Embed Size (px)
Citation preview

N° d’ordre 2004ISAL0036 Année 2004
Thèse
Etude de la mobilité des hydrocarbures aromatiques polycycliques (HAP) contenus
dans un sol industriel pollué
Présentée devant L’Institut National des Sciences Appliquées de Lyon
Pour obtenir
Le grade de docteur
Ecole doctorale de chimie de LYON
Spécialité : SCIENCES ET TECHNIQUES DU DECHET
Par Frédéric Jouannin
Soutenue le 9 juillet 2004 devant la Commission d’examen
Jury
Radu BARNA Directeur Jacques BOURGOIS Rapporteur Patrick SHAROCK Rapporteur Yves PERRODIN Emmanuel VERNUS Pierre MOSZCOWICZ
Cette thèse a été préparée au Laboratoire d’Analyse Environnementale des Procédés et des Systèmes (LAEPSI) Industriels de L’INSA de LYON
1

2
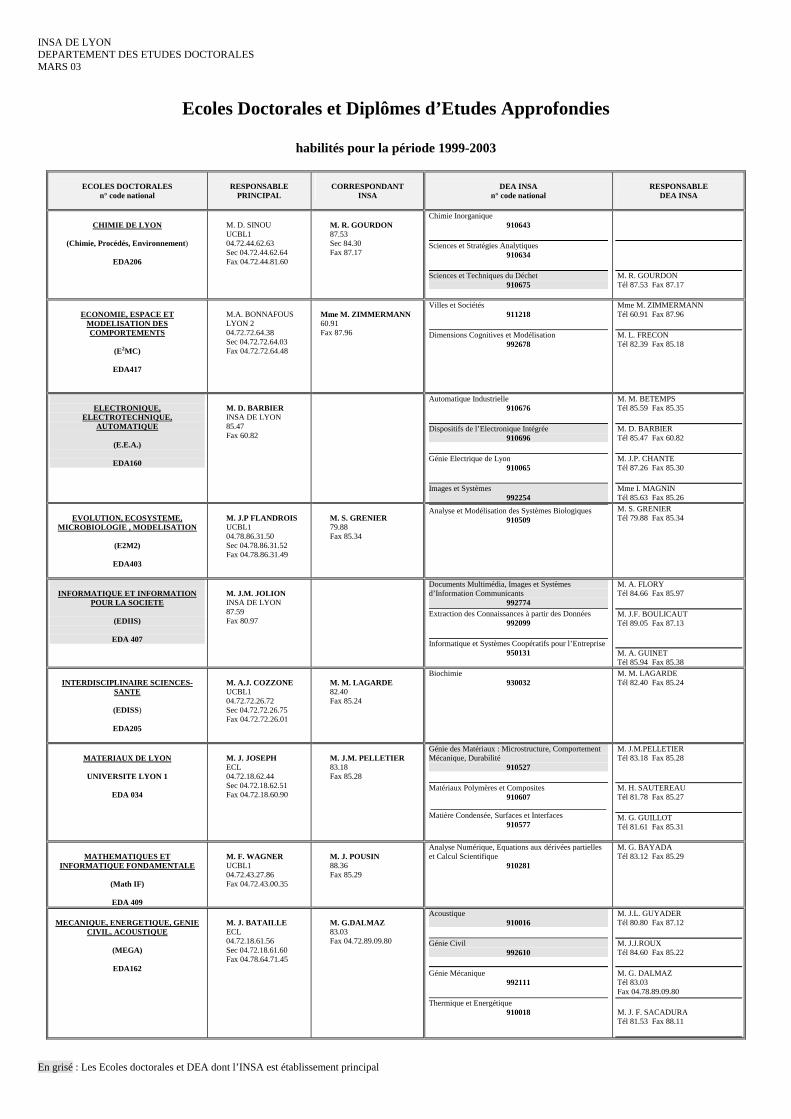
INSA DE LYON DEPARTEMENT DES ETUDES DOCTORALES MARS 03
Ecoles Doctorales et Diplômes d’Etudes Approfondies
habilités pour la période 1999-2003
ECOLES DOCTORALES
n° code national
RESPONSABLE
PRINCIPAL
CORRESPONDANT
INSA
DEA INSA
n° code national
RESPONSABLE
DEA INSA
CHIMIE DE LYON
(Chimie, Procédés, Environnement)
EDA206
M. D. SINOU UCBL1 04.72.44.62.63 Sec 04.72.44.62.64 Fax 04.72.44.81.60
M. R. GOURDON 87.53 Sec 84.30 Fax 87.17
Chimie Inorganique 910643
Sciences et Stratégies Analytiques
910634
Sciences et Techniques du Déchet 910675
M. R. GOURDON Tél 87.53 Fax 87.17
ECONOMIE, ESPACE ET
MODELISATION DES COMPORTEMENTS
(E2MC)
EDA417
M.A. BONNAFOUS LYON 2 04.72.72.64.38 Sec 04.72.72.64.03 Fax 04.72.72.64.48
Mme M. ZIMMERMANN 60.91 Fax 87.96
Villes et Sociétés 911218
Dimensions Cognitives et Modélisation
992678
Mme M. ZIMMERMANN Tél 60.91 Fax 87.96 M. L. FRECON Tél 82.39 Fax 85.18
ELECTRONIQUE,
ELECTROTECHNIQUE, AUTOMATIQUE
(E.E.A.)
EDA160
M. D. BARBIER INSA DE LYON 85.47 Fax 60.82
Automatique Industrielle 910676
Dispositifs de l’Electronique Intégrée
910696
Génie Electrique de Lyon 910065
Images et Systèmes
992254
M. M. BETEMPS Tél 85.59 Fax 85.35 M. D. BARBIER Tél 85.47 Fax 60.82 M. J.P. CHANTE Tél 87.26 Fax 85.30 Mme I. MAGNIN Tél 85.63 Fax 85.26
EVOLUTION, ECOSYSTEME,
MICROBIOLOGIE , MODELISATION
(E2M2)
EDA403
M. J.P FLANDROIS UCBL1 04.78.86.31.50 Sec 04.78.86.31.52 Fax 04.78.86.31.49
M. S. GRENIER 79.88 Fax 85.34
Analyse et Modélisation des Systèmes Biologiques 910509
M. S. GRENIER Tél 79.88 Fax 85.34
INFORMATIQUE ET INFORMATION
POUR LA SOCIETE
(EDIIS)
EDA 407
M. J.M. JOLION INSA DE LYON 87.59 Fax 80.97
Documents Multimédia, Images et Systèmes d’Information Communicants
992774 Extraction des Connaissances à partir des Données
992099
Informatique et Systèmes Coopératifs pour l’Entreprise 950131
M. A. FLORY Tél 84.66 Fax 85.97 M. J.F. BOULICAUT Tél 89.05 Fax 87.13 M. A. GUINET Tél 85.94 Fax 85.38
INTERDISCIPLINAIRE SCIENCES-
SANTE
(EDISS)
EDA205
M. A.J. COZZONE UCBL1 04.72.72.26.72 Sec 04.72.72.26.75 Fax 04.72.72.26.01
M. M. LAGARDE 82.40 Fax 85.24
Biochimie 930032
M. M. LAGARDE Tél 82.40 Fax 85.24
MATERIAUX DE LYON
UNIVERSITE LYON 1
EDA 034
M. J. JOSEPH ECL 04.72.18.62.44 Sec 04.72.18.62.51 Fax 04.72.18.60.90
M. J.M. PELLETIER 83.18 Fax 85.28
Génie des Matériaux : Microstructure, Comportement Mécanique, Durabilité
910527
Matériaux Polymères et Composites 910607
____________________________________________ Matière Condensée, Surfaces et Interfaces
910577
M. J.M.PELLETIER Tél 83.18 Fax 85.28 M. H. SAUTEREAU Tél 81.78 Fax 85.27 M. G. GUILLOT Tél 81.61 Fax 85.31
MATHEMATIQUES ET
INFORMATIQUE FONDAMENTALE
(Math IF)
EDA 409
M. F. WAGNER UCBL1 04.72.43.27.86 Fax 04.72.43.00.35
M. J. POUSIN 88.36 Fax 85.29
Analyse Numérique, Equations aux dérivées partielles et Calcul Scientifique
910281
M. G. BAYADA Tél 83.12 Fax 85.29
MECANIQUE, ENERGETIQUE, GENIE
CIVIL, ACOUSTIQUE
(MEGA)
EDA162
M. J. BATAILLE ECL 04.72.18.61.56 Sec 04.72.18.61.60 Fax 04.78.64.71.45
M. G.DALMAZ 83.03 Fax 04.72.89.09.80
Acoustique 910016
Génie Civil
992610 Génie Mécanique
992111
Thermique et Energétique 910018
M. J.L. GUYADER Tél 80.80 Fax 87.12 M. J.J.ROUX Tél 84.60 Fax 85.22 M. G. DALMAZ Tél 83.03 Fax 04.78.89.09.80 M. J. F. SACADURA Tél 81.53 Fax 88.11
En grisé : Les Ecoles doctorales et DEA dont l’INSA est établissement principal

MAI 2003
INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE LYON Directeur : STORCK A. Professeurs : AUDISIO S. PHYSICOCHIMIE INDUSTRIELLE BABOT D. CONT. NON DESTR. PAR RAYONNEMENTS IONISANTS BABOUX J.C. GEMPPM*** BALLAND B. PHYSIQUE DE LA MATIERE BAPTISTE P. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BARBIER D. PHYSIQUE DE LA MATIERE BASTIDE J.P. LAEPSI**** BAYADA G. MECANIQUE DES CONTACTS BENADDA B. LAEPSI**** BETEMPS M. AUTOMATIQUE INDUSTRIELLE BIENNIER F. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BLANCHARD J.M. LAEPSI**** BOISSON C. VIBRATIONS-ACOUSTIQUE BOIVIN M. (Prof. émérite) MECANIQUE DES SOLIDES BOTTA H. UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOTTA-ZIMMERMANN M. (Mme) UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOULAYE G. (Prof. émérite) INFORMATIQUE BOYER J.C. MECANIQUE DES SOLIDES BRAU J. CENTRE DE THERMIQUE DE LYON - Thermique du bâtiment BREMOND G. PHYSIQUE DE LA MATIERE BRISSAUD M. GENIE ELECTRIQUE ET FERROELECTRICITE BRUNET M. MECANIQUE DES SOLIDES BRUNIE L. INGENIERIE DES SYSTEMES D’INFORMATION BUREAU J.C. CEGELY* CAVAILLE J.Y. GEMPPM*** CHANTE J.P. CEGELY*- Composants de puissance et applications CHOCAT B. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine COMBESCURE A. MECANIQUE DES CONTACTS COUSIN M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures DAUMAS F. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et Thermique DOUTHEAU A. CHIMIE ORGANIQUE DUFOUR R. MECANIQUE DES STRUCTURES DUPUY J.C. PHYSIQUE DE LA MATIERE EMPTOZ H. RECONNAISSANCE DE FORMES ET VISION ESNOUF C. GEMPPM*** EYRAUD L. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE FANTOZZI G. GEMPPM*** FAVREL J. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS FAYARD J.M. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS FAYET M. MECANIQUE DES SOLIDES FERRARIS-BESSO G. MECANIQUE DES STRUCTURES FLAMAND L. MECANIQUE DES CONTACTS FLORY A. INGENIERIE DES SYSTEMES D’INFORMATIONS FOUGERES R. GEMPPM*** FOUQUET F. GEMPPM*** FRECON L. REGROUPEMENT DES ENSEIGNANTS CHERCHEURS ISOLES GERARD J.F. INGENIERIE DES MATERIAUX POLYMERES GERMAIN P. LAEPSI**** GIMENEZ G. CREATIS** GOBIN P.F. (Prof. émérite) GEMPPM*** GONNARD P. GENIE ELECTRIQUE ET FERROELECTRICITE GONTRAND M. PHYSIQUE DE LA MATIERE GOUTTE R. (Prof. émérite) CREATIS** GOUJON L. GEMPPM*** GOURDON R. LAEPSI****. GRANGE G. GENIE ELECTRIQUE ET FERROELECTRICITE GUENIN G. GEMPPM*** GUICHARDANT M. BIOCHIMIE ET PHARMACOLOGIE GUILLOT G. PHYSIQUE DE LA MATIERE GUINET A. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS GUYADER J.L. VIBRATIONS-ACOUSTIQUE GUYOMAR D. GENIE ELECTRIQUE ET FERROELECTRICITE HEIBIG A. MATHEMATIQUE APPLIQUEES DE LYON JACQUET-RICHARDET G. MECANIQUE DES STRUCTURES JAYET Y. GEMPPM*** JOLION J.M. RECONNAISSANCE DE FORMES ET VISION JULLIEN J.F. UNITE DE RECHERCHE EN GENIE CIVIL - Structures JUTARD A. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE KASTNER R. UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique KOULOUMDJIAN J. INGENIERIE DES SYSTEMES D’INFORMATION LAGARDE M. BIOCHIMIE ET PHARMACOLOGIE LALANNE M. (Prof. émérite) MECANIQUE DES STRUCTURES LALLEMAND A. CENTRE DE THERMIQUE DE LYON - Energétique et thermique LALLEMAND M. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et thermique LAUGIER A. PHYSIQUE DE LA MATIERE Mai 2003 LAUGIER C. BIOCHIMIE ET PHARMACOLOGIE

LAURINI R. INFORMATIQUE EN IMAGE ET SYSTEMES D’INFORMATION LEJEUNE P. UNITE MICROBIOLOGIE ET GENETIQUE LUBRECHT A. MECANIQUE DES CONTACTS MASSARD N. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE MAZILLE H. PHYSICOCHIMIE INDUSTRIELLE MERLE P. GEMPPM*** MERLIN J. GEMPPM*** MIGNOTTE A. (Mle) INGENIERIE, INFORMATIQUE INDUSTRIELLE MILLET J.P. PHYSICOCHIMIE INDUSTRIELLE MIRAMOND M. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine MOREL R. MECANIQUE DES FLUIDES ET D’ACOUSTIQUES MOSZKOWICZ P. LAEPSI**** NARDON P. (Prof. émérite) BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS NIEL E. AUTOMATIQUE INDUSTRIELLE NORTIER P. DREP ODET C. CREATIS** OTTERBEIN M. (Prof. émérite) LAEPSI**** PARIZET E. VIBRATIONS-ACOUSTIQUE PASCAULT J.P. INGENIERIE DES MATERIAUX POLYMERES PAVIC G. VIBRATIONS-ACOUSTIQUE PELLETIER J.M. GEMPPM*** PERA J. UNITE DE RECHERCHE EN GENIE CIVIL - Matériaux PERRIAT P. GEMPPM*** PERRIN J. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PINARD P. (Prof. émérite) PHYSIQUE DE LA MATIERE PINON J.M. INGENIERIE DES SYSTEMES D’INFORMATION PONCET A. PHYSIQUE DE LA MATIERE POUSIN J. MODELISATION MATHEMATIQUE ET CALCUL SCIENTIFIQUE PREVOT P. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PROST R. CREATIS** RAYNAUD M. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux REDARCE H. AUTOMATIQUE INDUSTRIELLE RETIF J-M. CEGELY* REYNOUARD J.M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures RIGAL J.F. MECANIQUE DES SOLIDES RIEUTORD E. (Prof. émérite) MECANIQUE DES FLUIDES ROBERT-BAUDOUY J. (Mme) (Prof. émérite) GENETIQUE MOLECULAIRE DES MICROORGANISMES ROUBY D. GEMPPM*** ROUX J.J. CENTRE DE THERMIQUE DE LYON – Thermique de l’Habitat RUBEL P. INGENIERIE DES SYSTEMES D’INFORMATION SACADURA J.F. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux SAUTEREAU H. INGENIERIE DES MATERIAUX POLYMERES SCAVARDA S. AUTOMATIQUE INDUSTRIELLE SOUIFI A. PHYSIQUE DE LA MATIERE SOUROUILLE J.L. INGENIERIE INFORMATIQUE INDUSTRIELLE THOMASSET D. AUTOMATIQUE INDUSTRIELLE THUDEROZ C. ESCHIL – Equipe Sciences Humaines de l’Insa de Lyon UBEDA S. CENTRE D’INNOV. EN TELECOM ET INTEGRATION DE SERVICES VELEX P. MECANIQUE DES CONTACTS VIGIER G. GEMPPM*** VINCENT A. GEMPPM*** VRAY D. CREATIS** VUILLERMOZ P.L. (Prof. émérite) PHYSIQUE DE LA MATIERE Directeurs de recherche C.N.R.S. : BAIETTO-CARNEIRO M-C. (Mme) MECANIQUE DES CONTACTS ET DES SOLIDES BERTHIER Y. MECANIQUE DES CONTACTS CONDEMINE G. UNITE MICROBIOLOGIE ET GENETIQUE COTTE-PATAT N. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE ESCUDIE D. (Mme) CENTRE DE THERMIQUE DE LYON FRANCIOSI P. GEMPPM*** MANDRAND M.A. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE POUSIN G. BIOLOGIE ET PHARMACOLOGIE ROCHE A. INGENIERIE DES MATERIAUX POLYMERES SEGUELA A. GEMPPM*** Directeurs de recherche I.N.R.A. : FEBVAY G. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS GRENIER S. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS RAHBE Y. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS Directeurs de recherche I.N.S.E.R.M. : PRIGENT A.F. (Mme) BIOLOGIE ET PHARMACOLOGIE MAGNIN I. (Mme) CREATIS** * CEGELY CENTRE DE GENIE ELECTRIQUE DE LYON ** CREATIS CENTRE DE RECHERCHE ET D’APPLICATIONS EN TRAITEMENT DE L’IMAGE ET DU SIGNAL ***GEMPPM GROUPE D'ETUDE METALLURGIE PHYSIQUE ET PHYSIQUE DES MATERIAUX ****LAEPSI LABORATOIRE D’ANALYSE ENVIRONNEMENTALE DES PROCEDES ET SYSTEMES INDUSTRIELS

TABLE DES MATIERES
INTRODUCTION .....................................................................................
CHAPITRE I: LA MOBILITE DES COMPOSES ORGANIQUES HYDROPHOBES DU TYPE HAP DANS LES MATRICES POREUSES DE TYPE SOL........................... 20
I.1 PROPRIETES DES SOLS ...................................................................................................... 20 I.1.1 Porosité .................................................................................................................... 21 I.1.2 Type d’eau dans la zone saturée.............................................................................. 22 I.1.3 Masse volumique...................................................................................................... 22 I.1.4 Teneur en eau........................................................................................................... 23 I.1.5 Perméabilité............................................................................................................. 23
I.2 ORIGINES ET PROPRIETES DES HAP.................................................................................. 24 I.2.1 Nomenclature des HAP............................................................................................ 24 I.2.2 Origine des HAP dans l'environnement................................................................... 25 I.2.3 Propriétés physico-chimiques.................................................................................. 26 I.2.4 Biodégradabilité ...................................................................................................... 27
I.3 ASPECTS RELATIFS A LA MOBILITE DES HAP DANS LES SOLS........................................... 29 I.3.1 Aspects hydrodynamiques – Transport d’un soluté non réactif en milieu poreux saturé................................................................................................................................ 29
I.3.1.1 Modélisation de l’écoulement convectif dispersif ............................................ 29 I.3.1.2 Modèle des réacteurs en cascade ...................................................................... 33 I.3.1.3 Comparaison entre les deux approches............................................................. 34
I.3.2 Aspects physico-chimiques relatifs à la mobilité des HAP dans un sol................... 34 I.3.2.1 Solubilité ........................................................................................................... 34 I.3.2.2 Processus de sorption/désorption...................................................................... 36 I.3.2.3 Processus de dissolution ................................................................................... 43 I.3.2.4 Influence des paramètres physico-chimiques sur les mécanismes de transfert des HAP en phase liquide et sur la solubilité des HAP................................................ 45 I.3.2.5 Transport particulaire........................................................................................ 50
I.4 LES TESTS DE LIXIVIATION ............................................................................................... 51 I.5 CONCLUSION.................................................................................................................... 52
CHAPITRE II: DESCRIPTION DES CARACTERISTIQUES DU SOL ........................54
II.1 GRANULOMETRIE ........................................................................................................... 54 II.2 HUMIDITE ....................................................................................................................... 55 II.3 TENEUR EN HAP ............................................................................................................ 55 II.4 TENEUR EN PHENOL........................................................................................................ 56
7

II.5 CARBONE ORGANIQUE TOTAL (COT) ............................................................................ 57 II.6 CARACTERISATION DE LA NATURE DU SOL AU MICROSCOPE ENVIRONNEMENTAL A
BALAYAGE ELECTRONIQUE (MEB) ....................................................................................... 57 II.7 LE POUVOIR TAMPON DU SOL.......................................................................................... 60 II.8 CONCLUSION .................................................................................................................. 61
CHAPITRE III: APPLICATIONS DES TESTS CLASSIQUES DE LIXIVIATION A L’ETUDE DE LA MOBILITE DES HAP..................................................................
III.1 PRINCIPAUX TESTS DE LABORATOIRE DANS LE DOMAINE DE LA LIXIVIATION ................ 62 III.2 LIXIVIATION EN MILIEU DISPERSE.................................................................................. 64
III.2.1 Cinétiques de mise en solution .............................................................................. 64 III.2.1.1 Protocole......................................................................................................... 64 III.2.1.2 Résultats et commentaires .............................................................................. 65
III.2.2 Isotherme de désorption par renouvellements séquentiels ................................... 66 III.2.2.1 Protocole......................................................................................................... 66 III.2.2.2 Résultats et commentaires .............................................................................. 67
III.3 PERCOLATION EN COLONNE........................................................................................... 73 III.3.1 Protocole ............................................................................................................... 73 III.3.2 Résultats et commentaires..................................................................................... 74
III.4 COMPARAISON ENTRE LES RESULTATS OBTENUS PAR LES TESTS DE LIXIVIATION
CLASSIQUES........................................................................................................................... 75 III.5 CONCLUSIONS ............................................................................................................... 77
CHAPITRE IV: CONCEPTION D’UN OUTIL EXPERIMENTAL POUR L’ETUDE DE LA MOBILITE DES POLLUANTS ORGANIQUES HYDROPHOBES (HAP)
DANS DES MILIEUX POREUX DE TYPE SOL ...........................................................
IV.1 OBJECTIFS ET SOLUTIONS TECHNIQUES PROPOSEES DANS LE CAHIER DES CHARGES...... 78 IV.1.1 Mode de contact solide/liquide.............................................................................. 78 IV.1.2 Contrôle du temps de contact solide/liquide ......................................................... 78 IV.1.3 Dispositifs de séparation solide/liquide ................................................................ 80 IV.1.4 Contrôle de la température ................................................................................... 80 IV.1.5 Protection de la dégradation des HAP par la lumière.......................................... 81 IV.1.6 Phase initiale ......................................................................................................... 81
IV.2 MISE EN ŒUVRE DU TEST .............................................................................................. 82 IV.2.1 Mise en place du sol .............................................................................................. 82 IV.2.2 Mise en route ......................................................................................................... 83 IV.2.3 Paramètres du batch percolant ............................................................................. 83
IV.3 DETERMINATION DE LA DISTRIBUTION DES TEMPS DE SEJOUR (DTS) .......................... 84 IV.4 DESCRIPTIONS DES PROTOCOLES................................................................................... 87
8

IV.4.1 Etude des vitesses de relargage dans le système................................................... 87 IV.4.2 Renouvellements séquentiels ................................................................................. 89 IV.4.3 Percolation ............................................................................................................ 89
IV.5 CONCLUSION................................................................................................................. 89
CHAPITRE V: APPLICATION DU BATCH PERCOLANT A L’ETUDE DE LA MOBILITE DES POLLUANTS ORGANIQUES HYDROPHOBES (HAP) PRESENTS
DANS DES SOLS INDUSTRIELS – APPROCHE METHODOLOGIQUE................... 91
V.1 ETUDE DE LA MOBILITE DES HAP................................................................................... 91 V.1.1 Identification des mécanismes de relargage vers la phase liquide ........................ 91 V.1.2 Influence de paramètres physico-chimiques sur les vitesses de relargage vers la phase liquide .................................................................................................................... 92 V.1.3 Evolution du relargage au cours du temps............................................................. 94
V.2 MISE EN EVIDENCE D’UNE FRACTION SOLUBILISEE ET D’UNE FRACTION PARTICULAIRE
DANS LES CONDITIONS EXPERIMENTALES DU BATCH PERCOLANT.......................................... 95 V.2.1 Résultats.................................................................................................................. 96 V.2.2 Conclusion .............................................................................................................. 99
V.3 MOBILITE DE LA FRACTION SOLUBILISEE........................................................................ 99 V.3.1 Identification du mécanisme contrôlant le relargage des HAP dans la phase liquide lors d'un contact avec le sol ................................................................................. 99
V.3.1.1 Concentrations expérimentales stationnaires (à l'équilibre).......................... 100 V.3.1.2 Calcul théoriques des concentrations à l'équilibre à 25°C dans l'eau............ 101 V.3.1.3 Modèle de dissolution du premier ordre pour le batch percolant.................. 105
V.3.2 Justification de l’utilisation du cosolvant méthanol ............................................. 107 V.3.2.1 Théorie sur la solubilisation des composés hydrophobes dans des mélanges binaires. ...................................................................................................................... 108 V.3.2.2 Application de la théorie de solubilisation des composés hydrophobes utilisant les mélanges binaires sur le sol étudié ....................................................................... 109
V.3.3 Influence des paramètres L/S, T°, f et q sur le relargage des HAP dans la phase liquide
c
............................................................................................................................. 112 V.3.3.1 Influence du rapport L/S et validation du modèle de dissolution.................. 113 V.3.3.2 Influence du cosolvant................................................................................... 117 V.3.3.3 Influence de la température ........................................................................... 119 V.3.3.4 Influence du débit .......................................................................................... 125 V.3.3.5 Conclusion : mèthode d’estimation graphique (abaque) de la concentration stationnaire ................................................................................................................. 126
V.3.4 Evolution du relargage suite à des renouvellements séquentiels des éluats ........ 128 V.3.4.1 Résultats ........................................................................................................ 129 V.3.4.2 Conclusion..................................................................................................... 132
9

V.3.5 Synthèse des résultats menées sur la fraction solubilisée..................................... 133 V.4 MOBILITE DE LA FRACTION PARTICULAIRE................................................................... 134
V.4.1 Détermination de la nature des particules au MEB environnemental ................. 135 V.4.2 Influence de paramètres physico-chimiques sur le relargage des HAP sous forme particulaire..................................................................................................................... 137
V.4.2.1 Influence de la bioréactivité .......................................................................... 138 V.4.2.2 Influence des sels dissous dans la phase liquide ........................................... 140 V.4.2.3 Influence de la masse de sol en contact avec la phase liquide ...................... 142 V.4.2.4 Influence du débit .......................................................................................... 147
V.4.3 Evolution du relargage des HAP particulaires au cours du temps ...................... 151 V.5 CONCLUSION ................................................................................................................ 152
CONCLUSION GENERALE .............................................................................
REFERENCES BIBLIOGRAPHIQUES....................................................................
ANNEXES.............................................................................................
10

LISTE DES FIGURES
Figure 1 : Structure des 16 HAP de la liste EPA ..................................................................... 25 Figure 2: exemple de la distribution de la matière organique dans un goudron de houille,
exprimée en pourcentage du Carbone Organique Total (COT), avec COT=900 g.Kg , [HAESELER, 1999].
-1
....................................................................................................... 26 Figure 3 : Dégradation aérobie d'un noyau benzénique ........................................................... 27 Figure 4 : Dégradation anaérobie du benzène en présence de nitrate ...................................... 27 Figure 5 : Clivage de la structure cyclique du naphtalène et du benzo(a)pyrène..................... 28 Figure 6 : Métabolites identifiés lors de la dégradation du pyrène par Mycobactérium sp. .... 28 Figure 7: Facteurs responsables de la dispersion longitudinal [FETTER, 1999]..................... 32 Figure 8 : Schéma des réacteurs en cascade pour le modèle M.C.E. ....................................... 33 Figure 9: Représentation schématique des enthalpies mises en jeu lors de la dissolution d'une
molécule organique hydrophobe ...................................................................................... 36 Figure 10 : Différents modèles d’isotherme de sorption [GRATHWHOL, 1998] .................. 39 Figure 11 : Système eau mobile et eau immobile .................................................................... 41 Figure 12: vue schématique du modèle à deux film stagnants: Le transfert de masse à travers
les deux films est supposé contrôlé par la seconde loi de Fick. A l'interface de chacun des films, l'équilibre est supposé atteint et les concentrations de chaque coté sont données par les coefficients de partage entre la phase organique et l'eau. δ et δ sont les épaisseurs de chacun des films. C et C sont respectivement les concentrations dans la phase organique et l'eau.
o e
i,PLNA i,e
............................................................................................................. 45 Figure 13 : Profil du contenu total moyen en HAP dans le sol ................................................ 56 Figure 14:Distribution de la matière organique dans les goudrons de houille en pourcentage
du COT [HAESELER, 1999]........................................................................................... 57 Figure 15: Photographie du sol réalisée au MEB environnemental......................................... 58 Figure 16: % atomique de C, O, Fe, Si dans le sol brut tamisé à 2 mm................................... 59 Figure 17: Evolution du pH pour des ajouts de base et d'acide................................................ 60 Figure 18: Principales expériences menées à l'INSA de Lyon dans le domaine de la lixiviation
des sols ............................................................................................................................. 63 Figure 19: représentation en pourcentage de la pollution organique totale ............................. 64 Figure 20:Cinétique de mise en solution des HAP à 3 cycles.................................................. 65 Figure 21: Cinétique de mise en solution des HAP à 4 cycles................................................. 65 Figure 22:Cinétique de mise en solution.................................................................................. 66 Figure 23 : évolution des concentrations et de la conductivité en fin de chacune des séquences
d'extractions avec la solution préparée (eau +NaN )3 ....................................................... 67 Figure 24: formation d’un film lors du prélèvement de la phase liquide centrifugée.............. 68 Figure 25: Quantité relarguée cumulée par rapport au contenu initial..................................... 69
11

Figure 26: évolution des concentrations à chaque extraction avec le mélange eau/méthanol . 71 Figure 27: Rapport des concentrations dans le mélange eau/méthanol sur les concentrations
dans l’eau.......................................................................................................................... 72 Figure 28: Quantité relarguée cumulée par rapport au contenu initial..................................... 72 Figure 29: Principe de la percolation ....................................................................................... 73 Figure 30:Evolution des concentrations en HAP dans les percolats ........................................ 74 Figure 31: Evoultion de la conductivité et de la turbidité dans les percolats........................... 75 Figure 32:Lixiviation en milieu dispersé – Profil des concentrations en HAP lors de la
cinétique avec l'eau permutée à t=50 heures .................................................................... 76 Figure 33:Lixiviation en milieu dispersé – Profil des concentrations en HAP lors de la
première et dernière extractions avec l'eau permutée....................................................... 76 Figure 34:Lixiviation en milieu dispersé - Profil des concentrations en HAP lors de la
première et dernière extractions avec le mélange eau/méthanol...................................... 76 Figure 35:Percolation -Profil des concentrations en HAP dans le 1 et dernier percolater ........ 76 Figure 36: Principe de fonctionnement du dispositif expérimental ......................................... 79 Figure 37: Principe du maintien de la température dans la colonne et dans le réacteur tampon
.......................................................................................................................................... 81 Figure 38 Schéma global de fon ctionnement .......................................................................... 82 Figure 39: Procédure de remplissage de la colonne ................................................................. 83 Figure 40:Distribution des temps de séjour dans la colonne.................................................... 85 Figure 41: vision schématique de l’évaluation de la mobilité des HAP dans les sols ............. 95 Figure 42: Séparation de l'échantillon R3 en trois fractions .................................................... 96 Figure 43 : Evolution des HAP relargués à chaque renouvellement pour 3 systèmes de
filtration différents............................................................................................................ 97 Figure 44: Evolution de la conductivité et de la turbidité à chaque renouvellement avec de
l’eau permutée et le sable [0-400] µm.............................................................................. 97 Figure 45: Fraction solubilisée et fraction particulaire dans R3 .............................................. 97 Figure 46: Répartition des fractions solubilisées et particulaires dans R3............................... 98 Figure 47 : Comportement d’un sol étudié au laboaratoire lors des renouvellements successifs
avec de l’eau permutée..................................................................................................... 98 Figure 48: vitesse de relargage du phénanthrène et de l'anthracène dans la phase liquide .. 100 Figure 49:Prédiction des concentrations en phase aqueuse à partir de résultats obtenus en
présence de méthanol ..................................................................................................... 108 Figure 50: Vitesse de relargage du phénanthrène et de l'anthracène dans la phase liquide pour fc=0,5 ...... 110 Figure 51: Vitesse de relargage du phénanthrène et de l'anthracène dans la phase liquide pour
fc=0,4.............................................................................................................................. 110 Figure 52: Vitesse de relargage du phénanthrène et de l'anthracène dans la phase liquide pour
fc=0,3.............................................................................................................................. 110
12

Figure 53: Vitesse de relargage du phénanthrène et de l'anthracène dans la phase liquide en fonction du temps pour fc=0 .......................................................................................... 110
Figure 54:Représentation en log des solubilités en fonction de la fraction de méthanol en volume dans la solution de lixiviation. (●) représente les point expérimentaux, (─) représente la régression linéaire ..................................................................................... 111
Figure 55: Influence de L/S sur les vitesses de relargage du phénanthrène et de l’anthracène avec fc=0.5. Les symboles (○) et(●) représentent les points expérimentaux; les lignes représentent les simulations pour K optimal. K est exprimé en cm .h .3 -1 ...................... 114
Figure 56: Sensibilité de K pour la courbe expérimentale L/S=6 représentée par le symbole (○)................................................................................................................................... 116
Figure 57:Simulation des vitesses de relargages pour différents L/S avec K=3,5.10 m.h-6 -1 116 Figure 58: Influence de la fraction de méthanol sur les vitesses de dissolution. Les symboles
(○)(●)(▲)(∆) représentent les points expérimentaux; les lignes continues représentent les simulations pour K optimal. K est exprimé en cm .h3 -1 ........................................... 118
Figure 59 Solubilité en fonction de la fraction de méthanol en volume dans la solution de lixiviation. (●) représente les point expérimentaux, (─) représente la courbe de tendance de forme exponentielle ................................................................................................... 118
Figure 61: Evolution du coefficient de transfert K [m .h ] en fonction de la fraction de méthanol dans la phase liquide
3 -1
....................................................................................... 119 Figure 62: Influence de la température sur les vitesses de dissolution. Les symboles
(○)(●)(▲)(∆) .................................................................................................................. 120 Figure 63 Solubilité en fonction de la température. (●) représente les point expérimentaux,
(─) représente la courbe de tendance de forme exponentielle ....................................... 121 Figure 64: Concentration à l’équilibre pour le phénanthrène et l’anthracène en fonction de la
température..................................................................................................................... 122 Figure 65: Evolution de K (m .h ) en fonction de la température3 -1 ......................................... 124 Figure 65:Influence du débit sur la vitesse de dissolution. . (●),(○) représentent les point
expérimentaux, les lignes pleines représentent la simulation pour K optimal. K est exprimé en cm .h3 -1.......................................................................................................... 125
Figure 67 : abaque du phénanthrène fournissant C pour une température donnée et une fraction de méthanol donnée
sat
.......................................................................................... 128 Figure 68: Evolution des concentrations après chaque renouvellement séquentiel R de 24 h à
22 et 40°C. Les symboles (•) et(∆) représentent les point expérimentaux. Les symboles (-
) et (▬) sont les valeurs théoriques, respectivement à 22 et 40 °C, des concentrations à l'équilibre calculées d'après la loi de Raoult .................................................................. 130
Figure 69:Représentation schématique de l'évolution des vitesses de dissolution au cours de renouvellements séquentiels........................................................................................... 131
13

Figure 70: Comparaison entre le pourcentage relargué par le sol (●) et les concentrations expérimentales obtenues après chaque renouvellement séquentiel de 24 heuresà 40°C (∆), pour le phénanthrène et l'anthracène ....................................................................... 132
Figure 71 : Sensibilité de K pour la courbe expérimentale L/S=6 représentée par le symbole (o) Résultats obtenus page 116....................................................................................... 134
Figure 72: Concentration en sortie de la colonne. (●) représente les points expérimentaux et les lignes pleines les simulation ..................................................................................... 134
Figure 72: % atomique de Carbone et Oxygène dans les particules mobilisées par le test des renouvellement successifs .............................................................................................. 136
Figure 74: Photographies des particules mobilisées ; réalisées au MEB environnemental ... 137 Figure 74 : Evaluation de la biodégradation dans les conditions de l’essai ........................... 139 Figure 75: Evolution de la conductivité et de la turbidité à chaque renouvellement séquentiel
avec l’eau permutée et la solution de NaN à 15 mM3 .................................................... 139 Figure 76 : Etude de l'influence du CaCl sur le relargage de particules par renouvellements
séquentiels2
...................................................................................................................... 141 Figure 77: Evolution des conductivités et de la turbidité avec le solution de CaCl2 ............. 142 Figure 79: Evolution des conductivités et de la turbidité avec l’eau permutée...................... 142 Figure 80: Evolution des concentrations lors des renouvellements séquentiels ; L/S=6 (150
ml d'eau et 25 g de sol) ; T=22°C ; Q=30 mL.h-1 et un lit de sable de granulométrie [0-400]µm. .......................................................................................................................... 143
Figure 81: Evolution de la conductivité et des concentrations en phénanthrène et anthracène pour 25 g de sol .............................................................................................................. 145
Figure 82 : Evolution de la conductivité et des concentrations en phénanthrène et anthracène pour 100 g de sol ............................................................................................................ 145
Figure 83: Evolution de la conductivité et des concentrations en phénanthrène et anthracène pour q=30 mL.h-1............................................................................................................ 149
Figure 84 : Evolution de la conductivité et des concentrations en phénanthrène et anthracène pour q=100 mL.h-1.......................................................................................................... 149
Figure 85: évolution en log népérien du potentiel de HAP particulaire relargable par le sol en fonction du débit............................................................................................................. 151
Figure 86 : Schéma du dispositif soxhlet ............................................................................... 168 Figure 87 : dispositif expérimental d’extraction en phase solide (SPE) ................................ 171
14

LISTE DES TABLEAUX Tableau 1: composition moyenne des substance humiques [KHAN, 1978]............................ 21 Tableau 2 : Propriétés physico-chimiques des HAP [US EPA, 1986]..................................... 26 Tableau 3 : répartition granulométrique................................................................................... 54 Tableau 4 : contenu total en HAP extractibles dans le sol ....................................................... 55 Tableau 5: caractéristiques des photos ..................................................................................... 59 Tableau 6: Comparaison des pourcentages atomiques mesurés au MEB E avec ceux de
[HAEELER, 1999] ........................................................................................................... 60 Tableau 7: Synthèse du cahier des charges .............................................................................. 81 Tableau 8: Liste des paramètres variables dans le batch percolant.......................................... 84 Tableau 9: synthèse des conditions opératoires ....................................................................... 95 Tableau 10: synthèse des conditions expérimentales ............................................................. 100 Tableau 11: Concentrations théoriques à l’équilibre pour un mécanisme de dissolution...... 102 Tableau 12: Calcul théorique des concentrations à l’équilibre selon des isothermes de
désorption ....................................................................................................................... 104 Tableau 13: Récapitulatif des concentrations à l’équilibre dans l’eau................................... 104 Tableau 14: synthèse des conditions expérimentales ............................................................. 109 Tableau 15:Valeur de σ obtenue lors de la thése et par d’autres auteurs............................... 111 Tableau 16: choix paramétriques pour les études du transfert en phase liquide en fonction du
temps .............................................................................................................................. 112 Tableau 17: synthèse des conditions expérimentales pour l’étude de l’influence du rapport L/S
........................................................................................................................................ 113 Tableau 18: synthèse des conditions expérimentales ............................................................. 117 Tableau 19: synthèse des conditions expérimentales ............................................................. 120 Tableau 20: comparaision des enthalpies de dissolution avec la littérature........................... 122 Tableau 21: synthèse des conditions expérimentales ............................................................. 125 Tableau 22: synthèse des études sur les vitesses de relargage de la fraction solubilisée....... 127 Tableau 23: Première étape de la construction de l'abaque.................................................... 127 Tableau 24 : 2 étape de la construction de l'abaqueieme .......................................................... 128 Tableau 25: synthèse des conditions expérimentales ............................................................. 129 Tableau 26: synthèse des conditions expérimentales imposées pour le test de percolation .. 133 Tableau 27: caractéristiques des photos prises au MEB environnemental ............................ 137 Tableau 28: Récapitulation des essais réalisés pour l’étude de la fraction particulaire ......... 138 Tableau 29: synthèse des conditions opératires ..................................................................... 138 Tableau 30: synthèse des conditions opératires ..................................................................... 141 Tableau 31: synthèse des conditions opératoires ................................................................... 144 Tableau 32:Concentrations moyenne d'origine particulaire................................................... 146
15

Tableau 33: Valeurs de L/S relative à l'apparition et la disparition du relargage de HAP particulaires au cours de la percolation .......................................................................... 146
Tableau 34: Potentiel de HAP particulaires relargables par le sol pour q=30 ml.h et T=22°C-1
........................................................................................................................................ 146 Tableau 35: synthèse des conditions opératoires ................................................................... 148 Tableau 36:Concentrations moyennes en phénanthrène et anthracène particulaire pour q=30
ml.h-1 .............................................................................................................................. 150 Tableau 37:Concentrations moyennes en phénanthrène et anthracène particulaire pour q=100
ml.h-1 .............................................................................................................................. 150 Tableau 38: Potentiel de HAP particulaires relargables par le sol pour m=25 g et T=22°C 150
16

INTRODUCTION La pollution des sols et des sous-sols résulte des conséquences des diverses activités humaines (industrielles, agricoles…) cumulées au cours des temps. Ces pollutions négligées jusqu’à une époque relativement récente deviennent aujourd’hui une préoccupation majeure en raison de leurs conséquences sanitaires, environnementales et socio-économiques. De nos jours, la réglementation environnementale de plus en plus contraignante, les améliorations des procédés et des pratiques de protection de l’environnement, l’application plus suivie des principes du développement durable et la gestion améliorée des déchets limitent de plus en plus l’impact polluant de ces diverses activités humaines. Comme la plupart des pays industriels, la France hérite des conséquences de son passé industriel. Un type de pollution très répandue en France est le cas des sites où des combustibles fossiles ou leurs produits dérivés ont été utilisés. C’est par exemple le cas des sites de traitement du bois où la créosote et l’huile d’anthracène, deux produits riches en HAP, servaient en tant que conservateur. Un autre cas à citer est celui des goudrons de houille obtenus lors de la pyrolyse du charbon (usines à gaz, cokeries). Ces sites pollués, très nombreux dans les pays industriels, sont aujourd’hui délaissés pour la plupart est constituent autant de friches industrielles. On compte en France de l’ordre de 700 à 800 anciennes usines à gaz. Le devenir de ces friches pose des problèmes environnementaux majeurs et notamment celui des risques de transfert de pollution. La problématique de la gestion et du traitement des sites pollués est complexe du point de vue scientifique, technique et financier. Une première étape consiste en l’identification de la pollution, de son ampleur et de sa répartition sur le site, y compris dans la nappe. L’instrumentation et le suivi en temps peut donner des informations importantes quant à l’urgence de la mise en œuvre du processus de réhabilitation/traitement. En effet, ce n’est pas tant la présence de polluants dans les sols qui est problématique, mais le fait que cette pollution soit mobilisable et donc qu’elle risque d’affecter une population. La connaissance des mécanismes de mobilisation et de transfert joue à cet égard un rôle largement aussi important que l’identification de la présence d’un contaminant à un endroit donné. De plus, dans le domaine de la réhabilitation des sols pollués, avant de décider objectivement d’un traitement efficace, il est important que des études sur la mobilité des polluants aient été réalisées. Cependant, à l’heure actuelle le processus de normalisation et d’élaboration de guides ou de méthodologies permettant d’évaluer et d’orienter ce types de travaux se trouve encore dans une phase de début. Ainsi, le guide intitulé « Diagnostic approfondi des sites et sols pollués »
17

édité par le BRGM, seul document français administratif à caractère réglementaire abordant le sujet, ne renvoi pas, dans sa version actuelle, à des méthodes, ni à des tests spécifiques à l’étude des mécanismes de transfert, bien que leur compréhension y soit recommandée « …la compréhension des mécanismes de transfert des polluants dans ces milieux, fortement dépendants des propriétés physico-chimiques des substances (viscosité, solubilité, densité,…) mais aussi des paramètres du sol (pH, potentiel redox, teneurs en matières organiques, …)… ». Une batterie importante de tests normalisés existe dans le domaine de la lixiviation des déchets. Ceux-ci fournissent notamment les outils nécessaires pour étudier la mobilité de leurs polluants. Certains de ces tests sont d’ailleurs abusivement employés, à notre avis, pour l'étude des sols pollués. Il n'existe pas encore de tests de lixiviations normalisés dans le domaine des sols pollués mais on attend prochainement la publication par l’ISO des premiers tests de conformité élaborés par le Comité technique 190 « Soil quality ». En France, le groupe AFNOR X31-E travaille actuellement sur une norme dont les aboutissants seraient, à travers une approche en scénario, d'orienter l'expérimentateur vers des tests adaptés aux sites étudiés, en fonction des objectifs poursuivis, par exemple pour l’identification des mécanismes de transfert, pour la classification du site etc. Aujourd’hui, la nécessité d’étudier les mécanismes de transfert dans les sols pollués est donc bien réelle. Cependant, il est primordial d’établir une méthodologie de travail, les outils expérimentaux spécifiques à la problématique des sols pollués ainsi que l’interprétation des résultats expérimentaux, notamment par modélisation. C’est dans ce contexte et dans le cadre d’un programme de recherche mené par l’ADEME, organisme Français chargé notamment de l’identification, de la caractérisation, du suivi et de la gestion des sites pollués, que se sont déroulés les travaux de thèse. L’objectif était d’étudier la mobilité des HAP d’un site industriel du Nord de la France. Ce travail a été réalisé en plusieurs grandes étapes :
1. La première partie a consisté à recueillir les connaissances sur la problématique de la mobilité des composés organiques dans les sols, afin d’orienter de la meilleure façon le déroulement du programme.
2. Le sol brut, après avoir été échantillonné de façon représentative, a tout d’abord était
caractérisé du point de vue de ses principales caractéristiques physico-chimiques : humidité, contenu total en polluant, Carbone Organique Total, composition élémentaire, teneur en phénol, granulométrie, humidité,...
18

3. Ensuite, une série d’expérimentation basée sur des tests de lixiviations classiques a été réalisée afin d’obtenir les premiers renseignements sur le comportement du sol au contact de l’eau. Suite à cette première phase, il a été décidé de poursuivre les lixiviations à l’aide d’un outil expérimental plus adapté à nos objectifs.
4. Conception et mise au point de l’outil expérimental adapté à l’étude de la mobilité des
HAP dans le sol étudié.
5. Enfin, une seconde série d’expérimentations a permis d’appréhender les principes de la mobilité des HAP dans le sol étudié, à travers l’identification des mécanismes majoritaires contrôlant le transfert ainsi que l’évaluation de l’influence de certains paramètres physico-chimiques sur ce transfert.
Outre le fait d’étudier la mobilité des polluants d’un site industriel donné, notre travail a aussi contribué à l’élaboration d’une méthodologie de caractérisation de la mobilité des HAP (et plus généralement des composés organiques hydrophobes) dans les sols/sites pollués.
19

Chapitre I: La mobilité des composés organiques hydrophobes du type HAP dans les matrices poreuses de type sol
I.1 Propriétés des sols
Un sol est une pellicule d'altération recouvrant une roche. Il est formé d'une fraction minérale et de matière organique (humus). Un sol prend naissance à partir de la roche puis il évolue sous l'action des facteurs du milieu, essentiellement le climat et la biosphère (végétation). La formation de la fraction minérale d'un sol provient de :
la désagrégation mécanique des roches qui donne des fragments, l'altération chimique des roches qui produit des ions solubles (cations, anions : silicates...).
Ce processus d'altération de la roche forme le complexe d'altération qui comprend les argiles et d'autres constituants comme les oxydes et hydroxydes, carbonates, phosphates, sulfates etc de Fe, Al, Mg, Si. La Kaolinite, l'argile la plus fréquemment présente dans les sols a comme formule générale: Si4Al4010(0H)8,nH2O. Les éléments traces (conc. <1 g/kg) jouent également un rôle important : Mn, Sr, V, Cr,… Les 10 éléments les plus présents dans les sols sont, dans l'ordre décroissant : O > Si >Al > Fe > C > Ca > K > Na > Mg . Environ la moitié à deux tiers du volume d'un sol est constitué de matière solide. Les composés minéraux représentent, plus de 90 % de cette matière solide. La matière organique peut être définie comme une matière carbonée provenant d'êtres vivants végétaux et animaux. Elle est composée d'éléments principaux (C, H, O, N) et d'éléments secondaires (S, P, K, Ca, Mg…). La matière organique se répartit en 4 groupes, selon son origine :
la matière organique vivante, animale et végétale, qui englobe la totalité de la biomasse en activité, les débris d'origine végétale (résidus végétaux, exsudats) et animale (déjections, cadavres) regroupés sous le nom de "matière organique fraîche", des composés organiques intermédiaires, appelés matière organique transitoire, provenant de l'évolution de la matière organique fraîche, des composés organiques stabilisés, les matières humiques (acides humiques et acides fulviques), provenant de l'évolution des matières précédentes.
20
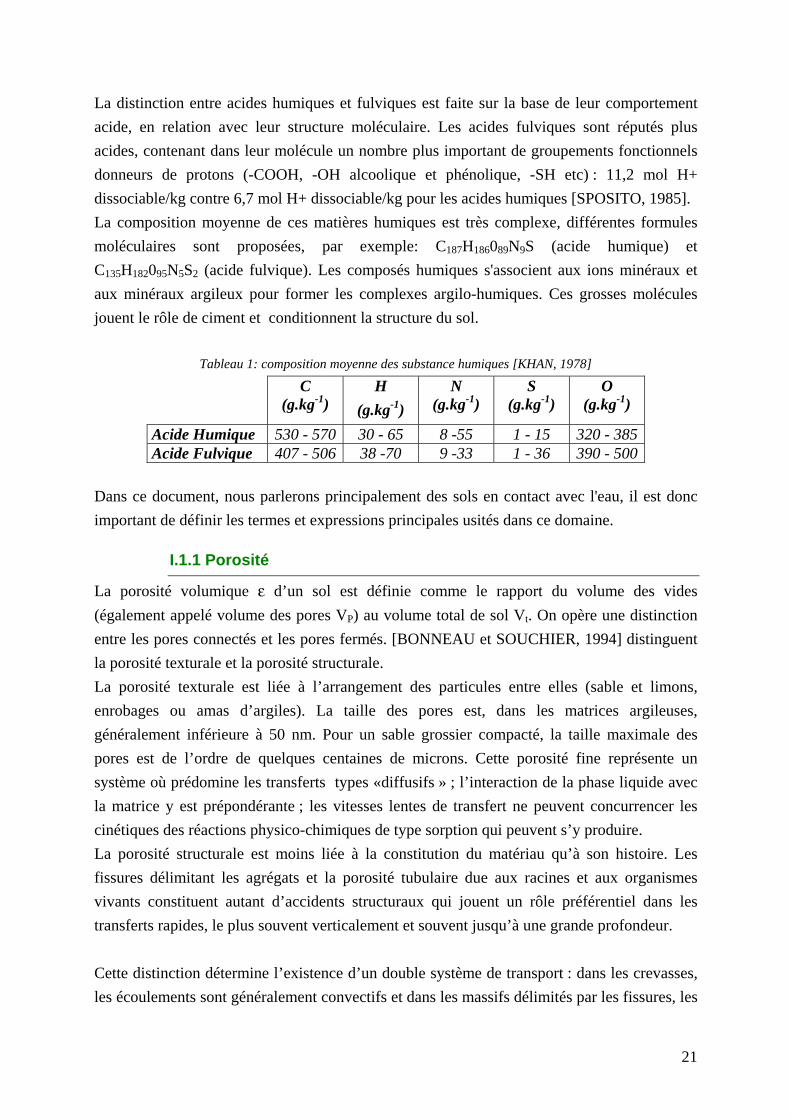
La distinction entre acides humiques et fulviques est faite sur la base de leur comportement acide, en relation avec leur structure moléculaire. Les acides fulviques sont réputés plus acides, contenant dans leur molécule un nombre plus important de groupements fonctionnels donneurs de protons (-COOH, -OH alcoolique et phénolique, -SH etc) : 11,2 mol H+ dissociable/kg contre 6,7 mol H+ dissociable/kg pour les acides humiques [SPOSITO, 1985]. La composition moyenne de ces matières humiques est très complexe, différentes formules moléculaires sont proposées, par exemple: C187H186089N9S (acide humique) et C135H182095N5S2 (acide fulvique). Les composés humiques s'associent aux ions minéraux et aux minéraux argileux pour former les complexes argilo-humiques. Ces grosses molécules jouent le rôle de ciment et conditionnent la structure du sol.
Tableau 1: composition moyenne des substance humiques [KHAN, 1978]
C (g.kg-1)
H (g.kg-1)
N (g.kg-1)
S (g.kg-1)
O (g.kg-1)
Acide Humique 530 - 570 30 - 65 8 -55 1 - 15 320 - 385 Acide Fulvique 407 - 506 38 -70 9 -33 1 - 36 390 - 500
Dans ce document, nous parlerons principalement des sols en contact avec l'eau, il est donc important de définir les termes et expressions principales usités dans ce domaine.
I.1.1 Porosité
La porosité volumique ε d’un sol est définie comme le rapport du volume des vides (également appelé volume des pores VP) au volume total de sol Vt. On opère une distinction entre les pores connectés et les pores fermés. [BONNEAU et SOUCHIER, 1994] distinguent la porosité texturale et la porosité structurale. La porosité texturale est liée à l’arrangement des particules entre elles (sable et limons, enrobages ou amas d’argiles). La taille des pores est, dans les matrices argileuses, généralement inférieure à 50 nm. Pour un sable grossier compacté, la taille maximale des pores est de l’ordre de quelques centaines de microns. Cette porosité fine représente un système où prédomine les transferts types «diffusifs » ; l’interaction de la phase liquide avec la matrice y est prépondérante ; les vitesses lentes de transfert ne peuvent concurrencer les cinétiques des réactions physico-chimiques de type sorption qui peuvent s’y produire. La porosité structurale est moins liée à la constitution du matériau qu’à son histoire. Les fissures délimitant les agrégats et la porosité tubulaire due aux racines et aux organismes vivants constituent autant d’accidents structuraux qui jouent un rôle préférentiel dans les transferts rapides, le plus souvent verticalement et souvent jusqu’à une grande profondeur. Cette distinction détermine l’existence d’un double système de transport : dans les crevasses, les écoulements sont généralement convectifs et dans les massifs délimités par les fissures, les
21

transferts hydriques et de solutés sont de natures diffusives. Ce double déplacement induit l’existence de plusieurs échelles temporelles pour l’écoulement.
I.1.2 Type d’eau dans la zone saturée
L’hydrodynamique considère deux types extrêmes d’eau en milieu poreux saturé :
• L’eau de gravité qui représente la fraction mobile de l’eau du sol et contribue activement aux transferts de matière en solution et en suspension.
• L’eau de rétention, immobile, qui constitue le principal milieu réactionnel des phénomènes de solubilisation et d’insolubilisation organo-minérales [BONNEAU et SOUCHIER, 1994].
L’eau de rétention est absorbée par la roche. Elle comprend l’eau hygroscopique et l’eau capillaire. L’eau gravitaire remplit l’espace libre des pores, obéit à la force de gravité et transmet la pression hydrostatique. La proportion entre les deux types d’eau peut varier au cours du temps.
I.1.3 Masse volumique
La masse volumique apparente humide ou totale, d’un milieu poreux, est exprimée par :
(1) t
t
VM
=ρ
avec
Mt = masse totale du solide humide, Vt = volume total de solide humide.
La masse volumique apparente sèche est le rapport de la masse du sol sec Ms et du volume total Vt :
(2) t
s
VM
=ρ
La masse volumique réelle du solide est définie par :
(3) s
s
VM
=ρ
avec Vs = volume de solide sec
I.1.4 Teneur en eau
Les teneurs en eau massique θp et volumique θ sont définies respectivement par :
22

(4) t
e
VV
=θ
(5) t
ep M
M=θ
où Me et Ve sont, respectivement, la masse et le volume total d’eau dans le milieu. Le degré de saturation S représente la fraction du volume des pores Vp occupée par l’eau Ve. On a :
(6) εθ
θθ ===satp
e
VV
S
ε représente la porosité, θsat est la teneur en eau à saturation. S tend vers 0 dans un sol sec et vers 100 % dans un sol totalement saturé.
I.1.5 Perméabilité
La perméabilité est l’aptitude d’un milieu à se laisser traverser par l’eau, sous l’action d’un gradient hydraulique. Elle exprime la résistance du milieu à l’écoulement de l’eau qui le traverse. Dans le cas d’un écoulement permanent saturé monodirectionnel la relation définissant la vitesse d’écoulement v (vitesse de Darcy) est :
(7) dxdhK
AQv −==
où K= coefficient de perméabilité ou conductivité hydraulique [m/s] h= charge hydraulique ou hauteur piézométrique [m] Le coefficient de perméabilité est défini par la loi de Darcy (7) : c’est le volume d’eau gravitaire (en m3) traversant en une unité de temps une unité de section (en m2) orthogonale à la direction de l’écoulement sous l’effet d’une unité de gradient hydraulique. Il a la dimension d’une vitesse. Le coefficient de perméabilité permet la mesure de la perméabilité :
(8) ][10 27 mKKg
P −≈= ν
où ν= viscosité cinétique [m2.s-1] g= accélération gravitationnelle [m.s-2]
23

I.2 Origines et propriétés des HAP
I.2.1 Nomenclature des HAP
Les HAP sont constitués d’au moins deux cycles aromatiques fusionnés. Au sens strict ils ne contiennent que des atomes de carbone et d’hydrogène. Les structures des 16 HAP retenus comme polluants prioritaires par l’American Environmental Protection Agency (EPA) sont présentés dans la Figure 1. Ces HAP appartiennent à une liste de 129 polluants d’origine et de nature variés, pris en compte notamment pour la fréquence de leur présence dans les eaux [KEITH et TEILLARD, 1979]. Ces HAP sont constitués de cycles aromatiques accolés en nombre croissant allant de deux cycles (naphtalène) jusqu’à six cycles (benzo(ghi)pérylène), l’agencement des cycles pouvant être linéaire (anthracène), angulaire (fluoranthène) ou groupé (pyrène). Parmi ces HAP, on distingue parfois ceux à bas poids moléculaire (deux et trois cycles) de ceux à haut poids moléculaire (quatre cycles et plus) >200 g/mol. Les structures présentées dans la Figure 1 illustrent également les principes de la nomenclature des HAP. Cette nomenclature semble un peu déconcertante mais il est en premier lieu difficile d’imaginer un système qui allie la simplicité d’emploi à la capacité de désigner les structures complexes rencontrées. Les principes de cette nomenclature, extraits de l'IUPAC (Internationnal Union of Pure and Applied Chemistry) peuvent être résumés comme suit :
• Les structures des HAP sont positionnées en orientant sur une base horizontale l’alignement de cycles le plus grand possible, avec le reste de la molécule disposée en majorité dans le quadrant supérieur droit.
• Les atomes de carbone extérieurs des structures sont numérotés dans le sens antitrigonométrique avec l’origine sur le sommet libre le plus à gauche du cycle supérieur le plus à droite.
• Les structures des HAP complexes sont exprimées comme dérivée des structures de
base par accolement d’autres systèmes élémentaires aussi simples que possible.
24

Naphtalène Acénaphtène Acénaphtylène Fluorène
Phénanthrène FluoranthèneAnthracène Pyrene
Benzo(a)anthracène Chrysène Benzo(b)fluoranthène Benzo(k)fluoranthène
Benzo(a)pyrène Dibenzo(a,h)anthracène Indéno(c,d)pyrène Benzo(g,h,i)pérylène
Figure 1 : Structure des 16 HAP de la liste EPA
I.2.2 Origine des HAP dans l'environnement
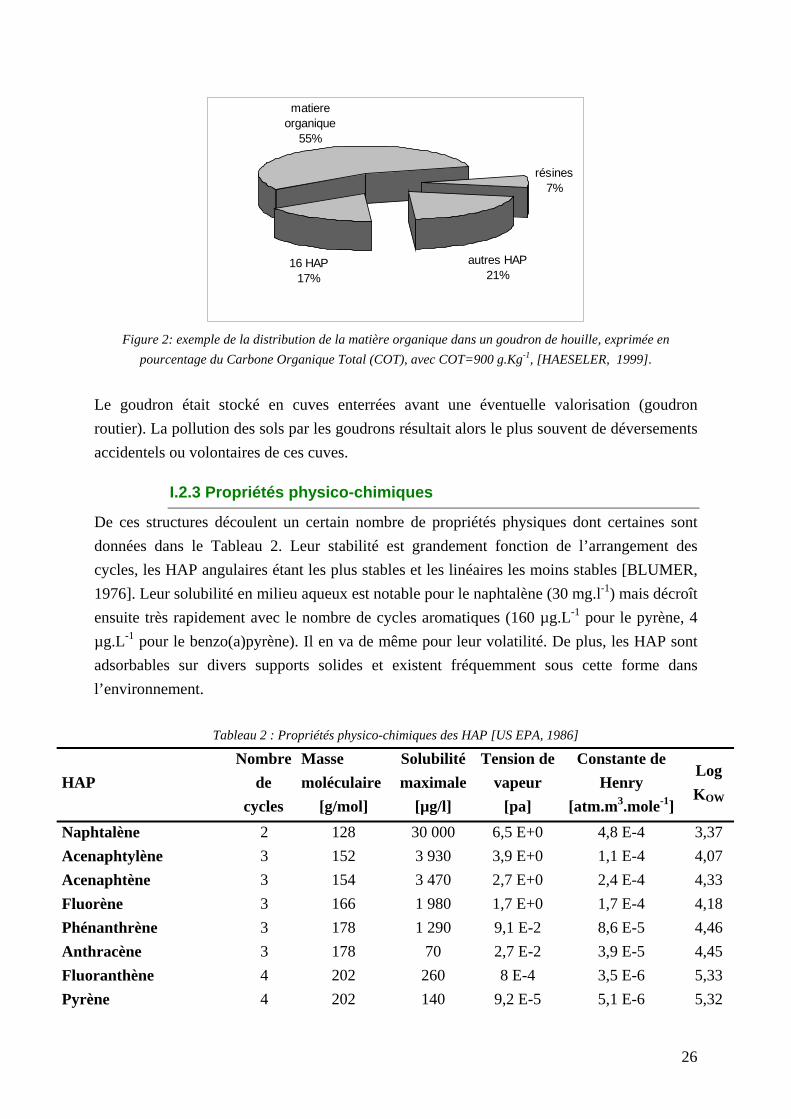
Naturellement présents dans les combustibles fossiles, les HAP se retrouvent dans tous les compartiments de l'écosystème terrestre. Les origines naturelles de ces composés sont extrêmement variées: feux de forêt, activité volcanique, processus géochimique, etc... Cependant, une quantité importante de ces composés est d'origine anthropique et résulte notamment des procédés de pyrolyse ou de combustion, utilisés dans l'industrie, les transports ou le chauffage. Parmi les procédés de pyrolyse, les anciennes usines à gaz et les anciennes cokeries ont systématiquement pollué par des HAP les sols de ces sites industriels. Ces activités consistaient en un traitement thermique à haute température du charbon de houille. Ce traitement thermique des charbons permettait d'obtenir le coke (utilisé longtemps comme combustible) et du gaz qui une fois épuré était utilisé pour l'éclairage des villes. Ce procédé thermique entraînait dans le même temps la formation de goudron (renfermant une fraction importante de HAP: Figure 2) et autres composés chimiques en quantités moindres (benzols, ammoniac, hydrogène sulfuré, phénols et acides cyanhydrique).
25
résines7%
matiere organique
55%
16 HAP17%
autres HAP21%
Figure 2: exemple de la distribution de la matière organique dans un goudron de houille, exprimée en
pourcentage du Carbone Organique Total (COT), avec COT=900 g.Kg-1, [HAESELER, 1999].
Le goudron était stocké en cuves enterrées avant une éventuelle valorisation (goudron routier). La pollution des sols par les goudrons résultait alors le plus souvent de déversements accidentels ou volontaires de ces cuves.
I.2.3 Propriétés physico-chimiques
De ces structures découlent un certain nombre de propriétés physiques dont certaines sont données dans le Tableau 2. Leur stabilité est grandement fonction de l’arrangement des cycles, les HAP angulaires étant les plus stables et les linéaires les moins stables [BLUMER, 1976]. Leur solubilité en milieu aqueux est notable pour le naphtalène (30 mg.l-1) mais décroît ensuite très rapidement avec le nombre de cycles aromatiques (160 µg.L-1 pour le pyrène, 4 µg.L-1 pour le benzo(a)pyrène). Il en va de même pour leur volatilité. De plus, les HAP sont adsorbables sur divers supports solides et existent fréquemment sous cette forme dans l’environnement.
Tableau 2 : Propriétés physico-chimiques des HAP [US EPA, 1986]
HAP Nombre
de cycles
Masse moléculaire
[g/mol]
Solubilité maximale
[µg/l]
Tension de vapeur
[pa]
Constante de Henry
[atm.m3.mole-1]
Log KOW
Naphtalène 2 128 30 000 6,5 E+0 4,8 E-4 3,37 Acenaphtylène 3 152 3 930 3,9 E+0 1,1 E-4 4,07 Acenaphtène 3 154 3 470 2,7 E+0 2,4 E-4 4,33 Fluorène 3 166 1 980 1,7 E+0 1,7 E-4 4,18 Phénanthrène 3 178 1 290 9,1 E-2 8,6 E-5 4,46 Anthracène 3 178 70 2,7 E-2 3,9 E-5 4,45 Fluoranthène 4 202 260 8 E-4 3,5 E-6 5,33 Pyrène 4 202 140 9,2 E-5 5,1 E-6 5,32
26

