Embed Size (px)
Citation preview

Die Interaktion zwischen dem Transkriptionsfaktor FoxO1aund der Proteinkinase DYRK1 und ihre Bedeutung bei der
Regulation des Glucose-6-Phosphatasegens
Von der Medizinischen Fakultätder Rheinisch-Westfälischen Technischen Hochschule Aachen
zur Erlangung des akademischen Gradeseines Doktors der Medizingenehmigte Dissertation
vorgelegt von
Florian Till von Groote-Bidlingmaier
aus
Neuss
Berichter: Herr UniversitätsprofessorDr. rer. nat. Dr. med. Hans Georg Joost
Herr UniversitätsprofessorDr. rer. nat. Peter C. Heinrich
Tag der mündlichen Prüfung: 19. Dezember 2007
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.


Wesentliche Teile der vorliegenden Arbeit wurden publiziert in:
Groote-Bidlingmaier, F., Schmoll, D., Orth, H.M., Joost, H.G., Becker, W., and
Barthel, A. (2003). DYRK1 is a co-activator of FKHR (FOXO1a)-dependent
glucose-6-phosphatase gene expression. Biochem. Biophys. Res. Commun.
300, 764-769.


Inhalt1 Einleitung..........................................................................................................1
1.1 Einführung in die Epidemiologie und Pathophysiologie des Typ 2-
Diabetes...................................................................................................1
1.2 Die Glucoseproduktion der Leber .....................................................2
1.3 Signaltransduktion des Insulinrezeptors............................................4
1.4 Der Transkriptionsfaktor FoxO1a und seine Rolle bei der Regulation
des Glucose-6-Phosphatasegens............................................................6
1.5 Die Familie der DYRK Kinasen und ihre Verbindung mit dem
Transkriptionsfaktor FoxO1a....................................................................9
1.7 Arbeitshypothese und Zielsetzung...................................................11
2 Materialien und Methoden............................................................................122.1 Konstrukte........................................................................................12
2.1.1 Der Promotor des Glucose-6-Phosphatasegens......................12
2.1.2 FoxO1a Wildtyp.........................................................................12
2.1.3 FoxO1a Ser329-Punktmutanten...............................................12
2.1.3.1 FoxO1a Ser329Ala-Mutante..................................................12
2.1.3.2 FoxO1a Ser329Asp-Mutante.................................................13
2.1.4 IRU-Mutante des Glucose-6-Phosphatasereportergens...........13
2.1.5 DYRK-Konstrukte......................................................................14
2.2 Zellbiologische Arbeitstechniken.....................................................15
2.2.1 Zelllinien....................................................................................15
2.2.1.1 Medien und allgemeine Kulturbedingungen..........................15
2.2.1.2 Subkultivierung.......................................................................15
2.2.2 Transfektion...............................................................................16
2.2.3 Retroviraler Gentransfer............................................................17
2.2.4 Selektionierung der infizierten Zellen........................................18
2.3 Proteinchemische Arbeitstechniken.................................................19
2.3.1 SDS-PAGE................................................................................19
2.3.1.1 Gelherstellung........................................................................19
2.3.1.2 Probenvorbereitung................................................................21
2.3.1.3 Proteinbestimmung (BCA-Methode)......................................21
2.3.1.4 Gelelektrophorese..................................................................22
2.3.2 Western-Blotting und ECL-System...........................................22
I

2.3.2.1 Transfer..................................................................................22
2.3.2.2 Inkubation mit Antikörpern.....................................................22
2.3.3 Ponceau-Färbung......................................................................23
2.4 Molekularbiologische Arbeitstechniken...........................................24
2.4.1 DNA-Präparation.......................................................................24
2.4.1.1 Minipräparation.......................................................................24
2.4.1.2 Präparation größerer DNA-Mengen ......................................24
2.4.2 Restriktionsverdau.....................................................................25
2.4.3 Herstellung kompetenter E. coli-Bakterien................................25
2.4.4 Transformation von E.coli-Bakterien.........................................26
2.4.4.1 Hitzeschocktransformation.....................................................26
2.4.4.2 Elektroporation.......................................................................27
2.4.5 DNA-Agarosegelelektrophorese...............................................27
2.4.6 Sequenzspezifische Mutagenese.............................................28
2.4.7 DNA-Sequenzierung.................................................................29
2.4.8 Northern Blotting ......................................................................31
2.4.8.1 RNA-Isolierung.......................................................................31
2.4.8.2 RNA-Agarosegelelektrophorese............................................31
2.4.8.3 Blotting....................................................................................32
2.4.8.4 Aufreinigen der DNA-Sonden nach Gelelektrophorese.........33
2.4.8.5 Markieren einer Sonde mit [32P]...........................................33
2.4.8.6 Hybridisierung der Northern Blots..........................................34
2.4.9 Reportergen-Assay (Luciferase)...............................................34
2.5 Statistische Analyse.........................................................................34
3 Ergebnisse......................................................................................................353.1 Effekte von überexprimiertem DYRK1A auf den Gehalt an
Glucose-6-Phosphatase-mRNA und die Aktivität des G6Pase-
Promotors in Hepatomzellen..................................................................35
3.1.1 Überexpression von DYRK1A in stabil transfizierten
Rattenhepatomzellen.........................................................................35
3.1.2 Der Einfluss von stabil transfiziertem DYRK1A auf die
endogene Glucose-6-Phosphatase-mRNA in Rattenhepatomzellen 36
3.1.3 Der Effekt von DYRK1A auf die Promotoraktivität des
Glucose-6-Phosphatasegens im Reportergen-Assay........................37
II

3.2 Der Effekt von DYRK1A auf die G6Pase-Promotoraktivität in
Abhängigkeit von der DYRK1A-Kinaseaktivität.....................................40
3.2.1 Der Einfluss von FoxO1a-Ser329-Punktmutanten auf die durch
DYRK1A vermittelte Expression des Glucose-6-Phosphatasegens..40
3.2.2 Der Effekt einer katalytisch inaktiven DYRK1A-Punktmutante
auf den Glucose-6-Phosphatase-Promotor.......................................43
3.3 Untersuchungen zu Effekten verschiedener DYRK-Isoformen auf
die Aktivität des Glucose-6-Phosphatase-Promotors............................44
3.4 Die Effekte unterschiedlicher DYRK-Isoformen auf die FoxO1a-
induzierte Aktivität des Glucose-6-Phosphatase-Promotors.................45
3.5 Der Effekt von DYRK1 auf den Glucose-6-Phosphatase-Promotor in
Abhängigkeit von FoxO1a-Bindungsstellen (IRUs)...............................47
4 Diskussion......................................................................................................495 Zusammenfassung........................................................................................556 Literaturverzeichnis.......................................................................................56
III


Abbildungsverzeichnis
Abb. 1: Darstellung der pathophysiologischen Komponenten des Typ-2-Diabetes mellitus
Abb. 2: Schematische Darstellung der Glucoseproduktion der Leber
Abb. 3: Darstellung des Insulinrezeptors und der Signaltransduktion des Insulins
Abb. 4: Strukturausschnitt des Promotors des G6Pase-Gens
Abb. 5: Vergleich der drei in dieser Arbeit behandelten DYRK-Isoformen
Abb. 6: Das Prinzip des retroviralen Gentransfers
Abb. 7: Prinzip der Sequenzspezifischen Mutagenese
Abb. 8: Überexpression von DYRK1A in H4IIEC3-Zellen
Abb. 9: Der Effekt von überexprimiertem DYRK1A auf den Gehalt an spezifischer G6Pase-
mRNA in H4-Zellen
Abb. 10: Der Effekt von überexprimiertem DYRK1A auf die Promotoraktivität der G6Pase in
stabil transfizierten Hepatomzellen (H4IIEC3)
Abb. 11: Der Effekt von überexprimiertem DYRK1A auf die Promotoraktivität der G6Pase in
transient transfizierten Hepatomzellen (HepG2)
Abb. 12: Aktivität der G6Pase-Promotors in Abhängigkeit von der Menge des transfizierten
DRK1A-Expressionsvektors
Abb.13: Strukturausschnitt aus dem Promotor der FoxO1a-Punktmutante S329A
Abb. 14: Restriktionsanalyse der FoxO1a-Punktmutante S329A
Abb. 15: Der Effekt der DYRK1A-Phosphorylierung an der Position 329 auf die Aktivität des
G6Pase-Promotors
Abb. 16: Der Effekt einer katalytisch inaktiven DYRK1A-Punktmutante auf die Aktivität des
G6Pase-Promotors
Abb. 17: Der Effekt der verschiedenen DYRK-Isoformen auf die Aktivität des G6Pase-
Promotors
Abb. 18: Struktur der FoxO1a-bindenden Region des G6Pase-Promotors und Veränderungen
der Promotor-Punktmutante
Abb.19: Der Effekt von DYRK1 auf den G6Pase-Promotor (schwarz) und den mutierten
G6Pase-Promotor (weiß).
Tab. 1: Der Effekt von DYRK1 auf den Aktivität des G6Pase-Promotors in Synergismus mit
FoxO1a-WT und einer konstitutiv im Kern lokalisierten FoxO1a-Mutante

Abkürzungen
A Adenin
A, Ala Alanin
AICAR 5-Aminoimidazol-4-Carboxamid Ribosid
APS Ammoniumpersulfat
BamH1 Restriktionsenzym
BCA bicinchoninic acid
BSA bovine serum albumine
BspH1 Restriktionsenzym
C Cytosin
CaCl2 Calciumchlorid
CAP Cbl associated protein
Cat catalytic domain
CBP CREB binding protein
cDNA complementary DNA
cm centimeter
CO2 Kohlendioxid
CREBP cAMP responsive element binding protein
CrkII CT 10 regulator of kinase II
CSF-1 colony stimulating factor-1
D, Asp Asparaginsäure
DAF 16 Bezeichnung für FoxO in C. elegans
dATP Desoxyadenosintriphosphat
dCTP Desoxycytosintriphosphat
ddNTP 2'-Didesoxynucleotid-5'-triphosphat
dGTP Desoxyguanosintriphosphat
DH5α Name eines Escherichia coli-Stammes
DMEM Dulbecco's Modified Eagle Medium
DMSO Dimethylsulfoxid
DNA desoxyribonucleic acid
DpnI Restriktionsenzym
DTT Dithiothreitol
dTTP Desoxythymidintriphosphat
DYRK dual tyrosine regulated kinase
ECL enhanced chemoluminescence
E. coli Escherichia coli
EcoRI Restriktionsenzym
EDTA Ethylendiamintetraessigsäure
EGFR epidermal growth factor receptor
ENV envelope, retrovirales Hüllprotein

ER estrogen receptor
ERK extracellular signal-regulated kinase
FAS fatty acid synthase
FCS fetal calf serum
FGFR fibroblast growth factor receptor
FKHR forkhead in rhabdomyosarcoma
FOXO Forkhead box protein
G Guanin
Gab-1 GRB2-associated binding protein 1
GAG group antigen, retrovirales Protein
GFP green fluorescent protein
GgPase Glycogenphosphatase
GLI1 glioma-associated oncoprotein
GLUT4 Glucosetransporter 4
G6Pase Glucose-6-Phosphatase
Grb2 growth factor receptor-bound protein 2
GRE Glucocorticoid-responsives Element
GSK-3 Glycogensynthase-Kinase-3
h Stunde
H2O Wasser
H4IIEC3 Hepatomzelllinie der Ratte
HBD Hormon-Bindungsdomäne
HBS HEPES buffered saline
HCl Salzsäure
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HepG2 Humane Hepatomzelllinie
HNF-4 hepatocyte nuclear factor 4
HNP Hexanucleotidprimer
IR Insulinrezeptor
IRS1 Insulinrezeptor Substrat 1
IRR insulin receptor-related Rezeptor
IRU insulin responsive unit
JAK Janus Kinase
JNK c-Jun-N-terminal kinase
K, Lys Lysin
kb kilo base
LB Nährmedium nach Luria Bertani
LiCl Lithiumchlorid
LSB Laemmli sample buffer
mA Milliampere
MAPK Mitogen-aktivierte Kinase

MgSO4 Magnesiumsulfat
MNB minibrain
MnCl2 Manganchlorid
MOPS 3-(4-Morpholino-)propansulfonsäure
mRNA messenger RNA
mTOR mammalian target of rapamycin
MUT Mutante
µM mikromolar
NaCl Natriumchlorid
NaF Natriumfluorid
NaN3 Natriumazid
Na4P2O7 Tetranatriumdiphosphat, Natriumpyrophosphat
Na3VO4 Trinatriumvanadattrihydrats
NFAT nuclear factor of activated T-cells
NLS nuclear localization sequence
OHT 4-OH Tamoxifen
OLB Oligo labelling buffer32P Radioaktives Phosphor-Isotop
pCDNA3.1 Expressionsvektor
PCR Polymerase Chain Reaction
PDGFR platelet derived growth factor receptor
PDK phosphatidylinositol-dependent kinase
pEGFP-C1 GFP-enthaltender Expresisonsvektor
PEPCK Phosphoenolpyruvatcarboxykinase
PEST Prolin, Glutamat, Serin, Threonin
PGC-1 PPARγ Coactivator -1
pGL2, pGL3 Expressionsvektoren
PH pleckstrin homology
PMSF Phenylmethylsulfonylfluorid
POL Polymerase
PPAR peroxysome proliferator-activator receptor
PEPCK Phosphoenolpyruvat-Carboxykinase
PIP2 Phosphatidyl-Inositol-3,4-Bisphosphat
PIP3 Phosphatidyl-Inositol-3,4,5-Trisphosphat
PI 3-K Phosphatidylinositol 3-Kinase
PKB/Akt Proteinkinase B
POL retrovirale reverse Transkriptase
pRLTK Renilla Luciferase Thymidin Kinase
pWZL Blasto Retroviraler Vektor
R, Arg Arginin
RbCl Rubiciumchlorid

RLU relative light units
RNA ribonucleic acid, Ribonukleinsäure
SDS-PAGE sodium dodecylsulfate polyacrylamide gel electrophoresis
Ser Serin
Shc Src-homology-2-containing protein
SOCS supressor of cytokine signalling
Shp-2 Src homology 2-containing tyrosine phosphatase
SRC1 steroid hormone receptor coactivator-1
SSC Standard Saline Citrate
STAT3 signal transducer and activator of transcription 3
STET Sucrose, Triton, EDTA, Tris
T Thymin
Ta Annealing-Temperatur
TAE Tris-Acetat-EDTA
TBE Tris-Borat-EDTA
TBS(-T) Tris buffered saline (-tween)
TE Tris-EDTA
TEMED Tetramethylethylendiamin
TfB I, TfB II Transformationbuffers I + II
Thr Threonin
UKPDS United Kingdom Prospective Diabetes Study
U/min Umdrehungen pro Minute
UV ultraviolett
WT Wildtyp
Yak1p Yet another kinase; Serin-/Threoninkinase in Sacc. cerevisiae
XhoI Restriktionsenzym

1 Einleitung
1 Einleitung
1.1 Einführung in die Epidemiologie und Pathophysiologie des Typ 2-Diabetes
Der Diabetes mellitus ist eine chronische Stoffwechselerkrankung, die den Fett-
und den Kohlenhydratstoffwechsel betrifft und auf einem absoluten oder
relativen Insulinmangel beruht. Weltweit leiden zur Zeit etwa 170 Millionen
Menschen an einem Diabetes mellitus, im Jahr 2030 wird sich die Zahl der
Patienten voraussichtlich mehr als verdoppelt haben (Wild et al., 2004). Etwa 5
Millionen Diabetiker leben in Deutschland. Über 90% der Diabetes-Patienten
leiden am so genannten Typ-2-Diabetes. Man beobachtet zum einen eine
steigende Prävalenz des Typ-2-Diabetes in den Wohlstandsgesellschaften, zum
anderen haben etwa 40% der Patienten mindestens ein Elternteil mit der
gleichen Erkrankung (Klein et al., 1996). Diese Form des Diabetes mellitus
kommt gehäuft im Rahmen des Metabolischen Syndroms vor. Das
metabolische Syndrom beschreibt die Assoziation von stammbetonter
Adipositas, Dyslipoproteinämie, arterieller Hypertonie und
Glucosetoleranzstörung (Reaven, 1988). Das anabole Hormon Insulin steht im
Mittelpunkt der pathophysiologischen Vorgänge bei beiden Formen des
Diabetes mellitus. Insulin sorgt für eine Aufnahme von Glucose in Skelettmuskel
und Fettgewebe und inhibiert die hepatische Glucoseproduktion. Durch diese
Mechanismen wird die Blutglucosekonzentration gesenkt. Die Auswirkungen
und Langzeitfolgen sind bei beiden Typen des Diabetes mellitus ähnlich, sie
unterscheiden sich aber pathophysiologisch und hinsichtlich ihrer Therapie
erheblich voneinander. Während beim Typ-1-Diabetes durch
Autoimmunreaktionen ß-Zellen des Pankreas zugrunde gehen und somit ein
absoluter Insulinmangel vorliegt, steht beim Typ-2-Diabetes eine Resistenz der
Zielgewebe gegenüber Insulin im Vordergrund. Die Insulinresistenz führt zur
Hyperglykämie aufgrund einer verminderten Glucoseaufnahme in Fett und
Muskel und einer gesteigerten hepatischen Glucoseproduktion.
Kompensatorisch kommt es zur reaktiven Hyperinsulinämie. Die gestörte
Glucoseaufnahme kann durch erhöhte Insulinkonzentration für eine gewisse
1

1 Einleitung
Zeit kompensiert werden, dieser Mechanismus verstärkt allerdings die
Insulinresistenz (Kahn, 2003). Mit fortschreitender Erkrankungsdauer nimmt die
Insulinempfindlichkeit der peripheren Zielorgane ab, was zu einer progredienten
Störung der insulinabhängigen Glucoseverwertung führt. Die Folge ist eine
manifeste Hyperglykämie mit einer Nüchtern-Plasmaglucosekonzentration ≥
126 mg/dl oder einer Plasmaglucosekonzentration ≥ 200 mg/dl zu irgendeinem
Zeitpunkt während des Tages (Expert Committee on the Diagnosis and
Classification of Diabetes Mellitus, 1997). Die pathophysiologischen
Zusammenhänge des Typ-2-Diabetes mellitus sind in Abbildung 1 dargestellt.
Adipositas
GenetikAlter
Hyperinsulinämie
erhöhte Fettsäuren
gesteigerteGlukosefreisetzung
verminderteInsulinsekretion
muskul.Insulinresistenz
β-Zell Kompensation
β-ZellDekompensation
gesteigerteLipolyse
gesteigerteGlukoneogenese
Diabetes
verminderteGlukosetoleranz
Abb. 1 Darstellung der pathophysiologischen Komponenten des Typ-2-Diabetes mellitus. Durch Faktoren wie Genetik, Adipositas und Alter kommt es zur Insulinresistenz, die über eine verminderte Glucosetoleranz zum manifesten Diabetes mellitus führt (nach Saltiel, Cell 2001).
1.2 Die Glucoseproduktion der Leber
Neben der Glycogenolyse, der Bereitstellung von Glucose aus
Kohlenhydratspeichern, ist die Gluconeogenese der Hauptmechanismus der
hepatischen Glucoseproduktion. Unter Gluconeogenese versteht man die
2

