Embed Size (px)
Citation preview

1
UNIVERSITE DE REIMS CHAMPAGNE-ARDENNE
ECOLE DOCTORALE SCIENCES, TECHNOLOGIES, SANTE
THESE DE DOCTORAT
Présentée par
Alexandre Alain VASSEUR
En vue d’obtenir le grade de
DOCTEUR DE L’UNIVERSITE DE REIMS CHAMPAGNE-ARDENNE
SPECIALITE : CHIMIE ORGANIQUE
FONCTIONNALISATION C-H D’HETEROCYCLES DERIVES DE LA BIOMASSE : REACTIONS PALLADO-CATALYSEES
DE HECK DESHYDROGENANTES
Soutenue le 08 novembre 2012 devant le jury :
M. Jean-Cyrille HIERSO, Professeur, Université de Bourgogne. Rapporteur
M. Jérôme WASER, Professeur, Ecole Polytechnique Fédérale de Lausanne. Rapporteur
M. Nicolas RABASSO, Maître de Conférences, Université Paris-Sud 11. Examinateur
M. Norbert HOFFMANN, Directeur de Recherche CNRS, Université de Reims. Examinateur
M. Jacques MUZART, Directeur de Recherche CNRS, Université de Reims. Invité
M. Jean LE BRAS, Chargé de Recherche CNRS, Université de Reims. Directeur de thèse

2
Ce mémoire est le résultat d’un travail réalisé au sein de l’équipe Catalyse de l’UMR CNRS
7312 sous la direction du Dr. Jean LE BRAS. Je tiens à lui exprimer ma sincère reconnaissance pour
m’avoir accueilli dans son laboratoire ainsi que pour ses conseils, sa disponibilité et la totale liberté
avec laquelle il m’a laissé évoluer au cours de ces trois années. Je tiens également à remercier M. le
Dr. Jacques MUZART pour ses conseils, ses remarques, les discussions scientifiques échangées et
l’aide précieuse qu’il m’a fourni lors de la rédaction du manuscrit.
Par ailleurs, j’exprime ma reconnaissance envers les Professeurs Jean-Cyrille HIERSO et
Jérôme WASER pour avoir accepté d’être les rapporteurs de cette thèse. Je suis également
reconnaissant envers MM. les Dr. Nicolas RABASSO et Norbert Hoffmann pour avoir accepté de
participer à ce jury.
Mes remerciements s’adressent aussi au personnel technique et administratif de l’unité et au
Dr. Dominique Harakat avec qui j’ai travaillé sur les études mécanistiques par spectrométrie de
masse à ionisation électrospay.
Enfin, je remercie le CNRS et la région Champagne-Ardenne pour le soutien financier dont ils
m’ont fait bénéficier.

3
Sommaire
Instrumentation ............................................................................................................. 9
Liste des abréviations ................................................................................................... 11
INTRODUCTION ............................................................................................................ 13
Chapitre 1 : Réactions de Heck Déshydrogénantes de furanes et thiophènes avec
des styrènes .................................................................................................................. 17
Introduction ................................................................................................................ 18
La réaction de Mizoroki - Heck .............................................................................. 18
La réaction de Heck oxydante ............................................................................... 20
La Réaction de Heck Déshydrogénante (RHD)...................................................... 21
Partie A : Réactions de Heck Déshydrogénantes de furanes avec des styrènes dans
des conditions douces .................................................................................................. 24
Introduction ................................................................................................................ 24
A.1. Bibliographie ....................................................................................................... 26
A.2. Résultats et discussion ....................................................................................... 36
A.2.1. Constat de départ ........................................................................................ 36
A.2.2. Effet du co-solvant ...................................................................................... 38
A.2.3. Synthèse de styrylfuranes........................................................................... 39
A.2.4. Limites de la méthode ................................................................................ 41
A.2.5. Conclusion ................................................................................................... 41
A.3. Partie expérimentale .......................................................................................... 42
A.3.1. Informations générales ............................................................................... 42
A.3.2. Préparation du (E)-3-furylacrylate de méthyle .......................................... 42
A.3.3. Synthèse de styrylfuranes........................................................................... 43
Partie B : Réactions de Heck Déshydrogénantes de thiophènes avec des styrènes
dans des conditions douces ......................................................................................... 48
Introduction ................................................................................................................ 48
B.1. Bibliographie ....................................................................................................... 49

4
B.2. Résultats et discussion ....................................................................................... 52
B.2.1. Constat de départ ........................................................................................ 52
B.2.2. Synthèse de styrylthiophènes..................................................................... 52
B.2.2.1. Avec les 2-alkylthiophènes .................................................................. 52
B.2.2.2. Avec les 2-halogénothiophènes .......................................................... 53
B.2.3. Limites de la méthode ................................................................................. 54
B.2.4. Valorisation de la réaction .......................................................................... 55
B.2.5. Conclusion ................................................................................................... 56
B.3. Partie expérimentale .......................................................................................... 56
B.3.1. Informations générales ............................................................................... 56
B.3.2. Synthèse de 5-alkyl-2-styrylthiophènes ..................................................... 57
B.3.2. Synthèse de 2-halogéno-5-styrylthiophènes ............................................. 61
Partie C : Influence de l’oxydant sur l’activité du catalyseur ..................................... 65
Introduction ................................................................................................................ 65
C.1. Bibliographie ....................................................................................................... 65
C.1.1. La benzoquinone, un réoxydant du Pd(0) en Pd(II) ................................... 65
C.1.2. La benzoquinone, un promoteur de l’activation de liaisons C-H .............. 68
C.1.3. La benzoquinone, un promoteur de l’élimination réductrice ................... 69
C.1.4. La benzoquinone, un promoteur de la transmetallation oxydante .......... 76
C.1.5. La benzoquinone, un promoteur de l’oxydation de l’hydrure de Palladium
H-Pd ............................................................................................................. 77
C.1.6. La benzoquinone, un inhibiteur de l’élimination ββββ-H ................................ 78
C.1.7. La benzoquinone, un promoteur de la migration de ligand ...................... 80
C.2. Résultats et discussion ........................................................................................ 80
C.2.1. Constat de départ ........................................................................................ 80
C.2.2. Etude de l’étape d’activation de liaison C-H .............................................. 81
C.2.3. Influence de la benzoquinone sur la vitesse initiale .................................. 82
C.2.4. Influence de la benzoquinone sur l’étape d’insertion de l’alcène ............ 83
C.2.5. Cycle catalytique.......................................................................................... 86

5
C.3. Conclusion ........................................................................................................... 87
C.4. Partie expérimentale .......................................................................................... 88
C.4.1. Préparation du 2-méthylthiophène-5d ...................................................... 88
C.4.2. Mode opératoire pour le suivi cinétique des réactions ............................. 88
Chapitre 2 : Etude SM-IES de la RHD de furanes avec des acrylates en utilisant la
benzoquinone comme oxydant et le DMSO comme solvant ..................................... 89
Introduction ................................................................................................................ 90
Partie D : Introduction à l’électrospray couplé masse, ............................................... 91
Introduction ................................................................................................................ 91
D.1. Schéma général du spectromètre de masse Q-TdV .......................................... 91
D.2. Source et interface ............................................................................................. 92
D.3. L’analyseur quadripolaire .................................................................................. 93
D.4. La cellule de collision .......................................................................................... 95
D.5. L’analyseur à Temps de Vol ................................................................................ 95
D.6. L’analyse SM/SM-IES .......................................................................................... 98
D.7. Conclusion ........................................................................................................... 98
Partie E : Etude SM-IES de la RHD de furanes et d’acrylates en utilisant la
benzoquinone comme oxydant et le DMSO comme solvant ..................................... 99
Introduction ................................................................................................................ 99
E.1. Bibliographie ....................................................................................................... 99
E.2. Résultats et discussion ...................................................................................... 110
E.2.1. Méthodologie ............................................................................................ 110
E.2.2. Réaction étudiée ........................................................................................ 110
E.2.3. Rôle du DMSO ............................................................................................ 111
E.2.3.1. Analyse par SM-IES(+) de Pd(OAc)2 et de BQ, dans un mélange
AcOH/DMSO ....................................................................................... 111
E.2.3.2. Effet positif du solvant sur l’activité du catalyseur .......................... 113
E.2.4. Etude de l’étape de l’activation de liaison C-H de l’hétéroarène ............ 114

6
E.2.4.1. Analyse par SM-IES(+) de Pd(OAc)2, BQ, et A1 dans un mélange
AcOH/DMSO ....................................................................................... 114
E.2.5. Etude de l’insertion de l’alcène et de l’élimination ββββ-H .......................... 120
E.2.5.1. Analyse par SM-IES(+) du mélange Pd(OAc)2, BQ, A1 et D1 dans un
mélange AcOH/DMSO ..................................................................... 120
E.2.5.2. Réaction secondaire : activation de l’acrylate de t-butyle D1 ......... 123
E.2.5.3. Insertion de l’alcène dans la liaison Pd-furyle .................................. 124
E.2.5.3. Réaction d’élimination ββββ-H et rôle complexant du produit généré 125
E.2.6. Evolution du milieu réactionnel ................................................................ 127
E.2.7. Cycles catalytiques .................................................................................... 129
E.3. Conclusion ......................................................................................................... 130
E.4. Partie expérimentale ........................................................................................ 131
E.4.1. Dispositif expérimental ............................................................................. 131
E.4.2. Mode opératoire ....................................................................................... 131
E.4.3. SMHR des espèces observées ................................................................... 131
E.4.4. Agrandissement des spectres présentées en partie E ............................. 132
E.4.5. Spectres SM/SM des espèces identifiées ................................................. 143
Chapitre 3 : RHD d’hétéroarènes avec des styrènes en milieu aérobie et effet négatif
des co-oxydants métalliques ..................................................................................... 148
Introduction .............................................................................................................. 149
Partie F : RHD d’hétéroarènes avec des styrènes en milieu aérobie ....................... 150
F.1. Bibliographie ..................................................................................................... 150
F.2. Résultats et discussion ...................................................................................... 161
F.2.1. Constat de départ ...................................................................................... 161
F.2.2. Etude du système oxydant ........................................................................ 161
F.2.3. Synthèse de styrylfuranes dans des conditions aérobies ........................ 162
F.2.3.1. A partir d’alkylfuranes ....................................................................... 162
F.2.3.2. A partir du 2-(méthoxyméthyl)furane ............................................... 164
F.2.3.3. A partir du furane ............................................................................... 164

7
F.2.4. Synthèse de styrylthiophènes dans des conditions aérobies .................. 165
F.2.5. Synthèse de styrylindoles dans des conditions aérobies ......................... 168
F.2.6. Limites de la méthode ............................................................................... 168
F.2.7. Conclusion .................................................................................................. 168
F.3. Partie expérimentale ........................................................................................ 169
F.3.1. Informations générales ............................................................................. 169
F.3.2. Synthèse de styrylfuranes, styrylthiophènes et styrylindoles ................. 169
F.3.3. Synthèse de styrylfuranes en milieu aérobie ........................................... 170
F.3.3.1. A partir d’alkylfuranes ....................................................................... 170
F.3.3.2. A partir du 2-(méthoxyméthyl)furane ............................................... 173
F.3.3.3. A partir du furane ............................................................................... 175
F.3.4. Synthèse de styrylthiophènes en milieu aérobie ..................................... 178
F.3.4.1. A partir d’alkylthiophènes ................................................................. 178
F.3.4.2. A partir du thiophène ......................................................................... 181
F.3.5. Synthèse de styrylindoles en milieu aérobie ............................................ 181
Partie G : Etude mécanistique de la réaction et de l’effet des co-oxydants
métalliques ................................................................................................................. 182
G.1. Etude du mécanisme ........................................................................................ 182
G.1.1. Activation de la liaison C-H ....................................................................... 182
G.1.2. Rôle de l’oxygène moléculaire ................................................................. 183
G.1.3. Analyse par SM-IES du mélange réactionnel ........................................... 184
G.1.4. Cycle catalytique ....................................................................................... 187
G.2. Influence du co-oxydant métallique ................................................................ 188
G.2.1. Influence sur le profil cinétique................................................................ 188
G.2.2. Analyse par SM-IES de la réaction en présence d’acétate d’argent ....... 190
G.2.3. Conclusion ................................................................................................. 192
G.3. Partie expérimentale ........................................................................................ 192
G.3.1. Mode opératoire pour les suivis cinétiques des réactions ..................... 192
G.3.2. Dispositif expérimental............................................................................. 193

8
G.3.3. SMHR des espèces observées ................................................................... 193
G.3.4. Spectres SM/SM des espèces identifiées ................................................. 194
CONCLUSION .............................................................................................................. 196

9
Instrumentation
Les solvants
La plupart des réactions sont réalisées avec les solvants commerciaux. Lorsque les traces
d’eau doivent être éliminées, ils sont purifiés par distillation sous argon : le tétrahydrofurane (THF) et
l’éther diéthylique sur sodium / benzophénone. L’eau utilisée comme solvant est à pH = 7.
Chromatographies
L’évolution des réactions a été suivie par chromatographie sur couche mince de silice (Merck
Art 5554 DC Alufolien Kieselgel 60 PF254). La purification des produits se fait par chromatographie
sur gel de silice (MERCK Kieselgel 60, 0,063-0,200 nm).
Caractérisation des produits :
Les spectres de résonance magnétique nucléaire (RMN) du proton 1H et du carbone
13C sont
enregistrés sur un appareil Brucker AC 250, respectivement à 250,13 et 62,89 MHz, à température
ambiante dans CDCl3 avec le tétraméthylsilane comme référence interne, ou dans l’acétone-d6. Pour
les spectres du fluor 19
F, enregistrés à 235,36 MHz, la référence utilisée est le
trichlorofluorométhane. Les déplacements chimiques sont exprimés en ppm, les couplages en hertz
et la multiplicité des signaux est symbolisée comme suit : s (singulet), sl (singulet large), d (doublet), t
(triplet), q (quadruplet), quint (quintuplet), sext (sextuplet), sept (septuplet), m (massif ou multiplet).
Les spectres NOEDIFF ont été enregistrés sur un appareil Brucker DRX 500 (1H : 500 MHz,
13C : 125,8
MHz).
Les spectres infrarouges (IR) sont enregistrés sur un appareil Avatar 320 FT-IR, en pastille de
KBr pour les solides ou en film pour les huiles.
Les spectres de masse basse et haute résolution ont été réalisés sur un appareil Q-TdV
(Micromass Manchester) possédant une source électrospray. Les spectres de masse théoriques sont
calculés à partir du programme « Masslynx ».

10
Les analyses élémentaires sont effectuées sur un appareil Perkin-Elmer CHN 2400.
Les chromatographies en phase gazeuse (CPG) sont réalisées sur un appareil THERMOQUEST
FOCUS GC équipé d’une colonne DB1 (30 m x 0,25 mm x 0,25 μm). La programmation utilisée est :
température initiale 50 °C, palier de 15 °C/min, température finale : 250 °C, débit de N2 : 1 mL/min.
Les spectres de chromatographie en phase gazeuse couplée masse (CPG/SM) ont été réalisés
sur un appareil THERMOQUEST TRACE GC série 2000 couplé à un spectromètre THERMOQUEST
TRACE MS. La colonne est une TR1-MS (30 m x 0,25 mm x 0,25 μm). La programmation utilisée est :
température initiale 50 °C, palier de 20 °C/min, température finale : 250 °C, débit d’Hélium : 1
mL/min. Les spectres de masse sont réalisés en impact électronique (I.E., 70 eV).

11
Liste des abréviations
A
Ac : acétyle
APTS : acide paratoluènesulfonique
Ar : aryle
atm : atmosphère
B
BBT : 5-(but-3-èn-1-yl)-2,2'-bithiophène
BINAP : (diphénylphosphino)-1,1’-binaphtyle
Bn : benzyle
Boc : tertiobutylocarbonyle
BQ : benzoquinone
Bu : butyle
Bz : benzoate
Bzq : benzoquinoline
C
CPG : chromatrographie en phase gazeuse
D
dba : dibenzylidéneacétone
DCM : dichlorométhane
DILAM : Désorption/ionisation laser assistée par matrice
DMAc : N,N-diméthylacétamide
DMF : N,N-diméthylformamide
dmphen : 2,9-diméthyl-1,10-phénanthroline
DMSO : diméthylsulfoxyde
E
e.e. : excès énantiomérique
EP : éther de pétrole
équiv. : équivalent Et : éthyle
eV : électronvolt
H
h : heure
I
IMes : 1,3-bis(2,4,6-triméthylphényl)imidazol-2-ylidène
IPA : ionisation à pression atmosphérique
IR : Infrarouge

12
M
Me : méthyle
min : minute
MW : volume/volume/heure
m/z : rapport masse sur charge
N
n- : normal
NBS : N-bromosuccinimide
P
Ph : phényle
PPV : poly(p-phénylènevinylène)
Pr : propyle
Q
Q : quadripôle
R
Rdt : rendement
RHD : réaction de Heck déshydrogénante
RMN : résonnance magnétique nucléaire
S
SCL : systèmes conjugués linéaires
SM/SM : spectrométrie de masse en tandem
SMHR : spectre de masse haute résolution
SM-IES : spectrométrie de masse à ionisation électrospray
T
t-amylOH : alcool tertio-amylique
t-Bu : tertio-butyl
TA : température ambiante
TDF : théorie de la fonctionnelle de la densité
TdR : taux de roulement
TdV : temps de vol
Tf : triflate
TFA : acide trifluoroacétique
THF : tétrahydrofurane
TIPS : triisopropylsilyle
TM : tamis moléculaire
TMBA : acide 2,4,6-triméthylbenzoique
U
u.m.a. : unité de masse atomique
13
INTRODUCTION
Les molécules organiques développées industriellement, sont utilisées dans des domaines
aussi variés que la chimie médicinale, la pharmacochimie, l’agrochimie, les cosmétiques, et les
matériaux organiques, d’où la nécessité de développer des méthodes de synthèse nouvelles et
efficaces. Par ailleurs, les exigences de plus en plus strictes du développement industriel, du contrôle
des autorités de tutelles et des autorisations de mise sur le marché nécessitent une évolution vers
une chimie plus performante et plus soucieuse de l’environnement.1
L’une des solutions à ces défis est le développement de réactions catalysées par des métaux
de transition. L’utilisation de catalyseurs issus de ces métaux a révolutionné la synthèse organique
classique en permettant l’accès à toute une gamme de réactions inédites, plus propres, jusqu’alors
impossibles ou indirectes. Ces outils de synthèse ont connu un essor important ces dernières années
et les complexes de palladium sont parmi les plus étudiés et utilisés.
En effet, depuis une trentaine d’années, leur utilisation comme catalyseur en synthèse
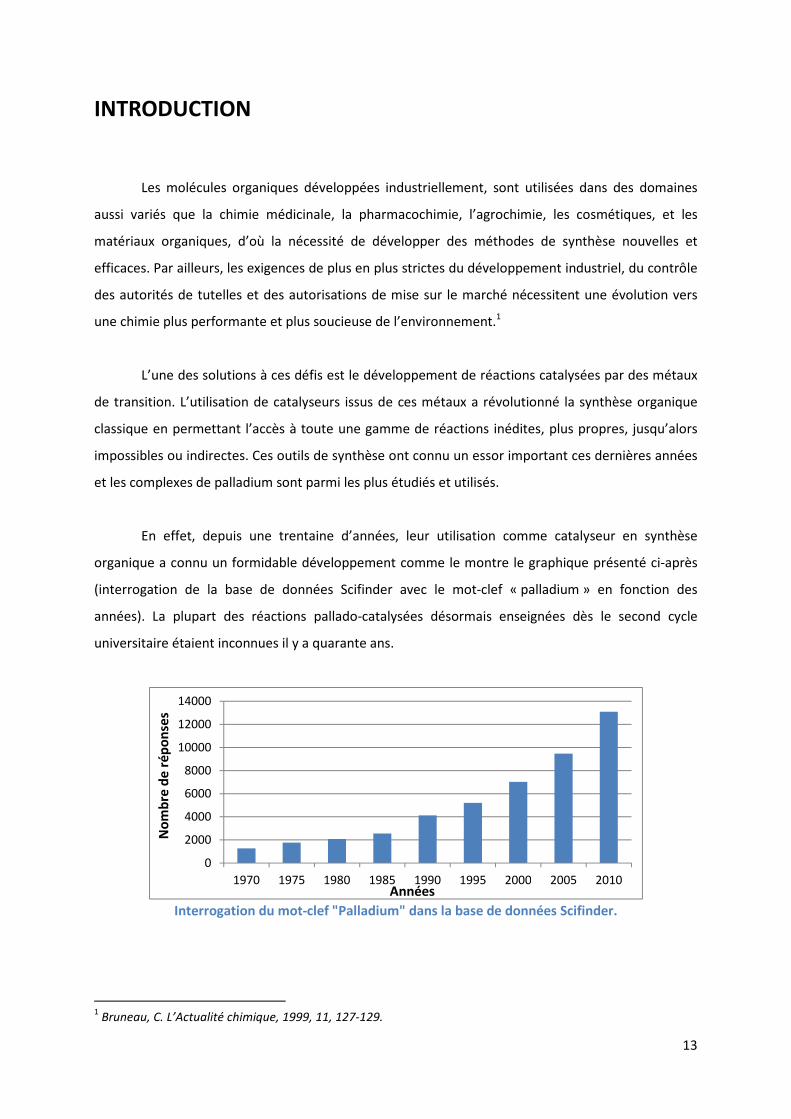
organique a connu un formidable développement comme le montre le graphique présenté ci-après
(interrogation de la base de données Scifinder avec le mot-clef « palladium » en fonction des
années). La plupart des réactions pallado-catalysées désormais enseignées dès le second cycle
universitaire étaient inconnues il y a quarante ans.
Interrogation du mot-clef "Palladium" dans la base de données Scifinder.
1 Bruneau, C. L’Actualité chimique, 1999, 11, 127-129.
0
2000
4000
6000
8000
10000
12000
14000
1970 1975 1980 1985 1990 1995 2000 2005 2010
No
mb
re d
e r
ép
on
ses
Années

14
Les catalyseurs au palladium sont devenus un outil de synthèse extrêmement performant
tant en recherche fondamentale qu’en milieu industriel.2 En effet, les modulations possibles des
complexes palladiés permettent une adaptation à un grand nombre de substrats et de réactions. De
plus, ces complexes peuvent être compatibles avec la présence dans les substrats de toute une
gamme de fonctionnalités (cétones, alcools, aldéhydes, groupements soufrés…), évitant ainsi parfois
des étapes lourdes de protection-déprotection. L’exemple suivant illustre l’intérêt ces nouvelles
réactions.
Schéma 1. Méthode traditionnelle de synthèse du précurseur de l’himbacine.
Le composé diénique 1 est un précurseur direct de l’himbacine, produit naturel et
antagoniste des récepteurs de la thrombine,3 actuellement en essais clinique. La voie
traditionnellement utilisée4 pour synthétiser ce diène est constituée de cinq étapes nécessitant des
purifications intermédiaires et l’élimination délicate d’un produit secondaire soufré. Une autre
stratégie,5 implique une réaction de Heck catalysée par du palladium (0) (schéma 2). Cette séquence
réactionnelle ne nécessite que deux étapes et ne requière pas la purification du triflate
intermédiaire. Elle présente donc le double avantage d’être à la fois plus économique et plus
écologique (moins de déchets sont produits).
2 Campagne, J-M.; Prim, D. Les complexes de palladium en synthèse organique, CNRS Edition : Paris, 2001.
3 Wang, Y.; Greenlee, W. J.; Hu, Z.; Xia, Y.; Ahn, H-S.; Boykow, G.; Hsieh, Y.; Palamanda, J.; Agans-Fantuzzi, J.;
Kurowski, S.; Graziano, M.; Chintala, M. J. Med. Chem. 2008, 51, 3061-3064. 4 (a) Chackalamannil, S.; Davies, R. J.; Asberom, T.; Doller, D.; Leone D. J. Am. Chem. Soc. 1996, 118, 9812-9813.
(b) Tanikaga, R.; Nozaki, Y.; Tamura, T. Kaji, A. Synthesis, 1983, 134-136. 5 Lai, G.; McAllister, T. Synth. Comm. 1999, 29, 409-413.

15
Schéma 2. Méthode industrielle de synthèse du précurseur de l’himbacine.
La catalyse au palladium (0) est aujourd’hui relativement bien maîtrisée et de nombreuses
méthodes de couplages ont été développées.6 Bien que plus ancienne, le procédé Wacker étant
utilisé de façon industrielle depuis les années 1960,7 la chimie du palladium (II) est moins connue et
donc moins utilisée industriellement que celle du Pd(0). Pourtant, elle est attrayante puisqu’elle
permet des conditions douces et de nouvelles perspectives de synthèse.
L’utilisation en synthèse organique des complexes de palladium (II) repose sur deux
propriétés :
- Leur propriété d’oxyder de nombreux substrats, impliquant de facto leur réduction en Pd(0).
En conséquence, la catalyse au palladium (II) requiert l’utilisation d’un oxydant en quantité
stœchiométrique, d’où l’incompatibilité de l’utilisation des ligands de type phosphine, qui
ont grandement contribué au succès du palladium (0) mais qui sont sensibles à l’air.
- La capacité du palladium (II) à se coordonner aux oléfines.
L’un des potentiels importants pour la mise au point de nouvelles voies de synthèse via des
complexes de palladium (II) réside dans la fonctionnalisation de liaisons C-H. C’est dans le cadre
d’une recherche fondamentale que nous nous sommes intéressés à ce concept dans des Réactions
de Heck Déshydrogénantes (RHD) impliquant des hétérocycles dérivés de la biomasse.
Le travail présenté s’articule autour de deux objectifs : la valorisation d’hétérocycles pouvant
être dérivés de la biomasse tels que les furanes et l’utilisation de conditions réactionnelles plus
respectueuses de l’environnement que celles traditionnellement utilisées.
6 Tsuji J. Palladium Reagents and Catalysts : New Perspectives for the 21st Century, Wiley and Sons : New York,
2004. 7 Smidt, J.; Hafner, W.; Jira, R.; Sedlmeier, J.; Sieber, R.; Ruttinger, R.; Kojer, H. Angew. Chem. 1959, 71, 176-182.

16
Le premier chapitre présentera tout d’abord des RHD mettant en jeu des furanes. Puis, nous
exposerons une nouvelle méthode de RHD de furanes avec des styrènes à température ambiante et
extensible aux thiophènes. L’influence de l’oxydant sur l’activité du catalyseur sera discutée.
Le second chapitre sera consacré à l’étude mécanistique de la RHD de furanes avec des acrylates
dans les conditions réactionnelles développées au chapitre précédent. Cette étude est réalisée par
« Spectrométrie de Masse - Ionisation par Electrospray » (SM-IES) dont le principe sera détaillé. Des
exemples d’études mécanistiques déjà réalisées à l’aide de cette technique seront fournis.
Le troisième chapitre s’intéressera aux RHD ayant recours à une réoxydation du palladium (0) par
l’oxygène en présence ou non d’une quantité catalytique de co-oxydant. Après un état des lieux de la
littérature, une méthode de couplage de Heck déshydrogénant de furanes, thiophènes et indoles
avec des styrènes faisant intervenir l’oxygène comme unique oxydant sera proposée, et le
mécanisme de la réaction sera étudié. Nous montrerons également que l’utilisation de co-oxydants
métalliques en association avec l’oxygène peut avoir un effet négatif sur la transformation.

17
Chapitre 1 :
Réactions de Heck Déshydrogénantes de
furanes et thiophènes avec des styrènes

18
Introduction
La réaction de Mizoroki - Heck
La réaction de Heck ou réaction de Mizoroki – Heck est une réaction de couplage pallado-
catalysée entre des oléfines et des halogénures ou triflates de dérivés aromatiques ou vinyliques, en
présence de bases telles que la triéthylamine et l’acétate ou carbonate de sodium.8 Ces réactions ont
été d'abord connues comme des vinylations d’halogénures d'aryle (ou arylation d’oléfines) en
présence de Pd(0) (Schéma I1).
Schéma I1. Vinylation d’un halogénure d’aryle selon la réaction de Heck.
Dans leurs travaux originels, Heck et Nolley ont cité la publication signée par Mizoroki, Mori et
Osaki. Les articles des équipes de Mizoroki et Heck ont été soumis respectivement le 20 octobre
19709 et le 13 janvier 1972,
10 mais, le 12 janvier 1971, Julia et Duteil déposèrent un pli cacheté
auprès de la Société Chimique de France révélant la même réaction. Ce pli cacheté, ouvert le 5 mai
1973 et ensuite publié,11
n’est pas mentionné dans la plupart des comptes-rendus et revues. Ce type
de réaction, qui aurait pu se nommer « réaction de Mizoroki-Julia-Heck », a conduit à pléthore
d’articles et, récemment à un livre.12
Avant de trouver les conditions catalytiques menant à l’addition du palladium dans la liaison
Arène – Halogène, Heck a réalisé, à la fin des années 1960, la synthèse in situ de complexes ArPdX à
partir de la réaction entre des sels de palladium et des aryles stanneux, mercuriques ou plombeux.13
8 Clayden J.; Greeves, N.; Warren, S. Organic Chemistry 1
st Ed., Oxford University Press: Oxford, 2000.
9 Mizoroki, T.; Mori, K.; Ozaki, A. Bull. Chem. Soc. Jpn. 1971, 44, 581.
10 Heck, R. F.; Nolley, J. P., Jr. J. Org. Chem. 1972, 37, 2320-2322.
11 Julia, M.; Duteil, M. Bull. Soc. Chim. Fr. 1973, 2790.
12 Oestreich, M. The Mizoroki-Heck Reaction Ed., Wiley: Chichester, 2009.
13 (a) Heck, R. F. J. Am. Chem. Soc. 1968, 90, 5518-5526. (b) Heck, R. F. J. Am. Chem. Soc. 1969, 91, 6707-6714.

19
Ces dernières années, cette transmétallation a été revisitée. Effectuée désormais en utilisant
des quantités catalytiques de palladium, elle est couramment nommée « réaction de Heck
oxydante »,14
« réaction de type Mizoroki – Heck »,15
ou « réaction de couplage croisé de Heck ».16
La réaction de Mizoroki-Heck est initiée par une espèce à base de Pd(0). Selon les conditions
réactionnelles, elle procède suivant deux mécanismes majeurs : le mécanisme neutre et le
mécanisme cationique. Le mécanisme cationique s'effectue en présence d’additifs, notamment des
sels d'argent. L'argent en fixant les anions halogénures favorise ainsi l'état à 16 électrons du
complexe de palladium cationique. Dans ces conditions, on assiste parfois à une plus grande
efficacité de la réaction accompagnée d'une augmentation des stéréosélectivités.
Schéma I2. Cycle catalytique du mécanisme neutre de la réaction de Mizoroki - Heck.
Le cycle débute par la génération du Pd(0) suite à la réduction du Pd(II) en présence de
ligands. Une addition oxydante de l’halogénure d’aryle au Pd(0) précède la syn-addition de l’oléfine
14
(a) Enquist, P.-A.; Nilsson, P.; Sjöberg, P.; Larhed, M. J. Org. Chem. 2006, 71, 8779-8786 et references citées.
(b) Gligorich, K. M.; Sigman, M. S. Chem. Commun. 2009, 3854–3867 15
Ye, Z.; Chen, F.; Luo, F.; Wang, W.; Lin, B.; Jia, X.; Cheng, J. Synlett 2009, 2198-2200. 16
Stefani, H. A.; Pena, J. M.; Gueogjian, K.; Petragnani, N.; Vaz, B. G.; Eberlin, M. N. Tetrahedron Lett. 2009, 50,
5589-5595.

20
au complexe organo-palladié précédemment formé. L’étape d’élimination de l’hydrure en β est
cruciale pour l'orientation future de la réaction. En effet, si elle ne se réalise pas, on assiste à d'autres
processus réactionnels (formant une suite en cascade avec la réaction de Heck). L’élimination β-H
génère la nouvelle oléfine substituée et l'hydrure de palladium. C'est alors qu'intervient la base qui
décompose cet hydrure de palladium en regénérant le Pd(0) et en piègeant l’acide HX formé. A
quelques exceptions près, dues notamment aux composés de départ, l’alcène formé est
généralement de stéréochimie E. L’étape de rotation est fondamentale car elle permet de placer un
hydrogène en β-syn du palladium, ce qui donne la possibilité d’une syn-élimination.17
Les réactions de Heck sont devenues aujourd’hui des méthodes versatiles de construction
asymétrique de liaison C-C et sont parmi les mieux éprouvées dans la construction des carbones
tertiaires et quaternaires chiraux. En effet, l'utilisation du BINAP en 1989 par Shabasaki et al.18
ainsi
que les travaux du groupe d’Overmann19
la même année, ont posé les fondements des réactions
asymétriques de Heck. Celles-ci ont connu une rapide expansion dans les années qui suivirent, et
sont largement utilisées en synthèse de produits naturels.20
Dans ce cadre, les travaux de Narasaka et
al.21
sur les réactions amino-Heck ont ouvert le chemin à la synthèse des hétérocycles azotés. La
réaction de Heck tolère une grande variété de substrats et réactifs conduisant ainsi à des produits
pouvant être densément fonctionnalisés. L’alcène peut en effet être mono- ou disubstitué, riche ou
pauvre en électrons.
La réaction de Heck oxydante
La réaction de Heck oxydante implique une catalyse au palladium (II) et des halogénures
d’aryles mercuriques, stanneux, ou encore plombeux, d’où une production de déchets plus
importante que dans la réaction de Mizoroki – Heck. La première étape du cycle catalytique consiste
dans ce cas en une transmetallation pour générer l’espèce ArPdX. Après la syn-addition de l’alcène et
l’élimination β-H conduisant à un hydrure de palladium et au produit de couplage, une élimination
17
Rabasso, N. Chimie Organique. Hétéroéléments, stratégies de synthèse et chimie organométallique 2e Ed., De
Boeck Université: Paris, 2009. 18
Sato, Y.; Sodeoka, M.; Shibasaki, M. J. Org. Chem. 1989, 54, 4738-4739. 19
Carpenter, N. E.; Kucera, D. J.; Overman, L. E. J. Org. Chem. 1989, 54, 5846-5848. 20
Amy B. D.; Overmann L. E. Chem. Rev. 2003, 103, 2945-2963. 21
Kitamura, M.; Narasaka, K. Chem. Rec. 2002, 2, 268-277; Kitamura, M.; Yanagisawa, H.; Yamane M.;
Narasaka, K. Heterocycles 2005, 62, 273-277.

21
réductrice fournit le Pd(0) qui est ensuite réoxydé pour régénérer les espèces catalytiques actives
(Schéma I3).
Schéma I3. Cycle catalytique du mécanisme de la réaction de Heck oxydante.
La Réaction de Heck Déshydrogénante (RHD)
Le couplage pallado-catalysé d’un arène ArH avec un alcène peut également conduire à un
alcène arylé. Comme la réaction de Heck oxydante présentée précédemment, celui-ci nécessite du
Pd(II) comme catalyseur et un réoxydant. Bien que cette réaction soit parfois appelée « réaction
oxydante de type Heck »,22,23
son mécanisme est différent puisqu’une activation de la liaison C-H de
l’arène se substitue à l’étape de trans-métallation (Schéma I4).
22
Rodriguez, A.; Moran, W. J. Eur J. Org. Chem. 2009, 1313-1316. 23
Ferreira, E. M.. Zhang, H.. Stoltz, B. M. dans The Mizoroki-Heck Reaction Oestreich, M. Ed Wiley : Chichester,
2009.

22
PdIIX2activation C-H
syn-insertion
élimination -H
oxydation
PdIIX
R
Ar
HPdIIX
Pd0
2 HX + oxydant
oxydant réduit
HX
HX
ArPdIIX
R
Ar
R
Ar-H
Commence ici
éliminationréductrice
X = OAc, OCOCF3...
Schéma I4. Cycle catalytique du mécanisme de la Réaction de Heck Déshydrogénante.
L’équipe de Fujiwara ayant, en 1967, révélé la synthèse de stilbène à partir de la réaction d’un
complexe PdCl2-styrène avec du benzène24
, puis en 1968, le couplage oxydant entre le styrène et le
benzène avec un taux de roulement (TdR) égal à deux25
, ces réactions pallado-catalysées sont parfois
appelées « réactions de Fujiwara »,26, 27
« réactions de Fujiwara – Moritani »,28,29
réactions de
Fujiwara – Moritani oxydantes de Heck ».30
Cependant, Fujiwara et Kitamura ont utilisé, en 2005, le
terme de « réaction de Fujiwara » pour les hydroarylations pallado-catalysées d’alcynes et
d’alcènes.31
Par conséquent, notre équipe a proposé en 2011 le terme de « Réaction de Heck
Déshydrogénante » (RHD) pour désigner les couplages pallado-catalysés oxydants d’arènes et
d’hétéroarènes avec des alcènes,32
terme plus approprié qui avait déjà été utilisé par le passé.33
Parmi les trois méthodes de synthèse d’alcènes arylés présentées ci-dessus, et si l’on considère
uniquement les substrats, c’est la RHD qui respecte le plus le principe de l’économie d’atome,
24
Moritani, I.; Fujiwara, Y. Tetrahedron Lett. 1967, 1119-1122. 25
Fujiwara, Y.; Moritani, I.; Matsuda, M.; Teranishi, S. Tetrahedron Lett. 1968, 3863-3865. 26
Dams, M.; De Vos, D. E.; Celen, S.; Jacobs, P. A. Angew. Chem. Int. Ed. 2003, 42, 3512-3515. 27
Zhang, Y.-H.; Shi, B.-F.; Yu, J.-Q. J. Am. Chem. Soc. 2009, 131, 5072-5074. 28
Mikami, K.; Hatano, M.; Terada, M. Chem. Lett. 1999, 55-56. 29
Zhang Zhang, H.; Ferreira, E. M.; Stoltz, B. M. Angew. Chem. Int. Ed. 2004, 43, 6144-6148. 30
Pan, D.; Yu, M.; Chen, W.; Jiao, N. Chem. Asian J. 2010, 5, 1090-1093. 31
Fujiwara, Y.; Kitamura, T. Handbook of C-H Transformations Dyker, G. Ed., Wiley: Darmstadt, 2005, 1, pp 194-
202. 32
Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170-1214. 33
Ferreira, E.M.;Zhang,H.; Stoltz,B.M. Tetrahedron 2008, 64, 5987-6001.

23
puisque son résultat est la formation d’une liaison C-C à partir de deux liaisons C-H. Par ailleurs, la
réaction de Heck et la réaction oxydante de type Heck génèrent des quantités stoechiométriques de
sels halogénés peu valorisables. Compte-tenu de ces observations, une recherche active sur les RHD
intermoléculaires se développe puisqu’elles excluent une étape préalable de fonctionnalisation de
l’arène et peuvent limiter la production de déchets.
Nous proposons dans ce chapitre une nouvelle méthode de RHD de furanes avec le styrène
dans des conditions douces. Nous montrerons ensuite sa compatibilité non seulement avec les
styrènes halogénés, mais aussi avec les thiophènes. Enfin, l’influence de l’oxydant sur l’activité du
catalyseur sera discutée.

24
Partie A : Réactions de Heck Déshydrogénantes de furanes
avec des styrènes dans des conditions douces
Introduction
La valorisation non alimentaire de la biomasse en chimie fine constitue un défi majeur du
21ème
siècle. Son utilisation comme source de matière première alternative suscite de nombreux
travaux puisqu’elle présente l’avantage d’être renouvelable.
L’aldéhyde pyromucique (souvent appelé furfural) est une molécule facilement accessible à
partir de la biomasse, par hydrolyse de sous-produits de l’agriculture comme la paille, les coques de
céréales et les poudres de bois.34
Ainsi, les hémicelluloses contenues dans la biomasse sont
hydrolysées en hexoses (glucose et fructose) et en pentoses (principalement le xylose). Ces derniers,
après cyclo-déshydratation,35,36
mènent à l’aldéhyde pyromucique (schéma A1). Avec une production
annuelle avoisinant les 250.000 tonnes, l’aldéhyde pyromucique apparait comme le seul composé
organique insaturé produit en grandes quantités à partir de sucres.33
Schéma A1. De la biomasse à l’aldéhyde pyromucique.
Cette molécule est utilisée comme solvant pour l’extraction sélective d’oléfines et de
composés aromatiques issus d’huile de lubrification. Elle est aussi utilisée comme intermédiaire pour
la chimie pharmaceutique, des parfums et des arômes. Par ailleurs, outre ses propriétés
thermodurcissantes et sa résistance à la corrosion lui valant d’être utilisé dans la composition de
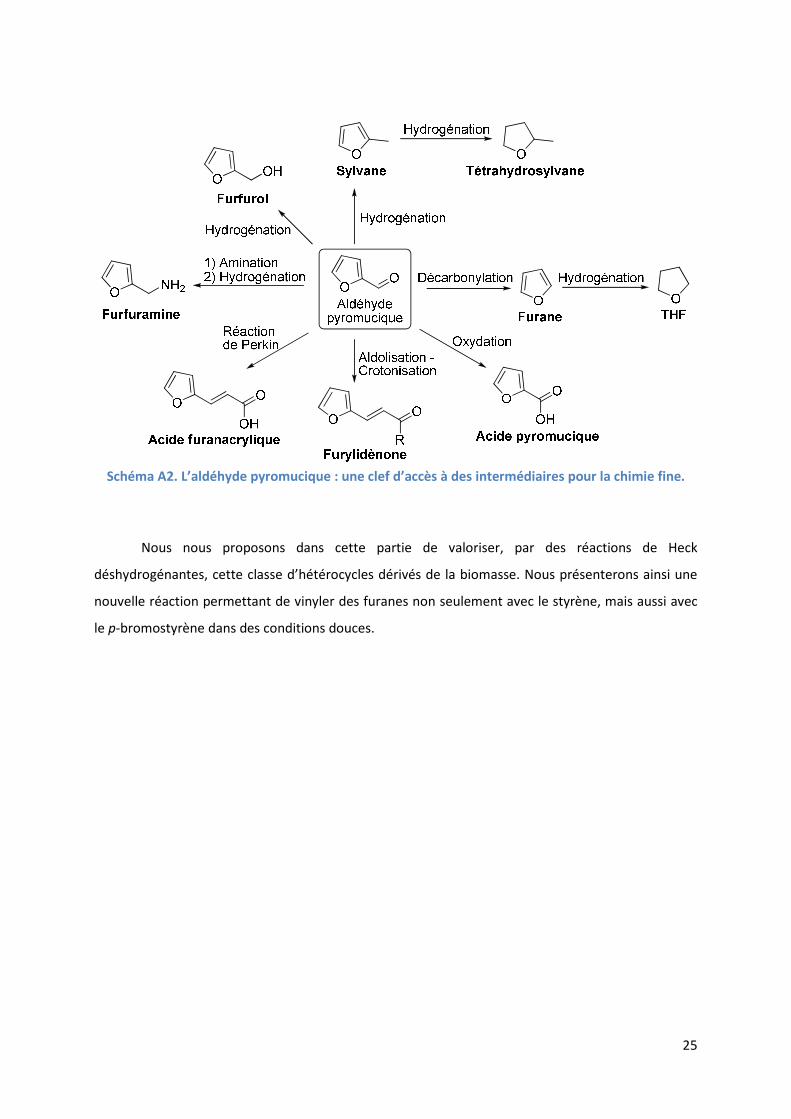
matériaux, l’aldéhyde pyromucique est une « molécule plateforme » offrant une clef d’accès à une
large variété de molécules par des procédés chimiques industriels simples (Schéma A2).
34
Mamman, A. S.; Lee, J.-M.; Kim, Y.-C.; Hwang, I. T.; Park, N.-J.; Hwang, Y. K.; Chang, J.-S.; Hwang, J.-S. Biofuels,
Bioprod. Bioref. 2008, 2, 438–454. 35
a) McKillip, W. J. Chem. Technol. 1981, 11, 501-527. b) Lichtenthaler F. W. Acc. Chem. Res. 2002, 35, 728-737. 36
Lichtenthaler, F. W.; Brust, A.; Cuny, E. Green Chem. 2001, 3, 201-209.
25
Schéma A2. L’aldéhyde pyromucique : une clef d’accès à des intermédiaires pour la chimie fine.
Nous nous proposons dans cette partie de valoriser, par des réactions de Heck
déshydrogénantes, cette classe d’hétérocycles dérivés de la biomasse. Nous présenterons ainsi une
nouvelle réaction permettant de vinyler des furanes non seulement avec le styrène, mais aussi avec
le p-bromostyrène dans des conditions douces.

26
A.1. Bibliographie
Parmi les arènes utilisés pour les RHD, les furanes font partie de ceux qui ont reçu le moins
d’attention, probablement en raison de leur sensibilité aux conditions acides généralement requises.
En 1976, Kozhevnikov révèle le premier couplage pallado-catalysé oxydant mettant en jeu un
furane et du Pd(II) en quantité catalytique. Le couplage du furane avec l’éthylène, dans le DMF à
96°C, en présence d’acétate de cuivre (II) comme réoxydant, d’acétate de palladium comme
catalyseur et de chlorure de calcium comme additif est alors réalisé avec un faible rendement
(Schéma A3). 37
Schéma A3. Première RHD mettant en jeu un furane.
En 1978, l’équipe de cet auteur a utilisé l’hétérophosphomolybdate de vanadium (IV)
H6PMo9V3O40 avec l’acétate de lithium à la place de l’association Cu(OAc)2/CaCl2 mais l’efficacité
reste faible38
(Schéma A4). Les auteurs ont précisé que cette réaction pouvait être effectuée avec des
quantités catalytiques d’hétéropolyacide, étant donné la présence d’oxygène dans le mélange.
Schéma A4. RHD du furane et de l’éthylène en présence d’une combinaison H6PMo9V3O40/LiOAc
Par la suite, des couplages déshydrogénants ont été réalisés entre des furanes substitués et
des alcènes porteurs de groupements électroattrateurs.
37
Kozhevnikov, I. V. React. Kinet. Catal. Lett. 1976, 5, 439-443; Chem. Abstr. 1977, 86, 106247. 38
Taraban'ko, V. E.; Kozhevnikov, I. V.; Matveev, K. I. Kinet. Katal. 1978, 19, 1160-1166.

27
Ainsi, plusieurs combinaisons d’acétate de cuivre (II)/oxygène dans un système dioxane/acide
acétique39,40
testées par les équipes de Fujiwara se sont révélées inefficaces avec de faibles quantités
de sel de cuivre (schéma A5).
Z Cu(OAc)2 Température Durée Rendement
CO2Me 0,1 équiv. 96 °C 3 h Mono : 7 % ; Di : 1 %
CO2Me 2,05 équiv. 96 °C 3 h Mono : 20 % ; Di : 9 %
CN 0,50 équiv. 100 °C 8 h Mono : 21 % ; Di : 13 %
CN 2,00 équiv. 100 °C 8 h Mono : 39 % ; Di : 21 %
Schéma A5. Résultats obtenus par Fujiwara en 1979 et 1981 pour les RHD du furane avec des
acrylates ou des acrylonitriles.
Les travaux de l’équipe de Miura confirmeront en 2008 cette conclusion par des expériences
réalisées à 120°C dans du DMF41
contenant de l’acétate de lithium (schéma A6), deux équivalents
d’acétate de cuivre (II) restant nécessaires pour obtenir des rendements moyens, y compris pour les
RHD impliquant des furanes substitués en position 2.
R Durée Rdt
t-Bu 2 h 57 %
1,3-dioxolan-2-yl 8 h 40 %
Schéma A6. Résultats obtenus par Miura en 2008 pour les RHD de furanes substitués en position 2
avec des acrylates en présence de Cu(OAc)2 et de LiOAc.
39
Maruyama, O.; Yoshidomi, M.; Fujiwara, Y.; Taniguchi, H. Chem. Lett. 1979, 1229-1230. 40
Fujiwara, Y.; Maruyama, O.; Yoshidomi, M.; Taniguchi, H. J. Org. Chem. 1981, 46, 851-855. 41
Maehara, A.; Satoh, T.; Miura, M. Tetrahedron 2008, 64, 5982-5986.

28
En 1984, Tsuji et Nagashima ont utilisé un perester en tant qu’oxydant pour des réactions de
couplage déshydrogénant réalisées dans l’acide acétique. Ces réactions font intervenir du benzoate
de palladium comme catalyseur à 100 °C42
(Schéma A7).
Schéma A7. Résultats obtenus par Tsuji en 1984 pour la RHD de furanes avec des acrylates en
utilisant un perester comme oxydant.
Quinze ans plus tard, Fujiwara et ses collaborateurs ont utilisé du t-BuOOH en association
avec des quantités catalytiques de benzoquinone (BQ)43
(Schéma A8). Ils ont ainsi montré que les
RHD étaient possibles entre le sylvane (nom trivial pour le 2-méthylfurane) et les acrylonitriles, les
énones ou encore l’acroléine.
Schéma A8. RHD impliquant le sylvane selon la méthode développée par Fujiwara en 1999.
D’autre part, cette équipe a aussi appliqué sa méthode aux RHD de benzofuranes (également
encore appelés coumarones) (Schéma A9).
Schéma A9. RHD impliquant le benzofurane selon la méthode développée par Fujiwara en 1999.
42
Tsuji, J.; Nagashima, H. Tetrahedron 1984, 40, 2699-2702. 43
Jia, C.; Lu, W.; Kitamura, T.; Fujiwara, Y. Org. Lett. 1999, 1, 2097-2100.

29
En 2003, l’équipe d’Ishii44
a développé un système proche de celui proposé par Kozhevnikov
et ses collaborateurs 25 ans plus tôt. Ainsi, un mélange de produits mono- et disubstitués a été
obtenu avec des rendements acceptables, sous oxygène et en présence de quantités catalytiques
d’acétylacétone et d’acétate de sodium comme additifs (Schéma A10).
Schéma A10. RHD du furane avec l’acrylate d’éthyle selon les conditions d’Ishii
développées en 2003.
La même équipe a ensuite observé que la vinylation de l’acide pyromucique ou de l’acide
benzofuroïque s’accompagnait de la décarboxylation45
(Schéma A11), ce qui contraste avec la
réactivité des acides benzoïques.46
Un mélange de deux régioisomères est obtenu, la réaction
pouvant s’opérer selon la séquence pallado-décarboxylation/activation de liaison C-H ou selon la
séquence activation de liaison C-H/pallado-décarboxylation.
R Durée Rendement Ratio
H 5 h 39 % 1 : 1
CH=CH-CH=CH 4 h 51 % > 95 : 5
Schéma A11. Vinylation – décarboxylation de l’acide pyromucique ou benzofuroique selon Ishii.
Les expériences sont parfois réalisées dans le DMF avec de l’acétate de lithium comme
additif. Selon Miura et ses collaborateurs, l’un des rôles de LiOAc est de fournir des ligands acétate
stabilisateurs des intermédiaires de palladium (0).41
Notons que Kozhevnikov, qui avait, 30 ans
auparavant, observé la favorisation du couplage oxydant par l’ion acétate, suspectait sa participation
à la formation du complexe ArPdOAc à partir de l’arène ArH.37
44
Yokota, T.; Tani, M.; Sakaguchi, S.; Ishii, Y. J. Am. Chem. Soc. 2003, 125, 1476-1478. 45
Maehara, A.; Tsurugi, H.; Satoh, T.; Miura, M. Org. Lett. 2008, 10, 1159-1162. 46
Miura, M.; Tsuda, T.; Satoh, T.; Pivsa-Art, S.; Nomura, M. J. Org. Chem. 1998, 63, 5211-5215.

30
En 2008, notre équipe a réalisé la synthèse de difurylalcanes en utilisant de la benzoquinone
en quantité stœchiométrique comme oxydant.47
Ainsi, l’utilisation du trifluoroacétate de palladium
(II) dans un mélange MeCN/AcOH fournit un mélange dans des rapports 85 : 15 de 2-phénylbis(5-
méthyl-2-furyl)éthane et de phénylbis(5-méthyl-2-furyl)éthane (Schéma A12) avec un rendement de
60 %, à partir du sylvane. L’emploi de différents alcènes a montré l’influence de leur substitution sur
la sélectivité. Avec des styrènes et des esters α,β−insaturés, la réaction principale a lieu en position β
alors qu’avec des allylalcènes, les composés majoritaires sont des 2-difuryles (Schéma A12).
R Z Rendement (%) ββββ/αααα Ratio
H Ph 60 85 : 15
" 4-MeC6H4 50 75 : 25
" 4-ClC6H4 62 83 : 17
" 4-BrC6H4 60 87 : 13
" 3-ClC6H4 62 89 : 11
" 2-naphthyl 30 84 : 16
" C6F5 76 75 : 25
" CO2Et 57 100 : 00
" CO2t-Bu 71 100 : 00
" Bn 94 15 : 85
" 4-MeOC6H4CH2 52 40 : 60
" 3,4-(MeO)2C6H3CH2 62 "
Me Ph 70 100 : 00
" 4-MeOC6H4 74 85 : 15
" 2-MeOC6H4 75 83 : 17
" 2-BnOC6H4 41 80 : 20
Et Ph 65 100 : 00
Schéma A12. Résultats obtenus en 2008 par notre laboratoire pour la synthèse de difurylalcanes.
La sélectivité envers la formation des difurylalcanes dépend également de la nature du
catalyseur, comme le montre le schéma A13. En effet, le couplage du sylvane procure avec
l’allylbenzène un mélange de 5,5'-(3-phénylpropane-1,1-diyl)bisylvane et de 5,5'-(1-phénylpropane-
47
Thiery, E.; Harakat, D.; Le Bras, J.; Muzart, J. Organometallics 2008, 27, 3996-4004.

31
2,2-diyl)bisylvane dans des proportions 1 : 1 avec de l’acétate de palladium (II) et dans des
proportions 15 : 85 avec du trifluoroacétate de palladium (II). Lorsque le styrène est mis en jeu à la
place de l’allylbenzène avec du trifluoroacétate de palladium (II), le couplage conduit à deux
composés (le 5,5'-(2-phényléthane-1,1-diyl)bisylvane et le 5,5'-(1-phényléthane-1,1-diyl)bisylvane
dans des proportions 85 : 15 alors que l’utilisation d’acétate de palladium fournit en plus le (E)-2-
styrylsylvane. Enfin, avec l’acrylate de t-butyle et l’acétate de palladium, le (E)-2-styrylsylvane et le
tert-butyl-3,3-bis(5-méthylfuran-2-yl)propanoate sont obtenus dans des proportions 90 : 10 alors que
l’utilisation de trifluoroacétate de palladium ne fournit que le tert-butyl-3,3-bis(5-méthylfuran-2-
yl)propanoate.
Schéma A13. Dépendance de la sélectivité vis-à-vis de la nature du catalyseur.
En 2009, notre laboratoire est parvenu à réaliser des couplages pallado-catalysés de type
Heck déshydrogénant de furanes avec des styrènes48
(Schéma A14) avec des rendements supérieurs
à ceux obtenus habituellement. Les styrènes sont une classe de composés riches en électrons
capables de se polymériser et de se rompre facilement en benzaldéhydes dans des conditions
pallado-oxydantes.49
De ce fait, seuls quelques exemples de couplages de Heck déshydrogénants
mettant en jeu ces composés avaient été décrits, mais avec des rendements modestes (<40
%)44,50,51,52
et une gamme de substrats peu étendue.53,54,55,56
En effet, les méthodes efficaces
rapportées faisaient intervenir généralement des alcènes pauvres en électrons.37-46,57
48
Aouf, C.; Thiery, E.; Le Bras, J.; Muzart, J. Org Lett. 2009, 11, 4096-4099. 49
Cornell, C. N.; Sigman, M. S. Inorg. Chem. 2007, 46, 1903-1909. 50
Cai, G. ; Fu, Y.; Li, Y.; Wan, X.; Shi, Z. J. Am. Chem. Soc. 2007, 129, 7666-7673. 51
Wang, J.-R.; Yang, C.-T. ; Liu, L. ; Guo, Q.-X. Tetrahedron Lett. 2007, 48, 5449-5453. 52
Amatore, C.; Cammoun, C.; Jutand, A. Adv. Synth. Catal. 2007, 349, 292-296. 53
Grimster, N. P.; Gauntlett, C.; Godfrey, C. R. A.; Gaunt, M. J. Angew. Chem., Int. Ed. 2005, 44, 3125. 54
Cho, S. H.; Hwang, S. J.; Chang, S. J. Am. Chem. Soc. 2008, 130, 9254. 55
Koubachi, J.; Berteina-Raboin, S.; Mouaddib, A.; Guillaumet, G. Synthesis 2009, 271. 56
Cheng, D.; Gallagher, T. Org. Lett. 2009, 11, 2639. 57
Zhao, J.; Huang, L.; Cheng, K.; Zhang, Y. Tetrahedron Lett. 2009, 50, 2758-2761.

32
Schéma A14. RHD de furanes avec des styrènes selon la méthode développée au laboratoire.
La méthode mise au point par le laboratoire fait intervenir l’acétate de palladium (II) comme
catalyseur, l’acide propionique comme solvant, l’éther diéthylique ou le THF comme co-solvant et
l’oxygène et des quantités catalytiques de benzoquinone et d’acétate de cuivre comme réoxydants.
Ces transformations s’opèrent à 40 - 60 °C. L’utilisation d’acide propionique comme solvant conduit à
des rendements plus élevés que ceux obtenus avec l’acide acétique et à des résultats reproductibles.
Ces conditions contrastent avec celles utilisées précédemment37,39,41,45
puisqu’elles fournissent de
bons résultats avec des quantités inférieures de sels de cuivre (II). Cette méthode semble générale
car elle fonctionne avec des furanes mono-, di- et trisubstitués ainsi qu’avec des styrènes porteurs de
groupements électroattrateurs et donneurs. Elle conduit toujours aux isomères E, montrant ainsi une
bonne stéréosélectivité. Comme cela a déjà été observé pour les réactions impliquant les furanes
substitués en 2 par un groupement électroattracteur ou électrodonneur avec les acrylates, le
couplage a lieu sur le carbone 5 démontrant également une bonne régiosélectivité. De plus, ces
transformations laissent certaines liaisons carbone-halogène intactes comme le montrent les
exemples mettant en jeu les chloro- et fluorostyrènes.

33
En outre, notre équipe a également remarqué que la benzoquinone peut, dans des
conditions acides,58
être utilisée en tant qu’unique oxydant48
(Schéma A15). Des résultats
sensiblement identiques ont été observés avec le trifluoroacétate de palladium.
Schéma A15. RHD du sylvane avec le styrène en présence
de BQ comme unique oxydant.
En 2009 également, Cheng et Zhang ont montré que l’utilisation d’acétate d’argent comme
oxydant et de pyridine comme additif permettait de réaliser des couplages oxydants pallado-
catalysés efficaces de furanes et d’esters ou amides α,β-insaturés,57
dans le DMF à 120 °C (Schéma
A16).
Schéma A16. RHD de furanes avec des acrylates ou des acrylamides en présence de pyridine et
d’acétate d’argent.
En 2010, Kar et ses collaborateurs ont utilisé de l’acétate de cuivre (II) dans le DMF et sous air
pour effectuer des réactions pallado-catalysées de Heck déshydrogénantes (Schéma A17)59
. Ils ont
remarqué qu’avec des substrats ayant pour substituants R2 = CO2Et et R
3 = Et, la réaction peut se
dérouler en l’absence d’oxydant. Ces dernières conditions conduisent néanmoins à la réaction de
Heck classique, c’est-à-dire à un couplage au niveau de l’unité ArBr quand R2 = H et R
3 = Me. Selon
58
Des conditions acides sont requises pour régénérer Pd(II) en Pd(0) avec la benzoquinone: (a) Bäckvall, J.-E.;
Byström, S. E.; Nordberg, R. E. J. Org. Chem. 1984, 49, 4619-4631. (b) Grennberg, A.; Bäckvall, J.-E.; Gogoll, A.
Organometallics 1993, 12, 1790-1793. (c) Nahra, F.; Liron, F.; Prestat, G.; Mealli, C.; Messaoudi, A.; Poli, G.
Chem. Eur. J. 2009, 15, 11078-11082. (d) Delcamp, J. H.; Brucks, A. P.; White, M. C. J. Am. Chem. Soc. 2008,
130, 11270-11271. Dans des conditions particulières, quelques régénérations ont lieu en absence de conditions
protiques : Babjak, M.; Zálupský, P.; Gracza, T. Arkivoc 2005, v, 45-57. 59
Patra, P.; Ray, J. K.; Kar, G. K. Tetrahedron Lett. 2010, 51, 3580-3582.

34
eux, l’absence de couplage de Heck quand R2 = CO2Et et R
3 = Et est due à l’encombrement stérique
causé par la présence des deux esters. Aucune explication n’est fournie quant aux réactions de Heck
déshydrogénantes en l’absence de réoxydant. Cependant, les rendements obtenus selon cette
méthode restent faibles (jamais plus de 55 %) et la récupération des composés de départ n’est pas
mentionnée. Nous pouvons supposer que la régénération des espèces palladiées actives s’effectue
via la participation d’unités de bromure d’aryle.32
Schéma A17. RHD selon les conditions développées par Kar en 2010.
En 2011, l’équipe de Tolstikov a appliqué la méthode développée par notre laboratoire48
en
2009 au phlomisoate de méthyle en vue de réaliser la synthèse de dérivés diterpènoïdes (schéma
A18). Ainsi, la réaction du phlomisoate de méthyle avec le styrène, catalysée par Pd(OAc)2, en
présence de Cu(OAc)2 et de BQ dans un mélange EtCOOH/Et2O donne stéréosélectivement le dérivé
15,16-distyryle correspondant et seulement quelques traces de furanolabsanoïdes monosubstitués
en position 15 ou 16.60
Le couplage réalisé avec l’acrylate de méthyle à la place du styrène dans des
conditions similaires a fourni un mélange de produits monosubstitués en position 15 ou 16 et
disubstiués en position 15 et 16, dont le ratio change au cours de la réaction.
Schéma A18. RHD du phlomisoate de méthyle avec le styrène selon la méthode développée par
notre équipe et appliquée par Tolstikov.
60
Kharitonov, Yu. V.; Shul’ts, E. E.; Shakirov. M. M. Tolstikov, G. A. Russ. J. Org. Chem. 2011, 47, 597-600.