Benzo(a)anthracène 4 228 14 6,7 E-7 1,2 E-6 5,61 Chrysène 4 228 2 8,4 E-5 1,1 E-6 5,61 Benzo(b)fluoranthène 5 252 1.2 6,7 E-5 1,2 E-5 6,57 Benzo(k)fluoranthène 5 252 0.6 6,7 E-5 3,9 E-5 6,84 Benzo(a)pyrène 5 252 3.8 6,7 E-5 4,9 E-7 6,04 Dibenzo(a,h)anthracène 5 278 0.5 1,3 E-8 7,3 E-8 5,97 Indeno(c,d)pyrène 6 276 0.3 1,3 E-8 7 E-8 7,66 Benzo(g,h,i)perylène 6 276 0.1 1,3 E-8 5,3 E-8 7,23
I.2.4 Biodégradabilité
Les hydrocarbures aromatiques sont susceptibles d’être biodégradés en conditions aérobies. La première étape lors du clivage du noyau est le remplacement de deux atomes d’hydrogène situés sur des carbones adjacents, par deux groupes hydroxyles. Le noyau benzénique est alors coupé entre les deux carbones portant les groupes hydroxyles pour donner un diacide carboxylique. La Figure 3 illustre le mécanisme de clivage d’un noyau benzénique.
OH
OH
O2 O2 CO2H
CO2H
Figure 3 : Dégradation aérobie d'un noyau benzénique
De plus, les hydrocarbures aromatiques peuvent subir une dégradation anaérobie [EVANS, 1977]. Le noyau benzénique peut être dégradé en présence de nitrate par Pseudomonas sp. et Moraxella sp. Le noyau benzénique est tout d’abord saturé en cyclohexane, puis oxydé en kétone, puis coupé par hydrolyse, pour donner finalement un acide carboxylique (Figure 4). Au cours de ce processus, le nitrate est réduit en azote.
+ 3H2 oxydation
O
+ H2O
CO2H
CH3
Figure 4 : Dégradation anaérobie du benzène en présence de nitrate
Il a été démontré que dans des conditions aérobies, les hydrocarbures aromatiques polycycliques (HAP) majeurs composants d'une huile d’anthracène (mixture complexe de HAP obtenue après distillation fractionnée du goudron de houille) peuvent être dégradés.
27
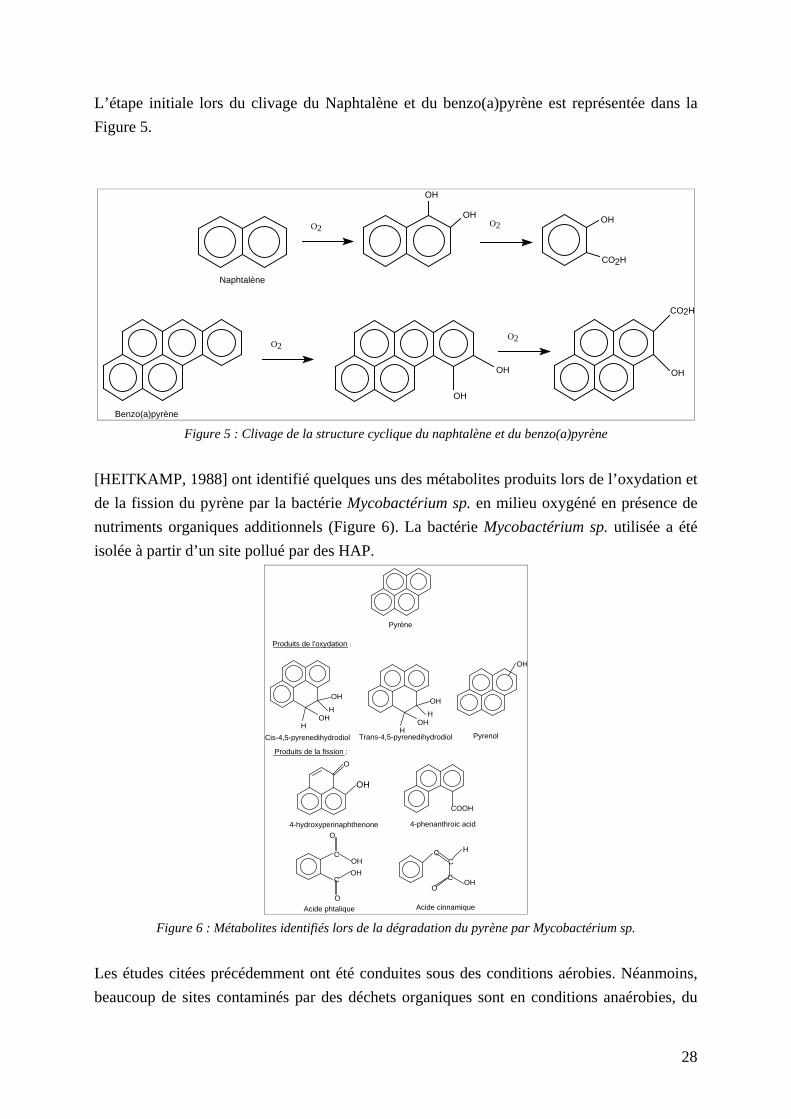
L’étape initiale lors du clivage du Naphtalène et du benzo(a)pyrène est représentée dans la Figure 5.
O2
OH
OHO2 OH
Naphtalène
CO2H
O2O2
OH
OH
CO2H
OH
Benzo(a)pyrène Figure 5 : Clivage de la structure cyclique du naphtalène et du benzo(a)pyrène
[HEITKAMP, 1988] ont identifié quelques uns des métabolites produits lors de l’oxydation et de la fission du pyrène par la bactérie Mycobactérium sp. en milieu oxygéné en présence de nutriments organiques additionnels (Figure 6). La bactérie Mycobactérium sp. utilisée a été isolée à partir d’un site pollué par des HAP.
OH
OH
HOH
HHOH
H
OH
Pyrène
Cis-4,5-pyrenedihydrodiol Trans-4,5-pyrenedihydrodiol Pyrenol
Produits de l'oxydation :
Produits de la fission :O
OH
COOH
4-hydroxyperinaphthenone 4-phenanthroic acid
C
C
O
O
OHOH
CC
CO
H
OH
Acide phtalique Acide cinnamique Figure 6 : Métabolites identifiés lors de la dégradation du pyrène par Mycobactérium sp.
Les études citées précédemment ont été conduites sous des conditions aérobies. Néanmoins, beaucoup de sites contaminés par des déchets organiques sont en conditions anaérobies, du
28

fait de la consommation de l’oxygène par les bactéries dégradant la matière organique. [MIHELCIC 1988] ont découvert que la population microbienne était capable de dégrader l’Acénaphtène et le Naphtalène en conditions anaérobies si la quantité de nitrates disponibles était suffisante. Lorsque la source de nitrate est épuisée, la minéralisation des HAP est stoppée.
I.3 Aspects relatifs à la mobilité des HAP dans les sols
Dans cette partie, le terme mobilité correspond à la capacité d'un élément à passer de la phase solide (le sol) à la phase liquide (l'eau de lixiviation), et à migrer dans l'espace. Sur le terrain, les mécanismes contrôlant la migration des polluants à travers un sol sont la percolation convective et la dispersion qui regroupe la diffusion moléculaire et la dispersion cinématique, ainsi que les modes d’échange entre le solide et la phase mobile.
I.3.1 Aspects hydrodynamiques – Transport d’un soluté non réactif en milieu poreux saturé
Depuis quelques années, de nombreux travaux ont été réalisés sur la théorie du transport de masse en réponse à l’intérêt croissant des problèmes liés à la contamination des eaux souterraines. Deux approches principales s’en dégagent. La première consiste à assimiler le milieu poreux à un milieu homogène dans lequel on applique les lois de la mécanique des milieux continus. La seconde propose de partager le milieu en éléments de taille finie et dans lesquels la composition est supposée uniforme.
• modèle de convection dispersion, • modèle des réacteurs en cascade.
Le soluté est dissout dans l’eau d’écoulement et il n’a pas d’interactions avec le milieu poreux. L’écoulement se fait à travers une colonne de sol en milieu saturé.
I.3.1.1 Modélisation de l’écoulement convectif dispersif
I.3.1.1.1 Transport par gradient de concentration
Un composé dans l’eau se déplacera de la zone la plus concentrée vers la zone de plus faible concentration. Ce processus est connu sous le terme de diffusion moléculaire. La diffusion a lieu tant qu’il existe un gradient de concentration, même s’il n’y a pas d’écoulement. Ce phénomène est décrit par la première loi de Fick, qui donne pour un cas monodirectionnel :
29

(9) dxdCDF −=
avec
F: Flux massique par unité de section, par unité de temps [M.L-2.t-1] D: Coefficient de diffusion [L2.t-1] C: Concentration du soluté [M.L-3] dC/dx: Gradient de concentration [M.L-3.L-1]
Pour les systèmes où la concentration change avec le temps, la seconde loi de Fick donne :
(10) 2
2
xCD
tC
∂∂=
∂∂
avec tC
∂∂ = changement de concentration avec le temps [M.L-3.T-1].
En milieu poreux, la diffusion ne peut pas se faire à la même vitesse que dans l’eau car le composé dissout doit suivre un chemin tortueux à travers le sol. Pour prendre en considération cela, un coefficient de diffusion effectif, De, doit être utilisé : (11) De = ω D
où ω est un coefficient lié à la tortuosité. La tortuosité (rapport entre le chemin linéaire et celui réellement parcouru) est une mesure de l’incidence de la forme du parcours sur le trajet suivi par les molécules d’eau en milieu poreux.
I.3.1.1.2 Transport par advection
Les composés dissous sont transportés à travers le milieu poreux saturé perméable par l’écoulement de l’eau. Ce processus est appelé transport par advection ou convection. La quantité de soluté transportée est fonction de sa concentration et de la quantité d’eau s’écoulant. Pour un écoulement dans une dimension à travers une unité de section, la quantité d’eau s’écoulant est égale à la vitesse moyenne linéaire multipliée par la porosité effective. La vitesse linéaire moyenne, νx, est la vitesse de l'eau à travers une unité de section de la porosité. Elle est différente de la vitesse moyenne d’une molécule d’eau se déplaçant à l’intérieur d’un pore, celle-ci étant plus grande que la vitesse linéaire moyenne du fait de la tortuosité. La porosité effective, θe, est la porosité à travers laquelle l’écoulement peut
30

réellement se produire. Les pores non connectés ainsi que les pores fermés ne font donc pas partie de la porosité effective.
(12) dldhKv
eex θθ
υ ==
avec
vx: Vitesse linéaire moyenne dans les pores [L.T-1] K: Conductivité hydraulique [L.T-1] θe: porosité effective dh/dl: Gradient hydraulique [L.L-1]
Le flux unidirectionnel attribué à l’advection est égal à la quantité d’eau s’écoulant multipliée par la concentration du soluté dissous :
(13) Fx = νx θe C L’équation du transport unidirectionnelle par advection est :
(14) xC
tC
x ∂∂−=
∂∂ ν
I.3.1.1.3 Dispersion mécanique
Les vitesses réelles de l’eau circulant dans un milieu poreux se situent autour de la vitesse linéaire moyenne. A l’échelle macroscopique, il existe trois causes à ce phénomène : (1) l’eau circulant au centre du pore possède une vitesse supérieure à l’eau circulant sur les bords. (2) Pour se rendre à une même distance linéaire, des particules du fluide parcourrons un trajet plus long dans le milieu poreux que d’autres particules. (3) Certains pores plus larges que d’autres, permettent à l’eau de circuler plus rapidement.
31

lent
rapidecourt
long
lent
lentrapide
Taille des pores Longueur du trajet Friction
Figure 7: Facteurs responsables de la dispersion longitudinal [FETTER, 1999]
Ces «différentes vitesses » de l’eau permettent le mélange des solutés en solution. Ce mélange est appelé dispersion mécanique. La dispersion se faisant selon l’axe de l’écoulement est appelée dispersion longitudinale. La dispersion selon une normale à l’écoulement est appelée dispersion transversale. Pour caractériser le mécanisme de dispersion, il a été introduit un coefficient de dispersion mécanique. Il est égal à une propriété du milieu, la dispersivité (α) multipliée par la vitesse linéaire moyenne : Coefficient de dispersion mécanique longitudinal = αl . νx
où αl à la dimension d’une longueur [L].
I.3.1.1.4 Dispersion hydrodynamique
Le processus de diffusion moléculaire ne peut pas être séparé de la dispersion mécanique. C’est pourquoi les deux sont combinés et définissent un paramètre appelé le coefficient de dispersion hydrodynamique D :
(15) exll DD += να .
où Dl est le coefficient de dispersion hydrodynamique longitudinal.
I.3.1.1.5 Expression analytique globale du transport unidirectionnel de soluté en milieu poreux
La combinaison des différentes formes de transport vues précédemment permet de construire une équation globale du transport en milieu poreux sans transformation bio-physico- chimique. Ecrite pour un écoulement unidirectionnel, elle donne :
32

(16) xC
xCD
tC
xL ∂∂−
∂∂=
∂∂ ν2
2
Si des processus physico-chimiques (sorption) interviennent, il faut ajouter le terme de sorption pour les espèces suivies :
tCss
∂∂ )(ρ
où : Cs est la concentration totale en phase solide [g/g] et ρs est la masse volumique de milieu poreux (ρ) par unité de volume de liquide contenu
consts =−=θ
θρρ 1
et l’équation (16) devient :
(17) xC
xCD
tC
tC
xLs
s ∂∂−
∂∂=
∂∂
+∂∂ νρ 2
2
I.3.1.2 Modèle des réacteurs en cascade
Ce modèle à l’origine conçu pour le génie chimique, propose de découper le système continu décrit précédemment en éléments de volume fini mis en série. Chaque élément de volume présente des concentrations uniformes par hypothèse. On obtient alors le modèle des mélangeurs en cascade. Le système de volume V et de porosité uniforme θ (volume de liquide contenu V0 = θV) est alors composé par une cascade de J réacteurs identiques en série contenant chacun les phases mobiles et stationnaires supposées de composition uniforme à chaque instant.
1 i2 J
1 CiCi-1C2C1 CjCj-1
Figure 8 : Schéma des réacteurs en cascade pour le modèle M.C.E.
Le bilan de matière pour la cellule i, pour un soluté n’interagissant pas avec la phase stationnaire s’écrit :
dtdC
qq i⋅⋅+= θJVCC i1-i
(18) ( )iii CC
VJq
dtdC
−= −10
.
avec
33

q : débit de percolation [L3.T-1] θ : porosité dans chacun des réacteurs.
Si des processus de sorption existent, le même terme d’accumulation que précédemment s’ajoute à chaque réacteur et la relation (17) devient :
(19) ( iiis
si CC
VJq
dtdC
dtdC −=+ −1
0
, .ρ )
I.3.1.3 Comparaison entre les deux approches
Il existe des similitudes entre ces deux approches mathématiques du transport [Molinari, 1976]. La convection dans le modèle continu serait équivalente au temps de séjour τ (τ=V0/q) dans le modèle des réacteurs en cascade. La dispersion DL du modèle continu serait, quant à elle, équivalente à J le nombre de réacteurs du modèle des réacteurs en cascade. Le modèle des réacteurs en cascade s'avère présenter de nombreux avantages au niveau de la mise en équation et du calcul numérique. En effet, il ne fait intervenir que des équations différentielles ordinaires, et, lors d'une intégration par une méthode numérique, moins de problèmes de "dispersion numérique" se posent, à l'opposé de ce qui se passe pour le modèle continu. Signalons, en outre, qu'aucun des deux modèles ne repose sur une approche microscopique de la dispersion. Il s'agit de modèles phénoménologiques qui n'ont de validité que par leur capacité à représenter les résultats expérimentaux [VILLERMAUX, 1993].
I.3.2 Aspects physico-chimiques relatifs à la mobilité des HAP dans un sol
Si des HAP sont présents initialement dans le sol, ceux ci peuvent être transférés, par relargage, de la phase solide vers la phase liquide (mobile). Selon [GRATHWOHL, 1998], les HAP présents dans le sol sous forme de gouttelettes de goudron de houille dispersées sont relargués dans la phase mobile par un mécanisme de dissolution. Par contre, lorsque les HAP sont fixés par un processus de sorption à la surface des particules ou bien à l'intérieur de la porosité intra particulaire, le relargage vers la phase mobile est contrôlé par un mécanisme de désorption. Ces deux mécanismes de relargage (dissolution et désorption), font appel aux notions de solubilité et de solubilisation, notions que nous allons préalablement définir. De plus, les HAP, comme tous les polluants hydrophobes sont susceptibles d'être entraînés sous forme particulaire, c'est à dire qu'ils sont fixés sur des particules mobiles. Cet aspect de la mobilité sera abordé sous le nom de transport particulaire.
I.3.2.1 Solubilité
La solubilité d'un composé chimique en phase aqueuse est communément définit comme la quantité maximale de composé par unité de volume dans la phase aqueuse, lorsque la solution
34

est en équilibre avec le composé pur à une température et une pression donnée (25°C, 1 atm). On parle alors d'une solution saturée (Csat). Parmi les molécules présentes dans l'environnement, les HAP ont une très faible solubilité en raison de leur fort caractère hydrophobe, ils sont considérés comme peu solubles. La solubilisation est le processus qui aboutit à la présence du composé en phase aqueuse. Par exemple lors de la dissolution dans l'eau d'un composé organique liquide pure, non miscible avec l'eau, l'atteinte de la solubilité peut être décrite de la manière suivante: Lorsque les molécules organiques, du liquide organique pur, sont mises en contact avec la phase aqueuse, des molécules organiques quittent la phase organique et se dissolvent dans l'eau et des molécules d'eau entrent dans la phase organique. Le processus est réversible. Lorsque le transfert entre les deux phases s'équilibre, on dit que le système est à l'équilibre thermodynamique. La quantité de molécules organiques dans l'eau correspond à la solubilité du composé organique liquide pur. De la même façon, la quantité de molécule d'eau dans la phase organique correspond à la solubilité de l'eau dans le liquide organique. La force qui contrôle le transfert de phase est quantifiée par l'énergie libre molaire de la solution (∆Gs).
(20) sss STHG ∆−∆=∆
Dans l'expression (19), représente l'enthalpie de dissolution et représente l'entropie
de dissolution et T, la température en Kelvin. sH∆ sS∆
Le schéma Figure 9 [SCHARZENBACH, 1993] représente une vision schématique du chemin enthalpique de la dissolution d'un composé organique liquide pur dans l'eau. La première étape lors de la dissolution est l'isolement d'une molécule organique (sans création d'une cavité dans la phase organique) et la création d'une cavité dans la phase aqueuse. Dans
les deux cas, c'est de l'énergie dépensée ( et >0) pour contrebalancer les attractions
dans la phase organique (o-o) et dans la phase aqueuse (e-e). Dans l'étape suivante, la molécule organique est insérée dans la cavité de la phase aqueuse. De cette façon, une partie de l'énergie dépensée est récupérée, puisque des attractions molaires vont se créer entre les molécules organiques et les molécules d'eau voisines ( <0). La nature de ces attractions
pour des molécules organiques hydrophobes sont de type Van der Waals et dipôles induits. La dernière étape consiste en une réorientation des molécules d'eau entourant la molécules organiques ( <0). Plus est grand, moins la molécule est soluble.
1H∆ 2H∆
3H∆
4H∆ sH∆
(21) 4321 HHHHHs ∆+∆+∆+∆=∆
35
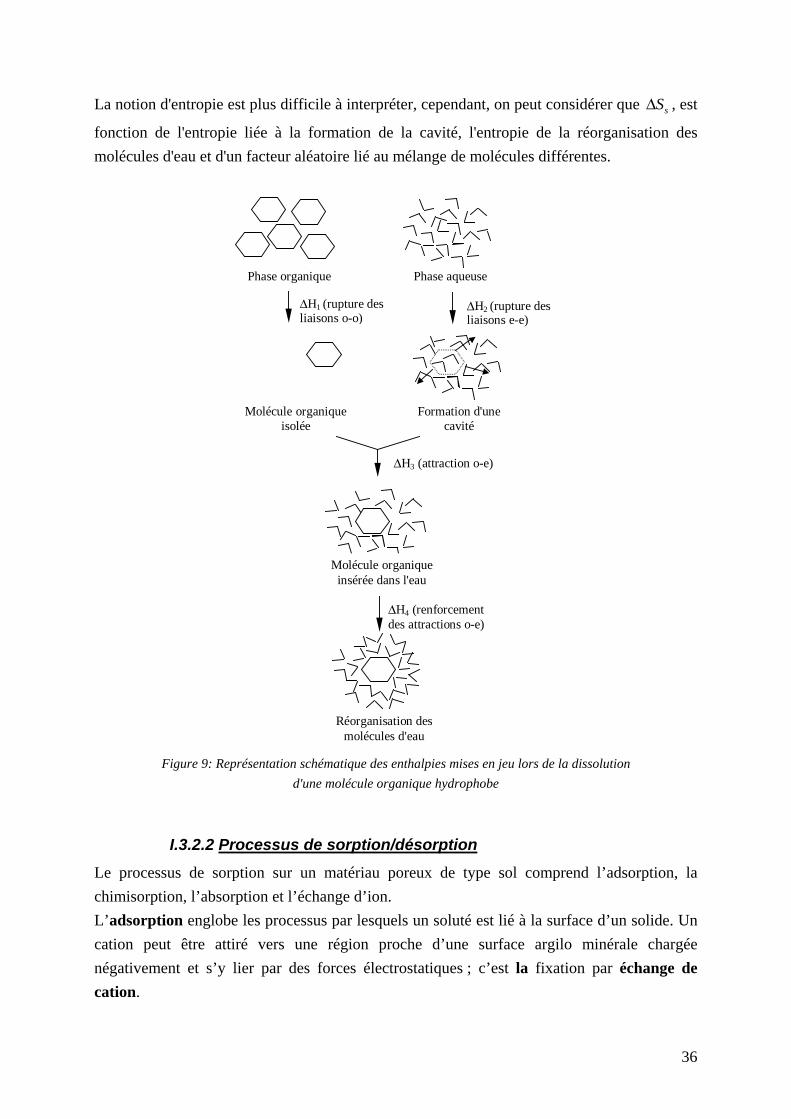
La notion d'entropie est plus difficile à interpréter, cependant, on peut considérer que , est
fonction de l'entropie liée à la formation de la cavité, l'entropie de la réorganisation des molécules d'eau et d'un facteur aléatoire lié au mélange de molécules différentes.
sS∆
Phase organique Phase aqueuse
∆H2 (rupture desliaisons e-e)
∆H1 (rupture desliaisons o-o)
Formation d'unecavité
∆H3 (attraction o-e)
Molécule organiqueinsérée dans l'eau
∆H4 (renforcementdes attractions o-e)
Réorganisation desmolécules d'eau
Molécule organiqueisolée
Figure 9: Représentation schématique des enthalpies mises en jeu lors de la dissolution
d'une molécule organique hydrophobe
I.3.2.2 Processus de sorption/désorption
Le processus de sorption sur un matériau poreux de type sol comprend l’adsorption, la chimisorption, l’absorption et l’échange d’ion. L’adsorption englobe les processus par lesquels un soluté est lié à la surface d’un solide. Un cation peut être attiré vers une région proche d’une surface argilo minérale chargée négativement et s’y lier par des forces électrostatiques ; c’est la fixation par échange de cation.
36

La chimisorption apparaît quand le soluté est incorporé sur le sol par réaction chimique. L’absorption intervient quand les particules poreuses de l’aquifère permettent au soluté, sa diffusion intra particulaire puis sa sorption sur les surfaces intérieures des particules. Dans cette partie, nous ne tenterons pas de différencier ces phénomènes, nous engloberons dans le terme de sorption l’ensemble des processus vus précédemment. Le processus, par lequel un contaminant se distribue entre la phase solide et la phase liquide, est appelé partage. In situ, si le processus de sorption est rapide par rapport à la vitesse d’écoulement de l’eau, la concentration sorbée sur le sol atteindra un équilibre local et le processus pourra être décrit par une isotherme de sorption à l’équilibre. Dans le cas inverse, où le soluté vis à vis de la surface solide sera dans un état de non équilibre locale, il faudra utiliser un modèle de sorption cinétique décrivant cet état de non équilibre.
I.3.2.2.1 Hypothèse de l’équilibre local ; isotherme de sorption à l’équilibre
Dans l’étude des transferts, une situation d’équilibre local correspond donc à une sorption considérée comme instantanée comparativement aux temps caractéristiques de la convection et de la dispersion. Les isothermes de sorption sont les représentations graphiques ou les modèles mathématiques des données à l’équilibre, à une température donnée, des concentrations du polluant en solution en fonction de la teneur en polluant sorbée dans la fraction solide du sol. La modélisation de la réaction est donc fondée sur des lois de la forme Cs,eq = f(Ce,eq), où Cs,eq est la concentration massique du contaminant dans la phase solide à l'équilibre (exprimée en fonction de la masse de solide sec) et Ce,eq la concentration du contaminant en phase liquide à l'équilibre (exprimée en fonction du volume de liquide). Dans le cas le plus simple la concentration dans le solide Cs,eq est directement proportionnelle à la concentration en solution (Ce,eq) à l’équilibre :
(22) Cs,eq = Kd .Ce,eq
Avec
Cs,eq = masse de soluté sorbé à l'équilibre par unité de masse sèche de sol [M.M-1] Ce,eq = Concentration du soluté en solution à l’équilibre avec la masse de soluté sorbée
sur le solide [M.L-3] Kd = coefficient de distribution [L3.M-1]
37

Lorsque la sorption est linéaire, Kd est appelé coefficient de partage et ce coefficient représente la pente de l’isotherme. C’est l’isotherme le plus simple et le plus utilisé pour décrire le partage entre la phase dissoute et la phase sorbée. Par contre, si l’isotherme de sorption n’est pas linéaire, le coefficient Kd dépend de la concentration du contaminant (Cs,eq Ce,eq). Pour représenter les isothermes non linéaires dans les sols, le modèle de Freundlich est souvent utilisé.
(23) neqefreqs CKC /1
,, =
Kfr est le coefficient de sorption de Freudlich et 1/n est un exposant empirique. Si 1/n=1, le modèle de Freudlich est équivalent à l’isotherme de sorption linéaire. Il est à noter que la valeur de Kfr dépend des unités de Cs,eq et Ce,eq, aussi des précautions doivent être prises si des valeurs de différentes études sont comparées. Les isothermes de Langmuir et BET sont d’autres modèles qui décrivent les processus de sorption non linéaire dans les sols. Les deux, à l’origine ont été développés pour l’adsorption des gaz sur solide. Le modèle de Langmuir prend en compte une concentration maximale (plateau) sorbée sur le sol (Cs,eq,max).
(24) eqel
eqeeqsleqs CK
CCKC
,
,max,,, 1+
=
Kl est le coefficient de sorption de Langmuir. Pour de très faibles concentrations et Kl.Ce,eq <<1 , le modèle de Langmuir représente une relation linéaire analogue à celle décrite par l’isotherme linéaire (Kl.Cs,eq,max =Kd). Pour des valeurs de Kl.Ce,eq >> 1, Cs,eq tend vers Cs,eq,max L’isotherme de type Langmuir permet de décrire des réactions (mécanismes) de complexation de surface entre les espèces suivies et les sites activés du sol. Le modèle BET a été développé par [BRUNAUER, 1938] pour l’adsorption des gaz sur la surface des solides. En plus de l’adsorption de couches multimoléculaires, il prend en considération la condensation capillaire du soluté dans les méso pores (2nm-50nm) :
(25)
( ) ⎥⎦⎤
⎢⎣⎡ −+−
=
sat
eqeeqesat
eqeeqseqs
CCKCC
CCKC
,,
,max,,,
11)(
Avec Csat = Concentration de vapeur saturante (ou solubilité dans l’eau en milieu aqueux). Si Ce,eq << Csat , on retrouve une relation linéaire entre les concentrations en phase aqueuse et solide. Si Ce,eq tend vers Csat, Cs,eq devient infiniment grand (condensation capillaire de la vapeur, précipitation ou agglomération) [GRATWHOL, 1998].
38
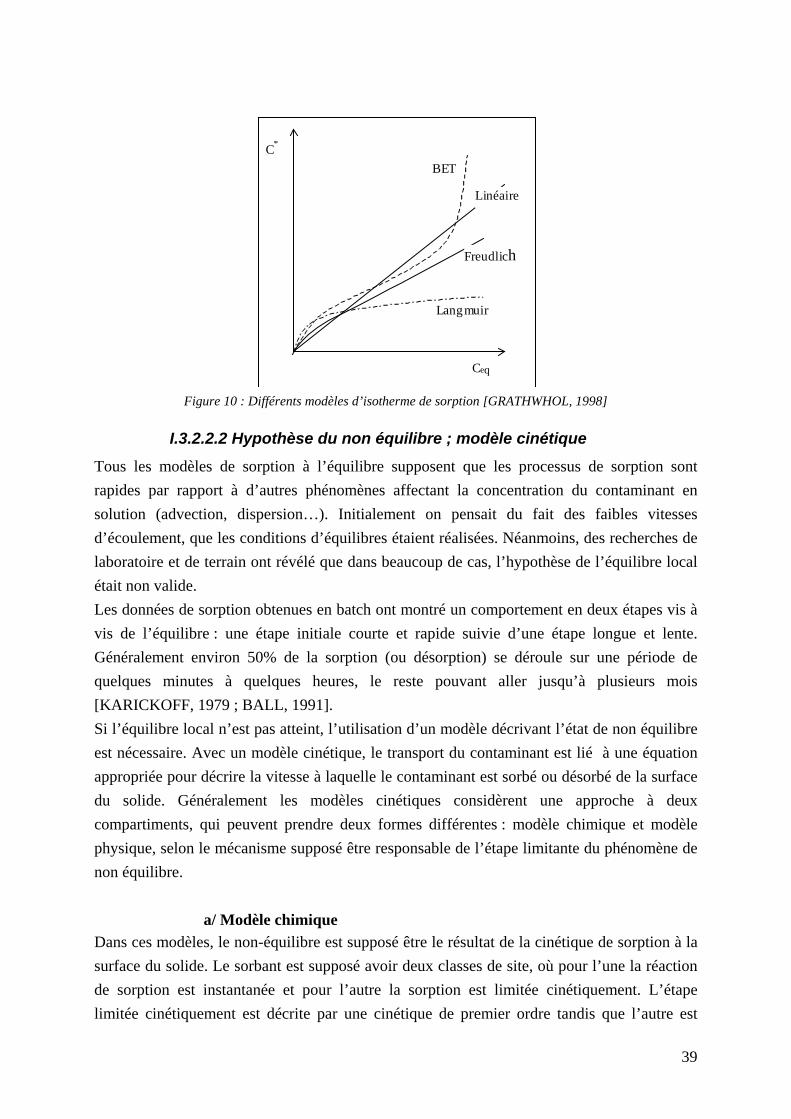
BET
Ceq
C*
Linéaire
Langmuir
Freudlich
Figure 10 : Différents modèles d’isotherme de sorption [GRATHWHOL, 1998]
I.3.2.2.2 Hypothèse du non équilibre ; modèle cinétique
Tous les modèles de sorption à l’équilibre supposent que les processus de sorption sont rapides par rapport à d’autres phénomènes affectant la concentration du contaminant en solution (advection, dispersion…). Initialement on pensait du fait des faibles vitesses d’écoulement, que les conditions d’équilibres étaient réalisées. Néanmoins, des recherches de laboratoire et de terrain ont révélé que dans beaucoup de cas, l’hypothèse de l’équilibre local était non valide. Les données de sorption obtenues en batch ont montré un comportement en deux étapes vis à vis de l’équilibre : une étape initiale courte et rapide suivie d’une étape longue et lente. Généralement environ 50% de la sorption (ou désorption) se déroule sur une période de quelques minutes à quelques heures, le reste pouvant aller jusqu’à plusieurs mois [KARICKOFF, 1979 ; BALL, 1991]. Si l’équilibre local n’est pas atteint, l’utilisation d’un modèle décrivant l’état de non équilibre est nécessaire. Avec un modèle cinétique, le transport du contaminant est lié à une équation appropriée pour décrire la vitesse à laquelle le contaminant est sorbé ou désorbé de la surface du solide. Généralement les modèles cinétiques considèrent une approche à deux compartiments, qui peuvent prendre deux formes différentes : modèle chimique et modèle physique, selon le mécanisme supposé être responsable de l’étape limitante du phénomène de non équilibre.
a/ Modèle chimique Dans ces modèles, le non-équilibre est supposé être le résultat de la cinétique de sorption à la surface du solide. Le sorbant est supposé avoir deux classes de site, où pour l’une la réaction de sorption est instantanée et pour l’autre la sorption est limitée cinétiquement. L’étape limitée cinétiquement est décrite par une cinétique de premier ordre tandis que l’autre est
39

représentée par une isotherme de sorption à l’équilibre. Le modèle à deux compartiments a été développé par [SELIM, 1976].
(26) tCFK
tS
dI
∂∂=
∂∂
(27) ( )[ ] IIdII SCKFt
S−−=
∂∂ 1α
Avec SI = concentration sorbée [M.M-1] sur les sites instantanés SII = concentration sorbée [M.M-1] sur les sites à cinétique limité F = représente la fraction de soluté pour laquelle la sorption est instantanée C = concentration en phase aqueuse [M.L-3] Kd = coefficient de distribution à l’équilibre [L3.M-1] α = coefficient de transfert de masse (1/T)
La discrétisation de la surface du sorbant en deux catégories de classes de sites se différenciant par leur temps de réaction de sorption peut être étendue à un plus grand nombre de classes, chacune avec sa propre constante de transfert. Le cas limite correspond à une distribution continue de sites, où la population des sites est représentée par une constante de distribution. La réaction de sorption des composés organiques hydrophobes montre de faibles valeurs d’énergie d’activation incompatibles avec l’utilisation d’un modèle chimique. Cependant, [BRUSSEAU, 1991] a interprété en terme de mécanismes physiques les modèles chimiques en reliant le coefficient (α) de transfert de masse aux paramètres de diffusion intra particulaire, introduits dans la partie suivante (coefficient de diffusion effectif, tortuosité, diamètre des particules). Ainsi, l’utilisation de modèle chimique, pour la description du non-équilibre local de sorption des composés organiques hydrophobes, devient cohérente.
b/ Modèle physique Avec ce modèle, le processus de sorption à l’interface solvant-sorbant est supposé instantané : c’est la vitesse de transport qui contrôle la cinétique de sorption. Le concept sur lequel repose ce modèle est basé sur l’existence de deux régions différentes de l’eau du sol (une région mobile et une région immobile ; Figure 11). La région immobile se situe dans la microporosité intra particulaire et l’eau pendulaire (film à la surface des particules). Le transport du soluté par advection-dispersion est supposé limité à la région mobile, tandis que le transport dans la région immobile est supposé se faire uniquement par diffusion [BRUSSEAU, 1989]. Le phénomène de non-équilibre est alors une conséquence du transfert entre ces deux régions. Il est important de noter que le phénomène de non équilibre attribué à un mécanisme physique de transport concerne à la fois les solutés sorbables et les non sorbables. Il faut souligner aussi que le mécanisme responsable du non équilibre physique est
40

le transport, et que la cinétique de sorption est négligée. Si la définition de la sorption se limite aux processus se déroulant à l’interface solide-liquide, il serait plus juste de l’appeler non équilibre de transport plutôt que non équilibre de sorption.
Porositéinterparticulaire
Porositéintraparticulaire
Figure 11 : Système eau mobile et eau immobile
Le plus souvent ce processus de transfert par diffusion est décrit par la loi de Fick et implique plusieurs mécanismes: (1) transport par advection-dispersion vers la couche limite (film) ; (2) transport par diffusion à travers le film et (3) diffusion intra particulaire. Généralement c’est la diffusion intra particulaire qui se révèle être l’étape limitante, les autres pouvant être le plus souvent négligées. La description du mécanisme à l’échelle de la particule considérée sphérique est alors décrite par la deuxième loi de Fick en coordonnée sphérique:
(28) ⎥⎦
⎤⎢⎣
⎡∂∂+
∂∂=
∂∂
rC
rrCD
tC
a2
2
2
Avec r = distance par rapport au centre de la particule [L] Da = coefficient de diffusion apparent (constant pour une sorption linéaire) [L2T-1] C = concentration dans la phase aqueuse immobile [ML-3] Le coefficient de diffusion apparent est inférieur au coefficient de diffusion effectif car dans le cas de la diffusion apparente, il y a un retard induit par le processus de sorption dans la porosité intra particulaire. Pour une distance r égale au rayon de la particule, on obtient la concentration présente à l’interface eau immobile / eau mobile. Les différentes solutions de l’équation (27) sont disponibles dans [GRATWHOL, 1998]. Ce modèle diffusionnel ne permet pas de décrire le processus de sorption ou désorption instantanée, c’est pourquoi [BALL et ROBERTS, 1991] ont introduit un terme correcteur dans la forme de la solution de
41

cette équation. Ce modèle est largement utilisé par [GRATHWOHL, 1991] pour prendre en compte l’hypothèse du non équilibre local lors de certaines études réalisées en Batch.
I.3.2.2.3 Sorption des composés organiques hydrophobes
Les composés organiques hydrophobes (COH) sont des molécules avec différents degrés de polarité. La solubilité de ces composés dans l’eau est fonction de leur degré d’attraction avec les molécules polaires de l’eau. Cette attraction dépend de la polarité du COH concerné. Les COH peuvent être dissous dans un grand nombre de solvants organiques mais ont une faible solubilité dans l’eau. Lorsqu’elles sont dissoutes dans l’eau, ces molécules tendent à être attirées par des surfaces moins polaires que l’eau. On retrouve peu de sorptions de COH sur les surfaces purement minérales [GRIFFIN, 1980]. Seules les surfaces minérales constituées d’au moins 1% de carbone organique (foc) sont concernées par le processus de sorption des COH, [KARICKOFF, 1979]. D’après [CHIOU, 1989] le processus de sorption des COH pourrait être interprété comme le partage du composé entre la phase organique du sol et la phase aqueuse, telle que la distribution du composé dans un système bi-phasique liquide (octanol/eau, KOW). Dans ces conditions la sorption pourrait être décrite par un simple coefficient de partage Koc [L3.M-1] :
(29) oc
doc f
KK =
On retrouve aussi dans la littérature un coefficient Kom basé sur la matière organique contrairement à Koc qui se réfère au carbone organique du sol. Etant donné que la masse de matière organique est plus élevée que la masse de carbone organique, Koc est plus grand que Kom. Il existe des équations empiriques permettant de relier Koc et Kom [OLSEN et DAVIS, 1990]. [KENAGA, 1980] a proposé une relation empirique qui permet d’estimer le coefficient Koc uniquement en fonction de la solubilité du composé dans l’eau :
(30) Log Koc = -0.55 log S + 3,64
S étant la solubilité dans l’eau [mg.L-1]. Le coefficient de partage Koc peut aussi être estimé à partir du coefficient Kow de partage octanol / eau, coefficient mesuré pour de nombreux COH. [KARICKOFF, 1979] propose la relation suivante pour les composés avec des Kow >2 :
42

(31) Log Koc = log Kow – 0.21 ⇒ Koc = 0.62 Kow
L’application de ces différentes relations empiriques construites à partir de Kow et de S peut poser des problèmes car certains composés organiques de même solubilité dans l’eau présentent de grandes différences de Kow. C’est pourquoi des auteurs ont proposés une approche plus fondamentale basée sur la structure moléculaire des composés [KOCH, 1983].
I.3.2.3 Processus de dissolution
Lorsque des HAP sont présents dans le sol sous forme dissoute dans une phase organique complexe, le relargage vers la phase liquide est régit par un mécanisme de dissolution. La phase organique complexe est souvent appelée Phase Liquide Non Aqueuse (PLNA). Si les conditions d'équilibre sont réalisées entre la phase mobile et la PLNA, les concentrations en solution sont égales à celles prévues par la loi de Raoult, dans le cas contraire il faut prendre en compte les aspects cinétiques du transfert.
I.3.2.3.1 Equilibre chimique : loi de Raoult
La loi de Raoult décrit le comportement à l’équilibre d’un soluté dans un mélange idéal de deux phases. A l’équilibre le potentiel chimique de chaque soluté (espèce) est uniforme dans chaque phase. La concentration d’un composé individuel i (Ci,e,eq) mis en solution à partir d’une PLNA, à l’équilibre avec l’eau, est donnée par la loi de Raoult :
(32) eiiieqei SC ,,, γχ=
où χi , γi et Si,e sont respectivement la fraction molaire [ni / ∑ni], le coefficient d’activité dans la PLNA et la solubilité dans l’eau pure du composé i [M.L-1]. Le coefficient d’activité dans la PLNA décrit la déviation par rapport à un comportement idéal, il est dans beaucoup de cas (molécules non polaires ou peu polaires) considéré égal à 1. La loi de Raoult est appliquée à l’équilibre. Son utilisation pour des mélanges complexes (PLNA) et avec des cinétiques individuelles différentes est courante dans la littérature. Lors de la phase de dissolution aboutissant à l’équilibre (loi de Raoult), ce sont par exemple les constituants de plus forte solubilité qui sont mis en solution majoritairement dans un premier temps. Par conséquent, la composition de la PLNA et donc les fractions molaires changent au cours du temps. Par exemple, si un constituant avec une forte solubilité est mis en solution relativement rapidement, parallèlement sa fraction molaire dans la PLNA diminue. Sa concentration molaire en phase aqueuse va donc baisser avec le temps, tandis que la
43

concentration molaire en phase aqueuse des constituants les moins solubles, donc les plus persistants de la PLNA, augmente avec le temps.
I.3.2.3.2 Cinétique de dissolution
La dissolution de la PLNA est généralement considérée comme un processus au transfert de masse limité cinétiquement. Un modèle simple suppose que la diffusion des constituants se fait à partir de l’interface PLNA/eau au travers d’un film d’eau immobile (épaisseur δ) adsorbé à la surface de la PLNA. Le flux de dissolution est alors dans ce cas décrit par l’équation du premier ordre:
(33) )( ,,, eioeiaq CC
DF −=
δ
où Daq est le coefficient de diffusion en milieu aqueux, Ci,e et Ci,e,0 sont respectivement la
concentration du composé i dans la phase mobile (aqueuse) et la concentration à l’interface.
Pour les liquides organiques pures (un seul constituant) et l’eau, Ci,e,0 est égale à la solubilité
dans l’eau. Lorsque la phase organique est une PLNA (plusieurs constituants), Ci,e,0 = Ci,e,eq =
Csat, calculée avec la loi de Raoult. Le rapport Daq/δ est généralement assimilé à un coefficient
de transfert de masse k [L.T-1].
La vitesse de dissolution peut s'écrire [YEOM, 1996] :
(34) )( ,,,,
eioeiei CCsk
dtdC
−⋅=
avec s la surface spécifique interfaciale [L2.L-3]. On trouve aussi dans la littérature, d’autres modèles plus complexes considérant la diffusion au travers deux films adjacents (modèle du double film). Un premier film à la périphérie de la PLNA et le second constitué par l’eau immobile.
44

PLNA Phase aqueuse
immobile mobile
δo δe
Ci,PLNA
Ci , o/ e
Ci , e/ o
Ci , e
Figure 12: vue schématique du modèle à deux film stagnants: Le transfert de masse à travers les deux films est supposé contrôlé par la seconde loi de Fick. A l'interface de chacun des films, l'équilibre est supposé atteint et les concentrations de chaque coté sont données par les coefficients de partage entre la phase organique et l'eau. δo et δe sont les épaisseurs de chacun des films. Ci,PLNA et Ci,e sont respectivement les concentrations dans la phase organique et l'eau.
La concentration du contaminant à travers le film organique diminue de la concentration initiale (Ci,PLNA,,t0) dans la PLNA jusqu’à la concentration à l’interface PLNA / eau (Ci,o/e). A la limite entre les deux films, le contaminant organique quitte la phase organique pour entrer dans la phase aqueuse [POWERS, 1991]. Le calcul du flux à travers les deux films nécessite les hypothèses suivantes :
• la concentration dans l’eau mobile est constante et proche de zéro ; Ci,e = 0,
• le flux de contaminant à travers les deux films est soumis à des conditions constantes,
• la concentration à l’interface PLNA/eau (Ci,e/o)est à l’équilibre (concentration à saturation dans l’eau, calculé selon la loi de Raoult).
I.3.2.4 Influence des paramètres physico-chimiques sur les mécanismes de transfert des HAP en phase liquide et sur la solubilité des HAP
Les paramètres environnementaux (scénario) peuvent avoir une influence sur la solubilisation des HAP et sur les mécanismes de sorption et de dissolution. Nous allons présenter successivement l’influence des paramètres cosolvant, température, de la matière organique naturelle (MON), du vieillissement, de la force ionique, du débit, et enfin du pH.
45

I.3.2.4.1 Cosolvant
La présence de molécules organiques dans l‘eau peut influencer le comportement des molécules organiques hydrophobes. En ce qui concerne la solubilité, des molécules codissoutes peuvent influencer la formation des cavités entre les molécules d’eau, réceptacles des composés organiques hydrophobes. Trois cas peuvent se produire : (1) lorsque les molécules organiques représentent plus de 10% en volume de la solution (pas assez d’eau pour toutes les hydrater), elles agissent elles-mêmes comme solvant (cosolvant). Elles participent aussi à la formation des cavités, en proportion de leur fraction dans la solution [YALKOWSKY, 1976]. La solubilité apparente du composé i peut être calculée d’après :
(35) ceimi fSS σ+= ,, loglog
avec Si,m la solubilité dans le mélange (eau / cosolvant), Si,e la solubilité dans l’eau pure, fc la fraction du cosolvant dans la solution et σ le pouvoir du solvant (pente du graphique représentant le log de la solubilité en fonction de fc). (2) Lorsque les molécules sont présentes dans de plus petites proportions, elles se logent dans les cavités créées par les molécules d’eau. Si cette hydratation (formation des cavités) est partagée avec les molécules hydrophobes, alors la solubilité de ces dernières sera diminuée [BANERJEE, 1988]. On parle alors de cosolutes. (3) Finalement, quand les molécules organiques sont présentes dans des proportions infimes, on peut s’attendre à ne voir aucun changement dans la solubilité du composé organique hydrophobe suivi. C’est le cas d’un mélange d’hydrocarbures de faible solubilité en solution, la solubilité de chacun n’est pas influencée par celle des autres [LEINONEN, 1973]. En raison de leur polarité et de leur solubilité infinie, les cosolvants tel que le méthanol ne présentent pas de sorption avec le sol [WOOD, 1990 et BRUSSEAU, 1991b]. La présence de cosolvants aurait une incidence sur le processus de sorption [RAO, 1985]: la présence de cosolvant influence le coefficient de partage Kd, le coefficient α, et la fraction F du modèle chimique de sorption (équations (26) et (27)). Le coefficient Kd diminue avec une augmentation de la fraction de cosolvant.
(36) cdm fKKd
σβ−= loglog
46

Avec est le coefficient de partage du contaminant dans la solution contenant le cosolvant,
K
md
K
d est le coefficient de partage dans l’eau, β est un paramètre empirique introduit pour prendre en compte l’augmentation de la solubilité du contaminant (équation (35).Le coefficient de transfert α, augmente avec la fraction volumique de cosolvant selon la relation [JI, 1998]:
(37) cm fa σααα −= loglog
(38) Fm = F 0 ≤ fc ≤ 0,2 (39) Fm
= F – b(fc – 0.2) 0.2 ≤ fc ≤ 1 Où, Fm représente la fraction de soluté pour laquelle la désorption est instantanée et a et b sont des paramètres empiriques. [PIGNATELLO, 1999] a montré une influence du pyrène sur la cinétique de désorption du phénanthrène, le pyrène défavoriserait la sorption du phénanthrène sur la matière organique naturelle du sol, autorisant ainsi le phénanthrène à se déplacer plus rapidement dans la matrice. Pour la dissolution, la présence de cosolvant ne semble pas influencer le coefficient de transfert de masse dans l'équation (34) [JI, 1998].
I.3.2.4.2 Température
La température des eaux naturelles peut varier entre 0 et 35°C. La solubilité des HAP est fortement dépendante de la température. En passant d'une température de 4°C à 30°C, la solubilité du phénanthrène et de l'anthracène est multipliée respectivement par 3,4 et 4,6 [SCHARZENBACH, 1993]. [REZA, 2002] a étudié la solubilité des HAP en fonction de la température à l'aide d'un dispositif d'extraction en phase solide dans une colonne, couplé à une analyse en ligne par chromatographie liquide. La solubilité de l'anthracène dans l'eau passe de 15,5 µg.L-1 pour une température de 9°C à 212 µg.L-1 pour une température de 50°C. En supposant l'enthalpie de dissolution d’un composé organique quelconque constante sur l'échelle de température considérée, la solubilité des HAP dans l'eau peut être décrite par l'équation de Van't Hoff :
47

(40) CRTHLn s +∆=χ
Avec χ la solubilité exprimée en fraction molaire, R la constante des gaz parfaits et T la température en Kelvin, C est une constante. La température influence aussi le processus de sorption. Le coefficient de partage Kd augmente de 1-1,6 fois avec une diminution de la température de 22°C [PIATT, 1996]. La fraction F (fraction instantanément désorbable définie dans le modèle chimique cinétique, équations (26) et (27)) ne semble pas être influencée par une variation de température de 22°C. La constante α quant à elle diminue de 1,2-2,6 fois avec une diminution de 22°C. En ce qui concerne l'effet de la température sur le mécanisme de dissolution, [MAHJOUB,
2000] relate une augmentation du coefficient de transfert k (équation (34)) lors de la
dissolution d'un goudron dans l'eau: le coefficient de transfert k du phénanthrène passe de
0,734 m.s-1 pour T=9°C à 1,253 m.s-1 pour T=35°C. Cet auteur suppose que cette
augmentation de k est due à une augmentation du coefficient de diffusion à l'interface entre la
phase organique et l'eau.
I.3.2.4.3 Matière Organique Naturelle
Sous le nom de matière organique naturelle (MON), on fait généralement référence à des substances organiques, telles que les macromolécules dissoutes ou particules de sols, sédiments en suspension dans l’eau. La caractéristique de ces substances est qu’elles possèdent un domaine hydrophobe et un domaine hydrophile. Lorsqu’elles sont dans l’eau, elles sont conceptualisées comme une phase à part, non aqueuse, qui peut retenir les composés organiques hydrophobes. Dans un milieu poreux comme un sol, la MON est mobile, donc participe au transport [JOHNSON, 1995]. La MON a une influence sur la solubilité. L’interaction entre un composé organique hydrophobe et la MON est supposée instantanée comparativement aux vitesses d’écoulement. De plus, la sorption est considérée comme linéaire. Il en résulte que la solubilité apparente est représentée par la fraction dissoute mais aussi la fraction associée à la MON [CHIOU, 1986] :
(41) MONKSS
miei
mi,
,
, 1+=
où Si,m est la solubilité apparente dans la solution de MON, Si,e est la solubilité dans l’eau pure, Ki,m est le coefficient de partage du composé organique hydrophobe entre l'eau et la MON et MON est la concentration de la matière organique naturelle dans l’eau.
48

Selon la littérature, une partie des MON est susceptible d’être sorbée sur le milieu poreux [JARDINE, 1992], ce qui augmenterait la sorption du composé organique hydrophobe. De plus le complexe MON/ contaminant peut aussi se sorber sur le milieu poreux. Il n'existe pas d'informations précises sur l'effet des MON sur la cinétique de dissolution des PLNA. Cependant, d’après [BRUSSEAU, 1998], la MON n’aurait pas d’influence directe sur le mécanisme de dissolution des gouttelettes de goudrons.
I.3.2.4.4 Vieillissement
Les contaminants hydrophobes sorbés sur le sol peuvent voir leurs interactions de sorption évoluer dans le temps. Certaines études ont montré que le prolongement du temps de contact entre le polluant et le sol entraînait une plus forte adsorption ainsi qu’une plus grande résistance à la désorption [HATZINGER, 1995]. De plus de nombreux auteurs ont observé un phénomène d’hystérésis se traduisant par un décalage significatif entre les équilibres d’adsorption, et de désorption [BECK, 1995]. Dans certains cas, ce phénomène est un artefact expérimental. Dans d’autres cas, l’hystérésis apparente peut être attribuée aux conditions expérimentales qui ne permettent pas d’atteindre les états d’équilibre réels d’adsorption et de désorption [HUANG, 1998]. [CONNAUGHTON, 1993] a observé que la fraction résistante était généralement plus importante dans les sols de sites réels. Dans de tels cas, l’atteinte de l’équilibre de désorption exigerait des durées de contact avec l’eau bien plus longues que celles qui sont classiquement utilisées au laboratoire. Dans un nombre relativement limité de cas, le phénomène d’hystérésis paraît bien réel, probablement dû à l’établissement d’interactions plus ou moins irréversibles entre le polluant et le sol. On assiste donc à la formation d’une fraction de polluants non extractible de la phase solide. Cette fraction est souvent désignée sous le terme de « résidus liés ». La modification de la désorbabilité et de l’extractabilité des polluants avec le temps et la formation de résidus liés sont très peu prises en compte dans les modèles de description du comportement des polluants dans les sols. En effet, de nombreuses réactions bio-physico-chimiques avec des cinétiques très variées peuvent avoir lieu dans/entre les différents compartiments du sol avec des conséquences complexes. En ce qui concerne la dissolution, le vieillissement provoquerait une transformation du film interfacial, ce qui diminuerait le coefficient de transfert k [m.s-1] (équation (34) [MAHJOUB, 1999].
I.3.2.4.5 Force ionique ou sels dissous
Défini en fonction de la concentration molaire C et la charge Z de l’ensemble des ions i
composant la solution :
∑=i
ii ZCI 2
21
(40)
49

la force ionique I (40) des solutions du sol est un paramètre intervenant à différents niveaux
dans les mécanismes de transfert/transport solide/liquide mais aussi dans la formation des
colloïdes, de la déstructuration du sol etc. La force ionique des eaux marines est d’environ 1
M, des eaux souterraines d’environ 0,1 M et des eaux de surface d’environ 0,01 M [TAN,
1993]
Il a été observé que les espèces ioniques prédominantes dans les eaux naturelles (Na+, K+,
Ca2+, Mg2+, Cl-, SO42-, HCO3
-) diminuaient souvent par effet de compétition, la solubilité
dans l’eau des molécules organiques hydrophobes. [MAHJOUB, 2000] a montré que les
solubilités du phénanthrène et du naphtalène étaient plus faibles lors de la dissolution d'un
goudron dans une solution de KCl à 1g.L-1 que dans l'eau permutée. La présence de cations
dissous dans la phase aqueuse diminuerait le coefficient de partage Koc lors de la sorption du
phénanthrène sur des acides humiques issus de sols [JONES, 1999]. Cet effet sur Koc serait
plus important avec un cation trivalent comme Al3+ qu'avec un cation monovalent comme
Na+. La conformation des acides humiques qui détermine l'accessibilité des composés
organiques hydrophobes aux sites de sorption, serait dépendante des cations en solution.
I.3.2.4.6 Débit
Le débit de percolation peut intervenir sur l’épaisseur du film d’eau situé à la surface de la PLNA et donc modifier le coefficient de transfert dans le mécanisme de dissolution. Une augmentation de la vitesse d’écoulement provoque une diminution de l’épaisseur du film, donc une augmentation du coefficient de transfert et, en conséquence la quantité de contaminant présent dans l’eau mobile [GRATHWOHL, 1998]. Le débit est en partie responsable des conditions d'équilibre ou de non équilibre local dans les mécanismes de désorption et de dissolution, puisqu'il détermine le temps de contact dans un dispositif percolant.
I.3.2.4.7 PH et potentiel
Le pH, s’il n’a pas d’influence directe sur les molécules organiques hydrophobes, peut en avoir sur les métabolites de dégradation. Le pH peut également contrôler les conditions de biodégradation. Le pH influe indirectement sur la force ionique de la solution du sol et, à travers elle, sur l’ensemble des mécanismes de répartition solide/liquide. Le potentiel-redox est un paramètre primordial lors des dégradations biotiques et abiotiques car il caractérise le pouvoir oxydant ou réducteur du milieu.
I.3.2.5 Transport particulaire
Les colloïdes sont des particules solides, de très faibles solubilités dans l'eau, dont le diamètre est compris entre quelques nanomètres et quelques micromètres. Parmi les colloïdes présents
50

dans les sols, on retrouve des macromolécules telles que les substances humiques, des microorganismes, des fines gouttelettes de liquides organiques insolubles, et des matières minérales [McCARTHY, 1989]. Lorsqu'une partie des polluants dissous se fixe sur ces colloïdes [ROY, 1998], il se crée une seconde phase mobile. Les colloïdes, pour participer au transport des polluants, doivent tout d'abord être relargués dans l'eau. Ceci se produit généralement par dissolution/précipitation chimique, activité biologique ou désagrégation d'agrégats stables. Les colloïdes sont mobiles, leur surface est telle que les colloïdes se repoussent entre eux. En effet ceci évite qu'ils se rapprochent les uns des autres pour former de plus grosses particules. Dans le même temps, la taille des pores du sol doit être suffisante pour éviter l'élimination des colloïdes, de la suspension, par effet filtrant. Les colloïdes possèdent généralement des charges négatives à leur surface, ce qui provoque la répulsion des colloïdes entre eux. Cependant, des cations dissous dans la solution peuvent modifier la répartition des charges à la surface et diminuer la répulsion des colloïdes entre eux. Les colloïdes sont alors susceptibles de se rapprocher pour former des plus grosses particules qui ne sont plus mobiles. La présence de sels dissous dans la solution de lixiviation permet donc de stabiliser le sol vis à vis du transport particulaire. [COMMANS, 2000] a montré par des tests Batch que le relargage des HAP était fortement lié au relargage de gros colloïdes, diamètre compris entre 0,45 et 12 µm. Lors de tests par percolation [BOUCHEZ, 1996] a montré que le relargage des HAP était plus important pendant les premières heures de lixiviation. Cet auteur suppose que ce relargage initial massif est en relation avec le relargage de colloïdes dans la phase mobile.
I.4 Les tests de lixiviation
La lixiviation est l'extraction par voie liquide d'éléments ou de molécules inorganiques ou organiques, contenus ou fixés sur une matrice solide, mettant en œuvre des mécanismes chimiques ou biologiques[Académie des Sciences, 1998]. La lixiviation ou la percolation d'un matériau peut se produire sur le terrain, par exposition du matériau à l'infiltration naturelle ou aux précipitations ou au laboratoire par la mise en œuvre de tests en batch de lixiviation ou de percolation en colonne. Deux grandes classes de tests de lixiviations peuvent être distinguées:
Les tests en batch Les tests en colonne.
Les caractéristiques du comportement en lixiviation/percolation déterminées par chacun de ces tests ne doivent pas être confondues, les phénomènes mis en évidence étant
51

qualitativement différents. Les tests en batch sont généralement utilisés pour étudier les cinétiques de mise en solution des composés à partir d'une phase solide et déterminer des isothermes d'adsorption et désorption. Ils sont aussi utilisés pour évaluer l'influence spécifique de certains facteurs sur le relargage comme l'étude des phénomènes de non-équilibre [BRUSSEAU, 1989], de vieillissement [MAJHOUB, 1999]. La plupart de ces essais rapportés dans la littérature sont réalisés sur des matrices (sols) artificiellement pollués. Deux types de tests doivent être distingués au sein des systèmes batch. Dans le cas d'un système batch parfaitement agité, l'hypothèse émise est celle d'un système particulaire complètement dispersé où toutes les surfaces des particules de sol en suspension sont exposées au lixiviant. A l'issue du temps de contact, la fraction liquide après séparation d'avec la suspension est analysée. Dans un système « batch renouvelé » le liquide surnageant peut être renouvelé de manière séquentielle. Ces renouvellements séquentiels peuvent être faits soit dans le cas d'un système particulaire parfaitement dispersé [BAYARD, 1997], soit dans un système où la phase solide poreuse est consolidée, dans ce dernier cas la diffusion moléculaire dans le solide joue un rôle prépondérant [Compact Granular Leaching Test, NVN 7347]. Dans les tests en colonne, les échantillons mis en œuvre sont des systèmes structurés. La solution lixiviante s'écoule à travers une colonne de sol de manière ascendante ou descendante, puis est collectée par fraction est analysée. Les particules de sols sont immobiles et la solution de lixiviation s'écoule par percolation entre ou à travers les particules de solides en entraînant les constituants dissous. En raison de la structure du sol et de la géométrie des pores, seule une fraction de la surface totale des particules entre en contact avec la solution lixiviante. Les tests en colonne sont généralement employés dans l'intention de simuler les conditions du terrain.
I.5 Conclusion
De nombreux travaux ont été réalisés sur les processus de désorption et de dissolution de phase liquide non aqueuse comme source de contamination des eaux souterraines. Une partie des résultats de ces études ont abouti à la formulation de modèles de transfert de masse. De telles expériences ont généralement été conduites pour des systèmes idéaux et homogènes. Fréquemment le milieu est contaminé artificiellement sous des conditions contrôlées de laboratoire. Cette contamination est généralement simple et maîtrisée puisque constituée de quelques polluants. Pour étudier la mobilité de la pollution dans un sol de site industriel, l’identification des mécanismes contrôlant le relargage est indispensable. On distingue deux situations extrêmes :
52

le cas où le relargage est contrôlé par la désorption des molécules organiques, respectivement le cas où le relargage est contrôlé par la dissolution de phase liquide non aqueuse. Néanmoins, dans les deux cas, les contaminants s’accumulent dans l’eau traversant le milieu poreux. Si la zone contaminée est assez longue, les conditions d’équilibre peuvent s’établir après une certaine distance de contact sol/nappe. Dans le cas de la dissolution, la concentration à l’équilibre correspond à la limite de solubilité, tandis que lors de la désorption, elle est déterminée d’après le coefficient de partage Kd [GRATHWOHL ,1998]. Dans des conditions d’équilibre, la concentration maximale possible dans l’eau est atteinte, ce qui permet un calcul simple en fonction du processus de relargage. Par contre dans des conditions de non équilibre, le relargage se fait avec un gradient de concentration maximum. La longueur à parcourir pour atteindre un état d’équilibre est fonction du processus de relargage (dissolution, désorption). D’après [EBERHARDT, 2002], dans les zones contaminées par des Phase Liquides Non Aqueuse (PLNA), l’équilibre est atteint rapidement et la concentration du contaminant est donnée par la solubilité du contaminant (loi de Raoult). Par contre, il semblerait que dans les zones où le relargage est contrôlé par la désorption, les conditions d’équilibre ne soient pas réalisées. Le calcul des concentrations en phase aqueuse doit donc se faire à partir des modèles de transport introduits précédemment. Relativement peu de travaux existent sur les phénomènes de transport particulaire lors de la lixiviation des sols, cependant l'idée de la prise en compte des polluants transportés sous forme particulaire est de plus en plus souvent évoquée. Notre objectif est d'étudier la mobilité des HAP dans un sol de site industriel. L’étude va consister à identifier le/les processus de relargage majoritaires (dissolution et/ou désorption, transport particulaire) dans un sol de site industriel et à évaluer l'influence de paramètres pertinents sur le/les processus de relargage majoritaires. Mais tout d'abord, commençons par décrire les caractéristiques de la pollution du sol étudié.
53

Chapitre II : Description des caractéristiques du sol
Chapitre II: Description des caractéristiques du sol Le sol étudié provient d'un site industriel du Nord de la France. Sur ce sol, il n'existe plus d'activité industrielle polluante. Cependant, en raison de la succession d'activités polluantes dans le passé, ce sol présente une pollution complexe, importante dont l'historique est mal connu. Le sol a été prélevé sur le site dans la zone non saturée. Le sol, après réception dans 4 fûts de 200 kg chacun, a été échantillonné par la méthode des pelletages alternés. L'échantillon de sol représentatif ainsi formé a ensuite été étalé pour séchage, à l'abri de la lumière, à température ambiante, en contact avec l'air ambiant, sur une bâche pendant 7 jours. C'est sur ce dernier échantillon représentatif que nous avons travaillé. Nous avons caractérisé en premier lieu la granulométrie et l'humidité du sol. Ensuite, sur la fraction la plus représentative, nous avons caractérisé la teneur en HAP, le carbone organique total (COT), la teneur en phénol, et le pouvoir tampon du sol. Nous travaillions sur un site pollué par des organiques majoritairement donc toutes les procédures de caractérisation établies et reconnues dans le domaine des pollutions métalliques (capacité d'échange...) n'étaient pas justifiées dans notre cas.
II.1 Granulométrie
La granulométrie du sol, obtenue par tamisage, est répartie principalement dans les tranches suivantes :
Tableau 1 : répartition granulométrique
Fraction du sol % massique du sol X < 2 mm
2 < X < 6 mm 6 < X < 20 mm
37,3 26,0 27,7
La fraction de sol supérieure à 20 mm correspond seulement à 9% de la masse sèche de sol. L’analyse a permis de conclure que la majeure partie de la pollution organique observée était concentrée dans la tranche granulométrique <2 mm. Cette fraction est la plus représentative pour la problématique environnementale posée par le sol, elle l'est également en pourcentage massique. Nous avons décidé de concentrer l’ensemble du programme expérimental sur cette tranche granulométrique de sol. En conséquence, en parlant de sol dans notre rapport nous nous référons implicitement à la tranche granulométrique inférieure à 2 mm du sol échantillonné sur le site industriel..
54
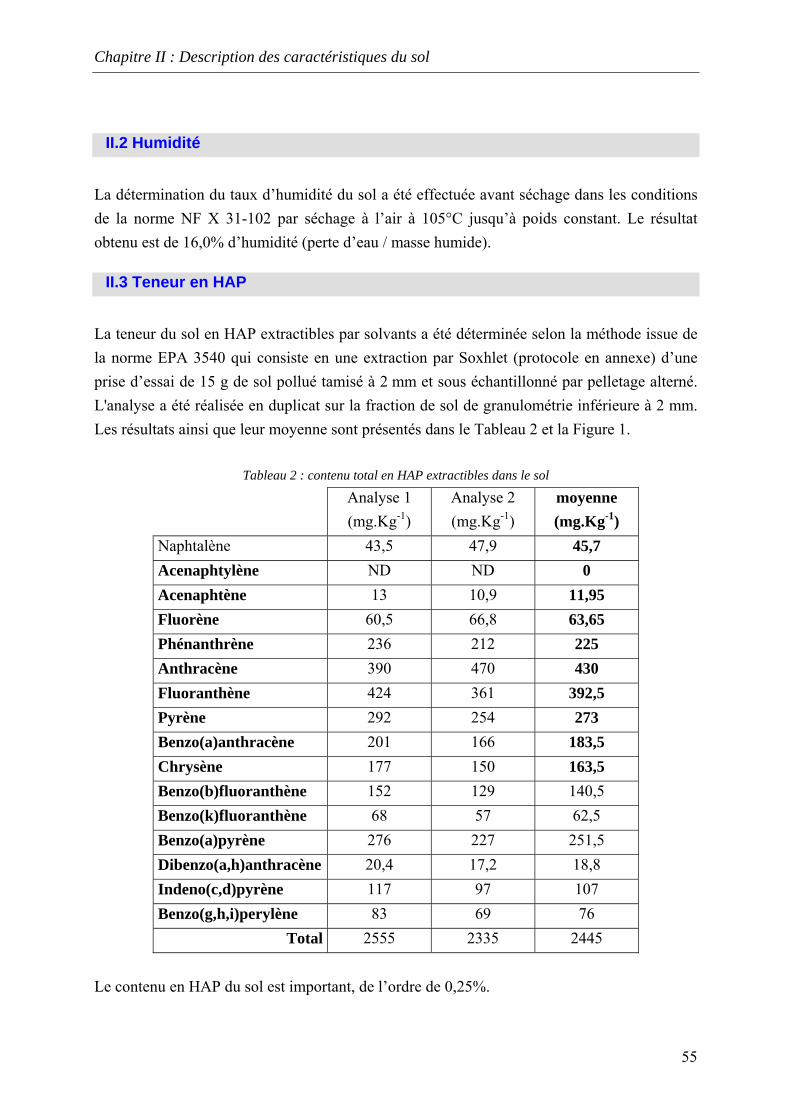
Chapitre II : Description des caractéristiques du sol
II.2 Humidité
La détermination du taux d’humidité du sol a été effectuée avant séchage dans les conditions de la norme NF X 31-102 par séchage à l’air à 105°C jusqu’à poids constant. Le résultat obtenu est de 16,0% d’humidité (perte d’eau / masse humide).
II.3 Teneur en HAP
La teneur du sol en HAP extractibles par solvants a été déterminée selon la méthode issue de la norme EPA 3540 qui consiste en une extraction par Soxhlet (protocole en annexe) d’une prise d’essai de 15 g de sol pollué tamisé à 2 mm et sous échantillonné par pelletage alterné. L'analyse a été réalisée en duplicat sur la fraction de sol de granulométrie inférieure à 2 mm. Les résultats ainsi que leur moyenne sont présentés dans le Tableau 2 et la Figure 1.
Tableau 2 : contenu total en HAP extractibles dans le sol
Analyse 1 (mg.Kg-1)
Analyse 2 (mg.Kg-1)
moyenne (mg.Kg-1)
Naphtalène 43,5 47,9 45,7 Acenaphtylène ND ND 0 Acenaphtène 13 10,9 11,95 Fluorène 60,5 66,8 63,65 Phénanthrène 236 212 225 Anthracène 390 470 430 Fluoranthène 424 361 392,5 Pyrène 292 254 273 Benzo(a)anthracène 201 166 183,5 Chrysène 177 150 163,5 Benzo(b)fluoranthène 152 129 140,5 Benzo(k)fluoranthène 68 57 62,5 Benzo(a)pyrène 276 227 251,5 Dibenzo(a,h)anthracène 20,4 17,2 18,8 Indeno(c,d)pyrène 117 97 107 Benzo(g,h,i)perylène 83 69 76
Total 2555 2335 2445
Le contenu en HAP du sol est important, de l’ordre de 0,25%.
55

