Embed Size (px)
Citation preview

ELSEVIER
3 October 1997
Chemical Physics Letters 277 (1997) 125-131
GHEMIGAL PHY$1G$ LETTERS
External electric field effect on interlayer vectorial electron transfer from photoexcited oxacarbocyanine to viologen in
Langmuir-Blodgett films
Takashi Ito, Iwao Yamazaki, Nobuhiro Ohta *
Department of Molecular Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo 060, Japan
Received 30 June 1997; in final form 4 August 1997
Abstract
Photoinduced interlayer electron transfer from the excited state of oxacarbocyanine (OCC) to viologen (VIO) is depressed or enhanced by an external electric field, depending on the direction of applied electric fields, in a donor-acceptor system composed of mixed monolayers of OCC and mixed monolayers of VIO separated by a spacer layer of fatty acid. As a result, field-induced increase and decrease of the quantum yield of OCC fluorescence are observed in the molecular assemblies of LB films. © 1997 Elsevier Science B.V.
1. Introduction
The energy of radical-ion pairs produced by elec- tron transfer may be to a great extent influenced by an external electric field because of their large dipole moment. Applied electric fields may also affect the electronic coupling between the initial and final states of the electron transfer, which depends on the over- lap of the wavefunctions of the donor and acceptor, since the applied fields may polarize the orbitals and change the overlap. These electric field effects seem to induce a change in electron wansfer rate. In fact, electric fields seem to play a significant physio- logical role in regulating various aspects of photo- synthesis [1].
When electron donor and acceptor molecules are
• Corresponding author.
distributed in a homogeneous system, there exists a distribution of the donor-acceptor distance with a random orientation in three dimensional space. In such a system, the analysis of the field effect is quite complicated, even if the field effect exists, since different pairs of donor and acceptor will show different magnitudes of field effect from each other. If the electron donor and acceptor can be arranged with a well defined molecular order, where photoin- duced electron transfer occurs with a definite direc- tion, the elucidation of not only the mechanism of electron transfer but also the field effect on the process seems to be developed extremely well since the molecular orientation and arrangement with re- spect to the direction of the applied electric field can be well defined. The Langmuir-Blodgett technique is one of the excellent methods to prepare such a molecular assembly [2-4].
0009-2614/97/$17.00 © 1997 Elsevier Science B.V. All rights reserved. PII S0009-2614(97)00926-3

126 T. /to et aL / Chemical Physics Letters 277 (1997) 125-131
In this letter, external electric field effects on the interlayer vectorial electron transfer between pho- toexcited oxacarbocyanine and viologen separated by a fatty acid monolayer are reported, which has been examined by comparing the field-induced change in fluorescence spectrum both in the absence and in the presence of viologen. By combining the electrofluo- rescence spectra with the fluorescence decay, field effects on the rate of electron transfer from photoex- cited state of oxacarbocyanine chromophore to violo- gen have been examined.
2. Experimental
N,N'-Dioctadecyloxacarbocyanine (Nippon Kanko Shikiso), hereafter denoted by OCC, was used with- out further purification. N,N'-Dioctadecyl-4,4'-bi- pyridinium dibromide, hereafter denoted by VIO, was synthesized according to Ref. [5]. Commercially available arachidic acid (AA) and methyl arachidate (MA) were purified by recrystallization from ace- tone. All the samples of the LB films were deposited as a cadmium salt.
A mixture of AA and MA whose ratio is I:1, denoted by AA/MA, was used as a matrix. In the present study, a mixing fraction of OCC to A A / M A was 0.5 mol%. Two kinds of stacked multilayer films were prepared: one includes a VIO monolayer (sample (I)), and the other does not include a VIO monolayer (sample (II)). Sample (II) was prepared as follows: At first, seven layers of A A were deposited on the quartz substrate coated by a semi-transparent aluminum film. Then, six mixed monolayers com- posed of OCC and A A / M A were deposited with a spacer composed of four layers of AA between the adjacent mixed layers. Finally, 10 layers of AA were postcoated. Only the monolayers of AA following the OCC mixed layers were deposited as X type, and other films were deposited as Y type. In sample (I), all the second monolayer films of AA deposited following the OCC layer and the first AA monolayer were replaced by a mixed monolayer composed of VIO (10 mol%) and AA/MA. A schematic illustra- tion of OCC, AA and VIO films in sample (I) is shown in Fig. 1. Note that LB films deposited on one side of the substrate were wiped off and that only the LB films deposited on the other side, whose total
AI--
~ , ~ ~ VIO
spacel (AA)
F
OCC
I:.~:":::~ ~.; :~:~::~:: ~:, " .~'...%~;~:~ ~; :~;,::~:~;~,;.,:~,:i : :~ :~ . . ,~ I -AI - - Fig. 1. Schematic illustration of LB multilayer films of OCC and VIO separated with a spacer layer of AA.
