gente A con B. Da questa formula segue che, in ogni trasformazione infinitesimareversibile, la variazione di S e data da
dS =dQ
T
che e un differenziale esatto.Evidenziamo ora le seguenti proprieta dell’entropia:
(a) Per un’arbitraria trasformazione,∫ B
A
dQ
T≤ S(B)− S(A)
Vale l’uguaglianza se la trasformazione e reversibile.
Dimostrazione. Siano rispettivamente R ed I un qualsiasi cammino rever-sibile ed un qualsiasi cammino irreversibile che connettono A e B. Per il
4
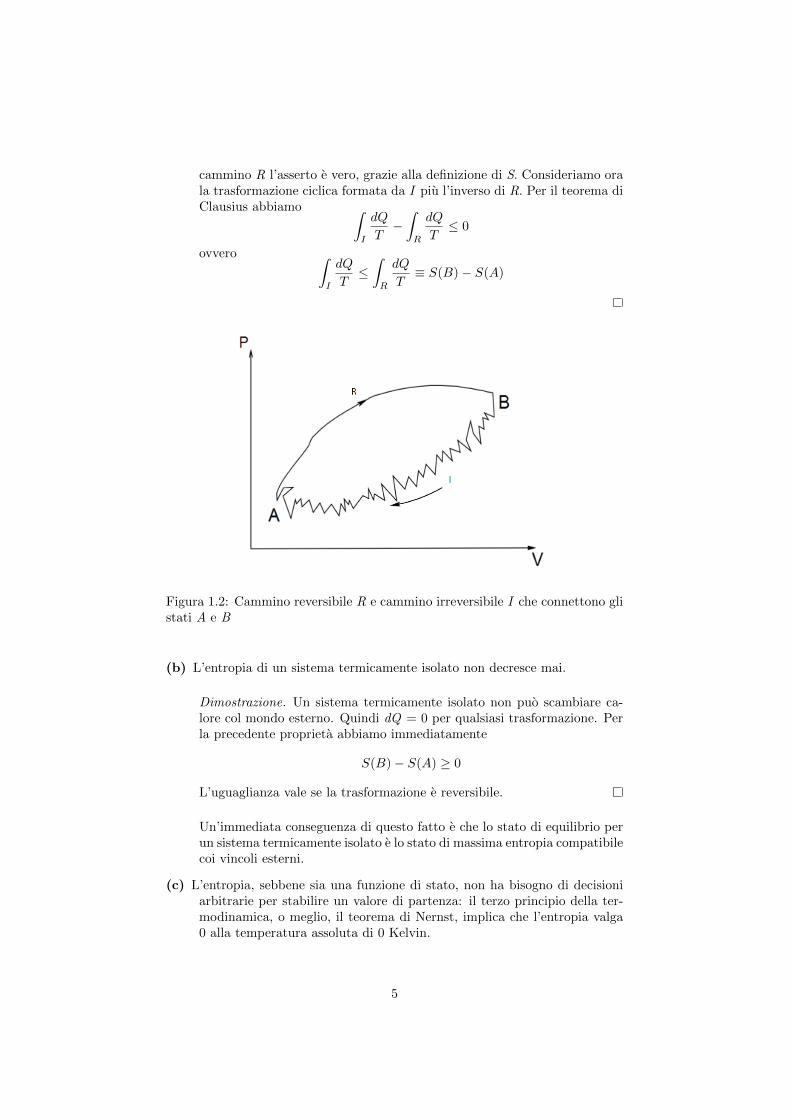
cammino R l’asserto e vero, grazie alla definizione di S. Consideriamo orala trasformazione ciclica formata da I piu l’inverso di R. Per il teorema diClausius abbiamo ∫
I
dQ
T−∫R
dQ
T≤ 0
ovvero ∫I
dQ
T≤∫R
dQ
T≡ S(B)− S(A)
Figura 1.2: Cammino reversibile R e cammino irreversibile I che connettono glistati A e B
(b) L’entropia di un sistema termicamente isolato non decresce mai.
Dimostrazione. Un sistema termicamente isolato non puo scambiare ca-lore col mondo esterno. Quindi dQ = 0 per qualsiasi trasformazione. Perla precedente proprieta abbiamo immediatamente
S(B)− S(A) ≥ 0
L’uguaglianza vale se la trasformazione e reversibile.
Un’immediata conseguenza di questo fatto e che lo stato di equilibrio perun sistema termicamente isolato e lo stato di massima entropia compatibilecoi vincoli esterni.
(c) L’entropia, sebbene sia una funzione di stato, non ha bisogno di decisioniarbitrarie per stabilire un valore di partenza: il terzo principio della ter-modinamica, o meglio, il teorema di Nernst, implica che l’entropia valga0 alla temperatura assoluta di 0 Kelvin.
5

1.2 Entropia in meccanica statistica
La termodinamica ha condotto le sue ricerche sia a livello macroscopico sia alivello microscopico. In questo secondo caso lo studio non ha piu come oggettoimmediato le grandezze osservabili di un sistema (volume, pressione, temperatu-ra, massa), ma piu direttamente il comportamento delle sue molecole e dei suoiatomi, dal momento che quelle grandezze sono la risultante degli stati in cui sitrovano le singole particelle, in termini di velocita, impulsi, energia e posizione(ad esempio, la temperatura di un corpo e riconducibile al valore medio dellavelocita con cui le molecole si muovono al suo interno). Cambiato l’oggetto distudio, e evidente che anche gli strumenti di analisi devono cambiare: poicheatomi e molecole sfuggono all’osservazione diretta, e il calcolo statistico che di-venta lo strumento principale del ricercatore. I risultati di questo secondo mododi affrontare i problemi hanno portato alla convalida dei principi gia dimostratia livello macroscopico, ma con formulazioni nuove. Nel caso dell’entropia nonsolo e stata confermata la validita del concetto, ma nel 1877 L. Boltzmann neha dato una definizione in termini statistici come misura del disordine del siste-ma a livello molecolare, fornendo nello stesso tempo la formula matematica percalcolarla.Bisogna quindi entrare nel campo della Meccanica Statistica la quale si occupadelle proprieta della materia in equilibrio in senso empirico usato in termodina-mica, tuttavia essa non descrive come un sistema raggiunga l’equilibrio, ne seun sistema possa mai essere trovato all’equilibrio ma dice semplicemente qual’ela situazione di equilibrio.Consideriamo un sistema classico composto da un numero N grande di parti-celle in un volume V. Il sistema verra considerato isolato nel senso che l’energiae una costante del moto. Questa e ovviamente un’idealizzazione dal momentoche in laboratorio non abbiamo mai a che fare con sistemi realmente isolati. Ilsemplice fatto che si possano fare misure sul sistema richiede qualche interazionetra esso e il mondo esterno; se, tuttavia, queste interazioni sono sufficientementedeboli, in modo che l’energia del sistema resti approssimativamente costante, losi puo considerare isolato.Uno stato di tale sistema e completamente ed univocamente definito dalle 3Ncoordinate canoniche q1, q2, . . . qN e dai 3N momenti canonici p1, p2, . . . pN .Queste 6N variabili sono indicate tutte insieme in maniera abbreviata con (p, q).La dinamica del sistema e determinata dalla hamiltoniana H(q, p) dalla qualepossiamo ottenere le equazioni canoniche del moto
∂H(q, p)
∂pi= qi
∂H(q, p)
∂qi= −pi
(1.1)
E utile introdurre a questo punto lo spazio 6N -dimensionale Γ, o spazio delle fasidel sistema, ogni punto del quale rappresenta uno stato del sistema e viceversa.Il luogo geometrico dei punti di Γ che soddisfano la condizione H(q, p) = Ee una superficie detta superficie di energia E. Nel tempo gli stati del sistemaevolvono secondo le (1.1) ed il punto rappresentativo descrive una traiettorianello spazio Γ. Questa traiettoria sta sempre sulla stessa superficie di energia,
6

essendo, per definizione, l’energia conservata.Di un sistema macroscopico non abbiamo ne mezzi ne motivi per accertare lostato ad ogni istante, ci interessano unicamente poche sue proprieta macroscopi-che. In particolare, richiediamo solamente che il sistema abbia N particelle, unvolume V ed un’energia tra E e E+∆. Queste condizioni sono soddisfatte daun numero infinito di stati, di conseguenza possiamo pensare di avere a che farenon con un solo sistema, ma con un numero infinito di sue copie, ognuna dellequali posta in uno dei possibili stati che soddisfano le condizioni date. L’insiemedi questi sistemi e rappresentato da una distribuzione di punti nello spazio Γcaratterizzata da una funzione di densita ρ(p, q, t) definita in modo che
ρ(p, q, t)d3Npd3Nq (1.2)
dia il numero di punti rappresentativi contenuti nell’elemento di volume d3Npd3Nqposto nel punto (p,q) dello spazio Γ all’istante t. Poiche siamo interessati allasituazione di equilibrio, limiteremo le nostre considerazioni a quegli ensemble lacui funzione di densita non dipende esplicitamente dal tempo.C’e un postulato che afferma che quando un sistema macroscopico e in equili-brio termodinamico, il suo stato puo essere con uguale probabilita ognuno diquelli che soddisfano le condizioni macroscopiche del sistema. Questo postulatoimplica che all’equilibrio termodinamico il sistema in esame appartiene ad unensemble, detto ensemble microcanonico, con funzione densita:
ρ(p, q, t) =
{cost. se E < H(q, p) < E + ∆
0 altrimenti(1.3)
In un ensemble microcanonico dunque ogni sistema ha N particelle, un volu-me V ed un’energia compresa tra E ed E+∆(E). L’entropia e la quantitafondamentale che fornisce la connsessione tra l’ensemble microcanonico e la ter-modinamica.Sia Γ(E) il volume occupato nello spazio Γ dall’ensemble microcanonico:
Γ(E) ≡∫E<H(q,p)<E+∆
d3Npd3Nq (1.4)
Si sottintende la dipendenza di Γ(E) da N,V e ∆. Sia Σ(E) il volume nellospazio Γ delimitato dalla superficie di nergia E :
Σ(E) =
∫H(q,p)<E
d3Npd3Nq (1.5)
Allora si haΓ(E) = Σ(E + ∆)− Σ(E) (1.6)
Se ∆ e tale che ∆� E, allora
Γ(E) = ω(E)∆ (1.7)
dove ω(E) e detta densita degli stati del sistema all’energia E ed e definita dalla
ω(E) =∂Σ(E)
∂E(1.8)
7

L’entropia e definita come
S(E,V) ≡ kB log Γ(E) (1.9)
dove kB e una costante universale che alla fine, nel dimostrare che la (1.9)possiede tutte le proprieta della funzione entropia in termodinamica, risultaessere la costante di Boltzmann. La forma piu nota della (1.9) e S = kB logWcon W numero di tutti i possibili microstati che danno origine ad un determinatomacrostato. Quest’ultima e un caso particolare di un’espressione piu generaledi entropia, valida anche fuori dagli stati di equilibrio, la cosiddetta entropia diGibbs:
S = −kB∑i
pi log pi
dove i pi sono le probabilita associate alla realizzazione di un certo microstato.
8

1.3 Entropia e disordine
Abbiamo dunque visto che l’entropia puo essere interpretata, oltre che dalle leg-gi della termodinamica classica, anche da un punto di vista statistico. Nota e larelazione tra entropia e disordine: il concetto intuitivo di disordine puo esseretrattato da un punto di vista matematico in termini di termodinamica statisticae viene definito come lo stato piu probabile in cui puo presentarsi un sistema.Ad esempio, se un gas contenuto in un recipiente viene messo a contatto conun altro recipiente vuoto, esso tendera ad occupare tutto lo spazio a sua dispo-sizione perche questo e lo stato “piu probabile” che esso puo assumere: infattie estremamente improbabile che le molecole gassose, animate da moti casuali,permangano nel primo contenitore o si addensino prevalentemente in uno deidue.Lo stato piu probabile e definito come quello stato o “configurazione” che puorealizzarsi nel maggior numero dei modi. In sintesi : massima entropia = massi-mo numero di modi in cui si puo realizzare un certo stato = massimo disordine.Ma vediamolo con un esempio semplice. Consideriamo un sistema costituito da4 particelle A,B,C e D che hanno a disposizione due recipienti uguali e valutia-mo quali sono le loro possibili disposizioni:
1 2ABCD
ABCDA BCDB ACDC ABDD ABC
BCD AACD BABD CABC DAB CDAC BDAD BCBC ADCD ABBD AC
Tabella 1.1: Tutte le possibili disposizioni delle 4 particelle nei 2 recipienti
Dal calcolo combinatorio risulta che il numero totale di disposizioni possibili e: W = 2N dove N e il numero di particelle (nel nostro caso N = 4). Di talidisposizioni, alcune corrisponderanno alla stessa configurazione (ad esempio laconfigurazione con 3 particelle a sinistra e una particella a destra puo essererealizzata in 4 modi : ABC/D , ABD/C, ACD/B, BCD/A). Si deduce che ilnumero di modi in cui si puo realizzare una configurazione corrispondente a N1
particelle in un recipiente, e N2 particelle nell’altro e dato da :
WN1,N2=
N !
N1!N2!(1.10)
9

in cui N e il numero totale di particelle. Nel caso del nostro sistema abbiamodunque:
1 2 N0 di combinazioni probabilita4 0 1 1/160 4 1 1/163 1 4 4/161 3 4 1/162 2 6 6/16
Tabella 1.2: Configurazioni e probabilita
Risulta pertanto che la configurazione in cui le particelle sono equidistribuitee la piu probabile perche si puo realizzare nel maggior numero di modi (6): laprobabilita di tale configurazione e data da 6/16 essendo 6 il numero di eventifavorevoli e 16 il numero di eventi totali. La probabilita di una configurazioneuniforme nei due recipienti aumenta con il numero di particelle ed e vicinaall’unita se ne consideriamo un numero enorme, quale e quello contenuto in unamassa anche minima di sostanza.Difatti, fissato il numero totale N di particelle, supposto molto grande, sia m ilnumero di particelle nel recipiente 1. Il numero di possibili combinazioni di mparticelle prese tra N totali risulta essere dato dal coefficiente binomiale(
N
m
)=
N !
m!(N −m)!(1.11)
Vogliamo ora ricavare il valore di m per cui tale numero di combinazioni, econ esso la probabilita che il sistema si trovi in quello stato, e massimo. E piusemplice studiare il logaritmo di questa quantita ed esprimere il comportamentodei fattoriali mediante la formula di Stirling:
log n! '(n+
1
2
)log n− n+
1
2log(2π) (1.12)
Tale approssimazione e tanto piu accurata quanto piu e grande il numero nconsiderato. Nel nostro caso, trattandosi di un sistema macroscopico, N e unnumero dell’ordine del numero di Avogadro ed m, essendo il numero di particelledello stato piu probabile, sara anche esso un numero molto grande. Ricordiamoche, essendo il logaritmo una funzione monotona, il massimo della quantitaoriginale, e quello del suo logaritmo coincidono. Risulta quindi
log
(N
m
)'(N +
1
2
)logN −N −
(m+
1
2
)logm+m
−(N −m+
1
2
)log(N −m) + (N −m) (1.13)
dove abbiamo trascurato il contributo del termine log 2π. Imponendo che laderivata in m sia nulla otteniamo
− log(m) + log(N −m)− 1
2m+
1
2(N −m)= 0 (1.14)
10

e nell’ipotesi di N ed m molto grandi possiamo trascurare i termini senza ilogaritmi, per cui otteniamo
logN −mm
= 0 (1.15)
che e soddisfatta per m = N/2.Abbiamo mostrato percio che lo stato piu probabile e quello in cui le particellesono distribuite ugualmente tra le due cavita del recipiente. Il concetto di di-sordine si ricollega proprio a questo. Se inizialmente le particelle fossero tutteordinate nella stessa parte del recipiente, nel rimuovere un ipotetico setto sepa-ratore il sistema avrebbe possibilita di evolvere spontaneamente portandosi aduno stato di entropia maggiore, in cui tutte le particelle sarebbero disposte ca-sualmente tra le due cavita, in uno stato visibilmente piu disordinato. Va fattonotare che, preso un certo sistema in equilibrio, sarebbe possibile, spendendoenergia, riportarlo in uno stato di ordine a minore entropia, tuttavia, il bilanciototale dell’entropia sistema-ambiente risulterebbe comunque in positivo.
11

