Embed Size (px)
Citation preview

Física Termodinámica
Parte 3

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.1 Presión 7.1 Presión
7. Teoría Cinética de los Gases
Considere N moléculas de un gas ideal al interior de un contendor de volumen V a una temperatura absoluta T.
Presión es el resultado de la fuerza promedio que actúa en las paredes del contenedor.
• las moléculas se mueven rápida y caóticamente colisionando con las paredes del contenedor.
•Las moléculas ejercen fuerza en las paredes del contendor durante la colisión.
•Durante la colisión la componente y de la velocidad de las moléculas no varia, pero la componente x de la velocidad cambia en dirección y magnitud.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.1 Presión 7.1 Presión
7. Teoría Cinética de los Gases
Considere la colisión de una molécula contra la pared del contenedor:
xvyv
xv
yv
El cambio de la velocidad durante un intervalo de tiempo ∆t es:
Por lo tanto, la fuerza de impacto de la molécula sobre la pared es:
x
xxx
vvvvv
22
=∆=−−
tvm
tvmF x
∆=
∆∆
=2

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.1 Presión 7.1 Presión
7. Teoría Cinética de los Gases
Durante un tiempo ∆t, las moléculas a una distancia menor o igual a de la pared, golpeara la pared. Por lo tanto el numero de colisiones será el numero de moléculas dentro del volumen elemental
Si hay N moléculas en el volumen total V, entonces el numero es:
tAvV x∆=∆
tvx∆
VVNV ∆
=∆
AV∆
tvx∆ VtvNN x
colisiones∆∆
=21
Numero total de moléculas
La mitad de las moléculas colisionan con la pared el resto viaja en otra dirección
Volumen del contenedor
Volumen de la mitad de las moléculas

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.1 Presión 7.1 Presión
7. Teoría Cinética de los Gases
La fuerza total sobre la pared en cualquier instante de tiempo Dt:
2
212
x
xx
total
mvVN
AF
tAvVN
tvmF
=
⎥⎦⎤
⎢⎣⎡ ∆⎥⎦⎤
⎢⎣⎡∆
=Pero esto sucede mientras asumamos que vx es el mismo para cada molécula.Las moléculas en el contenedor tiene un rango de velocidad. El valor promedio de la componenteda lugar a la fuerza promedio (y por lo tanto la presión) en la pared:
2xv
PvmVN
AF
Nvvvv
xprom
xxxx
==
+++=
2
23
22
212 ...

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.1 Presión 7.1 Presión
7. Teoría Cinética de los Gases
Seria mas conveniente dejar una expresión donde se incluya la velocidad total en vez de la componente x de la velocidad, por lo tanto:
2
22
2222
31
xprom
x
zyxx
vmVN
AF
vv
vvvv
=
=
++=Magnitud del promedio de la velocidad2 dada por la suma del promedio de las componentes
2
31 vm
VNP = 2vvrms =
Promedio de la raíz cuadrada de la velocidad

2
21
23 vmkT =
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.1 Presión 7.1 Presión
7. Teoría Cinética de los Gases
Promedio de la energía cinética de translación de una sola molécula
2
2
2
2
2
21
23
21
32
21
32
21
3231
vmTNR
vmnNnRT
vmNnRT
nRTPV
vmNPV
vNmPV
A
a
=⎥⎦
⎤⎢⎣
⎡
⎟⎠⎞
⎜⎝⎛=
⎟⎠⎞
⎜⎝⎛=
=
⎟⎠⎞
⎜⎝⎛=
=
AnNN =
Pero, R y NA son constantes. el cuociente entre ellos es una nueva constante llamada la constante de Boltzmann k
KJ23108.1 −⋅
El promedio de la energía cinética de translación de una molécula depende solo de la temperatura T
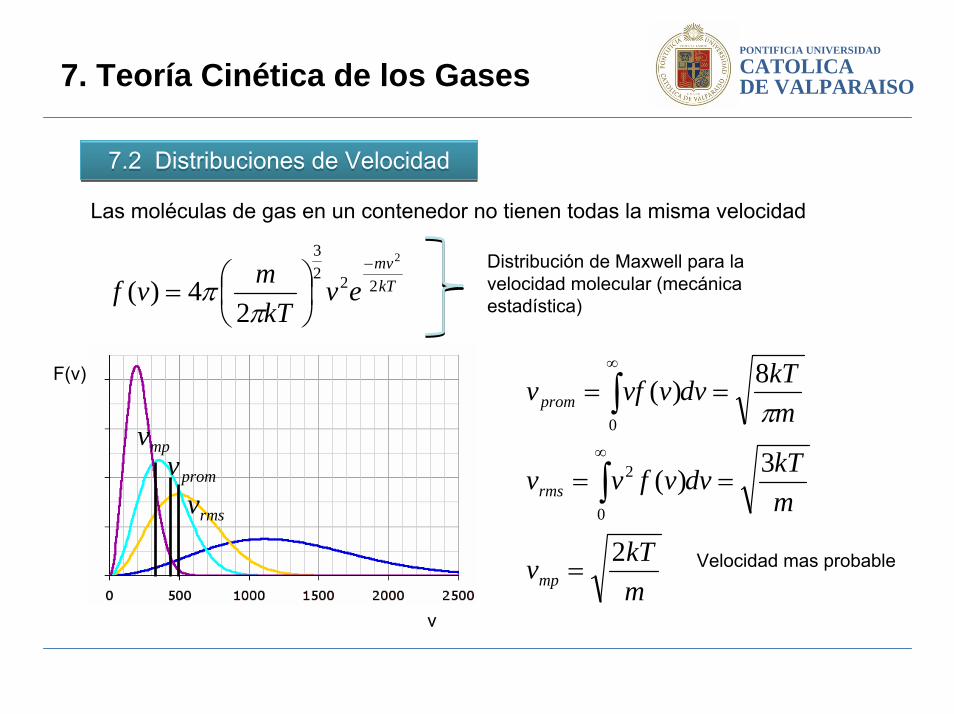
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.2 Distribuciones de Velocidad 7.2 Distribuciones de Velocidad
7. Teoría Cinética de los Gases
kTmv
evkT
mvf 2223 2
24)(
−
⎟⎠⎞
⎜⎝⎛=π
π
Las moléculas de gas en un contenedor no tienen todas la misma velocidad
Distribución de Maxwell para la velocidad molecular (mecánica estadística)
v
F(v)
mpvpromv
rmsv
mkTv
mkTdvvfvv
mkTdvvvfv
mp
rms
prom
2
3)(
8)(
0
2
0
=
==
==
∫
∫∞
∞
π
Velocidad mas probable