number of the monolayers were 43, were used for the optical measurements. All the monolayer films were deposite~, at 25 mN/m. The thickness of each layer is 27.3 A [6], yielding a total thickness of the LB film of 117.4 nm.
Following the deposition of the above-mentioned multilayer films on the aluminum coated plate, a semi-transparent aluminum film was again coated with evaporation. These aluminum films were used as electrodes. Electric fields were applied up to 1.0 MV cm- i.
All the optical measurements were carded out at room temperature under vacuum conditions. The amount of the field-induced change in fluorescence intensity and in absorption intensity was measured with a spectrometer equipped with an electric field modulation apparatus for the first harmonic (lf) and for the second harmonic (20 of the modulation frequency (typically 20 Hz). The procedures have been described elsewhere [7,8].
Fluorescence decays were measured by using a femtosecond pulsed laser combined with a single photon counting system equipped with a microchan- nel plate photomultiplier [8]. The laser system was a
T. lto et aL/ Chemical Physics Letters 277 (1997) 125-131 127
mode-locked titanium:sapphire laser pumped by an argon ion laser combined with a pulse picker. The second harmonic of the laser light was used for excitation.
3. Results and discussion
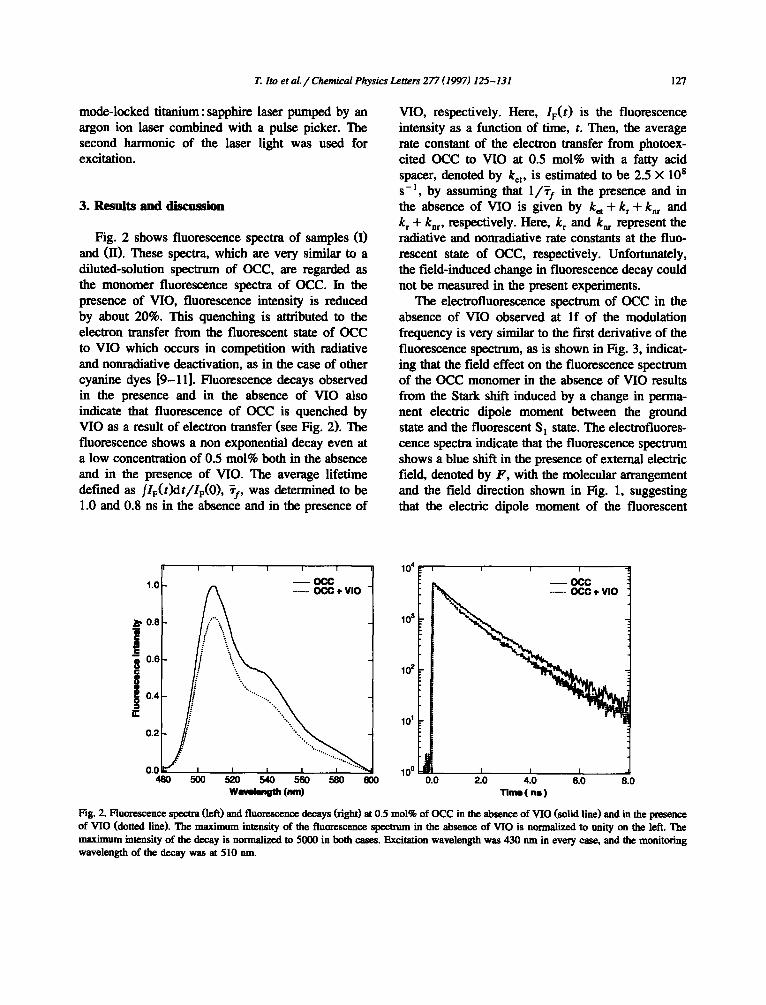
Fig. 2 shows fluorescence spectra of samples (I) and (II). These spectra, which are very similar to a diluted-solution spectrum of OCC, are regarded as the monomer fluorescence spectra of OCC. In the presence of VIO, fluorescence intensity is reduced by about 20%. This quenching is attributed to the electron transfer from the fluorescent state of OCC to VIO which occurs in competition with radiative and nonradiative deactivation, as in the case of other cyanine dyes [9-11]. Fluorescence decays observed in the presence and in the absence of VIO also indicate that fluorescence of OCC is quenched by VIO as a result of electron transfer (see Fig. 2). The fluorescence shows a non exponential decay even at a low concentration of 0.5 mol% both in the absence and in the presence of VIO. The average lifetime defined as fle(t)dt/IF(O), ~f, was determined to be 1.0 and 0.8 ns in the absence and in the presence of