Capitolo 2
Entropia nella teoriadell’informazione
Quando – negli anni ’40 – Claude E. Shannon riuscı a definire l’equazione concui calcolare il livello di imprevedibilita di una fonte d’informazione, non potenon constatare che la sua formula era praticamente uguale a quella con cuiBoltzmann aveva calcolato l’entropia di un sistema termodinamico. Fu JohnVon Neumann, uno dei padri del computer, a proporgli di adottare il termineentropia per indicare la complessita dell’informazione disponibile alla fonte inun qualsiasi sistema di comunicazione, evidenziando l’analogia esistente tra duecontesti scientifici cosı lontani tra loro. Dice infatti in una lettera a ClaudeShannon:“You should call it entropy, for two reasons. In the first place your uncertaintyfunction has been used in statistical mechanics under that name, so it alreadyhas a name. In the second place, and more important, no one really knows whatentropy really is, so in a debate you will always have the advantage.”Il consiglio venne accolto, anche perche Shannon nutriva una certa resistenzaa usare la parola informazione, consapevole che il termine nella sua teoria erautilizzato in un’accezione piuttosto disorientante rispetto a quella usuale. Nellateoria dell’informazione, infatti, l’informazione e la misura della prevedibilitadi un segnale. Se e ‘facilmente’ prevedibile, significa che le alternative in giocosono poche (bastano pochi bit per controllarle). Il segnale e invece altamenteimprevedibile quando il sistema e complesso e, quindi, sono necessari molti bitper individuarlo. Nel primo caso l’entropia della fonte e ridotta, nel secondo in-vece e alta (tanto piu alta, quanto piu numerosi ed equiprobabili sono i segnalipossibili).Il primo aspetto affrontato dalla teoria e quello di misurare la quantita d’in-formazione presente in un messaggio emesso da una sorgente. Ma prima dimisurare una grandezza e necessario definirla! Cosa si intende per quantitad’informazione? Nell’affrontare la codifica di sorgente e di canale, Shannon, nelsuo lavoro “A mathematical theory of communication” del 1948, propone chela quantita di informazione sia una funzione della probabilita. Alcuni sempliciesempi dovrebbero chiarire quanto da egli suggerito. Se si considerano le duefrasi “Domani il sole sorge” e “Domani il sole sorge alle 6 e 34 minuti”, allaprima non e associata alcuna informazione, dato il carattere di regolarita e di
12

certezza dell’evento, mentre la seconda frase presenta una quantita d’informa-zione diversa da zero. Si dice che fa notizia la frase “Un uomo morde un cane”e non il viceversa, perche tale evento si manifesta con una probabilita tenden-te a zero. In un testo scritto ci sono lettere che si manifestano con maggiorefrequenza di altre, per esempio la lettera “e” e piu frequente della lettera “q”.Lo stesso accade nella lingua parlata dove alcune parole sono piu probabili dialtre. Pertanto, seguendo tale criterio, si puo affermare che la quantita d’in-formazione associata a un messaggio e legata alla probabilita che il messaggioha di manifestarsi. Quanto piu alta e la sua probabilita di presentarsi a priori,tanto minore e l’informazione ad esso associata. Al contrario, se la probabilitadi un messaggio e piuttosto bassa la quantita d’informazione che esso trasportae notevole. Per la misura dell’informazione diventa, quindi, di fondamentaleimportanza l’incertezza a priori sul contenuto del messaggio.Passando alle sorgenti coinvolte nei sistemi di nostro interesse, queste possonoessere distinte in due categorie :
• sorgenti senza memoria, quando ciascun simbolo emesso e indipendentedai simboli precedenti;
• sorgenti con memoria, quando il simbolo emesso dipende da uno o piusimboli precedenti;
Una sorgente digitale senza memoria (DSM) e definita completamente quando:
• e noto l’alfabeto dei simboli utilizzati;
• sono note le probabilita con le quali si manifestano i simboli;
• e nota la frequenza con la quale la sorgente emette i simboli (numero disimboli/s).
Indichiamo con {x0, x1, . . . , xM−1} l’alfabeto tramite il quale la sorgente X siesprime e con P (xm) le probabilita con le quali i vari simboli si manifestano. Sela sorgente e stazionaria le probabilita rimangono costanti nel tempo. Ogni sim-bolo puo essere considerato come un messaggio che si presenta con la probabilitaP (xm). Per definizione deve valere la relazione
M−1∑m=0
P (xm) = 1 (2.1)
La quantita d’informazione fornita dal singolo simbolo, detta anche autoinfor-mazione, e definita dall’espressione
I(xm) = log2
1
P (xm)= − log2 P (xm) (2.2)
La (2.1) ha il grande pregio di razionalizzare quanto detto sul piano intuitivo.Infatti, se il generico simbolo xm fosse caratterizzato da una P (xm) = 1 (eventocerto), la quantita d’informazione sarebbe nulla. Viceversa, se P (xm) assumessevalori sempre piu vicini allo zero, la quantita d’informazione crescerebbe asin-toticamente verso l’infinito. La funzione I(xm), decrescente al crescere dellaprobabilita, e riportata in Fig.2.1, nell’intervallo [0,1] in cui sono definite leP (xm). Si puo osservare che la quantita d’informazione e sempre maggiore di
13
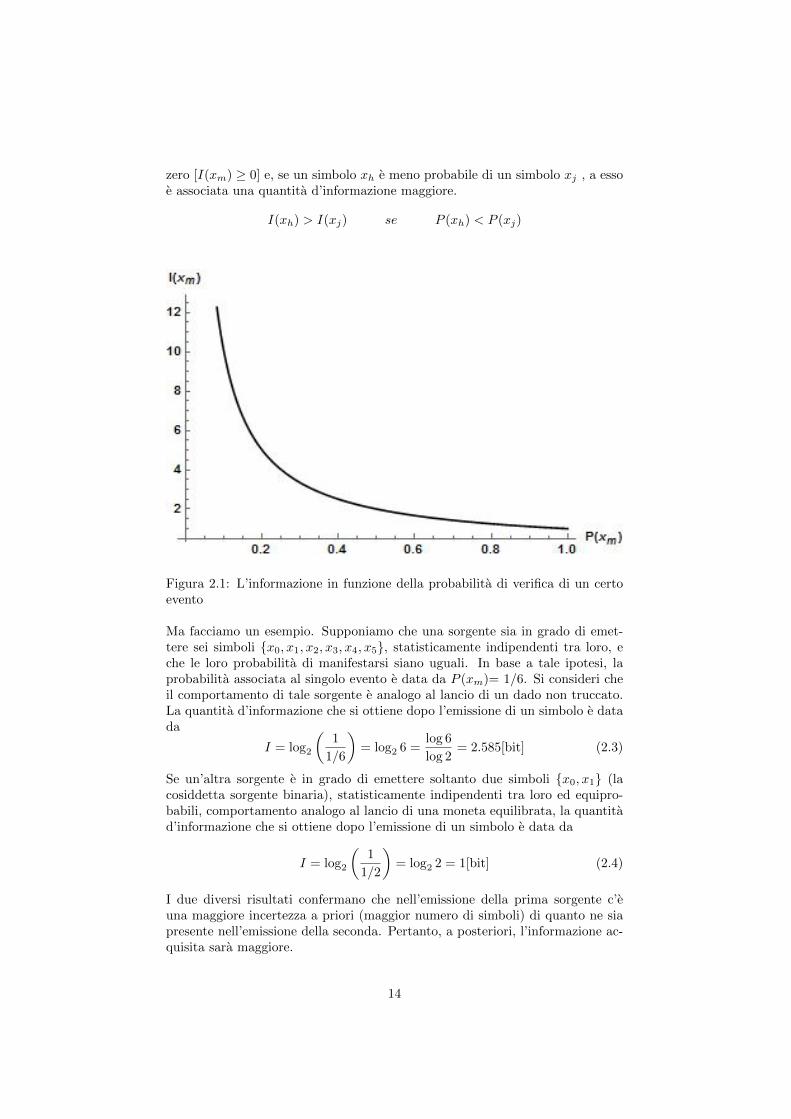
zero [I(xm) ≥ 0] e, se un simbolo xh e meno probabile di un simbolo xj , a essoe associata una quantita d’informazione maggiore.
I(xh) > I(xj) se P (xh) < P (xj)
Figura 2.1: L’informazione in funzione della probabilita di verifica di un certoevento
Ma facciamo un esempio. Supponiamo che una sorgente sia in grado di emet-tere sei simboli {x0, x1, x2, x3, x4, x5}, statisticamente indipendenti tra loro, eche le loro probabilita di manifestarsi siano uguali. In base a tale ipotesi, laprobabilita associata al singolo evento e data da P (xm)= 1/6. Si consideri cheil comportamento di tale sorgente e analogo al lancio di un dado non truccato.La quantita d’informazione che si ottiene dopo l’emissione di un simbolo e datada
I = log2
(1
1/6
)= log2 6 =
log 6
log 2= 2.585[bit] (2.3)
Se un’altra sorgente e in grado di emettere soltanto due simboli {x0, x1} (lacosiddetta sorgente binaria), statisticamente indipendenti tra loro ed equipro-babili, comportamento analogo al lancio di una moneta equilibrata, la quantitad’informazione che si ottiene dopo l’emissione di un simbolo e data da
I = log2
(1
1/2
)= log2 2 = 1[bit] (2.4)
I due diversi risultati confermano che nell’emissione della prima sorgente c’euna maggiore incertezza a priori (maggior numero di simboli) di quanto ne siapresente nell’emissione della seconda. Pertanto, a posteriori, l’informazione ac-quisita sara maggiore.
14
Generalizziamo ora il discorso e prendiamo una sorgente che emetta M simbolistatisticamente indipendenti descritti dalle probabilita P (xm). La quantita d’in-formazione, variando in base al simbolo emesso, diventa una variabile aleatoriadiscreta. Ed ecco che nel lavoro citato, Shannon definı entropia della sorgen-te il valore medio o valore atteso della quantita d’informazione, indicandolacon la lettera H. Alla sorgente che emette M simboli viene dunque associataun’entropia data da
H =
M−1∑m=0
P (xm) log2
1
P (xm)= −
M−1∑m=0
P (xm) log2 P (xm) bit/simbolo (2.5)
Si ha che
• se tutte le P (xi) sono nulle, tranne una sola, non c’e alcuna incertezza el’entropia e nulla.
• se tutte le P (xi) sono uguali l’entropia e massima, perche e massimal’incertezza su quale simbolo sara emesso.
Nel caso particolare di sorgente binaria (lancio di una moneta), se si indica conP la probabilita che sia emesso lo “0” (Testa) e con (1-P) la probabilita che siaemesso l’ “1” (Croce) si ha
H =
1∑m=0
P (xm) log2
(1
P (xm)
)= −P log2 P + (P − 1) log2(−P + 1) (2.6)
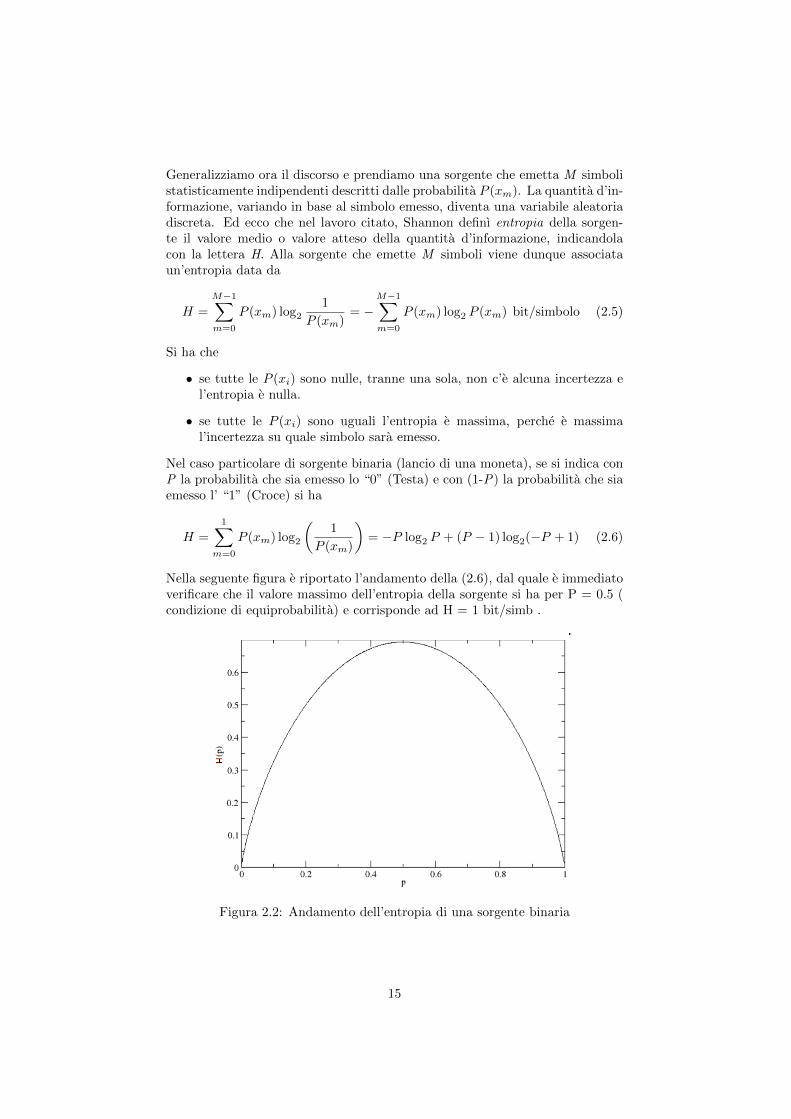
Nella seguente figura e riportato l’andamento della (2.6), dal quale e immediatoverificare che il valore massimo dell’entropia della sorgente si ha per P = 0.5 (condizione di equiprobabilita) e corrisponde ad H = 1 bit/simb .
Figura 2.2: Andamento dell’entropia di una sorgente binaria
15

L’entropia di Shannon di una certa variabile aleatoria X quantifica dunquel’ammontare di informazione che si guadagna, in media, quando si apprende ilvalore di X oppure l’incertezza che si ha su X prima di apprendere il suo valore,e quindi una funzione delle probabilita {p1, p2, . . . , pn} dei diversi possibili valoriche tale variabile puo assumere. E definita come:
H(X) ≡ H(p1, p2, . . . , pn) = −∑i
pi log pi (2.7)
Consideriamo le seguenti quantita:
• Entropia relativa: e un’utile misura di quanto siano “vicine” due distri-buzioni di probabilita pi e qi sullo stesso insieme. E definita come
H(pi‖qi) =∑i
pi logpiqi
= −H(X)−∑i
pi log qi
L’entropia relativa non e mai negativa: H(pi‖qi) ≥ 0
• Entropia congiunta: misura la totale incertezza che si ha sulla coppia divariabili aleatorie (X,Y ) ed e definita come:
H(X,Y ) = −∑ij
pij log pij
con i e j valori che possono assumere rispettivamente X e Y, e pij pro-babilita che questi due valori vengano assunti contemporaneamente dalledue variabili.
• Entropia condizionata: e una misura di quanta incertezza rimane su unavariabile X, una volta noto il valore di Y (e viceversa), ovvero di cio cheY non dice riguardo X :
H(X|Y ) ≡ H(X,Y )−H(Y )
• Informazione mutua: rappresenta l’ammontare di incertezza in X, me-no l’ammontare di incertezza di X che rimane dopo che Y e noto, ol’ammontare di incertezza in X che e eliminato conoscendo Y.
H(X : Y ) ≡ H(X) +H(Y )−H(X|Y )
Seguono le proprieta che caratterizzano l’entropia di Shannon:
1. H(X,Y ) = H(Y,X) H(X : Y ) = H(Y : X)
2. H(Y |X) ≥ 0 e quindi H(X : Y ) ≤ H(Y ), con l’ugualglianza⇔ Y = f(X)
3. H(X) ≤ H(X,Y ) con l’ugualglianza ⇔ Y = f(X)
4. Subadditivita: H(X,Y ) ≤ H(X) + H(Y ) con l’uguaglianza ⇔ H(X) eH(Y) sono indipendenti tra di loro.
5. Subadditivita forte: H(X,Y, Z) +H(Y ) ≤ H(X,Y ) +H(Y, Z)
A queste verra fatto riferimento in seguito in un confronto con le proprieta del-l’entropia di Von Neumman.
16

