Embed Size (px)
Citation preview


DEDICACES

C’est avec joie que je dédie ce travail......

A mes très chers parents
A qui je dois tout, et pour qui aucune dédicace ne saurait exprimer mon profond amour, ma gratitude, ni mon infinie reconnaissance pour l’ampleur des sacrifices que vous avez fait pour pouvoir nous éduquer et nous voir heureux.
Acceptez ce modeste travail qui n’est qu’un simple hommage de
dévouement, de respect et de piété filiale.
Que Dieu vous donnent santé et longue vie.
A mes très chers frères et sœurs et à ma très chère nièce
Pour l’affection qui nous lie, pour l’intérêt que vous portez à ma vie, pour votre soutien, votre compréhension et vos encouragements.
Veuillez trouver dans ce travail le témoignage de mes sentiments les plus sincères et les plus affectueux.
Que Dieu vous protège et vous procure santé et bonheur.
A mes grands-parents, mes tantes, mes oncles et leurs familles
En témoignage de ma grande affection, mon profond attachement et mon grand respect.
A mes ami(e)s et leurs familles
Trouvez dans ce travail le témoignage de mon amitié
Je vous souhaite tout le bonheur que vous méritez.
A tous ceux que j’aime et que j’ai omis involontairement de citer.

REMERCIEMENTS

Au professeur et président de thèse AMARTI RIFFI Afaf, Professeur et chef de service d’anatomie pathologique au CHU HASSAN II de Fès. Nous sommes infiniment sensibles à l’insigne honneur que vous nous faites en acceptant la présidence de notre thèse. Vous nous avez accueilli avec simplicité, bonté et gentillesse. Nous vous prions de trouver ici le témoignage de notre estime et notre profond respect. Au professeur et rapporteur de thèse BONO Wafae, Professeur et chef de service de médecine interne au CHU HASSAN II de Fès. Nous tenons à vous exprimer toute notre reconnaissance pour l’honneur que vous nous avez fait en nous confiant ce travail. Nous avons bénéficié de votre enseignement tant théorique que pratique et gardons de vous le souvenir d’une enseignante remarquable par sa modestie, ses qualités humaines et sa rigueur dans l’exercice de sa profession. Qu’il nous soit permis de vous témoigner notre admiration et notre grand respect. Au professeur HIDA Moustafa, juge de thèse, professeur et chef de service de pédiatrie au CHU HASSAN II de Fès C’est un grand honneur pour nous de vous voir juger ce travail. Nous vous prions de trouver dans ce travail, l’expression de notre profonde estime et respect. Au professeur ATMANI Samir, juge de thèse, professeur de pédiatrie au CHU HASSAN II de Fès Nous vous remercions d’avoir bien voulu nous faire l’honneur de juger ce modeste travail. Veuillez croire, cher maître, à notre grande estime et profond respect.
Au professeur KHATOUF Mohamed, juge de thèse, Professeur d’anesthésie-réanimation au CHU HASSAN II de Fès
Vous nous faites l’honneur de vous intéresser à notre travail et de bien vouloir siéger parmi le jury de notre thèse. Qu’il nous soit permis de vous exprimer notre reconnaissance et notre estime. A tous mes professeurs de la faculté de médecine et de pharmacie de Fès.

GLOSSAIRE DES ABREVIATIONS

GLOSSAIRE DES ABREVIATIONS
ADP : adénopathie
ALAT : alanine amino-transférase
BHAS : bacteria associated hemophagocytic syndrome)
BK : bacille de Koch
CCMH : concentration corpusculaire moyenne en hémoglobine
CFU-G : Colony Forming Unit-Granulocytic
CFU-GM : Colony Forming Unit Granulo-Monocyt
CFU-M : Colony Forming Unit-Monocytic
CIVD : coagulation intra-vasculaire disséminée
CMH : complexes majeurs d’histocompatibilité
CMV : cytomégalovirus
CRP : C reactiv protein
EBER : Epstein Barr early RNA
EBV : epstein barr virus
EBNA : Epstein- Barr Nuclear Antigen
ECBU : examen cytobactériologique des urines
F-actine : filamentous actin
Fc : fragment constant de l’immunglobuline
FOGD : fibroscopie œsogastroduodénale
G-CSF : Granulocytic Colony Stimulating Factor
g/l : gramme par litre

GM-CSF : Granulo- Monocytic Colony Stimulating Factor
GT : glutamy transférase
HAV : hépatitis A virus
Hb : hémoglobine
HCV : hépatitis C virus
HHV : human herpes virus
HLA : humain leucocyt antigen
HMG : hépatomégalie
HSV : herpes simplex virus
HTLV : Human T cell lymphotropic virus
Ig : immunoglobulines
IgIV : immunoglobulines intraveineuses.
IL : interleukine
INF : interféron
Kg : kilogramme
LAM4 : leucémie aigue myélocytaire
LAM5 : leucémie aigue monocytaire
LCR : liquide céphalorachidien
LDH : lactico-deshydrogénase
LHF : lymphohistiocytose familial
LMP : latent membran protein
LYST : LYSosomal Traffıcking regulator
M-CSF : Monocytic-Colony Stimulating Factor
NFS : numeration formule sanguine

NK : cellules naturel killer
PAF : Platelet Activating Factor
PCR : polymérase chain reaction
PDF : produits de dégradation de la fibrine
PHC : La panniculite histiocytaire cytophagique
PNN : polynucléaires neutrophiles.
PNP : purine nucléoside phosphorylase
RHM : réticulose histiocytaire médullaire
SAM : syndrome d’activation macrophagique
SAP : SLAM Associated Protein
sCD25(=sIL-2R): récepteur soluble de l’interleukine2
SCID : Syndrome d’immunodéficit combiné sévère
SGOT : Sérum GlutamoOxaloacetate Transférase
SGPT : Sérum GlutamoPyruvate Transférase
SH2-D1A : SH2- Domain containing protein 1A
SIS : Small Induced Secreted
SLAM : Signalling Lymphocytic Activation Molecule
SMG : splénomégalie
SNC : système nerveux central
sFasL : ligand soluble de Fas
SIS : Small Induced Secreted
TCA : temps de céphaline active
TDM : tomodensitométrie
TG : triglycérides

Th : lymphocytes T helper
TNF : tumor necrosis factor
TP : taux de prothrombine
UI/l : unités internationales par litre
VCA : viral capsid antigen
VGM : volume globulaire moyen
VHAS : virus associated hemophagocytic syndrome
VIH : virus de l’immunodéficience humaine
VLDL : Very Low Density Lipoprotein
VS : vitesse de sédimentation
VZV : varicelle zona virus
WASp : Wiskott-Aldrich syndrome protein
XLP : X-linked lymphoprolifération

1
TABLE DES MATIERES

2
TABLE DES MATIERES
INTRODUCTION : ................................................................................................ 8
PARTIE I: ETUDE THEORIQUE
I. DEFINITION ...................................................................................................... 11
II. HISTORIQUE ET CLASSIFICATION .................................................................... 13
III. ETIOPATHOGENIE ............................................................................................ 18
A. Rappels sur la cellule macrophagique ........................................................... 18
1. Origine des macrophages .......................................................................... 18
2. Fonction des macrophages ........................................................................ 24
3. L’activation des macrophages .................................................................... 29
B. Mécanisme du SAM ....................................................................................... 31
C. Conséquences de l’activation inappropriée des macrophages ......................... 34
IV. EPIDEMIOLOGIE ............................................................................................... 36
V. DIAGNOSTIC POSITIF ....................................................................................... 38
A. Manifestations cliniques ............................................................................... 38
1. Fièvre ........................................................................................................ 38
2. Organomégalie .......................................................................................... 38
3. Signes cutanés ........................................................................................... 38
4. Signes neurologiques ................................................................................. 39

3
5. Signes pulmonaires ................................................................................... 40
6. Signes digestifs ......................................................................................... 40
7. Autres signes ............................................................................................. 40
B. Examens biologiques ..................................................................................... 41
1. signes hématologiques .............................................................................. 42
a. Numération formule sanguine ................................................................. 42
b. Troubles de l’hémostase ......................................................................... 43
2. Bilan biochimique ...................................................................................... 43
a. Bilan hépatique ....................................................................................... 43
b. Bilan lipidique .......................................................................................... 44
c. Hyperferritinémie .................................................................................... 44
d. Bilan hydroéléctrolytique ......................................................................... 45
e. Autres ..................................................................................................... 45
C. Aspects cytologiques-histologiques ............................................................. 46
1. L’étude médullaire ..................................................................................... 47
a. Le myélogramme .................................................................................... 47
b. Biopsie ostéomédullaire .......................................................................... 50
2. Etude des autres tissus .............................................................................. 51
a. Biopsie ganglionnaire .............................................................................. 51
b. Biopsie hépatique ................................................................................... 51
c. Biopsie splénique .................................................................................... 51
d. Autres .................................................................................................... 51
D. CRITERES DIAGNOSTIQUES ............................................................................ 52

4
VI. DIAGNOSTIC DIFFERENTIEL ............................................................................ 60
1. La leucémie aiguë ......................................................................................... 60
2. L’histiocytose langerhansienne ..................................................................... 60
3. Troubles métaboliques ................................................................................. 60
4. Toute autre étiologie de fièvre prolongée ...................................................... 61
VII. ETIOLOGIES ................................................................................................... 64
A. Les SAM primitifs ......................................................................................... 64
1. La lymphohistiocytose familiale (LHF) ......................................................... 66
2. Le syndrome de Chediak-Higashi : .............................................................. 67
3. Le syndrome de Griscelli : ........................................................................... 67
4. Le syndrome de Purtilo : ............................................................................. 68
B. Les SAM secondaires : .................................................................................... 71
1. Les SAM post-infectieux : ........................................................................... 74
a. Les infections virales : .............................................................................. 76
b. Les infections bactériennes: .................................................................... 79
c. Les infections fongiques et parasitaires : .................................................. 80
2. Les SAM et affections malignes : ................................................................. 80
a. Les hémopathies : .................................................................................... 80
b. Les tumeurs solides : ............................................................................... 82
3. Les affections auto-immunes et les maladies de système: ........................... 82
4. Les SAM associées aux déficits immunitaires acquis : .................................. 84
a. Les SAM secondaires à la chimiothérapie et à la greffe des cellules souches
autologue ou allo-génique: .......................................................................... 84
b. Les SAM et transplantation d’organe : ...................................................... 85
5. Les SAM secondaires à autres étiologies : ................................................... 86
C. Le bilan étiologique: ...................................................................................... 88

5
VIII. TRAITEMENT : ................................................................................................ 93
A. Traitement symptomatique : .......................................................................... 93
B. Traitement étiologique :................................................................................. 94
1. Les moyens : .............................................................................................. 94
a. Moyens thérapeutiques contrôlant l’inflammation excessive du SAM : ....... 94
b. Moyens thérapeutiques supprimant la cause déclenchant le SAM : ............ 99
2. Les indications : .......................................................................................... 99
a. Les SAM primaires : .................................................................................. 99
b. Les SAM secondaires : ............................................................................ 100
IX. EVOLUTION ET PRONOSTIC : ........................................................................ 104
PARTIEII: ETUDE PRATIQUE
I. NOTRE OBSERVATION ...................................................................................... 112
II. DISCUSSION .................................................................................................... 119
A. Discussion de notre observation .................................................................. 119
B. Suggestion d’une conduite à tenir pratique devant un SAM .......................... 122
1. Suspecter le diagnostic du SAM ................................................................. 122
2. Affirmer le diagnostic du SAM .................................................................. 122
3. Evaluer la gravité du SAM ......................................................................... 124
4. Mener une enquête étiologique infectieuse afin de mettre en route un traitement anti-infectieux à large spectre .................................................... 124

6
5. Poursuivre l’enquête étiologique en réalisant un bilan à la recherche d’une pathologie néoplasique, auto-immune ou d’un déficit immunitaire .............. 126
6. Démarrer une stratégie thérapeutique dés le diagnostic ........................... 126
CONCLUSION ................................................................................................. 129
RESUMES .......................................................................................................... 133
REFERENCES .................................................................................................... 137

7
INTRODUCTION

8
INTRODUCTION :
Le syndrome d’activation macrophagique (SAM) est la traduction clinico-
biologique d’une activation macrophagique inappropriée avec hémophagocytose.
Sa physiopathologie ferait intervenir une dysrégulation des lymphocytes T
avec production excessive de cytokines.
Le SAM associe des signes cliniques et biologiques non spécifiques, un examen
cytologique ou histologique permet d’en confirmer le diagnostic.
Ce syndrome peut être primaire principalement dans le cadre d’une
lymphohistiocytose hémophagocytaire familiale ou secondaire à diverses affections:
un lymphome, maladie inflammatoire ou auto-immune, infections,…
Il s’agit d’une pathologie grave, dont le pronostic est sévère et le traitement
encore mal codifié.
Rare et souvent mal diagnostiqué, nous essaierons dans ce travail de mettre la
lumière sur le SAM avec ses différents aspects cliniques, para-cliniques, étiologiques
et thérapeutiques, afin de conclure à une conduite à tenir pratique en cas de
suspicion de SAM. A la fin, nous rapportons un cas de SAM, colligé dans le service
de médecine interne au CHU HASSAN II de Fès.

9
PARTIE I : ETUDE THEORIQUE

10
I. DEFINITION

11
I. DEFINITION :
Les termes du SAM ou syndrome d’activation monocyto-macrophagique ou
syndrome hémophagocytaire ou encore lymphohistiocytose hémophagocytaire
recouvrent la même entité.
La définition du SAM est clinique, biologique et cytohistologique :
Ø Les signes cliniques :
Certains d’entre eux sont constants : la fièvre, l’altération de l’état général,
l’hépato-splénomégalie, d’autres sont moins fréquents notamment des
adénopathies, des signes cutanés et neurologiques.
Ø Les signes biologiques :
Ils sont représentés par des cytopénies sanguines, des troubles de la
coagulation, une altération du bilan hépatique, une hypertriglycéridémie et une
hyperferritinémie.
Ø Les signes anatomo-pathologiques :
Ils consistant en une prolifération médullaire et systémique (foie, rate,
ganglions) de macrophages bénins phagocytant activement les éléments figurés du
sang.
Le SAM est à différencier des proliférations malignes des histiocytes-
macrophages : la leucémie aiguë monocytaire (LAM5), les leucémies
myélomonocytaires aiguës (LAM4) et chroniques, et l’histiocytose maligne vraie.

12
II. HISTORIQUE ET
CLASSIFICATION

13
II. HISTORIQUE ET CLASSIFICATION :
La définition du SAM est passée par plusieurs étapes avant qu’il ne soit conçu
tel qu’il est actuellement.
ü En effet, en 1939, Scott et Robb-Smith décrivent à partir de 10 observations
une entité anatomo-clinique qu’ils nomment « réticulose histiocytaire
médullaire »(RHM). Cette affection touchant l’adulte, sans prédominance de
sexe, est caractérisée par une fièvre, un amaigrissement, une hépato-
splénomégalie, des adénopathies disséminées, biologiquement par une
pancytopénie et sur le plan histopathologique par une prolifération
d’histiocytes érythrophagocytaires. Son évolution est rapidement fatale. [1,2]
ü En 1956 : Marshall A.H.E. constate une anomalie des tests hépatiques à type de
cholestase. [3]
ü En 1962 : Greenberg E. souligne l’intérêt de l’étude de la moelle osseuse dans
le diagnostic de cette affection. [4]
ü En 1966 : Rappaport introduit le terme général d’ « histiocytose maligne »
caractérisée par l’envahissement des tissus par des histiocytes
morphologiquement atypiques et de leurs précurseurs. Selon sa description,
l’affection peut aussi toucher l’enfant, et on peut retrouver une forme clinique
à début cutané et une forme viscérale. Histologiquement, la prolifération est
multifocale (ganglions, foie, rate, moelle osseuse et peau) et est constituée
d’histiocytes de différents degrés de différenciation. [5,6]
Par la suite, les termes de réticulose médullaire histiocytaire et histiocytose
maligne sont employés comme synonymes dans la littérature. Ils correspondent à
une prolifération systémique néoplasique d’histiocytes et de leurs précurseurs.
Par ailleurs, quelques observations permettaient de s’interroger sur une
étiologie possible. Ainsi :

14
ü En 1965, Boake décrit l’apparition de RMH chez un père et son fils à quelques
semaines d’intervalle. [7]
ü Zinkhan, quant à lui, rapporte en 1967, 21observations de nouveaux nés
atteints d’une rougeole congénitale associée à une RHM. [8]
ü En 1975, Chandra.P rapporte 2 observations de RHM, toutes deux réversibles,
ce qui n’avait encore jamais été décrit, l’une associée à une tuberculose et
l’autre sans infection associée. [9]
ü C’est en 1979 que Risdall R.J. identifie le syndrome hémophagocytaire et le
sépare de l’histiocytose maligne permettant ainsi de mieux expliquer les
pathogénies à proliférations histiocytaires. [10]
Selon cet auteur, le syndrome hémophagocytaire est du à une prolifération
macrophagique suite à un dysfonctionnement immunitaire non malin, ayant un
caractère réversible, le distinguant ainsi de l’histiocytose maligne. Risdall définie
alors une entité de nature réactionnelle à une infection virale ou bactérienne et
l’appelle « syndrome hémophagocytaire associé aux virus ou aux bactéries » (VHAS
=virus associated hemophagocytic syndrome, BHAS=bacteria associated
hemophagocytic syndrome). [10,11]
D’autres étiologies ont été décrites, infectieuses : virales, bactériennes,
parasitaires et mycosiques. Mais aussi non infectieuses: immunodéficience,
affections néoplasiques et maladies auto-immunes. Ainsi, en 1987 Chan J.K.C
introduit une appellation plus générale de « syndrome hémophagocytaire
réactionnel ».

15
ü En 1987, l’ « Histiocyte Society » propose une classification permettant,
selon des critères anatomopathologiques, de distinguer en 3 groupes les
pathologies prolifératives histiocytaires [12,13]:
v Classe I : les histiocytoses de langerhans (exprimant des protéines
S100 et Cd1a+).
v Classe II : les histiocytoses avec cellules phagocytaires mononuclées
autres que les cellules de langerhans ou histiocytoses non
langerhansiennes.
v Classe III : les histiocytoses malignes.
Le SAM appartenant au groupe des histiocytoses non langerhansiennes ou
histiocytoses hémophagocytaires.
ü Une classification contemporaine a été établie par FAVARA en 1997 [14,15]:
1) Affections de pronostic variable :
a. Concernant les cellules dendritiques :
§ Histiocytose langerhansienne.
§ Affections secondaires des cellules dendritiques.
§ Xanthogranulome juvénile.
§ Histiocytose solitaire de phénotype dendritique variable.
b. concernant les macrophages :
§ Syndromes hémophagocytaires :
« Lymphohistiocytose hémophagocytaire primaire (sous forme
familiale ou sous forme de cas sporadiques).
« Syndromes hémophagocytaires secondaires.

16
§ Maladie de Rosai-Dorfman [16]: Cette affection touche tous les âges
avec un pic de fréquence à 20 ans et une prédilection pour le sexe
masculin. La clinique est dominée essentiellement par la présence
d’une lymphadénopathie surtout cervicale et bilatérale dans un
contexte fébrile.
La biopsie ganglionnaire apporte le diagnostic de certitude de la
maladie de Rosaï-Dorfman en montrant au niveau des sinus des
ganglions atteints une fibrose avec prolifération d’histiocytes
phagocytant surtout des lymphocytes.
Son évolution est presque toujours spontanément favorable.
§ Histiocytoses solitaires avec phénotype de macrophages.
2) Affections malignes :
a. Concernant les monocytes :
§ Leucémies.
§ Sarcomes monocytaires extra-médullaires.
b. Sarcome histiocytaire provenant des cellules dendritiques :
§ Sarcome histiocytaire provenant des macrophages : ce sont des
tumeurs malignes composées de macrophages ou de cellules
dendritiques.

17
III.ETIOPATHOGENIE
SOMMAIRE :
A. Rappels sur la cellule macrophagique............18
1. Origine des macrophages...........................18
2. Fonctions des macrophages.......................24
3. L’activation des macrophages.....................29
B. Mécanisme du SAM........................................31
C. Conséquences de l’activation inappropriée des macrophages....................................................34

18
III. ETIOPATHOGENIE :
A. Rappels sur la cellule macrophagique :
1. Origine des macrophages : [17]
Le macrophage fait partie du système des phagocytes mononucléés.
Ce système comprend:
-Le macrophage et les cellules accessoires de l’immunité dans les tissus,
-Le monocyte : cellule circulante dans le sang,
-Leurs précurseurs dans la moelle osseuse : cellule souche, monoblaste, pro-
monocyte.
Le macrophage est donc l’équivalent intra-tissulaire du monocyte circulant.
[17]
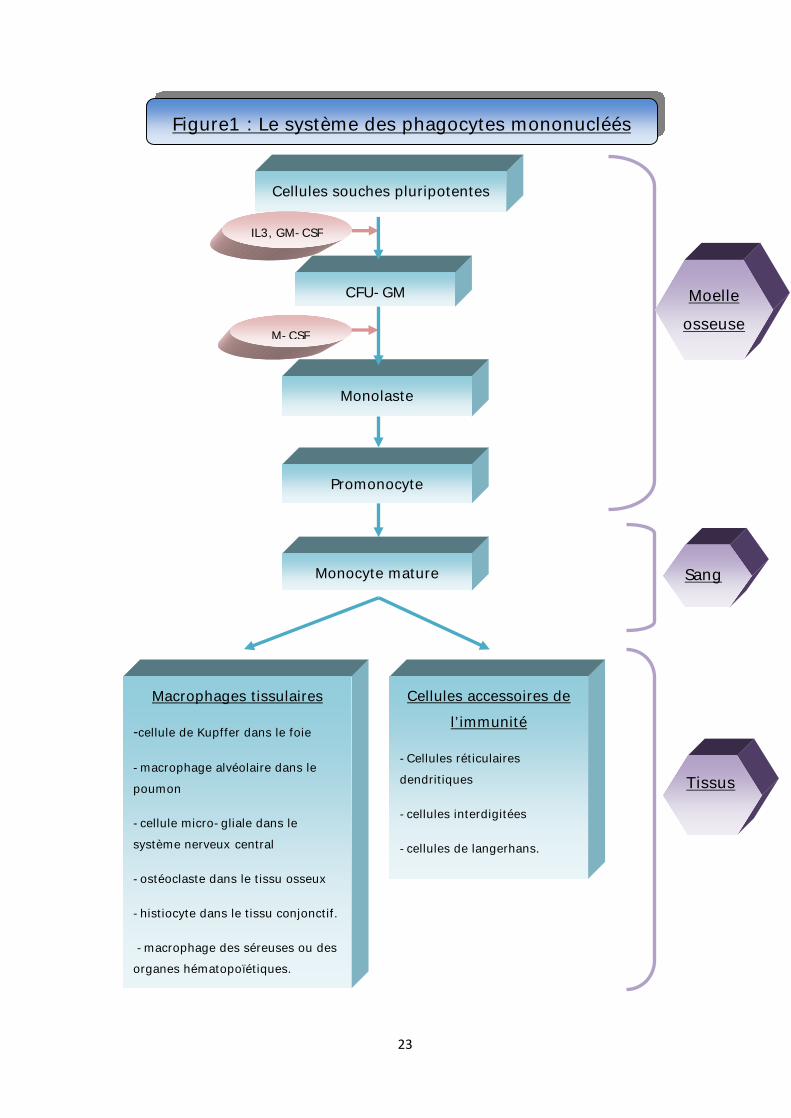
v Le système des phagocytes mononuclées: (figure 1)
a. Dans la moelle osseuse :
Comme pour toute l’hématopoïèse, la monocytopoïése se produit dans la
moelle osseuse. Elle dure environ deux jours. Il n’existe pas de réserves médullaires
importantes. [17]
Ø La cellule souche : CFU-GM :
La CFU-GM (Colony Forming Unit Granulo-Monocyt) est le progéniteur commun
aux lignées granulocytaires et monocytaire.
Ce progéniteur va proliférer sous l’effet des cytokines : IL-3 et GM-CSF
(Granulo- Monocytic Colony Stimulating Factor). Ensuite, la présence supplémentaire
de G-CSF (Granulocytic Colony Stimulating Factor) stimule l'orientation vers les
CFU-G (Colony Forming Unit-Granulocytic) qui donneront naissance à la lignée
neutrophile, alors que la présence supplémentaire de M-CSF (Monocytic-Colony

19
Stimulating Factor) différencie vers les progéniteurs CFU-M (Colony Forming Unit-
Monocytic) puis vers les précurseurs monocytaires. [17]
Ø Le monoblaste :
C’est une cellule de grande taille (25-40µ), son noyau est arrondi, son
cytoplasme est basophile. Le monoblaste se différencie en promonocyte. [18]
Ø Le promonocyte :
Il a un noyau ovoïde, replié sur lui-même ou déjà réniforme, son cytoplasme
est faiblement basophile.
Le promonocyte se divise deux fois pour donner quatre monocytes. [18]
b. Dans le sang :
Ø Le monocyte :
Cette cellule mononucléée est de grande taille (15-20µ). Son noyau est
irrégulier ou réniforme, avec une chromatine dense et filamenteuse. Son cytoplasme
est gris-bleu, semé de fines granulations azurophiles à peine visibles.
Le monocyte produit de nombreuses substances, son produit de sécrétion le
plus important est le lysozyme, mis en évidence par immuno-cytochimie ou par
dosage microbiologique dans le plasma.
Il passe dans le sang et y reste deux à trois jours. C’est une cellule encore
relativement immature, qui va se transformer dans les tissus. [18]
c. Dans les tissus :
Bien que cette différenciation soit artificielle, il est habituel à des fins de
compréhension, de séparer les cellules matures intra-tissulaires en deux
compartiments : les cellules phagocytaires et les cellules accessoires de l'immunité.
[6]

20
Ø Les cellules accessoires de l'immunité :
Elles sont concentrées dans le ganglion, les muqueuses, la peau et la rate. Les
cellules réticulaires dendritiques sont situées dans les centres germinatifs et les
follicules spléniques alors que les cellules réticulaires inter-digitées sont situées
dans les zones para-corticales et les autres zones T des ganglions et de la rate. Les
cellules de Langerhans sont localisées principalement dans la peau et les
muqueuses. Ces cellules ont principalement un rôle de présentation de l'antigène et
d'induction de la réaction mixte lymphocytaire et plus accessoirement des propriétés
de phagocytose. [6]
Ø Les cellules phagocytaires ou macrophages : [17,19]
Le macrophage se distingue des monocytes par une plus grande taille (de
diamètre parfois >70µ). Ils ont un contour irrégulier avec des expansions
cytoplasmiques qui forment de véritables pseudopodes (image 1). Leur survie est
prolongée (de plusieurs semaines au moins). Ils contiennent de grosses granulations
cytoplasmiques, voire des particules phagocytées.
Image 1 : un macrophage phagocytant des bactéries. [19’]

21
Les macrophages adoptent des morphologies différentes, et sont dénommées
de façon variable, en fonction de leur localisation :
*cellule de Küpffer dans le foie.
*macrophage alvéolaire dans le poumon.
*cellule microgliale dans le système nerveux central.
*ostéoclaste dans le tissu osseux.
*histiocyte dans le tissu conjonctif.
*macrophage des séreuses ou des organes hématopoïétiques.
Les macrophages présentent à leur surface membranaire des antigènes et des
récepteurs, indispensables à leurs fonctions et à leur identification
immunocytochimique :
-les marqueurs membranaires comprennent :
• les antigènes de membranes propres aux macrophages,
• les molécules des complexes majeurs d’histocompatibilité (CMH) de classe
II nécessaires à la présentation d’antigènes aux lymphocytes T,
• et les antigènes de différenciation.
-les récepteurs pour les opsonines du sérum :
• Récepteurs pour la portion constante des immunoglobulines. On retrouve
principalement des récepteurs pour les IgG et les IgE, ils permettent aux
macrophages de reconnaître et de détruire les particules recouvertes de ces
immunoglobulines, dites opsonisées.
• Récepteurs CR1 (complement receptor 1) et CR3 (complement receptor3)
reconnaissent les fragments du complément activés C3b et iC3B
respectivement. Les particules recouvertes de complément (opsonisées) se
fixent au macrophage via ces récepteurs.

