Embed Size (px)
Citation preview

Allegato I
Elenco dei nomi dei medicinali, della forma farmaceutica, del dosaggio, della via di somministrazione, dei titolari dell’autorizzazione all’immissione in commercio negli Stati Membri.
1
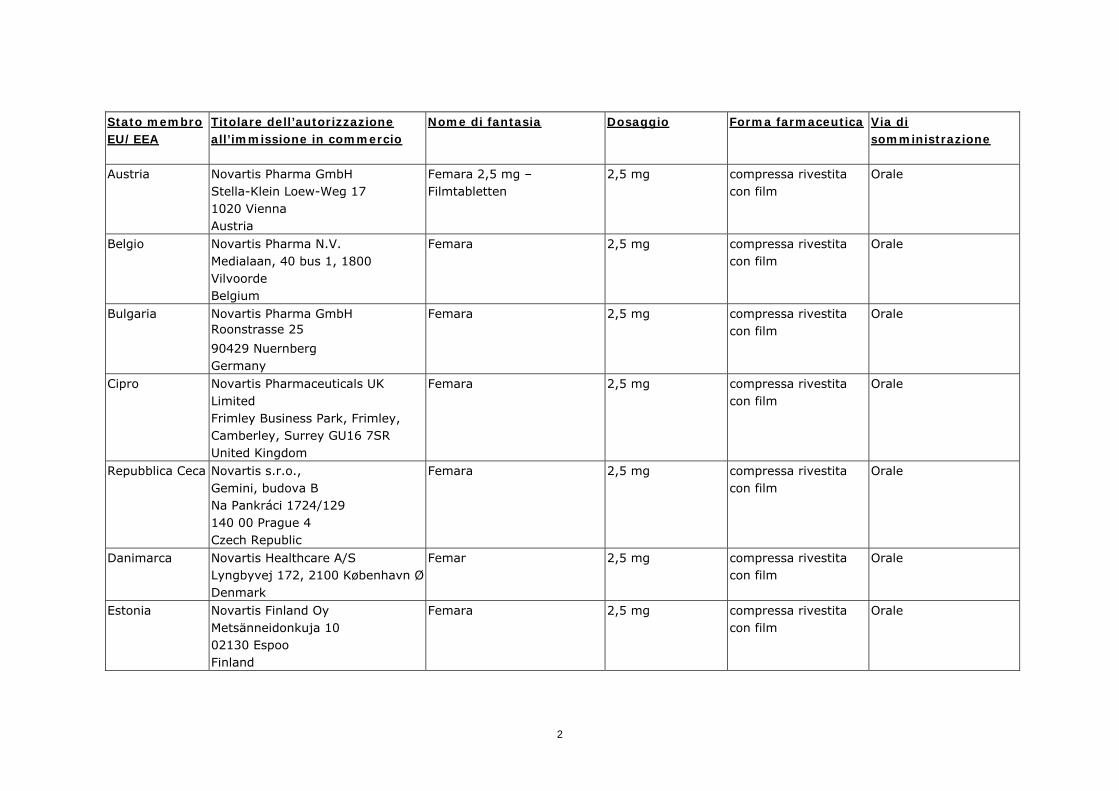
Stato membro EU/EEA
Titolare dell’autorizzazione all’immissione in commercio
Nome di fantasia Dosaggio Forma farmaceutica Via di somministrazione
Austria
Novartis Pharma GmbH Stella-Klein Loew-Weg 17 1020 Vienna Austria
Femara 2,5 mg –Filmtabletten
2,5 mg compressa rivestita con film
Orale
Belgio
Novartis Pharma N.V. Medialaan, 40 bus 1, 1800 Vilvoorde Belgium
Femara 2,5 mg compressa rivestita con film
Orale
Bulgaria
Novartis Pharma GmbH Roonstrasse 25
90429 Nuernberg Germany
Femara 2,5 mg compressa rivestita con film
Orale
Cipro
Novartis Pharmaceuticals UK Limited Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR United Kingdom
Femara 2,5 mg compressa rivestita con film
Orale
Repubblica Ceca
Novartis s.r.o., Gemini, budova B Na Pankráci 1724/129 140 00 Prague 4 Czech Republic
Femara 2,5 mg compressa rivestita con film
Orale
Danimarca
Novartis Healthcare A/S Lyngbyvej 172, 2100 København Ø Denmark
Femar 2,5 mg compressa rivestita con film
Orale
Estonia
Novartis Finland Oy Metsänneidonkuja 10 02130 Espoo Finland
Femara 2,5 mg compressa rivestita con film
Orale
2
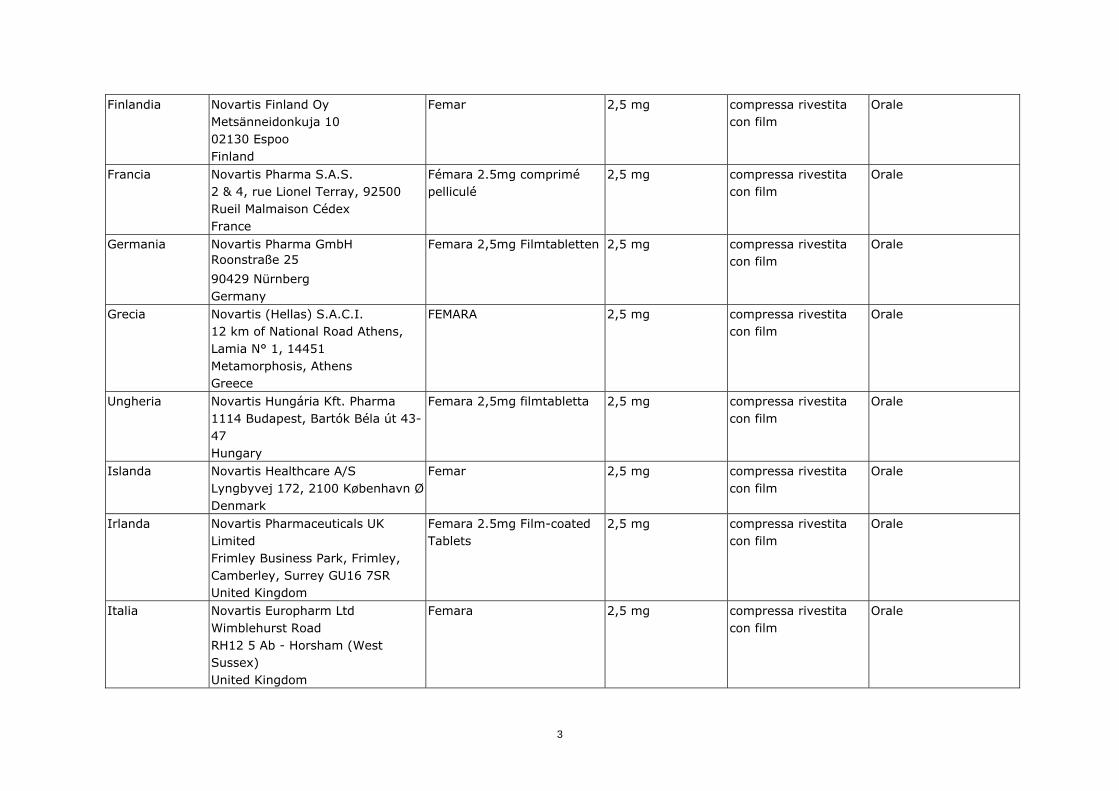
Finlandia
Novartis Finland Oy Metsänneidonkuja 10 02130 Espoo Finland
Femar 2,5 mg compressa rivestita con film
Orale
Francia
Novartis Pharma S.A.S. 2 & 4, rue Lionel Terray, 92500 Rueil Malmaison Cédex France
Fémara 2.5mg comprimé pelliculé
2,5 mg compressa rivestita con film
Orale
Germania
Novartis Pharma GmbH Roonstraße 25
90429 Nürnberg Germany
Femara 2,5mg Filmtabletten 2,5 mg compressa rivestita con film
Orale
Grecia
Novartis (Hellas) S.A.C.I. 12 km of National Road Athens, Lamia N° 1, 14451 Metamorphosis, Athens Greece
FEMARA 2,5 mg compressa rivestita con film
Orale
Ungheria
Novartis Hungária Kft. Pharma 1114 Budapest, Bartók Béla út 43-47 Hungary
Femara 2,5mg filmtabletta 2,5 mg compressa rivestita con film
Orale
Islanda
Novartis Healthcare A/S Lyngbyvej 172, 2100 København Ø Denmark
Femar 2,5 mg compressa rivestita con film
Orale
Irlanda
Novartis Pharmaceuticals UK Limited Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR United Kingdom
Femara 2.5mg Film-coated Tablets
2,5 mg compressa rivestita con film
Orale
Italia
Novartis Europharm Ltd Wimblehurst Road RH12 5 Ab - Horsham (West Sussex) United Kingdom
Femara 2,5 mg compressa rivestita con film
Orale
3
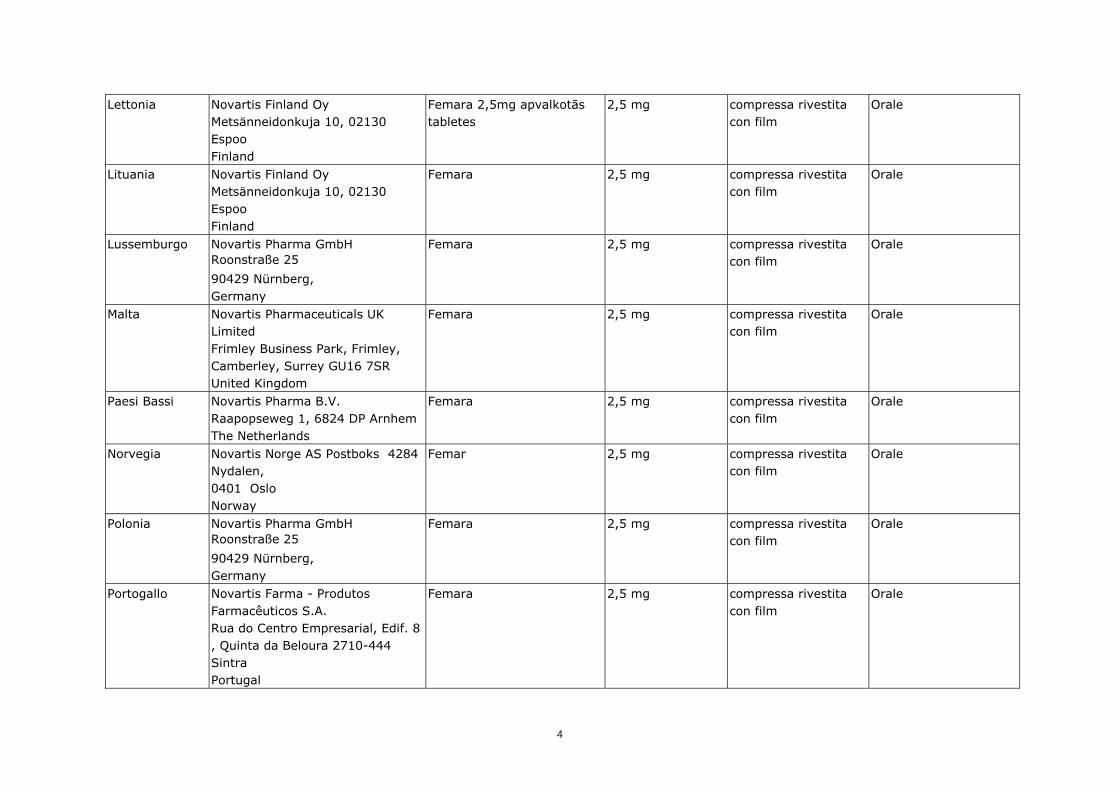
Lettonia
Novartis Finland Oy Metsänneidonkuja 10, 02130 Espoo Finland
Femara 2,5mg apvalkotās tabletes
2,5 mg compressa rivestita con film
Orale
Lituania
Novartis Finland Oy Metsänneidonkuja 10, 02130 Espoo Finland
Femara 2,5 mg compressa rivestita con film
Orale
Lussemburgo
Novartis Pharma GmbH Roonstraße 25
90429 Nürnberg, Germany
Femara 2,5 mg compressa rivestita con film
Orale
Malta
Novartis Pharmaceuticals UK Limited Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR United Kingdom
Femara 2,5 mg compressa rivestita con film
Orale
Paesi Bassi
Novartis Pharma B.V. Raapopseweg 1, 6824 DP Arnhem The Netherlands
Femara 2,5 mg compressa rivestita con film
Orale
Norvegia Novartis Norge AS Postboks 4284 Nydalen, 0401 Oslo Norway
Femar 2,5 mg compressa rivestita con film
Orale
Polonia
Novartis Pharma GmbH Roonstraße 25
90429 Nürnberg, Germany
Femara 2,5 mg compressa rivestita con film
Orale
Portogallo
Novartis Farma - Produtos Farmacêuticos S.A. Rua do Centro Empresarial, Edif. 8 , Quinta da Beloura 2710-444 Sintra Portugal
Femara 2,5 mg compressa rivestita con film
Orale
4
5
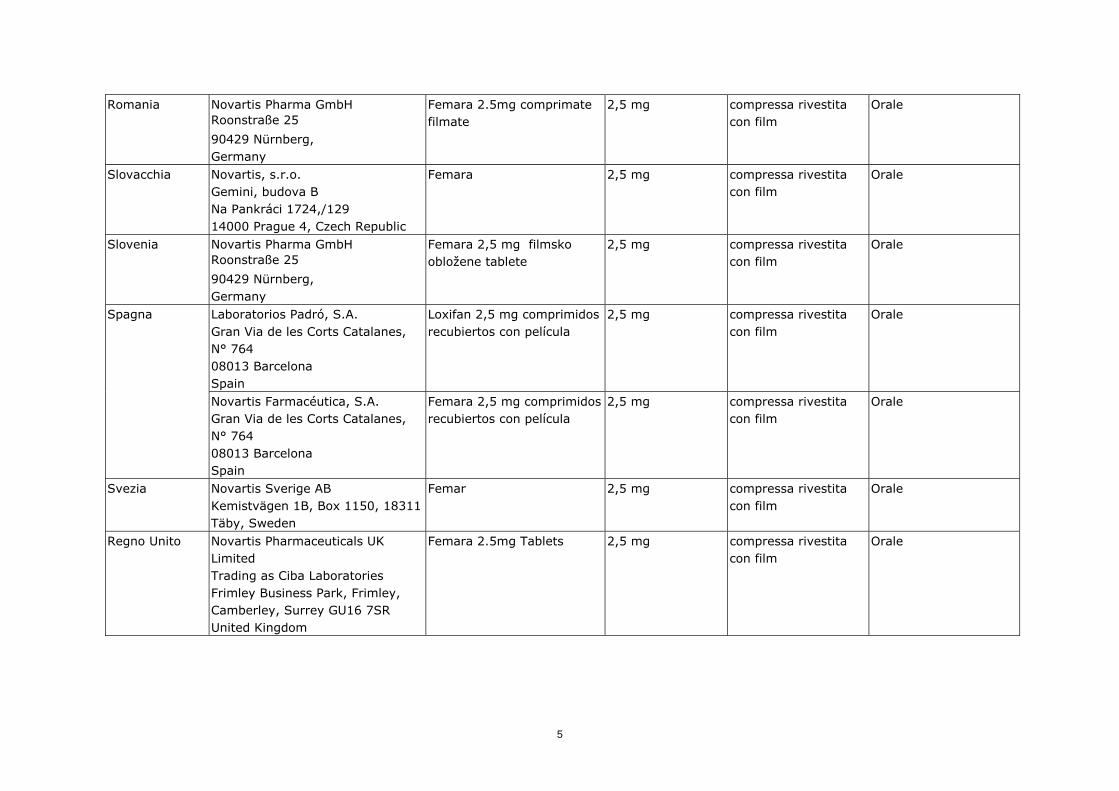
Romania
Novartis Pharma GmbH Roonstraße 25
90429 Nürnberg, Germany
Femara 2.5mg comprimate filmate
2,5 mg compressa rivestita con film
Orale
Slovacchia
Novartis, s.r.o. Gemini, budova B Na Pankráci 1724,/129 14000 Prague 4, Czech Republic
Femara 2,5 mg compressa rivestita con film
Orale
Slovenia
Novartis Pharma GmbH Roonstraße 25
90429 Nürnberg, Germany
Femara 2,5 mg filmsko obložene tablete
2,5 mg compressa rivestita con film
Orale
Laboratorios Padró, S.A. Gran Via de les Corts Catalanes, N° 764 08013 Barcelona Spain
Loxifan 2,5 mg comprimidos recubiertos con película
2,5 mg compressa rivestita con film
Orale
Spagna
Novartis Farmacéutica, S.A. Gran Via de les Corts Catalanes, N° 764 08013 Barcelona Spain
Femara 2,5 mg comprimidos recubiertos con película
2,5 mg compressa rivestita con film
Orale
Svezia
Novartis Sverige AB Kemistvägen 1B, Box 1150, 18311 Täby, Sweden
Femar 2,5 mg compressa rivestita con film
Orale
Regno Unito
Novartis Pharmaceuticals UK Limited Trading as Ciba Laboratories Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR United Kingdom
Femara 2.5mg Tablets 2,5 mg compressa rivestita con film
Orale