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.2 Distribuciones de Velocidad 7.2 Distribuciones de Velocidad
7. Teoría Cinética de los Gases
Se dijo previamente que el promedio de la energía cinética de una molécula de gas en un contenedor puede ser calculada sabiendo la temperatura del gas. Sin embargo a menudo es complicado decidir cual es la velocidad para esta situación.
Deberíamos usar la velocidad rms cuando tratamos con la energía cinética de las moléculas. Deberíamos usar la velocidad promedio cuando el proceso bajo consideraciones sea afectado por la velocidad de las moléculas.
Para calcular la velocidad rms, elevamos al cuadrado la velocidad, luego dividimos por el numero total de moléculas y luego extraemos la raíz cuadrada. Esto es diferente que hallar la velocidad promedio desde el acto de elevar al cuadrado las velocidades.Es por eso que la velocidad rms es un poco mas alta que la velocidad promedio
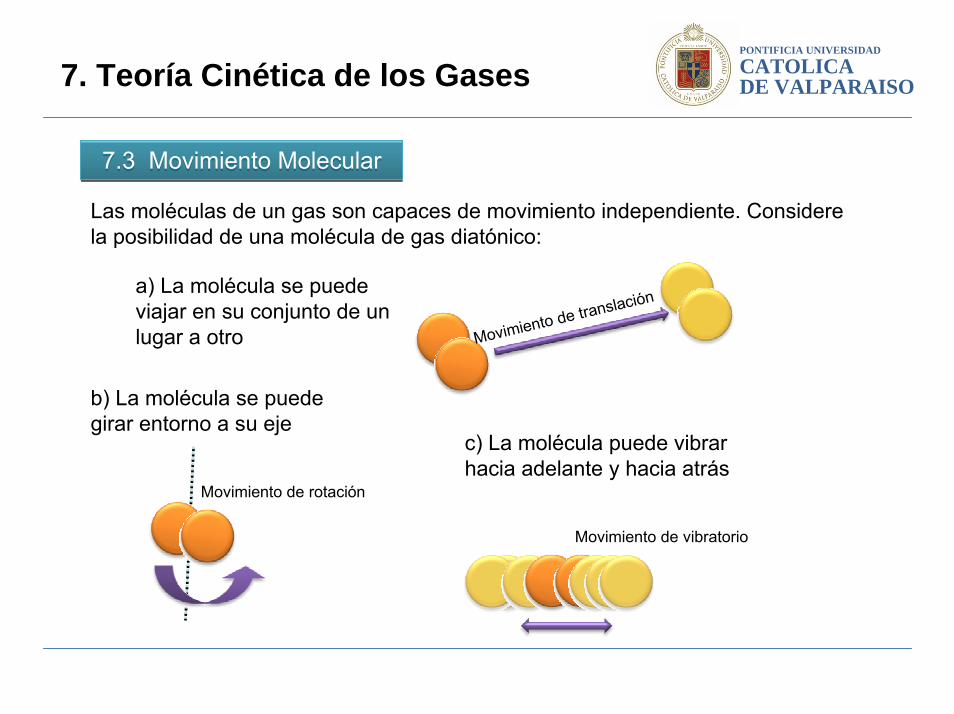
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.3 Movimiento Molecular 7.3 Movimiento Molecular
7. Teoría Cinética de los Gases
Las moléculas de un gas son capaces de movimiento independiente. Considere la posibilidad de una molécula de gas diatónico:
Movimiento de translacióna) La molécula se puede viajar en su conjunto de un lugar a otro
b) La molécula se puede girar entorno a su eje
Movimiento de rotación
c) La molécula puede vibrar hacia adelante y hacia atrás
Movimiento de vibratorio
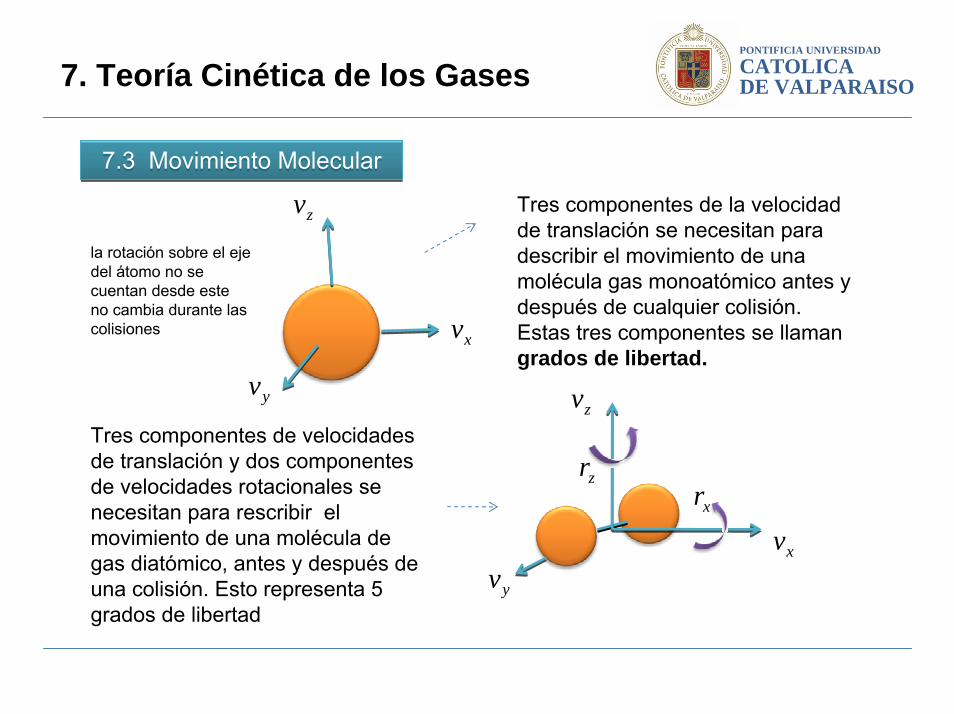
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.3 Movimiento Molecular 7.3 Movimiento Molecular
7. Teoría Cinética de los Gases
Tres componentes de la velocidad de translación se necesitan para describir el movimiento de una molécula gas monoatómico antes y después de cualquier colisión. Estas tres componentes se llaman grados de libertad.
yv
xv
zvla rotación sobre el eje del átomo no se cuentan desde este no cambia durante las colisiones
Tres componentes de velocidades de translación y dos componentes de velocidades rotacionales se necesitan para rescribir el movimiento de una molécula de gas diatómico, antes y después de una colisión. Esto representa 5 grados de libertad
yv
zv
xvxr
zr

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.4 Calor Especifico y coeficiente adiabático7.4 Calor Especifico y coeficiente adiabático
7. Teoría Cinética de los Gases
El promedio de la energía cinética de un gas es igualmente dividido entre las componentes de rotación y la traslación. Para cada grado de libertad, la energía cinética media viene dada por kT
21
Tipo de gas Grados de libertad Energía interna total
Monoatómico 3 3/2 kT
Diatómico 5 5/2 kT
Poliatómico 6* 6/2 kTEnergía de translación y rotación cinética
Toda la energía cinética de translación
* Puede ser mas o menos dependiendo del gas
Considere un cambio de temperatura para n moles de gas monoatómico. Para un determinado aumento de la temperatura, el cambio promedio en energía cinética de translación es igual al cambio en interior de la energía:
12 TTT −=∆
)( 12 TTnCU v −=∆
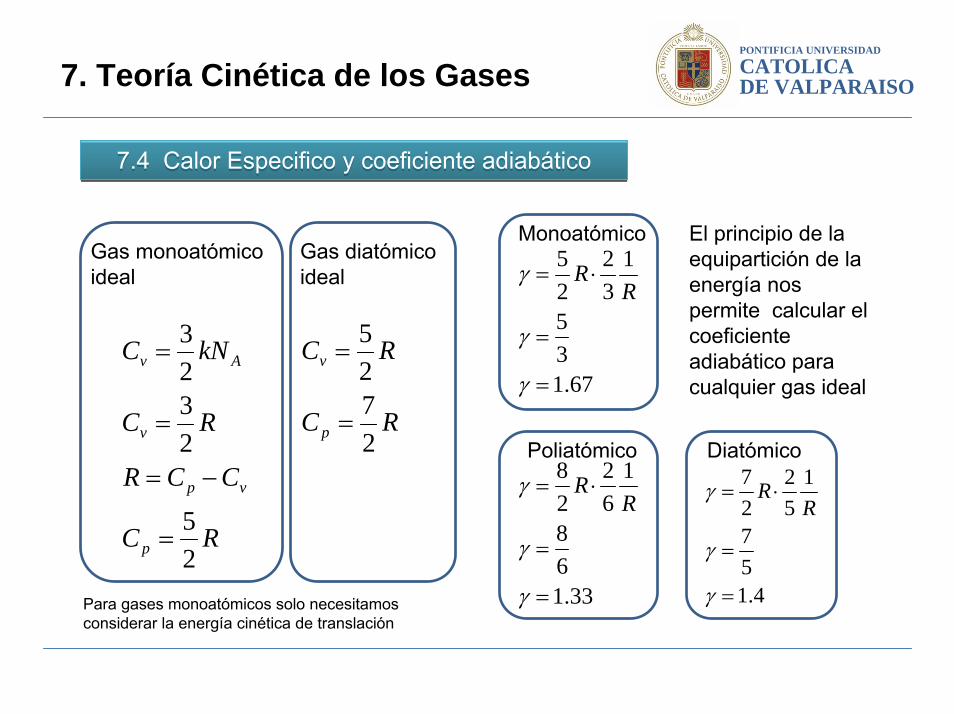
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.4 Calor Especifico y coeficiente adiabático7.4 Calor Especifico y coeficiente adiabático
7. Teoría Cinética de los Gases
RC
CCR
RC
kNC
p
vp
v
Av
25
2323
=
−=
=
=
Gas monoatómico ideal
RC
RC
p
v
2725
=
=
Gas diatómicoideal
67.135
132
25
=
=
⋅=
γ
γ
γR
R
Monoatómico
4.157
152
27
=
=
⋅=
γ
γ
γR
R
Diatómico
33.168
162
28
=
=
⋅=
γ
γ
γR
RPoliatómico
El principio de la equipartición de la energía nos permite calcular el coeficiente adiabático para cualquier gas ideal
Para gases monoatómicos solo necesitamos considerar la energía cinética de translación
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
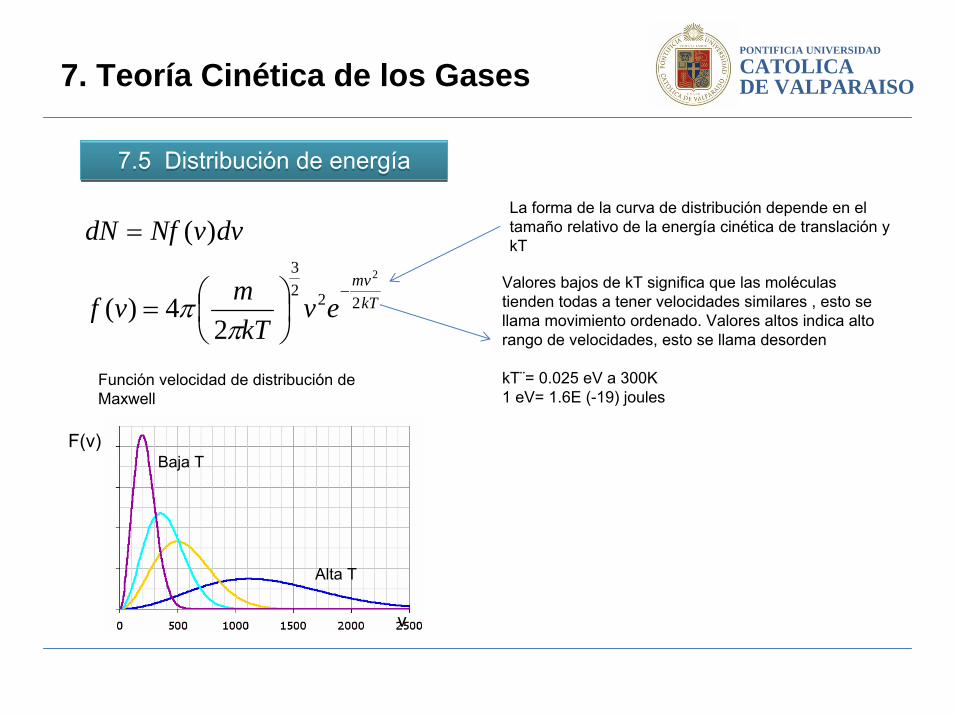
7.5 Distribución de energía7.5 Distribución de energía
7. Teoría Cinética de los Gases
La forma de la curva de distribución depende en el tamaño relativo de la energía cinética de translación y kT
kTmv
evkT
mvf
dvvNfdN
2223 2
24)(
)(
−⎟⎠⎞
⎜⎝⎛=
=
ππ
Valores bajos de kT significa que las moléculas tienden todas a tener velocidades similares , esto se llama movimiento ordenado. Valores altos indica alto rango de velocidades, esto se llama desorden
kT¨= 0.025 eV a 300K1 eV= 1.6E (-19) joules
Función velocidad de distribución de Maxwell
v
F(v)Baja T
Alta T
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
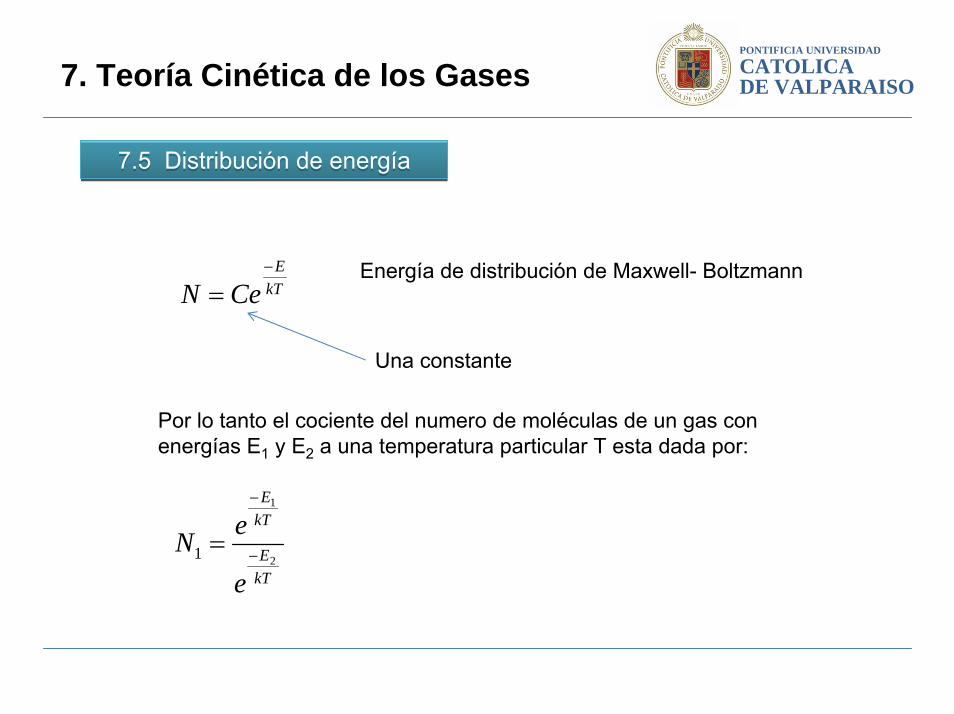
7.5 Distribución de energía7.5 Distribución de energía
7. Teoría Cinética de los Gases
Energía de distribución de Maxwell- BoltzmannkT
E
CeN−
=
Una constante
Por lo tanto el cociente del numero de moléculas de un gas con energías E1 y E2 a una temperatura particular T esta dada por:
kTE
kTE
e
eN2
1
1 −
−
=