35
Récemment, Liu et ses collaborteurs sont parvenus à oléfiner la liaison C-H vinylique d’un
hétéroarène au lieu de vinyler la liaison C-H en position 5 par catalyse à l’acétate de palladium et en
utilisant de l’acétate d’argent dans un mélange ClCH2CH2Cl/DMSO61
(schéma A19).
Schéma A19. Oléfination de liaisons C-H vinyliques d’hétéroarènes selon Liu.
En conclusion, la Réaction de Heck Déshydrogénante est une stratégie intéressante pour la
fonctionnalisation d’arènes. Des progrès ont été accomplis non seulement en matière de
régiosélectivité, de réoxydation, de fonctionnalisation d’arènes acido-sensibles ou d’arènes inactivés
mais aussi dans l’extension de ce type de couplage, aux oléfines riches en électrons tels que les
styrènes. Cependant, un certain nombre d’inconvénients demeurent. En effet, les méthodes les plus
efficaces font souvent appel à des conditions drastiques : des quantités importantes de catalyseur,
des temps réactionnels longs, des températures parfois élevées (jusqu’à 140 °C) et des réoxydations
conduisant à des quantités abondantes de sous-produits. Par ailleurs, notre équipe avait avancé que
les substrats bromés étaient peu disposés à réagir48
et les exemples mettant en jeu des alcènes
riches en électrons sont encore peu nombreux.
Par conséquent, notre intérêt s’est porté sur l’optimisation de la RHD de furanes avec des
alcènes riches en électrons, les styrènes. Dans un premier temps, l’objectif était de travailler dans
des conditions plus douces (température ambiante et sous air), tout en conservant des rendements
satisfaisants, et de rendre la méthode compatible avec l’utilisation de bromostyrènes.
61
Zhang, Y.; Cui, Z.; Li, Z.; Liu, Z-Q. Org. Lett. 2012, 14, 1838-1841.
36
A.2. Résultats et discussion
A.2.1. Constat de départ
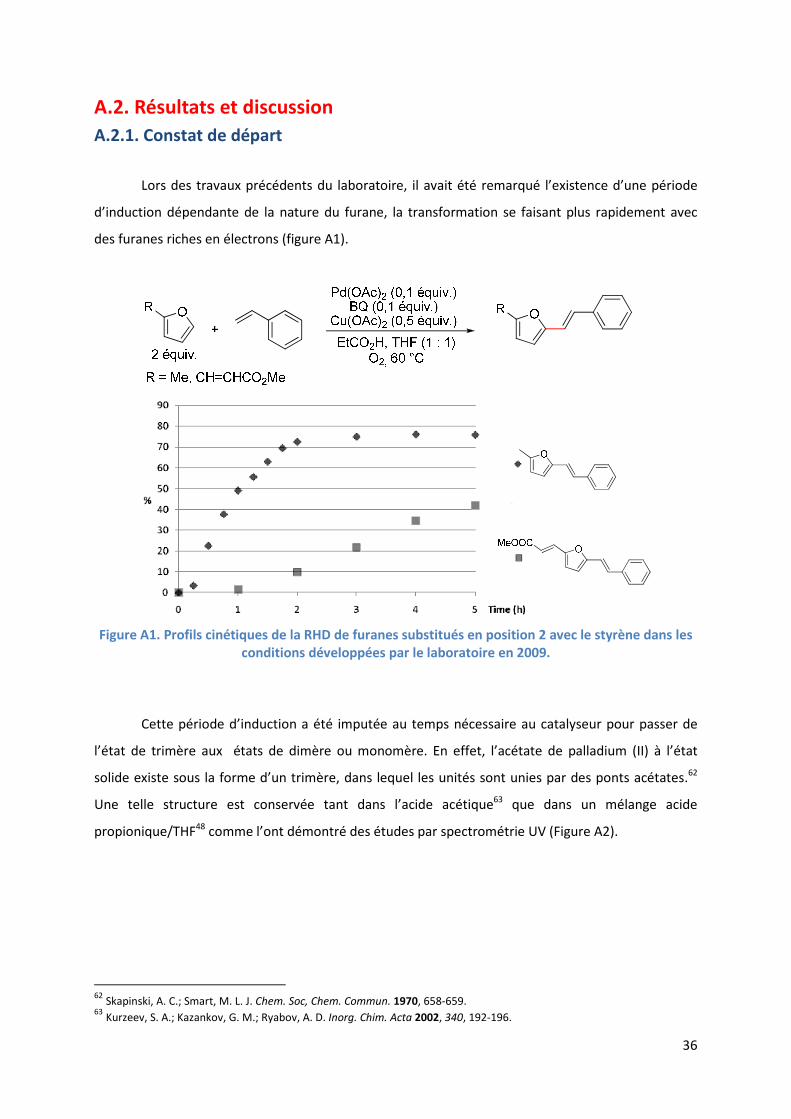
Lors des travaux précédents du laboratoire, il avait été remarqué l’existence d’une période
d’induction dépendante de la nature du furane, la transformation se faisant plus rapidement avec
des furanes riches en électrons (figure A1).
Figure A1. Profils cinétiques de la RHD de furanes substitués en position 2 avec le styrène dans les
conditions développées par le laboratoire en 2009.
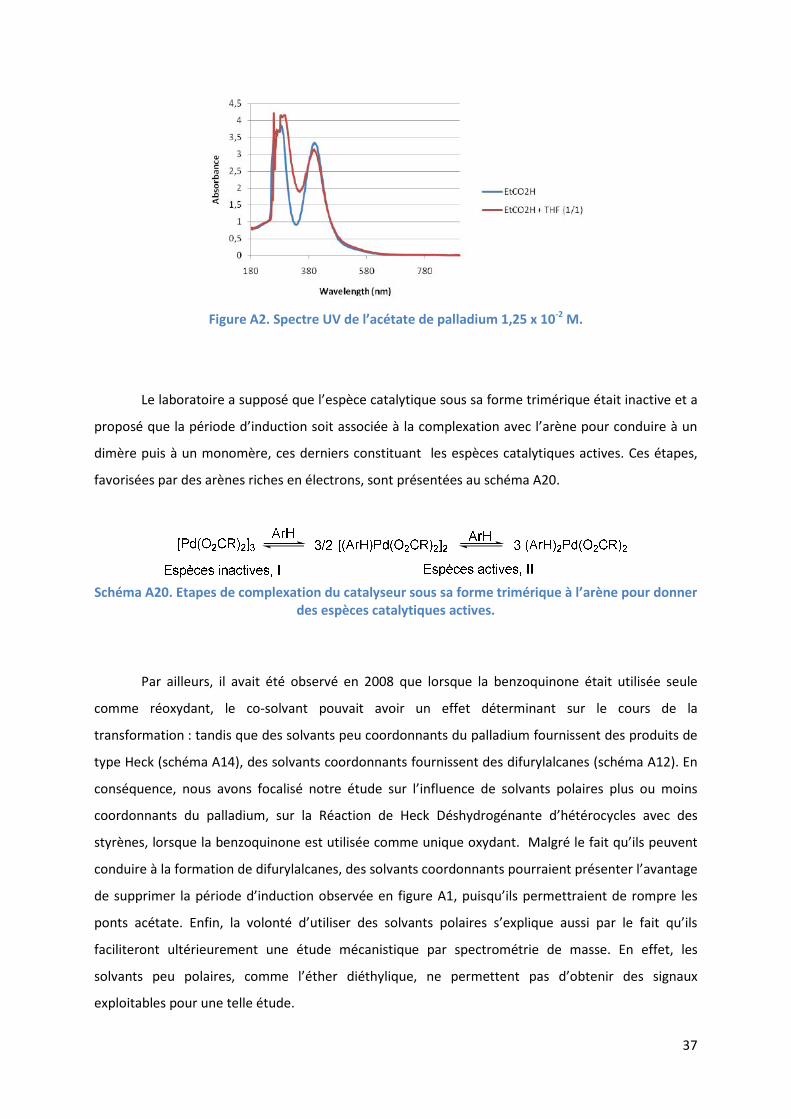
Cette période d’induction a été imputée au temps nécessaire au catalyseur pour passer de
l’état de trimère aux états de dimère ou monomère. En effet, l’acétate de palladium (II) à l’état
solide existe sous la forme d’un trimère, dans lequel les unités sont unies par des ponts acétates.62
Une telle structure est conservée tant dans l’acide acétique63
que dans un mélange acide
propionique/THF48
comme l’ont démontré des études par spectrométrie UV (Figure A2).
62
Skapinski, A. C.; Smart, M. L. J. Chem. Soc, Chem. Commun. 1970, 658-659. 63
Kurzeev, S. A.; Kazankov, G. M.; Ryabov, A. D. Inorg. Chim. Acta 2002, 340, 192-196.
37
Figure A2. Spectre UV de l’acétate de palladium 1,25 x 10-2
M.
Le laboratoire a supposé que l’espèce catalytique sous sa forme trimérique était inactive et a
proposé que la période d’induction soit associée à la complexation avec l’arène pour conduire à un
dimère puis à un monomère, ces derniers constituant les espèces catalytiques actives. Ces étapes,
favorisées par des arènes riches en électrons, sont présentées au schéma A20.
Schéma A20. Etapes de complexation du catalyseur sous sa forme trimérique à l’arène pour donner
des espèces catalytiques actives.
Par ailleurs, il avait été observé en 2008 que lorsque la benzoquinone était utilisée seule
comme réoxydant, le co-solvant pouvait avoir un effet déterminant sur le cours de la
transformation : tandis que des solvants peu coordonnants du palladium fournissent des produits de
type Heck (schéma A14), des solvants coordonnants fournissent des difurylalcanes (schéma A12). En
conséquence, nous avons focalisé notre étude sur l’influence de solvants polaires plus ou moins
coordonnants du palladium, sur la Réaction de Heck Déshydrogénante d’hétérocycles avec des
styrènes, lorsque la benzoquinone est utilisée comme unique oxydant. Malgré le fait qu’ils peuvent
conduire à la formation de difurylalcanes, des solvants coordonnants pourraient présenter l’avantage
de supprimer la période d’induction observée en figure A1, puisqu’ils permettraient de rompre les
ponts acétate. Enfin, la volonté d’utiliser des solvants polaires s’explique aussi par le fait qu’ils
faciliteront ultérieurement une étude mécanistique par spectrométrie de masse. En effet, les
solvants peu polaires, comme l’éther diéthylique, ne permettent pas d’obtenir des signaux
exploitables pour une telle étude.

38
A.2.2. Effet du co-solvant
Le couplage en 4 heures du sylvane (A1) et du 2-méthylthiophène (B1) avec le styrène (C1) a
été étudié en présence d’acétate de palladium, de benzoquinone (BQ), d’acide acétique et d’une
série de co-solvants (Tableau A1)
Entrée 1 Co-solvant 1-C1 (%)b
1 A1 Et2O 11
2 A1 CF3CH2OH 8
3 A1 MeOH 8
4 A1 t-BuOH 9
5 A1 CH2Cl2 11
6 A1 DMF 9
7 A1 DMAc 8
8 A1 DMSO 67 (64)
9 B1 DMSO 58
10c B1 DMSO (67)
11d B1 DMSO 45
a A1 ou B1 (2,0 mmol), C1 (1,0 mmol), Pd(OAc)2 (0,05 mmol), AcOH (2 mL), co-solvant (2 mL), TA, 4 h.
b
rendement determiné par CPG ; rendement isolé entre parenthèses. c La réaction est réalisée en 24 h.
d BQ (0,5
équiv.), Cu(OAc)2 (0,1 équiv.), O2 (1 atm), 24 h.
Tableau A1. Etude de l’influence du co-solvant sur le couplage de A1 et B1 avec le styrène C1.a
Le tableau A1 montre que la réaction s’avère inefficace lorsqu’elle est réalisée avec Et2O,
CF3CH2OH, MeOH, t-BuOH, CH2Cl2, DMF ou encore le DMAc comme co-solvant (entrées 1 à 7). Seul le
DMSO fournit de bons résultats permettant ainsi l’obtention du composé A1-C1 avec un rendement
isolé de 64 % (entrée 8). Cette observation est surprenante puisque que le DMSO est un solvant
complexant du palladium. Or, notre équipe avait montré, en 2008, que l’utilisation d’acétonitrile
comme co-solvant et de benzoquinone comme oxydant, pouvait mener à des difurylalcanes.47
D’autre part, avec le DMSO et le 2-méthylthiophène (entrées 9 et 10), le produit de couplage B1-C1
est isolé avec un rendement de 67 % en 24 heures (entrée 10). L’utilisation d’une quantité plus
importante de benzoquinone (2 équivalents) que celle utilisée par le laboratoire précédemment48
reste nécessaire pour élargir notre méthode aux thiophènes. En effet, l’utilisation d’une quantité

39
d’oxydant plus faible (une combinaison de 0,5 équivalent de benzoquinone et de 10 % d’acétate de
cuivre) ne conduit qu’à un rendement de 45 % (entrée 11).
A.2.3. Synthèse de styrylfuranes
Ces nouvelles conditions réactionnelles ont été utilisées pour la synthèse d’une variété de
styrylfuranes (tableau A2). Après quelques légères modifications, des rendements moyens à bons ont
été obtenus (de 40 à 79 %). De plus, la méthode est compatible avec l’utilisation du p-bromosytrène,
pour lesquel aucun exemple de RHD n’est rapporté (entrées 2, 5 et 9). Jusqu’à présent, les dérivés
bromés étaient réputés peu disposés à réagir en RHD.48
Seuls Glorius et son équipe étaient parvenus
à faire réagir deux exemples dans des réactions de couplage via une activation de liaison C-H tout en
laissant la liaison C-Br intacte64
grâce à une catalyse au rhodium (III).
Quelque soit la nature du groupement porté par les furanes en position 2, le couplage
s’effectue toujours en position 5 avec une bonne stéréosélectivité, les composés étant tous obtenus
sous forme d’isomères E.
Ce sont les furanes substitués en 2 par un groupement alkyle qui procurent les meilleurs
résultats (entrées 1 à 5). Quant aux furanes disubstitués en 2 et 5 (entrées 8 et 9), le système
apparaît moins sélectif. Leur réaction dans les mêmes conditions conduit à une conversion
importante à totale (selon la quantité de catalyseur utilisée) du styrène mais à un rendement très
faible en produit de couplage. L’augmentation à cinq équivalents de la quantité d’arène s’est alors
avérée nécessaire. Précédemment, les couplages réalisés avec ce type d’arène avaient d’ailleurs
nécessité des quantités de palladium (10 % molaire à la place de 5) et des températures plus élevées
(60 °C).48
Ainsi, les rendements plus modestes observés pour ces arènes montrent que la position 3
du furane réagit difficilement.
Quant au furane porteur d’un groupement électroattracteur en position 2 (entrées 6), il
nécessite une légère augmentation de température réactionnelle pour atteindre des rendements
moyens.
Soulignons enfin que le furfurol (ou alcool furfurylique) a pu être couplé avec un très bon
rendement (entrée 7). Malgré les conditions oxydantes utilisées, aucune trace d’aldéhyde
pyromucique ou d’acide pyromucique n’a été détecté en CPG, montrant que cette méthode est
hautement sélective vis-à-vis de l’activation de liaison C-H.
64
Patureau, F. W.; Glorius, F. J. Am. Chem. Soc. 2010, 132, 9982-9983.
40
entrée A C Pd (%) durée A-C, %b
1 A1 C1 5 24
2 A1 C2 5 24
3 A2 C1 5 24
4 A3 C1 5 24
5 A3 C2 5 24
6c A4 C1 5 48
7 A5 C1 5 24
8d A6 C1 5 24
9d A6 C2 10 48
a A (2,0 mmol), C (1,0 mmol), BQ (2,0 mmol) AcOH (2 mL), DMSO (2 mL), TA.
b rendement isolé.
c La réaction est
réalisée à 40 °C. d
A (5 mmol).
Tableau A2. Synthèse de styrylfuranes selon la méthode développée par le laboratoire.a

41
A.2.4. Limites de la méthode
La réaction s’avère compatible aussi bien avec les furanes porteurs de groupements
électroattracteurs qu’électrodonneurs. Les principales limites résident dans la difficulté à réaliser ces
réactions en absence de substitution du cycle furanique. En effet, la RHD via cette méthode, du
furane avec le styrène n’a conduit au 2-styrylfurane qu’avec un rendement de 14 %. Par ailleurs, la
méthode s’est révélée inefficace pour la réaction du furylacrylate de méthyle avec le p-
bromostyrène.
A.2.5. Conclusion
Les conditions réactionnelles mises au point permettent le couplage régiosélectif et
stéréosélectif dans des conditions douces d’une large gamme de furanes avec le styrène et le p-
bromostyrène en laissant pour ce dernier la liaison carbone-halogène intacte.
Cette méthode élargit les possibilités de valorisation des composés dérivés de la biomasse.
Ainsi, la vinylation de furanes peut ouvrir une voie pour la synthèse d’hétérocycles d’intérêt
biologique (schéma A21). Les vinylfuranes, peuvent mener, après fonctionlisation de la double liaison
vinylique, à des précurseurs de synthèse de la castanospermine, la goniothalamine, la
swainsonine65
… Notons également que le 2-hydroxyméthyl-5-styrylfurane est un intermédiaire clef
de molécules ayant montré une activité inhibitrice de l’artériosclérose.66
65
Mukherjee, D.; Yousuf, S. K.; Taneja, S. C. Org. Lett. 2008, 10, 4831-4834. 66
Takezawa, H.; Hayashi, M.; Iwasawa, Y.; Hosoi, M.; Lida, Y.; Tsuchiya, Y.; Horie, M.; Kamei, T. 1988, (Banyu Pharmaceutical
CO., LTD), Eur. Patent 0318860A2.

42
Schéma A21. Exemples de molécules d’intérêt biologique synthétisables à partir de motifs amines
α-furfuryliques ou encore de motifs alcools α-furfuryliques.
A.3. Partie expérimentale
A.3.1. Informations générales
La Benzoquinone (BQ), Cu(OAc)2, Pd(OAc)2, A1, A2, A3, A5, A6, C1 et C2 sont des produits
comerciaux et utilisés sans purification.
A.3.2. Préparation du (E)-3-furylacrylate de méthyle
Le (E)-3-furylacrylate de méthyle (A4) a été préparé selon une méthode standard : l’acide (E)-
3-furylacrylique est dissout dans du méthanol ; après ajout d’une quantité catalytique de H2SO4, le
milieu est porté à reflux pendant 24 h.

43
A.3.3. Synthèse de styrylfuranes
A l’acétate de palladium (11,2 – 24,4 mg, 0,05 – 0,1 mmol) et la benzoquinone (216 mg, 2
mmol) mis en solution dans un mélange solvant/co-solvant AcOH (2 mL)/DMSO (2 mL) sont ajoutés
l’arène (2 – 5 mmol) et l’alcène (1 mmol). Le mélange est agité à température ambiante (sauf pour la
synthèse de A4-C1 : 40 °C) pendant 24h – 48 h. Après ajout de Et2O (20 mL), la réaction est traitée
par une solution aqueuse saturée de NaHCO3 (20 mL). La phase aqueuse est extraite avec Et2O (20
mL × 2). Les phases organiques sont lavées avec une solution de NaOH 2M (20 mL x 2), avec H2O (20
mL x 2), séchées sur MgSO4, et filtrées sur coton. Le solvant est évaporé sous vide. Les produits sont
séparés sur colonne de gel de silice flash (éluant : éther de pétrole/acétate d’éthyle, 98-70 : 2-30).
(E)-2-styrylsylvane (A1-C1). Solide blanc ; 64 %.
Point de fusion : 59 – 61 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,31 (s, 3H, H7), 5,97 (d, 1H, J = 2,1 Hz, H4), 6,20 (d, 1H, J = 3,0 Hz,
H5), 6,80 (d, 1H, J = 16,2 Hz, H2), 6,95 (d, 1H, J = 16,2 Hz, H1), 7,19 (t, 1H, J = 7,1 Hz, HAr), 7,29 (t, 2H, J =
7,4 Hz, HAr), 7,41 (d, 2H, J = 7,2 Hz, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 13,7 (C7), 107,8 (C4), 109,9 (C5), 116,6 (C2), 125,4 (CH), 126,1
(2CH), 127,1 (2CH), 128,6 (C1), 137,2 (C), 151,7 (C6), 152,2 (C3).
IR (KBr) ν (cm-1
) : 2921, 1593, 1531, 1486, 1443, 1358, 1260, 1182, 1013, 958, 776, 751, 693.
Analyse élémentaire (%) pour C13H12O théorique : C 84,75 ; H 6,57 ; obtenue : C 84,42 ; H 6,68.
(E)-2-(4-bromostyryl)sylvane (A1-C2). Cristaux blancs ; 73 %.
Point de fusion : 91 - 93 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,24 (s, 3H, H7), 5,91 (d, J = 2,2 Hz, 1H, H4), 6,14 (d, J = 3,0 Hz, 1H,
H5), 6,68 (d, J = 16,2 Hz, 1H, H2), 6,78 (d, J = 16,2 Hz, 1H, H1), 7,17 (d, J = 8,5 Hz, 2H, HAr), 7,32 (d, J =
8,5 Hz, 2H, HAr),

44
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 14,2 (C7), 108,4 (C4), 111,0 (C5), 117,7 (C2), 121,2 (C-Br), 124,5
(2CH), 128,0 (C1), 132,2 (2CH), 136,7 (C), 151,9 (C6), 153,0 (C3).
IR (KBr) ν (cm-1
) : 2922, 1589, 1561, 1483, 1361, 1262, 1181, 957, 786, 761, 707
Analyse élémentaire (%) pour C13H11BrO théorique : C, 59,34 ; H, 4,21 ; obtenue : C, 59,67 ; H, 4,15.
(E)-2-éthyl-5-styrylfurane67
(A2-C1). Huile jaune ; 68 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,51 (t, J = 7,50 Hz, 3H, H8), 2,93 (q, J = 7,50, 2H, H7), 6,25 (d, J
= 2,9 Hz, 1H,H4), 6,47 (d, J = 3,1 Hz, 1H, H5), 7,07 (d, J = 16,2 Hz, 1H, H2), 7,22 (d, J = 16,2 Hz, 1H,
H1), 7,35-7,50 (m, 1H, HAr), 7,55 (t, J = 7,3 Hz, 2H, HAr), 7,68 (d, J = 7,4 Hz, 2H, HAr).
(E)- 5-styrylsylvan-3-oate de méthyle (A3-C1). Cristaux blancs ; 79 %.
Point de fusion : 90 - 94 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,46 (s, 3H, H6), 3,64 (s, 3H, H7), 6,39 (s, 1H, H4), 6,61 (d, 1H, J =
16,3 Hz, H2), 6,85 (d, 1H, J = 16,3 Hz, H1), 7,08 (m, 1H, HAr), 7,17 (t, 2H, J = 7,3 Hz, HAr), 7,28 (d, 2H, J =
7,6 Hz, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 13,9 (C6), 51,3 (C7), 108,8 (C4), 115,1 (CCO2), 115,6 (C2), 126,4
(2CH), 127,6 (CH), 127,7 (C1), 128,7 (2CH), 136,7 (C), 151,0 (C5), 159,0 (C3), 164,2 (CO2).
IR (KBr) ν (cm-1
) : 3024, 2953, 2920, 1708, 1639, 1598, 1437, 1396, 1310, 1227, 1201, 1123, 1088,
998, 964, 822, 779, 751, 691.
SMHR : pour C15H14O3Na théorique : 265,0841 ; obtenue : 265,0846.
67
Barluenga, J. ; Fanlo, H.; Lopez S.; Flórez J. Angew. Chem. Int. Ed. 2007, 46, 4136–4140.

45
(E)-5-(4-bromostyryl)sylvan-3-oate de méthyle (A3-C2). Cristaux blancs ; 61 %.
Point de fusion : 103 - 107 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,47 (s, 3H, H6), 3,69 (s, 3H, H7), 6,41 (s, 1H, H4), 6,57 (d, J = 16,2
Hz, 1H, H2), 6,75 (d, J = 16,2 Hz, 1H, H1), 7,12 (d, J = 8,4 Hz, 2H, HAr), 7,29 (d, J = 8,3 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 14,3 (C6), 51,8 (C7), 109,8 (C4), 115,6 (CCO2), 116,6 (C2), 121,8 (C-
Br), 126,6 (C1), 128,2 (2CH), 132,2 (2CH), 136,0 (C), 151,1 (C5), 159,6 (C3), 164,5 (CO2).
IR (KBr) ν (cm-1
) : 3051, 3033, 1705, 1640, 1598, 1439, 1395, 1307, 1228, 1124, 1081, 999, 970, 816,
773, 705.
Analyse élémentaire (%) pour C15H13BrO3 théorique : C, 56,10 ; H, 4,08 ; obtenue : C, 56,22 ; H,
4,03.
(E)- 2-acrylate de méthyle-5-styrylfurane (A4-C1). Cristaux jaunes ; 40 %.
Point de fusion : 130 – 134 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 3,68 (s, 3H, H9), 6,27 (m, 2H, H8 et H4), 6,49 (d, 1H, J = 3,4 Hz, H5),
6,72 (d, 1H, J =16,2 Hz, H2), 7,03 (d, 1H, J = 16,2 Hz, H1), 7,20 (m, 4H, H7, 3HAr), 7,35 (t, J = 6,8 Hz, 2H,
2HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 51,5 (C9), 111,2 (C5), 114,8 (C4), 115,5 (C8), 117,3 (C2), 126,5 (2CH),
128,0 (CH), 128,6 (2CH), 129,6 (C7), 130,6 (C1), 136,3(C), 150,1(C6), 155,3 (C3), 167,4 (CO2).
IR (KBr) ν (cm-1
) : 2964, 1714, 1620, 1504, 1432, 1383, 1308, 1239, 1176, 1025, 963, 802, 752, 692.
SMHR : pour C16H14O3Na théorique : 277,0841 ; obtenue : 277,0837.
(E)-5-styrylfurfurol (A5-C1). Gomme jaune pâle ; 78 %.

46
RMN 1H (250 MHz, acétone-d6) δ (ppm) : 4,58 (s, 2H, H7), 6,34 (d, J = 2,8 Hz, 1H, H4), 6,40 (d, J = 2,8
Hz, 1H, H5), 7,02 (m, 2H, H1 et H2), 7,23 (t, J = 7,1 Hz, 1H, HAr), 7,34 (t, J = 7,4 Hz, 2H, HAr), 7,52 (d, J =
7,4 Hz, 2H, HAr).
RMN 13
C (63 MHz, acétone-d6) δ (ppm) : 58,6 (C7), 111,3 (C4), 112,0 (C5), 118,8 (C2), 128,4 (2CH),
128,5 (C1), 129,7 (CH), 130,9 (2CH), 139,3 (C), 154,8 (C3), 157,7 (C6).
IR (KBr) ν (cm-1
) : 3256, 3030, 2932, 2863, 1635, 1596, 1490, 1448, 1359, 1262, 1193, 1014, 957, 779,
749.
SMHR : pour C13H12O2Na théorique : 223,0735 ; obtenue : 223,0738.
(E)- 5-methyl-4-styrylsylvane (A6-C1). Gomme jaune pâle ; 54 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,22 (s, 3H, H7), 2,30 (s, 3H, H8), 6,13 (s, 1H, H4), 6,62 (d, J = 16,1
Hz, 1H, H2), 6,85 (d, J = 16,1 Hz, 1H, H1), 7,17 (t, J = 7,2 Hz, 1H, HAr), 7,29 (t, J = 7,4, 2H, HAr), 7,41 (d, J =
7,6 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 11,9 (C7), 13,5 (C8), 103,7 (C4), 119,5 (C2), 119,8 (C3), 126,0 (2CH),
126,5 (CH), 127,0 (C1), 128,7 (2CH), 138,1 (C), 148,5 (C6), 150,6 (C5).
IR (film) ν (cm-1
) : 3028, 2961, 2918, 1644, 1581, 1447, 1260, 1224, 1095, 1009, 954, 800, 751, 693.
SMHR : pour C14H15O théorique : 199,1123 ; obtenue : 199,1125.
(E)- 5-methyl-4-(4-bromostyryl)sylvane (A6-C2). Semi-solide orange ; 40 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,25 (s, 3H, H7), 2,32 (s, 3H, H8), 6,13 (s, 1H, H4), 6,54 (d, J = 16,1
Hz, 1H, H2), 6,84 (d, J = 16,1 Hz, 1H, H1), 7,28 (d, J = 8,4 Hz, 2H, HAr), 7,42 (d, J = 8,3 Hz, 2H, HAr).

47
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 11,9 (C7), 13,6 (C8), 103,5 (C4), 119,5 (C3), 120,2 (C2), 120,5 (C-Br),
125,1 (C1), 127,5 (2CH), 131,8 (2CH), 137,0 (C), 148,9 (C6), 150,8 (C5).
IR (KBr) ν (cm-1
) : 2964, 2919, 1639, 1562, 1485, 1262, 1099, 1071, 954, 803, 751, 693.
SMHR : pour C14H13BrO théorique : 276,0150 ; obtenue : 276,0113.

48
Partie B : Réactions de Heck Déshydrogénantes de
thiophènes avec des styrènes dans des conditions douces
Introduction
Les thiophènes sont une classe d’hétérocycles très peu présente dans la nature. Ils peuvent
être extraits des tagettes, un genre de plantes de la famille des Asteraceae, dont font partie l’œillet
d’inde, la rose d’Inde, la tagette citron ou encore le souci français. Ces plantes synthétisent,
principalement au niveau de leurs racines, le 2,2’ : 5’,2’’-terthiophène et le 5-(but-3-èn-1-yl)-2,2'-
bithiophène (BBT), à partir desquels la plante biosynthétise plusieurs dérivés décrits au schéma B1.68
Ces composés présentent notamment des propriétés insecticides et antimicrobiennes.69,70
Schéma B1. Principaux thiophènes extraits des tagettes.
Un certain nombre de réactions permettant de convertir les furanes en thiophènes sont
connues. Elles s’effectuent à haute température (400 à 500 °C) en présence de H2S et de catalyseurs
alumino-supportés (schéma B2).71,72,73
68
Marotti, I.; Marotti, M.; Piccaglia, R.; Nastri, A.; Grandi, S.; Dinelli, G. J. Sci. Food Agric. 2010, 90, 1210-1217. 69
Green, M. M.; Singer, J. M. J. Am. Mosc Control Assoc. 1993, 7, 282-286. 70
Franz, Ch. Mathé, A. Buchbauer, G. Essential Oils: Basic and Applied Research, Allured Publishing: Vienne,
1997. 71
Li, Q.; Xu, Y.; Liu, C.; Kim, J. Catal. Lett. 2008, 122, 354-358. 72
Mashkina, A. V.; Khairulina, L. N. Kinet. Catal. 2008, 49, 245-252. 73
Mashkina, A. V. Kinet. Catal. 2011, 84, 1223-1228.

49
O S
Al2O3 calciné
500 °C
97 %
H2S (10 équiv.)
MW = 0,2 h-1
Schéma B2. Synthèse catalytique du thiophène à partir du furane.
Nous montrerons dans cette partie que la méthode de couplage présentée dans la partie
précédente est compatible avec une large gamme de thiophènes et avec des styrènes halogénés ou
non.
B.1. Bibliographie
Le thiophène est un cycle insaturé à 5 chaînons où un atome de soufre donne des électrons
aux systèmes π, jouant ainsi le rôle de substituant +E activateur du cycle. La conjugaison étant
importante (ER = 121 kJ.mol–1
), le thiophène est presque aussi stable que le benzène.74
En
conséquence, les réactions de couplage de Heck déshydrogénant avec les thiophènes n’ont bénéficié
que de peu d’études et requièrent des températures élevées. La plupart des résultats concernant ces
hétérocycles font partie d’études globales regroupant les O- et N-arènes.
En 1976, Kozhevnikov a réalisé le couplage du thiophène avec l’éthylène dans les mêmes
conditions réactionnelles que celles utilisées pour ses RHD entre le furane et cet alcène. A l’instar des
furanes, les rendements sont faibles (Schéma B3).37
Schéma B3. RHD du thiophène avec l’éthylène selon les conditions de Kozhevnikov.
La méthode élaborée en 1978 faisant appel à un hétéropolyacide comme oxydant et de
l’acétate de lithium comme additif dans l’acide acétique à 96 °C38
ne s’est pas révélée plus efficace
(schéma B4).
74
Joule, J. A.; Mills K. Heterocyclic Chemistry, John Wiley & Sons, Chichester, 2010.

50
Schéma B4. RHD du thiophène avec l’éthylène en présence d’une combinaison H6PMo9V3O40/LiOAc
Alors que les conditions de Fujiwara faisant intervenir l’acétate de cuivre comme oxydant
dans un mélange dioxane/AcOH à 100 °C avaient conduit à des vinylfuranes avec des rendements
moyens,39,40
elles ne mènent à des vinylthiophènes qu’avec des rendements très faibles (schéma B5).
Schéma B5. Résultats obtenus par Fujiwara en 1979 et 1981 pour les RHD entre le thiophène et des
acrylates ou des acrylonitriles.
L’équipe d’Ishii est la première qui, en 2004, est parvenue à réaliser une RHD impliquant un
thiophène avec un bon rendement.75
Le 2-méthylthiophène et l’acrylate d’éthyle ont été couplés
avec un rendement de 86 %, en utilisant de l’acétate de palladium sur charbon comme catalyseur,
une combinaison HPMo11V/O2 comme oxydant et de l’acétate de sodium comme additif dans l’acide
propionique à 60 °C (schéma B6).
Schéma B6. RHD entre le 2-méthylthiophène et l’acrylate d’éthyle selon les conditions
développées par Ishii.
En 2008, Miura a montré qu’avec le diphényl(thiéno)méthanol, l’emploi d’acétate de
palladium comme catalyseur, d’acétate de cuivre monohydrate comme oxydant et d’acétate de
75
Tani, M.; Sakaguchi, S.; Ishii, Y. J. Org Chem. 2004, 69, 1221-1226.

51
lithium comme additif dans le DMF à 96 °C pouvait se révéler plus efficace pour le couplage avec
l’acrylate d’éthyle qu’avec un alcène riche en électrons, tel que le styrène (schéma B7).41
Schéma B7. RHD impliquant le diphényl(thiéno)méthanol selon les conditions de Miura.
Le seul article à notre connaissance, principalement consacré aux thiophènes, a été publié
par Zhang et ses collaborateurs57
qui ont montré que l’utilisation de l’acétate d’argent au lieu
l’acétate de cuivre (II), de DMF et de pyridine, améliorait le rendement (schéma B8). La méthode
mise au point est également utilisable avec le furane et le sylvane.
Schéma B8. RHD mettant en jeu des thiophènes selon les conditions élaborées par Zhang en 2009.
D’une manière générale, de bons rendements avec une bonne régio- et stérosélectivité sont
observés en ce qui concerne les couplages avec les acrylates et les acrylamides. En revanche, les
résultats sont mitigés avec les acrylonitriles où les rendements sont inférieurs à 50 % et les
stéréosélectivités faibles.
Il convient de souligner en conclusion que les alcènes mis en jeu lors de ces transformations
sont des alcènes pauvres en électrons et requièrent des températures élevées. Un seul couplage de
Heck déshydrogénant faisant intervenir un alcène riche en électrons, le styrène, a été rapporté
(schéma B7), et le rendement obtenu est faible comparé à ceux obtenus avec des alcènes appauvris
en électrons. En conséquence, nous avons ambitionné d’étendre la méthode présentée en partie A

52
aux thiophènes et styrènes, et d’étudier sa compatibilité avec les styrènes halogénés pour lesquels
aucun exemple n’est connu.
B.2. Résultats et discussion
B.2.1. Constat de départ
Nous avions remarqué dans le paragraphe A.2.2. que dans un mélange AcOH/DMSO, avec la
benzoquinone comme oxydant et l’acétate de palladium comme catalyseur, la RHD du 2-
méthylthiophène avec le styrène fournissait le 2-styryl-5-méhtylthiophène avec un rendement de 67
% à température ambiante et en 24 h (schéma B9). Par conséquent, nous avons examiné la
compatibilité de cette méthode pour les 2-alkylthiophènes et les 2-halogénothiophènes avec le
styrène et des styrènes halogénés.
Schéma B9. RHD du 2-méthylthiophène avec le styrène selon la méthode développée en partie A.
B.2.2. Synthèse de styrylthiophènes
B.2.2.1. Avec les 2-alkylthiophènes
Les 2-alkylthiophènes ont pu être couplés avec des styrènes à température ambiante
(tableau B1). La RHD s’effectue non seulement avec les chlorostyrènes (entrée 3 à 6), mais également
avec les bromostyrènes (entrée 7 à 10). Les rendements en 24 h varient de 59 à 97 % avec une
quantité de catalyseur n’excèdant pas 5 % molaire, à l’exception de la RHD du 2-éthylthiophène avec
le 2-bromostyrène qui en requière 10 % molaire (entrée 8).

53
Entrée B C
Pd
(%) B-C, rdtb (%)
Entrée B C
Pd
(%) B-C, rdtb (%)
1 B1 C1 5
6 B2 C5 5
2 B2 C1 5
7 B1 C2 5
3 B1 C4 5
8 B2 C2 10
4 B2 C4 5
9 B1 C3 5
5 B1 C5 5
10 B2 C3 5
a B (2,0 mmol), C (1,0 mmol), BQ (2,0 mmol), AcOH (2 mL), DMSO (2 mL), TA.
b Rendement isolé.
Tableau B8. RHD d’alkylthiophènes avec des styrènes.a
B.2.2.2. Avec les 2-halogénothiophènes
La méthode a pu être appliquée aux 2-chloro- et 2-bromothiophènes sans affecter la liaison
Ar-X. Ainsi, ces composés ont été couplés de façon régiosélective et stéréosélective aussi bien avec le
styrène, qu’avec le chlorostyrène et le bromostyrène. Ces réactions réalisées à température
ambiante fournissent des rendements de 56 à 92 %. Des temps réactionnels plus longs que lors des
réactions avec les 2-alkylthiophènes ont été cependant nécessaires (48 h à la place de 24 h). Enfin,
les réactions mettant en jeu le 2-bromothiophène demandent une quantité de catalyseur plus élevée
que celle requise pour le 2-chlorothiophène (10 % molaire au lieu de 5 % molaire).

54
+
Pd(OAc)2 (0,05 - 0,1 équiv.)
BQ (2 équiv.)
AcOH/DMSO (1 : 1)
TA, 48 h
R2
S R2S
R2 = H (C1), p-Br (C2), o-Br (C3), p-Cl (C4), m-Cl (C5),
SBr
B4
R R
B C B-C
SCl
B3
2 équiv.
Entrée B C
Pd
(%) B-C, rdt (%)
Entrée B C
Pd
(%) B-C, rdt (%)
1 B3 C1 5
5 B3 C5 5
2 B4 C1 10
6 B4 C2 10
3 B3 C4 5
7 B3 C2 5
4 B4 C4 10
8 B4 C3 10
a B (2,0 mmol), C (1,0 mmol), BQ (2,0 mmol), AcOH (2 mL), DMSO (2 mL), TA.
b Rendement isolé.
Tableau B2. RHD de 2-halogénothiophènes avec des styrènes.
B.2.3. Limites de la méthode
A température ambiante, la diminution de la quantité en oxydant conduit à des résultats
nettement moins bons. Deux équivalents de benzoquinone restent donc nécessaires pour effectuer
ces couplages. Un prochain défi consistera à mettre au point une méthode permettant ces synthèses
à température ambiante et en utilisant un oxydant plus respectueux de l’environnement comme par
exemple l’oxygène moléculaire.
Comme pour le furane, le thiophène est peu disposé à réagir dans ces conditions. En effet, le
styrylthiophène n’a été isolé qu’avec un rendement de 28 %. Le constat est similaire pour les
thiophènes substitués en 3 par un groupement électroattracteur comme le 3-bromothiophène.
SCl
B3-C2, 65 %
Br

55
B.2.4. Valorisation de la réaction
Les thiophènes substitués en 2 sont utiles comme intermédiaires dans les colorants, les
parfums, les médicaments et les inhibiteurs.76
Certains produits de couplages synthétisés présentent
un intérêt comme intermédiaires de fabrication de polymères électroluminescents.77
Les
styrylthiophènes sont aussi reconnus pour entrer dans la composition de films polymériques semi-
conducteurs.78,79
Pour vinyler un thiophène substitué en position 2, la littérature met souvent en avant une
réaction de formylation suivie d’une réaction de Wittig.80
La méthode proposée ici offre donc une
possibilité d’accès simplifié à ces composés. Enfin, la réaction apparaît bien adaptée aux thiophènes
et styrènes halogénés où les rendements sont parfois supérieurs à 90 %. Ces résultats rendent cette
méthode intéressante pour fournir des intermédiaires de la synthèse d’oligothiophènes, une famille
de polymères conjugués qui deviennent conducteurs lorsqu’ils sont dopés, c’est-à-dire lorsque des
électrons sont ajoutés ou enlevés des orbitales p conjuguées. Les applications des polymères
conducteurs concernent des domaines variés comme le stockage d’énergie (batteries, piles), la
protection anti-statique et électromagnétique, les semi-conducteurs organiques et les capteurs
électrochimiques. La mise en évidence des propriétés d’électroluminescence du poly(p-
phénylènevinylène) (PPV) par le groupe de Friend en 199081
marque un tournant dans les recherches
sur les systèmes conjugués. En effet, depuis 1990, les propriétés semi-conductrices de la forme
neutre des systèmes conjugués linéaires (SCL) font l’objet de recherches visant le développement
d’applications dans des domaines tels que les transistors à effet de champ organiques, les diodes
électroluminescentes organiques et plus récemment les cellules photovoltaïques.
76
Campaigne, E. dans Comprehensive Heterocyclic Chemistry, Katritzky A. R., Rees, C. W., Eds.; Pergamon:
Oxford, 1984, p 910. 77
Heun, S.; Bad, S.; Ludemann, A.; Anémian, R. M.; Schulte, N. DE 10 2008 044 868 A1. 78
Videlot-Ackermann, C.; Ackermann, J.; Brisset, H.; Kawamura, K.; Ymoshimoto, N.; Raynal, P.; El Kassmi, A.;
Fages, F. J. Am. Chem. Soc. 2005, 127, 16346-16347. 79
Smith, J. R.; Ratcliffe, N. M.; Campbell, S. A. Synthetic Metals, 1995, 73, 171-182. 80
(a) Jiang, B.; Dou, Y.; Xu, X. Y.; Xu, M. Org Lett. 2008, 10, 593-596. (b) Molander, G. A.; Oliveira, R. A.
Tetrahedron Lett. 2008, 49, 1266-1268. (c) El-Batta, A.; Jiang, C. C.; Zhao, W.; Anness, R.; Coosky, A. L.;
Bergdahl, M. J. Org. Chem. 2007, 72, 5244-5259. (d) Thiemann, T.; Watanabe, M. ; Tanaka, Y.; Mataka, S. New J.
Chem. 2006, 30, 359-369. 81
Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.; Mackay, K.; Friend, R. H.; Burns, P. L.; Holmes,
A. B. Nature, 1990, 347, 539-541.

56
B.2.5. Conclusion
Des RHD stéréosélectives et régiosélectives de thiophènes substitués en position 2 par un
halogène ou un groupement alkyle avec des styrènes, notamment halogénés, ont été réalisées dans
des conditions douces et avec des rendements atteignant 92 %.
Ces réactions constituaient les seuls exemples de couplage entre des substrats bromés, sans
porter atteinte à la liaison carbone – halogène. Récemment l’équipe de Liu a décrit des couplages de
Heck déshydrogénants de thiophènes avec des acétates allyliques en faisant appel au carbonate
d’argent comme oxydant, dans un système dioxane/DMSO à 110 °C.82
Cependant, la méthode
s’avère peu régiosélective et ne permet pas de maintenir intacte les liaisons Ar-Br. Quant au 2-
chlorothiophène, les rendements globaux sont inférieurs à 45 % (tableau B3).
Entrée R Durée (h) I (E), rdt (%) I (Z), rdt (%) II, rdt (%) III, rdt (%)
1 2-Me 12 63 7 13
2 2-Et 12 56 6 11
3 2-Cl 21,5 32 9 4
4 2-Br 20 26 2 2 33
Tableau B3. Résultats obtenus par Liu pour la RHD de thiophènes substitués en position 2 avec
l’acétate allylique.
A.3. Partie expérimentale
A.3.1. Informations générales
La Benzoquinone (BQ), Pd(OAc)2, B1, B2, B3, B4, C1, C2, C3, C4 et C5 sont des produits
commerciaux et utilisés sans purification.
82
Zhang, Y.; Li, Z.; Liu, Z-Q. Org. Lett. 2012, 14, 226-229.

57
A.3.2. Synthèse de 5-alkyl-2-styrylthiophènes
A l’acétate de palladium (11,2 – 24,4 mg, 0,05 – 0,1 mmol) et la benzoquinone (216 mg, 2
mmol) en solution dans un mélange solvant/co-solvant AcOH (2 mL)/DMSO (2 mL) sont ajoutés
l’arène (2 mmol) et l’alcène (1 mmol). Le mélange est agité à température ambiante pendant 24 h.
Après ajout de Et2O (20 mL), la réaction est traitée par une solution aqueuse saturée de NaHCO3 (20
mL). La phase aqueuse est extraite avec Et2O (20 mL × 2). Les phases organiques sont lavées avec une
solution de NaOH 2M (20 mL x 2), avec H2O (20 mL x 2), séchées sur MgSO4, et filtrées sur coton. Le
solvant est évaporé sous vide. Les produits sont séparés sur colonne de gel de silice flash (éluant :
éther de pétrole/acétate d’éthyle, 100-0 : 98-2).
(E)-2-méthyl-5-styrylthiophène (B1-C1).83
Cristaux blancs ; 67 %.
Point de fusion : 86 - 89 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,42 (s, 3H, H7), 6,55-6,60 (m, 1H, H4), 6,76-6,79 (m, 2H, H5 et H2),
7,10 (d, J = 16,2 Hz, 1H, H1), 7,17 (m, 1H, HAr), 7,28 (t, J = 7,41, 2H, HAr), 7,38 (d, J = 7,54 Hz, 2H, HAr).
(E)-2-éthyl-5-styrylthiophène (B2-C1). Cristaux blancs cassés ; 72 %.
Point de fusion : 34 - 36 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,28 (t, J = 7,5 Hz, 3H, H8), 2,78 (q, J = 7,4 Hz, 2H, H7), 6,63 (d, J =
3,3 Hz, 1H, H4), 6,80 (m,2H, H5 et H2), 7,23-7,03 (m, 2H, H1 et HAr), 7,28 (t, J = 7,4 Hz, 2H, HAr), 7,39 (d,
J = 7,5 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,9 (C8), 23,9 (C7), 122,4 (C4), 124,0 (C5), 126,3 (2CH), 126,4 (CH),
127,2 (C2), 127,4 (C1), 128,8 (2CH), 137,3 (C), 140,6 (C6), 147,0 (C3).
IR (KBr) ν (cm-1
) : 3058, 3023, 2965, 1627, 1594, 1538, 1473, 1352, 1201, 951, 769, 747, 690.
SMHR : pour C14H14S théorique : 214,0816 ; obtenue : 214,0817.
83
Katritzky, A. R.; Serdyuk, L.; Xie, L.; Ghiviriga, I. J. Org. Chem. 1997, 62, 6215–6221.

58
(E)-2-(4-chlorostyryl)-5-méthylthiophène (B1-C4). Cristaux jaunes clairs ; 97 %.
Point de fusion : 111 - 116 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,40 (s, 3H, H7), 6,60 (m, 2H, H2 et H4), 6,76 (d, J = 3,3 Hz, 1H, H5),
7,01 (d, J = 16,0 Hz, 1H, H1), 7,32-7,14 (m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,7 (C7), 122,8 (C4), 125,7(C5), 126,0 (C2), 126,9 (C1) , 127,3 (2CH),
128,8 (2CH), 132,8 (C-Cl), 135,8 (C), 139,7 (C6), 140,5 (C3).
IR (KBr) ν (cm-1
) : 3015, 2917, 1623, 1540, 1470, 1345, 1236, 956, 797, 699.
Analyse élémentaire (%) pour C13H11ClS théorique : C, 66,51 ; H, 4,72 ; S, 13,66 ; obtenue : C, 66,66 ;
H, 4,38 ; S, 13,93.
(E)-2-(4-chlorostyryl)-5-éthylthiophène (B2-C4). Cristaux blancs cassés ; 70 %.
Point de fusion : 50 – 54 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,27 (t, J = 7.5 Hz, 3H, H8), 2,78 (q, J = 7.5, 2H, H7), 6,65 (m, 2H, H2
et H4), 6,80 (d, J = 3.4 Hz, 1H, H5), 7,05 (d, J = 16.0 Hz, 1H, H1), 7,24 (m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,9 (C7), 23,9 (C8), 122,9 (C4), 124,1 (C5), 125,7 (C2), 126,8 (C1),
127,4 (2CH), 128,9 (2CH), 132,8 (C-Cl), 135,8 (C), 140,1 (C6), 147,4 (C3).
IR (KBr) ν (cm-1
) : 3014, 2968, 2935, 1623, 1538, 1490, 1471, 1263, 95673, 751, 697.
SMHR : pour C14H13ClS théorique : 248,0426 ; obtenue : 248,0427.
(E)-2-(3-chlorostyryl)-5-méthylthiophène (B1-C5). Cristaux jaunes clairs ; 59 %.
Point de fusion : 77 - 81 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,44 (s, 3H, H7), 6,64 (m, 2H, H2 etH4), 6,82 (d, J = 3.4 Hz, 1H, H5),
7,01-7,28 (m, 4H, HAr et H1), 7,37 (s, 1H, HAr).

59
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,8 (C7), 123,0 (C4), 123,6 (C5), 124,5 (C2), 125,5 (C1), 126,0 (CH),
127,2 (2CH), 129,9 (CH), 134,7 (C-Cl), 139,2 (C), 140,0 (C6), 140,4 (C3).
IR (KBr) ν (cm-1
) : 3059, 3014, 2918, 1586, 1560, 1407, 1234, 1165, 948, 775, 708.
Analyse élémentaire (%) pour C13H11ClS théorique : C, 66,51 ; H, 4,72 ; S, 13,66 ; Obtenue : C, 66,12 ;
H, 4,72 ; S, 13,91.
(E)-2-(3-chlorostyryl)-5-éthylthiophène (B2-C5). Huile jaune ; 80 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,30 (t, J = 7,5 Hz, 3H, H8), 2,81 (q, J = 7,5 Hz, 2H, H7), 6,75-6,60
(m, 2H, H2 et H4), 6,86 (d, J = 3,4 Hz, 1H, H5), 7,30-7,06 (m, 4H, HAr et H1), 7,39 (s, 1H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,9 (C8), 23,9 (C7), 123,6 (C4), 124,1 (C5), 124,4 (C2), 125,5 (C1),
126,0 (CH), 127,1 (CH), 127,2 (CH), 129,9 (2CH), 134,7 (C-Cl), 139,2 (C), 139,9 (C6), 147,6 (C3).
IR (film) ν (cm-1
) : 3064, 3020, 2968, 1591, 1564, 1427, 1231, 1165, 946, 776, 682.
SMHR : pour C14H13ClS théorique : 248,0426 ; obtenue : 248,0424.
(E)-2-(4-bromostyryl)-5-méthylthiophène (B1-C2). Cristaux jaunes ; 90 %.
Point de fusion : 130 - 133 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,42 (s, 3H, H7), 6,63 (m, 2H, H2 et H4), 6,78 (d, J = 3,2 Hz, 1H, H5),
7,04 (d, J = 16,0 Hz, 1H, H1), 7,19 (d, J = 8,3 Hz, 2H, HAr), 7,37 (d, J = 8,3 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,7 (C7), 121,0 (C-Br), 122,9 (C4), 125,7 (C5), 126,0 (C2), 127,0
(C1), 127,7 (2CH), 131,8 (2CH), 136,2 (C), 139,8 (C6), 140,5 (C3).
IR (KBr) ν (cm-1
) : 3074, 3012, 2914, 1623, 1583, 1400, 1236, 1164, 956, 770, 698.
Analyse élémentaire (%) pour C13H11BrS théorique : C, 55,92 ; H, 3,97 ; S, 11,48 ; obtenue : C, 56,20 ;
H, 3,95 ; S, 11,74.

60
(E)-2-(4-bromostyryl)-5-éthylthiophene (B2-C2). Cristaux jaunes clairs ; 56 %.
Point de fusion : 125 - 128 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,19 (t, J = 7,5 Hz, 3H, H8), 2,69 (q, J = 7,5, 2H, H7), 6,57 (m, 2H, H2
et H4), 6,72 (d, J = 3,4 Hz, 1H, H5), 6,98 (d, J = 16,0 Hz, 1H, H1), 7,12 (d, J = 8,4 Hz, 2H, HAr), 7,28 (d, J =
8,4 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,9 (C8), 23,9 (C7), 121,0 (C-Br), 123,0 (C4), 124,1 (C5), 125,7 (C2),
126,8 (C1), 127,7 (2CH), 131,8 (2CH), 136,3 (C), 140,1 (C6), 147,5 (C3).
IR (KBr) ν (cm-1
) : 2965, 1622, 1582, 1470, 1434, 1235, 1178, 956, 770, 701.
Analyse élémentaire (%) pour C14H13BrS théorique : C, 57,35 ; H, 4,47 ; S, 10,94 ; obtenue : C, 57,56 ;
H, 4,24 ; S, 11,10.
(E)-2-(2-bromostyryl)-5-méthylthiophène (B1-C3). Cristaux jaunes ; 56 %.
Point de fusion : 43 - 47 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,44 (s, 3H, H7), 6,61 (d, J = 2,5 Hz, 1H, H4), 6,84 (d, J = 3,2 Hz, 1H,
H5), 6,95-7,25 (m, 4H, H1, H2 et HAr), 7,51 (m, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,8 (C7), 123,8 (C-Br), 124,8 (C4), 125,6 (C5), 126,0 (C2), 126,3
(C1), 127,3 (CH), 127,6 (CH), 128,5 (CH), 133,2 (CH), 137,0 (C), 140,2 (C6), 140,6 (C3).
IR (KBr) ν (cm-1
) : 3063, 3034, 2943, 1688, 1618, 1582, 1257, 1159, 943, 791, 701.
Analyse élémentaire (%) pour C13H11BrS théorique : C, 55,92 ; H, 3,97 ; S, 11,48 ; obtenue : C,
56,09 ; H, 3,99 ; S, 11,33.

61
(E)-2-(2-bromostyryl)-5-éthylthiophène (B2-C3). Cristaux jaunes pales ; 68 %.
Point de fusion : 103 - 107 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,30 (t, J = 7,5 Hz, 3H, H8), 2,81 (q, J = 7,6 Hz, 2H, H7), 6,66 (td, J =
3,5, 1,02 Hz, 1H, H4), 6,88 (d, J = 3,5 Hz, 1H, H5), 6,97-7,18 (m, 3H, H1, H2 et HAr), 7,22 (td, J = 7,7, 0,9
Hz, 1H, HAr), 7,53 (m, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,9 (C8), 23,9 (C7), 123,8 (C-Br), 124,1 (C4), 124,9 (C5), 125,5 (C2),
126,3 (C1), 127,1 (CH), 127,6 (CH), 128,5 (CH), 133,1, 137,0 (C), 140,1 (C6), 147,9 (C3).
IR (KBr) ν (cm-1
) : 3059, 3036, 2969, 1618, 1581, 1456, 1258, 1200, 941, 802.
Analyse élémentaire (%) pour C14H13BrS théorique : C, 57,35 ; H, 4,.47 ; S, 10,94 ; obtenue : C,
57,43 ; H, 4,44 ; S, 10,92.
A.3.2. Synthèse de 2-halogéno-5-styrylthiophènes
A l’acétate de palladium (11,2 – 24,4 mg, 0,05 – 0,1 mmol) et la benzoquinone (216 mg, 2
mmol) mis en solution dans un mélange solvant/co-solvant AcOH (2 mL)/DMSO (2 mL) sont ajoutés
l’arène (2 mmol) et l’alcène (1 mmol). Le mélange est agité à température ambiante pendant 48 h.
Après ajout de Et2O (20 mL), la réaction est traitée par une solution aqueuse saturée de NaHCO3 (20
mL). La phase aqueuse est extraite avec Et2O (20 mL × 2). Les phases organiques sont lavées avec une
solution de NaOH 2M (20 mL x 2), avec H2O (20 mL x 2), séchées sur MgSO4, et filtrées sur coton. Le
solvant est évaporé sous vide. Les produits sont séparés sur colonne de gel de silice flash (éluant :
éther de pétrole/acétate d’éthyle, 100-0 : 98-2).
(E)-2-chloro-5-styrylthiophene (B3-C1). Cristaux jaunes pale ; 72 %.
Point de fusion : 86 - 89 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,67-6,74 (m, 3H, H4, H5 et H2), 6,97 (d, J = 16,1 Hz, 1H, H1), 7,16-
7,36 (m, 5H, HAr).

62
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 121,4 (C4), 125,5 (C5), 126,5 (2CH), 126,8 (CH), 127,9 (C2), 128,6
(C1), 128,7 (C), 128,8 (2CH), 136,6 (C3), 141,8 (C6).
IR (KBr) ν (cm-1
) : 3021, 1623, 1593, 1224, 1193, 948, 795, 749, 689.
Analyse élémentaire (%) pour C12H9ClS théorique : C, 65,30 ; H, 4,11 ; S, 14,53 ; obtenue : C, 64,87 ;
H, 4,05 ; S, 14,04.
(E)-2-bromo-5-styrylthiophene (B4-C1). Cristaux jaunes ; 77 %.
Point de fusion : 84 - 87 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,64 (m, 2H, H4 et H2), 6,79 (d, J = 3,8 Hz, 1H, H5), 6,93 (d, J =
16,0 Hz, 1H, H1), 7,16 (m, 3H, HAr), 7,27 (d, J = 7,2 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 111,2 (C6), 121,3 (C4), 126,4 (C5), 126,5 (C2), 128,0 (3CH), 128,9
(C1), 130,6 (CH), 136,6 (C), 144,6 (C3).
IR (KBr) ν (cm-1
) : 3075, 3021, 2951, 1623, 1592, 1429, 1226, 1192, 948, 795, 749, 689.
Analyse élémentaire (%) pour C12H9BrS : C, 54,35 ; H, 3,42 ; S, 12,09 ; obtenue : C, 54,43 ; H, 3,40 ; S
12,04.
(E)-2-chloro-5-(4-chlorostyryl)thiophène (B3-C4). Cristaux blancs cassés ; 88 %.
Point de fusion : 94 - 97 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,50 (d, J = 16,1 Hz, 1H, H2), 6,62 (m, 2H, H4 et H5), 6,81 (d, J =
16,1 Hz, 1H, H1), 7,12 (m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 121,9 (C4), 125,8 (C5), 126,9 (C2), 127,1 (C1), 127,5 (2CH), 129,0
(2CH), 129,1 (C), 133,4 (C-Cl), 135,1 (C3), 141,4 (C6).
IR (KBr) ν (cm-1
) : 3094, 3014, 1623, 1589, 1261, 1230, 943, 794, 696.
Analyse élémentaire (%) pour C12H8Cl2S : C, 56,48 ; H, 3,16 ; S, 12,57 ; obtenue : C, 56,45; H, 3,22; S,
12,41.

63
(E)-2-bromo-5-(4-chlorostyryl)thiophène (B4-C4). Cristaux jaunes ; 76 %.
Point de fusion : 81 - 85 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,51-6,61 (m, 2H, H2 et H4), 6,77-6,89 (m, 2H, H1 et H5), 7,14
(m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 111,6 (C6), 121,8 (C4), 126,7 (C5), 127,4 (C2), 127,6 (C1), 129,0
(2CH), 130,6 (2CH), 133,5 (C-Cl), 135,1 (C), 144,2 (C3).
IR (KBr) ν (cm-1
) : 3083, 3013, 1622, 1587, 1433, 1230, 1093, 1055, 794, 748, 696.
Analyse élémentaire (%) pour C12H8BrClS : C, 48,10 ; H, 2,69; S,10,70 ; obtenue : C, 47,98 ; H, 2,66 ; S,
10,82.
(E)-2-chloro-5-(3-chlorostyryl)thiophène (B3-C5). Solide jaune ; 92 %.
Point de fusion : 45 - 48 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,57 (d, J = 16,1 Hz, 1H, H2), 6,72 (m, 2H, H4 et H5), 6,93 (d, J =
16,1 Hz, 1H, H1), 7,15 (m, 3H, HAr), 7,30 (m, 1H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 122,6 (C4), 124,6 (C5), 126,1 (C2), 126,8 (C1), 126,9 (2CH), 127,7
(CH), 129,4 (C-Cl), 129,9 (CH) , 134,7 (C), 138,5 (C3), 141,2 (C6).
IR (KBr) ν (cm-1
) : 3061, 3027, 1625, 1591, 1436, 1252, 1085, 783, 768, 675.
Analyse élémentaire (%) pour C12H8Cl2S : C, 56,48 ; H, 3,16 ; S, 12,57 ; obtenue : C, 56,62 ; H, 3,13 ; S,
12,84.
(E)-2-bromo-5-(4-bromostyryl)thiophène (B4-C2). Cristaux jaunes ; 56 %.
Point de fusion : 70 - 73 °C.

64
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,46-6,73 (m, 2H, H4 et H2), 6,73-7,02 (m, 2H, H5 et H1), 7,13 (d, J
= 8,0 Hz, 2H, HAr), 7,31 (d, J = 7,8 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 111,7 (C6), 121,7 (C4), 121,9 (C-Br), 126,8 (C5), 127,5 (C2), 127,9
(2CH), 130,7 (C1), 131,9 (2CH), 135,6 (C), 144, 2 (C3).
IR (KBr) ν (cm-1
) : 3082, 3012, 1622, 1582, 1433, 1419, 1074, 1056, 794, 768, 746, 704.
SMHR : pour C12H8Br2S théorique : 341,8713 ; obtenue : 341,8706.
(E)-5-(4-bromostyryl)-2-chlorothiophène (B3-C2). Cristaux jaunes ; 65 %.
Point de fusion : 32 - 35 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,63 (d, J = 16,1 Hz, 1H, H2), 6,76 (m, 2H, H4 et H5), 6,98 (d, J =
16,1 Hz, 1H, H1), 7,22 (d, J = 8,3 Hz, 2H, HAr), 7,40 (d, J = 8,3 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 121,7 (C-Br), 122,1 (C4), 125,9 (C5), 126,9 (C2), 127,2 (C1), 127,9
(2CH), 129,2 (C), 132,0 (2CH), 135,6 (C3), 141,4 (C6).
IR (KBr) ν (cm-1
) : 1652, 1621, 1609, 1582, 1563, 1526, 1439, 1425, 1231, 997, 956, 943, 793, 704.
Analyse élémentaire (%) pour C12H8BrClS : C, 48,10 ; H, 2,69 ; S, 10,70 ; obtenue : C, 47,94 ; H, 2,41 ;
S, 10,64.
(E)-2-bromo-5-(2-bromostyryl)thiophene (B4-C3). Cristaux jaunes ; 57 %.
Point de fusion : 68 - 71 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,91-6,97 (m, 1H), 7,02-7,52 (m, 5H), 7,84-7,57 (m, 2H);
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 112,1 (C6), 123,6 (C4), 124,0 (C-Br), 126,4 (C5), 127,0 (C2), 127,2
(C1), 127,6 (CH), 129,0 (CH), 130,6 (CH), 133,2 (CH), 136,3 (C), 144,1 (C3).
IR (KBr) ν (cm-1
) : 3022, 1617, 1520, 1465, 1426, 1251, 966, 939, 783, 699.
SMHR : pour C14H13ClS théorique 341,8713 ; obtenue : 341,8705.