Allegato II
Conclusioni scientifiche e motivi della modifica di riassunto delle caratteristiche del prodotto, etichettatura e foglio illustrativo presentati dall’Agenzia europea per i medicinali
6

Conclusioni scientifiche
Riassunto generale della valutazione scientifica di Femara e denominazioni associate (v. Allegato I)
Femara contiene letrozolo, un inibitore dell’aromatase che inibisce la conversione di androgeni in estrogeni. Femara è stato approvato per la prima volta nell’Unione europea (UE) nel 1996 ed è disponibile sotto forma di compresse rivestite con film da 2,5 mg. L’approvazione comprende una serie di indicazioni connesse al trattamento del carcinoma al seno in donne in post-menopausa con progressione della malattia. Alla luce delle divergenti decisioni nazionali adottate negli Stati membri riguardo all’autorizzazione del medicinale, Femara è stato incluso nell’elenco di prodotti per i quali si richiede un’armonizzazione del Riassunto delle caratteristiche del prodotto (RCP). Pertanto è stata avviata una procedura di deferimento ai sensi dell’articolo 30, paragrafo 2, della direttiva 2001/83/CE, e successive modifiche, diretta ad appianare le divergenze e armonizzare le informazioni sul prodotto in tutta l’UE.
Sezione 4.1 – Indicazioni terapeutiche
1) Trattamento adiuvante di donne in post-menopausa con tumore invasivo al seno in fase iniziale positivo per il recettore dell’ormone Il Comitato per i medicinali per uso umano (CHMP) ha preso atto dello studio cardine sull’indicazione del trattamento adiuvante, BIG 1-98, coordinato dal gruppo collaborativo IBCSG (International Breast Cancer Study Group, il gruppo internazionale di studio sul cancro al seno) e dall’organizzazione centrale BIG (Breast International Group), che ha escluso specificamente le pazienti senza una diagnosi confermata di carcinoma invasivo al seno. Parimenti, non è stato svolto alcuno studio su pazienti con CDIS (carcinoma duttale in situ) o con CLIS (carcinoma lobulare in situ). Inoltre, il CHMP ha notato che la dicitura era conforme alla maggior parte degli RCP approvati a livello nazionale. 2) Trattamento adiuvante esteso del carcinoma mammario ormonosensibile in donne in post-menopausa dopo trattamento adiuvante standard con tamoxifene della durata di 5 anni Il CHMP ha ritenuto giustificato il termine “ormonosensibile ”, dal momento che il letrozolo non ha alcuna efficacia nel carcinoma mammario negativo per il recettore ormonale. Il CHMP ha ritenuto del pari giustificate le informazioni concernenti la durata del trattamento con tamoxifene sulla base di studi in cui il letrozolo è stato somministrato dopo 5 anni di trattamento adiuvante con tamoxifene. Circa il termine “invasivo”, non è prassi comune prescrivere inibitori dell’aromatase a pazienti senza componente invasiva. Non è stato svolto alcuno studio su CDIS o su CLIS riguardante l’uso di inibitori dell’aromatase per il trattamento adiuvante esteso dopo tamoxifene e non sussiste motivo che suggerisca un diverso meccanismo di azione nello scenario adiuvante esteso rispetto a quello adiuvante. Il CHMP ha ritenuto quindi giustificato precisare l’indicazione con “invasivo”. Il CHMP ha osservato inoltre che la dicitura era conforme alla maggior parte degli RCP approvati a livello nazionale. 3) Trattamento di prima linea di donne in post-menopausa con carcinoma mammario avanzato ormonosensibile Il CHMP ha ritenuto consolidata questa indicazione e ha osservato che la dicitura fosse conforme alla maggior parte degli RCP approvati a livello nazionale. 4) Carcinoma mammario in fase avanzata in donne in post-menopausa naturale o artificialmente indotta, dopo recidiva o progressione della malattia che siano state trattate in precedenza con
7

antiestrogeni. Il CHMP ha ritenuto anzitutto consolidata l’indicazione e, inoltre, che la conferma dello stato di post-menopausa naturale delle donne prima dell’inizio del trattamento sia giustificata da motivi di efficacia e di sicurezza. Stando agli studi le donne in stato di menopausa indotta potrebbero non essere in stato di post-menopausa endocrina, di modo che l’efficacia risulterebbe sub-ottimale con un’incidenza elevata di sintomi menopausali come vampate di calore, lo stato di peri-menopausa delle pazienti non essendo sufficiente a eliminare il circolo della sintesi degli estrogeni. Inoltre, è essenziale evitare gravidanze nel corso della terapia con letrozolo a causa del rischio di embriotossicità e fetotossicità. Infine, il CHMP ha osservato che la dicitura era perlopiù conforme agli RCP approvati a livello nazionale. 5) Trattamento neoadiuvante di donne in post-menopausa con carcinoma mammario con recettore ormonale positivo, HER-2 negativo, in cui la chemioterapia non è adatta e l’intervento chirurgico immediato non è indicato. L’indicazione di trattamento neoadiuvante (pre-operatorio) costituiva la divergenza principale in tutti gli RCP approvati a livello nazionale: mentre è stata approvata in alcuni Stati membri nel 2001, in altri la richiesta è stata ritirata principalmente perché, da un lato, il trattamento endocrino neoadiuvante all’epoca non era un concetto convalidato e, dall’altro, i risultati dello studio cardine sull’adiuvante non erano ancora disponibili. Il CHMP ha esaminato tutti i dati disponibili e ritenuto che il letrozolo mostri una superiorità significativa rispetto al tamoxifene nei tassi di risposta clinica e mammografica, nell’ecografia del seno e nel tasso di chirurgia conservativa della mammella (breast-conserving surgery, BCS). Il CHMP ha concluso quindi che i dati attuali, comprese le informazioni di follow-up a lungo termine, siano sufficienti a sostenere l’indicazione di trattamento neoadiuvante (pre-operatorio) per Femara, in particolare per rimpicciolire tumori con recettore ormonale positivo in modo di consentire la BCS o per far sì che tumori inoperabili diventino operabili. Il CHMP ha definito come popolazione destinataria le donne in stato di post-menopausa accertato e pazienti con tumori ER positivi, pazienti i cui tumori sono HER-2 (Human Epidermal Growth Factor Receptor 2, recettore del fattore di crescita epiteliale 2) negativo e pazienti che non sono in grado di ricevere chemioterapia neoadiuvante, o che si rifiutano di riceverla, nonché pazienti per cui l’intervento chirurgico immediato non è una possibile alternativa. In conclusione, sono state adottate per Femara le seguenti indicazioni armonizzate: “Sezione 4.1 – Indicazioni terapeutiche Trattamento adiuvante di donne in post-menopausa con tumore invasivo al seno in fase
iniziale positivo per il recettore dell’ormone. Trattamento adiuvante esteso del carcinoma mammario invasivo ormonosensibile in donne in
post-menopausa che abbiano ricevuto un precedente trattamento adiuvante standard con tamoxifene della durata di 5 anni.
Trattamento di prima linea di donne in post-menopausa con carcinoma mammario avanzato ormonosensibile.
Carcinoma mammario in fase avanzata in donne in post-menopausa naturale o artificialmente indotta, in seguito a recidiva o progressione della malattia che siano state trattate in precedenza con antiestrogeni.
Trattamento neoadiuvante di donne in post-menopausa con carcinoma mammario con recettore ormonale positivo, HER-2 negativo, in cui la chemioterapia non è adatta e l’intervento chirurgico immediato non è indicato.
L’efficacia non è stata dimostrata nelle pazienti con carcinoma mammario negativo per il recettore dell’ormone.
8

Sezione 4.2 - Posologia e metodo di somministrazione
Il CHMP ha approvato una dose giornaliera di 2,5 mg in pazienti adulte, sulla base di diversi studi dosimetrici (dose-finding). Riguardo all’uso nelle persone anziane, il CHMP, stando alle analisi dell’efficacia e della sicurezza condotte per tutti gli studi cardine su Femara, non ha ritenuto necessario modificare la dose. Riguardo alla durata del trattamento, il CHMP ha ritenuto che in base agli studi il trattamento con Femara debba continuare fino a quando la progressione della malattia non risulti evidente. Circa il trattamento adiuvante e adiuvante esteso, dagli studi è emerso che l’efficacia permanga sulla durata media di 5 anni di trattamento. Riguardo al trattamento neoadiuvante, studi recenti sulla durata della terapia, hanno evidenziato tassi di riposta più elevati e un maggior numero di pazienti idonee per la BCS a 4-8 mesi, con un tenue beneficio incrementale nel caso di una durata maggiore, con un minimo di 4-6 mesi. Riguardo ai bambini, il carcinoma mammario invasivo è estremamente raro in bambini e adolescenti, pur essendo stato riferito. In assenza di sperimentazioni cliniche, il CHMP ha concluso che la sicurezza e l’efficacia di Femara in questa popolazione non sia definita. Riguardo all’insufficienza renale il CHMP ha preso atto dei dati farmacocinetici disponibili, osservando come il letrozolo venga eliminato principalmente mediante il metabolismo epatico con clearance (eliminazione) renale riferita inferiore al 5%. Inoltre il CHMP ha osservato che, sebbene l’eliminazione dei metaboliti sia in genere più lenta in pazienti con insufficienza renale, dai dati disponibili risulta che ciò non crea ripercussioni sul profilo di sicurezza e non altera la farmacocinetica in pazienti con grave insufficienza renale. Il CHMP ha concluso che non è necessaria alcuna avvertenza specifica o raccomandazione di modificare la dose in pazienti con CrCl 10 ml/min. Riguardo all’insufficienza epatica, il CHMP ha osservato che in base agli studi Femara risulta sicuro in caso di insufficienza epatica da lieve a moderata; peraltro sussiste limitata esperienza di uso nei pazienti con funzionalità epatica gravemente compromessa, così come in quelli non cancerosi con cirrosi epatica e/o insufficienza epatica Child-Pugh C. Dal momento che il raddoppio dell’esposizione a letrozolo non desta preoccupazioni, in termini di sicurezza, e poiché la sotto-esposizione deve essere evitata, essendo dimostrato che l’efficacia dipende dal dosaggio, il CHMP ha concluso che non è necessario alcun adeguamento della dose in pazienti con grave insufficienza epatica, ma che debba essere inserita un’avvertenza nella sezione 4.4.
Sezione 4.3 – Controindicazioni
Riguardo allo stato endocrino di pre-menopausa, il CHMP ha osservato che, sebbene non sia stata condotta alcuna sperimentazione clinica per verificare la sicurezza o l’efficacia di Femara in donne in pre-menopausa, sussistono motivi di sicurezza per non trattare con letrozolo donne in pre-menopausa o in peri-menopausa. Il letrozolo inibisce l’enzima coinvolto nella sintesi degli estrogeni che sono necessari per il corretto sviluppo di embrione e feto. Si prevede quindi che dal letrozolo possano derivare potenziali effetti avversi sull’embrione-feto, come confermato in studi su ratti e conigli in gravidanza. Il CHMP ha concluso che sussistono timori significativi sulla sicurezza che giustificano la controindicazione in donne in pre-menopausa, specialmente in donne in stato di pre-menopausa endocrina. Riguardo all’insufficienza epatica, il CHMP ha ritenuto non idonea una rigida controindicazione per il letrozolo, che è un trattamento potenzialmente salva-vita con un profilo di sicurezza relativamente benigno. Inoltre il letrozolo non è considerato avere un indice terapeutico ristretto. Per contro, il CHMP ha incluso un’avvertenza nella sezione 4.4 in cui si dichiara che mancano dati di sicurezza in pazienti con significativa compromissione di organi. Riguardo all’uso preoperatorio del letrozolo, se lo stato del recettore è negativo o sconosciuto, il CHMP ha ritenuto che non fosse necessaria una controindicazione poiché questo è già sufficientemente spiegato nella sezione 4.4. Il CHMP ha chiarito inoltre le controindicazioni in caso di gravidanza e allattamento al seno, rendendo le informazioni più visibili e ha inserito una controindicazione per ipersensibilità nota al letrozolo o a uno qualunque degli eccipienti.
9