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.6 Ejemplos7.6 Ejemplos
7. Teoría Cinética de los Gases
1. Compute el promedio de la energía cinética de una molécula de gas a temperatura ambiente (300K)
Solución
JkE
kE
kTkE
prom
prom
prom
21
23
102.6
1038.13002323
−
−
⋅=
⋅⋅⋅=
=

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.6 Ejemplos7.6 Ejemplos
7. Teoría Cinética de los Gases
2. Si en gas en la pregunta anterior fuera H2 calcule la velocidad rms de una sola molécula.
Solución
smv
v
mkTv
rms
rms
rms
1934
1032.3300)1038.1(3
3
27
23
=
⋅⋅
=
=
−
−

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
7.6 Ejemplos7.6 Ejemplos
7. Teoría Cinética de los Gases
3. Para un mol de gas a 300K, calcule el total de la energía cinética de translación.
Solución
( ) ( )JKE
KE
kTNKE
total
total
Atotal
3750
3001038.1231002.6
23
2323
=
⋅⋅=
=
−

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.1 Procesos cíclicos8.1 Procesos cíclicos
8. Maquinas de calor
1. Un gas va desde P1 a P2 en un proceso a volumen constante
P
VP1
P2
2. Luego va a P3 por una expansión isotérmica
P
V
P3
P2
2. Finalmente regresa a P1 por compresión de presión constante
P
VP1
P3
Todos estos procesos son termodinámicos, cuasi-estáticos, reversiblesCuasi-estático significa que la presión, temperatura y volumen están cambiando muy lentamente, lo suficientemente lento para que los efectos dinámicos tales como cambio de momento o viscosidad, sean insignificantes.
La palabra reversible significa que la dirección del proceso puede revertirse cambiando la temperatura, presión volumen, etc. Una expansión isotérmica debe ser reversible, revistiendo la dirección del trabajo W, donde el trabajo W se convierte en calor Q y transferida a regreso a la fuente de calor

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.1 Procesos cíclicos8.1 Procesos cíclicos
8. Maquinas de calor
Combinemos todos los procesos en un solo diagrama. Esta combinación, donde la presión, temperatura y volumen del fluido trabajando regresa a su estado inicial, es un ciclo termodinámico.
P
VP1
P2
P3
El área encerrada por el ciclo denota el trabajo hecho por o sobre el sistema
La naturaleza cíclica de esta configuración significa que puede obtenerse un continuo trabajo de salida.la continua conversión de calor a trabajo es la característica mas destacable de una maquina de calor. Otros dispositivos que convierte el calor en trabajo en una manera no cíclica no son maquinas de calor
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
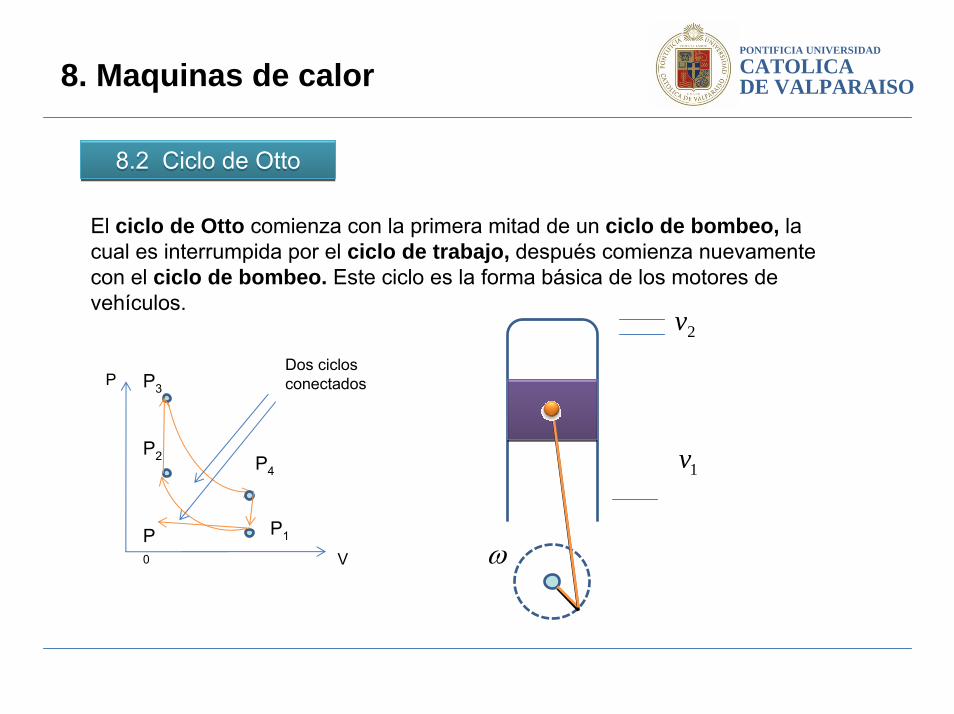
8.2 Ciclo de Otto8.2 Ciclo de Otto
8. Maquinas de calor
El ciclo de Otto comienza con la primera mitad de un ciclo de bombeo, la cual es interrumpida por el ciclo de trabajo, después comienza nuevamente con el ciclo de bombeo. Este ciclo es la forma básica de los motores de vehículos.
Dos ciclos conectados P
V
P1
P2
P3
P4
P0 ω
1v
2v
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
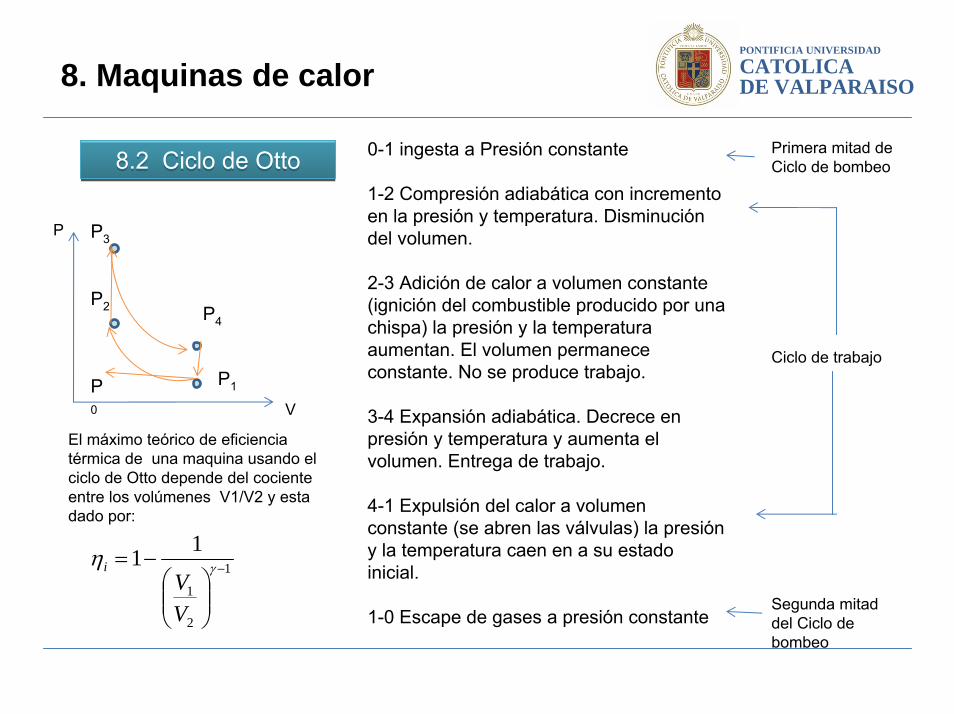
8.2 Ciclo de Otto8.2 Ciclo de Otto
8. Maquinas de calor
0-1 ingesta a Presión constante
1-2 Compresión adiabática con incremento en la presión y temperatura. Disminución del volumen.
2-3 Adición de calor a volumen constante (ignición del combustible producido por una chispa) la presión y la temperatura aumentan. El volumen permanece constante. No se produce trabajo.
3-4 Expansión adiabática. Decrece en presión y temperatura y aumenta el volumen. Entrega de trabajo.
4-1 Expulsión del calor a volumen constante (se abren las válvulas) la presión y la temperatura caen en a su estado inicial.
1-0 Escape de gases a presión constante
P
V
P1
P2
P3
P4
P0
Primera mitad de Ciclo de bombeo
Segunda mitad del Ciclo de bombeo
Ciclo de trabajo
El máximo teórico de eficiencia térmica de una maquina usando el ciclo de Otto depende del cociente entre los volúmenes V1/V2 y esta dado por:
1
2
1
11 −
⎟⎟⎠
⎞⎜⎜⎝
⎛−= γη
VV
i