Chapitre II : Description des caractéristiques du sol
050
100150200250300350400450500
Naphta
lène
Acéna
phtyl
ène
Acéna
phtèn
e
Fluorèn
e
Phéna
nthrèn
e
Anthrac
ène
Fluoran
thène
Pyrène
Benzo
(a)an
thrac
ène
Chrysè
ne
Benzo
(b)flu
oranth
ène
Benzo
(k)flu
oranth
ène
Benzo
(a)py
rène
Dibenz
o(a,h)
anthr
acèn
e
Benzo
(g,h,i
)pyrèn
e
Indén
o(1,2,
-
Tene
ur e
n m
g/kg
de
sol s
ec Teneur totale = 2445 mg/kg de sol sec
Figure 1 : Profil du contenu total moyen en HAP dans le sol
II.4 Teneur en phénol
La présence de phénols dans la solution de sol a été mise en évidence au moyen de la mesure de l’indice phénol sur une solution préparée par la mise en contact durant 24 h sous agitation de 100 g de sol avec 300 ml d’eau distillée. La solution prélevée après décantation du sol a fait l’objet d’une mesure de l’indice phénol (Iφ) selon la méthode colorimétrique issue de la norme NF T 90-109. Le résultat moyen de cette mesure est le suivant Iφ = 0,145 mg/l Remarque La représentativité de l’indice phénol doit être relativisée. En effet, la méthode de détermination de l’indice phénol décrite par la norme NFT90-109 est basée sur le développement d’une coloration entre les « phénols » et l’amino-4 antipyrine. Or tous les composés phénolés ne réagissent pas avec ce réactif et, par ailleurs, ceux qui réagissent conduisent chacun à une coloration dont les caractéristiques (longueur d’onde du maximum d’absorption, intensité) dépendent de la nature chimique du « phénol » concerné. En particulier, les composés phénolés, dans lesquels la position para- est occupée ne réagissent pas, ou ne réagissent pas quantitativement avec l’amino-4 antipyrine. De plus, la coloration formée avec certains « phénols » est extrêmement dépendante de la force ionique de la solution et de la présence d’acides humiques, lignines, etc…, et certaines amines aromatiques sont susceptibles d’entraîner des interférences. Cette mesure de l’indice phénol nous permet donc uniquement de révéler la présence de certains composés phénolés dans cette solution préparée en mettant en contact le sol pendant
56

Chapitre II : Description des caractéristiques du sol
24 heures sous agitation avec une quantité d’eau correspondant au rapport Liquide/Solide de 3, en présence d’un inhibiteur biologique (NaN3). La détermination de l’indice phénol ne nous permettra pas de connaître précisément (nature et quantité) des composés phénolés présents ni de suivre leur solubilité au cours des essais. La quantité de phénol trouvée dans l'échantillon est relativement faible. Les HAP, composés hydrophobes très peu réactifs, n'ont probablement pas d'interactions avec les phénols. Pour ces raisons nous avons choisi de ne pas suivre le paramètre phénol lors des études de mobilité des HAP.
II.5 Carbone Organique Total (COT)
Le sol contient 30.9 % de carbone organique total. L’analyse a été effectuée selon la Norme X31-409. En excluant la part du carbone organique issue des 16 HAP, le contenu en carbone organique est ramené 30.65 %. En général, le COT des sols se situe entre 0,5 et 5%. La forte teneur en carbone organique semble indiquer que nous sommes en présence d'un sol particulier. Le sol pourrait contenir par exemple de la matière organique non extractible (matrice riche en carbone) comme c'est le cas dans les goudrons de houille [HAESELER, 1999]. Ceci laisserait supposer qu'une partie de la pollution aurait pour origine des goudrons de houille. Cependant l'aspect visuel du sol est en contradiction avec cette hypothèse car il n'a pas l'odeur caractéristique des goudrons et il n'y a pas non plus de particules collantes noires comme dans les sols pollués par les goudrons de houille.
matiereOrganique non extractible
55%
résines7%
autre HAP21%
16 HAP17%
Figure 2:Distribution de la matière organique dans les goudrons de houille
en pourcentage du COT [HAESELER, 1999].
II.6 Caractérisation de la nature du sol au microscope environnemental à balayage électronique (MEB)
Une série de 8 photographies a été réalisée sur le sol tamisé à 2 mm. Ces photos (Figure 3) ont été prises au microscope environnemental à balayage électronique MEB E (voir description en annexe) à l'Ecole des Mines d'Albi. Les paramètres de chaque photo sont présentés dans le Tableau 3.
57
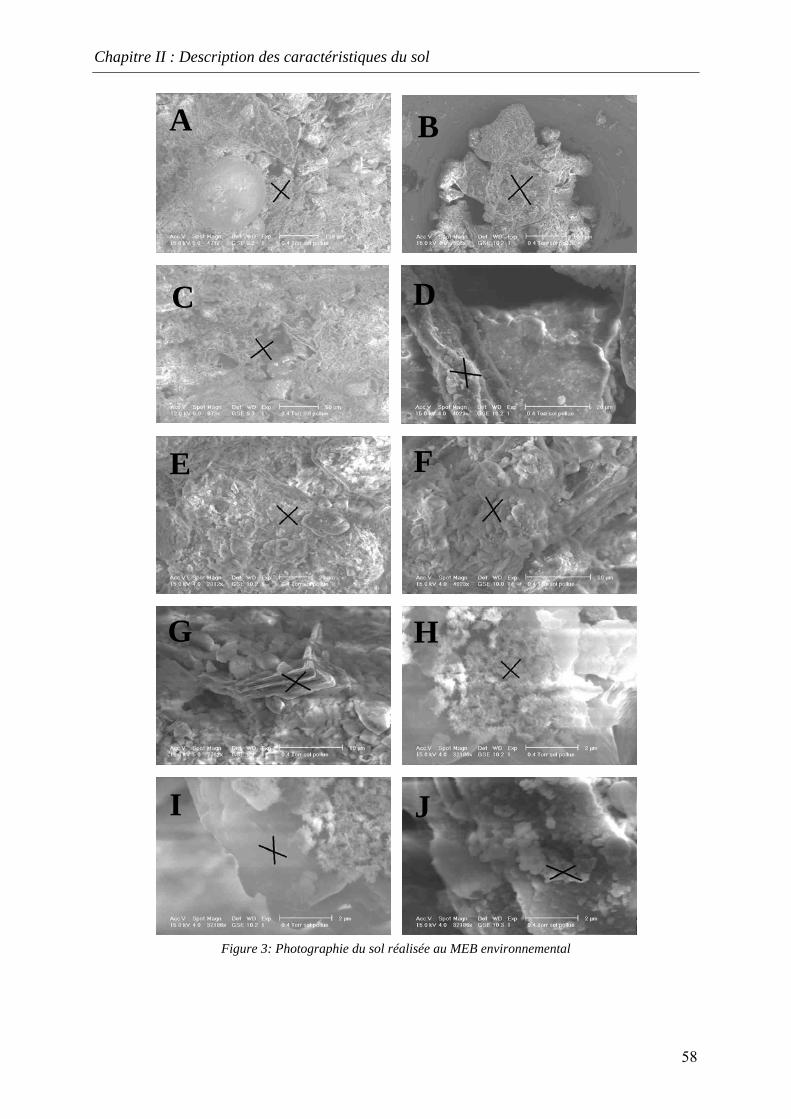
Chapitre II : Description des caractéristiques du sol
A
B
C
D
E
F
G
H
I
J
Figure 3: Photographie du sol réalisée au MEB environnemental
58
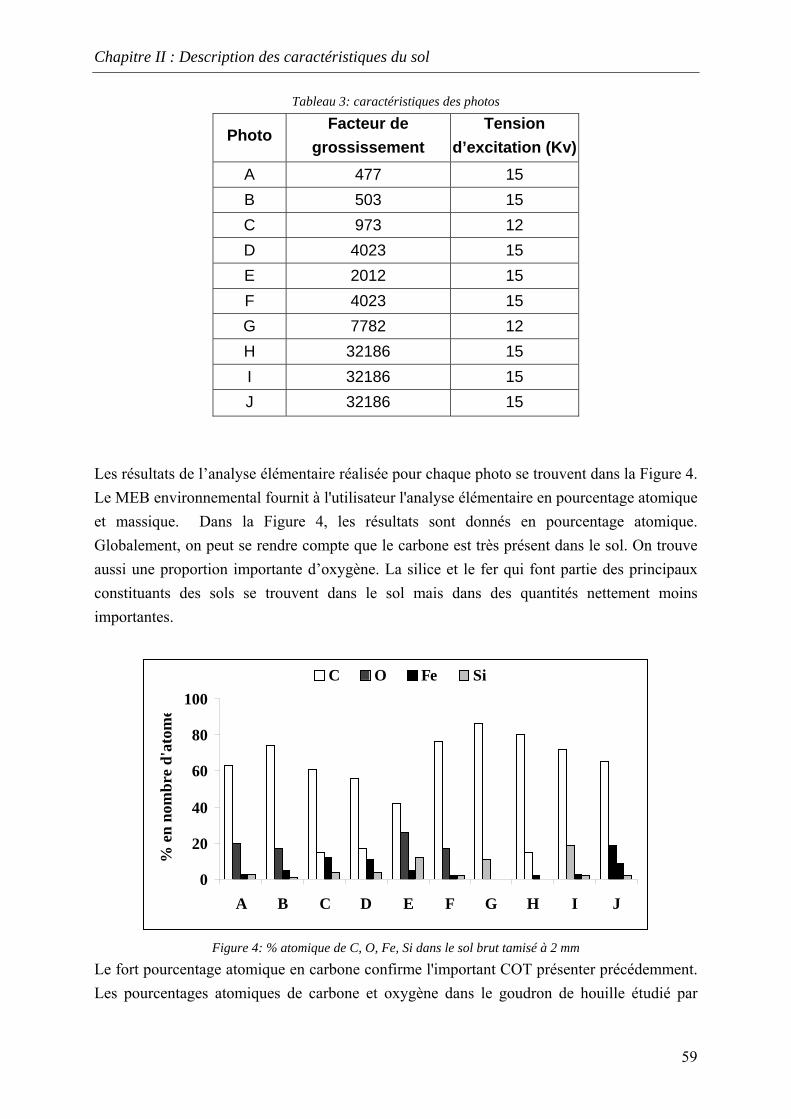
Chapitre II : Description des caractéristiques du sol
Tableau 3: caractéristiques des photos
Photo Facteur de
grossissement Tension
d’excitation (Kv) A 477 15 B 503 15 C 973 12 D 4023 15 E 2012 15 F 4023 15 G 7782 12 H 32186 15 I 32186 15 J 32186 15
Les résultats de l’analyse élémentaire réalisée pour chaque photo se trouvent dans la Figure 4. Le MEB environnemental fournit à l'utilisateur l'analyse élémentaire en pourcentage atomique et massique. Dans la Figure 4, les résultats sont donnés en pourcentage atomique. Globalement, on peut se rendre compte que le carbone est très présent dans le sol. On trouve aussi une proportion importante d’oxygène. La silice et le fer qui font partie des principaux constituants des sols se trouvent dans le sol mais dans des quantités nettement moins importantes.
0
20
40
60
80
100
A B C D E F G H I J
% e
n no
mbr
e d'
atom
e
C O Fe Si
Figure 4: % atomique de C, O, Fe, Si dans le sol brut tamisé à 2 mm
Le fort pourcentage atomique en carbone confirme l'important COT présenter précédemment. Les pourcentages atomiques de carbone et oxygène dans le goudron de houille étudié par
59
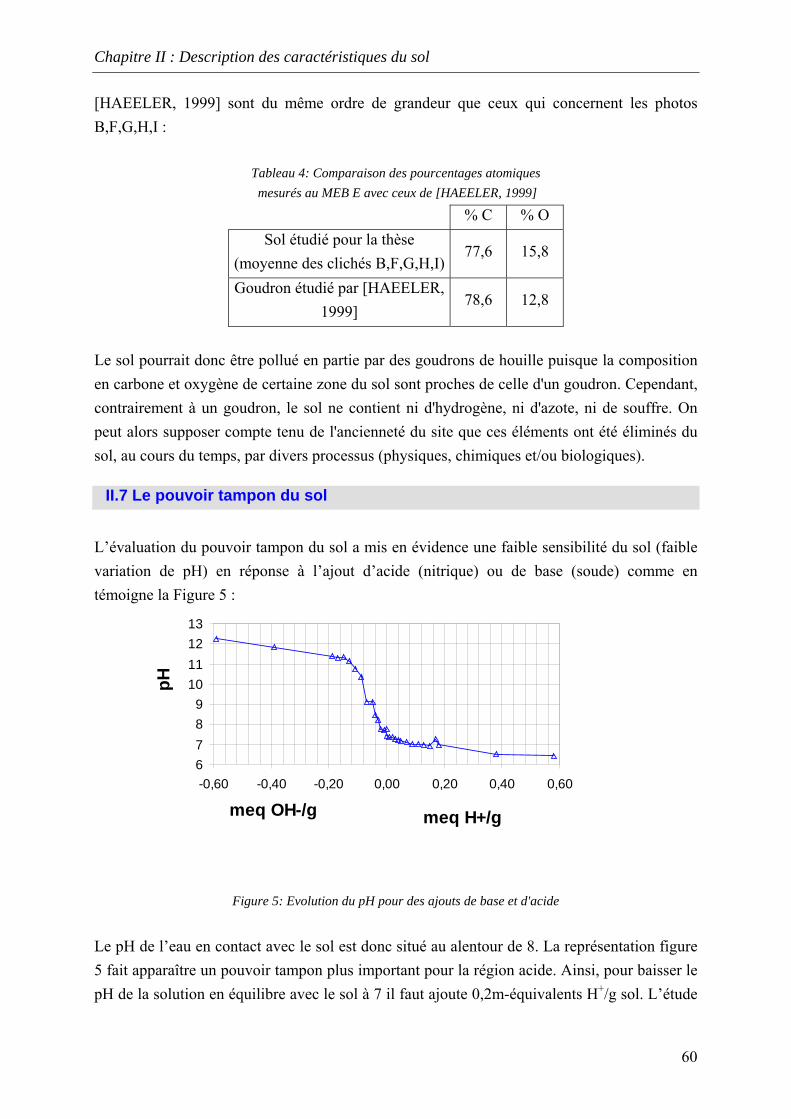
Chapitre II : Description des caractéristiques du sol
[HAEELER, 1999] sont du même ordre de grandeur que ceux qui concernent les photos B,F,G,H,I :
Tableau 4: Comparaison des pourcentages atomiques mesurés au MEB E avec ceux de [HAEELER, 1999]
% C % O Sol étudié pour la thèse
(moyenne des clichés B,F,G,H,I)77,6 15,8
Goudron étudié par [HAEELER, 1999]
78,6 12,8
Le sol pourrait donc être pollué en partie par des goudrons de houille puisque la composition en carbone et oxygène de certaine zone du sol sont proches de celle d'un goudron. Cependant, contrairement à un goudron, le sol ne contient ni d'hydrogène, ni d'azote, ni de souffre. On peut alors supposer compte tenu de l'ancienneté du site que ces éléments ont été éliminés du sol, au cours du temps, par divers processus (physiques, chimiques et/ou biologiques).
II.7 Le pouvoir tampon du sol
L’évaluation du pouvoir tampon du sol a mis en évidence une faible sensibilité du sol (faible variation de pH) en réponse à l’ajout d’acide (nitrique) ou de base (soude) comme en témoigne la Figure 5 :
6789
10111213
-0,60 -0,40 -0,20 0,00 0,20 0,40 0,60
meq OH-/g
pH
meq H+/g
Figure 5: Evolution du pH pour des ajouts de base et d'acide
Le pH de l’eau en contact avec le sol est donc situé au alentour de 8. La représentation figure 5 fait apparaître un pouvoir tampon plus important pour la région acide. Ainsi, pour baisser le pH de la solution en équilibre avec le sol à 7 il faut ajoute 0,2m-équivalents H+/g sol. L’étude
60

Chapitre II : Description des caractéristiques du sol
paramétrique du pH ne semble, a priori, pas un facteur prépondérant pour la détermination du comportement des HAP dans ces conditions.
II.8 Conclusion
Les résultats présentés dans ce chapitre, en particulier la teneur en HAP vont être utilisés tout au long de l’étude. La fraction granulométrique <2 mm a été considérée représentative du sol étudié. : elle est représentative de la masse du sol et concentre la majeur partie de la pollution organique du sol. Cette fraction granulométrique du sol est très riche en C organique (COT=30 %) ce qui éloigne le site étudié de la composition d’un sol pour l’approcher plus d’un déchet (matériau) riche en polluants organiques, notamment en HAP. La pollution du sol est probablement issue de goudrons de houille. La pollution du sol caractérisée, nous pouvons maintenant passer à l'étude expérimentale de lixiviation, c'est à dire à l'étude du comportement du sol lors de son contact avec l'eau.
61

Chapitre III : Application des tests classiques de lixiviation
Chapitre III: Applications des tests classiques de lixiviation à l’étude de la mobilité des HAP La plus part des essais dans le domaine des sols pollués sont réalisés le plus souvent sur des sols artificiellement pollués avec comme objectif d'étudier des points spécifiques du relargage. Malgré l’hypothèse qu’ils ne soient à priori pas ou peu adaptés à l’étude des pollutions réelles, nous avons choisi, dans un premier temps, de les utiliser. Notre étude expérimentale dont le but était d'étudier la mobilité des polluants organiques hydrophobes dans un sol industriel a débuté à partir du test expérimental le plus simple et le plus utilisé, le batch en milieu dispersé. Parallèlement un essai de percolation en colonne a été réalisé pour approcher le comportement du sol in situ vis à vis du relargage. Les premiers résultats obtenus par les tests batch en milieu dispersé et la percolation en colonne ont révélé, comme l’étude précédente de caractérisation du sol, la grande complexité du sol étudié et les limites des essais traditionnels de lixiviation pour étudier une telle complexité. Par contre l’analyse critique des résultats a permis de formuler des premières hypothèses sur les paramètres et les mécanismes intervenant lors du relargage.
III.1 Principaux tests de laboratoire dans le domaine de la lixiviation
Trois grandes catégories de tests de lixiviation des sols ressortent de la littérature et de l’expérience acquise dans le domaine à l'INSA de Lyon (LAEPSI – Polden): - Le test classique X31-210, test réglementaire français utilisé pour la caractérisation du
potentiel polluant d'un matériau granulaire. Ce test est, par exemple, fréquemment employé pour évaluer la fraction polluante mobilisable à l'eau, sous agitation, avant et après traitement d’une pollution.
- Le test de percolation en colonne pour simuler le comportement In situ d’une pollution
réelle. Ces deux premiers tests ont été utilisés majoritairement par Polden dans le cadre de pollutions réelles toutefois sans objectifs d’appréhender les mécanismes contrôlant le relargage. - L’étude menée initialement par [BAYARD, 1997] portant sur des sols artificiellement
pollués par du naphtalène et du phénanthrène a été réalisée avec des tests batch en milieu dispersé. Elle consistait en une étape de caractérisation des cinétiques d’adsorption et de désorption puis une étape de caractérisation des isothermes d’adsorption désorption.
62

Chapitre III : Application des tests classiques de lixiviation
L’objectif principal de l’étape cinétique était de connaître le temps nécessaire pour atteindre un état d’équilibre apparent dans le batch. Les isothermes permettaient quant à eux de définir l’ « affinité » du polluant envers le sol par l’intermédiaire d’une constante de partage (Kd).
Percolation en colonne
Lixiviation en milieu dispersé
X31-210- Caractérisation du potentiel polluant des
déchets granulaires
- Cinétique de mise en solution
- Isotherme d’adsorption-désorption
- Test de simulation
Figure 1: Principales expériences menées à l'INSA de Lyon dans le domaine de la lixiviation des sols
Parmi les procédures précédemment expérimentées au laboratoire, deux ont été retenues comme base de départ pour l’étude des sites industriels présentant des pollutions complexes: la lixiviation en milieu dispersé et la percolation en colonne. Ces essais devaient permettre d'obtenir des informations sur le comportement du sol afin de caractériser la mobilité des HAP dans le sol. Pour les raisons présentées ci-après, les résultats de lixiviation en milieu dispersé et par percolation avec l'eau permutée portent uniquement sur la série des HAP comprise entre le fluorène (3 cycles, M=166) et B(a)pyrène (5 cycles, M=252). La pollution organique comprenant la série des HAP du fluorène jusqu'au B(a)pyrène est
largement majoritaire, elle représente 89 % des HAP totaux contenus dans le sol (Figure 2).
63

Chapitre III : Application des tests classiques de lixiviation
autres HAP11%
HAP caractérisés
89%
Figure 2: représentation en pourcentage de la pollution organique totale
Le traitement des échantillons aqueux avant analyse, en particulier l'évaporation du
solvant d'extraction sous courant d'azote entraîne une perte difficilement quantifiable des HAP les plus volatils (naphtalène, acénaphtylène, acénaphtène). La procédure analytique (présentée en annexe) retenue et notamment la durée de l'analyse
a été optimisée pour la série des HAP allant du naphtalène au B(a)pyrène.
III.2 Lixiviation en milieu dispersé
Les tests de lixiviation en milieu dispersé ont été pratiqués suivant les protocoles mis en place par [BAYARD, 1997] pour ses travaux sur des sols artificiellement pollués par des HAP. Deux types d'essai ont été réalisés:
1. Cinétique de mise en solution 2. Isotherme de désorption
Les protocoles expérimentaux des lixiviations en milieu dispersé sont présentés succinctement puisqu'ils proviennent de ceux décrits par [BAYARD, 1997].
III.2.1 Cinétiques de mise en solution
L’étude cinétique permet de connaître le temps de contact nécessaire pour atteindre un état d’équilibre apparent dans la répartition des HAP lors du contact sol/eau en milieu dispersé (agitation).
III.2.1.1 Protocole
La solution utilisée pour les essais est de l’eau permutée rendue toxique pour l’activité bactérienne par ajout de NaN3 à 15.10-3M [LOTRARIO, 1995]. Un échantillon de sol est mis en contact avec la solution préparée dans un rapport massique égal à 3 pendant une durée
64
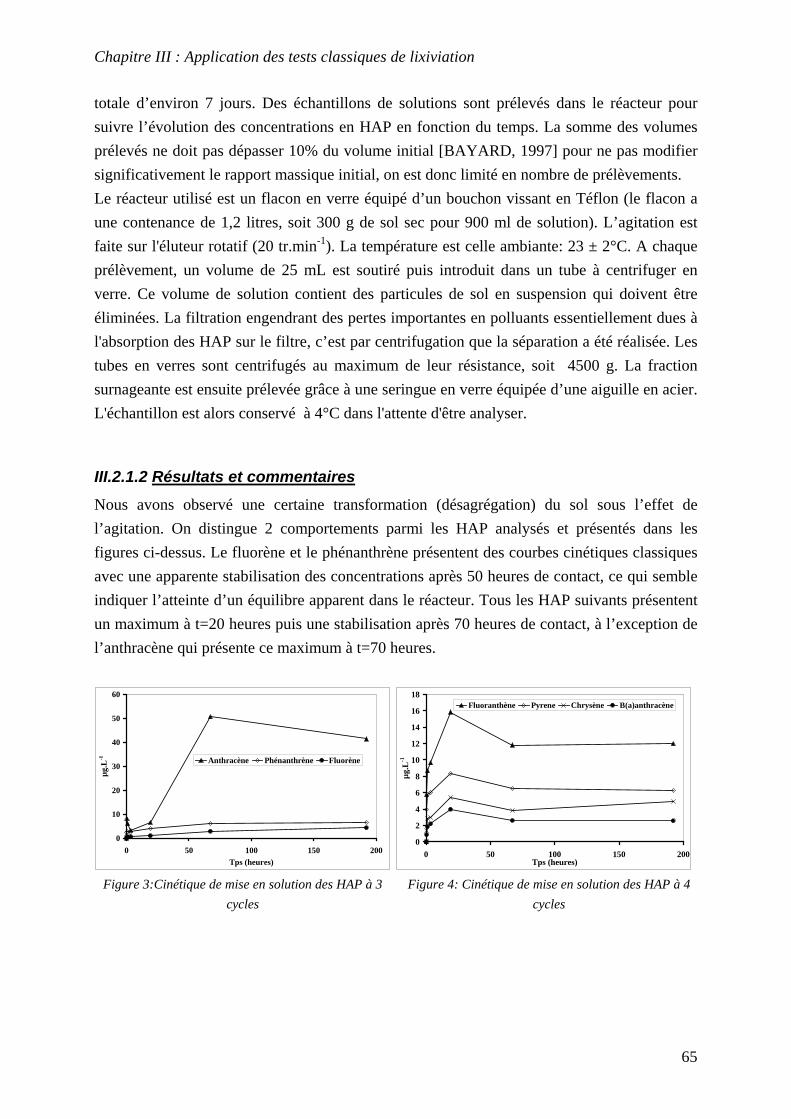
Chapitre III : Application des tests classiques de lixiviation
totale d’environ 7 jours. Des échantillons de solutions sont prélevés dans le réacteur pour suivre l’évolution des concentrations en HAP en fonction du temps. La somme des volumes prélevés ne doit pas dépasser 10% du volume initial [BAYARD, 1997] pour ne pas modifier significativement le rapport massique initial, on est donc limité en nombre de prélèvements. Le réacteur utilisé est un flacon en verre équipé d’un bouchon vissant en Téflon (le flacon a une contenance de 1,2 litres, soit 300 g de sol sec pour 900 ml de solution). L’agitation est faite sur l'éluteur rotatif (20 tr.min-1). La température est celle ambiante: 23 ± 2°C. A chaque prélèvement, un volume de 25 mL est soutiré puis introduit dans un tube à centrifuger en verre. Ce volume de solution contient des particules de sol en suspension qui doivent être éliminées. La filtration engendrant des pertes importantes en polluants essentiellement dues à l'absorption des HAP sur le filtre, c’est par centrifugation que la séparation a été réalisée. Les tubes en verres sont centrifugés au maximum de leur résistance, soit 4500 g. La fraction surnageante est ensuite prélevée grâce à une seringue en verre équipée d’une aiguille en acier. L'échantillon est alors conservé à 4°C dans l'attente d'être analyser.
III.2.1.2 Résultats et commentaires
Nous avons observé une certaine transformation (désagrégation) du sol sous l’effet de l’agitation. On distingue 2 comportements parmi les HAP analysés et présentés dans les figures ci-dessus. Le fluorène et le phénanthrène présentent des courbes cinétiques classiques avec une apparente stabilisation des concentrations après 50 heures de contact, ce qui semble indiquer l’atteinte d’un équilibre apparent dans le réacteur. Tous les HAP suivants présentent un maximum à t=20 heures puis une stabilisation après 70 heures de contact, à l’exception de l’anthracène qui présente ce maximum à t=70 heures.
0
10
20
30
40
50
60
0 50 100 150 200Tps (heures)
µg.L
-1 Anthracène Phénanthrène Fluorène
0
2
4
6
8
10
12
14
16
18
0 50 100 150 200Tps (heures)
µg.L
-1
Fluoranthène Pyrene Chrysène B(a)anthracène
Figure 3:Cinétique de mise en solution des HAP à 3
cycles Figure 4: Cinétique de mise en solution des HAP à 4
cycles
65

Chapitre III : Application des tests classiques de lixiviation
0
1
2
3
4
5
6
0 50 100 150 200Tps (heures)
µg.L
-1
B(b)fluoranthène B(k)fluoranthène B(a)pyrene
Figure 5:Cinétique de mise en solution
des HAP à 5 cycles
Le pic observait dans la Figure 4 et la Figure 5 pourrait être attribué à une redistribution des HAP initialement mis en solution. En effet, la désagrégation des particules de sol produite par l’agitation du réacteur pourrait entraîner la formation de nouvelles surfaces d’échange qui refixeraient les HAP initialement mis en solution. Selon les résultats du test, on peut considérer que 48 heures de contact suffisent pour atteindre des concentrations stables dans le réacteur. Les résultats, bien que correspondant à une situation stationnaire, n’ont pas été retenus comme points d’isothermes en raison de la transformation du sol et de l’évolution implicite de ses propriétés de transfert solide/liquide.
III.2.2 Isotherme de désorption par renouvellements séquentiels
L’objectif est de suivre les concentrations à l’équilibre apparent en fonction de l'évolution des HAP dans le sol. Ce suivi du relargage à l’équilibre est réalisé de façon discontinue (renouvellement total et régulier de la solution. Les concentrations en HAP, mesurées en solution après chaque lixiviation, rapportées à la masse de sol lixiviée (mg.kg-1) étaient insignifiantes comparativement au contenu total en HAP. Il n'a donc pas été possible de mettre en relation dans un graphique de type isotherme l'évolution des concentrations en solution avec les concentrations sur solide. C'est pourquoi, dans la suite du document, nous parlerons du test des renouvellements séquentiels en milieu dispersé plutôt que d'isothermes de désorption.
III.2.2.1 Protocole
Une solution d'azoture de sodium à 15 mM (I=0,015 M) est préparée préalablement aux renouvellements séquentiels. L'azoture de sodium dans la solution permet de bloquer l'activité biologique. L’essai est réalisé à température ambiante (23 ± 2°C) dans un tube à centrifuger en verre de 25 mL muni d’un bouchon vissant en Téflon. 19,5 mL de solution initialement
66
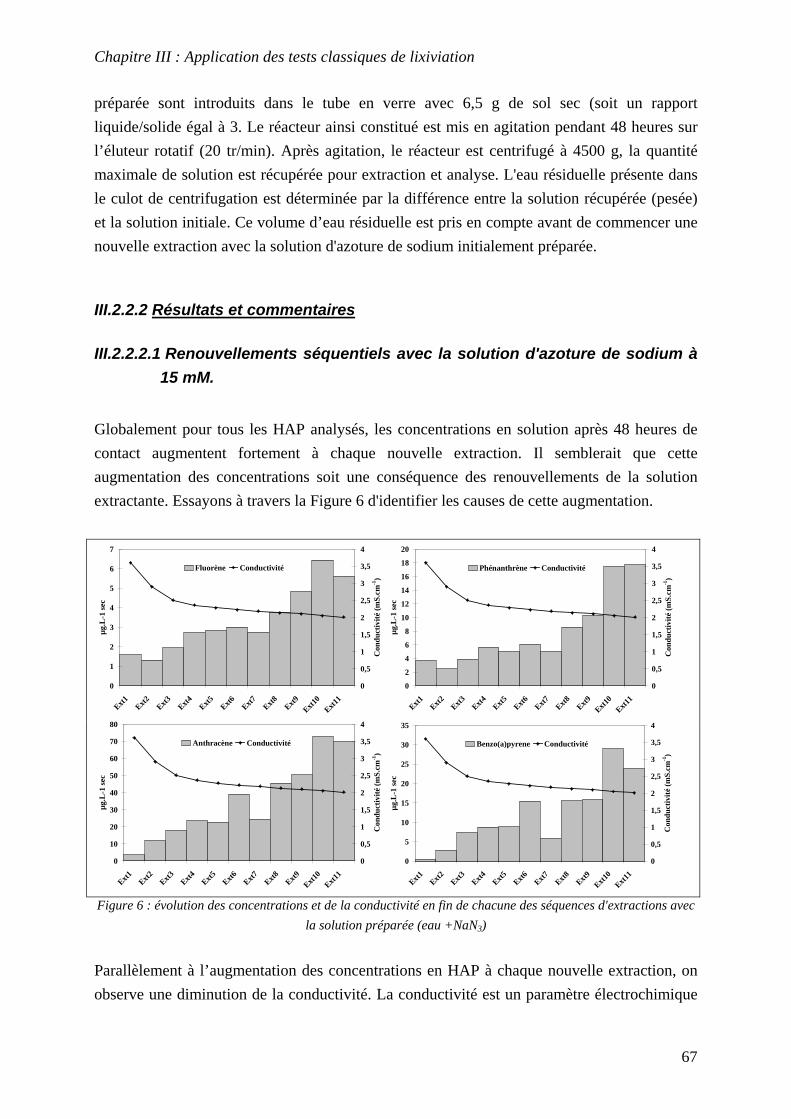
Chapitre III : Application des tests classiques de lixiviation
préparée sont introduits dans le tube en verre avec 6,5 g de sol sec (soit un rapport liquide/solide égal à 3. Le réacteur ainsi constitué est mis en agitation pendant 48 heures sur l’éluteur rotatif (20 tr/min). Après agitation, le réacteur est centrifugé à 4500 g, la quantité maximale de solution est récupérée pour extraction et analyse. L'eau résiduelle présente dans le culot de centrifugation est déterminée par la différence entre la solution récupérée (pesée) et la solution initiale. Ce volume d’eau résiduelle est pris en compte avant de commencer une nouvelle extraction avec la solution d'azoture de sodium initialement préparée.
III.2.2.2 Résultats et commentaires
III.2.2.2.1 Renouvellements séquentiels avec la solution d'azoture de sodium à 15 mM.
Globalement pour tous les HAP analysés, les concentrations en solution après 48 heures de contact augmentent fortement à chaque nouvelle extraction. Il semblerait que cette augmentation des concentrations soit une conséquence des renouvellements de la solution extractante. Essayons à travers la Figure 6 d'identifier les causes de cette augmentation.
0
1
2
3
4
5
6
7
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
0
0,5
1
1,5
2
2,5
3
3,5
4
Con
duct
ivité
(mS.
cm-1
)
Fluorène Conductivité
0
2
4
6
8
10
12
14
16
18
20
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
0
0,5
1
1,5
2
2,5
3
3,5
4
Con
duct
ivité
(mS.
cm-1
)
Phénanthrène Conductivité
0
10
20
30
40
50
60
70
80
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
0
0,5
1
1,5
2
2,5
3
3,5
4
Con
duct
ivité
(mS.
cm-1
)
Anthracène Conductivité
0
5
10
15
20
25
30
35
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
0
0,5
1
1,5
2
2,5
3
3,5
4
Con
duct
ivité
(mS.
cm-1
)
Benzo(a)pyrene Conductivité
Figure 6 : évolution des concentrations et de la conductivité en fin de chacune des séquences d'extractions avec
la solution préparée (eau +NaN3)
Parallèlement à l’augmentation des concentrations en HAP à chaque nouvelle extraction, on observe une diminution de la conductivité. La conductivité est un paramètre électrochimique
67

Chapitre III : Application des tests classiques de lixiviation
qui informe sur le contenu et la mobilité des ions dans la solution. La mobilité des ions dépend de nombreux facteurs, tel que la charge surfacique spécifique et le volume de l'ion hydraté, la température etc. Grossièrement on peut la relier à la force ionique. Dans la Figure 6, la conductivité baisse significativement entre la première et la dernière extraction, elle passe de 3,5 à 2 mS.cm-1. Cette baisse de conductivité est due à l'épuisement progressif des espèces ioniques du sol au cours des renouvellements séquentiels. Nous avons vu au chapitre I qu'une diminution de la force ionique augmente la solubilité des molécules hydrophobes. Par conséquent, il se pourrait que l'augmentation des concentrations en HAP (Figure 6) soit en partie corrélée à la diminution des espèces ioniques en solution. Par ailleurs, la formation de colloïdes dans la solution en contact avec le sol est augmentée suite à la diminution de la force ionique. Il est connu que les colloïdes peuvent « fixer » plus facilement des polluants hydrophobes de type HAP. Expérimentalement, on a pu observer la présence d'un film noirâtre (Figure 7), lors des prélèvements de la phase liquide. En effet l'aspiration de la phase liquide avec la seringue provoquait le déplacement d’un «film » ou «voile » en direction de l'aiguille. On peut supposer, suite à cette observation, que la phase liquide contient des particules, de types colloïdes, constitutifs du "film", non décantables par centrifugation à 4500 g. A chaque nouvelle extraction, l'aspect des phases liquides après le prélèvement était de moins en moins claire. En supposant que la turbidité de la phase liquide soit en relation avec le nombre de particules constituant le film, on peut dire que la quantité de particules dans la phase liquide augmente au cours des renouvellements séquentiels.
Culot
«Film » ou «voile»
La solution prélevée contient unecertaine quantité de film deconstitution inconnue.
Figure 7: formation d’un film lors du prélèvement de la phase liquide centrifugée
68
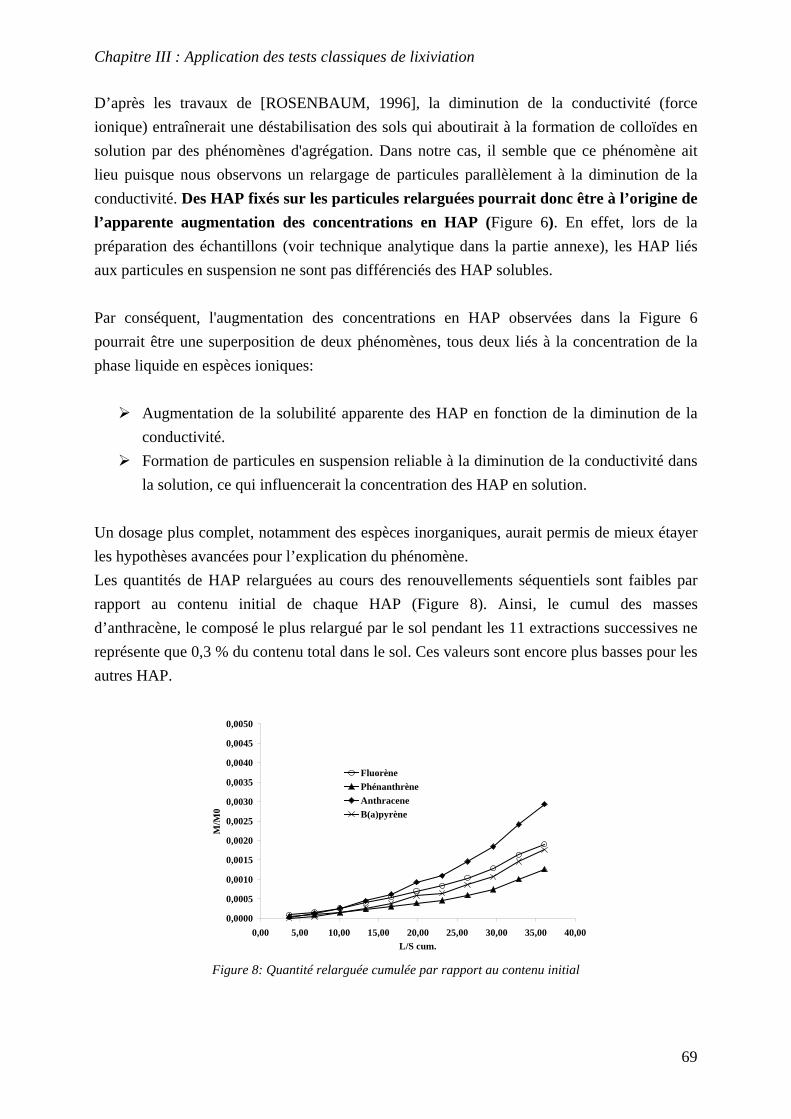
Chapitre III : Application des tests classiques de lixiviation
D’après les travaux de [ROSENBAUM, 1996], la diminution de la conductivité (force ionique) entraînerait une déstabilisation des sols qui aboutirait à la formation de colloïdes en solution par des phénomènes d'agrégation. Dans notre cas, il semble que ce phénomène ait lieu puisque nous observons un relargage de particules parallèlement à la diminution de la conductivité. Des HAP fixés sur les particules relarguées pourrait donc être à l’origine de l’apparente augmentation des concentrations en HAP (Figure 6). En effet, lors de la préparation des échantillons (voir technique analytique dans la partie annexe), les HAP liés aux particules en suspension ne sont pas différenciés des HAP solubles. Par conséquent, l'augmentation des concentrations en HAP observées dans la Figure 6 pourrait être une superposition de deux phénomènes, tous deux liés à la concentration de la phase liquide en espèces ioniques:
Augmentation de la solubilité apparente des HAP en fonction de la diminution de la conductivité. Formation de particules en suspension reliable à la diminution de la conductivité dans
la solution, ce qui influencerait la concentration des HAP en solution. Un dosage plus complet, notamment des espèces inorganiques, aurait permis de mieux étayer les hypothèses avancées pour l’explication du phénomène. Les quantités de HAP relarguées au cours des renouvellements séquentiels sont faibles par rapport au contenu initial de chaque HAP (Figure 8). Ainsi, le cumul des masses d’anthracène, le composé le plus relargué par le sol pendant les 11 extractions successives ne représente que 0,3 % du contenu total dans le sol. Ces valeurs sont encore plus basses pour les autres HAP.
0,0000
0,0005
0,0010
0,0015
0,0020
0,0025
0,0030
0,0035
0,0040
0,0045
0,0050
0,00 5,00 10,00 15,00 20,00 25,00 30,00 35,00 40,00L/S cum.
M/M
0
FluorènePhénanthrèneAnthraceneB(a)pyrène
Figure 8: Quantité relarguée cumulée par rapport au contenu initial
69

Chapitre III : Application des tests classiques de lixiviation
Pour tenter d’extraire des quantités de HAP plus importantes et suivre l’évolution du relargage, le même essai de renouvellements séquentiels a été réalisé avec un mélange constitué de 50% eau et 50% méthanol. Le choix s'est porté sur le méthanol, solvant classique, car il existe de nombreuses données bibliographiques sur la solubilité des HAP en présence de méthanol [Yalkowsky S.H.,1976] ; [Lane W.F, 1995] ; [Bouchard C.D., 1998]. Un pourcentage de méthanol égal à 50% a été choisi afin d'extraire des quantités significatives de HAP. En effet, la solution obtenue avec 50% de méthanol a un pouvoir extractant envers les HAP bien supérieur à celui de l'eau pure. Enfin, le dosage des HAP dans une solution contenant 50% de méthanol est beaucoup plus simple du point de vue expérimentale. En effet, les niveaux de concentration dans l'échantillon lixivié étant plus élevés, les analyses peuvent être réalisées directement en chromatographie liquide (pas d’étapes de concentration).
III.2.2.2.2 Renouvellements séquentiels avec un mélange eau/méthanol (50% V :V)
Le protocole expérimental est le même que lors des renouvellements séquentiels avec la solution eau + NaN3, seul diffère la technique analytique qui ne comporte pas de prétraitement de l’échantillon (voir annexe). Le temps de contact entre le sol et la phase liquide a été choisi égal à 48 heures, comme pour les renouvellements séquentiels à l'eau. Nous n'avons pas réalisé d'étude cinétique préalable pour déterminer précisément le temps nécessaire à l'atteinte de l'équilibre car l'objectif était ici de comparer le comportement avec la solution eau + NaN3 pour des conditions expérimentales proches, notamment lors de contacts solides/liquides. Nous avons choisi de garder le même temps d’extraction de 48 heures/séquence. La préparation des échantillons pour l'analyse en HPLC ne nécessitant pas d'évaporation sous courant d’azote, les résultats concernant le naphtalène ont pu être exploités convenablement (Figure 11).
70
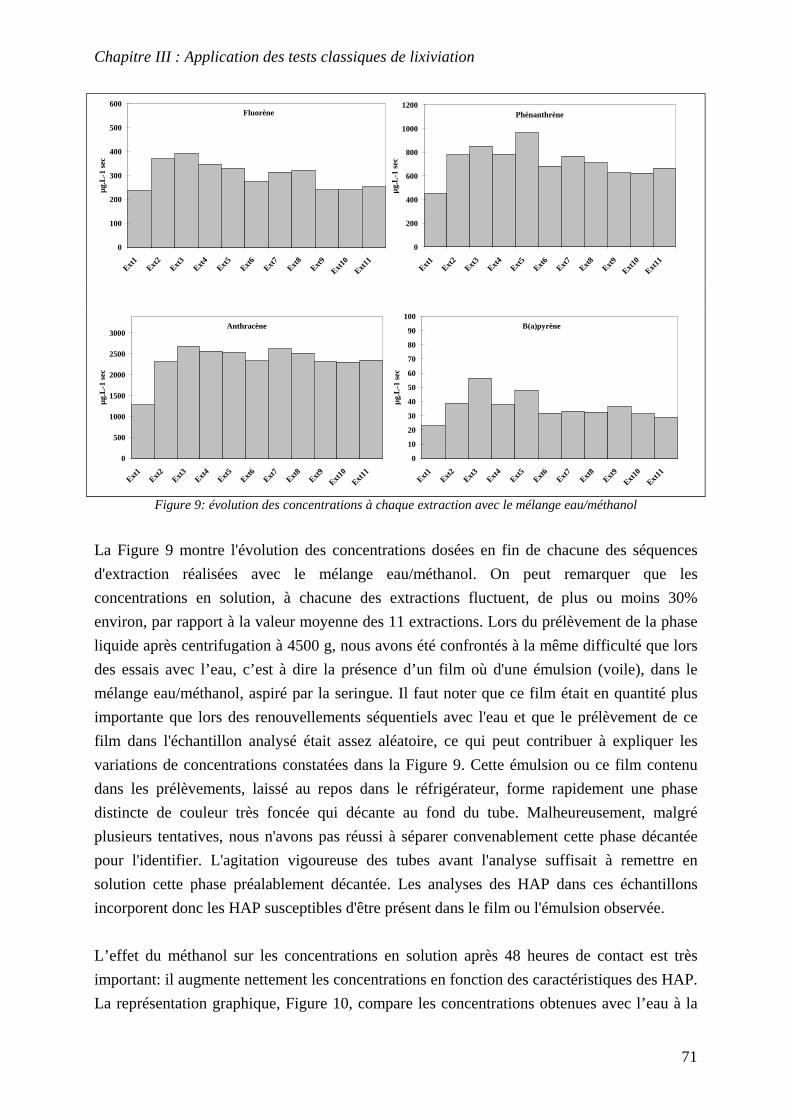
Chapitre III : Application des tests classiques de lixiviation
Fluorène
0
100
200
300
400
500
600
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
cPhénanthrène
0
200
400
600
800
1000
1200
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
Anthracène
0
500
1000
1500
2000
2500
3000
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
B(a)pyrène
0
10
20
30
40
50
60
70
80
90
100
Ext1 Ext2 Ext3 Ext4 Ext5 Ext6 Ext7 Ext8 Ext9Ext1
0Ext1
1
µg.L
-1 se
c
Figure 9: évolution des concentrations à chaque extraction avec le mélange eau/méthanol
La Figure 9 montre l'évolution des concentrations dosées en fin de chacune des séquences d'extraction réalisées avec le mélange eau/méthanol. On peut remarquer que les concentrations en solution, à chacune des extractions fluctuent, de plus ou moins 30% environ, par rapport à la valeur moyenne des 11 extractions. Lors du prélèvement de la phase liquide après centrifugation à 4500 g, nous avons été confrontés à la même difficulté que lors des essais avec l’eau, c’est à dire la présence d’un film où d'une émulsion (voile), dans le mélange eau/méthanol, aspiré par la seringue. Il faut noter que ce film était en quantité plus importante que lors des renouvellements séquentiels avec l'eau et que le prélèvement de ce film dans l'échantillon analysé était assez aléatoire, ce qui peut contribuer à expliquer les variations de concentrations constatées dans la Figure 9. Cette émulsion ou ce film contenu dans les prélèvements, laissé au repos dans le réfrigérateur, forme rapidement une phase distincte de couleur très foncée qui décante au fond du tube. Malheureusement, malgré plusieurs tentatives, nous n'avons pas réussi à séparer convenablement cette phase décantée pour l'identifier. L'agitation vigoureuse des tubes avant l'analyse suffisait à remettre en solution cette phase préalablement décantée. Les analyses des HAP dans ces échantillons incorporent donc les HAP susceptibles d'être présent dans le film ou l'émulsion observée. L’effet du méthanol sur les concentrations en solution après 48 heures de contact est très important: il augmente nettement les concentrations en fonction des caractéristiques des HAP. La représentation graphique, Figure 10, compare les concentrations obtenues avec l’eau à la
71
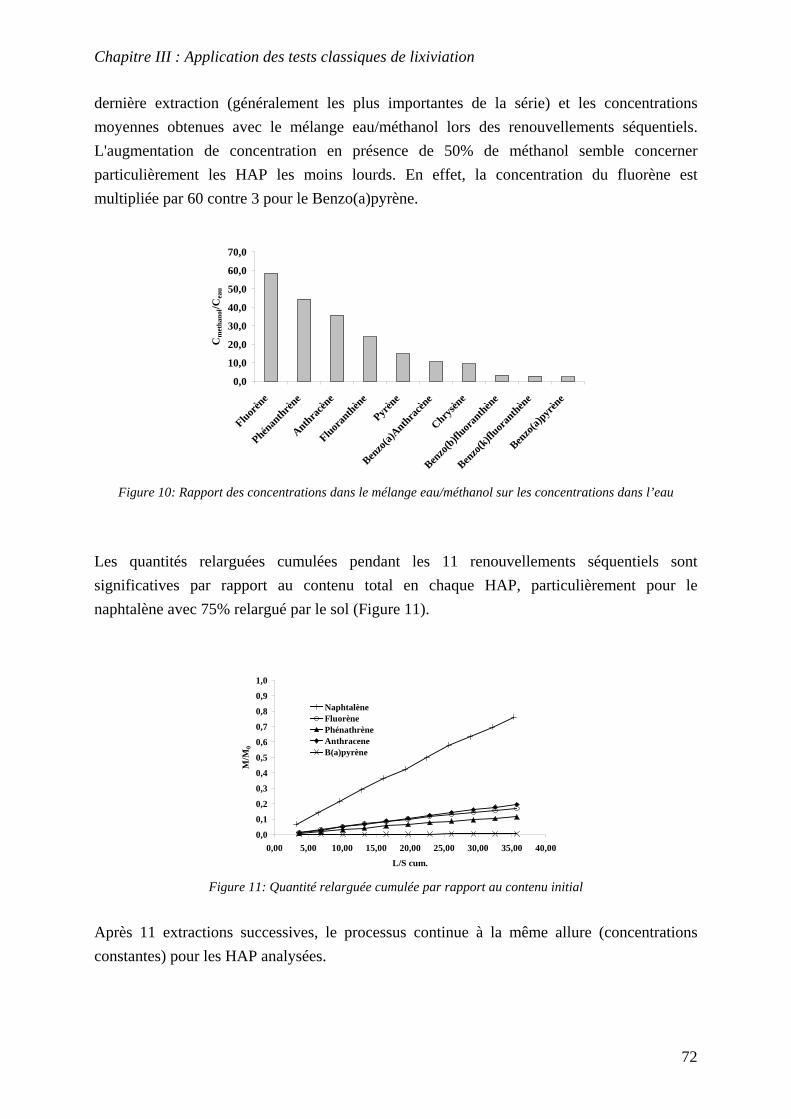
Chapitre III : Application des tests classiques de lixiviation
dernière extraction (généralement les plus importantes de la série) et les concentrations moyennes obtenues avec le mélange eau/méthanol lors des renouvellements séquentiels. L'augmentation de concentration en présence de 50% de méthanol semble concerner particulièrement les HAP les moins lourds. En effet, la concentration du fluorène est multipliée par 60 contre 3 pour le Benzo(a)pyrène.
0,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
Fluorèn
e
Phénan
thrène
Anthracèn
e
Fluoran
thène
Pyrèn
e
Benzo
(a)Anthra
cène
Chrysèn
e
Benzo
(b)fluora
nthène
Benzo(
k)fluor
anthèn
e
Benzo
(a)pyr
ène
Cm
etha
nol/C
eau
Figure 10: Rapport des concentrations dans le mélange eau/méthanol sur les concentrations dans l’eau
Les quantités relarguées cumulées pendant les 11 renouvellements séquentiels sont significatives par rapport au contenu total en chaque HAP, particulièrement pour le naphtalène avec 75% relargué par le sol (Figure 11).
0,0
0,10,2
0,3
0,4
0,5
0,60,7
0,8
0,9
1,0
0,00 5,00 10,00 15,00 20,00 25,00 30,00 35,00 40,00L/S cum.
M/M
0
NaphtalèneFluorènePhénathrèneAnthraceneB(a)pyrène
Figure 11: Quantité relarguée cumulée par rapport au contenu initial
Après 11 extractions successives, le processus continue à la même allure (concentrations constantes) pour les HAP analysées.
72

Chapitre III : Application des tests classiques de lixiviation
III.3 Percolation en colonne
III.3.1 Protocole
Le test de percolation en colonne en milieu saturé est utilisé ici pour comparer le comportement du sol par rapport aux essais de lixiviations en milieu dispersé (batch) L’intérêt du test vient du fait que :
la lixiviation est réalisée avec le sol en place, de la même façon à ce qui se passe in situ, le test est dynamique, l'échelle d'expérimentation est plus grande.
Le test appliqué correspond à la proposition de test de lixiviation en colonne en discussion pour la normalisation dans le cadre du WG6 du TC 292 du CEN et basé sur l’expérience de POLDEN dans le domaine.
Filtre en fibre deverre,1,2µm
Sol3 kg
Réservoird’eau
Pompe péristaltic
Réservoir
Figure 12: Principe de la percolation
La colonne est remplie avec 3 kg de sol. En haut et en bas de la colonne, un filtre en fibre de verre de diamètre égal à celui de la colonne est mis en contact avec le sol avant de refermer les extrémités. Un réservoir à percolat contient de l'eau permutée à laquelle on a ajouté de
73
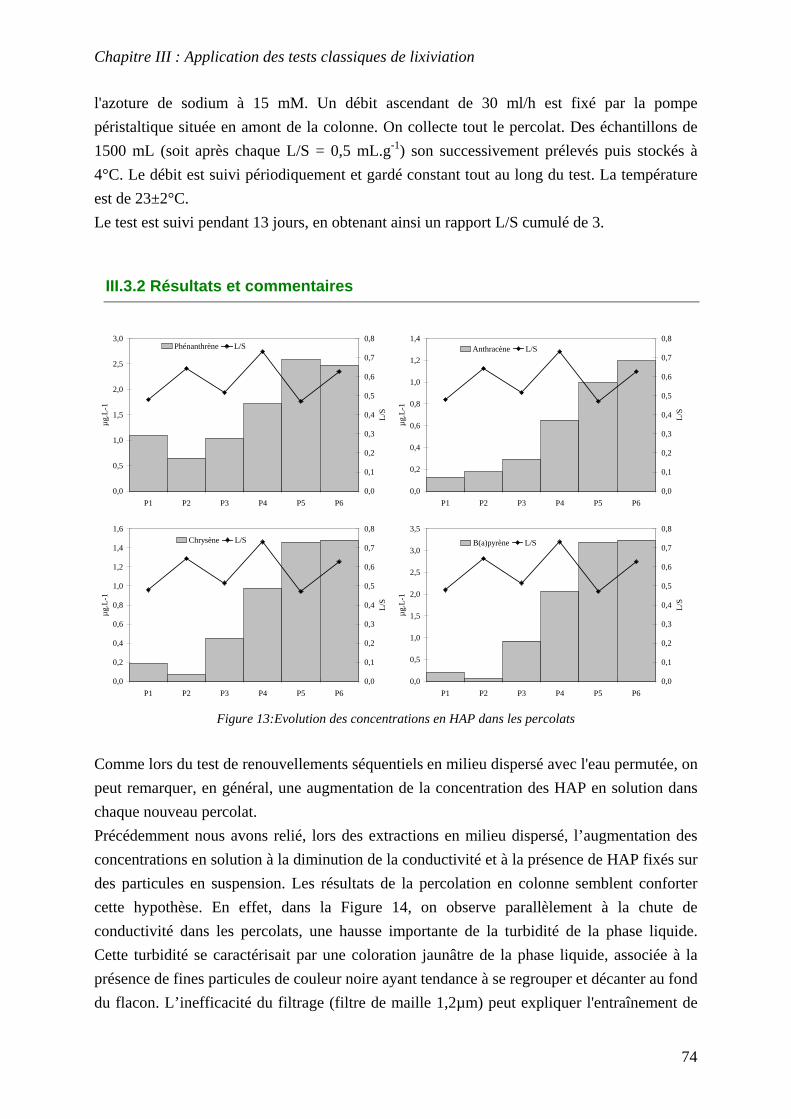
Chapitre III : Application des tests classiques de lixiviation
l'azoture de sodium à 15 mM. Un débit ascendant de 30 ml/h est fixé par la pompe péristaltique située en amont de la colonne. On collecte tout le percolat. Des échantillons de 1500 mL (soit après chaque L/S = 0,5 mL.g-1) son successivement prélevés puis stockés à 4°C. Le débit est suivi périodiquement et gardé constant tout au long du test. La température est de 23±2°C. Le test est suivi pendant 13 jours, en obtenant ainsi un rapport L/S cumulé de 3.
III.3.2 Résultats et commentaires
0,0
0,5
1,0
1,5
2,0
2,5
3,0
P1 P2 P3 P4 P5 P6
µg.L
-1
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
L/S
Phénanthrène L/S
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
P1 P2 P3 P4 P5 P6
µg.L
-1
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
L/S
Anthracène L/S
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
P1 P2 P3 P4 P5 P6
µg.L
-1
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
L/S
Chrysène L/S
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
P1 P2 P3 P4 P5 P6
µg.L
-1
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
L/S
B(a)pyrène L/S
Figure 13:Evolution des concentrations en HAP dans les percolats
Comme lors du test de renouvellements séquentiels en milieu dispersé avec l'eau permutée, on peut remarquer, en général, une augmentation de la concentration des HAP en solution dans chaque nouveau percolat. Précédemment nous avons relié, lors des extractions en milieu dispersé, l’augmentation des concentrations en solution à la diminution de la conductivité et à la présence de HAP fixés sur des particules en suspension. Les résultats de la percolation en colonne semblent conforter cette hypothèse. En effet, dans la Figure 14, on observe parallèlement à la chute de conductivité dans les percolats, une hausse importante de la turbidité de la phase liquide. Cette turbidité se caractérisait par une coloration jaunâtre de la phase liquide, associée à la présence de fines particules de couleur noire ayant tendance à se regrouper et décanter au fond du flacon. L’inefficacité du filtrage (filtre de maille 1,2µm) peut expliquer l'entraînement de
74
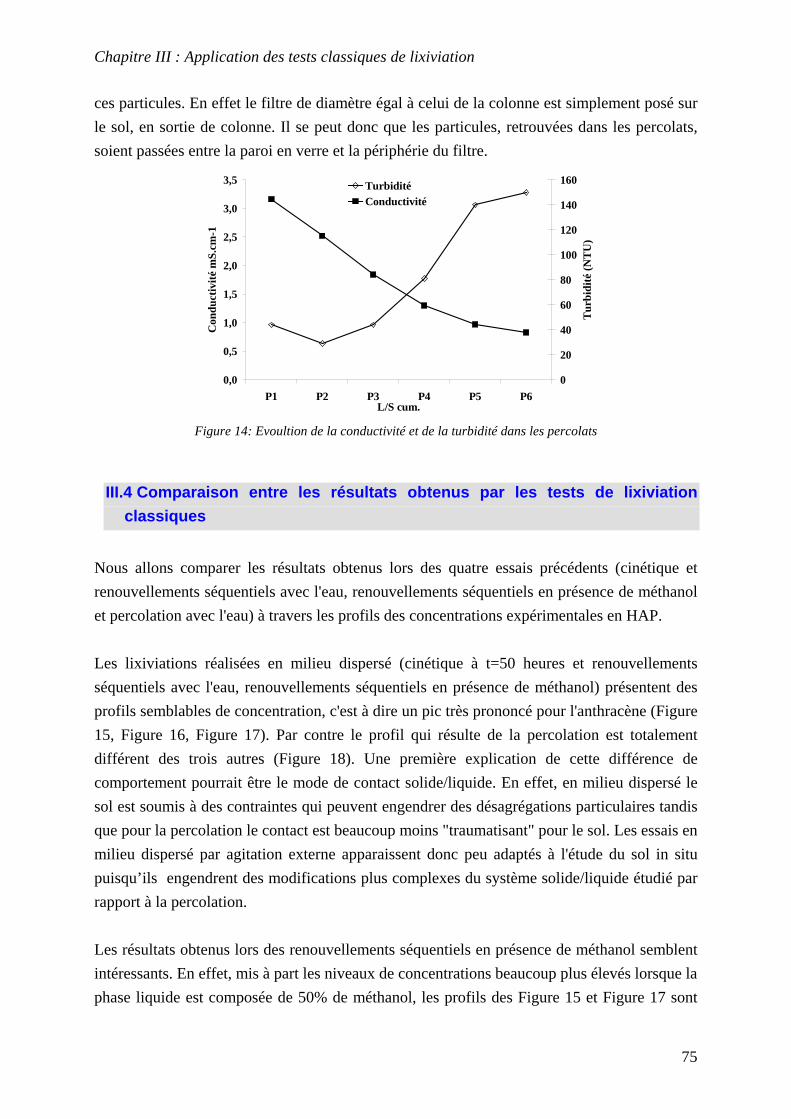
Chapitre III : Application des tests classiques de lixiviation
ces particules. En effet le filtre de diamètre égal à celui de la colonne est simplement posé sur le sol, en sortie de colonne. Il se peut donc que les particules, retrouvées dans les percolats, soient passées entre la paroi en verre et la périphérie du filtre.
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
P1 P2 P3 P4 P5 P6L/S cum.
Con
duct
ivité
mS.
cm-1
0
20
40
60
80
100
120
140
160
Tur
bidi
té (N
TU
)
TurbiditéConductivité
Figure 14: Evoultion de la conductivité et de la turbidité dans les percolats
III.4 Comparaison entre les résultats obtenus par les tests de lixiviation classiques
Nous allons comparer les résultats obtenus lors des quatre essais précédents (cinétique et renouvellements séquentiels avec l'eau, renouvellements séquentiels en présence de méthanol et percolation avec l'eau) à travers les profils des concentrations expérimentales en HAP. Les lixiviations réalisées en milieu dispersé (cinétique à t=50 heures et renouvellements séquentiels avec l'eau, renouvellements séquentiels en présence de méthanol) présentent des profils semblables de concentration, c'est à dire un pic très prononcé pour l'anthracène (Figure 15, Figure 16, Figure 17). Par contre le profil qui résulte de la percolation est totalement différent des trois autres (Figure 18). Une première explication de cette différence de comportement pourrait être le mode de contact solide/liquide. En effet, en milieu dispersé le sol est soumis à des contraintes qui peuvent engendrer des désagrégations particulaires tandis que pour la percolation le contact est beaucoup moins "traumatisant" pour le sol. Les essais en milieu dispersé par agitation externe apparaissent donc peu adaptés à l'étude du sol in situ puisqu’ils engendrent des modifications plus complexes du système solide/liquide étudié par rapport à la percolation. Les résultats obtenus lors des renouvellements séquentiels en présence de méthanol semblent intéressants. En effet, mis à part les niveaux de concentrations beaucoup plus élevés lorsque la phase liquide est composée de 50% de méthanol, les profils des Figure 15 et Figure 17 sont
75
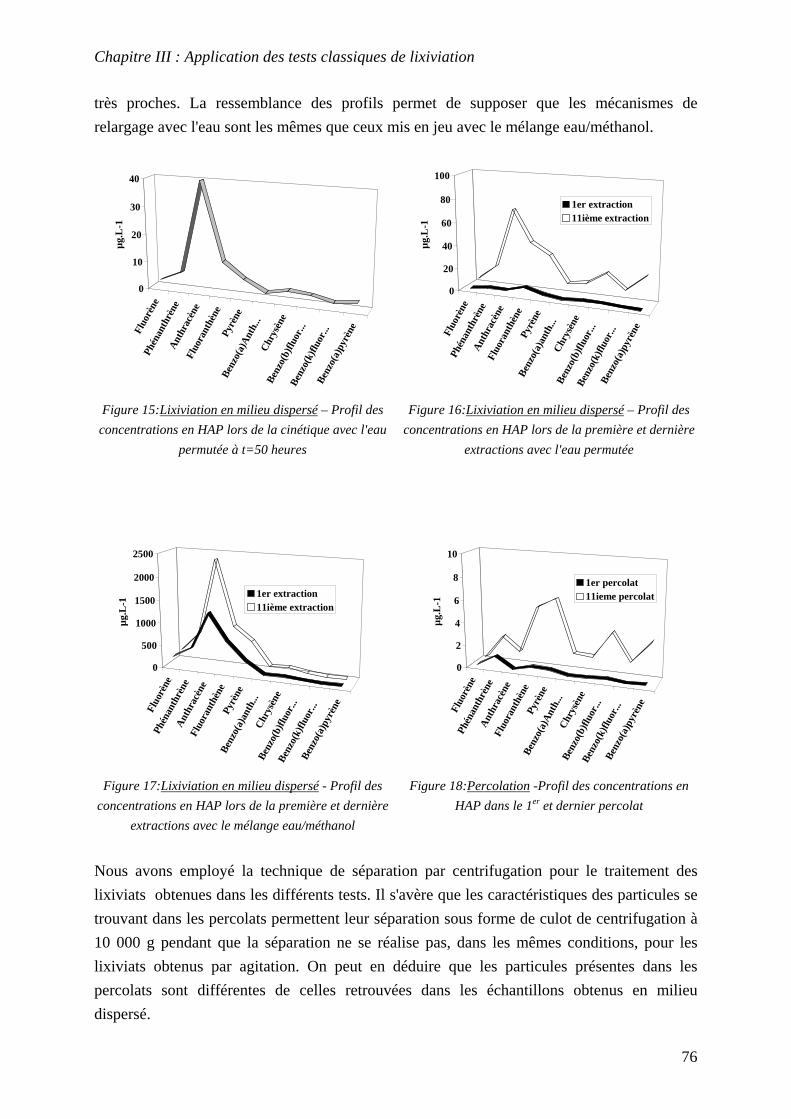
Chapitre III : Application des tests classiques de lixiviation
très proches. La ressemblance des profils permet de supposer que les mécanismes de relargage avec l'eau sont les mêmes que ceux mis en jeu avec le mélange eau/méthanol.
Fluo
rène
Phén
anth
rène
Anth
racè
neFl
uora
nthè
nePy
rène
Benz
o(a)
Anth
...Ch
rysè
neBe
nzo(
b)flu
or...
Benz
o(k)
fluor
...Be
nzo(
a)py
rène
0
10
20
30
40
µg.L
-1
Fluo
rène
Phén
anth
rène
Anth
racè
neFl
uora
nthè
nePy
rène
Benz
o(a)
anth
...Ch
rysè
neBe
nzo(
b)flu
or...
Benz
o(k)
fluor
...Be
nzo(
a)py
rène
0
20
40
60
80
100
µg.L
-1
1er extraction11ième extraction
Figure 15:Lixiviation en milieu dispersé – Profil des concentrations en HAP lors de la cinétique avec l'eau
permutée à t=50 heures
Figure 16:Lixiviation en milieu dispersé – Profil des concentrations en HAP lors de la première et dernière
extractions avec l'eau permutée
Fluo
rène
Phén
anth
rène
Anth
racè
neFl
uora
nthè
nePy
rène
Benz
o(a)
anth
...Ch
rysè
neBe
nzo(
b)flu
or...
Benz
o(k)
fluor
...Be
nzo(
a)py
rène
0
500
1000
1500
2000
2500
µg.L
-1
1er extraction11ième extraction
Fluo
rène
Phén
anth
rène
Anth
racè
neFl
uora
nthè
nePy
rène
Benz
o(a)
Anth
...Ch
rysè
neBe
nzo(
b)flu
or...
Benz
o(k)
fluor
...Be
nzo(
a)py
rène
0
2
4
6
8
10
µg.L
-1
1er percolat11ieme percolat
Figure 17:Lixiviation en milieu dispersé - Profil des
concentrations en HAP lors de la première et dernière extractions avec le mélange eau/méthanol
Figure 18:Percolation -Profil des concentrations en HAP dans le 1er et dernier percolat
Nous avons employé la technique de séparation par centrifugation pour le traitement des lixiviats obtenues dans les différents tests. Il s'avère que les caractéristiques des particules se trouvant dans les percolats permettent leur séparation sous forme de culot de centrifugation à 10 000 g pendant que la séparation ne se réalise pas, dans les mêmes conditions, pour les lixiviats obtenus par agitation. On peut en déduire que les particules présentes dans les percolats sont différentes de celles retrouvées dans les échantillons obtenus en milieu dispersé.
76