VIO, respectively. Here, IF(t) is the fluorescence intensity as a function of time, t. Then, the average rate constant of the electron transfer from photoex- cited OCC to VIO at 0.5 mol% with a fatty acid spacer, denoted by ket, is estimated to be 2.5 × l0 s s - l , by assuming that 1 /~ I in the presence and in the absence of VIO is given by k= + k r - t -knr and k r -t- knr , respectively. Here, k r and knr represent the radiative and nonradiative rate constants at the fluo- rescent state of OCC, respectively. Unfortunately, the field-induced change in fluorescence decay could not be measured in the present experiments.
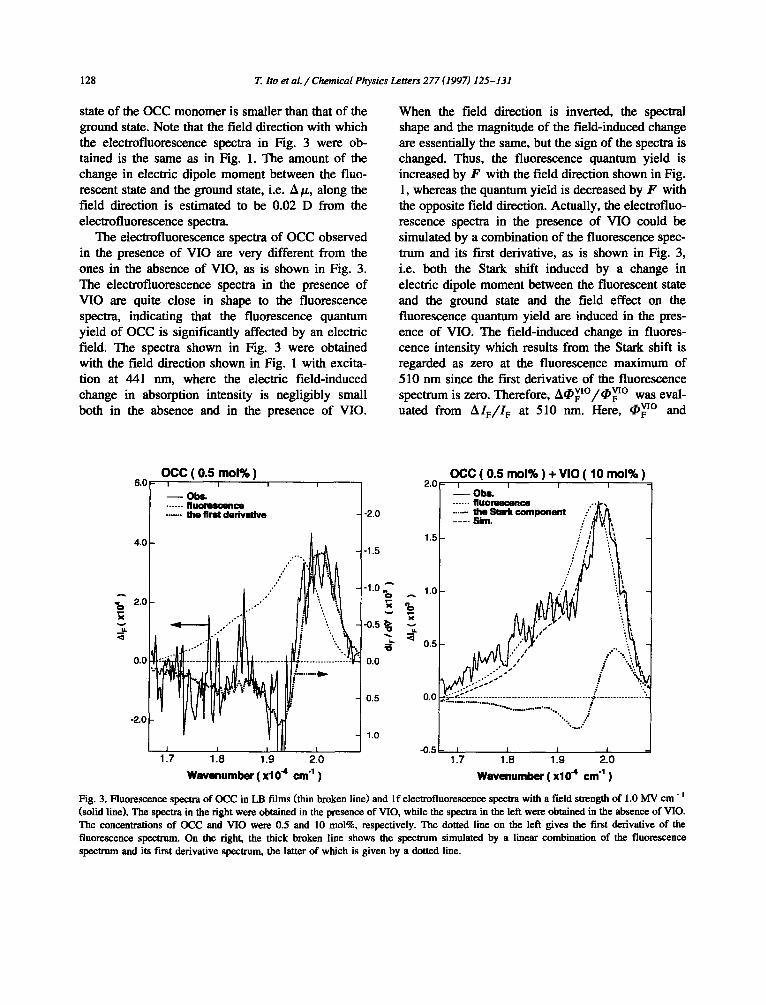
The electrofluorescence spectrum of OCC in the absence of VIO observed at I f of the modulation frequency is very similar to the first derivative of the fluorescence spectrum, as is shown in Fig. 3, indicat- ing that the fe ld effect on the fluorescence spectrum of the OCC monomer in the absence of VIO results from the Stark shift induced by a change in perma- nent electric dipole moment between the ground state and the fluorescent S~ state. The electrofluores- cence spectra indicate that the fluorescence spectrum shows a blue shift in the presence of external electric field, denoted by F , with the molecular arrangement and the field direction shown in Fig. 1, suggesting that the electric dipole moment of the fluorescent
1.0
i 0.8
i 0.6
0 0.4
,"r
0.2
0.0 48O
I I I I I
~OCC ........ OCC + VIO
500 520 540 560 580 600 ww~e~g~ (rim)
10 4
103
102
101 -
-
100 LJ I I ) 0.0 2.0 4.0 6.0 8.0
Time(ns)
Fig. 2. Fluorescence spectra (left) and fluorescence decays (right) at 0.5 mol% of OCC in the absence of VIO (solid line) and in the presence of VIO (dotted line). The maximum intensity of the fluorescence spectrum in the absence of VIO is normalized to unity on the left. The maximum intensity of the decay is normalized to 5000 in both cases. Excitation wavelength was 430 nm in every ease, and the monitoring wavelength of the decay was at 510 nm.