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.3 Eficiencia Térmica8.3 Eficiencia Térmica
8. Maquinas de calor
Una maquina de calor usa un ciclo termodinámico para producir trabajo. Luego de que la maquina de calor completa un ciclo, el sistema regresa a su estado inicial. La única diferencia observable es que el calor tomado de la fuente, una cantidad de este se ha convertido en trabajo , y el restante es expulsado
FQ∆RQ∆W∆
Fuente de calor a Tc
Disipador de calor a Tf
RQ
FQ
DQ
W
Representa el calor disipado (fricción, turbulencia, etc)

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.3 Eficiencia Térmica8.3 Eficiencia Térmica
8. Maquinas de calor
Una maquina de calor siempre tiene dos salidas:
La energía suministrada FQ
Maquina de calor
TrabajoSalida utilizable
W
Calor Calor expulsado + calor disipado
DR QQ +
Eficiencia térmica
F
R
F
RF
RF
RF
F
entrada
salida
QQQQQWWQQ
QWEnegiaEnergia
−=−
=
−=+=
=
=
10
0
0
η
η
η
La eficiencia máxima posible se llama eficiencia de Carnot y siempre tiene que ser menor que 1.
No hay cambios en la energía interna del fluido

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.4 Ciclo de Carnot8.4 Ciclo de Carnot
8. Maquinas de calor
Condiciones para la eficiencia máxima:•El calor es suministrado de manera isotérmica a Tc•El calor es expulsado de manera isotermica a Tf•No hay transferencia de calor en ninguna parte del ciclo.
P
V
P1
P2
P3
P4
cTTT == 43
fTTT == 21
fR TQ ,
cF TQ ,1-2 Compresión isotérmica, el calor se expulsa a Tf, mientras la presión aumenta.
2-3 compresión adiabática, la presión y la temperatura, aumentan sin ningún flujo de calor.
3-4 Expansión isotérmica, el calor es suministrado por la fuente a Tc y la presión decrece.
4-1 Expansión adiabática, la presión y la temperatura decrecen a los valores iníciales, no hay flujo de calor.
2211 VPVP =

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO8. Maquinas de calor
( )( )
c
f
c
f
fR
cF
F
F
R
TT
VVTVVT
VVnRTQ
VVnRTQ
VVnRTQ
nRTVPahoraVVVPWQ
VVVPWQ
−=
−=
=
=
=
=
==−
==−
−
−
1
lnln
1
ln
ln
ln
,
ln:43
ln:21
34
21
2
1
3
4
3
43
333
3
43343
2
11121
η
η
3
4
2
1
1
4
1
1
4
1
3
2
2
3
:14
:32
VV
VV
TT
VV
TT
TT
VV
TT
f
c
f
c
=
=⎟⎟⎠
⎞⎜⎜⎝
⎛=−
=⎟⎟⎠
⎞⎜⎜⎝
⎛=−
−
−
γ
γ
Procesos adiabáticos
Máxima eficiencia posible
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
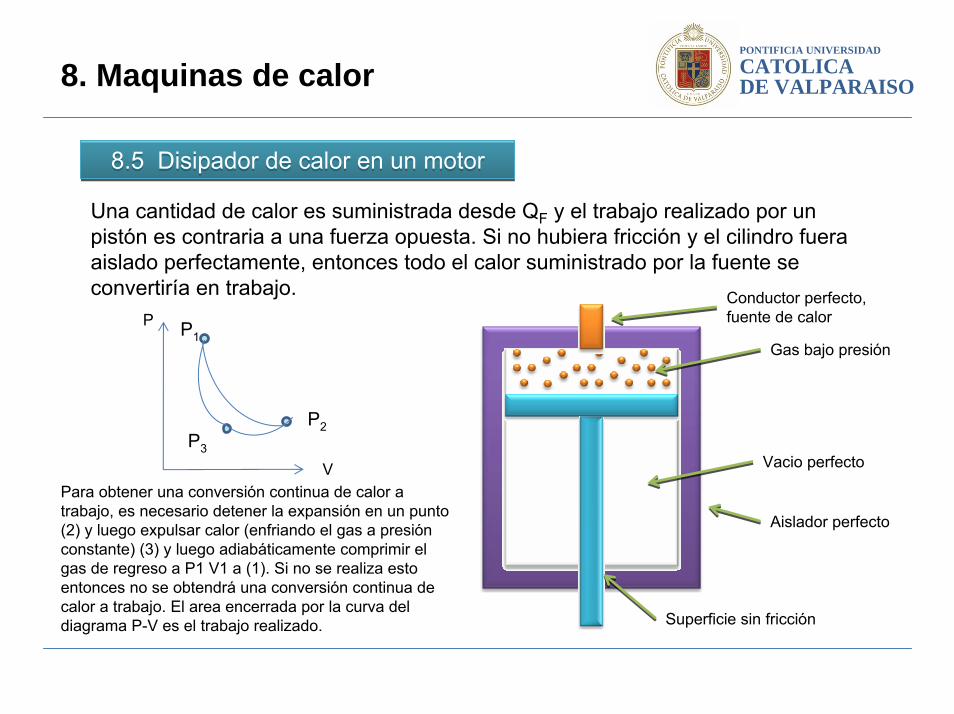
8.5 Disipador de calor en un motor8.5 Disipador de calor en un motor
8. Maquinas de calor
Una cantidad de calor es suministrada desde QF y el trabajo realizado por un pistón es contraria a una fuerza opuesta. Si no hubiera fricción y el cilindro fuera aislado perfectamente, entonces todo el calor suministrado por la fuente se convertiría en trabajo.
P
V
P1
P2P3
Gas bajo presión
Vacio perfecto
Superficie sin fricción
Aislador perfecto
Conductor perfecto, fuente de calor
Para obtener una conversión continua de calor a trabajo, es necesario detener la expansión en un punto (2) y luego expulsar calor (enfriando el gas a presión constante) (3) y luego adiabáticamente comprimir el gas de regreso a P1 V1 a (1). Si no se realiza esto entonces no se obtendrá una conversión continua de calor a trabajo. El area encerrada por la curva del diagrama P-V es el trabajo realizado.
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
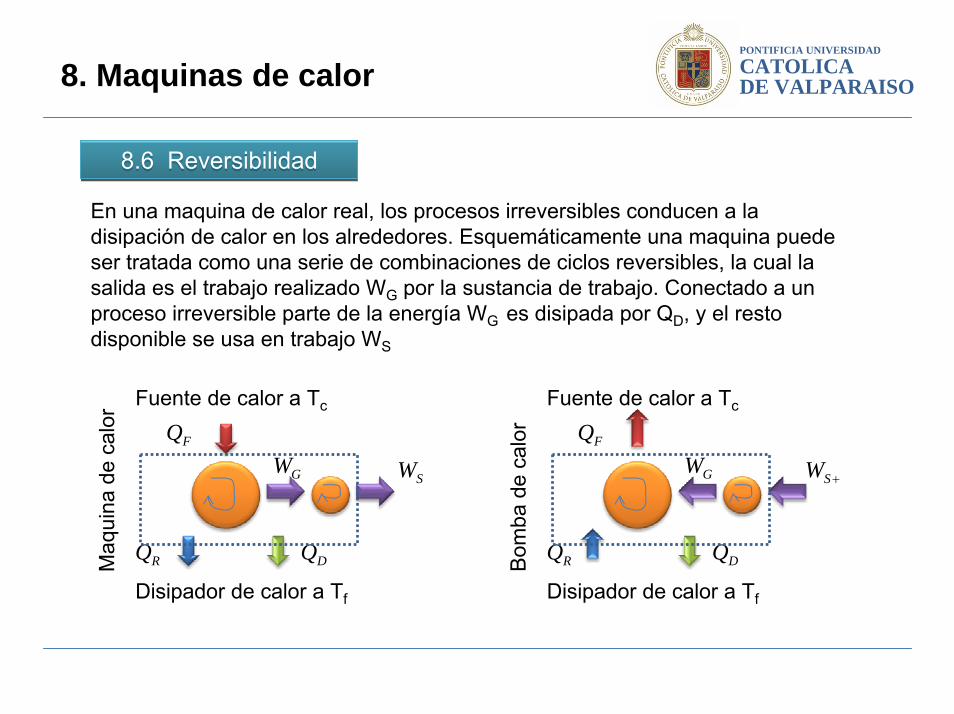
8.6 Reversibilidad8.6 Reversibilidad
8. Maquinas de calor
En una maquina de calor real, los procesos irreversibles conducen a la disipación de calor en los alrededores. Esquemáticamente una maquina puede ser tratada como una serie de combinaciones de ciclos reversibles, la cual la salida es el trabajo realizado WG por la sustancia de trabajo. Conectado a un proceso irreversible parte de la energía WG es disipada por QD, y el resto disponible se usa en trabajo WS
Fuente de calor a Tc
Disipador de calor a Tf
RQ
FQ
DQ
GWSW
Maq
uina
de
calo
r Fuente de calor a Tc
Disipador de calor a Tf
RQ
FQ
DQ
GW+SW
Bom
ba d
e ca
lor
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.6 Reversibilidad8.6 Reversibilidad
8. Maquinas de calor
Fuente de calor a Tc
Disipador de calor a Tf
RQ
FQ
DQ
GWSW
Maq
uina
de
calo
r Fuente de calor a Tc
Disipador de calor a Tf
RQ
FQ
DQ
GW+SW
Bom
ba d
e ca
lor
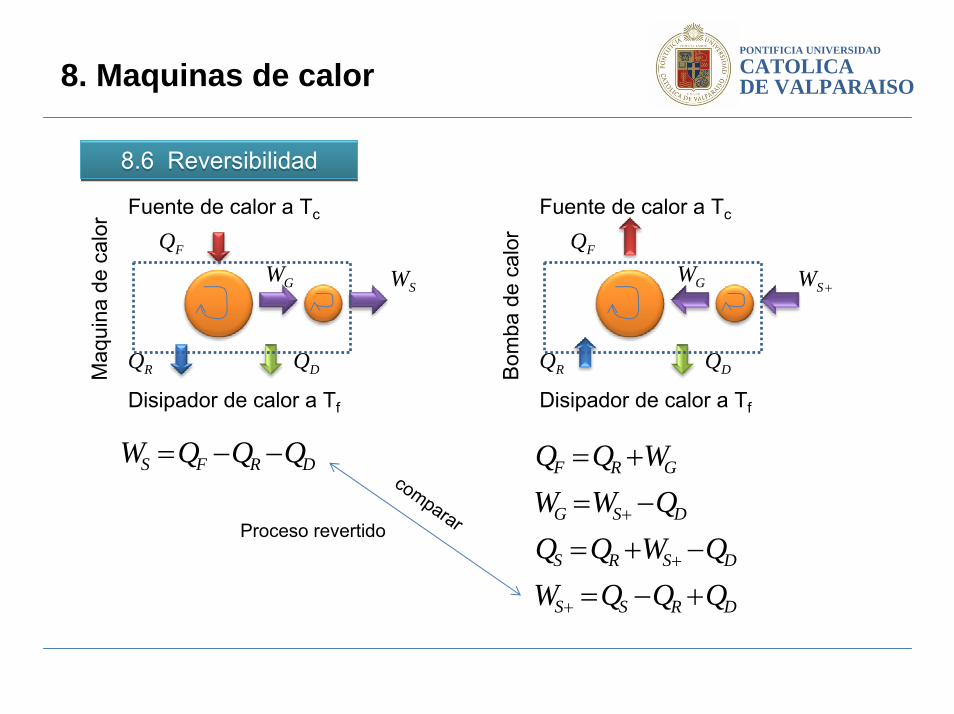
DRFS QQQW −−=
DRSS
DSRS
DSG
GRF
QQQWQWQQ
QWWWQQ
+−=−+=
−=+=
+
+
+
compararProceso revertido