Chapitre III : Application des tests classiques de lixiviation
En conclusion, les essais de lixiviation pour étudier le sol doivent se faire par des contacts solides/liquides de type percolation, pour limiter les conséquences de l’agitation externe sur l’intégrité structurale du sol.
III.5 Conclusions
L’application des essais traditionnels de lixiviation a révélé la grande complexité du sol du site étudié et de nombreuses difficultés expérimentales et d'interprétation. La partie mobile de la pollution semble se répartir entre une phase dissoute et une phase particulaire fine. Globalement, nous avons observé une augmentation des concentrations en HAP en solution lors des renouvellements séquentiels avec l'eau et de la percolation en colonne. Cette augmentation des concentrations a été reliée à deux phénomènes: la hausse de la solubilité des HAP et l'augmentation du nombre de particules en suspension. C'est deux phénomènes semblerait être reliés à la baisse d'espèces ioniques en solution, constatée par la mesure de la conductivité. Le comportement du sol vis à vis du relargage est différent si le contact sol/eau est réalisé en milieu maintenu dispersé par agitation externe ou s'il est réalisé par percolation du sol en place. Cette différence de comportement probablement induite par la désagrégation particulaire rend les tests en milieu dispersé par agitation externe, peu adaptée aux études du comportement in situ de ce type de sol. Le mode de contact obtenu en colonne semble plus approprié, mais l'amélioration du protocole de ce test est nécessaire. L’essai de renouvellements séquentiels en milieu dispersé effectué avec le mélange eau/méthanol a montré un grand intérêt puisque:
les concentrations en HAP sont bien supérieures, augmentant ainsi la précision des dosages,
les techniques préparatoires avant analyse sont simplifiées, il semblerait que seul l'intensité du relargage change par rapport à l'essai avec l'eau
permutée. Au cas où les mécanismes de relargage seraient similaires, il pourrait y avoir des perspectives intéressantes pour les études sur la mobilité des HAP.
77

Chapitre IV : Conception d’un outil expérimental
Chapitre IV: Conception d’un outil expérimental pour l’étude de la mobilité des polluants organiques hydrophobes (HAP) dans des milieux poreux de type sol Comme nous l'avons montré dans le chapitre précédent, les essais réalisés par l’application de tests « traditionnels », à savoir lixiviation en milieu maintenu dispersé par agitation externe et percolation ascendante en colonne sur le sol en place, n'ont pas permis d'arriver à des résultats concluants concernant l’observation de la mobilité des HAP. Nous avons ainsi pu montrer les limites de ces tests classiques de lixiviation et formuler quelques hypothèses sur les mécanismes contribuant à la mobilité des HAP. A partir de ces informations, nous nous sommes fixés, dans un cahier des charges, les objectifs à atteindre par un nouveau test de lixiviation et les solutions techniques correspondantes. L’objectif est de mettre au point un outil de lixiviation performant et facile à mettre en œuvre pour étudier la mobilité des polluants organiques hydrophobes (HAP) contenus dans les sols des sites industriels.
IV.1 Objectifs et solutions techniques proposées dans le cahier des charges
IV.1.1 Mode de contact solide/liquide
Il a été démontré dans le chapitre précédent que le principe de la percolation était mieux adapté que le contact en milieu dispersé agité puisque ce dernier provoque une désagrégation particulaire qui modifie la structure du sol et modifie son comportement par rapport au scénario environnemental considéré. Le contact entre le sol et le liquide lors de la percolation résulte du passage de la phase liquide, à un débit donné, à travers une quantité de sol compactée dans une colonne en verre. Le sol se trouve saturé en liquide. La circulation de la phase liquide à travers le sol se fait verticalement du bas vers le haut pour éviter au maximum la formation de chemins préférentiels. Le test de lixiviation devra donc respecter ce mode de contact solide/liquide. Ce mode de contact sera appelé dans la suite du document contact solide/liquide en milieu compacté par opposition au milieu maintenu dispersé par agitation externe.
IV.1.2 Contrôle du temps de contact solide/liquide
Le contrôle du temps de contact est nécessaire pour étudier les aspects "cinétiques" de la mobilité, à savoir la vitesse de réalisation des processus de solubilisation: désorption et dissolution des HAP dans la phase liquide. C'est habituellement le test en milieu dispersé (réacteur fermé parfaitement agité) qui permet de réaliser ce type d'étude, car on considère en
78

Chapitre IV : Conception d’un outil expérimental
général les mécanismes mis en jeu suffisamment lents pour être observables dans ce type d'expérimentation. En conséquence, en milieu dispersé, il suffit de stopper l'agitation à différent temps et de prélever un échantillon de la phase liquide (si la modification de volume de phase liquide est considérée négligeable), de l'analyser immédiatement pour obtenir les informations nécessaires à la caractérisation de la cinétique de relargage. Une autre solution expérimentale consiste en une série d’expérimentations en parallèle avec des durées de contact différentes. Cependant, nous avons écarté le contact solide/liquide en milieu dispersé car il modifie les caractéristiques du sol et interfère avec les mécanismes de la mobilité. Il fallait donc concevoir un dispositif expérimental qui permettrait d'obtenir des informations sur la cinétique tout en ayant un contact solide/liquide en milieu compacté. Ce type de contacteur doit avoir un fonctionnement identique ou proche d'un réacteur parfaitement agité, le contacteur solide/liquide le mieux adapté en génie chimique pour l’étude expérimentale de ce type de problème. La solution proposée est de faire circuler en boucle la phase liquide à travers le sol compacté et saturé, de bas en haut. Le volume de phase liquide utilisé est supérieur au volume poreux et au volume des tuyaux. La phase liquide est stockée dans un réacteur tampon, c'est dans ce compartiment que s'effectueront les prélèvements. Pour avoir une phase liquide homogène, un agitateur magnétique est placé dans le réservoir tampon. L'entraînement de la phase liquide se fait à l'aide d'une pompe péristaltique.
Agitateur magnétique
Pompe péristaltique
Contactsolide/liquide enmilieu compacté
Réacteur tampon
Figure 1: Principe de fonctionnement du dispositif expérimental
Le test expérimental proposé doit être polyvalent afin de mener les différentes études (cinétiques, renouvellements séquentiels, et percolations) à partir du même dispositif expérimental. Le fait de réaliser l'ensemble des essais à partir d'un même dispositif expérimental permet de comparer facilement les résultats des études cinétiques, renouvellements séquentiels et percolations puisqu'ils sont obtenus pour des conditions de contact solide/liquide identiques. Le dispositif choisi (Figure 1) est donc adapté puisque
79

Chapitre IV : Conception d’un outil expérimental
L’étude cinétique est réalisable en prélevant à différents temps des échantillons dans le réacteur tampon. Il aurait été préférable de pouvoir brancher un deuxième circuit (en boucle fermé avec des petits volumes) pour pouvoir mesurer en continu les variations des caractéristiques du percolat. Mais nous ne disposions malheureusement pas de ce type de système de dosage en continu de HAP. En conséquence, nous avons choisi de prélever à différents temps des échantillons de la phase liquide.
Les renouvellements séquentiels sont faisables en remplaçant la totalité de la phase
liquide par une nouvelle phase liquide. Il faudra bien sûr tenir compte du volume résiduel présent dans le sol et la tuyauterie (volume résiduel qu'il faudra limiter au maximum).
Pour réaliser une lixiviation par percolation, il suffit de récupérer la phase liquide en
sortie de colonne sans la recirculer.
IV.1.3 Dispositifs de séparation solide/liquide
Pour maintenir le sol en place dans la colonne et pour assurer la distribution uniforme du liquide, notamment en entrée, nous avons ajouté en entrée et en sortie de colonne un système de filtration. Cette filtration pourra être effectuée selon les objectifs, soit au travers d'un lit de sable de granulométrie définie, soit au travers d'une membrane en polypropylène de maille 10 µm. Des essais préalables ont montré, pour le sol étudié, que ces deux méthodes de filtration permettent de laisser circuler les particules fines à travers le dispositif de la Figure 1. Le dispositif expérimental doit permettre d'étudier séparément la mobilité des HAP sous formes dissoutes et la mobilité des HAP sous formes particulaires. Dans le chapitre précédent, des essais de centrifugation ont montré que les particules mises en suspension lors de la percolation pouvaient être décantées à 10 000 g dans des tubes en Teflon. L'analyse des échantillons non centrifugés donne la mobilité globale (forme dissoute et particulaire) des HAP. La mobilité de la fraction dissoute peut être étudiée par analyse de la phase liquide après centrifugation des échantillons à 10 000 g. Ensuite la mobilité des HAP sous forme particulaire peut être déduite des résultats de la mobilité globale (échantillons non centrifugés) et de la mobilité des HAP dissous (échantillons centrifugés).
IV.1.4 Contrôle de la température
La solubilité des HAP est fortement sensible aux variations de température [REZA, 2002], le dispositif expérimental proposé devait donc posséder un système pour maintenir la température à une valeur donnée. Différentes solutions étaient envisageables. La solution adoptée a consisté à placer le réacteur tampon et la colonne contenant le sol dans un bain
80

Chapitre IV : Conception d’un outil expérimental
thermostaté (Figure 2) et à constituer une protection thermique sur l'ensemble des tuyaux du dispositif.
Colonne
Réacteurtampon
Eauthermostatée
Figure 2: Principe du maintien de la température dans la colonne
et dans le réacteur tampon
IV.1.5 Protection de la dégradation des HAP par la lumière
Pour éviter un risque de dégradation des HAP par la lumière chaque partie du dispositif doit être protégée de la lumière à l'aide de feuilles d'aluminium.
IV.1.6 Phase initiale
Pour limiter les problèmes occasionnés par l'adsorption des HAP à la surface des matériaux, préalablement à chaque essai, nous avons choisi de réaliser une recirculation de la phase liquide au travers du sol compacté pendant 24 heures pour assurer des conditions initiales stables et saturer les différents matériaux susceptibles d'adsorber des HAP [JAYR, 2002]. La totalité de la phase liquide est alors analysée tandis que le sol reste en place dans la colonne. On connaît ainsi les conditions initiales du système mis en place. Cette première opération commune à tous les essais permet aussi le remplissage total de la porosité connectée par la phase liquide, ainsi qu'une mise à l'équilibre des écoulements.
Tableau 1: Synthèse du cahier des charges
Objectifs Solutions proposées
Contact solide/liquide Contact en milieu compacté
Contrôle du temps de contact Recirculation de la phase liquide à
travers le sol compacté
Séparation Liquide/solide Filtration sur sable ou membrane
Contrôle de la température Bain thermostaté
Protection contre la lumière Feuille d'aluminium sur les parties
exposées Limitation des pertes par adsorption à
la surface des matériaux Recirculation préalable
81

Chapitre IV : Conception d’un outil expérimental
IV.2 Mise en œuvre du test
Le test proposé que nous avons appelé « batch percolant » est un compromis entre le batch en milieu dispersé et la percolation en colonne. Le dispositif peut être assimilé à un « milieu fermé » puisqu'il n'y a pas d'entrées et pas de sorties pendant la durée du contact, d'où le terme batch. L’agitation est le résultat de la recirculation de la phase liquide dans le système. On emploi le terme percolant car le mode de contact solide/liquide retenu (contact en milieu compacté) est équivalent à celui d'une percolation. Principaux éléments composant le dispositif : • Colonne en verre (hauteur=4,5-15 cm et diamètre=2.6 cm) • Pompe péristaltique • Bain thermostaté • Agitateur magnétique • Tuyaux en Teflon
Systemes de contrôle de latemperature
Agitateur magnétique
Pompe péristaltique
Sol compacté
Filtration
Réacteur tampon àl’abri de la lumière
Figure 3 Schéma global de fon ctionnement
IV.2.1 Mise en place du sol
Le sol est mis en place manuellement dans la colonne. Afin de réaliser un compactage reproductible, une procédure de remplissage a été mise au point. Des portions de 5g sont successivement introduites dans la colonne. Entre chaque remplissage, le sol est compacté à l'aide d'un outil lesté à 100 g. Le compactage consiste à laisser tomber, 3 fois de suite, la masse de 100 g, d'une hauteur de 10 cm (Figure 4), sur toute la surface du sol.
82

Chapitre IV : Conception d’un outil expérimental
100 g 10 cmMasse de 100 g
Sol non compacté
Sol compacté
Colonne en verre
Figure 4: Procédure de remplissage de la colonne
IV.2.2 Mise en route
La procédure de mise en route du dispositif est commune aux diverses expérimentations. Elle permet le remplissage de la porosité ouverte par la phase liquide et une mise à l’équilibre de l’écoulement. Un volume (V) de phase liquide présent à t=0 dans le réacteur tampon est mis en circulation dans le système avec un débit constant (Q) par l’intermédiaire de la pompe péristaltique. Le dispositif expérimental est maintenu à une température (T) constante pendant toute la durée de l’essai. Après 24 heures de fonctionnement la totalité de la phase liquide, contenue dans le réacteur tampon, est retirée du réacteur, puis analysée. Le sol reste en place dans la colonne, ainsi que la phase liquide contenue dans les tuyau (Vt). Connaissant le volume de liquide contenu dans les tuyaux (diamètre et longueur), on peut estimer le volume poreux (Vp)du sol accessible par le liquide:
(1) Vp=Vinitial – Vt - Vrécolté
Une nouvelle phase liquide est mise dans le réacteur tampon puis le réacteur tampon est refermé. A partir de ce moment on peut réaliser soit une étude cinétique de mise en solution, soit des renouvellements séquentiels, soit une percolation avec renouvellement continu.
IV.2.3 Paramètres du batch percolant
On peut intervenir sur différentes variables du batch percolant afin d'étudier l'effet d'un paramètre spécifique sur la mobilité des HAP. En effet, chacun des paramètres présentés dans le Tableau 2 peut être considéré indépendant des autres, il est donc possible de faire varier un ou plusieurs paramètres tout en conservant les autres.
83
Chapitre IV : Conception d’un outil expérimental
Tableau 2: Liste des paramètres variables dans le batch percolant
Paramètres Symboles Débit Q
Rapport liquide/solide (ou volume éluat/volume poreux)
L/S (V/V0 )
Pourcentage de méthanol dans la phase liquide
fc
Température T°C Nature de la phase liquide -
Temps de contact t
IV.3 Détermination de la Distribution des Temps de Séjour (DTS)
Avant d'exploiter le batch percolant pour étudier la mobilité des HAP dans notre sol, il est nécessaire de caractériser l'écoulement dans la colonne. Cette caractérisation se fait grâce à la détermination de la Distribution du Temps de Séjour (DTS). La détermination de la DTS d’un traceur dans la colonne de percolation a comme premier objectif l’identification du régime hydrodynamique de la percolation et par la suite le choix du modèle le plus adapté à la simulation de l’écoulement. Selon la méthodologie traditionnelle, nous avons pratiqué à l’entrée de la colonne, sur le débit d’alimentation, une perturbation de type « échelon positif » (signal de Heaviside). La colonne est alimentée à débit constant (30,1 ml/h) de liquide. Pendant la première période le liquide est l’eau déminéralisée. Brusquement, à partir du temps t0, la colonne est alimentée en solution de NaCl à concentration constante en Cl- (385 mg/l). Pratiquement, l’opération se réalise par changement de réservoir d’alimentation. Elle ne modifie pas le régime d’écoulement dans la colonne. L'ion Cl- a été choisi pour ces propriétés de traceur d’écoulement pour le type de matériau étudié : il n’interfère et ne modifie pas les propriétés du lit percolé. Pour faire le « bilan de population », à savoir la restitution en sortie de la colonne du changement opéré à son entrée, nous avons prélevé périodiquement (tout les 3-5 minutes) le percolat pour dosage par chromatographie ionique des ions Cl-. Nous obtenons ainsi l’évolution échantillonnée de la concentration normalisée de Cl- dans le percolat, distribution « cumulée » notée F(t). A partir de celle-ci nous avons calculé: - La distribution des temps de séjour E(ts):
(2) tdtFdtE s)()( =
84
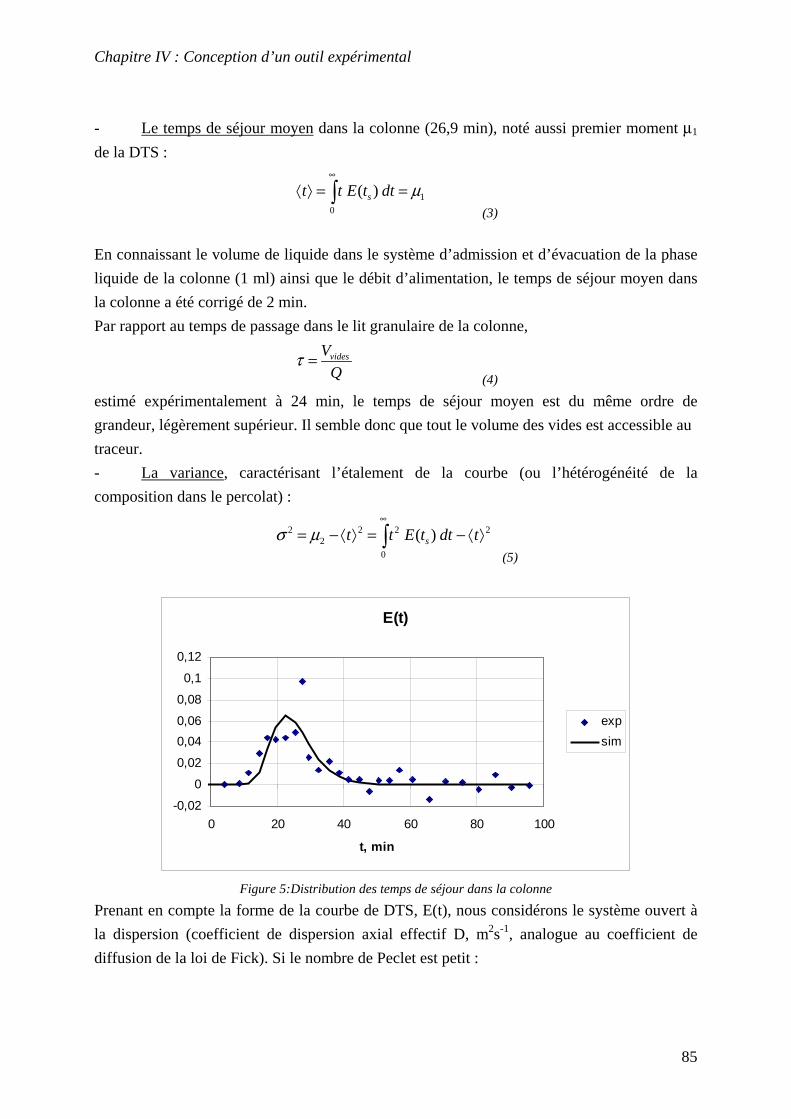
Chapitre IV : Conception d’un outil expérimental
- Le temps de séjour moyen dans la colonne (26,9 min), noté aussi premier moment µ1 de la DTS :
10
)( µ==⟩⟨ ∫∞
dttEtt s
(3)
En connaissant le volume de liquide dans le système d’admission et d’évacuation de la phase liquide de la colonne (1 ml) ainsi que le débit d’alimentation, le temps de séjour moyen dans la colonne a été corrigé de 2 min. Par rapport au temps de passage dans le lit granulaire de la colonne,
QVvides=τ
(4)
estimé expérimentalement à 24 min, le temps de séjour moyen est du même ordre de grandeur, légèrement supérieur. Il semble donc que tout le volume des vides est accessible au traceur. - La variance, caractérisant l’étalement de la courbe (ou l’hétérogénéité de la composition dans le percolat) :
2
0
222
2 )( ⟩⟨−=⟩⟨−= ∫∞
tdttEtt sµσ(5)
E(t)
-0,02
0
0,02
0,040,06
0,08
0,1
0,12
0 20 40 60 80 100
t, min
expsim
Figure 5:Distribution des temps de séjour dans la colonne
Prenant en compte la forme de la courbe de DTS, E(t), nous considérons le système ouvert à la dispersion (coefficient de dispersion axial effectif D, m2s-1, analogue au coefficient de diffusion de la loi de Fick). Si le nombre de Peclet est petit :
85

Chapitre IV : Conception d’un outil expérimental
(6) 50⟨=
DLuPe
où u (m s-1) est la vitesse du fluide et L (m) la longueur une solution analytique est donnée dans la littérature (Villermaux, 1993) qui permet le calcul de la distribution théorique des temps de séjour (simulation, figure 5) et respectivement, par intégration, la courbe F(t) :
(7)
( )⎥⎦
⎤⎢⎣
⎡ −−=−
τπτ
τπ 4exp
21)(
221 tPetPetE
Le nombre de Pe a été estimé à partir de l’expression des moments:
(8)
⎥⎦⎤
⎢⎣⎡ +=⟩⟨
Pet 21τ
(9) ⎥⎦⎤
⎢⎣⎡ += 2
22 82PePe
τσ
Dans notre cas, Pe=10 ce qui signifie une dispersion significative, l’écoulement s’approche du modèle « agitation parfaite ». L’écoulement peut être représenté par une série de « j » réacteurs unitaires parfaitement agités de volumes égaux et de volume total V, celui de la phase liquide contenue dans le lit percolé. L’équation du bilan de traceur donne pour chacun des j réacteurs de la série en régime permanent :
jssjee
j
cQcQdt
cj
Vd
SortieonConsommatiCréationEntréeonAccumulati
,, 00 −−+=⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛
−−+=
(10)
Pour la résolution du système de j équations différentielles nous prenons en compte l’hypothèse du volume et du débit constant ; les concentrations d’entrée dans le premier et respectivement de sortie dans le dernier de la série de j réacteurs sont connues. On utilise les résultats expérimentaux obtenus précédemment. Le paramètre j est à déterminer par recherche inverse, le critère étant l’adéquation entre les résultats expérimentaux en sortie de colonne et la simulation de la concentration du traceur. Le meilleur résultat est obtenu pour j=4 ; nous convenons d’assimiler l’écoulement dans la
86
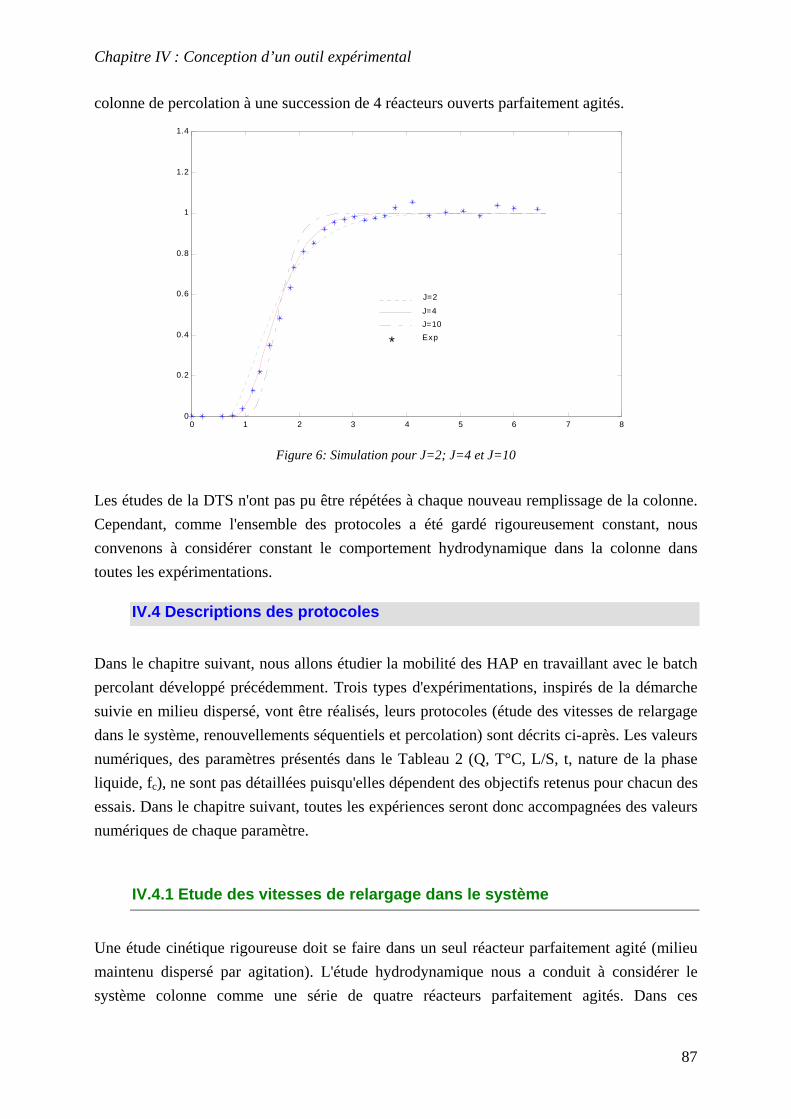
Chapitre IV : Conception d’un outil expérimental
colonne de percolation à une succession de 4 réacteurs ouverts parfaitement agités.
0 1 2 3 4 5 6 7 80
0.2
0.4
0.6
0.8
1
1.2
1.4
J=2
J=4 J=10
* Exp
Figure 6: Simulation pour J=2; J=4 et J=10
Les études de la DTS n'ont pas pu être répétées à chaque nouveau remplissage de la colonne. Cependant, comme l'ensemble des protocoles a été gardé rigoureusement constant, nous convenons à considérer constant le comportement hydrodynamique dans la colonne dans toutes les expérimentations.
IV.4 Descriptions des protocoles
Dans le chapitre suivant, nous allons étudier la mobilité des HAP en travaillant avec le batch percolant développé précédemment. Trois types d'expérimentations, inspirés de la démarche suivie en milieu dispersé, vont être réalisés, leurs protocoles (étude des vitesses de relargage dans le système, renouvellements séquentiels et percolation) sont décrits ci-après. Les valeurs numériques, des paramètres présentés dans le Tableau 2 (Q, T°C, L/S, t, nature de la phase liquide, fc), ne sont pas détaillées puisqu'elles dépendent des objectifs retenus pour chacun des essais. Dans le chapitre suivant, toutes les expériences seront donc accompagnées des valeurs numériques de chaque paramètre.
IV.4.1 Etude des vitesses de relargage dans le système
Une étude cinétique rigoureuse doit se faire dans un seul réacteur parfaitement agité (milieu maintenu dispersé par agitation). L'étude hydrodynamique nous a conduit à considérer le système colonne comme une série de quatre réacteurs parfaitement agités. Dans ces
87

Chapitre IV : Conception d’un outil expérimental
conditions, nous avons préféré parler de l'étude des vitesses de relargage dans le système plutôt que de cinétiques de relargage. Chaque étude de la vitesse de relargage dans le système doit être réalisée après la phase initiale de mise en route décrite précédemment. Cette phase initiale est effectuée pour assurer un remplissage total de la porosité par la phase liquide et une mise à l’équilibre de l’écoulement. Elle permet aussi de déterminer les caractéristiques initiales du système (concentrations dans l'eau résiduelle) nécessaires lors de la modélisation des phénomènes. La phase initiale de mise en route est réalisée avec la phase liquide et la température souhaitée pour la suite de l'étude des vitesses de relargage. Après la phase initiale de mise en route, une nouvelle phase liquide est placée dans le réacteur tampon, puis le liquide est mis en recirculation pour étudier les vitesses de relargage. La composition initiale de la nouvelle phase liquide correspond en réalité à une dilution du volume résiduel resté dans le système après la mise en route. Le bilan de matière doit donc prendre en compte cet effet de dilution. Nous avions deux possibilités pour étudier les vitesses de relargage:
Prélèvements successifs dans un même système solide/liquide: Utilisé lorsque le volume nécessaire pour l’analyse est faible, il est le plus facile à mettre en œuvre. Cette méthode consiste à prélever à différents temps dans le réacteur tampon en fonctionnement un volume suffisant pour doser la concentration en solution. La somme des volumes prélevés ne doit pas représenter plus de 10% du volume initial présent dans le réacteur tampon pour ne pas modifier de façon importante le rapport L/S. On est donc limité par le nombre de prélèvements. Cette méthode à été employée lorsque la phase liquide était constituée d’au moins 30% de méthanol car dans ce cas l’analyse directe de la solution par HPLC était possible sans passer par une phase de concentration de la solution à analyser.
Prélèvements en parallèle dans différents systèmes solide/liquide: Cette méthode est
utilisée lorsque le volume nécessaire à l’analyse est important. C’est le cas pour les phases liquides aqueuses puisqu’il est nécessaire de préparer l’échantillon avant analyse. Des prélèvements successifs effectués selon la méthode précédente engendreraient des modifications trop importantes du rapport L/S. Chaque prélèvement est donc associé à un dispositif expérimental. La pompe péristaltique utilisée permettait de lancer 4 essais simultanément (pompe à 4 têtes), ces études ont été limitées à 4 points afin que les réacteurs fonctionnent dans des conditions expérimentales proches, en particulier le débit. La fiabilité des résultats dépend, en priorité, de l'homogénéité de l'échantillon de sol utilisé pour la constitution des colonnes et de la reproductibilité du compactage des quatre colonnes.
88

Chapitre IV : Conception d’un outil expérimental
Les prélèvements sont effectués avec une seringue en verre directement dans le réacteur tampon. Un volume de 1 mL est retiré du réacteur tampon dans la méthode des prélèvements successifs dans un même système solide/liquide, ce qui permet de faire plusieurs mesures sans modifier significativement le rapport L/S initial. Tandis que 20 mL sont retirés de chaque réacteur tampon dans la méthode des prélèvements en parallèle dans différents systèmes solide/liquide. Les échantillons prélevés sont stockés à 4°C avant d'être analysés.
IV.4.2 Renouvellements séquentiels
Les renouvellements séquentiels doivent être réalisés après la phase initiale de mise en route décrite précédemment. Cette phase initiale est réalisée pour assurer un remplissage total de la porosité par la phase liquide et une mise à l’équilibre de l’écoulement. Elle permet aussi de déterminer les caractéristiques initiales du système (concentrations dans l'eau résiduelle) nécessaire lors de la modélisation des phénomènes. Le système est alors mis en recirculation pour réaliser les renouvellements séquentiels. Après un temps t de contact en milieu compacté, la totalité de la phase liquide est retirée (à l'exception bien sûr des volumes résiduels) et un échantillon de 20 ml est stocké à 4°C avant analyse. Le sol est laissé en place dans la colonne tandis qu'un volume de phase liquide égale au volume retiré est remis dans le réacteur tampon. Le système est remis en marche pour une même durée t. Cette opération de renouvellement est répétée autant de fois que nécessaire. Les compositions initiales des phases liquides renouvelées correspondent à des dilutions des volumes résiduels restés dans le système à chaque renouvellement, le bilan matière rendra compte du relargage dans chaque séquence de percolixiviation.
IV.4.3 Percolation
Après la mise en place du sol, décrite lors de la mise œuvre du test, la boucle de retour permettant la recirculation de la phase liquide est démontée. Le dispositif est donc équivalent à une colonne de percolation à débit vertical ascendant. Un réservoir de percolat est placé dans un bain thermostaté, de même que la colonne où se déroule les échanges solides/liquides. A différents L/S, le volume total percolé est prélevé puis stocké à 4°C avant d'être analysé. Les concentrations mesurées sont donc des concentrations moyennes entre deux prélèvements.
IV.5 Conclusion
Après avoir défini les objectifs d'un nouveau test de lixiviation, des solutions techniques ont été données. Elles ont abouti à la conception d'un test expérimental que nous avons appelé "batch percolant". Ce test doit nous conduire à évaluer, dans de meilleures conditions, la
89

Chapitre IV : Conception d’un outil expérimental
mobilité des HAP dans le sol à notre disposition. La mise en œuvre du batch percolant passe par plusieurs phases préliminaires, à savoir le remplissage de la colonne et la saturation en eau du milieu contenu. L'écoulement de la phase liquide à travers le sol compacté a été caractérisé via la Distribution du Temps de Séjour (DTS). La colonne ainsi élaborée se comporte comme une série de quatre réacteurs parfaitement agités en série. Pour des études spécifiques de mécanismes, notamment de cinétique, il serait préférable de réduire les dimensions des colonnes afin de diminuer encore plus le nombre de ROPA de la série (idéal 1 ROPA) tout en sachant qu’on augmente en même temps les problèmes de la représentativité de l’échantillon de sol testé. Les protocoles de vitesses de dissolution (cinétiques), renouvellements séquentiels et percolations ont été détaillés dans ce chapitre. Nous allons maintenant passer à l'évaluation de la mobilité des HAP présents dans le sol étudié en exploitant les résultats obtenus par l’application du batch percolant.
90

Chapitre V : Application du batch percolant – Approche méthodologique
Chapitre V: Application du Batch percolant à l’étude de la mobilité des polluants organiques hydrophobes (HAP) présents dans des sols industriels – Approche méthodologique Dans les chapitres précédents, nous avons montré que les tests classiques de lixiviation (milieu dispersé et percolation en colonne) n’étaient pas adaptés à l’étude du sol que nous avions à notre disposition. Cependant, ces tests classiques nous ont permis d’émettre quelques hypothèses sur la mobilité des HAP. Un test spécifique que nous avons appelé "batch percolant» a alors été conçu afin d’approfondir les hypothèses émises. La description de ce test ainsi que les protocoles expérimentaux ont été exposés. De plus, une étude hydrodynamique a été réalisée pour caractériser l’écoulement dans le "batch percolant".
V.1 Etude de la mobilité des HAP
L’objectif du chapitre suivant est d’évaluer la mobilité des HAP dans le sol à notre disposition, par le biais du "batch percolant". Pour ce faire, et sur la base des hypothèses émises dans le chapitre précédent, notamment la contribution probable d'une fraction particulaire, l’étude de la mobilité a été divisée en deux parties. Dans un premier temps, nous avons travaillé sur la fraction des HAP mobilisés sous forme dissoute dans la phase liquide lixiviante. Puis dans un second temps, la mobilité des HAP sous forme particulaire a été étudiée. L'approche de la mobilité des HAP de la fraction dissoute et de la fraction particulaire a été faite en trois étapes :
• Identification des mécanismes de relargage vers la phase liquide. • Influence de paramètres physico-chimiques pertinents (débits, températures…) sur les
mécanismes de relargage. • Evolution du relargage au cours du temps.
V.1.1 Identification des mécanismes de relargage vers la phase liquide
En fonction du type et du degré de pollution des sites pollués par des composés organiques hydrophobes, les mécanismes susceptibles de participer au transfert des HAP de la phase solide vers la phase liquide sont :
1. le transfert par dissolution, 2. le transfert par désorption, 3. le transfert sous forme particulaire.
91

Chapitre V : Application du batch percolant – Approche méthodologique
Les mécanismes 1 et 2 sont de types physico-chimiques et correspondent à une solubilisation des HAP dans la phase liquide. Cette fraction de la pollution est étudiée dans ce chapitre sous le nom de fraction solubilisée par opposition à la fraction particulaire. Dans le chapitre I, nous avons présenté le mode de calcul des concentrations à l’équilibre dans la phase aqueuse :
loi de Raoult pour la dissolution, Isotherme pour la désorption.
Dans le cadre des sols pollués par HAP, de nombreuses études ont montré que le transfert par dissolution ou par désorption n’est pas instantané. Des modèles phénoménologiques, décrits dans le chapitre I prennent en compte les aspects cinétiques du transfert. Les plus usités sont :
modèle de diffusion à travers un film (ou double film) pour la dissolution, modèle chimique à deux compartiments et modèle physique décrit par la loi de Fick pour la désorption.
Ces outils contribuent à l’identification des mécanismes de transfert qui contrôlent le relargage des polluants dans la phase liquide. Le transfert sous forme particulaire, décrit dans le chapitre I, résulte d'une déstabilisation du sol qui provoque un transfert dans la phase liquide de particules fines chargées en polluants. L’étude expérimentale avec le "batch percolant" permettra de mettre en évidence la présence d’un transfert particulaire. Si ce transfert est significatif et qu’il est susceptible d’avoir lieu in situ, il devra être intégrer à l'étude de la mobilité des HAP.
V.1.2 Influence de paramètres physico-chimiques sur les vitesses de relargage vers la phase liquide
Etudier le comportement d’un site industriel pollué au laboratoire est une étape incontournable de l'évaluation de son impact environnemental. Des conclusions qui découleraient d’une expérimentation réalisée avec des paramètres physico-chimiques maintenus constants (température, débit …) seraient difficilement transposables directement aux conditions rencontrées sur le terrain. En effet, les paramètres physico-chimiques rencontrés sur le terrain varient en fonction du temps et du lieu considéré, ils peuvent donc influencer significativement la mobilité des polluants. Cette variabilité des paramètres peut être naturelle (conditions climatiques, déversements accidentelles) ou résulter d’un traitement du sol (qu’il soit biologique, thermique, chimique, etc…). L’étude à l’échelle du laboratoire doit donc fournir des informations sur les effets de certains paramètres physico-chimiques pertinents pour le scénario environnemental, sur les mécanismes de relargage. Voici les
92

Chapitre V : Application du batch percolant – Approche méthodologique
paramètres retenus pour notre étude : L/S Le rapport L/S correspond au rapport du volume de liquide mis en contact avec la masse de solide. Dans les conditions du test batch percolant, deux situations sont rencontrées : - fonctionnement en système fermé, situation ou le rapport L/S est constant au cours du
temps, il dépend à la fois de la masse de sol dans la colonne et du volume de solution lixiviante.
- fonctionnement en système ouvert, avec alimentation et évacuation continue. Dans ce cas le rapport L/S évolue au cours de l'essai puisque le volume de solution ayant percolé augmente en fonction du débit imposé.
Il peut avoir une signification « instantanée », à savoir le volume de liquide en contact avec le sol mai aussi en cumulé, à savoir la somme des volumes qui ont été en contact avec le sol. Souvent il est exprimé par le rapport V liquide/V pores. Nous avons décidé de garder le rapport L/S par souci de cohésion avec le développement en cours des tests normalisés de perco-lixiviation. Le transfert vers la phase liquide peut être influencé par ce rapport suivant le mécanisme mis en jeu. Cosolvants La présence de certains produits organiques sur le site peut influencer la mobilité des HAP en raison de leur effet de cosolvant. En effet lors des remaniements du site, il peut se produire, dans la zone polluée, une mise en contact accidentelle de cosolvants avec le sol. Nous avons choisi d’étudier le méthanol parce que c’est un cosolvant qui a été largement étudié/utilisé et pour un souci de simplicité : en effet, la solution eau/méthanol est monophasique et le dosage des HAP est facilité. Température Les températures sur le site peuvent varier en fonction de la saison pour les zones polluées se trouvant à faible profondeur. De plus, certains procédés de traitement proposent de chauffer la zone à traiter pour améliorer le traitement: la température a une forte influence sur la solubilisation des HAP pures [REZA, 2002]. Débit Le débit de recirculation et d’alimentation de la phase liquide est un paramètre important. En effet, des précipitations importantes ou un traitement par pompage en aval de la zone polluée peuvent entraîner des variations importantes des débits de liquide en contact avec le sol. Ces variations de débit, en fonction du mécanisme de transfert considéré, peuvent jouer un rôle important dans la mobilité des polluants.
93

Chapitre V : Application du batch percolant – Approche méthodologique
Force ionique La force ionique d'une solution est définie par :
(1) 2
21
ii ZCI ∑=
où Ci est la concentration de l'ion i et Zi est la charge de l'ion i. Des études ont montré que la force ionique pouvait, d’une part, modifier les solubilités des molécules et, d’autre part, avoir une incidence sur la stabilité des particules du sol. La force ionique d’une eau traversant une zone polluée peut évoluer en fonction du temps (lent épuisement en éléments ioniques), du débit, de la température… pH Compte tenu du pouvoir tampon du sol vis à vis d’attaques acides ou basiques (voir chapitre II), le pH n’a pas été considéré comme un paramètre environnemental significatif pour le scénario. De plus le pH n'a pas une influence directe sur la solubilité des HAP. En conséquence, l’effet du pH sur la mobilité des HAP n’a pas été étudié Activité biologique Selon les informations bibliographiques, certains HAP seraient des molécules biodégradables, il fallait donc étudier l’impact de ce facteur sur leur mobilité dans les sols, milieux permettant une importante activité biologique. Les phénomènes de transformation biologique des polluants, et leur impact sur la mobilité des polluants, peuvent être bloqués et donc dissociés des processus physiques de transfert des polluants par l’emploi d’un inhibiteur de l’activité biologique, comme de l’azoture de sodium (NaN3). Par comparaison entre une lixiviation du sol avec un inhibiteur de l'activité bactérienne et une lixiviation du sol sans inhibiteur, il est possible de mettre en évidence des phénomènes de biodégradation pour les étudier ensuite spécifiquement si nécessaire.
V.1.3 Evolution du relargage au cours du temps
Les informations obtenues lors des deux étapes précédentes (identification des mécanismes relargage et influence de paramètres physico-chimiques sur les mécanismes de relargage) ne sont pas suffisantes pour conclure l'étude de laboratoire sur la mobilité. Il est nécessaire d'étudier l'évolution du relargage au cours du temps.
94

Chapitre V : Application du batch percolant – Approche méthodologique
Température L/S Cosolvant Débit Force ioniqueBioréactivité
Dissolution
Désorption
Destabilisation
Fraction solubilisée
Fraction particulaire
Mécanisme de transfert
Paramètres physico-chimiques
Mobilité des
Polluants
Evolution à long terme
Figure 1: vision schématique de l’évaluation de la mobilité des HAP dans les sols
V.2 Mise en évidence d’une fraction solubilisée et d’une fraction particulaire dans les conditions expérimentales du batch percolant
Pour confirmer la présence d’un mécanisme de transfert particulaire, des renouvellements séquentiels avec de l’eau permutée ont été réalisés avec différents systèmes de filtration en sortie de colonne: deux granulométries de sable et une membrane de maille 10 µm. La synthèse des conditions opératoires se trouve dans le Tableau 1.
Tableau 1: synthèse des conditions opératoires
Filt
ratio
n en
ent
rée
et
sort
ie d
e co
lonn
e
Nat
ure
de la
pha
se li
quid
e
Vol
ume
de la
pha
se
liqui
de (m
l)
Mas
se d
e so
l (g)
L/S
Q (m
l.h-1
)
T°C
Dur
ée d
e ch
aque
con
tact
(h
) N
br d
e re
nouv
elle
men
t (R
)
Sable [0-400] µm
Eau 150 25 6 30 22 24 9
Sable [0,5-1] mm
Eau 150 25 6 30 22 24 4
Membrane,maille 10µm
Eau 150 25 6 30 22 24 3
95

Chapitre V : Application du batch percolant – Approche méthodologique
A chaque renouvellement les solutions ont été caractérisées par dosage des HAP. De plus, lors de l'essai effectué avec comme technique de filtration la membrane de maille 10 µm, la phase liquide correspondante au troisième renouvellement (noté R3) a été séparée en 3 fractions (Figure 2). Une des fractions a été directement préparée et analysée (R3 brut). Une autre fraction a été centrifugée dans un tube en teflon à 10000 g avant préparation et analyse (R3 centrifugé). La troisième fraction a été mise 30 minutes en contact avec un tube en téflon pour évaluer les éventuelles pertes par adsorption sur les parois du tube (R3 témoin) lors de la centrifugation.
R3
Préparation deR3 brut puisanalyse des
HAP parHPLC Centrifugation
de R3 à 10000gdans un tube en
teflon
Préparation deR3 centrifugépuis analysedes HAP par
HPLC
Mise en contactde R3 avec untube en teflon
pendant 30minutes
Préparation deR3 puis
analyse desHAP parHPLC
R3 brut
R3 centrifugé R3 témoin
Figure 2: Séparation de l'échantillon R3 en trois fractions
V.2.1 Résultats
On peut voir dans la Figure 3 que les concentrations en HAP obtenues lors des renouvellements séquentiels ne dépendent pas de la technique de filtration utilisée en sortie de colonne. Le pic R3 (troisième renouvellement) observé pour le phénanthrène et l’anthracène (Figure 3) semble être corrélé au pic R3 de la Figure 4 qui représente l'évolution de la turbidité au cours des renouvellements séquentiels. La turbidité est utilisée ici comme indicateur de la quantité de particules en suspension dans la phase liquide. Par conséquent, le pic R3 observé expérimentalement semble être en relation avec la présence de particules en suspension.
96
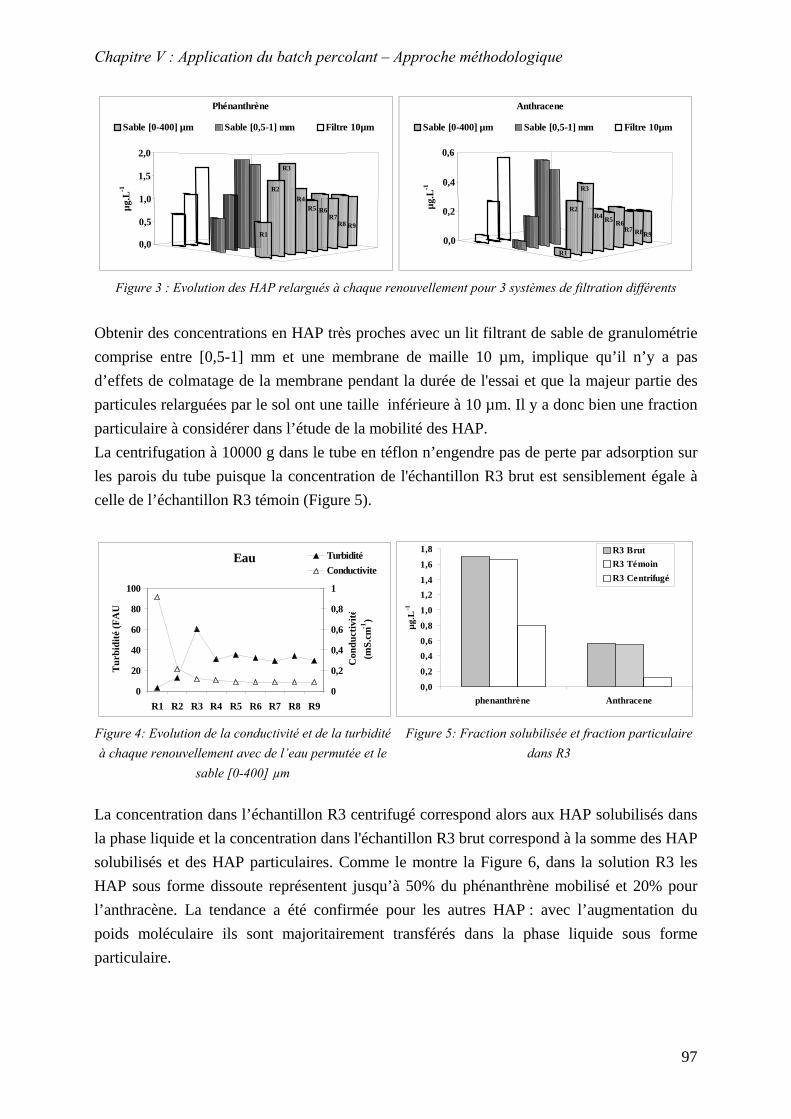
Chapitre V : Application du batch percolant – Approche méthodologique
R9R8R7
R6R5R4
R3
R2
R10,0
0,5
1,0
1,5
2,0
µg.L
-1
Phénanthrène
Sable [0-400] µm Sable [0,5-1] mm Filtre 10µm
R9R8R7R6R5R4
R3
R2
R1
0,0
0,2
0,4
0,6
µg.L
-1
Anthracene
Sable [0-400] µm Sable [0,5-1] mm Filtre 10µm
Figure 3 : Evolution des HAP relargués à chaque renouvellement pour 3 systèmes de filtration différents
Obtenir des concentrations en HAP très proches avec un lit filtrant de sable de granulométrie comprise entre [0,5-1] mm et une membrane de maille 10 µm, implique qu’il n’y a pas d’effets de colmatage de la membrane pendant la durée de l'essai et que la majeur partie des particules relarguées par le sol ont une taille inférieure à 10 µm. Il y a donc bien une fraction particulaire à considérer dans l’étude de la mobilité des HAP. La centrifugation à 10000 g dans le tube en téflon n’engendre pas de perte par adsorption sur les parois du tube puisque la concentration de l'échantillon R3 brut est sensiblement égale à celle de l’échantillon R3 témoin (Figure 5).
Eau
0
20
40
60
80
100
R1 R2 R3 R4 R5 R6 R7 R8 R9
Tur
bidi
té (F
AU
0
0,2
0,4
0,6
0,8
1
Con
duct
ivité
(mS.
cm-1
)
TurbiditéConductivite
0,0
0,2
0,40,60,8
1,0
1,21,4
1,61,8
phenanthrène Anthracene
µg.L
-1
R3 BrutR3 TémoinR3 Centrifugé
Figure 4: Evolution de la conductivité et de la turbidité à chaque renouvellement avec de l’eau permutée et le
sable [0-400] µm
Figure 5: Fraction solubilisée et fraction particulaire dans R3
La concentration dans l’échantillon R3 centrifugé correspond alors aux HAP solubilisés dans la phase liquide et la concentration dans l'échantillon R3 brut correspond à la somme des HAP solubilisés et des HAP particulaires. Comme le montre la Figure 6, dans la solution R3 les HAP sous forme dissoute représentent jusqu’à 50% du phénanthrène mobilisé et 20% pour l’anthracène. La tendance a été confirmée pour les autres HAP : avec l’augmentation du poids moléculaire ils sont majoritairement transférés dans la phase liquide sous forme particulaire.
97
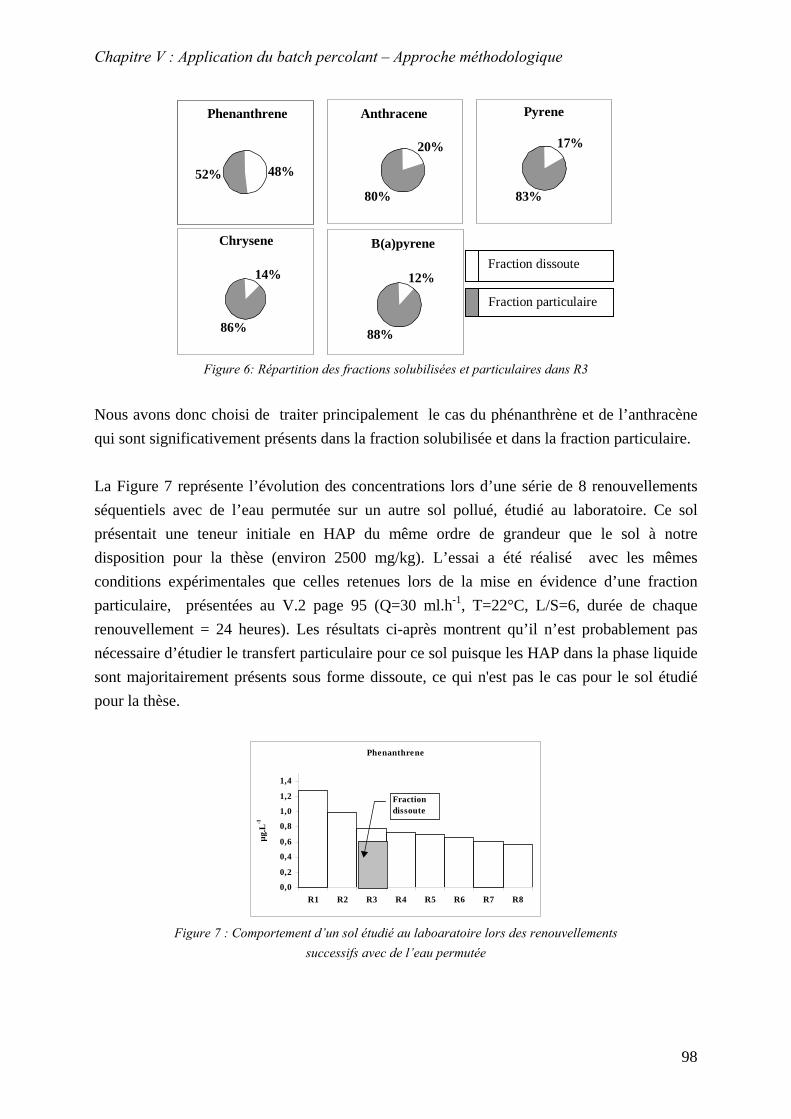
Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
48%52%
Anthracene
20%
80%
Pyrene
17%
83%
Chrysene
14%
86%
B(a)pyrene
12%
88%
Fraction dissoute
Fraction particulaire
Figure 6: Répartition des fractions solubilisées et particulaires dans R3
Nous avons donc choisi de traiter principalement le cas du phénanthrène et de l’anthracène qui sont significativement présents dans la fraction solubilisée et dans la fraction particulaire. La Figure 7 représente l’évolution des concentrations lors d’une série de 8 renouvellements séquentiels avec de l’eau permutée sur un autre sol pollué, étudié au laboratoire. Ce sol présentait une teneur initiale en HAP du même ordre de grandeur que le sol à notre disposition pour la thèse (environ 2500 mg/kg). L’essai a été réalisé avec les mêmes conditions expérimentales que celles retenues lors de la mise en évidence d’une fraction particulaire, présentées au V.2 page 95 (Q=30 ml.h-1, T=22°C, L/S=6, durée de chaque renouvellement = 24 heures). Les résultats ci-après montrent qu’il n’est probablement pas nécessaire d’étudier le transfert particulaire pour ce sol puisque les HAP dans la phase liquide sont majoritairement présents sous forme dissoute, ce qui n'est pas le cas pour le sol étudié pour la thèse.
Phenanthrene
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
R1 R2 R3 R4 R5 R6 R7 R8
µg.L
-1
Fraction dissoute
Figure 7 : Comportement d’un sol étudié au laboaratoire lors des renouvellements
successifs avec de l’eau permutée
98

Chapitre V : Application du batch percolant – Approche méthodologique
V.2.2 Conclusion
Le sol étudié en perco-lixiviation à l’eau dans le batch percolant a permis de distinguer deux formes (appelées aussi fractions) de relargage des HAP:
Fraction solubilisée Fraction particulaire
Dans les conditions données, la répartition du relargage en faveur du mécanisme particulaire augmente avec le poids moléculaire et la diminution de la solubilité des HAP. Les particules relarguées sont de tailles inférieures à 10 µm car elles ne colmatent pas le système de filtration en sortie de colonne. Deux polluants ont été ciblés pour leur répartition plus équilibrée entre les deux formes : le phénanthrène et l’anthracène. En effet, le poids du relargage particulaire représente respectivement 50 et 80 % du total mobilisé. Seul ces deux polluants seront suivis par la suite. Par la mise en évidence de la coexistence des deux formes de relargage des HAP contenus dans le sol étudié, nous allons développer et présenter séparément l’étude de la fraction solubilisée et respectivement particulaire.
V.3 Mobilité de la fraction solubilisée
L'étude de la fraction solubilisée, c'est à dire l'étude des HAP relargués sous forme solubilisée dans la solution lors du contact avec le sol pollué, a été réalisée en plusieurs points:
Identification du mécanisme majoritaire contrôlant la mise en solution des HAP Evaluation de l'influence de paramètres (T°C, L/S, q, fc) sur le mécanisme de mise en
solution à travers des études de vitesse de relargage. Evolution du relargage à travers des études dynamiques par renouvellements
séquentiels.
V.3.1 Identification du mécanisme contrôlant le relargage des HAP dans la phase liquide lors d'un contact avec le sol
Selon la littérature [Grathwohl, 1998], la mise en solution des HAP, à partir du sol étudié, devrait être contrôlée soit par un mécanisme de dissolution, soit par un mécanisme de
99

Chapitre V : Application du batch percolant – Approche méthodologique
désorption. A partir de la loi de Raoult et des isothermes de désorption, décrits dans le chapitre I, on peut calculer théoriquement des concentrations à l’équilibre dans l’eau. La comparaison entre ces valeurs théoriques et les valeurs expérimentales à l’équilibre permettra de déterminer la nature du mécanisme contrôlant le relargage de la fraction solubilisée des HAP vers la phase liquide.
V.3.1.1 Concentrations expérimentales stationnaires (à l'équilibre)
La connaissance du temps de contact nécessaire à l’obtention de l’état stationnaire du système est une information essentielle pour le protocole expérimental. Pour l’obtenir dans les conditions du contact entre le sol et l'eau déminéralisée, nous avons suivi la vitesse de relargage des HAP dans l'eau par la méthode des prélèvements en parallèle (voir protocole du batch percolant au chapitre IV). L'analyse des échantillons devait porter uniquement sur la fraction solubilisée, il fallait donc éliminer les HAP particulaires. Par la centrifugation des échantillons à 10000 g et la séparation de la phase surnageante nous avons réussi, comme démontré page 97, à séparer les HAP solubilisés. La phase surnageante a été ensuite concentrée et analysée par HPLC (voir protocole en annexe). Les conditions opératoires sont présentées dans le Tableau 2.
Tableau 2: synthèse des conditions expérimentales
Nat
ure
de la
ph
ase
liqui
de
Filt
ratio
n en
en
trée
et s
ortie
de
col
onne
Vol
ume
de
liqui
de (m
L)
Mas
se d
e so
l (g
)
L/S
Q (m
l.h-1
)
T°C
Eau Membrane 10µm 150 25 6 30 22 Dans la Figure 8, nous présentons l’évolution des concentrations des deux HAP cibles sélectionnés. L’état stationnaire (d'équilibre) est plus rapidement atteint dans le cas de l’anthracène.
0,0
0,2
0,4
0,6
0,8
1,0
0 50 100 150
time (h)
µg.L
-1 PhénanthrèneAnthracène
Figure 8: vitesse de relargage du phénanthrène
et de l'anthracène dans la phase liquide
100

Chapitre V : Application du batch percolant – Approche méthodologique
Les concentrations à l'équilibre sont donc approximativement de 0,9 µg.L-1 pour le phénanthrène et 0,1 µg.L-1 pour l'anthracène.
V.3.1.2 Calcul théoriques des concentrations à l'équilibre à 25°C dans l'eau
V.3.1.2.1 Première hypothèse : dissolution des HAP selon la loi de Raoult
[Lane,1992 et 1995], [Peters, 1993] et [Lee, 1992] ont développé et adapté la loi de Raoult pour prédire les concentrations à l’équilibre dans une phase aqueuse en contact avec un sol pollué par des HAP. La loi de Raoult établit que dans un mélange à l’équilibre de deux phases liquides idéales, la fraction molaire d’un composé dans un des deux liquides peut être estimée à partir de sa fraction molaire dans l’autre liquide. En supposant les HAP incorporés dans une phase organique plus ou moins liquide, distincte du sol et située dans la porosité inter particulaire, la loi de Raoult (2), détaillée dans le chapitre I, peut être employée pour décrire le partage entre les deux phases liquides considérées idéales. Nous appellerons la phase organique polluante: "mixture" car elle est composée d’une multitude de composés.
(2) eimixieqei SC ,,,, ⋅= χ
avec
Ci,e,eq : concentration du composé i à l’équilibre dans la phase aqueuse (mol.L-1)
mixi,χ : fraction molaire du composé i dans la mixture organique
Si,e : solubilité du composé i pur dans l’eau à 25 °C (mol.L-1)
(3) imix
imixi nn
n+
=,χ
avec
ni: nombre de mole du composé i dans la mixture organique (mol) nmix: nombre de mole composant la mixture organique (mol)
Dans l'équation (3) ni au dénominateur est négligeable devant nmix. Pour 1 kg de sol,
(4) i
osii M
Cn ,,= (5)
mix
mixmix MW
mn =
avec Ci,s,0: concentration initiale du composé i dans le sol (g.kg-1) Mi: masse moléculaire du composé i (g.mol-1)
101

Chapitre V : Application du batch percolant – Approche méthodologique
MWmix : masse moléculaire moyenne de la mixture organique (g.mol-1) mmix : masse de mixture dans 1 kg de sol (g)
La détermination de la masse moléculaire moyenne de la mixture organique, MWmix, n’a pas été expérimentalement réalisable pour le sol étudié. En partant des données disponibles dans la littérature sur la masse moléculaire moyenne des goudrons, nous avons choisi les valeurs extrêmes de la masse moléculaire (MWmix =200 et 1000 g.mol-1) pour le calcul des concentrations à l’équilibre.
[Meta Environnemental, 1990] 537 g.mol-1
[EPRI, 1993] 200-1000 g.mol-1
La masse de la mixture organique présente dans 1 kg de sol (mmix) a été estimée à partir des hypothèses developpées par [Lane, 1995] qui propose, à partir du carbone organique total (COT) dans le sol, de considérer que 71% de la mixture organique est composée de carbone organique et que toute la matière organique du sol est contenue dans la mixture supposée homogène. Nous avons alors:
(6) 71,0
COTmmix = avec COT=300 g.kg-1
L'équation (2) devient alors:
(7) eii
mixsieqei S
COTMMWC
C ,0,,
,,
71,0⋅
⋅⋅⋅
=
Les concentrations à l’équilibre calculées par l'application de la loi de Raoult (7) sont données dans le Tableau 3 pour quelques HAP.
Tableau 3: Concentrations théoriques à l’équilibre pour un mécanisme de dissolution
Composé Solubilité dans l’eau
du composé pure (µg.L-1) à 25 °C
Ci,s,0
mg.kg-1
Ci,e,eq (µg.L-1) pour MWmix=
200 – 1000 g.mol-1
Naphtalène 30000 45,7 5,1-25,3 Phénanthrène 1290 225 0,8-3,9 Anthracène 70 430 0,08-0,4
Pyrène 140 280 0,08-0,4
102

