Embed Size (px)
Citation preview

Anais do XVI Encontro de Iniciação Científica e Pós-Graduação do ITA – XVI ENCITA / 2010 Instituto Tecnológico de Aeronáutica, São José dos Campos, SP, Brasil, 20 de outubro de 2010
ESTUDO DA TEORIA DO FUNCIONAL DA DENSIDADE APLICADO ÀS SIMULAÇÕES DE PROPRIEDADES ELETRÔNICAS E ESTRUTURAI S DE
SEMICONDUTORES
Mayara Everlim Bonani Universidade Estadual “Júlio de Mesquita Filho” Bolsista PIBIC-CNPq [email protected]
Profa. Dra. Lara Kühl Teles Instituto Tecnológico da Aerounáutica [email protected]
Resumo. Neste trabalho, aplicamos cálculos de primeiros princípios baseados na Teoria do Funcional da Densidade (DFT)
para investigar propriedades eletrônicas e estruturais de alguns semicondutores. Foi realizado o estudo desta teoria bem como o estudo de teorias anteriores que servirão de base para esta, como a de Hartree e a de Hartree-Fock.
Utilizamos o método ab-initio de pseudopotenciais contido no código VASP (Vienna Ab-initio Simulation Package). Estas simulações foram realizadas usando primeiramente potenciais atômicos obtidos pelo método de Aproximação da Densidade Local (LDA – Local Density Approximation) e posteriormente, alguns resultados pelo método LDA-1/2 e GGA (GGA- Generalized Gradient Approximation).
Os resultados obtidos foram utilizados para entender principalmente propriedades como: estrutura de bandas, densidade de estados, gaps de energia em pontos de alta simetria da Zona de Brillouin.
As simulações foram realizadas para os seguintes compostos: silício (Si), arseneto de gálio (GaAs), sulfeto de chumbo (PbS), seleneto de chumbo (PbSe) e telureto de chumbo (PbTe).
Observamos que as aproximações LDA e GGA produzem resultados comparáveis com a experiência para o parâmetro de rede, porém não para o gap de energia. Desta forma, utilizamos o método LDA-1/2 para obtenção do gap, o que resultou em excelentes resultados quando comparados com a experiência.
Palavras chave: Teoria do Funcional da Densidade, método ab-initio, semicondutores.
1. Introdução
Dentre as várias áreas da Física da Matéria Condensada, a Física de Semicondutores está entre as que possuem
maior número de aplicações tecnológicas. Isto é devido à grande variedade de fenômenos que ocorrem nestes materiais e que proporcionam a confecção de uma enorme quantidade de dispositivos.
Atualmente o elemento semicondutor é primordial na indústria eletrônica e confecção de seus componentes. Nenhum aparelho eletrônico atual, desde um simples relógio digital ao mais avançado dos computadores, seria possível sem os mesmos. Dispositivos baseados em semicondutores incluem transistores, interruptores, diodos, células fotovoltaicas, detectores e termistores. Muitos destes são usados como elementos únicos em um circuito ou como componentes em circuitos integrados. Existem outros componentes eletrônicos de diversos graus de complexidade tecnológica como microprocessadores, e nanocircuitos usados em nanotecnologia.
Os principais materiais semicondutores utilizados na eletrônica são o germânio (Ge) e o silício (Si), sendo este último o mais utilizado. Recentemente há muito investimento em pesquisas com materiais semicondutores para aplicação na eletrônica fabricado a partir do carbono, pesquisas estas que já obtiveram sucesso.
Semicondutores são sólidos cristalinos de condutividade elétrica intermediária entre condutores e isolantes; são caracterizados pela extraordinária sensibilidade a impurezas atômicas, defeitos e ao controle externo de carga. Essa propriedade faz dos semicondutores a base da eletrônica.
Para descrever o comportamento de elétrons e núcleos em um sólido cristalino, como por exemplo, em um semicondutor, utilizamos o método ab-initio, também chamado de método dos primeiros princípios. Através dele podemos descrever de maneira rigorosa a natureza quântica de um sistema de maneira a obter resultados condizentes com os experimentais.

Anais do XVI ENCITA, ITA,20 de outubro de 2010
Este método é baseado em conhecimentos de mecânica quântica, tendo como parâmetros empíricos de entrada apenas os números atômicos dos elementos envolvidos e as constantes fundamentais da natureza.
Em Mecânica Quântica, resolver um sistema significa buscar uma solução para a equação de Schrödinger:
( ) ( )φ , tφ , t ,
ri H r
t
∂− =
∂h (1)
onde ℏ é a constante de Planck divida por 2�, H é o operador Hamiltoniano, e as variáveis independentes � e � são o vetor posição da partícula(s) do sistema e tempo, respectivamente. Devemos resolver a Eq. (1) e determinar a φ��, t�, chamada de função de onda, que descreve o estado físico deste sistema. Conhecendo de φ��, t� é possível determinar propriedades e obter as informações necessárias para caracterização do sistema.
Limitando nossa abordagem a casos onde a energia potencial não depende do tempo, a técnica de separação de variáveis permite escrever a função de onda como um produto de uma função dependente só das coordenadas e uma função dependente do tempo:
( ) ( ) ( )φ , t Ψ t ,χ=r r (2)
onde
( ) iEtt ,Aeχ
−= h (3)
sendo A é uma constante arbitrária e E a energia do sistema quântico em questão. Substituindo as Eq. (2) e Eq. (3) na Eq. (1) obtemos a chamada Equação de Schrödinger Independente do tempo,
ou também, Equação de Autovalores:
( ) ( )Ψ Ψ .H E=r r (4)
Infelizmente para a maioria dos sistemas quânticos a equação não pode ser resolvida analiticamente. Mesmo para
o átomo de Hidrogênio, problema de um único corpo, não é trivial resolvê-la. Semicondutores, como quase todos os compostos da natureza, são sólidos cristalinos formados por muitos átomos onde cada um tem Z (número atômico) elétrons que interagem entre si. Essa interação, também chamada de “perturbação”, torna impossível encontrar solução analítica para ���.