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.6 Reversibilidad8.6 Reversibilidad
8. Maquinas de calor
Un refrigerador es una maquina de calor invertida
0
0
12
)(QWQ
WQQUUWQ
i
i
=+−=+−−=−
Calor tomado de un deposito frio
Entrada de trabajo al refrigerador
Expulsión de calor e el deposito de calor
Suponiendo que no hay aumento en la temperatura ∆U=0
Signo ha sido incluido en la formula
La mejor nevera transfieren el calor de Tf, usando el mínimo de trabajo W. por lo tanto el coeficiente de rendimiento es:
fc
f
i
TTT
COP
WQCOP
−=
=

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.7 Ejemplo8.7 Ejemplo
8. Maquinas de calor
1. Un ciclo de Carnot es operado entre dos depósitos de calor a 800K y 300K. Si 600 J son retirados desde el deposito caliente en cada ciclo, calcular la cantidad de calor liberada al deposito frio.
Solución
Fuente de calor a Tc
Disipador de calor a Tf
RQ
FQ
DQ
WJ600
K800
K300
JQQ
JW
WQWQ
TT
R
R
RF
c
f
225375600
375
625.0600
%5.628003001
1
=−=
=
=
+==
−=
−=
η
η
η

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
8.7 Ejemplo8.7 Ejemplo
8. Maquinas de calor
2. La salida de una maquina a petróleo es de 50kW. La eficiencia termica es del 15%. Calcular el calor suministrado y expulsado en un minuto.
Solución
MJQWQQ
MJQQW
JW
W
R
RF
F
F
17
20
15.0
103
5000060
6
=+=
=
==
⋅=
=
η

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.1 Procesos reversibles e irreversibles9.1 Procesos reversibles e irreversibles
9. Entropía
Pregunta: Un kilogramo de agua a 0ºC es mezclado con un kilogramo de agua a 100ºC.
¿Qué sucederá?
Tendremos 2 kilogramos de agua a 50ºC
( )( ) ( )( )CT
TTTmcQ
º5010041861041861
2
22
=−−=−
∆=∆
caliente fríatibia
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.1 Procesos reversibles e irreversibles9.1 Procesos reversibles e irreversibles
9. Entropía
Pero ¿por qué sucede así?Sin dudas podemos calcular que el calor perdido por el agua caliente es igual al
calor ganado por el agua fría, pero por que no se puede obtener en el mismo recipiente 1kg de agua caliente y un kilogramo de agua fría. La energía se conservaría de todas maneras. ¿Por qué el calor fluye desde los objetos calientes hacia los mas fríos?¿por que el agua no se separa en regiones de agua caliente y agua fria?
La mezcla de agua es un ejemplo de proceso irreversible. El calor fluye desde el agua caliente al agua fría, no se realiza trabajo. Hay una preferencia o dirección natural para todos los procesos involucrados con el flujo de calor. La segunda ley esta relacionada con esta preferencia de dirección
caliente fría
Calor fluye
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.2 Entropía y reversibilidad9.2 Entropía y reversibilidad
9. Entropía
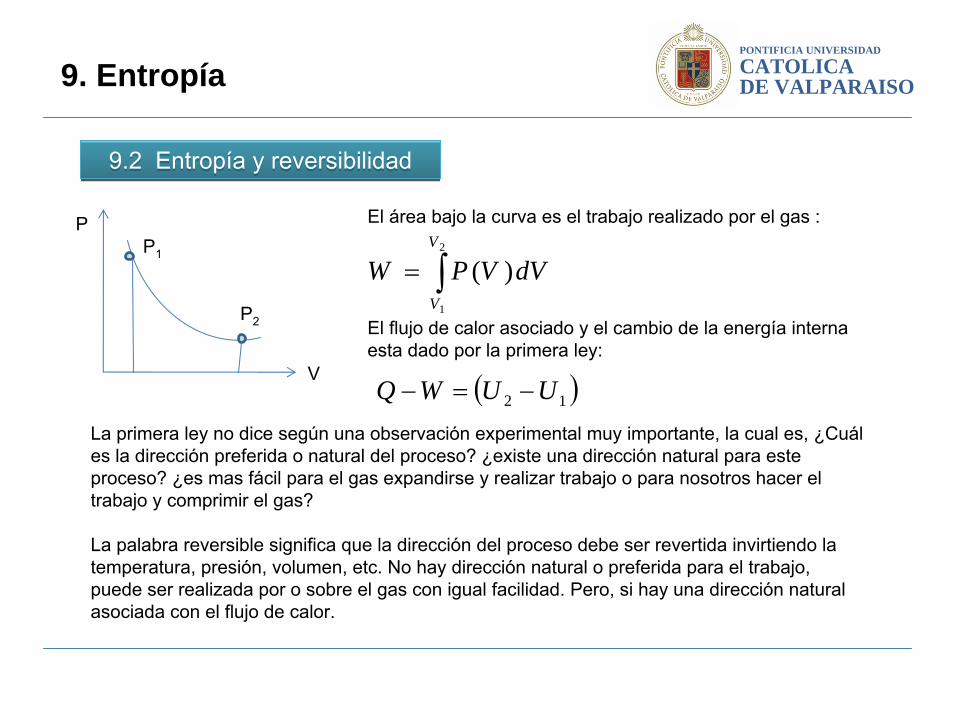
La primera ley no dice según una observación experimental muy importante, la cual es, ¿Cuál es la dirección preferida o natural del proceso? ¿existe una dirección natural para este proceso? ¿es mas fácil para el gas expandirse y realizar trabajo o para nosotros hacer el trabajo y comprimir el gas?
La palabra reversible significa que la dirección del proceso debe ser revertida invirtiendo la temperatura, presión, volumen, etc. No hay dirección natural o preferida para el trabajo, puede ser realizada por o sobre el gas con igual facilidad. Pero, si hay una dirección natural asociada con el flujo de calor.
P1
P2
P
V
El área bajo la curva es el trabajo realizado por el gas :
El flujo de calor asociado y el cambio de la energía interna esta dado por la primera ley:
∫=2
1
)(V
V
dVVPW
( )12 UUWQ −=−