Sezione 4.4 - Avvertenze speciali e precauzioni per l’uso
Il CHMP ha introdotto un’avvertenza riguardo allo stato endocrino di post-menopausa perché studi hanno mostrato efficacia sub-ottimale, oltre a maggiore frequenza e gravità di eventi avversi in donne in stato di peri-menopausa. Riguardo all’insufficienza renale, il CHMP ha osservato che sussistono informazioni molto limitate su pazienti con clearance della creatinina inferiore a 10 ml/min e quindi si è detto concorde su una precauzione per tali pazienti. Riguardo all’insufficienza epatica, il CHMP ha rilevato che non sono riportati segnali riguardo alla sicurezza di Femara in pazienti con grave compromissione della funzionalità epatica. Anche se non era assicurata una controindicazione in tali pazienti, il CHMP ha inserito una precauzione. Riguardo agli effetti sulle ossa, il CHMP ha esaminato degli studi da cui emerge che l’uso a lungo termine di Femara è associato a un tasso significativamente più alto sia di osteoporosi sia di fratture ossee rispetto al tamoxifene (scenario adiuvante) o al placebo (scenario adiuvante esteso). Donne con una storia di fratture e/o osteoporosi hanno riportato tassi di fratture ossee più alti rispetto alle altre pazienti, indipendentemente dal trattamento. Il CHMP ha quindi aggiunto una dichiarazione riguardo agli effetti sulle ossa associati all’uso di Femara. Riguardo al cancro alla mammella negli uomini il CHMP ha preso atto dell’assenza di sperimentazioni cliniche o di specifiche indagini sistematiche sull’uso di Femara in questo tipo di cancro. Il CHMP ha spostato la dichiarazione dalla sezione 4.4 alla sezione 5.1. Il CHMP inoltre ha aggiunto un’avvertenza secondo cui estrogeni e/o tamoxifene possono abbassare i livelli di letrozolo nel plasma, riducendone quindi l’azione farmacologica per cui deve essere evitata la somministrazione concomitante. È stata ugualmente aggiunta una dichiarazione contraria all’uso di Femara in pazienti con problemi di intolleranza al galattosio, grave deficienza di lattosio o cattivo assorbimento di glucosio-galattosio.
Sezione 4.5 - Interazione con altri prodotti medicinali e altre forme di interazione
Il CHMP ha esaminato il potenziale di interazione di una serie di sostanze tra cui cimetidina e agenti anti-tumorali nonché il ruolo svolto da CYP2A6 e CYP3A4 nel metabolismo del letrozolo. Il CHMP ha ritenuto che le revisioni dei database clinici siano poco idonee a valutare il rischio di interazioni o a valutare rapporti di mancanza di interazione e pertanto ha cancellato i riferimenti a questi esami. Infine, il CHMP ha inserito una dichiarazione che sconsiglia una somministrazione concomitante di Femara con tamoxifene, altri anti-estrogeni o estrogeni. Sezione 4.6 – Gravidanza e allattamento
Il CHMP ha rilevato l’assenza di adeguate sperimentazioni cliniche in donne in gravidanza e che casi isolati di difetti alla nascita sono stati riportati in donne in gravidanza esposte a Femara, mentre studi non clinici hanno mostrato che letrozolo è embriotossico e fetotossico. Inoltre il CHMP ha espresso il parere che sussistono timori sulla ridotta efficacia di letrozolo in donne in pre-menopausa e peri-menopausa. Il CHMP ha osservato inoltre le limitazioni nell’uso di contraccettivi e ritenuto pertanto che lo stato di post-menopausa debba essere completamente definito prima e durante il trattamento con Femara e che il medicinale non debba essere usato in donne in cui lo stato di post-menopausa non sia completamente definito.
Altre sezioni
Per le rimanenti sezioni, il CHMP ha adottato una dicitura armonizzata in linea con gli RCP approvati a livello nazionale, sebbene siano state apportate piccole revisioni. In particolare, per la sezione 4.8 – Effetti indesiderati, la dicitura è stata notevolmente abbreviata e sintetizzata. La tabella delle reazioni avverse correlate al farmaco è stata strutturata secondo le linee guida dell’RCP e diverse reazioni avverse sono state spostate in una classe sistemica organica più appropriata. Riguardo alla descrizione di reazioni avverse selezionate, il CHMP ha convenuto di tenere separate le indicazioni metastatico, adiuvante e adiuvante esteso. Per la sezione 5.1 – Proprietà farmacodinamiche, la dicitura è stata sostanzialmente abbreviata in modo di concentrarsi sulle informazioni pertinenti per il medico prescrivente rispetto alle indicazioni approvate. È stata aggiunta anche una dichiarazione sull’assenza di studi circa l’uso di Femara nel carcinoma alla mammella negli uomini.
10

Motivi per la modifica al riassunto delle caratteristiche del prodotto, etichettatura e foglio illustrativo
La presente procedura di deferimento si basava su di un’armonizzazione del riassunto delle caratteristiche del prodotto, dell’etichettatura e del foglio illustrativo. Avendo considerato tutti i dati presentati dal titolare dell’autorizzazione all’immissione in commercio, le relazioni di valutazione del relatore e del correlatore nonché le discussioni scientifiche in seno al Comitato, il CHMP si è detto del parere che il rapporto rischi/benefici di Femara e denominazioni associate sia favorevole.
Considerato quanto segue:
la procedura di deferimento verteva sull’armonizzazione del riassunto delle caratteristiche del
prodotto, dell’etichettatura e del foglio illustrativo;
il riassunto delle caratteristiche del prodotto, l’etichettatura e il foglio illustrativo proposti dai
titolari dell’autorizzazione all’immissione in commercio sono stati valutati sulla base della
documentazione presentata e della discussione scientifica in seno al Comitato,
il CHMP ha raccomandato la modifica alle autorizzazioni all’immissione di commercio per le quali il riassunto delle caratteristiche del prodotto, l’etichettatura e il foglio illustrativo sono previsti nell’Allegato III per Femara e denominazioni associate (v. Allegato I).
11

Allegato III
Riassunto delle caratteristiche del prodotto, etichettatura e foglio illustrativo Nota: Questa versione di RCP, etichettattura e foglio illustrativo è valida al momento della decisione della Commissione. Dopo la decisione della Commissione le autorità competenti dello Stato Membro, in collegamento con lo Stato Membro di riferimento, aggiornerà le informazioni del prodotto come richiesto. Pertanto, questo RCP, etichettatura e foglio illustrativo potrebbero non necessariamente rappresentare il testo corrente.
12

RIASSUNTO DELLE CARATTERISTICHE DEL PRODOTTO
13

1. DENOMINAZIONE DEL MEDICINALE {Nome (di fantasia)} e denominazioni associate (Vedere Allegato I) dosaggio forma farmaceutica [Vedere Allegato I – Completare con i dati nazionali] 2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA [Completare con i dati nazionali] Per l’elenco completo degli eccipienti, vedere paragrafo 6.1. 3. FORMA FARMACEUTICA [Completare con i dati nazionali] 4. INFORMAZIONI CLINICHE 4.1 Indicazioni terapeutiche Trattamento adiuvante del carcinoma mammario invasivo in fase precoce in donne in
postmenopausa con stato recettoriale ormonale positivo. Trattamento adiuvante del carcinoma mammario ormonosensibile invasivo in donne in
postmenopausa dopo trattamento adiuvante standard con tamoxifene della durata di 5 anni. Trattamento di prima linea del carcinoma mammario ormonosensibile, in fase avanzata, in
donne in postmenopausa. Trattamento del carcinoma mammario in fase avanzata in donne in postmenopausa naturale o
artificialmente indotta, dopo ripresa o progressione della malattia che siano state trattate in precedenza con antiestrogeni.
Trattamento neoadiuvante in donne in postmenopausa con carcinoma mammario con stato recettoriale ormonale positivo, HER-2 negativo in cui la chemioterapia non è possibile e un immediato intervento chirurgico non è indicato.
L’efficacia non è stata dimostrata in pazienti con stato recettoriale ormonale negativo. 4.2 Posologia e modo di somministrazione Posologia Pazienti adulte ed anziane La dose raccomandata di {Nome (di fantasia)} è di 2.5 mg una volta al giorno. Non è richiesta alcuna modifica della dose nelle pazienti anziane. Nelle pazienti con carcinoma mammario avanzato o metastatico, il trattamento con {Nome (di fantasia)} deve essere continuato finché la progressione tumorale risulta evidente. Nel trattamento adiuvante e nel trattamento adiuvante dopo terapia standard con tamoxifene, il trattamento con {Nome (di fantasia)} deve essere continuato per 5 anni o fino alla comparsa di recidiva del tumore, a seconda di cosa si verifichi per prima. Nel trattamento adiuvante può essere considerato anche uno schema di trattamento sequenziale (letrozolo per 2 anni seguito da tamoxifene per 3 anni) (vedere paragrafi 4.4 e 5.1). Nel trattamento neoadiuvante, il trattamento con {Nome (di fantasia)} deve essere continuato da 4 a 8 mesi in modo da stabilire una riduzione ottimale del tumore. Se la risposta non è adeguata, il trattamento con {Nome (di fantasia)} deve essere interrotto e deve essere programmato l’intervento
14

chirurgico e/o devono essere discusse con la paziente ulteriori alternative terapeutiche. Popolazione pediatrica {Nome (di fantasia)} non è raccomandato per l’uso nei bambini e negli adolescenti. La sicurezza e l’efficacia di {Nome (di fantasia)} nei bambini e negli adolescenti di età maggiore di 17 anni non sono state ancora stabilite. Sono disponibili dati limitati e non può essere fatta alcuna raccomandazione riguardante la posologia. Compromissione renale Non è richiesta alcuna modifica della dose di {Nome (di fantasia)} per le pazienti con insufficienza renale con clearance della creatinina ≥10 ml/min. Non sono disponibili dati sufficienti nei casi di insufficienza renale con clearance della creatinina inferiori a 10 ml/min (vedere paragrafi 4.4 e 5.2). Compromissione epatica Non è richiesta alcuna modifica della dose di {Nome (di fantasia)} per le pazienti con insufficienza epatica da lieve a moderata (Child-Pugh A o B). Non sono disponibili dati sufficienti per pazienti con insufficienza epatica grave. Pazienti con insufficienza epatica grave (Child-Pugh C) richiedono uno stretto controllo (vedere paragrafi 4.4 e 5.2). Modo di somministrazione {Nome (di fantasia)} deve essere assunto per via orale e può essere assunto con o senza cibo. 4.3 Controindicazioni Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1 Stato ormonale premenopausale Gravidanza (vedere paragrafo 4.6) Allattamento (vedere paragrafo 4.6) 4.4 Avvertenze speciali e precauzioni di impiego Stato menopausale Nelle pazienti dove lo stato menopausale non è chiaro prima di iniziare il trattamento con {Nome (di fantasia)}devono essere misurati l’ormone luteinizzante (LH), l’ormone follicolo-stimolante (FSH) e/o l’estradiolo. Solo le donne con stato ormonale postmenopausale possono ricevere {Nome (di fantasia)}. Compromissione renale {Nome (di fantasia)} non è stato studiato in un numero sufficiente di pazienti con una clearance della creatinina inferiore a 10 ml/min. In tali pazienti deve essere attentamente considerato il potenziale rapporto rischio/beneficio prima della somministrazione di {Nome (di fantasia)}. Compromissione epatica In pazienti con grave compromissione epatica (Child-Pugh C), l’esposizione sistemica e l’emivita terminale sono approssimativamente doppie rispetto ai volontari sani. Queste pazienti devono quindi essere tenute sotto stretto controllo (vedere paragrafo 5.2). Effetti sull’osso {Nome (di fantasia)} è un potente agente di riduzione degli estrogeni. Le pazienti con anamnesi di osteoporosi e/o di fratture, o con aumentato rischio di osteoporosi, devono essere sottoposte ad una valutazione della densità minerale ossea prima dell’inizio del trattamento adiuvante e del trattamento adiuvante dopo terapia standard con tamoxifene e devono essere monitorate durante e dopo il trattamento con letrozolo. Il trattamento o la profilassi dell’osteoporosi devono essere iniziati in modo appropriato e monitorati attentamente. Nel trattamento adiuvante potrebbe essere considerato anche uno schema di trattamento sequenziale (letrozolo per 2 anni seguito da tamoxifene per 3 anni) sulla base del profilo di sicurezza della paziente (vedere paragrafi 4.2, 4.8 e 5.1).
15