65
Partie C : Influence de l’oxydant sur l’activité du catalyseur
Introduction
Les réactions de Heck déshydrogénantes requièrent une quantité stœchiométrique
d’oxydant. Si certaines équipes concentrent leurs efforts sur l’utilisation de l’oxygène moléculaire
comme unique oxydant afin de parvenir à des réactions plus respectueuses de l’environnement,84
d’autres privilégient l’emploi d’oxydants organiques, comme par exemple la benzoquinone, parce
qu’ils peuvent excercer un rôle supplémentaire sur l’activité du catalyseur. Il est en effet établi que la
benzoquinone puisse intervenir sur certaines étapes réactionnelles du cycle catalytique.
Après avoir présenté les différents rôles que peut jouer la benzoquinone dans les réactions
pallado-catalysées, nous exposerons une étude mécanistique destinée à définir le rôle de la
benzoquinone dans nos conditions réactionnelles.
C.1. Bibliographie
C.1.1. La benzoquinone, un réoxydant du Pd(0) en Pd(II)
Bäckvall et al, ont montré l’oxydation du palladium (0) par la benzoquinone en milieu
acide.85
BQ se complexerait au Pd(0) et le complexe ainsi généré réagirait avec l’acide acétique pour
conduire au palladium (II) et à l’hydroquinone libre (schéma C1).
Schéma C1. Mécanisme de réoxydation du Pd(0) par la benzoquinone proposé par Bäckvall.
Des études mécanistiques menées par Stahl en 200686
soulignent les similarités entre les
réactions de O2 et d’alcènes déficients en électrons (BQ par exemple) avec Pd(0). Leurs études
84
Gligorich, K. M.; Sigman, M. S. Chem. Commun. 2009, 3854-3867. 85
Grennberg, H.; Gogoll, A; Bäckvall, J. E. Organometallics 1993, 12, 1790-1793. 86
Popp, B. V.; Thorman J. L.; Stahl. S. S. J. Mol. Catal-A Chem. 2006, 251, 2-7.
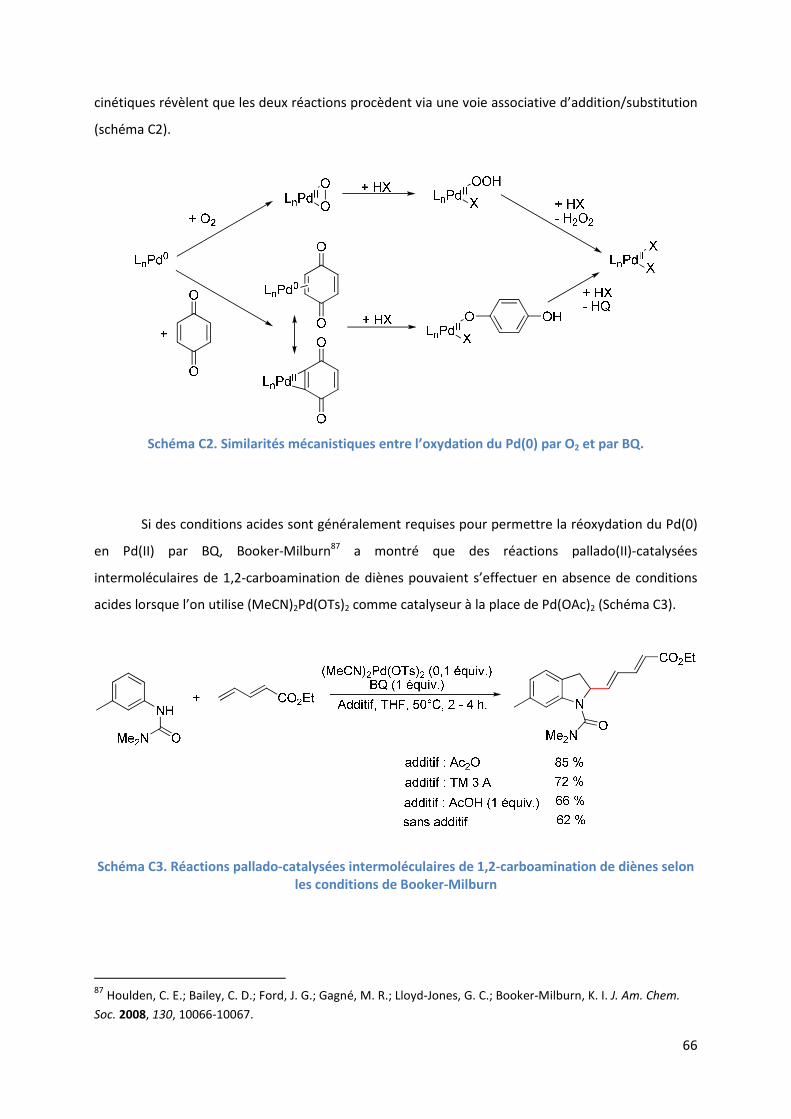
66
cinétiques révèlent que les deux réactions procèdent via une voie associative d’addition/substitution
(schéma C2).
Schéma C2. Similarités mécanistiques entre l’oxydation du Pd(0) par O2 et par BQ.
Si des conditions acides sont généralement requises pour permettre la réoxydation du Pd(0)
en Pd(II) par BQ, Booker-Milburn87
a montré que des réactions pallado(II)-catalysées
intermoléculaires de 1,2-carboamination de diènes pouvaient s’effectuer en absence de conditions
acides lorsque l’on utilise (MeCN)2Pd(OTs)2 comme catalyseur à la place de Pd(OAc)2 (Schéma C3).
Schéma C3. Réactions pallado-catalysées intermoléculaires de 1,2-carboamination de diènes selon
les conditions de Booker-Milburn
87
Houlden, C. E.; Bailey, C. D.; Ford, J. G.; Gagné, M. R.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. J. Am. Chem.
Soc. 2008, 130, 10066-10067.
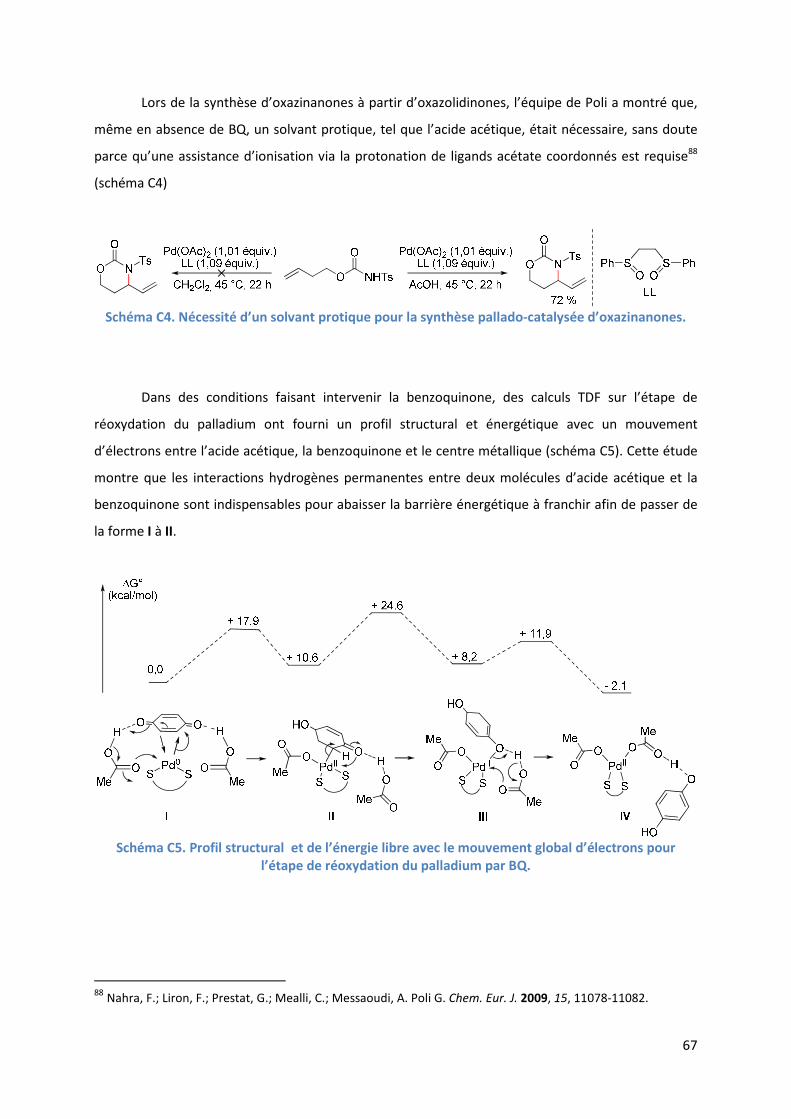
67
Lors de la synthèse d’oxazinanones à partir d’oxazolidinones, l’équipe de Poli a montré que,
même en absence de BQ, un solvant protique, tel que l’acide acétique, était nécessaire, sans doute
parce qu’une assistance d’ionisation via la protonation de ligands acétate coordonnés est requise88
(schéma C4)
Schéma C4. Nécessité d’un solvant protique pour la synthèse pallado-catalysée d’oxazinanones.
Dans des conditions faisant intervenir la benzoquinone, des calculs TDF sur l’étape de
réoxydation du palladium ont fourni un profil structural et énergétique avec un mouvement
d’électrons entre l’acide acétique, la benzoquinone et le centre métallique (schéma C5). Cette étude
montre que les interactions hydrogènes permanentes entre deux molécules d’acide acétique et la
benzoquinone sont indispensables pour abaisser la barrière énergétique à franchir afin de passer de
la forme I à II.
Schéma C5. Profil structural et de l’énergie libre avec le mouvement global d’électrons pour
l’étape de réoxydation du palladium par BQ.
88
Nahra, F.; Liron, F.; Prestat, G.; Mealli, C.; Messaoudi, A. Poli G. Chem. Eur. J. 2009, 15, 11078-11082.
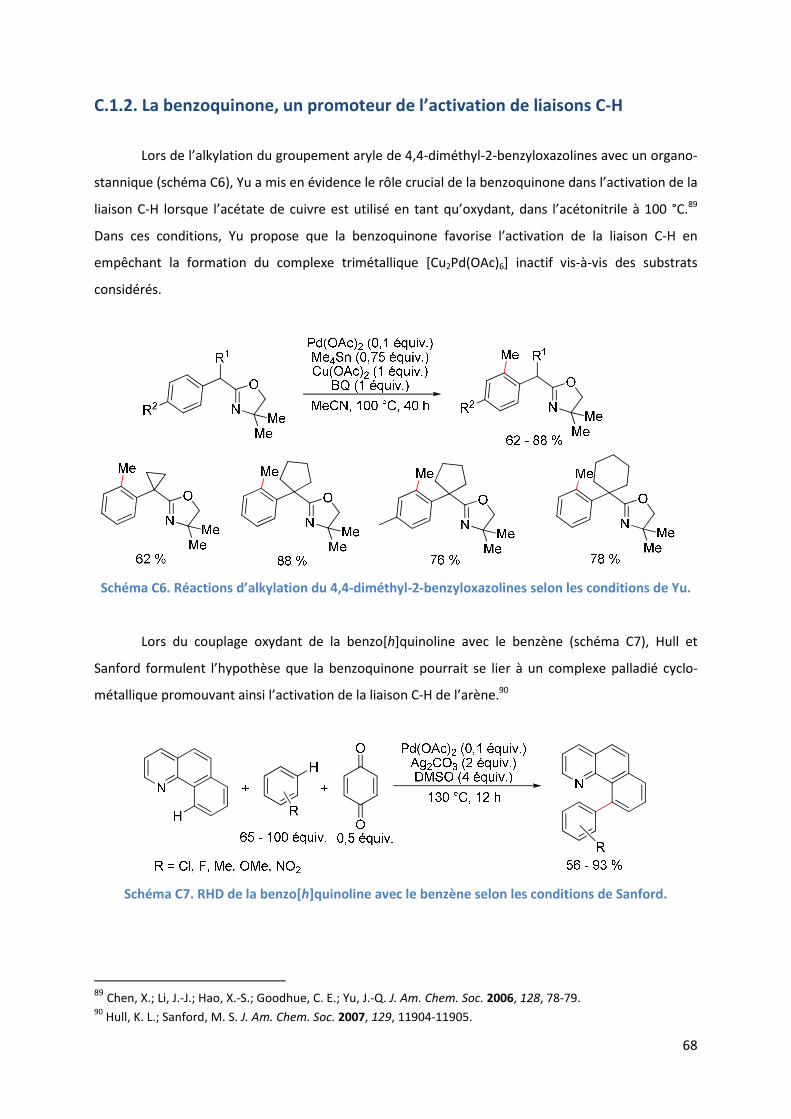
68
C.1.2. La benzoquinone, un promoteur de l’activation de liaisons C-H
Lors de l’alkylation du groupement aryle de 4,4-diméthyl-2-benzyloxazolines avec un organo-
stannique (schéma C6), Yu a mis en évidence le rôle crucial de la benzoquinone dans l’activation de la
liaison C-H lorsque l’acétate de cuivre est utilisé en tant qu’oxydant, dans l’acétonitrile à 100 °C.89
Dans ces conditions, Yu propose que la benzoquinone favorise l’activation de la liaison C-H en
empêchant la formation du complexe trimétallique [Cu2Pd(OAc)6] inactif vis-à-vis des substrats
considérés.
Schéma C6. Réactions d’alkylation du 4,4-diméthyl-2-benzyloxazolines selon les conditions de Yu.
Lors du couplage oxydant de la benzo[h]quinoline avec le benzène (schéma C7), Hull et
Sanford formulent l’hypothèse que la benzoquinone pourrait se lier à un complexe palladié cyclo-
métallique promouvant ainsi l’activation de la liaison C-H de l’arène.90
Schéma C7. RHD de la benzo[h]quinoline avec le benzène selon les conditions de Sanford.
89
Chen, X.; Li, J.-J.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 78-79. 90
Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2007, 129, 11904-11905.
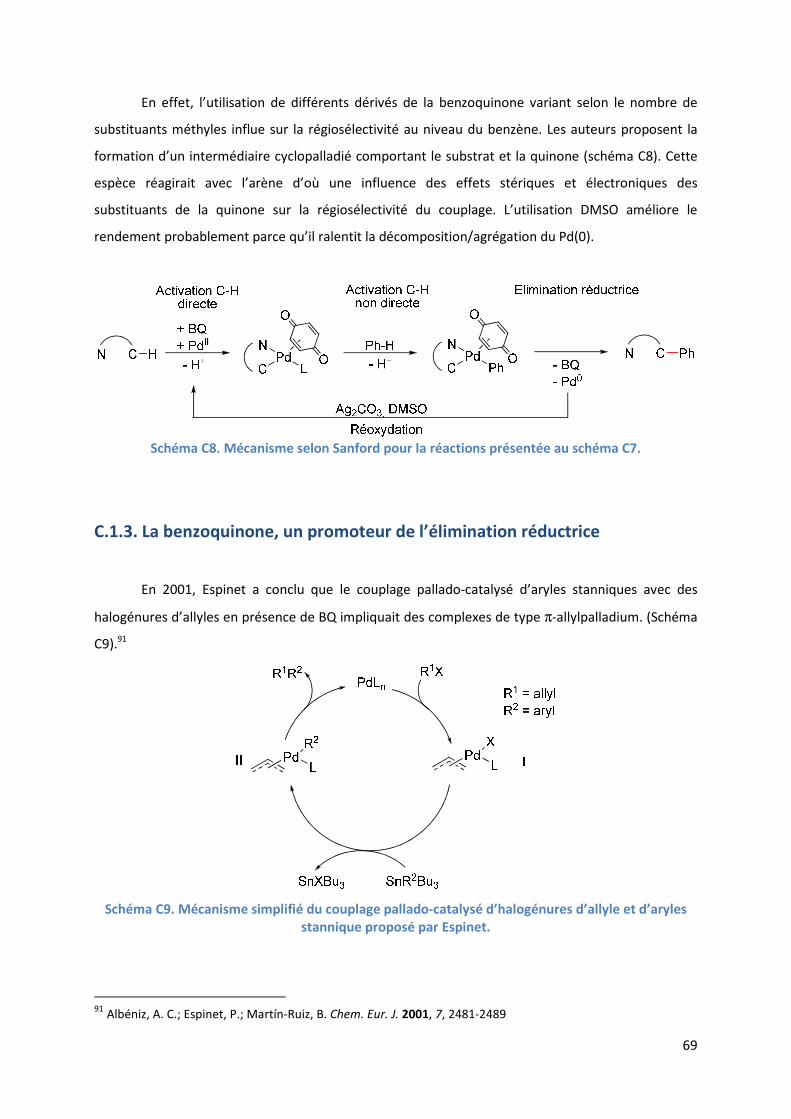
69
En effet, l’utilisation de différents dérivés de la benzoquinone variant selon le nombre de
substituants méthyles influe sur la régiosélectivité au niveau du benzène. Les auteurs proposent la
formation d’un intermédiaire cyclopalladié comportant le substrat et la quinone (schéma C8). Cette
espèce réagirait avec l’arène d’où une influence des effets stériques et électroniques des
substituants de la quinone sur la régiosélectivité du couplage. L’utilisation DMSO améliore le
rendement probablement parce qu’il ralentit la décomposition/agrégation du Pd(0).
Schéma C8. Mécanisme selon Sanford pour la réactions présentée au schéma C7.
C.1.3. La benzoquinone, un promoteur de l’élimination réductrice
En 2001, Espinet a conclu que le couplage pallado-catalysé d’aryles stanniques avec des
halogénures d’allyles en présence de BQ impliquait des complexes de type π-allylpalladium. (Schéma
C9).91
Schéma C9. Mécanisme simplifié du couplage pallado-catalysé d’halogénures d’allyle et d’aryles
stannique proposé par Espinet.
91
Albéniz, A. C.; Espinet, P.; Martín-Ruiz, B. Chem. Eur. J. 2001, 7, 2481-2489

70
Au cours de cette réaction, la benzoquinone se coordonnerait au centre métallique du
complexe II et favoriserait l’élimination réductrice au détriment d’une élimination β-H afin de fournir
sélectivement le produit de couplage (schéma C10). La capacité de la benzoquinone à promouvoir
cette élimination réductrice dépendrait de la nature du solvant utilisé. Tandis que dans un solvant
non coordonnant tel que le CDCl3, des quantités catalytiques de BQ suffisent, un excès de BQ dans un
solvant coordonnant comme l’acétonitile, est nécessaire.
Schéma C10. Rôle de la benzoquinone dans la réaction étudiée par Espinet en 2001.
En 2005, Christina White a observé un rôle similaire de la benzoquinone lors d’oxydations
allyliques de liaisons C-H faisant appel à un système Pd(OAc)2/BQ/AcOH/sulfoxyde. Un tel système
conduit dans des conditions douces à un allylacétate branché de façon sélective et efficace92
(schéma
C11).
Schéma C11. Réactions d’oxydation allylique de liaisons C-H selon les conditions de White.
92
Chen, M. S.; Prabagaran, N.; Labenz N. A.; White, M. C. J. Am. Chem. Soc., 2005, 127, 6970-6971.
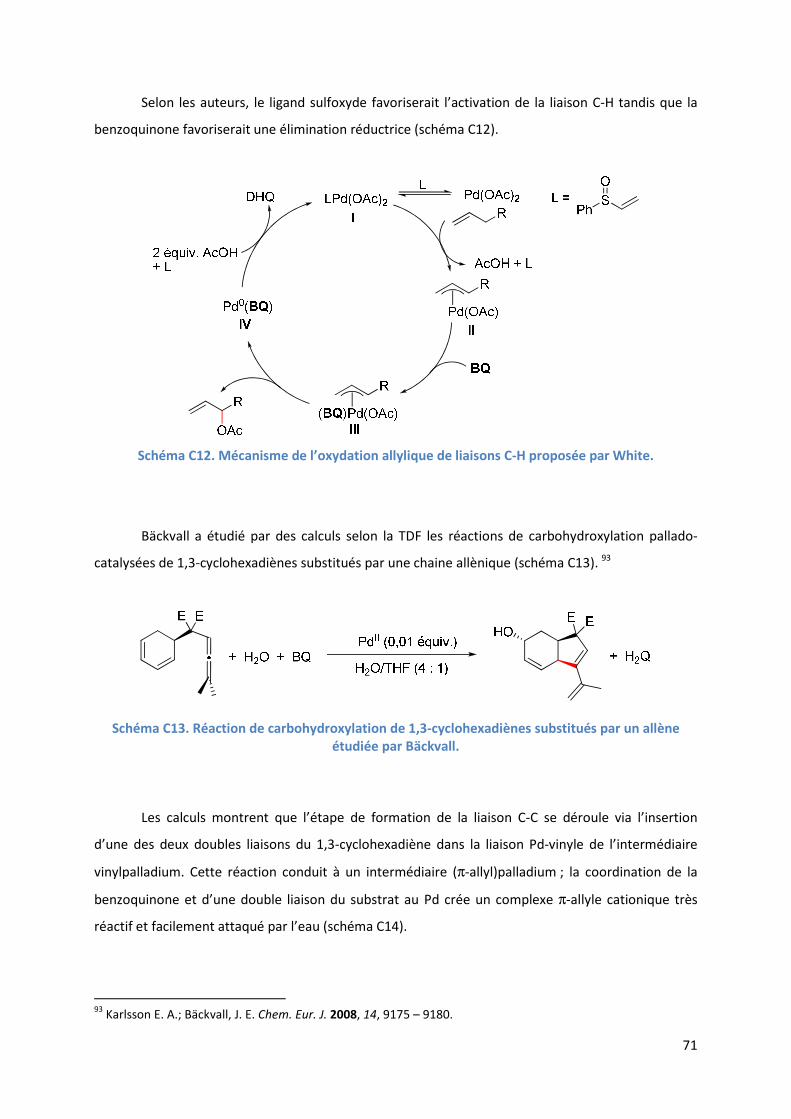
71
Selon les auteurs, le ligand sulfoxyde favoriserait l’activation de la liaison C-H tandis que la
benzoquinone favoriserait une élimination réductrice (schéma C12).
Schéma C12. Mécanisme de l’oxydation allylique de liaisons C-H proposée par White.
Bäckvall a étudié par des calculs selon la TDF les réactions de carbohydroxylation pallado-
catalysées de 1,3-cyclohexadiènes substitués par une chaine allènique (schéma C13). 93
Schéma C13. Réaction de carbohydroxylation de 1,3-cyclohexadiènes substitués par un allène
étudiée par Bäckvall.
Les calculs montrent que l’étape de formation de la liaison C-C se déroule via l’insertion
d’une des deux doubles liaisons du 1,3-cyclohexadiène dans la liaison Pd-vinyle de l’intermédiaire
vinylpalladium. Cette réaction conduit à un intermédiaire (π-allyl)palladium ; la coordination de la
benzoquinone et d’une double liaison du substrat au Pd crée un complexe π-allyle cationique très
réactif et facilement attaqué par l’eau (schéma C14).
93
Karlsson E. A.; Bäckvall, J. E. Chem. Eur. J. 2008, 14, 9175 – 9180.

72
Schéma C14. Mécanisme proposé par Bäckvall pour les réactions de carbohydroxylation de 1,3-
cyclohexadiène substitué par une chaîne allènique.
L’étude TDF de l’élimination réductrice R-R (R = Me, Ph, vinyle) se produisant à partir des
intermédiaires plausibles de réactions pallado-catalysées a été réalisée par Espinet en 2009.94
Celle-ci
inclue des systèmes tétracoordonnés cis-[PdR2(PMe3)2] plans carrés, eux-mêmes possibles
intermédiaires de systèmes cis-[PdR2(PMe)3L] formés en solution ou par addition de promoteurs de
couplage (L = acétonitrile, éthylène, anhydride maléique, lacune électronique) (schéma C15). Chaque
intermédiaire a été modélisé pour toutes les combinaisons de R et L. Les résultats indiquent que
l’énergie d’activation varie de 0,6 à 28,6 kcal/mol en phase gazeuse selon R (vinyl-vinyl < Ph-Ph < Me-
Me et selon L (acide maléique < lacune électronique < éthylène < PMe3 = MeCN).
Par ailleurs, l’étude de l’effet de l’ajout d’une série de composés éthylèniques a fourni l’ordre
d’énergie d’activation suivant : p-benzoquinone < acide maléique < trans-1,2-dicyanoethylène < 3,5-
diméthylcyclopent-1-ène < 2,5-dihydrofurane < ethylène < trans-2-butène. Ces résultats montrent
que la benzoquinone utilisée en tant que ligand permet d’abaisser la barrière énergétique
d’activation pour que le processus d’élimination réductrice puisse s’opérer.
94
Pérez-Rodriguez, M.; Braga, A. A. C.; Garcia-Melchor, M.; Pérez-Temprano, M. H.; Casares, J. A.; Ujacque, G.;
de Lera, A. R.; Alvarez, R.; Maseras, F.; Espinet, P. J. Am. Chem. Soc. 2009, 131, 3650-3657.

73
Schéma C15. Les quatre intermédiaires plausibles pour le profil réactionnel de l’élimination
réductrice étudiés par Espinet en TDF.
L’équipe de Sanford s’est concentrée sur le mécanisme du couplage pallado-catalysé de la
benzo[h]quinoline avec des arènes95
(schéma C16). Dans les conditions proposées, ces
transformations s’opèrent avec une grande régiosélectivité. Elles conduisent à des sélectivités
modestes à excellentes en faveur du couplage sur la position la moins encombrée stériquement des
arènes 1,2- et 1,3-disubstitués. Cependant, ce système est unique en ce sens où un très large excès
de benzoquinone est requis. De plus, la structure de la quinone a une influence considérable sur la
régiosélectivité du couplage avec certains arènes, suggérant que la quinone agit comme un ligand
durant l’étape sélectivement déterminante.
Schéma C16. Couplage de la benzo[h]quinoline avec des arènes selon Sanford.
Des études cinétiques ont défini davantage le rôle de la benzoquinone. Le mécanisme
impliquerait une activation de la liaison C-H de l’arène s’effectuant selon un équilibre suivie d’une
élimination réductrice favorisée par coordination de la benzoquinone conduisant ainsi au produit de
couplage (schéma C17).
95
Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2009, 131, 9651-9653.

74
Schéma C17. Mécanisme proposé par Sanford pour le couplage de la benzo[h]quinoline avec des
arènes.
Sakaki apporte des précisions quant à ce mécanisme grâce à une étude théorique.96
Une
première activation hétérolytique de la liaison C-H de la benzo[h]quinoline (HBzq) par Pd(OAc)2
conduirait à l’espèce Pd(Bzq)(OAc). Le centre palladié Pd(Bzq)(OAc) étant plus riche en électrons que
Pd(OAc)2, la benzoquinone se coordonne plus facilement à Pd(Bzq)(OAc) avec une barrière
d’activation plus basse pour mener au complexe plan carré distordu Pd(Bzq)(OAc)(BQ) qui est plus
stable que Pd(Bzq)(OAc). Une seconde activation C-H du benzene s’opère avec une barrière
d’activation modérée et une faible endothermicité. L’étape finale est une élimination réductrice,
s’opérant avec une faible barrière d’énergie. Dans ce mécanisme, la benzoquinone jouerait un rôle
clef dans l’accélération de la réaction :
• La benzoquinone, ligandée au palladium permet de changer la géométrie du complexe. Le
complexe plan carré Pd(Ph)(Bzq)(OAc) devient alors Pd(Ph)(Bzq)(OAc)(BQ) à géométrie
bipyramidale à base triangulaire plus favorable pour une élimination réductrice.
• La benzoquinone stabilise l’état de transition et le complexe ainsi formé par des interactions
électroniques de rétro-donnation. Ces effets contribuent ainsi à abaisser la barrière
énergétique d’activation correspondant à l’élimination réductrice.
L’étape cinétiquement déterminante serait la seconde activation C-H dont la barrière
d’activation est considérablement plus élevée que celle correspondant à la première activation C-H.
Pour cette étape, bien que la benzoquinone diminue la densité électronique du palladium dans le
complexe Pd(Bzq)(OAc), celle-ci ne jouerait pas de rôle majeur, la barrière d’énergie de cette étape
n’étant que très faiblement abaissée.
96
Ishikawa, A.; Nakao, Y.; Sato, H.; Sakaki, S. Dalton Trans. 2010, 39, 3279–3289.

75
En 2011, Sanford est revenue sur l’étude de cette réaction et s’est intéressée au changement
de sélectivité en fonction de la concentration de benzoquinone97
(schéma C18).
Schéma C18. Modification de la sélectivité selon la quantité de benzoquinone mise en jeu.
L’étape cinétiquement déterminante serait déterminée par la concentration de
benzoquinone. Pour la réaction du complexe [(bzq)Pd(OAc)]2 avec le 1,3-diméthoxybenzène, l’étape
cinétiquement déterminante serait la complexation de la benzoquinone (étape 2 du schéma C19)
lorsqu’elle est introduite en faible concentration (1 équivalent) tandis qu’en milieu saturé en
benzoquinone (20 équivalents), l’étape cinétiquement déterminante serait l’activation de la liaison C-
H (étape 1 du schéma C19). Le mécanisme exact de la complexation de la benzoquinone et de la
promotion de l’élimination réductrice reste à élucider. Selon les auteurs, la liaison avec la
benzoquinone pourrait se faire via un intermédiaire pentacoordonné (comme présentée au schéma
C19) ou via une réaction de substitution de ligand et dans ce cas, l’intermédiaire pentacoordonné
serait un état de transition.
97
Lyons, T. W.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2011, 133, 4455–4464.

76
Schéma C19. Mécanisme proposé par Sanford pour la réaction présentée au schéma C18.
C.1.4. La benzoquinone, un promoteur de la transmetallation oxydante
La méthoxycarbonylation de borures d’aryle et d’alcène, réalisée par Yamamoto en utilisant
une combinaison [Pd(OAc)2/PPh3] comme catalyseur et la benzoquinone comme oxydant
stoechiométrique dans du méthanol à température ambiante, fournit les esters de méthyle
correspondants avec de bons rendements (schéma C20).98
Schéma C20. Méthoxycarbonylation de borures d’aryle et d’alcène réalisée par Yamamoto.
Une étape de transmetalation assistée par la benzoquinone est proposée pour expliquer
l’efficacité de ces conditions (schéma C21).
98
Yamamoto, Y. Adv. Synth. Catal. 2010, 352, 478-492.

77
Pd(0)Ph3P
OO
B(OR)2
OMe
Htransmetalation
oxydantePd(II)
Ph3P
OHO
Ph
+ MeOB(OR)2
PPh3, - CO CO, - PPh3,
Pd(II)OC
OHO
PhPd(II)
OC
OHO
Ph
Pd(II)OC
OHO
Ph
MeOH
Pd(II)OC
OMe
Ph
L
Pd(II)
OMe
LO
Ph
ou/et Pd(II)
L
PhO
OMe
PhCO2Me + Pd(0)(BQ)PPh3
L = CO ou PPh3
BQ,PPh3
- HQ
Schéma C21. Mécanisme proposé pour la méthoxycarbonylation de borures d’aryle et d’alcène
réalisée par Yamamoto.
C.1.5. La benzoquinone, un promoteur de l’oxydation de l’hydrure de
Palladium H-Pd
Stahl a formulé l’hypothèse que la benzoquinone agisse comme un promoteur de l’oxydation
d’hydrure de palladium.99
En étudiant la réaction du complexe (IMes)2(H)(O2CPh) en présence
d’oxygène, Stahl propose que la benzoquinone se coordonne à l’hydrure de palladium ; ceci
favoriserait l’élimination réductrice de PhCO2H et donc la formation d’un complexe PdII-OOH (schéma
C22).
99
Decharin N.; Stahl S. S. J. Am. Chem. Soc. 2011, 133, 5732–5735.

78
Schéma C22. Mécanisme proposé par Stahl pour la réoxydation de l’hydrure de palladium par BQ,
O2 et une combinaison O2/BQ.
C.1.6. La benzoquinone, un inhibiteur de l’élimination ββββ-H
Larhed et son équipe100
se sont intéressés aux réactions domino pallado-catalysées de Heck-
Suzuki entre la N,N-diméthyl-2-(vinyloxy)éthanamine et des acides arylboroniques. Lorsque la
réaction catalysée par Pd(OAc)2 est effectuée sous oxygène, la formation de N,N-diméthyl-2-
(styryloxy)éthanamine est observée. Dans des conditions différentes avec notamment la
benzoquinone à la place de O2 en tant qu’oxydant, le cours de la réaction est modifié et donne accès
à des N,N-diméthyl-2-(éthoxy)éthanamine α,β-arylées (schéma C23).
100
Trejos, A.; Fardost, A.; Yahiaoui, S.; Larhed, M. Chem. Commun. 2009, 7587-7589
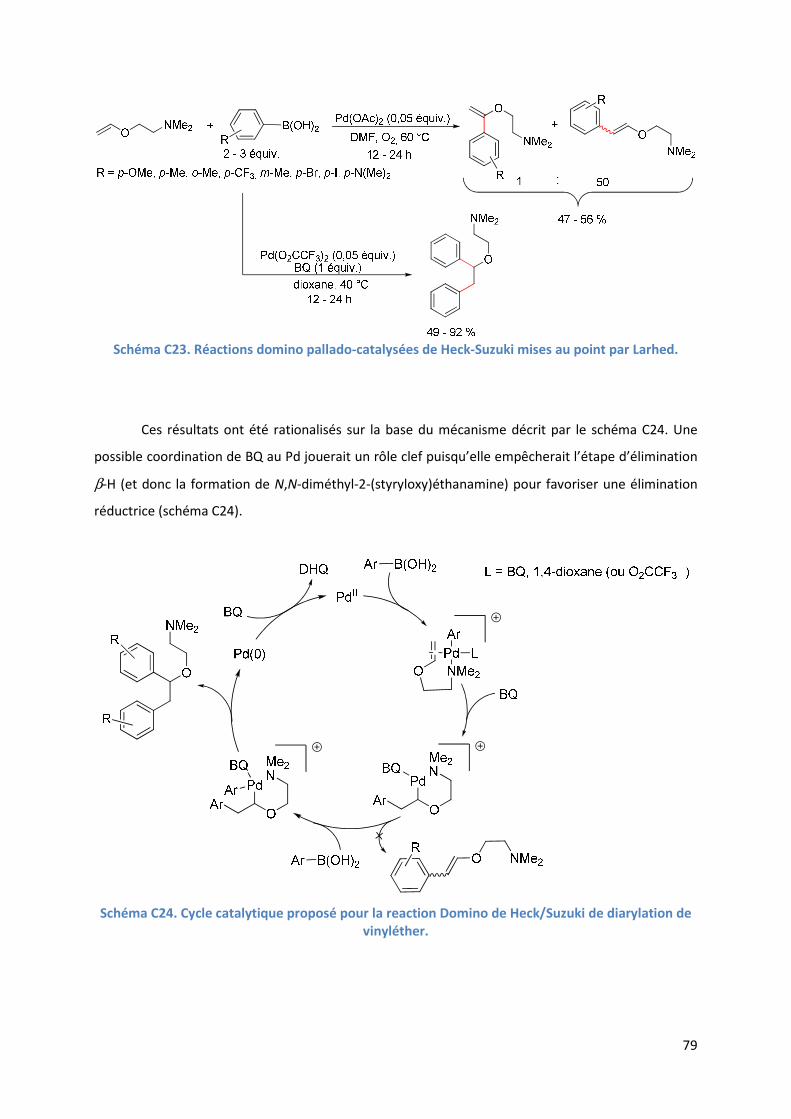
79
Schéma C23. Réactions domino pallado-catalysées de Heck-Suzuki mises au point par Larhed.
Ces résultats ont été rationalisés sur la base du mécanisme décrit par le schéma C24. Une
possible coordination de BQ au Pd jouerait un rôle clef puisqu’elle empêcherait l’étape d’élimination
β-H (et donc la formation de N,N-diméthyl-2-(styryloxy)éthanamine) pour favoriser une élimination
réductrice (schéma C24).
Schéma C24. Cycle catalytique proposé pour la reaction Domino de Heck/Suzuki de diarylation de
vinyléther.

80
C.1.7. La benzoquinone, un promoteur de la migration de ligand
Szabó a rapporté en 1998 une étude théorique de la migration de chlorures induite par la
benzoquinone dans les complexes (η3-allyl)palladium de BQ et du motif allylique.
101 La benzoquinone
et ses dérivés coordonneraient le palladium selon un mode η2. La benzoquinone, ligand encombrant,
oriente Cl- vers le motif allyle, facilitant la migration de ce dernier. Plusieurs types de coordination,
tels que endo et exo, sont possibles fournissant des complexes arylpalladium de stabilité semblable.
Szabó résume les effets des substituants de la benzoquinone de la façon suivante :
• Les substituants électro-attracteurs diminuent l’énergie d’activation pour la migration de
ligands.
• Les substituants présentant des paires d’électrons libres déstabilisent les complexes (η3-
allyl)palladium-quinone.
• Les substituants encombrants intéragissent avec le ligand Cl- et le motif allyle.
Pour conclure, outre son rôle d’oxydant, la benzoquinone est donc un ligand du palladium
pouvant induire des effets électroniques et/ou stériques facilitant certaines étapes du cycle
catalytique. Elle pourrait promouvoir l’activation de liaisons C-H, l’élimination réductrice, des
transmétalations oxydantes, l’oxydation d’hydrures de palladium (II), la migration de ligands ou
encore inhiber les éliminations β-H. Par le biais d’études cinétiques, nous montrerons que la
benzoquinone exerce un rôle positif sur l’insertion de l’alcène dans la liaison Ar-Pd.
C.2. Résultats et discussion
C.2.1. Constat de départ
L’étude du couplage de B1 avec C1 a montré que l’agent oxydant a une influence sur le
rendement de la réaction, ce qui laisse entendre qu’il intervient sur une ou plusieurs étapes du cycle
catalytique. En effet, alors que l’utilisation de 2 équivalents de BQ permet l’obtention du produit de
couplage avec un rendement de 67 %, l’utilisation d’une combinaison BQ (0,5 équiv.)/Cu(OAc)2 (0,1
équiv.)/O2 diminue le rendement à 45 % (tableau A1). Afin de déterminer plus précisément le rôle de
BQ, des études cinétiques ont été entreprises.
101
Szabó, K. J. Organometallics 1998, 17, 1677-1686
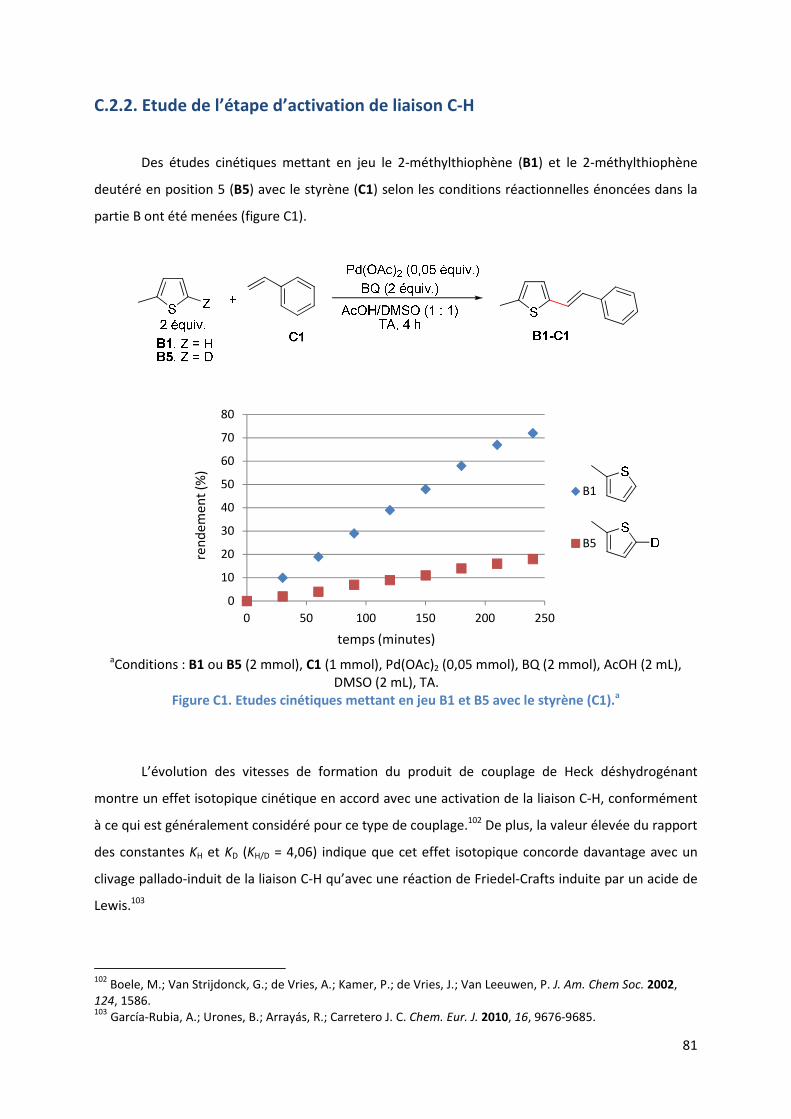
81
C.2.2. Etude de l’étape d’activation de liaison C-H
Des études cinétiques mettant en jeu le 2-méthylthiophène (B1) et le 2-méthylthiophène
deutéré en position 5 (B5) avec le styrène (C1) selon les conditions réactionnelles énoncées dans la
partie B ont été menées (figure C1).
aConditions : B1 ou B5 (2 mmol), C1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL),
DMSO (2 mL), TA.
Figure C1. Etudes cinétiques mettant en jeu B1 et B5 avec le styrène (C1).a
L’évolution des vitesses de formation du produit de couplage de Heck déshydrogénant
montre un effet isotopique cinétique en accord avec une activation de la liaison C-H, conformément
à ce qui est généralement considéré pour ce type de couplage.102
De plus, la valeur élevée du rapport
des constantes KH et KD (KH/D = 4,06) indique que cet effet isotopique concorde davantage avec un
clivage pallado-induit de la liaison C-H qu’avec une réaction de Friedel-Crafts induite par un acide de
Lewis.103
102
Boele, M.; Van Strijdonck, G.; de Vries, A.; Kamer, P.; de Vries, J.; Van Leeuwen, P. J. Am. Chem Soc. 2002,
124, 1586. 103
García-Rubia, A.; Urones, B.; Arrayás, R.; Carretero J. C. Chem. Eur. J. 2010, 16, 9676-9685.
0
10
20
30
40
50
60
70
80
0 50 100 150 200 250
B1
B5
temps (minutes)
ren
de
me
nt
(%)
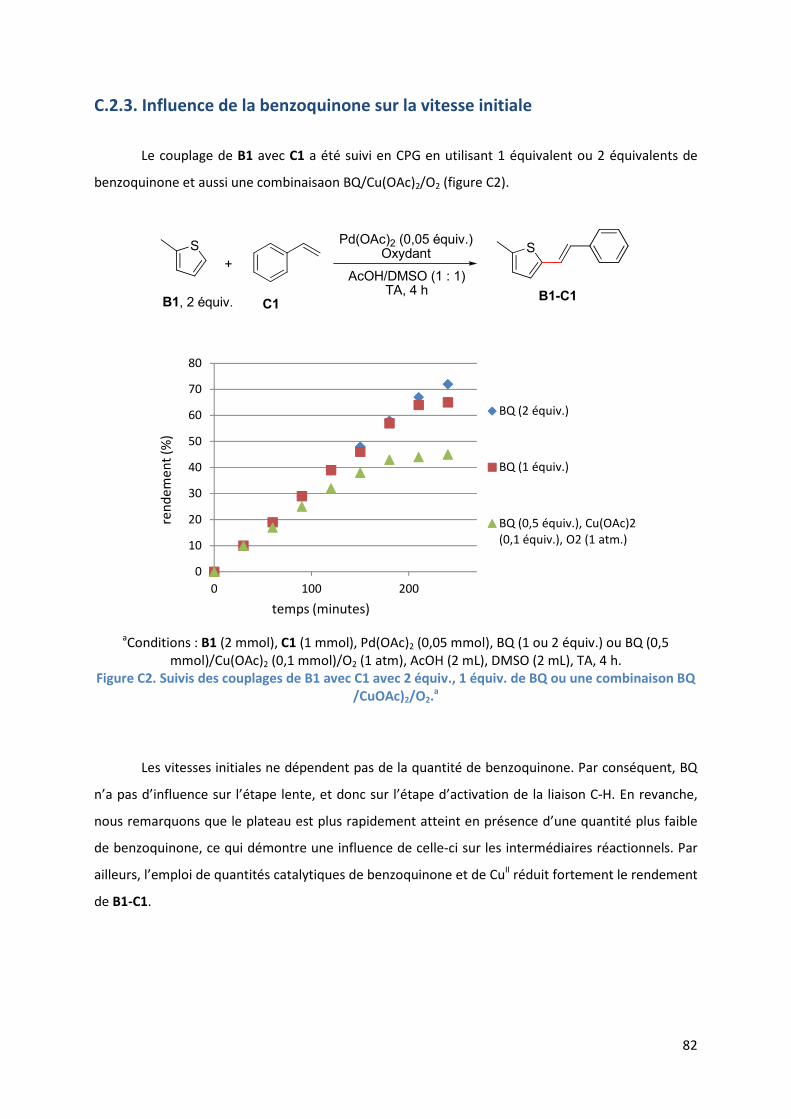
82
C.2.3. Influence de la benzoquinone sur la vitesse initiale
Le couplage de B1 avec C1 a été suivi en CPG en utilisant 1 équivalent ou 2 équivalents de
benzoquinone et aussi une combinaisaon BQ/Cu(OAc)2/O2 (figure C2).
S
+
Pd(OAc)2 (0,05 équiv.)Oxydant
AcOH/DMSO (1 : 1)TA, 4 h
S
B1, 2 équiv. C1B1-C1
aConditions : B1 (2 mmol), C1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (1 ou 2 équiv.) ou BQ (0,5
mmol)/Cu(OAc)2 (0,1 mmol)/O2 (1 atm), AcOH (2 mL), DMSO (2 mL), TA, 4 h.
Figure C2. Suivis des couplages de B1 avec C1 avec 2 équiv., 1 équiv. de BQ ou une combinaison BQ
/CuOAc)2/O2.a
Les vitesses initiales ne dépendent pas de la quantité de benzoquinone. Par conséquent, BQ
n’a pas d’influence sur l’étape lente, et donc sur l’étape d’activation de la liaison C-H. En revanche,
nous remarquons que le plateau est plus rapidement atteint en présence d’une quantité plus faible
de benzoquinone, ce qui démontre une influence de celle-ci sur les intermédiaires réactionnels. Par
ailleurs, l’emploi de quantités catalytiques de benzoquinone et de CuII réduit fortement le rendement
de B1-C1.
0
10
20
30
40
50
60
70
80
0 100 200
BQ (2 équiv.)
BQ (1 équiv.)
BQ (0,5 équiv.), Cu(OAc)2
(0,1 équiv.), O2 (1 atm.)
temps (minutes)
ren
de
me
nt
(%)
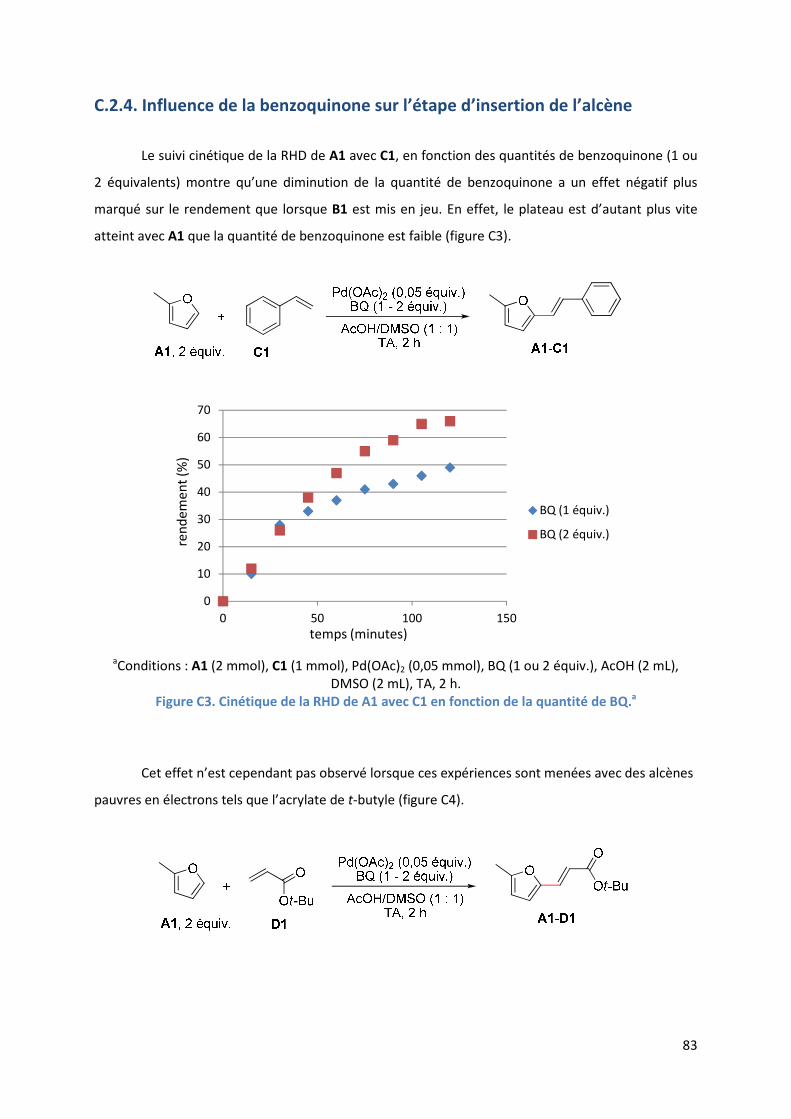
83
C.2.4. Influence de la benzoquinone sur l’étape d’insertion de l’alcène
Le suivi cinétique de la RHD de A1 avec C1, en fonction des quantités de benzoquinone (1 ou
2 équivalents) montre qu’une diminution de la quantité de benzoquinone a un effet négatif plus
marqué sur le rendement que lorsque B1 est mis en jeu. En effet, le plateau est d’autant plus vite
atteint avec A1 que la quantité de benzoquinone est faible (figure C3).
aConditions : A1 (2 mmol), C1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (1 ou 2 équiv.), AcOH (2 mL),
DMSO (2 mL), TA, 2 h.
Figure C3. Cinétique de la RHD de A1 avec C1 en fonction de la quantité de BQ.a
Cet effet n’est cependant pas observé lorsque ces expériences sont menées avec des alcènes
pauvres en électrons tels que l’acrylate de t-butyle (figure C4).
0
10
20
30
40
50
60
70
0 50 100 150
BQ (1 équiv.)
BQ (2 équiv.)
temps (minutes)
ren
de
me
nt
(%)
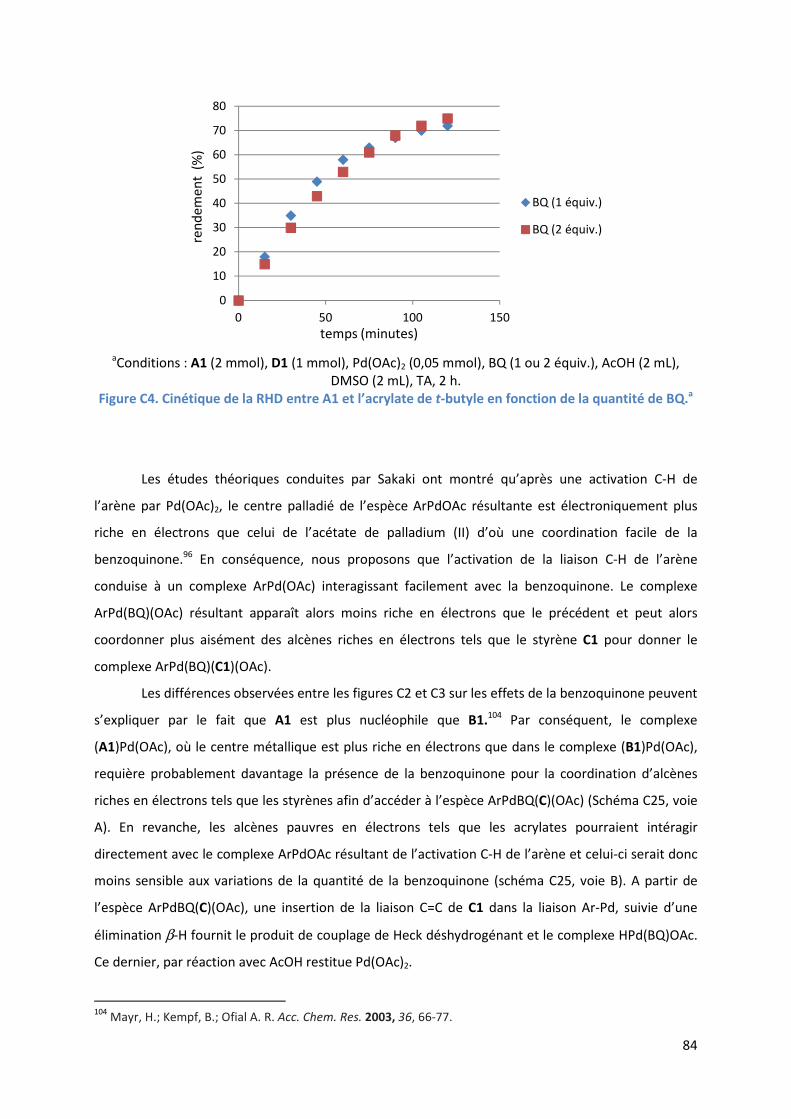
84
aConditions : A1 (2 mmol), D1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (1 ou 2 équiv.), AcOH (2 mL),
DMSO (2 mL), TA, 2 h.
Figure C4. Cinétique de la RHD entre A1 et l’acrylate de t-butyle en fonction de la quantité de BQ.a
Les études théoriques conduites par Sakaki ont montré qu’après une activation C-H de
l’arène par Pd(OAc)2, le centre palladié de l’espèce ArPdOAc résultante est électroniquement plus
riche en électrons que celui de l’acétate de palladium (II) d’où une coordination facile de la
benzoquinone.96
En conséquence, nous proposons que l’activation de la liaison C-H de l’arène
conduise à un complexe ArPd(OAc) interagissant facilement avec la benzoquinone. Le complexe
ArPd(BQ)(OAc) résultant apparaît alors moins riche en électrons que le précédent et peut alors
coordonner plus aisément des alcènes riches en électrons tels que le styrène C1 pour donner le
complexe ArPd(BQ)(C1)(OAc).
Les différences observées entre les figures C2 et C3 sur les effets de la benzoquinone peuvent
s’expliquer par le fait que A1 est plus nucléophile que B1.104
Par conséquent, le complexe
(A1)Pd(OAc), où le centre métallique est plus riche en électrons que dans le complexe (B1)Pd(OAc),
requière probablement davantage la présence de la benzoquinone pour la coordination d’alcènes
riches en électrons tels que les styrènes afin d’accéder à l’espèce ArPdBQ(C)(OAc) (Schéma C25, voie
A). En revanche, les alcènes pauvres en électrons tels que les acrylates pourraient intéragir
directement avec le complexe ArPdOAc résultant de l’activation C-H de l’arène et celui-ci serait donc
moins sensible aux variations de la quantité de la benzoquinone (schéma C25, voie B). A partir de
l’espèce ArPdBQ(C)(OAc), une insertion de la liaison C=C de C1 dans la liaison Ar-Pd, suivie d’une
élimination β-H fournit le produit de couplage de Heck déshydrogénant et le complexe HPd(BQ)OAc.
Ce dernier, par réaction avec AcOH restitue Pd(OAc)2.
104
Mayr, H.; Kempf, B.; Ofial A. R. Acc. Chem. Res. 2003, 36, 66-77.
0
10
20
30
40
50
60
70
80
0 50 100 150
BQ (1 équiv.)
BQ (2 équiv.)
temps (minutes)
ren
de
me
nt
(%
)
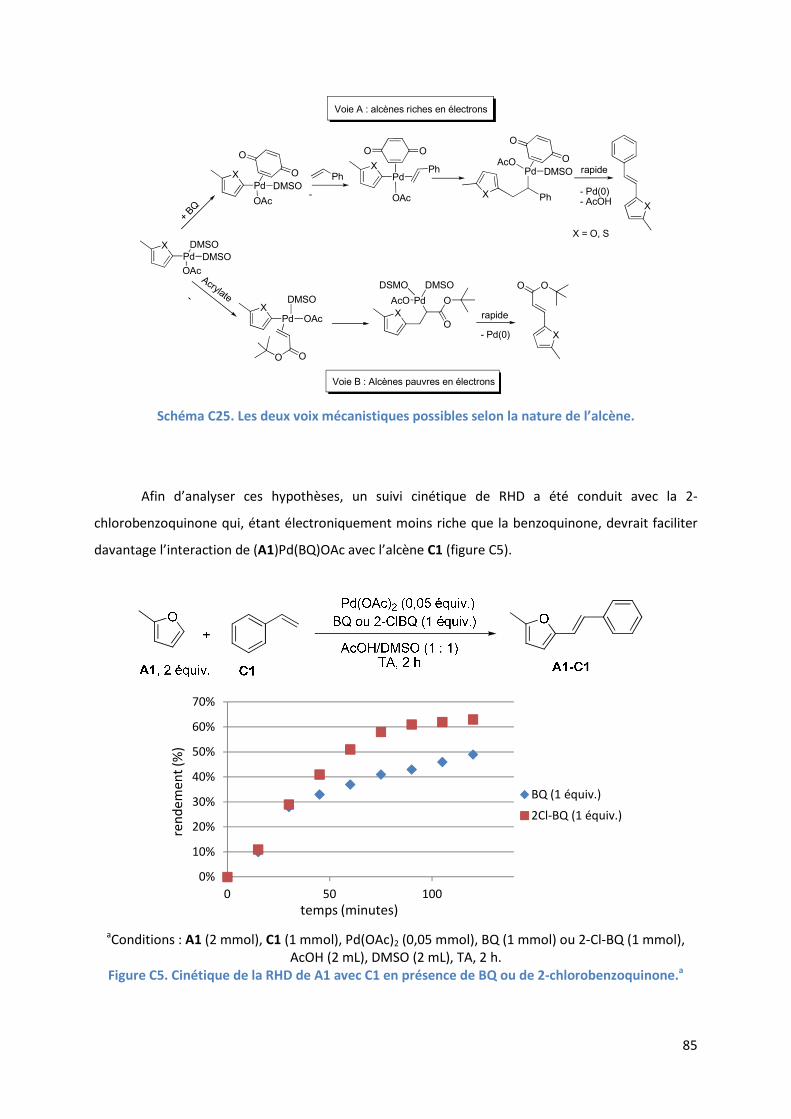
85
XPd
DMSO
DMSO
OAc
+BQ
XPd DMSO
O
O
-
Pd DMSO
O
O
X
- Pd(0)- AcOH
rapide
PhXOAc
AcOXPd
OAc
O O
PhPh
Voie A : alcènes riches en électrons
X = O, S
XPd
DMSO
OAc
X- Pd(0)
rapide
Acrylate-O O
OO
XO
OPdAcO
DSMO DMSO
Voie B : Alcènes pauvres en électrons
Schéma C25. Les deux voix mécanistiques possibles selon la nature de l’alcène.
Afin d’analyser ces hypothèses, un suivi cinétique de RHD a été conduit avec la 2-
chlorobenzoquinone qui, étant électroniquement moins riche que la benzoquinone, devrait faciliter
davantage l’interaction de (A1)Pd(BQ)OAc avec l’alcène C1 (figure C5).
aConditions : A1 (2 mmol), C1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (1 mmol) ou 2-Cl-BQ (1 mmol),
AcOH (2 mL), DMSO (2 mL), TA, 2 h.
Figure C5. Cinétique de la RHD de A1 avec C1 en présence de BQ ou de 2-chlorobenzoquinone.a
0%
10%
20%
30%
40%
50%
60%
70%
0 50 100
BQ (1 équiv.)
2Cl-BQ (1 équiv.)
temps (minutes)
ren
de
me
nt
(%)

86
Les résultats obtenus confirment l’hypothèse. En effet, la réaction de A1 avec C1 est plus
efficace en présence de 2-chlorobenzoquinone qu’en présence de benzoquinone. Les quinones
facilitent donc la complexation d’alcènes riches en électrons au centre palladié.
La possibilité pour la benzoquinone ligandée au palladium de s’insérer dans la liaison Ar-Pd a
été écartée. En effet, dans des réactions de Heck classiques mettant en jeu la benzoquinone, C1, le 4-
iodobenzoate de méthyle dans le THF en présence de Pd(OAc)2, PPh3 et de Et3N, seul le (E)-4-
styrylbenzoate de méthyle est observé, montrant que C1 est plus sujet que la benzoquinone a
réaliser une réaction d’insertion.
C.2.5. Cycle catalytique
La période d’induction observée dans le mélange Et2O/AcOH a été attribuée à la
complexation du furane A à l’espèce catalytique trimérique inactive menant aux espèces palladiées
monomériques et dimériques actives (schéma C26). Par contre, aucune période d’induction n’a été
observée lorsque le mélange DMSO/AcOH est utilisé comme solvant. Ceci s’explique par le fait que le
DMSO peut agir comme ligand du palladium pour fournir le complexe monomérique
Pd(OAc)2(DMSO)2.105
Schéma C26. Mécanisme de la formation des espèces catalytiques monomèriques et dimériques
actives à partir de l’acétate de palladium trimérique.
Les résultats obtenus et ces observations permettent de proposer le cycle catalytique du
schéma C27 pour les réactions conduites dans le mélange AcOH/DMSO. L’activation d’une liaison C-H
de l’arène par L2Pd(OAc)2 forme l’intermédiaire III. Après complexation de la benzoquinone et
coordination de l’alcène à cet intermédiaire, l’insertion de l’alcène dans la liaison Ar-Pd mène à
l’intermédiaire VI qui subit l’élimination β-H pour fournir le produit de couplage et l’hydrure de
palladium VII. Ce dernier, par réaction avec AcOH restitue l’espèce catalytique active.
105
(a) Zierkiewicz, W. Privalov T. Organometallics 2005, 24, 6019-6028. (b) Stash, A. I.; Perepelkova, T. I.;
Kravtsova, S. V.; Noskov, Yu. G.; P. Romm, I. Russ. J. Coord. Chem. 1998, 24, 36–39.

87
Schéma C27. Cycle catalytique proposé.
C.3. Conclusion
Il était connu que la benzoquinone, en sus de son rôle d’oxydant, pouvait avoir, un rôle de
promoteur de l’activation de la liaison C-H, de l’élimination réductrice, de l’oxydation de l’hydrure de
palladium, de la transmetallation oxydante, de la migration de ligand, ou encore un rôle d’inhibiteur
de l’élimination β-H. Nous avons montré que la benzoquinone pouvait favoriser l’étape d’insertion de
l’alcène dans la liaison Ar-Pd. En revanche, la benzoquinone n’a pas d’influence sur l’étape
d’activation de la liaison C-H de l’arène dans nos conditions réactionnelles.