22
- des sélectines et des intégrines (regroupées de façon plus générale sous le terme
adhésines) qui permettent l’adhésion des macrophages aux cellules environnantes
et à la matrice extracellulaire.
-par ailleurs, on retrouve des récepteurs pour :
• Des facteurs de croissance permettant la différenciation des macrophages.
• Des cytokines provoquant l’activation des macrophages.
• Des facteurs chimiotactiques.
• Des hormones.
23
Monolaste
Cellules souches pluripotentes
CFU-GM
Promonocyte
Monocyte mature
Macrophages tissulaires
-cellule de Kupffer dans le foie
-macrophage alvéolaire dans le poumon
-cellule micro-gliale dans le système nerveux central
-ostéoclaste dans le tissu osseux
-histiocyte dans le tissu conjonctif.
-macrophage des séreuses ou des organes hématopoïétiques.
Cellules accessoires de l’immunité
-Cellules réticulaires dendritiques
-cellules interdigitées
-cellules de langerhans.
IL3, GM-CSF
M-CSF
Moelle osseuse
Sang
Tissus
Figure1 : Le système des phagocytes mononucléés

24
2. Fonction des macrophages :
Le macrophage possède trois fonctions principales : [17, 18, 19,20]
-la phagocytose.
-La sécrétion.
-l’intervention dans la coopération cellulaire immunitaire.
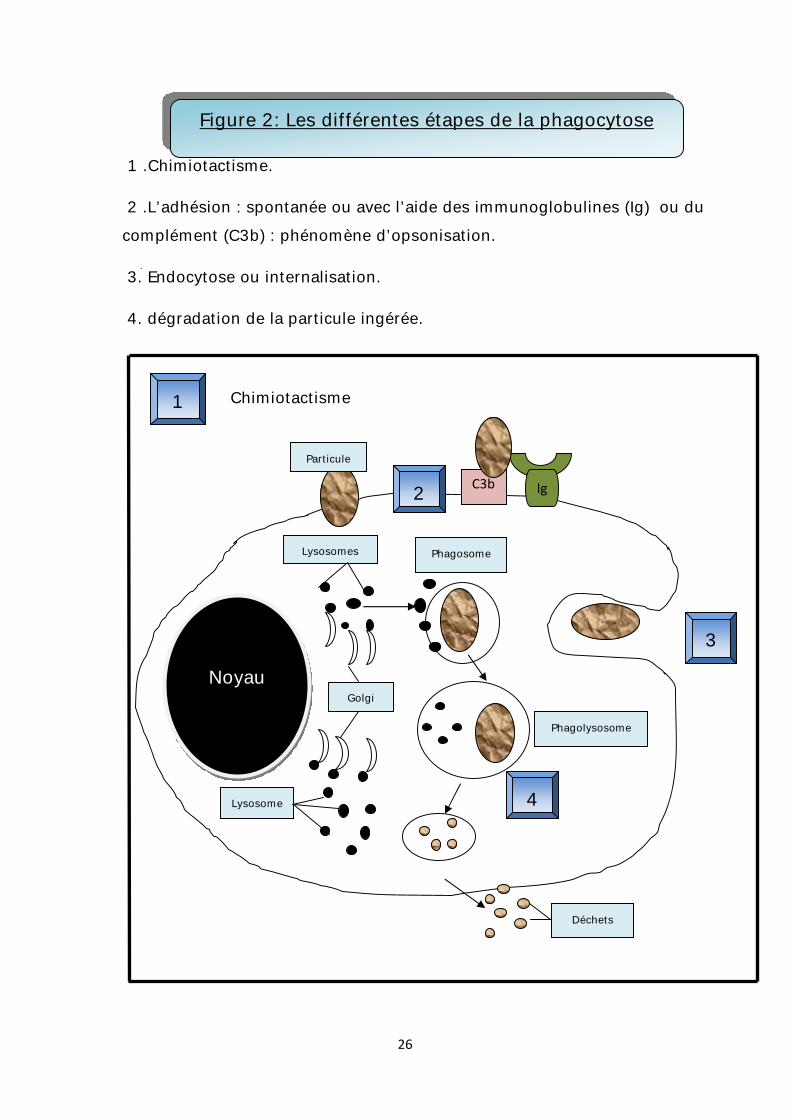
a. La phagocytose : Elle est une fonction essentielle qui permet une épuration et une
désintoxication en débarrassant l’organisme de particules étrangères, de débris
cellulaires, de particules chimiques ainsi que des cellules apoptotiques.
Le processus de phagocytose s’effectue classiquement en différentes étapes:
Ø Chimiotactisme :
Les macrophages se caractérisent par une mobilité extrême et une capacité de
développer considérablement leur membrane pour former des pseudopodes et de
quitter les vaisseaux sanguins (ou diapédèse).
Cette mobilité est orientée par des substances nommées facteurs
chimiotactiques, qui attirent de manière unidirectionnelle les macrophages et les
concentrent sur un territoire donné : c’est ce qu’on appelle le chimiotactisme.
Les facteurs chimiotactiques sont essentiellement le C5a, l’activateur du
plasminogène, des peptides d’origine bactérienne, le PAF (Platelet Activating Factor),
et la famille des cytokines SIS (Small Induced Secreted) récemment identifiée.
Ø L’adhésion :
L’adhésion de la particule à phagocyter est favorisée par son opsonisation, c’est
–à-dire son recouvrement par des immunoglobulines ou du complément, capables
d’interagir avec les récepteurs de surface spécifiques du macrophage.

25
Ø L’endocytose ou internalisation :
Le macrophage émet de grands voiles plasmiques qui entourent la particule à
phagocyter, l'englobe à l’intérieur d'une vésicule de phagocytose appelée :
phagosome.
Ø Dégradation du matériel ingéré :
Le phagosome fusionne avec le lysosome pour créer un phagolysosome où les
lysosomes déversent leur équipement enzymatique (Estérases, phosphatases,
peroxydases, catalases) qui permet la lyse du matériel ingéré.
La figure 2 représente les différentes phases de la phagocytose.
26
1 .Chimiotactisme.
2 .L’adhésion : spontanée ou avec l’aide des immunoglobulines (Ig) ou du complément (C3b) : phénomène d’opsonisation.
3. Endocytose ou internalisation.
4. dégradation de la particule ingérée.
Chimiotactisme
Noyau
Lysosomes
Lysosomes
Golgi
Phagosome
C3b Ig
Phagolysosome
3
2
4
Déchets
Particule
1
Figure 2: Les différentes étapes de la phagocytose

27
b. La sécrétion :
Les macrophages synthétisent et libèrent dans le milieu extracellulaire
plusieurs substances :
ü Les enzymes :
• Les enzymes hydrolytiques telle la phosphatase acide
• Les lysozymes
• L’activateur du plasminogène
ü Les cytokines ou monokines possédant une action à distance sur certaines
cellules cibles :
• L’IL-1 : secrété par le macrophage activé par les endotoxines
bactériennes ou par l’interféron (IFN ) lymphokine produite par
les lymphocytes T.
L’IL-1 a une action systémique : production de fièvre, induction de
la sécrétion hépatique de protéines inflammatoires, relargage de
polynucléaires neutrophiles par la moelle, enfin activation des
lymphocytes.
• L’IL-6 : induit la production par les hépatocytes des protéines de
la phase aiguë de l’inflammation.
• L’IL-8 qui est un facteur chimiotactique pour les polynucléaires,
elle facilite leur recrutement sur le site inflammatoire et en active
les fonctions.
• L’IL-12 et l’IL-18
ü Les interférons.
ü Le TNF-α (Tumor Necrosis Factor alpha) : il partage les mêmes actions
systémiques de l’IL-1 exceptée l’activation des lymphocytes. D’autre part il

28
induit la production d’IL-1 par les macrophages ou par les cellules
endothéliales.
ü Le GM-CSF (Granulo-Monocytic Colony Stimulating Factor) facteur de
croissance de l’hématopoïèse.
ü Fractions du complément.
ü Les facteurs de coagulations : V, VII, IX, X.
ü Molécules transporteuses : transferrine, transcobalamine.
ü Autres : somatomédine, fibronectine ......
c. La coopération des macrophages dans la défense immunitaire : Le macrophage est un des partenaires essentiels de la réaction immunologique,
en étroite coopération avec les lymphocytes.
• Présentation de l’antigène :
Le macrophage, après avoir capté l’antigène le dégrade et l’associe avec les
molécules HLA classe II. Le complexe antigène-HLA classe II est ensuite exprimé à
la surface du macrophage. Ainsi s’établit la reconnaissance spécifique de l’antigène
par le lymphocyte T CD4. De plus le macrophage exprime une IL-1 membranaire
dont la présence est nécessaire pour que la cellule CD4 activée (par l’interaction
avec le complexe antigène-HLA classe II) exprime des récepteurs solubles à l’IL2
(sIL-2R) et entame la synthèse d’IL2. Ces lymphocytes CD4+ secrètent alors l’IL2,
responsable de leur prolifération. [18,20]
• Macrophage et activité anti-tumorale :
Le macrophage est une cellule effectrice pouvant exercer directement un
pouvoir cytotoxique contre de cellules infectées par des micro-organismes mais
également contre des cellules tumorales. L’IFN est susceptible d’être le support
essentiel de cette activité anti-tumorale. La sécrétion de TNF-α serait un autre
mécanisme de cytotoxicité contre les cellules tumorales. [18]

29
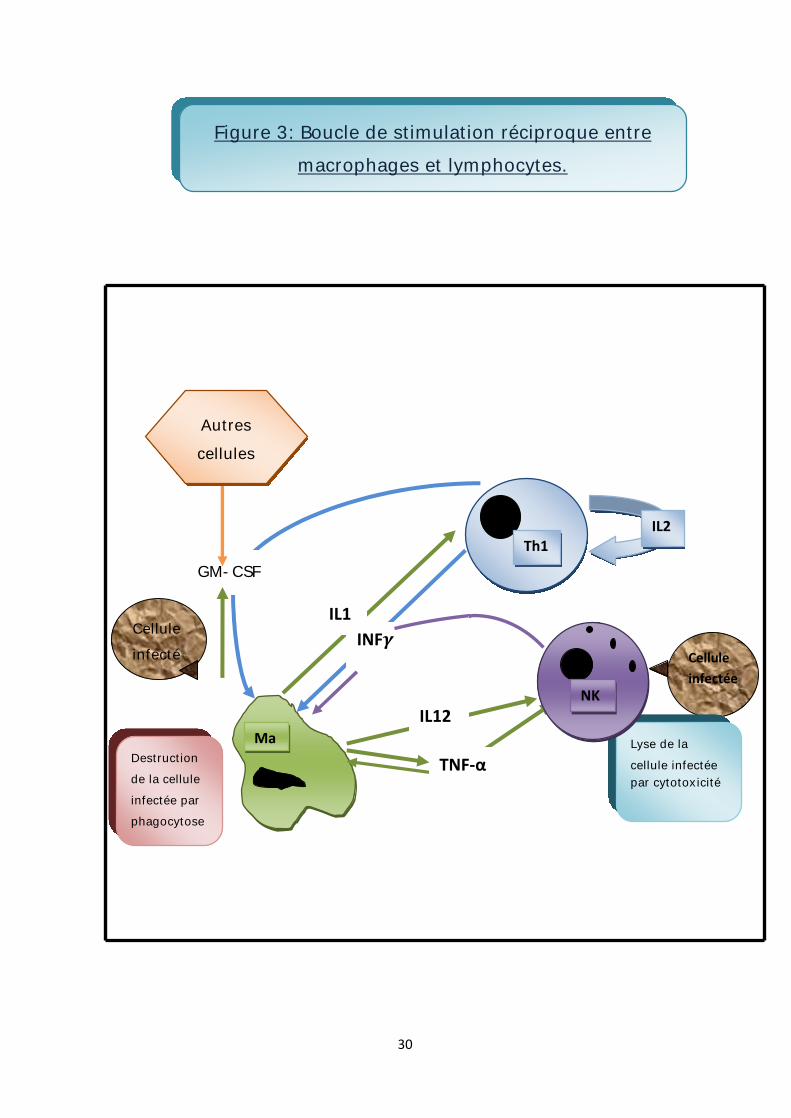
3. L’activation des macrophages :
L’activation des macrophages désigne l’intensification de leurs propriétés
physiologiques, avec un pouvoir important de sécrétion et de phagocytose
bactéricide et tumoricide.
Cette activation est sous la dépendance de plusieurs substances :
-IFN secrété par les lymphocytes Th1 (T helper) activés et par les cellules NK
(naturel killer).
-GM-CSF secrété par les cellules du microenvironnement (cellules endothéliales,
fibroblastes), les macrophages activés et les lymphocytes activés.
-TNF-α secrété par les macrophages.
-les endotoxines bactériennes.
Dans la mesure où l’activation du lymphocyte Th1 est sous le contrôle de l’IL1
produite par les macrophages, il s’établit ainsi un circuit amplificateur de la réponse
immunitaire (figure3). [17, 20,21]
30
Ma
Th1
IL1 INF
IL12
TNF-α
Cellule infecté Cellule
infectée
GM-CSF
IL2
Destruction de la cellule infectée par phagocytosee
Lyse de la cellule infectée par cytotoxicité
NK
Autres cellules
Figure 3: Boucle de stimulation réciproque entre macrophages et lymphocytes.

31
B. Mécanisme du SAM :
La physiopathologie du SAM reste encore en partie mystérieuse, mais l’étude
génétique des formes familiales apportent quelques éléments essentiels dans sa
compréhension. [22]
La coopération entre macrophages, lymphocytes Th1, lymphocytes TCD8
cytotoxiques et Naturel killer (NK), est l’élément central du mécanisme du SAM. Lors
d’une agression par un agent pathogène, s’établit une boucle de coopération entre
ces cellules afin d’augmenter l’efficience de cytotoxicité et la capacité de
macrophagie. Cette réponse s’amplifie en boucle jusqu’à l’élimination de l’agent
pathogène et disparition des cellules présentatrices d’antigène, puis elle s’éteint.
Au cours du SAM, tout se passe comme si cette réponse immunitaire ne pouvait
s’achever et ne cessait de s’amplifier. [1]
L’activation lymphocytaire Th1 se reflète dans l’augmentation des taux
plasmatiques de β2-microglobuline et de récepteur soluble de l’IL-2 (sIL-2R) ainsi
que d’interféron gamma (IFN ) circulant. Les taux plasmatiques de sIL2-R et d’IFN
sont d’ailleurs corrélés à la gravité de la maladie et au pronostic de l’affection. À
l’inverse, les taux plasmatiques d’IL-4 sont effondrés dans ce contexte, montrant
bien le déséquilibre de la balance Th1/Th2 au profit des lymphocytes Th1, impliqués
dans la réponse cellulaire et cytotoxique [23]. Les lymphocytes CD8 sont ainsi en
état d’activation excessive, comme en témoigne l’élévation des taux sanguins de
CD8 soluble et de ligand soluble de Fas (sFasL). [22]
Les monokines produites par les macrophages sont aussi retrouvées à des
titres très élevés : l’IL-1, l’IL-6, l’IL-12, l’IL-18 [24], le TNF-α, et le G-CSF, ainsi que
des facteurs de la coagulation (Facteurs V, VII, IX, X) et de la transferrine. [1] [25]
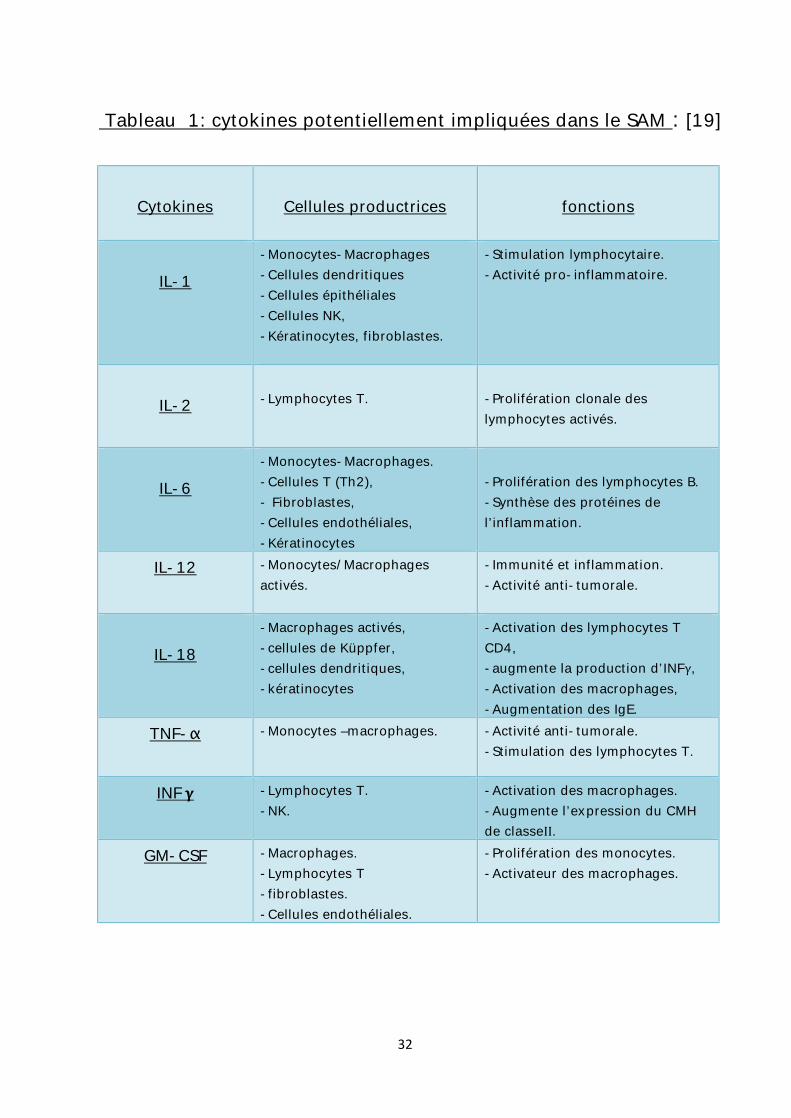
32
Tableau 1: cytokines potentiellement impliquées dans le SAM : [19]
Cytokines
Cellules productrices
fonctions
IL-1
-Monocytes-Macrophages -Cellules dendritiques -Cellules épithéliales -Cellules NK, -Kératinocytes, fibroblastes.
-Stimulation lymphocytaire. -Activité pro-inflammatoire.
IL-2
-Lymphocytes T.
-Prolifération clonale des lymphocytes activés.
IL-6
-Monocytes-Macrophages. -Cellules T (Th2), - Fibroblastes, -Cellules endothéliales, -Kératinocytes
-Prolifération des lymphocytes B. -Synthèse des protéines de l’inflammation.
IL-12 -Monocytes/Macrophages activés.
-Immunité et inflammation. -Activité anti-tumorale.
IL-18
-Macrophages activés, -cellules de Küppfer, -cellules dendritiques, -kératinocytes
-Activation des lymphocytes T CD4, -augmente la production d’INFγ, -Activation des macrophages, -Augmentation des IgE.
TNF-α -Monocytes –macrophages. -Activité anti-tumorale. -Stimulation des lymphocytes T.
INF -Lymphocytes T. -NK.
-Activation des macrophages. -Augmente l’expression du CMH de classeII.
GM-CSF -Macrophages. -Lymphocytes T -fibroblastes. -Cellules endothéliales.
-Prolifération des monocytes. -Activateur des macrophages.

33
Les SAM primaires ont permis de mieux connaitre le mécanisme de cette
pathologie. La découverte par génétique positionnelle des gènes responsables a
complètement modifié la compréhension de la physiopathologie du SAM. Ces
déficits génétiques ont en commun d’altérer la cytotoxicité des lymphocytes T CD8
et NK sans modifier leur capacité d’activation ni leur sécrétion de cytokines. La
plupart de ces déficits intéressent les granules de cytotoxicité, soit leur contenu
effecteur (perforine) soit leur capacité de migration à la membrane cellulaire. [1]
En présence d’un microorganisme le système immunitaire s’active
normalement mais reste inefficient aboutissant à la persistance de l’agent
pathogène dans l’organisme, responsable à son tour de l’activation et de la
prolifération continue des lymphocytes T CD8 produisant des concentrations élevées
d’IFN [26,27]. Le déficit de cytotoxicité entraînerait une perte de la régulation
négative exercée par les cellules cytotoxiques (cellules NK et/ou lymphocytes T CD8)
sur les macrophages, conduisant à un excès d’activation lymphocytaire Th1 et à une
hypersécrétion d’IFN [28]. L’IFN , en activant les macrophages, favorise
l’expansion et l’activation des lymphocytes T CD8 et NK via la sécrétion d’IL-12 et
TNF-α. La boucle s’auto-amplifie ainsi sans fin expliquant la prolifération
macrophagique responsable du syndrome tumoral et de l’hémophagocytose, et
«l’orage cytokinique» responsable des autres signes clinico-biologiques. [1]
La cytotoxicité CD8 n’a jamais été étudiée dans les formes secondaires du SAM
et la cytotoxicité NK ne l’a été que rarement, mais paraît déficitaire. Le déficit de la
fonction cytotoxique NK observé au cours des SAM secondaires semble être le plus
souvent non pas une conséquence du SAM, mais au contraire le facteur
prédisposant. C’est possiblement par le biais du déficit de la cytotoxicité NK, qui
leur est fréquemment associé, que les néoplasies, les hémopathies malignes, les

34
traitements immunosuppresseurs au long cours, le lupus érythémateux disséminé
ou l’arthrite juvénile chronique représentent les situations à risque de SAM
secondaire. [29, 30,31]
C. Conséquences de l’activation inappropriée des macrophages:
§ L’activation des macrophages est responsable à la fois d’un syndrome
inflammatoire général et de la fièvre par la production d’IL-1, de TNF-α et d’IL-
6 qui affectent le centre thermorégulateur de l’hypothalamus. [22]
§ La pancytopénie pourrait être expliquée par un double mécanisme : d’une part
par l’hémophagocytose et d’autre part par l’action myélo-suppressive du TNF-
α et l’IFN . Les cellules hématopoïétiques expriment le Fas sous stimulation de
l’IFN , ce qui les rend sensible à l’action cytotoxique du FasL (Fas Ligand). [22]
§ L’organomégalie est liée à l’infiltration tissulaire par des macrophages activés et
phagocytant les éléments figurés du sang. [22]
§ Les perturbations du bilan hépatique sont la conséquence à la fois de
l’activation macrophagique intra-hépatique (cellules de Küpffer) avec cytolyse
hépatique, et de l’action de l’IFN sur les hépatocytes. [32]
§ L’hypertriglycéridémie classique est, dans le SAM, liée à l’inhibition de la
lipoprotéine lipase par l’association TNF-α et IL-1. [32]
§ L’hyperferritinémie résulterait de l’érythrophagocytose, de l’inflammation
systémique et du dysfonctionnement hépatique engendré. [22]
§ La libération excessive de l’activateur du plasminogène par les macrophages
activés est à l’origine des troubles de coagulation et de fibrinopénie. [33]
§ L’hyponatrémie serait due à une probable sécrétion inappropriée de l’hormone
antidiurétique. [34]

35
IV. EPIDEMIOLOGIE

36
IV. EPIDEMIOLOGIE
Le syndrome d’activation macrophagique est une pathologie dont la prévalence
est probablement sous estimée. [22]
Il peut survenir à tout âge avec une légère prédominance masculine (sexe ratio
entre 1,5 et 2,5).Toutes les populations sont touchées, mais la fréquence des
affections associées peut être variable en fonction de la population considérée.
[35]
Son incidence globale au japon a été estimée en 1994 à 51,7 cas par an,
incluant les SAM pédiatriques et ceux de l’adulte. [22]
Les formes pédiatriques sont souvent mieux documentées et une série suédoise
note une incidence d’un cas annuel par million d’enfants [22].La fréquence du SAM
est moins bien connue chez l’adulte; bien qu’elle soit considérée comme plus rare,
cette notion de rareté est à reconsidérer en fonction de la fréquence de sa survenue
dans le cadre de sepsis sévères ou de pathologies rhumatismales. [29,36]
Dans une étude portant sur 2 634 prélèvements médullaires effectués entre
1982 et 1987 à l’hôpital Johns Hopkins à Baltimore aux États-Unis, 22 malades (0,8
%), âgés en moyenne de 47,9 ans (22-77 ans), avaient une activation
macrophagique. [2,37]
En l’absence de données épidémiologiques récentes et pour évaluer la
faisabilité d’un projet thérapeutique, un questionnaire a été adressé aux médecins
internistes, infectiologues et réanimateurs français pour évaluer le nombre de
patients atteints de SAM de l’adulte au cours de l’année 2000. Le nombre de SAM de
l’adulte, toutes étiologies confondues, s’élevait à 85, dont 55 d’étiologie infectieuse
(environ deux tiers) dans les 39 centres qui ont répondu. [37]

37
V. DIAGNOSTIC POSITIF
SOMMAIRE
A. Manifestations cliniques..........................................38
B. Examens biologiques...............................................42
1. Signes hématologiques........................................42
2. Bilan biochimique................................................44
C. Aspects cytologiques-histologiques........................47
1. Etude médullaire..................................................47
2. Etude des autres tissus........................................51
D. Critères diagnostiques............................................53

38
V. DIAGNOSTIC POSITIF :
A. Manifestations cliniques :
La présentation clinique du SAM est souvent bruyante avec un début assez
brutal, on trouve une fièvre précoce quasi-constante, une altération importante de
l’état général, une organomégalie, des signes cutanés, neurologiques et d’autres
(Tableau 2).
1. Fièvre :
Elément constant du tableau, la fièvre est présente dans 95% des cas et son
absence doit remettre en cause le diagnostic du SAM [6]. Souvent élevée pouvant
atteindre 40 °C, avec frissons, elle s’accompagne d’une altération profonde de l’état
général, conduisant parfois à la cachexie. [22]
2. Organomégalie :
• L’hépatomégalie et/ou la splénomégalie peuvent être notées au début ou
apparaître secondairement et devenir monstrueuses (40à 70%). Elles témoignent
de l’infiltration des organes hématopoïétiques par le contingent histiocytaire. [1]
• Les adénopathies sont retrouvées dans 30à 70% des cas [22]. Elles sont
disséminées dans plusieurs territoires ganglionnaires, aussi bien périphériques
que profonds, leur diamètre dépasse parfois 2 cm. Elles sont non inflammatoires
mais sensibles. Leur consistance est souple. [1,37’]
3. Signes cutanés :
Les signes cutanés sont présents dans 20% des cas. [1] On distingue des signes
non spécifiques tels un ictère lié à une atteinte hépatique, des éruptions

39
érythémateuses, purpuriques, des érosions muqueuses, des ulcères cutanés, des
nodules hypodermiques, des œdèmes localisés ou généralisés. [38]
La panniculite histiocytaire cytophagique (PHC) correspond à une manifestation
cutanée spécifique du SAM, survenant surtout chez l’adulte (20-30ans). Elle se
caractérise cliniquement, par des nodules hypodermiques qui peuvent évoluer vers
des ulcérations profondes, parfois de grande taille. Le diagnostic se fait par
l’examen histologique qui est caractéristique et montre une infiltration des lobules
graisseux par des histiocytes avec des images de macrophages en cytophagie. Ces
infiltrats macrophagiques sont d’allure bénigne par opposition à ceux des
histiocytoses malignes. [39,40]
4. Signes neurologiques :
L’atteinte du système nerveux central (SNC) est possible, surtout notable dans
les formes infantiles (lymphohistiocytose familiale), il s’agit de troubles divers :
irritabilité, somnolence, confusion voire coma, ataxie, troubles visuels, crises
convulsives, syndrome méningé, hémiplégie ou tétraplégie, des signes non
spécifiques d’hypertension intracrânienne, et retard de développement
psychomoteur. [41,42].
Des atteintes neurologiques périphériques, essentiellement par axonopathie
avec paralysies périphériques et/ou des paires crâniennes, ont également été
décrites. [41].
Des études ont montré que les manifestations neurologiques au cours du SAM
peuvent être expliquées par une mort neuronale et une nécrose des tissus. Ces
lésions sont dues à l’infiltration du SNC par des monocytes et lymphocytes activés,
et la sécrétion de cytokines neurotoxiques comme le TNF-α, de nombreux