Considerazioni
Innanzitutto osserviamo la forma identica dell’entropia di Boltzmann (1.9) all’entropia di Shannon (2.7), a meno della costante kB che emerge per conside-razioni relative alle unita di misura utilizzate in termodinamica classica. Inun articolo del 1957 [9], E.T. Jaynes espone una visione secondo cui l’entropiatermodinamica e un “applicazione” della teoria dell’informazione: essa risultaessere proporzionale alla quantita dell’informazione di Shannon necessaria a de-finire un dettagliato stato microscopico del sistema, che rimarrebbe ignota dauna descrizione unicamente in termini delle variabili macroscopiche della ter-modinamica classica. Consideriamo, infatti, un sistema fisico in date condizionidi temperatura, pressione e volume, e stabiliamone il valore dell’entropia; inconnessione e possibile definire il grado di ordine e quindi l’ammontare dellenostre informazioni (in senso microscopico). Supponiamo ora di abbassare latemperatura lasciando invariati gli altri parametri: osserviamo che la sua en-tropia diminuisce poiche il suo grado di ordine aumenta (ordine statico, checorrisponde alla mancanza di movimento) e con esso il nostro livello d’informa-zione. Al limite, alla temperatura prossima allo zero assoluto, tutte le molecolesono “quasi” ferme, l’entropia tende al minimo e l’ordine e il massimo possibilee con esso si ha la massima certezza d’informazione; infatti non esiste piu alcunaalternativa fra cui scegliere.Fu fatta in ogni caso molta attenzione a non generalizzare la relazione tra infor-mazione ed entropia a qualsiasi tipo di sistema, tale relazione vale infatti soloin un contesto dove essi possono essere legati per mezzo delle configurazionimicroscopiche del sistema considerato. Per Shannon, l’informazione, nel sensodella teoria dell’Informazione, e una caratteristica dei sistemi che vengono usatiper comunicare. L’informazione di un messaggio non puo mai aumentare oltre ilvalore che aveva al momento della sua trasmissione. Essa puo, invece, diminuireper colpa di svariati processi che conducono ad una perdita parziale o ad undeterioramento del messaggio (basta pensare ai disturbi nelle trasmissioni radioo nei cellulari). Il collegamento quindi e abbastanza immediato, in entrambi icasi l’entropia (termodinamica o di informazione) e una misura di quanto si siadegradato il contenuto utilizzabile del sistema, un contenuto energetico nel casotermodinamico, un contenuto di informazione nel caso di un messaggio. L’in-formazione gioca il ruolo dell’energia disponibile, che diminuisce con il passaredel tempo.Citiamo infine il famoso Principio di Landauer, secondo cui l’eliminazione di bitdi informazione a temperatura T produca una quantita di calore che non possaessere diminuita oltre un certo limite, che risulta essere ∆Q = kBT log 2 per bit,o in termini di entropia ∆S = kB log 2.Questo principio rappresenta il collegamento tra informazione e termodinamica.L’idea di tale corrispondenza puo essere fatta risalire al “diavoletto di Maxwell”,protagonista di un esperimento mentale proposto nel 1871 da James Clerk Max-well: attraverso la conversione in energia delle informazioni sulla posizione e lavelocita di ogni particella, il demone sarebbe stato in grado di diminuire l’en-tropia del sistema senza eseguire egli stesso un lavoro, in apparente violazionedella seconda legge della termodinamica.Il demone e una creatura intelligente in grado di monitorare singole molecoledi un gas contenuto in due camere adiacenti inizialmente alla stessa tempera-tura. Alcune delle molecole andranno piu velocemente della media, altre piu
17

lentamente. Aprendo e chiudendo in modo opportuno una saracinesca delledimensioni delle molecole presente nella parete divisoria, il demone potrebberaccogliere le molecole piu veloci (calde) in una delle camere e quelle piu lente(fredde) nell’altra. La differenza di temperatura cosı creata potrebbe quindiessere utilizzata per alimentare un motore e produrre lavoro utile. Un’elegante
Figura 2.3: Il demone all’opera
soluzione del paradosso della apparente violazione della seconda legge, sul qualesi sono esercitati numerosi brillanti scienziati, arrivo nel 1961 quando il fisicoRolf Landauer fece notare che, durante un ciclo termodinamico completo, lamemoria del diavoletto, necessaria a registrare coordinate e velocita di ciascunamolecola e quindi agire di conseguenza, deve essere periodicamente resettataal suo stato iniziale eseguendo un’operazione di cancellazione (a meno di nonpresupporre un’improbabile memoria infinita).Sviluppando con Charles Bennett un suggerimento di Leo Szilard, Rolf Landauerarrivo a formulare un principio che permetteva di salvare il secondo principiodella termodinamica: quello di cancellare informazioni e un processo dissipa-tivo, vale a dire che quando viene eliminato un bit (classico) di informazione,viene con cio stesso necessariamente prodotta una certa quantita di calore chenon puo essere ridotta al di sotto di una soglia, il cosiddetto limite di Landauer.Una diretta conseguenza di questa trasformazione irreversibile e che l’entropiadell’ambiente aumenta di una quantita finita.
18

Capitolo 3
L’operatore densita
3.1 Insiemi puri e miscele
Parlando di sistemi quantistici, si fa abitualmente riferimento, esplicito od im-plicito, ad un loro stato o alla funzione che descrive un tale stato. Nello spaziodelle configurazioni, questa e la funzione d’onda, ampiezza di probabilita di po-sizione da cui si estraggono tutte le informazioni che e possibile ottenere sulsistema da essa descritto, quali valori medi, possibili risultati per la misura diuna arbitraria grandezza e relative probabilita. Da un punto di vista pratico,pero, per ottenere informazioni di questo genere e necessario avere a disposizio-ne un gran numero di sistemi identici, tutti nello stesso stato completamentedefinito. Cosı, per esempio, se vogliamo studiare il processo di diffusione diun elettrone contro un bersaglio, per descrivere il sistema elettrone-bersaglioutilizziamo una funzione d’onda, associata ad un singolo elettrone, e la sua co-noscenza ci permette, tra l’altro, di valutare teoricamente la sezione d’urto delprocesso, ma per ottenere quest’ultima sperimentalmente dobbiamo servirci diun fascio di moltissimi elettroni, tutti nelle medesime condizioni iniziali (stessomodulo e stessa direzione della velocita iniziale), da spedire contro il bersaglio,analizzandone poi lo sparpagliamento seguıto all’interazione. E chiaro che, perquanto ben fatti siano gli apparati acceleratori, monocromatori e focalizzatoriutilizzati per preparare il fascio, questo non risultera mai costituito da elettroniche si trovino esattamente nelle stesse condizioni e quindi che il pensare un uni-co elettrone come rappresentativo di tutto il fascio implica comunque un certogrado di idealizzazione. Ora, per la maggior parte delle considerazioni, una taleidealizzazione e del tutto lecita e permette di ottenere previsioni in pieno accordocoi dati sperimentali, come dimostrato ampiamente dal trionfo della meccanicaquantistica. Ma non sempre cio e possibile. Supponiamo infatti, per esempio,di aver preparato un fascio di elettroni ben collimato e monocromatizzato senzapero prenderne in considerazione lo spin; se non sono stati utilizzati polariz-zatori, dobbiamo aspettarci che non esista alcuna direzione privilegiata per glispin degli elettroni ed e chiaro che facendo la misura di una sua componentelungo una direzione arbitraria, troveremo statisticamente meta degli spin in su emeta in giu. La funzione d’onda caratterizzante il fascio sara una combinazione
19

lineare degli spinori χ+ e χ− con lo stesso peso statistico e quindi della forma
ψ =1√2
[χ+ + eiαχ−]
Una tale funzione d’onda (con α fissato) o piu in generale , una funzione di ondadel tipo
ψ = aχ+ + bχ−
con a e b arbitrari numeri complessi prefissati e soddisfacenti la condizione dinormalizzazione
|a|2 + |b|2 = 1
descrive un elettrone avente uno spin orientato lungo una ben determinata di-rezione n = n(θ, ϕ) con θ = 2 arccos |a| ≡ 2 arcsin |b| e ϕ = arg(b) − arg(a).Essa puo dunque essere assunta come funzione rappresentativa di uno qualun-que degli elettroni di un fascio completamente polarizzato lungo la direzione ne costituito da una sovrapposizione coerente di stati. Nel caso, invece, di unfascio di elettroni senza nessuna polarizzazione o solo parzialmente polarizzatoci troviamo di fronte a una miscela o sovrapposizione incoerente di stati che,come tale, non puo essere ricondotta ad un singolo elemento della miscela equindi descritta da una singola funzione di stato.In una tale situazione, quando l’informazione che possediamo su un insieme disistemi singoli non e sufficiente a permetterci la descrizione completa di ognu-no di essi, possiamo ancora tentare di descrivere l’insieme, associando ad ognisingolo sistema delle proprieta medie, utilizzando quindi una analisi statisticadell’insieme. Cio viene fatto sfruttando un operatore statistico, introdotto daVon Neumann nel 1927, detto operatore densita, che permette di valutare valorimedi e probabilita di risultati di singole misure quando si ha a che fare con lemiscele, non descrivibili quindi come sovrapposizioni coerenti di stati quantisti-ci.Ad un insieme di sistemi preparati tutti nello stesso stato puro si puo dare allorail nome di insieme puro e per un insieme di tale genere vale ovviamente unadescrizione fatta tramite lo stato di un singolo sistema ovvero la sua funzioned’onda. Il nome di miscela viene invece riservato ad una collezione arbitraria disistemi singoli sulla preparazione dello stato dei quali l’informazione non e, ingenerale, completa.
3.2 Stati puri
Prendiamo dunque in esame un sistema quantistico che si trovi in un stato |s〉normalizzato all’unita che sia puro o costituito da una sovrapposizione coerentedi stati siffatti. La conoscenza dello stato |s〉 o equivalentemente della suafunzione d’onda ψs=〈x|s〉 ci permette di calcolare valori medi e probabilita dirisultati di singole misure relative ad una qualsivoglia osservabile fisica. Indicatocon F l’operatore hermitiano associato a tale osservabile, sia {|ϕn〉} un insiemeortonormale completo di suoi autostati, soddisfacenti l’equazione agli autovalori
F |ϕn〉 = fn |ϕn〉
20

Sappiamo che lo stato |s〉 e certo esprimibile come una sovrapposizione degliautostati di F, come
|s〉 =∑n
anϕn
dove i coefficienti di Fourieran = 〈ϕn|s〉
rappresentano l’ampiezza di probabilita di ottenere l’autovalore fn come risul-tato di una misura di F effettuata sul sistema che si trova nello stato |s〉. Laprobabilita
P (fn) = |an|2
di trovare fn utilizzando la forma esplicita dei coefficienti an ed introducendo ilproiettore Pn dello stato |ϕn〉,
Pn = |ϕn〉 〈ϕn|
puo essere riscritta come
P (fn) = 〈s|ϕn〉 〈ϕn|s〉 = 〈s|Pn|s〉 ≡ 〈Pn〉s (3.1)
e risulta quindi data dal valor medio, calcolato nello stato |s〉, del proiettoredell’autostato |ϕn〉 di F appartenente all’autovalore fn. Per quanto riguarda ilvalor medio di F nello stato |s〉,
〈F 〉s ≡ 〈s|F |s〉 =∑n
|an|2fn
esso puo essere formalmente riscritto come
〈F 〉s =∑n
〈ϕn|s〉 〈s|ϕn〉 fn =∑n
〈ϕn|s〉 〈s|F |ϕn〉
e quindi definendo l’operatoreρ = |s〉 〈s| (3.2)
come〈F 〉s =
∑n
〈ϕn| ρF |ϕn〉 (3.3)
L’operaroe ρ della (3.2) e l’operatore densita relativo al semplice caso di unostato puro. E importante osservare che, mentre uno stato |s〉, anche se nor-malizzato, e sempre definito solo a meno di un fattore di fase moltiplicativoarbitrario, l’operatore densita risulta definito in modo univoco, stante la suastessa definizione di prodotto di un ket per il suo corrispondente bra.La (3.3) mostra che il valor medio nello stato |s〉 di una grandezza fisica asso-ciata ad un operatore F puo essere ottenuto calcolando la traccia della matriceρF:
〈F 〉s = Tr(ρF ) (3.4)
e poiche la traccia di una matrice e invariante per cambiamento del sistemadi riferimento la (3.4) ha validita generale. Calcolando, infatti, la traccia dellamatrice ρF in una base ortonormale arbitraria |αk〉 e sfruttando la completezzadi quest’ultima, si ottiene effettivamente
Tr(ρF ) =∑k
〈αk|s〉 〈s|F |αk〉 =∑k
〈s|F |αk〉 〈αk|s〉 = 〈s|F |s〉 ≡ 〈F 〉s
21

Grazie alla (3.1), la probabilita di ottenere un particolare risultato fn effettuan-do una singola misura di F e esprimibile come la traccia, calcolata in una basearbitraria, del prodotto dell’operatore densita per l’operatore Pn di proiezionedello stato |ϕn〉 ∈ fn :
P (fn) = Tr(ρPn) = Tr(Pnρ)
3.3 Descrizione di una miscela statistica
Consideriamo ora un insieme che sia una miscela statistica di sistemi che possonostare in un certo numero n di stati |s(i)〉, i=1,2,. . . , n, con probabilita pi. Lostato che descrive un tale insieme e definito stato misto. E bene sottolineareche in quanto segue non viene assunto in alcun modo che gli stati |s(i)〉 sianotra loro ortogonali, il che vuol dire, in sostanza, che il loro numero puo superarequello delle dimensioni dello spazio in cui e definita l’azione degli operatori chesi considerano. Essenziale e invece l’ipotesi che essi siano normalizzati ad uno:
〈s(i)|s(i)〉 = 1
i pesi statistici pi sono poi soggetti alle ovvie restrizioni
0 ≤ pi ≤ 1∑i
pi = 1 (3.5)
A questo punto, il valor medio di un’osservabile fisica calcolato nello stato mistoe definito come
〈F 〉 =∑i
pi〈F 〉i
dove gli 〈F 〉i sono i valori medi di F negli stati |si〉. Analogamente, la probabi-lita P (fn) di trovare un certo autovalore fn di F effettuando una misura dellacorrispondente grandezza fisica sara data da
P (fn) =∑i
piP(i)(fn)
Se lo sviluppo del singolo stato puro |si〉 nella base degli autostati {|ρn〉} di Fviene scritto come
|si〉 =∑n
a(i)n |ϕ〉n con a(i)
n = 〈ϕn|s(i)〉
la probabilita di trovare il valore fn, effettuando una misura di F nello stato|si〉, e notoriamente data da
P (i)(fn) = |a(i)n |2 ≡ | 〈ϕn|s(i)〉 |2
Sottolineiamo dunque il fatto che, quando parliamo di insiemi che siano miscelestatistiche di stati quantistici, entrano in gioco due tipi di probabilita che nonbisogna confondere: le prime, indicate con pi, di tipo meramente statistico, sonole probabilita di trovare i vari stati puri nella miscela considerata; le seconde,P (i)(fn) sono legate alla natura quantistica dei sistemi considerati e alla generale
22

incertezza del risultato di una misura. Se ora teniamo conto del fatto che il valormedio di F nello stato |si〉 e dato da
〈F 〉i ≡ 〈s(i)|F |s(i)〉 =∑n
fn|a(i)n |2 ≡
∑n
fn| 〈ϕn|s(i)〉 |2
il valor medio di F mediato sull’insieme considerato, dato dalla (3.3), puo essereespresso come
〈F 〉 =∑i
pi∑n
fn 〈ϕn|s(i)〉 〈s(i)|ϕn〉 =∑n
〈ϕn|∑i
pi |s(i)〉 〈s(i)|F |ϕn〉
viene allora spontaneo definire l’operatore densita di uno stato misto come
ρ =∑i
pi |s(i)〉 〈s(i)| (3.6)
e scrivere quindi il valor medio di F sotto la forma
〈F 〉 =∑n
〈ϕn| ρF |ϕn〉 ≡ Tr(ρF ) = Tr(Fρ)
ottenendo per 〈F 〉 un’espressione formalmente semplicissima e coincidente conquella gia ottenuta (3.4) per il caso dell’insieme puro. E bene sottolineare cheanche in questo caso l’operatore densita risulta definito senza ambiguita datoche i fattori di fase arbitrari, contenuti implicitamente negli stati |si〉, scompa-iono nei proiettori |si〉 〈si| che intervengono nella definizione di ρ. Con questorisultato possiamo riscrivere le probabilita P (fn) come:
P (fn) =∑i
pi 〈s(i)|ϕn〉 〈ϕn|s(i)〉 =∑i
pi 〈s(i)|Pn |s(i)〉 ≡∑i
pi〈Pn〉i ≡ 〈Pn〉
3.4 Proprieta dell’operatore densita
Vediamo ora quali sono le proprieta che caratterizzano l’operatore densita sianel caso degli stati puri che in quello degli stati misti:
1. ρ† = ρ: palesemente mostrata dalla definizione (3.6) poiche i pi ∈ R ∀i;
2. Tr ρ = 1:considerando infatti una generica base ortonormale {|αk〉}, si ha
Tr ρ =∑k
〈αk|∑i
pi |s(i)〉 〈s(i)|αk〉 =∑i
pi∑k
| 〈αk|s(i)〉 |2;
ma l’equazione di Parceval ci dice che∑k
| 〈αk|s(i)〉 |2 = 1
e quindi, tenendo conto che la somma dei pesi statistici e a sua volta ugualea uno (eq. (3.5)), si ha che, anche per una miscela la traccia dell’operatoredensita e unitaria, come gia per un insieme puro.
23