Chapitre V : Application du batch percolant – Approche méthodologique
V.3.1.2.2 Deuxième hypothèse : désorption
Pour le cas d’un sol pollué avec des polluants organiques et relargués selon un mécanisme de désorption, [Lane, 1995] propose de calculer la concentration à l’équilibre à partir du coefficient de partage (8) :
(8) eqei
eqsiococp C
CfKK
,,
,,=⋅=
Avec
Koc : coefficient de partage entre la phase organique du sol et la phase aqueuse (L.M-1), foc : fraction de carbone organique présent dans le sol, Ci,s,eq et Ci,e,eq : concentrations à l’équilibre dans le sol et dans l’eau respectivement du
composé i.
En exprimant Ci,e,eq à partir des concentrations initiales en phase solide et des conditions expérimentales, .
(9) S
LCCC eqei
sieqsi
⋅−= ,,
0,,,,
avec
L : volume de liquide en contact avec le sol (L) S : masse de sol en contact avec la phase liquide (M) Ci,s,0 : concentration initiale du composé i en phase solide (M.M-1)
et en substituant l’équation (8) en (9), la concentration à l’équilibre en phase aqueuse peut être calculée à partir de la concentration initiale en phase solide (10).
(10)
SLfK
CC
ococ
sieqei
+⋅= 0,,
,,
Le Tableau 4 présente les résultats des calculs obtenus pour certains HAP contenus dans le sol étudié.
103
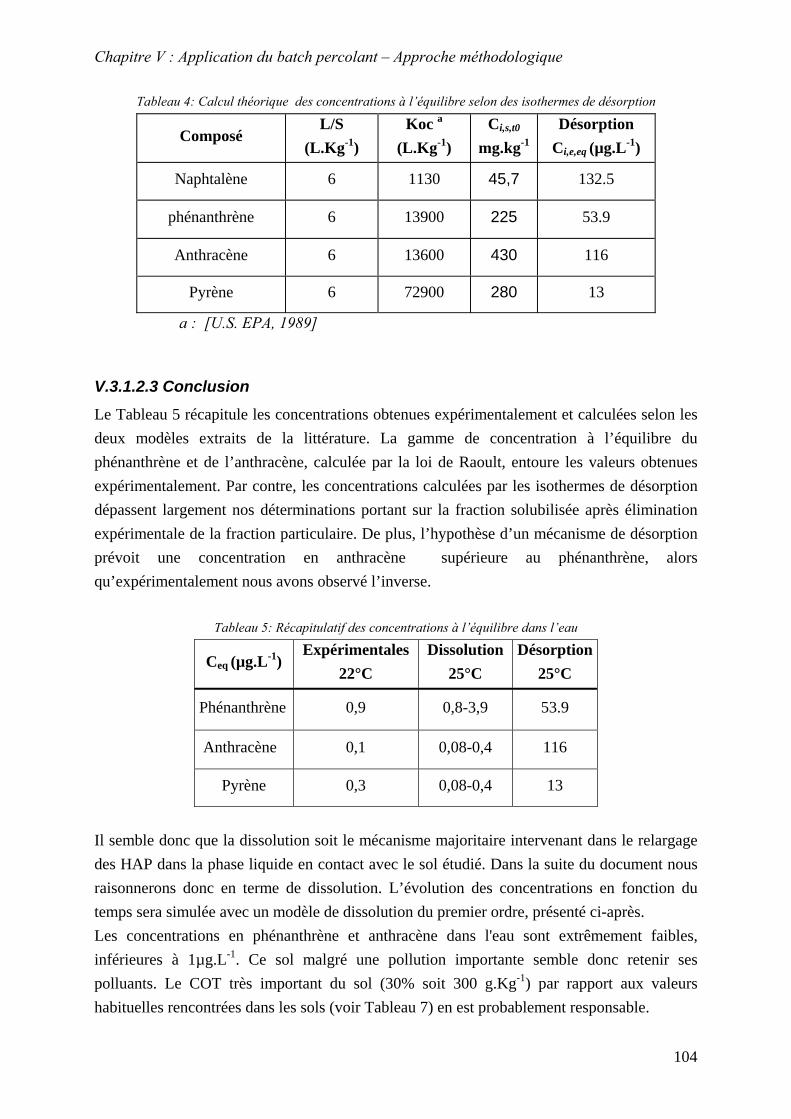
Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 4: Calcul théorique des concentrations à l’équilibre selon des isothermes de désorption
Composé L/S
(L.Kg-1) Koc a
(L.Kg-1) Ci,s,t0
mg.kg-1Désorption
Ci,e,eq (µg.L-1)
Naphtalène 6 1130 45,7 132.5
phénanthrène 6 13900 225 53.9
Anthracène 6 13600 430 116
Pyrène 6 72900 280 13
a : [U.S. EPA, 1989]
V.3.1.2.3 Conclusion
Le Tableau 5 récapitule les concentrations obtenues expérimentalement et calculées selon les deux modèles extraits de la littérature. La gamme de concentration à l’équilibre du phénanthrène et de l’anthracène, calculée par la loi de Raoult, entoure les valeurs obtenues expérimentalement. Par contre, les concentrations calculées par les isothermes de désorption dépassent largement nos déterminations portant sur la fraction solubilisée après élimination expérimentale de la fraction particulaire. De plus, l’hypothèse d’un mécanisme de désorption prévoit une concentration en anthracène supérieure au phénanthrène, alors qu’expérimentalement nous avons observé l’inverse.
Tableau 5: Récapitulatif des concentrations à l’équilibre dans l’eau
Ceq (µg.L-1) Expérimentales
22°C Dissolution
25°C Désorption
25°C
Phénanthrène 0,9 0,8-3,9 53.9
Anthracène 0,1 0,08-0,4 116
Pyrène 0,3 0,08-0,4 13
Il semble donc que la dissolution soit le mécanisme majoritaire intervenant dans le relargage des HAP dans la phase liquide en contact avec le sol étudié. Dans la suite du document nous raisonnerons donc en terme de dissolution. L’évolution des concentrations en fonction du temps sera simulée avec un modèle de dissolution du premier ordre, présenté ci-après. Les concentrations en phénanthrène et anthracène dans l'eau sont extrêmement faibles, inférieures à 1µg.L-1. Ce sol malgré une pollution importante semble donc retenir ses polluants. Le COT très important du sol (30% soit 300 g.Kg-1) par rapport aux valeurs habituelles rencontrées dans les sols (voir Tableau 7) en est probablement responsable.
104

Chapitre V : Application du batch percolant – Approche méthodologique
V.3.1.3 Modèle de dissolution du premier ordre pour le batch percolant
L’étude hydrodynamique présentée, au chapitre IV, a permis de définir que les écoulements dans la colonne pouvaient être simulés avec 4 réacteurs parfaitement agités en série. Précédemment, nous avons conclu que le mécanisme contrôlant le relargage des HAP par le sol lixivié était la dissolution. Nous avons choisi et appliqué un modèle cinétique de dissolution du premier ordre (11) pour la simulation des essais visant à étudier les vitesses de relargage des HAP en phase liquide. Conformément à l’identification de la colonne (série de 4 ROPA) à partir de la DTS, le schéma du système expérimental (batch percolant) est présenté Figure 7.
1 32
Ce C3C2C1 C4
4
T Figure 7 : Schèma des réacteurs caractérisant le batch percolant
Comme présenté dans la description du protocole expérimental (chapitre IV), les études des vitesses de relargage des HAP en phase liquide sont réalisées après une phase dite de "saturation" nécessaire pour l'atteinte d'un état homogène, soit un contact liquide/solide préalable de 24 heures. Par conséquent à t=0, les concentrations dans les 4 réacteurs sont supposées identiques et égales à la concentration mesurée à la fin du premier contact de 24 heures (R1). La concentration dans le réacteur tampon (T), dont le contenu est renouvelé, est égale à 0. Condition initiale : pour chaque HAP à t=0
C 1-4 = CR1
CT = 0Equation de bilan pour chaque HAP dans les réacteurs 1 à 4 : L'application de l'équation générale du bilan de masse à chacun des réacteurs considérés parfaitement agités, est réalisée pour chacun des HAP suivis:
Accumulation = entrée + création – consommation – sortie
105

Chapitre V : Application du batch percolant – Approche méthodologique
La vitesse de dissolution surfacique est considérée du premier ordre:
(11) )(1 CCkdtdm
Sv sat −×=×=
Dans cette relation, la surface de transfert S [m2] et la constante cinétique surfacique de premier ordre k [m.s-1] sont inconnues. En les considérant constantes lors du processus, nous regroupons les deux termes sous la forme d'un coefficient constant, K= k . S [m3.s-1] identique pour chacun des réacteurs et qui reste à identifier (méthode inverse). Ainsi les équations composant le modèle du batch percolant deviennent :
Réacteur 1 : )(4 11
1 CCKqCqCdt
dCVsate −⋅+−=⋅θ
(12)
Réacteur 2 : )(4 221
2 CCKqCqCdt
dCVsat −⋅+−=⋅θ
(13)
Réacteur 3 : )(4 332
3 CCKqCqCdt
dCVsat −⋅+−=⋅θ
(14)
Réacteur 4 : )(4 443
4 CCKqCqCdt
dCVsat −⋅+−=⋅θ
(15)
Equation bilan dans le réacteur T pour chaque HAP :
(16) eqCqCdt
dCL −=⋅ 44 )
L : volume de liquide dans le réacteur tampon (L) V : volume du sol contenu dans la colonne (L) C : concentration volumique en phase liquide (µg.L-1) Csat : concentration à l’équilibre ou à saturation dans le réacteur tampon (µg.L-1) q : débit constant d’alimentation de la colonne (m3.h-1) S : surface de transfert (m2) k: coefficient de transfert (m.h-1) K =k . S : coefficient de transfert global (m3.h-1) θ : porosité du sol dans la colonne t : temps (h)
106

Chapitre V : Application du batch percolant – Approche méthodologique
Ces systèmes d’équations différentielles écrits pour chacun des HAP suivis seront résolus sous Matlab pour chacune des expérimentations du programme de travail. Tous les paramètres nécessaires à l’alimentation du modèle sont mesurés ou donnés par les conditions opératoires, sauf le paramètre K qui est inconnu. Le coefficient global K, spécifique à chaque HAP et aux conditions expérimentales, regroupe un nombre important d’informations et hypothèses de fonctionnement et comportement : nombre et type de contacteurs solide/liquide, structure et surface d’échange, le mécanisme majeur de transfert de masse et sa cinétique etc. Bien que son identification numérique par méthode inverse ne pose pas, généralement, de problèmes difficiles, son utilisation n’est pas toujours automatique.
V.3.2 Justification de l’utilisation du cosolvant méthanol
Travailler avec des phases aqueuses présentant des concentrations de l’ordre du µg.L-1 est délicat et long du point de vue expérimental et analytique. Ce problème est commun à tous les polluants hydrophobes qui sont très faiblement solubles. Aussi, pour faciliter l’étude de ces polluants et sur la base des travaux scientifiques de [Yalkowsky 1976] ; [Lane, 1995] ; [Bouchard, 1998] nous avons effectué la majorité des essais expérimentaux dans le batch percolant avec des mélanges eau/méthanol comme phase liquide. Travailler en présence de méthanol simplifie l’étude de la mobilité de la fraction solubilisée pour les raisons suivantes: - Le méthanol est totalement miscible avec l’eau. - Le méthanol est moins polaire que l’eau en raison de l’effet +I du groupement CH3. Plus
on augmente la fraction de méthanol dans la phase aqueuse, plus la polarité du mélange diminue. Les solubilités des HAP augmentent donc en présence de méthanol et dépassent largement le seuil de détection du système d'analyse employé (HPLC) ce qui évite l’étape de concentration obligatoire lorsqu’on travaille avec de l’eau, étape qui est aussi source d’erreurs expérimentales.
- L'utilisation du méthanol a aussi pour effet de diminuer les transferts sous forme
particulaire. Cet effet stabilisant a été observé pour des pourcentages de méthanol de 20% à 50%. Il permet donc de travailler directement sur la fraction solubilisée en évitant l’étape de centrifugation.
- Chaque échantillon fabriqué est analysable directement par HPLC ce qui évite les
problèmes de préparations des échantillons en phase aqueuse. - Les études cinétiques peuvent se faire en série dans un seul réacteur (voir chapitre II). En
effet, les prélèvements successifs de 1 mL (volume nécessaire à l’analyse par HPLC en
107

Chapitre V : Application du batch percolant – Approche méthodologique
présence de méthanol) ne modifient pas significativement le rapport L/S contrairement à ce qui se passe avec l’eau.
- Précédemment au chapitre III, nous sommes arrivés à la conclusion que les mécanismes
de relargage était similaires lorsque la lixiviation du sol se faisait avec de l'eau ou avec un mélange eau/méthanol.
V.3.2.1 Théorie sur la solubilisation des composés hydrophobes dans des mélanges binaires.
L'étude des composés hydrophobes dans des mélanges binaires a permis à [Yalkowsky S.H., 1976] de formuler une loi sur la variation de la concentration à l'équilibre dans différents mélanges de deux solvants miscibles.
( 17) cm fCLogCLog σ+=
Avec Cm et C qui sont respectivement la solubilité dans le mélange eau/cosolvant et la solubilité dans l’eau, fc est la fraction du cosolvant dans la phase liquide (0-1). Le paramètre adimensionnel σ est dépendant des interactions entre le soluté et le cosolvant, il caractérise le pouvoir de solubilisation du cosolvant [RUBINO, 1987]. Dans la représentation graphique de la relation ( 17), σ est égal à la pente (Figure 9)
0 0,1 0,2 0,3 0,4 0,5 0,6
Fc
Log
Cm
σ
Figure 9:Prédiction des concentrations en phase aqueuse à partir de résultats obtenus en présence de méthanol
C'est sur sur principe que nous avons élaboré la phase expérimentale de l'étude la mobilité des HAP dans le sol étudié. Nous avons tout d'abord vérifié la validité de la théorie à notre sol (c'est à dire plusieurs lixiviations à l'équilibre avec différentes fractions de méthanol) et déterminé la valeur de σ. Ensuite, connaissant la valeur du coefficient σ pour notre système sol/eau/méthanol, il suffisait de réaliser les études paramétriques (température, débit...) avec
108
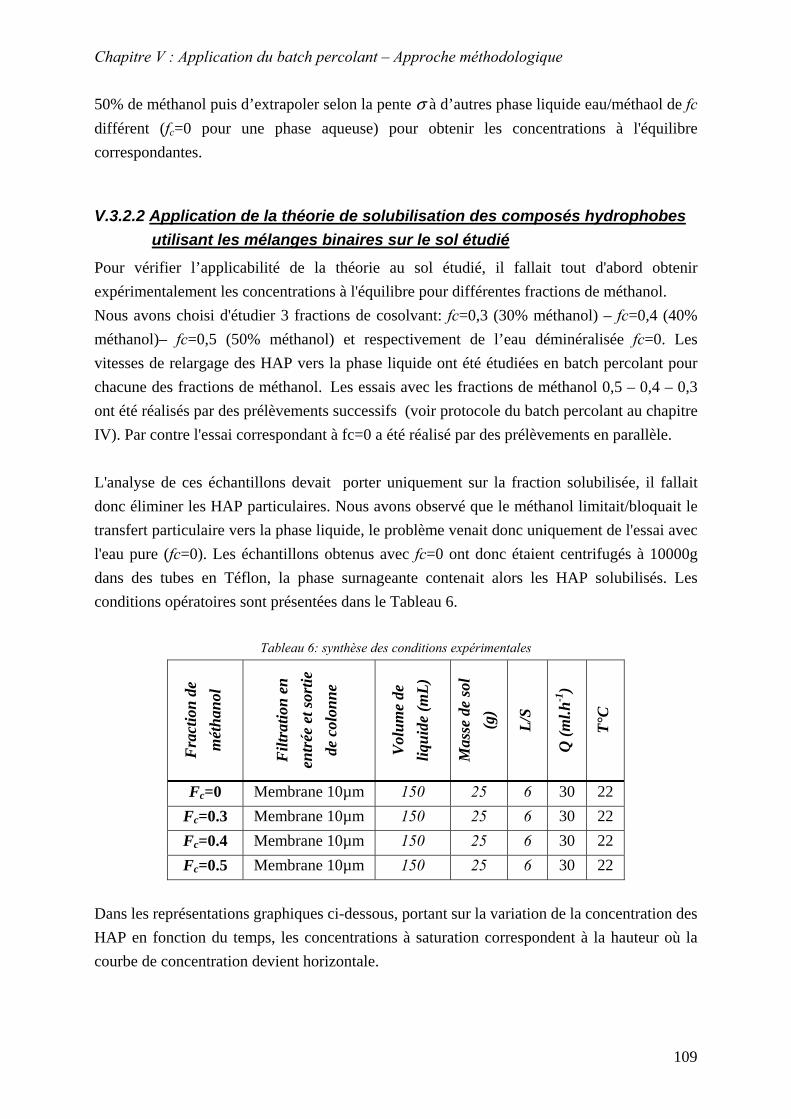
Chapitre V : Application du batch percolant – Approche méthodologique
50% de méthanol puis d’extrapoler selon la pente σ à d’autres phase liquide eau/méthaol de fc différent (fc=0 pour une phase aqueuse) pour obtenir les concentrations à l'équilibre correspondantes.
V.3.2.2 Application de la théorie de solubilisation des composés hydrophobes utilisant les mélanges binaires sur le sol étudié
Pour vérifier l’applicabilité de la théorie au sol étudié, il fallait tout d'abord obtenir expérimentalement les concentrations à l'équilibre pour différentes fractions de méthanol. Nous avons choisi d'étudier 3 fractions de cosolvant: fc=0,3 (30% méthanol) – fc=0,4 (40% méthanol)– fc=0,5 (50% méthanol) et respectivement de l’eau déminéralisée fc=0. Les vitesses de relargage des HAP vers la phase liquide ont été étudiées en batch percolant pour chacune des fractions de méthanol. Les essais avec les fractions de méthanol 0,5 – 0,4 – 0,3 ont été réalisés par des prélèvements successifs (voir protocole du batch percolant au chapitre IV). Par contre l'essai correspondant à fc=0 a été réalisé par des prélèvements en parallèle. L'analyse de ces échantillons devait porter uniquement sur la fraction solubilisée, il fallait donc éliminer les HAP particulaires. Nous avons observé que le méthanol limitait/bloquait le transfert particulaire vers la phase liquide, le problème venait donc uniquement de l'essai avec l'eau pure (fc=0). Les échantillons obtenus avec fc=0 ont donc étaient centrifugés à 10000g dans des tubes en Téflon, la phase surnageante contenait alors les HAP solubilisés. Les conditions opératoires sont présentées dans le Tableau 6.
Tableau 6: synthèse des conditions expérimentales
Fra
ctio
n de
m
étha
nol
Filt
ratio
n en
en
trée
et s
ortie
de
col
onne
Vol
ume
de
liqui
de (m
L)
Mas
se d
e so
l (g
)
L/S
Q (m
l.h-1
)
T°C
Fc=0 Membrane 10µm 150 25 6 30 22 Fc=0.3 Membrane 10µm 150 25 6 30 22 Fc=0.4 Membrane 10µm 150 25 6 30 22 Fc=0.5 Membrane 10µm 150 25 6 30 22
Dans les représentations graphiques ci-dessous, portant sur la variation de la concentration des HAP en fonction du temps, les concentrations à saturation correspondent à la hauteur où la courbe de concentration devient horizontale.
109

Chapitre V : Application du batch percolant – Approche méthodologique
Fc=0,5
050
100150200250300350400
0 50 100 150
time (h)
µg.L
-1 PhénanthrèneAnthracène
Fc=0,4
020406080
100
0 100
time (h)
µg.L
-1 PhénanthrèneAnthracène
Figure 10: Vitesse de relargage du phénanthrène et de
l'anthracène dans la phase liquide pour fc=0,5 Figure 11: Vitesse de relargage du phénanthrène et de
l'anthracène dans la phase liquide pour fc=0,4
Fc=0,3
0
2
4
6
8
10
0 50 100 150time (h)
µg.L
-1 PhénanthrèneAnthracène
Fc=0
0,0
0,2
0,4
0,6
0,8
1,0
0 50 100 150
time (h)
µg.L
-1 PhénanthrèneAnthracène
Figure 12: Vitesse de relargage du phénanthrène et de
l'anthracène dans la phase liquide pour fc=0,3 Figure 13: Vitesse de relargage du phénanthrène et de l'anthracène dans la phase liquide en fonction du temps
pour fc=0
Les concentrations stationnaires pour le phénanthrène et l'anthracène sont donc:
Fc 0 0,3 0,4 0,5
Phénanthrène (µg.L-1) 0,9 8 70 250 Anthracène (µg.L-1) 0,1 8 60 300
Dans les représentations graphiques ci-après, les concentrations sont exprimées en µmol.L-1
pour être homogènes avec les résultats publiés par [BOUCHARD, 2002] et [LANE, 1992].
110

Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
y = 4,85x - 2,43R2 = 0,94
-3-2-101
0 0,1 0,2 0,3 0,4 0,5 0,6
Fc
Log
Cm
(µm
ol.L-1
)
Anthracene
y = 6,72x - 3,20R2 = 0,99
-4-3-2-101
0 0,1 0,2 0,3 0,4 0,5 0,6
Fc
Log
Cm
(µm
ol..L
-1)
Figure 14:Représentation en log des solubilités en fonction de la fraction de méthanol en volume dans la
solution de lixiviation. (●) représente les point expérimentaux, (─) représente la régression linéaire
Les représentations (Figure 14) en log des solubilités en fonction de la fraction de cosolvant sont en accord avec les travaux de [YALKOWSKY, 1976] sur la solubilisation des composés hydrophobes dans des solvants binaires. Pour valider ces résultats voici une comparaison avec les résultats obtenus par d’autres auteurs.
Tableau 7:Valeur de σ obtenue lors de la thése et par d’autres auteurs.
σ Expérimentales
σ [BOUCHARD]
σ [LANE]
a b c d e f
Phénanthrène 4,85 3,32 4,23 4,33 5,05 4,24 Anthracène 6,7 - 5,46 4,63 5,35 5,29 a : étude réalisée sur un sol pollué b : étude réalisée sur le composé pure c,d,e,f : étude réalisée sur des sols pollués
c : HAP totaux = 354 mg/kg ; COT = 5,4 g/kg d : HAP totaux = 1907 mg/kg ; COT = 13 g/kg e : HAP totaux = 1108 mg/kg ; COT = 13 g/kg f : HAP totaux = 38 mg/kg ; COT = 28 g/kg
La valeur de σ déterminée expérimentalement à partir de 4 fractions de cosolvant semble cohérente avec les valeurs déterminées par [BOUCHARD, 2002] et [LANE, 1992]. Le pouvoir de solubilisation en présence de méthanol paraît plus important lorsque le composé est présent dans un sol pollué. En effet, le σ du phénanthrène obtenu dans cette étude est proche des valeurs de Lane qui ont été déterminées pour 4 sols pollués différents (c,d,e,f). Par contre Bouchard qui a travaillé sur les composés purs trouve une valeur de σ plus faible. L'augmentation de la valeur expérimentale de σ dans le cas du mélange de HAP par rapport au composé pur pourrait s'expliquer par un effet cosolvant du mélange des HAP dissous (contribution du mélange). En ce qui concerne σ de l’anthracène, les valeurs de Lane sont, comme pour nos travaux,
111
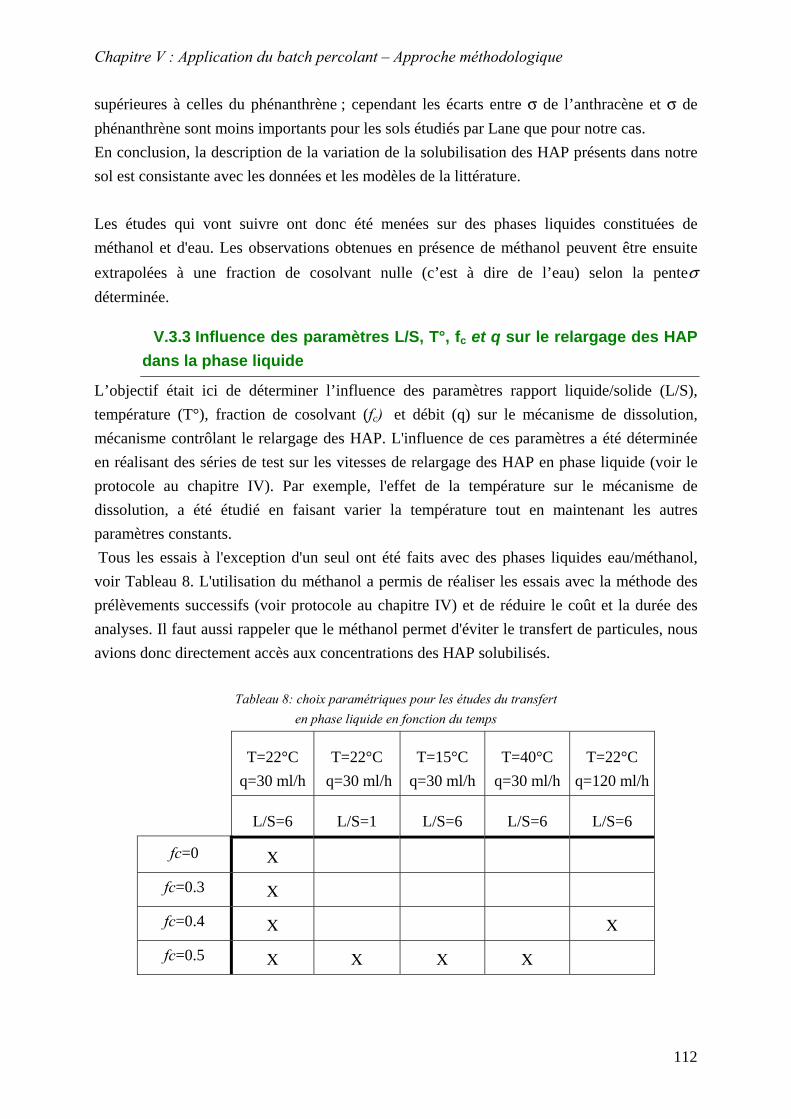
Chapitre V : Application du batch percolant – Approche méthodologique
supérieures à celles du phénanthrène ; cependant les écarts entre σ de l’anthracène et σ de phénanthrène sont moins importants pour les sols étudiés par Lane que pour notre cas. En conclusion, la description de la variation de la solubilisation des HAP présents dans notre sol est consistante avec les données et les modèles de la littérature. Les études qui vont suivre ont donc été menées sur des phases liquides constituées de méthanol et d'eau. Les observations obtenues en présence de méthanol peuvent être ensuite extrapolées à une fraction de cosolvant nulle (c’est à dire de l’eau) selon la penteσ déterminée.
V.3.3 Influence des paramètres L/S, T°, fc et q sur le relargage des HAP dans la phase liquide
L’objectif était ici de déterminer l’influence des paramètres rapport liquide/solide (L/S), température (T°), fraction de cosolvant (fc) et débit (q) sur le mécanisme de dissolution, mécanisme contrôlant le relargage des HAP. L'influence de ces paramètres a été déterminée en réalisant des séries de test sur les vitesses de relargage des HAP en phase liquide (voir le protocole au chapitre IV). Par exemple, l'effet de la température sur le mécanisme de dissolution, a été étudié en faisant varier la température tout en maintenant les autres paramètres constants. Tous les essais à l'exception d'un seul ont été faits avec des phases liquides eau/méthanol, voir Tableau 8. L'utilisation du méthanol a permis de réaliser les essais avec la méthode des prélèvements successifs (voir protocole au chapitre IV) et de réduire le coût et la durée des analyses. Il faut aussi rappeler que le méthanol permet d'éviter le transfert de particules, nous avions donc directement accès aux concentrations des HAP solubilisés.
Tableau 8: choix paramétriques pour les études du transfert en phase liquide en fonction du temps
T=22°C
q=30 ml/h T=22°C
q=30 ml/hT=15°C
q=30 ml/h T=40°C
q=30 ml/h T=22°C
q=120 ml/h
L/S=6 L/S=1 L/S=6 L/S=6 L/S=6
fc=0 Χ
fc=0.3 Χ
fc=0.4 Χ Χ
fc=0.5 Χ Χ Χ Χ
112
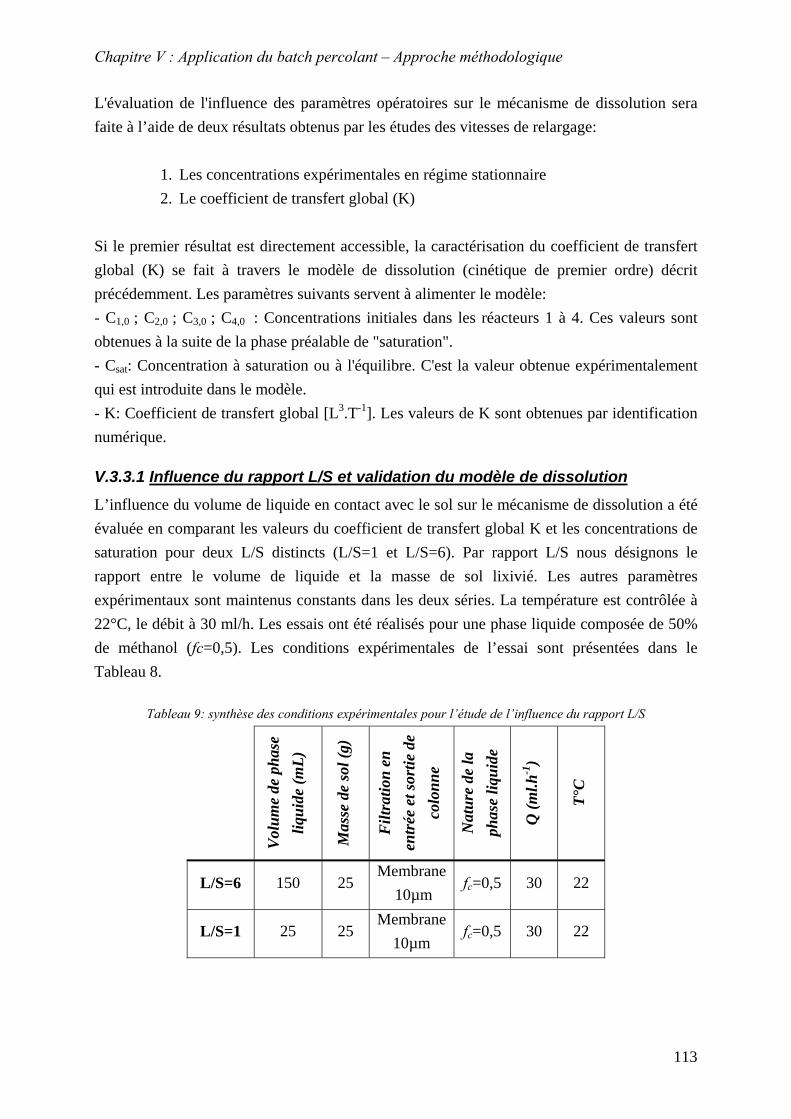
Chapitre V : Application du batch percolant – Approche méthodologique
L'évaluation de l'influence des paramètres opératoires sur le mécanisme de dissolution sera faite à l’aide de deux résultats obtenus par les études des vitesses de relargage:
1. Les concentrations expérimentales en régime stationnaire 2. Le coefficient de transfert global (K)
Si le premier résultat est directement accessible, la caractérisation du coefficient de transfert global (K) se fait à travers le modèle de dissolution (cinétique de premier ordre) décrit précédemment. Les paramètres suivants servent à alimenter le modèle: - C1,0 ; C2,0 ; C3,0 ; C4,0 : Concentrations initiales dans les réacteurs 1 à 4. Ces valeurs sont obtenues à la suite de la phase préalable de "saturation". - Csat: Concentration à saturation ou à l'équilibre. C'est la valeur obtenue expérimentalement qui est introduite dans le modèle. - K: Coefficient de transfert global [L3.T-1]. Les valeurs de K sont obtenues par identification numérique.
V.3.3.1 Influence du rapport L/S et validation du modèle de dissolution
L’influence du volume de liquide en contact avec le sol sur le mécanisme de dissolution a été évaluée en comparant les valeurs du coefficient de transfert global K et les concentrations de saturation pour deux L/S distincts (L/S=1 et L/S=6). Par rapport L/S nous désignons le rapport entre le volume de liquide et la masse de sol lixivié. Les autres paramètres expérimentaux sont maintenus constants dans les deux séries. La température est contrôlée à 22°C, le débit à 30 ml/h. Les essais ont été réalisés pour une phase liquide composée de 50% de méthanol (fc=0,5). Les conditions expérimentales de l’essai sont présentées dans le Tableau 8.
Tableau 9: synthèse des conditions expérimentales pour l’étude de l’influence du rapport L/S
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l (g)
Filt
ratio
n en
en
trée
et s
ortie
de
colo
nne
Nat
ure
de la
ph
ase
liqui
de
Q (m
l.h-1
)
T°C
L/S=6 150 25 Membrane
10µm fc=0,5 30 22
L/S=1 25 25 Membrane
10µm fc=0,5 30 22
113
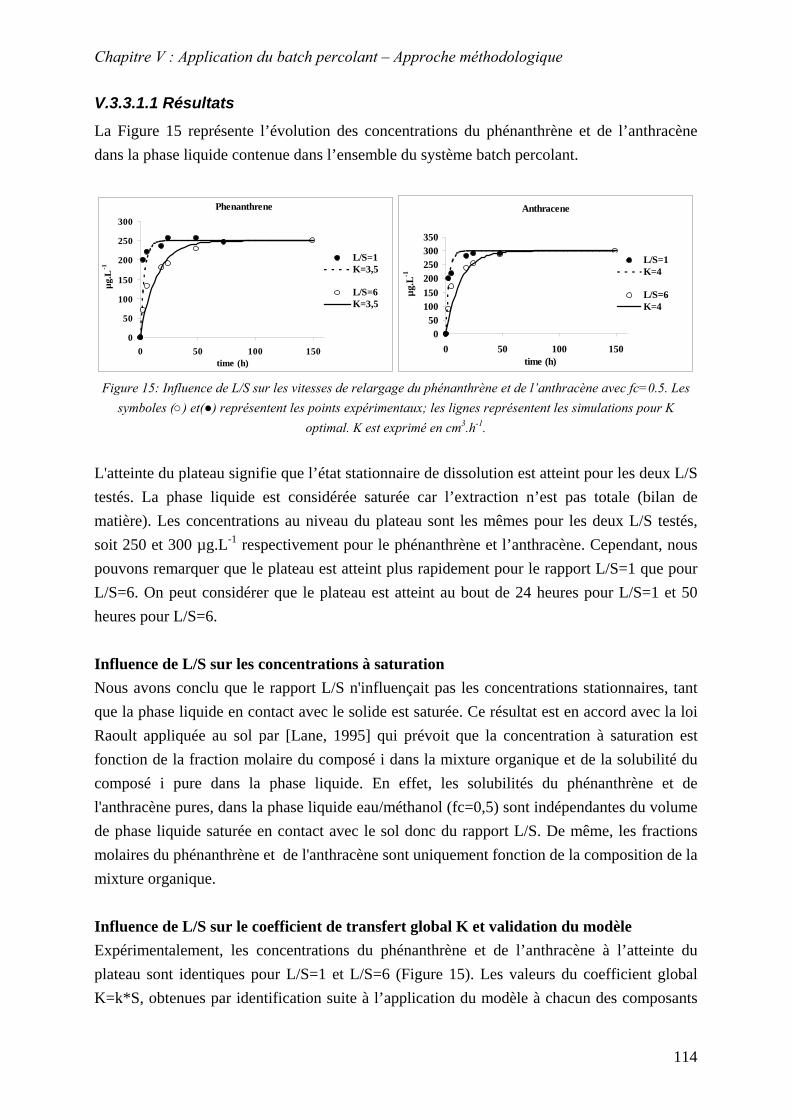
Chapitre V : Application du batch percolant – Approche méthodologique
V.3.3.1.1 Résultats
La Figure 15 représente l’évolution des concentrations du phénanthrène et de l’anthracène dans la phase liquide contenue dans l’ensemble du système batch percolant.
Phenanthrene
0
50
100
150
200
250
300
0 50 100 150time (h)
µg.L
-1
L/S=1K=3,5
L/S=6K=3,5
Anthracene
050
100150200250300350
0 50 100 150time (h)
µg.L
-1
L/S=1K=4
L/S=6K=4
Figure 15: Influence de L/S sur les vitesses de relargage du phénanthrène et de l’anthracène avec fc=0.5. Les
symboles (○) et(●) représentent les points expérimentaux; les lignes représentent les simulations pour K optimal. K est exprimé en cm3.h-1.
L'atteinte du plateau signifie que l’état stationnaire de dissolution est atteint pour les deux L/S testés. La phase liquide est considérée saturée car l’extraction n’est pas totale (bilan de matière). Les concentrations au niveau du plateau sont les mêmes pour les deux L/S testés, soit 250 et 300 µg.L-1 respectivement pour le phénanthrène et l’anthracène. Cependant, nous pouvons remarquer que le plateau est atteint plus rapidement pour le rapport L/S=1 que pour L/S=6. On peut considérer que le plateau est atteint au bout de 24 heures pour L/S=1 et 50 heures pour L/S=6. Influence de L/S sur les concentrations à saturation Nous avons conclu que le rapport L/S n'influençait pas les concentrations stationnaires, tant que la phase liquide en contact avec le solide est saturée. Ce résultat est en accord avec la loi Raoult appliquée au sol par [Lane, 1995] qui prévoit que la concentration à saturation est fonction de la fraction molaire du composé i dans la mixture organique et de la solubilité du composé i pure dans la phase liquide. En effet, les solubilités du phénanthrène et de l'anthracène pures, dans la phase liquide eau/méthanol (fc=0,5) sont indépendantes du volume de phase liquide saturée en contact avec le sol donc du rapport L/S. De même, les fractions molaires du phénanthrène et de l'anthracène sont uniquement fonction de la composition de la mixture organique. Influence de L/S sur le coefficient de transfert global K et validation du modèle Expérimentalement, les concentrations du phénanthrène et de l’anthracène à l’atteinte du plateau sont identiques pour L/S=1 et L/S=6 (Figure 15). Les valeurs du coefficient global K=k*S, obtenues par identification suite à l’application du modèle à chacun des composants
114

Chapitre V : Application du batch percolant – Approche méthodologique
sont très proches pour L/S=1 et L/S=6 : K=3,5.10-6 m3.h-1 pour le phénanthrène et K=4.10-6 m3.h-1 pour l’anthracène. Conformément aux hypothèses du modèle cinétique adopté, le coefficient de transfert (K) est théoriquement indépendant du rapport L/S. Les résultats obtenus sont donc en accord avec le modèle. Le K optimal choisi sous-estime légèrement les concentrations dans les premières heures, pour L/S=6. Ensuite la simulation se superpose aux points expérimentaux. Le mécanisme de transfert par dissolution est probablement plus complexe que le mécanisme retenu par le modèle de dissolution du premier ordre. Le modèle pourrait être amélioré pour que la vitesse de transfert (la pente) soit plus importante pendant les premières heures pour, ensuite, revenir aux pentes calculées par le modèle de dissolution du premier ordre. Les travaux portant sur les cinétiques de désorption des composés organiques hydrophobes dans les sols présentent des allures de courbes similaires a celles présentées dans la Figure 15. Pour prendre en compte des vitesses de désorption plus rapides au début du relargage, les auteurs utilisent deux catégories de modèles présentées dans le chapitre I. [Brusseau, 1989, 1991 et 1997] utilise un modèle chimique de désorption à deux compartiments, d’autres auteurs tel que [Grathwohl, 1994], ont opté pour des modèles dits physiques basés sur les phénomènes de diffusion couplés à la désorption. Dans le cas du mécanisme de dissolution des HAP, [Powers, 1991] a proposé de coupler le modèle de dissolution de premier ordre avec les variations de la surface interfaciale, (S). Pour un processus intense de dissolution et/ou à long terme, la prise en compte de l’épuisement du terme source s’impose. Un tel modèle demande une approche expérimentale encore plus élaborée. Sensibilité des paramètres L/S et K sur les vitesses de relargage L’étude de la sensibilité de la mise en solution en fonction de la variation de K et L/S est réalisée sur la base du modèle proposé pour le batch percolant et pour la cinétique de dissolution. La Figure 16 ci-après montre la simulation de l'évolution de la concentration du phénanthrène dans le réacteur tampon en fonction de la valeur du coefficient K (étude de sensibilité par rapport à K) pour L/S = 6. Comme attendu, la vitesse du processus approche un signal échelon pour K très grand (solubilisation très rapide).
115
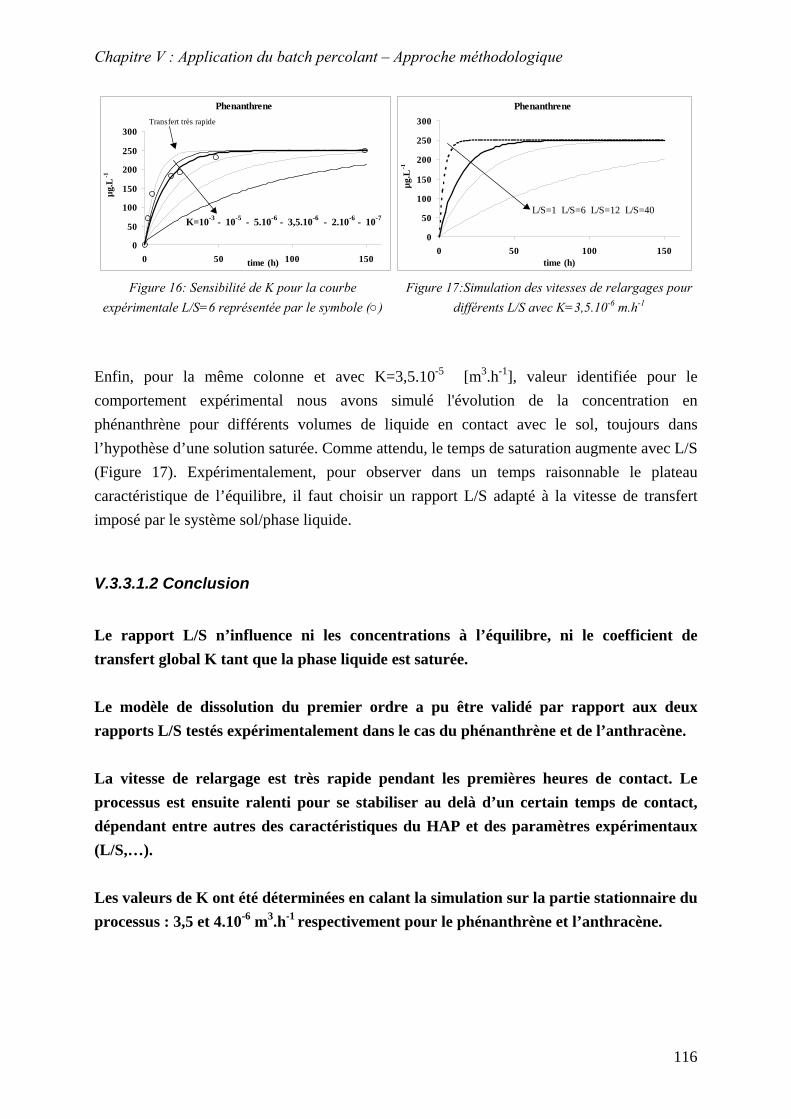
Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
0
50
100
150
200
250
300
0 50 100 150time (h)
µg.L
-1
K=10-3 - 10-5 - 5.10-6 - 3,5.10-6 - 2.10-6 - 10-7
Transfert très rapide
Phenanthrene
0
50
100
150
200
250
300
0 50 100 15time (h)
µg.L
-1
0
L/S=1 L/S=6 L/S=12 L/S=40
Figure 16: Sensibilité de K pour la courbe
expérimentale L/S=6 représentée par le symbole (○) Figure 17:Simulation des vitesses de relargages pour
différents L/S avec K=3,5.10-6 m.h-1
Enfin, pour la même colonne et avec K=3,5.10-5 [m3.h-1], valeur identifiée pour le comportement expérimental nous avons simulé l'évolution de la concentration en phénanthrène pour différents volumes de liquide en contact avec le sol, toujours dans l’hypothèse d’une solution saturée. Comme attendu, le temps de saturation augmente avec L/S (Figure 17). Expérimentalement, pour observer dans un temps raisonnable le plateau caractéristique de l’équilibre, il faut choisir un rapport L/S adapté à la vitesse de transfert imposé par le système sol/phase liquide.
V.3.3.1.2 Conclusion
Le rapport L/S n’influence ni les concentrations à l’équilibre, ni le coefficient de transfert global K tant que la phase liquide est saturée. Le modèle de dissolution du premier ordre a pu être validé par rapport aux deux rapports L/S testés expérimentalement dans le cas du phénanthrène et de l’anthracène. La vitesse de relargage est très rapide pendant les premières heures de contact. Le processus est ensuite ralenti pour se stabiliser au delà d’un certain temps de contact, dépendant entre autres des caractéristiques du HAP et des paramètres expérimentaux (L/S,…). Les valeurs de K ont été déterminées en calant la simulation sur la partie stationnaire du processus : 3,5 et 4.10-6 m3.h-1 respectivement pour le phénanthrène et l’anthracène.
116
Chapitre V : Application du batch percolant – Approche méthodologique
V.3.3.2 Influence du cosolvant
Dans cette partie nous comparons les résultats des études de vitesse de solubilisation réalisées à 22°C avec de l’eau et respectivement 3 fractions volumiques de méthanol : 0,3 - 0,4 - 0,5. L’objectif était d’évaluer l’influence du méthanol sur le mécanisme de dissolution en comparant les coefficients de transfert global K identifiés par la résolution du modèle de premier ordre et respectivement les concentrations à saturation dans les conditions expérimentales de l’essai, présentées dans le Tableau 9. L'étude des vitesses de relargage dans la phase aqueuse (fc=0) a posé quelques problèmes expérimentaux puisque le grand volume de phase liquide nécessaire pour l'analyse a imposé de réaliser plusieurs expérimentations en parallèle (voir le protocole au chapitre IV). Il a donc fallu réaliser 4 batch percolants les plus similaires possibles. Le protocole de remplissage des colonnes a été respecté rigoureusement dans tous les tests. Ceci nous a permis d'utiliser l'hypothèse que le régime hydrodynamique a été le même, pour notamment assimiler chaque colonne à une série de 4 réacteurs parfaitement agités.
Tableau 10: synthèse des conditions expérimentales
L/S
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l (g)
Filt
ratio
n en
en
trée
et s
ortie
de
colo
nne
Q (m
l.h-1
)
T°C
fc=0 6 150 25 Membrane
10µm 30 22
fc=0.3 6 150 25 Membrane
10µm 30 22
fc=0.4 6 150 25 Membrane
10µm 30 22
fc=0.5 6 150 25 Membrane
10µm 30 22
V.3.3.2.1 Résultats
L'évolution des vitesses de relargage du phénanthrène et de l'anthracène en phase liquide est présentée Figure 25. On observe l’atteinte d’un état stationnaire dans tous les cas, généralement dans les premières 50 heures de contact. Le bilan de matière a permis de considérer les solutions comme étant à saturation par rapport aux HAP suivis.
117
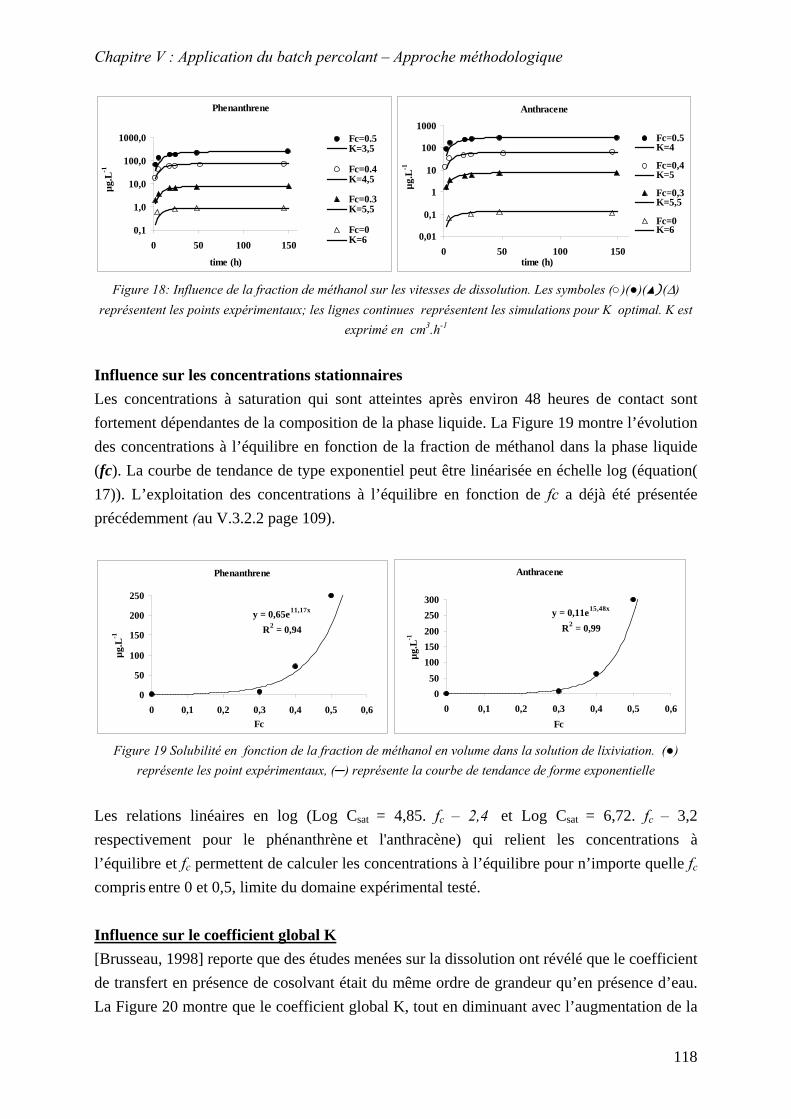
Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
0,1
1,0
10,0
100,0
1000,0
0 50 100 150
time (h)
µg.L
-1
Fc=0.5K=3,5
Fc=0.4K=4,5
Fc=0.3K=5,5
Fc=0K=6
Anthracene
0,01
0,1
1
10
100
1000
0 50 100 150time (h)
µg.L
-1
Fc=0.5K=4
Fc=0,4K=5
Fc=0,3K=5,5
Fc=0K=6
Figure 18: Influence de la fraction de méthanol sur les vitesses de dissolution. Les symboles (○)(●)(▲)(∆)
représentent les points expérimentaux; les lignes continues représentent les simulations pour K optimal. K est exprimé en cm3.h-1
Influence sur les concentrations stationnaires Les concentrations à saturation qui sont atteintes après environ 48 heures de contact sont fortement dépendantes de la composition de la phase liquide. La Figure 19 montre l’évolution des concentrations à l’équilibre en fonction de la fraction de méthanol dans la phase liquide (fc). La courbe de tendance de type exponentiel peut être linéarisée en échelle log (équation( 17)). L’exploitation des concentrations à l’équilibre en fonction de fc a déjà été présentée précédemment (au V.3.2.2 page 109).
Phenanthrene
y = 0,65e11,17x
R2 = 0,94
0
50
100
150
200
250
0 0,1 0,2 0,3 0,4 0,5 0,6Fc
µg.L
-1
Anthracene
y = 0,11e15,48x
R2 = 0,99
050
100150200250300
0 0,1 0,2 0,3 0,4 0,5 0,6Fc
µg.L
-1
Figure 19 Solubilité en fonction de la fraction de méthanol en volume dans la solution de lixiviation. (●)
représente les point expérimentaux, (─) représente la courbe de tendance de forme exponentielle
Les relations linéaires en log (Log Csat = 4,85. fc – 2,4 et Log Csat = 6,72. fc – 3,2 respectivement pour le phénanthrène et l'anthracène) qui relient les concentrations à l’équilibre et fc permettent de calculer les concentrations à l’équilibre pour n’importe quelle fc
compris entre 0 et 0,5, limite du domaine expérimental testé. Influence sur le coefficient global K [Brusseau, 1998] reporte que des études menées sur la dissolution ont révélé que le coefficient de transfert en présence de cosolvant était du même ordre de grandeur qu’en présence d’eau. La Figure 20 montre que le coefficient global K, tout en diminuant avec l’augmentation de la
118

Chapitre V : Application du batch percolant – Approche méthodologique
fraction de méthanol, reste du même ordre de grandeur comme l’affirme Brusseau. Une courbe de tendance établie pour chaque HAP suivis peut relier K au contenu en méthanol dans le domaine expérimental couvert. Cependant il faut noter les difficultés de simulation du régime transitoire de solubilisation des HAP dues d’une part à l’option de privilégier la phase stationnaire et d’autre part à la probable coexistence de plusieurs mécanismes de transfert.
0
2
4
6
8
0 0,1 0,2 0,3 0,4 0,5 0,6
Fc
K.1
0-6Phenanthrene Anthracene
Figure 20: Evolution du coefficient de transfert K [m3.h-1] en
fonction de la fraction de méthanol dans la phase liquide
V.3.3.2.2 Conclusion
La fraction méthanol influence fortement les concentrations à l’équilibre : elles augmentent avec le contenu en méthanol. La relation entre les concentrations à saturation et la fraction de cosolvant peut être décrite par une équation linéaire en échelle log. On peut considérer que le méthanol influence faiblement le coefficient de transfert des polluants contenus dans le sol étudié.
V.3.3.3 Influence de la température
L’objectif de cette partie de l’étude est d’évaluer l’influence de la température sur le mécanisme de dissolution par comparaison des coefficients de transfert global K et des concentrations à saturation (Csat) à 3 températures différentes (15°C, 22°C, 40°C). Les essais ont été réalisés avec une phase liquide composée de 50% de méthanol. Les conditions expérimentales sont présentées dans le Tableau 11.
119
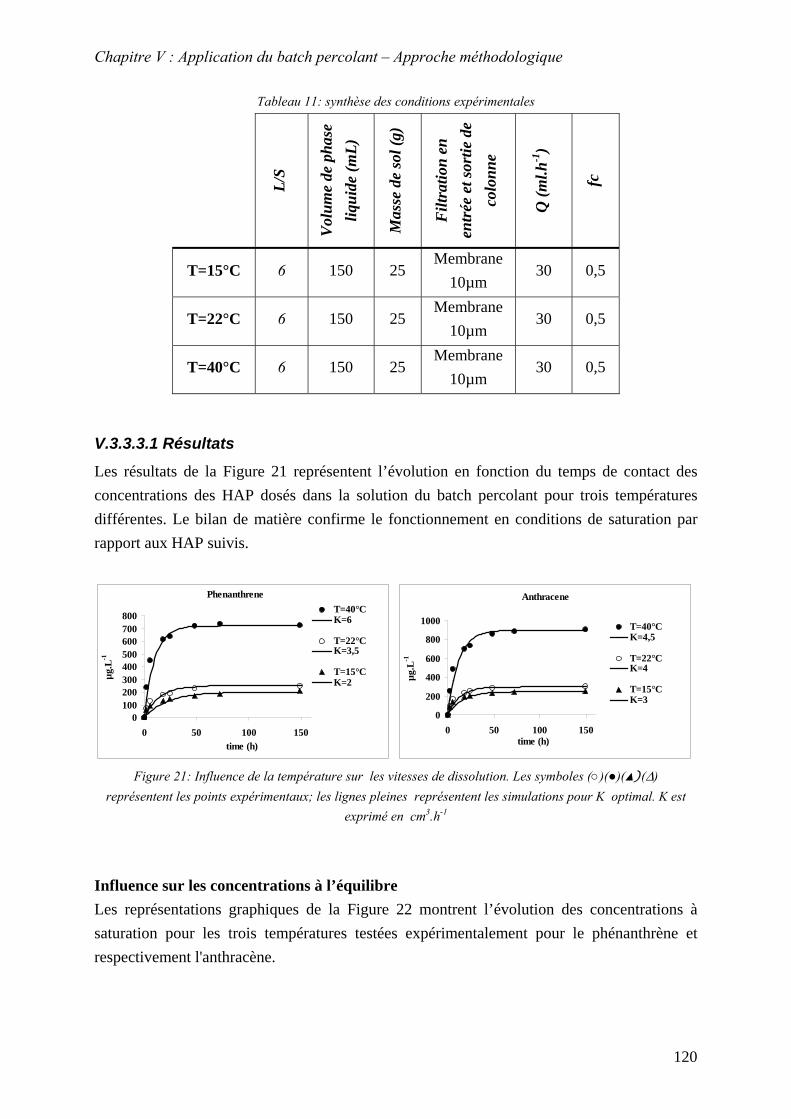
Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 11: synthèse des conditions expérimentales
L/S
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l (g)
Filt
ratio
n en
en
trée
et s
ortie
de
colo
nne
Q (m
l.h-1
)
fc
T=15°C 6 150 25 Membrane
10µm 30 0,5
T=22°C 6 150 25 Membrane
10µm 30 0,5
T=40°C 6 150 25 Membrane
10µm 30 0,5
V.3.3.3.1 Résultats
Les résultats de la Figure 21 représentent l’évolution en fonction du temps de contact des concentrations des HAP dosés dans la solution du batch percolant pour trois températures différentes. Le bilan de matière confirme le fonctionnement en conditions de saturation par rapport aux HAP suivis.
Phenanthrene
0100200300400500600700800
0 50 100 150time (h)
µg.L
-1
T=40°CK=6
T=22°CK=3,5
T=15°CK=2
Anthracene
0
200
400
600
800
1000
0 50 100 150time (h)
µg.L
-1
T=40°CK=4,5
T=22°CK=4
T=15°CK=3
Figure 21: Influence de la température sur les vitesses de dissolution. Les symboles (○)(●)(▲)(∆)
représentent les points expérimentaux; les lignes pleines représentent les simulations pour K optimal. K est exprimé en cm3.h-1
Influence sur les concentrations à l’équilibre Les représentations graphiques de la Figure 22 montrent l’évolution des concentrations à saturation pour les trois températures testées expérimentalement pour le phénanthrène et respectivement l'anthracène.
120

Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
y = 93,10e0,05x
R2 = 0,99
0
200
400
600
800
0 10 20 30 40 50
T (°C)
µg.L
-1
Anthracene
y = 103,45e0,05x
R2 = 0,98
0
200
400
600
800
1000
0 10 20 30 40 50
T (°C)
µg.L
-1
Figure 22 Solubilité en fonction de la température. (●) représente les point expérimentaux, (─) représente la
courbe de tendance de forme exponentielle
Les concentrations à l’équilibre des deux HAP augmentent exponentiellement avec la température dans la gamme 15°C à 40°C. La relation entre la température et la solubilité peut être décrite par l’équation de Van’t Hoff :
(18) 2
lnRT
HdT
d s∆=χ
avec
χ: solubilité du composé dans la phase liquide en fraction molaire, T : température en K, ∆Hs : enthalpie de dissolution (J.mol-1) R : constante des gaz parfaits= 8,31 (J.mol-1.K-1).
En supposant que ∆Hs est constant sur l’échelle de température considérée, l’intégration de (18) conduit à :
(19) CRT
H s +∆−
=χln
Les fractions molaires de l'anthracène et du phénanthrène ont été calculées à partir des concentrations à saturation et de la composition de la phase liquide constituée de 50 % eau et 50 % méthanol. Les concentrations molaires des composés mis en solution lors du contact avec le sol sont supposées négligeables par rapport au nombre de moles formant la solution. Les fractions molaires du phénanthrène et de l'anthracène peuvent être raisonnablement approximées :
(20) méthanoldemoléculedemoledenombreeaudmoléculedemoledenombre
nephénanthrèdemoledenombrenephénanthrè +
='
χ
121

Chapitre V : Application du batch percolant – Approche méthodologique
(21) méthanoldemoléculedemoledenombreeaudmoléculedemoledenombre
anthracènedmoledenombreanthracène +
='
'χ
Supposant l’additivité des volumes, pour un litre de solution (500 ml d'eau et 500 ml de méthanol), le nombre de moles est égal à:
OHCH
OHCH
OH
OHsolutionL M
dMd
n3
3
2
2
221 += (22)
Avec nsolution en mole, dH20 et dCH3OH les densités de l'eau et du méthanol exprimées en g.L-1, et MH20 et MCH3OH les masses molaires exprimées en g.mol-1. La Figure 23 représente en accord avec l’équation (19) les concentrations à saturation en fonction de la température. L’enthalpie de dissolution peut être calculée, pour les deux composés étudiés, à partir de la pente de la droite (ligne (a), Tableau 12).
Phenanthrene
y = -4604,1x - 1,415R2 = 0,9912
-18
-17,5
-17
-16,5
-16
3,10E-03 3,20E-03 3,30E-03 3,40E-03 3,50E-03
1/T (K-1)
Ln (x
)
Anthracene
y = -4827,8x - 0,4941R2 = 0,9739
-18
-17,5
-17
-16,5
-16
-15,5
3,10E-03 3,20E-03 3,30E-03 3,40E-03 3,50E-03
1/T (K-1)
Ln (x
)
Figure 23: Concentration à l’équilibre pour le phénanthrène et l’anthracène en fonction de la température.
Tableau 12: comparaision des enthalpies de dissolution avec la littérature
∆Hs (KJ.mol-1) Phénanthrène Anthracène Conditions exp.
Expérimental (a) 38,2 40,1 Sol, 50% méthanol [Reza, 2002] (b) - 48,8 Composé pur, eau
Le processus est endothermique (∆H<0) : l’augmentation de la température augmente la solubilité. Les conditions expérimentales des deux études comparées dans le tableau 12 sont bien différentes. La comparaison fait apparaître : - Les enthalpies de dissolution (a) du phénanthrène et de l’anthracène ont été calculées, au
cours de nos travaux, à partir d’essai de solubilisation (sol pollué et phase liquide composée de 50% de méthanol) à différentes températures dans le batch percolant.
- L’enthalpie de dissolution de l’anthracène (b) a été calculée à partir des résultats de [Reza,
122

Chapitre V : Application du batch percolant – Approche méthodologique
2002]. Cet auteur a mis au point un dispositif pour déterminer l’influence de la température sur les solubilités des composés purs. Ce dispositif appelé " generator column-on line solid-phase extraction-liquid chromatographic method » est basé sur un contact entre l’eau et des billes de verres enduites du composé pure. L’eau circule entre les billes jusqu’à ce que les concentrations soient constantes (saturation). Le système est thermostaté pour pouvoir réguler la température.
L’enthalpie de dissolution de l’anthracène pur dans l’eau est plus importante que l’enthalpie de dissolution de l’anthracène d’un sol pollué dans une phase liquide composée de 50 % de méthanol. Lorsque l’enthalpie de dissolution d’un composé augmente, les conditions sont moins favorables à la dissolution. ∆Hs, l’enthalpie de dissolution peut être interprétée comme la somme de 4 enthalpies (voir chapitre I). La somme des 4 enthalpies est donc différentes pour (a) et (b), ∆H1 et ∆H3 peuvent probablement expliquer cette différence :
- L’enthalpie ∆H1>0 nécessaire pour rompre les liaisons :anthracéne-anthracène pour le composé pure (cas b) est supérieure à celle nécessaire pour rompre les liaisons anthracène mixture organique dans le sol pollué. - L’enthalpie ∆H3<0 libérée par la solvatation de l’anthracène est plus importante dans une phase liquide composé de 50% de méthanol que dans une phase aqueuse pure.
En conclusion, les valeurs des enthalpies de dissolution calculées suite à notre expérimentation sont comparables aux valeurs publiées dans la littérature. Les corrélations linéaires déterminées dans la Figure 23 sont valables uniquement pour une phase liquide composée de 50% de méthanol. En effet la pente de la droite, ∆Hs (notamment ∆H3), peut varier selon la fraction de cosolvant. Influence de la température sur le coefficient global K Le coefficient K a été identifié par l’application du modèle de dissolution aux résultats expérimentaux (Figure 23). On peut voir dans la Figure 24 que, lorsque la température passe de 15 à 40°C, le coefficient de transfert global K (phénanthrène et anthracène) augmente jusqu’à 3 fois pour le phénanthrène, tout en restant dans le même ordre de grandeur. C’est probablement la vitesse de transfert au travers du film interfacial de dissolution qui est activée sous l’effet de la température.
123

Chapitre V : Application du batch percolant – Approche méthodologique
0
2
4
6
8
0 10 20 30 40 50
T°C
K.1
0-6
Phenanthrene Anthracene
Figure 24: Evolution de K (m3.h-1) en fonction de la température
En considérant le processus de dissolution de type Arrhenius, nous pouvons identifier les énergies d’activation pour les deux HAP suivis. L’application de la définition du coefficient global permet d’écrire :
(23) TREa
eASSkK−
== *
et en considérant la surface de transfert constante pour deux températures voisines, on obtient :
(24) ⎟⎟⎠
⎞⎜⎜⎝
⎛−
= 12
11
2
1 TTREa
eKK
et respectivement :
(25) ⎟⎟⎠
⎞⎜⎜⎝
⎛−
=2
1
21
21 lnKK
RTT
TTEa
En appliquant la relation (25) on calcule l’énergie d’activation du phénanthrène de 33 kJ mol-1 et respectivement de l’anthracène de 12 kJ mol-1, valeurs qui se situent dans le domaine énergétique des processus physiques.
V.3.3.3.2 Conclusion
La température influence fortement la concentration à l’équilibre : l'augmentation de la température entraîne la modification du coefficient de transfert global K (dépendance de type Arrhenius). L’énergie d’activation du processus de dissolution est plus importante pour le phénanthrène que pour l’anthracène, tout en restant dans les limites des processus physiques. L'évolution en fonction de la température des concentrations en HAP lors du contact sol pollué / solution eau/méthanol est décrite par la loi de van’t Hoff.
124

Chapitre V : Application du batch percolant – Approche méthodologique
V.3.3.4 Influence du débit
L’influence du débit sur le mécanisme de dissolution a été évaluée en comparant les valeurs du coefficient de transfert global K et les concentrations de saturation pour deux débits distincts (q=30 ml.h-1 et q=120 ml.h-1). Les essais ont été réalisés avec une phase liquide composée de 50% de méthanol. Les conditions expérimentales de l’essai sont présentées dans le Tableau 13.
Tableau 13: synthèse des conditions expérimentales
L/S
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l (g)
Filt
ratio
n en
ent
rée
et so
rtie
de
colo
nne
T°C
fc
Q=30 ml.h-1 6 150 25 Membrane
10µm 22 0,4
Q=120 ml.h-1 6 150 25 Membrane
10µm 22 0,4
V.3.3.4.1 Résultats
Les vitesses de relargage du phénanthrène et de l'anthracène pour les deux débits testés sont présentées Figure 25. Lors du premier prélèvement réalisé après 1,5 heures de recirculation, les concentrations dans le réacteur tampon sont nettement supérieures pour un débit de 120 ml.h-1.
Phenanthrene
01020304050607080
0 50 100 150time (h)
µg.L
-1
q=30 ml/hK=4,5
q=120 ml/hK=4,5
Anthracène
010203040506070
0 50 100 150time (h)
µg.L
-1
q=30 ml/hK=5
q=120 ml/hK=5
Figure 25:Influence du débit sur la vitesse de dissolution. . (●),(○) représentent les point
expérimentaux, les lignes pleines représentent la simulation pour K optimal. K est exprimé en cm3.h-1
125

