Embed Size (px)
Citation preview

Einfluss von Arsenverbindungen auf die
Funktion der DNA-Reparaturproteine
Fpg, XPA und PARP-1
vorgelegt von
Diplom-Lebensmittelchemiker
Ingo Walter aus Überlingen
Von der Fakultät III – Prozesswissenschaften
Der Technischen Universität Berlin (TU)
Zur Erlangung des akademischen Grades
DOKTOR DER NATURWISSENSCHAFTEN
- Dr. rer. nat. -
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. L. W. Kroh
Berichter: Prof. Dr. A. Hartwig
Berichter: Prof. Dr. B. Epe
Tag der wissenschaftlichen Aussprache: 4. Mai 2007
Berlin 2007
D 83


Inhaltsverzeichnis I
Inhaltsverzeichnis
1. Zusammenfassung............................................................................................ 1
2. Einleitung........................................................................................................... 3
2.1 Thematische Einführung .............................................................................. 3
2.2 Arsen............................................................................................................ 4 2.2.1 Vorkommen und Verwendung .......................................................................................... 5 2.2.2 Toxikokinetik ..................................................................................................................... 6 2.2.3 Toxische Wirkungen ......................................................................................................... 8 2.2.4 Genotoxizität von Arsenverbindungen.............................................................................. 9
2.3 Zinkfingerproteine in der DNA-Reparatur ................................................... 10 2.3.1 Die Basenexzisionsreparatur und das Fpg-Protein........................................................ 11 2.3.2 Die Nukleotidexzisionsreparatur und das XPA-Protein .................................................. 14 2.3.3 Poly(ADP-Ribose)polymerase........................................................................................ 16
3. Fragestellung................................................................................................... 22
4. Material und Methoden ................................................................................... 23
4.1 Zellkultur..................................................................................................... 23 4.1.1 Zellen .............................................................................................................................. 23 4.1.2 Koloniebildungsfähigkeit ................................................................................................. 23
4.2 Inkubationslösungen .................................................................................. 24
4.3 Präparation von PM2-DNA......................................................................... 25 4.3.1 Reaktivierung der Phagen .............................................................................................. 25 4.3.2 Plaque-Test zur Bestimmung des Phagentiters ............................................................. 26 4.3.3 PM2-Präparation............................................................................................................. 26
4.4 DNA-Relaxationstest .................................................................................. 29 4.4.1 Versuchsdurchführung.................................................................................................... 29
4.5 Zinkfreisetzung aus XPAzf ......................................................................... 31 4.5.1 Versuchsdurchführung.................................................................................................... 31
4.6 DNA-Bindungsaktivität von XPA mittels Gelshift ........................................ 32 4.6.1 Versuchsdurchführung.................................................................................................... 33
4.7 Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen .............................................. 34 4.7.1 Versuchsdurchführung.................................................................................................... 34 4.7.2 Auswertung am Fluoreszenzmikroskop.......................................................................... 35
4.8 PARP-1 Proteingehalt ................................................................................ 36 4.8.1 Versuchsdurchführung.................................................................................................... 36

II Inhaltsverzeichnis
4.9 Isolierung von PARP-1 ............................................................................... 38 4.9.1 Infektion der Sf9-Zellen................................................................................................... 38 4.9.2 Aufreinigung rekombinanter PARP-1 ............................................................................. 38
4.10 Poly(ADP-Ribosyl)ierung isolierter PARP-1 ............................................... 40 4.10.1 Versuchsdurchführung.................................................................................................... 40
5. Ergebnisse und Diskussion ........................................................................... 42
5.1 Eingesetzte Arsenverbindungen ................................................................ 42
5.2 Zytotoxizität ................................................................................................ 44
5.3 Einfluss auf die Aktivität von Fpg................................................................ 48
5.4 Einfluss auf XPA......................................................................................... 52
5.5 Einfluss auf die Poly(ADP-Ribosyl)ierung .................................................. 55 5.5.1 Poly(ADP-Ribosyl)ierung in HeLa S3-Zellen.................................................................. 55 5.5.2 Einfluss der Arsenverbindungen auf die DNA-Schadensinduktion durch H2O2 und auf
die Genexpression der PARP-1 ..................................................................................... 64 5.5.3 Bestimmung des Proteingehalts von PARP-1................................................................ 67 5.5.4 Poly(ADP-Ribosyl)ierung isolierter PARP-1 ................................................................... 73
6. Zusammenfassende Diskussion und Ausblick ............................................ 78
7. Literatur............................................................................................................ 86
8. Anhang............................................................................................................. 97
8.1 Abkürzungsverzeichnis .............................................................................. 97
8.2 Liste der verwendeten Chemikalien ......................................................... 101
8.3 Liste der verwendeten Geräte und Verbrauchsmatrialien ........................ 105
8.4 Verwendete Puffer und Lösungen............................................................ 108 8.4.1 Zellkultur ....................................................................................................................... 108 8.4.2 PM2-Isolierung.............................................................................................................. 108 8.4.3 DNA-Relaxationstest .................................................................................................... 110 8.4.4 XPA-Zinkfreisetztung.................................................................................................... 111 8.4.5 XPA-Gelshiftassay........................................................................................................ 111 8.4.6 Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen ................................................................ 112 8.4.7 PARP-1 Proteingehalt................................................................................................... 113 8.4.8 PARP-1 Isolierung ........................................................................................................ 114 8.4.9 Poly(ADP-Ribosyl)ierung isolierter PARP-1 ................................................................. 115
8.5 Vergleich Methyloxoarsin-Diiodomethylarsin............................................ 117
8.6 Qualitätsprüfung der isolierten PM2-DNA ................................................ 119
8.7 Hemmung isolierter PARP-1 durch CuSO4 .............................................. 120

Inhaltsverzeichnis III
Publikationsliste ................................................................................................... 121
Lebenslauf............................................................................................................. 126
Danksagung.......................................................................................................... 127




Zusammenfassung 1
1. Zusammenfassung
In weiten Teilen der Welt sind Millionen von Menschen einer Exposition von Arsen
über das Trinkwasser ausgesetzt. Nach chronischer Arsenexposition kann es neben
anderen Krankheiten zu einer Krebsentstehung in verschiedenen Organen kommen.
Es wird davon ausgegangen, dass vor allem die Induktion von oxidativen DNA-
Schäden und die Hemmung von DNA-Reparaturprozessen eine entscheidende Rolle
in der nach wie vor nicht vollständig verstandenen arseninduzierten Kanzerogenese
spielt. In den letzten Jahren wurden reaktive methylierte Metabolite im menschlichen
Urin nachgewiesen, die ein ähnliches oder sogar höheres genotoxisches Potential
als das anorganische Arsenit besitzen. Da Arsen eine hohe Affinität zu Thiolgruppen
besitzt, stellen so genannte Zinkfingerstrukturen, in denen Zink durch vier Cystein-
und/oder Histidin-Reste komplexiert wird einen potentiellen Angriffspunkt dar. In der
vorliegenden Arbeit werden daher die drei DNA-Zinkfinger-Reparaturproteine
Formamidopyrimidin-DNA-Glykosylase (Fpg), Xeroderma Pigmentosum Protein A
(XPA) und Poly(ADP-Ribose)polymerase-1 (PARP-1) auf ihre Wechselwirkungen mit
Arsenverbindungen untersucht.
In einem ersten Versuch konnte für die bakterielle Fpg gezeigt werden, dass alleine
die methylierten dreiwertigen Arsenverbindungen monomethylarsonige Säure
(MMA(III)) und dimethylarsinige Säure (DMA(III)) in mikromolaren bis millimolaren
Konzentrationen in der Lage sind eine Hemmung hervorzurufen, während Arsenit
und die fünfwertigen Metabolite Monomethylarsonsäure (MMA(V)) und
Dimethylarsinsäure (DMA(V)) keine Reaktion zeigten. Eine Koinkubation mit Zink
konnte die durch DMA(III) induzierte Fpg-Hemmung reduzieren, was einen ersten
Hinweis auf die Beteiligung der Zinkfingerstruktur von Fpg gibt.
Im Falle von menschlichem XPA wurde zunächst untersucht, inwieweit die
Arsenverbindungen in der Lage sind, Zink aus einer synthetischen Zinkfingerdomäne
freizusetzen. Auch hier zeigten ausschließlich die dreiwertigen Verbindungen eine
Zinkfreisetzung, wobei die methylierten Metabolite stärker reagierten als
anorganisches Arsenit. Für DMA(III) konnte zusätzlich gezeigt werden, dass die
Bindung von murinem XPA an DNA in hohen millimolaren Konzentrationen gestört
wird.
Ausgehend von diesen Versuchen mit den isolierten Zinkfingerproteinen Fpg und
XPA, wurde anschließend auch eine Hemmung eines Zinkfingerproteins unter

2 Zusammenfassung
zellulären Bedingungen untersucht. Als ein möglicher erster molekularer Ansatzpunkt
konnte kürzlich die zwei Zinkfingerstrukturen enthaltende PARP-1 identifiziert
werden, die zum Großteil für die Poly(ADP-Ribosyl)ierung von verschiedenen
Zielproteinen und damit für die Regulation wichtiger zellulärer Mechanismen wie der
DNA-Reparatur verantwortlich ist. Der Einfluss von Arsenit und seinen methylierten
Metaboliten auf die Poly(ADP-Ribosyl)ierung wurde in menschlichen HeLa S3 Zellen
mittels immunfluorimetrischer Quantifizierung der Poly(ADP-Ribose) nach vorheriger
Stimulierung durch H2O2 mikroskopisch bestimmt. Arsenit und seine dreiwertigen
methylierten Metabolite reduzieren die Poly(ADP-Ribosyl)ierung in niedrigen
nanomolaren, nicht zytotoxischen Konzentrationen, während die fünfwertigen
methylierten Metabolite bis zu einer Konzentration von
500 µM keinen Effekt zeigten. Die methylierten Metabolite verursachten die
Hemmung schon in 10-fach tieferen Konzentrationen verglichen mit Arsenit. In
weiteren Versuchen konnte gezeigt werden, dass die beobachtete Hemmung weder
auf eine zeitliche Verschiebung des Poly(ADP-Ribose)-Signals, noch auf eine
veränderte Aktivierung der PARP-1 durch H2O2-induzierte Strangbrüche oder eine
veränderte Gen- oder Proteinexpression zurückzuführen ist. Anschließend wurden
Versuche mit isolierter PARP-1 durchgeführt, um zu überprüfen, ob es wirklich zu
direkten Interaktionen der Arsenverbindungen mit dem Protein kommt. Die
festgestellte Hemmung durch die dreiwertigen Arsenverbindungen lag jedoch bei
höheren Konzentrationen als im zellulären System.
Die Hemmung der Poly(ADP-Ribosyl)ierung durch dreiwertige Arsenverbindungen ist
sowohl im zellulären, wie auch im isolierten System die bisher sensitivste
enzymatische Reaktion, die direkt mit der Reparatur der DNA verbunden ist.
Zusammen mit der Hemmung anderer DNA-Reparatur-Zinkfingerproteine wie XPA
oder möglicherweise menschlichen Homologen von Fpg speziell durch die
dreiwertigen methylierten Arsenmetabolite MMA(III) und DMA(III) könnte sie damit
einen entscheidenden Mechanismus der arseninduzierten Genotoxizität darstellen.

Einleitung 3
2. Einleitung
2.1 Thematische Einführung
Metallionen und Metallverbindungen sind in der Umwelt ubiquitär verbreitet. Viele
von ihnen, wie Zink, Kupfer, Eisen oder Selen spielen als essentielle
Spurenelemente eine wichtige Rolle in biologischen Systemen. Im Gegensatz dazu
konnten für Verbindungen von Arsen, Nickel, Cadmium oder Blei teilweise starke
toxische Effekte, wie eine erhöhte Tumorinzidenz festgestellt werden
(zusammengefasst in Hartwig, 2000). Eines der größten Umweltprobleme auf diesem
Gebiet ist die chronische Belastung von weltweit über 200 Millionen Menschen durch
arsenbelastetes Trinkwasser. Zahlreiche Studien belegen einen Zusammenhang
zwischen dem Arsengehalt im Trinkwasser und dem vermehrten Auftreten von
verschiedenen Krebsarten sowie anderen Krankheiten, wie z.B. der endemischen
blackfoot desease (Yoshida et al., 2004). Aufgrund dieser Studien wurde Arsen
international von der IARC und in Deutschland von der Senatskommission zur
Prüfung gesundheitlicher Arbeitsstoffe der DFG („MAK-Kommision“) als
Humankanzerogen eingestuft (IARC, 1980; DFG, 2004).
Die grundlegenden biochemischen Mechanismen der arseninduzierten
Kanzerogenese sind jedoch nach wie vor nicht vollständig geklärt. Als mögliche
Ursachen werden sowohl direkt genotoxische Effekte, wie die durch reaktive
Sauerstoffspezies (ROS) vermittelte Induktion von DNA-Schäden, aber auch
indirekte Effekte wie ein Einfluss auf die Genexpression oder die Hemmung von
DNA-Reparaturmechanismen diskutiert (Gebel, 2001). Im menschlichen Körper
existieren zur Entfernung endogener und exogener DNA-Schädigung eine Reihe von
Reparaturmechanismen, wie die Basenexzisionsreparatur (BER), die
Nukleotidexzissionsreparatur (NER), die Mismatchreparatur (MMR) oder die
Doppelstrangbruchreparatur. Während die BER für die Reparatur von oxidativen
DNA-Schäden, die NER für großräumige Helixverzerrungen, wie sie durch UVC-Licht
oder Benzo[a]pyren verursacht werden, die MMR für Basenfehlpaarungen aufgrund
von Replikationsfehlern zuständig sind, können Doppelstrangbrüche in der DNA nur
noch durch rekombinatorische Mechanismen oder nichthomologe End-zu-End-

4 Einleitung
Verknüpfung repariert werden (Friedberg et al., 2006). Die Aufrechterhaltung der
DNA-Reparatur ist essentiell für die genomische Stabilität.
Für Arsenit konnte die Hemmung von verschiedenen DNA-Reparatursystemen, wie
die Reparatur oxidativer, UVC- oder Benzo[a]pyren-induzierter DNA-Schäden
gezeigt werden (Hartwig et al., 1997; Gebel, 2001; Schwerdtle et al., 2003a). Als ein
möglicher molekularer Ansatzpunkt sind die an vielen DNA-Reparaturwegen
beteiligten Zinkfingerproteine identifiziert worden, in denen Zink durch 4 Cystein-
und/oder Histidin-Reste komplexiert wird. So konnte gezeigt werden, dass Arsenit die
an der BER beteiligte und hauptsächlich durch das Zinkfingerenzym Poly(ADP-
Ribose)polymarse-1 (PARP-1) katalysierte Poly(ADP-Ribosyl)ierung schon in extrem
niedrigen Konzentrationen hemmt (Hartwig et al., 2003).
In der vorliegenden Arbeit soll geklärt werden, ob alleine das anorganische Arsenit
oder auch die im menschlichen Körper gebildeten methylierten Metabolite, deren
Bildung lange Zeit als Detoxifizierung angesehen wurde, für die Hemmung von
Zinkfingerproteinen verantwortlich ist. Hierzu wurde die Interaktion der
Arsenverbindungen mit den drei Zinkfingerproteinen Formamidopyrimidin-DNA-
Glycosylase (Fpg), Xeroderma Pigmentosum Protein A (XPA) und Poly(ADP-
Ribose)polymerase-1 (PARP-1) in zellulären und in subzellulären Systemen
untersucht.
2.2 Arsen
Arsen mit der Ordnungszahl 33 steht in der 5. Hauptgruppe des Periodensystems
und besitzt sowohl metallischen als auch nicht-metallischen Charakter und wird
daher als Halbmetall bezeichnet. Elementares Arsen ist in der Natur selten zu finden,
jedoch kommt es in zahlreichen, vor allem sulfidischen und oxidischen Mineralien
(Arsenkiese) in der Erdkruste in den Oxidationsstufen -3, +3 und +5 vor (Hollemann
und Wieberg, 1995). Seine mittlere Konzentration in der Erdkruste wird auf 1-2 mg/kg
geschätzt, wobei es lokal zu extrem hohen Arsenkonzentrationen im Boden kommen
kann (Matschullat, 1999).

Einleitung 5
2.2.1 Vorkommen und Verwendung
Der Name des schon im Altertum in der Heilkunde, später im Mittelalter und der
Renaissance vor allem als Mordgift, verwendeten Arsens leitet sich vom persischen
Namen az-zarnikh (zar = Gold) bzw. vom griechischen Namen arsenikon für das
Mineral Auripigment (As2S3) ab (Hollemann und Wieberg, 1995).
Das im Boden natürlich vorkommende Arsen gelangt durch Verwitterung, Erosion
und Vulkanismus in das Grundwasser und die Luft. In der Luft tritt Arsen vor allem
nach Vulkanausbrüchen, durch Verhüttung von arsenhaltigen Metallerzen sowie
durch Verbrennen von arsenbelasteter Kohle in Form des partikulären Arsentrioxids
(As2O3) auf. Im Trinkwasser dominieren die anorganischen Formen Arsenit und
Arsenat, die in belasteten Gebieten der Erde wie Bangladesch, Indien, Taiwan, Chile,
Argentinien, Mexiko oder in den USA in Konzentationen bis zu 4 mg/l vorkommen
können. Die weltweite Arsenbelastung über das Trinkwasser gehört zu unseren
größten Umweltproblemen. In vielen Teilen der Erde wird der WHO-Grenzwert von
10 µg/l weit überschritten, was alleine in Indien zu 30 Millionen arsenbelasteten
Menschen führt (NRC, 1999; NRC, 2001). Aufgrund einer Konzentration von
anorganischem Arsen von ca. 1,5 µg/l im Meerwasser wird Arsen nach
Metabolisierung auch in Form von organischen Verbindungen als Arsenocholin,
Arsenobetain oder Arsenozucker in marinen Lebewesen wie Algen oder Fischen
gefunden. Durch Verstoffwechselung von anorganischen Arsen durch zahlreiche
Mikroorganismen können zudem methylierte Arsenmetabolite in die Natur
eingebracht werden (NRC, 1999; NRC, 2001; Chou und De Rosa, 2003).
Die „Giftigkeit“ von Arsen wurde in zahlreichen Anwendungen, wie z.B. als
Konservierungs- oder Unkrautvertilgungsmittel, Pestizid (DMA(V), Kakodylsäure)
oder zur Druckimprägnierung von Bauholz in Form von chromiertem Kupferarsenat
(CCA) eingesetzt. Solche Anwendungen sind heute jedoch für viele Bereiche
verboten und rückläufig. In der Industrie wird Arsen mittlerweile vor allem in der
Glasindustrie als Läuterungsmittel, sowie als Legierungsbestandteil in der
Halbleitertechnik als Gallium- oder Indiumarsenit eingesetzt (Chou und De Rosa,
2003). Traditionell wurde Arsen in der Medizin als Fowler’sche Lösung (1%ige
K2AsO3) eingesetzt und findet heute noch Anwendung zur Behandlung der
Schlafkrankheit und von Leukämie (Tallman, 2004).

6 Einleitung
2.2.2 Toxikokinetik
Hauptexpositionsquellen bei oraler Aufnahme sind für Arsenverbindungen das
Trinkwasser und die Nahrung. In Deutschland enthält das Trinkwasser nur geringe
Mengen an Arsen, so dass die Aufnahme über die Nahrung bei beruflich nicht
belasteten Menschen zu über 90% zu der Gesamtarsenexposition beiträgt (ATSDR,
1993). Arsenbelastete Lebensmittel sind vor allem Meeresfische, Krusten- und
Schalentiere oder auch Meeresalgen wie sie z.B. in Sushi verarbeitet werden. Bei
der oralen Aufnahme wird davon ausgegangen dass es zu einer fast vollständigen
Resorption kommt, so dass eine amerikanischen Studie auf eine tägliche
Gesamtarsenaufnahme von 27-92 mg kommt, darunter 4-14 µg anorganisches Arsen
(WHO, 2001).
Neben der oralen Exposition spielt bei der inhalativen Aufnahme von Arsen neben
dem Rauchen vor allem die berufliche Exposition eine wichtige Rolle. So findet man
in der Umgebung von Kohlekraftwerken oder Kupferhütten zwischen 1400 und
160000 ng/m3 Arsen, während in ländlichen Gebieten nur 1-5 ng/m3 nachgewiesen
wurden (LAI, 1993). Die Resorptionsrate der vor allem aus As2O3 bestehenden
Partikel ist schwer zu bestimmen und liegt zwischen 30 und 90% (WHO, 1987).
Arsen wird zunächst im Körper in allen Organen und Geweben verteilt, wobei Leber,
Niere, Milz und Lunge akut hohe Arsenspiegel aufweisen können. Die langfristige
Anreicherung von Arsen erfolgt vor allem im Skelett, den Haaren, den Nägeln und
der Haut. Für eine Abschätzung der chronischen Arsenexposition dient daher oft die
Arsenkonzentration in den Nägeln (WHO, 2001).
Anorganische Arsenverbindungen werden vom Menschen und den meisten
Säugetieren zu methylierten Metaboliten verstoffwechselt. Zunächst erfolgt im
Plasma nahezu quantitativ die schnelle Reduktion von fünfwertigem Arsenat zu
dreiwertigem Arsenit. Arsenit wird anschließend in der Leber zu den fünfwertigen
Hauptmetaboliten Monomethylarsonsäure (MMA(V)) und Dimethylarsinsäure
(DMA(V)) methyliert. Welchen Anteil die reaktiven dreiwertigen Zwischenprodukte
monomethylarsonige Säure (MMA(III)) und dimethylarsinige Säure (DMA(III))
ausmachen ist aufgrund ihrer komplizierten Analytik umstritten (Vahter, 1999). Die
einzelnen enzymatischen und evtl. nicht-enzymatischen Schritte des
Arsenmetabolismus sind nach wie vor nicht vollständig aufgeklärt (Abbildung 1). So
wird diskutiert, ob neben den genannten methylierten Metaboliten weitere GSH-

Einleitung 7
konjugierte (Hayakawa et al., 2005) oder Schwefelarsenmetabolite (Hansen et al.,
2004) entstehen können. Die organischen Arsenverbindungen, wie sie z.B. aus
fischreicher Nahrung stammen, werden im Gegensatz dazu nicht oder nur schwach
umgesetzt (NRC, 1999).
O-
OH
OHO AsV
Arsenat
Arsenat-Reduktase
GSH OH
O-
CH3O AsV
MMA(V)
Reduktase
GSSG
OH
OH
CH3AsIII
MMA(III)
MMA(V)-Methyltransferase
OH
CH3CH3 AsIII
DMA(III)
CH3
CH3
O-
O AsV
DMA(V)
OH
OH-O AsIII
Arsenit
SAHC
Arsenit-Methyltransferase
SAM
GSH
GSSG
SAM
SAHC
Ausscheidung über den Urin
10 - 20%
10 - 20%
? %
60 - 80%? %
O-
OH
OHO AsV
Arsenat
Arsenat-Reduktase
GSH OH
O-
CH3O AsV
MMA(V)
Reduktase
GSSG
OH
OH
CH3AsIII
MMA(III)
MMA(V)-Methyltransferase
OH
CH3CH3 AsIII
DMA(III)
CH3
CH3
O-
O AsV
DMA(V)
OH
OH-O AsIII
Arsenit
SAHC
Arsenit-Methyltransferase
SAM
GSH
GSSG
SAM
SAHC
Ausscheidung über den Urin
10 - 20%
10 - 20%
? %
60 - 80%? %
O-
OH
OHO AsV
O-
OH
OHO AsV
Arsenat
Arsenat-Reduktase
GSH OH
O-
CH3O AsV
OH
O-
CH3O AsV
MMA(V)
Reduktase
GSSG
OH
OH
CH3AsIII
OH
OH
CH3AsIII
MMA(III)
MMA(V)-Methyltransferase
OH
CH3CH3 AsIII
OH
CH3CH3 AsIII
DMA(III)
CH3
CH3
O-
O AsV CH3
CH3
O-
O AsV
DMA(V)
OH
OH-O AsIII
OH
OH-O AsIII
Arsenit
SAHC
Arsenit-Methyltransferase
SAM
GSH
GSSG
SAM
SAHC
Ausscheidung über den UrinAusscheidung über den Urin
10 - 20%
10 - 20%
? %
60 - 80%? %
Abbildung 1: Schematische Darstellung des Arsenmetabolismus beim Menschen (Pott et al., 2001) SAM: S-Adenosylmethionin, SAHC: S-Adenosylhomocystein, GSH: Glutathion (red.), GSSG:
Glutathion (ox.)
Die Ausscheidung von Arsen erfolgt in erster Linie über die Niere. Hauptmetabolite
im menschlichen Urin sind Arsenit (10- 20%), MMA(V) (10- 20%) sowie DMA(V) (60-
80%) (Vahter, 1999). Aufgrund kürzlich verbesserter Analyseverfahren konnten
zudem MMA(III) und DMA(III) nachgewiesen werden (Mandal et al., 2001). Die
genaue Speziation und die quantitative Bestimmung sind jedoch noch umstritten.
Fünfwertige Metabolite zeigen eine geringere Reaktivität gegenüber zellulären
Makromolekülen und werden daher schneller ausgeschieden als dreiwertige
Verbindungen (Vahter, 1999).

8 Einleitung
2.2.3 Toxische Wirkungen
Arsen wird aufgrund von Tierversuchen von manchen Autoren als
Ultraspurenelement bezeichnet, obwohl keine physiologischen Daten vom Menschen
vorliegen. Die tägliche Aufnahme an Arsen sollte den Tagesbedarf jedoch bei weitem
decken (Thornton, 1999). Weitaus relevanter sind die toxischen Wirkungen von
Arsen. Nach oraler Aufnahme größerer Mengen von anorganischem Arsen kommt es
zu Wirkungen wie Schädigungen der Schleimhäute im Gastrointestinaltrakt,
Erbrechen und Lähmung der Herz-Kreislauffunktion sowie des peripheren
Nervensystems. Eine hohe Dosis von 1-3 mg/kg Körpergewicht kann binnen 48
Stunden zum Tod im Kreislaufschock führen (Becher und Wahrendorf, 1998).
Die chronische Exposition von Arsen über die Atemluft oder das Trinkwasser geht
neben einer Vielzahl von Krankheiten wie z.B. Hautläsionen (endemische blackfoot
disease in Taiwan) vor allem mit einem erhöhten Krebsrisiko einher. So entsteht
nach inhalativer Aufnahme vor allem Lungenkrebs, während bei oraler Exposition
primär Hautkrebs gebildet wird. Epidemiologische Studien konnten jedoch auch
einen Zusammenhang zwischen einer Arsenbelasung über das Tinkwasser und dem
Entstehen von Lungen-, Nieren-, Blasen und Lebertumoren bereits ab einer
Konzentration von 50 µg/l zeigen (NRC, 1999; NRC, 2001; Yoshida et al., 2004). Für
die Entstehung von Lungenkrebs ergab sich bei chronischer Exposition von nur
1,8 ng/m3, wie sie in der Atemluft in ländlichen Räumen in Deutschland zu finden ist,
schon ein geschätztes Krebsrisiko von 10-5 (Hassauer und Kalberlah, 1999).
Obwohl die Krebsentstehung durch Arsen beim Menschen gut dokumentiert ist und
wichtige Gremien wie die Interational Agency for Reseach on Cancer (IARC),
Weltgesundheitsorganisation (WHO), Environmental Protection Agency (EPA) oder
die MAK Arsen als Humankanzerogen einstufen, dauerte es lange bis ein adäquates
Tiermodell insbesondere für die orale Aufnahme gefunden wurde (IARC, 1980).
Inzwischen ist gut belegt, dass anorganisches Arsen kanzerogene, kokanzerogene
und promovierende Eigenschaften besitzt (Waalkes et al., 2004). DMA(V) konnte als
komplettes Kanzerogen identifiziert werden (Hayashi et al., 1998).

Einleitung 9
2.2.4 Genotoxizität von Arsenverbindungen
Wie auch andere kanzerogene Metalle zeigt anorganisches Arsen kein oder nur ein
schwaches mutagenes Potential in bakteriellen Testsystemen und Säugerzellen. Im
Gegensatz dazu wurden für Arsenit komutagene und klastogene Eigenschaften
nachgewiesen. Zur in vitro Toxizität von Arsenverbindungen liegen umfangreiche
Daten vor. In vielen Studien konnte gezeigt werden, dass die dreiwertigen
Arsenverbindungen ein viel höheres genotoxisches Potential besitzen als die
fünfwertigen Substanzen. So induzieren Arsenit und auch seine dreiwertigen
methylierten Metabolite Chromosonenabberationen, Schwester-Chromatid-
Austausche und Mikrokerne (Gebel, 2001; Kligerman et al., 2003). Als Ursache für
diese Effekte werden unter anderem direkt genotoxische Effekte wie die Induktion
von oxidativen DNA-Schäden verantwortlich gemacht. Arsenit induziert sowohl
reaktive Sauerstoff- als auch Stickstoffspezies (Huang et al., 2004; Shi et al., 2004).
Als Folge dessen konnte in zellulären Systemen, wie auch unter isolierten
Bedingungen eine DNA-Schädigung durch verschiedene Arsenverbindungen gezeigt
werden (u.a Mass et al., 2001; Ahmad et al., 2002; Schwerdtle et al., 2003b).
Neben diesen direkten genotoxischen Wirkungen werden eine ganze Reihe von
indirekten Effekten für die Genotoxizität von Arsen verantwortlich gemacht. So wurde
eine Interaktion mit verschiedenen DNA-Reparatursystemen nachgewiesen.
Arsenverbindungen hemmen sowohl die Reparatur von oxidativen DNA-Schäden,
die durch die BER entfernt werden, wie auch die Reparatur von UVC-Schäden und
Benzo[a]pyren-Addukten, für die die NER verantwortlich ist (Hartwig et al., 1997; Bau
et al., 2001; Kitchin, 2001; Schwerdtle et al., 2003a; Huang et al., 2004). Aufgrund
ihrer hohen Affinität insbesondere zu vicinalen Thiolgruppen können vor allem
dreiwertige Arsenverbindungen zu einer Hemmung von einer Vielzahl von Proteinen
führen. Ein potentieller molekularer Angriffspunkt könnten daher so genannte
Zinkfingerstrukturen, wie sie in vielen DNA-Reparaturproteinen vorkommen (siehe
2.3), sein. So konnte gezeigt werden, dass die zelluläre Poly(ADP-Ribosyl)ierung, die
vor allem durch das Zinkfingerprotein PARP-1 katalysiert wird und in der BER eine
wichtige Rolle spielt, schon in extrem niedrigen nanomolaren Arsenit-
Konzentrationen gehemmt wird (Hartwig et al., 2003). Neben der Hemmung
verschiedener DNA-Reparaturwege gelten eine veränderte Genexpression oder eine

10 Einleitung
Beeinflussung der Signaltransduktion als mögliche indirekte genotoxische oder
kanzerogene Faktoren von Arsen (Gebel, 2001; Andrew et al., 2003).
Eine entscheidende Rolle in der arseninduzierten Kanzerogenese spielt zudem die
Metabolisierung. Aufgrund ihrer besseren Ausscheidung wurde die Bildung der
fünfwertigen methylierten Metabolite lange Zeit als Detoxifizierung angesehen. In den
letzten Jahren haben mehrere Studien gezeigt, dass die dreiwertigen metylierten
Metabolite ein ähnliches oder sogar stärkeres genotoxisches Potential als
anorganisches Arsenit haben. So konnte beispielsweise eine stärkere Zytotoxiziät
(Petrick et al., 2000; Styblo et al., 2000), DNA-Schädigung (Schwerdtle et al., 2003b),
Induktion von Apoptose (Chen et al., 2003) oder klastogene Wirkung (Kligerman et
al., 2003) von MMA(III) und DMA(III) im Vergleich zu Arsenit gezeigt werden. Die
methylierten Metabolite oder auch kürzlich beschriebene GSH-Konjugate oder
Schwefelarsenmetabolite können damit einen entscheidenden Beitrag zur
Genotoxizität und in Folge dessen zur kanzerogenen Wirkung von Arsen haben.
2.3 Zinkfingerproteine in der DNA-Reparatur
Zinkfingerproteine spielen als größte Klasse an DNA-bindenden Proteinen (Webster
et al., 2001) eine wichtige Rolle in vielen biologischen Prozessen der Zelle. Nach
Sequenzierung des menschlichen Genoms, geht man heute davon aus, dass 3%
aller Gene für Zinkfingerproteine kodieren (Maret, 2003). Zinkfingerstrukturen sind in
Proteinen vor allen für Wechselwirkungen mit anderen Molekülen der Zelle, wie DNA
oder RNA aber auch für Protein-Protein Interaktionen verantwortlich und sind daher
an der Regulation von Prozessen wie Transkription, Translation, DNA-Reparatur,
Zellproliferation oder Apoptose beteiligt (Laity et al., 2001; Krishna et al., 2003). Mit
dem Transkriptionsfaktor IIIA (TFIIIA) aus Xenopus leavis wurde 1985 das erste
Zinkfingerprotein identifiziert. Gemeinsames Strukturmerkmal der Zinkfingerproteine
ist ein Zinkion, das durch vier invariante Cystein- und/oder Histidin-Reste komplexiert
wird und so im Protein für die Stabilisierung der Tertiärstruktur sorgt. Im klassischen
Zinkfingermotiv von TFIIIA binden 2 Cysteine und 2 Histidine das zentrale Zinkion. In
den letzten 20 Jahren konnten jedoch weitere Zinkfingertypen wie das Cys4- und das
Cys3His1-Motiv identifiziert werden, die in Proteinen wie dem Östrogen- und
Gukokortidoidrezeptor, Transkriptionsfaktor SP1, breast cancer protein 1 (BRCA-1)
Replikationsprotein A (RPA), Poly(ADP-Ribose)Polymerase-1 (PARP-1),

Einleitung 11
Formamidopyrimidin-DNA-Glycosylase (Fpg) oder Xeroderma Pigmentosum Typ A
Protein (XPA) eine entscheidende Rolle bei der Funktion des Proteins spielen
(Webster et al., 2001; Wilcox et al., 2001). Die Unversehrtheit der Zinkfingerstruktur
ist in diesen Proteinen für ihre Funktion unerlässlich. Störungen im
Redoxgleichgewicht der Zelle oder auch toxische Metallverbindungen können zu
einer Zerstörung der Zinkfingerstruktur und damit zu einem Funktionsverlust des
ganzen Proteins führen. Besonders kritisch ist eine solche Hemmung bei
Zinkfingerproteinen, die an der Reparatur von DNA-Schäden beteiligt sind (Hartwig,
2001). Tabelle 1 gibt einen Überblick über diejenigen Zinkfingerproteine, die an
verschiedenen DNA-Reparaturwegen beteiligt sind und in der vorliegenden Arbeit auf
eine Wechselwirkung mit Arsenverbindungen untersucht wurden.
Tabelle 1: Überblick über die in der vorliegenden Arbeit untersuchten Zinkfingerproteine
Zinkfingermotiv Anzahl der Zinkfingermotive
DNA-Reparaturweg
Fpg Cys4 1 Basenexzisionsreparatur in E. coli
XPA Cys4 1 Nukleotidexzisionsreparatur
PARP-1 Cys3His1 2 Basenexzisionsreparatur DNA-Einzel- und Doppel-
Strangbruchreparatur
2.3.1 Die Basenexzisionsreparatur und das Fpg-Protein
2.3.1.1 Die Basenexzisionreparatur
Die Basenexzisionsreparatur (BER) ist einer der am häufigsten in der Natur
anzutreffenden DNA-Reparaturwege. Die BER ist unter anderem verantwortlich für
die Reparatur von oxidativ geschädigten Basen, wie sie durch reaktive
Sauerstoffspezies (ROS), die durch entzündliche Prozesse in der Zelle, ionisierende
Strahlung, langwelliges UV-Licht oder bei der Atmungskette in den Mitochondrien
entstehen können, gebildet werden. Das prämutagene 8-Oxoguanin stellt dabei
einen der wichtigsten oxidativen DNA-Schäden dar, da es in Folge einer Replikation
mit Adenin paaren und somit eine Mutation hervorrufen kann. Neben den oxidativen