3. ρ e un operatore definito positivo. Per qualsiasi stato |a〉, si ha infatti
〈a| ρ |a〉 =∑i
pi 〈a|s(i)〉 〈s(i)|a〉 =∑i
pi| 〈a|s(i)〉 |2 ≥ 0 (3.7)
4. la (3.7) contiene, in particolare, l’informazione che ρ e un operatore icui autovalori sono tutti positivi o nulli; infatti, essendo ρ hermitiano ,esiste certo una base ortonormale, quella dei suoi autostati, i suoi elementidiagonali sono proprio i suoi autovalori. Se indichiamo con λk un genericoautovalore di ρ e con |λk〉 l’autovettore ad esso appartenente, l’equazioneagli autovalori per ρ viene scritta come
ρ |λk〉 = λk |λk〉 (3.8)
Vale allora per ρ la rappresentazione spettrale
ρ =∑k
λk |λk〉 〈λk| (3.9)
Poiche nella base dei suoi autostati la traccia di ρ coincide con la sommadei suoi autovalori, grazie alla proprieta 2, si ha che∑
k
λk = 1 (3.10)
e quindi gli autovalori, oltre ad essere tutti non negativi, sono anche tuttiminori od uguali ad uno:
0 ≤ λk ≤ 1 ∀k (3.11)
Il caso di un autovalore uguale ad 1 e ovviamente del tutto particolare eimplica che tutti gli altri autovalori siano nulli e si verifica solo se l’insiemeconsiderato e in realta un insieme puro.
5. Tr ρ2 ≤ 1.Abbiamo che la rappresentazione spettrale di ρ2 e
ρ2 =∑k
λ2k |λk〉 〈λk| (3.12)
e in generale risultera ρ2 6= ρ; solo se uno degli autovalori e uguale ad unoe tutti gli altri sono nulli, cioe nel caso di un insieme puro, risulta ρ2 = ρ.La (3.12) mostra poi che
Tr ρ2 ≤ 1
dove il segno di uguaglianza vale solo nel caso particolare di un insiemepuro.
24

3.5 Evoluzione temporale di un insieme statisti-co
Supponiamo che sia nota l’hamiltoniana di un sistema quantistico che sia unamiscela statisica. Poiche l’operatore densita di una generica miscela e definitosecondo la (3.6), se guardiamo le cose dal punto di vista della formulazione diSchrodinger della meccanica quantistica, esso dipende dal tempo; dato che, unavolta definito l’insieme di cui si parla, i pesi statistici pi rimangono immutati neltempo, se l’insieme non interagisce col mondo esterno, la dipendenza dal tempodell’operatore ρ e conseguenza esclusiva del fatto che gli stati |s(i)〉 ≡ |s(i)(t)〉evolvono nel tempo secondo la legge
d
dt|s(i)(t)〉 =
1
i~H |s(i)(t)〉 (3.13)
Grazie alla (3.13), dalla (3.6) si vede che
dρ
dt=∑i
pi
[ 1
i~H |s(i)(t)〉 〈s(i)(t)| − 1
i~|s(i)(t)〉 〈s(i)(t)|H
](3.14)
cioe che ρ ubbidisce alla importante relazione
dρ
dt=
1
i~[H, ρ] (3.15)
La (3.15) , che e la forma differenziale della legge di evoluzione temporale del-l’operatore statistico ρ, viene citata nella letteratura sia come equazione di VonNeumann, sia come equazione di Liouville quantistica. E interessante osservareche la (3.15) puo effettivamente riguardarsi come l’analogo quantistico dell’e-quazione di Liouville , valida nella meccanica statistica classica, per la funzioneρcl(q, p) gia incontrata nel Capitolo 1, che e l’equazione
dρcldt
= [H, ρcl]P
La legge (3.15) per l’evoluzione temporale dell’operatore densita ricorda quellaper l’evoluzione di un operatore A di Heisenberg, che e
dA
dt=
1
i~[A,H] (3.16)
a meno di un segno. In realta le (3.15) e (3.16) sono due relazioni che non hannonulla a che fare l’una con l’altra. La prima vale nell’ambito della formulazionedi Schrodinger della meccanica quantisitca, dove gli operatori sono, in generaleindipendenti dal tempo; l’operatore densita, tuttavia, e un operatore del tuttoparticolare che, essendo costruito con i vettori di stato, ha una ben definitadipendenza temporale, governata dalla equazione di Liouville-Von Neumann(3.15), mentre nella formulazione di Heisenberg risulta indipendente dal tempo.Se guardiamo le cose alla Schrodinger, sfruttando l’invarianza della traccia perpermutazioni cicliche, si ha infatti
d〈A〉dt
= Tr(dρdtA)
=1
i~Tr([H, ρ]A) ≡ 1
i~Tr(HρA− ρHA)
=1
i~Tr(ρAH − ρHA) ≡ 1
i~Tr(ρ[A,H]) =
i
i~〈[A,H]〉
25

Se facciamo invece riferimento alla formulazione di Heisenberg, allora usando la(3.16), abbiamo direttamente
d〈A〉dt
= Tr
(ρdA
dt
)=
1
i~Tr(ρ[A,H]) =
1
i~〈[A,H]〉
Nel caso in cui l’hamiltoniana sia indipendente dal tempo, e facile vedere che laforma integrata della legge di Liouville-Von Neumann per l’operatore densita e
ρ(t) = e−i(H/~)tρ(0)ei(H/~)t (3.17)
infatti, nel formalismo alla Schrodinger, la legge di evoluzione degli stati e
|s(i)(t)〉 = e−i(H/~)t |s(i)(0)〉
e pertanto, esplicitando nella (3.6) la dipendenza temporale, si ha
ρ(t) =∑i
pi |s(i)(t)〉 〈s(i)(t)| =∑i
pie−i(H/~)t |s(i)(0)〉 〈s(i)(0)| ei(H/~)t
= e−i(H/~)tρ(0)ei(H/~)t
3.6 Elementi di matrice dell’operatore densita
L’analisi degli elementi di matrice dell’operatore statistico si presta ad alcuneconsiderazioni interessanti. Scelta una base ortonormale arbitraria {|αk〉}, ilgenerico elemento di matrice dell’operatore ρ e:
ρkl ≡ 〈αk| ρ |αl〉 =∑i
pi 〈αk|s(i)〉 〈s(i)|αl〉 (3.18)
dove i coefficienti dello sviluppo
c(i)k = 〈αk|s(i)〉
hanno significato di ampiezze di probabilita di trovare, effettuando un insiemeopportuno di misure, lo stato |αk〉 nello stato puro |s(i)〉, la (3.18) puo essereallora riscritta come
ρkl =∑i
pic(i)k c
(i)∗
l (3.19)
Gli elementi diagonali di ρ sono
ρkk
=∑i
pi|c(i)k |2 (3.20)
ed il singolo elemento di matrice ρkk rappresenta la probabilita, mediata sullamiscela statistica descritta da ρ, di trovare un generico elemento della miscelanello stato |αk〉. In base a queste considerazioni, gli elementi diagonali ρkk ven-gono indicati come le popolazioni (relative) degli stati |αk〉.A proposito degli elementi non diagonali di ρ, dati dalla (3.19) per k 6= l, i
coefficienti c(i)k c
(i)l dei pesi statistici sono ora dei termini incrociati che tengono
conto degli effetti di interferenza tra gli stati |αk〉 e |αl〉 che possono appari-re se lo stato puro |s(i)〉 e una sovrapposizione lineare coerente contente tali
26

stati. Viceversa, un elemento di matrice ρkl non nullo implica che nell’insiemesopravviva una qualche correlazione, cioe che si manifesti anche nella miscela uneffetto residuo di coerenza tra gli stati |αk〉 e |αl〉. Per questa ragione gli elemen-ti non diagonali di ρ vengono sovente indicati col nome di coerenze. Come giale popolazioni, anche le coerenze dipendono in modo essenziale dalla specificarappresentazione.Le coerenze ρkl tra due arbitrari stati della base scelta sono legate alle popola-zioni degli stati stessi dalle disuguaglianze
|ρkl|2 ≤ ρkkρll
Un cenno specifico va al caso in cui, essendo l’hamiltoniana H dell’insiemeindipendente dal tempo, gli stati della base {|αk〉} siano, in particolare, autostatidi H :
H |αk〉 = Ek |αk〉
le quantita |c(i)k |2 che compaiono nella (3.20) rappresentano allora la probabilitache si ha di trovare l’energia Ek, effettuando una misura di energia nello statopuro |s(i)〉, e gli elementi diagonali della matrice densita, cioe le popolazionidegli stati |αk〉, forniscono la probabilita di trovare un elemento della miscelacon energia Ek. A proposito dell’evoluzione temporale degli elementi di matricedi ρ in una base di autostati dell’hamiltoniana, dalla (3.17) si vede subito chesi ha
ρkk(t) = ρkk(0)
ρkl(t) = ei~ (El−Ek)tρkl(0)
e quindi le popolazioni degli stati stazionari |αk〉 rimangono immutate, mentrele coerenze oscillano nel tempo con frequenze di Bohr.E particolarmente interessante esaminare i possibili aspetti delle matrici densitanei due casi estremi di una miscela completamente caotica e di un insieme puro.
• Nel primo caso in cui nessuno stato puo essere privilegiato rispetto aglialtri, tutti gli stati devono risultare egualmente popolati, qualsiasi sia labase scelta per la rappresentazione. Pertanto, se N e la dimensionalitadello spazio , la popolazione di ogni stato risulta uguale ad 1/N in qual-siasi base, non esistono coerenze e la matrice ρ risulta uguale alla matriceidentita, moltiplicata per il fattore 1/N (ρkk = 1/N e ρkl = 0 per l 6= k)
• Nel caso, invece, di un insieme puro, identificato da uno stato |s〉, con-siderando la rappresentazione dell’operatore statistico ρ = |s〉 〈s| relativoal caso di un insieme puro, in una base arbitraria {|αk〉}, essendo |s〉esprimibile come
|s〉 =∑k
ak |αk〉 con ak = 〈αk|s〉
ne segue che vale l’identita
ρ =∑k,l
aka∗l |αk〉 〈αl|
27

che mostra che, nella base {|αk〉}, gli elementi di matrice di ρ risultano
ρkl ≡ 〈αk| ρ |αl〉 = aka∗l
e tutte le popolazioni e tutte le coerenze sono diverse da zero.Come caso particolare si ha ovviamente quello in cui lo stato puro |s〉faccia parte di una base in cui si va a rappresentare l’operatore ρ. In unatale base tutte le popolazioni, eccettuata una, sono nulle e pure nulle sonoovviamente tutte le coerenze.
3.7 Matrice densita ridotta
Un problema che si incontra sovente e quello relativo alle misure di una osser-vabile definita solo per un sottoinsieme di un dato insieme. Sia dunque A uninsieme facente parte di un piu vasto insieme C e B il complemento di A inC. Supposto di conoscere l’operatore statistico ρ relativo all’intero sistema C,vogliamo predire i risultati di eventuali misure sugli elementi di A ad esempio.Consideriamo separatamente lo spazio degli stati di A, in cui {|αk〉} sia una baseortonormale, e quello di B, in cui {|βl〉} sia a sua volta una base ortonorma-le; nello spazio degli stati di C costituisce allora una base ortonormale {|γkl〉}l’insieme dei vettori
|γkl〉 = |αk〉 |βl〉
Sia ora ρ l’operatore statistico relativo a C e sia FA un operatore hermitiano cheopera solo nel sottospazio degli stati di A, associato ad una osservabile relativaal solo sottoinsieme A; possiamo formalmente definire una estensione di FA cheoperi nell’intero spazio di C, come l’operatore
F = FA · IB
(dove IB indica, ovviamente, l’operatore identita che opera nello spazio di B)e, di conseguenza, definire il valor medio di FA in C, come coincidente colvalor medio di F in C. Usando la completezza della base {|γkl〉} nonche la suadefinizione fattorizzata (3.7), si ha allora
〈FA〉 ≡ 〈F 〉 = Tr ρF =∑kl
〈γkl| ρF |γkl〉 =∑kl
∑jm
〈γjm|FA · IB |γkl〉
=∑kljm
〈αk| 〈βl| ρ |βm〉 |αj〉 〈βm|βl〉 〈αj |FA |αk〉
=∑kj
〈αk|∑l
〈βl| ρ |βl〉 |αj〉 〈αj |FA |αk〉
Se ora battezziamo col nome di ρA la traccia parziale di ρ fatta nel sottospaziodegli stati di B,
ρA = TrB ρ =∑l
〈βl| ρ |βl〉 (3.21)
abbiamo
〈FA〉 =∑kj
〈αk| ρA |αj〉 〈αj |FA |αk〉 =∑k
〈αk| ρAFA |αk〉 = Tr ρAFA (3.22)
28

e il calcolo del valor medio di FA viene cosı ricondotto al calcolo di una traccianel sottospazio degli stati di A. L’operatore ρA definito dalla (3.21) agisce solonello spazio degli stati di A, gode di tutte le prpieta di un operatore statistico epuo essere chiamato operatore statistico ridotto o equivalentemente la sua rap-presentazione come matrice densita ridotta.In generale la matrice densita potra essere espressa nella propria auto-base{|i〉A}, in cui sara diagonale
ρA =∑i
pi |i〉A 〈i|A 0 ≤ pi ≤ 1 e∑i
pi = 1 (3.23)
Uno stato misto avra piu di un termine nella somma e diremo, in questo caso,che il sistema si trovera in una sovrapposizione incoerente di stati.
29

3.8 Postulati della MQ attraverso la matrice den-sita
I postulati della meccanica quantistica sono un insieme di asserti di base cherappresentano un punto di partenza nella formulazione della teoria quantistica informa assiomatica. Tali postulati, ovvero assunzioni accettate a priori, trovanola loro unica giustificazione nell’ abilita di predire e correlare i fatti sperimentalie nella loro applicabilita generale.In questa sezione essi verranno presentati sia in forma canonica sia utilizzandola definizione dell’operatore densita.Attraverso il primo postulato si definisce l’ambiente in cui agisce la MeccanicaQuantistica.
Postulato 1. (Gli stati quantici) A ogni sistema fisico isolato e associatouno spazio vettoriale complesso dotato di prodotto interno (i.e. uno spaziodi Hilbert) che prende il nome di spazio degli stati del sistema. Il sistema edescritto completamente dal suo vettore di stato, che e un vettore unitarionello spazio degli stati del sistema.Il secondo postulato fornisce una descrizione di come evolve lo stato di unsistema nel tempo:
Postulato 2. (Dinamica) L’evoluzione di un sistema quantistico chiuso e de-scritta da una trasformazione unitaria. Ovvero lo stato |ψ〉 del sistema altempo t1 e legato allo stato |ψ′〉 del sistema al tempo t2 da un operatoreunitario U che dipende solo dai tempi t1 e t2:
|ψ′〉 = U |ψ〉
Tale operatore U risulta della forma:
U(t1, t2) = e−iH(t2−t1)
~
dove H rappresenta l’hamiltoniana del sistema, supposta essere indipen-dente dal tempo.La richiesta che il sistema fisico descritto sia chiuso significa che non deveinteragire in alcun modo con altri sistemi. Cio in realta e un’idealizzazio-ne, poiche l’interazione, seppur minima, con altri sistemi c’e sempre.Gli effetti prodotti da un’operazione di misura su un sistema quantisticosono descritti dal terzo postulato:
Postulato 3. (Misura) Le misure quantistiche son descritte da un insieme{Mm} di operatori di misura. Tali operatori agiscono sullo spazio deglistati del sistema da misurare. L’indice m si riferisce al risultato che puoessere ottenuto con un esperimento. Se lo stato del sistema quantisticoe |ψ〉 immediatamente prima della misura, la probabilita di ottenere ilrisultato m e data da
p(m) = 〈ψ|M†mMm |ψ〉 (3.24)
e lo stato del sistema dopo la misura e
|ψm〉 =Mm |ψ〉√
〈ψ|M†mMm |ψ〉(3.25)
30