Como veremos a seguir, uma alternativa que se vislumbrou para contornar esse problema foi a substituição da função de onda pela densidade eletrônica no estudo de sistemas complexos, já que a densidade eletrônica, além de ser uma função real, depende apenas a posição espacial, tendo então uma forma muito mais simples que a função de onda para o mesmo sistema. Iremos explanar na próxima seção as aproximações que foram surgindo para resolver sistema de N átomos interagentes, desde a mais simples, que é considerar os núcleos fixos (Aproximação de Born-Oppenheimer), até chegar à chamada de Teoria do Funcional da Densidade (DFT). Apresentaremos também as propriedades e resultados obtidos para alguns semicondutores, bem como a importância deste estudo teórico.
Para simplificar a maioria das expressões, descreveremos as equações em unidades atômicas, onde ℏ, a carga eletrônica e, a massa do elétron me e a constante 4�� assumem valores unitários.
2. Teoria e Métodos de Cálculo
2.1 Introdução
Descrever quanticamente um sistema, como no caso, um sólido cristalino, consiste em resolver a equação de
Schrödinger na sua forma estacionária. A função de onda Ψ de sistemas quânticos como átomos, moléculas e sólidos, é

Anais do XVI ENCITA, ITA,20 de outubro de 2010
função das coordenadas nucleares R e eletrônicas r. Escrevemos então o Hamiltoniano completo para um sistema quântico de N elétrons, M núcleos:
2 2
1 1 1 1 1 1 1
1 1 1,
2 2
N M N M N N M MA A B
i Ai A i A i j A B AA iA ij AB
Z Z ZH
M r r R= = = = = > = >= − ∇ − ∇ − + +∑ ∑ ∑∑ ∑∑ ∑∑ (5)
onde MA e ZA representam a massa e o número atômico do núcleo A, riA a distância entre i-ésimo elétron e o a-ésimo núcleo, rij a distância entre os i-ésimo e j-ésimo elétrons e RAB a distância entre os a-ésimo e b-ésimo núcleos. Os
operadores 2i∇ e 2
A∇ envolvem diferenciações em relação às coordenadas eletrônicas e nucleares, de tal maneira que o
primeiro termo representa a energia cinética dos elétrons, o segundo termo a energia cinética dos núcleos. Os três últimos termos correspondem, ordenadamente, ao potencial coulombiano de atração entre os elétrons e núcleos, e de repulsão entre elétron-elétron e núcleo-núcleo.
O Hamiltoniano descrito acima representa um problema de muitos corpos que interagem entre si. Infelizmente, este sistema não pode ser resolvido de forma analítica devido principalmente ao acoplamento elétron-elétron via interação Coulombiana. Mesmo se fosse possível encontrar uma expressão para a função de onda, sua manipulação, a despeito do advento dos modernos computadores, seria demasiado laboriosa para sistemas com centenas de elétrons.
Impondo simplificações e aproximações ao Hamiltoniano completo, foram sendo introduzidos modelos para reduzir a complexidade matemática envolvida. Vários métodos foram desenvolvidos, porém a efetivação dos mesmos só se deu graças ao avanço computacional que permitiu uma maior eficiência na utilização desses métodos.
O mérito destes métodos reside no fato de que dos dados obtidos pela teoria podem ser validados experimentalmente.
Nas próximas seções serão mencionadas e discutidas as aproximações utilizadas na resolução da Equação de Schrödinger para o sistema físico abordado. Finalizado o estudo destas aproximações, ressaltaremos brevemente algumas das propriedades dos semicondutores que serão úteis para compreensão dos resultados.
2.2 Aproximação de Born-Oppenheimer Para uma parte significativa dos problemas normalmente estudados em mecânica quântica, uma primeira
simplificação consiste em assumir que a equação de Schrödinger pode ser separada em uma parte eletrônica (correspondente às informações dos elétrons) e outra nuclear (correspondente às informações dos núcleos). Esta simplificação é conhecida como Aproximação de Born-Oppenheimer. Ela é a primeira das aproximações que simplifica o Hamiltoniano da Eq. (5) por meio da desconsideração o movimento do núcleo como mostra (Born e Oppenheimer, 1927).
Esta simplificação decorre da grande diferença entre as massas do elétron e do núcleo (Mnúcleo ≈ (104 a 105) melétron). Como resultado desta o movimento dos elétrons é bem mais rápido que o dos núcleos, sendo que estes podem ser considerados fixos, não afetando o estado e a energia dos elétrons. Sendo assim, o termo de energia cinética do núcleo pode ser desprezado na Eq. (5) e o último termo, a energia de repulsão entre os núcleos pode ser considerada constante.
2.3 Teoria de Hartree: Aproximação do Campo Médio Devido à dificuldade da solução para o problema multieletrônico estar associada ao termo de repulsão
intereletrônico, os métodos de cálculos de estrutura eletrônica por primeiros princípios tentam mapear o sistema de muitos elétrons interagentes em um sistema de elétrons não interagentes. De acordo com a Teoria de Hartree, como mostra (Vianna et al., 2004) uma maneira de alcançar isto é considerando a interação elétron-elétron sentida por um elétron i como um potencial de campo médio devido a todos os outros elétrons.
As hipóteses iniciais são que os elétrons são distinguíveis, atribuindo um estado específico a cada um deles e que a função de onda total de um sistema de N elétrons será representada como um produto simples desses estados.