12 Einleitung
DNA-Schäden werden von der BER auch alkylierte DNA-Basen, Uracil oder AP-
Stellen (Apurin/Apyrimidin-Stellen) erkannt und repariert.
Initiiert wird die BER von einer Klasse von Enzymen, den DNA-Glykosylasen, die
geschädigte DNA-Basen erkennen und diese durch Hydrolyse der N-glykosidischen
Bindung entfernen. Beim Menschen sind bisher 11 verschiedene Glykosylasen
bekannt. Die entstandenen AP-Stellen werden anschließend durch Endonukleasen,
die 5’ von der AP-Stelle das Zuckerphosphatrückgrad einschneiden, sowie von
Desoxyribosephosphordiesterasen, die 3’ das Desoxyribosephosphat entfernen,
weiterprozessiert, so dass eine Lücke von einem Nukleotid in der DNA entsteht.
Manche DNA-Glykosylasen vereinigen zudem ihre Glykosylase- mit einer
Endonuklease- oder 3’-Lyase-Aktivität. Durch Einfügen eines Nukleotids durch
Polymerase ß wird die Lücke geschlossen. Im Falle der sogenannten short patch
BER erfolgt anschließend das Zusammenspiel von Ligase III, XRCC1, Polymerase ß
und PARP-1 um die Ligation zu katalysieren und die Reparatur zu vollenden. Bei der
long-patch BER hingegen werden durch die Polymerasen δ oder ε sowie PCNA und
RFC bis zu 10 weitere Nukleotide eingeführt. Im Zusammenspiel von Flap-
Endonukleose-1 (FEN-1) und PCNA wird das überhängige Deoxyribosephosphat
entfernt und die Reparatur durch Ligation mittels Ligase I, die mit PCNA und
Polymerase ß interagiert, beendet. Auch PARP-1 ist bei letzten Schritt der long-patch
BER am Aufbau des Proteinkomplexes beteiligt. (zusammengefasst in Friedberg et
al., 2006)
2.3.1.2 Die Formamidopyrimidin-DNA-Glykosylase
Die Formamidopyrimidin-DNA-Glykosylase (Fpg oder MutM) spielt eine wichtige
Rolle in der Reparatur oxidativer DNA-Schäden in E. coli. Das Enzym, das
ursprünglich als FaPy-DNA-Glykosylase identifiziert wurde, erkennt neben
ringoffenen Purin-Basen (FaPy) hauptsächlich 8-Oxopurine wie das prämutagene
8-Oxoguanin, welches beim Menschen durch die strukturell völlig verschiedene
8-OxoGuanin-DNA-Glykosylase (OGG1) erkannt wird (Friedberg et al., 2006).
8-Oxoguanin wird von Fpg vor allem in Paarung mit Cytosin oder Thymin repariert.
Die nach einer Replikation entstehende Paarung mit Adenin wird 200-fach schlechter
erkannt und in E. coli durch eine weitere Glykosylase (MutY) identifiziert (Tchou et
al., 1994). Neben seiner DNA-Glykosylase-Aktivität besitzt Fpg eine 3’-AP-Lyase-

Einleitung 13
sowie eine 5’-Desoxyribosephosphordiesterase-Aktivität, die über eine
ß,δ- Elimination eine Lücke von einem Nukleotid in der DNA erzeugen (Boiteux,
1993; Bhagwat und Gerlt, 1996).
Das Fpg-Protein ist ein relativ kleines Enzym mit einem Molekulargewicht von
30,2 kDa und besteht aus 269 Aminosäuren. Die starke Bindung an Duplex-DNA
wird durch zwei DNA-Bindungsmotive bewerkstelligt. Im N-terminalen Bereich ist ein
helix-two-turn-helix-Motiv und im C-terminalen ein Zinkfingermotiv zu finden
(Abbildung 2), in dem Zink durch vier Cystein-Reste (Cys244, Cys247, Cys264,
Cys267) komplexiert wird (Boiteux, 1993; Buchko et al., 2000). Die Bindung an die
DNA ist eng verknüpft mit der enzymatischen Funktion des Proteins. So hemmen
Punktmutationen eines der Cysteine des Zinkfingers die Funktion von Fpg,
wohingegen Mutationen von Cystein-Resten außerhalb des Zinkfingers keinen
Einfluss haben (O'Connor et al., 1993; Tchou et al., 1993). Cabrera et al. (1988)
konnten zudem zeigen, dass eine Inaktivierung von Fpg zu einer erhöhten Menge an
durch G → T Transversionen verursachten Mutationen führt.
(NH2) C CE•P
R
V
T
•
••
••
••••
•
••
•
•C
C
G•T•P•I•V•A
Q
R
QA
HK
•C
Y•F•T•A•R
K•
Q•C
(COOH)
Zn
147 195
244
247
267
264
(NH2) C CE•P
R
V
T
•
••
••
••••
•
••
•
•C
C
G•T•P•I•V•A
Q
R
QA
HK
•C
Y•F•T•A•R
K•
Q•C
(COOH)
Zn
147 195
244
247
267
264
(NH2) C CE•P
R
V
T
•
••
••
••••
•
••
•
•C
C
G•T•P•I•V•A
Q
R
QA
HK
•C
Y•F•T•A•R
K•
Q•C
(COOH)
Zn
147 195
244
247
267
264
Abbildung 2: Struktur des Zinkfingers von Fpg (modifiziert nach Boiteux, 1993)

14 Einleitung
2.3.2 Die Nukleotidexzisionsreparatur und das XPA-Protein
2.3.2.1 Die Nukleotidexzisionsreparatur
Im Gegensatz zu den oxidativen, kleinräumigen DNA-Schäden, die durch die BER
repariert werden, erkennt die Nukleotidexzisionsreparatur (NER) vor allem
großräumige DNA-Schäden, wie UV-induzierte 6-4-Photoprodukte und
Cyclobutandimere (CPD) sowie Addukte von z.B. Aflatoxin oder Benzo[a]pyren, die
zu Verzerrungen der DNA-Helix führen. Für das Funktionieren der NER ist ein
koordiniertes Zusammenspiel von mindestens 30 verschiedenen Proteinen
erforderlich. Gendefekte in einem der zentralen NER-Proteine führen zu einer
extremen Lichtempfindlichkeit und oft zum Entstehen von Hautkrebs bei betroffenen
Patienten. Diese Erbkrankheit wird Xeroderma Pigmentosum genannt und kann in
acht Komplementationsgruppen (XPA bis XPG und XPV) unterteilt werden, die auf
Funktionsstörungen von zentralen NER-Proteinen zurückzuführen sind.
Die NER kann in die globale genomische Reparatur (GGR), die Schäden aus dem
gesamten Genom entfernt, sowie die transkriptionsgekoppelte Reparatur (TCR), die
nur Schäden im transkribierten Strang aktiver Gene repariert, unterteilt werden. Die
beiden Reparaturwege unterscheiden sich nur im ersten Schritt der
Schadenserkennung. Bei der TCR erfolgt die Erkennung der Helixverzerrung der
DNA durch die RNA Polymerase II. Es kommt zu einem Stopp der Transkription, zu
einer Konformationsänderung der DNA und zur Anlagerung der NER-Proteine. In der
GGR wird ein Proteinkomplex bestehend aus XPC und hHR23B für den ersten
Schritt verantwortlich gemacht (Sugasawa et al., 1998). Vor allem bei der Erkennung
von CPDs scheint ein weiteres Protein, das damage DNA binding protein (DDB) die
Bindung von XPC-hHR23B zu vermitteln. Nach der Bindung von XPC-hHR23B
kommt es zu einer sequenziellen Anlagerung und Ablösung von weiteren NER-
Faktoren (Volker et al., 2001). An XPC-hHR23B bindet als nächstes der
Transkriptionsfaktor TFIIH, der aus 10 Untereinheiten besteht und als wichtige
Proteine die beiden Helikasen XPB und XPD enthält. Nach der lokalen Entwindung
der DNA kommt es zur Anlagerung der 3’-Nuklease XPG, von XPA, RPA und als
letztes von der 5’-Nuklease ERCC1-XPF, während XPC-hHR23B den so genannten
Präinzisionskomplex verlässt. Durch die Nukleasen XPG und ERCC1-XPF kommt es
nun zur Exzision eines 24 bis 30 Nukleotiden langen Oligonukleotids. Es wird

Einleitung 15
anschließend ein zweiter Multienzymkomplex, bestehend aus den Polymerasen δ
und ε, sowie PCNA, RPA und RFC ausgebildet, der die DNA-Neusynthese in der
entstandenen Lücke katalysiert. Der Reparaturprozess wird durch Ligation mittels
Ligase I abgeschlossen (Friedberg, 2001).
2.3.2.2 Das Xeroderma Pigmentosum Protein A
Mutationen des für das NER-Protein Xeroderma Pigmentosum Protein A (XPA)
kodierenden Gens führen zu einer sehr schwerwiegenden Form von Xeroderma
Pigmentosum mit verbundener Lichtempfindlichkeit und dem Entstehen von
Hautkrebs (in Sonnenlicht 1000-fach erhöht). Das Ausschalten von XPA führt zu
einer Absenkung der NER auf weniger als 2% (Vogelstein et al., 2002).
Lange Zeit wurde XPA für den ersten Schritt der NER-Schadenserkennung
verantwortlich gemacht. Sugasawa et al. (1998) konnten jedoch zeigen, dass zuerst
XPC-hHR23B an den Schaden bindet. Man geht heute davon aus, dass XPA eine
wichtige Rolle bei dem Aufbau des Präinzisionskomplexes der NER spielt und nach
Ablösen von XPC-hHR23B die Schadenserkennung verifiziert. Es konnte gezeigt
werden, dass XPA unabhängig von XPC an eine Vielzahl von DNA-Schäden wie
UVC- und cis-Platin-Addukte (Jones und Wood, 1993; Asahina et al., 1994) oder
Benzo[a]pyren- und Aflatoxinaddukte (zusammengefasst in Cleaver und States,
1997) bindet. Für XPA konnte neben der Bindung an geschädigte und ungeschädigte
DNA eine Interaktion mit verschiedenen Faktoren der NER nachgewiesen werden.
So bindet XPA an TFIIH sowie RPA und ist für die Aktivität der 5’-Nuklease ERCC1-
XPF unerlässlich, während die 3’-Nuklease XPG unabhängig von XPA funktioniert
(Sugasawa et al., 1998; Volker et al., 2001).
Das 36 kDa große und 273 Aminosäuren lange XPA besitzt wie auch Fpg eine
Zinkfingerdomäne, in der Zink durch vier Cysteine (Cys105, Cys108, Cys126 und
Cys129) komplexiert wird (Abbildung 3) (Buchko et al., 1998). Im Gegensatz zu Fpg
bindet der Zinkfinger nicht direkt an die DNA. Für die DNA-Bindung ist vielmehr die
loopreiche Domäne von XPA verantwortlich, deren Tertiärstruktur durch die
Wechselwirkungen mit der Zinkfingerstruktur aufrecht erhalten wird. Der Zinkfinger
wird darüber hinaus für die Wechselwirkung mit der 70 kDa Untereinheit von RPA
verantwortlich gemacht (Ikegami et al., 1998; Morikawa und Shirakawa, 2000). Durch

16 Einleitung
Substitution eines der Zink-bindenden Cysteine kommt es zu einem Funktionsverlust
von XPA und einer fast vollständigen Hemmung der NER (Miyamoto et al., 1992).
Abbildung 3: Links: Minimale Bindungsdomäne (MBD) (98-210) von XPA. Rechts: Zinkfingerdomöne (XPAzf), die zur Zinkfreisetztung in dieser Arbeit eingesetzt wurde (101-137) (Ikegami et al., 1998; Bal et al., 2003)
2.3.3 Poly(ADP-Ribose)polymerase
Die Poly(ADP-Ribosyl)ierung ist eine posttranslationale Modifikation von Proteinen,
die erstmals in den 60er Jahren des 20. Jahrhunderts Jahren beschrieben wurde
(Chambon et al., 1963). Sie wird durch eine Familie von Enzymen, den Poly(ADP-
Ribose)polymerasen (PARP) katalysiert. Nach heutigem Kenntnisstand besteht die
sogenannte PARP-Superfamilie aus 17 Mitgliedern, die nach Datenbankrecherche
alle die gleiche minimale katalytische Domäne aufweisen (Ame et al., 2004;
Schreiber et al., 2006). Durch die Poly(ADP-Ribosyl)ierung von Proteinen werden
negative Ladungen in das Molekül eingeführt, die in der Regel zu einem
Aktivitätsverlust der Enzyme führen. Zudem kann es zu sterischen Hemmungen von
Proteinen aufgrund der langen Addukte kommen. Auf diese Weise regulieren
Poly(ADP-Ribose)polymerasen eine Vielzahl von biochemischen Prozessen in der
Zelle, wie z.B. DNA-Reparatur, DNA-Replikation oder Apoptose. Zuletzt wurde PARP
in Zusammenhang mit dem Alterungsprozess gebracht (Bürkle et al., 2005b). Den
größten Anteil an der Poly(ADP-Ribosyl)ierung hat mit etwa 90% das nukleäre
Protein PARP-1. Das Enzym wird mit ca. 200.000 Molekülen pro HeLa- Zelle

Einleitung 17
kontinuierlich stark exprimiert und kann in pflanzlichen wie auch tierischen
Lebewesen, nicht jedoch in Hefen, hochkonserviert nachgewiesen werden
(zusammengefasst in Diefenbach und Bürkle, 2005).
2.3.3.1 Struktur der PARP-1
Das 113 kDa große und 1014 Aminosäuren lange PARP-1 Molekül kann in 3
Domänen unterteilt werden (Abbildung 4). In der 46 kDa großen N-terminalen DNA-
bindenden Domäne finden sich 2 Zinkfingermotive, in denen Zink durch 3 Cystein-
und einen Histidin-Rest komplexiert wird (Cys21, Cys24, His53, Cys56 sowie
Cys125, Cys129, His159, Cys162) (Ikejima et al., 1990). Im Vergleich zu anderen
Zinkfingerproteinen besitzt PARP-1 mit 28 bzw. 30 Aminosäuren sehr große
Zinkfinger, wie sie nur noch in Ligase III gefunden werden. Die beiden
Zinkfingerstrukturen sind für die katalytische Aktivierbarkeit von PARP-1 durch DNA-
Strangbrüche essentiell, wobei Zinkfinger 1 sowohl an Doppel- als auch an
Einzelstrangbrüche bindet, während Zinkfinger 2 nur mit Einzelstrangbrüchen
interagiert. Zudem sind die Zinkfinger an Protein-Protein-Wechselwirkungen beteiligt.
Wie Trucco et al. (1996) zeigen, sind jedoch nicht nur intakte Zinkfingermotive,
sondern auch andere Stukturen der N-terminalen Domäne für die Aktivierung der
katalytischen Aktivität verantwortlich (Trucco et al., 1996). Neben den beiden
Zinkfingern kann PARP-1 auch über eine helix-turn-helix-Domäne an die DNA
binden, in der sich neben einem Kernerkennungssignal (NLS) auch eine Schnittstelle
für die apoptotischen Enzyme Caspase 3 und 7 (siehe 2.3.3.4) befindet.
Die zentrale 22 kDa große Domäne der PARP-1 ist die basische
Automodifikationsdomäne. PARP-1 poly(ADP-ribosyl)iert sich hier vor allem an
Glutamatresten selbst. Zusätzlich befindet sich in dieser Domäne ein BRCT
Bindungsmotiv, das für Protein-Protein-Wechselwirkungen z.B. mit XRCC1
verantwortlich gemacht wird (siehe 2.3.3.3).
In der dritten C-terminalen katalytischen Domäne wird die eigentliche PARP-
Reaktion katalysiert. Durch Spaltung des Cofaktors NAD+ entsteht Nicotinamid und
ADP-Ribose, die dann zu Polymeren aufgebaut wird. Die minimale katalytische
Domäne lässt sich hochkonserviert in der cDNA von allen 17 Mitgliedern der PARP-
Familie wieder finden (zusammengefasst in D'Amours et al., 1999; Schreiber et al.,
2006).

18 Einleitung
N C
Katalytische Domäne
minimale katalytischeDomäne
524 1014
ß-NAD+Nicotinamid
ADPr
ZF1 ZF2 HTH
DNA Bindungsdomäne1
Automodifikations-domäne
372
NLS
Caspase-3/-7Schnittstelle
N CN C
Katalytische Domäne
minimale katalytischeDomäne
524 1014
ß-NAD+Nicotinamid
ADPr
ZF1 ZF2 HTH
DNA Bindungsdomäne1
Automodifikations-domäne
372
NLS
Caspase-3/-7Schnittstelle
Abbildung 4: Molekulare Struktur der PARP-1 (modifiziert nach D'Amours et al., 1999) ZF: Zinkfinger, HTH: Helix-turn-helix-Motif, NLS: Kernerkennungssequenz, ADPr: ADP-Ribose,
ß-NAD+: Nicotinamidadenindinukleotid
2.3.3.2 Metabolismus von Poly(ADP-Ribose)
PARP-1 bindet in dimerer Form (Pion et al., 2005) als eines der ersten Moleküle der
Zelle an einen DNA-Strangbruch und stabilisiert die charkteristische V-Form der
DNA. Durch die Bindung an die DNA wird PARP-1 bis zu 500fach aktiviert und
beginnt nun Zielmoleküle wie p53, NF-κB, Histone, DNA-Ligasen, DNA-
Topoisomerasen, DNA-Polymerasen und vor allem sich selbst zu poly(ADP-
ribosyl)ieren. Die Polymerisationsreaktion lässt sich in 3 Teilabschnitte unterteilen.
Durch die Veresterung der ersten ADP-Ribose-Einheit mit einem Aspartat- oder
Glutamat-Rest des Zielmoleküls wird die Reaktion initiiert. In der Elongationsphase
werden nun bis zu 200 weitere ADP-Ribose-Einheiten α-1’’-2’ glycosidisch verknüpft.
Alle ca. 40 Einheiten kann eine ADP-Ribose-Einheit α-1’’’-2’ glycosidisch angehängt
werden, so dass es zu einer Verzweigung des Polymers kommt. Durch die starken
negativen Ladungen des Polymers in Folge der Modifikation dissoziiert PARP-1 von
der DNA ab und inaktiviert sich so selbst. Poly(ADP-Ribose) hat nur eine kurze
Halbwertszeit von ca. 1 min und wird schnell von der Poly(ADP-
Ribose)glykohydrolase (PARG) abgebaut. PARG besitzt sowohl eine Endo- als auch
eine Exoglykosylase-Aktivität. Die Abspaltung der letzten ADP-Ribose-Einheit vom
Protein wird von der ADP-Ribose-Protein-Lyase katalysiert (Abbildung 5)
(zusammengefasst in Diefenbach und Bürkle, 2005).

Einleitung 19
Abbildung 5: M
etabolismus der Poly(A
DP-R
ibose) (modifiziert nach D
iefenbach und Bürkle, 2005)
NA
D+: N
icotinamidadenindinukleotid, P
AR
P-1: P
oly(ADP
-Ribose)polym
erase-1, PA
RG
: Poly(AD
P-R
ibose)glykohydrolase

20 Einleitung
2.3.3.3 PARP und DNA-Reparatur
Bisher konnte nur für zwei Mitglieder der PARP-Superfamilie, PARP-1 und PARP-2,
eine Aktivierung durch DNA-Strangbrüche gezeigt werden, die auf eine Rolle beider
Enzyme in der DNA-Einzelstrangbruchreparatur und in der BER (siehe 2.3.1.1)
schließen lässt. Knock-out Versuche bei Mäusen offenbarten, dass sowohl der
PARP-1 als auch der PARP-2 knock-out zu schweren Defekten in der BER und zu
einer Hypersensitivität gegenüber ionisierender Strahlung und alkylierenden
Agentien führt. PARP-1-PARP-2-doppel-knock-out Mäuse sind darüber hinaus nicht
lebensfähig (Menissier de Murcia et al., 2003). Auch in Zellkultursystemen mit
ausgeschalteter PARP-1-Aktivität konnten ähnliche Ergebnisse erhalten werden
(Küpper et al., 1995). Zudem konnte gezeigt werden, dass eine erhöhte Menge an
PARP-1 in der Zelle zu einer erhöhten genomischen Stabilität beiträgt (Meyer et al.,
2000). Obwohl die molekularen Mechanismen der Rolle von PARP in der BER noch
nicht vollständig geklärt sind, existiert eine Reihe von Erklärungsmodellen. Als eines
der wichtigsten Akzeptorproteine von PARP-1 gelten die Histone. Ihre lokale
Poly(ADP-Ribosyl)ierung in der Nähe des DNA-Strangbruchs bewirkt eine Relaxation
des Chromatins und ermöglicht damit einen besseren Zugang von anderen DNA-
Reparaturfaktoren an den Schaden (Bürkle et al., 2005a). Neben der Modifikation
von Histonen konnte auch die Poly(ADP-Ribosyl)ierung von wichtigen BER-
Proteinen wie XRCC1, Ligase III oder Polymerase ß nachgewiesen werden. Über die
Poly(ADP-Ribosyl)ierung könnte demnach die Aktivität dieser Proteine reguliert
werden. Außerdem konnte gezeigt werden, dass PARP-1 und PARP-2 direkt mit
BER Enzymen über Protein-Protein- oder aber über Wechselwirkungen der Proteine
mit dem Poly(ADP-Ribose)-Polymeren interagieren. PARP-1 könnte also dafür
verantwortlich sein, diese Proteine nach einem DNA-Strangbruch, wie er auch als
Zwischenprodukt bei der BER gebildet wird, vor allem für die long-patch BER zu
rekrutieren. Hier bildet sich ein Komplex bestehend aus DNA-Ligase III, Polymerase
ß, XRCC1, FEN-1 und PARP-1, die zusammen den letzten Schritt der BER
katalysieren (zusammengefasst in Bouchard et al., 2003; Malanga und Althaus,
2005). Allison et al. (2003) dagegen schlagen vor, dass nicht eine Interaktion mit
PARP-1 selbst, sondern Wechselwirkungen mit poly(ADP-ribosyl)ierten Proteinen die
entscheidende Rolle in der BER spielen (Allinson et al., 2003). Aus dem Abbau von
Poly(ADP-Ribose) könnte zudem an der geschädigten Stelle lokal ATP gebildet

Einleitung 21
werden, das dann z.B. für den Ligationsschritt der BER verwendet werden könnte
(Oei und Ziegler, 2000).
Neben ihrer bedeutsamen Rolle in der BER, konnte für PARP-1 auch eine Interaktion
mit p53 (zusammengefasst in Malanga und Althaus, 2005), mit der DNA-
Doppelstrangbruchreparatur (Süsse et al., 2004) oder auch der Reparatur von UV-
induzierten-Schäden (Flohr et al., 2003) gezeigt werden.
2.3.3.4 PARP und Apoptose
Unter der Apoptose versteht man den programmierten Zelltod, der unter
Enegieaufwand für das gezielte Absterben von stark geschädigten Zellen
verantwortlich ist. Während der Apoptose werden über eine Caspasekaskade
degradative Enzyme wie Endonukleasen oder Proteasen aktiviert, die DNA und
Proteine abbauen und damit zum programmierten Zelltod beitragen. Die Caspasen 3
und 7 sind in der Lage PARP-1 zu spalten und damit zu inaktivieren. Die Bildung des
typischen apoptoischen 85 kDa-Fragments der PARP-1 gilt als ein früher Nachweis
von Apoptose (Diefenbach und Bürkle, 2005).
Neben der Spaltung von PARP-1 kann es aufgrund starker DNA-Schädigung in den
frühren Phasen der Apoptose zu einem massiven Anstieg der Poly(ADP-
Ribosyl)ierung kommen. Diese Überaktivierung von PARP-1 führt zu einer
Depletierung von NAD+ und zu einer Translokation des Apoptose induzierenden
Faktors (AIF) aus den Mitochondrien in den Zellkern. Dieser Reaktionsweg stellt eine
PARP-1-abhängige, aber p53-unabhängige Form der Apoptose dar (Ame et al.,
2004).

22 Fragestellung
3. Fragestellung
Aufgrund epidemiologischer Studien wurde Arsen als Humankanzerogen eingestuft,
obwohl die grundlegenden molekularen Mechanismen der arseninduzierten
Kanzerogenese nach wie vor ungeklärt sind. Neben direkt genotoxischen Effekten,
wie der Induktion von DNA-Schäden, verursacht Arsen vor allem indirekte
genotoxische Effekte wie die Hemmung von DNA-Reparaturprozessen. Aufgrund
seiner hohen Affinität zu Thiolgruppen sind Zinkfingerstrukturen, wie sie auch in
DNA-Reparaturproteinen vorkommen, ein möglicher molekularer Ansatzpunkt für
eine Enzymhemung.
Anorganisches Arsen wird im menschlichen Körper zu methylierten Metaboliten
umgesetzt. Als Zwischenprodukte entstehen dabei die hochreaktiven dreiwertigen
methylierten Metabolite MMA(III) und DMA(III). Ziel der vorliegenden Arbeit ist es zu
klären inwieweit die methylierten Metabolite von anorganischem Arsen zur indirekten
Genotoxizität beitragen. Für die Untersuchungen wurden drei Zinkfingerproteine
ausgewählt, die an verschiedenen DNA-Reparaturprozessen beteiligt sind. Die
bakterielle Fpg ist an der Reparatur oxidativer DNA-Schäden in E. coli beteiligt. Hier
soll zudem geklärt werden, inwieweit eine Koinkubation mit Zink eine möglicherweise
auftretende Hemmung durch Arsen aufheben kann, um so Rückschlüsse auf eine
Beteiligung der Zinkfingerstruktur zu erhalten. Im Falle des zweiten
Zinkfingerproteins XPA, das an der menschlichen NER beteiligt ist, soll untersucht
werden, ob Arsenverbindungen Zink aus der zinkbindenden Domäne (XPAzf)
freisetzen können und ob in Folge dessen die Bindung an die DNA nicht mehr
möglich ist. Mit der Untersuchung der Poly(ADP-Ribosyl)ierung in HeLa S3 wird
geklärt, ob Arsenverbindungen in der Lage sind, auch im zellulären System eine
Reaktion zu hemmen, die zu ca. 90% von dem Zinkfingerenzym PARP-1 katalysiert
wird. Weitere Untersuchungen zur PARP-1 sollen zeigen, worauf eine mögliche
Hemmung der Poly(ADP-Ribosyl)ierung zurückzuführen ist. So wird mittels Western
Blot-Analyse die Proteinmenge der PARP-1 nach Arseninkubation bestimmt. Mit
diesem Versuch ist es zudem möglich, die Induktion von Apoptose über die
Entstehung eines typischen apoptotischen Fragments der PARP-1 nachzuweisen.
Abschließend soll auch für isolierte PARP-1 untersucht werden, ob speziell die
dreiwertigen Verbindungen in der Lage sind das Enzym zu hemmen.

Material und Methoden 23
4. Material und Methoden
Eine Auflistung von allen eingesetzten Chemikalien und Lösungen, deren
Konzentrationen sowie eine Liste der verwendeten Geräte ist im Anhang zu finden.
Anorganisches Arsen ist als humanes Kanzerogen eingestuft. Die folgenden
Chemikalien sind gefährlich und sollten mit äußerster Sorgfalt gehandhabt werden:
Natriumarsenit, Monomethyloxoarsin, Diiodomethylarsin, Iododimethylarsin,
Monomethylarsinsäure und Dimethylarsinsäure.
4.1 Zellkultur
Medien, Puffer, Lösungen und alle verwendeten Verbrauchsmaterialien für die
Zellkultur wurden sterilfiltriert oder autoklaviert bzw. hitzesterilisiert.
4.1.1 Zellen
In der vorliegenden Arbeit wurde die humane, reparaturkompetente Zelllinie HeLa S3
(Zervixkarzinomzellen) verwendet. Zur Lagerung wurden die Zellen in einem
Einfriermedium bestehend aus 90% FKS (fötales Kälberserum) und 10% DMSO bei
-196°C in flüssigem Stickstoff tiefgefroren. Die Zellen wurden in F12-Medium,
versetzt mit 10% FKS, 100 Einheiten/ml Penicillin und 100 µg/ml Streptomycin bei
37°C, 5% CO2-Atmosphäre und 100% Luftfeuchtigkeit kultiviert. HeLa S3-Zellen
wachsen als Monolayer mit einer Generationszeit von etwa 24 Stunden. Die Zellen
wurden bei Erreichen einer Konfluenz von ca. 70% abtrypsieniert, in neue
Zellkulturschalen ausgestreut und mit frischen Medium versetzt. Für die Versuche
wurden die Zellen maximal 25 Passagen lange kultiviert, bis eine neue Charge
aufgetaut wurde.
4.1.2 Koloniebildungsfähigkeit
Zur Ermittlung der Zytotoxizität der Arsenverbindungen wurde die
Koloniebildungsfähigkeit bestimmt. Nach Ablauf der Inkubationszeit wurde hierfür die
Zellzahl bestimmt und eine definierte Zahl von Zellen weitergesetzt und ohne
Substanzinkubation mehrere Tage weiterkultiviert. Anhand der Anzahl an gebildeten

24 Material und Methoden
Kolonien kann man, im Vergleich zu unbehandelten Zellen, Rückschlüsse auf
Langzeitschäden und die Wirkung auf die Reproduktionsfähigkeit des getesteten
Agens ziehen.
Um die Koloniebildungsfähigkeit zu ermitteln, wurden jeweils 1x106 Zellen in eine
Zellkulturschale (100 x 20 mm) mit 10 ml Kulturmedium ausgestreut und nach einem
Teilungszyklus von ca. 24 Stunden mit der zu untersuchenden Substanz für 18
Stunden inkubiert, indem die entsprechende Menge einer Stammlösung direkt in das
Zellkulturmedium pipettiert wurde. Nach Ende der Inkubationszeit wurde das Medium
abgesaugt und die Schalen wurden mit 5 ml frischem Medium gewaschen. Die Zellen
wurden abtrypsiniert, die Zellzahl mit einem automatischen Zellzählgerät (Casy)
gemessen und jeweils genau 300 Zellen in eine Zellkulturschale (60 x 15 mm) mit 5
ml frischem Medium überführt. Nach etwa einer Woche im Brutschrank hatten sich
Kolonien gebildet, die mit dem bloßen Auge sichtbar waren. Das Medium wurde
abgenommen, die Zellen mit PBS gewaschen und mit 2-3 ml vergälltem Ethanol
fixiert. Die Kolonien wurden mit Giemsa-Farbstoff gefärbt und ausgezählt.
4.2 Inkubationslösungen
Alle Inkubationslösungen wurden frisch am Versuchstag unmittelbar vor dem
Versuch in bidestilliertem Wasser angesetzt, um vor allem bei den dreiwertigen
methylierten Arsenverbindungen einer Oxidation vorzubeugen. Arsenit und die
beiden fünfwertigen Metabolite Monomethylarsonsäure und Dimethylarsinsäure
(Cacodylsäure) sind mit Reinheiten von >99,0%, 96,0% bzw. 98,8% kommerziell zu
erweben (siehe Anhang). Die Reinsubstanzen der dreiwertigen methylierten
Arsenverbindungen (Monomethyloxoarsin, Diiodomethylarsin und Iododimethylarsin)
wurden aliquotiert und bei -80°C gelagert. Diese Substanzen sind kommerziell nicht
erhältlich und wurden uns mit einer Reinheit von 99,0% freundlicherweise von Prof.
W. R. Cullen (University of British Columbia, Vancouver, Kanada) zur Verfügung
gestellt.

Material und Methoden 25
4.3 Präparation von PM2-DNA
Für die Untersuchung des Einflusses von Arsen auf die Aktivität des bakterielllen
Fpg-Proteins wird die DNA des Bakteriophagen PM2 als Substrat verwendet. Die
zirkulare, etwa 10.000 Basenpaare (bp) lange doppelstängige DNA, kommt in drei
verschiedenen Formen vor. In vivo liegt zu über 95% die überspriralisierte
(supercoiled) Form vor, die durch einen Einzelstrangbruch in eine relaxierte,
ringoffene (open circular) Form übergeht. Kommt es sogar zu einem
Doppelstrangbruch, liegt lineare DNA vor (Epe et al., 1993). Für den DNA-
Relaxationstest (Kapitel 4.4) ist es wichtig, dass die DNA möglichst wenige
Strangbrüche und oxidativen Schäden aufweist und dadurch der Anteil an ringoffener
und linearer Form auf ein Minimum reduziert wird.
Der Bakteriophage PM2 befällt das Meeresbakterium Alteromonas espeijana,
vermehrt sich in den Bakterienzellen und lysiert diese. Für die Präparation werden
die Phagen in den Wirtszellen vermehrt und nach der erfolgten Lyse aufkonzentriert
und gereinigt. Zum Schluss wird die DNA mittels Phenol-Chloroform-Extraktion
isoliert. Die Herstellung von PM2-DNA wurde mit einigen Modifikationen wie von
(Salditt et al., 1972) beschrieben durchgeführt. Das Wirtsbakterium Alteromonas
sowie die PM2-Phagen wurden uns von Prof. Bernd Epe (Mainz) zur Verfügung
gestellt.
4.3.1 Reaktivierung der Phagen
100 ml Nährmedium wurden mit einer Spatelspitze der bei -80°C gelagerten
Alteromonas-Kultur beimpft und bis zu einer OD600 nm= 0,4 (optische Dichte) auf
einem Schüttler bei Raumtemperatur (Optimum 28°C) angezogen. Bei Erreichen
einer OD von 0,4, die photometrisch kontrolliert wurde, wurde 1 ml Alteromonas-
Phagen-Lösung (gelagert bei -80°C) zugesetzt. Vor Eintritt der Lyse sollte die
Alteromonassuspension eine OD von 1 erreichen, die nach erfolgter Lyse der
Bakterien bis auf ca. 0,1 abfällt. Um Zelltrümmer und nicht lysierte Zellen zu
entfernen, wurde der Extrakt 40 min bei 10.000 g und 4°C zentrifugiert. Das
Phagenlysat im Überstand wurde bis zur weiteren Verarbeitung bei 4°C gelagert. Pro
Woche kommt es zu einem Verlust von ca. 30% der Phagenaktivität.