128 T. Ito et al. / Chemical Physics Letters 277 (1997) 125-131
state of the OCC monomer is smaller than that of the ground state. Note that the field direction with which the electrofluorescence spectra in Fig. 3 were ob- tained is the same as in Fig. 1. The amount o f the change in electric dipole moment between the fluo- rescent state and the ground state, i.e. A/z, along the field direction is est imated to be 0.02 D from the electrofluorescence spectra.
The electrofluorescence spectra of OCC observed in the presence of VIO are very different from the ones in the absence of VIO, as is shown in Fig. 3. The electrofluorescence spectra in the presence of VIO are quite close in shape to the fluorescence spectra, indicating that the fluorescence quantum yield of OCC is significantly affected by an electric field. The spectra shown in Fig. 3 were obtained with the field direction shown in Fig. 1 with excita- tion at 441 nm, where the electric f ield-induced change in absorption intensity is negligibly small both in the absence and in the presence of VIO.
When the field direction is inverted, the spectral shape and the magnitude of the f ield-induced change are essentially the same, but the sign of the spectra is changed. Thus, the fluorescence quantum yield is increased by F with the field direction shown in Fig. 1, whereas the quantum yield is decreased by F with the opposite field direction. Actually, the electrofluo- rescence spectra in the presence of VIO could be simulated by a combinat ion of the fluorescence spec- trum and its first derivative, as is shown in Fig. 3, i.e. both the Stark shift induced by a change in electric dipole moment between the fluorescent state and the ground state and the field effect on the fluorescence quantum yield are induced in the pres- ence of VIO. The f ield-induced change in fluores- cence intensity which results from the Stark shift is regarded as zero at the fluorescence maximum of 510 nm since the first derivative of the fluorescence spectrum is zero. Therefore, v[o vio ACF /¢F w a s e v a l - u a t e d from A I F / I F at 510 r im. Here, ¢VlO and
6.0
4.0
A ~:j 2.0
u,.