Gli operatori di misura soddisfano l’equazione di completezza∑m
M†mMm = 1 (3.26)
che deriva dalla normalizzazione delle probabilita
1 =∑m
p(m) =∑m
〈ψ|M†mMm |ψ〉 (3.27)
L’ultimo postulato si interessa dell’interazione fra piu sistemi quantisticiquando essi vanno a formare uno spazio piu ampio. In particolare defini-sce come costruire lo spazio degli stati dagli spazi degli stati dei sistemicomponenti il sistema composto.
Postulato 4. Lo spazio degli stati di un sistema fisico composito e il prodottotensore degli spazi degli stati dei sistemi fisici componenti il sistema totale.Inoltre, se si numerano i sistemi da 1 a n, e il sistema i e preparato nellostato |ψi〉, lo stato completo del sistema totale e |ψ1〉 ⊗ |ψ2〉 ⊗ · · · ⊗ |ψn〉.
E possibile fornire una formulazione alternativa, ma matematicamente equiva-lente, della MQ fondata sull’operatore densita o matrice densita. Benche le dueformulazioni siano del tutto equivalenti, in alcune circostanze, come in questolavoro di tesi, risulta piu conveniente riferirsi all’una piuttosto che all’altra.
Postulato 1 Ad ogni sistema fisico isolato e associato uno spazio vettorialecomplesso dotato di prodotto interno (i.e. uno spazio di Hilbert) definitospazio degli stati del sistema. Il sistema e completamente descritto dalsuo operatore densita, che e un operatore positivo ρ con traccia unitariaagente sullo spazio degli stati del sistema. Se un sistema quantistico sitrova nello stato ρi con probabilita pi, l’operatore densita per il sistema edefinito da
ρ =∑i
ρipi
Postulato 2 L’evoluzione di un sistema quantistico chiuso e descritto da unatrasformazione unitaria. Cioe lo stato ρ del sistema al tempo t1 e legatoallo stato ρ
′del sistema al tempo t2 da un operatore unitario U che dipende
solo dai tempi t1 e t2.ρ′
= UρU†
Postulato 3 Le misure quantistiche sono descritte da un insieme {Mm} dioperatori di misura che agiscono sullo spazio degli stati del sistema damisurare. L’indice m si riferisce al possibile risultato della misura durantegli esperimenti. Se il sistema si trova nello stato ρ immediatamente primadella misura , la probabilita di ottenere un valore m e dato da
p(m) = Tr[M†mMmρ] (3.28)
e il sistema decade nello stato
ρm =MmρM
†m
Tr[M†mMmρ](3.29)
31

Gli operatori di misura soddisfano la relazione di completezza∑m
M†mMm = I (3.30)
Postulato 4 Lo spazio degli stati di un sistema fisico composto e il prodottotensore degli spazi degli stati dei sistemi fisici componenti il sistema. Inol-tre, se si numerano i sistemi da 1 a n, e il sistema i e preparato nellostato ρi, lo stato completo del sistema e ρ1 ⊗ ρ2 ⊗ · · · ⊗ ρn.
32

Capitolo 4
Entanglement
Io considero l’entanglement nonuno, ma il tratto piu caratteristicodella MQ, quello che implica ilsuo completo distacco da qualsiasiconcezione classica.
E. Shrodinger
L’entanglement e una proprieta tipica dei sistemi quantistici che non haanalogo classico. Esso si manifesta in sistemi composti da due o piu sottosiste-mi che possono anche essere separati nello spazio da una distanza arbitraria.L’entanglement fa in modo che risultati di misure effettuate separatamente suisottosistemi siano tra loro correlati. Tutta la trattazione che verra fatta d’orain avanti riguardera solo sistemi bipartiti.
4.1 Sistemi bipartiti
I postulati della MQ definiscono uno schema formale perfettamente accetta-bile per la descrizione di fenomeni quantistici. Questi, pero, caratterizzano ilcomportamento dell’intero universo e non valgono quando limitiamo la nostraattenzione ad una fetta del sistema totale. Consideriamo un sistema bipartito,ovvero un sistema costituito da due sottosistemi A e B. Lo spazio di Hilbert delsistema totale sara dato dal prodotto tensoriale dei sottospazi di Hilbert HA eHB
H = HA ⊗HB (4.1)
Siano {|n〉A} e {|m〉B} basi ortonormali degli spazi di Hilbert HA e HB . Labase di H sara dunque data da {|n〉A ⊗ |m〉B} che per brevita verra scrittasemplicemente {|n〉A |m〉B}.Uno stato generico che vive nello spazio H sara
|ψ〉AB =∑nm
anm |n〉A |m〉B con∑nm
|anm|2 = 1 (4.2)
Lo stato si dira non entangled o separabile se lo stato |ψ〉AB puo essere scrittoin forma fattorizzata, ad esempio lo stato di tripletto ( S = 1, Sz = 1) di due
33

spin:|ψ1〉 = |↑↑〉 = |↑〉 ⊗ |↑〉
Lo stato e detto entangled (“aggrovigliato”, “intrecciato”) se non si puo scriverein forma fattorizzata, ad esempio lo stato di singoletto di due spin (S=0)
|ψ0〉 =1√2
(|↑↓〉 − |↓↑〉)
Considereremo osservabili per ognuno dei sottosistemi, che saranno rappresen-tati come operatori agenti sullo spazio corrispondente. Per esempio σA1 staraper σA1 ⊗ IB , che denota un operatore σ1 agente su A e I agente su B. Infat-ti, se abbiamo uno stato separabile |ψ〉 = |ψ1〉 |ψ2〉 , il valore di aspettazione〈ψ|σA1 ⊗ σB2 |ψ〉 si fattorizza in 〈ψ1|σA1 |ψ1〉 〈ψ2|σB2 |ψ2〉 , per cui il risultato diuna misura in entrambi i sistemi sara scorrelata.Per gli stati entangled, al contrario, esistera sempre una coppia di osservabiliin A e in B il cui valore di aspettazione non si puo fattorizzare e per i quali,quindi, i risultati di una misura saranno correlati. Esempi di stati entangledsono i cosiddetti stati di Bell :
|Φ±〉 =1√2
(|0, 0〉 ± |1, 1〉)
|Ψ±〉 =1√2
(|0, 1〉 ± |1, 0〉)
Per capire se un generico stato dello spazio di Hilbert H e separabile o entanglede molto utile un risultato noto come decomposizione di Schmidt.
4.1.1 Decomposizione di Schmidt
Consideriamo uno stato appartenente ad un generico sistema bipartito, espressodalla (4.2). Definendo |n〉B =
∑m anm |m〉B , lo stato |ψ〉AB potra allora essere
riscritto come|ψ〉AB =
∑n
|n〉A |n〉B (4.3)
Supponiamo che la base {|n〉A} sia stata scelta in modo che la matrice densitaρA sia diagonale e quindi esprimibile come nella (3.23). Per definizione, inoltre,sappiamo che la matrice densita ridotta sara ottenibile tracciando via i gradi diliberta del sistema B. Avremo quindi
ρA = TrB(|ψ〉AB AB〈ψ|) = TrB(∑nm
|n〉A |n〉B 〈m|A 〈m|B)
=∑k
〈k|B(∑nm
|n〉A |n〉B 〈m|A 〈m|B)|k〉B
=∑nm
(∑k
〈k|B |n〉B 〈m|B |k〉B)|n〉A 〈m|A
=∑nm
〈m|B(∑
k
|k〉B 〈k|B)|n〉B |n〉A 〈m|A
=∑nm
〈n|B |m〉B |n〉A 〈m|A
=∑n
pn |n〉A 〈n|A da cui che 〈n|B |m〉B = δnmpm
34

dove |k〉B e una base ortonormale di HB ed abbiamo utilizzato la relazione dicompletezza
∑k |k〉B 〈k|B = I. I vettori |n〉B sono dunque ortogonali tra loro.
Possiamo fare di questi un insieme ortonormale normalizzandoli,
|n′〉B =1√pn|n〉B con pn 6= 0 ∀n
Inserendo tale espressione in (4.3), possiamo riscrivere lo stato del sistema come
|ψ〉AB =∑n
√pn |n〉A |n
′B〉
Questa espressione prende il nome di decomposizione di Schmidt.La matrice densita ridotta del sistema B prendera la forma
ρB = TrA(|ψ〉AB AB〈ψ|) =∑n
pn |n′〉B 〈n′|B
Si noti che le due matrici ρA e ρB hanno gli stessi autovalori non nulli.Dato uno stato puro |ψ〉AB , possiamo quindi associare ad esso un numero inte-ro positivo, il numero di Schmidt ns ≤ min{dimHA, dimHB} che corrispondeal numero degli autovalori non nulli delle due matrici densita ridotte ρA e ρBnonche al numero di addendi presenti nella decomposizione di Shmidt dello sta-to |ψ〉AB
Il numero di Schmidt e una “misura” della non separabilita di uno stato:
• Se ns = 1 lo stato e separabile, ovvero fattorizzabile nel prodotto di duestati appartenenti ai due sottosistemi.
• Se ns > 1 lo stato e detto entangled e non potra essere esprimibile comeprodotto.
Risulta pertanto che:
1. Ogni stato puro che descrive un sistema composto bipartito puo esseredecomposto secondo Schmidt.
2. A ogni stato puro corrisponde il suo numero di Schmidt, ovvero il numerodegli autovalori non nulli comuni ai due operatori ρA e ρB , questo anchenel caso in cui le dimensioni di HA eHB fossero diverse (infatti e il numerodegli autovalori nulli che differirebbe).
3. Se lo stato puro che rappresenta il sistema composto e separabile allora ρAe ρB descrivono sottosistemi puri, se invece e entangled allora descrivonosottosistemi misti. Vediamolo con un esempio:Consideriamo il seguente stato puro in H:
|ψ〉 =|00〉+ |11〉√
2
che si traduce nel seguente operatore densita:
ρ = |ψ〉 〈ψ| = 1
2[|00〉 〈11|+ |00〉 〈00|+ |11〉 〈11|+ |11〉 〈00|]
35

Ora se tracciamo sul sottoinsieme B otteniamo:
ρA =1
2[|0〉 〈0|+ |1〉 〈1|]
che e uno stato misto. L’esempio ci insegna dunque che anche partendo dauno stato puro per il sistema complessivo, il calcolo dell’operatore densitaridotta per un sottoinsieme puo portare ad una miscela di stati.
L’entanglement descrive pertanto una correlazione tra A e B che e inaccessibilea livello locale. Agendo con una misura nel sottosistema A non siamo in gradodi ricevere informazione alcuna per quello che concerne B. La misura di A nonmodifichera inoltre il numero di Schmidt. L’entanglement, quindi, non puoessere creato localmente: se lo stato e inizialmente separabile, lo rimane. L’unicomodo per far sı che A e B diventino entangled e far interagire i due sistemi l’unocon l’altro.
Purificazione
Un altro concetto importante e quello di purificazione. Supponiamo che ci siadato lo stato ρA di un sistema A. E possibile introdurre un altro sistema, chedenoteremo R, e definire uno stato puro |ψ〉AR per il sistema composto AR taleche ρA = TrR(|ψ〉AR AR〈ψ|). Ovvero lo stato puro |ψAB〉 si riduce a ρA quandoponiamo l’attenzione solo sul sistema A. Questa e una procedura puramentematematica, nota come purificazione, che ci permette di associare stati puri astati misti. R in questo caso non ha alcun significato fisico, e un sistema fittiziopreso di riferimento. Per far vedere che la purificazione puo essere fatta per ognistato spieghiamo come si costruisce il sistema R e la purificazione |ψ〉AB per ρA.Supponiamo ρA abbia una decomposizione ortonormale ρA =
∑i pi |i〉A 〈i|A.
Per purificare ρA introduciamo il sistema R che ha lo stesso spazio degli statidi A, con una base di stati ortonormali |i〉R, e definiamo uno stato puro per ilsistema composto
|ψ〉AR =∑i
√pi |i〉A |i〉R
Adesso calcoliamo l’operatore densita ridotto per il sistema A corrispondenteallo stato |ψ〉AB
TrR(|ψ〉AR AR〈ψ|) =∑k
R〈k|(∑ij
√pipj |i〉A A〈j| |i〉R R〈j|
)|k〉R
=∑ij
√pipj |i〉A A〈j|δkiδjk =
∑k
pk |k〉A A〈k| = ρA
Pertanto |ψ〉AR e la purificazione di ρA.Da notare la stretta relazione tra la decomposizione di Schmidt e la purificazione.
36

4.2 Entropia di Von Neumann
Vogliamo ora quantificare il contenuto di entanglement di un sistema composto,ovvero introdurne una misura. L’entanglement non e un’osservabile fisica, mapiuttosto una proprieta dello stato di un sistema quantistico ripartito in piu sot-tosistemi; come tale non e direttamente associabile ad una quantita misurabilesperimentalmente. In tal senso il termine “misura”, pur essendo il piu usatoin letteratura, non e particolarmente preciso e sarebbe piu opportuno parlaredi estimatore. Inoltre, come si vedra anche in seguito, sono state introdottenel corso del tempo diverse misure, definite in modo assiomatico o operativo,ciascuna con opportune caratteristiche.Consideriamo un sistema quantistico nello spazio di Hilbert H e facciamo riferi-mento alla matrice densita ρ di un sistema bipartito, espressa nella sua auto-basecome ρ =
∑i pi |i〉 〈i|. Lo stato del sistema sara tanto piu entangled quanto piu
sparpagliata e la distribuzione delle probabilita pi all’interno di detta matrice.In analogia a quanto visto nel caso classico potremmo associare alla distribu-zione di probabilita pi l’entropia di Shannon (2.7) H(p1, p2, . . . ). Tale quantitapero non e caratteristica soltanto dello stato ρ, ma anche della particolare de-composizione spettrale che ne abbiamo scelto e non e quindi una estensionesoddisfacente al caso quantistico. E stato dimostrato da Von Neumann che unabuona generalizzazione della (2.7) e:
S(ρ) = −Tr(ρ log ρ) (4.4)
che prende il nome di entropia di Von Neumann. Si noti che quando ρ ediagonale l’espressione si riduce a quella di Shannon, ovvero l’entropia di VonNeumann di un operatore densita e l’entropia di Shannon dei suoi autovalori.Se infatti λi sono gli autovalori di ρ allora la condizione di Von Neumann puoessere riespressa come
S(ρ) = −∑i
λi log(λi)
dove si definisce 0 log(0) = 0. E chiaro che per i calcoli e molto piu utilequest’ultima equazione.Nel caso in cui il sistema si trovi in uno stato puro ρ = |ψ〉 〈ψ|, allora S(ρ) =H(p1 = 1) = 0. Viceversa, essendo tutti i termini della sommatoria (2.7) positiviper definizione, se S(ρ) = 0 deve esistere uno stato |α〉 tale che ρ = |α〉 〈α|, cioeil sistema si trova in uno stato puro.
4.2.1 Proprieta dell’entropia di Von Neumann
Prima di elencare tutte le proprieta che caratterizzano l’entropia di Von Neu-mann definiamo le seguenti quantita, analoghe a quelle gia scritte per l’entropiadi Shannon:
• L’entropia relativa di due operatori densita ρ e σ e
S(ρ ‖ σ) ≡ Tr(ρ log ρ)− Tr(ρ log σ) (4.5)
Per quest’ultima vale la disuguaglianza di Klein:
S(ρ ‖ σ) ≥ 0
con l’uguaglianza ⇔ ρ = σ
37

• L’entropia congiunta per un sistema composto AB e:
S(A,B) ≡ −Tr(ρAB log ρAB) (4.6)
dove ρAB e chiaramente la matrice densita associata al sistema AB. L’en-tropia congiunta misura pertanto la totale incertezza che si ha sullo statoρAB .
• L’entropia condizionata risulta essere:
S(A|B) ≡ S(A,B)− S(B) (4.7)
Rappresenta una misura di quanta incertezza si ha, in media, sullo statodel sistema A una volta che sia noto lo stato di B.
• La mutua informazione:
S(A : B) ≡ S(A) + S(B)− S(A,B) = S(A)− S(A|B) = S(B)− S(B|A)(4.8)
misura quanta informazione A e B hanno in comune.
Possiamo ora procedere con l’elenco di tutte le proprieta e con alcune dellerelative dimostrazioni.
1. L’entropia e non-negativa, S(ρ) ≥ 0. E nulla ⇐⇒ calcolata su uno statopuro.
2. L’entropia e invariante sotto trasformazioni unitarie, ovvero S(ρ) = S(UρU†)con U trasformazione unitaria.
3. In uno spazio di Hilbert d -dimensionale l’entropia e al massimo log d.E uguale a log d ⇐⇒ il sistema si trova in una miscela completamenteincoerente ρ = I/d.
4. Se il sistema composto AB si trova in uno stato puro allora S(A) = S(B).
5. Siano pi le probabilita e gli stati ρi abbiano supporto su sottospazi orto-gonali. Allora
S(∑
i
piρi
)= H(pi) +
∑i
piS(ρi) (4.9)
6. Siano pi le probabilita, |i〉 una base ortogonale per il sistema A, e ρi unqualunque set di operatori densita per un altro sistema, B. Allora
S(∑
i
pi |i〉 〈i| ⊗ ρi)
= H(pi) +∑i
piS(ρi) (4.10)
7. Se ρ e σ sono due operatori densita caratterizzanti rispettivamente ilsistema A e B, allora
S(ρ⊗ σ) = S(ρ) + S(σ) (4.11)
8. Sia |Ψ〉AB uno stato puro del sistema composto AB. Allora |Ψ〉AB eentangled ⇐⇒ S(B|A) < 0.
38