Sendo assim, supomos que o hamiltoniano total do sistema de N elétrons possa ser escrito de uma forma desacoplada, isto é, pelo somatório de hamiltonianos de cada elétron:

Anais do XVI ENCITA, ITA,20 de outubro de 2010
1
( ),N
i
H h i=
=∑ (6)
onde
( ) ( ) ( ),i i i i ih i εΦ = Φr r (7)
onde h(i), ϕ�����, � são o operador hamiltoniano, a função de onda e a autoenergia do i-ésimo elétron respectivamente. A solução mais simples da equação de Schroedinger para o hamiltoniano acima é obtida expressando-se a função
de onda total dos elétrons, ���, ��, ��, … , ���, como um produto simples das autofunções de cada elétron:
( ) ( ) ( ) ( ) ( )1 2 3 1 1 2 2 3 3Ψ , , , , Φ Φ Φ Φ .N N N… = …r r r r r r r r
(8)
Esta função de onda é conhecida pelo nome de produto de Hartree, e cada autofunção, Φ�����, é conhecida como orbital espacial.
Vamos considerar Φ normalizada. Aplicando o método variacional e adotando a função acima como função tentativa, fazemos uma pequena variação arbitrária em Ψ e procuramos o mínimo de energia. Isto nos resulta a chamada equação de Hartree:
( ) ( ) ( )2i i i
1
1,
2
MA
i H iA A
ii
hZ
Vr
ε=
Φ = − ∇ + + Φ = Φ
∑r r r
(9)
onde,
2| ( ') |
',| ' |
jH
j i
V d≠
Φ=
−∑∫r
rr r
(10)
é o potencial (efetivo) de Hartree. Este potencial indica que a interação coulombiana com os demais elétrons aparece de forma média, isto é, cada elétron interage com a distribuição eletrônica |Φ�|� de cada um dos demais elétrons.
Embora esta aproximação de campo médio tenha eliminado o termo de repulsão coulombiana entre os elétrons, esta teoria não contempla as características eletrônicas. Um desses aspectos corresponde a indistinguibilidade dos elétrons. Outro seria o fato de que os elétrons são caracterizados como férmions e a interação de troca de elétrons de mesmo spin não aparece no potencial efetivo VH. Além disso, a função de onda adotada não é anti-simétrica com relação à troca de coordenadas r de dois elétrons (não obedece ao princípio da exclusão de Pauli). A teoria que leva em conta tais aspectos é chamada de Hartree-Fock, conforme está descrita na próxima seção.
2.4 Teoria de Hartree-Fock
Para incluir os efeitos de spin a função total deve ser função da coordenada � que inclui as coordenadas espaciais
� e de spin �. Estas funções são denominadas de spin-orbitais e são dadas pela Eq. (11) ou pela Eq. (12):
( ) ( ), Φ ( ) ( ),i i iφ φ α= =x r s r s (11)
( ) ( ), Φ ( ) ( ).i i iφ φ ρ= =x r s r s (12)
Elas representam o spin-orbital do elétron i dado pelo produto do orbital espacial Φ���� com as funções de spin “up” ���� ou “down” ����, respectivamente; são completas e ortonormais.

Anais do XVI ENCITA, ITA,20 de outubro de 2010
Outra característica que deve ser incluída na função de onda é que, uma vez que ela representa férmions, ela deve ser antissimétrica, Uma maneira simples de construir funções de onda antissimétricas foi proposta por John Slater, mostrando que as propriedades exigidas para a função de onda são observadas escrevendo essa função como um determinante cujos elementos são os spin-orbitais. A função escrita nesse formato recebe o nome de determinante de Slater:
1 1 1 2 1
2 1 2 2 21 2
1 2
( ) ( ) ( )
( ) ( ) ( )1( , ,..., ) .
!( ) ( ) ( )
N
NN
N N N N
N
φ φ φφ φ φ
φ φ φ
Ψ =
x x x
x x xx x x
x x x
K
K
M M O M
K
(13) O primeiro termo da direita da equação (13) corresponde apenas a um fator de normalização.
Da mesma forma que na teoria de Hartree, devemos obter as equações que descrevem o elétron independente que se move no potencial efetivo de Hartree-Fock. Para isto vamos aplicar o princípio variacional. Minimizando a energia total E do sistema com relação aos spins-orbitais, obtêm-se um conjunto de equações, conhecidas como equações de Hartree-Fock:
2
*2
1
( ') ( ') ( ')1' ( ) ( ) ' (
' '),
2
Mj j iA
i i j i iA j jiA
Zd d
r
φ φ φφ φ ε φ
=
− ∇ + + + =
− −∑ ∑ ∑∫ ∫x x x
x xr r r r
x x x
(14)
que na forma de uma equação de autovalores:
( ) ( ) ( ),i i if i φ ε φ=x x (15)
onde
2
1
1( ) ( )
2
MA
i HFA iA
Zf i V i
r=
= − ∇ + +∑ (16)
é o operador de Fock, que é hermitiano, e VHF é o potencial efetivo de Hartree-Fock. Como o operador ���� depende dos �′s, ele só é conhecido quando os �′s forem determinados. Assim, a
solução da Eq. (14), também conhecida como Equação Canônica de Hartree-Fock, é encontrada de forma iterativa: Define-se um conjunto " �# inicial e calcula-se $%&; resolve-se a Eq. (14), obtendo um novo conjunto de spins-orbitais; repete-se o procedimento até que �′s determinam ���� que por sua vez determina �, ou seja, até que a autoconsistência seja alcançada.
Sobre o potencial VHF pode ser interpretado como sendo o potencial médio sentido por um elétron i devido a todos os outros elétrons. O potencial se divide em um termo local e um não local:
( ) ( ) ,HF H bV i V i k= + (17)
com $% sendo o potencial de Hartree (termo local) dada na equação (10). O ultimo termo '( , que é não local, é definido como potencial de exchange. Ele surge da antissimetria da função de onda e leva à correlação entre elétrons de spins paralelos.