1 Einleitung
Produktion von Glucose aus Vorstufen wie Laktat, gluconeogenen Aminosäuren
(z.B. Alanin) und Glycerol.
Leber und Niere sind die Organe, welche zur Gluconeogenese befähigt sind,
wobei die Niere als Glucosequelle des Gesamtorganismus eher eine
untergeordnete Rolle spielt. Die entscheidenden Regelgrößen für die
hepatische Glucoseproduktion sind die Konzentrationen der verfügbaren
Substrate und die Aktivität regulatorischer Enzyme. Die Glucose-6-
Phosphatase (G6Pase) ist eines der Schlüsselenzyme der Gluconeogenese.
Das Enzym wird hauptsächlich in Leber und Nieren exprimiert, zu einem
geringeren Grad auch in Darm und Pankreas. Es ist im endoplasmatischen
Retikulum lokalisiert und besteht aus mindestens vier Komponenten, der
katalytischen Untereinheit, einem bisher strukturell nicht bekannten
Glucosetransporter, einem G6Pase-Transporter und einem Transporter für
anorganisches Phosphat (Mithieux, 1997; Foster et al., 1997; van de Werve et
al., 2000; van Schaftingen and Gerin, 2002). Die G6Pase katalysiert den letzten
Schritt der Gluconeogenese, die Hydrolyse von Glucose-6-Phosphat in
Phosphat und freie Glucose, die dann ins Blut abgegeben wird. Im
Hungerzustand und bei Diabetes ist eine gesteigerte G6Pase-Aktivität in vivo
nachweisbar, die auf eine gesteigerte Expression des Enzyms zurückzuführen
ist (Segal and Washko, 1959; Arion and Nordlie, 1965; Argaud et al., 1996;
Minassian et al., 1996; Liu et al., 1994). Während die Gluconeogenese durch
Hormone wie Glucocorticoide (Garland, 1986) oder Glucagon (Chatelain et al.,
1998) gesteigert wird, vermindert Insulin den mRNA-Gehalt und die Aktivität der
G6Pase (Argaud et al., 1996; Liu et al., 1994; Massillon et al., 1996). Eine
Verminderung der Expression des G6Pase-Gens wird in vivo als auch in
Hepatomzellen beobachtet (Argaud et al., 1996; Hornbuckle et al., 2001). Auch
durch schnelle, posttranslationale Effekte wirkt Insulin auf die Aktivität von
Enzymen der Gluconeogenese und der Glycogensynthese.
Die Insulinresistenz beim Typ-2-Diabetes mellitus führt dazu, dass Insulin die
Aktivität der glucogenen Enzyme wie G6Pase oder Phosphoenolpyruvat-
Carboxykinase (PEPCK) nicht mehr ausreichend hemmen kann. Dies führt zu
gesteigerter hepatischer Glucoseproduktion und zu erhöhter
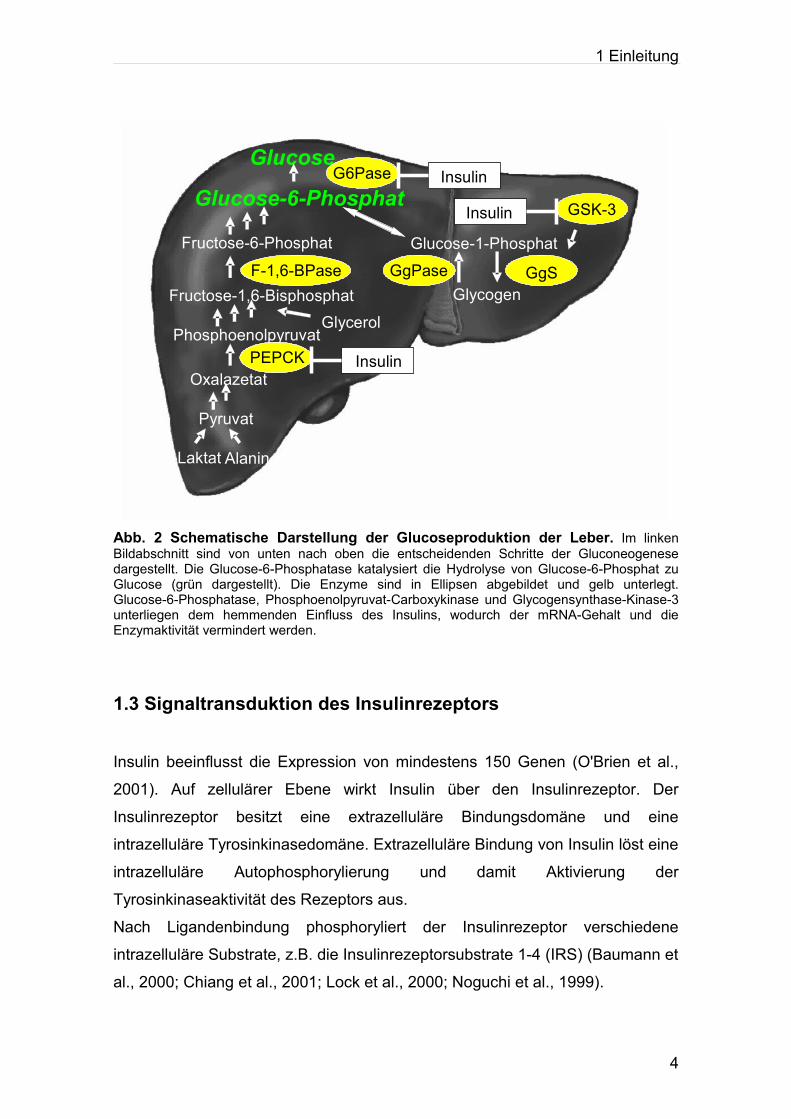
Blutglucosekonzentration. Abbildung 2 fasst die wesentlichen Schritte der
hepatischen Glucoseproduktion zusammen.
3
1 Einleitung
Pyruvat
Fructose-1,6-Bisphosphat
Glucose-6-Phosphat
Alanin
Fructose-6-Phosphat
Phosphoenolpyruvat
Oxalazetat
Glucose
Glucose-1-Phosphat
Glycerol
G6Pase
Glycogen
Laktat
F-1,6-BPase
PEPCK
GgPase GgS
Insulin
Insulin
GSK-3Insulin
Abb. 2 Schematische Darstellung der Glucoseproduktion der Leber. Im linken Bildabschnitt sind von unten nach oben die entscheidenden Schritte der Gluconeogenese dargestellt. Die Glucose-6-Phosphatase katalysiert die Hydrolyse von Glucose-6-Phosphat zu Glucose (grün dargestellt). Die Enzyme sind in Ellipsen abgebildet und gelb unterlegt. Glucose-6-Phosphatase, Phosphoenolpyruvat-Carboxykinase und Glycogensynthase-Kinase-3 unterliegen dem hemmenden Einfluss des Insulins, wodurch der mRNA-Gehalt und die Enzymaktivität vermindert werden.
1.3 Signaltransduktion des Insulinrezeptors
Insulin beeinflusst die Expression von mindestens 150 Genen (O'Brien et al.,
2001). Auf zellulärer Ebene wirkt Insulin über den Insulinrezeptor. Der
Insulinrezeptor besitzt eine extrazelluläre Bindungsdomäne und eine
intrazelluläre Tyrosinkinasedomäne. Extrazelluläre Bindung von Insulin löst eine
intrazelluläre Autophosphorylierung und damit Aktivierung der
Tyrosinkinaseaktivität des Rezeptors aus.
Nach Ligandenbindung phosphoryliert der Insulinrezeptor verschiedene
intrazelluläre Substrate, z.B. die Insulinrezeptorsubstrate 1-4 (IRS) (Baumann et
al., 2000; Chiang et al., 2001; Lock et al., 2000; Noguchi et al., 1999).
4

1 Einleitung
Eine Rezeptorstimulierung führt zur Aktivierung von zwei wichtigen
Signalkaskaden, der MAP-Kinase (Mitogen activated protein-Kinase)-Kaskade
und der PI-3-Kinase (Phosphatidyl-Inositol-3-Kinase)-Kaskade, wobei letztere
eine besondere Rolle bei der Vermittlung der Stoffwechseleffekte des Insulins
spielt. Phosphorylierung der IRS führt zu einer Aktivierung der PI-3-Kinase und
damit zur Bildung von Phosphatidyl-Inositol-3,4-Bisphosphat (PIP2) und
Phosphatidyl-Inositol-3,4,5-Trisphosphat (PIP3) in der Plasmamembran (Alessi
and Cohen, 1998). Dies hat eine gesteigerte Aktivität der Phosphatidyl-Inositol-
abhängigen Kinase 1 (PDK1) zur Folge (Lizcano and Alessi, 2002; Saltiel and
Pessin, 2002; Vanhaesebroeck and Alessi, 2000). Die Identität der PDK2 ist
nach wie vor unbekannt. PDK1 phosphoryliert wiederum die Proteinkinase B
(PKB, AKT). Es wurde gezeigt, dass PKB eine wichtige Rolle bei der
insulinabhängigen Hemmung der Expression des PEPCK- und G6Pase-Gens
spielt. So wurde beschrieben, dass Überexpression von PKB in Hepatomzellen
und primären Hepatozyten die Transkription von PEPCK und G6Pase
vermindert (Agati et al., 1998; Schmoll et al., 2000; Liao et al., 1998). Ein
direktes Substrat der PKB ist der insulinregulierte Transkriptionsfaktor FoxO1a,
der ein wichtiger Transaktivator des G6Pase-Gens ist (Schmoll et al., 2000;
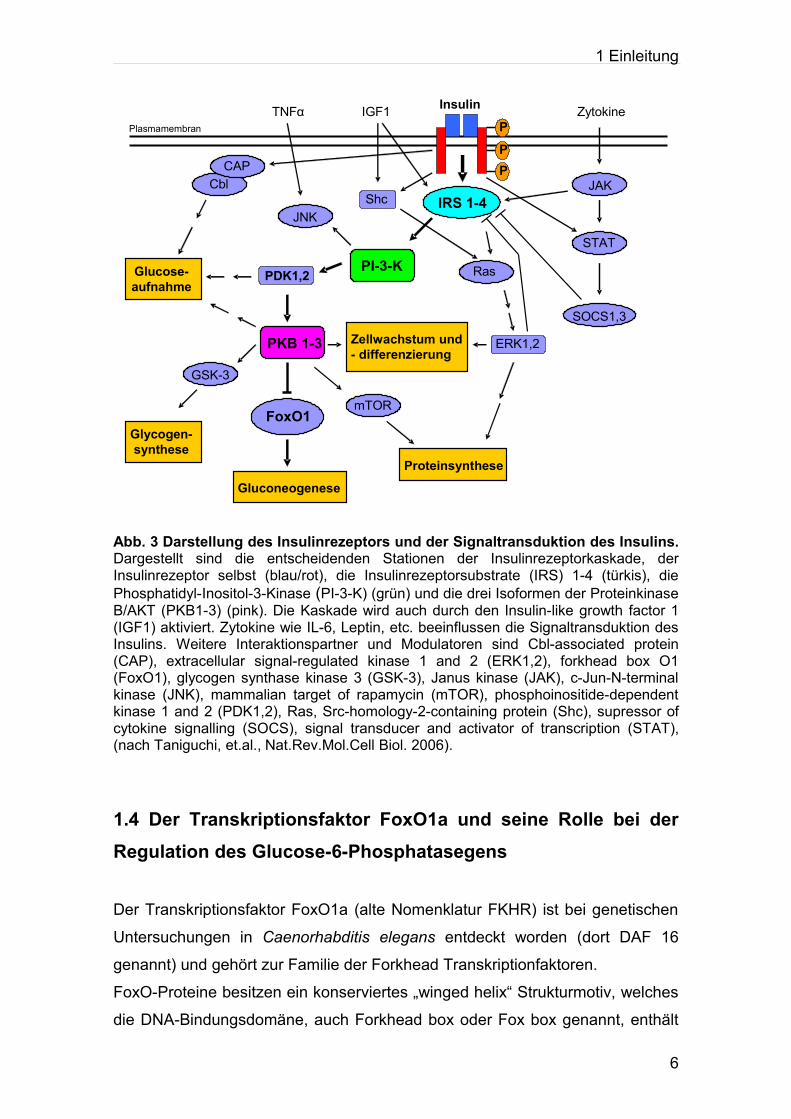
Barthel et al., 2001). Abbildung 3 zeigt eine Darstellung der Signaltransduktion
des Insulinrezeptors.
5
1 Einleitung
IRS 1-4
Gluconeogenese
PKB 1-3
PI-3-K
FoxO1
CblCAP
Proteinsynthese
Glycogen-synthese
Zellwachstum und- differenzierung
STAT
ERK1,2
Shc
PDK1,2 Ras
mTOR
GSK-3
PPP
Glucose-aufnahme
JAK
SOCS1,3
JNK
TNFα IGF1 Insulin ZytokinePlasmamembran
Abb. 3 Darstellung des Insulinrezeptors und der Signaltransduktion des Insulins. Dargestellt sind die entscheidenden Stationen der Insulinrezeptorkaskade, der Insulinrezeptor selbst (blau/rot), die Insulinrezeptorsubstrate (IRS) 1-4 (türkis), die Phosphatidyl-Inositol-3-Kinase (PI-3-K) (grün) und die drei Isoformen der Proteinkinase B/AKT (PKB1-3) (pink). Die Kaskade wird auch durch den Insulin-like growth factor 1 (IGF1) aktiviert. Zytokine wie IL-6, Leptin, etc. beeinflussen die Signaltransduktion des Insulins. Weitere Interaktionspartner und Modulatoren sind Cbl-associated protein (CAP), extracellular signal-regulated kinase 1 and 2 (ERK1,2), forkhead box O1 (FoxO1), glycogen synthase kinase 3 (GSK-3), Janus kinase (JAK), c-Jun-N-terminal kinase (JNK), mammalian target of rapamycin (mTOR), phosphoinositide-dependent kinase 1 and 2 (PDK1,2), Ras, Src-homology-2-containing protein (Shc), supressor of cytokine signalling (SOCS), signal transducer and activator of transcription (STAT), (nach Taniguchi, et.al., Nat.Rev.Mol.Cell Biol. 2006).
1.4 Der Transkriptionsfaktor FoxO1a und seine Rolle bei der Regulation des Glucose-6-Phosphatasegens
Der Transkriptionsfaktor FoxO1a (alte Nomenklatur FKHR) ist bei genetischen
Untersuchungen in Caenorhabditis elegans entdeckt worden (dort DAF 16
genannt) und gehört zur Familie der Forkhead Transkriptionfaktoren.
FoxO-Proteine besitzen ein konserviertes „winged helix“ Strukturmotiv, welches
die DNA-Bindungsdomäne, auch Forkhead box oder Fox box genannt, enthält
6

1 Einleitung
und zur Klassifizierung dient (Kaufmann and Knochel, 1996; Kaestner et al.,
2000). Im Gegensatz zu anderen Forkhead Proteinen besitzen FoxO-Proteine
eine besondere Aminosäuresequenz in der DNA-Bindungsdomäne. FoxO1a ist
an zahlreichen biologischen Prozessen wie Zellzykluskontrolle (Schmidt et al.,
2002), Apoptose (Gilley et al., 2003) sowie an der Regulation von
Stoffwechselprozessen in Leber, Muskel, Fett und Pankreas beteiligt. Ende der
Neunziger Jahre wurde in Studien mit Caenorhabditis elegans erstmals gezeigt,
dass FoxO-Proteine bei der Insulin-Signalkaskade eine Rolle spielen (Ogg et
al., 1997; Lin et al., 1997). In jüngeren Studien wurde eine Beteiligung von
FoxO-Proteinen an metabolischen Prozessen in Drosophila nachgewiesen
(Hwangbo et al., 2004).
Das humane FoxO1a enthält an den Aminosäureresten Threonin 24, Serin 256
und Serin 319 drei Konsensusmotive für die Phosphorylierung durch die
Serin/Threonin-Kinase PKB (RXRXXS/T) und ist ein direktes Substrat der PKB
(Rena et al., 1999; Nakae et al., 1999). Durch Phosphorylierung nimmt die
transkriptionelle Aktivität von FKHR ab und es erfolgt ein Transport aus dem
Zellkern ins Zytoplasma (Belham et al., 1999; Biggs, III et al., 1999; Brunet et
al., 1999; Lange et al., 1994; Liao et al., 1998; Lizcano and Alessi, 2002).
Weitere Phosphorylierungen an den Aminosäureresten Serin 322 und Serin 325
durch die Proteinkinase CK I (Rena et al., 2002) und am Aminosäurerest Serin
329 durch die Proteinkinase DYRK1A (Woods et al., 2001b) sind ebenfalls
bekannt.
In früheren Untersuchungen wurde gezeigt, dass Forkhead
Transkriptionsfaktoren an so genannte Insulin-responsive Elemente (IRUs)
binden, die erstmals im Gen der PEPCK identifiziert wurden (Unterman et al.,
1994; O'Brien et al., 1990). Die katalytische Untereinheit des G6Pase-
Promotors enthält drei solcher IRUs mit dem Konsensusmotiv T(G/A)TTT.
7

1 Einleitung
CAGGCTGTTTTTGTGTGCCTGTTTTTCTATTTTACGTAA
GRE
IRU IRU IRU
-186 -172 -164-180
Abb. 4. Strukturausschnitt des Promotors des G6Pase-Gens. Die Insulin-responsiven Elemente IRUs (rot), die als FoxO1a-Bindungsstellen dienen, liegen in einem Abschnitt, der auch ein glucocorticoidresponsives Element (GRE, grau) enthält.
FoxO1a ist in der Lage, mit hoher Affinität an die IRUs im G6Pase-Promotor zu
binden (Ayala et al., 1999). Dies führt zu gesteigerter Transkription des Gens
der G6Pase (Schmoll et al., 2000; Barthel et al., 2001; Nakae et al., 2001).
Durch Mutationsanalysen in Reportergensystemen wurde die entscheidende
Rolle dieser Strukturen bei der Regulation des G6Pase-Gens durch Insulin
nachgewiesen. Die Phosphorylierung von FoxO1a durch Wachstumsfaktoren,
somit auch die insulinabhängige Phosphorylierung, führt zur transkriptionellen
Inaktivierung und nukleären Exklusion des Transkriptionsfaktors (Biggs, III et
al., 1999; Durham et al., 1999; Guo et al., 1999; Tang et al., 1999; Schmoll et
al., 2000).
Phosphorylierung durch andere Kinasen wie z.B. JNK (Jun-N-terminal kinase)
als Folge von oxidativem Stress führen hingegen zu einer Translokation in den
Zellkern. Die Angriffspunkte in den FoxO-Proteinen unterscheiden sich von den
oben beschriebenen (Essers et al., 2004; Matsumoto and Accili, 2005; Lehtinen
et al., 2006). Bemerkenswert ist, dass die Reaktion auf oxidativen Streß den
Effekt der Wachstumsfaktoren überwiegt (Wang et al., 2005).
Neben der Phosphorylierung ist die Acetylierung/Deacetylierung ein weiterer
Regulationsmechanismus der FoxO-Transkriptionsfaktoren. SIRT1 deacetyliert
FoxO-Proteine und vermindert somit deren Aktivität (Motta et al., 2004; Brunet
et al., 2004; van der Horst et al., 2004). Eine Phosphatase, die FoxO-Proteine
dephosphoryliert, ist noch nicht identifiziert.
Bei Glucagon- und Glucocorticoid-induzierter Expression des G6Pase-Gens
spielt FoxO1a ebenfalls eine essentielle Rolle (Nakae et al., 2001). Der
Glucocorticoidrezeptor bindet an „Glucocorticoid-responsive Elemente“ (GREs).
Ein solches GRE liegt im Promotor des G6Pase-Gens im selben
Sequenzabschnitt wie die IRUs (siehe Abbildung 4). Diese Erkenntnisse legen
8

1 Einleitung
die Vermutung nahe, dass FoxO-Proteine bei der Regulation der hepatischen
Glucoseproduktion eine wichtige Rolle spielen.
1.5 Die Familie der DYRK Kinasen und ihre Verbindung mit dem Transkriptionsfaktor FoxO1a
Die Proteinkinasen DYRK1A (DYRK = dual-specificity tyrosine-phosphorylated
and regulated kinase), DYRK1B und DYRK2, die in dieser Arbeit im
Wesentlichen untersucht wurden, gehören zu einer Familie bestehend aus
mindestens sieben Säuger-Isoformen (DYRK1A, DYRK1B, DYRK1C, DYRK2,
DYRK3, DYRK4A und DYRK4B), dem Hefehomolog Yak1p und der Drosophila-
Kinase minibrain (MNB). Dual Specificity beschreibt ihre Fähigkeit sowohl
Serin-/Threonin- als auch Tyrosinreste zu phosphorylieren (Himpel et al., 2001;
Kentrup et al., 1996; Becker et al., 1998; Becker and Joost, 1999).
Während DYRK1A ubiquitär exprimiert wird und DYRK1B in Muskel- und
Hodengewebe vorkommt (Leder et al., 1999), wurden die anderen Isoformen
bisher nur in Hodengewebe nachgewiesen.
DYRK1A und DYRK1B sind subzellulär im Kern lokalisiert, DYRK2 hingegen ist
nur im Zytoplasma vorhanden. Dies ist auf eine Kernlokalisationssequenz
(nuclear localisation sequence, NLS) zurückzuführen, die alle DYRK1-
Isoformen aufweisen.
629 AS 92% Kern Hoden, Muskel ja
Größe Lokalisation ExpressionGemeinsamkeit
mit DYRK1A(katal. Domäne)
528 AS 46% Zytoplasma Hoden nein
763 AS 100% Kern ubiquitär jaDYRK1A
DYRK1B
DYRK2
NLS
Abb. 5 Vergleich der drei in dieser Arbeit behandelten DYRK-Isoformen. DYRK1A und DYRK1B sind im Gegensatz zu DYRK2 im Kern lokalisiert. Die Isoformen unterscheiden sich zudem in ihrer Expression (NLS = nuclear localisation sequence (Kernlokalisationssequenz).
9

1 Einleitung
Die verschiedenen Isoformen unterscheiden sich außerdem durch ihre
Substratspezifität, was ihre Beteiligung an unterschiedlichen zellulären
Prozessen vermuten lässt. Bereits länger bekannte Substrate von DYRK1A
sind Transkriptionsfaktoren wie FoxO1a, STAT3, CREB und Gli1 (Matsuo et al.,
2001; Woods et al., 2001b; Mao et al., 2002; Yang et al., 2001). Neuere
Untersuchungen haben Mitglieder der NFAT (nuclear factor of activated T-
cells)-Transkriptionsfaktorenfamilie als Substrate von DYRK1A und DYRK2
identifiziert (Gwack et al., 2006). Neben den genannten Transkriptionsfaktoren
wurden auch zytoplasmatische Proteine als in vitro-Substrate von DYRK1A
nachgewiesen. Es handelt sich hierbei unter anderem um Proteine wie elF2Bε,
Tau und Dynamin (Chen-Hwang et al., 2002; Woods et al., 2001a), wobei
elF2Bε und Tau in vitro auch durch DYRK2 und DYRK3 phosphoryliert werden,
die im Zytoplasma lokalisiert sind (Woods et al., 2001a). Die Phosphorylierung
des Spleißfaktors SF3b1 lässt eine Beteiligung von DYRK1A an mRNA-
Spleißprozessen vermuten (De Graaf et al., 2006). Kürzlich wurde eine
Interaktion von DYRK1A mit 14-3-3-Proteinen nachgewiesen, wodurch die
katalytische Aktivität von DYRK1A gesteigert wird (Alvarez et al., 2007). Aus
der Familie der Cycline wird Cyclin L2 durch DYRK1A und Cyclin D3 durch
DYRK1B phosphoryliert (De Graaf et al., 2004; Takahashi-Yanaga et al., 2006).
DYRK1B phosphoryliert und aktiviert außerdem HNF1α (Lim et al., 2002).
In aktuellen Untersuchungen wurde eine Phosphorylierung des
Tumorsuppressorproteins p53 durch DYRK2 und somit eine Beteiligung an
Prozessen der Apoptose nachgewiesen (Taira et al., 2007).
Die DYRK-Isoformen haben große Ähnlichkeit in der katalytischen Domäne,
unterscheiden sich jedoch im N- und C-terminalen Bereich. Die wahrscheinlich
größte Gemeinsamkeit aller DYRK-Kinasen ist ihre Fähigkeit zur
Autophosphorylierung.
Das Gen für DYRK1 liegt auf Chromosom 21 in einer Region, die als „Down´s
syndrome critical region“ bezeichnet wird (Becker et al., 1998). DYRK1A scheint
eine Rolle bei der Entwicklung des zentralen Nervensystems zu spielen.
Überexpression von DYRK1A in transgenen Mäusen hat zu Veränderungen der
Neuronenentwicklung, zu motorischen Störungen und Verhaltensänderungen
geführt (Altafaj et al., 2001). DYRK1A wird nicht nur in Verbindung mit der
10

1 Einleitung
Trisomie 21 (Down´s Syndrom) sondern auch mit der Alzheimer-
Demenzerkrankung gebracht (Kelly and Rahmani, 2005; Kimura et al., 2007).
Es wurde beschrieben, dass DYRK1A den Transkriptionsfaktor FoxO1a an der
Position Ser329 phosphoryliert. Darüber hinaus wurde gezeigt, dass FoxO1a
und DYRK1A in bestimmten Kernregionen co-lokalisiert sind („speckles“) und
aus Zellextrakten co-immunopräzipitiert werden können. Diese Daten lassen
vermuten, dass DYRK1 und FoxO1a auch funktionell miteinander interagieren
(Woods et al., 2001b).
1.7 Arbeitshypothese und Zielsetzung
Es war bekannt, dass FoxO1a an der Regulation des Glucose-6-
Phosphatasegens beteiligt ist. Außerdem wurde in vorausgehenden Arbeiten
eine Protein-Protein-Interaktion zwischen FoxO1a und DYRK1 beschrieben. Es
stellt sich somit die Frage nach der funktionellen Bedeutung dieser Interaktion.
Dieser Arbeit liegt die Hypothese zugrunde, dass DYRK1 eine Rolle bei der
Vermittlung FoxO1a-abhängig regulierter biologischer Funktionen wie z.B. der
Regulation des G6Pase-Gens spielt.
Ziel dieser Arbeit war es deshalb zu untersuchen,
1.) welchen Effekt DYRK1 auf die FoxO1a-abhängige Aktivierung des
Promotors der Glucose-6-Phosphatase hat und
2.) über welche molekularen Mechanismen (z. B. Phosphorylierung von
FoxO1a durch DYRK1) dieser Effekt vermittelt wird.
11