26 Material und Methoden
4.3.2 Plaque-Test zur Bestimmung des Phagentiters
Mit dem Plaque-Test lässt sich die Konzentration der reaktivierten Phagen
bestimmen, die für die Beimpfung der Hauptkultur wichtig ist. Hierfür wurde am
Vorabend eine Alteromonas-Übernachtkultur (ca. 10 Stunden) angesetzt. Am Testtag
wurden bei Erreichen einer OD von ca. 0,8 100 µl Übernachtkultur und 100 µl
Phagenlysat der Verdünnungsstufen 1:106 – 1:1012 30 min bei Raumtemperatur
inkubiert. 100 µl des Gemisches wurden mit 2 ml flüssigem, 46-48°C warmem Top-
Agar auf mit Bottom-Agar ausgegossenen Platten ausplattiert und über Nacht bei
Raumtemperatur inkubiert. Am nächsten Tag konnten die Plaques ausgezählt und
die Konzentration der aktiven Phagen wurde als plaque forming units (PFU)
berechnet.
4.3.3 PM2-Präparation
1.Tag:
Acht 2L-Schikanekolben mit je 1 l Medium wurden mit je 40 ml Alteromonas-
Übernachtkultur (siehe 4.3.2) und auf einem Taumler gut geschüttelt, so dass eine
gute Sättigung der Kultur mit Sauerstoff erreicht wurde. Nach ca. 2 Stunden und dem
Erreichen einer OD von 0,7 wurden pro Liter Bakerienmedium 1x1010 Phagen
zugegeben und der Anstieg der OD am Photometer stündlich kontrolliert. Nach
Erreichen eines Maximums von über 1,0 und vollständiger Lyse (OD ca. 0,1) wurden
die Lysate in zwei 5L-Erlenmeyerkolben verteilt und 43 g pro Liter Lysat
Polyethylenglycol 6000 (PEG) eingerührt. Nachdem sich das PEG gelöst hat wurde
2,36 g pro Liter Lysat Natriumdextransulfat zugegeben. Sobald die Lösungen
homogen waren wurden die Kolben in 45°-Position für ca. 36 Stunden bei 4°C
gelagert.
2. Tag:
Alle folgenden Schritte wurden auf Eis durchgeführt und die Zentrifugationen
erfolgten bei 4°C. In den Kolben hatten sich Sedimente abgesetzt. Der Überstand
wurde fast vollständig mit einer Pumpe abgesaugt, die Sedimente resuspendiert und
auf 40 ml-Zentrifugenröhrchen verteilt. Nach der Zentrifugation (25 min, 10.000 g)
befanden sich die Phagen im Sediment. Die Oberphase wurde vorsichtig abpipettiert,
die Pellets in einem Becherglas gewogen, mit der doppelten Menge NTC-Puffer

Material und Methoden 27
(1M NaCl, 9,7 mM CaCl2, 10,15 mM Tris/HCl) resuspendiert und 20 min bei 18.000 g
zentrifugiert. Der Überstand wurde abdekantiert, die Pellets in ein Pottergefäß
überführt und mit 3 ml/l Kulturlösung NTC-Puffer in einem Homogenisator nach
Potter homogenisiert (12 x bei 1.500 rpm). Nach einer weiteren Zentrifugation bei
12.000 g und 20 min befanden sich die PM2-Phagen im Überstand. Dieser wurde
daraufhin in ein Becherglas überführt und das Pellet mit 1,5 ml/l Kulturlösung NTC-
Puffer erneut extrahiert und abzentrifugiert. Die gewonnen Überstände wurden
vereinigt und 3 Stunden bei 30.000 g zentrifugiert. Der Überstand wurde abgezogen
und verworfen und die im Pellet befindlichen Phagen mit 2-3 ml
Flüssigkeitsüberstand über Nacht bei 4°C gelagert.
3.Tag:
Die Pellets wurden erneut in 27 ml NTC Puffer resuspendiert und noch einmal nach
Potter homogenisiert. Die Lösung wurde in einen Erlenmeyerkolben überführt und
pro ml Lösung 0,39 g CsCl eingewogen um eine Dichte von genau 1,27-1,29 g/ml
einzustellen. Die Lösung wurde anschließend 10 min bei 5.000 rpm abzentrifugiert
um Proteinreste zu entfernen. Nach erneuter Kontrolle der Dichte wurde die Lösung
in Ultrazentrifugen-Röhrchen überführt und für mindestens 12 Stunden über Nacht
bei 30.000 g zentrifugiert.
4.Tag:
Durch die Ultrazentrifugation wurden die Phagen im CsCl-Gradienten
aufkonzentriert, die als trübe Bande im oberen Drittel des Röhrchens gefunden
werden konnten. Die Bande wurde nun vorsichtig von oben mit einer
abgeschnittenen Pipettenspitzte abgenommen und in sterilen 2ml-Reaktionsgefäßen
aufgefangen; ihr Volumen betrug ca. 3 ml.
Anschließend wurde die Phagenlösung über eine sorfältig mit BE1-Puffer
vorgespülte Sepharosesäule (10 cm Länge, Duchmesser 2,5 cm, Material Sepharose
2B, Amersham) vom CsCl gereinigt. Das Eluat wurde in Fraktionen aufgefangen und
die Fraktionen am Biophotometer vermessen. Fraktionen mit einem 260/280 nm
Verhältnis von 1,35 – 1,43 wurden vereinigt (ca. 30 ml). Die Isolierung der DNA aus
den Phagen erfolgte mittels Phenol-Chloroform-Extraktion. In einem ersten Schritt
wurde dazu das gleiche Volumen an Phenol (Roti-Phenol, pH 7) mit 1% SDS zum
Aufspalten der Phagen und 0,1% Hydroxychinolin zum Schutz vor oxidativen
Schäden, zugesetzt. Das Gemisch wurde 20 min bei Raumtemperatur über Kopf
geschüttelt anschließend 5 min bei 10.000 g zentrifugiert und der Überstand

28 Material und Methoden
nochmals mit Phenol ohne SDS extrahiert. Zur Entfernung des Phenols wurde der
klare Überstand mit Chloroform ausgeschüttelt und erneut 5 min bei 10.000 g
zentrifugiert. Die DNA aus dem Überstand wurde mit dem 2,5-fachen Volumen
EtOH/125 mM Natriumacetat ausgefällt. Die DNA fiel nach ca. 1 h bei
Raumtemperatur aus. Falls dies nicht der Fall ist, kann das Gemisch über Nacht bei -
20°C gelagert werden. Die 10 min bei 10.000 g abzentrifugierte DNA wurde mit
eiskaltem 70%igem Ethanol gewaschen, erneut abzentrifugiert und in 1 ml BE1-
Puffer gelöst.
Die Konzentration der DNA wurde photometrisch am Biophotometer (1 OD260 nm =
50 µg DNA/ml) durchgeführt. Abschließend wurde das Verhältnis von
überspiralisierter zu ringoffener Form im Agarosegel (siehe 4.4) bestimmt, das
höchstens bei 20% ringoffener Form liegen sollte. Die gereinigte PM2-DNA wurde
aliquotiert und in BE1-Puffer bei -20°C gelagert.

Material und Methoden 29
4.4 DNA-Relaxationstest
Die Enzymaktivität des Fpg-Proteins wird anhand seiner Eigenschaft gemessen, in
oxidativ geschädigter PM2-DNA einen Einzelstrangbruch zu erzeugen. Durch den
Thiazinfarbstoff Methylenblau in Kombination mit sichtbarem Licht wird
Singulettsauerstoff erzeugt, der in isolierter PM2-DNA neben wenigen Strangbrüchen
fast ausschließlich Fpg-sensitive Stellen und darunter überwiegend 8-Oxoguanin
erzeugt (Floyd et al., 1989). Die Endonuklease Fpg ist in der Lage, in einer so
geschädigten DNA einzuschneiden. Die überspiralisierte PM2-DNA wird dadurch in
die realxierte, ringoffene Form überführt. Beide DNA-Formen können mittels einer
Agarose Gelelektophorese voneinander getrennt und nach Anfärbung mit
Ethidiumbromid am Geldokumentationsgerät quantifiziert werden. Aus dem
Verhältnis der beiden Banden lässt sich abschließend die DNA-Strangbruchrate und
die Fpg-Aktivität berechnen (Müller et al., 1990).
4.4.1 Versuchsdurchführung
PM2-DNA (20 µg/ml) wurde in einem 50 mM Natriumphosphatpuffer (pH 7,5) gelöst,
der zur Erzielung der geeigneten Ionenstärke zusätzlich 100 mM NaCl enthält. Die
Schadensinduktion erfolgte in einem abgedunkelten Raum mit Hilfe des Photo-
sensibilisators Methylenblau (10 µg/ml) und sichtbarem Licht (60 W, 50 cm
Entfernung = 5,25 J/m2/s). Nach der Schädigung wurde die DNA nach Zugabe eines
ethanolischen Fällungsreagenz (5% 2,5 M Natriumacetat, 95% Ethanol) 30 min bei
4°C im Dunkeln ausgefällt. Die 5 min bei 7000 g abzentrifugierte DNA wurde
vollständig vom Überstand abgetrennt. Noch vorhandene Reste des Überstandes
wurden sorgfältig mit einem Papiertuch entfernt. Anschließend wurde das erhaltene
DNA- Pellet in Enzympuffer (20 µg/ml) resuspendiert.
Parallel zur DNA-Schädigung wurde frisch bereitete Fpg-Lösung (1,3 µg/ml)
zusammen mit der zu untersuchenden Arsenverbindung in einem 100 µl-Ansatz für
30 min bei 37°C inkubiert (Vorinkubation). Es wurde für jede Konzentration der
Arsen- bzw. Zink-Verbindung oder Kombination jeweils ein Ansatz mit Fpg und ohne
Fpg mitgeführt, um zu unterscheiden, welche Strangbrüche durch die Fpg und
welche durch die Arsen-Verbindung selbst erzeugt wurden.

30 Material und Methoden
10 µl der oxidativ geschädigten DNA bzw. ungeschädigte Kontroll-DNA (300
ng/Ansatz) wurden zusammen mit 30 µl Vorinkubationslösung 30 min bei 37°C
inkubiert, so dass sich eine Fpg-Endkonzentration von 1 µg/ml ergibt
(Hauptinkubation). Während dieser Zeit detektierte die Fpg die zuvor erzeugten
oxidativen Basenschäden und setzte Einzelstrangbrüche. Die Reaktion wurde mit
7 µl Stoppreagenz beendet und die Proben auf ein 1%iges Agarose-Gel aufgetragen.
Nach 2,5 h bei 90 V waren die DNA-Formen von einander getrennt und das Gel
wurde für 30 min mit Ethidiumbromid (1,25 µg/ml) gefärbt. Abschließend wurde das
Gel für 10 min in Wasser entfärbt, und mit Hilfe eines HEROLAB Gel-
Dokumentationsgerätes und eines computergestützten Auswertungsprogramms
(Easy Win 32) wurden die Banden der beiden DNA-Formen bei einer Wellenlänge
von 254 nm densitometrisch detektiert. Aus dem Verhältnis der beiden Banden lies
sich eine Poisson-Verteilung der Strangbrüche nach Müller berechnen. Der
Korrekturfaktor von 1,4 gleicht die geringere Fluoreszenz der überspiralisierten
gegenüber der ringoffenen Form aus (Müller et al., 1990).
N = -ln[(1,4 x I)/(1,4 x I + II)]
N = Strangbruchrate/ PM2- Molekül
I = % der überspiralisierten Form
II = % der ringoffenen Form
Die Fpg-Enzymaktivität wurde berechnet als % der Enzymaktivität bezogen auf die
Kontrolle ohne Substanzzusatz, die gleich 100% gesetzt wurde. Aus der
Enzymaktivität lies sich eine eventuell vorhandene Reparaturhemmung des Fpg-
Proteins durch die eingesetzte Verbindung ableiten.

Material und Methoden 31
4.5 Zinkfreisetzung aus XPAzf
Die Reaktivität von Zink-gebundenen Cysteinen, wie sie in Zinkfingerstrukturen von
z.B. XPA vorkommen, gegenüber Metallverbindungen lässt sich durch die Messung
des freigesetzten Zinks ermitteln. Freie Zinkionen werden von dem Metallindikator 4-
2-Pyridylazoresorcinol mit hoher Aktivität gebunden. Der Indikator verändert sein
Absorptionsmaximum und der Zink-Farbstoff-Komplex kann bei 492 nm
photometrisch bestimmt werden (Abb. 6) (McCall und Fierke, 2000; Maret, 2002).
NN
N OH
ON
NN
O
OH
Zn2+
NN
N OH
ON
NN
O
OH
Zn2+
Abbildung 6: Komplex zwischen 4-2-Pyridylazoresorcinol und Zink (McCall und Fierke, 2000)
Für die Zinkfreisetzungsversuche wird ein kommerziell synthetisiertes Peptid
verwendet, das die Zinkfingerstruktur von humanem XPA repräsentiert (XPAzf) (101-
137). Es besitzt folgende Aminosäuresequenz, wobei das N-terminale Ende
acetyliert und das C-terminale Ende amidiert wurde, um zusätzliche
Metallbindungsstellen zu blockieren:
AcDYVICEECGKEFMDSYLMNHFDLPTCDNCRDADDKHKam
4.5.1 Versuchsdurchführung
XPAzf mit einer Molmasse von ca. 6800 g/mol wurde als erstes mit Zink abgesättigt,
um die Zinkfingerstruktur zu erzeugen. Dafür wurde eine 100 µM ZnCl2-Lösung in 20
mM Hepes/NaOH-Puffer hergestellt und pro 0,28 mg XPAzf 420 µl dieser Zink-
Lösung zugegeben. Ein Aliquot dieser XPAzf-Stammlösung wurde nun mit 4-2-
Pyridylazoresorcinol versetzt und optisch mit Vergleichslösungen verglichen, die 0, 2,

32 Material und Methoden
3 bzw. 4 µM Zink enthielten. Es wurde nun in kleinen Schritten so lange ZnCl2-
Lösung (100 µM) zugegeben bis ein Überschuss von Zink vorlag.
Für die Zinkfreisetzung wurde ein Mastermix hergestellt. Pro Probe wurden 53,1 µl
Hepes-Puffer sowie 13,4 µl XPAzf-Stammlösung vorgelegt und dann 0,672 µl der
Arsenverbindung zugegeben. Als Positivkontrolle wurde bei jedem Versuch 10 mM
H2O2 mitgeführt, das eine 100%ige Zinkfreisetzung bewirkt. Alle Inkubationslösungen
wurden in den Deckel des Reaktionsgefäßes pipettiert und dann gemeinsam
herunterzentrifugiert, um einen gleichzeitigen Beginn aller Reaktionen zu
gewährleisten. Nach einer Inkubationszeit von 30 min bei 37°C wurden 2,8 µl 4-2-
Pyridylazoresorcinol zugegeben. Aus einer Probe wurden 3 x 20 µl Lösung in eine
384-Loch-Mikrotiterplatte pipettiert und die Platte am Mikrotiterplattenlesegerät
(Tecan) bei 492 nm vermessen. Alle Werte wurden auf die Kontrolle mit 10 mM H2O2
bezogen, die definitionsgemäß 100% Zinkfreisetzung darstellt.
4.6 DNA-Bindungsaktivität von XPA mittels Gelshift
Mit Hilfe eines so genannten Gelshift-Assays ist es möglich die DNA-
Bindungsfähigkeit des XPA-Proteins nach Inkubation mit Arsen zu bestimmen. Man
macht sich hierbei die Eigenschaft zu nutzte, dass in einem nativen Polyacrylamidgel
ein Komplex aus einem Oligonukleotid und XPA langsamer wandert als freies
Oligonukleotid. Für die Experimente werden rekombinantes Maus-XPA und ein 70 bp
langes Oligonukleotid verwendet, das jeweils 5’ mit einem Digoxigeninderivat
endmarkiert war und folgende Sequenz besitzt (Blessing et al., 2004):
5’-ATA TGT GCA CAT GGC GCA CGT ATG TAT CTA TAG TCT
3’-TAT ACA CGT GTA CCG CGT GCA TAC ATA GAT ATC AGA
GCC ATC ACG CCA GTC AAT CGC TGT GGT ATA TGC A-3’
CGG TAG TGC GGT CAG TTA GCG ACA CCA TAT ACG T-5’

Material und Methoden 33
4.6.1 Versuchsdurchführung
Die beiden einzelsträngigen Oligonukleotide wuden zuerst getrennt von einander für
5 min und anschließend vereinigt für weitere 5 min bei 80°C aufgeschmolzen. Um ein
Annealing der beiden Stränge zu gewährleisten, wurden sie langsam bei
Raumtemperatur im Dunkeln ca. 15 min abgekühlt. Im Anschluss daran wurde der
Doppelstrang auf Eis mit einer Dosis von 18 KJ/m2 bei 254 nm (UVC) bestrahlt.
Durch die gewählte Sequenz werden hier vor allem 6-4-Photoprodukte induziert, an
die XPA bevorzugt bindet.
Pro Ansatz wurde nun 4 µl Gelshiftpuffer, 4,5 µl (Ansätze mit Arsen) bzw. 5,5 µl
(Ansätze ohne Arsen) bidestilliertes Wasser und 2 µl XPA (500 ng) vorgelegt. Als
nächstes wurde 1 µl Arsenlösung in den Deckel des Reaktionsgefäßes pipettiert, die
Lösung herunterzentrifugiert und 15 min bei Raumtemperatur inkubiert.
Anschließend wurden 2 µl Oligonukleotid (120 fmol/l) zupipettiert und die Proben
gemeinsam herunterzentrifugiert, damit die Bindung der Oligonukleotide an XPA
gleichzeitig startet. Nach einer Inkubationszeit von 30 min bei Raumtemperatur im
Dunkeln wurde jeder Probe 1,5 µl Ladepuffer (ohne Farbstoffe) zugesetzt. Die
Proben wurden mittels eines 5%igem nativem Polyacrylamidgel bei 90 V innerhalb
von ca. 75 min aufgetrennt. Zur Markierung der Lauffront wurden zusätzlich die
äußeren beiden Geltaschen mit Farbstoffen beladen.
Zur Vorbereitung des Semidry-Southern-Blots wurden zuerst eine Nylonmembran
(Hybond N+, Amersham) sowie 6 Blotpapiere (3MM-Whatman) einige Minuten in
0,5fachem TBE-Puffer äquilibriert. Zum Transfer der Oligonukleotide auf die
Membran wurde ein Stapel bestehend aus drei Blotpapieren, der Membran, dem Gel
und wiederum 3 Blotpapieren gebildet. Es ist darauf zu achten, dass sich keine
Luftblasen zwischen den einzelnen Schichten bilden. Zum Blotten der
Oligonukleotide wurde für 30 min eine Spannung von 20 V an die Apparatur angelegt
und die Oligonukleotide abschließend für 1,5 h bei 100°C im Trockenschrank fixiert.
Die Membran wurde für eine Minute in Maleinsäurepuffer und für 30 min in
Blockierlösung getaucht. Anschließend wurde die Membran mit einem Anti-
Digoxigenin-Antikörper, der mit Peroxidase gekoppelt war, in Blockierlösung 1:1000
verdünnt, in eine Folie eingeschweißt und 1 h bei Raumtemperatur unter Schütteln
inkubiert. Die Membran wurde für 15 min in Waschpuffer und weitere
15 min in Maleinsäurepuffer gewaschen. Für die Detektion wurde die Membran für

34 Material und Methoden
eine Minute mit 750 µl einer 1:1-Mischung der ECL-Lösungen inkubiert. Die
Detektionslösungen wurden von der Membran gestrichen und im Dunkeln ein
Autoradiographie-Film aufgelegt. Bei der Reaktion der Peroxidase mit ihrem Substrat
wird Licht emittiert, das den Fotofilm schwärzt (Chemilumineszenz). Zur Entwicklung
des Films wurde dieser nacheinander in Entwickler, Wasser, Fixierer und nochmals
in Wasser getaucht und getrocknet.
4.7 Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen
Der Nachweis der Poly(ADP-Ribosyl)ierung in HeLa S3 basiert auf einer in situ
immunflourimetrischen Detektion durch den primären monoklonalen Anti-PAR-
Antikörper 10H (Kawamitsu et al., 1984). Die Quantifizierung erfolgt am
Fluoreszenzmikroskop über die Detektion eines fluoreszenzmarkierten sekundären
Antikörpers. Die Poly(ADP-Ribosyl)ierung wird durch eine DNA-Strangbruchinduktion
mittels H2O2 aktiviert, so dass vorwiegend die an der DNA-Reparatur beteiligten
PARP-1 und PARP-2 für die Reaktion verantwortlich sind (Küpper et al., 1990; Burkle
et al., 1993; Hartwig et al., 2003). Der primäre Antikörper wurde uns
freundlicherweise von Prof. Alexander Bürkle (Universität Konstanz) zur Verfügung
gestellt.
4.7.1 Versuchsdurchführung
200.000 HeLa S3 Zellen wurden in Zellkulturschalen (40 x 10 mm) ausgestreut und
für ca. 2 Tage auf in den Schalen liegenden Deckgläschen (15 mm Durchmeser)
wachsen gelassen. Für die Versuche müssen die Zellen etwa halbkonfluent sein.
Dies wurde am Mikroskop überprüft wurde. Die Inkubation mit den
Arsenverbindungen erfolgte ca. 24 h nach dem Ausstreuen für weitere 18 h. Nach
Ende der Inkubationszeit wurden die Deckgläschen für 5 min bei 37°C mit 100 µM
H2O2 zur Induktion von DNA-Strangbrüchen inkubiert.
Anschließend wurden die Deckgläschen mit einer gebogenen Pinzette aus den
Schalen entfernt, kurz in eiskaltem PBS gewaschen und für mindestens 10 min in
eiskalter 10%iger (w/v) Trichloressigsäure fixiert. Anschließend wurden die
Deckgläschen für 5 min in auf -20°C vorgekühltem 70%igem Ethanol gewaschen,
gefolgt von zwei weiteren Waschschritten in 90% und 100% Ethanol. Die

Material und Methoden 35
Deckgläschen wurden auf Blotpapier gelegt und im Dunkeln an der Luft getrocknet.
Auf diese Weise fixiert sind die Präparate einige Tage lagerstabil.
Die Deckgläschen wurden zur Detektion mit den Antikörpern zuerst kurz in PBS
angefeuchtet und mit jeweils 50 µl der Antikörperlösung inkubiert (10H, 1:300
verdünnt in Blockierlösung, 5 µg/ml). Die Inkubation erfolgte in einer feuchten
Kammer im Dunkeln 30 min bei 37°C. Anschließend wurde überschüssige
Antikörperlösung 4 x 5 min mit PBS abgewaschen, wobei die Deckgläschen mit
einem starken Strahl aus der Spritzflasche abgespült wurden. Der sekundäre FITC-
gekoppelte Antikörper (1:50 in Blockierreagenz) wurde analog zum primären
Antikörper inkubiert. Hier ist darauf zu achten, dass bei gedämpftem Licht gearbeitet
wird, um ein Ausbleichen des Fluoreszenzfarbstoffs zu vermeiden. Nach weiteren
Waschschritten (s.o.) wurden die Deckgläschen mit der Oberseite nach unten auf mit
einem Tropfen Vectashield (Eindeckmedium mit DAPI) vorbereiteten Objekträgern
luftblasenfrei gelegt. DAPI bindet an DNA und färbt so die Zellkerne an. Vectashield-
Eindeckmedium bewahrt die Fluoreszenz des Objekts für mehrere Tage.
4.7.2 Auswertung am Fluoreszenzmikroskop
Die Auswertung erfolgte an einem Fluoreszenzmikroskop der Firma Nikon (Eclipse
E400). Dazu wurden mit einer SPOT RT (real time) Kamera (Visitron Systems,
Puchheim) sowohl für den DAPI- als auch für den FITC-Kanal pro Objektträger
mindestens 5 Bilder von verschiedenen Stellen in der Mitte des Objekts
aufgenommen. Über den DAPI-Kanal wurden nun mindestens 50 Zellen pro
Objektträger ausgewählt, die Regionen auf den jeweiligen FITC-Kanal übertragen
und die Fluoreszenzintensität mittels eines computergestützten
Auswertungsprogramms (MetaView, Version 4.1.5, Visitron Systems, Puchheim)
quantifiziert. Für die Auswertung wurden nur intakte Zellen ausgewählt, da z.B.
Zellen, die sich in der Mitose oder Apoptose befinden, ein unspezifisch hohes
Fluoreszenzsignal zeigen.

36 Material und Methoden
4.8 PARP-1 Proteingehalt
Zur Bestimmung des PARP-1 Proteingehalts nach Inkubation mit den
Arsenverbindungen werden zunächst die Proteine aus den Zellen isoliert. Die
Proteinextrakte werden anschließend denaturiert und über ein 10%iges
Polyacrylamidgel aufgetrennt. Nach dem Transfer der Proteine auf eine PVDF-
Membran (Western Blot) kann PARP-1 durch Inkubation mit dem primären
Antikörper CII-10 und einem sekundären Peroxidase-gekoppelten Antikörper über
Chemilumineszenz detektiert werden. CII-10 erkennt neben der 116 kDa großen
vollständigen PARP-1 auch das apoptosetypische 85 kDa Fragment.
4.8.1 Versuchsdurchführung
In eine Zellkulturschale (60 x 15 mm) wurden 400.000 HeLa S3-Zellen ausgestreut
und für ca. 24 Stunden anwachsen gelassen. Die Arsenverbindungen wurden für 18
Stunden direkt in das Kulturmedium inkubiert. Alle folgenden Schritte bis zur
Denaturierung der Proteine wurden auf Eis durchgeführt um eine zu starke
Automodifizierung der PARP-1 zu verhindern. Das gebrauchte Kulturmedium wurde
nach Ablauf der Inkubationszeit in ein 15 ml Zentrifugenröhrchen gegeben, die Zellen
mit Trypsin inkubiert und mit dem gebrauchten Medium von der Schale abgelöst.
Dies ist wichtig, da auf diese Weise auch sich vom Boden der Zellkulturschale schon
abgelöste Zellen, zur Ermittlung des apoptotischen Fragments der PARP-1 miterfasst
werden. Die Zellen wurden 5 min bei 1000 g und 4°C abzentrifugiert und das Medium
abgesaugt. Das Zellpellet wurde mit eiskaltem PBS resuspendiert und die Zellzahl
am automatischen Zellzählgerät (CASY) bestimmt. Die Zellen wurden ein weiteres
Mal 5 min bei 1000 g bei 4°C abzentrifugiert und der Überstand vollständig
abgesaugt. Das Zellpellet wurde anschließend in 50 µl Proteaseinhibitorlösung
resuspendiert und in ein Mikroreaktionsgefäß überführt. Sofort wurden 100 µl 95°C
heißer 1,5x Probenpuffer zugegeben und die Proteine 5 min bei 95°C denaturiert.
Der noch heiße und sehr viskose Zellextrakt wurde nun mehrfach mit einer Pipette
auf- und abgezogen um die DNA zu scheren und so eine pipettierbare Lösung zu
erhalten. Die Proteinextrakte können sofort auf ein Polyacrylamidgel aufgetragen
oder bei -20°C gelagert werden.

Material und Methoden 37
Die Extrakte (100.000 Zellen pro Bahn) wurden zusammen mit einem
Molekulargewichtsmarker (Peqgold, Peqlab) und isolierter PARP-1 (50 ng, Isolierung
siehe 4.9) auf ein denaturierendes SDS-PAGE Gel aufgetragen. Zur Detektion der
116 kDa- und 85 kDa-Banden wurde ein 10%iges Trenngel und ein 3%iges
Sammelgel verwendet. Die Elekrophorese erfolgte bei 80 V im Sammelgel und zur
Trennung der Proteine bei 100 V im Trenngel. Nach Beendigung der Elektrophorese
wurde das Gel für 20 min in eiskaltem Transferpuffer und eine PVDF-Membran
(Hybond P, GE Healthcare) zunächst 10 sec in Methanol, 5 min in Wasser und
abschießend 10 min in eiskaltem Transferpuffer äquilibriert. Der Aufbau des
Blotstapels erfolgte wie in 4.6.1 beschrieben. Der Transfer der Proteine wurde in der
Semidry-Blot-Apparatur bei 0,8 A/cm2 für eine Stunde durchgeführt.
Die Membran wurde nach Ende des Transfers kurz in PBS SDS-frei gewaschen und
zur Absättigung von unspezifischen Bindungsstellen für eine Stunde in
Magermilchpulver (5% in PBS-T) geschwenkt. Anschließend wurde die Membran
über Nacht bei 4°C mit dem primären Antikörper (CII-10, Alexis, 1:2.500 verdünnt in
Blockierlösung) in Folie eingeschweißt inkubiert. Am nächsten Tag wurde die
Membran 3 x 10 min in PBS-T gewaschen und für eine weitere Stunde mit dem
sekundären Peroxidase-gekoppelten Antikörper (Anti-Maus, Santa Cruz, 1:2.500
verdünnt in Blockierlösung) bei Raumtemperatur inkubiert. Die Membran wurde
wiederum 3 x 10 in PBS-T gewaschen, kurz in PBS getaucht und mit ca. 800 µl einer
1:1-Mischung von ECL-Advance Lösungen (Amersham) für 5 min inkubiert. Die
Signale wurden über ein Chemilumineszenz-Geldokumentationsgerät (LAS 3000,
Fuji) aufgenommen und die Banden über eine computergestützte
Auswertungssoftware (AIDA, Raytest) quantifiziert.
Die Membran wurde für mindestens eine Stunde in PBS gewaschen, mit primärem
Anti-Aktin-Antikörper (Kaninchen) für eine weitere Stunde inkubiert und wie oben
beschrieben gewaschen. Nach der Inkubation mit sekundärem Anti-Kaninchen-
Antikörper (HRP-markiert) und Waschschritten (s.o.) wurde Aktin als Ladekontrolle
wie oben beschrieben mittels Chemilumineszenzdetektionsgerät nachgewiesen.

38 Material und Methoden
4.9 Isolierung von PARP-1
Humane PARP-1 wird aus mit rekombinanten Bacculoviren infizierten Insektenzellen
(Sf9) isoliert. Mit Hilfe eines Baculo-Gold-Kits wurde ein rekombinanter Bacculovirus-
PARP-1-Klon erzeugt, der Sf9-Zellen infiziert, welche dadurch zu über 50% humane
PARP-1 exprimieren. Bei der Aufreinigung der Zellextrakte macht man sich zu Nutze,
dass PARP-1 aufgrund seiner hohen Affinität zur DNA an eine mit doppelsträngiger
DNA modifizierten Cellulosesäule bindet. Die Arbeiten wurden mit Unterstützung der
Arbeitsgruppe von Prof. Bürkle an der Universität Konstanz modifiziert nach der
Methode von (Beneke et al., 2000) durchgeführt. Recombinante Bacculovieren sowie
Sf9-Zellen wurden dort freundlicherweise zur Verfügung gestellt.
4.9.1 Infektion der Sf9-Zellen
2 x 107 Sf9 Zellen wurden in Zellkulturschalen ausgesät und nach 15minütigem
Anheften mit einer moi ≥ 3 (Verhältnis Viruszahl zu Zellzahl) infiziert. Die Schalen
wurden über 1 Stunde alle 20 min kurz geschwenkt, nachfolgend die Zellen
abgespült in 200 ml Aliquots, in 500 ml Erlenmeyerkolben überführt, mit 0,2%
Pluronsäure F-68 versetzt und 45 h in einem 26°C Schüttelwasserbad (90 rpm, 2 cm
Auslenkung) inkubiert. Anschließend wurden die Zellen 10 min bei Raumtemperatur
und 140 g zentrifugiert und das Zellsediment bei -70°C eingefroren (Miranda et al.,
1997).
4.9.2 Aufreinigung rekombinanter PARP-1
108 Zellen wurden in 7,5 ml Puffer A resuspendiert und Tween 20, Nonidet P-40
sowie NaCl zugegeben, so dass jeweils eine Enkonzentration von 0,2% bzw. 0,5 M
(NaCl) erreicht wurde. Diese Lösung wurde 20 min bei 4°C auf einem
Überkopfmischer inkubiert und nachfolgend 20 min bei 20.000 g und 4°C
zentrifugiert. Die DNA im Überstand wurde durch Zugabe von Protaminsulfat mit
einer Endkonzentration von 1 mg/ml ausgefällt und durch Zentrifugation wie oben
sedimentiert. Anschließend wurde eine fraktionierte Ammoniumsulfat-Fällung der
Proteine durchgeführt in Schritten von 30%iger (NH4)2SO4-Sättigung und 70%iger
(NH4)2SO4-Sättigung, wobei das Ammoniumsulfat jeweils langsam zugegeben

Material und Methoden 39
wurde. Nach einer Zentrifugation für 20 min bei 20.000 g und 4°C wurde im ersten
Schritt der Überstand weiterverwendet, im zweiten Schritt das Sediment, welches in
3 ml Puffer B resuspendiert wurde. Diese Lösung wurde nur kurzfristig bei 4°C
gelagert und anschließend zur einer Gelfiltration eingesetzt (Giner et al., 1992).
1 g Sephadex G100-50 superfine (Sigma) wurde mit 20 ml Puffer C versetzt und
30 min bei Raumtemperatur zur Quellung belassen. Eine 60 ml Säule wurde mit
Glaswatte gestopft und mit der Sephadex-Suspension beschickt. Nach Abtropfen der
Flüssigkeit bei 4°C wurde die Säule mit 3 ml Puffer B-Resuspendat beladen. Der
Durchfluss wurde verworfen und anschließend 30 ml Puffer D auf die Säule
gegeben. Die Eluat-ml 2-23 wurden aufgefangen und nachfolgend auf eine
dsDNA(Doppelstrang)-Cellulose-Säule gegeben.
0,3 g dsDNA-Cellulose wurden in 15 ml Puffer BII inklusive 0,1 M KCL
aufgenommen. Die Suspension wurde 30 min bei Raumtemperatur unter Schütteln
inkubiert und anschließend 5 min bei RT und 1100 g zentrifugiert. Das Sediment
wurde mit 7 ml Puffer BII aufgenommen, für weitere 30 min unter Schütteln inkubiert
und das Säulenmaterial nochmals wie oben beschrieben abzentrifugiert. Das
Sediment wurde wiederum in 6 ml BII-Puffer resuspendiert und auf eine dsDNA-
Cellulose-Säule in 10 ml Polyprep-Säulen gegeben, so dass sich nach dem
Abtropfen des Puffers ein Säulenvolumen von 2 ml ergab. Die weiteren
Arbeitsschritte wurden alle bei 4°C durchgeführt.
Je 4 ml des Sephadex-Eluats wurden auf die dsDNA-Säule gegeben und nach
Durchfluss die Säule mit 10 ml Puffer D gespült. Danach wurde die Säule mit 2 ml
Puffer D/0,28 M KCl beladen und anschließend mit 500 µl Puffer D/2,8 M KCl
gewaschen. Nach dieser Präelution wurde die an der DNA gebundene PARP-1 mit
1,5 ml Puffer D/2,8 M KCl von der Säule eluiert und sofort mit 20% Glycerin versetzt
(Kofler et al., 1993).
Die PARP-1 Proben wurden bei 4°C in Kollodium-Hülsen über Nacht gegen
PBS/20% Glycerin dialysiert. Die Bestimmung der Proteinkonzentration erfolgte
abschließend durch die Messung der Extinktion bei 235 und 280 nm. Die Differenz
dieser beiden Werte dividiert durch den Faktor 2,51 ergab dabei die Konzentration in
mg/ml. Als Vergleich und Kontrolle dienten entsprechende Lösungen von BSA in
PBS/20% Glycerin (Whitaker und Granum, 1980).

