Embed Size (px)
Citation preview

1
F-Praktikum Physikalische Chemie
V2
Fluoreszenzspektroskopie
Eigenschaften des
angeregten Zustands von 2-Naphtol
-- Überarbeitete Versuchsanleitung, Dr. Ludwig Kibler 21.05.15

2
Theoretische Grundlagen
- Absorption von elektromagnetischer Strahlung
- harmonischer Oszillator, Energie-Eigenwerte, Morsepotential
- elektronische, Vibrations- und Rotationsanregungen in Molekülen
- quantenmechanische Betrachtung von Anregungen in Atomen und Molekülen
- spontane und induzierte Emission
- Fluoreszenz, Phosphoreszenz
- Franck-Condon-Prinzip
- dipolerlaubte Übergänge (Auswahlregeln)
- metastabile Zustände, Singulett-, Triplettzustände
- Grundlagen der Reaktionskinetik
- Quasistationaritätsprinzip
Technische Hinweise (für Praktikumsteilnehmer und Versuchsbetreuer)
Die Napthol-Lösung (10-3
M) sollte im Dunkeln gelagert werden.
Lösemittel Ethanol (>99%)
Der Stenhagen-Puffer setzt sich aus 10 mM Phosphorsäure, 15 mM Borsäure, 7 mM
Zitronensäure, und 69 mM NaOH zusammen.
Bei diesem Versuch ist besonders darauf zu achten, dass sehr sauber gearbeitet wird!
Die Küvetten sollten natürlich transparent sein (keine Fingerabdrücke!), damit ein
Spektrum aufgenommen werden kann.
Die pH-Elektrode wird in 3M KCl-Lösung gelagert. Sie sollte regelmäßig kalibriert
werden (z.B. mit Pufferlösungen bei pH 7 und pH 9).
Damit die Spektren nicht verrauscht sind, sollte als scan mode „slow“ oder „medium“
gewählt werden.
Die Daten werden als ASCII gespeichert! (*.cvs). Am besten sollten diese Daten
direkt am Messrechner in ein *.xls-File umgewandelt werden, damit es anschließend
keine Probleme bei der Auswertung gibt.

3
1. Theoretische Grundlagen
1.1 Kurze Darstellung (Überblick) zu den Grundlagen der Fluoreszenz
Anmerkung:
Die hier gegebene Darstellung der Grundlagen soll nur einen Überblick über die
Anforderungen geben, die notwendig zum Verständnis der Fluoreszenz und zur Durchführung
des Versuches sind. Die Ausführungen sind keineswegs vollständig und detailliert. Hier wird
entsprechende Literatur empfohlen.
Unter Fluoreszenz steht man die spontane Emission von Licht, die nur solange andauert,
wie die fluoreszierenden Moleküle durch Lichtabsorption angeregt werden. Das
Fluoreszenzspektrum ist dabei dem Absorptionsspektrum gegenüber energetisch
verschoben zu kleineren Wellenzahlen. (Definition lt. Wedler)
Die Grundlage der Lichtabsorption von Molekülen (und auch Festkörpern) wird durch das
Lambert-Beersche Gesetz beschrieben. Tritt ein Lichtstrahl mit der Intensität I durch ein
Medium durch, so wird die Abnahme der Intensität dI beschrieben durch:
dxIkdI
Die Größe x bezeichnet dabei die Dicke des durchstrahlten Mediums, k ist die Absorptions-
konstante (quasi der Imaginärteil des komplexen Brechungsindex). Durch Integration erhält
man dann die Transmission T:
0
kxIT e
I
Dabei ist I0 die eingestrahlte Intensität des Lichtes und I die Intensität nach Durchstrahlen des
Mediums.
Bei Molekülen ist die Absorption von Licht verbunden mit einer energetischen Anregung des
Teilchens aus dem Zustand Em in den Zustand En. Bei dem Übergang spielen die energetische
Lage der beiden Zustände, die Temperatur T und die Besetzungsdichten Ni eine wichtige
Rolle. Die Population (Besetzungsdichte) eines bestimmten Zustandes mit der Energie Ei
hängt in der statistischen Thermodynamik von der Verteilungsfunktion nach Boltzmann ab:
TkE
iBieNN
/ (Boltzmann-Konstante kB)
Für eine effektive Absorption sollte die Besetzung im Anfangszustand hoch und im
Endzustand gering sein. Gleichzeitig spielt der energetische Abstand eine entscheidende
Rolle. Für resonante Absorption sollte die Differenz der Energien der beteiligten Zustände
gerade der Energie der eingestrahlten Photonen entsprechen.
n m PhotonE E E E h
Verschiedene Prozesse führen darüber hinaus zu energetischen Verbreiterungen der
beteiligten Niveaus (z.B. die natürliche Lebensdauer eines Zustands, Dopplerverbreiterung,
etc.).

4
In der quantenmechanischen Beschreibung wird dieser Sachverhalt genauer beleuchtet. Die
Anfangs- und Endzustände werden dabei durch die stationären Wellenfunktionen m und n
beschrieben. Der Prozess der Absorption wird mithilfe der zeitabhängigen Schrödinger-
Gleichung behandelt (siehe z.B. Wedler, Kap. 3.4), da die einfallende elektromagnetische
Strahlung durch einen zeitabhängigen Störoperator H dargestellt werden kann. Auf eine
detaillierte Darstellung der zeitabhängigen Störungsrechnung wird an dieser Stelle verzichtet,
sie ist in geeigneten Lehrbüchern zu finden.
Schlussendlich wird die Übergangswahrscheinlichkeit W für die Anregung durch das
Übergangsmoment Rnm beschrieben,
*
nm n mˆR ( x ) ( x ) dx
wobei das Dipolmoment
einen Teil des Störoperators darstellt ( ˆ EH , E
:
elektromagnetisches Feld der Strahlung). Die Wahrscheinlichkeit W, das System im Zustand
n zu finden, ist letztendlich proportional zum Quadrat der Amplitude des elektromagnetischen
Feldes und des Übergangsmomentes sowie linear in der Zeit t (vgl. auch Fermi´s Goldene
Regel). In der Physik wird oft eine geringfügig andere Beschreibung über das
Übergangsmatrixelement T verwendet: 2||ˆ|| mn HT
Diese Beschreibung ist natürlich äquivalent zur hier benutzten Darstellung.
1.2 Schwingungsspektroskopie:
Die Schwingungsspektroskopie lässt sich gut über den harmonischen Oszillator in der
Quantenmechanik beschreiben. Hierzu werden die Energieniveaus als Funktion der
Schwingungszahl v dargestellt. Der harmonische Oszillator zeichnet sich dabei durch
äquidistante Abstände der einzelnen Energieniveaus Ev aus:
0
1
2vE h v
Auch hier soll noch einmal darauf hingewiesen werden, dass eine Nullpunktsenergie existiert.
In der Abbildung 1 ist der harmonische Oszillator im linken Teilbild mit den Energieniveaus
dargestellt, in der Mitte sind die zugehörigen Wellenfunktionen und rechts die
Aufenthaltswahrscheinlichkeiten gezeigt. Die Wellenfunktionen ergeben sich aus der Lösung
der Schrödinger-Gleichung für den harmonischen Oszillator
02
12 2
22
2
DxE
dx
d
mit den zugehörigen Normierungsbedingungen Nv. Die Wellenfunktionen v lassen sich
durch Hermitesche Polynome Hv(x) darstellen, die allgemeine Form lautet:
2
2
x
v v vN H ( x ) e
mit 2/1)(1
D