40
macrophages cérébraux résidents (les cellules microgliales) et des astrocytes,
peuvent être aussi activés et sécréter à leur tour le glutamate neurotoxique et des
radicaux libres. [42,43].
5. Signes pulmonaires :
L’atteinte pulmonaire peut se traduire par une simple dyspnée avec une toux
sèche ou même par un syndrome de détresse respiratoire aigue. Il n’est pas rare de
mettre en évidence un infiltrat interstitiel diffus sur la radiographie des poumons.
[41]
6. Signes digestifs :
Ils sont inconstants et non spécifiques : nausées, vomissements, diarrhées,
douleur abdominale… [22]
7. Autres signes :
Des œdèmes et des épanchements séreux peuvent être observés : pleurésie,
ascite. Une atteinte oculaire à type d’œdème et d’hémorragies rétiniennes a été
rapportée. [29]
Des signes de défaillance multi-viscérale (hémorragie viscérale dans le cadre
d’une coagulation intra-vasculaire disséminée, ictère, insuffisance rénale, collapsus
et détresse respiratoire) pouvant émailler l’évolution de la maladie dans
l’hémophagocytose fulminante ou ne répondant pas au traitement. [22]
41
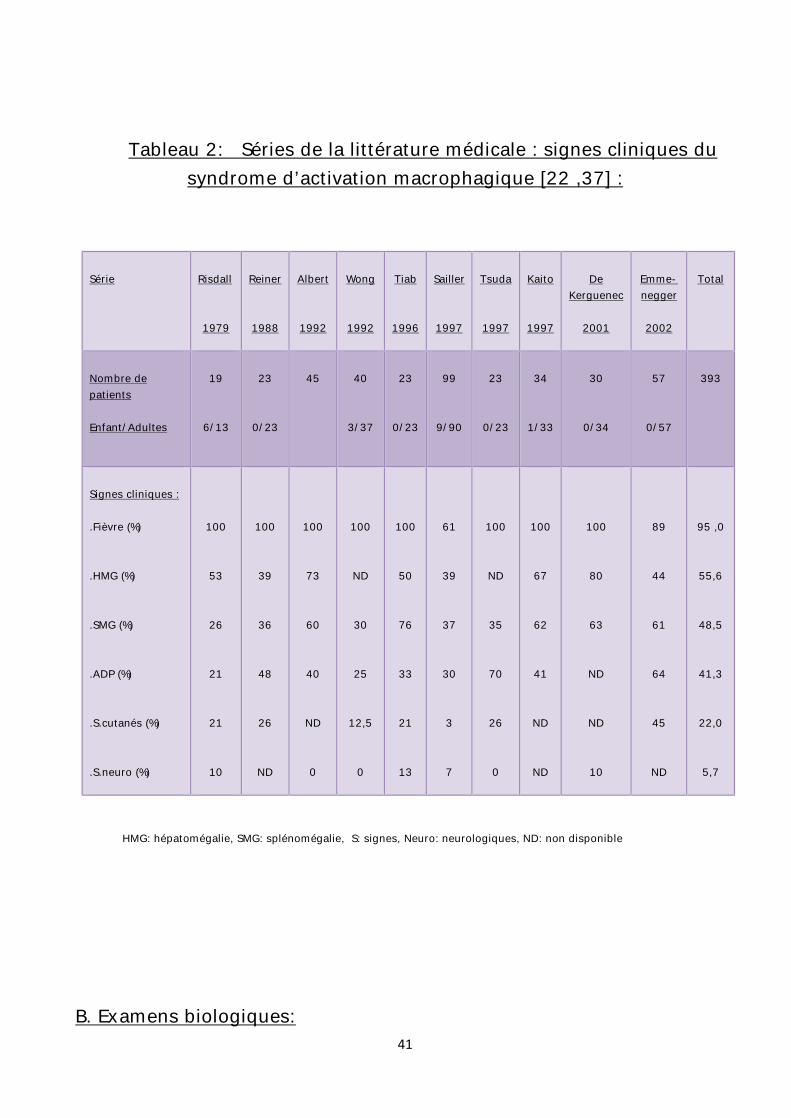
Tableau 2: Séries de la littérature médicale : signes cliniques du syndrome d’activation macrophagique [22 ,37] :
Série
Risdall
1979
Reiner
1988
Albert
1992
Wong
1992
Tiab
1996
Sailler
1997
Tsuda
1997
Kaito
1997
De
Kerguenec
2001
Emme-negger
2002
Total
Nombre de patients
Enfant/Adultes
19
6/13
23
0/23
45
40
3/37
23
0/23
99
9/90
23
0/23
34
1/33
30
0/34
57
0/57
393
Signes cliniques : .Fièvre (%)
.HMG (%) .SMG (%) .ADP (%) .S.cutanés (%) .S.neuro (%)
100
53
26
21
21
10
100
39
36
48
26
ND
100
73
60
40
ND 0
100
ND
30
25
12,5 0
100
50
76
33
21
13
61
39
37
30 3 7
100
ND
35
70
26 0
100
67
62
41
ND
ND
100
80
63
ND
ND
10
89
44
61
64
45
ND
95 ,0
55,6
48,5
41,3
22,0
5,7
HMG: hépatomégalie, SMG: splénomégalie, S: signes, Neuro: neurologiques, ND: non disponible
B. Examens biologiques:

42
Les anomalies biologiques sont nombreuses, souvent majeures, mais non
spécifiques. C’est leur association qui amène à évoquer le diagnostic du syndrome
d’activation macrophagique (Tableau 3).
1. Signes hématologiques :
a. Numération formule sanguine :
Une pancytopénie est observée dans environ 70% des cas, alors qu’une
bicytopénie est présente dans 100% des cas. [22]
Ø L’anémie : Est la perturbation la plus fréquente de l’hémogramme, retrouvée
dans 80% à 100% des cas, le taux de l’hémoglobine varie entre 8 et 9 g/dl mais
peut-être bien inférieur. [10]
Elle est à la fois centrale, par avortement intra médullaire lié au moins en partie à
la phagocytose des précurseurs érythroblastiques, et périphérique par
érythrophagocytose extra-hématopoïétique. Ceci rend compte de son caractère
particulier : elle est normocytaire, normochrome, arégénérative, mais associe des
stigmates d’anémie hémolytique intra-tissulaire avec érythroblastose, chute de
l’haptoglobine, augmentation des lacticodéshydrogénases (LDH) de la bilirubine
libre. Le test de Coombs érythrocytaire est habituellement négatif. [41]
Ø La thrombopénie : retrouvée dans 70 à 100% des cas [44], souvent inférieure à
100 000 éléments/mm3. Elle est précoce et profonde, peut être d’origine
centrale mais aussi périphérique par coagulation intra-vasculaire disséminée
(CIVD). [22]
Ø La leucopénie : présente dans 70% des cas, elle est plus inconstante et plus
tardive. Le déficit portant sur les lymphocytes mais aussi sur les polynucléaires
neutrophiles. [22]

43
b. Troubles de l’hémostase :
Ils sont présents dans 50 à 70 % des cas. On note essentiellement une
hypofibrinogénémie, soit isolée, soit associée à l’abaissement des taux de
thrombine, de prothrombine et à l’allongement du temps de céphaline activée,
témoignant d’une activation de la coagulation, voire d’une réelle CIVD, ce qui
constitue un facteur de mauvais pronostic étant donné la survenue de complications
hémorragiques pouvant être fatales. [22]
L’hypofibrinogénémie a pu être mise sur le compte de la sécrétion d’un
activateur de plasminogène par les macrophages activés, aboutissant à de hauts
niveaux de plasmine clivant le fibrinogène [35]. Le tableau de CIVD est lié à une
production excessive d’IFN ainsi que de TNF-α [1,10].
Ainsi, une hypofibrinémie est notée dans 5O à 100% des cas. Elle est souvent
inférieure à 1 g/dl. Le TP est fréquemment abaissé, le TCA peut être allongé. On
retrouve aussi des stigmates de CIVD avec l’augmentation des D-dimères et des
PDF (produits de dégradation de la fibrine). [45]
La diminution modérée des facteurs II, VII et X, peut être expliquée par
l’installation d’une insuffisance hépato-cellulaire. [46]
2. Bilan biochimique :
a. Bilan hépatique :

44
Il est habituel de retrouver des altérations du bilan hépatique (80% des cas) :
une cytolyse, précoce et parfois sévère, prédominant sur les ALAT, accompagnée de
signes d’insuffisance hépatocellulaire (hypoalbuminémie, diminution du facteur V).
La cholestase, souvent plus tardive, avec élévation de la bilirubinémie. Elle
semble plus fréquente et corrélée à un pronostic plus défavorable.
L’augmentation constante des LDH (lactico-déshydrogénase) plasmatiques,
reflète la lyse cellulaire. [22,47]
b. Bilan lipidique:
• L’hypertriglycéridémie souvent précoce, pouvant atteindre des taux à plus de 10
fois la normale [34]. En général, elle est supérieure à 2g/dl. Très caractéristique,
elle s’accompagne d’une augmentation des lipoprotéines de très basse densité
VLDL (Very Low Density Lipoprotein). Elle correspond à un déficit en lipoprotéine
lipase, inhibée par le TNF-α. [1] Elle permet de suivre l’activité de la maladie et se
normalise lors de la guérison. [6, 34]
• Le taux du cholestérol peut être normal [6], ou diminué [46’]. Par ailleurs, une
hypercholestérolémie a pu être constatée dans au moins 1 observation dans la
littérature associée à une connectivite infantile [47’].
c. Hyperferritinémie :
L’hyperferritinémie est quasi constante excédant le plus souvent 3000 μg/l. Les
taux de ferritine sérique semblent être corrélés avec l’activité de la maladie, en
particulier au cours de l’évolution sous traitement. [48]
La physiopathologie de cette hyperferritinémie n’est pas complètement élucidée
mais quelques hypothèses ont été envisagées [49] :

45
§ une diminution de la clairance de la ferritine liée à la diminution de ses
récepteurs,
§ un relargage accru par les macrophages après érythrophagocytose,
§ ou un relargage accru par les organes riches en fer, comme le foie et la rate.
d. Bilan hydroéléctrolytique :
Une hyponatrémie avec natriurèse conservée et hypoprotidémie liées à une
hémodilution suggérant l’existence d’une sécrétion inappropriée d’hormone
antidiurétique.
On peut également observer une insuffisance rénale aigue avec augmentation
des taux plasmatiques de l’urée et de la créatinine. [1,50]
e. Autres :
ü Une hypo- ou hypergammaglobulinémie polyclonale peut être notée. [1]
ü Le sCD25 (récepteur soluble de l’interleukine 2), synthétisé par les
lymphocytes T activés, est un marqueur très sensible du SAM puisque son
augmentation est constante [51,52]. Les très hauts niveaux dosés dans le
SAM ne sont en général pas observés lors d’affections bénignes mais
peuvent être présents dans des hémopathies lymphoïdes telles que les
leucémies aiguës lymphoblastiques, les leucémies liées au virus HTLV ou
bien les leucémies à tricholeucocytes [51]. Le taux du sCD25 diminue en cas
d’évolution favorable du SAM [51].
ü L’activité NK: rarement mesurée, elle serait diminuée au cours du SAM.
[52,53]
46
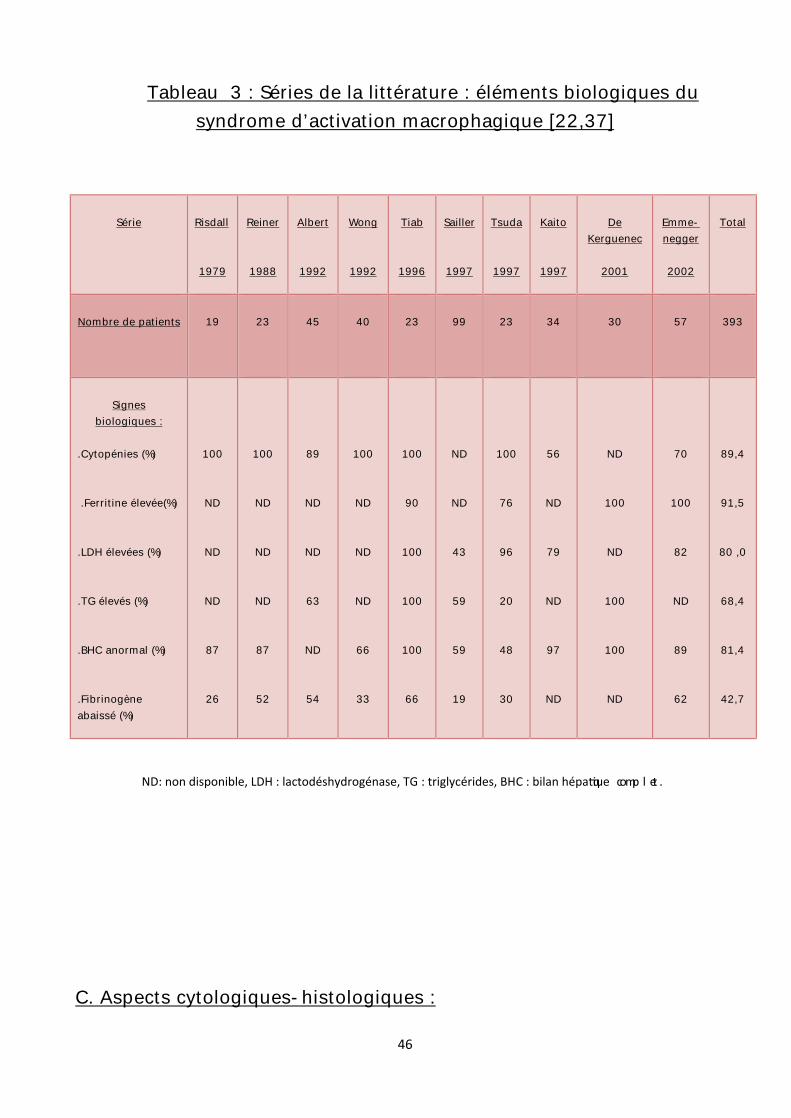
Tableau 3 : Séries de la littérature : éléments biologiques du syndrome d’activation macrophagique [22,37]
ND: non disponible, LDH : lactodéshydrogénase, TG : triglycérides, BHC : bilan hépa que comp l et .
C. Aspects cytologiques-histologiques :
Série
Risdall
1979
Reiner
1988
Albert
1992
Wong
1992
Tiab
1996
Sailler
1997
Tsuda
1997
Kaito
1997
De
Kerguenec
2001
Emme-negger
2002
Total
Nombre de patients
19
23
45
40
23
99
23
34
30
57
393
Signes
biologiques :
.Cytopénies (%)
.Ferritine élevée(%)
.LDH élevées (%) .TG élevés (%) .BHC anormal (%) .Fibrinogène abaissé (%)
100
ND
ND
ND
87
26
100
ND
ND
ND
87
52
89
ND
ND
63
ND
54
100
ND
ND
ND
66
33
100
90
100
100
100
66
ND
ND
43
59
59
19
100
76
96
20
48
30
56
ND
79
ND
97
ND
ND
100
ND
100
100
ND
70
100
82
ND
89
62
89,4
91,5
80 ,0
68,4
81,4
42,7

47
L’aspect histologique typique est celui d’une prolifération histiocytaire et/ou
macrophagique avec des images d’hémophagocytose [1]. Toutefois,
l’hémophagocytose cytologique est importante au diagnostic de SAM mais pas
obligatoire, comme nous allons le voir dans le sous-chapitre des critères
diagnostiques.
En effet, des images de phagocytose peuvent être observées chez des patients
infectés par le VIH, au cours de leucémies lymphoïdes ou myéloïdes, chez des
patients polytransfusés ou encore lors de syndromes hémolytiques d’origine
congénitale ou acquise [1,35].
Les signes d’hémophagocytose sont très souvent recherchés sur le
myélogramme mais de façon beaucoup moins fréquente dans les ganglions ou la
rate, puisque la pratique d’une biopsie ganglionnaire ou d’une splénectomie ne fait
pas partie, sauf complication, du bilan systématique ou du traitement du SAM [29].
1. Etude médullaire :
a. Le myélogramme :
Le myélogramme est l’examen de référence, il apporte les critères
morphologiques du diagnostic du SAM. [1]
Il montre une moelle riche avec une infiltration médullaire par des histiocytes
d’aspect cytologique bénin : il s’agit d’histiocytes matures, bien différenciés, sans
atypie cytologique [35], ce qui les différencie des histiocytoses malignes. Ces
histiocytes médullaires présentent de nombreuses vacuoles intracytoplasmiques,
contenant des éléments cellulaires sanguins (érythrocytes, érythroblastes,
granulocytes, plaquettes, lymphocytes) ou leurs précurseurs hématopoïétiques,
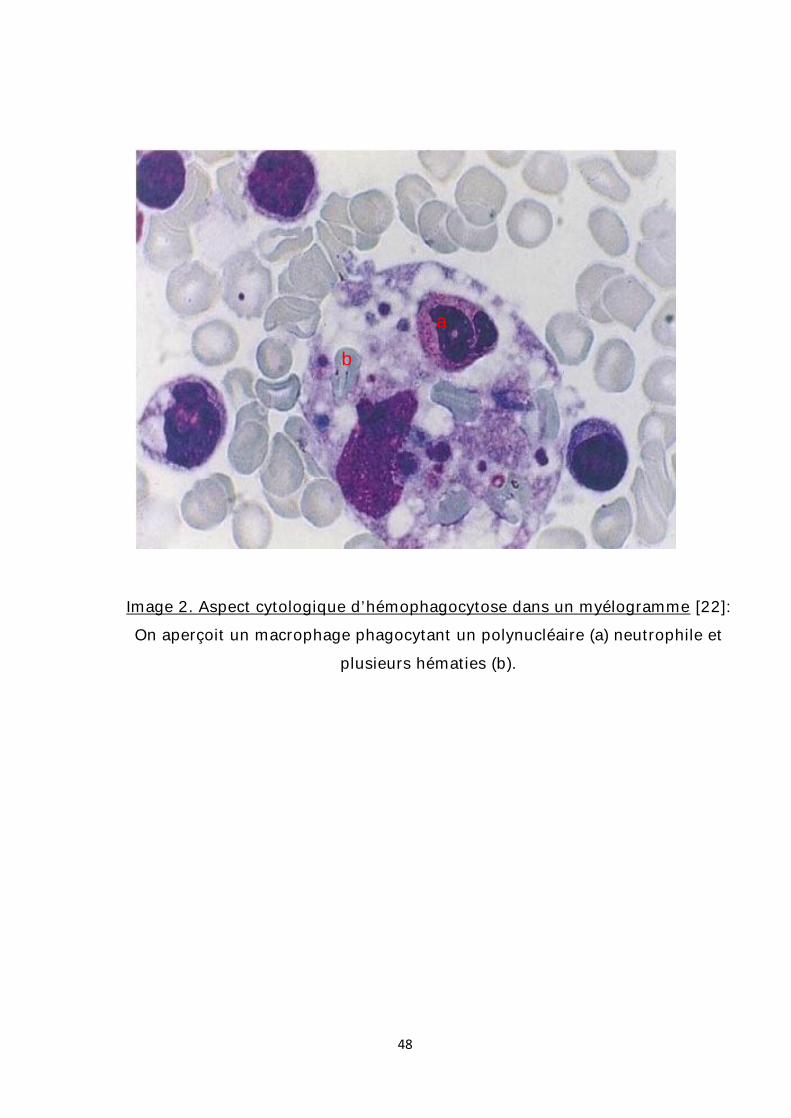
intacts ou partiellement digérés (images 1et 2). [22]
48
Image 2. Aspect cytologique d’hémophagocytose dans un myélogramme [22]: On aperçoit un macrophage phagocytant un polynucléaire (a) neutrophile et
plusieurs hématies (b).
a
b
49
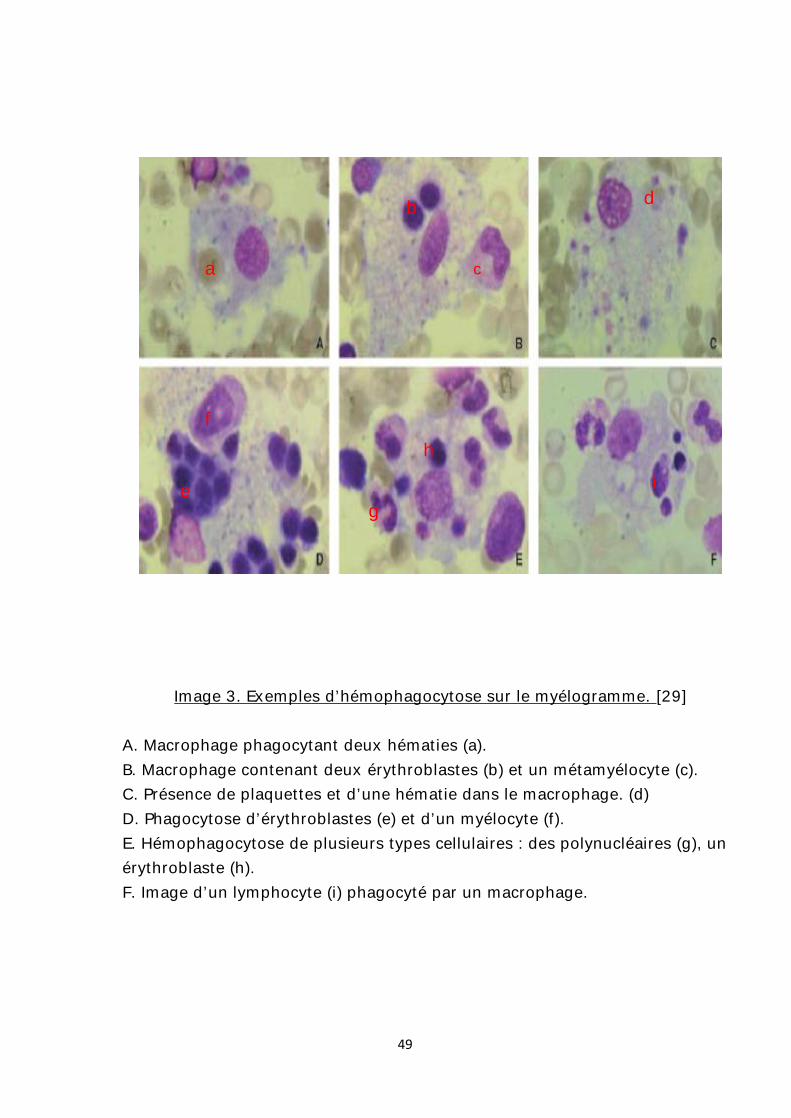
Image 3. Exemples d’hémophagocytose sur le myélogramme. [29]
A. Macrophage phagocytant deux hématies (a). B. Macrophage contenant deux érythroblastes (b) et un métamyélocyte (c). C. Présence de plaquettes et d’une hématie dans le macrophage. (d) D. Phagocytose d’érythroblastes (e) et d’un myélocyte (f). E. Hémophagocytose de plusieurs types cellulaires : des polynucléaires (g), un érythroblaste (h). F. Image d’un lymphocyte (i) phagocyté par un macrophage.
b
c
e
f h
g i
a
d

50
Le pourcentage des histiocytes-macrophages hémophages est, pour certains
auteurs, un critère diagnostic important (ils doivent représenter plus de 2% des
cellules nucléées pour Wong KF et al [54] et plus de 3 % pour Tsuda et al. [55]).
Cependant, aucune étude n’a prouvé la relation entre le nombre d’histiocytes
médullaires et la gravité de la maladie et son évolution [22].
Une érythroblastose est fréquente, témoin de l’érythropoïèse réactionnelle à
l’hémolyse intramédullaire [22]. La lignée mégacaryocytaire est quasiment toujours
hyperplasique au début avec une maturation qui s’effectue correctement [1]. La
lignée rouge apparaît parfois dysplasique, la lignée granuleuse préservée à la phase
initiale de la maladie, peut devenir déplétive au cours de l’évolution [6].
La présence de lymphocytes de types activés, identiques à ceux du sang
périphérique, peut être notée [35].
Le contexte étiologique peut être parfois évoqué sur le myélogramme lorsqu’il
existe un infiltrat hémopathique lymphomateux, en plus si le myélogramme met en
évidence des signes d’activation lymphoïdes, il permet d’évoquer une pathologie
virale ou une lymphohistiocytose familiale (LHF) [6].
b. Biopsie ostéomédullaire :
La biopsie ostéomédullaire paraît moins performante pour la mise en évidence
d’histiocytose médullaire et d’hémophagocytose active. Sa réalisation peut
néanmoins aider au diagnostic étiologique en montrant notamment un lymphome
sous-jacent au SAM, ou encore un processus infectieux (tuberculose par exemple)
[1].
La recherche du SAM peut être facilitée par les immunomarquages: les
macrophages sont identifiés par l’anticorps monoclonal anti-CD68. [35]

51
2. Etude des autres tissus :
a. Biopsie ganglionnaire :
Les adénopathies, lorsqu'elles sont périphériques, sont accessibles et méritent
d'être biopsiées car elles peuvent montrer, outre des images d'hémophagocytose
souvent sinusales, des anomalies lymphoïdes, un infiltrat lymphomateux ou des
stigmates d'infection virale [6].
b. Biopsie hépatique :
La biopsie hépatique est souvent difficile à réaliser du fait de la thrombopénie
et de la coagulopathie. Cependant, elle peut être très informative en montrant une
infiltration histiocytaire des capillaires sinusoïdes, des espaces portes (image 3) et
parfois une nécrose hépatocellulaire. [6, 27, 47]
c. Biopsie splénique :
C’est un geste inhabituel car très risqué, pratiqué selon une méthode
spécialisée. On retrouve à l’histologie une expansion des cordons de la pulpe rouge
avec prédominance de l’activité hémophagocytaire à ce niveau et une déplétion
lymphocytaire de la pulpe blanche [1]. L’hémophagocytose peut de toute manière
être identifiée après splénectomie si le diagnostic n’est pas porté précédemment.
[35]
d. Autres :
Plus rarement, l’hémophagocytose peut être mise en évidence dans d’autres
organes, tels la peau, le poumon, les reins, les surrénales, l’estomac…… [6]
Il est possible, de façon très rare, de retrouver des signes d’hémophagocytose
dans le liquide des épanchements séreux ou le liquide céphalorachidien. [29]
52
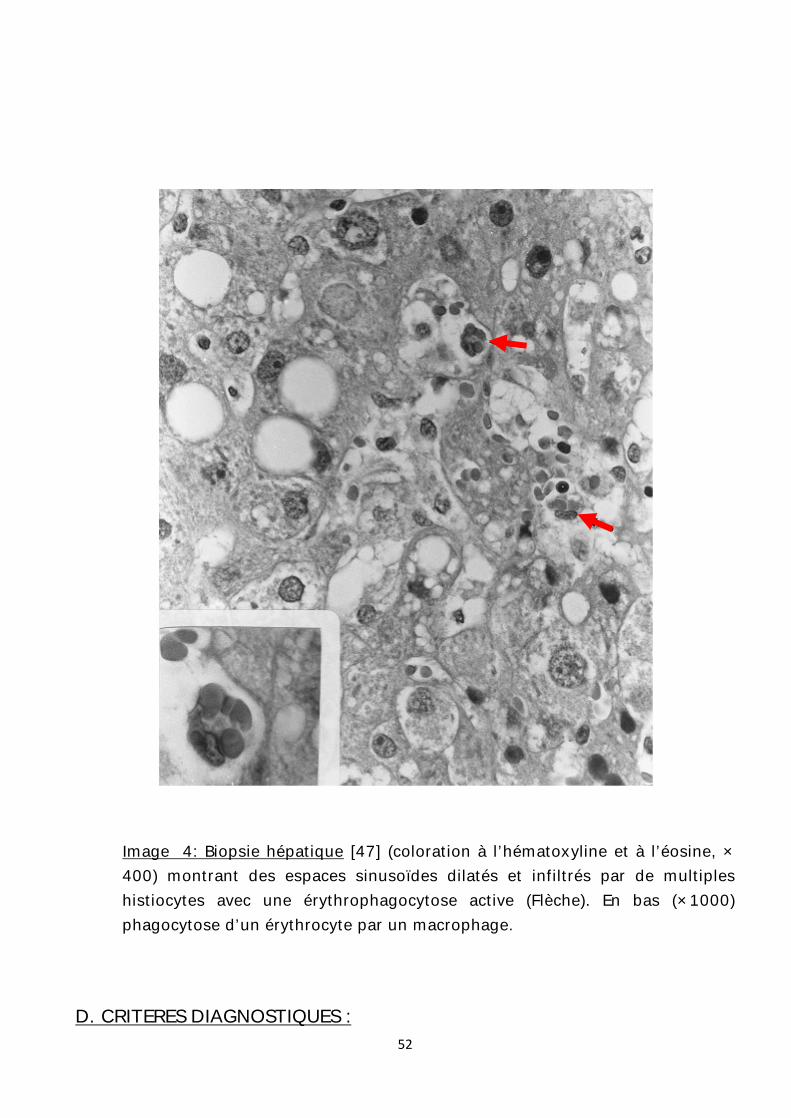
Image 4: Biopsie hépatique [47] (coloration à l’hématoxyline et à l’éosine, × 400) montrant des espaces sinusoïdes dilatés et infiltrés par de multiples histiocytes avec une érythrophagocytose active (Flèche). En bas (×1000) phagocytose d’un érythrocyte par un macrophage.
D. CRITERES DIAGNOSTIQUES :

53
L’existence d’un aspect d’hémophagocytose sur les prélèvements cytologiques
ou histologiques ne suffit pas pour porter le diagnostic de syndrome d’activation
macrophagique [56]. En effet, des images d’hémophagocytose sont rencontrées
dans des situations diverses. Il est donc nécessaire d’avoir une confrontation entre
les données cliniques, biologiques et cyto-histologiques. [22]
Ainsi, plusieurs auteurs ont proposé des critères diagnostiques s’appuyant sur
des faisceaux d’arguments cliniques, biologiques et cytohistologiques. Ci-dessous
sont mentionnées les trois propositions de critères diagnostiques formulées dans la
littérature, avec une remise à jour proposée par Henter et al. en 2006.