Chapitre V : Application du batch percolant – Approche méthodologique
Influence sur les concentrations à l’équilibre Les concentrations à l’équilibre du phénanthrène et de l’anthracène sont identiques pour un débit de 30 ml.h-1 ou 120 ml.h-1. Nous avons vu précédemment que les concentrations à l’équilibre peuvent être calculées par la loi de Raoult. Cette loi suppose que la concentration à l’équilibre est fonction de la fraction molaire du composé dans la phase polluée du sol (1) et de la solubilité du composé pur (2) dans la phase liquide. (1) et (2) sont indépendants du débit, il est donc normal que le débit n’influence pas les concentrations à l’équilibre. Influence sur le coefficient de transfert K Globalement (Figure 25), il n’y a pas d’influence significative du débit sur le coefficient de transfert K pendant les 150 heures de recirculation. Dans le chapitre I, nous avons vu qu’une augmentation significative du débit pouvait diminuer l’épaisseur du film de dissolution, ce qui aurait pour effet d’augmenter le transfert. Dans notre cas l’hydrodynamique de l’écoulement est différente au débit 3 fois plus fort et probablement que le modèle utilisé (série de 4 ROPA) est inadapté pour mettre en évidence ce phénomène.
V.3.3.4.2 Conclusion
L’augmentation du débit de recirculation n’influence pas les concentrations à l’équilibre mais augmente la vitesse du processus de transfert en solution dans la partie transitoire. Même si le débit n’influence pas fondamentalement la nature et la hiérarchie des mécanismes de solubilisation des HAP, des variations importantes de débit amènent des modifications de fonctionnement de la colonne, notamment au niveau de la diffusivité ou du nombre des ROPA.
V.3.3.5 Conclusion : mèthode d’estimation graphique (abaque) de la concentration stationnaire
Le Tableau 14 résume les informations principales obtenues lors des études d'influence des paramètres physico-chimiques sur les vitesses de relargage des HAP en phases liquides méthanol/eau dans le cadre de l’application du test batch percolant.
126
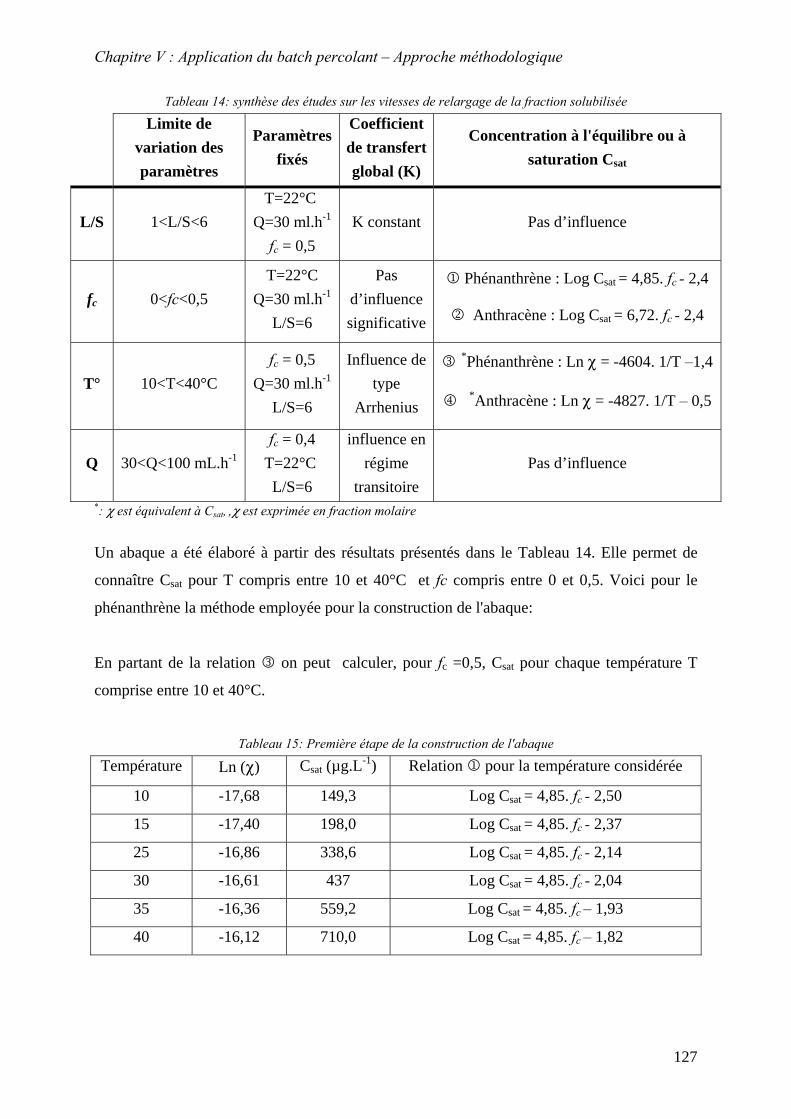
Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 14: synthèse des études sur les vitesses de relargage de la fraction solubilisée
Limite de variation des paramètres
Paramètres fixés
Coefficient de transfert global (K)
Concentration à l'équilibre ou à saturation Csat
L/S 1<L/S<6 T=22°C
Q=30 ml.h-1
fc = 0,5 K constant Pas d’influence
fc 0<fc<0,5 T=22°C
Q=30 ml.h-1
L/S=6
Pas d’influence significative
1 Phénanthrène : Log Csat = 4,85. fc - 2,4
2 Anthracène : Log Csat = 6,72. fc - 2,4
T° 10<T<40°C fc = 0,5
Q=30 ml.h-1
L/S=6
Influence de type
Arrhenius
3 *Phénanthrène : Ln χ = -4604. 1/T –1,4
4 *Anthracène : Ln χ = -4827. 1/T – 0,5
Q 30<Q<100 mL.h-1fc = 0,4
T=22°C L/S=6
influence en régime
transitoire Pas d’influence
*: χ est équivalent à Csat, ,χ est exprimée en fraction molaire
Un abaque a été élaboré à partir des résultats présentés dans le Tableau 14. Elle permet de
connaître Csat pour T compris entre 10 et 40°C et fc compris entre 0 et 0,5. Voici pour le
phénanthrène la méthode employée pour la construction de l'abaque:
En partant de la relation 3 on peut calculer, pour fc =0,5, Csat pour chaque température T
comprise entre 10 et 40°C.
Tableau 15: Première étape de la construction de l'abaque
Température Ln (χ) Csat (µg.L-1) Relation 1 pour la température considérée
10 -17,68 149,3 Log Csat = 4,85. fc - 2,50
15 -17,40 198,0 Log Csat = 4,85. fc - 2,37
25 -16,86 338,6 Log Csat = 4,85. fc - 2,14
30 -16,61 437 Log Csat = 4,85. fc - 2,04
35 -16,36 559,2 Log Csat = 4,85. fc – 1,93
40 -16,12 710,0 Log Csat = 4,85. fc – 1,82
127
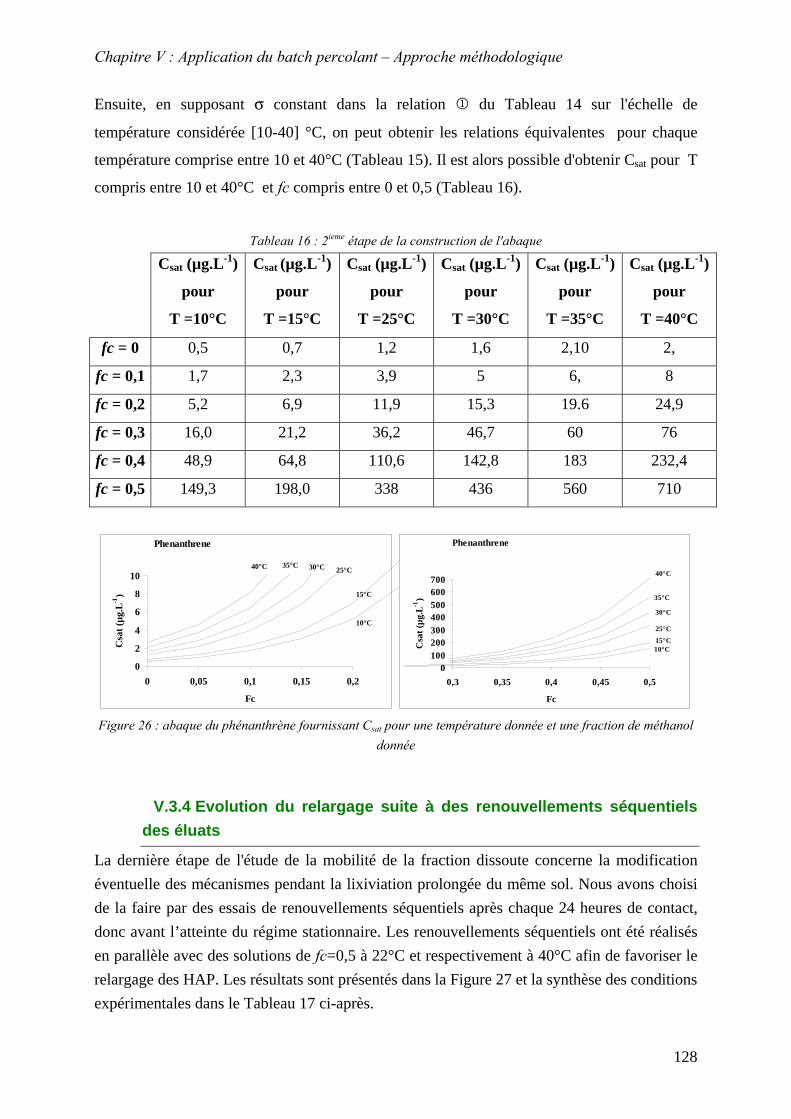
Chapitre V : Application du batch percolant – Approche méthodologique
Ensuite, en supposant σ constant dans la relation 1 du Tableau 14 sur l'échelle de
température considérée [10-40] °C, on peut obtenir les relations équivalentes pour chaque
température comprise entre 10 et 40°C (Tableau 15). Il est alors possible d'obtenir Csat pour T
compris entre 10 et 40°C et fc compris entre 0 et 0,5 (Tableau 16).
Tableau 16 : 2ieme étape de la construction de l'abaque
Csat (µg.L-1)
pour
T =10°C
Csat (µg.L-1)
pour
T =15°C
Csat (µg.L-1)
pour
T =25°C
Csat (µg.L-1)
pour
T =30°C
Csat (µg.L-1)
pour
T =35°C
Csat (µg.L-1)
pour
T =40°C
fc = 0 0,5 0,7 1,2 1,6 2,10 2,
fc = 0,1 1,7 2,3 3,9 5 6, 8
fc = 0,2 5,2 6,9 11,9 15,3 19.6 24,9
fc = 0,3 16,0 21,2 36,2 46,7 60 76
fc = 0,4 48,9 64,8 110,6 142,8 183 232,4
fc = 0,5 149,3 198,0 338 436 560 710
Phenanthrene
0
2
4
6
8
10
0 0,05 0,1 0,15 0,2
Fc
Csa
t (µg
.L-1
)
40°C 35°C 30°C 25°C
15°C
10°C
Phenanthrene
0100200300400500600700
0,3 0,35 0,4 0,45 0,5
Fc
Csa
t (µg
.L-1
)
40°C
35°C
30°C
25°C15°C10°C
Figure 26 : abaque du phénanthrène fournissant Csat pour une température donnée et une fraction de méthanol
donnée
V.3.4 Evolution du relargage suite à des renouvellements séquentiels des éluats
La dernière étape de l'étude de la mobilité de la fraction dissoute concerne la modification éventuelle des mécanismes pendant la lixiviation prolongée du même sol. Nous avons choisi de la faire par des essais de renouvellements séquentiels après chaque 24 heures de contact, donc avant l’atteinte du régime stationnaire. Les renouvellements séquentiels ont été réalisés en parallèle avec des solutions de fc=0,5 à 22°C et respectivement à 40°C afin de favoriser le relargage des HAP. Les résultats sont présentés dans la Figure 27 et la synthèse des conditions expérimentales dans le Tableau 17 ci-après.
128
Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 17: synthèse des conditions expérimentales
L/S
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l (g)
Filt
ratio
n en
en
trée
et s
ortie
de
col
onne
q (m
l.h-1
)
fc
T=22°C 6 150 25 Membrane
10µm 22 0,5
T=40°C 6 150 25 Membrane
10µm 22 0,5
V.3.4.1 Résultats
Les renouvellements séquentiels sont toujours effectués après 24 heures de recirculation. D’après les études précédentes sur les vitesses de relargage, ce temps de contact ne permet pas d’atteindre le régime stationnaire ni à T=22°C ni à T=40°C. Donc, chaque renouvellement est fait dans des conditions a priori transitoires (non stationnaires) de relargage des HAP par le sol. La Figure 27 présente les résultats des dosages de phénanthrène et anthracène dans les 9 éluats successifs (R1-R9) obtenus chacun après 24 h de recirculation avec un débit q=22 ml/h. Les concentrations en phénanthrène et en anthracène, avec la phase liquide à 22°C, sont pratiquement constantes lors de tous les renouvellements de R1 à R9: voisines 200 µg.L-1 (Figure 27). Les concentrations en régime stationnaire ou à saturation (Csat) obtenues expérimentalement à 22 °C lors de l'étude des vitesses de relargage étaient respectivement 250 et 300 µg.L-1 pour le phénanthrène et l'anthracène. Ceci permet de valider l’hypothèse du régime dynamique transitoire de relargage.
129

Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
0100200300400500600700800
R1 R2 R3 R4 R5 R6 R7 R8 R9
µg.L
-1T=40°CT=22°C
Anthracene
0
200
400
600
800
1000
R1 R2 R3 R4 R5 R6 R7 R8 R9
µg.L
-1
T=40°CT=22°C
Figure 27: Evolution des concentrations après chaque renouvellement séquentiel R de 24 h à 22 et 40°C. Les
symboles (•) et(∆) représentent les point expérimentaux. Les symboles (-) et (▬) sont les valeurs théoriques, respectivement à 22 et 40 °C, des concentrations à l'équilibre calculées d'après la loi de Raoult
Par contre on observe une nette diminution des concentrations en phénanthrène et de l’anthracène pour T=40°C lors des étapes de renouvellements successifs de 24 heures, surtout après R5. La concentration du phénanthrène passe d'une valeur légèrement inférieure à 700 µg.L-1 pour le deuxième renouvellement (la valeur de Csat à 40 °C était de 730 µg.L-1) à environ 300 µg.L-1 au neuvième renouvellement. En ce qui concerne l'anthracène (Csat=900 µg.L-1 à 40°C), la concentration passe de 840 µg.L-1 lors du deuxième renouvellement à une valeur voisine de 400 µg.L-1 au neuvième renouvellement. Afin de comprendre l'origine de la diminution significative des concentrations observée à 40°C, nous avons calculé avec la loi de Raoult, pour chaque renouvellement séquentiel, l'évolution théorique des concentrations à l'équilibre:
(26) 5,0,,,5,0, == ⋅= fcimixieqfci SC χ
avec
Ci,fc=0,5,eq : Concentration à l'équilibre du composé i obtenue par le contact du sol avec la phase liquide constituée, en volume, de 50% eau et 50% méthanol (fc=0,5).
mixi,χ : fraction molaire du composé i dans la mixture organique
Si,fc=0,5 : Solubilité du composé i pure dans un mélange eau/méthanol (fc=0,5) à 40 °C (mol.L-1)
Préalablement, il fallait déterminer la constante Si,fc=0,5. La valeur de Si,fc=0,5 a été obtenue à partir de l'étude des vitesses de relargage des HAP dans la phase liquide eau/méthanol (fc=0,5) à 40 °C. En effet, par le biais de cette étude, nous connaissions les valeurs de Ci,fc=0,5,eq à 40 °C respectivement 730 et 900 µg.L-1 pour le phénanthrène et l'anthracène. Nous avons alors calculer la fraction molaire du phénanthrène et de l'anthracène correspondante
130

Chapitre V : Application du batch percolant – Approche méthodologique
(voir méthode de calcul page 101) pour en déduire la valeur de la constante Si,fc=0,5 à 40°C. Nous avons obtenu: 0,007 mol.L-1 pour le phénanthrène et 0,0045 pour l'anthracène. Il était alors possible de calculer avec la relation (26) les concentrations à saturation dans chaque renouvellement en tenant compte de l'évolution des fractions molaires. Les valeurs théoriques des concentrations à l'équilibre calculées sont représentées dans la Figure 27. Pour information, la solubilité du phénanthrène pur à 25 °C dans une phase liquide composé de 50% de méthanol (fc=0,5) est de 0,004 mol.L-1 [Rezza, 2002]. La comparaison des concentrations calculées à l'équilibre et des concentrations expérimentales obtenues avec 50% de méthanol (fc=0,5) au cours des renouvellements successifs montre un décalage croissant : les concentrations calculées à l'équilibre sont logiquement supérieures aux concentrations expérimentales puisque ces dernières correspondent à un temps de contact avec le sol de 24 heures, temps insuffisant pour atteindre le régime stationnaire. On peut observer que ce décalage est plus important pour T=40°C, que pour 22°C. La représentation schématique suivante est un « zoom » du constat expérimental (après R5).
C eq
C exp
teq tR
Ri+1Ri Ri+2 Ri+n
tR tR tR teq
C eq C eq
C exp
C exp
C exp
1
2
Figure 28:Représentation schématique de l'évolution des vitesses de
dissolution au cours de renouvellements séquentiels
Dans ce schéma, sont représentées les vitesses de relargage des HAP dans la phase liquide pour n renouvellements séquentiels. La courbe 1 décrit la diminution des concentrations théoriques à l'équilibre suite à la diminution de la fraction molaire du composé dans la mixture organique du sol. La courbe 2 quant à elle décrit la tendance d'évolution des concentrations expérimentales, concentrations obtenues pour chaque renouvellement séquentiel de 24 heures. La pente de la droite 1 est inférieure à la pente de la droite 2: la décroissance de la concentration expérimentale est plus rapide. Ce comportement expérimental peut être expliqué par la modification du processus de relargage des HAP à partir du mélange contenu dans le sol, par exemple par l’augmentation du temps d’atteinte de
131
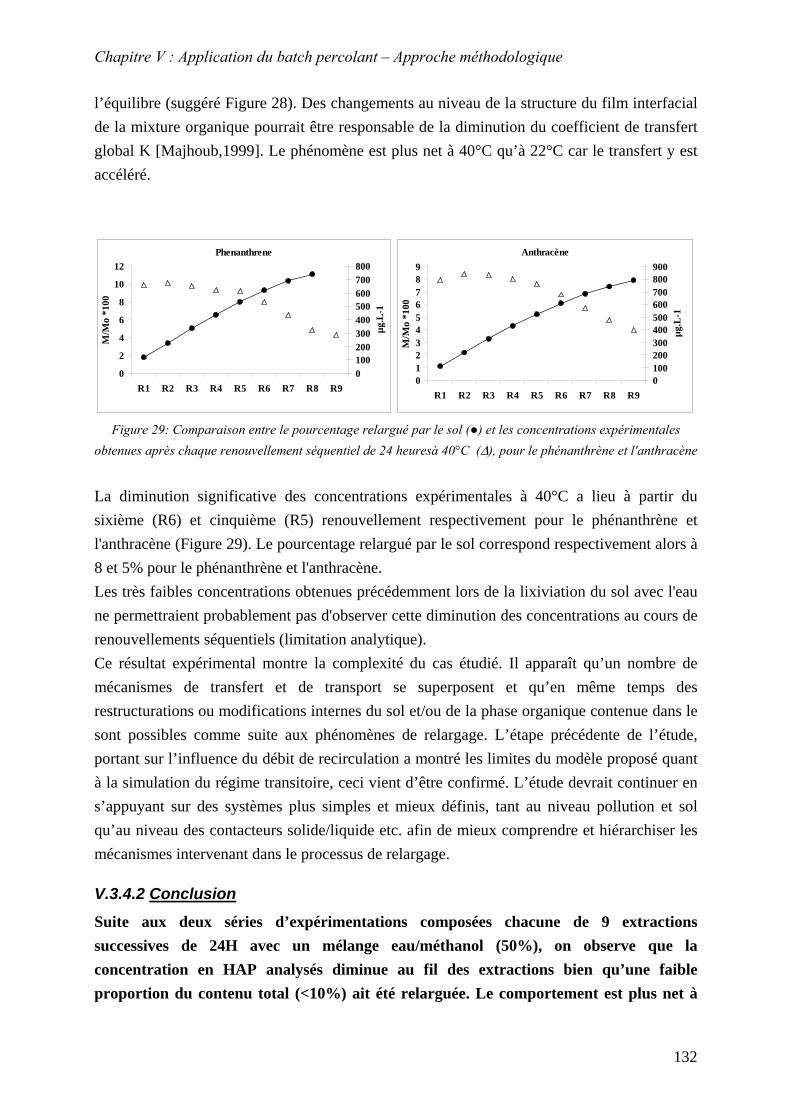
Chapitre V : Application du batch percolant – Approche méthodologique
l’équilibre (suggéré Figure 28). Des changements au niveau de la structure du film interfacial de la mixture organique pourrait être responsable de la diminution du coefficient de transfert global K [Majhoub,1999]. Le phénomène est plus net à 40°C qu’à 22°C car le transfert y est accéléré.
Phenanthrene
0
2
4
6
8
10
12
R1 R2 R3 R4 R5 R6 R7 R8 R9
M/M
o *1
00
0100200300400500600700800
µg.L
-1
Anthracène
0123456789
R1 R2 R3 R4 R5 R6 R7 R8 R9M
/Mo
*100
0100200300400500600700800900
µg.L
-1
Figure 29: Comparaison entre le pourcentage relargué par le sol (●) et les concentrations expérimentales
obtenues après chaque renouvellement séquentiel de 24 heuresà 40°C (∆), pour le phénanthrène et l'anthracène
La diminution significative des concentrations expérimentales à 40°C a lieu à partir du sixième (R6) et cinquième (R5) renouvellement respectivement pour le phénanthrène et l'anthracène (Figure 29). Le pourcentage relargué par le sol correspond respectivement alors à 8 et 5% pour le phénanthrène et l'anthracène. Les très faibles concentrations obtenues précédemment lors de la lixiviation du sol avec l'eau ne permettraient probablement pas d'observer cette diminution des concentrations au cours de renouvellements séquentiels (limitation analytique). Ce résultat expérimental montre la complexité du cas étudié. Il apparaît qu’un nombre de mécanismes de transfert et de transport se superposent et qu’en même temps des restructurations ou modifications internes du sol et/ou de la phase organique contenue dans le sont possibles comme suite aux phénomènes de relargage. L’étape précédente de l’étude, portant sur l’influence du débit de recirculation a montré les limites du modèle proposé quant à la simulation du régime transitoire, ceci vient d’être confirmé. L’étude devrait continuer en s’appuyant sur des systèmes plus simples et mieux définis, tant au niveau pollution et sol qu’au niveau des contacteurs solide/liquide etc. afin de mieux comprendre et hiérarchiser les mécanismes intervenant dans le processus de relargage.
V.3.4.2 Conclusion
Suite aux deux séries d’expérimentations composées chacune de 9 extractions successives de 24H avec un mélange eau/méthanol (50%), on observe que la concentration en HAP analysés diminue au fil des extractions bien qu’une faible proportion du contenu total (<10%) ait été relarguée. Le comportement est plus net à
132
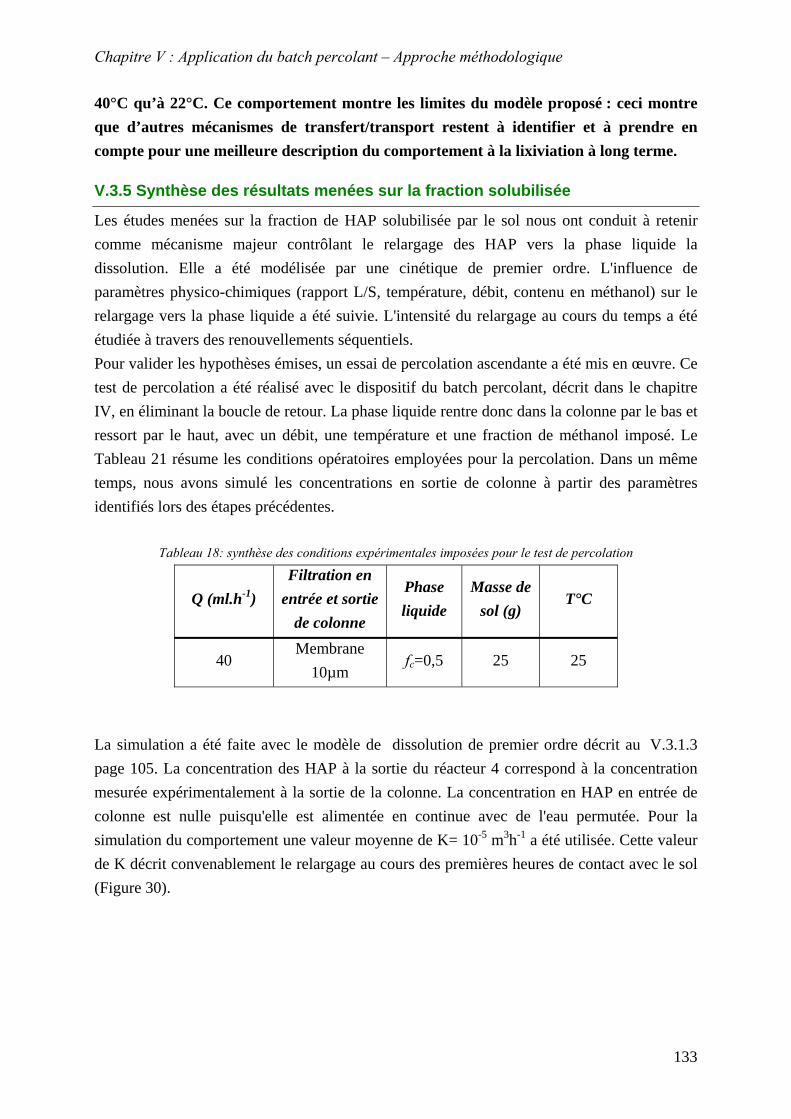
Chapitre V : Application du batch percolant – Approche méthodologique
40°C qu’à 22°C. Ce comportement montre les limites du modèle proposé : ceci montre que d’autres mécanismes de transfert/transport restent à identifier et à prendre en compte pour une meilleure description du comportement à la lixiviation à long terme.
V.3.5 Synthèse des résultats menées sur la fraction solubilisée
Les études menées sur la fraction de HAP solubilisée par le sol nous ont conduit à retenir comme mécanisme majeur contrôlant le relargage des HAP vers la phase liquide la dissolution. Elle a été modélisée par une cinétique de premier ordre. L'influence de paramètres physico-chimiques (rapport L/S, température, débit, contenu en méthanol) sur le relargage vers la phase liquide a été suivie. L'intensité du relargage au cours du temps a été étudiée à travers des renouvellements séquentiels. Pour valider les hypothèses émises, un essai de percolation ascendante a été mis en œuvre. Ce test de percolation a été réalisé avec le dispositif du batch percolant, décrit dans le chapitre IV, en éliminant la boucle de retour. La phase liquide rentre donc dans la colonne par le bas et ressort par le haut, avec un débit, une température et une fraction de méthanol imposé. Le Tableau 21 résume les conditions opératoires employées pour la percolation. Dans un même temps, nous avons simulé les concentrations en sortie de colonne à partir des paramètres identifiés lors des étapes précédentes.
Tableau 18: synthèse des conditions expérimentales imposées pour le test de percolation
Q (ml.h-1) Filtration en
entrée et sortie de colonne
Phase liquide
Masse de sol (g)
T°C
40 Membrane
10µm fc=0,5 25 25
La simulation a été faite avec le modèle de dissolution de premier ordre décrit au V.3.1.3 page 105. La concentration des HAP à la sortie du réacteur 4 correspond à la concentration mesurée expérimentalement à la sortie de la colonne. La concentration en HAP en entrée de colonne est nulle puisqu'elle est alimentée en continue avec de l'eau permutée. Pour la simulation du comportement une valeur moyenne de K= 10-5 m3h-1 a été utilisée. Cette valeur de K décrit convenablement le relargage au cours des premières heures de contact avec le sol (Figure 30).
133
Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
0
50
100
150
200
250
300
0 50 100 150time (h)
µg.L
-1
K=10-3 - 10-5 - 5.10-6 - 3,5.10-6 - 2.10-6 - 10-7
Transfert très rapidePhenanthrene : T=25°C ; fc=0,5 ---> Csat=350 µg.L-1
et K=10-5
0
100
200
300
400
0 20 40 60
L/S cum.
C m
oyen
(µg.
L-1
)
q = 2 ml.h-1
q = 40 ml.h-1
q = 100 ml.h-1
Figure 30 : Sensibilité de K pour la courbe
expérimentale L/S=6 représentée par le symbole (o) Résultats obtenus page 116
Figure 31: Concentration en sortie de la colonne. (●) représente les points expérimentaux et les lignes
pleines les simulation
L’abaque Figure 26 donne pour fc=0,5 et T=25°C, une concentration à saturation égale à 350 µg.L-1. Expérimentalement, la valeur de la concentration à la sortie de colonne dépend du débit imposé à la colonne, puisqu’il agit directement sur le temps de contact. La Figure 31 montre les simulations et les concentrations expérimentales obtenues. En abscisse est représenté le rapport L/Scum qui correspond au volume de phase liquide (mL) qui a percolé au travers des 25 g de sol. Les résultats expérimentaux sont en accord avec la simulation. Comme supposé, la phase liquide à la sortie de la colonne n’est pas saturée en phénanthrène. Le temps de contact avec un débit de 2 ml.h-1 est plus important ce qui laisse le temps à la phase liquide de se saturer, comme le montre la Figure 31. Un fort débit quant à lui engendre un faible temps de contact, la concentration en sortie de colonne est alors loin de la saturation. Selon les résultats, de l’étude de l'évolution du relargage au cours du temps réalisée précédemment, la concentration en phénanthrène à la sortie de la colonne devrait à long terme diminuer, phénomène expérimental observé pour L/S= 25 et T = 40°C. Rappelons dans ce contexte que les tests d’évaluation du comportement à long terme actuellement en cours de normalisation ne dépassent pas le rapport de L/S cumulé 10.
V.4 Mobilité de la fraction particulaire
En préalable à l'étude de la mobilité des HAP dans le sol, nous avons réalisé des essais pour mettre en évidence la participation d'une fraction particulaire dans le processus de relargage des HAP (voir page 95) . Cette première étape a consisté à réaliser des renouvellements séquentiels avec les conditions expérimentales suivantes : L/S=6 (150 ml d'eau et 25 g de sol) ; T=22°C ; Q=30 mL.h-1 et un lit de sable de granulométrie [0-400] µm en entrée et sortie de colonne. Ces premiers résultats ont permis de démontrer la nécessité de considérer la fraction particulaire comme une deuxième voie de relargage significatif des HAP. Les résultats analytiques présentés ci-après sur la fraction particulaire correspondent au
134

Chapitre V : Application du batch percolant – Approche méthodologique
relargage global vers la phase liquide (fraction solubilisée + fraction particulaire). Cependant, les études sur la fraction solubilisée, menées précédemment, permettent de calculer la contribution du relargage par solubilisation et ainsi d’obtenir des informations sur le relargage des HAP associés au transport particulaire. Dans cette partie, nous avons respecté le même schéma développé pour l’étude de la solubilité, à savoir les étapes:
• Description du mécanisme de relargage vers la phase liquide. • Influence de paramètres physico-chimiques pertinents (débits, températures…) sur les
mécanismes de relargage. • Evolution du relargage au cours du temps.
Le relargage des HAP associés aux formes particulaires est dû à la déstabilisation/déstructuration du sol. Nous n'avons pas étudié spécifiquement cet aspect, nous nous sommes plutôt orientés vers la détermination de la nature de ces particules. L'influence de paramètres jugés pertinents (bio-réactivité, sels dissous, débit et quantité de sol en contact avec la phase liquide) sur le relargage des HAP particulaires a été étudiée.
V.4.1 Détermination de la nature des particules au MEB environnemental
Un essai de renouvellements séquentiels avec de l’eau et un filtre en sortie et entrée de colonne constitué d'un lit de sable de granulométrie [0-400] µm a été réalisé selon un protocole identique à celui présenté au V.2 page 95 . A la fin du troisième renouvellement, renouvellement où le transfert particulaire est le plus significatif (R3), la solution du réacteur tampon a été mise en contact avec l’air, à l'abri de la lumière, jusqu’à l’évaporation totale de l’eau. Les particules déposées au fond du flacon, suite à l'évaporation totale de l'eau, ont été prélevées et analysées au MEB environnemental (les caractéristiques de cet appareil sont présentées en annexe). Les photos sont présentées Figure 33 et leurs caractéristiques dans le Tableau 19. Dans la Figure 32 se trouvent les résultats des analyses élémentaires de chaque photographie. L'analyse élémentaire des particules inclut les molécules peu volatiles (HAP…) initialement présentes en phase dissoute avant évaporation de la solution. Cependant, on suppose que l'analyse élémentaire (particules solides + molécules dissoutes peu volatiles) est représentative de la composition des particules. En effet, la contribution, à l'analyse élémentaire, des atomes constitutifs des molécules peu volatils est négligeable par rapport aux nombres d'atomes constitutifs des particules.
135
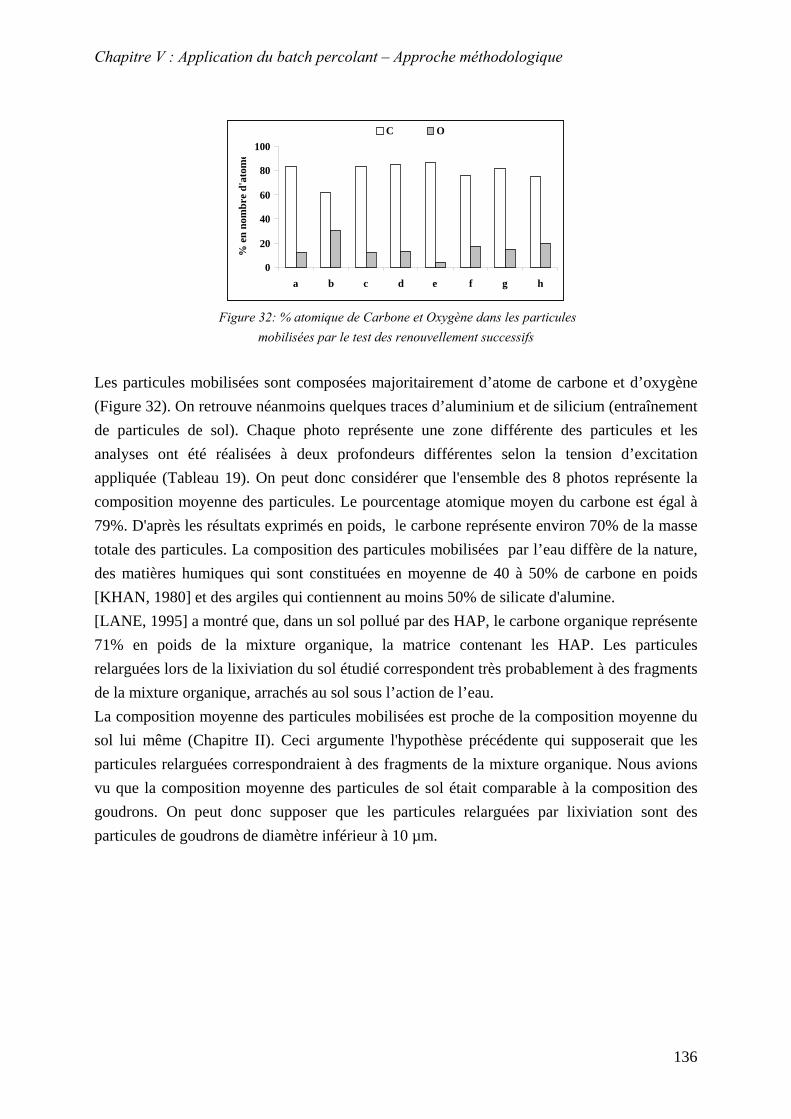
Chapitre V : Application du batch percolant – Approche méthodologique
0
20
40
60
80
100
a b c d e f g h
% e
n no
mbr
e d'
atom
e
C O
Figure 32: % atomique de Carbone et Oxygène dans les particules
mobilisées par le test des renouvellement successifs
Les particules mobilisées sont composées majoritairement d’atome de carbone et d’oxygène (Figure 32). On retrouve néanmoins quelques traces d’aluminium et de silicium (entraînement de particules de sol). Chaque photo représente une zone différente des particules et les analyses ont été réalisées à deux profondeurs différentes selon la tension d’excitation appliquée (Tableau 19). On peut donc considérer que l'ensemble des 8 photos représente la composition moyenne des particules. Le pourcentage atomique moyen du carbone est égal à 79%. D'après les résultats exprimés en poids, le carbone représente environ 70% de la masse totale des particules. La composition des particules mobilisées par l’eau diffère de la nature, des matières humiques qui sont constituées en moyenne de 40 à 50% de carbone en poids [KHAN, 1980] et des argiles qui contiennent au moins 50% de silicate d'alumine. [LANE, 1995] a montré que, dans un sol pollué par des HAP, le carbone organique représente 71% en poids de la mixture organique, la matrice contenant les HAP. Les particules relarguées lors de la lixiviation du sol étudié correspondent très probablement à des fragments de la mixture organique, arrachés au sol sous l’action de l’eau. La composition moyenne des particules mobilisées est proche de la composition moyenne du sol lui même (Chapitre II). Ceci argumente l'hypothèse précédente qui supposerait que les particules relarguées correspondraient à des fragments de la mixture organique. Nous avions vu que la composition moyenne des particules de sol était comparable à la composition des goudrons. On peut donc supposer que les particules relarguées par lixiviation sont des particules de goudrons de diamètre inférieur à 10 µm.
136

Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 19: caractéristiques des photos prises au MEB environnemental
PhotoFacteur de
grossissement Tension d’excitation
(Kv) a 2000 25b 4000 20c 4000 20d 8000 20e 8000 20f 8000 25
a
b
c
d
f
Figure 33: Photographies des particules mobilisées ; réalisées au MEB environnemental
V.4.2 Influence de paramètres physico-chimiques sur le relargage des HAP sous forme particulaire
L'influence de la bio-réactivité et des sels dissous (force ionique) sur le relargage des HAP associés à des particules a été étudiée à travers des essais de renouvellements séquentiels. Tandis que l'influence du débit et de la quantité de sol en contact avec la phase liquide a été
137
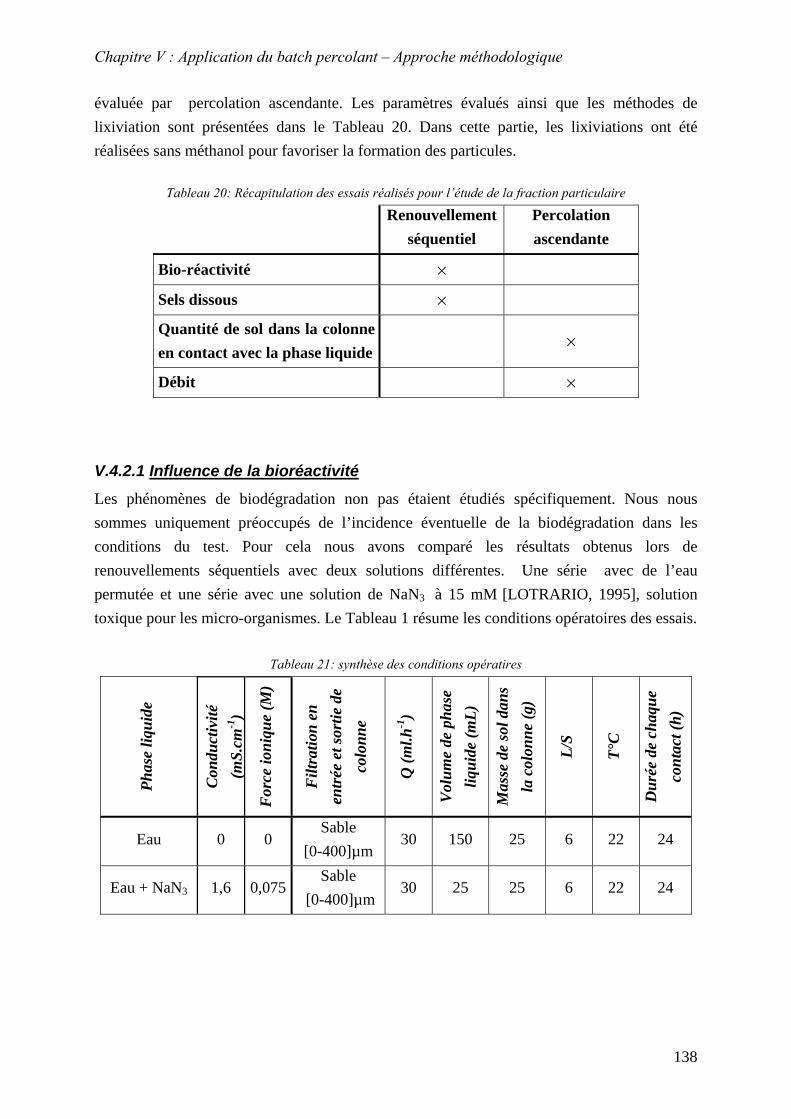
Chapitre V : Application du batch percolant – Approche méthodologique
évaluée par percolation ascendante. Les paramètres évalués ainsi que les méthodes de lixiviation sont présentées dans le Tableau 20. Dans cette partie, les lixiviations ont été réalisées sans méthanol pour favoriser la formation des particules.
Tableau 20: Récapitulation des essais réalisés pour l’étude de la fraction particulaire
Renouvellement séquentiel
Percolation ascendante
Bio-réactivité ×
Sels dissous ×
Quantité de sol dans la colonne en contact avec la phase liquide
×
Débit ×
V.4.2.1 Influence de la bioréactivité
Les phénomènes de biodégradation non pas étaient étudiés spécifiquement. Nous nous sommes uniquement préoccupés de l’incidence éventuelle de la biodégradation dans les conditions du test. Pour cela nous avons comparé les résultats obtenus lors de renouvellements séquentiels avec deux solutions différentes. Une série avec de l’eau permutée et une série avec une solution de NaN3 à 15 mM [LOTRARIO, 1995], solution toxique pour les micro-organismes. Le Tableau 1 résume les conditions opératoires des essais.
Tableau 21: synthèse des conditions opératires
Phas
e liq
uide
Con
duct
ivité
(m
S.cm
-1)
For
ce io
niqu
e (M
)
Filt
ratio
n en
en
trée
et s
ortie
de
colo
nne
Q (m
l.h-1
)
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l dan
s la
col
onne
(g)
L/S
T°C
Dur
ée d
e ch
aque
co
ntac
t (h)
Eau 0 0 Sable
[0-400]µm 30 150 25 6 22 24
Eau + NaN3 1,6 0,075 Sable
[0-400]µm 30 25 25 6 22 24
138
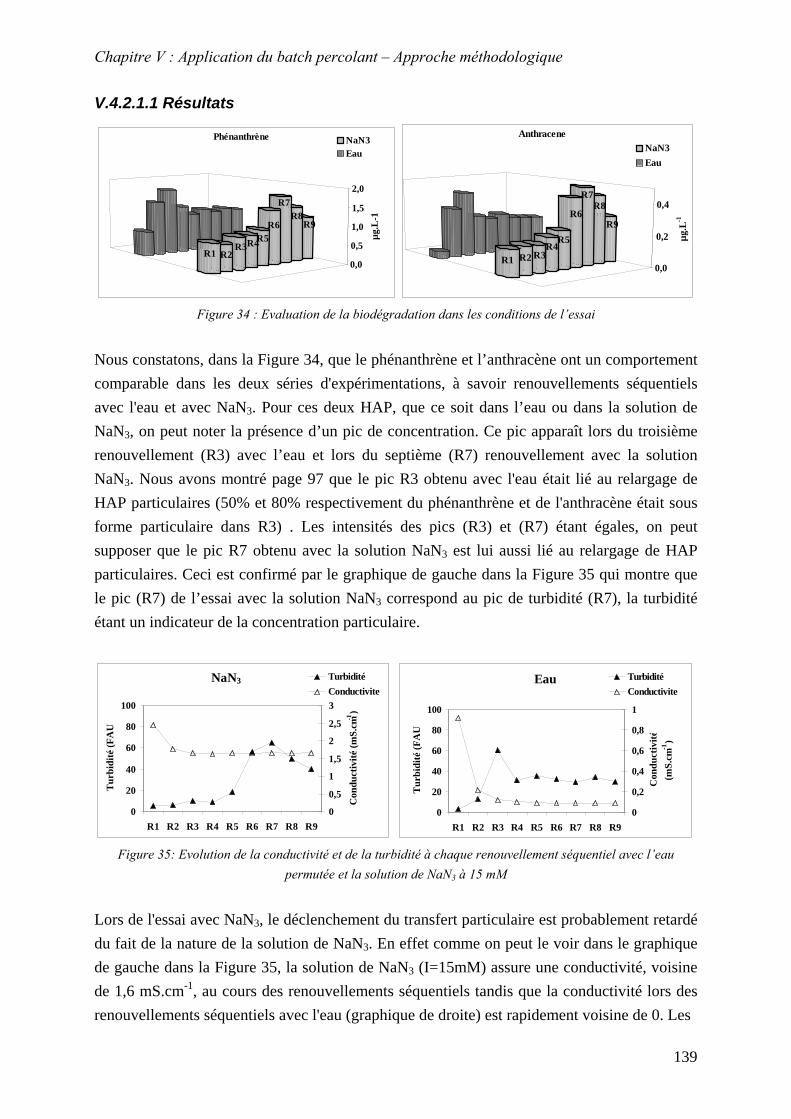
Chapitre V : Application du batch percolant – Approche méthodologique
V.4.2.1.1 Résultats
R9R8
R7
R6R5R4R3
R2R10,0
0,5
1,0
1,5
2,0
µg.L
-1
Phénanthrène NaN3Eau
R9
R8R7
R6
R5R4
R3R2R10,0
0,2
0,4
µg.L
-1
AnthraceneNaN3Eau
Figure 34 : Evaluation de la biodégradation dans les conditions de l’essai
Nous constatons, dans la Figure 34, que le phénanthrène et l’anthracène ont un comportement comparable dans les deux séries d'expérimentations, à savoir renouvellements séquentiels avec l'eau et avec NaN3. Pour ces deux HAP, que ce soit dans l’eau ou dans la solution de NaN3, on peut noter la présence d’un pic de concentration. Ce pic apparaît lors du troisième renouvellement (R3) avec l’eau et lors du septième (R7) renouvellement avec la solution NaN3. Nous avons montré page 97 que le pic R3 obtenu avec l'eau était lié au relargage de HAP particulaires (50% et 80% respectivement du phénanthrène et de l'anthracène était sous forme particulaire dans R3) . Les intensités des pics (R3) et (R7) étant égales, on peut supposer que le pic R7 obtenu avec la solution NaN3 est lui aussi lié au relargage de HAP particulaires. Ceci est confirmé par le graphique de gauche dans la Figure 35 qui montre que le pic (R7) de l’essai avec la solution NaN3 correspond au pic de turbidité (R7), la turbidité étant un indicateur de la concentration particulaire.
NaN3
0
20
40
60
80
100
R1 R2 R3 R4 R5 R6 R7 R8 R9
Tur
bidi
té (F
AU
0
0,5
1
1,5
2
2,5
3
Con
duct
ivité
(mS.
cm-1
)
TurbiditéConductivite
Eau
0
20
40
60
80
100
R1 R2 R3 R4 R5 R6 R7 R8 R9
Tur
bidi
té (F
AU
0
0,2
0,4
0,6
0,8
1
Con
duct
ivité
(mS.
cm-1
)TurbiditéConductivite
Figure 35: Evolution de la conductivité et de la turbidité à chaque renouvellement séquentiel avec l’eau
permutée et la solution de NaN3 à 15 mM
Lors de l'essai avec NaN3, le déclenchement du transfert particulaire est probablement retardé du fait de la nature de la solution de NaN3. En effet comme on peut le voir dans le graphique de gauche dans la Figure 35, la solution de NaN3 (I=15mM) assure une conductivité, voisine de 1,6 mS.cm-1, au cours des renouvellements séquentiels tandis que la conductivité lors des renouvellements séquentiels avec l'eau (graphique de droite) est rapidement voisine de 0. Les
139

Chapitre V : Application du batch percolant – Approche méthodologique
ions sodium et azoture en solution pourraient retarder le relargage des particules du sol pendant un temps de lixiviation équivalent à 7 renouvellements. L’influence de sels dissous dans la solution sur le transfert particulaire, a donc été étudiée dans la partie suivante. Etant donné que NaN3 bloque l'activité biologique et que la différence de comportement entre les deux tests, avec l'eau et respectivement avec la solution de NaN3 a été attribuée à la présence de sels dissous, on peut conclure qu'il n'y a pas d'activité biologique significative dans les conditions expérimentales imposées qui interférerait avec le phénomène de relargage par lixiviation.
V.4.2.1.2 Conclusion
La biodégradation des HAP n’est pas une contrainte dans les conditions du test. Nous pouvons considérer qu'il n’est pas nécessaire d’approfondir les études sur la bio-réactivité pour comprendre, à l'échelle du laboratoire, la mobilité des HAP dans le sol étudié.
V.4.2.2 Influence des sels dissous dans la phase liquide
Le chlorure de calcium permet de limiter l’entraînement de particules en stabilisant le sol. Ceci a été démontré lorsque les particules colloïdales transportées sont des argiles ou des matières humiques. Nous avons vu que les particules relarguées par le sol, composées à 70% de carbone, ne sont typiquement ni des argiles, ni des matières humiques. Nous avons donc testé l'effet stabilisant du chlorure de calcium sur ces particules en comparant les résultats obtenus lors de renouvellement séquentiels avec de l'eau et une solution de CaCl2 à 7,2 mmol.L-1 (800 mg.L-1, I=21,6 mM). Cette concentration a été déterminée de façon à obtenir une conductivité équivalente à celle d’une solution de NaN3 à 15 mM (I=15mM). Les conditions opératoires sont présentées dans le Tableau 22 et les résultats dans la Figure 36.
140
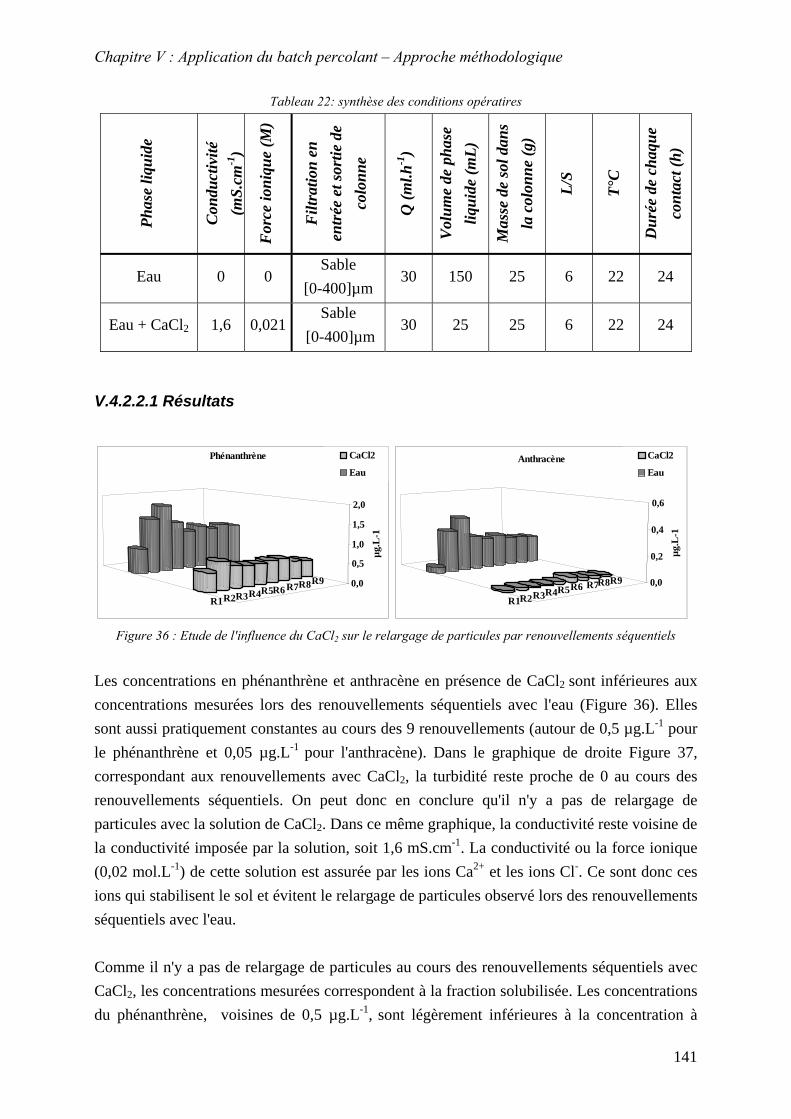
Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 22: synthèse des conditions opératires
Phas
e liq
uide
Con
duct
ivité
(m
S.cm
-1)
For
ce io
niqu
e (M
)
Filt
ratio
n en
en
trée
et s
ortie
de
colo
nne
Q (m
l.h-1
)
Vol
ume
de p
hase
liq
uide
(mL)
Mas
se d
e so
l dan
s la
col
onne
(g)
L/S
T°C
Dur
ée d
e ch
aque
co
ntac
t (h)
Eau 0 0 Sable
[0-400]µm 30 150 25 6 22 24
Eau + CaCl2 1,6 0,021 Sable
[0-400]µm 30 25 25 6 22 24
V.4.2.2.1 Résultats
R9R8R7R6R5R4R3R2R1
0,0
0,5
1,0
1,5
2,0
µg.L
-1
Phénanthrène CaCl2
Eau
R9R8R7R6R5R4R3R2R1
0,0
0,2
0,4
0,6
µg.L
-1
Anthracène CaCl2
Eau
Figure 36 : Etude de l'influence du CaCl2 sur le relargage de particules par renouvellements séquentiels
Les concentrations en phénanthrène et anthracène en présence de CaCl2 sont inférieures aux concentrations mesurées lors des renouvellements séquentiels avec l'eau (Figure 36). Elles sont aussi pratiquement constantes au cours des 9 renouvellements (autour de 0,5 µg.L-1 pour le phénanthrène et 0,05 µg.L-1 pour l'anthracène). Dans le graphique de droite Figure 37, correspondant aux renouvellements avec CaCl2, la turbidité reste proche de 0 au cours des renouvellements séquentiels. On peut donc en conclure qu'il n'y a pas de relargage de particules avec la solution de CaCl2. Dans ce même graphique, la conductivité reste voisine de la conductivité imposée par la solution, soit 1,6 mS.cm-1. La conductivité ou la force ionique (0,02 mol.L-1) de cette solution est assurée par les ions Ca2+ et les ions Cl-. Ce sont donc ces ions qui stabilisent le sol et évitent le relargage de particules observé lors des renouvellements séquentiels avec l'eau. Comme il n'y a pas de relargage de particules au cours des renouvellements séquentiels avec CaCl2, les concentrations mesurées correspondent à la fraction solubilisée. Les concentrations du phénanthrène, voisines de 0,5 µg.L-1, sont légèrement inférieures à la concentration à
141

Chapitre V : Application du batch percolant – Approche méthodologique
l'équilibre obtenue avec l'eau (voir page 100 (0,9 µg.L-1) . Il en va de même pour l'anthracène, 0,05 µg.L-1 lors des renouvellements séquentiels avec CaCl2 et 0,1 µg.L-1 pour la concentration à l'équilibre avec l'eau. Les renouvellements sont faits après 24 heures de contact, aussi l’équilibre n’est pas encore atteint. Ceci explique peut être en partie que les concentrations du phénanthrène et de l'anthracène soient inférieures aux concentrations à l'équilibre avec l'eau.
CaCl2
0
10
20
30
40
50
60
R1 R2 R3 R4 R5 R6 R7 R8 R9
Turb
idité
(NTU
)
0
0,5
1
1,5
2
2,5
Con
duct
ivité
(mS.
cm-1
)
TurbiditéConductivite Eau
0
20
40
60
80
100
R1 R2 R3 R4 R5 R6 R7 R8 R9
Tur
bidi
té (F
AU
0
0,2
0,4
0,6
0,8
1
Con
duct
ivité
(mS.
cm-1
)
TurbiditéConductivite
Figure 37: Evolution des conductivités et de la
turbidité avec le solution de CaCl2
Figure 38: Evolution des conductivités et de la turbidité avec l’eau permutée
V.4.2.2.2 Conclusion
La solution de CaCl2 à 7,2 mol.L-1 (800 mg.L-1, I= 21,6 mM) :
stabilise les particules relarguées par lixiviation à l'eau ; les HAP sont relargués sous forme dissoute uniquement,
a un effet stabilisant sur le système solide plus important que la solution 15 mM
NaN3, (I=15 mM), effet plutôt dû à l’action spécifique des ions (Ca en particulier) qu’à la force ionique.
.
V.4.2.3 Influence de la masse de sol en contact avec la phase liquide
Page 96, nous avons mis en évidence la nécessité de considérer la fraction particulaire pour l'étude de la mobilité des HAP. Pour cela nous avons effectué des renouvellements séquentiels avec les conditions expérimentales suivantes : L/S=6 (150 ml d'eau et 25 g de sol) ; T=22°C ; Q=30 mL.h-1 et un lit de sable de granulométrie [0-400]µm. Les résultats sont à nouveau présentés dans la Figure 39.
142
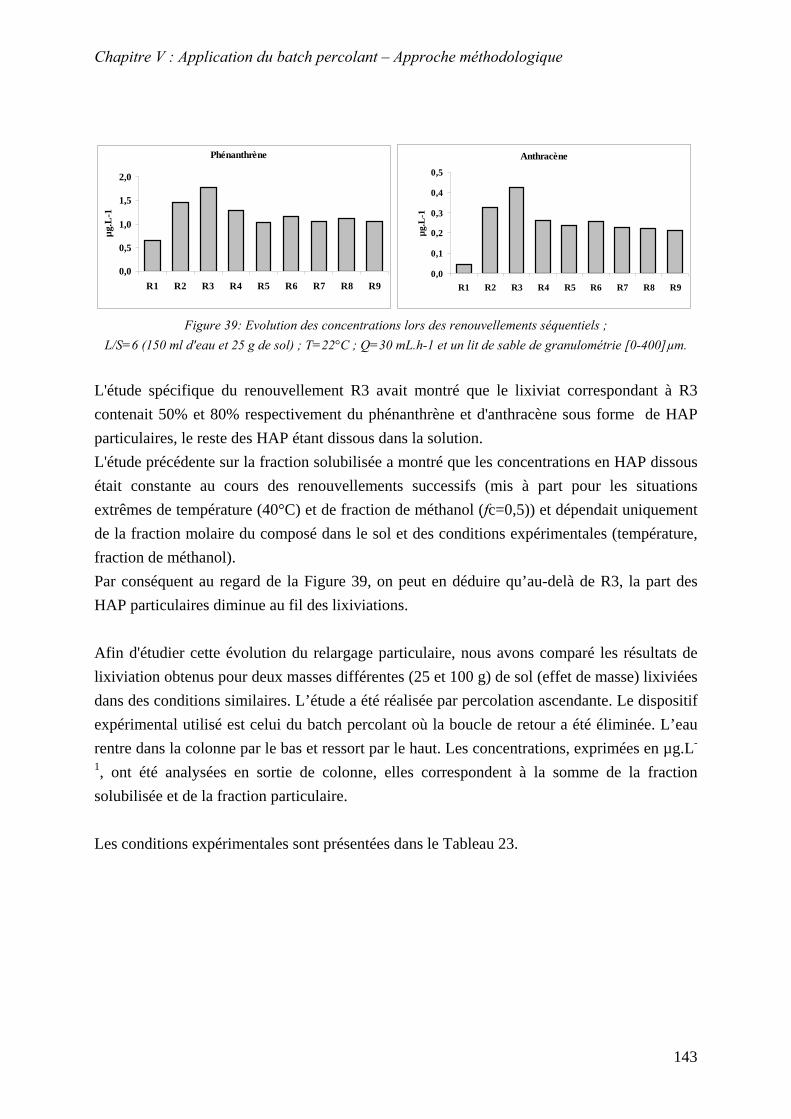
Chapitre V : Application du batch percolant – Approche méthodologique
Phénanthrène
0,0
0,5
1,0
1,5
2,0
R1 R2 R3 R4 R5 R6 R7 R8 R9
µg.L
-1
Anthracène
0,0
0,1
0,2
0,3
0,4
0,5
R1 R2 R3 R4 R5 R6 R7 R8 R9
µg.L
-1
Figure 39: Evolution des concentrations lors des renouvellements séquentiels ;
L/S=6 (150 ml d'eau et 25 g de sol) ; T=22°C ; Q=30 mL.h-1 et un lit de sable de granulométrie [0-400]µm.
L'étude spécifique du renouvellement R3 avait montré que le lixiviat correspondant à R3 contenait 50% et 80% respectivement du phénanthrène et d'anthracène sous forme de HAP particulaires, le reste des HAP étant dissous dans la solution. L'étude précédente sur la fraction solubilisée a montré que les concentrations en HAP dissous était constante au cours des renouvellements successifs (mis à part pour les situations extrêmes de température (40°C) et de fraction de méthanol (fc=0,5)) et dépendait uniquement de la fraction molaire du composé dans le sol et des conditions expérimentales (température, fraction de méthanol). Par conséquent au regard de la Figure 39, on peut en déduire qu’au-delà de R3, la part des HAP particulaires diminue au fil des lixiviations. Afin d'étudier cette évolution du relargage particulaire, nous avons comparé les résultats de lixiviation obtenus pour deux masses différentes (25 et 100 g) de sol (effet de masse) lixiviées dans des conditions similaires. L’étude a été réalisée par percolation ascendante. Le dispositif expérimental utilisé est celui du batch percolant où la boucle de retour a été éliminée. L’eau rentre dans la colonne par le bas et ressort par le haut. Les concentrations, exprimées en µg.L-
1, ont été analysées en sortie de colonne, elles correspondent à la somme de la fraction solubilisée et de la fraction particulaire. Les conditions expérimentales sont présentées dans le Tableau 23.
143
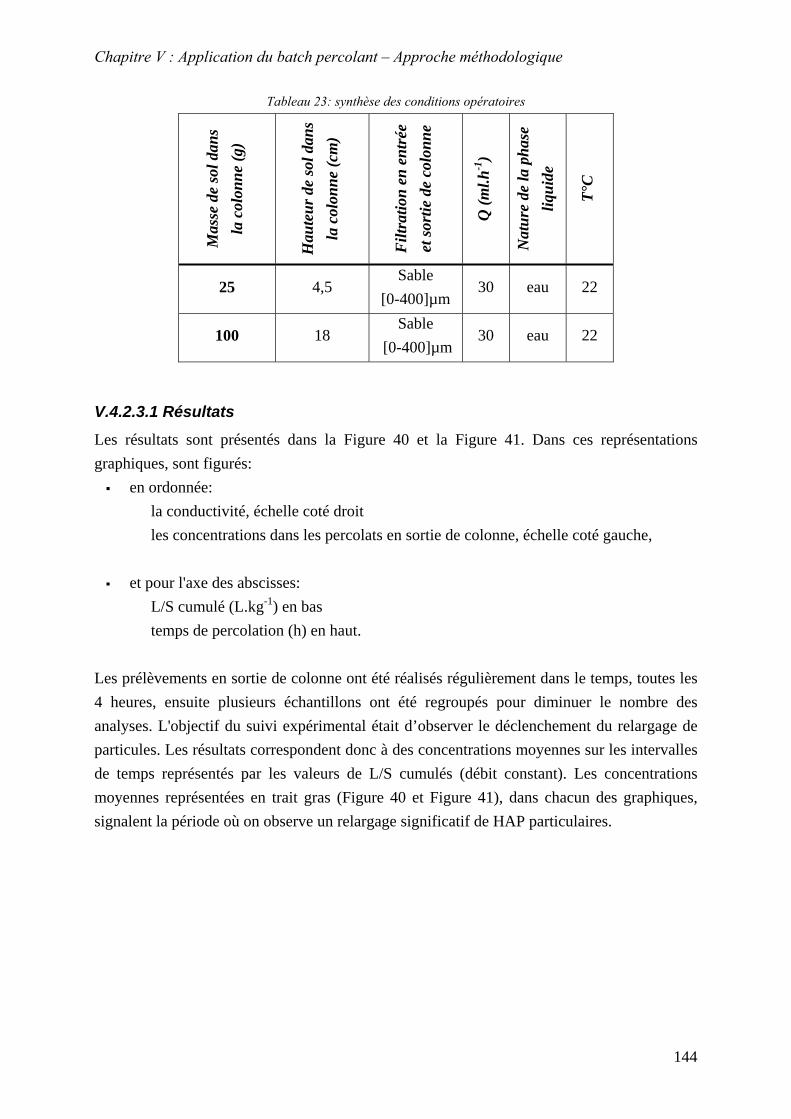
Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 23: synthèse des conditions opératoires
Mas
se d
e so
l dan
s la
col
onne
(g)
Hau
teur
de
sol d
ans
la c
olon
ne (c
m)
Filt
ratio
n en
ent
rée
et so
rtie
de
colo
nne
Q (m
l.h-1
)
Nat
ure
de la
pha
se
liqui
de
T°C
25 4,5 Sable
[0-400]µm 30 eau 22
100 18 Sable
[0-400]µm 30 eau 22
V.4.2.3.1 Résultats
Les résultats sont présentés dans la Figure 40 et la Figure 41. Dans ces représentations graphiques, sont figurés: en ordonnée:
la conductivité, échelle coté droit les concentrations dans les percolats en sortie de colonne, échelle coté gauche,
et pour l'axe des abscisses:
L/S cumulé (L.kg-1) en bas temps de percolation (h) en haut.
Les prélèvements en sortie de colonne ont été réalisés régulièrement dans le temps, toutes les 4 heures, ensuite plusieurs échantillons ont été regroupés pour diminuer le nombre des analyses. L'objectif du suivi expérimental était d’observer le déclenchement du relargage de particules. Les résultats correspondent donc à des concentrations moyennes sur les intervalles de temps représentés par les valeurs de L/S cumulés (débit constant). Les concentrations moyennes représentées en trait gras (Figure 40 et Figure 41), dans chacun des graphiques, signalent la période où on observe un relargage significatif de HAP particulaires.
144
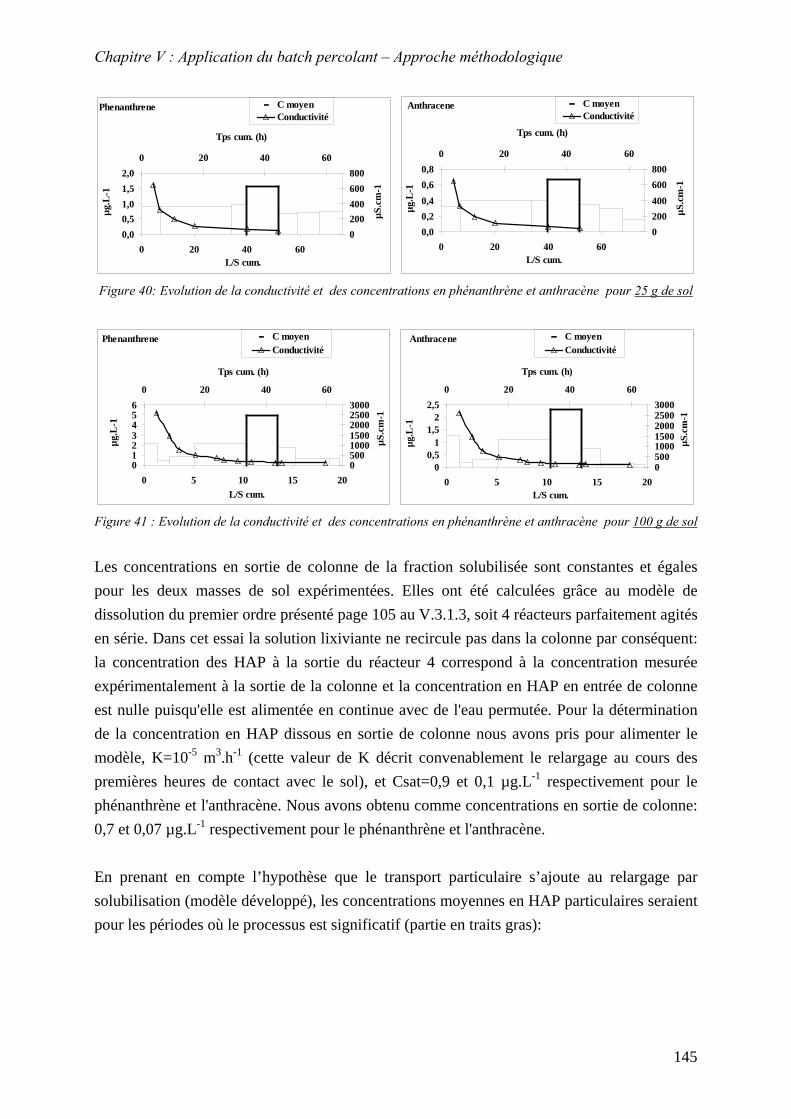
Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
0,00,51,01,52,0
0 20 40 60L/S cum.
µg.L
-1
0200400600800
0 20 40 60
Tps cum. (h)
µS.c
m-1
C moyenConductivité
Anthracene
0,00,20,40,60,8
0 20 40 60L/S cum.
µg.L
-1
0200400600800
0 20 40 60
Tps cum. (h)
µS.c
m-1
C moyenConductivité
Figure 40: Evolution de la conductivité et des concentrations en phénanthrène et anthracène pour 25 g de sol
Phenanthrene
0123456
0 5 10 15 20L/S cum.
µg.L
-1
050010001500200025003000
0 20 40 60
Tps cum. (h)µS
.cm
-1
C moyenConductivité
Anthracene
00,5
11,5
22,5
0 5 10 15 20L/S cum.
µg.L
-1
050010001500200025003000
0 20 40 60
Tps cum. (h)
µS.c
m-1
C moyenConductivité
Figure 41 : Evolution de la conductivité et des concentrations en phénanthrène et anthracène pour 100 g de sol
Les concentrations en sortie de colonne de la fraction solubilisée sont constantes et égales pour les deux masses de sol expérimentées. Elles ont été calculées grâce au modèle de dissolution du premier ordre présenté page 105 au V.3.1.3, soit 4 réacteurs parfaitement agités en série. Dans cet essai la solution lixiviante ne recircule pas dans la colonne par conséquent: la concentration des HAP à la sortie du réacteur 4 correspond à la concentration mesurée expérimentalement à la sortie de la colonne et la concentration en HAP en entrée de colonne est nulle puisqu'elle est alimentée en continue avec de l'eau permutée. Pour la détermination de la concentration en HAP dissous en sortie de colonne nous avons pris pour alimenter le modèle, K=10-5 m3.h-1 (cette valeur de K décrit convenablement le relargage au cours des premières heures de contact avec le sol), et Csat=0,9 et 0,1 µg.L-1 respectivement pour le phénanthrène et l'anthracène. Nous avons obtenu comme concentrations en sortie de colonne: 0,7 et 0,07 µg.L-1 respectivement pour le phénanthrène et l'anthracène. En prenant en compte l’hypothèse que le transport particulaire s’ajoute au relargage par solubilisation (modèle développé), les concentrations moyennes en HAP particulaires seraient pour les périodes où le processus est significatif (partie en traits gras):
145