2 Materialien und Methoden
2 Materialien und Methoden
2.1 Konstrukte
2.1.1 Der Promotor des Glucose-6-Phosphatasegens
Das in dieser Arbeit verwandte Promotorkonstrukt enthält den Glucose-6-
Phosphatase-Promotor der Ratte (-1227/+57), der in den promotorlosen
Luciferase-Reportergen-Vektor pGL3 basic kloniert wurde. Der G6Pase-
Promotor enthält drei sog. „Insulin responsive Elemente“ (T(G/A)TTT), die für
die Regulation des G6Pase-Gens durch Insulin entscheidend sind, da sie als
Bindungsstellen für FoxO1a dienen (Schmoll et al., 1996). Das Konstrukt wurde
freundlicherweise von PD Dr. Dieter Schmoll (Frankfurt) zur Verfügung gestellt
(siehe auch Abbildung 4).
2.1.2 FoxO1a Wildtyp
Dieses Plasmid wurde hauptsächlich zur Kontrolle und zum Vergleich mit
anderen FoxO1a-Konstrukten eingesetzt.
FoxO1a WT wurde in den Expressionsvektoren pCDNA3.1/His C (Fa.
Invitrogen, USA) und pEGFP-C1 (Fa. Clontech, USA) verwendet. Die cDNA
wurde freundlicherweise von Frank Rauscher (Pennsylvania, USA) zur
Verfügung gestellt.
2.1.3 FoxO1a Ser329-Punktmutanten
FoxO1a wird an der Position Serin 329 durch die Proteinkinase DYRK1A
phosphoryliert. Es wurden Experimente mit zwei FoxO1a-Mutanten
durchgeführt.
2.1.3.1 FoxO1a Ser329Ala-Mutante
Bei diesem Konstrukt wurde die Aminosäure Serin an der Position 329 durch
Alanin ersetzt. Durch diese Veränderung kann an dieser Position keine
Phosphorylierung mehr erfolgen.
12

2 Materialien und Methoden
2.1.3.2 FoxO1a Ser329Asp-Mutante
Durch den Austausch der Aminosäure Serin 329 gegen Asparaginsäure wurden
negative Ladungen eingefügt, die den Effekt einer permanenten
Phosphorylierung simulieren.
Diese Mutanten wurden nach dem Prinzip der „Sequenzspezifischen
Mutagenese“ hergestellt (siehe unten). Es wurden Pfu Turbo-DNA-Polymerase
(Fa. Promega) und die folgenden Primer (Fa. MWG Biotech) eingesetzt:
FoxO1 a Ser329Ala :
S G R L A P I M T E Q5´- AGT GGG AGA CTT GCT CCC ATA ATG ACA GAG CAG -3´3´- TCA CCC TCT GAA CGA GGG TAT TAC TGT CTC GTC -5´
FoxO1a Ser329Asp:
I S G R L D P I M T5´- ATC AGT GGG AGA CTG GA T CCC ATC ATG ACA -3´3´- TAG TCA CCC TCT GAC CTA GGG TAG TAC TGT -5´
Die fett gedruckten Buchstaben markieren die veränderten Nucleotide, die roten
die ausgetauschten Aminosäuren. Im Falle der Serin-Alanin-Mutante wurde
eine im Wildtyp enthaltene BspH1-Schnittstelle entfernt, bei der Serin-
Asparaginsäure-Mutante eine zusätzliche BamH1-Schnittstelle (unterstrichen)
eingefügt. Diese Schnittstellen dienen dazu, den Wildtyp von Mutanten und
Mutanten untereinander mittels einfacher Restriktionsverdaus zu unterscheiden.
Die Serin-Punktmutanten wurden in den Vektor pEGFP-C1 (Fa. Clontech, USA)
kloniert.
2.1.4 IRU-Mutante des Glucose-6-Phosphatasereportergens
Auch diese Mutante wurde, wie die anderen FoxO1a-Punktmutanten, nach dem
Prinzip der „Sequenzspezifischen Mutagenese“ hergestellt. Es wurden die drei
Phosphorylierungsstellen der PKB in FoxO1a Thr24, Ser256 und Ser319 durch
Alaninreste ersetzt. Dadurch ist diese Mutante resistent gegenüber einer
Stimulation mit Insulin und konstitutiv im Zellkern lokalisiert.
Für dieses Konstrukt wurden folgende Primer (Fa. MWG Biotech) verwendet:
13

2 Materialien und Methoden
Thr24Ala:
5´-CCC CGG CCG CGC TCG TGC GCA TGG CCG CTG CCC-3´
5´-GGG CAG CGG CCA TGC GCA CGA GCG CGG CCG GGG-3´
Ser256Ala:
5´-CCT AGG AGA AGA GCG GCC GCA ATG GAC AAC AAC-3´
5´-GTT GTT GTC CAT TGC GGC CGC TCT TCT CCT AGG-3´
Ser319Ala:
5´-CGC CCT CGA ACT AGT GCA AAT GCT AGT AC-3´
5´-GTA CTA GCA TTT GCA CTA GTT CGA GGG CG-3´
Die FoxO1a-Konstrukte wurden in die Vektoren pcDNA3.1/His C (Fa.
Invitrogen) und pEGFP-C1 (Fa. Clontech) kloniert. Sämtliche Konstrukte
wurden durch Sequenzierung des kompletten Leserahmens der FoxO1a-cDNA
überprüft.
2.1.5 DYRK-Konstrukte
Die verwendeten DYRK-Konstrukte DYRK1A, DYRK1B, DYRK2 und DYRK1A
K188R (alle in pEGFP-N1 (Fa. Clontech, USA)) wurden freundlicherweise von
Prof. Walter Becker (Aachen) zur Verfügung gestellt. Sie sind in
vorangegangenen Arbeiten beschrieben worden (Becker et al., 1998; Himpel et
al., 2001; Leder et al., 1999). Die Punktmutante DYRK1A K188R wurde, wie
auch die FoxO1a-Punktmutanten, nach dem Prinzip der „Sequenzspezifischen
Mutagenese“ hergestellt.
14

2 Materialien und Methoden
2.2 Zellbiologische Arbeitstechniken
2.2.1 Zelllinien
Bei den im Rahmen dieser Dissertation verwendeten Zellen handelt es sich um
die Hepatomzelllinien HepG2 (Mensch) und H4IIEC3 (Ratte) sowie um eine
retrovirale Verpackungszelllinie (Phoenix).
Mit HepG2-Zellen wurden ausschließlich transiente Transfektionen
durchgeführt.
Stabile Transfektionen wurden mit einer H4-Rattenhepatomzelllinie, die den
Promotor des G6Pasegens stabil exprimiert, durchgeführt. Diese Zelllinie wurde
freundlicherweise von PD Dr. Dieter Schmoll (Frankfurt) zur Verfügung gestellt.
Es wurde zusätzlich eine Zelllinie hergestellt, die DYRK1A stabil exprimiert.
Dafür wurde die in früheren Arbeiten beschriebene DYRK1A-cDNA (Kentrup et
al., 1996) in die EcoRI- und XhoI-Schnittstellen des retroviralen Vektors pWZL-
Blasto kloniert. Anschließend wurde das DYRK1A-Konstrukt mittels retroviralen
Gentransfers in H4-Hepatomzellen gebracht, die nun sowohl den Promotor des
G6Pase-Gens als auch DYRK1A stabil exprimieren. Als Kontrollzellen wurden
H4-Zellen mit dem leeren Vektor pWZL-Blasto infiziert.
2.2.1.1 Medien und allgemeine Kulturbedingungen
Die Zellen wurden in einer wasserdampfgesättigten Atmosphäre mit einem
Anteil von 5% CO2 bei 37°C kultiviert. Sie wurden in DMEM F-12 (HepG2) und
DMEM Medium (HAIIEC3) gehalten. Den Medien (Fa. PAA Laboratories GmbH,
A) wurden 10% FCS, 50 µg/ml Penicillin und 100 mg/ml Streptomycin (Fa.
GIBCO) zugesetzt. Die Kulturgefäße stammen von der Firma Nalge Nunc
International. Sämtliche Kulturmedien wurden über Nitrocellulosemembranen
(Fa. Becton Dickinson) steril filtriert.
2.2.1.2 Subkultivierung
Jeweils nach Erreichen der Konfluenz wurden die Zellen passagiert. Hierzu
wurden die Zellen mit PBS gewaschen und anschließend mit 1 ml 0,05%-igem
15

2 Materialien und Methoden
Trypsin-EDTA (Fa. GIBCO) für ca. 10 min inkubiert. Der Prozess wurde durch
Zugabe von frischem Medium gestoppt und die Zellen auf neue Gefäße verteilt.
2.2.2 Transfektion
HepG2-Zellen wurden mit FuGENETM 6 Transfection Reagent (Fa. Roche) nach
den Beschreibungen des Herstellers transient transfiziert.
Am Tag vor Transfektion wurden die Zellen trypsiniert und auf neue
Kulturgefäße verteilt, so dass sie zum Zeitpunkt der Transfektion eine
Konfluenz von 50-80% besaßen.
Für die Transfektion wurde ein Reagenz-DNA-Gemisch im Verhältnis 3:1 (µl
Reagenz: µg DNA) hergestellt. Diese Mischung wurde bei Raumtemperatur ca.
15 min lang inkubiert. Anschließend wurde der Reagenz-DNA-Komplex
tröpfchenweise zum Kulturmedium der zu transfizierenden Zellen gegeben.
Bis zum weiteren Experiment wurden die Zellen bei 37 °C inkubiert.
24 Stunden nach Transfektion wurden die Zellen geteilt und nach 36 Stunden
auf F12-Medium mit 0,01mg/ml BSA, 20mM Hepes, pH 7,4 und
Penicillin/Streptomycin gesetzt.
Phoenix-Zellen wurden transient mit der Calcium-Phosphat-Methode
transfiziert.
Materialien:
• Verpackungszelllinie (Phoenix-Zellen)
• Plasmid-DNA
• 2 M CaCl2
• 2x HBS
Durchführung:
30 µg der zu transfizierenden DNA wurden in einem sterilen 15 ml Kunststoff-
Röhrchen vorgelegt. Es wurden 1,5 ml Bidest und 183 µl der 2 M CaCl2-Lösung
hinzugefügt. Zur Transfektionskontrolle wurde außerdem 0,5 µg pEGFP-CF1-
Plasmid-DNA hinzu gegeben. Nach Durchmischen und kurzer Zentrifugation
16

2 Materialien und Methoden
wurde tropfen weise die 2x HBS-Lösung hinzu pipettiert. Mit einer
Pasteurpipette wurde für 20 s Luft in die Lösung geblasen, danach 15 min bei
Raumtemperatur inkubiert. An einer Trübung der Lösung kann man die Bildung
der Calciumphosphat-Kristalle erkennen. Das Gemisch wurde tropfen weise auf
die Zellkultur pipettiert und die Zellen über Nacht bei 37°C inkubiert. Der Erfolg
der Transfektion konnte anhand der Expression von GFP unter dem UV-
Mikroskop anhand grün fluoreszierender Zellen beurteilt werden.
2.2.3 Retroviraler Gentransfer
Die Retroviren enthalten die Gene GAG (gruppenspezifisches Antigen), POL
(reverse Transkriptase) und ENV (Hüllprotein). Im Vektor pWZL-Blasto ist GAG
enthalten, die Phoenixzellen exprimieren POL und ENV. Bei Transfektion von
Phoenixzellen mit pWZL Blasto und der enthaltenen Plasmid-DNA ergibt sich
ein retrovirales Genom. Die produzierten Retroviren können nun weitere Zellen
infizieren, die dann die durch das Plasmid kodierten Gene stabil exprimieren
können.
Infektion der H4IIEC3-Zellen
Retrovirusproduktion
Retrovirales Plasmid
pWZLblasto
blastogag
LTR
LTR
amp
DYRK1AEcoRI XhoI
Ф-Zellen
Transfektion Infektion
Abb. 6 Das Prinzip des retroviralen Gentransfers. Die Verpackungszellen (Phoenix-Zellen, Ф) werden mit dem retroviralen Plasmid transfiziert. Die produzierten Retroviren infizieren die Hepatomzelllinie H4IIEC3, die das enthaltene Plasmid stabil exprimiert.
Die Verpackungszelllinie zur Herstellung der Retroviren (Phoenix-Zellen) wurde
mit dem retroviralen Vektor (pWZL) transient transfiziert (s.o.).
17

2 Materialien und Methoden
Nach der Transfektion wurden die Zellen über Nacht bei 37°C inkubiert.
Anschließend wurden die Zellen nach einem Medienwechsel für weitere 48-72
h bei 32°C inkubiert, weil bei dieser Temperatur die von der transfizierten
Verpackungszelllinie produzierten Retroviren am stabilsten sind. Die
produzierten Viren akkumulierten so im Medium.
Der virushaltige Überstand wurde abgenommen, durch kurze Zentrifugation von
Zelldetritus gereinigt, steril filtriert (20µm Filter, Fa. Sartorius) und bis zur
weiteren Verwendung bei –20°C gelagert.
Das Zellsystem, das das gewünschte Protein stabil exprimieren soll, wurde
dann mit den gewonnenen Retroviren infiziert. Dazu wurden die Zellen
(H4IIIEC3) auf Kulturschalen ausgesät, so dass sie zu Beginn der Infektion
einen Konfluenzgrad von 30-50 % hatten. Das alte Medium wurde durch 1 ml
frisches ersetzt. Dazu wurden 3 µl 10-3 M Insulin und 3 µl Polybrene (Fa. Sigma)
pipettiert. Auf das Medium wurden 2 ml des virushaltigen Mediums pipettiert.
Die Kulturschale wurde 90 min bei Raumtemperatur und 3400 U/min
zentrifugiert.
Anschließend wurden die Zellen für mind. 48 h bei 32°C inkubiert, bevor mit der
Selektion begonnen wurde.
2.2.4 Selektionierung der infizierten Zellen
Materialien:
• Retroviral infizierte Zellen
• Antibiotikum (Blasticidin, Fa. Calbiochem, USA)
Durchführung:
Um ausschließlich diejenigen Zellen zu erhalten, die die gewünschte cDNA ins
Genom integriert hatten, wurde das Prinzip der Antibiotikaresistenz zur
Selektionierung genutzt. Das Retrovirus enthält zusätzlich zur gewünschten
cDNA ein Antibiotika-Resistenz-Gen, welches es den erfolgreich infizierten
Zellen ermöglicht, in Gegenwart von Antibiotika, in diesem Fall Blasticidin, zu
überleben. Zur Kontrolle werden nicht-infizierte Zellen eingesetzt.
18

2 Materialien und Methoden
Die Selektionierung erfolgte mit einer Antibiotika-Konzentration von 500 µg/ml.
Das Medium mit dem Antibiotikum wurde alle zwei Tage gewechselt, bis alle
Kontrollzellen abgestorben waren.
2.3 Proteinchemische Arbeitstechniken
2.3.1 SDS-PAGE
Materialien:
• 30% Acrylamid/Bisacrylamid, 37,5:1 (Fa. Bio-Rad)
• 10% Ammoniumpersulfat
• TEMED (Fa. Bio-Rad)
• 10% SDS
• 1,5 M Tris-HCl pH 8,8
• 0,5 M Tris-HCl pH 6,8
• Elektrodenpuffer : 25 mM Tris
250 mM Glycin
0,1% SDS
• Probenpuffer (nach Laemmli, 2X LSB) :
20 Vol% Glycerin
8% SDS
10 mM EDTA
0,25 M Tris
5 Vol% Bromphenolblau-Lsg.
• Kaleidoskop-Proteingrößen-Standard (Fa. Bio-Rad)
2.3.1.1 Gelherstellung
In Abhängigkeit vom Molekulargewicht des zu bestimmenden Proteins wurde
ein Polyacrylamidgel entsprechender Zusammensetzung polymerisiert.
1) Polyacrylamid-Trenngel
19
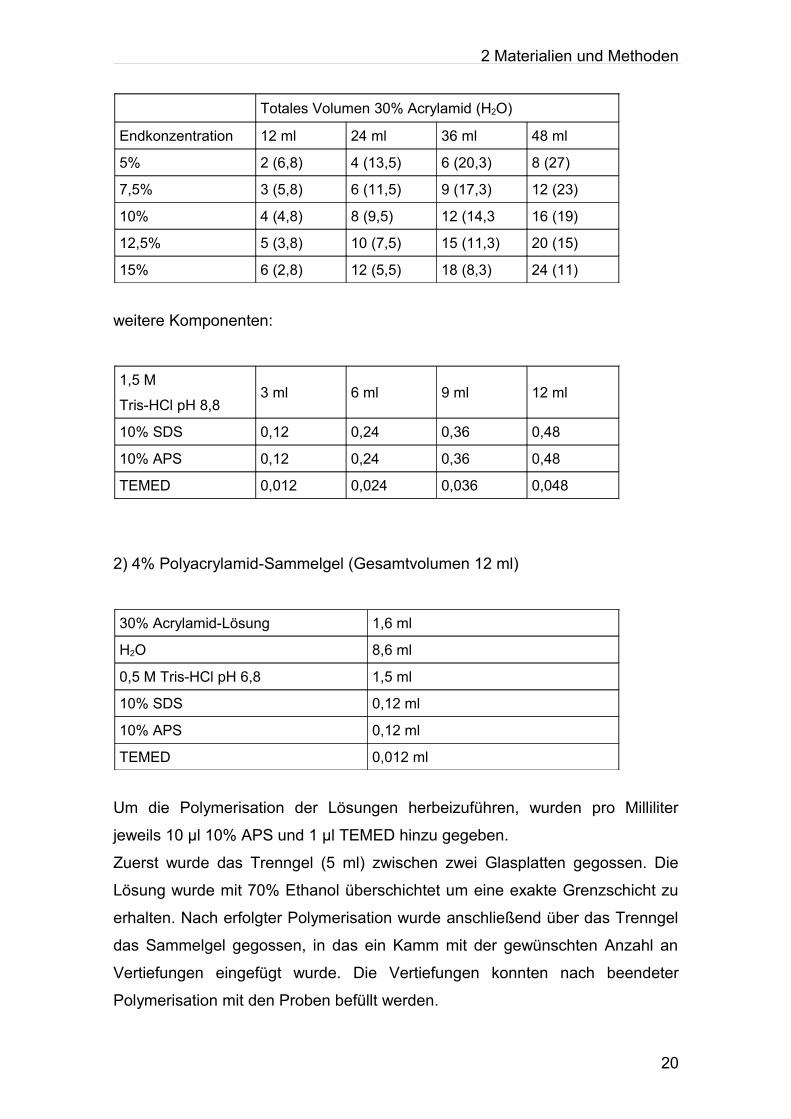
2 Materialien und Methoden
Totales Volumen 30% Acrylamid (H2O)
Endkonzentration 12 ml 24 ml 36 ml 48 ml
5% 2 (6,8) 4 (13,5) 6 (20,3) 8 (27)
7,5% 3 (5,8) 6 (11,5) 9 (17,3) 12 (23)
10% 4 (4,8) 8 (9,5) 12 (14,3 16 (19)
12,5% 5 (3,8) 10 (7,5) 15 (11,3) 20 (15)
15% 6 (2,8) 12 (5,5) 18 (8,3) 24 (11)
weitere Komponenten:
1,5 M
Tris-HCl pH 8,83 ml 6 ml 9 ml 12 ml
10% SDS 0,12 0,24 0,36 0,48
10% APS 0,12 0,24 0,36 0,48
TEMED 0,012 0,024 0,036 0,048
2) 4% Polyacrylamid-Sammelgel (Gesamtvolumen 12 ml)
30% Acrylamid-Lösung 1,6 ml
H2O 8,6 ml
0,5 M Tris-HCl pH 6,8 1,5 ml
10% SDS 0,12 ml
10% APS 0,12 ml
TEMED 0,012 ml
Um die Polymerisation der Lösungen herbeizuführen, wurden pro Milliliter
jeweils 10 µl 10% APS und 1 µl TEMED hinzu gegeben.
Zuerst wurde das Trenngel (5 ml) zwischen zwei Glasplatten gegossen. Die
Lösung wurde mit 70% Ethanol überschichtet um eine exakte Grenzschicht zu
erhalten. Nach erfolgter Polymerisation wurde anschließend über das Trenngel
das Sammelgel gegossen, in das ein Kamm mit der gewünschten Anzahl an
Vertiefungen eingefügt wurde. Die Vertiefungen konnten nach beendeter
Polymerisation mit den Proben befüllt werden.
20

2 Materialien und Methoden
2.3.1.2 Probenvorbereitung
Die Zelllysate wurden in eiskaltem Lysepuffer gewonnen und 10 min bei 12000g
zentrifugiert.
Pufferzusammensetzung: 50 mM Hepes, pH 7,6
1% Triton X-100
150 mM NaCl
1 mM EDTA
10% Glycerol
1 mM DTT
1 mM Benzamidin
0,5 mM PMSF
10 mM NaF
1 mM Na3VO4
30 mM Na4P2O7
2.3.1.3 Proteinbestimmung (BCA-Methode)
Materialien:
• BCA Protein Assay Reagent A (Fa. Pierce)
• BCA Protein Assay Reagent B (1:50 mit Reagenz A)
• Lyse-Puffer
Durchführung:
Von dem zuvor bereiteten Zelllysat wurden 1-5 µl eingesetzt und mit dem Lyse-
Puffer auf 5 µl Endvolumen in einer Mikrotiterplatte vorgelegt. Zu den einzelnen
Proben wurden 100 µl fertiges BCA-Reagenz hinzugefügt.
Als Referenz wurde eine Eichkurve mit 0, 1, 2, 5, 10 bzw. 20 µg/µl BSA
(Bovines Serum Albumin) erstellt, wobei die BSA-Stocklösung mit dem Lyse-
Puffer verdünnt wurde. Diese Proben wurden ebenfalls in die Mikrotiterplatte
pipettiert. Der Reaktionsansatz wurde dann 30 min bei 37°C inkubiert. Das
Fortschreiten der Reaktion ließ sich anhand einer zunehmenden Violettfärbung
verfolgen.
21