9. L’entropia e subadditiva. Se, infatti, due sistemi A e B sono descritti dauno stato ρAB allora l’entropia congiunta per i due sistemi soddisfa ledisuguaglianze:
S(A,B) ≤ S(A) + S(B) e S(A,B) ≥ |S(A)− S(B)|
Questa proprieta non ha una controparte nella teoria dell’informazioneclassica, in cui l’entropia di un sistema non puo mai essere piu bassa diquella delle sue componenti.
10. L’entropia di Von Neumann e anche fortemente subadditiva. Dati tre spazidi Hilbert A,B,C si ha:
S(ρABC) + S(ρB) ≤ S(ρAB) + S(ρBC)
11. L’entropia e una funzione concava dei suoi inputs, ovvero, date le probabi-lita pi tale che
∑i pi = 1 e i corrispondenti operatori densita ρi, l’entropia
soddisfa la disuguaglianza:
S(∑
i
piρi
)≥∑i
piS(ρi)
Vediamo dunque le dimostrazioni:
1. Chiara dalla definizione.
2. Omessa.
3. Il risultato segue dalla non-negativita dell’entropia relativa:
0 ≤ S(ρ‖I/d) = −S(ρ) + log d
4. Dalla decomposizione di Schmidt sappiamo che gli autovalori degli opera-tori densita dei sistemi A e B sono gli stessi. Essendo l’entropia definitacompletamente dagli autovalori si ha che S (A)=S (B)
5. Siano λji e |eji 〉 gli autovalori e i corrispondenti autovettori di ρi. Risulta
che piλji e |eji 〉 sono gli autovalori e gli autovettori di
∑i piρi, e pertanto:
S(∑
i
piρi
)= −
∑ij
piλji log piλ
ji
= −∑i
pi log pi −∑i
pi∑j
λji log λji
= H(pi) +∑i
piS(ρi)
come richiesto.
6. Immediata dalla precedente dimostrazione.
39

7. Siano pi le probabilita, ρi abbiano supporto su sottospazi ortogonali diuno spazio HA e σi su quelli di un altro spazio HB . Siano piλ
ji e |eji 〉A gli
autovalori e i corrispondenti autovettori di piρi, mentre ηki e |gki 〉B auto-
valori e autovettori di σi. Da cui che piλjiηki sono autovalori dell’operatore∑
i ρi ⊗ σi corrispondenti agli autovettori |eji 〉A |gki 〉B . Dalla definizione
dell’entropia di Von Neumann si ha che:
S(∑
i
piρi ⊗ σi)
= −∑ijk
piλjiηki log
(piλ
jiηki
)= −
∑I
pi log pi −∑i
pi∑jk
λjiηki log
(λjiη
ki
)= H(pi)−
∑i
pi
(∑j
λji log λji +∑k
ηki log ηki
)= H(pi) +
∑i
pi(S(ρi) + S(σi))
(4.12)
Definendo Γi ≡ ρi ⊗ σi e usando la proprieta (2), si ha:
S(∑
i
piρi ⊗ σi)
= S(∑
i
piΓi
)= H(pi) +
∑i
piS(Γi)
Confrontando le ultime due equazoni scritte abbiamo che:
S(Γi) = S(ρi) + S(σi)
che prova la proprieta.
8. S(A,B) = S(B,A) = 0 poiche |Ψ〉AB e uno stato puro. Se |Ψ〉AB fos-se entangled allora ρA sarebbe uno stato misto, per cui S(A) > 0, diconseguenza S(B|A) = S(B,A)− S(A) = −S(A) < 0
9. La dimostrazione della subadditivita e una semplice applicazione delladisuguaglianza di Klein, S(ρ) ≤ −Tr(ρ log σ). Ponendo ρ ≡ ρAB e σ ≡ρA ⊗ ρB , osserviamo che
−Tr(ρ log σ) = −Tr(ρAB(log ρA + log ρB))
= −Tr(ρA log ρA)− Tr(ρB log ρB) = S(A) + S(B)
La disuguaglianza di Klein ci da poi S(A,B) ≤ S(A)+S(B). La condizioneρ = σ per la disuguaglianza di Klein fornisce la condizione di uguaglianzaρAB = ρA ⊗ ρB per la subadditivita.Per provare la disuguaglianza triangolare si introduce un sistema R perpurificare i sistemi A e B. Applicando la subadditivita abbiamo che
S(R) + S(A) ≥ S(A,R)
Dal momento che ABR e in uno stato puro, S(A,R) = S(B) e S(R) =S(A,B). La precedente disuguaglianza puo essere riarrangiata per dare
S(A,B) ≥ S(B)− S(A)
Dalla simmetria tra il sistema A e B abbiamo anche che S(A,B) ≥ S(A)−S(B). Combinando quest’ultima con S(A,B) ≥ S(B)−S(A) si ottiene ladisuguaglianza triangolare.
40

10. La dimostrazione sulla subadditivita forte si basa sul teorema di Lieb.Usando la disuguaglianza triangolare si puo far vedere che la disugua-glianza della subadditivita forte e equivalente alla seguente:
S(ρA) + S(ρC) ≤ S(ρAB) + S(ρBC)
dove ρAB e ρBC sono le matrici densita ridotte di ρABC . Se applichia-mo l’ordinaria subadditivita alla parte sinistra di questa disuguaglianza econsideriamo tutte le permutazioni di A,B,C otteniamo la disuguaglianzatriangolare per ρABC . Ognuno dei tre numeri S(ρAB), S(ρaC), S(ρBC) eminore o uguale alla somma degli altri due.
11. L’intuizione e che∑i piρi esprima lo stato di un sistema che puo trovarsi
negli stati ρi con probabilita pi, e la nostra incertezza su questa miscela distati potrebbe essere maggiore dell’incertezza media sugli stati ρi. In altreparole, una maggiore ignoranza sulla preparazione dello stato si traducein valori maggiori di S(ρ).Supponiamo dunque che i ρi siano gli stati di un sistema A. Introduciamoun sistema ausiliario B il cui spazio degli stati abbia una base ortonormale|i〉. Definiamo uno stato composto di AB come
ρAB ≡∑i
piρi ⊗ |i〉 〈i|
Per provare la concavita usiamo la subadditivita dell’entropia. Osserviamoche per la matrice densita ρAB si ha
S(A) = S(∑
i
piρi
)S(B) = S
(∑i
pi |i〉 〈i|)
= H(pi)
S(A,B) = H(pi) +∑i
piS(ρi)
e usando la S(A,B) ≤ S(A) + S(B) otteniamo∑i
piS(ρi) ≤ S(∑
i
piρi
)che caratterizza la concavita. L’uguaglianza si ottiene ⇐⇒ tutti gli sta-ti ρi per i quali pi > 0 sono identici, ovvero l’entropia e una funzionastrettamente concava dei suoi inputs.
Considerazioni
Dopo aver elencato le piu importanti proprieta che caratterizzano l’entropia diVon Neumann, vediamo subito come esse si ricollegano al concetto di infor-mazione trattato nel secondo capitolo e in cosa differiscono dalla definizioneclassica. Innanzitutto si e constatato che l’entropia di Shannon cade in difettoin una sua applicazione nel caso quantistico quando gli stati della miscela nonsono tra di loro ortogonali. L’entropia di Von Neumann non dipende dalla basescelta, cosı non e per quella di Shannon. In generale quindi SV N ≤ SShannon.
41

Si e vista inoltre la proprieta subadditiva dell’entropia che risulta al piu parialla somma delle entropie delle parti nel caso in cui queste non siano correlate,sottolineandone quindi l’evidente non estensivita. La proprieta (11) corrispon-de a tutti gli effetti alla nozione di perdita d’informazione nel mescolamento distati diversi e quindi ad un incremento dell’entropia. Fisicamente abbiamo piuinformazione dalla conoscenza di tutte le quantita {pi, ρi} piuttosto che di ρ.Infatti, conoscendo solo ρ non possiamo ricostruire l’insieme originario {pi, ρi}poiche ovviamente ci sono molti modi in cui ρ potrebbe essere decomposto nellesotto-miscele.Concludiamo, infine, questo paragrafo con una breve digressione sulla connessio-ne fra il formalismo dell’operatore densita e la meccanica statistica. Vediamo diricavare l’espressione della matrice densita per un insieme in equilibrio termico.L’assunzione di base che facciamo e che la natura tenda a massimizzare ρ, conil vincolo che la media di insieme della hamiltoniana abbia un valore prescritto.Facciamo quindi vedere che fisicamente ρ puo essere considerata come una mi-sura del disordine.Imponendo che sia massima l’entropia e usando il metodo dei moltiplicatori diLagrange, si ha:
∂∂pi
[S + λ(
∑j pj − 1)
]= 0
∂∂λ
[S + λ(
∑j pj − 1)
]= 0
dove λ e il moltiplicatore di Lagrange inteso ad assicurare la condizione che∑i pi = 1, da cui {
log pi + 1 + λ = 0∑j pj = 1
e quindi ∀i pi = e−(λ+1) = 1N . Tutti gli stati sono quindi equiprobabili e si ha
Smax = −∑ 1
Nlog
1
N= logN
Un sistema privo di vincoli ha pertanto entropia massima quando la matricedensita che lo descrive e proporzionale all’identita, si parla del sistema come diuna miscela completamente incoerente.Ora ricaviamo l’entropia di un sistema all’equilibrio termodinamico con il vin-colo di avere un preciso valore dell’energia. Posto 〈H〉 = U ed essendo il sistemain equilibrio risulta
∂ρ
∂t= 0
dall’equazione (3.15) si ha che [H, ρ] = 0, esiste percio una base comune diautovettori per ρ e H ; essendo anche 〈H〉 = Tr(ρH) e detti pi ed Ei gli autovaloridi ρ ed H risulta
U =∑i
piEi
che, assieme a∑i pi = 1, e un vincolo per l’equazione ai moltiplicatori di
Lagrange
∂
∂pi
[−∑j
pj log pj − λ(∑
j
pj − 1)− β
(∑j
pjEj − U)]
= 0
42

si ricava che log pi = −βEi − λ− 1, da cui
pi = ce−βEi
con c = e−λ−1 e β = 1/kBT , inoltre dalla condizione di normalizzazione
pi =e−βEi∑j e−βEj
La matrice densita risulta allora
ρ =
∑i e−βEi |ψi〉 〈ψi|∑j e−βEj
per cui, nota ρ possiamo ricavare l’entropia del sistema, essendo:
ρ = 1Z
e−βE1 0 . . . 0
0 e−βE2 . . . 0...
.... . .
...0 0 . . . e−βEn
con n numero totali di stati e Z =
∑i e−βEi , la funzione di partizione del siste-
ma. Calcoliamo la quantita ρ log ρ:
ρ log ρ = − 1
Z
e−βE1(βE1 + logZ) 0 . . . 0
0 e−βE2(βE2 + logZ) . . . 0...
.... . .
...0 0 . . . e−βEn(βEn + logZ)
per cui, essendo S(ρ) = −Tr(ρ log ρ) risulta
S(ρ) =1
Z
[ n∑i
e−βEi(βEi + logZ)]
separando la somma e ricordando la definizione di Z si ha
S(ρ) =β
Z
n∑i
Eie−βEi + logZ
ma la somma∑ni Eie
−βEi rappresenta il valor medio U dell’energia, quindi
S(ρ) =U
kBT+ logZ
A questo punto, ricordando che l’energia libera di Helmotz e F = −kBT logZsi ha che S(ρ) = U−F
kBTed essendo F = U − TSTd si ha
STd = kBS(ρ)
Abbiamo quindi mostrato che, a meno di una costante, l’entropia di Von Neu-mann di un insieme canonico corrisponde all’entropia termodinamica.
43

4.3 Entanglement di stati puri
Consideriamo ora un sistema quantistico bipartito definito nello spazio di Hil-bert H = HA ⊗HB e supponiamo che si trovi in uno stato puro |ψ〉AB . Comeabbiamo visto precedentemente, conoscere con certezza lo stato del sistema com-plessivo non implica la conoscenza dello stato dei sistemi che lo compongono.Nel caso in cui A sia entangled con B abbiamo infatti che A si trova nello statomisto ρA = TrB |ψ〉AB AB〈ψ|, quindi S(ρA) 6= 0. Se invece A non e entangledcon B allora ρA e puro e S(ρA) = 0.Si definisce entropia di entanglement di uno stato puro che descrive un siste-ma bipartito l’entropia di Von Neumann di uno dei due sottosistemi che locompongono, formalmente:
E(A,B) = S(A) = S(B)
dove E (A,B), che dipende dallo stato e dalla partizione scelta, esprime l’entan-glement tra i sottosistemi A e B. Si ha quindi
E(|ψ〉AB) = S(ρA) = −TrA(ρA log ρA) (4.13)
Facciamo un esempio di calcolo dell’entropia di entanglement, e lo facciamo peril seguente stato di Bell
|Ψ−〉 =1√2
(|01〉 − |10〉)
Si ha la seguente matrice densita
ρAB = |Ψ−〉 〈Ψ−| = 12
0 0 0 00 +1 −1 00 −1 +1 00 0 0 0
La matrice densita ridotta per questo (e per ogni altro) stato di Bell risulta
ρA = ρB =
(1/2 00 1/2
)da cui che S(ρA) = S(ρB) = 1. Quindi anche partendo da uno stato puro sucui si ha massima informazione, quando consideriamo lo stato di una delle dueparticelle vediamo che c’e una miscela statistica di stati “up” e “down”, ovveroabbiamo perso informazione.
L’ entropia statistica e proporzionale al logaritmo del numero di stati compatibilicon i parametri termodinamici e l’entropia di entanglement ha un’interpretazio-ne analoga in quanto e proporzionale al logaritmo del numero di stati puri incui puo trovarsi il sottosistema A se non si ha alcuna informazione sullo stato diB. Tale numero e anche lo stesso degli stati in cui, a sua volta, puo trovarsi B senon si ha alcuna informazione sullo stato di A. Si noti comunque la differenzaimportante per cui l’entropia statistica e una proprieta dell’ intero sistema atemperatura T mentre l’entropia di entanglement e specifica di un singolo statodel sistema (diviso in due parti).
44

4.4 Stati misti
Nella realta e difficile trovare sistemi quantistici che si trovino in stati puri acausa dell’inevitabile interazione che si instaura alquanto velocemente con altrisistemi. Pertanto, anche se inizialmente il sistema viene preparato in uno statopuro, con il passare del tempo, a causa della decoerenza si perviene ad un me-scolamento di stati.Considerando sempre sistemi bipartiti, riprendiamo innanzitutto alcune impor-tanti definizioni:
• Diciamo che ρ rappresenta uno stato prodotto ogni volta che riusciamo atrovare due stati ρA e ρB negli spazi di Hilbert HA e HB tale che:
ρ = ρA ⊗ ρB
• Uno stato misto e separabile se puo essere scritto come una miscela statisti-ca di stati prodotto, ovvero come una combinazione convessa dei prodottitra gli stati dei due sottosistemi A e B :
ρ =∑i
piρ(i)A ⊗ ρ
(i)B con
∑i
pi = 1
altrimenti si dice che ρ rappresenti uno stato entangled.Dalla precedente si evince che, ad eccezione del caso con un solo valoredi i a probabilita 1, gli stati non siano indipendenti gli uni dagli altri. Acausa del vincolo imposto da
∑i pi = 1 i diversi stati risultano legati tra
di loro, tra i diversi sistemi c’e una correlazione classica.
Cosa importante da osservare e che, mentre ogni stato misto puo essere scrittocome
ρ =∑k
pk |Ψk〉 〈Ψk|
se si richiede che ρ rappresenti uno stato separabile allora necessariamente ogni
|Ψk〉 〈Ψk| deve essere uno stato prodotto, ovvero |Ψk〉 〈Ψk| = ρ(k)A ⊗ ρ
(k)B , o
equivalentemente |Ψk〉 = |ak〉 |bk〉, con |ak〉 e |bk〉 stati in cui possono trovarsirispettivamente A e B. Non sempre pero risulta facile dire se uno stato misto siaseparabile oppure no. Questo e noto come il problema della separabilita. Nellaprossima sezione verranno elencati alcuni risultati che forniscono delle condizio-ni per verificare se uno stato misto sia separabile oppure entangled.
45