5
Beispiele für diese Funktionen sind in der Mitte der Abb. 1 gezeigt, rechts daneben dann die
Wahrscheinlichkeitsdichten, die man durch Quadrieren der Wellenfunktionen erhält.
Abb.1: Energieniveaus des harmonischen Oszillators (links) und zugehörige Wellenfunktionen (Mitte) und
Wahrscheinlichkeitsdichten (rechts)
Schwingungsspektrum:
Aus dem harmonischen Oszillator ergeben sich Schwingungsterme G(v):
0
1
2
E( v )G( v ) v
hc
wobei die Auswahlregel 1v für die Übergänge gilt. Da alle Niveaus den gleichen
Abstand haben, sollte man nur eine Absorptionslinie erwarten, die Wellenzahl~ des
absorbierten Lichtes hängt dann mit der Schwingungsfrequenz 0~
der Schwingungsmode
zusammen. Daraus kann man dann die Kraftkonstante einer chemischen Bindung berechnen.
Ein genaueres Modell muss einige Forderungen erfüllen:
(i) bei hoher Schwingungsanregungen dürfen sich die Atome nicht zu nahe kommen,
d.h., der linke Teil der Potentialkurve muss steiler ansteigen
(ii) der rechte Teil der Kurve muss in eine Horizontale übergehen, die
Energiedifferenz zwischen dem Minimum der Kurve und der Horizontalen muss
der Dissoziationsenergie entsprechen
Das empirisch gefundene Morsepotential V(r) in Abbildung 2 entspricht diesen Forderungen
näherungsweise, die Potentialkurve lässt sich folgendermaßen formulieren:
2
1 g( r r )
eV( r ) D e
mit 2 1 2
0 2 /
e( c / D h )
Die Konstante ist hier anders definiert als bei den Wellenfunktionen für den harmonischen
Oszillator. Die in Abb. 2 eingezeichneten Dissoziationsenergie D0 und De unterscheiden sich
gerade durch die Nullpunktsenergie.
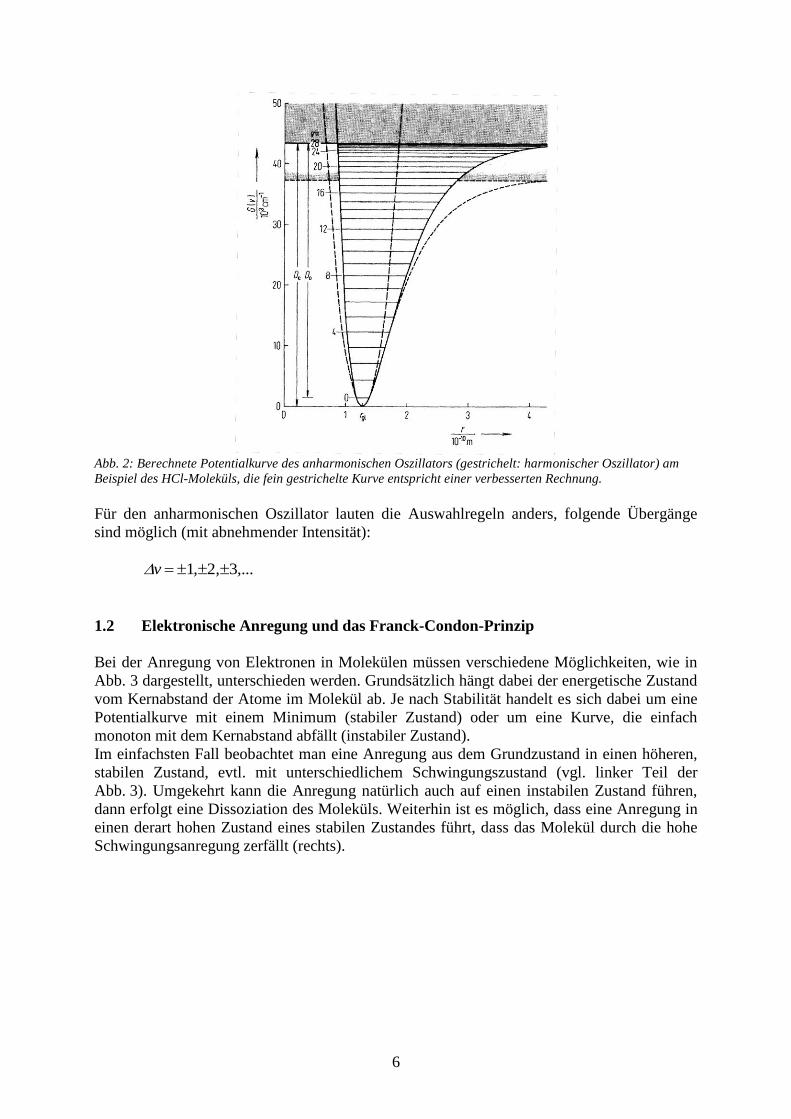
6
Abb. 2: Berechnete Potentialkurve des anharmonischen Oszillators (gestrichelt: harmonischer Oszillator) am
Beispiel des HCl-Moleküls, die fein gestrichelte Kurve entspricht einer verbesserten Rechnung.
Für den anharmonischen Oszillator lauten die Auswahlregeln anders, folgende Übergänge
sind möglich (mit abnehmender Intensität):
1 2 3v , , ,...
1.2 Elektronische Anregung und das Franck-Condon-Prinzip
Bei der Anregung von Elektronen in Molekülen müssen verschiedene Möglichkeiten, wie in
Abb. 3 dargestellt, unterschieden werden. Grundsätzlich hängt dabei der energetische Zustand
vom Kernabstand der Atome im Molekül ab. Je nach Stabilität handelt es sich dabei um eine
Potentialkurve mit einem Minimum (stabiler Zustand) oder um eine Kurve, die einfach
monoton mit dem Kernabstand abfällt (instabiler Zustand).
Im einfachsten Fall beobachtet man eine Anregung aus dem Grundzustand in einen höheren,
stabilen Zustand, evtl. mit unterschiedlichem Schwingungszustand (vgl. linker Teil der
Abb. 3). Umgekehrt kann die Anregung natürlich auch auf einen instabilen Zustand führen,
dann erfolgt eine Dissoziation des Moleküls. Weiterhin ist es möglich, dass eine Anregung in
einen derart hohen Zustand eines stabilen Zustandes führt, dass das Molekül durch die hohe
Schwingungsanregung zerfällt (rechts).
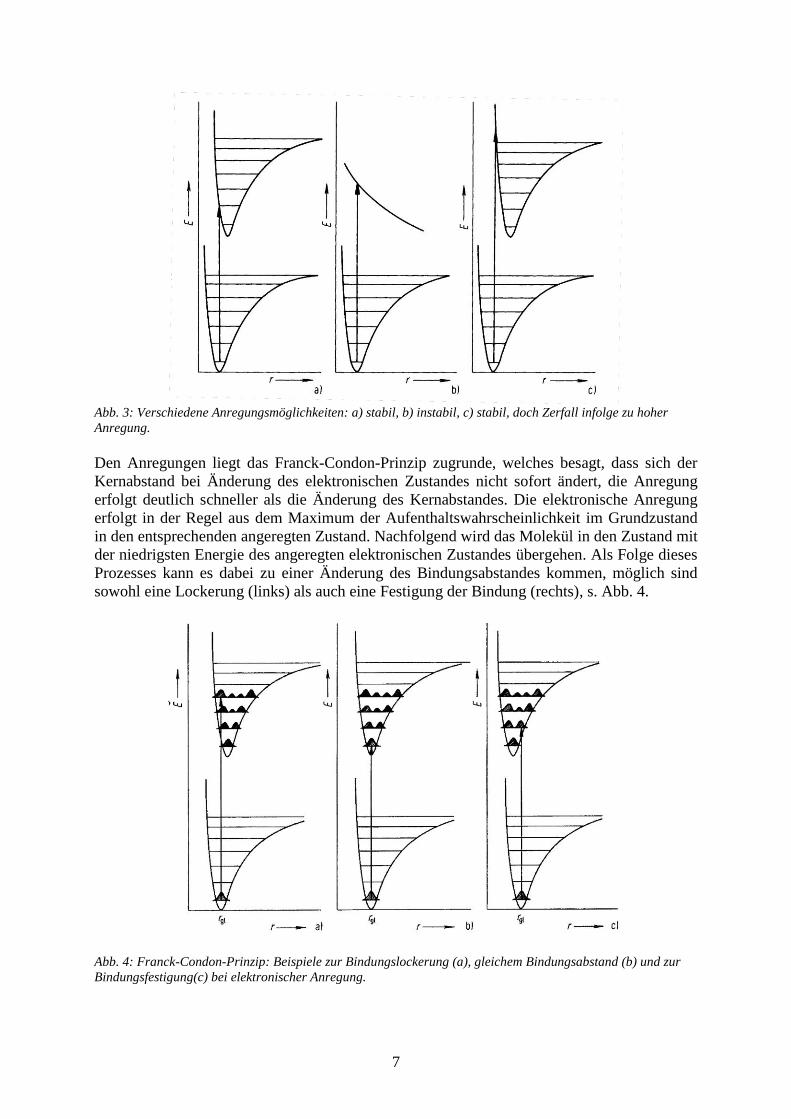
7
Abb. 3: Verschiedene Anregungsmöglichkeiten: a) stabil, b) instabil, c) stabil, doch Zerfall infolge zu hoher
Anregung.
Den Anregungen liegt das Franck-Condon-Prinzip zugrunde, welches besagt, dass sich der
Kernabstand bei Änderung des elektronischen Zustandes nicht sofort ändert, die Anregung
erfolgt deutlich schneller als die Änderung des Kernabstandes. Die elektronische Anregung
erfolgt in der Regel aus dem Maximum der Aufenthaltswahrscheinlichkeit im Grundzustand
in den entsprechenden angeregten Zustand. Nachfolgend wird das Molekül in den Zustand mit
der niedrigsten Energie des angeregten elektronischen Zustandes übergehen. Als Folge dieses
Prozesses kann es dabei zu einer Änderung des Bindungsabstandes kommen, möglich sind
sowohl eine Lockerung (links) als auch eine Festigung der Bindung (rechts), s. Abb. 4.
Abb. 4: Franck-Condon-Prinzip: Beispiele zur Bindungslockerung (a), gleichem Bindungsabstand (b) und zur
Bindungsfestigung(c) bei elektronischer Anregung.