54
Critères diagnostiques du SAM selon le FHL study group of Histiocyte Society (1991) [57] : tous les critères sont exigés :
Critères cliniques :
v fièvre > 7 jours, avec pics > 38,5 °C v splénomégalie.
Critères biologiques :
v cytopénie sur 2 ou 3 lignées non expliquée par une
moelle pauvre ou dysplasique : ü Hémoglobine < 9 g/dl, ü neutrophiles < 100/mm³, ü plaquettes< 100 000/mm³.
v hypertriglycéridémie > 2 mmol/l et/ou
hypofibrinogénémie < 1,5 g/l.
Critères histologiques : v hémophagocytose (médullaire, splénique ou
ganglionnaire) ; v absence de signe de malignité.

55
Critères diagnostiques du SAM selon Tsuda (1997) [55]: tous les critères sont exigés :
Critères cliniques :
v fièvre > 7 jours
Critères biologiques :
v cytopénie inexpliquée sur deux ou trois lignées.
Critères histologiques :
v hémophagocytose médullaire avec histiocytose > 3 p. 100
(ou > 2 500/ml) ou présence d’hémophagocytose hépatique, splénique ou ganglionnaire.

56
Critères diagnostiques du SAM selon Imashuku (1997 ) [58] : tous les critères sont exigés :
Critères cliniques : v fièvre > 7 jours, avec pics > 38,5 °C.
Critères biologiques : v cytopénie sur 2 ou 3 lignées non expliquée par une moelle
pauvre ou dysplasique : ü Hémoglobine inférieure à 9 g/dl, ü neutrophiles inférieurs à 100/mm³, ü plaquettes inférieurs à 100 000/mm³,
v augmentation de la ferritine plasmatique (> 3 fois la
normale ou > 1000 µg/l) ;
v augmentation de la LDH (> 3 fois la normale ou > 1000 UI/l).
Critères histologiques : v hémophagocytose (médullaire, splénique ou ganglionnaire).

57
Critères diagnostiques du syndrome d’activation macrophagique d’après Henter et al. (2006) [59]. Le diagnostic est retenu en présence du critère1 ou du critère 2 :
Critère 1 :
ü Diagnostic moléculaire de lymphohistiocytose hémophagocytaire.
Critères 2 : 5 des 8 critères suivants :
ü Fièvre. ü Splénomégalie. ü cytopénies affectant 2 lignées ou plus :
-hémoglobine < 9g/dl, -plaquettes < 100 000/mm³, -neutrophiles < 100/mm³. ü hypertriglycéridémie et/ou hypofibrinogénémie :
triglycérides >3mmol/l, fibrinogène < 1,5 g/l. ü hémophagocytose dans la moelle osseuse, la rate ou les
ganglions lymphatiques. ü activité natural killer diminuée ou absente. ü ferritine > 500 µg/l. ü sCD25 ≥ 2400 U/ml.

58
On remarque que les cinq critères de la classification de 1991 persistent (fièvre,
splénomégalie, bicytopénie, hémophagocytose au niveau de la moelle osseuse, des
ganglions lymphatiques ou de la rate), mais que viennent s’y ajouter trois nouveaux
critères qui sont une activité natural killer (NK) altérée, l’hyperferritinémie ainsi que
des niveaux élevés du récepteur soluble de l’interleukine 2 (sCD25). Il faut préciser
que ces critères ont été élaborés essentiellement dans un contexte pédiatrique du
syndrome d’activation macrophagique primaire [29]. La fréquence à laquelle on
retrouve ces divers signes au moment du début de la symptomatologie du syndrome
d’activation macrophagique est la suivante [29]:
ü la fièvre et la splénomégalie sont présentes dans 70 % des cas,
ü une bicytopénie et des triglycérides augmentés dans à peu près 50 %
des cas,
ü une diminution du fibrinogène dans un peu plus de 20 % des cas,
ü des signes d’hémophagocytose dans 35 % des cas environ,
ü une ferritine augmentée dans un peu plus de 35 % des cas,
ü une augmentation du sCD25 dans pratiquement 90 % des cas,
ü une diminution de l’activité NK dans 100 % des cas.
En raison du délai entre les premiers symptômes et le moment du diagnostic du
syndrome d’activation macrophagique, certains signes cardinaux deviennent quasi
constants au moment du diagnostic: c’est le cas de la fièvre, de la splénomégalie, de
la bicytopénie ainsi que de l’augmentation du sCD25 [29].
En pratique, ces différents critères ne pourront pas tout le temps être satisfaits
en entier. D’ailleurs les séries rapportées dans ce travail ne se sont pas toutes
appuyées sur ces critères à 100%. [2, 10, 36, 47, 54]

59
VI.DIAGNOSTIC DIFFERENTIEL
SOMMAIRE :
1. La leucémie aiguë..............................................60
2. L’histiocytose langerhansienne .........................60
3. Troubles métaboliques......................................60
4. Toute autre étiologie de fièvre prolongée..........61

60
VI. DIAGNOSTIC DIFFERENTIEL :
Le diagnostic de SAM est difficile dans la mesure où de nombreuses
circonstances paraissent pouvoir favoriser son apparition (lymphome, cancer,
maladie systémique, infection, etc..) et que les symptômes correspondant à ces
circonstances peuvent être au premier plan masquant, ou ne suggérant pas
cliniquement, ceux du SAM [45].
Le SAM peut prêter confusion avec certaines affections :
1. La leucémie aiguë :
L’hépato-splénomégalie, la fièvre et la cytopénie sont des signes de leucémie
aiguë, cette symptomatologie est retrouvée également dans le SAM, cependant une
étude de moelle osseuse peut facilement exclure une leucémie aiguë. [60]
2. L’histiocytose langerhansienne :
Le SAM et l’histiocytose langerhansienne peuvent partager des points
communs. Néanmoins, l'éruption cutanée caractéristique, les lésions osseuses, la
rareté de l’atteinte méningée et l’image histologique distincte de l’histiocytose
langerhansienne, séparent clairement cette affection du SAM. [60]
3. Troubles métaboliques :
L’organomégalie, l’altération du bilan hépatique et l’augmentation des
triglycérides, retrouvées au cours du SAM peuvent suggérer un trouble de
métabolisme surtout chez les nourrissons, cependant, les cytopénies progressives,
la fièvre prolongée et les anomalies immunologiques caractéristiques redressent le
diagnostic. [60]

61
4. Toute autre étiologie de fièvre prolongée :
Le diagnostic de fièvre prolongée est souvent complexe et repose avant tout
sur l’interrogatoire et la recherche des circonstances de survenue, la connaissance
des antécédents en tenant compte particulièrement de ceux à type de lymphome ou
de maladie hématologique, de cancer, de chimiothérapie, de maladie systémique,
etc., dans lesquels se développe, avant tout, le SAM. Outre ces éléments
d’interrogatoire, l’examen clinique minutieux, l’interprétation des examens
biologiques, l’aide des examens radiologiques et immunologiques et finalement la
ponction médullaire permettront le diagnostic. Le dosage de la ferritine paraît
également d’un bon apport. [45]
Palazzi et al. [61] ont proposé un modèle d’investigation de SAM dans le cadre
d’une fièvre apparemment d’origine inconnue (figure 4).
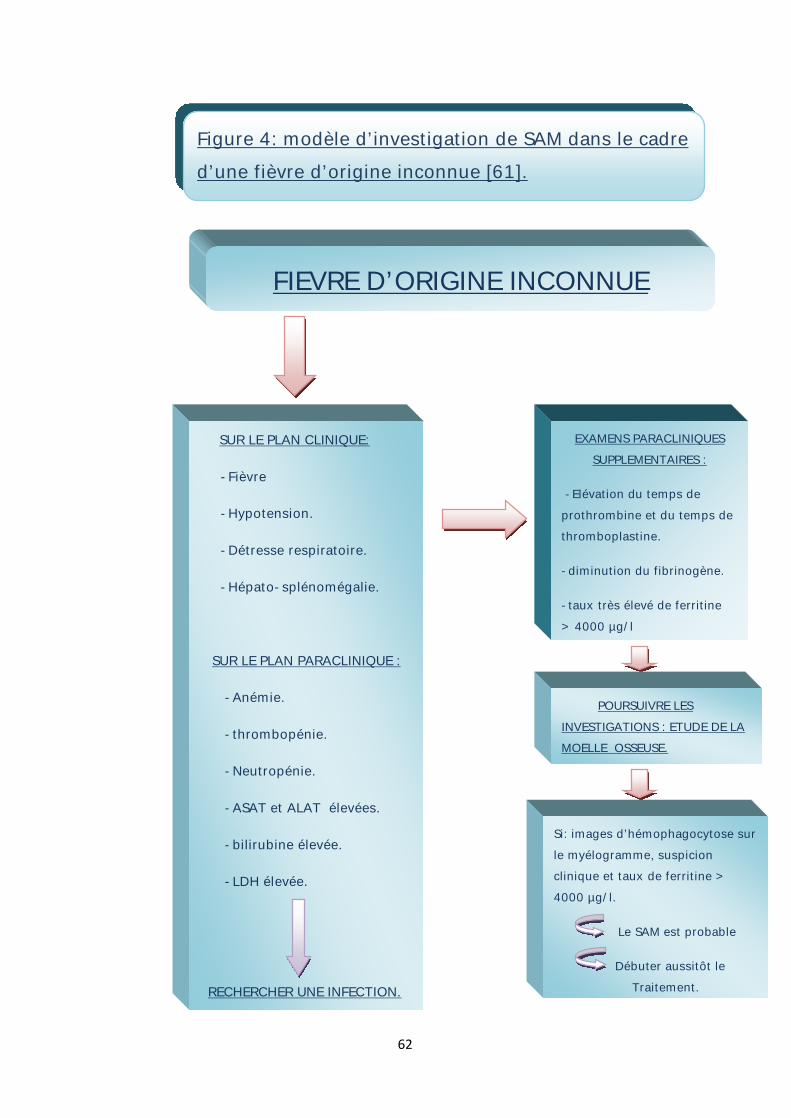
62
SUR LE PLAN CLINIQUE:
-Fièvre
-Hypotension.
-Détresse respiratoire.
-Hépato-splénomégalie.
SUR LE PLAN PARACLINIQUE :
-Anémie.
-thrombopénie.
-Neutropénie.
-ASAT et ALAT élevées.
-bilirubine élevée.
-LDH élevée.
RECHERCHER UNE INFECTION.
EXAMENS PARACLINIQUES SUPPLEMENTAIRES :
-Elévation du temps de prothrombine et du temps de thromboplastine.
-diminution du fibrinogène.
-taux très élevé de ferritine > 4000 µg/l
POURSUIVRE LES INVESTIGATIONS : ETUDE DE LA MOELLE OSSEUSE.
Si: images d’hémophagocytose sur le myélogramme, suspicion clinique et taux de ferritine > 4000 µg/l.
Le SAM est probable
Débuter aussitôt le Traitement.
FIEVRE D’ORIGINE INCONNUE
Figure 4: modèle d’investigation de SAM dans le cadre d’une fièvre d’origine inconnue [61].

63
VII. ETIOLOGIES
SOMMAIRE :
A. Les SAM primitifs...................................................................64
1. La lymphohistiocytose familiale...........................................66
2. Le syndrome de Chediak-Higashi........................................67
3. Le syndrome de Gricelli.......................................................67
4. Le syndrome de Purtilo........................................................68
B. Les SAM secondaires...............................................................71
1. Les SAM post-infectieux......................................................74
2. Les SAM et affections malignes............................................80
3. Les affections auto-immunes et les maladies de système....82
4. Les SAM associées aux déficits immunitaires acquis............85
5. Les SAM secondaires à autres étiologies..............................87
C. Le bilan étiologique...............................................................89

64
VII. ETIOLOGIES :
Il est classique d’opposer deux grandes formes du SAM [60]:
ü Une primitive héréditaire : c’est la lymphohistiocytose familiale ou sporadique de
l’enfant.
ü L’autre est secondaire souvent associée à une pathologie sous jacente.
A. Les SAM primitifs :
Plusieurs pathologies héréditaires du système immunitaire sont caractérisées
par une activation macrophagique et lymphocytaire T appelée lymphohistiocytose
hémophagocytaire, souvent déclenchée par une infection intercurrente. Ces
maladies sont surtout décrites chez l’enfant et l’adulte jeune [22]. (Tableau 4)
Dans la plupart de ces formes primaires, des anomalies moléculaires bien
précises ont été identifiées [62], qui font dans ce cas partie des critères
diagnostiques du SAM [29].

65
Tableau 4: Principaux syndromes de déficit immunitaire primitif et SAM [29]:
v Lymphohistiocytose hémophagocytaire isolée :
Lymphohistiocytose hémophagocytaire familiale (LHF)
ü Type 1 : anomalie génétique non connue. ü Type 2 : gène de la perforine PRF1. ü Type 3 : gène UNC13D. ü Type 4 : gène de la syntaxine STX11.
v Déficits immunitaires primaires associés au SAM :
ü Syndrome de Griscelli de type 2 (gène RAB27). ü Syndrome de Chediak-Higashi (gène LYST). ü Syndrome lymphoprolifératif lié à l’X (syndrome
de Purtilo).
v Déficits immunitaires primaires pouvant se compliquer
de SAM ü Syndrome de Wiskott-Aldrich. ü Syndrome de Di George. ü Syndrome d’immunodéficit combiné sévère
(SCID). ü Syndrome de déficit en purine nucléoside
phosphorylase (PNP).

66
1. La lymphohistiocytose familiale (LHF) :
La lymphohistiocytose familiale est la plus fréquente des SAM primaires. Elle
s’exprime le plus souvent avant l’âge de 18 mois de vie selon un mode autosomique
récessif [63]. Les premiers cas ont été décrits par Farquhar et Claireaux en 1952
[22].
Cette maladie semble être induite, le plus souvent, par une infection virale, les
organes lymphoïdes sont alors infiltrés par une population cellulaire polyclonale
mixte, faite de lymphocytes T et de macrophages, avec évolution secondaire quasi-
constante vers le décès en l’absence de traitement. [22]
Lorsqu’un cas d’activation macrophagique survient chez l’enfant, la
lymphohistiocytose héréditaire doit être évoquée s’il existe des antécédents
familiaux (50 % dans le registre international) ou une consanguinité (10% des cas).
[22]
De façon plus spécifique ont été décrits plusieurs types d’anomalies
moléculaires liés au syndrome d’hémophagocytose lymphohistiocytaire familiale
(HLF) [29]. Un premier locus de susceptibilité (9q21.3-22 pour FHL1) a été localisé
mais le gène muté n’est pas encore identifié [1]. Trois autres anomalies ont été
identifiées. La première est en rapport avec des mutations du gène de la perforine
(FHL2), protéine effectrice majeure de la cytotoxicité [64, 65, 66]. Une seconde
anomalie est induite par des mutations du gène UNC13D, qui code Munc (FHL3),
protéine intervenant dans les étapes précoces de sécrétion des granules
cytotoxiques [67]. Une troisième anomalie moléculaire a été identifiée, intéressant le
gène STX11, qui code la syntaxine 11(FHL4), protéine impliquée dans les transferts
intracellulaires [68]. Le défaut de cytotoxicité des lymphocytes CD8 résultant des
mutations de ces gènes empêcherait la lyse des cellules présentatrices d’antigènes
exprimant à leur surface un antigène viral ou bactérien, et par conséquent

67
entretiendrait une activation permanente d’une population lymphocytaire dirigée
vis-à-vis de cet antigène. [22]
Les signes cliniques et biologiques sont identiques à ceux observés dans tout
SAM en dehors d’une fréquence accrue d’atteinte du système nerveux central (50 %
des patients), ce qui représente un facteur de mauvais pronostic. [22]
2. Le syndrome de Chediak-Higashi :
Le syndrome de Chediak-Higashi est une maladie rare de transmission
autosomique récessive, caractérisée par un albinisme partiel, cutané et oculaire
(reconnaissable par la couleur gris argentée des cheveux) associé à un déficit
immunitaire T cytotoxique et NK. [22, 69, 70]
Le syndrome d’activation macrophagique peut très souvent survenir chez ces
enfants, avec des atteintes neurologiques graves et un mauvais pronostic vital. [1]
Le gène muté dans cette maladie code pour une protéine appelée LYST
(LYSosomal Traffıcking regulator), impliquée dans l’adressage des protéines
intracellulaires. Certaines protéines membranaires lymphocytaires (perforine,
CTLA4…), jouant un rôle clé dans la régulation du système immunitaire, semblent
dans ce cas déviées de leur destination primitive et sont adressées vers les
lysosomes cellulaires, expliquant la présence des grandes granulations intra-
cytoplasmiques non fonctionnels, aussi bien dans les cellules mélaniques et les
synapses que dans les cellules cytotoxiques (TCD8 et NK) [71]. Ces granulations
géantes sont très caractéristiques de la maladie [22].
3. Le syndrome de Griscelli :
Le syndrome de Griscelli est une pathologie assez voisine du syndrome de
Chediak-Higashi, et seule l’absence des grandes granulations intracytoplasmiques
peut distinguer les deux maladies [22].

68
De transmission autosomique récessive, cette maladie est caractérisée par une
dépigmentation de la peau et des phanères et une susceptibilité aux infections
virales. Certains patients développent des manifestations neurologiques précoces
[1].
Deux gènes semblent pouvoir être mutés dans ce syndrome, MYO-VA et RAB-
27A, codant respectivement pour la myosine 5A et la protéine RAB27A. Ces
protéines impliquées dans le trafic intracellulaire [72,73]. Les patients présentant
une mutation de la protéine RAB27A [72] présentent également un déficit
lymphocytaire T cytotoxique et quelquefois un syndrome d’activation
macrophagique déclenché par un épisode infectieux. Cette protéine ayant une
fonction importante dans la liaison de la vésicule d’exocytose avec la membrane
cellulaire, on peut supposer que sa mutation conduit à un mauvais routage de
certaines molécules (ex. CTLA4) contrôlant l’activation lymphocytaire T [22].
4. Le syndrome de Purtilo :
Le syndrome de Purtilo ou syndrome d’immuno-prolifération lié à l’X ou XLP-
syndrome (X-linked lymphoprolifération), est un déficit immunitaire héréditaire rare
caractérisé par une susceptibilité accrue à l’infection par l’EBV [74]. Près de la moitié
des patients présentent des manifestations avant toute rencontre avec l’EBV
(hypogammaglobulinémie, lymphome B souvent de localisation intestinale ou iléo-
cæcale) mais l’évolution est marquée, en l’absence de traitement, par la survenue
d’une mononucléose infectieuse gravissime et fatale après l’infection par ce virus.
Les autres manifestations possibles sont une aplasie médullaire, une vascularite
nécrosante du système nerveux central, une granulomatose lymphomatoïde
pulmonaire ou une hépatite fulminante. [22]

69
Biologiquement ces patients ne développent pas d’anticorps anti–EBNA
(Epstein- Barr Nuclear Antigen) et présentent histologiquement une infiltration
tissulaire massive par des lymphocytes T cytotoxiques (CD8+) responsables des
lésions nécrotiques. [22]
La mortalité spontanée de cette maladie (fatale dans 100 % des cas avant l’âge
de 40 ans) rend nécessaire une greffe de moelle osseuse. [22]
Quant à la génétique de cette maladie, elle a été élucidée en 1998, avec la mise
en évidence de mutations portant sur un gène localisé sur le bras long du
chromosome X [22]. Ce gène SH2-D1A (SH2- Domain containing protein 1A) code
pour la protéine SAP (SLAM Associated Protein), qui est indispensable pour le signal
intracellulaire d’une famille de récepteurs SLAM (Signaling Lymphocytic Activation
Molecule) présents sur les lymphocytes T [75], NK et les monocytes–macrophages.
La mutation transforme la SAP, protéine normalement activatrice, en protéine
d’inhibition des fonctions cytotoxiques des lymphocytes T et des cellules NK [29].
Lors d’une infection par le virus d’Epstein-Barr (EBV), le patient est alors incapable
de développer une réponse cytotoxique antivirale appropriée et succombe le plus
souvent à une lymphoprolifération liée à l’EBV avec une augmentation de la
production de l’INF [1], ce qui pourrait expliquer la fréquence du syndrome
d’activation macrophagique dans cette pathologie. [1,29]
Toutes ces pathologies ont donc en commun une activation primitive
lymphocytaire T, souvent déclenchée par une infection opportuniste, le plus souvent
virale, avec production majeure de cytokines pro-inflammatoires (IFN , IL–1, IL–6,
TNF-α), puis une activation macrophagique qui participe aux lésions tissulaires
disséminées.

70
D’autres déficits immunitaires primaires peuvent se compliquer de SAM, parmi
lesquels on trouve :
v Le syndrome de Wiskott-Aldrich [29] : c’est une immunodéficience liée à l’X
touchant à la fois l’immunité humorale et cellulaire [76]. Il se manifeste, dans
sa forme habituelle, par des infections récurrentes (bactériennes, virales et
fongiques), de l’eczéma, une thrombopénie avec microplaquettes. Les patients
atteints du syndrome de Wiskott-Aldrich ont une propension au
développement de pathologies auto-immunes ainsi que d’hémopathies
malignes. Des aspects d’hémophagocytose, en particulier sur biopsie
ganglionnaire, ont été identifiés de longue date dans ce syndrome. En plus du
déficit immunitaire dans sa globalité, les cellules NK des patients atteints de
syndrome de Wiskott-Aldrich présentent un défaut de cytotoxicité associé au
déficit d’expression de la protéine WASp (Wiskott-Aldrich syndrome protein) et
au défaut d’accumulation de F-actine (filamentous actin) dans la synapse
immunologique [77]. Cette anomalie entrave les fonctions des cellules NK et
pourrait participer au développement du SAM, puisque les anomalies des
cellules NK sont probablement impliquées dans sa physiopathologie.
v Le déficit en purine phosphorylase (PNP) est une affection autosomique
récessive caractérisée par un déficit immunitaire combiné sévère et des
anomalies neurologiques complexes, incluant retard de développement, ataxie
et spasticité, ainsi que des anomalies hématologiques avec des aspects de
dysmyélopoïèse [29].

71
B. Les SAM secondaires :
À côté des SAM primaires liés à un déficit immunitaire, le SAM secondaire (ou
réactionnel) est relié à différentes situations pathologiques ayant en commun une
stimulation importante du système immunitaire [29].
Les SAM secondaires s’opposent aux SAM primitifs par le fait [45] :
§ qu’ils surviennent habituellement chez des enfants plus âgés ou des adultes
jeunes, voire des sujets âgés (par exemple, SAM associé à des lymphomes, à
des cancers) ;
§ qu’il n’y a pas de notion familiale retrouvée ;
§ qu’actuellement au moins, aucune anomalie héréditaire au niveau des gènes
ou des processus de l’immunité n’a été démontrée.
Les différentes associations décrites concernent des hémopathies malignes, des
infections virales ou bactériennes, voire parasitaires, des pathologies rhumatismales,
auto-immunes ou bien des réactions médicamenteuses [29]. Les affections les plus
souvent associées au SAM sont les infections et les lymphomes. Le Tableau 5 est tiré
de l’article fort documenté de Karras et Hermine. Ces auteurs ont établi les facteurs
étiologiques des SAM secondaires à partir de l’analyse des huit plus grandes séries
publiées de ce syndrome.
Le diagnostic étiologique demeure souvent négatif car le spectre des
pathologies associées à ce syndrome est extrêmement large et le tableau clinique
est habituellement dominé par les manifestations secondaires au SAM, occultant les
signes spécifiques de la pathologie causale. [22]
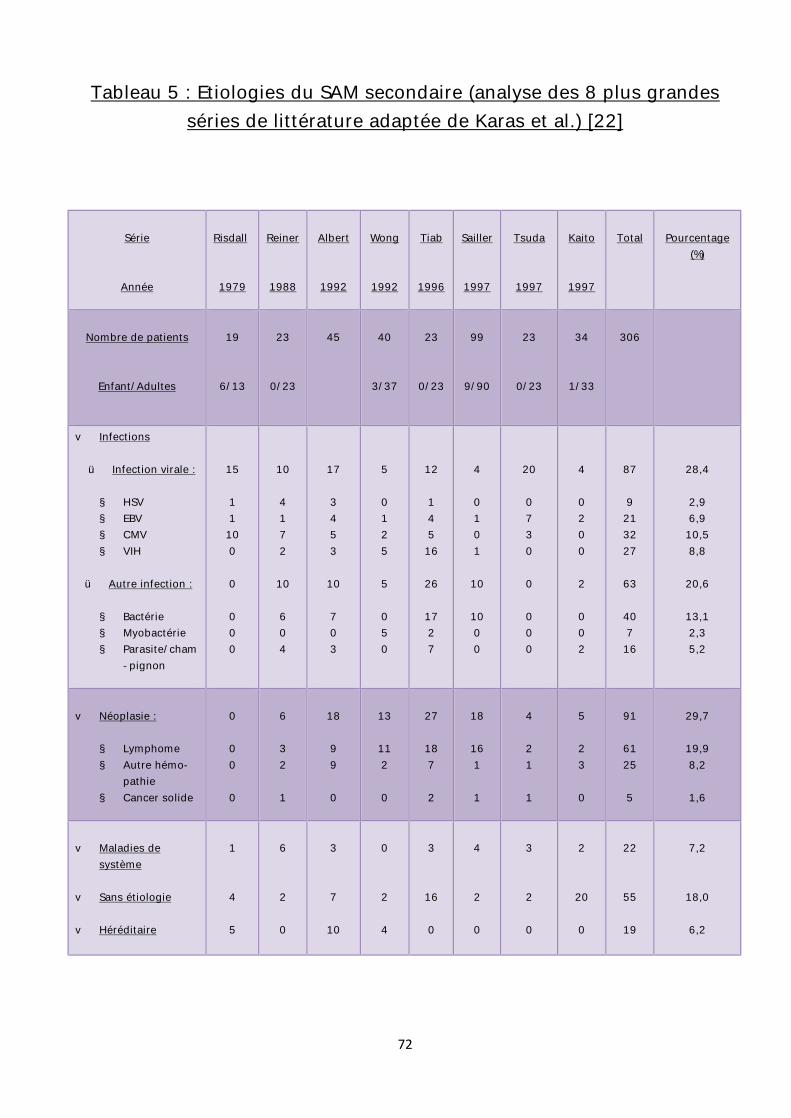
72
Tableau 5 : Etiologies du SAM secondaire (analyse des 8 plus grandes séries de littérature adaptée de Karas et al.) [22]
Série
Année
Risdall
1979
Reiner
1988
Albert
1992
Wong
1992
Tiab
1996
Sailler
1997
Tsuda
1997
Kaito
1997
Total
Pourcentage
(%)
Nombre de patients
Enfant/Adultes
19
6/13
23
0/23
45
40
3/37
23
0/23
99
9/90
23
0/23
34
1/33
306
v Infections
ü Infection virale :
§ HSV § EBV § CMV § VIH
ü Autre infection :
§ Bactérie § Myobactérie § Parasite/cham
-pignon
15 1 1 10 0 0 0 0 0
10 4 1 7 2
10 6 0 4
17 3 4 5 3
10 7 0 3
5 0 1 2 5 5 0 5 0
12 1 4 5 16
26
17 2 7
4 0 1 0 1
10
10 0 0
20 0 7 3 0 0 0 0 0
4 0 2 0 0 2 0 0 2
87 9 21 32 27
63
40 7 16
28,4
2,9 6,9 10,5 8,8
20,6
13,1 2,3 5,2
v Néoplasie :
§ Lymphome § Autre hémo-
pathie § Cancer solide
0 0 0 0
6 3 2 1
18 9 9 0
13
11 2 0
27
18 7 2
18
16 1 1
4 2 1 1
5 2 3 0
91
61 25 5
29,7
19,9 8,2
1,6
v Maladies de
système
v Sans étiologie
v Héréditaire
1 4 5
6 2 0
3 7
10
0 2 4
3
16 0
4 2 0
3 2 0
2
20 0
22
55
19
7,2
18,0
6,2