Altre avvertenze La somministrazione concomitante di {Nome (di fantasia)} con tamoxifene, altri anti-estrogeni o terapie contenenti estrogeni deve essere evitata in quanto queste sostanze possono diminuire l’azione farmacologica del letrozolo (vedere paragrafo 4.5). Poiché le compresse contengono lattosio, {Nome (di fantasia)} non è raccomandato per pazienti affette da rari problemi ereditari di intolleranza al galattosio, da gravi deficit di lattasi o da malassorbimento di glucosio-galattosio. 4.5 Interazioni con altri medicinali ed altre forme di interazione Il metabolismo del letrozolo è mediato in parte dal CYP2A6 e dal CYP3A4. La cimetidina, un inibitore debole aspecifico degli enzimi CYP450, non ha influenzato le concentrazioni plasmatiche del letrozolo. L’effetto degli inibitori potenti di CYP450 non è noto. Ad oggi non vi è esperienza clinica sull’uso di {Nome (di fantasia)} in combinazione con estrogeni o altri agenti antineoplastici, oltre che il tamoxifene. Il tamoxifene, altri anti-estrogeni o terapie contenenti estrogeni possono diminuire l’azione farmacologica del letrozolo. In aggiunta la somministrazione concomitante di tamoxifene con il letrozolo ha dimostrato di ridurre sostanzialmente le concentrazioni plasmatiche del letrozolo. La somministrazione concomitante di letrozolo con tamoxifene, altri agenti anti-estrogeni o estrogeni deve essere evitata. In vitro, il letrozolo inibisce gli isoenzimi 2A6 e, moderatamente il 2C19 del citocromo P450, ma la rilevanza clinica non è nota. Pertanto, deve essere usata cautela qualora sia necessario somministrare il letrozolo in concomitanza con medicinali la cui eliminazione dipende soprattutto da questi isoenzimi e il cui indice terapeutico è ristretto (es.. fenitoina, clopidrogel). 4.6 Fertilità, gravidanza e allattamento Donne in stato perimenopausale o in età fertile {Nome (di fantasia)} deve essere usato solo in donne con uno stato di post-menopausa definito chiaramente (vedere paragrafo 4.4). Poiché vi sono segnalazioni di donne che hanno recuperato la funzione ovarica durante il trattamento con {Nome (di fantasia)} nonostante un chiaro stato postmenopausale all’inizio della terapia, il medico deve discutere di una contraccezione adeguata in caso di necessità. Gravidanza Sulla base dei dati sull’uomo in cui ci sono stati casi isolati di difetti alla nascita (fusione labiale, genitali ambigui), {Nome (di fantasia)} può causare malformazioni congenite quando somministrato durante la gravidanza. Gli studi su animali hanno mostrato una tossicità riproduttiva (vedere paragrafo 5.3). {Nome (di fantasia)} è controindicato durante la gravidanza (vedere paragrafi 4.3 e 5.3). Allattamento Non è noto se il letrozolo/metaboliti siano escreti nel latte materno. Il rischio per i neonati/lattanti non può essere escluso. {Nome (di fantasia)} è controindicato durante l’allattamento (vedere paragrafo 4.3). Fertilità L’azione farmacologica del letrozolo è di ridurre la produzione di estrogeni attraverso l’inibizione dell’aromatasi. Nelle donne in premenopausa, l’inibizione della sintesi degli estrogeni determina come risposta aumenti nei livelli di gonadotropine (LH, FSH). Gli aumentati livelli di FSH stimolano a loro volta la crescita follicolare e possono indurre l’ovulazione.
16

4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari {Nome (di fantasia)} altera lievemente la capacità di guidare veicoli o di usare macchinari. Deve essere usata cautela quando si guidano o si usano macchinari dal momento che con l’uso di {Nome (di fantasia)}sono stati riportati stanchezza e capogiri e non comunemente sonnolenza. 4.8 Effetti indesiderati Riassunto del profilo di sicurezza Le frequenze delle reazioni avverse per {Nome (di fantasia)} sono principalmente basate su dati raccolti da studi clinici. Fino a circa un terzo delle pazienti trattate con {Nome (di fantasia)} nella fase metastatica e circa l’80% delle pazienti in trattamento adiuvante, così come nel trattamento adiuvante dopo terapia standard con tamoxifene, hanno manifestato delle reazioni avverse. La maggior parte delle reazioni avverse si sono manifestate durante le prime settimane di trattamento. Le reazioni avverse riportate con maggiore frequenza negli studi clinici sono state vampate, ipercolesterolemia, artralgia, affaticamento, aumento della sudorazione e nausea. Ulteriori reazioni avverse importanti che si possono manifestare con {Nome (di fantasia)} sono: eventi scheletrici come osteoporosi e/o fratture ossee ed eventi cardiovascolari (comprendenti eventi cerebrovascolari e tromboembolici). La categoria di frequenza per queste reazioni avverse è descritta in Tabella 1. Elenco in tabella delle reazioni avverse Le frequenze delle reazioni avverse per {Nome (di fantasia)} sono principalmente basate su dati raccolti da studi clinici. Le seguenti reazioni avverse, elencate in Tabella 1, sono state segnalate dagli studi clinici e dall’esperienza successiva alla commercializzazione di {Nome (di fantasia)}: Tabella 1 Le reazioni avverse sono classificate all’interno di ciascuna classe di frequenza, in ordine decrescente di frequenza, usando la seguente convenzione: molto comune 10%, comune da 1% a 10%, non comune da 0.1% a 1%, raro da 0.01% a 0.1%, molto raro 0.01%, non nota (la frequenza non può essere definita sulla base dei dati disponibili). Infezioni ed infestazioni Non comune: Infezione del tratto urinario Tumori benigni, maligni e non specificati (cisti e polipi compresi) Non comune: Dolore tumorale1 Patologie del sistema emolinfopoietico Non comune: Leucopenia Disturbi del sistema immunitario Non nota: Reazioni anafilattiche Disturbi del metabolismo e della nutrizione Molto comune: Ipercolesterolemia Comune: Anoressia, aumento dell’appetito Disturbi psichiatrici Comune: Depressione Non comune: Ansietà (incluso nervosismo), irritabilità Patologie del sistema nervoso Comune: Cefalea, capogiri Non comune: Sonnolenza, insonnia, compromissione della memoria, disestesia (incluse
parestesia, ipoestesia), alterazione del gusto, accidente cerebrovascolare
17
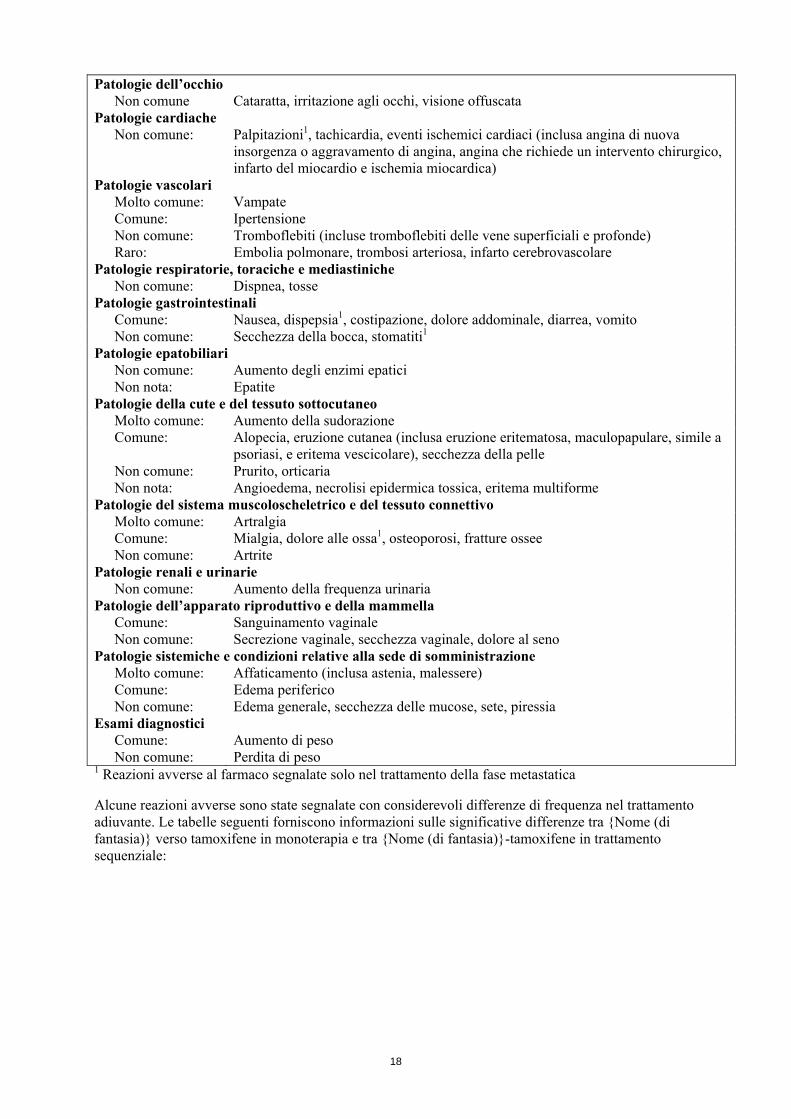
Patologie dell’occhio Non comune Cataratta, irritazione agli occhi, visione offuscata Patologie cardiache Non comune: Palpitazioni1, tachicardia, eventi ischemici cardiaci (inclusa angina di nuova
insorgenza o aggravamento di angina, angina che richiede un intervento chirurgico, infarto del miocardio e ischemia miocardica)
Patologie vascolari Molto comune: Vampate Comune: Ipertensione Non comune: Tromboflebiti (incluse tromboflebiti delle vene superficiali e profonde) Raro: Embolia polmonare, trombosi arteriosa, infarto cerebrovascolare Patologie respiratorie, toraciche e mediastiniche Non comune: Dispnea, tosse Patologie gastrointestinali Comune: Nausea, dispepsia1, costipazione, dolore addominale, diarrea, vomito Non comune: Secchezza della bocca, stomatiti1 Patologie epatobiliari Non comune: Aumento degli enzimi epatici Non nota: Epatite Patologie della cute e del tessuto sottocutaneo Molto comune: Aumento della sudorazione Comune: Alopecia, eruzione cutanea (inclusa eruzione eritematosa, maculopapulare, simile a
psoriasi, e eritema vescicolare), secchezza della pelle Non comune: Prurito, orticaria Non nota: Angioedema, necrolisi epidermica tossica, eritema multiforme Patologie del sistema muscoloscheletrico e del tessuto connettivo Molto comune: Artralgia Comune: Mialgia, dolore alle ossa1, osteoporosi, fratture ossee Non comune: Artrite Patologie renali e urinarie Non comune: Aumento della frequenza urinaria Patologie dell’apparato riproduttivo e della mammella Comune: Sanguinamento vaginale Non comune: Secrezione vaginale, secchezza vaginale, dolore al seno Patologie sistemiche e condizioni relative alla sede di somministrazione Molto comune: Affaticamento (inclusa astenia, malessere) Comune: Edema periferico Non comune: Edema generale, secchezza delle mucose, sete, piressia Esami diagnostici Comune: Aumento di peso Non comune: Perdita di peso 1 Reazioni avverse al farmaco segnalate solo nel trattamento della fase metastatica Alcune reazioni avverse sono state segnalate con considerevoli differenze di frequenza nel trattamento adiuvante. Le tabelle seguenti forniscono informazioni sulle significative differenze tra {Nome (di fantasia)} verso tamoxifene in monoterapia e tra {Nome (di fantasia)}-tamoxifene in trattamento sequenziale:
18
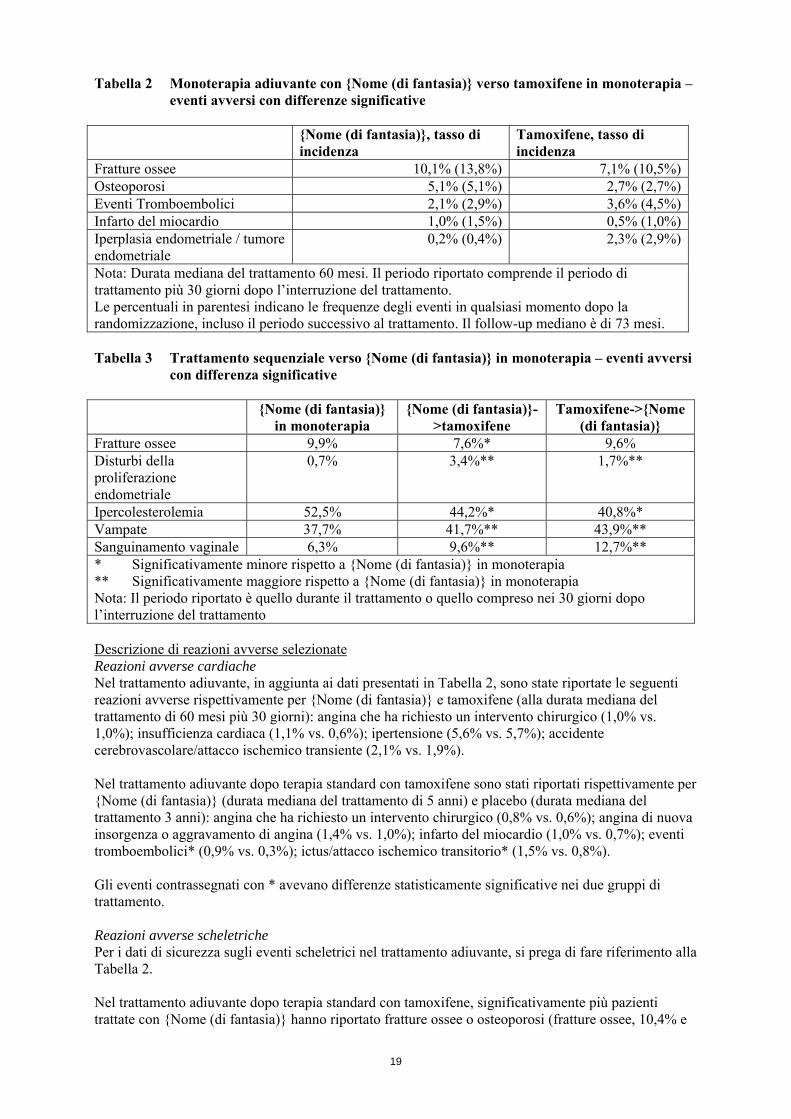
Tabella 2 Monoterapia adiuvante con {Nome (di fantasia)} verso tamoxifene in monoterapia – eventi avversi con differenze significative
{Nome (di fantasia)}, tasso di
incidenza Tamoxifene, tasso di incidenza
Fratture ossee 10,1% (13,8%) 7,1% (10,5%)Osteoporosi 5,1% (5,1%) 2,7% (2,7%)Eventi Tromboembolici 2,1% (2,9%) 3,6% (4,5%)Infarto del miocardio 1,0% (1,5%) 0,5% (1,0%)Iperplasia endometriale / tumore endometriale
0,2% (0,4%) 2,3% (2,9%)
Nota: Durata mediana del trattamento 60 mesi. Il periodo riportato comprende il periodo di trattamento più 30 giorni dopo l’interruzione del trattamento. Le percentuali in parentesi indicano le frequenze degli eventi in qualsiasi momento dopo la randomizzazione, incluso il periodo successivo al trattamento. Il follow-up mediano è di 73 mesi. Tabella 3 Trattamento sequenziale verso {Nome (di fantasia)} in monoterapia – eventi avversi
con differenza significative {Nome (di fantasia)}
in monoterapia {Nome (di fantasia)}-
>tamoxifene Tamoxifene->{Nome
(di fantasia)} Fratture ossee 9,9% 7,6%* 9,6% Disturbi della proliferazione endometriale
0,7% 3,4%** 1,7%**
Ipercolesterolemia 52,5% 44,2%* 40,8%* Vampate 37,7% 41,7%** 43,9%** Sanguinamento vaginale 6,3% 9,6%** 12,7%** * Significativamente minore rispetto a {Nome (di fantasia)} in monoterapia ** Significativamente maggiore rispetto a {Nome (di fantasia)} in monoterapia Nota: Il periodo riportato è quello durante il trattamento o quello compreso nei 30 giorni dopo l’interruzione del trattamento Descrizione di reazioni avverse selezionate Reazioni avverse cardiache Nel trattamento adiuvante, in aggiunta ai dati presentati in Tabella 2, sono state riportate le seguenti reazioni avverse rispettivamente per {Nome (di fantasia)} e tamoxifene (alla durata mediana del trattamento di 60 mesi più 30 giorni): angina che ha richiesto un intervento chirurgico (1,0% vs. 1,0%); insufficienza cardiaca (1,1% vs. 0,6%); ipertensione (5,6% vs. 5,7%); accidente cerebrovascolare/attacco ischemico transiente (2,1% vs. 1,9%). Nel trattamento adiuvante dopo terapia standard con tamoxifene sono stati riportati rispettivamente per {Nome (di fantasia)} (durata mediana del trattamento di 5 anni) e placebo (durata mediana del trattamento 3 anni): angina che ha richiesto un intervento chirurgico (0,8% vs. 0,6%); angina di nuova insorgenza o aggravamento di angina (1,4% vs. 1,0%); infarto del miocardio (1,0% vs. 0,7%); eventi tromboembolici* (0,9% vs. 0,3%); ictus/attacco ischemico transitorio* (1,5% vs. 0,8%). Gli eventi contrassegnati con * avevano differenze statisticamente significative nei due gruppi di trattamento. Reazioni avverse scheletriche Per i dati di sicurezza sugli eventi scheletrici nel trattamento adiuvante, si prega di fare riferimento alla Tabella 2. Nel trattamento adiuvante dopo terapia standard con tamoxifene, significativamente più pazienti trattate con {Nome (di fantasia)} hanno riportato fratture ossee o osteoporosi (fratture ossee, 10,4% e
19