0.0
-2.0
O C C ( 0 .5 m o l % ) I I I
- - Obo. ...... fluoremcence
i 2.0
....... the first derivative -2.0
1.5 -1.5
. . - . ,
-1.0 ~ 1.0
.~ 0.5
....... 0.0
0.5 0.0
1.0 I I I - 0 • 5
.7 1.8 1.9 2.0
W n v e n u m b e r ( x lO 4 c m "1 )
O C C ( 0 .5 m o l % ) + V l O ( 10 m o l % i I I I
- - O ~ ...... f l U O ~ C O ..•j-~ ....... the Stnrk component / . ,~. /~ . . . . Sim• / ,~ ~.. ~l
: i i / i i / ,
: e
.' ,." #
• I
:::::::::::::::::::::::::::::::::::::::::::::::::::::::::: ............................ %.,,"
i I I I
1.7 1.8 1.9 2.0
W a v e n u m b e r ( x10 4 cm 4 )
Fig. 3. Fluorescence spectra of OCC in LB films (thin broken line) and If electrofiuorescence spectra with a field strength of 1.0 MV cm- =
(solid line)• The spectra in the right were obtained in the presence of VIO, while the spectra in the left were obtained in the absence of VIO. The concentrations of OCC and VIO were 0.5 and 10 tool%, respectively. The dotted line on the left gives the first derivative of the fluorescence spectrum. On the right, the thick broken line shows the spectrum simulated by a linear combination of the fluorescence spectrum and its first derivative spectrum, the latter of which is given by a dotted line•

T. Ito et al. / Chemical Physics Letters 277 (1997) 125-131 129
2.0
~ 0 0 ........................................................ _ . L ~ ........... , ...........................
o ° ° i
-1.0
-2.0 -~ 0 -~s 0o 015 10
F ( u v c m "~)
Fig. 4. Plots of A I F/1F of the OCC fluorescence at 510 nm in the presence of VIO as a function of the field strength. AI F was observed at. If, and the field direction shown in Fig. 1 was taken as the positive one.
AO v[° represent the fluorescence quantum yield of OCC and its field-induced change in the presence of VIO, respectively, while I F and A 1 F give the fluo- rescence intensity and its field-induced change, re- spectively. Fig. 4 shows the plots of A I F / 1 F at 510 nm observed at I f as a function of the strength of external electric field. Note that the field direction given in Fig. 1 is taken as the positive one. The present results at I f show that A ¢ ~ F V I O / ~ F w O is nearly given by 1.7 X 10 -9 17 with the units of V cm-~ for F. When the signal intensity was observed at 2f of the modulation frequency, the electrofluores- cence spectra were essentially the same in shape as the fluorescence spectrum both in the absence and in the presence of VIO and show a negative sign irrespective of the field direction, indicating that the quadratic term corresponds to the decrease of the fluorescence quantum yield. The magnitude of A I J I F at 2f was estimated to be 2 × 10 -4 with a field strength of 1 MV c m - ~ both in the absence and in the presence of VIO. Thus, the field-induced change in fluorescence intensity at 2f is very small, and so the quadratic term is ignored in the following discussion.
The field effect of AO vx° can be interpreted in terms of the field effect on the rate of electron transfer which occurs from the photoexcited OCC to VIO. With a simple kinetic scheme, • vI° and O vI°
+ A ~ vl° can be expressed by k r / ( k r + knr + ket) and k r / ( k r + knr + ket + Aket) , respectively, where A ket represents the field-induced change in electron t rans fe r rate. Then , A ket is g iven by
VIO --~ VlO = r i o - ( a ~ I ° / ~ ° ) / ( 1 + ( a ~ F / Y ' F ) } / ~ f , where ~.vio corresponds to the average lifetime ob- served in the presence of VIO [8]. By employing the average fluorescence lifetime of 0.8 ns as ~vlo, Aket is determined to be - 2 . 1 F in units of s -1 with units of V c m - ] for F . The negative value indicates that the electron transfer becomes slower as the applied electric field increases with the same direc- tion as in Fig. 1. Note that the electron transfer is enhanced in the presence of electric field with the opposite direction and the sign of the above-men- tioned A ket changes.
Electron transfer rate depends on various parame- ters: the free energy change (AG) between the initial and final states; the magnitude of the electronic coupling between the donor and acceptor, which depends on the mutual distance and orientation; the overall reorganization energy involving both the ex- ternal and internal reorganization [12,13]. External electric field with the direction shown in Fig. 1 will make unstable the radical-ion pairs produced by electron transfer from the excited state of OCC to VIO, whereas the ion-pair states are stabilized by electric fields whose direction is opposite to the one shown in Fig. 1. As a result, a change in DG as large as 0.273 eV may be induced by an electric field with a strength of 1 MV cm-1 with a spacer of AA.