On remarque que le SAM est secondaire à des infections dans presque la moitié
des cas. La cause néoplasique est retrouvée dans environ 30% des cas et les
maladies de système dans 7,2% des cas. Par ailleurs, aucune cause n’a été décelée
dans 18% des cas. (Figure 5
27%
7%6%
16%
Figure 5: Distribution des étiologies du SAM selon
la méta
73
que le SAM est secondaire à des infections dans presque la moitié
La cause néoplasique est retrouvée dans environ 30% des cas et les
maladies de système dans 7,2% des cas. Par ailleurs, aucune cause n’a été décelée
Figure 5)
44%
16%
infec ons
néoplasies
maladies de système
héréditaire
sans é ol ogi es
: Distribution des étiologies du SAM selon
méta-analyse de Karras et Hermine
que le SAM est secondaire à des infections dans presque la moitié
La cause néoplasique est retrouvée dans environ 30% des cas et les
maladies de système dans 7,2% des cas. Par ailleurs, aucune cause n’a été décelée
infec ons
néoplasies
maladies de système
héréditaire
sans é ol ogi es
: Distribution des étiologies du SAM selon
Karras et Hermine

74
1. les SAM post-infectieux :
Pratiquement toutes les infections, bactériennes, virales, fongiques ou
parasitaires peuvent être associées au SAM [78] (Tableau 6).
Tableau 6 : Infections associées au SAM [22]
Infections virales :
Infections bactériennes :
Infections parasitaires et
fungiques :
• HSV • VZV • EBV • CMV • HHV6 • HHV8 • Parvovirus B19 • Adénovirus • Entérovirus • HAV, HCV • HIV • Oreillons • Rubéole • Myxovirus
parainfluenzae • Dengue
• Mycobacterium tuberculosis • Mycobacterium avium • Salmonella typhi • Borrelia burgdorferi • Leptospirose • Brucellose • Chlamydia psittaci • Mycoplasma pneumoniae • Coxiella burnetii • Ehrlichiose • Rickettsiose • Syphilis • Legionella pneumophila • Pneumocoque
• Staphylocoque • Bacilles à Gram négatif
• Babésiose • Leishmaniose • Toxoplasmose • Paludisme • Strongyloidiase • Pneumocystose
• Candida albicans • Aspergillus fumigatus • Cryptococcus neoformans • Histoplasma capsulatum • Penicillium marneffei

75
L’analyse des quelques séries publiées (Tableau 5) montre que les infections
virales sont responsables de près de la moitié des cas de SAM post-infectieux.
Suivent par ordre décroissant les mycobactéries, les bactéries intracellulaires et
pyogènes, puis les parasites. Il n’est pas rare de retrouver deux agents infectieux
pathogènes chez le même patient au cours d’une hémophagocytose. [22]
Il faut souligner qu’il est parfois difficile d’imputer la survenue d’un syndrome
hémophagocytaire à une infection. D’une part, l’agent infectieux pourrait juste jouer
le rôle de facteur déclenchant sur un terrain immunologique particulier, comme c’est
le cas chez les enfants ayant une anomalie génétique particulière du système
immunitaire ou chez les patients sous traitement immunosuppresseur chronique.
D’autre part, l’immunosuppression qui résulte du SAM (secondaire à la neutropénie
et parfois aux traitements administrés) peut favoriser les surinfections secondaires
rendant parfois impossible le diagnostic étiologique. [22]
Un grand nombre des cas rapportés dans la littérature surviennent chez des
patients immunodéprimés de façon chronique (infection par le VIH, traitement
immunosuppresseur pour une transplantation ou une maladie systémique,
chimiothérapie anticancéreuse, splénectomie). Il est de ce fait difficile de savoir si
l’immunodépression favorise l’activation macrophagique en soi ou plus simplement
si c’est l’infection qui est à l’origine du SAM. On peut cependant supposer que
l’immunosuppression empêche dans certains cas l’élimination de l’agent infectieux,
entraînant une stimulation et une activation anormale des lymphocytes et/ou des
macrophages impliqués dans la réponse anti-infectieuse et souvent infectés par ces
agents pathogènes intracellulaires, viraux ou bactériens. [22]

76
a. les infections virales :
Le SAM peut être associé à toute infection virale avec une prédominance pour
les virus du groupe Herpes qui en constituent plus de la moitié des cas. [1]
Ø Le SAM associé à EBV [79, 80, 81] se rencontre à tout âge mais prédomine
chez les jeunes enfants ; les formes graves sont plus fréquentes chez
l’immunodéprimé [1], ce virus peut être impliqué dans le développement
d’un SAM lié au syndrome lymphoprolifératif lié à l’X [82]. La recherche des
mutations de SH2D1A correspondant à ce syndrome doit faire partie du
bilan à proposer aux patients ayant une forme grave d’infection à EBV car
elle est positive chez un quart des patients dans la série de Sumazaki et al.
[83]. À côté de cette circonstance particulière de déficit immunitaire, des
formes chez l’adulte immunocompétent peuvent également être mortelles.
Le SAM survient fréquemment au cours de primo-infections EBV, et plus
rarement au cours de réactivation EBV. La mise en évidence de l’ADN viral
par PCR est nécessaire au diagnostic [1]. L’infection par l’EBV associée au
SAM est particulière car elle présente une prolifération oligo ou monoclonale
de lymphocytes T ou de cellules NK infectées par l’EBV, alors que ces
populations, dans la mononucléose infectieuse, sont réactionnelles à
l’infection des lymphocytes B et polyclonales [29]. L’infection des
lymphocytes T serait rendue possible par l’expression sur les thymocytes
immatures d’une molécule apparentée au CD21 [84] et peut aboutir au
développement d’un lymphome T [29].
Ø Le cytomégalovirus (CMV), impliqué dans 30 à 50 % des causes virales, est à
rechercher systématiquement, un traitement spécifique étant disponible
[85].

77
Ø L’HHV6, l’herpes virus simplex (HSV), ainsi que le parvovirus sont
fréquemment rapportés. Les infections à adénovirus, HBV, HAV, rubéole,
VRS, rougeole, VZV [86], coxsackie sont plus anecdotiques. [1]
Ø L’infection par le VIH est connue pour être associée à des syndromes
hémophagocytaires depuis les années 1980. Dans une étude rétrospective la
fréquence est évaluée à 0,6 % des patients infectés par le VIH [87]. Les liens
entre le virus de l’immunodéficience humaine (VIH) et le SAM sont
complexes et variables dans l’évolution de l’infection, avec une implication à
la fois de l’infection virale mais aussi et surtout du déficit immunitaire
induit. Le SAM peut apparaître en même temps que la primo-infection par le
VIH, mais il s’agit d’une situation rare. Dans ce contexte, le SAM peut
répondre favorablement aux immunoglobulines intraveineuses et
l’instauration d’une thérapie antirétrovirale peut prévenir sa rechute. À
l’inverse, dans le cadre du syndrome de reconstitution immunitaire, un SAM
peut apparaître lors de l’institution du traitement antirétroviral. Par la suite,
les diverses infections opportunistes et les lymphomes pouvant émailler
l’évolution de l’infection par le VIH sont autant de facteurs favorisant le
développement d’un SAM [29]. En reprenant les 27 patients recensés par
Grateau et al. en 1997 [87] et les 21 autres cas décrits par Sailler et al. [88]
et Tiab et al. [89], nous pouvons analyser les facteurs déclenchants
potentiels. (Tableau 7)

78
Tableau 7 : Étiologies des syndromes d’activation macrophagique chez les patients infectés par le VIH (48 observations [87, 88, 89]) :
v Infection virale : 8 cas
ü 3 CMV, ü 2 VZV, ü 1 HSV, ü 1 EBV, ü 1 adénovirus.
v Infection parasitaire :
7 cas
ü 3 toxoplasmoses, ü 1 pneumocystose, ü 1 cryptococcose, ü 1 leishmaniose, ü 1 candidose.
v Infection à
mycobactéries:7 cas
ü 4 atypiques, ü 3 tuberculoses.
v Lymphomes:11 cas
ü 6 lymphomes B, ü 5 lymphomes T.
v Pas d’étiologie (sauf
VIH) :15 cas

79
b. Les infections bactériennes:
Des infections bactériennes peuvent être associées au SAM. La série de Dhote
et al. [90] rapporte 10 cas d’infections bactériennes sur 26 cas de SAM (en excluant
les SAM liées à une pathologie cancéreuse). La série pédiatrique de Veerakul et al.
[91] retrouve une étiologie bactérienne pour 9 des 27 SAM (hors pathologies
néoplasiques) avec des germes très variés : staphylocoque, salmonelles,
Enterobacter, Serratia, Penicillium. Les salmonelloses et la fièvre typhoïde [92] en
particulier sont régulièrement rapportées en association avec le SAM mais d’autres
germes ont été décrits, comme Peptostreptocccus ou Pseudomonas aeruginosa ou
Escherichia coli [93]. La relation de causalité est difficile à établir. Étant donné que la
neutropénie et le déficit immunitaire font partie du SAM, il n’est pas étonnant que
soient rapportés chez ces patients de nombreux épisodes infectieux, dont la
responsabilité dans le SAM est difficile à affirmer. Il est donc difficile de pouvoir
établir la relation de cause à effet. C’est pourquoi la notion de « bacteria-associated
hemophagocytic syndrome » de Risdall et al. [11] doit être retenue avec une grande
prudence. Une association particulièrement fréquente avec le SAM est néanmoins
indiscutable avec les infections à mycobactéries (M. tuberculosis [94, 95] ou autres)
qui nécessitent un traitement spécifique, en particulier devant la possibilité
d’infection par une mycobactérie du complexe avium [29].
Les infections bactériennes associées au SAM sont généralement très sévères,
prenant dans ce contexte, la forme d’une défaillance multi-viscérale [22]. Une étude
prospective en réanimation a montré que le myélogramme systématique chez des
patients thrombopéniques au cours d’un choc septique montrait une activation
macrophagique dans 60 % des cas [44]. Cela montre que l’hémophagocytose est
probablement sous-estimée, surtout au cours des syndromes septiques sévères et

80
qu’elle pourrait expliquer en partie les pancytopénies observées dans cette
pathologie [22].
c. Les infections fongiques et parasitaires :
L’histoplasmose est parmi les infections fongiques, la plus fréquemment
compliquée de SAM. La leishmaniose [96], quant à elle, constitue presque un modèle
expérimental d’hémophagocytose. [1]
Des syndromes d’activation ont aussi été notés au cours d’accès palustres [97].
Plus rarement et le plus souvent sur terrain immunodéprimé, ont été rapportées
d’autres infections : anguillulose disséminée, pneumocystose, aspergillose,
toxoplasmose, cryptococcose, candidose [98].
2. SAM et affections malignes :
L’hémophagocytose peut s’associer, compliquer ou même révéler une néoplasie
évolutive. Le pourcentage de syndromes hémophagocytaires attribuables à une
maladie néoplasique est difficile à préciser, variable selon les séries. L’analyse
cumulée des 8 plus grandes séries publiées (Tableau 5), totalisant plus de 300
patients, montre 20 % de lymphomes et 10 % d’autres néoplasies (hémopathies ou
tumeurs solides). [22]
a. Les hémopathies :
L’étiologie la plus fréquente dans ce groupe d’affections est le lymphome de
haut grade de malignité [99,100], pouvant être de différents types, mais
essentiellement (70 % des cas) non Hodgkinien de phénotype T ou NK.
Dans la série de Su et al. [101], sur les 23 patients atteints de lymphome avec
activation macrophagique, 15 présentaient des lymphomes T associés à l’EBV. Cette

81
entité hématologique désormais bien connue, est marquée par une
hémophagocytose fréquente et extrêmement sévère puisque sur les 22 lymphomes
T induits par l’EBV rapportés par Yao et al. [102], 15 patients sont décédés d’un SAM
d’évolution fulminante. Dans les lymphomes B, la fréquence du SAM est bien
moindre : 7 cas de SAM sur 105 lymphomes B dans la série de Miyahara et al. [99]. À
signaler cependant que dans la population d’Extrême- Orient, le pourcentage de
lymphomes B parmi les lymphomes compliqués d’hémophagocytose est plus élevé
qu’en Occident, atteignant 48 % dans une série japonaise [103].
Dans les lymphomes, le SAM pourrait résulter d’une synthèse anormale de
cytokines par les cellules tumorales, stimulant directement les macrophages ou
inhibant les fonctions cytotoxiques des lymphocytes T, avec pour conséquence
l’augmentation de la charge virale EBV et leur activation secondaire (comme cela est
constaté dans les lymphohistiocytoses familiales de l’enfant) [22]. Les lymphomes B
et T se distinguent, du point de vue du SAM, par des profils cytokiniens différents,
avec en particulier des niveaux plus élevés d’IL-6, d’IL-10 et de TNF-α pour les
lymphomes B [29].
D’autres hémopathies diverses, ont été associées à la survenue d’un syndrome
hémophagocytaire [22] :
• les leucémies aiguës myéloblastiques,
• les leucémies aiguës lymphoblastiques,
• les syndromes myéloprolifératifs,
• le myélome multiple.

82
b. Les tumeurs solides :
Le SAM a été décrit, de façon assez rare et souvent à des stades avancés, en
association avec de multiples cancers solides tels que: [22, 29,41]
Ø Mélanome,
Ø cancer de la prostate [104],
Ø cancer du pancréas [105],
Ø carcinome gastrique ou colique,
Ø thymome,
Ø sarcomes divers : rhabdomyosarcome, angiosarcome,
Ø cancer pulmonaire à petites cellules,
Ø cancer de l’ovaire,
Ø cancer du nasopharynx,
Ø tumeurs germinales………
3. Les affections auto-immunes et les maladies de système:
L’analyse des 8 plus grandes séries publiées (Tableau 5) trouve une maladie
systémique chez 7,2 % des malades. Il est souvent difficile de préciser si l’activation
macrophagique est secondaire à la maladie auto-immune ou à une infection latente
survenant chez des patients recevant très souvent un traitement
immunosuppresseur au long cours. Cependant, dans plusieurs cas décrits dans la
littérature, l’hémophagocytose révèle la maladie auto-immune et répond de façon
étonnante aux immunosuppresseurs, ce qui semble écarter un SAM réactionnel à
une infection secondaire. [22]
Plusieurs cas de lupus érythémateux systémique avec hémophagocytose sont
signalés dans la littérature, avec une fréquence de 2,4 % dans la série de Wong et al.
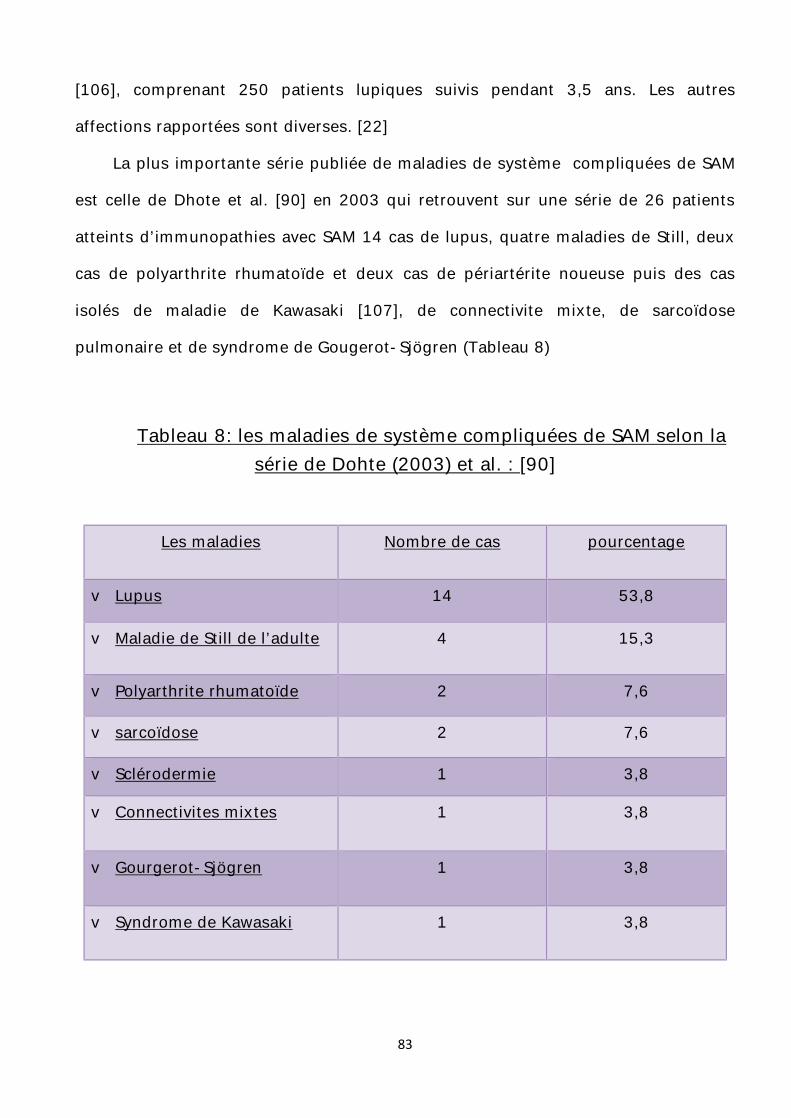
83
[106], comprenant 250 patients lupiques suivis pendant 3,5 ans. Les autres
affections rapportées sont diverses. [22]
La plus importante série publiée de maladies de système compliquées de SAM
est celle de Dhote et al. [90] en 2003 qui retrouvent sur une série de 26 patients
atteints d’immunopathies avec SAM 14 cas de lupus, quatre maladies de Still, deux
cas de polyarthrite rhumatoïde et deux cas de périartérite noueuse puis des cas
isolés de maladie de Kawasaki [107], de connectivite mixte, de sarcoïdose
pulmonaire et de syndrome de Gougerot-Sjögren (Tableau 8)
Tableau 8: les maladies de système compliquées de SAM selon la
série de Dohte (2003) et al. : [90]
Les maladies Nombre de cas pourcentage
v Lupus 14 53,8
v Maladie de Still de l’adulte 4 15,3
v Polyarthrite rhumatoïde 2 7,6
v sarcoïdose 2 7,6
v Sclérodermie 1 3,8
v Connectivites mixtes 1 3,8
v Gourgerot-Sjögren 1 3,8
v Syndrome de Kawasaki 1 3,8

84
La polyarthrite rhumatoïde est fréquemment associée au SAM, surtout dans sa
forme juvénile ou dans la forme adulte de la maladie de Still [108, 109, 110]. Avec
selon l’expérience de Janka : une protéine C réactive très élevée, une cytopénie
modérée, des taux de ferritine extrêmement élevés, une érythropoïèse diminuée sur
le myélogramme, des taux plus élevés d’IL-1, de TNF-α et d’IL-6 [29].
L’association du SAM avec le lupus érythémateux disséminé présente aussi
certaines caractéristiques : une protéine C réactive peu élevée, la rareté de l’hépato-
splénomégalie et surtout la fréquence de l’atteinte cardiaque, qui touche deux
patients sur trois, à type de péricardite ou de myocardite. [29]
4. Les SAM associées aux déficits immunitaires acquis :
Ces déficits immunitaires acquis peuvent être en rapport avec [29] :
Ø Chimiothérapie intensive.
Ø Auto-/allogreffe de cellules souches.
Ø Transplantation d’organes.
Ø Traitement immunosuppresseur au cours des maladies systémiques.
Ø Infection par le VIH.
a. SAM secondaire à la chimiothérapie et à la greffe des cellules souches
autologue ou allo-génique:
De nombreux cas de développement de SAM après chimiothérapie ont été
rapportés, par exemple dans le myélome [111], la leucémie aiguë lymphoblastique
ou myéloblastique, le lymphome de Burkitt [112].
Il est difficile de faire la part entre ce qui revient à la pathologie néoplasique à
proprement parler, aux complications infectieuses qui lui sont liées et à la
chimiothérapie [111]. Néanmoins, il faut savoir penser au développement d’un SAM
devant un tableau de neutropénie fébrile persistant, car dans ce cas un traitement
par corticoïdes peut amener à une récupération hématologique rapide [112].

85
Certaines drogues semblent plus particulièrement aptes à déclencher un SAM,
comme le méthotrexate [113].
À côté des chimiothérapies par elles-mêmes, la transplantation de cellules
souches hématopoïétiques, autologue ou allo-génique, peut être un facteur
déclenchant du SAM [111, 114, 115], alors même qu’il peut s’agir par ailleurs d’une
option thérapeutique dans le SAM. Une hypothèse physiopathologique serait la
déplétion des lymphocytes T régulateurs par le conditionnement à la greffe [114].
L’intérêt de la reconnaissance précoce du SAM réside dans les possibilités
thérapeutiques ouvertes par ce diagnostic dans un contexte de fièvre inexpliquée,
d’absence de récupération hématologique ou même de rejet du greffon [29]. Un
traitement par corticoïdes ou par immunoglobulines intraveineuses donne des
résultats favorables dans certains cas [29]. Bien que l’on retrouve souvent un
contexte infectieux pour ces SAM, un mécanisme allo-immun pourrait être impliqué
[115].
b. SAM et transplantation d’organe :
Le SAM constitue une complication rare de la transplantation d’organe et a été
décrite dans la greffe hépatique [116], rénale [117], cardiaque [118] ou intestinale
[29].
La série de Karras et al. [117] rapporte 17 observations chez des patients
greffés rénaux et permet de préciser les caractéristiques du SAM post-
transplantation d’organe. Les deux tiers des patients avaient reçu un
conditionnement à la greffe par sérum anti-lymphocytaire. Le délai moyen entre la
transplantation et le SAM était de 52 jours. La présentation clinique correspondait
aux critères classiques de définition du SAM. Une cause infectieuse était retrouvée
dans la majorité des cas : virale chez neuf patients (CMV, EBV, HSV6 ou HSV8),

86
bactérienne chez trois patients (tuberculose ou infection à Bartonella), parasitaire
chez deux autres (toxoplasmose ou infection à Pneumocystis carinii). Un traitement
immunosuppresseur ou une infection pouvant par eux-mêmes déclencher un SAM, il
est difficile de faire la part de ce qui revient à la greffe elle-même mais il n’est pas
déraisonnable de penser que le conflit immunologique hôte-greffon puisse faciliter
le développement du SAM. [29]
5. Les SAM secondaires à autres étiologies :
Divers facteurs ont été incriminés dans la survenue d’une activation
macrophagique, sans que leur rôle ne soit reconnu de façon certaine. On note ainsi
plusieurs cas d’hémophagocytose associée à [22] :
Ø la prise de certains médicaments (phénytoïne, acide valproïque, glycopeptides,
triméthoprime-sulfaméthoxazole [119]),
Ø la nutrition parentérale avec des solutés lipidiques [120],
Ø une transfusion sanguine,
Ø une vaccination,
Ø certaines anomalies innées du métabolisme, telles les glycogénoses,
Ø La splénectomie [41],
Ø Intoxication éthylique chronique [41].
Enfin, dans de rares cas, aucune étiologie n’est mise en évidence : syndrome
d’activation macrophagique « idiopathique » [41].
Le tableau 9 résume les principales étiologies du SAM secondaire.

87
Tableau 9: Principales étiologies du SAM secondaire. [29]
v Déficits immunitaires acquis ü Chimiothérapie intensive. ü Auto-/allogreffe de cellules souches. ü Transplantation d’organes. ü Traitement immunosuppresseur. ü Infection par le VIH.
v Infections ü virales en particulier EBV, ü bactéries, mycobactéries, ü parasites (leishmanioses), ü infection fongique.
v Affections néoplasiques ü Lymphomes, en particulier de sous-type T ou NK. ü Autres hémopathies néoplasiques. ü Tumeurs solides.
v Maladies de système/auto-immunes ü Polyarthrite rhumatoïde, en particulier dans sa
forme pédiatrique (maladie de Still). ü Lupus érythémateux disséminé. ü Sarcoïdose. ü Sclérodermie, dermatomyosites. ü Maladie de Kawasaki. ü Maladie de Crohn. ü Glomérulonéphrites. ü Thyroïdite d’Hashimoto.

88
C. Le bilan étiologique:
Le rôle potentiel de certains agents infectieux, du déficit immunitaire, de
certaines maladies auto-immunes et d’hémopathies lymphomateuses, dans la
survenue d'une activation inappropriée du système monocyte-macrophage, a été
envisagé. Il apparaît, au terme de cette énumération, nécessaire d'insister sur la
possible et fréquente intrication des phénomènes. L'enquête étiologique doit être
systématique et exhaustive et ne pas s'arrêter lorsqu'un facteur étiologique a été
mis en évidence car il peut en masquer un autre. Le diagnostic étiologique est
urgent car il conditionne la prise en charge thérapeutique et le pronostic. L'enquête
étiologique doit comporter ainsi systématiquement les quatre versants essentiels :
[6]
v recherche d'un déficit immunitaire
v bilan de maladies auto-immunes
v recherche d'hémopathie en particulier un lymphome.
v enquête microbiologique en particulier centrée sur l'enquête virale.
La démarche diagnostique peut se concevoir comme suit [37] :
ü un examen clinique quotidien centré sur la recherche d’adénopathies
périphériques et d’une hépato-splénomégalie, sur un examen neurologique
complet et sur un examen cutané notant la présence d’un rash ou de
manifestations hémorragiques cutanéo-muqueuses ;
ü des examens biologiques comportant initialement : numération formule sanguine
(NFS), plaquettes, protéine C réactive (CRP), fibrinogène, hémostase complète (TP,
TCA, facteurs de coagulation, D dimères latex), urée, créatinine, ionogramme
sanguin, électrophorèse des protéines plasmatiques, bilan hépatique complet,
lacticodéshydrogénase (LDH), β-2 microglobuline, triglycérides, ferritinémie,

89
ü des examens microbiologiques comportant : hémocultures et examen
cytobactériologique des urines, recherche de bacille de Koch (BK) dans les
expectorations, les urines, le sang et la moelle. Les sérologies du virus Epstein-
Barr (EBV) avec la recherche d’IgM VCA ( viral capsid antigen), du virus de
l’immunodéficience humaine (VIH), de l’herpes virus 8 (HHV8) si la sérologie VIH
est positive, du cytomégalovirus (CMV), de l’herpes virus 6 (HHV6) avec les IgM
en cas de défaillance multiviscérale, de l’herpès simplex virus, du parvovirus B19,
des hépatites A, B, et C sont systématiquement demandées. Il faut d’emblée
rechercher les génomes viraux par PCR (polymerase chain reaction) pour l’EBV
(PCR quantitative), l’HHV8 si le patient est VIH positif, la charge virale VIH si le
patient est VIH positif en sérologie, et la PCR HHV6 en cas de défaillance
multiviscérale. Une infection à CMV est recherchée par l’antigénémie CMV pp65
ou la PCR. Les autres sérologies, PCR, leucoconcentration dépendent de
l’anamnèse du patient et de son terrain (immunodépression, etc...). Le diagnostic
de SAM infectieux est en général assez facile, en n’oubliant pas que le patient
peut être infecté avec plusieurs agents et qu’une infection peut compliquer un
lymphome ou une maladie inflammatoire ou auto-immune.
ü le myélogramme est réalisé par ponction sternale ou en crête iliaque, avec une
myéloculture BK systématique. Il est fondamental de prévenir les cytologistes et
de leur expliquer l’urgence diagnostique. Les cellules atypiques doivent être
recherchées soigneusement de même que les bactéries, parasites ou
champignons en intracellulaire. La présence de cellules lymphoïdes atypiques
doit conduire très rapidement à la recherche d’un lymphome.
Le SAM est une entité dynamique qui évolue avec le temps. Il est donc
indispensable de surveiller le patient cliniquement et biologiquement. Une

90
surveillance biologique est proposée tous les deux jours, voire tous les jours en
fonction de la gravité du patient avec NFS, plaquettes, CRP, fibrinogène, TP, TCA,
urée, créatinine, ionogramme sanguin, bilan hépatique complet, LDH, β-2
microglobuline, et triglycérides. La ferritinémie peut être demandée une fois par
semaine. Les examens bactériologiques standards seront répétés, d’autant plus si le
patient est hospitalisé dans un service de réanimation.
Si le cytologiste trouve une ou des cellules atypiques sur le frottis médullaire ou
si l’étiologie du SAM n’est pas évidente après 10 jours de fièvre, il faut compléter le
bilan avec la recherche d’une hémopathie lymphoïde. La réalisation d’une biopsie
médullaire avec des immunomarquages (EBER et/ou LMP doivent être faits
systématiquement étant donné la fréquence des lymphomes liés à l’EBV), et la
recherche d’une clonalité T et B et, au mieux, un caryotype semblent très utiles. Si le
bilan hépatique est anormal et si le geste est techniquement réalisable, la biopsie
hépatique montre une dilatation sinusoïdale et une hémophagocytose dans 100 %
des cas : elle permet de faire le diagnostic étiologique dans 50 % des cas [47].
Le Tableau 10 récapitule cette démarche diagnostique.