8
1.3 Fluoreszenz
Bei der Fluoreszenz erfolgt eine elektronische Anregung aus dem Grundzustand in einen
angeregten Zustand unter Ausnutzung des Franck-Condon-Prinzips. Somit endet die
Anregung fast immer in einem angeregten Schwingungszustand (an dieser Stelle wollen wir
uns auf Vibrationszustände beschränken, Rotationszustände werden vernachlässigt). Durch
die Wechselwirkung mit anderen Molekülen (Stöße) kann das Molekül Schwingungsenergie
verlieren und geht strahlungslos in den Schwingungsgrundzustand über wie im linken Teil der
Abb. 5 dargestellt. Dieser Prozess ermöglicht aber keine Änderung des elektronischen
Zustandes, hierzu ist die Emission von Strahlung notwendig. Bei dem Übergang in den
elektronischen Grundzustand unter Emission von Strahlung (Fluoreszenz) muss wiederum das
Franck-Condon-Prinzip beachtet werden, die Übergänge enden in der Regel in höheren
Vibrationsniveaus im elektronischen Grundzustand. Aus diesem Grund ist das
Fluoreszenzspektrum gegenüber dem Absorptionsspektrum zu kleineren Frequenzen (bzw.
größeren Wellenlängen) verschoben. Der rechte Teil der Abb. 5 zeigt den Zusammenhang
zwischen den einzelnen Absorptions- und Emissionslinien und dem jeweiligen Spektrum.
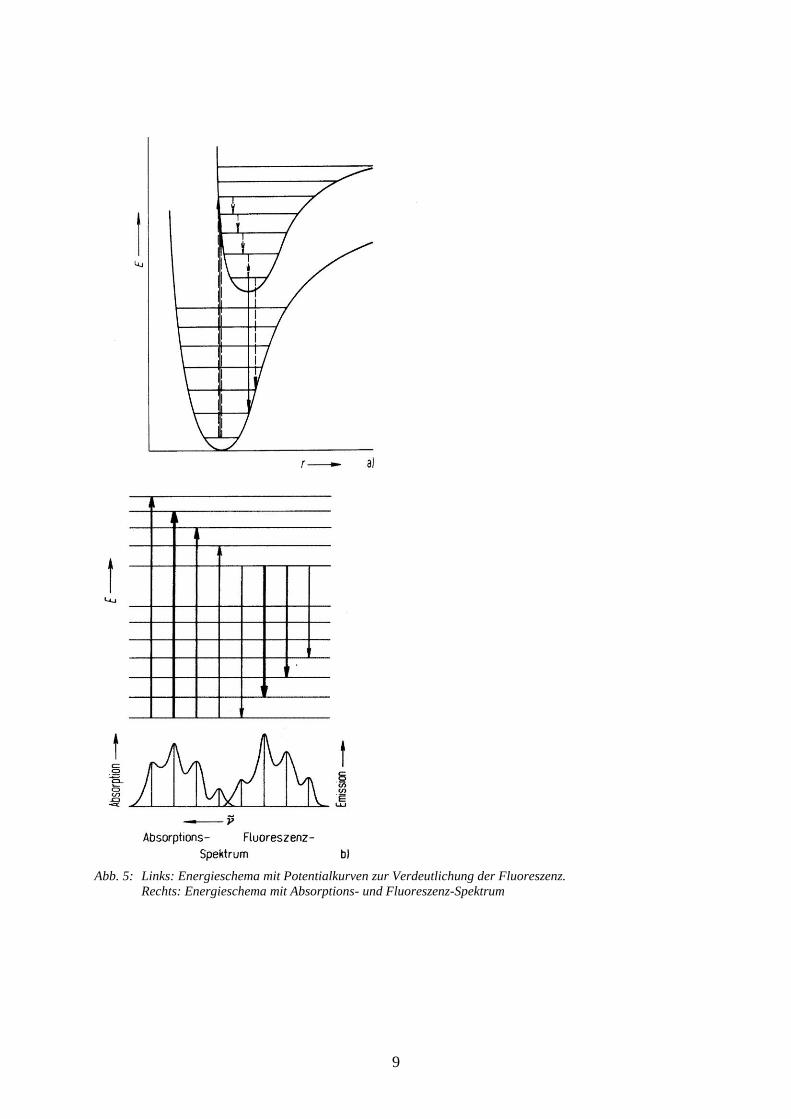
9
Abb. 5: Links: Energieschema mit Potentialkurven zur Verdeutlichung der Fluoreszenz.
Rechts: Energieschema mit Absorptions- und Fluoreszenz-Spektrum
10
1.4 Phosphoreszenz
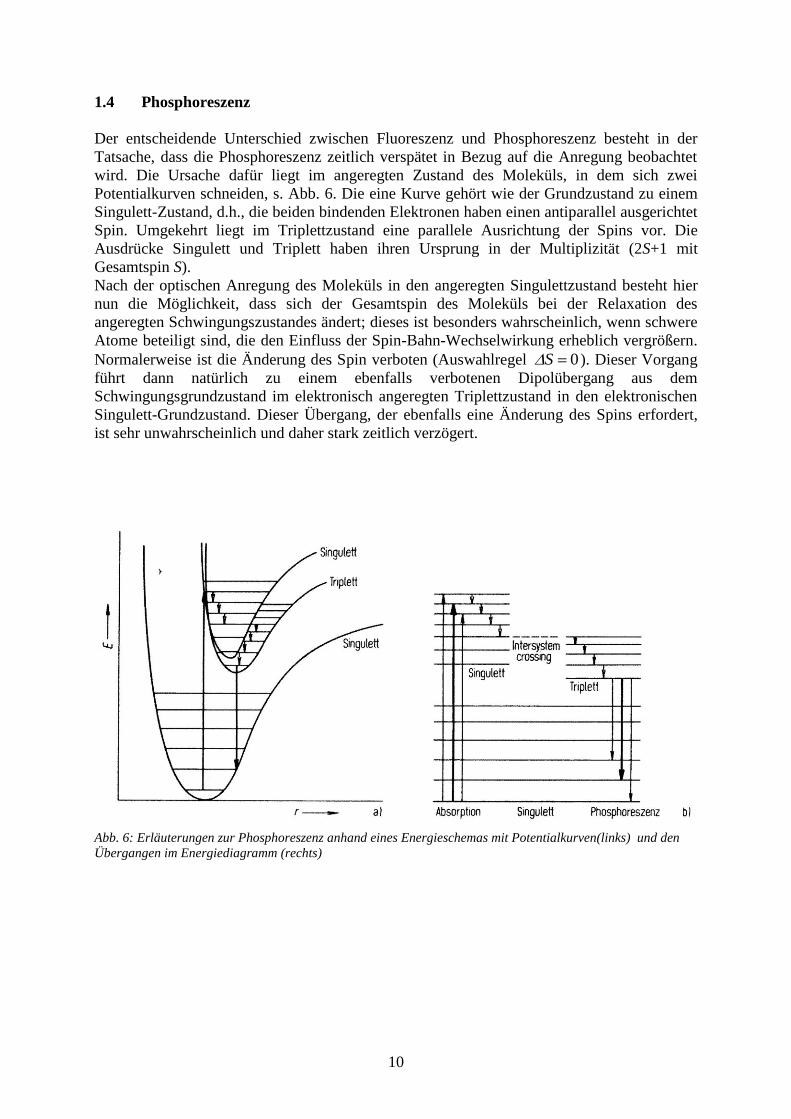
Der entscheidende Unterschied zwischen Fluoreszenz und Phosphoreszenz besteht in der
Tatsache, dass die Phosphoreszenz zeitlich verspätet in Bezug auf die Anregung beobachtet
wird. Die Ursache dafür liegt im angeregten Zustand des Moleküls, in dem sich zwei
Potentialkurven schneiden, s. Abb. 6. Die eine Kurve gehört wie der Grundzustand zu einem
Singulett-Zustand, d.h., die beiden bindenden Elektronen haben einen antiparallel ausgerichtet
Spin. Umgekehrt liegt im Triplettzustand eine parallele Ausrichtung der Spins vor. Die
Ausdrücke Singulett und Triplett haben ihren Ursprung in der Multiplizität (2S+1 mit
Gesamtspin S).
Nach der optischen Anregung des Moleküls in den angeregten Singulettzustand besteht hier
nun die Möglichkeit, dass sich der Gesamtspin des Moleküls bei der Relaxation des
angeregten Schwingungszustandes ändert; dieses ist besonders wahrscheinlich, wenn schwere
Atome beteiligt sind, die den Einfluss der Spin-Bahn-Wechselwirkung erheblich vergrößern.
Normalerweise ist die Änderung des Spin verboten (Auswahlregel 0S ). Dieser Vorgang
führt dann natürlich zu einem ebenfalls verbotenen Dipolübergang aus dem
Schwingungsgrundzustand im elektronisch angeregten Triplettzustand in den elektronischen
Singulett-Grundzustand. Dieser Übergang, der ebenfalls eine Änderung des Spins erfordert,
ist sehr unwahrscheinlich und daher stark zeitlich verzögert.
Abb. 6: Erläuterungen zur Phosphoreszenz anhand eines Energieschemas mit Potentialkurven(links) und den
Übergangen im Energiediagramm (rechts)