2 Materialien und Methoden
Die Auswertung und damit die Bestimmung der Proteinkonzentration erfolgte
mit dem Spektrometer LAMBDA E (Fa. MWG Biotech) bei 562 nm und der
Software KC4-Kineticalc for Windows (Fa. Bio-Tec Instruments Inc.).
2.3.1.4 Gelelektrophorese
Die Elektrophorese wurde in einer Elektrodenkammer durchgeführt, die
vollständig mit Elektrodenpuffer gefüllt wurde. Die Proteine wurden bei 30-40
mA über eine Laufstrecke von 5 cm elektrophoretisch aufgetrennt.
2.3.2 Western-Blotting und ECL-System
Materialien:
• PROTRAN® Nitrocellulose Transfer Membran (Fa. Schleicher & Schuell)
• Whatman-Papier
• Transfer-Puffer 2,5 mM Tris
192 mM Glycin
20 Vol% Methanol
• TBS-T TrisCl 20 mM, pH 7,5
NaCl 150 mM
Triton 0,1%
2.3.2.1 Transfer
Nach der Elektrophorese wurde das Gel auf eine Nitrocellulosemembran gelegt
und zwischen 2 Lagen Whatman-Papier in einer Elektrophoresekammer fixiert.
Die Proteine wurden elektrophoretisch bei 400 mA auf die Membran transferiert.
Der Blot wurde anschließend in einer 3%-igen BSA-TBS-T-Lösung 15 min lang
geblockt, danach mit TBS-T gewaschen.
2.3.2.2 Inkubation mit Antikörpern
Die Inkubation mit dem entsprechenden Primärantikörper (Anti-DYRK1A-
Antikörper, Fa. BD Transduction Labs, USA) erfolgte über Nacht in einer 3%
BSA-Lösung in TBS-T mit 0,02% NaN3.
22

2 Materialien und Methoden
Am darauf folgenden Tag wurde der Blot gründlich mit TBST gewaschen.
Anschließend wurde der Blot für eine Stunde mit dem Peroxidase-konjugierten
Sekundärantikörper (Fa. Amersham Pharmacia, Schweden) (1:5000 einer 3%
(w/v) Milchpulver-TBS-T-Lösung) inkubiert. Danach wurde der Blot erneut
intensiv mit TBS-T gewaschen.
Die Antikörperreaktion mit dem Antigen wurde durch die Peroxidaseaktivität
mittels eines Lumineszenssubstrates detektiert.
Zusammensetzung der Substratlösung:
500 µl Tris-Cl 3M, pH 8,8
200 µl Luminol (Stocklsg. : 0,44 g/10 ml DMSO)
100 µl p-Coumarinsäure (Stocklsg.: 0,15 g/10 ml DMSO)
19,2 ml H2O
20 µl 30% H2O2
Der Blot wurde einige Sekunden lang in der Lösung inkubiert. Die emittierte
Lumineszenz wurde mit einem Film (Fa. Kodak) detektiert.
2.3.3 Ponceau-Färbung
Der Nachweis des erfolgreichen Proteintransfers vom Gel auf die
Nitrocellulosemembran erfolgte durch Färbung der Membran mit Ponceau S.
Durch reversible Bindung der Sulfonsäuregruppen an Proteine können die
Proteinbanden auf einem Western Blot sichtbar gemacht werden.
Materialien:
• Ponceau-Lösung 0,25 g Ponceau S (Fa. Sigma)
40 ml Methanol
15 ml Eisessig
45 ml H20
23

2 Materialien und Methoden
2.4 Molekularbiologische Arbeitstechniken
2.4.1 DNA-Präparation
2.4.1.1 Minipräparation
Materialien:
• Bakterienkolonien
• LB-Medium mit Ampicillin (100 µg/ml)
• STET-Puffer: 50 mM Tris-HCl, pH 8,8
50 mM EDTA
0,5% Triton X 100
8% Glucose
• Lysozym (10 mg/ml in H2O)
• TE-Puffer: TrisCl 10 mM
EDTA 0,5 mM
• Isopropanol
Durchführung:
Zur Isolierung kleinerer Mengen Plasmid-DNA diente die Methode nach
HOLMES und QUIGLEY (1981). Einzelkolonien wurden von Kolonieplatten
geerntet und über Nacht in 1,5 ml LB-Medium mit Ampicillin (100 µg/ml) bei
37°C angezüchtet. Die Suspension wurde in Probengefäße (Fa. Eppendorf)
überführt und bei 12.000 g abzentrifugiert. Das Pellet wurde in 250 µl STET-
Puffer und 10 µl Lysozym (10 mg/ml) resuspendiert. Anschließend wurden die
Bakterien durch Kochen (50 sec) lysiert. Es wurde 15 min bei 12.000 g
zentrifugiert und anschließend das Pellet verworfen. Der verbleibende
Überstand wurde mit 260 µl Isopropanol gefällt und das getrocknete Pellet in
TE-Puffer aufgenommen.
2.4.1.2 Präparation größerer DNA-Mengen
Größere Mengen Plasmid-DNA wurden mit Hilfe des Plasmid Maxi Kits (Fa.
QIAgen) nach den Angeben des Herstellers präpariert. 250 ml LB-Medium mit
24

2 Materialien und Methoden
Ampicillin (100 µg/ml) wurden mit einer Kolonie inokuliert und über Nacht bei
37°C inkubiert. Die so gezüchteten E. coli-Bakterien wurden abzentrifugiert (5
min, 8.000 g) und das Pellet zur Präparation von Plasmid DNA eingesetzt.
2.4.2 Restriktionsverdau
Restriktionsverdaus wurden nach den Angaben der Hersteller mit den
empfohlenen Puffern durchgeführt. Die Restriktionsenzyme wurden von den
Firmen NEB, Pharmacia und Gibco bezogen.
2.4.3 Herstellung kompetenter E. coli-Bakterien
Materialien:
• TfB I-Puffer (pH 5,8) 90 mM Kaliumacetat
10 mM CaCl2xH2O
0,5 mM LiCl
100 mM RbCl
50 mM MnCl2xH2O
15% (w/v) Glycerin
• TfB II-Puffer (pH 7,0) 10 mM MOPS
10 mM RbCl
75 mM CaCl2xH2O
• LB-Medium
Beide Puffer wurden steril filtriert.
Durchführung:
Aus einer Gefrierkultur wurden E. coli-Zellen des Stammes DH5α auf LB-
Platten ausplattiert und über Nacht bei 37°C inkubiert.
Eine einzelne Kolonie wurde am nächsten Tag in 2 ml LB-Medium
aufgenommen und über Nacht bei 37°C unter Schütteln bei 200 U/min inkubiert.
25

2 Materialien und Methoden
Zu 150 ml LB-Medium wurden 800 µl dieser Kultur gegeben. Unter Kontrolle
der optischen Dichte (Eppendorf Photometer 1101M) wurde die Kultur bei 37°C
auf dem Schüttler (200 U/min) bis zu einer OD578 = 0,5 herangezogen .
Die Zellsuspension wurde in einem 250 ml-Zentrifugenbecher überführt und 20
min auf Eis gestellt. Anschließend erfolgte die Pelletierung der Zellen (5000 U/
min/4°C/10 min/Rotor JA-14 (Fa. Beckmann)) Das Pellet wurde vorsichtig in 45
ml TfB I-Puffer resuspendiert und für zwei Stunden auf Eis gekühlt. Die Zellen
wurden erneut pelletiert und in 6 ml TfB II-Puffer resuspendiert. Nach dem
Schockgefrieren in flüssigem Stickstoff erfolgte die Lagerung der kompetenten
Zellen bei –80 °C.
2.4.4 Transformation von E.coli-Bakterien
2.4.4.1 Hitzeschocktransformation
Materialien:
• Plasmid-DNA
• Kompetente E. coli-Bakterien
• LB-Medium
Durchführung:
Zu 80-100 µl kompetenter E.coli-Bakterien wurden 1-2 µl der durch Mini-Preps
(s.o.) isolierten Plasmid-DNA in ein Probengefäß pipettiert. Dieses wurde 15
min auf Eis gestellt und anschließend 1-2 min bei 42°C erwärmt. Es wurden 900
µl LB-Medium hinzugefügt und das Reaktionsgefäß bei 37°C und 200 U/min für
eine Stunde in den Schüttler gestellt. Danach wurde 2 min bei 3000 U/min
zentrifugiert, 900 µl des Überstandes wurden verworfen und die sedimentierten
E. coli-Bakterien wurden durch Auf- und Abpipettieren in die verbleibenden 100
µl des LB-Mediums verteilt. Diese wurden dann auf eine mit Ampicillin versetzte
Agarplatte ausplattiert. Die Agarplatte wurde bei 37°C über Nacht inkubiert und
am nächsten Tag auf amplifizierte Klone überprüft.
26

2 Materialien und Methoden
2.4.4.2 Elektroporation
Materialien:
• Plasmid
• Elektroporationsküvetten (Fa. Eurogentec, Belgien)
• Elektrokompetente E. coli
• Antibiotikahaltige Agarplatten
Durchführung:
Das aufgereinigte Plasmid wurde mit 100 µl Bakteriensuspension in eine
Elektroporationsküvette gegeben. Die Bakterien wurden bei 2,5 kV, 25 µF und
200 Ω elektroporiert (Gene Pulser, Fa. Bio-Rad). Anschließend wurde die
Suspension mit LB-Medium ca. 1 h bei 37°C inkubiert und auf Agarplatten,
welche ein Antibiotikum enthielten, ausplattiert.
2.4.5 DNA-Agarosegelelektrophorese
Materialien:
• Agarose (Fa. Seakem)
• Ethidiumbromid, Stock-Lsg.: 10 µg/µl (Fa. ICN Biomedicals)
• DNA-Leiter (Fa. Gibco)
• TAE-Puffer: 40 mM Tris-Acetat, pH 8,0
1 mM EDTA
• Orange-G-Probenpuffer: 50% Glycerin
0,25% Orange-G Stock-Lsg. in TAE-Puffer
Durchführung:
DNA-Fragmente wurden in 0,7-1,5% Agarosegelen (Agarose in TAE mit 0,5 µg/
ml Ethidiumbromid) aufgetrennt. Zu den Proben wurde vor dem Auftragen 1/5
Volumen Orange-G-Probenpuffer zugegeben. Als Größenstandard diente die 1
27

2 Materialien und Methoden
kb DNA Leiter, als Laufpuffer TAE. Die Elektrophorese wurde bei 100 V
durchgeführt. Die Banden wurden im UV-Durchlicht sichtbar gemacht.
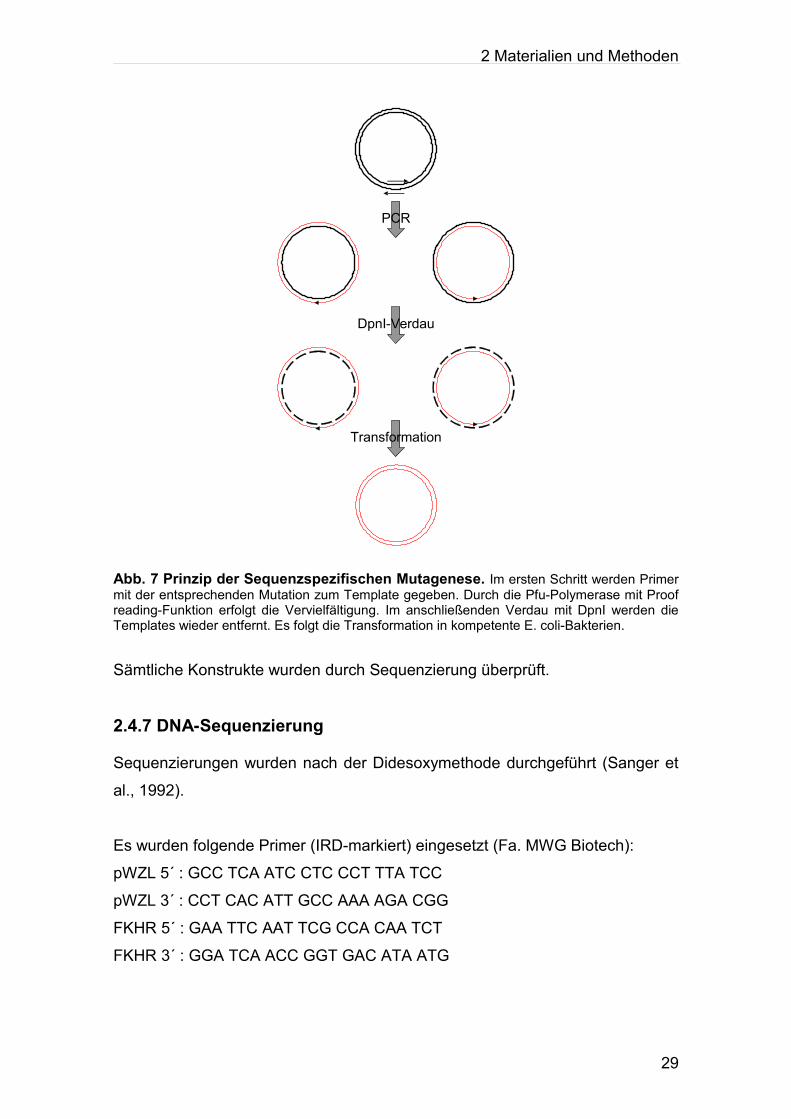
2.4.6 Sequenzspezifische Mutagenese
Die FoxO1a Ser329-Punktmutanten wurden mit Hilfe des QuickChange™ Site-
Directed Mutagenesis Kits (Fa. Stratagene) nach den Angeben des Herstellers
generiert.
Prinzip der Sequenzspezifischen Mutagenese (nach WEINER et.al.1994):
Man benötigt für jede Mutation zwei Primer, einen Strang- und einen
Gegenstrangprimer, die beide die gewünschte Mutation enthalten. Mit
PfuTurbo-DNA-Polymerase (Fa. Promega) (besitzt Proofreading-Affinität) führt
man 16 Zyklen PCR durch, um das gesamte Plasmid komplett zu amplifizieren.
Anschließend verdaut man das Gemisch aus PCR-Produkt und
Ausgangsplasmid (Template) mit DpnI. DpnI schneidet spezifisch methylierte
und hemimethylierte DNA, so dass nur die Templates abgebaut werden. Nun
transformiert man die verbleibenden PCR-Produkte in superkompetente XL1-
Blue-Zellen, um das Plasmid mit der Mutation zu vermehren.
28
2 Materialien und Methoden
PCR
Transformation
DpnI-Verdau
Abb. 7 Prinzip der Sequenzspezifischen Mutagenese. Im ersten Schritt werden Primer mit der entsprechenden Mutation zum Template gegeben. Durch die Pfu-Polymerase mit Proof reading-Funktion erfolgt die Vervielfältigung. Im anschließenden Verdau mit DpnI werden die Templates wieder entfernt. Es folgt die Transformation in kompetente E. coli-Bakterien.
Sämtliche Konstrukte wurden durch Sequenzierung überprüft.
2.4.7 DNA-Sequenzierung
Sequenzierungen wurden nach der Didesoxymethode durchgeführt (Sanger et
al., 1992).
Es wurden folgende Primer (IRD-markiert) eingesetzt (Fa. MWG Biotech):
pWZL 5´ : GCC TCA ATC CTC CCT TTA TCC
pWZL 3´ : CCT CAC ATT GCC AAA AGA CGG
FKHR 5´ : GAA TTC AAT TCG CCA CAA TCT
FKHR 3´ : GGA TCA ACC GGT GAC ATA ATG
29

2 Materialien und Methoden
Materialien:
• DNA/Primer Premix: 0,5 µg DNA
2 pmol Primer
1 µl DMSO
ad 26 µl H2O
• Terminations-Mischung
• Sequenzgel 18 g Harnstoff
3,74 ml 10x TBE
4,2 ml 40% Acrylamid (Fa. BIO-RAD)
12,4 ml H2O
186 µl APS (10% Stock-Lsg.)
20 µl TEMED
• Stop Lösung
• TBE-Puffer
Durchführung
Für jede der zu sequenzierenden DNAs wurden vier Ansätze vorbereitet, einer
für jedes Didesoxynucleotid. Zuerst wurden 2 µl der Terminations-Mischung
(enthält die ddNTPs) in PCR-Gefäße vorgelegt. Dazu wurden 6 µl des
jeweiligen DNA/Primer Mixes gegeben.
Die PCR-Reaktion wurde mit dem Hybaid Thermo Cycler (Fa. MWG Biotech)
nach folgendem Protokoll durchgeführt.
1. Schritt 94°C 2´ 1 Zyklus
2. Schritt 94°C 15´´
Ta (°C) 15´´ 30 Zyklen
70°C 30´´
3. Schritt 4°C
(Ta=Annealing-Temperatur)
30

2 Materialien und Methoden
Zu jedem Ansatz wurden dann 4 µl der Stop-Lösung gegeben, anschließend 1
µl des fertigen Ansatzes auf das Acrylamidgel geladen.
Die Sequenzanalyse wurde mit dem LiCor Sequenzierautomaten (Fa. MWG
Biotech) bei 50°C über Nacht durchgeführt und am darauf folgenden Tag
ausgewertet.
2.4.8 Northern Blotting
2.4.8.1 RNA-Isolierung
Die Präparation der RNA wurde mit dem RNeasy Kit (Fa. QIAgen) nach den
Angaben des Herstellers durchgeführt.
2.4.8.2 RNA-Agarosegelelektrophorese
Materialien:
• RNA
• Puffer (200 µl): 96 µl Formamid
20 µl 10x MOPS
35 µl Formaldehyd
29 µl Glycerol (37%)
20 µl Bromphenol Blau (0,1%)
• Formaldehyd-Agarose-Gel (100 ml):
10 ml 10x MOPS
75 ml bidestilliertes Wasser
0,8 g Agarose (Fa. Seakem)
Durchführung:
40 µg RNA einer jeden Probe wurden mit RNase freiem Wasser auf 40 µl
Volumen aufgefüllt und mit 20 µl des oben beschriebenen Puffers versetzt.
Zusätzlich wurden zu jeder Probe 3 µg Ethidiumbromid in 5 µl des gleichen
Puffers pipettiert. Diese wurden dann 5 min bei 65°C erhitzt. Anschließend
wurden sie auf ein Formaldehyd-Agarose-Gel aufgetragen.
31

2 Materialien und Methoden
2.4.8.3 Blotting
Materialien:
• Glasschüssel
• 20x SSC (1l): 175,3 g NaCl
88,2 g Trinatriumcitrat
• Whatman-Pappe
• Nylonmembran
• Parafilm
Durchführung:
Eine rechtwinklige Glasschüssel wurde mit 20x SSC etwa zur Hälfte gefüllt.
Darin wurde ein Quader eingelegt und auf diesen wiederum eine Glasscheibe.
Drei ca. 60 cm lange Whatman-Pappstreifen in der Breite der Glasschüssel
wurden übereinander gelegt, mit dem Puffer getränkt, an ihren beiden Enden
jeweils eingerollt und so auf die Glasscheibe gelegt, dass ihre beiden Enden in
den Puffer eintauchen und somit durch Kapillarkräfte die gesamte Pappe
ständig von Puffer benetzt wurde. Auf diese Pappstreifen wurde nun das Gel so
aufgelegt, dass seine Oberseite nach unten zeigt. Auf des Gel wurde die
Nylonmembran und auf diese ein genau so großes Stück Whatman-Pappe
gelegt.
Diese Anordnung wurde bis auf das bedeckte Gel selbst komplett mit Parafilm
überzogen, damit die Kapillarkräfte den Puffer ausschließlich durch das Gel auf
die Membran übertragen.
Auf die jetzt oben liegende Whatman-Pappe wurden dicht nebeneinander zwei
Stapel Falthandtücher aus Papier gelegt, auf diese jeweils ein Ziegelstein (ca.
1-2 kg), damit der Flüssigkeitssog durch die Kapillarkräfte ausreichend stark ist.
Der Transfer der RNA auf die Membran erfolgte über Nacht. Die Membran
wurde getrocknet und die RNA mittels UV-Licht (Fa. Biometra) kovalent an die
Membran gebunden („Crosslinking“).
32

2 Materialien und Methoden
2.4.8.4 Aufreinigen der DNA-Sonden nach Gelelektrophorese
Nach elektrophoretischer Trennung von Plasmid und Insert (Sonde)
(Agarosegelelektrophorese, s.o.), wurde die Sonde mit einem Skalpell unter
UV-Bestrahlung aus dem Gel herausgeschnitten und in ein Eppendorf-Gefäß
gegeben.
Die weitere Aufreinigung der Sonde erfolgte mit dem QIAquick Gel Extraction
Kit (Fa. QIAgen) nach den Angaben des Herstellers.
2.4.8.5 Markieren einer Sonde mit [32P]
Materialien:
• 10x Oligo labelling buffer: 500 mM Tris-HCL, pH 6,9
100 mM MgSO4
1 mM DTT
0,6 mM dATP
0,6 mM dGTP
0,6 mM dTTP
• DNA-Sonde
• HNP (Hexanucleotidprimer) (Fa. Pharmacia)
• [γ-32P]-dCTP (30 µCi) (Fa. Hartmann)
• Klenow-Fragment (10 U/µl) (Fa. NEB)
• Micro Spin-Säule (Fa. Pharmacia)
Durchführung:
10-20 ng einer DNA-Sonde wurden in einem Eppendorfgefäß vorgelegt. Dazu
wurden 2 µl OLB, 1 µl HNP (0,2 µg) gegeben und mit Wasser auf 16 µl
aufgefüllt. Das Eppendorfgefäß wurde 7 min in kochendem Wasser erhitzt und
anschließend sofort auf Eis gelagert. Es wurden dann 3 µl [γ-32P]-dCTP (30 µCi)
und 1 µl Klenow-Fragment dazu gegeben und 30 min bei 37°C inkubiert. Das
nichtinkorporierte [γ -32P]-dCTP wurde anschließend durch Gelfiltration über
eine Micro Spin Säule von der markierten Sonde getrennt. Um die Einbaurate
33