A aproximação de Hartree-Fock não inclui efeitos de correlação entre elétrons de spins opostos. Para isso, é necessário considerar a função de onda exata, pelo fato de um único determinante de Slater não ser o suficiente para

Anais do XVI ENCITA, ITA,20 de outubro de 2010
expandir a função de onda multieletrônica. Na próxima seção iremos descrever outro método que engloba os efeitos de exchange-correlação e é aplicável a um maior numero de sistemas.
2.5 Teoria do Funcional da Densidade
A Teoria do Funcional da Densidade (DFT, do inglês, Density Functional Theory) é a teoria base de todos os
cálculos de estrutura eletrônica que realizamos neste trabalho. Atualmente é também a mais empregada em cálculos de estrutura eletrônica de primeiros princípios, principalmente, por causa dos bons resultados fornecidos frente à eficiência computacional que apresenta.
Em DFT a quantidade fundamental é a densidade eletrônica do sistema �� r � em detrimento da função de onda multieletrônica. Enquanto a função de onda necessita de 3N variáveis para a sua descrição, onde N é o número de elétrons do sistema, a densidade é uma função real da posição (3 variáveis espaciais).
Os artigos responsáveis pela formulação desta teoria são: Inhomogeneous electron gás de (Hohenberg e Kohn, 1964), que rendeu a Kohn o prêmio Nobel de 1998; Self-consistent equations including Exchange and correlation effects, de (Kohn e Sham, 1965). Segue adiante uma explanação das aproximações e cálculos contidos nestes artigos.
Para um sistema contendo muitos elétrons em um potencial externo )� r �, a função de onda Ψ, a densidade
eletrônica �* r + = ∑ Ψ�∗�
�/� � r �Ψ�� r � e demais propriedades do estado fundamental desse sistema, são obtidas pelas
solução da equação de Schrodinger de muitas partículas. Sendo que )� r � define o Hamiltoniano do sistema, todas as propriedades anteriores são funcionais deste potencial.
No trabalho Inhomogeneous electron gas, Hohenberg e Kohn demonstram um teorema importante: �� r � é
determinada de forma unívoca, a menos de uma constante aditiva, a partir de um potencial externo )� r �. Portanto a
energia total do sistema pode ser definida como um funcional de �� r �:
[ ] 1 '( ) ( )' [ ] ( ),
2( (
') ) xcd d TE v Ed
ρ ρρρ ρ ρ+ + +−
= ∫ ∫r r
r r r r rr r
(18)
onde
* 21[ ] ( ) ( )
2
N
i ii
T dρ = − Ψ ∇ Ψ∑∫ r r r (19)
Explicando então a equação acima temos que: os dois primeiros termos acima correspondem respectivamente à
contribuição do potencial externo �� r � e à interação coulombiana clássica (termo de Hartree); 0[�] é a energia cinética de um sistema fictício de N elétrons não interagentes, com a mesma densidade eletrônica do sistema original; 345[�] é a
energia chamada energia de exchange-correlação do sistema original com densidade �� r � , que é a energia restante que completa 3[�].
Para obter as equações que descrevem o estado fundamental do sistema, aplica-se o princípio variacional
como foi feito para obter as equações de Hartree-Fock, porém, considerando a densidade eletrônica �� r � como variável básica. Assim, devemos procurar o mínimo de energia 3[�], com a restrição de manter fixo o número N de partículas. Este problema é equivalente a encontrar o extremo do funcional de Lagrange 6[�]. Obtemos as equações de Kohn-Sham:
2 [ ]1 ( )( )
2 '' xc
i i i i
Ev dr
δ ρρ εδρ
− ∇ + + + Ψ = Ψ −∫
rr
r r e 1,2,...,i N= (20)
A equação acima foi obtida de maneira formalmente exata. Nota-se que essa é análoga a equação de Schroedinger de uma partícula, sob um potencial efetivo dado por:

Anais do XVI ENCITA, ITA,20 de outubro de 2010
[ ]( )( ) ( .
') ' xc
ef
EV v dr
δ ρρδρ−
= + +∫r
r rr r
(21)
Portanto, as Equações de Kohn-Sham nos permitem considerar um sistema de muitos elétrons interagentes em um sistema em que eles não interagem, mas que cada um se move em um potencial efetivo devido a todos os demais elétrons.
Essas equações devem ser resolvidas auto-consistentemente: parte-se de uma densidade inicial, obtêm-se o Hamiltoniano de Kohn-Sham (KS), que é diagonalizado para a obtenção dos autovalores e autovetores; então uma nova densidade eletrônica é obtida, e o processo continua até que a convergência seja alcançada.
Para que a equação de Kohn-Sham possa ser resolvida é necessário se conhecer o 345. Devido o fato desse funcional não ser conhecido, não temos uma solução exata para o problema de muitos copos. Portanto, para tornar este formalismo aplicável ao estudo de sistemas físicos, necessitamos realizar aproximações em 345.
Existem várias aproximações para 345. Duas delas são conhecidas como LDA (Local Density-functional Approximation) e GGA (Generalized Gradient Approximation).
LDA é a tão conhecida aproximação da densidade local. Este funcional é o resultado de uma abordagem simples do problema de correlação eletrônica, pois supõe-se que um gás de elétrons real pode ser aproximado por um gás homogêneo. Já o funcional da Aproximação do Gradiente Generalizado, GGA, representa uma melhoria na abordagem do problema da correlação em sistemas não homogêneos (Vianna et al., 2004).
Não iremos abordar como estas aproximações são realizadas, mas iremos na Seção 3.0, comparar alguns resultados obtidos para semicondutores quando se utiliza uma ou outra.