4.5 Criteri di separabilita degli stati misti
4.5.1 Criterio PPT o della trasposta parziale positiva
Il criterio di Peres-Horodecki definisce una condizione necessaria affinche unostato ρAB di un sistema bipartito composto AB sia separabile. Nei casi dimen-sionali 2⊗ 2 e 2⊗ 3 tale condizione risulta essere anche sufficiente.Se abbiamo un operatore densita generico ρ che agisce sullo spazioH = HA⊗HB:
ρ =∑ijkl
pijkl |i〉 〈j| ⊗ |k〉 〈l|
la sua trasposta parziale (rispetto a B) e definita come:
ρTB : = I ⊗ ρT =∑ijkl
pijkl |i〉 〈j| ⊗ (|k〉 〈l|)T
=∑ijkl
pijkl |i〉 〈j| ⊗ |l〉 〈k|
Con il termine parziale ci si riferisce al fatto che solo una parte dello stato vienetrasposta.Questa definizione puo essere vista piu chiaramente se scriviamo lo stato comela seguente matrice:
ρ =
A11 A12 . . . A1n
A21 A22 . . . A2n
.... . .
An1 . . . Ann
dove n = dim(HA) e ogni blocco e una matrice di dimensione m = dim(HB).La trasposta parziale si fa semplicemente trasponendo ogni blocco.Il criterio stabilisce che se ρ e separabile allora ρTB possiede autovalori nonnegativi. In altre parole, se ρTB ha un autovalore negativo, e garantito che ρ siauno stato entangled. Se gli autovalori non sono negativi ma la dimensione dellospazio e piu grande di 6 allora tale test e da considerarsi inconcludente.Il risultato e indipendente dalla parte che viene trasposta, poiche ρTA = (ρTB )T .Consideriamo ad esempio la famiglia degli stati di Werner :
ρW = p |Φ+〉 〈Φ+|+ (1− p)4
I con |Φ+〉 =|00〉+ |11〉√
2e p ∈ [0, 1]
Quest’ultimo puo essere visto come una combinazione convessa di |Φ+〉, unostato massimamente entangled, e l’identita, uno stato massimamente misto. Lasua matrice densita e la rispettiva trasposta parziale risultano:
ρ = 14
1− p 0 0 0
0 p+ 1 −2p 00 −2p p+ 1 00 0 0 1− p
ρTB = 14
1− p 0 0 −2p
0 p+ 1 0 00 0 p+ 1 0−2p 0 0 1− p
46

Il suo autovalore piu piccolo risulta (1 − 3p)/4, percio lo stato e entangled perp > 1/4.
DimostrazioneSe ρ e uno stato separabile allora puo essere scritto come
ρ =∑i
piρAi ⊗ ρBi
In questo caso l’effetto della trasposta parziale e banale:
ρTB = I ⊗ ρT =∑i
piρAi ⊗ (ρBi )T
Poiche l’operazione di trasposizione conserva gli autovalori, lo spettro di (ρBi )T
e lo stesso di ρBi , e in particolare (ρBi )T deve essere ancora semidefinita positiva.Pertanto lo deve essere anche ρTB . Questo prova la condizione necessaria delcriterio PPT. Si tralascia la dimostrazione della condizione sufficiente.
4.5.2 Entanglement witness
Gli entanglement witnesses sono operatori che distinguono specifici stati entan-gled dagli stati separabili. Abbiamo visto la definizione di stati separabili (4.4)ed e chiaro che questi formano un insieme convesso, ovvero ogni combinazionelineare di due stati ρA e ρB
aρA + (1− a)ρB con a ∈ [0, 1]
e ancora dentro l’insieme.Useremo la seguente variante del teorema di Hahn-Banach :
Teorema: Siano S1 e S2 due insiemi disgiunti in uno spazio reale di Banache sia uno di essi compatto, allora esiste un funzionale lineare F che separa i dueinsiemi.Questa e una generalizzazione del fatto che, in uno spazio reale euclideo, datoun insieme convesso e un punto fuori da esso, esiste sempre un sottospazio affineche separa i due. Nel nostro caso, dato l’insieme convesso degli stati separabilie uno stato ρE entangled (che giace fuori da esso), dal teorema scritto primadeve esistere un funzionale che separa ρE dagli stati separabili. Ed e proprioquesto funzionale che noi chiamiamo entanglement witness. Il fatto che esistepiu di un iperpiano che separa l’insieme convesso dal punto che giace fuori daesso implica che per un certo stato entangled ci siano piu di uno entanglementwitnesses. Identificando F con un operatore hermitiano abbiamo il seguenteteorema:Teorema: Per ogni stato entangled ρE esiste un operatore hermitiano W taleche:
Tr(WρE) < 0 e Tr(Wσ) ≥ 0
per tutti gli stati separabili σ. Abbiamo immediatamente uno stato σ e separa-bile ⇔ Tr(Wσ) ≥ 0 per ogni operatore limitato W.Se uno stato e separabile chiaramente vale l’implicazione del teorema. D’altra
47

parte, dato uno stato entangled, uno dei suoi entanglement witnesses potrebbeviolare la condizione. Quindi anche se il valore d’aspettazione dell’operatoresuddetto e positivo, cio non implica che lo stato sia non entangled.E stato poi dimostrato da Horodecki che per ogni stato entangled esiste sempreun entanglement witness che lo rileva. Purtroppo non esiste un modo semplicedi trovare un tale witness, il che rende la rilevazione di entanglement in statimisti piuttosto non bananle.
4.5.3 Criterio di riduzione
Il criterio di riduzione consiste nell’applicare la mappa positiva
Λ(σ) = ITr(σ)− σ
ad uno dei sottosistemi. Se si verifica che
(ΛA ⊗ IB)(ρAB) = I⊗ ρB − ρAB ≥ 0
(IA ⊗ ΛB)(ρAB) = ρA ⊗ I− ρAB ≥ 0
allora ρAB e uno stato misto separabile.Cosı come il criterio della trasposta parziale il criterio di riduzione rappresentauna condizione di separabilita necessaria e sufficiente solo nelle dimensioni 2⊗2e 2⊗ 3 , e una condizione necessaria altrimenti.
4.5.4 Criterio di maggiorazione
Un altro criterio sufficiente ma non necessario e il cosiddetto criterio di maggio-razione. Consideriamo le distribuzioni classiche {pj} e {qj} ordinate in modoche per i < j pi ≥ pj e qi ≥ qj . Diremo che la distribuzione {pj} e piu ordinata
della distribuzione {qj} 7−→ {pj} ≺ {qj} se ∀k risulta che∑kj=1 pj ≤
∑kj=1 qj .
Ora, con ρAB =∑j pj |ψj〉AB AB〈ψj | e ρA = TrB(ρAB) =
∑j qj |φj〉A A〈φ| ∗, si
ha che se ρAB e separabile, allora {pj} ≺ {qj} per cui {pj}{qj} implica che ρABe entangled.Gli stati classici, dunque, sono piu ordinati localmente di quanto non lo sia-no globalmente, mentre gli stati entangled possono essere molto ordinati nelcomplesso ma disordinati localmente.
∗e possibile aggiungere zeri all’insieme {qj} in modo che esso abbia lo stesso numero dielementi di {pj}.
48

4.6 Misura di entanglement di stati misti
Una volta che si e capaci di identificare gli stati entangled, e naturale volerquantificare il loro grado di entanglement e definire pertanto una misura diquest’ultima. Una prima domanda che, quindi, potrebbe sorgere e che tipo diproprieta deve possedere tale misura per essere utile. E chiaro che non ci puoessere una risposta completamente oggettiva. Mentre alcune proprieta sembranonecessarie, di altre magari ci si serve solo per preservare una certa immagineintuitiva che non necessariamente e sempre corretta.Elenchiamo dunque una serie di requisiti, o anche assiomi, che si presume unabuona misura E(ρ) di Entanglement di un sistema debba avere:
1. L’entanglement di uno stato separabile deve essere nullo:†
E(ρ =
∑k ckρ
(k)A ⊗ ρ
(k)B
)= 0
2. Poiche gli stati entangled non possono essere creati con operazioni localie comunicazioni classiche (LOCC) sembra ragionevole che nemmeno laquantita di Entanglement di uno stato quantico possa essere aumentatadalle LOCC: E(ρ) ≥ E(ΛLOCC(ρ)). Cio chiaramente non e vero per lecorrelazioni classiche.
3. Invarianza sotto operazioni locali unitarie:
E(UA ⊗ UBρU†A ⊗ U†B) = E(ρ) (4.14)
Dal momento che operazioni unitarie possono essere viste come cambia-menti di base, cio e equivalente a richiedere che l’entanglement non dipen-da dalla scelta della base. Se tale richiesta e soddisfatta, essa induce classidi equivalenza di stati che contengono la stessa quantita di entanglemente che quindi possono essere collegati tramite trasformazioni unitarie.
4. Qualsiasi misura di Entanglement dovrebbe essere continua sullo spaziodegli stati di Hilbert: ‖ρ1 − ρ2‖ → 0⇒ E(ρ1)− E(ρ2)→ 0
5. Una misura di Entanglement deve essere una funzione convessa: E(λρ1 +(1− λ)ρ2) ≤ λE(ρ1) + (1− λ)E(ρ2), dal momento che una combinazioneconvessa ‡ di matrici densita e puramente una sovrapposizione statisticaclassica di stati.
6. Una misura di Entanglement deve essere additiva: E(ρ1 ⊗ ρ2) = E(ρ1) +E(ρ2). Spesso pero e richiesta una forma piu debole di questa proprietache si chiama additivita per stati uguali e subadditivita per quelli diversi:
E(ρ⊗n) = nE(ρ) e E(ρA ⊗ ρB) ≤ E(ρA) + E(ρB) (4.15)
Il problema di fondo nella scelta di una “buona” misura di entanglement e chein genere pochi assiomi non sono sufficienti per determinarla in modo univoco.
†e una condizione necessaria ma non sufficiente. Esistono infatti i cosiddetti ”bound en-tangled states”, che possono avere E = 0 ma non essere distillabili, cio perche violano alcunedisuguaglianze di Bell.‡una combinazione convessa e una combinazione lineare di elementi i cui coefficienti sono
tutti non negativi e sommano a 1.
49

Con questo intendiamo dire che in alcuni contesti, esistono misure distinte, adesempio E ed F che soddisfano le proprieta sopra elencate, che pero non clas-sificano alcuni stati quantici nello stesso modo; ovvero e possibile scegliere duestati ρ e σ tale che E(ρ) > E(σ), ma F (ρ) < F (σ). Per esempio, benche perdue particelle l’idea di uno “stato massimamente entangled” e ben definita, essanon e piu cosı chiara nel caso di piu particelle.Questa potrebbe sembrare un’aggravante ma allo stesso tempo implica che ci siaspazio per esplorare varie misure di entanglement, e quindi che molti problemie questioni aperte saranno oggetto di studio per ancora molto tempo. General-mente, tuttavia, per una data applicazione, vi sono altri assiomi che restringonola natura di una misura di entanglement.
Cio che ci accingiamo a fare in questa sezione e trovare una misura di entan-glement per stati misti. Gia da subito, confrontando le proprieta sopra elencatecon quelle dell’entropia di Von-Neumann, ci si rende conto che quest’ultima nonvada bene a tale scopo poiche e una funzione concava, mentre la (5) richiedeche E(ρ) sia convessa. Possiamo verificare cio considerando un generico statomisto ρ =
∑i pi |Ψi〉 〈Ψi| e valutando il suo entanglement tramite l’entropia di
Von Neumann
ES(ρ) = S(ρA) dove ρA = TrB(ρ) =∑i
pi TrB(|Ψi〉 〈Ψi|) ≡∑i
piρiA
⇒ S(ρA) = S(∑
i
piρiA
)e vista la concavita dell’entropia di Von Neumann risulta:
ES(ρ) = S(ρA) >∑i
piS(ρiA) (4.16)
Quest’ultima disuguaglianza e chiaramente in contraddizione con l’assioma dellaconvessita:
E(ρ) = E(∑
i
pi |Ψi〉 〈Ψi|)≤∑i
piE(|Ψi〉)
e poiche E(|Ψi〉) = S(ρiA), si ha
E(ρ) ≤∑i
piS(ρiA) (4.17)
Le disuguaglianze (4.16) e (4.17) mostrano che ES(ρ) 6= E(ρ), da cui la conclu-sione dell’inefficienza dell’entropia di Von Neumann nel caso di stati misti.
4.6.1 Entanglement of formation & Concurrence
Esso rappresenta una naturale estensione dell’entropia di Von Neumann al casodegli stati misti.Data una matrice densita ρ che descrive un sistema bipartito, consideriamo tuttele sue possibili decomposizioni in stati puri, ossia tutti gli insiemi {|ψk〉 , pk taliche
ρ =∑k
pk |ψk〉 〈ψk| con pk ≥ 0 e∑k
pk = 1
50

Per ognuno degli stati |ψk〉 possiamo costruire la matrice ridotta
ρ(k)A = TrB(|ψk〉 〈ψk|)
per cui l’entropia di entanglement sara
E(|ψk〉) ≡ S(ρ(k)A ) = Tr
(ρ
(k)A log ρ
(k)A
)L’entanglement di formazione dello sato ρ e allora definito come
EF (ρ) := min{∑
k
pkE(|ψk〉)}
(4.18)
In generale la minimizzazione richiesta nella valutazione di (4.18) e estrema-mente difficile da risolvere, tuttavia, nel caso in cui i due sottosistemi sianobidimensionali Wootters nel 1998 e riuscito a trovare un’espressione calcolabilein termini di una grandezza nota come concurrence.Quest’ultima e data da
C = max{0,√λ1 −
√λ2 −
√λ3 −
√λ4} (4.19)
con λ1 ≥ λ2 ≥ λ3 ≥ λ4 autovalori della matrice 4× 4 ρρ′
ρ′
= (σy ⊗ σy)ρ∗(σy ⊗ σy) con σy =(o −ii 0
)dove ρ∗ rappresenta la complessa coniugata di ρ nella base standard {|00〉 , |01〉 , |10〉 , |11〉}.Per un generico stato misto a due qubit si ha un entanglement di formazionedefinito nel seguente modo
EF (ρ) = −x log x− (1− x) log(1− x)
con x = 12 (1 +
√1− C2).
Una importante osservazione e che esiste una corrispondenza uno ad uno trala concurrence e l’entanglement di formazione. La funzione EF (ρ) e mono-tona crescente nell’intevallo [0, 1] e dipendera dalla misura della concurrenceC, anch’essa con valori in [0, 1]. Pertanto potremo usare la concurrence comemisura dell’entanglement, dove 0 indichera uno stato separabile e 1 uno statomassimamente entangled.
4.6.2 Entanglement distillation
Il processo di distillazione consiste nell’aumentare la quantita di entanglementassociata ad uno stato di un certo sistema quantistico composto. Tale processoprevede la trasformazione di un certo numero di coppie debolmente entangled inun numero piu piccolo di coppie massimamente entangled (stati di Bell), usandosolo operazioni locali e comunicazione classica.L’entanglement distillation di uno stato ρ e definito come il massimo rendimentoasintotico di stati massimamente entangled distillati per ogni copia dell’input ρ.
ED(ρ) = max limn→+∞
m(n)
n
51

dove n e il numero degli stati input ρ, m e il numero degli stati massimamenteentangled in output e il massimo viene preso su tutti i possibili protocolli didistillazione (ovvero sequenze di operazioni LOCC).Per definizione tale misura e normalizzata, 0 < ED < 1 e il limite superiore siraggiunge quando ρ e entangled.Se lo stato iniziale e separabile la definizione precedente restituisce correttamen-te zero, mentre si ha 1 se lo stato di partenza e gia massimamente entangled.Esistono tuttavia stati non distillabili, ovvero non separabili, ma con entangle-ment di distillazione nullo. E stato provato che tali stati, detti bound entangledstates, hanno come sottoinsieme gli stati non separabili PPT, ma non e noto sevale il viceversa. Dal momento che si sa molto poco sui protocolli di distillazio-ne, l’entanglement di distillazione e difficilmente computabile. Pero, visto che edefinito come un massimo, limiti inferiori possono essere facilmente ottenuti ap-plicando qualsiasi protocollo si voglia. In questa ottica l’entanglement di forma-zione fornisce un limite superiore all’entanglement di distillazione (EF > ED).Per gli stati misti ρAB e anche utile considerare la seguente differenza:
B(ρAB) ≡ EF (ρAB)− ED(ρAB) (4.20)
nota come bound entanglement che e chiaramente non negativa, B(ρAB) ≥ 0.
4.6.3 Entanglement cost
Siano n gli stati massimamente entangled e m(n) il numero massimo di copie diun certo stato ρAB ottenibile dagli n, si definisce entanglement cost la seguentequantita
EC(ρAB) = sup limn→+∞
n
m(n)
Si prova che EC(ρAB) ≥ ED(ρAB). Risulta poi che EC(ρAB) > 0 ⇔ ρAB eentangled; se ρAB e uno stato puro allora EC(ρAB) = ED(ρAB). E stato poiprovato che
EC(ρAB) = limn→+∞
EF (ρ⊗nAB)
n
Questo e il valore asintotico dell’entanglement di formazione medio. EC ingenerale e difficile da calcolare.A questo punto si potrebbe vedere la differenza EC − ED come “entanglementnon distillabile”.ED e una quantita difficile da calcolare ma l’entropia relativa di Von NeumannER che ora definiremo fornisce un limite superiore ad ED ed e piu rapidamentecalcolabile. Quest’ultima risulta
ER(ρ) ≡ minσ∈F
Tr(ρ log ρ− ρ log σ)
dove F rappresenta l’insieme convesso di tutti gli operatori densita σ separabili.In un certo modo l’entropia relativa di entanglement puo essere vista come unadistanza D(ρ‖σ) tra lo stato entangled ρ e lo stato separabile piu vicino σ.Si ha poi che per gli stati puri ER si riduce all’entropia di Von Neumann delsottosistema. Un calcolo analitico esplicito dell’entropia relativa e possibile soloin casi speciali di alta simmetria.Osserviamo infine che per gli stati puri si ha EF = ED = EC = ER, mentre ingenerale vale EF ≥ EC ≥ ER ≥ ED.
52