2 Materialien und Methoden
der Reaktion zu bestimmen, wurde die Radioaktivität von einem Mikroliter des
Eluats mit dem Flüssigkeitsszintilations-Messgerät (Fa. Beckman) bestimmt.
2.4.8.6 Hybridisierung der Northern Blots
Materialien:
• Northern Blot
• radioaktiv markierte Sonde
• Prähybridisierungslösung (Fa. Clontech)
• 2x SSC-Puffer, pH 7,0
Durchführung
Der feuchte Blot wurde in einen Glaszylinder gelegt und bei 72°C mit 10 ml der
Prähybridisierungslösung 30 min lang unter Rotation inkubiert. Danach wurde
die zuvor bei 95°C für 5 min aufgekochte Sonde hinzu gegeben. Der Blot wurde
nun mit der Sonde über Nacht bei 42°C hybridisiert.
Am darauf folgenden Tag wurde der Blot zweimal mit 200 ml 2x SSC/0,1%SDS
bei 42°C gewaschen. Der Blot wurde dann 24 lang mit einem Screen inkubiert,
welcher dann mit dem Phospho-Imager (Fa. Packard) gelesen wurde.
2.4.9 Reportergen-Assay (Luciferase)
Die Luciferase-Aktivität wurde mit dem Luciferase Assay System und dem Dual
Luciferase System (beide Fa. Promega) nach den Angaben des Herstellers
gemessen. Die Daten wurden auf die co-exprimierte Renilla (pRLTK)-Aktivität
(HepG2) oder die Proteinkonzentration (H4) bezogen.
2.5 Statistische Analyse
Die Mittelwerte und Standardabweichungen der einzelnen Experimente wurden
mittels eines zweiseitigen, ungepaarten (Student´s) t-Tests auf signifikante
Unterschiede hin überprüft. Als Signifikanzniveau wurde α= 0,05 festgelegt.
34

3 Ergebnisse
3 Ergebnisse
3.1 Effekte von überexprimiertem DYRK1A auf den Gehalt an Glucose-6-Phosphatase-mRNA und die Aktivität des G6Pase-Promotors in Hepatomzellen
3.1.1 Überexpression von DYRK1A in stabil transfizierten Rattenhepatomzellen
Um zu untersuchen, ob DYRK1A die Expression von FoxO1a-abhängig
regulierten Genen beeinflusst, wurden Zellen einer Rattenhepatomlinie
(H4IIEC3) hergestellt, die DYRK1A stabil überexprimieren. H4IIEC3-Zellen
wurden zum einen ausgewählt, weil sie gut differenziert sind und
leberspezifische Funktionen wie Gluconeogenese erhalten sind. Zum anderen
sind sie in Bezug auf die Signaltransduktion des Insulins gut charakterisiert.
Die Zellen wurden mittels retroviralen Gentransfers mit einem Vektor infiziert,
der für DYRK1A codiert. Im SDS-PAGE-Verfahren wurden Proteine aus totalem
Zelllysat aufgetrennt. Mittels der Western Blot-Technik wurden die Proteine auf
eine Nitrocellulosemembran transferiert, auf der sie mit spezifischen
Antikörpern detektiert wurden. In diesem Experiment kam ein α-DYRK1A-
Antikörper (Fa. BD Transduction Labs, USA) zur Anwendung.
Im Vergleich zu H4-Zellen, die mit einem Kontrollvektor pWZL Blasto
transfiziert wurden, zeigten die mit DYRK1A infizierten Zellen eine deutliche
Überexpression von DYRK1A (Abbildung 8).
35

3 Ergebnisse
Kontro
lle
DYRK1A
Western Blotα-DYRK1A
Ponceau
Abb. 8 Überexpression von DYRK1A in H4IIEC3-Zellen. Jeweils 20µg Protein von Kontrollzellen (links) und von solchen, die DYRK1A überexprimieren (rechts) wurden elektrophoretisch getrennt und auf eine Nitrocellulosemembran transferiert. Im ersten Schritt wurde die Membran mit einem α-DYRK1A-Antikörper (Fa. BD Transduction Labs, USA), im zweiten mit einem Peroxidase-gekoppelten Antikörper inkubiert. Das Signal wurde mit der Chemilumineszenzmethode sichtbar gemacht. Die Doppelbande im Western Blot ist möglicherweise auf unterschiedliche Phosphorylierungszustände von DYRK1A zurückzuführen.Im unteren Teil der Abbildung ist eine Ponceau-Färbung der Membran als Beladungskontrolle dargestellt.
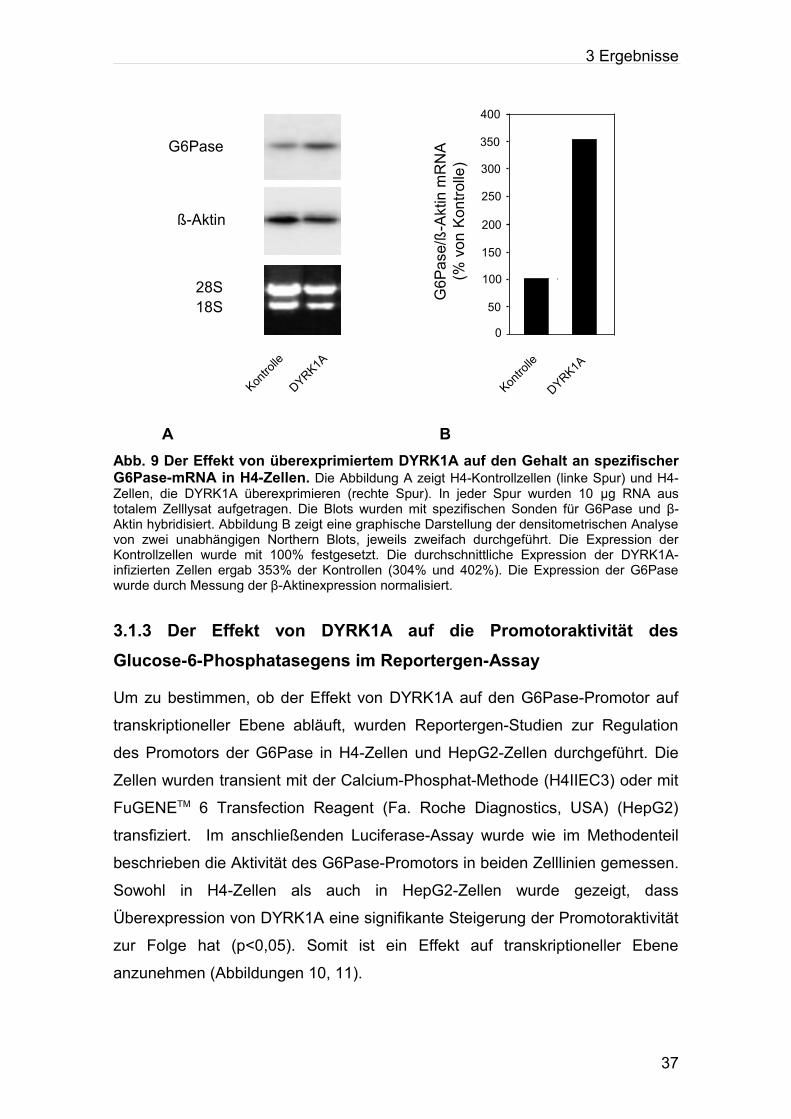
3.1.2 Der Einfluss von stabil transfiziertem DYRK1A auf die endogene Glucose-6-Phosphatase-mRNA in Rattenhepatomzellen
In weiteren Experimenten wurde untersucht, ob Überexpression von DYRK1A
einen Einfluss auf den mRNA-Gehalt endogener G6Pase in H4-Zellen hat. Zu
diesem Zweck wurde aus H4-Zellen, die DYRK1A überexprimieren, und aus
Zellen, die mit einem leeren Kontrollvektor infiziert waren, RNA isoliert. Die
RNA-Proben wurden elektrophoretisch getrennt und im Northern Blot-Verfahren
auf eine Membran transferiert. Diese Membran wurde dann mit einer radioaktiv
markierten spezifischen Glucose-6-Phosphatase-Sonde inkubiert. Es wurde
gezeigt, dass die Zellen, die DYRK1A überexprimieren, einen ca. 3,5-fach
erhöhten mRNA-Gehalt an endogener G6Pase im Vergleich zu Kontrollzellen
haben (Abbildung 9). Diese Ergebnisse lassen vermuten, dass DYRK1A an der
Regulation der G6Pase beteiligt ist.
36
3 Ergebnisse
G6Pase
ß-Aktin
28S18S
Kontro
lle
DYRK1A
0
50
100
150
200
250
300
350
400
Kontro
lle
DYRK1A
G6P
ase/
ß-Ak
tin m
RN
A(%
von
Kon
trolle
) A BAbb. 9 Der Effekt von überexprimiertem DYRK1A auf den Gehalt an spezifischer G6Pase-mRNA in H4-Zellen. Die Abbildung A zeigt H4-Kontrollzellen (linke Spur) und H4-Zellen, die DYRK1A überexprimieren (rechte Spur). In jeder Spur wurden 10 µg RNA aus totalem Zelllysat aufgetragen. Die Blots wurden mit spezifischen Sonden für G6Pase und β-Aktin hybridisiert. Abbildung B zeigt eine graphische Darstellung der densitometrischen Analyse von zwei unabhängigen Northern Blots, jeweils zweifach durchgeführt. Die Expression der Kontrollzellen wurde mit 100% festgesetzt. Die durchschnittliche Expression der DYRK1A-infizierten Zellen ergab 353% der Kontrollen (304% und 402%). Die Expression der G6Pase wurde durch Messung der β-Aktinexpression normalisiert.
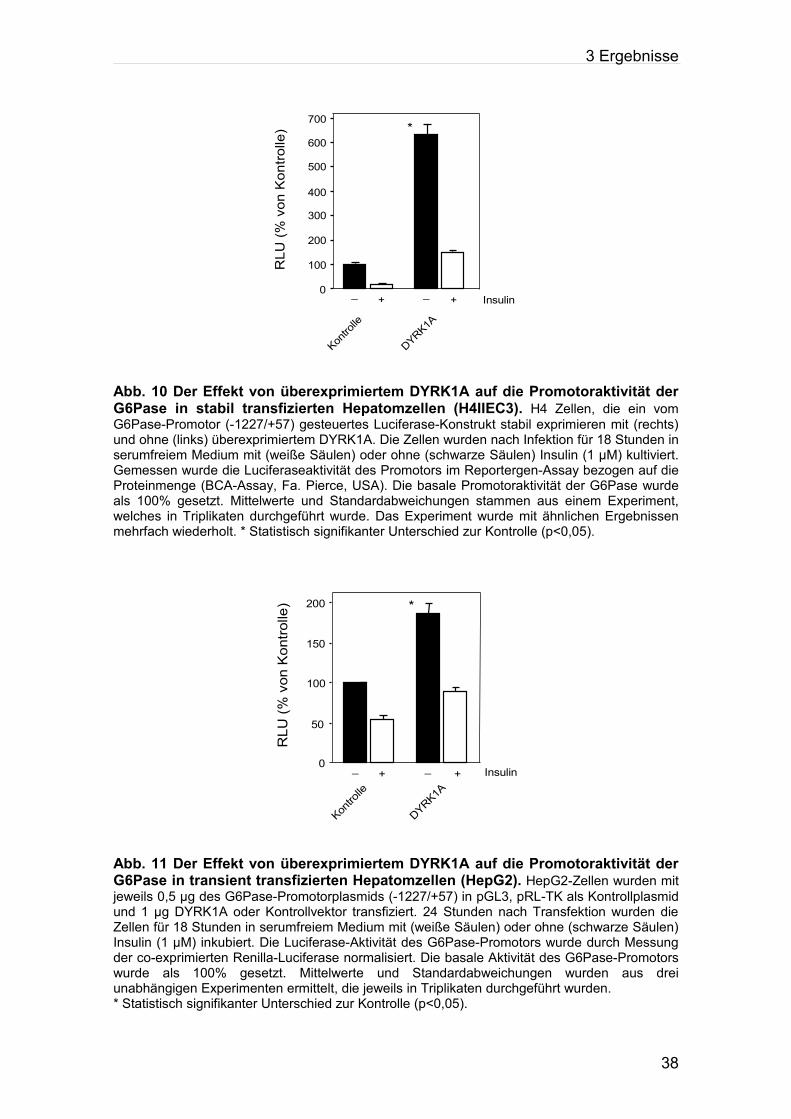
3.1.3 Der Effekt von DYRK1A auf die Promotoraktivität des Glucose-6-Phosphatasegens im Reportergen-Assay
Um zu bestimmen, ob der Effekt von DYRK1A auf den G6Pase-Promotor auf
transkriptioneller Ebene abläuft, wurden Reportergen-Studien zur Regulation
des Promotors der G6Pase in H4-Zellen und HepG2-Zellen durchgeführt. Die
Zellen wurden transient mit der Calcium-Phosphat-Methode (H4IIEC3) oder mit
FuGENETM 6 Transfection Reagent (Fa. Roche Diagnostics, USA) (HepG2)
transfiziert. Im anschließenden Luciferase-Assay wurde wie im Methodenteil
beschrieben die Aktivität des G6Pase-Promotors in beiden Zelllinien gemessen.
Sowohl in H4-Zellen als auch in HepG2-Zellen wurde gezeigt, dass
Überexpression von DYRK1A eine signifikante Steigerung der Promotoraktivität
zur Folge hat (p<0,05). Somit ist ein Effekt auf transkriptioneller Ebene
anzunehmen (Abbildungen 10, 11).
37
3 Ergebnisse
Kontro
lle
DYRK1A
Insulin0
100
200
300
400
500
600
700
+_ +_R
LU (%
von
Kon
trolle
) *
Abb. 10 Der Effekt von überexprimiertem DYRK1A auf die Promotoraktivität der G6Pase in stabil transfizierten Hepatomzellen (H4IIEC3). H4 Zellen, die ein vom G6Pase-Promotor (-1227/+57) gesteuertes Luciferase-Konstrukt stabil exprimieren mit (rechts) und ohne (links) überexprimiertem DYRK1A. Die Zellen wurden nach Infektion für 18 Stunden in serumfreiem Medium mit (weiße Säulen) oder ohne (schwarze Säulen) Insulin (1 µM) kultiviert. Gemessen wurde die Luciferaseaktivität des Promotors im Reportergen-Assay bezogen auf die Proteinmenge (BCA-Assay, Fa. Pierce, USA). Die basale Promotoraktivität der G6Pase wurde als 100% gesetzt. Mittelwerte und Standardabweichungen stammen aus einem Experiment, welches in Triplikaten durchgeführt wurde. Das Experiment wurde mit ähnlichen Ergebnissen mehrfach wiederholt. * Statistisch signifikanter Unterschied zur Kontrolle (p<0,05).
0
50
100
150
200
Kontro
lle
DYRK1AInsulin+_+_
RLU
(% v
on K
ontro
lle) *
Abb. 11 Der Effekt von überexprimiertem DYRK1A auf die Promotoraktivität der G6Pase in transient transfizierten Hepatomzellen (HepG2). HepG2-Zellen wurden mit jeweils 0,5 µg des G6Pase-Promotorplasmids (-1227/+57) in pGL3, pRL-TK als Kontrollplasmid und 1 µg DYRK1A oder Kontrollvektor transfiziert. 24 Stunden nach Transfektion wurden die Zellen für 18 Stunden in serumfreiem Medium mit (weiße Säulen) oder ohne (schwarze Säulen) Insulin (1 µM) inkubiert. Die Luciferase-Aktivität des G6Pase-Promotors wurde durch Messung der co-exprimierten Renilla-Luciferase normalisiert. Die basale Aktivität des G6Pase-Promotors wurde als 100% gesetzt. Mittelwerte und Standardabweichungen wurden aus drei unabhängigen Experimenten ermittelt, die jeweils in Triplikaten durchgeführt wurden. * Statistisch signifikanter Unterschied zur Kontrolle (p<0,05).
38
3 Ergebnisse
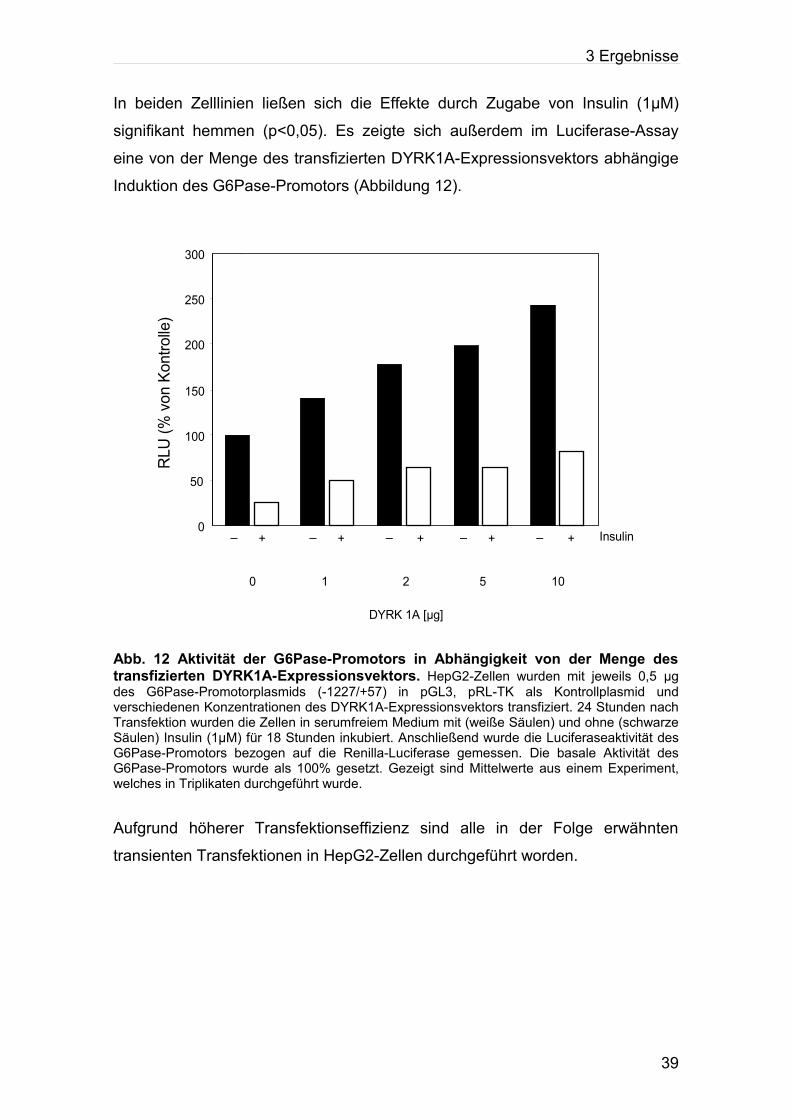
In beiden Zelllinien ließen sich die Effekte durch Zugabe von Insulin (1μM)
signifikant hemmen (p<0,05). Es zeigte sich außerdem im Luciferase-Assay
eine von der Menge des transfizierten DYRK1A-Expressionsvektors abhängige
Induktion des G6Pase-Promotors (Abbildung 12).
0
50
100
150
200
250
300
RLU
(% v
on K
ontro
lle)
0 1 2 5 10
DYRK 1A [µg]
Insulin+_+_+_+_+_
Abb. 12 Aktivität der G6Pase-Promotors in Abhängigkeit von der Menge des transfizierten DYRK1A-Expressionsvektors. HepG2-Zellen wurden mit jeweils 0,5 µg des G6Pase-Promotorplasmids (-1227/+57) in pGL3, pRL-TK als Kontrollplasmid und verschiedenen Konzentrationen des DYRK1A-Expressionsvektors transfiziert. 24 Stunden nach Transfektion wurden die Zellen in serumfreiem Medium mit (weiße Säulen) und ohne (schwarze Säulen) Insulin (1µM) für 18 Stunden inkubiert. Anschließend wurde die Luciferaseaktivität des G6Pase-Promotors bezogen auf die Renilla-Luciferase gemessen. Die basale Aktivität des G6Pase-Promotors wurde als 100% gesetzt. Gezeigt sind Mittelwerte aus einem Experiment, welches in Triplikaten durchgeführt wurde.
Aufgrund höherer Transfektionseffizienz sind alle in der Folge erwähnten
transienten Transfektionen in HepG2-Zellen durchgeführt worden.
39

3 Ergebnisse
3.2 Der Effekt von DYRK1A auf die G6Pase-Promotoraktivität in Abhängigkeit von der DYRK1A-Kinaseaktivität
3.2.1 Der Einfluss von FoxO1a-Ser329-Punktmutanten auf die durch DYRK1A vermittelte Expression des Glucose-6-Phosphatasegens
Es galt nun herausfinden, ob die bekannte Phosphorylierung von FoxO1a durch
DYRK1A am Serinrest 329 funktionell wichtig für den Effekt von DYRK1A auf
die Aktivität des G6Pase-Promotors ist. Aus diesem Grunde wurden nach dem
Prinzip der Sequenzspezifischen Mutagenese wie im Methodenteil beschrieben
FoxO1a-Punktmutanten hergestellt, bei denen der Serinrest an Position 329
entweder durch einen Alanin- oder durch einen Asparaginsäurerest ersetzt
wurde (Abbildung 13). Durch den Austausch mit Alanin ist FoxO1a an diesem
Rest nicht mehr phosphorylierbar. Da Asparaginsäure unter physiologischen
pH-Bedingungen negativ geladen ist, wird durch diesen Aminosäureaustausch
eine Dauerphosphorylierung simuliert. Im Restriktionsverdau konnten Wildtyp
und Mutanten anhand des unterschiedlichen Verdaus unterschieden werden
(Abbildung 14).
FoxO1 S329ApWZL Blasto
1000
2000800
3700
I S G R L A P I M TATC AGT GGG AGA CTT GCT CCC ATA ATG ACAATC AGT GGG AGA CTT TCT CCC ATC ATG ACA
I S G R L S P I M T
BspH1-Schnittstelle (in Mutante entfernt)
Abb. 13 Strukturausschnitt aus dem Promotor der FoxO1a-Punktmutante S329A. Im großen Kasten sind die mutierten Basen und die daraus resultierenden Aminosäuren rot dargestellt. Das kleine Kästchen innerhalb des großen Kastens stellt die veränderte Restriktionsschnittstelle dar. Im Fall der Alaninmutante ist eine BspH1-Schnittstelle entfernt worden, bei der Asparaginsäuremutante (nicht abgebildet) wurde eine zusätzliche BamH1-Schnittstelle eingefügt. Die veränderten Schnittstellen dienen zur Unterscheidung der Mutanten vom Wildtyp.
40