3. Resultados
Para a resolução das equações de Khon-Sham utiliza-se um código computacional, dada a complexidade
envolvida nesse processo. Existem diversos códigos computacionais baseados nos mais diversos métodos. Cada código, com seu respectivo método apresenta um bom desempenho para certo conjunto de aplicações.
Há vários métodos para se resolver estas equações: pseudopotenciais, PAW (Projector Augmented Wave), LAPW (Linearized Augmented Plane Waves), LMTO (Linear muffin-tin orbital), os quais utilizam aproximações diferentes para as funções de onda e potenciais.
No presente trabalho utilizou-se o pacote de simulação computacional VASP (Vienna Ab-initio Simulation Package), que nos permitiu estudar os materiais semicondutores através do método ab-initio. Este pacote resolve a equação de Kohn-Sham e pode-se escolher dentro dois métodos para isto: o método do pseudopotencial e o PAW. Quanto aos funcionais de troca e correlação, o pacote permite a utilização da aproximação LDA assim como varias aproximações GGA.
Neste trabalho, os cálculos de primeiros princípios foram realizados dentro do formalismo da DFT utilizando as aproximações, LDA, GGA e LDA-1/2.
As simulações foram realizadas para os seguintes compostos: silício (Si), arseneto de gálio (GaAs), sulfeto de chumbo (PbS), seleneto de chumbo (PbSe) e telureto de chumbo (PbTe).
3.1 Propriedades Estruturais Como mencionado anteriormente utilizamos o potencial de troca e correlação LDA e GGA. Compararemos a seguir o resultado obtido utilizando estas aproximações. Vale ressaltar que os cálculos para os compostos Si e GaAs foram efetuados na estrutura zinc-blende e os compostos PbS, PbSe e PbTe, na estrutura rocksalt.
3.1.1 Parâmetro de Rede a
Primeiramente, antes de obtermos a estrutura de bandas para cada elemento estudado, obtivemos os respectivos
parâmetros de rede mínimos, para isto variamos a energia total, Etot (também chamada de energia livre pelo programa, uma vez que Etot=F se T=0K) em função do parâmetro de rede.
Anais do XVI ENCITA, ITA,20 de outubro de 2010
Utilizamos o método LDA e GGA e consideramos nos cálculos dois átomos por célula. Realizamos como teste, simulações com oito átomos por célula e, como o esperado, o parâmetro de rede não se alterou.
Na Tab.1, nós mostramos os resultados para os parâmetros de rede obtidos e comparamos com outros cálculos e com a experiência.
Tabela 1- Parâmetro de rede para os compostos Si, GaAs, PbS, PbSe e PbTe. Os valores experimentais para o Si e o GaAs são mostrados em (Kittel, 2006) e os para os calcogênios em (Dantas et al., 2008)
Parâmetros de rede a (Å) Método Si GaAs PbS PbSe PbTe LDA 5,39 5,60 5,85 6,05 6,39 GGA 5,46 5,73 6,01 6,22 6,56 Valor Experimetal 5,43 5,65 5,936 6,117 6,462
Observamos que os cálculos LDA e GGA concordam de forma excelente entre si, bem como os resultados
experimentais. A aproximação LDA é conhecida por superestimar a ligação entre átomos livres resultando assim, em valores menores para o volume da célula de cerca de 1-2%. Já pela aproximação GGA, percebemos que o valor para o parâmetro de rede sempre é maior que o experimental.
3.1.2 Módulo de Compressibilidade/ Bulk Modulus
Segue abaixo a Tabela 2 com os resultados obtidos para o Bulk Modulus através dos métodos LDA e GGA.
Nestas simulações foram considerados os parâmetros de rede encontrados. Tabela 2- Bulk Modulus para os compostos PbS, PbSe e PbTe. Os valores experimentais são mostrados em (Wei, S.-H., Zunger, A., 1997)
Módulo de Compressibilidade/ Bulk Modulus (GPa) Método PbS PbSe PbTe LDA 64,14 57,80 48,32 GGA 53,40 47,05 39,40 Valor Experimetal 53-70 41-61 40
Comparando com os valores experimentais, o método GGA foi o que resultou em menores erros. Comparando os
dois métodos, o GGA resultou em valores menores para B. Percebemos que para o PbTe, o método LDA resultou em quase 2% de erro.