91
Tableau 10 : récapitulatif de la démarche diagnostique devant un SAM : [37]
J0
v Examen clinique complet. v NFS, plaquettes, CRP, fibrinogène, hémostase
complète, urée, créatinine, ionogramme sanguin, électrophorèse des protéines plasmatiques, bilan hépatique complet, LDH, β-2 microglobuline, triglycérides, ferritinémie, hémocultures, ECBU, recherche de BK. Sérologies : EBV, HHV8, CMV, HSV, HHV6, VIH, parvovirus B19, hépatites A B, et C. PCR quantitative EBV, HHV8, HHV6, charge virale VIH. Antigénémie CMV pp65 ou PCR CMV.
v Myélogramme ± biopsie médullaire. v ± Clonalité sang + moelle, caryotype.
J1, J2, J4, J6, J8, J10
v Examen clinique complet. v NFS, plaquettes, CRP, fibrinogène, TP, TCA,
D dimères (latex), bilan hépatique complet, LDH, triglycérides, urée, créatinine, ionogramme sanguin, ferritinémie.
v ± Myélogramme, biopsie médullaire, biopsie hépatique.
v ± Clonalité sang + moelle, caryotype.
J15, J20, J25, ≥30
v Examen clinique complet. v NFS, plaquettes, CRP, fibrinogène, TP, TCA,
D dimères (latex), bilan hépatique complet, LDH, triglycérides, urée, créatinine, ionogramme sanguin, ferritinémie.

92
VIII. TRAITEMENT
SOMMAIRE :
A. Traitement symptomatique............................................................................93
B. Traitement étiologique...................................................................................94
1. Moyens.......................................................................................................94
a. Moyens thérapeutiques contrôlant l’inflammation excessive du SAM.......94
b. Moyens thérapeutiques supprimant la cause déclenchant le SAM.............99
2. Indications.................................................................................................99
a. SAM primaires..........................................................................................99
b. SAM secondaires....................................................................................100

93
VIII. TRAITEMENT :
Tout clinicien confronté à un patient atteint de SAM doit garder en mémoire
trois maximes qui s’appliquent à la majorité des cas de SAM [37]:
v le SAM est une urgence diagnostique, il faut tout faire pour trouver rapidement le
ou les étiologies du SAM ;
v le SAM est une urgence thérapeutique, il faut traiter le ou les étiologies du SAM
dans les meilleurs délais ;
v le problème majeur reste le délai diagnostique du SAM : Dans les formes sévères
(avec facteurs de mauvais pronostic), le traitement est urgent. Dans les cas moins
graves, l’enquête étiologique peut être menée avant, afin de ne pas gêner
l’interprétation des prélèvements histologique; les prélèvements doivent
néanmoins être réalisés rapidement, l’aggravation clinique pouvant être rapide et
brutale [1].
Le traitement des syndromes hémophagocytaires est assez mal codifié et
aucune étude thérapeutique n’a été réalisée, en dehors du cadre des
hémophagocytoses héréditaires de l’enfant. Le traitement associe: un traitement
symptomatique toujours nécessaire, un traitement pathogénique et un traitement
spécifique chaque fois que l’étiologie de la maladie a pu être élucidée.
A. Traitement symptomatique : [35]
Le traitement symptomatique constitue le premier temps de la stratégie
thérapeutique et doit être débuté rapidement.
Ce traitement vise à palier l’hémodilution, les troubles de l’hémostase, l’anémie
et les infections associées. Il consiste en :
ü une restriction hydrique,
ü des antipyrétiques,

94
ü un support transfusionnel fait de plaquettes, d’érythrocytes et de fibrinogène,
ü un traitement anti-infectieux empirique nécessaire devant tout épisode de
leucopénie fébrile,
ü parfois des anticonvulsivants.
Une splénectomie peut être envisagée, en dernier recours si la cytopénie
sanguine est profonde et surtout s’il existe un hypersplénisme. Mais son efficacité
est transitoire.
L’évolution est favorable en 1à 8 semaines dans 46 à 70% des cas avec un
traitement anti-infectieux adapté et des mesures symptomatiques.
B. Traitement étiologique :
1. Les moyens :
Le principe de traitement du SAM est de contrôler d’une part l’inflammation
excessive et d’autre part de supprimer la cause déclenchante quand il s’agit de
forme secondaire.
a. Moyens thérapeutiques contrôlant l’inflammation excessive du SAM :
ü Les corticoïdes :
Le traitement anti-inflammatoire repose essentiellement sur les corticoïdes, qui
inhibent les fonctions lymphocytaires cytotoxiques, la sécrétion de cytokines ainsi
que les fonctions des cellules dendritiques. [29]
Étant donné que la dexaméthasone traverse mieux la barrière hémato-
encéphalique que d’autres corticoïdes, son utilisation est à privilégier dans les cas
où existe une atteinte neurologique. Néanmoins, il pourrait être utile d’effectuer des
injections intrathécales de dexaméthasone chez les patients présentant des signes
neurologiques non rapidement résolutifs par le traitement systémique. [29,49]

95
L’intérêt des formes liposomiques de corticoïdes, qui auraient l’avantage
théorique de mieux pénétrer dans les macrophages, reste à démontrer. [29]
ü Les immunoglobulines :
Les gammaglobulines intraveineuses sont souvent utilisées dans les SAM, bien
que l’on ne dispose pas d’essai randomisé démontrant leur réelle efficacité, mais un
taux de réponse global de près de 60 % est avancé. [29]
Larroche et al. [121] ont ainsi rapporté 17 cas de SAM avant tout associés à
une affection virale traités avec bénéfice par gammaglobulines à forte dose (dose
moyenne utilisée 1,6 mg/kg —1 ou 2 cycles). Le taux de réponse était de 78 % pour
les SAM d’origine infectieuse et de 39% dans les autres étiologies, avec une
inefficacité notable dans les SAM associées aux lymphomes ou autres pathologies
malignes.
Il est nécessaire d’instituer le traitement par immunoglobulines pendant la
phase précoce d’installation du SAM, correspondant à la période d’augmentation des
taux de ferritine, dont la diminution sert de marqueur d’efficacité du traitement
[122].
Les mécanismes présumés de l’efficacité des immunoglobulines intraveineuses
sont multiples : clairance des agents pathogènes ayant déclenché le SAM ou de
superantigènes, régulation du réseau anti-idiotypique et cytokinique, saturation des
récepteurs Fc [29].
ü La ciclosporine A :
La ciclosporine est un agent immunosuppresseur, elle intervient dans les
premières étapes de l’activation des lymphocytes T entraînant une défaillance de la
transcription des gènes responsables de l’activation dite « précoce » tels les gènes
codant pour les cytokines [123], elle agit également en inhibant l’expression de

96
l’IL-6, l’IL-1, TNF-α, la nitrite-oxyde synthétase et la cyclo-oxyénase 2 [49].
La posologie utilisée est de 3 à 7 mg/kg/j avec une résolution rapide des
symptômes : disparition de la fièvre dans les 24h et normalisation des anomalies
biologiques dans les jours qui suivent [123,124]. La durée du traitement par la
ciclosporine A n’est pas encore bien définie, la majorité des auteurs l’utilisent
jusqu’à normalisation des marqueurs biologiques [33].
ü L’étoposide (VP16) : [29]
Une drogue cytotoxique joue un rôle majeur dans les SAM, l’étoposide.
L’étoposide présente un effet cytostatique en particulier sur les lymphoproliférations
T, et son utilisation, en association à la ciclosporine, est naturellement justifiée dans
ce contexte. De plus, l’étoposide peut avoir un effet antiviral sur l’EBV en bloquant
l’expression de l’EBNA (Epstein Barr Nuclear Antigen). Ces données in vitro se
traduisent in vivo par une mortalité très élevée des patients atteints de SAM liée à
l’EBV et ne recevant pas d’étoposide de façon précoce [125].
Ce produit est généralement administré par voie veineuse, dans un premier
temps, à des doses généralement de 100 à 150 mg/m² par 24 heures, puis par voie
orale, le plus souvent à des doses de l’ordre de 50 mg/m² par jour. [45, 126]
Cependant, les effets secondaires gonadotoxiques, myélotoxiques,
cardiotoxiques et l’incapacité de traverser la barrière hématoméningée de ce
produit, en limitent l’utilisation, mais ne sauraient être des obstacles à sa
prescription en urgence dans les formes graves, compte tenu de sa rapidité d’action
qui est de 24 à 48 heures. [126]

97
ü Les globulines anti-thymocytes :
Bien que les globulines anti-thymocytes puissent être l’équivalent de
l’étoposide dans des situations où la maladie est réfractaire, le coût et les effets
secondaires potentiels (comme les réactions allergiques, et l’immunodépression
sévère) limitent leur utilisation. [49,127]
ü Les échanges plasmatiques et les plasmaphérèses :
Les échanges plasmatiques on été décrits dans plusieurs séries avec des
résultats positifs dans la plupart des cas. Ils sont actifs sur l’hypercytokinémie
qu’ils peuvent atténuer. [49,128]
ü Traitement anti-TNF α :
Divers autres types de traitements ont été appliqués au SAM, en particulier le
blocage du système du TNF par des anticorps anti-TNF α (infliximab Remicade*)
[129] et par le récepteur soluble de TNF-α (etanercept Enbrel*).
L’utilisation de l’Etanercept à la dose de 0,4 mg deux fois par semaine
pendant quatre semaines après le diagnostic de SAM a montré son efficacité avec
une diminution des symptômes dans les 24 heures suivant l’injection [130].
Si des succès ont été obtenus, la plus grande circonspection doit être de mise
dans ce type de traitement dont on a pu montrer, dans la polyarthrite rhumatoïde,
qu’il pouvait au contraire déclencher un SAM, peut-être par le biais d’un effet
facilitant sur le développement d’infections, d’où l’importance de n’appliquer ce
genre de traitement que si toute infection est exclue et notamment une tuberculose
dans notre pays. [29, 53, 131,138]

98
ü Greffe allo-génique de la moelle osseuse :
Représente le traitement de choix dans la lymphohistiocytose familiale et dans
les autres formes héréditaires du SAM. Cependant, elle est rarement indiquée chez
l’adulte : en cas de SAM réfractaire associé à l’EBV ou dans un contexte de
transplantation pour une néoplasie hématologique sous-jacente. [49,126]
ü Autres :
L'administration de fludarabine, un antimétabolite purinique, entraîne une
immunosuppression profonde en agissant en particulier sur les lymphocytes T et les
cellules NK. Ce produit a démontré son efficacité dans la lymphohistiocytose
familiale. [132]
Le Méthotrexate a été employé par voie intra-thécale dans le protocole HLH-94
[133] et pourrait être une option thérapeutique dans les maladies rhumatismales
associées au SAM, étant donné que le méthotrexate est un moyen thérapeutique
standard dans les arthrites inflammatoires chroniques. Cependant, ce produit peut
potentiellement déclencher un SAM. [113,134]
La chimiothérapie par le 2-CdA (2 chlorodéoxydénosine) peut être un moyen
thérapeutique du SAM. C’est un dérivé chloré de la déoxydénosine, résistant à
l’adénosine-désaminase et transformé en chlorodésoxy-ATP entrainant une
apoptose des lymphocytes. [6,22]
Daclizumab, un anticorps anti-CD25, et l’interféron α, doivent encore trouver
leur place dans le traitement du SAM, bien qu’il existe de fortes raisons pour qu’ils
soient utilisés. [49]
Finalement, une thérapie dirigée contre la cellule B via un anticorps Anti-CD20
(rituximab*) pourrait être une approche prometteuse dans quelques cas de SAM
secondaire à une infection par l’EBV. [49]

99
b. Moyens thérapeutiques supprimant la cause déclenchant le SAM :
Bien entendu, le traitement spécifique de l’affection ayant déclenché le SAM
doit être institué sans délai.
Ø Traitement anti-infectieux :
Avant même les résultats de l’enquête infectieuse, il est utile d’instituer un
traitement antibiotique à large spectre, en veillant aussi à couvrir plus
particulièrement les germes intracellulaires. Les traitements antiviraux,
antirétroviraux, antifongiques ou antiparasitaires sont rapidement introduits au
moindre doute en fonction du contexte clinique. Ceci est d’autant plus vrai que les
traitements spécifiques du SAM ont une action immunosuppressive qui pourrait
aggraver l’évolution de la pathologie infectieuse.
Ø Traitement anti néoplasique :
Le contexte de néoplasie avérée relève du traitement spécifique de la tumeur.
Ø Traitement immunosuppresseur :
En cas de maladie systémique.
2. Les indications :
a. Les SAM primaires :
Le traitement des formes du SAM associées à un déficit immunitaire primitif
relève essentiellement de la greffe de cellules souches allo-géniques.
Les résultats du protocole HLH-94, destiné à des enfants ayant une forme
familiale du SAM, mais aussi une forme récurrente ou persistante [133,135],
permettent de disposer d’une base rationnelle dans la gestion des SAM de l’enfant.
Le protocole comprenait une phase de chimiothérapie par étoposide et

100
dexaméthasone d’une durée de 8 semaines, suivie d’un entretien comprenant des
bolus de dexaméthasone alternant avec de l’étoposide, associés à de la ciclosporine
A [133]. Ce protocole de chimiothérapie et son entretien ont permis d’obtenir un
taux de réponse complète de 78%, les décès constatés (25 patients sur 113) étant
dus dans leur grande majorité (20 sur 25) à la progression du SAM [133]. Cette
phase de chimiothérapie est indispensable avant la greffe, même autologue, car
« l’orage cytokinique » déclenché par cette procédure peut exacerber le SAM.
L’allogreffe de cellules souches, réservée aux patients ayant une forme familiale
ou réfractaire, a donné dans l’étude de Henter un taux de survie sans maladie à 3
ans de 62 %, la majorité des décès (17 sur 25) étant dus aux complications de
l’allogreffe.
Étant donné que la distinction entre SAM familial primitif par déficit
immunitaire et SAM secondaire est parfois difficile, Horne et al. [136] ont essayé de
prévoir, en se fondant sur le type de déficit des cellules NK, quels sont les patients
qui auront a priori besoin d’une allogreffe. Parmi les quatre types définis par
Schneider et al. [52], le type 3 correspond à un déficit complet de la cytotoxicité. Sur
les 22 patients de type 3 allo-greffés, 14 ont survécu alors qu’à l’opposé sur les 14
patients de type 3 non allo-greffés, 11 sont décédés, suggérant la nécessité de
l’allogreffe plus particulièrement pour ce sous-type de gravité majeure [136]
b. Les SAM secondaires :
Ø SAM et infections : [37]
Dans les SAM d’étiologie infectieuse, à coté d’un traitement anti-infectieux à
large spectre (antibactérien, antiviral, antifungique et antiparasitaire), le traitement
par des immunoglobulines intraveineuses (Ig IV) a été proposé en première intention
sur la base d’une étude rétrospective réalisée avec l’aide du groupe d’experts sur les

101
Ig IV du Comité d’évaluation et de diffusion des innovations technologiques de
l’Assistance Publique-Hôpitaux de Paris [121].
La dose préconisée est de 2 grammes par kilo et par cure, en réalisant une
seule cure. La réponse aux Ig IV est rapide avec disparition de la fièvre et
amélioration des cytopénies, en moyenne dans les 8 jours suivant la perfusion des Ig
IV.
En cas d’inefficacité, une corticothérapie générale est recommandée par des
injections quotidiennes de méthylprednisolone habituellement à fortes doses (500 à
1 000 mg/j). Si le SAM est réfractaire, le pronostic du patient est engagé et
l’utilisation de l’étoposide seul ou associé à d’autres thérapeutiques paraît légitime.
Les patients atteints de SAM lié au virus Epstein- Barr doivent impérativement
recevoir de l’étoposide dans le régime thérapeutique, comme l’ont parfaitement
démontré Imashuku et al. [125].
Ø SAM et hémopathies :
Dans les SAM d’étiologie lymphomateuse, le pronostic du patient est
malheureusement très réservé. En attente de la caractérisation exacte de la
prolifération lymphoïde, il paraît justifié d’utiliser d’emblée l’association de
corticoïdes et de l’étoposide. [37]
Les anticorps monoclonaux anti-CD20 (rituximab) peuvent être d’un grand
apport dans les lymphoproliférations B. [37]
Ø SAM et maladies auto-immunes ou maladies de système:
Dans les SAM compliquant les maladies auto-immunes ou inflammatoires
(principalement le lupus érythémateux systémique et la maladie de Still en poussée)
en l’absence d’infection à l’origine du SAM, la corticothérapie générale est
habituellement suffisante.

102
Les Ig IV peuvent être une alternative thérapeutique intéressante dans ces
situations.
Les anticorps monoclonaux anti-TNF semblent être prometteurs dans la
maladie de Still [137]. Un cas d’aggravation d’un SAM après deux injections
d’étanercept a été rapporté chez une jeune femme de 22 ans ayant une maladie de
Still d’après les auteurs, mais surtout une primo-infection EBV qui explique cet
échec. [37]
Ainsi, il n’y a pas de protocole définitivement établi pour
le traitement du SAM. La prise en charge des syndromes
hémophagocytaires doit être précoce. Le traitement de l’affection
déclenchante (infection virale, hémopathie maligne, cancer solide) est
nécessaire mais non suffisant. En effet le traitement du SAM doit
interrompre la réaction hyper-inflammatoire qui s’auto-entretient. Ceci
amène à utiliser, sous couvert du contrôle d’éventuels processus
infectieux, différents traitements immunosuppresseurs ou immuno-
modulateurs (corticoïdes, ciclosporine, immunoglobulines
intraveineuses), voire, en particulier dans les cas où l’EBV est impliqué,
une chimiothérapie par l’étoposide.

103
IX. EVOLUTION ET PRONOSTIC

104
IX. EVOLUTION ET PRONOSTIC :
Sous traitement, l’évolution peut être favorable, une rémission ou une guérison
du SAM est alors obtenue, la résolution des symptômes et des anomalies
biologiques s’effectue assez rapidement, en moyenne entre 1 et 8 semaines. La
disparition totale des signes d’hémophagocytose au niveau médullaire peut être plus
tardive et persister plusieurs semaines ou mois sans que cela ait une signification
particulière. Les rechutes, une fois la guérison obtenue, sont possibles en particulier
au cours de la maladie lupique ou de certains lymphomes. Les évolutions
chroniques sont possibles, notamment au cours du sida. [35,41]
Non traité, l’évolution du SAM est fatale. Le décès est précoce dans les quatre à
huit semaines, souvent en rapport avec une défaillance multi-viscérale, une
hémorragie, ou un sepsis. [1]
Le pronostic vital du SAM reste globalement réservé, et est essentiellement lié à
la maladie associée.
Dans les SAM héréditaires, la greffe de moelle allo-génique a totalement
modifié le pronostic. Ainsi, selon le registre international de lymphohistiocytose
familiale (122 patients) la survie à 5 ans est à peine de 10 % pour les patients traités
par chimiothérapie conventionnelle (VP16- corticoïdes–méthotrexate intrathécal),
alors qu’elle est de 66 % pour les patients ayant pu bénéficier d’une allogreffe [139].
Le schéma de conditionnement idéal pour l’allogreffe n’étant pas encore établi, des
avancées peuvent encore être espérées dans ce domaine.

105
Dans les SAM réactionnels, le pronostic dépend de plusieurs paramètres : [22]
• précocité du diagnostic,
• positivité du bilan étiologique,
• mise en route précoce d’un traitement anti-infectieux adapté,
• étiologie néoplasique associée,
• statut immunitaire antérieur (HIV, immunodéprimé).
Dans la méta-analyse cumulant les principales séries publiées, le pronostic est
défavorable dans environ 48 % des cas, montrant bien la gravité de cette pathologie
[22].
Le Tableau 11 montre la mortalité du SAM selon ces différentes séries.

106
Tableau 11: mortalité du SAM selon différentes études. [1,22]
Nombre de
cas
Nombre de décès
Pourcentage de décès(%)
v Risdall, 1979
19
5
26
v Reiner, 1988
23
7
30
v Albert, 1992
45
28
62
v Tiab, 1996
23
17
74
v Wong, 1992
40
18
45
v Sailler, 1997
99
49
49
v Tsuda, 1997
23
5
22
v Kaito, 1997
34
20
59
v Dhote, 2002
26
10
38
v Total
332
159
48

107
En essayant d’analyser les différents sous groupes de patients, il apparaît que
les patients dont le pronostic est le plus réservé sont ceux qui sont infectés par le
VIH ou atteints d’une hémopathie maligne [22]. Ainsi, sur les 26 patients infectés
par le VIH étudiés rétrospectivement par Grateau et al. [87], 19 étaient décédés en
moins d’un an.
Les lymphomes associés à une hémophagocytose ont également un pronostic
extrêmement péjoratif : la médiane de survie est de 44 jours pour les 12 lymphomes
T induits par l’EBV rapportés par Yao et al. [102], et de 9 mois pour les 25
lymphomes B repris par Shimazaki et al. [140]. Ces chiffres seraient confirmés par
l’analyse rétrospective japonaise de 134 cas de lymphome avec activation
macrophagique [103], où la médiane de survie est de 69 jours pour les lymphomes
T et 242 jours pour les lymphomes B. De façon générale, la survenue d’un SAM est
un facteur de très mauvais pronostic dans les hémopathies malignes [100]. Seule la
possibilité d’une allogreffe de moelle semble pouvoir offrir un traitement efficace
dans ces cas, tout en s’exposant aux risques inhérents à cette transplantation à haut
risque. [22]
Ø Facteurs de pronostic : [22,37]
L’analyse de quelques grandes séries a permis de dégager certains autres
facteurs de mauvais pronostic, en plus de la pathologie associée. Ainsi Kaito et al.
[87] ont élaboré des facteurs de risque corrélés au décès des patients dans une série
de 34 malades âgés de plus de 15 ans.
v Les facteurs corrélés au décès à l’admission du patient sont :
ü un âge de plus de 30 ans,
ü la nature de la pathologie sous-jacente,
ü une hémoglobine inférieure à 10 g/dL,
ü des plaquettes à moins de 100 000/mm³,

108
ü une ferritinémie supérieure à 500 μg/L,
ü des produits de dégradation de la fibrine supérieurs à 10μg/mL et
ü une β-2 microglobuline supérieure à 3 μg/mL.
v Pendant l’hospitalisation les facteurs de mauvais pronostic sont :
ü une chute de l’hémoglobine,
ü une baisse des plaquettes et
ü l’apparition d’une cholestase (bilirubine > 22 μmol/l, phosphatases
alcalines > 740 UI/l).
La sévérité de la cholestase (et non de la cytolyse hépatique) est également
corrélée à un pronostic fatal pour Kerguenec et al. [47] dans leur série comprenant
30 patients avec SAM et atteinte hépatique, tout comme l’hypofibrinogénémie et la
diminution du facteur V plasmatique
Dans des plus petites séries, l’augmentation d’autres paramètres, non dosés
de façon usuelle semble liée à une gravité plus importante: taux plasmatiques de
TNF-α, d’IFN , et le récepteur soluble à l’IL-2.

109
PARTIE II : ETUDE PRATIQUE

110
PARTIE II: ETUDE PRATIQUE
Nous rapportons dans ce chapitre un cas de SAM secondaire à une maladie
cœliaque, colligé dans le service de médecine interne au CHU HASSAN II de Fès.
A travers l’analyse de cette observation, nous comparerons les éléments
diagnostiques, pronostiques et thérapeutiques retrouvés chez notre patiente aux
données actuellement disponibles dans la littérature.
Nous proposerons une conduite à tenir pratique en cas de suspicion de SAM.

111
NOTRE OBSERVATION

112
I. NOTRE OBSERVATION
Madame Hayat Z. âgée de 42 ans, divorcée et nullipare, originaire et habitant
Fès, est hospitalisée le 21 septembre 2007, au service de médecine interne au CHU
HASSAN II de Fès, pour un syndrome hémorragique.
Elle est suivie depuis 12 ans pour une anémie ferriprive, secondaire à une
géophagie, mise sous traitement martial qu’elle prend toujours.
Elle rapporte une notion d’hémorroïdes traitées, elle n’a pas d’antécédents
d’ictère ni de prise médicamenteuse ni de transfusion. Depuis 2 ans la patiente
présente une notion de diarrhées liquidiennes chroniques à raison de 6 selles par
jour sans glaire ni sang ni autre signe digestif. Le 10 septembre 2007 un syndrome
hémorragique cutané s’est installé de façon brutale chez notre patiente, fait de
grosses ecchymoses intéressant les membres supérieurs et un grand placard
hémorragique au niveau de la cuisse gauche avec des œdèmes des membres
inférieurs surtout à gauche.
Le tout évolue dans un contexte de fièvre et d’altération de l’état général.
A l’admission l’examen clinique a noté :
§ Un état général altéré, avec un subictère conjonctival et une pâleur cutanéo-
muqueuse. Une fièvre à T°=38,5°C.
§ Des œdèmes des membres inférieurs surtout à gauche prenant le godet.
§ L’examen cutanéo-muqueux trouve des ecchymoses étendues au niveau des
membres supérieurs et inférieurs sans d’autres lésions.
§ L’abdomen est souple légèrement augmenté de volume, sans masse palpable. Il
n’y avait pas d’hépatomégalie ni de splénomégalie. A la percussion on retrouve
une ascite de moyenne abondance.
§ Par ailleurs, il n’y a pas de signes respiratoires, l’examen cardiovasculaire et
neurologique sont sans particularités.

113
Un bilan paraclinique a été réalisé
• Une numération formule sanguine montrant :
-Une anémie normochrome normocytaire avec Hb=7,8g/dl,
VGM=96µm ³, CCMH=30,8% et un taux de réticulocytes à2%,
-Une hyperleucocytose à 19 400 éléments /mm³ avec une prédominance des
PNN.
-Une thrombocytose à 591 000 éléments /mm³.
-Le frottis est sans constat de schizocytes.
• Le bilan d’hémostase a montré un taux de prothrombine abaissé à 25%. Le TCA
est allongé à 55/34’’. Le taux de fibrinogène est bas à 2,42g/l.
• Le bilan hépatique :
-Transaminases augmentées:
SGOT=70UI/l (1,75 fois la valeur normale)
SGPT=121UI/l (3 fois la valeur normale)
-Bilirubine : Totale augmentée à 72,5 µmol/l
Directe=46,17 µmol/l
Indirecte= 26,3 µmol /l
-Gamma glutamyl transférase augmentée à 242U/l
- LDH augmenté à 1439UI/l.
• Le bilan lipidique :
-Les triglycérides sont augmentés à 2,63g/l (soit 3mmol/l).
-le cholestérol total est élevé à 4 g/l, avec un taux de LDL cholestérol à
3,5g/l, et un taux d’HDL cholestérol à 0,1g/l.
• Le bilan inflammatoire :
-VS=27 mm à la première heure, 63mm à la deuxième heure.
-CRP négative à 4,8 mg/l.