88
C.4. Partie expérimentale
C.4.1. Préparation du 2-méthylthiophène-5d
Le 2-méthylthiophène-5d a été préparé selon une méthode standard. Dans un schlenk mis
sous argon, le 2-méthylthiophène est mis en solution dans le THF (20 mL). Le n-butyllithium en
solution dans l’hexane (1,4 équiv.) est ensuite ajouté au milieu réactionnel placé à 0 °C. Après 2 h
d’agitation à température ambiante, l’eau deutérée (1,4 équiv.) est additionnée au mélange
réactionnel remis à 0 °C, lequel est ensuite laissé sous agitation à température ambiante pendant 10
min. Après ajout de Et2O (20 mL) et un traitement avec une solution aqueuse saturée de NH4Cl (20
mL), la phase organique est extraite et séchée sur MgSO4. Le 2-méthylthiophène-5d est ensuite
obtenu par évaporation sous vide (pression de 160 mbar et bain à 40 °C).
C.4.2. Mode opératoire pour le suivi cinétique des réactions
L’acétate de palladium (11,2 mg, 0,05 mmol), la benzoquinone (54 - 216 mg, 0,5 - 2 mmol) ou
la 2-chlorobenzoquinone (142,5 mg , 1 mmol), et l’acétate de cuivre (II) (0 – 18,2 mg, 0 – 0,1 mmol)
sont mis en solution dans AcOH (2 mL) et DMSO (2 mL). Après ajout de l’arène (2 mmol), de l’alcène
(1 mmol) et du nitrobenzène (103 µL, 1 mmol), le mélange est agité à température ambiante. Des
échantillons sont prélevés toutes les 15 min (environ 0,2 mL) pendant 2 h ou 4 h, filtrés avec Et2O sur
silice et analysés par CPG. Lorsque la réaction est effectuée oxygène, un ballon rempli de O2 est
adapté au dispositif réactionnel.

89
Chapitre 2 :
Etude SM-IES de la RHD de furanes avec
des acrylates en utilisant la benzoquinone
comme oxydant et le DMSO comme
solvant

90
Introduction
La spectrométrie de masse est une méthode analytique qui permet l'étude d'objets isolés en
phase gazeuse. Elle peut être appliquée à tous les domaines scientifiques qui relèvent de l’étude de
la matière dès lors que celle-ci peut être, d’une part volatilisée et, d’autre part ionisée. Ces domaines
s'étendent de la chimie et la biochimie à l'analyse de particules cosmiques, en passant par la
protéomique, l’environnement, l’archéologie ...
Au cours des deux dernières décennies, la spectrométrie de masse a connu un
développement très important, dû essentiellement à l’amélioration des techniques d’ionisation à
pression atmosphérique (IPA), en particulier l’ionisation par électrospray (IES), qui permet l’étude
d’espèces ioniques en solution. Ce mode d’ionisation a ouvert un accès direct à l’investigation de
réactions chimiques en solution. En principe, l’IES rend possible la détection et l’étude, non
seulement des réactifs et des produits, mais aussi, des intermédiaires réactionnels de courte durée
de vie, ce qui apporte de nouvelles connaissances sur les mécanismes.
L’IES et sa version tandem SM/SM-IES, sont rapidement devenues des techniques de choix
pour l’étude mécanistique de réactions en solution.
Dans un premier temps, ce chapitre sera consacré à la présentation de généralités sur la
technique et dans un deuxième temps à l’étude par spectrométrie de masse - ionisation par
électrospray (SM-IES) de la Réaction de Heck Déshydrogénante de furanes avec des acrylates selon
les conditions développées par notre équipe au chapitre 1.

91
Partie D : Introduction à l’électrospray couplé masse106, 107
Introduction
La spectrométrie de masse est une technique d'analyse, dont les fondements ont été posés à
la fin du XIXème siècle. Elle consiste à séparer des molécules, préalablement ionisées et mises dans
leur état gazeux, en fonction de leur masse et de leur charge. Les premières analyses de
spectrométrie de masse ont été décrites en 1912 par Joseph J. Thompson. Cette technique a
longtemps été employée sur des petites et moyennes molécules, et reste d’actualité, car elle tire
avantage de sa grande sensibilité.
Malgré les années de développement intensif de la spectrométrie de masse, l'analyse des
macromolécules est restée difficile pendant plus de 70 ans car ces composés sont facilement
dégradés par l’ionisation. A la fin des années 80, deux méthodes décrites par J. Fenn (IES : ionisation
electrospray) et par K. Tanaka (DILAM : desorption/ionisation laser assistée par matrice),
récompensées par le prix Nobel de chimie 2002, permettent d'éviter la dégradation des
macromolécules au cours de la phase d'ionisation. L’ionisation électrospray (IES) est une technique
douce d’ionisation, à pression atmosphérique, qui permet le transfert des ions d’une solution vers
une phase gazeuse.
D.1. Schéma général du spectromètre de masse Q-TdV
L’appareil utilisé par notre laboratoire est schématisé ci-après (schéma D1). Il est constitué
de deux analyseurs : un quadripolaire et un à temps de vol (TdV). Le spray est formé à pression
atmosphérique, puis la pression diminue au fur et à mesure que les ions parcourent les différents
compartiments du spectromètre. Une pompe à palette et deux pompes turbo permettent d’obtenir
le vide. Les différentes étapes, de la formation du spray à la détection, sont détaillées par la suite.
106
de Hoffmann, E.; Charete J.; Stroobant, V. Spectrométrie de masse 2e Ed., Dunod: Paris, 1999. 107
Gross, J. H. Mass Spectrometry: A Textbook, Springer-Verlag: Berlin Heidelberg, 2004.
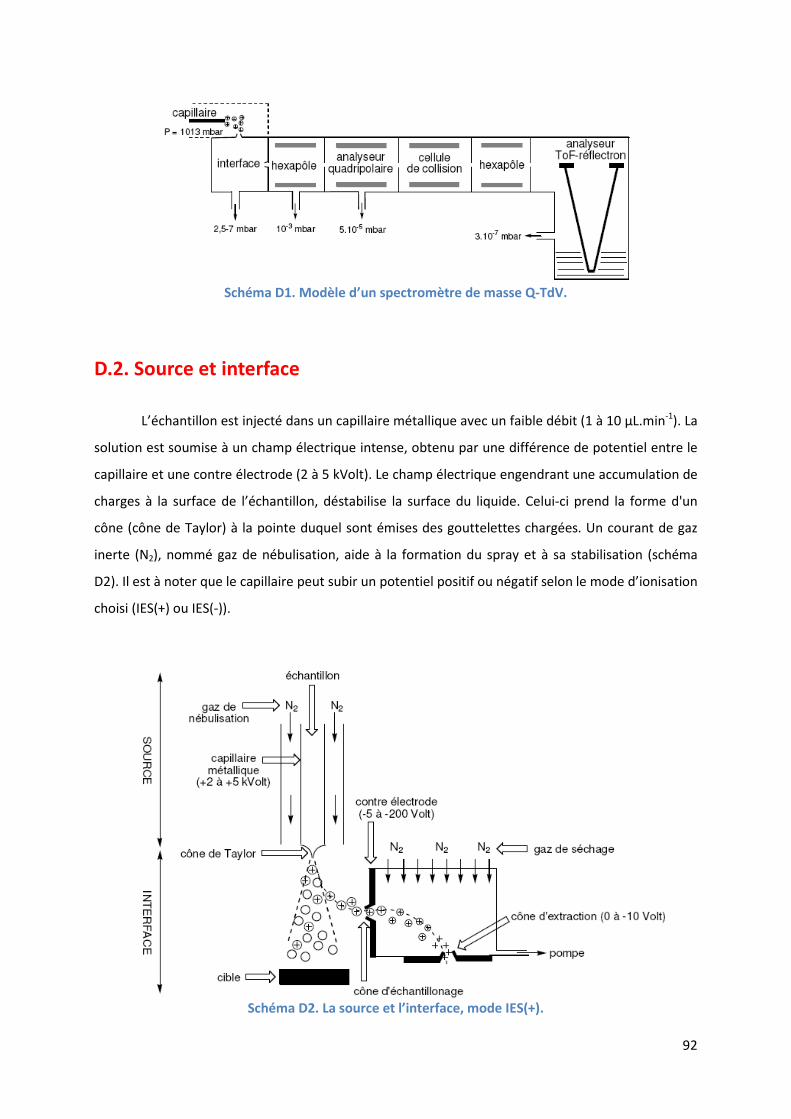
92
Schéma D1. Modèle d’un spectromètre de masse Q-TdV.
D.2. Source et interface
L’échantillon est injecté dans un capillaire métallique avec un faible débit (1 à 10 μL.min-1
). La
solution est soumise à un champ électrique intense, obtenu par une différence de potentiel entre le
capillaire et une contre électrode (2 à 5 kVolt). Le champ électrique engendrant une accumulation de
charges à la surface de l’échantillon, déstabilise la surface du liquide. Celui-ci prend la forme d'un
cône (cône de Taylor) à la pointe duquel sont émises des gouttelettes chargées. Un courant de gaz
inerte (N2), nommé gaz de nébulisation, aide à la formation du spray et à sa stabilisation (schéma
D2). Il est à noter que le capillaire peut subir un potentiel positif ou négatif selon le mode d’ionisation
choisi (IES(+) ou IES(-)).
Schéma D2. La source et l’interface, mode IES(+).

93
Les gouttelettes chargées sont attirées par le cône d’échantillonnage (tension accélératrice, 5
à 200 Volt), alors que les neutres sont récupérés sur une cible. Les premières subissent alors un flux
de gaz de séchage (N2) afin d’évaporer le solvant et d’obtenir des ions en phase gazeuse (schéma D2).
Il est déjà possible, dans l’interface, de fragmenter les ions en variant la tension d’accélération et la
pression du gaz de séchage.
Deux modèles ont été proposés pour former un ion en phase gazeuse à partir d’une
gouttelette d’électrolyte. Le premier, le modèle de la charge résiduelle, a été proposé par Dol en
1968. L’évaporation du solvant entraîne une tension de charge trop importante dans la gouttelette
qui éclate, jusqu’à l’obtention d’un ion solvaté qui passera en phase gazeuse après l’évaporation des
dernières molécules de solvant (Schéma D3).
Schéma D3. Le modèle de la charge résiduelle.
Le modèle de l’évaporation des ions a été proposé en 1976 par Thomson et Iribarne.108
Ils
proposent que lorsque les gouttelettes atteignent une taille de l’ordre de 10 nm, des ions sont
expulsés, se retrouvant en phase gazeuse.
Lors du séchage des gouttelettes d’électrolyte, les ions peuvent entrer en collision ce qui
provoque une dispersion angulaire. Les ions arrivent donc dans le spectromètre de masse par un
hexapôle, qui permet de refocaliser le faisceau d’ions et ainsi d’augmenter la sensibilité de l’appareil.
D.3. L’analyseur quadripolaire
Le faisceau d’ions arrive alors dans le premier analyseur : le quadripôle, constitué de quatre
électrodes cylindriques, de section hyperbolique ou circulaire, parfaitement parallèles (schéma D4).
108
Iribarne, J.V.; Thomson, B. A. J. Chem. Phys. 1976, 64, 2287–2294.

94
Les électrodes opposées, distantes de 2r0, sont soumises au même potentiel, alors que les électrodes
adjacentes sont portées à des potentiels de même valeur, mais opposés.
Schéma D4. L’analyseur quadripolaire.
Les ions, soumis à un champ quadripolaire, suivent alors une trajectoire selon l’axe z. Le
champ résulte de l’application de potentiels Φ0 sur les électrodes tel que : Φ0 = U – V cosωt et – Φ0 =
– (U – V cosωt), où ω est la fréquence angulaire, U le potentiel pouvant aller de 500 à 2000 volts, et V
l’amplitude du voltage de la radio fréquence compris entre 0 et 3000 volts.
Pour être détecté, un ion doit avoir une trajectoire stable selon l’axe z, c’est-à-dire que ses
coordonnées en fonction de x et y doivent être strictement inférieures à r0, sinon il se déchargera
lors de sa collision avec les parois du quadripôle et ne pourra pas être analysé. Il est possible de
définir, en fonction des valeurs U et V, une zone de stabilité pour des ions de même masse (schéma
D5).
Schéma D5. Diagramme de stabilité pour différentes masses en fonction de U et V.

95
La droite de fonctionnement de l'analyseur, définie par le constructeur, correspond au
potentiel U en fonction de l’amplitude du voltage de la radio fréquence V tel que U/V = constante.
Un balayage de U et V, suivant cette droite, permet l'observation successive de tous les ions dont la
zone de stabilité est coupée par la droite de fonctionnement. La résolution entre ces ions est
d'autant plus grande que la pente de la droite est élevée.
Il est à noter que l’analyseur quadripolaire nécessite une source d’ions continue pour réaliser
le balayage, et que la perte d’ions (trajectoire instable) entraîne une perte de sensibilité. Cet
analyseur a une résolution unitaire et permet donc de travailler uniquement en basse résolution.
D.4. La cellule de collision
Le spectromètre de masse est ensuite équipé d’une cellule de collision qui permet de
fragmenter les ions. Pour ce faire, un gaz (He) est envoyé dans la cellule et entre en collision avec les
ions provoquant leur fragmentation. Il est possible de choisir la pression du gaz injecté dans la cellule
qui correspondra à une énergie définie exprimée en eV DIC (dissociation induite par collission). Cette
cellule servira en analyse SM/SM-IES
D.5. L’analyseur à Temps de Vol
Un deuxième hexapôle suit la cellule de collision afin de refocaliser le faisceau d’ions avant
l’entrée dans l’analyseur à Temps de Vol (TdV pour tube à temps de vol). Il existe deux types
d’appareil : le TdV mode linéaire, et le mode réflectron dont notre spectromètre est équipé.
Dans un premier temps, les ions sont accélérés par un champ électrique (tension
accélératrice, V = 10-25 kV), et acquièrent ainsi la même énergie cinétique (EC = z.e.V). Les ions sont
ensuite envoyés au même moment vers le tube de vol, au moyen de deux électrodes (pusher et
puller) auxquelles un potentiel sera appliqué à un instant donné (schéma D6).

96
Schéma D6. Accélération et expulsion des ions en fonction du temps.
En mode linéaire, le tube de vol est libre de tout champ et la séparation des ions ne va
dépendre que de leur vitesse, induite par l’énergie cinétique acquise lors de la phase d’accélération.
Comme EC = ½ m.v2 et que EC est la même pour tous les ions, leur vitesse dans le tube de vol va être
inversement proportionnelle à leur masse (schéma D7).
Schéma D7. Analyseur Temps de Vol mode linéaire.
La résolution de l’analyseur TdV permet de distinguer deux ions de masse proche. La
résolution est d’autant élevée que tous les ions, de rapport m/z égal, arrivent dans le laps de temps
∆t le plus court au niveau du détecteur. Expérimentalement, les ions de même rapport m/z vont
atteindre celui-ci avec une dispersion dans les temps d’arrivée, due notamment à une incertitude sur
l’énergie initiale des ions. Le TdV en mode linéaire ne corrige pas les écarts d’énergie (schéma D8) et
ne permet pas de travailler en haute résolution.
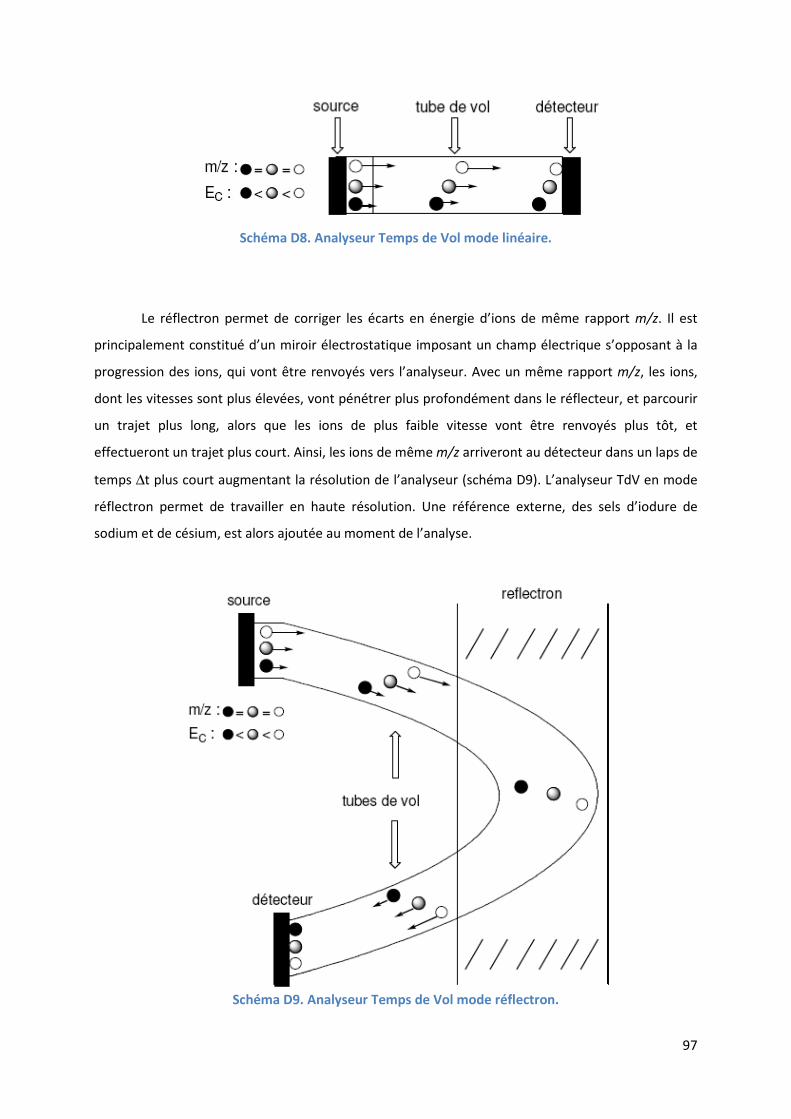
97
Schéma D8. Analyseur Temps de Vol mode linéaire.
Le réflectron permet de corriger les écarts en énergie d’ions de même rapport m/z. Il est
principalement constitué d’un miroir électrostatique imposant un champ électrique s’opposant à la
progression des ions, qui vont être renvoyés vers l’analyseur. Avec un même rapport m/z, les ions,
dont les vitesses sont plus élevées, vont pénétrer plus profondément dans le réflecteur, et parcourir
un trajet plus long, alors que les ions de plus faible vitesse vont être renvoyés plus tôt, et
effectueront un trajet plus court. Ainsi, les ions de même m/z arriveront au détecteur dans un laps de
temps ∆t plus court augmentant la résolution de l’analyseur (schéma D9). L’analyseur TdV en mode
réflectron permet de travailler en haute résolution. Une référence externe, des sels d’iodure de
sodium et de césium, est alors ajoutée au moment de l’analyse.
Schéma D9. Analyseur Temps de Vol mode réflectron.

98
D.6. L’analyse SM/SM-IES
Pour réaliser des analyses SM/SM-IES, il est nécessaire d’utiliser un spectromètre de masse
possédant deux analyseurs. L’analyseur quadripolaire va permettre le choix des ions d’un rapport
m/z défini. Ces ions seront fragmentés dans la cellule de collision puis les fragments seront analysés
par l’analyseur TdV. La sélection d’une seule masse au niveau de l’analyseur quadripolaire ne permet
pas l’ajout de la référence et les spectres SM/SM-IES sont donc réalisés en basse résolution.
D.7. Conclusion
Les développements récents de la technique de spectrométrie de masse, et en particulier du
mode d’ionisation électrospray, ont ouvert l’accès à l’étude directe des réactions chimiques en
solution.
Nous allons voir que la technique permet l’identification d’intermédiaires réactionnels y
compris ceux de courte durée de vie. En fonction des différentes versions de spectromètres, on peut
accéder aux informations telles que la SM/SM et la SMHR.
La technique permet des études qualitatives et quantitatives. A titre d’exemple,
l’appréciation du ratio des abondances d’intermédiaires dans un mélange de catalyseurs reflète dans
certains cas la stéréosélectivité intrinsèque des catalyseurs.
La spectrométrie de masse en mode d’ionisation électrospray et la version tandem SM/SM-
IES sont donc rapidement devenues des techniques de choix pour l’étude mécanistique des réactions
chimiques en milieu liquide.

99
Partie E : Etude SM-IES de la RHD de furanes et d’acrylates
en utilisant la benzoquinone comme oxydant et le DMSO
comme solvant
Introduction
La spectroscopie de masse est basée sur la séparation des espèces ioniques en fonction de
leur masse mono-isotopique sur leur charge (m/z). L’identification d’espèces détectées est réalisée
par comparaison des modèles de distribution des isotopes calculés et observés. Le palladium possède
six isotopes, les ions contenant cet atome seront détectés en masse sous forme d’amas isotopiques
centrés sur l’isotope le plus abondant (106) (schéma E1).
Schéma E1. Amas isotopiques des ions contenant du palladium.
E.1. Bibliographie
Différents travaux relatent l’utilisation de la technique SM/SM-IES pour identifier des
intermédiaires des réactions pallado-catalysées et proposer des mécanismes réactionnels. En 2004,
l’équipe d’Eberlin a étudié, par analyse SM-IES, le couplage du tétrafluoroborate de 4-
méthoxybenzènediazonium avec le 2,3-dihydrofurane (schéma E2). Les auteurs ont proposé un
équilibre entre les espèces [p-MeOC6H4Pd(MeCN)3]+, [p-MeOC6H4Pd(MeCN)2]
+, [p-
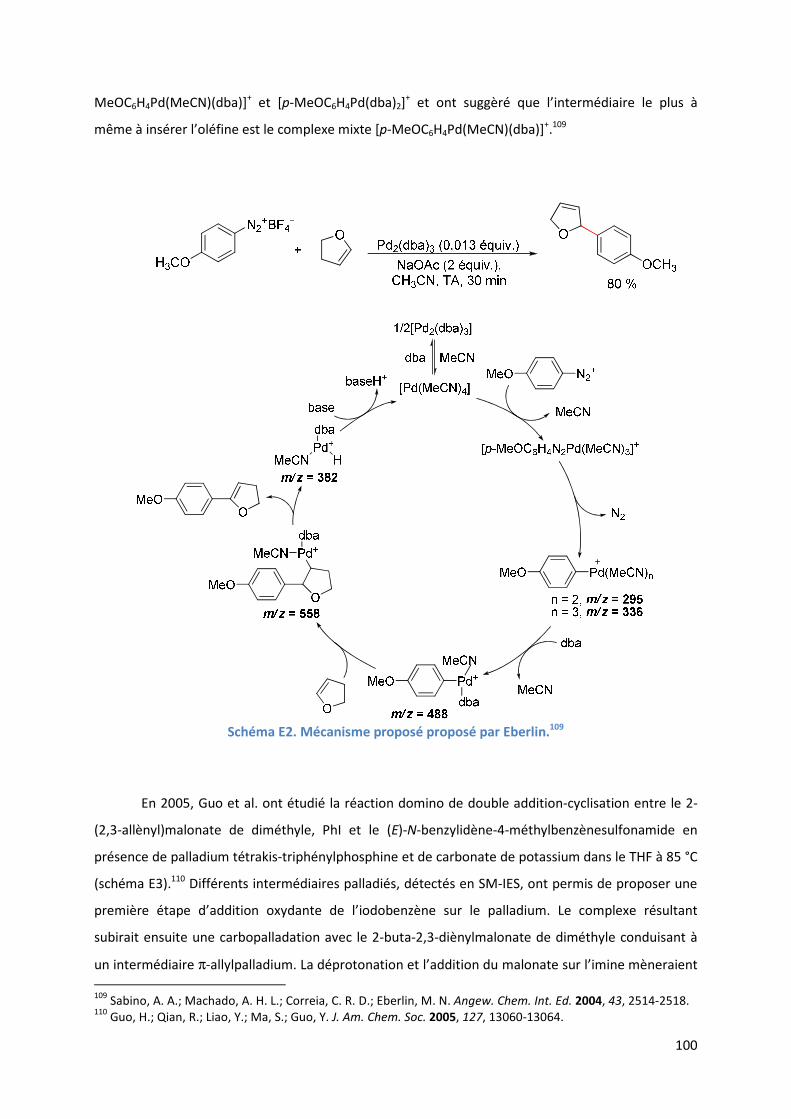
100
MeOC6H4Pd(MeCN)(dba)]+ et [p-MeOC6H4Pd(dba)2]
+ et ont suggèré que l’intermédiaire le plus à
même à insérer l’oléfine est le complexe mixte [p-MeOC6H4Pd(MeCN)(dba)]+.109
Schéma E2. Mécanisme proposé proposé par Eberlin.
109
En 2005, Guo et al. ont étudié la réaction domino de double addition-cyclisation entre le 2-
(2,3-allènyl)malonate de diméthyle, PhI et le (E)-N-benzylidène-4-méthylbenzènesulfonamide en
présence de palladium tétrakis-triphénylphosphine et de carbonate de potassium dans le THF à 85 °C
(schéma E3).110
Différents intermédiaires palladiés, détectés en SM-IES, ont permis de proposer une
première étape d’addition oxydante de l’iodobenzène sur le palladium. Le complexe résultant
subirait ensuite une carbopalladation avec le 2-buta-2,3-diènylmalonate de diméthyle conduisant à
un intermédiaire π-allylpalladium. La déprotonation et l’addition du malonate sur l’imine mèneraient
109
Sabino, A. A.; Machado, A. H. L.; Correia, C. R. D.; Eberlin, M. N. Angew. Chem. Int. Ed. 2004, 43, 2514-2518. 110
Guo, H.; Qian, R.; Liao, Y.; Ma, S.; Guo, Y. J. Am. Chem. Soc. 2005, 127, 13060-13064.
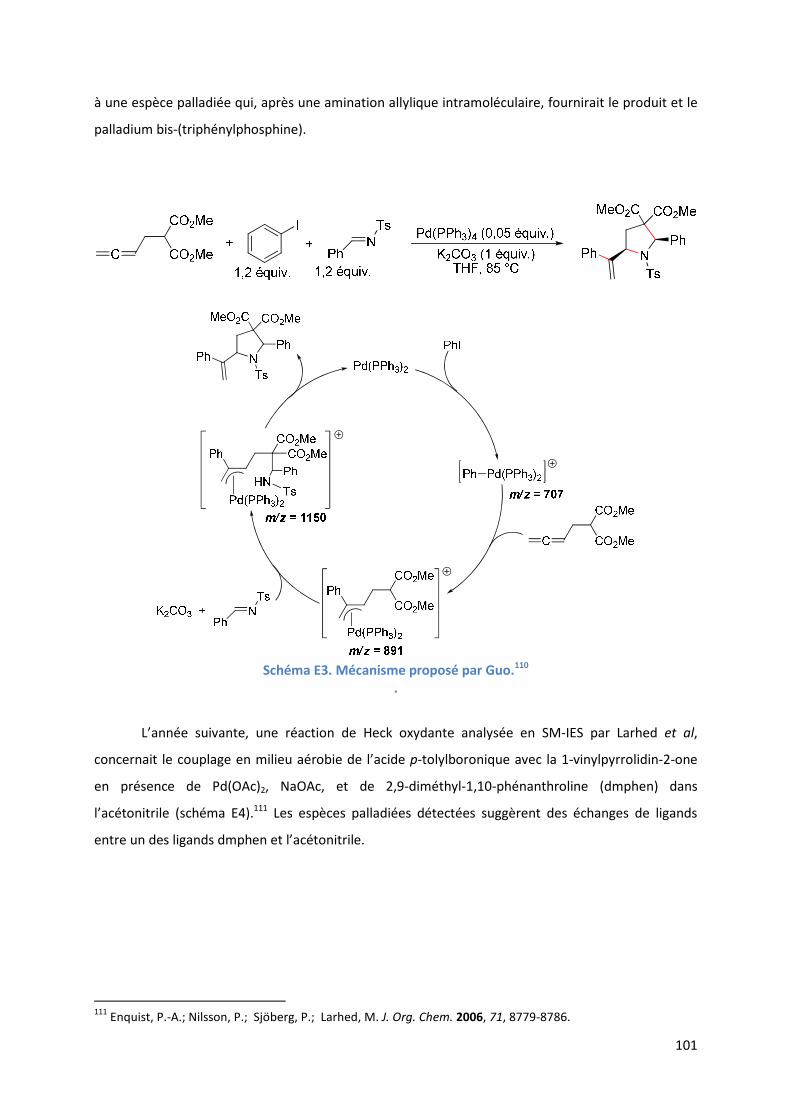
101
à une espèce palladiée qui, après une amination allylique intramoléculaire, fournirait le produit et le
palladium bis-(triphénylphosphine).
Schéma E3. Mécanisme proposé par Guo.
110
.
L’année suivante, une réaction de Heck oxydante analysée en SM-IES par Larhed et al,
concernait le couplage en milieu aérobie de l’acide p-tolylboronique avec la 1-vinylpyrrolidin-2-one
en présence de Pd(OAc)2, NaOAc, et de 2,9-diméthyl-1,10-phénanthroline (dmphen) dans
l’acétonitrile (schéma E4).111
Les espèces palladiées détectées suggèrent des échanges de ligands
entre un des ligands dmphen et l’acétonitrile.
111
Enquist, P.-A.; Nilsson, P.; Sjöberg, P.; Larhed, M. J. Org. Chem. 2006, 71, 8779-8786.
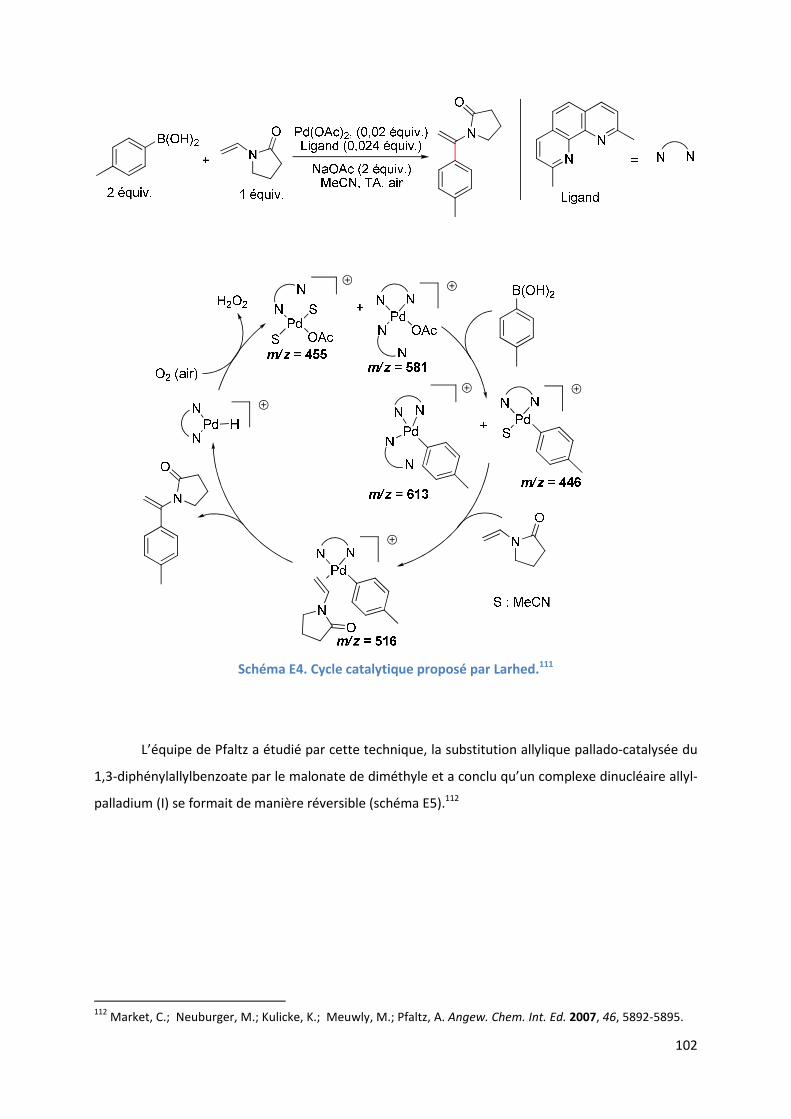
102
Schéma E4. Cycle catalytique proposé par Larhed.111
L’équipe de Pfaltz a étudié par cette technique, la substitution allylique pallado-catalysée du
1,3-diphénylallylbenzoate par le malonate de diméthyle et a conclu qu’un complexe dinucléaire allyl-
palladium (I) se formait de manière réversible (schéma E5).112
112
Market, C.; Neuburger, M.; Kulicke, K.; Meuwly, M.; Pfaltz, A. Angew. Chem. Int. Ed. 2007, 46, 5892-5895.
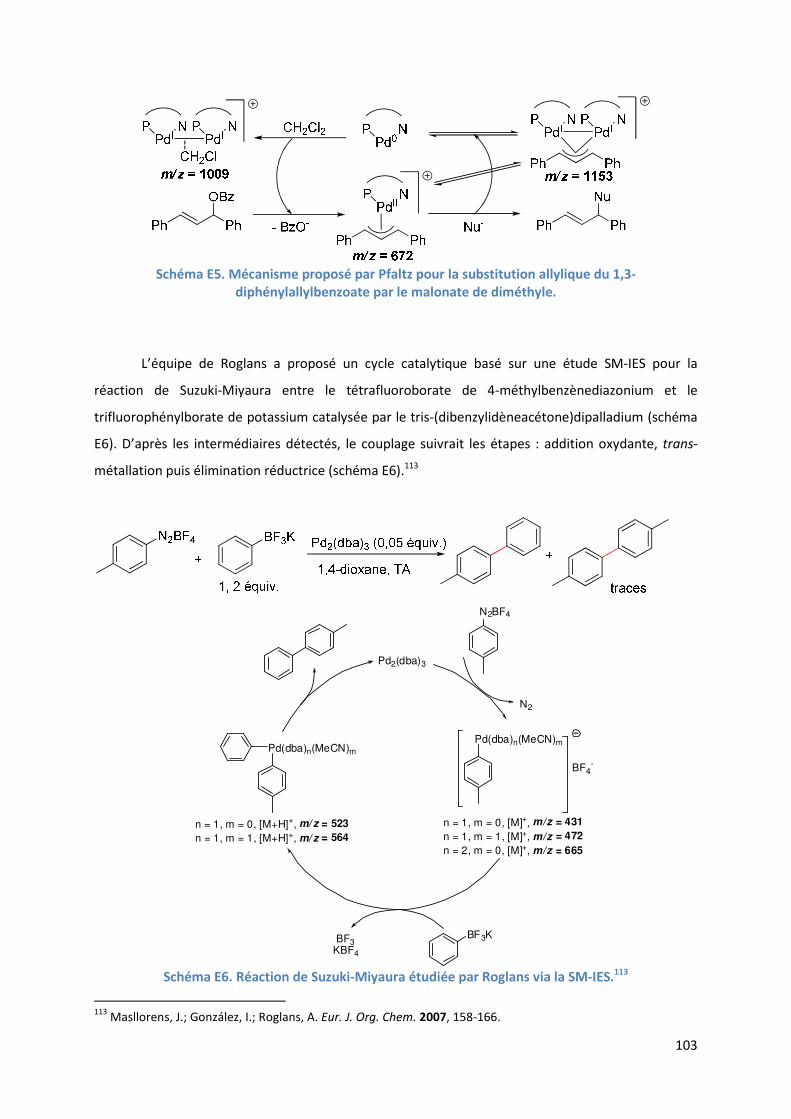
103
Schéma E5. Mécanisme proposé par Pfaltz pour la substitution allylique du 1,3-
diphénylallylbenzoate par le malonate de diméthyle.
L’équipe de Roglans a proposé un cycle catalytique basé sur une étude SM-IES pour la
réaction de Suzuki-Miyaura entre le tétrafluoroborate de 4-méthylbenzènediazonium et le
trifluorophénylborate de potassium catalysée par le tris-(dibenzylidèneacétone)dipalladium (schéma
E6). D’après les intermédiaires détectés, le couplage suivrait les étapes : addition oxydante, trans-
métallation puis élimination réductrice (schéma E6).113
Schéma E6. Réaction de Suzuki-Miyaura étudiée par Roglans via la SM-IES.113
113
Masllorens, J.; González, I.; Roglans, A. Eur. J. Org. Chem. 2007, 158-166.
Pd2(dba)3
N2BF4
N2
Pd(dba)n(MeCN)m
BF4-
BF3KBF3KBF4
Pd(dba)n(MeCN)m
n = 1, m = 0, [M+H]+, m/ z = 523
n = 1, m = 1, [M+H]+, m/ z = 564
n = 1, m = 0, [M]+, m/z = 431
n = 1, m = 1, [M]+, m/z = 472
n = 2, m = 0, [M]+, m/z = 665
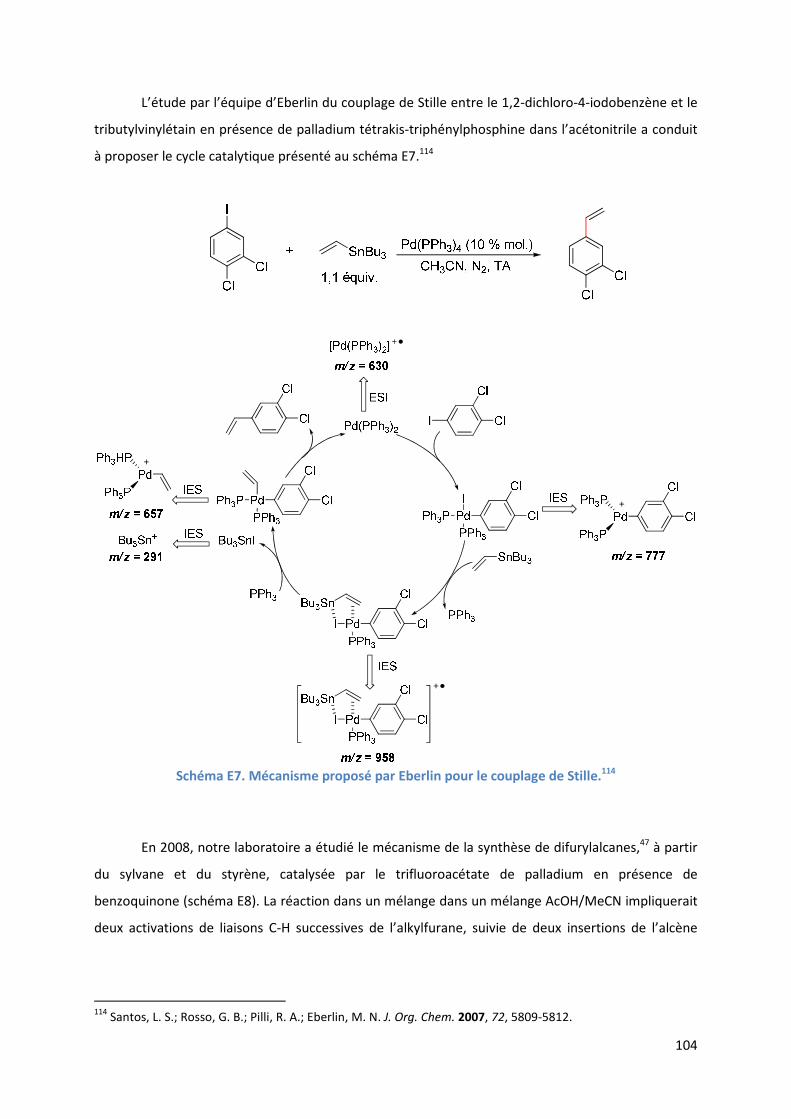
104
L’étude par l’équipe d’Eberlin du couplage de Stille entre le 1,2-dichloro-4-iodobenzène et le
tributylvinylétain en présence de palladium tétrakis-triphénylphosphine dans l’acétonitrile a conduit
à proposer le cycle catalytique présenté au schéma E7.114
Schéma E7. Mécanisme proposé par Eberlin pour le couplage de Stille.114
En 2008, notre laboratoire a étudié le mécanisme de la synthèse de difurylalcanes,47
à partir
du sylvane et du styrène, catalysée par le trifluoroacétate de palladium en présence de
benzoquinone (schéma E8). La réaction dans un mélange dans un mélange AcOH/MeCN impliquerait
deux activations de liaisons C-H successives de l’alkylfurane, suivie de deux insertions de l’alcène
114
Santos, L. S.; Rosso, G. B.; Pilli, R. A.; Eberlin, M. N. J. Org. Chem. 2007, 72, 5809-5812.
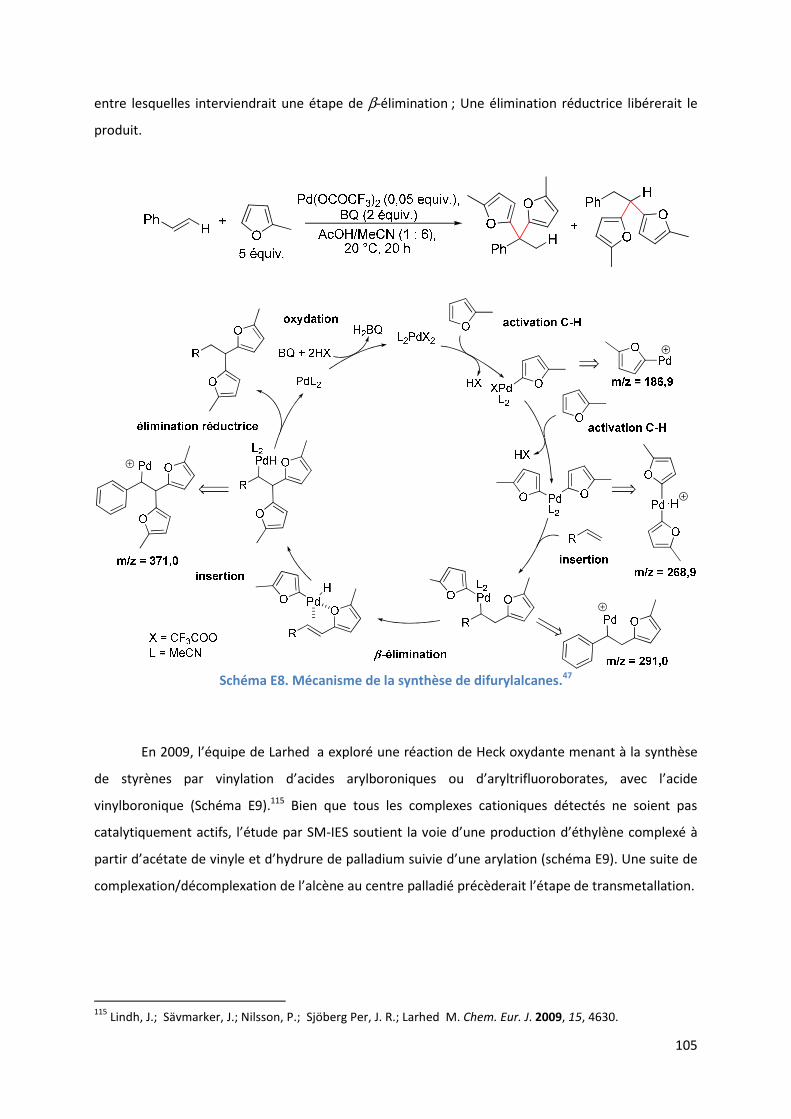
105
entre lesquelles interviendrait une étape de β-élimination ; Une élimination réductrice libérerait le
produit.
Schéma E8. Mécanisme de la synthèse de difurylalcanes.47
En 2009, l’équipe de Larhed a exploré une réaction de Heck oxydante menant à la synthèse
de styrènes par vinylation d’acides arylboroniques ou d’aryltrifluoroborates, avec l’acide
vinylboronique (Schéma E9).115
Bien que tous les complexes cationiques détectés ne soient pas
catalytiquement actifs, l’étude par SM-IES soutient la voie d’une production d’éthylène complexé à
partir d’acétate de vinyle et d’hydrure de palladium suivie d’une arylation (schéma E9). Une suite de
complexation/décomplexation de l’alcène au centre palladié précèderait l’étape de transmetallation.
115
Lindh, J.; Sävmarker, J.; Nilsson, P.; Sjöberg Per, J. R.; Larhed M. Chem. Eur. J. 2009, 15, 4630.
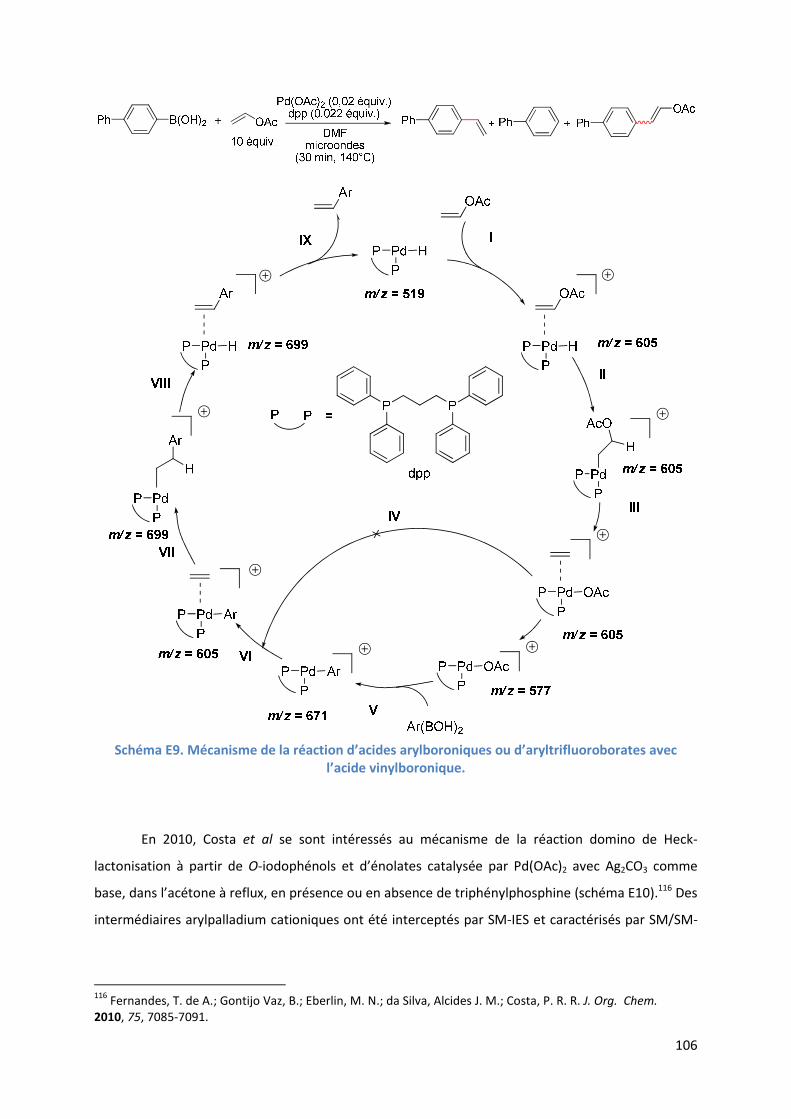
106
Schéma E9. Mécanisme de la réaction d’acides arylboroniques ou d’aryltrifluoroborates avec
l’acide vinylboronique.
En 2010, Costa et al se sont intéressés au mécanisme de la réaction domino de Heck-
lactonisation à partir de O-iodophénols et d’énolates catalysée par Pd(OAc)2 avec Ag2CO3 comme
base, dans l’acétone à reflux, en présence ou en absence de triphénylphosphine (schéma E10).116
Des
intermédiaires arylpalladium cationiques ont été interceptés par SM-IES et caractérisés par SM/SM-
116
Fernandes, T. de A.; Gontijo Vaz, B.; Eberlin, M. N.; da Silva, Alcides J. M.; Costa, P. R. R. J. Org. Chem.
2010, 75, 7085-7091.
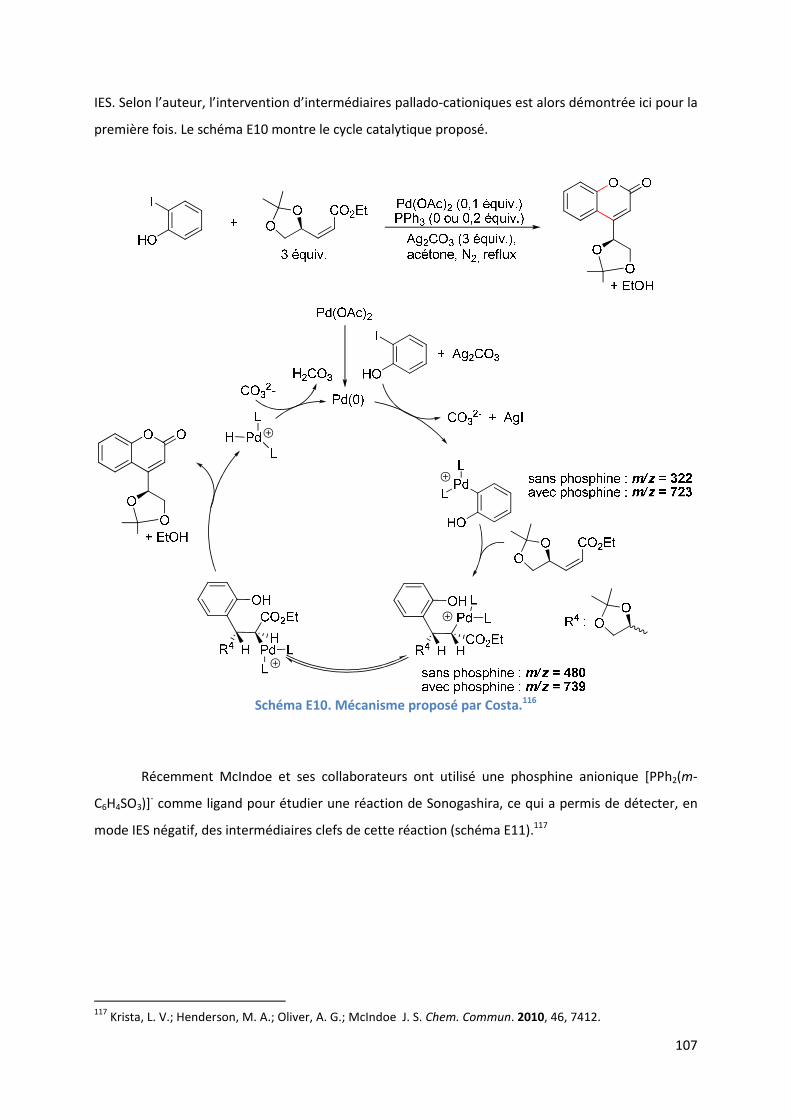
107
IES. Selon l’auteur, l’intervention d’intermédiaires pallado-cationiques est alors démontrée ici pour la
première fois. Le schéma E10 montre le cycle catalytique proposé.
Schéma E10. Mécanisme proposé par Costa.
116
Récemment McIndoe et ses collaborateurs ont utilisé une phosphine anionique [PPh2(m-
C6H4SO3)]- comme ligand pour étudier une réaction de Sonogashira, ce qui a permis de détecter, en
mode IES négatif, des intermédiaires clefs de cette réaction (schéma E11).117
117
Krista, L. V.; Henderson, M. A.; Oliver, A. G.; McIndoe J. S. Chem. Commun. 2010, 46, 7412.
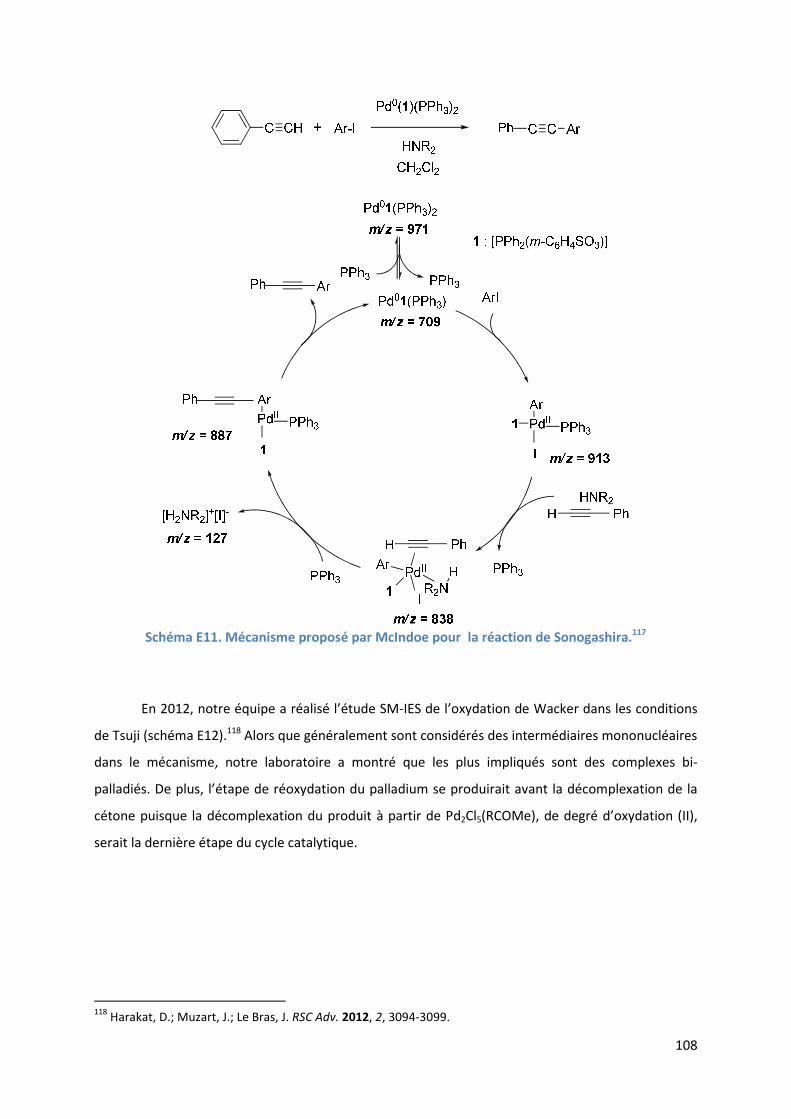
108
Schéma E11. Mécanisme proposé par McIndoe pour la réaction de Sonogashira.
117
En 2012, notre équipe a réalisé l’étude SM-IES de l’oxydation de Wacker dans les conditions
de Tsuji (schéma E12).118
Alors que généralement sont considérés des intermédiaires mononucléaires
dans le mécanisme, notre laboratoire a montré que les plus impliqués sont des complexes bi-
palladiés. De plus, l’étape de réoxydation du palladium se produirait avant la décomplexation de la
cétone puisque la décomplexation du produit à partir de Pd2Cl5(RCOMe), de degré d’oxydation (II),
serait la dernière étape du cycle catalytique.
118
Harakat, D.; Muzart, J.; Le Bras, J. RSC Adv. 2012, 2, 3094-3099.
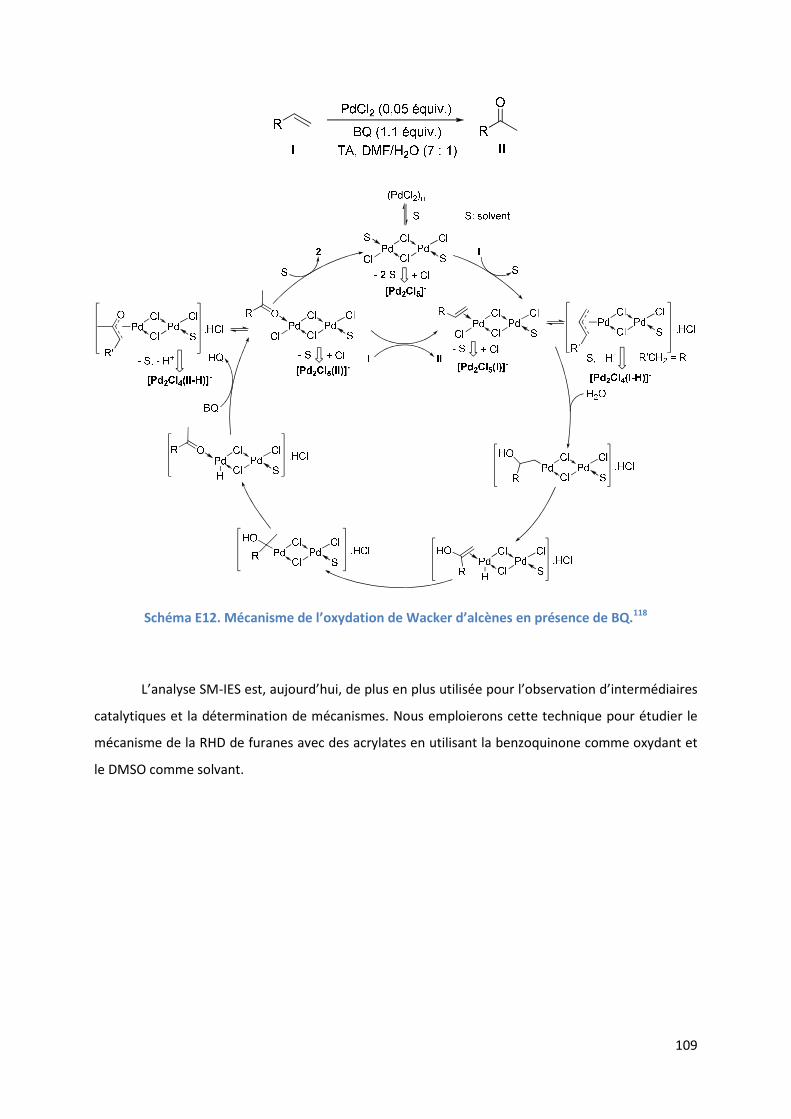
109
Schéma E12. Mécanisme de l’oxydation de Wacker d’alcènes en présence de BQ.118
L’analyse SM-IES est, aujourd’hui, de plus en plus utilisée pour l’observation d’intermédiaires
catalytiques et la détermination de mécanismes. Nous emploierons cette technique pour étudier le
mécanisme de la RHD de furanes avec des acrylates en utilisant la benzoquinone comme oxydant et
le DMSO comme solvant.

110
E.2. Résultats et discussion
E.2.1. Méthodologie
Il existe deux façons d’étudier une réaction en utilisant les techniques IPA : « online » et
« offline ». L’étude en « mode offline » comprend plusieurs étapes : mise au point des conditions en
mélangeant les réactifs pour produire les différents intermédiaires, puis détermination par SM-IES
de la composition de la solution au cours de la transformation des réactifs en produits, avec
caractérisation éventuelle par SM/SM. Cet objectif est réalisable s’il y a une concentration
raisonnable d’intermédiaires, et si ces espèces ne dégradent pas trop vite. Une des limites inhérentes
à cette approche, est l’impossibilité d’analyser des espèces de courte durée de vie.
En mode « online », le réacteur est directement couplé à la source d’ionisation du
spectromètre de masse, permettant la détection de la masse des produits stables et des
intermédiaires de durée de vie de l’ordre de la milliseconde. Ainsi, des changements de composition
du milieu réactionnel peuvent être observés. Le réacteur « online » le plus simple, est la seringue
d’injection même. Plusieurs groupes ont développé des appareils adaptés à l’étude de réactions
radicalaires, photochimiques, électrochimiques, ou organométalliques.
C’est le mode « online » que nous utiliserons. L’analyse du milieu réactionnel s’effectue par
injection directe du brut réactionnel prélevé à l’aide de la seringue utilisée pour l’infusion
électrospray. Pour chaque massif, une hypothèse sera formulée quant à sa nature. L’observation de
décalages du rapport m/z lors de l’utilisation d’arènes ou d’acrylates différents ainsi que les SM/SM
permettront d’appréhender ces hypothèses. De plus, les SM/SM permettront d’apporter des
informations quant aux types de liaisons engagées entre les substrats présents et le centre
métallique. Enfin, les analyses en Haute Résolution pourront confirmer les formules brutes
proposées.
E.2.2. Réaction étudiée
Puisque l’efficacité de l’ionisation par SM-IES dépend de la polarité de la molécule, nous
avons choisi les acrylates comme alcènes. La réaction du sylvane (A1) avec l’acrylate de t-butyle (D1)
en présence de Pd(OAc)2 et de benzoquinone dans un mélange AcOH/DMSO à température
ambiante mène en 4 heures au produit de type Heck avec un rendement de 91 % et au 2,2'-bisylvane
avec un rendement de 7 % (schéma E13).