3.2 Propriedades Eletrônicas
3.2.1 Densidade de Estados (DOS)
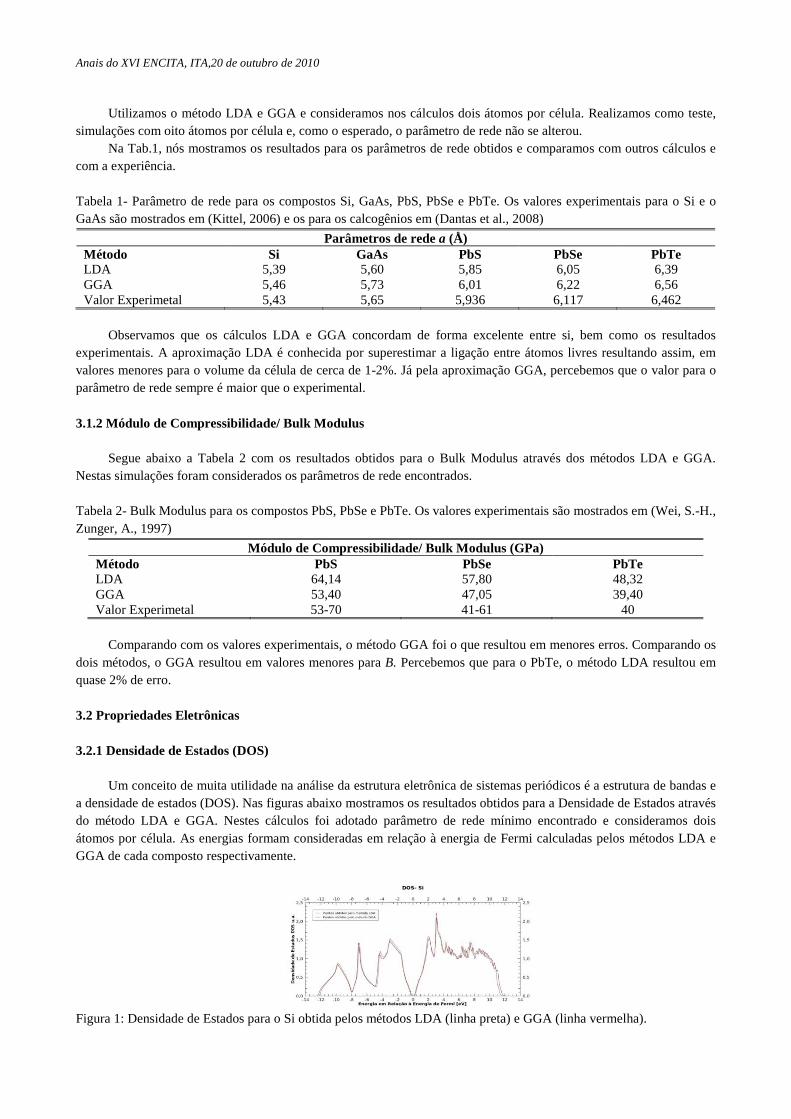
Um conceito de muita utilidade na análise da estrutura eletrônica de sistemas periódicos é a estrutura de bandas e a densidade de estados (DOS). Nas figuras abaixo mostramos os resultados obtidos para a Densidade de Estados através do método LDA e GGA. Nestes cálculos foi adotado parâmetro de rede mínimo encontrado e consideramos dois átomos por célula. As energias formam consideradas em relação à energia de Fermi calculadas pelos métodos LDA e GGA de cada composto respectivamente.
Figura 1: Densidade de Estados para o Si obtida pelos métodos LDA (linha preta) e GGA (linha vermelha).

Anais do XVI ENCITA, ITA,20 de outubro de 2010
Figura 2: Densidade de Estados para o GaAs obtida pelos métodos LDA (linha preta) e GGA (linha vermelha).
Figura 3: Densidade de Estados para o PbS obtida pelos métodos LDA (linha preta) e GGA (linha vermelha).
Figura 4: Densidade de Estados para o PbSe obtida pelos métodos LDA (linha preta) e GGA (linha vermelha).
Figura 5: Densidade de Estados para o PbTe obtida pelos métodos LDA (linha preta) e GGA (linha vermelha).
3.2.2 Estruturas de Bandas
Um conceito importante na física do estado sólido são as chamadas estruturas de bandas. Essas estruturas são calculadas a partir da primeira zona de Brillouin do material.
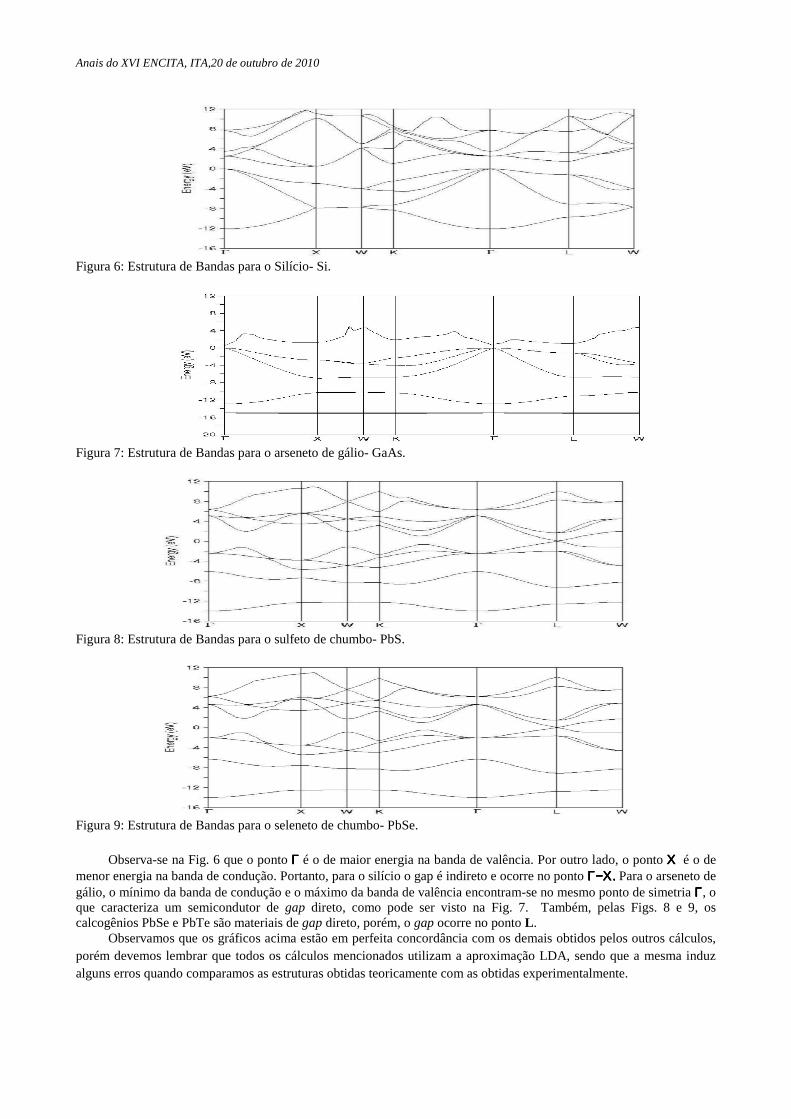
Nas Figs. 6, 7, 8 e 9 mostramos as estruturas de bandas dos semicondutores Si, GaAs, PbS e PbSe obtidas pelo método LDA. Adotamos nos cálculos o parâmetro de rede mínimo encontrado. Nos gráficos considerou-se como energia de referência a energia do topo das bandas de valência dos materiais.