osteoporosi 12,2%) rispetto ai pazienti nel gruppo (rispettivamente 5,8% e 6,4%). La durata mediana del trattamento era di 5 anni per {Nome (di fantasia)}, rispetto a 3 anni per il placebo. 4.9 Sovradosaggio Sono stati segnalati isolati casi di sovradosaggio con {Nome (di fantasia)}. Non è noto alcun trattamento specifico per il sovradosaggio; il trattamento deve essere sintomatico e di supporto. 5. PROPRIETÀ FARMACOLOGICHE 5.1 Proprietà farmacodinamiche Categoria farmacoterapeutica: Terapie endocrine. Ormone antagonista e agenti correlati: inibitore dell’aromatasi, codice ATC: L02BG04. Effetti farmacodinamici L’inibizione della stimolazione della crescita cellulare mediata da estrogeni è un prerequisito per la risposta tumorale nei casi in cui la crescita del tessuto tumorale dipenda dalla presenza di estrogeni e sia utilizzata la terapia endocrina. Nelle donne in postmenopausa, gli estrogeni derivano principalmente dall’azione dell’enzima dell’aromatasi, che converte gli estrogeni surrenalici - principalmente l’androstenedione ed il testosterone - in estrone e estradiolo. La soppressione della biosintesi di estrogeni nei tessuti periferici e nel tessuto neoplastico stesso può pertanto essere ottenuta mediante l’inibizione specifica dell’enzima aromatasi. Il letrozolo è un inibitore non steroideo dell’aromatasi. Esso inibisce l’enzima aromatasi legandosi completamente all’eme del citocromo P450, con conseguente riduzione della biosintesi di estrogeni in tutti i tessuti dove è presente. Nelle donne sane in postmenopausa la somministrazione di dosi singole di 0,1 mg, 0,5 mg, e 2,5 mg di letrozolo sopprimono i livelli serici di estrone e di estradiolo rispettivamente del 75%-78% e del 78% rispetto ai valori basali. La soppressione massima viene raggiunta entro 48-78 ore. Nelle pazienti in postmenopausa con carcinoma mammario in fase avanzata, dosi giornaliere di 0,1-5 mg sopprimono le concentrazioni plasmatiche di estradiolo, estrone ed estrone solfato del 75-95% rispetto ai valori basali in tutte le pazienti trattate. A dosi pari a 0,5 mg e oltre, molti valori di estrone ed estrone solfato risultano inferiori alla soglia di sensibilità del saggio; il che significa che, a queste dosi, si ottiene una maggiore soppressione della produzione estrogenica. Tale soppressione è stata mantenuta per tutta la durata del trattamento in tutte le pazienti. L'inibizione dell'attività dell'aromatasi da parte del letrozolo è altamente specifica. Non è stata rilevata alcuna compromissione della steroidogenesi surrenalica. Non sono stati rilevati cambiamenti clinicamente rilevanti delle concentrazioni plasmatiche di cortisolo, aldosterone, 11-deossicortisolo, 17-idrossi-progesterone e ACTH, nonché dell’attività della renina plasmatica nelle pazienti in postmenopausa trattate con una dose giornaliera di 0,1-5 mg di letrozolo. Il test di stimolazione con ACTH, eseguito dopo 6 e 12 settimane di trattamento con somministrazioni giornaliere di 0,1 mg, 0,25 mg, 0,5 mg, 1 mg, 2,5 mg e 5 mg, non ha indicato alcuna riduzione della produzione di aldosterone o di cortisolo. Conseguentemente, non è stato necessario somministrare integratori a base di glucocorticoidi e mineralcorticoidi. Non sono stati osservati cambiamenti nelle concentrazioni plasmatiche di androgeni (androstenedione e testosterone) tra donne sane in postmenopausa dopo singole dosi di 0,1 mg, 0,5 mg, e 2,5 mg di letrozolo o nelle concentrazioni plasmatiche di androstenedione tra pazienti in postmenopausa trattate con dosi giornaliere da 0,1 mg a 5 mg, indicando che il blocco della biosintesi di estrogeni non determina accumulo di precursori androgenici. Né i livelli plasmatici di LH e FSH, né la funzione
20
tiroidea, valutata in base al TSH e al test dell’uptake di T3 e T4, vengono influenzati dal letrozolo. Trattamento adiuvante Studio BIG 1-98 BIG 1-98 è uno studio multicentrico, in doppio cieco in cui più di 8.000 donne in postmenopausa con carcinoma della mammella in fase precoce con recettori ormonali positivi sono state randomizzate ad uno dei seguenti trattamenti: A. tamoxifene per 5 anni; B. {Nome (di fantasia)} per 5 anni; C. tamoxifene per 2 anni seguito da {Nome (di fantasia)} per 3 anni; D. {Nome (di fantasia)} per 2 anni seguito da tamoxifene per 3 anni. L’endpoint primario era la sopravvivenza libera da malattia (DFS); gli endpoints secondari di efficacia erano tempo alle metastasi a distanza (TDM), sopravvivenza libera da malattia a distanza (DDFS), sopravvivenza globale (OS), sopravvivenza libera da malattia sistemica (SDFS), tasso di carcinoma invasivo mammario controlaterale e tempo alla recidiva di carcinoma mammario. Risultati di efficacia al follow-up mediano di 26 e 60 mesi I dati in Tabella 4 riflettono i risultati del Primary Core Analysis (PCA) basati sui dati dei gruppi in monoterapia (A and B) e sui dati dei due gruppi in cui era previsto lo switch (C and D) ad un trattamento della durata mediana di 24 mesi ed un follow-up mediano di 26 mesi e ad un trattamento della durata mediana di 32 mesi ed un follow-up mediano di 60 mesi. Le frequenze per la DFS a 5-anni erano dell’84% per {Nome (di fantasia)} e dell’81,4% per il tamoxifene. Tabella 4 Primary Core Analysis: Sopravvivenza libera da malattia e globale, ad un follow-up
mediano di 26 mesi ed ad un follow-up mediano di 60 mesi (Popolazione ITT) Primary Core Analysis Follow-up mediano 26 mesi Follow-up mediano 60 mesi {Nome (di
fantasia)}N=4003
TamoxifeneN=4007
HR1 (95% IC)
P
{Nome (di fantasia)} N=4003
TamoxifeneN=4007
HR1 (95% IC)
P
Sopravvivenza libera da malattia (endpoint primario) - eventi (definizione da protocollo2)
351 428 0,81 (0,70, 0,93)
0,003
585 664 0,86 (0,77, 0,96)
0,008
Sopravvivenza globale (endpoint secondario) decessi
166 192 0,86
(0,70, 1,06)
330 374 0,87
(0,75, 1,01) HR = Hazard ratio; IC = intervallo di confidenza 1 Logrank test, stratificato per randomizzazione e uso di chemioterapia (si/no) 2 Eventi DFS: recidiva loco-regionale, metastasi a distanza, cancro invasivo mammario controlaterale,
secondo tumore maligno primario(non al seno), decesso per qualsiasi causa senza un precedente evento tumorale.
Risultati ad un follow-up mediano di 73 mesi (solo gruppi in monoterapia) L’analisi dei gruppi in monoterapia (MAA) con aggiornamento a lungo termine dell’efficacia della monoterapia con {Nome (di fantasia)} rispetto alla monoterapia con tamoxifene (durata mediana del trattamento adiuvante: 5 anni) e presentata nella Tabella 5.
21
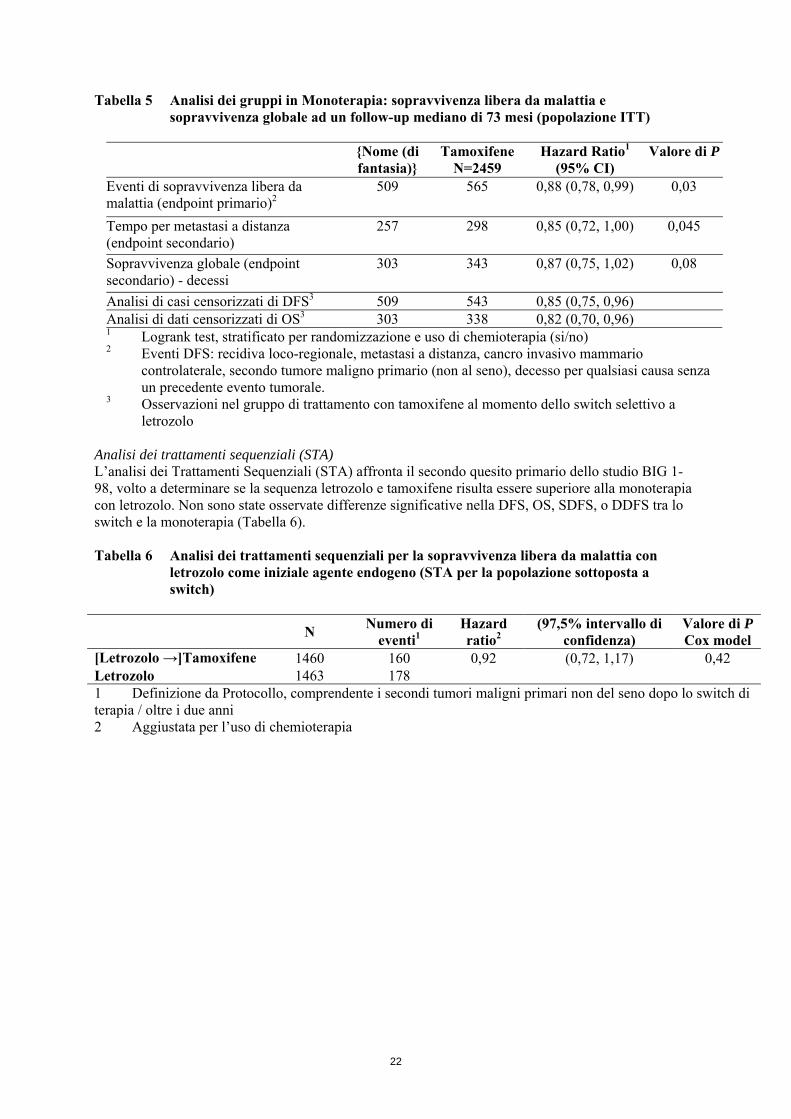
Tabella 5 Analisi dei gruppi in Monoterapia: sopravvivenza libera da malattia e
sopravvivenza globale ad un follow-up mediano di 73 mesi (popolazione ITT)
{Nome (di fantasia)}
Tamoxifene N=2459
Hazard Ratio1 (95% CI)
Valore di P
Eventi di sopravvivenza libera da malattia (endpoint primario)2
509 565 0,88 (0,78, 0,99) 0,03
Tempo per metastasi a distanza (endpoint secondario)
257 298 0,85 (0,72, 1,00) 0,045
Sopravvivenza globale (endpoint secondario) - decessi
303 343 0,87 (0,75, 1,02) 0,08
Analisi di casi censorizzati di DFS3 509 543 0,85 (0,75, 0,96) Analisi di dati censorizzati di OS3 303 338 0,82 (0,70, 0,96) 1 Logrank test, stratificato per randomizzazione e uso di chemioterapia (si/no) 2 Eventi DFS: recidiva loco-regionale, metastasi a distanza, cancro invasivo mammario
controlaterale, secondo tumore maligno primario (non al seno), decesso per qualsiasi causa senza un precedente evento tumorale.
3 Osservazioni nel gruppo di trattamento con tamoxifene al momento dello switch selettivo a letrozolo
Analisi dei trattamenti sequenziali (STA) L’analisi dei Trattamenti Sequenziali (STA) affronta il secondo quesito primario dello studio BIG 1-98, volto a determinare se la sequenza letrozolo e tamoxifene risulta essere superiore alla monoterapia con letrozolo. Non sono state osservate differenze significative nella DFS, OS, SDFS, o DDFS tra lo switch e la monoterapia (Tabella 6). Tabella 6 Analisi dei trattamenti sequenziali per la sopravvivenza libera da malattia con
letrozolo come iniziale agente endogeno (STA per la popolazione sottoposta a switch)
N Numero di eventi1
Hazard ratio2
(97,5% intervallo di confidenza)
Valore di P Cox model
[Letrozolo →]Tamoxifene 1460 160 0,92 (0,72, 1,17) 0,42 Letrozolo 1463 178 1 Definizione da Protocollo, comprendente i secondi tumori maligni primari non del seno dopo lo switch di terapia / oltre i due anni 2 Aggiustata per l’uso di chemioterapia
22
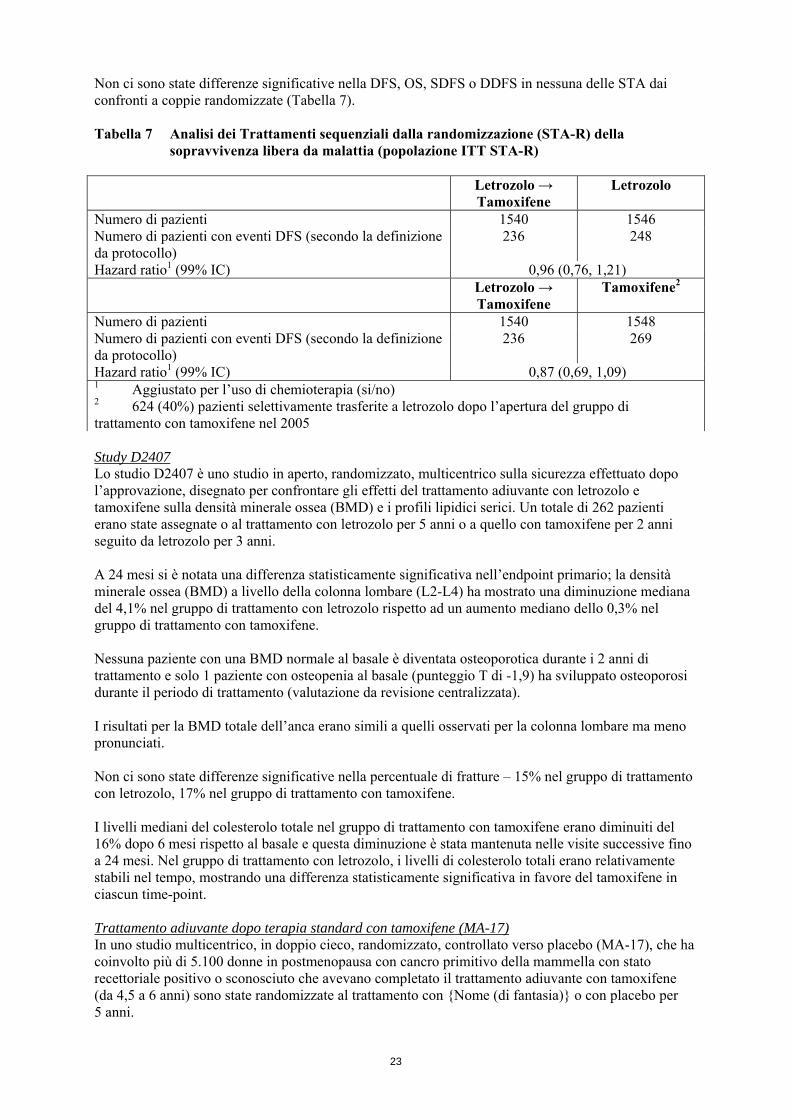
Non ci sono state differenze significative nella DFS, OS, SDFS o DDFS in nessuna delle STA dai confronti a coppie randomizzate (Tabella 7). Tabella 7 Analisi dei Trattamenti sequenziali dalla randomizzazione (STA-R) della
sopravvivenza libera da malattia (popolazione ITT STA-R) Letrozolo →
Tamoxifene Letrozolo
Numero di pazienti 1540 1546 Numero di pazienti con eventi DFS (secondo la definizione da protocollo)
236 248
Hazard ratio1 (99% IC) 0,96 (0,76, 1,21) Letrozolo →
Tamoxifene Tamoxifene2
Numero di pazienti 1540 1548 Numero di pazienti con eventi DFS (secondo la definizione da protocollo)
236 269
Hazard ratio1 (99% IC) 0,87 (0,69, 1,09) 1 Aggiustato per l’uso di chemioterapia (si/no) 2 624 (40%) pazienti selettivamente trasferite a letrozolo dopo l’apertura del gruppo di trattamento con tamoxifene nel 2005 Study D2407 Lo studio D2407 è uno studio in aperto, randomizzato, multicentrico sulla sicurezza effettuato dopo l’approvazione, disegnato per confrontare gli effetti del trattamento adiuvante con letrozolo e tamoxifene sulla densità minerale ossea (BMD) e i profili lipidici serici. Un totale di 262 pazienti erano state assegnate o al trattamento con letrozolo per 5 anni o a quello con tamoxifene per 2 anni seguito da letrozolo per 3 anni. A 24 mesi si è notata una differenza statisticamente significativa nell’endpoint primario; la densità minerale ossea (BMD) a livello della colonna lombare (L2-L4) ha mostrato una diminuzione mediana del 4,1% nel gruppo di trattamento con letrozolo rispetto ad un aumento mediano dello 0,3% nel gruppo di trattamento con tamoxifene. Nessuna paziente con una BMD normale al basale è diventata osteoporotica durante i 2 anni di trattamento e solo 1 paziente con osteopenia al basale (punteggio T di -1,9) ha sviluppato osteoporosi durante il periodo di trattamento (valutazione da revisione centralizzata). I risultati per la BMD totale dell’anca erano simili a quelli osservati per la colonna lombare ma meno pronunciati. Non ci sono state differenze significative nella percentuale di fratture – 15% nel gruppo di trattamento con letrozolo, 17% nel gruppo di trattamento con tamoxifene. I livelli mediani del colesterolo totale nel gruppo di trattamento con tamoxifene erano diminuiti del 16% dopo 6 mesi rispetto al basale e questa diminuzione è stata mantenuta nelle visite successive fino a 24 mesi. Nel gruppo di trattamento con letrozolo, i livelli di colesterolo totali erano relativamente stabili nel tempo, mostrando una differenza statisticamente significativa in favore del tamoxifene in ciascun time-point. Trattamento adiuvante dopo terapia standard con tamoxifene (MA-17) In uno studio multicentrico, in doppio cieco, randomizzato, controllato verso placebo (MA-17), che ha coinvolto più di 5.100 donne in postmenopausa con cancro primitivo della mammella con stato recettoriale positivo o sconosciuto che avevano completato il trattamento adiuvante con tamoxifene (da 4,5 a 6 anni) sono state randomizzate al trattamento con {Nome (di fantasia)} o con placebo per 5 anni.
23