11
1.5. Reaktionen von angeregten Zuständen
Häufig untersucht man mit der Fluoreszenz-Spektroskopie die Wechselwirkungen von
Fluorophoren mit ihrer chemischen Umgebung, welche das Verhalten der angeregten
Zustände beeinflussen kann. Angeregte Moleküle können aber auch selbst Reaktionen
eingehen. Durch die Lichtabsorption kann sich die Elektronenverteilung so stark ändern, dass
chemische und physikalische Eigenschaften sowie die Reaktivität verändert werden.
In dieser Hinsicht sind Phenole die am besten untersuchten Moleküle, denn im angeregten
Zustand kann das Proton der Phenol-Gruppe leicht abgespalten werden. Die OH-Gruppe ist
stärker sauer, weil die Elektronen im angeregten Zustand in höherem Maß in den
aromatischen Ring verschoben werden. Deshalb ist der pK*-Wert im angeregten Zustand (*)
kleiner als der pK des Grundzustands. Umgekehrt wird der pK-Wert von Acridin im
angeregten Zustand größer.
Tabelle: pKa-Werte für Moleküle im Grundzustand und im angeregten Zustand.
pKa pKa*
Phenol 10,0 4,1
1-Naphthol 9,2 2,0
2-Naphthol 9,5 2,8
Acridin 5,1 10,6
Im Fall von 2-Naphthol sinkt der pK-Wert im angeregten Zustand [6]. Emissionsspektren in
zeigen Maxima bei 357 nm (saure Lösung) und 409 nm (alkalische Lösung). Bei mittleren
pH-Werten (z.B. pH 3) erkennt man beide Beiträge, obwohl im Grundzustand nur Naphthol
vorliegt. Das Naphtholat-Signal deutet auf die Deprotonierung im angeregten Zustand.
Charakteristisch für Reaktionen im angeregten Zustand sind demnach die unterschiedlichen
Spektren für Absorption und Emission. Je nach Fluorophor und Lösungsmittel verläuft die
Reaktion in der Lebensdauer des angeregten Zustands vollständig oder nur teilweise.
Der Dissoziationsgrad hängt auch von der jeweiligen Pufferkonzentration ab. Bei konstantem
pH-Wert wird der Dissoziationsgrad mit steigender Pufferkonzentration ansteigen, da die
Konzentration der schwachen Pufferbase größer ist.
Mithilfe eines Förster-Zyklus kann aus der Verschiebung der Spektralbanden im angeregten
Zustand die Änderung des pK–Werts berechnet werden [9].
Für die Freie Standardreaktionsenthalpie im angeregten Zustand und im Grundzustand gilt:
* ln * * *
ln
r r r
r r r
G RT K H T S
G RT K H T S
Abbildung: Elektronische Energieniveaus der Säure AH und der konjugierten Base A
- im
Grundzustand und im angeregten Zustand.