111
Schéma E13. RHD du sylvane avec l’acrylate de t-butyle.
E.2.3. Rôle du DMSO
E.2.3.1. Analyse par SM-IES(+) de Pd(OAc)2 et de BQ, dans un mélange AcOH/DMSO
Nous avons d’abord analysé par SM-IES(+) un mélange de Pd(OAc)2 et de benzoquinone dans
le mélange AcOH/DMSO (1 : 1) dans le but de connaître le comportement du palladium, et de
visualiser les espèces présentes avant le début de la réaction (spectre E1). Après 5 minutes
d’agitation, cinq massifs contenant du palladium ont été détectés à m/z = 242,9, 260,9, 320,9, 466,9,
544,9. Ces massifs ont été respectivement attribués aux espèces cationiques [Pd(OAc)(DMSO)]+,
[Pd(DMSO-H)(DMSO)]+, [Pd(OAc)(DMSO)2]
+, [Pd2(OAc)3(DMSO)]
+, et [Pd2(OAc)3(DMSO)2]
+.
AV 667
200 250 300 350 400 450 500 550 600m/z0
100
%
11BR1075 120 (2.230) Cm (99:128) 1: TOF MS ES+ 4.52e4x2 320.9
320.0
319.0
262.9259.9
241.9264.9
325.0
546.9
544.9
468.9
465.9
542.9
541.9472.9
548.9
550.9
AV 667
460 465 470 475m/z0
100
%
11BR1075 120 (2.230) Cm (91:123)7.55e3468.9
466.9
464.9
463.9
470.9
472.9
474.9
AV 667
460 465 470 475mass0
100
%
11BR1075 (0.019) Is (1.00,1.00) C8H15O7Pd2S1.64e12468.9
466.9
464.9
463.9
462.9
470.9
472.9
474.9
aConditions : Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2,0 mL), DMSO (2 mL), TA, 5 min.
Spectre E1. SM-IES(+) d’une solution AcOH/DMSO de Pd(OAc)2 et BQ après 5 min.a
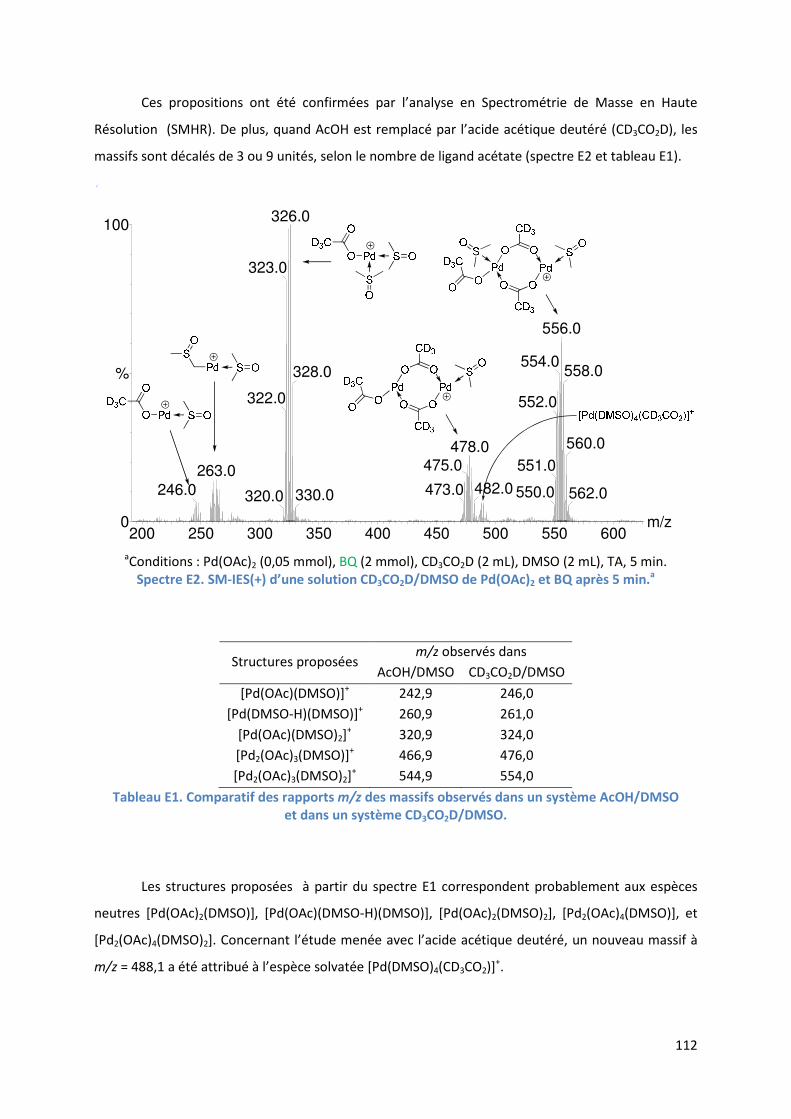
112
Ces propositions ont été confirmées par l’analyse en Spectrométrie de Masse en Haute
Résolution (SMHR). De plus, quand AcOH est remplacé par l’acide acétique deutéré (CD3CO2D), les
massifs sont décalés de 3 ou 9 unités, selon le nombre de ligand acétate (spectre E2 et tableau E1).
AV 761
200 250 300 350 400 450 500 550 600m/z0
100
%
12HR81 94 (2.964) Cm (1:166) 1: TOF MS ES+ 2.02e3326.0
323.0
322.0
263.0
246.0 320.0
556.0
554.0328.0
552.0
478.0475.0
473.0330.0
551.0
482.0 550.0
558.0
560.0
562.0
aConditions : Pd(OAc)2 (0,05 mmol), BQ (2 mmol), CD3CO2D (2 mL), DMSO (2 mL), TA, 5 min.
Spectre E2. SM-IES(+) d’une solution CD3CO2D/DMSO de Pd(OAc)2 et BQ après 5 min.a
Structures proposées m/z observés dans
AcOH/DMSO CD3CO2D/DMSO
[Pd(OAc)(DMSO)]+ 242,9 246,0
[Pd(DMSO-H)(DMSO)]+ 260,9 261,0
[Pd(OAc)(DMSO)2]+ 320,9 324,0
[Pd2(OAc)3(DMSO)]+ 466,9 476,0
[Pd2(OAc)3(DMSO)2]+ 544,9 554,0
Tableau E1. Comparatif des rapports m/z des massifs observés dans un système AcOH/DMSO
et dans un système CD3CO2D/DMSO.
Les structures proposées à partir du spectre E1 correspondent probablement aux espèces
neutres [Pd(OAc)2(DMSO)], [Pd(OAc)(DMSO-H)(DMSO)], [Pd(OAc)2(DMSO)2], [Pd2(OAc)4(DMSO)], et
[Pd2(OAc)4(DMSO)2]. Concernant l’étude menée avec l’acide acétique deutéré, un nouveau massif à
m/z = 488,1 a été attribué à l’espèce solvatée [Pd(DMSO)4(CD3CO2)]+.

113
E.2.3.2. Effet positif du solvant sur l’activité du catalyseur
L’acétate de palladium (II) à l’état solide existe sous la forme d’un trimère,62
et une telle
structure est maintenue dans l’acide acétique.63
Comme le montre l’étude SM-IES(+), l’utilisation de
DMSO comme co-solvant conduit à la formation d’espèces mono- et dinucléaires (schéma E14).
Schéma E14. Comportement de l’acétate de palladium trimérique dans un système AcOH/DMSO.
Cela permet de comprendre l’effet positif du solvant constaté dans ces réactions. En effet,
Davies et Macgregor, dans une récente revue d’études computationnelles sur les activations de
liaisons C-H métallo-induites, supposent que l’activation pallado-induite du benzène se fasse par
assistance d’un ligand acétate (schéma E15).119
Dans l’espèce [Pd(OAc)2]3, les unités Pd(OAc)2 sont
jointes par des doubles ponts acétates et par conséquent, aucun ligand acétate ne peut participer à
l’activation d’une liaison C-H. En revanche, les espèces mononucléaires et dinucléaires présentées au
schéma E14 possèdent un ligand acétate disponible. Ceci explique la période d’induction observée
pour les RHD de furanes dans un système Et2O/RCO2H (R = Me, Et), dans lequel la structure du
trimère est plutôt stable.48
Il a été observé que cette période d’induction dépendait de la nature du
furane, la complexation de [Pd(OAc)2]3 par des furanes électroniquement riches menant
probablement plus facilement à des espèces palladiées monomériques et dimériques actives.48
Par
contre, aucune période d’induction n’est observée dans un mélange AcOH/DMSO comme solvant,
puisque le DMSO agit comme un ligand du palladium fournissant des espèces mononucléaires et
dinucléaires pouvant participer à la RHD.120
119
Boutadla, Y.; Davies, D. L.; Macgregor, S. A.; Poblador-Bahamonde, A. I. Dalton Trans. 2009, 5820-5831. 120
Le DMSO est un ligand ambident. D’après des études théoriques, le mode de coordination du DMSO au
Pd(II) peut se faire par le soufre ou l’oxygène selon l’environnement de ligands, alors que pour Pd(0) elle se
ferait principalement par l’atome de soufre : Zierkiewicz, W.; Privalov, T. Organometallics 2005, 24, 6019-6028.

114
Schéma E15. Mécanisme proposé par Davies et Macgregor pour l’activation C-H du benzène par
l’acétate de palladium.
Enfin, l’observation du massif correspondant à [Pd(DMSO-H)(DMSO)]+ suggère que l’espèce
[Pd(OAc)2(DMSO)2] puisse aussi réagir avec une des liaisons C(sp3)-H du DMSO.
E.2.4. Etude de l’étape de l’activation de liaison C-H de l’hétéroarène
E.2.4.1. Analyse par SM-IES(+) de Pd(OAc)2, BQ, et A1 dans un mélange AcOH/DMSO
Nous avons analysé par SM-IES(+) un mélange de Pd(OAc)2, BQ, et A1 en solution dans
AcOH/DMSO (1 : 1). L’ajout de A1 au milieu précédemment analysé se traduit par l’apparition de 5
nouveaux massifs à m/z = 343,0, 345,0, 455,0, 488,9, et 566,9 après 5 minutes d’agitation. Ces
massifs ont été attribués respectivement aux espèces [Pd(A1-H)(DMSO)2]+, [Pd(A1)(DMSO)2H]
+,
[Pd(A1-H)2(BQ)(DMSO)H]+, [Pd2(A1-H)(OAc)2(DMSO)]
+, et [Pd2(A1-H)(OAc)2(DMSO)2]
+ (spectre E3).
Ces formules brutes ont été confirmées par analyse SMHR. De plus, lorsque le 2-éthylfurane (A2) est
utilisé à la place de A1, les rapports m/z correspondant aux espèces [Pd(A-H)(DMSO)2]+,
[Pd(A)(DMSO)2H]+, [Pd(A-H)2(BQ)(DMSO)H]
+, [Pd2(A-H)(OAc)2(DMSO)]
+ sont décalés de 14 ou de 28
unités de masse selon le nombre de ligands furyles (spectre E4 et tableau E2).
Tableau E2. Comparatif des rapports m/z des massifs observés lorsque A1 ou A2 sont utilisés
comme substrats.
Structures proposées m/z observés avec
A1 comme substrat A2 comme substrat
[Pd(A-H)(DMSO)2]+ 343,0 357,1
[Pd(A)(DMSO)2H]+ 345,0 359,1
[Pd(A-H)2(BQ)(DMSO)H]+ 455,0 483,2
[Pd2(A-H)(OAc)2(DMSO)]+ 488,9 503,1
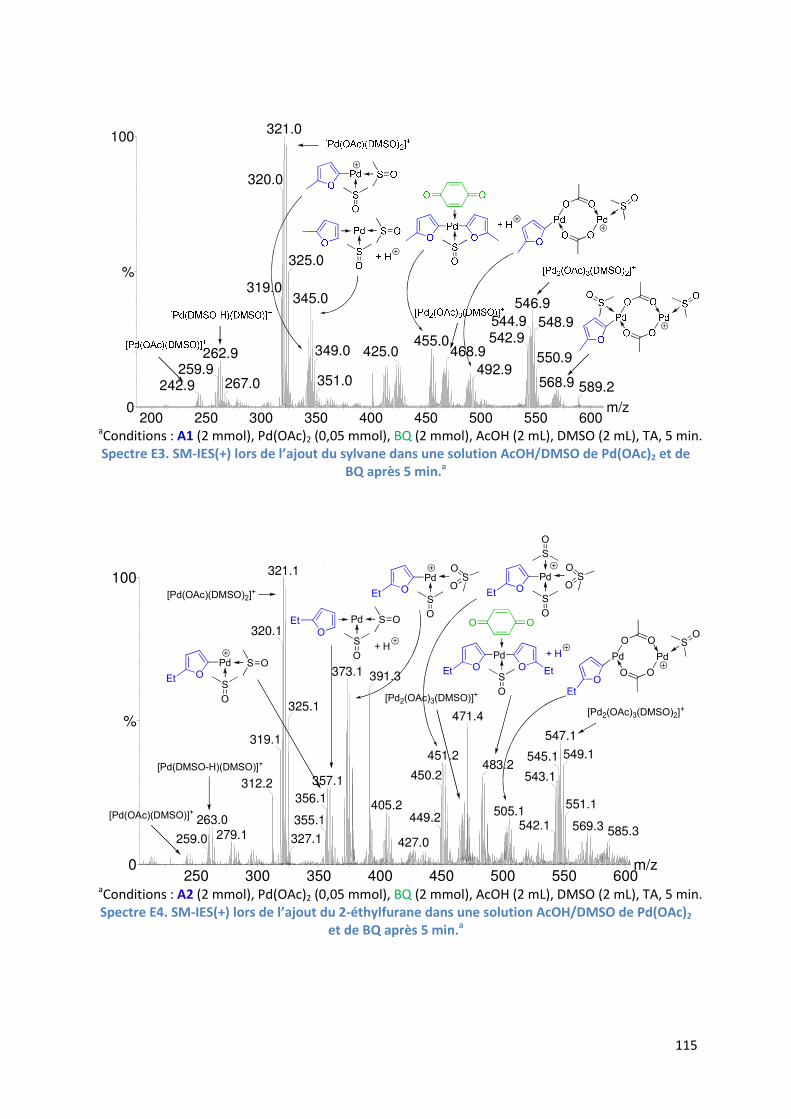
115
AV 667
200 250 300 350 400 450 500 550 600m/z0
100
%
11BR1077 1 (0.019) Cm (1:40) 1: TOF MS ES+ 2.79e4321.0
320.0
319.0
262.9259.9
242.9 267.0
325.0
345.0 546.9
544.9542.9455.0
349.0 425.0
351.0
468.9
492.9
548.9
550.9
568.9 589.2
aConditions : A1 (2 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2 mL), TA, 5 min.
Spectre E3. SM-IES(+) lors de l’ajout du sylvane dans une solution AcOH/DMSO de Pd(OAc)2 et de
BQ après 5 min.a
AV 736
250 300 350 400 450 500 550 600m/z0
100
%
12BR63 131 (2.442) Cm (82:145) 1: TOF MS ES+ 1.54e4321.1
320.1
319.1
312.2
263.0
259.0 279.1
373.1
325.1
357.1
356.1
355.1
327.1
391.3
471.4
451.2
450.2
405.2449.2
427.0
547.1
545.1483.2
543.1
505.1542.1
549.1
551.1
569.3 585.3
[Pd(OAc)(DMSO)]+
[Pd(DMSO-H)(DMSO)]+
[Pd(OAc)(DMSO)2]+
Pd S O
S
O
OEt
Pd
S
O
O OEt Et
OO
+ H
[Pd2(OAc)3(DMSO)]+
[Pd2(OAc)3(DMSO)2]+
Pd
OO
SO
Pd
O O
OEt
Pd SO
S
O
OEtO
Pd SO
S
O
OEtO
S
O
Pd S O
S
O
O
Et
+ H
aConditions : A2 (2 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2 mL), TA, 5 min.
Spectre E4. SM-IES(+) lors de l’ajout du 2-éthylfurane dans une solution AcOH/DMSO de Pd(OAc)2
et de BQ après 5 min.a
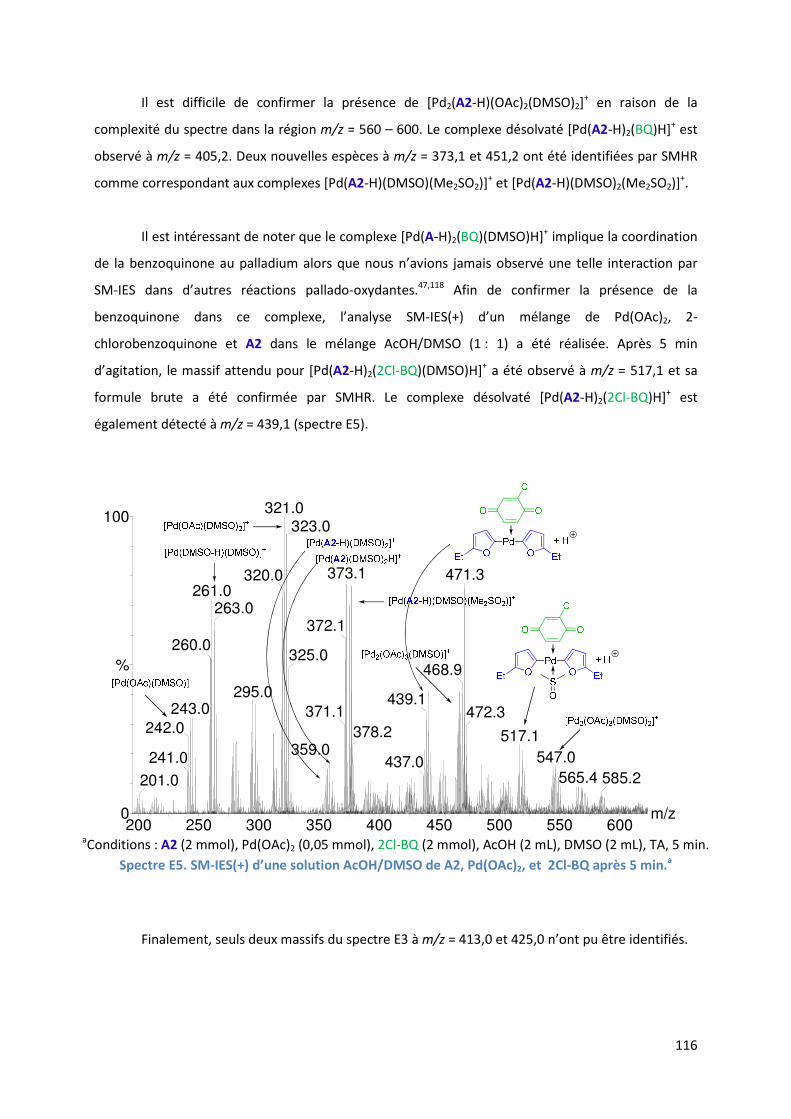
116
Il est difficile de confirmer la présence de [Pd2(A2-H)(OAc)2(DMSO)2]+ en raison de la
complexité du spectre dans la région m/z = 560 – 600. Le complexe désolvaté [Pd(A2-H)2(BQ)H]+ est
observé à m/z = 405,2. Deux nouvelles espèces à m/z = 373,1 et 451,2 ont été identifiées par SMHR
comme correspondant aux complexes [Pd(A2-H)(DMSO)(Me2SO2)]+ et [Pd(A2-H)(DMSO)2(Me2SO2)]
+.
Il est intéressant de noter que le complexe [Pd(A-H)2(BQ)(DMSO)H]+ implique la coordination
de la benzoquinone au palladium alors que nous n’avions jamais observé une telle interaction par
SM-IES dans d’autres réactions pallado-oxydantes.47,118
Afin de confirmer la présence de la
benzoquinone dans ce complexe, l’analyse SM-IES(+) d’un mélange de Pd(OAc)2, 2-
chlorobenzoquinone et A2 dans le mélange AcOH/DMSO (1 : 1) a été réalisée. Après 5 min
d’agitation, le massif attendu pour [Pd(A2-H)2(2Cl-BQ)(DMSO)H]+ a été observé à m/z = 517,1 et sa
formule brute a été confirmée par SMHR. Le complexe désolvaté [Pd(A2-H)2(2Cl-BQ)H]+ est
également détecté à m/z = 439,1 (spectre E5).
AV 759
200 250 300 350 400 450 500 550 600m/z0
100
%
12BR136 95 (1.770) Cm (86:99) 1: TOF MS ES+ 405321.0
320.0261.0
260.0
243.0
242.0
241.0
201.0
263.0
295.0
323.0
373.1
372.1
325.0
371.1
359.0
471.3
468.9
439.1
378.2
437.0
472.3
517.1
547.0
565.4 585.2
aConditions : A2 (2 mmol), Pd(OAc)2 (0,05 mmol), 2Cl-BQ (2 mmol), AcOH (2 mL), DMSO (2 mL), TA, 5 min.
Spectre E5. SM-IES(+) d’une solution AcOH/DMSO de A2, Pd(OAc)2, et 2Cl-BQ après 5 min.a
Finalement, seuls deux massifs du spectre E3 à m/z = 413,0 et 425,0 n’ont pu être identifiés.

117
La SM/SM-IES(+) de [Pd(A1-H)(DMSO)2]+ identifié à m/z = 343,0 montre la perte de DMSO et
de A1 (tableau E3). Celle de A1 est assez surprenante mais cette observation est probablement due à
la superposition du signal avec celui à m/z = 345,0 correspondant à [Pd(A1)(DMSO)2H]+ et montrant
une fragmentation similaire (tableau E3). [Pd(A1-H)2(BQ)(DMSO)H]+ est dissocié par SM/SM-IES(+)
pour produire le DMSO, BQ, et le Pd. La perte du palladium est une fragmentation classique connue
comme la résultante d’une élimination réductrice de l’espèce R2Pd.121
La perte de benzoquinone
confirme donc qu’un tel composé se coordonne réellement au palladium. Les SM/SM-IES(+) de
[Pd2(A1-H)(OAc)2(DMSO)]+, and [Pd2(A1-H)(OAc)2(DMSO)2]
+ fournissent des fragmentations similaires
et montrent les pertes de DMSO, AcOH et de Pd. Enfin, la perte d’acide organique a été confirmée
par utilisation de CD3CO2D comme solvant.122
Structures proposées Pertes observées par ESI(+)-MS/MS
[Pd(A1-H)(DMSO)2]+ DMSO, A1
[Pd(A1)(DMSO)2H]+ DMSO, A1
[Pd(A1-H)2(BQ)(DMSO)H]+ DMSO, BQ, Pd,
[Pd2(A1-H)(OAc)2(DMSO)]+ DMSO, AcOH, Pd
[Pd2(A1-H)(OAc)2(DMSO)2]+ DMSO, AcOH, Pd
Tableau E3. Fragmentations observées lors de l’analyse SM/SM des massifs du spectre E2.
[Pd(A-H)(DMSO)2]+, [Pd2(A-H)(OAc)2(DMSO)]
+, et [Pd2(A-H)(OAc)2(DMSO)2]
+ peuvent être
reliées aux espèces neutres Pd(A-H)(OAc)(DMSO)2, Pd2(A-H)(OAc)3(DMSO), et Pd2(A-
H)(OAc)3(DMSO)2, tandis que [Pd(A-H)2(BQ)(DMSO)H]+ peut correspondre à la forme protonée de
[Pd(A-H)2(BQ)(DMSO)].
E.2.4.2. Espèces catalytiques actives vis-à-vis de liaisons C-H
Le complexe Pd(A1-H)(OAc)(DMSO)2 résulte de la réaction de A1 avec l’espèce
monopalladiée Pd(OAc)2(DMSO)2 présente sur le spectre E1. Cette dernière constitue donc une
espèce intervenant dans l’étape d’activation de la liaison C-H de l’hétéroarène (schéma E16).
121
Qian, R.; Guo, Y.; Lü, L. Chin. J. Chem. 2008, 26, 123-129. 122
Les fragmentations de [Pd(A1-H)(DMSO)2]+, [Pd(A1)(DMSO)2H]
+ et de [Pd(A1-H)2(BQ)(DMSO)H]
+ ont montré
la perte d’un fragment de 60 unités de masse, ce qui aurait pu être attribué à la perte de AcOH. Cependant,
cette perte est également observée quand CD3CO2D est employé comme solvant et viendrait donc
probablement de la fragmentation de A1 ou du DMSO.
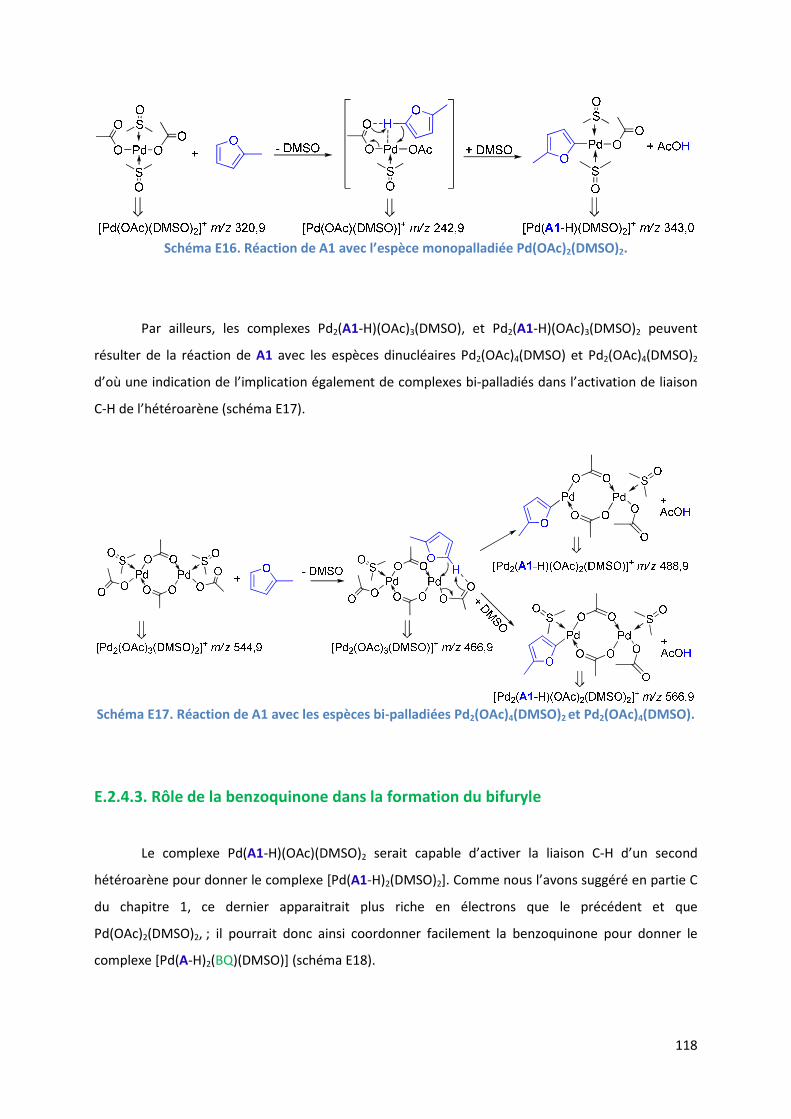
118
Schéma E16. Réaction de A1 avec l’espèce monopalladiée Pd(OAc)2(DMSO)2.
Par ailleurs, les complexes Pd2(A1-H)(OAc)3(DMSO), et Pd2(A1-H)(OAc)3(DMSO)2 peuvent
résulter de la réaction de A1 avec les espèces dinucléaires Pd2(OAc)4(DMSO) et Pd2(OAc)4(DMSO)2
d’où une indication de l’implication également de complexes bi-palladiés dans l’activation de liaison
C-H de l’hétéroarène (schéma E17).
Schéma E17. Réaction de A1 avec les espèces bi-palladiées Pd2(OAc)4(DMSO)2 et Pd2(OAc)4(DMSO).
E.2.4.3. Rôle de la benzoquinone dans la formation du bifuryle
Le complexe Pd(A1-H)(OAc)(DMSO)2 serait capable d’activer la liaison C-H d’un second
hétéroarène pour donner le complexe [Pd(A1-H)2(DMSO)2]. Comme nous l’avons suggéré en partie C
du chapitre 1, ce dernier apparaitrait plus riche en électrons que le précédent et que
Pd(OAc)2(DMSO)2, ; il pourrait donc ainsi coordonner facilement la benzoquinone pour donner le
complexe [Pd(A-H)2(BQ)(DMSO)] (schéma E18).

119
Schéma E18. Réaction du complexe [Pd(A1-H)(OAc)(DMSO)2] avec A1 et BQ.
Le complexe [Pd(A1-H)2(BQ)(DMSO)] pourrait être un intermédiaire de la formation de
bifuryle A1-A1. A la suite d’une élimination réductrice favorisée par la présence de benzoquinone,91-
97 l’espèce Pd(A1-H)2(BQ)(DMSO) pourrait fournir le produit d’homocouplage A1-A1. Notons que lors
des travaux précédents réalisés en 2008 par notre équipe, aucune trace de A1-A1 n’avait été
observée quand le couplage de A1 avec C1 était réalisé dans CH3CN.47
Néanmoins, le complexe de
bifuryle [Pd(A1-H)2]+ fut observé mais sans coordination de la benzoquinone. L’acétonitrile, solvant
coordonnant, empêcherait donc probablement la complexation de la benzoquinone et donc
l’élimination réductrice.
La réaction d’élimination réductrice doit s’accompagner de la formation de palladium (0).
Cette présence de Pd(0) est suggérée par le complexe [Pd(A1)(DMSO)2] identifié au spectre E3 sous
la forme cationique [Pd(A1)(DMSO)2H]+ (schéma E19). Le Pd(0) serait alors maintenu en solution par
ligandation avec le DMSO et le substrat A1. Cependant, les similarités très proches au niveau des
SM/SM-IES entre Pd(A1-H)(OAc)(DMSO)2 et [Pd(A1)(DMSO)2] suggèrent aussi que ce dernier peut
être obtenu par réduction du précédent via des processus électrochimiques ayant lieu au cours du
procédé électrospray (notamment au cours de l’étape de désolvatation). Néanmoins, ce phénomène
n’est probablement pas significatif puiqu’il n’est pas observé pour d’autres amas.
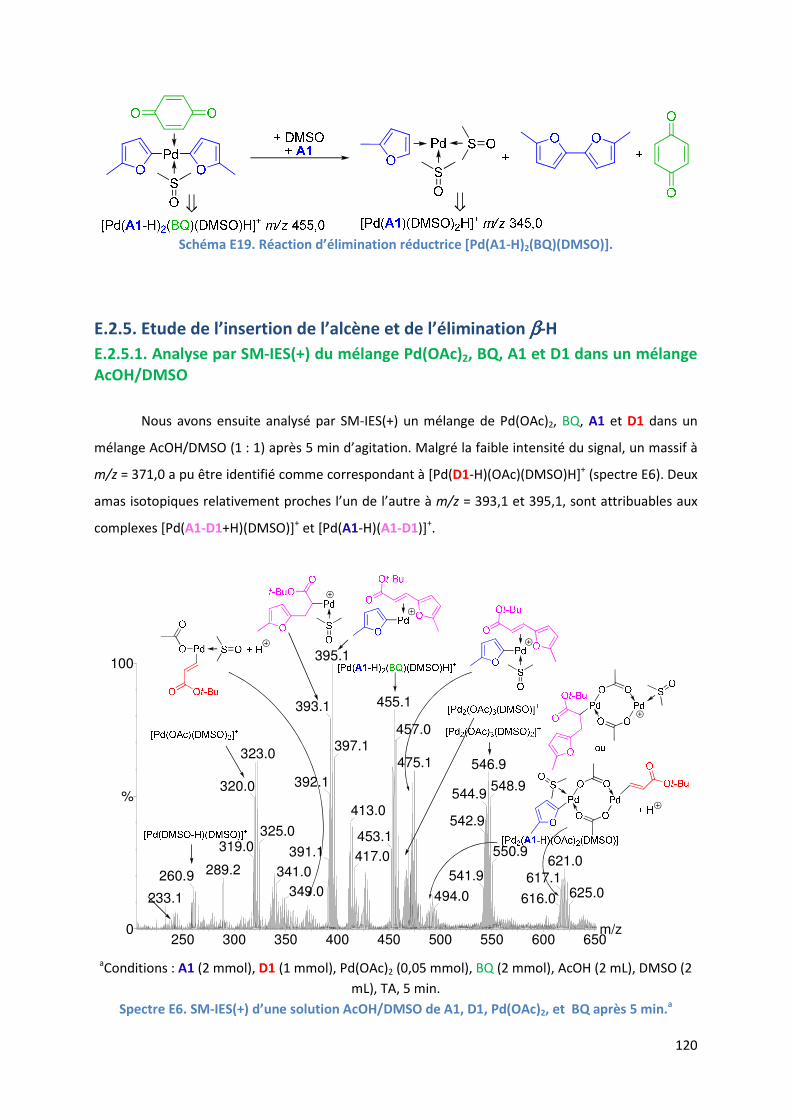
120
Schéma E19. Réaction d’élimination réductrice [Pd(A1-H)2(BQ)(DMSO)].
E.2.5. Etude de l’insertion de l’alcène et de l’élimination ββββ-H
E.2.5.1. Analyse par SM-IES(+) du mélange Pd(OAc)2, BQ, A1 et D1 dans un mélange
AcOH/DMSO
Nous avons ensuite analysé par SM-IES(+) un mélange de Pd(OAc)2, BQ, A1 et D1 dans un
mélange AcOH/DMSO (1 : 1) après 5 min d’agitation. Malgré la faible intensité du signal, un massif à
m/z = 371,0 a pu être identifié comme correspondant à [Pd(D1-H)(OAc)(DMSO)H]+ (spectre E6). Deux
amas isotopiques relativement proches l’un de l’autre à m/z = 393,1 et 395,1, sont attribuables aux
complexes [Pd(A1-D1+H)(DMSO)]+ et [Pd(A1-H)(A1-D1)]
+.
AV 688
250 300 350 400 450 500 550 600 650m/z0
100
%
11HR743 105 (3.295) Cm (97:142) 1: TOF MS ES+ 2.26e3395.1
393.1
323.0
320.0
319.0
289.2260.9
233.1
392.1
325.0
391.1
341.0
349.0
455.1
397.1
413.0
453.1
417.0
457.0
475.1 546.9
544.9
542.9
541.9
494.0
548.9
550.9621.0
617.1
616.0 625.0
aConditions : A1 (2 mmol), D1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2
mL), TA, 5 min.
Spectre E6. SM-IES(+) d’une solution AcOH/DMSO de A1, D1, Pd(OAc)2, et BQ après 5 min.a

121
Le complexe [Pd(A1-D1+H)(DMSO)]+ est également observé à m/z = 473,1 correspondant
ainsi à l’espèce solvatée [Pd(A1-D1+H)(DMSO)2]+. Cependant, il est contaminé par un complexe à m/z
= 475,1 lequel, selon l’analyse SMHR, pourrait être attribué à [Pd(A1)(A1-D1)(DMSO)H]+. Néanmoins,
la SM/SM-IES(+) ne met pas en évidence la perte de A1 et de A1-D1 comme attendu et cette
proposition ne peut donc pas être confirmée. Enfin, un autre amas a été détecté à m/z = 617,0 et
attribué à [Pd2(A1-D1+H)(OAc)2(DMSO)]+ ou [Pd2(A1-H)(D1-H)(OAc)2(DMSO)H]
+ (schéma E20).
Schéma E20. Structures possibles pour l’amas observé à m/z = 617,0.
Toutes les propositions formulées sont en accord avec les analyses par SMHR. Par ailleurs,
l’étude SM-IES(+) du couplage de A2 avec D1 et de A1 avec l’acrylate d’éthyle (D2) révèle des
décalages de 14 ou 28 unités de masse comme attendu, à l’exception de [Pd(D2-
H)(OAc)(DMSO)H]+123
qui présente une rapport m/z proche de celui de [Pd(A1-H)(DMSO)2]+ et
[Pd2(A1-D2+H)(OAc)2(DMSO)]+/[Pd2(A1-H)(D2-H)(OAc)2(DMSO)H]
+ dont la structure ne peut donc pas
être confirmé en raison de la complexité du spectre (tableau E4 et spectres E7 et E8). Soulignons que
le massif a m/z = 481 n’a pas pu être identifié.
Tableau E4. Comparatif des rapports m/z des massifs observés lorsque A1 ou A2 sont utilisés avec
D1 ou D2 comme substrats.
123
Si la présence de [Pd(D2-H)(OAc)(DMSO)H]+ n’a pas pu être confirmé, il faut noter que le signal à m/z = 371
attribué à Pd(D1-H)(OAc)(DMSO)H]+ n’est pas observé lorsque D2 est utilisé comme alcène.
Structures proposées m/z observés pour couplage de
A1 avec D1 A2 avec D1 A1 avec D2
[Pd(D-H)(OAc)(DMSO)H]+ 371,0 371,0 -
[Pd(A-D+H)(DMSO)]+ 393,1 407,2 365,1
[Pd(A-H)(A-D)]+ 395,1 423,2 367,3
[Pd(A-H)(A-D)(DMSO)]+ 473,1 501,3 445,2
[Pd2(A-D+H)(OAc)2(DMSO)]+ ou
[Pd2(A-H)(D-H)(OAc)2(DMSO)H]+
617,0 631,4 -
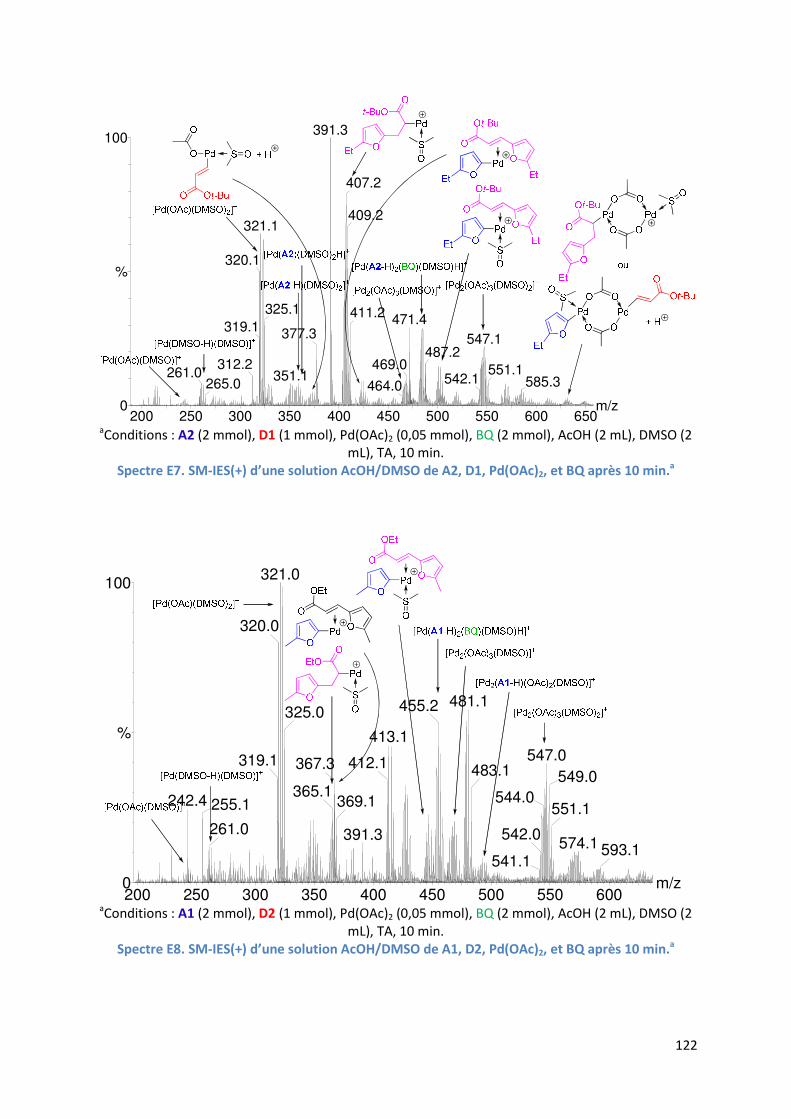
122
AV 736
200 250 300 350 400 450 500 550 600 650m/z0
100
%
12BR65 224 (4.174) Cm (163:289) 1: TOF MS ES+ 3.04e4391.3
321.1
320.1
319.1
312.2261.0
265.0
325.1
377.3
351.1
407.2
409.2
411.2471.4
469.0
464.0
547.1487.2
542.1551.1
585.3
aConditions : A2 (2 mmol), D1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2
mL), TA, 10 min.
Spectre E7. SM-IES(+) d’une solution AcOH/DMSO de A2, D1, Pd(OAc)2, et BQ après 10 min.a
AV 737
200 250 300 350 400 450 500 550 600m/z0
100
%
12BR67 196 (3.649) Cm (195:283) 1: TOF MS ES+ 2.38e3x2321.0
320.0
319.1
242.4 255.1
261.0
481.1455.2325.0
413.1
412.1367.3
365.1369.1
391.3
547.0483.1
544.0
542.0
541.1
549.0
551.1
574.1 593.1
aConditions : A1 (2 mmol), D2 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2
mL), TA, 10 min.
Spectre E8. SM-IES(+) d’une solution AcOH/DMSO de A1, D2, Pd(OAc)2, et BQ après 10 min.a

123
La SM/SM-IES de [Pd(D1-H)(OAc)(DMSO)H]+ montre la perte de DMSO, AcOH et C4H8 (tableau
E5), cette dernière confirmant la présence d’un groupement t-butyle. [Pd(A1-D1+H)(DMSO)]+ est
dissocié en DMSO et C4H8. La SM-IES(+) de [Pd(A1-H)(A1-D1)(DMSO)]+ montre également une perte
de C4H8. De plus, la perte de A1-D1 est observée, ce qui indique la coordination du produit au centre
palladié. Enfin, la fragmentation par SM/SM-IES(+) de [Pd2(A1-D1+H)(OAc)2(DMSO)]+/[Pd2(A1-H)(D1-
H)(OAc)2(DMSO)H]+ révèle la perte de DMSO, AcOH, Pd et C4H8. Parmi les deux structures possibles,
[Pd2(A1-D1+H)(OAc)2(DMSO)]+ ou [Pd2(A1-H)(D1-H)(OAc)2(DMSO)H]
+, la première semble la plus
probable, avec une perte du groupement acétate de t-butyle (m/z = 116) (schéma E20).
Structures proposées Pertes observées par SM/SM-IES(+)
[Pd(D1-H)(OAc)(DMSO)H]+ DMSO, AcOH, C4H8
[Pd(A1-D1+H)(DMSO)]+ DMSO, C4H8
[Pd(A1-H)(A1-D1)(DMSO)]+ DMSO, C4H8, A1-D1
[Pd2(A1-D1+H)(OAc)2(DMSO)]+ ou
[Pd2(A1-H)(D1-H)(OAc)2(DMSO)H]+
DMSO, AcOH, Pd, C4H8, CH3CO2t-Bu
Tableau E5. Fragmentations oberservées lors de l’analyse SM/SM des massifs du spectre E4.
Les structures proposées au tableau E4 sont probablement reliables aux espèces neutres
Pd(D-H)(OAc)(DMSO), Pd(A-D+H)(OAc)(DMSO), Pd(A-H)(A-D)(OAc), Pd(A-H)(A-D)(OAc)(DMSO), et
[Pd2(A-D+H)(OAc)3(DMSO)]/Pd2(A-H)(D-H)(OAc)2(DMSO).
E.2.5.2. Réaction secondaire : activation de l’acrylate de t-butyle D1
L’espèce Pd(OAc)(DMSO)(D1-H) résulte de la réaction de D1 avec les complexes
mononucléaires de Pd(II) (schéma E21). Les SM/SM ne révèlent pas de perte de D1, indiquant ainsi
que l’alcène est lié au palladium par une liaison σ. Ainsi, les espèces mono-palladiées sont efficaces
pour l’activation de liaison C-H de l’acrylate de t-butyle.
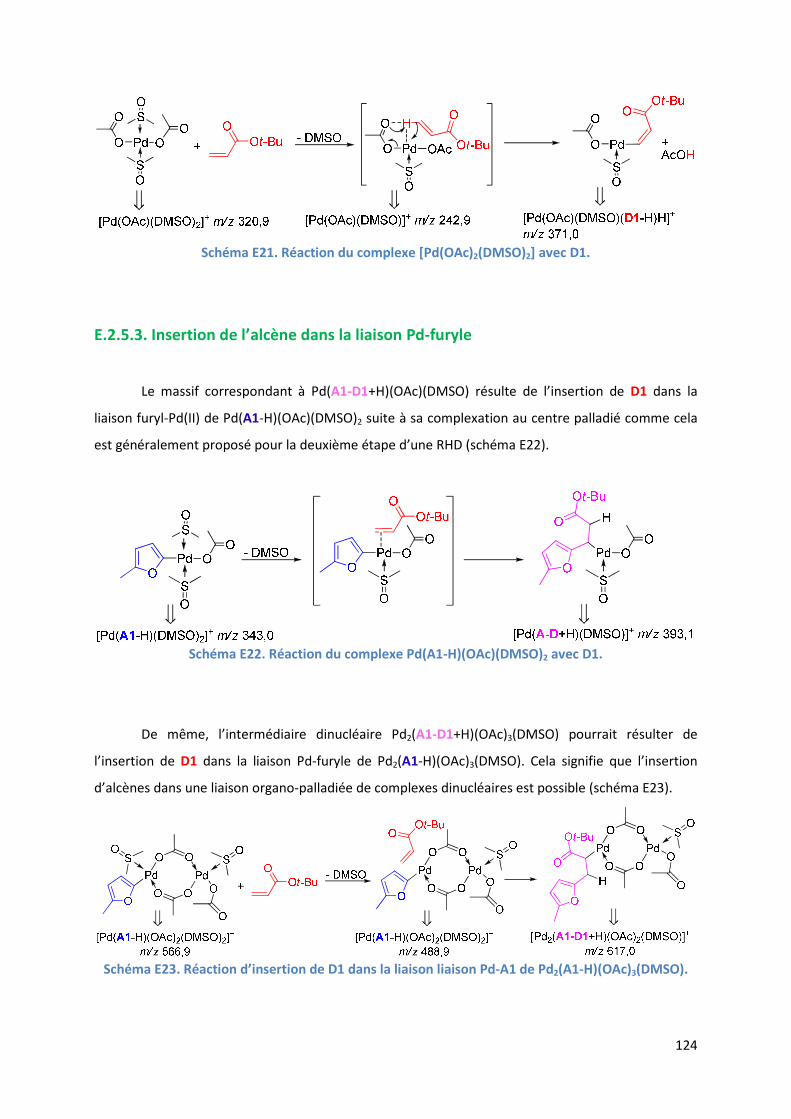
124
Schéma E21. Réaction du complexe [Pd(OAc)2(DMSO)2] avec D1.
E.2.5.3. Insertion de l’alcène dans la liaison Pd-furyle
Le massif correspondant à Pd(A1-D1+H)(OAc)(DMSO) résulte de l’insertion de D1 dans la
liaison furyl-Pd(II) de Pd(A1-H)(OAc)(DMSO)2 suite à sa complexation au centre palladié comme cela
est généralement proposé pour la deuxième étape d’une RHD (schéma E22).
Schéma E22. Réaction du complexe Pd(A1-H)(OAc)(DMSO)2 avec D1.
De même, l’intermédiaire dinucléaire Pd2(A1-D1+H)(OAc)3(DMSO) pourrait résulter de
l’insertion de D1 dans la liaison Pd-furyle de Pd2(A1-H)(OAc)3(DMSO). Cela signifie que l’insertion
d’alcènes dans une liaison organo-palladiée de complexes dinucléaires est possible (schéma E23).
Schéma E23. Réaction d’insertion de D1 dans la liaison liaison Pd-A1 de Pd2(A1-H)(OAc)3(DMSO).

125
L’autre complexe dinucléaire Pd2(A1-H)(D1-H)(OAc)2(DMSO) pourrait provenir de
l’association des deux espèces monucléaires Pd(A1-H)(OAc)(DMSO)2 et Pd(D1-H)(OAc)(DMSO) ou de
l’activation C-H de D1 par Pd2(A1-H)(OAc)3(DMSO). La double activation C-H d’arènes et d’alcènes par
le palladium (II) suivie d’une élimination réductrice pour fournir le produit de Heck correspondant a
été discutée dans les premières études concernant les RHD.124
Il a été proposé que l’étape
cinétiquement déterminante puisse correspondre à la formation d’une liaison σ entre le palladium et
l’alcène.119b
Cependant, la comparaison de la réactivité de PhH vis-à-vis de PhCH=CD2 et PhCH=CH2
permet de conclure que la rupture de la liaison hydrogène en β du styrène n’est pas impliquée dans
l’étape cinétiquement déterminante.125
Les études ultérieures s’accordent sur la formation des
espèces ArPd(II) à partir d’un mélange CH2=CHR/ArH/Pd(OAc)2 comme étape cinétiquement
déterminante, laquelle est suivie par l’insertion de l’alcène.32
Il convient cependant de souligner que
la détection par SM-IES(+) de Pd(D1-H)(OAc)2(DMSO) suggère que l’activation C-H de D1 par Pd(II) est
possible et peut donc jouer un rôle dans le mécanisme.
E.2.5.3. Réaction d’élimination ββββ-H et rôle complexant du produit généré
Le produit de couplage de Heck est formé après une étape d’élimination β-H que subissent
les complexes organo-palladiés Pd2(A-D+H)(OAc)3(DMSO) et Pd(A-D+H)(OAc)(DMSO). Cette étape
génère également un hydrure de palladium [HPdOAc], qui après une élimination réductrice de l’acide
acétique, fournit du palladium (0) (schéma E24 et E25)
Schéma E24. Réaction d’élimination ββββ-H subie par le complexe Pd(A-D+H)(OAc)(DMSO).
124
(a) Danno, S.; Moritani, I.; Fujiwara, Y. Tetrahedron 1969, 25, 4819-4823; (b) Fujiwara, Y.; Moritani, I.; Asano,
R.; Tanaka, H.; Teranishi, S. Tetrahedron 1969, 25, 4815-4818; (c) Danno, S.; Moritani, I.; Fujiwara, Y.; Teranishi,
S. J. Chem. Soc. B 1971, 196-198. 125
Shue, R. S. J. Am. Chem. Soc. 1971, 93, 7116-7117.
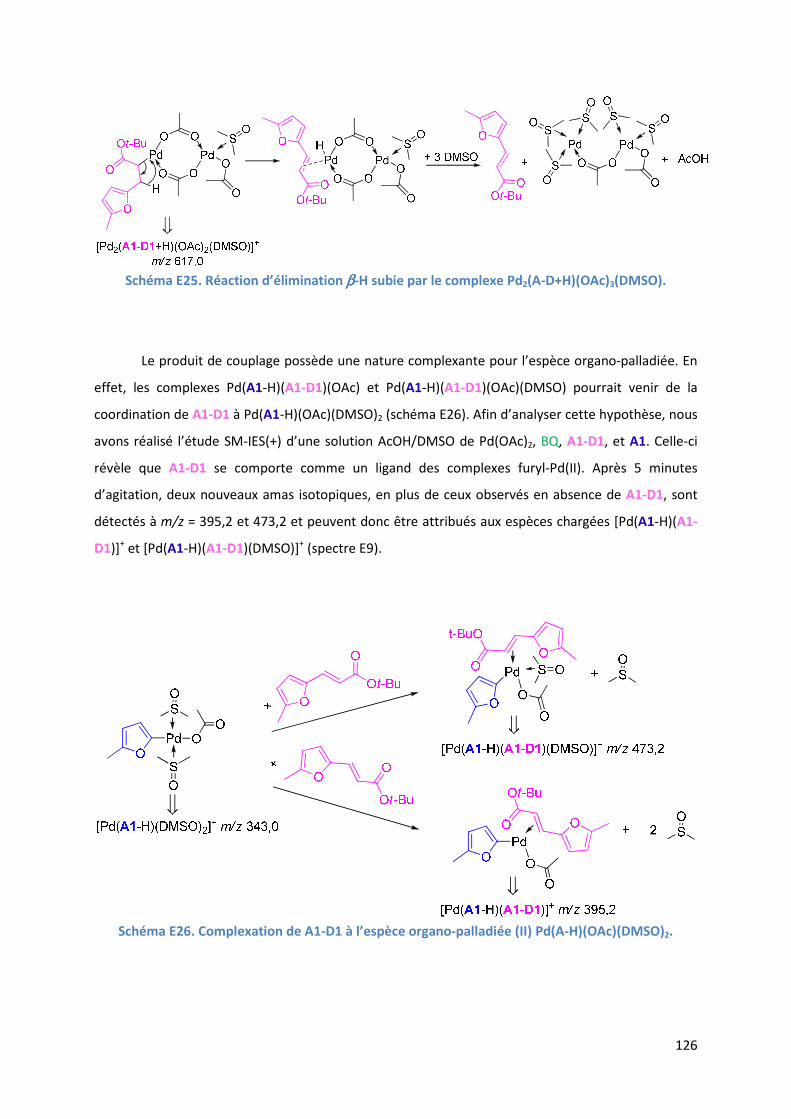
126
Schéma E25. Réaction d’élimination ββββ-H subie par le complexe Pd2(A-D+H)(OAc)3(DMSO).
Le produit de couplage possède une nature complexante pour l’espèce organo-palladiée. En
effet, les complexes Pd(A1-H)(A1-D1)(OAc) et Pd(A1-H)(A1-D1)(OAc)(DMSO) pourrait venir de la
coordination de A1-D1 à Pd(A1-H)(OAc)(DMSO)2 (schéma E26). Afin d’analyser cette hypothèse, nous
avons réalisé l’étude SM-IES(+) d’une solution AcOH/DMSO de Pd(OAc)2, BQ, A1-D1, et A1. Celle-ci
révèle que A1-D1 se comporte comme un ligand des complexes furyl-Pd(II). Après 5 minutes
d’agitation, deux nouveaux amas isotopiques, en plus de ceux observés en absence de A1-D1, sont
détectés à m/z = 395,2 et 473,2 et peuvent donc être attribués aux espèces chargées [Pd(A1-H)(A1-
D1)]+ et [Pd(A1-H)(A1-D1)(DMSO)]
+ (spectre E9).
Schéma E26. Complexation de A1-D1 à l’espèce organo-palladiée (II) Pd(A-H)(OAc)(DMSO)2.
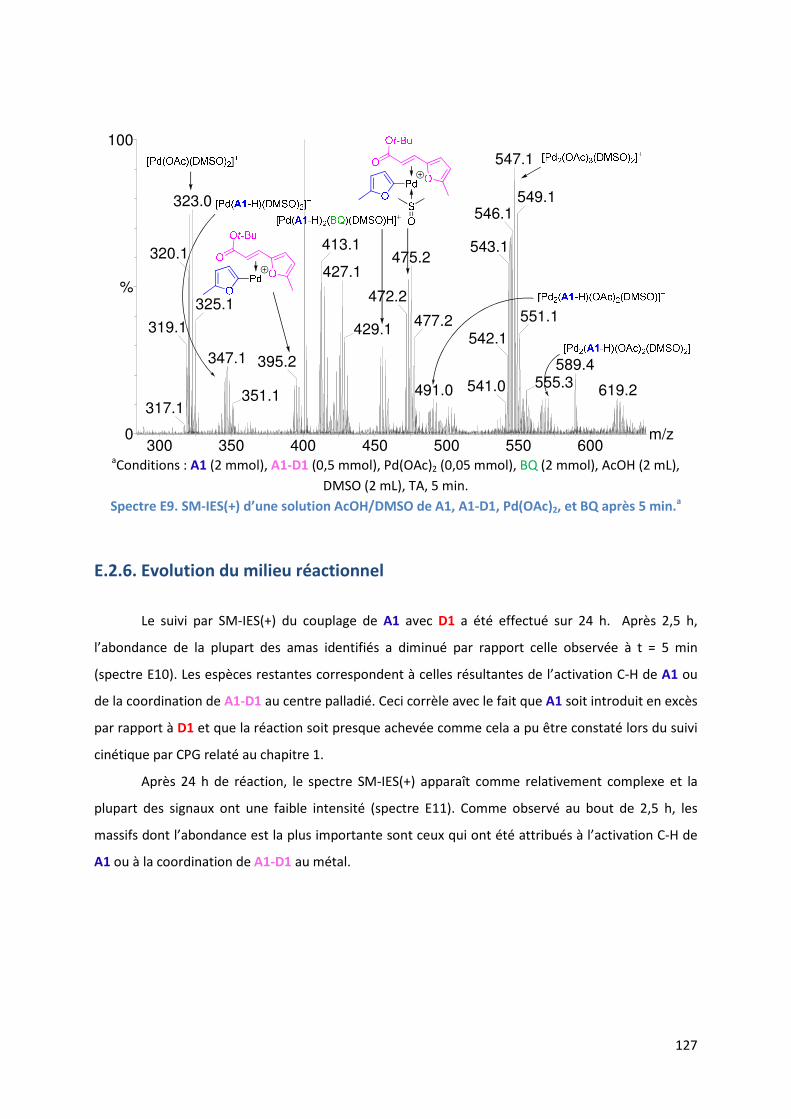
127
AV 740
300 350 400 450 500 550 600m/z0
100
%
12BR70A 123 (2.281) Cm (108:123) 1: TOF MS ES+ 266x2
323.0
320.1
319.1
317.1
325.1
347.1 395.2
351.1
547.1
546.1
413.1 543.1475.2
427.1
472.2
429.1477.2
542.1
491.0 541.0
549.1
551.1
589.4555.3
619.2
aConditions : A1 (2 mmol), A1-D1 (0,5 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL),
DMSO (2 mL), TA, 5 min.
Spectre E9. SM-IES(+) d’une solution AcOH/DMSO de A1, A1-D1, Pd(OAc)2, et BQ après 5 min.a
E.2.6. Evolution du milieu réactionnel
Le suivi par SM-IES(+) du couplage de A1 avec D1 a été effectué sur 24 h. Après 2,5 h,
l’abondance de la plupart des amas identifiés a diminué par rapport celle observée à t = 5 min
(spectre E10). Les espèces restantes correspondent à celles résultantes de l’activation C-H de A1 ou
de la coordination de A1-D1 au centre palladié. Ceci corrèle avec le fait que A1 soit introduit en excès
par rapport à D1 et que la réaction soit presque achevée comme cela a pu être constaté lors du suivi
cinétique par CPG relaté au chapitre 1.
Après 24 h de réaction, le spectre SM-IES(+) apparaît comme relativement complexe et la
plupart des signaux ont une faible intensité (spectre E11). Comme observé au bout de 2,5 h, les
massifs dont l’abondance est la plus importante sont ceux qui ont été attribués à l’activation C-H de
A1 ou à la coordination de A1-D1 au métal.
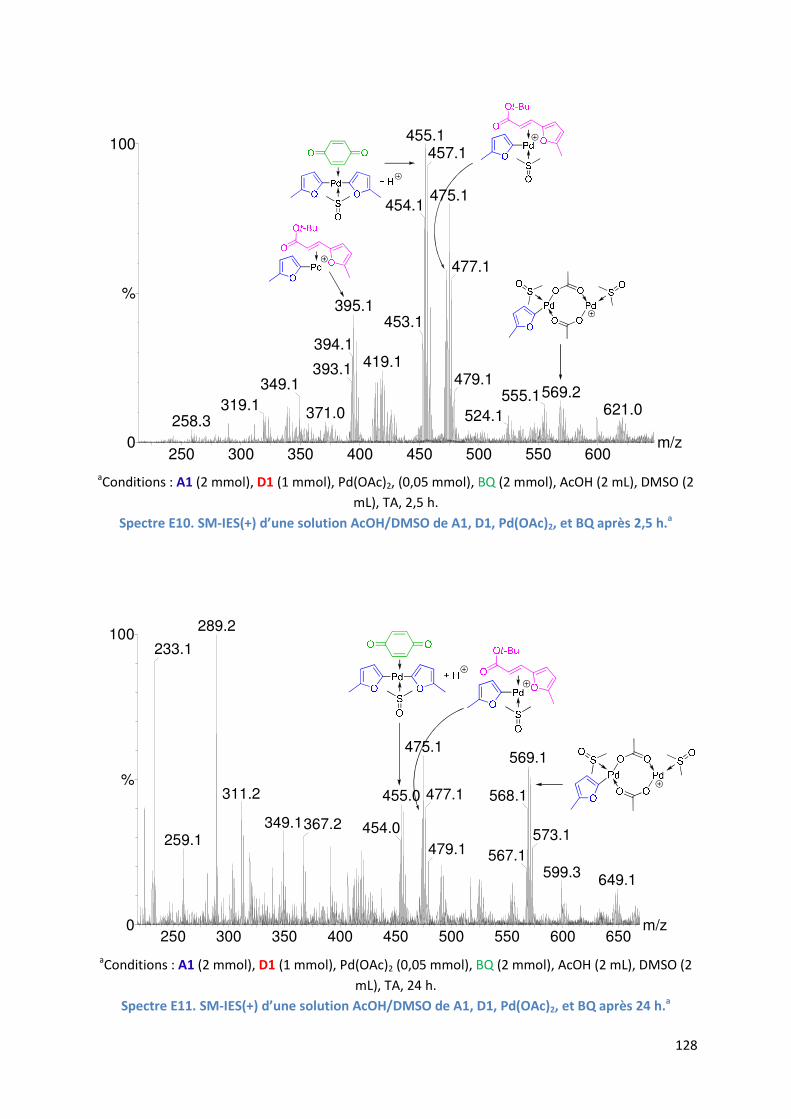
128
AV 688
250 300 350 400 450 500 550 600m/z0
100
%
11HR746 81 (2.537) Cm (14:135) 1: TOF MS ES+ 1.02e3455.1
454.1
395.1
394.1
393.1349.1
319.1258.3
371.0
453.1
419.1
457.1
475.1
477.1
479.1569.2555.1
524.1 621.0
aConditions : A1 (2 mmol), D1 (1 mmol), Pd(OAc)2, (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2
mL), TA, 2,5 h.
Spectre E10. SM-IES(+) d’une solution AcOH/DMSO de A1, D1, Pd(OAc)2, et BQ après 2,5 h.a
AV 666
250 300 350 400 450 500 550 600 650m/z0
100
%
11BR1074 158 (2.937) Cm (106:159) 1: TOF MS ES+ 452289.2
233.1
259.1
475.1
311.2 455.0
349.1367.2 454.0
569.1
477.1 568.1
479.1 567.1
573.1
599.3649.1
aConditions : A1 (2 mmol), D1 (1 mmol), Pd(OAc)2 (0,05 mmol), BQ (2 mmol), AcOH (2 mL), DMSO (2
mL), TA, 24 h.
Spectre E11. SM-IES(+) d’une solution AcOH/DMSO de A1, D1, Pd(OAc)2, et BQ après 24 h.a

129
E.2.7. Cycles catalytiques
Les différents résultats et observations découlant de ces études SM-IES(+) permettent de
proposer les cycles catalytiques des schémas E27 et E28. Le trimère inactif [Pd(OAc)2]3 réagit avec le
DMSO pour donner des dérivés monomériques et dimériques I et I’ (schéma E27). I’ active une liaison
C-H du DMSO et de D1, tandis que I’ et I activent une liaison C-H de A1 pour donner les complexes
Pd-furyle. Après quelques roulements de cycle, le produit de couplage A1-D1 est présent dans le
milieu réactionnel et peut par conséquent agir comme ligand de l’intermédiaire II. L’insertion de D1
mène à l’intermédiaire III, lequel subit une élimination β-H pour donner l’intermédiaire IV. Une
élimination réductrice fournit ensuite le complexe Pd(0) V ; une réoxydation de Pd(0) par BQ/AcOH
clôture le cycle.
Concernant la formation de A1-A1, le complexe II active la liaison C-H de A1 pour fournir
l’intermédiaire VI (schéma E28). Ce dernier, riche en électrons coordonne la benzoquinone pour
donner l’intermédiaire VII, lequel après une élimination réductrice libère le produit de couplage A1-
A1 et le Pd(0). Le Pd(0) est ensuite réoxydé par l’association BQ/AcOH.
Schéma E27. Cycle catalytique pour la RHD de A1 avec D1.

130
Schéma E28. Cycle catalytique pour l’homocouplage déshydrogénant de A1.
E.3. Conclusion
Les propositions mécanistiques concernant les RHD impliquent généralement des
intermédiaires mononucléaires. En faisant appel à la SM-IES, nous avons montré que les espèces
dinucléaires peuvent jouer un rôle. Les intermédiaires mononucléaires et dinucléaires furyl-Pd(II)
obtenus par activation de liaisons C-H, ainsi que des complexes résultant de l’insertion de l’alcène
dans la liaison furyl-Pd(II) ont été interceptés et caractérisés.
Des réactions secondaires, telles que l’activation d’une des liaisons C-H du DMSO ou encore
de l’alcène ont été identifiées. Enfin, un intermédiaire impliqué dans l’homocouplage de furanes via
une activation C-H a été également détecté et caractérisé. De plus, la coordination de la
benzoquinone au palladium a été mise en évidence pour la première fois par SM-IES.