40 Material und Methoden
4.10 Poly(ADP-Ribosyl)ierung isolierter PARP-1
Isolierte PARP-1 modifiziert sich selbst und zugesetzte Histone, wenn sie durch ein
kurzes Oligonukleotid aktiviert wird. Nach Abstoppen der Reaktion kann das
Gemisch mit Hilfe einer Slot-Blot-Apparatur auf eine Membran aufgesaugt werden.
Die poly(ADP-ribosyl)ierten Proteine werden nachfolgend mit dem gegen Poly(ADP-
Ribose) gerichteten Antikörper 10H und einem sekundären Peroxidase-gekoppelten
Antikörper mittels Chemilumineszenz detektiert.
4.10.1 Versuchsdurchführung
27 µl isolierte PARP-1 (0,5 µg/ml) wurden in Reaktionsgefäße vorgelegt und 3 µl
Arsenlösung in den Deckel pipettiert. Nachdem die Proben gemeinsam
herunterzentrifugiert wurden, wurde die PARP-1 30 min bei 37°C vorinkubiert. In der
Zwischenzeit wurde ein Mastermix bereitet, bestehend (pro Probe) aus 10 µl
Reaktionspuffer (10x), 20 µl Histone Typ IIa (0,1 mg/ml), 5 µl Oligonukleotid (8mer,
GGAATTCC, 1mg/ml), 5 µl DTT (20 mM), 10 µl ß-NAD+ (2mM) und 45 µl H2O.
Histone, DTT und ß-NAD+ wurden frisch angesetzt. Aus dem Vorinkubationsansatz
wurden 3 x 5 µl in den Deckel eines mit Mastermix vorgelegten Reaktionsgefäßes
pipettiert, so dass eine Dreifachbestimmung entsteht. Es wurde wiederum
gemeinsam herunterzentrifugiert und die PARP-1-Reaktion für 10 min bei 37°C
ablaufen gelassen. Die Reaktion wird durch Zugabe von 200 µl eiskaltem PARP
Inhibitor 3-Aminobenzamid (12,5 mM) gestoppt. Die Proben wurden nach ca. 20 min
auf Eis mit einer Slot-Blot-Apparatur (Roth) auf eine Membran (Hybond N+,
Amersham) aufgesaugt. Mit Hilfe einer Multisteppipette wurden anschließend 150 µl
eiskalte 10%ige TCA/2%ige Na4P2O7 in jedes Loch pipettiert und mit 200 µl 70%igem
Ethanol überschichtet. Die Mischung wurde bis zur Trockne abgesaugt und die
Membran für eine Stunde im Trockenschrank bei 90°C fixiert. Die Membran wurde im
Anschluss eine Stunde mit Blockierlösung (5% Magermilchpulver in PBS-T)
abgesättigt und eine weitere Stunde mit primärem Antikörper (10 H, 1:300 verdünnt
in Blockierlösung) gegen Poly(ADP-Ribose) bei Raumtemperatur inkubiert.
Anschließend wurde die Membran 4 x 5 min mit PBS-T gewaschen und für eine
Stunde mit einem sekundären Peroxidase-gekoppelten Antikörper (Anti-Maus, Santa
Cruz) bei Raumtemperatur behandelt. Nach weiteren vier fünfminütigen

Material und Methoden 41
Waschschritten mit PBS-T wurde die Membran kurz in PBS getaucht und wie unter
4.8.1 beschrieben mit ECL detektiert.

42 Ergebnisse und Diskussion
5. Ergebnisse und Diskussion
Im folgenden Abschnitt der Arbeit werden die Effekte von Arsenit und seinen
methylierten Metaboliten auf die Zinkfingerproteine Fpg, XPA und PARP-1
nacheinander beschrieben. Zunächst wird jedoch auf die eingesetzten
Arsenverbindungen und deren zytotoxische Wirkungen auf HeLa S3 Zellen
eingegangen.
5.1 Eingesetzte Arsenverbindungen
Für die Untersuchungen wurden neben anorganischem Arsenit die fünfwertigen
Metabolite MMA(V) und DMA(V) (Kakodylsäure) eingesetzt. Die beiden fünfwertigen
Metabolite sind im Gegensatz zu den dreiwertigen Metaboliten kommerziell
erhältlich. Zur Untersuchung der dreiwertigen Metabolite wurden Methyloxoarsin,
Diiodomethylarsin und Iododimethylarsin eingesetzt. Für die beiden
monomethylierten Verbindungen konnte von Petrick et al. (2001) gezeigt werden,
dass sie in wässriger Lösung MMA(III) (CH3AsIII(OH)2) bilden (Petrick et al., 2001).
Man geht davon aus, dass aus Iododimethylarsin unter ähnlichen Bedingungen
DMA(III) ((CH3)2AsIIIOH) entsteht (Mass et al., 2001). Um auszuschließen, dass
mögliche Effekte auf das Iod in den Verbindungen zurückzuführen sind, wurden für
Monomethyloxoarsin und Diiodomethylarsin vergleichende Untersuchungen zum
Einfluss auf Fpg und zur Zinkfreisetztung aus XPAzf durchgeführt. Es konnte hierbei
kein Unterschied zwischen der iodhaltigen und der nicht-iodhaltigen Verbindung
festgestellt werden (Daten siehe Anhang). Zudem konnte in unserer Arbeitsgruppe
gezeigt werden, dass auch im zellulären System bezogen auf die Zytotoxizität sowie
auf die Benzo[a]pyrendiolepoxid-induzierte DNA-Adduktbildung und Reparatur in
A549 Zellen kein Unterschied zwischen den beiden Verbindungen zu sehen ist
(Schwerdtle et al., 2003a). Für die weiteren Versuche zur Poly(ADP-Ribosyl)ierung
wurde auf vergleichende Untersuchungen verzichtet und nur Methyloxoarsin
eingesetzt. Alle in der Arbeit dargestellten Ergebnisse beziehen sich daher auf
Methyloxoarsin, das im Weiteren mit MMA(III) abgekürzt wird. Man kann zudem
davon ausgehen, dass auch im Falle der dimethylierten Verbindung

Ergebnisse und Diskussion 43
Iododimethylarsin, die als DMA(III) bezeichet wird, Iod keine Effekte auf die
untersuchten Parameter ausübt.

44 Ergebnisse und Diskussion
5.2 Zytotoxizität
Für die Untersuchungen im zellulären System wurde die humane
Zervixkarzinomzelllinie HeLa S3 eingesetzt, die nur eine geringe
Methylierungskapazität für Arsenverbindungen besitzt (Peel et al., 1991). Die
Ergebnisse für die Zytotoxizität und die Poly(ADP-Ribosyl)ierung sowie der PARP-1
Proteingehalt können daher für die einzelnen Arsenverbindungen unabhängig
voneinander betrachtet werden. Die verschiedenen Arsenverbindungen wurden bei
allen Zellkultur-Versuchen 18 Stunden inkubiert, da für Arsenit in dieser Zeit eine
maximale intrazelluläre Konzentration erreicht wird (Hartwig et al., 1997).
Zur Ermittlung der Zytotoxizität von Arsenit, MMA(III), DMA(III), MMA(V) und DMA(V)
wurde sowohl ihr Einfluss auf die Zellzahl wie auch auf die Koloniebildungsfähigkeit
untersucht. Bezogen auf beide Parameter zeigten die dreiwertigen Verbindungen um
ein Vielfaches höhere Effekte als die fünfwertigen Metabolite. Die beiden
dreiwertigen Metabolite waren zudem stärker zytotoxisch als anorganisches Arsenit.
So reduzierten MMA(III) und DMA(III) bei einer Konzentration von 5 µM die Zellzahl
auf 33% bzw. 34%, während Arsenit bei der gleichen Konzentration nur eine
Reduzierung auf 43% bewirkt. Die fünfwertigen Metabolite MMA(V) und DMA(V)
zeigten erst in 100fach höherer Konzentration von 500 µM einen Effekt auf die
Zellzahl mit 69% bzw. 45% verbliebenen Zellen (Abbildung 7).
Im Vergleich zu den Effekten auf die Zellzahl zeigten die dreiwertigen Verbindungen
stärkere Effekte auf die Koloniebildungsfähigkeit. Hier wird der Unterschied zwischen
Arsenit und den dreiwertigen Metaboliten besonders deutlich. Während MMA(III) und
DMA(III) die Koloniebildungsfähigkeit bei 5 µM auf 20% bzw. 2% reduzierten,
erzeugte Arsenit nur einen Effekt von 53% bei 20 µM. Für die beiden fünfwertigen
Verbindungen konnten bis in Konzentrationen von 500 µM nur geringe Effekte auf die
Koloniebildungsfähigkeit festgestellt werden (Abbildung 8). Für die beiden
Zytotoxizitätsparameter Zellzahl und Koloniebildungsfähigkeit ergibt sich in HeLa S3
Zellen dann folgende Reihenfolge der Reaktivität: MMA(V) < DMA(V) << Arsenit <
DMA(III) < MMA(III). Die Unterschiede bezüglich der Zytotoxizität der dreiwertigen
Verbindungen wurden auch anhand morphologischer Veränderungen der Zellen
deutlich. Lichtmikroskopische Aufnahmen zeigten, dass sich die Zellen im Falle von

Ergebnisse und Diskussion 45
10
100
0 100 200 300 400 500
Arsenverbindung [µM]
Zellz
ahl (
% d
er K
ontr
olle
)
Arsenit
MMA(III)DMA(III)
MMA(V)
DMA(V)10
100
0 2 4 6 8 10
10
100
0 100 200 300 400 500
Arsenverbindung [µM]
Zellz
ahl (
% d
er K
ontr
olle
)
ArsenitArsenit
MMA(III)MMA(III)DMA(III)DMA(III)
MMA(V)MMA(V)
DMA(V)DMA(V)10
100
0 2 4 6 8 10
Abbildung 7: Einfluss der Arsenverbindungen auf die Zellzahl nach 18 h Inkubation von
HeLa S3 Zellen. Dargestellt sind Mittelwerte aus mindestens 4 Bestimmungen ± SD.
10
100
0 100 200 300 400 500
Arsenverbindung [µM]
Kol
onie
bild
ungs
fähi
gkei
t (%
der
Kon
trol
le)
ArsenitMMA(III)DMA(III)MMA(V)DMA(V)
1
10
100
0 2 4 6 8 10
10
100
0 100 200 300 400 500
Arsenverbindung [µM]
Kol
onie
bild
ungs
fähi
gkei
t (%
der
Kon
trol
le)
ArsenitArsenitMMA(III)MMA(III)DMA(III)DMA(III)MMA(V)MMA(V)DMA(V)DMA(V)
1
10
100
0 2 4 6 8 10
Abbildung 8: Einfluss der Arsenverbindungen auf die Koloniebildungsfähigkeit nach 18 h
Inkubation von HeLa S3 Zellen. Gezeigt sind Mittelwerte aus mindestens 12 Bestimmungen ± SD.

46 Ergebnisse und Diskussion
Arsenit und MMA(III) in zytotoxischen Konzentrationen abrundeten und auch vom
Boden der Zellkulturschale ablösten, während bei DMA(III) die Umrisse der Zellen
eine Art „Verfransung“ aufwiesen und die Zellen sich im Vergleich zu den anderen
Verbindungen nicht so leicht vom Boden der Schale lösten (siehe Abbildung 9).
20 µM MMA(III) 7,5 µM DMA(III)
Kontrollzellen
15 µm
10 µM Arsenit
20 µM MMA(III) 7,5 µM DMA(III)
Kontrollzellen
15 µm15 µm
10 µM Arsenit
Abbildung 9: Lichtmikroskopische Aufnahmen von HeLa S3 Zellen nach 3 h Inkubation mit den Arsenverbindungen.
Die viel stärkeren zytotoxischen Effekte der dreiwertigen Arsenspezies gegenüber
den fünfwertigen Verbindungen sind in der Literatur oft beschrieben worden. So
konnte sowohl in unserem Labor in menschlichen A549-Lungenzellen (Schwerdtle et
al., 2003b), in menschlichen Rattenhepatozyten (Petrick et al., 2000), primären
Rattenhepatozyten und verschiedenen menschlichen Zellen (Styblo et al., 2000),
menschlichen Keratinozyten (Vega et al., 2001) sowie menschlichen NB4-Zellen
(Chen et al., 2003) eine erhöhte Zytotoxizität der dreiwertigen gegenüber den
fünfwertigen Spezies festgestellt werden. In diesen Studien besitzen die dreiwertigen
Metabolite mindestens das gleiche zytotoxische Potential wie Arsenit. In den meisten
Fällen zeigt vor allem MMA(III) stärkere zelltoxische Effekte als das anorganische
Arsenit. Es ist jedoch schwierig eine generelle Reihenfolge für die Zytotoxizität
bezogen auf den Methylierungsgrad festzulegen. Die Ergebnisse variieren von

Ergebnisse und Diskussion 47
Zelllinie zu Zelllinie und sind von Faktoren wie z.B. dem intrazellulären Glutathion
(GSH)-Gehalt abhängig.
Die geringere Zytotoxizität der fünfwertigen Metabolite gegenüber den dreiwertigen
Verbindungen lässt sich durch deren viel schlechtere Aufnahme in die Zellen
erklären. Während in Rattenhepatozyten nach 2 h Inkubation 22% Arsenit und nach
8 h Inkubation 50% MMA(III) aufgenommmen werden, sind in nach 24 h Inkubation
mit DMA(V) weniger als 5% intrazellulär nachzuweisen (Styblo et al., 2000). Dies
kann einerseits an einer verschlechterten Aufnahme aus dem Zellkulturmedium
aufgrund von Konkurrenzreaktionen mit extrazellulärem Phosphat (Huang und Lee,
1996) oder aber auch an den unterschiedlichen Ladungsverhältnissen der
Arsenverbindungen liegen. So liegt die dreiwertige Verbindung Arsenit unter
physiologischen Bedingungen zu 99% ungeladen vor, während MMA(V) und DMA(V)
vorwiegend geladen sind. Diese Ladung könnte einerseits die Aufnahme
verlangsamen, jedoch auch dafür sorgen, dass die fünfwertigen Metabolite schneller
aus den Zellen ausgeschleust werden. Buchet et al. (1981) konnten zeigen, dass
nach oraler Gabe von MMA(V), DMA(V) und Arsenit jeweils 78%, 75% bzw. 45% des
Arsens nach 4 h im Urin gefunden wurden. In metabolisch kompetenten Zellen wird
der gebildete Hauptmetabolit DMA(V) schnell in das Medium abgegeben (Styblo et
al., 2000). Es ergiebt sich folgende Reihenfolge in der Menge der aufgenommenen
Verbindungen: DMA(III) > MMA(III) > Arsenit > Arsenat > MMA(V), DMA(V) (Dopp et
al., 2004; Dopp et al., 2005). Hughes et al. (2005) zeigten in Mäuseversuchen für
MMA(V) und MMA(III) eine schnelle Aufnahme und Verteilung in allen Organen,
wobei MMA(III) schneller aufgenommen wurde als MMA(V) (Hughes et al., 2005).
Die Aufnahme der dreiwetigen Verbindungen Arsenit und MMA(III) erfolgt vermutlich
u.a. durch Aquaglyceroporine, die normalerweise für den Transport von Glycerin
verantwortlich sind (Liu et al., 2006).
Die stärkeren zytotoxischen Effekte der dreiwertigen Metabolite gegenüber
anorganischem Arsenit sind noch nicht geklärt. Es ist jedoch zu vermuten, dass die
Metabolite eine erhöhte Reaktivität gegenüber Makromolekülen der Zelle, und vor
allem Thiolgruppen von Proteinen haben, die eine wichtige Rolle bei der Einleitung
zytotoxischer Mechanismen der Zelle spielen. Auf die Reaktivität der dreiwertigen
methylierten Metabolite gegenüber SH-haltigen Proteinen wird in den nächsten
Kapiteln dieser Arbeit genauer eingegangen.

48 Ergebnisse und Diskussion
5.3 Einfluss auf die Aktivität von Fpg
Zur Untersuchung des Einflusses von Arsenverbindungen auf Zinkfingerproteine, die
an der DNA-Reparatur beteiligt sind, wurde als erstes die Fpg ausgewählt. Die
Aktivität isolierter Fpg wurde mit Hilfe eines DNA-Relaxationstests bestimmt.
Zunächst wurde für den Versuch PM2-DNA aus mit dem Bakteriophagen PM2
infizierten Bakterienzellen gewonnen. Zum Abschluss der Isolierung wurde die
Ausbeute und die Qualität der frisch isolierten DNA bestimmt (Daten siehe Anhang).
Für die Aktivitätsbestimmung wurde Fpg zunächst mit den Arsenverbindungen
vorinkubiert und anschließend mit oxidativ geschädigter DNA koinkubiert. Fpg setzt
in der geschädigten DNA einen Strangbruch, die daraufhin aus einer
superspiralisierten in eine offen ringförmige Form übergeht. Abbildung 10 zeigt
beispielhaft für eine Inkubation mit DMA(III), inwieweit Fpg noch in der Lage ist in
PM2-DNA einzuschneiden. So nimmt mit steigenden Konzentrationen der Anteil an
eingeschnittener, offen ringförmiger DNA ab, während die Intensität der Bande für
superspiralisierte DNA zunimmt.
0 50 100 200 400
DMA(III) [µM]
offen ringförmige DNA
superspiralisierte DNA
lineare DNA
0 50 100 200 400
DMA(III) [µM]
offen ringförmige DNA
superspiralisierte DNA
lineare DNA
Abbildung 10: Einfluss von DMA(III) auf die Aktivität von isolierter Fpg. Isolierte Fpg wurde
30 min bei 37°C mit den Arsenverbindungen inkubiert und die Aktivität gegenüber oxidativ
geschädigter DNA mittels DNA-Relaxationstest bestimmt. Dargestellt ist ein repräsentatives Agarose-
Gel.
Aus dem Verhältnis der beiden Banden lässt sich nun der Einfluss der
Arsenverbindungen auf die Aktivität isolierter Fpg berechnen. Dreiwertiges
anorganisches Arsenit und seine fünfwertigen methylierten Metabolite MMA(V) und
DMA(V) zeigten bis zu Konzentrationen von 10 mM keinen Effekt, wohingegen
MMA(III) und DMA(III) eine konzentrationsabhängige und signifikante Hemmung der
Fpg verursachten. So hemmte DMA(III) Fpg bereits ab 100 µM um etwa 20% und

Ergebnisse und Diskussion 49
reduzierte die Aktivität bei 1 mM auf ca. 10%, während MMA(III) erst bei 2 mM eine
Hemmung auf 20% verursachte (Abbbildung 11).
0
20
40
60
80
100
120
140
0 1 2 3 4 5
Arsenverbindung [mM]
Fpg
Akt
ivitä
t (%
der
Kon
trol
le)
ArsenitMMA(III)DMA(III)
MMA(V)DMA(V)
0
20
40
60
80
100
120
140
0 1 2 3 4 5
Arsenverbindung [mM]
Fpg
Akt
ivitä
t (%
der
Kon
trol
le)
ArsenitMMA(III)DMA(III)
MMA(V)DMA(V)
Abbildung 11: Einfluss der Arsenverbindungen auf die Aktivität von isolierter Fpg. Isolierte Fpg
wurde 30 min bei 37°C mit den Arsenverbindungen inkubiert und die Aktivität gegenüber oxidativ
geschädigter DNA mittels DNA-Relaxationstest bestimmt. Dargestellt sind Mittelwerte aus mindestens
5 Bestimmungen ± SD.
Als ein möglicher molekularer Ansatzpunkt zur Erklärung der Fpg-Hemmung durch
die dreiwertigen Metabolite gilt deren hohe Affinität zu Thiolgruppen. Die
Zinkfingerstruktur, deren Funktion essentiell für die Aktivität der Fpg ist, könnte daher
durch die Arsenverbindungen zerstört werden. Um eine Beteiligung dieser Domäne
nachzuweisen, wurden Koinkubationsversuche von DMA(III) und ZnCl2 durchgeführt.
Wie in Abbildung 12 zu sehen ist, war ZnCl2 in der Lage die Fpg-Hemmung durch
DMA(III) konzentrationsabhängig aufzuheben. So wurde die Hemmung schon ab
kleiner als äquimolaren Konzentrationen teilweise aufgehoben und konnte bei einer
ZnCl2 Konzentration von 1000 µM auf 45 % reduziert werden, wobei zu beachten ist,
dass 1000 µM ZnCl2 alleine schon eine Fpg-Hemmung um 20% verursachten. Für
MMA(III) konnten leider keine Koinkubationsversuche durchgeführt werden, da die
nötigen höheren ZnCl2-Konzentrationen von 2-10 mM selbst schon eine zu starke
Fpg-Hemmung hervorriefen.

50 Ergebnisse und Diskussion
0
10
20
30
40
50
60
70
80
90
400 400 400 400
Fpg-
Akt
ivitä
t(%
der
Kon
trol
le)
0DMA(III) [µM]
0 40 400 1000 1000ZnCl2 [µM]
0
10
20
30
40
50
60
70
80
90
400 400 400 400
Fpg-
Akt
ivitä
t(%
der
Kon
trol
le)
0DMA(III) [µM]
0 40 400 1000 1000ZnCl2 [µM]
Abbildung 12: Einfluss von ZnCl2 auf die Fpg-Hemmung durch DMA(III). Isolierte Fpg wurde
30 min bei 37°C mit den Arsenverbindungen und ZnCl2 inkubiert und die Aktivität gegenüber oxidativ
geschädigter DNA mittels DNA-Relaxationstest bestimmt. Dargestellt sind Mittelwerte aus mindestens
5 Bestimmungen + SD.
Es ist wahrscheinlich, dass die Hemmung der Fpg durch die dreiwertigen
methylierten Arsenmetabolite mit einer Interaktion mit dem Zinkfingermotiv des
Enzyms zusammenhängt. Durch Bindung der Metabolite an die Cysteine des
Zinkfingers könnte es zu einer Freisetzung von Zink und damit zur Zerstörung der
Domäne kommen. So konnten O’Connor et al. (1993) zeigen, dass der
Funktionsverlust von einem Cystein-Rest des Zinkfingers zu einer Hemmung des
ganzen Enzyms führt. Die Koinkubationsversuche mit ZnCl2 haben einen weiteren
Hinweis auf den Zinkfinger als molekularen Angriffspunkt geliefert, jedoch weisen sie
auch darauf hin, dass es unter diesen Bedingungen wohl nicht zu einer kovalenten
Bindung der Metabolite an den Zinkfinger kommt, die durch eine Zinkzugabe nicht
rückgängig gemacht werden könnte. Der fehlende Effekt von anorganischem Arsenit,
der auch schon von Asmuss et al. (2000) gezeigt werden konnte, ist wohl auf die
geringere Thiolreaktivität im Vergleich zu MMA(III) und DMA(III) zurückzuführen, die
sich auch in anderen Systemen zeigt. Zudem ist es denkbar, dass die

Ergebnisse und Diskussion 51
Zinkfingerstruktur für Arsenit im Vergleich zu den methylierten Metaboliten eine
unterschiedliche Zugänglichkeit zeigt (siehe folgende Kapitel und
zusammenfassende Diskussion). Mit diesen Versuchen ist es zum ersten Mal
gelungen eine Hemmung eines isolierten DNA-Reparaturproteins, wenn auch in
relativ hohen Konzentrationen, durch die dreiwertigen methylierten Arsenmetabolite
zu zeigen. Eine Hemmung von Fpg bewirkt in E. coli, vor allem aufgrund der
gestörten Reparatur des prämutagenen DNA-Schadens 8-Oxoguanin, eine Zunahme
von G → T Transversionen und damit von Mutationen (Cabrera et al., 1988). Eine
Übertragung dieser Ergebnisse von E. coli auf den Menschen ist schwierig. Die
Entdeckung der menschlichen Homologen von Fpg, den DNA-Glykosylasen Neil I,II
und III gibt jedoch einen ersten Hinweis auf eine Relevanz dieser Ergebnisse auch in
Säugerzellen (Hazra et al., 2003).

52 Ergebnisse und Diskussion
5.4 Einfluss auf XPA
Ausgehend von den Versuchen mit dem BER-Enzym Fpg wurden weitere Versuche
mit dem Zinkfingerprotein XPA als ein Vertreter für die NER durchgeführt. Für XPA
sollte zunächst gezeigt werden, ob Arsenverbindungen in der Lage sind Zink aus
seiner Zinkfingerstruktur freizusetzen. Hierzu wurde ein synthetisches Peptid,
bestehend aus 37 Aminosäuren, so mit Zink gesättigt, dass sich die
Zinkfingerstruktur von humanem XPA ausbildete.
Abbildung 13 zeigt, dass alle dreiwertigen Arsenverbindungen in der Lage waren
nach 30minütiger Inkubation bei 37°C Zink aus XPAzf konzentrationsabhängig
freizusetzen. Auch hier konnte nachgewiesen werden, dass die methylierten
Metabolite MMA(III) und DMA(III) eine höhere Reaktivität als anorganisches Arsenit
besitzen. Während für MMA(III) eine Zinkfreisetzung ab 25 µM, die mit 100 µM 100%
erreichte, nachgewiesen wurde, begann für DMA(III) die Freisetzung ab 50 µM und
0
20
40
60
80
100
120
1 10 100 1000 10000
Arsenverbindung [µM]
Zink
frei
setz
ung
aus
XPA
zf(%
der
H2O
2K
ontr
olle
)
Arsenit
MMA(III)
DMA(III)
MMA(V)
DMA(V)
0
20
40
60
80
100
120
1 10 100 1000 10000
Arsenverbindung [µM]
Zink
frei
setz
ung
aus
XPA
zf(%
der
H2O
2K
ontr
olle
)
Arsenit
MMA(III)
DMA(III)
MMA(V)
DMA(V)
Abbildung 13: Einfluss der Arsenverbindungen auf die Zinkfreisetzung aus XPAzf. XPAzf wurde
30 min bei 37°C mit den Arsenverbindungen inkubiert und die Zinkfreisetzung mittels 4-2-
Pyridylazoresorcinol quantifiziert. Dargestellt sind Mittelwerte aus 6 Bestimmungen ± SD.

Ergebnisse und Diskussion 53
erreichte 100% bei einer Konzentration von 1000 µM. Für Arsenit konnte eine
Zinkfreisetzung ab 100 µM festgestellt werden, die bei 10 mM einen Wert von 86%
erreichte. Für die beiden fünfwertigen Metabolite konnte nur im Falle von DMA(V)
eine sehr schwache Zinkfreisetzung von 18% bei 10 mM nachgewiesen werden.
Ausgehend von den Ergebnissen der Zinkfreisetzung aus XPAzf stellte sich nun die
Frage, welche Folgen dies für das ganze XPA-Protein hat. Hierzu wurde die DNA-
Bindungsfähigkeit von murinem XPA an ein UV-geschädigtes Oligonukleotid mittels
eines Gelshift-Versuchs untersucht. Die Untersuchungen zur DNA-Bindungsfähigkeit
konnten leider nur für DMA(III) durchgeführt werden, da aufgrund eines Ausfalls des
Biofreezer (-80°C Tiefkühler) das XPA-Protein vor Beginn der Versuche mit den
anderen Metaboliten irreversiebel geschädigt wurde. So konnte nur für DMA(III)
gezeigt werden, dass die XPA-Bindung an das UV-bestrahlte Oligonukleotid ab
Konzentrationen von 5 mM vollständig verloren geht (Abbildung 14). Auf anderen
Photofilmen ist zu sehen, dass bereits ab 2,5 mM eine leichte Reduzierung der DNA-
Bindungsfähigkeit festzustellen ist.
10.00010 50 100 500 1.000 5.000K + 1 5 K -
DMA(III) [µM]
Gebundenes Oligonukleotid
FreiesOligonukleotid
10.00010 50 100 500 1.000 5.000K + 1 5 K -
DMA(III) [µM]
Gebundenes Oligonukleotid
FreiesOligonukleotid
Abbildung 14: Einfluss von DMA(III) auf die DNA-Bindungsfähigkeit von XPA. XPA wurde 15 min
bei Raumtemperatur mit DMA(III) vorinkubiert, 30 min bei Raumtemperatur an das Oligonukleotid
binden gelassen und dann auf ein natives Polyacrylamidgel aufgetragen. Dargestellt ist ein
repäsentativer Photofilm. K+: Kontrolle mit XPA und Oligonukleotid, K-: Kontrolle mit Oligonukleotid.

54 Ergebnisse und Diskussion
Frühere Versuche aus unserem Labor zeigen, dass anorganisches Arsenit bis
Konzentrationen von 1 mM keine Reduzierung der DNA-Bindungsfähigkeit bewirkt.
Höhere Konzentrationen wurden damals nicht eingesetzt (Asmuss et al., 2000). Im
Gegensatz zu den Zinkfreisetzungsversuchen zeigt DMA(III) bei der Untersuchung
der DNA-Bindungsfähigkeit erst in sehr viel höheren milimolaren Konzentrationen
Effekte auf XPA. Dieser Unterschied kann einerseits durch die bessere
Zugänglichkeit für die Arsenverbindungen von XPAzf gegenüber murinem XPA
herrühren, oder aber auch davon kommen, dass bei dem Gelshift-Versuch hohe
Konzentrationen von 1 mM an Dithiotreitol (DTT) im Gelshift-Puffer verwendet
werden müssen, um eine oxidative Schädigung des XPA-Proteins zu verhindern.
Arsen hat eine hohe Affinität zu Ditholen wie DTT, so dass man davon ausgehen
kann, dass ein Teil der eingesetzten Arsenverbindung mit DTT abreagiert und gar
nicht die Möglichkeit hat mit XPA zu interagieren.
Die Zinkfreisetzungsversuche geben wie auch beim Fpg einen Hinweis darauf, dass
Zinkfingerstrukturen empfindlich gegenüber Arsenverbindungen sind und in Folge
einer Schädigung zur Inaktivierung des Proteins führen können. Hier konnten wir
erstmals zeigen, dass dreiwertige Arsenverbindungen Zink aus einem für die DNA-
Reparatur essentiellen Protein freisetzen können. Im Gegensatz zur Fpg ist der
Zinkfinger von XPA nicht direkt an der Wechselwirkung mit der DNA beteiligt. Er
stabilisiert jedoch die loopreiche Domäne, die für die Interaktion mit der DNA
verantwortlich ist (Morikawa und Shirakawa, 2000). Eine Schädigung des Zinkfingers
könnte also einen Verlust der DNA-Bindungsfähigkeit bewirken. Daneben ist der
Zinkfinger von XPA an der Interaktion mit anderen NER-Proteinen wie z.B. RPA
beteiligt und trägt so zum Aufbau des Präinzisionskomplexes bei. So kommt es durch
Substitutuion eines der zinkbindenden Cysteine zum fast vollständigen Erliegen der
NER (Miyamoto et al., 1992).
Inwieweit diese Ergebnisse aus isolierten Systemen in Zusammenhang gebracht
werden können mit Zellkulturversuchen, die eine Hemmung der NER durch
Arsenverbindungen nach UV-Bestrahlung (Hartwig et al., 1997; Bau et al., 2001)
oder Benzo[a]pyren-Schädigung (Schwerdtle et al., 2003a) nachweisen, müssten
weitere Versuche mit XPA im zellulären System zeigen.

Ergebnisse und Diskussion 55
5.5 Einfluss auf die Poly(ADP-Ribosyl)ierung
Ausgehend von den Ergebnissen mit den isolierten DNA-Reparaturproteinen Fpg
und XPA stellten wir uns die Frage, ob es möglich ist eine Arsen-induzierte
Hemmung einer Reaktion, die vorwiegend von einem Zinkfingerprotein katalysiert
wird, auch im zellulären System nachzuweisen. Die Poly(ADP-Ribosyl)ierung spielt
eine wichtige Rolle in der BER und wird zu ca. 90% durch das Zinkfingerenzym
PARP-1 katalysiert.
5.5.1 Poly(ADP-Ribosyl)ierung in HeLa S3-Zellen
Die Poly(ADP-Ribosyl)ierung in intakten HeLa S3-Zellen wurde nach Aktivierung
mittels H2O2-induzierten DNA-Strangbrüchen über immunologische Detektion des
Enzymprodukts Poly(ADP-Ribose) (PAR) mittels eines hochspezifischen
monoklonalen Antikörpers 10H und eines FITC-konjugierten sekundären Antikörpers
am Fluoreszenzmikroskop quantifiziert. Zusätzlich zur PAR wurde die nukleäre DNA
mittels DAPI eingefärbt.
Abbildung 15 zeigt zwei fluoreszenzmikroskopische Aufnahmen. Links ist auf einer
dreidimensionalen Aufnahme zu sehen, dass das grüne FITC-Signal nur im
20 µm20 µm20 µm
Abbildung 15: Mikroskopische Aufnahmen von poly(ADP-Ribosyl)ierenden HeLa S3 Zellen. Links: Dreidimensionale Darstellung mittels Apotome-Technik (Zeiss). grüne Fluoreszenz: PAR, blaue
Fluoreszenz: DNA (Zellkern)
Rechts: Phasenkontrastaufnahme einer HeLa S3 Zelle. In der Mitte der Zelle ist die fluoreszierende
DNA und die markierte PAR zu erkennen (Mikroskop: Axioimager M1).

56 Ergebnisse und Diskussion
Bereich der blaue DAPI-Fluoreszenz und damit im Zellkern lokalisiert ist. Das rechte
Bild zeigt eine Phasenkontrast-Aufnahme einer HeLa S3 Zelle. In der Mitte der Zelle
ist der Zellkern mit den PAR-Signalen zu erkennen.
5.5.1.1 Einfluss der Arsenverbindungen auf die Poly(ADP-Ribosyl)ierung
Um die Effekte der methylierten Arsenmetabolite auf die Poly(ADP-Ribosyl)ierung zu
untersuchen, wurden logarithmisch wachsende HeLa S3 Zellen für 18 h mit den
Arsenverbindungen inkubiert. Wie zuvor in unserer Arbeitsgruppe für Arsenit
(Hartwig et al., 2003) gezeigt, konnte für MMA(III), DMA(III), MMA(V) und DMA(V)
keine Induktion der Poly(ADP-Ribosyl)ierung festgestellt werden. Die Zellen zeigten
hier nur ein konstant schwaches Hintergrundsignal (Daten nicht dargestellt).
Nach einer Induktion der Poly(ADP-Ribosyl)ierung durch fünfminütige Inkubation mit
H2O2 ist es möglich, den Einfluss der Arsenverbindungen zu untersuchen. Für das
dreiwertige anorganische Arsenit konnte zuvor in unserem Labor eine
konzentrationsabhängige Hemmung der Reaktion schon in sehr niedrigen nicht-
zytotoxischen nanomolaren Konzentrationen gezeigt werden (Hartwig et al., 2003).
Der Vollständigkeit halber und zur besseren Vergleichbarkeit mit den Ergebnissen
der methylierten Metabolite, wird hier auch das Ergebnis für Arsenit dargestellt
(Abbildung 16).
Abbildung 17 zeigt immunfluorimetrische Aufnahmen des FITC-Kanals, der die
Poly(ADP-Ribosyl)ierung repräsentiert, nach Inkubation mit den methylierten
Arsenmetaboliten. Es ist zu sehen, dass die beiden dreiwertigen Verbindungen
MMA(III) und DMA(III) die Poly(ADP-Ribosyl)ierung in nanomolaren Konzentrationen
hemmten, während MMA(V) und DMA(V) bis in hohe mikromolare Konzentrationen
keinen Effekt zeigten.
Nach Quantifizierung der Fluoreszenzintensität mit einem Auswertungsprogramm
zeigte sich, dass MMA(III) und DMA(III) die Poly(ADP-Ribosyl)ierung
konzentrationsabhängig und signifikant schon in sehr niedrigen nanomolaren und
nichtzytotoxischen Konzentrationen hemmten. Eine erste Reduktion war schon ab
1 nM zu sehen, die bei 1000 nM für MMA(III) und 100 nM für DMA(III) ein Minimum
bei 59% bzw. 49% erreicht (Abbildung 18). In höheren und beginnend zytotoxischen

Ergebnisse und Diskussion 57
0
20
40
60
80
100
120
0 10 100 500 1000
Arsenit [nM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100K
oloniebildungsfähigkeit (% der K
ontrolle)
*** ***
***
***
0
20
40
60
80
100
120
0 10 100 500 1000
Arsenit [nM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100K
oloniebildungsfähigkeit (% der K
ontrolle)
*** ***
***
***
Abbildung 16: Effekt von Arsenit auf die Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen. Logarithmisch wachsende Zellen wurden für 18 h mit Arsenit vorinkubiert und die Poly(ADP-
Ribosyl)ierung 5 min mit 100 µM H2O2 induziert. PAR wurde immunfluorimetrisch am
Fluoreszenzmikroskop quantifiziert. Gezeigt sind Mittelwerte aus mind. 4 Bestimmungen mit jeweils
mindestens 100 Zellen + 95% Konfidenzintervall. Statistisch unterschiedlich von alleine H2O2-
behandelten Zellen: *** P < 0,001 (Student’s t-Test) (Hartwig et al., 2003).
Konzentrationen war keine weitere Hemmung und im Fall von DMA(III) für 1000 nM,
wie auch für Arsenit (Hartwig et al., 2003) sogar ein leichter Anstieg der Poly(ADP-
Ribosyl)ierung zu erkennen (Daten nicht dargestellt), was auf eine Sättigung des
inhibitorischen Effekts hinweist. Im Vergleich zu anorganischem Arsenit erwiesen
sich die dreiwertigen methylierten Metabolite auch in diesem Testsystem als
toxischer. Für Arsenit ist ein Beginn der Hemmung ab 10 nM zu sehen, während für
MMA(III) und DMA(III) schon in 10-fach niedrigeren Konzentrationen eine erste
Reduktion der Poly(ADP-Ribosyl)ierung auftrat.
Im Gegensatz zu den dreiwertigen Verbindungen konnte weder für MMA(V) noch für
DMA(V) bis in beginnend zytotoxische Konzentrationen von 500 µM bzw. 250 µM
keine Hemmung der Poly(ADP-Ribosyl)ierung festgestellt werden (Abbildung 19). So
konnte auch in diesem Versuch gezeigt werden, dass die Wertigkeit der
Arsenverbindungen entscheidend für deren Toxizität ist.