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.2 Entropía y reversibilidad9.2 Entropía y reversibilidad
9. Entropía
Veremos que el flujo de calor a los alrededores lleva a un proceso irreversible. flujo de trabajo realizada por un sistema es reversible.
Una medida de la reversibilidad es la Entropía y puede ser calculada usando:
S es una función de Q y T ya que hay una tendencia natural de Q al viajar de cuerpos calientes a T2 a cuerpos fríos a T1.Es por eso que la temperatura y el calor son variables importantes
TQS =
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.3 Procesos Reversibles9.3 Procesos Reversibles
9. Entropía
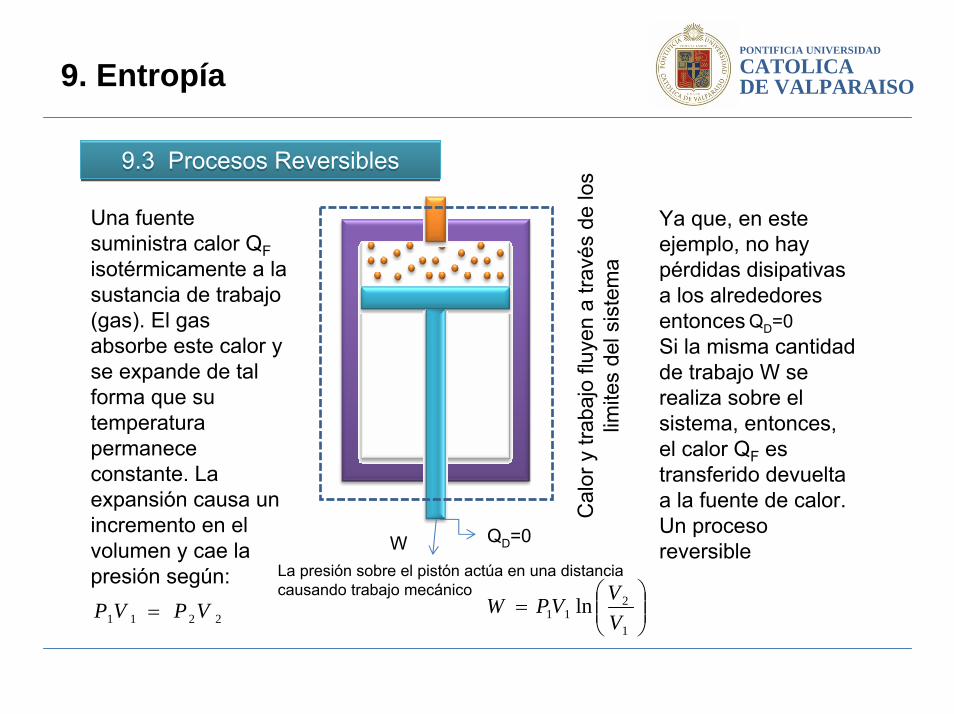
Una fuente suministra calor QFisotérmicamente a la sustancia de trabajo (gas). El gas absorbe este calor y se expande de tal forma que su temperatura permanece constante. La expansión causa un incremento en el volumen y cae la presión según:
2211 VPVP =
W QD=0
La presión sobre el pistón actúa en una distancia causando trabajo mecánico
⎟⎟⎠
⎞⎜⎜⎝
⎛=
1
211 ln
VVVPW
Cal
or y
trab
ajo
fluye
n a
travé
s de
los
limite
s de
l sis
tem
a
Ya que, en este ejemplo, no hay pérdidas disipativasa los alrededores entoncesSi la misma cantidad de trabajo W se realiza sobre el sistema, entonces, el calor QF es transferido devuelta a la fuente de calor. Un proceso reversible
QD=0

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.3 Procesos Reversibles9.3 Procesos Reversibles
9. Entropía
Pregunta:Un proceso reversible de gas ocurre en el sistema de frontera. ¿Cuál es el cambio de entropía del sistema debido a este evento?
Respuesta:en el cálculo de los cambios en la entropía, sólo tenemos que considerar los elementos o componentes del sistema que aceptar o rechazar el calor. Trabajo transferido a un entorno no afecta a la entropía (no hay una dirección preferida para el trabajo)
componente Flujo de calor
Sustancia de trabajo + QF y Tc
Cambiar la entropía del sistema
c
F
TQS
TQS
+=∆
∆=∆
Si este es un proceso reversible, entonces tal vez podría pensar que el cambio de entropía debe ser cero. vamos a ver por qué esto no es tan poco

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.4 Cambios de entropía del universo9.4 Cambios de entropía del universo
9. Entropía
Hasta ahora solo hemos considerado el cambio de entropía de un sistema. Ahora debemos incluir los cambio de entropía de cualquier cosa que absorbe o cede calor en las vecindades del sistema. Por lo tanto para un proceso reversible la entropía del sistema cambia y las cosas que son afectadas directamente por el sistema se encuentran:
componentes Flujo de calor
Fuente de calor -QF y Tc
Sustancia de trabajo +QF y Tc
Disipador de calor +QD=0 y TF
El sistema y los alrededores (fuente de calor y disipadores) juntos deben ser nombrados como universo
0+−
++
=∆
=∆c
F
c
F
TQ
TQ
TQS
Cambio de la entropía del sistema
Cambio de la entropía de la fuente
Cambio de la entropía del disipador
El cambio de la entropía del universo es un proceso reversible y este es cero