Chapitre V : Application du batch percolant – Approche méthodologique
Tableau 24:Concentrations moyenne d'origine particulaire
25 g de sol dans la colonne
100 g de sol dans la colonne
Concentration moyenne en phénanthrène particulaire (µg.L-1)
0,9 4,3
Concentration moyenne en anthracène particulaire (µg.L-1)
0,5 2,3
Dans le Tableau 25, sont présentés les L/S cumulés lus à l'apparition et à la disparition du relargage de HAP particulaires, ainsi que les ∆L/S correspondant.
Tableau 25: Valeurs de L/S relative à l'apparition et la disparition du relargage de HAP particulaires au cours de la percolation
Masse de sol dans la colonne (g) 25 g 100 g L/S cumulé relatif à l'apparition du
relargage de HAP particulaire (L.Kg-1) 40 10,5
L/S cumulé relatif à la disparition du relargage de HAP particulaire (L.Kg-1)
52 13
∆L/S (L.Kg-1) 12 2,5 Nous définissons le potentiel de HAP relargables sous forme particulaire par le sol dans les conditions d’expérimentation, selon la relation :
(27) SLCm ∆⋅=
avec m : Potentiel de HAP particulaires relargables par le sol (µg.Kg-1) C : Concentration moyenne (Tableau 24) en HAP particulaires (µg.L-1) en sortie de
colonne.
Tableau 26: Potentiel de HAP particulaires relargables par le sol pour q=30 ml.h-1 et T=22°C
Masse de sol dans la colonne (25 g)
Masse de sol dans la colonne (100 g)
Potentiel de phénanthrène particulaire relargable (µg/kg)
10,8 10,7
Potentiel d'anthracène particulaire relargable (µg/kg)
6 6,9
146

Chapitre V : Application du batch percolant – Approche méthodologique
On peut se rendre compte dans le Tableau 26 que les potentiels de phénanthrène particulaire et d'anthracène particulaire relargables par le sol, exprimées en µg par kg de sol, sont très proches pour les deux masses de sols expérimentées (25 et 100 g). Par conséquent, pour un débit de 30 mL.h-1, une température de 22°C, un lit de sable de granulométrie [0-400] µm en sortie de colonne et pour différentes masses de sol introduites dans la colonne, le potentiel de phénanthrène particulaire relargable par le sol est d'environ 11 µg.Kg-1, et de 6,5 µg.Kg-1 pour l'anthracène. Les conductivités initiales sont de l’ordre de 2500 µS.cm-1 et 650 µs.cm-1 respectivement pour les essais avec 100 et 25 g de sol (Figure 41 et Figure 40). Nous observons que dans les deux cas expérimentaux le relargage des particules intervient approximativement vers 100 µScm-1, valeur seuil pour le relargage de HAP particulaires pour le sol étudié, indépendante de la masse de sol lixiviée. L'apparition du relargage de HAP particulaires se fait après environ 33 heures de percolation pour les 2 masses de sol expérimentées, soit après qu'environ 1000 mL d'eau aient percolé à travers les 2 colonnes. Remarques : Le pic observé (2µg.L-1 pour le phénanthrène) en début de percolation à travers la colonne contenant 100 g de sol pourrait être aussi attribué à un relargage de HAP particulaire dont les particules seraient de natures différentes de celles observées précédemment. En effet, cet échantillon était fortement coloré en jaune, ce qui correspondrait à la présence d’autres matières en solution (humiques et/ou argiles). Cet effet, déclenché dès le début de la percolation et épuisé rapidement a été signalé pour d’autres sols pollués [Bouchez, 1996].
V.4.2.3.2 Conclusion
Pour un débit égale à 30 mL.h-1 et une température de 22°C:
Le potentiel de phénanthrène et d'anthracène particulaire relargable par le sol (exprimé en µg de HAP sous forme particulaire.kg-1 de sol) est constant et indépendant de la masse de sol lixivié. Le relargage de HAP particulaires intervient lorsque que la conductivité passe en dessous de 100 µS.cm-1.
147
Chapitre V : Application du batch percolant – Approche méthodologique
V.4.2.4 Influence du débit
L’influence du débit de lixiviant a été observée dans le dispositif de percolation ascendant sans recirculation de l’éluat. L'influence du débit sur le relargage de HAP sous forme particulaire a été évaluée en comparant les résultats obtenus pour deux débits différents (30 et 100 ml.h-1) dans les conditions expérimentales présentées dans le Tableau 27.
Tableau 27: synthèse des conditions opératoires
Q (m
l.h-1
)
Filt
ratio
n
Mas
se d
e so
l dan
s la
colo
nne
(g)
Nat
ure
de la
pha
se
liqui
de
T°C
30 Sable
[0-400]µm 25 eau 22
100 Sable
[0-400]µm 25 eau 22
V.4.2.4.1 Résultats
Les résultats sont présentés dans la Figure 42 et la Figure 43. Dans ces représentations graphiques, sont figurés: en ordonnée:
la conductivité, échelle coté droit les concentrations dans les percolats en sortie de colonne, échelle coté gauche,
et pour l'axe des abscisses:
L/S cumulé (L.kg-1) en bas temps de percolation (h) en haut.
Les prélèvements en sortie de colonne ont été réalisés régulièrement dans le temps, ensuite certains échantillons ont été regroupés pour diminuer le nombre des analyses, l'objectif était de visualiser le déclenchement du relargage de particules. Les résultats correspondent donc à des concentrations moyennes pour les intervalles de L/S figurés. Les concentrations moyennes représentées en gras (Figure 42 et Figure 43), dans chacun des graphiques, signalent les périodes où l'on observe un relargage significatif de HAP particulaires.
148
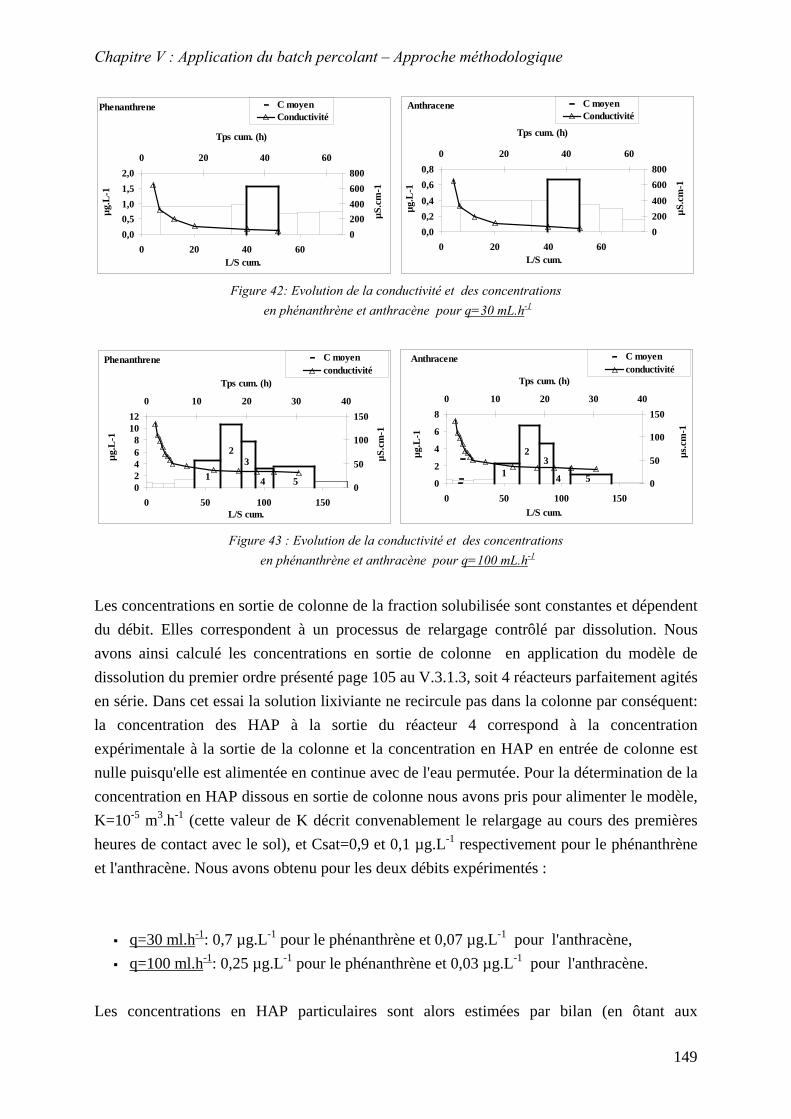
Chapitre V : Application du batch percolant – Approche méthodologique
Phenanthrene
0,00,51,01,52,0
0 20 40 60L/S cum.
µg.L
-1
0200400600800
0 20 40 60
Tps cum. (h)
µS.c
m-1
C moyenConductivité
Anthracene
0,00,20,40,60,8
0 20 40 60L/S cum.
µg.L
-1
0200400600800
0 20 40 60
Tps cum. (h)
µS.c
m-1
C moyenConductivité
Figure 42: Evolution de la conductivité et des concentrations
en phénanthrène et anthracène pour q=30 mL.h-1
Phenanthrene
02468
1012
0 50 100 150L/S cum.
µg.L
-1
0
50
100
1500 10 20 30 40
Tps cum. (h)µS
.cm
-1
C moyenconductivité
1
23
4 5
Anthracene
0
2
4
6
8
0 50 100 150L/S cum.
µg.L
-1
0
50
100
1500 10 20 30 40
Tps cum. (h)
µs.c
m-1
C moyenconductivité
2
13
4 5
Figure 43 : Evolution de la conductivité et des concentrations
en phénanthrène et anthracène pour q=100 mL.h-1
Les concentrations en sortie de colonne de la fraction solubilisée sont constantes et dépendent du débit. Elles correspondent à un processus de relargage contrôlé par dissolution. Nous avons ainsi calculé les concentrations en sortie de colonne en application du modèle de dissolution du premier ordre présenté page 105 au V.3.1.3, soit 4 réacteurs parfaitement agités en série. Dans cet essai la solution lixiviante ne recircule pas dans la colonne par conséquent: la concentration des HAP à la sortie du réacteur 4 correspond à la concentration expérimentale à la sortie de la colonne et la concentration en HAP en entrée de colonne est nulle puisqu'elle est alimentée en continue avec de l'eau permutée. Pour la détermination de la concentration en HAP dissous en sortie de colonne nous avons pris pour alimenter le modèle, K=10-5 m3.h-1 (cette valeur de K décrit convenablement le relargage au cours des premières heures de contact avec le sol), et Csat=0,9 et 0,1 µg.L-1 respectivement pour le phénanthrène et l'anthracène. Nous avons obtenu pour les deux débits expérimentés :
q=30 ml.h-1: 0,7 µg.L-1 pour le phénanthrène et 0,07 µg.L-1 pour l'anthracène, q=100 ml.h-1: 0,25 µg.L-1 pour le phénanthrène et 0,03 µg.L-1 pour l'anthracène.
Les concentrations en HAP particulaires sont alors estimées par bilan (en ôtant aux
149
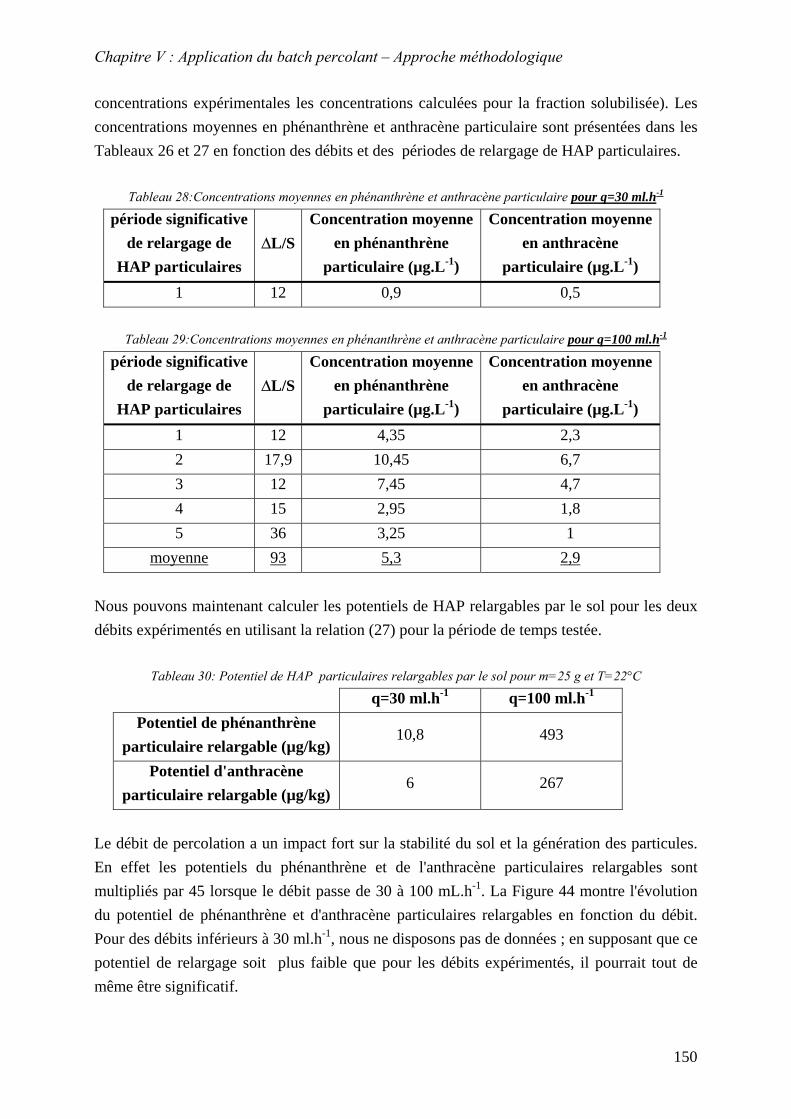
Chapitre V : Application du batch percolant – Approche méthodologique
concentrations expérimentales les concentrations calculées pour la fraction solubilisée). Les concentrations moyennes en phénanthrène et anthracène particulaire sont présentées dans les Tableaux 26 et 27 en fonction des débits et des périodes de relargage de HAP particulaires.
Tableau 28:Concentrations moyennes en phénanthrène et anthracène particulaire pour q=30 ml.h-1
période significative de relargage de
HAP particulaires ∆L/S
Concentration moyenne en phénanthrène
particulaire (µg.L-1)
Concentration moyenne en anthracène
particulaire (µg.L-1) 1 12 0,9 0,5
Tableau 29:Concentrations moyennes en phénanthrène et anthracène particulaire pour q=100 ml.h-1
période significative de relargage de
HAP particulaires ∆L/S
Concentration moyenne en phénanthrène
particulaire (µg.L-1)
Concentration moyenne en anthracène
particulaire (µg.L-1) 1 12 4,35 2,3 2 17,9 10,45 6,7 3 12 7,45 4,7 4 15 2,95 1,8 5 36 3,25 1
moyenne 93 5,3 2,9 Nous pouvons maintenant calculer les potentiels de HAP relargables par le sol pour les deux débits expérimentés en utilisant la relation (27) pour la période de temps testée.
Tableau 30: Potentiel de HAP particulaires relargables par le sol pour m=25 g et T=22°C
q=30 ml.h-1 q=100 ml.h-1
Potentiel de phénanthrène particulaire relargable (µg/kg)
10,8 493
Potentiel d'anthracène particulaire relargable (µg/kg)
6 267
Le débit de percolation a un impact fort sur la stabilité du sol et la génération des particules. En effet les potentiels du phénanthrène et de l'anthracène particulaires relargables sont multipliés par 45 lorsque le débit passe de 30 à 100 mL.h-1. La Figure 44 montre l'évolution du potentiel de phénanthrène et d'anthracène particulaires relargables en fonction du débit. Pour des débits inférieurs à 30 ml.h-1, nous ne disposons pas de données ; en supposant que ce potentiel de relargage soit plus faible que pour les débits expérimentés, il pourrait tout de même être significatif.
150

Chapitre V : Application du batch percolant – Approche méthodologique
0
2
4
6
8
10
0 50 100
Débit de percolation (ml.h-1)ln
m
Phénanthrène
Anthracène
Figure 44: évolution en log népérien du potentiel de HAP
particulaire relargable par le sol en fonction du débit
Le déclenchement du relargage de HAP particulaire, intervient lorsque la conductivité passe sous le seuil des 100 µS.cm-1.De plus, il intervient pour un L/S cumulé de 40 pour les 2 débits expérimentés, soit après qu'environ 1000 mL d'eau aient percolé à travers les 2 colonnes.
V.4.2.4.2 Conclusion
Pour une température de 22°C, et une masse de sol dans la colonne de 25 g : Le potentiel de phénanthrène et d'anthracène particulaire relargable est fortement dépendant du débit. Lorsque que le débit passe de 100 mLh-1 à 30 mL.h-1, les potentiels de relargage du phénanthrène et de l'anthracène particulaire sont multipliés par 45. Le relargage de HAP particulaires intervient quand la conductivité passe en dessous de 100 µS.cm-1. Le relargage de HAP particulaires intervient pour un L/S cumulé de 40 pour les 2 débits expérimentés, soit après qu'environ 1000 mL d'eau aient percolés à travers les 2 colonnes.
V.4.3 Evolution du relargage des HAP particulaires au cours du temps
Nous avons montré que le potentiel de phénanthrène et d'anthracène relargables sous forme particulaire est constant quelle que soit la masse de sol présente dans la colonne, pour des conditions opératoires constantes. Il a aussi été montré qu'il était dépendant du débit imposé : il augmente fortement avec le débit. Par conséquent, lorsque le relargage des HAP particulaires est amorcé (probablement en relation avec la force ionique/conductivité et action spécifique de certains ions), le sol va progressivement relarguer le phénanthrène et l'anthracène particulaire jusqu'à l'épuisement total de leur potentiel particulaire relargables respectifs. Ensuite les concentrations en sortie de colonne seront imposées par le mécanisme majeur, à savoir la solubilisation dans le cas du sol étudié.
151

Chapitre V : Application du batch percolant – Approche méthodologique
V.5 Conclusion
Le test développé au chapitre IV appelé "batch percolant" nous a permis de comprendre les mécanismes intervenant dans la mobilité des HAP. Nous avons ainsi pu distinguer d'une part, la mobilité des HAP transférés dans la phase liquide lixiviante sous forme dissoute (notée fraction solubilisée) et d'autre part, la mobilité des HAP transférés dans la phase liquide lixiviante sous forme particulaire (notée fraction particulaire). Voici, pour le sol étudié, les principales conclusions portant sur les mécanismes de la mobilité des HAP: La fraction solubilisée (fraction de la pollution qui correspond aux HAP mobilisés sous forme dissoute): Le mécanisme de dissolution semble contrôler le transfert des HAP vers la phase liquide. En contact avec une phase liquide lixiviante, les HAP présents dans le sol passent en solution. Les concentrations dans la phase liquide augmentent alors, avec une vitesse donnée jusqu'à ce que l'équilibre soit atteint. Lorsque cet équilibre est atteint, la solution est saturée (les concentrations à l'équilibre peuvent être calculées à partir de la loi de Raoult). Le modèle de dissolution du premier ordre qui a été utilisé, décrit convenablement ces cinétiques de mise en solution. L'influence de paramètres physico-chimiques, variables sur le terrain, a été évaluée en terme d'influence sur les concentrations à saturation et en terme d'influence sur les vitesses de transfert vers la phase liquide. Les paramètres retenus était la fraction de cosolvant (méthanol), la température, le débit et le rapport L/S. Cette étude paramétrique a montré que la température et la fraction de méthanol influencent fortement les concentrations à saturation. Par contre, le débit et le rapport L/S ne semble pas influencer les concentrations à saturation. Aucun des paramètres variables étudiés ne semble avoir un impact important sur les vitesses de transfert des HAP vers la phase liquide. L'étude dynamique a révélé que les concentrations à l'équilibre avaient tendance à diminuer avec le temps. Cette évolution des concentrations a été attribuée en grande partie à la diminution du coefficient de transfert global K et dans une moindre mesure à la diminution des fractions molaires des polluants dans le sol. Cependant, cet effet n'a été visible que pour des conditions d'extraction forte, c'est à dire une fraction de cosolvant de 0,5 et une température de 40°C. Un abaque a été élaboré, il donne la concentration à saturation pour une température et une fraction de cosolvant choisie. L'utilisation de cet abaque combinée au modèle de dissolution permet de prédire les concentrations en sortie d'une colonne de percolation en fonction du débit dans la colonne. La fraction particulaire L'eau en circulant au travers du sol arrache des particules chargées en HAP. Suite à l'étude réalisée à Albi au microscope environnemental à balayage électronique (MEB), nous avons émis l'hypothèse que les particules mobilisées(composées de plus de 70% de carbone) n'étaient ni des matières humiques ni des argiles mais plutôt des fragments de polluants
152

Chapitre V : Application du batch percolant – Approche méthodologique
insolubles dans l'eau. Nous avons aussi montré que ces fragments insolubles provenaient probablement de goudrons vieillis dans le sol. Dans les conditions de l'essai, la biodégradation ne semble pas influencer ni le relargage de HAP particulaires ni le relargage de HAP sous forme dissoute. Nous avons montré que la quantité de sol introduite dans la colonne n'influençait pas le potentiel de HAP particulaires relargables mais qu'il était fortement dépendant du débit imposé au système (le potentiel de HAP particulaires relargables représente la quantité maximale de HAP particulaire relargable dans l'eau par unité de masse de sol, dans des conditions de températures et de débits données). Un des facteurs susceptibles de déclencher le transfert particulaire est la force ionique. La présence de chlorure de calcium à 7,2 mM bloque le transfert particulaire. On peut donc dire que pour cette concentration de CaCl2 le potentiel de HAP particulaires relargables est nul. Il serait intéressant de connaître la concentration critique en CaCl2 qui provoque l'apparition des premiers HAP particulaires. Mobilité globale des HAP A partir des études menées sur la fraction dissoute et sur la fraction particulaire nous pouvons décrire, pour une lixiviation du sol en colonne, la mobilité globale des HAP : La lixiviation du sol avec une solution de chlorure de calcium de force ionique égale à 0,02 M ou une solution constituée d'un mélange eau/méthanol (0,3<fc<0,5) ne concerne que la fraction dissoute. Les concentrations en sortie de colonne peuvent être considérées constantes dans le temps et inférieures ou égales aux concentrations à l'équilibre (à l'exception des conditions d'extraction fortes: fc=0,5 et T=40°C). Les concentrations à l'équilibre sont dépendantes de la température, de la force ionique et de la fraction de méthanol de la solution. L'écart entre les concentrations à l'équilibre et les concentrations mesurées en sortie de colonne sont fonction du débit imposé au système.
153

Conclusion générale
CONCLUSION GENERALE L’objectif principal de ce travail de thèse était d’étudier la mobilité des HAP contenus
dans un sol industriel pollué situé au nord de la France. Très vite la problématique abordée a montré une très grande complexité. Nous voulions, à l’échelle du laboratoire, répondre à des questions précises sur le transfert des HAP dans l’eau (vecteur principal du transfert/transport de la pollution vers les différentes cibles) :
1. Quels sont les mécanismes qui contrôlent le transfert des HAP du sol saturé vers la phase liquide ?
2. Quels sont les paramètres physico-chimiques qui influent sur ce transfert ? 3. S’il a lieu, comment ce transfert peut-il évoluer avec le temps ?
Il n’existe pas à l’heure actuelle de normalisations, ni de méthodologies adaptées qui
permettent d’orienter ce type de travaux. En effet, le seul guide disponible (Diagnostic approfondi et évaluation détaillée des risques des sites et sols pollués [BRGM, 2000]) ne renvoie pas à des tests spécifiques pour étudier les mécanismes de transfert des polluants, bien que la compréhension de ces mécanismes y soit recommandée.
Notre travail a donc consisté à confronter et le cas échéant élaborer une série d’expérimentation à l’échelle du laboratoire pour obtenir le maximum d’informations sur la mobilité des HAP contenus dans le sol à étudier et de proposer une méthodologie d’étude.
Les premières données proviennent de la caractérisation du site étudié. Sur la base de la
répartition granulométrique du sol et du contenu en polluants dans chacune des tranches, il a été décidé de garder pour l’étude la fraction granulométrique inférieure à 2 mm, la plus représentative de la pollution. Le sol prélevé sur le site est une matrice extrêmement riche en carbone organique (COT>30%) ce qui l’éloigne de la composition d’un sol pour l’approcher plus d’un déchet. La composition en carbone et oxygène se rapproche de celle des goudrons, ce qui laisse supposer que le sol est pollué en partie par des goudrons de houille (confirmé par ailleurs par certaines informations historiques). Le contenu total en HAP est voisin de 2500 mg/kg avec une prédominance des HAP à 3 et 4 cycles aromatiques : phénanthrène, anthracène, fluoranthène et pyrène.
La première phase d’essai de lixiviation/percolation basée sur l’expérience acquise dans notre laboratoire a révélé la grande complexité du sol étudié. En effet, l’analyse des résultats des tests a montré que les lixiviations en milieu dispersé agité (réacteur de type
154

Conclusion générale
batch), couramment usitées, ne sont pas adaptées au sol étudié, ceci du fait de la désagrégation des particules de sol au cours de l’agitation. Ce phénomène conduit à la transformation du sol ayant comme conséquence la modification des propriétés de relargage du sol, donc du terme source de la pollution. Cette transformation du terme source a été confirmée lors de la comparaison des résultats des lixiviations en milieu dispersé avec ceux obtenus dans une colonne classique de percolation. Cependant, les colonnes de percolation ne sont pas un outil suffisamment adapté à l’identification au laboratoire des mécanismes de mobilisation des polluants, elles sont plus généralement employées comme test de simulation à l’échelle du laboratoire. Nous avons donc été amenés à mettre au point un outil spécifique de lixiviation pour maîtriser les principaux problèmes expérimentaux, notamment celui de la désagrégation du sol et celui de la durée d’observation expérimentale. De plus, au cours de cette première phase, nous avons supposé que le transport particulaire puisse être un acteur non négligeable de la mobilité des HAP lixiviés à l’eau, il était donc nécessaire de l’étudier spécifiquement.
L’outil de lixiviation élaboré et mis au point a été appelé « batch percolant ». L’origine
du nom vient de son principe de fonctionnement : batch puisque le système est assimilable à un système fermé et percolant car le contact solide/liquide est assuré selon la technique de la colonne de percolation fonctionnant à grand débit en boucle fermée, la recirculation interne assurant l’agitation. De plus, le même dispositif peut être utilisé en système ouvert avec alimentation ascendante (ou descendante), obtenant ainsi une colonne de percolation classique. Le même dispositif peut par conséquent, assurer, plusieurs types d’études : isothermes, cinétiques et de perco-lixiviations. Une gamme assez large de paramètres fonctionnels a été accessible : le dispositif peut contenir une masse variable (de préférence petite) de sol. Le volume de solution à recirculer, au débit choisi, peut être fixé et changé ou échantillonné à tout moment dans le récipient tampon. Un mécanisme de régulation de la température du système a été prévu. Le protocole de remplissage de la colonne et de saturation du sol a été élaboré et appliqué. Enfin, l’utilisation de la distribution du temps de séjour d’un traceur a permis la modélisation du comportement hydrodynamique de la colonne « type » par une série de quatre réacteurs ouverts parfaitement agités. Pour affiner les études spécifiques des mécanismes de mobilisation, notamment des cinétiques, il serait préférable de réduire encore plus la masse de sol par rapport au débit de la phase liquide pour s’approcher encore plus d’un seul réacteur parfaitement agité. Par l’intermédiaire de ce test, la mobilité du phénanthrène et de l’anthracène, considérés représenter le comportement des principaux HAP contenus dans le sol a pu être mieux étudiée par la prise en compte des paramètres suivants :
le volume de liquide en contact avec le sol lors de séquences de lixiviation en batch ou en système ouvert, la composition initiale de l’éluant (eau, mélange eau/méthanol, solution saline),
155

Conclusion générale
le débit d’alimentation de la colonne et la température du système.
Les étapes précédentes et les conditions du site (eau à faible contenu salin) nous ont
amené à prendre en compte les deux formes de mobilité de la pollution HAP observées, à savoir la part dissoute, appelée aussi fraction solubilisée et respectivement celle par entraînement, appelée aussi fraction particulaire. La fraction solubilisée :
Dans le cas du site étudié, les concentrations des HAP dissous en phase aqueuse sont extrêmement faibles (de l’ordre de 1µg.L-1 ), ce qui rend difficile le travail expérimental et en particulier le protocole d’analyse des HAP puisqu’il est nécessaire de passer par une phase délicate de concentration des échantillons. Afin de limiter ces problèmes analytiques, nous avons choisi de travailler majoritairement avec des solutions contenant du méthanol. Des essais préalables ont montré que des lixiviations réalisées avec des solutions contenant du méthanol permettaient d’accéder directement aux informations sur la fraction solubilisée. En effet, nous avons constaté que pour des fractions de méthanol allant de 30 à 50%, il n’y avait pas de particules mobilisées contrairement à ce qui se passe lors des lixiviations à l’eau. De plus son utilisation ne change pas le mécanisme de transfert et augmente beaucoup les concentrations dans les lixiviats, ce qui facilite le travail expérimental et notamment le protocole d’analyse des HAP et diminue fortement le coût des analyses. La validité de la théorie sur la solubilisation des composés hydrophobes dans des mélanges binaires [Yalkowsky S.H., 1976], nous permettant ensuite d’extrapoler les résultats à des lixiviations à l’eau.
Sur la base des données de la littérature et par comparaison des concentrations
expérimentales à l’équilibre avec les valeurs théoriques calculées à partir de la loi de Raoult, nous avons conclu que le mécanisme principal responsable du transfert des HAP du sol vers la phase aqueuse en contact est la dissolution. A ce stade de l’étude, nous avons émis l’hypothèse que les très faibles concentrations de saturation des HAP en phase aqueuse (voisine de 1 µg.L-1), était probablement en relation avec la valeur importante du Carbone Organique Total (environ 30%) contenu dans le sol. Les études paramétriques ont montré que la température et la fraction de méthanol (utilisé comme cosolvant) influencent fortement les concentrations à saturation. Par contre, le débit de recirculation et le rapport L/S n’influencent pas significativement les concentrations tant que l’état stationnaire est atteint et que le mélange ne se trouve pas modifié. Cependant, la vitesse d’atteinte de l’état stationnaire (intensité du processus de transfert) se trouve augmentée avec le débit. La température influence le processus de dissolution, généralement endotherme et par conséquent la mobilité des HAP. A partir des informations obtenues, nous avons pu élaborer un abaque qui donne
156

Conclusion générale
pour le phénanthrène la concentration à saturation pour des températures comprises entre 10 et 40° et des fractions de cosolvant comprises entre 0 et 50%. L’étude d’une succession de 9 séquences de lixiviation en batch percolant du même sol, à une température de 40°C et avec une solution contenant 50% de méthanol, sans changement des paramètres opératoires, a montré une tendance de diminution des concentrations de l’anthracène et du phénanthrène à la fin de chaque séquence. Bien que seulement 5-8% du contenu total soit ainsi lixivié, ces changements montrent des modifications dans les mécanismes de mobilisation, dues probablement à une évolution des caractéristiques de la structure du film interfacial au cours des lixiviations successives. Ce comportement montre les limites du modèle proposé et que d’autres mécanismes de transfert/transport restent à identifier et à prendre en compte pour une meilleure description du comportement à la lixiviation à long terme. Enfin, le modèle élaboré sur la base des essais en batch a été transposé et comparé, pour le phénanthrène, avec le comportement de la colonne lixiviée (application en système ouvert) pendant 15 heures (solution 50% méthanol, débit 40 ml/h, température 25°C). Nous atteignions un ratio de lixiviation L/S cumulé de 25 et les prévisions de la simulation concordent très bien avec les résultats expérimentaux. Fraction particulaire :
En parallèle de l’étude sur la fraction solubilisée, nous avons exploré l’autre composante de la mobilité, la fraction de la pollution qui correspond aux HAP mobilisés sous forme particulaire. Les particules mobilisées de diamètre inférieur à 10 µm, sont probablement des particules constituées de goudrons vieillis associées à des colloïdes. Ce phénomène d’entraînement particulaire n’a été quantifiable que pour des lixiviations à l’eau permutée, les lixiviations avec les solutions ioniques (NaN3 et surtout CaCl2) limitant considérablement ce phénomène, même à faible concentration (force ionique de l’ordre de 20 mM). On retrouve ainsi l’effet stabilisant de l’ion Ca2+ pour empêcher la formation des colloïdes. On prouve ainsi que l’évaluation du comportement d’un sol pollué doit tenir compte de la force ionique (salinité) de l’eau avec laquelle il peut entrer en contact et que celle-ci peut contrôler d’une façon importante la mobilité des polluants contenus, en intensité (transport particulaire ou non) et implicitement en durée du processus. Le rôle de la force ionique dans le transport particulaire est confirmé par la corrélation avec la diminution progressive de la conductivité de la solution : pour les ratios L/S grands l’épuisement de la réserve en ions solubles du sol se manifeste, la conductivité et la force ionique de la solution diminuent. Ainsi, pour le sol étudié le transport particulaire est maximal pour des conductivités<100 µScm-1. Il connaît une atténuation de son intensité par la suite. Nous avons proposé la définition d’un potentiel de HAP particulaires relargables (quantité maximale de HAP particulaires relargables dans l’eau par unité de masse de sol). Nos expérimentations laissent apparaître notamment sa dépendance vis à vis des débits de percolation.
157

Conclusion générale
En conclusion, ce mécanisme de transport/transfert peut correspondre au contact avec
une eau peu minéralisée et peut devenir critique pour la gestion d’un site pollué. En effet, Les concentrations en HAP, faibles lorsque le relargage n’est composé que de la fraction dissoute, deviennent significatives au moment du pic de relargage de HAP particulaires.
Les résultats de notre étude ouvrent des nombreuses questions dans le domaine extrêmement complexe de l’évaluation et prévision du comportement des sols pollués par des HAP. D’autres investigations seraient nécessaires pour confirmer nos résultats et notamment des études à plus grandes échelles : en lysimètre et in situ ainsi que sur d’autres sols pollués par des HAP ou autres polluants organiques.
L’expérience acquise au cours de ces travaux peut contribuer à l’élaboration d’une méthodologie relative à l’étude des mécanismes de transfert dans les sites pollués par des HAP. L’utilisation d’un batch percolant (encore à améliorer) pour les études cinétiques et/ou d’équilibre, en parallèle à une colonne de percolation s’avère une solution expérimentale bien adaptée à ce type de problématique. Quelle solution de percolation utiliser : solution avec cosolvants, eau ou solution de force ionique donnée ? La hiérarchie des mécanismes responsables de la mobilisation des polluants (transfert/transport) peut en dépendre largement. Une partie des choix pourraient être imposés par les conditions de disposition environnementale du site étudié.
158

REFERENCES BIBLIOGRAPHIQUES
REFERENCES BIBLIOGRAPHIQUES
1. Academie des sciences. Contamination des sols par les éléments en traces : les risques
et leur gestion. Rapport n°42. Lavoisier Tec&Doc, Paris, 1998, 440 p.
2. Ball, W.P., Roberts, P.V. Long term sorption of halogenated organic chemicals by aquifer material. 1. Intraparticle diffusion. Environmental Science and Technology, 1991, 25(7) : 1237-1249.
3. Barnajee, S., Yalkowsky, S.H. Cosolvent-induced solubilisation of hydrophobic
compounds into water. Anal. Chem., 1988, 60 : 2153-2155.
4. Bayard, R. Etude de l'adsorption/désorption de polluants organiques hydrophobes dans les sols. Approche méthodologique et application au pentachlorophènol et aux hydrocarbures aromatiques polycycliques. Thèse : Institut National des Sciences Appliquées de Lyon. 1997. 231 p.
5. Beck, A.J., Wilson, S.C., Alcock, R.E., Jones, K.C. Kinetic constraints on the loss of
organic chemical from contaminated soils: implications for soil-quality limits. Critical Review in Environmental Science and Technology, 1995, 25(1) : 1-43.
6. Blumer, M. Polycyclic aromatic compouds in nature. Sci. Am. 1976, 234 : 35-45.
7. Bonneau, M., Souchier, B. Pédologie. 2. Constituants et propriétés du sol. Deuxième
édition. Paris : Masson, 1994, 479 p.
8. Bouchard, D.C. Cosolvent effects on sorption isotherm linearity. Journal of Contaminant Hydrology, 2002, 56 : 159-174.
9. Bouchard, D.C. Organic cosolvent effects on the sorption and transport of neutral
organic chemicals. Chemosphere, 1998, 36 : 1883-1892.
10. Bouchez, M., Blanchet, D., Haeseler, F., Vandecasteele, J.P. Les hydrocarbures aromatiques polycycliques dans l'environnement - Première partie : propriétés, origines, devenir. Revue de l'institut français du pétrole, 1996, 51(3) : 407-419.
159

REFERENCES BIBLIOGRAPHIQUES
11. Brunauer, S., Emmett, P.H., Teller, E. Adsorption of gases in multimolecular layers. Journal of the American Chemical Society, 1938, 60 : 309-319.
12. Brusseau, M.L, Rao P.S.C. Sorption nonideality during organic contaminant
transport in porous media. Critical Reviews in Environmental Control, 1989, 19 : 33-99.
13. Brusseau, M.L., Jessup, R.E., Rao P.S.C. Non equilibrium sorption of organic
chemical : elucidation of rate-limiting processes. Environmental Science and Technology ,1991a, 25 : 134-142.
14. Brusseau, M.L., Wood, A.L., Rao, P.S.C. Influence of organic cosolvents on the
sorption kinetics of hydrophobic organic chemical. Environmental Science and Technology. 1991b, 25(5) : 903-910.
15. Chiou, C.T. Reaction and movement of organic chemicals in soils - Theoritical
considerations of the partition of non ionic organic compounds by soil organic, Sawhney, edition Brown, 1989, 1-30.
16. Chiou, C.T., Malcolm, R.L., Brinton, T.I., Kile, D.E. Water solubilty enhancement
of some organic pollutants and pesticides by dissolved humic and fulvic acids, Environmental Science and Technology. 1986, 24 : 502-508.
17. Commans, R.N.J. Development of standard leaching tests for organic pollutants in
soils, sediments, and granular waste materials. Draft Progress report ECN-C-00-094, 2000, 6p.
18. Connaughton, D.F., Stedlinger, J.R., Lion, L.W., Shuler, M.L. Description of time-
varying desorption kinetics : release of naphtalene from contaminated soils. Environmental Science and Technology, 1993, 29 : 2397-2403.
19. Eberhardt, C., Grathwohl, P. Time sclales of organic contaminant dissolution from
complex source zones: coal tar pools vs. blobs. Journal of Contaminant Hydrology, 2002, 59(1-2) : 45-66.
20. Epri (Electric power research institute), Chemical and physical characteristics of tar
sample from selected manufactered gas plant sites. Project No. 2879. Final report. Colchester (connecticust) : Atlantic Environmental Services Inc, 1993.
160

REFERENCES BIBLIOGRAPHIQUES
21. Evans, W.C. Biochemistry of the bacterial catabolism of aromatic compounds in anaerobic environments. Applied and Environmental Microbiology, 1977, 270 : 17-22.
22. Ewardt, N.T. Polycyclic Aromatic Hydrocarbons (PAHs) in the terrestrial
environment. Journal of Enviromental Quality, 1983, 12 : 427-441.
23. Fetter, C.W. Contaminant Hydrology. 2. Mass transport in satured media. 2nd édition. New Jersey : Prentice Hall, 1999, 497 p.
24. Grathwohl, P. Diffusion in natural porous media: contaminant transport, sorption/
desorption and dissolution kinetics. Boston : Kluwer Academic Publishers, 1998, 224 p.
25. Griffin, R.A., Chian, E.S.K. Attenuation of water soluble polychlorinated biphenyl
by earth materials, Rapport final EPA-600/2-80-0207, Washington : United States Environmental Protection Agency, 1980, 169-181.
26. Gupta, S.K., Vollmer, M.K. Krebs, R. The importance of mobile, mobilisable and
pseudo-total heavy metal fractions in soil for three-level risk assessement and risk management. The Science of the Total Environment, 1996, 178 : 11-20.
27. Haeseler, F., Blanchet, D., Druelle, V., Werner, P., Vandecasteele, J.P. Analytical
Characterization of contaminated soils from former manufactured gaz plants. Environmental Science and Technology, 1999, 33(6) : 825-830.
28. Hatzinger, P., Alexander, M. Effect of aging of chemicals in soil on their
biodegradabilty and extractability. Environmental Science and Technology, 1995, 29 : 537-545.
29. Heitkamp, M.A., Cerniglia, C.E. Mineralization of polycyclic aromatic
hydrocarbons by a bacterium isolated from sediment below an oil field. Applied and Environmental Microbiology, 1988, 54 : 1612-1614.
30. Huang W., Yu, H., Weber Jr, W.J. hysteresis in the sorption and desorption of
hydrophobic organic contaminants by soils and sediment : 1. A comparative analysis of experimental protocols. Journal of contaminant hydrology, 1998, 31 : 129-148.
161

REFERENCES BIBLIOGRAPHIQUES
31. Jardine, P.M., Dunnivant, F.M., Selim, H.M., Mccarthy, J.F. Comparaison of models for describing the transport of dissolved organic carbon in aquifer columns. 1992, Soil Science Society of America Journal, 1992, 56 : 393-401.
32. Jayr, E. Devenir des hydrocarbures aromatiques polycycliques (HAP) en milieu
poreux crayeux : sorption et biodégradation. Thèse : Institut National des Sciences Appliquées de Lyon. 2001. 190 p.
33. Ji, W. Brusseau, M.L. A general mathematical model for chemical-enhanced flushing
of soil contaminated by organic compounds. Water Ressources Research, 1998, 34(7) : 1635-1648.
34. Johnson, W.P., Amy, G.L. Facilitated Transport and enhanced desoprtion of
Polycyclic Aromatic Hydrocarbons (PAH) by Natural Organic Matter (NOM) in aquifer sediments. Environmental Science and Technology, 1995, 29 : 807-817.
35. Jones, D.K., Tiller, C.L. Effect of solution chemistry on the extent of binding of
phenanthrene by a soil humic acid: A comparaison of dissolved and clay bound humic. Environmental Science and Technology, 1999, 33 : 55-80.
36. Kao, P.S.C., Hornsby, A.G., Kilcrease, D.P. Sorption and transport of hydrophobic
organic chemicals in aqueous and mixed solvent systems: Model development and primilary evaluation. Journal of Environmental Quality, 1985, 14 : 376-383.
37. Karickoff, S.N., Brown, D.S., Scott, T.A. Sorption of hydrophobic pollutants on
natural sediments and soils. Water Research, 1979, 13 : 241-248.
38. Keith, L.H., Teillard, W.A. Priority pollutants. 1. A perspective view. Environmental Science and Technology. 1979, 13 : 416-423.
39. Kenaga, E.E. Predicted bioconcentration factors and soil sorption coefficients of
pesticides and other chemicals. Ecotoxicology and Environmental Safety, 1980, 4: 26-38.
40. Koch, R. Molecular connectivity index for assessing ecotoxicological behaviour of
organic compounds. Toxicological and environmental chemistry, 1983, 6 : 87-96.
162

REFERENCES BIBLIOGRAPHIQUES
41. Lane, W.F., Loehr, R.C. Estimating the equilibrium aqueous concentrations of polynuclear aromatic hydrocarbons in complex mixtures. Environmental Science and Technology, 1992, 26 : 983-990.
42. Lane, W.F., Loehr, R.C. Predicting aqueous concentrations of polynuclear aromatic
hydrocarbons in complex mixture. Water Environment Research, 1995, 67(2) : 169-173.
43. Lee .L.S., Rao, P.S.C., Okuda, I. Estimating equilibrium partitioning of polycyclic
aromatic hydrocarbons from coal tar into water. Environmental Science and Technology, 1992, 26 : 2110-2115.
44. Leinonen, P.J., Mackay, D. The multicomponent solubilty of hydrocarbons in water.
Canadian Journal of Chemical engineering, 1973, 51 : 230-233.
45. Lotrario, J.B., Stuart, B.J., Lam, T., Arands, R.R., O'Connor, O.A., Kosson, D.S. Effect of sterilization methods on the physical characteristics of soil: Implication for sorption isotherm analyses. Bull. Environ. Contam. Toxicol., 1995, 54 : 668-675.
46. Mahjoub, B. Comportement dans le sol de polluants aromatiques issus du goudron de
houille. Etude du partage goudron/eau et de l'effet du vieillissement sur la mobilité des polluants. Thèse : Institut National des Sciences Appliquées de Lyon. 1999. 262 p.
47. Mahjoub, B., Jayr, E., Bayard, R., Gourdon, R. Phase partition of organic
pollutants between coal tar and water under variable experimental conditions. Water Research, 2000, 34(14) : 3551-3560.
48. McCarthy, J.F., Zachara, J.M. Subsurface transport of contaminants. Environmental
Science and Technology, 1989, 23(5) : 496-502.
49. Meta Environmental. Draft report prepared for electric Power research institute, RP2879-01, 1990.
50. Mihelcik, J.R., Luthy, R.G. Degradation of polycyclic aromatic compounds under
various redox conditions in soil-water systems. Applied and Environmental Microbiology, 1988, 54 : 1182-1187.
51. Molinari, J., Rochon, J. Mesure des paramètres de transport de l'eau et des
substances en solution en zone saturée. La houille blanche, 1976, 3/4 : 223-242.
163

REFERENCES BIBLIOGRAPHIQUES
52. Peters, C.A., Luthy, R.G. Coal tar dissolution in water-miscible solvents:
experimental evaluation. Environmental Science and Technology, 1993, 27 : 2831-2843.
53. Piatt, J.J., Backhus, D.A., Capel, P.D., Einsenreich, S.J. Temperature dependent
sorption of naphtalene, phenenthrene, and pyrene to low organic carbon aquifer sediment. Environmental Science and Technology, 1906, 30 : 751-760.
54. Pignatella, J.J., White, J.C. Influence of bisolute competition on the desorption
kinetics of polycyclic aromatic hydrocarbons in soil. Environmental Science and Technology, 1999, 33(23) : 4292-4298.
55. Powers, S.E., C.O. Loureiro, Abriola, L.M. Theoritical study of the significance of
non equilibrium dissolution of non aqueous phase liquids in subsurface systems. Water Ressources Research, 1991, 27(4) : 463-477.
56. Reza, J., Trejo, A., Elena, L. Determination of the temperature dependance of water
solubilies of polycyclic aromatic hydrocarbons by a generator column-on-line solid phase extraction-liquid chromatographic method. Chemosphere, 2002, 47 : 933-945.
57. Rosenbaum, D., Zamora, P.C., Zukoski, C.F. Phase behavior of small attractive
colloidal particles. Physical Review Letters, 1996, 71(1) : 150-153.
58. Roy, S.B., Dzombak, A.D. Sorption non equilibrium effects on colloid-enhanced transport of hydrophobic organic compounds in porous media. Journal of Contaminant Hydrology, 1998, 30 : 179-200.
59. Rubino, J.T., Yalkowsky, S.H. Cosolvency and deviations from log-linear
solubilization. Pharmacological Research,1987, 4 : 231-236.
60. Scharzenbach, R.P., Gschwend, P.M., Imboden, D.M. Environmental organic chemistry. 5. Solubilty and activity coefficient in water. New York : John Wiley & Sons, 1993, 681 p.
61. Schnitzer, M., Khan S.U. Soil Organic Matter. Amsterdam: Elsevier, 1978), 250 p.
164

REFERENCES BIBLIOGRAPHIQUES
62. Selim, H.M., Davidson, J.M., Mansel, R.S. Evaluation of a two site adsorption-desorption model for describing solute transport in soils, In proceedings of the summer computer simulation conference, Washington : Simulation Councils, 1976, 444-448.
63. Sposito, G. The chemistry of soils. New York: Oxford University Press, 1985, 400 p.
64. Tan, K.H. Principles of soil chemistry. 2nd edition.New York : Marcel Decker, 1993,
362 p.
65. Villermaux, J. Génie de la réaction chimique, conception et fonctionnement des réacteurs. 2nd édition. Paris : Lavoisier Tec & doc, 1993, 250 p.
66. Wood, A.L., Bouchard, D.C., Brusseau, M.L., Rao, P.S.C. Cosolvent effects on
sorption and mobility of organic contaminants in soils. Chemosphere, 1990, 21 : 575-587.
67. Yalkowsky, S.H., Valvani, S.C., Amidon, G.L. Solubilty of non electrolytes in polar
solvents. IV. Non polar drugs in mixed solvents. J. Pharm. Sci., 1976, 65 : 1488-1494.
68. Yeom, I.T., Ghosh, M.M., Cox C.D., Ahn, K.H. Dissolution of polycyclic aromatic hydrocarbons from weathered contaminated soil. Water Science Technology, 1996, 34(7-6) : 335-342.
165

Annexes
ANNEXES
Annexe 1 : Extraction des HAP en phase solide
Annexe 2 : Extraction des HAP en phase aqueuse
Annexe 3 : Caractéristiques de L’HPLC Waters
Annexe 4 : Caractéristiques du MEB
Annexe 5 : Cinétique en batch avec contact en milieu dispersé avec l’eau
Annexe 6 : Renouvellements séquentiels en batch avec contact en milieu dispersé avec l’eau
Annexe 7 : Renouvellements séquentiels en batch avec contact en milieu dispersé avec le
méthanol
Annexe 8 : Percolation en colonne
Annexe 9 : Renouvellements en batch percolant avec l’eau
Annexe 10 : Renouvellements en batch percolant avec le méthanol
Annexe 11 : Cinétique en bath percolant avec l’eau
Annexe 12 : Cinétiques en bath percolant avec le méthanol
Annexe 13 : Percolation à travers le dispositif du batch percolant avec le méthanol
Annexe 14 : Percolation à travers le dispositif du batch percolant avec de l’eau permutée
ANNEXE 15 : CONTRIBUTION A L’ELABORATION D’UNE METHODOLOGIE RELATIVE A L’ETUDE
DE LA MOBILITE DES COMPOSES ORGANIQUES HYDROPHOBES TELS QUE LES HAP
166

Annexes
Extraction des HAP en phase solide
Annexe 1
167

Annexes
L’extraction en phase solide correspond dans notre étude à la mesure du contenu total en
polluant dans le sol. La technique utilisée au laboratoire est l’extraction par le dispositif
soxhlet :
Figure 1 : Schéma du dispositif soxhlet
Le solvant hydrophobe est porté à ébulition, les vapeurs montent jusqu’au réfrigérant où elles
se condensent et tombent sur la cartouche en cellulose contenant le sol. A ce stade, le solvant
extrait les polluants présents dans le sol. Lorsque le solvant atteint un certain niveau, un
système de siphon provoque le retour du volume total de solvant au contact de la cartouche
cellulose vers le ballon. La cartouche se remplit de nouveau avec du solvant propre puisque la
pollution extraite précédemment reste dans le ballon. L’extraction est terminée lorsque le
condensas retombant sur la cartouche devient claire (24 heures).
.
Le protocole présenté est une adaptation de la norme NF ISO 13877 de avril 1999.
• Remplir la cartouche avec 5 g de sol sec
• Introduire 300 ml de solvant (150 ml d’acetone + 150 ml de dichlorométhane) dans le
ballon et ajouter quelques morceaux de pierre ponce et peser l’ensemble.
• Amener le solvant à ébulition, veillez à ce que la condensation ait lieu dans les premiers
cm du réfrigérant.
• Lorsque l’extraction est terminée, peser à nouveau le ballon.
• Evaporer 10 ml de solvant sous courant d’azote
168

Annexes
• Reprendre dans 10 ml d’acétonitrile puis diluer 10 fois
L’échantillon ainsi préparé est disponible pour l’analyse.
169

Annexes
Extraction des HAP en phase aqueuse
Annexe 2
170

Annexes
L’objectif de cette technique est de mesurer la concentration en polluants dans la phase
aqueuse. Cependant, l’analyse immédiate à partir de la phase aqueuse n’est pas possible
puisque les concentrations obtenues sont trop proches du seuil de détection de l’appareil de
mesure. Elle est basée sur le transfert des polluants de la phase aqueuse vers un solvant
organique. L’extraction liquide-liquide n’étant pas satisfaisante (du fait de la formation d’une
émulsion), c’est l’extraction sur cartouche SPE qui a été retenue :
Pompe à vide
DispositifVACMASTER
CartoucheSPE
TubeTEFLON
Flacond’échantillonage
Figure 2 : dispositif expérimental d’extraction en phase solide (SPE)
Le modèle de cartouche utilisé pour l’extraction des HAP en solution est ISOLUTE PAH
1,5g/6mL.
• Prétraitement : Pour 100 ml d’échantillon aqueux, ajouter 10% isopropanol (V/V) et
agiter vigoureusement.
• Solvatation : solvater la cartouche avec de l’isopropanol (5ml) à un débit de 5 ml/min.
• Equilibrage : Equilibrer la cartouche avec de l’eau permutée contenant 2% (V/V)
isopropanol (5 ml) à un débit de 5 ml/min.
• Application de l’échantillon : charger la cartouche avec 3 ml d’échantillon puis
installer le dispositif (voir Figure 2). Utiliser un débit de 20 ml/min. Rincer le flacon avec
10 ml d’acetone puis diluer dans 100 ml d’eau permutée et appliquer le solvant de
rinçage à la cartouche.
171

Annexes
• Elimination des interférences : Rincer la colonne avec 5 ml d’un mélange eau
permutée/isopropanol V/V (90/10). Sécher la colonne pendant 20 min en créant un vide
de 20’’ de Hg.
• Elution des HAP : éluer avec 2*3 ml du mélange THF/hexane (50/50, V/V), débit de 5
ml/min. Pour maximiser la récupération des HAP, laisser baigner 10 min avant chaque
élution.
Ensuite il reste à évaporer l’extrait à basse température sous courant d’azote (attention de ne
pas assécher complètement l’extrait). Reprendre l’extrait dans 5 ml d’acetonitrile
(l’échantillon aqueux a donc été concentré 20 fois). L’échantillon est prêt pour l’analyse.
172

Annexes
Caractéristiques de L’HPLC
Annexe 3
173

Annexes
Les caractéristiques de l’HPLC (Waters LC moldule I) sont les suivantes :
• Injection automatique
• Détecteur UV à 254 nm
• Détecteur Fluorimètre programmable
• Précolonne SUPELGUARD LC-18 d’une longueur de 2 cm
• colonne YMC de 250 mm de longueur et de 4 mm de diamètre ;
• phase éluante constituée d’un mélange eau/acétonitrile;
• débit d’élution 1,0 ml/min
174

Annexes
Caractéristiques du MEB environnemental
Annexe 4
175

Annexes
PRINCIPE DE LA MICROSCOPIE ELECTRONIQUE
Image en contraste topographique
information
Image en contraste chimique
Spectre X (analyse élémentaire)
électrons secondaires
électrons rétrodiffusés
photons X
intéraction faisceaud ’électrons- matière
Faisceau d ’électrons incidents
échantillon
détecteur
GSE
BSE
EDS
GSE : Gazeous secondary electronsBSE : Back- Scattered electronsEDS : Energy Dispersive Spectroscopy poire d ’intéraction
Caractéristiques du microscope XL30 ESEM FEG: ESEM : Environnemental Scanning Electron Microscope
FEG : Field Emission Gun
source : canon à émission de champ (pointe FEG) donnant une très haute résolution et
permettant l’observation non destructive d’échantillons fragiles (source très brillante
autorisant une diminution de la tension d’accélération).
mode environnemental : la possibilité d’introduire un gaz dans la chambre
( pression maximale : 15 torrs ) permet l’observation de tout type d’échantillons (non
conducteurs, humides, graisseux…) dans un environnement gazeux choisi.
accessoires :
176

Annexes
- platine refroidie par effet Peltier :
refroidissement de l’échantillon jusqu’à – 20°C
permet, en ajustant la température et la pression de vapeur d’eau dans la
chambre, de contrôler le séchage et l’humidification de l’échantillon.
- platine chauffante : chauffage de l’échantillon jusqu’à 1500°C, avec possibilité
de programmer des rampes de chauffage.
- microanalyse dispersive en énergie : permet de déterminer la composition
chimique élémentaire de l’échantillon (détection des éléments à partir du bore).
177

Annexes
Cinétique en batch avec contact en milieu dispersé avec l’eau
Annexe 5
178
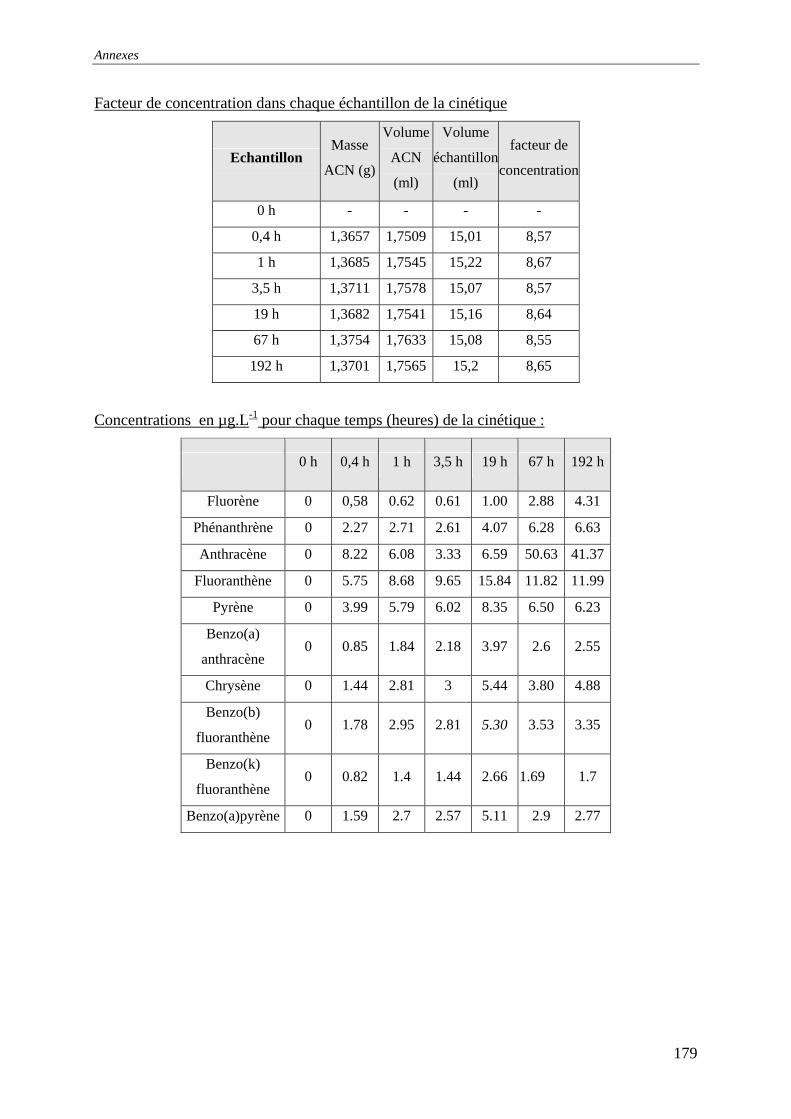
Annexes
Facteur de concentration dans chaque échantillon de la cinétique
Echantillon Masse
ACN (g)
Volume
ACN
(ml)
Volume
échantillon
(ml)
facteur de
concentration
0 h - - - -
0,4 h 1,3657 1,7509 15,01 8,57
1 h 1,3685 1,7545 15,22 8,67
3,5 h 1,3711 1,7578 15,07 8,57
19 h 1,3682 1,7541 15,16 8,64
67 h 1,3754 1,7633 15,08 8,55
192 h 1,3701 1,7565 15,2 8,65
Concentrations en µg.L-1 pour chaque temps (heures) de la cinétique :
0 h 0,4 h 1 h 3,5 h 19 h 67 h 192 h
Fluorène 0 0,58 0.62 0.61 1.00 2.88 4.31
Phénanthrène 0 2.27 2.71 2.61 4.07 6.28 6.63
Anthracène 0 8.22 6.08 3.33 6.59 50.63 41.37
Fluoranthène 0 5.75 8.68 9.65 15.84 11.82 11.99
Pyrène 0 3.99 5.79 6.02 8.35 6.50 6.23
Benzo(a)
anthracène 0 0.85 1.84 2.18 3.97 2.6 2.55
Chrysène 0 1.44 2.81 3 5.44 3.80 4.88
Benzo(b)
fluoranthène 0 1.78 2.95 2.81 5.30 3.53 3.35
Benzo(k)
fluoranthène 0 0.82 1.4 1.44 2.66 1.69 1.7
Benzo(a)pyrène 0 1.59 2.7 2.57 5.11 2.9 2.77
179