58 Ergebnisse und Diskussion
MMA(III) [nM] 0 0,1 1 10 100
DMA(III) [nM] 0 0,1 1 10 100
MMA(V) [µM] 0 1 10 100 500
DMA(V) [µM] 0 1 10 100 250
MMA(III) [nM] 0 0,1 1 10 100MMA(III) [nM] 0 0,1 1 10 100MMA(III) [nM] 0 0,1 1 10 100
DMA(III) [nM] 0 0,1 1 10 100DMA(III) [nM] 0 0,1 1 10 100DMA(III) [nM] 0 0,1 1 10 100
MMA(V) [µM] 0 1 10 100 500MMA(V) [µM] 0 1 10 100 500MMA(V) [µM] 0 1 10 100 500
DMA(V) [µM] 0 1 10 100 250DMA(V) [µM] 0 1 10 100 250DMA(V) [µM] 0 1 10 100 250
Abbildung 17: Effekt von MMA(III), DMA(III), MMA(V) und DMA(V) auf die Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen. Logarithmisch wachsende Zellen wurden für 18 h mit Arsenit
vorinkubiert und die Poly(ADP-Ribosyl)ierung 5 min mit 100 µM H2O2 induziert. PAR wurde
immunfluorimetrisch am Fluoreszenzmikroskop quantifiziert. Gezeigt sind repräsentative
Immunfluoreszenzaufnahmen des FITC-Kanals.
Die vorliegenden Ergebnisse zeigen für die dreiwertigen methylierten
Arsenmetabolite MMA(III) und DMA(III) zum ersten Mal einen Zusammenhang
zwischen der Hemmung der DNA-Reparatur durch Arsen und einer konkreten
enzymatischen Reaktion unter zellulären Bedingungen. Die Hemmung der
Poly(ADP-Ribosyl)ierung durch die dreiwertigen Arsenverbindungen ist damit die
sensitivste Reaktion, die mit der DNA-Reparaturhemmung verbunden ist, die jemals
beschrieben wurde. Für MMA(III) und DMA(III) existieren in der Literatur keine
Vergleichswerte zu einer Interaktion mit der Poly(ADP-Ribosyl)ierung, während es zu
anorganischem Arsenit widersprüchliche Studien gibt. So beschreiben Yager und
Wiencke (1997) eine Hemmung der Poly(ADP-Ribosyl)ierung in Konzentrationen
über 5 µM Arsenit in menschlichen Molt-3 Zellen, während Lynn et al. (1998) eine
Stimulierung der Poly(ADP-Ribosyl)ierung in hohen Konzentrationen über 40 µM

Ergebnisse und Diskussion 59
0
20
40
60
80
100
120
140
160
0 0,1 1 10 100 1000
MMA(III) [nM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100Zytotoxizität(%
der Kontrolle)
ZellzahlKoloniebildungsfähigkeit
A
****
******
0
20
40
60
80
100
120
140
160
0 0,1 1 10 100 1000
MMA(III) [nM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100Zytotoxizität(%
der Kontrolle)
ZellzahlKoloniebildungsfähigkeit
A
****
******
0
20
40
60
80
100
120
140
160
0 0,1 1 10 100
DMA(III) [nM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100
Zytotoxizität(% der K
ontrolle)
B
**
*** ***
ZellzahlKoloniebildungsfähigkeit
0
20
40
60
80
100
120
140
160
0 0,1 1 10 100
DMA(III) [nM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100
Zytotoxizität(% der K
ontrolle)
B
**
*** ***
ZellzahlKoloniebildungsfähigkeit
Abbildung 18: Effekt von MMA(III) (A) und DMA(III) (B) auf die Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen. Logarithmisch wachsende Zellen wurden für 18 h mit Arsenit vorinkubiert und die
Poly(ADP-Ribosyl)ierung 5 min mit 100 µM H2O2 induziert. PAR wurde immunfluorimetrisch am
Fluoreszenzmikroskop quantifiziert. Gezeigt sind Mittelwerte aus mind. 5 Bestimmungen mit jeweils
mindestens 50 Zellen + SD. Statistisch unterschiedlich von alleine H2O2-behandelten Zellen:
** P < 0,01; *** P < 0,001 (Student’s t-Test).
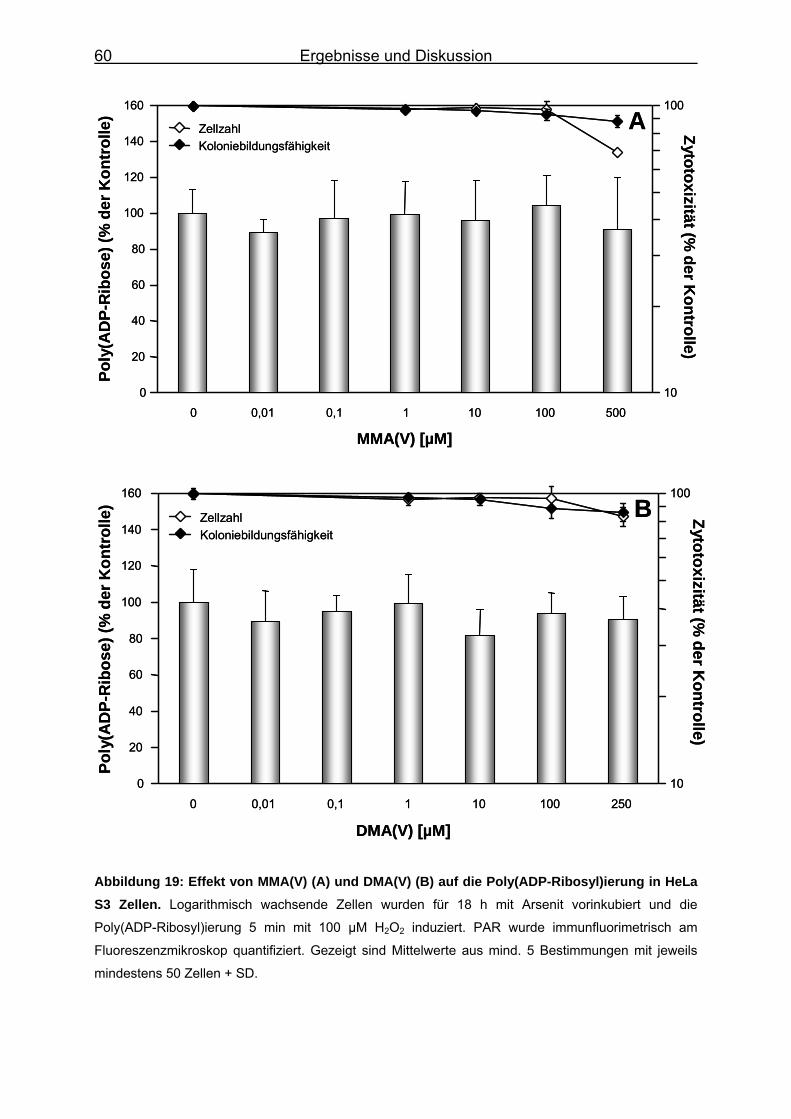
60 Ergebnisse und Diskussion
0
20
40
60
80
100
120
140
160
0 0,01 0,1 1 10 100 500
MMA(V) [µM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100Zytotoxizität(%
der Kontrolle)
AZellzahlKoloniebildungsfähigkeit
0
20
40
60
80
100
120
140
160
0 0,01 0,1 1 10 100 500
MMA(V) [µM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100Zytotoxizität(%
der Kontrolle)
AZellzahlKoloniebildungsfähigkeit
0
20
40
60
80
100
120
140
160
0 0,01 0,1 1 10 100 250
DMA(V) [µM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100
Zytotoxizität(% der K
ontrolle)
BZellzahlKoloniebildungsfähigkeit
0
20
40
60
80
100
120
140
160
0 0,01 0,1 1 10 100 250
DMA(V) [µM]
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
10
100
Zytotoxizität(% der K
ontrolle)
BZellzahlKoloniebildungsfähigkeit
Abbildung 19: Effekt von MMA(V) (A) und DMA(V) (B) auf die Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen. Logarithmisch wachsende Zellen wurden für 18 h mit Arsenit vorinkubiert und die
Poly(ADP-Ribosyl)ierung 5 min mit 100 µM H2O2 induziert. PAR wurde immunfluorimetrisch am
Fluoreszenzmikroskop quantifiziert. Gezeigt sind Mittelwerte aus mind. 5 Bestimmungen mit jeweils
mindestens 50 Zellen + SD.

Ergebnisse und Diskussion 61
Arsenit in CHO-K1 Zellen zeigen. Im Gegensatz zu den niedrigen nanomolaren
Konzentrationen, die in den Versuchen in unserem Labor eine Hemmung durch
Arsenit zeigten (Hartwig et al., 2003), wurden in beiden oben genannten Studien
jedoch sehr hohe zytotoxische Konzentrationen eingesetzt.
Auch wenn die Poly(ADP-Ribosyl)ierung durch die dreiwertigen Arsenverbindungen
nicht komplett gehemmt wird, stellt sie einen relevanten Parameter für die
genomische Stabilität der Zelle dar. So konnten Bürkle et al. (1987) zeigen, dass
eine Reduzierung der Poly(ADP-Ribosyl)ierung durch 3-Aminobenzamid in
ähnlichem Umfang wie durch die dreiwertigen methylierten Arsenmetabolite zu einer
erhöhten MNNG-induzierten DNA-Amplifikation führte, was auf eine erniedrigte
genomische Stabilität hindeutet. Eine Hemmung von PARP-1 hat weitreichende
Konsequenzen für die Zelle. Sie ist an verschiedenen DNA- Reparaturprozessen wie
der BER oder der Rekombinationsreparatur beteiligt und wirkt als negativer
Regulator der genomischen Stabilität (zusammengefasst in Bürkle et al., 2005a). So
kommt es beispielsweise in PARP-1-/--Zellen zu einer erhöhten Menge an
klastogenen Schäden, wie Chromosomenabberationen (CA) und
Schwesterchromatidaustauschen (SCE) (Le Rhun et al., 1998), während PARP-1
überexprimierende Zellen einen verbesserten Schutz vor SCE’s, die durch
Alkylierung induziert wurden, zeigen (Meyer et al., 2000). Ganz ähnliche klastogene
Schäden werden auch für Arsenit (zusammengefasst in Gebel, 2001) und seine
dreiwertigen methylierten Metabolite (Kligerman et al., 2003) beschrieben, so dass
hier über einen Zusammenhang der Hemmung der Poly(ADP-Ribosyl)ierung und der
Klastogenität von Arsen spekuliert werden kann. Eine mögliche Rolle von PARP-1 in
der arseninduzierten Genotoxizität zeigten vor kurzem Poonepalli et al. (2005)
(Poonepalli et al., 2005). So induziert Arsenit in PARP-1-/--Zellen im Vergleich zu
PARP-1+/+-Zellen eine erhöhte Anzahl an Mikrokernen.
Die Poly(ADP-Ribosyl)ierung ist eine komplexe zelluläre Reaktion, die auf ganz
verschiedene Art und Weise gestört werden kann. Eine mögliche Erkärung für die
reduzierte Poly(ADP-Ribosyl)ierung nach Inkubation mit den dreiwertigen
Arsenverbindungen könnte beispielsweise eine Reduzierung der zellulären
Konzentration von NAD+, dem Substrat der PARP, sein. So berichten Lynn et al.
(1998) von einer leichten Reduktion der NAD+-Level nach Inkubation mit
mikromolaren Konzentrationen von Arsenit. In unserem System ist jedoch nicht
davon auszugehen, dass es zu einem Abfall der NAD+-Konzentration kommt. Eine

62 Ergebnisse und Diskussion
Reduktion des NAD+-Levels wäre verbunden mit einer Wachstumshemmung der
Zellen, da NAD+ an fast allen metabolischen Reaktionen der Zelle beteiligt ist. Solche
Effekte wurden in den niedrigen nanomolaren Arsenkonzentrationen, bei denen die
Effekte auf die Poly(ADP-Ribosyl)ierung auftraten, jedoch nicht beobachtet.
Im Folgenden wurden weitere mögliche Gründe für die Hemmung der Poly(ADP-
Ribosyl)ierung untersucht. Zudem soll geklärt werden, ob die Reduzierung der
Poly(ADP-Ribosyl)ierung auf eine Hemmung von PARP-1 zurückzuführen ist.
5.5.1.2 Zeitverlauf der Poly(ADP-Ribosyl)ierung nach Arseninkubation
Der Aufbau der Poly(ADP-Ribose) zu 200 bis 400 Einheiten großen Polymeren wird
vor allem durch PARP-1 und PARP-2 während der fünfminütigen Inkubation mit H2O2
katalysiert, welche sich in Vorversuchen als optimal herausstellte (Hartwig et al.,
2003). Die Poly(ADP-Ribose) wird dann jedoch schnell durch PARG abgebaut. Die
Halbwertszeit der Poly(ADP-Ribose) liegt bei ca. 1 min. Um festzustellen, ob es sich
bei der Reduzierung der Poly(ADP-Ribosyl)ierung nach fünfminütiger H2O2-
Inkubation durch die dreiwertigen Metabolite MMA(III) und DMA(III) wirklich um eine
Hemmung handelt und nicht nur die Reaktion zeitlich nach hinten verschoben wird,
wurde die Poly(ADP-Ribosyl)ierung nach verschiedenen H2O2-Inkubationszeiten
quantifiziert.
Wie Abbildung 20 zeigt, konnte, wie auch für Arsenit (Hartwig et al., 2003), weder für
MMA(III) noch für DMA(III) eine zeitliche Verschiebung der Poly(ADP-Ribosyl)ierung
festgestellt werden. Die Signale der arseninkubierten Zellen waren in keinem Fall
stärker als die parallel mitgeführten Kontrollzellen, so dass davon ausgegangen
werden kann, dass es sich um eine wirkliche Hemmung der Poly(ADP-Ribosyl)ierung
handelt.

Ergebnisse und Diskussion 63
0
20
40
60
80
100
120
140
0 5 10 15 30
H2O2- Behandlung (min)
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
100 µM H2O2
100 µM H2O2 + 100 nM MMA(III)
A
***
0
20
40
60
80
100
120
140
0 5 10 15 30
H2O2- Behandlung (min)
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
100 µM H2O2
100 µM H2O2 + 100 nM MMA(III)
A
***
0
20
40
60
80
100
120
140
0 5 10 15 30
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
H2O2- Behandlung (min)
100 µM H2O2
100 µM H2O2 + 100 nM DMA(III)
B
***
0
20
40
60
80
100
120
140
0 5 10 15 30
Poly
(AD
P-R
ibos
e) (%
der
Kon
trol
le)
H2O2- Behandlung (min)
100 µM H2O2
100 µM H2O2 + 100 nM DMA(III)
B
***
Abbildung 20: Zeitabhängigkeit der Poly(ADP-Ribosyl)ierung nach Inkubation mit MMA(III) (A) und DMA(III) (B). Logarithmisch wachsende Zellen wurden für 18 h mit den Arsenverbindungen
vorinkubiert und die Poly(ADP-Ribosyl)ierung 5, 10, 15 bzw. 30 min mit 100 µM H2O2 induziert. PAR
wurde immunfluorimetrisch am Fluoreszenzmikroskop quantifiziert. Gezeigt sind Mittelwerte aus mind.
5 Bestimmungen mit jeweils mindestens 50 Zellen + SD. Statistisch unterschiedlich von den
dazugehörigen Kontrollzellen: *** P < 0,001 (Student’s t-Test).

64 Ergebnisse und Diskussion
5.5.2 Einfluss der Arsenverbindungen auf die DNA-Schadens-induktion durch H2O2 und auf die Genexpression der PARP-1
Zur Klärung des Wirkungsmechanismus der Hemmung der Poly(ADP-Ribosyl)ierung
wurden in unserem Labor zwei weitere Parameter untersucht. Diese Ergebnisse
wurden in Zusammenarbeit mit Dr. Tanja Schwerdtle und Dr. Christina Thuy
durchgeführt.
Im ersten Versuch sollte geklärt werden, ob die Arsenverbindungen während der
Schadensinduktion mit H2O2 interagieren und möglicherweise weniger DNA-
Strangbrüche induzieren. Eine Reduzierung der Strangbruchrate hätte eine geringere
PARP-1 und PARP-2 Aktivierung und damit einen geringeren Grad an Poly(ADP-
Ribose) zur Folge. Aus diesem Grund wurde mit Hilfe der Alkalischen Entwindung
(AU) (Hartwig et al., 1997) die Menge an DNA-Strangbrüchen nach Koinkubation der
Arsenverbindungen mit H2O2 bestimmt. In Abbildung 21 ist zu sehen, dass es für
0,0
0,5
1,0
1,5
2,0
2,5
3,0
DN
A-S
tran
gbrü
che
/ 106
Bas
enpa
are 0 µM H2O2
100 µM H2O2
Kontrolle 0,1 µM
MMA(III)100 µMMMA(V)
0,1 µMDMA(III)
100 µMDMA(V)
*** ***
0,0
0,5
1,0
1,5
2,0
2,5
3,0
DN
A-S
tran
gbrü
che
/ 106
Bas
enpa
are 0 µM H2O2
100 µM H2O2
Kontrolle 0,1 µM
MMA(III)100 µMMMA(V)
0,1 µMDMA(III)
100 µMDMA(V)
*** ***
Abbildung 21: Effekt der Arsenverbindungen auf die DNA-Strangbruchinduktion durch H2O2. Logarithmisch wachsende HeLa S3 Zellen wurden für 18 h mit den Arsenverbindungen vorinkubiert,
dann 5 min mit 100 µM H2O2 koinkubiert, und die DNA-Strangbrüche mittels Alkalischer Entwindung
(AU) bestimmt. Dargestellt sind Mittelwerte aus 6 Bestimmungen + SD. Statistisch unterschiedlich von
der nur mit H2O2 inkubierten Kontrolle: *** P < 0,001 (Student’s t-Test).

Ergebnisse und Diskussion 65
keinen der methylierten Arsenmetabolite MMA(III), DMA(III), MMA(V) und DMA(V) zu
einer reduzierten DNA-Strangbruchrate nach Koinkubation mit 100 µM H2O2 im
Vergleich zu H2O2 allein kam. Die monomethylierten Verbindungen MMA(III) und
MMA(V) zeigen sogar einen leichten Anstieg der Menge an DNA-Strangbrüchen
(Walter et al., 2007). Auch für anorgansiches Arsenit konnte ein leichter Anstieg der
DNA-Strangbruchrate festgestellt werden (Hartwig et al., 2003). Die
unterschiedlichen Effekte der Arsenverbindungen bezüglich ihres Methylierungs-
grades und ihrer Wertigkeit deuten auf komplexe Interaktionen mit H2O2 hin. So
könnte die Induktion eines redox cyclings oder die Hemmung der Reparatur der
H2O2-induzierten DNA-Schäden eine Rolle spielen.
In einem weiteren Versuch zur Klärung der Hemmung der Poly(ADP-Ribosyl)ierung
wurde die Genexpression der PARP-1, die den größten Teil der Poly(ADP-
Ribosyl)ierung katalysiert, nach Arseninkubation untersucht. Es konnte bereits für
eine Reihe von DNA-Reparaturgenen gezeigt werden, dass sie nach Inkubation mit
0,0
0,4
0,8
1,2
1,6
2,0
Kontrolle1 µM
Arsenit0,1 µM
MMA(III)0,1 µM
DMA(III)100 µMMMA(V)
100 µMDMA(V)
PAR
P-1
Gen
expr
essi
on
0,0
0,4
0,8
1,2
1,6
2,0
Kontrolle1 µM
Arsenit0,1 µM
MMA(III)0,1 µM
DMA(III)100 µMMMA(V)
100 µMDMA(V)
PAR
P-1
Gen
expr
essi
on
Abbildung 22: Effekt der Arsenverbindungen auf die Genexpression von PARP-1. Logarithmisch
wachsende HeLa S3 Zellen wurden für 18 h mit den Arsenverbindungen inkubiert und die
Genexpression mit Real-Time RT-PCR bestimmt. Dargestellt sind Mittelwerte aus drei unabhängigen
Versuchen mit jeweils 3 Messungen bezogen auf die Kontrolle und normiert auf das housekeeping
Gen GAPDH + SD.

66 Ergebnisse und Diskussion
Arsenit in menschlichen Keratinozyten aber auch in arsenexponierten Menschen
eine erniedrigte Genexpression aufweisen (Hamadeh et al., 2002; Andrew et al.,
2003). Die Messung der Genexpression wurde mit Real-Time RT-PCR im Falle der
dreiwertigen Arsenverbindungen für die Konzentrationen durchgeführt, die die größte
Hemmung der Poly(ADP-Ribosyl)ierung hervorriefen. Nach einer Inkubation von 18 h
mit Arsenit und den methylierten Metaboliten zeigte sich, dass keine der fünf
Verbindungen die Genexpression von PARP-1 signifikant veränderte. Es kann daher
ausgeschlossen werden, dass die Hemmung der Poly(ADP-Ribosyl)ierung durch
eine reduzierte Genexpression von PARP-1 hervorgerufen wird (Abbildung 22)
(Walter et al., 2007).

Ergebnisse und Diskussion 67
5.5.3 Bestimmung des Proteingehalts von PARP-1
Eine mögliche Erklärung für die gehemmte Poly(ADP-Ribosyl)ierung könnte eine
Reduzierung der Proteinmenge von PARP-1 sein. Aus diesem Grund wurde mittels
Western Blot Analyse der Proteingehalt von PARP-1 in HeLa S3 Zellen nach
18stündiger Inkubation mit den Arsenverbindungen bestimmt. Zur Detektion der
PARP-1 wird der monoklonale Antikörper CII-10 verwendet, der neben dem intakten
Protein auch das apoptotische Fragment der PARP-1 erkennt. So ist mit diesem
Versuch zusätzlich auch eine Aussage über die Induktion von Apoptose durch die
Arsenverbindungen möglich.
Abbildung 23 zeigt PARP-1 Western Blots mit Aktin als Ladekontrolle für Arsenit und
seine drei- und fünfwertigen Verbindungen. Es ist zu erkennen, dass es nur in hohen
zytotoxischen Konzentrationen zu einer Reduzierung der PARP-1 Menge durch
Arsenit, MMA(III) und DMA(III) kommt. Für Arsenit war in hohen zytotoxischen
Konzentrationen die Bildung des Apoptose-Fragments zu sehen. Auch MMA(III) und
DMA(III) bilden ab Konzentrationen von 10 µM das Apoptose-Fragment, wenn auch
in viel geringerem Ausmaß als Arsenit (Western Blots nicht dargestellt). MMA(V) und
DMA(V) zeigen bis zu zytotoxischen Konzentrationen von 1000 µM keine
Reduzierung der PARP-1 Bande.
Nach Quantifizierung der Western Blots mit Hilfe eines Auswertungsprogramms,
zeigte sich für Arsenit eine signifikante Reduktion des PARP-1-Proteingehalts erst ab
der zytotoxischen Konzentration von 10 µM. Der Proteingehalt der PARP-1 fällt bei
100 µM Arsenit bis auf 32% der Kontrolle ab und es kommt mit 29% der Kontrolle zur
Bildung des Apoptose-Fragments (Abbildung 24). Auch für MMA(III) und DMA(III)
kam es erst in beginnend zytotoxischen Konzentrationen von 1 µM zu einem Abfall
der PARP-1- Menge auf 58% bzw. 43%. Bei einer Konzentration von 50 µM bildeten
sich nur geringe Mengen von 2,3% bzw. 10,7% des Apoptose-Fragments (Abbildung
25). Der Beginn der Abnahme der Proteinmege variirte etwas von Versuch zu
Versuch, worauf auch die großen Fehlerbalken in den Graphiken zurückzuführen
sind. Für die beiden fünfwertigen Verbindungen MMA(V) und DMA(V) konnte bis zu
Konzentrationen von 1000 µM keine Reduzierung des PARP-1-Gehalts und keine
Bildung des Apoptose-Fragments festgestellt werden (Abbildung 26). Zusammen mit
den Ergebnissen zur Genexpression zeigt die Quantifizierung des PARP-1-
Proteingehalts, dass die Hemmung der Poly(ADP-Ribosyl)ierung in den niedrigen

68 Ergebnisse und Diskussion
PARP-1
Aktin
0,5 1 5 10 50 100
isol.
PARP-10Arsenit [µM]
PARP-1
Aktin
0,5 1 5 10 50 100
isol.
PARP-10Arsenit [µM]
PARP-1
Aktin
0,05 0,1 0,5 1,0 2,5 5,0
isol.
PARP-10MMA(III) [µM]
PARP-1
Aktin
0,05 0,1 0,5 1,0 2,5 5,0
isol.
PARP-10MMA(III) [µM]
PARP-1
Aktin
0,05 0,1 0,5 1,0 2,5 5,0
isol.
PARP-10DMA(III) [µM]
PARP-1
Aktin
0,05 0,1 0,5 1,0 2,5 5,0
isol.
PARP-10DMA(III) [µM]
PARP-1
Aktin
10 50 100 250 500 1000
isol.
PARP-10MMA(V) [µM]
PARP-1
Aktin
10 50 100 250 500 1000
isol.
PARP-10MMA(V) [µM]
PARP-1
Aktin
10 50 100 250 500 1000
isol.
PARP-10DMA(V) [µM]
PARP-1
Aktin
10 50 100 250 500 1000
isol.
PARP-10DMA(V) [µM]
Abbildung 23: PARP-1 Western Blots nach Inkubation mit Arsenit, MMA(III), DMA(III), MMA(V) und DMA(V). HeLa S3 Zellen wurden für 18 h mit den Arsenverbindungen inkubiert, ein Zellextrakt
präpariert und auf ein Polyacrylamidgel aufgetragen. Dargestellt ist je ein repräsentativer Western
Blot.

Ergebnisse und Diskussion 69
0
20
40
60
80
100
120
140
160
0 0,5 1 5 10 50 100
Arsenit [µM]
PAR
P-1
(% d
er K
ontr
olle
)
******
*
PARP-1 (ganzes Protein)PARP-1 Apoptose-Fragment
0
20
40
60
80
100
120
140
160
0 0,5 1 5 10 50 100
Arsenit [µM]
PAR
P-1
(% d
er K
ontr
olle
)
******
*
PARP-1 (ganzes Protein)PARP-1 Apoptose-Fragment
Abbildung 24: PARP-1-Proteingehalt nach Inkubation mit Arsenit. HeLa S3 Zellen wurden für
18 h mit Arsenit inkubiert, ein Zellextrakt präpariert und auf ein Polyacrylamidgel aufgetragen. Die
Banden wurden mit einem computergestützten Auswertungsprogramm quantifiziert. Dargestellt sind
Mittelwerte aus mind. 3 Versuchen ± SD. Statistisch unterschiedlich von der Kontrolle: * P < 0,05; ***
P < 0,001 (Student’s t-Test).
nanomolaren Konzentrationen nicht auf eine Veränderung der Gen- oder
Proteinexpression der PARP-1 zurückzuführen ist. Neben der Quantifizierung des
PARP-1-Proteingehalts konnte für Arsenit und die dreiwertigen methylierten
Metabolite eine Bildung des apoptotischen PARP-1- Fragments nachgewiesen
werden. Für alle drei Verbindungen konnte das apoptotische Fragment jedoch erst in
sehr hohen zytotoxischen Konzentrationen festgestellt werden. Das typische 85 kDa-
Fragment bildete sich bei den methylierten Metaboliten jedoch sowohl in niedrigeren
Konzentrationen als auch in gerigerem Umfang verglichen mit anorganischem
Arsenit.
In der Literatur existieren eine Vielzahl von Studien zur Induktion von Apoptose durch
die verschiedenen Arsenverbindungen. So konnten Chen et al. (2003) durch
durchflusszytometrische Messungen wie auch durch Nachweis des apoptotischen
PARP-1-Fragments die Induktion von Apoptose in menschlichen NB4-Zellen für
Arsenit, MMA(III) und DMA(III) zeigen. Im Vergleich zu unseren Ergebnissen in HeLa
S3-Zellen konnte das PARP-1-Fragment in NB4-Zellen nach

70 Ergebnisse und Diskussion
0
20
40
60
80
100
120
0 0,05 0,1 0,5 1 2,5 5 10 50
MMA(III) [µM]
PAR
P-1
(% d
er K
ontr
olle
)
******
******
**
PARP-1 (ganzes Protein)PARP-1 Apoptose-Fragment
0
20
40
60
80
100
120
0 0,05 0,1 0,5 1 2,5 5 10 50
MMA(III) [µM]
PAR
P-1
(% d
er K
ontr
olle
)
******
******
**
PARP-1 (ganzes Protein)PARP-1 Apoptose-Fragment
0
20
40
60
80
100
120
0 0,05 0,1 0,5 1 2,5 5 10 50
DMA(III) [µM]
PAR
P-1
(% d
er K
ontr
olle
)
******
******
***
PARP-1 (ganzes Protein)PARP-1 Apoptose-Fragment
0
20
40
60
80
100
120
0 0,05 0,1 0,5 1 2,5 5 10 50
DMA(III) [µM]
PAR
P-1
(% d
er K
ontr
olle
)
******
******
***
PARP-1 (ganzes Protein)PARP-1 Apoptose-Fragment
Abbildung 25: PARP-1-Proteingehalt nach Inkubation mit MMA(III) (A) und DMA(III) (B). HeLa S3
Zellen wurden für 18 h mit den Arsenverbindungen inkubiert, ein Zellextrakt präpariert und auf ein
Polyacrylamidgel aufgetragen. Die Banden wurden mit einem computergestützten Auswertungs-
programm quantifiziert. Dargestellt sind Mittelwerte aus mind. 3 Versuchen ± SD. Statistisch
unterschiedlich von der Kontrolle: *** P < 0,001 (Student’s t-Test).

Ergebnisse und Diskussion 71
0
20
40
60
80
100
120
140
0 10 50 100 250 500 1000
MMA(V) [µM]
PAR
P-1
(% d
er K
ontr
olle
) A
0
20
40
60
80
100
120
140
0 10 50 100 250 500 1000
MMA(V) [µM]
PAR
P-1
(% d
er K
ontr
olle
) A
0
20
40
60
80
100
120
140
0 10 50 100 250 500 1000
DMA(V) [µM]
PAR
P-1
(% d
er K
ontr
olle
)
B
0
20
40
60
80
100
120
140
0 10 50 100 250 500 1000
DMA(V) [µM]
PAR
P-1
(% d
er K
ontr
olle
)
B
Abbildung 26: PARP-1-Proteingehalt nach Inkubation mit MMA(V) (A) und DMA(V) (B). HeLa S3
Zellen wurden für 18 h mit den Arsenverbindungen inkubiert, ein Zellextrakt präpariert und auf ein
Polyacrylamidgel aufgetragen. Die Banden wurden mit einem computergestützten Auswertungs-
programm quantifiziert. Dargestellt ist sind Mittelwerte aus mind. 3 Versuchen ± SD.

72 Ergebnisse und Diskussion
24 h Inkubation schon in sehr niedrigen Konzentrationen von 1 µM nachgewiesen
werden, wobei sich MMA(III) und DMA(III) als stärker Apoptose-auslösend verglichen
mit Arsenit erwiesen. Man geht davon aus, dass anorganisches Arsenit vor allem
Nekrose und weniger Apoptose induziert (Sakurai et al., 1998).
Für die fünfwertigen Verbindungen konnte in HeLa S3 Zellen kein Apoptose-
Fragment detektiert werden. Die Induktion von Apoptose durch die fünfwertigen
Metabolite scheint stark vom GSH-Gehalt der Zellen abzuhängen. Während GSH für
die Induktion von Apoptose im Falle von DMA(V) essentiell ist (Ochi et al., 1996;
Sakurai et al., 2002), konnte für MMA(V) Apoptose nur nach Depletion von GSH
nachgewiesen werden (Sakurai et al., 2005). Der GSH-Gehalt von HeLa Zellen, der
verglichen mit anderen Zelllinien relativ gering ist, liegt bei 16,45 nmol/106 Zellen was
bei einem Volumen von ca. 2 x 10-12 l einer Konzentration von 8,2 mM entspricht
(Yamauchi et al., 1990).
Ein weiterer wichtiger Parameter ist der richtige Zeitpunkt der Apoptose-Messung. So
findet die Spaltung von PARP-1 durch die Caspasen 3 und 7 früh im Verlauf der
Apoptose statt. Um eine bessere Aussage über die Induktion des programmierten
Zelltods durch die Arsenverbindungen zu treffen, sind weitere Western Blot-Versuche
mit verschiedenen Inkubationszeiten sowie andere Apoptosetests nötig.
99% der menschlichen Zervixcarcinomzelllinien, wie auch HeLa S3, entstanden
durch Infektion mit dem humanen Papillomavirus HPV, was zur Folge hat, dass die
Onkogene E6 und E7 aktiviert werden, die unter anderem den Ubiquitin-vermittelten
Abbau von p53 beschleunigen. Dies ist wohl ein Gund für die geringere Induktion von
Apoptose und die beschleunigte Proliferationsrate in HeLa S3 Zellen. In
verschiedenen Studien und auch in unserem Labor (Daten nicht gezeigt) konnte
jedoch gezeigt werden, dass HeLa S3 Zellen nach wie vor eine basale p53-
Expression besitzen. Inwieweit die reduzierte p53-Menge Einfluss auf die
arseninduzierte Apoptose hat ist unklar. Andere Substanzen wie Staurosporin
induzieren in HeLa S3 Zellen jedoch nach wie vor Apoptose und weisen evtl. auf eine
p53 unabhängige Aktivierung der Apoptose hin, wie sie auch für Arsenverbindungen
nicht ausgeschlossen werden kann (Bernard et al., 2003).

Ergebnisse und Diskussion 73
5.5.4 Poly(ADP-Ribosyl)ierung isolierter PARP-1
Die bisherigen Ergebnisse im zellulären System deuten darauf hin, dass die
Hemmung der Poly(ADP-Ribosyl)ierung durch die Arsenverbindungen auf eine
Wechselwirkung mit PARP-1 zurückzuführen ist. Aus diesem Grund wurde als
nächstes die Hemmung isolierter PARP-1 untersucht. Zunächst wurde für diesen
Versuch PARP-1 aus mit rekombinanten Bacculoviren infizierten Sf9- Insektenzellen
isoliert. Diese Arbeiten wurden an der Universität Konstanz im Arbeitskreis von Prof.
Bürkle durchgeführt. Die Methode zur Bestimmung der PARP-1 Aktivität konnte
folglich für unser Labor etabliert werden. Zunächst wurde isolierte PARP-1 mit den
Arsenverbindungen 30 min bei 37°C vorinkubiert, und dann die PARP-Reaktion 10
min bei 37°C durchgeführt. Nach Aufsaugen des Gemischs auf eine Membran mittels
Slot-Blot konnte die Poly(ADP-Ribose) immunologisch detektiert werden.
Arsenit
DMA(III)
MMA(III)
DMA(V)
MMA(V)
Kontrolle
0,01
0,5
0,1
1 2,5
5 10
Arsenverbindung [mM]
- - --
- - --
Arsenit
DMA(III)
MMA(III)
DMA(V)
MMA(V)
Kontrolle
0,01
0,5
0,1
1 2,5
5 10
Arsenverbindung [mM]
- - --
- - --
- - --
- - --
Abbildung 27: Effekt der Arsenverbindungen auf die Aktivität isolierter PARP-1. Isolierte
PARP-1 wurde für 30 min bei 37°C mit den Arsenverbindungen vorinkubiert, die PARP-Reaktion mit
einem Oligonukleotid als Aktivator-DNA 10 min bei 37°C durchgeführt, die Reaktion gestoppt und der
Reaktionsmix auf eine Membran mittels Slot-Blot aufgesaugt. Die Poly(ADP-Ribose) wurde
anschließend immunologisch bestimmt. Gezeigt ist für jede Arsenverbindung ein repräsentativer Slot-
Blot.