114
• La fonction rénale est normale avec une urée sanguine à 0,18g/l.
• L’ECBU a montré une leucocyturie à 240.000 éléments/ml avec culture positive à
E.coli.
• Les sérologies des hépatites A, B et C sont négatives.
• La protéinurie de 24h est à 638mg/24h (expliquée par l’infection).
• Sur l’ionogramme sanguin on note une glycémie normale à 1,02 g/l, une
hypocalcémie à 69ng/l, une hypoprotidémie à 56,4g/l. La natrémie et la kaliémie
sont normales.
• L’échographie abdomino-pelvienne retrouve :
-Une stéatose du foie,
-un épanchement intrapéritonéal libre de moyenne abondance.
• Une tomodensitométrie thoraco-abdomino-pelvienne a montré un
épaississement digestif étendu circonférentiel et asymétrique, avec de multiples
adénopathies intrapéritonéales dont les plus volumineuses siégeant au niveau de
la fosse iliaque droite, faisant 15 mm de diamètre.
Vu le syndrome de malabsorption :
• une FOGD a été réalisée avec biopsie jéjunale, l’étude histologique a été
compatible avec une maladie cœliaque (image 5),
• les taux des anticorps anti-gliadine demandés se sont révélés fortement positifs
confortant ainsi le diagnostic de maladie cœliaque.
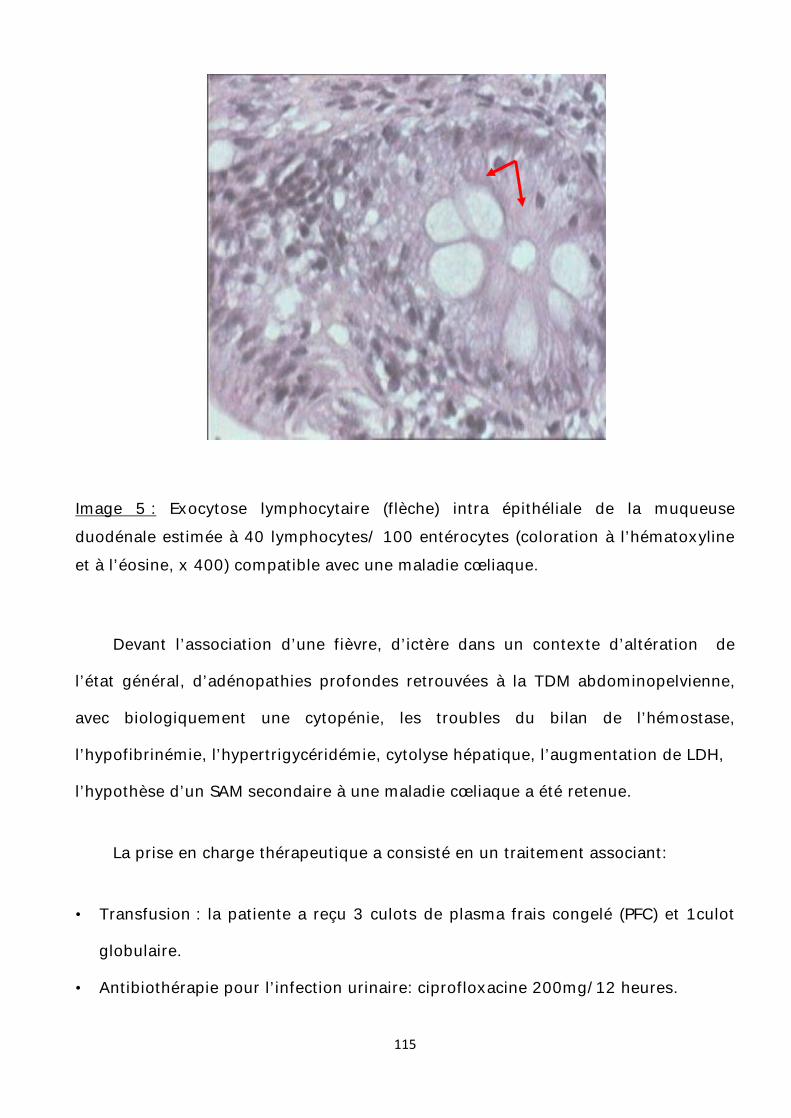
115
Image 5 : Exocytose lymphocytaire (flèche) intra épithéliale de la muqueuse duodénale estimée à 40 lymphocytes/ 100 entérocytes (coloration à l’hématoxyline et à l’éosine, x 400) compatible avec une maladie cœliaque.
Devant l’association d’une fièvre, d’ictère dans un contexte d’altération de
l’état général, d’adénopathies profondes retrouvées à la TDM abdominopelvienne,
avec biologiquement une cytopénie, les troubles du bilan de l’hémostase,
l’hypofibrinémie, l’hypertrigycéridémie, cytolyse hépatique, l’augmentation de LDH,
l’hypothèse d’un SAM secondaire à une maladie cœliaque a été retenue.
La prise en charge thérapeutique a consisté en un traitement associant:
• Transfusion : la patiente a reçu 3 culots de plasma frais congelé (PFC) et 1culot
globulaire.
• Antibiothérapie pour l’infection urinaire: ciprofloxacine 200mg/12 heures.

116
• Traitement de l’hyperlipémie :
-régime hypolipidique.
-fibrates : une gélule par jour.
• traitement de la maladie cœliaque :
-régime sans gluten.
L’évolution a été marquée par une amélioration spectaculaire sur le plan
clinique (patiente est apyrétique avec amélioration de l’état général et disparition du
syndrome hémorragique cutané) et paraclinique : diminution de la VS, SGOT, SGPT
et normalisation du bilan lipidique et du bilan de la crase.

TABLEAU 12 : TABLEAU RECAPITULATIF DE L’OBSERVATION MEDICALE J1 J3 J7 J10 J14 J18 J19 J32
clinique Fièvre+AEG ecchymose sub-ictère
OMI
Apyrexie Subictère
Régression des OMI
Amélioration de l’état général
Apyrexie
Bon état général
NFS Hb en g/dl 7,8 12,9
10,1
11 11
GB/ml 19400
17200
12300
11000 9500
Plaquettes/ml 591000 560 000 577 000 537 000 410 000 TP% 25 34 70 100 100 Fibrinogène g/l 2 ,4 Ionogramme glycémie 1 0, 8
Na 139 Urée 0,18 0,3 Protides 56,4 calcémie 69
Autres SGOT/SGPT 70/121 244/75 185/77 131/71 Bilirubine totale µmol/l 72,5 α-GT U/l 242 LDH U/l 1439 Triglycérides g/l 2,63 2,52 2 Choles total/LDLch g/l 4/3,54 3,97/3,36 1,61/0,95 VS(1ére heure/2éme h) 27/63 57/72 17/47 ECBU positif à E.coli échographie Épanchement moyen TDM ADP intra-péritonéales
Traitement transfusion+antibiothérapie+ traitement de la maladie cœliaque

118
DISCUSSION
SOMMAIRE :
A. Notre observation..................................................................119
B. Suggestion d’une conduite à tenir pratique devant un SAM....122

119
II. DISCUSSION
A. Discussion de notre observation:
Le syndrome d’activation macrophagique (SAM) est une entité anatomoclinique
caractérisée par une symptomatologie associant des anomalies cliniques,
biologiques et anatomopathologiques. Il peut être primaire essentiellement chez
l’enfant, ou secondaire à diverses affections : hématologiques, infectieuses ainsi
qu’à des maladies auto-immunes variées.
Le SAM survient le plus souvent sur un terrain d’immunodépression, ou plus
largement de dysrégulation immunitaire. Le mécanisme physiopathologique serait
au départ une anomalie de la collaboration lymphocyte T-macrophage. Il en
résulterait une hypersécrétion de cytokines avec une phagocytose anormale des
éléments figurés du sang.
Dans notre observation, la patiente avait une infection urinaire confirmée par
l’ECBU qui s’est révélé positif à E.coli. Ceci rejoint les données de la littérature qui
suggèrent que le facteur déclenchant du SAM est le plus souvent une infection
intercurrente.
Notre patiente a présenté un ensemble de signes cliniques et biologiques
définissant le SAM qui est dans notre cas secondaire à une maladie cœliaque. Nous
retrouvons les principaux signes du syndrome : la fièvre, l’altération de l’état
général, qui sont quasi-constants dans tous les cas rapportés dans la littérature.
L’ictère quant à lui n’est pas constant. Les manifestations cutanées sont
essentiellement des ecchymoses secondaires au trouble de l’hémostase, et des
œdèmes. Par ailleurs l’absence de signes neurologiques, qui peuvent parfois être
inauguraux, témoigne de la gravité moyenne du SAM.

120
L’ascite de moyenne abondance retrouvée chez notre patiente sur le plan
clinique et radiologique, peut être observée au cours du SAM.
L’organomégalie faite d’hépato-splénomégalie et d’adénopathies, était absente
chez notre patiente, néanmoins des adénopathies profondes ont été objectivées sur
la tomodensitométrie abdomino-pelvienne.
Le tableau biologique est fortement évocateur du SAM. La cytopénie,
l’hypertrigycéridémie, l’hypofibrinémie, l’augmentation de LDH, les perturbations du
bilan hépatique, les troubles de l’hémostase, sont des signes rencontrés
habituellement dans le SAM, et ont été retrouvés chez notre patiente. Cependant,
nous ne disposons pas du taux de ferritine dans notre observation, dont
l’interprétation risquait d’être difficile à cause de la malabsorption.
La preuve histologique mettant en évidence des images d’hémophagocytose
manque dans notre cas. Toutefois, l’hémophagocytose n’est pas obligatoire pour
faire le diagnostic : un tableau clinico-biologique cohérent est par contre primordial.
Signalons que les images d’hémophagocytose peuvent être inconstantes. La
répétition du myélogramme est indispensable s’il est d’abord négatif. Ceci donc
peut engendrer un retard du diagnostic voire l’absence du diagnostic.
Le degré de gravité du SAM dans notre observation peut être évalué en
confrontant les paramètres retrouvés chez notre patiente aux données de la
littérature. Sans oublier de signaler que certains éléments dits pronostiques dans la
littérature ne sont pas dosés de façon usuelle et n’ont pas été recherchés chez notre
patiente, il s’agit du taux de bêta 2 microglobuline, les taux plasmatiques de :
l’IFN , de TNF-α et du récepteur soluble de l’IL-2. Dans notre cas on trouve :
• L’âge plus de 30 ans est facteur de mauvais pronostic.

121
• La pathologie sous jacente est une maladie auto-immune ce qui est de bon
pronostic.
• Sur le plan clinique :
-l’altération de l’état général dont le terrain clinique est fragilisé par le
syndrome de malabsorption due a la maladie cœliaque.
-la présence d’un subictère d’intensité moyenne sans autres signes de
défaillance viscérale.
-la présence d’une ascite de moyenne abondance.
-l’absence de signes neurologiques.
• Sur le plan paraclinique :
-l’hémoglobine inférieure à 10g/dl mais sans thrombopénie.
-la perturbation du bilan de l’hémostase avec hypofibrinémie.
-la présence d’une cholestase qui est corrélée à un pronostic fatal selon
Kerguenec et al. [47]
-L’hypercholestérolémie qui peut être considérée comme un facteur de
mauvais pronostic comme le rapportent Wada Y. et al. [47‘]
Au bout de cette confrontation des paramètres pronostiques aux données
recensées de la littérature, ce SAM secondaire à la maladie cœliaque chez notre
patiente peut être qualifié de moyennement grave.
Les options thérapeutiques décrites dans la littérature, ont été utilisées chez
notre patiente, elle a bénéficié d’un traitement symptomatique avec une
antibiothérapie pour l’infection urinaire, et d’un traitement étiologique spécifique de
la maladie cœliaque. Signalons qu’on n’a pas eu recours à un traitement immuno-
modulateur.
En effet, la patiente s’est améliorée d’une façon spectaculaire sur le plan
clinique (apyrexie et amélioration de l’état général) et paraclinique: diminution de la

122
VS, SGOT, SGPT et normalisation du bilan lipidique et du bilan de la crase. Ce qui
suggère qu’une utilisation rapide du traitement adéquat confère au SAM une issue
favorable.
Il est nécessaire de rappeler que le SAM est une pathologie grave, souvent
méconnue, pouvant mettre en cause le pronostic vital, d’où l’importance de cerner
les éléments permettant une orientation diagnostique rapide, et donc la mise en
route d’une thérapeutique efficace le plus rapidement possible dans le but
d’améliorer l’issue de ce syndrome.
B. Suggestion d’une conduite à tenir pratique devant un SAM :
1. Suspecter le diagnostic du SAM:
Devant l’association des signes suivants, le diagnostic du SAM doit être
suspecté :
ü Fièvre.
ü Organomégalie.
ü Cytopénie inexpliquée touchant une ou plusieurs lignées.
ü Une perturbation du bilan de coagulation avec une hypofibrinémie.
ü Une hyperferritinémie.
ü Une hypertriglycéridémie.
ü Une augmentation du taux de LDH.
La présence d’une cytolyse hépatique appuie l’hypothèse du diagnostic du SAM.
2. Affirmer le diagnostic du SAM :
Le diagnostic est habituellement affirmé par une étude anatomopathologique
qui est nécessaire pour la mise en évidence d’images d’hémophagocytose.

123
Le myélogramme est l’examen de référence apportant les critères
morphologiques du diagnostic du SAM, il sera réalisé en premier lieu. Néanmoins, il
peut être normal ce qui n’élimine pas le diagnostic.
Si le contexte clinique et biologique est fortement suggestif, la répétition du
myélogramme s’impose même s’il avait été considéré comme négatif.
Il faut savoir que le diagnostic des syndromes d’hémophagocytose a
récemment évolué avec l’introduction en particulier de nouveaux critères en rapport
direct avec la physiopathologie, à savoir la recherche d’anomalies génétiques en cas
de SAM primaire, l’augmentation des taux du récepteur soluble de l’IL-2 (sCD25),
reflet de l’hyper-activation du système immunologique, et la diminution des
fonctions de cytotoxicité des cellules de l’immunité innée appelées « natural killer ».
Des critères de diagnostic ont été proposés par Henter et al. en 2006 :
Ø Critère de type 1
Diagnostic moléculaire en cas SAM primaire, la présence de ce critère à lui seul
permet de retenir le diagnostic.
Ø Critère de type 2
La présence de 5 des 8 critères suivants permet de retenir le diagnostic :
- fièvre
- splénomégalie
- cytopénies affectant 2 lignées ou plus : hémoglobine < 90 g/l,
plaquettes < 100 000/ml, neutrophiles < 100/ml.
- hypertriglycéridémie et/ou hypofibrinogénémie :
triglycérides >3mmol/l, fibrinogène < 1,5 g/l
- hémophagocytose dans lamoelle osseuse, la rate ou les ganglions
lymphatiques.
- activité natural killer diminuée ou absente

124
- ferritine > 500 µg/l
- sCD25 ≥ 2400 U/ml.
3. Evaluer la gravité du SAM :
La synthèse des différents signes cliniques et paracliniques permet de repérer
certains éléments qui peuvent être considérés comme des facteurs de mauvais
pronostic permettant ainsi d’évaluer le degré de gravité du SAM et de décider de la
stratégie thérapeutique à mettre en œuvre.
4. Mener une enquête étiologique infectieuse afin de mettre en route un
traitement anti-infectieux à large spectre :
Un bilan infectieux complet doit être réalisé, comprenant :
• D’une manière systématique :
Ø Hémocultures.
Ø ECBU.
Ø Radiographie thoracique
Ø Sérologies EBV, CMV et HIV.
• Selon l’orientation :
Ø Intradermoréaction à la tuberculine avec recherche de BK dans les
expectorations.
Ø Sérologies à la recherche d’une infection virale : VZV, HSV.
Ø Recherche d’une infection fongique : en particulier une candidose, une
aspergillose, une histoplasmose, ou une cryptococcose, des examens
peuvent alors être utilisés :
ü Sérologies d’aspergillose, d’histoplasmose, ou de cryptococcose.
ü Recherche d’aspergillus sur un prélèvement bronchique distal, un
lavage broncho-alvéolaire ou dans les expectorations.

125
ü Recherche d’histoplasmose par examen direct des expectorations,
d’hémoculture, des selles, de biopsies de peau ou d’organes
profonds.
ü Recherche du cryptocoque à l’examen direct ou après coloration à
l’encre de chine ou en culture dans le sang ou dans le LCR.
Ø Recherche de parasitose surtout la toxoplasmose, la leishmaniose, la
pneumocystose et l’anguillulose grâce à :
ü La sérologie de la toxoplasmose.
ü Un examen parasitologique des selles à la recherche de candidose
ou d’anguillulose.
ü Mise en évidence de kystes de Pneumocystis carnii dans le lavage
broncho-alvéolaire après coloration de Gomori-Grocott ou en
immunofluorescence.
ü Recherche de leishmaniose par la sérologie de leishmaniose ainsi que
la mise en évidence de corps de leishmanies par ponction médullaire.
Après la réalisation de ce bilan infectieux, un traitement anti-infectieux à large
spectre peut alors être mis en route :
v Une antibiothérapie de façon systématique.
v Si infection virale : débuter un traitement antiviral (vidarabine ou aciclovir à
titre d’exemple) puis démarrer les immunoglobulines qui semblent être
efficaces dans ce contexte, associées à une corticothérapie générale et en
dernier recours l’étoposide en cas de SAM réfractaire.
v Un traitement antifongique systématique en cas d’infection fungique.
Cette première étape de l’attitude thérapeutique contribue dans le contrôle de
certains SAM d’origine infectieuse.

126
5. Poursuivre l’enquête étiologique en réalisant un bilan à la recherche d’une
pathologie néoplasique, auto-immune ou d’un déficit immunitaire :
Les examens pouvant faire partie de ce bilan sont :
• Une biopsie médullaire à la recherche de stigmates d’une hémopathie sous-
jacente, une ponction d’une adénopathie peut aussi être utile si elle n’a pas été
déjà faite.
• Un bilan d’imagerie à la recherche d’une néoplasie profonde: une radiographie
thoracique, une échographie abdominopelvienne, une tomodensitométrie
thoraco-abdomino-pelvienne.
• Une électrophorèse des protides sériques, recherche des anticorps anti-
nucléaires ainsi que de marqueurs de maladies systémiques. 6. Démarrer une stratégie thérapeutique dés le diagnostic :
Les formes graves ayant des facteurs de mauvais pronostic constituent des
urgences thérapeutiques.
Dans les formes moins graves, l’enquête étiologique peut être menée avant,
afin de ne pas gêner l’interprétation des prélèvements histologiques, cependant la
réalisation des prélèvements doit être précoce permettant ainsi une prise en charge
thérapeutique dans les meilleurs délais.
La stratégie thérapeutique comprend trois volets :
1. Un traitement symptomatique systématique et précoce.
2. Un traitement pathogénique.
3. Un traitement à visée étiologique sera envisagé après avoir déterminé le facteur
déclenchant l’activation inappropriée des macrophages.
La figure 6 représente un résumé de la prise en charge diagnostique et
thérapeutique du SAM sous forme d’un arbre décisionnel.
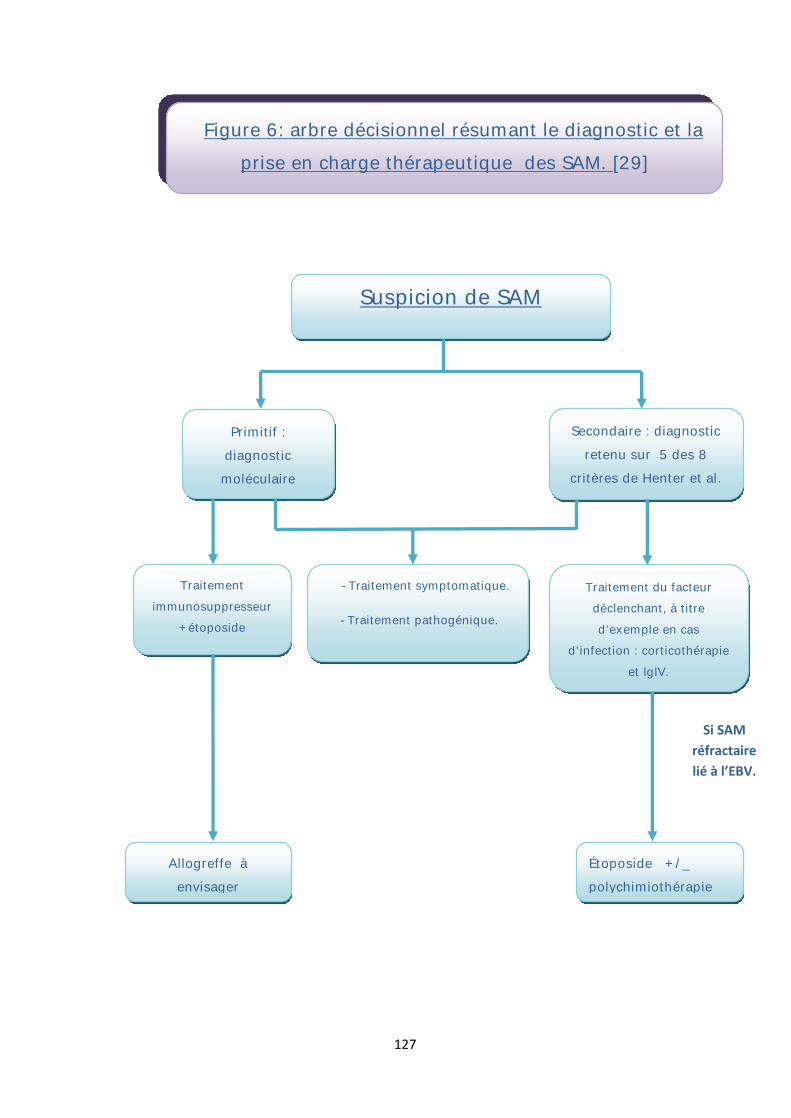
127
Suspicion de SAM
Secondaire : diagnostic retenu sur 5 des 8
critères de Henter et al.
Primitif : diagnostic moléculaire
-Traitement symptomatique.
-Traitement pathogénique.
Traitement immunosuppresseur
+étoposide
Traitement du facteur déclenchant, à titre d’exemple en cas
d’infection : corticothérapie et IgIV.
Si SAM réfractaire lié à l’EBV.
Figure 6: arbre décisionnel résumant le diagnostic et la prise en charge thérapeutique des SAM. [29]
Allogreffe à envisager
Étoposide +/_ polychimiothérapie

128
CONCLUSION

129
CONCLUSION :
Le SAM est une entité clinique et biologique caractérisée par l’activation non
spécifique du système monocyte-macrophage dont la traduction est une infiltration
tissulaire par des macrophages activés.
Il peut être classé en deux catégories :
ü le SAM primaire associé à des déficits immunitaires primitifs.
ü Le SAM secondaire réactionnel à une infection, une pathologie maligne ou
auto-immune, ou un médicament.
L’incidence du SAM demeure sous estimée, ceci s’expliquerait, en partie, par la
méconnaissance de ce syndrome, l’absence de spécificité des signes cliniques et
biologiques, étiologies multiples, et l’évolution fulminante sans preuve
diagnostique.
Les mécanismes physiopathologiques, mieux explorés depuis les découvertes
des molécules impliquées dans les formes héréditaires de ce syndrome, semblent
incriminer plutôt un dérèglement initial des lymphocytes T, qui déclenche une
«tempête» cytokinique et stimule les monocytes-macrophages.
Sur le plan clinique, une fièvre précoce et quasi-constant, présentant des
clochers à 39–40°C, une altération de l’état général, un syndrome tumoral associant
une hépatomégalie, une splénomégalie et des adénopathies périphériques, des
signes neurologiques, des signes cutanés. D’autres signes peuvent s’ajouter au
tableau clinique donnant parfois un tableau de défaillance multi-viscérale.

130
La para-clinique peu spécifique mais caractéristique redresse le diagnostic.
L’hémogramme montre une bi- ou pancytopénie (75 % des cas). L’hémostase décèle
un taux de TP abaissé, un allongement de TCA, une hypofibrinogénémie, une baisse
modérée des facteurs II, VII et X, qui peut traduire une insuffisance hépatocellulaire
parfois une CIVD. Les signes biochimiques sont une augmentation des lactate
déshydrogénases (LDH), une hypertriglycéridémie sans hypercholestérolémie, une
cytolyse, une cholestase et/ou une insuffisance hépatocellulaire, une hyponatrémie
avec natriurèse conservée et hypoprotidémie, une augmentation de la créatinine
dans le sang et enfin une hyperferritinémie. Après confrontation des données
cliniques et biologiques, le myélogramme est indiqué, ce dernier est caractéristique
en montrant une moelle riche avec une prolifération médullaire et systémique
d’histiocytes bénins activement macrophagiques, et surtout l’englobement par les
macrophages médullaires de débris cellulaires ou de cellules figurés de sang,
plaquettes, érythroblastes ou de cellules lymphoïdes.
Dans la littérature, des critères diagnostiques du SAM ont été élaborés. Une
remise à jour a été récemment établie avec l’introduction en particulier de nouveaux
critères en rapport direct avec la physiopathologie, à savoir l’augmentation des taux
du récepteur soluble de l’IL-2 (sCD25), reflet de l’hyperactivation du système
immunologique, et la diminution des fonctions de cytotoxicité des cellules de
l’immunité innée appelées « Natural Killer ».
Le pronostic du SAM reste compromis dans environ la moitié des cas,
principalement dans les SAM d’origine lymphomateuse.

131
La difficulté réside dans la caractérisation rapide de ou des étiologies et dans la
prise en charge thérapeutique. Celle-ci associe trois volets :
v un traitement symptomatique qui vise à pallier l’hémodilution, les troubles
de l’hémostase, l’anémie et les infections associées.
v Le traitement de la cause du SAM : traitement anti-infectieux en cas
d’infection, traitement spécifique d’un lymphome ou d’une maladie de
système.
v Le traitement du SAM lui-même : corticoïdes, immunoglobulines
intraveineuses, cyclosporine A.
Cependant il n’existe pas de protocole définitivement établi pour le traitement
du SAM. Les avancées récentes dans la compréhension de l’origine cellulaire du SAM
permettront peut être de mieux adapter les traitements et d’améliorer la survie de
ces patients.

132
RESUMES

133
RESUME
Le syndrome d’activation macrophagique (SAM) est une entité anatomo-
clinique due à une stimulation inappropriée des macrophages.
Il demeure une pathologie rare dont la prévalence est probablement sous
estimée.
Le SAM associe des signes cliniques peu spécifiques (fièvre, altération de
l’état général, hépatosplénomégalie, adénopathies) et des éléments biologiques
évocateurs (bi- ou pancytopénie, altération du bilan hépatique, coagulopathie,
augmentation des LDH, de la ferritine et des triglycérides). Le diagnostic est
confirmé par un examen cytologique ou histologique retrouvant
l’hémophagocytose.
Ce syndrome peut être primaire essentiellement chez l’enfant, ou secondaire
à diverses affections.
Les progrès récents réalisés en matière des études génétiques des formes
héréditaires, ont permis de mieux comprendre la physiopathologie du SAM, ils
ont ouvert aussi la voie à de nouvelles perspectives thérapeutiques dont le
développement pourrait améliorer le pronostic encore sombre du SAM.
En pratique, l’hypothèse du SAM doit inciter à l’obtention d’une preuve
histologique (données du médullogramme). Une enquête étiologique doit être
démarrée recherchant une infection, une néoplasie, ou une maladie de système.
Le traitement est avant tout étiologique sans oublier un traitement
symptomatique précoce adapté à la sévérité des troubles cliniques et
biologiques.

134
SUMMARY
Macrophage activation syndrome (MAS) is a clinicopathological entity due to an
inappropriate stimulation of macrophages. It remains a rare pathology, its
prevalence is probably under estimated.
This life–threatening disease combines non-specific clinical signs (fever,
cachexia, hepatomegaly, enlargement of spleen and lymph nodes) as well as typical
laboratory findings (bi- or pancytopenia, abnormal hepatic tests, hypofibrinemia,
elevation of serum LDH, ferritinemia and triglyceride levels). Diagnosis is confirmed
by cytological or pathological examination of bone marrow or tissue specimens.
Hemophagocytosis may be primitive, essentially in pediatric population, or
secondary, related to various diseases.
Recent advances, essentially due to genetic studies of familial hemophagocytic
syndrome, allowed to understand better the pathogenesis of HS, they also open the
way to the emergence of new therapeutic options which their development could
improve the still dark prognosis of HS.
In practice, the hypothesis of HS has to lead to the obtaining of a histological
proof (given by the myelogram). An etiologic inquiry must be started looking for an
infection, a neoplasia, or a systemic disease. The treatment aims essentially to
control the etiology and to provide an early supportive care adapted to the severity
of the clinical and biological disorders.