131
E.4. Partie expérimentale
E.4.1. Dispositif expérimental
Toute les expériences (SM, SMHR et SM/SM) ont été réalisées avec un spectromètre tandem
Quadripôle-Temps-de-vol (Q-Tdv micro Waters « Micromass » Manchester UK), équipé d’une source
d’ionisation électrospray (Z-Spray), la désolvatation des ions est assistée par l’arrivée à contre
courant d’un gaz de séchage (flux d’azote) chauffé à une température de 50 à 80 °C. La tension du
cône d’extraction variait entre 20 et 40 V, la tension du capillaire IES est comprise entre 3000 et 3500
V. La dissociation induite par collision (DIC) a été réalisée par introduction d’argon dans la cellule de
collision.
L’injection des échantillons a été réalisée à un débit de 3 à 6 µL/min, la quantité d’échantillon
dont nous disposions n’imposait pas de travailler à des débits plus faibles.
E.4.2. Mode opératoire
Nous avons suivi le mode opératoire mis au point dans le chapitre 1. L’acétate de palladium
(11,2 mg, 0,05 mmol) et la benzoquinone (216 mg, 2 mmol) sont mis en solution dans un mélange
AcOH (2 mL)/DMSO (2 mL). Après ajout de l’arène A (2 mmol) et de l’alcène D (1 mmol), le mélange
est agité à température ambiante. Le prélèvement du mélange réactionnel se fait directement à
l’aide de la seringue utilisée pour l’injection dans le spectromètre de masse. Le brut de réaction est
ainsi analysé sans modification.
E.4.3. SMHR des espèces observées
Le 106
Pd a été utilisé pour la détermination des masses exactes.
[Pd(OAc)(DMSO)]+ : C4H9O3PdS, théorique : 242,9307 ; obtenue : 242,9308.
[Pd(DMSO-H)(DMSO)]+ : C4H11O2PdS2, théorique : 260,9235 ; obtenue : 260,9235.
[Pd(OAc)(DMSO)2]+ : C6H15O4PdS2, théorique : 320,9447 ; obtenue : 320,9451.
[Pd2(OAc)3(DMSO)]+ : C8H15O7Pd2S, théorique : 466,8608 ; obtenue : 466,8600.
[Pd2(OAc)3(DMSO)2]+ : C10H21O8Pd2S2, théorique : 544,8747 ; obtenue : 544,8739.
[Pd(A1-H)(DMSO)2]+ : C9H17O3PdS2, théorique : 342,9654 ; obtenue : 342,9657.

132
[Pd(A1)(DMSO)2H]+ : C9H19O3PdS2, théorique : 344,9810 ; obtenue : 344,9806.
[Pd(A1-H)2(BQ)(DMSO)H]+ : C18H21O5PdS, théorique : 455,0144 ; obtenue : 455,0142.
[Pd2(A1-H)(OAc)2(DMSO)]+ : C11H17O6Pd2S, théorique : 488,8815 ; obtenue : 488,8810.
[Pd2(A1-H)(OAc)2(DMSO)2]+ : C13H23O7Pd2S2, théorique : 566,8955 ; obtenue : 566,8951.
[Pd(A2-H)(DMSO)(Me2SO2)]+ : C10H19O4PdS2, théorique : 372,9760 ; obtenue : 372,9746.
[Pd(A2-H)(DMSO)2(Me2SO2)]+ : C12H25O5PdS3, théorique : 450,9899 ; obtenue : 450,9908.
[Pd(A2-H)2(2-ClBQ)(DMSO)H]+ : C20H24ClO5PdS, théorique : 517,0068 ; obtenue : 517,0079.
[Pd(D1-H)(OAc)(DMSO)H]+ : C11H21O5PdS, théorique : 371,0139 ; obtenue : 371,0140.
[Pd(A1-D1+H)(DMSO)]+, C14H23O4PdS, théorique : 393,0352 ; obtenue : 393,0347.
[Pd(A1-H)(A1-D1)]+ : C17H21O4Pd, théorique : 395,0475 ; obtenue : 395,0473.
[Pd(A1-H)(A1-D1)(DMSO)]+ : C19H27O5PdS, théorique : 473,0609 ; obtenue : 473,0598.
[Pd2(A1-D1+H)(OAc)2(DMSO)]+ or [Pd2(A1-H)(D1-H)(OAc)2(DMSO)H]
+, C18H29O8Pd2S : théorique :
616,9647 ; obtenue : 616,9646.
E.4.4. Agrandissement des spectres présentées en partie E
E.4.4.1. Agrandissement du spectre E1 entre m/z = 200-400 et m/z = 400-600.
AV 667
200 220 240 260 280 300 320 340 360 380 400m/z0
100
%
11BR1075 120 (2.230) Cm (99:128) 1: TOF MS ES+ 4.52e4x2 320.9
320.0
319.0
262.9259.9
242.9264.9
325.0
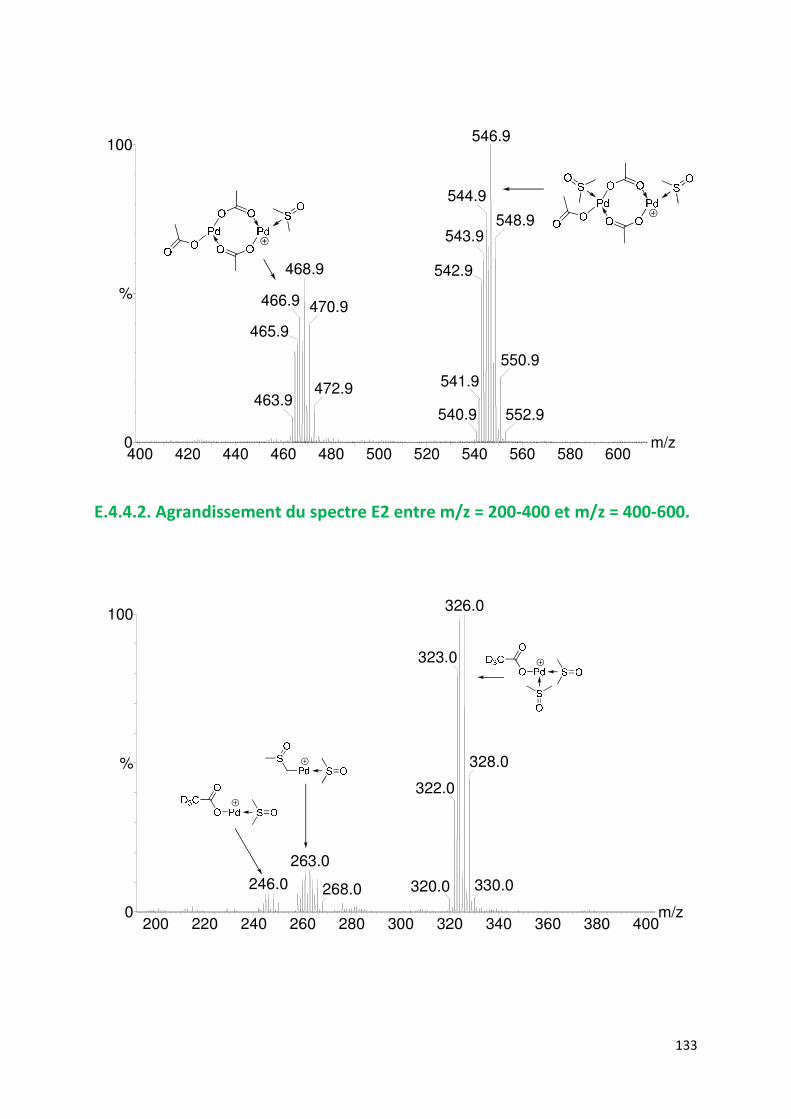
133
AV 667
400 420 440 460 480 500 520 540 560 580 600m/z0
100
%
11BR1075 120 (2.230) Cm (99:128) 1: TOF MS ES+ 1.46e4546.9
544.9
543.9
468.9
466.9
465.9
463.9
542.9
470.9
541.9472.9
540.9
548.9
550.9
552.9
E.4.4.2. Agrandissement du spectre E2 entre m/z = 200-400 et m/z = 400-600.
AV 761
200 220 240 260 280 300 320 340 360 380 400m/z0
100
%
12HR81 94 (2.964) Cm (1:166) 1: TOF MS ES+ 2.02e3326.0
323.0
322.0
263.0
246.0 320.0268.0
328.0
330.0
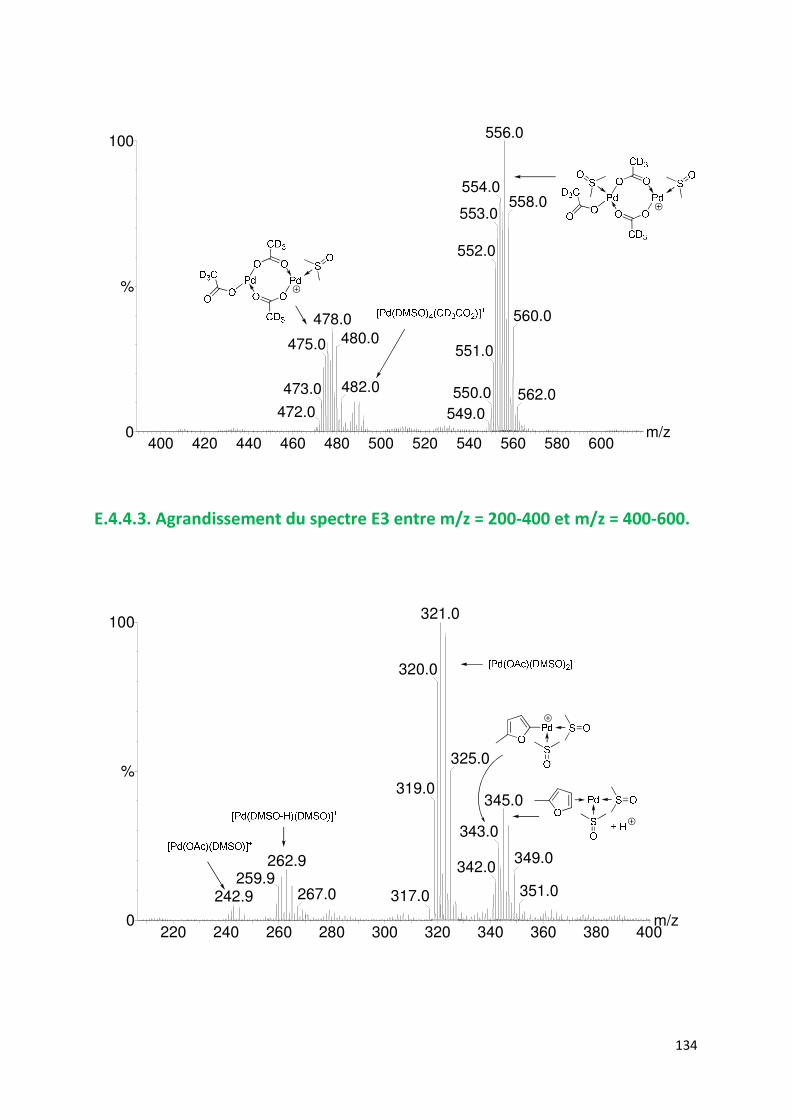
134
AV 761
400 420 440 460 480 500 520 540 560 580 600m/z0
100
%
12HR81 94 (2.964) Cm (1:166) 1: TOF MS ES+ 1.25e3556.0
554.0
553.0
552.0
478.0
475.0
473.0
472.0
480.0551.0
482.0 550.0
549.0
558.0
560.0
562.0
E.4.4.3. Agrandissement du spectre E3 entre m/z = 200-400 et m/z = 400-600.
AV 667
220 240 260 280 300 320 340 360 380 400m/z0
100
%
11BR1077 1 (0.019) Cm (1:40) 1: TOF MS ES+ 2.79e4321.0
320.0
319.0
262.9259.9
242.9 267.0 317.0
325.0
345.0
343.0
342.0349.0
351.0
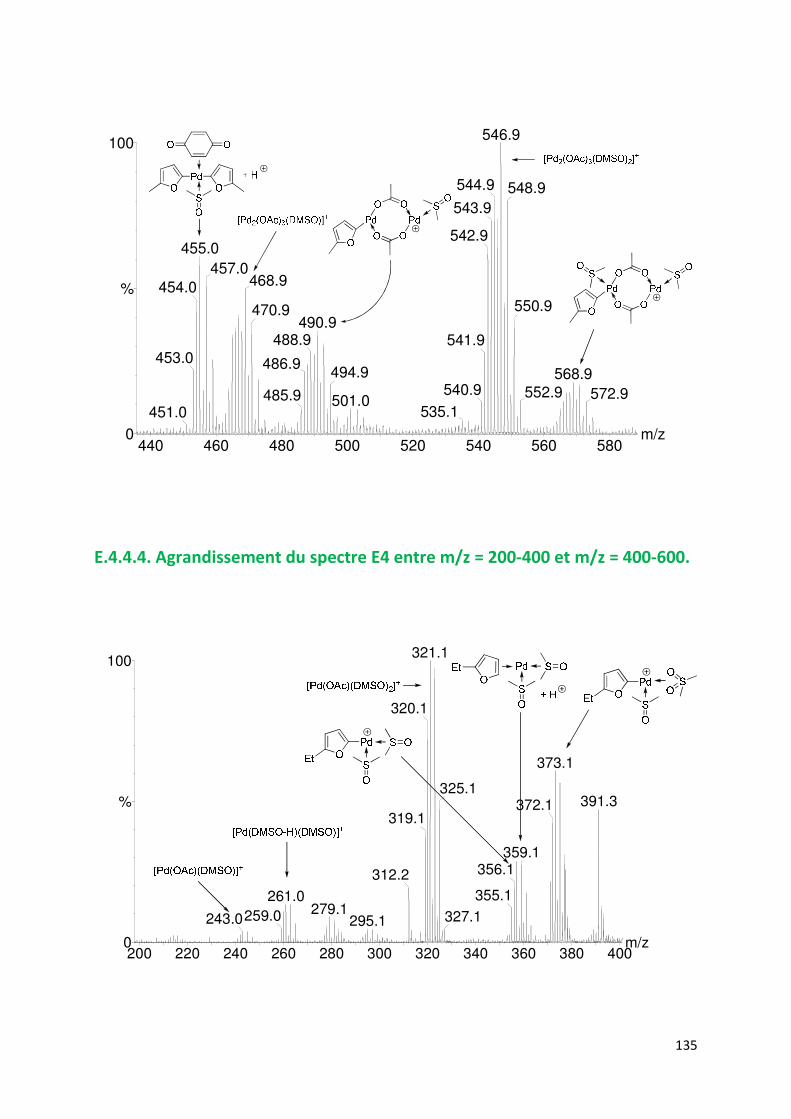
135
AV 667
440 460 480 500 520 540 560 580m/z0
100
%
11BR1077 1 (0.019) Cm (1:40) 1: TOF MS ES+ 9.91e3546.9
544.9
543.9
542.9455.0
454.0
453.0
451.0
457.0468.9
470.9490.9
488.9
486.9
485.9
541.9
494.9540.9
501.0535.1
548.9
550.9
568.9
552.9 572.9
E.4.4.4. Agrandissement du spectre E4 entre m/z = 200-400 et m/z = 400-600.
AV 736
200 220 240 260 280 300 320 340 360 380 400m/z0
100
%
12BR63 4 (0.075) Cm (1:112) 1: TOF MS ES+ 3.14e4321.1
320.1
319.1
312.2
261.0
259.0243.0279.1
295.1
373.1
325.1372.1
359.1356.1
355.1
327.1
391.3
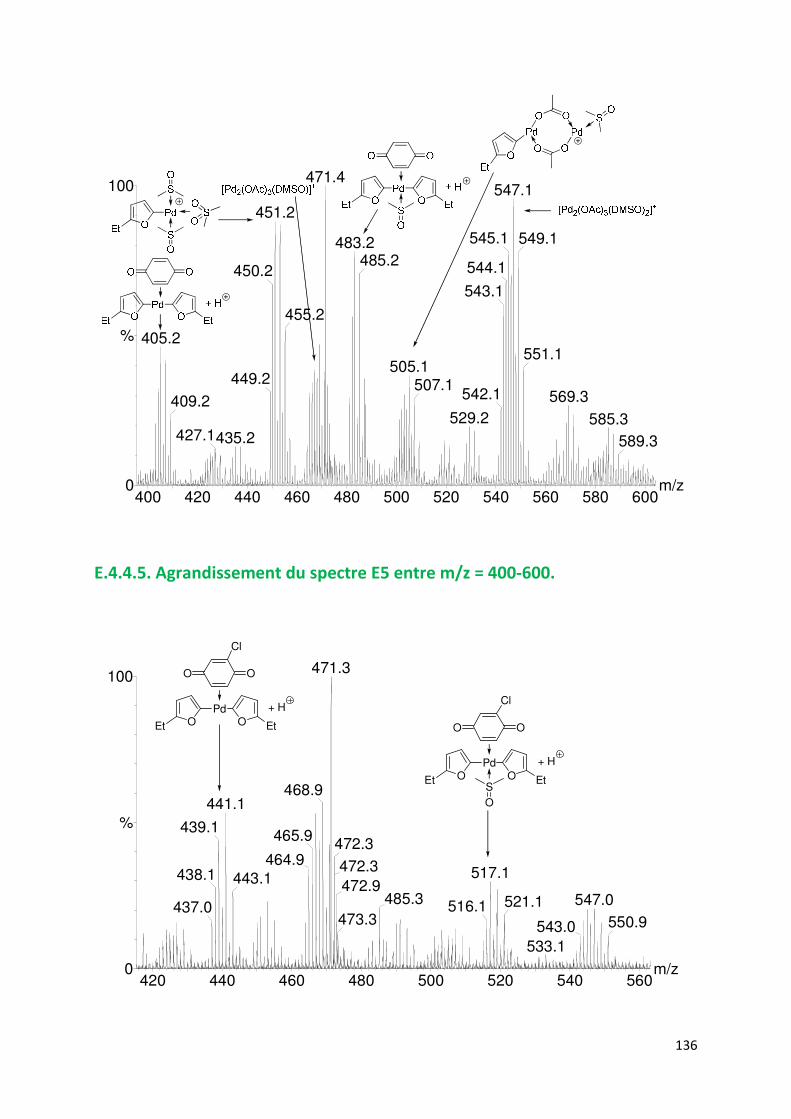
136
AV 736
400 420 440 460 480 500 520 540 560 580 600m/z0
100
%
12BR63 4 (0.075) Cm (1:112) 1: TOF MS ES+ 1.19e4471.4
451.2
450.2
405.2
449.2
409.2
435.2427.1
455.2
547.1
483.2 545.1
485.2 544.1
543.1
505.1507.1
542.1
529.2
549.1
551.1
569.3
585.3
589.3
E.4.4.5. Agrandissement du spectre E5 entre m/z = 400-600.
AV 759
420 440 460 480 500 520 540 560m/z0
100
%
12BR136 95 (1.770) Cm (86:99) 1: TOF MS ES+ 314471.3
468.9441.1
439.1
438.1
437.0
465.9
464.9
443.1
472.3
472.3 517.1472.9
485.3
473.3516.1 547.0521.1
543.0
533.1
550.9
PdO OEt Et
OO
+ H
Cl
Pd
S
O
O OEt Et
OO
+ H
Cl
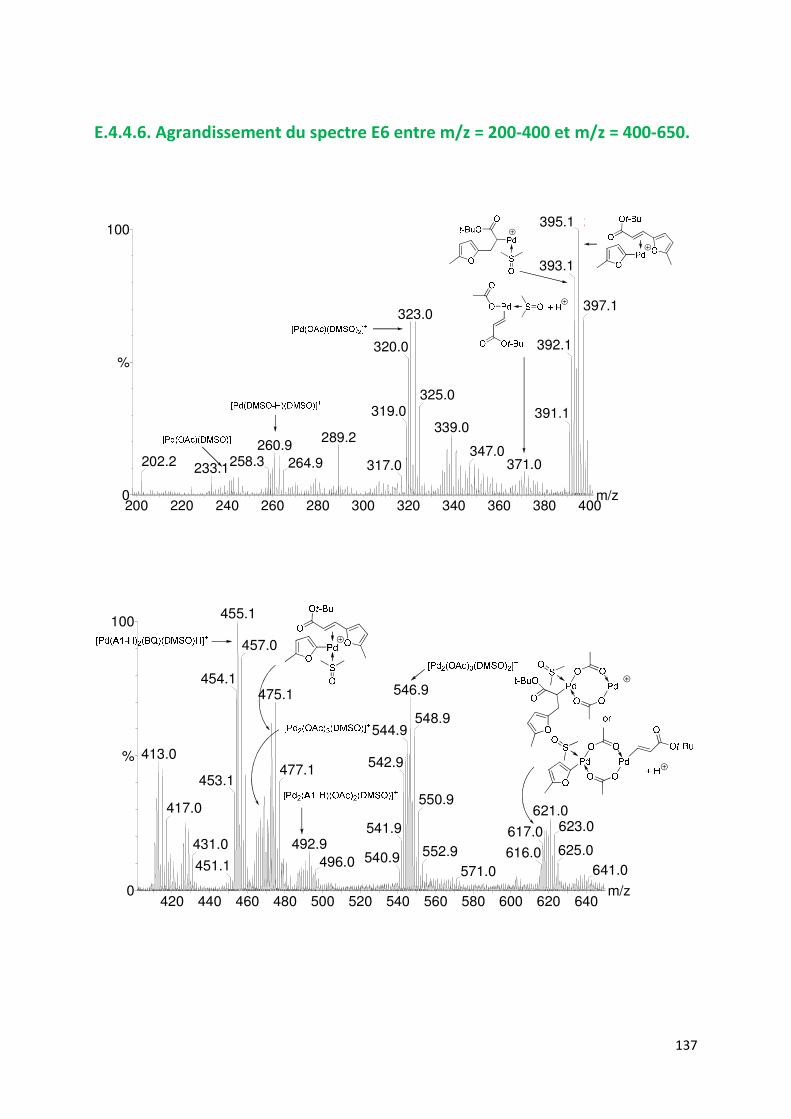
137
E.4.4.6. Agrandissement du spectre E6 entre m/z = 200-400 et m/z = 400-650.
AV 688
200 220 240 260 280 300 320 340 360 380 400m/z0
100
%
11HR743 93 (2.917) Cm (93:147) 1: TOF MS ES+ 2.88e3395.1
393.1
323.0
320.0
319.0
289.2260.9
258.3202.2233.1 264.9 317.0
392.1
325.0
391.1339.0
347.0371.0
397.1
AV 688
420 440 460 480 500 520 540 560 580 600 620 640m/z0
100
%
11HR743 93 (2.917) Cm (93:147) 1: TOF MS ES+ 2.50e3455.1
454.1
413.0
453.1
417.0
431.0
451.1
457.0
546.9475.1
544.9
542.9477.1
541.9492.9
540.9496.0
548.9
550.9621.0
617.0
552.9 616.0
571.0
623.0
625.0
641.0
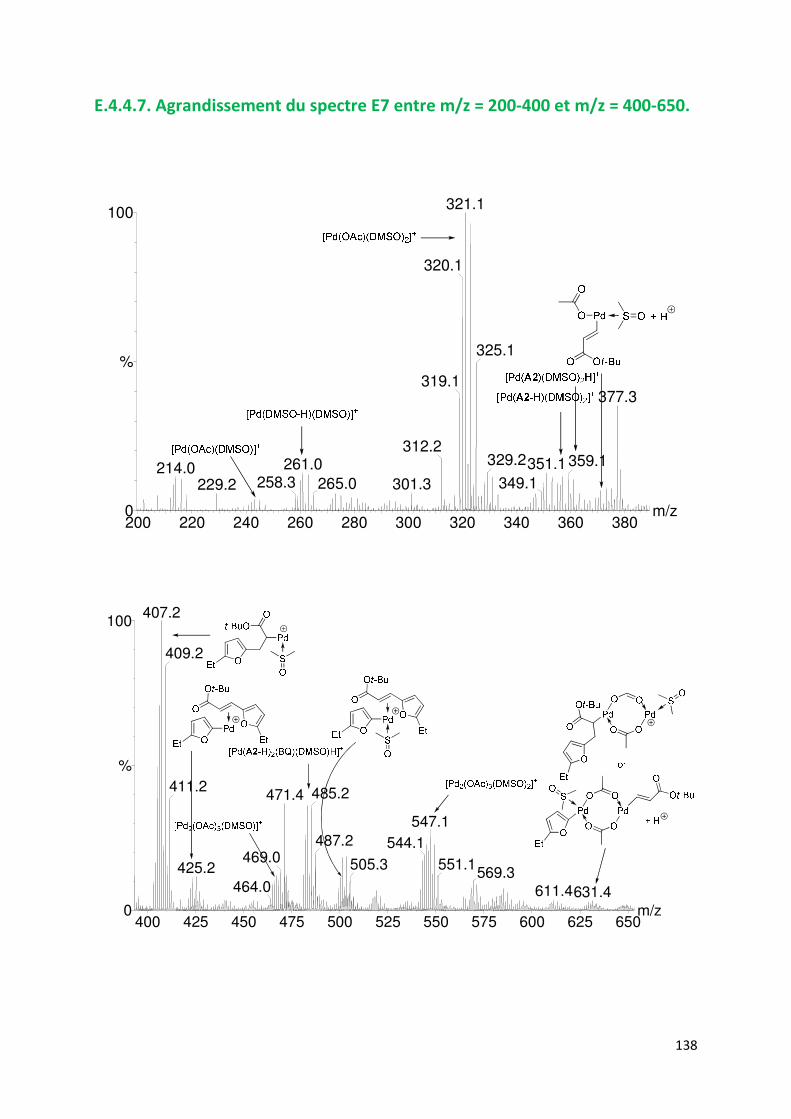
138
E.4.4.7. Agrandissement du spectre E7 entre m/z = 200-400 et m/z = 400-650.
AV 736
200 220 240 260 280 300 320 340 360 380m/z0
100
%
12BR65 224 (4.174) Cm (163:289) 1: TOF MS ES+ 1.95e4321.1
320.1
319.1
312.2261.0214.0
229.2 258.3 265.0 301.3
325.1
377.3
351.1329.2
349.1
359.1
AV 736
400 425 450 475 500 525 550 575 600 625 650m/z0
100
%
12BR65 224 (4.174) Cm (163:289) 1: TOF MS ES+ 2.40e4407.2
409.2
411.2471.4
469.0425.2
464.0
485.2
547.1
487.2 544.1
505.3 551.1569.3
611.4631.4
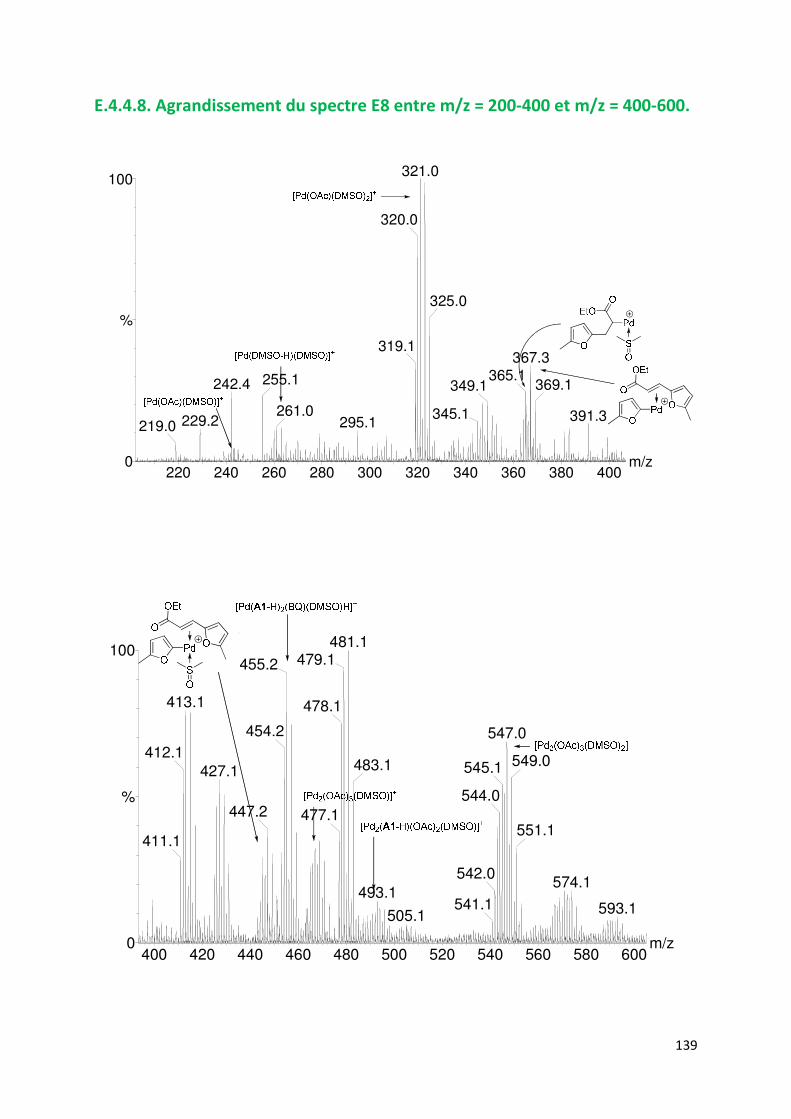
139
E.4.4.8. Agrandissement du spectre E8 entre m/z = 200-400 et m/z = 400-600.
AV 737
220 240 260 280 300 320 340 360 380 400m/z0
100
%
12BR67 196 (3.649) Cm (195:283) 1: TOF MS ES+ 2.38e3321.0
320.0
319.1
242.4
229.2219.0
255.1
261.0295.1
325.0
367.3365.1
349.1
345.1
369.1
391.3
AV 737
400 420 440 460 480 500 520 540 560 580 600m/z0
100
%
12BR67 196 (3.649) Cm (195:283) 1: TOF MS ES+ 1.37e3x2 481.1
479.1455.2
413.1
412.1
411.1
454.2
427.1
447.2
478.1
477.1
547.0
483.1 545.1
544.0
542.0
493.1541.1
505.1
549.0
551.1
574.1
593.1
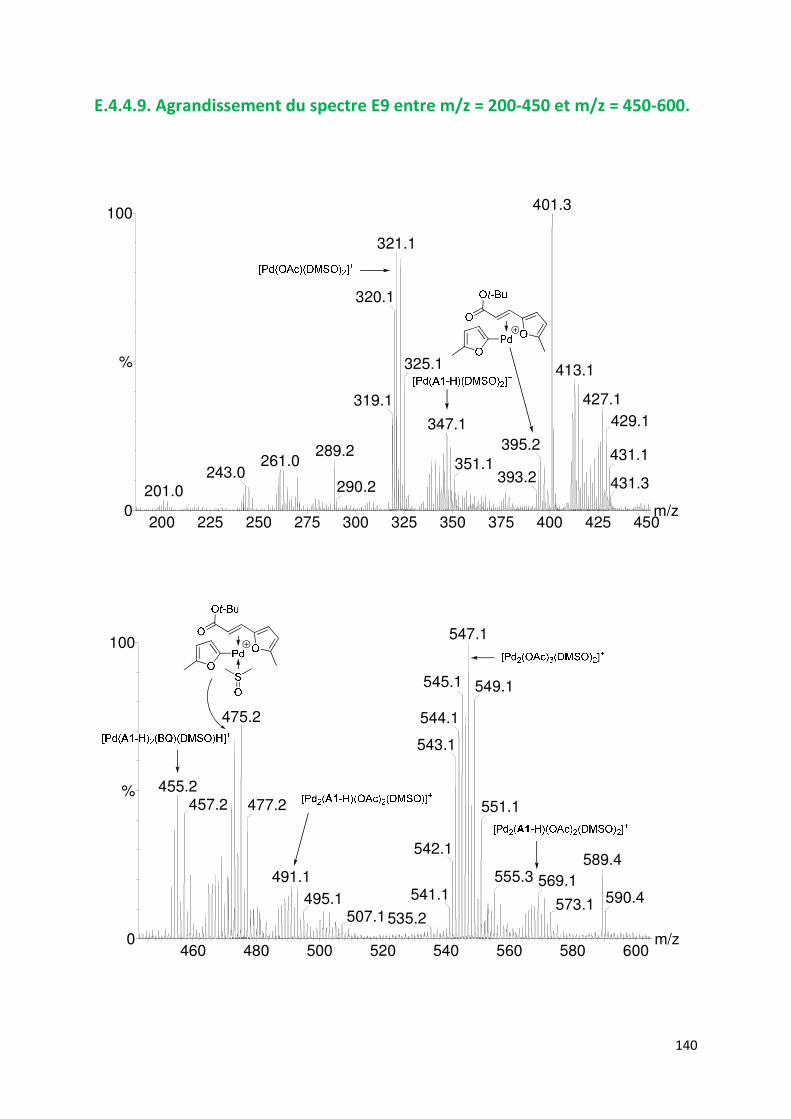
140
E.4.4.9. Agrandissement du spectre E9 entre m/z = 200-450 et m/z = 450-600.
AV 740
200 225 250 275 300 325 350 375 400 425 450m/z0
100
%
12BR70A 203 (3.768) Cm (1:237) 1: TOF MS ES+ 7.12e3401.3
321.1
320.1
319.1
289.2261.0
243.0201.0 290.2
325.1
347.1
395.2
351.1393.2
413.1
427.1
429.1
431.1
431.3
AV 740
460 480 500 520 540 560 580 600m/z0
100
%
12BR70A 203 (3.768) Cm (1:237) 1: TOF MS ES+ 4.79e3547.1
545.1
475.2
455.2457.2
544.1
543.1
477.2
542.1
491.1541.1495.1
507.1535.2
549.1
551.1
589.4555.3 569.1
573.1 590.4
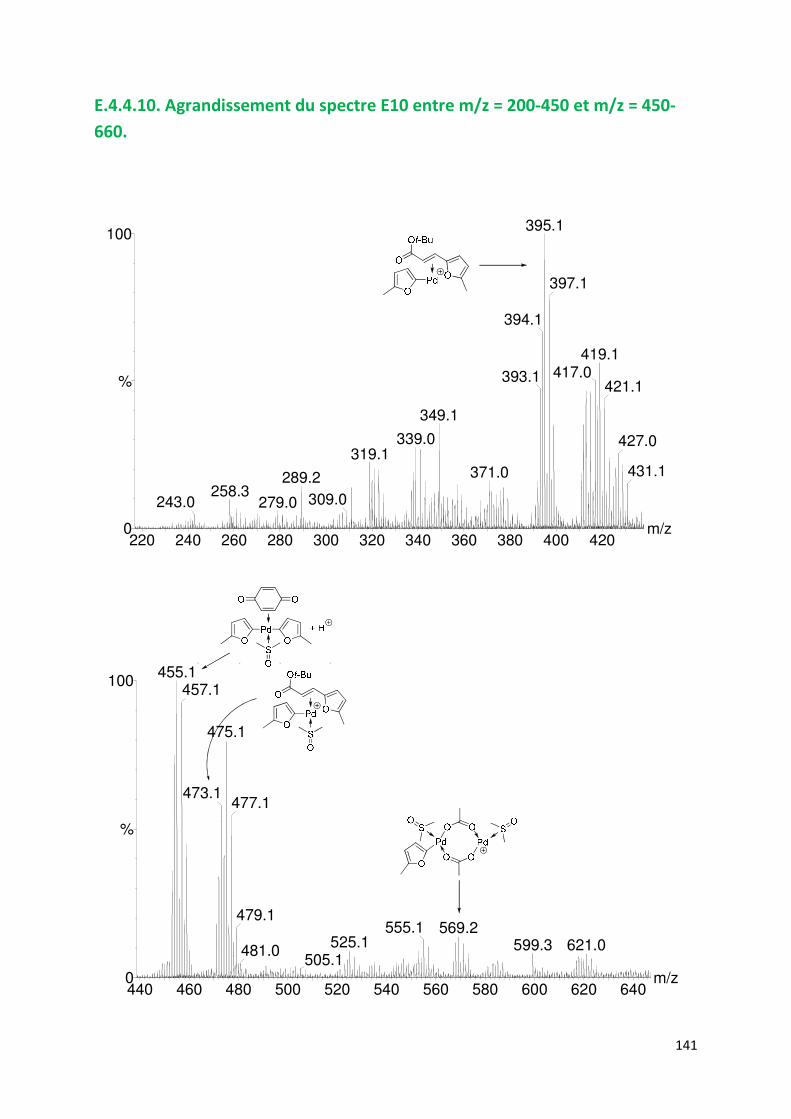
141
E.4.4.10. Agrandissement du spectre E10 entre m/z = 200-450 et m/z = 450-
660.
AV 688
220 240 260 280 300 320 340 360 380 400 420m/z0
100
%
11HR746 81 (2.537) Cm (14:135) 1: TOF MS ES+ 439395.1
394.1
393.1
349.1
339.0319.1
289.2258.3
243.0 279.0 309.0
371.0
397.1
419.1417.0
421.1
427.0
431.1
AV 688
440 460 480 500 520 540 560 580 600 620 640m/z0
100
%
11HR746 81 (2.537) Cm (14:135) 1: TOF MS ES+ 1.02e3455.1
457.1
475.1
473.1477.1
479.1569.2555.1
525.1481.0
505.1621.0599.3
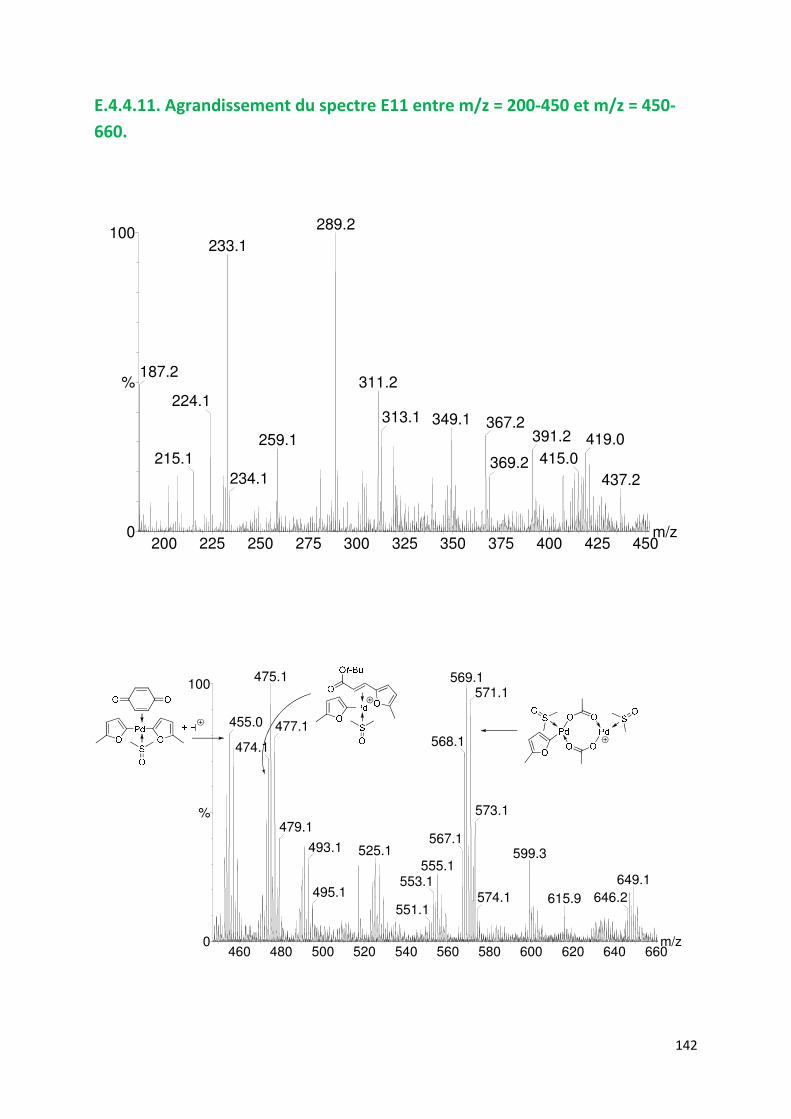
142
E.4.4.11. Agrandissement du spectre E11 entre m/z = 200-450 et m/z = 450-
660.
AV 666
200 225 250 275 300 325 350 375 400 425 450m/z0
100
%
11BR1074 158 (2.937) Cm (71:159) 1: TOF MS ES+ 607289.2
233.1
187.2
224.1
215.1259.1
234.1
311.2
349.1313.1 367.2391.2
369.2
419.0
415.0
437.2
AV 666
460 480 500 520 540 560 580 600 620 640 660m/z0
100
%
11BR1074 158 (2.937) Cm (71:159) 1: TOF MS ES+ 355475.1
455.0
474.1
569.1
477.1568.1
479.1567.1
525.1493.1
495.1
555.1553.1
551.1
571.1
573.1
599.3
574.1
649.1
615.9 646.2
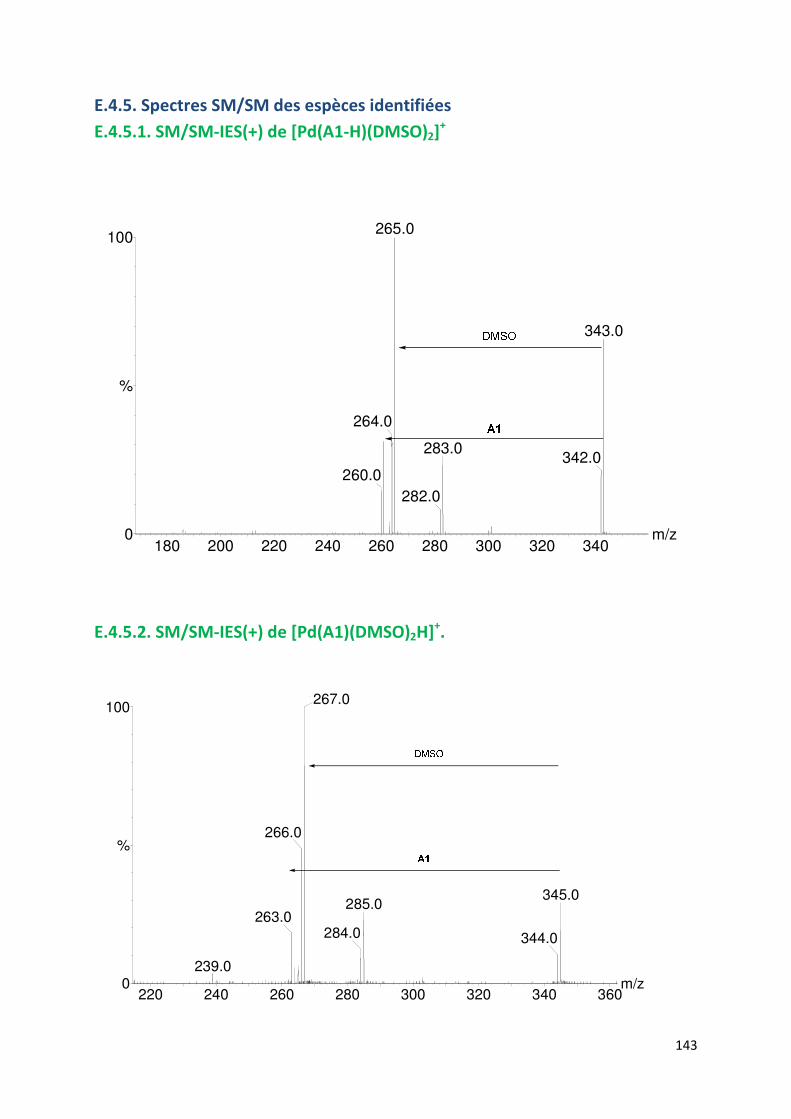
143
E.4.5. Spectres SM/SM des espèces identifiées
E.4.5.1. SM/SM-IES(+) de [Pd(A1-H)(DMSO)2]+
AV 446
180 200 220 240 260 280 300 320 340m/z0
100
%
11BR248 157 (2.929) Cm (60:299) 1: TOF MSMS 343.00ES+ 756265.0
264.0
260.0
343.0
283.0
282.0
342.0
E.4.5.2. SM/SM-IES(+) de [Pd(A1)(DMSO)2H]+.
AV 852
220 240 260 280 300 320 340 360m/z0
100
%
12BR226 64 (2.251) Cm (64:82) 1: TOF MSMS 345.00ES+ 209x2 267.0
266.0
263.0
239.0
345.0285.0
284.0 344.0
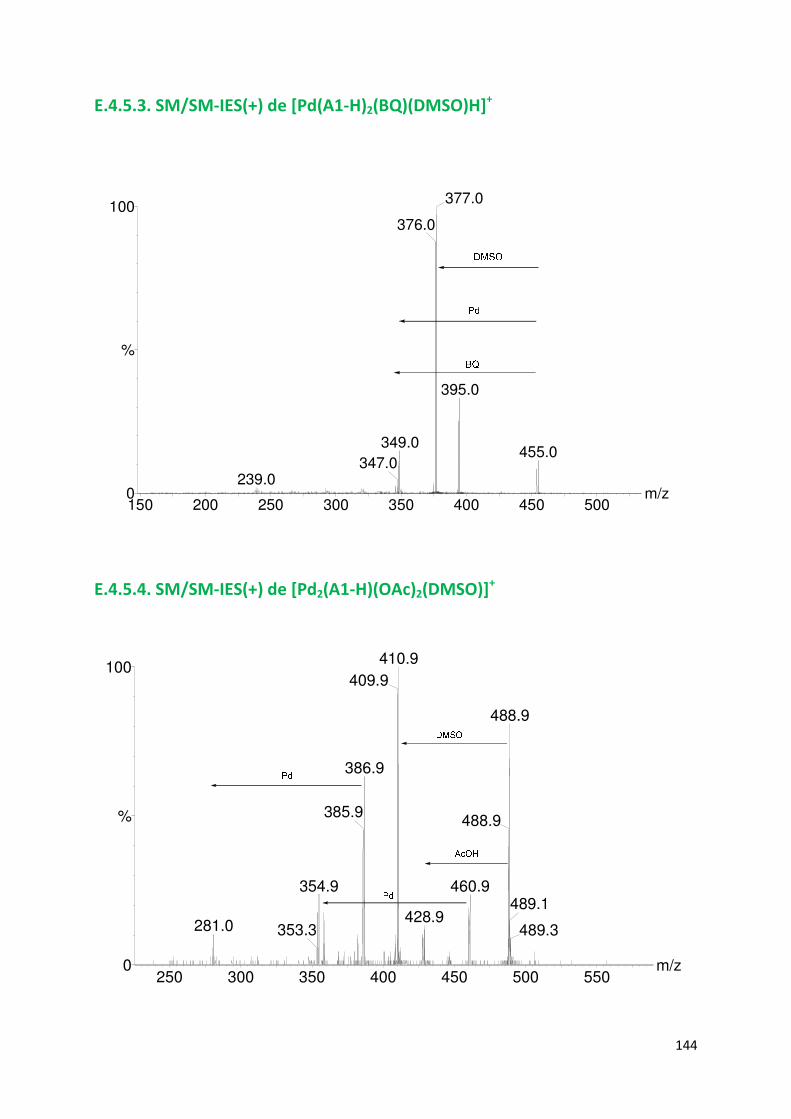
144
E.4.5.3. SM/SM-IES(+) de [Pd(A1-H)2(BQ)(DMSO)H]+
AV 667
150 200 250 300 350 400 450 500m/z0
100
%
11BR1079 92 (1.717) Cm (79:92) 1: TOF MSMS 455.00ES+ 1.02e3x4 377.0
376.0
349.0
347.0239.0
395.0
455.0
E.4.5.4. SM/SM-IES(+) de [Pd2(A1-H)(OAc)2(DMSO)]+
AV 734
250 300 350 400 450 500 550m/z0
100
%
12BR60 66 (1.229) Cm (1:218) 1: TOF MSMS 488.90ES+ 68x4 410.9
409.9
386.9
385.9
354.9
281.0 353.3
488.9
488.9
460.9
428.9489.1
489.3
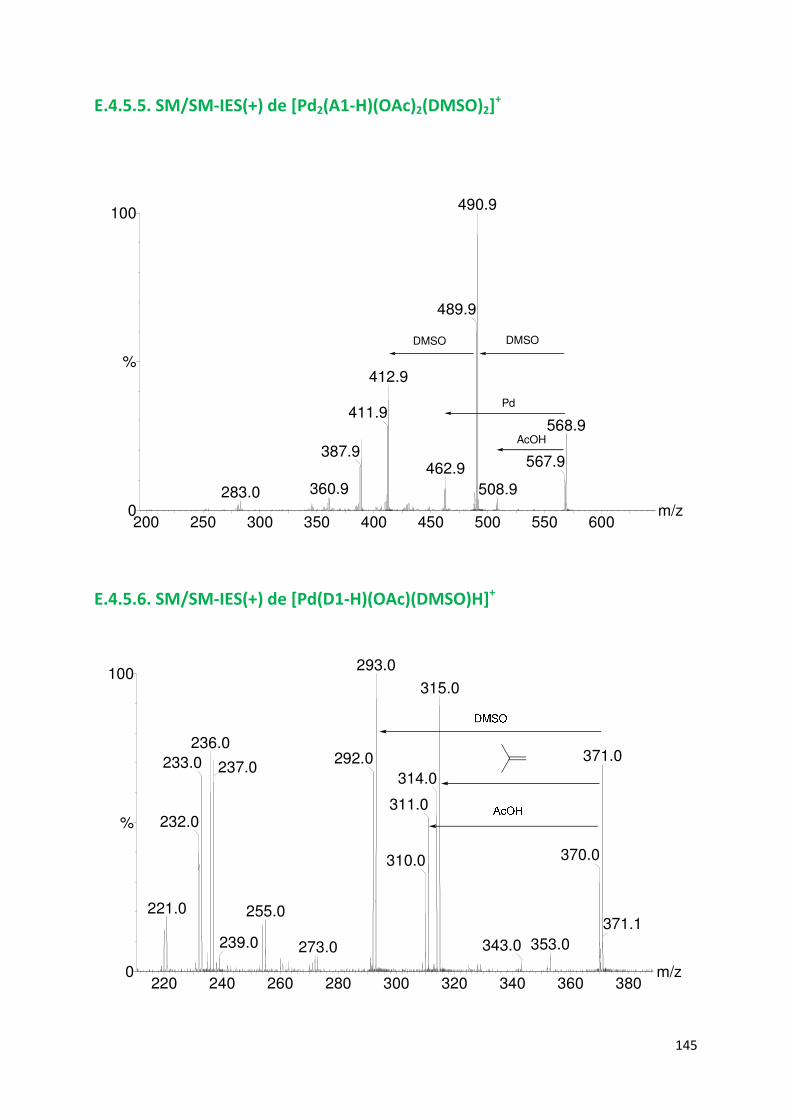
145
E.4.5.5. SM/SM-IES(+) de [Pd2(A1-H)(OAc)2(DMSO)2]+
AV 667
200 250 300 350 400 450 500 550 600m/z0
100
%
11BR1078 56 (1.047) Cm (56:160) 1: TOF MSMS 569.00ES+ 2.44e3490.9
489.9
412.9
411.9
387.9
360.9283.0
462.9
568.9
567.9
508.9
AcOH
DMSO
Pd
DMSO
E.4.5.6. SM/SM-IES(+) de [Pd(D1-H)(OAc)(DMSO)H]+
AV 668
220 240 260 280 300 320 340 360 380m/z0
100
%
11BR1086 40 (0.745) Cm (40:188) 1: TOF MSMS 371.00ES+ 207293.0
236.0
233.0
232.0
221.0
292.0237.0
255.0
239.0 273.0
315.0
314.0
311.0
310.0
371.0
370.0
353.0343.0
371.1
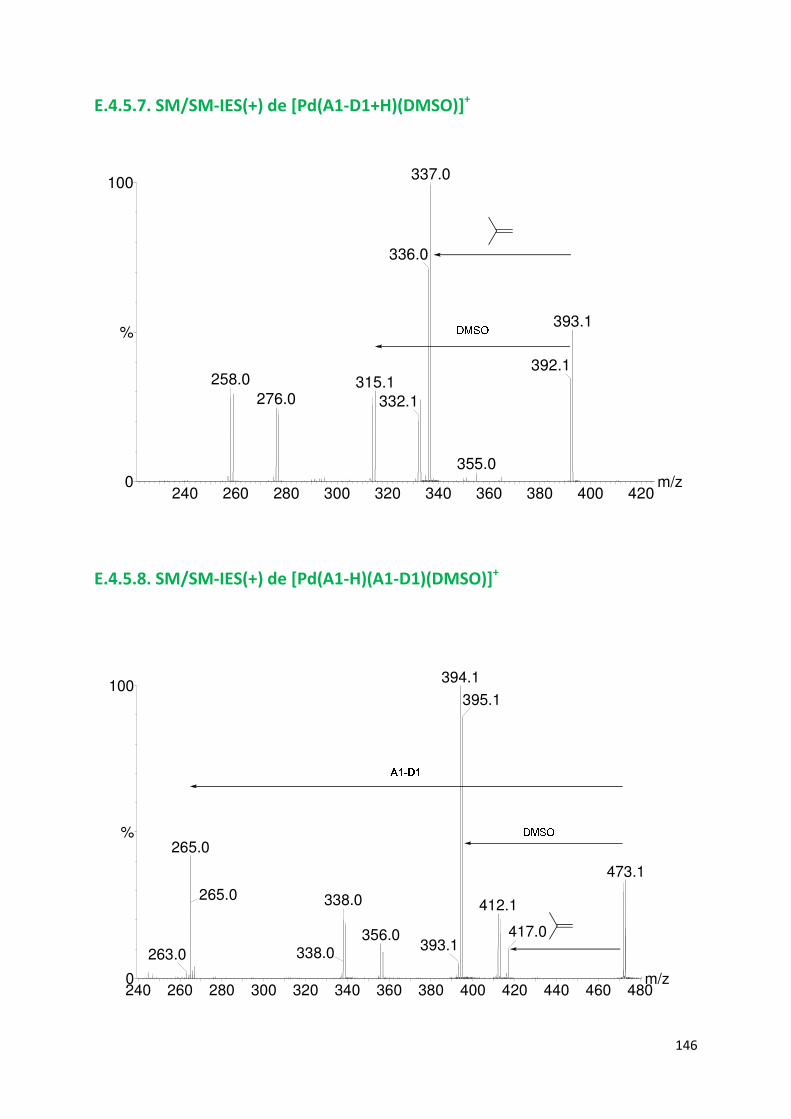
146
E.4.5.7. SM/SM-IES(+) de [Pd(A1-D1+H)(DMSO)]+
AV 668
240 260 280 300 320 340 360 380 400 420m/z0
100
%
11BR1085 106 (1.977) Cm (42:147) 1: TOF MSMS 393.00ES+ 2.27e3337.0
336.0
258.0 315.1276.0 332.1
393.1
392.1
355.0
E.4.5.8. SM/SM-IES(+) de [Pd(A1-H)(A1-D1)(DMSO)]+
AV 668
240 260 280 300 320 340 360 380 400 420 440 460 480m/z0
100
%
11BR1088 131 (2.443) Cm (50:238) 1: TOF MSMS 473.00ES+ 1.18e3x16 394.1
265.0
263.0
265.0 338.0
338.0356.0
393.1
395.1
473.1
412.1
417.0
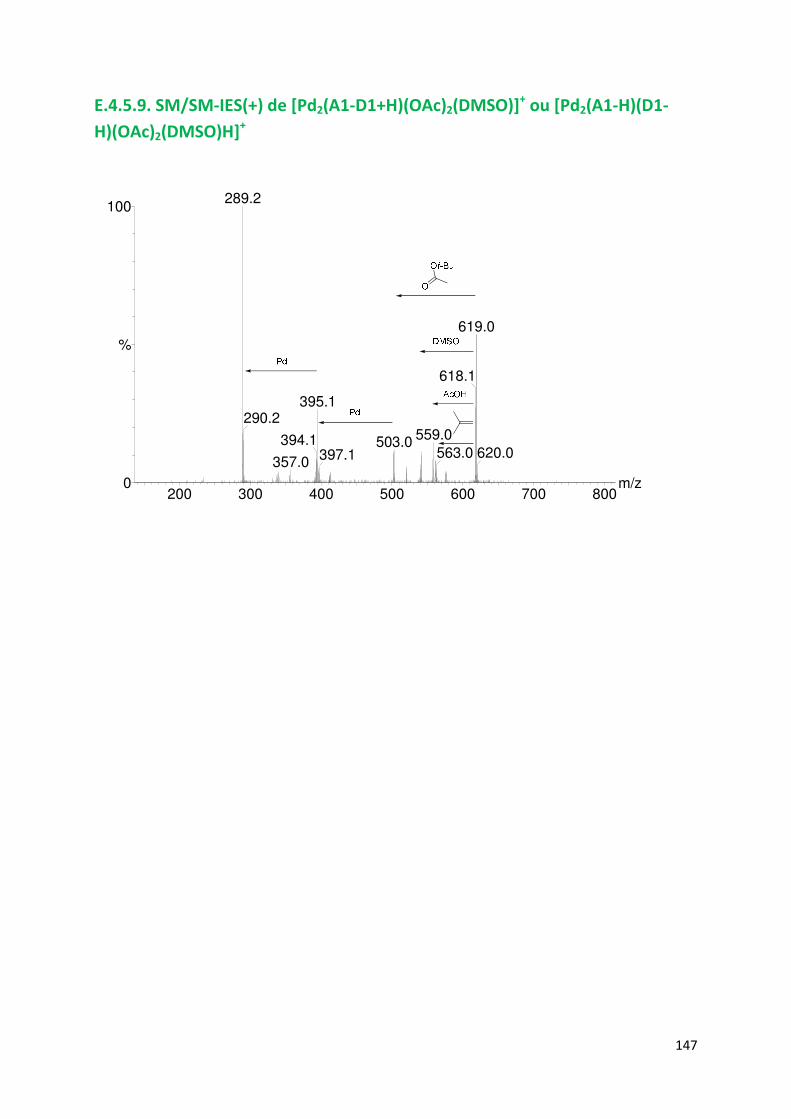
147
E.4.5.9. SM/SM-IES(+) de [Pd2(A1-D1+H)(OAc)2(DMSO)]+ ou [Pd2(A1-H)(D1-
H)(OAc)2(DMSO)H]+
AV 668
200 300 400 500 600 700 800m/z0
100
%
11BR1089 110 (2.047) Cm (58:127) 1: TOF MSMS 619.00ES+ 151289.2
619.0
618.1
395.1290.2
394.1
357.0
559.0503.0
397.1 563.0 620.0

148
Chapitre 3 :
RHD d’hétéroarènes avec des styrènes en
milieu aérobie et effet négatif des co-
oxydants métalliques

149
Introduction
Si la méthode présentée précédemment a permis les RHD de furanes et thiophènes avec des
styrènes dans des conditions douces et avec de bons rendements, celle-ci requiert une quantité
stoechiométrique en benzoquinone. La benzoquinone ou encore certains sels métalliques comme le
cuivre (II), l’argent (I) ou le manganèse (III) sont les oxydants les plus couramment utilisés pour
régénérer l’espèce palladiée active. Encore souvent utilisés en quantités stoechiométriques pour ces
réactions, ils présentent l’inconvénient de conduire à des sous-produits nocifs pour l’environnement
et la santé. D’une part, la benzoquinone et ses dérivés sont des métabolites du foie formés à partir
du benzène et responsables de troubles du système nerveux, d’anémies, de leucopénie ou encore de
thrombocytopénie, d’où sa nocivité pour la santé lorsqu’elle est inhalée.126, 127, 128
D’autre part, les
métaux posent le problème de ne pas être biodégradables et par conséquent, tout le long de la
chaîne alimentaire, ils se concentrent dans les organismes vivants et peuvent ainsi atteindre des taux
très élevés chez certaines espèces consommées par l’homme. Cette bio-accumulation explique leur
forte toxicité.
C’est dans ce contexte que s’inscrit la nécessité de développer des méthodes plus
respectueuses de l’environnement et moins couteuses. Ainsi, l’utilisation de l’oxygène moléculaire
comme réoxydant est de plus en plus étudiée. Notre atmosphère étant constituée d’environ 20 %
d’oxygène, O2 utilisé comme oxydant du Pd(0) représente l’oxydant idéal non seulement d’un point
de vue économique mais aussi d’un point de vue environnemental.129
Ce chapitre présentera un état des lieux des avancées en matière de RHD ayant recours à une
réoxydation de l’espèce palladium (0) par l’oxygène. Puis après avoir décrit une nouvelle méthode de
RHD d’hétéroarènes avec des styrènes, nous proposerons un mécanisme sur la base d’investigations
par SM-IES et nous montrerons l’impact négatif de la présence de co-oxydants métalliques dans
certaines conditions réactionnelles.
126
WHO, benzene, in Who Air quality guidelines for Europe, 2nd
edition, World Health Organization, Regional
Office for Europe, Copenhagen, 1998. 127
IPCS, Benzene, Environmental Health Criteria 150. International Programme on Chemical Safety, World
Health Organization, Geneva, 1993. 128
Paustenbach, D. J.; Bass, R. D.; Price, P. Environ. Health Perpec. 1993, 101, 77. 129
Boisvert, Luc.; Denney, M. C.; Hanson, S. K.; Goldberg, K. I. J. Am. Chem. Soc. 2009, 131, 15802-15814.

150
Partie F : RHD d’hétéroarènes avec des styrènes en milieu
aérobie
F.1. Bibliographie
La réaction décrite par Fujiwara et al. en 1969 est le premier exemple d’arylation d’oléfine via
l’activation par le palladium de liaisons C-H aromatiques. 130
Le couplage du styrène avec le benzène,
est effectué avec une quantité stoechiométrique d’acétate de palladium, dans l’acide acétique à
reflux (Schéma F1). Le chlorure de palladium peut également être utilisé mais requiert l’addition
d’acétate de sodium ou de potassium. L’acide acétique serait le solvant le plus approprié car il
permet d’obtenir un milieu réactionnel homogène.
Schéma F1. RHD pallado-induite du benzène avec le styrène.130
Cette équipe a également réalisé, le même couplage avec des quantités catalytiques
d’acétate de palladium (0,1 équiv.) et d’acétate de cuivre en présence d’oxygène (Schéma F2), mais
avec toutefois la formation de trans, trans-diphénylbutadiène et de traces de β-acétoxystyrène.
Notons que le couplage peut également utiliser de l’acétate d’argent (I) à la place de l’acétate de
cuivre (II) mais avec des rendements inférieurs (25 % avec 0,1 équiv. de AgOAc contre 44 % avec 0,1
équiv. de Cu(OAc)2).
Ph PhPh
PhH +
Cu(OAc)2 (0,1 équiv.)
O2 (50 atm), 80 °C, 8 h
PhH/AcOH (4 : 1)
Pd(OAc)2 (0,1 équiv.)
44 %45 équiv.
+Ph
Ph
18 %
AcOPh+
traces
Schéma F2. RHD pallado-induite du benzène avec le styrène en présence de Cu(OAc)2.130
130
Fujiwara,Y.; Moritani, I.; Danno, S.; Asano, R.; Teranishi, S. J. Am. Chem. Soc. 1969, 91, 7166-7169.