74 Ergebnisse und Diskussion
Abbildung 27 zeigt Slot-Blots für alle fünf eingesetzten Arsenverbindungen. Es ist zu
sehen, dass nur die dreiwertigen Verbindungen Arsenit, MMA(III) und DMA(III) in der
Lage sind die Aktivität isolierter PARP-1 zu hemmen, während für MMA(V) und
DMA(V) keine Abnahme der Bandenintensität zu erkennen war. Die Hemmung durch
die dreiwertigen Verbindungen konnte jedoch erst in sehr hohen millimolaren
Konzentrationen beobachtet werden.
Nach Quantifizierung der Banden der Slot-Blots ergab sich für die dreiwertigen
Arsenverbindungen eine signifikante, konzentrationsabhängige Hemmung der
PARP-1 in millimolaren Konzentrationen. Während für Arsenit und DMA(III) eine
signifikante Abnahme ab 2,5 mM festzustellen ist, begann die Abnahme der
Bandenintensität für MMA(III) erst ab 5 mM (Signifikanzdaten nicht dargestellt). Bei
einer Konzentration von 10 mM ist für Arsenit, MMA(III) und DMA(III) nur noch eine
PARP-1-Aktivität von 27%, 37% bzw. 31% detektierbar. Für MMA(V) und DMA(V) ist
keine Abnahme der PARP-1-Aktivität festzustellen (Abbildung 28).
0
20
40
60
80
100
120
140
0 2 4 6 8 10Arsenverbindung [mM]
PAR
P-1
Akt
ivitä
t (%
der
Kon
trol
le)
Arsenit
MMA(III)DMA(III)
MMA(V)
DMA(V)
0
20
40
60
80
100
120
140
0 2 4 6 8 10Arsenverbindung [mM]
PAR
P-1
Akt
ivitä
t (%
der
Kon
trol
le)
Arsenit
MMA(III)DMA(III)
MMA(V)
DMA(V)
Abbildung 28: Effekt der Arsenverbindungen auf die Aktivität isolierter PARP-1. Isolierte
PARP-1 wurde für 30 min bei 37°C mit den Arsenverbindungen vorinkubiert, die PARP-Reaktion mit
einem Oligonukleotid als Aktivator-DNA 10 min bei 37°C durchgeführt, die Reaktion gestoppt und der
Reaktionsmix auf eine Membran mittels Slot-Blot aufgesaugt. Die Poly(ADP-Ribose) wurde
anschließend immunologisch bestimmt. Gezeigt sind Mittelwerte aus mindestens 3 Bestimmungen.

Ergebnisse und Diskussion 75
Im Gegensatz zu den anderen Parametern, die in dieser Arbeit untersucht wurden,
zeigte Arsenit hier sogar eine etwas stärkere Hemmung als die beiden methylierten
Metabolite MMA(III) und DMA(III). Die Hemmung der Poly(ADP-Ribosyl)ierung tritt im
Vergleich zu den nanomolaren Konzentrationen im zellulären System hier erst in
millimolaren Konzentrationen auf.
Aus diesem Grund wurden von unserem Arbeitskreis in Zusammenarbeit mit Prof.
Dianov von der Universität Oxford die dreiwertigen Arsenverbindungen in einem
zweiten System zur Messung der Aktivität isolierter PARP-1 eingesetzt.
Hochaufgereinigte PARP-1 wurde 10 min bei Raumtemperatur mit den
Arsenverbindungen vorinkubiert, 5 min bei 37°C durch ein eingeschnittenes Plasmid
aktiviert, die Reaktion gestoppt und schließlich das Reaktionsgemisch auf ein
PAR
P-1
Akt
ivitä
t (%
der
Kon
trol
le)
0
20
40
60
80
100
0 10 50 100 200 500
Arsenverbindung [µM]
Arsenit
MMA(III)
DMA(III)***
*** **
**
***
***
***
***
***
******
***
***
***
PAR
P-1
Akt
ivitä
t (%
der
Kon
trol
le)
0
20
40
60
80
100
0 10 50 100 200 500
Arsenverbindung [µM]
Arsenit
MMA(III)
DMA(III)***
*** **
**
***
***
***
***
***
******
***
***
***
Abbildung 29: Effekt der Arsenverbindungen auf die Aktivität isolierter PARP-1. Isolierte
PARP-1 wurde für 10 min bei Raumtempertur mit den Arsenverbindungen vorinkubiert, die PARP-
Reaktion mit einem eingeschnittenen Plasmid als Aktivator-DNA 5 min bei 37°C durchgeführt, die
Reaktion gestoppt und der Reaktionsmix auf ein Polyacrylamidgel aufgetragen. Die Poly(ADP-Ribose)
wurde anschließend immunfluorimetrisch bestimmt. Gezeigt sind Mittelwerte aus mindestens 4
Bestimmungen + SD. Statistisch unterschiedlich von der Kontrolle: * P < 0,05; ** P < 0,01;
*** P < 0,001 (Student’s t-Test).

76 Ergebnisse und Diskussion
Polyacrylamidgel aufgetragen. Die Quantifizierung erfolgte immunologisch über den
primären Anti-PAR-Antikörper 10 H sowie einen sekundären fluoreszenzgekoppelten
Antikörper. Auch in diesem zweiten System ist zu sehen, dass die dreiwertigen
Arsenverbindungen die Aktivität der PARP-1 konzentrationsabhängig hemmen. Die
Hemmung konnte hier allerdings schon ab einer Konzentration von 10 µM festgestellt
werden. MMA(III) und DMA(IIII) erwiesen sich hier als toxischer verglichen mit
anorganischem Arsenit (Abbildung 29) (Walter et al., 2007). Im Gegensatz zu den
Ergebnissen des Slot-Blot-Testsystems konnte hier, wie auf für Fpg und XPA eine
höhere Reaktivität von MMA(III) und DMA(III) verglichen mit Arsenit festgestellt
werden. Worauf dieser Unterschied zurückzuführen ist, bleibt ungeklärt.
Im Folgenden sollte geklärt werden, worauf die unterschiedliche Sensitivität der
beiden Testsysteme beruht. Zunächst wurde mit beiden Versuchen auch die
Hemmung von PARP-1 durch CuSO4 untersucht. Für Cu2+ ist bekannt, wenn auch in
höheren Konzentrationen als Arsen, dass es die Poly(ADP-Ribosyl)ierung in HeLa
S3 Zellen hemmt (Hartwig et al., 2002; Schwerdtle et al., 2007). Vergleicht man nun
die Hemmung isolierter PARP-1 in beiden Testsystemen, ergaben sich nahezu
identische Kurvenverläufe mit einer signifikanten Hemmung ab 50 µM CuSO4 (Daten
siehe Anhang) (Schwerdtle et al., 2007). Diese Ergebnisse lassen auf einen für
Arsen spezifischen Grund für die unterschiedliche Sensitivität schließen. Daher ist es
unwahrscheinlich, dass die unterschiedliche PARP-1-Aktivierung, die
unterschiedliche Aufreinigung der isolierten PARP-1, verschiedene Inkubationszeiten
oder die unterschiedliche Detektion einen großen Einfluss haben. Viel
wahrscheinlicher ist, dass Pufferkomponenten, wie der Gehalt an Thiolreagenzien,
einen entscheidenden Einfluss auf die Reaktivität der Arsenverbindungen haben. In
beiden Versuchen wird DTT als Oxidationsschutz zur Stabilisierung der PARP-1
eingesetzt. Im Slot-Blot System liegt die Konzentration mit 1 mM um mehr als den
Faktor 100 über der verwendeten Dosis im PAGE-Testsystem. Wie schon für den
XPA-Gelshift (Kapitel 5.4) beschrieben, ist es möglich, dass ein Großteil der
Arsenverbindungen aufgrund ihrer hohen Affinität zu vicinalen Thiolgruppen
bevorzugt mit DTT abreagieren. Eine solch hohe Affinität zu Thiolgruppen ist für
Kupfer nicht beschrieben und könnte daher den Unterschied erklären.
Die Konzentrationen, in denen Arsen die Poly(ADP-Ribosyl)ierung isolierter PARP-1
hemmt, liegen jedoch in beiden Testsystemen weit über den Konzentrationen die
nötig sind, um die zelluläre Poly(ADP-Ribosyl)ierung zu hemmen. Auch hier könnte

Ergebnisse und Diskussion 77
der hohe Gehalt von DTT eine mögliche Erklärung für die Unterschiede sein. Des
weiteren konnte für Arsenit gezeigt werden, dass es in der Zelle akkumuliert und
extrazelluläre Konzentrationen um ungefähr das 15-fache übersteigt (Hartwig et al.,
1997). Ähnliche Effekte könnten auch für MMA(III) und DMA(III) denkbar sein. Ein
weiterer Unterschied zwischen zellulärem umd isoliertem System könnte darin
liegen, dass es unter zellulären Bedingungen zur Bildung von noch reaktiveren
Arsenverbindungen, wie z.B. Arsen-GSH-Komplexen, kommt.
Als möglicher molekularer Ansatzpunkt zur Hemmung von PARP-1 durch Arsen
gelten, wie auch schon für Fpg und XPA, die Zinkfingerstrukturen von PARP-1. Mit
den bisherigen Daten lässt sich jedoch keine Aussage zur Beteiligung des
Zinkfingers machen. So hat die DNA-bindende Domäne mit 11 Cystein-Resten eine
hohe Dichte an Thiolgruppen, jedoch auch in der Automodifikationsdomäne und der
katalytischen Domäne mit einem bzw. zwei Cysteinen finden sich mögliche
Angriffspunkte für Arsenverbindungen (Ame et al., 1999). Hier sind weitere Studien,
wie Untersuchungen der DNA-Bindung von arsengeschädigter PARP-1 oder Arsen-
PARP-1 Bindungsversuche nötig.

78 Zusammenfassende Diskussion und Ausblick
6. Zusammenfassende Diskussion und Ausblick
Die vorliegende Arbeit liefert einen Beitrag zum Verständnis der genotoxischen
Wirkung von Arsen. Es konnte gezeigt werden, dass neben der bekannten Induktion
von oxidativen DNA-Schäden durch Arsenverbindungen, auch indirekte Effekte, wie
Hemmungen von DNA-Reparaturproteinen, eine wichtige Rolle spielen. Aufgrund der
hohen Affinität von vor allem dreiwertigen Arsenverbindungen zu vicinalen
Thiolgruppen konnten Zinkfingerstrukturen als ein möglicher molekularer
Angriffspunkt identifiziert werden.
Die Hemmung der Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen erwies sich dabei als
empfindlichste Reaktion. Die Poly(ADP-Ribosyl)ierung wird zu über 90% von dem
Zinkfingerenzym PARP-1 katalysiert, das an vielen zellulären Reaktion beteiligt ist
und entscheidenden Einfluss auf die genomische Stabilität hat. Man geht heute
davon aus, dass PARP-1 als Regulator der zellulären Stabilität wirkt. Die Hemmung
der Poly(ADP-Ribosyl)ierung konnte spezifisch für die dreiwertigen
Arsenverbindungen schon in extrem niedrigen nanomolaren Konzentrationen
festgestellt werden. Bisher ist in der Literatur keine andere mit der DNA-Reparatur
verbundene, zelluläre Reaktion bekannt, auf die Arsen in ähnlich niedrigen
Konzentrationen einen Einfluss ausübt. In der vorliegenden Arbeit ist es zudem
gelungen, zum ersten Mal eine Hemmung einer zellulären Reaktion durch die
dreiwertigen Arsenmetabolite MMA(III) und DMA(III) zu zeigen, die direkt mit der
DNA-Reparatur und der Aufrechterhaltung der genomischen Stabilität in
Zusammenhang gebracht werden kann. Inwieweit die Hemmung der Poly(ADP-
Ribosyl)ierung spezifisch für Arsenverbindungen ist und damit einen wichtigen
Mechanismus der arseninduzierten Kanzerogenese darstellt, muss weiter untersucht
werden. Die Untersuchung der Poly(ADP-Ribosyl)ierung kann zudem in Zukunft
einen wichtigen Biomarker für eine Arsenexposition darstellen. Epidemiologische
Studien könnten zeigen, dass eine berufliche Arsenexposition oder eine Aufnahme
von Arsen über das Trinkwasser mit einer reduzierten Aktivität der Poly(ADP-
Ribosyl)ierung in Lymphozyten einhergeht.
In umfangreichen Untersuchungen konnte in der vorliegenden Arbeit gezeigt werden,
dass die Hemmung der Poly(ADP-Ribosyl)ierung wahrscheinlich auf eine Interaktion

Zusammenfassende Diskussion und Ausblick 79
der Arsenverbindungen mit PARP-1 zurückzuführen ist. Wie bereits ausführlich
dargestellt, konnte keine zeitliche Verschiebung der Poly(ADP-Ribosyl)ierung zu
längeren H2O2-Inkubationszeiten, keine Reduzierung der DNA-Strangbruchinduktion
nach Koinkubation der Arsenverbindungen mit H2O2, keine Veränderung der PARP-
1-Genexpression sowie keine Veränderungen der PARP-1-Proteinmenge im
niedrigen Konzentrationsbereich beobachtet werden. Zudem ist eine NAD+-
Depletierung der Zelle in den niedrigen nanomolaren Konzentrationen aufgrund nicht
auftretender zytotoxischer Reaktionen auszuschließen. Aufgrund dieser Daten ist
anzunehmen, dass es zu direkten Wechselwirkungen der Arsenverbindungen mit
PARP-1 kommt, wobei nicht ganz ausgeschlossen werden kann, dass weitere
Reaktionen, wie z.B. eine Phosphorylierung der PARP-1 oder eine reduzierte PAR-
Polymerlänge eine Rolle spielen.
Die Hemmung isolierter PARP-1, wenn auch in hohen Konzentrationen, zeigt dass
eine solche Interaktion durchaus möglich ist. Eine Hemmung der PARP-1 konnte im
PAGE-Testsystem schon in mikromolaren Konzentrationen festgestellt. Verglichen
mit anderen Hemmungen von DNA-Reparaturproteinen stellt auch hier PARP-1 das
sensitivste Zielmolekül für Arsenverbindungen dar. Aufgrund der vorliegenden
Datenlage ist es jedoch nicht möglich, eine Aussage über den Mechanismus einer
möglichen PARP-1-Enzymhemmung zu treffen. Neben den Thiolgruppen der beiden
Zinkfingerstrukturen, könnten auch SH-Gruppen aus der Automodifikations- oder der
katalytischen Domäne der PARP-1 Ziel für die Arsenverbindungen sein.
Unersuchungen mit den beiden Zinkfingerproteinen Fpg und XPA zeigen jedoch,
dass Zinkfingerstrukturen einen molekularen Angriffspunkt vor allem für dreiwertige
Arsenverbindungen darstellen können. So weisen die Ergebnisse zur Koinkubation
mit ZnCl2 im Falle von Fpg und zur Zinkfreisetzung aus XPAzf auf den Zinkfinger als
Zielstruktur hin. Tabelle 2 gibt einen Überblick über die in dieser Arbeit
durchgeführten Untersuchungen isolierter Zinkfingerproteine bzw. –peptide. Für Fpg
konnte neben PARP-1 zum ersten Mal eine Enzymhemmung eines isolierten DNA-
Reparaturproteins durch die dreiwertigen Metabolite MMA(III) und DMA(III) gezeigt
werden.
In der Literatur werden eine Vielzahl von Enzymhemmungen für Arsen beschrieben.
Eine sensitive Hemmung von isolierten DNA-Reparaturproteinen konnte bisher
jedoch noch nicht beschrieben werden. So konnten weder Asmuss et al. (2000) für

80 Zusammenfassende Diskussion und Ausblick
Tabelle 2: Wechselwirkungen der Arsenverbindungen mit den isolierten Proteinen bzw. Peptiden Fpg, XPAzf und PARP-1. + : Wechselwirkung, - : keine Wechselwirkung
Arsenit MMA(III) DMA(III) MMA(V) DMA(V)
Fpg -
bis 10 mM +
≤ 1 mM +
≤ 200 µM -
bis 10 mM -
bis 10 mM
XPAzf +
≤ 100 µM +
≤ 25 µM +
≤ 50 µM -
bis 10 mM -
bis 10 mM
PARP-1 +
≤ 2,5 mM +
≤ 5 mM +
≤ 2,5 mM -
bis 10 mM -
bis 10 mM
XPA bis 1 mM Arsenit noch Hu et al. (1998) im Falle von Ligase I, Ligase III und
Polymerase ß bis 5 mM Arsenit bzw. 30 mM Arsenat eine Hemmung zeigen. Für alle
Versuche mit isolierten Proteinen ist jedoch zu beachten, dass meistens hohe
Konzentrationen an DTT verwendet wurden, das, wie schon beschrieben, selbst mit
Arsen interagieren kann. Für in vitro Experimente wurde bisher nur für DNA-Ligase
eine Hemmung in sehr hohen millimolaren Konzentrationen berichtet (Li und
Rossman, 1989). Hemmungen isolierter Enzyme durch MMA(III) und DMA(III) sind
nur selten beschrieben worden. Es konnte gezeigt werden, dass die
Glutathionreduktase durch MMA(III) und DMA(III) in mikromolaren Konzentrationen
gehemmt wird, während Arsenit erst in 100-fach höheren Konzentrationen hemmt.
Auch die Thioredoxinreduktase wird durch MMA(III) stärker gehemmt als durch
Arsenit oder fünfwertige Arsenverbindungen (zusammengefasst in Thomas et al.,
2001).
Eine Hemmung von Zinkfingerproteinen konnte schon für eine ganze Reihe von
Metallen und Zinkfingerstrukturen gezeigt werden. Die Art und Weise, wie die
Interaktion zwischen Metall und Proteindomäne abläuft, ist dabei von den
Eigenschaften des Metalls wie auch von der Struktur des Zinkfingers abhängig. So
ist denkbar, dass es zu einem Austausch der Metalle, zu gemischten Komplexen mit
Zink oder auch zur Oxidation der Cysteine des Zinkfingers kommt. Alle
Wechselwirkungen führen in der Regel zu einem Funktionsverlust des Zinkfingers
und damit in den meisten Fällen zur Hemmung des gesamten Proteins (Hartwig,
2001). Aufgrund seiner hohen Affinität zu Thiolgruppen ist für dreiwertiges Arsen eine
direkte Interaktion mit den Cysteinen des Zinkfingers wahrscheinlich. So ist bekannt,

Zusammenfassende Diskussion und Ausblick 81
dass Arsenit Gluthation-Komplexe bildet (Hayakawa et al., 2005) und es konnte
gezeigt werden, dass Arsenit, MMA(III) und DMA(III) an Metallothionein binden. Bei
Metallothionein handelt es sich um ein metallspeicherndes Polypeptid mit 20
Cysteinresten. Während Arsenit drei Bindungsstellen für SH-Gruppen besitzt, können
MMA(III) und DMA(III) nur eine bzw. zwei Thiolgruppen binden. Ein Molekül
Metallothionein ist also in der Lage 6 Moleküle Arsenit, 10 Moleküle MMA(III) und 20
Moleküle DMA(III) zu binden (Jiang et al., 2003). Während diese Studien auf eine
kovalente Bindung von Arsen an Thiole auch in Zinkfingerstrukturen hindeuten,
weisen die Koinkubationsversuche von Zink und DMA(III) im Falle für Fpg eher auf
eine reversible Hemmung und damit auf eine nicht-kovalente Bindung an den
Zinkfinger hin. Arsen könnte Zinkfingerstrukturen allerdings auch indirekt schädigen.
So könnten durch Arsenverbindungen erzeugte reaktive Sauerstoff- oder
Zn
Zn
AsAs
As
S
S
S
S
+ Arsen
ROS
S
S
AsHS
HS
RNS
ZnZn
Zn
AsAs AsAs
AsAs
S
S
S
S
+ Arsen
ROS
S
S
AsAsHS
HS
RNS
Abbildung 30: Mögliche Wechselwirkungen von Arsenverbindungen mit Zinkfingerstrukturen. ROS: reaktive Sauerstoffspezies (modifiziert nach Hartwig, 2001)

82 Zusammenfassende Diskussion und Ausblick
Stickstoffspezies (Huang et al., 2004; Shi et al., 2004) eine Oxidation der SH-
Gruppen des Zinkfingers zu Disulfitbrücken und damit einen Funktionsverlust des
Proteins bewirken. Die möglichen Wechselwirkungen von Arsen mit
Zinkfingerstrukturen werden in Abbildung 30 zusammengefasst. Hier wird
beispielshaft davon ausgegangen, dass ein Molekül einer Arsenverbindung, wie z.B.
MMA(III) zwei Cysteine binden kann, so dass ein Zinkfinger bis zu zwei
Arsenmoleküle binden könnte. Zudem ist es denkbar, dass es neben einer Bindung
von Arsen an das Protein gleichzeitig zu Oxidationsreaktionen und zur Ausbildung
von Disulfitbrücken kommen kann.
Die Hemmung wichtiger DNA-Reparaturproteine könnte die bekannten hemmenden
Effekte auf die BER, NER oder auch im Falle von PARP-1 auch die klastogenen
Wirkungen von Arsen erklären. Diese Befunde sind scheinbar wiedersprüchlich zu
einer Arbeit von Snow et al. (2005). In dieser Studie konnte gezeigt werden, dass es
in Fibroblasten und Keratinozyten nach 24stündiger Inkubation mit 0,5 µM Arsenit zu
einer geringeren ROS-Induktion verglichen mit der Kontrolle kam. Zudem fanden die
Autoren eine Stimulierung der Promotor-Aktivität von Polymerase ß und erhöhte
Polymerase ß-Gehalte nach Inkubation mit 0,1-0,5 µM Arsenit. Die Stimulierung von
DNA-Reparaturenzymen in niedrigen Arsenit-Konzentrationen deutet, so die
Interpretation der Autoren, damit eher auf protektiven Mechanismen durch Arsen hin.
Eine solche Induktion der Genexpression von DNA-Reparaturproteinen lässt sich
jedoch auch als Reaktion der Zelle auf genotoxische Effekte, wie die Induktion von
oxidativen DNA-Schäden oder Hemmungen von anderen DNA-Reparaturereignissen
z.B. der Poly(ADP-Ribosyl)ierung, interpretieren.
Die Konzentrationen, die nötig sind, die zelluläre Poly(ADP-Ribosyl)ierung zu
hemmen, liegen, wie schon ausführlich dargelegt, in extrem niedrigen nanomolaren
Konzentrationen. Diese Konzentrationen liegen im gleichen Bereich, wie
Urinkonzentrationen von über das Trinkwasser arsenexponierten Personen. In
verschiedenen Studien konnte gezeigt werden, dass bei einer Arsenaufnahme über
das Trinkwasser (30-1100 µg/l) die Konzentrationen von Arsenit (64-828 nM),
MMA(III) (9-242 nM) und DMA(III) (47-937 nM) im Bereich der beobachteten
Enzymhemmung liegen (Aposhian et al., 2000; Del Razo et al., 2001; Mandal et al.,
2001). Le et al (2000) konnten in zwei Studien für MMA(III) bei einer Arsen-
Trinkwasserkonzentration von 550-610 µg/l sogar Urinkonzentrationen über 1 µM
nachweisen. Für die beiden fünfwertigen Verbindungen MMA(V) und DMA(V)

Zusammenfassende Diskussion und Ausblick 83
konnten Spitztenwerte von bis zu 4 µM festgestellt werden (Le et al., 2000a; Le et al.,
2000b; Del Razo et al., 2001). Die Arsenspeziation im Blut ist schwieriger als im Urin,
so dass hier nur Gesamtarsengehalte angegeben werden können. Während in der
nichtexponierten Allgemeinbevölkerung Gesamtarsengehalte von 4-27 nM gefunden
wurden, zeigen mit belastetem Trinkwasser (100-400 µg/l) exponierte Personen
Blutkonzentrationen zwischen 55 und 172 nM Gesamtarsen (NRC, 1999; NRC,
2001). Es ist durchaus möglich, dass die Arsenverbindungen in der Zelle
akkumulieren und damit die intrazellulären Arsenkonzentrationen noch höher liegen
(Hartwig et al., 1997).
Vergleicht man die dreiwertigen mit den fünfwertigen Arsenverbindungen ist zudem
zu beachten, dass dreiwertige Verbindungen vorwiegend ungeladen vorliegen,
während fünfwertige Arsenspezies geladen sind und damit schlechter in die Zelle
aufgenommen werden. Dies ist eine Erklärungsmöglichkeit für die teilweise um
mehrere Größenordnungen geringere Reaktivität der fünfwertigen gegenüber den
dreiwertigen Verbindungen. Die Reaktivitätsunterschiede je nach Wertigkeit konnten
in allen Untersuchungen dieser Arbeit gezeigt werden und bestätigen damit eine
Vielzahl von Genotoxizitätsprüfungen wie z.B. die Induktion von DNA-Schäden
(Schwerdtle et al., 2003b) oder die Hemmung Benzo[a]pyren-induzierter DNA-
Reparatur (Schwerdtle et al., 2003a). Während es in Bezug auf die Wertigkeit leicht
ist eine Reaktivitätsreihenfolge zu erkennen, fällt es schwer eine generelle
Reihenfolge bezogen auf den Methylierungsgrad der Arsenverbindungen
festzulegen. In den meisten Testsystemen zeigen die methylierten dreiwertigen
Arsenverbindungen ein höheres genotoxisches Potential verglichen mit ihrer
Ausgangssubstanz Arsenit. Vergleicht man allerdings MMA(III) mit DMA(III) ergibt
sich kein klarer Reaktivitätsunterschied. So setzt beispielsweise MMA(III) in
niedrigeren Konzentrationen Zink aus XPAzf frei, während DMA(III) den stärkeren
hemmenden Effekt auf Fpg ausübt. Wichtig festzuhalten ist allerdings, dass in fast
allen Versuchen dieser Arbeit gezeigt werden konnte, dass die dreiwertigen
Metaboliten die stärksten Effekte aller Arsenverbindungen verursachten. Die Einzige
Ausnahme bildet die Hemmung isolierter PARP-1, bestimmt über das Slot-Blot-
Testsystem, wo Arsenit, MMA(III) und DMA(III) in etwa gleiche Effekte zeigten.
Ausgehend von diesen Ergebnissen zeigt sich, dass die Biomethylierung von Arsen
keine detoxifizierende Reaktion darstellt. Vielmehr ist es wahrscheinlich, dass vor
allem die dreiwertigen Arsenmetabolite MMA(III) und DMA(III) entscheidend zur

84 Zusammenfassende Diskussion und Ausblick
arseninduzierten Genotoxizität und damit auch zur Kanzerogenität von
anorganischem Arsen beitragen.
Es ist anzunehmen, dass die unterschiedlichen Arsenverbindungen verschiedene
Schädigungsmechanismen und/oder Schädigungsspektren besitzen. So zeigten die
dreiwertigen Verbindungen in zytotxischen Konzentrationen beispielsweise
unterschiedliche morphologische Veränderungen an HeLa S3 Zellen. Während
Arsenit und MMA(III) eine Abrundung der Zellen bewirkten, zeigte sich im Falle von
DMA(III) eine Ausfransung der Zellumrisse. Es wird zudem spekuliert, dass die
verschiedenen Arsenverbindungen unterschiedliche Zielorgane schädigen können. In
der Leber werden alle Arsenverbindungen gefunden, in der Niere vor allem MMA(V)
und DMA(V). In der Blase werden die fünfwertigen Verbindungen schnell
ausgeschieden. Obwohl hier keine Metabolisierung stattfindet kann es sein, dass
sich dreiwertige Arsenverbindungen durch Reduktion von fünfwertigen Spezies
bilden (Sampayo-Reyes et al., 2000) und damit zur Entstehung von Blasenkrebs
beitragen (zusammengefasst in Ahmad et al., 2002). In anderen Organen wie der
Haut, in der keine Metabolisierungsreaktionen ablaufen, werden synergistische
Effekte mit anderen genotoxischen Effekten wie z.B. UV-Strahlung diskutiert
(Rossman et al., 2001). Betrachtet man vor allem die berufliche Exposition
gegenüber Arsen, zeigt sich, dass es oft zu einer Mischexposition mit anderen
Metallen oder Umweltgiften wie z.B. Benzo[a]pyren kommen kann, die die Wirkung
der einzelnen Verbindungen noch erhöhen kann. Die Neubewertung und Einstufung
der Toxizität von Umweltkanzerogenen wie Arsen ist daher eine wichtige Aufgabe, zu
der die Ergebnisse dieser Arbeit beitragen können.
Die Daten dieser Arbeit liefern zudem einen wichtigen Beitrag zum Verständnis der
DNA-Reparatur-Hemmung durch Arsenverbindungen. Zinkbindende Proteine, die an
wichtigen DNA-Reparaturwegen beteiligt sind könnten sensitive Angriffspunkte vor
allem für dreiwertige Arsenverbindungen darstellen. Neben den in dieser Arbeit
untersuchten Zinkfingerproteinen spielen noch weitere zinkbindende Proteine eine
wichtige Rolle an der Aufrechtherhaltung der genomischen Stabilität der Zelle. So ist
es beispielsweise denkbar, dass auch p53, welches Zink über drei Cyteine und ein
Histidin bindet, durch Arsen gestört wird und damit wichtige Funktionen wie z.B. die
Regulation des Zellzyklus oder das Auslösen von Apoptose nicht mehr erfüllen kann
(Levine, 1997). Die möglichen Auswirkungen auf wichtige Mechanismen zur
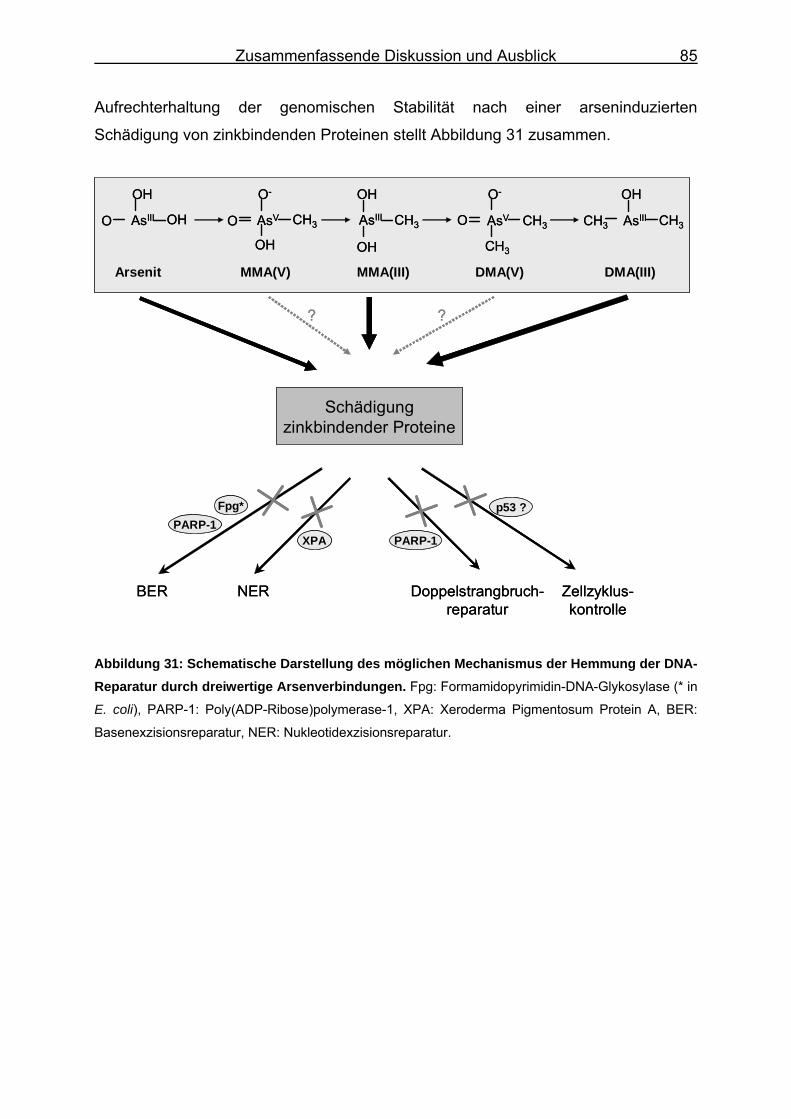
Zusammenfassende Diskussion und Ausblick 85
Aufrechterhaltung der genomischen Stabilität nach einer arseninduzierten
Schädigung von zinkbindenden Proteinen stellt Abbildung 31 zusammen.
OH
O-
CH3O AsV
MMA(V)
OH
OH
CH3AsIII
MMA(III)
CH3
CH3
O-
O AsV
DMA(V)
OH
CH3CH3 AsIII
DMA(III)
OH
OHO AsIII
Arsenit
BER NER Doppelstrangbruch-reparatur
Zellzyklus-kontrolle
XPA
Fpg*PARP-1
PARP-1
p53 ?
Schädigungzinkbindender Proteine
? ?
OH
O-
CH3O AsV
OH
O-
CH3O AsV
MMA(V)
OH
OH
CH3AsIII
OH
OH
CH3AsIII
MMA(III)
CH3
CH3
O-
O AsV CH3
CH3
O-
O AsV
DMA(V)
OH
CH3CH3 AsIII
OH
CH3CH3 AsIII
DMA(III)
OH
OHO AsIII
OH
OHO AsIII
Arsenit
BER NER Doppelstrangbruch-reparatur
Zellzyklus-kontrolle
XPA
Fpg*PARP-1
PARP-1
p53 ?
Schädigungzinkbindender Proteine
? ?
Abbildung 31: Schematische Darstellung des möglichen Mechanismus der Hemmung der DNA-Reparatur durch dreiwertige Arsenverbindungen. Fpg: Formamidopyrimidin-DNA-Glykosylase (* in
E. coli), PARP-1: Poly(ADP-Ribose)polymerase-1, XPA: Xeroderma Pigmentosum Protein A, BER:
Basenexzisionsreparatur, NER: Nukleotidexzisionsreparatur.