135
ملخص
اعتیادي غیر تحفیز عن ناتجة مجموعة أعراض عن عبارة يھ البلعمي التنشیط متالزمة إن ٠اانتشارھ رتقدی ھناك نقص في كونی أن المحتمل من و نادرا مرضاتعتبر زالت ال ٠للبلعمیات ،العامة الصحیة الحالة تدھور ، مىح( سریریة أعراض بین تجمع البلعمي التنشیط متالزمة إن
تدھور ،مالد كریات عدد انخفاض( بیولوجیة أعراضو ) ویةاللمفا العقد و الكبد و الطحال تضخم
مع ارتفاع نسبة الفیریتین و ثالثي ،LDH ةنزیمأ نسبة ارتفاع ،رقاءإلا حصیلة و الكبد حصیلة
٠و یتأكد تشخیص المرض من خالل دراسة خلویة أو نسیجیة تظھر بلعمة خالیا الدم ٠ )الغلیسیرید
٠أن تكون أولیة خصوصا لدى األطفال أو ثانویة ألمراض متعددة متالزمةلھذه ال یمكن
و قد ساعدت التطورات الحدیثة في مجال الدراسات الجینیة لألشكال الوراثیة في فھم آلیة الجیة جدیدة حیث من الممكن أن یؤدي تطویر ھذه األخیرة إلى و فتحت آفاق ع البلعمي التنشیط متالزمة ٠القاتم لھذا المرض آلتحسین الم
إلى الحصول على دلیل من خالل تخطیط البلعمي التنشیط متالزمةعملیا یجب أن تؤدي فرضیة
أورام أو أمراض ،كما یجب بدء عملیة تنقیب عن أسباب المرض بحثا عن تعفنات•النخاع
توفیر عالج أولي مناسب ألعراض المرض العالج یرتكز أساسا على معالجة السبب المنشط مع٠باطنیة ٠بیولوجیةوال سریریةال

136
REFERENCES

137
REFERENCES
1. Créput C, Galicier L, Oksenhendler E, Azoulay E. Syndrome d’activation lymphohistiocytaire: revue de la littérature, implications en réanimation. Réanimation 2005 ; 14 :604-13.
2. Reiner AP., Spivak JL.
Hematophagic histiocytosis. A report of 23 new patients and review of the literature. Med(Baltimore) 1988; 67: 369-88.
3. Marshall AHE.
Histiocytic medullary reticulosis. J Pathol Bacter.1965; 71:61-71.
4. Greenberg E., Cohen D.M., Pease G.L., Kyle R.A. Histiocytic medullary reticulosis. Proc.Mayo.Clin.1962; 37: 271-83.
5. Rappaort H. Tumors of hematopoietic system. Atlas of tumor pathology. Washington DC: Armed Forces Institute of Pathology, 1966, 3(8): 49-63.
6. Méchinaud-Lacroix F, Gaillard F, Harousseau JL.
Syndrome d'activation monocytomacrophagique. Encyclopédie médicochirurgicale: Hématologie 1996; 13-012-G-10: 10.

138
7. Boake WC., Card WH., Kimmey JF. Histiocytic medullary reticulosis: concurrence in father and son. Arch.Intern.Med.1965; 116:245-52.
8. Zinkham WH., Medearis DN., Osborn JE.
Blood and bone marrow findings in congenital rubella. J.Pediat.1967; 71:512-24.
9. Chandra P., Chaudherry SA., Rosner F.
Transcient histiocytosis with striking phagocytosis of platelets, leukocytes and erythrocytes. Arch.Intern.Med.1975; 135:989-91.
10. Risdall RJ. , McKenna RW. , Nesbit ME. & AL.
Virus associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer1979; 44:993-1002.
11. Risdall RJ. , Bruning RD., Hernandez JL. &AL. Bacteria-Associated Hemophagocytic Syndrome Cancer1984; 54:2968-72.
12. Chan JFK., NG CS., Law CK. & AL. Reactive hemphagocytic syndrome: a study of 7 fatal cases. Pathology.1987; 19: 43-50.
13. Malone M.
The histiocytoses of childhood. Histopathology 1991; 19: 105–19.
14. Favara B E., Feller AC., Paulli M. & AL.
Contemporary classification of histiocytic disorders. Medical and Pediatric Oncology1997; 29:157–66.

139
15. Ronald J., M.B., B.Ch. The Histiocytoses . Clinics in laboratory medicine 1999 march; 19, Number 1.
16. Laboudi A., Haouazine N., Benabdallah L., Arzouk N. & AL. Maladie de Rosaï-Dorfman révélée par une insuffisance rénale: à propos d’un cas. Néphrologie 2001 ; 22 (2) : 53-6.
17. Colombat Ph. Hématologie pratique Paris : Doin, 1991. 39-41.
18. SEBAHOUN G. Hématologie clinique et biologique Arnette. 1998,125-127.
19. Bach JF. Traité d’immunologie. Paris : Flammarion. 1993, 133-55.
19’. Gene Mayer .
Microbiology and immunology University of South Carolina, School of Medicine http://pathmicro.med.sc.edu/ghaffar/innate.htm
20. Reyes F, Bernaudin JF
Différenciation monocytaire : origine, morphologie et fonctions des macrophages. L’hématologie de Bernard Dreyfus. 1992, 138-45.

140
21. BERGAMINI A., F. BOLACCHI, B. BONGIOVANNI, & AL. Granulocyte±macrophage colony-stimulating factor regulates cytokine production in cultured macrophages through CD14 dependent and independent mechanisms. Immunology .2000 Oct; 101(2): 256-61.
22. Karras A, Hermine O. Syndrome d’activation macrophagique. Rev. Med. Intern 2002 ; 23 :768-778.
23. Osugi Y.
Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood1997; 89:4100–3.
24. Mazodier M., Marin V., Novick D., Farnarier C.& AL. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood2005; 106 (10): 3483-3489.
25. Sawhney S., Woo P., Murray KJ.
Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis CHILD 2001 Nov; 85(5): 421-6.
26. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood2004; 104: 735-43.

141
27. BilliauAD, Roskams T, Damme-Lombaerts R, & AL. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma producing lymphocytes and IL-6- andTNF-alpha producing macrophages. Blood2005; 105: 1648-51.
28. De Saint BG, Fischer A. The role of cytotoxicity in lymphocyte homeostasis. Curr Opin Immunol2001; 13: 549-54.
29. Costello R., Baccini V., Mazodier K., Kaplanski G., & AL. Lymphohistiocytose hémophagocytaire Encyclopédie médicochirurgicale: hématologie 2007 ; 13-012-G-10.
30. Fauriat C, Just-Landi S,Mallet F,Arnoulet C, Sainty D & AL. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCR dull phenotype induction. Blood 2007; 109: 323-30.
31. Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, & AL. Defective expression and function of natural killer cell triggering receptors in patients with acute myeloid leukemia. Blood 2002;99:3661-7.
32. Henter JI. , Elinder G., Soder O. &AL. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood1991; 78:2918-22.
33. Mouy R., Stephan JL. , Pillet P. & AL. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: Report of five cases. J Pediatr 1996 Nov; 129(5): 750-4.

142
34. KARRAS A., THAUNAT O., NOËL L.H., & DELAHOUSSE M. Syndrome d’activation macrophagique: implications pour le néphrologue. FLAMMARION MÉDECINE-SCIENCES — ACTUALITÉS NÉPHROLOGIQUES 2005 :59-80.
35. Fléchaire A., Colle B., Bernard P., Dupuy o., & Philippe P.
Les syndromes hémophagocytaires. Rev Med Interne 1996 ; 17 (2) : 157-62.
36. Emmenegger U, Reimers A, Frey U, Fux C, Bihl F, Semela D, & AL.
Reactive macrophage activation syndrome: a simple screening Strategy and its potential in early treatment initiation. Swiss Med Wkly 2002; 132:230-6.
37. Laroche C. Syndrome d’activation macrophagique de l’adulte : état des connaissances en 2003. Mini-revue: Sang Thrombose Vaisseaux 2003 ; 15, n° 3: 135–42.
37’. Berthou Ch. Adénopathie superficielle. Maladies du sang. Disponible à partir de : http://www.leucemie-espoir.org/spip/article26.html
38. Morrell DS. , Pepping MA. , Scott JP. , Esterly NB., Drolet BA. Cutaneous manifestations of hemophagocytic lymphohistiocytosis Arch Dermatol 2002; 138: 1208-1212.
39. LIPSKER D.
Hypodermites: panniculiyte histiocytaire cytophagique. Thérapeutique dermatologique, Médecine-Sciences Flammarion 2001.
40. Lipsker D., Grosshans E. Panniculite histiocytaire cytophagique. La presse médicale 1997; 26 (40) :1987.

143
41. Papo T. Syndrome d’activation des macrophages. Syndromes hémophagocytaires. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés), AKOS Encyclopédie Pratique de Médecine, 4-0300, 2000,3p.
42. Haddad E., Sulis ML., Jabado N., Blanche S., & AL.
Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood1997; 89: 794-800.
43. Akiyoshi K., Hamada Y., Yamada H., Kojo M., Izumi T. Acute Necrotizing Encephalopathy Associated With Hemophagocytic Syndrome. Pediatr Neurol 2006; 34:315-318.
44. Stephan F., Thioliere B., Verdy E., & Tulliez M. Role of hemophagocytic histiocytosis in the etiology of thrombocytopenia in patients with sepsis syndrome or septic shock. Clin. Infect. Diseases1997; 25: 1159–64.
45. Pradalier A., Teillet F., Molitor J.L., Drappier JC. Syndrome d’activation macrophagique (syndrome d’hémophagocytose) Macrophage activation syndrome, hemophagocytic syndrome. Pathologie Biologie 2004 ; 52 :407-14.
46. Stéphan J.L., Galambrun C. Syndrome d’activation lymphohistiocytaire chez l’enfant. Arch Pédiatr 2000 ; 7 :278-86.
46’. Onaga M., Hayashi K., Nishimagi T., Nagata K., Uto H., Kubuki Y. & AL.
A case of acute hepatitis A with marked hemophagocytosis in bone marrow. Hepatol Res. 2000 Jun;17(3):205-211.

144
47. De Kerguenec C., Hillaire S., Molinie V. & AL. Hepatic manifestations of hemophagocytic syndrome: a study of 30 Cases. Am J Gastroenterology 2001; 96:852-57.
47’. Wada Y., Kitajima H., Kubo M. Seven cases of hemophagocytic syndrome complicated with childhood collagen diseases. Ryumachi. 1996 Aug;36(4):637-43.
48. Esumi N., Ikushima S. , Hibi S., Todo S. , Imashuku S.
High serum ferritin level as a marker of malignant histiocytosis and virus-associated hemophagocytic syndrome. Cancer1988; 6: 2071-76.
49. Emmenegger U., Schaer D. J., Larroche C., Neftel K. A. Haemophagocytic syndromes in adults:current concepts and challenges ahead. Swiss Med Wkly 2005; 135:299–314.
50. Bouskraoui M.
Une observation exceptionnelle associant une hépatite aiguë virale A, une arthrite chronique juvénile et un syndrome d’activation macrophagique. Médecine et maladies infectieuses 2003 ; 33 : 358–60.
51. Komp DM, McNamara J, Buckley P. Elevated soluble interleukin-2 receptor in childhood hemophagocytic histiocytic syndromes. Blood1989; 73:2128-32.
52. Schneider EM., Lorenz I., Muller-Rosenberger M., & AL.
Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer-cell-induced apoptosis. Blood 2002; 100:2891-8.

145
53. Grom AA. Natural killer cell dysfunction: A common pathway in systemic onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis? Arthritis Rheum 2004; 50:689–98.
54. WongKF, Chan JK. Reactive hemophagocytic syndrome : a clinicopathologic study of 40 patients in an Oriental population. Am J Med.1992; 93: 177-80.
55. Tsuda H.
Hemophagocytic syndrome in children and adults. Int J Hematol.1997; 65: 215-26.
56. Grom AA. Macrophage activation syndrome and reactive hemophagocytic lymphohistiocytosis: the same entities? Curr Opin Rheumatol 2003; 15: 587–90.
57. HENTER JI. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. Semin Oncol, 1991,18: 29-33.
58. IMASHUKU S. Differential diagnosis of hemophagocytic syndrome : underlying disordrers and selection of the most effective treatment. Int J Haematol, 1997, 66: 135-151.
59. Henter JI, HorneA,AricoM, Egeler RM, FilipovichAH, & AL. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48: 124-31.

146
60. Gritta E. Janka Hemophagocytic syndromes. Blood Reviews (2007), doi:10.1016/j.blre.2007.05.001
61. Palazzi D, Mac Clain K, Kaplan S. Hemophagocytic syndrome in children: an important diagnostic consideration in fever of unknown origin. Clin Inf Dis 2003; 36:306–12.
62. Grunebaum E, Roifman CM. Gene abnormalities in patients with hemophagocytic lymphohistiocytosis. Isr Med Assoc J 2002; 4:366-9.
63. Imashuku S, Hibi S, Todo S. Hemophagocytic lymphohistiocytosis in infancy and childhood. J Pediatr1997; 130: 352–7.
64. Goransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, & AL.
Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet 2001; 68:590-7.
65. Feldmann J, Le Deist F, Ouachee-Chardin M, Certain S, & AL. Functional consequences of perforin genemutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br J Haematol 2002; 117:965-72.
66. De Saint Basile G. Altération du gène codant pour la perforine dans la lymphohistiocytose familiale. médecine/sciences 1999 ; 15 : 1479-81.

147
67. Feldmann J,Callebaut I,RaposoG,Certain S,BacqD,DumontC, & AL.
Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003; 115: 461-73.
68. Rudd E., Goransdotter Ericson K., Zheng C., Uysal Z., & AL. Spectrum and clinical implications of syntaxin 11 gene mutations in familial haemophagocytic lymphohistiocytosis: association with disease-free remissions and haematopoietic malignancies. J Med Genet 2006; 43:e14.
69. Stéphan J.L. Syndromes hémophagocytaires et deficits immunitaires primitifs. Hemophagocytic syndromes and primary immunodeficiency. Archives de pédiatrie 2003; 10 (Suppl.4):517-20.
70. Blanche S. Lymphohistiocytose familiale et autres syndromes d'activation macrophagique. Encyclopédie médicochirurgicale 1994 ; Pédiatrie/Maladies infectieuses [4-082-J-40].
71. Karim MA, Nagle DL, Kandil HH, Burger J, Moore KJ, Spritz RA. Mutations in the Chediak-Higashi syndrome gene (CHS1) indicate requirement for the complete 3801 amino acid CHS protein. Hum Mol Genet 1997; 6:1087–9.
72. Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, & AL. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000; 25:173–6.

148
73. Pastural E., Ersoy F., Yalman N. &AL. Two Genes Are Responsible for Griscelli Syndrome at the Same 15q21 Locus. Genomics 2000; 63: 299–306.
74. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, & AL.
Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet 1998;20: 129–35.
75. Howie J, Sayos C, Terhorst M, Morra.
The gene defective in X–linked lymphoproliferative disease controls T cell dependent immune surveillance against EBV. Curr Opin Immunol 2000;12:474–8.
76. Ochs HD., Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol 2006; 117:725-38.
77. Orange JS, RameshN, Remold-O’Donnell E, SasaharaY, & AL. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci USA 2002; 99:11351-6.
78. Fisman D. Hemophagocytic syndromes and infections. Emerg Infect Dis 2000; 6: 601–8.
79. Kikuta H., Sakiyama H., Matsumoto S. & AL. Fatal Epstein-Barr Virus associated hemophagocytic syndrome. Blood 1993; 82 (11): 3259-64.

149
80. Lay JD. , Tsao CJ., Chen JY, Kadin ME. & Su IJ. Upregulation of tumor necrosis factor a gene by Epstein-Barr-Virus and activation of macrophages in Epstein - Barr virus infected T Cells in the pathogenesis of Hemophagocytic Syndrome. J. Clin. Invest. 1997; 100 (8): 1969-79.
81. Ohga S., Nomura A. , Takada H. & AL. Epstein-Barr Virus (EBV) load and cytokine gene expression in activated T cells of chronic active EBV infection. The Journal of Infectious Diseases 2001; 183:1–7.
82. Dupre L,AndolfiG,Tangye SG,ClementiR, Locatelli F,AricoM, & AL.
SAP controls the cytolytic activity of CD8+ T cells against EBV infected cells. Blood 2005; 105:4383-9.
83. Sumazaki R, Kanegane H., Osaki M., Fukushima T., & AL. SH2D1A mutations in Japanese males with severe Epstein-Barr virus associated illnesses. Blood 2001; 98:1268-70.
84. Paterson RL, Kelleher C,Amankonah TD, Streib JE, & AL. Model of Epstein-Barr virus infection of human thymocytes: expression of viral genome and impact on cellular receptor expression in the T-lymphoblastic cell line, HPB-ALL. Blood1995; 85:456-64.
85. Charles P., Hot A., Girard Madouxb M.-H., Simonb M. & AL. Succès d’un traitement par immunoglobulines intraveineuses au cours d’un syndrome d’activation macrophagique associé à une primo-infection à cytomégalovirus. La Revue de médecine interne2006 ; 27 : S336–S418

150
86. Mercya G., Zidi S., Le Corguille M., Ziol M., Roulot D. & AL. Varicelle multiviscérale compliquée d’un syndrome d’activation macrophagique, une situation rare mais grave. La Revue de médecine interne 2007 28 : S83–S160.
87. Grateau G, Bachmeyer C, Blanche P, Jouanne M, Tulliez M, & AL. Haemophagocytic syndrome in patients infected with HIV: 9 cases and a review.
J Infect 1997; 34:219–25. 88. Sailler L, Duchayne E, Marchou B, Brousset P, Pris J, & AL.
Aspects étiologiques des hemophagocytoses réactionnelles : étude rétrospective chez 99 patients.
Rev Med Interne1997;18:855–64.
89. Tiab M, Mechinaud F, Hamidou M, Gaillard F, Raffi F, & AL. Syndromes hémophagocytaires, une série de 23 observations. Ann Med Interne 1996;147:138–44.
90. Dhote R, Simon J, Papo T, Detournay B, Sailler L, Andre MH, et AL. Reactive hemophagocytic syndrome in adult systemic disease: Report of twenty-six cases and literature review. Arthritis Rheum 2003; 49: 633-9.
91. Veerakul G, Sanpakit K, Tanphaichitr VS, Mahasandana C, &AL. Secondary hemophagocytic lymphohistiocytosis in children: an analysis of etiology and outcome. J Med Assoc Thai 2002; 85(suppl2):S530-S541.
92. SinayaL., Corne P. , Combes N., Amalric-Diop A. and Jonquet O. Syndrome d’activation macrophagique et fièvre typhoïde sévère. Severe typhoid feverand hemophagocytic syndrome. La Revue de Médecine Interne 2004 ; 25 (2) :163-165.

151
93. El Khoury N., Lassoued K., Pellé G., Foucher A., Costa M.A., & AL. Syndrome d’activation macrophagique associé à une septicémie à Escherichia coli : à propos d’un cas. La revue de médecine interne2003 ; 24 : 688–691.
94. Arlet J.B., Lafaurie M., Furco A., Neuville S., Bani-Sadr F. & AL. Syndrome d'activation macrophagique et tuberculose. Rev Med Interne 2002; 23 (2) : S662.
95. Bonnotte B., Rossignoii N., Goegebawr G., Chauffert B. & AL. Tuberculose hépatique et syndrome hémophagocytaire. Rev Med Interne 1997 ; 18 (suppl2) : S208.
96. Charfeddine B., Laradi S., Kassab A., Ferchichi S., & AL. Syndrome d’activation monocytomacrophagique : à propos d’un cas. Annales de biologie clinique 2003; 61 (1) : 81-83.
97. Retornaz F., Seux V., Arnoulet C., Kadjo Ouattara K., &AL. Syndrome d’activation macrophagique associé à une infection à plasmodium falciparum. Rev med interne 2000; 21(suppl 4): S587.
98. Bhatia S, Bauer F, Bilgrami SA. Candidiasis-associated hemophagocytic lymphohistiocytosis in a patient infected with human immunodeficiency virus. Clin Infect Dis 2003;37:e161–e166.
99. Miyahara M., Sano M., Shibata K., Matsuzaki M., & AL. B-cell lymphoma-associated hemophagocytic syndrome: clinicopathological characteristics. Ann Hematol 2000; 79:378–88.

152
100. Majluf-Cruz A, Sosa-Camas R, Perez-Ramirez O, & AL. Hemophagocytic syndrome associated with haematological neoplasias. Leuk Res 1998; 22:893–8.
101. Su IJ, Wang CH, Cheng AL, Chen RL.
Hemophagocytic syndrome in EBV-associated T lymphoproliferative disorders: disease spectrum, pathogenesis and management. Leuk Lymphoma1995; 19:401–6.
102. Yao M, Cheng AL, Su IJ, Lin MT, Uen WC, Tien HF, & AL. Clinicopathological spectrum of haemophagocytic syndrome in EBV-associated peripheral T-cell lymphoma. Br J Haematol1994; 87: 535–43.
103. Takahashi R, ChubatiA,Miura I, Nakamura S,Miura T. Lymphoma-associated hemophagocytic syndrome in Japan. Japn J Clin Hematol 1999; 40: 542–9
104. Koizumi K, HaseyamaY,Machino R, SatoY, Sawada K, Koike T. The hemophagocytic syndrome in prostate cancer revealed by disseminated carcinomatosis of the bone marrow. J Urol 2002; 168: 1101-2.
105. Chinen K, OhkuraY,Matsubara O, Tsuchiya E. Hemophagocytic syndrome associated with clostridial infection in a pancreatic carcinoma patient. Pathol Res Pract 2004; 200: 241-5.
106. Wong KF, Hui PK, Chan J, Chan YW, Ha SY. The acute lupus hemophagocytic syndrome. Ann Int Med1991; 114: 387–90.

153
107. Muise A., Tallett S.E. and Silverman E.D. Are Children With Kawasaki Disease and Prolonged Fever at Risk for Macrophage Activation Syndrome? Pediatrics 2003; 112 (6): e495-e497.
108. Avcin T, Tse SM, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr 2006; 148: 683-6.
109. Morris J.A., Adamson A.R., Holt J.P. and Davson J.
Still's disease and the virus-associated haemophagocytic syndrome. Ann Rheum Dis1985; 44: 349-353.
110. Hur M., Kim Y.C., Lee K.M., Kim K.N. Macrophage Activation Syndrome in a Child with Systemic Juvenile Rheumatoid Arthritis J Korean Med Sci 2005; 20: 695-8.
111. Ostronoff M, Ostronoff F, Coutinho M, Calixto R, & AL. Hemophagocytic syndrome after autologous peripheral blood stem cell transplantation for multiple myeloma; successful treatment with high-dose intravenous immunoglobulin.
Bone Marrow Transplant 2006; 37:797-8.
112. Bertozzi AI, Suc A, Rubie H, Duchayne E, Demur C, Robert A. Syndromes hémophagocytaires en phase d’aplasie post chimiothérapie.
Arch Pediatr 2002; 9:125-9.
113. SterbaG, Rodriguez C, Sifontes S,Vigilanza P. Macrophage activation syndrome due to methotrexate in a 12- year-old boy with dermatomyositis. J Rheumatol 2004; 31:1014-5.

154
114. Kishi Y, Kami M, Murashige N, Tanaka Y, Haraguchi K., & AL. Hyperacute GVHD and emergence of peripheral CD3+CD56+ T cells and activated natural killer cells are useful markers for early diagnosis of post- transplant hemophagocytic syndrome.
Bone Marrow Transplant 2005; 35: 415-7.
115. Abe Y, Choi I, Hara K, Matsushima T, Nishimura J, Inaba S, & AL. Hemophagocytic syndrome: a rare complication of allogeneic nonmyeloablative hematopoietic stem cell transplantation. Bone Marrow Transplant 2002; 29:799-801.
116. Akamatsu N, Sugawara Y, Tamura S, Matsui Y, & AL. Hemophagocytic syndrome after adult-to-adult living donor liver transplantation.
Transplant Proc 2006; 38:1425-8.
117. KarrasA, Thervet E, Legendre C. Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation 2004; 77:238-43.
118. Masri K, Mahon N, Rosario A, Mirza I, Keys TF, Ratliff NB, & AL. Reactive hemophagocytic syndrome associated with disseminated histoplasmosis in a heart transplant recipient. J Heart Lung Transplant 2003; 22:487-91.
119. Lambotte O, Costedoat-Chalumeau N, Amoura Z & AL. Drug-induced hemophagocytosis. Am J Med. 2002; 112: 592-3.

155
120. Roth B, Grande PO, Nilsson-Ehle P, Eliasson I. Possible role of short-term parenteral nutrition with fat emulsions for development of haemophagocytosis with multiple organ failure in a patient with traumatic brain injury. Intensive Care Med 1993; 19:111–4.
121. Larroche C, Bruneel F, Andre MH, Bader-Meunier B, & AL. Les immunoglobulines intraveineuses dans les syndromes d’activation macrophagique secondaires. Ann Med Interne 2000;151:533–9.
122. Emmenegger U, Frey U, Reimers A, Fux C, Semela D, &AL. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol 2001; 68:4-10.
123. Grom A.A., Passo M. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis. J Pediatr1996; 129: 630-2. 124. Chen H.H., Kuo H.C., Wang L., Yu H-R., Shen J-M., & AL.
Childhood macrophage activation syndrome differs from infection-associated hemophagocytosis syndrome in etiology and outcome in Taiwan. Microbiol Immunol Infect. 2007;40:265-271.
125. Imashuku S, Kuriyama K, Teramura T, Ishii E, & AL.
Requirement for etoposide in the treatment of Epstein-Barr Virus associated hemophagocytic lymphohistiocytosis. J Clin Oncol 2001; 19:2665-73.
126. Blanche S., Caniglia M., Girault D., Landman J., Griscelli C., & AL. Treatment of hemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation: a single center study of 22 cases. Blood 1991.78 (1): 51-54.

156
127. Mahlaoui N., Ouachée-Chardin M., de Saint Basile G. & AL. Immunotherapy of Familial Hemophagocytic Lymphohistiocytosis With Antithymocyte Globulins: A Single-Center Retrospective Report of 38 Patients. PEDIATRICS2007; 120 (3): e622-8.
128. Song K.S., & Sung H.J. Effect of plasma exchange on the circulating IL-6 levels in a patient with fatal hemophagocytic syndrome associated with bile ductopenia. Therapeutic Apheresis and Dialysis 2006; 10(1):87–89.
129. Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, & AL. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol2006; 81:59-61.
130. Prahalad S, Bove KE, Dickens D, Lovell DJ, GromAA. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol 2001; 28:2120–4.
131. Aouba A., de Bandt M., Perrot S., Genty I., Aslangul E.1, & AL. Syndrome hémophagocytaire réactionnel au cours d'une polyarthrite rhumatoïde traitée par infliximab : effet lateral surprenant inédit d'un traitement anti-TNFα! Rev Med Interne 2002; 23 (Suppl 1): S177.
132. Schneider P, Greene V, Kanold J, Vannier JP. Fludarabine in the treatment of an active phase of a familial haemophagocytic lymphohistiocytosis. Arch Dis Child 2001; 84:373.
133. Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, & AL.
Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 2002; 100:2367-73.

157
134. Ravelli A, Caria MC, Buratti S, Malattia C, Temporini F, Martini A.
Methotrexate as a possible trigger of macrophage activation Syndrome in systemic juvenile idiopathic arthritis. J Rheumatol 2001; 28:865–7.
135. Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku & AL. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol 2005; 129: 622-30.
136. Horne A, Zheng C, Lorenz I, Lofstedt M, & AL. Subtyping of natural killer cell cytotoxicity deficiencies in haemophagocytic lymphohistocytosis provides therapeutic guidance. Br J Haematol 2005; 129:658-66.
137. Husni ME, Maier AL, Mease PJ, & AL.
Etanercept in the treatment of adult patients with Still’s disease. Arthritis Rheum 2002; 46: 1171-6.
138. Sandhu C, Chesney A, Piliotis E, Buckstein R, Koren S.
Macrophage activation syndrome after etanercept treatment. J Rheumatol. 2007; 34(1): 241-2.
139. Arico M, Janka G, Fischer A, Henter JL, Blanche S,& AL. for the FHL Study Group of the Histiocyte Society.
Hemophagocytic Lymphohistiocytosis. Report of 122 children From the International Registry. Leukemia1996; 10:197–203.
140. Shimazaki C, Inaba T, Nakagawa M. B-cell lymphoma-associated hemophagocytic syndrome. Leuk Lymphoma 2000; 38: 121-30.

158





