Anais do XVI ENCITA, ITA,20 de outubro de 2010
Figura 6: Estrutura de Bandas para o Silício- Si.
Figura 7: Estrutura de Bandas para o arseneto de gálio- GaAs.
Figura 8: Estrutura de Bandas para o sulfeto de chumbo- PbS.
Figura 9: Estrutura de Bandas para o seleneto de chumbo- PbSe.
Observa-se na Fig. 6 que o ponto ΓΓΓΓ é o de maior energia na banda de valência. Por outro lado, o ponto ΧΧΧΧ é o de menor energia na banda de condução. Portanto, para o silício o gap é indireto e ocorre no ponto ΓΓΓΓ−Χ−Χ−Χ−Χ.... Para o arseneto de gálio, o mínimo da banda de condução e o máximo da banda de valência encontram-se no mesmo ponto de simetria ΓΓΓΓ, o que caracteriza um semicondutor de gap direto, como pode ser visto na Fig. 7. Também, pelas Figs. 8 e 9, os calcogênios PbSe e PbTe são materiais de gap direto, porém, o gap ocorre no ponto L .
Observamos que os gráficos acima estão em perfeita concordância com os demais obtidos pelos outros cálculos, porém devemos lembrar que todos os cálculos mencionados utilizam a aproximação LDA, sendo que a mesma induz alguns erros quando comparamos as estruturas obtidas teoricamente com as obtidas experimentalmente.
Anais do XVI ENCITA, ITA,20 de outubro de 2010
3.2.3 Energia de Gap Eg
Os cálculos que utilizam LDA apesar de fornecerem com certa precisão as propriedades estruturais, os mesmos tendem a subestimar os valores para os gaps de energia, o que está relacionado à omissão do termo referente à interação de caráter não-local. Desta forma, os valores obtidos com o cálculo LDA são subestimados de 1,5 – 2,0 eV com relação aos resultados experimentais. No entanto, o valor do gap e a obtenção dos estados excitados com precisão possui grande importância tecnológica. Existem diversas tentativas de se resolver esse problema, principalmente usando teoria de perturbação, como é feito por métodos como GW e SIC (Self-interaction correction). Apesar de fornecerem bons resultados para o gap de compostos simples, esses métodos, além de complicados, exigem grande capacidade computacional, o que os torna inviáveis para o estudo de sistemas complexos como ligas e heteroestruturas. Para contornar esse problema, foi desenvolvido recentemente pelo nosso grupo no ITA em colaboração com o professor Luiz Guimarães Ferreira da USP um método que consiste na correção do potencial de troca e correlação LDA chamado LDA-1/2.
O método LDA−1/2 é especialmente útil para o estudo de materiais com gap pequeno, para os quais o valor calculado com o potencial LDA pode ser negativo. É importante que se diga que praticamente não há aumento do tempo computacional entre os cálculos LDA e LDA−1/2, o que é extraordinário e torna este método promissor para o estudo do gap e de outras propriedades dos estados excitados de sistemas complexos.
A aproximação LDA-1/2 prevê gaps de energia em excelente acordo com os resultados. Neste período, aprendemos como utilizar o método LDA-1/2 em conjunto com o VASP. Iniciamos então os cálculos utilizando este método para os semicondutores silício (Si) e arseneto de gálio (GaAs).
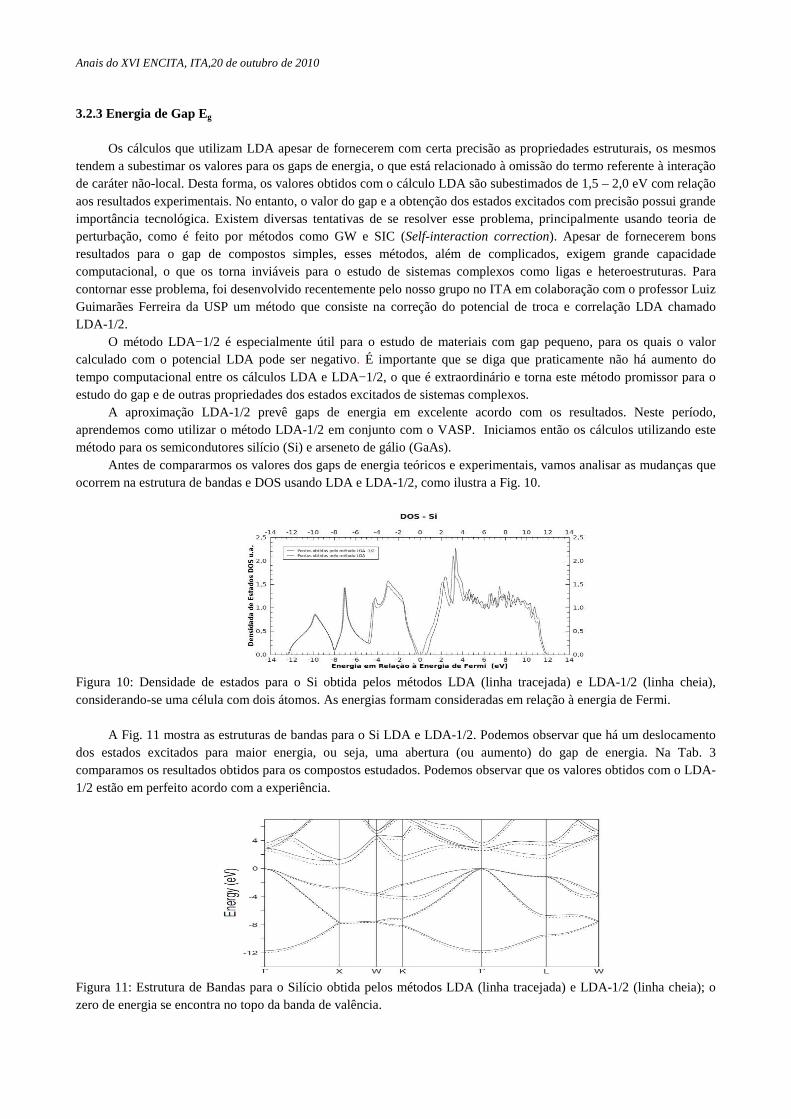
Antes de compararmos os valores dos gaps de energia teóricos e experimentais, vamos analisar as mudanças que ocorrem na estrutura de bandas e DOS usando LDA e LDA-1/2, como ilustra a Fig. 10.