86 Literatur
7. Literatur
Ahmad, S., Kitchin, K. T. and Cullen, W. R. (2002). Plasmid DNA damage caused by methylated arsenicals, ascorbic acid and human liver ferritin. Toxicol Lett 133(1): 47-57. Allinson, S. L., Dianova, II and Dianov, G. L. (2003). Poly(ADP-ribose) polymerase in base excision repair: always engaged, but not essential for DNA damage processing. Acta Biochim Pol 50(1): 169-179. Ame, J. C., Rolli, V., Schreiber, V., Niedergang, C., Apiou, F., Decker, P., Muller, S., Hoger, T., Menissier-de Murcia, J. and de Murcia, G. (1999). PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274(25): 17860-17868. Ame, J. C., Spenlehauer, C. and de Murcia, G. (2004). The PARP superfamily. Bioessays 26(8): 882-893. Andrew, A. S., Karagas, M. R. and Hamilton, J. W. (2003). Decreased DNA repair gene expression among individuals exposed to arsenic in United States drinking water. Int J Cancer 104(3): 263-268. Aposhian, H. V., Gurzau, E. S., Le, X. C., Gurzau, A., Healy, S. M., Lu, X., Ma, M., Yip, L., Zakharyan, R. A., Maiorino, R. M., Dart, R. C., Tircus, M. G., Gonzalez-Ramirez, D., Morgan, D. L., Avram, D. and Aposhian, M. M. (2000). Occurrence of monomethylarsonous acid in urine of humans exposed to inorganic arsenic. Chem Res Toxicol 13(8): 693-697. Asahina, H., Kuraoka, I., Shirakawa, M., Morita, E. H., Miura, N., Miyamoto, I., Ohtsuka, E., Okada, Y. and Tanaka, K. (1994). The XPA protein is a zinc metalloprotein with an ability to recognize various kinds of DNA damage. Mutat Res 315(3): 229-237. Asmuss, M., Mullenders, L. H., Eker, A. and Hartwig, A. (2000). Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis 21(11): 2097-2104. ATSDR (1993). Toxicological profile for arsenic, update. Department of Health and Human Services, Public Health Service, Atlanta, Georgia. Bal, W., Schwerdtle, T. and Hartwig, A. (2003). Mechanism of nickel assault on the zinc finger of DNA repair protein XPA. Chem Res Toxicol 16(2): 242-248. Bau, D. T., Gurr, J. R. and Jan, K. Y. (2001). Nitric oxide is involved in arsenite inhibition of pyrimidine dimer excision. Carcinogenesis 22(5): 709-716. Becher, K. and Wahrendorf, J. (1998). Metalle/Arsen, in H.E. Wichmann, H.W. Schlipköter and G. Füllgraff (Hrsg.). Handbuch der Umweltmedizin, ecomed, Landsberg/Lech.

Literatur 87
Beneke, S., Alvarez-Gonzalez, R. and Bürkle, A. (2000). Comparative characterisation of poly(ADP-ribose) polymerase-1 from two mammalian species with different life span. Exp Gerontol 35(8): 989-1002. Bernard, B., Pretet, J. L., Charlot, J. F. and Mougin, C. (2003). Human papillomaviruses type 16+ and 18+ cervical carcinoma cells are sensitive to staurosporine-mediated apoptosis. Biol Cell 95(1): 17-26. Bhagwat, M. and Gerlt, J. A. (1996). 3'- and 5'-strand cleavage reactions catalyzed by the Fpg protein from Escherichia coli occur via successive beta- and delta-elimination mechanisms, respectively. Biochemistry 35(2): 659-665. Blessing, H., Kraus, S., Heindl, P., Bal, W. and Hartwig, A. (2004). Interaction of selenium compounds with zinc finger proteins involved in DNA repair. Eur J Biochem 271(15): 3190-3199. Boiteux, S. (1993). Properties and biological functions of the NTH and FPG proteins of Escherichia coli: two DNA glycosylases that repair oxidative damage in DNA. J Photochem Photobiol B 19(2): 87-96. Bouchard, V. J., Rouleau, M. and Poirier, G. G. (2003). PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 31(6): 446-454. Buchet, J. P., Lauwerys, R. and Roels, H. (1981). Comparison of the urinary excretion of arsenic metabolites after a single oral dose of sodium arsenite, monomethylarsonate, or dimethylarsinate in man. Int Arch Occup Environ Health 48(1): 71-79. Buchko, G. W., Hess, N. J., Bandaru, V., Wallace, S. S. and Kennedy, M. A. (2000). Spectroscopic studies of Zinc(II)- and Cobalt(II)-associated escherichia coli formamidopyrimidine-DNA glycosylase: extended X-ray absorption fine structure evidence for a metal-binding domain [In Process Citation]. Biochemistry 39(40): 12441-12449. Buchko, G. W., Ni, S., Thrall, B. D. and Kennedy, M. A. (1998). Structural features of the minimal DNA binding domain (M98-F219) of human nucleotide excision repair protein XPA. Nucleic Acids Res 26(11): 2779-2788. Bürkle, A., Brabeck, C., Diefenbach, J. and Beneke, S. (2005a). The emerging role of poly(ADP-ribose) polymerase-1 in longevity. Int J Biochem Cell Biol 37(5): 1043-1053. Bürkle, A., Chen, G., Kupper, J. H., Grube, K. and Zeller, W. J. (1993). Increased poly(ADP-ribosyl)ation in intact cells by cisplatin treatment. Carcinogenesis 14(4): 559-561. Bürkle, A., Diefenbach, J., Brabeck, C. and Beneke, S. (2005b). Ageing and PARP. Pharmacol Res 52(1): 93-99.

88 Literatur
Bürkle, A., Meyer, T., Hilz, H. and zur Hausen, H. (1987). Enhancement of N-methyl-N'-nitro-N-nitrosoguanidine-induced DNA amplification in a Simian virus 40-transformed Chinese hamster cell line by 3-aminobenzamide. Cancer Res 47(14): 3632-3636. Cabrera, M., Nghiem, Y. and Miller, J. H. (1988). mutM, a second mutator locus in Escherichia coli that generates G.C----T.A transversions. J Bacteriol 170(11): 5405-5407. Chambon, P., Weill, J. D. and Mandel, P. (1963). Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun 11: 39-43. Chen, G. Q., Zhou, L., Styblo, M., Walton, F., Jing, Y., Weinberg, R., Chen, Z. and Waxman, S. (2003). Methylated metabolites of arsenic trioxide are more potent than arsenic trioxide as apoptotic but not differentiation inducers in leukemia and lymphoma cells. Cancer Res 63(8): 1853-1859. Chou, C. H. and De Rosa, C. T. (2003). Case studies--arsenic. Int J Hyg Environ Health 206(4-5): 381-386. Cleaver, J. E. and States, J. C. (1997). The DNA damage-recognition problem in human and other eukaryotic cells: the XPA damage binding protein. Biochem J 328(Pt 1): 1-12. D'Amours, D., Desnoyers, S., D'Silva, I. and Poirier, G. G. (1999). Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342 ( Pt 2): 249-268. Del Razo, L. M., Styblo, M., Cullen, W. R. and Thomas, D. J. (2001). Determination of trivalent methylated arsenicals in biological matrices. Toxicol Appl Pharmacol 174(3): 282-293. DFG (2004). Arsen und anorganische Arsenverbindungen, in: H.Greim (Hrsg), Gesundheitliche Arbeitsstoffe. Toxikologisch -arbeitsmedizinische Begründung von MAK-Werten. Deutsche Forschungsgemeinschaft, Weinheim: 1-50. Diefenbach, J. and Bürkle, A. (2005). Introduction to poly(ADP-ribose) metabolism. Cell Mol Life Sci 62(7-8): 721-730. Dopp, E., Hartmann, L. M., Florea, A. M., von Recklinghausen, U., Pieper, R., Shokouhi, B., Rettenmeier, A. W., Hirner, A. V. and Obe, G. (2004). Uptake of inorganic and organic derivatives of arsenic associated with induced cytotoxic and genotoxic effects in Chinese hamster ovary (CHO) cells. Toxicol Appl Pharmacol 201(2): 156-165. Dopp, E., Hartmann, L. M., von Recklinghausen, U., Florea, A. M., Rabieh, S., Zimmermann, U., Shokouhi, B., Yadav, S., Hirner, A. V. and Rettenmeier, A. W. (2005). Forced uptake of trivalent and pentavalent methylated and inorganic arsenic and its cyto-/genotoxicity in fibroblasts and hepatoma cells. Toxicol Sci 87(1): 46-56.

Literatur 89
Epe, B., Pflaum, M. and Boiteux, S. (1993). DNA damage induced by photosensitizers in cellular and cell-free systems. Mutat Res 299(3-4): 135-145. Flohr, C., Burkle, A., Radicella, J. P. and Epe, B. (2003). Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res 31(18): 5332-5337. Floyd, R. A., West, M. S., Eneff, K. L. and Schneider, J. E. (1989). Methylene blue plus light mediates 8-hydroxyguanine formation in DNA. Arch Biochem Biophys 273(1): 106-111. Friedberg, E. C. (2001). How nucleotide excision repair protects against cancer. Nat Rev Cancer 1(1): 22-33. Friedberg, E. C., Walker, G. C., Siede, W., Wood, R. D., Schultz, R. A. and Ellenberger, T. (2006). DNA Repair and Mutagenesis. ASM Press, Washington, D. C.(2. Edition). Gebel, T. W. (2001). Genotoxicity of arsenical compounds. Int J Hyg Environ Health 203(3): 249-262. Giner, H., Simonin, F., de Murcia, G. and Menissier-de Murcia, J. (1992). Overproduction and large-scale purification of the human poly(ADP-ribose) polymerase using a baculovirus expression system. Gene 114(2): 279-283. Hamadeh, H. K., Trouba, K. J., Amin, R. P., Afshari, C. A. and Germolec, D. (2002). Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicol Sci 69(2): 306-316. Hansen, H. R., Raab, A., Jaspars, M., Milne, B. F. and Feldmann, J. (2004). Sulfur-containing arsenical mistaken for dimethylarsinous acid [DMA(III)] and identified as a natural metabolite in urine: major implications for studies on arsenic metabolism and toxicity. Chem Res Toxicol 17(8): 1086-1091. Hartwig, A. (2000). Metallionen zwischen Essenzialität und Toxizität. Chemie in unserer Zeit 34(4): 224-231. Hartwig, A. (2001). Zinc finger proteins as potential targets for toxic metal ions: differential effects on structure and function. Antioxid Redox Signal 3(4): 625-634. Hartwig, A., Asmuss, M., Blessing, H., Hoffmann, S., Jahnke, G., Khandelwal, S., Pelzer, A. and Bürkle, A. (2002). Interference by toxic metal ions with zinc-dependent proteins involved in maintaining genomic stability. Food Chem Toxicol 40(8): 1179-1184. Hartwig, A., Groblinghoff, U. D., Beyersmann, D., Natarajan, A. T., Filon, R. and Mullenders, L. H. (1997). Interaction of arsenic(III) with nucleotide excision repair in UV- irradiated human fibroblasts. Carcinogenesis 18(2): 399-405.

90 Literatur
Hartwig, A., Pelzer, A., Asmuss, M. and Bürkle, A. (2003). Very low concentrations of arsenite suppress poly(ADP-ribosyl)ation in mammalian cells. Int J Cancer 104(1): 1-6. Hassauer, M. and Kalberlah, F. (1999). Arsen und Verbindungen. Gefährdungsabschätzungen von Umweltschadstoffen: 1-28. Hayakawa, T., Kobayashi, Y., Cui, X. and Hirano, S. (2005). A new metabolic pathway of arsenite: arsenic-glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch Toxicol 79(4): 183-191. Hayashi, H., Kanisawa, M., Yamanaka, K., Ito, T., Udaka, N., Ohji, H., Okudela, K., Okada, S. and Kitamura, H. (1998). Dimethylarsinic acid, a main metabolite of inorganic arsenics, has tumorigenicity and progression effects in the pulmonary tumors of A/J mice. Cancer Lett 125(1-2): 83-88. Hazra, T. K., Izumi, T., Kow, Y. W. and Mitra, S. (2003). The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis 24(2): 155-157. Hollemann, A. F. and Wieberg, E. (1995). Lehrbuch der anorganischen Chemie. Berlin, New York, de Gruyter-Verlag. Hu, Y., Su, L. and Snow, E. T. (1998). Arsenic toxicity is enzyme specific and its affects on ligation are not caused by the direct inhibition of DNA repair enzymes. Mutat Res 408(3): 203-218. Huang, C., Ke, Q., Costa, M. and Shi, X. (2004). Molecular mechanisms of arsenic carcinogenesis. Mol Cell Biochem 255(1-2): 57-66. Huang, R. N. and Lee, T. C. (1996). Cellular uptake of trivalent arsenite and pentavalent arsenate in KB cells cultured in phosphate-free medium. Toxicol Appl Pharmacol 136(2): 243-249. Hughes, M. F., Devesa, V., Adair, B. M., Styblo, M., Kenyon, E. M. and Thomas, D. J. (2005). Tissue dosimetry, metabolism and excretion of pentavalent and trivalent monomethylated arsenic in mice after oral administration. Toxicol Appl Pharmacol 208(2): 186-197. IARC (1980). Arsenic and Arsenic compounds. IARC monographs on the evaluation of the carcinogenic risk of chemicals and Humans 32: 39-141. Ikegami, T., Kuraoka, I., Saijo, M., Kodo, N., Kyogoku, Y., Morikawa, K., Tanaka, K. and Shirakawa, M. (1998). Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat Struct Biol 5(8): 701-706. Ikejima, M., Noguchi, S., Yamashita, R., Ogura, T., Sugimura, T., Gill, D. M. and Miwa, M. (1990). The zinc fingers of human poly(ADP-ribose) polymerase are differentially required for the recognition of DNA breaks and nicks and the consequent enzyme activation. Other structures recognize intact DNA. J Biol Chem 265(35): 21907-21913.

Literatur 91
Jiang, G., Gong, Z., Li, X. F., Cullen, W. R. and Le, X. C. (2003). Interaction of trivalent arsenicals with metallothionein. Chem Res Toxicol 16(7): 873-880. Jones, C. J. and Wood, R. D. (1993). Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry 32(45): 12096-12104. Kawamitsu, H., Hoshino, H., Okada, H., Miwa, M., Momoi, H. and Sugimura, T. (1984). Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry 23(16): 3771-3777. Kitchin, K. T. (2001). Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol 172(3): 249-261. Kligerman, A. D., Doerr, C. L., Tennant, A. H., Harrington-Brock, K., Allen, J. W., Winkfield, E., Poorman-Allen, P., Kundu, B., Funasaka, K., Roop, B. C., Mass, M. J. and DeMarini, D. M. (2003). Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: Induction of chromosomal mutations but not gene mutations. Environ Mol Mutagen 42(3): 192-205. Kofler, B., Wallraff, E., Herzog, H., Schneider, R., Auer, B. and Schweiger, M. (1993). Purification and characterization of NAD+:ADP-ribosyltransferase (polymerizing) from Dictyostelium discoideum. Biochem J 293 ( Pt 1): 275-281. Krishna, S. S., Majumdar, I. and Grishin, N. V. (2003). Structural classification of zinc fingers: survey and summary. Nucleic Acids Res 31(2): 532-550. Küpper, J. H., de Murcia, G. and Burkle, A. (1990). Inhibition of poly(ADP-ribosyl)ation by overexpressing the poly(ADP- ribose) polymerase DNA-binding domain in mammalian cells. J Biol Chem 265(31): 18721-18724. Küpper, J. H., van Gool, L. and Burkle, A. (1995). Molecular genetic systems to study the role of poly(ADP-ribosyl)ation in the cellular response to DNA damage. Biochimie 77(6): 450-455. LAI (1993). Krebsrisiko durch Luftverunreinigungen. Materialband, Band I und II. Raumordnung und Landwirtschaft des Landes Nordrhein-westfalen, Länderausschuss für Emissionsschutz. Laity, J. H., Lee, B. M. and Wright, P. E. (2001). Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol 11(1): 39-46. Le Rhun, Y., Kirkland, J. B. and Shah, G. M. (1998). Cellular responses to DNA damage in the absence of Poly(ADP-ribose) polymerase. Biochem Biophys Res Commun 245(1): 1-10. Le, X. C., Lu, X., Ma, M., Cullen, W. R., Aposhian, H. V. and Zheng, B. (2000a). Speciation of key arsenic metabolic intermediates in human urine. Anal Chem 72(21): 5172-5177.

92 Literatur
Le, X. C., Ma, M., Cullen, W. R., Aposhian, H. V., Lu, X. and Zheng, B. (2000b). Determination of monomethylarsonous acid, a key arsenic methylation intermediate, in human urine. Environ Health Perspect 108(11): 1015-1018. Levine, A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88(3): 323-331. Li, J. H. and Rossman, T. G. (1989). Inhibition of DNA ligase activity by arsenite: a possible mechanism of its comutagenesis. Mol Toxicol 2(1): 1-9. Liu, Z., Styblo, M. and Rosen, B. P. (2006). Methylarsonous acid transport by aquaglyceroporins. Environ Health Perspect 114(4): 527-531. Lynn, S., Shiung, J. N., Gurr, J. R. and Jan, K. Y. (1998). Arsenite stimulates poly(ADP-ribosylation) by generation of nitric oxide. Free Radic Biol Med 24(3): 442-449. Malanga, M. and Althaus, F. R. (2005). The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol 83(3): 354-364. Mandal, B. K., Ogra, Y. and Suzuki, K. T. (2001). Identification of dimethylarsinous and monomethylarsonous acids in human urine of the arsenic-affected areas in West Bengal, India. Chem Res Toxicol 14(4): 371-378. Maret, W. (2002). Optical methods for measuring zinc binding and release, zinc coordination environments in zinc finger proteins, and redox sensitivity and activity of zinc-bound thiols. Methods Enzymol 348: 230-237. Maret, W. (2003). Cellular zinc and redox states converge in the metallothionein/thionein pair. J Nutr 133(5 Suppl 1): 1460S-1462S. Mass, M. J., Tennant, A., Roop, B. C., Cullen, W. R., Styblo, M., Thomas, D. J. and Kligerman, A. D. (2001). Methylated trivalent arsenic species are genotoxic. Chem Res Toxicol 14(4): 355-361. Matschullat, J. (1999). Arsen in der Geosphäre. Schriftenreihe Deutsche Geologische Gesellschaft 6: 5-20. McCall, K. A. and Fierke, C. A. (2000). Colorimetric and fluorimetric assays to quantitate micromolar concentrations of transition metals. Anal Biochem 284(2): 307-315. Menissier de Murcia, J., Ricoul, M., Tartier, L., Niedergang, C., Huber, A., Dantzer, F., Schreiber, V., Ame, J. C., Dierich, A., LeMeur, M., Sabatier, L., Chambon, P. and de Murcia, G. (2003). Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. Embo J 22(9): 2255-2263.

Literatur 93
Meyer, R., Müller, M., Beneke, S., Küpper, J. H. and Bürkle, A. (2000). Negative regulation of alkylation-induced sister-chromatid exchange by poly(ADP-ribose) polymerase-1 activity. Int J Cancer 88(3): 351-355. Miranda, E. A., de-Murcia, G. and Menissier-de-Murcia, J. (1997). Large-scale production and purification of recombinant protein from an insect cell/baculovirus system in Erlenmeyer flasks: application to the chicken poly(ADP-ribose) polymerase catalytic domain. Braz J Med Biol Res 30(8): 923-928. Miyamoto, I., Miura, N., Niwa, H., Miyazaki, J. and Tanaka, K. (1992). Mutational analysis of the structure and function of the xeroderma pigmentosum group A complementing protein. Identification of essential domains for nuclear localization and DNA excision repair. J Biol Chem 267(17): 12182-12187. Morikawa, K. and Shirakawa, M. (2000). Three-dimensional structural views of damaged-DNA recognition: T4 endonuclease V, E. coli Vsr protein, and human nucleotide excision repair factor XPA. Mutat Res 460(3-4): 257-275. Müller, E., Boiteux, S., Cunningham, R. P. and Epe, B. (1990). Enzymatic recognition of DNA modifications induced by singlet oxygen and photosensitizers. Nucleic Acids Res 18(20): 5969-5973. NRC (1999). Arsenic in drinking water. National Acadamy Press, Washington D.C. NRC (2001). Arsenic in drinking water 2001 update. National Acadamy Press, Washington D.C. Ochi, T., Nakajima, F., Sakurai, T., Kaise, T. and Oya-Ohta, Y. (1996). Dimethylarsinic acid causes apoptosis in HL-60 cells via interaction with glutathione. Arch Toxicol 70(12): 815-821. O'Connor, T. R., Graves, R. J., de Murcia, G., Castaing, B. and Laval, J. (1993). Fpg protein of Escherichia coli is a zinc finger protein whose cysteine residues have a structural and/or functional role. J Biol Chem 268(12): 9063-9070. Oei, S. L. and Ziegler, M. (2000). ATP for the DNA ligation step in base excision repair is generated from poly(ADP-ribose). J Biol Chem 275(30): 23234-23239. Peel, A. E., Brice, A., Marzin, D. and Erb, F. (1991). Cellular uptake and biotransformation of arsenic(V) in transformed human cell lines HeLa S3 and Hep G2. Toxicol. in Vitro 5(2): 165-168. Petrick, J. S., Ayala-Fierro, F., Cullen, W. R., Carter, D. E. and Vasken Aposhian, H. (2000). Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol 163(2): 203-207. Petrick, J. S., Jagadish, B., Mash, E. A. and Aposhian, H. V. (2001). Monomethylarsonous Acid (MMAIII) and Arsenite: LD50 in Hamsters and In Vitro Inhibition of Pyruvate Dehydrogenase. Chemical Research in Toxicology 14(6): 651-656.

94 Literatur
Pion, E., Ullmann, G. M., Ame, J. C., Gerard, D., de Murcia, G. and Bombarda, E. (2005). DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry 44(44): 14670-14681. Poonepalli, A., Balakrishnan, L., Khaw, A. K., Low, G. K., Jayapal, M., Bhattacharjee, R. N., Akira, S., Balajee, A. S. and Hande, M. P. (2005). Lack of poly(ADP-ribose) polymerase-1 gene product enhances cellular sensitivity to arsenite. Cancer Res 65(23): 10977-10983. Pott, W. A., Benjamin, S. A. and Yang, R. S. (2001). Pharmacokinetics, metabolism, and carcinogenicity of arsenic. Rev Environ Contam Toxicol 169: 165-214. Rossman, T. G., Uddin, A. N., Burns, F. J. and Bosland, M. C. (2001). Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: an animal model for arsenic carcinogenesis. Toxicol Appl Pharmacol 176(1): 64-71. Sakurai, T., Kaise, T. and Matsubara, C. (1998). Inorganic and methylated arsenic compounds induce cell death in murine macrophages via different mechanisms. Chem Res Toxicol 11(4): 273-283. Sakurai, T., Ochiai, M., Kojima, C., Ohta, T., Sakurai, M. H., Takada, N. O., Qu, W., Waalkes, M. P., Himeno, S. and Fujiwara, K. (2005). Preventive mechanism of cellular glutathione in monomethylarsonic acid-induced cytolethality. Toxicol Appl Pharmacol 206(1): 54-65. Sakurai, T., Qu, W., Sakurai, M. H. and Waalkes, M. P. (2002). A major human arsenic metabolite, dimethylarsinic acid, requires reduced glutathione to induce apoptosis. Chem Res Toxicol 15(5): 629-637. Salditt, M., Braunstein, S. N., Camerini-Otero, R. D. and Franklin, R. M. (1972). Structure and synthesis of a lipid-containing bacteriophage. X. Improved techniques for the purification of bacteriophage PM2. Virology 48(1): 259-262. Sampayo-Reyes, A., Zakharyan, R. A., Healy, S. M. and Aposhian, H. V. (2000). Monomethylarsonic acid reductase and monomethylarsonous acid in hamster tissue. Chem Res Toxicol 13(11): 1181-1186. Schreiber, V., Dantzer, F., Ame, J. C. and de Murcia, G. (2006). Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7(7): 517-528. Schwerdtle, T., Hamann, I., Jahnke, G., Walter, I., Richter, C., Parsons, J. L., Dianov, G. L. and Hartwig, A. (2007). Impact of copper on the induction and repair of oxidative DNA damage, poly(ADP-ribosyl)ation and PARP-1 activity. Molecular Nutrition and Food Research 51: 201-210. Schwerdtle, T., Walter, I. and Hartwig, A. (2003a). Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair (Amst) 2(12): 1449-1463.

Literatur 95
Schwerdtle, T., Walter, I., Mackiw, I. and Hartwig, A. (2003b). Induction of oxidative DNA damage by arsenite and its trivalent and pentavalent methylated metabolites in cultured human cells and isolated DNA. Carcinogenesis 24(5): 967-974. Shi, H., Shi, X. and Liu, K. J. (2004). Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem 255(1-2): 67-78. Snow, E. T., Sykora, P., Durham, T. R. and Klein, C. B. (2005). Arsenic, mode of action at biologically plausible low doses: What are the implications for low dose cancer risk? Toxicol Appl Pharmacol 207(2S): 557-564. Styblo, M., Del Razo, L. M., Vega, L., Germolec, D. R., LeCluyse, E. L., Hamilton, G. A., Reed, W., Wang, C., Cullen, W. R. and Thomas, D. J. (2000). Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol 74(6): 289-299. Sugasawa, K., Ng, J. M., Masutani, C., Iwai, S., van der Spek, P. J., Eker, A. P., Hanaoka, F., Bootsma, D. and Hoeijmakers, J. H. (1998). Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell 2(2): 223-232. Süsse, S., Scholz, C. J., Bürkle, A. and Wiesmuller, L. (2004). Poly(ADP-ribose) polymerase (PARP-1) and p53 independently function in regulating double-strand break repair in primate cells. Nucleic Acids Res 32(2): 669-680. Tallman, M. S. (2004). Acute promyelocytic leukemia as a paradigm for targeted therapy. Semin Hematol 41(2 Suppl 4): 27-32. Tchou, J., Bodepudi, V., Shibutani, S., Antoshechkin, I., Miller, J., Grollman, A. P. and Johnson, F. (1994). Substrate specificity of Fpg protein. Recognition and cleavage of oxidatively damaged DNA. J Biol Chem 269(21): 15318-15324. Tchou, J., Michaels, M. L., Miller, J. H. and Grollman, A. P. (1993). Function of the zinc finger in Escherichia coli Fpg protein. J Biol Chem 268(35): 26738-26744. Thomas, D. J., Styblo, M. and Lin, S. (2001). The cellular metabolism and systemic toxicity of arsenic. Toxicol Appl Pharmacol 176(2): 127-144. Thornton, I. (1999). Arsenic in the global environment: Looking towards the millenium, in: W.R. Chappell, C.O. Abernathy and R.L. Calderon (Hrsg),. Arsenic exposure and health effects, Elsevier, Oxford, 1-7. Trucco, C., Flatter, E., Fribourg, S., de Murcia, G. and Menissier-de Murcia, J. (1996). Mutations in the amino-terminal domain of the human poly(ADP-ribose) polymerase that affect its catalytic activity but not its DNA binding capacity. FEBS Lett 399(3): 313-316. Vahter, M. (1999). Methylation of inorganic arsenic in different mammalian species and population groups. Sci Prog 82(( Pt 1)): 69-88.

96 Literatur
Vega, L., Styblo, M., Patterson, R., Cullen, W., Wang, C. and Germolec, D. (2001). Differential effects of trivalent and pentavalent arsenicals on cell proliferation and cytokine secretion in normal human epidermal keratinocytes. Toxicol Appl Pharmacol 172(3): 225-232. Vogelstein, B., Alberts, B. and Shine, K. (2002). Genetics. Please don't call it cloning! Science 295(5558): 1237. Volker, M., Mone, M. J., Karmakar, P., van Hoffen, A., Schul, W., Vermeulen, W., Hoeijmakers, J. H., van Driel, R., van Zeeland, A. A. and Mullenders, L. H. (2001). Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell 8(1): 213-224. Waalkes, M. P., Liu, J., Ward, J. M. and Diwan, B. A. (2004). Animal models for arsenic carcinogenesis: inorganic arsenic is a transplacental carcinogen in mice. Toxicol Appl Pharmacol 198(3): 377-384. Walter, I., Schwerdtle, T., Thuy, C., Parsons, J. L., Dianov, G. L. and Hartwig, A. (2007). Impact of arsenite and its methylated metabolites on PARP-1 activity, PARP-1 gene expression and poly(ADP-ribosyl)ation in cultured human cells. DNA Repair (Amst) 6(1): 61-70. Webster, K. A., Prentice, H. and Bishopric, N. H. (2001). Oxidation of zinc finger transcription factors: physiological consequences. Antioxid Redox Signal 3(4): 535-548. Whitaker, J. R. and Granum, P. E. (1980). An absolute method for protein determination based on difference in absorbance at 235 and 280 nm. Anal Biochem 109(1): 156-159. WHO (1987). Air quality guidelines for Europe. WHO Regional Publications European Series No. 23, Kopenhagen. WHO (2001). Arsenic and arsenic compounds. IPCS, International Programme on Chemical Safety Environmental Health Criteria 224, Genf. Wilcox, D. E., Schenk, A. D., Feldman, B. M. and Xu, Y. (2001). Oxidation of zinc-binding cysteine residues in transcription factor proteins. Antioxid Redox Signal 3(4): 549-564. Yager, J. W. and Wiencke, J. K. (1997). Inhibition of poly(ADP-ribose) polymerase by arsenite. Mutat Res 386(3): 345-351. Yamauchi, N., Watanabe, N., Kuriyama, H., Neda, H., Maeda, M., Himeno, T., Tsuji, Y. and Niitsu, Y. (1990). Suppressive effects of intracellular glutathione on hydroxyl radical production induced by tumor necrosis factor. Int J Cancer 46(5): 884-888. Yoshida, T., Yamauchi, H. and Fan Sun, G. (2004). Chronic health effects in people exposed to arsenic via the drinking water: dose-response relationships in review. Toxicol Appl Pharmacol 198(3): 243-252.

Anhang 97
8. Anhang
8.1 Abkürzungsverzeichnis
10H monoklonaler Antikörper gegen PAR
A Adenin
ADP Adenosindiphosphat
AIF apoptosis inducing factor
ATP Adenosintriphosphat
AP- Stelle Apurin-/Apyrimidin-Stelle
ATSDR Agency for Toxic Substances and Disease Registry
AU Alkalische Entwindung
BER Basenexzisionsreparatur
bp Basenpaare
BPDE Benzo[a]pyrendiolepoxid
BRCA-1 breast cancer protein 1
BRCT BRACA-1 homologe Domöne
BSA Rinderserumalbumin
C Cytosin
CA Chromosomenabberation
CCA Chromiertes Kupferarsenat
cDNA komplementäre DNA
CII-10 monoklonaler Antikörper gegen PARP-1
CPD Cyclopyriminindimer
Cys Cystein
DAPI 4',6-Diamidino-2-Phenylindol
DDB damage DNA binding protein
DFG Deutsche Forschungsgemeinschaft
DMA(III) dimethylarsinige Säure
DMA(V) Dimethylarsinsäure
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
DTT Dithiotreitol

98 Anhang
E. coli Escherichia coli
EDTA Ethylendiamintetraessigsäure
EPA Environmental Protection Agency
ERCC1 excision repair cross-complementing factor 1
F 12 F 12 Kulturmedium
FaPy Formamidopyrimidin
FEN-1 Flapendonuklease-1
FITC Flurescin-5-isothicyanat
FKS Fötales Kälberserum
Fpg Formamidopyrimidin-DNA-Glykosylase
G Guanin
GAPDH Glycerinaldehyd-3-Phosphatdehydrogenase
GGR globale genomische Reapratur
GSSG Glutathion (ox.)
GSH Glutathion (red.)
HeLa S3 menschliche Zervixkarzinomzellinie
HEPES 2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethansulfonsäure
His Histidin
hHR23B UV-Exzisionsreparaturprotein Rad 23 Homolog B
HPV humaner Papillomavirus
HRP Meerrettichperoxidase
HTH helix-turn-helix-Motiv
IARC Interational Agency for Reseach on Cancer
kDa Kilodalton
LAI Länderausschuss für Emmisionsschutz
MAK Maximale Arbeitsplatz Konzentration
MBD minimale Bindungsdomäne
MMA(III) monomethylarsonige Säure
MMA(V) Monomethylasonsäure
MMR Mismatch- Reparatur
MNNG N-methyl-N'-nitro-N-nitrosoguanidin
moi multiplicity of infection = Verhältnis Viruszahl zu Zellzahl
NAD+ Nicotinamidadenindinukleotid
Neil I, II und III Endonuklease-VIII-like-DNA-glykosylase I,II und III

Anhang 99
NER Nukleotidexzisionsreparatur
NF-κB Nukleärer Faktor κB
NLS Kernerkennungssignal
NRC National Research Council
NTC-Puffer NaCl-CaCl2-TRIS-Puffer
OGG1 8-Oxoguanin-DNA-glycosylase-1
OD optische Dichte
PAGE Polyacrylamidgelelektrophorese
PAR Poly(ADP-Ribose)
PARG Poly(ADP-Ribose)glycohydrolase
PARP Poly(ADP-Ribose)polymerase
PBS phosphatgepupperte Salzlösung
PBS-T PBS mit Tween 20
PBS-MT PBS mit Tween 20 und Magermilchpulver
PCNA proliferating cell nuclear antigen
PEG Polyethylenglycol
PFU plaque forming unit
PM2 Bakteriophage
PMSF Phenylmethansulfonylfluorid
PVDF Polyvinylidenfluorid
RFC DNA Replications Faktor
RNA Ribonukleinsäure
ROS reaktive Sauerstoffspezies
RPA Replikationsprotein A
RT- PCR Reverse Transkriptase Polymerasekettenreaktion
SAHC S-Adenosylhomocystein
SAM S-Adenosylmethionin
SCE Schwesterchromatidaustausch
SD Standardabweichung
SDS Natriumdodecylsulfat
Sf9 Spodoptera frugiperda Insektenzellen
Sp1 Transkriptionsfaktor Sp1
T Thymin
TBE-Puffer Tris-Borsäure-EDTA-Puffer

100 Anhang
TCA Trichloressigsäure
TCR transkriptionsgekoppelte Reparatur
TEMED N,N,N',N'-Tetramethylethylenediamine
TFIIH Transkriptionsfaktor II H
TFIIIA Transkriptionsfaktor III A
TRIS Tris-(hydroxymethyl)-aminomethan
UV ultraviolet
WHO Weltgesundheitsorganisation
XPA bis XPG, XPV Xeroderma Pigmentosum Protein A bis G und V
XPAzf Zinkfingerdomäne von XPA
XRCC1 X-ray repair cross-complementing faktror 1
ZF Zinkfinger

Anhang 101
8.2 Liste der verwendeten Chemikalien
Bezeichnung Lieferant
Acryamid-Bisacrylamid-Lösung 37,5:1 (40%) Merck, Darmstadt
Agarose NEEO Ultra-Qualität Roth, Karlsruhe
3-Aminobenzamid Alrich, Steinheim
Ammoniumperoxodisulfat Merck, Darmsatdt
Ammoniumsulfat Merck, Darmstadt
Anti-Actin-Antikörper (Kaninchen) Santa Cruz, Santa Cruz (USA)
Anti-Dig-POD-Antikörper Roche, Mannheim
Anti-Kaninchen-Antikörper-HRP Santa Cruz, Santa Cruz (USA)
Anti-Maus-Antikörper-FITC Sigma, Deisenhofen
Anti-Maus-Antikörper-HRP Santa Cruz, Santa Cruz (USA)
Anti-PAR-Antikörper, 10H (Maus) A. Bürkle, Konstanz
Anti-PARP-1-Antikörper (Maus) Alexis, Lausen (Schweiz)
ß-Mercaptoethanol Merck, Darmstadt
Bacto Agar BD, Le Pont de Claix (Frankr.)
Bacto Trypton BD Le Pont de Claix (Frankr.)
Blockierreagenz Roche, Mannheim
Bromphenolblau Merck, Darmsadt
BSA (≥ 96%) Sigma, Deisenhofen
Borsäure p.a. Merck, Darmstadt
Calciumchlorid Merck, Darmstadt
Cäsiumchlorid Fisher, Loughbrorough (UK)
Casy-Ton Schärfe System, Reutlingen
Casy-Clean Schärfe System, Reutlingen
Chloroform Roth, Karlsruhe
Diiodomethylarsin (99,0 %) W.R. Cullen, University of
British Columbia (Kanada)
Dimethylarsinsäure (Cacodylsäure) 98,8% Riedel- de Haën, Seelze
Dimethylsulfoxid (DMSO) Sigma, Deisenhofen
Dithiotreitol (DTT) Roth, Karlsruhe
Ds-DNA Cellulose Sigma, Deisenhofen