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.4 Cambios de entropía del universo9.4 Cambios de entropía del universo
9. Entropía
Para un proceso irreversible, QD no es cero, por lo tanto, la variación de entropía del sistema y las cosas que son afectadas directamente por el sistema esta dada por:componentes Flujo de calor
Fuente de calor -QF y Tc
Sustancia de trabajo +QF y Tc
Disipador de calor +QD y TF
Balance de energía:
WQQWQ
F
DF
≠+= No realiza trabajo y por lo tanto
puede ser solamente recuperado (volver a la fuente) si se suministra trabajo adicional
Cabio total en la entropía:
F
D
c
F
c
F
TQ
TQ
TQ
TQS +
−+
+=
∆=∆
isotérmico
Cambio de la entropía del la sustancia de trabajo
Cambio de la entropía de la fuente
Cambio de la entropía del disipador
F
D
TQS =∆
Esto es mas grande que cero, por lo tanto, existe un incremento neto en la entropía del universo en un proceso irreversible
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.5 Entropía en un ciclo9.5 Entropía en un ciclo
9. Entropía
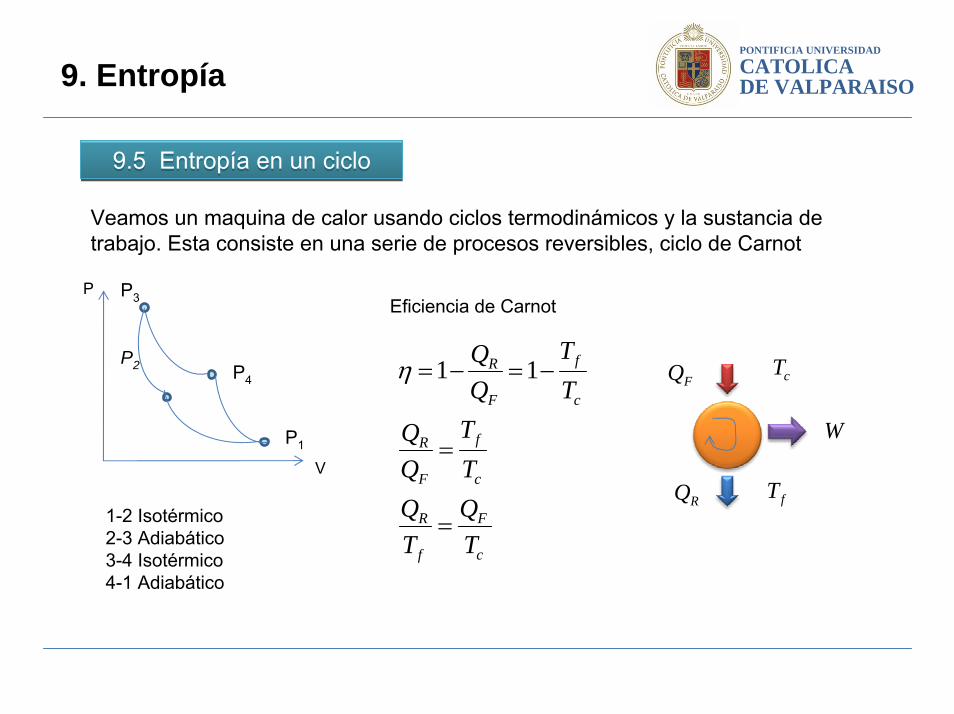
Veamos un maquina de calor usando ciclos termodinámicos y la sustancia de trabajo. Esta consiste en una serie de procesos reversibles, ciclo de Carnot
P
V
P1
P2
P3
P4
1-2 Isotérmico2-3 Adiabático3-4 Isotérmico4-1 Adiabático
c
F
f
R
c
f
F
R
c
f
F
R
TQ
TQ
TT
TT
=
=
−=−= 11η
Eficiencia de Carnot
RQ
FQ
fT
W
cT
PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.5 Entropía en un ciclo9.5 Entropía en un ciclo
9. Entropía
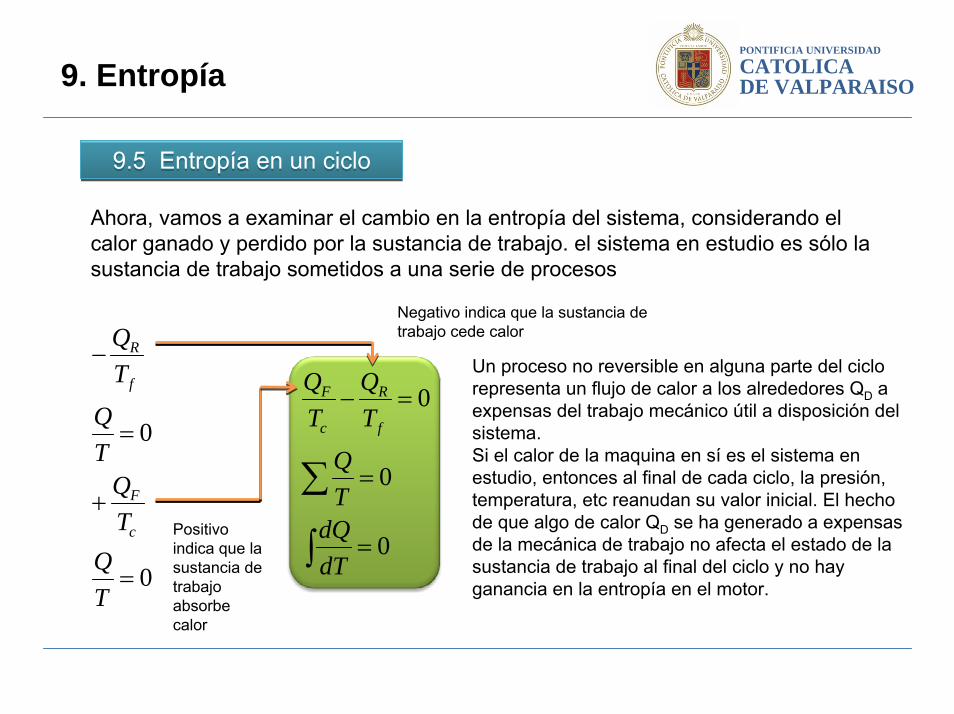
Ahora, vamos a examinar el cambio en la entropía del sistema, considerando el calor ganado y perdido por la sustancia de trabajo. el sistema en estudio es sólo la sustancia de trabajo sometidos a una serie de procesos
0
0
=
+
=
−
TQ
TQ
TQ
TQ
c
F
f
R
0
0
0
=
=
=−
∫
∑
dTdQ
TQ
TQ
TQ
f
R
c
F
Negativo indica que la sustancia de trabajo cede calor
Positivo indica que la sustancia de trabajo absorbe calor
Un proceso no reversible en alguna parte del ciclo representa un flujo de calor a los alrededores QD a expensas del trabajo mecánico útil a disposición del sistema. Si el calor de la maquina en sí es el sistema en estudio, entonces al final de cada ciclo, la presión, temperatura, etc reanudan su valor inicial. El hecho de que algo de calor QD se ha generado a expensas de la mecánica de trabajo no afecta el estado de la sustancia de trabajo al final del ciclo y no hay ganancia en la entropía en el motor.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.6 Entropía 9.6 Entropía
9. Entropía
Para el sistema y el entorno (las fuentes y sumideros), no hay cambio neto en la entropía, si todos los procesos dentro del sistema son reversibles. La entropía total aumenta si todos los procesos dentro del sistema son irreversibles.
•El calor de la fuente pierde entropía –QF/Tc•El calor de los sumideros ganan entropía +QR/Tf•El calor de la maquina en sí es devuelto al mismo estado al final de cada ciclo el calor fluye dentro y fuera de la sustancia de trabajo no se registra ningún cambio neto en la entropía en el motor.•El trabajo es hecho por la maquina y debe dejar el sistema, pero este no conduce a ninguna modificación en la entropía.•Para una maquina reversible, QF/Tc=QR/Tf•Si fuera un proceso disipativo, entonces menos trabajo mecánico se realizaría. El calor de los sumideros ganaría entropía adicional +QD/Tf

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.6 Entropía 9.6 Entropía
9. Entropía
Considere la mezcla de agua fría y caliente. La diferencia de temperatura entre los dos pueden haber sido utilizados como TH y TL para una maquina de calor. Tenemos la oportunidad de realizar trabajo mecánico. Una vez mezclados y a la misma temperatura, la oportunidad de realizar trabajo mecánico se pierde. La entropía es una medida de la perdida de oportunidad.
Entropía es la medida del desorden. energía térmica se plantea debido al movimiento al azar de las moléculas. Hay una tendencia natural para que las cosas se vuelvan más desordenada. Por lo tanto, todo tratamiento de calor implica la participación de alguna tendencia natural al desorden. Esta tendencia hacia el desorden hace todos los procesos reales irreversibles ya que la pérdida de energía para el estado desordenado no puede recuperarse a menos que se aplique un trabajo adicional que es donde está a expensas de otros trastorno en algún otro lugar del universo. Entropía es una medida cuantitativa de la cantidad de desorden asociado con un proceso real.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.7 La Segunda Ley de la Termodinámica 9.7 La Segunda Ley de la Termodinámica
9. Entropía
Cuando un sistema sufre un proceso, la variación de entropía del sistema, sumada al cambio de entropía de los alrededores de la fuente y sumideros, es el total de lavariación de entropía del universo. ∆S (variación total de la entropía)
∆S > 0 Proceso irreversible∆S = 0 Proceso reversible∆S < 0 Proceso imposible
Todo proceso real es irreversible. A mayor irreversibilidad del proceso, mayor seráel incremento de entropía.
Nota: la entropía no es energía, no existe ninguna ley sobre la conservación de la entropía.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.7 La Segunda Ley de la Termodinámica 9.7 La Segunda Ley de la Termodinámica
9. Entropía
Las segunda ley de la termodinámica puede ser expresada en variadas formas. No hay una sola ecuación como en la primera ley. Existen 3 postulados populares para la segunda ley:
1.Ninguna maquina de calor puede continuamente convertir calor en trabajo debido a la necesidad de rechazar el calor.2.El calor no fluirá espontáneamente desde una temperatura baja a una temperatura alta3.La entropía total en cualquier sistema aislado no puede disminuir con el tiempo.
La energía no se puede destruir o crear, pero la oportunidad de utilizarla si puede. La energía se vuelve inutilizable. La entropía es la medida de las perdidas de oportunidad.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.8 Ejemplos9.8 Ejemplos
9. Entropía
1. Un kilogramo de hielo a 0ºC se derrite y se convierte en agua a 0ºC. ¿Cuál es la variación de la entropía del sistema?
SoluciónTemperatura constante a 273KCalor suministrado= +335kJ/kg (calor latente de fusión)
KJS
dQT
S
dQT
S
Q
Q
Q
Q
1227273
335000
1
1
2
1
2
1
==∆
=∆
=∆
∫
∫

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.8 Ejemplos9.8 Ejemplos
9. Entropía
2. Un kilogramo de hielo a 0ºC se calienta a 100ºC. ¿Cuál es la variación de la entropía del sistema?
SoluciónLa temperatura no es constante pero Q debe ser expresado en términos de T
KJmcS
mcdTT
S
dQT
S
mcdTdQ
T
T
Q
Q
1306273373ln
1
1
2
1
2
1
==∆
=∆
=∆
=
∫
∫

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
9.8 Ejemplos9.8 Ejemplos
9. Entropía
2. Un kilogramo de hielo a 0ºC se mezcla con un kilogramo de agua a 100ºC. ¿Cuál es la variación total de la entropía?
SoluciónAntes de mezclar, la entropía total es 1306J/K + 0Luego de mezclar, tenemos 2 kg de agua a 50ºC (323K)
KJmcS
mcdTT
S
dQT
S
mcdTdQ
T
T
Q
Q
1406273323ln
1
1
2
1
2
1
==∆
=∆
=∆
=
∫
∫
KJSS despuesantes 100=−

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
Hemos estudiado la segunda ley para sistemas aislados.Supongamos ahora que el sistema a considerar puede intercambiarcon el medio exterior calor y trabajo (pero no materia: nosrestringimos a un sistema cerrado). ¿Cómo podemos formular lasegunda ley de sistemas?
Supongamos, por ejemplo, que el sistema se halla en contacto conla atmosfera, la cual lo mantiene a temperatura T y presión p fijas. La segunda ley se puede formular si consideramos el conjunto delambiente más el sistema en estudio como un gran sistema aislado.En este caso tendremos
[°]