L’endpoint primario era la sopravvivenza libera da malattia, definita come l’intervallo tra la randomizzazione e il primo evento di recidiva loco-regionale, metastasi a distanza o carcinoma mammario controlaterale. La prima analisi ad interim programmata ad un follow-up mediano di circa 28 mesi (il 25% dei pazienti era seguito per almeno 38 mesi), ha dimostrato che {Nome (di fantasia)} ha significativamente ridotto il rischio di recidiva di cancro mammario del 42% rispetto al placebo (HR 0,58; 95% IC 0,45, 0,76; P=0,00003). Il beneficio in favore di letrozolo è stato osservato indipendentemente dallo stato linfonodale. Non ci sono state differenze significative nella sopravvivenza globale: ({Nome (di fantasia)} 51 decessi; placebo 62; HR 0,82; 95% IC 0,56, 1,19). Di conseguenza, dopo la prima analisi ad interim lo studio è continuato in aperto e, i pazienti nel gruppo di trattamento con placebo sono stati autorizzati al passaggio a {Nome (di fantasia)} per 5 anni. Oltre il 60% delle pazienti eleggibili (libere da malattia all’apertura dello studio) ha scelto di passare a {Nome (di fantasia)}. L’analisi finale ha incluso 1.551 donne che sono passate dal placebo a {Nome (di fantasia)} in un periodo mediano di 31 mesi (intervallo da 12 a 106 mesi) dopo il completamento della terapia adiuvante con tamoxifene. La durata mediana del trattamento con {Nome (di fantasia)}era di 40 mesi. Le analisi finali condotte ad un follow-up mediano di 62 mesi hanno confermato la significativa riduzione del rischio di recidiva di carcinoma mammario con {Nome (di fantasia)}.
24
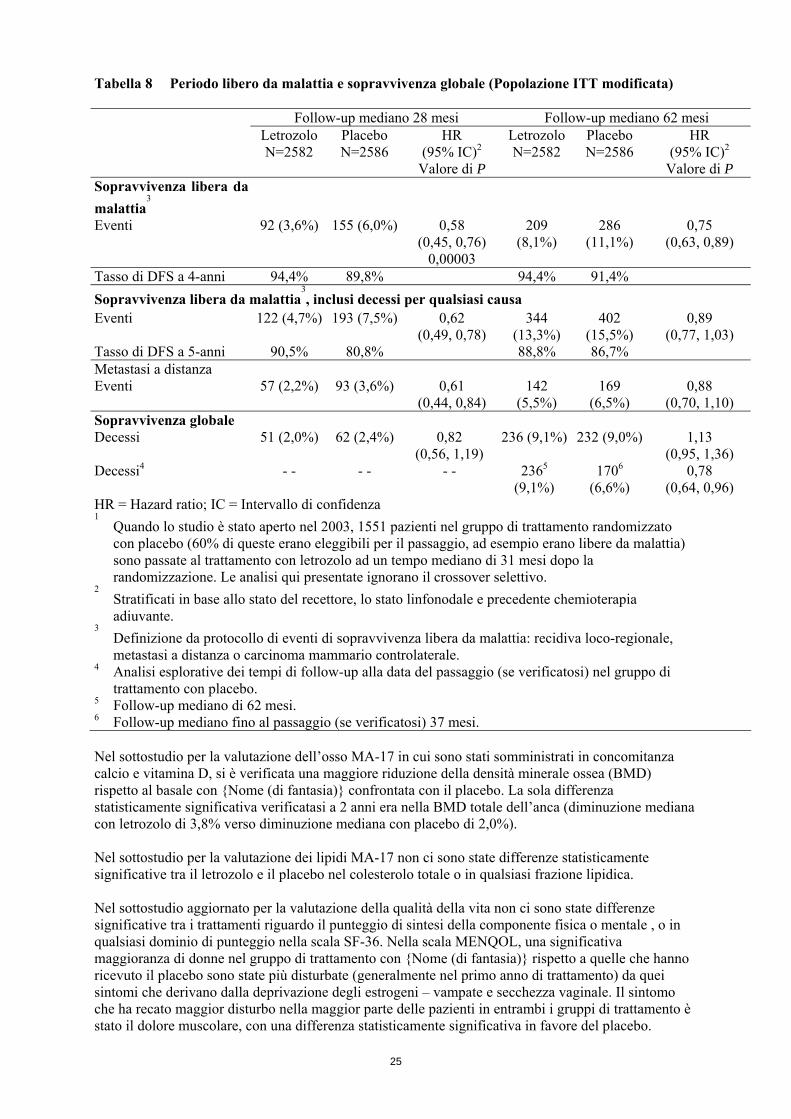
Tabella 8 Periodo libero da malattia e sopravvivenza globale (Popolazione ITT modificata) Follow-up mediano 28 mesi Follow-up mediano 62 mesi Letrozolo
N=2582 Placebo N=2586
HR (95% IC)2
Valore di P
Letrozolo N=2582
Placebo N=2586
HR (95% IC)2
Valore di P Sopravvivenza libera da
malattia3
Eventi 92 (3,6%) 155 (6,0%) 0,58 (0,45, 0,76)
0,00003
209 (8,1%)
286 (11,1%)
0,75 (0,63, 0,89)
Tasso di DFS a 4-anni 94,4% 89,8% 94,4% 91,4% Sopravvivenza libera da malattia
3, inclusi decessi per qualsiasi causa
Eventi 122 (4,7%) 193 (7,5%) 0,62 (0,49, 0,78)
344 (13,3%)
402 (15,5%)
0,89 (0,77, 1,03)
Tasso di DFS a 5-anni 90,5% 80,8% 88,8% 86,7% Metastasi a distanza Eventi 57 (2,2%) 93 (3,6%) 0,61
(0,44, 0,84) 142
(5,5%) 169
(6,5%) 0,88
(0,70, 1,10) Sopravvivenza globale Decessi 51 (2,0%) 62 (2,4%) 0,82
(0,56, 1,19) 236 (9,1%) 232 (9,0%) 1,13
(0,95, 1,36) Decessi4 - - - - - - 2365
(9,1%) 1706
(6,6%) 0,78
(0,64, 0,96) HR = Hazard ratio; IC = Intervallo di confidenza 1 Quando lo studio è stato aperto nel 2003, 1551 pazienti nel gruppo di trattamento randomizzato
con placebo (60% di queste erano eleggibili per il passaggio, ad esempio erano libere da malattia) sono passate al trattamento con letrozolo ad un tempo mediano di 31 mesi dopo la randomizzazione. Le analisi qui presentate ignorano il crossover selettivo.
2 Stratificati in base allo stato del recettore, lo stato linfonodale e precedente chemioterapia adiuvante.
3 Definizione da protocollo di eventi di sopravvivenza libera da malattia: recidiva loco-regionale, metastasi a distanza o carcinoma mammario controlaterale.
4 Analisi esplorative dei tempi di follow-up alla data del passaggio (se verificatosi) nel gruppo di trattamento con placebo.
5 Follow-up mediano di 62 mesi. 6 Follow-up mediano fino al passaggio (se verificatosi) 37 mesi. Nel sottostudio per la valutazione dell’osso MA-17 in cui sono stati somministrati in concomitanza calcio e vitamina D, si è verificata una maggiore riduzione della densità minerale ossea (BMD) rispetto al basale con {Nome (di fantasia)} confrontata con il placebo. La sola differenza statisticamente significativa verificatasi a 2 anni era nella BMD totale dell’anca (diminuzione mediana con letrozolo di 3,8% verso diminuzione mediana con placebo di 2,0%). Nel sottostudio per la valutazione dei lipidi MA-17 non ci sono state differenze statisticamente significative tra il letrozolo e il placebo nel colesterolo totale o in qualsiasi frazione lipidica. Nel sottostudio aggiornato per la valutazione della qualità della vita non ci sono state differenze significative tra i trattamenti riguardo il punteggio di sintesi della componente fisica o mentale , o in qualsiasi dominio di punteggio nella scala SF-36. Nella scala MENQOL, una significativa maggioranza di donne nel gruppo di trattamento con {Nome (di fantasia)} rispetto a quelle che hanno ricevuto il placebo sono state più disturbate (generalmente nel primo anno di trattamento) da quei sintomi che derivano dalla deprivazione degli estrogeni – vampate e secchezza vaginale. Il sintomo che ha recato maggior disturbo nella maggior parte delle pazienti in entrambi i gruppi di trattamento è stato il dolore muscolare, con una differenza statisticamente significativa in favore del placebo.
25