151
En 1971, Shue et ses collaborateurs parviennent à utiliser uniquement l’oxygène comme
réoxydant (schéma F3).131
Ce procédé catalytique peut être considéré comme le premier à n’utiliser
que de l’oxygène pour régénérer les espèces palladiées actives.132
Schéma F3. RHD impliquant le benzène.131
En 1997, Miura et ses collaborateurs ont couplé le biphényl-2-ol au styrène via une catalyse
par l’acétate de palladium avec l’air associé à une quantité catalytique d’acétate de cuivre comme
oxydant, dans du DMF, à 80 °C pendant 5 h. (schéma F4).133
Miura n’expose cependant qu’un seul
exemple, l’étude portant essentiellement sur des couplages intramoléculaires de 2-phénylphénols
avec des acrylates et des arylamides pour donner des dérivés 6H-benzo[c]chromènes substitués en
position 6.
Schéma F4. RHD du biphényl-2-ol avec un styrène.
Cette même année, Park et Hwang ont accompli les mêmes couplages que ceux réalisés par
Fujiwara en 1969, mais en ayant recours à des phosphines (Schéma F5) et des températures et
pressions en oxygène plus faibles.134
Cependant, leurs conditions s’avèrent moins efficaces et des
131
Shue, R. S. Chem. Commun. 1971, 1510-1511. 132
(a) Gligorich, K. M.; Sigman, M. S. Angew. Chem. Int. Ed. 2006, 45, 6612-6615. (b) Muzart, J. Chem. Asian J.
2006, 1, 508-515. 133
Miura, M.; Tsuda, T.; Satoh, T.; Nomura, M. Chem. Lett. 1997, 1103-1104. 134
Park, Y. W.; Hwang, S. W. Bull. Korean Chem. Soc. 1997, 18, 218-221.

152
composés de type stilbènique sont formés à partir de la réaction entre le styrène et les phosphines
arylées.
Schéma F5. RHD du benzène avec le styrène.134
En 2003, l’équipe de Jacobs décrit le couplage du benzène avec l’acrylate de n-butyle en
employant des conditions catalytiques similaires à celles de Shue. Le 3-phénylacrylate de n-butyle est
obtenu avec des sélectivités dépendantes du temps (Schéma F6).135
La diminution de la quantité de
catalyseur explique l’accroissement des temps de réactions.
Schéma F6. RHD du benzène avec l’acrylate de n-butyle.135
Le triacétate de manganèse (III) comme co-oxydant de l’oxygène s’est avéré efficace lorsque
le 4-phénylbut-3-èn-2-one était mis en jeu (Schéma F7).
Schéma F7. RHD du benzène avec le 4-phénylbut-3-èn-2-one.135
La même équipe a réalisé le couplage pallado-catalysé de l’anisole avec le 4-phenylbut-3-èn-
2-one, via une activation C-H, catalysée par le palladium, en présence d’oxygène comme unique
oxydant et d’acide benzoïque pour accélérer la réaction (Schéma F8). Par la suite, un catalyseur
135
Dams, M.; De Vos, D. E.; Celen, S.; Jacobs, P. A. Angew. Chem. Int. Ed. 2003, 42, 3512-3515.

153
hétérogène, constitué d’un support polymérique modifié par l’acide benzoïque et imprégné
d’acétate de palladium a été utilisé pour le couplage de l’anisole avec le trans-cinnamate d’éthyle.136
Schéma F8. RHD du méthoxybenzène avec le 4-phénylbut-3-èn-2-one.135
En 2003, le groupe d’Ishii a décrit le couplage de l’acrylate d’éthyle avec le benzène catalysé
par l’acétate de palladium en présence de petites quantités d’acétylacétone et d’acide
molybdovanadophophorique (H7PMo8V4O40) sous 1 atm d’oxygène (schéma F9).137
Ces conditions
conduisent à un mélange de produits de couplage et de double couplage de type Heck ainsi qu’à des
traces de produits de type Wacker. Ces mêmes réactions effectuées dans l’acide acétique ont donné
des résultats moins sélectifs et nécessitent des temps réactionnels plus longs. Il convient de souligner
que l’efficacité de ces réactions dépend grandement de la structure de l’hétéropolyacide.
Schéma F9. RHD du benzène avec l’acrylate d’éthyle.137
L’équipe a développé des couplages d’hétéroarènes tels que le furane (Schéma F10) ou le 2-
méthylthiophène138
avec des acrylates.
Schéma F10. RHD du furane et de l’acrylate d’éthyle.138
136
Hajek, J.; Dams, M.; Detrembleur, C; Jérôme, R.; Jacobs, P. A.; De Vos, D. E. Catal. Commun. 2007, 8, 1047-
1051. 137
Yokota, T.; Tani, M.; Sakaguchi, S.; Ishii, Y. J. Am. Chem. Soc. 2003, 125, 1476-1478. 138
Tani, M.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2004, 69, 1221-1226.

154
La réaction permettant le couplage du 2-méthylthiophène et de l’acrylate d’éthyle a été
réalisée avec un bon rendement dans l’acide propionique et avec de l’acétate de palladium (II) sur
charbon comme catalyseur à 60 °C. La présence d’acétate de sodium reste requise pour cette
réaction, mais celle d’acétylacétone n’est plus nécessaire (Schéma F11). Le système
Pd(OAC)2/hétéropolyacide/O2 a également été utilisé pour la RHD du benzène avec différents alcènes
activés (acrylonitriles, acryladéhydes…)44,139 et l’éthylène.
140
Schéma F11. RHD du 2-méthylthiophène et de l’acrylate d’éthyle.138
Gaunt et ses collaborateurs ont décrit en 2006 une méthode efficace pour les RHD de
pyrroles avec des acrylates via une catalyse par l’acétate de palladium dans le mélange
AcOH/dioxane/DMSO, à 35 °C avec de l’oxygène comme unique oxydant (Schéma F12).141
Schéma F12. RHD de pyrroles avec des acrylates.141
Par ailleurs, les auteurs font remarquer que la nature du groupement protecteur détermine
la régiosélectivité de la transformation. En effet, alors qu’avec un pyrrole N-Boc, la réaction a lieu en
position C2, elle se produit en position C3 avec un pyrrole N-TIPS (Schéma F13).
Schéma F13. Dépendance de la régiosélectivité avec la nature du groupement protecteur.141
139
Yamada, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2005, 70, 5471-5474. 140
Yamada, T.; Sakakura, A.; Sakaguchi, S.; Obora, Y.; Ishii, Y. New J. Chem. 2008, 32, 738-742. 141
Beck, E. M.; Grimster, N. P.; Hatley, R.; Gaunt, M. J. J. Am. Chem. Soc. 2006, 128, 2528-2529.

155
En 2007, Djakovitch et Rouge ont rapporté la vinylation d’indoles dans le DMF/DMSO, en
utilisant de l’air et des quantités catalytiques d’acétate de cuivre (II) ou de chlorure de cuivre (II)
comme réoxydant (Schéma F14).142
La nature de l’anion du cuivre influence l’efficacité et la
régiosélectivité de la réaction (Schéma F14). Pd(OAc)2/Cu(OAc)2 fournit un seul produit indolique
vinylé en C3 tandis que PdCl2/CuCl2 conduit à un mélange de produits.
Pd((II) (0,1 équiv)Cu (II) (0,1 équiv.)
DMF/DMSO (10 : 1)air (1 atm), 70 °C, 20 h
NH
+ CO2nBu
NH
CO2nBu
2 équiv.
68 %82 %63 %58 %
R
R = HR = MeR = C6H5R = CO2Me
R
NH
CO2nBu
100
NH
CO2nBu+
0:
67 33:
X = OAc
X = Cl
PdX2 (0,1 équiv)CuX2 (0,1 équiv.)
DMF/DMSO (10 : 1)air (1 atm), 70 °C, 20 h
R = H
Schéma F14. Vinylation de NH-indoles et influence de l’anion sur la régiosélectivité.142
Cette même année, Guo et Liu parviennent à coupler des anilides avec des acrylates dans des
conditions douces et en utilisant de l’oxygène moléculaire associé à 3 % molaire d’acétate de cuivre
en guise d’oxydant.143
Les rendements varient entre 57 et 91 % (schéma F15) mais leurs conditions
s’avèrent inéfficaces dès qu’un dérivé halogéné est mis en jeu. Il en est de même lorsque l’acrylate
est remplacé par un alcène riche en électrons tel que le styrène.
Schéma F15. RHD d’anilides avec des acrylates.143
142
Djakovitch, L.; Rouge, P. J. Mol. Catal. A: Chem. 2007, 273, 230-239. Djakovitch, L.; Rouge, P. Catal. Today
2009, 140, 90-99. 143
Wang, J. -R.; Yang, C. –T.; Liu, L.; Guo, Q. –X. Tetrahedron Lett. 2007, 48, 5449-5453.

156
Signalons également que l’équipe de Jutand a effectué l’ortho-fonctionnalisation
régiosélective d’acétanilides, catalysée par l’acétate de palladium avec une quantité catalytique en
benzoquinone qui est recyclée par voie électrochimique, offrant ainsi une alternative intéressante
sur le plan environnemental144
(Schéma F16).
Schéma F16. Ortho-fonctionnalisation régiosélective d’acétanilides.144
En 2009, Yu et ses collègues décrivent la RHD de benzènes mono- ou disubstitués avec des
acrylates en utilisant de l’acétate de palladium avec un ligand azoté comme catalyseur et de
l’anhydride acétique à 90 °C dans des conditions aérobies145
(schéma F17). Quelque soit la nature des
substituants, le couplage à lieu majoritairement en position méta du cycle et l’ajout d’acétate
d’éthyle permet parfois d’améliorer le rendement, notamment lorsque la quantité d’arène est
diminuée. Ces conditions réactionnelles se révèlent compatibles avec des alcènes disubstitués tels
que le cinnamate de méthyle ou d’éthyle.
Schéma F17. RHD de benzènes mono- ou disubstitués.
145
144
Amatore, C.; Cammoun, C.; Jutand, A. Adv. Synth. Catal. 2007, 349, 292. 145
Zhang, Y. –H.; Shi, B. –F.; Yu, J. –Q. J. Am. Chem. Soc. 2009, 131, 5072-5074.

157
Cette équipe a également synthétisé des divinylbenzènes à partir de l’acide 2-
phénylacétique, via deux RHD successives dans des conditions aérobies, en utilisant de l’acétate de
palladium et un ligand dérivé de la valine dans du tAmylOH à 90 °C146
(schéma F18). Les alcènes
généralement impliqués sont pauvres en électrons ; seuls deux exemples mettent en jeu des alcènes
riches en électrons. Le remplacement du ligand Ac-Val-OH par 10 ou 20 % molaire de benzoquinone
ne permet d’effectuer qu’une seule RHD.
Schéma F18. RHD de l’acide 2-phénylacétique avec des acrylates.146
La même année, Ishii et ses collaborateurs ont présenté la synthèse de cinnamotriles à partir
d’acrylonitriles et de benzènes en recourant au système catalytique Pd(OAc)2/HPMoV/O2 (schéma
F19).147
Rendement (%)
Entrée Ar-H total (MONO + DI) MONO (o : m : p) DI
1 benzène 47 47 2
2 Toluène 47 43 (23 : 29 : 48) 4
3 Anisole 67 43 (22 : 15 : 63) 24
4 Chlorobenzène 28 28 (36 : 27 : 37) indéterminé
5 p-xylène 37 37 traces
Schéma F19. Synthèse de cinnamonitriles.147
146
Engle, K. M.; Wang, D. –H.; Yu, J. –Q. Angew. Chem. Int. Ed. 2010, 49, 6169-6173. 147
Obora, Y.; Okabe, Y.; Ishii, Y. Org. Biomol. Chem. 2010, 8, 4071-4073.

158
Recemment, Zakrzewski et son équipe ont développé l’oléfination pallado-catalysée et
aérobie de férrocènes avec des alcènes pauvres en électrons et des diènes (acrylate d’éthyle, (E)-
cinnamate de méhtyle, fumarate de diéthyle, acide sorbique, sorbate d’éthyle).148
Dans le cas des
acrylates et du cinnamate, la réaction conduit à un mélange de férrocènes mono- et disubstitués
(schéma F22).
Schéma F20. Oléfination de ferrocènes.148
Zhu et al ont rapporté une séquence d’annellation/oléfination via une activation C-H menant
à des isoindolinones à partir du N-tosylbenzamide.149
La transformation s’opère dans le toluène à 110
°C avec de l’acétate de palladium comme catalyseur, un ligand de type phénantroline et de l’oxygène
moléculaire comme unique oxydant (schéma F21). Ces conditions s’avèrent compatibles non
seulement avec les alcènes aliphatiques, mais aussi avec les alcènes conjugués.
Schéma F21. Synthèse d’isoindolinones à partir du N-tosylbenzamide.149
148
Piotrowicz, M.; Zakrzewski, J.; Makal, A.; Bak, J.; Malinska, M.; Wozniak, K. J. Organomet. Chem. 2011, 696,
3499-3506. 149
Zhu, C.; Falck, J. R. Org. Lett. 2011, 13, 1214-1217.

159
Booker-Milburn et Lloyd-Jones ont également proposé une méthode pallado-catalysée de
synthèse directe d’isoindolinones mais à partir de N-méthoxybenzamides et d’acrylates ou encore
d’acrylamides.150
La formation d’isoindolinones se déroule selon la séquence d’activation de liaisons
C-H/réaction de Heck/réaction d’aza-Wacker. Des quantités catalytiques de benzoquinone associées
à l’oxygène moléculaire sont utilisées comme réoxydant (schéma F22).
Schéma F22. Méthode pallado-catalysée de synthèse directe d’isoindolinones à partir de N-
méthoxybenzamides.150
Très récemment, Cheng et al ont décrit une méthode d’ortho-oléfination pallado-catalysée
sélective d’arènes assistée par une double liaison C-C allylique à température ambiante et dans des
conditions aérobies (schéma F23).151
Les auteurs ont proposé un mécanisme impliquant une
coordination initiale de la liaison C=C allylique au palladium suivie d’une métallation de la liaison o-C-
H.
Schéma F23. Méthode d’ortho-oléfination pallado-catalysée sélective d’arènes.151
150
Wrigglesworth, J. W.; Cox, B.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. Org. Lett. 2011, 13, 5326-5329. 151
Gandeepan, P.; Cheng, C-H. J. Am. Chem. Soc. 2012, 134, 5738-5741.

160
L’équipe d’Ishii a réalisé l’oléfination pallado-catalysée régio- et stéréosélective via une
activation C-H de l’aminobenzène avec des acrylates grâce au système PdII/HPMoV/TMBA/O2 dans du
DMF à 60 °C (schéma F24).152
Schéma F24. RHD d’aminobenzène avec des acrylates.152
En conclusion, les exemples ci-dessus mettent en lumière les progrès concernant la
réoxydation du palladium (0). Il est possible d’effectuer des RHD en utilisant uniquement l’oxygène
comme oxydant terminal. Cependant, la capacité de l’oxygène moléculaire ou atmosphérique à
oxyder n’est généralement pas suffisante et il est nécessaire d’y associer de petites quantités de
métaux.
Par ailleurs, la gamme de substrats mise en jeu dans ces réactions est généralement
restreinte aux alcènes porteurs de groupements électroattracteurs et peu d’exemples sont décrits
avec des hétérocycles, ce qui limite les applications.
En conséquence, notre intérêt s’est porté sur la mise au point d’une méthode permettant les
RHD de styrènes avec des furanes, des thiophènes et des indoles en utilisant l’oxygène comme
oxydant terminal, en association éventuelle avec une quantité catalytique de co-oxydant.
152
Mizuta, Y.; Obora, Y.; Shimizu, Y.; Ishii Y. ChemCatChem 2012, 4, 187-191.

161
F.2. Résultats et discussion
F.2.1. Constat de départ
La synthèse via des RHD de styrylfuranes dans des conditions aérobies a déjà été effectuée
au sein de notre laboratoire48
mais en présence de 10 % molaire de benzoquinone, de 50 % molaire
de sels de cuivre (II) et à une température de 40 – 60 °C pour être réaliser de manière efficace.
Nous avons présenté les RHD de furanes et de thiophènes avec des styrènes à température
ambiante en utilisant le DMSO comme co-solvant et la benzoquinone comme oxydant
stoechiométrique.
Par conséquent, nous avons choisi d’explorer la possibilité de réaliser ces RHD en utilisant le
DMSO sous oxygène atmosphérique sans avoir recours à l’emploi d’oxydant organique. Le système
catalytique Pd(OAc)2/DMSO est connu pour être efficace pour l’oxydation des fonctions alcools et de
nombreuses réactions induites par Pd(II) avec uniquement l’utilisation de l’oxygène moléculaire pour
régénérer l’espèce active. 153,154
F.2.2. Etude du système oxydant
Puisque des co-oxydants métalliques en quantités catalytiques sont souvent utiles pour une
réoxydation efficace du palladium lors des RHD et pour demeurer dans des conditions douces en
température et en pression, nous avons étudié leur influence sur l’efficacité du couplage du sylvane
(A1) avec le styrene (C1) en présence de Pd(OAc)2 comme catalyseur, d’une combinaison
DMSO/AcOH comme solvant et d’oxygène moléculaire comme oxydant stoechiométrique (Tableau
F1).
153
(a) Larock, R. C.; Hightower, T. R. J. Org. Chem. 1993, 58, 5298-5300. (b) van Benthem, R. A. T. M.; Hiemstra,
H.; Michels, J. J.; Speckamp, W. N. J. Chem. Soc., Chem. Commun. 1994, 357-359. (c) van Benthem, R. A. T. M.;
Hiemstra, H.; Longarela, G. R.; Speckamp, W. N. Tetrahedron Lett. 1994, 35, 9281-9284. (d) Larock, R. C.;
Hightower, T. R.; Kraus, G. A.; Hahn, P.; Zheng, D. Tetrahedron Lett. 1995, 36, 2423-2426. (e) van Benthem, R. A.
T. M.; Hiemstra, H.; van Leeuwen, P. W. N. M.; Geus, J. W.; Speckamp, W. N. Angew. Chem., Int. Ed. Engl. 1995,
34, 457-460. (f) Larock, R. C.; Hightower, T. R.; Hasvold, L. A.; Peterson, K. P. J. Org. Chem. 1996, 61, 3584-3585.
(g) Peterson, K. P.; Larock, R. C. J. Org. Chem. 1998, 63, 3185-3189. 154
Steinhoff B. A.; Stahl S. S. J. Am. Chem. Soc. 2006, 128, 4348-4355

162
Entrée Co-oxydant Rendements
b
4 h 24 h
1 AgOAc 8 % 9 %
2 Cu(OAc)2 28 % 50 %
3 Cu2O 17 % 23 %
4 Fe3O4 18 % 33 %
5 Mn(OAc)3 41 % 66 %
6 Mn(OAc)2 45 % 79 %
7 MnO 42 % 79 %
8 MnO2 39 % 78 %
9 Mn2O3 45 % (64 %)
10 aucun 51 % (68 %)
aSylvane (2 mmol), styrène (1 mmol), Pd(OAc)2 (0,05
mmol), co-oxydant (0 - 0,1 mmol), AcOH (2 mL),
DMSO (2 mL), O2 (1 atm), TA, 4-24 h. b Rendement en
CPG. Rendement isolé entre parenthèse
Tableau F1. Influence du co-oxydant sur la RHD du sylvane avec le styrène.a
L’utilisation de AgOAc ou de Cu(OAc)2 mènent à de faibles rendements en 4 heures (tableau
F1, entrées 1 et 2). Le rendement n’est pas amélioré après 24 h lorsque la réaction est réalisée avec
AgOAc (entrée 1) alors qu’il devient moyen avec Cu(OAc)2 (entrée 2). Tandis que les oxydes
métalliques de cuivre ou de fer n’augmentent pas le rendement (entrées 3 et 4), les sels et oxydes de
manganèse fournissent 39 à 45 % du produit de couplage en 4 h (entrées 5 à 9). De manière
surprenante, en l’absence de co-oxydant, le rendement croît à 51 % en 4 heures et le composé A1-C1
est isolé avec un rendement de 68 % lorsque le temps de réaction est de 24 h. Vu ces résultats,
l’étude a été poursuivie avec un système Pd(OAc)2/AcOH/DMSO et O2 comme réoxydant.
F.2.3. Synthèse de styrylfuranes dans des conditions aérobies
F.2.3.1. A partir d’alkylfuranes
Le 2-styrylsylvane ou encore le 4-méthyl-2-stryrylsylvane ont été obtenus avec des
rendements respectifs de 68 % et de 61 % (Tableau F2, entrées 1 et 2).

163
Entrée A C T
(°C)
Durée
(h) Rdt (%)
b
Entrée A C
T
(°C)
Durée
(h) Rdt (%)
b
1 A1 C1 TA 24
6c A7 C5 40 48
2 A7 C1 40 24
7 A1 C6 TA 24
3 A1 C4 40 24
8 A7 C6 40 24
4 A7 C4 40 24
9 A1 C7 TA 24
5 A1 C5 40 24
10 A7 C7 40 48
a A (2,0 mmol), C (1,0 mmol), Pd(OAc)2 (0,05 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), 24 h
b Rendement
isolé. C Pd(OAc)2 (0,1 mmol).
Tableau F2. RHD de 2-alkylfuranes avec des styrènes en milieu aérobie.a
Lorsque le cycle aromatique du système styrénique est substitué par un chlore, un fluor, ou
un groupement acétoxyle, une température de 40 °C est souvent nécessaire. Les produits de
couplage sont obtenus avec des rendements de 50 % et 88 % (entrées 3 à 10) en conservant la liaison
carbone-hétéroatome.

164
F.2.3.2. A partir du 2-(méthoxyméthyl)furane
Comme attendu, le furfurol est peu disposé à donner les produits de couplage dans ces
conditions, celui-ci s’oxydant facilement en aldéhyde pyromucique, à partir duquel les RHD sont
difficilement réalisables. En conséquence, l’alkylation de la fonction alcool s’est avèrée nécessaire
afin d’éviter cette oxydation. La RHD du 2-(méthoxyméthyl)furane avec le styrène dans nos
conditions conduit au (E)-2-(méthoxyméthyl)-5-styrylfurane avec un rendement de 57 % en 24 h
(entrée 1). Les réactions menées avec les chlorostyrènes et le 4-fluorostyrène (entrée 2 à 4)
fournissent également de bons rendements (jusqu’à 95 %), avec toutefois des quantités de
catalyseurs et des temps réactionnels plus importants en ce qui concerne les chlorostyrènes.
Entrée A C Pd
(%)
Durée
(h) Rdt (%)
b
Entrée A C
Pd
(%)
Durée
(h) Rdt (%)
b
1 A8 C1 5 24
3 A8 C5 10 48
2 A8 C4 10 48
4 A8 C6 5 24
a A8 (2,0 mmol), C (1,0 mmol), Pd(OAc)2 (0,05 – 0,10 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), 40 °C, 24 –
48 h. b Rendement isolé.
Tableau F3. RHD du methoxyméthylfurane avec les styrènes en milieu aérobie.a
F.2.3.3. A partir du furane
Les réactions menées avec le furane conduisent à un mélange de 2-styrylfurane et de 2,5-
distyrylfurane avec une conversion totale du styrène (tableau F4). Quelque soit le substituant porté
par le cycle aromatique du styrène, le 2-styrylfurane est toujours obtenu majoritairement. La RHD du
furane avec les styrènes selon la méthode présentée au chapitre 1 n’avait conduit qu’à un
rendement de 14 % malgré une conversion totale du styrène. Cette transformation est désormais
possible lorsque l’on se place à 40 °C (entrée 1). Un excès de furane (8 équivalents) demeure
toutefois nécessaire afin de contrecarrer d’autres réactions du styrène.

165
+
A9, 8 équiv.
Pd(OAc)2 (0,05 - 0,1 équiv.)
AcOH/DMSO (1 : 1)O2 (1 atm)
TA - 40 °C, 24 - 48 h
O
R1
R1
A9-C, 35 - 41 %
O
R1
C-A9-C, 12 - 28 %
R1 = H (C1), p-Cl (C4), m-Cl (C5), p-F (C6)
O
+
R1
C
f
a A9 (8,0 mmol), C (1,0 mmol), Pd(OAc)2 (0,05 – 0,10 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), TA – 40 °C,
24 – 48 h. b Rendement isolé.
Tableau F4. RHD du furane avec les styrènes en milieu aérobie.a
F.2.4. Synthèse de styrylthiophènes dans des conditions aérobies
Les alkylthiophènes fournissent également de bons résultats avec les styrènes lorsque la
réaction est conduite à 60 °C et qu’ils sont introduits dans les mêmes proportions (tableau F5). En
effet, l’emploi de 2 équiv. d’alkylthiophène empoisonne le catalyseur.
Entrée A C Pd
(%)
Durée
(h)
Temp
(°C) A9-C, Rdt (%) C-A9-C, Rdt (%)
1 A9 C1 10 24 TA
2 A9 C4 10 24 40
3 A9 C5 10 24 40
4 A9 C6 5 48 40
O
C1-A9-C1, 28 %

166
Entrée B C Durée
(h) B1-C, Rdt (%)
b
1 B1 C1 24
2 B1 C4 48
3 B1 C5 48
4 B1 C6 48
a B1 (1,0 mmol), C (1,0 mmol), Pd(OAc)2 (0,10 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), 60 °C, 24 – 48 h.
b
Rendement isolé.
Tableau F5. RHD du 2-méthylthiophène avec les styrènes en milieu aérobie.a
Ce problème d’empoisonnement n’est pas rencontré lorsque le 3-méthylthiophène est mis
en jeu. Au contraire, nous sommes alors confrontés à une conversion très rapide du styrène, même à
40 °C, d’où la nécessité d’introduire 5 équivalents de 3-méthylthiophène (entrées 1 à 4, tableau F6).
Le substituant méthyle en position 3 du thiophène influe sur la régiosélectivité de la transformation ;
un mélange difficilement séparable de 3-méthyl-5-styrylthiophène et de 3-méthyl-2-styrylthiophène
dans des proportions 9 : 1 est obtenu. Zhang et ses collaborateurs avaient d’ailleurs été confrontés à
ce problème de régiosélectivité au cours de leurs travaux.57
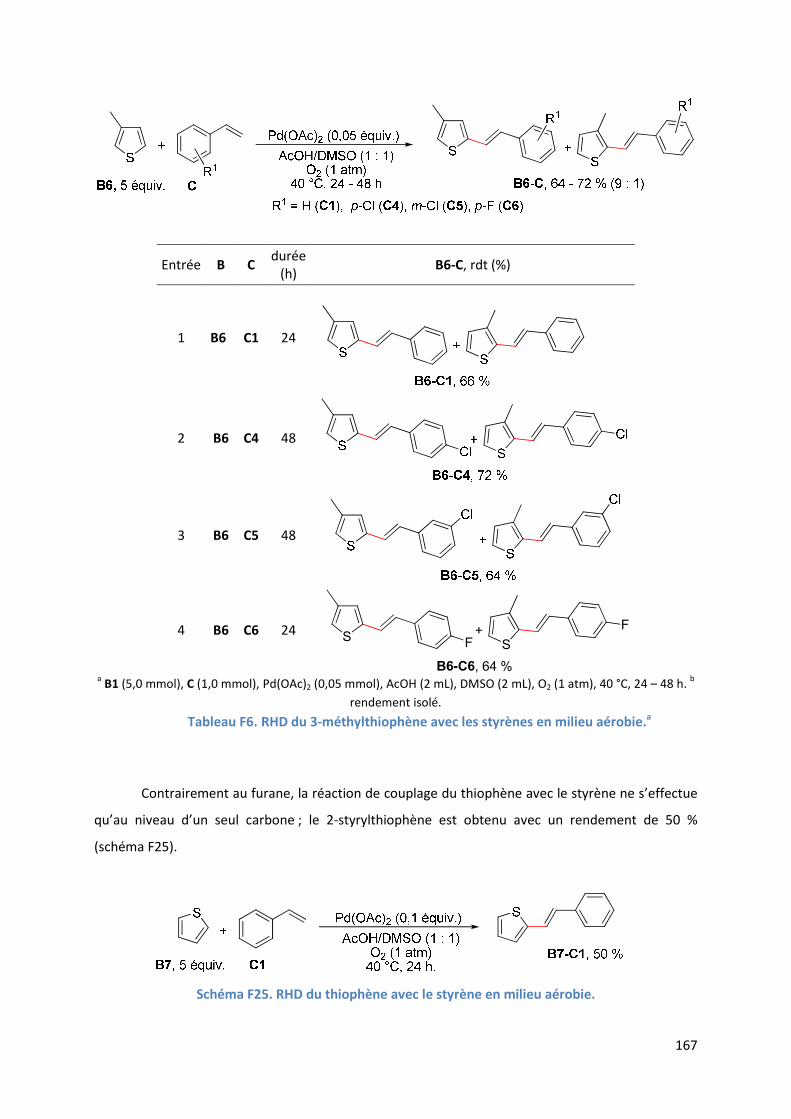
167
Entrée B C durée
(h) B6-C, rdt (%)
1 B6 C1 24
2 B6 C4 48
3 B6 C5 48
4 B6 C6 24
a B1 (5,0 mmol), C (1,0 mmol), Pd(OAc)2 (0,05 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), 40 °C, 24 – 48 h.
b
rendement isolé.
Tableau F6. RHD du 3-méthylthiophène avec les styrènes en milieu aérobie.a
Contrairement au furane, la réaction de couplage du thiophène avec le styrène ne s’effectue
qu’au niveau d’un seul carbone ; le 2-styrylthiophène est obtenu avec un rendement de 50 %
(schéma F25).
Schéma F25. RHD du thiophène avec le styrène en milieu aérobie.
S
B6-C6, 64 %
S+ F
F

168
F.2.5. Synthèse de styrylindoles dans des conditions aérobies
A température ambiante et avec 0,05 équiv. de catalyseur, la méthode est également
applicable à la synthèse de styryl-N-méthylindole. Cependant, la gamme d’indole demeure limitée
aux N-alkylindoles non substitués ou substitués par un groupement alkyle (schéma F26).
Schéma F26. RHD de N-alkylindoles avec le styrène en milieu aérobie.
F.2.6. Limites de la méthode
Les principales limites de la méthode résident dans la restriction de sa compatibilité aux
hétéroaarènes porteurs principalement de groupements électrodonneurs. En effet, les 2-
halogénothiophènes ou les bromostyrènes, par exemple, réagissent difficilement dans ces
conditions. Des problèmes de régiosélectivité peuvent également intervenir avec certains substrats,
notamment avec le furane et le 3 méthylthiophène.
F.2.7. Conclusion
En conclusion, des furanes, thiophènes et indoles ont été couplés avec des styrènes via une
activation C-H pour donner des produits de type Heck avec des rendements moyens à bons. Si
certaines réactions requièrent une charge en catalyseur de 10 % molaire pour atteindre une
conversion totale du styrène, elles sont réalisées dans des conditions douces en température (60 °C
au maximum) et en pression (1 atm). Comme observé au chapitre 1, les furanes et thiophènes
subsitués en position 2 réagissent en position C5. Quant au 3-méthylthiophène, la position C5 est
très largement la plus réactive. Si les furanes conduisent à un mélange de styrylfuranes (composés
majoritaires) et de bis(styryl)furanes, les réactions menées avec le thiophène fournissent seulement
le produit monosubstitué. Les RHD avec le p-acétoxystyrène et les chloro- et fluorostyrènes laissent
les liaisons C-O et C-halogènes intactes. Enfin, la méthode est applicable à certains indoles.

169
Par ailleurs, nous avons montré que des co-oxydants standards de réactions pallado-
catalysées ne sont pas toujours bénéfiques pour les RHD. En effet, l’oxygène moléculaire utilisé
comme seul oxydant peut conduire à des systèmes catalytiques plus actifs que ceux constitués par
l’utilisation d’une combinaison O2/Cu(OAc)2 ou O2/AgOAc.
F.3. Partie expérimentale
F.3.1. Informations générales
Le DMSO, AcOH, BQ, PhNO2, Mn2O3, Cu(OAc)2, MnO2, MnO, Cu2O, Mn(OAC)3, Mn(OAc)2,
Ag(OAc), Pd(OAc)2, tous les arènes (sauf A8) et styrènes sont disponibles commercialement et ont
été utilisés sans purification. Le Fe3O4155
et le 2-(methoxymethyl)furane156
ont été préparés selon la
litérature. PhNO2 a servi de référence pour les etudes cinétiques et la détermination de rendements
par CPG.
F.3.2. Synthèse de styrylfuranes, styrylthiophènes et styrylindoles
A l’acétate de palladium (11,2 – 24,4 mg, 0,05 – 0,1 mmol) en solution dans un mélange
AcOH (2 mL)/DMSO (2 mL) sont ajoutés l’arène (1 - 8 mmol) et l’alcène (1 mmol). Le mélange est
agité sous un atmosphère d’oxygène, à température ambiante ou à une température de 40 – 60 °C
pendant 24 à 48 h. Après ajout de Et2O (20 mL), la réaction est traitée par une solution aqueuse
saturée de NaHCO3 (20 mL). La phase aqueuse est extraite avec Et2O (20 mL × 2). Les phases
organiques sont lavées avec une solution de NaOH 2M (20 mL x 2), avec H2O (20 mL x 2), séchées sur
MgSO4, et filtrées sur coton. Le solvant est évaporé sous vide. Les produits sont séparés sur colonne
de gel de silice flash (éluant : éther de pétrole/acétate d’éthyle, 100-0 : 70-30).
155
Wang, N.; Zhu, L.; Wang, M.; Wang, D.; Tang, H. Ultrason. Sonochem. 2010, 17, 78-83. 156
Greeves, N.; Torode, J. S. Synthesis, 1993, 1109-1112

170
F.3.3. Synthèse de styrylfuranes en milieu aérobie
F.3.3.1. A partir d’alkylfuranes
(E)-2-styrylsylvane (A1-C1).
48 Solide blanc ; 68 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,31 (s, 3H, H7), 5,97 (d, 1H, J = 2,1 Hz, H4), 6,20 (d, 1H, J = 3,0 Hz,
H5), 6,80 (d, 1H, J = 16,2 Hz, H2), 6,95 (d, 1H, J = 16,2 Hz, H1), 7,19 (t, 1H, J = 7,1 Hz, HAr), 7,29 (t, 2H, J =
7,4 Hz, HAr), 7,41 (d, 2H, J = 7,2 Hz, HAr).
(E)- 4-méthyl-2-styrylsylvane (A7-C1).157
Huile jaune ; 61 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,84 (s, 3H, H8), 2,15 (s, 3H, H7), 6,03 (s, 1H, H4), 6,69 (d, J = 16,2
Hz, 1H, H2), 6,84 (d, J = 16,2 Hz, 1H, H1), 7,10 (t, J = 7,1 Hz, 1H, HAr), 7,21 (t, J = 7,4, 2H, HAr), 7,33 (d, J =
7,3 Hz, 2H, HAr).
(E)-2-(4-chlorostyryl)sylvane (A1-C4).48
Solide blanc ; 88 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,21 (s, 3H, H7), 5,88 (d, J = 2,2 Hz, 1H, H4), 6,11 (d, J = 2,9 Hz, 1H,
H5), 6,63 (d, J = 16,2 Hz, 1H, H2), 6,77 (d, J = 16,2 Hz, 1H, H1), 7,10-7,22 (m, 4H, HAr).
(E)-2-(4-chlorostyryl)-4-méthylsylvane (A7-C4). Cristaux jaunes ; 56 %.
Point de fusion : 93 - 95 °C.
157
Kikas, I.; Skoric, I.; Maric, Z.; Sindler-Kulyk, M. Tetrahedron 2010, 66, 9405-9414.

171
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,80 (s, 3H, H8), 2,11 (s, 3H, H7), 5,98 (s, 1H, H4), 6,58 (d, J = 16,2
Hz, 1H, H2), 6,72 (d, J = 16,2 Hz, 1H, H1), 7,10-7,20 (m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 10,9 (C8), 12,6 (C7), 114,1 (C4), 117,5 (C5), 118,2 (C2), 124,7 (C1),
128,3 (2CH), 129,8 (2CH), 133,6 (C-Cl), 137,0 (C), 149,1 (C6), 151,2 (C3).
IR (KBr) ν (cm-1
) : 508, 620, 704, 822, 859, 953, 1003, 1090, 1152, 1177, 1300, 1361, 1403, 1443,
1487, 1532, 1615, 1639, 1861, 2918, 2954, 3033.
SMHR : pour C14H13ClO théorique : 232,0655 ; Obtenue : 232,0658.
(E)-2-(3-chlorostyryl)sylvane (A1-C5).48
Huile incolore ; 74 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,22 (s, 3H, H7), 5,89 (d, J = 2,3 Hz, 1H, H4), 6,13 (d, J = 3,03 Hz,
1H, H5), 6,66 (d, J = 16,2 Hz, 1H, H2), 6,76 (d, J = 16,2 Hz, 1H, H1), 7,20-6,99 (m, 3H, HAr), 7,29 (s, 1H,
HAr).
(E)-2-(3-chlorostyryl)-4-méthylsylvane (A7-C5). semi-solide jaune ; 51 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 1,89 (s, 3H, H8), 2,20 (s, 3H, H7), 6,10 (s, 1H, H4), 6,70 (d, J = 16,2
Hz, 1H, H2), 6,80 (d, J = 16,2 Hz, 1H, H1), 7,28-7,03 (m, 3H, HAr), 7,35 (s, 1H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 9,5 (C8), 11,3 (C7), 113,1 (C4), 116,2 (C5), 117,6 (C2), 123,1 (C1),
124,0 (CH), 125,5 (CH), 126,6 (CH), 129,5 (CH), 134,3 (C-Cl), 139,1 (C), 147,9 (C6), 149,7 (C3).
IR (KBr) ν (cm-1
) : 511, 565, 624, 684, 684, 777, 807, 873, 902, 951, 998, 1083, 1152, 1200, 1306,
1363, 1427, 1468, 1533, 1564, 1591, 1615, 2865, 2919, 2952, 3022.
SMHR : pour C14H13ClO théorique : 232,0655 ; obtenue : 248,0659.

172
(E)-2-(4-fluorostyryl)sylvane (A1-C6). Solide blanc ; 82 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,23 (s, 3H, H7), 5,89 (d, J = 2,1 Hz, 1H, H4), 6,10 (d, J = 2,8 Hz, 1H,
H5), 6,61 (d, J = 16,2 Hz, 1H, H2), 6,81 (d, J = 16,2 Hz, 1H, H1), 6,89 (t, J = 8,6 Hz, 2H, HAr), 7,27 (dd, J =
8,3, 5,6 Hz, 2H, HAr).
(E)-2-(4-fluorostyryl)-4-méthylsylvane (A7-C6). Cristaux jaunes ; 58 %.
Point de fusion : 67 – 70 °C
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,04 (s, 3H, H8), 2,36 (s, 3H, H7), 6,22 (s, 1H, H4), 6,79 (d, J = 16,2
Hz, 1H, H2), 7,00 (d, J = 16,2 Hz, 1H, H1), 7,10 (t, J = 8,6 Hz, 2H, HAr), 7,47 (dd, J = 8,3, 5,6 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 10,8 (C8), 12,5 (C7), 113,5 (C4), 116,6 (2CH), 117,3 (C5), 117,5 (C2),
124,9 (C1), 128,5 (2CH), 134,6 (C), 148,7 (C6), 151,2 (C3), 163,0 (C-F).
IR (KBr) ν (cm-1
) : 516, 622, 658, 769, 819, 857, 952, 1001, 1097, 1154, 1223, 1301, 1444, 1502, 1536,
1596, 1619, 2862, 2920, 3037.
SMHR : pour C14H13FO théorique : 216,0950 ; obtenue : 216,0954.
(E)-2-(4-acétoxystyryl)sylvane (A1-C7).
48 Solide blanc ; 77 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,20 (s, 3H, H8), 2,25 (s, 3H, H7), 5,92 (d, J = 2,1 Hz, 1H, H4), 6,14
(d, J = 2,9 Hz, 1H, H5), 6,68 (d, J = 16,2 Hz, 1H, H2), 6,85 (d, J = 16,2 Hz, 1H, H1), 6,96 (d, J = 8,5 Hz, 2H,
HAr), 7,35 (d, J = 8,5 Hz, 2H, HAr).

173
(E) 2-(4-acétoxystyryl) -4-méthylsylvane (A7-C7). Cristaux blancs ; 50 %.
Point de fusion : 114 - 116 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,18 (s, 3H, H8), 2,49 (s, 3H, H9), 2,51 (s, 3H, H7), 6,37 (s, 1H, H4),
6,97 (d, J = 16,2 Hz, 1H, H2), 7,14 (d, J = 16,2 Hz, 1H, H1), 7,28 (d, J = 8,5 Hz, 2H, HAr), 7,66 (d, J = 8,5
Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 8,1 (C8), 9,8 (C7), 19,4 (C9), 110,9 (C4), 114,6 (C5), 115,1 (C2), 120,0
(2CH), 122,3 (C1), 125,2 (2CH), 133,5 (C), 146,1 (C6), 147,9 (C3), 148,5 (C-O-C=O), 167,7 (O-C=O).
IR (KBr) ν (cm-1
) : 520, 636, 831, 863, 910, 946, 1013, 1194, 1301, 1368, 1441, 1501, 1537, 1618,
1761, 2920.
SMHR : pour C16H16O3 théorique : 256,1099 ; obtenue : 256,1109.
F.3.3.2. A partir du 2-(méthoxyméthyl)furane
(E)-2-(méthoxyméthyl)-5-styrylfurane (A8-C1). Huile jaune ; 57 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 3,32 (s, 3H, H8), 4,34 (s, 2H, H7), 6,21 (d, J = 3,1 Hz, 1H, H4), 6,27
(d, J = 3,1 Hz, 1H, H5), 6,77 (d, J = 16,3 Hz, 1H, H2), 6,99 (d, J = 16,3 Hz, 1H, H1), 7,15 (m, 1H, HAr), 7,25
(t, J = 7,4 Hz, 2H, HAr), 7,37 (d, J = 7,3 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 56,5 (C8), 65,1 (C7), 107,8 (C4), 110,1 (C5), 114,9 (C2), 124,9
(2CH), 126,1 (CH), 126,2 (C1), 127,3 (2CH), 135,6 (C), 149,9 (C3), 152,2 (C6).
IR (film) ν (cm-1
) : 578, 693, 726, 752, 789, 851, 903, 957, 1000, 1020, 1089, 1190, 1258, 1359, 1448,
1495, 1529, 1576, 1598, 1638, 2822, 2928, 2987, 3027.
SMHR : pour C14H14O2 théorique : 214,0994 ; obtenue : 214,0997.

174
(E)-2-(4-chlorostyryl)-5-(méthoxyméthyl)furane (A8-C4). Cristaux jaunes ; 95 %.
Point de fusion : 57 - 59 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 3,19 (s, 3H, H8), 4,20 (s, 2H, H7), 6,08 (d, J = 2,9 Hz, 1H, H4), 6,14
(d, J = 3,0 Hz, 1H, H5), 6,57 (d, J = 16,2 Hz, 1H, H2), 6,78 (d, J = 16,2 Hz, 1H, H1), 7,09 (m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 57,3 (C8), 65,9 (C7), 109,3 (C4), 111,0 (C5), 116,3 (C2), 125,5 (C1),
127,0 (2CH), 128,3 (2CH), 132,5 (C-Cl), 135,0 (C), 151,1 (C3), 152,7 (C6).
IR (KBr) ν (cm-1
) : 507, 661, 710, 808, 854, 955, 1024, 1090, 1187, 1266, 1369, 1404, 1446, 1489,
1524, 1589, 1636, 2819, 2891, 2922, 2895, 3117
SMHR : pour C14H13ClO2 théorique : 248,0604 ; Obtenue : 248,0603.
(E)-2-(3-chlorostyryl)-5-(méthoxyméthyl)furane (A8-C5). Semi-solide jaune ; 69 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 3,25 (s, 3H, H8), 4,27 (s, 2H, H7), 6,17 (d, J = 3,1 Hz, 1H, H4), 6,21
(d, J = 3,0 Hz, 1H, H5), 6,67 (d, J = 16,2 Hz, 1H, H2), 6,83 (d, J = 16,2 Hz, 1H, H1), 7,19-6,98 (m, 3H, HAr),
7,28 (s, 1H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 60,6 (C8), 69,2 (C7), 112,9 (C4), 114,3 (C5), 120,3 (C2), 127,3 (C1),
128,5 (CH), 128,7 (CH), 130,1 (CH), 132,6 (CH), 137,3 (C-Cl), 141,6 (C), 154,5 (C3), 155,8 (C6).
IR (KBr) ν (cm-1
) : 582, 622, 687, 736, 785, 838, 904, 953, 999, 1019, 1090, 1187, 1259, 1363, 1427,
1472, 1528, 1555, 1593, 1638, 2820, 2895, 2928, 2985, 3058.
SMHR : pour C14H13ClO2 théorique 248,0604 ; Obtenue : 248,0605.
(E)-2-(4-fluorostyryl)-5-(méthoxyméthyl)furane (A8-C6). Semi-solide jaune ; 64 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 3,60 (s, 3H, H8), 4,62 (s, 2H, H7), 6,48 (d, J = 2,9 Hz, 1H, H4), 6,56
(d, J = 3,0 Hz, 1H, H5), 6,96 (d, J = 16,3 Hz, 1H, H2), 7,33-7,13 (m, 3H, H1 et HAr), 7,60 (m, 2H, HAr).

175
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 58,6 (C8), 67,2 (C7), 110,1 (C4), 112,3 (C5), 116,4 (C2), 116,9 (2CH),
127,0 (C1), 128,6 (2CH), 133,9 (C), 152,1 (C3), 154,2 (C6), 163,1 (C-F).
IR (KBr) ν (cm-1
) : 517, 661, 719, 794, 821, 862, 905, 960, 1020, 1092, 1159, 1193, 1226, 1264, 1367,
1413, 1451, 1504, 1530, 1598, 1637, 2824, 2898, 2930, 2996, 3041.
SMHR : pour C14H13FO2 théorique: 233,0900 ; Obtenue : 233,0901.
F.3.3.3. A partir du furane
(E)-2-styrylfurane (A9-A1).
158 Solide blanc ; 35 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,35 (d, J = 3,1 Hz, 1H, H4), 6,49-6,39 (m, 1H, H5), 6,89 (d, J = 16,3
Hz, 1H, H2), 7,04 (d, J = 16,3 Hz, 1H, H1), 7,24 (m, 1H, HAr), 7,34 (t, J = 7,3 Hz, 2H, HAr), 7,46 (d, J = 7,4
Hz, 2H, HAr et H1).
2,5-di(E)-styrylfurane (C1-A9-C1).48
Solide jaune ; 28 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,38 (s, 2H, H4 et H5), 6,88 (d, J = 16,2 Hz, 2H, H2 et H2’), 7,14 (d, J
= 16,2 Hz, 2H, H1 et H1’), 7,29-7,19 (m, 3H, HAr), 7,35 (t, J = 7,4, 4H, HAr), 7,50 (d, J = 7,4 Hz, 4H, HAr).
O
2
1
3
45
6Cl
(E)-2-(4-chlorostyryl)furane (C9-C4). Solide blanc ; 41 %.
Point de fusion : 95 - 97 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,40 (m, 1H, H4), 6,46 (m, 1H, H5), 6,88 (d, J = 16,2 Hz, 1H, H2),
7,02 (d, J = 16,2 Hz, 1H, H1), 7,30-7,45 (m, 5H, HAr et H6).
158
Cella, R.; Stefani, A. H. Tetrahedron 2006, 62, 5656-5662.

176
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 110,3 (C4), 113,0 (C5), 118,3 (C2), 127,0 (C1), 128,7 (2CH), 130,1
(2CH), 134,3 (C-Cl), 136,8 (C), 143,6 (C6), 154,2 (C3).
IR (KBr) ν (cm-1
) : 516, 592, 737, 794, 826, 862, 925, 965, 1015, 1096, 1154, 1222, 1412, 1487, 1508,
1556, 1595, 1635, 3037.
SMHR : pour C12H9ClO théorique : 204,0342 ; Obtenue : 204,0345.
O
2
1
3
45
3'
2'
1'
Cl
Cl
2,5-bis(E)-(4-chlorostyryl)furane (C4-A9-C4). Solide jaune ; 20 %.
Point de fusion : 154 - 157 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,31 (s, 2H, H4 et H5), 6,75 (d, J = 16,2 Hz, 2H, H2 et H2’), 6,98 (d, J
= 16,2 Hz, 2H, H1 et H1’), 7,23 (d, J = 8,4 Hz, 4H, HAr), 7,33 (d, J = 8,4 Hz, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 113,9 (C4 et C5), 118,7 (C2 et C2’), 128,2 (C1 et C1’), 129,6 4(4CH),
131,0 (4CH), 135,3 (C-Cl), 137,6 (C), 155,0 (C3 et C3’).
IR (KBr) ν (cm-1
) : 515, 747, 791, 819, 857, 959, 1019, 1096, 1155, 1226, 1377, 1506, 1556, 1595,
1631, 2923, 3041.
SMHR : pour C20H14Cl2O théorique : 340,0422 ; Obtenue : 340,0424.
(E)-2-(3-chlorostyryl)furane (A9-C5). Cristaux jaunes ; 37 %.
Point de fusion : 57 – 59 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,27 (m, 1H), 6,32 (m, 1H), 6,76 (d, J = 16,2 Hz, 1H), 6,85 (d, J =
16,3 Hz, 1H), 7,21-7,11 (m, 3H), 7,31 (d, J = 7,2 Hz, 2H).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 111,6 (C4), 113,9 (C5), 119,9 (C2), 126,7 (C1), 127,6 (CH), 128,2
(CH), 129,5 (CH), 132,0 (CH), 136,7 (C-Cl), 141,0 (C), 144,6 (C6), 154,9 (C3).
IR (KBr) ν (cm-1
) : 488, 591, 689, 740, 786, 800, 881, 925, 960, 1012, 1076, 1147, 1198, 1258, 1408,
1476, 1554, 1638, 2924, 3058.
SMHR : pour C12H9ClO théorique : 204,0342 ; Obtenue : 204,0344.

177
2,5-bis(E)-(3-chlorostyryl)furane (C5-A9-C5). Solide jaune ; 12 %.
Point de fusion : 115 - 117 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,35 (s, 2H, H4 et H5), 6,79 (d, J = 16,2 Hz, 2H, H2 et H2’), 6,98 (d, J
= 16,2 Hz, 2H, H1 et H1’), 7,15-7,30 (m, 7H, HAr), 7,41 (s, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 112,8 (C4 et C5), 117,9 (C2 et C2’), 125,3 (2CH), 126,7 (2CH), 126,8
(2CH), 128,2 (C1 et C1’), 130,6 (2CH), 135,4 (C-Cl), 139,5 (C), 153,5 (C3 et C3’).
IR (KBr) ν (cm-1
) : 431, 684, 776, 869, 903, 951, 1020, 1086, 1258, 1423, 1468, 1567, 1588, 1632,
2924.
SMHR : pour C20H14Cl2O théorique : 340,0422 ; Obtenue : 340,0419.
(E)-2-(4-fluorostyryl)furane (A9-C6). Solide blanc ; 35 %.
Point de fusion : 73 - 74 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,21 (m, 1H, H4), 6,29 (m, 1H, H5), 6,67 (d, J = 16,3 Hz, 1H, H2),
6,95-6,95 (m, 3H, H1, 2HAr), 7,26-7,31 (m, 2H, 2HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 109,7 (C4), 112,7 (C5), 116,5 (C2), 116.9 (CH), 117,4 (CH), 127,0
(C1), 128,8 (2CH), 134,2 (C), 143,2 (C6), 154,1 (C3), 163,3 (C-F).
IR (KBr) ν (cm-1
) : 505, 590, 662, 738, 816, 859, 880, 924, 962, 1012, 1082, 1105, 1146, 1175, 1201,
1259, 1402, 1484, 1551, 1632, 1727, 3030.
SMHR : pour C12H9FO théorique : 188,0637 ; Obtenue : 188,0639.
2,5-bis(E)- (4-fluorostyryl)furane (C6-A9-C6). Solide jaune ; 23 %.
Point de fusion : 145 - 148 °C.

178
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,28 (m, 2H, H4 et H5), 6,69 (d, J = 16,2 Hz, 2H, H2 et H2’), 6,93-
7,03 (m, 6H, H1, H1’ et 4HAr), 7,34-7,40 (m, 4H, 4HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 112,2 (C4 et C5), 116,6 (C2 et C2’), 116,9 (2CH), 127,0 (C1 et C1’),
128,8 (2CH), 134,1 (C), 153,7 (C3 et C3’), 163,3 (2 C-F).
IR (KBr) ν (cm-1
) : 502, 668, 709, 734, 771, 809, 857, 959, 986, 1012, 1086, 1158, 1265, 1380, 1404,
1489, 1545, 1633, 1890, 3043.
SMHR : pour C20H14F2O théorique : 308,1013 ; Obtenue : 308,1021.
F.3.4. Synthèse de styrylthiophènes en milieu aérobie
F.3.4.1. A partir d’alkylthiophènes
(E)-2-méthyl-5-styrylthiophène (B1-C1). Cristaux blancs ; 69 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,40 (s, 3H, H7), 6,58 (m, 1H, H4), 6,72-6,78 (m, 2H, H5 et H2), 7,08
(d, J = 16,2 Hz, 1H, H1), 7,16 (m, 1H, HAr), 7,26 (t, J = 7,41, 2H, HAr), 7,36 (d, J = 7,54 Hz, 2H, HAr).
S
2
1
3
45
6
7Cl
(E)-2-(4-chlorostyryl)-5-méthylthiophène (B1-C4). Cristaux jaunes clairs ; 60 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,38 (s, 3H, H7), 6,55 (m, 1H, H4), 6,62 (d, J = 16,1 Hz, 1H, H2), 6,75
(d, J = 3,2 Hz, 1H, H5), 7,00 (d, J = 16,0 Hz, 1H, H1), 7,14-7,27 (m, 4H, HAr).
(E)-2-(3-chlorostyryl)-5-méthylthiophène (B1-C5). Cristaux jaunes clairs ; 71 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,44 (s, 3H, H7), 6,61-6,68 (m, 2H, H4 et H2), 6,82 (d, J = 3,3 Hz, 1H,
H5), 7,08 (d, J = 16,1 Hz, 1H, H1), 7,15-7,21 (m, 3H, HAr), 7,31 (s, 1H, HAr).

179
(E)-2-(4-fluorostyryl)-5-méthylthiophène (B1-C6). Cristaux jaunes ; 55 %.
Point de fusion : 94 - 96 °C.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,35 (s, 3H, H7), 6,52 (m, 1H, H4), 6,62 (d, J = 16,0 Hz, 1H, H2), 6,70
(d, J = 3,3 Hz, 1H, H5), 6,90 (m, 3H, H1 et HAr), 7,25 (dd, J = 8,3, 5,57 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 16,1 (C7), 116,1 (C4), 122,4 (C5), 126,3 (C2), 126,9 (C1), 128,0 (2CH),
128,1 (2CH), 133,9 (C), 139,8 (C6), 141,1 (C3), 162,6 (C-F).
IR (KBr) ν (cm-1
) : 528, 577, 793, 817, 854, 953, 1040, 1096, 1158, 1232, 1470, 1507, 1597, 1627,
2369, 2920, 3066.
SMHR : pour C13H11FS théorique : 218,0566 ; Obtenue : 218,0566.
(E)-4-méthyl-2-styrylthiophène (B6-C1). Cristaux blancs ; 66 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,57 (s, 3H, H7), 7,07 (d, J = 5,1 Hz, 1H, H4), 7,14 (d, J = 16,0 Hz,
1H, H2), 7,32 (d, J = 5,0 Hz, 1H, H6), 7,45-7,55 (m, 2H, H1 et HAr), 7,60 (t, J = 7,5 Hz, 2H, HAr), 7,73 (d, J =
7,5 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 14,5 (C7), 120,8 (C6), 123,4 (C4), 126,8 (2CH), 128,0 (C2), 128,3 (C1),
129,2 (2CH), 131,4 (CH), 136,1 (C), 137,0 (C5), 137,9 (C3).
IR (KBr) ν (cm-1
) : 521, 590, 618, 713, 751, 811, 955, 1015, 1184, 1384, 1424, 1490, 1531, 1593, 1621,
2921, 3024.
SMHR : pour C13H12S théorique : 200,0660 ; Obtenue : 200,0661.
(E)-2-(4-chlorostyryl)-4-méthylthiophène (B6-C4). Cristaux blancs ; 72 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,20 (s, 3H, H7), 6,66-6,73 (m, 2H, H4 et H2), 6,99 (d, J = 5,0 Hz, 1H,
H6), 7,09 (d, J = 16,0 Hz, 1H, H1), 7,17-7,28 (m, 4H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 13,1 (C7), 119,9 (C6), 122,3 (C4), 125,4 (C2), 126,5 (2CH) 127,9
(2CH), 130,0 (C1), 132,0 (C-Cl), 135,0 (C), 135,1 (C5), 135,3 (C3).

180
IR (KBr) ν (cm-1
) : 521, 616, 713, 808, 831, 851, 954, 1008, 1088, 1183, 1242, 1290, 1401, 1427, 1490,
1618, 2340, 2369, 2862, 2922, 3020.
SMHR : pour C13H11ClS théorique : 234,0268 ; Obtenue : 234,0269.
(E)-2-(3-chlorostyryl)-4-methylthiophene (B6-C5). Cristaux jaunes ; 67 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,25 (s, 3H, H7), 6,67-6,81 (m, 2H, H4 et H2), 7,04 (d, J = 4,9 Hz, 1H,
H6), 7,30-7,09 (m, 4H, H1 et HAr), 7,38 (s, 1H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 15,0 (C7), 122,5 (C6), 124,4 (C4), 125,4 (C2), 126,9 (CH), 127,0
(CH), 128,1 (C1), 130,8 (CH) 131,8 (CH), 135,5 (C-Cl), 136,9 (C), 137,3 (C5), 140,2 (C3).
IR (KBr) ν (cm-1
) : 520, 615, 706, 739, 775, 828, 880, 947, 1082, 1216, 1248, 1424, 1476, 1562, 1590,
1623, 2961, 3018.
SMHR : pour C13H11ClS théorique : 234,0268 ; Obtenue : 234,0269.
(E)-2-(4-fluorostyryl)-4-méthylthiophène (B6-C6). Cristaux blancs ; 64 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,33 (s, 3H, H7), 6,81-6,88 (m, 2H, H4 et H2), 7,10-7,20 (m, 4H, H6,
H1 et HAr), 7,44 (dd, J = 8,4, 5,5 Hz, 2H, HAr).
RMN 13
C (63 MHz, CDCl3) δ (ppm) : 14,2 (C7), 115,9 (C6), 120,4 (C4), 123,1 (C2), 127,9 (CH), 128,0 (C1),
128,8 (CH), 131,1 (2CH), 133,8 (C), 135,9 (C5), 136,6 (C3), 162,5 (C-F).
IR (KBr) ν (cm-1
) : 524, 594, 614, 705, 722, 778, 814, 851, 927, 946, 954, 1097, 1155, 1228, 1295,
1407, 1439, 1596, 1621, 2928, 3022.
SMHR : pour C13H11FS théorique : 218,0566 ; Obtenue : 218,0567.