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
Esta formulación es incómoda, ya que en ella intervienen no sololas propiedades del sistema, sino también las del ambiente exterior.Interesaría obtener una formulación en que solo aparecieranparámetros del propio sistema. Ello es posible si consideramos la primera ley de la termodinámica, por un lado, y la definición de laentropía, por otro. Según la primera ley se tiene
, según la expresión del trabajo realizado sobre el sistema, , podemos encontrar el calor ganado por el
ambiente a partir del calor , ya que ; es decir, el calor ganado por el ambiente es igual al calor perdido por el sistema. Asípues,

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
Como la temperatura del ambiente es constante aunque reciba oceda calor, tendremos, según la definición de entropía, en el casoisotermo
Donde hemos utilizado la formula anterior para . El criterio [°] de la segunda ley se puede escribir, aplicando la expresión para que acabamos de obtener, como
O bien, en forma más compacta,
[°°]

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
Así pues, solo serán espontáneos aquellos procesos que satisfaganla condición [°°], que como hemos visto, es totalmente equivalente ala segunda ley en su expresión con la ventaja de que solo aparecenen ella las variables del propio sistema, ya que T y p son comunes al ambiente y al sistema.
Se define la energía libre de Gibbs o entalpia libre G como G = U +pV – TS, en función de la cual el criterio [°°] se escribe como
Donde los subíndices T y p indican que la variación de G se lleva a término a T y p constantes. Esta es la expresión de la segunda ley para sistemas cerrados sometidos a temperatura y presión fijas.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
En el caso en que V y T (el volumen y la temperatura) semantengan fijos, , por lo cual [°°] queda reducida a
Esta relación se puede escribir en forma más elegante si se definela energía libre de helmholtz, o energía libre, como F = U – TS, en función de la cual [°°°] se escribe como
Cuyos subíndices indican que T y V se mantienen constantes
Observemos finalmente que para un sistema aislado (energía yvolumen constantes, es decir, ) el criterio [°°°] se reduce a T , es decir se recupera el criterio según el cual en un sistema aislado solo son posibles aquellos procesos queaumenten la entropía.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
La relación [°°°] manifiesta un compromiso entre las tendencias aminimizar la energía y a maximizar la entropía. A temperaturasbajas predominara la tendencia a la mínima energía mientras que atemperaturas elevadas predomina la tendencia a maximizar laentropía, como se pone en manifiesto en el siguiente ejemplo.
Ejemplo
Calcular la temperatura por encima de la cual tendrá lugarespontáneamente un determinado proceso termodinámico atemperatura y volumen constantes (por ejemplo, transición de solidoa liquido en un cierto metal) si la variación de energías asociadacon el proceso es y si la variaciónde entropía correspondiente es .
Según el criterio [°°], un proceso a T y V constantes seráespontaneo si

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN SISTEMAS NO
AISLADOS
La temperatura a las que el proceso será espontaneo vendrándadas por la condición
Para temperaturas superiores a 1666,7 K el metal será liquido, y aratemperaturas inferiores será solido.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOINTERPRETACION MICROSCOPICA DE
LA ENTROPIA
El físico austriaco Ludwig Boltzmann propuso hacia 1870 unainterpretación microscópica de la entropía, es decir, unainterpretación en términos de propiedades del movimientomolecular. Hasta ahora todas nuestras afirmaciones podíanprescindir de la naturaleza atómica de los sistemas: las leyes de latermodinámica resultarían validas tanto para un continuo como parau conjunto discreto de moléculas. Boltzmann relaciono la entropíacon S de un estado cualquiera del sistema macroscópico con el numero W de microestados compatibles con el macroestado en cuestión, según la relación
S = k ln W
Donde k, la constante Boltzmann, vale . Esta constante es universal: no depende del sistema ni de ningunacircunstancia concreta, por lo cual tiene una gran importancia en Física.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOINTERPRETACION MICROSCOPICA DE
LA ENTROPIA
Para aclarar que significan los macroestados y microestados, acudiremos a unejemplo sencillo. Consideremos a un gas formado por cuatro moléculas as cualespueden hallarse en un cualquiera de dos recipientes en contacto. Los macroestados(estado que estudia la termodinámica, es decir, apreciables a simple vista) son, si denominamos el numero de moléculas en el recipiente A y el numero de las que se encuentran en el recipiente B.
Macroestados

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOINTERPRETACION MICROSCOPICA DE
LA ENTROPIA
Tenemos, pues cinco posibles macroestados, los cuales especifican tan solo cuantas moléculas hay en cada recipiente, pero no cuales de ellas. El macroestado podría ser determinado por métodos puramente macroscópicos, como por ejemplo, pesando cada recipiente por separado. Un microestado, en cambio, es más detallado y explicita cuales son las moléculas que hay a cada lado. El numero de microestados correspondientes a cada microestado viene dado en este caso, por combinatoria elemental, por
Así, el número de microestados correspondientes a cada macroestado es:
Macroestado Número de microestados W
0 4 1 1 3 4 2 2 6 3 1 4 4 0 1

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
Ahora bien, sabemos por experiencia que si tenemos cuatro moléculas (o cuatro moles) de gas en el recipiente B y abrimos el conducto que lo comunica con el recipiente A, el estado final de equilibrio será la distribución homogénea; es decir, aquella con dos moléculas en cada recipiente. ¡Pero este es precisamente el estado con mayor numero de microestados!
En términos probabilísticos, la probabilidad de encontrar un sistema en un macroestado dado es proporcional al número de microestados correspondientes y, por tanto, el estado estacionario final será el de máxima probabilidad, o de máximo número de microestados. Así pues, la tendencia de la entropía a aumentar y la tendencia del sistema a presentar el macroestado más probable quedan relacionadas por la interpretación de la entropía debida a Boltzmann.
INTERPRETACION MICROSCOPICA DE LA ENTROPIA

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN BIOLOGIA
El paso del mundo microscópico al macroscópico presenta diversas
paradojas. Una de las más llamativas y sorprendentes se refiere a la
aplicación de la segunda ley a los sistemas biológicos. Según la Física –
según la termodinámica- la naturaleza tiende hacia la máxima entropía o,
según Boltzmann, al máximo desorden, ya que un sistema esta tanto más
desordenado cuantos más microestados resultan compatibles con su
macroestado. En cambio, los sistemas biológicos tienden hacia el orden y
la estructuración. ¿Obedecen, pues, los sistemas biológicos a las leyes de
termodinámica? ¿Son reductibles los sistemas biológicos a las leyes de la
física?

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISO
Esta contradicción, que preocupo mucho a los físicos y biólogos, es solo
aparente. En primer lugar, los sistemas biológicos no son temas aislados,
sino que intercambian energía y materia con el mundo exterior: comen,
respiran, excretan, etc. Un sistema biológico muere poco después de ser
aislado. En sistemas no aislados, según acabamos de ver en la sección 4.4,
la entropía puede disminuir, a condición de que aumente suficientemente la
entropía del ambiente. Esto resuelve la paradoja y permite afirmar que no es
incompatible con la segunda ley. Así, muchos seres ingieren sustancias de
gran peso molecular y excretan moléculas sencillas de poco peso molecular.
Como un conjunto de moléculas pequeñas pueden disponerse de muchas
más maneras diferentes que cuando dichas moléculas pequeñas están
ligadas formando una sola molécula, se ingiere poca entropía interior del ser
vivo puede disminuir, ya que la del exterior aumenta.
LA SEGUNDA LEY EN BIOLOGIA

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOLA SEGUNDA LEY EN BIOLOGIA
Es importante notar además que los sistemas vivos se mantienen fuera del
equilibrio. (Un sistema en equilibrio es un sistema muerto.) Se ha podido
comprobar que en muchos sistemas aparecen estructuras espontáneas
cuando están suficientemente alejados del equilibrio, estructuras que se
mantienen mientras se suministra al sistema una potencia suficiente para
mantenerlo lejos del equilibrio. Si se deja de alimentar al sistema con cantidad
suficiente de energía, la estructura desaparece. Este tipo de estructura se
denominan estructuras disipativas, ya que se necesita disipar energía para
mantenerlas. Esto permite afirmar que no sólo la Física y Biología son
compatibles, sino que la termodinámica puede proporcionar una explicación,
en ciertos casos, a los fenómenos de estructuración tan frecuentes en los
seres vivos, que se hallan permanentemente fuera del equilibrio.

PONTIFICIA UNIVERSIDAD
CATOLICA DE VALPARAISOREFERENCIASREFERENCIAS
FisherFisher, A. (2004). , A. (2004). TheThe PhysicsPhysics CompanionCompanion. . PhiladelphiaPhiladelphia: : InstituteInstitute ofof PhysicsPhysics PublishingPublishing, , CapCap. 1.. 1.
JouJou, D.; , D.; LlebotLlebot J.EJ.E.; P.; Péérezrez--GarcGarcíía, C. (1999). a, C. (1999). FFíísica para sica para Ciencias de la VidaCiencias de la Vida. . McMc GrawGraw--Hill, Hill, CapCap. 4.. 4.
Gargallo, L.; Gargallo, L.; RadicRadic, D. (1997). , D. (1997). QuQuíímica Fmica Fíísica Bsica Báásica. sica. TermodinTermodináámica Qumica Quíímicamica. . EdEd. Universidad Cat. Universidad Catóólica de lica de Chile, Chile, CapCap. 1. 1--4.4.
GarcGarcííaa--ColinColin, L. (1986). , L. (1986). IntroducciIntroduccióón a la Termodinn a la Termodináámica mica ClCláásicasica. . EdEd. Trillas, . Trillas, CapCap. 1. 1--7.7.





