3 Ergebnisse
FoxO1a
WT
FoxO1a
S329A
60004000
2000
3000
1500
1000750
Abb. 14 Restriktionsanalyse der FoxO1a-Punktmutante S329A. Durch Restriktionsverdau mit BspH1 erhält man beim Wildtyp vier Fragmente (linke Spalte), während aufgrund der Deletion einer BspH1-Schnittstelle in der Mutante drei Fragmente entstehen (rechte Spalte). Somit können nach Klonierung der Wildtyp und die Mutante unterschieden werden.
Im Reportergen-Assay wurden dann die verschiedenen FoxO1a-Konstrukte mit
und ohne Co-Expression von DYRK1A auf ihren Einfluss auf die Aktivität des
G6Pase-Promotors hin untersucht. In gleicher Weise wie zuvor beschrieben
wurden HepG2-Zellen transient mit den verschiedenen FoxO1a-Konstrukten
transfiziert, im einer zweiten Versuchsreihe wurde zusätzlich DYRK1A co-
transfiziert. Die Aktivität des G6Pase-Promotors wurde jeweils anhand der
relativen Luciferaseaktivität gemessen. Alle drei Konstrukte zeigten eine
gesteigerte Promotoraktivität im Vergleich zur Kontrolle (p≤0,05), es fanden sich
allerdings keine signifikanten Unterschiede zwischen den einzelnen
Konstrukten. In den Experimenten mit co-transfiziertem DYRK1A erhält man
ähnliche Ergebnisse (im Fall der S329D-Mutante + DYRK1A statistisch nicht
signifikant (p=0,0795)), so dass davon auszugehen ist, dass eine
Phosphorylierung an der Position 329 keine funktionelle Bedeutung bei der
Regulation der Glucose-6-Phosphatase hat (Abbildung 15).
41
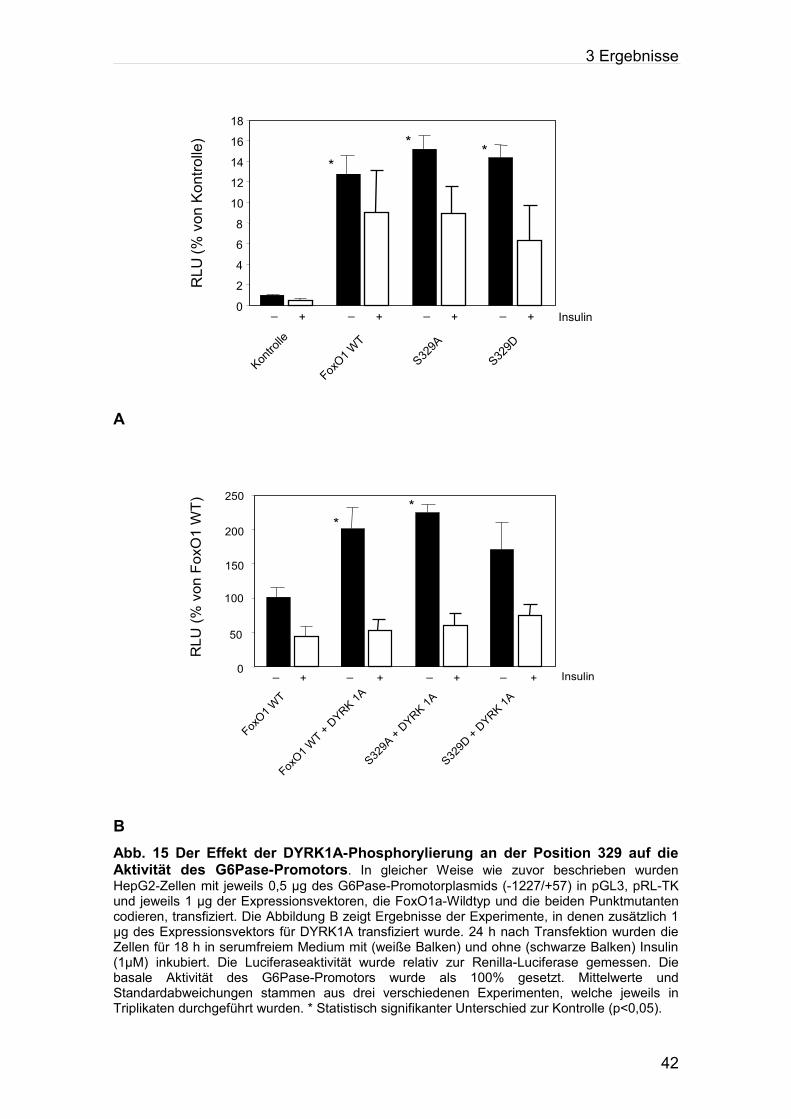
3 Ergebnisse
0
2
4
6
8
10
12
14
16
18
Kontro
lle
FoxO1 W
T
S329A
S329D
RLU
(% v
on K
ontro
lle)
+_+_+_+_ Insulin
** *
A
0
50
100
150
200
250
FoxO1 W
T
FoxO1 W
T + DYRK 1A
S329D
+ DYRK 1A
S329A
+ DYRK 1A
RLU
(% v
on F
oxO
1 W
T)
Insulin+_+_+_+_
**
BAbb. 15 Der Effekt der DYRK1A-Phosphorylierung an der Position 329 auf die Aktivität des G6Pase-Promotors. In gleicher Weise wie zuvor beschrieben wurden HepG2-Zellen mit jeweils 0,5 µg des G6Pase-Promotorplasmids (-1227/+57) in pGL3, pRL-TK und jeweils 1 µg der Expressionsvektoren, die FoxO1a-Wildtyp und die beiden Punktmutanten codieren, transfiziert. Die Abbildung B zeigt Ergebnisse der Experimente, in denen zusätzlich 1 µg des Expressionsvektors für DYRK1A transfiziert wurde. 24 h nach Transfektion wurden die Zellen für 18 h in serumfreiem Medium mit (weiße Balken) und ohne (schwarze Balken) Insulin (1µM) inkubiert. Die Luciferaseaktivität wurde relativ zur Renilla-Luciferase gemessen. Die basale Aktivität des G6Pase-Promotors wurde als 100% gesetzt. Mittelwerte und Standardabweichungen stammen aus drei verschiedenen Experimenten, welche jeweils in Triplikaten durchgeführt wurden. * Statistisch signifikanter Unterschied zur Kontrolle (p<0,05).
42

3 Ergebnisse
3.2.2 Der Effekt einer katalytisch inaktiven DYRK1A-Punktmutante auf den Glucose-6-Phosphatase-Promotor
In weiteren Experimenten wurde eine DYRK1A-Punktmutante (DYRK1A
Lys188Arg) (Kentrup et al., 1996; Himpel et al., 2001) untersucht, die keine
katalytische Aktivität besitzt. Ziel dieser Versuchsreihe war es, die
Unabhängigkeit der DYRK1A-Effekte auf den Promotor der G6Pase von der
Kinaseaktivität zu bestätigen. In der Mutante ist ein Lysinrest gegen einen
Argininrest ausgetauscht, was zu einer Inaktivität der katalytischen Einheit führt.
Diese Punktmutante wurde nach dem gleichen Prinzip wie die FoxO1a-
Punktmutanten hergestellt und uns freundlicherweise von Prof. Walter Becker
(Aachen) zur Verfügung gestellt.
Es wurden Reportergen-Assays durchgeführt, bei denen der Effekt dieser
Mutante mit dem Effekt von DYRK1A WT auf den Promotor des G6Pase-Gens
verglichen wurde. HepG2-Zellen wurden wie oben beschrieben transfiziert.
Auch bei diesen Versuchen zeigte sich kein signifikanter Unterschied in der
Aktivität des G6Pase-Promotors zwischen der Punktmutante K188R und
DYRK1A WT (p=0,4414) (Abbildung 16). Es ist deshalb davon auszugehen,
dass weder die Phosphorylierung von FoxO1a durch DYRK1A an der Position
Ser329 noch die katalytische Aktivität von DYRK1A eine Rolle bei der
Aktivierung des Promotors der G6Pase spielt.
43
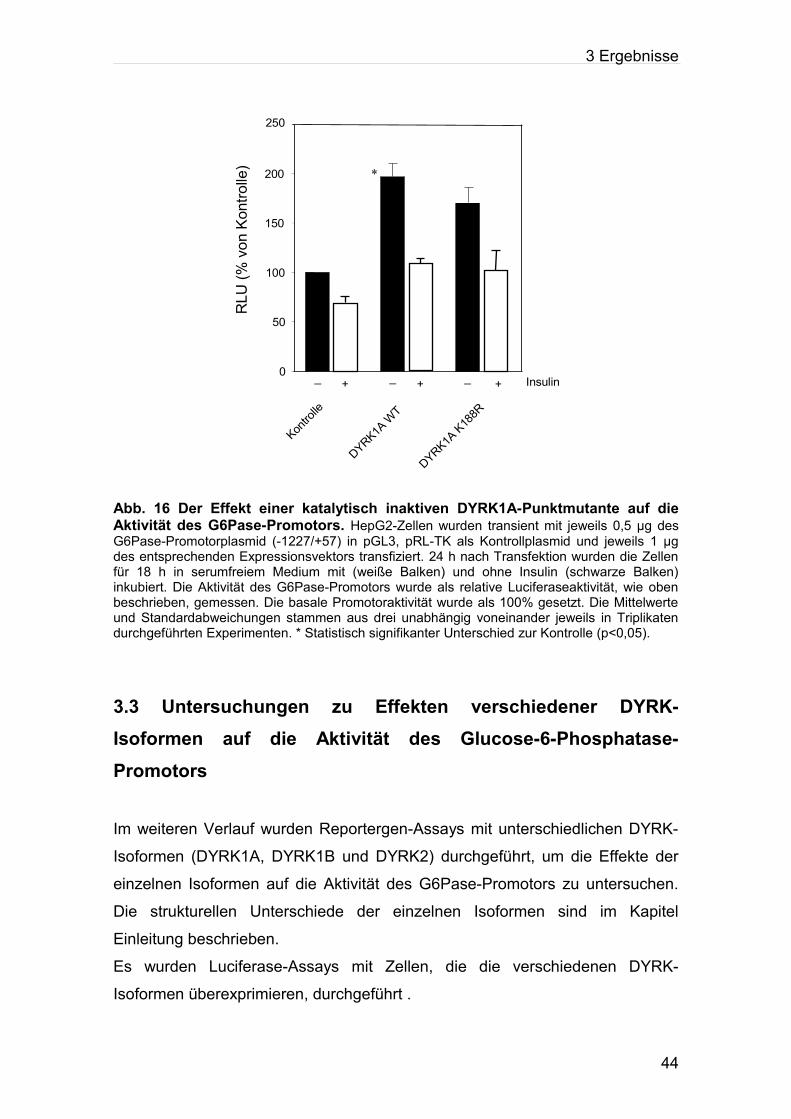
3 Ergebnisse
0
50
100
150
200
250
Kontro
lle
DYRK1A W
T
DYRK1A K18
8R
Insulin
RLU
(% v
on K
ontro
lle)
+ ++_ __
*
Abb. 16 Der Effekt einer katalytisch inaktiven DYRK1A-Punktmutante auf die Aktivität des G6Pase-Promotors. HepG2-Zellen wurden transient mit jeweils 0,5 µg des G6Pase-Promotorplasmid (-1227/+57) in pGL3, pRL-TK als Kontrollplasmid und jeweils 1 µg des entsprechenden Expressionsvektors transfiziert. 24 h nach Transfektion wurden die Zellen für 18 h in serumfreiem Medium mit (weiße Balken) und ohne Insulin (schwarze Balken) inkubiert. Die Aktivität des G6Pase-Promotors wurde als relative Luciferaseaktivität, wie oben beschrieben, gemessen. Die basale Promotoraktivität wurde als 100% gesetzt. Die Mittelwerte und Standardabweichungen stammen aus drei unabhängig voneinander jeweils in Triplikaten durchgeführten Experimenten. * Statistisch signifikanter Unterschied zur Kontrolle (p<0,05).
3.3 Untersuchungen zu Effekten verschiedener DYRK-Isoformen auf die Aktivität des Glucose-6-Phosphatase-Promotors
Im weiteren Verlauf wurden Reportergen-Assays mit unterschiedlichen DYRK-
Isoformen (DYRK1A, DYRK1B und DYRK2) durchgeführt, um die Effekte der
einzelnen Isoformen auf die Aktivität des G6Pase-Promotors zu untersuchen.
Die strukturellen Unterschiede der einzelnen Isoformen sind im Kapitel
Einleitung beschrieben.
Es wurden Luciferase-Assays mit Zellen, die die verschiedenen DYRK-
Isoformen überexprimieren, durchgeführt .
44

3 Ergebnisse
DYRK1A und DYRK1B zeigten den schon vorher beschriebenen signifikanten
Effekt auf die Aktivität des G6Pase-Promotors (p<0,05). Die Promotoraktivität
ist bei Überexpression von DYRK2 im Gegensatz dazu jedoch nicht signifikant
erhöht (p=0,0921) (Abbildung 17). Diese Ergebnisse lassen schlussfolgern,
dass die Struktur der DYRK-Isoform entscheidend für die beschriebenen
Effekte ist. Da DYRK2 keine Kernlokalisationssequenz (NLS) besitzt und damit
extranukleär lokalisiert ist, liegt die Vermutung nah, dass FoxO1a und DYRK
gemeinsam im Kern lokalisiert sein müssen, um die G6Pase-Promotoraktivität
zu beeinflussen.
0
50
100
150
200
250
300
350
Insulin
DYRK 1A
Kontro
lle
DYRK 1B
RLU
(% v
on K
ontro
lle)
+
DYRK 2
_ _ _ _ ++ +
**
Abb. 17 Der Effekt der verschiedenen DYRK-Isoformen auf die Aktivität des G6Pase-Promotors. HepG2-Zellen wurden transient mit jeweils 0,5 µg des G6Pase-Promotorplasmid (-1227/+57) in pGL3, pRL-TK als Kontrollplasmid und jeweils 1 µg des entsprechenden Expressionsvektors oder Kontrollvektors transfiziert. 24 h nach Transfektion wurden die Zellen für 18 h in serumfreiem Medium mit (weiße Balken) und ohne Insulin (1µM) (schwarze Balken) inkubiert. Die Luciferaseaktivität wurde relativ zur co-exprimierten Renilla-Luciferase gemessen. Die basale Promotoraktivität wurde als 100% gesetzt. Mittelwerte und Standardabweichungen stammen aus drei unabhängigen jeweils in Triplikaten durchgeführten Experimenten. * Statistisch signifikanter Unterschied zur Kontrolle (p<0,05).
3.4 Die Effekte unterschiedlicher DYRK-Isoformen auf die FoxO1a-induzierte Aktivität des Glucose-6-Phosphatase-Promotors
Die Mitglieder der DYRK-Familie unterscheiden sich hinsichtlich ihrer
Gewebeexpression und subzellulären Verteilung (Becker et al., 1998; Leder et
al., 1999). Das vorherige Experiment legt den Schluss nahe, dass die
45

3 Ergebnisse
Lokalisation von FoxO1a und DYRK1 entscheidend für den Effekt auf die
G6Pase-Promotoraktivität ist. Es wurden zu diesem Zweck Reportergen-
Assays mit verschiedenen FoxO1a- und DYRK1-Konstrukten durchgeführt. Wir
haben zum einen den Effekt von DYRK1A und DYRK1B allein, zum anderen
den Effekt mit co-exprimiertem Wildtyp-FoxO1a oder einer konstitutiv im Kern
lokalisierten FoxO1a-Mutante, hier FoxO1a MUT (Thr24Ala; Ser256Ala;
Ser319Ala) genannt, verglichen.
Der Effekt von DYRK1A ist sowohl basal als auch bei FoxO1a-induzierter
G6Pase-Promotoraktivität vergleichbar mit dem von DYRK1B, DYRK1 und
FoxO1a wirken dabei synergistisch. Wie erwartet, erhöht die Expression der im
Kern lokalisierten FoxO1a-Mutante (FoxO1a MUT) die Promotoraktivität des
G6Pase-Promotors. Dieser Effekt ist sowohl bei überexprimiertem DYRK1A und
DYRK1B als auch bei deren Abwesenheit zu sehen (Tabelle 1) Diese Resultate
geben weiteren Anhalt dafür, dass die Lokalisation von FoxO1a wichtig für
seine Funktion ist.
Expressionsvektor - Insulin + InsulinKontrolle 100 62 (+/-6)DYRK1A 187 (+/-13) 89 (+/-6)DYRK1B 210 (+/-16) 91 (+/-18)FoxO1a WT 9241 (+/-2589) 4182 (+/-1400)FoxO1a MUT 13108 (+/- 3097) 10351 (+/-3859)FoxO1a WT + DYRK1A 25919 (+/-6581) 9187 (+/-2601)FoxO1a MUT + DYRK1A 32229 (+/-10496) 30998 (+/-10632)FoxO1a WT + DYRK1B 29209 (+/-7391) 10277 (+/-2805)FoxO1a MUT + DYRK1B 60063 (+/-17983) 47805 (+/-13241)
Tab. 1 Der Effekt von DYRK1 auf den Aktivität des G6Pase-Promotors in Synergismus mit FoxO1a-WT und einer konstitutiv im Kern lokalisierten FoxO1a-Mutante. HepG2-Zellen wurden transient mit jeweils 0,5 µg des G6Pase-Promotorplasmid (-1227/+57) in pGL3, pRL-TK als Kontrollplasmid und jeweils 1 µg des entsprechenden DYRK1- bzw. FoxO1a-Expressionsvektors oder Kontrollvektors transfiziert. 24 h nach Transfektion wurden die Zellen für 18 h in serumfreiem Medium mit (rechte Spalte) und ohne Insulin (1µM) (linke Spalte) inkubiert. Die Luciferaseaktivität wurde relativ zur co-exprimierten Renilla-Luciferase gemessen. Die basale Promotoraktivität wurde als 100% gesetzt. Mittelwerte und Standardabweichungen stammen aus drei unabhängig voneinander durchgeführten Experimenten.
46

3 Ergebnisse
3.5 Der Effekt von DYRK1 auf den Glucose-6-Phosphatase-Promotor in Abhängigkeit von FoxO1a-Bindungsstellen (IRUs)
Weder DYRK1A noch DYRK1B besitzen eine DNA-Bindungsdomäne. Deshalb
wurde untersucht, ob der Effekt von DYRK1 auf den G6Pase-Promotor direkt
abläuft oder durch den Transkriptionsfaktor FoxO1a vermittelt wird. Es wurden
Experimente durchgeführt, bei denen ein mutierter Promotor des G6Pase-Gens
verwendet wurde, in welchem die die drei Insulinresponsiven Elemente deletiert
waren. Dieser Promotor besitzt Punktmutationen in allen drei FoxO1a-
Bindungsstellen und ist somit durch FoxO1a nicht mehr regulierbar (Abbildung
18).
CAGGCTGTTTTTGTGTGCCTGTTTTTCTATTTTACGTAA
CGAG C C
Konsensus IRU: T(G/A)TTT
G6Pase WTG6Pase IRU MUT
Abb. 18 Struktur der FoxO1a-bindenden Region des G6Pase-Promotors und Veränderungen der Promotor-Punktmutante. Mittels Sequenzspezifischer Mutagenese (siehe Material und Methoden) wurden die drei FoxO1a-Bindungsstellen (Kästen obere Zeile) (Konsensusmotiv der Insulinresponsiven Elemente T(G/A)TTT) verändert (untere Zeile).
Im Luciferase-Assay zeigte sich, dass die Fähigkeit von DYRK1, den G6Pase-
Promotor zu aktivieren, um ca. 70% niedriger ist, wenn die FoxO1a-
Bindungsstellen im Promotor mutiert sind. Dieser deutliche Unterschied zeigte
sich auch bei co-exprimiertem FoxO1a (Abbildung 19). DYRK1B steigert die
Aktivität des G6Pase-Promotors signifikant (p<0,05), wobei die Aktivität der
G6Pase-Promotor-Mutante nicht signifikant induziert wird (p=0,1555). Diese
Ergebnisse legen den Schluss nahe, dass der Effekt von DYRK1 auf den
Promotor des G6Pase-Gens ein FoxO1a-vermittelter Prozess ist.
47
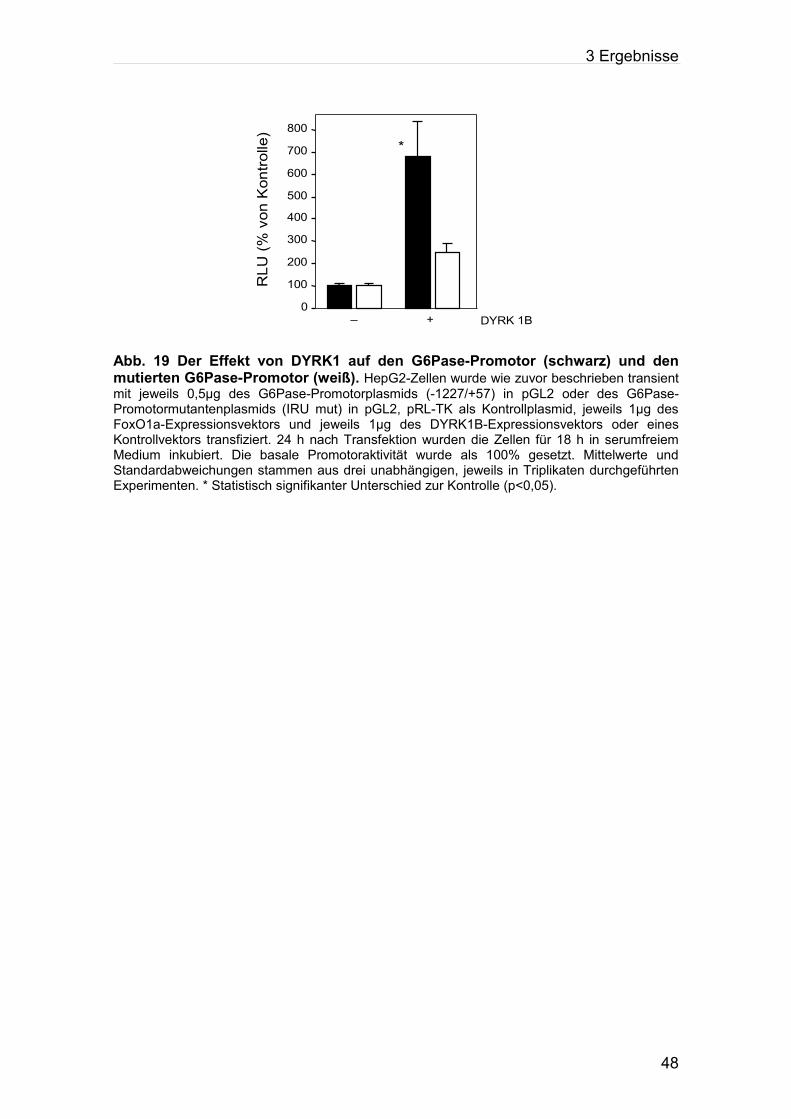
3 Ergebnisse
0
100
200
300
400
500
600
700
800
RLU
(% v
on K
ontro
lle)
DYRK 1B_ +
*
Abb. 19 Der Effekt von DYRK1 auf den G6Pase-Promotor (schwarz) und den mutierten G6Pase-Promotor (weiß). HepG2-Zellen wurde wie zuvor beschrieben transient mit jeweils 0,5µg des G6Pase-Promotorplasmids (-1227/+57) in pGL2 oder des G6Pase-Promotormutantenplasmids (IRU mut) in pGL2, pRL-TK als Kontrollplasmid, jeweils 1µg des FoxO1a-Expressionsvektors und jeweils 1µg des DYRK1B-Expressionsvektors oder eines Kontrollvektors transfiziert. 24 h nach Transfektion wurden die Zellen für 18 h in serumfreiem Medium inkubiert. Die basale Promotoraktivität wurde als 100% gesetzt. Mittelwerte und Standardabweichungen stammen aus drei unabhängigen, jeweils in Triplikaten durchgeführten Experimenten. * Statistisch signifikanter Unterschied zur Kontrolle (p<0,05).
48