12
Unter der Annahme, dass sich die Standardreaktionsentropien nicht unterscheiden, ergibt sich:
* ln10 * ln10
**
ln10 ln10
r r
HAr r A
H RT pK H RT pK
E EH HpK pK pK
RT RT
Die Anregungsenergien können aus den Spektren erhalten werden.

13
2. Aufbau des Spektrometers:
Eine Übersicht zu dem verwendeten Fluoreszenzspektrometer ist im linken Teil der Abb. 7
gegeben, der rechte Teil zeigt den optischen Strahlengang im Spektrometer, beginnend oben
links mit der Blitzlampe zur Erzeugung der Strahlung. Das Gerät besteht aus einem
Probenbereich (grün), in dem die zu untersuchende Substanz (in einer Küvette) in der Mitte
des Bereiches in einem Halter befestigt wird. Der vordere untere Bereich zeigt eine Übersicht
über den Strahlengang (lila) bis zur Probe, der hintere Bereich gehört zur Analyse der
emittierten Strahlung (orange), die 90° versetzt zur einfallenden Strahlung spektroskopiert
wird. Der Strahlengang und die optischen Elemente sind detaillierter im rechten Teil der
Abb. 7 zu sehen. Die Erzeugung erfolgt in einer Blitzlampe (oben links), die Strahlung wird
dann vor und hinter dem dispersiven Element zur Monochromatisierung (Gitter) durch eine
Blende räumlich eingeengt. Die Größe der Blende hinter dem Gitter wird für die Intensität
und damit gleichzeitig auch für die energetische Auflösung der einfallenden Strahlung
gewählt (Werte zwischen 1.5 und 20). Eine große Blende bedeutet natürlich hohe Intensität
bei gleichzeitig schlechter Energieauflösung. Zusätzlich steht im Strahlengang noch ein Filter
zur Verfügung, der automatisch vom Programm angepasst wird. Ein Teil der Strahlung wird
über eine Strahlteiler als Referenz ausgekoppelt, der größte Teil wird nach dem
Umlenkspiegel in die Küvette fokussiert.
Die von der Probe emittierte Strahlung wird dann analog durch eine Filter und eine Blende
auf eine optisch dispersives Element (Gitter) fokussiert und spektral aufgelöst. Ein
Photomultiplier misst dann die Intensität der elektromagnetischen Strahlung.
Abb. 7: Aufbau des Spektrometers und Strahlengang mit optischen Elementen (schematisch)
Zugänglicher Messbereich des Spektrometers: 190 – 900 nm
Messmodi: Es stehen verschiedene Programme zur Verfügung, von denen einige kurz
erwähnt werden sollten:
- Messung der Fluoreszenz (spektral aufgelöst) bei fester Anregungswellenlänge
(Exzitation)
- Messung der Fluoreszenz bei fester Emissionswellenlänge als Funktion der
Anregungswellenlänge
- Gleichzeitiges Scannen der Anregungsenergie und der Emissionsenergie mit
vorgewähltem Energieabstand