4.6.4 Negativity
Tale misura di entanglement risulta essere strettamente legata al criterio PPTdi Horodecki. Abbiamo visto infatti che se uno stato stato e separabile allora latrasposta parziale della sua matrice densita e semidefinita positiva. Sia ρTB latrasposta parziale e ‖ρTB‖1 = Tr
√(ρTB )†ρTB la norma . Se ρTB ha autovalori
{λi} allora questa norma puo essere scritta come∑i |λi|. Sia {µi} l’insieme
degli autovalori negativi di ρTB , esso non e vuoto solo se ρ e entangled. Si haquindi che per uno stato separabile ρ, Tr ρ = ‖ρ‖1 = 1. Questo vale anche perla trasposta parziale, Tr
(ρTB)
= 1, ma dal momento che ρTB puo anche avereautovalori negativi µi la sua norma puo essere scritta nel seguente modo:
‖ρTB‖1 = 1 + 2|∑i
µi|
Da cui la definizione di negativity di uno stato come
N(ρ) :=‖ρTB‖1 − 1
2
che da una misura di quanto ρTB fallisca nell’avere solo autovalori positivi. N(ρ)puo essere positiva solo se abbiamo uno stato entangled, pertanto abbiamo unacondizione sufficiente ma non necessaria: esistono infatti stati entangled per cuitale quantita fa zero.
Considerazioni
In quest’ultima parte del capitolo abbiamo dunque fatto una rassegna di alcunedelle innumerevoli misure di entanglement degli stati misti proposte negli ul-timi anni. Da un punto di vista operazionale abbiamo costruito misure comel’ “entanglement distillation” e l’ “entanglement cost” che quantificano in uncerto senso l’efficienza del riuscire a preparare un insieme di particelle in unparticolare stato. D’altra parte, utilizzando un approccio assiomatico, quindiselezionando in maniera giudiziosa poche proprieta fisicamente motivate di unabuona misura di entanglement, abbiamo definito ad esempio l’ “entanglementof formation” e la “Negativity”.Vogliamo ora soffermarci brevemente sulla corrispondenza che e stata esploratacon successo tra la teoria dell’entanglement e la termodinamica. In [7] Popescu eRohrlich hanno mostrato che ogni processo che usi operazioni locali e comunica-zioni classiche (che preservano il grado di entanglement) deve essere reversibile.Questo e stato stabilito da un argomento analogo alla ben conosciuta caratte-rizzazione termodinamica dell’efficienza ideale di una macchina termica in unciclo di Carnot. Sappiamo infatti che le leggi della natura sono tali che sia im-possibile realizzare un moto perpetuo. Questo principio ( seconda legge dellatermodinamica) permise a Carnot di mostrare che due macchine termiche cheoperino tra due determinate temperature T1 e T2 abbiano lo stesso rendimento.La condizione centrale di una buona misura di entanglement, ovvero E(ρ) ≥∑i piE(ρi) con l’insieme {pi, ρi} prodotto da una LOCC a partire da uno stato
ρ, a volte considerata anche come postulato fondamentale dell’entanglement, sie visto essere analoga alla seconda legge della termodinamica: non puo avvenirealcun aumento netto di entanglement tra sistemi spazialmente separati che siadovuto alle sole operazioni locali e comunicazioni classiche. Considerando anche
53

la proprieta additiva (4.15), e stato dimostrato che l’entropia di Von Neumann el’unica misura di un ordinario entanglement bipartito. Hanno fatto il seguenteragionamento: le trasformazioni locali permesse di stati quantici di un sistemabipartito, con un sottosistema nel laboratorio A e l’altro nel laboratorio B, sonoreversibili solo nel limite di un numero estremamente grande di copie di stati dipartenza. D’altra parte, non c’e alcun modo di definire l’entanglement totale perun numero infinito di stati copie poiche chiaramente potrebbe assumere valoriinfiniti, cosa che non ha senso fisico. Risulta pertanto necessaria una definizioneintensiva e cio richiede che una misura di entanglement di n singoletti sia pro-porzionale a n. In questo caso, l’entanglement di una collezione di k sistemi inun certo stato puro |Ψ〉AB approssima quello di n sistemi ognuno nello stato disingoletto |Ψ−〉, ovvero
E(|Ψ〉AB) = limn,k→+∞
(nk
)E(|Ψ−〉AB)
che viene identificato come “entropia di entanglement” dello stato |Ψ〉AB . Ognimisura dell’entanglement di uno stato puro e allora definita a meno di una co-stante, vale a dire che allo stato di singoletto, preso come unita fondamentale,e associato un e-bit.. Sulla scia di questo risultato ci sono stati i tentativi dicostruire una “termodinamica di entanglement”. L’entanglement gioca dunqueil ruolo analogo al calore in termodinamica mentre la distillazione di stati purientangled corrisponde all’estrazione di lavoro dal calore e viene identificata conl’energia libera.Il “bound entanglement” definito nella (4.20) risulta essere in quest’ottica for-malmente simile all’equazione di Gibbs-Helmotz in termodinamica TS = U−A,dove U e l’energia interna, A e l’energia libera e TS funge da “bound energy”.Richiamando le tre leggi della termodinamica: 1) Il calore e una forma di ener-gia. 2) Per un qualsiasi processo ciclico e impossibile, come unico risultato,convertire il calore , prelevato da un’unica fonte, in lavoro. 3) L’entropia di unsistema tende ad una costante quando T tende a zero. Queste leggi permettonouna trasformazione reversibile di lavoro in calore e viceversa.Adesso consideriamo due agenti nei laboratori A e B che condividono inizial-mente una collezione di coppie di sottosistemi descritti dal prodotto tensorialedi n stati ρ, ρ⊗n = ρ ⊗ · · ·⊗, con n grande, che operano sulle rispettive parti,comunicano usando un canale classico e possono organizzare i loro sottosistemilocali in sottoinsiemi ρi con probabilita pi. Il “postulato fondamentale” dellateoria dell’entanglement afferma che l’entanglement rimanente alla fine di unatale trasformazione LOCC , in media, non puo eccedere quello di partenza. Lacombinazione tra il processo di distribuzione di stati entangled e il processodi distillazione, che forma stati entangled puri a partire da quelli misti, vienevista come un’analogia con il processo di funzionamento di una macchina ter-mica che trasforma calore in lavoro; gli stati bound entangled vengono inveceassociati a quei sistemi termodinamici dai quali non si puo trarre alcun lavoroe sono quelli che, pertanto, contengono un “entanglement completamente di-sordinato”. Dall’analogia con le leggi della termodinamica vengono spontaneele seguenti leggi della “termodinamica di entanglement”: 1) L’entanglement diformazione e conservato. 2) Il disordine di entanglement puo solo aumenta-
54

re. 3) Non si puo distillare uno stato di singoletto con perfetta fidelity§, latrasformazione di entanglement puro in entanglement misto e irreversibile. La(1) corrisponde alla proprieta (4.14). C’e un’analogia per l’estrazione di lavororeversibile: in generale c’e bisogno di piu entanglement (in e-bits) per creareuno stato rispetto a quello che ne potrebbe essere “estratto”. Per la secondalegge della termodinamica ogni sistema possiede piu energia di quella che nepotrebbe essere prelevata, eccetto il caso in cui uno dei due serbatoi si trovia temperatura nulla; lo stesso avviene nella “termodinamica di entanglement”dove per un generale stato misto ρ, ED(ρ) < EF (ρ). Bisogna poi trovare unacorrispondenza tra tutte le altre quantita fondamentali nelle due teorie. Adesempio, se un sistema termodinamico guadagna una quantita ∆Q di energiatermica, allora ci sara un incremento di entropia di ∆S = ∆Q/T . Considerandoche il ruolo dell’entropia e assunto da S(ρ) nella teoria quantistica di entangle-ment non e ad esempio chiaro quale quantita giochi il ruolo della temperatura,T. In ogni caso, una buona “temperatura di entanglement”, T (ρ), per gli statimisti (S(ρ) > 0) della forma T (ρ) = B(ρ)/S(ρ), e richiesta se l’“entropia dientanglement” e proprio S(ρ) e la mancanza di una tale quantita pone questoapproccio fortemente in discussione.In aggiunta a questa difficolta di completare la “termodinamica di entangle-ment” si e visto che l’argomento sull’unicita di una misura di entanglementinduce un’ingiustificata dipendenza dalla scelta dell’unita (e stato scelto il sin-goletto di Bell come portatore di un e-bit di entanglement). C’e infatti unproblema del rapporto di misure: il rapporto ad esempio dell’entanglement diformazione o dell’entanglemnt di distillazione tra due stati differenti, dipendein generale dallo stato scelto portatore dell’unita base di entanglement di rife-rimento. Questa definizione dell’unita di entanglement risulta particolarmenteproblematica nel contesto ad esempio dei sistemi multipartiti, che in questa tesinon sono stati trattati. Per di piu e stato mostrato che non esiste una misu-ra unica di entanglement nel caso di stati misti, problema che non si riscontraper l’entropia termodinamica, con un’unica misura ben definita. Pertanto, laquestione di trovare una perfetta analogia tra la teoria di entanglement e latermodinamica rimane ancora aperta.
§Fidelity e una misura della distinguibilita tra due distribuzioni di probabilita ρ e σ definitacome:
F (ρ, σ) =(
Tr[√√
ρσ√ρ])2
∈ [0, 1]
oppure per il teorema di Uhlmann F (ρ, σ) = max|ψ〉|φ〉
| 〈ψ|φ〉 |2 dove |ψ〉 e |φ〉 sono purificazioni
di ρ e σ rispettivamente
55

Conclusioni
In questo lavoro di tesi si e cercato, come gia preannunciato nell’introduzione,di fare un percorso il piu possibile coerente nella trattazione del concetto dientropia.La si e definita classicamente come funzione di stato, introdotta con l’obiettivodi fornire una espressione quantitativa per una legge fenomenologica generale(la seconda legge della termodinamica), almeno per certi tipi di sistemi, relativaal degrado dell’energia durante le sue trasformazioni, con implicazioni sul suoutilizzo a fini pratici. Tale definizione si riferisce pero alle sole variabili macro-scopiche, come le quantita di calore e la temperatura, e non richiede quindi, inlinea di principio, alcuna ipotesi sulla costituzione microscopica del sistema os-servato. Essendo pero l’entropia espressione di una proprieta intrinseca, non sie mai abbandonata la ricerca di una sua derivazione da proprieta microscopiche.Fu J.C. Maxwell ad approfondire una tale derivazione utilizzando un approcciostatistico.Fu L.Boltzmann, sulla scia di Maxwell, a sottolineare la connessione intima trala seconda legge e la teoria della probabilita. Perseguendo tale prospettiva ar-rivo a concludere che lo stato macroscopico verso cui evolve spontaneamenteun sistema e quello che corrisponde al maggior numero di stati microscopici,ed e dunque il piu probabile a priori. Boltzmann ha sviluppato cosı in terminiquantitativi l’interpretazione probabilistica dell’entropia, e ha ricavato la famo-sa espressione statistica S = k logW . La geniale intuizione di Boltzmann e stataquella di far corrispondere l’equilibrio macroscopico alla distribuzione energeti-ca realizzata dal massimo numero di configurazioni microscopiche, esprimendol’entropia in funzione di tale numero. Noi l’abbiamo visto nella formulazioneche fa utilizzo dello spazio delle fasi in cui viene definita una densita di distri-buzione microcanonica degli stati che permette di ricavare tutte le informazionimacroscopiche del sistema. Nel caso specifico se n’e fatto uso per calcolare ilvolume occupato dall’ensemble microcanonico. L’entropia e risultata essere pro-porzionale proprio a tale volume.Siamo dunque passati a definire l’entropia di Shannon come misura della quan-tita di incertezza o mancanza di informazione sulla struttura effettiva di unsistema. Questo rapporto tra entropia ed informazione ha permesso tra l’altrodi “esorcizzare” il demone di Maxwell.Nel terzo capitolo abbiamo poi definito gli stati puri e gli stati misti: i primicome quegli stati perfettamente noti con un’indeterminazione quantistica sullamisura e i secondi, non del tutto noti, con un’indeterminazione sia classica chequantistica sulla misura. A questo punto abbiamo visto che si potesse adotta-re un unico formalismo per i sistemi quantistici introducendo una definizionegenerale della matrice densita. Se ne sono elencate le proprieta, si e definita
56

la matrice densita ridotta, che sarebbe stata utile in seguito nello studio deisistemi composti, e infine si e mostrato come la meccanica quantistica potesseessere riformulata in termini di questo formalismo. Due sono infatti i vantagginell’usare l’approccio degli operatori densita: permettono la descrizione di si-stemi il cui stato e ignoto e di sottosistemi di un sistema composto noto.Nel quarto capitolo si e introdotto il concetto di entanglement per i sistemibipartiti. Nel caso di stati puri si e visto che uno strumento utile per capirequando essi sono separabili e la decomposizione di Schmidt, che pero non e ingenerale possibile nel caso di sistemi multipartiti. Dopodiche si e definita l’en-tropia di Von Neumann con le relative proprieta e si e visto che essa si riduceall’entropia di Shannon solo nel caso di una miscela di stati ortogonali. Quandol’ortogonalita non e verificata, quindi gli stati non sono piu distinguibili concertezza, l’informazione che si ha su una tale miscela e chiaramente minore. Daqui si e passati a ricavare l’espressione della matrice densita per un insieme inequilibrio termico e si e arrivati a definire il legame diretto tra l’entropia di VonNeumann e quella termodinamica. A questo punto si e introdotto il concetto dimisura del grado di entanglement di un sistema composto e nel caso di stati purisi e visto che una buona misura e rappresentata dall’entropia di entanglement ,definita come entropia di Von Neumann di una delle due matrici densita ridot-ta. Dopo aver caratterizzato gli stati misti, si sono elencate una serie di criteriutili per capire quando quest’ultimi sono separabili, dopodiche si e procedutoin maniera assiomatica ad elencare tutti i requisiti indispensabili richiesti peravere una buona misura di entanglement degli stati misti. Infine si e fatto untentativo di ricollegare con delle analogie la teoria dell’entanglement alla teoriatermodinamica.Si chiude qui questa piccola finestra aperta sul mondo dell’entropia e dell’en-tanglement, con la speranza di essere stata chiara e con la piena consapevolezzadi non esser stata esauriente. Gli aspetti che si sarebbero potuti affrontare inquesto contesto sono innumerevoli ma e stata fatta una scelta strutturale che diconseguenza si e cercato di rispettare.
57

Bibliografia
[1] Rossetti, Cesare, Rudimenti di Meccanica Quantistica,Levrotto&Bella, Torino.
[2] Michael A.Nielsen & Isaac L.Chuang, Quantum Computation andQuantum Information, Cambridge
[3] Kerson Huang, Meccanica Statistica, Zanichelli
[4] Gregg Jaeger, Entanglement, Information, and the Interpretationof Quantum Mechanics. Springer, 2009.
[5] Tzu Chieh Wei, Kae Nemoto, Paul M.Goldbart, William Munro,Frank Verstraete, Maximal entanglement versus entropy for mixedquantum states, Physical Review A67, 022110 , 2003
[6] V.Vedral, M.B.Plenio, M.A.Rippin, P.L.Knight Quantifyingentanglement, [arXiv:quant-ph/9702027] 11 Feb 1997
[7] Sandu Popescu, Daniel Rohrlich, Thermodynamics and theMeasure of Entanglement, [arXiv:quant-ph/9610044], 11 Sept1997
[8] William K.Wootters, Entanglement of formation and concurrence,Quantum Information and Computation, Vol. 1, No.1 (2001)
[9] E.T.Jaynes, Information Theory and Statistical Mechanics,Physical Review, Vol. 106, No.4, 620-630, May 15 1957
58





