Trattamento neoadiuvante Uno studio in doppio cieco (P024) è stato condotto in 337 pazienti in postmenopausa con carcinoma mammario randomizzate per ricevere ognuna {Nome (di fantasia)} 2,5 mg per 4 mesi o tamoxifene per 4 mesi. Al basale tutte le pazienti avevano tumori allo stadio T2-T4c, N0-2, M0, ER e/o PgR positivo e nessuna delle pazienti poteva essere eleggibile per la chirurgia conservativa del seno. Sulla base della valutazione clinica sono state registrate risposte obiettive nel 55% del gruppo di trattamento con {Nome (di fantasia)} verso il 36% del gruppo di trattamento con tamoxifene (P<0,001). Questo risultato è stato consistentemente confermato dall’ecografia ({Nome (di fantasia)} 35% verso tamoxifene 25%, P=0,04) e dalla mammografia ({Nome (di fantasia)} 34% verso tamoxifene 16%, P<0,001). In totale il 45% delle pazienti nel gruppo di trattamento con {Nome (di fantasia)} verso il 35% delle pazienti nel gruppo di trattamento con tamoxifene (P=0,02) sono state sottoposte a chirurgia conservativa del seno. Durante i 4-mesi del periodo di trattamento preoperatorio, il 12% delle pazienti trattate con {Nome (di fantasia)} e il 17% delle pazienti trattate con tamoxifene hanno avuto una progressione della malattia alla valutazione clinica. Trattamento di prima linea Uno studio controllato in doppio cieco è stato condotto per confrontare {Nome (di fantasia)} (letrozolo) 2,5 mg e tamoxifene 20 mg come terapia di prima linea in donne in postmenopausa con cancro mammario avanzato. In 907 donne, letrozolo è risultato superiore a tamoxifene per il tempo alla progressione (endpoint primario) ed il tasso di risposte obiettive, il tempo al fallimento del trattamento ed il beneficio clinico. I risultati ottenuti sono riassunti nella Tabella 9: Tabella 9 Risultati ad un follow-up mediano di 32 mesi Variabile Statistica {Nome (di fantasia)}
N=453 Tamoxifene N=454
Tempo alla progressione
Mediana 9,4 mesi 6,0 mesi
(95% IC per mediana) (8,9, 11,6 mesi) (5,4, 6,3 mesi) Hazard ratio (HR) 0,72 (95% IC per HR) (0,62, 0,83) P <0,0001 Tasso di risposta (ORR)
RC+PR 145 (32%) 95 (21%)
(95% IC per tasso) (28, 36%) (17, 25%) Odds ratio 1,78 (IC 95% per odds ratio) (1,32, 2,40) P 0,0002 Il tempo alla progressione è stato significativamente più lungo e il tasso di risposta significativamente più elevato per letrozolo indipendentemente dal fatto che fosse stata somministrata una terapia adiuvante antiestrogenica o meno. Il tempo alla progressione è stato significativamente più lungo per letrozolo indipendentemente dal sito dominante di malattia. Il tempo mediano alla progressione è stato di 12,1 mesi per {Nome (di fantasia)} e di 6,4 mesi per tamoxifene nelle pazienti con sede di malattia solo nei tessuti molli e una mediana di 8,3 mesi per {Nome (di fantasia)} e di 4,6 mesi per tamoxifene nelle pazienti con metastasi viscerali. Il disegno dello studio permetteva alle pazienti di effettuare alla progressione di malattia il cross over alla terapia alternativa o l’interruzione dello studio. Approssimativamente il 50% delle pazienti ha eseguito il cross over al gruppo di trattamento opposto e il cross over è stato di fatto completato entro 36 mesi. Il tempo mediano al crossover è stato di 17 mesi (da {Nome (di fantasia)} a tamoxifene) e 13 mesi (da tamoxifene a {Nome (di fantasia)}). Il trattamento di prima linea del cancro mammario in stadio avanzato è risultato in una sopravvivenza
26

globale mediana per {Nome (di fantasia)} 34 mesi rispetto a 30 mesi per tamoxifene (logrank test P=0,53, non significativo). La mancanza di un vantaggio per {Nome (di fantasia)} sulla sopravvivenza globale può essere spiegata dal disegno con cross over dello studio. Trattamento di seconda linea In donne in postmenopausa con carcinoma mammario in fase avanzata, precedentemente trattate con anti estrogeni, sono state condotte due sperimentazioni cliniche , ben controllate, di confronto tra due dosi di letrozolo (0,5 mg e 2,5 mg) e, rispettivamente, megestrolo acetato e aminoglutetimide. Il tempo alla progressione non è stato significativamente differente tra letrozolo 2,5 mg e megestrolo acetato (P=0,07). Si sono osservate differenze statisticamente significative a favore del letrozolo 2,5 mg rispetto al megestrolo acetato per quanto riguarda il tasso complessivo di risposta obiettiva del tumore (24% verso 16%, P=0,04), e il tempo al fallimento del trattamento (P=0,04). La sopravvivenza globale non è risultata significativamente differente tra i 2 gruppi (P=0,2). Nel secondo studio, il tasso di risposta non è risultato significativamente differente tra letrozolo 2,5 mg e aminoglutetimide (P=0,06). Il letrozolo 2,5 mg è risultato statisticamente superiore all’aminoglutetimide per quanto riguarda il tempo alla progressione (P=0,008), il tempo al fallimento del trattamento (P=0,003) e la sopravvivenza globale (P=0,002). Cancro mammario maschile L’uso di {Nome (di fantasia)} nell’uomo con cancro mammario non è stato studiato. 5.2 Proprietà farmacocinetiche Assorbimento Il letrozolo viene assorbito rapidamente e completamente dal tratto gastroenterico (biodisponibilità media assoluta: 99,9%). Il cibo riduce lievemente la velocità di assorbimento (tmax mediano 1 ora a digiuno verso 2 ore dopo il pasto; e Cmax media 129 ± 20,3 nmol/litro a digiuno verso 98,7 ± 18,6 nmol/litro dopo il pasto) ma l’entità dell’assorbimento (AUC) non varia. Si ritiene che tale modesto effetto sulla velocità di assorbimento non abbia rilevanza clinica e pertanto il letrozolo può essere assunto indipendentemente dai pasti. Distribuzione Il legame del letrozolo alle proteine plasmatiche è di circa 60%, di cui la maggior parte (55%) è legata all’albumina. La concentrazione di letrozolo negli eritrociti è pari a circa l’80% del livello plasmatico. Dopo la somministrazione di 2,5 mg di letrozolo marcato con 14C, circa l’82% della radioattività plasmatica è rappresentata dal composto immodificato. L’esposizione sistemica ai metaboliti è bassa. Il letrozolo si distribuisce rapidamente e diffusamente nei tessuti. Il suo volume di distribuzione apparente allo steady state è di circa 1,87 0,47 l/kg. Biotrasformazione La principale via di eliminazione del letrozolo è rappresentata dalla clearance metabolica con formazione di un metabolita farmacologicamente inattivo, il carbinolo CLm = 2.1 l/h ma è relativamente lenta rispetto al flusso sanguigno epatico (circa 90 l/h). Gli isoenzimi 3A4 e 2A6 del citocromo P450 sono in grado di convertire il letrozolo in questo metabolita. La formazione di questi metaboliti minori non identificati e l’escrezione diretta per via renale e fecale hanno un ruolo di secondo piano nell’ambito dell’eliminazione globale del letrozolo. Dopo la somministrazione di 2.5 mg di letrozolo marcato con 14C a volontarie sane in postmenopausa, l’88,2 ± 7,6% della radioattività è stata recuperata nelle urine ed il 3,8 ± 0,9% nelle feci entro 2 settimane. Almeno il 75% della radioattività recuperata nelle urine fino a 216 ore (84,7 ± 7,8% della dose) è stato attribuito al glucuronide del metabolita carbinolo, circa il 9% a due metaboliti non identificati ed il 6% a letrozolo immodificato. L’apparente emivita plasmatica di eliminazione terminale è di circa 2 giorni. Dopo la somministrazione giornaliera di 2,5 mg lo steady-state è stato raggiunto entro 2-6 settimane. Le concentrazioni plasmatiche allo steady-state sono approssimativamente 7 volte più elevate delle
27

concentrazioni rilevate dopo una singola dose di 2,5 mg, mentre sono da 1,5 a 2 volte più alte rispetto ai valori allo steady-state previsti in base alle concentrazioni rilevate dopo una dose unica, questo suggerisce che vi è una lieve mancanza di linearità della farmacocinetica del letrozolo dopo la somministrazione giornaliera di 2,5 mg. Dato che i livelli allo steady-state vengono mantenuti nel tempo, si può concludere che non vi è accumulo continuo di letrozolo. Popolazioni speciali Pazienti anziani L’età non ha effetti sulla farmacocinetica del letrozolo. Compromissione renale In uno studio che ha coinvolto 19 volontarie con vari gradi di funzionalità renale (clearance della creatinina a 24-ore 9-116 ml/min) non sono state rilevate modificazioni sulla farmacocinetica del letrozolo dopo una singola dose di 2,5 mg. Compromissione epatica In uno studio simile su soggetti con vari gradi di funzionalità epatica, i valori medi di AUC nelle volontarie con una moderata compromissione della funzionalità epatica (classe B secondo la scala Child-Pugh) sono stati superiori del 37% rispetto a quelli dei soggetti normali, ma ancora entro i limii osservati in soggetti senza compromissione della funzionalità epatica. La farmacocinetica di letrozolo è stata valutata in uno studio di confronto in cui, dopo somministrazione di una dose singola orale in otto soggetti di sesso maschile con cirrosi epatica e insufficienza epatica grave (classe C secondo la scala Child-Pugh) e in volontari sani (N=8), l’area sotto la curva AUC e l’emivita t½ sono aumentate rispettivamente del 95 e 187%. Pertanto, {Nome (di fantasia)} deve essere somministrato in queste pazienti con cautela e dopo attenta considerazione del potenziale rapporto/rischio beneficio. 5.3 Dati preclinici di sicurezza Nell’ambito di una serie di studi tossicologici preclinici condotti con specie animali standard non vi è stata alcuna evidenza di tossicità sistemica o a carico di organi bersaglio. La tossicità acuta del letrozolo è stata bassa nei roditori esposti a dosi fino a 2000 mg/kg. Nei cani il letrozolo ha indotto segni di tossicità moderata a dosi fino 100 mg/kg. Nell’ambito di studi tossicologici per somministrazione ripetuta nel ratto e nel cane , aventi una durata fino a 12 mesi, i principali risultati osservati possono essere attribuiti all’attività farmacologica del composto. La dose priva di eventi avversi è risultata pari a 0.3 mg/kg in entrambe le specie. Studi sul potenziale mutageno del letrozolo condotti sia in vitro che in vivo non hanno documentato alcuna evidenza di genotossicità. In uno studio di carcinogenesi in ratti maschi della durata di 104 settimane, non sono stati rilevati tumori correlati al trattamento. In ratti femmina, è stata riscontrata una riduzione dell’incidenza di tumori mammari di natura sia benigna che maligna a tutte le dosi impiegate di letrozolo. In ratte e coniglie gravide il letrozolo si è dimostrato embriotossico e fetotossico a seguito di somministrazione orale a dosi clinicamente rilevanti. Nelle ratte che hanno partorito feti vivi, c’è stato un aumento dell’incidenza di malformazioni fetali comprendenti testa a cupola e fusione vertebrale cervicale/centrale. Non è stato osservato nel coniglio nessun aumento di malformazioni fetali. Non è noto se queste malformazioni siano state una conseguenza indiretta delle proprietà farmacologiche (inibizione della biosintesi degli estrogeni) o di un effetto diretto del farmaco (vedere paragrafi 4.3 e 4.6). Le osservazioni emerse dagli studi preclinici sono limitate a quelle associate all’attività farmacologica nota, che rappresenta l’unico ambito di preoccupazione in termini di sicurezza per l’impiego nell’uomo derivante dall’estrapolazione da studi condotti negli animali.
28