14
3. Versuchsbeschreibung
3.1 Grundlegendes
Im Folgenden soll die Dissoziation von 2-Naphthol im Grundzustand und angeregten Zustand
betrachtet werden.
+ H2O + H3O+
kurz: ROH + H2O RO- + H3O
+
Aufgrund der Resonanzstabilisierung durch chinoide Strukturen kann bei 2-Naphtol das
Proton der OH-Gruppe abgespalten werden. Im angeregten Zustand ist die Ladungsdichte am
Sauerstoff herabgesetzt. Dies sollte zur Folge haben, dass das Proton leichter abgespalten
werden kann und somit die Säurestärke höher als im Grundzustand ist.
3.2 Bestimmung der Gleichgewichtkonstanten und pK-Werte
Das Naphthol-Naphtholat-System kann durch das folgende Reaktionsschema beschrieben
werden:
Bei einer Wellenlänge von 360 nm wird das Naphtholat-Anion selektiv zur Fluoreszenz
angeregt, während Naphthol erst bei kleineren Wellenlängen absorbiert. Die gemessene
Fluoreszenzintensität ist äquivalent zur Konzentration des Naphtholat-Anions. Dies kann man
nutzen, um die Dissoziationskonstante zu bestimmen.
Im Experiment wird zum einen die Fluoreszenzintensität des Naphtholat-Anions im pH-
Bereich von 12 bis 6 gemessen. Die Fluoreszenzintensität bei pH=12 und größer entspricht
der maximalen Fluoreszenzintensität des Naphtholat-Anions (das bei pH=12 praktisch
ausschließlich vorliegt). Bei Absenken des pH-Werts verringert sich die Konzentration des
Naphtholat-Anions und somit auch die Fluoreszenzintensität, bis bei pH=6 so gut wie
ausschließlich Naphthol vorliegt und die Fluoreszenzintensität verschwindet. Aus der
Auftragung der Fluoreszenzintensität gegen pH-Wert kann die Gleichgewichtskonstante K
und der pK-Wert ermittelt werden. Am Wendepunkt der Kurve gilt pH = pK.
Zum anderen nutzt man die Tatsache aus, das auch Naphthol zur Fluoreszenz angeregt wird,
allerdings mit einer kleineren Wellenlänge von 320 nm. Bestimmt man wieder die
Fluoreszenzintensität in Abhängigkeit vom pH-Wert im Bereich von pH 12 bis 1, lässt sich
zunächst wieder, wie oben beschrieben, die Abnahme der Fluoreszenzintensität des
Naphtholat-Anions im Grundzustand beobachten. Allerdings nimmt die Fluoreszenzintensität

15
bei pH = 6 nicht den Wert null an, da Naphthol im angeregten Zustand eine höhere
Säurestärke hat und daher auch noch bei einem pH-Wert von 6 dissoziiert vorliegt.
Bei dem angeregten Molekül können zwei Fluoreszenzbanden – für Naphthol und das
Naptholat-Anionion – beobachtet werden, da die Dissoziation von ROH* innerhalb der
Lebensdauer erfolgt. Bei kleinem pH-Wert (<4) wird das Naphtolat-Anion zu Naphthol
umgesetzt. Dies führt dazu, dass sich die Intensität der Naphtholat-Bande verringert. Bei
einem pH-Wert von 1 liegt schließlich nur noch ROH* vor und die die Fluoreszenzintensität
dieser Bande ist maximal. Trägt man wieder die Fluoreszenzintensität der Bande für die
Naphtholat-Ionen gegen den pH-Wert auf, so erhält man am Wendepunkt der Kurve den pK*-
Wert.
Arbeitet man in einer gepufferten Lösung, muss beachtet werden, dass als Konkurrenz zu der
Dissoziation auch eine Reaktion mit dem Puffer erfolgen kann. Dies führt dazu, dass der
bestimmte pK*-Wert um 0,4 Einheiten zu niedrig ist.
3.2 Bestimmung der Geschwindigkeitskonstanten für die Dissoziation des
Moleküls im angeregten Zustand
Für das Molekül im angeregten Zustand, gilt im pH-Bereich zwischen 1 und 4 folgendes
Reaktionsschema:
Das angeregte Naphthol bzw. Naphtholat-Ion kann durch Fluoreszenz oder durch
strahlungslose Übergänge wieder in den Grundzustand zurückfallen. kF und kF- sind die
Geschwindigkeits-konstanten des Abklingens der Fluoreszenz, kQ und kQ- sind die
Geschwindigkeitskonstanten der strahlungslosen Deaktivierung und kA ist die
Geschwindigkeitskonstante der Anregung.
Aus dem Bodenstein’schen Quasistationaritätsprinzip ergibt sich:
2 2 0
*
* * * *
F Q A
d ROHk k k ROH k RO H k ROH
dt
(3.1)
2 2 0
*
* * *
F Q
d ROk k k H RO k ROH *
dt
(3.2)
Aus den beiden Gleichungen erhält man:
2
22
*
F Q*
A * *
F Q F Q F Q
k k k H ROHROH k
k H ( k k ) ( k k k )( k k )
(3.3)
Bei pH = 1 ist 2
*
F Qk H k k
, somit ergibt sich:
0
*
A
F Q
ROHROH k
k k
(3.4)
Dasselbe Ergebnis erhält man, wenn man nur die Reaktionen zu kA, kF und kQ berücksichtigt.

16
Die Fluoreszenzintensität ist proportional zur Konzentration und somit gilt *
0 0ROH I
(I0 = I bei pH=1) und *ROH I . Des Weiteren definiert man wie folgt:
22
2
1 1 **
F Q F Q *
kk k , k k , K
k
und erhält somit:
* * *2 20 2
* *
2 2
11
1 1
k k HI k
I k H k H
(3.5)
Daraus folgt:
*
2
* * *0 2 2 2
11 1
1
k H H
I k k K
I
(3.6)
Trägt man 0
1
1I
I
gegen H auf, so erhält man die Größen *
2K , *
2k und *
2k durch lineare
Regression.
Die Werte von und sind laut Literatur in der Größenordnung
810 s[2-7].
Die folgende Tabelle zeigt Daten für den Abfall der Fluoreszenz von -Naphtol in 0.10M
H2SO4 bei 25 °C [8]. Zeit t / ns Intensität I
0 21753
1 18907
2 16380
3 14171
4 12432
5 10757
6 9288
7 8138
8 7083
9 6014
10 5350
Auf dem hier gezeigten Weg können allgemein aus Messungen der Fluoreszenzintensität
Gleichgewichts- und Geschwindigkeitskonstanten berechnet werden. Diese Methode ist
besonders vorteilhaft, da die zu untersuchenden Reaktionen diffusionskontrolliert, also extrem
schnell sein können, ohne dass eine schnelle Beobachtungsmethode erforderlich ist.
Allerdings muss mindestens ein Reaktionspartner fluoreszieren.

17
4. Versuchsdurchführung
Stellen sie zunächst folgende Lösungen her:
Lösung 1: 1mL NaOH (1N), 20 mL Naphthol-Lsg. 10-3
M, 42 mL demin. H2O, 2 mL
Stenhagenpuffer (enthält: 10 mM Phosphorsäure, 15 mM Borsäure, 7 mM
Citronensäure, 69 mM NaOH)
Lösung 2: 10ml HCl (1N), 20 mL Naphthol-Lsg. 10-3
M, 35 mL demin. H2O
Benutzen Sie die dazu bereitgestellten Lösungen, Vollpipetten, und Bechergläser bzw.
Kolben.
Zunächst wird der pH-Wert von Lösung 1 bestimmt und ein Fluoreszenzsspektrum mit der
Anregungswellenlänge 360nm und 322nm aufgenommen. Man titriert nun die Lösung 1 mit
Lösung 2 und nimmt alle 0,3 pH-Einheiten (der pH-Wert wird mit einer Glaselektrode
gemessen) ein Spektrum mit 360nm (bis zum pH-Wert 6) bzw. 322nm (bis zum pH-Wert 0,5)
auf. Man gibt jedes Mal nach der Aufnahme eines Spektrums die Lösung aus der Küvette
zurück zu Lösung 1 und spült einige Male mit der Lösung mit dem neuen pH-Wert.