4 Diskussion
4 Diskussion
Die Erforschung der Signaltransduktion des Insulin ist in Bezug auf die dem
Diabetes mellitus Typ 2 zugrunde liegende Insulinresistenz und die hepatische
Glucoseproduktion von zentralem Interesse und eine Voraussetzung für die
Entwicklung neuer antidiabetisch wirksamer Substanzen. Zur Behandlung des
Diabetes mellitus Typ 2 stehen derzeit diverse Medikamente zur Verfügung,
deren Wirkmechanismen sich voneinander unterscheiden. So führen die
Sulfonylharnstoffe und die so genannten „Glinide“ zu einer verstärkten
Sekretion von Insulin. Eine weitere Substanzklasse, die Thiazolidindione wirken
als Aktivatoren des Transkriptionsfaktors PPARγ und verbessern somit auf
bislang noch ungeklärte Weise die Signaltransduktion des Insulins. Ein Co-
Aktivator dieses Transkriptionsfaktors ist das Protein PPARγ Coaktivator-1
(PGC-1). PGC-1 spielt sowohl bei der Regulation der GLUT4-Genexpression
als auch bei der Regulation der hepatischen Glucoseproduktion eine
entscheidende Rolle (Michael et al., 2001; Vidal-Puig and O'Rahilly, 2001).
Andere Medikamente wie die Glucosidasehemmer vermindern die enterale
Resorption von Glucose.
In der UKPD-Studie hat sich die Substanz Metformin, die zu den Biguaniden
zählt, als günstigstes orales Antidiabetikum gerade für übergewichtige
Diabetiker herausgestellt (UK Prospective Diabetes Study (UKPDS) Group,
1998). Die relativ alte Substanz verstärkt die Aufnahme von Glucose in die
Muskulatur, führt aber auch zu einer Hemmung der hepatischen
Gluconeogenese. Der molekulare Mechanismus ist hierbei noch nicht endgültig
geklärt.
Eine Verminderung der hepatischen Glucoseproduktion durch Insulin geschieht
hauptsächlich durch Verringerung der Expression der G6Pase- und PEPCK-
Gene über die Aktivierung von Insulinrezeptorsubstraten. Dabei spielen IRS 1
und 2 offenbar bei der Regulation des Kohlenhydratstoffwechsels eine
bedeutende Rolle (White, 2002). Studien an knock-out-Modellen haben gezeigt,
dass Insulin die hepatische Glucoseproduktion hauptsächlich IRS-2-vermittelt
vermindert (Kubota et al., 2000; Withers et al., 1998). Es wird daher
49

4 Diskussion
angenommen, dass diese Isoform eine bedeutende Rolle bei der
Signaltransduktion zur Regulation der Gluconeogenese spielt. In der Folge wird
die PI3-Kinase/PKB-Signalkaskade aktiviert. Es wurde gezeigt, dass eine
Aktivierung der PI3-Kinase-Kaskade die Expression der Gene der PEPCK und
G6Pase entscheidend verringert. Studien mit einer dominant negativen Mutante
der regulatorischen Untereinheit der PI3-Kinase p85α hatten eine vermehrte
Expression der Gene und damit eine gesteigerte Glucoseproduktion zur Folge
(Miyake et al., 2002). Eine Hemmung der PI-3- Kinase durch Substanzen wie
z.B. Wortmannin verringert die insulinvermittelte Glucoseaufnahme (Cheatham
et al., 1994; Okada et al., 1994).
Eine Überexpression von PKB in Hepatomzellen und primären Hepatozyten hat
eine verminderte Transkription der PEPCK- und G6Pase-Gene zur Folge (Agati
et al., 1998; Liao et al., 1998; Schmoll et al., 2000). In Studien mit PKB-knock
out-Mäusen hat sich gezeigt, dass ein knock out der Isoform PKB β zu einer
Insulinresistenz und zu einem diabetischen Phänotyp führt, wobei ein knock out
der PKB α keine Stoffwechselveränderungen verursachte (Cho et al., 2001b;
Cho et al., 2001a). In einer Familie mit ausgeprägter Insulinresistenz ist eine
Mutation in der katalytischen Domäne der PKB β beschrieben worden, die eine
Phosphorylierung der PEPCK zur Folge hat und damit eine Hemmung der
hepatischen Glucoseproduktion unmöglich macht (Michael et al., 2001). Die
Aktivierung der PI3-Kinase/PKB-Kaskade ist von großer Bedeutung für die
Regulation der Glucose-6-Phosphatase. Als einer der Mediatoren dieses
Effektes wurde der Transkriptionsfaktor FoxO1a identifiziert.
Die Beteiligung von FoxO1a an Stoffwechselprozessen, insbesondere der
Regulation der hepatischen Glucoseproduktion, ist somit bereits bekannt. In
Studien mit einer heterozygoten FoxO1a-knock out-Maus wurde eine
Besserung der Insulinresistenz bei diabetischen Mäusen mit einer
Haploinsuffizienz des Insulinrezeptors beobachtet. Die Tiere hatten im
Vergleich zu den Kontrolltieren eine verbesserte Glucosetoleranz (Nakae et al.,
2002) Dies könnte mit einer verminderten Expression des G6Pase-Gens
zusammen hängen. In transgenen Mäusen mit einer konstitutiv aktiven FoxO1a
findet man eine gesteigerte Expression des G6Pase-Gens mit Insulinresistenz,
gesteigerter hepatischer Glucoseproduktion und konsekutiver Erhöhung der
50

4 Diskussion
Nüchternblutglucose (Nakae et al., 2002). In Studien mit einer dominant
negativen Form von FoxO1a zeigten die Tiere eine verminderte Expression der
G6Pase sowie eine verminderte hepatische Glucoseproduktion (Altomonte et
al., 2003). Diese Daten unterstützen die Bedeutung der FoxO Proteine bei der
Regulation des Glucosestoffwechsels.
Neben dem Transkriptionsfaktor STAT3 (Kortylewski et al., 2003) sowie dem
Östrogenrezeptor (Schuur et al., 2001; Zhao et al., 2001) konnte die
Proteinkinase DYRK1A als Interaktionspartner einer Protein-Protein-Interaktion
von FoxO1a identifiziert werden (Woods et al., 2001b). Darüber hinaus wurde
FoxO1a als direktes Substrat (Phosphorylierung an Serin 329) der DYRK-
Kinase beschrieben. In der vorliegenden Arbeit wurde der Einfluss der
Interaktion dieser beiden Proteine auf die Regulation der Glucose-6-
Phosphatase untersucht.
Ein Teil der Experimente wurde mit H4IIEC3-Zellen durchgeführt. Diese
Rattenhepatomzelle zeichnet sich durch eine sehr hohe Insulinempfindlichkeit
und eine erhaltene Regulation gluconeogener Gene durch Insulin aus. Deshalb
ist dieses Zellmodell sehr gut für die Untersuchung insulinabhängiger
Stoffwechselprozesse geeignet (Liao et al., 1998).
Es wurde gezeigt, dass DYRK1 einen stimulatorischen Effekt auf die FoxO1a-
abhängige Transaktivierung des G6Pase-Promotors hat. In Northern Blot-
Untersuchungen wurde gezeigt, dass nach stabiler Transfektion von DYRK1A
in Hepatomzellen der Gehalt an G6Pase-mRNA deutlich erhöht ist (Groote-
Bidlingmaier et al., 2003). In der vorliegenden Arbeit konnte mittels
Reportergenanalysen gezeigt werden, dass der Effekt von DYRK1A auf die
Menge des G6Pase-Transkriptes auf eine gesteigerte Aktivität des G6Pase-
Promotors zurückzuführen ist und damit auf transkriptioneller Ebene abläuft.
Studien haben gezeigt, dass der Transkriptionsfaktor FoxO1a ein
entscheidender Transaktivator des G6Pase-Gens ist (Schmoll et al., 2000;
Barthel et al., 2001) und die G6Pase-Gentranskription durch Bindung an die
IRUs im Promotor auslöst (Schmoll et al., 2000; Ayala et al., 1999). Die drei
IRUs im G6Pase-Promotor sind funktionell verschieden und binden FoxO1a in
unterschiedlichem Maße (Vander Kooi et al., 2003). In der vorliegenden Arbeit
wurde gezeigt, dass die Transaktivierung des G6Pase-Promotorgens deutlich
51

4 Diskussion
herabgesetzt wird, wenn die IRUs im Promotor Mutationen enthalten, die eine
Bindung von FoxO1a verhindern (Groote-Bidlingmaier et al., 2003). Damit
wurde gezeigt, dass die Transaktivierung des Promotors des G6Pase-Gens von
intakten FoxO1a-Bindungsstellen in den IRUs des Promotors abhängig ist.
Diese Daten lassen den Schluss zu, dass der Effekt von DYRK1A auf die
Expression des G6Pase-Gens indirekt über den Transkriptionsfaktor FoxO1a
vermittelt wird. Offenbar erfüllt DYRK1A die Funktion eines Co-Aktivators für
den Transkriptionsfaktor FoxO1a.
In Experimenten mit FoxO1a-Mutanten wurde in dieser Arbeit gezeigt, dass der
Effekt auf die Transaktivierung des G6Pase-Gens unabhängig von der
Phosphorylierung von FoxO1a durch DYRK1 ist. Experimente mit FoxO1a-
Punktmutanten, an denen der Serinrest 329 zu Alanin oder Asparaginsäure
mutiert ist, zeigen keine signifikante Änderung der G6Pase-Promotoraktivität. In
Versuchen mit einer DYRK-Mutante, die keine Kinaseaktivität mehr besitzt,
konnte ebenfalls kein signifikanter Unterschied bei der Transaktivierung des
G6Pase-Promotors festgestellt werden. Diese Ergebnisse legen den Schluss
nahe, dass der regulatorische Effekt von DYRK1A auf die transkriptionelle
Aktivität von FoxO1a unabhängig von der FoxO1a-Phosphorylierung durch
DYRK1A ist (Groote-Bidlingmaier et al., 2003). Der untersuchte Effekt ist somit
wahrscheinlich Ausdruck weiterer Mechanismen, bzw. Protein-Protein-
Interaktionen. Über Art und Weise kann hier jedoch nur spekuliert werden. In
Frage kommen Prozesse wie de novo-Proteinsynthese oder
Proteinstabilisierung. Für letzteres spricht die PEST-Domäne in DYRK1, eine
Struktur, die an Prozessen der Proteinstabilisierung beteiligt zu sein scheint
(Kentrup et al., 1996). PEST-Domänen sind beispielsweise an der Regulation
von IκBα über Phosphorylierung durch die Casein Kinase II beteiligt (Lin et al.,
1996). Die hier durchgeführten Experimente deuten jedoch darauf hin, dass die
gemeinsame Lokalisation von FoxO1a und DYRK1A im Zellkern für die
Interaktion von entscheidender Bedeutung ist. Es ist bekannt, dass das
Homolog von DYRK aus Hefe, Yak1, im Zustand des Glucosemangels in den
Zellkern transportiert wird (Moriya et al., 2001). Denkbar ist, dass DYRK1
ähnlich reguliert wird. Der Einfluss von FoxO1a auf die Aktivität der G6Pase
und damit auf die hepatische Glucoseproduktion wird ebenfalls unter anderem
durch den Lokalisationszustand zwischen Zellkern und Zytosol reguliert. Eine
52

4 Diskussion
Phosphorylierung von FoxO1a durch die Proteinkinase B und eine Interaktion
mit 14-3-3-Proteinen führen zu einer transkriptionellen Inaktivität und zur
nukleären Exklusion von FoxO1a (Brunet et al., 1999; Brunet et al., 2001;
Nakae et al., 1999; Kops et al., 1999; Guo et al., 1999; Rena et al., 2002; Biggs,
III et al., 1999). In dieser Arbeit wurde gezeigt, dass DYRK1, aber nicht die
Isoform DYRK2 die G6Pase-Promotoraktivität induzieren kann (Groote-
Bidlingmaier et al., 2003). Dies könnte auf die fehlende
Kernlokalisationssequenz in DYRK2 zurück zu führen sein, welche für die im
wesentlichen zytoplasmatische Lokalisation von DYRK2 verantwortlich ist
(Becker et al., 1998). Dies unterstützt die Vermutung, dass die Lokalisation von
DYRK1 im Zellkern entscheidend für die Transkription des G6Pase-Gens ist.
Diese These wird zusätzlich durch die Tatsache gefestigt, dass eine konstitutiv
im Kern lokalisierte FoxO1a-Mutante zu einer zusätzlichen Steigerung des
Effektes von DYRK1 auf die G6Pase-Promotor-Aktivität führt (Groote-
Bidlingmaier et al., 2003). Die fehlende Induzierbarkeit des G6Pase-
Promotorgens durch DYRK2 ist ein wichtiger Hinweis auf die Spezifität des
Effekts durch DYRK1.
FoxO-Transkriptionsfaktoren gehen mit diversen Mediatoren Protein-Protein-
Interaktionen ein, so werden z.B. die Effekte von CBP, p300, SRC1 und
PGC-1α über FoxO-Bindungsstellen vermittelt (Mahmud et al., 2002; Nasrin et
al., 2000; Puigserver et al., 2003). PGC-1α interagiert beispielsweise mit
Abschnitten der DNA-Bindungsdomäne und ist zusammen mit FoxO1a an der
Regulation der Glucose-6-Phosphatase beteiligt (Herzig et al., 2001). Durch
Phosphorylierung von FoxO1a wird diese Interaktion unterbrochen. FoxO-
Proteine können aber auch ohne FoxO-Bindungsstellen an der Regulation von
Promotoren teilnehmen, in dem sie an andere Transkriptionsfaktoren wie z.B.
STAT3 binden (Dowell et al., 2003; Schuur et al., 2001; Zhao et al., 2001; Li et
al., 2003; Kortylewski et al., 2003; Hirota et al., 2003; Seoane et al., 2004;
Nakae et al., 2003). In ähnlicher Form könnten DYRK1A und DYRK1B als
Zusatzfaktoren die FoxO1a-abhängige Transkription beeinflussen. Die
erhobenen Daten zeigen, dass DYRK1 neben der beschriebenen
Phosphorylierung auch weitere funktionelle Interaktionen mit FoxO-Proteinen
eingeht und somit als Coaktivator von FoxO1a einen Einfluss auf die Regulation
der hepatischen Glucoseproduktion hat.
53

4 Diskussion
Für ein genaueres Verständnis der Interaktion zwischen DYRK1 und FoxO1a
auf molekularer Ebene sind weitere Untersuchungen erforderlich. Aufgrund der
hier vorgestellten Ergebnisse ist es aber durchaus denkbar, dass durch eine
gezielte Hemmung der Interaktion zwischen DYRK1 und FoxO1a die
hepatische Glucoseproduktion über eine Hemmung der G6Pase-
Promotoraktivierung vermindert werden kann. Dies könnte zu einer
Verbesserung der diabetischen Stoffwechsellage bei Patienten mit Typ-2-
Diabetes führen. Die in dieser Arbeit beschriebenen Daten sind somit von
potenziellem Interesse für die Entwicklung neuer therapeutischer Strategien in
der Behandlung des Typ-2-Diabetes.
54

5 Zusammenfassung
5 Zusammenfassung
In der vorgelegten Arbeit wurde der funktionelle Zusammenhang zwischen der
Kinase DYRK1 und dem Transkriptionsfaktor FoxO1a detailliert charakterisiert.
Es wurde untersucht, welche Rolle DYRK1 bei der insulinabhängigen
Regulation der hepatischen Glucoseproduktion spielt. Dies geschah anhand der
Glucose-6-Phosphatase, eines insulinabhängig regulierten Enzyms der
Gluconeogenese.
Es wurde durch mehrere unabhängige experimentelle Ansätze gezeigt, dass
die Proteinkinase DYRK1A einen stimulatorischen Effekt auf die Aktivität des
Glucose-6-Phosphatase-Promotors im Leberzellmodell ausübt. In Northern-
Blot-Untersuchungen fand sich in stabil mit DYRK1A transfizierten
Hepatomzellen eine erhöhte Menge an endogener G6Pase-mRNA im Vergleich
zu Kontrollzellen. In Luciferase-Reportergen-Assays wurde ein synergistischer
Effekt von DYRK1A mit dem Transkriptionsfaktor FoxO1a gefunden. Darüber
hinaus wurde ausgeschlossen, dass eine zuvor beschriebene Phosphorylierung
von FoxO1a durch DYRK1 (Ser329) für den Effekt von DYRK1A auf den
G6Pase-Promotor verantwortlich ist. In weiteren Experimenten wurde eine
Abhängigkeit des Effekts von DYRK1 auf die Aktivität des G6Pase-Promotors
von intakten FoxO1a-Bindungsstellen im G6Pase-Promotor nachgewiesen.
Dies legt den Schluss nahe, dass der Effekt von DYRK1 auf den G6Pase-
Promotor nicht direkt, sondern über FoxO1a vermittelt wird. Weiterhin zeigten
verschiedene DYRK-Isoformen unterschiedliche Effekte auf die G6Pase-
Promotoraktivität. Dies ist wahrscheinlich auf die unterschiedliche subzelluläre
Lokalisation der einzelnen DYRK-Isoformen zurückzuführen.
Diese Arbeit liefert somit den ersten Bericht der funktionellen Bedeutung einer
Interaktion zwischen DYRK1A und DYRK1B mit einem Transkriptionsfaktor.
Außerdem wird der Einfluss auf die Expression eines bestimmten Gens, der
Glucose-6-Phosphatase, beschrieben.
55

6 Literaturverzeichnis
6 Literaturverzeichnis
Agati,J.M., Yeagley,D., and Quinn,P.G. (1998). Assessment of the roles of mitogen-activated protein kinase, phosphatidylinositol 3-kinase, protein kinase B, and protein kinase C in insulin inhibition of cAMP-induced phosphoenolpyruvate carboxykinase gene transcription. J. Biol. Chem. 273, 18751-18759.
Alessi,D.R. and Cohen,P. (1998). Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev. 8, 55-62.
Altafaj,X., Dierssen,M., Baamonde,C., Marti,E., Visa,J., Guimera,J., Oset,M., Gonzalez,J.R., Florez,J., Fillat,C., and Estivill,X. (2001). Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum. Mol. Genet. 10, 1915-1923.
Altomonte,J., Richter,A., Harbaran,S., Suriawinata,J., Nakae,J., Thung,S.N., Meseck,M., Accili,D., and Dong,H. (2003). Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am. J. Physiol Endocrinol. Metab 285, E718-E728.
Alvarez,M., Altafaj,X., Aranda,S., and de la,L.S. (2007). DYRK1A autophosphorylation on serine residue 520 modulates its kinase activity via 14-3-3 binding. Mol. Biol. Cell 18, 1167-1178.
Argaud,D., Zhang,Q., Pan,W., Maitra,S., Pilkis,S.J., and Lange,A.J. (1996). Regulation of rat liver glucose-6-phosphatase gene expression in different nutritional and hormonal states: gene structure and 5'-flanking sequence. Diabetes 45, 1563-1571.
Arion,W.J. and Nordlie,R.C. (1965). Liver glucose-6-phosphatase and pyrophosphate-glucose phosphotransferase: effects of fasting. Biochem. Biophys. Res. Commun. 20, 606-610.
Ayala,J.E., Streeper,R.S., Desgrosellier,J.S., Durham,S.K., Suwanichkul,A., Svitek,C.A., Goldman,J.K., Barr,F.G., Powell,D.R., and O'Brien,R.M. (1999). Conservation of an insulin response unit between mouse and human glucose-6-phosphatase catalytic subunit gene promoters: transcription factor FKHR binds the insulin response sequence. Diabetes 48, 1885-1889.
Barthel,A., Schmoll,D., Kruger,K.D., Bahrenberg,G., Walther,R., Roth,R.A., and Joost,H.G. (2001). Differential regulation of endogenous glucose-6-phosphatase and phosphoenolpyruvate carboxykinase gene expression by the forkhead transcription factor FKHR in H4IIE-hepatoma cells. Biochem. Biophys. Res. Commun. 285, 897-902.
Baumann,C.A., Ribon,V., Kanzaki,M., Thurmond,D.C., Mora,S., Shigematsu,S., Bickel,P.E., Pessin,J.E., and Saltiel,A.R. (2000). CAP defines a second
56

6 Literaturverzeichnis
signalling pathway required for insulin-stimulated glucose transport. Nature 407, 202-207.
Becker,W. and Joost,H.G. (1999). Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid Res. Mol. Biol. 62, 1-17.
Becker,W., Weber,Y., Wetzel,K., Eirmbter,K., Tejedor,F.J., and Joost,H.G. (1998). Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J. Biol. Chem. 273, 25893-25902.
Belham,C., Wu,S., and Avruch,J. (1999). Intracellular signalling: PDK1--a kinase at the hub of things. Curr. Biol. 9, R93-R96.
Biggs,W.H., III, Meisenhelder,J., Hunter,T., Cavenee,W.K., and Arden,K.C. (1999). Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. U. S. A 96, 7421-7426.
Brunet,A., Bonni,A., Zigmond,M.J., Lin,M.Z., Juo,P., Hu,L.S., Anderson,M.J., Arden,K.C., Blenis,J., and Greenberg,M.E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857-868.
Brunet,A., Park,J., Tran,H., Hu,L.S., Hemmings,B.A., and Greenberg,M.E. (2001). Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell Biol. 21, 952-965.
Brunet,A., Sweeney,L.B., Sturgill,J.F., Chua,K.F., Greer,P.L., Lin,Y., Tran,H., Ross,S.E., Mostoslavsky,R., Cohen,H.Y., Hu,L.S., Cheng,H.L., Jedrychowski,M.P., Gygi,S.P., Sinclair,D.A., Alt,F.W., and Greenberg,M.E. (2004). Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011-2015.
Chatelain,F., Pegorier,J.P., Minassian,C., Bruni,N., Tarpin,S., Girard,J., and Mithieux,G. (1998). Development and regulation of glucose-6-phosphatase gene expression in rat liver, intestine, and kidney: in vivo and in vitro studies in cultured fetal hepatocytes. Diabetes 47, 882-889.
Cheatham,B., Vlahos,C.J., Cheatham,L., Wang,L., Blenis,J., and Kahn,C.R. (1994). Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol. Cell Biol. 14, 4902-4911.
Chen-Hwang,M.C., Chen,H.R., Elzinga,M., and Hwang,Y.W. (2002). Dynamin is a minibrain kinase/dual specificity Yak1-related kinase 1A substrate. J. Biol. Chem. 277, 17597-17604.
Chiang,S.H., Baumann,C.A., Kanzaki,M., Thurmond,D.C., Watson,R.T., Neudauer,C.L., Macara,I.G., Pessin,J.E., and Saltiel,A.R. (2001). Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 410, 944-948.
57