6. INFORMAZIONI FARMACEUTICHE 6.1 Elenco degli eccipienti [Completare con i dati nazionali] 6.2 Incompatibilità [Completare con i dati nazionali] 6.3 Periodo di validità [Completare con i dati nazionali] 6.4 Precauzioni particolari per la conservazione [Completare con i dati nazionali] 6.5 Natura e contenuto del contenitore [Completare con i dati nazionali] 6.6 Precauzioni particolari per lo smaltimento [Completare con i dati nazionali] 7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO [Vedere Allegato I - Completare con i dati nazionali] 8. NUMERO(I) DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO [Completare con i dati nazionali] 9. DATA DELLA PRIMA AUTORIZZAZIONE/RINNOVO DELL’AUTORIZZAZIONE [Completare con i dati nazionali] 10. DATA DI REVISIONE DEL TESTO [Completare con i dati nazionali]
29

FOGLIO ILLUSTRATIVO
30

Foglio illustrativo: Informazioni per l’utilizzatore
{Nome (di fantasia)} e denominazioni associate (Vedere Allegato I) dosaggio e forma farmaceutica
[Vedere Allegato I – Completare con i dati nazionali]
Letrozolo
Legga attentamente questo foglio prima di prendere questo medicinale perchè contiene importanti informazioni per lei. - Conservi questo foglio. Potrebbe aver bisogno di leggerlo di nuovo. - Se ha qualsiasi dubbio, si rivolga al medico o al farmacista. - Questo medicinale è stato prescritto soltanto per lei. Non lo dia ad altre persone, anche se i
sintomi della malattia sono uguali ai suoi, perché potrebbe essere pericoloso. - Se si manifesta un qualsiasi effetto indesiderato, compresi quelli non elencati in questo foglio, si
rivolga al medico o al farmacista. Contenuto di questo foglio 1. Che cos’è {Nome (di fantasia)} e a che cosa serve 2. Che cosa deve sapere prima di prendere {Nome (di fantasia)} 3. Come prendere {Nome (di fantasia)} 4. Possibili effetti indesiderati 5. Come conservare {Nome (di fantasia)} 6. Contenuto della confezione e altre informazioni 1. Che cos’è {Nome (di fantasia)} e a che cosa serve Che cos’è {Nome (di fantasia)} e come agisce {Nome (di fantasia)} contiene una sostanza attiva chiamata letrozolo. Appartiene ad un gruppo di medicinali chiamati inibitori dell’aromatasi. È’ un trattamento ormonale (o “endocrino”) del tumore della mammella. La crescita del tumore della mammella è frequentemente stimolata dagli estrogeni che sono ormoni sessuali femminili. {Nome (di fantasia)} riduce la quantità di estrogeni mediante il blocco di un enzima (“aromatasi”) che è coinvolto nella produzione di estrogeni e pertanto può bloccare la crescita di tumori al seno che hanno bisogno di estrogeni per crescere. Come conseguenza si rallenta o si interrompe la crescita delle cellule tumorali e/o la loro diffusione verso altre parti del corpo. Per cosa viene utilizzato {Nome (di fantasia)} {Nome (di fantasia)} è utilizzato per il trattamento del tumore della mammella in donne in menopausa cioè che non hanno più il ciclo mestruale. È usato per prevenire il ripresentarsi del tumore della mammella. Può essere utilizzato come un primo trattamento prima di un intervento chirurgico al seno nel caso in cui l’intervento immediato non sia possibile o come primo trattamento dopo intervento chirurgico al seno o dopo il trattamento di cinque anni con tamixofene. {Nome (di fantasia)} è anche utilizzato per prevenire la diffusione del tumore della mammella in altre parti del corpo in pazienti con tumore della mammella in uno stadio avanzato. Se ha una qualsiasi domanda su come agisce {Nome (di fantasia)} o perché le è stato prescritto questo medicinale, si rivolga al medico. 2. Cosa deve sapere prima di prendere {Nome (di fantasia)} Segua attentamente le istruzioni del medico. Esse potrebbero essere diverse dalle informazioni generali riportate in questo foglio.
31

Non prenda {Nome (di fantasia)} - se è allergico al letrozolo o ad uno qualsiasi degli altri componenti di questo medicinale
(elencati al paragrafo 6), - se ha ancora il ciclo, cioè se non è ancora in menopausa, - se è in gravidanza, - se sta allattando. Se una di queste condizioni la riguarda, non prenda questo medicinale ed informi il medico. Avvertenze e precauzioni Si rivolga al medico o al farmacista prima di prendere {Nome (di fantasia)} - se ha una grave malattia renale, - se ha una grave malattia epatica, - se ha una storia di osteoporosi o di fratture ossee (vedere anche “Monitoraggio del trattamento
con {Nome (di Fantasia)} nel paragrafo 3). Se una di queste condizioni la riguarda, informi il medico. Questo verrà tenuto in considerazione dal medico durante il trattamento con {Nome (di fantasia)}. Bambini e adolescenti (età inferiore a 18 yanni) I bambini e gli adolescenti non devono usare questo medicinale. Anziani (età pario superiore a 65 anni) Le donne di età pari o superiore a 65 anni possono usare questo medicinale alla stessa dose prevista per le donne adulte. Altri medicinali e {Nome (di fantasia)} Informi il medico o il farmacista se sta assumendo, ha recentemente assunto o potrebbe assumere qualsiasi altro medicinale, inclusi quelli senza prescrizione medica. Gravidanza allattamento e fertilità - Deve assumere {Nome (di fantasia)} solo quando è entrata in menopausa. Tuttavia, il medico
discuterà con lei la necessità di usare un efficace sistema contraccettivo in quanto potrebbe potenzialmente essere in gravidanza durante il trattamento con {Nome (di fantasia)}.
- Non deve prendere {Nome (di fantasia)} se è in stato di gravidanza o sta allattando perché può essere dannoso per il bambino.
Guida di veicoli e utilizzo di macchinari Se ha una sensazione di vertigini, di stanchezza, di sonnolenza o di malessere generale, non guidi e non utilizzi macchinari finché non si sente di nuovo normale. {Nome (di fantasia)} contiene lattosio {Nome (di fantasia)} contiene lattosio (zucchero del latte). Se il medico le ha diagnosticato una intolleranza ad alcuni zuccheri, lo contatti prima di prendere questo medicinale. 3. Come prendere {Nome (di fantasia)} Prenda questo medicinale seguendo sempre esattamente le istruzioni del medico o del farmacista. Se ha dubbi consulti il medico o il farmacista. La dose usuale è di una compressa di {Nome (di fantasia)} da prendere una volta al giorno. Prendere {Nome (di fantasia)} ogni giorno alla stessa ora la aiuterà a ricordare quando prendere la compressa. La compressa deve essere assunta con o senza cibo e deve essere deglutita intera con un bicchiere d’acqua o di un’altra bevanda. Per quanto tempo prendere {Nome (di fantasia)} Continui prendere {Nome (di fantasia)} ogni giorno per la durata che il medico le ha detto. Può aver bisogno di prenderlo per mesi o anche per anni. Se ha domande in merito a quanto tempo prendere
32

{Nome (di fantasia)}, parli con il medico. Monitoraggio durante il trattamento con {Nome (di fantasia)} Deve prendere questo medicinale sotto stretto controllo del medico. Il medico controllerà regolarmente il suo stato di salute per verificare che il trattamento abbia il giusto effetto. {Nome (di fantasia)} può causare fragilità o perdita della massa ossea (osteoporosi) per la diminuzione di estrogeni nel corpo. Il medico può decidere di effettuare la misurazione della densità ossea (un modo per controllare l’osteoporosi) prima, durante e dopo il trattamento. Se prende più {Nome (di fantasia)} di quanto deve Se ha preso troppo {Nome (di fantasia)}, o se qualche altra persona ha preso accidentalmente le sue compresse, contatti immediatamente il medico o l’ospedale per un consiglio. Mostri loro la confezione di compresse. Potrebbe essere necessario un trattamento medico. Se dimentica di prendere {Nome (di fantasia)} - Se è vicino il momento di prendere la dose successiva (per esempio entro 2 o 3 ore), salti la
dose che ha dimenticato e prenda la dose successiva quando è previsto che la prenda. - Altrimenti, prenda la dose appena si ricorda e quindi prenda la compressa successiva come
farebbe normalmente. - Non prenda una dose doppia per compensare la dimenticanza della dose. Se interrompe il trattamento con {Nome (di fantasia)} Non interrompa l’assunzione di {Nome (di fantasia)} a meno che non le venga detto dal medico. Vedere anche sopra il paragrafo “Per quanto tempo prendere {Nome (di fantasia)}”. 4. Possibili effetti indesiderati Come tutti i medicinali, questo medicinale può causare effetti indesiderati sebbene non tutte le persone li manifestino. La maggior parte degli effetti indesiderati è da lieve a moderato e di solito scomparirà un periodo di trattamento compreso tra pochi giorni e poche settimane. Alcuni di questi effetti indesiderati come vampate di calore, perdita di capelli o sanguinamento vaginale, possono essere causati dalla mancanza di estrogeni nel corpo. Non si preoccupi per questo elenco di possibili effetti indesiderati. Potrebbe non esserne soggetta. Alcuni effetti indesiderati possono essere gravi: Effetti rari o non comuni (cioè possono colpire tra 1 e 100 pazienti ogni 10.000): - Debolezza, paralisi o perdita di sensibilità in una qualsiasi altra parte del corpo (in particolare
braccio o gamba), perdita di coordinazione, nausea, o difficoltà nel parlare o di respirazione (sintomo di una patologia cerebrale, ad esempio ictus).
- Improvviso dolore al torace con senso di oppressione (sintomo di una patologia cardiaca). - Difficoltà di respirazione, dolore al torace, svenimento, battito cardiaco accelerato, colorazione
bluastra della pelle, o dolore improvviso al braccio, alla gamba o al piede (sintomi di una possibile formazione di un coagulo di sangue).
- Gonfiore ed arrossamento in corrispondenza di una vena che risulta estremamente sensibile e anche dolorosa al tatto.
- Febbre alta, brividi o ulcerazione della bocca causate da infezioni (mancanza di globuli bianchi).
- Visione offuscata in modo grave e persistente. Se uno di questi effetti si manifesta, informi il medico immediatamente. Deve informare immediatamente il medico se si manifesta uno dei seguenti sintomi durante il
33

trattamento con {Nome (di fantasia)}: - Gonfiore principalmente del viso e della gola (segni di reazione allergica). - Pelle ed occhi giallastri, nausea, perdita dell’appetito, urine scure (segni di epatite). - Eruzione cutanea, pelle arrossata, vesciche alle labbra, occhi o labbra, esfoliazione della pelle,
febbre (segni di patologie cutanee). Alcuni effetti indesiderati sono molto comuni. Questi effetti indesiderati possono colpire più di 10 pazienti ogni 100. - Vampate di calore - Aumento dei livelli di colesterolo (ipercolesterolemia) - Affaticamento - Aumento della sudorazione - Dolore alle ossa e alle giunture (artralgia) Se uno di questi effetti si manifesta in modo grave, informi il medico. Alcuni effetti indesiderati sono comuni. Questi effetti indesiderati possono colpire tra 1 e 10 pazienti ogni 100. - Eruzione cutanea - Cefalea - Capogiri - Malessere (di solito sensazione di malessere) - Patologie gastrointestinali come nausea, vomito, indigestione, stitichezza, diarrea - Aumento o perdita dell’appetito - Dolore muscolare - Fragilità o perdita della massa ossea (osteoporosi), che in alcuni casi porta a fratture ossee
(vedere anche “Monitoraggio durante il trattamento con {Nome(di fantasia)}” nel paragrafo 3) - Gonfiore delle braccia, delle mani, dei piedi, delle caviglie (edema) - Depressione - Aumento di peso - Perdita dei capelli - Aumento della pressione arteriosa (ipertensione) - Dolore addominale - Secchezza della pelle - Sanguinamento vaginale Se uno di questi effetti si manifesta in modo grave, informi il medico. Altri effetti indesiderati sono non comuni. Questi effetti indesiderati possono colpire tra 1 e 10 pazienti ogni 1.000. - Patologia del sistema nervoso come ansia, nervosismo, irritabilità, sopore, problemi di memoria,
sonnolenza, insonnia - Compromissione della sensibilità, specialmente al tatto - Patologie dell’occhio come visione offuscata, irritazione agli occhi - Palpitazioni, battito cardiaco veloce - Patologie cutanee come prurito (orticaria) - Perdite o secchezza vaginale - Rigidità articolare (artriti) - Dolore al seno - Febbre - Sete, disturbi del gusto, secchezza della bocca - Secchezza delle mucose - Perdita di peso - Infezioni del tratto urinario, aumento della frequenza urinaria - Tosse - Aumento dei livelli degli enzimi nel fegato Se uno di questi effetti si manifesta in modo grave, informi il medico. Se si manifesta un qualsiasi effetto indesiderato, compresi quelli non elencati in questo foglio, si
34

35
rivolga al medico o al farmacista. 5. Come conservare {Nome (di fantasia)} [Completare con i dati nazionali] 6. Contenuto della confezione e altre informazioni Cosa contiene {Nome (di fantasia)} - Il principio attivo è il letrozolo. Ogni compressa rivestita con film contiene 2,5 mg di letrozolo. - Gli altri component sono [Completare con i dati nazionali] Descrizione dell’aspetto di {Nome (di fantasia)} e contenuto della confezione [Completare con i dati nazionali] Titolare dell’autorizzazione all’immissione in commercio e produttore [Vedere Allegato I – Completare con i dati nazionali] Questo medicinale è autorizzato negli Stati Membri dello Spazio Economico Europeo con le seguenti denominazioni: [Vedere Allegato I – Completare con i dati nazionali] Questo foglio illustrativo è stato aggiornato il {MM/YYYY}. [Completare con i dati nazionali]




