18
5. Auswertung
Folgende Werte sollen bestimmt werden:
- Gleichgewichtskonstanten für die Dissoziation des Protons sowohl für den
angeregten Zustand als auf für den Grundzustand
- pK-Werte für angeregten Zustand und Grundzustand
- Geschwindigkeitskonstanten für Dissoziation des Protons im angeregten Zustand
Literatur zum Versuch
[1] G. Wedler, Lehrbuch der Physikalischen Chemie, Wiley-VCH Verlag
Das Lehrbuch gibt in den Kapiteln 3.1-3.4 eine gute Einführung in das Thema, vom Aufbau
der Materie in der quantenmechanischen Behandlung bis zur Wechselwirkung von Atomen
und Molekülen mit elektromagnetischer Strahlung.
[2] Der Reaktionsmechanismus der Säuredissoziation am Beispiel der protolytischen
Reaktion des angeregten ß-Naphthols
A. Weller, Z. Phys. Chem., Neue Folge 3 (1955) 238–254.
[3] Allgemeine Basenkatalyse bei der elektrolytischen Dissoziation angeregter Naphthole
A. Weller, Z. Elektrochem. 58 (1954) 849–853.
[4] Quantitative Untersuchungen der Fluoreszenzumwandlung bei Naphtholen
A. Weller, Z. Elektrochem. 56 (1952) 662-668.
[5] The protolysis of singlet excited ß-naphthol - a two-day laboratory experiment to
introduce photophysics
J. van Stam, J.-E. Lofroth, J. Chem. Educ. 63 (1986) 181-184.
[6] Analysis of Two-State Excited-State Reactions. The Fluorescence Decay of 2-Naphthol.
W.R. Laws, L. Brand, J. Phys. Chem. 83 (1979) 795.
[7] Excited-State Deprotonation of 2-Naphthol by Anions
M. Lawrence, Ch.J. Marzzacco, C. Morton, C. Schwab, A.M. Halpern, J. Phys. Chem. 95
(1991) 10294.
[8] A.M. Halpern, Experimental Physical Chemistry, 2nd
edition, Prentice Hall 1997
[9] Estimation of pKa* in the first excited singlet state. A physical chemistry experiment that
explores acid-base properties in the excited state
B. Marciniak , H. Kozubek and S. Paszyc, J. Chem. Educ. 69 (3) (1992) 247