102 Anhang
ECL-Detektionslösung Amersham, Freiburg
ECL-Advanced-Detektionslösung Amersham, Freiburg
Entwickler für Photofilm Amersham, Freiburg
Ethidiumbromid Fluka, Buchs
Ethanol (Rotipuran > 99,8%) Roth, Karlsruhe
EDTA Riedel de Haën, Seelze
Ficoll 400 Sigma, Deisenhofen
Formamidopyrimidine-DNA Glykosylase (Fpg) Dr. Serge Boiteux, Fontenay
aux Roses (Frankreich)
Fötales Kälberserum (FKS) Sigma, Deisenhofen
Fixierer für Photofilm Amersham, Freiburg
Giemsa- Farbstoff Merck, Darmstadt
Glycerol Merck, Darmstadt
Glycin Roth, Karlsruhe
Harnstoff Roth, Karlsruhe
HEPES Merck, Darmstadt
Histone Typ IIa Sigma, Deisenhofen
2-Hydroxychinolin Aldrich, Steinheim
Inversionsöl für Fluoreszenzmikroskopie Zeiss, Oberkochen
Iododimethylarsin (99,0%) W.R. Cullen, University of
British Columbia (Kanada)
Isopropanol Sigma, Deisenhofen
Kaliumchlorid, p.a. Merck, Darmstadt
Kaliumdihxdrogenphosphat, p.a. Merck, Darmstadt
Di-Kaliumhydrogenphosphat p.a. Merck, Darmstadt
Kaliumhydroxid Merck, Darmstadt
Laborsand Bernd Kraft GmbH, Duisburg
Magermilchpulver Roth, Karlsruhe
Magnesiumsulfat Roth, Karlsruhe
Magnesiumchlorid Sigma, Deisenhofen
Maleinsäure Roth, Karlsruhe
Methanol Riedel de Haën, Seelze
Methylenblau p.a. Merck, Darmstadt

Anhang 103
Methyloxoasin (99,0%) W.R. Cullen, University of
British Columbia (Kanada)
Molekulargewichtsmarker RPN 800 Amersham, Freiburg MMA(V) (Natriumsalz, sesquihydrat, 96,0 %) Greyhound Chromatography
and allied Chemicals
(Birkenhead, UK)
ß-NAD+ Sigma, Deisenhofen
Natriumacetat p.a. Merck, Darmstadt
Natriumbisulfit Sigma, Deisenhofen
Natriumchlorid p.a. Merck, Darmstadt
Natriumdextransulfat Amersham, Freiburg
Natrium-Dihydrogenphosphat p.a. Merck, Darmstadt
Di-Natriumhydrogenphosphat Merck, Darmstadt
Natriumdodecylsulfat (SDS) Merck, Darmastadt
Natrium(meta)arsenit p.a. (≥ 99,0%) Fluka, Buchs
Natriumpyrophosphat Roth, Karlsruhe
Natronlauge Merck, Darmstadt
Nährmedium F12 Sigma, Deisenhofen
Nonidet P-40 Sigma, Deisenhofen
Nutrient broth Fluka, Buchs
Oligonukleotide MWG Biotech, Ebersberg
Penicilin/Streptomycin Sigma, Deisenhofen
Pepstatin A Sigma, Deisenhofen
Phenol (Roti-Phenol) Roth, Karlsruhe
Pluronsäure F-68 Sigma, Deisenhofen
PMSF Serva, Heidelberg
Polyethylenglycol 6000 Serva, Heidelberg
Protaminsulfat Sigma, Deisenhofen
4-2-Pyridylazoresorcinol Riedel de Häen, Seelze
Salzsäure Merck, Darmstadt
Sephadex G100-50sf Sigma, Deisenhofen
Sepharose 2B (für Gelfiltration) Amersham, Freiburg
TEMED Roth, Karlsruhe
Trichloressigsäure Roth, Karlsruhe

104 Anhang
TRIS-Base p.a. Sigma, Deisenhofen
Trypsin Sigma, Deisenhofen
Tween 20 Roth, Karlsruhe
Vectashield mit DAPI Vector, Burlingame (USA)
Wasserstoffperoxid Merck, Darmstadt
Xylencyanol FF Fluka, Buchs
XPAzf, Peptid Schafer-N, Kopenhagen
(Dänemark)
XPA-Protein (Maus) Dr. André Ecker, Universität
Rotterdamm (Niederlande)
Zinkchlorid Merck, Darmstadt

Anhang 105
8.3 Liste der verwendeten Geräte und Verbrauchs-matrialien
Bezeichnung Hersteller
Auflichtmikroskop, Axoivert 25 Zeiss, Oberkochen
Bildauswerteprogramm (Aida Image Analyzer) Raytest, Straubenhardt
Biophotometer Eppendorf, Köln
Blotmembran (Hybond P) Amersham, Freiburg
Blotmembran (Nylon Hybond N+) Amersham, Freiburg
Blotpapier Whatman, Dassel
Bottle Top Filter (0,2 µm) Nalgene, Hamburg
Brutschrank, Heracell Heraeus, Hanau
Elektrophorese-Kammer GNA 200 (Agarose Gel) Amersham, Freiburg
Elektrophorese-Kammer (PAGE) Hoefer, Wiesloch
Entwicklungskammer für Chemilumineszenz Siemens, München
Deckgläschen, 15 mm ∅ Marienfeld, Lauda Königshofen
Dialysekammer Pierce, Rockford (USA)
Fluoreszenzmikroskop, Eclipse E400 Nikon, Düsseldorf
Fluoreszenzmikroskop, Axioimager Zeiss, Oberkochen
Geldokumentationssystem Easy Win 32 Herolab, Wiesloch
Geldokumentationssystem Fuji LAS 3000 Raytest, Straubenhardt
H2O-Aufbereitungsanlage Ultra Clear SG, Barsbüttel
Glaspipetten Roth, Karlsruhe
Glaswolle Merck, Darmstadt
Homogenisator nach Potter Roth, Karlsruhe
Inkubator Heraeus. Hanau
Kolbenhubpipetten Eppendorf, Köln
Koloniezählgerät Colony counter BZG 30 WTW,Weilheim
Kryoröhrchen Greiner, Frickenhausen
Kühlschrank und Tiefkühler Liebherr, Ochsenhausen
Küvetten (UVetten) Eppendorf, Köln
Magnetrührer Heidolph, Schwabach

106 Anhang
MetaView (Version 4.1.5) Visitron Systems, Puchheim
Mikroreaktionsgefäße 1,5 ml Roth, Karlsruhe
Mikrotiterplatten Sarstedt, Nümbrecht
Mikrotiterplattenlesegerät Tecan, Crailsheim
Mulisteppipette Brand, Wertheim
Netzgerät Power Pac 200 Bio-Rad, München
Neubauer-Zählkammer Marienfeld, Lauda-Königshofen
Objektträger Marienfeld, Lauda-Königshofen
Petrischalen Amicrin, Scorze (Italien)
pH-Meter pH 320 WTW, Weilheim
Photometer Analytik Jena, Jena
Photofilm für Chemilumineszenzdetektion Amersham, Freiburg
Pipettierhilfe, Pipettus Hirschmann, Eberstadt
Pipettenspitzen Roth, Karlsruhe
Eppendorf, Köln
Polyprep-Säulen Bio Rad, München
Rotator Neolab, Heidelberg
Schikanekolben 2l TH Geyer, Renningen
Schüttelinkubator Vortemp 56 EVC Labnet, Ried im Innkreis
(Österreich)
Semidry-Blotter Amersham, Freiburg
Slot-Blot-Apperatur Roth, Karlsruhe
Spot RT Kamera für Fluoreszenzmikroskop Visitron Systems, Puchheim
Sterilbank, Hera safe Heraeus, Hanau
Stickstoffbehälter MVE, Brunsville (USA)
Taumler GFL, Burgwedel
Tiekühlschrank (-80°C) GFL, Burgwedel
Trockenschrank Heraeus, Hanau
Ultra-Zentrifuge (Typ!) Beckman-Coulter, Krefeld
UV-Lampe Bioblock Scientific, Illkirch
Cedex (Frankreich)
Vakuumpumpe Vacubrand, Wertheim
Vortex Genie 2 Scientific Industries, USA
Waage BP 61S Sartorius, Göttingen

Anhang 107
Wasserbad Julabo, Seelbach
Zentrifuge Biofuge pico Heraeus, Hanau
Zentrifuge Eppendorf, Köln
Zentrifuge Sorvall, Langenselbold
Zellzählgerät (CASY 1) Schärfe System, Reutlingen
Zellkulturschalen Ø 40, 60, 100 und 150 mm Biochrom, Trasadingen
(Schweiz)
Zentrifugenröhrchen, 15 und 50 ml Sarstedt. Nümbrecht
Zentrifügenröhrchen für Ultra-Zentrifuge TH Geyer, Renningen

108 Anhang
8.4 Verwendete Puffer und Lösungen
Alle Lösungen wurden mit über Austauschkanülen aufgereinigtem destilliertem
Wasser angesetzt. Die Medien und Lösungen für die Zellkultur und PM2-Isolierung
wurden sterilfiltiriert.
8.4.1 Zellkultur
F 12 Kulturmedium F12, pH 6,8
10% FKS
100 U/ml Penicillin
100 µg/ml Streptomycin
PBS, pH7,4 100 mM NaCl 7 mM Na2HPO4
4,5 mM KCl
3 mM KH2PO4
PBS-EDTA, pH 7,4 100 mM NaCl 7 mM Na2HPO4
4,5 mM KCl
3 mM KH2PO4
3 mM EDTA
Trypsin-Lösung 0,25% Trypsin in PBS-EDTA
8.4.2 PM2-Isolierung
AMS-Lösung 47 mM KCl
51 mM CaCl2 x 2 H2O
2,2 M NaCl
MgSO4 –Lösung 234 mM MgSO4

Anhang 109
Nutrient broth 8 g Nutrient broth in 0,6 l Wasser
Nährmedium 600 ml Nutrient broth
200 ml AMS-Lösung
200 ml MgSO4-Lösung
Lösungen getrennt ansetzten und
dann mischen
Bottom-Agar 4,5 g Nutrient broth
5 g Bacto-Agar
ad 300 ml Wasser
nach dem Autoklavieren zufügen:
100 ml AMS
100 ml MgSO4-Lösung
Top-Agar 0,8 g Bacto-Pepton
0,5 g Bacto Agar ab 60 ml H2O
nach dem Autoklavieren zufügen:
20 ml AMS
20 ml MgSO4 -Lösung
NTC-Puffer, pH 7,5 1 M NaCl
9,7 mM CaCl2 x 2 H2O
10,15 mM Tris/HCl
BE1-Puffer, pH 7,5 100 mM NaCl
20 mM Tris/HCl
1 mM EDTA
Phenollösung für die DNA-Fällung Roti-Phenol pH 7
1% SDS
0,1% Hydroxychinolin

110 Anhang
Ethanol/Na-Acetat-Lösung (DNA-Fällung) 95% Ethanol (-20°C)
5% Na-Acetat (125mM)
8.4.3 DNA-Relaxationstest
Elektrophoresepuffer (TBE), pH 8,2 445 mM Tris-Base
445 mM Borsäure
5 mM EDTA
Gebrauchslösung 1:5 verd.
Agarose-Gel (1 %) 2 g Agarose in 200 ml
Elektrophoresepuffer (1x)
Enzympuffer, pH 7,5 85 ml 50 mM Na2HPO4
15 ml 50 mM NaH2PO4
davon 80 ml zu 10 ml 1 M NaCl
und 10ml H2Obidest geben
Methylenblau- Lösung 40 mg in 10 ml H2Obidest
Gebrauchslösung 1:100 verd.
stets frisch ansetzten
Ethanol/Na-Acetat-Lösung (DNA-Fällung) 95% Ethanol (-20°C)
5% Na-Acetat (125mM)
Stoppreagenz 0,25% Bromphenolblau
0,25% Xylencyanol
15% Ficoll 400
Ethidiumbromid 10 mg/ml in H2Obidest

Anhang 111
8.4.4 XPA-Zinkfreisetztung
HEPES/NaOH-Puffer, pH 7,4 20 mM HEPES
pH mit NaOH einstellen
ZnCl2-Lösung 100 µM ZnCl2 in HEPES/NaOH
4-2 Pyridylazoresorcinol-Lösung 2,5 mM 4-2 Pyridylazoresorcinol
8.4.5 XPA-Gelshiftassay
Gelshift-Puffer, pH 8,3 25 mM HEPES-KOH
30 mM KCl
4 mM MgCl2
1 mM EDTA
10% Glycerol
45 µg/ml BSA
0,9 mM DTT (frisch am Versuchtag)
TBE-Puffer (5x) 0,5 M Tris
0,5 M Borsäure
0,01 M EDTA
Für Elekrophorese: pH 8
Für Blot: pH nicht einstellen
TE-Puffer (zur Verdünnung der Oligos) 100 mM Tris
10 mM EDTA
Ladepuffer TBE-Puffer (0,25x), pH 8
40% Glycerol
Oligonukleotidlösung 120 fmol/µl in TE-Puffer

112 Anhang
Ladepuffer mit Farbstoffen TBE-Puffer (0,25x), pH 8
40% Glycerol
0,2% Bromphenolblau
0,2% Xylencyanol
Polyacrylamid-Gel (5%) 5% Acrylamid
0,5 x TBE, pH 8
2,6% Glycerol
0,1% APS
1:1000 TEMED
Maleinsäurepuffer, pH 7,5 100 mM Maleinsäure
150 mM NaCl
pH mit ca. 40 NaOH-Plätzchen einst.
Blockiersubstanz-Stammlösung 10 g Blockiersunstanz in Maleinsäure-
Puffer, autoklavieren und aliquotieren
Blockier-Gebrauchslösung 90 ml Maleinsäurepuffer
10 ml Blockiersubstanz-Stammlösung
Waschpuffer Maleinsäurepuffer mit
0,3% Tween 20
8.4.6 Poly(ADP-Ribosyl)ierung in HeLa S3 Zellen
PBS, pH 7,4 siehe 8.4.1
Blockierlösung (PBS-MT) 5% Magermilchpuffer
0,05% Tween 20
im PBS, pH 7,4

Anhang 113
8.4.7 PARP-1 Proteingehalt
1,5 x Probenpuffer 93,75 mM Tris-HCl, pH 6,8
9 M Harnstoff
15% Glycerol
3% SDS
0,0045% Bromphenolblau
7,5% ß-Mercaptoethanol (frisch)
Proteaseinhibitorlösung 1 mM EDTA
10 mM Natriumbisulfit
10 µM Pepstatin A (frisch)
100 µM PMSF (frisch)
in PBS lösen
PBS, pH 7,4 siehe 8.4.1
Elekrophoresepuffer 25 mM Tris
0,1% SDS
192 mM Glycin (frisch)
Trenn- und Sammelgelpuffer 1,5 M Tris, pH 8,8
Trenngel (10%) 10% Acrylamid
375 µM Tris
0,1% SDS
0,1% APS
1:1000 TEMED
Sammelgel (3%) 5% Acrylamid
125 µM Tris
0,1% SDS
0,1% APS
1:1000 TEMED

114 Anhang
Transferpuffer 192 mM Glycin
20 mM Tris
0,01% SDS
10% Methanol
Blockierlösung (PBS-MT) 5% Magermilchpulver
0,3% Tween 20
in PBS, pH 7,4
Waschpuffer (PBS-T) 0,3% Tween 20
in PBS, pH 7,4
8.4.8 PARP-1 Isolierung
Puffer A 25 mM Tris-HCl, pH 8
50 mM Glucose
10 mM EDTA
1 mM ß-Mercaptoethanol
1 mM PMSF
Puffer B 100 mM Tris-HCl, pH 7,4
10% Glycerin
0,5 mM EDTA
12 mM ß-Mercaptoethanol
1 mM PMSF
Puffer BII 50 mM Tris-HCl pH 8.0
0,5 mM EDTA
5 mM MgCl2
5% Glycerol
12 mM β-Mercaptoethanol
1mM PMSF

Anhang 115
Puffer C 50 mM Tris-HCl, pH 8
1 mM EDTA
200 mM KCl
1 mM DTT
10 mM ß-Mercaptoethanol
Puffer D 50 mM Tris-HCl, pH 8
0,5 mM EDTA
5 mM MgCl2
5% Glycerin
12 mM ß-Mercaptoethanol
1 mM PMSF
PBS, pH 7,4 siehe 8.4.1
8.4.9 Poly(ADP-Ribosyl)ierung isolierter PARP-1
10 x Reaktionspuffer, pH 7,8 100 mM Tris-HCl
100 mM MgCl2
Oligonukleotidlösung 1 mg/ml in 15 mM NaCl
PBS, pH 7,4 siehe 8.4.1
Puffer zur Enzymverdünnung 20% Glycerin
in PBS, pH 7,4
Stopppuffer 10% TCA
2% Na2P2O7
Blockierlösung (PBS-MT) 5% Magermilchpulver
0,3% Tween 20
in PBS, pH 7,4

116 Anhang
Waschpuffer (PBS-T) 0,3% Tween 20
in PBS, pH 7,4

Anhang 117
8.5 Vergleich Methyloxoarsin-Diiodomethylarsin
Um festzustellen, ob die beiden Verbindungen Methyloxoarsin und Diiodomethlarsin,
die beide in wässriger Lösung MMA(III) bilden, die gleichen genotoxischen Effekte
verursachen, wurden vergleichende Untersuchungen zur Hemmung der Fpg und zur
Zinkfreisetzung aus XPAzf durchgeführt (Abbildung 32 und 33). Für beide Parameter
war kein großer Unterschied zwischen den beiden Verbindungen festzustellen, so
dass davon ausgegangen werden kann, dass das Iod in Diiodomethylarsin keinen
Effekt ausübt.
0
20
40
60
80
100
120
140
0 0,1 0,5 1 2 5 10Arsenverbindung [mM]
Fpg
Akt
ivitä
t (%
der
Kon
trol
le)
Methyloxoarsin
Diiodomethylarsin
0
20
40
60
80
100
120
140
0 0,1 0,5 1 2 5 10Arsenverbindung [mM]
Fpg
Akt
ivitä
t (%
der
Kon
trol
le)
Methyloxoarsin
Diiodomethylarsin
Abbildung 32: Vergleich des Einflusses von Methyloxoarsin und Diiodomethylarsin auf die Aktivität von Fpg. Isolierte Fpg wurde 30 min bei 37°C mit den Arsenverbindungen inkubiert und die
Aktivität gegenüber oxidativ geschädigter DNA mittels DNA-Relaxationstest bestimmt. Dargestellt sind
Mittelwerte aus mindestens 5 Bestimmungen ± SD.

118 Anhang
0
20
40
60
80
100
120
5 10 25 50 75 100
Arsenverbindung [µM]
Zink
frei
setz
ung
aus
XPA
zf(%
der
H2O
2-K
ontr
olle
)Methyloxoarsin
Diiodomethylarsin
0
20
40
60
80
100
120
5 10 25 50 75 100
Arsenverbindung [µM]
Zink
frei
setz
ung
aus
XPA
zf(%
der
H2O
2-K
ontr
olle
)Methyloxoarsin
Diiodomethylarsin
Abbildung 33: Vergleich des Einflusses von Methyloxoarsin und Diiodomethylarsin auf die Zinkfreisetzung aus XPAzf. XPAzf wurde 30 min bei 37°C mit den Arsenverbindungen inkubiert und
anschließend das freigesetzte Zink mittels 2-4-Pyridylazoresorcinol quantifiziert. Dargestellt sind
Mittelwerte aus mindestens 6 Bestimmungen ± SD.

Anhang 119
8.6 Qualitätsprüfung der isolierten PM2-DNA
Die Ausbeute an isolierter PM2-DNA bei den beiden durchgeführten Versuchen lag
ausgehend von 8 l Bakteriensuspension bei 0,8 bzw. 1 mg. Dies entspricht den
Vorgaben aus dem Versuchsprotokoll, wo die Ausbeute mit 0,5 – 1,5 mg angegeben
wird. Zur Bestimmung der Qualität der DNA wurde sie zunächst oxidativ geschädigt
(Kapitel 4.4) und dann sowohl mit als auch ohne Fpg für 30 min inkubiert. Abbildung
34 zeigt einen Vergleich von zuvor in unserem Labor isolierter mit frisch gewonnener
DNA. Es ist zu sehen, dass der Anteil an offen ringförmiger DNA bei beiden Aliquots
mit ca. 20% gleich groß ist. Auch nach einer Inkubation mit Fpg wurde bei beiden
Proben ein Einschnitt auf ca. 63% offen ringförmige DNA erreicht.
früher isolierte DNA
Fpg +Fpg - Fpg +Fpg -
frisch isolierte DNA
offen ringförmige DNA
superspiralisierte DNA
lineare DNA
früher isolierte DNA
Fpg +Fpg - Fpg +Fpg -
frisch isolierte DNA
offen ringförmige DNA
superspiralisierte DNA
lineare DNA
Abbildung 34: Vergleich zwischen frisch und früher isolierter PM2-DNA. Dargestellt ist ein
repräsentatives Agarose-Gel.

120 Anhang
8.7 Hemmung isolierter PARP-1 durch CuSO4
Zum Vergleich der beiden Testsysteme zur Messung der Aktivität isolierter PARP-1
mittels Slot-Blot bzw. PAGE (Schwerdtle et al., 2007) wurde zusätzlich CuSO4
untersucht. Es ist zu sehen, dass beide Testsysteme die Hemmung der PARP-1 im
gleichen Konzentrationsbereich zeigen (Abbildung 35).
0
20
40
60
80
100
120
140
0 10 50 75 100 250
CuSO4 [µM]
PAR
P-1
Akt
ivitä
t (%
der
Kon
trol
le) Slot-Blot
PAGE
***
***
***
***
******
***
*
0
20
40
60
80
100
120
140
0 10 50 75 100 250
CuSO4 [µM]
PAR
P-1
Akt
ivitä
t (%
der
Kon
trol
le) Slot-Blot
PAGE
***
***
***
***
******
***
*
Abbildung 35: Effekt von CuSO4 auf die Aktivität isolierter PARP-1. Slot-Blot: Isolierte
PARP-1 wurde für 30 min bei 37°C mit den Arsenverbindungen vorinkubiert, die PARP-Reaktion mit
einem Oligonukleotid als Aktivator-DNA 10 min bei 37°C durchgeführt, die Reaktion gestoppt und der
Reaktionsmix mittels Slot-Blot auf eine Membran aufgesaugt. Die Poly(ADP-Ribose) wurde
anschließend immunologisch bestimmt. Gezeigt sind Mittelwerte aus mindestens 3 Bestimmungen ±
SD. PAGE (Schwerdtle et al., 2007): Isolierte PARP-1 wurde für 10 min bei Raumtempertur mit den
Arsenverbindungen vorinkubiert, die PARP-Reaktion mit einem eingeschnittenen Plasmid als
Aktivator-DNA 5 min bei 37°C durchgeführt, die Reaktion gestoppt und der Reaktionsmix auf ein
Polyacrylamidgel aufgetragen. Die Poly(ADP-Ribose) wurde anschließend immunfluorimetrisch
bestimmt. Gezeigt sind Mittelwerte aus mindestens 4 Bestimmungen ± SD. Statistisch unterschiedlich
von der Kontrolle: * P < 0,05; *** P < 0,001 (Student’s t-Test).

Publikationsliste 121
Publikationsliste
Publikationen in Fachzeitschriften
Tanja Schwerdtle, Ingo Walter, Iris Mackiw and Andrea Hartwig (2003) Induction of
oxidative DNA damage by arsenite and its trivalent and pentavalent methylated
metabolites in cultured human cells and isolated DNA. Carcinogenesis, 24(5), 967-
974.
Andrea Hartwig, Holger Blessing, Tanja Schwerdtle, Ingo Walter (2003) Modulation
of DNA repair processes by arsenic and selenium compounds. Toxicology, 193(1-2),
161-9.
Andrea Hartwig, Tanja Schwerdtle and Ingo Walter (2003) Current aspects on the
genotoxicity of arsenite and its methylated metabolites: Oxidative stress and
interactions with the cellular response to DNA damage. Proceedings Workshop
“Metall(oid)organische Verbindungen in der Umwelt”, Springer Verlag 2003, Berlin /
Heidelberg, 221-233.
Tanja Schwerdtle, Ingo Walter and Andrea Hartwig (2003) Arsenite and its
biomethylated metabolites interfere with the formation and repair of stable BPDE-
induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair, 2(12),
1449-63.
Ingo Walter, Tanja Schwerdtle, Christina Thuy, Jason L. Parsons, Grigory L. Dianov
and Andrea Hartwig (2007) Impact of arsenite and its methylated metabolites on
PARP-1 activity, PARP-1 gene expression and poly(ADP-ribosyl)ation in cultured
human cells. DNA Repair, 6(1), 61-70.
Tanja Schwerdtle, Ingrit Hamann, Gunnar Jahnke, Ingo Walter, Constanze Richter,
Jason L. Parsons, Grigory L. Dianov and Andrea Hartwig (2007) Impact of copper on
the induction and repair of oxidative DNA damage, poly(ADP-ribosyl)ation and
PARP-1 activity. Molecular Nutrition and Food Research, 51, 201-210

122 Publikationsliste
Veröffentlichte Abstracts
Tanja Schwerdtle, Iris Mackiw, Ingo Walter, Andrea Hartwig (2002) Genotoxizität
von Arsenit und seinen methylierten Metaboliten I: kultivierte menschliche Zellen.
Lebensmittelchemie, 56 (6): 136-137.
Ingo Walter, Tanja Schwerdtle, Holger Blessing, Andrea Hartwig (2002)
Genotoxizität von Arsenit und seinen methylierten Metaboliten II: subzelluläre
Systeme. Lebensmittelchemie, 57 (1): 4.
Tanja Schwerdtle, Ingo Walter, Iris Mackiw, Andrea Hartwig (2003) Comparative genotoxicity of arsenite and its trivalent and pentavalent methylated metabolites. Archives of Pharmacology, 367 (1): R138 (537)
Ingo Walter, Tanja Schwerdtle, Andrea Hartwig (2003) Interference by inorganic arsenic and its methylated metabolites with zinc finger proteins. Archives of Pharmacology, 367 (1): R146 (571)
Vorträge auf Fachtagungen
1st PARP Regio Meeting, 20.2.2004 Konstanz
Ingo Walter, Anke Pelzer, Tanja Schwerdtle, Christina Thuy und Andrea Hartwig
Inhibtion of PARP by very low-dose arsenic compounds.
21. GUM Tagung, 5. – 8.10.2004 Würzburg
Ingo Walter, Anke Pelzer, Tanja Schwerdtle, Christina Thuy und Andrea Hartwig
Inhibierung der Poly(ADP-ribosy)lierung durch Arsenverbindungen in kultivierten
menschlichen Zellen
3rd PARP Regio Meeting, 19.5.2006 Strasbourg Ingo Walter, Tanja Schwerdtle, Christina Thuy und Andrea Hartwig Impact of trivalent arsenicals on cellular poly(ADP)ribose levels and on the activity of isolated PARP-1

Publikationsliste 123
Posterbeiträge auf Fachtagungen
32nd Annual Meeting of the EEMS, 3. - 7. 9. 2002, Warsaw, Poland
Tanja Schwerdtle, Ingo Walter, Iris Mackiw and Andrea Hartwig
Genotoxicity of arsenite and its trivalent and pentavalent methylated metabolites.
7. Tagung des DNA-Reparaturnetzwerks, 17 – 20. 9. 2002, Karlsruhe
Ingo Walter, Tanja Schwerdtle and Andrea Hartwig
Genotoxicity of trivalent arsenic metabolites.
31. Deutscher Lebensmittelchemikertag, 7. – 11. 9. 2002, Frankfurt
Ingo Walter, Tanja Schwerdtle und Andrea Hartwig
Einfluss von methylierten Arsenverbindungen auf die Induktion und Reparatur
oxidativer DNA-Schäden.
ESCODD plenary meeting 2003, 12. – 13. 1. 2003, Firenze, Italy
Tanja Schwerdtle, Ingo Walter and Andrea Hartwig
Genotoxicity of biomethylated arsenicals in cultured human cells.
44. Tagung der DGPT und 20. Tagung der GUM, 17 - 20. 3. 2003, Mainz
Ingo Walter, Tanja Schwerdtle and Andrea Hartwig
Einfluss von Arsenit und seinen methylierten Metaboliten auf Zinkfingerproteine.
33rd Annual Meeting of the EEMS, 24.-28.8. 2003, Aberdeen, Scotland
Tanja Schwerdtle, Ingo Walter and Andrea Hartwig
Arsenite and its biomethylated metabolites induce oxidative DNA damage and
interfere with DNA repair in cultured human cells and subcellular systems.
33. Deutscher Lebensmittelchemikertag, 13. – 15. 9. 2004, Bonn
Ingo Walter, Anke Pelzer, Tanja Schwerdtle, Christina Thuy und Andrea Hartwig
Arsenit und seine dreiwertigen methylierten Metabolite MMA(III) und DMA(III)
reduzieren die Poly(ADP-Ribosyl)ierung in kultivierten menschlichen Zellen.

124 Publikationsliste
33. Deutscher Lebensmittelchemikertag, 13. – 15. 9. 2004, Bonn
Tanja Schwerdtle,. Ingrit Lohter, Gunnar Jahnke, Ingo Walter, Andrea Hartwig,
Genotoxizität von Cu(II) in kultivierten menschlichen Zellen.
8. Tagung des DNA-Reparaturnetzwerks, 28.9. – 1.10. 2004, Ulm
Ingo Walter, Anke Pelzer, Tanja Schwerdtle, Christina Thuy and Andrea Hartwig
Impact of arsenic compounds on the poly(ADP-ribosyl)ation in cultured human cells
35th Annual Meeting of the EEMS, 3 .- 7.7. 2005, Kos, Greece
Ingo Walter, Tanja Schwerdtle, Christina Thuy, and Andrea Hartwig
Biomethylated arsenicals interfere with poly(ADP-ribosyl)ation in cultured human
cells.
22. Tagung der GUM, 21. – 24.2. 2006, Darmstadt
Ingo Walter, Tanja Schwerdtle, Christina Thuy, Grigory L. Dianov und Andrea
Hartwig
Hemmunng von PARP-1 durch dreiwertige Arsenverbindungen
9. Tagung des DNA-Reparaturnetzwerks, 12.9. – 15.9. 2006, Hamburg
Ingo Walter, Tanja Schwerdtle, Christina Thuy, Grigory L. Dianov and Andrea
Hartwig
Impact of trivalent arsenicals on poly(ADP-ribosyl)ation in cellular and sub-cellular
systems
9. Tagung des DNA-Reparaturnetzwerks, 12.9. – 15.9. 2006, Hamburg
Claudia Großkopf, Chiara Corso, Ingo Walter and Andrea Hartwig
Antimony – impact on zinc finger structures and DNA repair
Preise und Stipendien
Landesgraduiertenförderung Baden-Würtemberg, 1.10.2002 – 30.6.2004
Ingo Walter Promotionsstipendium

Publikationsliste 125
Karlsruher chemische Gesellschaft, 10.6.2003
Ingo Walter Procter & Gamble Förderpreis für ausgezeichnete Diplomarbeit
8. Tagung des DNA-Reparaturnetzwerks, 28.9. – 1.10. 2004, Ulm
Ingo Walter Meeting Fellowship Award 2004
21. GUM Tagung, 5. – 8.10.2004 Würzburg
Ingo Walter Tagungsstipendium
35th Annual Meeting of the EEMS, 3.-7.7. 2005, Kos, Greece
Ingo Walter EU Network of Excellence (ECNIS) fellowship

126 Lebenslauf
Lebenslauf
Name Ingo Walter
Persönliche Daten Gebutsdatum: 14.09.1976 in Konstanz
Staatsangehörigkeit: deutsch
Familienstand: ledig
Schulbildung 1983 - 1987 Wiestor-Grundschule in Überlingen
1987 -1996 Gymnasium Überlingen
Abschluss: 25.6.1996 Algemeine Hochschulreife
Studium 04/1997 - 07/2002 Studium der Lebensmittelchemie an der Univerität Karlsruhe
29.10.1999 Vorprüfung der Staatsprüfung für Lebensmittelchemiker
8.11.2001 2. Prüfungsabschnitt der Staatsprüfung für Lebensmittel-
chemiker
01/2002 - 07/2002 Wissenschaftliche Abschlussarbeit am Institut für
Lebensmittelchemie und Toxikologie der Universität Karlsruhe:
Thema: Genotoxizität von methylierten Arsenverbindungen
Abschluss: Diplom-Lebensmittelchemiker
Dissertation 10/2002 Promotion am Institut für Lebensmittelchemie und Toxikologie
der Universität Karlsruhe (Prof. Dr. A. Hartwig).
Thema: Einfluss von Arsenverbindungen auf die DNA-
Reparaturproteine Fpg, XPA und PARP-1
Seit 07/2004 Fortsetzung der Promotion am Institut für
Lebensmitteltechnologie und Lebensmittelchemie der TU Berlin
(Prof. Dr. A. Hartwig).

Danksagung 127
Danksagung
Mein besonderer Dank gilt Frau Prof. Dr. Andrea Hartwig für die immer freundliche
Betreuung und fachliche Unterstützung mit vielen hilfreichen Diskussionen. Zudem möchte
ich ihr dafür danken mir den Umzug nach Berlin ermöglicht zuhaben, wodurch ich viele neue
Erfahrungen in einer faszinierenden Stadt machen konnte.
Besonders bedanken möchte ich mich auch bei Dr. Tanja Schwerdtle, die mich begonnen
mit der Diplomarbeit auch während der Promotion immer in praktischen und theoretischen
Fragen unterstützt hat.
Auch bei meinem Arbeitskreis sowohl in Karlsruhe, wie auch in Berlin möchte ich mich für
den guten Zusammenhalt während, aber auch außerhalb der Arbeitszeit bedanken.
Besonders erwähnen möchte ich Dr. Christina Thuy und Gunnar Jahnke mit denen ich mich
nach dem Umzug nach Berlin in der neuen Stadt erst zurechtfinden musste. Bei Dr. Holger
Blessing möchte ich mich speziell für die praktische Unterstützung im Labor bedanken. Die
Arbeit von Dr. Christina Thuy zur Genexpression von PARP-1 ist für die Interpretation meiner
Ergebnisse wichtig. Dafür bedanke ich mich herzlich.
Neben dem eigenen Arbeitskreis gilt mein Dank auch den Arbeitskreisen von Prof. Metzler in
Karlsruhe und Prof. Kroh in Berlin für die nette kollegiale Atmosphäre danken.
Ein besonderer Dank gilt zudem Prof. Dr. Alexander Bürkle und seinem Arbeitskreis und
dabei besonders Jörg Fahrer und Dr. Sascha Beneke, die mich bei der Isolierung der PARP-
1 und der Messung der PARP-1 Aktivität sehr gut unterstützt haben.
Zudem möchte ich mich bei Prof. Dr. W. Cullen für die freundliche Überlassung der
dreiwertigen methylierten Arsenmetabilote, bei Dr. S. Boiteux für das Fpg Protein und bei Dr.
André Ecker für das murine XPA bedanken.
Für die finanzielle Unterstützung während der Promotionszeit in Karlsruhe bedanke ich mich
bei der Landesgraduiertenförderung Baden-Würtemberg.
Zudem möchte ich mich bei meinen lieben Eltern dafür bedanken mir den Weg zu dieser
Arbeit ermöglicht zu haben.




















![Einfluss der DNA-Glykosylase OGG1 auf die TGF vermittelte ... · Benzo[a]pyren), Naturstoffen (z.B. Aflatoxin B1) und Arzneistoffen (z.B. Temozolomid) auch verschiedenen Formen der](https://img.pdfslide.tips/doc/110x75/5e140af2cd6c5f157520ef8e/einfluss-der-dna-glykosylase-ogg1-auf-die-tgf-vermittelte-benzoapyren-naturstoffen.jpg)