6 Literaturverzeichnis
Cho,H., Mu,J., Kim,J.K., Thorvaldsen,J.L., Chu,Q., Crenshaw,E.B., III, Kaestner,K.H., Bartolomei,M.S., Shulman,G.I., and Birnbaum,M.J. (2001a). Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292, 1728-1731.
Cho,H., Thorvaldsen,J.L., Chu,Q., Feng,F., and Birnbaum,M.J. (2001b). Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 276, 38349-38352.
De Graaf,K., Czajkowska,H., Rottmann,S., Packman,L.C., Lilischkis,R., Luscher,B., and Becker,W. (2006). The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC. Biochem. 7, 7.
De Graaf,K., Hekerman,P., Spelten,O., Herrmann,A., Packman,L.C., Bussow,K., Muller-Newen,G., and Becker,W. (2004). Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: phosphorylation by DYRK1A and colocalization with splicing factors. J. Biol. Chem. 279, 4612-4624.
Dowell,P., Otto,T.C., Adi,S., and Lane,M.D. (2003). Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J. Biol. Chem. 278, 45485-45491.
Durham,S.K., Suwanichkul,A., Scheimann,A.O., Yee,D., Jackson,J.G., Barr,F.G., and Powell,D.R. (1999). FKHR binds the insulin response element in the insulin-like growth factor binding protein-1 promoter. Endocrinology 140, 3140-3146.
Essers,M.A., Weijzen,S., Vries-Smits,A.M., Saarloos,I., de Ruiter,N.D., Bos,J.L., and Burgering,B.M. (2004). FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 23, 4802-4812.
Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (1997). Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 20, 1183-1197.
Foster,J.D., Pederson,B.A., and Nordlie,R.C. (1997). Glucose-6-phosphatase structure, regulation, and function: an update. Proc. Soc. Exp. Biol. Med. 215, 314-332.
Garland,R.C. (1986). Induction of glucose 6-phosphatase in cultured hepatoma cells by dexamethasone. Biochem. Biophys. Res. Commun. 139, 1130-1134.
Gilley,J., Coffer,P.J., and Ham,J. (2003). FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162, 613-622.
Groote-Bidlingmaier,F., Schmoll,D., Orth,H.M., Joost,H.G., Becker,W., and Barthel,A. (2003). DYRK1 is a co-activator of FKHR (FOXO1a)-dependent glucose-6- phosphatase gene expression. Biochem. Biophys. Res. Commun. 300, 764-769.
58

6 Literaturverzeichnis
Guo,S., Rena,G., Cichy,S., He,X., Cohen,P., and Unterman,T. (1999). Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274, 17184-17192.
Gwack,Y., Sharma,S., Nardone,J., Tanasa,B., Iuga,A., Srikanth,S., Okamura,H., Bolton,D., Feske,S., Hogan,P.G., and Rao,A. (2006). A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 441, 646-650.
Herzig,S., Long,F., Jhala,U.S., Hedrick,S., Quinn,R., Bauer,A., Rudolph,D., Schutz,G., Yoon,C., Puigserver,P., Spiegelman,B., and Montminy,M. (2001). CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179-183.
Himpel,S., Panzer,P., Eirmbter,K., Czajkowska,H., Sayed,M., Packman,L.C., Blundell,T., Kentrup,H., Grotzinger,J., Joost,H.G., and Becker,W. (2001). Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J. 359, 497-505.
Hirota,K., Daitoku,H., Matsuzaki,H., Araya,N., Yamagata,K., Asada,S., Sugaya,T., and Fukamizu,A. (2003). Hepatocyte nuclear factor-4 is a novel downstream target of insulin via FKHR as a signal-regulated transcriptional inhibitor. J. Biol. Chem. 278, 13056-13060.
Hornbuckle,L.A., Edgerton,D.S., Ayala,J.E., Svitek,C.A., Oeser,J.K., Neal,D.W., Cardin,S., Cherrington,A.D., and O'Brien,R.M. (2001). Selective tonic inhibition of G-6-Pase catalytic subunit, but not G-6-P transporter, gene expression by insulin in vivo. Am. J. Physiol Endocrinol. Metab 281, E713-E725.
Hwangbo,D.S., Gersham,B., Tu,M.P., Palmer,M., and Tatar,M. (2004). Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429, 562-566.
Kaestner,K.H., Knochel,W., and Martinez,D.E. (2000). Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 14, 142-146.
Kahn,S.E. (2003). The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46, 3-19.
Kaufmann,E. and Knochel,W. (1996). Five years on the wings of fork head. Mech. Dev. 57, 3-20.
Kelly,P.A. and Rahmani,Z. (2005). DYRK1A enhances the mitogen-activated protein kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol. Biol. Cell 16, 3562-3573.
Kentrup,H., Becker,W., Heukelbach,J., Wilmes,A., Schurmann,A., Huppertz,C., Kainulainen,H., and Joost,H.G. (1996). Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J. Biol. Chem. 271, 3488-3495.
59

6 Literaturverzeichnis
Kimura,R., Kamino,K., Yamamoto,M., Nuripa,A., Kida,T., Kazui,H., Hashimoto,R., Tanaka,T., Kudo,T., Yamagata,H., Tabara,Y., Miki,T., Akatsu,H., Kosaka,K., Funakoshi,E., Nishitomi,K., Sakaguchi,G., Kato,A., Hattori,H., Uema,T., and Takeda,M. (2007). The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 16, 15-23.
Klein,B.E., Klein,R., Moss,S.E., and Cruickshanks,K.J. (1996). Parental history of diabetes in a population-based study. Diabetes Care 19, 827-830.
Kops,G.J., de Ruiter,N.D., Vries-Smits,A.M., Powell,D.R., Bos,J.L., and Burgering,B.M. (1999). Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398, 630-634.
Kortylewski,M., Feld,F., Kruger,K.D., Bahrenberg,G., Roth,R.A., Joost,H.G., Heinrich,P.C., Behrmann,I., and Barthel,A. (2003). Akt modulates STAT3-mediated gene expression through a FKHR (FOXO1a)-dependent mechanism. J. Biol. Chem. 278, 5242-5249.
Kubota,N., Tobe,K., Terauchi,Y., Eto,K., Yamauchi,T., Suzuki,R., Tsubamoto,Y., Komeda,K., Nakano,R., Miki,H., Satoh,S., Sekihara,H., Sciacchitano,S., Lesniak,M., Aizawa,S., Nagai,R., Kimura,S., Akanuma,Y., Taylor,S.I., and Kadowaki,T. (2000). Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 49, 1880-1889.
Lange,A.J., Argaud,D., el Maghrabi,M.R., Pan,W., Maitra,S.R., and Pilkis,S.J. (1994). Isolation of a cDNA for the catalytic subunit of rat liver glucose-6-phosphatase: regulation of gene expression in FAO hepatoma cells by insulin, dexamethasone and cAMP. Biochem. Biophys. Res. Commun. 201, 302-309.
Leder,S., Weber,Y., Altafaj,X., Estivill,X., Joost,H.G., and Becker,W. (1999). Cloning and characterization of DYRK1B, a novel member of the DYRK family of protein kinases. Biochem. Biophys. Res. Commun. 254, 474-479.
Lehtinen,M.K., Yuan,Z., Boag,P.R., Yang,Y., Villen,J., Becker,E.B., DiBacco,S., de,l., I, Gygi,S., Blackwell,T.K., and Bonni,A. (2006). A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125, 987-1001.
Li,P., Lee,H., Guo,S., Unterman,T.G., Jenster,G., and Bai,W. (2003). AKT-independent protection of prostate cancer cells from apoptosis mediated through complex formation between the androgen receptor and FKHR. Mol. Cell Biol. 23, 104-118.
Liao,J., Barthel,A., Nakatani,K., and Roth,R.A. (1998). Activation of protein kinase B/Akt is sufficient to repress the glucocorticoid and cAMP induction of phosphoenolpyruvate carboxykinase gene. J. Biol. Chem. 273, 27320-27324.
Lim,S., Jin,K., and Friedman,E. (2002). Mirk protein kinase is activated by MKK3 and functions as a transcriptional activator of HNF1alpha. J. Biol. Chem. 277, 25040-25046.
60

6 Literaturverzeichnis
Lin,K., Dorman,J.B., Rodan,A., and Kenyon,C. (1997). daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319-1322.
Lin,R., Beauparlant,P., Makris,C., Meloche,S., and Hiscott,J. (1996). Phosphorylation of IkappaBalpha in the C-terminal PEST domain by casein kinase II affects intrinsic protein stability. Mol. Cell Biol. 16, 1401-1409.
Liu,Z., Barrett,E.J., Dalkin,A.C., Zwart,A.D., and Chou,J.Y. (1994). Effect of acute diabetes on rat hepatic glucose-6-phosphatase activity and its messenger RNA level. Biochem. Biophys. Res. Commun. 205, 680-686.
Lizcano,J.M. and Alessi,D.R. (2002). The insulin signalling pathway. Curr. Biol. 12, R236-R238.
Lock,L.S., Royal,I., Naujokas,M.A., and Park,M. (2000). Identification of an atypical Grb2 carboxyl-terminal SH3 domain binding site in Gab docking proteins reveals Grb2-dependent and -independent recruitment of Gab1 to receptor tyrosine kinases. J. Biol. Chem. 275, 31536-31545.
Mahmud,D.L., Amlak,M., Deb,D.K., Platanias,L.C., Uddin,S., and Wickrema,A. (2002). Phosphorylation of forkhead transcription factors by erythropoietin and stem cell factor prevents acetylation and their interaction with coactivator p300 in erythroid progenitor cells. Oncogene 21, 1556-1562.
Mao,J., Maye,P., Kogerman,P., Tejedor,F.J., Toftgard,R., Xie,W., Wu,G., and Wu,D. (2002). Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 277, 35156-35161.
Massillon,D., Barzilai,N., Chen,W., Hu,M., and Rossetti,L. (1996). Glucose regulates in vivo glucose-6-phosphatase gene expression in the liver of diabetic rats. J. Biol. Chem. 271, 9871-9874.
Matsumoto,M. and Accili,D. (2005). All roads lead to FoxO. Cell Metab 1, 215-216.
Matsuo,R., Ochiai,W., Nakashima,K., and Taga,T. (2001). A new expression cloning strategy for isolation of substrate-specific kinases by using phosphorylation site-specific antibody. J. Immunol. Methods 247, 141-151.
Michael,L.F., Wu,Z., Cheatham,R.B., Puigserver,P., Adelmant,G., Lehman,J.J., Kelly,D.P., and Spiegelman,B.M. (2001). Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl. Acad. Sci. U. S. A 98, 3820-3825.
Minassian,C., Zitoun,C., and Mithieux,G. (1996). Differential time course of liver and kidney glucose-6 phosphatase activity during long-term fasting in rat correlates with differential time course of messenger RNA level. Mol. Cell Biochem. 155, 37-41.
Mithieux,G. (1997). New knowledge regarding glucose-6 phosphatase gene and protein and their roles in the regulation of glucose metabolism. Eur. J. Endocrinol. 136, 137-145.
61

6 Literaturverzeichnis
Miyake,K., Ogawa,W., Matsumoto,M., Nakamura,T., Sakaue,H., and Kasuga,M. (2002). Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3-kinase signaling in the liver. J. Clin. Invest 110, 1483-1491.
Moriya,H., Shimizu-Yoshida,Y., Omori,A., Iwashita,S., Katoh,M., and Sakai,A. (2001). Yak1p, a DYRK family kinase, translocates to the nucleus and phosphorylates yeast Pop2p in response to a glucose signal. Genes Dev. 15, 1217-1228.
Motta,M.C., Divecha,N., Lemieux,M., Kamel,C., Chen,D., Gu,W., Bultsma,Y., McBurney,M., and Guarente,L. (2004). Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551-563.
Nakae,J., Biggs,W.H., III, Kitamura,T., Cavenee,W.K., Wright,C.V., Arden,K.C., and Accili,D. (2002). Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 32, 245-253.
Nakae,J., Kitamura,T., Kitamura,Y., Biggs,W.H., III, Arden,K.C., and Accili,D. (2003). The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 4, 119-129.
Nakae,J., Kitamura,T., Silver,D.L., and Accili,D. (2001). The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest 108, 1359-1367.
Nakae,J., Park,B.C., and Accili,D. (1999). Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J. Biol. Chem. 274, 15982-15985.
Nasrin,N., Ogg,S., Cahill,C.M., Biggs,W., Nui,S., Dore,J., Calvo,D., Shi,Y., Ruvkun,G., and Alexander-Bridges,M.C. (2000). DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells. Proc. Natl. Acad. Sci. U. S. A 97, 10412-10417.
Noguchi,T., Matozaki,T., Inagaki,K., Tsuda,M., Fukunaga,K., Kitamura,Y., Kitamura,T., Shii,K., Yamanashi,Y., and Kasuga,M. (1999). Tyrosine phosphorylation of p62(Dok) induced by cell adhesion and insulin: possible role in cell migration. EMBO J. 18, 1748-1760.
O'Brien,R.M., Lucas,P.C., Forest,C.D., Magnuson,M.A., and Granner,D.K. (1990). Identification of a sequence in the PEPCK gene that mediates a negative effect of insulin on transcription. Science 249, 533-537.
O'Brien,R.M., Streeper,R.S., Ayala,J.E., Stadelmaier,B.T., and Hornbuckle,L.A. (2001). Insulin-regulated gene expression. Biochem. Soc. Trans. 29, 552-558.
Ogg,S., Paradis,S., Gottlieb,S., Patterson,G.I., Lee,L., Tissenbaum,H.A., and Ruvkun,G. (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994-999.
62

6 Literaturverzeichnis
Okada,T., Kawano,Y., Sakakibara,T., Hazeki,O., and Ui,M. (1994). Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J. Biol. Chem. 269, 3568-3573.
Puigserver,P., Rhee,J., Donovan,J., Walkey,C.J., Yoon,J.C., Oriente,F., Kitamura,Y., Altomonte,J., Dong,H., Accili,D., and Spiegelman,B.M. (2003). Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 423, 550-555.
Reaven,G.M. (1988). Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37, 1595-1607.
Rena,G., Guo,S., Cichy,S.C., Unterman,T.G., and Cohen,P. (1999). Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 274, 17179-17183.
Rena,G., Woods,Y.L., Prescott,A.R., Peggie,M., Unterman,T.G., Williams,M.R., and Cohen,P. (2002). Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 21, 2263-2271.
Saltiel,A.R. and Pessin,J.E. (2002). Insulin signaling pathways in time and space. Trends Cell Biol. 12, 65-71.
Sanger,F., Nicklen,S., and Coulson,A.R. (1992). DNA sequencing with chain-terminating inhibitors. 1977. Biotechnology 24, 104-108.
Schmidt,M., Fernandez,d.M., van der,H.A., Klompmaker,R., Kops,G.J., Lam,E.W., Burgering,B.M., and Medema,R.H. (2002). Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell Biol. 22, 7842-7852.
Schmoll,D., Allan,B.B., and Burchell,A. (1996). Cloning and sequencing of the 5' region of the human glucose-6- phosphatase gene: transcriptional regulation by cAMP, insulin and glucocorticoids in H4IIE hepatoma cells. FEBS Lett. 383, 63-66.
Schmoll,D., Walker,K.S., Alessi,D.R., Grempler,R., Burchell,A., Guo,S., Walther,R., and Unterman,T.G. (2000). Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J. Biol. Chem. 275, 36324-36333.
Schuur,E.R., Loktev,A.V., Sharma,M., Sun,Z., Roth,R.A., and Weigel,R.J. (2001). Ligand-dependent interaction of estrogen receptor-alpha with members of the forkhead transcription factor family. J. Biol. Chem. 276, 33554-33560.
Segal,H.L. and Washko,M.E. (1959). Studies of liver glucose 6-phosphatase. III. Solubilization and properties of the enzyme from normal and diabetic rats. J. Biol. Chem. 234, 1937-1941.
63

6 Literaturverzeichnis
Seoane,J., Le,H.V., Shen,L., Anderson,S.A., and Massague,J. (2004). Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211-223.
Taira,N., Nihira,K., Yamaguchi,T., Miki,Y., and Yoshida,K. (2007). DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 25, 725-738.
Takahashi-Yanaga,F., Mori,J., Matsuzaki,E., Watanabe,Y., Hirata,M., Miwa,Y., Morimoto,S., and Sasaguri,T. (2006). Involvement of GSK-3beta and DYRK1B in differentiation-inducing factor-3-induced phosphorylation of cyclin D1 in HeLa cells. J. Biol. Chem. 281, 38489-38497.
Tang,E.D., Nunez,G., Barr,F.G., and Guan,K.L. (1999). Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741-16746.
UK Prospective Diabetes Study (UKPDS) Group (1998). Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352, 854-865.
Unterman,T.G., Fareeduddin,A., Harris,M.A., Goswami,R.G., Porcella,A., Costa,R.H., and Lacson,R.G. (1994). Hepatocyte nuclear factor-3 (HNF-3) binds to the insulin response sequence in the IGF binding protein-1 (IGFBP-1) promoter and enhances promoter function. Biochem. Biophys. Res. Commun. 203, 1835-1841.
van de Werve,G., Lange,A., Newgard,C., Mechin,M.C., Li,Y., and Berteloot,A. (2000). New lessons in the regulation of glucose metabolism taught by the glucose 6-phosphatase system. Eur. J. Biochem. 267, 1533-1549.
van der Horst,A., Tertoolen,L.G., Vries-Smits,L.M., Frye,R.A., Medema,R.H., and Burgering,B.M. (2004). FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 279, 28873-28879.
van Schaftingen,E. and Gerin,I. (2002). The glucose-6-phosphatase system. Biochem. J. 362, 513-532.
Vander Kooi,B.T., Streeper,R.S., Svitek,C.A., Oeser,J.K., Powell,D.R., and O'Brien,R.M. (2003). The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J. Biol. Chem. 278, 11782-11793.
Vanhaesebroeck,B. and Alessi,D.R. (2000). The PI3K-PDK1 connection: more than just a road to PKB. Biochem. J. 346, 561-576.
Vidal-Puig,A. and O'Rahilly,S. (2001). Metabolism. Controlling the glucose factory. Nature 413, 125-126.
Wang,M.C., Bohmann,D., and Jasper,H. (2005). JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 121, 115-125.
64

6 Literaturverzeichnis
White,M.F. (2002). IRS proteins and the common path to diabetes. Am. J. Physiol Endocrinol. Metab 283, E413-E422.
Wild,S., Roglic,G., Green,A., Sicree,R., and King,H. (2004). Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047-1053.
Withers,D.J., Gutierrez,J.S., Towery,H., Burks,D.J., Ren,J.M., Previs,S., Zhang,Y., Bernal,D., Pons,S., Shulman,G.I., Bonner-Weir,S., and White,M.F. (1998). Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391, 900-904.
Woods,Y.L., Cohen,P., Becker,W., Jakes,R., Goedert,M., Wang,X., and Proud,C.G. (2001a). The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J. 355, 609-615.
Woods,Y.L., Rena,G., Morrice,N., Barthel,A., Becker,W., Guo,S., Unterman,T.G., and Cohen,P. (2001b). The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 355, 597-607.
Yang,E.J., Ahn,Y.S., and Chung,K.C. (2001). Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem. 276, 39819-39824.
Zhao,H.H., Herrera,R.E., Coronado-Heinsohn,E., Yang,M.C., Ludes-Meyers,J.H., Seybold-Tilson,K.J., Nawaz,Z., Yee,D., Barr,F.G., Diab,S.G., Brown,P.H., Fuqua,S.A., and Osborne,C.K. (2001). Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor-interacting protein with both coactivator and corepressor functions. J. Biol. Chem. 276, 27907-27912.
65

Danksagung
Herrn Prof. Dr. Dr. Hans Georg Joost danke ich für die Überlassung des
Dissertationsthemas und für die ausgezeichneten Arbeitsbedingungen im
Labor.
Herrn Prof. Dr. Peter C. Heinrich danke ich für die Übernahme des Koreferats.
Herrn PD Dr. Andreas Barthel danke ich für die intensive Betreuung und seine
ständige Bereitschaft, Fragen zu diskutieren und Hilfestellung zu geben. Ich
möchte ihm außerdem für die Durchsicht und Korrektur des Manuskripts und für
viele Anregungen danken.
Herrn Prof. Dr. Walter Becker danke ich für die Bereitstellung der DYRK-
Konstrukte und für interessante Diskussionen.
Allen Mitarbeitern des Institutes für Pharmakologie und Toxikologie danke ich
für die freundliche Arbeitsatmosphäre und die gute Zusammenarbeit. Herrn
Dipl.-Chemiker Klaus-Dieter Krüger und meinem Kommilitonen Herrn Hans-
Martin Orth gilt hierbei mein besonderer Dank.
Meiner Schwester Carolin danke ich für die Lösung technischer Probleme bei
der Textverarbeitung.
Nicht zuletzt gilt mein größter Dank meinen Eltern für Ihre bedingungslose
Unterstützung und Ihr Vertrauen.

Lebenslauf
Persönliche Daten Name Florian Till von Groote-Bidlingmaier
Geburtsdatum/-ort 08.10.1976, Neuss
Staatsangehörigkeit deutsch
Familienstand ledig
Schulbildung 08/1987 - 06/1993 Albert-Einstein-Gymnasium Kaarst
08/1993 - 06/1994 Pebblebrook High School Mableton, USA
08/1994 - 06/1996 Albert-Einstein-Gymnasium Kaarst
Abschluss: Allgemeine Hochschulreife
Zivildienst 09/1996 - 08/1997 Malteser Hilfsdienst Neuss
Hochschulbildung 10/1997 - 11/2004 Studium der Humanmedizin an der RWTH Aachen
08/2000 Erster Abschnitt der Ärztlichen Prüfung
08/2003 Zweiter Abschnitt der Ärztlichen Prüfung
10/2003 - 10/2004 Praktisches Jahr, University of Pretoria, Universität
Zürich, RWTH Aachen
11/2004 Dritter Abschnitt der Ärztlichen Prüfung
Berufsausbildung 04/2005 - heute Assistenzarzt, Medizinische Klinik, Evangelisches
Krankenhaus Düsseldorf




























 Algorithm Design and Analysis](https://img.pdfslide.tips/doc/110x75/568168b2550346895ddf78d4/csie-213602-algorithm-design-and-analysis-56cefc5905aac.jpg)