According to the Marcus theory [13,14], the rate constant of electron transfer is given by
ket - - - -Kexp[- ( AG-I- Xo)~(4ksTX0)- I]. (1)
Here, k n and T are the Boltzrnann constant and temperature, and A 0 is the so-called reorganization energy given by the following equation: A 0 = ( e 2 / 2 X ~ p I - ~- IXR~I + R ~ l - 2 R - ] ) , where e is the electronic charge, Cop and es are the optical and static dielectric constants of the matrix, respectively, R a and R a are the radii of the donor and acceptor, respectively, and R is the donor-acceptor distance. On photoinduced vectorial electron transfer through fatty acid rnonolayer in molecular architecture of LB assemblies, the donor-acceptor distance dependence of the fluorescence quenching suggests that electron

130 T. Ito et aL / Chemical Physics Letters 277 (1997) 125-131
tunneling through the potential barrier of a fatty acid occurs from the initial state [ 9 - 1 1 ] . In such a case, K
in Eq. (1) may be expressed as
K = K o exp( - 2kd0),
k 2 = 2radp/h 2. (2)
Here, m is the electron mass, h is the Planck's constant divided by 21r, $ is a height of barrier represented by the fatty acid spacer, d o is the donor-acceptor distance [ 13,15].
In the presence of external field, AG may be replaced by AG o - - / zF , where AGo is the free en- ergy change at zero field and /~ is the electric dipole moment of the produced radical-ion pair. External electric field may also play a role to change the barrier height of tunneling. When external electric field is applied with a direction in Fig. 1, the barrier height may become higher, while the barrier may become lower in the opposite direction. Then, may be replaced by O0 + $ ' F in the presence of F , where O0 is the height at zero field and $ ' is the first derivative of the potential barrier with respect to the field strength. Then, Aket(F), which is defined as k~(F -- 0) - ket(F), divided by ket(F = 0) is ap- proximately given by
ako,( F ) lko,( F = O)
= [2tudor $_ l (2m$o) -1/2((~'F)
- 2 ( I~F) (AG o + X o ) / ( 4 k s T X o ) - ' ] • (3)
At firsL the field dependence of the potential barrier of electron tunneling is considered. For the simplifi- cation, AG o + A 0 is assumed to be zero, and only the first term of Eq. (1) is considered. In the present donor-acceptor system, ket and A ket were deter- mined to be 2.5 × 10 8 s -1 and -2 .1 F s - l , respe c- tively; Aket/ket is - 8 . 4 × 10 -9 F with units of V era-~ for F. It is worth mentioning that the nega- tive sign shows the de-enhancement of the electron transfer rate with the field direction in Fig. 1. When 0.7 eV is employed as ~b 0 [15], the above value of Aket/ket can be reproduced with (k' of 3.2 × 10 -9 e V / V era- 1.
The bell-shaped dependence of the electron trans- fer rate constant on AG covering both the inverted and normal regions, which is predicted by F-xl. (I),
has been confirmed in charge-shift and also charge- recombination reactions [16-20]. Similar AG depen- dence may be able to be considered in the present charge separation reaction. In the normal region, the electron transfer rate becomes slower with the same field direction as in Fig. 1, while the rate becomes faster with the opposite direction. Then, it looks that the present results of the field dependence of the electron transfer rate can also be interpreted in terms of the field dependence of AG in the normal region; A o > - A G . Then, the data was tried to analyze with the assumption that the field effect comes from the field dependence of AG and that O' = 0. By combin- ing the second term of Eq. (1) with the above value of Aket/ket, AG o + A 0 is given by 1.6 × 10 -3 A o in units of eV at 295 K (4knT= 0.102 eV) with the
o