Figura 10: Densidade de estados para o Si obtida pelos métodos LDA (linha tracejada) e LDA-1/2 (linha cheia), considerando-se uma célula com dois átomos. As energias formam consideradas em relação à energia de Fermi.
A Fig. 11 mostra as estruturas de bandas para o Si LDA e LDA-1/2. Podemos observar que há um deslocamento
dos estados excitados para maior energia, ou seja, uma abertura (ou aumento) do gap de energia. Na Tab. 3 comparamos os resultados obtidos para os compostos estudados. Podemos observar que os valores obtidos com o LDA-1/2 estão em perfeito acordo com a experiência.
Figura 11: Estrutura de Bandas para o Silício obtida pelos métodos LDA (linha tracejada) e LDA-1/2 (linha cheia); o zero de energia se encontra no topo da banda de valência.

Anais do XVI ENCITA, ITA,20 de outubro de 2010
Energia de Gap Eg [eV] Método Si GaAs LDA 0,45 0,41 LDA-1/2 1,13 1,41 Valor Experimental 1,12 1,43
Tabela 3: Valores de Eg obtidos por ambos os métodos, LDA e/ou LDA-1/2, bem como os valores experimentais mostrados por (Rezende, 2004).
Também calculamos os valores do gap através dos métodos LDA e GGA, como apresenta a Tab. 4. Percebemos que o método LDA subestima o gap,enquanto o GGA, superestima este valor. Percebemos que estes dois métodos não são tão eficientes quanto o método LDA-1/2.
Energia de Gap Eg [eV] Método PbS PbSe PbTe
0,71 GGA 0,34 0,25 LDA 0,21 0,15 0,63 Valor Experimental 0,29 0,17 0,19
Tabela 4: Valores de Eg obtidos para os calcogênios pelos métodos LDA e GGA, bem como os valores experimentais mostrados por (Wei, S.-H., Zunger, A., 1997). 4.0 Conclusão
Estudando a teoria no qual se baseiam as simulações, pode-se perceber a dificuldade de ser resolvida a Eq. de
Schrodinger para um sistema de N átomos interagentes, e então, estudar as aproximações realizadas sobre o Hamiltoniano de tais sistemas físicos. Estudamos desde a Aproximação de Born-Oppenheimer, que considera os núcleos atômicos fixos, até a Teoria do Funcional da Densidade (DFT) que substitui a variável função de onda pela densidade eletrônica, tornando então uma sistema de 3N graus de liberdade em um sistema de apenas 3.
Concluímos que simulações realizadas através da Teoria DFT fornecem resultados de boa qualidade quando comparados aos resultados obtidos na literatura. Utilizamos o código VASP, no qual está embutida esta teoria. Obtivemos resultados para alguns semicondutores, analisando as propriedades estruturais, como parâmetro de rede e Bulk Modulus, e também propriedades eletrônicas, como gaps de energia, densidades de estados e estrutura de bandas.
O método VASP utiliza quatro arquivos de entrada para sua execução. Os parâmetros escolhidos para cada arquivo envolvem a caracterização do composto correspondente a simulação, como por exemplo, sua estrutura cristalina, parâmetro de rede, a descrição da posição dos átomos na célula em coordenadas cartesianas ou em função dos vetores da base, entre outros. Portanto todo estudo realizado sobre estado sólido foi de essencial aplicação nas simulações.
Observamos que as aproximações LDA e GGA produzem resultados comparáveis com a experiência para o parâmetro de rede, embora o primeiro resulte em valores menores e o segundo em valores maiores que os da literatura. O mesmo já não ocorre para o gap de energia. Desta forma, utilizamos o método LDA-1/2 para obtenção do gap, o que resultou em excelentes resultados quando comparados com a experiência.
5. Agradecimentos
A professora Lara, pela idealização do projeto, e por todo apoio e dedicação e à CNPq pelo apoio financeiro. 6. Referências
BORN, D; OPPENHEIMER, J. R. Ann. Phys. Leipzig 84, 457. DANTAS, N. S.; SILVA, A. F.; PERSSON, C. Electronic band-edge properties of rock salt PbY and SnY (Y= S, Se,
and Te). Optical Materials, v. 30, n. 9, p. 1451-1460, May 2008. HOHENBERG P.; KOHN, W.Phys. Rev. 136, B864 (1964). KITTEL, Charles. Introdução à Física do Estado Sólido. Ed. LTC. 8ª Ed. 2006

Anais do XVI ENCITA, ITA,20 de outubro de 2010
KOHN W., SHAM, L. J. Phys. Rev. 140, A1133 (1965). REZENDE, Sérgio. Materiais e dispositivos eletrônicos. Ed. Livraria da Física. 2ª Ed. 2004 VIANNA, J.D., FAZZIO, A., CANUTO, S. Teoria Quântica de Moléculas e Sólidos-Simulação Computacional. Ed.
Livraria da Física,2004. Wei, S.-H., Zunger, A. Phys. Rev. B, v. 55, p.13506, 1997.





