181
F.3.4.2. A partir du thiophène
(E)-2-styrylthiophène (B7-C1).159
Solide jaune clair ; 50 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 6,75-6,93 (m, 2H, H4 et H2), 6,96 (d, J = 3,1 Hz, 1H, H6), 7,02-7,20
(m, 3H, H5, H1 et HAr), 7,24 (t, J = 7,3, 2H, HAr), 7,36 (d, J = 7,2 Hz, 2H, HAr).
F.3.5. Synthèse de styrylindoles en milieu aérobie
(E)-1-méthyl-3-styryl-1H-indole (E1-C1).160
Solide blanc ; 57 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 3,84 (s, 3H, H5), 7,34 (d, J = 14,2 Hz, 2H, H2 et H1), 7,40-7,66 (m,
7H, HAr), 7,75 (d, J = 7,2 Hz, 2H, HAr et H4), 8,24 (d, J = 6,3 Hz, 1H, HAr).
(E)-1,2-diméthyl-3-styryl-1H-indole (E2-C1).161
Cristaux jaunes ; 42 %.
RMN 1H (250 MHz, CDCl3) δ (ppm) : 2,64 (s, 3H, H6), 3,76 (s, 3H, H5), 7,22-7,91 (m, 10H, H2, H1 et HAr),
8,22 (s, 1H, HAr).
159
Ren, G.; Cui, X.; Yang, E.; Yang, F.; Wu, Y. Tetrahedron, 2010, 66, 4022-4028. 160
Gao, H.; Wu, X.; Zhang, J. Chem. Eur. J. 2011, 17, 2838-2841. 161
Adam, W.; Takayama, K. J. Org. Chem. 1980, 45, 447-452.
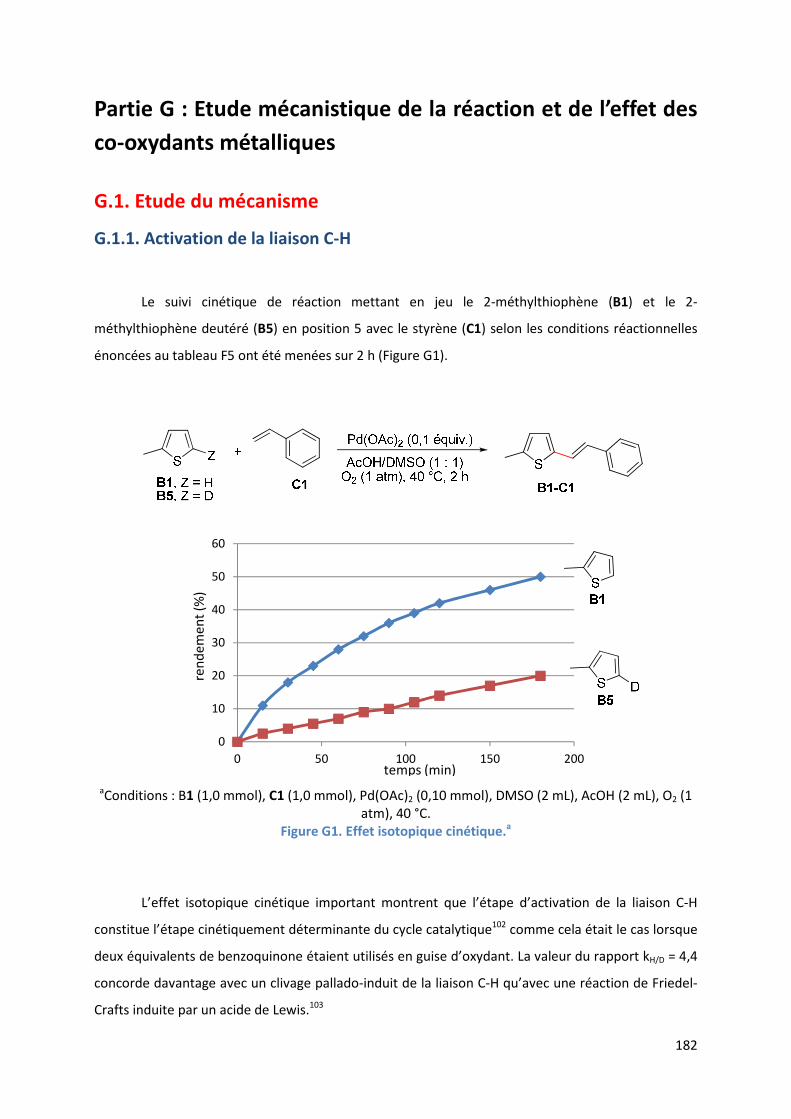
182
Partie G : Etude mécanistique de la réaction et de l’effet des
co-oxydants métalliques
G.1. Etude du mécanisme
G.1.1. Activation de la liaison C-H
Le suivi cinétique de réaction mettant en jeu le 2-méthylthiophène (B1) et le 2-
méthylthiophène deutéré (B5) en position 5 avec le styrène (C1) selon les conditions réactionnelles
énoncées au tableau F5 ont été menées sur 2 h (Figure G1).
aConditions : B1 (1,0 mmol), C1 (1,0 mmol), Pd(OAc)2 (0,10 mmol), DMSO (2 mL), AcOH (2 mL), O2 (1
atm), 40 °C.
Figure G1. Effet isotopique cinétique.a
L’effet isotopique cinétique important montrent que l’étape d’activation de la liaison C-H
constitue l’étape cinétiquement déterminante du cycle catalytique102
comme cela était le cas lorsque
deux équivalents de benzoquinone étaient utilisés en guise d’oxydant. La valeur du rapport kH/D = 4,4
concorde davantage avec un clivage pallado-induit de la liaison C-H qu’avec une réaction de Friedel-
Crafts induite par un acide de Lewis.103
0
10
20
30
40
50
60
0 50 100 150 200temps (min)
ren
de
me
nt
(%)
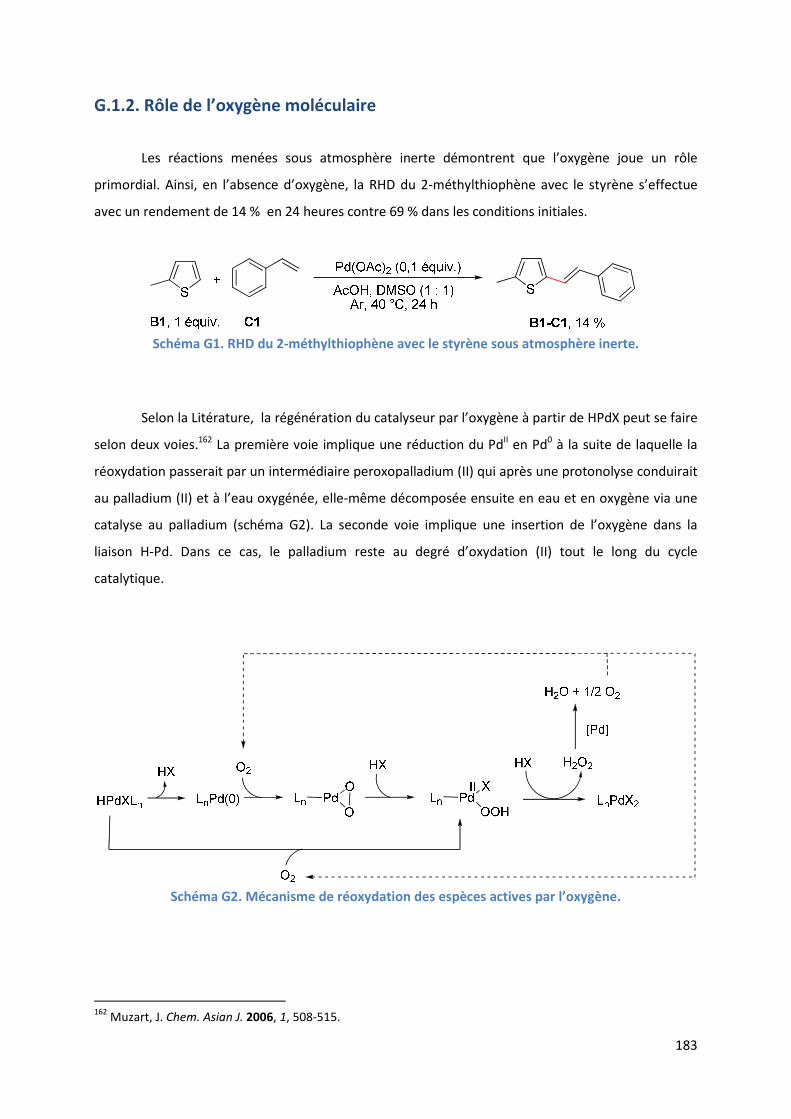
183
G.1.2. Rôle de l’oxygène moléculaire
Les réactions menées sous atmosphère inerte démontrent que l’oxygène joue un rôle
primordial. Ainsi, en l’absence d’oxygène, la RHD du 2-méthylthiophène avec le styrène s’effectue
avec un rendement de 14 % en 24 heures contre 69 % dans les conditions initiales.
Schéma G1. RHD du 2-méthylthiophène avec le styrène sous atmosphère inerte.
Selon la Litérature, la régénération du catalyseur par l’oxygène à partir de HPdX peut se faire
selon deux voies.162
La première voie implique une réduction du PdII en Pd
0 à la suite de laquelle la
réoxydation passerait par un intermédiaire peroxopalladium (II) qui après une protonolyse conduirait
au palladium (II) et à l’eau oxygénée, elle-même décomposée ensuite en eau et en oxygène via une
catalyse au palladium (schéma G2). La seconde voie implique une insertion de l’oxygène dans la
liaison H-Pd. Dans ce cas, le palladium reste au degré d’oxydation (II) tout le long du cycle
catalytique.
Schéma G2. Mécanisme de réoxydation des espèces actives par l’oxygène.
162
Muzart, J. Chem. Asian J. 2006, 1, 508-515.
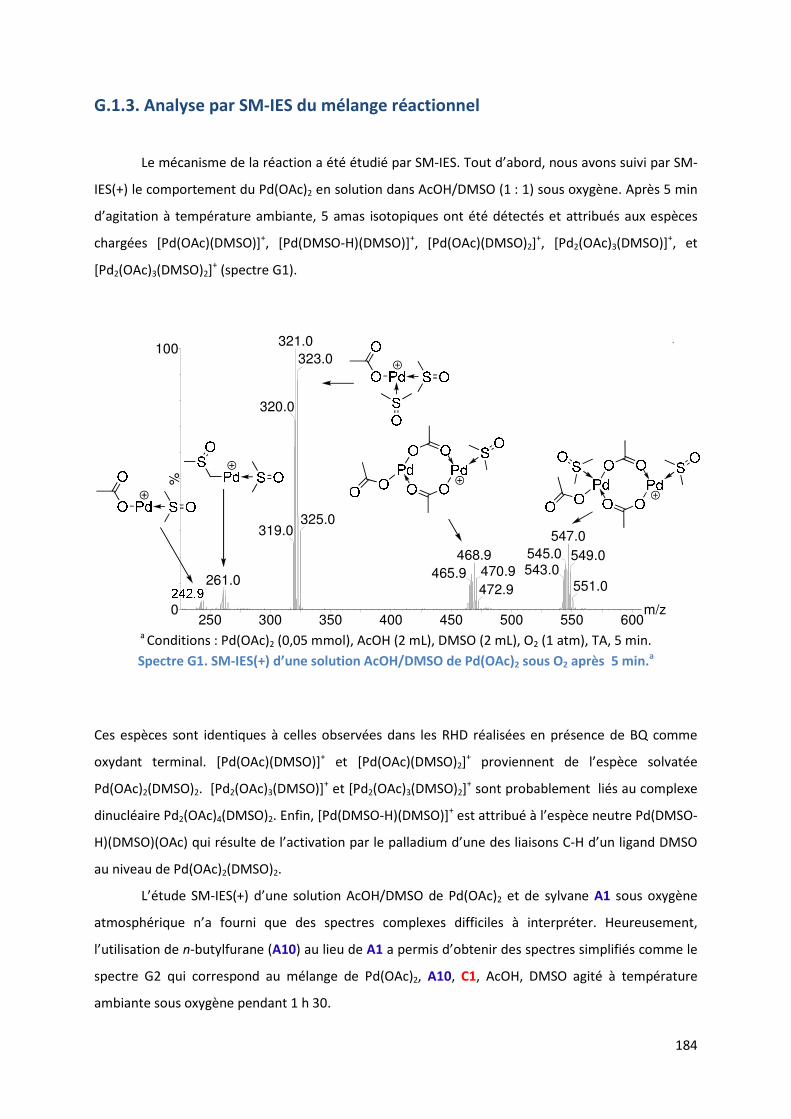
184
G.1.3. Analyse par SM-IES du mélange réactionnel
Le mécanisme de la réaction a été étudié par SM-IES. Tout d’abord, nous avons suivi par SM-
IES(+) le comportement du Pd(OAc)2 en solution dans AcOH/DMSO (1 : 1) sous oxygène. Après 5 min
d’agitation à température ambiante, 5 amas isotopiques ont été détectés et attribués aux espèces
chargées [Pd(OAc)(DMSO)]+, [Pd(DMSO-H)(DMSO)]
+, [Pd(OAc)(DMSO)2]
+, [Pd2(OAc)3(DMSO)]
+, et
[Pd2(OAc)3(DMSO)2]+ (spectre G1).
AV 875
m/z250 300 350 400 450 500 550 600
%
0
100
12HR417 510 (19.993) Cm (372:559) 1: TOF MS ES+ 8.21e4321.0
320.0
319.0
261.0
323.0
325.0
547.0
545.0468.9
465.9 543.0470.9
472.9
549.0
551.0
a Conditions : Pd(OAc)2 (0,05 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), TA, 5 min.
Spectre G1. SM-IES(+) d’une solution AcOH/DMSO de Pd(OAc)2 sous O2 après 5 min.a
Ces espèces sont identiques à celles observées dans les RHD réalisées en présence de BQ comme
oxydant terminal. [Pd(OAc)(DMSO)]+ et [Pd(OAc)(DMSO)2]
+ proviennent de l’espèce solvatée
Pd(OAc)2(DMSO)2. [Pd2(OAc)3(DMSO)]+ et [Pd2(OAc)3(DMSO)2]
+ sont probablement liés au complexe
dinucléaire Pd2(OAc)4(DMSO)2. Enfin, [Pd(DMSO-H)(DMSO)]+ est attribué à l’espèce neutre Pd(DMSO-
H)(DMSO)(OAc) qui résulte de l’activation par le palladium d’une des liaisons C-H d’un ligand DMSO
au niveau de Pd(OAc)2(DMSO)2.
L’étude SM-IES(+) d’une solution AcOH/DMSO de Pd(OAc)2 et de sylvane A1 sous oxygène
atmosphérique n’a fourni que des spectres complexes difficiles à interpréter. Heureusement,
l’utilisation de n-butylfurane (A10) au lieu de A1 a permis d’obtenir des spectres simplifiés comme le
spectre G2 qui correspond au mélange de Pd(OAc)2, A10, C1, AcOH, DMSO agité à température
ambiante sous oxygène pendant 1 h 30.
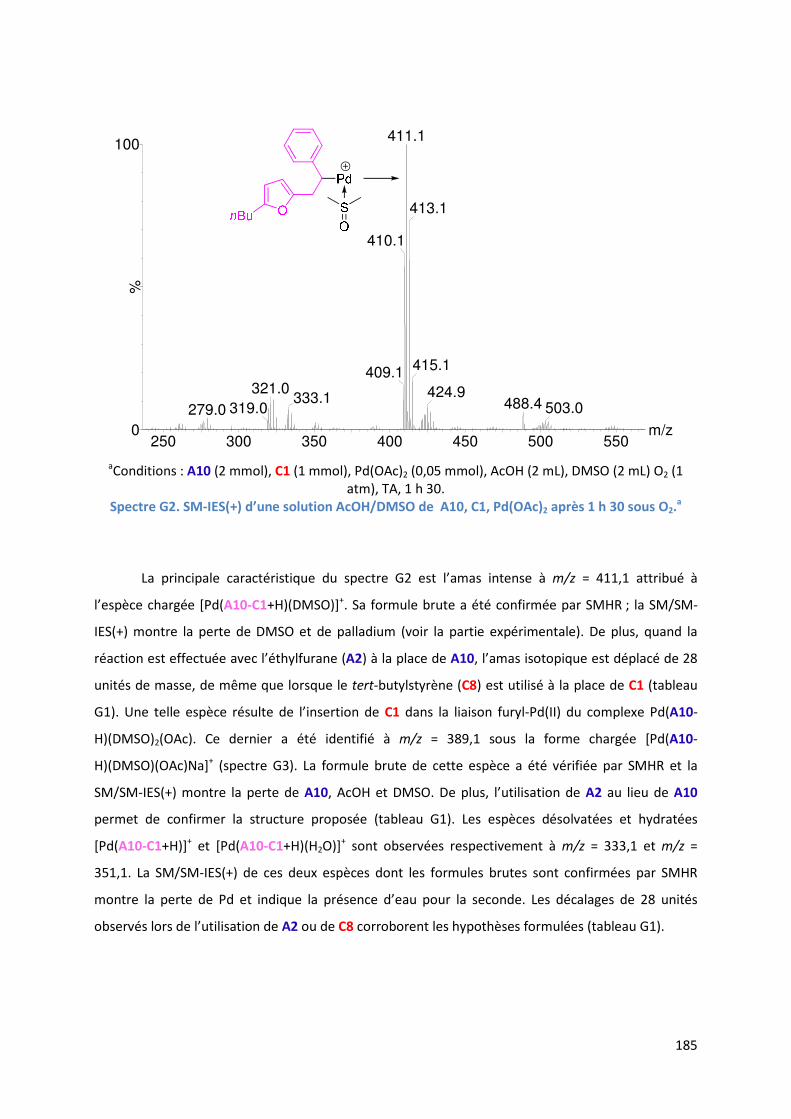
185
AV 873
m/z250 300 350 400 450 500 550
%
0
100
12HR414 55 (2.170) Cm (38:169) 1: TOF MS ES+ 1.36e4411.1
410.1
409.1321.0
279.0 319.0333.1
413.1
415.1
424.9488.4 503.0
aConditions : A10 (2 mmol), C1 (1 mmol), Pd(OAc)2 (0,05 mmol), AcOH (2 mL), DMSO (2 mL) O2 (1
atm), TA, 1 h 30.
Spectre G2. SM-IES(+) d’une solution AcOH/DMSO de A10, C1, Pd(OAc)2 après 1 h 30 sous O2.a
La principale caractéristique du spectre G2 est l’amas intense à m/z = 411,1 attribué à
l’espèce chargée [Pd(A10-C1+H)(DMSO)]+. Sa formule brute a été confirmée par SMHR ; la SM/SM-
IES(+) montre la perte de DMSO et de palladium (voir la partie expérimentale). De plus, quand la
réaction est effectuée avec l’éthylfurane (A2) à la place de A10, l’amas isotopique est déplacé de 28
unités de masse, de même que lorsque le tert-butylstyrène (C8) est utilisé à la place de C1 (tableau
G1). Une telle espèce résulte de l’insertion de C1 dans la liaison furyl-Pd(II) du complexe Pd(A10-
H)(DMSO)2(OAc). Ce dernier a été identifié à m/z = 389,1 sous la forme chargée [Pd(A10-
H)(DMSO)(OAc)Na]+ (spectre G3). La formule brute de cette espèce a été vérifiée par SMHR et la
SM/SM-IES(+) montre la perte de A10, AcOH et DMSO. De plus, l’utilisation de A2 au lieu de A10
permet de confirmer la structure proposée (tableau G1). Les espèces désolvatées et hydratées
[Pd(A10-C1+H)]+ et [Pd(A10-C1+H)(H2O)]
+ sont observées respectivement à m/z = 333,1 et m/z =
351,1. La SM/SM-IES(+) de ces deux espèces dont les formules brutes sont confirmées par SMHR
montre la perte de Pd et indique la présence d’eau pour la seconde. Les décalages de 28 unités
observés lors de l’utilisation de A2 ou de C8 corroborent les hypothèses formulées (tableau G1).
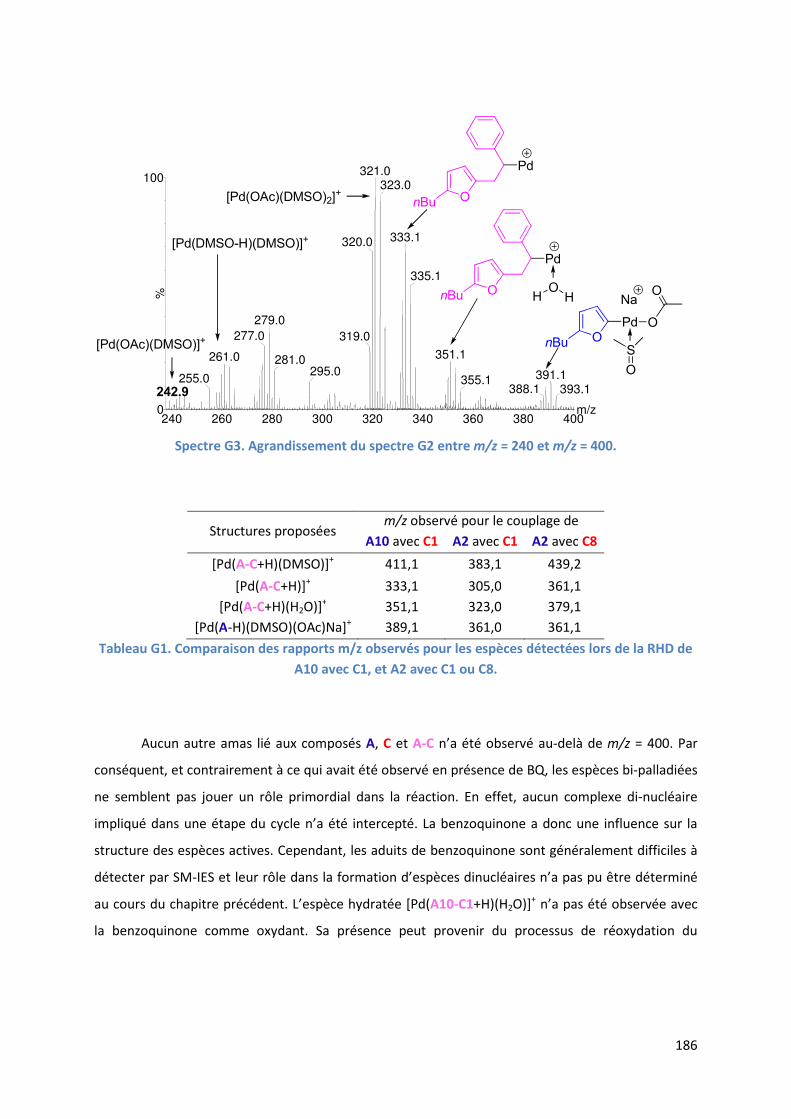
186
AV 873
m/z240 260 280 300 320 340 360 380 400
%
0
100
12HR414 55 (2.170) Cm (38:169) 1: TOF MS ES+ 1.57e3321.0
320.0
279.0
277.0
261.0
255.0
319.0
281.0295.0
323.0
333.1
335.1
351.1
391.1355.1388.1 393.1
Pd
OnBu
Pd
OnBuOHH
PdOnBu
O
O
S
O
Na
[Pd(OAc)(DMSO)2]+
[Pd(DMSO-H)(DMSO)]+
242.9
[Pd(OAc)(DMSO)]+
Spectre G3. Agrandissement du spectre G2 entre m/z = 240 et m/z = 400.
Structures proposées m/z observé pour le couplage de
A10 avec C1 A2 avec C1 A2 avec C8
[Pd(A-C+H)(DMSO)]+ 411,1 383,1 439,2
[Pd(A-C+H)]+ 333,1 305,0 361,1
[Pd(A-C+H)(H2O)]+ 351,1 323,0 379,1
[Pd(A-H)(DMSO)(OAc)Na]+ 389,1 361,0 361,1
Tableau G1. Comparaison des rapports m/z observés pour les espèces détectées lors de la RHD de
A10 avec C1, et A2 avec C1 ou C8.
Aucun autre amas lié aux composés A, C et A-C n’a été observé au-delà de m/z = 400. Par
conséquent, et contrairement à ce qui avait été observé en présence de BQ, les espèces bi-palladiées
ne semblent pas jouer un rôle primordial dans la réaction. En effet, aucun complexe di-nucléaire
impliqué dans une étape du cycle n’a été intercepté. La benzoquinone a donc une influence sur la
structure des espèces actives. Cependant, les aduits de benzoquinone sont généralement difficiles à
détecter par SM-IES et leur rôle dans la formation d’espèces dinucléaires n’a pas pu être déterminé
au cours du chapitre précédent. L’espèce hydratée [Pd(A10-C1+H)(H2O)]+ n’a pas été observée avec
la benzoquinone comme oxydant. Sa présence peut provenir du processus de réoxydation du
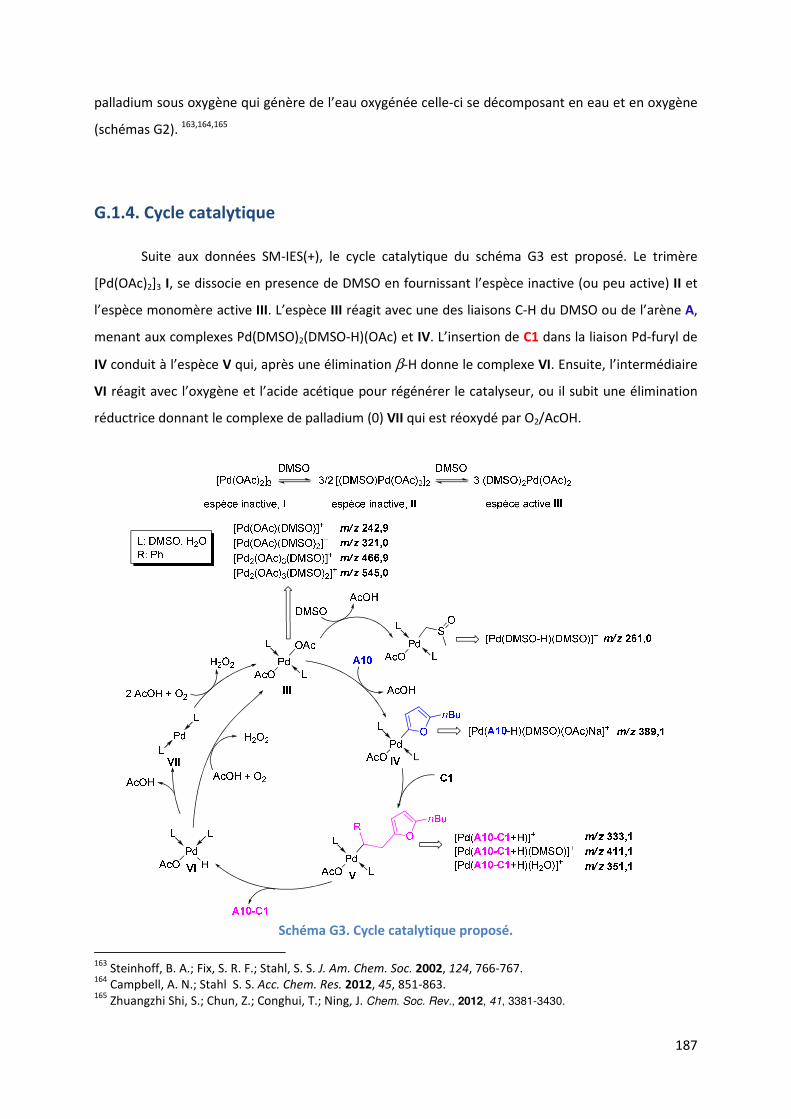
187
palladium sous oxygène qui génère de l’eau oxygénée celle-ci se décomposant en eau et en oxygène
(schémas G2). 163,164,165
G.1.4. Cycle catalytique
Suite aux données SM-IES(+), le cycle catalytique du schéma G3 est proposé. Le trimère
[Pd(OAc)2]3 I, se dissocie en presence de DMSO en fournissant l’espèce inactive (ou peu active) II et
l’espèce monomère active III. L’espèce III réagit avec une des liaisons C-H du DMSO ou de l’arène A,
menant aux complexes Pd(DMSO)2(DMSO-H)(OAc) et IV. L’insertion de C1 dans la liaison Pd-furyl de
IV conduit à l’espèce V qui, après une élimination β-H donne le complexe VI. Ensuite, l’intermédiaire
VI réagit avec l’oxygène et l’acide acétique pour régénérer le catalyseur, ou il subit une élimination
réductrice donnant le complexe de palladium (0) VII qui est réoxydé par O2/AcOH.
Schéma G3. Cycle catalytique proposé.
163
Steinhoff, B. A.; Fix, S. R. F.; Stahl, S. S. J. Am. Chem. Soc. 2002, 124, 766-767. 164
Campbell, A. N.; Stahl S. S. Acc. Chem. Res. 2012, 45, 851-863. 165
Zhuangzhi Shi, S.; Chun, Z.; Conghui, T.; Ning, J. Chem. Soc. Rev., 2012, 41, 3381-3430.
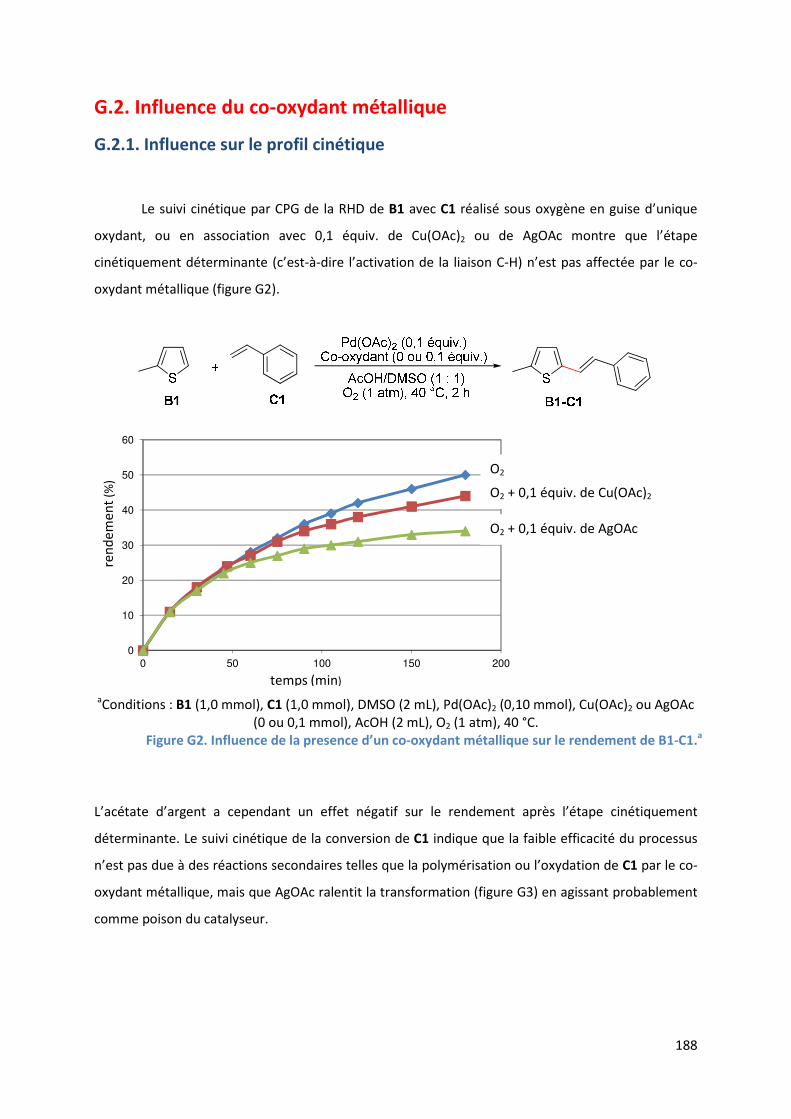
188
G.2. Influence du co-oxydant métallique
G.2.1. Influence sur le profil cinétique
Le suivi cinétique par CPG de la RHD de B1 avec C1 réalisé sous oxygène en guise d’unique
oxydant, ou en association avec 0,1 équiv. de Cu(OAc)2 ou de AgOAc montre que l’étape
cinétiquement déterminante (c’est-à-dire l’activation de la liaison C-H) n’est pas affectée par le co-
oxydant métallique (figure G2).
aConditions : B1 (1,0 mmol), C1 (1,0 mmol), DMSO (2 mL), Pd(OAc)2 (0,10 mmol), Cu(OAc)2 ou AgOAc
(0 ou 0,1 mmol), AcOH (2 mL), O2 (1 atm), 40 °C.
Figure G2. Influence de la presence d’un co-oxydant métallique sur le rendement de B1-C1.a
L’acétate d’argent a cependant un effet négatif sur le rendement après l’étape cinétiquement
déterminante. Le suivi cinétique de la conversion de C1 indique que la faible efficacité du processus
n’est pas due à des réactions secondaires telles que la polymérisation ou l’oxydation de C1 par le co-
oxydant métallique, mais que AgOAc ralentit la transformation (figure G3) en agissant probablement
comme poison du catalyseur.
0
10
20
30
40
50
60
0 50 100 150 200
temps (min)
ren
de
me
nt
(%)
O2
O2 + 0,1 équiv. de Cu(OAc)2
O2 + 0,1 équiv. de AgOAc
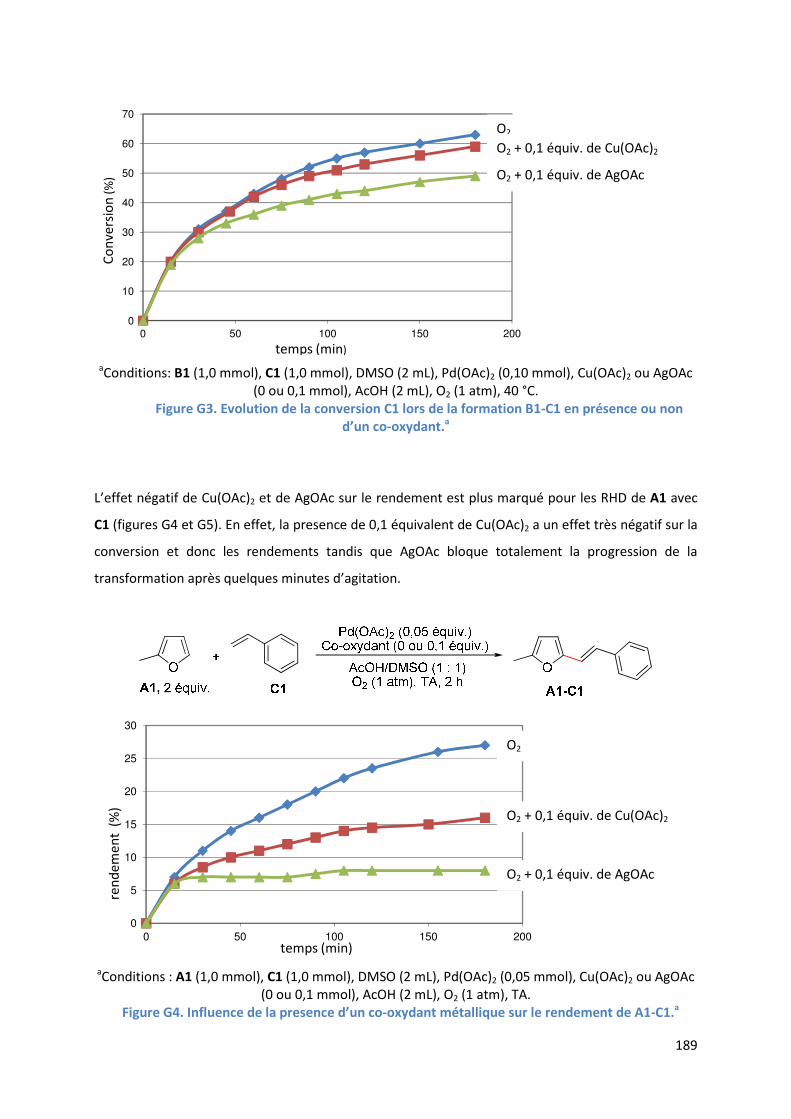
189
aConditions: B1 (1,0 mmol), C1 (1,0 mmol), DMSO (2 mL), Pd(OAc)2 (0,10 mmol), Cu(OAc)2 ou AgOAc
(0 ou 0,1 mmol), AcOH (2 mL), O2 (1 atm), 40 °C.
Figure G3. Evolution de la conversion C1 lors de la formation B1-C1 en présence ou non
d’un co-oxydant.a
L’effet négatif de Cu(OAc)2 et de AgOAc sur le rendement est plus marqué pour les RHD de A1 avec
C1 (figures G4 et G5). En effet, la presence de 0,1 équivalent de Cu(OAc)2 a un effet très négatif sur la
conversion et donc les rendements tandis que AgOAc bloque totalement la progression de la
transformation après quelques minutes d’agitation.
aConditions : A1 (1,0 mmol), C1 (1,0 mmol), DMSO (2 mL), Pd(OAc)2 (0,05 mmol), Cu(OAc)2 ou AgOAc
(0 ou 0,1 mmol), AcOH (2 mL), O2 (1 atm), TA.
Figure G4. Influence de la presence d’un co-oxydant métallique sur le rendement de A1-C1.a
0
10
20
30
40
50
60
70
0 50 100 150 200
temps (min)
Co
nv
ers
ion
(%
)
0
5
10
15
20
25
30
0 50 100 150 200
ren
de
me
nt
(%
)
temps (min)
O2
O2 + 0,1 équiv. de Cu(OAc)2
O2 + 0,1 équiv. de AgOAc
O2
O2 + 0,1 équiv. de Cu(OAc)2
O2 + 0,1 équiv. de AgOAc
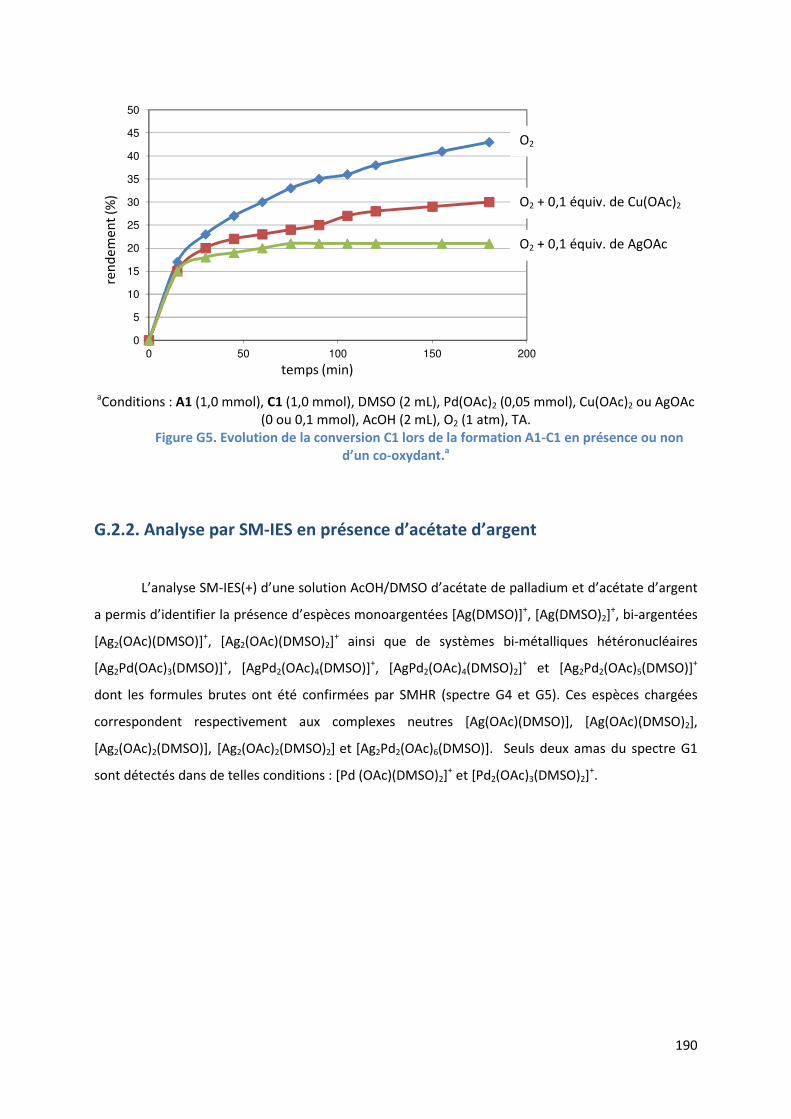
190
aConditions : A1 (1,0 mmol), C1 (1,0 mmol), DMSO (2 mL), Pd(OAc)2 (0,05 mmol), Cu(OAc)2 ou AgOAc
(0 ou 0,1 mmol), AcOH (2 mL), O2 (1 atm), TA.
Figure G5. Evolution de la conversion C1 lors de la formation A1-C1 en présence ou non
d’un co-oxydant.a
G.2.2. Analyse par SM-IES en présence d’acétate d’argent
L’analyse SM-IES(+) d’une solution AcOH/DMSO d’acétate de palladium et d’acétate d’argent
a permis d’identifier la présence d’espèces monoargentées [Ag(DMSO)]+, [Ag(DMSO)2]
+, bi-argentées
[Ag2(OAc)(DMSO)]+, [Ag2(OAc)(DMSO)2]
+ ainsi que de systèmes bi-métalliques hétéronucléaires
[Ag2Pd(OAc)3(DMSO)]+, [AgPd2(OAc)4(DMSO)]
+, [AgPd2(OAc)4(DMSO)2]
+ et [Ag2Pd2(OAc)5(DMSO)]
+
dont les formules brutes ont été confirmées par SMHR (spectre G4 et G5). Ces espèces chargées
correspondent respectivement aux complexes neutres [Ag(OAc)(DMSO)], [Ag(OAc)(DMSO)2],
[Ag2(OAc)2(DMSO)], [Ag2(OAc)2(DMSO)2] et
[Ag2Pd2(OAc)6(DMSO)]. Seuls deux amas du spectre G1
sont détectés dans de telles conditions : [Pd (OAc)(DMSO)2]+ et [Pd2(OAc)3(DMSO)2]
+.
0
5
10
15
20
25
30
35
40
45
50
0 50 100 150 200
ren
de
me
nt
(%)
temps (min)
O2
O2 + 0,1 équiv. de Cu(OAc)2
O2 + 0,1 équiv. de AgOAc
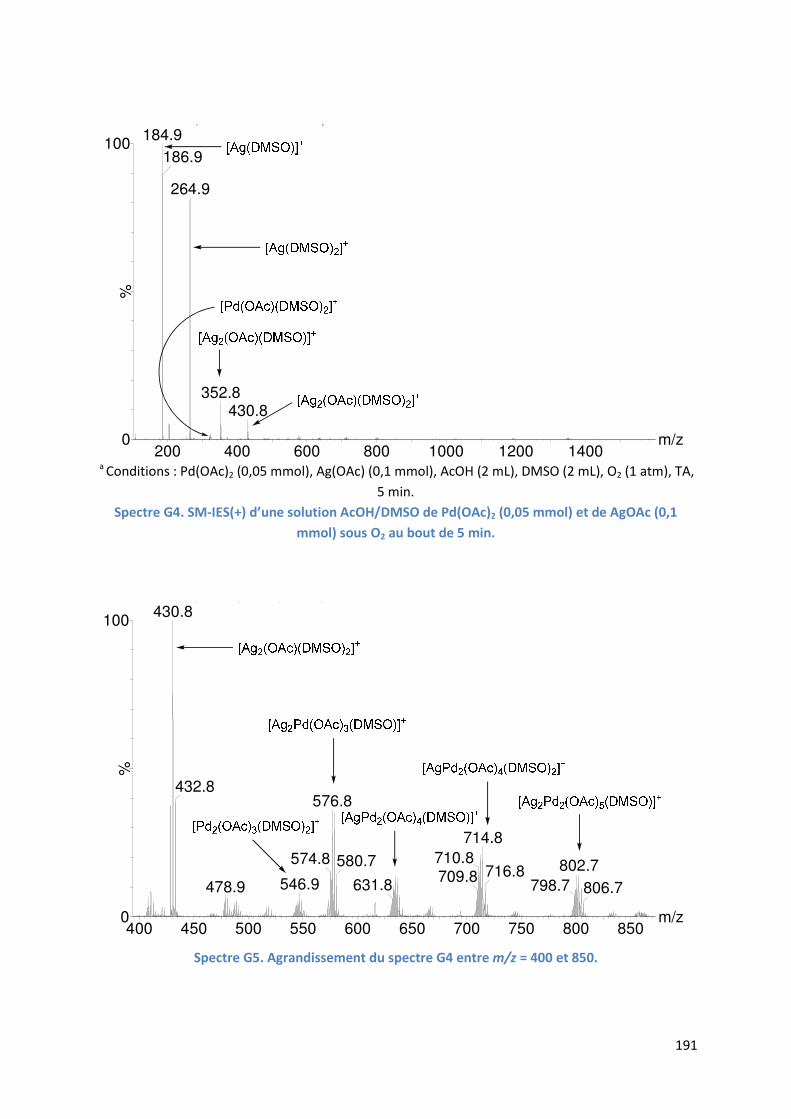
191
AV 662
m/z200 400 600 800 1000 1200 1400
%
0
100
11HR682 98 (3.079) Cm (90:98) 1: TOF MS ES+ 2.88e4x2184.9
186.9
264.9
352.8430.8
a Conditions : Pd(OAc)2 (0,05 mmol), Ag(OAc) (0,1 mmol), AcOH (2 mL), DMSO (2 mL), O2 (1 atm), TA,
5 min.
Spectre G4. SM-IES(+) d’une solution AcOH/DMSO de Pd(OAc)2 (0,05 mmol) et de AgOAc (0,1
mmol) sous O2 au bout de 5 min.
AV 662
m/z400 450 500 550 600 650 700 750 800 850
%
0
100
11HR682 98 (3.079) Cm (90:98) 1: TOF MS ES+ 969430.8
432.8576.8
574.8
546.9478.9
714.8
710.8580.7
631.8709.8
802.7716.8798.7 806.7
Spectre G5. Agrandissement du spectre G4 entre m/z = 400 et 850.

192
Nous avions proposé au chapitre 2 que le complexe trinucléaire [Pd(OAc)2]3 était inactif à
cause des ponts acétates. De façon similaire, nous suspectons que des complexes
(Ag)x(Pd)y(OAc)n(DMSO)m (x, y, m = 1, 2 ; n = 1, 2, 3, 4, 5) soient également inactifs à cause des ponts
formés par les acétates entre le palladium et l’argent.
G.2.3. Conclusion
L’étude par SM-IES(+) du milieu réactionnel a permis l’identification de l’intermédiaire
réactionnel Pd(A-H)(DMSO)(OAc)
résultant de l’activation de la liaison C-H de l’arène et des
intermédiaires Pd(A-C+H)(OAc)(DMSO),
Pd(A-C+H)(OAc) et
Pd(A-C+H)(OAc)(H2O) résultant de
l’insertion de l’alcène dans la liaison Pd-Ar. Contrairement à ce qui avait été observé au chapitre 2,
les complexes dinucléaires de palladium ne semblent pas jouer de rôle, ce qui laisse entendre que
précédemment, la présence de la benzoquinone les rendait actifs.
L’étude par SM-IES de la RHD en présence d’acétate argent a permis de proposer que
l’impact négatif de Cu(OAc)2 et AgOAc pourrait être dû à la formation de systèmes bi-métalliques
[Ag]x[Pd]y et [Cu]x[Pd]y (x, y = 1, 2), dans lesquels aucun ligand acétate n’est disponible pour
participer à l’activation de liaisons C-H.
G.3. Partie expérimentale
G.3.1. Mode opératoire pour les suivis cinétiques des réactions
A l’acétate de palladium (11,2 - 22,4 mg, 0,05 – 0,1 mmol) et au co-oxydant (0 ou 0,1 mmol)
mis en solution dans un mélange solvant/co-solvant AcOH (2 mL)/DMSO (2 mL) sont ajoutés l’arène
(1 mmol – 97 µL pour B1 ou 2 mmol – 180 µL pour A1), l’alcène C1 (115 µL - 1 mmol) et le
nitrobenzène (103 µL – 1 mmol). Un ballon d’oxygène est adapté au dispositif et le mélange est agité
à température ambiante ou à 40 °C. Des échantillons sont prélevés toutes les 15 min (environ 0,2 mL)
pendant 3 h, filtrés sur silice avec Et2O et analysés par CPG.

193
G.3.2. Dispositif expérimental
Toutes les expériences (SM, SMHR et SM/SM) ont été réalisées avec un spectromètre
tandem Quadripôle-Temps-de-vol (Q-TdV micro Waters « Micromass » Manchester UK), équipé
d’une source d’ionisation électrospray (Z-Spray), la désolvatation des ions est assistée par l’arrivée à
contre courant d’un gaz de séchage (flux d’azote) chauffé à une température de 50 à 80 °C. La
tension du cône d’extraction variait entre 20 et 40 V, la tension du capillaire IES était comprise entre
3000 et 3500 V. La dissociation induite par collision (DIC) a été réalisée par introduction d’argon
dans la cellule de collision.
L’injection des échantillons a été réalisée à un débit de 3 à 6 µL/min, la quantité d’échantillon
dont nous disposions n’imposait pas de travailler à des débits plus faibles.
G.3.3. SMHR des espèces observées
[Pd(A10-C1+H)]+ : C16H19OPd, théorique : 333,0471 ; obtenue : 333,0469.
[Pd(A10-C1+H)(H2O)]+ : C16H21O2Pd, théorique : 351,0576 ; obtenue : 351,0568.
[Pd(A10-C1+H)(DMSO)]+ : C18H25O2PdS, théorique : 411,0610 ; obtenue : 411,0617.
[Pd(A10-H)(DMSO)(OAc)Na]+ : C12H20NaO4PdS, théorique : 389,0015 ; obtenue : 389,0019.
[Ag(DMSO)]+ : C2H6AgOS, théorique : 184,9190 ; obtenue : 184,9193.
[Ag(DMSO)2]+ : C4H12AgO2S2, théorique : 264,9330 ; obtenue : 264,9330.
[Ag2(OAc)(DMSO)]+ : C4H9Ag2O3S, théorique : 350,8374 ; obtenue : 350,8366.
[Ag2(OAc)(DMSO)2]+ : C6H15Ag2O4S2, théorique : 428,8514 ; obtenue : 428,8520.
[Ag2Pd(OAc)3(DMSO)]+ : C8H15Ag2O7PdS théorique : 574,7675 ; obtenue : 574,7665.
[AgPd2(OAc)4(DMSO)]+ : C10H18AgO9Pd2S théorique : 632,7792 ; obtenue : 632,7803.
[AgPd2(OAc)4(DMSO)2]+ : C12H24AgO10Pd2S2 théorique : 710,7931 ; obtenue : 710,7935.
[Ag2Pd2(OAc)5(DMSO)]+ : C12H21Ag2O11Pd2S théorique : 798,6976 ; obtenue : 798,6971.
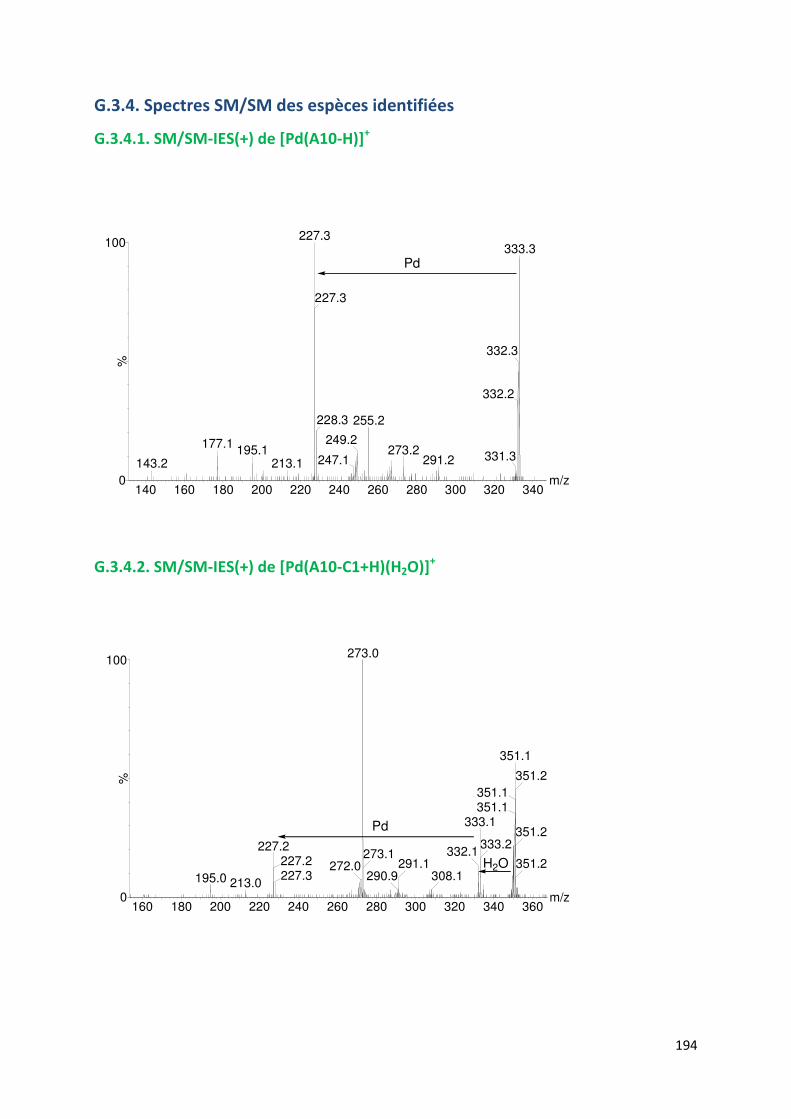
194
G.3.4. Spectres SM/SM des espèces identifiées
G.3.4.1. SM/SM-IES(+) de [Pd(A10-H)]+
AV 876
m/z140 160 180 200 220 240 260 280 300 320 340
%
0
100
12BR408 10 (0.161) Cm (10:39) 1: TOF MSMS 333.20ES+ 72227.3
177.1
143.2195.1
213.1
333.3
227.3
332.3
332.2
255.2228.3
249.2
247.1273.2
331.3291.2
Pd
G.3.4.2. SM/SM-IES(+) de [Pd(A10-C1+H)(H2O)]+
AV 876
m/z160 180 200 220 240 260 280 300 320 340 360
%
0
100
12BR409A 61 (1.132) Cm (27:155) 1: TOF MSMS 351.20ES+ 97x6 273.0
227.2
195.0 213.0
227.2 272.0227.3
351.1
351.1351.1
333.1
332.1273.1291.1
290.9 308.1
333.2
351.2
351.2
351.2H2O
Pd
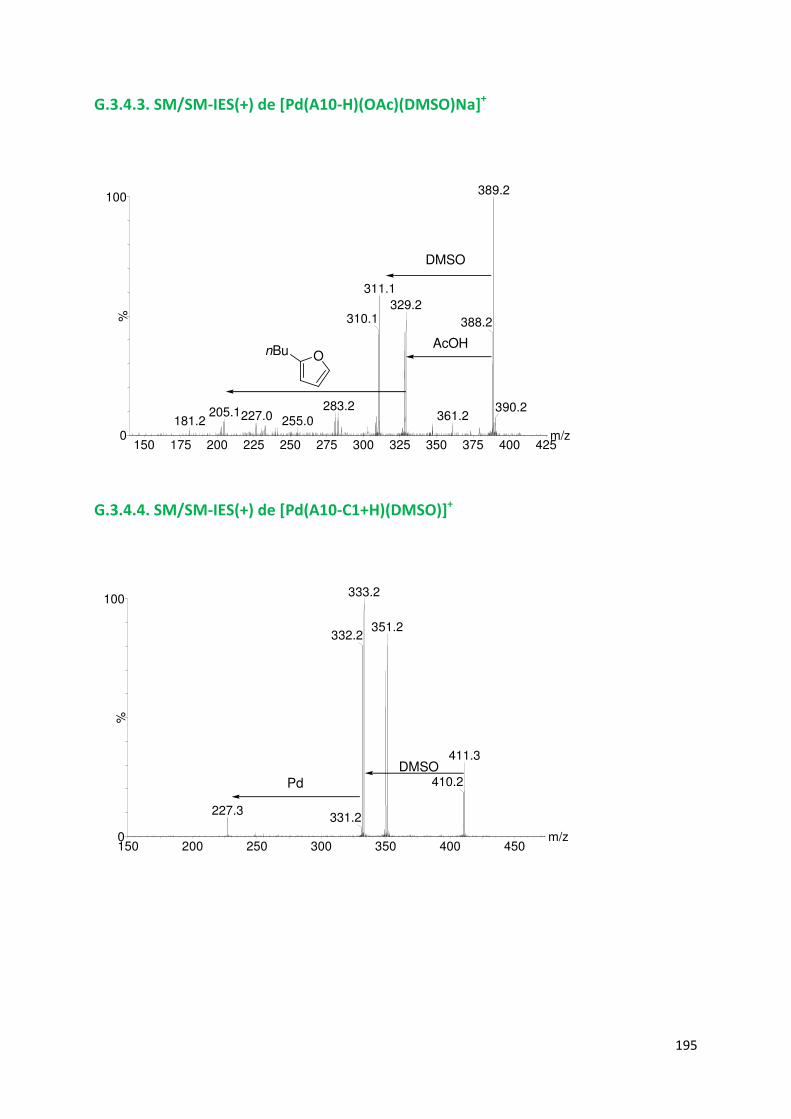
195
G.3.4.3. SM/SM-IES(+) de [Pd(A10-H)(OAc)(DMSO)Na]+
AV 876
m/z150 175 200 225 250 275 300 325 350 375 400 425
%
0
100
12BR410 111 (2.062) Cm (1:290) 1: TOF MSMS 389.10ES+ 191389.2
311.1
310.1
283.2205.1
181.2 227.0 255.0
329.2
388.2
361.2390.2
AcOH
DMSO
OnBu
G.3.4.4. SM/SM-IES(+) de [Pd(A10-C1+H)(DMSO)]+
AV 876
m/z150 200 250 300 350 400 450
%
0
100
12BR412 75 (1.392) Cm (18:81) 1: TOF MSMS 411.20ES+ 333333.2
332.2
227.3331.2
351.2
411.3
410.2DMSO
Pd

196
CONCLUSION
Depuis la découverte de la réaction de Heck déshydrogénante par Fujiwara en 1967,
d’importants progrès ont été réalisés. Alors que ces réactions étaient en premier lieu accomplies en
ayant recours à des quantités stoechiométriques de palladium, des méthodes efficaces ne faisant
intervenir que des quantités catalytiques sont aujourd’hui disponibles. La plupart des RHD
catalytiques utilise cependant des conditions drastiques, avec par exemple une température
relativement élevée, et requière un oxydant terminal autre que l’oxygène moléculaire, cet oxydant
étant parfois couteux. Ces conditions, qui constituent un problème pour des applications à grande
échelle, diminuent considérablement l’aspect vert de ces procédés ainsi que leur compatibilité avec
le principe d’économie d’atome. Par ailleurs, des méthodes permettant l’amélioration ou le contrôle
de la régiosélectivité ainsi que la formation de liaison C-C impliquant une variété d’hétéroarènes et
d’alcènes, notamment les alcènes riches en électrons restaient à mettre au point.
C’est avec l’objectif de contribuer à la résolution de ces problématiques que ces travaux de
thèse ont été effectués. Dans un premier temps, des furanes et des thiophènes ont pu être couplés
dans des conditions douces en utilisant la benzoquinone comme réoxydant dans un mélange
AcOH/DMSO. La méthode décrite au cours du premier chapitre est compatible avec des substrats
halogénés, incluant les styrènes et les thiophènes bromés.166
En comparaison avec les réactions
catalysées par Rh(III) précédemment rapportées par l’équipe de Glorius, cette méthode apparaît
avantageuse puisqu’elle concerne une plus large gamme de substrats et met en jeu un catalyseur
moins couteux. En se coordonnant au Pd, le DMSO permet la formation d’espèces catalytiques
monomériques et dimériques actives. Nous avons attribué à BQ un rôle original : celle-ci agit dans
nos conditions comme un promoteur de l’insertion d’alcènes électroniquement riches dans la liaison
Ar-Pd.
Pour étudier et proposer le mécanisme de cette réaction, l’électrospray couplé à la
spectrométrie de masse a été un outil précieux. Quinze complexes de palladium ont été interceptés,
et caractérisés,167
ce qui conduit à en retenir douze, dont des complexes dinucléaires ou contenant la
benzoquinone, comme pouvant être des intermédiaires de la réaction. Trois complexes sont le
résultat de réactions secondaires, dont un identifié comme étant un intermédaire impliqué dans
l’homocouplage de furanes via une activation C-H.
L’oxygène est le réactif de choix pour régénérer des espèces palladiées catalytiquement
actives afin d’aboutir à des procédés plus verts et plus compatibles avec le principe d’économie
166
Vasseur, A.; Muzart, J.; Le Bras, J. Chem. Eur. J. 2011, 17, 12556-12560. 167
Vasseur, A.; Harakat, D.; Muzart, J.; Le Bras, J. J. Org. Chem. 2012, 77, 5751-5758.

197
d’atome et les contraintes économiques. Des résultats intéressants ont été obtenus avec le mélange
AcOH/DMSO.168
Des furanes, thiophènes et indoles ont été ainsi couplés avec le styrène, des styrènes
halogénés ou encore le p-acétoxystyrène avec des rendements convenables. Dans ces conditions,
l’utilisation de co-oxydants métalliques, pourtant souvent employés pour mener à bien de telles
réactions, peut avoir un effet négatif qui serait dû à la formation de complexes bi-métalliques
hétéronucléaires inéfficaces pour activer la liaison C-H de l’arène. Il s’agit là d’une des seules études
mettant en évidence l’avantage d’utiliser l’oxygène comme seul oxydant, plutôt que de l’utiliser en
association avec d’autres co-oxydants pour achever ces tranformations.
Notre équipe poursuit l’étude de la fonctionnalisation de liaisons C-H, l’objectif étant le
couplage de Heck déshydrogénant d’hétéroarènes avec des alcènes disubsitués dans des conditions
douces. Des premiers résultats prometteurs ont déjà été obtenus à l’aide d’un ligand original
(schéma 3).
Schéma 3. Résultats préliminaires de RHD d’hétéroarènes avec des alcènes disubstitués.
Nous nous sommes également engagés dans la valorisation de ces réactions, les RHD étant
encore très peu utilisées pour des synthèses organiques ciblées. Notre équipe a ainsi réalisé la
synthèse d’un analogue de la goniothalamine dont la réaction de Heck déshydrogénante est une
étape clef.
168
Vasseur, A.; Harakat, D.; Muzart, J.; Le Bras, J. Adv. Synth. Catal. 2012, soumis.
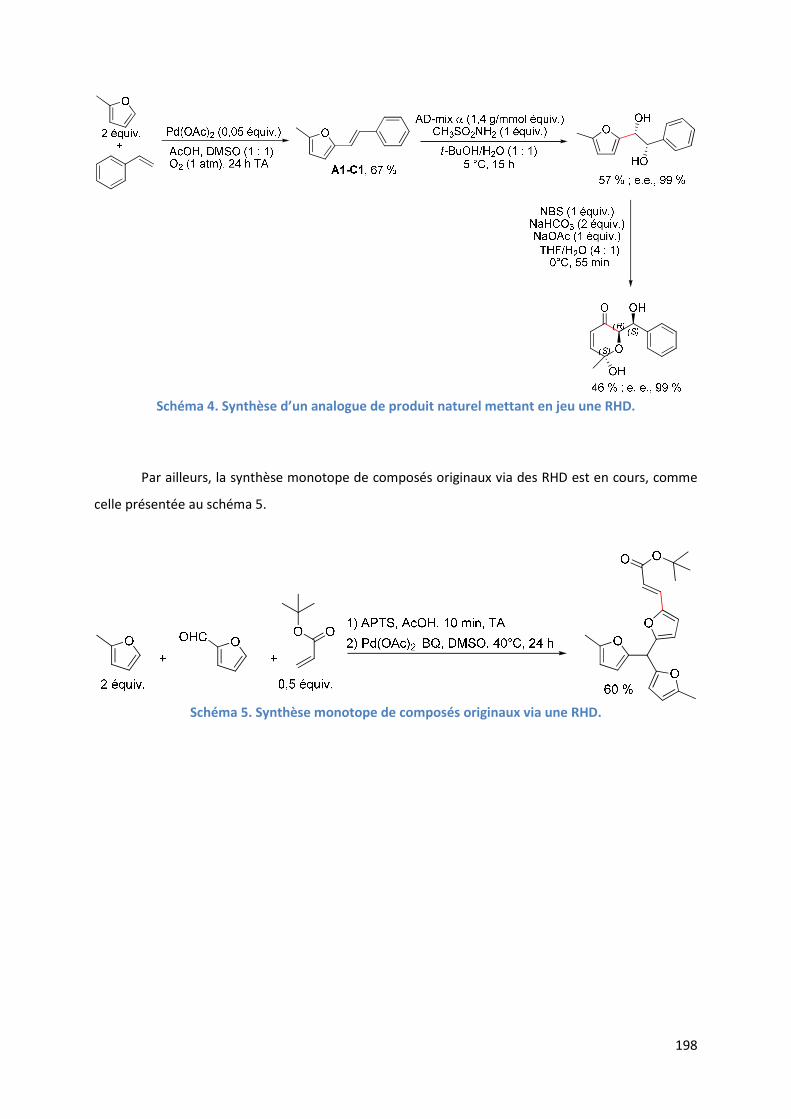
198
Schéma 4. Synthèse d’un analogue de produit naturel mettant en jeu une RHD.
Par ailleurs, la synthèse monotope de composés originaux via des RHD est en cours, comme
celle présentée au schéma 5.
Schéma 5. Synthèse monotope de composés originaux via une RHD.

199
Résumé
Ce mémoire décrit des réactions pallado-catalysées de Heck déshydrogénantes d’hétéroarènes
pouvant être dérivés de la biomasse avec des alcènes riches en électrons tels que les styrènes.
Le premier chapitre, présente une méthodologie permettant le couplage croisé de Heck
désydrogénant de furanes et thiophènes avec des styrènes dans des conditions douces et discute de
l’influence de l’agent oxydant sur l’activité du catalyseur.
Le second chapitre est consacré à l’étude mécanistique par SM-IES de réactions de Heck
déshydrogénantes de furanes avec des acrylates en présence de benzoquinone comme agent
oxydant et de DMSO comme solvant.
La présentation d’une nouvelle méthodologie pour les réactions de Heck déshydrogénantes
d’hétérocyles avec des styrènes dans des conditions aérobies et l’explication de l’effet négatif de co-
oxydants métalliques représentent le dernier chapitre.
Abstract
This thesis describes Pd-catalyzed dehydrogenative Heck reactions of heteroarenes that could be
derived from the biomass with electron-rich alkenes such as styrenes.
The first chapter presents a new methodology enabling cross coupling dehydrogenative Heck
Reactions of furans and thiophenes with styrenes under Mild conditions and discusses the Influence
of the oxidizing agent on the reaction rate.
The second chapter focuses on ESI-MS studies of the dehydrogenative Heck reactions of furans with
acrylates using benzoquinone as reoxidant and DMSO as solvent.
The presentation of a new methodology for aerobic dehydrogenative Heck reactions of heterocycles
with styrenes and the explanation about the negative effect of metallic co-oxidants represent the
third chapter.





