donor-acceptor distance of 27.3 A. Cop of the LB films, which is given by 1.05 n 2 with the refractive index n, may be assumed to be 2.36, while E~ may be regarded as the same as the value of cadmium salt of arachidic acid, i.e., 2.52 [21]. R is regarded as the same as the thickness of theoSPacer, i.e., 27.3 A. By assuming that R d = R a = 3 A, A o is calculated to be 0.12 eV. Then, AG o is regarded as a little larger than - 1.2 eV, and the difference between A 0 and AGo is as small as 2 × 10 -4 eV. In this case, the relation between A o and - AG is considered to change when the field is varied from - 1 MV cm - l to 1 MV cm - l ; both the inverted region and the normal re- gion are expected to appear in the presence of elec- tric field. In the present experiments, however, such a field effect was not observed (see Fig. 4). Even when much larger values of A 0 are employed as the total reorganization energy, the difference between A 0 and the derived value of - A G o is too small to interpret the lack of the field-induced inverted re- gion. Then, it may be said for the present that the observed field effect on vectorial electron transfer from photoexcited OCC to VIO in LB films comes from the field effect on potential barrier of electron tunneling rather than the field dependence of AG. However, the present results remind us of the Rehm-WeUer plots for electron transfer fluorescence quenching [22], where the inverted region was not observed in the charge separation reaction in spite of - A G o > A 0. In order to elucidate the mechanism of the present field effect in more detail, further studies of the donor-acceptor distance dependence as well

7". lto et al. /Chemical Physics Letters 277 (1997) 125-131 131
as the temperature dependence of the field effects are now in progress.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
References
[1] S.G. Boxer, in: J. Deisenhofer, J.R. Norris (Eds.), The Photosynthetic Reaction Center, Vol. H, Academic Press, San Diego, 1993, p. 179.
[2] A. Ulman, An Introduction to Ultrathin Organic Films from Langmuir-Blodgett to Self-Assembly, Academic Press, San Diego, 1991.
[3] Z.D. Popovic, G.J. Kovacs, P.S. VincetL G. Alegria, P.L. Dutton, Biochim. Biophys. Acta 851 (1986) 38.
[4] Y. Hsu, T.L. Penner, D.G. Whitten, J. Phys. Chem. 96 (1992) 2790.
[5] X.R. Xiao, C.B. Wang, T. Ti Tien, J. Mol. Catal. 23 (1984) 9.
[6] P. Fromherz, G. Reinbold, Thin Solid Films 160 (1988) 347.
[7] N. Ohm, S. Okazaki, I. Yamazaki, Chem. Phys. Lett. 229 (1994) 394.
[8] N. Ohm, T. Nomura, L Yamazaki, J. Photochem. Photobiol., to be published.
[9] H. Kulm, J. Photocbem. 10 (1979) 111. [10] D. M~bius, Ace. Chem. Res. 14 (1981) 63. [11] H. Kuhn, in: Proc. Robert A. Welch Found. Conf. Chem.
Res. XXX, Advances in Electrochemistry, Houston, TX, 1986, p. 338.
[12] N. Mataga, Pure Appl. Chem. 56 (1984) 1255. [13] R.A. Marcus, N. Sutin, Biochim. Biophys. Acta 811 (1985)
265. [14] R.A. Marcus, J. Chem. Phys. 24 (1956) 966. [15] H. Kulm and D. M~bius, Investigations of surfaces and
interfaces, Part B, in: B.W. Rossiter, R.C. Baetzold (Eds.), Physical Methods of Chemistry Series, Vol. IXB, Wiley, New York, 1993, Chap. 6, p. 375.
[16] J.V. Beitz, J.R. Miller, J. Chem. Phys. 71 (1979) 4579. [17] J.R. Miller, J.V. Beitz, R.K. Huddleston, J. Am. Chem. Soc.
106 (1984) 5057. [18] J.R. Miller, L.T. Calcaterra, G.L. Closs, J. Am. Chem. Soc.
I06 (1984) 3047. [19] M.R. Wasielewski, M.P. Niernczyk, W.A. Svec, E.B. Pewitt,
J. Am. Chem. Soc. 107 (1985) 1080. [20] N. Mataga, T. Asahi, Y. Kanda, T. Okada, T. Kakitani,
Chem. Phys. 127 (1988) 249. [21] B. Mann, H. Kulm, J. Appl. Phys. 42 0971) 4398. [22] D. Rehm, A. Weller, Isr. J. Chem. 8 (1970) 259.





