Annexes
Renouvellements séquentiels en batch avec contact en milieu
dispersé avec l’eau
Annexe 6
180
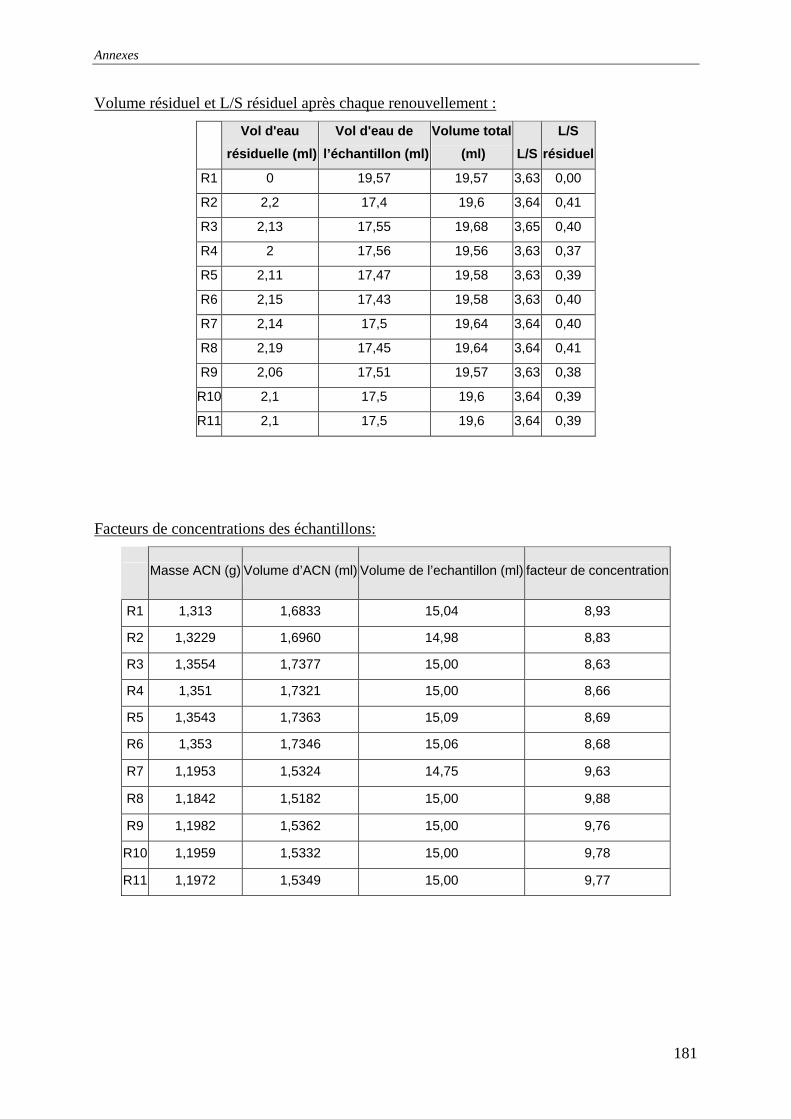
Annexes
Volume résiduel et L/S résiduel après chaque renouvellement :
Vol d'eau résiduelle (ml)
Vol d'eau de l’échantillon (ml)
Volume total (ml) L/S
L/S résiduel
R1 0 19,57 19,57 3,63 0,00
R2 2,2 17,4 19,6 3,64 0,41
R3 2,13 17,55 19,68 3,65 0,40
R4 2 17,56 19,56 3,63 0,37
R5 2,11 17,47 19,58 3,63 0,39
R6 2,15 17,43 19,58 3,63 0,40
R7 2,14 17,5 19,64 3,64 0,40
R8 2,19 17,45 19,64 3,64 0,41
R9 2,06 17,51 19,57 3,63 0,38
R10 2,1 17,5 19,6 3,64 0,39
R11 2,1 17,5 19,6 3,64 0,39
Facteurs de concentrations des échantillons:
Masse ACN (g) Volume d’ACN (ml) Volume de l’echantillon (ml) facteur de concentration
R1 1,313 1,6833 15,04 8,93
R2 1,3229 1,6960 14,98 8,83
R3 1,3554 1,7377 15,00 8,63
R4 1,351 1,7321 15,00 8,66
R5 1,3543 1,7363 15,09 8,69
R6 1,353 1,7346 15,06 8,68
R7 1,1953 1,5324 14,75 9,63
R8 1,1842 1,5182 15,00 9,88
R9 1,1982 1,5362 15,00 9,76
R10 1,1959 1,5332 15,00 9,78
R11 1,1972 1,5349 15,00 9,77
181
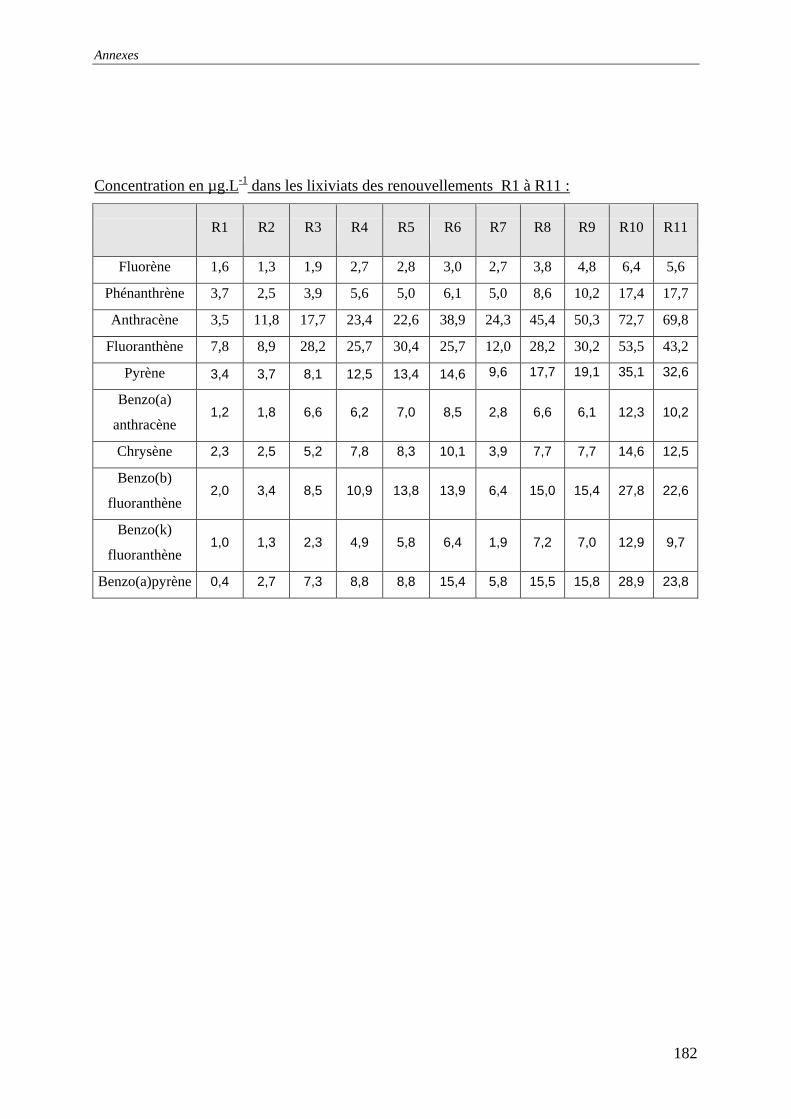
Annexes
Concentration en µg.L-1 dans les lixiviats des renouvellements R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8 R9 R10 R11
Fluorène 1,6 1,3 1,9 2,7 2,8 3,0 2,7 3,8 4,8 6,4 5,6
Phénanthrène 3,7 2,5 3,9 5,6 5,0 6,1 5,0 8,6 10,2 17,4 17,7
Anthracène 3,5 11,8 17,7 23,4 22,6 38,9 24,3 45,4 50,3 72,7 69,8
Fluoranthène 7,8 8,9 28,2 25,7 30,4 25,7 12,0 28,2 30,2 53,5 43,2
Pyrène 3,4 3,7 8,1 12,5 13,4 14,6 9,6 17,7 19,1 35,1 32,6
Benzo(a)
anthracène 1,2 1,8 6,6 6,2 7,0 8,5 2,8 6,6 6,1 12,3 10,2
Chrysène 2,3 2,5 5,2 7,8 8,3 10,1 3,9 7,7 7,7 14,6 12,5
Benzo(b)
fluoranthène 2,0 3,4 8,5 10,9 13,8 13,9 6,4 15,0 15,4 27,8 22,6
Benzo(k)
fluoranthène 1,0 1,3 2,3 4,9 5,8 6,4 1,9 7,2 7,0 12,9 9,7
Benzo(a)pyrène 0,4 2,7 7,3 8,8 8,8 15,4 5,8 15,5 15,8 28,9 23,8
182

Annexes
Renouvellements séquentiels en batch avec contact en milieu
dispersé avec le méthanol
Annexe 7
183
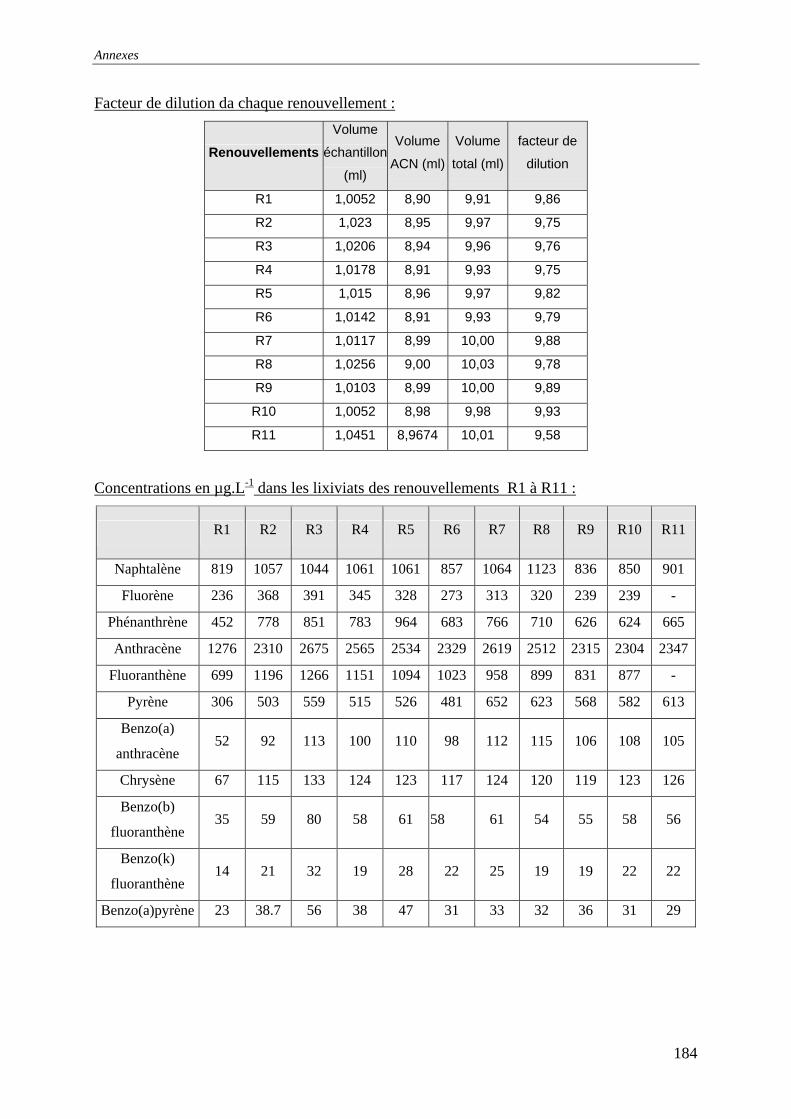
Annexes
Facteur de dilution da chaque renouvellement :
Renouvellements Volume
échantillon
(ml)
Volume
ACN (ml)
Volume
total (ml)
facteur de
dilution
R1 1,0052 8,90 9,91 9,86
R2 1,023 8,95 9,97 9,75
R3 1,0206 8,94 9,96 9,76
R4 1,0178 8,91 9,93 9,75
R5 1,015 8,96 9,97 9,82
R6 1,0142 8,91 9,93 9,79
R7 1,0117 8,99 10,00 9,88
R8 1,0256 9,00 10,03 9,78
R9 1,0103 8,99 10,00 9,89
R10 1,0052 8,98 9,98 9,93
R11 1,0451 8,9674 10,01 9,58
Concentrations en µg.L-1 dans les lixiviats des renouvellements R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8 R9 R10 R11
Naphtalène 819 1057 1044 1061 1061 857 1064 1123 836 850 901
Fluorène 236 368 391 345 328 273 313 320 239 239 -
Phénanthrène 452 778 851 783 964 683 766 710 626 624 665
Anthracène 1276 2310 2675 2565 2534 2329 2619 2512 2315 2304 2347
Fluoranthène 699 1196 1266 1151 1094 1023 958 899 831 877 -
Pyrène 306 503 559 515 526 481 652 623 568 582 613
Benzo(a)
anthracène 52 92 113 100 110 98 112 115 106 108 105
Chrysène 67 115 133 124 123 117 124 120 119 123 126
Benzo(b)
fluoranthène 35 59 80 58 61 58 61 54 55 58 56
Benzo(k)
fluoranthène 14 21 32 19 28 22 25 19 19 22 22
Benzo(a)pyrène 23 38.7 56 38 47 31 33 32 36 31 29
184

Annexes
Percolation en colonne
Annexe 8
185
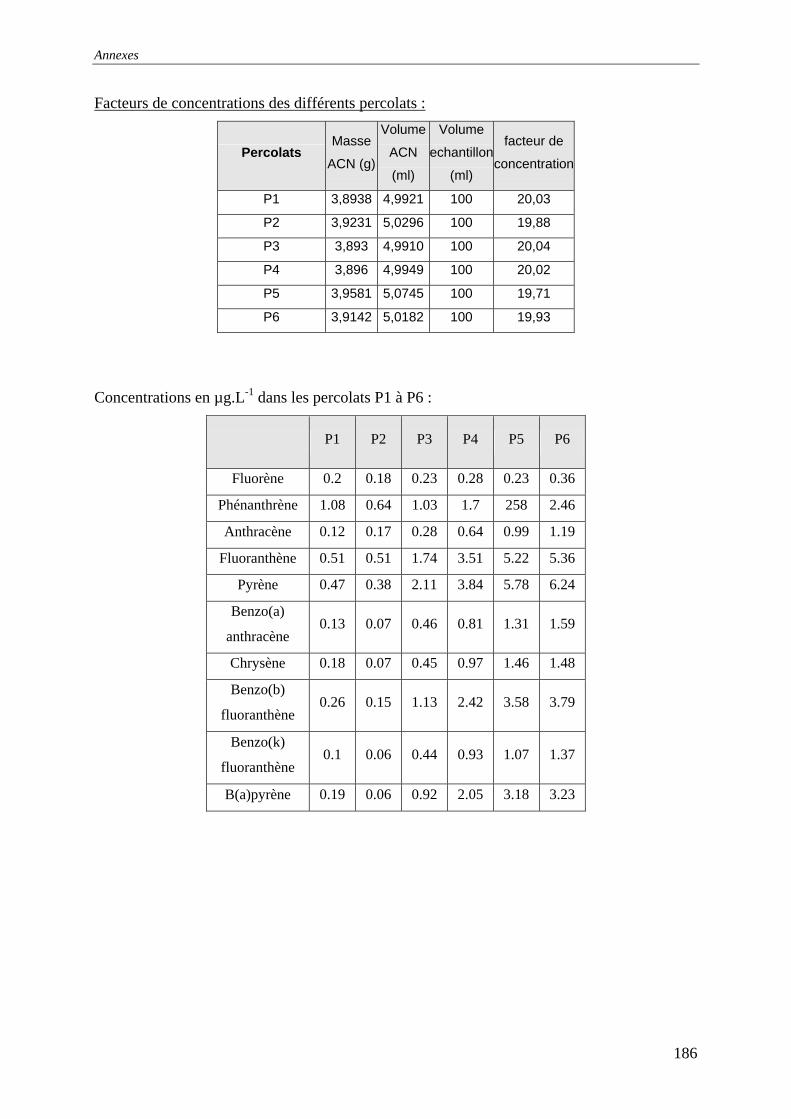
Annexes
Facteurs de concentrations des différents percolats :
Percolats Masse
ACN (g)
Volume
ACN
(ml)
Volume
echantillon
(ml)
facteur de
concentration
P1 3,8938 4,9921 100 20,03
P2 3,9231 5,0296 100 19,88
P3 3,893 4,9910 100 20,04
P4 3,896 4,9949 100 20,02
P5 3,9581 5,0745 100 19,71
P6 3,9142 5,0182 100 19,93
Concentrations en µg.L-1 dans les percolats P1 à P6 :
P1 P2 P3 P4 P5 P6
Fluorène 0.2 0.18 0.23 0.28 0.23 0.36
Phénanthrène 1.08 0.64 1.03 1.7 258 2.46
Anthracène 0.12 0.17 0.28 0.64 0.99 1.19
Fluoranthène 0.51 0.51 1.74 3.51 5.22 5.36
Pyrène 0.47 0.38 2.11 3.84 5.78 6.24
Benzo(a)
anthracène 0.13 0.07 0.46 0.81 1.31 1.59
Chrysène 0.18 0.07 0.45 0.97 1.46 1.48
Benzo(b)
fluoranthène 0.26 0.15 1.13 2.42 3.58 3.79
Benzo(k)
fluoranthène 0.1 0.06 0.44 0.93 1.07 1.37
B(a)pyrène 0.19 0.06 0.92 2.05 3.18 3.23
186

Annexes
Renouvellements en batch percolant avec l’eau
Annexe 9
187
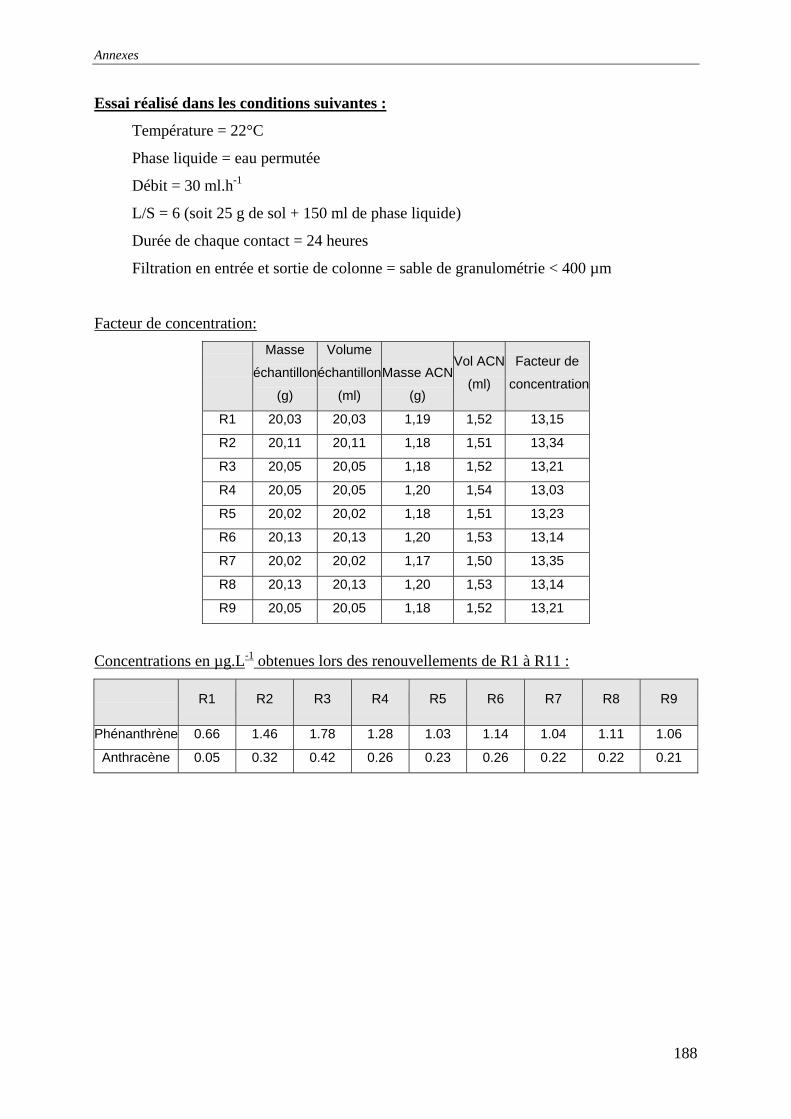
Annexes
Essai réalisé dans les conditions suivantes :
Température = 22°C
Phase liquide = eau permutée
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Facteur de concentration:
Masse
échantillon
(g)
Volume
échantillon
(ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
R1 20,03 20,03 1,19 1,52 13,15
R2 20,11 20,11 1,18 1,51 13,34
R3 20,05 20,05 1,18 1,52 13,21
R4 20,05 20,05 1,20 1,54 13,03
R5 20,02 20,02 1,18 1,51 13,23
R6 20,13 20,13 1,20 1,53 13,14
R7 20,02 20,02 1,17 1,50 13,35
R8 20,13 20,13 1,20 1,53 13,14
R9 20,05 20,05 1,18 1,52 13,21
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8 R9
Phénanthrène 0.66 1.46 1.78 1.28 1.03 1.14 1.04 1.11 1.06
Anthracène 0.05 0.32 0.42 0.26 0.23 0.26 0.22 0.22 0.21
188
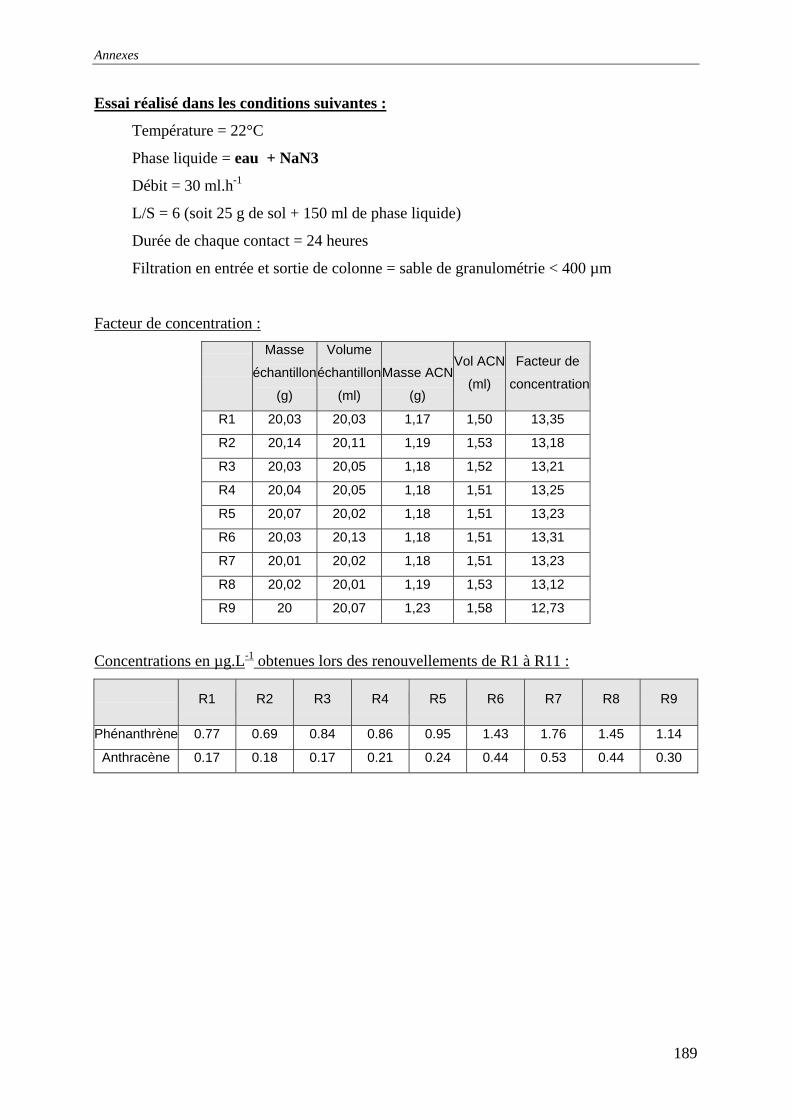
Annexes
Essai réalisé dans les conditions suivantes :
Température = 22°C
Phase liquide = eau + NaN3
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Facteur de concentration :
Masse
échantillon
(g)
Volume
échantillon
(ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
R1 20,03 20,03 1,17 1,50 13,35
R2 20,14 20,11 1,19 1,53 13,18
R3 20,03 20,05 1,18 1,52 13,21
R4 20,04 20,05 1,18 1,51 13,25
R5 20,07 20,02 1,18 1,51 13,23
R6 20,03 20,13 1,18 1,51 13,31
R7 20,01 20,02 1,18 1,51 13,23
R8 20,02 20,01 1,19 1,53 13,12
R9 20 20,07 1,23 1,58 12,73
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8 R9
Phénanthrène 0.77 0.69 0.84 0.86 0.95 1.43 1.76 1.45 1.14
Anthracène 0.17 0.18 0.17 0.21 0.24 0.44 0.53 0.44 0.30
189
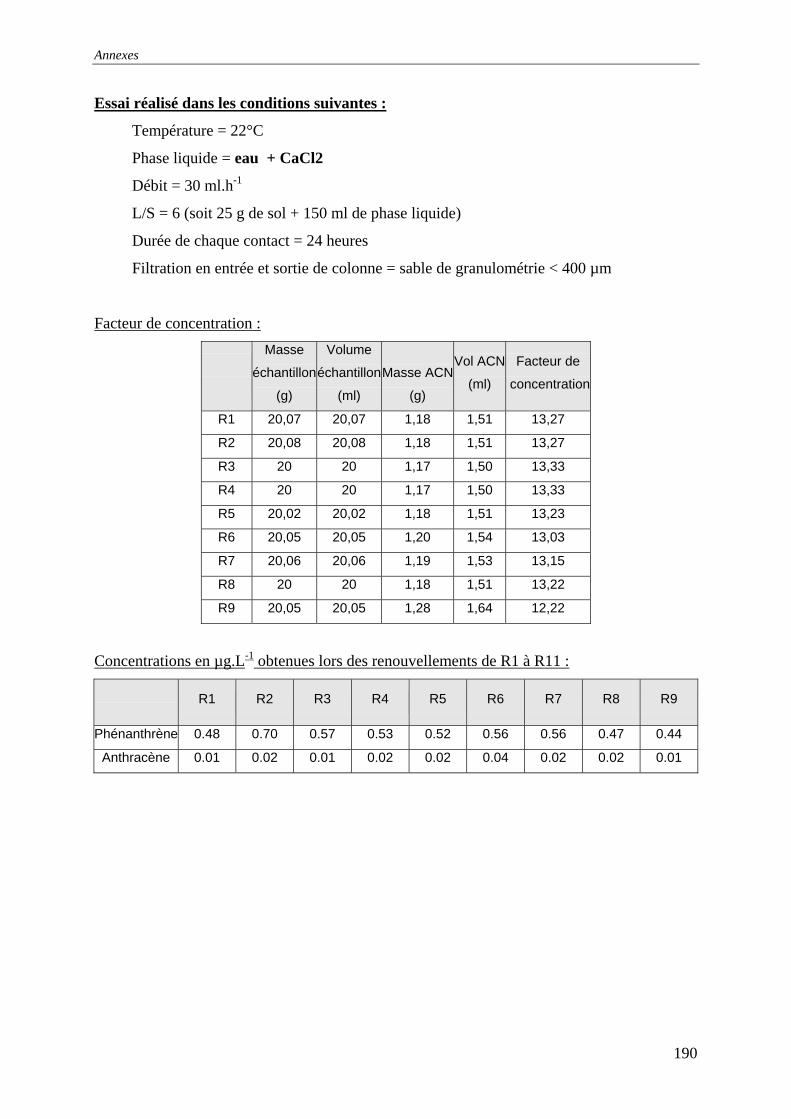
Annexes
Essai réalisé dans les conditions suivantes :
Température = 22°C
Phase liquide = eau + CaCl2
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Facteur de concentration :
Masse
échantillon
(g)
Volume
échantillon
(ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
R1 20,07 20,07 1,18 1,51 13,27
R2 20,08 20,08 1,18 1,51 13,27
R3 20 20 1,17 1,50 13,33
R4 20 20 1,17 1,50 13,33
R5 20,02 20,02 1,18 1,51 13,23
R6 20,05 20,05 1,20 1,54 13,03
R7 20,06 20,06 1,19 1,53 13,15
R8 20 20 1,18 1,51 13,22
R9 20,05 20,05 1,28 1,64 12,22
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8 R9
Phénanthrène 0.48 0.70 0.57 0.53 0.52 0.56 0.56 0.47 0.44
Anthracène 0.01 0.02 0.01 0.02 0.02 0.04 0.02 0.02 0.01
190
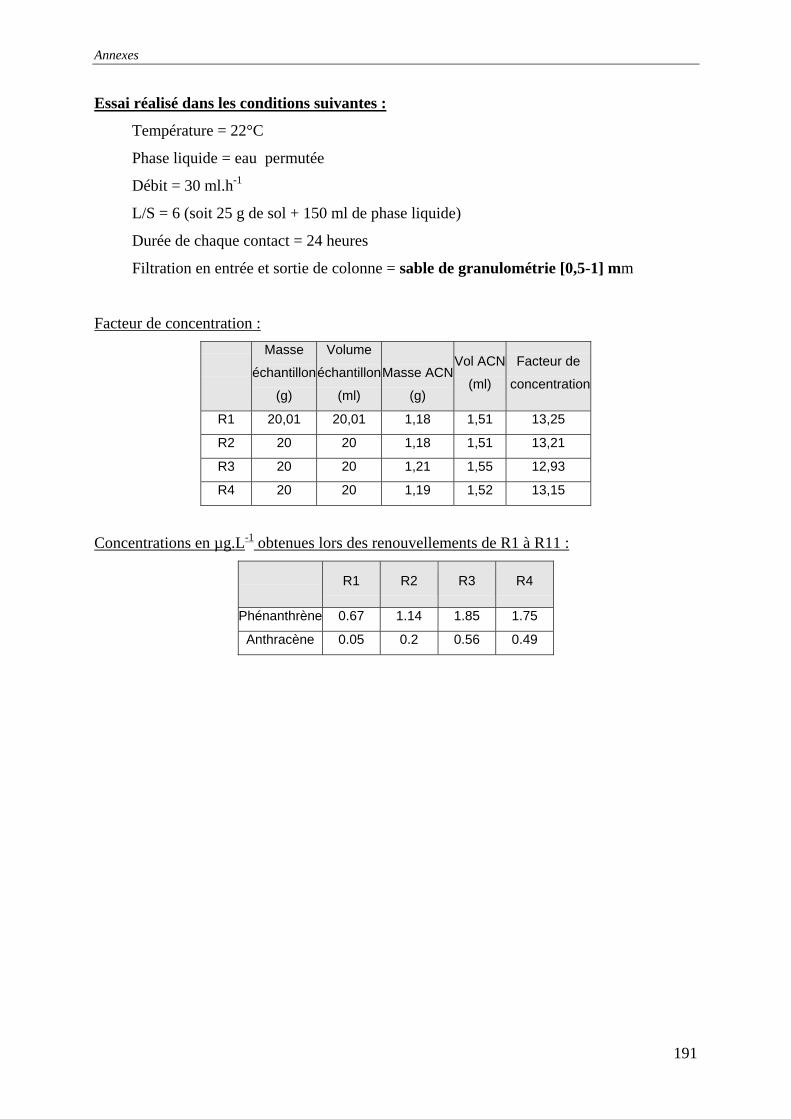
Annexes
Essai réalisé dans les conditions suivantes :
Température = 22°C
Phase liquide = eau permutée
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = sable de granulométrie [0,5-1] mm
Facteur de concentration :
Masse
échantillon
(g)
Volume
échantillon
(ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
R1 20,01 20,01 1,18 1,51 13,25
R2 20 20 1,18 1,51 13,21
R3 20 20 1,21 1,55 12,93
R4 20 20 1,19 1,52 13,15
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R4
Phénanthrène 0.67 1.14 1.85 1.75
Anthracène 0.05 0.2 0.56 0.49
191
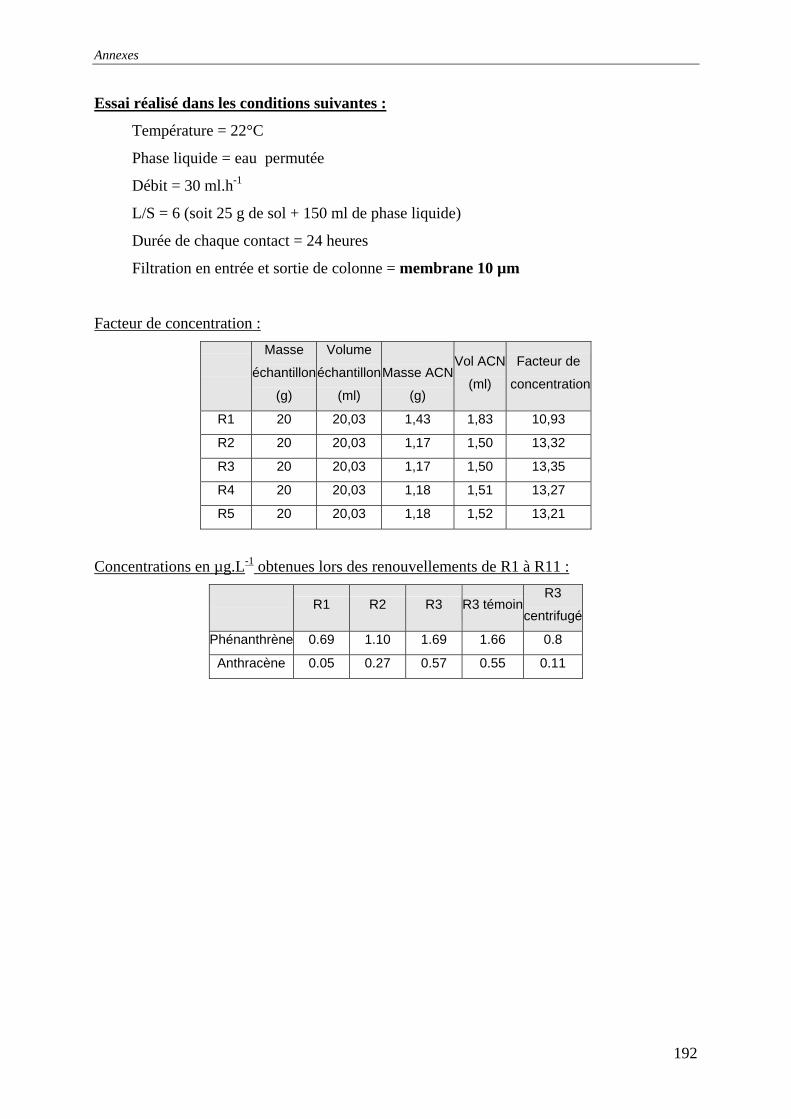
Annexes
Essai réalisé dans les conditions suivantes :
Température = 22°C
Phase liquide = eau permutée
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = membrane 10 µm
Facteur de concentration :
Masse
échantillon
(g)
Volume
échantillon
(ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
R1 20 20,03 1,43 1,83 10,93
R2 20 20,03 1,17 1,50 13,32
R3 20 20,03 1,17 1,50 13,35
R4 20 20,03 1,18 1,51 13,27
R5 20 20,03 1,18 1,52 13,21
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R3 témoinR3
centrifugé
Phénanthrène 0.69 1.10 1.69 1.66 0.8
Anthracène 0.05 0.27 0.57 0.55 0.11
192

Annexes
Renouvellements en batch percolant avec le méthanol
Annexe 10
193
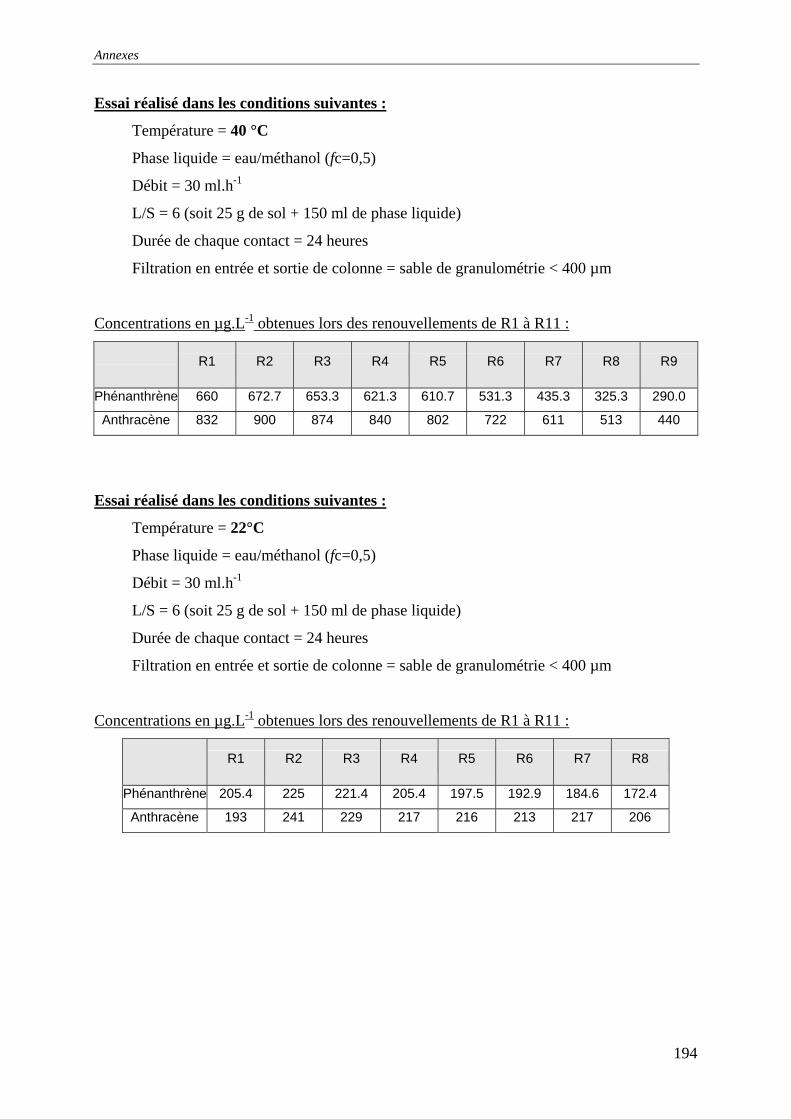
Annexes
Essai réalisé dans les conditions suivantes :
Température = 40 °C
Phase liquide = eau/méthanol (fc=0,5)
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8 R9
Phénanthrène 660 672.7 653.3 621.3 610.7 531.3 435.3 325.3 290.0
Anthracène 832 900 874 840 802 722 611 513 440
Essai réalisé dans les conditions suivantes :
Température = 22°C
Phase liquide = eau/méthanol (fc=0,5)
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Durée de chaque contact = 24 heures
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
R1 R2 R3 R4 R5 R6 R7 R8
Phénanthrène 205.4 225 221.4 205.4 197.5 192.9 184.6 172.4
Anthracène 193 241 229 217 216 213 217 206
194

Annexes
Cinétique en bath percolant avec l’eau
Annexe 11
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Phase liquide = eau permutée
195
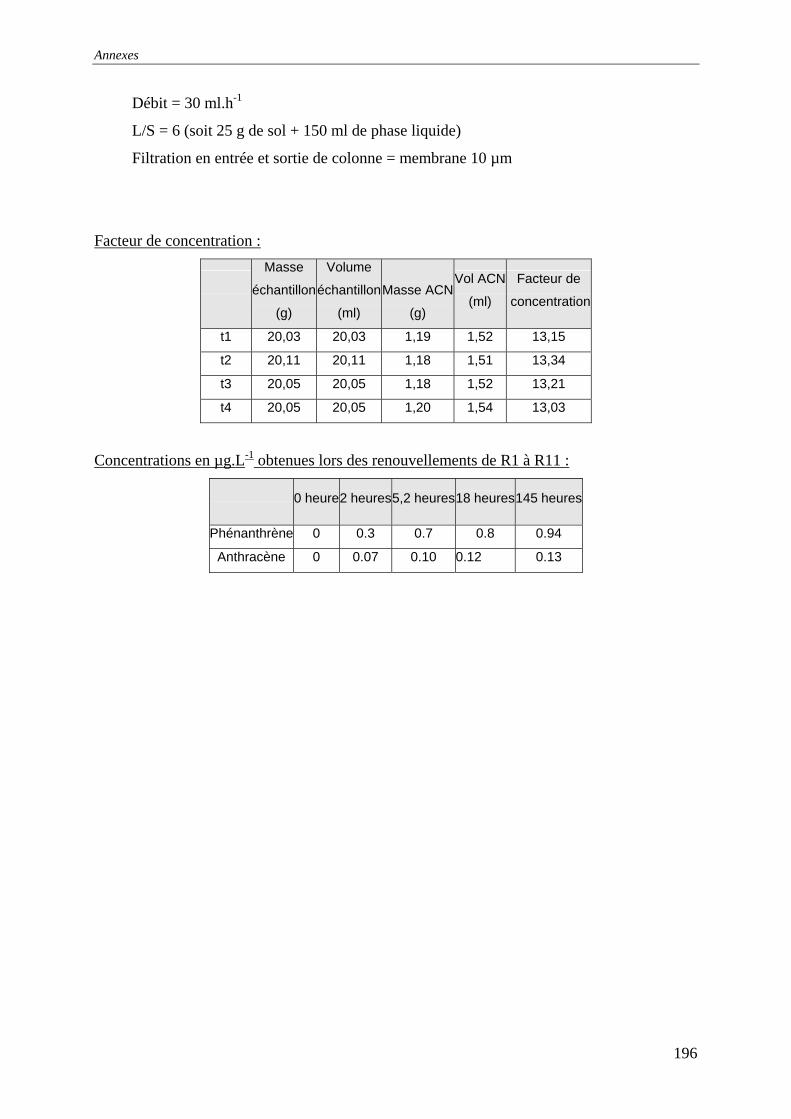
Annexes
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Facteur de concentration :
Masse
échantillon
(g)
Volume
échantillon
(ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
t1 20,03 20,03 1,19 1,52 13,15
t2 20,11 20,11 1,18 1,51 13,34
t3 20,05 20,05 1,18 1,52 13,21
t4 20,05 20,05 1,20 1,54 13,03
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure2 heures5,2 heures18 heures145 heures
Phénanthrène 0 0.3 0.7 0.8 0.94
Anthracène 0 0.07 0.10 0.12 0.13
196

Annexes
Cinétiques en bath percolant avec le méthanol
Annexe 12
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Phase liquide = eau /méthanol (fc=0,3)
197

Annexes
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure2 heures5,2 heures18 heures24 heures48 heures150 heures
Phénanthrène 0 1.94 3.72 6.3 6.65 7.28 7.69
Anthracène 0 1.75 3.47 5.34 5.82 7 7.5
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Phase liquide = eau /méthanol (fc=0,4)
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure 1,3 heures5 heures17 heures23,5 heures51 heures145 heures
Phénanthrène 0 18.27 45.06 57 59 69.3 72.3
Anthracène 0 13.73 33.4 46.6 50.17 58.8 64.95
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Phase liquide = eau /méthanol (fc=0,5)
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure2 heures5,2 heures18 heures24 heures48 heures72 heures149 heures
198

Annexes
Phénanthrène 0 70.8 133.7 182 191 231 250
Anthracène 0 90 171 240.2 256 287.4 300
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Phase liquide = eau /méthanol (fc=0,4)
Débit = 120 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure 2 heures5 heures18 heures24 heures48 heures96 heures
Phénanthrène 0 34.6 44.7 58.9 64 71.6 69.5
Anthracène 0 31 43 52 53.6 61.5 63
Essai réalisé dans les conditions suivantes :
Température = 10 °C
Phase liquide = eau /méthanol (fc=0,5)
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure2 heures5,2 heures18 heures24 heures48 heures72 heures149 heures
Phénanthrène 0 58.5 93 134 148.15 171.8 186 208.5
Anthracène 0 88.6 138.6 192 207 229.2 243.8 250
Essai réalisé dans les conditions suivantes :
Température = 40 °C
Phase liquide = eau /méthanol (fc=0,5)
Débit = 30 ml.h-1
L/S = 6 (soit 25 g de sol + 150 ml de phase liquide)
199
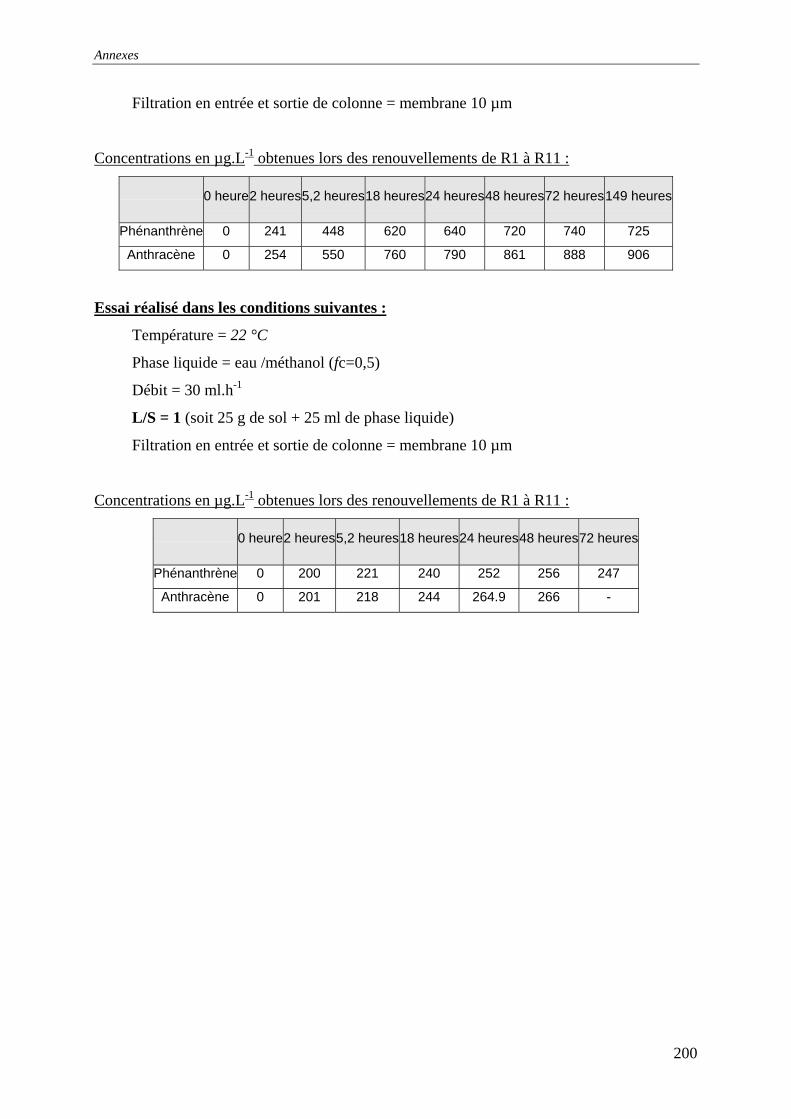
Annexes
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure2 heures5,2 heures18 heures24 heures48 heures72 heures149 heures
Phénanthrène 0 241 448 620 640 720 740 725
Anthracène 0 254 550 760 790 861 888 906
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Phase liquide = eau /méthanol (fc=0,5)
Débit = 30 ml.h-1
L/S = 1 (soit 25 g de sol + 25 ml de phase liquide)
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations en µg.L-1 obtenues lors des renouvellements de R1 à R11 :
0 heure2 heures5,2 heures18 heures24 heures48 heures72 heures
Phénanthrène 0 200 221 240 252 256 247
Anthracène 0 201 218 244 264.9 266 -
200

Annexes
Percolation à travers le dispositif du batch percolant avec le
méthanol
Annexe 13
Essai réalisé dans les conditions suivantes :
Température = 25 °C
Phase liquide = eau/méthanol (fc=0,5)
201
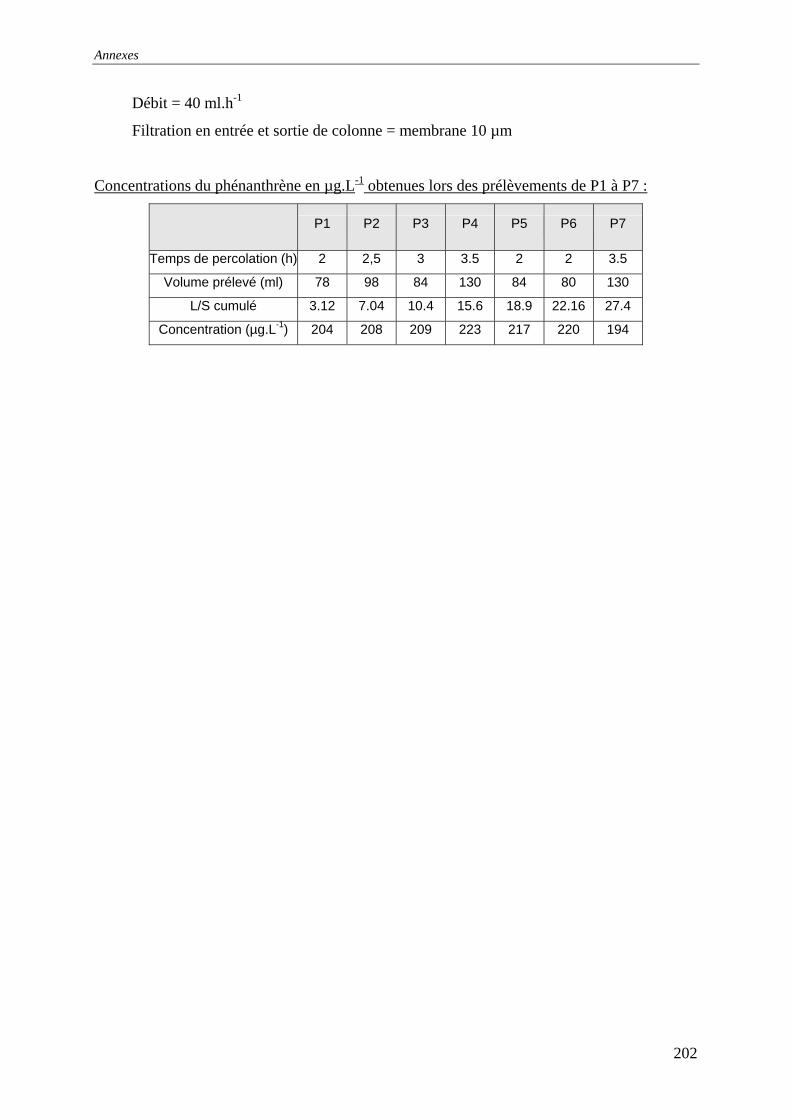
Annexes
Débit = 40 ml.h-1
Filtration en entrée et sortie de colonne = membrane 10 µm
Concentrations du phénanthrène en µg.L-1 obtenues lors des prélèvements de P1 à P7 :
P1 P2 P3 P4 P5 P6 P7
Temps de percolation (h) 2 2,5 3 3.5 2 2 3.5
Volume prélevé (ml) 78 98 84 130 84 80 130
L/S cumulé 3.12 7.04 10.4 15.6 18.9 22.16 27.4
Concentration (µg.L-1) 204 208 209 223 217 220 194
202

Annexes
Percolation à travers le dispositif du batch percolant avec de l’eau
permutée
Annexe 14
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Masse de sol = 25 g
203
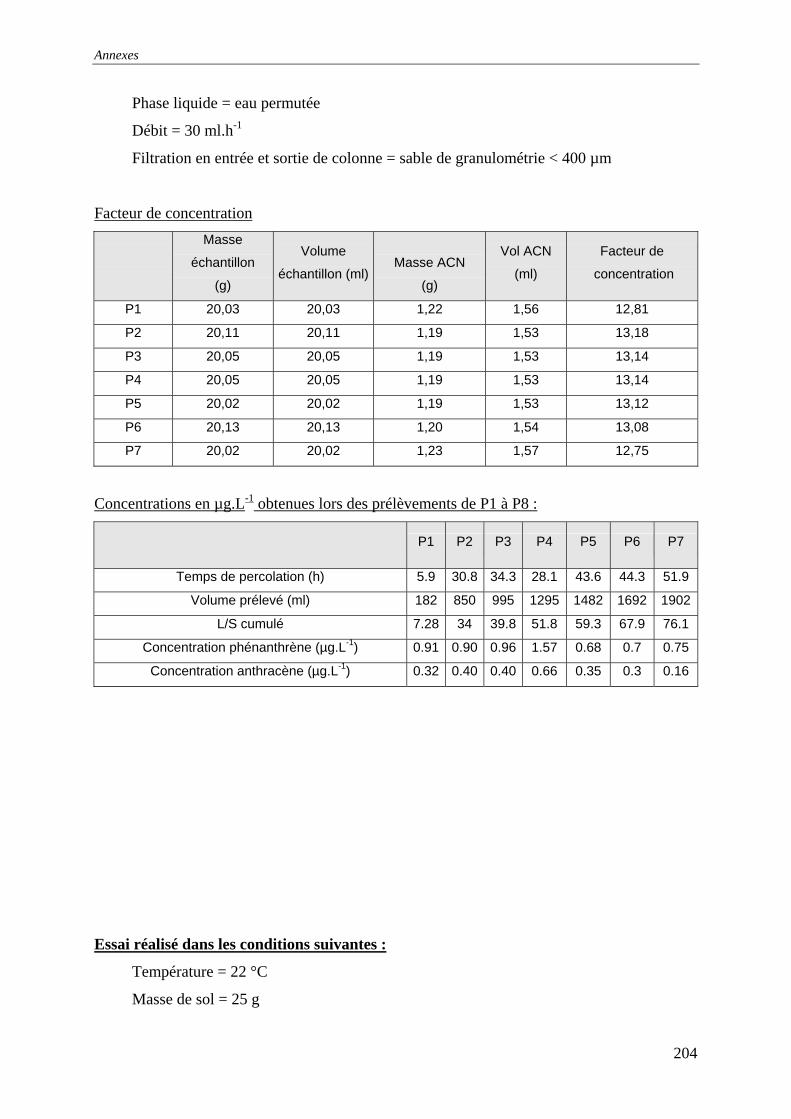
Annexes
Phase liquide = eau permutée
Débit = 30 ml.h-1
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Facteur de concentration
Masse
échantillon
(g)
Volume
échantillon (ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
P1 20,03 20,03 1,22 1,56 12,81
P2 20,11 20,11 1,19 1,53 13,18
P3 20,05 20,05 1,19 1,53 13,14
P4 20,05 20,05 1,19 1,53 13,14
P5 20,02 20,02 1,19 1,53 13,12
P6 20,13 20,13 1,20 1,54 13,08
P7 20,02 20,02 1,23 1,57 12,75
Concentrations en µg.L-1 obtenues lors des prélèvements de P1 à P8 :
P1 P2 P3 P4 P5 P6 P7
Temps de percolation (h) 5.9 30.8 34.3 28.1 43.6 44.3 51.9
Volume prélevé (ml) 182 850 995 1295 1482 1692 1902
L/S cumulé 7.28 34 39.8 51.8 59.3 67.9 76.1
Concentration phénanthrène (µg.L-1) 0.91 0.90 0.96 1.57 0.68 0.7 0.75
Concentration anthracène (µg.L-1) 0.32 0.40 0.40 0.66 0.35 0.3 0.16
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Masse de sol = 25 g
204
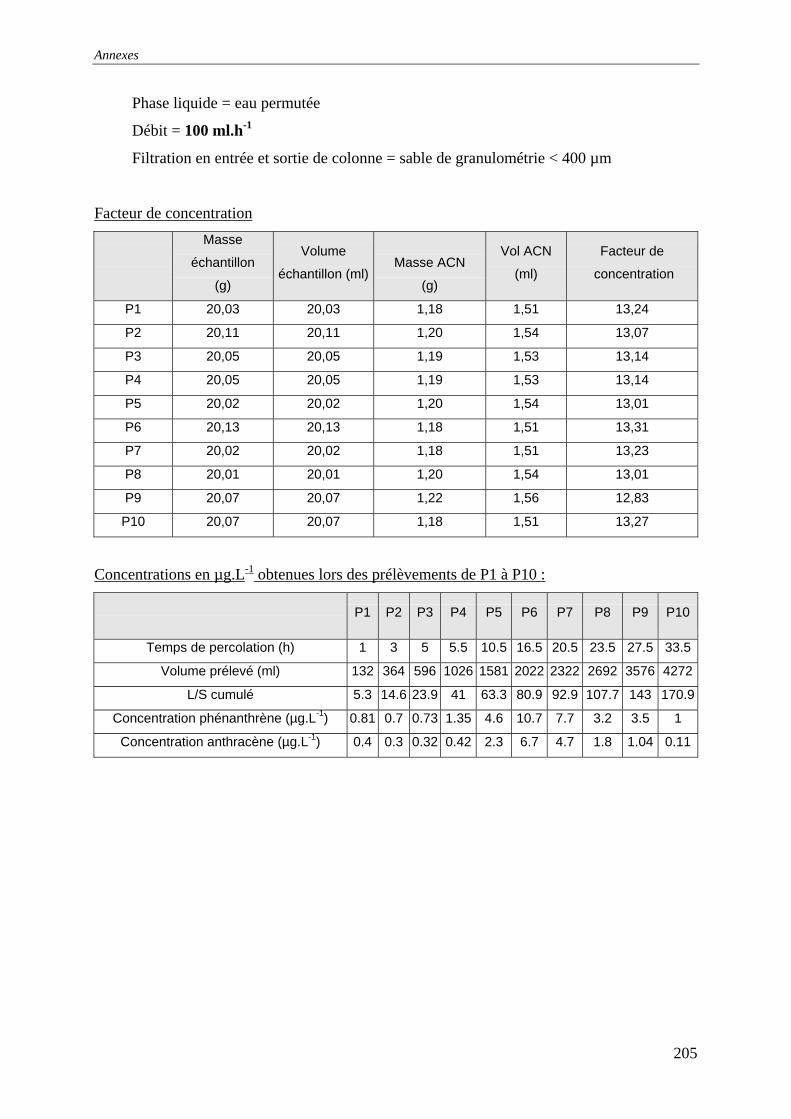
Annexes
Phase liquide = eau permutée
Débit = 100 ml.h-1
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Facteur de concentration
Masse
échantillon
(g)
Volume
échantillon (ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
P1 20,03 20,03 1,18 1,51 13,24
P2 20,11 20,11 1,20 1,54 13,07
P3 20,05 20,05 1,19 1,53 13,14
P4 20,05 20,05 1,19 1,53 13,14
P5 20,02 20,02 1,20 1,54 13,01
P6 20,13 20,13 1,18 1,51 13,31
P7 20,02 20,02 1,18 1,51 13,23
P8 20,01 20,01 1,20 1,54 13,01
P9 20,07 20,07 1,22 1,56 12,83
P10 20,07 20,07 1,18 1,51 13,27
Concentrations en µg.L-1 obtenues lors des prélèvements de P1 à P10 :
P1 P2 P3 P4 P5 P6 P7 P8 P9 P10
Temps de percolation (h) 1 3 5 5.5 10.5 16.5 20.5 23.5 27.5 33.5
Volume prélevé (ml) 132 364 596 1026 1581 2022 2322 2692 3576 4272
L/S cumulé 5.3 14.6 23.9 41 63.3 80.9 92.9 107.7 143 170.9
Concentration phénanthrène (µg.L-1) 0.81 0.7 0.73 1.35 4.6 10.7 7.7 3.2 3.5 1
Concentration anthracène (µg.L-1) 0.4 0.3 0.32 0.42 2.3 6.7 4.7 1.8 1.04 0.11
205
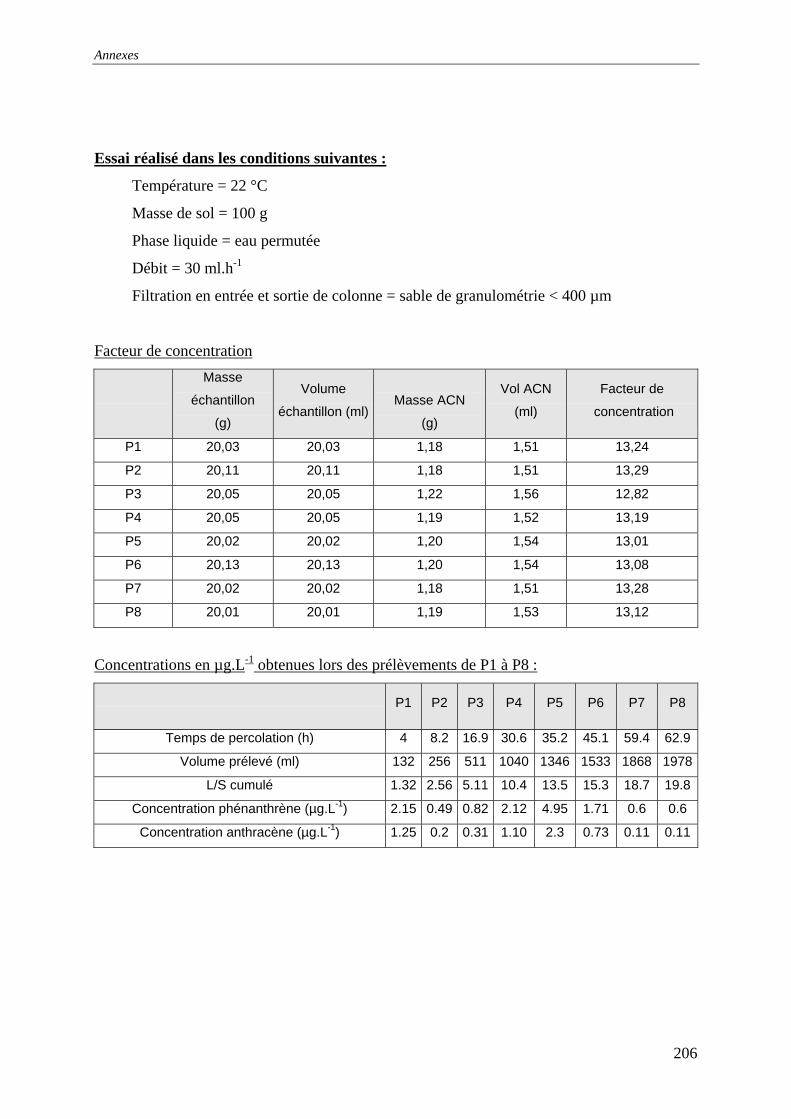
Annexes
Essai réalisé dans les conditions suivantes :
Température = 22 °C
Masse de sol = 100 g
Phase liquide = eau permutée
Débit = 30 ml.h-1
Filtration en entrée et sortie de colonne = sable de granulométrie < 400 µm
Facteur de concentration
Masse
échantillon
(g)
Volume
échantillon (ml)
Masse ACN
(g)
Vol ACN
(ml)
Facteur de
concentration
P1 20,03 20,03 1,18 1,51 13,24
P2 20,11 20,11 1,18 1,51 13,29
P3 20,05 20,05 1,22 1,56 12,82
P4 20,05 20,05 1,19 1,52 13,19
P5 20,02 20,02 1,20 1,54 13,01
P6 20,13 20,13 1,20 1,54 13,08
P7 20,02 20,02 1,18 1,51 13,28
P8 20,01 20,01 1,19 1,53 13,12
Concentrations en µg.L-1 obtenues lors des prélèvements de P1 à P8 :
P1 P2 P3 P4 P5 P6 P7 P8
Temps de percolation (h) 4 8.2 16.9 30.6 35.2 45.1 59.4 62.9
Volume prélevé (ml) 132 256 511 1040 1346 1533 1868 1978
L/S cumulé 1.32 2.56 5.11 10.4 13.5 15.3 18.7 19.8
Concentration phénanthrène (µg.L-1) 2.15 0.49 0.82 2.12 4.95 1.71 0.6 0.6
Concentration anthracène (µg.L-1) 1.25 0.2 0.31 1.10 2.3 0.73 0.11 0.11
206

Annexes
Contribution à l’élaboration d’une méthodologique relative à
l’étude de la mobilité des HAP dans les sols industriels pollués
Annexe 15
207

Annexes
Proposition méthodologique relative à l’étude de la mobilité des composés organiques
hydrophobes tels que les HAP dans les sols industriels pollués, à l’échelle du laboratoire
(Le déroulement des étapes de cette proposition est inspiré de la norme ENV 12-920 (CEN,
1997) relative à l’évaluation du comportement environnemental des matériaux contenant des
déchets).
Etape 1 : Préparation des échantillons et caractérisation du sol
1. Préparation des échantillons de sol à partir du sol brut prélevé sur le site.
2. Caractérisation du sol :
- Contenu total en HAP
- COT
- Granulométrie et humidité
Etape 2 : Détermination des mécanismes de relargage de la fraction solubilisée et mise en
évidence d’une fraction particulaire
1. A ce stade, une première idée de la fraction solubilisée mobilisée peut être déduite du
COT : les concentrations en HAP retrouvées en solution étant inversement liées au
COT du sol.
2. Une cinétique réalisée en batch percolant avec une phase liquide composée d’un
mélange eau-méthanol (50/50 en volume) permet d’estimer assez précisément la
valeur des concentrations à l’équilibre dans l’eau (voir méthode au V.3.2 du chapitre
V). Cette technique permet d’éviter les difficultés rencontrées lorsqu’on travaille avec
une phase aqueuse et des composés hydrophobes.
3. Calcul théorique des concentrations à l’équilibre pour les deux mécanismes :
désorption et dissolution (voir méthode V.3.I.2 du chapitre V). La comparaison avec la
valeur expérimentale trouvée au 2. permet de conclure sur le mécanisme majoritaire
contrôlant le relargage.
208

Annexes
4. Réalisation d’une série de 10 renouvellements séquentiels avec de l’eau permutée avec
suivi de la conductivité afin de mettre en évidence une éventuelle fraction particulaire.
Etape 3 : Etude paramétrique (influence de paramètres physico-chimiques)
La fraction particulaire et la fraction solubilisée doivent être étudiées séparément
1. Fraction solubilisée :
Réalisation d’une série de cinétiques en batch percolant en variant les paramètres séparément
(ratio L/S, Température, cosolvant, pH, débit, force ionique….). Ces paramètres doivent être
choisis en fonction de la situation du site (se reporté au travaux du groupe AFNOR X31-E
pour faciliter le choix des paramètres à évaluer). Ces résultats permettent d’interpréter
l’influence des paramètres sur les concentrations à l’équilibre ainsi que sur les vitesses de
relargage. Ces essais sont réalisés avec un mélange eau méthanol (50/50 en volume), ce qui
permet d’éviter les difficultés rencontrées lorsqu’on travaille avec une phase aqueuse et des
composés hydrophobes.
2. Fraction Particulaire
Réalisation de renouvellements séquentiels avec de l’eau permutée en faisant varier les
paramètres séparément (ratio L/S, Température, cosolvant, pH, débit, force ionique…).
Déterminer dans chaque cas les valeurs des potentiels de HAP particulaires relargables afin de
pouvoir établir des comparaisons entre les paramètres (voir méthode au V.4.2.3 du chapitre
V).
Etape 3 : Evolution de la mobilité à long terme
1. Fraction solubilisée :
Réalisation de renouvellements séquentiels avec des conditions d’extraction forte (exemple
50% de méthanol et température = 40°C). l’évolution des concentrations au cours des
renouvellements fournit des informations sur l’évolution de la mobilité à long terme.
2. Fraction Particulaire
Les potentiels de HAP particulaires déterminés lors de l’étape 3.2 informe sur l’évolution de
la mobilité de la fraction particulaire.
209

Annexes
210