19

20
Anhang
Bestimmung der Geschwindigkeitskonstanten für die Dissoziation von -Naphtol im
angeregten Zustand
Die Intensität I der Fluoreszenzstrahlung wird bei verschiedenen pH-Werten gemessen. Der
Wert I0 bezieht sich auf niedrige pH-Werte, bei denen praktisch keine Dissoziation
vorkommt.
*
0 2
* *
2 2
1
1
I k HI
k k H
Diese Gleichung kann nach entsprechender Umstellung für eine lineare Regression verwendet
werden:
* *0 2 2
1 1
1
H
I k K
I
Mit moderner Software ist eine nichtlineare Regression möglich. Dies hat den Vorteil, dass
Messgrößen direkt gefittet werden können, die immer gleich stark fehlerbehaftet sind.
Außerdem ist der Zusammenhang zwischen theoretischer Beschreibung und experimentellen
Messwerten in einer Graphik I vs. pH offensichtlich.
Beispiel: Nichtlinearer Fit mit OriginPro 9.0.
Origin ist an der Universität Ulm als Netzwerkversion auch für eigene Rechner verfügbar.
Diese Version ist für Institutsangehörige im Bereich Forschung und Lehre kostenlos. Die
Netzwerkversion von Origin kann über das Hochschuldiensteportal der Universität Ulm
heruntergeladen werden. Melden Sie sich hierzu mit Ihrem kiz-Account im Portal an. Unter
'Software zum Herunterladen' finden Sie neben der eigentlichen Software auch eine
Installationsanleitung.
Kurzanleitung am Beispiel PCFP_V2
In die Arbeitsmappe werden die Messdaten eingefügt.
Mit Strg+D können weitere Spalten hinzugefügt werden.
Durch Klicken auf die Spaltenbezeichnung, z.B. C(Y), kann eine ganze Spalte markiert
werden.
Über Spalte – Setzen als können den Spalten x- und y-Fehler zugeordnet werden. Die Werte
werden nach Auswählen einer Spalte mit Strq-Q (Spaltenwerte errechnen) zugewiesen.
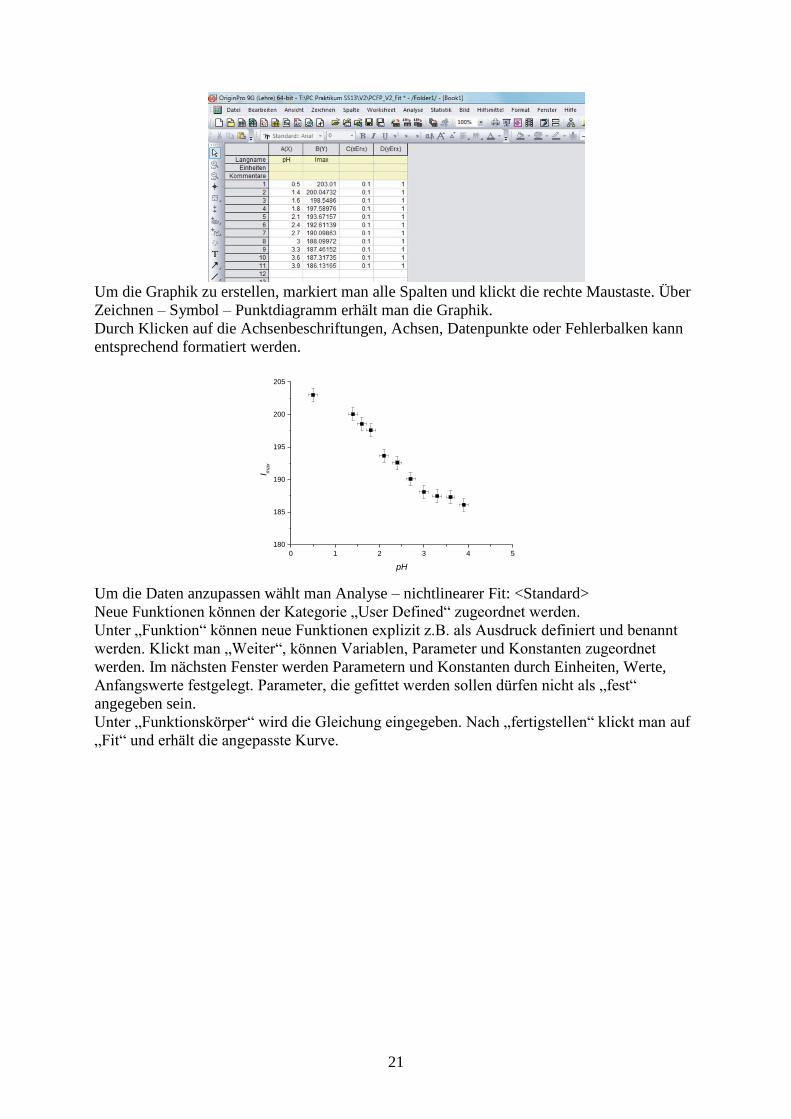
21
Um die Graphik zu erstellen, markiert man alle Spalten und klickt die rechte Maustaste. Über
Zeichnen – Symbol – Punktdiagramm erhält man die Graphik.
Durch Klicken auf die Achsenbeschriftungen, Achsen, Datenpunkte oder Fehlerbalken kann
entsprechend formatiert werden.
0 1 2 3 4 5
180
185
190
195
200
205
I ma
x
pH
Um die Daten anzupassen wählt man Analyse – nichtlinearer Fit: <Standard>
Neue Funktionen können der Kategorie „User Defined“ zugeordnet werden.
Unter „Funktion“ können neue Funktionen explizit z.B. als Ausdruck definiert und benannt
werden. Klickt man „Weiter“, können Variablen, Parameter und Konstanten zugeordnet
werden. Im nächsten Fenster werden Parametern und Konstanten durch Einheiten, Werte,
Anfangswerte festgelegt. Parameter, die gefittet werden sollen dürfen nicht als „fest“
angegeben sein.
Unter „Funktionskörper“ wird die Gleichung eingegeben. Nach „fertigstellen“ klickt man auf
„Fit“ und erhält die angepasste Kurve.
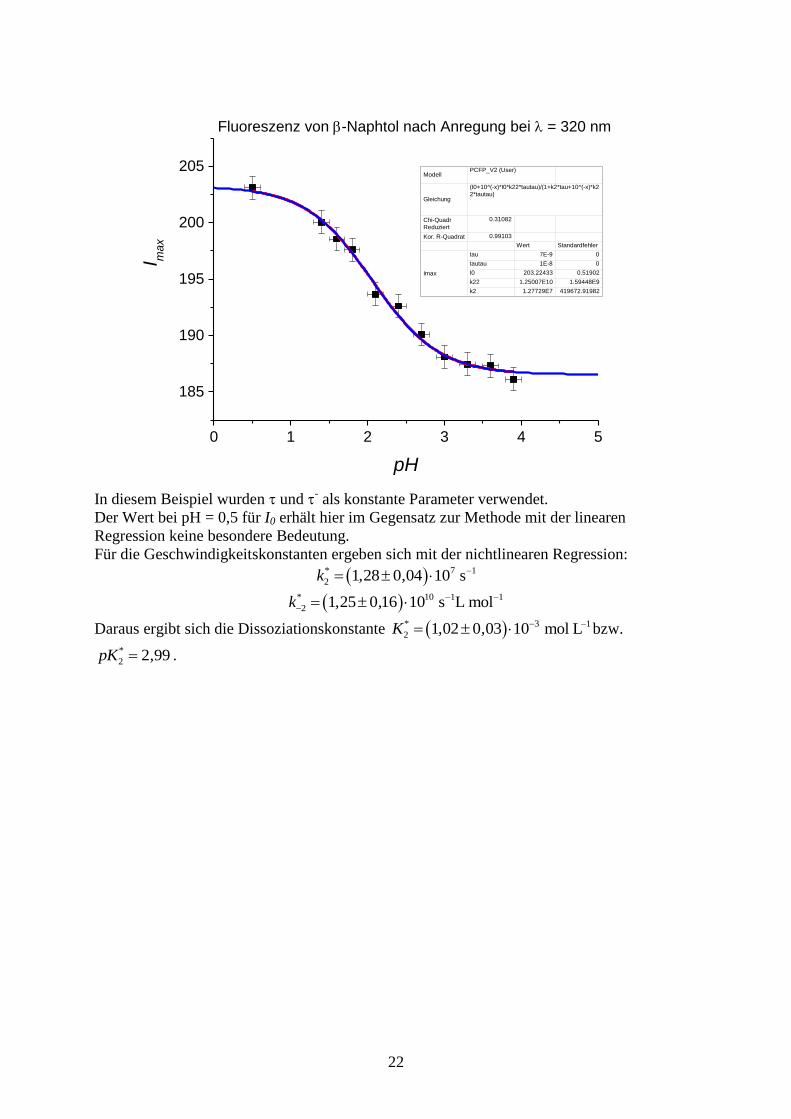
22
0 1 2 3 4 5
185
190
195
200
205I m
ax
pH
Fluoreszenz von -Naphtol nach Anregung bei = 320 nm
ModellPCFP_V2 (User)
Gleichung
(I0+10^(-x)*I0*k22*tautau)/(1+k2*tau+10^(-x)*k2
2*tautau)
Chi-Quadr
Reduziert
0.31082
Kor. R-Quadrat 0.99103
Wert Standardfehler
Imax
tau 7E-9 0
tautau 1E-8 0
I0 203.22433 0.51902
k22 1.25007E10 1.59448E9
k2 1.27729E7 419672.91982
In diesem Beispiel wurden und
- als konstante Parameter verwendet.
Der Wert bei pH = 0,5 für I0 erhält hier im Gegensatz zur Methode mit der linearen
Regression keine besondere Bedeutung.
Für die Geschwindigkeitskonstanten ergeben sich mit der nichtlinearen Regression:
7
2 1 28 0 04 10*k , , 1s
10
2 1 25 0 16 10*k , , 1 1s L mol
Daraus ergibt sich die Dissoziationskonstante 3
2 1 02 0 03 10*K , , 1mol L bzw.
2 2 99*pK , .




![Heterocyclische Siebenring-Verbindungen, XXX [1] Synthese ...zfn.mpdl.mpg.de/data/Reihe_B/42/ZNB-1987-42b-0217.pdf · -1, im 'H-NMR-Spektrum das Singulett der O CH3-Gruppe bei ö](https://img.pdfslide.tips/doc/110x75/5e1de407443159751c398534/heterocyclische-siebenring-verbindungen-xxx-1-synthese-zfnmpdlmpgdedatareiheb42znb-1987-42b-0217pdf.jpg)
















